WO2011081099A1 - ポリアミド化合物 - Google Patents
ポリアミド化合物 Download PDFInfo
- Publication number
- WO2011081099A1 WO2011081099A1 PCT/JP2010/073371 JP2010073371W WO2011081099A1 WO 2011081099 A1 WO2011081099 A1 WO 2011081099A1 JP 2010073371 W JP2010073371 W JP 2010073371W WO 2011081099 A1 WO2011081099 A1 WO 2011081099A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyamide compound
- mol
- unit
- polyamide
- acid
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/36—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from amino acids, polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/06—Polyamides derived from polyamines and polycarboxylic acids
Definitions
- the present invention relates to a polyamide compound (including a polyamide resin and a polyamide oligomer) that exhibits oxygen absorption performance.
- thermoplastic resins Conventionally, metal cans, glass bottles, or containers and molded articles made of thermoplastic resins have been used as packaging materials for pharmaceuticals, beverages, foods, chemicals, and the like.
- containers and molded bodies made of thermoplastic resins are superior in terms of light weight, moldability, packaging productivity such as heat sealing, and cost, and are used in the largest quantities.
- containers and molded bodies made of thermoplastic resin are generally excellent as packaging materials, but oxygen permeation through the container wall occurs in an order that cannot be ignored, and there remains a problem in terms of storage stability of contents. .
- the container wall or molded body of the thermoplastic resin has a multi-layer structure on the container wall, and at least one of them is polymetaxylylene adipamide (hereinafter referred to as “N-MXD6”). ), An oxygen barrier layer such as an ethylene-vinyl alcohol copolymer, polyacrylonitrile, or aluminum foil is provided.
- N-MXD6 polymetaxylylene adipamide
- An oxygen barrier layer such as an ethylene-vinyl alcohol copolymer, polyacrylonitrile, or aluminum foil is provided.
- Patent Documents 1 and 2 describe an oxygen-absorbing multilayer body and an oxygen-absorbing film in which an oxygen absorbent such as iron powder is dispersed in a resin.
- Patent Document 3 describes a packaging oxygen collection barrier that absorbs oxygen inside and outside a container obtained by adding a metal catalyst such as cobalt to a polymer material such as polyamide.
- Patent Document 4 describes a product having an oxygen scavenging layer containing an ethylenically unsaturated compound such as polybutadiene and a transition metal catalyst such as cobalt, and an oxygen barrier layer such as polyamide.
- Japanese Patent Laid-Open No. 2-72851 Japanese Patent Laid-Open No. 4-90848 Japanese Patent No. 2991437 Japanese Patent Laid-Open No. 5-115776
- Oxygen-absorbing multilayers and oxygen-absorbing films in which an oxygen absorbent such as iron powder is dispersed in the resin are opaque because the resin is colored by the oxygen absorbent such as iron powder. There is an application restriction that it cannot be used in the field.
- an oxygen scavenging resin composition containing a transition metal such as cobalt has an advantage that it can be applied to packaging containers that require transparency, but is not preferred because the resin composition is colored by a transition metal catalyst. . In these resin compositions, the resin is oxidized by absorbing oxygen by the transition metal catalyst.
- the generation of radicals due to the extraction of hydrogen atoms from the methylene chain adjacent to the arylene group of the polyamide resin by transition metal atoms the generation of peroxy radicals by the addition of oxygen molecules to the radicals, the peroxy radicals It is thought to occur by each reaction such as extraction of hydrogen atoms by. Since the resin is oxidized by oxygen absorption by such a mechanism, a decomposition product is generated and an unpleasant odor is generated in the contents of the container, or the color tone or strength of the container is impaired due to oxidative degradation of the resin. is there.
- An object of the present invention is to provide a polyamide compound exhibiting sufficient oxygen absorption performance without containing a metal, having no unpleasant odor, and having extremely good transparency.
- the present invention provides the following polyamide compounds. 25 to 50 mol% of a diamine unit containing 50 mol% or more of an aromatic diamine unit represented by the following general formula (I), a linear aliphatic dicarboxylic acid unit represented by the following general formula (II-1) and / or Or 25 to 50 mol% of dicarboxylic acid units containing a total of 50 mol% or more of aromatic dicarboxylic acid units represented by the following general formula (II-2), and structural units represented by the following general formula (III): A polyamide compound containing 1 to 50 mol%.
- an aromatic diamine unit represented by the following general formula (I) a linear aliphatic dicarboxylic acid unit represented by the following general formula (II-1) and / or Or 25 to 50 mol% of dicarboxylic acid units containing a total of 50 mol% or more of aromatic dicarboxylic acid units represented by the following general formula (II-2), and structural units represented by the following general formula (III): A poly
- n represents an integer of 2 to 18.
- Ar represents an arylene group.
- R represents a substituted or unsubstituted alkyl group or a substituted or unsubstituted aryl group.
- the polyamide compound of the present invention is excellent in oxygen absorption. Therefore, for example, the polyamide compound of the present invention is suitable for use as an oxygen absorbent by filling a sachet or the like.
- use in a packaging material or a packaging container is exemplified.
- the packaging material or packaging container using the polyamide compound of the present invention exhibits sufficient oxygen absorption performance without containing metal, does not generate unpleasant odor, has very good transparency, and contains Can be stored in good condition.
- FIG. 2 is a 1 H-NMR chart of polyamide compound 101 produced in Example 101.
- FIG. 2 is a 1 H-NMR chart of polyamide compound 201 produced in Example 201.
- Polyamide compound The polyamide compound of the present invention is represented by 25 to 50 mol% of a diamine unit containing 50 mol% or more of an aromatic diamine unit represented by the following general formula (I), and represented by the following general formula (II-1). 25 to 50 mol% of dicarboxylic acid units containing a total of 50 mol% or more of linear aliphatic dicarboxylic acid units and / or aromatic dicarboxylic acid units represented by the following general formula (II-2), and tertiary hydrogen-containing carboxylic acids And 0.1 to 50 mol% of an acid unit (preferably a structural unit represented by the following general formula (III)).
- an acid unit preferably a structural unit represented by the following general formula (III)
- n represents an integer of 2 to 18.
- Ar represents an arylene group.
- R represents a substituted or unsubstituted alkyl group or a substituted or unsubstituted aryl group.
- the total of the diamine unit, the dicarboxylic acid unit, and the tertiary hydrogen-containing carboxylic acid unit shall not exceed 100 mol%.
- the polyamide compound of the present invention may further contain structural units other than those described above as long as the effects of the present invention are not impaired.
- the polyamide compound of the present invention includes a polyamide resin and a polyamide oligomer.
- the “polyamide resin” of the present invention means a polymer having a relative viscosity of 1.8 or more in the polyamide compound of the present invention.
- Polyamide resin is a material that can be molded independently, and can be processed into a packaging material or a packaging container. If necessary, other resins and additives may be added to and mixed with the polyamide resin of the present invention, and the polyamide composition thus obtained may be molded.
- the polyamide resin of the present invention exhibits sufficient oxygen absorption performance without containing a metal, has no unpleasant odor, and has extremely good transparency.
- polyamide oligomer of the present invention means a polymer having a relative viscosity of less than 1.8 in the polyamide compound of the present invention.
- Polyamide oligomers are materials that cannot normally be molded by themselves.
- an oligomer often refers to a polymer having a number average molecular weight of 1000 or less, but the polyamide oligomer of the present invention includes not only such a general oligomer but also a polymer having a number average molecular weight of less than 10,000. Combinations can also be included.
- the polyamide oligomer of the present invention is suitable for filling into a sachet or the like and used as an oxygen absorbent. Moreover, the polyamide oligomer of this invention can be used conveniently as a resin raw material or a resin additive. When the polyamide oligomer of the present invention is used as a resin raw material, the polyamide oligomer can be copolymerized with another resin raw material to obtain a copolymer resin, and the copolymer resin is molded and processed into a packaging material or a packaging container. can do.
- a resin composition obtained by adding a polyamide oligomer to a resin can be molded and processed into a packaging material or a packaging container. At this time, sufficient oxygen absorption performance can be expressed without deteriorating the transparency and mechanical strength of the resin.
- the copolymer resin or resin composition obtained using the polyamide oligomer of the present invention exhibits sufficient oxygen absorption performance without containing a metal, and does not generate unpleasant odor.
- the content of the tertiary hydrogen-containing carboxylic acid unit is 0.1 to 50 mol%. If the content of the tertiary hydrogen-containing carboxylic acid unit is less than 0.1 mol%, sufficient oxygen absorption performance is not exhibited. On the other hand, if the content of the tertiary hydrogen-containing carboxylic acid unit exceeds 50 mol%, the tertiary hydrogen content is too high, so that the physical properties such as gas barrier properties and mechanical properties of the polyamide compound are deteriorated, especially the tertiary hydrogen content.
- the carboxylic acid is an amino acid
- the peptide bond is continuous, so that the heat resistance is not sufficient, and a cyclic product consisting of a dimer of amino acids is formed, which inhibits polymerization.
- the content of the tertiary hydrogen-containing carboxylic acid unit is preferably 0.2 mol% or more, more preferably 1 mol% or more, and preferably 40 mol% from the viewpoint of oxygen absorption performance and properties of the polyamide compound. Or less, more preferably 30 mol% or less.
- the content of diamine units is 25 to 50 mol%, and preferably 30 to 50 mol% from the viewpoint of oxygen absorption performance and polymer properties.
- the content of dicarboxylic acid units is 25 to 50 mol%, preferably 30 to 50 mol%.
- the proportion of the content of the diamine unit and the dicarboxylic acid unit is preferably substantially the same from the viewpoint of the polymerization reaction, and the content of the dicarboxylic acid unit is ⁇ 2 mol% of the content of the diamine unit. More preferred.
- the degree of polymerization of the polyamide compound becomes difficult to increase, so it takes a lot of time to increase the degree of polymerization and thermal degradation occurs. It becomes easy.
- the diamine unit in the polyamide compound of the present invention has the above general formula (I) from the viewpoint of improving transparency and color tone and facilitating moldability in addition to imparting excellent gas barrier properties to the polyamide compound.
- the diamine unit contains 50 mol% or more of the aromatic diamine unit, and the content is preferably 70 mol% or more, more preferably 80 mol% or more, still more preferably 90 mol% or more, Preferably, it is 100 mol% or less.
- Examples of the compound that can constitute the aromatic diamine unit represented by the general formula (I) include orthoxylylenediamine, metaxylylenediamine, and paraxylylenediamine. These can be used alone or in combination of two or more.
- the diamine unit in the polyamide compound of the present invention contains at least 50 mol% of a metaxylylenediamine unit from the viewpoint of facilitating moldability of a general-purpose thermoplastic resin in addition to exhibiting excellent gas barrier properties.
- the content is preferably 70 mol% or more, more preferably 80 mol% or more, still more preferably 90 mol% or more, and preferably 100 mol% or less.
- Examples of compounds that can constitute a diamine unit other than the aromatic diamine unit represented by the formula (I) include aromatic diamines such as paraphenylenediamine, 2-methyl-1,5-pentanediamine, and 1-amino-3.
- aromatic diamines such as paraphenylenediamine, 2-methyl-1,5-pentanediamine, and 1-amino-3.
- examples include aliphatic diamines such as aminomethyl-3,5,5-trimethylcyclohexane, polyether diamines having an ether bond represented by Huntsman's Jeffamine and Elastamine (both trade names), It is not limited to these. These can be used alone or in combination of two or more.
- the dicarboxylic acid unit in the polyamide compound of the present invention is a linear aliphatic group represented by the general formula (II-1) from the viewpoints of reactivity during polymerization and crystallinity and moldability of the polyamide compound.
- the dicarboxylic acid unit and / or the aromatic dicarboxylic acid unit represented by the general formula (II-2) is contained in the dicarboxylic acid unit in a total of 50 mol% or more, and the content is preferably 70 mol% or more, more Preferably it is 80 mol% or more, More preferably, it is 90 mol% or more, Preferably it is 100 mol% or less.
- Examples of the compound that can constitute a dicarboxylic acid unit other than the dicarboxylic acid unit represented by the general formula (II-1) or (II-2) include oxalic acid, malonic acid, fumaric acid, maleic acid, 1,3- Examples thereof include, but are not limited to, dicarboxylic acids such as benzenediacetic acid and 1,4-benzenediacetic acid.
- the content ratio of the linear aliphatic dicarboxylic acid unit to the aromatic dicarboxylic acid unit is particularly limited. Rather, it is determined appropriately according to the application. For example, when the purpose is to increase the glass transition temperature of the polyamide compound to reduce the crystallinity of the polyamide compound, the total of both units of the linear aliphatic dicarboxylic acid unit / aromatic dicarboxylic acid unit is 100. Sometimes 0/100 to 60/40, more preferably 0/100 to 40/60, still more preferably 0/100 to 30/70.
- the linear aliphatic dicarboxylic acid unit / aromatic dicarboxylic acid unit is 100 It is preferably 40/60 to 100/0, more preferably 60/40 to 100/0, still more preferably 70/30 to 100/0.
- Linear aliphatic dicarboxylic acid unit The polyamide compound of the present invention, in addition to imparting an appropriate glass transition temperature and crystallinity to the polyamide compound, in the case of providing the necessary flexibility as a packaging material or packaging container, It preferably contains a linear aliphatic dicarboxylic acid unit represented by the general formula (II-1).
- n represents an integer of 2 to 18, preferably 3 to 16, more preferably 4 to 12, and still more preferably 4 to 8.
- Examples of compounds that can constitute the linear aliphatic dicarboxylic acid unit represented by the general formula (II-1) include succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, 1, Examples thereof include, but are not limited to, 10-decanedicarboxylic acid, 1,11-undecanedicarboxylic acid, 1,12-dodecanedicarboxylic acid, and the like. These can be used alone or in combination of two or more.
- the type of the linear aliphatic dicarboxylic acid unit represented by the general formula (II-1) is appropriately determined according to the application.
- the linear aliphatic dicarboxylic acid unit in the polyamide compound of the present invention provides adipic acid in addition to imparting excellent gas barrier properties to the polyamide compound and maintaining heat resistance after heat sterilization of packaging materials and packaging containers.
- At least one selected from the group consisting of a unit, a sebacic acid unit, and a 1,12-dodecanedicarboxylic acid unit is contained in a total of 50 mol% or more in the linear aliphatic dicarboxylic acid unit, and the content is More preferably, it is 70 mol% or more, more preferably 80 mol% or more, particularly preferably 90 mol% or more, and preferably 100 mol% or less.
- the linear aliphatic dicarboxylic acid unit in the polyamide compound of the present invention is a linear aliphatic dicarboxylic acid unit from the viewpoint of thermal properties such as gas barrier properties and appropriate glass transition temperature and melting point of the polyamide compound. It is preferable to contain 50 mol% or more.
- the linear aliphatic dicarboxylic acid unit in the polyamide compound of the present invention has 50 sebacic acid units in the linear aliphatic dicarboxylic acid unit from the viewpoint of imparting appropriate gas barrier properties and molding processability to the polyamide compound.
- 1,12-dodecanedicarboxylic acid units be added to 50% of the linear aliphatic dicarboxylic acid units. It is preferable to contain more than mol%.
- Aromatic dicarboxylic acid unit The polyamide compound of the present invention has the above general formula (II-2) for the purpose of facilitating the molding processability of packaging materials and packaging containers in addition to imparting further gas barrier properties to the polyamide compound. It is preferable that the aromatic dicarboxylic acid unit represented by this is included.
- Ar represents an arylene group.
- the arylene group is preferably an arylene group having 6 to 30 carbon atoms, more preferably 6 to 15 carbon atoms, and examples thereof include a phenylene group and a naphthylene group.
- Examples of the compound that can constitute the aromatic dicarboxylic acid unit represented by the general formula (II-2) include terephthalic acid, isophthalic acid, and 2,6-naphthalenedicarboxylic acid, but are not limited thereto. is not. These can be used alone or in combination of two or more.
- the kind of the aromatic dicarboxylic acid unit represented by the general formula (II-2) is appropriately determined according to the use.
- the aromatic dicarboxylic acid unit in the polyamide compound of the present invention is a total of at least one selected from the group consisting of an isophthalic acid unit, a terephthalic acid unit, and a 2,6-naphthalenedicarboxylic acid unit in the aromatic dicarboxylic acid unit.
- the content is preferably 70 mol% or more, more preferably 80 mol% or more, particularly preferably 90 mol% or more, and preferably 100 mol% or less. is there. Among these, it is preferable to contain isophthalic acid and / or terephthalic acid in the aromatic dicarboxylic acid unit.
- the content ratio of the isophthalic acid unit to the terephthalic acid unit is not particularly limited and is appropriately determined according to the application.
- the total of both units is preferably 100/100 to 100/0, more preferably 0/100 to 60/40, and even more preferably 0. / 100 to 40/60, more preferably 0/100 to 30/70.
- tertiary hydrogen-containing carboxylic acid unit has at least one amino group and one carboxyl group or two carboxyl groups from the viewpoint of polymerization of the polyamide compound. Have more. Specific examples include structural units represented by any of the following general formulas (III), (IV), or (V).
- R, R 1 and R 2 each represent a substituent, and A 1 to A 3 each represent a single bond or a divalent linking group. However, the case where both A 1 and A 2 in the general formula (IV) are single bonds is excluded. ]
- the polyamide compound of the present invention contains a tertiary hydrogen-containing carboxylic acid unit.
- a tertiary hydrogen-containing carboxylic acid unit as a copolymerization component, the polyamide compound of the present invention can exhibit excellent oxygen absorption performance without containing a transition metal.
- the mechanism by which the polyamide compound having a tertiary hydrogen-containing carboxylic acid unit exhibits good oxygen absorption performance has not yet been clarified, but is estimated as follows.
- a compound that can constitute a tertiary hydrogen-containing carboxylic acid unit an electron-withdrawing group and an electron-donating group are bonded to the same carbon atom.
- a very stable radical is generated by a phenomenon called a stabilized captodative effect. That is, the carboxyl group is an electron withdrawing group, and the carbon to which the adjacent tertiary hydrogen is bonded becomes electron deficient ( ⁇ + ), so the tertiary hydrogen also becomes electron deficient ( ⁇ + ) and dissociates as a proton.
- radicals When oxygen and water are present here, it is considered that oxygen reacts with this radical to show oxygen absorption performance. It has also been found that the higher the humidity and temperature, the higher the reactivity.
- R, R 1 and R 2 each represent a substituent.
- substituent represented by R, R 1 and R 2 in the present invention include a halogen atom (eg, chlorine atom, bromine atom, iodine atom), alkyl group (1 to 15, preferably 1 to 6).
- Linear, branched or cyclic alkyl groups having the following carbon atoms for example, methyl group, ethyl group, n-propyl group, isopropyl group, t-butyl group, n-octyl group, 2-ethylhexyl group, cyclopropyl group, cyclopentyl Group), an alkenyl group (a linear, branched or cyclic alkenyl group having 2 to 10, preferably 2 to 6 carbon atoms, such as a vinyl group, an allyl group), an alkynyl group (2 to 10, preferably Alkynyl groups having 2 to 6 carbon atoms, such as ethynyl groups, propargyl groups), aryl groups (aryls having 6 to 16, preferably 6 to 10 carbon atoms) 1 to 12 groups obtained by removing one hydrogen atom from a group, for example, phenyl group, naphthyl group, heterocyclic group (5-membered or 6-
- An alkylthio group an alkylthio group having 1 to 10, preferably 1 to 6 carbon atoms, such as a methylthio group, an ethylthio group
- an arylthio group (6 to 12, preferably 6 to 8 carbon atoms).
- heterocyclic thio groups for example, heterocyclic thio groups having 2 to 10, preferably 1 to 6 carbon atoms, such as - benzothiazolylthio group
- an imido group (2 to 10, preferably an imido group having 4 to 8 carbon atoms, for example, N- succinimido group, N- phthalimido group.
- those having a hydrogen atom may be further substituted with the above groups, for example, an alkyl group substituted with a hydroxyl group (for example, hydroxyethyl group), an alkyl group substituted with an alkoxy group (For example, a methoxyethyl group), an alkyl group substituted with an aryl group (for example, a benzyl group), an aryl group substituted with an alkyl (for example, a p-tolyl group), an aryloxy group substituted with an alkyl group (for example, , 2-methylphenoxy group) and the like, but is not limited thereto.
- a hydroxyl group for example, hydroxyethyl group
- an alkoxy group for example, a methoxyethyl group
- an alkyl group substituted with an aryl group for example, a benzyl group
- an aryl group substituted with an alkyl for example, a p-tolyl group
- a functional group when a functional group is further substituted, the carbon number mentioned above shall not include the carbon number of the further substituent.
- a benzyl group is regarded as a C 1 alkyl group substituted with a phenyl group, and is not regarded as a C 7 alkyl group substituted with a phenyl group.
- the following description of the number of carbon atoms shall be similarly understood unless otherwise specified.
- a 1 to A 3 each represents a single bond or a divalent linking group.
- the divalent linking group include linear, branched or cyclic alkylene groups (C 1-12, preferably C 1-4 alkylene groups such as methylene and ethylene groups), aralkylene groups (carbon numbers). Examples thereof include an aralkylene group having 7 to 30 carbon atoms, preferably 7 to 13 carbon atoms, such as a benzylidene group, and an arylene group (arylene group having 6 to 30 carbon atoms, preferably 6 to 15 carbon atoms such as a phenylene group).
- substituents represented by R, R 1 and R 2 examples include the functional groups exemplified above as substituents represented by R, R 1 and R 2 .
- substituents represented by R, R 1 and R 2 examples include, but are not limited to, an arylene group (for example, a xylylene group) substituted with alkyl.
- the polyamide compound of the present invention preferably contains at least one structural unit represented by any one of the general formulas (III), (IV) or (V).
- a carboxylic acid unit having tertiary hydrogen on the ⁇ -carbon (carbon atom adjacent to the carboxyl group) is more preferable, and is represented by the general formula (III).
- the structural unit is particularly preferred.
- R in the general formula (III) is as described above.
- a substituted or unsubstituted alkyl group and a substituted or unsubstituted aryl group are more preferable, and a substituted or unsubstituted C 1-6 carbon atom is more preferable.
- An alkyl group and a substituted or unsubstituted aryl group having 6 to 10 carbon atoms are more preferred, and a substituted or unsubstituted alkyl group having 1 to 4 carbon atoms and a substituted or unsubstituted phenyl group are particularly preferred.
- R examples include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, t-butyl group, 1-methylpropyl group, 2-methylpropyl group, hydroxymethyl group, 1- Examples thereof include, but are not limited to, a hydroxyethyl group, a mercaptomethyl group, a methylsulfanylethyl group, a phenyl group, a naphthyl group, a benzyl group, and a 4-hydroxybenzyl group. Among these, a methyl group, an ethyl group, a 2-methylpropyl group, and a benzyl group are more preferable.
- Examples of the compound that can constitute the structural unit represented by the general formula (III) include alanine, 2-aminobutyric acid, valine, norvaline, leucine, norleucine, tert-leucine, isoleucine, serine, threonine, cysteine, methionine, 2 -Alpha-amino acids such as phenylglycine, phenylalanine, tyrosine, histidine, tryptophan, proline and the like can be exemplified, but are not limited thereto.
- examples of the compound that can constitute the structural unit represented by the general formula (IV) include ⁇ -amino acids such as 3-aminobutyric acid, which constitute the structural unit represented by the general formula (V).
- examples of the compound that can be used include, but are not limited to, dicarboxylic acids such as methylmalonic acid, methylsuccinic acid, malic acid, and tartaric acid. These may be any of D-form, L-form and racemate, or allo-form. Moreover, these can be used individually or in combination of 2 or more types.
- ⁇ -amino acids having tertiary hydrogen in the ⁇ carbon are particularly preferable from the viewpoint of availability of raw materials and improvement of oxygen absorption.
- alanine is most preferable from the viewpoints of ease of supply, inexpensive price, ease of polymerization, and low yellowness (YI) of the polymer. Since alanine has a relatively low molecular weight and a high copolymerization rate per 1 g of the polyamide compound of the present invention, oxygen absorption performance per 1 g of the polyamide compound is good.
- the purity of the compound that can constitute the tertiary hydrogen-containing carboxylic acid unit is 95% or more from the viewpoint of the influence on the polymerization such as the delay of the polymerization rate and the influence on the quality such as the yellowness of the polymer. Preferably, it is 98.5% or more, more preferably 99% or more.
- sulfate ions and ammonium ions contained as impurities are preferably 500 ppm or less, more preferably 200 ppm or less, and still more preferably 50 ppm or less.
- ⁇ -aminocarboxylic acid unit The polyamide compound of the present invention has the following general formula in addition to the diamine unit, the dicarboxylic acid unit and the tertiary hydrogen-containing carboxylic acid unit when the polyamide compound needs flexibility or the like.
- the ⁇ -aminocarboxylic acid unit represented by (A) may be further contained.
- p represents an integer of 2 to 18.
- the content of the ⁇ -aminocarboxylic acid unit is preferably from 0.1 to 49.9 mol%, more preferably from 3 to 40 mol%, still more preferably from 5 to 35 mol% in all the structural units of the polyamide compound. is there.
- the total of the diamine unit, dicarboxylic acid unit, tertiary hydrogen-containing carboxylic acid unit, and ⁇ -aminocarboxylic acid unit does not exceed 100 mol%.
- p represents an integer of 2 to 18, preferably 3 to 16, more preferably 4 to 14, and still more preferably 5 to 12.
- Examples of compounds that can constitute the ⁇ -aminocarboxylic acid unit represented by the general formula (A) include ⁇ -aminocarboxylic acids having 5 to 19 carbon atoms and lactams having 5 to 19 carbon atoms.
- Examples of the ⁇ -aminocarboxylic acid having 5 to 19 carbon atoms include 6-aminohexanoic acid and 12-aminododecanoic acid, and examples of the lactam having 5 to 19 carbon atoms include ⁇ -caprolactam and laurolactam. However, it is not limited to these. These can be used alone or in combination of two or more.
- the ⁇ -aminocarboxylic acid unit preferably contains 6-aminohexanoic acid units and / or 12-aminododecanoic acid units in a total of 50 mol% or more in the ⁇ -aminocarboxylic acid unit, and the content is More preferably, it is 70 mol% or more, More preferably, it is 80 mol% or more, More preferably, it is 90 mol% or more, Preferably it is 100 mol% or less.
- Relative viscosity is used for the degree of polymerization of the polyamide compound of the present invention.
- the preferred relative viscosity of the polyamide compound of the present invention is preferably 1.01 to 4.2.
- the relative viscosity is preferably from 1.8 to 4.2, more preferably from 1.9 to 4.0, from the viewpoint of the appearance of the molded product and molding processability. It is preferably 2.0 to 3.8.
- the polyamide resin of the present invention is used as an additive or modifier for other thermoplastic resins, it is not limited to this range.
- the relative viscosity is preferably 1.01 or more and less than 1.8, more preferably 1.1 to 1.1, from the viewpoints of handleability, reactivity, and thermal stability. 75, more preferably 1.2 to 1.65, particularly preferably 1.3 to 1.6.
- the oxygen absorption rate of the polyamide compound and the oxidative degradation of the polyamide compound due to oxygen absorption can be controlled by changing the terminal amino group concentration of the polyamide compound.
- the terminal amino group concentration is preferably in the range of 5 to 150 eq / 10 6 g, more preferably 10 to 100 eq / 10 6 g, from the viewpoint of the balance between oxygen absorption rate and oxidative degradation. It is preferably 15 to 80 eq / 10 6 g.
- the polyamide compound of the present invention comprises a diamine component that can constitute the diamine unit, a dicarboxylic acid component that can constitute the dicarboxylic acid unit, and a tertiary that can constitute the tertiary hydrogen-containing carboxylic acid unit. It can be produced by polycondensation of a hydrogen-containing carboxylic acid component and, if necessary, the ⁇ -aminocarboxylic acid component that can constitute the ⁇ -aminocarboxylic acid unit.
- the polymerization degree can be adjusted by adjusting the polycondensation conditions and the like. Can be controlled. A small amount of monoamine or monocarboxylic acid may be added as a molecular weight modifier during polycondensation. Further, in order to suppress the polycondensation reaction and obtain a desired degree of polymerization, the ratio (molar ratio) between the diamine component and the carboxylic acid component constituting the polyamide compound may be adjusted by shifting from 1.
- Examples of the polycondensation method of the polyamide compound of the present invention include, but are not limited to, a reactive extrusion method, a pressurized salt method, an atmospheric pressure dropping method, and a pressure dropping method. Moreover, the one where reaction temperature is as low as possible can suppress the yellowing and gelatinization of a polyamide compound, and the polyamide compound of the stable property is obtained.
- a polyamide composed of a diamine component and a dicarboxylic acid component (polyamide corresponding to the precursor of the polyamide compound of the present invention) or a polyamide composed of a diamine component, a dicarboxylic acid component and an ⁇ -aminocarboxylic acid component (this The polyamide corresponding to the precursor of the polyamide compound of the invention) and a tertiary hydrogen-containing carboxylic acid component are reacted by melting and kneading with an extruder.
- This is a method of incorporating a tertiary hydrogen-containing carboxylic acid component into a polyamide skeleton by an amide exchange reaction.
- a screw suitable for reactive extrusion is used, and a twin screw extruder having a large L / D is used. It is preferable to use it.
- a polyamide compound containing a small amount of a tertiary hydrogen-containing carboxylic acid component this is a simple method and suitable.
- the pressure salt method is a method of performing melt polycondensation under pressure using nylon salt as a raw material. Specifically, after preparing an aqueous nylon salt solution comprising a diamine component, a dicarboxylic acid component, a tertiary hydrogen-containing carboxylic acid component, and an ⁇ -aminocarboxylic acid component as necessary, the aqueous solution is concentrated, Next, the temperature is raised under pressure, and polycondensation is performed while removing condensed water. While the inside of the can is gradually returned to normal pressure, the temperature is raised to about the melting point of the polyamide compound + 10 ° C.
- the pressurized salt method is useful when a volatile component is used as a monomer, and is a preferred polycondensation method when the copolymerization rate of the tertiary hydrogen-containing carboxylic acid component is high. It is suitable when the acid component is contained in an amount of 15 mol% or more in all components constituting the polyamide compound.
- Normal pressure dropping method In the normal pressure dropping method, a diamine component is added to a mixture obtained by heating and melting a dicarboxylic acid component, a tertiary hydrogen-containing carboxylic acid component, and, if necessary, an ⁇ -aminocarboxylic acid component under normal pressure. It is continuously dropped and polycondensed while removing condensed water. The polycondensation reaction is performed while raising the temperature of the reaction system so that the reaction temperature does not fall below the melting point of the polyamide compound to be produced. Compared with the pressurized salt method, the atmospheric pressure dropping method does not use water to dissolve the salt, so the yield per batch is large, and the reaction rate is not required for vaporization / condensation of raw material components. The process time can be shortened.
- a dicarboxylic acid component, a tertiary hydrogen-containing carboxylic acid component, and, if necessary, an ⁇ -aminocarboxylic acid component are charged into a polycondensation can, and each component is stirred. Melt mix to prepare the mixture.
- the diamine component is continuously dropped into the mixture while the inside of the can is preferably pressurized to about 0.3 to 0.4 MPaG, and polycondensation is performed while removing condensed water. At this time, the polycondensation reaction is performed while raising the temperature of the reaction system so that the reaction temperature does not fall below the melting point of the polyamide compound to be produced.
- the pressure dropping method is useful when a volatile component is used as a monomer, and is a preferred polycondensation method when the copolymerization rate of the tertiary hydrogen-containing carboxylic acid component is high. .
- the tertiary hydrogen-containing carboxylic acid component is contained in an amount of 15 mol% or more in all components constituting the polyamide compound.
- the pressure dropping method By using the pressure dropping method, the transpiration of the tertiary hydrogen-containing carboxylic acid component can be prevented, and further, the polycondensation between the tertiary hydrogen-containing carboxylic acid components can be suppressed, and the polycondensation reaction can proceed smoothly. Therefore, a polyamide compound having excellent properties can be obtained.
- the pressure drop method does not use water for dissolving the salt compared to the pressure salt method, the yield per batch is large, and the reaction time can be shortened as in the atmospheric pressure drop method. A polyamide compound having a low yellowness can be obtained.
- Step of increasing the degree of polymerization The polyamide compound produced by the above polycondensation method can be used as it is, but may be subjected to a step of further increasing the degree of polymerization. Further examples of the step of increasing the degree of polymerization include reactive extrusion in an extruder and solid phase polymerization.
- the heating device used in the solid phase polymerization includes a continuous heating drying device, a tumble dryer, a conical dryer, a rotary drum heating device called a rotary dryer, etc., and a rotary blade inside a nauta mixer.
- a conical heating device can be preferably used, but a known method and device can be used without being limited thereto.
- the heating device of the rotating drum type in the above-mentioned apparatus can seal the inside of the system and facilitate polycondensation in a state where oxygen that causes coloring is removed.
- oxygen that causes coloring is removed.
- Phosphorus atom-containing compound, alkali metal compound In the polycondensation of the polyamide compound of the present invention, it is preferable to add a phosphorus atom-containing compound from the viewpoint of promoting the amidation reaction.
- the phosphorus atom-containing compound include phosphinic acid compounds such as dimethylphosphinic acid and phenylmethylphosphinic acid; hypophosphorous acid, sodium hypophosphite, potassium hypophosphite, lithium hypophosphite, magnesium hypophosphite, Diphosphite compounds such as calcium hypophosphite and ethyl hypophosphite; phosphonic acid, sodium phosphonate, potassium phosphonate, lithium phosphonate, potassium phosphonate, magnesium phosphonate, calcium phosphonate, phenylphosphonic acid, ethylphosphone Phosphonic acid compounds such as acid, sodium phenylphosphonate, potassium phenylphosphonate, lithium phenyl
- Phosphonic acid compounds Phosphonic acid compounds; phosphorous acid, sodium hydrogen phosphite, sodium phosphite, lithium phosphite, potassium phosphite, magnesium phosphite, calcium phosphite, triethyl phosphite, triphenyl phosphite, pyro-subite
- Examples thereof include phosphorous acid compounds such as phosphoric acid.
- hypophosphite metal salts such as sodium hypophosphite, potassium hypophosphite, lithium hypophosphite and the like are particularly preferable because they are highly effective in promoting amidation reaction and excellent in anti-coloring effect.
- sodium hypophosphite is preferred.
- the phosphorus atom containing compound which can be used by this invention is not limited to these compounds.
- the addition amount of the phosphorus atom-containing compound is preferably 0.1 to 1000 ppm, more preferably 1 to 600 ppm, still more preferably 5 to 400 ppm in terms of phosphorus atom concentration in the polyamide compound. If it is 0.1 ppm or more, the polyamide compound is difficult to be colored during polymerization, and transparency is increased. If it is 1000 ppm or less, the polyamide compound is less likely to be gelled, and it is possible to reduce the mixing of fish eyes considered to be caused by the phosphorus atom-containing compound into the molded product, thereby improving the appearance of the molded product.
- an alkali metal compound in combination with the phosphorus atom-containing compound in the polycondensation system of the polyamide compound.
- an alkali metal compound alkali metal hydroxide, alkali metal acetate, alkali metal carbonate, alkali metal alkoxide, and the like are preferable.
- Sodium methoxide, sodium ethoxide, sodium propoxide, sodium butoxide, potassium methoxide, lithium methoxide, sodium carbonate and the like but can be used without being limited to these compounds.
- the range of 1.5 is preferable, more preferably 1.0 / 0.1 to 1.0 / 1.2, and still more preferably 1.0 / 0.2 to 1.0 / 1.1.
- Polyamide composition According to the required use and performance of the polyamide compound of the present invention, a lubricant, a crystallization nucleating agent, a whitening inhibitor, a matting agent, a heat stabilizer, a weather stabilizer, an ultraviolet absorber, a plasticizer, It is good also as a polyamide composition by adding additives, such as a flame retardant, an antistatic agent, a coloring inhibitor, antioxidant, and an impact resistance improving material. These additives can be added as needed within a range not impairing the effects of the present invention.
- the polyamide compound of the present invention may be mixed with various resins according to required applications and performances to form a polyamide composition. In the polyamide composition, the polyamide resin or polyamide oligomer may react with the added additive or resin.
- a conventionally known method can be used, but dry mixing that is low in cost and does not receive heat history is preferably performed.
- a method in which a polyamide compound and the above-mentioned additive are put in a tumbler and mixed by rotating is mentioned.
- a method in order to prevent the classification of the polyamide compound and the additive after dry mixing a method may be adopted in which a viscous liquid is attached to the polyamide compound as a spreading agent, and then the additive is added and mixed. it can.
- the spreading agent include a surfactant and the like, but a known one can be used without being limited thereto.
- Anti-whitening agent In the polyamide composition containing the polyamide compound of the present invention, it is preferable to add a diamide compound and / or a diester compound to the polyamide compound as a whitening inhibitor after hot water treatment or after a long period of time.
- the diamide compound and / or diester compound is effective in suppressing whitening due to oligomer precipitation.
- the diamide compound and the diester compound may be used alone or in combination.
- the diamide compound is preferably a diamide compound obtained from an aliphatic dicarboxylic acid having 8 to 30 carbon atoms and a diamine having 2 to 10 carbon atoms.
- a whitening prevention effect can be expected.
- the aliphatic dicarboxylic acid has 30 or less carbon atoms and the diamine has 10 or less carbon atoms, uniform dispersion in the resin composition is good.
- the aliphatic dicarboxylic acid may have a side chain or a double bond, but a linear saturated aliphatic dicarboxylic acid is preferred.
- One kind of diamide compound may be used, or two or more kinds may be used in combination.
- Examples of the aliphatic dicarboxylic acid include stearic acid (C18), eicosanoic acid (C20), behenic acid (C22), montanic acid (C28), and triacontanoic acid (C30).
- Examples of the diamine include ethylenediamine, butylenediamine, hexanediamine, xylylenediamine, and bis (aminomethyl) cyclohexane. A diamide compound obtained by combining these is preferred.
- a diamide compound obtained from an aliphatic dicarboxylic acid mainly composed of stearic acid and a diamine mainly composed of ethylenediamine is particularly preferred.
- the diester compound is preferably a diester compound obtained from an aliphatic dicarboxylic acid having 8 to 30 carbon atoms and a diol having 2 to 10 carbon atoms.
- a whitening prevention effect can be expected.
- the aliphatic dicarboxylic acid has 30 or less carbon atoms and the diol has 10 or less carbon atoms, uniform dispersion in the resin composition is good.
- the aliphatic dicarboxylic acid may have a side chain or a double bond, but a linear saturated aliphatic dicarboxylic acid is preferred.
- One type of diester compound may be used, or two or more types may be used in combination.
- Examples of the aliphatic dicarboxylic acid include stearic acid (C18), eicosanoic acid (C20), behenic acid (C22), montanic acid (C28), and triacontanoic acid (C30).
- Examples of the diol component of the diester compound used in the present invention include ethylene glycol, propanediol, butanediol, hexanediol, xylylene glycol, and cyclohexanedimethanol. A diester compound obtained by combining these is preferred. Particularly preferred are diester compounds obtained from an aliphatic dicarboxylic acid mainly composed of montanic acid and a diol mainly composed of ethylene glycol and / or 1,3-butanediol.
- the amount of the diamide compound and / or diester compound added is 0.005 to 0.5 parts by mass, preferably 0.05 to 0.5 parts by mass, particularly preferably 0.12 to 0 parts per 100 parts by mass of the polyamide compound. .5 parts by mass.
- Addition of 0.005 parts by mass or more with respect to 100 parts by mass of the polyamide compound and use in combination with the crystallization nucleating agent can be expected to produce a synergistic effect for preventing whitening.
- Crystallization nucleating agent In the polyamide composition containing the polyamide compound of the present invention, it is preferable to add a crystallization nucleating agent from the viewpoint of improving transparency. In addition to improving transparency, it is also effective for whitening due to crystallization after hydrothermal treatment and after a long period of time. By adding a crystallization nucleating agent to the polyamide compound, the spherulite size can be reduced to It can be suppressed by setting it to 1 ⁇ 2 or less of the wavelength. In addition, when a diamide compound and / or a diester compound and a crystallization nucleating agent are used in combination, whitening suppression far superior to the level expected from the respective whitening suppression effects can be obtained by their synergistic effect.
- glass fillers glass fibers, crushed glass fibers (milled fibers), glass flakes, glass beads, etc.
- calcium silicate fillers wollastonite, etc.
- mica mica
- Talc powder talc and granular talc with rosin as binder
- kaolin potassium titanate whisker
- boron nitride layered silicate clay
- nanofiller carbon fiber, etc.
- the maximum diameter of the inorganic crystallization nucleating agent is preferably 0.01 to 5 ⁇ m.
- powdered talc with a particle size of 3.0 ⁇ m or less is preferable, powdered talc with a particle size of about 1.5 to 3.0 ⁇ m is more preferable, and powdered talc with a particle size of 2.0 ⁇ m or less is particularly preferable.
- powdered talc with a particle size of 3.0 ⁇ m or less is preferable, powdered talc with a particle size of about 1.5 to 3.0 ⁇ m is more preferable, and powdered talc with a particle size of 2.0 ⁇ m or less is particularly preferable.
- granular talc using rosin as a binder in this powdered talc is particularly preferable because the dispersed state in the polyamide composition is good.
- Organic crystallization nucleating agents include crystallization nucleating agents, capsules consisting of bilayer films of micro to nano level size, bis (benzylidene) sorbitol-based or phosphorus-based transparent crystal nucleating agents, rosinamide-based Gelling agents are preferred, and bis (benzylidene) sorbitol crystallization nucleating agents are particularly preferred.
- the amount of the crystallization nucleating agent added is preferably 0.005 to 2.0 parts by mass, more preferably 0.01 to 1.5 parts by mass with respect to 100 parts by mass of the polyamide compound.
- the inorganic crystallization nucleating agent such as talc is 0.05 to 1.5 parts by mass with respect to 100 parts by mass of the polyamide compound
- the organic crystallization nucleating agent such as bis (benzylidene) sorbitol crystallization nucleating agent is polyamide. It is particularly preferable to use 0.01 to 0.5 parts by mass with respect to 100 parts by mass of the compound.
- the bis (benzylidene) sorbitol-based crystallization nucleating agent is selected from bis (benzylidene) sorbitol and bis (alkylbenzylidene) sorbitol, and is a condensation product (diacetal compound) produced by acetalization reaction of sorbitol and benzaldehyde or alkyl-substituted benzaldehyde. And can be conveniently prepared by various synthetic methods known in the art.
- the alkyl may be linear or cyclic, and may be saturated or unsaturated.
- a common synthesis method uses the reaction of 1 mole of D-sorbitol with about 2 moles of aldehyde in the presence of an acid catalyst.
- the reaction temperature varies widely depending on the characteristics (melting point, etc.) of the aldehyde used as the starting material for the reaction.
- the reaction medium may be an aqueous medium or a non-aqueous medium.
- One preferred method that can be used to prepare the diacetal is described in US Pat. No. 3,721,682. Although this disclosure is limited to benzylidene sorbitols, the bis (alkylbenzylidene) sorbitols used in the present invention can also be conveniently prepared by the methods described therein.
- bis (benzylidene) sorbitol crystallization nucleating agent examples include bis (p-methylbenzylidene) sorbitol, bis (p-ethylbenzylidene) sorbitol, bis (n-propylbenzylidene) sorbitol, bis (p -Isopropylbenzylidene) sorbitol, bis (p-isobutylbenzylidene) sorbitol, bis (2,4-dimethylbenzylidene) sorbitol, bis (3,4-dimethylbenzylidene) sorbitol, bis (2,4,5-trimethylbenzylidene) sorbitol, Examples thereof include bis (2,4,6-trimethylbenzylidene) sorbitol, bis (4-biphenylbenzylidene) sorbitol and the like.
- alkyl-substituted benzaldehydes suitable for preparing bis (benzylidene) sorbitol crystallization nucleating agents include p-methylbenzaldehyde, n-propylbenzaldehyde, p-isopropylbenzaldehyde, 2,4-dimethylbenzaldehyde, 3,4 -Dimethylbenzaldehyde, 2,4,5-trimethylbenzaldehyde, 2,4,6-trimethylbenzaldehyde, 4-biphenylbenzaldehyde.
- the bis (benzylidene) sorbitol-based crystallization nucleating agent not only suppresses whitening but also improves the oxygen barrier property when added to the polyamide compound. It is particularly preferable to use a crystallization nucleating agent of bis (benzylidene) sorbitol (A) that can obtain both effects of suppressing whitening and improving oxygen barrier properties.
- the polyamide composition containing the polyamide compound of the present invention can be used as a gas barrier layer to which a layered silicate is added. Not only the oxygen barrier property of the molded body but also the barrier property against other gases such as carbon dioxide gas. Can also be improved.
- the layered silicate is a 2-octahedron or 3-octahedral layered silicate having a charge density of 0.25 to 0.6.
- Examples of the 2-octahedron type include montmorillonite, beidellite, and the like.
- Examples of the octahedron type include hectorite and saponite. Among these, montmorillonite is preferable.
- the layered silicate is obtained by expanding an interlayer of the layered silicate by previously bringing an organic swelling agent such as a polymer compound or an organic compound into contact with the layered silicate.
- an organic swelling agent such as a polymer compound or an organic compound
- a quaternary ammonium salt can be preferably used.
- a quaternary ammonium salt having at least one alkyl group or alkenyl group having 12 or more carbon atoms is used.
- organic swelling agents include trimethyl dodecyl ammonium salt, trimethyl tetradecyl ammonium salt, trimethyl hexadecyl ammonium salt, trimethyl octadecyl ammonium salt, trimethyl alkyl decyl ammonium salt, trimethyl alkyl decyl ammonium salt; trimethyl octadecenyl ammonium salt Trimethylalkenylammonium salts such as trimethyloctadecadienylammonium salt; triethylalkylammonium salts such as triethyldodecylammonium salt, triethyltetradecylammonium salt, triethylhexadecylammonium salt, triethyloctadecylammonium salt; tributyldodecylammonium salt, tributyltetradecyl Ammonium salt, tributyl
- hydroxyl group and / or ether group-containing ammonium salts among them, methyl dialkyl (PAG) ammonium salt, ethyl dialkyl (PAG) ammonium salt, butyl dialkyl (PAG) ammonium salt, dimethyl bis (PAG) ammonium salt, diethyl bis (PAG) ) Ammonium salt, dibutyl bis (PAG) ammonium salt, methyl alkyl bis (PAG) ammonium salt, ethyl alkyl bis (PAG) ammonium salt, butyl alkyl bis (PAG) ammonium salt, methyl tri (PAG) ammonium salt, ethyl tri (PAG) ammonium Salt, butyltri (PAG) ammonium salt, tetra (PAG) ammonium salt (wherein alkyl has 12 carbon atoms such as dodecyl, tetradecyl, hexadecyl, oct
- trimethyldodecyl ammonium salt trimethyl tetradecyl ammonium salt, trimethyl hexadecyl ammonium salt, trimethyl octadecyl ammonium salt, dimethyl didodecyl ammonium salt, dimethyl ditetradecyl ammonium salt, dimethyl dihexadecyl ammonium salt, dimethyl dioctadecyl ammonium salt, dimethyl A ditallow ammonium salt is preferred.
- organic swelling agents can be used alone or as a mixture of a plurality of types.
- those obtained by adding 0.5 to 8 parts by mass of a layered silicate treated with an organic swelling agent to 100 parts by mass of the polyamide compound are preferably used, more preferably 1 to 6 parts by mass, still more preferably. 2 to 5 parts by mass. If the amount of layered silicate added is less than 0.5 parts by mass, the effect of improving gas barrier properties is small, which is not preferable. On the other hand, if it is more than 8 parts by mass, the gas barrier layer becomes cloudy and the transparency of the container is impaired, which is not preferable.
- the layered silicate is preferably uniformly dispersed without locally agglomerating.
- the uniform dispersion means that the layered silicate is separated into a flat plate shape in the polyamide, and 50% or more of them have an interlayer distance of 5 nm or more.
- the interlayer distance refers to the distance between the centers of gravity of the flat objects. The larger the distance, the better the dispersion state, the better the appearance such as transparency, and the better the gas barrier properties such as oxygen and carbon dioxide.
- the polyamide compound contains sodium acetate, calcium acetate, magnesium acetate, calcium stearate, magnesium stearate, sodium stearate and derivatives thereof. It is preferable to add one or more carboxylates selected from: Examples of the derivatives include 12-hydroxystearic acid metal salts such as calcium 12-hydroxystearate, magnesium 12-hydroxystearate, and sodium 12-hydroxystearate. By adding the carboxylates, it is possible to prevent the gelation of the polyamide compound that occurs during the molding process, and to reduce fish eyes in the molded body, thereby improving the suitability of the molding process.
- the addition amount of the carboxylates is preferably 400 to 10,000 ppm, more preferably 800 to 5000 ppm, and still more preferably 1000 to 3000 ppm as a concentration in the polyamide composition. If it is 400 ppm or more, the thermal deterioration of the polyamide compound can be suppressed and gelation can be prevented. Moreover, if it is 10,000 ppm or less, a polyamide composition does not raise
- the carboxylates described above are excellent in handling properties, and among them, metal stearate is preferable because it is inexpensive and has an effect as a lubricant, and can stabilize the molding process. Furthermore, the shape of the carboxylates is not particularly limited, but when the powder and the smaller particle size are dry-mixed, it is easy to uniformly disperse in the polyamide composition, so the particle size is 0.2 mm or less is preferable.
- the polyamide composition containing the polyamide compound of the present invention preferably contains an antioxidant from the viewpoint of controlling oxygen absorption performance and suppressing deterioration of mechanical properties.
- the antioxidant include copper-based antioxidants, hindered phenol-based antioxidants, hindered amine-based antioxidants, phosphorus-based antioxidants, and thio-based antioxidants. Antioxidants and phosphorus antioxidants are preferred.
- hindered phenol antioxidant examples include triethylene glycol-bis [3- (3-tert-butyl-5-methyl-4-hydroxyphenyl) propionate, 4,4′-butylidenebis (3-methyl- 6-t-butylphenol), 1,6-hexanediol-bis [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionate, 2,4-bis- (n-octylthio) -6- (4-Hydroxy-3,5-di-t-butylanilino) -1,3,5-triazine, pentaerythrityl-tetrakis [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionate] 2,2-thio-diethylenebis [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionate], octadecyl-3- ( , 5-di-t-butyl-4
- phosphorus antioxidants include triphenyl phosphite, trioctadecyl phosphite, tridecyl phosphite, trinonylphenyl phosphite, diphenylisodecyl phosphite, bis (2,6-di-tert-butyl- 4-methylphenyl) pentaerythritol diphosphite, bis (2,4-di-tert-butylphenyl) pentaerythritol diphosphite, tris (2,4-di-tert-butylphenyl) phosphite, distearyl pentaerythritol And organic phosphorus compounds such as diphosphite, tetra (tridecyl-4,4′-isopropylidene diphenyl diphosphite, 2,2-methylenebis (4,6-di-tert-butylphenyl)
- the content of the antioxidant in the polyamide composition can be used without particular limitation as long as it does not impair the various performances of the composition, but from the viewpoint of controlling oxygen absorption performance and suppressing deterioration of mechanical properties,
- the amount is preferably 0.001 to 3 parts by mass, more preferably 0.01 to 1 part by mass with respect to 100 parts by mass of the polyamide compound.
- Impact resistance improving material In the polyamide composition containing the polyamide compound of the present invention, an impact resistance improving material may be added to improve impact resistance, pinhole resistance and flexibility of the film.
- Impact modifiers include polyolefins, polyamide elastomers, hydrogenated styrene-butadiene copolymer resins, ionomers, ethylene-ethyl acrylate copolymer resins, ethylene-ethyl acrylate copolymer resins modified with maleic anhydride, ethylene -Methacrylic acid copolymer resin, nylon 6, 66, 12, nylon 12, nylon 12 elastomer, ethylene-propylene copolymer elastomer, polyester elastomer, etc. can be added.
- the addition amount of the impact resistance improving material is preferably 1 to 10% by mass, more preferably 1 to 5% by mass, and particularly preferably 2 to 3% by mass. When the amount added is large, transparency and gas barrier properties are lowered. If the amount added is small, impact resistance, pinhole resistance and flexibility of the film are not improved so much.
- the polyamide compound of the present invention can be used for any application that requires oxygen barrier properties and oxygen absorption performance.
- the polyamide compound of the present invention may be filled alone in a sachet or the like and used as an oxygen absorbent.
- Representative examples of the use of the polyamide compound of the present invention include, but are not limited to, molded materials such as packaging materials and packaging containers.
- the polyamide compound of the present invention can be processed and used as at least a part of the molded body.
- the polyamide compound of the present invention can be used as at least a part of a film-like or sheet-like packaging material, and packaging containers such as bottles, trays, cups, tubes, flat bags, standing pouches and the like. Can be used as at least part of.
- the thickness of the layer made of the polyamide compound or polyamide composition of the present invention is not particularly limited, but preferably has a thickness of 1 ⁇ m or more.
- a molded product such as a packaging material or packaging container
- any method can be used.
- a polyamide compound or a polyamide composition melted through a T die, a circular die or the like is extruded from an attached extruder. Can do.
- the film-form molded object obtained by the above-mentioned method can also be processed into a stretched film by extending
- a bottle-shaped packaging container can be obtained by injecting a melted polyamide compound or polyamide composition into a mold from an injection molding machine to produce a preform, and then heating to a stretching temperature and blow-drawing.
- Containers such as trays and cups are manufactured by injecting a molten polyamide compound or polyamide composition into a mold from an injection molding machine, or by forming a sheet-shaped packaging material by a molding method such as vacuum molding or pressure molding. It can be obtained by molding.
- the packaging material and the packaging container can be manufactured through various methods regardless of the above-described manufacturing method.
- the packaging material and packaging container obtained using the polyamide compound of the present invention are suitable for storing and storing various articles.
- Store and store various items such as beverages, seasonings, cereals, liquids and solid processed foods that require aseptic filling or heat sterilization, chemicals, liquid daily necessities, pharmaceuticals, semiconductor integrated circuits, and electronic devices be able to.
- the component composition, relative viscosity, number average molecular weight, glass transition temperature and melting point of the polyamide compounds obtained in Examples and Comparative Examples were measured by the following methods. Further, oxygen absorption rate, oxygen absorption amount, oxygen permeability coefficient and haze measurement, sensory test and tensile test were performed by the following methods.
- Component composition 1 H-NMR 400 MHz, manufactured by JEOL Ltd., trade name: JNM-AL400, measurement mode: NON ( 1 H) was used to quantify the component composition of the copolymer. . Specifically, a 5 mass% solution of a polyamide compound was prepared using formic acid-d as a solvent, and 1 H-NMR measurement was performed.
- Oxygen Absorption Rate and Oxygen Absorption A powder sample of 2 g obtained by finely pulverizing a polyamide compound pellet or pulverized product with a pulverizer, wrapped in a medicine wrapping paper, 25 cm ⁇ 18 cm 3 consisting of an aluminum foil laminated film
- the side-sealed bag was charged with cotton containing 10 ml of water and sealed so that the air amount in the bag was 400 ml.
- the humidity in the bag was 100% RH (relative humidity).
- the oxygen concentration in the bag was measured with an oxygen concentration meter (trade name: LC-700F, manufactured by Toray Engineering Co., Ltd.), and the oxygen absorption amount (cc / g) was calculated from this oxygen concentration.
- Examples 101 to 113 and 116 to 121 and Comparative Examples 101 to 107 a film sample having a thickness of about 100 ⁇ m was cut into a 400 cm 2 sample and sealed in a bag in the same manner as described above, at 40 ° C. After storing for 28 days, the oxygen concentration in the bag was measured with an oxygen concentration meter (trade name: LC-700F, manufactured by Toray Engineering Co., Ltd.), and the amount of oxygen absorbed per 1 m 2 of the film sample was calculated from this oxygen concentration. The amount of oxygen absorbed per day was determined as the oxygen absorption rate (cc / (m 2 ⁇ day)). The higher the value, the better the oxygen absorption performance.
- Oxygen permeability coefficient Using an oxygen permeability measuring device (Mocon, model: OX-TRAN 2 / 21SH) according to ASTM D3985, the oxygen permeability of a 100 ⁇ m film at 23 ° C. and 60% RH was measured. The oxygen permeability coefficient was converted to (cc ⁇ mm / (m 2 ⁇ day ⁇ atm)). The lower the value, the less oxygen permeation is preferable.
- the oxygen permeation coefficients of N-MXD6, N-MXD10 and N-MXD12 to which no other component is added or copolymerized are 0.08 cc ⁇ mm / (m 2 ⁇ day ⁇ atm) and 1.6 cc ⁇ mm, respectively. / (M 2 ⁇ day ⁇ atm) and 2.9 cc ⁇ mm / (m 2 ⁇ day ⁇ atm).
- the humidity in the bag was 100% RH. After storage at 40 ° C. for 28 days, the air in the bag was taken out with a syringe, and 10 panelists sniffed and evaluated.
- a blank without a film sample and a powder sample was also prepared as a comparative blank.
- the sensory test evaluation was performed according to the following criteria, and the average value of the evaluation of 10 people was calculated. The lower the value, the less the smell and the better. (Evaluation criteria) 0: No odor is felt (blank). 1: A slight difference from the blank is felt. 2: I feel the difference from the blank. 3: I feel a significant difference from the blank.
- Tensile test A film sample having a thickness of about 100 ⁇ m stored in a constant temperature bath at 40 ° C. and 100% RH for 28 days to absorb oxygen was conditioned at 23 ° C. and 50% RH for 1 week. The film sample was cut into a width of 10 mm and a length of 100 mm, and was pulled with a tensile tester (manufactured by Toyo Seiki Co., Ltd., trade name: Strograph V1-C) at a pulling speed of 50 mm / min. The load at the time was measured, and the tensile strength at break was determined by the following formula.
- Tensile strength at break (MPa) Load at cutting (N) / Cross-sectional area of sample film (mm 2 )
- the tensile strength at break obtained from this test was used as an index for maintaining mechanical properties.
- an unstored film sample (before oxygen absorption) was conditioned at 23 ° C. and 50% RH for 1 week, and then a tensile test was performed in the same manner. The higher the value after oxygen absorption, the less the deterioration of the resin is preferable.
- Example 101 Melt polymerization by the atmospheric pressure dropping method of polyamide compound 13000 g of adipic acid (manufactured by Asahi Kasei Chemicals Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die ( 88.96 mol), DL-alanine (manufactured by Musashino Chemical Laboratory), 880.56 g (9.88 mol), sodium hypophosphite 11.7 g (0.11 mol), sodium acetate 6.06 g (0.074 mol) ), And after sufficiently purging with nitrogen, the system was heated to 170 ° C.
- adipic acid manufactured by Asahi Kasei Chemicals Co., Ltd.
- Metaxylylenediamine (Mitsubishi Gas Chemical Co., Ltd.) 12075.4 g (88.66 mol) was added dropwise thereto with stirring, and the inside of the system was continuously heated while removing the condensed water produced. After completion of the addition of metaxylylenediamine, the reaction is continued for 40 minutes at an internal temperature of 260 ° C., and then the inside of the system is pressurized with nitrogen, the polymer is taken out from the strand die and pelletized to obtain about 23 kg of polyamide compound. Obtained.
- the polyamide compound is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity of the tumble dryer is 99 volumes while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 200 ° C. over about 70 minutes, and then held at 200 ° C. for 30 minutes.
- nitrogen gas having a purity of 99% by volume or more was introduced into the system, and the tumble dryer was rotated while cooling to obtain DL-alanine copolymer N-MXD6 (polyamide compound 101).
- FIG. 1 A 1 H-NMR chart of the polyamide compound 101 is shown in FIG.
- the absorption peak near 1.5 to 1.7 ppm in FIG. 1 is the absorption peak a1 derived from hydrogen of the methylene group not adjacent to the carbonyl group of adipic acid and the absorption peak derived from hydrogen of the methyl group of DL-alanine. b1 and absorption peaks a1 and b1 appear overlapping.
- FIG. 1 shows the integrated intensity when the absorption peak a1 and the absorption peak b1 are added.
- the absorption peak near 2.5 ppm is an absorption peak c1 derived from hydrogen of a methylene group adjacent to the carbonyl group of adipic acid.
- the absorption peak around 7.0 to 7.47 ppm is an absorption peak d1 derived from hydrogen of the aromatic ring of metaxylylenediamine (MXDA).
- the amount of DL-alanine units in the polyamide compound is calculated from the integrated intensity ratio of each peak according to the following formula.
- Polyamide compound 101 pellets were formed into a non-stretched film having a width of 200 mm and a thickness of 95 to 105 ⁇ m using a 25 mm ⁇ single-screw extruder at an extrusion temperature of 260 ° C., a screw speed of 60 rpm, and a take-off speed of 1.2 m / min.
- Example 102 A D-alanine copolymer N-MXD6 (polyamide compound 102) and an unstretched film were prepared in the same manner as in Example 101 except that the ⁇ -amino acid was changed to D-alanine (produced by Musashino Chemical Laboratory). Obtained.
- Example 103 An L-alanine copolymer N-MXD6 (polyamide compound 103) and an unstretched film were prepared in the same manner as in Example 101, except that the ⁇ -amino acid was changed to L-alanine (manufactured by Sinogel Amino Acid Co., Ltd.). Obtained.
- Example 104 Implemented except that the ⁇ -amino acid was changed to DL-2-aminobutyric acid (manufactured by Nippon Finechem Co., Ltd., refined product), and the amount added was changed so that the content in the polyamide compound was 2.6 mol%.
- DL-2-aminobutyric acid copolymer N-MXD6 polyamide compound 104
- an unstretched film were obtained.
- Example 105 DL-2-aminobutyric acid copolymer N-MXD6 was prepared in the same manner as in Example 104, except that the amount of DL-2-aminobutyric acid was changed so that the content in the polyamide compound was 5.3 mol%. (Polyamide compound 105) and an unstretched film were obtained.
- Example 106 DL-2-aminobutyric acid copolymer N-MXD6 was prepared in the same manner as in Example 104 except that the content of DL-2-aminobutyric acid was changed so that the content in the polyamide compound was 11.1 mol%. (Polyamide compound 106) and an unstretched film were obtained.
- Example 107 DL-2-aminobutyric acid copolymer N-MXD6 was prepared in the same manner as in Example 104, except that the amount of DL-2-aminobutyric acid was changed so that the content in the polyamide compound was 25.0 mol%. (Polyamide compound 107) and an unstretched film were obtained.
- Example 108 A DL-valine copolymer N-MXD6 (polyamide compound 108) and an unstretched film were prepared in the same manner as in Example 104 except that the ⁇ -amino acid was changed to DL-valine (manufactured by Sinogel Amino Acid Co., Ltd.). Obtained.
- Example 109 DL-leucine copolymerized N-MXD6 (polyamide compound 109) and an unstretched film were obtained in the same manner as in Example 104 except that the ⁇ -amino acid was changed to DL-leucine (manufactured by Ningbo Haishu Bio-technology). .
- Example 110 DL-tert-leucine copolymerized N-MXD6 (polyamide compound 110) and DL-tert-leucine copolymerized in the same manner as in Example 104 except that the ⁇ -amino acid was changed to DL-tert-leucine (a refined product manufactured by Nippon Finechem Co., Ltd.) An unstretched film was obtained.
- Example 111 A DL-phenylalanine copolymer N-MXD6 (polyamide compound 111) and an unstretched film were prepared in the same manner as in Example 104 except that the ⁇ -amino acid was changed to DL-phenylalanine (manufactured by Sinogel Amino Acid Co., Ltd.). Obtained.
- Example 112 The same as Example 101 except that adipic acid was changed to sebacic acid (manufactured by Ito Oil Co., Ltd.) and the amount of DL-alanine was changed so that the content in the polyamide compound was 2.6 mol%.
- DL-alanine copolymer N-MXD10 polyamide compound 112
- an unstretched film were obtained.
- Example 113 DL-alanine copolymer N-MXD12 (polyamide compound 113) and an unstretched film were obtained in the same manner as in Example 112 except that adipic acid was changed to dodecanedioic acid (manufactured by Ube Industries).
- Example 114 DL-alanine copolymer N-MXD6 (polyamide compound 114) was obtained in the same manner as in Example 101 except that solid phase polymerization was not performed. In addition, since molecular weight did not fully increase in the polymerization, the measurement of the oxygen transmission coefficient and Haze in the above-mentioned unstretched film and the tensile test were not performed.
- Example 115 DL-alanine copolymerization N was carried out in the same manner as in Example 101 except that the final internal temperature after completion of the dropwise addition of metaxylylenediamine was 230 ° C. and solid phase polymerization was not performed. -MXD6 (polyamide compound 115) was obtained. In addition, since molecular weight did not fully increase in the polymerization, the measurement of the oxygen transmission coefficient and Haze in the above-mentioned unstretched film and the tensile test were not performed.
- Example 116 DL-alanine copolymer N-MXD6 polyamide compound 116) and an unstretched film were obtained in the same manner as in Example 101 except that the holding time after heating at 200 ° C. was changed to 150 minutes during solid phase polymerization. .
- Example 117 At the time of melt polymerization, DL- Alanine copolymerized N-MXD6 (polyamide compound 117) and an unstretched film were obtained.
- Example 118 Melting polymerization of polyamide compounds by pressure salt method 13000 g of adipic acid (manufactured by Asahi Kasei Chemicals Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die ( 88.96 mol), DL-alanine (manufactured by Musashino Chemical Laboratory) 880.56 g (9.88 mol), metaxylylenediamine (manufactured by Mitsubishi Gas Chemical Co., Inc.) 12075.4 g (88.66 mol), distillation 10000 g of water, 11.7 g (0.11 mol) of sodium hypophosphite and 6.06 g (0.074 mol) of sodium acetate were added, and after sufficiently purging with nitrogen, a nylon salt was prepared under 0.2 MP
- the polyamide compound is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity of the tumble dryer is 99 volumes while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 180 ° C. over about 60 minutes, and then held at 180 ° C. for 60 minutes.
- nitrogen gas having a purity of 99% by volume or more was introduced into the system, and the tumble dryer was rotated while cooling to obtain DL-alanine copolymer N-MXD6 (polyamide compound 118).
- Example 119 DL-alanine acid copolymerized N-MXD6 (polyamide compound 119) was the same as in Example 118 except that the amount of DL-alanine added was changed so that the content in the polyamide compound was 11.1 mol%. And the unstretched film was obtained.
- Example 120 Polymer compound melt polymerization by pressure drop method 13000 g of adipic acid (manufactured by Asahi Kasei Chemicals Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die ( 88.96 mol), DL-alanine (manufactured by Musashino Chemical Laboratory), 880.56 g (9.88 mol), sodium hypophosphite 11.7 g (0.11 mol), sodium acetate 6.06 g (0.074 mol) ) And sufficiently purged with nitrogen, and then heated and melted to 170 ° C.
- adipic acid manufactured by Asahi Kasei Chemicals Co., Ltd.
- the polyamide compound is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity of the tumble dryer is 99 volumes while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 180 ° C. over about 60 minutes, and then held at 180 ° C. for 60 minutes.
- nitrogen gas having a purity of 99% by volume or more was introduced into the system, and the tumble dryer was rotated while cooling to obtain DL-alanine copolymer N-MXD6 (polyamide compound 120).
- Example 121 DL-alanine acid copolymerized N-MXD6 (polyamide compound 121) was the same as in Example 120 except that the amount of DL-alanine added was changed so that the content in the polyamide compound was 11.1 mol%. And the unstretched film was obtained.
- Comparative Example 101 A glycine copolymerized N-MXD6 (polyamide compound 122) was prepared in the same manner as in Example 104, except that the ⁇ -amino acid was changed to glycine having secondary hydrogen at the ⁇ -position (manufactured by Tokyo Chemical Industry Co., Ltd., reagent). And the unstretched film was obtained.
- Comparative Example 102 The same method as in Example 104, except that the ⁇ -amino acid was changed to 2-aminoisobutyric acid (2-amino-2-methylpropanoic acid, manufactured by Nippon Finechem Co., Ltd.), which does not have hydrogen at the ⁇ -position.
- 2-aminoisobutyric acid copolymer N-MXD6 polyamide compound 123
- an unstretched film were obtained.
- Comparative Example 103 With respect to N-MXD6 (trade name: S6007, manufactured by Mitsubishi Gas Chemical Co., Ltd.), which is a polyamide composed of metaxylylenediamine and adipic acid, cobalt stearate becomes 400 ppm in the resin composition. And dry blended. The obtained blend was formed into a film by a 30 mm ⁇ twin screw extruder at an extrusion temperature of 260 ° C., a screw rotation speed of 60 rpm, a feed screw rotation speed of 12 rpm, and a take-off speed of 1.8 m / min. A stretched film was produced.
- Comparative Example 104 Maleic acid-modified polybutadiene (PB) (Nippon Petrochemical Co., Ltd.) with respect to 100 parts by mass of N-MXD6 (Mitsubishi Gas Chemical Co., Ltd., trade name: S6007), which is a polyamide composed of metaxylylenediamine and adipic acid (Product name: M-2000-20) 5 parts by mass and cobalt stearate were added so that the cobalt content in the resin composition was 400 ppm, and dry blended.
- PB Polybutadiene
- N-MXD6 Mitsubishi Gas Chemical Co., Ltd., trade name: S6007
- the obtained blend was formed into a film by a 30 mm ⁇ twin screw extruder at an extrusion temperature of 260 ° C., a screw rotation speed of 60 rpm, a feed screw rotation speed of 14 rpm, a take-off speed of 2.0 m / min, and a width of 300 mm and a thickness of 95 to 105 ⁇ m.
- a stretched film was produced.
- Comparative Example 105 Melt polymerization by the atmospheric pressure dropping method of polyamide compound
- adipic acid manufactured by Asahi Kasei Chemicals Co., Ltd.
- a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die 100 mol
- 12.7 g (0.120 mol) of sodium hypophosphite, 6.60 g (0.0805 mol) of sodium acetate and after sufficiently purging with nitrogen, stirring the system under a small amount of nitrogen stream Heated to 170 ° C.
- Metaxylylenediamine (manufactured by Mitsubishi Gas Chemical Co., Inc.) 13539.2 g (99.4 mol) was added dropwise with stirring, and the inside of the system was continuously heated while removing the condensed water produced. After completion of the dropwise addition of metaxylylenediamine, the internal temperature was 260 ° C. and the reaction was continued for 40 minutes. Thereafter, the inside of the system was pressurized with nitrogen, and the polymer was taken out from the strand die and pelletized to obtain about 25 kg of a polyamide compound.
- the polyamide compound is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity of the tumble dryer is 99 volumes while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 200 ° C. over about 70 minutes, and then held at 200 ° C. for 30 minutes.
- nitrogen gas having a purity of 99% by volume or more was introduced into the system, and the tumble dryer was rotated while cooling to obtain N-MXD6 (polyamide compound 124).
- Comparative Example 106 Melt polymerization by the atmospheric pressure dropping method of polyamide compound 17,000 g of sebacic acid (manufactured by Ito Oil Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die ( 84.1 mol), 13.1 g (0.124 mol) of sodium hypophosphite, 6.81 g (0.0830 mol) of sodium acetate, and after sufficient nitrogen substitution, the system was stirred under a small amount of nitrogen. While heating to 170 ° C.
- the polyamide compound is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity of the tumble dryer is 99 volumes while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 170 ° C. over about 15 minutes, and then kept at 170 ° C. for 240 minutes.
- nitrogen gas having a purity of 99% by volume or more was introduced into the system, and the tumble dryer was rotated while cooling to obtain N-MXD10 (polyamide compound 125).
- Comparative Example 107 Melt polymerization by the atmospheric pressure dropping method of polyamide compound 17500 g of dodecanedioic acid (manufactured by Ube Industries Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die (76.0 mol), 12.9 g (0.122 mol) of sodium hypophosphite, 6.73 g (0.0820 mol) of sodium acetate, and after sufficiently purging with nitrogen, the system was further purged with a small amount of nitrogen. Heat to 170 ° C. with stirring.
- the polyamide compound is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity of the tumble dryer is 99 volumes while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 160 ° C. over about 10 minutes, and then held at 160 ° C. for 420 minutes.
- nitrogen gas with a purity of 99% by volume or more was introduced into the system, and the tumble dryer was rotated while cooling to obtain N-MXD12 (polyamide compound 126).
- Polyamide compounds copolymerized with ⁇ -amino acids having no tertiary hydrogen had insufficient oxygen absorption performance (Comparative Examples 101 and 102).
- the conventional polyamide composition mixed with a cobalt compound exhibits oxygen absorption performance and a good oxygen permeability coefficient, but although it has good transparency, it has a bluish color due to the addition of the cobalt compound.
- many odors using polybutadiene were generated.
- the film after oxygen absorption is deteriorated and cannot retain its shape, and is not necessarily preferable for use in packaging containers (Comparative Examples 103 and 104).
- the polyamide composition in which the ⁇ -amino acid having tertiary hydrogen was mixed without copolymerization did not show oxygen absorption performance (Comparative Examples 105 to 107).
- a polyamide compound copolymerized with an ⁇ -amino acid having tertiary hydrogen can exhibit sufficient oxygen absorption performance without using a metal, and does not generate an unpleasant odor (Examples) 101-121).
- the oxygen permeability coefficient of the film is good, the transparency is good, and the mechanical properties after oxygen absorption are also retained (Examples 101 to 113 and 116 to 121).
- Examples 101 to 103 From the comparison of Examples 101 to 103, it can be seen that the optical isomer of ⁇ -amino acid has no influence on the oxygen absorption performance. Further, it can be seen from the comparison with Examples 104 to 107 that the oxygen absorption performance is improved by increasing the copolymerization ratio of ⁇ -amino acid. Further, from the comparison of Examples 101 and 114 to 116, it can be seen that the relative viscosity has no influence on the oxygen absorption performance. Furthermore, it can be seen that Examples 115 and 117, in which the melt polymerization temperature is lowered, can reduce YI as compared with Examples 101 to 114. It can also be seen that YI can be reduced in Examples 118 to 119 polymerized by the pressurized salt method and Examples 120 to 121 polymerized by the pressurized drop method.
- Examples 122-131 (Biaxially stretched film) Using the polyamide compounds 101, 104-107 and 117-121 pellets obtained in Examples 101, 104 to 107 and 117 to 121, an unstretched film having a thickness of about 250 ⁇ m was obtained in the same manner as in Example 101. It was. This film was stretched 4 times in the MD direction and 4 times in the TD direction at a stretching temperature of 130 ° C. using a biaxial stretching device (tenter method) manufactured by Toyo Seiki Seisakusho, and heat-fixed at 200 ° C. for 30 seconds A biaxially stretched film of about 15 ⁇ m was obtained. This biaxially stretched film was evaluated as the standard for ease of molding by evaluating the number of breaks in the trial production of 20 continuous films. In addition, the initial oxygen transmission coefficient of the biaxially stretched film, the period during which the initial oxygen transmission coefficient was completely maintained, and the haze of the film were measured. The results are shown in Table 2.
- Example 201 (Melt polymerization by the atmospheric pressure dropping method of polyamide compound) 13000 g of adipic acid (manufactured by Asahi Kasei Chemicals Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die ( 88.9 mol), high-purity isophthalic acid (manufactured by ADI International Chemical Co., Ltd.) 777.8 g (4.7 mol), DL-alanine as an ⁇ -amino acid (manufactured by Musashino Chemical Laboratory) 1472 .1 g (16.52 mol), 12.57 g (0.119 mol) of sodium hypophosphite, 6.52 g (0.0795 mol) of sodium acetate, and after sufficiently purging with nitrogen, the system was added under
- the polyamide compound is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity of the tumble dryer is 99 volumes while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 200 ° C. over about 70 minutes, and then held at 200 ° C. for 30 minutes.
- nitrogen gas having a purity of 99% by volume or more was introduced into the system, and the tumble dryer was rotated and cooled to obtain DL-alanine copolymer N-MXD6I (polyamide compound 201).
- a 1 H-NMR chart of the polyamide compound 201 is shown in FIG.
- the absorption peak near 1.5 to 1.7 ppm in FIG. 2 is the absorption peak a2 derived from hydrogen of the methylene group not adjacent to the carbonyl group of adipic acid and the absorption peak derived from hydrogen of the methyl group of DL-alanine.
- b2 and absorption peaks a2 and b2 appear overlapping.
- FIG. 2 shows the integrated intensity when the absorption peak a2 and the absorption peak b2 are added.
- the absorption peak near 2.5 ppm is an absorption peak c2 derived from hydrogen of a methylene group adjacent to carbonyl of adipic acid.
- the absorption peak near 4.5 ppm is an absorption peak d2 derived from hydrogen of the benzylmethylene group of metaxylylenediamine when adipic acid is adjacent, and the absorption peak near 4.7 ppm is isophthalic acid.
- FIG. 2 shows the integrated intensity when the absorption peak d2 and the absorption peak e2 are added.
- the absorption peak near 7.4 ppm is an absorption peak f2 derived from metaxylylenediamine and hydrogen of the benzene ring of isophthalic acid.
- the amount of DL-alanine units in the polyamide compound is calculated from the integrated intensity ratio of each peak according to the following formula. Moreover, the quantity of the isophthalic acid unit in a polyamide compound is computed by a following formula.
- Polyamide compound 201 pellets were formed into a non-stretched film having a width of 200 mm and a thickness of 95 to 105 ⁇ m using a 25 mm ⁇ single screw extruder at an extrusion temperature of 260 ° C., a screw speed of 60 rpm, and a take-off speed of 1.2 m / min.
- DL-AABA DL-2-aminobutyric acid
- Example 203 DL-valine copolymerized N-MXD6I (polyamide compound 203: MXDA unit / adipic acid) in the same manner as in Example 202 except that the ⁇ -amino acid was changed to DL-valine (manufactured by Sinogel Amino Acid Co., Ltd).
- Unit / isophthalic acid unit / DL-valine unit 48.6 / 46.4 / 2.4 / 2.6 (mol%)) and an unstretched film were obtained.
- Example 205 DL-phenylalanine copolymer N-MXD6I (polyamide compound 205: MXDA) was prepared in the same manner as in Example 202 except that the ⁇ -amino acid was changed to DL-phenylalanine (DL-Phe, manufactured by Sinogel Amino Acid Co., Ltd.).
- Unit / adipic acid unit / isophthalic acid unit / DL-phenylalanine unit 48.6 / 46.4 / 2.4 / 2.6 (mol%)) and an unstretched film were obtained.
- PTA high-purity isophthalic acid to high-purity terephthalic acid
- NDCA 2,6-naphthalenedicarboxylic acid
- Example 208 DL-alanine copolymer N-MXD6I (polyamide compound 208: MXDA) was prepared in the same manner as in Example 201 except that the addition amount of DL-alanine was changed so that the content in the polyamide compound was 2.6 mol%.
- Unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 48.6 / 46.4 / 2.4 / 2.6 (mol%)) and an unstretched film were obtained.
- Example 209 DL-alanine copolymer N-MXD6I (polyamide compound 209: the same as in Example 208, except that the amount of high-purity isophthalic acid added was changed so that the content in the polyamide compound was 4.9 mol%.
- MXDA unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 48.6 / 43.9 / 4.9 / 2.6 (mol%)) and an unstretched film were obtained.
- Example 210 DL-alanine copolymer N-MXD6I polyamide compound 210: polyamide compound 210: the same as in Example 208, except that the amount of high-purity isophthalic acid added was changed so that the content in the polyamide compound was 7.3 mol%.
- MXDA unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 48.6 / 41.5 / 7.3 / 2.6 (mol%)) and an unstretched film were obtained.
- Example 211 DL-alanine copolymer N-MXD6I polyamide compound 211: polyamide compound 211: the same as in example 208 except that the amount of high-purity isophthalic acid added was changed so that the content in the polyamide compound was 14.6 mol%.
- MXDA unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 48.6 / 34.2 / 14.6 / 2.6 (mol%)) and an unstretched film were obtained.
- Example 212 DL-alanine copolymer N-MXD6I (polyamide compound 212: the same as in Example 201) except that the amount of high-purity isophthalic acid added was changed so that the content in the polyamide compound was 4.6 mol%.
- MXDA unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 45.9 / 41.4 / 4.6 / 8.1 (mol%)) and an unstretched film were obtained.
- Example 214 DL-alanine copolymer N-MXD6I (polyamide compound 214: polyamide compound 214: the same as in Example 201, except that the amount of high-purity isophthalic acid added was changed so that the content in the polyamide compound was 13.8 mol%.
- MXDA unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 45.9 / 32.2 / 13.8 / 8.1 (mol%)) was obtained.
- molecular weight did not fully increase in the polymerization, the measurement of the oxygen transmission coefficient and Haze in the above-mentioned unstretched film and the tensile test were not performed.
- Example 215 DL-alanine copolymer N-MXD6I (polyamide compound 215: MXDA) was prepared in the same manner as in Example 201 except that the addition amount of DL-alanine was changed so that the content in the polyamide compound was 17.7 mol%.
- Unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 41.1 / 39.1 / 2.1 / 17.7 (mol%)).
- molecular weight did not fully increase in the polymerization, the measurement of the oxygen transmission coefficient and Haze in the above-mentioned unstretched film and the tensile test were not performed.
- Example 216 DL-alanine copolymer N-MXD6I (polyamide compound 216: 216), except that the amount of high-purity isophthalic acid added was changed so that the content in the polyamide compound was 4.1 mol%.
- MXDA unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 41.1 / 37.1 / 4 / 17.7 (mol%)) were obtained.
- molecular weight did not fully increase in the polymerization, the measurement of the oxygen transmission coefficient and Haze in the above-mentioned unstretched film and the tensile test were not performed.
- Example 217 DL-alanine copolymer N-MXD6I (polyamide compound 217: polyamide compound 217), except that the amount of high-purity isophthalic acid added was changed so that the content in the polyamide compound was 6.2 mol%.
- MXDA unit / adipic acid unit / isophthalic acid unit / DL-alanine unit 41.1 / 35.0 / 6.2 / 17.7 (mol%)) was obtained.
- molecular weight did not fully increase in the polymerization, the measurement of the oxygen transmission coefficient and Haze in the above-mentioned unstretched film and the tensile test were not performed.
- Example 218 DL-alanine copolymer N-MXD10I (polyamide compound 218: MXDA unit / sebacic acid unit / isophthalic acid) in the same manner as in Example 208 except that adipic acid was changed to sebacic acid (manufactured by Ito Oil Co., Ltd.).
- Unit / DL-alanine unit 48.6 / 46.4 / 2.4 / 2.6 (mol%)) and an unstretched film were obtained.
- Comparative Example 201 Melt polymerization by the atmospheric pressure dropping method of polyamide compound 13000 g of adipic acid (manufactured by Asahi Kasei Chemicals Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die 88.9 mol), high-purity isophthalic acid (manufactured by ADI International Chemical Co., Ltd.) 777.9 g (4.68 mol)), sodium hypophosphite 11.96 g (0.113 mol), sodium acetate 6 .21 g (0.0756 mol) was added, and after sufficient nitrogen substitution, the system was heated to 170 ° C.
- adipic acid manufactured by Asahi Kasei Chemicals Co., Ltd.
- the polyamide is charged into a tumble dryer with a jacket provided with a nitrogen gas introduction tube, a vacuum line, a vacuum pump, and a thermocouple for measuring the internal temperature, and the purity inside the tumble dryer is 99% by volume while rotating at a constant speed.
- the tumble dryer was heated under the same nitrogen gas stream, and the pellet temperature was raised to 150 ° C. over about 150 minutes.
- the pressure in the system was reduced to 1 torr or less.
- the temperature was further raised, and the pellet temperature was raised to 200 ° C. over about 70 minutes, and then held at 200 ° C. for 30 minutes.
- Polyamide compound 221 pellets were formed into a non-stretched film having a width of 200 mm and a thickness of 95 to 105 ⁇ m using a 25 mm ⁇ single-screw extruder at an extrusion temperature of 260 ° C., a screw speed of 60 rpm, and a take-off speed of 1.2 m / min.
- Comparative Example 205 The same as Example 201 except that the ⁇ -amino acid was changed to 2-aminoisobutyric acid having no hydrogen at the ⁇ -position (2-amino-2-methylpropanoic acid, AIB, manufactured by Nihon Finechem Co., Ltd.).
- an unstretched film is
- Comparative Example 206 Cobalt stearate was added to the polyamide compound 221 so that the cobalt content in the resin composition was 400 ppm, and dry blended.
- the obtained blend was formed into a film by a 30 mm ⁇ twin screw extruder at an extrusion temperature of 260 ° C., a screw rotation speed of 60 rpm, a feed screw rotation speed of 12 rpm, and a take-off speed of 1.8 m / min, and having a width of 200 mm and a thickness of 95 to 105 ⁇ m.
- a stretched film was prepared.
- Comparative Example 207 For 100 parts by mass of the polyamide compound 221, 5 parts by mass of maleic acid-modified polybutadiene (PB) (manufactured by Nippon Petrochemical Co., Ltd., trade name: M-2000-20) and cobalt stearate in the resin composition The cobalt content was added to 400 ppm and dry blended. The obtained blend was formed into a film by a 30 mm ⁇ twin screw extruder at an extrusion temperature of 260 ° C., a screw rotation speed of 60 rpm, a feed screw rotation speed of 14 rpm, and a take-off speed of 2.0 m / min. A stretched film was produced.
- PB maleic acid-modified polybutadiene
- Comparative Example 208 DL-alanine (manufactured by Musashino Chemical Laboratory Co., Ltd.) was added to 100 parts by mass of the polyamide compound 221 so that the DL-alanine content in the resin composition was 5 parts by mass, and dry blended.
- the extrusion temperature is 240 ° C.
- the screw speed is 30 rpm
- the feed screw speed is 14 rpm
- the take-up speed is 1.0 m / min.
- An unstretched film having a width of 110 mm and a thickness of 95 to 105 ⁇ m was produced.
- Polyamide compounds obtained by copolymerizing only aromatic dicarboxylic acids had insufficient oxygen absorption performance (Comparative Examples 201 to 203).
- the polyamide compound copolymerized with an ⁇ -amino acid having no tertiary hydrogen also had insufficient oxygen absorption performance (Comparative Examples 204 and 205).
- the conventional polyamide composition mixed with a cobalt compound exhibits oxygen absorption performance and a good oxygen permeability coefficient, but although it has good transparency, it has a bluish color due to the addition of the cobalt compound. In particular, many odors using polybutadiene were generated.
- the film after absorbing oxygen is deteriorated and cannot retain its shape, and is not necessarily preferable for packaging container applications (Comparative Examples 206 and 207).
- the polyamide composition in which the ⁇ -amino acid having tertiary hydrogen was mixed without copolymerization did not show oxygen absorption performance (Comparative Example 208).
- a polyamide compound obtained by copolymerizing an ⁇ -amino acid having tertiary hydrogen and an aromatic dicarboxylic acid can exhibit sufficient oxygen absorption performance without using a metal, and generates an unpleasant odor. (Examples 201 to 220).
- Example 201 to 213, 218 and 219 the oxygen permeability coefficient of the film is good, the transparency is good, and the mechanical properties after oxygen absorption are also retained (Examples 201 to 213, 218 and 219).
- the polyamide compound of Example 220 showed very good oxygen absorption performance although the molecular weight did not increase.
- Examples 218 and 219 using sebacic acid or dodecanedioic acid as the linear aliphatic dicarboxylic acid component have lower oxygen absorption performance than the examples using adipic acid, but ⁇ -having tertiary hydrogen. It can be seen that the oxygen absorption performance is superior to that of N-MXD10I (Comparative Example 202) or N-MXD12I (Comparative Example 203) that does not copolymerize amino acids.
- Example 301 Melt polymerization by the atmospheric pressure dropping method of polyamide compound 13000 g of adipic acid (manufactured by Asahi Kasei Chemicals Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die 89.0 mol), DL-alanine (manufactured by Musashino Chemical Laboratory) 880.56 g (9.88 mol), ⁇ -caprolactam (manufactured by Ube Industries) 4314.10 g (38.1 mol), hypophosphorous acid 13.75 g (0.13 mol) of sodium and 7.13 g (0.087 mol) of sodium acetate were added, and after sufficient nitrogen substitution, the system was heated to 170 ° C.
- adipic acid manufactured by Asahi Kasei Chemicals Co.,
- Example 302 D-alanine copolymer N-MXD6,6 (polyamide compound 302: MXDA unit / adipic acid unit / D-alanine unit / ⁇ ) in the same manner as in Example 301 except that DL-alanine was changed to D-alanine.
- -Caprolactam unit 39.3 / 39.4 / 4.4 / 16.9 (mol%)) was obtained.
- Example 303 L-alanine copolymer N-MXD6,6 (polyamide compound 303: MXDA unit / adipic acid unit / L-alanine unit / ⁇ ) in the same manner as in Example 301 except that DL-alanine was changed to L-alanine.
- -Caprolactam unit 39.3 / 39.4 / 4.4 / 16.9 (mol%)) was obtained.
- Example 311 DL-2-aminobutyric acid copolymer N— was prepared in the same manner as in Example 301 except that the ⁇ -amino acid was changed to DL-2-aminobutyric acid (DL-AABA, manufactured by Nippon Finechem Co., Ltd., purified product).
- DL-AABA DL-2-aminobutyric acid
- Example 312 DL-leucine copolymerized N-MXD6,6 (polyamide compound 312: MXDA unit / adipic acid) in the same manner as in Example 301 except that the ⁇ -amino acid was changed to DL-leucine (manufactured by Ningbo Haishu Bio-technology).
- Unit / DL-leucine unit / ⁇ -caprolactam unit 39.3 / 39.4 / 4.4 / 16.9 (mol%)) was obtained.
- Comparative Example 301 Melt polymerization by the atmospheric pressure dropping method of polyamide compound 13000 g of adipic acid (manufactured by Asahi Kasei Chemicals Co., Ltd.) precisely weighed in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, a partial condenser, a full condenser, a thermometer, a dropping funnel and a nitrogen introduction tube, and a strand die 89.0 mol), ⁇ -caprolactam (manufactured by Ube Industries, Ltd.) 4314.1 g (38.1 mol), sodium hypophosphite 13.4 g (0.13 mol), sodium acetate 6.9 g (0.084 mol) Then, after sufficiently purging with nitrogen, the system was heated to 170 ° C.
- adipic acid manufactured by Asahi Kasei Chemicals Co., Ltd.
- N-MXD6,6 polyamide compound 316: MXDA unit / adipic acid unit / ⁇ -caprolactam unit
- Comparative Example 303 The same as Example 301 except that DL-alanine was changed to 2-aminoisobutyric acid having no hydrogen at the ⁇ -position (2-amino-2-methylpropanoic acid, AIB, manufactured by Nippon Finechem Co., Ltd.).
- DL-alanine (manufactured by Musashino Chemical Laboratory Co., Ltd.) was added to the polyamide compound 316 obtained in Comparative Example 301 and dry blended to obtain a mixture of the polyamide compound and DL-alanine (DL- in the mixture). Alanine content: 5 mass%) was obtained.
- DL-alanine-containing N-MXD6 at a extrusion temperature of 240 ° C., a screw speed of 30 rpm, and a feed screw speed of 14 rpm using a small 15 mm ⁇ single screw extruder so that the blends in the mixture do not copolymerize with each other. 6 pellets were prepared.
- the polyamide compound copolymerized with an ⁇ -amino acid having no tertiary hydrogen had insufficient oxygen absorption performance (Comparative Examples 302 and 303). Also, polyamide compounds in which ⁇ -amino acids having tertiary hydrogen are not copolymerized or polyamide compositions in which ⁇ -amino acids having tertiary hydrogen are mixed without copolymerization did not exhibit oxygen absorption performance. (Comparative Examples 301, 304 to 306 and 307). In contrast, a polyamide compound copolymerized with an ⁇ -amino acid having tertiary hydrogen can exhibit sufficient oxygen absorption performance without using a metal. (Examples 301-315).
- Examples 401 to 416 and Comparative Examples 401 to 407 (Melt polymerization by the atmospheric pressure dropping method of polyamide oligomer) Aliphatic dicarboxylic acids of the types and blending amounts shown in Table 5 in a reaction vessel having an internal volume of 50 liters equipped with a stirrer, partial condenser, total condenser, thermometer, dropping funnel and nitrogen introduction tube, and strand die Aromatic dicarboxylic acid, ⁇ -amino acid, ⁇ -aminocarboxylic acid, sodium hypophosphite and sodium acetate were charged, and after sufficient nitrogen substitution, the system was stirred under a small amount of nitrogen flow up to 170 ° C. Heated.
- metaxylylenediamine which is an aromatic diamine having a blending amount shown in Table 5, was added dropwise with stirring, and the inside of the system was continuously heated while removing the produced condensed water. After completion of dropwise addition of the diamine, the reaction was continued for 40 to 60 minutes while keeping the internal temperature at 240 ° C. and paying attention to the increase in stirring torque. Thereafter, the inside of the system was pressurized with nitrogen, and the polyamide oligomer was taken out from the strand die. Those that can be taken out in a strand state were pelletized to obtain a pellet-like polyamide oligomer. Moreover, the thing which cannot be taken out in a strand state with low molecular weight was grind
- polyamide oligomers that are not copolymerized with ⁇ -amino acids having tertiary hydrogen did not show oxygen absorption performance.
- polyamide oligomer copolymerized with ⁇ -amino acid having tertiary hydrogen exhibited sufficient oxygen absorption performance without using metal (Examples 401 to 416). Therefore, the polyamide compound of the present invention can be used as an oxygen absorbent.
- the polyamide compound of the present invention is excellent in oxygen absorption performance.
- the polyamide compound of the present invention for a packaging material or packaging container, it exhibits sufficient oxygen absorption performance without containing metal, does not cause unpleasant odor, has very good transparency, It is possible to provide a packaging material and a packaging container capable of storing the contents in a good state.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Polyamides (AREA)
Abstract
Description
一方、コバルト等の遷移金属を含有する酸素捕捉性樹脂組成物は、透明性が必要な包装容器にも適用可能である利点を有するが、遷移金属触媒によって樹脂組成物が着色されるため好ましくない。また、これらの樹脂組成物では、遷移金属触媒によって、酸素を吸収することで樹脂が酸化される。具体的には、遷移金属原子によるポリアミド樹脂のアリーレン基に隣接するメチレン鎖から水素原子の引き抜きに起因するラジカルの発生、前記ラジカルに酸素分子が付加することによるパーオキシラジカルの発生、パーオキシラジカルによる水素原子の引き抜き等の各反応により起こるものと考えられている。このような機構による酸素吸収により樹脂が酸化されるため、分解物が発生して容器内容物に好ましくない臭気が発生したり、樹脂の酸化劣化により容器の色調や強度等が損なわれるという問題がある。
下記一般式(I)で表される芳香族ジアミン単位を50モル%以上含むジアミン単位25~50モル%と、下記一般式(II-1)で表される直鎖脂肪族ジカルボン酸単位及び/又は下記一般式(II-2)で表される芳香族ジカルボン酸単位を合計で50モル%以上含むジカルボン酸単位25~50モル%と、下記一般式(III)で表される構成単位0.1~50モル%とを含有する、ポリアミド化合物。

本発明のポリアミド化合物は、下記一般式(I)で表される芳香族ジアミン単位を50モル%以上含むジアミン単位25~50モル%と、下記一般式(II-1)で表される直鎖脂肪族ジカルボン酸単位及び/又は下記一般式(II-2)で表される芳香族ジカルボン酸単位を合計で50モル%以上含むジカルボン酸単位25~50モル%と、3級水素含有カルボン酸単位(好ましくは下記一般式(III)で表される構成単位)0.1~50モル%とを含有する。

ただし、前記ジアミン単位、前記ジカルボン酸単位、前記3級水素含有カルボン酸単位の合計は100モル%を超えないものとする。本発明のポリアミド化合物は、本発明の効果を損なわない範囲で、前記以外の構成単位をさらに含んでいてもよい。
本発明の「ポリアミド樹脂」は、本発明のポリアミド化合物において、相対粘度が1.8以上の重合体を意味する。ポリアミド樹脂は、単独で成形加工可能な材料であり、包包装材料や包装容器に加工することができる。本発明のポリアミド樹脂に、必要により、他の樹脂や添加剤を添加、混合してもよく、そのようにして得たポリアミド組成物を成型加工してもよい。本発明のポリアミド樹脂は、金属を含有せずとも十分な酸素吸収性能を発現し、かつ不快な臭気が発生せず、極めて良好な透明性を有する。
ジアミン単位とジカルボン酸単位との含有量の割合は、重合反応の観点から、ほぼ同量であることが好ましく、ジカルボン酸単位の含有量がジアミン単位の含有量の±2モル%であることがより好ましい。ジカルボン酸単位の含有量がジアミン単位の含有量の±2モル%の範囲を超えると、ポリアミド化合物の重合度が上がりにくくなるため重合度を上げるのに多くの時間を要し、熱劣化が生じやすくなる。
本発明のポリアミド化合物中のジアミン単位は、ポリアミド化合物に優れたガスバリア性を付与することに加え、透明性や色調の向上や、成形性を容易にする観点から、前記一般式(I)で表される芳香族ジアミン単位をジアミン単位中に50モル%以上含み、当該含有量は、好ましくは70モル%以上、より好ましくは80モル%以上、更に好ましくは90モル%以上であり、また、好ましくは100モル%以下である。
本発明のポリアミド化合物中のジカルボン酸単位は、重合時の反応性、並びにポリアミド化合物の結晶性及び成形性の観点から、前記一般式(II-1)で表される直鎖脂肪族ジカルボン酸単位及び/又は前記一般式(II-2)で表される芳香族ジカルボン酸単位を、ジカルボン酸単位に合計で50モル%以上含み、当該含有量は、好ましくは70モル%以上、より好ましくは80モル%以上、更に好ましくは90モル%以上であり、また、好ましくは100モル%以下である。
本発明のポリアミド化合物は、ポリアミド化合物に適度なガラス転移温度や結晶性を付与することに加え、包装材料や包装容器として必要な柔軟性を付与する目的の場合、前記一般式(II-1)で表される直鎖脂肪族ジカルボン酸単位を含むことが好ましい。
前記一般式(II-1)中、nは2~18の整数を表し、好ましくは3~16、より好ましくは4~12、更に好ましくは4~8である。
前記一般式(II-1)で表される直鎖脂肪族ジカルボン酸単位を構成しうる化合物としては、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、1,10-デカンジカルボン酸、1,11-ウンデカンジカルボン酸、1,12-ドデカンジカルボン酸等を例示できるが、これらに限定されるものではない。これらは単独で又は2種以上を組み合わせて用いることができる。
本発明のポリアミド化合物は、ポリアミド化合物に更なるガスバリア性を付与することに加え、包装材料や包装容器の成形加工性を容易にする目的の場合、前記一般式(II-2)で表される芳香族ジカルボン酸単位を含むことが好ましい。
前記一般式(II-2)中、Arはアリーレン基を表す。前記アリーレン基は、好ましくは炭素数6~30、より好ましくは炭素数6~15のアリーレン基であり、例えば、フェニレン基、ナフチレン基などが挙げられる。
前記一般式(II-2)で表される芳香族ジカルボン酸単位を構成しうる化合物としては、テレフタル酸、イソフタル酸、2,6-ナフタレンジカルボン酸等を例示できるが、これらに限定されるものではない。これらは単独で又は2種以上を組み合わせて用いることができる。
本発明における3級水素含有カルボン酸単位は、ポリアミド化合物の重合の観点から、アミノ基及びカルボキシル基を少なくとも1つずつ有するか、又はカルボキシル基を2つ以上有する。具体例としては、下記一般式(III)、(IV)又は(V)のいずれかで表される構成単位が挙げられる。

なお、官能基が更に置換されている場合、上述した炭素数には、更なる置換基の炭素数は含まれないものとする。例えば、ベンジル基は、フェニル基で置換された炭素数1のアルキル基と見なし、フェニル基で置換された炭素数7のアルキル基とは見なさない。以降の炭素数に記載についても、特に断りが無い限り、同様に解するものとする。
好ましいRの具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、t-ブチル基、1-メチルプロピル基、2-メチルプロピル基、ヒドロキシメチル基、1-ヒドロキシエチル基、メルカプトメチル基、メチルスルファニルエチル基、フェニル基、ナフチル基、ベンジル基、4-ヒドロキシベンジル基等を例示できるが、これらに限定されるものではない。これらの中でも、メチル基、エチル基、2-メチルプロピル基、及びベンジル基がより好ましい。
また、前記一般式(IV)で表される構成単位を構成しうる化合物としては、3-アミノ酪酸等のβ-アミノ酸を例示でき、前記一般式(V)で表される構成単位を構成しうる化合物としては、メチルマロン酸、メチルコハク酸、リンゴ酸、酒石酸等のジカルボン酸を例示できるが、これらに限定されるものではない。
これらはD体、L体、ラセミ体のいずれであってもよく、アロ体であってもよい。また、これらは単独で又は2種以上を組み合わせて用いることができる。
本発明のポリアミド化合物は、ポリアミド化合物に柔軟性等が必要な場合には、前記ジアミン単位、前記ジカルボン酸単位及び前記3級水素含有カルボン酸単位に加えて、下記一般式(A)で表されるω-アミノカルボン酸単位を更に含有してもよい。

前記ω-アミノカルボン酸単位の含有量は、ポリアミド化合物の全構成単位中、好ましくは0.1~49.9モル%、より好ましくは3~40モル%、更に好ましくは5~35モル%である。ただし、前記のジアミン単位、ジカルボン酸単位、3級水素含有カルボン酸単位、及びω-アミノカルボン酸単位の合計は100モル%を超えないものとする。
前記一般式(A)中、pは2~18の整数を表し、好ましくは3~16、より好ましくは4~14、更に好ましくは5~12である。
本発明のポリアミド化合物の重合度については、相対粘度が使われる。本発明のポリアミド化合物の好ましい相対粘度は、好ましくは1.01~4.2である。
本発明のポリアミド化合物がポリアミド樹脂である場合、相対粘度は、成形品の外観や成形加工性の観点から、好ましくは1.8~4.2、より好ましくは1.9~4.0、更に好ましくは2.0~3.8である。但し、本発明のポリアミド樹脂を他の熱可塑性樹脂への添加剤や改質剤等に使用する場合、この範囲に限定されない。
本発明のポリアミド化合物がポリアミドオリゴマーである場合、相対粘度は、取扱い性、反応性及び熱安定性等の観点から、好ましくは1.01以上1.8未満、より好ましくは1.1~1.75、更に好ましくは1.2~1.65、特に好ましくは1.3~1.6である。
なお、ここでいう相対粘度は、ポリアミド化合物1gを96%硫酸100mLに溶解し、キャノンフェンスケ型粘度計にて25℃で測定した落下時間(t)と、同様に測定した96%硫酸そのものの落下時間(t0)の比であり、次式で示される。
相対粘度=t/t0
ポリアミド化合物の酸素吸収速度、及び酸素吸収によるポリアミド化合物の酸化劣化は、ポリアミド化合物の末端アミノ基濃度を変えることで制御することが可能である。ポリアミド化合物がポリアミド樹脂である場合、酸素吸収速度と酸化劣化のバランスの観点から、末端アミノ基濃度は5~150eq/106gの範囲が好ましく、より好ましくは10~100eq/106g、さらに好ましくは15~80eq/106gである。
本発明のポリアミド化合物は、前記ジアミン単位を構成しうるジアミン成分と、前記ジカルボン酸単位を構成しうるジカルボン酸成分と、前記3級水素含有カルボン酸単位を構成しうる3級水素含有カルボン酸成分と、必要により前記ω-アミノカルボン酸単位を構成しうるω-アミノカルボン酸成分とを重縮合させることで製造することができ、重縮合条件等を調整することで重合度を制御することができる。重縮合時に分子量調整剤として少量のモノアミンやモノカルボン酸を加えてもよい。また、重縮合反応を抑制して所望の重合度とするために、ポリアミド化合物を構成するジアミン成分とカルボン酸成分との比率(モル比)を1からずらして調整してもよい。
反応押出法では、ジアミン成分及びジカルボン酸成分からなるポリアミド(本発明のポリアミド化合物の前駆体に相当するポリアミド)又はジアミン成分、ジカルボン酸成分及びω-アミノカルボン酸成分からなるポリアミド(本発明のポリアミド化合物の前駆体に相当するポリアミド)と、3級水素含有カルボン酸成分とを押出機で溶融混練して反応させる方法である。3級水素含有カルボン酸成分をアミド交換反応により、ポリアミドの骨格中に組み込む方法であり、十分に反応させるためには、反応押出に適したスクリューを用い、L/Dの大きい2軸押出機を用いるのが好ましい。少量の3級水素含有カルボン酸成分を含むポリアミド化合物を製造する場合に、簡便な方法であり好適である。
加圧塩法では、ナイロン塩を原料として加圧下にて溶融重縮合を行う方法である。具体的には、ジアミン成分と、ジカルボン酸成分と、3級水素含有カルボン酸成分と、必要に応じてω-アミノカルボン酸成分とからなるナイロン塩水溶液を調製した後、該水溶液を濃縮し、次いで加圧下にて昇温し、縮合水を除去しながら重縮合させる。缶内を徐々に常圧に戻しながら、ポリアミド化合物の融点+10℃程度まで昇温し、保持した後、更に、0.02MPaGまで徐々に減圧しつつ、そのままの温度で保持し、重縮合を継続する。一定の撹拌トルクに達したら、缶内を窒素で0.3MPaG程度に加圧してポリアミド化合物を回収する。
加圧塩法は、揮発性成分をモノマーとして使用する場合に有用であり、3級水素含有カルボン酸成分の共重合率が高い場合には好ましい重縮合方法である、特に、3級水素含有カルボン酸成分がポリアミド化合物を構成する全成分中に15モル%以上含まれる場合に、好適である。加圧塩法を用いることで、3級水素含有カルボン酸成分の蒸散を防ぎ、さらには、3級水素含有カルボン酸成分同士の重縮合を抑制でき、重縮合反応をスムーズに進めることが可能であるため、性状に優れたポリアミド化合物が得られる。
常圧滴下法では、常圧下にて、ジカルボン酸成分と、3級水素含有カルボン酸成分と、必要に応じてω-アミノカルボン酸成分とを加熱溶融した混合物に、ジアミン成分を連続的に滴下し、縮合水を除去しながら重縮合させる。なお、生成するポリアミド化合物の融点よりも反応温度が下回らないように、反応系を昇温しながら重縮合反応を行う。
常圧滴下法は、前記加圧塩法と比較すると、塩を溶解するための水を使用しないため、バッチ当たりの収量が大きく、また、原料成分の気化・凝縮を必要としないため、反応速度の低下が少なく、工程時間を短縮できる。
加圧滴下法では、まず、重縮合缶にジカルボン酸成分と、3級水素含有カルボン酸成分と、必要に応じてω-アミノカルボン酸成分とを仕込み、各成分を撹拌して溶融混合し混合物を調製する。次いで、缶内を好ましくは0.3~0.4MPaG程度に加圧しながら混合物にジアミン成分を連続的に滴下し、縮合水を除去しながら重縮合させる。この際、生成するポリアミド化合物の融点よりも反応温度が下回らないように、反応系を昇温しながら重縮合反応を行う。設定モル比に達したらジアミン成分の滴下を終了し、缶内を徐々に常圧に戻しながら、ポリアミド化合物の融点+10℃程度まで昇温し、保持した後、更に、0.02MPaGまで徐々に減圧しつつ、そのままの温度で保持し、重縮合を継続する。一定の撹拌トルクに達したら、缶内を窒素で0.3MPaG程度に加圧してポリアミド化合物を回収する。
加圧滴下法は、加圧塩法と同様に、揮発性成分をモノマーとして使用する場合に有用であり、3級水素含有カルボン酸成分の共重合率が高い場合には好ましい重縮合方法である。特に、3級水素含有カルボン酸成分がポリアミド化合物を構成する全成分中に15モル%以上含まれる場合に、好適である。加圧滴下法を用いることで3級水素含有カルボン酸成分の蒸散を防ぎ、さらには、3級水素含有カルボン酸成分同士の重縮合を抑制でき、重縮合反応をスムーズに進めることが可能であるため、性状に優れたポリアミド化合物が得られる。さらに、加圧滴下法は、加圧塩法に比べて、塩を溶解するための水を使用しないため、バッチ当たりの収量が大きく、常圧滴下法と同様に反応時間を短くできることから、ゲル化等を抑制し、黄色度が低いポリアミド化合物を得ることができる。
上記重縮合方法で製造されたポリアミド化合物は、そのまま使用することもできるが、更に重合度を高めるための工程を経てもよい。更に重合度を高める工程としては、押出機内での反応押出や固相重合等が挙げられる。固相重合で用いられる加熱装置としては、連続式の加熱乾燥装置やタンブルドライヤー、コニカルドライヤー、ロータリードライヤー等と称される回転ドラム式の加熱装置およびナウタミキサーと称される内部に回転翼を備えた円錐型の加熱装置が好適に使用できるが、これらに限定されることなく公知の方法、装置を使用することができる。特にポリアミド化合物の固相重合を行う場合は、上述の装置の中で回転ドラム式の加熱装置が、系内を密閉化でき、着色の原因となる酸素を除去した状態で重縮合を進めやすいことから好ましく用いられる。
本発明のポリアミド化合物の重縮合においては、アミド化反応を促進する観点から、リン原子含有化合物を添加することが好ましい。
リン原子含有化合物としては、ジメチルホスフィン酸、フェニルメチルホスフィン酸等のホスフィン酸化合物;次亜リン酸、次亜リン酸ナトリウム、次亜リン酸カリウム、次亜リン酸リチウム、次亜リン酸マグネシウム、次亜リン酸カルシウム、次亜リン酸エチル等のジ亜リン酸化合物;ホスホン酸、ホスホン酸ナトリウム、ホスホン酸カリウム、ホスホン酸リチウム、ホスホン酸カリウム、ホスホン酸マグネシウム、ホスホン酸カルシウム、フェニルホスホン酸、エチルホスホン酸、フェニルホスホン酸ナトリウム、フェニルホスホン酸カリウム、フェニルホスホン酸リチウム、フェニルホスホン酸ジエチル、エチルホスホン酸ナトリウム、エチルホスホン酸カリウム等のホスホン酸化合物;亜ホスホン酸、亜ホスホン酸ナトリウム、亜ホスホン酸リチウム、亜ホスホン酸カリウム、亜ホスホン酸マグネシウム、亜ホスホン酸カルシウム、フェニル亜ホスホン酸、フェニル亜ホスホン酸ナトリウム、フェニル亜ホスホン酸カリウム、フェニル亜ホスホン酸リチウム、フェニル亜ホスホン酸エチル等の亜ホスホン酸化合物;亜リン酸、亜リン酸水素ナトリウム、亜リン酸ナトリウム、亜リン酸リチウム、亜リン酸カリウム、亜リン酸マグネシウム、亜リン酸カルシウム、亜リン酸トリエチル、亜リン酸トリフェニル、ピロ亜リン酸等の亜リン酸化合物等が挙げられる。
これらの中でも特に次亜リン酸ナトリウム、次亜リン酸カリウム、次亜リン酸リチウム等の次亜リン酸金属塩が、アミド化反応を促進する効果が高くかつ着色防止効果にも優れるため好ましく用いられ、特に次亜リン酸ナトリウムが好ましい。なお、本発明で使用できるリン原子含有化合物はこれらの化合物に限定されない。
リン原子含有化合物の添加量は、ポリアミド化合物中のリン原子濃度換算で0.1~1000ppmであることが好ましく、より好ましくは1~600ppmであり、さらに好ましくは5~400ppmである。0.1ppm以上であれば、重合中にポリアミド化合物が着色しにくく透明性が高くなる。1000ppm以下であれば、ポリアミド化合物がゲル化しにくく、また、リン原子含有化合物に起因すると考えられるフィッシュアイの成形品中への混入も低減でき、成形品の外観が良好となる。
アルカリ金属化合物としては、アルカリ金属水酸化物やアルカリ金属酢酸塩、アルカリ金属炭酸塩、アルカリ金属アルコキシド等が好ましい。本発明で用いることのできるアルカリ金属化合物の具体例としては、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化ルビジウム、水酸化セシウム、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、酢酸ルビジウム、酢酸セシウム、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウムプロポキシド、ナトリウムブトキシド、カリウムメトキシド、リチウムメトキシド、炭酸ナトリウム等が挙げられるが、これらの化合物に限定されることなく用いることができる。なお、リン原子含有化合物とアルカリ金属化合物の比率は、重合速度制御の観点や、黄色度を低減する観点から、リン原子含有化合物/アルカリ金属化合物=1.0/0.05~1.0/1.5の範囲が好ましく、より好ましくは、1.0/0.1~1.0/1.2、さらに好ましくは、1.0/0.2~1.0/1.1である。
本発明のポリアミド化合物に、要求される用途や性能に応じて、滑剤、結晶化核剤、白化防止剤、艶消剤、耐熱安定剤、耐候安定剤、紫外線吸収剤、可塑剤、難燃剤、帯電防止剤、着色防止剤、酸化防止剤、耐衝撃性改良材等の添加剤を添加させてポリアミド組成物としてもよい。これらの添加剤は、本発明の効果を損なわない範囲で、必要に応じて添加することができる。また、本発明のポリアミド化合物を、要求される用途や性能に応じて、種々の樹脂と混合してポリアミド組成物としてもよい。ポリアミド組成物において、ポリアミド樹脂やポリアミドオリゴマーは、添加した添加剤や樹脂と反応していてもよい。
本発明のポリアミド化合物を含有するポリアミド組成物においては、熱水処理後や長時間の経時後の白化抑制として、ジアミド化合物及び/又はジエステル化合物をポリアミド化合物に添加することが好ましい。ジアミド化合物及び/又はジエステル化合物は、オリゴマーの析出による白化の抑制に効果がある。ジアミド化合物とジエステル化合物は単独で用いてもよいし、併用してもよい。
炭素数8~30の脂肪族ジカルボン酸と主としてエチレンジアミンから成るジアミンから得られるジアミド化合物、または主としてモンタン酸から成る脂肪族ジカルボン酸と炭素数2~10のジアミンから得られるジアミド化合物が好ましく、特に好ましくは主としてステアリン酸から成る脂肪族ジカルボン酸と主としてエチレンジアミンから成るジアミンから得られるジアミド化合物である。
特に好ましくは主としてモンタン酸から成る脂肪族ジカルボン酸と主としてエチレングリコール及び/又は1,3-ブタンジオールから成るジオールから得られるジエステル化合物である。
本発明のポリアミド化合物を含有するポリアミド組成物においては、透明性を改善する観点から、結晶化核剤を添加することが好ましい。透明性を改善するだけでなく、熱水処理後や長時間の経時後の結晶化による白化にも効果があり、結晶化核剤をポリアミド化合物に添加することにより、球晶サイズを可視光の波長の1/2以下にすることで抑制できる。また、ジアミド化合物および/またはジエステル化合物と結晶化核剤を併用すると、これらの相乗効果により、それぞれの白化抑制効果から予想される程度よりはるかに優れた白化抑制が得られる。
本発明のポリアミド化合物を含有するポリアミド組成物においては、ポリアミド化合物中に、酢酸ナトリウム、酢酸カルシウム、酢酸マグネシウム、ステアリン酸カルシウム、ステアリン酸マグネシウム、ステアリン酸ナトリウムおよびそれらの誘導体から選択される1種以上のカルボン酸塩類を添加することが好ましい。ここで該誘導体としては、12-ヒドロキシステアリン酸カルシウム、12-ヒドロキシステアリン酸マグネシウム、12-ヒドロキシステアリン酸ナトリウム等の12-ヒドロキシステアリン酸金属塩等が挙げられる。前記カルボン酸塩類を添加することで、成形加工中に起こるポリアミド化合物のゲル化防止や成形体中のフィッシュアイを低減することができ、成形加工の適性が向上する。
なお、前述のカルボン酸塩類はハンドリング性に優れ、この中でもステアリン酸金属塩は安価である上、滑剤としての効果を有しており、成形加工をより安定化することができるため好ましい。さらに、カルボン酸塩類の形状に特に制限はないが、粉体でかつその粒径が小さい方が乾式混合する場合、ポリアミド組成物中に均一に分散させることが容易であるため、その粒径は0.2mm以下が好ましい。
本発明のポリアミド化合物を含有するポリアミド組成物は、酸素吸収性能を制御する観点や機械物性低下を抑える観点から酸化防止剤を含有することが好ましい。酸化防止剤としては、銅系酸化防止剤、ヒンダードフェノール系酸化防止剤、ヒンダードアミン系酸化防止剤、リン系酸化防止剤、チオ系酸化防止剤等を例示することができ、中でもヒンダードフェノール系酸化防止剤、リン系酸化防止剤が好ましい。
本発明のポリアミド化合物を含有するポリアミド組成物においては、耐衝撃性、フィルムの耐ピンホール性、柔軟性を改善するため耐衝撃性改良材を添加してもよい。耐衝撃性改良材としては、ポリオレフィン、ポリアミドエラストマー、スチレン-ブタジエン共重合樹脂の水素添加処理物、アイオノマー、エチレン-エチルアクリレート共重合樹脂、エチレン-エチルアクリレート共重合樹脂の無水マレイン酸変性品、エチレン-メタクリル酸共重合樹脂、ナイロン6,66,12、ナイロン12、ナイロン12エラストマー、エチレン-プロピレン共重合エラストマー、ポリエステルエラストマー等を添加することができる。耐衝撃性改良材の添加量は1~10質量%が好ましく、1~5質量%が更に好ましく、2~3質量%が特に好ましい。添加量が多いと、透明性、ガスバリア性が低下する。添加量が少ないと、耐衝撃性、フィルムの耐ピンホール性、柔軟性があまり改善されない。
本発明のポリアミド化合物は、酸素バリア性や酸素吸収性能が要求されるあらゆる用途に利用できる。例えば、本発明のポリアミド化合物を単独で小袋などに充填して酸素吸収剤として利用してもよい。
本発明のポリアミド化合物の代表的な利用例としては包装材料や包装容器等の成型体が挙げられるが、これらに限定されるものではない。本発明のポリアミド化合物を、その成型体の少なくとも一部として加工して使用することができる。例えば、本発明のポリアミド化合物をフィルム状又はシート状の包装材料の少なくとも一部として使用することができ、また、ボトル、トレイ、カップ、チューブ、平袋やスタンディングパウチ等の各種パウチ等の包装容器の少なくとも一部として使用することができる。本発明のポリアミド化合物又はポリアミド組成物からなる層の厚みは、特に制限はないが、1μm以上の厚みを有することが好ましい。
また、トレイやカップ等の容器は射出成形機から金型中に溶融したポリアミド化合物又はポリアミド組成物を射出して製造する方法や、シート状の包装材料を真空成形や圧空成形等の成形法によって成形して得ることができる。包装材料や包装容器は上述の製造方法によらず、様々な方法を経て製造することが可能である。
なお、以下の実施例において、
ポリメタキシリレンアジパミドを「N-MXD6」、
ポリメタキシリレンセバカミドを「N-MXD10」、
ポリメタキシリレンドデカナミドを「N-MXD12」、
イソフタル酸共重合ポリメタキシリレンアジパミドを「N-MXD6I」、
イソフタル酸共重合ポリメタキシリレンセバカミドを「N-MXD10I」、
イソフタル酸共重合ポリメタキシリレンドデカナミドを「N-MXD12I」、
テレフタル酸共重合ポリメタキシリレンアジパミドを「N-MXD6T」、
2,6-ナフタレンジカルボン酸共重合ポリメタキシリレンアジパミドを「N-MXD6N」、
ε-カプロラクタム共重合ポリメタキシリレンアジパミドを「N-MXD6,6」、
ε-カプロラクタム共重合ポリメタキシリレンセバカミドを「N-MXD10,6」、
ε-カプロラクタム共重合ポリメタキシリレンドデカナミドを「N-MXD12,6」、
ラウロラクタム共重合ポリメタキシリレンアジパミドを「N-MXD6,12」という。
1H-NMR(400MHz,日本電子(株)製、商品名:JNM-AL400、測定モード:NON(1H))を用いて、共重合体の成分組成の定量を実施した。具体的には、溶媒としてギ酸-dを用いてポリアミド化合物の5質量%の溶液を調製し、1H-NMR測定を実施した。
ペレット状サンプル0.2gを精秤し、96%硫酸100mlに20~30℃で撹拌溶解した。完全に溶解した後、速やかにキャノンフェンスケ型粘度計に溶液5mlを取り、25℃の恒温漕中で10分間放置後、落下時間(t)を測定した。また、96%硫酸そのものの落下時間(t0)も同様に測定した。t及びt0から次式により相対粘度を算出した。
相対粘度=t/t0
まず、ポリアミド化合物を精秤し、フェノール/エタノール=4/1容量溶液に20~30℃で撹拌溶解させ、完全に溶解した後、撹拌しつつ、メタノール5mlで容器内壁を洗い流し、0.01mol/L塩酸水溶液で中和滴定して末端アミノ基濃度〔NH2〕を求めた。
また、ポリアミド化合物を精秤し、ベンジルアルコールに窒素気流下160~180℃で撹拌溶解させ、完全に溶解した後、窒素気流下80℃以下まで冷却し、撹拌しつつメタノール10mlで容器内壁を洗い流し、0.01mol/L水酸化ナトリウム水溶液で中和滴定して末端カルボキシル基濃度〔COOH〕を求めた。
測定した末端アミノ基濃度〔NH2〕及び末端カルボキシル基濃度〔COOH〕から、次式によって数平均分子量を求めた。
数平均分子量=2/(〔NH2〕+〔COOH〕)
〔NH2〕:末端アミノ基濃度(当量/g)
〔COOH〕:末端カルボキシル基濃度(当量/g)
示差走査熱量計((株)島津製作所製、商品名:DSC-60)を用い、昇温速度10℃/分で窒素気流下にDSC測定(示差走査熱量測定)を行い、ガラス転移温度(Tg)及び融点(Tm)を求めた。
なお、参考として、他成分を添加又は共重合していないN-MXD6、N-MXD10及びN-MXD12の融点は、それぞれ237℃、192℃及び187℃であった。また、他成分を添加又は共重合していないN-MXD6Iの融点は229℃であった。
ポリアミド化合物のペレット又は粉砕物を粉砕機で細かくして得られた粉状サンプル2gを薬包紙に包んだものを、アルミ箔積層フィルムからなる25cm×18cmの3方シール袋に、水10mlを含ませた綿と共に仕込み、袋内空気量が400mlとなるようにして密封した。袋内の湿度は100%RH(相対湿度)とした。40℃下で28日保存後に、袋内の酸素濃度を酸素濃度計(東レエンジニアリング(株)製、商品名:LC-700F)で測定し、この酸素濃度から酸素吸収量(cc/g)を計算した。数値が高いほど酸素吸収性能に優れ好ましい。
なお、実施例101~113及び116~121並びに比較例101~107では、厚み約100μmのフィルムサンプルを400cm2に切り出した試験片を用いて、上記と同様に袋に封入して、40℃下で28日保存後に、袋内の酸素濃度を酸素濃度計(東レエンジニアリング(株)製、商品名:LC-700F)で測定し、この酸素濃度からフィルムサンプル1m2当たりの酸素吸収量を計算し、1日当たりの酸素吸収量を酸素吸収速度(cc/(m2・day))として求めた。数値が高いほど酸素吸収性能に優れ好ましい。
ASTM D3985に準じた酸素透過率測定装置(Mocon社製、型式:OX-TRAN 2/21SH)を使用して100μmフィルムの23℃、60%RHにおける酸素透過率を測定し、酸素透過係数(cc・mm/(m2・day・atm))に換算した。数値が低いほど酸素の透過量が少なく好ましい。
なお、他成分を添加又は共重合していないN-MXD6、N-MXD10及びN-MXD12の酸素透過係数は、それぞれ0.08cc・mm/(m2・day・atm)、1.6cc・mm/(m2・day・atm)及び2.9cc・mm/(m2・day・atm)であった。
フィルムサンプルを400cm2に切り出した試験片、又はポリアミド化合物のペレットもしくは粉砕物を粉砕機で細かくして得られた粉状サンプル2gを薬包紙に包んだものを、アルミ箔積層フィルム(最内層のシーラントは、無臭グレードを使用)からなる25cm×18cmの3方シール袋に、水10mlを含ませた綿と共に仕込み、袋内空気量が400mlとなるようにして密封した。袋内の湿度は100%RHとした。40℃下で28日保存後に、注射器で袋内の空気を取り出し、10人のパネラーが臭気を嗅いで評価した。比較用のブランクとして、フィルムサンプル及び粉状サンプルを入れないものも用意した。官能試験評価は以下の基準により実施され、10人の評価の平均値を算出した。数値が低いほどにおいが少なく好ましい。
(評価基準)
0:臭いを感じない(ブランク)。
1:ブランクとの差をわずかに感じる。
2:ブランクとの差を感じる。
3:ブランクとかなりの差を感じる。
40℃100%RHの恒温槽に厚み約100μmのフィルムサンプルを28日間保存して酸素を吸収させた後のフィルムを23℃50%RHにて、1週間調湿した。このフィルムサンプルを、幅10mm、長さ100mmにカットして、引張試験機(東洋精機(株)製、商品名:ストログラフ V1-C)にて、引張速度50mm/minにて引っ張り、フィルム破断時の荷重を測定し、下記式により引張破断強度を求めた。
引張破断強度(MPa)=切断時の荷重(N)/試料フィルムの断面積(mm2)
本試験から得られる引張破断強度を機械物性保持の指標とした。なお、比較として、未保存(酸素吸収前)のフィルムサンプルを23℃50%RHにて1週間調湿後、同様に引張試験を実施した。酸素吸収後の数値が高いほど樹脂の劣化が少なく好ましい。
ポリアミド化合物のペレットの黄色度を、カラーメーター(日本電色工業(株)製、型式:ZE2000)を用いて測定した。
JIS-K-7105に準じて、厚み約100μmのフィルムサンプルについて、曇値測定装置(日本電色工業(株)製、型式:COH-300A)にて測定し、100μmあたりのHazeとして換算した。数値が低いほど着色が少なく好ましい。
(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)13000g(88.96mol)、DL-アラニン((株)武蔵野化学研究所製)880.56g(9.88mol)、次亜リン酸ナトリウム11.7g(0.11mol)、酢酸ナトリウム6.06g(0.074mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(三菱ガス化学(株)製)12075.4g(88.66mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。メタキシリレンジアミンの滴下終了後、内温を260℃として40分反応を継続し、その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約23kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド化合物を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約70分かけてペレット温度を200℃まで昇温した後、200℃で30分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してDL-アラニン共重合N-MXD6(ポリアミド化合物101)を得た。

以下の実施例及び比較例においても、同様の手法により、調製したポリアミド化合物の成分組成の定量を実施した。
ポリアミド化合物101のペレットを25mmφ単軸押出機により、押出温度260℃、スクリュー回転数60rpm、引き取り速度1.2m/minで製膜し、幅200mm、厚み95~105μmの無延伸フィルムを作製した。
α-アミノ酸をD-アラニン((株)武蔵野化学研究所製)に変更したこと以外は実施例101と同様の方法で、D-アラニン共重合N-MXD6(ポリアミド化合物102)及び無延伸フィルムを得た。
α-アミノ酸をL-アラニン(Sinogel Amino Acid Co.,Ltd製)に変更したこと以外は実施例101と同様の方法で、L-アラニン共重合N-MXD6(ポリアミド化合物103)及び無延伸フィルムを得た。
α-アミノ酸をDL-2-アミノ酪酸((株)日本ファインケム製、精製品)に変更し、その添加量をポリアミド化合物中の含有率が2.6mol%となるように変更したこと以外は実施例101と同様の方法で、DL-2-アミノ酪酸共重合N-MXD6(ポリアミド化合物104)及び無延伸フィルムを得た。
DL-2-アミノ酪酸の添加量をポリアミド化合物中の含有率が5.3mol%となるように変更したこと以外は実施例104と同様の方法で、DL-2-アミノ酪酸共重合N-MXD6(ポリアミド化合物105)及び無延伸フィルムを得た。
DL-2-アミノ酪酸の添加量をポリアミド化合物中の含有率が11.1mol%となるように変更したこと以外は実施例104と同様の方法で、DL-2-アミノ酪酸共重合N-MXD6(ポリアミド化合物106)及び無延伸フィルムを得た。
DL-2-アミノ酪酸の添加量をポリアミド化合物中の含有率が25.0mol%となるように変更したこと以外は実施例104と同様の方法で、DL-2-アミノ酪酸共重合N-MXD6(ポリアミド化合物107)及び無延伸フィルムを得た。
α-アミノ酸をDL-バリン(Sinogel Amino Acid Co.,Ltd製)に変更したこと以外は実施例104と同様の方法で、DL-バリン共重合N-MXD6(ポリアミド化合物108)及び無延伸フィルムを得た。
α-アミノ酸をDL-ロイシン(Ningbo Haishuo Bio-technology製)に変更したこと以外は実施例104と同様の方法で、DL-ロイシン共重合N-MXD6(ポリアミド化合物109)及び無延伸フィルムを得た。
α-アミノ酸をDL-tert-ロイシン((株)日本ファインケム製精製品)に変更したこと以外は実施例104と同様の方法で、DL-tert-ロイシン共重合N-MXD6(ポリアミド化合物110)及び無延伸フィルムを得た。
α-アミノ酸をDL-フェニルアラニン(Sinogel Amino Acid Co.,Ltd製)に変更したこと以外は実施例104と同様の方法で、DL-フェニルアラニン共重合N-MXD6(ポリアミド化合物111)及び無延伸フィルムを得た。
アジピン酸をセバシン酸(伊藤製油(株)製)に変更し、DL-アラニンの添加量をポリアミド化合物中の含有率が2.6mol%となるように変更したこと以外は実施例101と同様の方法で、DL-アラニン共重合N-MXD10(ポリアミド化合物112)及び無延伸フィルムを得た。
アジピン酸をドデカン二酸(宇部興産(株)製)に変更したこと以外は実施例112と同様の方法で、DL-アラニン共重合N-MXD12(ポリアミド化合物113)及び無延伸フィルムを得た。
固相重合を実施しなかったこと以外は実施例101と同様の方法で、DL-アラニン共重合N-MXD6(ポリアミド化合物114)を得た。なお、重合において分子量が十分に上がらなかったため、前述の無延伸フィルムでの酸素透過係数及びHazeの測定、並びに引張試験は行わなかった。
溶融重合時において、メタキシリレンジアミンの滴下終了後の内温最終到達温度を230℃にし、固相重合を実施しなかったこと以外は実施例101と同様の方法で、DL-アラニン共重合N-MXD6(ポリアミド化合物115)を得た。なお、重合において分子量が十分に上がらなかったため、前述の無延伸フィルムでの酸素透過係数及びHazeの測定、並びに引張試験は行わなかった。
固相重合時に200℃昇温後の保持時間を150分に変更したこと以外は実施例101と同様の方法で、DL-アラニン共重合N-MXD6(ポリアミド化合物116)及び無延伸フィルムを得た。
溶融重合時において、メタキシリレンジアミンの滴下終了後の内温最終到達温度を240℃にし、固相重合時の温度を180℃にしたこと以外は、実施例101と同様の方法で、DL-アラニン共重合N-MXD6(ポリアミド化合物117)及び無延伸フィルムを得た。
(ポリアミド化合物の加圧塩法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)13000g(88.96mol)、DL-アラニン((株)武蔵野化学研究所製)880.56g(9.88mol)、メタキシリレンジアミン(三菱ガス化学(株)製)12075.4g(88.66mol)、蒸留水10000g、次亜リン酸ナトリウム11.7g(0.11mol)、酢酸ナトリウム6.06g(0.074mol)を投入し、十分に窒素置換した後、0.2MPa加圧下で、ナイロン塩を調製した。その後、系内を撹拌しながら昇温し、内圧が1.0MPaになった時点で、水を抜きながら、1.0MPaを保持し、190℃まで加熱した。理論水量の90%を排出した後、内温を230℃として30分反応を継続した。その後、600mmHgまで減圧し、十分トルク上昇を確認した後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約23kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド化合物を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約60分かけてペレット温度を180℃まで昇温した後、180℃で60分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してDL-アラニン共重合N-MXD6(ポリアミド化合物118)を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が11.1mol%となるように変更したこと以外は実施例118と同様の方法で、DL-アラニン酸共重合N-MXD6(ポリアミド化合物119)及び無延伸フィルムを得た。
(ポリアミド化合物の加圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)13000g(88.96mol)、DL-アラニン((株)武蔵野化学研究所製)880.56g(9.88mol)、次亜リン酸ナトリウム11.7g(0.11mol)、酢酸ナトリウム6.06g(0.074mol)を入れ、十分に窒素置換した後、0.2MPaの加圧下で、系内を撹拌しながら170℃まで加熱溶融した。その後、メタキシリレンジアミン(三菱ガス化学(株)製)12075.4g(88.66mol)を滴下した後、系内を撹拌しながら、昇温し、内圧が0.4MPaになった時点で、水を抜きながら、0.4MPaを保持し、230℃まで加熱して30分反応を継続した。その後、600mmHgまで減圧し、十分トルク上昇を確認した後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約23kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド化合物を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約60分かけてペレット温度を180℃まで昇温した後、180℃で60分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してDL-アラニン共重合N-MXD6(ポリアミド化合物120)を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が11.1mol%となるように変更したこと以外は実施例120と同様の方法で、DL-アラニン酸共重合N-MXD6(ポリアミド化合物121)及び無延伸フィルムを得た。
α-アミノ酸をα位に2級水素を持つグリシン((株)東京化成工業製、試薬)に変更したこと以外は実施例104と同様の方法で、グリシン共重合N-MXD6(ポリアミド化合物122)及び無延伸フィルムを得た。
α-アミノ酸をα位に水素を持たない2-アミノイソ酪酸(2-アミノ-2-メチルプロパン酸、(株)日本ファインケム製、精製品)に変更したこと以外は実施例104と同様の方法で、2-アミノイソ酪酸共重合N-MXD6(ポリアミド化合物123)及び無延伸フィルムを得た。
メタキシリレンジアミンとアジピン酸とからなるポリアミドであるN-MXD6(三菱ガス化学(株)製、商品名:S6007)に対し、ステアリン酸コバルトを、樹脂組成物中のコバルト含有量が400ppmとなるように添加し、ドライブレンドした。得られたブレンド物を、30mmφ2軸押出機により、押出温度260℃、スクリュー回転数60rpm、フィードスクリュー回転数12rpm、引き取り速度1.8m/minで製膜し、幅300mm、厚み95~105μmの無延伸フィルムを作製した。
メタキシリレンジアミンとアジピン酸とからなるポリアミドであるN-MXD6(三菱ガス化学(株)製、商品名:S6007)100質量部に対し、マレイン酸変性ポリブタジエン(PB)(日本石油化学(株)製、商品名:M-2000-20)5質量部、及びステアリン酸コバルトを、樹脂組成物中のコバルト含有量が400ppmとなるように添加し、ドライブレンドした。得られたブレンド物を、30mmφ2軸押出機により、押出温度260℃、スクリュー回転数60rpm、フィードスクリュー回転数14rpm、引き取り速度2.0m/minで製膜し、幅300mm、厚み95~105μmの無延伸フィルムを作製した。
(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)14615g(100mol)、次亜リン酸ナトリウム12.7g(0.120mol)、酢酸ナトリウム6.60g(0.0805mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(三菱ガス化学(株)製)13539.2g(99.4mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。メタキシリレンジアミンの滴下終了後、内温を260℃として40分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約25kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド化合物を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約70分かけてペレット温度を200℃まで昇温した後、200℃で30分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してN-MXD6(ポリアミド化合物124)を得た。
ポリアミド化合物124の100質量部に対し、DL-アラニン((株)武蔵野化学研究所製)を5質量部となるようにドライブレンドし添加した。得られたブレンド物同士が共重合しないように、15mmφの小型の単軸押出機を用いて、押出温度260℃、スクリュー回転数30rpm、フィードスクリュー回転数14rpm、引き取り速度1.0m/minで製膜し、幅110mm、厚み95~105μmの無延伸フィルムを作製した。
(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したセバシン酸(伊藤製油(株)製)17000g(84.1mol)、次亜リン酸ナトリウム13.1g(0.124mol)、酢酸ナトリウム6.81g(0.0830mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(三菱ガス化学(株)製)11391.0g(83.6mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。メタキシリレンジアミンの滴下終了後、内温を260℃として40分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約25kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド化合物を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約15分かけてペレット温度を170℃まで昇温した後、170℃で240分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してN-MXD10(ポリアミド化合物125)を得た。
ポリアミド化合物125の100質量部に対し、DL-アラニン((株)武蔵野化学研究所製)を5質量部となるようにドライブレンドし添加した。得られたブレンド物同士が共重合しないように、15mmφの小型の単軸押出機を用いて、押出温度240℃、スクリュー回転数30rpm、フィードスクリュー回転数14rpm、引き取り速度1.0m/minで製膜し、幅110mm、厚み95~105μmの無延伸フィルムを作製した。
(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したドデカン二酸(宇部興産(株)製)17500g(76.0mol)、次亜リン酸ナトリウム12.9g(0.122mol)、酢酸ナトリウム6.73g(0.0820mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(三菱ガス化学(株)製)10302.3g(75.6mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。メタキシリレンジアミンの滴下終了後、内温を260℃として40分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約24kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド化合物を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約10分かけてペレット温度を160℃まで昇温した後、160℃で420分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してN-MXD12(ポリアミド化合物126)を得た。
ポリアミド化合物126の100質量部に対し、DL-アラニン((株)武蔵野化学研究所製)を5質量部となるようにドライブレンドし添加した。得られたブレンド物同士が共重合しないように、15mmφの小型の単軸押出機を用いて、押出温度230℃、スクリュー回転数30rpm、フィードスクリュー回転数12rpm、引き取り速度1.0m/minで製膜し、幅110mm、厚み95~105μmの無延伸フィルムを作製した。


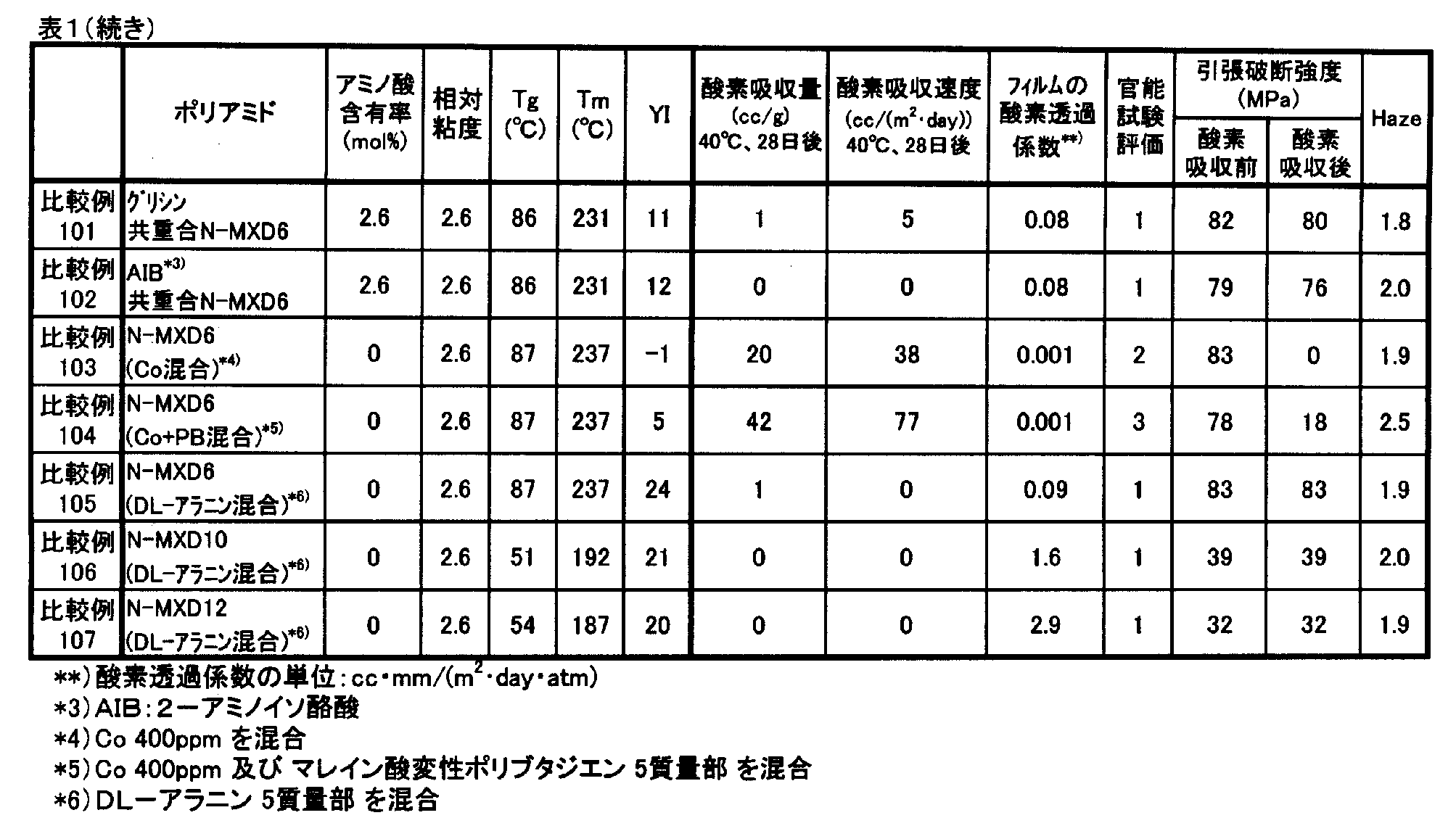
これらに対し、3級水素を有するα-アミノ酸を共重合したポリアミド化合物は、金属を用いることなく十分な酸素吸収性能を発現することができ、しかも不快な臭気を発生することがない(実施例101~121)。特に、フィルムサンプルの場合、フィルムの酸素透過係数も良好で、透明性も良好であり、更に酸素吸収後の機械物性も保持できている(実施例101~113及び116~121)。
なお、実施例101~103の対比からα-アミノ酸の光学異性体が酸素吸収性能に及ぼす影響はないことがわかる。また、実施例104~107の対比からα-アミノ酸の共重合率を増加することで酸素吸収性能が向上することがわかる。さらに、実施例101及び114~116の対比から、相対粘度が酸素吸収性能に及ぼす影響はないことがわかる。さらに、実施例101~実施例114に比べ、溶融重合温度を下げた実施例115及び117は、YIを低減できることがわかる。また、加圧塩法にて重合した実施例118~119及び加圧滴下法にて重合した実施例120~121においても、YIを低減できることがわかる。
(2軸延伸フィルム)
実施例101、104~107及び117~121で得られたポリアミド化合物101、104~107及び117~121のペレットを用いて、実施例101と同様の方法により厚さ約250μmの無延伸フィルムを得た。このフィルムを(株)東洋精機製作所製の二軸延伸装置(テンター法)を用いて、延伸温度130℃でMD方向に4倍、TD方向に4倍延伸し、200℃で30秒熱固定し、約15μmの2軸延伸フィルムを得た。この2軸延伸フィルムを連続20枚試作時の破断回数を評価し成形し易さの基準とした。また、2軸延伸フィルムの初期の酸素透過係数、初期の酸素透過係数を完全に保持した期間、及びフィルムのHazeを測定した。結果を表2に示す。

(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)13000g(88.9mol)、高純度イソフタル酸(エイ・ジイ・インタナショナル・ケミカル(株)製)777.8g(4.7mol)、α-アミノ酸としてDL-アラニン((株)武蔵野化学研究所製)1472.1g(16.52mol)、次亜リン酸ナトリウム12.57g(0.119mol)、酢酸ナトリウム6.52g(0.0795mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(MXDA、三菱ガス化学(株)製)12710g(93.3mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。メタキシリレンジアミンの滴下終了後、内温を260℃として40分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約24.3kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド化合物を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約70分かけてペレット温度を200℃まで昇温した後、200℃で30分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してDL-アラニン共重合N-MXD6I(ポリアミド化合物201)を得た。


以下の実施例及び比較例においても、同様の手法により、調製したポリアミド化合物の成分組成の定量を実施した。
ポリアミド化合物201のペレットを25mmφ単軸押出機により、押出温度260℃、スクリュー回転数60rpm、引き取り速度1.2m/minで製膜し、幅200mm、厚み95~105μmの無延伸フィルムを作製した。
α-アミノ酸をDL-2-アミノ酪酸(DL-AABA、(株)日本ファインケム製、精製品)に変更し、その添加量をポリアミド化合物中の含有率が2.6mol%となるように変更したこと以外は実施例201と同様の方法で、DL-2-アミノ酪酸共重合N-MXD6I(ポリアミド化合物202:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アミノ酪酸単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
α-アミノ酸をDL-バリン(Sinogel Amino Acid Co.,Ltd製)に変更したこと以外は実施例202と同様の方法で、DL-バリン共重合N-MXD6I(ポリアミド化合物203:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-バリン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
α-アミノ酸をDL-ロイシン(Ningbo Haishuo Bio-technology製)に変更したこと以外は実施例202と同様の方法で、DL-ロイシン共重合N-MXD6I(ポリアミド化合物204:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-ロイシン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
α-アミノ酸をDL-フェニルアラニン(DL-Phe、Sinogel Amino Acid Co.,Ltd製)に変更したこと以外は実施例202と同様の方法で、DL-フェニルアラニン共重合N-MXD6I(ポリアミド化合物205:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-フェニルアラニン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が2.6mol%となるように変更し、高純度イソフタル酸を高純度テレフタル酸(PTA、三菱ガス化学(株)製)に変更したこと以外は実施例201と同様の方法で、DL-アラニン共重合N-MXD6T(ポリアミド化合物206:MXDA単位/アジピン酸単位/テレフタル酸単位/DL-アラニン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
高純度イソフタル酸を2,6-ナフタレンジカルボン酸(NDCA、三菱ガス化学(株)製)に変更したこと以外は実施例206と同様の方法で、DL-アラニン共重合N-MXD6N(ポリアミド化合物207:MXDA単位/アジピン酸単位/NDCA単位/DL-アラニン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が2.6mol%となるように変更したこと以外は実施例201と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物208:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が4.9mol%となるように変更したこと以外は実施例208と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物209:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=48.6/43.9/4.9/2.6(mol%))及び無延伸フィルムを得た。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が7.3mol%となるように変更したこと以外は実施例208と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物210:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=48.6/41.5/7.3/2.6(mol%))及び無延伸フィルムを得た。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が14.6mol%となるように変更したこと以外は実施例208と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物211:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=48.6/34.2/14.6/2.6(mol%))及び無延伸フィルムを得た。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が4.6mol%となるように変更したこと以外は実施例201と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物212:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=45.9/41.4/4.6/8.1(mol%))及び無延伸フィルムを得た。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が6.9mol%となるように変更したこと以外は実施例201と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物213:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=45.9/39.1/6.9/8.1(mol%))及び無延伸フィルムを得た。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が13.8mol%となるように変更したこと以外は実施例201と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物214:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=45.9/32.2/13.8/8.1(mol%))を得た。なお、重合において分子量が十分に上がらなかったため、前述の無延伸フィルムでの酸素透過係数及びHazeの測定、並びに引張試験は行わなかった。
DL-アラニンの添加量をポリアミド化合物中の含有率が17.7mol%となるように変更したこと以外は実施例201と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物215:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=41.1/39.1/2.1/17.7(mol%))を得た。なお、重合において分子量が十分に上がらなかったため、前述の無延伸フィルムでの酸素透過係数及びHazeの測定、並びに引張試験は行わなかった。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が4.1mol%となるように変更したこと以外は実施例215と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物216:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=41.1/37.1/4.1/17.7(mol%))を得た。なお、重合において分子量が十分に上がらなかったため、前述の無延伸フィルムでの酸素透過係数及びHazeの測定、並びに引張試験は行わなかった。
高純度イソフタル酸の添加量をポリアミド化合物中の含有率が6.2mol%となるように変更したこと以外は実施例215と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物217:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=41.1/35.0/6.2/17.7(mol%))を得た。なお、重合において分子量が十分に上がらなかったため、前述の無延伸フィルムでの酸素透過係数及びHazeの測定、並びに引張試験は行わなかった。
アジピン酸をセバシン酸(伊藤製油(株)製)に変更したこと以外は実施例208と同様の方法で、DL-アラニン共重合N-MXD10I(ポリアミド化合物218:MXDA単位/セバシン酸単位/イソフタル酸単位/DL-アラニン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
アジピン酸をドデカン二酸(宇部興産(株)製)に変更したこと以外は実施例208と同様の方法で、DL-アラニン共重合N-MXD12I(ポリアミド化合物219:MXDA単位/ドデカン二酸単位/イソフタル酸単位/DL-アラニン単位=48.6/46.4/2.4/2.6(mol%))及び無延伸フィルムを得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が31.6mol%となるように変更し、イソフタル酸の添加量をポリアミド化合物中の含有率が、15.4mol%となるように変更したこと以外は実施例201と同様の方法で、DL-アラニン共重合N-MXD6I(ポリアミド化合物220:MXDA単位/アジピン酸単位/イソフタル酸単位/DL-アラニン単位=34.1/18.9/15.4/31.6(mol%))を得た。なお、重合において分子量が十分に上がらなかったため、前述の無延伸フィルムでの酸素透過係数及びHazeの測定、並びに官能試験及び引張試験は行わなかった。
(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)13000g(88.9mol)、高純度イソフタル酸(エイ・ジイ・インタナショナル・ケミカル(株)製)777.9g(4.68mol))、次亜リン酸ナトリウム11.96g(0.113mol)、酢酸ナトリウム6.21g(0.0756mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(三菱ガス化学(株)製)12710g(93.4mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。メタキシリレンジアミンの滴下終了後、内温を260℃として40分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約23.2kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミドを仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約70分かけてペレット温度を200℃まで昇温した後、200℃で30分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却して、N-MXD6I(ポリアミド化合物221:MXDA単位/アジピン酸単位/イソフタル酸単位=49.9/47.6/2.5(mol%))を得た。
ポリアミド化合物221のペレットを25mmφ単軸押出機により、押出温度260℃、スクリュー回転数60rpm、引き取り速度1.2m/minで製膜し、幅200mm、厚み95~105μmの無延伸フィルムを作製した。
アジピン酸をセバシン酸(伊藤製油(株)製)に変更したこと以外は比較例201と同様の方法で、N-MXD10I(ポリアミド化合物222:MXDA単位/セバシン酸単位/イソフタル酸単位=49.9/47.6/2.5(mol%))及び無延伸フィルムを得た。
アジピン酸をドデカン二酸(宇部興産(株)製)に変更したこと以外は比較例201と同様の方法で、N-MXD12I(ポリアミド化合物223:MXDA単位/ドデカン二酸単位/イソフタル酸単位=49.9/47.6/2.5(mol%))及び無延伸フィルムを得た。
α-アミノ酸をα位に2級水素を持つグリシン((株)東京化成工業製、試薬)に変更したこと以外は実施例201と同様の方法で、グリシン共重合N-MXD6I(ポリアミド化合物224:MXDA単位/アジピン酸単位/イソフタル酸単位/グリシン単位=45.9/43.7/2.3/8.1(mol%))及び無延伸フィルムを得た。
α-アミノ酸をα位に水素を持たない2-アミノイソ酪酸(2-アミノ-2-メチルプロパン酸、AIB、(株)日本ファインケム製、精製品)に変更したこと以外は実施例201と同様の方法で、2-アミノイソ酪酸共重合N-MXD6I(ポリアミド化合物225:MXDA単位/アジピン酸単位/イソフタル酸単位/AIB単位=45.9/43.7/2.3/8.1(mol%))及び無延伸フィルムを得た。
ポリアミド化合物221に対し、ステアリン酸コバルトを、樹脂組成物中のコバルト含有量が400ppmとなるように添加し、ドライブレンドした。得られたブレンド物を、30mmφ2軸押出機により、押出温度260℃、スクリュー回転数60rpm、フィードスクリュー回転数12rpm、引き取り速度1.8m/minで製膜し、幅200mm、厚み95~105μmの無延伸フィルムを作製した。
ポリアミド化合物221の100質量部に対し、マレイン酸変性ポリブタジエン(PB)(日本石油化学(株)製、商品名:M-2000-20)5質量部、及びステアリン酸コバルトを、樹脂組成物中のコバルト含有量が400ppmとなるように添加し、ドライブレンドした。得られたブレンド物を、30mmφ2軸押出機により、押出温度260℃、スクリュー回転数60rpm、フィードスクリュー回転数14rpm、引き取り速度2.0m/minで製膜し、幅200mm、厚み95~105μmの無延伸フィルムを作製した。
ポリアミド化合物221の100質量部に対し、DL-アラニン((株)武蔵野化学研究所製)を、樹脂組成物中のDL-アラニン含有量が5質量部となるように添加し、ドライブレンドした。得られたブレンド物同士が共重合しないように、15mmφの小型の単軸押出機を用いて、押出温度240℃、スクリュー回転数30rpm、フィードスクリュー回転数14rpm、引き取り速度1.0m/minで製膜し、幅110mm、厚み95~105μmの無延伸フィルムを作製した。
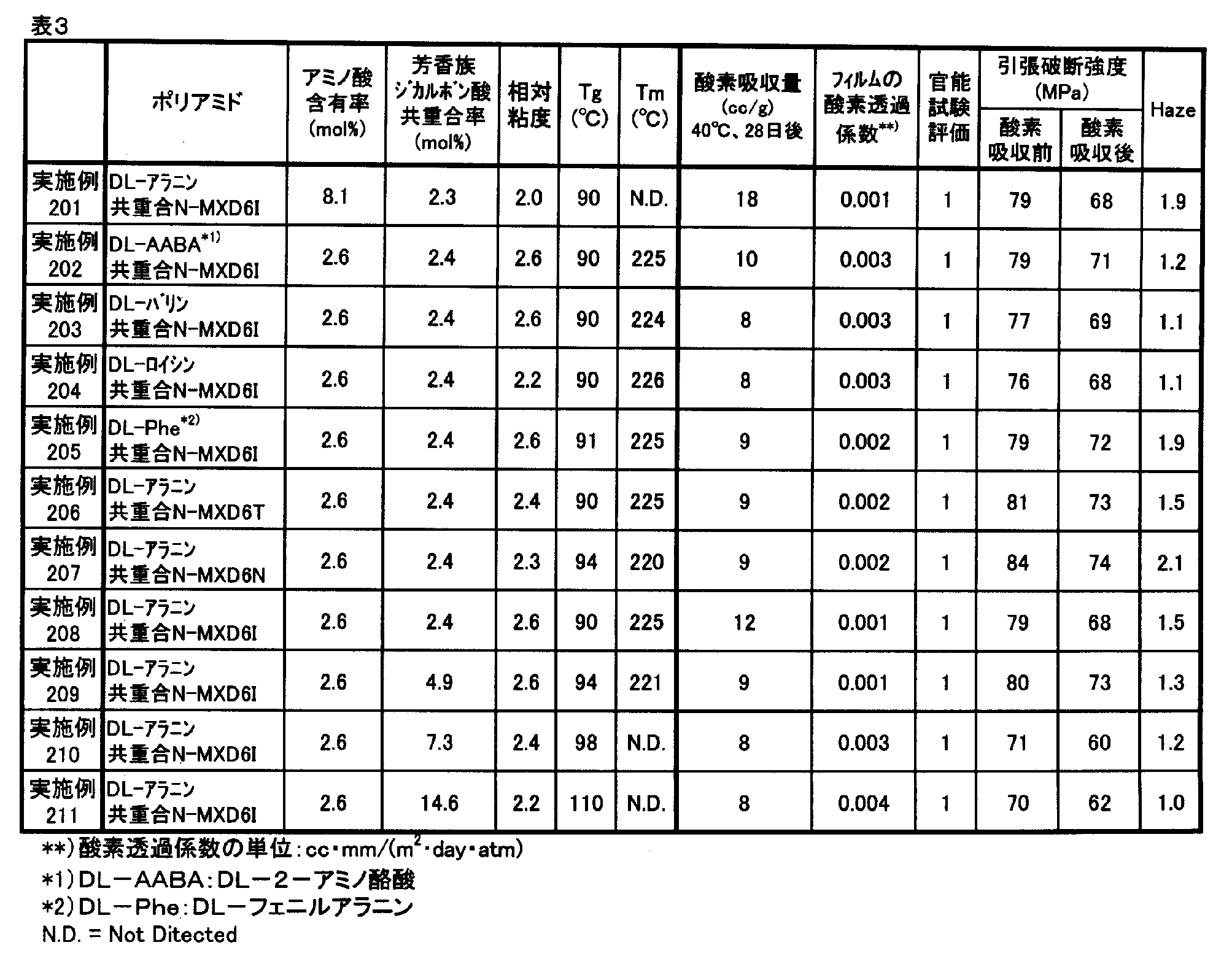

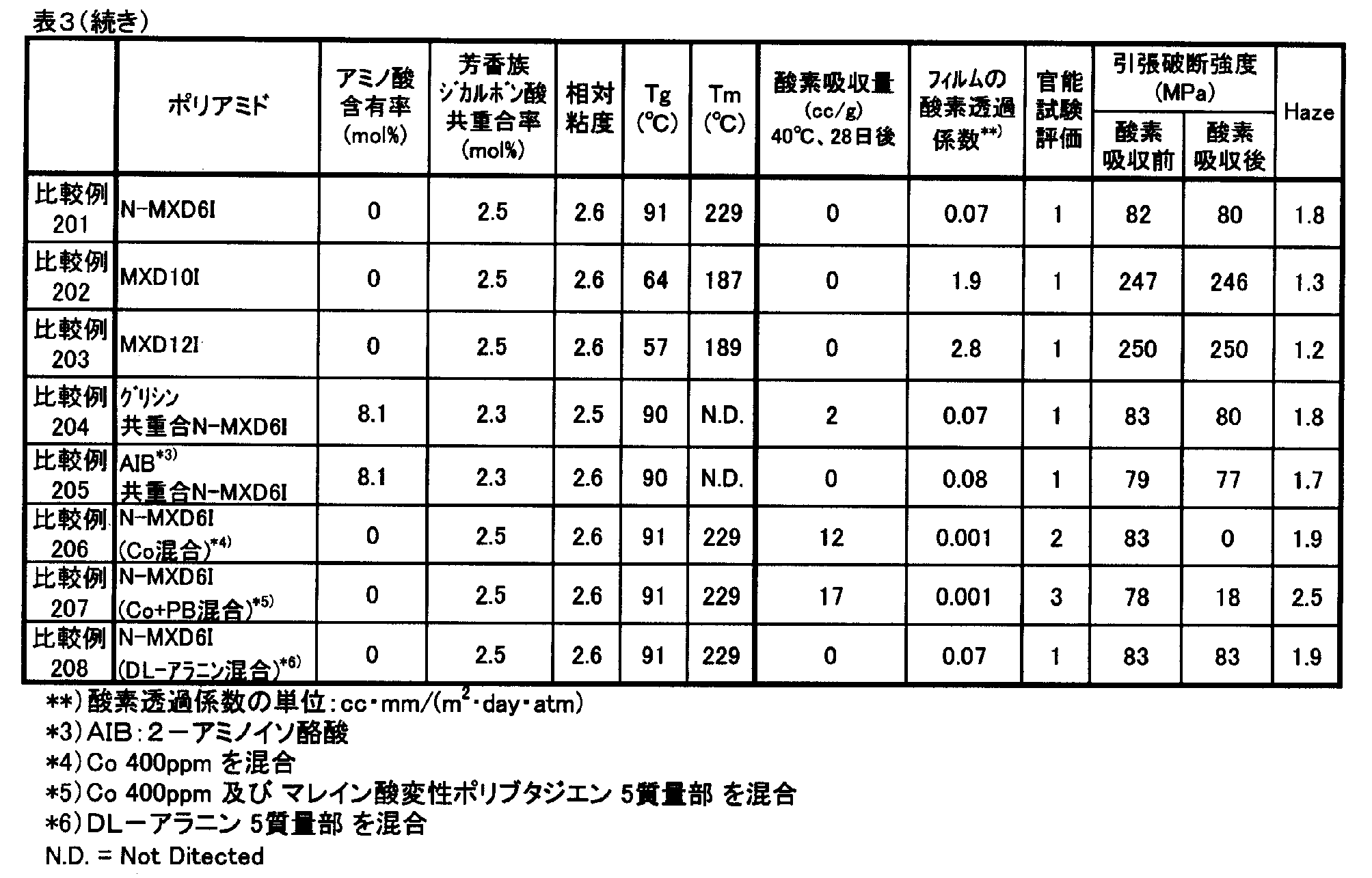
これらに対し、3級水素を有するα-アミノ酸及び芳香族ジカルボン酸を共重合したポリアミド化合物は、金属を用いることなく十分な酸素吸収性能を発現することができ、しかも不快な臭気を発生することがない(実施例201~220)。特に、フィルムサンプルの場合、フィルムの酸素透過係数も良好で、透明性も良好であり、更に酸素吸収後の機械物性も保持できている(実施例201~213、218及び219)。また、実施例220のポリアミド化合物は、分子量は上昇しなかったものの、非常に良好な酸素吸収性能を示した。
なお、直鎖脂肪族ジカルボン酸成分としてセバシン酸又はドデカン二酸を用いた実施例218及び219は、アジピン酸を用いた実施例に比べると酸素吸収性能が低いが、3級水素を有するα-アミノ酸を共重合しないN-MXD10I(比較例202)又はN-MXD12I(比較例203)に比べると優れた酸素吸収性能を有することがわかる。
(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)13000g(89.0mol)、DL-アラニン((株)武蔵野化学研究所製)880.56g(9.88mol)、ε-カプロラクタム(宇部興産株式会社製)4314.10g(38.1mol)、次亜リン酸ナトリウム13.75g(0.13mol)、酢酸ナトリウム7.13g(0.087mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(三菱ガス化学(株)製)12433g(88.7mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。メタキシリレンジアミンの滴下終了後、内温を260℃として40分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約26kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド及び水0.5質量%を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約70分かけてペレット温度を180℃まで昇温した後、180℃で30分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してDL-アラニン共重合N-MXD6,6(ポリアミド化合物301:MXDA単位/アジピン酸単位/DL-アラニン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
DL-アラニンをD-アラニンに変更したこと以外は実施例301と同様の方法で、D-アラニン共重合N-MXD6,6(ポリアミド化合物302:MXDA単位/アジピン酸単位/D-アラニン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
DL-アラニンをL-アラニンに変更したこと以外は実施例301と同様の方法で、L-アラニン共重合N-MXD6,6(ポリアミド化合物303:MXDA単位/アジピン酸単位/L-アラニン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が0.8mol%となるように変更し、ε-カプロラクタムの添加量をポリアミド化合物中の含有率が17.5mol%となるように変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD6,6(ポリアミド化合物304:MXDA単位/アジピン酸単位/DL-アラニン単位/ε-カプロラクタム単位=40.8/40.9/0.8/17.5(mol%))を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が15.0mol%となるように変更し、ε-カプロラクタムの添加量をポリアミド化合物中の含有率が15.0mol%となるように変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD6,6(ポリアミド化合物305:MXDA単位/アジピン酸単位/DL-アラニン単位/ε-カプロラクタム単位=34.8/35.2/15.0/15.0(mol%))を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が29.2mol%となるように変更し、ε-カプロラクタムの添加量をポリアミド化合物中の含有率が12.5mol%となるように変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD6,6(ポリアミド化合物306:MXDA単位/アジピン酸単位/DL-アラニン単位/ε-カプロラクタム単位=28.9/29.3/29.3/12.5(mol%))を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が49.2mol%となるように変更し、ε-カプロラクタムの添加量をポリアミド化合物中の含有率が9.0mol%となるように変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD6,6(ポリアミド化合物307:MXDA単位/アジピン酸単位/DL-アラニン単位/ε-カプロラクタム単位=20.7/21.1/49.2/9.0(mol%))を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が3.6mol%となるように変更し、ε-カプロラクタムの添加量をポリアミド化合物中の含有率が32.2mol%となるように変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD6,6(ポリアミド化合物308:MXDA単位/アジピン酸単位/DL-アラニン単位/ε-カプロラクタム単位=32.0/32.2/3.6/32.2(mol%))を得た。
DL-アラニンの添加量をポリアミド化合物中の含有率が5.0mol%となるように変更し、ε-カプロラクタムの添加量をポリアミド化合物中の含有率が5.0mol%となるように変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD6,6(ポリアミド化合物309:MXDA単位/アジピン酸単位/DL-アラニン単位/ε-カプロラクタム単位=44.9/45.1/5.0/5.0(mol%))を得た。
ε-カプロラクタムをラウロラクタム(宇部興産(株)製)に変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD6,12(ポリアミド化合物310:MXDA単位/アジピン酸単位/DL-アラニン単位/ラウロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
α-アミノ酸をDL-2-アミノ酪酸(DL-AABA、(株)日本ファインケム製、精製品)に変更したこと以外は実施例301と同様の方法で、DL-2-アミノ酪酸共重合N-MXD6,6(ポリアミド化合物311:MXDA単位/アジピン酸単位/DL-AABA単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
α-アミノ酸をDL-ロイシン(Ningbo Haishuo Bio-technology製)に変更したこと以外は実施例301と同様の方法で、DL-ロイシン共重合N-MXD6,6(ポリアミド化合物312:MXDA単位/アジピン酸単位/DL-ロイシン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
α-アミノ酸をDL-フェニルアラニン(DL-Phe、Sinogel Amino Acid Co.,Ltd製)に変更したこと以外は実施例301と同様の方法で、DL-フェニルアラニン共重合N-MXD6,6(ポリアミド化合物313:MXDA単位/アジピン酸単位/DL-フェニルアラニン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
アジピン酸をセバシン酸(伊藤製油(株)製)に変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD10,6(ポリアミド化合物314:MXDA単位/セバシン酸単位/DL-アラニン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
アジピン酸をドデカン二酸(宇部興産(株)製)に変更したこと以外は実施例301と同様の方法で、DL-アラニン共重合N-MXD12,6(ポリアミド化合物315:MXDA単位/ドデカン二酸単位/DL-アラニン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
(ポリアミド化合物の常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、精秤したアジピン酸(旭化成ケミカルズ(株)製)13000g(89.0mol)、ε-カプロラクタム(宇部興産(株)製)4314.1g(38.1mol)、次亜リン酸ナトリウム13.4g(0.13mol)、酢酸ナトリウム6.9g(0.084mol)を入れ、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これにメタキシリレンジアミン(MXDA)(三菱ガス化学(株)製)12437g(88.7mol)を撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。1,3-ビス(アミノメチル)シクロヘキサンの滴下終了後、内温を260℃として40分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリマーを取り出してこれをペレット化し、約26kgのポリアミド化合物を得た。
次いで、窒素ガス導入管、真空ライン、真空ポンプ、内温測定用の熱電対を設けたジャケット付きのタンブルドライヤーに前記ポリアミド及び、水0.5質量%を仕込み、一定速度で回転させつつ、タンブルドライヤー内部を純度が99容量%以上の窒素ガスで十分に置換した後、同窒素ガス気流下でタンブルドライヤーを加熱し、約150分かけてペレット温度を150℃に昇温した。ペレット温度が150℃に達した時点で系内の圧力を1torr以下に減圧した。さらに昇温を続け、約70分かけてペレット温度を180℃まで昇温した後、180℃で30分保持した。次いで、系内に純度が99容量%以上の窒素ガスを導入して、タンブルドライヤーを回転させたまま冷却してN-MXD6,6(ポリアミド化合物316:MXDA単位/アジピン酸単位/ε-カプロラクタム単位=41.1/41.2/17.7(mol%)))を得た。
DL-アラニンをα位に2級水素を持つグリシン((株)東京化成工業製)に変更したこと以外は実施例301と同様の方法で、グリシン共重合N-MXD6,6(ポリアミド化合物317:MXDA単位/アジピン酸単位/グリシン単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
DL-アラニンをα位に水素を持たない2-アミノイソ酪酸(2-アミノ-2-メチルプロパン酸、AIB、(株)日本ファインケム製、精製品)に変更したこと以外は実施例301と同様の方法で、2-アミノイソ酪酸共重合N-MXD6,6(ポリアミド化合物318:MXDA単位/アジピン酸単位/AIB単位/ε-カプロラクタム単位=39.3/39.4/4.4/16.9(mol%))を得た。
ε-カプロラクタムをラウロラクタム(宇部興産(株)製)に変更したこと以外は比較例301と同様の方法で、N-MXD6,12(ポリアミド化合物319:MXDA単位/アジピン酸単位/ラウロラクタム単位=41.1/41.2/17.7(mol%))を得た。
アジピン酸をセバシン酸(伊藤製油(株)製)に変更したこと以外は比較例301と同様の方法で、N-MXD10,6(ポリアミド化合物320:MXDA単位/セバシン酸単位/ε-カプロラクタム単位=41.1/41.2/17.7(mol%))を得た。
アジピン酸をドデカン二酸(宇部興産(株)製)に変更したこと以外は、比較例301と同様の方法でN-MXD12,6(ポリアミド化合物321:MXDA単位/ドデカン二酸単位/ε-カプロラクタム単位=41.1/41.2/17.7(mol%))を得た。
比較例301で得たポリアミド化合物316に対し、DL-アラニン((株)武蔵野化学研究所製)を添加し、ドライブレンドすることで、ポリアミド化合物とDL-アラニンとの混合物(混合物中のDL-アラニン含有量:5質量%)を得た。混合物中のブレンド物同士が共重合しないように、15mmφの小型の単軸押出機を用いて、押出温度240℃、スクリュー回転数30rpm、フィードスクリュー回転数14rpmにて、DL-アラニン含有N-MXD6,6ペレットを作製した。

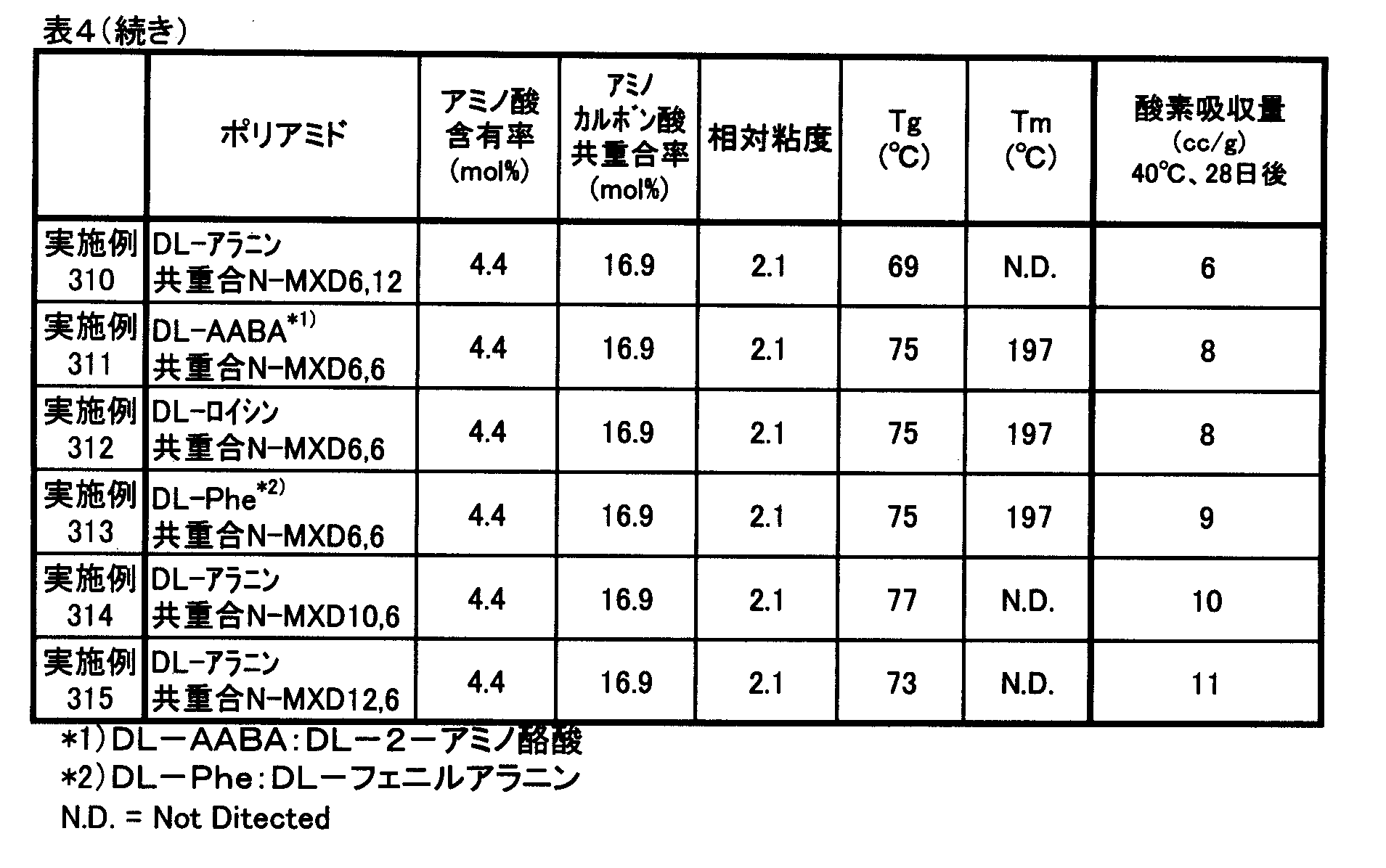

これらに対し、3級水素を有するα-アミノ酸を共重合したポリアミド化合物は、金属を用いることなく十分な酸素吸収性能を発現することができる。(実施例301~315)。
(ポリアミドオリゴマーの常圧滴下法による溶融重合)
撹拌機、分縮器、全縮器、温度計、滴下ロート及び窒素導入管、ストランドダイを備えた内容積50リットルの反応容器に、表5に記載された種類及び配合量の脂肪族ジカルボン酸、芳香族ジカルボン酸、α-アミノ酸、ω-アミノカルボン酸、次亜リン酸ナトリウム、酢酸ナトリウムを仕込み、十分に窒素置換した後、さらに少量の窒素気流下で系内を撹拌しながら170℃まで加熱した。これに、表5に記載された配合量の芳香族ジアミンであるメタキシリレンジアミンを撹拌下に滴下し、生成する縮合水を系外へ除きながら系内を連続的に昇温した。ジアミンの滴下終了後、内温を240℃として撹拌トルクの上昇度に注意しながら40~60分反応を継続した。その後、系内を窒素で加圧し、ストランドダイからポリアミドオリゴマーを取り出した。ストランド状態で取り出せるものはペレタイズし、ペレット状のポリアミドオリゴマーを得た。また、分子量が低くストランド状態で取り出せないものは、別途、粉砕機にて粉砕し、ポリアミドオリゴマー粉砕物を得た。


Claims (9)
- 下記一般式(I)で表される芳香族ジアミン単位を50モル%以上含むジアミン単位25~50モル%と、下記一般式(II-1)で表される直鎖脂肪族ジカルボン酸単位及び/又は下記一般式(II-2)で表される芳香族ジカルボン酸単位を合計で50モル%以上含むジカルボン酸単位25~50モル%と、下記一般式(III)で表される構成単位0.1~50モル%とを含有する、ポリアミド化合物。

- 前記一般式(III)におけるRが、置換もしくは無置換の炭素数1~6のアルキル基又は置換もしくは無置換の炭素数6~10のアリール基である、請求項1に記載のポリアミド化合物。
- 前記ジアミン単位が、メタキシリレンジアミン単位を50モル%以上含む、請求項1又は2に記載のポリアミド化合物。
- 前記直鎖脂肪族ジカルボン酸単位が、アジピン酸単位、セバシン酸単位、及び1,12-ドデカンジカルボン酸単位からなる群から選ばれる少なくとも1つを合計で50モル%以上含む、請求項1~3のいずれかに記載のポリアミド化合物。
- 前記芳香族ジカルボン酸単位が、イソフタル酸単位、テレフタル酸単位、及び2,6-ナフタレンジカルボン酸単位からなる群から選ばれる少なくとも1つを合計で50モル%以上含む、請求項1~4のいずれかに記載のポリアミド化合物。
- 更に、下記一般式(A)で表されるω-アミノカルボン酸単位を、ポリアミド化合物の全構成単位中0.1~49.9モル%含有する、請求項1~5のいずれかに記載のポリアミド化合物。

- 前記ω-アミノカルボン酸単位が、6-アミノヘキサン酸単位及び/又は12-アミノドデカン酸単位を合計で50モル%以上含む、請求項6に記載のポリアミド化合物。
- 相対粘度が1.8以上4.2以下である、請求項1~7のいずれかに記載のポリアミド化合物。
- 相対粘度が1.01以上1.8未満である、請求項1~7のいずれかに記載のポリアミド化合物。
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2012006869A MX2012006869A (es) | 2009-12-28 | 2010-12-24 | Compuesto de poliamida. |
| US13/519,205 US8835595B2 (en) | 2009-12-28 | 2010-12-24 | Polyamide compound |
| CA2785673A CA2785673C (en) | 2009-12-28 | 2010-12-24 | Polyamide compound |
| JP2011547654A JP5257522B2 (ja) | 2009-12-28 | 2010-12-24 | ポリアミド化合物 |
| BR112012015989A BR112012015989A2 (pt) | 2009-12-28 | 2010-12-24 | composto de poliamida |
| AU2010337546A AU2010337546B2 (en) | 2009-12-28 | 2010-12-24 | Polyamide compound |
| RU2012132427/05A RU2561075C2 (ru) | 2009-12-28 | 2010-12-24 | Полиамидное соединение |
| KR1020127016551A KR101442104B1 (ko) | 2009-12-28 | 2010-12-24 | 폴리아미드 화합물 |
| EP10840955.8A EP2520604B1 (en) | 2009-12-28 | 2010-12-24 | Polyamide compound |
| CN201080059872.5A CN102762636B (zh) | 2009-12-28 | 2010-12-24 | 聚酰胺化合物 |
| ZA2012/04794A ZA201204794B (en) | 2009-12-28 | 2012-06-27 | Polyamide compound |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-298756 | 2009-12-28 | ||
| JP2009298756 | 2009-12-28 | ||
| JP2010-070340 | 2010-03-25 | ||
| JP2010070340 | 2010-03-25 | ||
| JP2010-120893 | 2010-05-26 | ||
| JP2010120893 | 2010-05-26 | ||
| JP2010-127969 | 2010-06-03 | ||
| JP2010127969 | 2010-06-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011081099A1 true WO2011081099A1 (ja) | 2011-07-07 |
Family
ID=44226505
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/073371 WO2011081099A1 (ja) | 2009-12-28 | 2010-12-24 | ポリアミド化合物 |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US8835595B2 (ja) |
| EP (1) | EP2520604B1 (ja) |
| JP (1) | JP5257522B2 (ja) |
| KR (1) | KR101442104B1 (ja) |
| CN (1) | CN102762636B (ja) |
| AU (1) | AU2010337546B2 (ja) |
| BR (1) | BR112012015989A2 (ja) |
| CA (1) | CA2785673C (ja) |
| CL (1) | CL2012001780A1 (ja) |
| MX (1) | MX2012006869A (ja) |
| PE (1) | PE20130056A1 (ja) |
| RU (1) | RU2561075C2 (ja) |
| TW (1) | TWI443126B (ja) |
| WO (1) | WO2011081099A1 (ja) |
| ZA (1) | ZA201204794B (ja) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013002069A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層フィルム及びフィルム包装容器 |
| WO2013002070A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | フィルム及びフィルム包装容器 |
| WO2013002079A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 積層材及び紙容器 |
| WO2013002071A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層インジェクション成形体 |
| WO2013002074A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | ダイレクトブロー多層ボトル |
| WO2013002076A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層シート |
| WO2013002078A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 積層材及び紙容器 |
| WO2013002073A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層インジェクション成形体 |
| EP2660292A1 (en) * | 2010-12-27 | 2013-11-06 | Mitsubishi Gas Chemical Company, Inc. | Polyamide composition |
| JP2014037248A (ja) * | 2012-08-15 | 2014-02-27 | Mitsubishi Gas Chemical Co Inc | ガスバリア性キャップ及びスパウト |
| WO2014034624A1 (ja) | 2012-08-31 | 2014-03-06 | 三菱瓦斯化学株式会社 | ポリアミド樹脂組成物及びその製造方法 |
| JP2014068767A (ja) * | 2012-09-28 | 2014-04-21 | Mitsubishi Gas Chemical Co Inc | 医療用多層容器 |
| JPWO2013002080A1 (ja) * | 2011-06-27 | 2015-02-23 | 三菱瓦斯化学株式会社 | チューブ状容器 |
| JPWO2013002081A1 (ja) * | 2011-06-27 | 2015-02-23 | 三菱瓦斯化学株式会社 | チューブ状容器 |
| JP2020200405A (ja) * | 2019-06-11 | 2020-12-17 | 三菱瓦斯化学株式会社 | 延伸フィルムおよび多層体 |
| WO2021049266A1 (ja) | 2019-09-12 | 2021-03-18 | 宇部興産株式会社 | 共重合ポリアミドの製造方法 |
| WO2024024330A1 (ja) * | 2022-07-25 | 2024-02-01 | 三菱瓦斯化学株式会社 | 樹脂組成物、および、成形品 |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8653225B2 (en) | 2010-04-20 | 2014-02-18 | Mitsubishi Gas Chemical Company, Inc. | Polyamide compound |
| MX2012014661A (es) | 2010-06-29 | 2013-02-11 | Mitsubishi Gas Chemical Co | Compuesto de poliamida. |
| EP2725065B1 (en) | 2011-06-27 | 2018-06-06 | Mitsubishi Gas Chemical Company, Inc. | Injection-molded body |
| WO2014198770A1 (de) * | 2013-06-12 | 2014-12-18 | Basf Se | Verfahren zur herstellung aliphatischer oder teilaromatischer polyamide umfassend eine festphasenpolymerisation |
| EP3064527B1 (en) * | 2013-10-31 | 2018-12-12 | Mitsubishi Gas Chemical Company, Inc. | Xylylenediamine composition and method for producing polyamide resin |
| KR102192621B1 (ko) * | 2013-10-31 | 2020-12-17 | 미쯔비시 가스 케미칼 컴파니, 인코포레이티드 | 자일릴렌디아민 조성물 및 폴리아미드 수지의 제조방법 |
| CN105683253B (zh) * | 2013-10-31 | 2017-10-20 | 三菱瓦斯化学株式会社 | 苯二甲胺组合物和聚酰胺树脂的制造方法 |
| CN105683251B (zh) | 2013-10-31 | 2017-10-24 | 三菱瓦斯化学株式会社 | 苯二甲胺组合物和聚酰胺树脂的制造方法 |
| KR20160130786A (ko) * | 2014-03-07 | 2016-11-14 | 인비스타 테크놀러지스 에스.에이 알.엘. | 광물 첨가제를 가지는 폴리아마이드 수지 |
| CN108698730B (zh) * | 2016-02-16 | 2021-07-13 | 三菱瓦斯化学株式会社 | 多层容器、包含其的注射器和物品、该多层容器的制造方法及其应用 |
| AU2017326287B2 (en) | 2016-09-14 | 2021-12-09 | Basf Se | Polymer film comprising a co-polyamide of a diamine, a dimer acid and a lactam |
| CN110591082B (zh) * | 2019-08-22 | 2021-09-17 | 惠生(泰州)新材料科技有限公司 | 共聚尼龙树脂及其制备方法和用途 |
| ES2837489B2 (es) * | 2019-12-31 | 2022-02-28 | Primalchit Solutions S L | Mezcla de componentes organicos no polimericos con capacidad retardante de llama, metodo de preparacion y uso |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3721682A (en) | 1969-10-06 | 1973-03-20 | New Japan Chem Co Ltd | Manufacture of benzylidene sorbitols |
| JPH0272851A (ja) | 1988-09-08 | 1990-03-13 | Mitsubishi Gas Chem Co Inc | フィルム状脱酸素剤 |
| JPH0490848A (ja) | 1990-08-03 | 1992-03-24 | Toyo Seikan Kaisha Ltd | 酸素吸収剤及び該酸素吸収剤を用いた樹脂組成物並びに樹脂組成物からなるフィルム又はシート,包装用容器 |
| JPH05115776A (ja) | 1991-04-02 | 1993-05-14 | W R Grace & Co | 酸素掃去のための配合物、製品及び方法 |
| JP2991437B2 (ja) | 1987-07-27 | 1999-12-20 | カヌードメタルボックス パブリック リミテド カンパニー | 包装に関する改良 |
| JP2008274288A (ja) * | 2007-05-03 | 2008-11-13 | Ems-Patent Ag | 半芳香族ポリアミド成形組成物及びその使用 |
| JP2010248322A (ja) * | 2009-04-13 | 2010-11-04 | Asahi Kasei Chemicals Corp | ポリアミド組成物 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE791468A (fr) * | 1971-11-16 | 1973-05-16 | Hoechst Ag | Polyamides transparents et leur preparation |
| SU559933A1 (ru) * | 1974-03-18 | 1977-05-30 | Ордена Ленина Институт Элементоорганических Соединений Ан Ссср | Способ получени ароматических полиамидов |
| JPS63275629A (ja) * | 1987-05-07 | 1988-11-14 | Mitsubishi Kasei Corp | ポリアミドおよびポリヒドラジドの製造法 |
| JPH0445152A (ja) * | 1990-06-11 | 1992-02-14 | Toppan Printing Co Ltd | 酸素吸収性樹脂組成物 |
| DE4040056A1 (de) | 1990-12-14 | 1992-06-17 | Merck Patent Gmbh | Saeureamide |
| PT1065232E (pt) | 1999-06-29 | 2004-02-27 | Degussa | Copolimero de excerto de poliamida |
| EP1156073B1 (en) | 2000-05-19 | 2003-08-20 | Mitsubishi Gas Chemical Company, Inc. | Shaped article of polyamide resin and production thereof |
| AU2003252855B2 (en) * | 2002-10-22 | 2008-09-18 | Mitsubishi Gas Chemical Company, Inc. | Gas-barrier multi-layer structure |
| DE102004029935B4 (de) * | 2004-06-21 | 2007-08-09 | Pe Polymer Engineering Gmbh & Co Forschungs Kg | Verfahren zur kontinuierlichen Herstellung von Copolyamiden mit Schmelzpunkten oberhalb von 265 C |
| FR2884518B1 (fr) * | 2005-04-14 | 2007-09-21 | Arkema Sa | Structure barriere a base de polyamide mxd.10 |
-
2010
- 2010-12-24 JP JP2011547654A patent/JP5257522B2/ja active Active
- 2010-12-24 WO PCT/JP2010/073371 patent/WO2011081099A1/ja active Application Filing
- 2010-12-24 RU RU2012132427/05A patent/RU2561075C2/ru not_active IP Right Cessation
- 2010-12-24 US US13/519,205 patent/US8835595B2/en active Active
- 2010-12-24 EP EP10840955.8A patent/EP2520604B1/en active Active
- 2010-12-24 CA CA2785673A patent/CA2785673C/en not_active Expired - Fee Related
- 2010-12-24 PE PE2012000847A patent/PE20130056A1/es not_active Application Discontinuation
- 2010-12-24 AU AU2010337546A patent/AU2010337546B2/en not_active Ceased
- 2010-12-24 KR KR1020127016551A patent/KR101442104B1/ko active IP Right Grant
- 2010-12-24 BR BR112012015989A patent/BR112012015989A2/pt not_active Application Discontinuation
- 2010-12-24 MX MX2012006869A patent/MX2012006869A/es active IP Right Grant
- 2010-12-24 CN CN201080059872.5A patent/CN102762636B/zh not_active Expired - Fee Related
- 2010-12-28 TW TW099146208A patent/TWI443126B/zh not_active IP Right Cessation
-
2012
- 2012-06-27 CL CL2012001780A patent/CL2012001780A1/es unknown
- 2012-06-27 ZA ZA2012/04794A patent/ZA201204794B/en unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3721682A (en) | 1969-10-06 | 1973-03-20 | New Japan Chem Co Ltd | Manufacture of benzylidene sorbitols |
| JP2991437B2 (ja) | 1987-07-27 | 1999-12-20 | カヌードメタルボックス パブリック リミテド カンパニー | 包装に関する改良 |
| JPH0272851A (ja) | 1988-09-08 | 1990-03-13 | Mitsubishi Gas Chem Co Inc | フィルム状脱酸素剤 |
| JPH0490848A (ja) | 1990-08-03 | 1992-03-24 | Toyo Seikan Kaisha Ltd | 酸素吸収剤及び該酸素吸収剤を用いた樹脂組成物並びに樹脂組成物からなるフィルム又はシート,包装用容器 |
| JPH05115776A (ja) | 1991-04-02 | 1993-05-14 | W R Grace & Co | 酸素掃去のための配合物、製品及び方法 |
| JP2008274288A (ja) * | 2007-05-03 | 2008-11-13 | Ems-Patent Ag | 半芳香族ポリアミド成形組成物及びその使用 |
| JP2010248322A (ja) * | 2009-04-13 | 2010-11-04 | Asahi Kasei Chemicals Corp | ポリアミド組成物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2520604A4 * |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2660292A1 (en) * | 2010-12-27 | 2013-11-06 | Mitsubishi Gas Chemical Company, Inc. | Polyamide composition |
| EP2660292A4 (en) * | 2010-12-27 | 2014-05-21 | Mitsubishi Gas Chemical Co | POLYAMIDE COMPOSITION |
| JPWO2013002081A1 (ja) * | 2011-06-27 | 2015-02-23 | 三菱瓦斯化学株式会社 | チューブ状容器 |
| JPWO2013002080A1 (ja) * | 2011-06-27 | 2015-02-23 | 三菱瓦斯化学株式会社 | チューブ状容器 |
| WO2013002074A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | ダイレクトブロー多層ボトル |
| WO2013002076A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層シート |
| WO2013002078A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 積層材及び紙容器 |
| WO2013002073A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層インジェクション成形体 |
| WO2013002079A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 積層材及び紙容器 |
| US9731482B2 (en) | 2011-06-27 | 2017-08-15 | Mitsubishi Gas Chemical Company, Inc. | Multilayer injection-molded body |
| US9718259B2 (en) | 2011-06-27 | 2017-08-01 | Mitsubishi Gas Chemical Company, Inc. | Multilayer injection-molded body |
| WO2013002069A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層フィルム及びフィルム包装容器 |
| WO2013002070A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | フィルム及びフィルム包装容器 |
| WO2013002071A1 (ja) * | 2011-06-27 | 2013-01-03 | 三菱瓦斯化学株式会社 | 多層インジェクション成形体 |
| JPWO2013002074A1 (ja) * | 2011-06-27 | 2015-02-23 | 三菱瓦斯化学株式会社 | ダイレクトブロー多層ボトル |
| JP2014037248A (ja) * | 2012-08-15 | 2014-02-27 | Mitsubishi Gas Chemical Co Inc | ガスバリア性キャップ及びスパウト |
| CN104520381A (zh) * | 2012-08-31 | 2015-04-15 | 三菱瓦斯化学株式会社 | 聚酰胺树脂组合物及其制造方法 |
| WO2014034624A1 (ja) | 2012-08-31 | 2014-03-06 | 三菱瓦斯化学株式会社 | ポリアミド樹脂組成物及びその製造方法 |
| JP2014068767A (ja) * | 2012-09-28 | 2014-04-21 | Mitsubishi Gas Chemical Co Inc | 医療用多層容器 |
| JP2020200405A (ja) * | 2019-06-11 | 2020-12-17 | 三菱瓦斯化学株式会社 | 延伸フィルムおよび多層体 |
| JP7342439B2 (ja) | 2019-06-11 | 2023-09-12 | 三菱瓦斯化学株式会社 | 延伸フィルムおよび多層体 |
| WO2021049266A1 (ja) | 2019-09-12 | 2021-03-18 | 宇部興産株式会社 | 共重合ポリアミドの製造方法 |
| WO2024024330A1 (ja) * | 2022-07-25 | 2024-02-01 | 三菱瓦斯化学株式会社 | 樹脂組成物、および、成形品 |
Also Published As
| Publication number | Publication date |
|---|---|
| US8835595B2 (en) | 2014-09-16 |
| TWI443126B (zh) | 2014-07-01 |
| EP2520604A4 (en) | 2013-08-14 |
| EP2520604A1 (en) | 2012-11-07 |
| KR20120097387A (ko) | 2012-09-03 |
| CN102762636A (zh) | 2012-10-31 |
| AU2010337546A1 (en) | 2012-07-05 |
| EP2520604B1 (en) | 2015-07-29 |
| JPWO2011081099A1 (ja) | 2013-05-09 |
| JP5257522B2 (ja) | 2013-08-07 |
| PE20130056A1 (es) | 2013-02-11 |
| AU2010337546B2 (en) | 2013-11-21 |
| CA2785673C (en) | 2015-07-21 |
| CN102762636B (zh) | 2014-02-19 |
| RU2012132427A (ru) | 2014-02-10 |
| KR101442104B1 (ko) | 2014-09-18 |
| CL2012001780A1 (es) | 2013-08-30 |
| CA2785673A1 (en) | 2011-07-07 |
| BR112012015989A2 (pt) | 2016-08-16 |
| TW201134854A (en) | 2011-10-16 |
| US20120302723A1 (en) | 2012-11-29 |
| RU2561075C2 (ru) | 2015-08-20 |
| ZA201204794B (en) | 2013-09-25 |
| MX2012006869A (es) | 2012-07-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5257522B2 (ja) | ポリアミド化合物 | |
| JP5880446B2 (ja) | ポリアミド組成物 | |
| JP5648683B2 (ja) | ポリアミド化合物 | |
| WO2014034624A1 (ja) | ポリアミド樹脂組成物及びその製造方法 | |
| WO2012002000A1 (ja) | ポリアミド化合物 | |
| JP5867388B2 (ja) | ポリアミド化合物 | |
| JP5928254B2 (ja) | ポリアミド樹脂組成物及びその製造方法 | |
| WO2015115148A1 (ja) | ポリアミドまたはポリアミド組成物の造粒方法 | |
| JP5983396B2 (ja) | 着色ポリアミド樹脂組成物及びその製造方法 | |
| JP5954326B2 (ja) | 積層材及び紙容器 | |
| JP6011530B2 (ja) | チューブ状容器 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080059872.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10840955 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011547654 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010337546 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/006869 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 000847-2012 Country of ref document: PE |
|
| ENP | Entry into the national phase |
Ref document number: 20127016551 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2785673 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13519205 Country of ref document: US Ref document number: 2010840955 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5620/CHENP/2012 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201003244 Country of ref document: TH |
|
| ENP | Entry into the national phase |
Ref document number: 2010337546 Country of ref document: AU Date of ref document: 20101224 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13519205 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012132427 Country of ref document: RU |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012015989 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012015989 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120627 |