WO2012029794A1 - 分岐鎖含有芳香族化合物 - Google Patents
分岐鎖含有芳香族化合物 Download PDFInfo
- Publication number
- WO2012029794A1 WO2012029794A1 PCT/JP2011/069624 JP2011069624W WO2012029794A1 WO 2012029794 A1 WO2012029794 A1 WO 2012029794A1 JP 2011069624 W JP2011069624 W JP 2011069624W WO 2012029794 A1 WO2012029794 A1 WO 2012029794A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- protected
- formula
- peptide
- hydrogen atom
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
- C07K1/061—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups
- C07K1/066—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups for omega-amido functions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/20—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring
- C07C43/23—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B41/00—Formation or introduction of functional groups containing oxygen
- C07B41/04—Formation or introduction of functional groups containing oxygen of ether, acetal or ketal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B43/00—Formation or introduction of functional groups containing nitrogen
- C07B43/04—Formation or introduction of functional groups containing nitrogen of amino groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B43/00—Formation or introduction of functional groups containing nitrogen
- C07B43/06—Formation or introduction of functional groups containing nitrogen of amide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B51/00—Introduction of protecting groups or activating groups, not provided for in the preceding groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/04—Preparation of compounds containing amino groups bound to a carbon skeleton by substitution of functional groups by amino groups
- C07C209/06—Preparation of compounds containing amino groups bound to a carbon skeleton by substitution of functional groups by amino groups by substitution of halogen atoms
- C07C209/08—Preparation of compounds containing amino groups bound to a carbon skeleton by substitution of functional groups by amino groups by substitution of halogen atoms with formation of amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/58—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms with amino groups and the six-membered aromatic ring, or the condensed ring system containing that ring, bound to the same carbon atom of the carbon chain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/14—Preparation of carboxylic acid amides by formation of carboxamide groups together with reactions not involving the carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/16—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
- C07C233/24—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring
- C07C233/25—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring having the carbon atom of the carboxamide group bound to a hydrogen atom or to a carbon atom of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/09—Preparation of ethers by dehydration of compounds containing hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/20—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring
- C07C43/225—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring containing halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
- C07K1/061—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
- C07K1/061—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups
- C07K1/062—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups for alpha- or omega-carboxy functions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06086—Dipeptides with the first amino acid being basic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
- C07K5/06165—Dipeptides with the first amino acid being heterocyclic and Pro-amino acid; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0819—Tripeptides with the first amino acid being acidic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0821—Tripeptides with the first amino acid being heterocyclic, e.g. His, Pro, Trp
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/1008—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1021—Tetrapeptides with the first amino acid being acidic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/06—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members
- C07C2603/10—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members containing five-membered rings
- C07C2603/12—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members containing five-membered rings only one five-membered ring
- C07C2603/18—Fluorenes; Hydrogenated fluorenes
Definitions
- the present invention relates to a specific branched chain-containing aromatic compound.
- the present invention further relates to a protecting reagent containing the compound or an adduct thereof.
- the present invention also relates to a method for producing a peptide using the compound, and further to an organic synthesis method including a method for producing the peptide.
- the peptide production methods have been roughly divided into a solid phase method and a liquid phase method.
- the solid-phase method is advantageous in that isolation and purification after the reaction can be performed simply by washing the resin, but it is essentially a heterogeneous phase reaction, and excess reagents and reagents are used to compensate for low reactivity.
- the liquid phase method has good reactivity and has the advantage that the intermediate peptide can be purified by extraction washing, isolation, etc. after the condensation reaction, but in each step of the coupling reaction and deprotection.
- Patent Document 1 and Non-Patent Document 1 each disclose a method using a 3,4,5-tris (n-octadecyloxy) benzyl alcohol type compound as a protecting reagent for a carboxyl group or the like.
- Patent Documents 2 to 4 disclose protecting reagents such as 3,5-di (docosyloxy) benzyl alcohol type compound, 2,4-di (docosyloxy) benzyl alcohol type compound, and trityl type compound, respectively. .
- the reaction can be carried out in a uniform liquid phase, and after the reaction, precipitation is performed by changing the solvent composition, and isolation and purification can be carried out only by filtration and washing. it can.
- the use of these protecting reagents requires a reaction solvent evaporation step for precipitation, requires a long time for the filtration step of the precipitate, or these protecting reagents are converted into acetate ester or toluene. Has a problem that it is insoluble or hardly soluble, and the technique disclosed in the above document is not necessarily a universal method.
- Patent Document 5 introduces an example of peptide synthesis reaction using 3,4,5-tris (n-octadecyloxy) benzyl alcohol type compound as a protecting reagent.
- this document merely discloses a special layer separation example of organic solvents under dilute conditions, and this case is not necessarily an industrially versatile method.
- An object of the present invention is to produce a peptide or the like that is easily soluble in isopropyl acetate with excellent liquid separation operability and leads to the final product only through extraction and separation without crystallizing and isolating each intermediate in each step It is to provide a novel compound that can be used in (also referred to as a one-pot synthesis method).
- the present invention includes the following aspects.
- k Q's represent a single bond or —O—, —S—, —C ( ⁇ O) O—, —C ( ⁇ O) NH— or —NH—;
- k R a s independently have at least one aliphatic hydrocarbon group having one or more branched chains, the total number of branched chains is 3 or more, and the total number of carbon atoms is 14 or more and 300 or less.
- k represents an integer of 1 to 4;
- Ring A includes, in addition to R 1 , k QR a , and C (X) (Y) Z, a halogen atom, a C 1-6 alkyl group which may be substituted with one or more halogen atoms, And may have a substituent selected from the group consisting of a C 1-6 alkoxy group which may be substituted with one or more halogen atoms;
- X represents a hydrogen atom or a phenyl group;
- Y represents a hydroxyl group or —NHR group (R represents a hydrogen atom, an alkyl group or an aralkyl group);
- Z represents a hydrogen atom or a formula (a):
- m represents an integer of 0 to 4; m Qs are as defined above; m R b each independently has at least one aliphatic hydrocarbon group having one or more branched chains, the total number of branched chains is 3 or more, and the total number of carbon atoms is 14 or more and 300 or less.
- R 2 represents a hydrogen atom, or together with R 1 , may represent a single bond to form a fluorene ring together with ring A; and ring B is m QR b and R
- a halogen atom, a C 1-6 alkyl group which may be substituted with one or more halogen atoms, and a C 1-6 alkoxy group which may be substituted with one or more halogen atoms A group represented by the formula (which may have a substituent selected from the group consisting of: An organic group having at least one aliphatic hydrocarbon group having one or more branched chains in R a and R b , a total number of branched chains of 3 or more, and a total number of carbon atoms of 14 or more and 300 or less; Formula (b):
- * represents a bonding position with Q; R 5 and R 6 together represent a hydrogen atom or together represent ⁇ O; n 0 represents an integer of 2 to 40; n 0 R 7 and R 8 each independently represents a hydrogen atom or a C 1-4 alkyl group; n 0 X 2 each independently represents a single bond or a C 1-4 alkylene group; and R 9 represents a hydrogen atom or a C 1-4 alkyl group; R 10 represents a C 1-4 alkyl group or formula (I ′):
- ring A ′ further includes a halogen atom, a C 1-6 alkyl group that may be substituted with one or more halogen atoms, And a substituent selected from the group consisting of a C 1-6 alkoxy group which may be substituted with one or more halogen atoms. ).
- R 9 represents a C 1-4 alkyl group. Is a group represented by The branched chain-containing aromatic compound according to [1].
- the branched chain-containing aromatic compound according to [2].
- Organic having at least one aliphatic hydrocarbon group having at least one branched chain in R a and R b , a total number of branched chains of 3 or more, and a total number of carbon atoms of 14 or more and 300 or less A group of formula (d):
- * represents a bonding position with O; R 5 and R 6 together represent a hydrogen atom or together represent ⁇ O; n 0 represents an integer of 2 to 40; n 0 R 7 and R 8 each independently represents a hydrogen atom or a C 1-4 alkyl group; n 0 X 2 each independently represents a single bond or a C 1-4 alkylene group; and R 9 represents a hydrogen atom or a C 1-4 alkyl group; R 10 represents a C 1-4 alkyl group or formula (I ′):
- ring A ′ further includes a halogen atom, a C 1-6 alkyl group that may be substituted with one or more halogen atoms, And a substituent selected from the group consisting of a C 1-6 alkoxy group which may be substituted with one or more halogen atoms.
- R 7 and R 8 are not both hydrogen atoms and n 0 is 2, R 9 represents a C 1-4 alkyl group.
- a hydroxyl group substituted by a group represented by: m 1 represents an integer of 1 to 3.
- [5] Organic having at least one aliphatic hydrocarbon group having one or more branched chains in R a and R b , a total number of branched chains of 3 or more, and a total number of carbon atoms of 14 or more and 300 or less
- the group is of formula (e):
- n 1 represents an integer of 1 to 10
- n 2 represents an integer of 1 to 10
- n 1 R 15 and R 16 each independently represents a hydrogen atom or a C 1-4 alkyl group
- n 1 X 3 represents a single bond or a C 1-4 alkylene group
- n two R 17 and R 18 each independently represents a hydrogen atom or a C 1-4 alkyl group
- n 2 X 5 represent a single bond or a C 1-4 alkylene group
- X 4 represents a single bond or a C 1-4 alkylene group
- R 12 , R 13 , R 14 , R 19 , R 20 and R 21 each independently represents a hydrogen atom or a C 1-4 alkyl group.
- R 15 and R 16 and / or R 17 and R 18 are not both hydrogen atoms and n 1 + n 2 is 2, two or more of R 12 , R 13 and R 14 Each independently represents a C 1-4 alkyl group, or two or more of R 19 , R 20 and R 21 each independently represent a C 1-4 alkyl group.
- n 1 R 15 and R 16 are each independently a hydrogen atom or a methyl group
- n 1 X 3 is a single bond or a methylene group
- n 2 R 17 and R 18 are each independently a hydrogen atom or a methyl group
- n 2 X 5 are a single bond or a methylene group
- X 4 is a single bond or a methylene group
- R 12 , R 13 , R 14 , R 19 , R 20 and R 21 are methyl groups.
- R a and R b are each independently 3,7,11,15-tetramethylhexadecyl group, 3,7,11-trimethyldodecyl group, 2,2,4,8,10,10 -Hexamethyl-5-dodecanoyl group, 3,4,5-tri (3 ', 7', 11 ', 15'-tetramethylhexadecyloxy) benzyl group, 3,5-di (3', 7 ', 11 ', 15'-tetramethylhexadecyloxy) benzyl group, formula (f):
- ring A ′ further includes a halogen atom, a C 1-6 alkyl group that may be substituted with one or more halogen atoms, And a substituent selected from the group consisting of a C 1-6 alkoxy group which may be substituted with one or more halogen atoms. ).
- Q is —O—.
- the branched chain-containing aromatic compound according to [1] which is selected from the group consisting of: [16] The branched chain-containing aromatic compound according to any one of [1] to [15], wherein a saturated solubility in 100 g of isopropyl acetate at 20 ° C. is 1 to 95% by weight. [17] The branched chain-containing aromatic compound according to any one of [1] to [15], wherein a saturated solubility in 100 g of isopropyl acetate at 20 ° C. is 10 to 95% by weight.
- a reagent for protecting a carboxyl group or amide group of an amino acid or peptide comprising the branched chain-containing aromatic compound according to any one of [1] to [17].
- the branched chain-containing aromatic compound according to any one of [1] to [17] is condensed with the C-terminus of an N-protected amino acid or N-protected peptide in a soluble solvent of the compound.
- Obtaining an N-protected C-protected amino acid or N-protected C-protected peptide whose C-terminus is protected with an anchor which is a protecting group derived from the compound (2) removing the N-terminal protecting group of the obtained N-protected C-protected amino acid or N-protected C-protected peptide to obtain a C-protected amino acid or C-protected peptide; (3) a step of condensing the N-protected amino acid or N-protected peptide to the N-terminus of the obtained C-protected amino acid or C-protected peptide to obtain an N-protected C-protected peptide, and (4) Removing the N-terminal protecting group
- k Q's represent a single bond or —O—, —S—, —C ( ⁇ O) O—, —C ( ⁇ O) NH— or —NH—;
- k R a s independently have at least one aliphatic hydrocarbon group having one or more branched chains, the total number of branched chains is 3 or more, and the total number of carbon atoms is 14 or more and 300 or less.
- k represents an integer of 1 to 4;
- Ring A includes, in addition to R 1 , k QR a , and C (X) (Y) Z, a halogen atom, a C 1-6 alkyl group which may be substituted with one or more halogen atoms, And may have a substituent selected from the group consisting of a C 1-6 alkoxy group which may be substituted with one or more halogen atoms;
- X represents a hydrogen atom or a phenyl group;
- Y represents a hydroxyl group, —NHR group (R represents a hydrogen atom, an alkyl group or an aralkyl group) or a halogen atom (preferably a halogen atom);
- Z represents a hydrogen atom or formula (a):
- m represents an integer of 0 to 4; m Qs are as defined above; m R b each independently has at least one aliphatic hydrocarbon group having one or more branched chains, the total number of branched chains is 3 or more, and the total number of carbon atoms is 14 or more and 300 or less.
- R 2 represents a hydrogen atom, or together with R 1 , may represent a single bond to form a fluorene ring together with ring A; and ring B is m QR b and R
- a halogen atom, a C 1-6 alkyl group which may be substituted with one or more halogen atoms, and a C 1-6 alkoxy group which may be substituted with one or more halogen atoms A group represented by the formula (which may have a substituent selected from the group consisting of: An organic group having at least one aliphatic hydrocarbon group having one or more branched chains in R a and R b , a total number of branched chains of 3 or more, and a total number of carbon atoms of 14 or more and 300 or less; Formula (b):
- a method for producing a peptide comprising the steps (1) to (4).
- (1) The branched chain-containing aromatic compound described in [25] above is condensed with the C-terminus of an N-protected amino acid or N-protected peptide in a soluble solvent of the compound, and a protecting group derived from the compound is used.
- N-protected C-protected amino acid or N-protected C-protected peptide whose C-terminus is protected by an anchor (2) removing the N-terminal protecting group of the obtained N-protected C-protected amino acid or N-protected C-protected peptide to obtain a C-protected amino acid or C-protected peptide; (3) a step of condensing the N-protected amino acid or N-protected peptide to the N-terminus of the obtained C-protected amino acid or C-protected peptide to obtain an N-protected C-protected peptide, and (4) Removing the N-terminal protecting group and the C-terminal anchor of the obtained N-protected C-protected peptide to obtain a peptide.
- the branched chain-containing aromatic compound of the present invention is easily soluble in isopropyl acetate having excellent liquid separation operability. Therefore, if the branched chain-containing aromatic compound of the present invention is used, a method for producing a peptide or the like that leads to a final product only through extraction and separation can be performed without crystallizing and isolating each intermediate in each step. .
- the branched chain-containing aromatic compound of the present invention (hereinafter sometimes abbreviated as the present compound) is represented by the following formula (I).
- the compound of the present invention is a specific benzyl compound (in the formula (I), both X and Z are hydrogen atoms and R 1 is a hydrogen atom); a specific diphenylmethane compound (in the formula (I), X is hydrogen)
- k Q's represent a single bond or —O—, —S—, —C ( ⁇ O) O—, —C ( ⁇ O) NH— or —NH—;
- k R a s independently have at least one aliphatic hydrocarbon group having one or more branched chains, the total number of branched chains is 3 or more, and the total number of carbon atoms is 14 or more and 300 or less.
- k represents an integer of 1 to 4;
- Ring A includes, in addition to R 1 , k QR a , and C (X) (Y) Z, a halogen atom, a C 1-6 alkyl group which may be substituted with one or more halogen atoms, And may have a substituent selected from the group consisting of a C 1-6 alkoxy group which may be substituted with one or more halogen atoms;
- X represents a hydrogen atom or a phenyl group;
- Y represents a hydroxyl group, —NHR group (R represents a hydrogen atom, an alkyl group or an aralkyl group) or a halogen atom;
- Z represents a hydrogen atom or formula (a):
- m represents an integer of 0 to 4; m Qs are as defined above; m R b each independently has at least one aliphatic hydrocarbon group having one or more branched chains, the total number of branched chains is 3 or more, and the total number of carbon atoms is 14 or more and 300 or less.
- R 2 represents a hydrogen atom, or together with R 1 , may represent a single bond to form a fluorene ring together with ring A; and ring B is m QR b and R
- a halogen atom, a C 1-6 alkyl group which may be substituted with one or more halogen atoms, and a C 1-6 alkoxy group which may be substituted with one or more halogen atoms Group which may have a substituent selected from the group consisting of: ].
- the compound represented by the formula (I) of the present invention and the compound intended to be protected are a condensation reaction between a hydroxyl group, NHR group or halogen atom which is a Y group and a carboxyl group or the like of the compound intended to be protected. To combine.
- examples of the “alkyl group” represented by R include a C 1-30 alkyl group, preferably a C 1-10 alkyl group, more preferably a C 1-6 alkyl group.
- Preferable specific examples include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl and the like, and methyl and ethyl are particularly preferable.
- examples of the “aralkyl group” represented by R include a C 7-30 aralkyl group, preferably a C 7-20 aralkyl group, more preferably a C 7-16 aralkyl group (C 6-10 aryl group). -C 1-6 alkyl group).
- Preferred examples include benzyl, 1-phenylethyl, 2-phenylethyl, 1-phenylpropyl, ⁇ -naphthylmethyl, 1- ( ⁇ -naphthyl) ethyl, 2- ( ⁇ -naphthyl) ethyl, 1- ( Examples include ⁇ -naphthyl) propyl, ⁇ -naphthylmethyl, 1- ( ⁇ -naphthyl) ethyl, 2- ( ⁇ -naphthyl) ethyl, 1- ( ⁇ -naphthyl) propyl, and benzyl is particularly preferable.
- R is preferably a hydrogen atom, a C 1-6 alkyl group or a C 7-16 aralkyl group, more preferably a hydrogen atom, methyl, ethyl or benzyl, and particularly preferably a hydrogen atom.
- halogen atom is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- halogen atom for Y, a chlorine atom, a bromine atom and an iodine atom are preferable, and a bromine atom is more preferable.
- organic group has at least one aliphatic hydrocarbon group having one or more branched chains in its molecular structure, the total number of branched chains is 3 or more, and the total number of carbon atoms is 14 or more and 300
- the “branched chain” in the “aliphatic hydrocarbon group having one or more branched chains” is a linear or branched saturated aliphatic hydrocarbon group, preferably a C 1-6 alkyl group, and a C 1-4 alkyl group.
- a group is more preferable, and a methyl group or an ethyl group is more preferable.
- the “branched chain” may be substituted with one or more halogen atoms.
- the “aliphatic hydrocarbon group” in the “aliphatic hydrocarbon group having one or more branched chains” is a linear saturated or unsaturated aliphatic hydrocarbon group, preferably a C 2 -C 300 alkyl group (preferably Is a C 3 -C 100 alkyl group, more preferably a C 3 -C 60 alkyl group), a C 2 -C 300 alkenyl group (preferably a C 3 -C 100 alkenyl group, more preferably a C 3 -C 60 alkenyl groups) or C 2 -C 300 alkynyl groups (preferably C 3 -C 100 alkynyl groups, more preferably C 3 -C 60 alkynyl groups).
- part of "the aliphatic hydrocarbon group to have” is not specifically limited, It may exist in the terminal (monovalent group), and may exist in other site
- aliphatic hydrocarbon group having one or more branched chains include a propyl group, a butyl group, a pentyl group, a hexyl group, a heptyl group, an octyl group, a nonyl group, a decyl group, an undecyl group, and a dodecyl group.
- Organic groups having at least one aliphatic hydrocarbon group having one or more branched chains, a total number of branched chains of 3 or more, and a total number of carbon atoms of 14 to 300 When there are a plurality of “aliphatic hydrocarbon groups”, each of them may be the same or different.
- Sites other than the “aliphatic hydrocarbon group possessed” can be arbitrarily set. For example, it has a moiety such as —O—, —S—, —CO—, —NH—, —COO—, —OCONH—, —CONH—, —NHCO—, a hydrocarbon group (monovalent group or divalent group). You may do it.
- hydrocarbon group examples include an aliphatic hydrocarbon group, an araliphatic hydrocarbon group, a monocyclic saturated hydrocarbon group, an aromatic hydrocarbon group, and the like.
- an alkyl group Monovalent groups such as alkenyl group, alkynyl group, cycloalkyl group, aryl group, aralkyl group and the like and divalent groups derived therefrom are used.
- alkyl group for example, a C 1-6 alkyl group is preferable, and examples thereof include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, hexyl and the like.
- alkenyl group for example, a C 2-6 alkenyl group is preferable, and examples thereof include vinyl, 1-propenyl, allyl, isopropenyl, butenyl, isobutenyl and the like.
- alkynyl group is preferably, for example, a C 2-6 alkynyl group, and examples thereof include ethynyl, propargyl, 1-propynyl and the like.
- cycloalkyl group for example, a C 3-6 cycloalkyl group is preferable, and examples thereof include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- aryl group is preferably, for example, a C 6-14 aryl group, and examples thereof include phenyl, 1-naphthyl, 2-naphthyl, biphenylyl, 2-anthryl and the like.
- a C 6-10 aryl group is more preferable, and phenyl is particularly preferable.
- the “aralkyl group” for example, a C 7-20 aralkyl group is preferable, and examples thereof include benzyl, 1-phenylethyl, 2-phenylethyl, 1-phenylpropyl, naphthylmethyl, 1-naphthylethyl, 1-naphthylpropyl and the like. Is mentioned.
- a C 7-16 aralkyl group (C 6-10 aryl-C 1-6 alkyl group) is more preferable, and benzyl is particularly preferable.
- the “hydrocarbon group” may be substituted with a substituent selected from a halogen atom (chlorine atom, bromine atom, fluorine atom, iodine atom), oxo group and the like.
- the compound of the present invention has k QR a groups.
- Q is a single bond, or —O—, —S—, —C ( ⁇ O) O—, —C ( ⁇ O) NH— or —NH—, preferably O. .
- the k QR a groups may be the same or different.
- the total number of carbon atoms in the “organic group of 300 or less” is 14 or more, preferably 16 or more, and more preferably 18 or more.
- an organic group having an aliphatic hydrocarbon group having at least one aliphatic hydrocarbon group having one or more branched chains and a total number of branched chains of 3 or more represented by R a and R b
- the total number of carbon atoms is 300 or less, preferably 200 or less, and more preferably 160 or less.
- an “aliphatic hydrocarbon group having at least one aliphatic hydrocarbon group having one or more branched chains and a total number of branched chains of 3 or more, represented by R a and R b is represented.
- the total number of branched chains in the “organic group having” is 3 or more, preferably 4 or more, more preferably 8 or more, and still more preferably 10 or more. As the total number of branched chains increases, the compound protected by the compound of the present invention becomes an oily substance having better solubility in various organic solvents even when the peptide chain becomes a long chain.
- R 3 and R 4 each independently represent a hydrogen atom or a C 1-4 alkyl group
- X 1 represents a single bond, a C 1-4 alkylene group or an oxygen atom.
- R 3 and R 4 are not both hydrogen atoms.
- * represents a bonding position with Q; R 5 and R 6 together represent a hydrogen atom or together represent ⁇ O; n 0 represents an integer of 2 to 40; n 0 R 7 and R 8 each independently represents a hydrogen atom or a C 1-4 alkyl group; n 0 X 2 each independently represents a single bond or a C 1-4 alkylene group; and R 9 represents a hydrogen atom or a C 1-4 alkyl group; R 10 represents a C 1-4 alkyl group or formula (I ′):
- ring A ′ further includes a halogen atom, a C 1-6 alkyl group that may be substituted with one or more halogen atoms, And a substituent selected from the group consisting of a C 1-6 alkoxy group which may be substituted with one or more halogen atoms. ).
- R 7 and R 8 are not both hydrogen atoms and n 0 is 2, R 9 represents a C 1-4 alkyl group.
- More preferred groups of the formula (c) are branched isomers having 14 to 160 carbon atoms such as a myristyl group, a cetyl group, a stearyl group, an aralkyl group, and a behenyl group, and a group having a total number of branched chains of 3 or more.
- 2,3-dihydrophytyl group, 3,7,11-trimethyldodecyl group and 2,2,4,8,10,10-hexamethyl-5-dodecanoyl group are particularly preferable.
- R 11 is a branched isomer having 14 to 30 carbon atoms such as a myristyl group, a cetyl group, a stearyl group, an aralkyl group, or a behenyl group, and the total number of branched chains is 3 or more.
- the group is more preferable, among which 2,3-dihydrophytyl group and 3,7,11-trimethyldodecyl group are particularly preferable.
- n 1 represents an integer of 1 to 10
- n 2 represents an integer of 1 to 10
- n 1 R 15 and R 16 each independently represents a hydrogen atom or a C 1-4 alkyl group
- n 1 X 3 represents a single bond or a C 1-4 alkylene group
- n two R 17 and R 18 each independently represents a hydrogen atom or a C 1-4 alkyl group
- n 2 X 5 represent a single bond or a C 1-4 alkylene group
- X 4 represents a single bond or a C 1-4 alkylene group
- R 12 , R 13 , R 14 , R 19 , R 20 and R 21 each independently represents a hydrogen atom or a C 1-4 alkyl group.
- R 15 and R 16 and / or R 17 and R 18 are not both hydrogen atoms and n 1 + n 2 is 2, two or more of R 12 , R 13 and R 14 Each independently represents a C 1-4 alkyl group, or two or more of R 19 , R 20 and R 21 each independently represent a C 1-4 alkyl group.
- R 15 and R 16 , and / or R 17 and R 18 are not both hydrogen atoms, and when n 1 + n 2 is 2, R 12 , R 13 and Two or more of R 14 each independently represent a C 1-4 alkyl group, or two or more of R 19 , R 20 and R 21 each independently represent a C 1-4 alkyl group. ).
- Particularly preferred groups of formula (e) include n 1 is an integer from 1 to 5; n 2 is an integer from 1 to 5; n 1 R 15 and R 16 are each independently a hydrogen atom or a methyl group; n 1 X 3 is a single bond or a methylene group; n 2 R 17 and R 18 are each independently a hydrogen atom or a methyl group; n 2 X 5 are a single bond or a methylene group; X 4 is a single bond or a methylene group; and R 12 , R 13 , R 14 , R 19 , R 20, and R 21 include a group that is a methyl group (provided that R 15 and R 16 , and / Or R 17 and R 18 are not both hydrogen atoms).
- R a or R b an organic group having at least one aliphatic hydrocarbon group having one or more branched chains, a total number of branched chains of 3 or more, and a total number of carbon atoms of 14 or more and 300 or less, represented by R a or R b
- R a or R b As specific examples, the following groups may be mentioned. * In each group represents a bonding position, n 3 in the formula represents an integer of 3 or more, and n 4 can be appropriately set so that the total carbon number of the group is 14 or more and 300 or less.
- R a and R b having a total number of branched chains of 3 or more and a total number of carbon atoms of 14 or more and 300 or less.
- the following groups may be mentioned. * In each group indicates a bonding position.
- n 5 to n 9 can be appropriately set so that the total carbon number of each group is 14 or more and 300 or less.
- Preferred examples of "" include the following groups: 3,7,11,15-tetramethylhexadecyl group; 3,7,11-trimethyldodecyl group; 2,2,4,8,10,10-hexamethyl-5-dodecanoyl group; 3,4,5-tri (3 ′, 7 ′, 11 ′, 15′-tetramethylhexadecyloxy) benzyl group; 3,5-di (3 ′, 7 ′, 11 ′, 15′-tetramethylhexadecyloxy) benzyl group; Formula (f):
- Preferred examples of the compound of the present invention include the following benzyl compound, diphenylmethane compound or fluorene compound, but the present invention is not limited to these: 2,4-di (2 ′, 3′-dihydrophytyloxy) benzyl alcohol; 3,5-di (2 ′, 3′-dihydrophytyloxy) benzyl alcohol; 4- (2 ′, 3′-dihydrophytyloxy) benzyl alcohol; 1-[(2-chloro-5- (2 ′, 3′-dihydrophytyloxy) phenyl)]-1-phenylmethanamine; 3,4,5-tri (2 ′, 3′-dihydrophytyloxy) benzyl alcohol; 3,4,5-tri (2 ′, 3′-dihydrophytyloxy) benzylamine; 4- (2 ′, 3′-dihydrophytyloxy) benzylamine; 2- [3 ′, 4 ′, 5′
- n 18 represents 1 to 10; and the formula:
- n 19 represents 2 to 10
- Method for producing compound of the present invention Although it does not specifically limit as a manufacturing method of this invention compound, For example, it can synthesize
- the raw material compounds can be easily obtained as commercial products, or can be produced according to a method known per se or a method analogous thereto.
- the yields of the compounds obtained by the following methods may vary depending on the reaction conditions used, but these products are isolated and purified by ordinary means (recrystallization, column chromatography, etc.), and then the solution temperature is changed. It can be precipitated by a means for causing or a means for changing the solution composition.
- the raw material compound has a hydroxyl group, an amino group, a carboxyl group, a carbonyl group or the like, a protective group generally used in peptide chemistry or the like may be introduced into these groups,
- the target compound can be obtained by removing the protecting group as necessary after the reaction.
- the compound of the present invention can be produced, for example, by the following steps.
- [Q ′ in the formula represents —O—, —S—, —C ( ⁇ O) O— or —NH—, wherein R c represents a hydrogen atom, an OR d group (where R d represents C 1 -6 represents an alkyl group such as an alkyl group, an aralkyl group such as a benzyl group, etc.) or formula (a):
- Y 1 represents a leaving group such as a halogen atom, and the other symbols are as defined above.
- Examples of the base include alkali metal salts such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate, sodium hydride, potassium hydride, potassium tert-butoxide; pyridine, triethylamine, N, N-dimethylaniline, 1,8-diazabicyclo
- Examples thereof include amines such as [5.4.0] -7-undecene, among which sodium carbonate, potassium carbonate, sodium hydride and the like are preferable.
- the solvent examples include aromatic hydrocarbons such as toluene and xylene; ethers such as tetrahydrofuran and dioxane; amides such as dimethylformamide and dimethylacetamide; halogenated hydrocarbons such as chloroform and dichloromethane; nitriles such as acetonitrile. N-methylpyrrolidone and the like, or a mixture thereof. Among them, dimethylformamide, tetrahydrofuran, toluene, N-methylpyrrolidone and the like are preferable.
- the reaction temperature is usually 50 to 150 ° C., preferably 60 to 130 ° C.
- the reaction time is usually 2 to 30 hours, preferably 3 to 10 hours.
- Step (b) This step is a step for producing a compound represented by formula (Ia) (hereinafter abbreviated as compound (Ia)) by reducing compound (IIa).
- the reduction reaction can be performed by a method using a reducing agent.
- Examples of the reducing agent used in the reduction reaction include metal hydrides (sodium borohydride, lithium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride, dibutylaluminum hydride, aluminum hydride, hydrogenation) Among them, sodium borohydride, dibutylaluminum hydride and the like are preferable.
- the reaction is performed in a solvent that does not affect the reaction.
- the solvent include alcohols such as methanol and ethanol; ethers such as diethyl ether, tetrahydrofuran and dioxane; aromatic hydrocarbons such as toluene and xylene; or a mixture thereof, among which tetrahydrofuran, toluene and the like are mentioned.
- the reaction temperature is usually 0 to 100 ° C., preferably 30 to 70 ° C.
- the reaction time is usually 1 to 24 hours, preferably 2 to 5 hours.
- Step (c) In this step, compound (IIa) (wherein R c is not a hydrogen atom or an OR d group in formula (IIa)) is reduced by the same method as in step (b) above, or a phenyl group is obtained by Grignard reaction. (Corresponding to the Z group).
- Grignard reaction a commercially available Grignard reagent (eg, phenylmagnesium bromide, phenylmagnesium chloride, etc.) is used, or magnesium and halobenzene (chlorobenzene, bromobenzene, iodobenzene) are reacted in the presence of iodine or dibromoethane. Can be used.
- a commercially available Grignard reagent eg, phenylmagnesium bromide, phenylmagnesium chloride, etc.
- magnesium and halobenzene chlorobenzene, bromobenzene, iodobenzene
- the Grignard reaction is performed in a solvent that does not affect the reaction.
- the solvent include ethers such as diethyl ether, tetrahydrofuran and 1,2-dimethoxyethane; aromatic hydrocarbons such as toluene and xylene; or a mixture thereof, among which tetrahydrofuran, 1,2-dimethoxyethane, and the like. Etc. are preferred.
- the reaction temperature is usually ⁇ 20 to 100 ° C., preferably 0 to 70 ° C., and the reaction time is usually 1 to 24 hours, preferably 2 to 10 hours.
- Step (d-1) This step involves oximation of compound (IIa) (in formula (IIa), wherein R c is a hydrogen atom) to form a compound represented by formula (I′-a) (hereinafter referred to as compound (I ′) Abbreviated as -a)).
- the oximation reaction is carried out by reacting compound (IIa) with an acid addition salt of hydroxylamine in the presence of a base in a solvent that does not affect the reaction.
- the acid addition salt of hydroxylamine include mineral acid salts such as hydrochloride, sulfate, and nitrate, acetate, trifluoroacetate, methanesulfonate, trifluoromethanesulfonate, p-toluenesulfonate, and the like.
- the organic acid salt and the like can be mentioned, and the hydrochloride is particularly preferable.
- Examples of such bases include alkali metal salts such as potassium hydroxide, sodium hydroxide, sodium hydrogen carbonate, potassium carbonate; pyridine, triethylamine, diisopropylethylamine, N, N-dimethylaniline, 1,8-diazabicyclo [5.4. 0.0] undec-7-ene and the like, among which triethylamine, diisopropylethylamine and the like are preferable.
- Examples of the solvent include halogen solvents such as chloroform and dichloromethane; aromatic hydrocarbons such as toluene and xylene; ethers such as tetrahydrofuran and dioxane; and / or mixtures thereof.
- the reaction temperature is usually 10 to 100 ° C., preferably 20 to 60 ° C.
- the reaction time is usually 0.5 to 30 hours, preferably 2 to 20 hours.
- Step (d-2) In this step, compound (I′-a) is reduced by a catalytic hydrogenation reaction in the presence of a metal catalyst such as palladium-carbon or Raney nickel, or a reducing agent such as a metal hydride similar to step (b).
- a metal catalyst such as palladium-carbon or Raney nickel
- a reducing agent such as a metal hydride similar to step (b).
- Compound (Ib) can also be produced through steps (d-3) to (d-4) and (d-5).
- Step (d-3) the compound (Ia) is halogenated using a chlorinating agent such as acetyl chloride or thionyl chloride or a brominating agent such as acetyl bromide, phosphorus tribromide or diphenylphosphine / bromine.
- a chlorinating agent such as acetyl chloride or thionyl chloride
- a brominating agent such as acetyl bromide, phosphorus tribromide or diphenylphosphine / bromine.
- the solvent examples include halogenated hydrocarbons such as chloroform and dichloromethane; aromatic hydrocarbons such as toluene and xylene; ethers such as tetrahydrofuran and dioxane; and mixtures thereof, among which chloroform, tetrahydrofuran and toluene. Etc. are preferred.
- the reaction temperature is usually 10 to 150 ° C., preferably 30 to 80 ° C.
- the reaction time is usually 0.5 to 30 hours, preferably 2 to 20 hours.
- Step (d-4) In this step, compound (I′-b) is azidated by using an azidating agent such as sodium azide to give a compound represented by formula (I′-c) (hereinafter referred to as compound (I′-c) Abbreviated as))).
- This reaction is carried out by reacting compound (I′-b) with an azidating agent in a solvent that does not affect the reaction.
- the solvent examples include halogenated hydrocarbons such as chloroform and dichloromethane; aromatic hydrocarbons such as toluene and xylene; ethers such as tetrahydrofuran and dioxane; amides such as N, N-dimethylformamide; and mixtures thereof Among them, chloroform, N, N-dimethylformamide and the like are preferable.
- the reaction temperature is usually 10 to 150 ° C., preferably 20 to 100 ° C.
- the reaction time is usually 0.5 to 30 hours, preferably 2 to 20 hours.
- Step (d-5) This step is a step of producing compound (Ib) by amination of compound (I′-c).
- the reaction is carried out by reacting compound (I′-c) with triphenylphosphine in a solvent that does not affect the reaction in the presence of water or by catalytic hydrogenation reduction.
- the amount of triphenylphosphine to be used is preferably 1 to 10 mol, particularly preferably 1 to 5 mol, per 1 mol of compound (I′-c).
- the amount of water to be used is preferably 1 to 10 mol, particularly preferably 1 to 5 mol, per 1 mol of compound (I′-c).
- the solvent examples include aromatic hydrocarbons such as toluene and xylene; ethers such as tetrahydrofuran and dioxane; and mixtures thereof, among which toluene and tetrahydrofuran are preferable.
- the reaction temperature is usually 10 to 150 ° C., preferably 20 to 100 ° C., and the reaction time is usually 0.5 to 30 hours, preferably 2 to 20 hours.
- Step (d-6) This step is represented by the formula (Ic) wherein Y is a —NHR group in the compound of the present invention by reacting compound (I′-b) with RNH 2 (R is as defined above).
- R is as defined above.
- the compound (I′-b) is represented by R—NH 2 in a solvent that does not affect the reaction, if necessary, for example, in the presence of a base such as a tertiary amine such as triethylamine or diisopropylethylamine.
- a base such as a tertiary amine such as triethylamine or diisopropylethylamine.
- the solvent examples include aromatic hydrocarbons such as toluene and xylene; ethers such as tetrahydrofuran and dioxane; and halogen solvents such as chloroform and dichloromethane or a mixture thereof, among which toluene, tetrahydrofuran and chloroform Etc. are preferred.
- the reaction temperature is usually 10 to 100 ° C., preferably 20 to 60 ° C., and the reaction time is usually 0.5 to 30 hours, preferably 2 to 20 hours.
- Step (d-7) is a step of producing compound (Ie) by reacting compound (Id) with a compound having —CONH 2 group or —OCONH 2 group and then treating with a base.
- the reaction of compound (Id) with a compound having a —CONH 2 group or —OCONH 2 group is carried out in an acid catalyst in a solvent that does not affect the reaction.
- the acid catalyst include methanesulfonic acid, trifluoromethanesulfonic acid, toluenesulfonic acid and the like, and methanesulfonic acid and toluenesulfonic acid are particularly preferable.
- the amount of the acid catalyst to be used is preferably 0.05 to 0.5 mol, particularly preferably 0.1 to 0.3 mol, per 1 mol of compound (Id).
- Examples of the compound having —CONH 2 group or —OCONH 2 group include Fmoc-NH 2 , HCONH 2 , CF 3 CONH 2 , AcNH 2 , EtOCONH 2 , Cbz-NH 2, etc., among which Fmoc-NH 2 EtOCONH 2 and the like are preferable.
- Fmoc- means a 9-fluorenylmethoxycarbonyl group (hereinafter also referred to as Fmoc group)
- Cbz- means a benzyloxycarbonyl group (hereinafter also referred to as Cbz group). Means.
- the R a reagent used as the starting compound in the step (a) that is, a hydroxide, halide, alkylsulfonyloxyd (for example, methanesulfonyloxygen etc.) or arylsulfonyloxyd corresponding to the R a group
- a hydroxide, halide, alkylsulfonyloxyd for example, methanesulfonyloxygen etc.
- arylsulfonyloxyd corresponding to the R a group
- Commercially available products can be used for (eg, p-toluenesulfonyloxylate).
- the Ra- forming reagent is, for example, (1) halogenation of a hydroxide corresponding to the R a group, the alkylsulfonyloxy reduction or arylsulfonyloxy reduction, or (2) reduction of unsaturated hydroxide corresponding to R a group (e.g., platinum - Carbon (Pt / C), palladium-carbon (Pd / C), rhodium-carbon (Rh / C), catalytic hydrogenation reaction in the presence of a metal catalyst such as Raney nickel), and subsequent halogenation, alkylsulfonyl By oxidation or arylsulfonyloxylation, Can be manufactured.
- a hydroxide corresponding to the R a group e.g., platinum - Carbon (Pt / C), palladium-carbon (Pd / C), rhodium-carbon (Rh / C), catalytic hydrogenation reaction in the presence of a metal catalyst such as Raney nickel
- examples of the reagent used for converting the hydroxyl group to the leaving group include chlorinating agents such as thionyl chloride and N-chlorosuccinimide (NCS), hydrobromic acid, and acetyl bromide.
- halogenating agents such as brominating agents such as N-bromosuccinimide (NBS), phosphorus tribromide and diphenylphosphine / bromine
- alkylsulfonylating agents such as methanesulfonyl chloride and trifluoromethanesulfonyl chloride, benzenesulfonyl chloride
- alkylsulfonylating agents such as methanesulfonyl chloride and trifluoromethanesulfonyl chloride
- benzenesulfonyl chloride examples include arylsulfonylating agents such as p-toluenesulfonyl chloride, and among them, halogenating agents such as thionyl chloride and hydrobromic acid are preferable.
- the reaction is performed in a solvent that does not affect the reaction.
- the solvent include water; halogenated hydrocarbons such as chloroform and dichloromethane; aromatic hydrocarbons such as benzene, toluene and xylene; acetonitrile, Nitriles such as propionitrile; ethers such as tetrahydrofuran, 1,4-dioxane, diethyl ether and the like.
- halogenated hydrocarbons such as water and chloroform are preferable.
- the reaction temperature is usually 10 to 120 ° C., preferably 50 to 100 ° C.
- the reaction time is usually 1 to 72 hours, preferably 3 to 24 hours.
- the compound of the present invention (compound represented by formula (I) wherein Q is a single bond) can also be produced, for example, by the following method. That is, the introduction of the R a group on the benzene ring is (1) R a group to the corresponding halide (chloride, bromide or iodide), Friedel-Crafts reaction using a carboxylic acid or acid halide corresponding to R a group, (2) A method corresponding to the above compound (II) (provided that the compound in which the Q′H group is replaced with a —CHO group) is carbonized by Wittig reaction, followed by catalytic hydrogenation, or (3) metal The reaction can be performed by a conventional organic synthesis reaction such as cross coupling using a catalyst.
- R a group to the corresponding halide chloride, bromide or iodide
- Friedel-Crafts reaction using a carboxylic acid or acid halide corresponding to R a group
- the compound of the present invention can be used as a protecting reagent in organic synthesis reactions of peptides, oligonucleic acids, and other organic compounds.
- the compound of the present invention is preferably used as an amino acid or peptide protecting reagent in peptide synthesis and the like.
- the C-terminal carboxyl group, the carboxamide group (also referred to as an amide group) of the amino acid forming the C-terminal that is, —CONHR ′ group
- R ′ represents a hydrogen atom, an alkyl group or an aralkyl group
- R ′ is preferably a hydrogen atom.
- the compound of the present invention When used as a protecting reagent, the compound of the present invention may be activated or converted to an equivalent and then reacted with a protected substituent.
- the “organic compound protected by the branched chain-containing aromatic compound of the present invention” is referred to as “branched chain-containing aromatic compound adduct”.
- the compound of the present invention can be used as a protecting reagent for various organic synthesis reactions.
- an organic synthesis reaction can be performed by the following steps: Step (i) : Step of dissolving the compound of the present invention in a soluble solvent (dissolution step), Step (ii) : a step of binding the compound of the present invention dissolved in the soluble solvent obtained in the above step and a reaction substrate (binding step), Step (iii) : A step of adding water to the reaction solution containing the bound product obtained in the above step for washing, separating the layer, and removing the aqueous layer (a layer separation step), Step (iv) : The solution after washing with water containing the conjugate obtained in the above step is subjected to the reaction, water is added to the reaction solution containing the product after the reaction to wash, and the aqueous layer is separated.
- Step (v) A step of removing the protecting group derived from the compound of the present invention and other protecting groups from the product in the solution after washing with water containing the product obtained in the above step (deprotecting step).
- protecting group derived from the compound of the present invention and “other protecting group”, these may be referred to as “anchor” and “temporary protecting group”, respectively.
- Step (i) (dissolution step)
- This step is a step of dissolving the compound of the present invention in a soluble solvent.
- a soluble solvent a general organic solvent can be used for the reaction. Since the compound of the present invention has a long-chain branched aliphatic hydrocarbon group, it has high solubility in various organic solvents, and thus excellent reactivity can be expected.
- the soluble solvent include ethers such as diethyl ether, tetrahydrofuran, 1,4-dioxane, methyl-t-butyl ether and cyclopentyl methyl ether (CPME); acetates such as ethyl acetate and isopropyl acetate; chloroform And halogenated hydrocarbons such as dichloromethane; aromatic hydrocarbons such as toluene and xylene; hydrocarbons such as hexane, heptane and cyclohexane. Two or more of these solvents may be mixed and used at an appropriate ratio.
- ethers such as diethyl ether, tetrahydrofuran, 1,4-dioxane, methyl-t-butyl ether and cyclopentyl methyl ether (CPME); acetates such as ethyl acetate and isopropyl acetate; chloroform And halogenated hydro
- ethyl acetate, isopropyl acetate, dichloromethane, cyclopentyl methyl ether, and toluene are preferable, and ethyl acetate, isopropyl acetate, cyclopentyl methyl ether, and toluene are more preferable.
- Ethyl acetate, isopropyl acetate, and cyclopentyl methyl ether are more preferable, and isopropyl acetate and cyclopentyl methyl ether are still more preferable.
- the “solubility in the organic solvent required for the compound of the present invention” is originally “the solubility of the combined product of these substrate and the compound of the present invention in the organic solvent when each raw material and each product in each reaction is used as a substrate.
- isopropyl acetate is exemplified as a representative soluble solvent, and the characteristics of the compound of the present invention will be shown.
- the lower limit of the saturation solubility of the compound of the present invention in 100 g of isopropyl acetate at 20 ° C. is not particularly limited as long as the binding with the reaction substrate and the subsequent reaction proceed, but it is stable to any substrate industrially. From the viewpoint of allowing the reaction to proceed, 1% by weight is preferred, 2% by weight is more preferred, 5% by weight is more preferred, 10% by weight is even more preferred, 25% by weight is even more preferred, and 50% by weight is more preferred. Particularly preferred.
- the upper limit of the saturated solubility of the compound of the present invention in 100 g of isopropyl acetate at 20 ° C. is not particularly limited as long as a sufficiently high concentration reaction solution can be obtained, but it is possible to react stably regardless of the degree of progress of the reaction industrially. 80 wt% is preferable, 85 wt% is more preferable, 90 wt% is still more preferable, and 95 wt% is still more preferable.
- soluble solvent in order to improve the solubility of the substrate at the time of reaction; in order to improve the solubility of the unreacted product and by-products in the aqueous layer at the time of extraction (that is, removal of unreacted products and by-products).
- Various hydrophilic organic solvents may be added to improve the layer separation.
- hydrophilic organic solvents may be used instead of water in order to remove and wash unreacted substances and by-products at the time of extraction.
- heptane when used as a reaction solvent, it may be extracted and washed with acetonitrile.
- hydrophilic organic solvents include nitriles such as acetonitrile and propionitrile; ketones such as acetone, methyl ethyl ketone and 2-butanone; amides such as N, N-dimethylformamide and N-methylpyrrolidone.
- a sulfoxide such as dimethyl sulfoxide; Acetonitrile, N, N-dimethylformamide, and N-methylpyrrolidone are preferable, and N, N-dimethylformamide, and N-methylpyrrolidone are more preferable from the viewpoint of assisting solubility and not affecting the layer separation. More preferred is N-methylpyrrolidone.
- Step (ii) (joining step) This step is a step of binding the compound of the present invention dissolved in the soluble solvent obtained in the above step (i) and the reaction substrate.
- the reaction substrate has a carboxyl group such as a protected amino acid, and the use amount of the reaction substrate is 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of the compound of the present invention.
- an ester bond is formed by adding a condensing agent under a dimethylaminopyridine catalyst in a solvent that does not affect the reaction.
- Y is a —NHR group
- 1-hydroxybenzotriazole (HOBt) 1-hydroxy-1H-1,2,3-triazole-5-carboxylic acid ethyl ester (HOCt)
- HOAt triazole
- HOOBt 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine
- an ester bond is formed by adding a base such as diisopropylethylamine in a solvent that does not affect the reaction.
- the amount of the condensation additive is not particularly limited as long as the reaction proceeds, but it is preferably 0.05 to 1.5 mol with respect to 1 mol of the compound of the present invention.
- the condensing agent is not particularly limited as long as the reaction proceeds, and specifically, dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIC), N-ethyl-N′-3-dimethylaminopropylcarbodiimide and its Hydrochloride (EDC.HCl), hexafluorophosphoric acid (benzotriazol-1-yloxy) tripyrrolidinophosphonium (PyBop), O- (benzotriazol-1-yl) -N, N, N ′, N′-tetra Methyluronium tetrafluoroborate (TBTU), 1- [bis (dimethylamino) methylene] -5-chloro-1H-benzotriazolium 3-oxide hexafluorophosphate (HCTU), O-benzotriazole-N, N, N ', N'-tetramethyluronium hexafluoro
- the amount of the condensing agent to be used can be 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of the compound of the present invention.
- the solvent for example, the above-mentioned soluble solvents are suitable.
- the reaction temperature is usually ⁇ 10 to 30 ° C., preferably 0 ° C. to 20 ° C., and the reaction time is usually 1 to 30 hours.
- the same method as that for a general liquid phase organic synthesis reaction can be applied. That is, the reaction can be followed using thin layer silica gel chromatography, high performance liquid chromatography or the like.
- Step (iii) (layer separation step)
- water (and / or a hydrophilic organic solvent such as acetonitrile) is added to the reaction solution containing the conjugate obtained in the above step (ii), and the mixture is stirred and washed to obtain a water-soluble reaction residue (and / or / Alternatively, a reaction residue soluble in a hydrophilic organic solvent) is removed by phase separation (separation operation).
- Step (iv) (Reaction and Layer Separation Step)
- water is added to the reaction solution in which the product is dissolved, followed by stirring and washing, and then the water-soluble reaction residue is removed by phase separation (separation operation).
- Step (v) (deprotection step)
- the step finally removes only the protecting group (anchor) derived from the compound of the present invention or simultaneously the anchor and the temporary protecting group from the product contained in the solution after the layer separation step of the step (iv). This is a process for obtaining the target product.
- the anchor removed here has the formula (If):
- each group in the formula is as defined above
- It is group represented by these.
- Y is a hydroxyl group or a halogen atom
- the compound of the present invention reacts with the carboxyl group of the first reaction substrate to form an ester bond.
- anchor deprotection the C-terminus of the peptide becomes a carboxyl group.
- Y is a —NHR group
- the compound of the present invention reacts with the carboxyl group of the first reaction substrate to form an amide bond.
- anchor deprotection converts the C-terminus of the peptide to a -CONHR group. In this step, it is possible to selectively remove only the anchor without removing the temporary protecting group.
- X and Z are hydrogen atoms
- Y is a hydroxyl group
- the group QR a (especially OR a ) on the benzene ring is present at the 2-position and 4-position, or the 2-position, 4-position, and 6
- the temporary protecting group such as a peptide is an Fmoc group or a Cbz group
- deprotection is preferably performed by acid treatment.
- the acid to be used include trifluoroacetic acid (hereinafter referred to as TFA), hydrochloric acid, sulfuric acid, methanesulfonic acid, p-toluenesulfonic acid, etc.
- TFA is preferable, and the concentration of these acids is 0.1. It can be carried out under a solution condition of 5% to 5% chloroform, dichloromethane or THF solution.
- the protecting group (anchor) derived from the compound of the present invention can be removed simultaneously with the temporary protecting group.
- a conventional method used in this field, particularly peptide synthesis is used, but a method performed under hydrogen reduction conditions, acidic conditions, or the like is preferably employed.
- the acid TFA, hydrochloric acid, sulfuric acid, mesylic acid, tosylic acid, trifluoroethanol, hexafluoroisopropanol and the like are used. Of these, TFA is particularly preferable.
- the acid is appropriately set depending on the type of acid used, and an appropriate amount is used for removing the anchor.
- the amount of the acid used is, for example, 3 to 100 mol, preferably 5 to 50 mol, relative to 1 mol of the binder.
- trifluoromethanesulfonic acid, trimethylsilyl trifluoromethanesulfonate, boron trifluoride etherate (BF 3 ⁇ Et 2 O), or the like can be added as a further strong acid source.
- the reaction temperature is usually 0 ° C. to 80 ° C., preferably 0 ° C. to 30 ° C.
- the reaction time is usually 0.5 to 24 hours.
- a peptide can be produced by using the above process.
- the compound of the present invention can be mainly used as a protecting reagent for the C-terminus of amino acids or peptides, but is not limited thereto.
- the compound of the present invention in which Y is a hydroxyl group can be converted to the corresponding chloroformate by a conventional method in the art (for example, reaction with phosgene), the chloroformate is converted to N It can also be used as a protecting reagent for the terminal.
- the method for producing a peptide using the above steps specifically includes the following steps: (1) A compound of the present invention is condensed with an N-protected amino acid or the C-terminus of an N-protected peptide in a soluble solvent of the compound, and the N-protected C-protected amino acid or N-protected at the C-terminus with an anchor -A step of obtaining a protected C-protected peptide (protection step of C-terminal etc.), (2) Step of removing the N-terminal protecting group of the obtained N-protected C-protected amino acid or N-protected C-protected peptide to obtain a C-protected amino acid or C-protected peptide (N-terminal deprotection) Process), (3) A step of condensing an N-protected amino acid or N-protected peptide to the N-terminus of the obtained C-protected amino acid or C-protected peptide to obtain an N-protect
- N-protected amino acid and “N-protected peptide” refer to amino acids and peptides in which the N-terminal amino group is protected with a temporary protecting group and the C-terminal carboxyl group is not protected, respectively. means. These may be referred to as “P-AA-OH” or the like in the following (P is an N-terminal protecting group).
- the “N-protected C-protected amino acid” and the “N-protected C-protected peptide” each have an N-terminal amino group protected with a temporary protecting group and a C-terminal carboxyl group as an anchor. By protected amino acids and peptides.
- C-protected amino acid and “C-protected peptide” mean amino acids and peptides in which the N-terminal amino group is not protected and the C-terminal carboxyl group is protected with an anchor, respectively. To do.
- Step (1) C-terminal etc. protection step
- the compound of the present invention is condensed with an N-protected amino acid or the C-terminus of an N-protected peptide in a soluble solvent of the compound to obtain an N-protected C-protected amino acid or an N-protected C-protected peptide.
- It is a process.
- the said process can be implemented according to the said process (ii) and process (iii), for example.
- P 1 represents an amino-terminal protecting group for N-terminal
- AA 1 represents an amino acid-derived group
- Y ′ represents O or NR
- other symbols have the same meanings as described above.
- the condensation reaction at the C-terminus of the compound of the present invention with an N-protected amino acid or N-protected peptide is preferably carried out in a solvent that does not affect the reaction.
- a solvent that does not affect the reaction.
- Y is a hydroxyl group or —NHR group
- the condensation reaction is performed in the presence of a condensing agent
- Y is a halogen atom
- Y is a hydroxyl or halogen atom
- an ester bond is formed
- Y is a —NHR group
- an amide bond is formed.
- the condensing agent include dicyclohexylcarbodiimide, diisopropylcarbodiimide, N-ethyl-N′-3-dimethylaminopropylcarbodiimide and its hydrochloride (EDC ⁇ HCl).
- EDC ⁇ HCl hydrochloride
- the ester bond forming reaction is carried out in the presence of dimethylaminopyridine, and the amide bond forming reaction is carried out using a condensation additive such as HOBt or HOCt.
- the solvent used in the step the above-mentioned soluble solvent is suitable.
- the amount of the solvent to be used is preferably 2 to 50 ml with respect to 1 g of the compound of the present invention.
- toluene, cyclopentyl methyl ether, chloroform or the like can be selected as a solvent according to the length and type of the peptide chain. A mixture of two or more of these solvents may be used.
- the reaction temperature is usually ⁇ 10 ° C. to 40 ° C., preferably 0 ° C. to 30 ° C.
- the reaction time is usually 1 to 70 hours. After completion of the reaction, water is added, washed and separated to obtain a solution containing the desired C-protected amino acid or C-protected peptide, which is used as it is in the next step without isolation. Is possible.
- the compound of the present invention in which Y is a hydroxyl group and an amide compound represented by P 1 -AA 1 -NHR for example, Fmoc-Ala-NH 2 , Fmoc-Gly-NH 2, etc.
- an acid catalyst for example, Methanesulfonic acid, trifluoromethanesulfonic acid, p-toluenesulfonic acid, etc.
- a solvent that does not affect the reaction preferably 50 ° C. to 150 ° C., more preferably 60 ° C. to 120 ° C.
- Step (2) N-terminal deprotection step
- the N-protected C-protected amino acid or N-protected C-protected peptide obtained in step (1) is removed at the N-terminal protecting group to obtain a C-protected amino acid or C-protected peptide. It is.
- N-terminal protecting group an amino group protecting group described later generally used in the technical field such as peptide chemistry can be used.
- a tert-butoxycarbonyl group hereinafter also referred to as a Boc group.
- Cbz group and / or Fmoc group is preferably used.
- Deprotection conditions are appropriately selected depending on the type of N-terminal protecting group, but deprotection conditions different from anchor removal are preferred.
- the N-terminal protecting group is an Fmoc group
- treatment with a base eg, dimethylamine, diethylamine, piperidine, morpholine, DBU, diethylenetriamine, aminomethylpiperidine, triethylenetetramine, tetraethylenepentamine, etc.
- a base eg, dimethylamine, diethylamine, piperidine, morpholine, DBU, diethylenetriamine, aminomethylpiperidine, triethylenetetramine, tetraethylenepentamine, etc.
- a base eg, dimethylamine, diethylamine, piperidine, morpholine, DBU, diethylenetriamine, aminomethylpiperidine, triethylenetetramine, tetraethylenepentamine, etc.
- Cbz group in the case of a Cbz group
- it is carried out by treatment with an acid WO2009 / 014177).
- the reaction is performed in a solvent that does not affect the reaction (for example, the above
- Step (3) Peptide chain extension step
- the N-protected amino acid or N-protected peptide is condensed to the N-terminus of the C-protected amino acid or C-protected peptide obtained in step (2) to obtain an N-protected C-protected peptide.
- step (iv) the N-protected amino acid or N-protected peptide is condensed to the N-terminus of the C-protected amino acid or C-protected peptide obtained in step (2) to obtain an N-protected C-protected peptide.
- step (2) is performed under the conditions of peptide synthesis generally used in the field of peptide chemistry, using the condensing agent, condensing additive and the like described in step (1). After completion of the reaction, water and / or a hydrophilic organic solvent (acetonitrile, DMF, etc.) is added, washed and separated to obtain a solution containing an N-protected C-protected peptide.
- the N-protected C-protected peptide can be used as it is in the next step without isolation.
- This step is a step of removing the N-terminal protecting group and the C-terminal anchor from the N-protected C-protected peptide obtained in step (3) to obtain the target peptide.
- the above step (2) The deprotection step of the N-terminal protecting group is performed according to step (v).
- steps (5), (6), and (7) or (7 ′) are repeated one or more times for the N-protected C-protected peptide obtained in step (3). After that, step (4) can also be performed.
- N-terminal deprotecting step removing the N-terminal protecting group of the obtained N-protected C-protected peptide to obtain a C-protected peptide
- step (6) a step of condensing an N-protected amino acid or N-protected peptide to the N-terminus of the obtained C-protected peptide to obtain an N-protected C-protected peptide (peptide chain elongation step), and (7 )
- water is added to the reaction system, and the step of extracting and separating impurities into the aqueous layer (extraction and separation step), or (7 ′)
- step (6) a hydrophilic organic solvent is added to the reaction system And a step of extracting and separating impurities into the hydrophilic organic solvent layer (extraction separation step).
- Step (5) N-terminal deprotection step
- the said process is performed like the said process (2).
- Step (6) (Peptide chain extension step) The said process is performed like the said process (3).
- Step (7), (7 ') extraction separation step
- the N-protected C-protected peptide obtained in step (6) is left in the organic layer by separation, and impurities and the like caused by the condensation reaction are removed from the aqueous layer and / or hydrophilic organic solvent (acetonitrile, DMF, etc.) This is done by expelling to the layer.
- a step of isolation in the middle step may be added as appropriate, and extraction washing and the like may be performed as long as the reaction of the next step is not affected.
- the layer separation step can be omitted as appropriate.
- Examples of the protecting group for the hydroxyl group include (C 1 -C 6 ) alkyl groups (eg, methyl, ethyl, propyl, isopropyl, butyl, tert-butyl), phenyl groups, trityl groups, (C 7 -C 10 ) Aralkyl group (eg, benzyl), formyl group, (C 1 -C 6 ) alkyl-carbonyl group (eg, acetyl, propionyl), benzoyl group, (C 7 -C 10 ) aralkyl-carbonyl group (eg, benzylcarbonyl) 2-tetrahydropyranyl group, 2-tetrahydrofuranyl group, silyl group (eg, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), (
- These groups include halogen atoms (eg, fluorine, chlorine, bromine, iodine), (C 1 -C 6 ) alkyl groups (eg, methyl, ethyl, propyl), (C 1 -C 6 ) alkoxy groups (eg, (Methoxy, ethoxy, propoxy), a nitro group and the like may be substituted with 1 to 3 substituents.
- halogen atoms eg, fluorine, chlorine, bromine, iodine
- C 1 -C 6 alkyl groups eg, methyl, ethyl, propyl
- C 1 -C 6 alkoxy groups eg, (Methoxy, ethoxy, propoxy
- a nitro group and the like may be substituted with 1 to 3 substituents.
- amino-protecting groups include formyl group, (C 1 -C 6 ) alkyl-carbonyl group (eg, acetyl, propionyl), (C 1 -C 6 ) alkoxy-carbonyl group (eg, methoxycarbonyl, ethoxy) Carbonyl, Boc group), benzoyl group, (C 7 -C 10 ) aralkyl-carbonyl group (eg, benzylcarbonyl), (C 7 -C 14 ) aralkyloxy-carbonyl group (eg, CBz group, chlorobenzyloxycarbonyl group) , Bromobenzyloxycarbonyl group, Fmoc group), trityl group, phthaloyl group, N, N-dimethylaminomethylene group, silyl group (eg, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, e
- These groups have 1 to 3 substituents selected from halogen atoms (eg, fluorine, chlorine, bromine, iodine), (C 1 -C 6 ) alkoxy groups (eg, methoxy, ethoxy, propoxy), nitro groups, etc. It may be substituted with a group.
- halogen atoms eg, fluorine, chlorine, bromine, iodine
- C 1 -C 6 alkoxy groups eg, methoxy, ethoxy, propoxy
- nitro groups etc. It may be substituted with a group.
- Examples of the protecting group for the carboxyl group include (C 1 -C 6 ) alkyl groups (eg, methyl, ethyl, propyl, isopropyl, butyl, tert-butyl), (C 7 -C 10 ) aralkyl groups (eg, benzyl).
- These groups have 1 to 3 substituents selected from halogen atoms (eg, fluorine, chlorine, bromine, iodine), (C 1 -C 6 ) alkoxy groups (eg, methoxy, ethoxy, propoxy), nitro groups, etc. It may be substituted with a group.
- halogen atoms eg, fluorine, chlorine, bromine, iodine
- C 1 -C 6 alkoxy groups eg, methoxy, ethoxy, propoxy
- nitro groups etc. It may be substituted with a group.
- Examples of the protecting group for the carbonyl group include cyclic acetals (eg, 1,3-dioxane), acyclic acetals (eg, di- (C 1 -C 6 ) alkylacetal) and the like.
- the method for removing these protecting groups may be carried out according to a method known per se, for example, the method described in Protective Groups in Organic Synthesis, published by John Wiley and Sons (1980).
- a reduction method or the like is used.
- the present invention also provides a kit for producing a peptide comprising the compound of the present invention as an essential component.
- the kit may contain other components necessary for carrying out the method of producing the peptide, for example, various solvents used in the reaction, amino acids (or peptides) used as raw materials, and the like. Good. If desired, a manual for producing a peptide using the compound of the present invention can be attached.
- LC / MS Electrospray ionization liquid chromatography / mass spectrometry
- 2,3-dihydrophytol (33.7 mmol) was suspended in 48% hydrobromic acid (100 ml), concentrated sulfuric acid (0.17 ml) was added dropwise, and the mixture was stirred at 100 ° C. overnight.
- the reaction mixture was cooled to room temperature, extracted with hexane (200 ml), and washed twice with 5% aqueous sodium hydrogen carbonate solution (70 ml) and once with 20% brine (70 ml).
- GI-1000 (Nippon Soda Co., Ltd.) (5.02 g) was dissolved in heptane (50 ml) and washed twice with 80% acetonitrile aqueous solution (25 ml). The residue obtained by concentrating the heptane layer was suspended in 48% hydrobromic acid (50 ml), concentrated sulfuric acid (100 ⁇ l) was added dropwise, and the mixture was stirred at 120 ° C. overnight.
- the reaction mixture was cooled to room temperature, extracted with heptane (100 ml), twice with 5% aqueous sodium bicarbonate solution (25 ml), once with 20% brine (25 ml), and 2 times with 90% aqueous acetonitrile (40 ml). Washed twice.
- the residue obtained by evaporating the solvent of the filtrate was purified by silica gel column chromatography (short column, hexane only) to obtain GI-1000 dibromide (GI-1000 (Br)) (2.63 g).
- 1 H-NMR 300 MHz: ⁇ 0.90-1.06 (m), 1.83-1.86 (m, 4H), 3.38-3.44 (m, 4H).
- GI-2000 (manufactured by Nippon Soda Co., Ltd.) (5.33 g) was dissolved in heptane (50 ml) and washed twice with 80% acetonitrile aqueous solution (25 ml). The residue obtained by concentrating the heptane layer was suspended in 48% hydrobromic acid (50 ml), concentrated sulfuric acid (100 ⁇ l) was added dropwise, and the mixture was stirred at 120 ° C. overnight.
- reaction mixture was cooled to room temperature, extracted with heptane (100 ml), twice with 5% aqueous sodium bicarbonate solution (25 ml), once with 20% brine (25 ml), and 2 times with 90% aqueous acetonitrile (40 ml). Washed twice.
- the residue obtained by distilling off the solvent of the filtrate was purified by silica gel column chromatography (short column, hexane only) to obtain GI-2000 dibromide (GI-2000 (Br)) (2.83 g).
- 1 H-NMR 300 MHz: ⁇ 0.91-1.54 (m), 1.81-1.88 (m, 4H), 3.38-3.42 (m, 4H).
- TERGITOL-TMN6 (7.42 g) was dissolved in chloroform (70 ml) and washed with water (35 ml). The extracted organic layer was dried over sodium sulfate, concentrated to dryness, dissolved in chloroform (70 ml), and PBr 3 (1148 ⁇ l, 12.1 mmol, 1.0 eq), pyridine (1074 ⁇ l, 12. 1 mmol, 1.0 eq) was added dropwise and stirred at room temperature for 4 h.
- methyl 3,4,5-tri (2 ′, 3′-dihydrophytyloxy) benzoate (29.3 g, 30.0 mmol) is dissolved in THF (400 ml) and hydrogenated at 0 ° C. in a nitrogen atmosphere.
- Diisobutylaluminum (DIBAL) (1.0 mol / l toluene solution, 96 ml, 96 mmol) was added dropwise over 30 minutes. After stirring at room temperature overnight, 0.2N hydrochloric acid (50 ml) was added dropwise at 0 ° C. to stop the reaction.
- 2,3-dihydrophytyl bromide (895 mg, 2.48 mmol), methyl 3,5-dihydroxybenzoate (204 mg, 1.21 mmol) and potassium carbonate (513 mg, 3.71 mmol) were suspended in DMF (10 ml). , And stirred at 100 ° C. for 7 hours. The reaction mixture was extracted with ethyl acetate (30 ml), washed 3 times with 1N hydrochloric acid (10 ml), washed with 20% brine (10 ml) and dried over sodium sulfate. , 4,5-Tri (2 ′, 3′-dihydrophytyloxy) benzoic acid methyl ester (0.78 g, yield 92%) was obtained.
- Methyl 3,4,5-di (2 ′, 3′-dihydrophytyloxy) benzoate (0.70 g, 1.00 mmol) was dissolved in THF (10 ml), and aluminum hydride was added at 0 ° C. under nitrogen atmosphere. Lithium (2.0 mol / l THF solution, 1.2 ml, 2.4 mmol) was added dropwise. After stirring at room temperature for 5 hours, water was added dropwise at 0 ° C. to stop the reaction. The solution was dissolved in ethyl acetate (30 ml), washed 3 times with 1N hydrochloric acid (10 ml), once with 20% brine (20 ml) and dried over sodium sulfate.
- 2,3-dihydrophytyl bromide 600 mg, 1.66 mmol
- 4-hydroxybenzaldehyde (223 mg, 1.83 mmol)
- potassium carbonate 344 mg, 2.49 mmol
- the reaction mixture was cooled to room temperature, extracted with ethyl acetate (30 ml), 3 times with 1N hydrochloric acid (6 ml), 3 times with 5% aqueous sodium hydrogen carbonate solution (6 ml), and once with 20% brine (6 ml). Washed and dried over sodium sulfate.
- the 2-methoxy-4- (2 ′, 3′-dihydrophytyloxy) benzaldoxime is dissolved in a methanol-THF mixed solvent (20 + 10 ml), 10% palladium-carbon (K) (200 mg) is added, and hydrogen is added. Stir overnight at room temperature under atmosphere.
- thionyl chloride (1.92 ml, 26.3 mmol) was added dropwise to methanol (10 ml), and 4-hydroxy-2-methylbenzoic acid (2.00 g, 13.1 mmol) was added. Stir overnight. After completion of the reaction, the solvent was distilled off, and the residue was dissolved in ethyl acetate (20 ml), twice with 5% aqueous sodium hydrogen carbonate solution (10 ml), once with 1N hydrochloric acid (10 ml) and once with water (10 ml). Washing and evaporation of the solvent gave methyl 4-hydroxy-2-methylbenzoate (2.24 g, 100% yield).
- Example 18 Synthesis of di (2-hydroxymethyl-5-methoxyphenoxy) form of GI-1000 (Nippon Soda Co., Ltd.) (GI-1000 (2-hydroxymethyl-5-methoxyphenoxy))
- GI-1000 (Br) (1.70 g) obtained in Reference Example 5 was dissolved in DMF (20 ml), and 2-hydroxy-4-methoxybenzaldehyde (1.00 g, 6.57 mmol) and potassium carbonate (1. 17 g, 8.47 mmol) was added and the mixture was stirred at 120 ° C. overnight.
- the reaction mixture was cooled to room temperature, extracted with ethyl acetate / hexane (1: 1, 20 ml), 3 times with 1N hydrochloric acid (10 ml), once with 5% aqueous sodium hydrogen carbonate (30 ml), and purified water (30 ml). ) Once.
- GI-1000 (2-formyl-5-methoxyphenoxy) (1.60 g) was dissolved in a mixed solution of chloroform (16 ml) / methanol (1.6 ml), and sodium borohydride (0.38 g, 9.99 mmol) was dissolved. And stirred at 60 ° C. for 2.5 hours. The reaction mixture was cooled to room temperature and washed 3 times with 1N hydrochloric acid (16 ml) three times with 5% aqueous sodium hydrogen carbonate solution (16 ml) and once with 20% brine (16 ml). The solvent of the organic layer was distilled off, decanted with methanol, and concentrated to obtain GI-1000 (2-hydroxymethyl-5-methoxyphenoxy) (1.84 g).
- Example 19 Synthesis of di (3-hydroxymethylphenoxy) form of GI-1000 (Nippon Soda Co., Ltd.) (Bzl (3-O-GI-1000) -OH)
- GI-1000 (Br) (563 mg) obtained in Reference Example 5 was dissolved in DMF (5 ml), and 3-hydroxybenzyl alcohol (280 mg, 2.26 mmol) and potassium carbonate (389 mg, 2.81 mmol) were added. Stir at 100 ° C. overnight. The reaction mixture was cooled to room temperature, extracted with ethyl acetate / hexane (1: 1, 25 ml), 3 times with 1N hydrochloric acid (10 ml), once with 5% aqueous sodium hydrogen carbonate (10 ml), and methanol (10 ml). Wash once with. The solvent of the organic layer was distilled off to obtain Bzl (3-O-GI-1000) -OH (580 mg).
- Example 20 Synthesis of di (3-hydroxymethylphenoxy) form of GI-2000 (Nihon Soda Co., Ltd.) (Bzl (3-O-GI-2000) -OH)
- GI-2000 (Br) (503 mg) obtained in Reference Example 6 was dissolved in DMF (5 ml), and 3-hydroxybenzyl alcohol (125 mg, 1.00 mmol) and potassium carbonate (174 mg, 1.26 mmol) were added. Stir at 100 ° C. overnight. The reaction mixture was cooled to room temperature, extracted with ethyl acetate / hexane (1: 1, 25 ml), 3 times with 1N hydrochloric acid (10 ml), once with 5% aqueous sodium hydrogen carbonate (10 ml), and methanol (10 ml). Wash once with. The solvent of the organic layer was distilled off to obtain Bzl (3-O-GI-2000) -OH (512 mg).
- TERGITOL registered trademark
- TMN-6 Br form 5.21 g
- potassium carbonate (2.93 g, 21.2 mmol, 2.5 eq) obtained in Reference Example 7
- 3-hydroxy-benzyl alcohol (1.16 g) , 9.32 mmol, 1.1 eq) was suspended in DMF (50 ml) and stirred at 80 ° C. overnight.
- the reaction solution was extracted with ethyl acetate (500 ml), washed three times with 0.5N hydrochloric acid (250 ml), three times with 5% aqueous sodium bicarbonate (250 ml), and once with 20% brine (250 ml). It was.
- Surfonamine B-30 (2.0 g) was dissolved in chloroform (20 ml), washed 3 times with water (10 ml), the organic layer was dried over sodium sulfate, and concentrated to dryness. The residue was dissolved in chloroform (20 ml), monomethyl terephthalate (1.0 g) was added, EDC.HCl (1.15 g) and HOBt (72 mg) were added and reacted at room temperature for 3 hours. The mixture was washed with an aqueous sodium hydrogen carbonate solution and saturated brine, and concentrated to give an oil. Tetrahydrofuran was added to the oil, and 4.2 equivalents of 1M DIBAL toluene solution was added and allowed to react under ice cooling.
- Peptide synthesis was performed using the compounds of Examples 1, 3, 5, 19, and 20 as protecting reagents.
- Example 23 Fmoc-Ser (tBu) -OBzl (2,4-OPhy) by condensation of 2,4- (2 ', 3'-dihydrophytyloxy) benzyl alcohol with Fmoc-Ser (tBu) -OH synthesis
- Fmoc-Ser (tBu) -OH 3.28g, 8.56mmol
- isopropyl acetate 30ml
- EDC ⁇ HCl 1.80g, 9.39mmol
- DMAP Dimethyl-4-aminopyridine
- reaction mixture was washed 3 times with 0.1N hydrochloric acid (15 ml), 3 times with 5% aqueous sodium hydrogen carbonate solution (15 ml) and once with 20% brine (15 ml) to give a condensate ( Fmoc-Ser (tBu) -OBzl (2,4-OPhy)) and used in the subsequent reaction.
- Example 24 Decomposition of Fmoc-Ser (tBu) -OBzl (2,4-OPhy) Synthesis of H-Ser (tBu) -OBzl (2,4-OPhy) by Fmoc Fmoc-Ser (tBu) -OBzl (2 , 4-OPhy) (4.28 mmol) in isopropyl acetate (45 ml) was added 4-aminomethylpiperidine (1.47 g, 12.9 mmol) at 0 ° C., and the mixture was stirred at room temperature for 4 hours.
- Example 25 Fmoc-Lys (Boc) -Ser (tBu) -OBzl (2,4-) by condensation of H-Ser (tBu) -OBzl (2,4-OPhy) and Fmoc-Lys (Boc) -OH Synthesis of OPhy)
- H-Ser (tBu) -OBzl (2,4-OPhy) 4.18 mmol
- isopropyl acetate 55 ml
- HOBt 57 mg, 0.42 mmol
- Fmoc-Lys (Boc) -OH 2.16 g, 4.61 mmol
- EDC.HCl 970 mg, 5.06 mmol
- reaction mixture was washed three times with 0.1N hydrochloric acid (30 ml), three times with 5% aqueous sodium hydrogen carbonate solution (30 ml), and once with 20% brine (30 ml) to obtain Fmoc-Lys in the reaction system. Conversion to (Boc) -Ser (tBu) -OBzl (2,4-OPhy) was used in the subsequent reaction.
- Example 27 Fmoc-Glu (OtBu) -Lys (Boc) -Ser by condensation of H-Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) and Fmoc-Glu (OtBu) -OH Synthesis of (tBu) -OBzl (2,4-OPhy) HOBt (57 mg) in isopropyl acetate solution (105 ml) of H-Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) (4.18 mmol) , 0.42 mmol), Fmoc-Glu (OtBu) -OH (2.32 g, 5.23 mmol) was dissolved, and EDC ⁇ HCl (1.09 g, 5.69 mmol) was added at 0 ° C.
- Example 28 Fmoc-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) De-Fmoc to H-Glu (OtBu) -Lys (Boc) -Ser (tBu)- Synthesis of OBzl (2,4-OPhy) Fmoc-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) in isopropyl acetate solution (50 ml) at 0 ° C. Methylpiperidine (1.43 g, 12.5 mmol) was added and stirred at room temperature for 4 hours.
- Example 29 Fmoc-Ala-Glu (OtBu) -Lys by condensation of H-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) and Fmoc-Ala-OH Synthesis of Boc) -Ser (tBu) -OBzl (2,4-OPhy) H-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) in isopropyl acetate (150 ml) HOBt (57 mg, 0.42 mmol) and Fmoc-Ala (1.52 g, 4.62 mmol) were dissolved in EDC ⁇ HCl (970 mg, 5.06 mmol) at 0 ° C.
- Example 30 Fmoc-Ala-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) De-Fmoc to H-Ala-Glu (OtBu) -Lys (Boc) -Ser Synthesis of (tBu) -OBzl (2,4-OPhy) To an isopropyl acetate solution (100 ml) of Fmoc-Ala-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) 4-Aminomethylpiperidine (1.43 g, 12.5 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 3 hours.
- Example 31 H-Ala-Glu-Lys-Ser-OH ⁇ 2TFA salt by deprotection of H-Ala-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy)
- Chloroform (10 ml) is 1/20 of the total weight of the oil of H-Ala-Glu (OtBu) -Lys (Boc) -Ser (tBu) -OBzl (2,4-OPhy) obtained in Synthesis Example 30. And concentrated, and the remaining isopropyl acetate was distilled off.
- Example 32 1-[(2-chloro-5- (2 ′, 3′-dihydrophytyloxy) phenyl)]-1-phenylmethanamine (hereinafter abbreviated as NH 2 -Dpm (COP)) Of Boc-Cys (Acm) -NH-Dpm (COP) by Condensation of Boc-Cys (Acm) -OH with NH 2 -Dpm (COP) (950 mg, 1.85 mmol) obtained in Example 3 After dissolving in isopropyl acetate (20 ml) and separating and washing with 10% aqueous sodium carbonate solution and 20% brine, the organic layer was washed with Boc-Cys (Acm) -OH (594 mg, 2.03 mmol) at room temperature.
- Boc-Cys (Acm) -OH 594 mg, 2.03 mmol
- Example 33 Synthesis of Boc-Pro-Cys (Acm) -NH-Dpm (COP) by de-Boc of Boc-Cys (Acm) -NH-Dpm (COP) and subsequent condensation with Boc-Pro-OH Boc-Cys (Acm) -NH-Dpm (COP) (1.85 mmol) obtained in Example 32 was dissolved in isopropyl acetate (20 ml), and methanesulfonic acid (600 ⁇ l, 9.2 mmol) was added in an ice bath. It was dripped. After returning to room temperature and stirring for 3 hours, a 10% aqueous sodium carbonate solution (20 ml) was added again in an ice bath and stirred.
- Example 34 Boc-Trp (CHO) -Pro-Cys (Acm) by Boc-Pro-Cys (Acm) -NH-Dpm (COP) de-Boc followed by condensation with Boc-Trp (CHO) -OH ) -NH-Dpm (COP) Synthesis Boc-Pro-Cys (Acm) -NH-Dpm (COP) (1.55 mmol) obtained in Example 33 was dissolved in isopropyl acetate (15 ml) and methanesulfonic acid was dissolved. (500 ⁇ l, 7.7 mmol) was added dropwise in an ice bath.
- Example 35 Boc-Trp (CHO) -Pro-Cys (Acm) -NH-Dpm (COP) De-Boc, followed by Boc-Asp (OBzl) -OH by condensation with Boc-Asp (OBzl) -OH Synthesis of Trp (CHO) -Pro-Cys (Acm) -NH-Dpm (COP) Boc-Trp (CHO) -Pro-Cys (Acm) -NH-Dpm (COP) obtained in Example 34 (1.
- Boc-Asp (OBzl) -OH (451 mg, 1.39 mmol)
- HOBt (19 mg, 0.14 mmol)
- EDC ⁇ HCl (297 mg, 1.55 mol) were added in an ice bath, and the mixture was returned to room temperature and stirred overnight.
- Example 36 Boc-Gly-Asp by de-Boc of Boc-Asp (OBzl) -Trp (CHO) -Pro-Cys (Acm) -NH-Dpm (COP) followed by condensation with Boc-Gly-OH Synthesis of (OBzl) -Trp (CHO) -Pro-Cys (Acm) -NH-Dpm (COP) Boc-Asp (OBzl) -Trp (CHO) -Pro-Cys (Acm)-obtained in Example 35 NH-Dpm (COP) (1.29 mmol) was dissolved in isopropyl acetate (20 ml), methanesulfonic acid (400 ⁇ l, 6.2 mmol) was added dropwise in an ice bath, and the mixture was stirred at room temperature for 3 hours.
- COP NH-Dpm
- Methanesulfonic acid 300 ⁇ l, 4.6 mmol was added, and the mixture was further stirred for 2 hours. Then, 10% aqueous sodium carbonate solution (25 ml) was added in an ice bath, and the mixture was stirred briefly. The aqueous layer was separated and discarded, and the organic layer was washed once with 20% brine (20 ml), whereby Asp (OBzl) -Trp (CHO) -Pro-Cys (Acm) -NH— Conversion to Dpm (COP).
- Example 37 Synthesis of Boc-Leu-OBzl (3-O-GI-1000) by condensation of Bzl- (3-O-GI-1000) -OH and Boc-Leu-OH
- Example 38 Boc-Leu-OBzl (3-O-GI-1000) Boc-Tyr (Bzl) -Leu-OBzl (3-O-GI-1000) by de-Boc followed by condensation with Boc-Tyr (Bzl) -OH O-GI-1000)
- Boc-Leu-OBzl (3-O-GI-1000) (100 mg) obtained in Example 37 was dissolved in isopropyl acetate (1 ml), and methanesulfonic acid (32 ⁇ l, 0.49 mmol) was added dropwise for 5 hours. Stir. After completion of the reaction, the reaction system was converted to H-Leu-OBzl (3-O-GI-1000) by washing 3 times with 10% aqueous sodium carbonate solution and once with 20% brine.
- Example 39 Boc-Glu (OBzl) -Tyr by de-Boc of Boc-Tyr (Bzl) -Leu-OBzl (3-O-GI-1000) followed by condensation with Boc-Glu (OBzl) -OH Synthesis of (Bzl) -Leu-OBzl (3-O-GI-1000)
- Boc-Tyr (Bzl) -Leu-OBzl (3-O-GI-1000) 100 mg was dissolved in isopropyl acetate (1 ml), and methanesulfonic acid (32 ⁇ l, 0.49 mmol) was dissolved. The solution was added dropwise and stirred for 6 hours. After completion of the reaction, it was washed 3 times with 10% aqueous sodium carbonate solution and once with 20% saline. To this organic layer, HOBt (5 mg, 0.04 mmol), Boc-Glu (OBzl) -OH (140 mg, 0.38 mmol) and EDC ⁇ HCl (79 mg, 0.41 mmol) were added and stirred at 40 ° C. overnight. .
- the reaction solution was quenched by adding DMPDA (5 ⁇ l, 0.04 mmol), washed 3 times with 1N hydrochloric acid, the organic layer was concentrated, and Boc-Glu (OBzl) -Tyr (Bzl) -Leu-OBzl (3-O -GI-1000) (100 mg) was obtained.
- Example 40 Synthesis of Boc-Leu-OBzl (3-O-GI-2000) by condensation of Bzl (3-O-GI-2000) -OH and Boc-Leu-OH Bzl obtained in Example 20 (3-O-GI-2000) -OH (402 mg) was dissolved in isopropyl acetate (4 ml) and DMAP (5 mg, 0.06 mmol), Boc-Leu-OH (151 mg, 0.65 mmol) and EDC.HCl ( 128 mg, 0.70 mmol) was added, and the mixture was stirred at 40 ° C. for 4 hours.
- Example 41 Boc-Leu-OBzl (3-O-GI-2000) De-Boc, followed by Boc-Tyr (Bzl) -OH by condensation with Boc-Tyr (Bzl) -Leu-OBzl (3- Synthesis of O-GI-2000) Boc-Leu-OBzl (3-O-GI-2000) (100 mg) obtained in Example 40 was dissolved in isopropyl acetate (1 ml), and methanesulfonic acid (16 ⁇ l, 0. 25 mmol) was added dropwise and stirred for 3 hours. After completion of the reaction, it was washed 3 times with 10% aqueous sodium carbonate solution and once with 20% saline.
- Example 42 Boc-Glu (OBzl) -Tyr by de-Boc of Boc-Tyr (Bzl) -Leu-OBzl (3-O-GI-2000) followed by condensation with Boc-Glu (OBzl) -OH Synthesis of (Bzl) -Leu-OBzl (3-O-GI-2000) Boc-Tyr (Bzl) -Leu-OBzl (3-O-GI-2000) (100 mg) was dissolved in isopropyl acetate (1 ml). Methanesulfonic acid (32 ⁇ l, 0.49 mmol) was added dropwise and stirred for 6 hours.
- Example 43 Preparation of Fmoc-Leu-OBzl (3,4,5-OPhy) by condensation of 3,4,5-tri (2 ', 3'-dihydrophytyloxy) benzyl alcohol with Fmoc-Leu-OH Synthesis and subsequent synthesis of H-Leu-OBzl (3,4,5-OPhy) by de-Fmoc 3,4,5-tri (2 ′, 3′-dihydrophytyloxy) benzyl alcohol (5 g, 5.
- the solvent in the reaction solution was distilled off under reduced pressure to 25 ml, CPME (50 ml) was added to the residue, the solvent in the reaction solution was again distilled off under reduced pressure to 25 ml, and then CPME (25 ml) was added to the residue.
- the amount of solvent in the reaction solution was 50 ml. Nitrogen bubbling was performed on this reaction solution for 5 minutes, and then diethylenetriamine (2.71 ml, 25.1 mmol) was added in an ice bath in a nitrogen atmosphere, followed by stirring at room temperature for 5 hours.
- reaction solution was separated and washed twice with 10% aqueous sodium carbonate solution (50 ml), 20% brine (50 ml) was added to the resulting organic layer, and the organic layer and aqueous layer (20% brine) were added.
- 1N hydrochloric acid was added dropwise until the pH of the aqueous layer reached 6.8, and then the organic layer and the aqueous layer were transferred to a separatory funnel to remove the aqueous layer.
- the obtained organic layer was separated and washed once with 20% brine (50 ml), once with 10% aqueous sodium carbonate (50 ml), and once with 20% brine (50 ml).
- the obtained organic layer was dried over sodium sulfate, and then the sodium sulfate was removed by filtration to obtain a CPME solution (50 ml) of H-Leu-OBzl (3,4,5-OPhy) (5.01 mmol). .
- Example 44 Fmoc-Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) by condensation of H-Leu-OBzl (3,4,5-OPhy) with Fmoc-Tyr (tBu) -OH And synthesis of H-Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) by subsequent de-Fmoc of H-Leu-OBzl (3,4,5-OPhy) (5.01 mmol) HOBt (68 mg, 0.50 mmol) and Fmoc-Tyr (tBu) -OH (2.53 g, 5.52 mmol) were added to the CPME solution (50 ml), and then EDC ⁇ HCl (1 .16 g, 6.05 mmol) was added and stirred at room temperature overnight.
- the solvent in the reaction solution was distilled off under reduced pressure to 25 ml, CPME (50 ml) was added to the residue, the solvent in the reaction solution was again distilled off under reduced pressure to 25 ml, and then CPME (25 ml) was added to the residue.
- the amount of solvent in the reaction solution was 50 ml. Nitrogen bubbling was performed on the reaction solution for 5 minutes, and then diethylenetriamine (2.71 ml, 25.1 mmol) was added in an ice bath in a nitrogen atmosphere, followed by stirring at room temperature for 1.5 hours.
- reaction solution was separated and washed twice with 10% aqueous sodium carbonate solution (50 ml), 20% brine (50 ml) was added to the resulting organic layer, and the organic layer and aqueous layer (20% brine) were added.
- 1N hydrochloric acid was added dropwise until the pH of the aqueous layer reached 6.8, and then the organic layer and the aqueous layer were transferred to a separatory funnel to remove the aqueous layer.
- the same operation was performed again, and the organic layer was stirred and washed with an aqueous layer (20% saline) having a pH of 6.7, and the aqueous layer was removed.
- the obtained organic layer was separated and washed once with 20% brine (50 ml), once with 10% aqueous sodium carbonate (50 ml), and once with 20% brine (50 ml).
- the obtained organic layer was dried over sodium sulfate, sodium sulfate was removed by filtration, and a CPME solution of H-Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) (5.01 mmol) ( 50 ml) was obtained.
- Example 45 Fmoc-Glu (OtBu) -Tyr (tBu) -Leu- by condensation of H-Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) with Fmoc-Glu (OtBu) -OH Synthesis of OBzl (3,4,5-OPhy) and subsequent synthesis of H-Glu (OtBu) -Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) by de-Fmoc H-Tyr (tBu ) -Leu-OBzl (3,4,5-OPhy) (5.01 mmol) in CPME solution (50 ml), HOBt (68 mg, 0.50 mmol), Fmoc-Glu (OtBu) -OH (2.35 g, 5 Then, EDC ⁇ HCl (1.16 g, 6.05 mmol) was added thereto in an ice bath and stirred overnight at room temperature.
- Example 46 Fmoc-Glu (OtBu) -Glu by condensation of H-Glu (OtBu) -Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) and Fmoc-Glu (OtBu) -OH Synthesis of OtBu) -Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) and subsequent de-Fmoc H-Glu (OtBu) -Glu (OtBu) -Tyr (tBu) -Leu-OBzl ( Synthesis of 3,4,5-OPhy) To a CPME solution (50 ml) of H-Glu (OtBu) -Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) (5.01 mmol), HOBt (68 mg , 0.50 mmol), Fmoc-Glu (OtBu) -OH (2.35 g, 5.52 mmol) was added to the solution under ice bath.
- Example 47 H-Asp (OtBu) -Phe-Glu (OtBu) -Glu (OtBu) -Ile-Pro-Glu (OtBu) -Glu (OtBu) -Tyr (tBu) -Leu-OBzl (3,4, Synthesis of 5-OPhy) H-Glu (OtBu) -Glu (OtBu) -Tyr (tBu) -Leu-OBzl (3,4,5-OPhy) and the following protected amino acids: Fmoc-Pro, Fmoc-Ile, Fmoc -Glu (OtBu), Fmoc-Glu (OtBu), Fmoc-Phe and Fmoc-Asp (OtBu) were used in this order in the same manner as in Examples 43 to 46, followed by condensation reaction, deprotection reaction and After repeating the treatment operation to elongate the peptide chain, the product is precipitated with an acetonitrile aqueous solution, and H-Asp
- the final product is produced only through extraction and separation without crystallization and isolation of each intermediate in each step. It has become possible to provide a method for producing peptides and the like leading to products. Furthermore, organic synthesis reactions and practical industrial processes can be provided. In addition, the method for producing a peptide of the present invention can be stably dissolved in a solvent regardless of the sequence and chain length of the peptide, compared to the conventional liquid phase method. Has the advantage that high purity and high yield can be secured comprehensively.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Life Sciences & Earth Sciences (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Peptides Or Proteins (AREA)
Abstract
本発明は、特定の分岐鎖含有芳香族化合物を提供する。本発明の分岐鎖含有芳香族化合物は、分液操作性の優れた酢酸イソプロピルに易溶であり、各工程で各中間体を晶析単離することなく抽出分離のみを経て最終生成物へと導くペプチド等の製造方法に使用することができる。
Description
本発明は、特定の分岐鎖含有芳香族化合物に関する。本発明は、更に当該化合物を含む保護化試薬またはその付加体に関する。また、本発明は、当該化合物を用いたペプチドの製造方法、および更に当該ペプチドの製造方法を含む有機合成方法に関する。
ペプチドの製造方法としては、これまで概ね固相法と液相法に大別されてきた。固相法は、反応後の単離・精製をレジンの洗浄だけで行える点で有利ではあるが、本質的に不均一相の反応であり、低い反応性を補うために反応試剤・試薬を過剰量用いる必要があったり、反応の追跡、および担体に担持された状態での反応生成物の解析が困難であったりという問題点があった。一方、液相法は、反応性も良好で、縮合反応の後に抽出洗浄、単離等により中間体ペプチドの精製を行えるという利点を有しているが、カップリング反応および脱保護の各工程において、残留試薬および/または副生成物を除去するため、非極性有機溶媒および酸性または塩基性水溶液による抽出洗浄工程、および/または結晶化などの単離・精製工程が必要であるなど、製造工程が複雑化するという問題点があった。
近年、上述2法の問題点を改善する取組みが行われてきた。
特許文献1および非特許文献1には、それぞれ3,4,5-トリス(n-オクタデシロキシ)ベンジルアルコール型化合物をカルボキシル基等の保護化試薬とする手法が開示されている。また、特許文献2~4には、それぞれ3,5-ジ(ドコシルオキシ)ベンジルアルコール型化合物、2,4-ジ(ドコシルオキシ)ベンジルアルコール型化合物、トリチル型化合物等の保護化試薬が開示されている。これらの保護化試薬を使用すれば、反応を均一な液相中で行うことができ、その反応後に、溶媒組成を変化させることにより沈殿させ、単離・精製を濾過および洗浄だけで行うことができる。しかしながら、これらの保護化試薬の使用では、沈殿化のために反応溶媒留去工程が必要である、沈殿物の濾過工程に多大な時間を要する、或いはこれらの保護化試薬が酢酸エステルまたはトルエンには不溶または難溶であるという問題があり、上記文献に開示の手法は、必ずしも万能な方法であるとは言い難かった。
特許文献1および非特許文献1には、それぞれ3,4,5-トリス(n-オクタデシロキシ)ベンジルアルコール型化合物をカルボキシル基等の保護化試薬とする手法が開示されている。また、特許文献2~4には、それぞれ3,5-ジ(ドコシルオキシ)ベンジルアルコール型化合物、2,4-ジ(ドコシルオキシ)ベンジルアルコール型化合物、トリチル型化合物等の保護化試薬が開示されている。これらの保護化試薬を使用すれば、反応を均一な液相中で行うことができ、その反応後に、溶媒組成を変化させることにより沈殿させ、単離・精製を濾過および洗浄だけで行うことができる。しかしながら、これらの保護化試薬の使用では、沈殿化のために反応溶媒留去工程が必要である、沈殿物の濾過工程に多大な時間を要する、或いはこれらの保護化試薬が酢酸エステルまたはトルエンには不溶または難溶であるという問題があり、上記文献に開示の手法は、必ずしも万能な方法であるとは言い難かった。
また、特許文献5には、3,4,5-トリス(n-オクタデシロキシ)ベンジルアルコール型化合物を保護化試薬としたペプチド合成反応例が紹介されている。しかしながら、該文献には、希薄条件での有機溶媒同士の特殊な分層分離事例が開示されているに過ぎず、この事例は、必ずしも工業的に万能な方法であるとは言い難かった。
Bull.Chem.Soc.Jpn 74,733-738(2001)
本発明の課題は、分液操作性の優れた酢酸イソプロピルに易溶で、各工程で各中間体を晶析単離することなく抽出分離のみを経て最終生成物へと導くペプチド等の製造方法(ワンポット合成方法ともいう)に使用しうる、新規化合物を提供することである。
本発明者は、鋭意検討の結果、特定の分岐鎖含有芳香族化合物によって前記課題を解決できることを見出し、本発明を完成した。本発明は以下の態様を含む。
[1] 式(I):
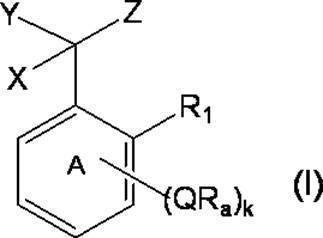
[式中、
k個のQは、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基または-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)を示し;かつ
Zは、水素原子または式(a):
k個のQは、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基または-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)を示し;かつ
Zは、水素原子または式(a):
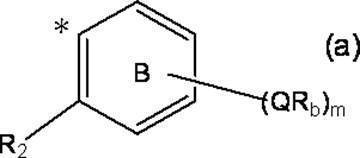
(式中、*は結合位置を示し;
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい)で表される基を示し;
前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(b):
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい)で表される基を示し;
前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(b):

(式中、*は、隣接原子との結合位置を示し;
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基である。]で表される分岐鎖含有芳香族化合物。
[2] 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(c):
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基である。]で表される分岐鎖含有芳香族化合物。
[2] 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(c):

[式中、*は、Qとの結合位置を示し;
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):
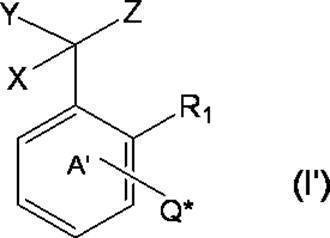
(式中、*は、結合位置を示し;
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]で表される基である、
前記[1]に記載の分岐鎖含有芳香族化合物。
[3] 前記式(c)中、
R5およびR6は、共に水素原子であり;
n0個のR7およびR8は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n0個のX2は、独立してそれぞれ、単結合、メチレン基またはエチレン基であり;かつ
R9は、水素原子、メチル基またはエチル基である、
前記[2]に記載の分岐鎖含有芳香族化合物。
[4] 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(d):
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]で表される基である、
前記[1]に記載の分岐鎖含有芳香族化合物。
[3] 前記式(c)中、
R5およびR6は、共に水素原子であり;
n0個のR7およびR8は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n0個のX2は、独立してそれぞれ、単結合、メチレン基またはエチレン基であり;かつ
R9は、水素原子、メチル基またはエチル基である、
前記[2]に記載の分岐鎖含有芳香族化合物。
[4] 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(d):
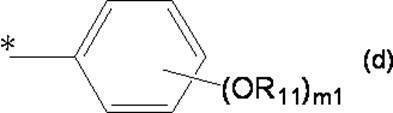
(式中、*は、Qとの結合位置を示し;
m1個のOR11は、式(c’):
m1個のOR11は、式(c’):

[式中、*は、Oとの結合位置を示し;
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):
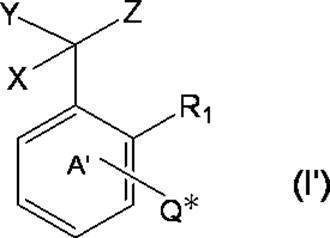
(式中、*は、結合位置を示し;
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)で表される基を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]で表される基により置換されたヒドロキシル基を示し;
m1は、1~3の整数を示す。)で表される基である、
前記[1]に記載の分岐鎖含有芳香族化合物。
[5] 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(e):
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)で表される基を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]で表される基により置換されたヒドロキシル基を示し;
m1は、1~3の整数を示す。)で表される基である、
前記[1]に記載の分岐鎖含有芳香族化合物。
[5] 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(e):
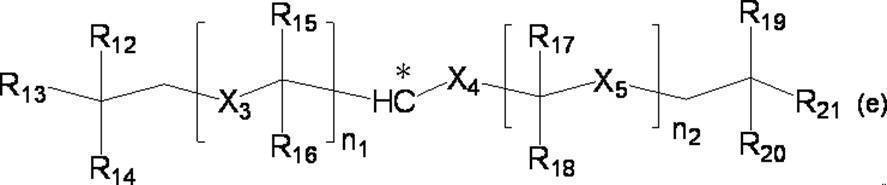
(式中、*は、Qとの結合位置を示し;
n1は、1~10の整数を示し;
n2は、1~10の整数を示し;
n1個のR15およびR16は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n1個のX3は、単結合またはC1-4アルキレン基を示し;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n2個のX5は、単結合またはC1-4アルキレン基を示し;
X4は、単結合またはC1-4アルキレン基を示し;かつ
R12、R13、R14、R19、R20およびR21は、独立してそれぞれ、水素原子またはC1-4アルキル基を示す。
但し、R15およびR16、および/またはR17およびR18が共に水素原子であることはなく、かつn1+n2が2の場合には、R12、R13およびR14の2個以上が独立してそれぞれ、C1-4アルキル基を示すか、またはR19、R20およびR21の2個以上が独立してそれぞれ、C1-4アルキル基を示す。)で表される基である、
前記[1]に記載の分岐鎖含有芳香族化合物。
[6] 前記式(e)中、
n1は、1~5の整数であり;
n2は、1~5の整数であり;
n1個のR15およびR16は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n1個のX3は、単結合、メチレン基またはエチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n2個のX5は、単結合、メチレン基またはエチレン基であり;
X4は、単結合、メチレン基またはエチレン基である、
前記[5]に記載の分岐鎖含有芳香族化合物。
[7] 前記式(e)中、
n1個のR15およびR16は、独立してそれぞれ、水素原子またはメチル基であり;
n1個のX3は、単結合またはメチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはメチル基であり;
n2個のX5は、単結合またはメチレン基であり;
X4は、単結合またはメチレン基であり;かつ
R12、R13、R14、R19、R20およびR21は、メチル基である、
前記[6]に記載の分岐鎖含有芳香族化合物。
[8] RaおよびRbが、独立してそれぞれ、3,7,11,15-テトラメチルヘキサデシル基、3,7,11-トリメチルドデシル基、2,2,4,8,10,10-ヘキサメチル-5-ドデカノイル基、3,4,5-トリ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基、3,5-ジ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基、式(f):
n1は、1~10の整数を示し;
n2は、1~10の整数を示し;
n1個のR15およびR16は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n1個のX3は、単結合またはC1-4アルキレン基を示し;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n2個のX5は、単結合またはC1-4アルキレン基を示し;
X4は、単結合またはC1-4アルキレン基を示し;かつ
R12、R13、R14、R19、R20およびR21は、独立してそれぞれ、水素原子またはC1-4アルキル基を示す。
但し、R15およびR16、および/またはR17およびR18が共に水素原子であることはなく、かつn1+n2が2の場合には、R12、R13およびR14の2個以上が独立してそれぞれ、C1-4アルキル基を示すか、またはR19、R20およびR21の2個以上が独立してそれぞれ、C1-4アルキル基を示す。)で表される基である、
前記[1]に記載の分岐鎖含有芳香族化合物。
[6] 前記式(e)中、
n1は、1~5の整数であり;
n2は、1~5の整数であり;
n1個のR15およびR16は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n1個のX3は、単結合、メチレン基またはエチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n2個のX5は、単結合、メチレン基またはエチレン基であり;
X4は、単結合、メチレン基またはエチレン基である、
前記[5]に記載の分岐鎖含有芳香族化合物。
[7] 前記式(e)中、
n1個のR15およびR16は、独立してそれぞれ、水素原子またはメチル基であり;
n1個のX3は、単結合またはメチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはメチル基であり;
n2個のX5は、単結合またはメチレン基であり;
X4は、単結合またはメチレン基であり;かつ
R12、R13、R14、R19、R20およびR21は、メチル基である、
前記[6]に記載の分岐鎖含有芳香族化合物。
[8] RaおよびRbが、独立してそれぞれ、3,7,11,15-テトラメチルヘキサデシル基、3,7,11-トリメチルドデシル基、2,2,4,8,10,10-ヘキサメチル-5-ドデカノイル基、3,4,5-トリ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基、3,5-ジ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基、式(f):
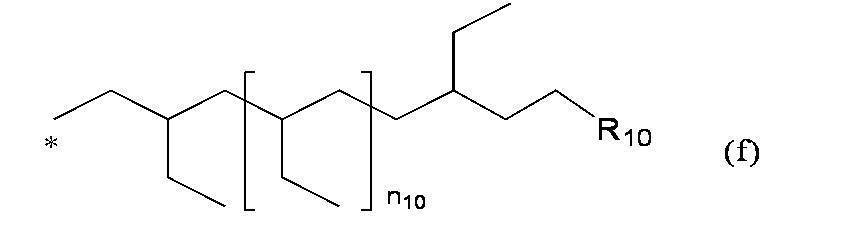
[式中、*は、Qとの結合位置であり、n10は、23~34であり、R10は、式(I’):
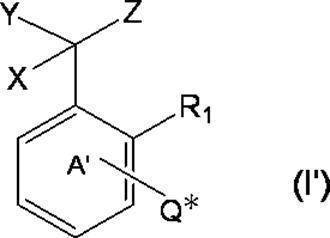
(式中、*は、結合位置を示し;
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)で表される基である。]で表される基、
式(g):
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)で表される基である。]で表される基、
式(g):

(式中、*は、Qとの結合位置であり、n11は、1~10である。)で表される基、
式(h):
式(h):
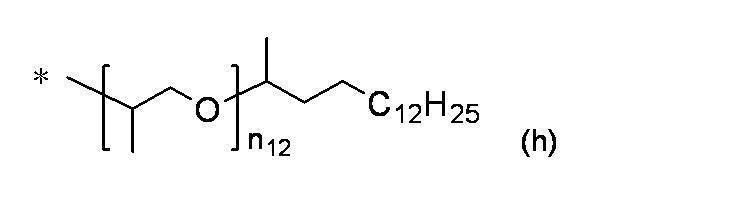
(式中、*は、Qとの結合位置であり、n12は、2~10である。)で表される基、
式(i):
式(i):

(式中、*は、Qとの結合位置であり、n13およびn14は、独立してそれぞれ、1~10である。)で表される基、または
式(j):
式(j):
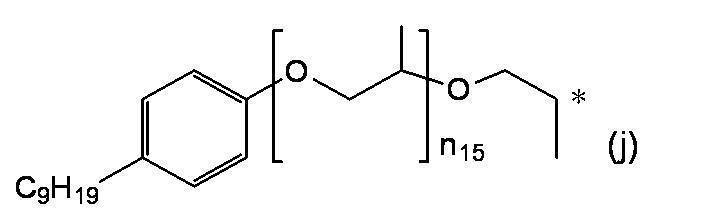
(式中、*は、Qとの結合位置であり、n15は、2~20である。)で表される基、
である、前記[1]に記載の分岐鎖含有芳香族化合物。
[9] XとZが共に水素原子であり、かつR1が水素原子である、前記[1]~[8]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[10] Xが水素原子であり、R1が水素原子であり、kが1であり、かつZが式(a)(式中、R2が水素原子であり、mが0である。)で表される基である、前記[1]~[8]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[11] Xがフェニル基であり、kが1であり、Zが式(a)(式中、mが0である。)で表される基であり、かつR2がR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成する、前記[1]~[8]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[12] Qが-O-である、前記[1]~[11]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[13] Yがヒドロキシル基である、前記[1]~[12]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[14] Yが-NHR基である、前記[1]~[12]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[15] 2,4-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
2-[3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ]-4-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン;
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド;
4-(3,7,11-トリメチルドデシルオキシ)ベンジルアルコール;
2-(3,7,11-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール;
式:
である、前記[1]に記載の分岐鎖含有芳香族化合物。
[9] XとZが共に水素原子であり、かつR1が水素原子である、前記[1]~[8]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[10] Xが水素原子であり、R1が水素原子であり、kが1であり、かつZが式(a)(式中、R2が水素原子であり、mが0である。)で表される基である、前記[1]~[8]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[11] Xがフェニル基であり、kが1であり、Zが式(a)(式中、mが0である。)で表される基であり、かつR2がR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成する、前記[1]~[8]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[12] Qが-O-である、前記[1]~[11]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[13] Yがヒドロキシル基である、前記[1]~[12]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[14] Yが-NHR基である、前記[1]~[12]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[15] 2,4-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
2-[3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ]-4-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン;
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド;
4-(3,7,11-トリメチルドデシルオキシ)ベンジルアルコール;
2-(3,7,11-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール;
式:

(式中、n16は、23または34を示す。)で表される化合物;
式:
式:
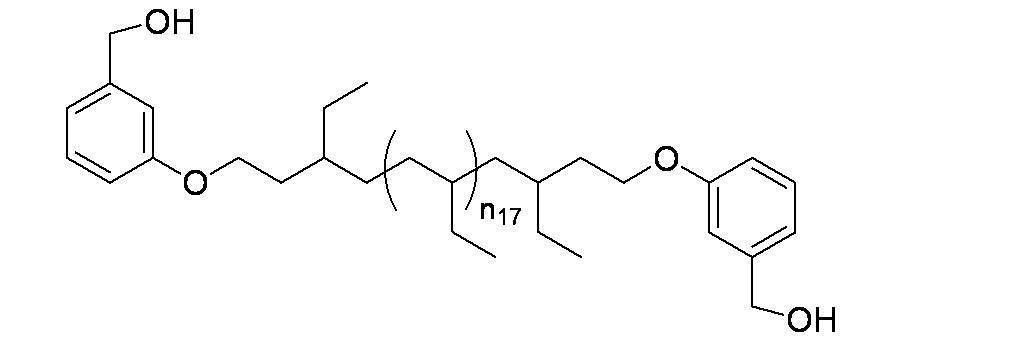
(式中、n17は、23または34を示す。)で表される化合物;
式:
式:

(式中、n18は、5~7を示す。)で表される化合物;および
式:
式:
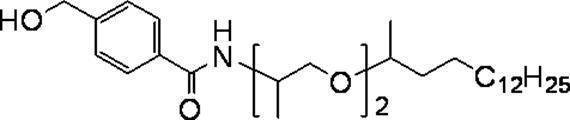
で表される化合物、からなる群から選択される、前記[1]に記載の分岐鎖含有芳香族化合物。
[16] 20℃における酢酸イソプロピル100g中の飽和溶解度が、1~95重量%である、前記[1]~[15]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[17] 20℃における酢酸イソプロピル100g中の飽和溶解度が、10~95重量%である、前記[1]~[15]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[18] 前記[1]~[17]のいずれか1つに記載の分岐鎖含有芳香族化合物を含む、アミノ酸またはペプチドのカルボキシル基またはアミド基の保護化試薬。
[19] アミノ酸またはペプチドの保護箇所がC末端である、前記[18]に記載の保護化試薬。
[20] 前記[1]~[17]のいずれか1つに記載の分岐鎖含有芳香族化合物によって保護された、分岐鎖含有芳香族化合物付加体。
[21] 工程(1)~(4)を含む、ペプチドの製造方法。
(1)前記[1]~[17]のいずれか1つに記載の分岐鎖含有芳香族化合物を、該化合物の可溶性溶媒中で、N-保護アミノ酸またはN-保護ペプチドのC末端と縮合させて、該化合物由来の保護基であるアンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、ペプチドを得る工程。
[22] さらに工程(5)~(7)の繰り返しを1以上含む、前記[21]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程。
[23] さらに工程(5)~(7’)の繰り返しを1以上含む、前記[21]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程。
[24] 前記[21]~[23]のいずれか1つに記載のペプチド製造方法を含む、有機合成方法。
[25] 式(I):
[16] 20℃における酢酸イソプロピル100g中の飽和溶解度が、1~95重量%である、前記[1]~[15]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[17] 20℃における酢酸イソプロピル100g中の飽和溶解度が、10~95重量%である、前記[1]~[15]のいずれか1つに記載の分岐鎖含有芳香族化合物。
[18] 前記[1]~[17]のいずれか1つに記載の分岐鎖含有芳香族化合物を含む、アミノ酸またはペプチドのカルボキシル基またはアミド基の保護化試薬。
[19] アミノ酸またはペプチドの保護箇所がC末端である、前記[18]に記載の保護化試薬。
[20] 前記[1]~[17]のいずれか1つに記載の分岐鎖含有芳香族化合物によって保護された、分岐鎖含有芳香族化合物付加体。
[21] 工程(1)~(4)を含む、ペプチドの製造方法。
(1)前記[1]~[17]のいずれか1つに記載の分岐鎖含有芳香族化合物を、該化合物の可溶性溶媒中で、N-保護アミノ酸またはN-保護ペプチドのC末端と縮合させて、該化合物由来の保護基であるアンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、ペプチドを得る工程。
[22] さらに工程(5)~(7)の繰り返しを1以上含む、前記[21]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程。
[23] さらに工程(5)~(7’)の繰り返しを1以上含む、前記[21]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程。
[24] 前記[21]~[23]のいずれか1つに記載のペプチド製造方法を含む、有機合成方法。
[25] 式(I):
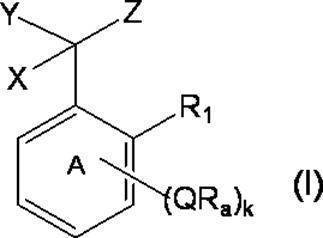
[式中、
k個のQは、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基、-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)またはハロゲン原子(好ましくはハロゲン原子)を示し;かつ
Zは、水素原子または式(a):
k個のQは、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基、-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)またはハロゲン原子(好ましくはハロゲン原子)を示し;かつ
Zは、水素原子または式(a):

(式中、*は結合位置を示し;
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい)で表される基を示し;
前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(b):
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい)で表される基を示し;
前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(b):
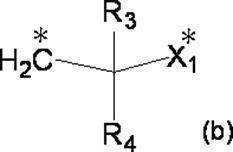
(式中、*は、隣接原子との結合位置を示し;
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基である。]で表される分岐鎖含有芳香族化合物。
[26] 上記[25]に記載の分岐鎖含有芳香族化合物を含む、アミノ酸またはペプチドのカルボキシル基またはアミド基の保護化試薬。
[27] 上記[25]に記載の分岐鎖含有芳香族化合物によって保護された、分岐鎖含有芳香族化合物付加体。
[28] 工程(1)~(4)を含む、ペプチドの製造方法。
(1)上記[25]に記載の分岐鎖含有芳香族化合物を、該化合物の可溶性溶媒中で、N-保護アミノ酸またはN-保護ペプチドのC末端と縮合させて、該化合物由来の保護基であるアンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、ペプチドを得る工程。
[29] さらに工程(5)~(7)の繰り返しを1以上含む、上記[28]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程。
[30] さらに工程(5)~(7’)の繰り返しを1以上含む、上記[28]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程。
[31] 上記[28]~[30]のいずれか一つに記載のペプチド製造方法を含む、有機合成方法。
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基である。]で表される分岐鎖含有芳香族化合物。
[26] 上記[25]に記載の分岐鎖含有芳香族化合物を含む、アミノ酸またはペプチドのカルボキシル基またはアミド基の保護化試薬。
[27] 上記[25]に記載の分岐鎖含有芳香族化合物によって保護された、分岐鎖含有芳香族化合物付加体。
[28] 工程(1)~(4)を含む、ペプチドの製造方法。
(1)上記[25]に記載の分岐鎖含有芳香族化合物を、該化合物の可溶性溶媒中で、N-保護アミノ酸またはN-保護ペプチドのC末端と縮合させて、該化合物由来の保護基であるアンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、ペプチドを得る工程。
[29] さらに工程(5)~(7)の繰り返しを1以上含む、上記[28]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程。
[30] さらに工程(5)~(7’)の繰り返しを1以上含む、上記[28]に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程。
[31] 上記[28]~[30]のいずれか一つに記載のペプチド製造方法を含む、有機合成方法。
本発明の分岐鎖含有芳香族化合物は、分液操作性の優れた酢酸イソプロピルに易溶である。そのため、本発明の分岐鎖含有芳香族化合物を使用すれば、各工程で各中間体を晶析単離することなく、抽出分離のみを経て最終生成物へと導くペプチド等の製造方法を実施できる。
文中で特に断らない限り、本明細書で用いるすべての技術用語および科学用語は、本発明が属する技術分野の当業者に一般に理解されるのと同じ意味をもつ。本明細書に記載されたものと同様または同等の任意の方法および材料は、本発明の実施または試験において使用することができるが、好ましい方法および材料を以下に記載する。本明細書で言及したすべての刊行物および特許は、例えば、記載された発明に関連して使用されうる刊行物に記載されている、構築物および方法論を記載および開示する目的で、参照として本明細書に組み入れられる。
〔本発明化合物〕
本発明の分岐鎖含有芳香族化合物(以下、本発明化合物と略称することもある。)は、下記式(I)で表される。本発明化合物は、特定のベンジル化合物(式(I)中、XとZが共に水素原子であり、かつR1が水素原子である);特定のジフェニルメタン化合物(式(I)中、Xが水素原子であり、R1が水素原子であり、kが1であり、かつZが式(a)(式中、R2が水素原子であり、mが0である。)で表される基である);および特定のフルオレン化合物(式(I)中、Xがフェニル基であり、kが1であり、Zが式(a)(式中、mが0である。)で表される基であり、かつR2がR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成する)を包含する。
式(I):
本発明の分岐鎖含有芳香族化合物(以下、本発明化合物と略称することもある。)は、下記式(I)で表される。本発明化合物は、特定のベンジル化合物(式(I)中、XとZが共に水素原子であり、かつR1が水素原子である);特定のジフェニルメタン化合物(式(I)中、Xが水素原子であり、R1が水素原子であり、kが1であり、かつZが式(a)(式中、R2が水素原子であり、mが0である。)で表される基である);および特定のフルオレン化合物(式(I)中、Xがフェニル基であり、kが1であり、Zが式(a)(式中、mが0である。)で表される基であり、かつR2がR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成する)を包含する。
式(I):

[式中、
k個のQは、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基、-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)またはハロゲン原子を示し;かつ
Zは、水素原子または式(a):
k個のQは、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基、-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)またはハロゲン原子を示し;かつ
Zは、水素原子または式(a):
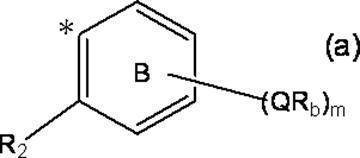
(式中、*は結合位置を示し;
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい)で表される基を示す。]。
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい)で表される基を示す。]。
本発明の式(I)で表される化合物と保護化を意図する化合物とは、Y基であるヒドロキシル基、NHR基またはハロゲン原子と、保護化を意図する化合物のカルボキシル基等との縮合反応によって、結合する。
本明細書中、Rで示される「アルキル基」としては、C1-30アルキル基が挙げられ、好ましくはC1-10アルキル基、より好ましくはC1-6アルキル基である。好適な具体例としては、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル等が挙げられ、特にメチル、エチルが好ましい。
本明細書中、Rで示される「アラルキル基」としては、C7-30アラルキル基が挙げられ、好ましくはC7-20アラルキル基、より好ましくはC7-16アラルキル基(C6-10アリール-C1-6アルキル基)である。好適な具体例としては、ベンジル、1-フェニルエチル、2-フェニルエチル、1-フェニルプロピル、α-ナフチルメチル、1-(α-ナフチル)エチル、2-(α-ナフチル)エチル、1-(α-ナフチル)プロピル、β-ナフチルメチル、1-(β-ナフチル)エチル、2-(β-ナフチル)エチル、1-(β-ナフチル)プロピル等が挙げられ、特にベンジルが好ましい。
Rとしては、水素原子、C1-6アルキル基またはC7-16アラルキル基が好ましく、水素原子、メチル、エチルまたはベンジルがより好ましく、水素原子が特に好ましい。
本明細書中、「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子またはヨウ素原子である。Yの「ハロゲン原子」としては、塩素原子、臭素原子、ヨウ素原子が好ましく、臭素原子がより好ましい。
本明細書中、Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」とは、その分子構造中に分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基である。
「分岐鎖を1以上有する脂肪族炭化水素基」における「分岐鎖」としては、直鎖または分岐状の飽和脂肪族炭化水素基であり、C1-6アルキル基が好ましく、C1-4アルキル基がより好ましく、メチル基またはエチル基が一層好ましい。また、該「分岐鎖」は、1個以上のハロゲン原子で置換されていてもよい。
「分岐鎖を1以上有する脂肪族炭化水素基」における「脂肪族炭化水素基」とは、直鎖状の飽和または不飽和の脂肪族炭化水素基であり、C2-C300アルキル基(好ましくは、C3-C100アルキル基、より好ましくは、C3-C60アルキル基)、C2-C300アルケニル基(好ましくは、C3-C100アルケニル基、より好ましくは、C3-C60アルケニル基)またはC2-C300アルキニル基(好ましくは、C3-C100アルキニル基、より好ましくは、C3-C60アルキニル基)である。
「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」における「分岐鎖を1以上有する脂肪族炭化水素基」の部位は、特に限定されず、末端に存在しても(1価基)、それ以外の部位に存在してもよい(例えば2価基)。
「分岐鎖を1以上有する脂肪族炭化水素基」としては、具体的には、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基(ラウリル基)、トリデシル基、ミリスチル基、セチル基、ステアリル基、アラキル基、ベヘニル基、オレイル基、リノリル基、リグノセリル基等の分岐異性体であって、1以上の分岐鎖を有する1価基およびそれらから誘導される2価基が挙げられ、好ましくは、3,7,11-トリメチルドデシル基、3,7,11,15-テトラメチルヘキサデシル基(以下、2,3-ジヒドロフィチル基ということもある。)、2,2,4,8,10,10-ヘキサメチルウンデカン-5-イル基、式:
「分岐鎖を1以上有する脂肪族炭化水素基」における「分岐鎖」としては、直鎖または分岐状の飽和脂肪族炭化水素基であり、C1-6アルキル基が好ましく、C1-4アルキル基がより好ましく、メチル基またはエチル基が一層好ましい。また、該「分岐鎖」は、1個以上のハロゲン原子で置換されていてもよい。
「分岐鎖を1以上有する脂肪族炭化水素基」における「脂肪族炭化水素基」とは、直鎖状の飽和または不飽和の脂肪族炭化水素基であり、C2-C300アルキル基(好ましくは、C3-C100アルキル基、より好ましくは、C3-C60アルキル基)、C2-C300アルケニル基(好ましくは、C3-C100アルケニル基、より好ましくは、C3-C60アルケニル基)またはC2-C300アルキニル基(好ましくは、C3-C100アルキニル基、より好ましくは、C3-C60アルキニル基)である。
「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」における「分岐鎖を1以上有する脂肪族炭化水素基」の部位は、特に限定されず、末端に存在しても(1価基)、それ以外の部位に存在してもよい(例えば2価基)。
「分岐鎖を1以上有する脂肪族炭化水素基」としては、具体的には、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基(ラウリル基)、トリデシル基、ミリスチル基、セチル基、ステアリル基、アラキル基、ベヘニル基、オレイル基、リノリル基、リグノセリル基等の分岐異性体であって、1以上の分岐鎖を有する1価基およびそれらから誘導される2価基が挙げられ、好ましくは、3,7,11-トリメチルドデシル基、3,7,11,15-テトラメチルヘキサデシル基(以下、2,3-ジヒドロフィチル基ということもある。)、2,2,4,8,10,10-ヘキサメチルウンデカン-5-イル基、式:

(式中、*は、Qとの結合位置を示す。)で表される基等である。
「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」中に「分岐鎖を1以上有する脂肪族炭化水素基」が複数存在する場合には、その各々は同一のものであっても異なるものであってもよい。
「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」中の「分岐鎖を1以上有する脂肪族炭化水素基」以外の部位は任意に設定することができる。例えば-O-、-S-、-CO-、-NH-、-COO-、-OCONH-、-CONH-、-NHCO-、炭化水素基(1価基または2価基)等の部位を有していてもよい。「炭化水素基」としては、例えば、脂肪族炭化水素基、芳香脂肪族炭化水素基、単環式飽和炭化水素基および芳香族炭化水素基等が挙げられ、具体的には、例えば、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、アリール基、アラルキル基等の1価基およびそれらから誘導される2価基が用いられる。「アルキル基」としては、例えば、C1-6アルキル基等が好ましく、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ヘキシル等が挙げられる。「アルケニル基」としては、例えば、C2-6アルケニル基等が好ましく、例えば、ビニル、1-プロペニル、アリル、イソプロペニル、ブテニル、イソブテニル等が挙げられる。「アルキニル基」としては、例えば、C2-6アルキニル基等が好ましく、例えば、エチニル、プロパルギル、1-プロピニル等が挙げられる。「シクロアルキル基」としては、例えば、C3-6シクロアルキル基等が好ましく、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルが挙げられる。「アリール基」は、例えば、C6-14アリール基等が好ましく、例えば、フェニル、1-ナフチル、2-ナフチル、ビフェニリル、2-アンスリル等が挙げられる。中でもC6-10アリール基がより好ましく、フェニルが特に好ましい。「アラルキル基」としては、例えば、C7-20アラルキル基が好ましく、例えば、ベンジル、1-フェニルエチル、2-フェニルエチル、1-フェニルプロピル、ナフチルメチル、1-ナフチルエチル、1-ナフチルプロピル等が挙げられる。中でも、C7-16アラルキル基(C6-10アリール-C1-6アルキル基)がより好ましく、ベンジルが特に好ましい。当該「炭化水素基」は、ハロゲン原子(塩素原子、臭素原子、フッ素原子、ヨウ素原子)、オキソ基等から選択される置換基で置換されていてもよい。
本発明化合物は、k個のQRa基を有する。ここで、Qは、単結合であるか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-であり、好ましくはOである。k個のQRa基は、それぞれ同一のものであっても異なるものであってもよい。
本発明化合物においては、Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」における、炭素数合計は、14以上であり、16以上が好ましく、18以上がより好ましい。一方、Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上である脂肪族炭化水素基を有する有機基」における、炭素数合計は、300以下であり、200以下が好ましく、160以下がより好ましい。また、本発明化合物においては、Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上である脂肪族炭化水素基を有する有機基」における、総分岐鎖数は3以上であり、4以上が好ましく、8以上がより好ましく、10以上が更に好ましい。当該総分岐鎖数が多いほど、ペプチド鎖が長鎖になった場合でも本発明化合物により保護された化合物は、各種有機溶媒に対する溶解性が良好な油状物となる。
Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」としては、式(b):

(式中、*は、隣接原子との結合位置を示し;
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基が好ましく、例えば、下記式(c)~(e)のいずれかで表される基が挙げられる。
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基が好ましく、例えば、下記式(c)~(e)のいずれかで表される基が挙げられる。
なお、式(c)~(e)における各記号の定義中の炭素数、繰り返し単位の数(m1、n0~n9)等は便宜上示されたものであって、総炭素数が14以上(好ましくは16以上、より好ましくは18以上)、300以下(好ましくは200以下、より好ましくは160以下)になるよう上記した定義の範囲内で適宜変更することができる。以下、式(c)~(e)について、順に説明する
式(c)は、以下の通りである。

[式中、*は、Qとの結合位置を示し;
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):
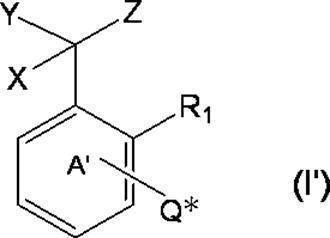
(式中、*は、結合位置を示し;
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる置換基を有していてもよい。)を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]
式(c)の基において、
R5およびR6は、共に水素原子であり;
n0は、2~40の整数であり;
n0個のR7およびR8は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n0個のX2は、独立してそれぞれ、単結合、メチレン基またはエチレン基であり;かつ
R9は、水素原子、メチル基またはエチル基である基が好ましい(但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はメチルまたはエチル基を示す。)。
R5およびR6は、共に水素原子であり;
n0は、2~40の整数であり;
n0個のR7およびR8は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n0個のX2は、独立してそれぞれ、単結合、メチレン基またはエチレン基であり;かつ
R9は、水素原子、メチル基またはエチル基である基が好ましい(但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はメチルまたはエチル基を示す。)。
より好適な式(c)の基は、ミリスチル基、セチル基、ステアリル基、アラキル基、ベヘニル基等の炭素数14~160の分岐異性体であって、総分岐鎖数が3以上である基であり、中でも2,3-ジヒドロフィチル基、3,7,11-トリメチルドデシル基、2,2,4,8,10,10-ヘキサメチル-5-ドデカノイル基が特に好ましい。
式(d)は、以下の通りである。
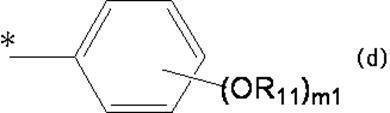
(式中、*は、Qとの結合位置を示し;
m1個のOR11は、式(c’)で表される基により置換されたヒドロキシル基または総分岐鎖数が3以上であるポリアルキレングリコール基を有する基(例えば、ポリプロピレングリコール基、ポリネオペンチルグリコール基)により置換されたヒドロキシル基を示し;
m1は、1~3の整数を示す。)
なお、上記式(c’)で表される基の説明は、*が、Qとの結合位置ではなく、Oとの結合位置を示すこと以外は、上記式(c)で表される基の説明と同じである。
m1個のOR11は、式(c’)で表される基により置換されたヒドロキシル基または総分岐鎖数が3以上であるポリアルキレングリコール基を有する基(例えば、ポリプロピレングリコール基、ポリネオペンチルグリコール基)により置換されたヒドロキシル基を示し;
m1は、1~3の整数を示す。)
なお、上記式(c’)で表される基の説明は、*が、Qとの結合位置ではなく、Oとの結合位置を示すこと以外は、上記式(c)で表される基の説明と同じである。
式(d)の基において、R11は、ミリスチル基、セチル基、ステアリル基、アラキル基、ベヘニル基等の炭素数14~30の分岐異性体であって、総分岐鎖数が3以上である基がより好ましく、中でも2,3-ジヒドロフィチル基、3,7,11-トリメチルドデシル基が特に好ましい。
式(e)は、以下の通りである。

(式中、*は、Qとの結合位置を示し;
n1は、1~10の整数を示し;
n2は、1~10の整数を示し;
n1個のR15およびR16は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n1個のX3は、単結合またはC1-4アルキレン基を示し;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n2個のX5は、単結合またはC1-4アルキレン基を示し;
X4は、単結合またはC1-4アルキレン基を示し;かつ
R12、R13、R14、R19、R20およびR21は、独立してそれぞれ、水素原子またはC1-4アルキル基を示す。
但し、R15およびR16、および/またはR17およびR18が共に水素原子であることはなく、かつn1+n2が2の場合には、R12、R13およびR14の2個以上が独立してそれぞれ、C1-4アルキル基を示すか、またはR19、R20およびR21の2個以上が独立してそれぞれ、C1-4アルキル基を示す。)
n1は、1~10の整数を示し;
n2は、1~10の整数を示し;
n1個のR15およびR16は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n1個のX3は、単結合またはC1-4アルキレン基を示し;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n2個のX5は、単結合またはC1-4アルキレン基を示し;
X4は、単結合またはC1-4アルキレン基を示し;かつ
R12、R13、R14、R19、R20およびR21は、独立してそれぞれ、水素原子またはC1-4アルキル基を示す。
但し、R15およびR16、および/またはR17およびR18が共に水素原子であることはなく、かつn1+n2が2の場合には、R12、R13およびR14の2個以上が独立してそれぞれ、C1-4アルキル基を示すか、またはR19、R20およびR21の2個以上が独立してそれぞれ、C1-4アルキル基を示す。)
式(e)の基において、
n1は、1~5の整数であり;
n2は、1~5の整数であり;
n1個のR15およびR16は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n1個のX3は、単結合、メチレン基またはエチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n2個のX5は、単結合、メチレン基またはエチレン基であり;
X4は、単結合、メチレン基またはエチレン基であり;かつ
R12、R13、R14、R19、R20およびR21は、独立してそれぞれ、水素原子またはC1-4アルキル基である基がより好ましい(但し、R15およびR16、および/またはR17およびR18が共に水素原子であることはなく、かつn1+n2が2の場合には、R12、R13およびR14の2個以上が独立してそれぞれ、C1-4アルキル基を示すか、またはR19、R20およびR21の2個以上が独立してそれぞれ、C1-4アルキル基を示す。)。
n1は、1~5の整数であり;
n2は、1~5の整数であり;
n1個のR15およびR16は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n1個のX3は、単結合、メチレン基またはエチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n2個のX5は、単結合、メチレン基またはエチレン基であり;
X4は、単結合、メチレン基またはエチレン基であり;かつ
R12、R13、R14、R19、R20およびR21は、独立してそれぞれ、水素原子またはC1-4アルキル基である基がより好ましい(但し、R15およびR16、および/またはR17およびR18が共に水素原子であることはなく、かつn1+n2が2の場合には、R12、R13およびR14の2個以上が独立してそれぞれ、C1-4アルキル基を示すか、またはR19、R20およびR21の2個以上が独立してそれぞれ、C1-4アルキル基を示す。)。
特に好適な式(e)の基としては、
n1は、1~5の整数であり;
n2は、1~5の整数であり;
n1個のR15およびR16は、独立してそれぞれ、水素原子またはメチル基であり;
n1個のX3は、単結合またはメチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはメチル基であり;
n2個のX5は、単結合またはメチレン基であり;
X4は、単結合またはメチレン基であり;かつ
R12、R13、R14、R19、R20およびR21は、メチル基である基が挙げられる(但し、R15およびR16、および/またはR17およびR18が、共に水素原子であることはない)。
n1は、1~5の整数であり;
n2は、1~5の整数であり;
n1個のR15およびR16は、独立してそれぞれ、水素原子またはメチル基であり;
n1個のX3は、単結合またはメチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはメチル基であり;
n2個のX5は、単結合またはメチレン基であり;
X4は、単結合またはメチレン基であり;かつ
R12、R13、R14、R19、R20およびR21は、メチル基である基が挙げられる(但し、R15およびR16、および/またはR17およびR18が、共に水素原子であることはない)。
Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」としては、上記式(c)~(e)のいずれかで表される基の他、上記式(b)におけるX1が酸素原子である基を3以上有する基、すなわち、総分岐鎖数が3以上であるポリプロピレングリコール基、ポリネオペンチルグリコール基等のポリアルキレングリコール基を含有する基であってもよい。
Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」の具体例として、以下の基が挙げられる。各基中の*は結合位置を示し、式中のn3は、3以上の整数を示し、n4は、該基の総炭素数が14以上300以下になるように適宜設定され得る。

また、Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」の別の態様として、以下の基が挙げられる。各基中の*は結合位置を示す。

式中、n5~n9は、各基の総炭素数が14以上、300以下になるよう適宜設定し得る。
Ra、Rbとして示される「分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基」の好ましい具体例として、以下の基が挙げられる:
3,7,11,15-テトラメチルヘキサデシル基;
3,7,11-トリメチルドデシル基;
2,2,4,8,10,10-ヘキサメチル-5-ドデカノイル基;
3,4,5-トリ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基;
3,5-ジ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基;
式(f):
3,7,11,15-テトラメチルヘキサデシル基;
3,7,11-トリメチルドデシル基;
2,2,4,8,10,10-ヘキサメチル-5-ドデカノイル基;
3,4,5-トリ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基;
3,5-ジ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基;
式(f):
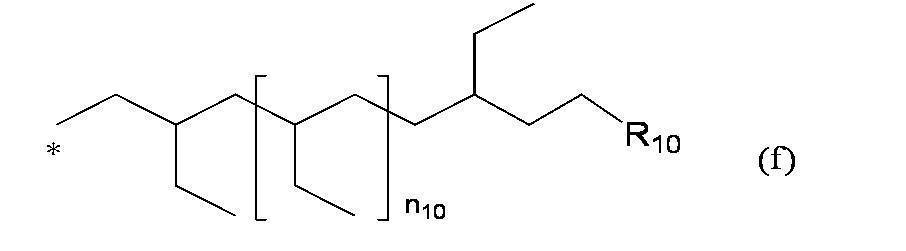
(式中、*は、Qとの結合位置であり、n10は、23~34であり、R10は、式(I’)で表される基である。)で表される基;
式(g):
式(g):
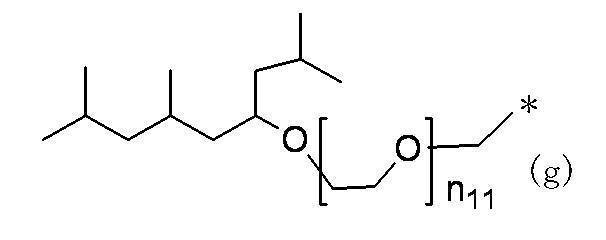
(式中、*は、Qとの結合位置であり、n11は、1~10である。)で表される基;
式(h):
式(h):
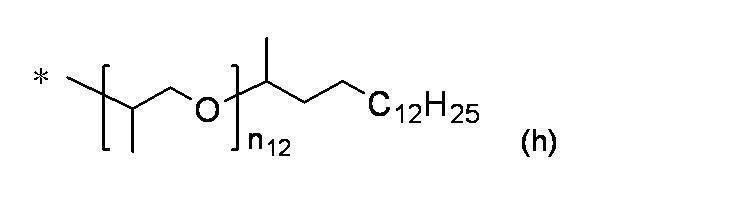
(式中、*は、Qとの結合位置であり、n12は、2~10である。)で表される基;
式(i):
式(i):
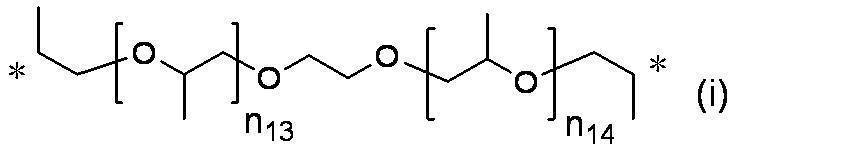
(式中、*は、Qとの結合位置であり、n13およびn14は、独立してそれぞれ、1~10である。)で表される基;および
式(j):
式(j):

(式中、*は、Qとの結合位置であり、n15は、2~20である。)で表される基。
本発明化合物の好ましい例として、以下のベンジル化合物、ジフェニルメタン化合物またはフルオレン化合物が挙げられるが、本発明は、これらに限定されるわけではない:
2,4-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
2-[3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ]-4-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン;
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド;
4-(3,7,11-トリメチルドデシルオキシ)ベンジルアルコール;
2-(3,7,11-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール;
式:
2,4-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
2-[3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ]-4-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン;
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド;
4-(3,7,11-トリメチルドデシルオキシ)ベンジルアルコール;
2-(3,7,11-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール;
式:

(式中、n16は、23~34を示す。)で表される化合物;
式:
式:
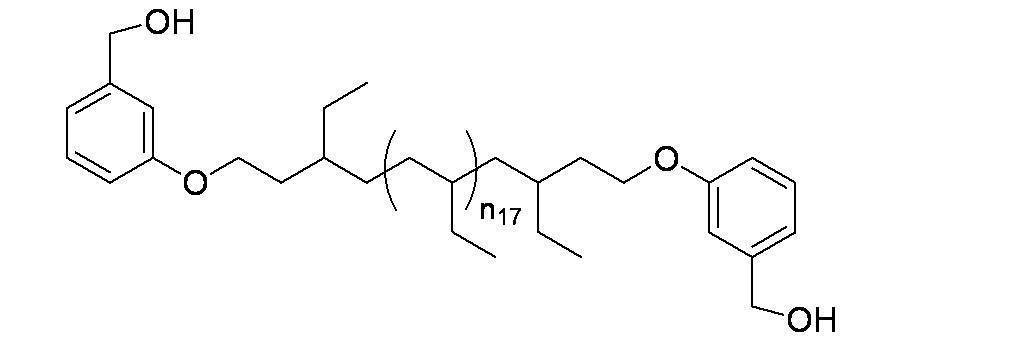
(式中、n17は、23~34を示す。)で表される化合物;
式:
式:

(式中、n18は、1~10を示す。)で表される化合物;および
式:
式:

(式中、n19は、2~10を示す。)で表される化合物。
中でも、
2,4-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
2-[3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ]-4-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン;
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド;
4-(3,7,11-トリメチルドデシルオキシ)ベンジルアルコール;
2-(3,7,11-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール;
式:
2,4-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
2-[3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ]-4-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン;
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド;
4-(3,7,11-トリメチルドデシルオキシ)ベンジルアルコール;
2-(3,7,11-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール;
式:

(式中、n16は、23または34を示す。)で表される化合物;
式:
式:

(式中、n17は、23または34を示す。)で表される化合物;
式:
式:

(式中、n18は、5~7を示す。)で表される化合物;および
式:
式:
で表される化合物が、特に好ましい。
〔本発明化合物の製造方法〕
本発明化合物の製造方法としては、特に限定されないが、例えば次のような反応を経て合成することができる。
原料化合物は、特に述べない限り、市販品として容易に入手できるか、あるいは、自体公知の方法またはこれらに準ずる方法に従って製造することができる。
以下の各方法で得られる化合物の収率は用いる反応条件によって異なりうるが、これらの生成物から通常の手段(再結晶、カラムクロマトグラフィー等)によって単離・精製し、次いで、溶液温度を変化させる手段や溶液組成を変化させる手段等によって沈殿化することができる。
また、各反応において、原料化合物がヒドロキシル基、アミノ基、カルボキシル基、カルボニル基等を有する場合、これらの基にペプチド化学等で一般的に用いられるような保護基が導入されていてもよく、反応後に必要に応じて保護基を除去することにより目的化合物を得ることができる。
本発明化合物の製造方法としては、特に限定されないが、例えば次のような反応を経て合成することができる。
原料化合物は、特に述べない限り、市販品として容易に入手できるか、あるいは、自体公知の方法またはこれらに準ずる方法に従って製造することができる。
以下の各方法で得られる化合物の収率は用いる反応条件によって異なりうるが、これらの生成物から通常の手段(再結晶、カラムクロマトグラフィー等)によって単離・精製し、次いで、溶液温度を変化させる手段や溶液組成を変化させる手段等によって沈殿化することができる。
また、各反応において、原料化合物がヒドロキシル基、アミノ基、カルボキシル基、カルボニル基等を有する場合、これらの基にペプチド化学等で一般的に用いられるような保護基が導入されていてもよく、反応後に必要に応じて保護基を除去することにより目的化合物を得ることができる。
本発明化合物は、例えば、以下の工程により製造することができる。

[式中のQ’は、-O-、-S-、-C(=O)O-または-NH-を示し、Rcは、水素原子、ORd基(ここで、RdはC1-6アルキル基等のアルキル基、ベンジル基等のアラルキル基等を示す。)または式(a):
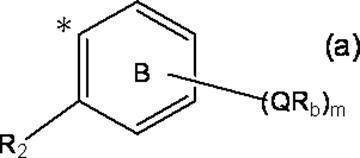
(式中の各記号は、前記と同意義である。)で表される基を示し、Y1はハロゲン原子等の脱離基を示し、他の記号は、前記と同意義である。]
工程(a)
当該工程は、式(II)で表される化合物(以下、化合物(II)と略称する。)のQ’H基(ここで、Q’は、-O-、-S-、-C(=O)O-または-NH-を示す。)にRa基を導入することにより、式(IIa)で表される化合物(以下、化合物(IIa)と略称する。)を製造する工程である。
当該反応は、Q’が、-O-、-S-または-NH-の場合、反応に影響を及ぼさない溶媒中、塩基の存在下または非存在下で、Ra基に対応するハロゲン化物(塩化物、臭化物またはヨウ化物)、Ra基に対応するカルボン酸若しくは酸ハロゲン化物またはRa基に対応するアルキルスルホニルオキシ化物(例えば、メタンスルホニルオキシ化物等)若しくはアリールスルホニルオキシ化物(例えば、p-トルエンスルホニルオキシ化物等)を用いて行われる。また、Q’が-O-の場合、化合物(II)とRa基に対応する水酸化物をトリフェニルホスフィンおよびアゾジカルボン酸ジイソプロピル存在下で反応させる光延反応条件下で、当該反応を行うこともできる。さらにQ’が-C(=O)O-の場合、例えば、化合物(II)とRa基に対応するアミン若しくは水酸化物を後述する縮合剤の存在下で反応させることにより化合物(IIa)を合成することができる。
塩基としては、例えば、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、水素化ナトリウム、水素化カリウム、カリウム tert-ブトキシド等のアルカリ金属塩;ピリジン、トリエチルアミン、N,N-ジメチルアニリン、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン等のアミン類等が挙げられ、中でも炭酸ナトリウム、炭酸カリウム、水素化ナトリウム等が好ましい。
溶媒としては、例えば、トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;ジメチルホルムアミド、ジメチルアセトアミド等のアミド類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;アセトニトリル等のニトリル類、N-メチルピロリドン等あるいはそれらの混合物が挙げられ、中でも、ジメチルホルムアミド、テトラヒドロフラン、トルエン、N-メチルピロリドン等が好ましい。
反応温度は、通常50~150℃であり、好ましくは60~130℃である。反応時間は、通常2~30時間であり、好ましくは3~10時間である。
当該工程は、式(II)で表される化合物(以下、化合物(II)と略称する。)のQ’H基(ここで、Q’は、-O-、-S-、-C(=O)O-または-NH-を示す。)にRa基を導入することにより、式(IIa)で表される化合物(以下、化合物(IIa)と略称する。)を製造する工程である。
当該反応は、Q’が、-O-、-S-または-NH-の場合、反応に影響を及ぼさない溶媒中、塩基の存在下または非存在下で、Ra基に対応するハロゲン化物(塩化物、臭化物またはヨウ化物)、Ra基に対応するカルボン酸若しくは酸ハロゲン化物またはRa基に対応するアルキルスルホニルオキシ化物(例えば、メタンスルホニルオキシ化物等)若しくはアリールスルホニルオキシ化物(例えば、p-トルエンスルホニルオキシ化物等)を用いて行われる。また、Q’が-O-の場合、化合物(II)とRa基に対応する水酸化物をトリフェニルホスフィンおよびアゾジカルボン酸ジイソプロピル存在下で反応させる光延反応条件下で、当該反応を行うこともできる。さらにQ’が-C(=O)O-の場合、例えば、化合物(II)とRa基に対応するアミン若しくは水酸化物を後述する縮合剤の存在下で反応させることにより化合物(IIa)を合成することができる。
塩基としては、例えば、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、水素化ナトリウム、水素化カリウム、カリウム tert-ブトキシド等のアルカリ金属塩;ピリジン、トリエチルアミン、N,N-ジメチルアニリン、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン等のアミン類等が挙げられ、中でも炭酸ナトリウム、炭酸カリウム、水素化ナトリウム等が好ましい。
溶媒としては、例えば、トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;ジメチルホルムアミド、ジメチルアセトアミド等のアミド類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;アセトニトリル等のニトリル類、N-メチルピロリドン等あるいはそれらの混合物が挙げられ、中でも、ジメチルホルムアミド、テトラヒドロフラン、トルエン、N-メチルピロリドン等が好ましい。
反応温度は、通常50~150℃であり、好ましくは60~130℃である。反応時間は、通常2~30時間であり、好ましくは3~10時間である。
工程(b)
当該工程は、化合物(IIa)を還元することにより、式(I-a)で表される化合物(以下、化合物(I-a)と略称する。)を製造する工程である。当該還元反応は、還元剤を用いる方法により行うことができる。
当該工程は、化合物(IIa)を還元することにより、式(I-a)で表される化合物(以下、化合物(I-a)と略称する。)を製造する工程である。当該還元反応は、還元剤を用いる方法により行うことができる。
当該還元反応に用いる還元剤としては、例えば、金属水素化物(水素化ホウ素ナトリウム、水素化ホウ素リチウム、シアノ水素化ホウ素ナトリウム、トリアセトキシ水素化ホウ素ナトリウム、水素化ジブチルアルミニウム、水素化アルミニウム、水素化アルミニウムリチウム等)等が挙げられ、中でも、水素化ホウ素ナトリウム、水素化ジブチルアルミニウム等が好ましい。
当該反応は、反応に影響を及ぼさない溶媒中で行われる。溶媒としては、例えば、メタノール、エタノール等のアルコール類;ジエチルエーテル、テトラヒドロフラン、ジオキサン等のエーテル類;トルエン、キシレン等の芳香族炭化水素類;あるいはそれらの混合物が挙げられ、中でもテトラヒドロフラン、トルエン等が好ましい。
反応温度は、通常0~100℃であり、好ましくは30~70℃であり、反応時間は、通常1~24時間であり、好ましくは2~5時間である。
反応温度は、通常0~100℃であり、好ましくは30~70℃であり、反応時間は、通常1~24時間であり、好ましくは2~5時間である。
工程(c)
当該工程は、化合物(IIa)(式(IIa)中、Rcが水素原子でもORd基でもない。)を、上記工程(b)と同様の方法により還元するか、またはグリニャール反応によりフェニル基(上記Z基に該当)を導入する工程である。
当該工程は、化合物(IIa)(式(IIa)中、Rcが水素原子でもORd基でもない。)を、上記工程(b)と同様の方法により還元するか、またはグリニャール反応によりフェニル基(上記Z基に該当)を導入する工程である。
当該グリニャール反応では、市販のグリニャール試薬(例えば、フェニルマグネシウムブロマイド、フェニルマグネシウムクロライド等)を使用するか、またはマグネシウムとハロベンゼン(クロロベンゼン、ブロモベンゼン、ヨードベンゼン)とを、ヨウ素またはジブロモエタン存在下で反応させることにより調製した試薬を使用し得る。
当該グリニャール反応は、反応に影響を及ぼさない溶媒中で行われる。溶媒としては、例えば、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等のエーテル類;トルエン、キシレン等の芳香族炭化水素類;あるいはそれらの混合物が挙げられ、中でもテトラヒドロフラン、1,2-ジメトキシエタン等が好ましい。
反応温度は、通常-20~100℃であり、好ましくは0~70℃であり、反応時間は、通常1~24時間であり、好ましくは2~10時間である。
反応温度は、通常-20~100℃であり、好ましくは0~70℃であり、反応時間は、通常1~24時間であり、好ましくは2~10時間である。
工程(d-1)
当該工程は、化合物(IIa)(式(IIa)中、Rcが水素原子である)を、オキシム化することにより、式(I’-a)で表される化合物(以下、化合物(I’-a)と略称する。)を製造する工程である。
当該工程は、化合物(IIa)(式(IIa)中、Rcが水素原子である)を、オキシム化することにより、式(I’-a)で表される化合物(以下、化合物(I’-a)と略称する。)を製造する工程である。
当該オキシム化反応は、反応に影響を及ぼさない溶媒中、塩基存在下で化合物(IIa)とヒドロキシルアミンの酸付加塩とを反応させることにより行われる。
ヒドロキシルアミンの酸付加塩としては、例えば、塩酸塩、硫酸塩、硝酸塩等の鉱酸塩、酢酸塩、トリフルオロ酢酸塩、メタンスルホン酸塩、トリフルオロメタンスルホン酸塩、p-トルエンスルホン酸塩等の有機酸塩等が挙げられるが、塩酸塩が特に好ましい。
かかる塩基としては、例えば、水酸化カリウム、水酸化ナトリウム、炭酸水素ナトリウム、炭酸カリウムなどのアルカリ金属塩;ピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N,N-ジメチルアニリン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エンなどの有機アミン類等が挙げられ、中でも、トリエチルアミン、ジイソプロピルエチルアミン等が好ましい。
溶媒としては、例えば、クロロホルム、ジクロロメタン等のハロゲン溶媒;トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;および/または、それらの混合物が挙げられ、中でも、ジクロロメタン、クロロホルム、トルエン等が好ましい。
反応温度は、通常10~100℃、好ましくは20~60℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
ヒドロキシルアミンの酸付加塩としては、例えば、塩酸塩、硫酸塩、硝酸塩等の鉱酸塩、酢酸塩、トリフルオロ酢酸塩、メタンスルホン酸塩、トリフルオロメタンスルホン酸塩、p-トルエンスルホン酸塩等の有機酸塩等が挙げられるが、塩酸塩が特に好ましい。
かかる塩基としては、例えば、水酸化カリウム、水酸化ナトリウム、炭酸水素ナトリウム、炭酸カリウムなどのアルカリ金属塩;ピリジン、トリエチルアミン、ジイソプロピルエチルアミン、N,N-ジメチルアニリン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エンなどの有機アミン類等が挙げられ、中でも、トリエチルアミン、ジイソプロピルエチルアミン等が好ましい。
溶媒としては、例えば、クロロホルム、ジクロロメタン等のハロゲン溶媒;トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;および/または、それらの混合物が挙げられ、中でも、ジクロロメタン、クロロホルム、トルエン等が好ましい。
反応温度は、通常10~100℃、好ましくは20~60℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
工程(d-2)
当該工程は、化合物(I’-a)を、パラジウム-炭素、ラネーニッケル等の金属触媒存在下の接触水素添加反応、または前記工程(b)と同様の金属水素化物等の還元剤により還元することにより、本発明化合物である式(I-b)で表される化合物(以下、化合物(I-b)と略称する。)を製造する工程である。
当該工程は、化合物(I’-a)を、パラジウム-炭素、ラネーニッケル等の金属触媒存在下の接触水素添加反応、または前記工程(b)と同様の金属水素化物等の還元剤により還元することにより、本発明化合物である式(I-b)で表される化合物(以下、化合物(I-b)と略称する。)を製造する工程である。
化合物(I-b)は、工程(d-3)から工程(d-4)および工程(d-5)を経て製造することもできる。
工程(d-3)
当該工程は、化合物(I-a)を、例えば塩化アセチル、塩化チオニル等のクロル化剤、または、例えば臭化アセチル、三臭化リン、ジフェニルホスフィン/臭素等のブロム化剤を用いてハロゲン化することにより、式(I’-b)で表される化合物(以下、化合物(I’-b)と略称する。)を製造する工程である。
溶媒としては、例えば、クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;それらの混合物が挙げられ、中でも、クロロホルム、テトラヒドロフラン、トルエン等が好ましい。
反応温度は、通常10~150℃、好ましくは30~80℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
当該工程は、化合物(I-a)を、例えば塩化アセチル、塩化チオニル等のクロル化剤、または、例えば臭化アセチル、三臭化リン、ジフェニルホスフィン/臭素等のブロム化剤を用いてハロゲン化することにより、式(I’-b)で表される化合物(以下、化合物(I’-b)と略称する。)を製造する工程である。
溶媒としては、例えば、クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;それらの混合物が挙げられ、中でも、クロロホルム、テトラヒドロフラン、トルエン等が好ましい。
反応温度は、通常10~150℃、好ましくは30~80℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
工程(d-4)
当該工程は、化合物(I’-b)をアジ化ナトリウム等のアジド化剤を用いてアジド化することにより、式(I’-c)で表される化合物(以下、化合物(I’-c)と略称する。)を製造する工程である。
当該反応は、反応に影響を及ぼさない溶媒中、化合物(I’-b)をアジド化剤と反応させることにより行われる。
溶媒としては、例えば、クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;N,N-ジメチルホルムアミド等のアミド類;それらの混合物が挙げられ、中でも、クロロホルム、N,N-ジメチルホルムアミド等が好ましい。
反応温度は、通常10~150℃、好ましくは20~100℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
当該工程は、化合物(I’-b)をアジ化ナトリウム等のアジド化剤を用いてアジド化することにより、式(I’-c)で表される化合物(以下、化合物(I’-c)と略称する。)を製造する工程である。
当該反応は、反応に影響を及ぼさない溶媒中、化合物(I’-b)をアジド化剤と反応させることにより行われる。
溶媒としては、例えば、クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;N,N-ジメチルホルムアミド等のアミド類;それらの混合物が挙げられ、中でも、クロロホルム、N,N-ジメチルホルムアミド等が好ましい。
反応温度は、通常10~150℃、好ましくは20~100℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
工程(d-5)
当該工程は、化合物(I’-c)をアミノ化することにより、化合物(I-b)を製造する工程である。
当該反応は、反応に影響を及ぼさない溶媒中、水存在下、化合物(I’-c)をトリフェニルホスフィンと反応させるか、接触水素化還元により行われる。
トリフェニルホスフィンの使用量としては、化合物(I’-c)1モルに対して、好ましくは1~10モル、特に好ましくは1~5モルである。
水の使用量は、化合物(I’-c)1モルに対して、好ましくは1~10モル、特に好ましくは1~5モルである。
溶媒としては、例えば、トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;および、それらの混合物が挙げられ、中でも、トルエン、テトラヒドロフラン等が好ましい。
反応温度は、通常10~150℃、好ましくは20~100℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
当該工程は、化合物(I’-c)をアミノ化することにより、化合物(I-b)を製造する工程である。
当該反応は、反応に影響を及ぼさない溶媒中、水存在下、化合物(I’-c)をトリフェニルホスフィンと反応させるか、接触水素化還元により行われる。
トリフェニルホスフィンの使用量としては、化合物(I’-c)1モルに対して、好ましくは1~10モル、特に好ましくは1~5モルである。
水の使用量は、化合物(I’-c)1モルに対して、好ましくは1~10モル、特に好ましくは1~5モルである。
溶媒としては、例えば、トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;および、それらの混合物が挙げられ、中でも、トルエン、テトラヒドロフラン等が好ましい。
反応温度は、通常10~150℃、好ましくは20~100℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
工程(d-6)
当該工程は、化合物(I’-b)をRNH2(Rは前記と同義である)と反応させることにより、本発明化合物においてYが-NHR基である式(I-c)で表される化合物(以下、化合物(I-c)と略称する。)を製造する工程である。
当該工程は、反応に影響を及ぼさない溶媒中、必要により、例えば、トリエチルアミン、ジイソプロピルエチルアミン等の第3級アミン等の塩基の存在下、化合物(I’-b)をR-NH2で表されるアミンと反応させることにより行われる。
溶媒としては、例えば、トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;および、クロロホルム、ジクロロメタン等のハロゲン溶媒または、それらの混合物が挙げられ、中でも、トルエン、テトラヒドロフラン、クロロホルム等が好ましい。
反応温度は、通常10~100℃、好ましくは20~60℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
当該工程は、化合物(I’-b)をRNH2(Rは前記と同義である)と反応させることにより、本発明化合物においてYが-NHR基である式(I-c)で表される化合物(以下、化合物(I-c)と略称する。)を製造する工程である。
当該工程は、反応に影響を及ぼさない溶媒中、必要により、例えば、トリエチルアミン、ジイソプロピルエチルアミン等の第3級アミン等の塩基の存在下、化合物(I’-b)をR-NH2で表されるアミンと反応させることにより行われる。
溶媒としては、例えば、トルエン、キシレン等の芳香族炭化水素類;テトラヒドロフラン、ジオキサン等のエーテル類;および、クロロホルム、ジクロロメタン等のハロゲン溶媒または、それらの混合物が挙げられ、中でも、トルエン、テトラヒドロフラン、クロロホルム等が好ましい。
反応温度は、通常10~100℃、好ましくは20~60℃であり、反応時間は、通常0.5~30時間、好ましくは2~20時間である。
工程(d-7)
当該工程は、化合物(I-d)を-CONH2基または-OCONH2基を有する化合物と反応させた後、塩基で処理することにより、化合物(I-e)を製造する工程である。
化合物(I-d)と-CONH2基または-OCONH2基を有する化合物との反応は、反応に影響を及ぼさない溶媒中、酸触媒下で行われる。
酸触媒としては、例えば、メタンスルホン酸、トリフルオロメタンスルホン酸、トルエンスルホン酸等が挙げられ、中でもメタンスルホン酸、トルエンスルホン酸が好ましい。
酸触媒の使用量は、化合物(I-d)1モルに対して、好ましくは0.05~0.5モル、特に好ましくは0.1~0.3モルである。
-CONH2基または-OCONH2基を有する化合物としては、例えば、Fmoc-NH2、HCONH2、CF3CONH2、AcNH2、EtOCONH2、Cbz-NH2等が挙げられ、中でもFmoc-NH2、EtOCONH2等が好ましい。
ここで、「Fmoc-」とは、9-フルオレニルメトキシカルボニル基(以下、Fmoc基ともいう。)を意味し、「Cbz-」は、ベンジルオキシカルボニル基(以下、Cbz基ともいう。)を意味する。
当該工程は、化合物(I-d)を-CONH2基または-OCONH2基を有する化合物と反応させた後、塩基で処理することにより、化合物(I-e)を製造する工程である。
化合物(I-d)と-CONH2基または-OCONH2基を有する化合物との反応は、反応に影響を及ぼさない溶媒中、酸触媒下で行われる。
酸触媒としては、例えば、メタンスルホン酸、トリフルオロメタンスルホン酸、トルエンスルホン酸等が挙げられ、中でもメタンスルホン酸、トルエンスルホン酸が好ましい。
酸触媒の使用量は、化合物(I-d)1モルに対して、好ましくは0.05~0.5モル、特に好ましくは0.1~0.3モルである。
-CONH2基または-OCONH2基を有する化合物としては、例えば、Fmoc-NH2、HCONH2、CF3CONH2、AcNH2、EtOCONH2、Cbz-NH2等が挙げられ、中でもFmoc-NH2、EtOCONH2等が好ましい。
ここで、「Fmoc-」とは、9-フルオレニルメトキシカルボニル基(以下、Fmoc基ともいう。)を意味し、「Cbz-」は、ベンジルオキシカルボニル基(以下、Cbz基ともいう。)を意味する。
なお、工程(a)の原料化合物として使用するRa化試薬[すなわち、Ra基に対応する水酸化物、ハロゲン化物、アルキルスルホニルオキシ化物(例えば、メタンスルホニルオキシ化物等)またはアリールスルホニルオキシ化物(例えば、p-トルエンスルホニルオキシ化物等)]は、市販品を用いることができる。また、Ra化試薬は、例えば、
(1)Ra基に対応する水酸化物のハロゲン化、アルキルスルホニルオキシ化またはアリールスルホニルオキシ化により、或いは
(2)Ra基に対応する不飽和水酸化物の還元反応(例えば、白金-炭素(Pt/C)、パラジウム-炭素(Pd/C)、ロジウム-炭素(Rh/C)、ラネーニッケル等の金属触媒の存在下での接触水素添加反応等)、およびそれに続くハロゲン化、アルキルスルホニルオキシ化またはアリールスルホニルオキシ化により、
製造することができる。
(1)Ra基に対応する水酸化物のハロゲン化、アルキルスルホニルオキシ化またはアリールスルホニルオキシ化により、或いは
(2)Ra基に対応する不飽和水酸化物の還元反応(例えば、白金-炭素(Pt/C)、パラジウム-炭素(Pd/C)、ロジウム-炭素(Rh/C)、ラネーニッケル等の金属触媒の存在下での接触水素添加反応等)、およびそれに続くハロゲン化、アルキルスルホニルオキシ化またはアリールスルホニルオキシ化により、
製造することができる。
当該Ra化試薬の製造において、ヒドロキシル基から脱離基への変換に用いる試薬としては、例えば、塩化チオニル、N-クロロスクシンイミド(NCS)等のクロロ化剤、臭化水素酸、臭化アセチル、N-ブロモスクシンイミド(NBS)、三臭化リン、ジフェニルホスフィン/臭素等のブロモ化剤等のハロゲン化剤の他、塩化メタンスルホニル、塩化トリフルオロメタンスルホニル等のアルキルスルホニル化剤、塩化ベンゼンスルホニル、塩化p-トルエンスルホニル等のアリールスルホニル化剤等が挙げられ、中でも、ハロゲン化剤である塩化チオニル、臭化水素酸等が好ましい。
当該反応は、反応に影響を及ぼさない溶媒中で行われ、溶媒としては、例えば、水;クロロホルム、ジクロロメタンなどのハロゲン化炭化水素類;ベンゼン、トルエン、キシレンなどの芳香族炭化水素類;アセトニトリル、プロピオニトリルなどのニトリル類;テトラヒドロフラン、1,4-ジオキサン、ジエチルエーテルなどのエーテル類が挙げられ、中でも水、クロロホルム等のハロゲン化炭化水素類が好ましい。
反応温度は、通常10~120℃、好ましくは50~100℃であり、反応時間は、通常1~72時間、好ましくは3~24時間である。
反応温度は、通常10~120℃、好ましくは50~100℃であり、反応時間は、通常1~72時間、好ましくは3~24時間である。
本発明化合物(前記Qが単結合である式(I)で表される化合物)は、例えば、以下の方法によっても製造することができる。すなわち、ベンゼン環上へのRa基の導入は、
(1)Ra基に対応するハロゲン化物(塩化物、臭化物、またはヨウ化物)、Ra基に対応するカルボン酸若しくは酸ハロゲン化物を用いるフリーデルクラフツ反応、
(2)上記化合物(II)に対応する化合物(但し、Q’H基が-CHO基に置き換わった化合物)をWittig反応により増炭させた後に、接触水素添加等する方法、または
(3)金属触媒を使用したクロスカップリング等の慣用の有機合成反応
によって行うことができる。
(1)Ra基に対応するハロゲン化物(塩化物、臭化物、またはヨウ化物)、Ra基に対応するカルボン酸若しくは酸ハロゲン化物を用いるフリーデルクラフツ反応、
(2)上記化合物(II)に対応する化合物(但し、Q’H基が-CHO基に置き換わった化合物)をWittig反応により増炭させた後に、接触水素添加等する方法、または
(3)金属触媒を使用したクロスカップリング等の慣用の有機合成反応
によって行うことができる。
なお、上記各スキーム中、Ra基で示される有機基の炭素数やハロゲン原子の種類、反応試薬等は便宜上示されたものであって、上記した定義の範囲内で適宜変更することができる。
〔有機合成反応〕
本発明化合物は、ペプチド、オリゴ核酸、その他の有機化合物の有機合成反応における保護化試薬として使用することができる。本発明化合物を、ペプチド合成等において、アミノ酸またはペプチドの保護化試薬として使用することが好ましい。具体的には、C末端のカルボキシル基、C末端を形成するアミノ酸が有するカルボキサミド基(アミド基ともいう)、即ち-CONHR’基(R’は水素原子、アルキル基またはアラルキル基を示す)、-SH基などの官能基、および側鎖官能基(以下、C末端等という。)の保護基として、本発明化合物をアミノ酸またはペプチドに導入することが好ましい。R’のアルキル基およびアラルキル基の説明は、上述したRの説明と同じである。R’は、好ましくは水素原子である。保護化試薬として使用する場合には、本発明化合物を活性化したり、等価体に変換してから、保護される置換基と反応させても構わない。なお、「本発明の分岐鎖含有芳香族化合物によって保護された有機化合物」を、「分岐鎖含有芳香族化合物付加体」と呼ぶ。
本発明化合物は、ペプチド、オリゴ核酸、その他の有機化合物の有機合成反応における保護化試薬として使用することができる。本発明化合物を、ペプチド合成等において、アミノ酸またはペプチドの保護化試薬として使用することが好ましい。具体的には、C末端のカルボキシル基、C末端を形成するアミノ酸が有するカルボキサミド基(アミド基ともいう)、即ち-CONHR’基(R’は水素原子、アルキル基またはアラルキル基を示す)、-SH基などの官能基、および側鎖官能基(以下、C末端等という。)の保護基として、本発明化合物をアミノ酸またはペプチドに導入することが好ましい。R’のアルキル基およびアラルキル基の説明は、上述したRの説明と同じである。R’は、好ましくは水素原子である。保護化試薬として使用する場合には、本発明化合物を活性化したり、等価体に変換してから、保護される置換基と反応させても構わない。なお、「本発明の分岐鎖含有芳香族化合物によって保護された有機化合物」を、「分岐鎖含有芳香族化合物付加体」と呼ぶ。
本発明化合物は、各種有機合成反応用の保護化試薬として使用できる。例えば、以下の工程により、有機合成反応を実施することができる:
工程(i):本発明化合物を可溶性溶媒に溶解する工程(溶解工程)、
工程(ii):上記工程で得られた可溶性溶媒に溶解された本発明化合物と反応基質を結合させる工程(結合工程)、
工程(iii):上記工程で得られた結合物を含む反応液に水を加えて洗浄し、分層して、水層を除去する工程(分層工程)、
工程(iv):上記工程で得られた結合物を含む水洗後の溶液を反応に供し、当該反応後の生成物を含む反応液に水を加えて洗浄し、分層して、水層を除去する工程(反応および分層工程)、
工程(v):上記工程で得られた生成物を含む水洗後の溶液中の生成物から、本発明化合物由来の保護基および他の保護基を除去する工程(脱保護工程)。
なお、本明細書では、「本発明化合物由来の保護基」および「他の保護基」を区別するために、これらを、それぞれ「アンカー」および「一時保護基」と呼ぶことがある。
工程(i):本発明化合物を可溶性溶媒に溶解する工程(溶解工程)、
工程(ii):上記工程で得られた可溶性溶媒に溶解された本発明化合物と反応基質を結合させる工程(結合工程)、
工程(iii):上記工程で得られた結合物を含む反応液に水を加えて洗浄し、分層して、水層を除去する工程(分層工程)、
工程(iv):上記工程で得られた結合物を含む水洗後の溶液を反応に供し、当該反応後の生成物を含む反応液に水を加えて洗浄し、分層して、水層を除去する工程(反応および分層工程)、
工程(v):上記工程で得られた生成物を含む水洗後の溶液中の生成物から、本発明化合物由来の保護基および他の保護基を除去する工程(脱保護工程)。
なお、本明細書では、「本発明化合物由来の保護基」および「他の保護基」を区別するために、これらを、それぞれ「アンカー」および「一時保護基」と呼ぶことがある。
上記各工程について、以下に詳細に説明する。
工程(i)(溶解工程)
当該工程は、本発明化合物を可溶性溶媒に溶解する工程である。
可溶性溶媒としては、一般的な有機溶媒を反応に用いることができる。本発明化合物は、長鎖の分岐鎖脂肪族炭化水素基を有することから各種有機溶媒に対する溶解度が高く、これにより優れた反応性が期待できる。
可溶性溶媒としては、具体的には、ジエチルエーテル、テトラヒドロフラン、1,4-ジオキサン、メチル-t-ブチルエーテル、シクロペンチルメチルエーテル(CPME)等のエーテル類;酢酸エチル、酢酸イソプロピル等の酢酸エステル類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;トルエン、キシレン等の芳香族炭化水素類;ヘキサン、ヘプタン、シクロヘキサン等の炭化水素類が挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。良好な抽出操作が期待でき、工業的に使用可能であるという観点から、酢酸エチル、酢酸イソプロピル、ジクロロメタン、シクロペンチルメチルエーテル、トルエンが好ましく、酢酸エチル、酢酸イソプロピル、シクロペンチルメチルエーテル、トルエンがより好ましく、酢酸エチル、酢酸イソプロピル、シクロペンチルメチルエーテルが更に好ましく、酢酸イソプロピル、シクロペンチルメチルエーテルが更に一層好ましい。
当該工程は、本発明化合物を可溶性溶媒に溶解する工程である。
可溶性溶媒としては、一般的な有機溶媒を反応に用いることができる。本発明化合物は、長鎖の分岐鎖脂肪族炭化水素基を有することから各種有機溶媒に対する溶解度が高く、これにより優れた反応性が期待できる。
可溶性溶媒としては、具体的には、ジエチルエーテル、テトラヒドロフラン、1,4-ジオキサン、メチル-t-ブチルエーテル、シクロペンチルメチルエーテル(CPME)等のエーテル類;酢酸エチル、酢酸イソプロピル等の酢酸エステル類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;トルエン、キシレン等の芳香族炭化水素類;ヘキサン、ヘプタン、シクロヘキサン等の炭化水素類が挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。良好な抽出操作が期待でき、工業的に使用可能であるという観点から、酢酸エチル、酢酸イソプロピル、ジクロロメタン、シクロペンチルメチルエーテル、トルエンが好ましく、酢酸エチル、酢酸イソプロピル、シクロペンチルメチルエーテル、トルエンがより好ましく、酢酸エチル、酢酸イソプロピル、シクロペンチルメチルエーテルが更に好ましく、酢酸イソプロピル、シクロペンチルメチルエーテルが更に一層好ましい。
なお、「本発明化合物に求められる有機溶剤に対する溶解度」は、本来は「各反応における各原料および各生成物を基質とする場合の、これら基質と本発明化合物との結合物の有機溶剤に対する溶解度」として評価されるべきであるが、様々な基質毎に、基質と本発明化合物との結合物の溶解度を全て想定して確認することは極めて困難であるため、「本発明化合物自体の有機溶剤に対する溶解度」として評価した。
以下、可溶性溶媒の代表として、酢酸イソプロピルを例示して、本発明化合物の特徴を示す。
20℃における酢酸イソプロピル100g中の本発明化合物の飽和溶解度の下限値は、反応基質との結合やその後の反応が進行しさえすれば特に制限はないが、工業的にあらゆる基質に対しても安定的に反応を進行させられるという観点から、1重量%が好ましく、2重量%がより好ましく、5重量%が更に好ましく、10重量%が更に一層好ましく、25重量%が殊更好ましく、50重量%が特に好ましい。
20℃における酢酸イソプロピル100g中の本発明化合物の飽和溶解度の下限値は、反応基質との結合やその後の反応が進行しさえすれば特に制限はないが、工業的にあらゆる基質に対しても安定的に反応を進行させられるという観点から、1重量%が好ましく、2重量%がより好ましく、5重量%が更に好ましく、10重量%が更に一層好ましく、25重量%が殊更好ましく、50重量%が特に好ましい。
20℃における酢酸イソプロピル100g中の本発明化合物の飽和溶解度の上限値は、十分に高濃度な反応溶液が得られれば特に制限はないが、工業的に反応の進行度合いによらず安定的に反応を進行させられるという観点から、80重量%が好ましく、85重量%がより好ましく、90重量%が更に好ましく、95重量%が更に一層好ましい。
なお、上記可溶性溶媒には、反応時点における基質の溶解性を向上させるため;抽出時点における未反応物および副生物の水層への溶解度を向上させるため(すなわち、未反応物および副生物の除去を容易にするため);または分層性を向上させるために、各種親水性有機溶媒を添加しても構わない。
また、抽出時点で未反応物や副生物を除去・洗浄するために、水の代わりに、各種親水性有機溶媒を使用しても構わない。具体的には、ヘプタンを反応溶媒として使用した場合に、アセトニトリルを用いて抽出・洗浄しても構わない。
各種親水性有機溶媒としては、具体的には、アセトニトリル、プロピオニトリル等のニトリル類;アセトン、メチルエチルケトン、2-ブタノン等のケトン類;N,N-ジメチルホルムアミド、N-メチルピロリドン等のアミド類;ジメチルスルホキシド等のスルホキシド類が挙げられる。溶解性を補助しつつ、分層性に影響を与えないという観点から、アセトニトリル、N,N-ジメチルホルムアミド、N-メチルピロリドンが好ましく、N,N-ジメチルホルムアミド、N-メチルピロリドンがより好ましく、N-メチルピロリドンが更に好ましい。
工程(ii)(結合工程)
当該工程は、上記工程(i)で得られた可溶性溶媒に溶解された本発明化合物と反応基質を結合させる工程である。
当該工程は、上記工程(i)で得られた可溶性溶媒に溶解された本発明化合物と反応基質を結合させる工程である。
ここで反応基質とは、保護アミノ酸等のカルボキシル基等を有するものであり、反応基質の使用量は、本発明化合物1モルに対して、1~10モル、好ましくは1~5モルである。
Yがヒドロキシル基である場合は、反応に影響を及ぼさない溶媒中、ジメチルアミノピリジン触媒下、縮合剤を添加することによりエステル結合が形成される。
Yが-NHR基である場合、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-1H-1,2,3-トリアゾール-5-カルボン酸エチルエステル(HOCt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)、3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(HOOBt)等の縮合添加剤の存在下、縮合剤を添加してアミド結合が形成される。
Yがハロゲン原子である場合、反応に影響を及ぼさない溶媒中、ジイソプロピルエチルアミンなどの塩基を添加することにより、エステル結合が形成される。
縮合添加剤の使用量は、反応が進行しさえすれば特に制限はないが、本発明化合物1モルに対して、好ましくは0.05~1.5モルである。
縮合剤としては、反応が進行しさえすれば特に制限はないが、具体的には、ジシクロヘキシルカルボジイミド(DCC)、ジイソプロピルカルボジイミド(DIC)、N-エチル-N’-3-ジメチルアミノプロピルカルボジイミドおよびその塩酸塩(EDC・HCl)、ヘキサフルオロリン酸(ベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウム(PyBop)、O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム テトラフルオロボレート(TBTU)、1-[ビス(ジメチルアミノ)メチレン]-5-クロロ-1H-ベンゾトリアゾリウム3-オキシド ヘキサフルオロホスフェート(HCTU)、O-ベンゾトリアゾール-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロボレート(HBTU)等が挙げられる。
縮合剤の使用量は、本発明化合物1モルに対して、1~10モル使用することができ、好ましくは1~5モルである。
溶媒としては、例えば、上述の可溶性溶媒が好適である。
反応温度は、通常-10~30℃、好ましくは0℃~20℃であり、反応時間は、通常1~30時間である。
反応の進行の確認は一般的な液相有機合成反応と同様の方法を適用できる。すなわち、薄層シリカゲルクロマトグラフィー、高速液体クロマトグラフィー等を用いて反応を追跡することができる。
Yが-NHR基である場合、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-1H-1,2,3-トリアゾール-5-カルボン酸エチルエステル(HOCt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)、3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(HOOBt)等の縮合添加剤の存在下、縮合剤を添加してアミド結合が形成される。
Yがハロゲン原子である場合、反応に影響を及ぼさない溶媒中、ジイソプロピルエチルアミンなどの塩基を添加することにより、エステル結合が形成される。
縮合添加剤の使用量は、反応が進行しさえすれば特に制限はないが、本発明化合物1モルに対して、好ましくは0.05~1.5モルである。
縮合剤としては、反応が進行しさえすれば特に制限はないが、具体的には、ジシクロヘキシルカルボジイミド(DCC)、ジイソプロピルカルボジイミド(DIC)、N-エチル-N’-3-ジメチルアミノプロピルカルボジイミドおよびその塩酸塩(EDC・HCl)、ヘキサフルオロリン酸(ベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウム(PyBop)、O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム テトラフルオロボレート(TBTU)、1-[ビス(ジメチルアミノ)メチレン]-5-クロロ-1H-ベンゾトリアゾリウム3-オキシド ヘキサフルオロホスフェート(HCTU)、O-ベンゾトリアゾール-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロボレート(HBTU)等が挙げられる。
縮合剤の使用量は、本発明化合物1モルに対して、1~10モル使用することができ、好ましくは1~5モルである。
溶媒としては、例えば、上述の可溶性溶媒が好適である。
反応温度は、通常-10~30℃、好ましくは0℃~20℃であり、反応時間は、通常1~30時間である。
反応の進行の確認は一般的な液相有機合成反応と同様の方法を適用できる。すなわち、薄層シリカゲルクロマトグラフィー、高速液体クロマトグラフィー等を用いて反応を追跡することができる。
工程(iii)(分層工程)
当該工程は、上記工程(ii)で得られた結合物を含む反応液に水(および/またはアセトニトリルなどの親水性有機溶媒)を添加し、攪拌、洗浄し、水溶性の反応残渣(および/または親水性有機溶媒に可溶の反応残渣)を分層(分液操作)により除去する工程である。
当該工程は、上記工程(ii)で得られた結合物を含む反応液に水(および/またはアセトニトリルなどの親水性有機溶媒)を添加し、攪拌、洗浄し、水溶性の反応残渣(および/または親水性有機溶媒に可溶の反応残渣)を分層(分液操作)により除去する工程である。
工程(iv)(反応および分層工程)
当該工程は、上記工程(iii)で得られた結合物を含む水洗後の有機溶液中で所望の有機合成反応を行う工程、および当該有機合成反応後に得られる生成物を粗精製するために、該生成物が溶解している反応液に水を添加し、攪拌、洗浄し、水溶性の反応残渣を分層(分液操作)により除去する工程である。
当該工程は、上記工程(iii)で得られた結合物を含む水洗後の有機溶液中で所望の有機合成反応を行う工程、および当該有機合成反応後に得られる生成物を粗精製するために、該生成物が溶解している反応液に水を添加し、攪拌、洗浄し、水溶性の反応残渣を分層(分液操作)により除去する工程である。
工程(v)(脱保護工程)
当該工程は、上記工程(iv)の分層工程後の溶液に含まれる該生成物から、最終的に本発明化合物由来の保護基(アンカー)のみ、または同時に該アンカーおよび一時保護基を除去し、目的物を得る工程である。
ここで除去されるアンカーは、式(I-f):
当該工程は、上記工程(iv)の分層工程後の溶液に含まれる該生成物から、最終的に本発明化合物由来の保護基(アンカー)のみ、または同時に該アンカーおよび一時保護基を除去し、目的物を得る工程である。
ここで除去されるアンカーは、式(I-f):
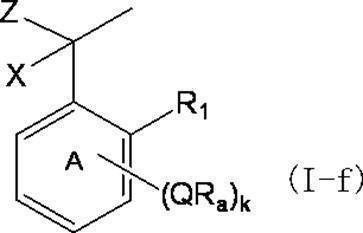
(式中の各基は、前記と同義である)
で表される基である。
Yがヒドロキシル基またはハロゲン原子である場合、本発明化合物は、最初の反応基質のカルボキシル基と反応し、エステル結合を形成している。この場合、アンカーの脱保護では、ペプチドのC末端がカルボキシル基となる。
一方、Yが-NHR基である場合、本発明化合物は、最初の反応基質のカルボキシル基と反応し、アミド結合を形成している。この場合、アンカーの脱保護では、ペプチドのC末端が-CONHR基に変換される。
当該工程では、一時保護基を除去することなく、アンカーのみを選択的に除去することが可能である。例えば、XおよびZが水素原子であり、Yがヒドロキシル基であり、かつベンゼン環上の基QRa(特にORa)が2位および4位に存在するか、または2位、4位および6位に存在する本発明化合物(アンカー)を使用し、ペプチド等の一時保護基がFmoc基またはCbz基である場合には、脱保護は好適には酸処理により行われる。
使用する酸としては、トリフルオロ酢酸(以下、TFAという。)、塩酸、硫酸、メタンスルホン酸、p-トルエンスルホン酸等が挙げられ、中でも、TFAが好ましく、これらの酸の濃度が0.1%~5%のクロロホルムやジクロロメタンまたはTHF溶液の溶液条件下に行うことができる。
で表される基である。
Yがヒドロキシル基またはハロゲン原子である場合、本発明化合物は、最初の反応基質のカルボキシル基と反応し、エステル結合を形成している。この場合、アンカーの脱保護では、ペプチドのC末端がカルボキシル基となる。
一方、Yが-NHR基である場合、本発明化合物は、最初の反応基質のカルボキシル基と反応し、アミド結合を形成している。この場合、アンカーの脱保護では、ペプチドのC末端が-CONHR基に変換される。
当該工程では、一時保護基を除去することなく、アンカーのみを選択的に除去することが可能である。例えば、XおよびZが水素原子であり、Yがヒドロキシル基であり、かつベンゼン環上の基QRa(特にORa)が2位および4位に存在するか、または2位、4位および6位に存在する本発明化合物(アンカー)を使用し、ペプチド等の一時保護基がFmoc基またはCbz基である場合には、脱保護は好適には酸処理により行われる。
使用する酸としては、トリフルオロ酢酸(以下、TFAという。)、塩酸、硫酸、メタンスルホン酸、p-トルエンスルホン酸等が挙げられ、中でも、TFAが好ましく、これらの酸の濃度が0.1%~5%のクロロホルムやジクロロメタンまたはTHF溶液の溶液条件下に行うことができる。
本発明化合物に由来する保護基(アンカー)を、一時保護基と同時に除去することも可能である。その場合には、当該分野、特にペプチド合成において行われている慣用の方法が用いられるが、水素還元条件や酸性条件等で行う方法が好適に採用される。酸としてTFA、塩酸、硫酸、メシル酸、トシル酸、トリフルオロエタノール、ヘキサフルオロイソプロパノール等が使用される。中でもTFAが特に好ましい。
酸は、用いる酸の種類によって適宜設定され、アンカーを除去するのに適当な量が用いられる。酸の使用量は、結合物1モルに対して、例えば3~100モル、好ましくは5~50モルである。上記TFA等の使用とともに、更なる強酸源として、トリフルオロメタンスルホン酸、トリフルオロメタンスルホン酸トリメチルシリル、三フッ化ホウ素・エーテラート(BF3・Et2O)などを加えることもできる。
反応温度は、通常0℃~80℃、好ましくは0℃~30℃である。
反応時間は、通常0.5~24時間である。
酸は、用いる酸の種類によって適宜設定され、アンカーを除去するのに適当な量が用いられる。酸の使用量は、結合物1モルに対して、例えば3~100モル、好ましくは5~50モルである。上記TFA等の使用とともに、更なる強酸源として、トリフルオロメタンスルホン酸、トリフルオロメタンスルホン酸トリメチルシリル、三フッ化ホウ素・エーテラート(BF3・Et2O)などを加えることもできる。
反応温度は、通常0℃~80℃、好ましくは0℃~30℃である。
反応時間は、通常0.5~24時間である。
上記工程を利用することによりペプチドを製造することができる。本発明化合物は、アミノ酸またはペプチドのC末端等の保護化試薬として主に使用することができるが、それに限定されるわけではない。また、Yがヒドロキシル基である本発明化合物は、当該分野における慣用の方法(例えば、ホスゲンとの反応)により対応するクロロホルメート体へと変換することができるので、当該クロロホルメート体をN末端等の保護化試薬として使用することも可能である。
上記工程を利用したペプチドの製造方法は、具体的には以下の工程を含む:
(1)本発明化合物を、該化合物の可溶性溶媒中でN-保護アミノ酸またはN-保護ペプチドのC末端等と縮合させて、アンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程(C末端等の保護工程)、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程(N末端の脱保護工程)、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程(ペプチド鎖の伸長工程)、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、目的のペプチドを得る工程(脱保護工程)。
(1)本発明化合物を、該化合物の可溶性溶媒中でN-保護アミノ酸またはN-保護ペプチドのC末端等と縮合させて、アンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程(C末端等の保護工程)、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程(N末端の脱保護工程)、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程(ペプチド鎖の伸長工程)、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、目的のペプチドを得る工程(脱保護工程)。
本発明において「N-保護アミノ酸」および「N-保護ペプチド」とは、それぞれ、N末端のアミノ基が一時保護基で保護されており、C末端のカルボキシル基が保護されていないアミノ酸およびペプチドを意味する。これらを、以下では「P-AA-OH」等と表示することがある(PはN末端の保護基である)。
本発明において「N-保護C-保護アミノ酸」および「N-保護C-保護ペプチド」とは、それぞれ、N末端のアミノ基が一時保護基で保護されており、C末端のカルボキシル基がアンカーで保護されているアミノ酸およびペプチドを意味する。
本発明において「C-保護アミノ酸」および「C-保護ペプチド」とは、それぞれ、N末端のアミノ基が保護されておらず、C末端のカルボキシル基がアンカーで保護されているアミノ酸およびペプチドを意味する。
本発明において「N-保護C-保護アミノ酸」および「N-保護C-保護ペプチド」とは、それぞれ、N末端のアミノ基が一時保護基で保護されており、C末端のカルボキシル基がアンカーで保護されているアミノ酸およびペプチドを意味する。
本発明において「C-保護アミノ酸」および「C-保護ペプチド」とは、それぞれ、N末端のアミノ基が保護されておらず、C末端のカルボキシル基がアンカーで保護されているアミノ酸およびペプチドを意味する。
工程(1)(C末端等の保護工程)
当該工程は、本発明化合物を、該化合物の可溶性溶媒中でN-保護アミノ酸またはN-保護ペプチドのC末端等と縮合し、N-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程である。当該工程は、例えば、上記工程(ii)および工程(iii)に準じて実施することができる。
当該工程は、本発明化合物を、該化合物の可溶性溶媒中でN-保護アミノ酸またはN-保護ペプチドのC末端等と縮合し、N-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程である。当該工程は、例えば、上記工程(ii)および工程(iii)に準じて実施することができる。

(式中、P1はN末端のアミノ基の保護基を示し、AA1はアミノ酸由来の基を示し、Y’はOまたはNRを示す。他の記号は前記と同意義を示す。)
本発明化合物とN-保護アミノ酸またはN-保護ペプチドとのC末端での縮合反応は、好適には反応に影響を及ぼさない溶媒中で行われる。例えば、Yがヒドロキシル基または-NHR基である場合には、縮合剤の存在下で縮合反応が行われ、Yがハロゲン原子である場合には、塩基の存在下で縮合反応が行われる。その結果、Yがヒドロキシルまたはハロゲン原子である場合は、エステル結合が形成され、Yが-NHR基である場合は、アミド結合が形成される。縮合剤としては、例えば、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、N-エチル-N’-3-ジメチルアミノプロピルカルボジイミドおよびその塩酸塩(EDC・HCl)等が挙げられる。エステル結合形成反応の際には、ジメチルアミノピリジン存在下で、アミド結合形成反応の際には、HOBtやHOCt等の縮合添加剤を用いて実施される。
当該工程に用いる溶媒としては、上述の可溶性溶媒が好適である。溶媒の使用量は、本発明化合物1gに対し、好適には、2~50mlである。
更にペプチド鎖の長さや種類に応じて、溶媒として、トルエン、シクロペンチルメチルエーテル、クロロホルム等を選択することができる。これら溶媒は、2種以上の混合物を使用してもよい。
反応温度は、通常-10℃~40℃、好ましくは0℃~30℃である。反応時間は、通常1~70時間である。
反応終了後、水を添加し、洗浄し、分層することにより、目的のC-保護アミノ酸またはC-保護ペプチドを含む溶液が得られ、単離することなく、そのまま次の工程に使用することが可能である。
また、Yがヒドロキシル基である本発明化合物とP1-AA1-NHRで表されるアミド化合物(例えば、Fmoc-Ala-NH2、Fmoc-Gly-NH2等)とを酸触媒(例えば、メタンスルホン酸、トリフルオロメタンスルホン酸、p-トルエンスルホン酸等)下、反応に影響を及ぼさない溶媒中で、高温処理(好ましくは、50℃~150℃、より好ましくは60℃~120℃)することによりアミド結合によりアンカー保護された化合物を得ることも可能である。
本発明化合物とN-保護アミノ酸またはN-保護ペプチドとのC末端での縮合反応は、好適には反応に影響を及ぼさない溶媒中で行われる。例えば、Yがヒドロキシル基または-NHR基である場合には、縮合剤の存在下で縮合反応が行われ、Yがハロゲン原子である場合には、塩基の存在下で縮合反応が行われる。その結果、Yがヒドロキシルまたはハロゲン原子である場合は、エステル結合が形成され、Yが-NHR基である場合は、アミド結合が形成される。縮合剤としては、例えば、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、N-エチル-N’-3-ジメチルアミノプロピルカルボジイミドおよびその塩酸塩(EDC・HCl)等が挙げられる。エステル結合形成反応の際には、ジメチルアミノピリジン存在下で、アミド結合形成反応の際には、HOBtやHOCt等の縮合添加剤を用いて実施される。
当該工程に用いる溶媒としては、上述の可溶性溶媒が好適である。溶媒の使用量は、本発明化合物1gに対し、好適には、2~50mlである。
更にペプチド鎖の長さや種類に応じて、溶媒として、トルエン、シクロペンチルメチルエーテル、クロロホルム等を選択することができる。これら溶媒は、2種以上の混合物を使用してもよい。
反応温度は、通常-10℃~40℃、好ましくは0℃~30℃である。反応時間は、通常1~70時間である。
反応終了後、水を添加し、洗浄し、分層することにより、目的のC-保護アミノ酸またはC-保護ペプチドを含む溶液が得られ、単離することなく、そのまま次の工程に使用することが可能である。
また、Yがヒドロキシル基である本発明化合物とP1-AA1-NHRで表されるアミド化合物(例えば、Fmoc-Ala-NH2、Fmoc-Gly-NH2等)とを酸触媒(例えば、メタンスルホン酸、トリフルオロメタンスルホン酸、p-トルエンスルホン酸等)下、反応に影響を及ぼさない溶媒中で、高温処理(好ましくは、50℃~150℃、より好ましくは60℃~120℃)することによりアミド結合によりアンカー保護された化合物を得ることも可能である。
工程(2)(N末端の脱保護工程)
当該工程は、工程(1)で得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程である。
当該工程は、工程(1)で得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程である。

(式中の記号は前記と同意義を示す。)
N末端の保護基としては、ペプチド化学等の技術分野で一般的に用いられる後述のアミノ基の保護基が使用可能であるが、本発明においては、tert-ブトキシカルボニル基(以下、Boc基ともいう。)、Cbz基、および/またはFmoc基が好適に用いられる。
脱保護条件としては、N末端の保護基の種類により適宜選択されるが、アンカーの除去とは異なる脱保護条件が好ましい。例えば、N末端の保護基がFmoc基である場合は、塩基(例えば、ジメチルアミン、ジエチルアミン、ピペリジン、モルホリン、DBU、ジエチレントリアミン、アミノメチルピペリジン、トリエチレンテトラミン、テトラエチレンペンタミン等)で処理することにより行なわれ(国際公開第2009/014177号参照);Cbz基である場合は、接触還元に付すことにより行われ;Boc基である場合は、酸で処理することにより行われる(国際公開第2009/014176号参照)。当該反応は、反応に影響を及ぼさない溶媒中(例えば、上述の可溶性溶媒中)で行われる。反応終了後は分層抽出することにより、目的の脱保護体を含む溶液が得られ、単離することなく、そのまま次の工程に使用することが可能である。
N末端の保護基としては、ペプチド化学等の技術分野で一般的に用いられる後述のアミノ基の保護基が使用可能であるが、本発明においては、tert-ブトキシカルボニル基(以下、Boc基ともいう。)、Cbz基、および/またはFmoc基が好適に用いられる。
脱保護条件としては、N末端の保護基の種類により適宜選択されるが、アンカーの除去とは異なる脱保護条件が好ましい。例えば、N末端の保護基がFmoc基である場合は、塩基(例えば、ジメチルアミン、ジエチルアミン、ピペリジン、モルホリン、DBU、ジエチレントリアミン、アミノメチルピペリジン、トリエチレンテトラミン、テトラエチレンペンタミン等)で処理することにより行なわれ(国際公開第2009/014177号参照);Cbz基である場合は、接触還元に付すことにより行われ;Boc基である場合は、酸で処理することにより行われる(国際公開第2009/014176号参照)。当該反応は、反応に影響を及ぼさない溶媒中(例えば、上述の可溶性溶媒中)で行われる。反応終了後は分層抽出することにより、目的の脱保護体を含む溶液が得られ、単離することなく、そのまま次の工程に使用することが可能である。
工程(3)(ペプチド鎖の伸長工程)
当該工程は、工程(2)で得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程であり、例えば工程(iv)に準じて実施することができる。
当該工程は、工程(2)で得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程であり、例えば工程(iv)に準じて実施することができる。
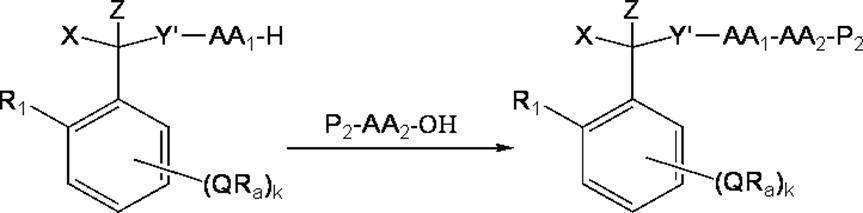
(式中、P2はN末端のアミノ基の保護基を示し、AA2はアミノ酸由来の基を示し、他の記号は前記と同意義を示す。)
当該工程は、工程(1)に記載の縮合剤、縮合添加剤等を使用し、ペプチド化学の分野において一般的に用いられるペプチド合成条件下で行われる。
反応終了後、水および/または親水性有機溶媒(アセトニトリル、DMF等)を添加し、洗浄し、分層することにより、N-保護C-保護ペプチドを含む溶液が得られる。N-保護C-保護ペプチドを単離することなく、そのまま次の工程に使用することが可能である。
当該工程は、工程(1)に記載の縮合剤、縮合添加剤等を使用し、ペプチド化学の分野において一般的に用いられるペプチド合成条件下で行われる。
反応終了後、水および/または親水性有機溶媒(アセトニトリル、DMF等)を添加し、洗浄し、分層することにより、N-保護C-保護ペプチドを含む溶液が得られる。N-保護C-保護ペプチドを単離することなく、そのまま次の工程に使用することが可能である。
工程(4)(脱保護工程)
当該工程は、工程(3)で得られたN-保護C-保護ペプチドから、N末端の保護基およびC末端のアンカーを除去して、目的のペプチドを得る工程であり、上記工程(2)のN末端の保護基の脱保護工程、および工程(v)に準じて行われる。
当該工程は、工程(3)で得られたN-保護C-保護ペプチドから、N末端の保護基およびC末端のアンカーを除去して、目的のペプチドを得る工程であり、上記工程(2)のN末端の保護基の脱保護工程、および工程(v)に準じて行われる。

(式中の記号は前記と同意義を示す。)
本発明のペプチドの製造方法において、工程(3)で得られたN-保護C-保護ペプチドに対して、工程(5)、(6)、および(7)または(7’)を1以上繰返した後に、工程(4)を行うこともできる。
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程(N末端の脱保護工程)、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程(ペプチド鎖の伸長工程)、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程(抽出分離工程)、または
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程(抽出分離工程)。
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程(N末端の脱保護工程)、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程(ペプチド鎖の伸長工程)、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程(抽出分離工程)、または
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程(抽出分離工程)。
工程(5)(N末端の脱保護工程)
当該工程は、上記工程(2)と同様にして行われる。
当該工程は、上記工程(2)と同様にして行われる。
工程(6)(ペプチド鎖の伸長工程)
当該工程は、上記工程(3)と同様にして行われる。
当該工程は、上記工程(3)と同様にして行われる。
工程(7)、(7’)(抽出分離工程)
当該工程は、工程(6)で得られたN-保護C-保護ペプチドを分層によって有機層に残し、縮合反応により生じる不純物等を水層および/または親水性有機溶媒(アセトニトリル、DMF等)層へと追い出すことにより行われる。
当該工程は、工程(6)で得られたN-保護C-保護ペプチドを分層によって有機層に残し、縮合反応により生じる不純物等を水層および/または親水性有機溶媒(アセトニトリル、DMF等)層へと追い出すことにより行われる。
本発明の有機合成反応またはペプチド合成反応が多工程を含む場合には、途中工程で単離する工程を適宜加えてもよく、また、次工程の反応に影響を及ぼさない範囲で抽出洗浄等の前記分層工程を適宜省略することも可能である。
各反応において、原料化合物がヒドロキシル基、アミノ基、カルボキシル基、カルボニル基を有する場合(特にアミノ酸またはペプチドの側鎖に官能基を有する場合)、これらの基にペプチド化学等で一般的に用いられるような保護基が導入されていてもよく、反応後に必要に応じて保護基を除去することにより目的化合物を得ることができる。
ヒドロキシル基の保護基としては、例えば、(C1-C6)アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、tert-ブチル)、フェニル基、トリチル基、(C7-C10)アラルキル基(例、ベンジル)、ホルミル基、(C1-C6)アルキル-カルボニル基(例、アセチル、プロピオニル)、ベンゾイル基、(C7-C10)アラルキル-カルボニル基(例、ベンジルカルボニル)、2-テトラヒドロピラニル基、2-テトラヒドロフラニル基、シリル基(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル)、(C2-C6)アルケニル基(例、1-アリル)、N-(アセチル)アミノメチル基(Acm)等が挙げられる。これらの基は、ハロゲン原子(例、フッ素、塩素、臭素、ヨウ素)、(C1-C6)アルキル基(例、メチル、エチル、プロピル)、(C1-C6)アルコキシ基(例、メトキシ、エトキシ、プロポキシ)、ニトロ基等から選ばれる1ないし3個の置換基で置換されていてもよい。
アミノ基の保護基としては、例えば、ホルミル基、(C1-C6)アルキル-カルボニル基(例、アセチル、プロピオニル)、(C1-C6)アルコキシ-カルボニル基(例、メトキシカルボニル、エトキシカルボニル、Boc基)、ベンゾイル基、(C7-C10)アラルキル-カルボニル基(例、ベンジルカルボニル)、(C7-C14)アラルキルオキシ-カルボニル基(例、CBz基、クロロベンジルオキシカルボニル基、ブロモベンジルオキシカルボニル基、Fmoc基)、トリチル基、フタロイル基、N,N-ジメチルアミノメチレン基、シリル基(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル)、(C2-C6)アルケニル基(例、1-アリル)等が挙げられる。これらの基は、ハロゲン原子(例、フッ素、塩素、臭素、ヨウ素)、(C1-C6)アルコキシ基(例、メトキシ、エトキシ、プロポキシ)、ニトロ基等から選ばれる1ないし3個の置換基で置換されていてもよい。
カルボキシル基の保護基としては、例えば、(C1-C6)アルキル基(例、メチル、エチル、プロピル、イソプロピル、ブチル、tert-ブチル)、(C7-C10)アラルキル基(例、ベンジル、ブロモベンジル、クロロベンジル、ニトロベンジル)、フェニル基、トリチル基、シリル基(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル、tert-ブチルジフェニルシリル)、(C2-C6)アルケニル基(例、1-アリル)等が挙げられる。これらの基は、ハロゲン原子(例、フッ素、塩素、臭素、ヨウ素)、(C1-C6)アルコキシ基(例、メトキシ、エトキシ、プロポキシ)、ニトロ基等から選ばれる1ないし3個の置換基で置換されていてもよい。
カルボニル基の保護基としては、例えば、環状アセタール(例、1,3-ジオキサン)、非環状アセタール(例、ジ-(C1-C6)アルキルアセタール)等が挙げられる。
また、これらの保護基の除去方法は、自体公知の方法、例えば、Protective Groups in Organic Synthesis,John Wiley and Sons刊(1980)に記載の方法等に準じて行えばよい。例えば、酸、塩基、紫外光、ヒドラジン、フェニルヒドラジン、N-メチルジチオカルバミン酸ナトリウム、テトラブチルアンモニウムフルオリド、酢酸パラジウム、トリアルキルシリルハライド(例、トリメチルシリルヨージド、トリメチルシリルブロミド等)等を使用する方法、還元法等が用いられる。
また、これらの保護基の除去方法は、自体公知の方法、例えば、Protective Groups in Organic Synthesis,John Wiley and Sons刊(1980)に記載の方法等に準じて行えばよい。例えば、酸、塩基、紫外光、ヒドラジン、フェニルヒドラジン、N-メチルジチオカルバミン酸ナトリウム、テトラブチルアンモニウムフルオリド、酢酸パラジウム、トリアルキルシリルハライド(例、トリメチルシリルヨージド、トリメチルシリルブロミド等)等を使用する方法、還元法等が用いられる。
〔ペプチドの製造用キット〕
本発明は、また、本発明化合物を必須の構成成分として含む、ペプチドの製造用キットを提供する。当該キットには、本発明化合物に加えて、ペプチドの製造方法反応を実施するのに必要な他の成分、例えば反応に用いる各種溶媒、原料となるアミノ酸(またはペプチド)等が含められていてもよい。所望により本発明化合物を用いたペプチドの製造の為のマニュアルを添付することもできる。
本発明は、また、本発明化合物を必須の構成成分として含む、ペプチドの製造用キットを提供する。当該キットには、本発明化合物に加えて、ペプチドの製造方法反応を実施するのに必要な他の成分、例えば反応に用いる各種溶媒、原料となるアミノ酸(またはペプチド)等が含められていてもよい。所望により本発明化合物を用いたペプチドの製造の為のマニュアルを添付することもできる。
以下、実施例に沿って本発明をさらに詳細に説明するが、これら実施例は本発明の範囲を何ら限定するものではない。また、本発明において使用する試薬や装置、材料は特に言及されない限り、商業的に入手可能である。また、本明細書において、アミノ酸等を略号で表示する場合、各表示は、IUPAC-IUB Commission on Biochemical Nomenclatureによる略号あるいは当該分野における慣用略号に基づくものである。
以下の参考例および実施例中の収率は、mol/mol%を示す。特段の定義がない限り、本明細書中における「%」は、「重量%」を表す。また、以下の参考例および実施例中の溶媒の比率は、体積比を示す。1H-NMRスペクトルは内部標準としてテトラメチルシランを用い、測定溶媒としてCDCl3を使用した。NMRスペクトルは、Bruker AVANCE AV300(300MHz)核磁気共鳴装置を用いて測定した。
エレクトロスプレーイオン化液体クロマトグラフィー/質量分析(以下、LC/MSと略す。)は、LC-MSD(liquid chromatography) system 1100 Series(Agilent Technologies)を用い、フローインジェクション分析(FIA)した(溶媒:0.05%TFA THF水、イオン化モード:ESI、イオンモード:ポジティブ、質量分析部:四重極、フラグメンター電圧:100V)。
以下の参考例および実施例中の収率は、mol/mol%を示す。特段の定義がない限り、本明細書中における「%」は、「重量%」を表す。また、以下の参考例および実施例中の溶媒の比率は、体積比を示す。1H-NMRスペクトルは内部標準としてテトラメチルシランを用い、測定溶媒としてCDCl3を使用した。NMRスペクトルは、Bruker AVANCE AV300(300MHz)核磁気共鳴装置を用いて測定した。
エレクトロスプレーイオン化液体クロマトグラフィー/質量分析(以下、LC/MSと略す。)は、LC-MSD(liquid chromatography) system 1100 Series(Agilent Technologies)を用い、フローインジェクション分析(FIA)した(溶媒:0.05%TFA THF水、イオン化モード:ESI、イオンモード:ポジティブ、質量分析部:四重極、フラグメンター電圧:100V)。
参考例1:2,3-ジヒドロフィトールの合成
フィトール(10.00g,33.7mmol)をメタノールに溶解させ、Pt/C(2%,1.00g)を懸濁させて水素雰囲気下で一晩攪拌した。反応終了後、濾過してPt/Cを除去し、濾液を濃縮して2,3-ジヒドロフィトールを得た。これは精製することなく次の反応に用いた。
1H-NMR(300MHz):δ0.80-0.93(15H,m,Me),0.98-1.70(24H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-OH),3.62-3.75(2H,-CH 2 -OH).
1H-NMR(300MHz):δ0.80-0.93(15H,m,Me),0.98-1.70(24H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-OH),3.62-3.75(2H,-CH 2 -OH).
参考例2:2,3-ジヒドロフィチルブロミドの合成
2,3-ジヒドロフィトール(33.7mmol)を48%臭化水素酸(100ml)に懸濁し、濃硫酸(0.17ml)を滴下して100℃で一晩攪拌した。反応混合液を室温に冷却後ヘキサン(200ml)で抽出し、5%炭酸水素ナトリウム水溶液(70ml)で2回、20%食塩水(70ml)で1回洗浄した。有機層を硫酸ナトリウムで乾燥し、濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ショートカラム、ヘキサンのみ)で精製し、2,3-ジヒドロフィチルブロミド(「2,3-ジヒドロフィチル基」を、以下「Phy」と称することもある。)(10.41g,28.8mmol,85% vs. フィトール)を得た。
1H-NMR(300MHz):δ0.79-0.92(15H,m,Me),0.95-1.95(24H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-Br),3.35-3.52(2H,-CH 2 -Br).
1H-NMR(300MHz):δ0.79-0.92(15H,m,Me),0.95-1.95(24H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-Br),3.35-3.52(2H,-CH 2 -Br).
参考例3:3,7,11-トリメチルドデカン-1-オールの合成
ファルネソール(3.00g,13.5mmol)をメタノール(30ml)に溶解させ、Pt/C(2%,0.30g)を懸濁させて水素雰囲気下で一晩攪拌した。反応終了後、濾過してPt/Cを除去し、濾液を濃縮して、3,7,11-トリメチルドデカン-1-オールを得た。これは精製することなく次の反応に用いた。
1H-NMR(300MHz):δ1.09-1.43(m,24H),1.48-1.66(m,5H),3.63-3.70(m,2H).
1H-NMR(300MHz):δ1.09-1.43(m,24H),1.48-1.66(m,5H),3.63-3.70(m,2H).
参考例4:1-ブロモ-3,7,11-トリメチルドデカンの合成
参考例3で得られた3,7,11-トリメチルドデカン-1-オールを48%臭化水素酸(31ml)に懸濁し、濃硫酸(57μl)を滴下して120℃で一晩攪拌した。反応混合液を室温に冷却後、ヘキサン(45ml)で抽出し、5%炭酸水素ナトリウム水溶液(20ml)で2回、20%食塩水(20ml)で1回洗浄した。有機層を硫酸ナトリウムで乾燥し、濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ショートカラム、ヘキサンのみ)で精製し、1-ブロモ-3,7,11-トリメチルドデカン(2.98g,10.2mmol,76% vs. ファルネソール)を得た。
1H-NMR(300MHz):δ1.12-1.43(m,24H),1.48-1.70(m,4H),1.84-1.90(m,1H),3.36-3.49(m,2H).
1H-NMR(300MHz):δ1.12-1.43(m,24H),1.48-1.70(m,4H),1.84-1.90(m,1H),3.36-3.49(m,2H).
参考例5:GI-1000(日本曹達株式会社製)(末端ジオール体;数平均分子量:約1500;n=約23)からジブロミド体(GI-1000(Br))への変換
GI-1000(日本曹達株式会社製)(5.02g)をヘプタン(50ml)に溶解させ、80%アセトニトリル水溶液(25ml)で2回洗浄した。ヘプタン層を濃縮して得られた残渣を48%臭化水素酸(50ml)に懸濁し、濃硫酸(100μl)を滴下して120℃で一晩攪拌した。反応混合液を室温に冷却後、ヘプタン(100ml)で抽出し、5%炭酸水素ナトリウム水溶液(25ml)で2回、20%食塩水(25ml)で1回、90%アセトニトリル水溶液(40ml)で2回洗浄した。濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ショートカラム、ヘキサンのみ)で精製し、GI-1000のジブロミド(GI-1000(Br))(2.63g)を得た。
1H-NMR(300MHz):δ0.90-1.06(m),1.83-1.86(m,4H),3.38-3.44(m,4H).
1H-NMR(300MHz):δ0.90-1.06(m),1.83-1.86(m,4H),3.38-3.44(m,4H).
参考例6:GI-2000(日本曹達株式会社製)(末端ジオール体;数平均分子量:約2100;n=約34)からジブロミド体(GI-2000(Br))への変換

GI-2000(日本曹達株式会社製)(5.33g)をヘプタン(50ml)に溶解させ、80%アセトニトリル水溶液(25ml)で2回洗浄した。ヘプタン層を濃縮して得られた残渣を48%臭化水素酸(50ml)に懸濁し、濃硫酸(100μl)を滴下して120℃で一晩攪拌した。反応混合液を室温に冷却後、ヘプタン(100ml)で抽出し、5%炭酸水素ナトリウム水溶液(25ml)で2回、20%食塩水(25ml)で1回、90%アセトニトリル水溶液(40ml)で2回洗浄した。濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ショートカラム、ヘキサンのみ)で精製し、GI-2000のジブロミド(GI-2000(Br))(2.83g)を得た。
1H-NMR(300MHz):δ0.91-1.54(m),1.81-1.88(m,4H),3.38-3.42(m,4H).
1H-NMR(300MHz):δ0.91-1.54(m),1.81-1.88(m,4H),3.38-3.42(m,4H).
参考例7:TERGITOL(登録商標)-TMN6(シグマ・アルドリッチ社製)(市販品(数平均分子量:543;n=約5)を以下に示す前処理した後(n:約7)に使用した。)からブロミド体(TERGITOL(Br))への変換
TERGITOL-TMN6(7.42g)をクロロホルム(70ml)に溶解させ、水(35ml)で分液洗浄を行った。抽出した有機層を硫酸ナトリウムで乾燥させて、濃縮乾固させ、クロロホルム(70ml)に溶解させて、氷冷下でPBr3(1148μl,12.1mmol,1.0eq)、ピリジン(1074μl,12.1mmol,1.0eq)を滴下し、室温で4h攪拌した。溶媒を除去し、酢酸エチル(100ml)に溶解させ、0.5N塩酸(50ml)で3回、5%炭酸水素ナトリウム水溶液(50ml)で3回、20%食塩水(50ml)で1回分液洗浄を行った。溶媒を除去し、シリカゲルカラムクロマトグラフィー(酢酸エチル→酢酸エチル:メタノール=1:1)で精製した。溶媒を除去し、酢酸エチル(100ml)で溶解させ、濾過でシリカゲルを除去してTERGITOL-TMN6のBr置換体(5.21g)を得た。
1H-NMR(300MHz):δ0.80-0.90(m,15H,CH3),1.00-1.60(m,11H,CH,CH2),3.37-3.47(m,3H,CH-O,CH2-Br),3.50-3.68(m,(OCH 2 CH 2 ) n ),3.79-3.83.(t,2H,OCH 2 CH2-Br).
13C-NMR:δ20.42-27.39(CH3,CH),30.81(CH2-Br),42.78-47.67(CH2),68.04(OCH2),70.79-71.55(OCH 2 CH 2 O).
1H-NMR(300MHz):δ0.80-0.90(m,15H,CH3),1.00-1.60(m,11H,CH,CH2),3.37-3.47(m,3H,CH-O,CH2-Br),3.50-3.68(m,(OCH 2 CH 2 ) n ),3.79-3.83.(t,2H,OCH 2 CH2-Br).
13C-NMR:δ20.42-27.39(CH3,CH),30.81(CH2-Br),42.78-47.67(CH2),68.04(OCH2),70.79-71.55(OCH 2 CH 2 O).
実施例1:2,4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコールの合成

2,3-ジヒドロフィチルブロミド(4.50g,12.5mmol)、2,4-ジヒドロキシベンズアルデヒド(851mg,6.16mmol)、炭酸カリウム(2.58g,18.7mmol)をDMF(45ml)中に懸濁し、90℃で一晩攪拌した。反応混合液を室温に冷却後、酢酸エチル(150ml)で抽出し、1N塩酸(50ml)で3回、5%炭酸水素ナトリウム水溶液(50ml)で3回、20%食塩水(50ml)で1回洗浄した。有機層を硫酸ナトリウムで乾燥し、濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ショートカラム、ヘキサン:酢酸エチル=20:1)で精製し、2,4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルデヒド(3.65g,5.22mmol,85% vs. 2,4-ジヒドロキシベンズアルデヒド)を得た。
前記2,4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルデヒド(3.65g,5.22mmol)をTHF-メタノール混合溶液(40+2ml)に溶解させ、0℃で水素化ホウ素ナトリウム(263mg,90%,6.26mmol)を加え、室温で1時間攪拌した。反応混合液を0℃に冷却後1N塩酸(5ml)で反応を停止し、酢酸エチル(100ml)を加えて、1N塩酸(30ml)で2回、5%炭酸水素ナトリウム水溶液(30ml)で1回、20%食塩水(30ml)で1回洗浄した。有機層を硫酸ナトリウムで乾燥し、濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ショートカラム、ヘキサン:酢酸エチル=10:1)で精製し、2,4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(3.29g,4.69mmol,90%)を得た。
1H-NMR(300MHz):δ0.81-0.90(24H,m,Me(Phytyl)),0.94(6H,dd,J=2.1,6.3Hz,Me(Phytyl)),1.00-1.95(48H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.23(1H,t,J=6.6Hz,OH),3.93-4.07(4H,m,-CH 2 -O-Ar),4.61(2H,d,J=6.6Hz,Ar-CH 2 -OH),6.43(1H,dd,J=2.1,8.1Hz,C5-H),6.46(1H,d,J=2.1Hz,C3-H),7.13(1H,d,J=8.1Hz,C6-H).
1H-NMR(300MHz):δ0.81-0.90(24H,m,Me(Phytyl)),0.94(6H,dd,J=2.1,6.3Hz,Me(Phytyl)),1.00-1.95(48H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.23(1H,t,J=6.6Hz,OH),3.93-4.07(4H,m,-CH 2 -O-Ar),4.61(2H,d,J=6.6Hz,Ar-CH 2 -OH),6.43(1H,dd,J=2.1,8.1Hz,C5-H),6.46(1H,d,J=2.1Hz,C3-H),7.13(1H,d,J=8.1Hz,C6-H).
実施例2:2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)ベンズヒドロールの合成
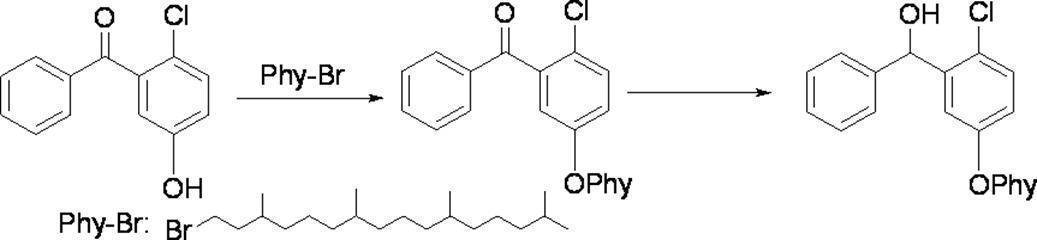
ジヒドロフィチルブロミド(1.02g,2.82mmol)にDMF(15ml)、2-クロロ-5-ヒドロキシベンゾフェノン(0.99g,4.23mmol)、K2CO3(0.78g,5.64mmol)を加え、90℃で3時間攪拌した。反応液を室温に戻し、酢酸エチル(25ml),1N塩酸(25ml)を加えて攪拌し、分層させ水層を分離・廃棄した。有機層を精製水(25ml)で2回洗浄し、有機層を減圧留去させ、2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)ベンゾフェノンを得た。
1H-NMR(300MHz):δ0.75-0.90(15H,m,Me),0.95-1.70(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.82-3.92(2H,br,-O-CH 2 -C19H39),6.89(1H,d,J=8.3Hz,C3-H),7.35-7.80(7H,m,C4,6-H,Ph-H).
1H-NMR(300MHz):δ0.75-0.90(15H,m,Me),0.95-1.70(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.82-3.92(2H,br,-O-CH 2 -C19H39),6.89(1H,d,J=8.3Hz,C3-H),7.35-7.80(7H,m,C4,6-H,Ph-H).
前記2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)ベンゾフェノンにクロロホルム(20ml)、メタノール(2ml)、水素化ホウ素ナトリウム(440mg,11.6mmol)を加え、50℃で一晩攪拌した。反応液を室温に戻し、さらに氷浴下で1N塩酸(15ml)を滴下して未反応の水素化ホウ素ナトリウムを分解させた後、水層を捨て、有機層を精製水(10ml)で2回洗浄した。有機層を減圧留去し、さらにアセトニトリルで水分を共沸させ、2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)ベンズヒドロールを得た。
1H-NMR(300MHz):δ0.82-0.90(15H,m,Me),1.00-1.90(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.88-4.00(2H,br,-O-CH 2 -C19H39),5.98(1H,s,Ar-CHOH-Ph),6.75-6.90(1H,m,C3-H),7.10-7.45(7H,m,C4,6-H,Ph-H).
1H-NMR(300MHz):δ0.82-0.90(15H,m,Me),1.00-1.90(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.88-4.00(2H,br,-O-CH 2 -C19H39),5.98(1H,s,Ar-CHOH-Ph),6.75-6.90(1H,m,C3-H),7.10-7.45(7H,m,C4,6-H,Ph-H).
実施例3:1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミンの合成
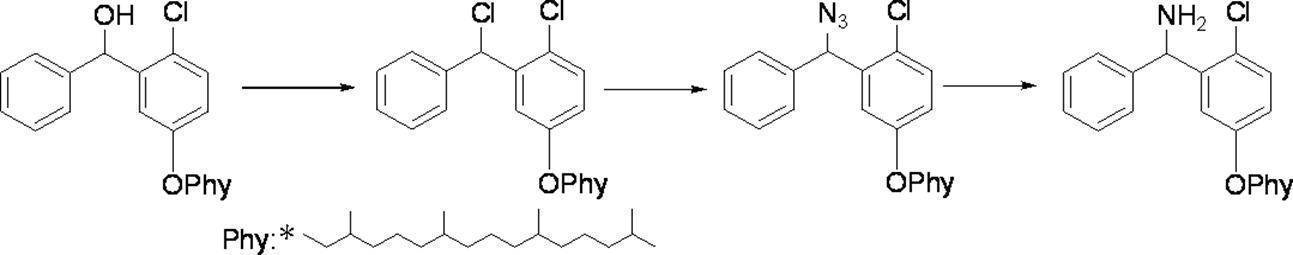
実施例2で得られた2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)ベンズヒドロールにクロロホルム(20ml)、DMF(43μl,559μmol)、塩化チオニル(1.03ml,14.1mmol)を加え、50℃で4時間攪拌した。反応液を室温に戻して溶媒を減圧留去し、残った塩化チオニルをトルエンで共沸させ、1-クロロ-1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)フェニルメタンを得た。
1H-NMR(300MHz):δ0.80-0.90(15H,m,Me),1.00-1.90(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.88-4.05(2H,m,-O-CH 2 -C19H39),6.48(1H,d,J=1.6Hz,Ar-CHCl-Ph),6.77(1H,d,J=8.7Hz,C3-H),7.10-7.55(7H,m,C4,6-H,Ph-H).
1H-NMR(300MHz):δ0.80-0.90(15H,m,Me),1.00-1.90(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.88-4.05(2H,m,-O-CH 2 -C19H39),6.48(1H,d,J=1.6Hz,Ar-CHCl-Ph),6.77(1H,d,J=8.7Hz,C3-H),7.10-7.55(7H,m,C4,6-H,Ph-H).
前記1-クロロ-1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)フェニルメタンにDMF(15ml)、アジ化ナトリウム(786mg,12.1mmol)を加え、80℃で一晩攪拌した。反応液を室温に戻して酢酸エチル(20ml)、ヘキサン(20ml)を加え、精製水(30ml)で1回、精製水(15ml)で2回分液洗浄した後、有機層を減圧留去し、1-アジド-1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)フェニルメタンを得た。
1H-NMR(300MHz):δ0.85-0.95(15H,m,Me),0.95-1.85(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.75-4.02(2H,m,-O-CH 2 -C19H39),5.90-6.10(1H,m,Ar-CHN3-Ph),6.79(1H,d,J=9.0Hz,C3-H),7.10-7.50(7H,m,C4,6-H,Ph-H).
1H-NMR(300MHz):δ0.85-0.95(15H,m,Me),0.95-1.85(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.75-4.02(2H,m,-O-CH 2 -C19H39),5.90-6.10(1H,m,Ar-CHN3-Ph),6.79(1H,d,J=9.0Hz,C3-H),7.10-7.50(7H,m,C4,6-H,Ph-H).
前記1-アジド-1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)フェニルメタンにTHF(20ml)、精製水(2ml)、トリフェニルホスフィン(813mg,3.10mmol)を加え、50℃で2時間攪拌した。反応液を室温に戻してTHFを留去し、ヘプタン(30ml)-50%アセトニトリル水溶液(15ml)で3回分液させ、ヘプタン層を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=100:0→5:1)で精製し、1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミンの油状物(1.35g,2.63mmol,収率93% vs. 2,3-ジヒドロフィチルブロミド)を得た。
1H-NMR(300MHz):δ0.85-0.95(15H,m,Me),0.95-1.85(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.85-4.00(2H,br,-O-CH 2 -C19H39),5.43(1H,s,Ar-CHNH2-Ph),6.75(1H,d,J=8.7Hz,C3-H),7.10-7.50(7H,m,C4,6-H,Ph-H).
1H-NMR(300MHz):δ0.85-0.95(15H,m,Me),0.95-1.85(24H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.85-4.00(2H,br,-O-CH 2 -C19H39),5.43(1H,s,Ar-CHNH2-Ph),6.75(1H,d,J=8.7Hz,C3-H),7.10-7.50(7H,m,C4,6-H,Ph-H).
実施例4:(4’,4’-ビスジヒドロフィチルオキシ)ベンズヒドロールの合成
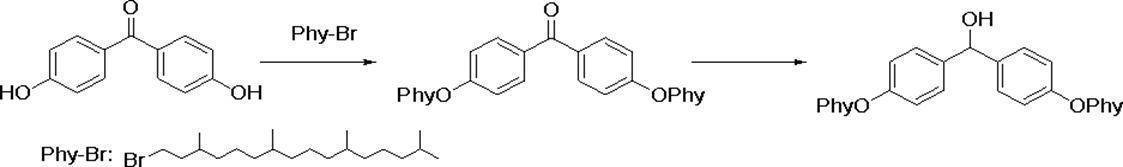
ジヒドロフィチルブロミド(14.3g,39.6mmol)にDMF(120ml)、4,4-ビスヒドロキシベンゾフェノン(4.04g,18.9mmol)、炭酸カリウム(7.82g,56.6mmol)を加え、80℃で5時間攪拌した。反応液を室温に戻し、酢酸エチル(300ml),1N塩酸(100ml)を加えて攪拌し、分層させ、水層を分離・廃棄した。有機層を精製水(100ml)で2回洗浄し、有機層を減圧留去させ、4,4-ビスジヒドロフィチルオキシベンゾフェノン油状物を得た。これをクロロホルム(60ml)とメタノール(10ml)に溶解させて水素化ホウ素ナトリウム(4.49g,119mmol)を加え、60℃で3時間攪拌した。反応液に1M塩酸(80ml)を添加し、濃縮し、酢酸エチル(100ml)を加え、1M塩酸と水にて順次洗浄した。有機層を濃縮し、(4’,4’-ビスジヒドロフィチルオキシ)ベンズヒドロール油状物を得た。
1H-NMR(300MHz):δ0.86-0.90(24H,m,Me),1.10-1.40(48H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.03(1H,s,OH),3.90-3.94(4H,m,-O-CH 2 -C19H39),5.76(1H,s,Ar-CHN3-Ph),6.85(4H,m,C3-H),7.20-7.26(4H,m,C4,6-H,Ph-H).
1H-NMR(300MHz):δ0.86-0.90(24H,m,Me),1.10-1.40(48H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.03(1H,s,OH),3.90-3.94(4H,m,-O-CH 2 -C19H39),5.76(1H,s,Ar-CHN3-Ph),6.85(4H,m,C3-H),7.20-7.26(4H,m,C4,6-H,Ph-H).
実施例5:3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコールの合成

2,3-ジヒドロフィチルブロミド(40.6g,112mmol)、没食子酸メチル(5.90g,32.0mmol)、炭酸カリウム(22.14g,160mmol)をDMF(400ml)に懸濁させ、110℃で一晩攪拌した。反応混合液をヘキサン(800ml)で抽出し、1N塩酸(400ml)、5%炭酸水素ナトリウム水溶液(400ml)、20%食塩水(400ml)で洗浄し、硫酸ナトリウムで乾燥した後濾液の溶媒を留去して、3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)安息香酸メチル(29.3g,収率93%)を得た。
前記3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)安息香酸メチル(29.3g,30.0mmol)をTHF(400ml)に溶解させ、窒素雰囲気下、0℃で水素化ジイソブチルアルミニウム(DIBAL)(1.0mol/l トルエン溶液,96ml,96mmol)を30分間かけて滴下した。室温で一晩攪拌した後、0℃で0.2N塩酸(50ml)を滴下して反応を停止した。溶媒を半分程度留去したものを、酢酸エチル(600ml)に溶解させ、1N塩酸(300ml)で3回、5%炭酸水素ナトリウム水溶液(300ml)で1回、20%食塩水(300ml)で1回洗浄し、硫酸ナトリウムで乾燥した後、濾液の溶媒を留去して、3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(26.8g,収率94%)を得た。
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.94(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.93-4.07(6H,m,-CH 2 -O-Ar),4.60(2H,s,-O-CH 2 -OH),6.57(2H,s,C2,6-H).
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.94(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.93-4.07(6H,m,-CH 2 -O-Ar),4.60(2H,s,-O-CH 2 -OH),6.57(2H,s,C2,6-H).
実施例6:2-(3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ)-4-メトキシベンジルアルコールの合成

3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(5.00g,4.87mmol)をクロロホルム(20ml)に溶解させ、0℃で塩化チオニル(1.16g,9.74mmol)を添加して室温で1時間攪拌した。溶媒を除去して3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルクロリド(4.88g,4.82mmol,収率96%)を得た。
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.94(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-),3.93-4.07(6H,m,CH2-O-),4.40(2H,s,CH2-Cl),6.57(2H,s,C2,6-H).
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.94(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-),3.93-4.07(6H,m,CH2-O-),4.40(2H,s,CH2-Cl),6.57(2H,s,C2,6-H).
前記3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルクロリド(4.88g,4.82mmol)、炭酸カリウム(1.68g,12.2mmol,2.5eq)、2-ヒドロキシ-4-メトキシベンズアルデヒド(0.82g,5.36mmol,1.1eq)をDMF(50ml)に懸濁させ、80℃で一晩攪拌した。反応液をヘキサン(500ml)で抽出し、1N塩酸(250ml)で3回、5%炭酸水素ナトリウム水溶液(250ml)で3回、20%食塩水(250ml)で1回分液洗浄を行った。溶媒を除去し、シリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:12)で精製して、2-(3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ)-4-メトキシベンズアルデヒド(5.53g,4.77mmol,収率99%)を得た。
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.94(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-),3.85(3H,s,OMe),3.93-4.13(6H,m,CH2-O-),5.05(2H,s,CH 2 -CHO),6.51(1H,s,C3-H),6.56(1H,d,J=9Hz,C6-H),6.63(2H,s,C2’,6’-H),7.83-7.86(1H,d,J=9Hz,C6-H),10.20(1H,s,CHO).
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.94(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-),3.85(3H,s,OMe),3.93-4.13(6H,m,CH2-O-),5.05(2H,s,CH 2 -CHO),6.51(1H,s,C3-H),6.56(1H,d,J=9Hz,C6-H),6.63(2H,s,C2’,6’-H),7.83-7.86(1H,d,J=9Hz,C6-H),10.20(1H,s,CHO).
2-(3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ)-4-メトキシベンズアルデヒド(32.0mmol)をTHF-メタノール(400ml+20ml)に溶解させ、0℃で水素化ホウ素ナトリウム(1.45g,38mmol)を加えた。室温で5.5時間攪拌した後、0℃で0.2N塩酸(20ml)を加えて反応を停止した。溶媒を半分程度留去し、酢酸エチル(600ml)に溶解させて、0.1N塩酸(300ml)で2回、5%炭酸水素ナトリウム水溶液(300ml)で1回、20%食塩水(300ml)で1回洗浄した。溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム→ヘキサン:酢酸エチル=7:1)で精製して2-(3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ)-4-メトキシベンジルアルコール(30.41g,26.8mmol,収率97%)を得た。
1H-NMR(300MHz):δ0.85(45H,t,J=6.3Hz,Me(Phytol)),1.00-1.95(72H,br,C3’,4’,5’-O-CH2CH 2 CHMe-[C3 H 6 CHMe]3-Me),2.18(1H,br,OH),3.80(3H,s,C4-OMe),3.90-4.08(6H,m,C3’,4’,5’-O-CH 2 -C19H39),4.65(2H,d,J=3.6Hz,Ar-CH 2 -OH),4.98(2H,s,Ar-O-CH 2 -Ar),6.48(1H,dd,J=2.1,8.1Hz,C5-H),6.54(1H,d,J=2.1Hz,C3-H),6.61(2H,s,C2’,6’-H),7.19(1H,d,J=8.1Hz,C6-H).
1H-NMR(300MHz):δ0.85(45H,t,J=6.3Hz,Me(Phytol)),1.00-1.95(72H,br,C3’,4’,5’-O-CH2CH 2 CHMe-[C3 H 6 CHMe]3-Me),2.18(1H,br,OH),3.80(3H,s,C4-OMe),3.90-4.08(6H,m,C3’,4’,5’-O-CH 2 -C19H39),4.65(2H,d,J=3.6Hz,Ar-CH 2 -OH),4.98(2H,s,Ar-O-CH 2 -Ar),6.48(1H,dd,J=2.1,8.1Hz,C5-H),6.54(1H,d,J=2.1Hz,C3-H),6.61(2H,s,C2’,6’-H),7.19(1H,d,J=8.1Hz,C6-H).
実施例7:3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミンの合成

3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルクロリド(6.46g,6.63mmol)をDMF-クロロホルム(60+20ml)に溶解させ、アジ化ナトリウム(861mg,13.2mmol)を加えて70℃で2時間攪拌した。反応混合液を室温に冷却後、酢酸エチル(160ml)を加えて、水(80ml)で2回、20%食塩水(50ml)で3回洗浄し、硫酸ナトリウムで乾燥した。濾液の溶媒を留去して、3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアジド油状物を得て、そのまま次工程に移行させた。
前記4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアジド油状物をTHF(80ml)に溶解させ、水(1.19ml,66.1mmol)、トリフェニルホスフィン(1.91g,7.28mmol)を加えて70℃で1時間攪拌した。室温まで冷却後、溶媒を留去し、残渣をヘプタン(160ml)に溶解させ、50%アセトニトリル水溶液(50ml)で3回、20%食塩水(50ml)で2回洗浄し、硫酸ナトリウムで乾燥した。濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1→クロロホルム:メタノール:アンモニア水=100:10:1)で精製して3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン(5.31g,5.32mmol,収率80% vs.クロル体)を得た。
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.93(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.79(2H,s,benzyl-H),3.85-4.10(6H,m,-CH 2 -O-Ar),6.52(2H,s,C2,6-H).
1H-NMR(300MHz):δ0.85(36H,t,J=6.3Hz,Me),0.93(9H,t,J=6.3Hz,Me),1.00-2.00(72H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.79(2H,s,benzyl-H),3.85-4.10(6H,m,-CH 2 -O-Ar),6.52(2H,s,C2,6-H).
実施例8:3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコールの合成

2,3-ジヒドロフィチルブロミド(895mg,2.48mmol)、3,5-ジヒドロキシ安息香酸メチル(204mg,1.21mmol)、炭酸カリウム(513mg,3.71mmol)をDMF(10ml)に懸濁させ、100℃で7時間攪拌した。反応混合液を酢酸エチル(30ml)で抽出し、1N塩酸(10ml)で3回、20%食塩水(10ml)で洗浄し、硫酸ナトリウムで乾燥した後、濾液の溶媒を留去して、3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)安息香酸メチル(0.78g,収率92%)を得た。
前記3,4,5-ジ(2’,3’-ジヒドロフィチルオキシ)安息香酸メチル(0.70g,1.00mmol)をTHF(10ml)に溶解させ、窒素雰囲気下0℃で水素化アルミニウムリチウム(2.0mol/l THF溶液,1.2ml,2.4mmol)を滴下した。室温で5時間攪拌した後、0℃で水を滴下して反応を停止した。溶液を酢酸エチル(30ml)に溶解させ、1N塩酸(10ml)で3回、20%食塩水(20ml)で1回洗浄し、硫酸ナトリウムで乾燥した。濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサンのみ→ヘキサン:酢酸エチル=5:1)で精製して、3,4,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(0.61g,収率90%)を得た。
1H-NMR(300MHz):δ0.80-0.90(24H,m,Me),0.93(6H,d,J=6.3Hz,Me),1.00-1.90(48H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.92-4.02(4H,m,C19H39-CH 2 -O-Ar),4.62(2H,s,Ar-CH 2 -OH),6.38(1H,t,J=2.1Hz,C4-H),6.50(2H,d,J=2.0Hz,C2,6-H).
1H-NMR(300MHz):δ0.80-0.90(24H,m,Me),0.93(6H,d,J=6.3Hz,Me),1.00-1.90(48H,br,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.92-4.02(4H,m,C19H39-CH 2 -O-Ar),4.62(2H,s,Ar-CH 2 -OH),6.38(1H,t,J=2.1Hz,C4-H),6.50(2H,d,J=2.0Hz,C2,6-H).
実施例9:4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコールの合成

2,3-ジヒドロフィチルブロミド(600mg,1.66mmol)、4-ヒドロキシベンズアルデヒド(223mg,1.83mmol)、炭酸カリウム(344mg,2.49mmol)をDMF(6ml)中に懸濁し、60℃で3日間攪拌した。反応混合液を室温に冷却後、酢酸エチル(30ml)で抽出し、1N塩酸(6ml)で3回、5%炭酸水素ナトリウム水溶液(6ml)で3回、20%食塩水(6ml)で1回洗浄し、硫酸ナトリウムで乾燥した。濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=15:1→5:1)で精製して、4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルデヒド(640mg,収率100% vs. 2,3-ジヒドロフィチルブロミド)を得た。
1H-NMR(300MHz):δ0.82-0.89(12H,m,Me),0.95(3H,d,J=6.4Hz,Me),1.00-1.95(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),4.03-4.13(2H,m,-O-CH 2 -C19H39),4.62(2H,s,Ar-CH 2 -OH),6.99(2H,m,C3,5-H),7.83(2H,m,C2,6-H),9.88(1H,s,CHO).
1H-NMR(300MHz):δ0.82-0.89(12H,m,Me),0.95(3H,d,J=6.4Hz,Me),1.00-1.95(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),4.03-4.13(2H,m,-O-CH 2 -C19H39),4.62(2H,s,Ar-CH 2 -OH),6.99(2H,m,C3,5-H),7.83(2H,m,C2,6-H),9.88(1H,s,CHO).
前記4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルデヒド(640mg,1.66mmol)をTHF-メタノール混合溶液(7+0.3ml)に溶解させ、0℃で水素化ホウ素ナトリウム(110mg,90%,2.62mmol)を加え、室温で30分間攪拌した。反応混合液を0℃に冷却後、1N塩酸で反応を停止し、酢酸エチル(30ml)を加えて1N塩酸(5ml)で3回、5%炭酸水素ナトリウム水溶液(5ml)で3回、20%食塩水(5ml)で1回洗浄し、硫酸ナトリウムで乾燥した。濾液の溶媒を留去して、4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(619mg,1.53mmol,収率92% vs. 2,3-ジヒドロフィチルブロミド)を得た。
1H-NMR(300MHz):δ0.81-0.90(12H,m,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.94-4.05(2H,m,-O-CH 2 -C19H39),6.89(2H,m,C3,5-H),7.28(2H,m,C2,6-H).
1H-NMR(300MHz):δ0.81-0.90(12H,m,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.94-4.05(2H,m,-O-CH 2 -C19H39),6.89(2H,m,C3,5-H),7.28(2H,m,C2,6-H).
実施例10:4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミンの合成
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(619mg,1.53mmol)をクロロホルム(6ml)に溶解させ、塩化チオニル(167μl,2.29mmol)を加えて5時間攪拌した。反応終了後、溶媒を留去して、4-(2’,3’-ジヒドロフィチルオキシ)-ベンジルクロリドの油状物を得て、そのまま次工程に移行させた。
前記4-(2’,3’-ジヒドロフィチルオキシ)ベンジルクロリド(1.53mmol)をDMF-CHCl3混合溶媒(6+3ml)に溶解させ、アジ化ナトリウム(298mg,4.58mmol)を加えて70℃で一晩攪拌した。反応混合液を室温に冷却後、酢酸エチル(20ml)を加えて、水(10ml)で5回洗浄し、硫酸ナトリウムで乾燥した。濾液の溶媒を留去して、4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアジド(632mg,収率96% vs. 4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール)を得た。
1H-NMR(300MHz):δ0.81-0.90(12H,m,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.94-4.04(2H,m,-O-CH 2 -C19H39),4.26(2H,s,Ar-CH 2 -N3),6.90(2H,d,J=8.6Hz,C3,5-H),7.23(2H,d,J=8.6Hz,C2,6-H).
1H-NMR(300MHz):δ0.81-0.90(12H,m,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.94-4.04(2H,m,-O-CH 2 -C19H39),4.26(2H,s,Ar-CH 2 -N3),6.90(2H,d,J=8.6Hz,C3,5-H),7.23(2H,d,J=8.6Hz,C2,6-H).
前記4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアジド(632mg,1.47mmol)をTHF(6ml)に溶解させ、水(265μl,14.7mmol)、トリフェニルホスフィン(424mg,1.62mmol)を加えて70℃で一晩攪拌した。室温まで冷却後、溶媒を留去し、残渣をヘキサン(10ml)に溶解、50%アセトニトリル水溶液(5ml)で3回洗浄した。溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1→クロロホルム:メタノール:アンモニア水=50:5:1)で精製して、4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン(555mg,1.37mmol,収率94%)を得た。
1H-NMR(300MHz):δ0.86(12H,t,J=6.0Hz,Me),0.94(3H,d,J=6.6Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.80(2H,s,Ar-CH 2 -NH2),3.92-4.04(2H,m,-O-CH 2 -C19H39),6.87(2H,d,J=8.6Hz,C3,5-H),7.21(2H,d,J=8.6Hz,C2,6-H).
1H-NMR(300MHz):δ0.86(12H,t,J=6.0Hz,Me),0.94(3H,d,J=6.6Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.80(2H,s,Ar-CH 2 -NH2),3.92-4.04(2H,m,-O-CH 2 -C19H39),6.87(2H,d,J=8.6Hz,C3,5-H),7.21(2H,d,J=8.6Hz,C2,6-H).
実施例11:2-メトキシ-4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミンの合成

2,3-ジヒドロフィチルブロミド(2.00g,5.53mmol)、2-メトキシ-4-ヒドロキシベンズアルデヒド(884mg,5.81mmol)、炭酸カリウム(1.15g,8.32mmol)をDMF(20ml)中に懸濁し、80℃で一晩攪拌した。反応混合液を室温に冷却後、酢酸エチル(50ml)で抽出し、1N塩酸(20ml)で3回、5%炭酸水素ナトリウム水溶液(20ml)で3回、20%食塩水(20ml)で1回洗浄し、硫酸ナトリウムで乾燥した。濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=20:1)で精製して、2-メトキシ-4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルデヒド油状物を得て、次工程に移行させた。
前記2-メトキシ-4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルデヒド、ヒドロキシルアミン塩酸塩(1.15g,16.5mmol)をジクロロメタン(25ml)に懸濁し、0℃でトリエチルアミン(3.84ml,27.7mmol)を加えて室温で3時間攪拌した。反応混合液にクロロホルム(30ml)を加え1N塩酸(15ml)で3回、5%炭酸水素ナトリウム水溶液(15ml)で3回、20%食塩水(15ml)で1回洗浄し、溶媒を留去して、2-メトキシ-4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルドキシムを得、NMRで構造を確認後、次工程に移行させた。
1H-NMR(300MHz):δ0.82-0.92(12H,m,Me),0.95(3H,d,J=6.4Hz,Me),1.00-1.95(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.83(3H,s,OMe),3.97-4.10(2H,m,-O-CH 2 -C19H39),6.44(1H,d,J=2.2Hz,C3-H),6.49(1H,dd,J=2.2,8.6Hz,C5-H),7.15(1H,s,-CHNOH),7.62(1H,d,J=8.6Hz,C6-H),8.41(1H,s,-CHNOH).
1H-NMR(300MHz):δ0.82-0.92(12H,m,Me),0.95(3H,d,J=6.4Hz,Me),1.00-1.95(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.83(3H,s,OMe),3.97-4.10(2H,m,-O-CH 2 -C19H39),6.44(1H,d,J=2.2Hz,C3-H),6.49(1H,dd,J=2.2,8.6Hz,C5-H),7.15(1H,s,-CHNOH),7.62(1H,d,J=8.6Hz,C6-H),8.41(1H,s,-CHNOH).
前記2-メトキシ-4-(2’,3’-ジヒドロフィチルオキシ)ベンズアルドキシムをメタノール-THF混合溶媒(20+10ml)に溶解させ、10%パラジウム-炭素(K)(200mg)を加えて水素雰囲気下室温で一晩攪拌した。濾液の溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:アンモニア水=100:10:1)で精製して、2-メトキシ-4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン(1.87g,4.31mmol,収率78% vs. 2,3-ジヒドロフィチルブロミド)を得た。
1H-NMR(300MHz):δ0.80-0.90(12H,m,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.74(2H,s,Ar-CH 2 -NH2),3.82(3H,s,OMe),3.90-4.05(2H,m,-O-CH 2 -C19H39),6.42(1H,dd,J=2.3,8.1Hz,C5-H),6.46(1H,d,J=2.1Hz,C3-H),7.09(1H,d,J=8.1Hz,C6-H).
1H-NMR(300MHz):δ0.80-0.90(12H,m,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),3.74(2H,s,Ar-CH 2 -NH2),3.82(3H,s,OMe),3.90-4.05(2H,m,-O-CH 2 -C19H39),6.42(1H,dd,J=2.3,8.1Hz,C5-H),6.46(1H,d,J=2.1Hz,C3-H),7.09(1H,d,J=8.1Hz,C6-H).
実施例12:4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコールの合成

0℃で、メタノール(10ml)に塩化チオニル(1.92ml,26.3mmol)を滴下し、4-ヒドロキシ-2-メチル安息香酸(2.00g,13.1mmol)を加えて、60℃で一晩攪拌した。反応終了後、溶媒を留去し、残渣を酢酸エチル(20ml)に溶解させて5%炭酸水素ナトリウム水溶液(10ml)で2回、1N塩酸(10ml)で1回、水(10ml)で1回洗浄し、溶媒を留去して、4-ヒドロキシ-2-メチル安息香酸メチル(2.24g,収率100%)を得た。
1H-NMR(300MHz):δ2.57(3H,s,C2-Me),3.86(3H,s,-COOMe),5.68(1H,s,br,-OH),6.66-6.72(2H,m,C3,5-H),7.89(1H,dd,J=2.4,6.9Hz,C6-H).
1H-NMR(300MHz):δ2.57(3H,s,C2-Me),3.86(3H,s,-COOMe),5.68(1H,s,br,-OH),6.66-6.72(2H,m,C3,5-H),7.89(1H,dd,J=2.4,6.9Hz,C6-H).
前記4-ヒドロキシ-2-メチル安息香酸メチル(269mg,1.62mmol)、2,3-ジヒドロフィチルブロミド(389mg,1.08mmol)、炭酸カリウム(297mg,2.15mmol)をDMF(5ml)中に懸濁し、90℃で5時間攪拌した。反応混合液を室温に冷却後、ヘキサン-酢酸エチル(10+10ml)で抽出し、1N塩酸(15ml)で1回、水(10ml)で2回洗浄し、溶媒を留去して、4-(2’,3’-ジヒドロフィチルオキシ)-2-メチル安息香酸メチルを得、NMRで構造を確認後、次工程に移行させた。
1H-NMR(300MHz):δ0.85(12H,t,J=6.6Hz,Me),0.94(3H,d,J=6.6Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.59(3H,s,C2-Me),3.85(3H,s,-COOMe),4.02(2H,dt,J=3.0,6.7Hz,O-CH 2 -C19H39),6.65-6.76(2H,m,C3,5-H),7.85-7.96(1H,m,C6-H).
1H-NMR(300MHz):δ0.85(12H,t,J=6.6Hz,Me),0.94(3H,d,J=6.6Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.59(3H,s,C2-Me),3.85(3H,s,-COOMe),4.02(2H,dt,J=3.0,6.7Hz,O-CH 2 -C19H39),6.65-6.76(2H,m,C3,5-H),7.85-7.96(1H,m,C6-H).
前記4-(2’,3’-ジヒドロフィチルオキシ)-2-メチル安息香酸メチル(1.08mmol)をTHF(6ml)に溶解させ、DIBAL(1.0M,4.9ml,4.9mmol)を加え、室温で100分間攪拌した。反応混合液を0℃に冷却後、1N塩酸(15ml)で反応を停止し、ヘキサン(10ml)、酢酸エチル(10ml)を加えて分液し、0.5N塩酸(10ml)で1回、水(10ml)で1回洗浄し、溶媒を留去して、4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコールを得た。
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.36(3H,s,C2-Me),3.98(2H,dt,J=3.0,6.7Hz,O-CH 2 -C19H39),4.63(2H,d,J=4.7Hz,Ar-CH 2 -OH),6.60-6.77(2H,m,C3,5-H),7.15-7.25(1H,m,C6-H).
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.94(3H,d,J=6.4Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.36(3H,s,C2-Me),3.98(2H,dt,J=3.0,6.7Hz,O-CH 2 -C19H39),4.63(2H,d,J=4.7Hz,Ar-CH 2 -OH),6.60-6.77(2H,m,C3,5-H),7.15-7.25(1H,m,C6-H).
実施例13:4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミンの合成
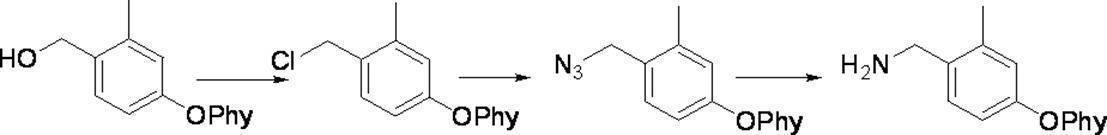
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチル-ベンジルアルコール(1.08mmol)をクロロホルム(8ml)に溶解させ、チオニルクロリド(393μl,5.38mmol)を加えて50℃で4.5時間攪拌した。反応終了後、溶媒を留去して、4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルクロリドを得、NMRで構造を確認後、次工程に移行させた。
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.93(3H,d,J=6.3Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.40(3H,s,C2-Me),3.97(2H,dt,J=2.7,6.7Hz,O-CH 2 -C19H39),4.59(2H,s,Ar-CH 2 -Cl),6.69(1H,dd,J=2.4,8.3Hz,C5-H),6.74(1H,d,J=2.3Hz,C3-H),7.21(1H,d,J=8.3Hz,C6-H).
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.93(3H,d,J=6.3Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.40(3H,s,C2-Me),3.97(2H,dt,J=2.7,6.7Hz,O-CH 2 -C19H39),4.59(2H,s,Ar-CH 2 -Cl),6.69(1H,dd,J=2.4,8.3Hz,C5-H),6.74(1H,d,J=2.3Hz,C3-H),7.21(1H,d,J=8.3Hz,C6-H).
前記4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルクロリド(1.08mmol)をDMF(6ml)に溶解させ、アジ化ナトリウム(350mg,5.38mmol)を加えて70℃で一晩攪拌した。反応混合液を室温に冷却後、ヘキサン(10ml)、酢酸エチル(5ml)を加えて、水(10ml)で3回洗浄した。濾液の溶媒を留去して、4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアジドを得、NMRで構造を確認後、次工程に移行させた。
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.94(3H,d,J=6.3Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.34(3H,s,C2-Me),3.98(2H,dt,J=3.0,6.6Hz,O-CH 2 -C19H39),4.28(2H,s,Ar-CH 2 -N3),6.71(1H,dd,J=2.6,8.2Hz,C5-H),6.77(1H,d,J=2.3Hz,C3-H),7.15(1H,d,J=8.3Hz,C6-H).
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.94(3H,d,J=6.3Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.34(3H,s,C2-Me),3.98(2H,dt,J=3.0,6.6Hz,O-CH 2 -C19H39),4.28(2H,s,Ar-CH 2 -N3),6.71(1H,dd,J=2.6,8.2Hz,C5-H),6.77(1H,d,J=2.3Hz,C3-H),7.15(1H,d,J=8.3Hz,C6-H).
前記4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアジド(1.08mmol)をTHF(10ml)に溶解させ、水(2ml)、トリフェニルホスフィン(565mg,2.15mmol)を加えて60℃で3時間攪拌した。室温まで冷却後、溶媒を留去し、残渣をヘプタン(10ml)に溶解させ、50%アセトニトリル水溶液(10ml)で3回洗浄した。溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=5:1→クロロホルム:メタノール:アンモニア水=50:5:1)で精製して、4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン(281mg,0.67mmol,収率62% vs. 2,3-ジヒドロフィチルブロミド)を得た。
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.93(3H,d,J=6.6Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.32(3H,s,C2-Me),3.79(2H,s,Ar-CH 2 -NH2),3.97(2H,dt,J=3.0,6.7Hz,O-CH 2 -C19H39),6.71(1H,dd,J=2.6,8.2Hz,C5-H),6.68-6.75(2H,br,C3,5-H),7.17(1H,d,J=8.7Hz,C6-H).
1H-NMR(300MHz):δ0.86(12H,t,J=6.3Hz,Me),0.93(3H,d,J=6.6Hz,Me),1.00-1.90(24H,m,Me2CH-[C3 H 6 -CHMe]3-CH 2 CH2-O-Ar),2.32(3H,s,C2-Me),3.79(2H,s,Ar-CH 2 -NH2),3.97(2H,dt,J=3.0,6.7Hz,O-CH 2 -C19H39),6.71(1H,dd,J=2.6,8.2Hz,C5-H),6.68-6.75(2H,br,C3,5-H),7.17(1H,d,J=8.7Hz,C6-H).
実施例14:2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミドの合成
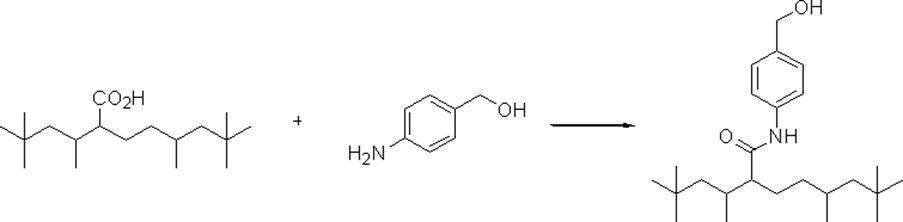
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(2.81g,9.88mmol)、4-アミノベンジルアルコール(1.00g,8.12mmol)、3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン(HOOBt)(133mg,0.812mmol)を、クロロホルム(10ml)に懸濁させて、0℃でEDC・HCl(2.05g,10.7mmol)加え、室温で一晩攪拌した。溶媒を除去し、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1)で精製して、2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド(2.69g,6.67mmol,収率82%)を得た。
1H-NMR(300MHz):δ0.92-0.99(m,24H),1.01-1.09(m,6H),1.19-1.23(m,4H),4.58(s,2H),7.21(d,2H,J=6Hz),7.43(d,2H,J=9Hz),7.53-7.66(b,1H).
1H-NMR(300MHz):δ0.92-0.99(m,24H),1.01-1.09(m,6H),1.19-1.23(m,4H),4.58(s,2H),7.21(d,2H,J=6Hz),7.43(d,2H,J=9Hz),7.53-7.66(b,1H).
実施例15:4-(3’,7’,11’-トリメチルドデシルオキシ)ベンジルアルコールの合成

参考例4で得られた1-ブロモ-3,7,11-トリメチルドデカン(1.00g,3.43mmol)をDMF(5ml)に溶解させ、4-ヒドロキシベンジルアルコール(0.85g,6.85mmol)と炭酸カリウム(1.42g,10.3mmol)を加えて120℃で一晩攪拌した。反応混合液を室温に冷却後、クロロホルム(50ml)で抽出し、1N塩酸(30ml)で3回、5%炭酸水素ナトリウム水溶液(30ml)で1回、精製水(30ml)で1回洗浄した。有機層の溶媒を留去して、4-(3’,7’,11’-トリメチルドデシルオキシ)ベンジルアルコール(1.09g,3.26mmol,収率95% vs. 1-ブロモ-3,7,11-トリメチルドデカン)を得た。
1H-NMR(300MHz):δ0.81-0.89(m,12H),1.08-1.37(m,12H),1.48-1.83(m,5H),3.96-4.02(m,2H),4.62(s,2H),6.89(d,2H,J=9Hz),7.29(d,2H,J=9Hz).
1H-NMR(300MHz):δ0.81-0.89(m,12H),1.08-1.37(m,12H),1.48-1.83(m,5H),3.96-4.02(m,2H),4.62(s,2H),6.89(d,2H,J=9Hz),7.29(d,2H,J=9Hz).
実施例16:2-(3’,7’,11’-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オールの合成

参考例4で得られた1-ブロモ-3,7,11-トリメチルドデカン(780mg,2.68mmol)をDMF(5ml)に溶解させ、2-ヒドロキシ-9-フルオレノン(1.05g,5.35mmol)と炭酸カリウム(1.06g,7.67mmol)を加えて100℃で5時間攪拌した。反応混合液を室温に冷却後、ヘプタン(30ml)で抽出し、1N塩酸(15ml)で3回、5%炭酸水素ナトリウム水溶液(15ml)で1回、メタノール(15ml)で3回洗浄した。有機層の溶媒を留去して、2-(3’,7’,11’-トリメチルドデシルオキシ)-9-フルオレノン(650mg,1.60mmol,収率60% vs. 1-ブロモ-3,7,11-トリメチルドデカン)を得た。
1H-NMR(300MHz):δ0.84-0.96(m,12H),1.08-1.30(m,12H),1.50-1.70(m,5H),4.04-4.07(m,2H),6.98(d,1H,J=9Hz),7.17-7.22(m,1H),7.28-7.30(m,1H),7.38-7.44(m,3H),7.60(d,1H,J=9Hz).
1H-NMR(300MHz):δ0.84-0.96(m,12H),1.08-1.30(m,12H),1.50-1.70(m,5H),4.04-4.07(m,2H),6.98(d,1H,J=9Hz),7.17-7.22(m,1H),7.28-7.30(m,1H),7.38-7.44(m,3H),7.60(d,1H,J=9Hz).
窒素雰囲気下、マグネシウム(280mg,11.5mmol)を脱水THF(2ml)に懸濁し、ヨウ素(45mg,0.18mmol)を加えた後、ブロモベンゼン(390μl,3.70mmol)をゆっくり滴下してから、40℃で4時間攪拌した。反応混合液に前記2-(3’,7’,11’-トリメチルドデシルオキシ)-9-フルオレノン(300mg,0.74mmol)を加え、50℃で一晩攪拌した。反応混合液を室温に冷却後、1N塩酸(30ml)で反応を停止した。クロロホルム(40ml)で抽出し、1N塩酸(20ml)で3回、5%炭酸水素ナトリウム水溶液(20ml)で3回、20%食塩水(20ml)で1回洗浄した。有機層の溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィーで分離・精製し(ヘキサン:酢酸エチル=10:1)、2-(3’,7’,11’-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール(181mg,0.37mmol,収率51%)を得た。
1H-NMR(300MHz):δ0.86-0.92(m,12H),1.04-1.25(m,12H),1.50-1.59(m,5H),2.49(s,1H),3.92-3.96(m,2H),6.80-6.91(m,3H),7.16-40(m,7H),7.52-7.63(m,2H).
1H-NMR(300MHz):δ0.86-0.92(m,12H),1.04-1.25(m,12H),1.50-1.59(m,5H),2.49(s,1H),3.92-3.96(m,2H),6.80-6.91(m,3H),7.16-40(m,7H),7.52-7.63(m,2H).
実施例17:2-(3’,7’,11’-トリメチルドデシルオキシ)-9-ブロモ-9-フェニルフルオレンの合成
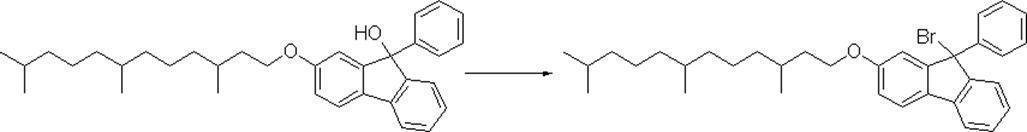
前記2-(3’,7’,11’-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール(181mg,0.37mmol)をクロロホルム(2ml)に溶解させ、臭化アセチル(55μl,0.74mmol)を滴下して6時間攪拌した。反応終了後溶媒を留去し、残渣をヘプタンに溶解させ、アセトニトリルで3回洗浄し、ヘプタン層の溶媒を留去し、2-(3’,7’,11’-トリメチルドデシルオキシ)-9-ブロモ-9-フェニルフルオレン(126mg,0.23mmol,収率62%)を得た。
1H-NMR(300MHz):δ0.83-0.94(m,12H),1.09-1.36(m,12H),1.50-1.59(m,5H),3.96-4.01(m,2H),6.90-6.93(m,1H),7.03(s,1H),7.21-7.35(m,5H),7.44-7.48(m,2H),7.54-7.58(m,3H).
1H-NMR(300MHz):δ0.83-0.94(m,12H),1.09-1.36(m,12H),1.50-1.59(m,5H),3.96-4.01(m,2H),6.90-6.93(m,1H),7.03(s,1H),7.21-7.35(m,5H),7.44-7.48(m,2H),7.54-7.58(m,3H).
実施例18:GI-1000(日本曹達株式会社製)のジ(2-ヒドロキシメチル-5-メトキシフェノキシ)体(GI-1000(2-ヒドロキシメチル-5-メトキシフェノキシ))の合成

参考例5で得られたGI-1000(Br)(1.70g)をDMF(20ml)に溶解させ、2-ヒドロキシ-4-メトキシベンズアルデヒド(1.00g,6.57mmol)と炭酸カリウム(1.17g,8.47mmol)を加えて120℃で一晩攪拌した。反応混合液を室温に冷却後、酢酸エチル/ヘキサン(1:1,20ml)で抽出し、1N塩酸(10ml)で3回、5%炭酸水素ナトリウム水溶液(30ml)で1回、精製水(30ml)で1回洗浄した。有機層の溶媒を留去し、メタノールでデカントし、濃縮して、GI-1000(2-ホルミル-5-メトキシフェノキシ)(1.60g)を得た。
1H-NMR(300MHz):δ0.98-1.49(m),1.82-1.87(m,4H),3.87(s,6H),4.01-4.06(m,4H),6.43(s,2H),6.53(d,2H,J=7.8Hz),7.82(d,2H,J=7.8Hz),10.34(s,2H)
1H-NMR(300MHz):δ0.98-1.49(m),1.82-1.87(m,4H),3.87(s,6H),4.01-4.06(m,4H),6.43(s,2H),6.53(d,2H,J=7.8Hz),7.82(d,2H,J=7.8Hz),10.34(s,2H)
前記GI-1000(2-ホルミル-5-メトキシフェノキシ)(1.60g)をクロロホルム(16ml)/メタノール(1.6ml)混合溶液に溶解させ、水素化ホウ素ナトリウム(0.38g,9.99mmol)を加えて60℃で2時間半攪拌した。反応混合液を室温に冷却後、1N塩酸(16ml)で3回、5%炭酸水素ナトリウム水溶液(16ml)で3回、20%食塩水(16ml)で1回洗浄した。有機層の溶媒を留去し、メタノールでデカントし、濃縮して、GI-1000(2-ヒドロキシメチル-5-メトキシフェノキシ)(1.84g)を得た。
1H-NMR(300MHz):δ1.06-1.43(m),1.71-1.83(m,4H),3.80(s,6H),3.98-4.01(m,4H),4.61(s,2H),6.42-6.45(m,4H),7.16(d,2H,J=7.9Hz).
1H-NMR(300MHz):δ1.06-1.43(m),1.71-1.83(m,4H),3.80(s,6H),3.98-4.01(m,4H),4.61(s,2H),6.42-6.45(m,4H),7.16(d,2H,J=7.9Hz).
実施例19:GI-1000(日本曹達株式会社製)のジ(3-ヒドロキシメチルフェノキシ)体(Bzl(3-O-GI-1000)-OH)の合成
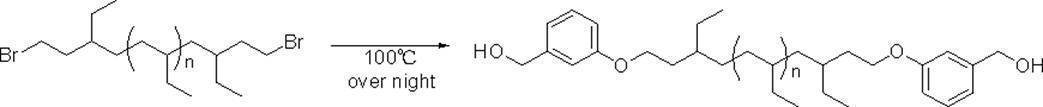
参考例5で得られたGI-1000(Br)(563mg)をDMF(5ml)に溶解させ、3-ヒドロキシベンジルアルコール(280mg,2.26mmol)と炭酸カリウム(389mg,2.81mmol)を加えて100℃で一晩攪拌した。反応混合液を室温に冷却後、酢酸エチル/ヘキサン(1:1,25ml)で抽出し、1N塩酸(10ml)で3回、5%炭酸水素ナトリウム水溶液(10ml)で1回、メタノール(10ml)で1回洗浄した。有機層の溶媒を留去して、Bzl(3-O-GI-1000)-OH(580mg)を得た。
1H-NMR(300MHz):δ0.85-1.28(m),3.94-3.98(m,4H),4,67(s,4H),6.83(d,2H,J=9Hz),6.91-6,93(m,4H),7.23-7.26(m,2H).
1H-NMR(300MHz):δ0.85-1.28(m),3.94-3.98(m,4H),4,67(s,4H),6.83(d,2H,J=9Hz),6.91-6,93(m,4H),7.23-7.26(m,2H).
実施例20:GI-2000(日本曹達株式会社製)のジ(3-ヒドロキシメチルフェノキシ)体(Bzl(3-O-GI-2000)-OH)の合成
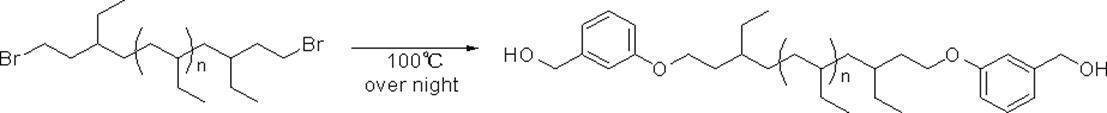
参考例6で得られたGI-2000(Br)(503mg)をDMF(5ml)に溶解させ、3-ヒドロキシベンジルアルコール(125mg,1.00mmol)と炭酸カリウム(174mg,1.26mmol)を加えて100℃で一晩攪拌した。反応混合液を室温に冷却後、酢酸エチル/ヘキサン(1:1,25ml)で抽出し、1N塩酸(10ml)で3回、5%炭酸水素ナトリウム水溶液(10ml)で1回、メタノール(10ml)で1回洗浄した。有機層の溶媒を留去して、Bzl(3-O-GI-2000)-OH(512mg)を得た。
1H-NMR(300MHz):δ0.88-1.31(m),3.94-4.01(m,4H),4,67(s,4H),6.83(d,2H,J=9Hz),6.91-6.93(m,4H),7.23-7.28(m,2H).
1H-NMR(300MHz):δ0.88-1.31(m),3.94-4.01(m,4H),4,67(s,4H),6.83(d,2H,J=9Hz),6.91-6.93(m,4H),7.23-7.28(m,2H).
実施例21:3-ヒドロキシメチルフェノールのTERGITOL化体の合成

参考例7で得られたTERGITOL(登録商標) TMN-6のBr体(5.21g)、炭酸カリウム(2.93g,21.2mmol,2.5eq)、3-ヒドロキシ-ベンジルアルコール(1.16g,9.32mmol,1.1eq)をDMF(50ml)に懸濁させ、80℃で一晩攪拌した。反応液を酢酸エチル(500ml)で抽出し、0.5N塩酸(250ml)で3回、5%炭酸水素ナトリウム水溶液(250ml)で3回、20%食塩水(250ml)で1回分液洗浄を行った。溶媒を除去し、シリカゲルカラムクロマトグラフィー(酢酸エチル→酢酸エチル:メタノール=1:1)で精製した。溶媒を除去し、酢酸エチル(100ml)で溶解させ、濾過でシリカゲルを除去してBzl(3-TERGITOL)-OH(3.93g)を得た。
1H-NMR(300MHz):δ0.80-0.90(m,15H,CH3),1.00-1.60(m,11H,CH,CH2),3.50-3.68(m,(OCH 2 CH 2 ) n ),4.55-4.59.(s,2H,CH 2 -OH),6.70-6.79(m,2H,C2,C4-H),6.87(s,1H,C6-H),7.08-7.13(t,1H,C5-H).
13C-NMR:δ20.42-27.39(CH3,CH),42.78-47.67(CH2),68.04(OCH2,CH2OH),70.79-71.55(OCH 2 CH 2 O),71.07(CH 2 O-BzlOH),114.21(C4),114.62(C2),118.19(C6),129.47(C5),143.19(C1),157.29(C3).
1H-NMR(300MHz):δ0.80-0.90(m,15H,CH3),1.00-1.60(m,11H,CH,CH2),3.50-3.68(m,(OCH 2 CH 2 ) n ),4.55-4.59.(s,2H,CH 2 -OH),6.70-6.79(m,2H,C2,C4-H),6.87(s,1H,C6-H),7.08-7.13(t,1H,C5-H).
13C-NMR:δ20.42-27.39(CH3,CH),42.78-47.67(CH2),68.04(OCH2,CH2OH),70.79-71.55(OCH 2 CH 2 O),71.07(CH 2 O-BzlOH),114.21(C4),114.62(C2),118.19(C6),129.47(C5),143.19(C1),157.29(C3).
実施例22:サーフォナミンB-30(三井化学ファイン社製またはハンツマンコーポレーション製)(数平均分子量:約325;n=約2)の(4-ヒドロキシメチル)カルボキサミド体の合成
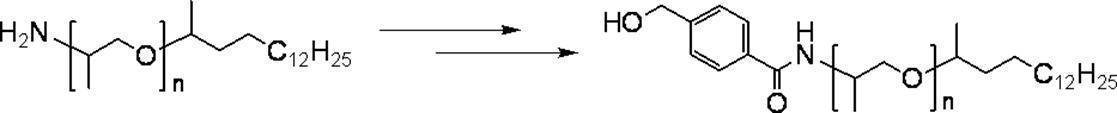
サーフォナミンB-30(2.0g)をクロロホルム(20ml)に溶解させ、水(10ml)で3回洗浄して有機層を硫酸ナトリウムで乾燥させて、濃縮乾固した。残査をクロロホルム(20ml)にて溶解させ、テレフタル酸モノメチル(1.0g)を加えて、EDC・HCl(1.15g)とHOBt(72mg)を加えて室温下、3時間反応させた後、炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄し、濃縮して油状物を得た。該油状物にテトラヒドロフランを加えて、氷冷下で1M DIBALトルエン溶液を4.2当量加えて反応させた。1M塩酸を加えて反応を停止させ、クロロホルムを加えて、1M塩酸、炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄し、ベンジルアルコール体(1.30g)を得た。
実施例1、3、5、19、20の化合物を保護化試薬として使用し、ペプチド合成を行った。
実施例23:2,4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコールとFmoc-Ser(tBu)-OHとの縮合によるFmoc-Ser(tBu)-OBzl(2,4-OPhy)の合成
Fmoc-Ser(tBu)-OH(3.28g,8.56mmol)を酢酸イソプロピル(30ml)に溶解させ、0℃でEDC・HCl(1.80g,9.39mmol)を加え、室温で30分攪拌後、実施例1で得られた2,4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(3.00g,4.28mmol)の酢酸イソプロピル(15ml)溶液、N,N-ジメチル-4-アミノピリジン(DMAP)(105mg,0.86mmol)を加えて一晩攪拌した。反応混合液を0.1N塩酸(15ml)で3回、5%炭酸水素ナトリウム水溶液(15ml)で3回、20%食塩水(15ml)で1回洗浄することにより、反応系中で縮合体(Fmoc-Ser(tBu)-OBzl(2,4-OPhy))へと変換し、続く反応に用いた。
Fmoc-Ser(tBu)-OH(3.28g,8.56mmol)を酢酸イソプロピル(30ml)に溶解させ、0℃でEDC・HCl(1.80g,9.39mmol)を加え、室温で30分攪拌後、実施例1で得られた2,4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(3.00g,4.28mmol)の酢酸イソプロピル(15ml)溶液、N,N-ジメチル-4-アミノピリジン(DMAP)(105mg,0.86mmol)を加えて一晩攪拌した。反応混合液を0.1N塩酸(15ml)で3回、5%炭酸水素ナトリウム水溶液(15ml)で3回、20%食塩水(15ml)で1回洗浄することにより、反応系中で縮合体(Fmoc-Ser(tBu)-OBzl(2,4-OPhy))へと変換し、続く反応に用いた。
実施例24:Fmoc-Ser(tBu)-OBzl(2,4-OPhy)の脱FmocによるH-Ser(tBu)-OBzl(2,4-OPhy)の合成
Fmoc-Ser(tBu)-OBzl(2,4-OPhy)(4.28mmol)の酢酸イソプロピル溶液(45ml)に、0℃で4-アミノメチルピペリジン(1.47g,12.9mmol)を加え、室温で4時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液をpH=5.5に調整した1Mリン酸ナトリウム水溶液(30ml)で3回、10%炭酸ナトリウム水溶液(30ml)で1回、20%食塩水(30ml)で1回洗浄することにより、反応系中で脱Fmoc体(H-Ser(tBu)-OBzl(2,4-OPhy))へと変換し、続く反応に用いた。
Fmoc-Ser(tBu)-OBzl(2,4-OPhy)(4.28mmol)の酢酸イソプロピル溶液(45ml)に、0℃で4-アミノメチルピペリジン(1.47g,12.9mmol)を加え、室温で4時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液をpH=5.5に調整した1Mリン酸ナトリウム水溶液(30ml)で3回、10%炭酸ナトリウム水溶液(30ml)で1回、20%食塩水(30ml)で1回洗浄することにより、反応系中で脱Fmoc体(H-Ser(tBu)-OBzl(2,4-OPhy))へと変換し、続く反応に用いた。
実施例25:H-Ser(tBu)-OBzl(2,4-OPhy)とFmoc-Lys(Boc)-OHとの縮合によるFmoc-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の合成
H-Ser(tBu)-OBzl(2,4-OPhy)(4.18mmol)の酢酸イソプロピル溶液(55ml)にHOBt(57mg,0.42mmol)、Fmoc-Lys(Boc)-OH(2.16g,4.61mmol)を溶解させ、0℃でEDC・HCl(970mg,5.06mmol)を加え、室温で一晩攪拌した。反応混合液を0.1N塩酸(30ml)で3回、5%炭酸水素ナトリウム水溶液(30ml)で3回、20%食塩水(30ml)で1回洗浄することにより、反応系中でFmoc-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
H-Ser(tBu)-OBzl(2,4-OPhy)(4.18mmol)の酢酸イソプロピル溶液(55ml)にHOBt(57mg,0.42mmol)、Fmoc-Lys(Boc)-OH(2.16g,4.61mmol)を溶解させ、0℃でEDC・HCl(970mg,5.06mmol)を加え、室温で一晩攪拌した。反応混合液を0.1N塩酸(30ml)で3回、5%炭酸水素ナトリウム水溶液(30ml)で3回、20%食塩水(30ml)で1回洗浄することにより、反応系中でFmoc-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
実施例26:Fmoc-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の脱FmocによるH-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の合成
Fmoc-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)(4.18mmol)の酢酸イソプロピル溶液(55ml)に、0℃で4-アミノメチルピペリジン(1.91g,16.7mmol)を加え、室温で4時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液を、pH=5.5のリン酸ナトリウム1M水溶液(30ml)で3回、10%炭酸ナトリウム水溶液(30ml)で1回、20%食塩水(30ml)で1回洗浄することにより、反応系中でH-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
Fmoc-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)(4.18mmol)の酢酸イソプロピル溶液(55ml)に、0℃で4-アミノメチルピペリジン(1.91g,16.7mmol)を加え、室温で4時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液を、pH=5.5のリン酸ナトリウム1M水溶液(30ml)で3回、10%炭酸ナトリウム水溶液(30ml)で1回、20%食塩水(30ml)で1回洗浄することにより、反応系中でH-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
実施例27:H-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)とFmoc-Glu(OtBu)-OHとの縮合によるFmoc-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の合成
H-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)(4.18mmol)の酢酸イソプロピル溶液(105ml)にHOBt(57mg,0.42mmol)、Fmoc-Glu(OtBu)-OH(2.32g,5.23mmol)を溶解させ、0℃でEDC・HCl(1.09g,5.69mmol)を加え、室温で一晩攪拌した。反応混合液を0.1N塩酸(30ml)で3回、5%炭酸水素ナトリウム水溶液(30ml)で3回、20%食塩水(30ml)で1回洗浄することにより、反応系中でFmoc-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
H-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)(4.18mmol)の酢酸イソプロピル溶液(105ml)にHOBt(57mg,0.42mmol)、Fmoc-Glu(OtBu)-OH(2.32g,5.23mmol)を溶解させ、0℃でEDC・HCl(1.09g,5.69mmol)を加え、室温で一晩攪拌した。反応混合液を0.1N塩酸(30ml)で3回、5%炭酸水素ナトリウム水溶液(30ml)で3回、20%食塩水(30ml)で1回洗浄することにより、反応系中でFmoc-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
実施例28:Fmoc-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の脱FmocによるH-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の合成
Fmoc-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の酢酸イソプロピル溶液(50ml)に、0℃で4-アミノメチルピペリジン(1.43g,12.5mmol)を加え、室温で4時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液を、pH=6.86のリン酸ナトリウム1M水溶液(20ml)で3回、10%炭酸ナトリウム水溶液(20ml)で1回、20%食塩水(20ml)で1回洗浄することにより、反応系中でH-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
Fmoc-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の酢酸イソプロピル溶液(50ml)に、0℃で4-アミノメチルピペリジン(1.43g,12.5mmol)を加え、室温で4時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液を、pH=6.86のリン酸ナトリウム1M水溶液(20ml)で3回、10%炭酸ナトリウム水溶液(20ml)で1回、20%食塩水(20ml)で1回洗浄することにより、反応系中でH-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
実施例29:H-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)とFmoc-Ala-OHとの縮合によるFmoc-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の合成
H-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の酢酸イソプロピル溶液(150ml)にHOBt(57mg,0.42mmol)、Fmoc-Ala(1.52g,4.62mmol)を溶解させ、0℃でEDC・HCl(970mg,5.06mmol)を加え、室温で一晩攪拌した。反応混合液を0.1N塩酸(30ml)で3回、5%炭酸ナトリウム水溶液(30ml)で3回、20%食塩水(30ml)で1回洗浄することにより、反応系中でFmoc-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
H-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の酢酸イソプロピル溶液(150ml)にHOBt(57mg,0.42mmol)、Fmoc-Ala(1.52g,4.62mmol)を溶解させ、0℃でEDC・HCl(970mg,5.06mmol)を加え、室温で一晩攪拌した。反応混合液を0.1N塩酸(30ml)で3回、5%炭酸ナトリウム水溶液(30ml)で3回、20%食塩水(30ml)で1回洗浄することにより、反応系中でFmoc-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)へと変換し、続く反応に用いた。
実施例30:Fmoc-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の脱FmocによるH-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の合成
Fmoc-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の酢酸イソプロピル溶液(100ml)に、0℃で4-アミノメチルピペリジン(1.43g,12.5mmol)を加え、室温で3時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液を、pH=6.86の1Mリン酸ナトリウム水溶液(20ml)で3回、10%炭酸ナトリウム水溶液(20ml)で1回、20%食塩水(20ml)で1回洗浄し、濃縮することにより、H-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)を油状物として得た。
Fmoc-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の酢酸イソプロピル溶液(100ml)に、0℃で4-アミノメチルピペリジン(1.43g,12.5mmol)を加え、室温で3時間攪拌した。反応混合液に二酸化炭素を通気し、生成した炭酸塩を濾過により除去し、濾液を、pH=6.86の1Mリン酸ナトリウム水溶液(20ml)で3回、10%炭酸ナトリウム水溶液(20ml)で1回、20%食塩水(20ml)で1回洗浄し、濃縮することにより、H-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)を油状物として得た。
実施例31:H-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の脱保護によるH-Ala-Glu-Lys-Ser-OH・2TFA塩の合成
実施例30で得られたH-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の油状物の全重量の1/20をクロロホルム(10ml)に溶解させ、濃縮し、残留する酢酸イソプロピルを留去した。その後に氷冷下でトリフルオロ酢酸(TFA):H2O:トリイソプロピルシランの95:2.5:2.5にした溶液中で脱保護し、反応完結後に濃縮し、残査にジエチルエーテル(5ml)を加えて攪拌し、沈殿物を濾過して乾燥することにより、H-Ala-Glu-Lys-Ser-OH・2TFA塩を101mg取得した。これは、全9工程を通して、収率71%(vs. HOBzl(2,4-OPhy))に相当する。
ESIMS MH+ 434.0
実施例30で得られたH-Ala-Glu(OtBu)-Lys(Boc)-Ser(tBu)-OBzl(2,4-OPhy)の油状物の全重量の1/20をクロロホルム(10ml)に溶解させ、濃縮し、残留する酢酸イソプロピルを留去した。その後に氷冷下でトリフルオロ酢酸(TFA):H2O:トリイソプロピルシランの95:2.5:2.5にした溶液中で脱保護し、反応完結後に濃縮し、残査にジエチルエーテル(5ml)を加えて攪拌し、沈殿物を濾過して乾燥することにより、H-Ala-Glu-Lys-Ser-OH・2TFA塩を101mg取得した。これは、全9工程を通して、収率71%(vs. HOBzl(2,4-OPhy))に相当する。
ESIMS MH+ 434.0
実施例32:1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン(以下、NH 2 -Dpm(COP)と略称する。)とBoc-Cys(Acm)-OHとの縮合によるBoc-Cys(Acm)-NH-Dpm(COP)の合成
実施例3で得られたNH2-Dpm(COP)(950mg,1.85mmol)を酢酸イソプロピル(20ml)に溶解させ、10%炭酸ナトリウム水溶液、および20%食塩水で分液洗浄した後、有機層に室温下で、Boc-Cys(Acm)-OH(594mg,2.03mmol)、HOBt(27mg,0.20mmol)、氷浴下でEDC・HCl(428mg,2.24mmol)をそれぞれ加え、室温で3時間攪拌させた。ジメチルプロパンジアミン(46μl,0.37mmol)を加えて10分間攪拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去し、Boc-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例3で得られたNH2-Dpm(COP)(950mg,1.85mmol)を酢酸イソプロピル(20ml)に溶解させ、10%炭酸ナトリウム水溶液、および20%食塩水で分液洗浄した後、有機層に室温下で、Boc-Cys(Acm)-OH(594mg,2.03mmol)、HOBt(27mg,0.20mmol)、氷浴下でEDC・HCl(428mg,2.24mmol)をそれぞれ加え、室温で3時間攪拌させた。ジメチルプロパンジアミン(46μl,0.37mmol)を加えて10分間攪拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去し、Boc-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例33:Boc-Cys(Acm)-NH-Dpm(COP)の脱Boc、およびそれに続くBoc-Pro-OHとの縮合によるBoc-Pro-Cys(Acm)-NH-Dpm(COP)の合成
実施例32で得られたBoc-Cys(Acm)-NH-Dpm(COP)(1.85mmol)を酢酸イソプロピル(20ml)に溶解させ、メタンスルホン酸(600μl,9.2mmol)を氷浴下で滴下した。室温に戻して3時間攪拌した後、再度氷浴下で10%炭酸ナトリウム水溶液(20ml)を加え、攪拌した。水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中でH-Cys(Acm)-NH-Dpm(COP)へと変換した。
Cys(Acm)-NH-Dpm(COP)(1.55mmol)を含む上記反応液に室温下でBoc-Pro-OH(367mg,1.70mmol)、HOBt(23mg,0.17mmol)、氷浴下でEDC・HCl(360mg,1.88mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間攪拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例32で得られたBoc-Cys(Acm)-NH-Dpm(COP)(1.85mmol)を酢酸イソプロピル(20ml)に溶解させ、メタンスルホン酸(600μl,9.2mmol)を氷浴下で滴下した。室温に戻して3時間攪拌した後、再度氷浴下で10%炭酸ナトリウム水溶液(20ml)を加え、攪拌した。水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中でH-Cys(Acm)-NH-Dpm(COP)へと変換した。
Cys(Acm)-NH-Dpm(COP)(1.55mmol)を含む上記反応液に室温下でBoc-Pro-OH(367mg,1.70mmol)、HOBt(23mg,0.17mmol)、氷浴下でEDC・HCl(360mg,1.88mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間攪拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例34:Boc-Pro-Cys(Acm)-NH-Dpm(COP)の脱Boc、およびそれに続くBoc-Trp(CHO)-OHとの縮合によるBoc-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)の合成
実施例33で得られたBoc-Pro-Cys(Acm)-NH-Dpm(COP)(1.55mmol)を酢酸イソプロピル(15ml)に溶解させ、メタンスルホン酸(500μl,7.7mmol)を氷浴下で滴下した。室温に戻して3時間攪拌した後、再度氷浴下で10%炭酸ナトリウム水溶液(20ml)、精製水(10ml)を加え、少しの間攪拌した。水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中でPro-Cys(Acm)-NH-Dpm(COP)へと変換した。
Pro-Cys(Acm)-NH-Dpm(COP)を含む上記反応液に室温下で、Boc-Trp(CHO)-OH(566mg,1.69mmol)、HOBt(23mg,0.17mmol)、氷浴下でEDC・HCl(360mg,1.88mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間撹拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例33で得られたBoc-Pro-Cys(Acm)-NH-Dpm(COP)(1.55mmol)を酢酸イソプロピル(15ml)に溶解させ、メタンスルホン酸(500μl,7.7mmol)を氷浴下で滴下した。室温に戻して3時間攪拌した後、再度氷浴下で10%炭酸ナトリウム水溶液(20ml)、精製水(10ml)を加え、少しの間攪拌した。水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中でPro-Cys(Acm)-NH-Dpm(COP)へと変換した。
Pro-Cys(Acm)-NH-Dpm(COP)を含む上記反応液に室温下で、Boc-Trp(CHO)-OH(566mg,1.69mmol)、HOBt(23mg,0.17mmol)、氷浴下でEDC・HCl(360mg,1.88mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間撹拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例35:Boc-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)の脱Boc、およびそれに続くBoc-Asp(OBzl)-OHとの縮合によるBoc-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)の合成
実施例34で得られたBoc-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)(1.29mmol)を酢酸イソプロピル(20ml)に溶解させ、メタンスルホン酸(400μl,6.2mmol)を氷浴下で滴下し、室温で3時間攪拌した。メタンスルホン酸(100μl,1.5mmol)を追加し、さらに3時間攪拌した後、メタンスルホン酸(200μl,3.1mmol)を加え、2時間攪拌した。氷浴下で10%炭酸ナトリウム水溶液(25ml)を加え、攪拌した後、水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中で、Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)へと変換した。
Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を含む上記反応液に室温下でBoc-Asp(OBzl)-OH(451mg,1.39mmol)、HOBt(19mg,0.14mmol)、氷浴下でEDC・HCl(297mg,1.55mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間撹拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例34で得られたBoc-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)(1.29mmol)を酢酸イソプロピル(20ml)に溶解させ、メタンスルホン酸(400μl,6.2mmol)を氷浴下で滴下し、室温で3時間攪拌した。メタンスルホン酸(100μl,1.5mmol)を追加し、さらに3時間攪拌した後、メタンスルホン酸(200μl,3.1mmol)を加え、2時間攪拌した。氷浴下で10%炭酸ナトリウム水溶液(25ml)を加え、攪拌した後、水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中で、Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)へと変換した。
Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を含む上記反応液に室温下でBoc-Asp(OBzl)-OH(451mg,1.39mmol)、HOBt(19mg,0.14mmol)、氷浴下でEDC・HCl(297mg,1.55mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間撹拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例36:Boc-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)の脱Boc、およびそれに続くBoc-Gly-OHとの縮合によるBoc-Gly-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)の合成
実施例35で得られたBoc-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)(1.29mmol)を酢酸イソプロピル(20ml)に溶解させ、メタンスルホン酸(400μl,6.2mmol)を氷浴下で滴下し室温で3時間攪拌した。メタンスルホン酸(300μl,4.6mmol)を追加しさらに2時間攪拌した後、氷浴下で10%炭酸ナトリウム水溶液(25ml)を加え、少しの間攪拌した。水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中で、Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)へと変換した。
Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を含む上記反応液に室温下でBoc-Gly-OH(245mg,1.40mmol)、HOBt(19mg,0.14mmol)、氷浴下でEDC・HCl(297mg,1.55mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間撹拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Gly-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例35で得られたBoc-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)(1.29mmol)を酢酸イソプロピル(20ml)に溶解させ、メタンスルホン酸(400μl,6.2mmol)を氷浴下で滴下し室温で3時間攪拌した。メタンスルホン酸(300μl,4.6mmol)を追加しさらに2時間攪拌した後、氷浴下で10%炭酸ナトリウム水溶液(25ml)を加え、少しの間攪拌した。水層を分離・廃棄し、有機層を20%食塩水(20ml)で1回洗浄することにより、反応系中で、Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)へと変換した。
Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を含む上記反応液に室温下でBoc-Gly-OH(245mg,1.40mmol)、HOBt(19mg,0.14mmol)、氷浴下でEDC・HCl(297mg,1.55mol)をそれぞれ加え、室温に戻し一晩攪拌した。ジメチルプロパンジアミン(39μl,0.31mmol)を加えて10分間撹拌した後、反応液を5%炭酸水素ナトリウム水溶液(20ml)で2回、0.1N塩酸(20ml)で2回、20%食塩水(20ml)で1回分液洗浄した。有機層を減圧留去することにより、Boc-Gly-Asp(OBzl)-Trp(CHO)-Pro-Cys(Acm)-NH-Dpm(COP)を油状物として得た。
実施例37:Bzl-(3-O-GI-1000)-OHとBoc-Leu-OHとの縮合によるBoc-Leu-OBzl(3-O-GI-1000)の合成
実施例19で得られたBzl-(3-O-GI-1000)-OH(450mg)を酢酸イソプロピル(4ml)に溶解させ、DMAP(13mg,0.11mmol)とBoc-Leu・H2O(321mg,1.29mmol)とEDC・HCl(270mg,1.41mmol)を加えて40℃で6時間攪拌した。反応混合液を室温に冷却後、ヘプタン(30ml)を加え、90%アセトニトリル水溶液(15ml)で6回洗浄した。有機層の溶媒を留去して、Boc-Leu-OBzl(3-O-GI-1000)(376mg)を得た。
1H-NMR(300MHz):δ0.86-1.57(m),2.01-2.04(m,3H),3.93-3.97(m,2H),4.88(t,1H),5.11(s,2H),6.84-6.91(m,3H),7.23-7.26(m,1H).
1H-NMR(300MHz):δ0.86-1.57(m),2.01-2.04(m,3H),3.93-3.97(m,2H),4.88(t,1H),5.11(s,2H),6.84-6.91(m,3H),7.23-7.26(m,1H).
実施例38:Boc-Leu-OBzl(3-O-GI-1000)の脱Boc、およびそれに続くBoc-Tyr(Bzl)-OHとの縮合によるBoc-Tyr(Bzl)-Leu-OBzl(3-O-GI-1000)の合成
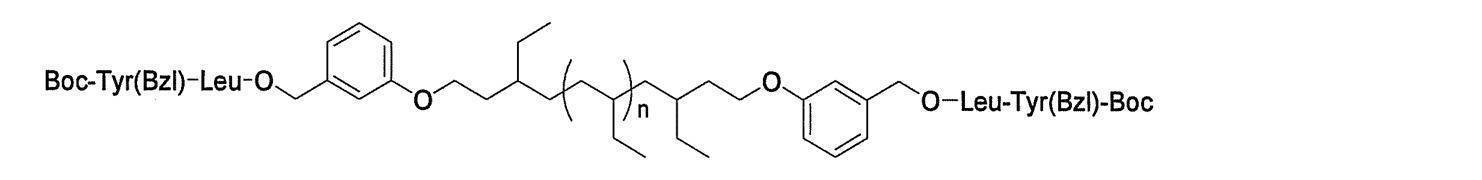
実施例37で得られたBoc-Leu-OBzl(3-O-GI-1000)(100mg)を酢酸イソプロピル(1ml)に溶解させ、メタンスルホン酸(32μl,0.49mmol)を滴下し、5時間攪拌した。反応終了後、10%炭酸ナトリウム水溶液で3回、20%食塩水で1回洗浄することにより、反応系中で、H-Leu-OBzl(3-O-GI-1000)へと変換した。
H-Leu-OBzl(3-O-GI-1000)を含む上記反応液に、HOBt(5mg,0.04mmol)とBoc-Tyr(Bzl)-OH(140mg,0.38mmol)とEDC・HCl(79mg,0.41mmol)を加え、一晩攪拌した。反応液にN,N-ジメチル-1,3-プロパンジアミン(DMPDA)(5μl,0.04mmol)を加えて反応を停止し、1N塩酸で3回洗浄し、有機層を濃縮することにより、Boc-Tyr(Bzl)-Leu-OBzl(3-O-GI-1000)(120mg)を得た。
1H-NMR(300MHz):δ0.85-1.59(m),2.02-2.10(m,3H),2.98-3.12(m,2H),3.93-3.96(m,2H),4.51-4.62(m,1H),4.39-5.09(m,5H),6.87-6.93(m,5H),7.09-7.12(m,3H),7.29-7.43(m,5H).
H-Leu-OBzl(3-O-GI-1000)を含む上記反応液に、HOBt(5mg,0.04mmol)とBoc-Tyr(Bzl)-OH(140mg,0.38mmol)とEDC・HCl(79mg,0.41mmol)を加え、一晩攪拌した。反応液にN,N-ジメチル-1,3-プロパンジアミン(DMPDA)(5μl,0.04mmol)を加えて反応を停止し、1N塩酸で3回洗浄し、有機層を濃縮することにより、Boc-Tyr(Bzl)-Leu-OBzl(3-O-GI-1000)(120mg)を得た。
1H-NMR(300MHz):δ0.85-1.59(m),2.02-2.10(m,3H),2.98-3.12(m,2H),3.93-3.96(m,2H),4.51-4.62(m,1H),4.39-5.09(m,5H),6.87-6.93(m,5H),7.09-7.12(m,3H),7.29-7.43(m,5H).
実施例39:Boc-Tyr(Bzl)-Leu-OBzl(3-O-GI-1000)の脱Boc、およびそれに続くBoc-Glu(OBzl)-OHとの縮合によるBoc-Glu(OBzl)-Tyr(Bzl)-Leu-OBzl(3-O-GI-1000)の合成

実施例38で得られたBoc-Tyr(Bzl)-Leu-OBzl(3-O-GI-1000)(100mg)を酢酸イソプロピル(1ml)に溶解させ、メタンスルホン酸(32μl,0.49mmol)を滴下し、6時間攪拌した。反応終了後、10%炭酸ナトリウム水溶液で3回、20%食塩水で1回洗浄した。この有機層に、HOBt(5mg,0.04mmol)とBoc-Glu(OBzl)-OH(140mg,0.38mmol)とEDC・HCl(79mg,0.41mmol)を加え、40℃で一晩攪拌した。反応液にDMPDA(5μl,0.04mmol)を加えてクエンチし、1N塩酸で3回洗浄し、有機層を濃縮してBoc-Glu(OBzl)-Tyr(Bzl)-Leu-OBzl(3-O-GI-1000)(100mg)を得た。
実施例40:Bzl(3-O-GI-2000)-OHとBoc-Leu-OHとの縮合によるBoc-Leu-OBzl(3-O-GI-2000)の合成
実施例20で得られたBzl(3-O-GI-2000)-OH(402mg)を酢酸イソプロピル(4ml)に溶解させ、DMAP(5mg,0.06mmol)とBoc-Leu-OH(151mg,0.65mmol)とEDC・HCl(128mg,0.70mmol)を加えて40℃で4時間攪拌した。反応混合液を室温に冷却後、ヘプタン(30ml)を加え、90%アセトニトリル水溶液(15ml)で6回洗浄した。有機層の溶媒を留去して、Boc-Leu-OBzl(3-O-GI-2000)(310mg)を得た。
1H-NMR(300MHz):δ0.88-1.51(m),2.01-2.04(m,3H),3.93-3.99(m,2H),4.88-4.90(m,1H),5.13(s,2H),6.84-6.91(m,3H),7.23-7.24(m,1H).
実施例20で得られたBzl(3-O-GI-2000)-OH(402mg)を酢酸イソプロピル(4ml)に溶解させ、DMAP(5mg,0.06mmol)とBoc-Leu-OH(151mg,0.65mmol)とEDC・HCl(128mg,0.70mmol)を加えて40℃で4時間攪拌した。反応混合液を室温に冷却後、ヘプタン(30ml)を加え、90%アセトニトリル水溶液(15ml)で6回洗浄した。有機層の溶媒を留去して、Boc-Leu-OBzl(3-O-GI-2000)(310mg)を得た。
1H-NMR(300MHz):δ0.88-1.51(m),2.01-2.04(m,3H),3.93-3.99(m,2H),4.88-4.90(m,1H),5.13(s,2H),6.84-6.91(m,3H),7.23-7.24(m,1H).
実施例41:Boc-Leu-OBzl(3-O-GI-2000)の脱Boc、およびそれに続くBoc-Tyr(Bzl)-OHとの縮合によるBoc-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)の合成
実施例40で得られたBoc-Leu-OBzl(3-O-GI-2000)(100mg)を酢酸イソプロピル(1ml)に溶解させ、メタンスルホン酸(16μl,0.25mmol)を滴下し、3時間攪拌した。反応終了後、10%炭酸ナトリウム水溶液で3回、20%食塩水で1回洗浄した。この有機層に、HOBt(3mg,0.02mmol)とBoc-Tyr(Bzl)-OH(70mg,0.19mmol)とEDC・HCl(40mg,0.20mmol)を加え、40℃で一晩攪拌した。反応液にDMPDA(5μl,0.04mmol)を加えて反応を停止させ、1N塩酸で3回洗浄し、有機層を濃縮してBoc-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)(120mg)を得た。
1H-NMR(300MHz):δ0.85-1.59(m),2.04-2.10(m,3H),2.99-3.15(m,2H),3.92-3.99(m,2H),4.50-4.63(m,1H),4.41-5.08(m,5H),6.86-6.95(m,5H),7.09-7.15(m,3H),7.27-7.44(m,5H)
実施例40で得られたBoc-Leu-OBzl(3-O-GI-2000)(100mg)を酢酸イソプロピル(1ml)に溶解させ、メタンスルホン酸(16μl,0.25mmol)を滴下し、3時間攪拌した。反応終了後、10%炭酸ナトリウム水溶液で3回、20%食塩水で1回洗浄した。この有機層に、HOBt(3mg,0.02mmol)とBoc-Tyr(Bzl)-OH(70mg,0.19mmol)とEDC・HCl(40mg,0.20mmol)を加え、40℃で一晩攪拌した。反応液にDMPDA(5μl,0.04mmol)を加えて反応を停止させ、1N塩酸で3回洗浄し、有機層を濃縮してBoc-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)(120mg)を得た。
1H-NMR(300MHz):δ0.85-1.59(m),2.04-2.10(m,3H),2.99-3.15(m,2H),3.92-3.99(m,2H),4.50-4.63(m,1H),4.41-5.08(m,5H),6.86-6.95(m,5H),7.09-7.15(m,3H),7.27-7.44(m,5H)
実施例42:Boc-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)の脱Boc、およびそれに続くBoc-Glu(OBzl)-OHとの縮合によるBoc-Glu(OBzl)-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)の合成
Boc-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)(100mg)を酢酸イソプロピル(1ml)に溶解させ、メタンスルホン酸(32μl,0.49mmol)を滴下し、6時間攪拌した。反応終了後、10%炭酸ナトリウム水溶液で3回、20%食塩水で1回洗浄した。この有機層に、HOBt(5mg,0.04mmol)とBoc-Glu(OBzl)-OH(140mg,0.38mmol)とEDC・HCl(79mg,0.41mmol)を加え、40℃で一晩攪拌した。反応液にDMPDA(5μl,0.04mmol)を加えてクエンチし、1N塩酸で3回洗浄し、有機層を濃縮してBoc-Glu(OBzl)-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)(100mg)を得た。
Boc-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)(100mg)を酢酸イソプロピル(1ml)に溶解させ、メタンスルホン酸(32μl,0.49mmol)を滴下し、6時間攪拌した。反応終了後、10%炭酸ナトリウム水溶液で3回、20%食塩水で1回洗浄した。この有機層に、HOBt(5mg,0.04mmol)とBoc-Glu(OBzl)-OH(140mg,0.38mmol)とEDC・HCl(79mg,0.41mmol)を加え、40℃で一晩攪拌した。反応液にDMPDA(5μl,0.04mmol)を加えてクエンチし、1N塩酸で3回洗浄し、有機層を濃縮してBoc-Glu(OBzl)-Tyr(Bzl)-Leu-OBzl(3-O-GI-2000)(100mg)を得た。
実施例43:3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコールとFmoc-Leu-OHとの縮合によるFmoc-Leu-OBzl(3,4,5-OPhy)の合成、およびそれに続く脱FmocによるH-Leu-OBzl(3,4,5-OPhy)の合成
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(5g,5.01mmol)をシクロペンチルメチルエーテル(CPME)(50ml)に溶解させ、Fmoc-Leu-OH(1.95g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)、DMAP(61mg,0.50mmol)を加え、室温で3時間攪拌し、その後クロロホルム(50ml)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を25mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度25mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を50mlとした。この反応液に5分間窒素バブリングを行った後、窒素雰囲気の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で5時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.8である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、20%食塩水(50ml)で1回、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除いて、H-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)を得た。
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール(5g,5.01mmol)をシクロペンチルメチルエーテル(CPME)(50ml)に溶解させ、Fmoc-Leu-OH(1.95g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)、DMAP(61mg,0.50mmol)を加え、室温で3時間攪拌し、その後クロロホルム(50ml)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を25mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度25mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を50mlとした。この反応液に5分間窒素バブリングを行った後、窒素雰囲気の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で5時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.8である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、20%食塩水(50ml)で1回、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除いて、H-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)を得た。
実施例44:H-Leu-OBzl(3,4,5-OPhy)とFmoc-Tyr(tBu)-OHとの縮合によるFmoc-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)の合成、およびそれに続く脱FmocによるH-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)の合成
H-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)に、HOBt(68mg,0.50mmol)、Fmoc-Tyr(tBu)-OH(2.53g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を25mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度25mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を50mlとした。この反応液に5分間窒素バブリングを行った後、窒素雰囲気の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で1.5時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.7である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、20%食塩水(50ml)で1回、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除いて、H-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)を得た。
H-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)に、HOBt(68mg,0.50mmol)、Fmoc-Tyr(tBu)-OH(2.53g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を25mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度25mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を50mlとした。この反応液に5分間窒素バブリングを行った後、窒素雰囲気の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で1.5時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.7である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、20%食塩水(50ml)で1回、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除いて、H-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)を得た。
実施例45:H-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)とFmoc-Glu(OtBu)-OHとの縮合によるFmoc-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)の合成、およびそれに続く脱FmocによるH-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)の合成
H-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)に、HOBt(68mg,0.50mmol)、Fmoc-Glu(OtBu)-OH(2.35g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を25mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度25mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を50mlとした。この反応液の少量を、トリフルオロ酢酸(TFA)/H2O/トリイソプロピルシラン溶液に加えて脱保護して、LC/MSで分析したところ、Fmoc-Glu-Tyr-Leu-OHのESIMS MH+ 646.2を確認した。
前記反応液に5分間窒素バブリングを行った後、窒素雰囲気の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で2時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.0である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除き、有機層の溶媒を減圧留去して、H-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)を得た。
H-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)に、HOBt(68mg,0.50mmol)、Fmoc-Glu(OtBu)-OH(2.35g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を25mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度25mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を50mlとした。この反応液の少量を、トリフルオロ酢酸(TFA)/H2O/トリイソプロピルシラン溶液に加えて脱保護して、LC/MSで分析したところ、Fmoc-Glu-Tyr-Leu-OHのESIMS MH+ 646.2を確認した。
前記反応液に5分間窒素バブリングを行った後、窒素雰囲気の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で2時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.0である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除き、有機層の溶媒を減圧留去して、H-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)を得た。
実施例46:H-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)とFmoc-Glu(OtBu)-OHとの縮合によるFmoc-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)の合成、およびそれに続く脱FmocによるH-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)の合成
H-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)に、HOBt(68mg,0.50mmol)、Fmoc-Glu(OtBu)-OH(2.35g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を35mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度30mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を55mlとした。この反応液を少量採取し、それを脱保護して分析したところ、Fmoc-Glu-Glu-Tyr-Leu-OHのESIMS MH+ 775.2を確認した。
前記反応液に5分間窒素バブリングを行った後、窒素雰囲気下の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で1.5時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.7である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、20%食塩水(50ml)で1回、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除いて、H-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(55ml)を得た。
H-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(50ml)に、HOBt(68mg,0.50mmol)、Fmoc-Glu(OtBu)-OH(2.35g,5.52mmol)を加えた後、これらに、氷浴下、EDC・HCl(1.16g,6.05mmol)を加え、室温で一晩攪拌した。反応終了後、反応液中の溶媒を35mlまで減圧留去し、その残渣にCPME(50ml)を加え、反応液中の溶媒を再度30mlまで減圧留去した後、その残渣にCPME(25ml)を加えて、反応液中の溶媒量を55mlとした。この反応液を少量採取し、それを脱保護して分析したところ、Fmoc-Glu-Glu-Tyr-Leu-OHのESIMS MH+ 775.2を確認した。
前記反応液に5分間窒素バブリングを行った後、窒素雰囲気下の氷浴下にて、ジエチレントリアミン(2.71ml,25.1mmol)を加え、室温で1.5時間攪拌した。反応終了後、反応液を10%炭酸ナトリウム水溶液(50ml)で2回分液洗浄し、得られた有機層へ20%食塩水(50ml)を加え、有機層および水層(20%食塩水)を攪拌しながら、水層のpHが6.8になるまで1N塩酸を滴下した後、有機層および水層を分液漏斗へ移し、水層を除いた。再度同様の操作を行い、pH=6.7である水層(20%食塩水)で有機層を撹拌洗浄し、水層を除いた。得られた有機層を、20%食塩水(50ml)で1回、10%炭酸ナトリウム水溶液(50ml)で1回、20%食塩水(50ml)で1回分液洗浄した。得られた有機層を硫酸ナトリウムで乾燥させた後、硫酸ナトリウムを濾過で除いて、H-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(5.01mmol)のCPME溶液(55ml)を得た。
実施例47:H-Asp(OtBu)-Phe-Glu(OtBu)-Glu(OtBu)-Ile-Pro-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)の合成
H-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)並びに次の保護アミノ酸:Fmoc-Pro、Fmoc-Ile、Fmoc-Glu(OtBu)、Fmoc-Glu(OtBu)、Fmoc-PheおよびFmoc-Asp(OtBu)をこの順序で用いて、実施例43~46と同様の操作にて、縮合反応、脱保護反応および後処理操作を繰り返してペプチド鎖を伸張させた後、生成物をアセトニトリル水溶液にて沈殿化させて、H-Asp(OtBu)-Phe-Glu(OtBu)-Glu(OtBu)-Ile-Pro-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(10.8g,4.15mmol,収率82% vs. 3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール,ESIMS MH+ 2598.5)を得た。
この化合物を少量用い、トリフルオロ酢酸(TFA)にて脱保護して分析したところ、H-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Tyr-Leu-OHのESIMS MH+ 1283.5を確認した。
H-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)並びに次の保護アミノ酸:Fmoc-Pro、Fmoc-Ile、Fmoc-Glu(OtBu)、Fmoc-Glu(OtBu)、Fmoc-PheおよびFmoc-Asp(OtBu)をこの順序で用いて、実施例43~46と同様の操作にて、縮合反応、脱保護反応および後処理操作を繰り返してペプチド鎖を伸張させた後、生成物をアセトニトリル水溶液にて沈殿化させて、H-Asp(OtBu)-Phe-Glu(OtBu)-Glu(OtBu)-Ile-Pro-Glu(OtBu)-Glu(OtBu)-Tyr(tBu)-Leu-OBzl(3,4,5-OPhy)(10.8g,4.15mmol,収率82% vs. 3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール,ESIMS MH+ 2598.5)を得た。
この化合物を少量用い、トリフルオロ酢酸(TFA)にて脱保護して分析したところ、H-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Tyr-Leu-OHのESIMS MH+ 1283.5を確認した。
比較実験例:本発明の分岐鎖含有芳香族化合物および直鎖含有芳香族化合物の各種溶媒に対する溶解度(20℃)
実験方法
本発明の分岐鎖含有芳香族化合物(実施例4、5、19、20および21の化合物)、および比較例として直鎖含有芳香族化合物(比較例1~3)を20℃で溶媒に飽和させ、その溶解度(単位:重量%)を測定した(表1、2)。
実験方法
本発明の分岐鎖含有芳香族化合物(実施例4、5、19、20および21の化合物)、および比較例として直鎖含有芳香族化合物(比較例1~3)を20℃で溶媒に飽和させ、その溶解度(単位:重量%)を測定した(表1、2)。


実験結果
本発明化合物(すなわち、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を1個以上有する特定の分岐鎖含有芳香族化合物)はいずれも、対応する有機基が直鎖である比較例化合物1~3と比べて、酢酸イソプロピルや酢酸エチルのみならず、様々な有機溶媒に対する溶解度が格段に高く、ほとんどの場合において、10~1000倍以上、場合により5000倍以上の溶解性を示すことが分かった(表1、2参照)。このことから、ペプチドの製造方法において、本発明化合物がアミノ酸および/またはペプチドの優れた保護化試薬として機能し得ることを見出した。
本発明化合物(すなわち、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を1個以上有する特定の分岐鎖含有芳香族化合物)はいずれも、対応する有機基が直鎖である比較例化合物1~3と比べて、酢酸イソプロピルや酢酸エチルのみならず、様々な有機溶媒に対する溶解度が格段に高く、ほとんどの場合において、10~1000倍以上、場合により5000倍以上の溶解性を示すことが分かった(表1、2参照)。このことから、ペプチドの製造方法において、本発明化合物がアミノ酸および/またはペプチドの優れた保護化試薬として機能し得ることを見出した。
分液操作性に優れた酢酸イソプロピルに易溶である、本発明の特定の分岐鎖含有芳香族化合物によって、各工程で各中間体を晶析単離することなく、抽出分離のみを経て最終生成物へと導くペプチド等の製造方法を提供できるようになった。更には、有機合成反応および実用的な工業的プロセスをも提供できるようになった。
また、本発明のペプチドの製造方法は、従来の液相法に比べても、ペプチドの配列および鎖長によらず、溶媒中に安定的に溶解させられるため、工程上では単離・精製工程を簡便化でき、総合的には高純度・高収率を確保できるという利点を有する。
また、本発明のペプチドの製造方法は、従来の液相法に比べても、ペプチドの配列および鎖長によらず、溶媒中に安定的に溶解させられるため、工程上では単離・精製工程を簡便化でき、総合的には高純度・高収率を確保できるという利点を有する。
本願は、日本の特願2010-192961号を基礎としており、その内容は本明細書に全て包含される。
Claims (31)
- 式(I):

[式中、
k個のQは、独立してそれぞれ、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる1以上の置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基または-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)を示し;かつ
Zは、水素原子または式(a):
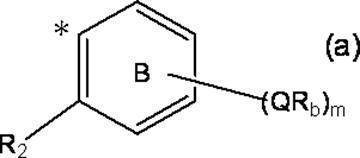
(式中、*は結合位置を示し;
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる1以上の置換基を有していてもよい)で表される基を示し;
前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(b):
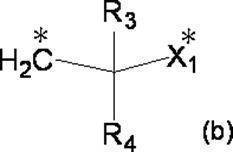
(式中、*は、隣接原子との結合位置を示し;
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基である。]で表される分岐鎖含有芳香族化合物。 - 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(c):

[式中、*は、Qとの結合位置を示し;
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):

(式中、*は、結合位置を示し;
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる1以上の置換基を有していてもよい。)で表される基を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]で表される基である、
請求項1に記載の分岐鎖含有芳香族化合物。 - 前記式(c)中、
R5およびR6は、共に水素原子であり;
n0個のR7およびR8は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n0個のX2は、独立してそれぞれ、単結合、メチレン基またはエチレン基であり;かつ
R9は、水素原子、メチル基またはエチル基である、
請求項2に記載の分岐鎖含有芳香族化合物。 - 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(d):

(式中、*は、Qとの結合位置を示し;
m1個のOR11は、式(c’):

[式中、*は、Oとの結合位置を示し;
R5およびR6は、共に水素原子を示すか、または一緒になって=Oを示し;
n0は、2~40の整数を示し;
n0個のR7およびR8は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n0個のX2は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;かつ
R9は、水素原子またはC1-4アルキル基を示し;
R10は、C1-4アルキル基または式(I’):

(式中、*は、結合位置を示し;
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる1以上の置換基を有していてもよい。)で表される基を示す。
但し、R7およびR8が共に水素原子であることはなく、かつn0が2の場合には、R9はC1-4アルキル基を示す。]で表される基により置換されたヒドロキシル基を示し;
m1は、1~3の整数を示す。)で表される基である、
請求項1に記載の分岐鎖含有芳香族化合物。 - 前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(e):
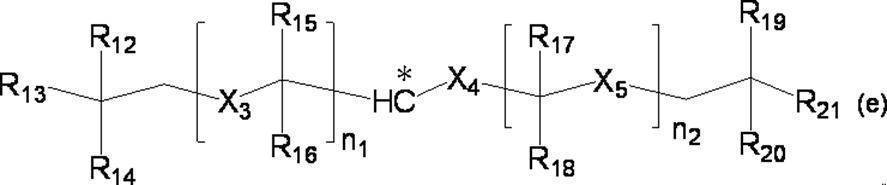
(式中、*は、Qとの結合位置を示し;
n1は、1~10の整数を示し;
n2は、1~10の整数を示し;
n1個のR15およびR16は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n1個のX3は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
n2個のX5は、独立してそれぞれ、単結合またはC1-4アルキレン基を示し;
X4は、単結合またはC1-4アルキレン基を示し;かつ
R12、R13、R14、R19、R20およびR21は、独立してそれぞれ、水素原子またはC1-4アルキル基を示す。
但し、R15およびR16、および/またはR17およびR18が共に水素原子であることはなく、かつn1+n2が2の場合には、R12、R13およびR14の2個以上が独立してそれぞれ、C1-4アルキル基を示すか、またはR19、R20およびR21の2個以上が独立してそれぞれ、C1-4アルキル基を示す。)で表される基である、
請求項1に記載の分岐鎖含有芳香族化合物。 - 前記式(e)中、
n1は、1~5の整数であり;
n2は、1~5の整数であり;
n1個のR15およびR16は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n1個のX3は、独立してそれぞれ、単結合、メチレン基またはエチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子、メチル基またはエチル基であり;
n2個のX5は、独立してそれぞれ、単結合、メチレン基またはエチレン基であり;
X4は、単結合、メチレン基またはエチレン基である、
請求項5に記載の分岐鎖含有芳香族化合物。 - 前記式(e)中、
n1個のR15およびR16は、独立してそれぞれ、水素原子またはメチル基であり;
n1個のX3は、独立してそれぞれ、単結合またはメチレン基であり;
n2個のR17およびR18は、独立してそれぞれ、水素原子またはメチル基であり;
n2個のX5は、独立してそれぞれ、単結合またはメチレン基であり;
X4は、単結合またはメチレン基であり;かつ
R12、R13、R14、R19、R20およびR21は、メチル基である、
請求項6に記載の分岐鎖含有芳香族化合物。 - RaおよびRbが、独立してそれぞれ、3,7,11,15-テトラメチルヘキサデシル基、3,7,11-トリメチルドデシル基、2,2,4,8,10,10-ヘキサメチル-5-ドデカノイル基、3,4,5-トリ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基、3,5-ジ(3’,7’,11’,15’-テトラメチルヘキサデシルオキシ)ベンジル基、式(f):

[式中、*は、Qとの結合位置であり、n10は、23~34であり、R10は、式(I’):
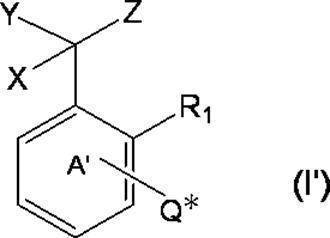
(式中、*は、結合位置を示し;
他の記号は、前記と同意義を示す。ここで、環A’は、R1、Q、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる1以上の置換基を有していてもよい。)で表される基である。]で表される基、
式(g):
(式中、*は、Qとの結合位置であり、n11は、1~10である。)で表される基、
式(h):

(式中、*は、Qとの結合位置であり、n12は、2~10である。)で表される基、
式(i):
(式中、*は、Qとの結合位置であり、n13およびn14は、独立してそれぞれ、1~10である。)で表される基、または
式(j):

(式中、*は、Qとの結合位置であり、n15は、2~20である。)で表される基、
である、請求項1に記載の分岐鎖含有芳香族化合物。 - XとZが共に水素原子であり、かつR1が水素原子である、請求項1~8のいずれか1項に記載の分岐鎖含有芳香族化合物。
- Xが水素原子であり、R1が水素原子であり、kが1であり、かつZが式(a)(式中、R2が水素原子であり、mが0である。)で表される基である、請求項1~8のいずれか1項に記載の分岐鎖含有芳香族化合物。
- Xがフェニル基であり、kが1であり、Zが式(a)(式中、mが0である。)で表される基であり、かつR2がR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成する、請求項1~8のいずれか1項に記載の分岐鎖含有芳香族化合物。
- Qが-O-である、請求項1~11のいずれか1項に記載の分岐鎖含有芳香族化合物。
- Yがヒドロキシル基である、請求項1~12のいずれか1項に記載の分岐鎖含有芳香族化合物。
- Yが-NHR基である、請求項1~12のいずれか1項に記載の分岐鎖含有芳香族化合物。
- 2,4-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,5-ジ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
1-[(2-クロロ-5-(2’,3’-ジヒドロフィチルオキシ)フェニル)]-1-フェニルメタンアミン;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアルコール;
3,4,5-トリ(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)ベンジルアミン;
2-[3’,4’,5’-トリ(2’’,3’’-ジヒドロフィチルオキシ)ベンジルオキシ]-4-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メトキシベンジルアミン;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアルコール;
4-(2’,3’-ジヒドロフィチルオキシ)-2-メチルベンジルアミン;
2,2,4,8,10,10-ヘキサメチル-5-ドデカン酸(4-ヒドロキシメチル)フェニルアミド;
4-(3,7,11-トリメチルドデシルオキシ)ベンジルアルコール;
2-(3,7,11-トリメチルドデシルオキシ)-9-フェニルフルオレン-9-オール;
式:
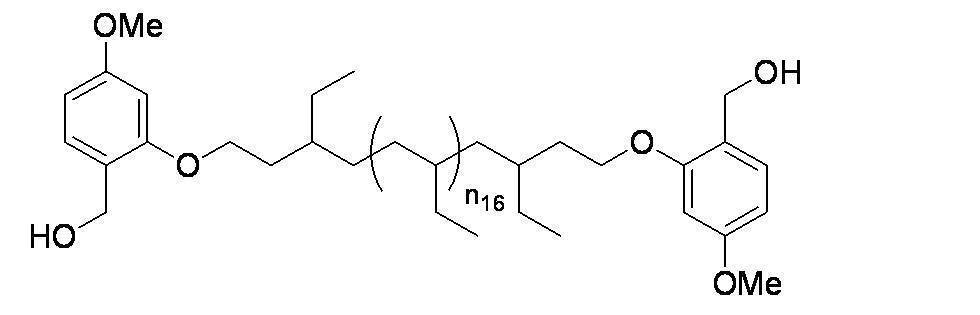
(式中、n16は、23または34を示す。)で表される化合物;
式:
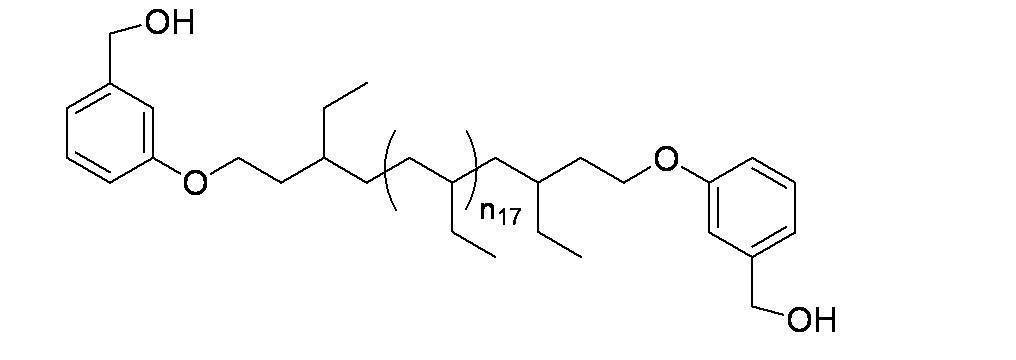
(式中、n17は、23または34を示す。)で表される化合物;
式:
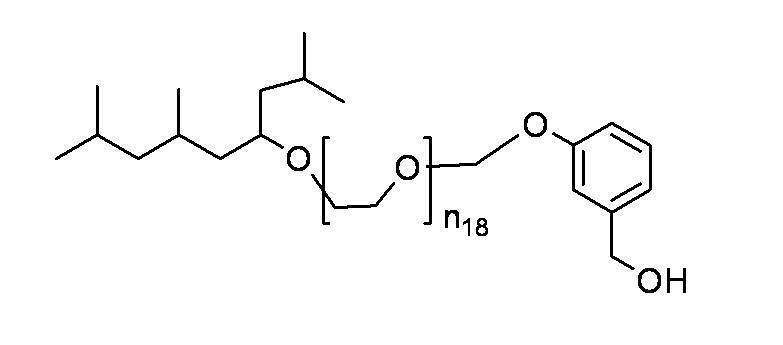
(式中、n18は、5~7を示す。)で表される化合物;および
式:

で表される化合物、
からなる群から選択される、請求項1に記載の分岐鎖含有芳香族化合物。 - 20℃における酢酸イソプロピル100g中の飽和溶解度が、1~95重量%である、請求項1~15のいずれか1項に記載の分岐鎖含有芳香族化合物。
- 20℃における酢酸イソプロピル100g中の飽和溶解度が、10~95重量%である、請求項1~15のいずれか1項に記載の分岐鎖含有芳香族化合物。
- 請求項1~17のいずれか1項に記載の分岐鎖含有芳香族化合物を含む、アミノ酸またはペプチドのカルボキシル基またはアミド基の保護化試薬。
- アミノ酸またはペプチドの保護箇所がC末端である、請求項18に記載の保護化試薬。
- 請求項1~17のいずれか1項に記載の分岐鎖含有芳香族化合物によって保護された、分岐鎖含有芳香族化合物付加体。
- 工程(1)~(4)を含む、ペプチドの製造方法。
(1)請求項1~17のいずれか1項に記載の分岐鎖含有芳香族化合物を、該化合物の可溶性溶媒中で、N-保護アミノ酸またはN-保護ペプチドのC末端と縮合させて、該化合物由来の保護基であるアンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、ペプチドを得る工程。 - さらに工程(5)~(7)の繰り返しを1以上含む、請求項21に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程。 - さらに工程(5)~(7’)の繰り返しを1以上含む、請求項21に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程。 - 請求項21~23のいずれか1項に記載のペプチド製造方法を含む、有機合成方法。
- 式(I):

[式中、
k個のQは、独立してそれぞれ、単結合を示すか、あるいは-O-、-S-、-C(=O)O-、-C(=O)NH-または-NH-を示し;
k個のRaは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
kは、1~4の整数を示し;
R1は、水素原子であるか、あるいはZが下記式(a)で表される基である場合には、R2と一緒になって単結合を示して、環Bと共にフルオレン環を形成していてもよく;
環Aは、R1、k個のQRa、およびC(X)(Y)Zに加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる1以上の置換基を有していてもよく;
Xは、水素原子またはフェニル基を示し;
Yは、ヒドロキシル基、-NHR基(Rは水素原子、アルキル基またはアラルキル基を示す)またはハロゲン原子を示し;かつ
Zは、水素原子または式(a):

(式中、*は結合位置を示し;
mは、0~4の整数を示し;
m個のQは、前記と同意義を示し;
m個のRbは、独立してそれぞれ、分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基を示し;
R2は、水素原子を示すか、またはR1と一緒になって単結合を示して、環Aと共にフルオレン環を形成していてもよく;かつ
環Bは、m個のQRb、およびR2に加えて、さらにハロゲン原子、1個以上のハロゲン原子により置換されていてもよいC1-6アルキル基、および1個以上のハロゲン原子により置換されていてもよいC1-6アルコキシ基からなる群から選ばれる1以上の置換基を有していてもよい)で表される基を示し;
前記RaおよびRbにおける分岐鎖を1以上有する脂肪族炭化水素基を少なくとも1つ有し、総分岐鎖数が3以上であって、かつ総炭素数14以上300以下である有機基が、式(b):

(式中、*は、隣接原子との結合位置を示し;
R3およびR4は、独立してそれぞれ、水素原子またはC1-4アルキル基を示し;
X1は、単結合、C1-4アルキレン基または酸素原子を示す。
但し、R3およびR4が共に水素原子であることはない。)で表される同一または異なる2価の基を3以上有する基である。]で表される分岐鎖含有芳香族化合物。 - 請求項25に記載の分岐鎖含有芳香族化合物を含む、アミノ酸またはペプチドのカルボキシル基またはアミド基の保護化試薬。
- 請求項25に記載の分岐鎖含有芳香族化合物によって保護された、分岐鎖含有芳香族化合物付加体。
- 工程(1)~(4)を含む、ペプチドの製造方法。
(1)請求項25に記載の分岐鎖含有芳香族化合物を、該化合物の可溶性溶媒中で、N-保護アミノ酸またはN-保護ペプチドのC末端と縮合させて、該化合物由来の保護基であるアンカーでC末端が保護されたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドを得る工程、
(2)得られたN-保護C-保護アミノ酸またはN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護アミノ酸またはC-保護ペプチドを得る工程、
(3)得られたC-保護アミノ酸またはC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(4)得られたN-保護C-保護ペプチドのN末端の保護基およびC末端のアンカーを除去して、ペプチドを得る工程。 - さらに工程(5)~(7)の繰り返しを1以上含む、請求項28に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7)工程(6)後に、反応系に水を添加し、不純物を水層に抽出分離する工程。 - さらに工程(5)~(7’)の繰り返しを1以上含む、請求項28に記載の方法;
(5)得られたN-保護C-保護ペプチドのN末端の保護基を除去して、C-保護ペプチドを得る工程、
(6)得られたC-保護ペプチドのN末端に、N-保護アミノ酸またはN-保護ペプチドを縮合させて、N-保護C-保護ペプチドを得る工程、および
(7’)工程(6)後に、反応系に親水性有機溶媒を添加し、不純物を親水性有機溶媒層に抽出分離する工程。 - 請求項28~30のいずれか1項に記載のペプチド製造方法を含む、有機合成方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11821809.8A EP2612845B1 (en) | 2010-08-30 | 2011-08-30 | Aromatic compound containing specific branch |
| JP2012531896A JP5929756B2 (ja) | 2010-08-30 | 2011-08-30 | 分岐鎖含有芳香族化合物 |
| DK11821809.8T DK2612845T3 (da) | 2010-08-30 | 2011-08-30 | Aromatisk forbindelse som indeholder specifik forgrening |
| CN201180041916.6A CN103080058B (zh) | 2010-08-30 | 2011-08-30 | 含有支链的芳香族化合物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-192961 | 2010-08-30 | ||
| JP2010192961 | 2010-08-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012029794A1 true WO2012029794A1 (ja) | 2012-03-08 |
Family
ID=45771169
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/069624 WO2012029794A1 (ja) | 2010-08-30 | 2011-08-30 | 分岐鎖含有芳香族化合物 |
Country Status (6)
| Country | Link |
|---|---|
| US (3) | US8546534B2 (ja) |
| EP (1) | EP2612845B1 (ja) |
| JP (1) | JP5929756B2 (ja) |
| CN (4) | CN107011132B (ja) |
| DK (1) | DK2612845T3 (ja) |
| WO (1) | WO2012029794A1 (ja) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012165545A1 (ja) * | 2011-05-31 | 2012-12-06 | 味の素株式会社 | ペプチドの製造方法 |
| WO2017038650A1 (ja) * | 2015-08-28 | 2017-03-09 | 積水メディカル株式会社 | ベンジル化合物 |
| WO2017221889A1 (ja) | 2016-06-20 | 2017-12-28 | 積水メディカル株式会社 | 新規ジフェニルメタン保護剤 |
| WO2018155669A1 (ja) * | 2017-02-27 | 2018-08-30 | 積水メディカル株式会社 | 新規四環式保護剤 |
| WO2019123994A1 (ja) | 2017-12-19 | 2019-06-27 | 積水メディカル株式会社 | 新規アルキルジフェニルメタン保護剤 |
| WO2019198833A1 (ja) | 2018-04-13 | 2019-10-17 | Jitsubo株式会社 | ペプチド合成方法 |
| US10508124B2 (en) | 2016-07-25 | 2019-12-17 | Sekisui Medical Co., Ltd. | Xanthene protective agent |
| WO2020101032A1 (ja) | 2018-11-16 | 2020-05-22 | 味の素株式会社 | 分子内s-s結合を有する環化ペプチドの製造方法 |
| WO2020218497A1 (ja) * | 2019-04-25 | 2020-10-29 | 味の素株式会社 | ペプチドの連続的製造方法 |
| JP2022516932A (ja) * | 2019-01-07 | 2022-03-03 | 広州同雋医薬科技有限公司 | ジフェニルメタン構造を含有する化合物及びその応用 |
| EP4049991A1 (en) | 2015-01-21 | 2022-08-31 | Ajinomoto Co., Inc. | Precipitation promoter and precipitation method in which same is used |
| WO2023277186A1 (ja) * | 2021-07-02 | 2023-01-05 | ペプチスター株式会社 | 液相ペプチド合成用担体結合ペプチドの分析方法 |
| WO2023127331A1 (ja) * | 2021-12-27 | 2023-07-06 | 株式会社トクヤマ | ペプチド製造方法、保護基の除去方法、除去剤、及びベンジル化合物 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2415745A4 (en) | 2009-03-30 | 2014-06-11 | Ajinomoto Kk | DIPHENYLMETHANE COMPOUND |
| EP3539968B1 (en) | 2016-11-11 | 2021-07-14 | Sekisui Medical Co., Ltd. | Novel trityl protecting agent |
| CN110317130B (zh) * | 2018-03-29 | 2021-12-21 | 深圳翰宇药业股份有限公司 | 化合物及其制备方法和应用 |
| FR3090636B1 (fr) | 2018-12-24 | 2021-01-01 | Strainchem | Procédé de synthèse de peptides |
| CN110183347B (zh) * | 2019-02-02 | 2021-03-23 | 广州同隽医药科技有限公司 | 一种含有苯甲基结构的化合物及其应用 |
| CN110256277B (zh) * | 2019-03-19 | 2020-12-15 | 广州同隽医药科技有限公司 | 一种含有芴环结构的化合物及其应用 |
| FR3095646B1 (fr) * | 2019-05-02 | 2024-02-23 | Strainchem | Methode de production de peptides ou proteines ou peptidomimetiques |
| JP7301965B2 (ja) * | 2019-06-28 | 2023-07-03 | 富士フイルム株式会社 | ペプチド化合物の製造方法、保護基形成用試薬、及び、縮合多環化合物 |
| JPWO2020262590A1 (ja) * | 2019-06-28 | 2020-12-30 | ||
| CN113999130B (zh) * | 2021-11-26 | 2023-09-29 | 湖北工业大学 | 一种邻硝基酰基苯胺类化合物的制备方法 |
| CN114920629B (zh) * | 2022-05-11 | 2023-06-02 | 广东微控生物科技有限公司 | 一种双子焦性没食子酸化合物及其制备方法与应用 |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000044493A (ja) | 1998-07-27 | 2000-02-15 | Asahi Chem Ind Co Ltd | 化合物ライブラリー合成用保護基 |
| WO2003018188A1 (fr) | 2001-08-24 | 2003-03-06 | Japan Science And Technology Agency | Systeme de solvant multiphase compatible |
| JP2004059509A (ja) * | 2002-07-30 | 2004-02-26 | Nokodai Tlo Kk | 液相ペプチド合成用アミノ酸試薬 |
| JP2004067555A (ja) * | 2002-08-05 | 2004-03-04 | Nokodai Tlo Kk | 液相ペプチド合成用担体、液相ペプチド合成法 |
| JP2006015283A (ja) * | 2004-07-02 | 2006-01-19 | Nokodai Tlo Kk | 化合物の分離用担体および化合物の分離方法 |
| WO2006104166A1 (ja) | 2005-03-29 | 2006-10-05 | National University Corporation, Tokyo University Of Agriculture And Technology | 晶析分離用担体及び化合物の分離方法 |
| WO2007034812A1 (ja) | 2005-09-20 | 2007-03-29 | National University Corporation, Tokyo University Of Agriculture And Technology | 分離用担体、化合物の分離方法、及びこれを用いたペプチド合成方法 |
| WO2007122847A1 (ja) | 2006-03-24 | 2007-11-01 | National University Corporation, Tokyo University Of Agriculture And Technology | 有機合成用試薬、及び当該試薬を用いた有機合成反応方法 |
| WO2009014177A1 (ja) | 2007-07-25 | 2009-01-29 | Ajinomoto Co., Inc. | ジベンゾフルベン誘導体の淘汰方法 |
| WO2009014176A1 (ja) | 2007-07-25 | 2009-01-29 | Ajinomoto Co., Inc. | ペプチドの製造方法 |
| JP2010106084A (ja) * | 2008-10-28 | 2010-05-13 | Univ Of Tokyo | 液晶材料、化合物、ピエゾクロミック発光材料、液晶材料膜、コーティング材、及び、液晶材料膜製造方法 |
| JP2010192961A (ja) | 2009-02-16 | 2010-09-02 | Epson Toyocom Corp | Sawデバイス |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1226814A (en) * | 1916-03-08 | 1917-05-22 | William J Ruff | Shipping-barrel. |
| FR2652581B1 (fr) | 1989-10-02 | 1991-12-13 | Rhone Poulenc Chimie | Procede de solubilisation de peptides et procede de synthese de peptides. |
| JP2579699Y2 (ja) | 1993-04-06 | 1998-08-27 | オート株式会社 | 筆記具用キャップ |
| RU2144374C1 (ru) * | 1998-11-23 | 2000-01-20 | Закрытое акционерное общество "ВАМ" | Способ получения композита окисленного глутатиона с cis-диаминодихлорплатиной и фармацевтических композиций на его основе, регулирующих метаболизм, пролиферацию, дифференцировку и механизмы апоптоза нормальных и трансформированных клеток |
| CN1298732C (zh) * | 2005-05-17 | 2007-02-07 | 南京工业大学 | 多肽微波固相合成法 |
| CN101463072B (zh) * | 2008-11-11 | 2011-09-28 | 吉尔生化(上海)有限公司 | 一种八肽胆囊收缩素的制备方法 |
| EP2415745A4 (en) | 2009-03-30 | 2014-06-11 | Ajinomoto Kk | DIPHENYLMETHANE COMPOUND |
-
2011
- 2011-08-30 CN CN201710165998.4A patent/CN107011132B/zh active Active
- 2011-08-30 CN CN201610569960.9A patent/CN106220482B/zh active Active
- 2011-08-30 US US13/220,841 patent/US8546534B2/en active Active
- 2011-08-30 WO PCT/JP2011/069624 patent/WO2012029794A1/ja active Application Filing
- 2011-08-30 CN CN201710165359.8A patent/CN107011131B/zh active Active
- 2011-08-30 CN CN201180041916.6A patent/CN103080058B/zh active Active
- 2011-08-30 EP EP11821809.8A patent/EP2612845B1/en active Active
- 2011-08-30 DK DK11821809.8T patent/DK2612845T3/da active
- 2011-08-30 JP JP2012531896A patent/JP5929756B2/ja active Active
-
2013
- 2013-08-29 US US14/013,590 patent/US9499579B2/en active Active
-
2016
- 2016-09-23 US US15/274,463 patent/US10711033B2/en active Active
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000044493A (ja) | 1998-07-27 | 2000-02-15 | Asahi Chem Ind Co Ltd | 化合物ライブラリー合成用保護基 |
| WO2003018188A1 (fr) | 2001-08-24 | 2003-03-06 | Japan Science And Technology Agency | Systeme de solvant multiphase compatible |
| JP2004059509A (ja) * | 2002-07-30 | 2004-02-26 | Nokodai Tlo Kk | 液相ペプチド合成用アミノ酸試薬 |
| JP2004067555A (ja) * | 2002-08-05 | 2004-03-04 | Nokodai Tlo Kk | 液相ペプチド合成用担体、液相ペプチド合成法 |
| JP2006015283A (ja) * | 2004-07-02 | 2006-01-19 | Nokodai Tlo Kk | 化合物の分離用担体および化合物の分離方法 |
| WO2006104166A1 (ja) | 2005-03-29 | 2006-10-05 | National University Corporation, Tokyo University Of Agriculture And Technology | 晶析分離用担体及び化合物の分離方法 |
| WO2007034812A1 (ja) | 2005-09-20 | 2007-03-29 | National University Corporation, Tokyo University Of Agriculture And Technology | 分離用担体、化合物の分離方法、及びこれを用いたペプチド合成方法 |
| WO2007122847A1 (ja) | 2006-03-24 | 2007-11-01 | National University Corporation, Tokyo University Of Agriculture And Technology | 有機合成用試薬、及び当該試薬を用いた有機合成反応方法 |
| WO2009014177A1 (ja) | 2007-07-25 | 2009-01-29 | Ajinomoto Co., Inc. | ジベンゾフルベン誘導体の淘汰方法 |
| WO2009014176A1 (ja) | 2007-07-25 | 2009-01-29 | Ajinomoto Co., Inc. | ペプチドの製造方法 |
| JP2010106084A (ja) * | 2008-10-28 | 2010-05-13 | Univ Of Tokyo | 液晶材料、化合物、ピエゾクロミック発光材料、液晶材料膜、コーティング材、及び、液晶材料膜製造方法 |
| JP2010192961A (ja) | 2009-02-16 | 2010-09-02 | Epson Toyocom Corp | Sawデバイス |
Non-Patent Citations (2)
| Title |
|---|
| "Protective Groups in Organic Synthesis", 1980, JOHN WILEY AND SONS |
| TAMIAKI H. ET AL.: "A Novel Protecting Group for Constructing Combinatorial Peptide Libraries", BULL. CHEM. SOC. JPN., vol. 74, 2001, pages 733 - 738, XP002989508 * |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012165545A1 (ja) * | 2011-05-31 | 2012-12-06 | 味の素株式会社 | ペプチドの製造方法 |
| US9353148B2 (en) | 2011-05-31 | 2016-05-31 | Ajinomoto Co., Inc. | Method for producing peptide |
| EP4049991A1 (en) | 2015-01-21 | 2022-08-31 | Ajinomoto Co., Inc. | Precipitation promoter and precipitation method in which same is used |
| WO2017038650A1 (ja) * | 2015-08-28 | 2017-03-09 | 積水メディカル株式会社 | ベンジル化合物 |
| JP6116782B1 (ja) * | 2015-08-28 | 2017-04-19 | 積水メディカル株式会社 | ベンジル化合物 |
| JP6201076B1 (ja) * | 2015-08-28 | 2017-09-20 | 積水メディカル株式会社 | ベンジル化合物 |
| JP2018002700A (ja) * | 2015-08-28 | 2018-01-11 | 積水メディカル株式会社 | ベンジル化合物 |
| KR20180044305A (ko) | 2015-08-28 | 2018-05-02 | 세키스이 메디칼 가부시키가이샤 | 벤질 화합물 |
| US10822357B2 (en) | 2015-08-28 | 2020-11-03 | Sekisui Medical Co., Ltd. | Benzyl compound |
| US11591351B2 (en) | 2015-08-28 | 2023-02-28 | Sekisui Medical Co., Ltd. | Benzyl compound |
| KR20190018626A (ko) | 2016-06-20 | 2019-02-25 | 세키스이 메디칼 가부시키가이샤 | 신규 디페닐메탄 보호제 |
| US11485747B2 (en) | 2016-06-20 | 2022-11-01 | Sekisui Medical Co., Ltd. | Diphenylmethane protective agent |
| US10851120B2 (en) | 2016-06-20 | 2020-12-01 | Sekisui Medical Co., Ltd. | Diphenylmethane protective agent |
| WO2017221889A1 (ja) | 2016-06-20 | 2017-12-28 | 積水メディカル株式会社 | 新規ジフェニルメタン保護剤 |
| US10508124B2 (en) | 2016-07-25 | 2019-12-17 | Sekisui Medical Co., Ltd. | Xanthene protective agent |
| JP6393857B1 (ja) * | 2017-02-27 | 2018-09-19 | 積水メディカル株式会社 | 新規四環式保護剤 |
| WO2018155669A1 (ja) * | 2017-02-27 | 2018-08-30 | 積水メディカル株式会社 | 新規四環式保護剤 |
| WO2019123994A1 (ja) | 2017-12-19 | 2019-06-27 | 積水メディカル株式会社 | 新規アルキルジフェニルメタン保護剤 |
| US11542287B2 (en) | 2017-12-19 | 2023-01-03 | Sekisui Medical Co., Ltd. | Alkyldiphenylmethane protective agent |
| KR20200100048A (ko) | 2017-12-19 | 2020-08-25 | 세키스이 메디칼 가부시키가이샤 | 신규 알킬디페닐메탄 보호제 |
| US10870667B2 (en) | 2017-12-19 | 2020-12-22 | Sekisui Medical Co., Ltd. | Alkyldiphenylmethane protective agent |
| US11420997B2 (en) | 2018-04-13 | 2022-08-23 | Jitsubo Co., Ltd. | Peptide synthesis method |
| WO2019198833A1 (ja) | 2018-04-13 | 2019-10-17 | Jitsubo株式会社 | ペプチド合成方法 |
| WO2020101032A1 (ja) | 2018-11-16 | 2020-05-22 | 味の素株式会社 | 分子内s-s結合を有する環化ペプチドの製造方法 |
| JP2022516932A (ja) * | 2019-01-07 | 2022-03-03 | 広州同雋医薬科技有限公司 | ジフェニルメタン構造を含有する化合物及びその応用 |
| JP7418027B2 (ja) | 2019-01-07 | 2024-01-19 | 広州同雋医薬科技有限公司 | ジフェニルメタン構造を含有する化合物及びその応用 |
| WO2020218497A1 (ja) * | 2019-04-25 | 2020-10-29 | 味の素株式会社 | ペプチドの連続的製造方法 |
| WO2023277186A1 (ja) * | 2021-07-02 | 2023-01-05 | ペプチスター株式会社 | 液相ペプチド合成用担体結合ペプチドの分析方法 |
| WO2023127331A1 (ja) * | 2021-12-27 | 2023-07-06 | 株式会社トクヤマ | ペプチド製造方法、保護基の除去方法、除去剤、及びベンジル化合物 |
| JP2023097442A (ja) * | 2021-12-27 | 2023-07-07 | 株式会社トクヤマ | ペプチド製造方法、及びベンジル化合物 |
| JP7356607B2 (ja) | 2021-12-27 | 2023-10-04 | 株式会社トクヤマ | ペプチド製造方法、及びベンジル化合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| US9499579B2 (en) | 2016-11-22 |
| US20120059149A1 (en) | 2012-03-08 |
| JP5929756B2 (ja) | 2016-06-08 |
| CN107011132A (zh) | 2017-08-04 |
| US8546534B2 (en) | 2013-10-01 |
| CN103080058A (zh) | 2013-05-01 |
| JPWO2012029794A1 (ja) | 2013-10-31 |
| EP2612845B1 (en) | 2020-08-05 |
| US20170008922A1 (en) | 2017-01-12 |
| DK2612845T3 (da) | 2020-11-02 |
| EP2612845A1 (en) | 2013-07-10 |
| CN106220482A (zh) | 2016-12-14 |
| CN107011131B (zh) | 2020-11-10 |
| CN107011131A (zh) | 2017-08-04 |
| EP2612845A4 (en) | 2015-08-05 |
| US20140005359A1 (en) | 2014-01-02 |
| CN107011132B (zh) | 2020-11-10 |
| US10711033B2 (en) | 2020-07-14 |
| CN103080058B (zh) | 2017-04-12 |
| CN106220482B (zh) | 2020-01-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5929756B2 (ja) | 分岐鎖含有芳香族化合物 | |
| US8722934B2 (en) | Diphenylmethane compound | |
| JP5803674B2 (ja) | ベンジル化合物 | |
| JP6350632B2 (ja) | ペプチドの製造方法 | |
| US8569453B2 (en) | Fluorene compound | |
| WO2012165545A1 (ja) | ペプチドの製造方法 | |
| CN113508103A (zh) | 肽化合物的制造方法、保护基形成用试药及稠合多环芳香族烃化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180041916.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11821809 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012531896 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011821809 Country of ref document: EP |