WO2013047813A1 - 1,2,4-トリアジン-6-カルボキサミド誘導体 - Google Patents
1,2,4-トリアジン-6-カルボキサミド誘導体 Download PDFInfo
- Publication number
- WO2013047813A1 WO2013047813A1 PCT/JP2012/075204 JP2012075204W WO2013047813A1 WO 2013047813 A1 WO2013047813 A1 WO 2013047813A1 JP 2012075204 W JP2012075204 W JP 2012075204W WO 2013047813 A1 WO2013047813 A1 WO 2013047813A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- amino
- triazine
- methyl
- carboxamide
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to novel 1,2,4-triazine-6-carboxamide derivatives having a Syk (Spleen tyrosine kinase) inhibitory action and pharmaceutical compositions containing these as active ingredients.
- Syk Stpleen tyrosine kinase
- Syk is a non-receptor tyrosine kinase that constitutes the Syk family together with ZAP70, and is expressed in a wide range of immune-related cells such as B cells, macrophages, neutrophils, and mast cells, and is involved in their functions. Syk binds to the ITAM domain of immune receptors such as the Fc receptor family (FcR) and B cell receptor (BCR) expressed in these cells, and plays a role in transmitting signals from those receptors downstream. Plays.
- FcR Fc receptor family
- BCR B cell receptor
- Syk is activated by BCR after antigen stimulation and activates various downstream signal transduction pathways such as PI3K pathway, Ca 2+ -NFAT pathway, RAS-MAPK pathway, and finally B cell activity It plays an important role in transformation and differentiation and maturation.
- B cell lymphoma B cell lymphoma and chronic lymphocytic leukemia (CLL).
- CLL chronic lymphocytic leukemia
- Non-patent Documents 1 and 2 it has been reported that treatment of a compound having a Syk inhibitory action on these blood cancer cells can provide effects of growth inhibition and cell death induction (Non-patent Documents 1 and 2). Therefore, it is expected to obtain a therapeutic effect on B cell lymphoma and CLL by inhibiting Syk from these information.
- Syk is also suggested to be involved in canceration of non-B cell blood cancers such as peripheral T-cell lymphoma (PTCL), myelodysplastic syndrome (MDS), and acute myeloid leukemia (AML). It is expected that Syk inhibitors can be effective therapeutic agents not only for B cell-derived cancer but also for T cell lymphoma and AML.
- PTCL peripheral T-cell lymphoma
- MDS myelodysplastic syndrome
- AML acute myeloid leukemia
- Syk inhibitors include autoimmune diseases (rheumatoid arthritis, systemic lupus erythematosus, nephrotic syndrome, etc.), allergic diseases (bronchial asthma, allergic rhinitis, atopic dermatitis, contact dermatitis, hives) It has also been reported that it can be a therapeutic agent for food allergy, conjunctivitis, etc.) and chronic obstructive pulmonary disease (COPD) (Non-patent Document 3).
- autoimmune diseases rheumatoid arthritis, systemic lupus erythematosus, nephrotic syndrome, etc.
- allergic diseases bronchial asthma, allergic rhinitis, atopic dermatitis, contact dermatitis, hives
- COPD chronic obstructive pulmonary disease
- Non-Patent Document 4 As a currently developed Syk inhibitor, there is R406 (Rigel) (Non-Patent Document 4), but the selectivity for Syk is low, and side effects caused by inhibiting kinases other than Syk have been reported. (Non-Patent Document 5). As other Syk inhibitors, heteroaromatic-carboxamide derivatives have been reported (Patent Document 1), but Syk inhibitory activity was not sufficient. Patent Document 2 reports 1,2,4-triazine-6-carboxamide derivatives, but does not describe Syk inhibitory activity.
- R 1 represents a hydrogen atom or a C 1 -C 6 alkyl group optionally substituted by R a ;
- A is, R a is substitutable C 1 -C be the 8 alkyl group, R a optionally substituted with C 2 -C 6 alkenyl group, R a is optionally C 2 -C be substituted 6 alkynyl group, R b is optionally substituted C 3 -C 10 cycloalkyl group, R b is an optionally substituted C 6 -C 14 aromatic hydrocarbon group, even R b is substituted A 4- to 10-membered saturated heterocyclic group or a 4- to 10-membered unsaturated heterocyclic group optionally substituted by R b, or a combination of R 1 and the nitrogen atom to which they are bonded; May form a 10-membered saturated heterocycle or a 4- to 10-membered unsaturated heterocycle; R a is a deuterium atom, a halogen atom, a cyano group
- 1 to 3 groups may be substituted, a C 3 -C 10 cycloalkyl group, a C 6 -C 14 aromatic hydrocarbon group, a 4 to 10 membered saturated heterocyclic group and a 4 to 10 membered group.
- the unsaturated heterocyclic group includes R d , an oxo group, an oxide group, a C 1 -C 6 alkyl group.
- the present invention also provides a pharmaceutical such as a Syk inhibitor and an antitumor agent comprising the compound represented by the general formula (I) as an active ingredient. Moreover, this invention provides the pharmaceutical containing the compound or its salt represented by the said General formula (1).
- the present invention also provides a pharmaceutical composition comprising the compound represented by the above general formula (1) or a salt thereof and a pharmaceutically acceptable carrier. Moreover, this invention provides the compound represented by the said General formula (1) or its salt for tumor treatment.
- the present invention also provides use of the compound represented by the general formula (1) or a salt thereof for producing an antitumor agent.
- the present invention also provides a method for treating a tumor, comprising administering a compound represented by the general formula (1) or a salt thereof.
- a novel compound represented by the above general formula (I) or a salt thereof useful as a Syk inhibitor is provided. It has been clarified that the compound of the present invention or a salt thereof has excellent Syk inhibitory activity and exhibits a growth inhibitory effect on cancer cell lines. Moreover, it has the advantage that there are few side effects by other kinases from the outstanding selectivity with respect to Syk. Therefore, the compound of the present invention or a salt thereof is useful as a preventive and / or therapeutic agent for tumors, particularly cancer.
- the compound represented by the general formula (I) of the present invention is a compound having a 1,2,4-triazine skeleton and having an unsaturated heterocyclic group via N at the 5-position thereof. Yes, it is a novel compound not described in any of the above prior art documents.
- the “halogen atom” specifically includes a chlorine atom, a bromine atom, a fluorine atom, and an iodine atom.
- the “C 1 -C 8 alkyl group” means a linear or branched alkyl group having 1 to 8 carbon atoms, specifically a methyl group, an ethyl group, an n-propyl group, Examples include isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, hexyl group, heptyl group, octyl group and the like.
- the “C 1 -C 6 alkyl group” means a linear or branched alkyl group having 1 to 6 carbon atoms, specifically, a methyl group, an ethyl group, an n-propyl group, Examples include isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, hexyl group and the like.
- the “C 1 -C 6 haloalkyl group” refers to a group in which one to all of the hydrogen atoms of the C 1 -C 6 alkyl group are substituted with the above halogen atoms.
- C 1 -C 6 deuterated alkyl group means a group in which one to all of the C 1 -C 6 alkyl groups are substituted with deuterium atoms, specifically Examples include methyl-d1, methyl-d2, methyl-d3, ethyl-d1, ethyl-d2, ethyl-d3, ethyl-d4, and ethyl-d5 groups.
- C 1 -C 6 alkylamino group refers to a group in which one or two hydrogen atoms of the amino group are substituted with the above C 1 -C 6 alkyl group, specifically, methyl Examples thereof include an amino group, an ethylamino group, a dimethylamino group, a diethylamino group, and an ethylmethylamino group.
- the “C 2 -C 6 alkenyl group” refers to a linear or branched alkenyl group having 2 to 6 carbon atoms and containing at least one carbon-carbon double bond, specifically, A vinyl group, an allyl group, a methyl vinyl group, a propenyl group, a butenyl group, a pentenyl group, a hexenyl group, and the like can be given.
- the “C 2 -C 6 alkynyl group” means a linear or branched alkynyl group having 2 to 6 carbon atoms containing at least one carbon-carbon triple bond, specifically ethynyl. Group, 2-propynyl group and the like.
- C 1 -C 6 alkoxy group refers to a linear or branched alkoxy group having 1 to 6 carbon atoms, specifically, a methoxy group, an ethoxy group, an n-propoxy group, An isopropoxy group, n-butoxy group, isobutoxy group, tert-butoxy group and the like can be mentioned.
- C 2 -C 7 alkanoyl group refers to a group in which a hydrogen atom of a carbonyl group is substituted with the above C 1 -C 6 alkyl group, specifically an acetyl group, an n-propanoyl group, An isopropanoyl group, an n-butyroyl group, a tert-butyroyl group and the like can be mentioned.
- the “C 3 -C 10 cycloalkyl group” means a monocyclic or polycyclic cycloalkyl group having 3 to 10 carbon atoms, specifically, a cyclopropyl group, a cyclobutyl group, a cyclopentyl group. Cyclohexyl group, cycloheptyl group, decalyl group and the like.
- the “C 6 -C 14 aromatic hydrocarbon group” means a monocyclic or polycyclic aromatic hydrocarbon group having 6 to 14 carbon atoms, specifically a phenyl group or a naphthyl group. Etc.
- the “4- to 10-membered saturated heterocyclic group” refers to a 4- to 10-membered monocyclic or polycyclic fully saturated heterocyclic group, specifically, an azetidinyl group, a pyrrolidinyl group, Examples include a piperidinyl group, a piperazinyl group, a hexamethyleneimino group, a morpholino group, a thiomorpholino group, a homopiperazinyl group, a tetrahydrofuranyl group, and a tetrahydropyranyl group.
- “4- to 10-membered unsaturated heterocyclic group” refers to a 4- to 10-membered monocyclic or polycyclic fully unsaturated or partially saturated heterocyclic group, specifically, Unsaturated unsaturated heterocyclic groups include imidazolyl, thienyl, furyl, pyrrolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyrazolyl, triazolyl, tetrazolyl, pyridyl, pyrazyl Group, pyrimidinyl group, pyridazinyl group, indolyl group, isoindolyl group, indazolyl group, triazolopyridyl group, benzoimidazolyl group, benzoxazolyl group, benzothiazolyl group, benzothienyl group, benzofuranyl group, purinyl group, quinolyl group,
- haloalkyl groups C 1 -C 6 deuterated alkyl group, C 1 -C 6 alkoxycarbonyl Shi group, C 2 -C 6 alkenyl, and C 2 -C 6 same or different groups selected from the group consisting of alkynyl groups may be substituted.
- the number of these substituents is not particularly limited 1 to 3 are preferred).
- R d represents a deuterium atom, a halogen atom, a cyano group, a nitro group, —C ( ⁇ O) R x , —C ( ⁇ O) OR x , —C ( ⁇ O) N (R x ).
- R x , R y and R z in the present specification are the same or different and each represents a hydrogen atom, a C 1 -C 6 alkyl group, a C 1 -C 6 haloalkyl group, a C 1 -C 6 deuterated alkyl group, C 2 -C 6 alkenyl group, C 2 -C 6 alkynyl group, C 3 -C 10 cycloalkyl group, C 6 -C 14 aromatic hydrocarbon group, 4-10 membered unsaturated heterocyclic group or 4-10 Membered saturated heterocyclic group.
- R xx and R yy are the same or different and are a hydrogen atom, a C 1 -C 6 alkyl group, a C 1 -C 6 haloalkyl group, or a C 3 -C 10 cycloalkyl group.
- R 1 is preferably a hydrogen atom or a C 1 -C 6 alkyl group, and particularly preferably a hydrogen atom.
- the “C 1 -C 8 alkyl group” in the “C 1 -C 8 alkyl group optionally substituted by R a ” represented by A includes a methyl group, an ethyl group, n -Propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, hexyl group, heptyl group and octyl group are preferred.
- R in the "R a is substitutable C 1 -C be the 8 alkyl group" represented by A a, halogen atom, -N (R x) (R y), —OR x , C 3 -C 10 cycloalkyl group, C 6 -C 14 aromatic hydrocarbon group, 4 to 10 membered unsaturated heterocyclic group or 4 to 10 membered saturated heterocyclic group (where C 3 — C 10 cycloalkyl group, C 6 -C 14 aromatic hydrocarbon group, 4-10 membered unsaturated heterocyclic group and 4-10 membered saturated heterocyclic group are deuterium atom, halogen atom, hydroxyl group, cyano Group, nitro group, amino group, oxo group, oxide group, imino group, C 1 -C 6 alkyl group, C 1 -C 6 haloalkyl group, C 1 -C 6 deuterated alkyl group, C 1 -C 6 alkoxy
- the “C 2 -C 6 alkynyl group” in the “C 2 -C 6 alkynyl group optionally substituted with R a ” represented by A is a C 2 -C 4 alkynyl group. Is preferred.
- R a in the "R a optionally substituted with C 2 -C 6 alkynyl group” represented by A halogen atoms are preferred.
- the number of Ra is not particularly limited and is preferably 1 to 3.
- the “C 3 -C 10 cycloalkyl group” in the “C 3 -C 10 cycloalkyl group optionally substituted by R b ” represented by A is C 4 -C 7
- a cycloalkyl group is preferable, a cyclopentyl group, a cyclohexyl group, and a cycloheptyl group are more preferable, and a cyclohexyl group is particularly preferable.
- R b in the "R b is -C C 3 may be substituted by 10 cycloalkyl group" represented by A, -N (R x) ( R y) or -OR x is preferred, —N (R xx ) (R yy ) or —OR xx is more preferred, hydroxyl group, amino group or C 1 -C 6 alkylamino group is more preferred, amino group or C 1 -C 6 alkylamino is preferred A group is further preferred, and an amino group is particularly preferred.
- the number of R b is not particularly limited and is preferably 1 to 3.
- the 4- to 10-membered saturated heterocyclic group in the “4- to 10-membered saturated heterocyclic group which R b may be substituted” represented by A is a 4- to 6-membered saturated heterocyclic group.
- Monocyclic saturated heterocyclic groups are preferred, 4- to 6-membered monocyclic saturated heterocyclic groups having 1 to 3 heteroatoms selected from N, S and O are more preferred, selected from N and O
- a 4- to 6-membered monocyclic saturated heterocyclic group having one hetero atom is more preferable, and an azetidinyl group, a pyrrolidinyl group, a piperidinyl group, a tetrahydrofuranyl group, or a tetrahydropyranyl group is more preferable.
- R b in the "saturated heterocyclic group R b are members also may 4 to have 10 substituted" represented by A, C 1 -C 6 alkyl group, -N (R x ) (R y ) or —OR x is preferred, and C 1 -C 6 alkyl group, —N (R xx ) (R yy ) or —OR xx is more preferred, C 1 -C 6 alkyl group, hydroxyl group or Amino groups are more preferred, hydroxyl groups or amino groups are more preferred, and amino groups are particularly preferred.
- the number of R b is not particularly limited and is preferably 1 to 3.
- the 4- to 10-membered unsaturated heterocyclic group in the “4- to 10-membered unsaturated heterocyclic group which R b may be substituted” represented by A is 4 to 6
- R b in the "unsaturated heterocyclic group R b is membered to 10 4 which may be substituted" represented by A, -N (R x) ( R y) is -N (R xx ) (R yy ) is more preferable, and an amino group is particularly preferable.
- the number of R b is not particularly limited and is preferably 1 to 3.
- A is a C 1 -C 8 alkyl group optionally substituted by 1 to 5 R a s that are the same or different; and 1 to 3 R b s that are the same or different from each other.
- Is preferably a C 3 -C 10 cycloalkyl group which may be substituted; or a 4- to 10-membered saturated heterocyclic group which may be substituted with the same or different 1 to 3 R b ,
- the groups are deuterium
- R 1 is a hydrogen atom or a C 1 -C 6 alkyl group, and A is the same or different and 1 to 5 R a are substituted.
- An optionally substituted C 1 -C 8 alkyl group; the same or different 1 to 3 R b optionally substituted C 3 -C 10 cycloalkyl groups; or the same or different 1 to 3 R b is an optionally substituted 4- to 10-membered saturated heterocyclic group, or a 4- to 10-membered unsaturated heterocyclic ring together with R 1 , A and the nitrogen atom to which they are bonded, or
- R 1 is a hydrogen atom or a C 1 -C 6 alkyl
- C 1 -C 8 alkyl group which may be substituted by 1 to 5 groups which are the same or different, and the same or different groups selected from the group consisting of an amino group or a C 1 -C 6 alkylamino group
- a C 3 -C 10 cycloalkyl group optionally substituted with 1 to 3 groups; the same or different 1 to 3 groups selected from a hydroxyl group or an amino group may be substituted; 10-membered saturated heterocyclic group Or when R 1 , A and the nitrogen atom to which they are attached form a 4-10 membered saturated heterocycle (wherein the 4-10 membered saturated heterocycle is an amino group and a hydroxyl group) More preferably, the same or different 1 to 3 groups selected from the group consisting of may be substituted)
- A is represented by a structural formula, it is the following (1) to (9), and (1), (3), (4), (5), (8), (9) are more preferable. , (1), (4) and (8) are particularly preferred.
- R 2 is preferably a hydrogen atom or a C 1 -C 6 alkyl group, particularly preferably a hydrogen atom.
- the “unsaturated heterocyclic group” in the “unsaturated heterocyclic group which R c may be substituted” represented by B is 1 to 4 selected from N, S and O 4- to 10-membered monocyclic or bicyclic fully unsaturated or partially saturated unsaturated heterocyclic group (excluding 3,4-methylenedioxyphenyl group) having a heteroatom of 5 to 6-membered monocyclic fully unsaturated or partially saturated unsaturated heterocyclic group having 1 to 3 heteroatoms selected from S, O, or 1 to 3 selected from N, S and O More preferably, it is a 9-10 membered bicyclic fully unsaturated or partially saturated unsaturated heterocyclic group having 3 heteroatoms (excluding 3,4-methylenedioxyphenyl group), N, 5- to 6-membered monocyclic having 1 to 3 heteroatoms selected from S and O A fully unsaturated unsaturated heterocyclic group or a 9-10 membered bicyclic fully unsaturated unsaturated heterocyclic group having 1 to
- Specific preferred unsaturated heterocyclic rings of B include imidazolyl, thienyl, furyl, pyrrolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyrazolyl, triazolyl, tetrazolyl, pyridyl Group, pyrazyl group, pyrimidinyl group, pyridazinyl group, indolyl group, isoindolyl group, indazolyl group, imidazopyridyl group, triazolopyridyl group, thienopyridyl group, benzoimidazolyl group, benzoxazolyl group, benzothiazolyl group, benzothienyl group, benzofuranyl group , Purinyl group, quinolyl group, isoquinolyl group, quinazolinyl group, quinoxalyl group, naphthyridy
- R c in the “unsaturated heterocyclic group which R c may be substituted” represented by B is a halogen atom, a cyano group, an oxide group, a C 1 -C 6 alkyl group, —C ( ⁇ O) R x , —C ( ⁇ O) OR x , —C ( ⁇ O) N (R x ) (R y ), C 3 -C 10 cycloalkyl group, C 6 -C 14 aromatic A hydrocarbon group, a 4- to 10-membered unsaturated heterocyclic group, or a 4- to 10-membered saturated heterocyclic group (wherein the C 1 -C 6 alkyl group may be substituted by R d , C 3- C 10 cycloalkyl group, C 6 -C 14 aromatic hydrocarbon group, 4- to 10-membered unsaturated heterocyclic group and 4- to 10-membered saturated heterocyclic group are R d , oxo group, oxide group, C 1 -C 6 alky
- 3 -C 10 cycloalkyl group; C 6 -C 14 aromatic hydrocarbon group; C 1 -C 6 alkyl group, the same or are selected from C 1 -C 6 alkoxy and C 2 -C 7 group consisting alkanoyl group 1 to 3 different groups may be substituted 4 to 1 A 0-membered unsaturated heterocyclic group; or a 4- to 10-membered saturated heterocyclic group optionally substituted by an oxo group is more preferable: 1 to 5 identical or different groups selected from the group consisting of a halogen atom; a cyano group; a deuterium atom, a halogen atom, a hydroxyl group, a C 1 -C 6 alkoxy group and a 4- to 10-membered unsaturated heterocyclic group An optionally substituted C 1 -C 6 alkyl group; —C ( ⁇ O) R x ; —C ( ⁇ O) OR x ; —C ( ⁇ O) N (R x
- the “unsaturated heterocyclic group which R c may be substituted” represented by B is 1 to 4 selected from N, S and O which R c may be substituted.
- a 4-10 membered monocyclic or bicyclic fully unsaturated or partially saturated unsaturated heterocyclic group having 1 heteroatom is preferred: 5- to 6-membered monocyclic fully unsaturated or partially saturated unsaturated heterocyclic group having 1 to 3 heteroatoms selected from N, S and O, optionally substituted by R c
- it is a 9 to 10-membered bicyclic fully unsaturated or partially saturated group having 1 to 3 heteroatoms selected from N, S and O, except 3,4-methylenedioxyphenyl group)
- Unsaturated heterocyclic groups (excluding 3,4-methylenedioxyphenyl groups) are more preferred: R c may be substituted, a 5- to 6-membered monocyclic fully unsaturated unsaturated heterocyclic group having 1 to 3 heteroatoms selected from N, S and O, or N, A
- the saturated heterocyclic group includes R d , an oxo group, an oxide group, a C 1 -C 6 alkyl group, a C 1 -C 6 haloalkyl group, a C 1 -C 6 deuterated alkyl group, a C 1 -C 6 alkoxy group, same or different from selected from C 2 -C 6 alkenyl group, and C 2 -C 6 group consisting of alkynyl groups 1 to 3 hetero groups selected from N, S and O, which may be substituted with the same or different 1 to 5 groups selected from the group consisting of 5- to 6-membered monocyclic fully unsaturated unsaturated heterocyclic group having an atom, or a 9 to 10-membered bicyclic group having 1 to 3 heteroatoms selected from N, S and O More preferred are unsaturated heterocyclic groups which are fully unsaturated: A halogen atom; a cyano group; an oxide group; a deuterium atom, a halogen atom,
- C 3 -C 10 cycloalkyl group C 6 -C 14 aromatic hydrocarbon group; selected from the group consisting of C 1 -C 6 alkyl group, C 1 -C 6 alkoxy group and C 2 -C 7 alkanoyl group 1 to 3 identical or different groups are substituted
- the same or different 1 to 5 groups selected from the group consisting of a 4 to 10-membered unsaturated heterocyclic group which may be substituted; or a 4 to 10-membered saturated heterocyclic group which may be substituted by an oxo group;
- C 6 -C 14 aromatic hydrocarbon group C 1 -C 6 alkyl group, the same or are selected from C 1 -C 6 alkoxy and C 2 -C 7 group consisting alkanoyl group 1 to 3 different groups may be substituted 4 to 1 1 to 5 groups of the same or different groups selected from the group consisting of a 0-membered unsaturated heterocyclic group; or a 4- to 10-membered saturated heterocyclic group optionally substituted by an oxo group are substituted.
- Preferred embodiments of “unsaturated heterocyclic group” of B can be represented by the structural formula as follows. As described above, the “unsaturated heterocyclic group” may have the above-described substituent.
- R 1 is a hydrogen atom or a C 1 -C 6 alkyl group, and 1 to 5 R a s where A is the same or different may be substituted.
- a saturated heterocyclic ring is formed (in this case, the 4- to 10-membered unsaturated heterocyclic ring and the 4- to 10-membered saturated heterocyclic ring may be substituted with R b ),
- R 2 is a hydrogen atom
- a 10-membered saturated heterocyclic group (wherein the C 1 -C 6 alkyl group may be substituted by R d , a C 3 -C 10 cycloalkyl group, a C 6 -C 14 aromatic hydrocarbon group, The 4- to 10-membered unsaturated heterocyclic group and the
- R 1 is a hydrogen atom or a C 1 -C 6 alkyl group
- A is a halogen atom, an amino group, a C 1 -C 6 alkylamino group, a hydroxyl group, or a C 1 -C 6 alkoxy group
- Specific preferred compounds of the present invention include the following. (1) 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2-cyclobutyl-2H-indazol-5-ylamino) -1,2,4-triazine-6-carboxamide (Examples) 11 compounds) (2) 3-((((3R, 4R) -3-aminotetrahydro-2H-pyran-4-yl) amino) -5-((2- (tert-butyl) -7-methyl-2H-indazole-5 -Yl) amino) -1,2,4-triazine-6-carboxamide (Compound of Example 61) (3) 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2- (difluoromethyl) -7-methyl-2H-indazol-5-yl) amino) -1,2 , 4-Triazine-6-carboxamide (Compound of Example 62)
- the compound (I) of the present invention can be produced, for example, by the following production methods or the methods shown in the examples. However, the production method of the compound (I) of the present invention is not limited to these reaction examples.
- L 1 and L 2 each independently represent a leaving group
- R 3 represents hydrogen or a protecting group
- A, B, R 1 and R 2 are as defined above.
- Step 1 a compound represented by the general formula (IV) is produced by reacting a compound represented by the general formula (II) with a compound represented by the general formula (III) or a salt thereof. Is the method.
- the removal or conversion of the protecting group and the conversion of the leaving groups L 1 and L 2 can be performed as appropriate.
- Examples of the leaving group represented by L 1 and L 2 include halogen atoms such as chlorine atom, bromine atom or iodine atom, organic sulfonyl groups such as methylsulfinyl group, methylsulfonyl group, ethylsulfonyl group and phenylsulfonyl group, or methyl Organic sulfonyloxy groups such as sulfonyloxy group, trifluoromethylsulfonyloxy group and p-tolylsulfonyloxy group, organic thio groups such as methylthio group, ethylthio group, phenylthio group and benzylthio group, 1H-benzotriazol-1-yloxy, etc.
- halogen atoms such as chlorine atom, bromine atom or iodine atom
- organic sulfonyl groups such as methylsulfinyl group, methyls
- This step is usually carried out using 0.5 mol to excess mol, preferably 1 to 3 mol of the compound represented by general formula (III) with respect to 1 mol of compound represented by general formula (II). Is called.
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- isopropanol, tert-butyl alcohol, toluene, benzene, methylene chloride, chloroform, tetrahydrofuran, dioxane, dimethylformamide, N-methylpyrrolidinone, dimethyl A sulfoxide or the like or a mixed solvent thereof is preferred.
- a base or an acid can be used as necessary.
- the base examples include organic bases such as triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine, or sodium hydrogen carbonate, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, potassium tert-butylate, etc.
- the acid for example, hydrochloric acid, p-toluenesulfonic acid, trifluoroacetic acid, acetic acid, phosphoric acid, phenol and the like can be used.
- the amount of the base to be used is generally 0.1 mol to excess mol, preferably 1 to 3 mol, per 1 mol of the compound represented by the general formula (II).
- the amount of the acid to be used is generally 0.01 mol to excess mol, preferably 0.1 to 3 mol, per 1 mol of the compound represented by the general formula (II).
- the reaction temperature is usually 0 ° C. to 200 ° C., preferably 0 ° C. to 150 ° C.
- the reaction time is usually 1 minute to 7 days, preferably 5 minutes to 24 hours.
- the compound (IV) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or without isolation and purification. It can be attached to the next process.
- Step 2 This step is a method for producing a compound represented by the general formula (VI) by reacting a compound represented by the general formula (IV) with ammonia or a salt thereof.
- the amount of ammonia or a salt thereof used in this step is usually equimolar to excess molar relative to 1 mol of the compound represented by the general formula (IV).
- the reaction solvent is not particularly limited as long as it does not hinder the reaction. For example, water, methanol, ethanol, isopropanol, tert-butyl alcohol, tetrahydrofuran, dioxane, dimethylformamide, N-methylpyrrolidone, dimethyl sulfoxide and the like or A mixed solvent thereof is preferred.
- the reaction temperature is generally 0 ° C. to 200 ° C., preferably room temperature to 150 ° C.
- the reaction time is usually 5 minutes to 7 days, preferably 1 minute to 24 hours.
- the compound (VI) thus obtained is isolated or purified by a known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or without isolation and purification. It can be attached to the next process.
- a known separation and purification means for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or without isolation and purification. It can be attached to the next process.
- Step 3 the compound represented by the general formula (VI) is reacted with the compound represented by the general formula (VII) or a salt thereof to produce the compound represented by the general formula (I). Is the method.
- This step is usually carried out using 0.5 mol to excess mol, preferably 1 to 3 mol of the compound represented by general formula (VII) with respect to 1 mol of compound represented by general formula (VI). Is called.
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- isopropanol, tert-butyl alcohol, toluene, benzene, methylene chloride, chloroform, tetrahydrofuran, dioxane, dimethylformamide, N-methylpyrrolidinone, dimethyl A sulfoxide or the like or a mixed solvent thereof is preferred.
- a base can be used for the said reaction as needed.
- the base examples include organic bases such as triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine, or sodium hydrogen carbonate, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, potassium tert-butylate, etc.
- organic bases such as triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine, or sodium hydrogen carbonate, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, potassium tert-butylate, etc.
- Inorganic bases can be used.
- the amount of the base to be used is generally 0.1 mol to excess mol, preferably 1 to 3 mol, per 1 mol of the compound represented by the general formula (VI).
- the reaction temperature is generally 0 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time is usually 1 minute to 7 days, preferably 5 minutes to 24 hours.
- the leaving group of the compound represented by the general formula (IV) or (VI) is an organic thio group, it may be oxidized and converted to an organic sulfinyl group or an organic sulfonyl group by a known method and used for the reaction. it can.
- an inert solvent such as benzene, toluene, methylene chloride, chloroform, tetrahydrofuran, acetonitrile, dimethylformamide, N-methylpyrrolidone
- an inert solvent such as benzene, toluene, methylene chloride, chloroform, tetrahydrofuran, acetonitrile, dimethylformamide, N-methylpyrrolidone
- desiccants such as anhydrous sodium sulfate, anhydrous magnesium sulfate, calcium sulfate, and molecular sieves, or inorganic bases such as sodium bicarbonate and sodium carbonate can be added.
- the leaving group of the compound represented by formula (IV) or (VI) is an organic thio group, it can be converted into a halogen group and used for the reaction by a known method.
- the thus obtained compound having an organic sulfinyl group, organic sulfonyl group or halogen group is isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like. Alternatively, it can be subjected to the next step without isolation and purification.
- the compound represented by the general formula (II) is a method described in the literature [Patent Publication No. 2009-007341, European Journal of Medicinal Chemistry, Volume 15 (No. 3). ) Pp. 269-273 etc.] or a method according to these methods.
- the compounds represented by the general formula (III) and the general formula (V) for example, commercially available products are used, described in the literature, methods according to these methods, or described in the following methods, examples and production examples. It can be produced by appropriately combining methods and the like as necessary.
- the compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- Step 4 the compound represented by the general formula (VIII) is reacted with the compound represented by the general formula (VII) or a salt thereof to produce the compound represented by the general formula (VIII).
- This step can be carried out by the same method as in step 3, a method according to this, or a combination of these with conventional methods.
- This step is usually carried out using 0.5 mol to excess mol, preferably equimolar to 3 mol of compound (VII) with respect to 1 mol of compound (IV).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- methanol, ethanol, isopropanol, tert-butyl alcohol, toluene, benzene, methylene chloride, chloroform, tetrahydrofuran, dioxane, dimethylformamide, acetonitrile, N -Methyl pyrrolidone, dimethyl sulfoxide or the like or a mixed solvent thereof is preferred.
- a base can be used for the said reaction as needed.
- the base examples include organic bases such as triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine, or sodium hydrogen carbonate, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydroxide, sodium hydride, potassium tert-butylate, etc.
- Inorganic bases can be used.
- the amount of the base used is usually equimolar to excess molar, preferably 1 to 3 molar relative to 1 molar of the compound represented by the general formula (IV).
- the reaction temperature is generally 0 ° C. to 200 ° C., preferably room temperature to 130 ° C.
- the reaction time is usually 1 minute to 7 days, preferably 5 minutes to 24 hours.
- Step 5 This step is a method for producing a compound represented by the general formula (I) by reacting a compound represented by the general formula (VIII) with ammonia or a salt thereof. This step can be carried out by the same method as in step 2, a method according to this, or a combination of these with conventional methods.
- the amount of the ammonia or a salt thereof is usually equimolar to excess mole, preferably excess mole relative to 1 mole of the compound represented by the general formula (VIII).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- methanol, ethanol, isopropanol, tert-butyl alcohol, toluene, benzene, methylene chloride, chloroform, tetrahydrofuran, dioxane, dimethylformamide, acetonitrile, N -Methylpyrrolidinone, dimethyl sulfoxide or the like, or a mixed solvent thereof is preferred.
- the reaction temperature is generally 0 ° C. to 200 ° C., preferably room temperature to 160 ° C.
- the reaction time is usually 10 minutes to 12 hours, preferably 5 minutes to 2 hours.
- the compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- amino groups, imino groups, hydroxyl groups, carboxyl groups, carbonyl groups and amide groups, and functional groups having active protons such as indoles are protected at appropriate steps in each production method.
- the protecting group can be removed after introducing a protecting group into the functional group using a conventional reagent or according to a conventional method.
- the “protecting group for amino group or imino group” is not particularly limited as long as it has the function.
- benzyl group, p-methoxybenzyl group, 3,4-dimethoxybenzyl group, o-nitrobenzyl group, Aralkyl groups such as p-nitrobenzyl group, benzhydryl group, trityl group and cumyl group; for example, lower alkanoyl groups such as formyl group, acetyl group, propionyl group, butyryl group, pivaloyl group, trifluoroacetyl group and trichloroacetyl group;
- Benzoyl group for example, arylalkanoyl group such as phenylacetyl group and phenoxyacetyl group; for example, lower alkoxycarbonyl group such as methoxycarbonyl group, ethoxycarbonyl group, propyloxycarbonyl group, tert-butoxycarbonyl group;
- the “hydroxyl-protecting group” is not particularly limited as long as it has the function, but for example, a lower alkyl group such as methyl group, ethyl group, propyl group, isopropyl group, tert-butyl group; A lower alkylsilyl group such as a butyldimethylsilyl group; a lower alkoxymethyl group such as a methoxymethyl group or a 2-methoxyethoxymethyl group; a tetrahydropyranyl group; a trimethylsilylethoxymethyl group; a benzyl group, a p-methoxybenzyl Aralkyl groups such as a group, 2,3-dimethoxybenzyl group, o-nitrobenzyl group, p-nitrobenzyl group, trityl group; for example, acyl groups such as formyl group, acetyl group, trifluoroacetyl group, etc.
- the “carboxyl-protecting group” is not particularly limited as long as it has the function, but for example, a lower alkyl group such as methyl group, ethyl group, propyl group, isopropyl group, tert-butyl group; Halo lower alkyl groups such as 1,2-trichloroethyl group; for example, lower alkenyl groups such as allyl group; for example, trimethylsilylethoxymethyl group; for example, benzyl group, p-methoxybenzyl group, p-nitrobenzyl group, benzhydryl group, trityl group, etc.
- a lower alkyl group such as methyl group, ethyl group, propyl group, isopropyl group, tert-butyl group
- Halo lower alkyl groups such as 1,2-trichloroethyl group
- lower alkenyl groups such as allyl group
- trimethylsilylethoxymethyl group for example
- methyl group ethyl group, tert-butyl group, allyl group, benzyl group, p-methoxybenzyl group, trimethylsilylethoxymethyl group, and the like.
- the “carbonyl-protecting group” is not particularly limited as long as it has the function, and examples thereof include acetals such as ethylene ketal, trimethylene ketal and dimethyl ketal, and ketal.
- the method for removing the protecting group varies depending on the kind of the protecting group and the stability of the target compound (I).
- the compound of the present invention can be easily isolated and purified by ordinary separation means.
- Examples of such means include solvent extraction, recrystallization, preparative reverse phase high performance liquid chromatography, column chromatography, preparative thin layer chromatography and the like.
- any isomers and mixtures are included in the compound of the present invention.
- an optical isomer exists in the compound of the present invention
- an optical isomer resolved from a racemate is also included in the compound of the present invention.
- Each of these isomers can be obtained as a single compound by synthetic methods and separation methods known per se (concentration, solvent extraction, column chromatography, recrystallization, etc.).
- R1 and R2 are hydrogen, the following tautomers exist, and any isomer is included in the compound of the present invention.
- the compound of the present invention or a salt thereof may be a crystal, and it is included in the compound of the present invention or a salt thereof regardless of whether the crystal form is single or polymorphic.
- the crystal can be produced by crystallization by applying a crystallization method known per se.
- the compound of the present invention or a salt thereof may be a solvate (such as a hydrate) or a non-solvate, and both are included in the compound of the present invention or a salt thereof.
- Compounds labeled with isotopes eg, 3H, 14C, 35S, 125I, etc. are also encompassed in the compounds of the present invention or salts thereof.
- a prodrug of the compound of the present invention or a salt thereof is a compound that is converted into the compound of the present invention or a salt thereof by a reaction with an enzyme, gastric acid or the like under physiological conditions in vivo, that is, enzymatically oxidized, reduced, hydrolyzed, etc.
- the prodrug of the compound of the present invention or a salt thereof can be converted into the compound of the present invention or a salt thereof under physiological conditions as described in Hirokawa Shoten 1990, “Development of Drugs”, Volume 7, pages 163 to 198. It may change.
- the salt of the compound of the present invention means a conventional salt used in the field of organic chemistry.
- a base addition salt in the carboxyl group or an amino group or a basic heterocyclic group.
- acid addition salts of the amino group or basic heterocyclic group include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt and magnesium salt; for example ammonium salt;
- organic amine salts such as salts, diethanolamine salts, triethanolamine salts, procaine salts, and N, N′-dibenzylethylenediamine salts.
- the acid addition salt examples include inorganic acid salts such as hydrochloride, sulfate, nitrate, phosphate and perchlorate; for example, acetate, formate, maleate, fumarate, tartrate, citric acid Organic acid salts such as salts, ascorbate and trifluoroacetate; for example, sulfonates such as methanesulfonate, isethionate, benzenesulfonate, and p-toluenesulfonate.
- inorganic acid salts such as hydrochloride, sulfate, nitrate, phosphate and perchlorate
- Organic acid salts such as salts, ascorbate and trifluoroacetate
- sulfonates such as methanesulfonate, isethionate, benzenesulfonate, and p-toluen
- the compound of the present invention or a salt thereof has excellent Syk inhibitory activity and is useful as an antitumor agent. In addition, it has an excellent selectivity for Syk and has the advantage of fewer side effects from other kinases.
- the target cancer is not particularly limited, but for example, head and neck cancer, esophageal cancer, stomach cancer, colon cancer, rectal cancer, liver cancer, gallbladder / bile duct cancer, biliary tract cancer, pancreatic cancer, lung cancer, breast cancer, ovarian cancer, uterus
- Examples include cervical cancer, endometrial cancer, renal cancer, bladder cancer, prostate cancer, testicular cancer, bone / soft tissue sarcoma, blood cancer, multiple myeloma, skin cancer, brain tumor, mesothelioma, etc.
- Hematological cancers such as cell lymphoma, chronic lymphocytic leukemia, peripheral T-cell lymphoma, myelodysplastic syndrome, acute myeloid leukemia, acute lymph
- a pharmaceutically acceptable carrier is blended as necessary, and various administration forms can be adopted depending on the purpose of prevention or treatment.
- any of oral preparations, injections, suppositories, ointments, patches and the like may be used, and oral preparations are preferably employed.
- Each of these dosage forms can be produced by a conventional formulation method known to those skilled in the art.
- the pharmaceutical carrier various organic or inorganic carrier substances commonly used as pharmaceutical materials are used, and excipients, binders, disintegrants, lubricants, coloring agents in solid preparations, solvents in liquid preparations, dissolution aids, It is blended as a suspending agent, isotonic agent, buffer, soothing agent and the like.
- formulation additives such as preservatives, antioxidants, colorants, sweeteners, stabilizers and the like can be used as necessary.
- an excipient When an oral solid preparation is prepared, an excipient, if necessary, an excipient, a binder, a disintegrant, a lubricant, a coloring agent, a flavoring / flavoring agent, and the like are added to the compound of the present invention. Tablets, coated tablets, granules, powders, capsules and the like can be produced by the method.
- a pH adjuster, buffer, stabilizer, isotonic agent, local anesthetic, etc. are added to the compound of the present invention, and subcutaneous, intramuscular and intravenous injections are prepared by conventional methods. Can be manufactured.
- the NMR spectrum was measured using AL400 (400 MHz; JEOL) or Mercury 400 (400 MHz; Varian) type spectrometer. When tetramethylsilane was included in the heavy solvent, tetramethylsilane was used as an internal standard. In other cases, measurement was performed using an NMR solvent as an internal standard, and all ⁇ values were expressed in ppm.
- the microwave reaction was performed using Initiator (registered trademark) manufactured by Biotage.
- the LCMS spectrum was measured using SQD manufactured by Waters under the following conditions.
- Reverse phase preparative HPLC purification was performed under the following conditions.
- Example 2 3-((1R, 2S) -2-aminocyclohexylamino) -5- (benzo [b] thiophen-4-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) To a solution of 70 mg of ethyl 5-chloro-3- (methylthio) -1,2,4-triazine-6-carboxylate and 70 mg of benzo [b] thiophen-4-amine in 1 ml of THF was added 0.10 ml of diisopropylethylamine, The reaction was stirred at room temperature for 35 minutes.
- the reaction mixture was diluted with ethyl acetate and washed successively with aqueous sodium hydrogen carbonate solution and saturated brine. After drying over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: hexane / ethyl acetate) to give ethyl 5- (benzo [b] thiophene-4- Ilamino) -3-((1R, 2S) -2- (tert-butoxycarbonylamino) cyclohexylamino) -1,2,4-triazine-6-carboxylate was obtained.
- Example 3 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-methyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) 500 ⁇ l of sulfuryl chloride was added to 466 mg of ethyl 5-chloro-3- (methylthio) -1,2,4-triazine-6-carboxylate, and the reaction solution was stirred at 70 ° C. for 2 hours, and then 150 ⁇ l of sulfuryl chloride was added. The mixture was further stirred for 30 minutes. After the reaction solution was diluted with THF, the solvent was concentrated under reduced pressure.
- the obtained residue was dissolved in 10 ml of THF, 470 mg of 1-methyl-1H-indole-4-amine and 0.85 ml of diisopropylethylamine were sequentially added, and the reaction solution was stirred at room temperature for 15 minutes.
- the reaction mixture was diluted with ethyl acetate, washed successively with aqueous sodium hydrogen carbonate solution and saturated brine, and dried over anhydrous sodium sulfate.
- Example 4 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2- (ethyl-d5) -2H-indazol-5-ylamino) -1,2,4-triazine-6-carboxamide (step 1) 60 mg of 60% sodium hydride (40% liquid paraffin added) was added to a solution of 210 mg of 5-nitro-1H-indazole and 170 mg of ethyl bromide-d5 in DMF, and the reaction solution was stirred at room temperature overnight.
- Example 5 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (ethyl-d5) -1H-indazol-5-ylamino) -1,2,4-triazine-6-carboxamide (step 1)
- Step 2 instead of 5-nitro-2- (ethyl-d5) -2H-indazole, 5-nitro-1- (ethyl-d5) obtained in Example 4 (Step 1) was used.
- -1H-indazole was used to give 1- (ethyl-d5) -1H-indazole-5-amine as a yellow solid.
- Example 6 3-((1R, 2S) -2- (dimethylamino) cyclohexylamino) -5- (1- (ethyl-d5) -1H-indazol-5-ylamino) -1,2,4-triazine-6- Carboxamide diformate 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (ethyl-d5) -1H-indazol-5-ylamino) -1 obtained in Example 5 , 2,4-Triazine-6-carboxamide To a solution of 2 mg of methanol in 1 ml of methanol was added 0.01 ml of 50% aqueous formaldehyde solution and 0.1 ml of 0.3 M Zn (BH 3 CN) 2 -methanol solution prepared by a known method, The reaction was stirred at room temperature for 15 minutes. The solvent was distilled off under reduced pressure, the residue was purified by reverse phase preparative HPLC
- Example 7 (S) -5- (1- (Ethyl-d5) -1H-indazol-5-ylamino) -3- (piperidin-3-ylamino) -1,2,4-triazine-6-carboxamide diformate 3- (Methylthio) -5- (1- (ethyl-d5) -1H-indazol-5-ylamino) -1,2,4-triazine-6-carboxamide obtained in Example 5 (Step 2) 4 mg of THF To 0.5 mL solution, 3.3 mg of m-CPBA was added, and the reaction solution was stirred at room temperature for 1 hour.
- Example 8 3-((3R, 4R) -3-aminotetrahydro-2H-pyran-4-ylamino) -5- (benzo [b] thiophen-4-ylamino) -1,2,4-triazine-6-carboxamide (step 1) To 234 mg of ethyl 5-chloro-3- (methylthio) -1,2,4-triazine-6-carboxylate was added 0.30 ml of sulfuryl chloride, and the reaction solution was stirred at room temperature for 10 hours.
- Example 10 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-cyclopropyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) To a solution of 0.96 g of 4-nitro-1H-indole in 60 ml of 1,2-dichloroethane, 1.0 g of cyclopropylboronic acid, 0.99 g of sodium bicarbonate, 1.1 g of copper (II) acetate, 2,2′-bipyridine 0.92 g was added, and the reaction solution was stirred at room temperature for 7 days, and further stirred at 60 ° C. for 2 hours.
- Step 1 To a solution of 0.96 g of 4-nitro-1H-indole in 60 ml of 1,2-dichloroethane, 1.0 g of cyclopropylboronic acid, 0.99 g of sodium bicarbonate, 1.1 g
- Example 11 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2-cyclobutyl-2H-indazol-5-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) To a solution of 490 mg of 5-nitro-1H-indazole in 10 ml of THF, 260 mg of cyclobutanol, 1.1 g of triphenylphosphine and 790 mg of diisopropyl azodicarboxylate were sequentially added at room temperature, and the reaction solution was stirred at room temperature overnight.
- Example 12 (3-((1R, 2S) -2-aminocyclohexylamino) 6-carbamoyl-1,2,4-triazin-5-ylamino) quinoline 1-oxide according to Example 1 (steps 3 to 4) , M-CPBA was used in excess to give the title compound as a white solid.
- Example 13 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-methyl-6- (1-methyl-1H-pyrazol-4-yl) -1H-indazol-4-ylamino) -1 , 2,4-Triazine-6-carboxamide (Step 1) 6-Bromo-4-nitro-1H-indazole To a solution of 901 mg of DMF in 19 ml of DMF was added 213 mg of 60% sodium hydride (40% liquid paraffin added). After stirring the reaction solution at 0 ° C. for 15 minutes, 0.35 ml of methyl iodide was added, and the reaction solution was allowed to react at room temperature for 11 hours.
- the reaction solution was poured into water, and the deposited precipitate was collected by filtration to obtain a crude product.
- the crude product was purified by silica gel column chromatography (developing solvent: hexane / ethyl acetate), and 6-bromo-1-methyl-4-nitro-1H-indazole and 6-bromo-2-methyl-4-nitro- Each 2H-indazole was obtained as a yellow solid.
- Example 14 5- (5-acetyl-2,3′-bithiophen-5′-ylamino) -3-((1R, 2S) -2-aminocyclohexylamino) -1,2,4-triazine-6-carboxamide
- Examples 13 (Steps 2 to 4), using tert-butyl 4-bromothiophen-2-ylcarbamate instead of 6-bromo-1-methyl-4-nitro-1H-indazole, By substituting 5-acetylthiophen-2-ylboronic acid for (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole, the title compound Obtained as a yellow solid.
- Example 15 5- (5- (5-acetylthiophen-2-yl) pyridin-3-ylamino) -3-((1R, 2S) -2-aminocyclohexylamino) -1,2,4-triazine-6-carboxamide
- Steps 2 to 4 instead of 6-bromo-1-methyl-4-nitro-1H-indazole, 5-bromopyridin-3-amine was used, and 1-methyl-4- (4 , 4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole in place of 5-acetylthiophen-2-ylboronic acid to give the title compound as a yellow solid Obtained.
- Example 16 3-((1R, 2S) -2-aminocyclohexylamino) -5- (5-cyclopropylpyridin-3-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) 5-bromopyridin-3-amine 500 mg, cyclopropylboronic acid 497 mg, palladium (II) acetate 65 mg, tricyclohexylphosphine 162 mg, potassium phosphate 2.5 g in toluene 9 mL, water 0.45 mL in solution at 100 ° C. for 11 hours Stir. Ethyl acetate and water were added, and the organic layer was separated and washed with saturated brine.
- Example 17 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-ethyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Step 1 the title compound was obtained as a white solid by using ethyl bromide instead of ethyl bromide-d5.
- Example 18 3-((1R, 2S) -2-aminocyclohexylamino) -5- (6- (cyclopropylcarbamoyl) pyridin-3-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) Ethyl 5-chloro-3- (methylthio) -1,2,4-triazine-6-carboxylate To a solution of 70 mg of dimethyl sulfoxide in 1.5 ml, 44 mg of 5-aminopicolinic acid was added and the reaction solution was allowed to stand at room temperature for 10 minutes.
- Example 20 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2-methyl-6- (1-methyl-1H-pyrazol-4-yl) -2H-indazol-4-ylamino) -1 , 2,4-Triazine-6-carboxamide
- Step 2 to 4 instead of 6-bromo-1-methyl-4-nitro-1H-indazole, obtained in Example 13 (Step 1)
- the title compound was obtained as a yellow solid.
- Example 21 3-((1R, 2S) -2-aminocyclohexylamino) -5- (6-methylbenzo [b] thiophen-4-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) To a solution of 2,6-dibromo-4-methylbenzaldehyde (153 mg) in dimethyl sulfoxide (1.7 mL) were added methyl thioglycolate (0,060 ml) and potassium carbonate (190 mg), and the reaction solution was stirred at 120 ° C. for 7 hours. The reaction mixture was neutralized with 2M hydrochloric acid, extracted with ethyl acetate, and washed with saturated brine.
- Example 22 Ethyl 4- (3-((1R, 2S) -2-aminocyclohexylamino) -6-carbamoyl-1,2,4-triazin-5-ylamino) benzo [b] thiophene-6-carboxylate (Step 1) ) 400 mg of potassium carbonate was added to 6 ml of ethanol solution of 760 mg of ethyl 4-acetoxybenzo [b] thiophene-6-carboxylate synthesized by the method described in WO2005 / 007635, and the reaction solution was stirred at room temperature for 1 hour.
- Example 23 3-((1R, 2S) -2-aminocyclohexylamino) -5- (6- (methylcarbamoyl) benzo [b] thiophen-4-ylamino) -1,2,4-triazine-6-carboxamide (step 1) To 110 mg of ethyl 4- (tert-butoxycarbonylamino) benzo [b] thiophene-6-carboxylate obtained in Example 22 (Step 3), 3 ml of a 40% methylamine-methanol solution was added, and the reaction solution was stirred at 90 ° C. At rt overnight.
- Example 24 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2- (difluoromethyl) -2H-indazol-6-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) ) To a solution of 6.90 g of 6-nitro-1H-indazole and 9.15 g of sodium chlorodifluoroacetate in 100 ml of N-methylpyrrolidinone, 1.2 g of 60% sodium hydride (40% liquid paraffin added) was added, and the reaction solution was allowed to cool to room temperature. The mixture was stirred at 15 ° C. for 15 minutes, and further stirred at 100 ° C. for 30 minutes.
- reaction mixture was diluted with ethyl acetate and washed successively with water and saturated brine. It dried with the anhydrous sodium sulfate and the solvent was depressurizingly distilled. The residue was purified by silica gel column chromatography (developing solvent: hexane-ethyl acetate) to give 1- (difluoromethyl) -6-nitro-1H-indazole and 2- (difluoromethyl) -6-nitro-2H-indazole. Obtained as a mixture.
- Example 25 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (1-methyl-1H-pyrazol-3-yl) -1H-indol-4-ylamino) -1,2,4 -Triazine-6-carboxamide (Step 1) To a mixture of 660 mg of 4-nitro-1H-indole, 853 mg of 3-iodo-1-methyl-1H-pyrazole, 864 mg of tripotassium phosphate and 775 mg of copper (I) iodide, trans-N, N′-dimethylcyclohexane-1 , 2-diamine (racemate) 0.643 ml and toluene 10 ml were added, and the reaction solution was stirred at 100 ° C.
- reaction solution was returned to room temperature, diluted with ethyl acetate, and washed successively with 28% aqueous ammonia and saturated brine. After drying over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (developing solvent: hexane-ethyl acetate) to give 1- (1-methyl-1H-pyrazol-3-yl ) -4-Nitro-1H-indole was obtained.
- Example 26 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (difluoromethyl) -1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Example 24 According to (Steps 1 to 3), 4-nitro-1H-indole was used in place of 6-nitro-1H-indazole to give the title compound as a white solid.
- Example 27 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-cyclobutyl-1H-indazol-5-ylamino) -1,2,4-triazine-6-carboxamide
- Step 2 According to 3), 5-nitro-1- (cyclobutyl) -1H-indazole obtained in Example 11 (Step 1) was used instead of 5-nitro-2- (ethyl-d5) -2H-indazole. Used to give the title compound as a yellow solid.
- Example 28 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2-ethyl-2H-indazol-4-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) According to Example 4 (Steps 1-2), 4-nitro-1H-indazole is used instead of 5-nitro-1H-indazole, and 2-iodoethane is used instead of ethyl bromide-d5. 2-Ethyl-2H-indazole-4-amine was obtained.
- Example 29 3- (5-acetylthiophen-2-yl) -5- (3-((1R, 2S) -2-aminocyclohexylamino) -6-carbamoyl-1,2,4-triazine-5ylamino) pyridine 1 - obtained in oxide example 15 3- (methylthio) -5- (5- (5-acetyl-2-yl) pyridin-3-ylamino) to 1,2,4-triazine-6-carboxamide Using an excess amount of m-CPBA, the title compound was converted into a white solid by the same method as in Example 1 (Step 3) and (Step 4), a method analogous thereto, or a combination thereof with a conventional method. Obtained.
- Example 30 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-ethyl-1H-indol-6-ylamino) -1,2,4-triazine-6-carboxamide
- Example 9 (Step 1) According to ⁇ 4), 6-nitro-1H-indole was used instead of 4-nitro-1H-indole, and ethyl bromide was used instead of ethyl bromide-d5 to obtain the title compound.
- Example 31 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-cyclopropyl-1H-indazol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Example 10 (Step According to 1-3), the title compound was obtained by using 4-nitro-1H-indazole instead of 4-nitro-1H-indole.
- Example 32 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-ethyl-2-methyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide (step 1) To a solution of 1.38 g of dimethyl sulfoxide in 20 ml of 3-nitroaniline, 1.0 mL of acetone and 2.7 g of tert-butoxy potassium were added, and the reaction solution was stirred at room temperature for 3 hours. The reaction mixture was diluted with ethyl acetate and washed successively with aqueous ammonium chloride solution and saturated brine.
- Example 33 Implementation of 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-cyclopropyl-2-methyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Step 1 instead of 4-nitro-1H-indole, 2-methyl-4-nitro-1H-indole obtained in Example 32 (Step 1) was used to obtain the title. A compound was obtained.
- Example 34 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (oxetan-3-yl) -1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide (Process 1)
- 1- (Oxetan-3-yl) -1H-indole-4-amine was obtained by using 3-oxetane bromide instead of ethyl bromide-d5 according to Example 9 (Steps 1 and 2). It was.
- Example 35 Implementation of 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (2-fluoroethyl) -1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- the title compound was obtained according to Example 34 (Steps 1-2) by using 1-iodo-2-fluoroethane instead of 3-oxetane bromide.
- Example 36 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (pyridin-2-yl) -1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- the title compound was obtained according to Example 25 (Steps 1 to 3) by using 2-bromopyridine in place of 3-iodo-1-methyl-1H-pyrazole.
- Example 37 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-phenyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Example 25 The title compound was obtained by using iodobenzene instead of 3-iodo-1-methyl-1H-pyrazole according to ⁇ 2).
- Example 38 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (difluoromethyl) -1H-indazol-4-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) ) According to Example 24 (Steps 1-2), 1- (difluoromethyl) -1H-indazole-4-amine and 4-nitro-1H-indazole were used instead of 6-nitro-1H-indazole. 2- (Difluoromethyl) -2H-indazol-4-amine was obtained respectively.
- Example 39 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (2-hydroxy-2-methylpropyl) -1H-indol-4-ylamino) -1,2,4-triazine- According to 6-carboxamide Example 34 (Steps 1 and 2), 1-iodo-2-methylpropan-2-ol was used instead of oxetane bromide to give the title compound.
- Example 40 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (2,2-difluoroethyl) -1H-indol-4-ylamino) -1,2,4-triazine-6- According to carboxamide example 34 (steps 1-2), 1,1-difluoro-2-iodoethane was used instead of oxetane bromide to give the title compound.
- Example 42 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (methyl-d3) -1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- the title compound was obtained as a white solid by using iodomethane-d3 in place of 3-oxetane bromide according to 34 (Steps 1-2).
- Example 43 3-((1R, 2S) -2-aminocyclohexylamino) -5- (thieno [2,3-c] pyridin-4-ylamino) -1,2,4-triazine-6-carboxamide (Step 1) To a solution of 1.6 g of 3,5-dibromo-4-pyridinecarboxaldehyde in 12 ml of DMF, 0.653 ml of methyl thioglycolate and 2.1 g of potassium carbonate were added, and the reaction solution was stirred at 120 ° C. for 2 hours.
- Example 44 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-propyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Step 1 the title compound was obtained by using 1-iodopropane instead of oxetane bromide.
- Example 45 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-isopropyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Step 1 the title compound was obtained by using 2-iodopropane instead of 3-bromooxetane.
- Example 46 Implementation of 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (2-methoxyethyl) -1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide According to Example 34 (Steps 1-2), 1-bromo-2-methoxyethane was used in place of 3-bromooxetane to give the title compound.
- Example 47 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-cyclobutyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Step 1 the title compound was obtained by using cyclobutane bromide instead of 3-bromooxetane.
- Example 48 3-((1R, 2S) -2-aminocyclohexylamino) -5- (5-methoxy-3-methylbenzo [b] thiophen-2-ylamino) -1,2,4-triazine-6-carboxamide (step 1) Methyl 3-chloro-5-methoxy-1-benzothiophene-2-carboxylate In a solution of 257 mg of 1,4-dioxane in 3.0 ml, methylboronic acid 120 mg, potassium phosphate 849 mg, water 0.30 ml, palladium acetate 22 mg, 2 -95 mg of dicyclohexylphosphino-2 ′, 4 ′, 6′-triisopropylbiphenyl was added, and the reaction solution was stirred at 100 ° C.
- reaction solution was diluted with ethyl acetate and washed successively with an aqueous ammonium chloride solution, an aqueous sodium hydrogen carbonate solution and saturated brine. After drying over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: hexane-ethyl acetate) to obtain methyl 5-methoxy-3-methylbenzo [b] thiophene. -2-Carboxylate was obtained.
- Example 50 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-ethyl-3-methyl-1H-indazol-6-ylamino) -1,2,4-triazine-6-carboxamide
- Examples 4 (Steps 1 to 3), using 6-nitro-3-methyl-1H-indazole instead of 5-nitro-1H-indazole and using iodoethane instead of ethyl bromide-d5, The title compound was obtained.
- Example 51 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1,3-dimethyl-1H-indol-4-ylamino) -1,2,4-triazine-6-carboxamide
- Example 34 ( According to steps 1-2), 3-methyl-4-nitro-1H-indole is used instead of 4-nitro-1H-indole, and methyl iodide is used instead of 3-oxetane bromide, The title compound was obtained.
- Example 52 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1-isopropyl-1H-indazol-6-ylamino) -1,2,4-triazine-6-carboxamide
- Example 4 (Step 1) To 3) by using 6-nitro-1H-indazole instead of 5-nitro-1H-indazole and 2-iodopropane instead of ethyl bromide-d5 to give the title compound as a yellow solid Got as.
- Example 53 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2-isopropyl-2H-indazol-6-ylamino) -1,2,4-triazine-6-carboxamide
- Step 2 According to 3), instead of 5-nitro-2- (ethyl-d5) -1H-indazole, 6-nitro-2-isopropyl-2H-indazole obtained as a by-product in Example 52 was used. The compound was obtained as a yellow solid.
- Example 54 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (2,2-difluoroethyl) -1H-indazol-6-ylamino) -1,2,4-triazine-6- Carboxamide according to Example 4 (steps 1 to 3), using 6-nitro-1H-indazole instead of 5-nitro-1H-indazole and 1,1-difluoro-2 instead of ethyl bromide-d5 -Using iodoethane gave the title compound as a yellow solid.
- Example 55 Implementation of 3-((1R, 2S) -2-aminocyclohexylamino) -5- (1- (2-fluoroethyl) -1H-indazol-6-ylamino) -1,2,4-triazine-6-carboxamide
- 6-nitro-1H-indazole is used instead of 5-nitro-1H-indazole
- 1-fluoro-2-iodoethane is used instead of ethyl bromide-d5. This gave the title compound as a yellow solid.
- Example 57 3-((1R, 2S) -2-aminocyclohexylamino) -5- (2- (difluoromethyl) -2H-indazol-5-ylamino) -1,2,4-triazine-6-carboxamide
- Example 24 The title compound was obtained as a yellow solid by using 5-nitro-1H-indazole instead of 6-nitro-1H-indazole according to (Steps 1 to 3).
- Example 62 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2- (difluoromethyl) -7-methyl-2H-indazol-5-yl) amino) -1,2,4- Triazine-6-carboxamide
- Step 1 5-Bromo-7-methyl-1H-indazole 2.11 g of potassium acetate 2.76 g and 2,2-difluoro-2- (fluorosulfonyl) acetic acid 2.06 mL were added to a solution of 2.11 g of ethyl acetate in 50 mL, and the reaction solution was brought to room temperature. And stirred for 3 hours.
- Example 64 3-(((2R, 3R) -3-Amino-4-methoxybutan-2-yl) amino) -5-((1-isopropyl-1H-indol-4-yl) amino) -1,2,4 -Triazine-6-carboxamide (Step 1)
- tert-butyl ((2R, 3R) -3-amino-1-methoxybutane-2 was obtained by using iodomethane instead of iodoethane of Example 63 (Step 1).
- -Yl) Carbamate was obtained as a white amorphous.
- Example 2 In accordance with Example 3 (Steps 1 to 4), 1-isopropyl-1H-indole-4-amine obtained in Example 45 was used instead of 1-methyl-1H-indole-4-amine, and tert-butyl was used. Instead of ((1S, 2R) -2-aminocyclohexyl) carbamate, use tert-butyl ((2R, 3R) -3-amino-1-methoxybutan-2-yl) carbamate obtained in Step 1 above. Gave the title compound as a pale yellow solid.
- Example 65 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-methyl-7-phenyl-2H-indazol-5-yl) amino) -1,2,4-triazine-6 Carboxamide (step 1) 7-Bromo-5-nitro-1H-indazole 1.5 g of trimethyloxonium tetrafluoroborate was added to a solution of 1.21 g of ethyl acetate in 12 mL, and the reaction solution was stirred at room temperature for 6 hours. The reaction mixture was diluted with ethyl acetate, washed successively with water and saturated brine, and dried over anhydrous sodium sulfate.
- Example 66 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2- (tert-butyl) -7- (1H-pyrazol-1-yl) -2H-indazol-5-yl) Amino) -1,2,4-triazine-6-carboxamide (Step 1) To 484 mg of 7-bromo-5-nitro-1H-indazole, 5 mL of 2-methyl-2-propanol and 0.1 mL of concentrated sulfuric acid were added, and the reaction solution was reacted at 100 ° C. for 1 hour using a microwave reactor.
- Example 67 3-((((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (1-methyl-1H-pyrazol-4-yl) -1H-indol-4-yl) amino) -1 , 2,4-Triazine-6-carboxamide
- 4-iodo-1-methyl-1H-pyrazole is used instead of 3-iodo-1-methyl-1H-pyrazole Gave the title compound as a pale yellow solid.
- Example 68 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-((R) -tetrahydrofuran-3-yl) -1H-indol-4-yl) amino) -1,2 , 4-Triazine-6-carboxamide (Step 1) To a solution of (S) -tetrahydrofuran-3-ol 1.0 g in toluene 10 mL were added triethylamine 2.05 mL and methanesulfonyl chloride 1.05 mL, and the reaction solution was stirred at room temperature for 2 hours. Water was added to the reaction solution, followed by extraction with toluene and washing with saturated brine.
- Example 69 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (2-methylpyridin-4-yl) -1H-indol-4-yl) amino) -1,2, 4-Triazine-6-carboxamide
- 4-iodo-2-picoline was used instead of 3-iodo-1-methyl-1H-pyrazole to give the title compound a pale yellow Obtained as a solid.
- Example 70 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-ethyl-7-methyl-2H-indazol-5-yl) amino) -1,2,4-triazine-6 Carboxamide (step 1) To a solution of 844 mg of 5-bromo-7-methyl-1H-indazole in 844 mg of DMF, 240 mg of sodium hydride (60% in oil) and 0.64 mL of iodoethane were added, and the reaction solution was stirred at room temperature for 15 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- Example 71 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (6-methylpyridazin-3-yl) -1H-indol-4-yl) amino) -1,2, 4-Triazine-6-carboxamide
- Steps 1 to 3 substituting 3-chloro-6-methylpyridazine instead of 3-iodo-1-methyl-1H-pyrazole gave the title compound Obtained as a yellow solid.
- Example 72 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (pyrimidin-5-yl) -1H-indol-4-yl) amino) -1,2,4-triazine According to -6-carboxamide Example 25 (Steps 1 to 3), 5-bromopyrimidine was used in place of 3-iodo-1-methyl-1H-pyrazole to obtain the title compound as a pale yellow solid.
- Example 73 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (trans-4-hydroxycyclohexyl) -1H-indol-4-yl) amino) -1,2,4- Triazine-6-carboxamide
- (S) -tetrahydrofuran-3-yl methanesulfonate can be synthesized according to the method described in WO2004 / 050024 instead of 4- ⁇ [ The title compound was obtained as a pale yellow solid by using tert-butyl (dimethyl) silyl] oxy ⁇ cyclohexyl methanesulfonate.
- Example 74 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-isopropyl-7-methyl-2H-indazol-5-yl) amino) -1,2,4-triazine-6 - according to carboxamide example 70 (step 1-3) by using 2-iodopropane instead of iodoethane, the title compound was obtained as a pale yellow solid.
- Example 75 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (tetrahydro-2H-pyran-4-yl) -1H-indol-4-yl) amino) -1,2 , 4-Triazine-6-carboxamide
- Steps 1 to 3 the title compound was prepared by substituting tetrahydro-2H-pyran-4-ol for (S) -tetrahydrofuran-3-ol. Obtained as a yellow solid.
- Example 76 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((4- (pyrimidin-2-yl) thiophen-2-yl) amino) -1,2,4-triazine-6- Carboxamide (Step 1) Tris (dibenzylideneacetone) dipalladium (0) 460 mg, 2- (dicyclohexylphosphino) -2 ′, 4 ′, 6′-triisopropyl-1,1′-biphenyl 960 mg, 3-thiopheneboronic acid 9.60 g, To 5.72 g of 2-chloropyrimidine, 21.2 g of potassium phosphate, and 12.5 g of molecular sieve 4A were added 100 mL of tert-amyl alcohol, and the reaction solution was stirred at 100 ° C.
- the solvent was distilled off under reduced pressure, diluted with ethyl acetate, and washed successively with water and saturated brine. After drying over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: hexane / ethyl acetate) to give tert-butyl (4- (4,4,5 , 5-tetramethyl-1,3,2-dioxaborolan-2-yl) thiophen-2-yl) carbamate.
- Example 78 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (trans-4-hydroxycyclohexyl) -6-methyl-1H-indol-4-yl) amino) -1, 2,4-Triazine-6-carboxamide
- Example 9 using 6-methyl-4-nitro-1H-indole instead of 4-nitro-1H-indole, ethyl bromide-d5
- the title compound was obtained as a pale yellow solid by using 4- ⁇ [tert-butyl (dimethyl) silyl] oxy ⁇ cyclohexyl methanesulfonate, which can be synthesized according to the method described in WO2004 / 050024, instead of.
- Example 79 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-ethyl-7- (1H-pyrazol-1-yl) -2H-indazol-5-yl) amino) -1 , 2,4-Triazine-6-carboxamide (Step 1) According to Example 65 (Step 1), 7-bromo-2-ethyl-5-nitro-2H-indazole was obtained by using triethyltyloxonium tetrafluoroborate instead of trimethyloxonium tetrafluoroborate.
- Example 80 5-((1- (1,3,4-thiadiazol-2-yl) -1H-indol-4-yl) amino) -3-(((1R, 2S) -2-aminocyclohexyl) amino) -1 , 2,4-Triazine-6-carboxamide
- 2-bromo-1,3,4-thiadiazole is used instead of 3-iodo-1-methyl-1H-pyrazole Gave the title compound as a pale yellow solid.
- Example 81 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((7-chloro-2-ethyl-2H-indazol-5-yl) amino) -1,2,4-triazine-6 Carboxamide (step 1) 5-Bromo-7-chloro-1H-indazole 2.5 g of triethyloxonium tetrafluoroborate was added to a solution of 1.38 g of ethyl acetate in 15 mL, and the reaction solution was stirred at room temperature overnight.
- the reaction mixture was diluted with ethyl acetate, washed successively with water and saturated brine, and dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (developing solvent: hexane / ethyl acetate) to obtain 5-bromo-7-chloro-2-ethyl-2H-indazole.
- Example 82 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2,7-dimethyl-2H-indazol-5-yl) amino) -1,2,4-triazine-6-carboxamide
- steps 1 to 3 5-bromo-7-methyl-1H-indazole was used instead of 5-bromo-7-chloro-1H-indazole and trimethyloxonium tetrafluoroborate was replaced with trimethyl
- the title compound was obtained as a yellow solid by using oxonium tetrafluoroborate.
- Example 83 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((7-methyl-2- (1-methylcyclobutyl) -2H-indazol-5-yl) amino) -1,2 , 4-Triazine-6-carboxamide (Step 1) To 633 mg of 5-bromo-7-methyl-1H-indazole, 3 mL of 1-methyl-1-cyclobutanol and 0.1 mL of concentrated sulfuric acid were added, and the reaction solution was reacted at 100 ° C. for 30 minutes using a microwave reactor. Thereafter, the mixture was further reacted at 120 ° C. for 30 minutes.
- Example 84 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (pyrazin-2-yl) -1H-indol-4-yl) amino) -1,2,4-triazine According to -6-carboxamide Example 25 (Steps 1 to 3), 2-bromopyrazine was used instead of 3-iodo-1-methyl-1H-pyrazole to obtain the title compound as a pale yellow solid.
- Example 85 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((7-cyclopropyl-2-methyl-2H-indazol-5-yl) amino) -1,2,4-triazine- 6-Carboxamide (Step 1) 361 mg of 7-bromo-2-methyl-5-nitro-2H-indazole obtained in Example 65 (Step 1), 347 mg of cyclopropylboronic acid, 31 mg of palladium (II) acetate, 99 mg of tricyclohexylphosphine tetrafluoroborate, Potassium phosphate 1 g of toluene 20 mL and water 1 mL were added, and the reaction solution was stirred at 100 ° C.
- Example 86 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (difluoromethyl) -4-methyl-1H-indazol-6-yl) amino) -1,2,4- Triazine-6-carboxamide (Step 1) 1- (Difluoromethyl) -4-methyl-1H by using 934 mg of 4-methyl-6-nitro-1H-indazole instead of 6-nitro-1H-indazole according to Example 24 (Steps 1-2) 112 mg of indazole-6-amine and 2- (difluoromethyl) -4-methyl-2H-indazole-6-amine were obtained.
- Example 87 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (difluoromethyl) -1H-indazol-5-yl) amino) -1,2,4-triazine-6- Carboxamide (Step 1) According to Example 24 (Steps 1-2), the title compound 1- (difluoromethyl) -1H-indazole-5-amine and 5-nitro-1H-indazole was used instead of 6-nitro-1H-indazole 2- (Difluoromethyl) -4-methyl-2H-indazol-5-amine was obtained.
- Example 88 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-ethyl-7-methyl-1H-indazol-5-yl) amino) -1,2,4-triazine-6 Carboxamide (step 1) According to Example 70 (Step 2), 5-bromo-1-ethyl-7-methyl obtained in Example 70 (Step 1) instead of 5-bromo-2-ethyl-7-methyl-2H-indazole By using 374 mg of -1H-indazole, 1-ethyl-7-methyl-1H-indazole-5-amine was obtained.
- Example 89 3-(((2S, 3R) -2-amino-5-methylhexane-3-yl) amino) -5-((2-cyclobutyl-2H-indazol-5-yl) amino) -1,2,4 -Triazine-6-carboxamide (Step 1) To 5.00 g of methyl 3-formyl-4-nitrobenzoate were added 30 mL of toluene and 2.25 mL of cyclobutylamine, and the mixture was stirred at room temperature for 5 minutes. After distilling off the solvent under reduced pressure, 11.3 mL of triethyl phosphite was added to the residue, and the reaction solution was stirred at 100 ° C. for 7 hours.
- Example 90 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (3-methylpyrazin-2-yl) -1H-indol-4-yl) amino) -1,2, 4-Triazine-6-carboxamide
- 2-bromo-3-methylpyrazine was used in place of 3-iodo-1-methyl-1H-pyrazole to give the title compound lightly. Obtained as a yellow solid.
- Example 91 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2,4-dimethyl-2H-indazol-6-yl) amino) -1,2,4-triazine-6-carboxamide 2-formate (step 1)
- 4-methyl-6-nitro-1H-indazole is used instead of 6-nitro-1H-indazole
- iodomethane is used instead of sodium chlorodifluoroacetate to give 1 4-dimethyl-1H-indazole-6-amine and 2,4-dimethyl-2H-indazole-6-amine were obtained.
- Process 2 The title compound was obtained as a yellow solid by using 2,4-dimethyl-2H-indazole-6-amine instead of 6-aminoquinoline according to Example 1 (Steps 1 to 4).
- Example 92 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((6-methyl-1- (tetrahydro-2H-pyran-4-yl) -1H-indol-4-yl) amino) -1,2,4-triazine-6-carboxamide
- steps 1 to 3 instead of 4-nitro-1H-indole used, 6-methyl-4-nitro-1H-indole was Substituting tetrahydro-2H-pyran-4-ol for S) -tetrahydrofuran-3-ol gave the title compound as a pale yellow solid.
- Example 93 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (2-hydroxyethyl) -1H-indol-4-yl) amino) -1,2,4-triazine- 6-carboxamide
- 2- (tert-butyldimethylsiloxy) ethanol was used in place of (S) -tetrahydrofuran-3-ol to give the title compound as a pale yellow solid. It was.
- Example 94 3-(((1R, 2S) -2-aminocycloheptyl) amino) -5-((1- (2-methoxyethyl) -1H-indol-4-yl) amino) -1,2,4-triazine -6-Carboxamide (Step 1) 2.16 g of di-tert-butyl dicarbonate was added to 20 mL of methanol of 1 g of (1S, 2S) -aminocycloheptanol, and the reaction solution was stirred at room temperature for 10 minutes, and then the solvent was distilled off under reduced pressure.
- Example 95 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1,7-dimethyl-1H-indazol-5-yl) amino) -1,2,4-triazine-6-carboxamide (Process 1)
- Example 70 Step 1
- 5-bromo-2,7-dimethyl-2H-indazole was obtained by using iodomethane instead of iodoethane.
- Step 2 In accordance with Example 88 (Steps 1 and 2), 5-bromo-2,7-dimethyl-2H-indazole obtained in the above step was used instead of 5-bromo-1-ethyl-7-methyl-1H-indazole. Used to obtain the title compound as a yellow solid.
- Example 96 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((7-methyl-2- (2,2,2-trifluoroethyl) -2H-indazol-5-yl) amino) -1,2,4-triazine-6-carboxamide (Step 1)
- Step 1 5-bromo-7-methyl-1- (2,2,2-trimethyl) was obtained by using 1-iodo-2,2,2-trifluoroethane instead of iodoethane.
- Fluoroethyl) -1H-indazole and 5-bromo-7-methyl-2- (2,2,2-trifluoroethyl) -2H-indazole were obtained.
- Example 97 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-ethyl-4-methyl-1H-indazol-6-yl) amino) -1,2,4-triazine-6 Carboxamide (step 1) 4-methyl-6-nitro-1H-indazole, which can be synthesized by the method described in the patent (WO2009 / 084695), 1.2 ml of iodoethane and 600 mg of sodium hydride (60% in oil) are added to a solution of 1.77 g of DMF in 17 ml. The reaction solution was stirred at room temperature for 15 minutes.
- reaction mixture was diluted with ethyl acetate, washed twice with water, and then washed with saturated brine. After drying over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: hexane / ethyl acetate) to give 1-ethyl-4-methyl-6-nitro-1H. -Indazole and 2-ethyl-4-methyl-6-nitro-2H-indazole were each obtained as a pale yellow solid.
- the title compound was obtained by using tert-butyl ((1S, 2R) -2-aminocyclohexyl) carbamate instead of tert-butyl ((3R, 4R) -4-aminotetrahydro-2H-pyran-3-yl) carbamate.
- tert-butyl ((1S, 2R) -2-aminocyclohexyl) carbamate instead of tert-butyl ((3R, 4R) -4-aminotetrahydro-2H-pyran-3-yl) carbamate.
- Example 98 3-(((3R, 4R) -3-Aminotetrahydro-2H-pyran-4-yl) amino) -5-((1-ethyl-6- (2-hydroxypropan-2-yl) -1H-indazole) -4-yl) amino) -1,2,4-triazine-6-carboxamide (Step 1)
- Step 1 methyl 4-bromo-1H-indazole-6-carbochelate was used instead of 5-nitro-1H-indazole, and 2-bromo-5-
- methyl 4-bromo-1-ethyl-1H-indazole-6-carbochelate was obtained.
- Step 2 Methyl 4-bromo-1-ethyl-1H-indazole-6-carbochelate obtained in the above Step 1 was added with 2.5 ml of methylmagnesium chloride (3M THF solution) to a solution of 0.70 g of THF in 20 mL, and the reaction solution was mixed. Stir at room temperature for 3 hours. A saturated aqueous ammonium chloride solution was added to the reaction solution, and the mixture was extracted with ethyl acetate.
- 3M THF solution methylmagnesium chloride
- Example 99 3-((((3R, 4R) -3-Aminotetrahydro-2H-pyran-4-yl) amino) -5-((2- (1-hydroxy-2-methylpropan-2-yl) -7-methyl -2H-indazol-5-yl) amino) -1,2,4-triazine-6-carboxamide (Step 1) 5-Bromo-7-methyl-1H-indazole To a solution of 2.11 g of DMF in 20 mL was added 6.52 g of cesium carbonate and 2.2 mL of ethyl 2-bromoisobutyrate, and the reaction solution was stirred at room temperature for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- Example 100 3-(((3R, 4R) -3-Aminotetrahydro-2H-pyran-4-yl) amino) -5-((2- (tert-butyl) -7-chloro-2H-indazol-5-yl) Amino) -1,2,4-triazine-6-carboxamide (Step 1)
- Step 1 5-bromo-7-chloro-1H-indazole was used instead of 5-bromo-7-methyl-1H-indazole, and instead of 1-methyl-1-cyclobutanol 2- (tert-butyl) -7-chloro-2H-indazol-5-amine was obtained by using 2-methyl-2-propanol.
- Example 101 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-ethyl-4-methyl-2H-indazol-6-yl) amino) -1,2,4-triazine-6 Carboxamide
- 2-ethyl-4-methyl obtained in Example 97 (Step 1) instead of 1-ethyl-4-methyl-6-nitro-1H-indazole
- the title compound was obtained as a yellow solid by using -6-nitro-2H-indazole.
- Example 104 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2- (tert-butyl) -2H-indazol-5-yl) amino) -1,2,4-triazine-6 Carboxamide
- tert-butylamine instead of cyclobutylamine
- tert-butyl ((2S, 3R) -3-amino-5-methylhexane-2-yl) carbamate The title compound was obtained as a pale yellow solid by using tert-butyl ((1S, 2R) -2-aminocyclohexyl) carbamate instead.
- Example 107 3-((((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-ethyl-1H-indazol-4-yl) amino) -1,2,4-triazine-6-carboxamide (step 1) According to Example 68 (Step 2), using 4-nitro-1H-indazole instead of 4-nitro-1H-indole and using iodoethane instead of (S) -tetrahydrofuran-3-yl methanesulfonate. 1-Ethyl-1H-indazole-4-amine was obtained.
- Example 108 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-cyclopentyl-2H-indazol-5-yl) amino) -1,2,4-triazine-6-carboxamide
- steps 1 to 3 using cyclopentylamine in place of cyclobutylamine and tert-butyl in place of tert-butyl ((2S, 3R) -3-amino-5-methylhexane-2-yl) carbamate
- the title compound was obtained as a pale yellow solid by using ((1S, 2R) -2-aminocyclohexyl) carbamate.
- Example 109 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-ethyl-4-methoxy-2H-indazol-6-yl) amino) -1,2,4-triazine-6 Carboxamide (step 1)
- Step 1 instead of 5-bromo-7-methyl-1H-indazole, 329 mg of 6-bromo-4-methoxy-1H-indazole was used to give 6-bromo-1-ethyl-4 210 mg of -methoxy-1H-indazole and 6-bromo-2-ethyl-4-methoxy-2H-indazole were obtained.
- Example 110 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-ethyl-4-methoxy-1H-indazol-6-yl) amino) -1,2,4-triazine-6 Carboxamide
- Step 2 instead of 5-bromo-2-ethyl-7-methyl-2H-indazole, 6-bromo-1-- obtained in Example 109 (Step 1) Ethyl-4-methoxy-1H-indazole was used to give the title compound as a pale yellow solid.
- Example 111 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (2,6-dimethoxypyrimidin-4-yl) -1H-indol-4-yl) amino) -1, 2,4-Triazine-6-carboxamide
- Example 25 steps 1 to 3
- 4-chloro-2,6-dimethoxypyrimidine instead of 3-iodo-1-methyl-1H-pyrazole
- Example 112 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((5- (pyrimidin-2-yl) thiophen-3-yl) amino) -1,2,4-triazine-6- Carboxamide (Step 1) To a solution of 9.8 g of thiophene-3-carboxylic acid in 40 mL of acetic acid was added a solution of 4.4 mL of bromine in 30 mL of acetic acid at room temperature over 30 minutes.
- Example 113 3-(((3R, 4R) -3-Aminotetrahydro-2H-pyran-4-yl) amino) -5-((7-methyl-2- (tert-pentyl) -2H-indazol-5-yl) Amino) -1,2,4-triazine-6-carboxamide (Step 1)
- Step 83 Steps 1-2
- 7-methyl-2- (tert-pentyl) -2H-indazole was obtained by using 2-methylbutan-2-ol instead of 1-methyl-1-cyclobutanol.
- -5-amine was obtained.
- Example 114 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (4-methoxypyrimidin-2-yl) -1H-indol-4-yl) amino) -1,2, 4-Triazine-6-carboxamide
- 2-chloro-4-methoxypyrimidine was used in place of 3-iodo-1-methyl-1H-pyrazole to give the title compound lightly. Obtained as a yellow solid.
- Example 115 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-methyl-4- (trifluoromethyl) -2H-indazol-6-yl) amino) -1,2,4 -Triazine-6-carboxamide (Step 1) According to Example 24 (Steps 1-2), instead of 6-nitro-1H-indazole, 4- (trifluoromethyl) -6-nitro-1H-indazole obtained in the patent (WO2009 / 064695) was used.
- Example 116 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((7-methyl-1- (2,2,2-trifluoroethyl) -1H-indazol-5-yl) amino) -1,2,4-Triazine-6-carboxamide
- Example 70 instead of 5-bromo-2-ethyl-7-methyl-2H-indazole in Example 96 (Step 1)
- the title compound was obtained as a pale yellow solid.
- Example 117 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2- (tetrahydro-2H-pyran-4-yl) -2H-indazol-5-yl) amino) -1,2 , 4-Triazine-6-carboxamide (Step 1)
- 1- (Tetrahydro-2H-pyran-4-yl) -1H-indazole-5 was obtained by using tetrahydro-2H-pyran-4-ol instead of cyclobutanol according to Example 11 (Steps 1-2).
- -Amine and 2- (tetrahydro-2H-pyran-4-yl) -2H-indazol-5-amine were obtained.
- Example 120 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-phenyl-2H-indazol-5-yl) amino) -1,2,4-triazine-6-carboxamide
- aniline is used instead of cyclobutylamine, and tert-butyl ((2S, 3R) -3-amino-5-methylhexane-2-yl) carbamate is replaced with tert-butyl (
- (1S, 2R) -2-aminocyclohexyl) carbamate the title compound was obtained as a pale yellow solid.
- Example 121 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (pyridin-3-yl) -1H-indazol-4-yl) amino) -1,2,4-triazine -6-carboxamide
- 4-nitro-1H-indazole was used instead of 4-nitro-1H-indole
- 3-iodo-1-methyl-1H-pyrazole was used instead The title compound was obtained as a pale yellow solid by using 3-iodopyridine.
- Example 122 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (1-methyl-1H-pyrazol-4-yl) -1H-indazol-4-yl) amino) -1 , 2,4-Triazine-6-carboxamide
- 4-nitro-1H-indazole was used instead of 4-nitro-1H-indole, and 3-iodo-1-methyl-
- the title compound was obtained as a pale yellow solid by using 4-iodo-1-methyl-1H-pyrazole instead of 1H-pyrazole.
- Example 123 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-cyclobutyl-4-methyl-1H-indazol-6-yl) amino) -1,2,4-triazine-6 Carboxamide dihydrochloride (Step 1) According to Example 24 (Steps 1-2), using 4-methyl-6-nitro-1H-indazole instead of 6-nitro-1H-indazole and bromocyclobutane instead of sodium chlorodifluoroacetate, 1-cyclobutyl-4-methyl-1H-indazole-6-amine and 2-cyclobutyl-4-methyl-2H-indazole-6-amine were obtained.
- Example 125 3-((((3R, 4R) -3-aminotetrahydro-2H-pyran-4-yl) amino) -5-((2- (difluoromethyl) -2H-indazol-5-yl) amino) -1, 2,4-Triazine-6-carboxamide (Step 1) 2- (Difluoromethyl) -5-nitro-2H obtained according to Example 62 (Step 1) by using 5-nitro-1H-indazole instead of 5-bromo-7-methyl-1H-indazole -To 429 mg of indazole, 20 mL of ethyl acetate was added, 300 mg of 10% palladium carbon was added, and the reaction solution was stirred at room temperature in a hydrogen atmosphere for 4 hours.
- Example 126 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-((S) -tetrahydrofuran-3-yl) -1H-indazol-4-yl) amino) -1,2 , 4-Triazine-6-carboxamide
- 4-nitro-1H-indazole was used instead of 4-nitro-1H-indole to give the title compound as a pale yellow solid It was.
- Example 130 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (tetrahydro-2H-pyran-4-yl) -1H-indazol-4-yl) amino) -1,2 , 4-Triazine-6-carboxamide
- tetrahydro-2H-pyran-4-ol was used instead of (S) -tetrahydrofuran-3-ol, and 4-nitro-1H-
- the title compound was obtained as a pale yellow solid by using 4-nitro-1H-indazole instead of indole.
- Example 133 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((2- (tert-butyl) -2H-indazol-6-yl) amino) -1,2,4-triazine-6 Carboxamide (step 1)
- 2,4-Dinitrobenzaldehyde 2 mL of tert-butylamine was added to 10 mL of 1.96 g of ethanol, and the reaction solution was stirred at 40 ° C. for 30 minutes. After the solvent was distilled off under reduced pressure, 1 mL of tert-butylamine was added to the resulting residue, and the mixture was heated at 50 ° C. for 30 minutes.
- Example 134 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (pyridin-4-ylmethyl) -1H-indazol-4-yl) amino) -1,2,4-triazine -6-carboxamide
- 4-nitro-1H-indazole was used instead of 4-nitro-1H-indole
- (S) -tetrahydrofuran-3-yl methanesulfonate was used instead.
- the title compound was obtained as a pale yellow solid by using 4- (chloromethyl) pyridine hydrochloride.
- Example 135 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (2-ethoxyethyl) -1H-indazol-4-yl) amino) -1,2,4-triazine- According to 6-carboxamide Example 68 (steps 2 to 3), 4-nitro-1H-indazole was used instead of 4-nitro-1H-indole, and (S) -tetrahydrofuran-3-yl methanesulfonate was used instead. By using bromoethyl ethyl ether, the title compound was obtained as a pale yellow solid.
- Example 136 3-((((3R, 4R) -3-aminotetrahydro-2H-pyran-4-yl) amino) -5-((2,7-diethyl-3-methyl-2H-indazol-5-yl) amino) -1,2,4-Triazine-6-carboxamide
- Example 81 Steps 1 to 3
- 5-bromo-7-ethyl-in place of 5-bromo-7-chloro-2-ethyl-2H-indazole The title compound was obtained as a pale yellow solid by using 3-methyl-1H-indazole.
- Example 139 3-(((3R, 4R) -3-Aminotetrahydro-2H-pyran-4-yl) amino) -5-((7-chloro-1-isobutyl-1H-indazol-5-yl) amino) -1 , 2,4-Triazine-6-carboxamide (Step 1)
- 5-bromo-7-chloro-1H-indazole was used instead of 5-bromo-7-methyl-1H-indazole, and 1-iodo-2- By using methylpropane, 7-chloro-1-isobutyl-1H-indazole-5-amine was obtained.
- Example 141 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (pyrazin-2-yl) -1H-indazol-4-yl) amino) -1,2,4-triazine -6-carboxamide
- 4-nitro-1H-indazole was used instead of 4-nitro-1H-indole
- 3-iodo-1-methyl-1H-pyrazole was used instead The title compound was obtained as a pale yellow solid by using 2-bromopyrazine.
- Example 142 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (2-oxotetrahydrofuran-3-yl) -1H-indol-4-yl) amino) -1,2, 4-Triazine-6-carboxamide dihydrochloride According to Example 68 (Steps 1-3), substituting 2-oxotetrahydrofuran-3-ol for (S) -tetrahydrofuran-3-ol gave the title The compound was obtained as a pale yellow solid.
- Example 144 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (pyridin-3-yl) -1H-indol-4-yl) amino) -1,2,4-triazine According to -6-carboxamide Example 25 (Steps 1 to 3), 3-iodopyridine was used in place of 3-iodo-1-methyl-1H-pyrazole to obtain the title compound as a pale yellow solid.
- Example 145 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (thiazol-2-yl) -1H-indol-4-yl) amino) -1,2,4-triazine According to -6-carboxamide Example 25 (Steps 1 to 3), 2-bromothiazole was used in place of 3-iodo-1-methyl-1H-pyrazole to obtain the title compound as a pale yellow solid.
- Example 147 3-((((1R, 2S) -2-aminocyclohexyl) amino) -5-((2-phenyl-2H-indazol-6-yl) amino) -1,2,4-triazine-6-carboxamide (step 1) According to Example 133 (Steps 1-2), 2-phenyl-2H-indazol-6-amine was obtained as a white solid by using aniline instead of tert-butylamine.
- Example 148 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-cyclopentyl-1H-indazol-4-yl) amino) -1,2,4-triazine-6-carboxamide
- Examples 68 (Steps 2 to 3), 4-nitro-1H-indazole is used instead of 4-nitro-1H-indole, and bromocyclopentane is used instead of (S) -tetrahydrofuran-3-yl methanesulfonate This gave the title compound as a pale yellow solid.
- Example 149 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-cyclohexyl-1H-indazol-4-yl) amino) -1,2,4-triazine-6-carboxamide
- Examples 68 (Steps 2 and 3), using 4-nitro-1H-indazole instead of 4-nitro-1H-indole and using bromocyclohexane instead of (S) -tetrahydrofuran-3-yl methanesulfonate Gave the title compound as a pale yellow solid.
- Example 150 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1-benzyl-1H-indazol-4-yl) amino) -1,2,4-triazine-6-carboxamide
- Examples 68 (Steps 2 and 3), using 4-nitro-1H-indazole instead of 4-nitro-1H-indole and using benzyl bromide instead of (S) -tetrahydrofuran-3-yl methanesulfonate Gave the title compound as a pale yellow solid.
- Example 151 3-(((1R, 2S) -2-aminocyclohexyl) amino) -5-((1- (pyridin-3-ylmethyl) -1H-indazol-4-yl) amino) -1,2,4-triazine -6-carboxamide
- 4-nitro-1H-indazole was used instead of 4-nitro-1H-indole
- (S) -tetrahydrofuran-3-yl methanesulfonate was used instead.
- the title compound was obtained as a pale yellow solid by using 3-picolyl chloride hydrochloride.
- Tables 12 to 94 show chemical structural formulas and physical property values of the compounds of Examples 1 to 414.
- Test Example 1 Syk Kinase Inhibition Test
- the purified human Syk protein used for the test was purchased from Carna Biosciences.
- DMSO dimethyl sulfoxide
- purified human Syk protein, FL- in reaction buffer (20 mM Tris-HCl pH 7.5, 20 mM NaCl, 2.5 mM DTT, 0.02% Tween-20, 1% Glycerol, 0.01 mM Pefabloc).
- Peptide 22 (final concentration is 1 uM) (caliper life science), magnesium chloride (final concentration is 5 mM), ATP (final concentration is 30 uM) and the present compound DMSO solution (DMSO final concentration is 5%) and 25
- the kinase reaction was performed by incubating at 30 ° C. for 30 minutes.
- EDTA final concentration is 30 mM diluted with Separation Buffer (Caliper Life Science) was added thereto to stop the kinase reaction.
- the amount of each phosphorylated peptide and non-phosphorylated peptide was measured with the LabChip TM 3000 system (Caliper Life Sciences, excitation wavelength 488 nm, detection wavelength 530 nm), and the amount of each phosphorylation reaction amount was measured.
- the compound concentration capable of suppressing the phosphorylation reaction by 50% was defined as the IC 50 value ( ⁇ M).
- known 1,2,4-triazine-6-carboxamide derivatives A, B and C (WO2000 / 075113 (A: Compound No. 121, B: Compound No. 154) and WO2000 / 076980 (C: Compound No. 39)) was synthesized, and the IC 50 value ( ⁇ M) was similarly calculated and compared with the compound of the present invention.
- Test Example 2 Cell Proliferation Inhibition Test
- SU-DHL-6 cells a diffuse large cell lymphoma cell line
- SU-DHL-6 cells ATCC, Cat #: CRL-2959
- RPMI 1640 medium ATCC, Cat #: 30-2001
- RPMI 1640 medium ATCC, Cat #: 30-2001
- FBS fetal bovine serum
- NUNC 96-well flat bottom microplates
- the compound of the present invention was serially diluted with dimethyl sulfoxide and added to RPMI 1640 medium containing 10% FBS.
- IC 50 value ( ⁇ M) of the above control compound was also calculated and compared with the compound of the present invention.
- Cell growth inhibition rate (%) (C ⁇ T) / C ⁇ 100 T: luminescence amount of well added with test compound (count per second)
- Tables 95 and 96 show the results of the Syk kinase inhibition test of the representative compound and the control compound in the present invention and the cell growth inhibition test on SU-DHL6 cells.
- the compound of the present invention has stronger Syk inhibitory activity and cell growth inhibitory effect than the control compounds A, B, and C. From this result, it was proved that the compound represented by the general formula (I) found in the present invention is a stronger Syk inhibitor than known triazine derivatives.
- Test Example 3 Confirmation of KDR Kinase Activity Inhibitory Action
- FL-Peptide 22 was added to the Caliper Life Science LabChip TM series reagent consumables price list. Since it was described that it corresponds as a substrate peptide in the activity measurement, it was referred.
- the purified recombinant human KDR protein used for the test is an in-house purified product.
- the compound of the present invention was serially diluted with dimethyl sulfoxide (DMSO).
- a buffer solution for reaction 100 mM HEPES pH 7.5, 1 mM
- a dephosphorase inhibitor cocktail PhosSTOP, Roche
- a protease inhibitor cocktail Complete Mini, EDTA-free, Roche
- Purified human KDR protein, FL-Peptide 22 final concentration is 1.5 uM
- magnesium chloride final concentration is 10 mM
- DTT 0.003% Briji35, 0.04% Tween-20
- CHAPSO ATP
- DMSO solution of the present invention final concentration of DMSO is 5%
- EDTA final concentration 30 mM
- Separation Buffer of Caliper Life Sciences was added to stop the kinase reaction.
- the amount of each phosphorylated peptide and non-phosphorylated peptide was measured with the LabChip TM 3000 system (Caliper Life Sciences, excitation wavelength 488 nm, detection wavelength 530 nm), and the amount of each phosphorylation reaction amount was measured.
- the compound concentration capable of suppressing the phosphorylation reaction by 50% was defined as an IC 50 value (nM) and shown in Table 97 below.
- the IC 50 value ( ⁇ M) of R406 (Rigel) which is an existing Syk inhibitor, was also calculated as a control compound and compared with the compound of the present invention.
- Test Example 4 Confirmation of Aurora B Kinase Activity Inhibitory Action
- the method described in the published patent publication Japanese Patent Application Laid-Open No. 2008-81492 was referred to for the in vitro inhibitory activity measurement method of the above compound for Aurora B kinase activity.
- the purified recombinant human Aurora B protein used for the test was purchased from Carna Bioscience.
- the compound of the present invention was serially diluted with dimethyl sulfoxide (DMSO).
- DMSO dimethyl sulfoxide
- the amount of phosphorylation reaction is determined from the degree of fluorescence polarization obtained by measurement with PHERAstar (BMG LABTECH, excitation wavelength 485 nm, detection wavelength 520 nm), and the phosphorylation reaction is suppressed by 50%.
- the compound concentration that can be defined is defined as IC 50 value (nM) and is shown in Table 97 below.
- IC 50 value ( ⁇ M) of R406 (Rigel), which is an existing Syk inhibitor was also calculated as a control compound and compared with the compound of the present invention.
- the compound of the present invention does not inhibit other kinases that may cause side effects as compared with the control compound, and has a remarkable Syk selectivity. From this result, it was shown that the compound represented by the general formula (I) found in the present invention has less side effects than the known Syk inhibitor.
Abstract
Description
なお、Syk阻害剤は、がんの他に、自己免疫疾患(関節リウマチ、全身性エリテマトーデス、ネフローゼ症候群等)、アレルギー疾患(気管支喘息、アレルギー性鼻炎、アトピー性皮膚炎、接触性皮膚炎、じんましん、食物アレルギー、結膜炎等)や慢性閉塞性肺疾患(COPD)などに対する治療剤となり得ることも報告されている(非特許文献3)。
また、その他のSyk阻害剤として、ヘテロ芳香族-カルボキサミド誘導体が報告されているが(特許文献1)、Syk阻害活性は十分なものではなかった。また、特許文献2には、1,2,4-トリアジン-6-カルボキサミド誘導体が報告されているが、Syk阻害活性は記載がない。
従って、本発明の課題は、選択的かつ強力にSykを阻害する新規化合物又はその塩を提供することにある。

Aは、Raが置換していてもよいC1-C8アルキル基、Raが置換していてもよいC2-C6アルケニル基、Raが置換していてもよいC2-C6アルキニル基、Rbが置換していてもよいC3-C10シクロアルキル基、Rbが置換していてもよいC6-C14芳香族炭化水素基、Rbが置換していてもよい4~10員の飽和複素環基又はRbが置換していてもよい4~10員の不飽和複素環基を示すか、R1及びそれらが結合する窒素原子と一緒になって4~10員の飽和複素環若しくは4~10員の不飽和複素環を形成してもよく;
Raは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、-C(=O)ORx、-C(=O)SRx、-C(=S)ORx、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、-SO2N(Rx)(Ry)、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の飽和複素環基又は4~10員の不飽和複素環基を示し(ここでC3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の飽和複素環基及び4~10員の不飽和複素環基は、重水素原子、ハロゲン原子、ヒドロキシル基、シアノ基、ニトロ基、アミノ基、オキソ基、オキシド基、イミノ基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C1-C6アルコキシ基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれるそれぞれ独立した1~3個の基が置換していてもよい);
Rbは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、オキソ基、オキシド基、-C(=O)ORx、-C(=O)SRx、-C(=S)ORx、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、-SO2N(Rx)(Ry)、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、又はC2-C6アルキニル基を示し;
R2は、水素原子又はRaが置換していてもよいC1-C6アルキル基を示し;
Bは、Rcが置換していてもよい不飽和複素環基を示し;
Rcは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、オキソ基、オキシド基、-C(=O)Rx、-C(=O)ORx、-C(=O)N(Rx)(Ry)、-C(=O)SRx、-C(=S)ORx、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、-SO2N(Rx)(Ry)、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、C2-C6アルキニル基、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の飽和複素環基又は4~10員の不飽和複素環基を示し(ここでC1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基及びC2-C6アルキニル基は、シアノ基、ニトロ基、-N(Rx)(Ry)、及び-ORxからなる群から選ばれるれぞれ独立した1~3個の基が置換していてもよく、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の飽和複素環基及び4~10員の不飽和複素環基は、Rd、オキソ基、オキシド基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれるそれぞれ独立した1~3個の基が置換していてもよい);
Rdは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、-C(=O)Rx、-C(=O)ORx、-C(=O)N(Rx)(Ry)、-C(=O)SRx、-C(=S)ORx、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、又は-SO2N(Rx)(Ry)を示し;
Rx、Ry及びRzは、同一又は相異なって、水素原子、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、C2-C6アルキニル基、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基4~10員の飽和複素環基又は4~10員の不飽和複素環基を示す。)で表される化合物又はその塩を提供するものである。
また、本発明は、上記一般式(I)で表される化合物を有効成分とするSyk阻害剤、抗腫瘍剤等の医薬を提供するものである。
また、本発明は、上記一般式(1)で表される化合物又はその塩を含有する医薬を提供するものである。
また、本発明は、上記一般式(1)で表される化合物又はその塩及び薬学的に許容される担体を含有する医薬組成物を提供するものである。
また、本発明は、腫瘍治療用の、上記一般式(1)で表される化合物又はその塩を提供するものである。
また、本発明は、抗腫瘍剤製造のための、一般式(1)で表される化合物又はその塩の使用を提供するものである。
又、本発明は、一般式(1)で表される化合物又はその塩を投与することを特徴とする腫瘍の治療方法を提供するものである。
本発明化合物又はその塩は、優れたSyk阻害活性を有し、且つ癌細胞株に対する増殖抑制効果を示すことが明らかとなった。また、Sykに対する優れた選択性から他のキナーゼによる副作用が少ないという利点を有する。従って、本発明化合物又はその塩は、腫瘍、特に癌の予防及び/又は治療剤として有用である。
本願明細書において「C1-C8アルキル基」とは、炭素数1~8の直鎖状若しくは分枝状のアルキル基を示し、具体的にはメチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基等が挙げられる。
本願明細書において「C1-C6アルキル基」とは、炭素数1~6の直鎖状若しくは分枝状のアルキル基を示し、具体的にはメチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、ヘキシル基等が挙げられる。
本願明細書において「C1-C6ハロアルキル基」とは、前記のC1-C6アルキル基の1個~全ての水素原子が前記のハロゲン原子で置換した基を示し、具体的にはモノフルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、1-フルオロエチル基、2-フルオロエチル基、1,1-ジフルオロエチル基、1,2-ジフルオロエチル基、2,2-ジフルオロエチル基等が挙げられる。
本願明細書において「C1-C6アルキルアミノ基」とは、アミノ基の1個又は2個の水素原子が前記のC1-C6アルキル基で置換した基を示し、具体的にはメチルアミノ基、エチルアミノ基、ジメチルアミノ基、ジエチルアミノ基、エチルメチルアミノ基等が挙げられる。
本願明細書において「C2-C6アルキニル基」とは、少なくとも一つの炭素-炭素三重結合を含む炭素数2~6の直鎖状若しくは分枝状のアルキニル基を示し、具体的にはエチニル基、2-プロピニル基等が挙げられる。
本願明細書において「C1-C6アルコキシ基」とは、炭素数1~6の直鎖状若しくは分枝状のアルコキシ基を示し、具体的にはメトキシ基、エトキシ基、n-プロポキシ基、イソプロポキシ基、n-ブトキシ基、イソブトキシ基、tert-ブトキシ基等が挙げられる。
本願明細書において「C2-C7アルカノイル基」とは、カルボニル基の水素原子が前記のC1-C6アルキル基で置換した基を示し、具体的にはアセチル基、n-プロパノイル基、イソプロパノイル基、n一ブチロイル基、tert-ブチロイル基等が挙げられる。
本願明細書において「C6-C14芳香族炭化水素基」とは、炭素数6~14の単環式若しくは多環式の芳香族炭化水素基を示し、具体的にはフェニル基、ナフチル基等が挙げられる。
一般式(I)中、Aで表される「Raが置換していてもよいC1-C8アルキル基」におけるRaとしては、ハロゲン原子、-N(Rx)(Ry)、-ORx、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基又は4~10員の飽和複素環基(ここでC3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基は、重水素原子、ハロゲン原子、ヒドロキシル基、シアノ基、ニトロ基、アミノ基、オキソ基、オキシド基、イミノ基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C1-C6アルコキシ基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい)が好ましく:
ハロゲン原子、アミノ基、C1-C6アルキルアミノ基、ヒドロキシル基、C1-C6アルコキシ基、C3-C10シクロアルキル基)、C6-C14芳香族炭化水素基、又は4~10員の飽和複素環基(ここでC3-C10シクロアルキル基及び4~10員の飽和複素環基は、アミノ基で置換されていてもよい)がさらに好ましく:
ハロゲン原子、アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)がさらに好ましく:
アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)が特に好ましい。
ここでRaの個数は、特に制限されず、1~5個が好ましい。
ハロゲン原子、-N(Rx)(Ry)、-ORx、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基(ここでC3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基は、重水素原子、ハロゲン原子、ヒドロキシル基、シアノ基、ニトロ基、アミノ基、オキソ基、オキシド基、イミノ基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C1-C6アルコキシ基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;-N(Rx)(Ry)又は-ORxからなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C1-C6アルキル基、-N(Rx)(Ry)又は-ORxからなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基がより好ましく、
ハロゲン原子、アミノ基、C1-C6アルキルアミノ基、ヒドロキシル基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基及び4~10員の飽和複素環基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;ヒドロキシル基、アミノ基又はC1-C6アルキルアミノ基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C1-C6アルキル基、ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基がより好ましく:
ハロゲン原子、アミノ基、C1-C6アルキルアミノ基、ヒドロキシル基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基及び4~10員の飽和複素環基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基又はC1-C6アルキルアミノ基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基がより好ましく:
ハロゲン原子、アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基がより好ましく:
アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;アミノ基が置換していてもよい4~10員の飽和複素環基がより好ましく:
アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC5-C7シクロアルキル基;アミノ基が置換していてもよいN及びOから選ばれる1個のヘテロ原子を有する4~6員の単環式の飽和複素環基がより好ましく:
アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC5-C7シクロアルキル基;アミノ基が置換していてもよいテトラヒドロピラニル基又はピペリジニル基が特に好ましい。
R1が水素原子又はC1-C6アルキル基であり、Aがハロゲン原子、-N(Rx)(Ry)、-ORx、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基(ここでC3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基は、重水素原子、ハロゲン原子、ヒドロキシル基、シアノ基、ニトロ基、アミノ基、オキソ基、オキシド基、イミノ基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C1-C6アルコキシ基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;-N(Rx)(Ry)又は-ORxからなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C1-C6アルキル基、-N(Rx)(Ry)又は-ORxからなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基であるか、R1、A及びそれらが結合する窒素原子と一緒になって4~10員の飽和複素環を形成する場合(ここで4~10員の飽和複素環は、-N(Rx)(Ry)及び-ORxからなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい)がより好ましく:
R1が水素原子又はC1-C6アルキル基であり、Aがハロゲン原子、アミノ基、C1-C6アルキルアミノ基、ヒドロキシル基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基及び4~10員の飽和複素環基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;ヒドロキシル基、アミノ基又はC1-C6アルキルアミノ基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C1-C6アルキル基、ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基であるか、R1、A及びそれらが結合する窒素原子と一緒になって4~10員の飽和複素環を形成する場合(ここで4~10員の飽和複素環は、アミノ基及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい)がより好ましく:
R1が水素原子又はC1-C6アルキル基であり、Aがハロゲン原子、アミノ基、C1-C6アルキルアミノ基、ヒドロキシル基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基及び4~10員の飽和複素環基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基又はC1-C6アルキルアミノ基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基であるか、R1、A及びそれらが結合する窒素原子と一緒になって4~10員の飽和複素環を形成する場合(ここで4~10員の飽和複素環は、アミノ基及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい)がより好ましく:
R1が水素原子又はC1-C6アルキル基であり、Aがハロゲン原子、アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基であるか、R1、A及びそれらが結合する窒素原子と一緒になって4~10員の飽和複素環を形成する場合がより好ましく:
R1が水素原子であり、Aがアミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;アミノ基が置換していてもよい4~10員の飽和複素環基である場合がより好ましく:
R1が水素原子であり、Aがアミノ基又はC1-C6アルコキシ基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;アミノ基が置換していてもよい4~10員の飽和複素環基である場合がより好ましく:
R1が水素原子であり、Aがアミノ基又はC1-C6アルコキシ基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC5-C7シクロアルキル基;アミノ基が置換していてもよいN及びOから選ばれる1個のヘテロ原子を有する4~6員の単環式の飽和複素環基である場合がより好ましく:
R1が水素原子であり、Aがアミノ基又はC1-C6アルコキシ基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC5-C7シクロアルキル基;アミノ基が置換していてもよいテトラヒドロピラニル基又はピペリジニル基である場合が特に好ましい。

ハロゲン原子;シアノ基;オキシド基;重水素原子、ハロゲン原子、-ORx、C6-C14芳香族炭化水素基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;C1-C6アルキル基及び-ORxからなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、-C(=O)Rx及び-ORxからなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基がより好ましく:
ハロゲン原子;シアノ基;オキシド基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基、C6-C14芳香族炭化水素基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基がより好ましく:
ハロゲン原子;シアノ基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基、C6-C14芳香族炭化水素基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基がより好ましく:
ハロゲン原子;シアノ基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基がより好ましく:
ハロゲン原子;重水素原子、ハロゲン原子及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又は4~10員の飽和複素環基がより好ましく:
ハロゲン原子;重水素原子、ハロゲン原子及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;1~3個のヒドロキシル基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;1~3個のC1-C6アルキル基が置換していてもよい4~10員の不飽和複素環基;又は4~10員の飽和複素環基が特に好ましい。
Rcが置換していてもよい、N、S及びOから選ばれる1~3個のヘテロ原子を有する5~6員の単環性の完全不飽和又は部分飽和である不飽和複素環基(ただし、3,4-メチレンジオキシフェニル基を除く)、又はN、S及びOから選ばれる1~3個のヘテロ原子を有する9~10員の2環性の完全不飽和又は部分飽和である不飽和複素環基(ただし、3,4-メチレンジオキシフェニル基を除く)がさらに好ましく:
Rcが置換していてもよい、N、S及びOから選ばれる1~3個のヘテロ原子を有する5~6員の単環性の完全不飽和である不飽和複素環基、又はN、S及びOから選ばれる1~3個のヘテロ原子を有する9~10員の2環性の完全不飽和である不飽和複素環基(ただし、3,4-メチレンジオキシフェニル基を除く)がさらに好ましく:
ハロゲン原子、シアノ基、オキシド基、C1-C6アルキル基、-C(=O)Rx、-C(=O)ORx、-C(=O)N(Rx)(Ry)、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基、又は4~10員の飽和複素環基(ここでC1-C6アルキル基は、Rdが置換していてもよく、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基は、Rd、オキソ基、オキシド基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C1-C6アルコキシ基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれる同一又は相異なった基が置換していてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、N、S及びOから選ばれる1~3個のヘテロ原子を有する5~6員の単環性の完全不飽和である不飽和複素環基、又はN、S及びOから選ばれる1~3個のヘテロ原子を有する9~10員の2環性の完全不飽和である不飽和複素環基がさらに好ましく:
ハロゲン原子;シアノ基;オキシド基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基、C6-C14芳香族炭化水素基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、チアゾリル基、イソチアゾリル基、チアジアゾリル基、ピラゾリル基、ピリジル基、インドリル基、インダゾリル基、イミダゾピリジル基、チエノピリジル基、ベンゾイミダゾリル基、ベンゾオキサゾリル基、ベンゾチアゾリル基、ベンゾチエニル基、ベンゾフラニル基、キノリル基、又はナフチリジル基がさらに好ましく:
ハロゲン原子;シアノ基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基、C6-C14芳香族炭化水素基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、チアゾリル基、イソチアゾリル基、チアジアゾリル基、ピラゾリル基、ピリジル基、インドリル基、インダゾリル基、イミダゾピリジル基、チエノピリジル基、ベンゾイミダゾリル基、ベンゾチアゾリル基、ベンゾチエニル基、ベンゾフラニル基又はキノリル基がさらに好ましく:
ハロゲン原子;シアノ基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、チアゾリル基、イソチアゾリル基、ピラゾリル基、ピリジル基、インドリル基、インダゾリル基、チエノピリジル基、ベンゾイミダゾリル基、ベンゾチアゾリル基、ベンゾチエニル基、ベンゾフラニル基又はキノリル基がさらに好ましく:
ハロゲン原子;重水素原子、ハロゲン原子及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;1~3個のヒドロキシル基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;1~3個のC1-C6アルキル基が置換していてもよい4~10員の不飽和複素環基;又は4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、インドリル基、インダゾリル基、ベンゾチエニル基、キノリル基が特に好ましい。
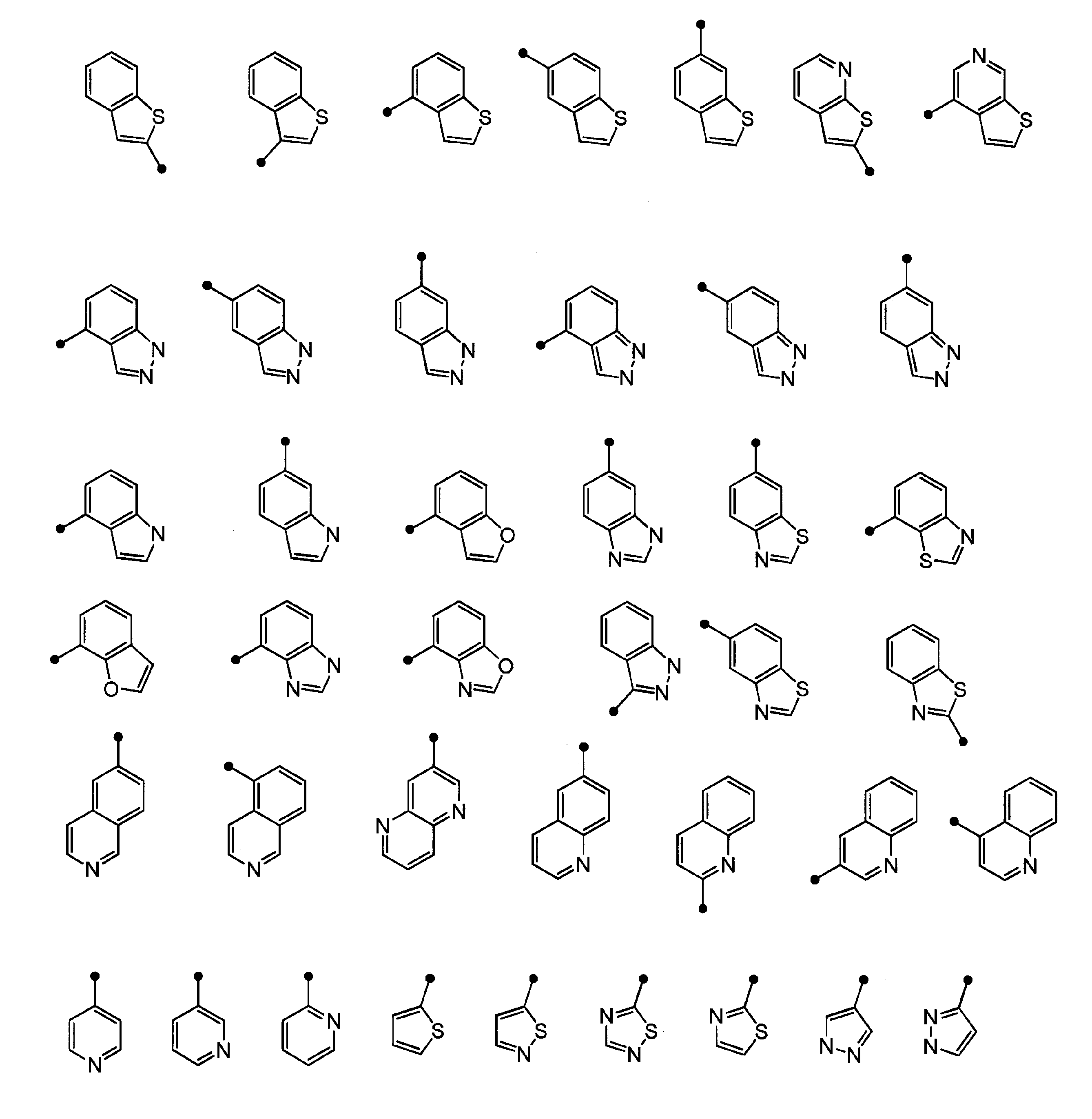
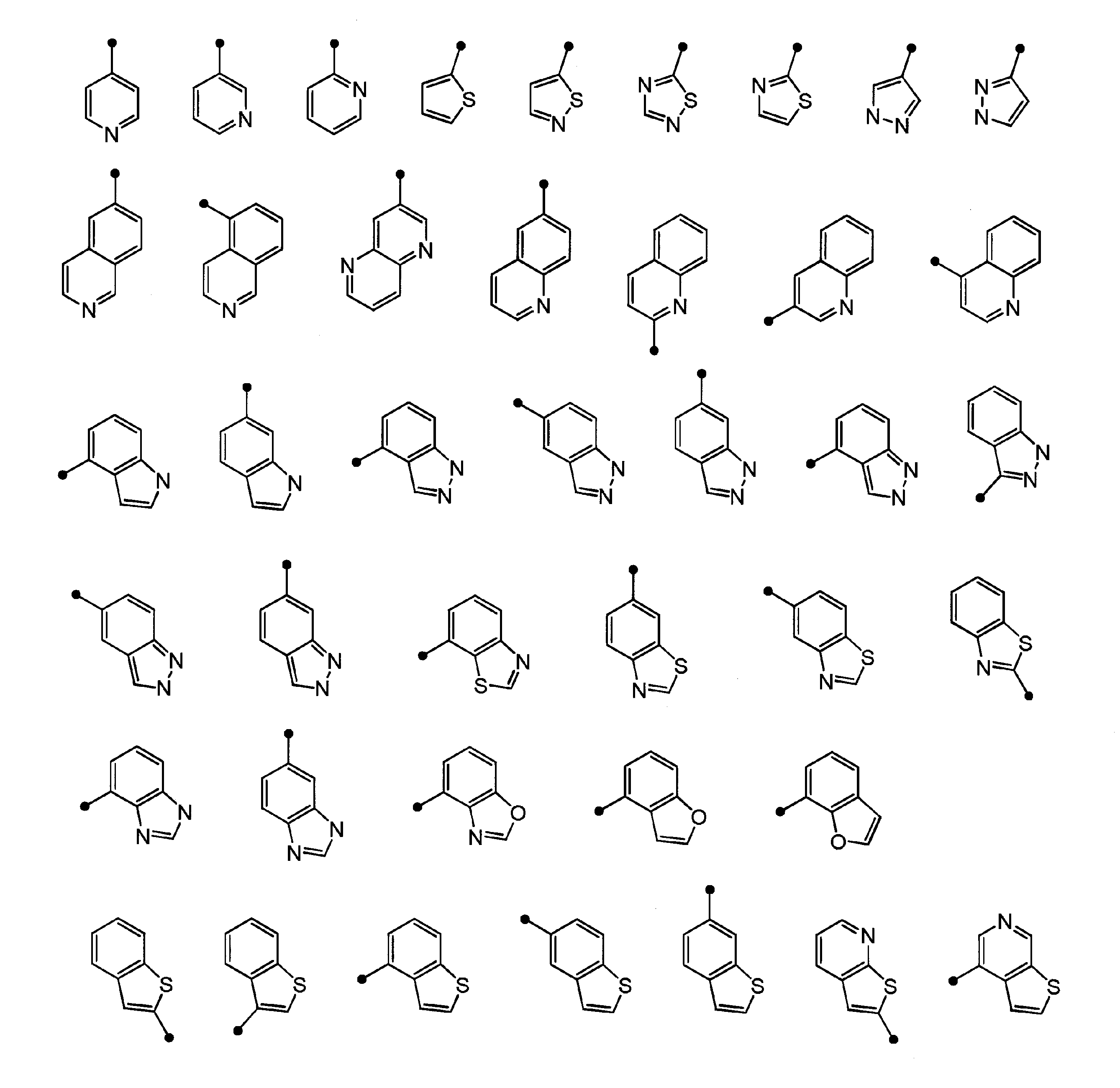
一般式(I)中、R1が水素原子又はC1-C6アルキル基であり、Aがハロゲン原子、アミノ基、C1-C6アルキルアミノ基、ヒドロキシル基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基及び4~10員の飽和複素環基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;ヒドロキシル基、アミノ基又はC1-C6アルキルアミノ基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C1-C6アルキル基、ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基であるか、R1、A及びそれらが結合する窒素原子と一緒になって4~10員の飽和複素環を形成し(ここで4~10員の飽和複素環は、アミノ基及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい)、R2が水素原子であり、Bがハロゲン原子;シアノ基;オキシド基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基、C6-C14芳香族炭化水素基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、チアゾリル基、イソチアゾリル基、チアジアゾリル基、ピラゾリル基、ピリジル基、インドリル基、インダゾリル基、イミダゾピリジル基、チエノピリジル基、ベンゾイミダゾリル基、ベンゾオキサゾリル基、ベンゾチアゾリル基、ベンゾチエニル基、ベンゾフラニル基、キノリル基、又はナフチリジル基である化合物が好ましく:
一般式(I)中、R1が水素原子又はC1-C6アルキル基であり、Aがハロゲン原子、アミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;ヒドロキシル基又はアミノ基から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の飽和複素環基であるか、R1、A及びそれらが結合する窒素原子と一緒になって4~10員の飽和複素環を形成し、R2が水素原子であり、Bがハロゲン原子;シアノ基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基、C6-C14芳香族炭化水素基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、チアゾリル基、イソチアゾリル基、チアジアゾリル基、ピラゾリル基、ピリジル基、インドリル基、インダゾリル基、イミダゾピリジル基、チエノピリジル基、ベンゾイミダゾリル基、ベンゾチアゾリル基、ベンゾチエニル基、ベンゾフラニル基又はキノリル基である化合物が好ましく:
一般式(I)中、R1が水素原子であり、Aがアミノ基、C1-C6アルコキシ基、C3-C10シクロアルキル基又はC6-C14芳香族炭化水素基(ここでC3-C10シクロアルキル基は、アミノ基で置換されていてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;アミノ基が置換していてもよい4~10員の飽和複素環基であり、R2が水素原子であり、Bがハロゲン原子;シアノ基;重水素原子、ハロゲン原子、ヒドロキシル基、C1-C6アルコキシ基及び4~10員の不飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;-C(=O)Rx;-C(=O)ORx;-C(=O)N(Rx)(Ry);-ORx;ヒドロキシル基及びC1-C6アルキル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;C1-C6アルキル基、C1-C6アルコキシ基及びC2-C7アルカノイル基からなる群から選ばれる同一又は相異なった1~3個の基が置換していてもよい4~10員の不飽和複素環基;又はオキソ基が置換していてもよい4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、チアゾリル基、イソチアゾリル基、ピラゾリル基、ピリジル基、インドリル基、インダゾリル基、チエノピリジル基、ベンゾイミダゾリル基、ベンゾチアゾリル基、ベンゾチエニル基、ベンゾフラニル基又はキノリル基である化合物が好ましく:
一般式(I)中、R1が水素原子であり、Aがアミノ基又はC1-C6アルコキシ基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;アミノ基が置換していてもよい4~10員の飽和複素環基であり、R2が水素原子であり、Bがハロゲン原子;重水素原子、ハロゲン原子及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;1~3個のヒドロキシル基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;1~3個のC1-C6アルキル基が置換していてもよい4~10員の不飽和複素環基;又は4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、インドリル基、インダゾリル基、ベンゾチエニル基、キノリル基である化合物が特に好ましい。
(1)3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-シクロブチル-2H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例11化合物)
(2)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例61化合物)
(3)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(ジフルオロメチル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例62化合物)
(4)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(1-メチル-1H-ピラゾール-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例67化合物)
(5)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((4-(ピリミジン-2-イル)チオフェン-2-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例76化合物)
(6)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(trans-4-ヒドロキシシクロヘキシル)-6-メチル-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例78化合物)
(7)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(2-ヒドロキシエチル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例93化合物)
(8)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-エチル-4-メチル-1H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例97化合物)
(9)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-クロロ-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例100化合物)
(10)3-(((2R,3R)-3-アミノ-4-メトキシブタン-2-イル)アミノ)-5-((1,6-ジメチル-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例256化合物)
(11)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-メチル-7-フェニル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例257化合物)
(12)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((8-メチルキノリン-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例259化合物)
(13)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(ジフルオロメチル)-4-メチル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例265化合物)
(14)3-(((2R,3R)-3-アミノ-4-メトキシブタン-2-イル)アミノ)-5-((1-エチル-7-メチル-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例311化合物)
(15)3-(((1R,2S)-2-アミノシクロヘプチル)アミノ)-5-((2-エチル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド(実施例316化合物)
(16)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((7-メチルベンゾ[b]チオフェン-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド 2塩酸塩(実施例359化合物)
本発明化合物(I)は、例えば、下記の製造法又は実施例に示す方法等により製造することができる。ただし、本発明化合物(I)の製造法はこれら反応例に限定されるものではない。
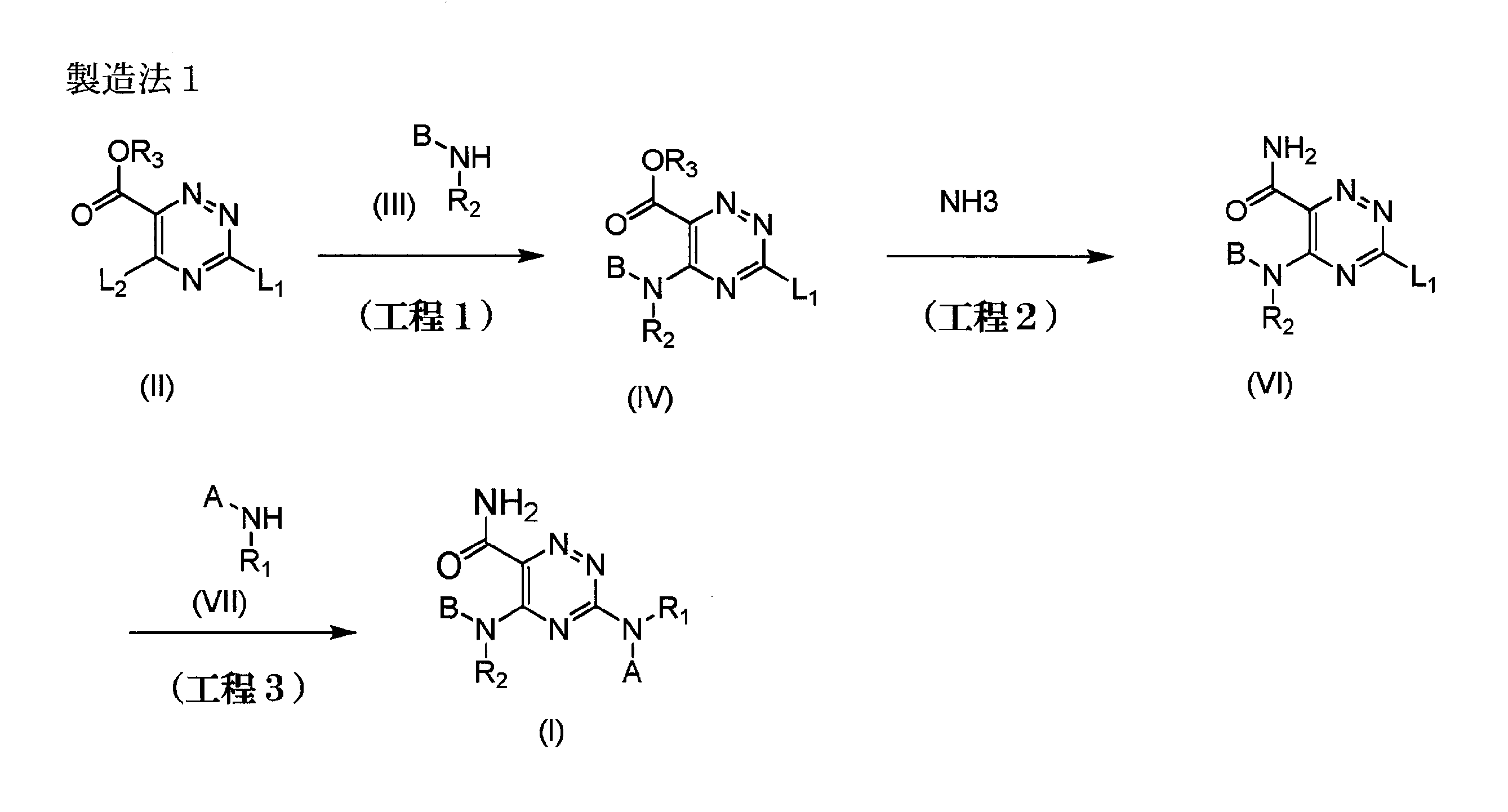
また、上記反応は必要に応じて塩基あるいは酸を用いることができる。当該塩基としては、例えばトリエチルアミン、ジイソプロピルエチルアミン、ピリジン、4-ジメチルアミノピリジン等の有機塩基又は炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水素化ナトリウム、カリウム-tert-ブチラート等の無機塩基を使用することができ、当該酸としては、例えば塩酸、p-トルエンスルホン酸、トリフルオロ酢酸、酢酸、リン酸、フェノール等を使用することができる。
当該酸の使用量は、通常、一般式(II)で表される化合物1モルに対して、0.01モルないし過剰モル、好ましくは0.1ないし3モルである。
反応温度は、通常、0℃ないし200℃、好ましくは0℃ないし150℃である。
反応時間は、通常、1分間ないし7日間、好ましくは5分間ないし24時間である。
このようにして得られる化合物(IV)は、公知の分離精製手段、例えば濃縮、減圧濃縮、結晶化、溶媒抽出、再沈殿、クロマトグラフィーなどにより単離精製するか又は単離精製することなく、次工程に付すことができる。
本工程において用いられるアンモニア又はその塩の量は、一般式(IV)で表される化合物1モルに対して、通常、等モルないし過剰モルである。反応溶媒は、反応に支障のないものであれば、特に限定されないが、例えば、水、メタノール、エタノール、イソプロパノール、tert-ブチルアルコール、テトラヒドロフラン、ジオキサン、ジメチルホルムアミド、N-メチルピロリドン、ジメチルスルホキシド等又はその混合溶媒等が好適である。
反応温度は、通常、0℃ないし200℃、好ましくは室温ないし150℃である。
反応時間は、通常、5分間ないし7日間、好ましくは1分間ないし24時間である。
また、上記反応は必要に応じて塩基を用いることができる。当該塩基としては、例えばトリエチルアミン、ジイソプロピルエチルアミン、ピリジン、4-ジメチルアミノピリジン等の有機塩基又は炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水素化ナトリウム、カリウム-tert-ブチラート等の無機塩基を使用することができる。
反応温度は、通常、0℃ないし200℃、好ましくは0℃ないし100℃である。
反応時間は、通常、1分間ないし7日間、好ましくは5分間ないし24時間である。
もしくは、一般式(IV)又は(VI)で表される化合物の脱離基が有機チオ基の場合、公知の方法により、ハロゲン基に変換し反応に用いることができる。例えば、無溶媒かベンゼン、トルエン、塩化メチレン、クロロホルム、テトラヒドロフラン、アセトニトリル、ジメチルホルムアミド、N-メチルピロリドン等の不活性溶媒中、例えば一般式(IV)又は(VI)で表される化合物の1モルに対して、0.5モルないし過剰モル、好ましくは3モルないし10モルの塩化スルフリル等のクロル化剤を用いて行うことができる。
このようにして得られる有機スルフィニル基、有機スルホニル基もしくはハロゲン基を有する化合物は、公知の分離精製手段、例えば濃縮、減圧濃縮、結晶化、溶媒抽出、再沈殿、クロマトグラフィーなどにより単離精製するか又は単離精製することなく、次工程に付すことができる。

本工程は、前記工程3と同様の方法、これに準じた方法又はこれらと常法とを組み合わせることにより行うことができる。
また、上記反応は必要に応じて塩基を用いることができる。当該塩基としては、例えばトリエチルアミン、ジイソプロピルエチルアミン、ピリジン、4-ジメチルアミノピリジン等の有機塩基又は炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水素化ナトリウム、カリウム-tert-ブチラート等の無機塩基を使用することができる。
当該塩基の使用量は、通常、一般式(IV)で表される化合物1モルに対して、等モルないし過剰モル、好ましくは1ないし3モルである。
反応温度は、通常、0℃ないし200℃、好ましくは室温ないし130℃である。
反応時間は、通常、1分間ないし7日間、好ましくは5分間ないし24時間である。
(工程5)本工程は、一般式(VIII)で表される化合物と、アンモニア又はその塩とを反応させて、一般式(I)で表される化合物を製造する方法である。
本工程は、前記工程2と同様の方法、これに準じた方法又はこれらと常法とを組み合わせることにより行うことができる。
当該アンモニア又はその塩の量は、通常、一般式(VIII)で表される化合物1モルに対して、等モルないし過剰モル、好ましくは過剰モルである。
反応溶媒は、反応に支障のないものであれば、特に限定されないが、例えばメタノール、エタノール、イソプロパノール、tert-ブチルアルコール、トルエン、ベンゼン、塩化メチレン、クロロホルム、テトラヒドロフラン、ジオキサン、ジメチルホルムアミド、アセトニトリル、N-メチルピロリジノン、ジメチルスルホキシド等又はその混合溶媒等が好適である。
反応温度は、通常、0℃ないし200℃、好ましくは室温ないし160℃である。
反応時間は、通常、10分間ないし12時間、好ましくは5分間ないし2時間である。

該塩基付加塩としては、例えばナトリウム塩、カリウム塩等のアルカリ金属塩;例えばカルシウム塩、マグネシウム塩等のアルカリ土類金属塩;例えばアンモニウム塩;例えばトリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、プロカイン塩、N,N’-ジベンジルエチレンジアミン塩等の有機アミン塩等が挙げられる。
該酸付加塩としては、例えば塩酸塩、硫酸塩、硝酸塩、リン酸塩、過塩素酸塩等の無機酸塩;例えば酢酸塩、ギ酸塩、マレイン酸塩、フマール酸塩、酒石酸塩、クエン酸塩、アスコルビン酸塩、トリフルオロ酢酸塩等の有機酸塩;例えばメタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩等のスルホン酸塩等が挙げられる。
注射剤を調製する場合は、本発明化合物にpH調節剤、緩衝剤、安定化剤、等張化剤、局所麻酔剤等を添加し、常法により皮下、筋肉内及び静脈内用注射剤を製造することができる。
また、上記投与形態を有する薬剤の1日あたりの投与量は、患者の症状、体重、年齢、性別等によって異なり一概には決定できないが、本発明化合物として通常成人(体重50kg)1日あたり約0.05~5000mg、好ましくは0.1~1000mgとすればよく、これを1日1回又は2~3回程度に分けて投与するのが好ましい。
またLCMSスペクトルはWaters社製SQDを用いて下記条件にて測定した。
MS検出:ESI positive
UV検出:254及び210nm
カラム流速:0.5mL/min
移動相:水/アセトニトリル(0.1%ギ酸)
インジェクション量:1μL
A法
グラディエント
Time(min) Water Acetonitrile
0 95 5
0.1 95 5
1.1 5 95
2.0 STOP
B法
グラディエント
Time(min) Water Acetonitrile
0 95 5
0.1 95 5
2.1 5 95
3.0 STOP
カラム:YMC社製YMC-Actus Triart C18,30X50mm,5μm
UV検出:254nm
カラム流速:40mL/min
移動相:水/アセトニトリル(0.1%ギ酸)
インジェクション量:1.0mL
グラディエント 水/アセトニトリル 10%→60%(7分)
s:シングレット
d:ダブレット
t:トリプレット
q:カルテット
dd:ダブル ダブレット
dt:ダブル トリプレット
td:トリプル ダブレット
tt:トリプル トリプレット
ddd:ダブル ダブル ダブレット
ddt:ダブル ダブル トリプレット
dtd:ダブル トリプル ダブレット
tdd:トリプル ダブル ダブレット
m:マルチプレット
br:ブロード
DMSO-d6:重ジメチルスルホキシド
CDCl3:重クロロホルム
CD3OD:重メタノール
THF:テトラヒドロフラン
DMF:N,N-ジメチルホルムアミド
NMP:N-メチル-ピロリジノン
DMSO:ジメチルスルホキシド
TFA:トリフルオロ酢酸
WSC:1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩
HOBt:1-ヒドロキシベンゾトリアゾール1水和物
HATU:O-(7-アザベンゾトリアゾ-1-イル)-N,N,N’,N’-テトラメチルヘキサウロニウム ヘキサフルオロホスフェート
m-CPBA:3-クロロ過安息香酸
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 234mg、6-アミノキノリン 173mgのアセトニトリル 3ml溶液に、ジイソプロピルエチルアミン 0.53mlを加え、反応液をマイクロウェーブ反応装置中、100℃にて1時間撹拌した。溶媒を減圧留去した後、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)で精製し、エチル 3-(メチルチオ)-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-6-カルボキシレートを白色固体として得た。
上記工程1で得られたエチル 3-(メチルチオ)-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-6-カルボキシレート 150mgのTHF 0.5ml溶液に、2M アンモニア-メタノール溶液 3mlを加えた後、反応液をマイクロウェーブ反応装置中、130℃にて30分間撹拌した。沈殿物を濾取することで、3-(メチルチオ)-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミドを白色固体の粗精製物として得た。
上記工程2で得られた3-(メチルチオ)-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド 45mgのDMF 1.5ml溶液に、m-CPBA 55mgのTHF 0.5ml溶液を加え、反応液を室温にて20分間撹拌した。さらに反応液にm-CPBA 55mgのTHF 0.5ml溶液を加え、反応液を室温にて20分間撹拌した後、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメート 60mg、ジイソプロピルエチルアミン 0.20mlを加え、反応液をマイクロウェーブ反応装置中、100℃で10分間加熱した。溶媒を減圧留去した後、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)で精製し、tert-ブチル(1S,2R)-2-(6-カルバモイル-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-3-イルアミノ)シクロヘキシルカーバメートを白色固体として得た。
上記工程3で得られたtert-ブチル(1S,2R)-2-(6-カルバモイル-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-3-イルアミノ)シクロヘキシルカーバメート 40mgに、トリフルオロ酢酸 0.6mLを加えた後、反応液を室温にて5分間撹拌した。溶媒を減圧留去し、得られた残渣を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)で精製し、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(ベンゾ[b]チオフェン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 70mg、及びベンゾ[b]チオフェン-4-アミン 70mgのTHF 1ml溶液に、ジイソプロピルエチルアミン 0.10mlを加え、反応液を室温にて35分間撹拌した。溶媒を減圧留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)で精製し、白色固体のエチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートを得た。
上記工程1で得られたエチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 33mgのクロロホルム 1.5ml溶液に、m-CPBA 65mgのクロロホルム 1.5ml溶液を加え、反応液を室温にて10分間撹拌した後、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメート 60mg、ジイソプロピルエチルアミン 0.10mlを加え、反応液を室温にて3時間撹拌した。反応液を酢酸エチルで希釈し、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、エチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-((1R,2S)-2-(tert-ブトキシカルボニルアミノ)シクロヘキシルアミノ)-1,2,4-トリアジン-6-カルボキシレートを得た。
上記工程2で得られたエチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-((1R,2S)-2-(tert-ブトキシカルボニルアミノ)シクロヘキシルアミノ)-1,2,4-トリアジン-6-カルボキシレート 71.8mgに7M アンモニア-メタノール溶液2mlを加え、反応液をマイクロウェーブ反応装置を用いて130℃にて1時間反応させた。溶媒を減圧留去した後、得られた残渣にトリフルオロ酢酸 0.5mlを加え、反応液を室温にて5分間撹拌した。溶媒を減圧留去した後、得られた残渣を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)で精製し、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 466mgに塩化スルフリル 500μlを加え、反応液を70℃にて2時間撹拌した後、塩化スルフリル150μlを追加し、さらに30分間撹拌した。反応液をTHFで希釈した後、溶媒を減圧下濃縮した。得られた残渣をTHF 10mlに溶解し、1-メチル-1H-インドール-4-アミン 470mg、ジイソプロピルエチルアミン 0.85mlを順次加え、反応液を室温にて15分間撹拌した。反応液を酢酸エチルで希釈し、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、エチル 3-クロロ-5-(1-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレートを白色固体として得た。
上記工程1で得られたエチル 3-クロロ-5-(1-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレート 390mgのTHF 10ml溶液に、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメート 302mg、ジイソプロピルエチルアミン 0.40mlを順次加え、反応液を70℃にて2時間反応させた。反応液を酢酸エチルに希釈し、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した後、無水硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、エチル 3-((1R,2S)-2-(tert-ブトキシカルボニルアミノ)シクロヘキシルアミノ)-5-(1-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレートを黄色固体として得た。
上記工程2で得られたエチル 3-((1R,2S)-2-(tert-ブトキシカルボニルアミノ)シクロヘキシルアミノ)-5-(1-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレート 596mgに、7M アンモニア-メタノール溶液 10mlを加え、反応液をマイクロウェーブ反応装置を用いて130℃にて90分間反応させた。溶媒を減圧留去し、得られた残渣にエタノールを加え、析出した沈殿物を濾取することにより、tert-ブチル (1S,2R)-2-(6-カルバモイル-5-(1-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-3-イルアミノ)シクロヘキシルカーバメートを黄色固体として得た。
上記工程3で得られたtert-ブチル (1S,2R)-2-(6-カルバモイル-5-(1-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-3-イルアミノ)シクロヘキシルカーバメート 575mgのエタノール 3ml溶液に、濃塩酸 3mlを加え、反応液を室温にて30分間撹拌した。反応液を水で希釈し、5N 水酸化ナトリウム水溶液を加え、塩基性にした後、クロロホルムにて抽出した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去した。得られた残渣にエタノールを加え、析出した沈殿物を濾取することにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-(エチル-d5)-2H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ニトロ-1H-インダゾール 210mg及び臭化エチル-d5 170mgのDMF 4.2ml溶液に、60%水素化ナトリウム(40%流動パラフィン添加) 60mgを加え、反応液を室温にて終夜攪拌した。反応液に水を加え、析出した沈殿物を濾取し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、5-ニトロ-1-(エチル-d5)-1H-インダゾール及び5-ニトロ-2-(エチル-d5)-2H-インダゾールをそれぞれ白色固体として得た。
上記工程1で得られた5-ニトロ-2-(エチル-d5)-2H-インダゾール 65mgをメタノール 3mlに溶解し、展開ラネーニッケル 20mg、及びヒドラジン一水和物 0.15mlを加え、反応液を室温にて15分間撹拌した。不溶物をセライトにより濾去し、溶媒を減圧留去することで、2-(エチル-d5)-2H-インダゾール-5-アミンを黄色固体として得た。
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに、上記工程2で得られた2-(エチル-d5)-2H-インダゾール-5-アミンを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(エチル-d5)-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例4(工程2)に準じ、5-ニトロ-2-(エチル-d5)-2H-インダゾールの代わりに、実施例4(工程1)で得られた5-ニトロ-1-(エチル-d5)-1H-インダゾールを用いることにより、1-(エチル-d5)-1H-インダゾール-5-アミンを黄色固体として得た。
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 60mgのTHF 1ml溶液に、上記工程1で得られた1-(エチル-d5)-1H-インダゾール-5-アミン 40mgを加え、反応液を50℃にて15分間反応させた。溶媒を減圧留去した後、残渣に水を加え、沈殿物を濾取することにより、エチル 3-(メチルチオ)-5-(1-(エチル-d5)-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキシレートを黄色固体として得た。
実施例1(工程3~4)に準じ、3-(メチルチオ)-5-(キノリン-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミドの代わりに、上記工程2で得られた3-(メチルチオ)-5-(1-(エチル-d5)-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミドを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-(ジメチルアミノ)シクロへキシルアミノ)-5-(1-(エチル-d5)-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド・二ギ酸塩
実施例5で得られた3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(エチル-d5)-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド 2mgのメタノール 1ml溶液に、50%ホルムアルデヒド水溶液 0.01ml及び既知の方法で調製した0.3M Zn(BH3CN)2-メタノール溶液 0.1mlを加え、反応液を室温にて15分間撹拌した。溶媒を減圧留去し、残渣を逆相分取HPLCにて精製し、得られたフラクションを減圧濃縮することで、表題化合物を得た。
(S)-5-(1-(エチル-d5)-1H-インダゾール-5-イルアミノ)-3-(ピペリジン-3-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド・二ギ酸塩
実施例5(工程2)で得られた3-(メチルチオ)-5-(1-(エチル-d5)-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド 4mgのTHF 0.5mL溶液に、m-CPBA 3.3mgを加え、反応液を室温にて1時間撹拌した。(S)-tert-ブチル 3-アミノピペリジン-1-カルボキシレート 5mgを加え、50℃にて終夜反応させた後、反応液の溶媒を留去した。残渣にトリフルオロ酢酸 0.2mlを加え、反応液を室温にて10分間撹拌した後、逆相分取HPLCにて精製し、得られたフラクションを減圧濃縮することで、表題化合物を白色アモルファスとして得た。
3-((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イルアミノ)-5-(ベンゾ[b]チオフェン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 234mgに塩化スルフリル 0.30mlを加え、反応液を室温にて10時間撹拌した。溶媒を減圧留去した後、得られた残渣をTHF 5mlに溶解し、ベンゾ[b]チオフェン-4-アミン 179mg、ジイソプロピルエチルアミン 0.51mlを加え、反応液を室温にて5分間撹拌した。反応液を酢酸エチルで希釈し、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、エチル 3-クロロ-5-(ベンゾ[b]チオフェン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレートを白色固体として得た。
上記工程1で得られたエチル 3-クロロ-5-(ベンゾ[b]チオフェン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレート 34mgのTHF 1mL溶液に、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメート 26mg、ジイソプロピルエチルアミン 0.050mlを加え、反応液を80℃にて30分間撹拌した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、エチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-((3R,4R)-3-(tert-ブトキシカルボニルアミノ)テトラヒドロ-2H-ピラン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレートを得た。
上記工程2で得られたエチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-((3R,4R)-3-(tert-ブトキシカルボニルアミノ)テトラヒドロ-2H-ピラン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキシレート 50mgに、7M アンモニア-メタノール溶液 2mLを加え、反応液をマイクロウェーブ反応装置中130℃にて1時間撹拌した。反応液を室温まで冷却した後、析出した沈殿物を濾取することで、tert-ブチル(3R,4R)-4-(5-(ベンゾ[b]チオフェン-4-イルアミノ)-6-カルバモイル-1,2,4-トリアジン-3-イルアミノ)テトラヒドロ-2H-ピラン-3-イルカーバメートを得た。
上記工程3で得られたtert-ブチル(3R,4R)-4-(5-(ベンゾ[b]チオフェン-4-イルアミノ)-6-カルバモイル-1,2,4-トリアジン-3-イルアミノ)テトラヒドロ-2H-ピラン-3-イルカーバメート 46mgにトリフルオロ酢酸 0.5mLを加え、反応液を室温にて5分間撹拌した。溶媒を減圧留去した後、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)で精製し、表題化合物を淡黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(エチル-d5)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
4-ニトロ-1H-インドール 320mg及び臭化エチル-d5 250mgのDMF 2ml溶液に、60%水素化ナトリウム(40%流動パラフィン添加) 100mgを加え、反応液を室温にて10分攪拌した。反応液を酢酸エチルで希釈し、水、飽和食塩水で順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、4-ニトロ-1-(エチル-d5)-1H-インドールを白色固体として得た。
上記工程1で得られた4-ニトロ-1-(エチル-d5)-1H-インドール 100mgをメタノール 5mlに溶解し、展開ラネーニッケル 50mg、及びヒドラジン一水和物 0.2mlを加え、反応液を室温にて15分間撹拌した。不溶物をセライトにより濾去し、溶媒を減圧留去することで、1-(エチル-d5)-1H-インドール-4-アミンを黄色油状物質として得た。
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 60mgのTHF 2ml溶液に、上記工程2で得られた1-(エチル-d5)-1H-インドール-4-アミン 40mgを加え、反応液を室温にて15分間撹拌した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、エチル 5-(1-(エチル-d5)-1H-インドール-4-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートを黄色固体として得た。
上記工程3で得られたエチル 5-(1-(エチル-d5)-1H-インドール-4-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 47mgに7M アンモニア-メタノール溶液 1mlを加え、反応液を80℃にて5分間撹拌した後、溶媒を減圧留去した。得られた残渣のTHF 2ml溶液に、m-CPBA 45mgを加え、反応液を室温にて30分間撹拌した後、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメート 50mgを加え、反応液を50℃にて2時間撹拌した。溶媒を減圧留去した後、残渣に4M 塩酸-ジオキサン 2mlを加え、反応液を室温にて10分間撹拌した後、溶媒を減圧留去し、残渣を逆相分取HPLCにて精製し、得られたフラクションを減圧濃縮することで、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-シクロプロピル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
4-ニトロ-1H-インドール 0.96gの1,2-ジクロロエタン 60ml溶液に、シクロプロピルボロン酸 1.0g、炭酸水素ナトリウム 0.99g、酢酸銅(II) 1.1g、2,2’-ビピリジン 0.92gを加え、反応液を室温にて7日間撹拌した後、さらに60℃にて2時間撹拌した。不溶物をセライトにより濾去した後、水及び飽和食塩水で順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、1-シクロプロピル-4-ニトロ-1H-インドールを黄色固体として得た。
上記工程1で得られた1-シクロプロピル-4-ニトロ-1H-インドール 190mgをメタノール 5mlに溶解し、展開ラネーニッケル 100mg、及びヒドラジン一水和物 0.2mlを加え、反応液を室温にて20分間撹拌した。不溶物をセライトにより濾去し、溶媒を減圧留去することで、1-シクロプロピル-1H-インドール-4-アミンを淡黄色固体として得た。
実施例9(工程3~4)に準じ、1-(エチル-d5)-1H-インドール-4-アミンの代わりに、上記工程2で得られた1-シクロプロピル-1H-インドール-4-アミンを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-シクロブチル-2H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ニトロ-1H-インダゾール 490mgのTHF 10ml溶液に、シクロブタノール 260mg、トリフェニルホスフィン 1.1g、ジイソプロピルアゾジカルボキシレート 790mgを室温にて順次加え、反応液を室温にて終夜撹拌した。溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、5-ニトロ-1-(シクロブチル)-1H-インダゾール及び5-ニトロ-2-(シクロブチル)-2H-インダゾールをそれぞれ黄色油状物質として得た。
実施例4(工程2~3)に準じ、5-ニトロ-2-(エチル-d5)-2H-インダゾールの代わりに、5-ニトロ-2-(シクロブチル)-2H-インダゾールを用いることにより、表題化合物を黄色固体として得た。
6-(3-((1R,2S)-2-アミノシクロへキシルアミノ)6-カルバモイル-1,2,4-トリアジン-5-イルアミノ)キノリン 1-オキシド
実施例1(工程3~4)に準じ、m-CPBAを過剰量用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-1H-インダゾール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
6-ブロモ-4-ニトロ-1H-インダゾール 901mgのDMF19ml溶液に、氷浴下、60%水素化ナトリウム(40%流動パラフィン添加) 213mgを加えた。反応液を0℃にて15分攪拌した後、ヨウ化メチル 0.35mlを加え、反応液を室温にて11時間反応させた。反応液を水に注ぎ、析出した沈殿物を濾取することで粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、6-ブロモ-1-メチル-4-ニトロ-1H-インダゾール及び6-ブロモ-2-メチル-4-ニトロ-2H-インダゾールをそれぞれ黄色固体として得た。
上記工程1で得られた6-ブロモ-1-メチル-4-ニトロ-1H-インダゾール 51.0mg、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール 65mgに、2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル 4.9mg、クロロ(2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピル-1,1’-ビフェニル)[2-(2-アミノエチル)フェニル)]パラジウム(II) 10mg、2M 炭酸ナトリウム水溶液 0.2ml、およびジオキサン 0.8mlを加え、反応液をマイクロウェーブ反応装置を用いて110℃にて1.5時間反応させた。溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、黄色固体の1-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-4-ニトロ-1H-インダゾールを得た。
上記工程2で得られた1-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-4-ニトロ-1H-インダゾール 46mgをメタノール-酢酸エチル混合溶媒(1:2) 13.5mlに溶解し、20%水酸化パラジウム-炭素 10mgを加え、反応液を水素雰囲気下、室温で5時間反応させた。不溶物をセライトにより濾去し、溶媒を減圧留去することで、1-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-1H-インダゾール-4-アミンを黄色油状物として得た。
実施例2(工程1~3)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程3で得られた1-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-1H-インダゾール-4-アミンを用いることにより、表題化合物を黄色固体として得た。
5-(5-アセチル-2,3’-ビチオフェン-5’ -イルアミノ)-3-((1R,2S)-2-アミノシクロへキシルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例13(工程2~4)に準じ、6-ブロモ-1-メチル-4-ニトロ-1H-インダゾールの代わりに、tert-ブチル 4-ブロモチオフェン-2-イルカーバメートを用い、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾールの代わりに、5-アセチルチオフェン-2-イルボロン酸を用いることにより、表題化合物を淡黄色固体として得た。
5-(5-(5-アセチルチオフェン-2-イル)ピリジン-3-イルアミノ)-3-((1R,2S)-2-アミノシクロへキシルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例13(工程2~4)に準じ、6-ブロモ-1-メチル-4-ニトロ-1H-インダゾールの代わりに、5-ブロモピリジン-3-アミンを用い、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾールの代わりに、5-アセチルチオフェン-2-イルボロン酸を用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(5-シクロプロピルピリジン-3-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ブロモピリジン-3-アミン 500mgにシクロプロピルボロン酸 497mg、酢酸パラジウム(II) 65mg、トリシクロヘキシルホスフィン 162mg、リン酸カリウム 2.5gのトルエン 9mL、水 0.45mL溶液を100℃にて11時間攪拌した。酢酸エチルおよび水を加え、有機層を分離し、飽和食塩水にて洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、3-アミノ-5-シクロプロピルピリジンを得た。
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 70mgのDMF溶液 1mlに、上記工程1で得られた3-アミノ-5-シクロプロピルピリジン 67mg、パラトルエンスルホン酸1水和物 10mgを加え、反応液をマイクロウェーブ反応装置中、100℃にて30分間撹拌した。反応液を酢酸エチルで希釈し、炭酸水素ナトリウム水溶液、水および飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、エチル 5-(5-シクロプロピルピリジン-3-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートを白色固体として得た。
実施例2(工程3~4)に準じ、エチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートの代わりに、エチル 5-(5-シクロプロピルピリジン-3-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートを用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-エチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例9(工程1~4)に準じ、臭化エチル-d5の代わりに、臭化エチルを用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(6-(シクロプロピルカルバモイル)ピリジン-3-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
エチル 5-クロロ-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレート 70mgのジメチルスルホキシド 1.5ml溶液に、5-アミノピコリン酸 44mgを加え、反応液を室温にて10分間撹拌した後、シクロプロピルアミン 0.017ml、ジイソプロピルエチルアミン 0.079ml、HATU 125mgを順次加え、反応液を室温にて終夜撹拌した。反応液を酢酸エチルで希釈し、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、エチル 5-(6-(シクロプロピルカルバモイル)ピリジン-3-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートを得た。
実施例2(工程2~3)に準じ、エチル 5-(ベンゾ[b]チオフェン-4-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートの代わりに、上記工程1で得られたエチル 5-(6-(シクロプロピルカルバモイル)ピリジン-3-イルアミノ)-3-(メチルチオ)-1,2,4-トリアジン-6-カルボキシレートを用いることにより、表題化合物を淡黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(6-(メチルカルバモイル)ピリジン-3-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例18(工程1~2)に準じ、シクロプロピルアミンの代わりに、メチルアミンを用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-2H-インダゾール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例13(工程2~4)に準じ、6-ブロモ-1-メチル-4-ニトロ-1H-インダゾールの代わりに、実施例13(工程1)で得られた6-ブロモ-2-メチル-4-ニトロ-2H-インダゾールを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(6-メチルベンゾ[b]チオフェン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
2,6-ジブロモ-4-メチルベンズアルデヒド 153mgのジメチルスルホキシド 1.7mL溶液に、チオグリコール酸メチル 0,060ml、炭酸カリウム 190mgを加え、反応液を120℃にて7時間撹拌した。反応液を2M 塩酸にて中和した後、酢酸エチルにて抽出し、飽和食塩水にて洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去した。得られた残渣のTHF-メタノール混合溶液(1:1) 3.2mlに、5M 水酸化ナトリウム水溶液 0.8mlを加え、反応液を室温にて2時間撹拌した。反応液を酢酸エチルで希釈し、2M 塩酸、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去した。得られた残渣のジメチルスルホキシド 1.1mL溶液に、炭酸銀 30mg、酢酸 0.005mlを加え、反応液を120℃にて6時間攪拌した。反応液を酢酸エチルで希釈し炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、4-ブロモ-6-メチルベンゾ[b]チオフェンを得た。
上記工程1で得られた4-ブロモ-6-メチルベンゾ[b]チオフェン 106mgの1,4-ジオキサン 2.3mL溶液に、tert-ブチル カーバメート 109mg、炭酸セシウム 456mg、酢酸パラジウム 5.0mg、2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル 22mgを加え、反応液を100℃にて2時間攪拌した。反応液を酢酸エチルで希釈し、水、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、tert-ブチル 6-メチルベンゾ[b]チオフェン-4-イルカーバメートを得た。
上記工程2で得られたtert-ブチル 6-メチルベンゾ[b]チオフェン-4-イルカーバメート 100mgにトリフルオロ酢酸 2.0mLを加え、反応液を室温にて30分間攪拌した。溶媒を減圧留去した後、残渣を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、6-メチルベンゾ[b]チオフェン-4-アミンを得た。
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程3で得られた6-メチルベンゾ[b]チオフェン-4-アミンを用いることにより、表題化合物を淡黄色固体として得た。
エチル 4-(3-((1R,2S)-2-アミノシクロへキシルアミノ)-6-カルバモイル-1,2,4-トリアジン-5-イルアミノ)ベンゾ[b]チオフェン-6-カルボキシレート
(工程1)
WO2005/007635記載の方法にて合成したエチル 4-アセトキシベンゾ[b]チオフェン-6-カルボキシレート 760mgのエタノール 6ml溶液に、炭酸カリウム 400mgを加え、反応液を室温にて1時間撹拌した。不溶物を濾去した後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、エチル 4-ヒドロキシベンゾ[b]チオフェン-6-カルボキシレートを白色固体として得た。
上記工程1で得られたエチル 4-ヒドロキシベンゾ[b]チオフェン-6-カルボキシレート 100mg及びトリエチルアミン 0.160mlの塩化メチレン 2.3ml溶液に、氷浴下、無水トリフルオロメタンスルホン酸 0.091mlを加え、反応液を0℃にて2時間撹拌した。反応液を酢酸エチルで希釈し、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、エチル 4-(トリフルオロメチルスルホニルオキシ)ベンゾ[b]チオフェン-6-カルボキシレートを得た。
上記工程2で得られたエチル 4-(トリフルオロメチルスルホニルオキシ)ベンゾ[b]チオフェン-6-カルボキシレート 120mgのジオキサン 2ml溶液に、tert-ブチル カーバメート 81mg、酢酸パラジウム 4mg、2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル 17mg、炭酸セシウム 340mgを加え、反応液を100℃にて3時間撹拌した。反応液を酢酸エチルで希釈し、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、エチル 4-(tert-ブトキシカルボニルアミノ)ベンゾ[b]チオフェン-6-カルボキシレートを得た。
実施例21(工程3~4)に準じ、tert-ブチル 6-メチルベンゾ[b]チオフェン-4-イルカーバメートの代わりに、上記工程3で得られたエチル 4-(tert-ブトキシカルボニルアミノ)ベンゾ[b]チオフェン-6-カルボキシレートを用いることにより、表題化合物を淡黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(6-(メチルカルバモイル)ベンゾ[b]チオフェン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例22(工程3)で得られたエチル 4-(tert-ブトキシカルボニルアミノ)ベンゾ[b]チオフェン-6-カルボキシレート 110mgに、40%メチルアミン-メタノール溶液 3mlを加え、反応液を90℃にて終夜撹拌した。得られた残渣にトリフルオロ酢酸 0.5mlを加え、反応液を室温にて15分間撹拌した後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、4-アミノ-N-メチルベンゾ[b]チオフェン-6-カルボキサミドを得た。
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程1で得られた4-アミノ-N-メチルベンゾ[b]チオフェン-6-カルボキサミドを用いることにより、表題化合物を淡黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-(ジフルオロメチル)-2H-インダゾール-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
6-ニトロ-1H-インダゾール 5.90g、及びクロロジフルオロ酢酸ナトリウム 9.15gのN-メチルピロリジノン 100ml溶液に、60%水素化ナトリウム(40%流動パラフィン添加) 1.2gを加え、反応液を室温にて15分間撹拌し、さらに100℃にて30分間撹拌した。反応液を酢酸エチルで希釈し、水および飽和食塩水にて順次洗浄した。無水硫酸ナトリウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、1-(ジフルオロメチル)-6-ニトロ-1H-インダゾールおよび2-(ジフルオロメチル)-6-ニトロ-2H-インダゾールを混合物として得た。
上記工程1で得られた1-(ジフルオロメチル)-6-ニトロ-1H-インダゾールおよび2-(ジフルオロメチル)-6-ニトロ-2H-インダゾールの混合物 7.05gの酢酸エチル 60ml溶液に、窒素気流下、10%パラジウム-炭素 4.0gを加え、反応液を、水素雰囲気下、室温にて終夜撹拌した。不溶物をセライトにて濾去後、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、1-(ジフルオロメチル)-1H-インダゾール-6-アミン及び2-(ジフルオロメチル)-2H-インダゾール-6-アミンをそれぞれ白色固体として得た。
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程2で得られた2-(ジフルオロメチル)-2H-インダゾール-6-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(1-メチル-1H-ピラゾール-3-イル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
4-ニトロ-1H-インドール 660mg、3-ヨード-1-メチル-1H-ピラゾール 853mg、リン酸三カリウム 864mg、ヨウ化銅(I) 775mgの混合物に、trans-N,N’-ジメチルシクロヘキサン-1,2-ジアミン (ラセミ体) 0.643ml、及びトルエン 10mlを加え、反応液を100℃にて8時間撹拌した。反応液を室温に戻した後、酢酸エチルで希釈し、28% アンモニア水および飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥した後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、1-(1-メチル-1H-ピラゾール-3-イル)-4-ニトロ-1H-インドールを得た。
上記工程1で得られた1-(1-メチル-1H-ピラゾール-3-イル)-4-ニトロ-1H-インドール 625mgの酢酸エチル 20ml溶液に、窒素気流下、10%パラジウム-炭素 500mgを加えた後、反応液を、水素雰囲気下、室温にて2時間半撹拌した。不溶物をセライトにより濾去後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製することで、1-(1-メチル-1H-ピラゾール-3-イル)-1H-インドール-4-アミンを得た。
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程2で得られた1-(1-メチル-1H-ピラゾール-3-イル)-1H-インドール-4-アミンを用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(ジフルオロメチル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例24(工程1~3)に準じ、6-ニトロ-1H-インダゾールの代わりに、4-ニトロ-1H-インドールを用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-シクロブチル-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例4(工程2~3)に準じ、5-ニトロ-2-(エチル-d5)-2H-インダゾールの代わりに、実施例11(工程1)で得られた5-ニトロ-1-(シクロブチル)-1H-インダゾールを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-エチル-2H-インダゾール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例4(工程1~2)に準じ、5-ニトロ-1H-インダゾールの代わりに、4-ニトロ-1H-インダゾールを用い、臭化エチル-d5の代わりに、2-ヨードエタンを用いることにより、2-エチル-2H-インダゾール-4-アミンを得た。
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに、上記工程1で得られた2-エチル-2H-インダゾール-4-アミンを用いることにより、3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-エチル-2H-インダゾール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミドを黄色固体として得た。
3-(5-アセチルチオフェン-2-イル)-5-(3-((1R,2S)-2-アミノシクロへキシルアミノ)-6-カルバモイル-1,2,4-トリアジン-5イルアミノ)ピリジン 1-オキシド
実施例15で得られた3-(メチルチオ)-5-(5-(5-アセチルチオフェン-2-イル)ピリジン-3-イルアミノ)-1,2,4-トリアジン-6-カルボキサミドに対し、過剰量のm-CPBAを用いて、実施例1(工程3)及び(工程4)と同様の方法、これに準じた方法又はこれらと常法とを組み合わせることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-エチル-1H-インドール-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例9(工程1~4)に準じ、4-ニトロ-1H-インドールの代わりに、6-ニトロ-1H-インドールを用い、臭化エチル-d5の代わりに、臭化エチルを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-シクロプロピル-1H-インダゾール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例10(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに、4-ニトロ-1H-インダゾールを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-エチル-2-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
3-ニトロアニリン 1.38gのジメチルスルホキシド 20ml溶液に、アセトン 1.0mL、tert-ブトキシカリウム 2.7gを加え、反応液を室温にて3時間攪拌した。反応液を酢酸エチルで希釈し、塩化アンモニウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、2-メチル-4-ニトロ-1H-インドールを得た。
実施例9(工程1~4)に準じ、4-ニトロ-1H-インドールの代わりに、上記工程1で得られた2-メチル-4-ニトロ-1H-インドールを用い、臭化エチル-d5の代わりに、臭化エチルを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-シクロプロピル-2-メチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例10(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに、実施例32(工程1)で得られた2-メチル-4-ニトロ-1H-インドールを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(オキセタン-3-イル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例9(工程1~2)に準じ、臭化エチル-d5の代わりに、3-臭化オキセタンを用いることにより、1-(オキセタン-3-イル)-1H-インドール-4-アミンを得た。
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程1で得られた1-(オキセタン-3-イル)-1H-インドール-4-アミンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(2-フルオロエチル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、1-ヨード-2-フルオロエタンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(ピリジン-2-イル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに、2-ブロモピリジンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-フェニル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~2)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに、ヨードベンゼンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(ジフルオロメチル)-1H-インダゾール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例24(工程1~2)に準じ、6-ニトロ-1H-インダゾールの代わりに、4-ニトロ-1H-インダゾールを用いることにより、1-(ジフルオロメチル)-1H-インダゾール-4-アミン及び2-(ジフルオロメチル)-2H-インダゾール-4-アミンをそれぞれ得た。
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程1で得られた1-(ジフルオロメチル)-1H-インダゾール-4-アミンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(2-ヒドロキシ-2-メチルプロピル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、1-ヨード-2-メチルプロパン-2-オールを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(2,2-ジフルオロエチル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、1,1-ジフルオロ-2-ヨードエタンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(1,1-ジフルオロ-2-ヒドロキシエチル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、2,2-ジフルオロ-ヨードエタノールを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(メチル-d3)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、ヨウ化メタン-d3を用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(チエノ[2,3-c]ピリジン-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
3,5-ジブロモ-4-ピリジンカルボキシアルデヒド 1.6gのDMF 12ml溶液に、チオグリコール酸メチル 0.653ml、炭酸カリウム 2.1gを加え、反応液を120℃にて2時間撹拌した。反応液に2M 塩酸を加え、弱酸性にした後、酢酸エチルにて抽出し、有機相を飽和食塩水にて洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、粗生成物を得た。得られた粗生成物のTHF-メタノール混合溶液(1:1) 24mlに、5M 水酸化ナトリウム水溶液 2.4mlを加え、反応液を室温にて4時間撹拌した。溶媒を減圧留去した後、2M 塩酸を加え、析出した沈殿物を濾取することで、4-ブロモチエノ[2,3-c]ピリジン-2-カルボン酸を得た。
上記工程1で得られた4-ブロモチエノ[2,3-c]ピリジン-2-カルボン酸 258mgのDMF 2.0ml溶液に、炭酸銀 28mg、酢酸 0.006mlを加え、反応液を120℃にて24時間攪拌した。反応液を酢酸エチルで希釈し、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、4-ブロモチエノ[2,3-c]ピリジンを得た。
実施例21(工程2~4)に準じ、4-ブロモ-6-メチルベンゾ[b]チオフェンの代わりに、上記工程2で得られた4-ブロモチエノ[2,3-c]ピリジンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-プロピル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、1-ヨードプロパンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-イソプロピル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、2-ヨードプロパンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(2-メトキシエチル)-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、1-ブロモ-2-メトキシエタンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-シクロブチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、3-臭化オキセタンの代わりに、臭化シクロブタンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(5-メトキシ-3-メチルベンゾ[b]チオフェン-2-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
メチル 3-クロロ-5-メトキシ-1-ベンゾチオフェン-2-カルボキシレート 257mgの1,4-ジオキサン 3.0ml溶液に、メチルボロン酸 120mg、リン酸カリウム 849mg、水 0.30ml、酢酸パラジウム 22mg、2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル 95mgを加え、反応液を100℃にて12時間攪拌した。反応液を酢酸エチルで希釈し、塩化アンモニウム水溶液、炭酸水素ナトリウム水溶液および飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、メチル 5-メトキシ-3-メチルベンゾ[b]チオフェン-2-カルボキシレートを得た。
上記工程1で得られたメチル 5-メトキシ-3-メチルベンゾ[b]チオフェン-2-カルボキシレート 250mgのTHF-メタノール(1:1)混合溶液 6.0mlに、5M 水酸化ナトリウム水溶液 1.5mlを加え、反応液を室温にて2時間撹拌した。反応液を2N 塩酸にて中和した後、酢酸エチルにて抽出し、飽和食塩水にて洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去した。得られた残渣のtert-ブタノール 2.0ml溶液に、N,N-ジイソプロピルエチルアミン 0.184ml、ジフェニルリン酸アジド 0.248mlを加え、反応液を100℃にて5時間攪拌した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-酢酸エチル)にて精製し、tert-ブチル 5-メトキシ-3-メチルベンゾ[b]チオフェン-2-イルカーバメートを得た。
上記工程2で得られたtert-ブチル 5-メトキシ-3-メチルベンゾ[b]チオフェン-2-イルカーバメート 210mgに4M 塩酸-1,4-ジオキサン溶液を加え、反応液を室温にて3時間攪拌した。反応液にジエチルエーテルを加え、析出した沈殿物を濾取することで、5-メトキシ-3-メチルベンゾ[b]チオフェン-2-アミン 一塩酸塩を得た。
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程3で得られた5-メトキシ-3-メチルベンゾ[b]チオフェン-2-アミン塩酸塩を用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-メチル-6-フェニル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例13(工程1~4)に準じ、6-ブロモ-4-ニトロ-1H-インダゾールの代わりに、6-ブロモ-4-ニトロ-1H-インドールを用い、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾールの代わりに、フェニルボロン酸を用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-エチル-3-メチル-1H-インダゾール-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例4(工程1~3)に準じ、5-ニトロ-1H-インダゾールの代わりに、6-ニトロ-3-メチル-1H-インダゾールを用い、臭化エチル-d5の代わりに、ヨードエタンを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1,3-ジメチル-1H-インドール-4-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例34(工程1~2)に準じ、4-ニトロ-1H-インドールの代わりに、3-メチル-4-ニトロ-1H-インドールを用い、3-臭化オキセタンの代わりに、ヨウ化メチルを用いることにより、表題化合物を得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-イソプロピル-1H-インダゾール-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例4(工程1~3)に準じ、5-ニトロ-1H-インダゾールの代わりに、6-ニトロ-1H-インダゾールを用い、臭化エチル-d5の代わりに、2-ヨードプロパンを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-イソプロピル-2H-インダゾール-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例4(工程2~3)に準じ、5-ニトロ-2-(エチル-d5)-1H-インダゾールの代わりに、実施例52において副生成物として得られる6-ニトロ-2-イソプロピル-2H-インダゾールを用い、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(2,2-ジフルオロエチル)-1H-インダゾール-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例4(工程1~3)に準じ、5-ニトロ-1H-インダゾールの代わりに、6-ニトロ-1H-インダゾールを用い、臭化エチル-d5の代わりに、1,1-ジフルオロ-2-ヨードエタンを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-(2-フルオロエチル)-1H-インダゾール-6-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例4(工程1~3)に準じ、5-ニトロ-1H-インダゾールの代わりに、6-ニトロ-1H-インダゾールを用い、臭化エチル-d5の代わりに、1-フルオロ-2-ヨードエタンを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(1-エチル-3-メチル-1H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例4(工程1~3)に準じ、5-ニトロ-1H-インダゾールの代わりに、5-ニトロ-3-メチル-1H-インダゾールを用い、臭化エチル-d5の代わりに、ヨードエタンを用いることにより、表題化合物を黄色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-(ジフルオロメチル)-2H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例24(工程1~3)に準じ、6-ニトロ-1H-インダゾールの代わりに、5-ニトロ-1H-インダゾールを用いることにより、表題化合物を黄色固体として得た。
5-(2-(2H-1,2,3-トリアゾール-2-イル)ピリジン-4-イルアミノ)-3-((1R,2S)-2-アミノシクロへキシルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
1H-1,2,3-トリアゾール 518mg、2-フルオロ-4-ヨードピリジン 892mg、炭酸セシウム 1.63mgのNMP 10mL溶液を100℃にて、4時間撹拌した。反応液を水に注ぎ、ジエチルエーテルにて抽出した。有機層を水及び飽和食塩水で洗浄し、硫酸ナトリウムにて乾燥した。濃縮後、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、4-ヨード-2-(2H-1,2,3-トリアゾール-2-イル)ピリジンを白色固体として得た。
(工程2)
上記工程1で得られた4-ヨード-2-(2H-1,2,3-トリアゾール-2-イル)ピリジン 394mg、酸化銅(I) 40mgに濃アンモニア水溶液 2mLとNMP 2mLの混合溶液を加え、マイクロウェーブ反応装置を用い、100℃にて12時間反応させた。反応液を水に注ぎ、酢酸エチルにて抽出した。有機層を水及び飽和食塩水で洗浄し、硫酸ナトリウムにて乾燥した。濃縮後、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(2H-1,2,3-トリアゾール-2-イル)ピリジン-4-アミンを白色固体として得た。
(工程3)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、2-(2H-1,2,3-トリアゾール-2-イル)ピリジン-4-アミンを用いることにより、表題化合物を白色固体として得た。
3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-(ジフルオロメチル)-3-メチル-2H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例24(工程1~3)に準じ、6-ニトロ-1H-インダゾールの代わりに、3-メチル-5-ニトロ-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1,6-ジメチル-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例9(工程1~2)に準じ、4-ニトロ-1H-インドールの代わりに6-メチル-4-ニトロ-1H-インドールを用い、臭化エチル-d5の代わりにヨウ化メチルを用いることにより、1,6-ジメチル-1H-インドール-4-アミンを得た。
(工程2)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程1で得られた1,6-ジメチル-1H-インドール-4-アミンを用いることにより、表題化合物を淡黄褐色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ブロモ-7-メチル-1H-インダゾール 35.9gにトルエン 150mL、酢酸tert-ブチルエステル 360mL、メタンスルホン酸 22mLを加え、反応液を80℃にて28時間撹拌した。反応液を室温まで冷却し、水 500mLを加え、トルエン 500mLと200mLで抽出した。有機層を分離後、水にて2回洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣にヘキサンを加えて不溶物を濾別した。濾液の溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、5-ブロモ-2-(tert-ブチル)-7-メチル-2H-インダゾールを淡赤色油状物として得た。
(工程2)
上記工程1で得られた5-ブロモ-2-(tert-ブチル)-7-メチル-2H-インダゾール 40.3gに酸化銅(I) 2.7g、NMP 150mL、濃アンモニア水 150mLを加え、反応液をポータブルリアクターにて95℃にて10時間攪拌した。反応液を室温まで冷却し、酢酸エチル 500mLとヘキサン 200mLおよび水 300mLを加えた。有機層を分離後、水にて4回、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、2-(tert-ブチル)-7-メチル-2H-インダゾール-5-アミンを紫色固体として得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程で得られた2-(tert-ブチル)-7-メチル-2H-インダゾール-5-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(ジフルオロメチル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ブロモ-7-メチル-1H-インダゾール 2.11gの酢酸エチル 50mL溶液に炭酸カリウム 2.76gと2,2-ジフルオロ-2-(フルオロスルホニル)酢酸 2.06mLを加え、反応液を室温にて3時間攪拌した。炭酸水素ナトリウム水溶液を加え、有機層を分離後、飽和食塩水にて洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製した。得られた生成物に酸化銅(I) 143mg、NMP 6mL、濃アンモニア水 6mLを加え、反応液をマイクロウェーブ反応装置にて100℃にて5時間反応させた。酢酸エチル 500mLおよび水 300mLを加え、有機層を分離後、水にて4回、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(ジフルオロメチル)-7-メチル-2H-インダゾール-5-アミンを白色固体として得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程1で得られた2-(ジフルオロメチル)-7-メチル-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((2R,3R)-3-アミノ-4-エトキシブタン-2-イル)アミノ)-5-((2-(ジフルオロメチル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
Bioorganic Chemistry 36(2008)4-15の記載の方法で合成可能な(4S,5S)-tert-ブチル 4-(ヒドロキシメチル)-2,2,5-トリメチルオキサゾリジン-3-カルボキシレート 1.23gのDMF 10mL溶液にヨードエタン 0.80mLを加え、氷冷下にて水素化ナトリウム(60% in oil) 300mgを加え、氷浴を外し、反応液を室温にて1時間攪拌した。反応液に水と酢酸エチルを加え有機層を分離後、水にて2回洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、(4S,5S)-tert-ブチル 4-(エトキシメチル)-2,2,5-トリメチルオキサゾリジン-3-カルボキシレートを淡黄色アモルファスとして得た。
(工程2)
上記工程1で得られた(4S,5S)-tert-ブチル 4-(エトキシメチル)-2,2,5-トリメチルオキサゾリジン-3-カルボキシレート 1.34gにメタノール 130mL、パラトルエンスルホン酸1水和物 134mgを加え、反応液を室温にて終夜攪拌した。反応液に炭酸水素ナトリウム水溶液を加え、メタノールを減圧留去し、水と酢酸エチルを加え有機層を分離後、飽和食塩水にて洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、tert-ブチル ((2S,3S)-1-エトキシ-3-ヒドロキシブタン-2-イル)カーバメートを無色アモルファスとして得た。
(工程3)
上記工程2で得られたtert-ブチル ((2S,3S)-1-エトキシ-3-ヒドロキシブタン-2-イル)カーバメート 1.06gのTHF 20mL溶液にトリエチルアミン 1.25mL、メタンスルホニルクロライド 0.457mLを加え、反応液を室温にて20分間攪拌した。反応液に10%リン酸水溶液を加えた後、酢酸エチルにて抽出し、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣にDMF 10mL、テトラブチルアンモニウムアジド 2gを加え、反応液を70℃にて2時間攪拌した。反応液を室温に戻した後、ジエチルエーテルで希釈し、水および飽和食塩水にて洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、溶媒を減圧留去した。得られた残渣にTHF 5mL、エタノール 5mLを加え、窒素雰囲気下、10%パラジウム炭素 600mgを加えた。反応液を室温、水素雰囲気下にて終夜攪拌した。不溶物を濾去し、溶媒を減圧留去することで、tert-ブチル ((2R,3R)-3-アミノ-1-エトキシブタン-2-イル)カーバメートを無色油状物質として得た。
(工程4)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに実施例62(工程1)で得られた2-(ジフルオロメチル)-7-メチル-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりに上記工程3で得られたtert-ブチル ((2R,3R)-3-アミノ-1-エトキシブタン-2-イル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((2R,3R)-3-アミノ-4-メトキシブタン-2-イル)アミノ)-5-((1-イソプロピル-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例63(工程1~3)に準じ、実施例63(工程1)のヨードエタンの代わりにヨードメタンを用いることにより、tert-ブチル ((2R,3R)-3-アミノ-1-メトキシブタン-2-イル)カーバメートを白色アモルファスとして得た。
(工程2)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに実施例45で得られた1-イソプロピル-1H-インドール-4-アミンを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりに上記工程1で得られたtert-ブチル ((2R,3R)-3-アミノ-1-メトキシブタン-2-イル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-メチル-7-フェニル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
7-ブロモ-5-ニトロ-1H-インダゾール 1.21gの酢酸エチル 12mL溶液にトリメチルオキソニウムテトラフルオロボレート 1.5gを加え、反応液を室温にて6時間攪拌した。反応液を酢酸エチルで希釈した後、水、飽和食塩水にて順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた残渣に酢酸エチルとイソプロピルエーテルを加え、析出した沈殿物を濾取することにより、7-ブロモ-2-メチル-5-ニトロ-2H-インダゾールを淡黄色固体としてを得た。
(工程2)
上記工程1で得られた7-ブロモ-2-メチル-5-ニトロ-2H-インダゾール 256mgにフェニルボロン酸 244mg、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(1:1) 40mg、ジオキサン 4mL、2M炭酸ナトリウム水溶液 1mL、反応液をを加えマイクロウェーブ反応装置を用い130℃にて1時間反応させた。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-メチル-5-ニトロ-7-フェニル-2H-インダゾールを得た。
(工程3)
上記工程2で得られた2-メチル-5-ニトロ-7-フェニル-2H-インダゾール 260mgに酢酸エチル 5mL、THF 2mLを加えた後、窒素雰囲気下、10%パラジウム炭素 300mgを加え、反応液を室温、水素雰囲気下にて終夜攪拌した。不溶物を濾去し、溶媒を減圧留去後、得られた残渣を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-メチル-7-フェニル-2H-インダゾール-5-アミンを褐色アモルファスとして得た。
(工程4)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程3で得られた2-メチル-7-フェニル-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(tert-ブチル)-7-(1H-ピラゾール-1-イル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
7-ブロモ-5-ニトロ-1H-インダゾール 484mgに2-メチル-2-プロパノール 5mL、濃硫酸 0.1mLを加え、反応液をマイクロウェーブ反応装置を用いて100℃にて1時間反応させた。反応液を酢酸エチルで希釈後、炭酸水素ナトリウム水、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、7-ブロモ-2-(tert-ブチル)-5-ニトロ-2H-インダゾールを淡黄色固体として得た。
(工程2)
上記工程1で得られた7-ブロモ-2-(tert-ブチル)-5-ニトロ-2H-インダゾール 438mg、ピラゾール 200mg、酸化銅(I) 42mg、炭酸セシウム 960mgにDMF 6mLを加え、反応液を120℃にて終夜攪拌した。反応液を室温まで冷却し、水と酢酸エチルを加えた後、不溶物を濾去した。有機層を分離した後、水および飽和食塩水にて洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(tert-ブチル)-5-ニトロ-7-(1H-ピラゾール-1-イル)-2H-インダゾールを無色アモルファスとして得た。
(工程3)
上記工程2で得られた2-(tert-ブチル)-5-ニトロ-7-(1H-ピラゾール-1-イル)-2H-インダゾール 255mgの酢酸エチル 5mLに、10%パラジウム炭素 300mgを加え、反応液を室温、水素雰囲気下にて終夜攪拌した。不溶物を濾去し、溶媒を減圧留去後、得られた残渣を塩基性シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(tert-ブチル)-7-(1H-ピラゾール-1-イル)-2H-インダゾール-5-アミンを褐色アモルファスとして得た。
(工程4)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに、上記工程3で得られた2-(tert-ブチル)-7-(1H-ピラゾール-1-イル)-2H-インダゾール-5-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(1-メチル-1H-ピラゾール-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに4-ヨード-1-メチル-1H-ピラゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-((R)-テトラヒドロフラン-3-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
(S)-テトラヒドロフラン-3-オ-ル 1.0gのトルエン 10mL溶液にトリエチルアミン 2.05mL、メタンスルホニルクロライド 1.05mLを加え、反応液を室温にて2時間攪拌した。反応液に水を加えた後、トルエンにて抽出し、飽和食塩水にて洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、(S)-テトラヒドロフラン-3-イル メタンスルホナートを無色オイル状物質として得た。
(工程2)
4-ニトロ-1H-インドール 980mgのDMF 10mL溶液に炭酸カリウム 2.5g、上記工程で得られた(S)-テトラヒドロフラン-3-イル メタンスルホナート 1.01g加え、反応液を100℃にて24時間攪拌した。反応液に水を加え、酢酸エチルにて2回抽出した後、水および飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製した。得られた残渣にTHF 10mLを加え、10%パラジウム炭素 100mgを加え、反応液を室温、水素雰囲気下にて14時間攪拌した。不溶物をセライトにて濾去し、溶媒を減圧留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、1-((R)-テトラヒドロフラン-3-イル)-1H-インドール-4-アミンを白色固体として得た。
(工程3)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに上記工程で得られた1-((R)-テトラヒドロフラン-3-イル)-1H-インドール-4-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(2-メチルピリジン-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに4-ヨード-2-ピコリンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-エチル-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ブロモ-7-メチル-1H-インダゾール 844mgのDMF 10mL溶液に水素化ナトリウム(60% in oil) 240mg、ヨードエタン 0.64mLを加え、反応液を室温にて15分間攪拌した。反応液に水を加えた後、酢酸エチルにて抽出した。有機層を水、飽和食塩水にて順次洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、5-ブロモ-1-エチル-7-メチル-1H-インダゾールおよび5-ブロモ-2-エチル-7-メチル-2H-インダゾールを得た。
(工程2)
上記工程1で得られた5-ブロモ-2-エチル-7-メチル-2H-インダゾール 446mgに酸化銅(I) 60mg、NMP 2mL、濃アンモニア水 2mLを加え、反応液をマイクロウェーブ反応装置を用いて100℃にて10時間攪拌した。反応液を室温まで冷却し、酢酸エチルで希釈後、水にて4回、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-エチル-7-メチル-2H-インダゾール-5-アミンを得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程2で得られた2-エチル-7-メチル-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(6-メチルピリダジン-3-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに3-クロロ-6-メチルピリダジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピリミジン-5-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに5-ブロモピリミジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(trans-4-ヒドロキシシクロヘキシル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにWO2004/050024に記載されている方法に準じて合成できる4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキシル メタンスルホナートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-イソプロピル-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例70(工程1~3)に準じ、ヨードエタンの代わりに2-ヨードプロパンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(テトラヒドロ-2H-ピラン-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程1~3)に準じ、(S)-テトラヒドロフラン-3-オールの代わりにテトラヒドロ-2H-ピラン-4-オールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((4-(ピリミジン-2-イル)チオフェン-2-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
トリス(ジベンジリデンアセトン)ジパラジウム(0) 460mg、2‐(ジシクロヘキシルホシフィノ)‐2’,4’,6’‐トリイソプロピル‐1,1’‐ビフェニル 960mg、3-チオフェンボロン酸 9.60g、2-クロロピリミジン 5.72g、リン酸カリウム 21.2g、モレキュラーシーブ4A 12.5gにtert-アミルアルコール 100mLを加え、反応液を100℃にて終夜攪拌した。不溶物を濾去し、溶媒を減圧留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(チオフェン-3-イル)ピリミジンを白色固体として得た。
(工程2)
濃硫酸 35mLに氷冷下、上記の工程1で得られた2-(チオフェン-3-イル)ピリミジン 6.99gを加え、さらに発煙硝酸(1.5) 1.81mLと濃硫酸 35mLの混合液を氷冷下でゆっくりと加えた後、氷浴を外しゆっくりと室温まで昇温した。反応液を室温にて1時間攪拌後、氷に注ぎ込み、析出した固体を濾取することで2-(5-ニトロチオフェン-3-イル)ピリミジンを得た。
(工程3)
上記の工程2で得られた2-(5-ニトロチオフェン-3-イル)ピリミジン 7.23g、二炭酸ジ-tert-ブチル 9.90g、炭酸水素ナトリウム 3.52gのTHF 100mLとエタノール 50mLにパールマン触媒(Pd20%)(約50%水湿潤品) 6.0gを加え、反応液を室温、水素雰囲気下にて終夜攪拌した。反応液に、二炭酸ジ-tert-ブチル 5.5gのTHF 50mLを加えた後、不溶物を濾去し、濾液を60℃に加温し30分間攪拌した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン-クロロホルム(1:1)混合溶媒/酢酸エチル)にて精製し、tert-ブチル (4-(ピリミジン-2-イル)チオフェン-2-イル)カーバメートを得た。
(工程4)
上記の工程3で得られたtert-ブチル (4-(ピリミジン-2-イル)チオフェン-2-イル)カーバメートに4N塩酸-ジオキサン溶液 20mLを加え、反応液を室温にて2時間攪拌した。溶媒を減圧留去した後、イソプロピルエーテルを加えた。析出した沈殿物を濾取し、4-(ピリミジン-2-イル)チオフェン-2-アミン塩酸塩を得た。
(工程5)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程4で得られた4-(ピリミジン-2-イル)チオフェン-2-アミン 塩酸塩を用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を褐色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((4-(ピリジン-2-イル)チオフェン-2-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
tert-ブチル (4-ブロモチオフェン-2-イル)カーバメート 5.4g、ビス(ピナコラト)ジボロン 9.86g、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(1:1) 800mg、酢酸カリウム 3.39gのDMF 40mLの反応液を80℃にて90分間攪拌した。溶媒を減圧留去後、酢酸エチルで希釈し、水、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、tert-ブチル (4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)チオフェン-2-イル)カーバメートを得た。
(工程2)
上記工程で得られたtert-ブチル (4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)チオフェン-2-イル)カーバメート 2.33gのジオキサン 30mLの溶液に、2-ブロモピリジン 2.26g、テトラキス(トリフェニルホスフィン)パラジウム(0) 414mg、2M 炭酸ナトリウム水溶液 7mLを加え、反応液を100℃にて終夜攪拌した。反応液を室温まで冷却し、酢酸エチルで希釈した後、水、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、得られた残渣に4N 塩酸-ジオキサン 8mLを加え、反応液を室温にて3時間放置した。溶媒を減圧留去した後、残渣にイソプロピルエーテルを加え、析出した沈殿物を濾取し、4-(ピリジン-2-イル)チオフェン-2-アミン 2塩酸塩を得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた4-(ピリジン-2-イル)チオフェン-2-アミン 2塩酸塩を用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(trans-4-ヒドロキシシクロヘキシル)-6-メチル-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例9(工程1~4)に準じ、4-ニトロ-1H-インドールの代わりに6-メチル-4-ニトロ-1H-インドールを用い、臭化エチル-d5の代わりにWO2004/050024に記載されている方法に準じて合成できる4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキシル メタンスルホナートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-エチル-7-(1H-ピラゾール-1-イル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例65(工程1)に準じ、トリメチルオキソニウムテトラフルオロボレートの代わりにトリエチルチルオキソニウムテトラフルオロボレートを用いることにより、7-ブロモ-2-エチル-5-ニトロ-2H-インダゾールを得た。
(工程2)
実施例66(工程2~3)に準じ、7-ブロモ-2-(tert-ブチル)-5-ニトロ-2H-インダゾールの代わりに上記工程1で得られた7-ブロモ-2-エチル-5-ニトロ-2H-インダゾールを用いることにより、2-エチル-7-(1H-ピラゾール-1-イル)-2H-インダゾール-5-アミンを得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程2で得られた2-エチル-7-(1H-ピラゾール-1-イル)-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
5-((1-(1,3,4-チアジアゾール-2-イル)-1H-インドール-4-イル)アミノ)-3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-ブロモ-1,3,4-チアジアゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((7-クロロ-2-エチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ブロモ-7-クロロ-1H-インダゾール 1.38gの酢酸エチル 15mL溶液にトリエチルオキソニウムテトラフルオロボレート 2.5gを加え、反応液を室温にて終夜攪拌した。反応液を酢酸エチルで希釈した後、水、飽和食塩水にて順次洗浄、無水硫酸ナトリウムで乾燥し、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、5-ブロモ-7-クロロ-2-エチル-2H-インダゾール得た。
(工程2)
上記工程1で得られた5-ブロモ-7-クロロ-2-エチル-2H-インダゾール 1.43gに酸化銅(I) 200mg、NMP 7mL、濃アンモニア水 7mLを加え、反応液をマイクロウェーブ反応装置を用いて80℃にて10時間攪拌した。反応液を室温まで冷却し、酢酸エチルで希釈した後、水にて4回、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、7-クロロ-2-エチル-2H-インダゾール-5-アミンを得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた7-クロロ-2-エチル-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2,7-ジメチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例81(工程1~3)に準じ、5-ブロモ-7-クロロ-1H-インダゾールの代わりに5-ブロモ-7-メチル-1H-インダゾールを用い、トリエチルオキソニウムテトラフルオロボレートの代わりにトリメチルオキソニウムテトラフルオロボレートを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((7-メチル-2-(1-メチルシクロブチル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ブロモ-7-メチル-1H-インダゾール 633mgに1-メチル-1-シクロブタノール 3mL、濃硫酸 0.1mLを加え、反応液をマイクロウェーブ反応装置を用いて100℃にて30分間反応させた後、さらに120℃にて30分間反応させた。反応溶液に炭酸水素ナトリウム水溶液および酢酸エチルを加え、有機層を分離後、飽和食塩水にて洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、5-ブロモ-7-メチル-2-(1-メチルシクロブチル)-2H-インダゾールを得た。
(工程2)
上記工程1で得られた5-ブロモ-7-メチル-2-(1-メチルシクロブチル)-2H-インダゾール 536mgに酸化銅(I) 50mg、NMP 2.5mL、濃アンモニア水 2.5mLを加え、反応液をマイクロウェーブ反応装置を用いて100℃にて5時間攪拌した。反応液を室温まで冷却し、酢酸エチルで希釈した後、水にて3回、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、7-メチル-2-(1-メチルシクロブチル)-2H-インダゾール-5-アミンを得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程2で得られた7-メチル-2-(1-メチルシクロブチル)-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピラジン-2-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-ブロモピラジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((7-シクロプロピル-2-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例65(工程1)で得られた7-ブロモ-2-メチル-5-ニトロ-2H-インダゾール 361mgにシクロプロピルボロン酸 347mg、酢酸パラジウム(II) 31mg、トリシクロヘキシルホスフィンテトラフルオロボラート 99mg、リン酸カリウム 1gのトルエン 20mL、水 1mLを加え、反応液を100℃にて11時間攪拌した。酢酸エチルで希釈した後、水、飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、7-シクロプロピル-2-メチル-5-ニトロ-2H-インダゾールを得た。
(工程2)
上記工程1で得られた7-シクロプロピル-2-メチル-5-ニトロ-2H-インダゾール 311mgの酢酸エチル 5mL溶液に窒素雰囲気下、10%パラジウム炭素 300mgを加え、反応液を室温、水素雰囲気下にて終夜攪拌した。不溶物を濾去し、溶媒を減圧留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、7-シクロプロピル-2-メチル-2H-インダゾール-5-アミンを褐色アモルファスとして得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた7-シクロプロピル-2-メチル-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ジフルオロメチル)-4-メチル-1H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例24(工程1~2)に準じ、6-ニトロ-1H-インダゾールの代わりに4-メチル-6-ニトロ-1H-インダゾール 934mgを用いることにより1-(ジフルオロメチル)-4-メチル-1H-インダゾール-6-アミン 112mgおよび2-(ジフルオロメチル)-4-メチル-2H-インダゾール-6-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた1-(ジフルオロメチル)-4-メチル-1H-インダゾール-6-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を白色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ジフルオロメチル)-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例24(工程1~2)に準じ、6-ニトロ-1H-インダゾールの代わりに5-ニトロ-1H-インダゾールを用いることにより表題化合物1-(ジフルオロメチル)-1H-インダゾール-5-アミンおよび2-(ジフルオロメチル)-4-メチル-2H-インダゾール-5-アミンを得た。
(工程2)
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに上記工程で得られた1-(ジフルオロメチル)-1H-インダゾール-5-アミンを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-エチル-7-メチル-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例70(工程2)に準じ、5-ブロモ-2-エチル-7-メチル-2H-インダゾールの代わりに実施例70(工程1)で得られた5-ブロモ-1-エチル-7-メチル-1H-インダゾール 374mgを用いることにより1-エチル-7-メチル-1H-インダゾール-5-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた1-エチル-7-メチル-1H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た
3-(((2S,3R)-2-アミノ-5-メチルヘキサン-3-イル)アミノ)-5-((2-シクロブチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
メチル 3-ホルミル-4-ニトロベンゾエート 5.00gにトルエン 30mL、シクロブチルアミン 2.25mLを加え、室温にて5分間攪拌した。溶媒を減圧留去した後、残渣に亜リン酸トリエチル 11.3mLを加え、反応液を100℃にて7時間攪拌した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製した。得られた粗生成物にエタノール 2mL、5N水酸化ナトリウム水溶液 5.0mLを加え、反応液を室温にて1時間攪拌した。10%リン酸水溶液を加え反応溶液を酸性にし、さらに水を加え、析出した沈殿物を濾取し、2-シクロブチル-2H-インダゾール-5-カルボン酸を白色固体として得た。
(工程2)
上記工程1で得られた2-シクロブチル-2H-インダゾール-5-カルボン酸 3.26gの1,4-ジオキサン50mL溶液にジフェニルホスホリルアジド 4.15mL、トリエチルアミン 3.76mLを加え、反応液を室温にて2時間20分間攪拌した後、反応液に1N 塩酸 100mLを加えた後、100℃にて30分間攪拌した。反応液を室温まで冷却し、炭酸カリウム 14gをゆっくりと加え、酢酸エチルにて抽出した後、有機層を飽和食塩水にて洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-シクロブチル-2H-インダゾール-5-アミンを淡黄色固体として得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた2-シクロブチル-2H-インダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりに特許(WO2012/002577)に記載されている方法にて合成したtert-ブチル ((2S,3R)-3-アミノ-5-メチルヘキサン-2-イル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(3-メチルピラジン-2-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-ブロモ-3-メチルピラジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2,4-ジメチル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド 2ギ酸塩
(工程1)
実施例24(工程1~2)に準じ、6-ニトロ-1H-インダゾールの代わりに4-メチル-6-ニトロ-1H-インダゾールを用い、クロロジフルオロ酢酸ナトリウムの代わりにヨードメタンを用いることにより、1、4-ジメチル-1H-インダゾール-6-アミンおよび2、4-ジメチル-2H-インダゾール-6-アミンを得た。
(工程2)
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに2,4-ジメチル-2H-インダゾール-6-アミンを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((6-メチル-1-(テトラヒドロ-2H-ピラン-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程1~3)に準じ、用いる4-ニトロ-1H-インドールの代わりに6-メチル-4-ニトロ-1H-インドールを用い、(S)-テトラヒドロフラン-3-オールの代わりにテトラヒドロ-2H-ピラン-4-オールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(2-ヒドロキシエチル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程1~3)に準じ、(S)-テトラヒドロフラン-3-オールの代わりに2-(tert-ブチルジメチルシロキシ)エタノールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘプチル)アミノ)-5-((1-(2-メトキシエチル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
(1S,2S)-アミノシクロヘプタノール1gのメタノール 20mLに、二炭酸ジ-tert-ブチル 2.16gを加え、反応液を室温にて10分撹拌した後、溶媒を減圧留去した。得られた残渣にヘキサンを加え、沈殿物を濾取し、tert-ブチル ((2S,3S)-(2-ヒドロキシシクロヘプチル))カーバメートを白色固体として得た。
(工程2)
実施例63(工程3)に準じ、tert-ブチル ((2S,3S)-1-エトキシ-3-ヒドロキシブタン-2-イル)カーバメートの代わりに、上記工程で得られたtert-ブチル ((2S,3S)-(2-ヒドロキシシクロヘプチル))カーバメート1.74gを用いることにより、tert-ブチル ((1S,2R)-2-アミノシクロヘプチル)カーバメートを得た。
(工程3)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに実施例46で得られた1-(2-メトキシエチル)-1H-インドール-4-アミンを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりに上記工程で得られたtert-ブチル ((1S,2R)-2-アミノシクロヘプチル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1,7-ジメチル-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例70(工程1)に準じ、ヨードエタンの代わりにヨードメタンを用いることにより、5-ブロモ-2、7-ジメチル-2H-インダゾールを得た。
(工程2)
実施例88(工程1~2)に準じ、5-ブロモ-1-エチル-7-メチル-1H-インダゾールの代わりに上記工程で得られた5-ブロモ-2、7-ジメチル-2H-インダゾールを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((7-メチル-2-(2,2,2-トリフルオロエチル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例70(工程1)に準じ、ヨードエタンの代わりに1-ヨード-2,2,2-トリフルオロエタンを用いることにより、5-ブロモ-7-メチル-1-(2,2,2-トリフルオロエチル)-1H-インダゾールおよび5-ブロモ-7-メチル-2-(2,2,2-トリフルオロエチル)-2H-インダゾールを得た。
(工程2)
実施例70(工程2、3)に準じ、5-ブロモ-2-エチル-7-メチル-2H-インダゾールの代わりに、上記工程で得られた5-ブロモ-7-メチル-2-(2,2,2-トリフルオロエチル)-2H-インダゾールを用いることにより表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-エチル-4-メチル-1H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
特許(WO2009/084695)に記載の方法で合成可能な4-メチル-6-ニトロ-1H-インダゾール 1.77gのDMF 17mL溶液にヨードエタン 1.2mL、水素化ナトリウム(60% in oil) 600mgを加え、反応液を室温にて15分間攪拌した。反応液を酢酸エチルで希釈した後、水にて2回洗浄後、飽和食塩水にて洗浄した。無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、1-エチル-4-メチル-6-ニトロ-1H-インダゾールおよび2-エチル-4-メチル-6-ニトロ-2H-インダゾールをそれぞれ淡黄色固体として得た。
(工程2)
上記の工程1で得られた1-エチル-4-メチル-6-ニトロ-1H-インダゾール 975mgの酢酸エチル 10mLに10%パラジウム炭素 1.0gを加え、反応液を室温、水素雰囲気下にて終夜攪拌した。不溶物を濾去後、溶媒を減圧留去し、1-エチル-4-メチル-1H-インダゾール-6-アミンを得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程2で得られた1-エチル-4-メチル-1H-インダゾール-6-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((1-エチル-6-(2-ヒドロキシプロパン-2-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例4(工程1~2)に準じ、5-ニトロ-1H-インダゾールの代わりに、メチル 4-ブロモ-1H-インダゾール-6-カルボキレートを用い、臭化エチル-d5の代わりに、2-ヨードエタンを用いることにより、メチル 4-ブロモ-1-エチル-1H-インダゾール-6-カルボキレートを得た。
(工程2)
上記工程1で得られたメチル 4-ブロモ-1-エチル-1H-インダゾール-6-カルボキレート 0.70gのTHF 20mL溶液にメチルマグネシウムクロリド (3M THF溶液)を2.5mlを加え、反応液を室温にて3時間撹拌した。反応液に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルにて抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(4-ブロモ-1H-インダゾール-6-イル)プロパン-2-オールを得た。
(工程3)
実施例58(工程2)に準じ、4-ヨード-2-(2H-1,2,3-トリアゾール-2-イル)ピリジンの代わりに、上記工程2で得られた2-(4-ブロモ-1H-インダゾール-6-イル)プロパン-2-オールを用いることにより、1-エチル-6-(2-ヒドロキシプロパン-2-イル)-1H-インダゾール-4-アミンを得た。
(工程4)
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに上記工程3で得られた1-エチル-6-(2-ヒドロキシプロパン-2-イル)-1H-インダゾール-4-アミンを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりにtert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートを用いることにより、表題化合物を黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(1-ヒドロキシ-2-メチルプロパン-2-イル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
5-ブロモ-7-メチル-1H-インダゾール 2.11gのDMF 20mL溶液に炭酸セシウム 6.52g、エチル 2-ブロモイソブチレート 2.2mLを加え、反応液を室温にて2時間攪拌した。反応液に水を加えた後、酢酸エチルにて抽出した。有機層を水、飽和食塩水にて順次洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、エチル 2-(5-ブロモ-7-メチル-2H-インダゾール-2-イル)-2-メチルプロパノエートを得た。
(工程2)
上記工程で得られたエチル 2-(5-ブロモ-7-メチル-2H-インダゾール-2-イル)-2-メチルプロパノエート 2.77gのTHF 30mLに、氷冷下、0.99M水素化ジイソブチルアルミニウムのトルエン溶液を34.4mL加えた。反応液を氷冷下にて30分間攪拌した後、反応液に5N塩酸 30mLをゆっくりと加えた。反応液を水で希釈した後、ジエチルエーテルにて抽出した。有機層を2N塩酸、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(5-ブロモ-7-メチル-2H-インドール-2-イル)-2-メチルプロパン-1-オールを得た。
(工程3)
実施例61(工程2~3)に準じ、5-ブロモ-2-(tert-ブチル)-7-メチル-2H-インダゾールの代わりに上記工程2で得られた2-(5-ブロモ-7-メチル-2H-インドール-2-イル)-2-メチルプロパン-1-オールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-クロロ-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例83(工程1~2)に準じ、5-ブロモ-7-メチル-1H-インダゾール代わりに5-ブロモ-7-クロロ-1H-インダゾールを用い、1-メチル-1-シクロブタノールの代わりに2-メチル-2-プロパノールを用いることにより2-(tert-ブチル)-7-クロロ-2H-インダゾール-5-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた2-(tert-ブチル)-7-クロロ-2H-インダゾール-5-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-エチル-4-メチル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例97(工程2~3)に準じ、1-エチル-4-メチル-6-ニトロ-1H-インダゾールの代わりに実施例97(工程1)で得られた2-エチル-4-メチル-6-ニトロ-2H-インダゾールを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((4-(4-メチルピリミジン-2-イル)チオフェン-2-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例77(工程2~3)に準じ、2-ブロモピリジンの代わりに2-クロロ-4-メチルピリミジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-イソプロピル-7-メチル-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例70(工程1)に準じ、ヨードエタンの代わりに2-ヨードプロパンを用いることにより5-ブロモ-1-イソプロピル-7-メチル-1H-インダゾール450mgおよび5-ブロモ-2-イソプロピル-7-メチル-2H-インダゾール479mgを得た。
(工程2)
実施例70(工程2~3)に準じ、5-ブロモ-2-エチル-7-メチル-2H-インダゾールの代わりに5-ブロモ-1-イソプロピル-7-メチル-1H-インダゾールを用いることにより、表題化合物をオレンジ色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(tert-ブチル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例89(工程1~3)に準じ、シクロブチルアミンの代わりにtert-ブチルアミンを用い、tert-ブチル ((2S,3R)-3-アミノ-5-メチルヘキサン-2-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(3-ヒドロキシ-2,2-ジメチルプロピル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにJournal of Organic Chemistry;1988;53(3);507-15で合成される (3-ブロモ-2,2-ジメチルプロポキシ)(tert-ブチル)ジメチルシランを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1-アミノシクロブチル)メチル)アミノ)-5-((1-(1-メチル-1H-ピラゾール-3-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例63(工程3)に準じ、tert-ブチル ((2S,3S)-1-エトキシ-3-ヒドロキシブタン-2-イル)カーバメートの代わりにtert-ブチル (1-(ヒドロキシメチル)シクロブチル)カーバメート 2.01gを用いることによりtert-ブチル (1-(アミノメチル)シクロブチル)カーバメートを得た。
(工程2)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに実施例25(工程2)で得られた1-(1-メチル-1H-ピラゾール-3-イル)-1H-インドール-4-アミンを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりに上記工程で得られたtert-ブチル (1-(アミノメチル)シクロブチル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-エチル-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例68(工程2)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにヨードエタンを用いることにより1-エチル-1H-インダゾール-4-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた1-エチル-1H-インダゾール-4-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-シクロペンチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例89(工程1~3)に準じ、シクロブチルアミンの代わりにシクロペンチルアミンを用い、tert-ブチル ((2S,3R)-3-アミノ-5-メチルヘキサン-2-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-エチル-4-メトキシ-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例70(工程1)に準じ、5-ブロモ-7-メチル-1H-インダゾールの代わりに6-ブロモ-4-メトキシ-1H-インダゾール 329mgを用いることにより、6-ブロモ-1-エチル-4-メトキシ-1H-インダゾール 210mgおよび6-ブロモ-2-エチル-4-メトキシ-2H-インダゾールを得た。
(工程2)
実施例70(工程2~3)に準じ、5-ブロモ-2-エチル-7-メチル-2H-インダゾールの代わりに、上記工程で得られた6-ブロモ-2-エチル-4-メトキシ-2H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-エチル-4-メトキシ-1H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例70(工程2~3)に準じ、5-ブロモ-2-エチル-7-メチル-2H-インダゾールの代わりに、実施例109(工程1)で得られた6-ブロモ-1-エチル-4-メトキシ-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(2,6-ジメトキシピリミジン-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに4-クロロ-2,6-ジメトキシピリミジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((5-(ピリミジン-2-イル)チオフェン-3-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
チオフェン-3-カルボン酸 9.8gの酢酸 40mL溶液に、臭素 4.4mLの酢酸 30mL溶液を室温にて30分間かけて加えた。反応液を室温にて7時間攪拌後、水 200mLを加えて析出した沈殿物を濾取し、2-ブロモチオフェン-4-カルボン酸と2.5-ジブロモチオフェン-3-カルボン酸とのおよそ5対1混合物を得た。
(工程2)
上記工程で得られた2-ブロモチオフェン-4-カルボン酸の混合物 14.48gのトルエン 250mL溶液に室温にてトリエチルアミン 15mL、ジフェニルホスホリルアジド 16.5mLを加え2時間攪拌した。反応液に2-メチル-2-プロパノール 40mLを加え、100℃にて2時間攪拌した。反応液を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、tert-ブチル (5-ブロモチオフェン-3-イル)カーバメートを白色固体として得た。
(工程3)
上記工程で得られたtert-ブチル (5-ブロモチオフェン-3-イル)カーバメート 1.4g、ビス(ピナコラト)ジボロン 2.54gに、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(1:1) 204mg、酢酸カリウム 3.39g、DMF 10mLを加え、反応液を80℃にて1時間攪拌した。反応液にジエチルエーテルおよび水を加え、不溶物を濾去した後、有機層を分離し、水にて2回および飽和食塩水にて順次洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣に2-クロロピリミジン 1g、テトラキストリフェニルホスフィンパラジウム 300mg、ジオキサン 50mL、2M炭酸ナトリウム水溶液 5mLを加え、反応液を85℃にて終夜攪拌した。反応液に酢酸エチルおよび水を加え、有機層を分離し、飽和食塩水にて洗浄した。無水硫酸ナトリウムにて乾燥後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、tert-ブチル (5-(ピリミジン-2-イル)チオフェン-3-イル)カーバメート を得た。
(工程4)
上記工程で得られたtert-ブチル (5-(ピリミジン-2-イル)チオフェン-3-イル)カーバメート 544mgに4N塩酸-ジオキサン溶液 8mLを加え、反応液を室温にて90分間攪拌した。溶媒を減圧留去し、5-(ピリミジン-2-イル)チオフェン-3-アミン 塩酸塩を褐色固体として得た。
(工程5)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた5-(ピリミジン-2-イル)チオフェン-3-アミン 塩酸塩を用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((7-メチル-2-(tert-ペンチル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例83(工程1~2)に準じ、1-メチル-1-シクロブタノールの代わりに、2-メチルブタン-2-オールを用いることにより7-メチル-2-(tert-ペンチル)-2H-インダゾール-5-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程で得られた7-メチル-2-(tert-ペンチル)-2H-インダゾール-5-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(4-メトキシピリミジン-2-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-クロロ-4-メトキシピリミジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-メチル-4-(トリフルオロメチル)-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例24(工程1~2)に準じ、6-ニトロ-1H-インダゾールの代わりに特許(WO2009/064695)で得られた4-(トリフルオロメチル)-6-ニトロ-1H-インダゾールを用い、クロロジフルオロ酢酸ナトリウムの代わりにヨードメタンを用いることにより、1-メチル-4-(トリフルオロメチル)-1H-インダゾール-6-アミンおよび2-メチル-4-(トリフルオロメチル)-2H-インダゾール-6-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた2-メチル-4-(トリフルオロメチル)-2H-インダゾール-6-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を白色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((7-メチル-1-(2,2,2-トリフルオロエチル)-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例70(工程2、3)に準じ、5-ブロモ-2-エチル-7-メチル-2H-インダゾールの代わりに実施例96(工程1)で得られた5-ブロモ-7-メチル-1-(2,2,2-トリフルオロエチル)-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(テトラヒドロ-2H-ピラン-4-イル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例11(工程1~2)に準じ、シクロブタノールの代わりにテトラヒドロ-2H-ピラン-4-オールを用いることにより、1-(テトラヒドロ-2H-ピラン-4-イル)-1H-インダゾール-5-アミンおよび2-(テトラヒドロ-2H-ピラン-4-イル)-2H-インダゾール-5-アミンを得た。
(工程2)
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに上記工程で得られた2-(テトラヒドロ-2H-ピラン-4-イル)-2H-インダゾール-5-アミンを用いることにより、表題化合物を黄色固体として得た。
3-(((2R,3R)-3-アミノ-4-メトキシブタン-2-イル)アミノ)-5-((2-エチル-4-メチル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例97(工程2~3)に準じ、1-エチル-4-メチル-6-ニトロ-1H-インダゾールの代わりに実施例97(工程1)で得られた2-エチル-4-メチル-6-ニトロ-2H-インダゾールを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりに、実施例64(工程1)で得られたtert-ブチル ((2R,3R)-3-アミノ-1-メトキシブタン-2-イル)カーバメートを用いることにより表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピリミジン-2-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-クロロピリミジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-フェニル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例89(工程1~3)に準じ、シクロブチルアミンの代わりにアニリンを用い、tert-ブチル ((2S,3R)-3-アミノ-5-メチルヘキサン-2-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピリジン-3-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、3-ヨード-1-メチル-1H-ピラゾールの代わりに3-ヨードピリジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(1-メチル-1H-ピラゾール-4-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、3-ヨード-1-メチル-1H-ピラゾールの代わりに4-ヨード-1-メチル-1H-ピラゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-シクロブチル-4-メチル-1H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド 2塩酸塩
(工程1)
実施例24(工程1~2)に準じ、6-ニトロ-1H-インダゾールの代わりに4-メチル-6-ニトロ-1H-インダゾールを用い、クロロジフルオロ酢酸ナトリウムの代わりにブロモシクロブタンを用いることにより、1-シクロブチル-4-メチル-1H-インダゾール-6-アミンおよび2-シクロブチル-4-メチル-2H-インダゾール-6-アミンを得た。
(工程2)
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに上記工程で得られた1-シクロブチル-4-メチル-1H-インダゾール-6-アミンを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-エチル-1H-ベンゾ[d]イミダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例68(工程2)に準じ、4-ニトロ-1H-インドールの代わりに5-ニトロベンゾイミダゾ-ルを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにヨードエタンを用いることにより、1-エチル-1H-ベンゾ[d]イミダゾール-5-アミン 317mgおよび1-エチル-1H-ベンゾ[d]イミダゾール-6-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた1-エチル-1H-ベンゾ[d]イミダゾール-5-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(ジフルオロメチル)-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例62(工程1)に準じ、5-ブロモ-7-メチル-1H-インダゾールの代わりに5-ニトロ-1H-インダゾールを用いることによって得られた2-(ジフルオロメチル)-5-ニトロ-2H-インダゾール 429mgに酢酸エチル20mLを加え、10%パラジウム炭素 300mgを加え、反応液を室温、水素雰囲気下にて4時間攪拌した。不溶物を濾去した後、溶媒を減圧留去し、2-(ジフルオロメチル)-2H-インダゾール-5-アミンを白色固体として得た。
(工程2)
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに上記工程で得られた2-(ジフルオロメチル)-2H-インダゾール-5-アミンを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりにtert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-((S)-テトラヒドロフラン-3-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-((S)-テトラヒドロフラン-3-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程1~3)に準じ、(S)-テトラヒドロフラン-3-オールの代わりに(R)-テトラヒドロフラン-3-オールを用い、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-エチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例83(工程1~2)に準じ、5-ブロモ-7-メチル-1H-インダゾール代わりに5-ブロモ-7-エチル-1H-インダゾールを用い、1-メチル-1-シクロブタノールの代わりに2-メチル-2-プロパノールを用いることにより2-(tert-ブチル)-7-エチル-2H-インダゾール-5-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程で得られた2-(tert-ブチル)-7-エチル-2H-インダゾール-5-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(1-メチル-1H-ピラゾール-3-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(テトラヒドロ-2H-ピラン-4-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程1~3)に準じ、(S)-テトラヒドロフラン-3-オールの代わりにテトラヒドロ-2H-ピラン-4-オールを用い、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-イソプロピル-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程1~3)に準じ、(S)-テトラヒドロフラン-3-オールの代わりに2-ヨードプロパンを用い、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピリジン-2-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-ブロモピリジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(tert-ブチル)-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
2,4-ジニトロベンズアルデヒド 1.96gのエタノール 10mLにtert-ブチルアミン 2mLを加え、反応液を40℃にて30分間攪拌した。溶媒を減圧留去した後、得られた残渣にtert-ブチルアミン 1mLを加え、50℃にて30分間加熱した。溶媒を減圧留去した後、亜リン酸トリメチル6mLを加え、反応液を110℃にて1時間攪拌した。溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、得られた残渣にイソプロピルエーテルを加え、沈殿物を濾取し、2-(tert-ブチル)-6-ニトロ-2H-インダゾールを白色固体として得た。
(工程2)
上記工程で得られた2-(tert-ブチル)-6-ニトロ-2H-インダゾール 1.1gの酢酸エチル 12mLの溶液に10%パラジウム炭素 1gを加え、反応液を室温、水素雰囲気下にて4時間攪拌した。不溶物を濾去し、溶媒を減圧留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン/酢酸エチル)にて精製し、2-(tert-ブチル)-2H-インダゾール-6-アミンを白色固体として得た。
(工程3)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた2-(tert-ブチル)-2H-インダゾール-6-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピリジン-4-イルメチル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりに4-(クロロメチル)ピリジン 塩酸塩を用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(2-エトキシエチル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにブロモエチル エチル エーテルを用いることにより、表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2,7-ジエチル-3-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例81(工程1~3)に準じ、5-ブロモ-7-クロロ-2-エチル-2H-インダゾールの代わりに5-ブロモ-7-エチル-3-メチル-1H-インダゾールを用いることにより表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((6-メチル-1-(テトラヒドロ-2H-ピラン-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例68(工程1~2)に準じ、(S)-テトラヒドロフラン-3-オールの代わりにテトラヒドロ-2H-ピラン-4-オールを用い、4-ニトロ-1H-インドールの代わりに6-メチル-4-ニトロ-1H-インドールを用いることにより、6-メチル-1-(テトラヒドロ-2H-ピラン-4-イル)-1H-インドール-4-アミンを得た。
(工程2)
実施例3(工程1~4)に準じ、1-メチル-1H-インドール-4-アミンの代わりに上記工程で得られた6-メチル-1-(テトラヒドロ-2H-ピラン-4-イル)-1H-インドール-4-アミンを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりtert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-メトキシ-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例128(工程1~2)に準じ、5-ブロモ-7-メチル-1H-インダゾール代わりに5-ブロモ-7-メトキシ-1H-インダゾールを用いることにより表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((7-クロロ-1-イソブチル-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例70(工程1~2)に準じ、5-ブロモ-7-メチル-1H-インダゾールの代わりに5-ブロモ-7-クロロ-1H-インダゾールを用い、ヨードエタンの代わりに1-ヨード-2-メチルプロパンを用いることで、7-クロロ-1-イソブチル-1H-インダゾール-5-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程で得られた7-クロロ-1-イソブチル-1H-インダゾール-5-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((1-(ジフルオロメチル)-7-メチル-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例70(工程1~2)に準じ、ヨードエタンの代わりにクロロジフルオロ酢酸ナトリウムを用いることにより、1-(ジフルオロメチル)-7-メチル-1H-インダゾール-5-アミンを得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに、上記工程で得られた1-(ジフルオロメチル)-7-メチル-1H-インダゾール-5-アミンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピラジン-2-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-ブロモピラジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(2-オキソテトラヒドロフラン-3-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド 2塩酸塩
実施例68(工程1~3)に準じ、(S)-テトラヒドロフラン-3-オールの代わりに2-オキソテトラヒドロフラン-3-オ-ルを用いることにより、表題化合物を淡黄色固体として得た。
3-((2-アミノエチル)(メチル)アミノ)-5-((1-エチル-d5-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例4(工程2)に準じ、5-ニトロ-2-(エチル-d5)-2H-インダゾールの代わりに、実施例4(工程1)で得られた5-ニトロ-1-(エチル-d5)-1H-インダゾールを用いることにより、1-エチル-d5-1H-インダゾール-5-アミンを白色固体として得た。
(工程2)
実施例1(工程1~4)に準じ、6-アミノキノリンの代わりに上記工程で得られた1-エチル-d5-1H-インダゾール-5-アミンを用い、tert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートの代わりにtert-ブチル (2-(メチルアミノ)エチル)カーバメートを用いることにより、表題化合物を黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピリジン-3-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに3-ヨードピリジンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(チアゾール-2-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-ブロモチアゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(チアゾール-2-イル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例25(工程1~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、3-ヨード-1-メチル-1H-ピラゾールの代わりに2-ブロモチアゾールを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-フェニル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(工程1)
実施例133(工程1~2)に準じ、tert-ブチルアミンの代わりにアニリンを用いることにより、2-フェニル-2H-インダゾール-6-アミンを白色固体として得た。
(工程2)
実施例8(工程1~4)に準じ、ベンゾ[b]チオフェン-4-アミンの代わりに上記工程で得られた2-フェニル-2H-インダゾール-6-アミンを用い、tert-ブチル ((3R,4R)-4-アミノテトラヒドロ-2H-ピラン-3-イル)カーバメートの代わりにtert-ブチル ((1S,2R)-2-アミノシクロヘキシル)カーバメートを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-シクロペンチル-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにブロモシクロペンタンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-シクロヘキシル-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにブロモシクロヘキサンを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-ベンジル-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりにベンジルブロミドを用いることにより、表題化合物を淡黄色固体として得た。
3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(ピリジン-3-イルメチル)-1H-インダゾール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
実施例68(工程2~3)に準じ、4-ニトロ-1H-インドールの代わりに4-ニトロ-1H-インダゾールを用い、(S)-テトラヒドロフラン-3-イル メタンスルホナートの代わりに3-ピコリルクロライド 塩酸塩を用いることにより、表題化合物を淡黄色固体として得た。
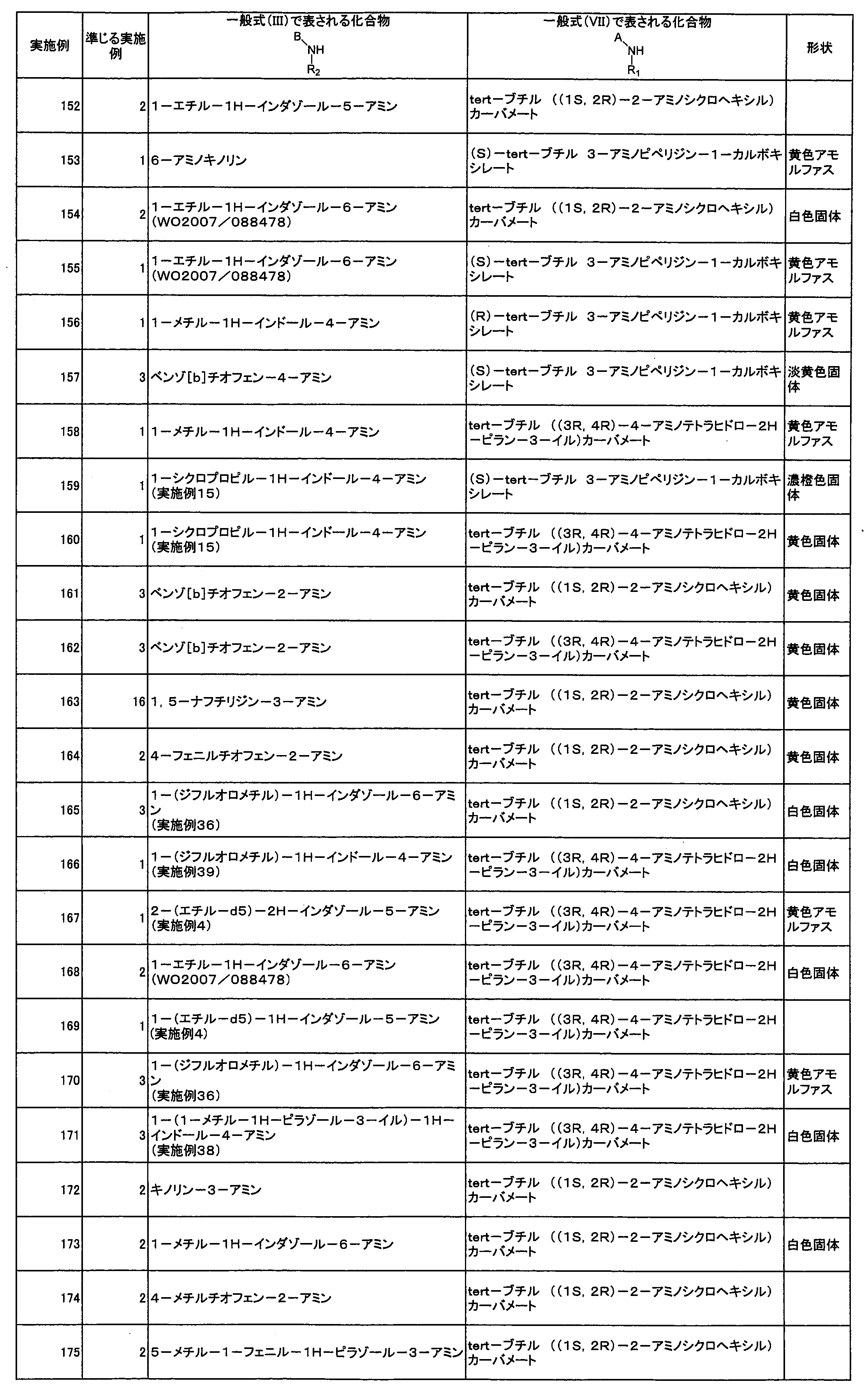










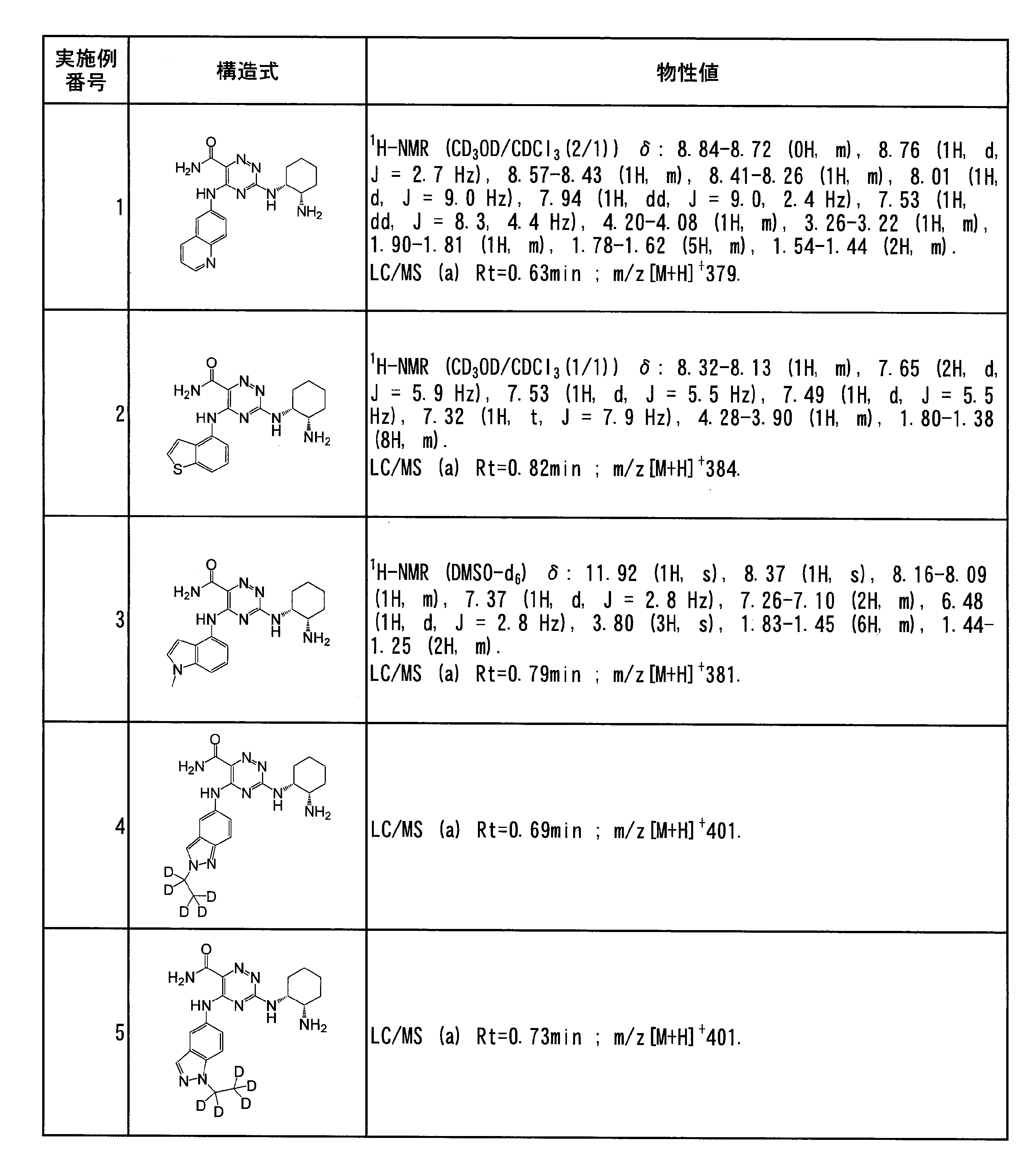

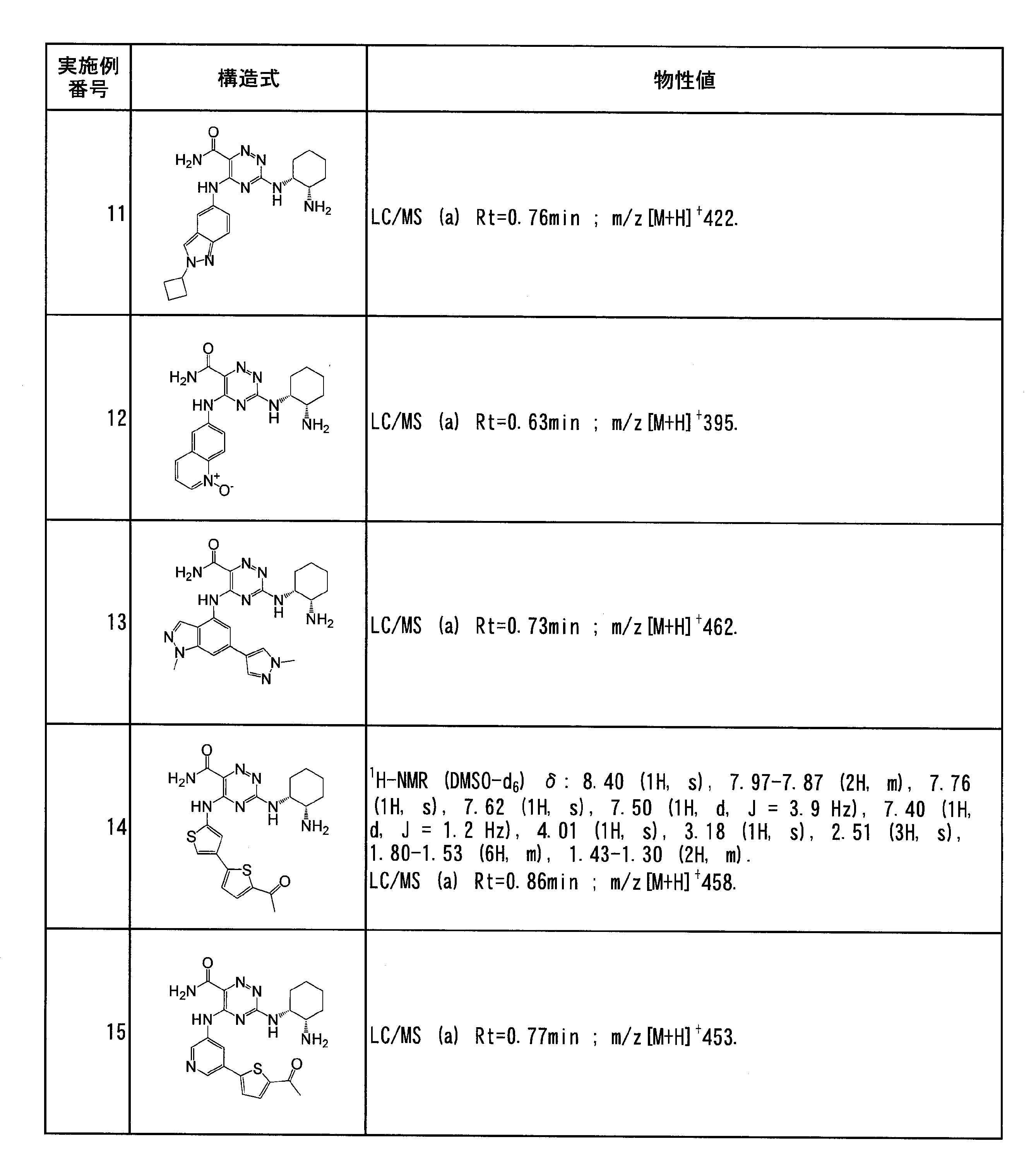







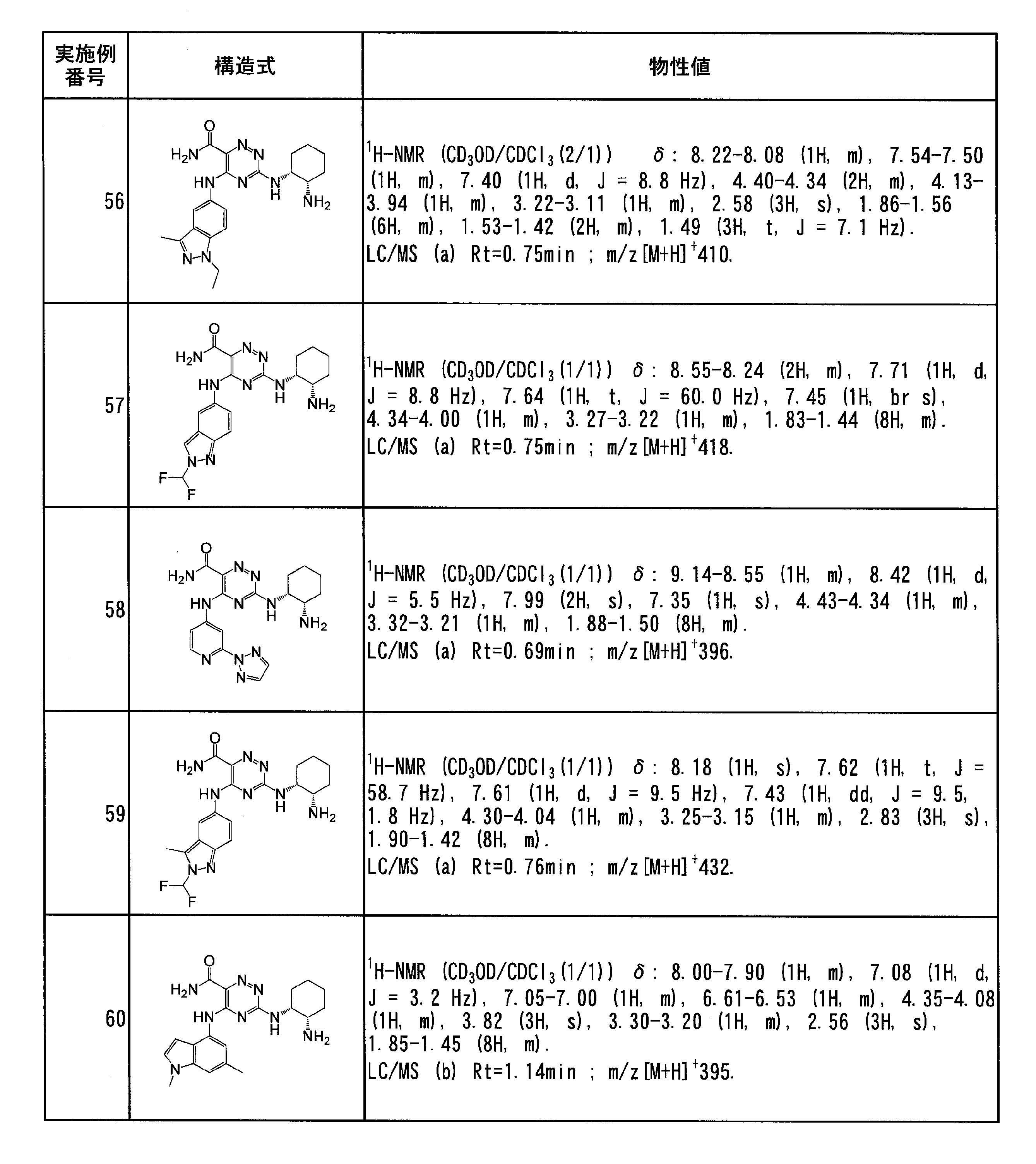
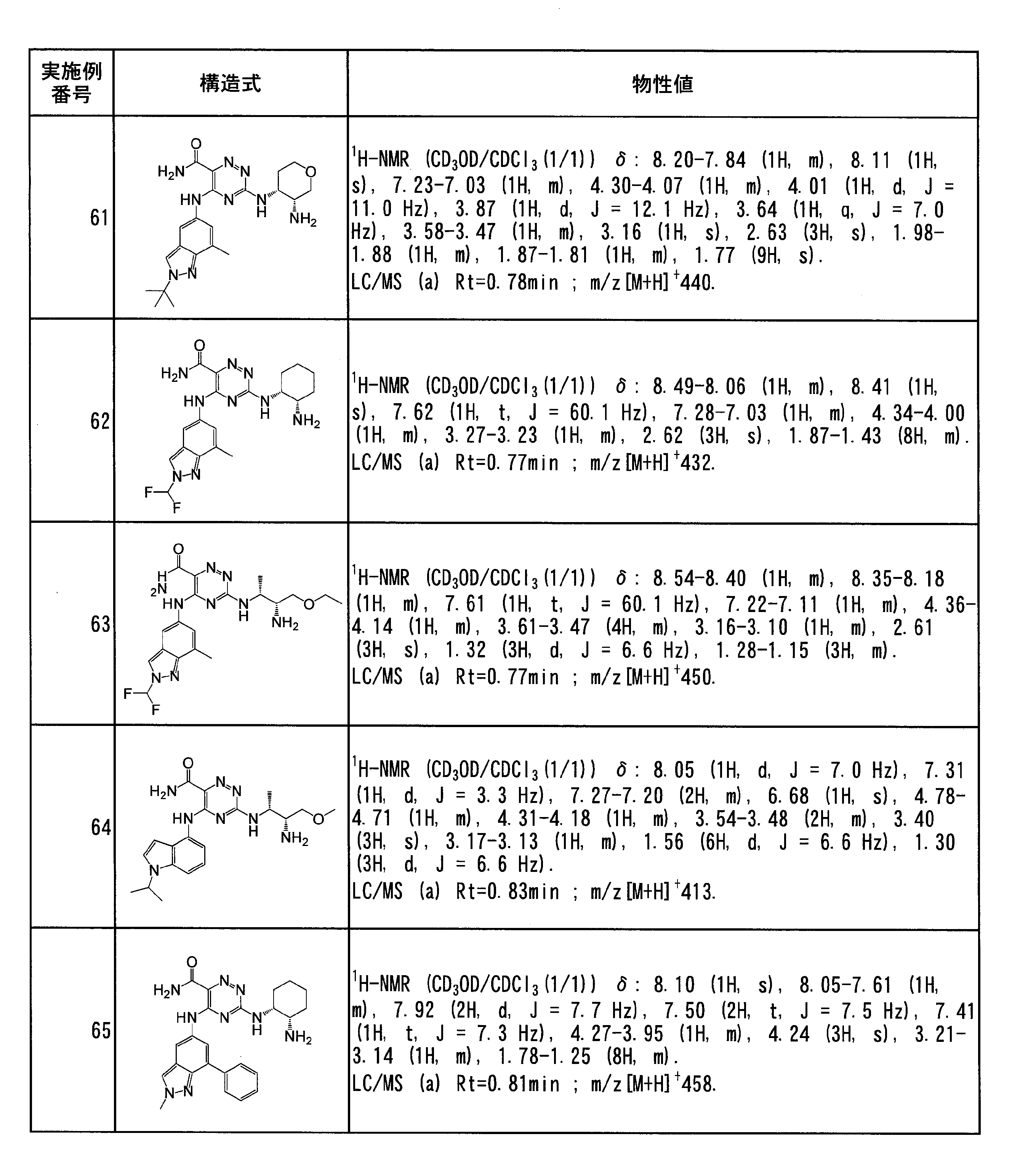
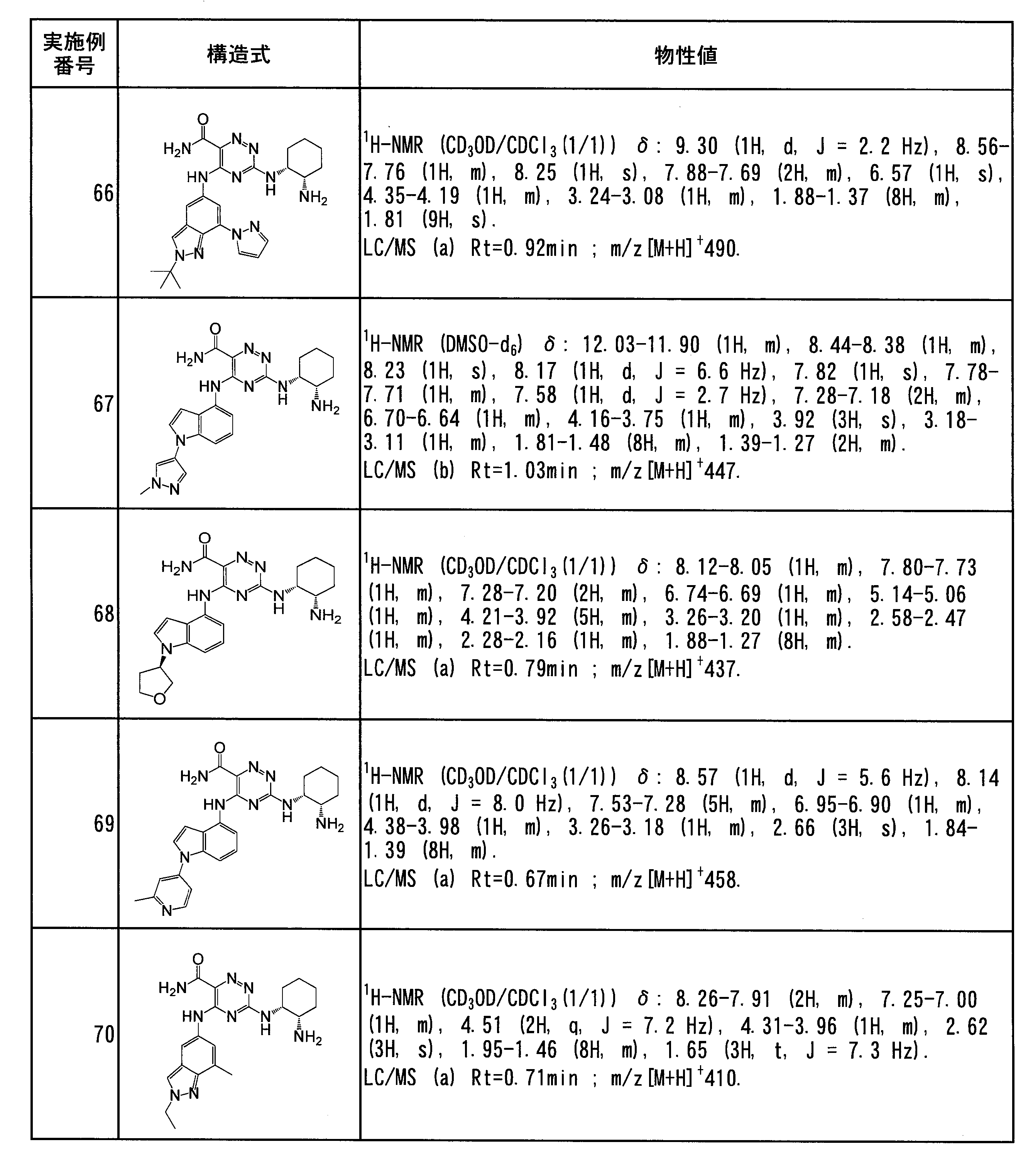

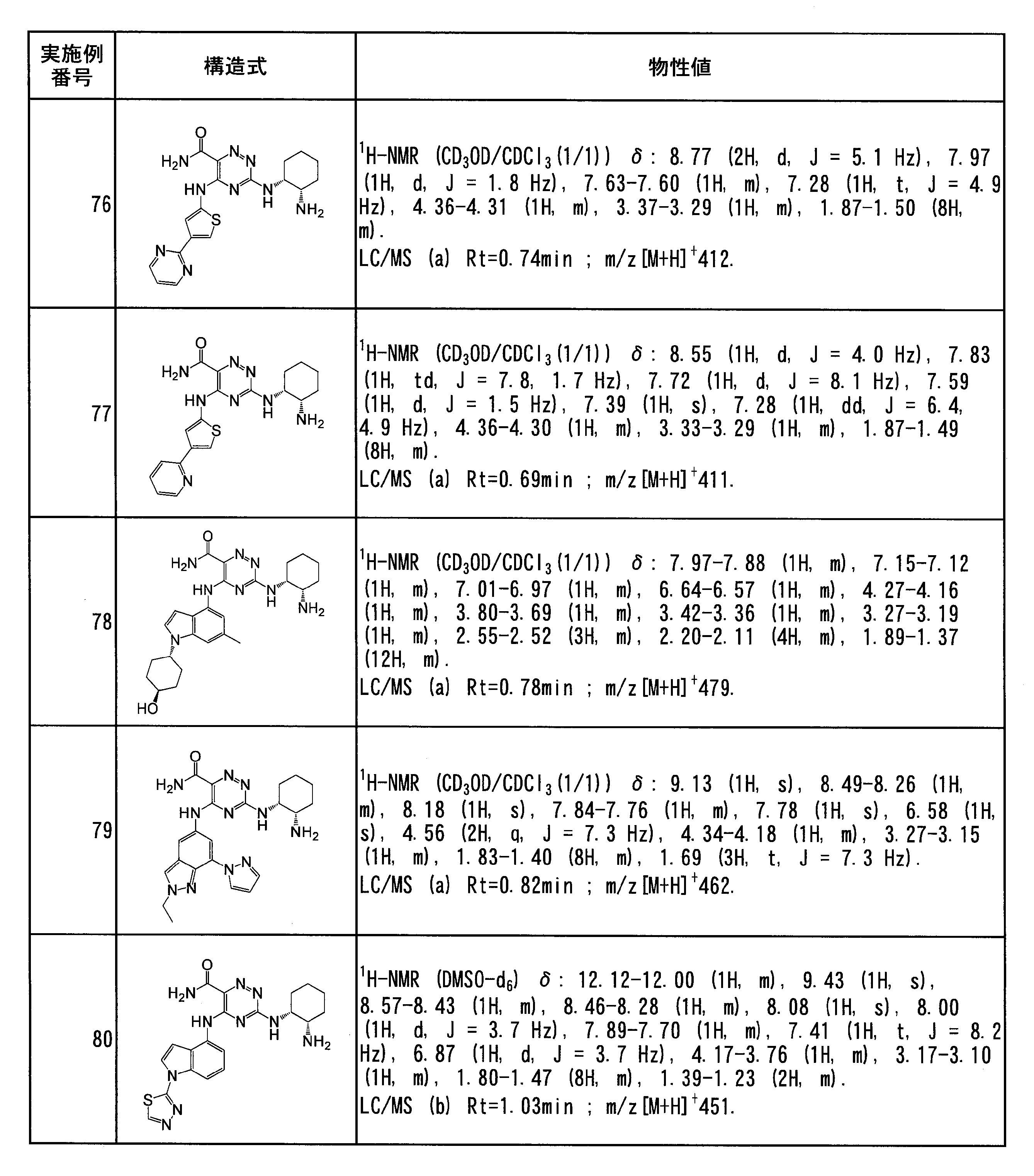



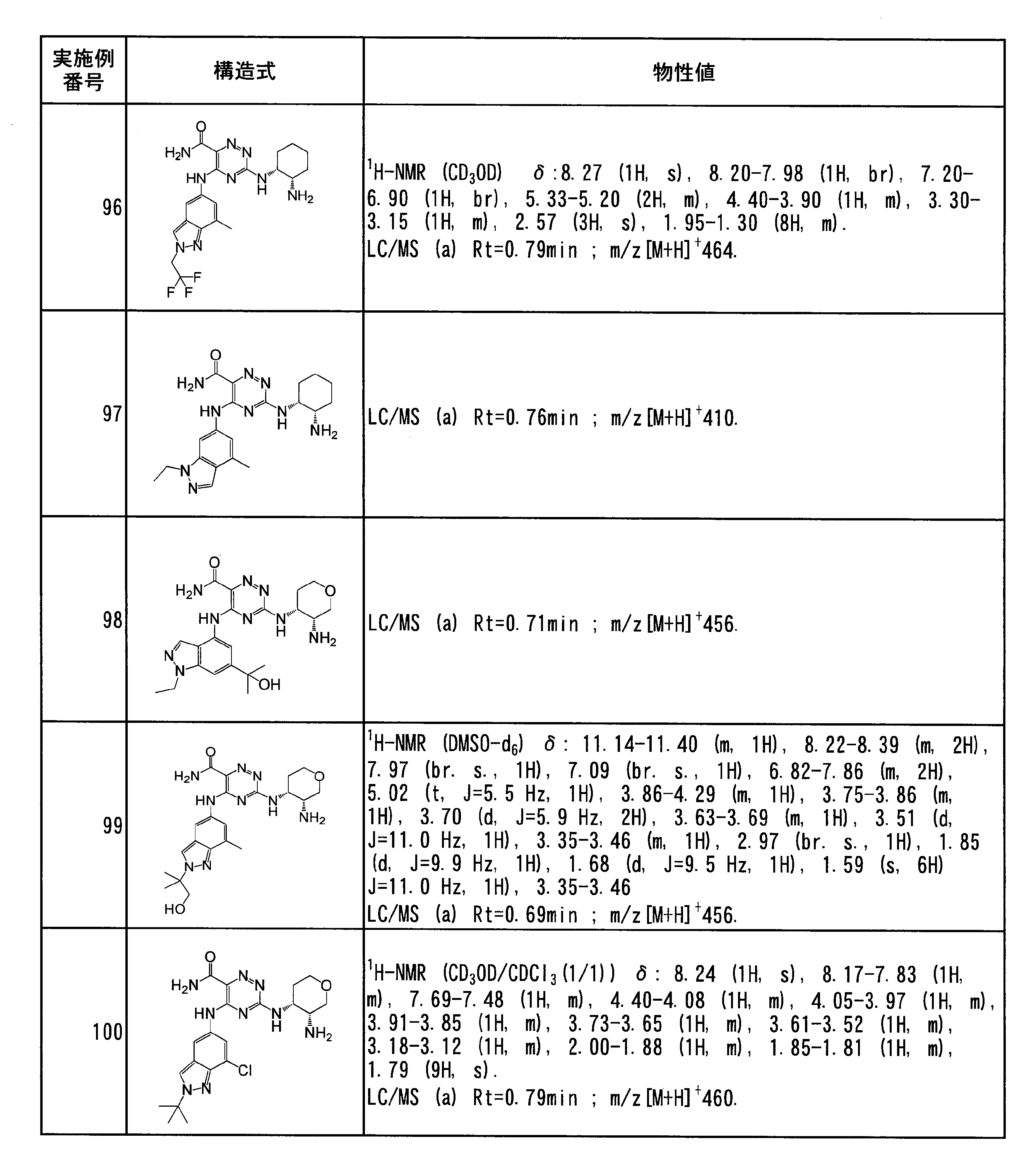

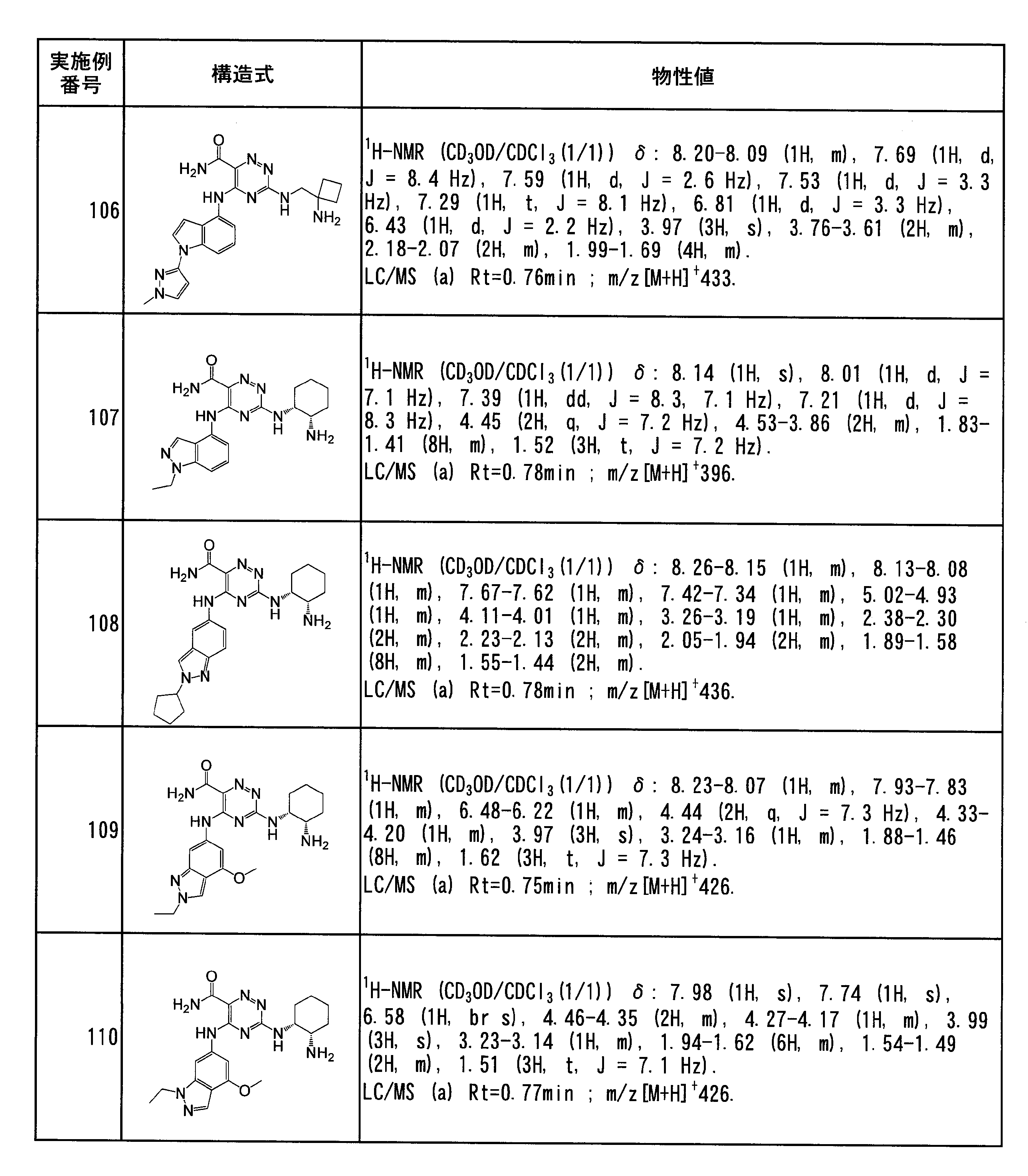

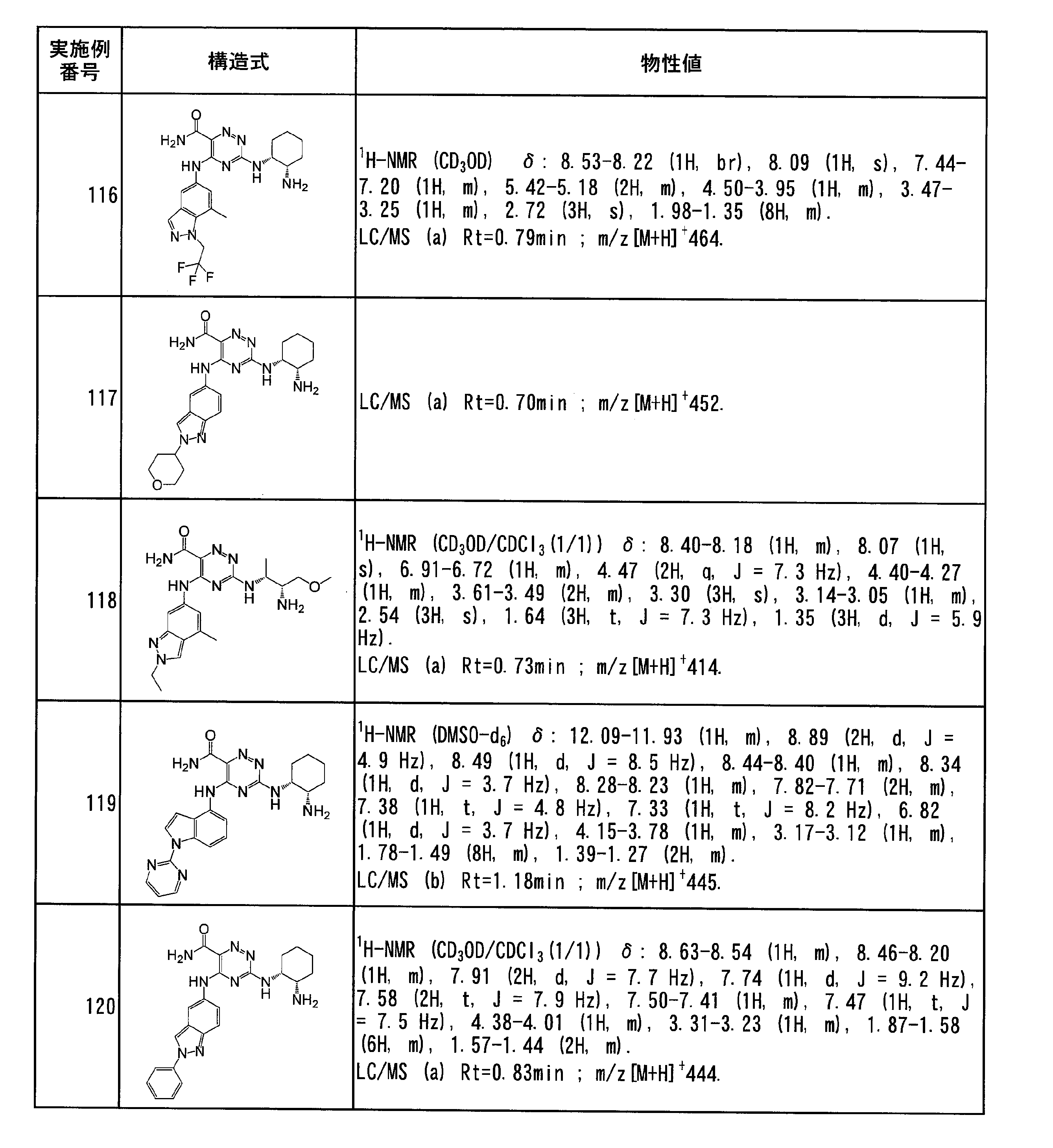

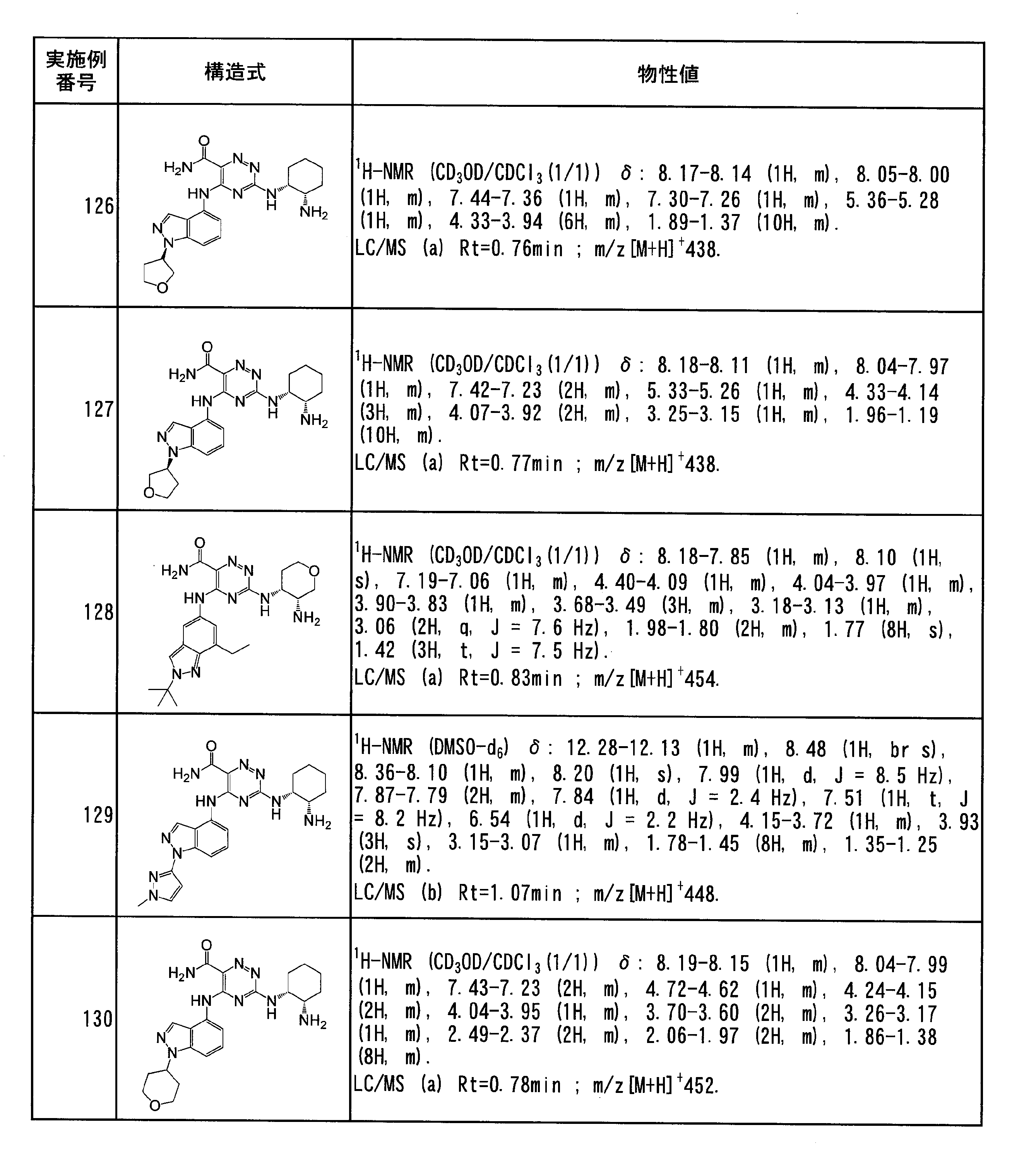



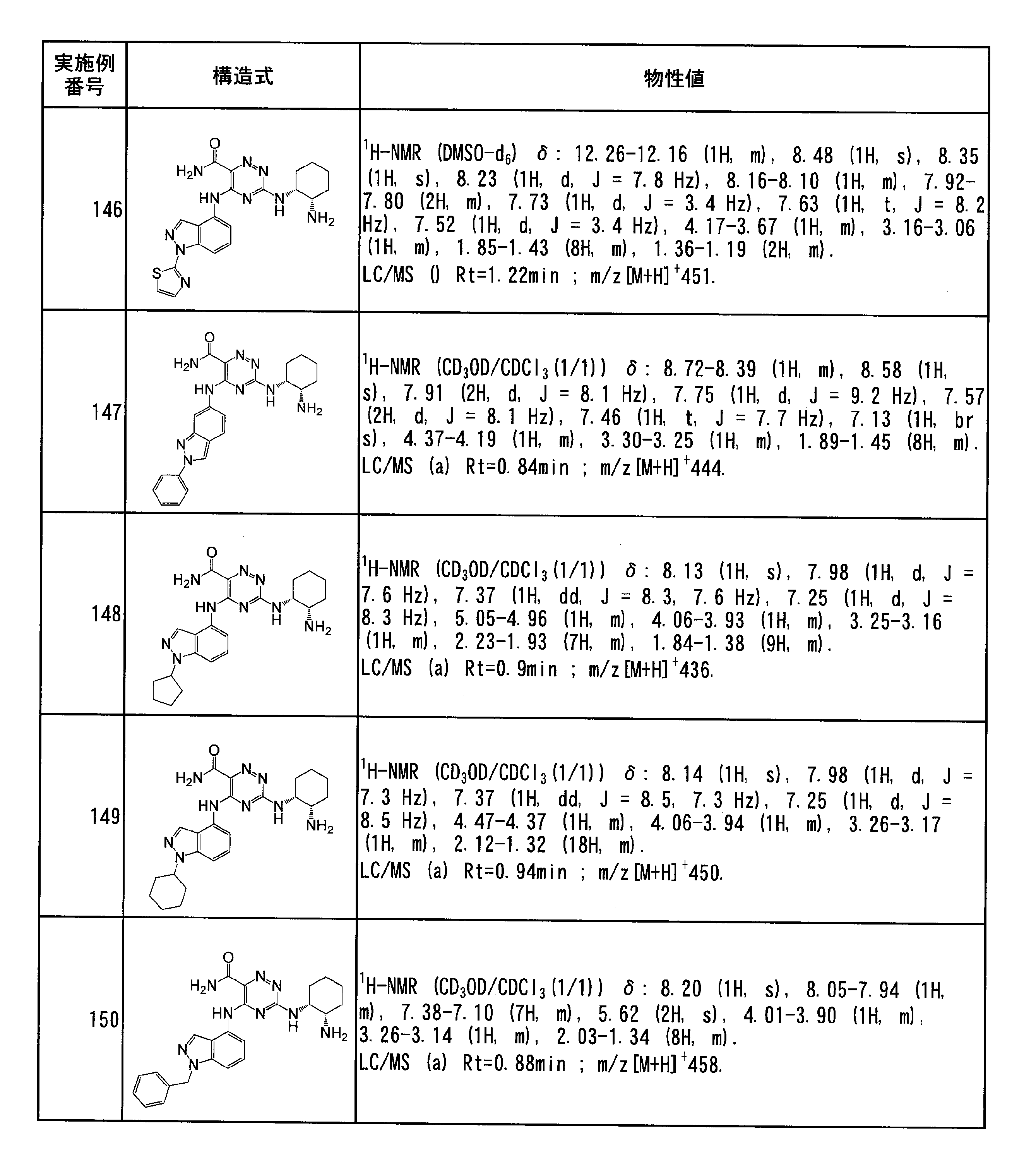
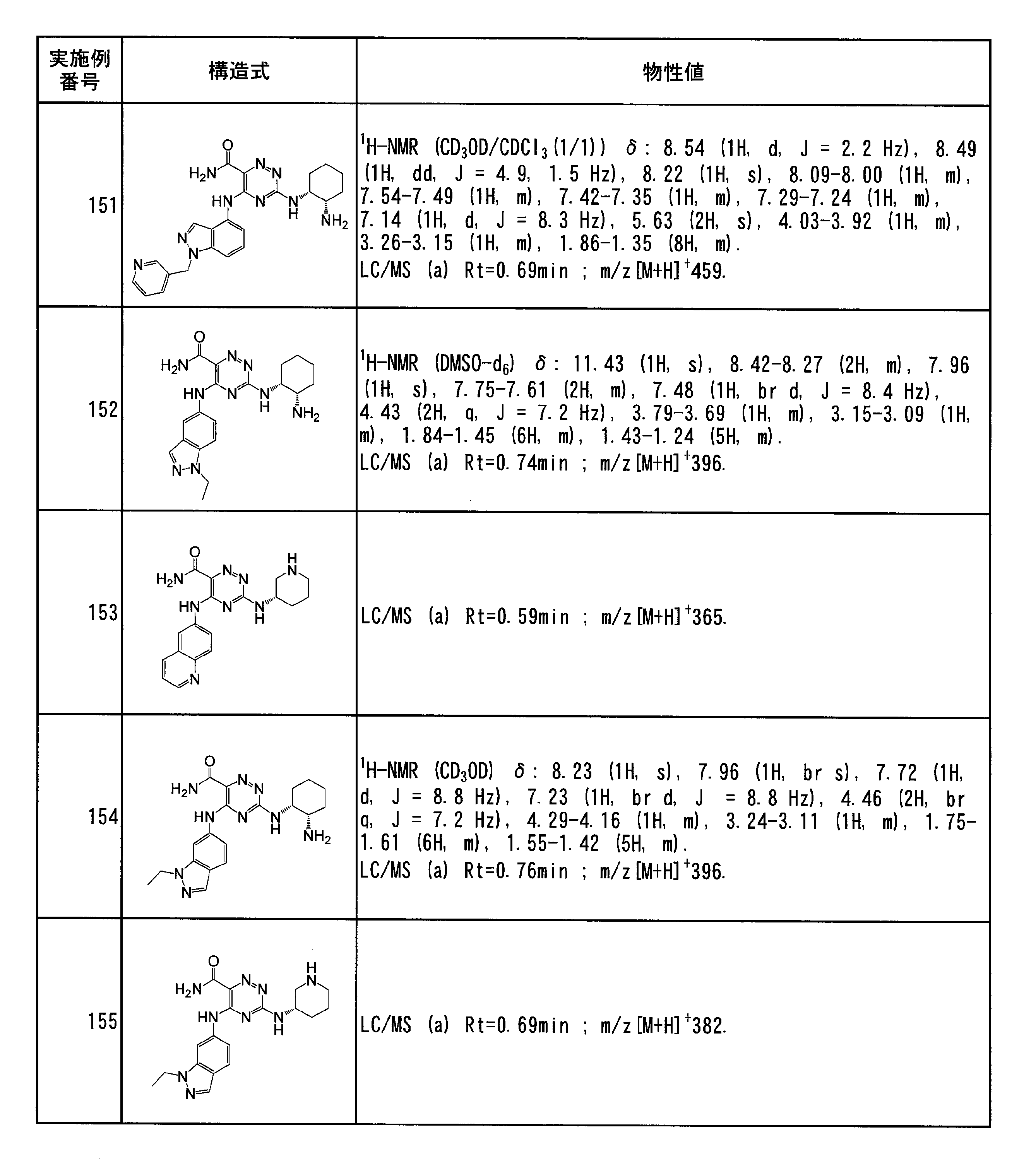

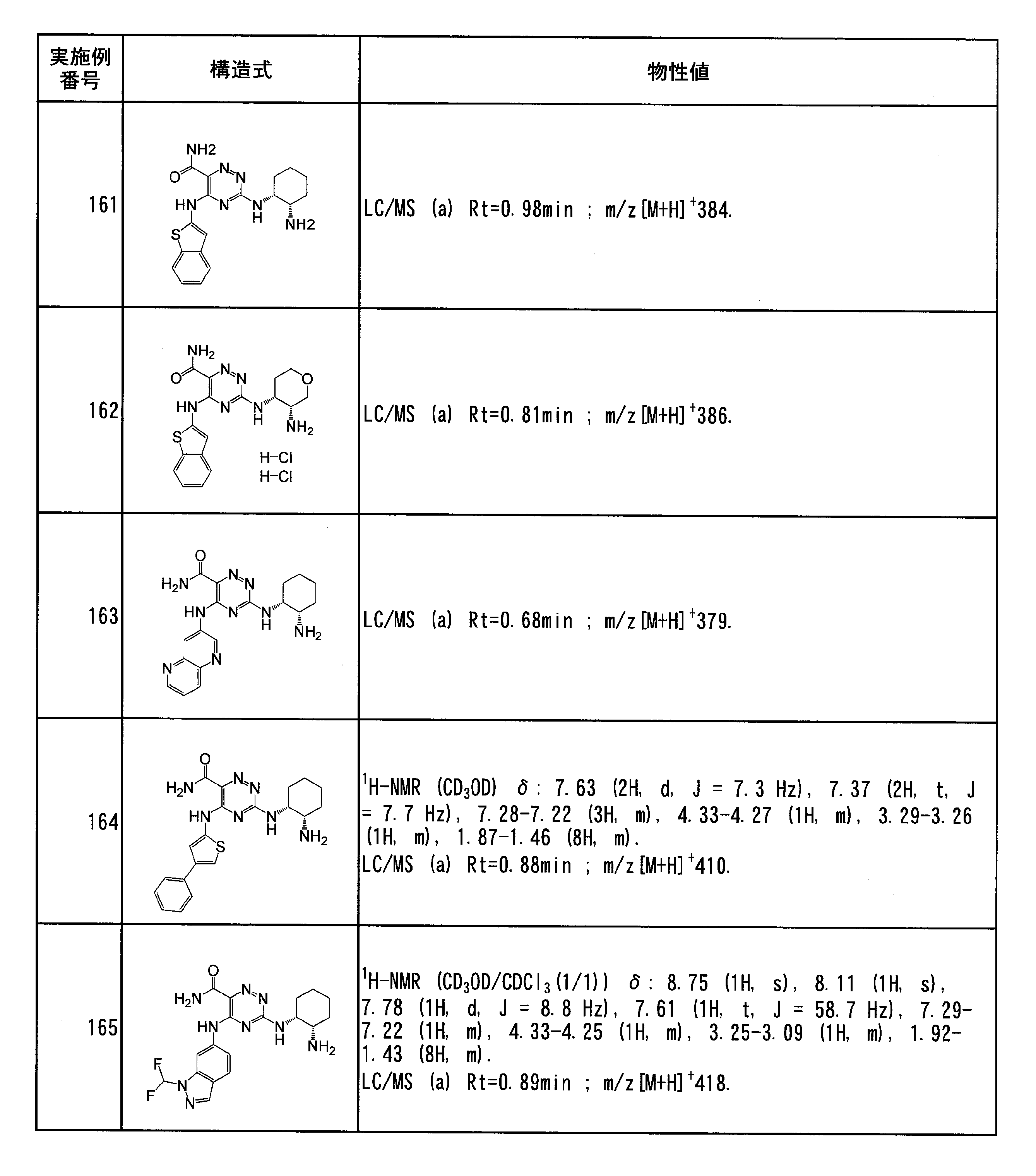

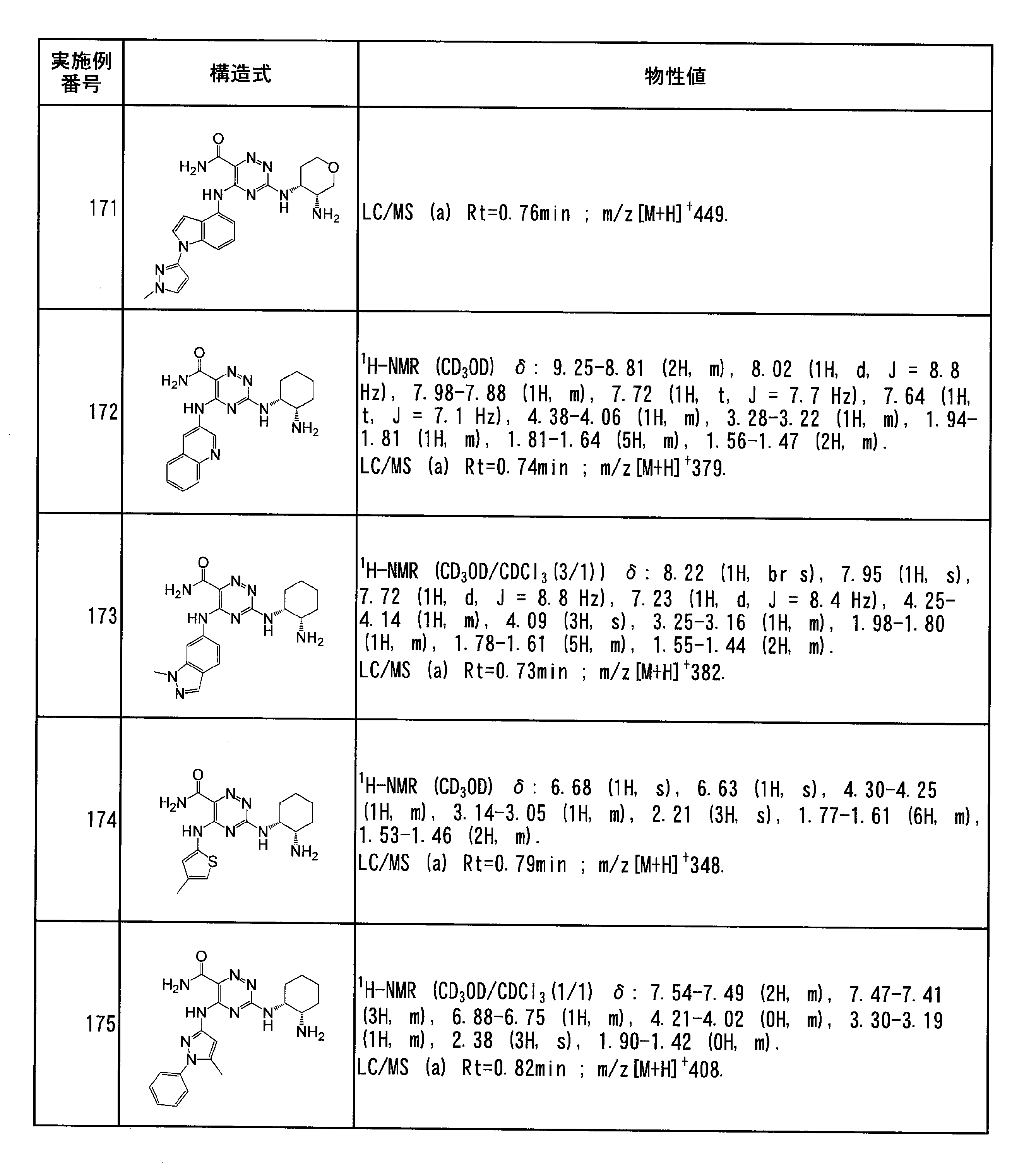



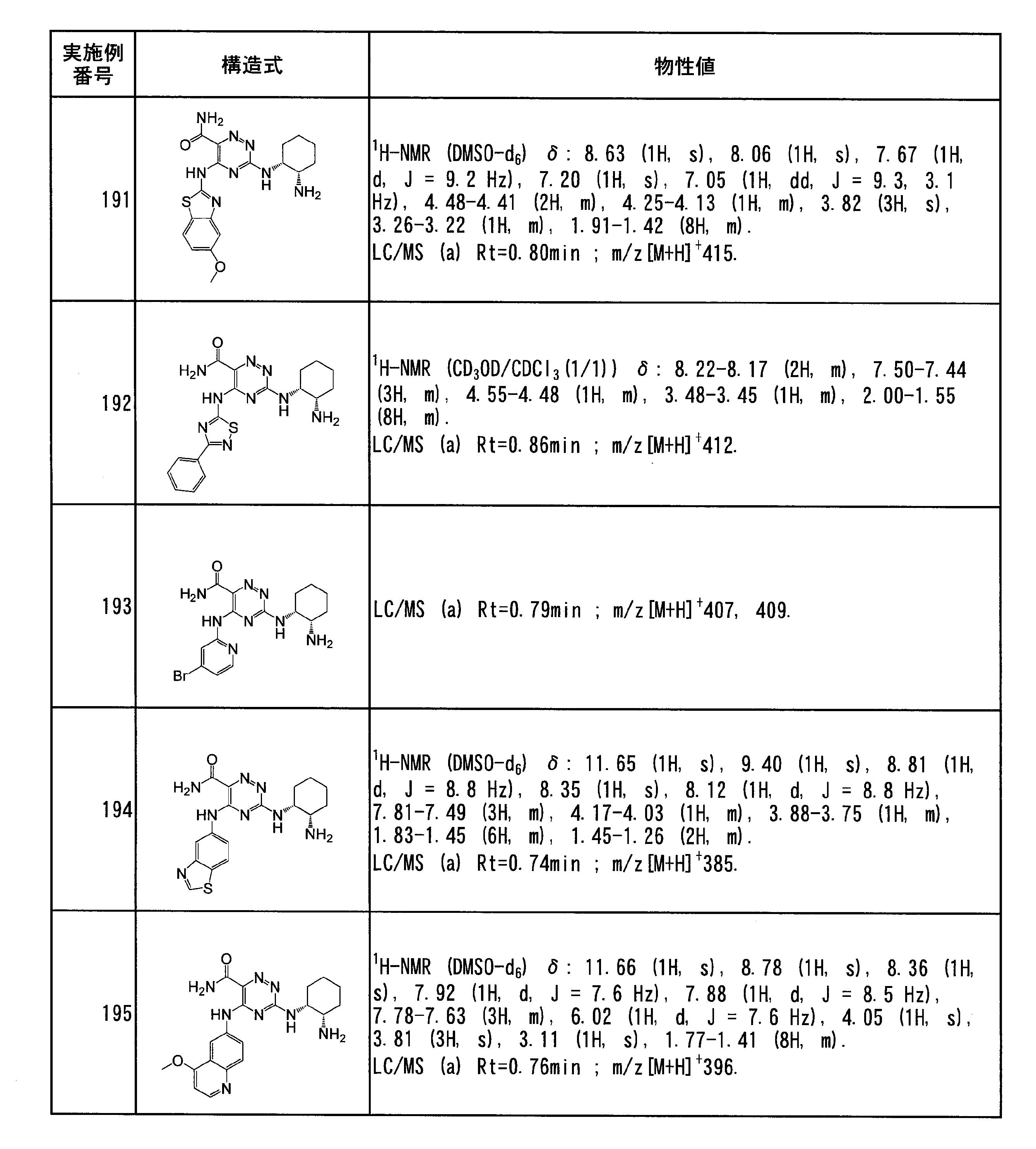

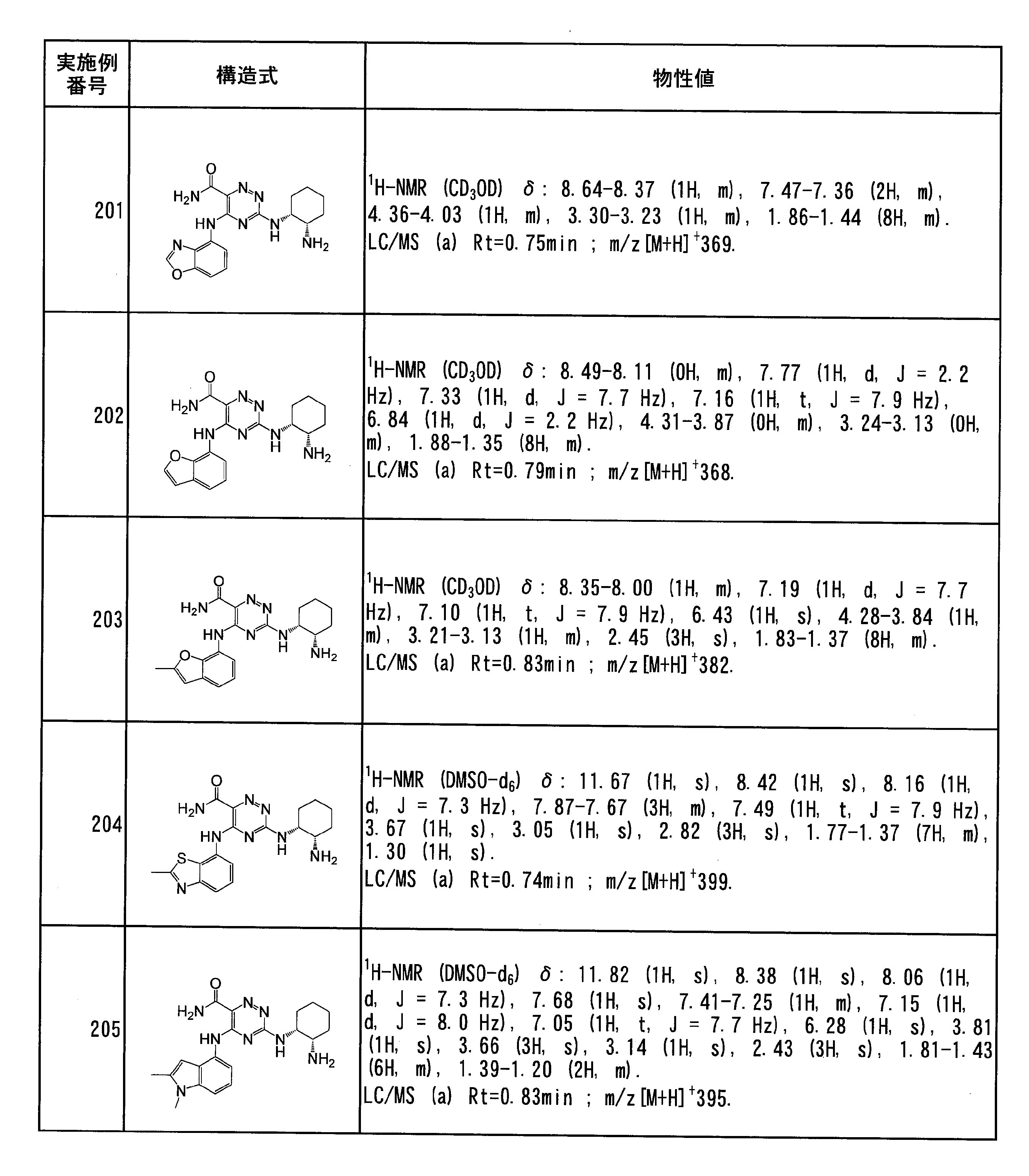




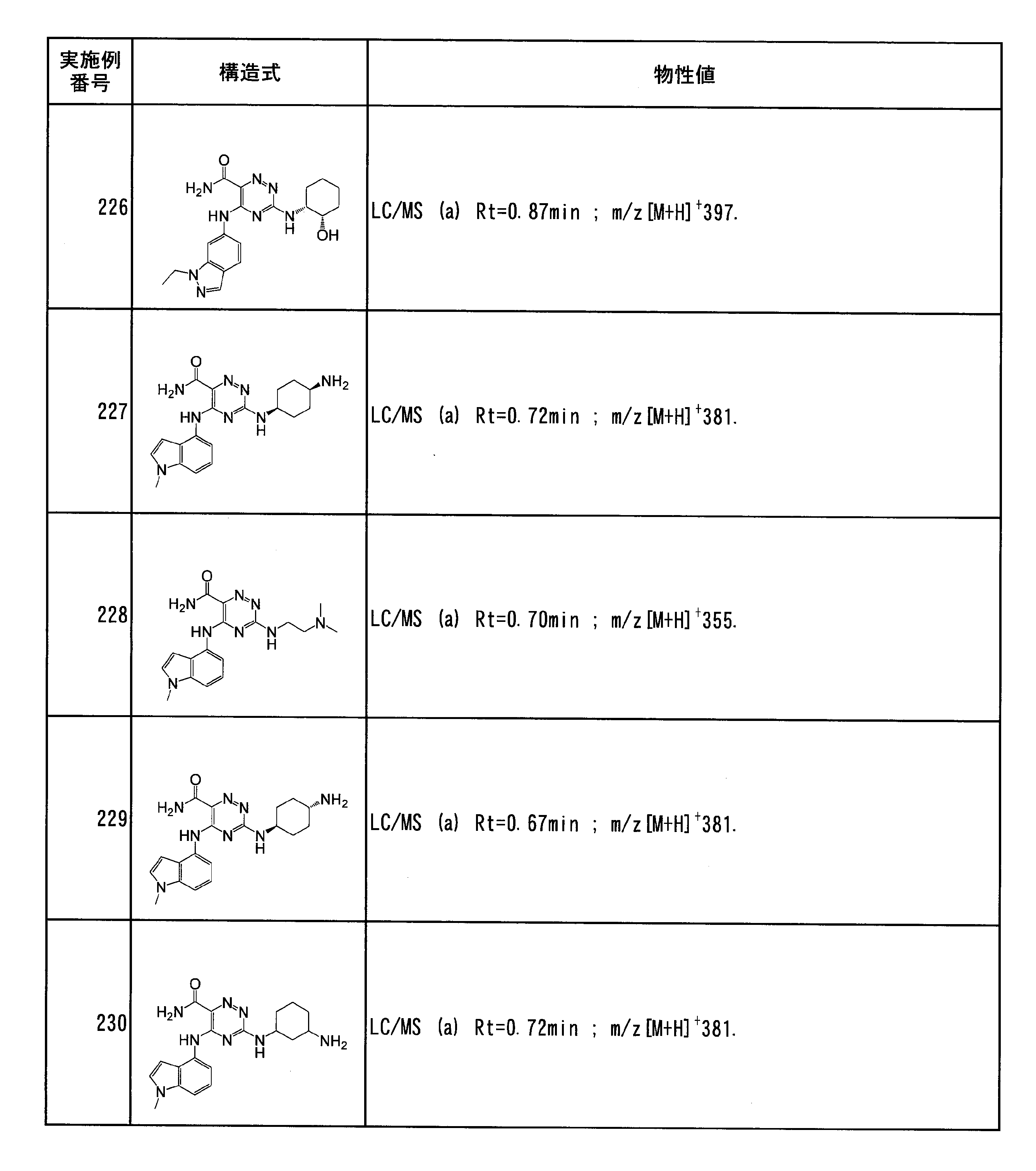
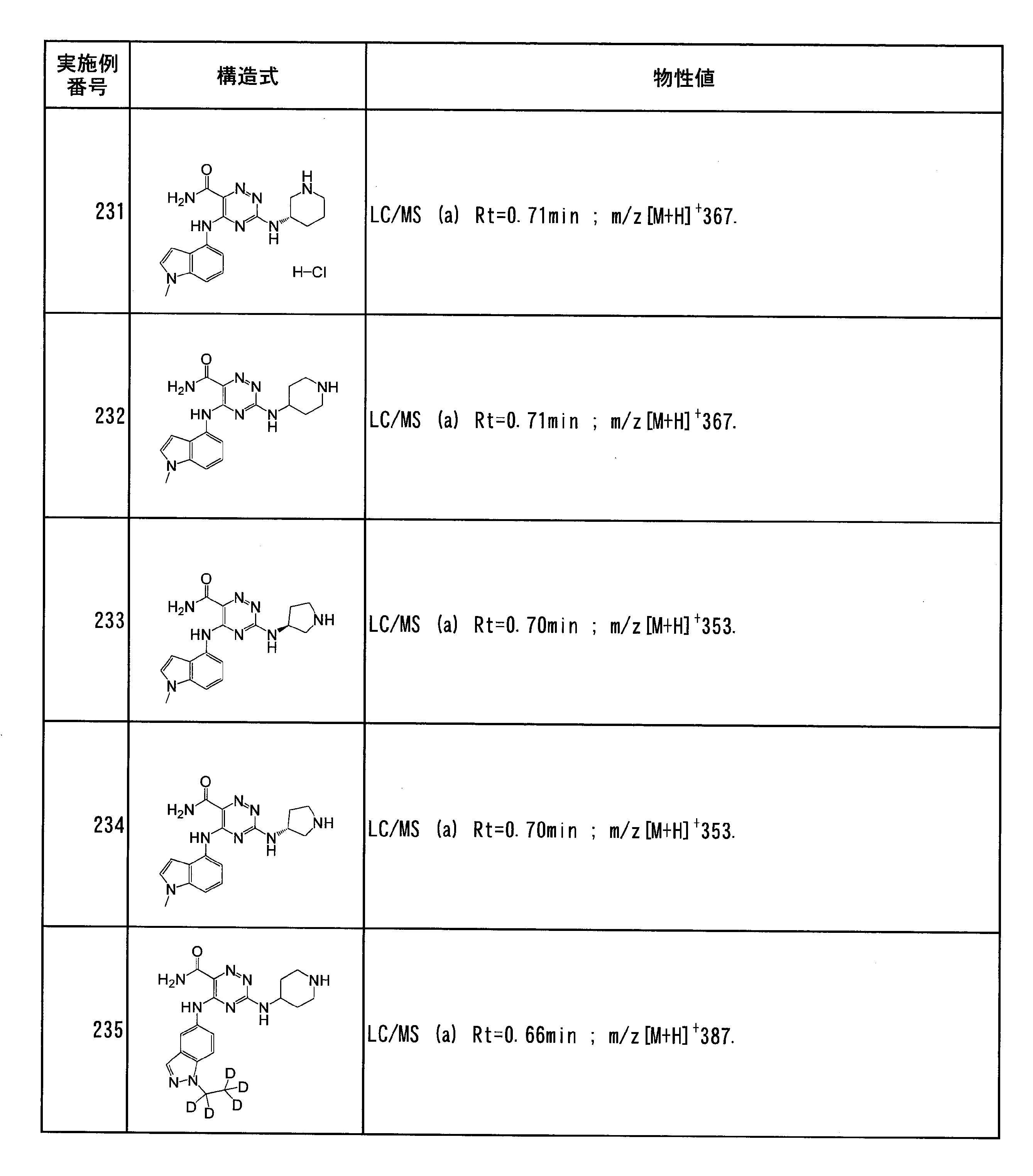

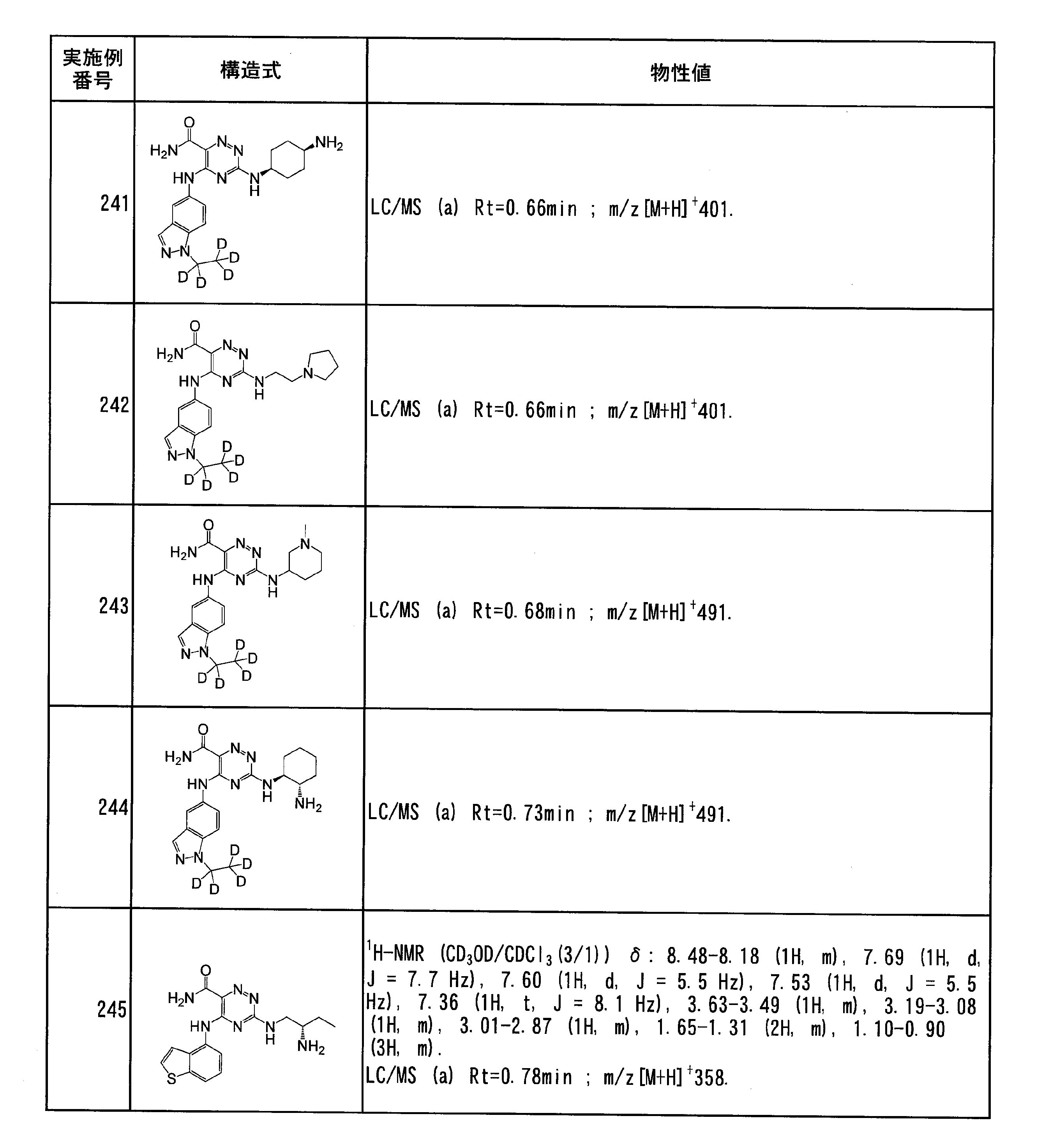
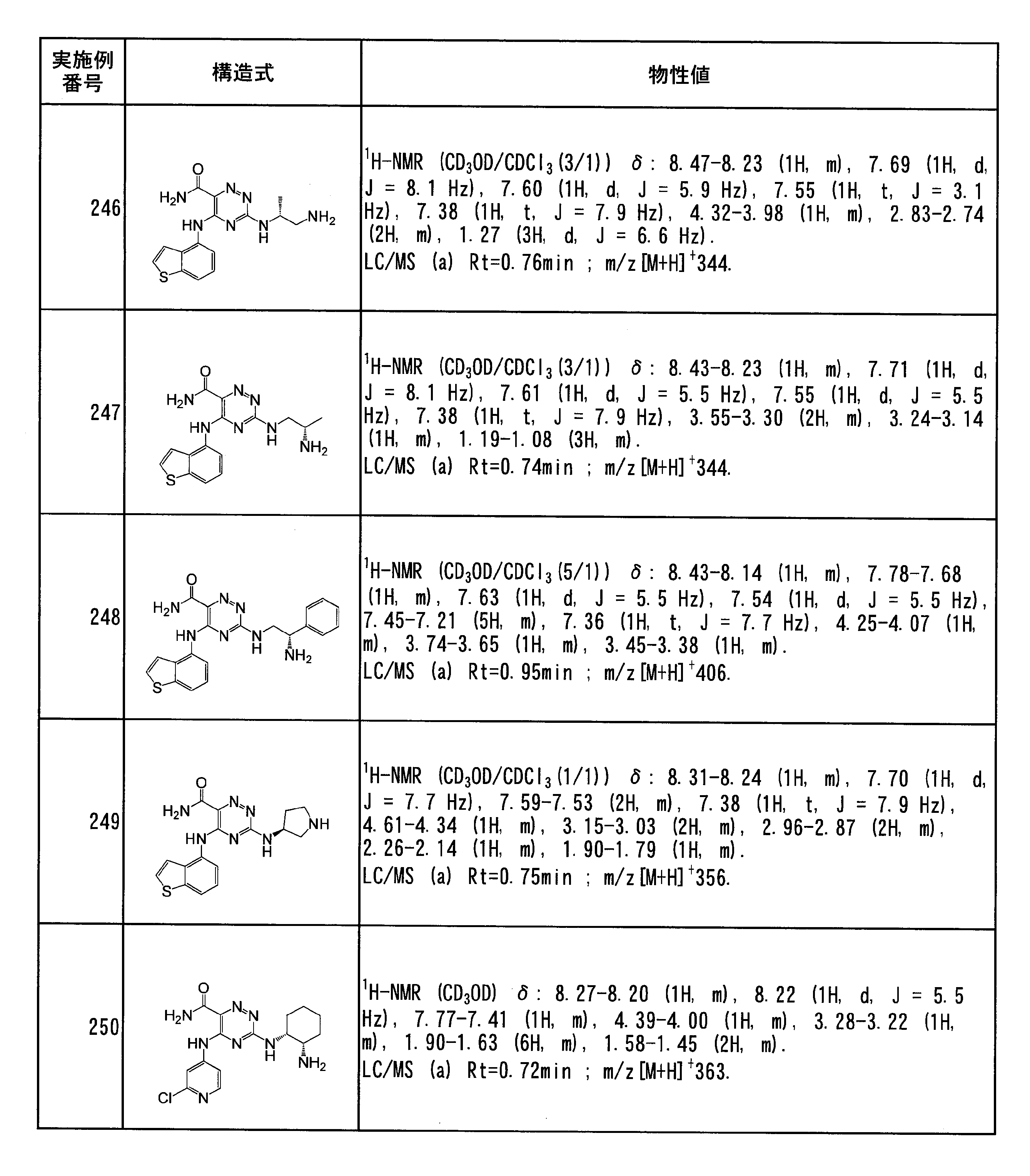
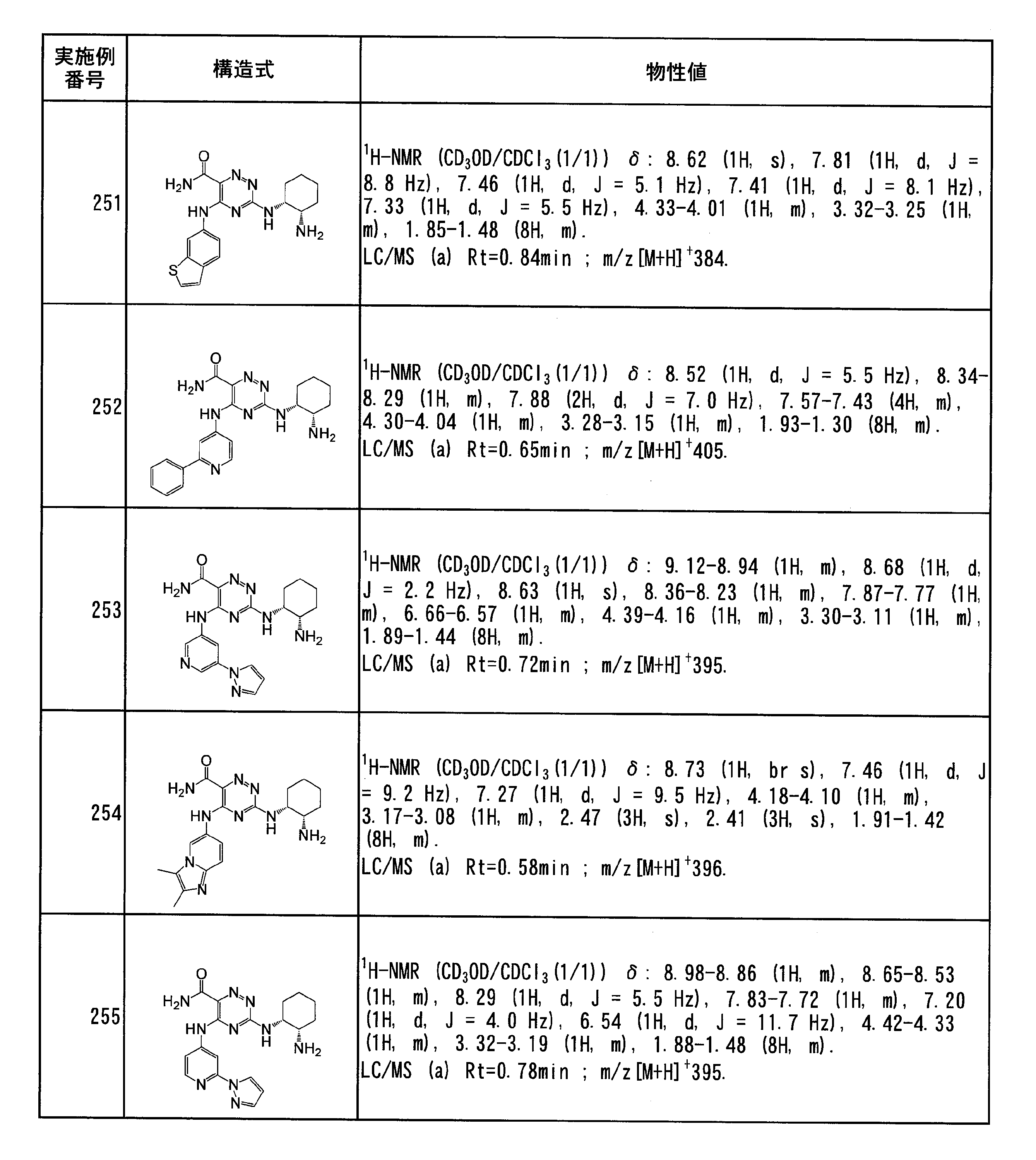



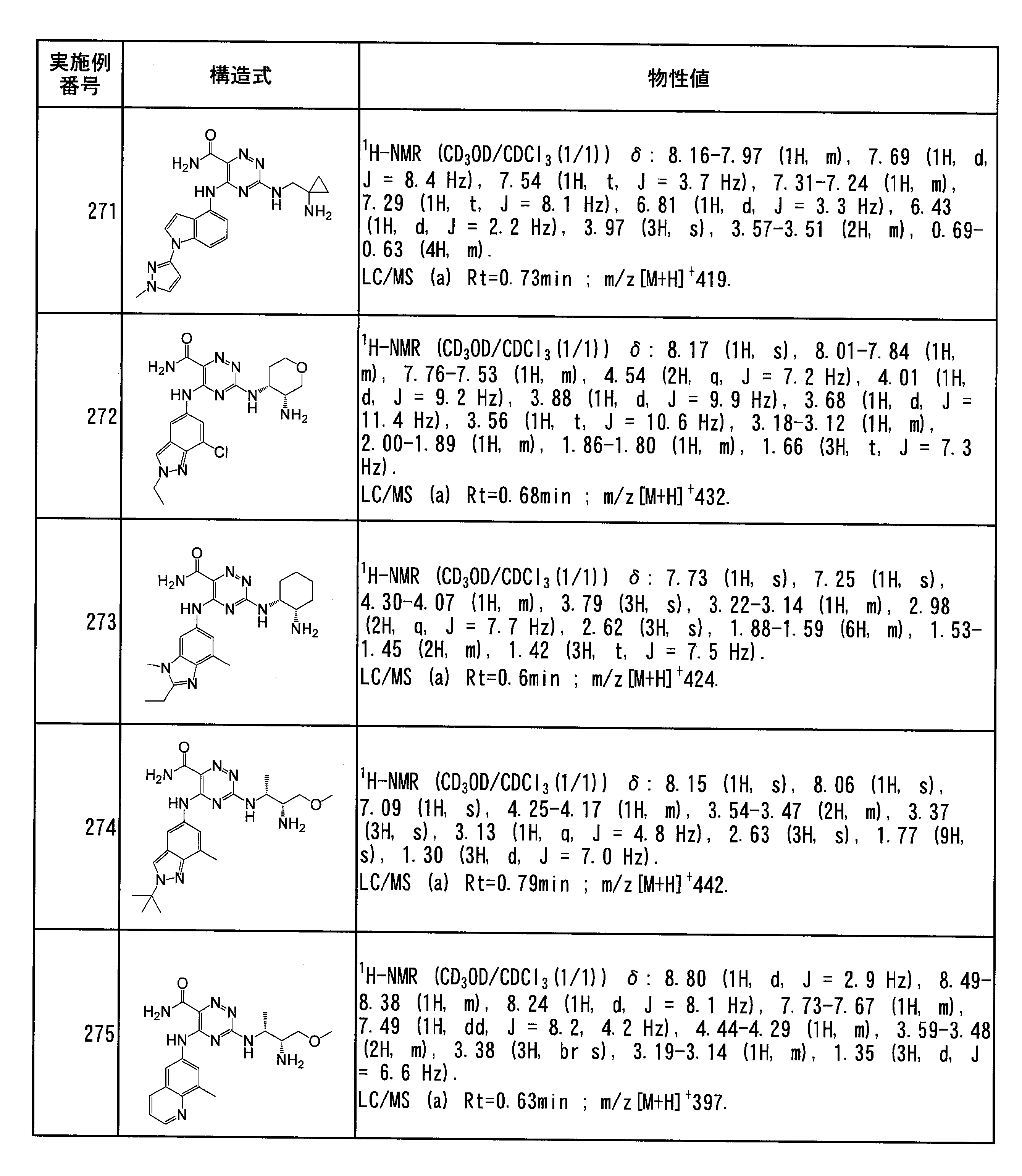
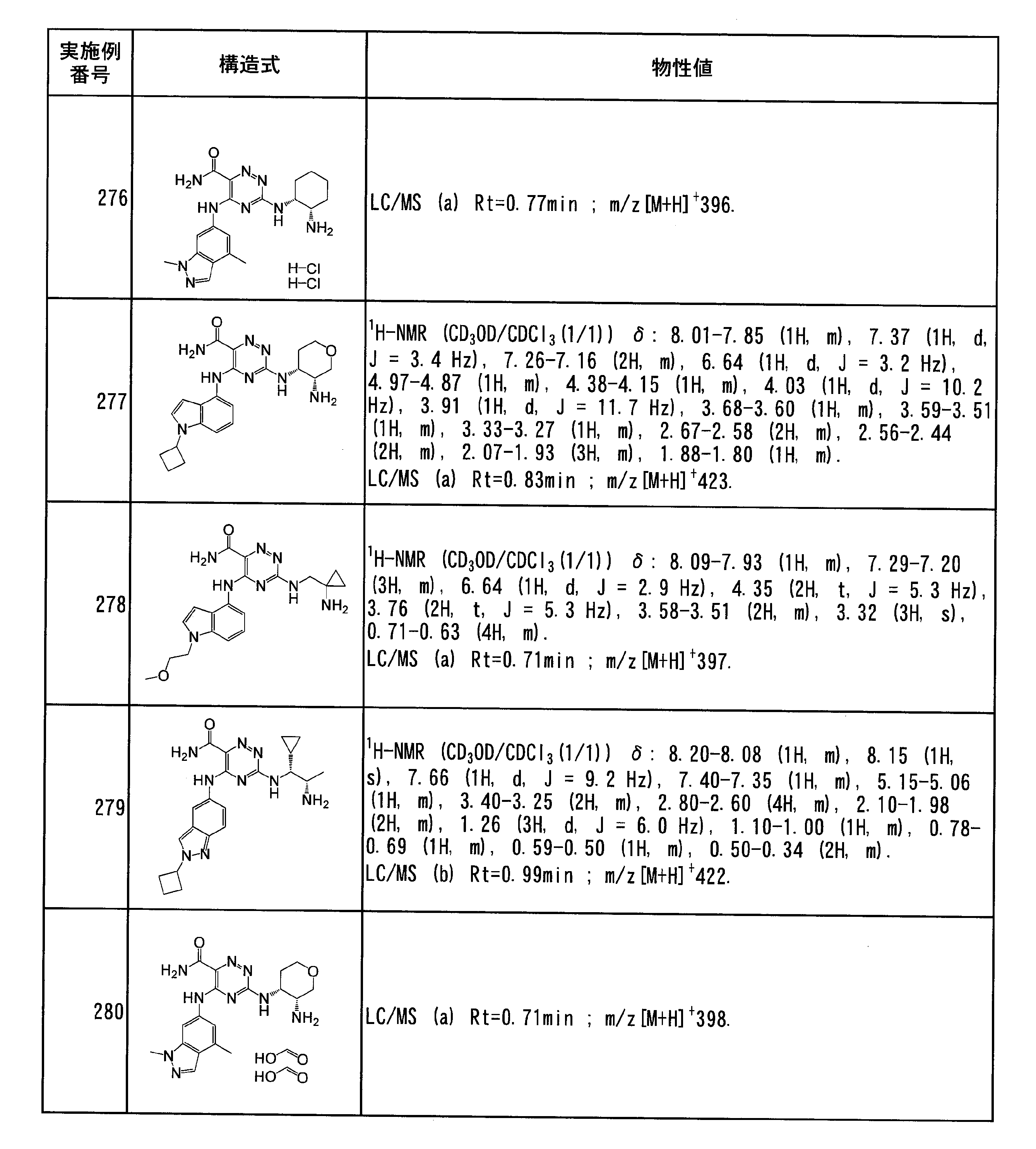
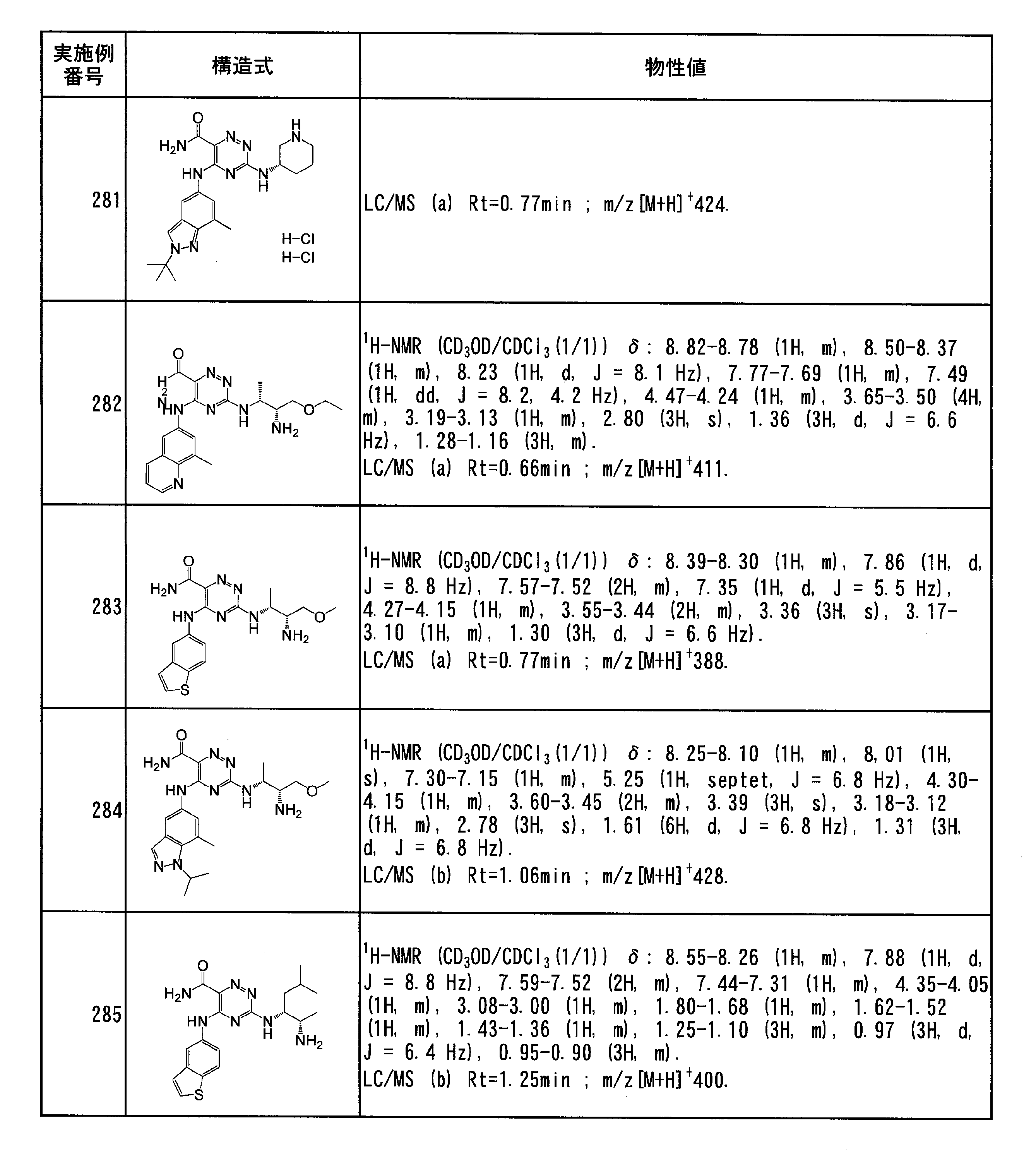
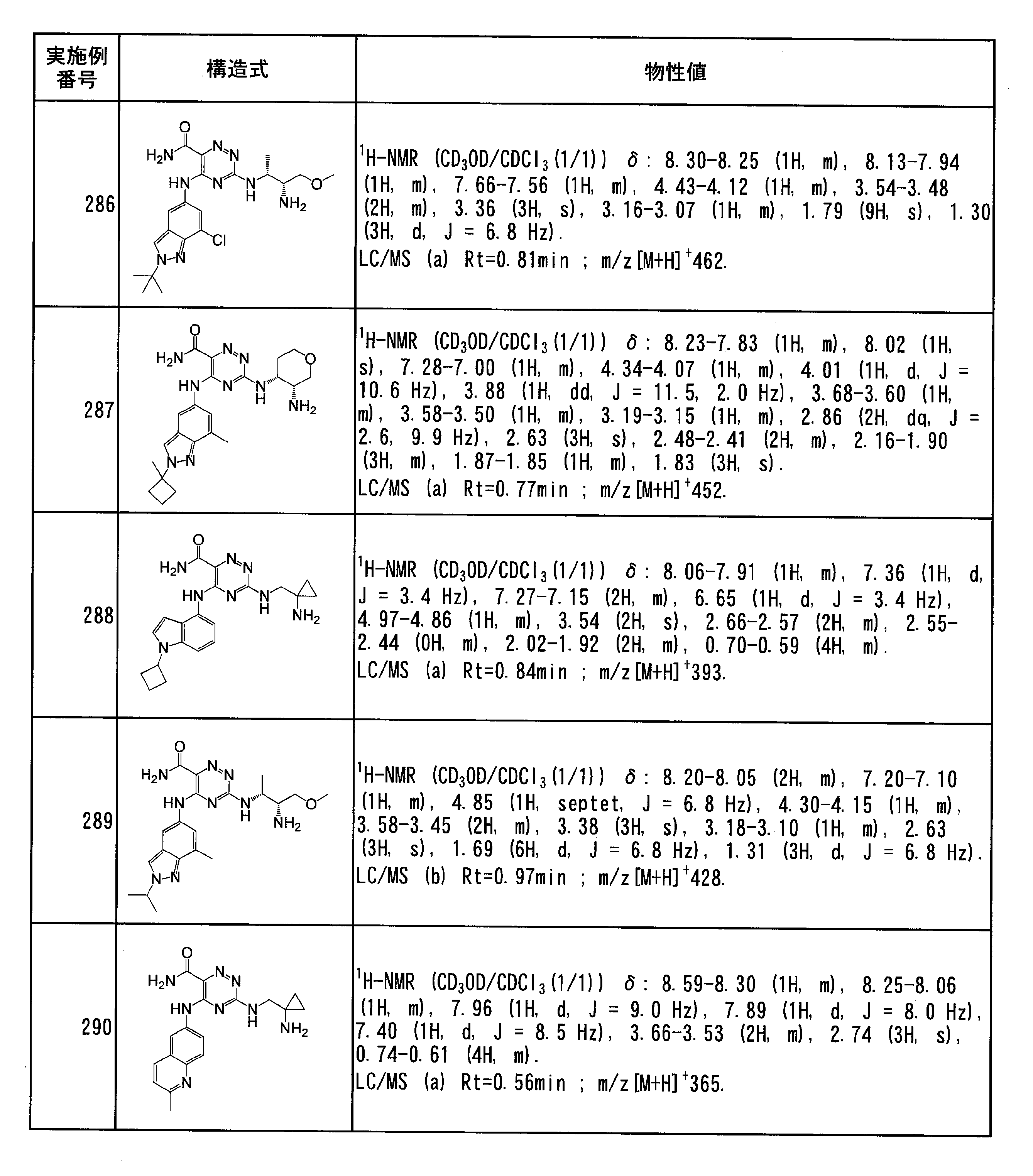

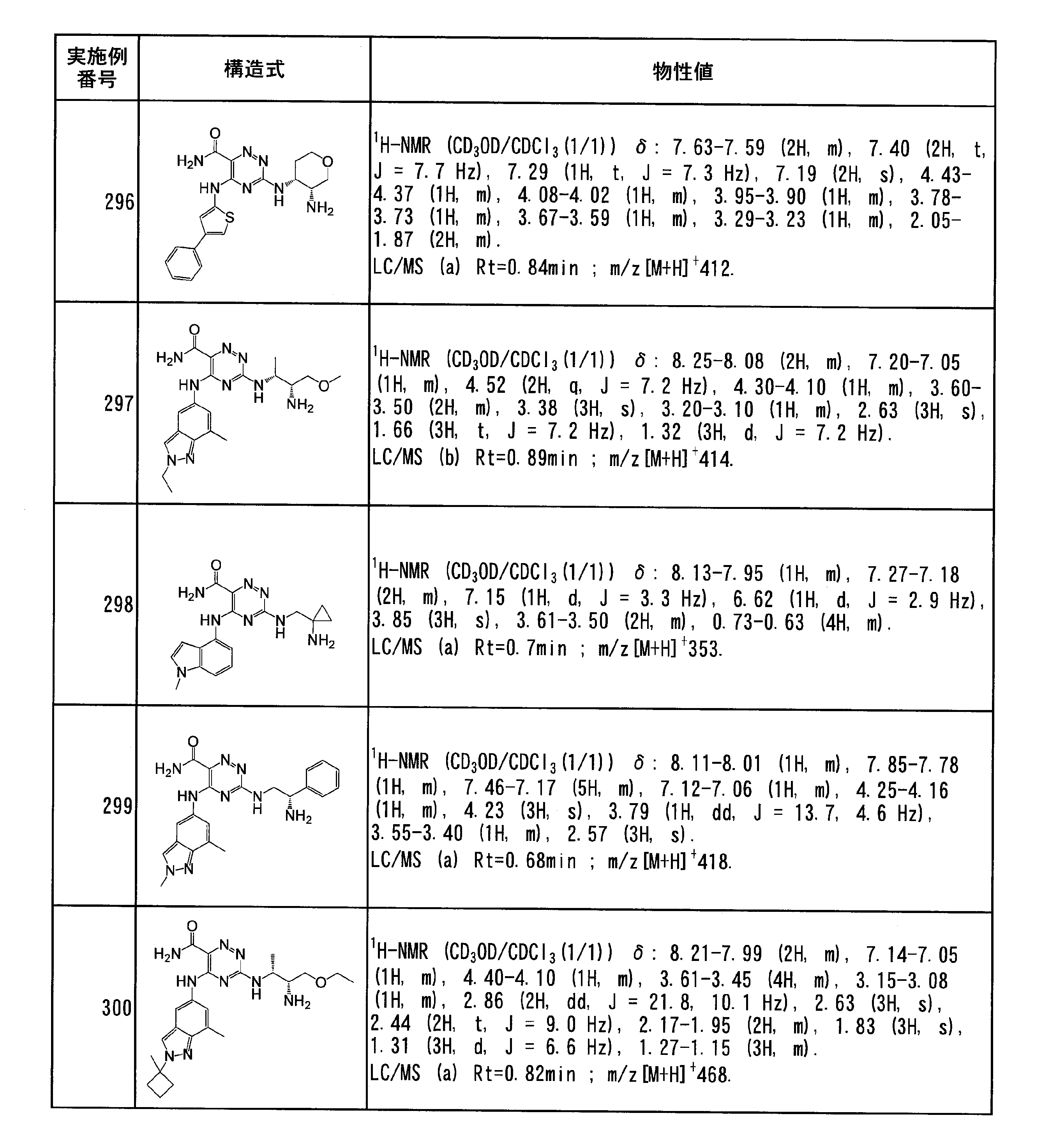




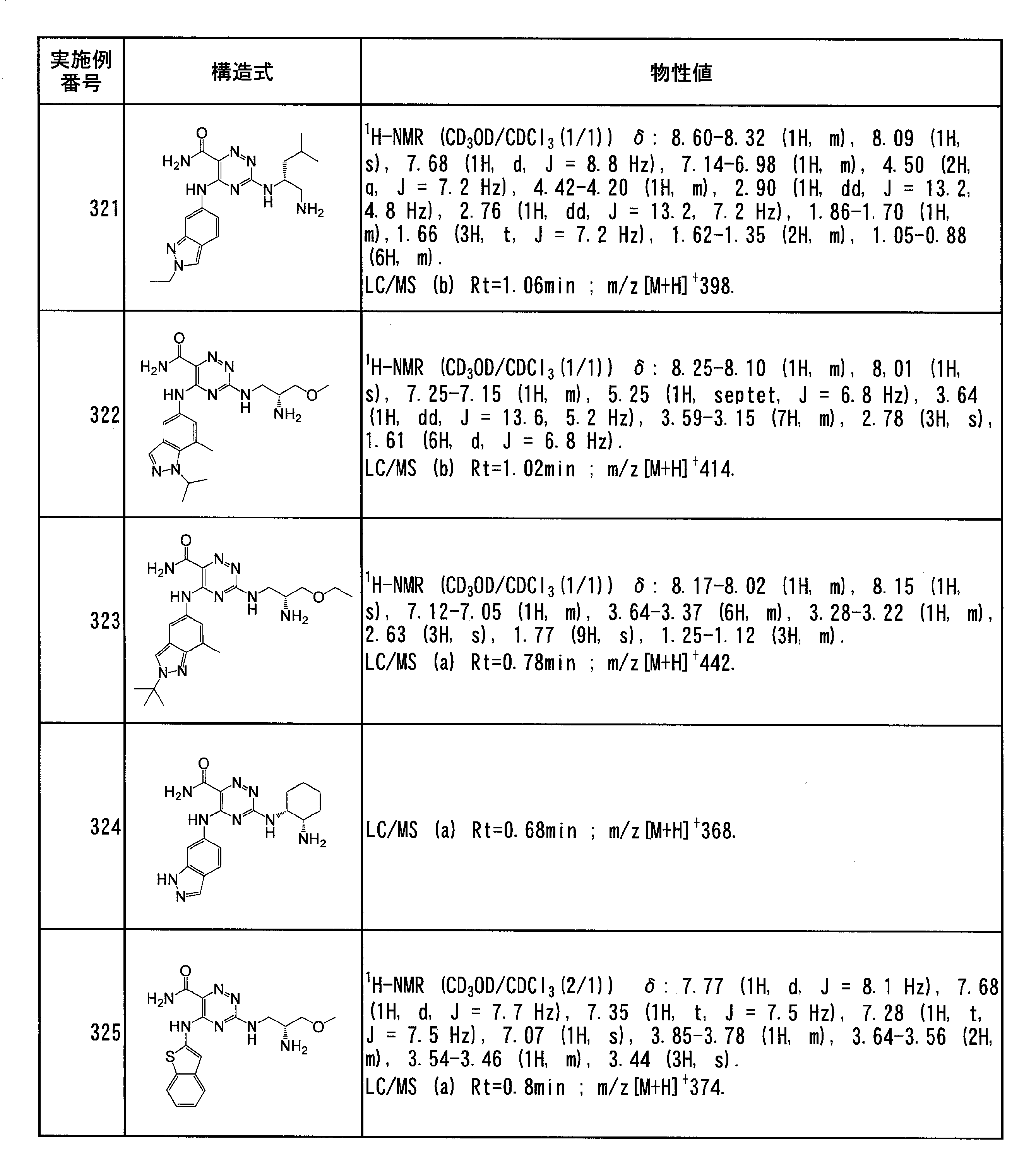
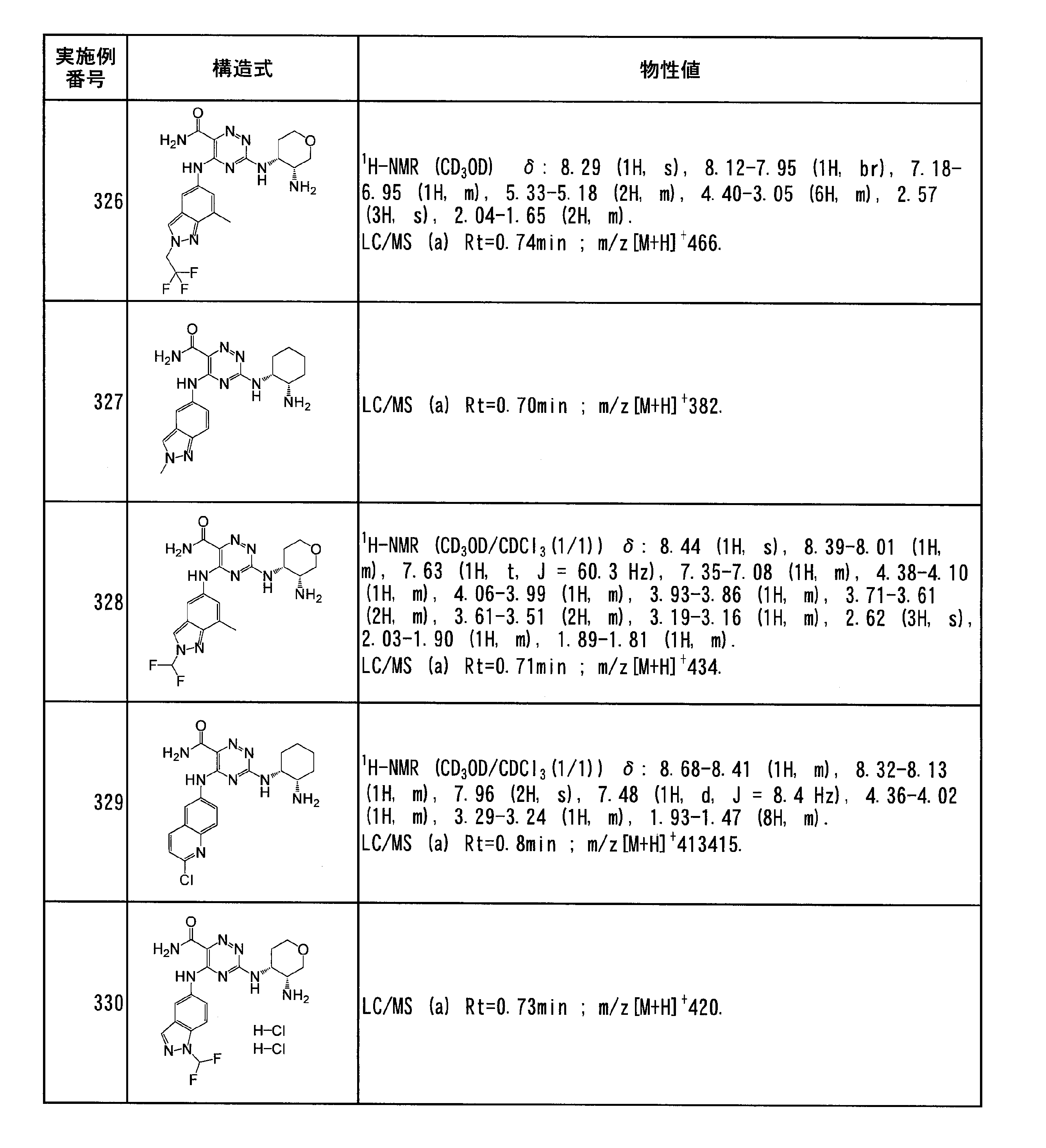

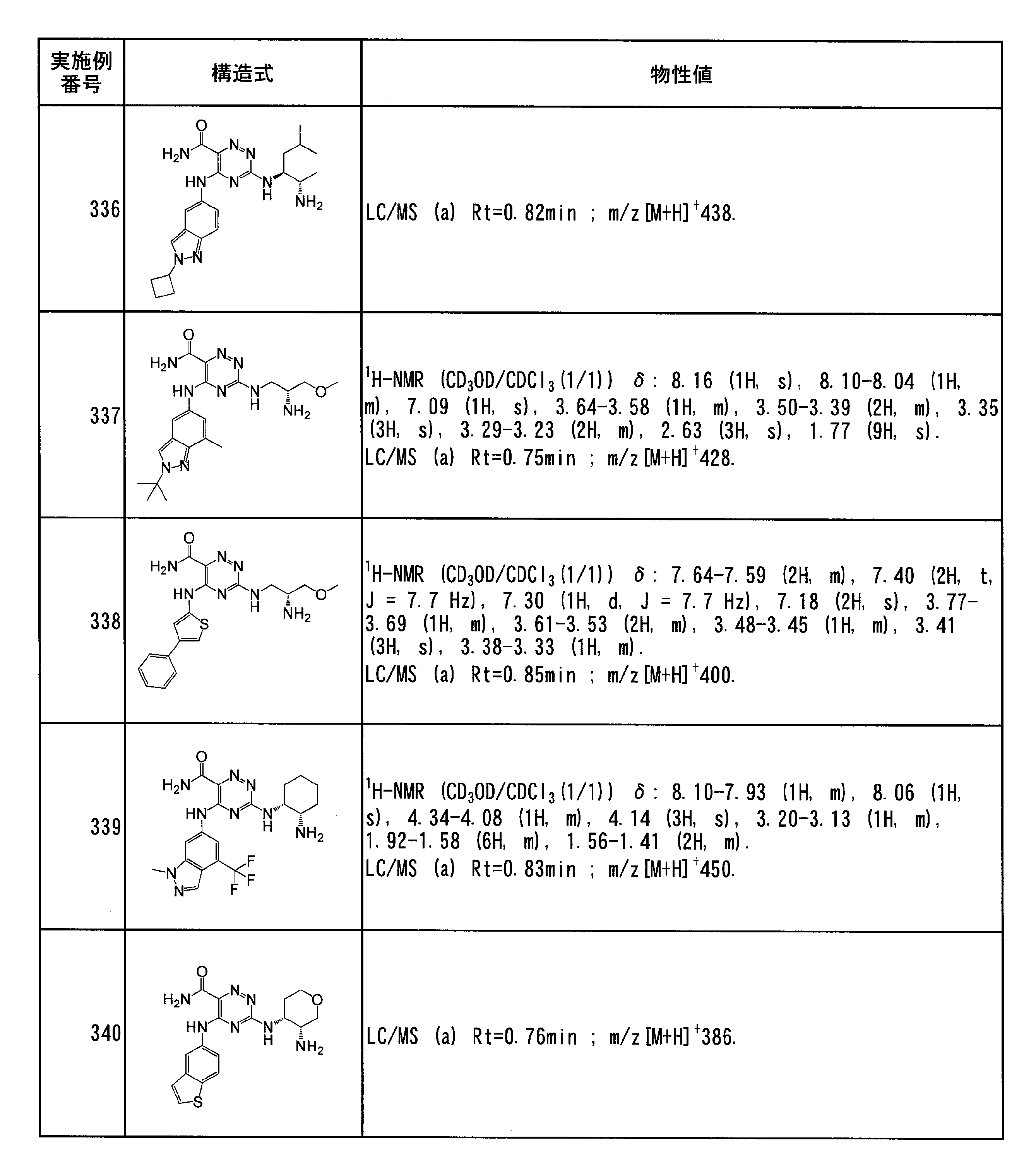



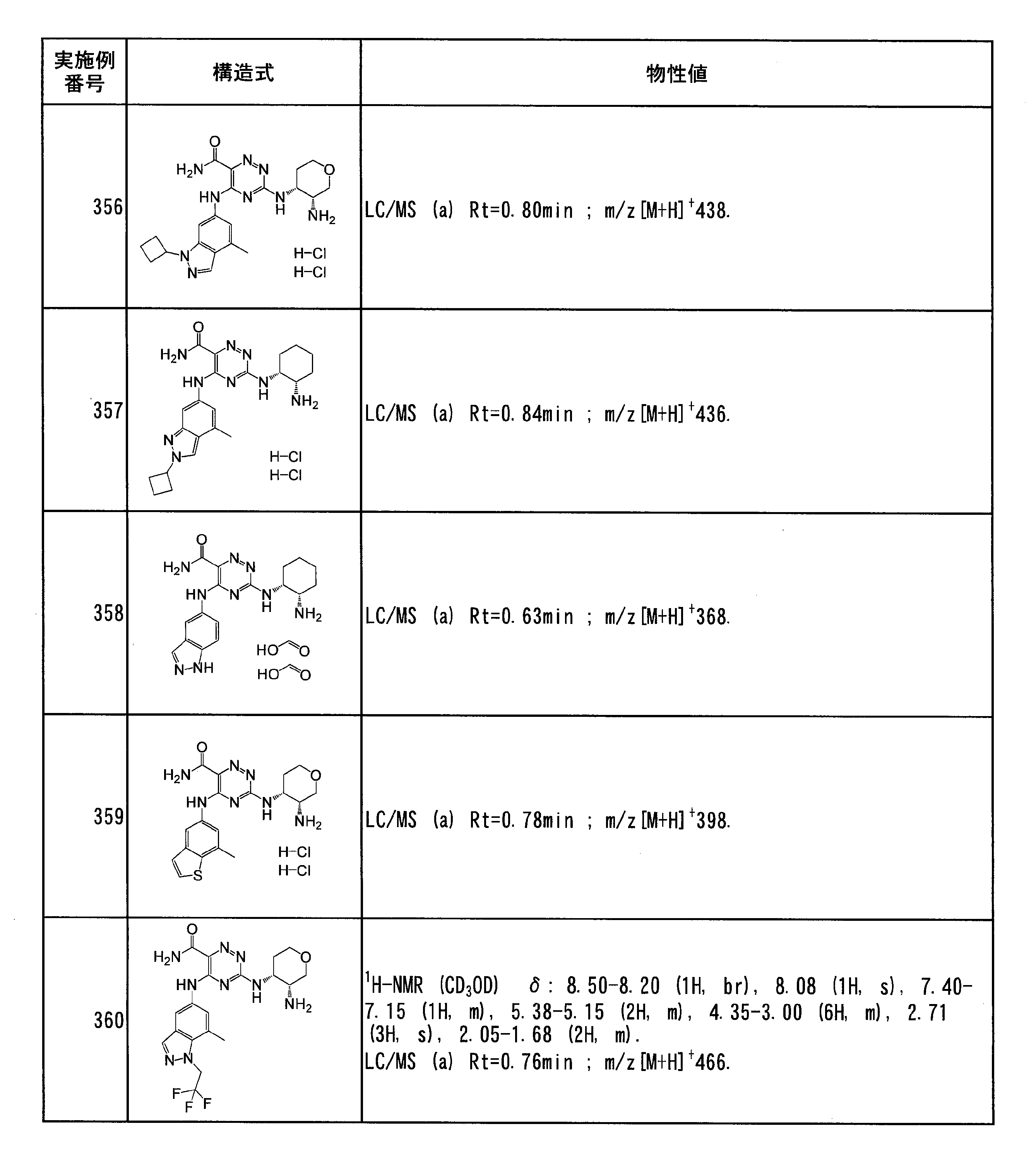
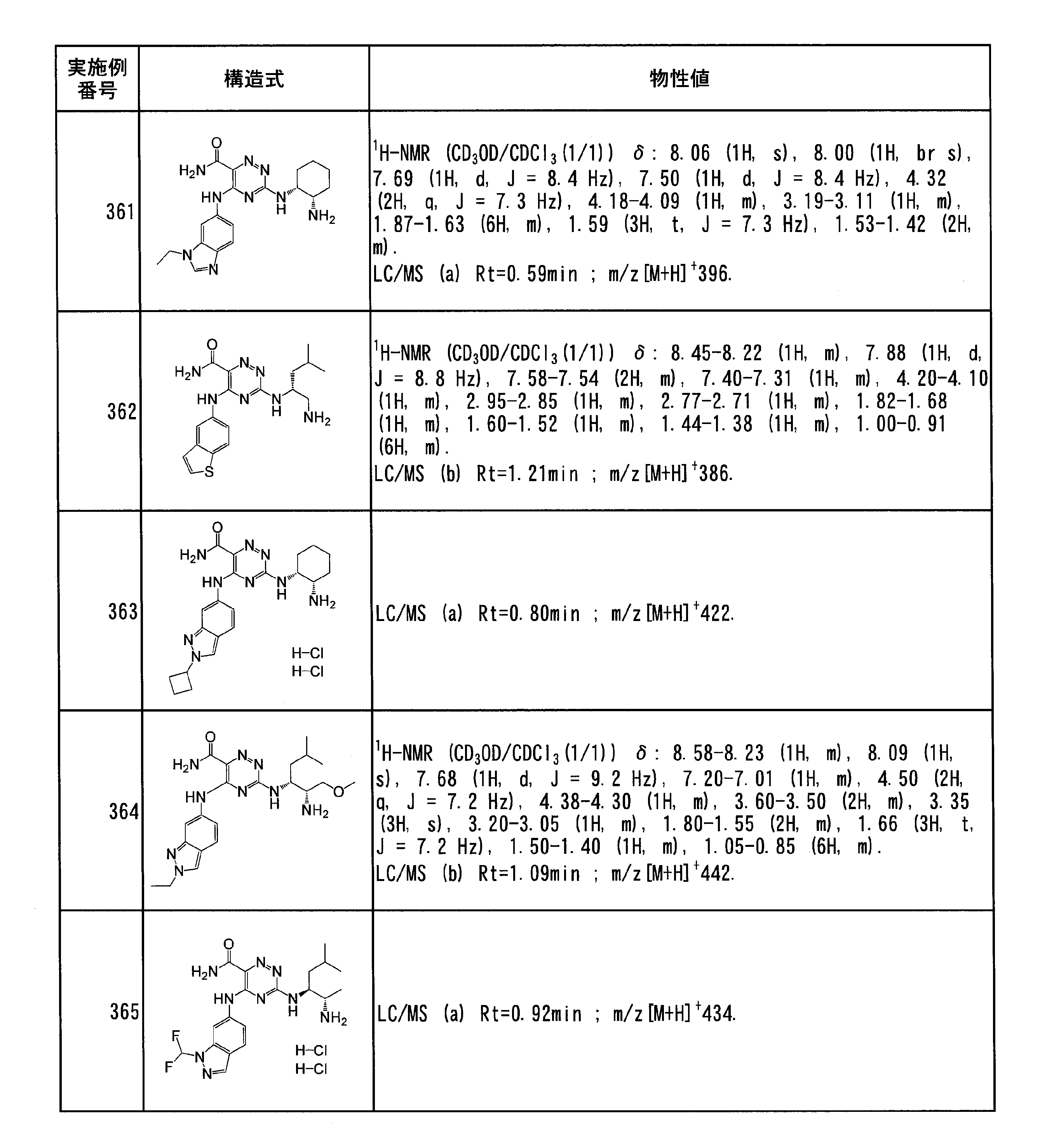
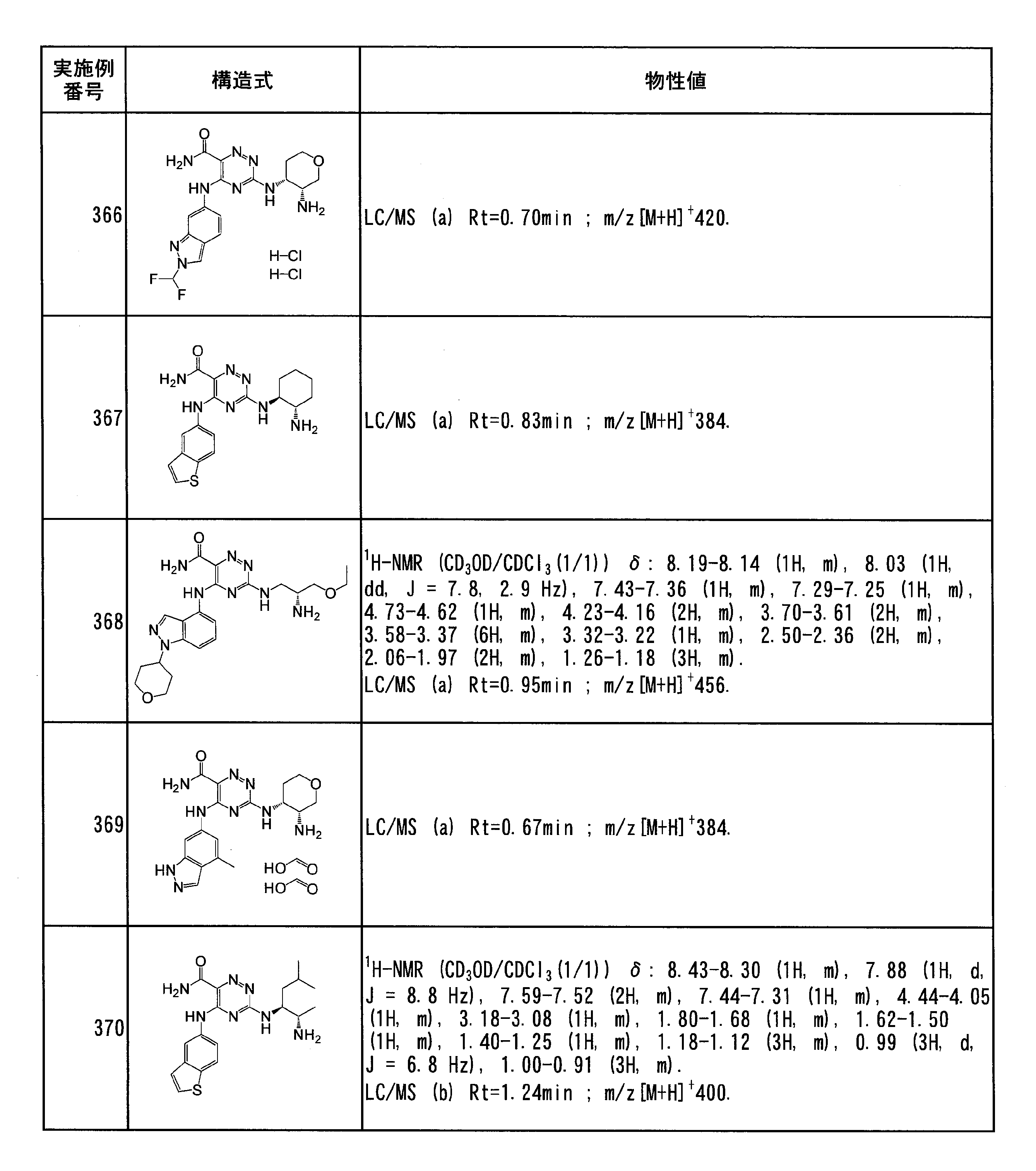


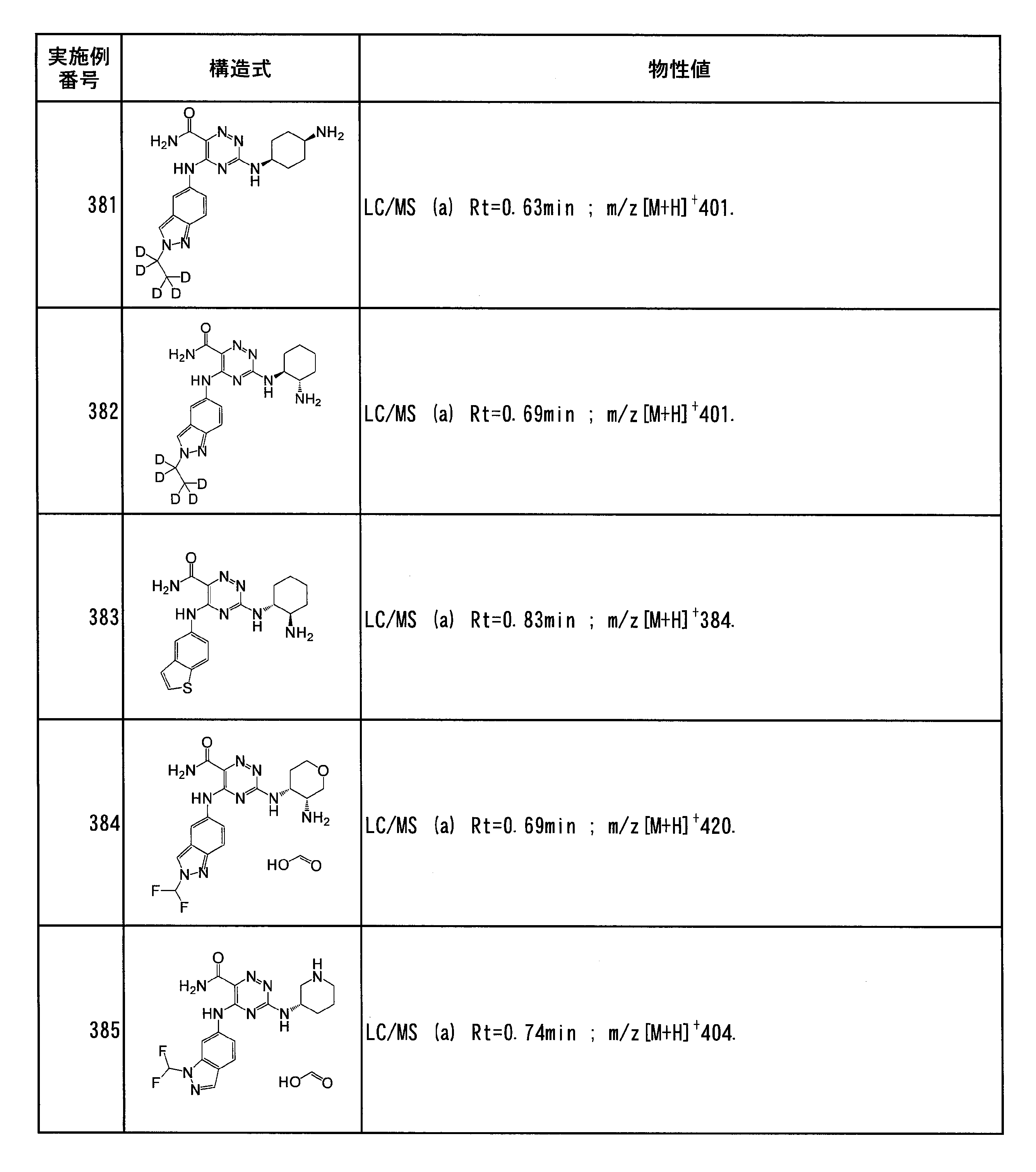



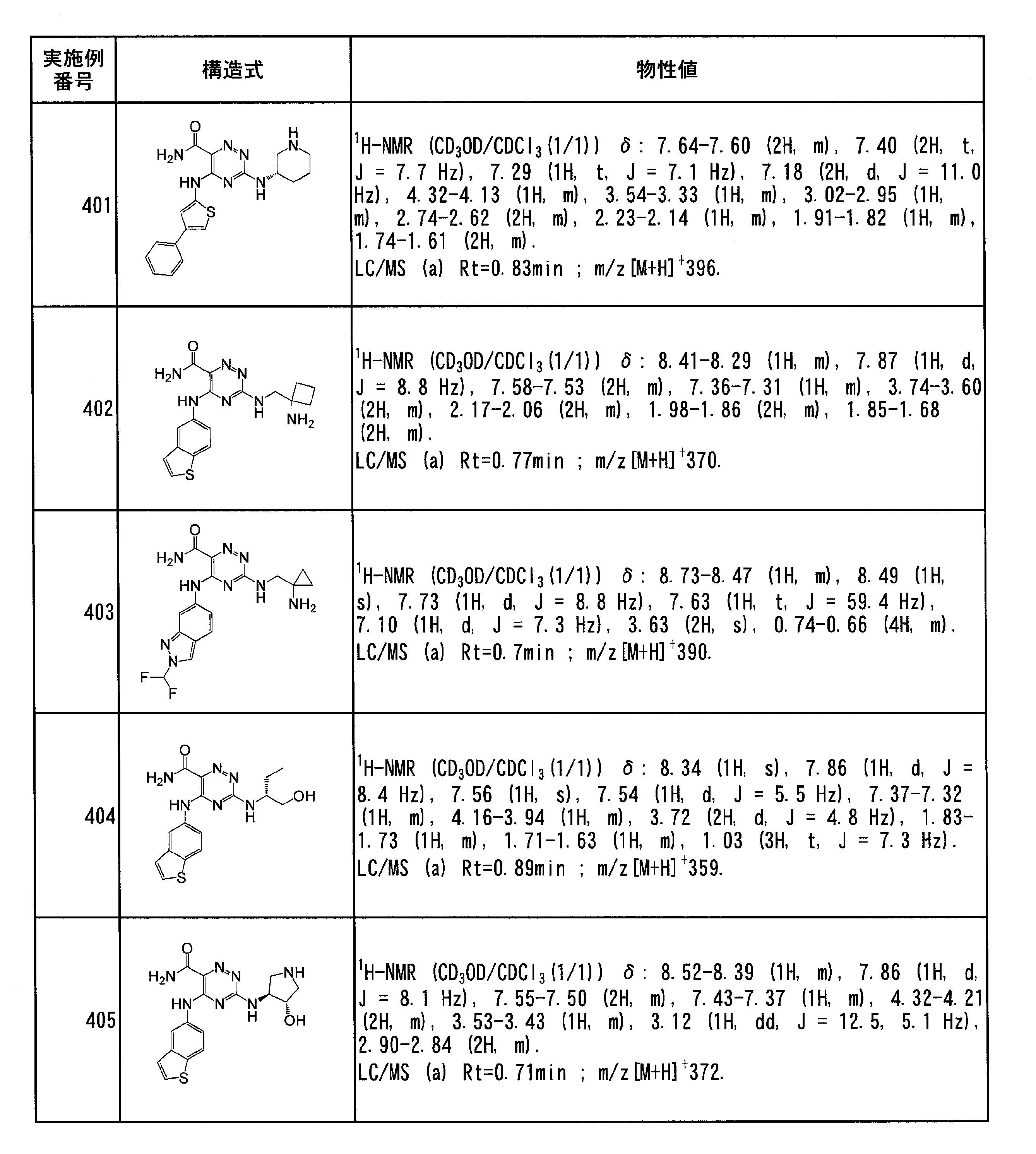
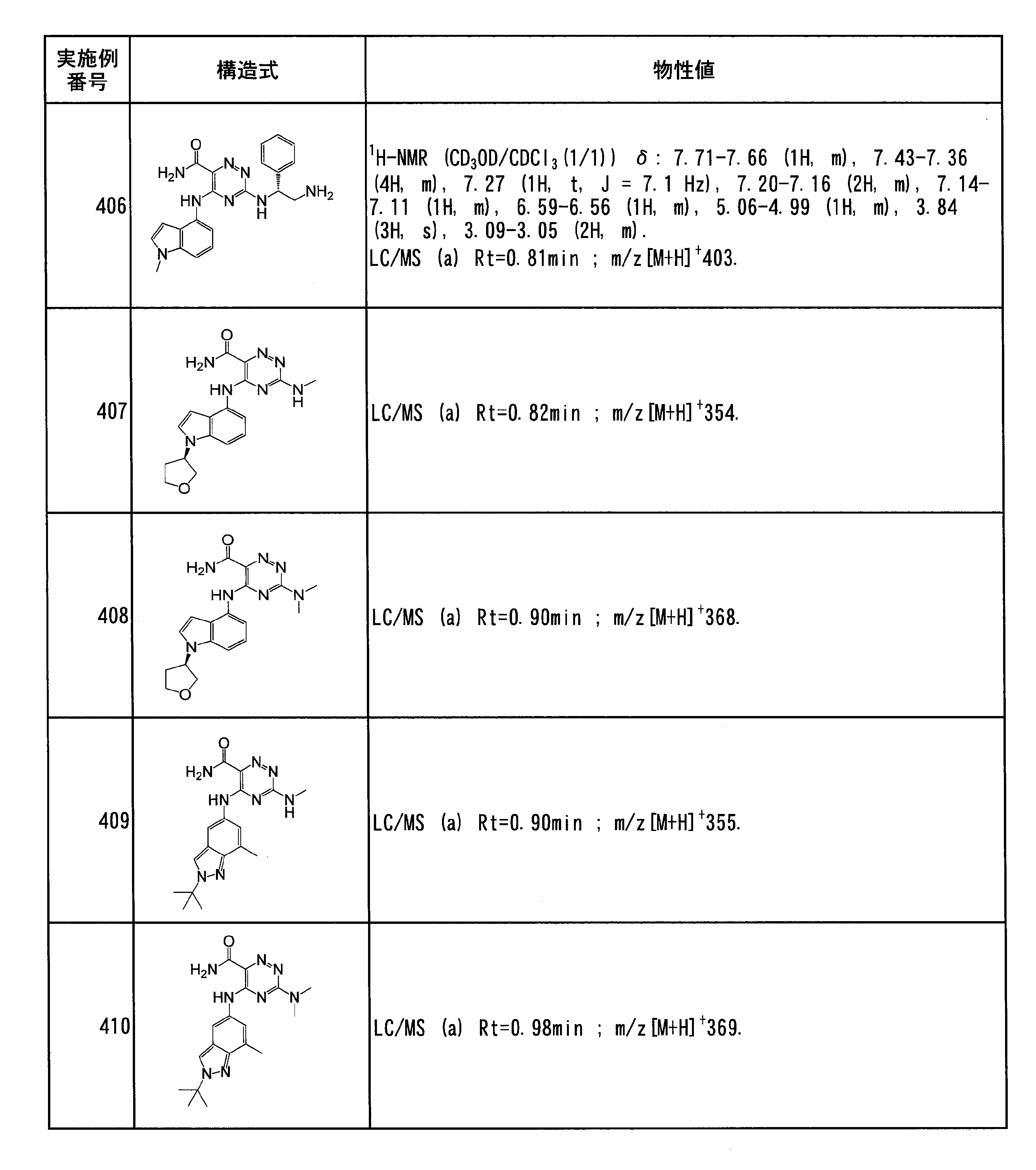
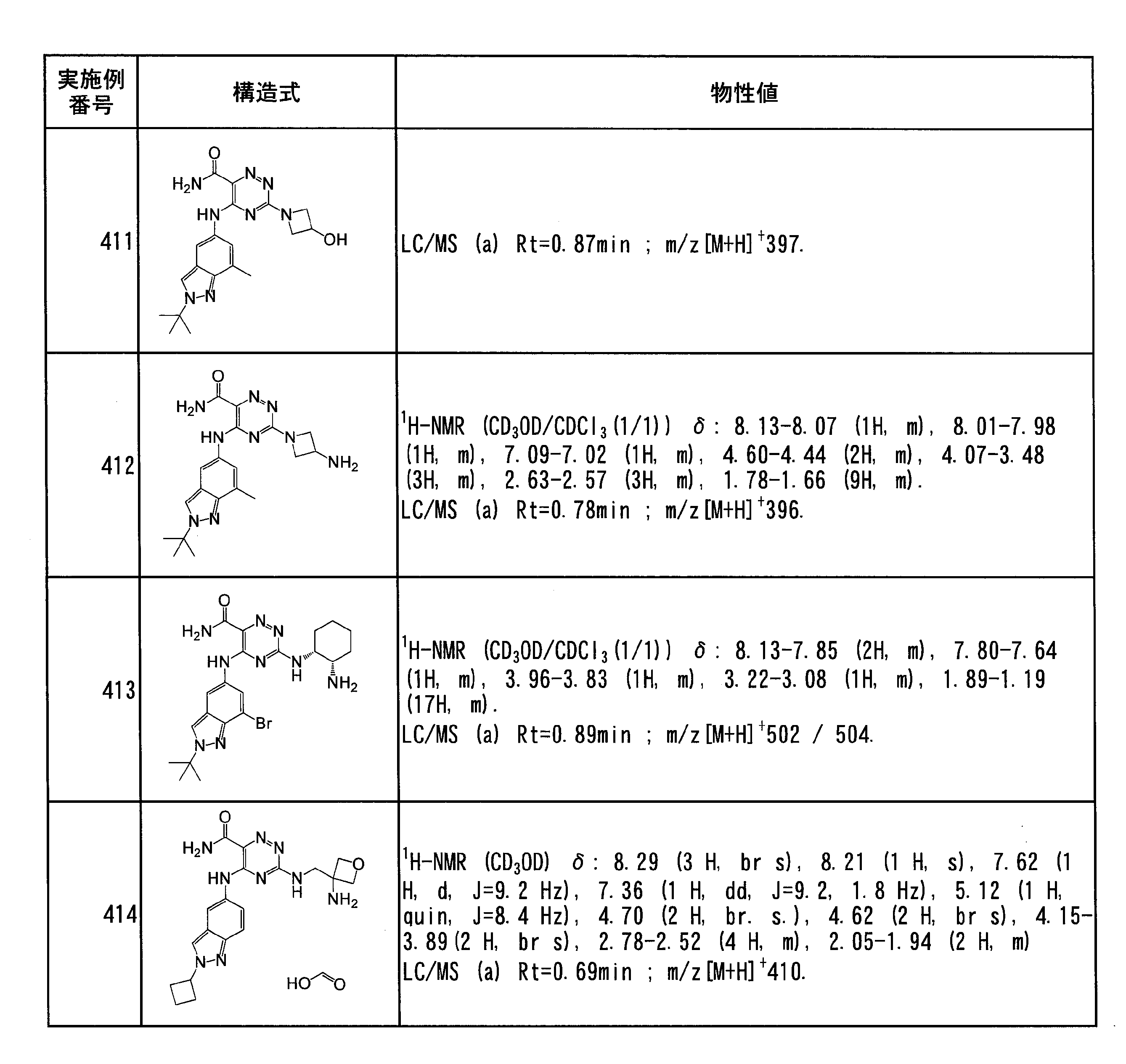
本発明の生物活性は以下の試験法を用いて評価した。
試験例1 Sykキナーゼ阻害試験
Sykキナーゼ活性に対する本発明化合物のインビトロでの阻害活性測定を以下の条件で実施した。試験に用いた精製ヒトSyk蛋白質はカルナバイオサイエンス株式会社より購入した。化合物の阻害活性測定においては、まず、本発明化合物をジメチルスルホキシド(DMSO)で段階希釈した。次に、反応用緩衝液(20mM Tris-HCl pH7.5,20mM NaCl,2.5mM DTT,0.02% Tween-20,1% Glycerol,0.01mM Pefabloc)中に精製ヒトSyk蛋白質、FL-Peptide 22(終濃度は1uM)(キャリパーライフサイエンス社)、塩化マグネシウム(終濃度は5mM)、ATP(終濃度は30uM)と本発明化合物DMSO溶液(DMSOの終濃度は5%)を加えて25℃で30分間インキュベーションしキナーゼ反応を行った。そこへSeparation Buffer(キャリパーライフサイエンス社)にて希釈したEDTA(終濃度は30mM)を加えてキナーゼ反応を停止させた。最後に、LabChipTM3000システム(キャリパーライフサイエンス社、励起波長488nm、検出波長530nm)でリン酸化体ペプチドと非リン酸化体ペプチドを分離し各々の量を測定し、その量比からリン酸化反応量を求め、リン酸化反応を50%抑制することのできる化合物濃度をIC50値(μM)と定義した。
また、対照化合物として、既知の1,2,4-トリアジン-6-カルボキサミド誘導体 A、B及びC(それぞれWO2000/075113(A:化合物番号121,B:化合物番号154)及びWO2000/076980(C:化合物番号39))を合成し、同様にIC50値(μM)を算出し、本発明化合物と比較した。

SU-DHL-6細胞(びまん性大細胞型リンパ腫細胞株)に対するin vitro細胞増殖抑制試験を以下の条件で実施した。
10%のFBSを含むRPMI1640培地(ATCC、Cat#:30-2001)中に浮遊させたSU-DHL-6細胞(ATCC、Cat#:CRL-2959)を、96ウェル平底マイクロプレート(NUNC、Cat#:165305)の各ウェルに8×103個(100μl)ずつ播種し、37℃、5%炭酸ガス含有の培養器中で1日培養した。本発明化合物をジメチルスルホキシドにて段階稀釈し、10%のFBSを含むRPMI1640培地に添加した。これを先に述べたSU-DHL-6細胞の培養プレートの各ウェルに100μlずつ加えて化合物の最終濃度がそれぞれ10、3、1、0.3、0.1、0.03、0.01、0.003μMになるようにし、37℃、5%炭酸ガス含有の培養器中で3日間培養した。培養後、室温に30分間放置し、遠心機(日立、himac CF7D2)にて1500rpmで3分間遠心し、各ウェルから上清を100μlずつ除き、100μlの細胞培養液が残るようにした。残った細胞培養液100μlに対し、等量のCellTiter-Glo Luminescent Cell Viability Assay(Promega、Cat#:G7573)を添加し、1分間プレートミキサーで振とうした。10分間暗所で放置した後、マイクロプレートリーダー(PerkinElmer、ARVOsx)にて各ウェルの生細胞由来発光量を測定した。以下の式より細胞増殖抑制率を算出し、50%抑制する本発明化合物の濃度(IC50値(μM))を求めた。
細胞増殖抑制率(%)=(C-T)/C×100
T:被検化合物を添加したウェルの発光量(count per second)
C:被検化合物を添加しなかったウェルの発光量(count per second)


KDRキナーゼ活性に対する上記化合物のインビトロでの阻害活性測定法の条件設定において、キャリパーライフサイエンス社のLabChipTMシリーズ試薬消耗品価格表にFL-Peptide 22がKDRキナーゼ活性測定において基質ペプチドとして対応していることが記載されていたので参考にした。試験に用いた精製リコンビナントヒトKDR蛋白質は自社精製品である。化合物の阻害活性測定においては、まず、本発明化合物をジメチルスルホキシド(DMSO)で段階希釈した。次に、脱リン酸化酵素阻害剤カクテル(PhosSTOP,Roche)及びタンパク分解酵素阻害剤カクテル(Complete Mini,EDTA-free,Roche)を推奨濃度で添加した反応用緩衝液(100mM HEPES pH7.5,1mM DTT, 0.003% Briji35,0.04% Tween-20,0.05% CHAPSO)中に精製ヒトKDR蛋白質、FL-Peptide 22(終濃度は1.5uM)、塩化マグネシウム(終濃度は10mM)、ATP(終濃度は200uM)と本発明化合物DMSO溶液(DMSOの終濃度は5%)を加えて30℃で180分間インキュベーションしキナーゼ反応を行った。そこへキャリパーライフサイエンス社のSeparation Bufferにて希釈したEDTA(終濃度は30mM)を加えてキナーゼ反応を停止させた。最後に、LabChipTM3000システム(キャリパーライフサイエンス社、励起波長488nm、検出波長530nm)でリン酸化体ペプチドと非リン酸化体ペプチドを分離し各々の量を測定し、その量比からリン酸化反応量を求め、リン酸化反応を50%抑制することのできる化合物濃度をIC50値(nM)と定義し以下の表97に示した。また、同様に、対照化合物として既存のSyk阻害剤であるR406(Rigel社)のIC50値(μM)も算出し、本発明化合物と比較した。
AuroraBキナーゼ活性に対する上記化合物のインビトロでの阻害活性測定法は、公開特許公報(特開2008-81492)に記載されている方法を参考にした。試験に用いた精製リコンビナントヒトAuroraB蛋白質はカルナバイオサイエンス社から購入した。化合物の阻害活性測定においては、まず、本発明化合物をジメチルスルホキシド(DMSO)で段階希釈した。次に、反応用緩衝液(20mM HEPES pH7.4,2mM DTT, 0.01% Tween-20)中に精製ヒトAuroraB蛋白質、FL-Peptide 21(キャリパーライフサイエンス社、終濃度は100nM)、塩化マグネシウム(終濃度は1mM)、ATP(終濃度は40uM)と本発明化合物DMSO溶液(DMSOの終濃度は5%)を加えて25℃で60分間インキュベーションしキナーゼ反応を行った。そこへモレキュラーデバイス社のIMAPTMProgressive Binding Buffer Aにて500倍希釈したIMAPTMProgressive Binding Reagentを加えてキナーゼ反応を停止させた。室温で暗所に120分間静置後に、PHERAstar(BMG LABTECH社、励起波長485nm、検出波長520nm)で測定して得られた蛍光偏光度よりリン酸化反応量を求め、リン酸化反応を50%抑制することのできる化合物濃度をIC50値(nM)と定義し以下の表97に示した。また、同様に、対照化合物として既存のSyk阻害剤であるR406(Rigel社)のIC50値(μM)も算出し、本発明化合物と比較した。

Claims (14)
- 下記一般式(I)

Aは、Raが置換していてもよいC1-C8アルキル基、Raが置換していてもよいC2-C6アルケニル基、Raが置換していてもよいC2-C6アルキニル基、Rbが置換していてもよいC3-C10シクロアルキル基、Rbが置換していてもよいC6-C14芳香族炭化水素基、Rbが置換していてもよい4~10員の不飽和複素環基又はRbが置換していてもよい4~10員の飽和複素環基を示すか、R1及びそれらが結合する窒素原子と一緒になって4~10員の不飽和複素環若しくは4~10員の飽和複素環を形成してもよく(ここで4~10員の不飽和複素環及び4~10員の飽和複素環は、Rbが置換していてもよい);
Raは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、-C(=O)ORx、-C(=O)SRx、-C(=S)ORx、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、-SO2N(Rx)(Ry)、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基又は4~10員の飽和複素環基を示し(ここでC3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基は、重水素原子、ハロゲン原子、ヒドロキシル基、シアノ基、ニトロ基、アミノ基、オキソ基、オキシド基、イミノ基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C1-C6アルコキシ基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれるそれぞれ独立した基が置換していてもよい);
Rbは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、オキソ基、オキシド基、-C(=O)ORx、-C(=O)SRx、-C(=S)ORx、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、-SO2N(Rx)(Ry)、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、又はC2-C6アルキニル基を示し;
R2は、水素原子又はRaが置換していてもよいC1-C6アルキル基を示し;
Bは、Rcが置換していてもよい不飽和複素環基を示し;
Rcは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、オキソ基、オキシド基、-C(=O)Rx、-C(=O)ORx、-C(=O)N(Rx)(Ry)、-C(=O)SRx、-C(=S)ORx、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、-SO2N(Rx)(Ry)、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、C2-C6アルキニル基、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基又は4~10員の飽和複素環基を示し(ここでC1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基及びC2-C6アルキニル基は、シアノ基、ニトロ基、-N(Rx)(Ry)、及び-ORxからなる群から選ばれるれぞれ独立した基が置換していてもよく、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基は、Rd、オキソ基、オキシド基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれるそれぞれ独立した基が置換していてもよい);
Rdは、重水素原子、ハロゲン原子、シアノ基、ニトロ基、-C(=O)Rx、-C(=O)ORx、-C(=O)N(Rx)(Ry)、-C(=O)SRx、-C(=S)ORx、-C(=O)ON(Rx)(Ry)、-N(Rx)(Ry)、-NRxC(=O)Ry、-NRxSO2Ry、-NRxC(=O)ORy、-NRxC(=O)N(Ry)(Rz)、-NRxSO2N(Ry)(Rz)、-N(Rx)-ORy、=NRx、=N-ORx、-ORx、-OC(=O)Rx、-OC(=S)Rx、-OC(=O)ORx、-OC(=O)N(Rx)(Ry)、-OC(=S)ORx、-SRx、-SO2Rx、又は-SO2N(Rx)(Ry)を示し;
Rx、Ry及びRzは、同一又は相異なって、水素原子、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C2-C6アルケニル基、C2-C6アルキニル基、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基又は4~10員の飽和複素環基を示す。
ただし、Bの不飽和複素環基が、3,4-メチレンジオキシフェニル基である場合を除く)で表される化合物又はその塩。 - R1が水素原子又はC1-C6アルキル基であり、R2が水素原子である請求項1記載の化合物又はその塩。
- Aが、同一又は相異なった1~5個のRaが置換していてもよいC1-C8アルキル基;同一又は相異なった1~3個のRbが置換していてもよいC3-C10シクロアルキル基;又は同一又は相異なった1~3個のRbが置換していてもよい4~10員の飽和複素環基であるか、又はR1及びそれらが結合する窒素原子と一緒になって4~10員の不飽和複素環又は4~10員の飽和複素環を形成するものである(ここで4~10員の不飽和複素環及び4~10員の飽和複素環は、Rbが置換していてもよい)である請求項1又は2記載の化合物又はその塩。
- Bが、Rcが置換していてもよい、N、S及びOから選ばれる1~3個のヘテロ原子を有する5~6員の単環性の不飽和複素環基、又はN、S及びOから選ばれる1~3個のヘテロ原子を有する9~10員の2環性の不飽和複素環基である請求項1~3のいずれか1項記載の化合物又はその塩。
- Bが、ハロゲン原子、シアノ基、オキシド基、C1-C6アルキル基、-C(=O)Rx、-C(=O)ORx、-C(=O)N(Rx)(Ry)、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基、又は4~10員の飽和複素環基(ここでC1-C6アルキル基は、Rdが置換していてもよく、C3-C10シクロアルキル基、C6-C14芳香族炭化水素基、4~10員の不飽和複素環基及び4~10員の飽和複素環基は、Rd、オキソ基、オキシド基、C1-C6アルキル基、C1-C6ハロアルキル基、C1-C6重水素化アルキル基、C1-C6アルコキシ基、C2-C6アルケニル基、及びC2-C6アルキニル基からなる群から選ばれる同一又は相異なった基が置換していてもよい)からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、N、S及びOから選ばれる1~3個のヘテロ原子を有する5~6員の単環性の不飽和複素環基、又はN、S及びOから選ばれる1~3個のヘテロ原子を有する9~10員の2環性の不飽和複素環基である請求項1~4のいずれか1項記載の化合物又はその塩。
- R1が水素原子であり、
Aがアミノ基又はC1-C6アルコキシ基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C8アルキル基;アミノ基が置換していてもよいC3-C10シクロアルキル基;アミノ基が置換していてもよい4~10員の飽和複素環基であり、
R2が水素原子であり、
Bがハロゲン原子;重水素原子、ハロゲン原子及びヒドロキシル基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよいC1-C6アルキル基;1~3個のヒドロキシル基が置換していてもよいC3-C10シクロアルキル基;C6-C14芳香族炭化水素基;1~3個のC1-C6アルキル基が置換していてもよい4~10員の不飽和複素環基;又は4~10員の飽和複素環基からなる群から選ばれる同一又は相異なった1~5個の基が置換していてもよい、チエニル基、インドリル基、インダゾリル基、ベンゾチエニル基、キノリル基である請求項1~4のいずれか1項記載の化合物又はその塩。 - 化合物が、以下の化合物群から選択されるものである請求項1~6のいずれか1項記載の化合物又はその塩。
(1)3-((1R,2S)-2-アミノシクロへキシルアミノ)-5-(2-シクロブチル-2H-インダゾール-5-イルアミノ)-1,2,4-トリアジン-6-カルボキサミド
(2)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(3)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(ジフルオロメチル)-7-メチル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(4)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(1-メチル-1H-ピラゾール-4-イル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(5)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((4-(ピリミジン-2-イル)チオフェン-2-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(6)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(trans-4-ヒドロキシシクロヘキシル)-6-メチル-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(7)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-(2-ヒドロキシエチル)-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(8)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((1-エチル-4-メチル-1H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(9)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-(tert-ブチル)-7-クロロ-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(10)3-(((2R,3R)-3-アミノ-4-メトキシブタン-2-イル)アミノ)-5-((1,6-ジメチル-1H-インドール-4-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(11)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((2-メチル-7-フェニル-2H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(12)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((8-メチルキノリン-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(13)3-(((1R,2S)-2-アミノシクロヘキシル)アミノ)-5-((2-(ジフルオロメチル)-4-メチル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(14)3-(((2R,3R)-3-アミノ-4-メトキシブタン-2-イル)アミノ)-5-((1-エチル-7-メチル-1H-インダゾール-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(15)3-(((1R,2S)-2-アミノシクロヘプチル)アミノ)-5-((2-エチル-2H-インダゾール-6-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド
(16)3-(((3R,4R)-3-アミノテトラヒドロ-2H-ピラン-4-イル)アミノ)-5-((7-メチルベンゾ[b]チオフェン-5-イル)アミノ)-1,2,4-トリアジン-6-カルボキサミド - 請求項1~7のいずれか1項記載の化合物又はその塩を有効成分とするSyk阻害剤。
- 請求項1~7のいずれか1項記載の化合物又はその塩及び薬学的に許容される担体を含有する医薬組成物。
- 請求項1~7のいずれか1項記載の化合物又はその塩及び薬学的に許容される担体を含有する医薬組成物。
- 請求項1~7のいずれか1項記載の化合物又はその塩を有効成分とする抗腫瘍剤。
- 腫瘍治療用の請求項1~7のいずれか1項記載の化合物又はその塩。
- 抗腫瘍剤製造のための、請求項1~7のいずれか1項記載の化合物又はその塩の使用。
- 請求項1~7のいずれか1項記載の化合物又はその塩を投与することを特徴とする腫瘍の治療方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/348,339 US9145414B2 (en) | 2011-09-30 | 2012-09-28 | 1,2,4-triazine-6-carboxamide derivative |
| EP20120836932 EP2762476A4 (en) | 2011-09-30 | 2012-09-28 | 1,2,4-triazine-6-carboxamide derivative |
| JP2013536457A JP5878178B2 (ja) | 2011-09-30 | 2012-09-28 | 1,2,4−トリアジン−6−カルボキサミド誘導体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011217750 | 2011-09-30 | ||
| JP2011-217750 | 2011-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013047813A1 true WO2013047813A1 (ja) | 2013-04-04 |
Family
ID=47995836
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/075204 WO2013047813A1 (ja) | 2011-09-30 | 2012-09-28 | 1,2,4-トリアジン-6-カルボキサミド誘導体 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9145414B2 (ja) |
| EP (1) | EP2762476A4 (ja) |
| JP (1) | JP5878178B2 (ja) |
| TW (1) | TW201317226A (ja) |
| WO (1) | WO2013047813A1 (ja) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013171690A1 (en) * | 2012-05-16 | 2013-11-21 | Novartis Ag | Monocyclic heteroaryl cycloalkyldiamine derivatives |
| US20130345191A1 (en) * | 2012-06-22 | 2013-12-26 | Portola Pharmaceuticals, Inc. | 1,2,4-triazine-6-carboxamide kinase inhibitors |
| WO2014060371A1 (en) * | 2012-10-19 | 2014-04-24 | F. Hoffmann-La Roche Ag | Inhibitors of syk |
| WO2014074661A1 (en) * | 2012-11-08 | 2014-05-15 | Bristol-Myers Squibb Company | AMIDE-SUBSTITUTED HETEROCYCLIC COMPOUNDS USEFUL AS MODULATORS OF IL-12, IL-23 AND/OR IFN ALPHα RESPONSES |
| WO2014111031A1 (zh) * | 2013-01-17 | 2014-07-24 | 四川恒康发展有限责任公司 | 三嗪化合物、其药用盐、异构体或水合物及其药物组合物 |
| WO2014157687A1 (ja) * | 2013-03-29 | 2014-10-02 | 大鵬薬品工業株式会社 | アセトアミド基を有する1,2,4-トリアジン-6-カルボキサミド誘導体 |
| WO2015084998A1 (en) * | 2013-12-05 | 2015-06-11 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| CN105683195A (zh) * | 2013-08-22 | 2016-06-15 | 大鹏药品工业株式会社 | 新的喹啉取代的化合物 |
| JP2018522823A (ja) * | 2015-06-02 | 2018-08-16 | ファーマサイクリックス エルエルシー | ブルトン型チロシンキナーゼの阻害剤 |
| JP2018529679A (ja) * | 2015-09-11 | 2018-10-11 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | ピラゾリル置換ヘテロアリール及び医薬としてのその使用 |
| JP2019052178A (ja) * | 2013-06-21 | 2019-04-04 | ゼニス・エピジェネティクス・リミテッドZenith Epigenetics Ltd. | ブロモドメイン阻害剤としての新規の置換された二環式化合物 |
| CN110183440A (zh) * | 2014-03-19 | 2019-08-30 | 勃林格殷格翰国际有限公司 | 杂芳基syk抑制剂 |
| KR20190133726A (ko) * | 2017-03-30 | 2019-12-03 | 브리스톨-마이어스 스큅 컴퍼니 | 6-(시클로프로판카르복스아미도)-4-((2-메톡시-3-(1-메틸-1H-1,2,4-트리아졸-3-일)페닐)아미노)-N-(메틸-d3)피리다진-3-카르복스아미드의 결정 형태 |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| US11701359B2 (en) | 2017-09-01 | 2023-07-18 | Taiho Pharmaceutical Co., Ltd. | Exon 18 and/or exon 21 mutant EGFR selective inhibitor |
| US11857513B2 (en) | 2016-10-31 | 2024-01-02 | Taiho Pharmaceutical Co., Ltd. | Selective inhibitor of exon 20 insertion mutant EGFR |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103450085B (zh) * | 2013-08-15 | 2015-11-18 | 凯莱英医药集团(天津)股份有限公司 | 一种盐酸帕唑帕尼关键中间体的制备方法 |
| CN104628648A (zh) * | 2015-01-22 | 2015-05-20 | 湖南华腾制药有限公司 | 一种吲唑衍生物的制备方法 |
| US10100015B2 (en) * | 2015-03-24 | 2018-10-16 | Shanghai Yingli Pharmaceutical Co., Ltd. | Condensed ring derivative, and preparation method, intermediate, pharmaceutical composition and use thereof |
| JP7399122B2 (ja) | 2018-07-05 | 2023-12-15 | サンフォード バーンハム プレビス メディカル ディスカバリー インスティテュート | ウレア構造を有する縮合環化合物 |
| CN116589410A (zh) * | 2023-05-29 | 2023-08-15 | 济南国鼎医药科技有限公司 | 一种5-氯-2-甲基-2h-吲唑-6-胺的合成方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999031073A1 (fr) * | 1997-12-15 | 1999-06-24 | Yamanouchi Pharmaceutical Co., Ltd. | Nouveaux derives de pyrimidine-5-carboxamide |
| WO2000075113A1 (fr) | 1999-06-09 | 2000-12-14 | Yamanouchi Pharmaceutical Co., Ltd. | Nouveaux derives carboxamide heterocycliques |
| WO2000076980A1 (fr) | 1999-06-10 | 2000-12-21 | Yamanouchi Pharmaceutical Co., Ltd. | Nouveaux derives heterocycliques azotes ou leurs sels |
| WO2004050024A2 (en) | 2002-11-27 | 2004-06-17 | Incyte Corporation | 3-aminopyrrolidine derivatives as modulators of chemokine receptors |
| WO2005007635A2 (en) | 2003-07-18 | 2005-01-27 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Combretastatin derivatives with cytotoxic action |
| JP2008081492A (ja) | 2006-08-31 | 2008-04-10 | Banyu Pharmaceut Co Ltd | オーロラa選択的阻害作用を有する新規アミノピリジン誘導体 |
| JP2009007341A (ja) | 2007-06-01 | 2009-01-15 | Mitsubishi Tanabe Pharma Corp | 医薬組成物 |
| WO2009064695A1 (en) | 2007-11-12 | 2009-05-22 | Igt | Discounted wagering game devices and methods |
| WO2009084695A1 (ja) | 2007-12-28 | 2009-07-09 | Carna Biosciences Inc. | 2-アミノキナゾリン誘導体 |
| WO2012002577A1 (ja) | 2010-06-30 | 2012-01-05 | 富士フイルム株式会社 | 新規なニコチンアミド誘導体またはその塩 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9040530B2 (en) | 2012-06-22 | 2015-05-26 | Portola Pharmaceuticals, Inc. | 1,2,4-triazine-6-carboxamide kinase inhibitors |
-
2012
- 2012-09-28 US US14/348,339 patent/US9145414B2/en not_active Expired - Fee Related
- 2012-09-28 EP EP20120836932 patent/EP2762476A4/en not_active Withdrawn
- 2012-09-28 JP JP2013536457A patent/JP5878178B2/ja not_active Expired - Fee Related
- 2012-09-28 WO PCT/JP2012/075204 patent/WO2013047813A1/ja active Application Filing
- 2012-10-01 TW TW101136275A patent/TW201317226A/zh unknown
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999031073A1 (fr) * | 1997-12-15 | 1999-06-24 | Yamanouchi Pharmaceutical Co., Ltd. | Nouveaux derives de pyrimidine-5-carboxamide |
| WO2000075113A1 (fr) | 1999-06-09 | 2000-12-14 | Yamanouchi Pharmaceutical Co., Ltd. | Nouveaux derives carboxamide heterocycliques |
| WO2000076980A1 (fr) | 1999-06-10 | 2000-12-21 | Yamanouchi Pharmaceutical Co., Ltd. | Nouveaux derives heterocycliques azotes ou leurs sels |
| WO2004050024A2 (en) | 2002-11-27 | 2004-06-17 | Incyte Corporation | 3-aminopyrrolidine derivatives as modulators of chemokine receptors |
| WO2005007635A2 (en) | 2003-07-18 | 2005-01-27 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Combretastatin derivatives with cytotoxic action |
| JP2008081492A (ja) | 2006-08-31 | 2008-04-10 | Banyu Pharmaceut Co Ltd | オーロラa選択的阻害作用を有する新規アミノピリジン誘導体 |
| JP2009007341A (ja) | 2007-06-01 | 2009-01-15 | Mitsubishi Tanabe Pharma Corp | 医薬組成物 |
| WO2009064695A1 (en) | 2007-11-12 | 2009-05-22 | Igt | Discounted wagering game devices and methods |
| WO2009084695A1 (ja) | 2007-12-28 | 2009-07-09 | Carna Biosciences Inc. | 2-アミノキナゾリン誘導体 |
| WO2012002577A1 (ja) | 2010-06-30 | 2012-01-05 | 富士フイルム株式会社 | 新規なニコチンアミド誘導体またはその塩 |
Non-Patent Citations (11)
| Title |
|---|
| "Development of Medicines", MOLECULAR DESIGN, vol. 7, 1990, pages 163 - 198 |
| ARTHRITIS RES THER., vol. 12, no. 6, 2010, pages 222 |
| BIOORGANIC CHEMISTRY, vol. 36, 2008, pages 4 - 15 |
| BLOOD, vol. 111, no. 4, 2008, pages 2230 - 7 |
| BLOOD, vol. 115, no. 13, 2010, pages 2578 - 85 |
| CANCER RES., vol. 69, no. 13, 2009, pages 5424 - 32 |
| EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 15, no. 3, pages 269 - 273 |
| J PHARMACOL EXP THER., vol. 319, no. 3, 2006, pages 998 - 1008 |
| JOURNAL OF ORGANIC CHEMISTRY, vol. 53, no. 3, 1988, pages 507 - 15 |
| See also references of EP2762476A4 * |
| T.W. GREENE: "Protective Groups in Organic Synthesis", 1999, JOHN WILEY & SONS, INC. |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9290481B2 (en) | 2012-05-16 | 2016-03-22 | Novartis Ag | Monocyclic heteroaryl cycloalkyldiamine derivatives |
| CN104302641A (zh) * | 2012-05-16 | 2015-01-21 | 诺华股份有限公司 | 单环杂芳基环烷基二胺衍生物 |
| WO2013171690A1 (en) * | 2012-05-16 | 2013-11-21 | Novartis Ag | Monocyclic heteroaryl cycloalkyldiamine derivatives |
| US20130345191A1 (en) * | 2012-06-22 | 2013-12-26 | Portola Pharmaceuticals, Inc. | 1,2,4-triazine-6-carboxamide kinase inhibitors |
| WO2013192049A2 (en) | 2012-06-22 | 2013-12-27 | Portola Pharmaceuticals, Inc. | 1,2,4-triazine-6-carboxamide kinase inhibitors |
| AU2013277476B2 (en) * | 2012-06-22 | 2016-12-15 | Alexion Pharmaceuticals, Inc. | 1,2,4-triazine-6-carboxamide kinase inhibitors |
| EP2863748A4 (en) * | 2012-06-22 | 2015-12-09 | Portola Pharm Inc | 1,2,4-triazine-6-CARBOXAMIDKINASEINHIBITOREN |
| US9040530B2 (en) * | 2012-06-22 | 2015-05-26 | Portola Pharmaceuticals, Inc. | 1,2,4-triazine-6-carboxamide kinase inhibitors |
| JP2015520232A (ja) * | 2012-06-22 | 2015-07-16 | ポートラ ファーマシューティカルズ, インコーポレイテッド | 1,2,4−トリアジン−6−カルボキサミドキナーゼ阻害剤 |
| CN104955805A (zh) * | 2012-10-19 | 2015-09-30 | 霍夫曼-拉罗奇有限公司 | Syk的抑制剂 |
| WO2014060371A1 (en) * | 2012-10-19 | 2014-04-24 | F. Hoffmann-La Roche Ag | Inhibitors of syk |
| JP2015534959A (ja) * | 2012-10-19 | 2015-12-07 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | Sykの阻害剤 |
| CN104884454A (zh) * | 2012-11-08 | 2015-09-02 | 百时美施贵宝公司 | 用作IL-12、IL-23和/或IFNα应答调节剂的酰胺取代的杂环化合物 |
| WO2014074661A1 (en) * | 2012-11-08 | 2014-05-15 | Bristol-Myers Squibb Company | AMIDE-SUBSTITUTED HETEROCYCLIC COMPOUNDS USEFUL AS MODULATORS OF IL-12, IL-23 AND/OR IFN ALPHα RESPONSES |
| EA028814B1 (ru) * | 2012-11-08 | 2018-01-31 | Бристол-Майерс Сквибб Компани | АМИДЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ОТВЕТОВ, ОПОСРЕДУЕМЫХ IL-12, IL-23 И/ИЛИ IFNα |
| JP2016506369A (ja) * | 2012-11-08 | 2016-03-03 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | IL−12、IL−23および/またはIFNα応答のモジュレーターとして有用なアミド置換ヘテロ環式化合物 |
| US10000480B2 (en) | 2012-11-08 | 2018-06-19 | Bristol-Myers Squibb Company | Amide-substituted heterocyclic compounds useful as modulators of IL-12, IL-23 and/or IFN alpha responses |
| US10526321B2 (en) | 2012-11-08 | 2020-01-07 | Bristol-Myers Squibb Company | Amide-substituted heterocyclic compounds useful as modulators of IL-12, IL-23 and/or IFN alpha responses |
| US11021475B2 (en) | 2012-11-08 | 2021-06-01 | Bristol-Myers Squibb Company | Amide-substituted heterocyclic compounds useful as modulators of IL-12, IL-23 and/or IFN alpha responses |
| USRE47929E1 (en) | 2012-11-08 | 2020-04-07 | Bristol-Myers Squibb Company | Amide-substituted heterocyclic compounds useful as modulators of IL-12, IL-23 and/or IFNα responses |
| US9505748B2 (en) | 2012-11-08 | 2016-11-29 | Bristol-Myers Squibb Company | Amide-substituted heterocyclic compounds useful as modulators of IL-12, IL-23 and/or IFNα responses |
| WO2014111031A1 (zh) * | 2013-01-17 | 2014-07-24 | 四川恒康发展有限责任公司 | 三嗪化合物、其药用盐、异构体或水合物及其药物组合物 |
| WO2014157687A1 (ja) * | 2013-03-29 | 2014-10-02 | 大鵬薬品工業株式会社 | アセトアミド基を有する1,2,4-トリアジン-6-カルボキサミド誘導体 |
| JP2019052178A (ja) * | 2013-06-21 | 2019-04-04 | ゼニス・エピジェネティクス・リミテッドZenith Epigenetics Ltd. | ブロモドメイン阻害剤としての新規の置換された二環式化合物 |
| CN105683195A (zh) * | 2013-08-22 | 2016-06-15 | 大鹏药品工业株式会社 | 新的喹啉取代的化合物 |
| US9650386B2 (en) | 2013-08-22 | 2017-05-16 | Taiho Pharmaceutical Co., Inc. | Quinoline-substituted compound |
| US9758526B2 (en) | 2013-08-22 | 2017-09-12 | Taiho Pharmaceutical Co., Ltd. | Quinoline-substituted compound |
| CN105683195B (zh) * | 2013-08-22 | 2017-11-10 | 大鹏药品工业株式会社 | 喹啉取代的化合物 |
| JP2020121988A (ja) * | 2013-12-05 | 2020-08-13 | ファーマサイクリックス エルエルシー | ブルトン型チロシンキナーゼの阻害剤 |
| WO2015084998A1 (en) * | 2013-12-05 | 2015-06-11 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US9382246B2 (en) | 2013-12-05 | 2016-07-05 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9656988B2 (en) | 2013-12-05 | 2017-05-23 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| AU2019202507B2 (en) * | 2013-12-05 | 2020-08-13 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| CN105960404B (zh) * | 2013-12-05 | 2019-09-03 | 药品循环有限责任公司 | 布鲁顿氏酪氨酸激酶的抑制剂 |
| CN105960404A (zh) * | 2013-12-05 | 2016-09-21 | 药品循环有限责任公司 | 布鲁顿氏酪氨酸激酶的抑制剂 |
| JP2016539152A (ja) * | 2013-12-05 | 2016-12-15 | ファーマサイクリックス エルエルシー | ブルトン型チロシンキナーゼの阻害剤 |
| CN110183440A (zh) * | 2014-03-19 | 2019-08-30 | 勃林格殷格翰国际有限公司 | 杂芳基syk抑制剂 |
| CN110183440B (zh) * | 2014-03-19 | 2022-04-22 | 勃林格殷格翰国际有限公司 | 杂芳基syk抑制剂 |
| JP2018522823A (ja) * | 2015-06-02 | 2018-08-16 | ファーマサイクリックス エルエルシー | ブルトン型チロシンキナーゼの阻害剤 |
| JP2018529679A (ja) * | 2015-09-11 | 2018-10-11 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | ピラゾリル置換ヘテロアリール及び医薬としてのその使用 |
| US11857513B2 (en) | 2016-10-31 | 2024-01-02 | Taiho Pharmaceutical Co., Ltd. | Selective inhibitor of exon 20 insertion mutant EGFR |
| KR20190133726A (ko) * | 2017-03-30 | 2019-12-03 | 브리스톨-마이어스 스큅 컴퍼니 | 6-(시클로프로판카르복스아미도)-4-((2-메톡시-3-(1-메틸-1H-1,2,4-트리아졸-3-일)페닐)아미노)-N-(메틸-d3)피리다진-3-카르복스아미드의 결정 형태 |
| KR102642407B1 (ko) | 2017-03-30 | 2024-02-28 | 브리스톨-마이어스 스큅 컴퍼니 | 6-(시클로프로판카르복스아미도)-4-((2-메톡시-3-(1-메틸-1H-1,2,4-트리아졸-3-일)페닐)아미노)-N-(메틸-d3)피리다진-3-카르복스아미드의 결정 형태 |
| US11701359B2 (en) | 2017-09-01 | 2023-07-18 | Taiho Pharmaceutical Co., Ltd. | Exon 18 and/or exon 21 mutant EGFR selective inhibitor |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2762476A4 (en) | 2015-03-25 |
| JP5878178B2 (ja) | 2016-03-08 |
| JPWO2013047813A1 (ja) | 2015-03-30 |
| EP2762476A1 (en) | 2014-08-06 |
| US20140343038A1 (en) | 2014-11-20 |
| TW201317226A (zh) | 2013-05-01 |
| US9145414B2 (en) | 2015-09-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5878178B2 (ja) | 1,2,4−トリアジン−6−カルボキサミド誘導体 | |
| CA3042960C (en) | Fgfr4 inhibitor, preparation method therefor and pharmaceutical use thereof | |
| JP5389785B2 (ja) | キナーゼ阻害剤として有用なチアゾールおよびピラゾール | |
| KR101744607B1 (ko) | 신규인 니코틴아미드 유도체 또는 그 염 | |
| JP6791979B2 (ja) | 含窒素ヘテロ環化合物、製造方法、中間体、組成物及び応用 | |
| CA2900748C (en) | Novel pyrimidine and pyridine compounds and their usage | |
| JP5389786B2 (ja) | キナーゼ阻害として有用なアミノピリミジン | |
| JP6035423B2 (ja) | 新規な縮合ピリミジン化合物又はその塩 | |
| JP5572087B2 (ja) | キナーゼ阻害として有用なアミノピリミジン | |
| KR102379517B1 (ko) | 세린/트레오닌 키나아제 억제제 | |
| JP5575799B2 (ja) | 7−フェノキシクロマンカルボン酸誘導体 | |
| KR20080052630A (ko) | Raf 억제제 화합물 및 그의 사용 방법 | |
| KR20110045688A (ko) | 단백질 키나아제 저해활성을 갖는 신규의 1,6-치환된 인돌 화합물 | |
| AU2014250836A1 (en) | Quinazolines and azaquinazolines as dual inhibitors of RAS/RAF/MEK/ERK and PI3K/AKT/PTEN/mTOR pathways | |
| WO2015022073A1 (en) | Annelated pyrroles and their use as crac inhibitors | |
| WO2018214867A9 (zh) | N-(氮杂芳基)环内酰胺-1-甲酰胺衍生物及其制备方法和应用 | |
| WO2014157687A1 (ja) | アセトアミド基を有する1,2,4-トリアジン-6-カルボキサミド誘導体 | |
| JP2013256532A (ja) | キナーゼインヒビターとして有用なアミノピリミジン | |
| WO2019180628A1 (en) | Imidazolidin-2-one compounds as prmt5 modulators | |
| KR20150015501A (ko) | 방향족 복소환 유도체 및 의약 | |
| WO2023067546A1 (en) | Novel bicyclic heteroaryl derivatives as sos1:kras proteinprotein interaction inhibitors | |
| US20150166505A1 (en) | Pyridinyl-substituted pyrazolyl carboxamides | |
| JP2014198693A (ja) | 免疫疾患の予防及び/又は治療剤 | |
| AU2016213030A1 (en) | Preventive and/or therapeutic agent of immune disease | |
| CN117777103A (zh) | 酰胺类化合物及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12836932 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013536457 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012836932 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012836932 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14348339 Country of ref document: US |