WO2011030824A1 - 徐放性粒子およびその製造方法 - Google Patents
徐放性粒子およびその製造方法 Download PDFInfo
- Publication number
- WO2011030824A1 WO2011030824A1 PCT/JP2010/065521 JP2010065521W WO2011030824A1 WO 2011030824 A1 WO2011030824 A1 WO 2011030824A1 JP 2010065521 W JP2010065521 W JP 2010065521W WO 2011030824 A1 WO2011030824 A1 WO 2011030824A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- polymer
- suspension
- term
- solubility parameter
- Prior art date
Links
Images
















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
Definitions
- the present invention relates to sustained release particles and a method for producing the same, and more particularly to sustained release particles for sustained release of an antibiotic compound and a method for producing the same.
- antibiotic active compounds such as bactericides, preservatives, and fungicides are microencapsulated to gradually release the antibiotic active compounds to ensure sustained efficacy.
- An object of the present invention is to provide a sustained-release particle that is simple, low-cost, and excellent in sustained-release properties, and a method for producing the same.
- a hydrophobic solution is prepared by dissolving a hydrophobic antibiotic compound having a specific range of h and compound with a hydrophobic polymerizable vinyl monomer in the absence of a solvent, and the hydrophobic solution is dispersed in water.
- a polymerizable vinyl monomer is radically polymerized to produce a polymer having a dipole force term ⁇ p, polymer and a hydrogen bonding force term ⁇ h, polymer with a solubility parameter ⁇ in a specific range,
- the inventors have found that low-cost, sustained-release particles excellent in sustained-release properties can be obtained, and have further advanced research. As a result, the present invention has been completed. .
- the present invention (1) The melting point is 100 ° C. or lower, the dipole force term ⁇ p, compound of the solubility parameter ⁇ defined by Hansen and calculated by the van Klevelen and Hoftyzer method is 2 to 8 [(J / cm 3 ) 1 / 2 ], a hydrophobic antibiotic compound having a hydrogen bond strength term ⁇ h, compound of the solubility parameter ⁇ of 5.5 to 9.5 [(J / cm 3 ) 1/2 ], A hydrophobic solution is prepared by dissolving with a hydrophobic polymerizable vinyl monomer in the presence, the hydrophobic solution is dispersed in water, and the polymerizable vinyl monomer is radically polymerized in the presence of an oil-soluble initiator.
- sustained release particles according to the above (1) (3) The sustained release particles according to (1) above, wherein the blending ratio of the antibiotic compound to the polymerizable vinyl monomer is 0.11 to 1.5 on a weight basis, (4) The melting point is 100 ° C.
- the dipole force term ⁇ p, compound of the solubility parameter ⁇ defined by Hansen and calculated by the van Klevelen and Hoftyzer method is 2 to 8 [(J / cm 3 ) 1 / 2 ], a hydrophobic antibiotic compound having a hydrogen bond strength term ⁇ h, compound of the solubility parameter ⁇ of 5.5 to 9.5 [(J / cm 3 ) 1/2 ],
- the hydrophobicity in which the antibiotic compound having the solubility parameter ⁇ having the dipole force term ⁇ p, compound and the hydrogen bond force term ⁇ h, compound is in a specific range is dissolved.
- the polymerized monomer is polymerized to produce a polymer in which the dipole force term ⁇ p, polymer and the hydrogen bond term ⁇ h, polymer of the solubility parameter ⁇ are in a specific range, so that the preparation of the raw material becomes simple, Since the manufacturing process is simplified and the raw material cost is reduced, the manufacturing cost can be reduced.
- sustained-release particles that are simple and low-cost, have excellent sustained-release properties, and can exhibit excellent efficacy sustaining effects.
- the hydrophobic solution is prepared without using a solvent, the environmental load can be reduced.
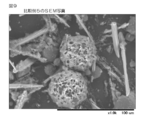
- FIG. 1 shows an image processing diagram of an SEM photograph of sustained-release particles of Example 1.
- FIG. 2 shows an image processing diagram of an SEM photograph of sustained-release particles of Example 5.
- FIG. 3 shows an image processing diagram of an SEM photograph of sustained-release particles of Example 8.
- FIG. 4 shows an image processing diagram of an SEM photograph of sustained-release particles of Example 14.
- FIG. 5 shows an image processing diagram of an SEM photograph of sustained-release particles of Example 15.
- FIG. 6 shows an image processing diagram of an SEM photograph of sustained-release particles of Example 16.
- FIG. 7 shows an image processing diagram of an SEM photograph of the particles of Comparative Example 2.
- FIG. 8 shows an image processing diagram of an SEM photograph of the particles of Comparative Example 4.
- FIG. 9 shows an image processing diagram of an SEM photograph of the particles of Comparative Example 5.
- FIG. 10 shows an image processing diagram of the SEM photograph of the particles of Comparative Example 6.
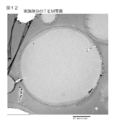
- FIG. 11 shows an image processing diagram of a TEM photograph of the sustained release particles of Example 1.
- FIG. 12 shows an image processing diagram of a TEM photograph of sustained-release particles of Example 9.
- FIG. 13 shows an image processing diagram of a TEM photograph of sustained-release particles of Example 10.
- FIG. 14 shows an image processing diagram of a TEM photograph of sustained-release particles of Example 11.
- FIG. 15 shows an image processing diagram of a TEM photograph of sustained-release particles of Example 12.
- FIG. 16 shows an image processing diagram of a TEM photograph of sustained-release particles of Example 13.
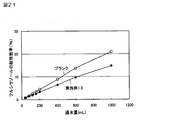
- FIG. 17 shows a graph of sustained release tests of Examples 1 to 3.
- FIG. 18 shows graphs of sustained release tests of Examples 14 to 16.
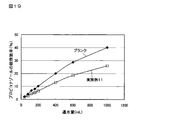
- FIG. 19 shows a graph of sustained release test of Example 11.
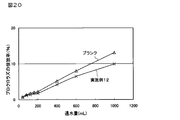
- 20 shows a graph of sustained release test of Example 12.
- FIG. 21 shows a graph of sustained release test of Example 13.
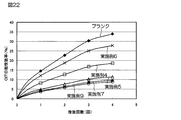
- FIG. 22 shows a graph of sustained release test of Examples 4 to 7 and 9.
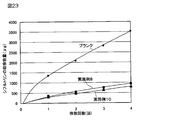
- FIG. 23 shows a graph of sustained release tests of Examples 8 and 10.
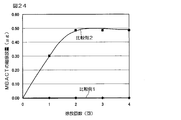
- FIG. 24 shows a graph of the sustained release test of Comparative Examples 1 and 2.
- the sustained-release particles of the present invention are prepared by dissolving a hydrophobic antibiotic compound with a hydrophobic polymerizable vinyl monomer to prepare a hydrophobic solution, dispersing the hydrophobic solution in water, The monomer is obtained by radical polymerization to produce a polymer.
- the antibiotic compound has, for example, at least two functional moieties that can interact with a polymer of a polymerizable vinyl monomer.
- Such functional moieties include polar functional groups such as carbonyl group, nitro group, amino group, cyano group, phosphate ester group, carboxyl group, such as carboxylate bond, phosphate bond, urea bond, carbon-halogen.
- polar functional groups such as carbonyl group, nitro group, amino group, cyano group, phosphate ester group, carboxyl group, such as carboxylate bond, phosphate bond, urea bond, carbon-halogen.
- Examples include a polar bond including a polar group such as a bond, such as a benzene ring, and a conjugated cyclic moiety such as a conjugated heterocycle such as a triazine ring, an imidazole ring, and an isothiazoline ring.
- the molecular weight of the antibiotic compound is, for example, 200 to 600, preferably 200 to 500.
- the compatibility of the antibiotic compound with the polymer may decrease.
- the molecular weight of the antibiotic compound is less than the above range, the antibiotic compound remains in the aqueous phase during suspension polymerization (described later). May precipitate and the suspension may solidify.
- the melting point of the antibiotic compound is 100 ° C. or less, preferably 90 ° C. or less, more preferably 80 ° C. or less.
- the antibiotic compound may be difficult to encapsulate in the sustained-release particles and may precipitate out of the sustained-release particles. Even when encapsulated in the sustained-release particles, the antibiotic compound may not be released from the sustained-release particles.
- the antibiotic compound is an antibacterial agent, antibacterial agent, antiseptic agent, antifungal agent, antifungal agent, herbicidal agent having antibacterial activity such as bactericidal, antibacterial, antiseptic, algae, fungicide, insecticide, etc. Selected from agents, insecticides, attractants, repellents and rodenticides.
- the compounds having antibiotic activity include bactericidal antiseptic and algal fungicides such as iodine compounds, triazole compounds, carbamoylimidazole compounds, dithiol compounds, isothiazoline compounds, nitroalcohol compounds, and paraoxybenzoic acid esters.
- anticides anticides
- pyrethroid compounds such as pyrethroid compounds, neonicotinoid compounds, organochlorine compounds, organophosphorus compounds, carbamate compounds, and oxadiazine compounds.
- iodine compounds include 3-iodo-2-propynylbutylcarbamate (IPBC), 1-[[(3-iodo-2-propynyl) oxy] methoxy] -4-methoxybenzene, 3-bromo-2, And 3-diiodo-2-propenyl ethyl carbonate.
- triazole compound examples include 1- [2- (2,4-dichlorophenyl) -4-n-propyl-1,3-dioxolan-2-ylmethyl] -1H-1,2,4-triazole (propico Nazole), bis (4-fluorophenyl) methyl (1H-1,2,4-triazol-1-ylmethylsilane (also known as flusilazole, 1-[[bis (4-fluorophenyl) methylsilyl] methyl] -1H- 1,2,4-triazole) and the like.
- carbamoylimidazole compound examples include N-propyl-N- [2- (2,4,6-trichloro-phenoxy) ethyl] imidazole-1-carboxamide (prochloraz).
- dithiol-based compound examples include 4,5-dichloro-1,2-dithiol-3-one.
- isothiazoline compound examples include 2-n-octyl-4-isothiazolin-3-one (OIT), 5-chloro-2-methyl-4-isothiazolin-3-one (Cl-MIT), and the like.
- nitroalcohol compound examples include 2,2-dibromo-2-nitro-1-ethanol (DBNE).
- paraoxybenzoic acid ester examples include butyl paraoxybenzoate and propyl paraoxybenzoate.
- pyrethroid compounds include pyrethrin, cineline, jasmolin and the like obtained from Shirovanamushiyogiiku, and arelesrin, bifenthrin, acrinathrin, alpha cypermethrin, tralomethrin, cyfluthrin ((RS) - ⁇ -cyano-4 derived from these compounds.
- neonicotinoid compounds include (E) -N 1 -[(6-chloro-3-pyridyl) methyl] -N 2 -cyano-N 1 -methylacetamidine (acetamipride).
- organochlorine compounds examples include Kelsen.
- organophosphorus compounds examples include oxime, pyridafenthion, fenitrothion, tetrachlorbinphos, diclofenthion, propetanephos, and the like.
- carbamate compounds examples include fenocarb and propoxur.
- Examples of the oxadiazine compound include indoxacarb.
- herbicides examples include pyraclonyl, pendimethalin, indanophan and the like.
- insecticide examples include pyriproxyfen.
- repellents examples include diet.
- Antibiotic active compounds are substantially hydrophobic and have, for example, very little solubility in water at room temperature (20-30 ° C., more specifically 25 ° C.). Solubility is 1 part by weight / 100 parts by weight of water (10000 ppm) or less, preferably 0.5 parts by weight / 100 parts by weight of water (5000 ppm) or less, more preferably 0.1 parts by weight / 100 water. Parts by weight (1000 ppm) or less, and on a volume basis, for example, 1 g / 100 mL or less of water, preferably 0.5 g / 100 mL or less of water, and more preferably 0.1 g / 100 mL or less of water.
- the antibiotic compound When the solubility of the antibiotic compound in water exceeds the above-mentioned range, the antibiotic compound is out of the sustained-release particles (that is, the aqueous phase) when the polymerizable vinyl monomer is polymerized (suspension polymerization). Since the antibiotic active compound dissolved in the aqueous phase is precipitated after the polymerization, it becomes difficult to synthesize sustained release particles sufficiently containing the antibiotic active compound.
- antibiotic compounds can be used alone or in combination of two or more.
- the above-mentioned antibiotic compound may contain, for example, impurities having a melting point outside the above range at an appropriate ratio during the production process.
- impurities having a melting point outside the above range at an appropriate ratio during the production process.
- a mixture of isomer I (melting point: 57 ° C.), isomer II (melting point: 74 ° C.) and isomer III (melting point: 66 ° C.) of cyfluthrin is, for example, isomer IV (impurity isomer IV (melting point: 66 ° C.)). Melting point 102 ° C.).
- the polymerizable vinyl monomer is, for example, a monomer having at least one vinyl group in the molecule.
- polymerizable vinyl monomer examples include (meth) acrylic acid ester monomers, (meth) acrylic acid monomers, aromatic vinyl monomers, vinyl ester monomers, maleic acid ester monomers, and halogenated monomers.
- examples thereof include vinyl, vinylidene halide, and nitrogen-containing vinyl monomer.
- Examples of (meth) acrylic acid ester monomers include methacrylic acid esters and // acrylic acid esters, and specifically include methyl (meth) acrylate, ethyl (meth) acrylate, and (meth) acrylic acid n.
- -Propyl iso-propyl (meth) acrylate, n-butyl (meth) acrylate, iso-butyl (meth) acrylate, tert-butyl (meth) acrylate, cyclohexyl (meth) acrylate, (meth) acryl
- Examples include 2-methoxyethyl acid.
- Examples of the (meth) acrylic acid monomer include methacrylic acid and acrylic acid.
- aromatic vinyl monomer examples include styrene, p-methylstyrene, ⁇ -methylstyrene, vinyltoluene and the like.
- vinyl ester monomers examples include vinyl acetate and vinyl propionate.
- maleate ester monomers examples include dimethyl maleate, diethyl maleate, and dibutyl maleate.
- Examples of the vinyl halide include vinyl chloride and vinyl fluoride.
- Examples of the vinylidene halide include vinylidene chloride and vinylidene fluoride.
- nitrogen-containing vinyl monomer examples include (meth) acrylonitrile, N-phenylmaleimide, vinylpyridine, and the like.
- the polymerizable vinyl monomer is substantially hydrophobic and has, for example, extremely low solubility in water at room temperature.
- the solubility at room temperature is, for example, 10 parts by weight / 100 parts by weight or less of water, preferably 8 parts by weight / 100 parts by weight or less of water.
- the above-mentioned antibiotic active compound compatible monomer (hereinafter, simply referred to as “compatible monomer”) that is highly compatible with the antibiotic active compound and can dissolve the antibiotic active compound. .) Is selected.
- These compatible monomers can be used alone or in combination of two or more.
- the compatible monomer preferably, the same type of (meth) acrylic acid ester monomers are used alone, different types of (meth) acrylic acid ester monomers are used together, or (meth) acrylic acid ester monomers and ( The combined use with a (meth) acrylic-acid type monomer is mentioned.
- methyl methacrylate is used alone, methyl methacrylate and C2-4 alkyl (meth) acrylate are used together, or methyl methacrylate and methacrylic acid are used together.
- C2-4 alkyl (meth) acrylate used in combination with methyl methacrylate examples include ethyl (meth) acrylate, n-propyl (meth) acrylate, iso-propyl (meth) acrylate, and (meth) acrylic. Examples thereof include n-butyl acid, iso-butyl (meth) acrylate, and tert-butyl (meth) acrylate.
- the blending ratio of methyl methacrylate is, for example, 20 weights with respect to 100 parts by weight of a compatible monomer (including a crosslinkable monomer described later). Part or more, preferably 40 parts by weight or more, for example, 99 parts by weight or less.
- the blending ratio of the (meth) acrylic acid monomer is 100 parts by weight of the compatible monomer including the crosslinkable monomer. On the other hand, for example, it is less than 30 parts by weight and 20 parts by weight or less, for example, 1 part by weight or more, preferably 3 parts by weight or more.
- the antibiotic active compound and the compatible monomer are selected such that the polymer of the polymerizable vinyl monomer and the antibiotic active compound are compatible at the polymerization temperature (heating temperature) described later.
- the polymerizable vinyl monomer can contain a crosslinkable monomer as a compatible monomer.
- the crosslinkable monomer is blended as necessary in order to adjust the sustained release property of the sustained release particles.
- mono- or polyethylene glycol di (meth) such as ethylene glycol di (meth) acrylate and diethylene glycol di (meth) acrylate.
- Acrylates for example alkanediol di (meth) acrylates such as 1,3-propanediol di (meth) acrylate, 1,4-butanediol di (meth) acrylate, 1,5-pentanediol di (meth) acrylate, for example , Alkane polyol poly (meth) acrylates such as trimethylolpropane tri (meth) acrylate, pentaerythritol tetra (meth) acrylate, for example, allyl monomers such as allyl (meth) methacrylate, triallyl (iso) cyanurate, Eg to such divinyl monomers such as divinylbenzene.
- alkanediol di (meth) acrylates such as 1,3-propanediol di (meth) acrylate, 1,4-butanediol di (meth) acrylate, 1,5-pentanedio
- the crosslinkable monomer a monomer having a molecular structure similar to the molecular structure of the compatible monomer excluding the crosslinkable monomer is selected in order to ensure compatibility with the compatible monomer excluding the crosslinkable monomer.
- the compatible monomer excluding the crosslinkable monomer contains methyl methacrylate, ethylene glycol dimethacrylate (EGDMA) or trimethylolpropane methacrylate (TMPTMA) is preferably selected as the crosslinkable monomer.
- the blending ratio of the crosslinkable monomer is, for example, 1 to 100 parts by weight, preferably 10 to 80 parts by weight with respect to 100 parts by weight of the polymerizable vinyl monomer (compatible monomer).
- the dipole force term ⁇ p, compound of the solubility parameter ⁇ defined by Hansen and calculated by the van Klevelen and Hoftyzer method is 2 to 8 [( J / cm 3 ) 1/2 ] and a hydrogen bonding term ⁇ h, compound of the solubility parameter ⁇ is 5.5 to 9.5 [(J / cm 3 ) 1/2 ]
- the dipole force term ⁇ p, polymer of the solubility parameter ⁇ is 5 to 7 [(J / cm 3 ) 1/2 ]
- the hydrogen bond term ⁇ h, polymer of the solubility parameter ⁇ is 8 to 10 [( J / cm 3 ) 1/2 ] is selected in combination with a polymerizable vinyl monomer that produces a polymer.
- ⁇ ( ⁇ p and ⁇ h ) indicate an antibiotic compound and a polymer, respectively.
- the dipole force term ⁇ p and the hydrogen bond force term ⁇ h of the solubility parameter ⁇ defined by Hansen and calculated by the van Klevelen and Hoftyzer method are the type and number of atomic groups (including chemical bonds or substituents). Specifically, it is represented by the following formulas (1) and (2), respectively.
- F p is a dipole force element of intermolecular force (polar component of the molar attraction function), and V is a molar volume.)
- the F p of the substituents> Si ⁇ , ⁇ N— and ⁇ C— is also calculated by the same calculation process as described above.
- E h of the substituents —I,> Si ⁇ , ⁇ N—, and ⁇ C— are also calculated by performing the same calculation processing as described above.
- the above calculation processing is recorded on a computer as a program and optimized.
- n represents the degree of polymerization.
- ⁇ p Dipole force term
- PMMA In the monomer unit (—CH 2 —C (CH 3 ) COOCH 3 —) of the above formula (3), F p and V corresponding to each atomic group are described below.
- the above-described monomer unit dipole force term ⁇ p, monomer unit is the polymethyl methacrylate dipole force term ⁇ p, PMMA which is a repeating structure of the monomer unit.
- Hydrogen bond strength term ⁇ h, PMMA In the monomer unit (—CH 2 —C (CH 3 ) COOCH 3 —) of the above formula (3), E h corresponding to each atomic group is described below.
- the hydrogen bond strength term ⁇ h, polymer of the monomer unit described above is used as the hydrogen bond strength term ⁇ h, PMMA of polymethyl methacrylate which is a repeating structure of the monomer unit. 2.
- Co-dipole force term ⁇ p and hydrogen bond force term ⁇ h of the copolymer Next, a dipole force term ⁇ p and a hydrogen bond force term ⁇ h of the copolymer are calculated.
- the dipole force term ⁇ p, monomer unit of each monomer unit by the monomer weight ratio and adding them together the dipole force term ⁇ p, copolymer of the solubility parameter ⁇ of the copolymer is calculated.
- the hydrogen bond strength term ⁇ h, copolymer of the solubility parameter ⁇ of the copolymer is calculated by multiplying the hydrogen bond strength term ⁇ h, monomer unit of each monomer unit by the weight ratio of the monomers and adding them together. To do.
- a polymethyl methacrylate-ethylene glycol dimethacrylate copolymer (PMMA-EGDMA), which is a copolymer of monomers containing methyl methacrylate and ethylene glycol dimethacrylate in a weight ratio of 90:10, is used.
- the dipole force term ⁇ p, PMMA-EGDMA and the hydrogen bond force term ⁇ h, PMMA-EGDMA of the solubility parameter ⁇ are calculated.
- Dipole force term ⁇ p, PMMA-EGDMA The dipole force term ⁇ p, MMA unit of the monomer unit of methyl methacrylate is 5.98 [(J / cm 3 ) 1/2 ] as calculated above.
- dipole force term ⁇ p, EDGMA of the monomer unit of ethylene glycol dimethacrylate is 5.37 [(J / cm 3 ) 1/2 ] by calculation in the same manner as described above.
- the dipole force term ⁇ p, PMMA-EGDMA of this copolymer is calculated as in the following formula (6).
- ⁇ p, PMMA-EGDMA (90/100) ⁇ p
- Hydrogen bond term ⁇ h, PMMA-EGDMA The hydrogen bond term ⁇ h, MMA unit of the monomer unit of methyl methacrylate is 9.25 [(J / cm 3 ) 1/2 ].
- the hydrogen bond term ⁇ h, EGDMA of the monomer unit of ethylene glycol dimethacrylate is 10.42 [(J / cm 3 ) 1/2 ].
- the hydrogen bonding force term ⁇ h, PMMA-EGDMA of this copolymer is calculated as in the following formula (7).
- the dipole force term ⁇ p, polymer of the solubility parameter ⁇ of the polymer is preferably 5 to 6.5 [(J / cm 3 ) 1/2 ], and the hydrogen of the solubility parameter ⁇ of the polymer is
- the binding force term ⁇ h, polymer is preferably 9 to 10 [(J / cm 3 ) 1/2 ].
- the dipole force term ⁇ p, polymer and / or the hydrogen bond force term ⁇ h, polymer of the polymer is less than the above range , the polymer is excessively hydrophobic and sufficient with the antibiotic compound. In some cases, the compatibility cannot be obtained, and even if the compatibility can be obtained, the antibiotic compound is leaked out of the sustained release particles during the polymerization (suspension polymerization), and the antibiotic compound Therefore, it becomes difficult to synthesize sustained-release particles that sufficiently encapsulate.
- Dipole force term ⁇ p, compound and hydrogen bond term ⁇ h, compound of solubility ⁇ of antibiotic compound The dipole force term ⁇ p, compound and the hydrogen bond term ⁇ h, compound of the solubility ⁇ of the antibiotic compound are also calculated in the same manner as the monomer unit described above.
- the dipole force term ⁇ p, compound of the solubility parameter ⁇ of the antibiotic compound is preferably 3-7 [(J / cm 3 ) 1/2 ], and the hydrogen bond force term ⁇ h, compound is Preferably, it is 5.8 to 9.5 [(J / cm 3 ) 1/2 ].
- the dipole force term ⁇ p, compound and / or the hydrogen bond force term ⁇ h, compound of the antibiotic compound is not within the above range, the hydrophobic property of the antibiotic compound is excessively high, In some cases, sufficient compatibility cannot be obtained.
- the dipole force term ⁇ p, compound and hydrogen bond force term ⁇ h, compound of the antibiotic compound are within the above-mentioned range, and the dipole force term ⁇ p, polymer and hydrogen bond force term of the polymer If ⁇ h , polymer is in the above range, the antibiotic compound is defined as being compatible with the polymer without leaking from the suspended particles during radical polymerization.
- the hydrophobic antibiotic compound is dissolved in a hydrophobic polymerizable vinyl monomer in the absence of a solvent to form a hydrophobic solution.
- a hydrophobic solution can be obtained by blending an antibiotic compound and a polymerizable vinyl monomer, and stirring uniformly without blending a solvent (a hydrophobic organic solvent such as hexane, toluene, ethyl acetate). .
- a solvent a hydrophobic organic solvent such as hexane, toluene, ethyl acetate.
- the blending ratio of the antibiotic compound to the polymerizable vinyl monomer is, for example, 10/90 to 60/40 (that is, 0 parts by weight) (that is, part by weight of antibiotic compound / part by weight of polymerizable vinyl monomer). .11 to 1.5).
- the proportion of the antibiotic compound to the polymerizable vinyl monomer is determined by polymerization of the antibiotic compound. For example, 1/99 to 60/40, and preferably 5/95 to 50/50 on a weight basis.
- the sustained release rate is slower than the diffusion rate of the antibiotic compound at room temperature, so that, for example, 10/90 to 70 on a weight basis. / 30, preferably 10/90 to 60/40.
- the preparation of the hydrophobic solution may be carried out at room temperature, for example, or may be carried out by heating as necessary.
- the heating temperature is, for example, 30 to 100 ° C., preferably 40 to 80 ° C.
- hydrophobic solution is dispersed (suspended) in water.
- hydrophobic solution and water are blended and stirred uniformly to disperse (suspend) the hydrophobic solution in water. Thereby, an aqueous dispersion (suspension) solution of the hydrophobic solution is obtained.
- the conditions for water dispersion are not particularly limited, and may be carried out, for example, at room temperature or by heating.
- the heating is also performed during the water dispersion.
- the heating temperature is, for example, not less than the heating temperature at the time of water dispersion described above, specifically, 30 to 100 ° C., preferably 40 to 80 ° C.
- the mixing ratio of water is, for example, 100 to 1000 parts by weight, preferably 150 to 500 parts by weight with respect to 100 parts by weight of the hydrophobic solution.
- a dispersant and a surfactant are preferably blended.
- dispersant examples include polyvinyl alcohol (PVA), polyvinyl pyrrolidone, gelatin, gum arabic, hydroxyethyl cellulose, hydroxypropyl cellulose, carboxymethyl cellulose, cationized starch, polyacrylic acid and its sodium salt, styrene maleic acid copolymer and its sodium Water-soluble polymers such as salts, for example, inorganic dispersants such as tricalcium phosphate, colloidal silica, montmorillonite, magnesium carbonate, aluminum hydroxide, zinc white, and the like.
- polyvinyl alcohol (PVA) and tricalcium phosphate are preferable.
- the mixing ratio of the dispersing agent is, for example, 0.01 to 10 parts by weight, preferably 0.1 to 5 parts by weight with respect to 100 parts by weight of the hydrophobic solution.
- the surfactant is preferably used in combination with the above-described dispersant, specifically, sodium dodecylbenzenesulfonate, sodium lauryl sulfate, di-2- Anionic surfactants such as sodium ethylhexyl sulfosuccinate, sodium dodecyl diphenyl ether disulfonate, sodium nonyl diphenyl ether sulfonate, sodium salt of naphthalene sulfonic acid formaldehyde, such as polyoxyethylene lauryl ether, polyoxyethylene nonyl phenyl ether, polyoxy Nonionic surfactants such as ethylene monostearate, polyoxyethylene sorbitan monooleate, and polyoxyethylene polyoxypropylene block copolymerPreferably, an anionic surfactant is used.
- sodium dodecylbenzenesulfonate sodium lauryl sulfate
- di-2- Anionic surfactants such as sodium e
- the blending ratio of the surfactant is, for example, 0.0001 to 1.0 part by weight, preferably 0.001 to 0.1 part by weight with respect to 100 parts by weight of the hydrophobic solution.
- dispersants and surfactants can be blended, for example, before or after blending the hydrophobic solution and water, and are preferably blended in water before blending with the hydrophobic solution. Thereby, an aqueous solution of the dispersant and the surfactant is prepared.
- a disperser such as a homomixer, an ultrasonic homogenizer, a pressure homogenizer, a milder, or a porous membrane press-in disperser is used. Is used.
- the polymerizable vinyl monomer in the hydrophobic solution dispersed in water is radically polymerized in the presence of an oil-soluble initiator to produce a polymer.
- oil-soluble initiators include dilauroyl peroxide, 1,1,3,3-tetramethylbutylperoxy-2-ethylhexanoate, t-hexylperoxy-2-ethylhexanoate, and diisopropyl peroxide.
- Organic peroxides such as oxydicarbonate and benzoyl peroxide, such as 2,2′-azobisisobutyronitrile, 2,2′-azobis (2,4-dimethylvaleronitrile), 2,2′-azobis And azo compounds such as (2-methylbutyronitrile).
- the oil-soluble initiator can be blended, for example, in a hydrophobic solution before blending with water, or can be blended in an aqueous dispersion after blending them. Preferably, it mix
- the blending ratio of the oil-soluble initiator is, for example, 0.01 to 2 parts by weight, preferably 0.1 to 1 part by weight with respect to 100 parts by weight of the polymerizable vinyl monomer.
- this radical polymerization is carried out while stirring the suspension so as to maintain the suspension state of the suspension, and is therefore suspension polymerization.
- the raw material monomer is only in the hydrophobic phase (oil phase), in situ polymerization is performed.
- the reaction is started by heating the aqueous dispersion, for example.
- the heating conditions are appropriately selected depending on the type of the oil-soluble initiator, the heating temperature is, for example, 30 to 100 ° C., preferably 50 to 100 ° C., and the heating time is, for example, 3 to 24 hours, preferably 5 to 12 hours. Furthermore, after heating to a predetermined temperature, the temperature can be maintained for a predetermined time, and then heating and temperature maintenance can be repeated to heat in stages.
- the pressure at the time of radical polymerization is not particularly limited and is a normal pressure.
- radical polymerization is performed at normal pressure, but it can also be performed under high pressure, for example.
- the reaction system can be set to a temperature exceeding 100 ° C., and the antibiotic compound which is solid at room temperature can be easily liquefied.
- the polymer of the polymerizable vinyl monomer is preferably compatible with the antibiotic compound. That is, the polymer is dissolved in the antibiotic compound to form an antibiotic compound solution of the polymer, and the antibiotic compound solution is dispersed in water.
- the polymerizable vinyl monomer preferably has a combination in which the polymer of the polymerizable vinyl monomer and the antibiotic compound are compatible as described above at the polymerization temperature (heating temperature) during the radical polymerization described above. Since it is selected, phase separation is prevented from occurring during radical polymerization (suspension polymerization), and the polymer (polymer during the reaction) dissolves the antibiotic compound, or the polymer (reaction) The reaction proceeds in a state where the intermediate polymer) is swollen with respect to the antibiotic compound, so that sustained-release particles in which a uniform phase is formed can be obtained. In addition, if an antibiotic compound is liquid at normal temperature, the state of a uniform phase will be maintained as it is at normal temperature.
- the aqueous dispersion after polymerization is cooled, for example, by cooling.
- the cooling temperature is, for example, room temperature (20 to 30 ° C., more specifically 25 ° C.).
- the antibiotic compound After cooling, if the antibiotic compound is liquid at room temperature, it is compatible with the polymer of the polymerizable vinyl monomer.
- the compatible state is frozen and dispersed as very fine solid particles in the polymer particles of the polymerizable vinyl monomer.
- the sustained-release particles are formulated as a powder (described later) or a granule (described later), preferably at a room temperature
- the hard-release glass is used to prevent the sustained-release particles from fusing together.
- a polymerizable vinyl monomer is selected.
- the particle diameter of the sustained release particles is not particularly limited, and is an average particle diameter (median diameter), for example, 500 nm to 1 mm, preferably 1 ⁇ m to 100 ⁇ m.
- the average particle size of sustained-release particles obtained from antibiotic active compounds that are liquid at normal temperature is, for example, 5 to 100 ⁇ m, and the average particle size of sustained-release particles obtained from antibiotic active compounds that are solid at normal temperatures. Is, for example, 0.5 to 30 ⁇ m.
- aqueous dispersion in which sustained-release particles in which the antibiotic compound is uniformly present are dispersed (suspended) in water.
- aqueous dispersion containing the sustained-release particles. Is appropriately blended.
- the sustained-release particles thus obtained may be used as they are (aqueous dispersion or suspension), that is, as a water dispersion or suspension, or by filtration and / or centrifugation. After solid-liquid separation, for example, it may be formulated into a known dosage form such as powder or granule. Further, if necessary, water washing and / or acid washing can be performed. Furthermore, the aqueous dispersion (suspension) can be spray-dried or air-dried as it is, and can be formulated into a dosage form such as powder or granule.
- the powder is excellent in fluidity, especially when tricalcium phosphate is used as a dispersant.
- an aqueous dispersion or suspension can be re-prepared by dispersing or suspending the powder again in water. Therefore, such a powder is excellent in re-water dispersibility or re-suspension.
- sustained release particles are prepared as a powder at the time of transportation, and prepared (reformulation, regeneration) as an aqueous dispersion or suspension at the time of use, thereby reducing transportation costs and further improving the use. Can be enlarged.
- the melting point of the antibiotic compound is as low as 100 ° C. or less
- the sustained-release particles containing the antibiotic compound, in particular, the powder of the sustained-release particles are excellent in handleability. Yes.
- an antibiotic compound having a solubility parameter ⁇ having dipole force terms ⁇ p, compound and hydrogen bond force terms ⁇ h, compound in a specific range is obtained. Since the dissolved hydrophobic polymerizable monomer is polymerized to produce a polymer in which the dipole force term ⁇ p, polymer and the hydrogen bond force term ⁇ h, polymer of the solubility parameter ⁇ are in a specific range.
- the production process can be simplified, the production process can be simplified, and the raw material cost can be reduced. Therefore, the production cost can be reduced.
- sustained-release particles that are simple and low-cost, have excellent sustained-release properties, and can exhibit excellent efficacy sustaining effects.
- the sustained-release particles can be applied to various industrial products, for example, indoor and outdoor paints, rubber, fibers, resins, plastics, adhesives, jointing agents, sealing agents, building materials, caulking agents, soils. It can be blended into a processing agent, wood, white water in a papermaking process, pigment, printing plate processing liquid, cooling water, ink, cutting oil, cosmetics, nonwoven fabric, spinning oil, leather, and the like.
- the blending amount of the antibiotic compound in the sustained release particles for these industrial products is, for example, 10 to 100,000 mg / kg (product weight).
- the hydrophobic solution is prepared without using a solvent, the environmental load can be reduced.
- the sustained-release particles can be suitably blended in indoor and outdoor water-based paints.
- water-based paints include acrylic, acrylic-styrene, styrene, vinyl acetate, and vinyl acetate-acrylic.
- IPBC Trade name “Fangitrol 400”, 3-iodo-2-propynylbutylcarbamate, molecular weight 281, melting point: 60 ° C., solubility in water: 150 ppm, dipole force term ⁇ p, compound of solubility parameter ⁇ : 3 .23 [(J / cm 3 ) 1/2 ], hydrogen bond strength term ⁇ h, compound of solubility parameter ⁇ : 7.83 [(J / cm 3 ) 1/2 ], OIT manufactured by International Specialty Products : Trade name “Caisson 893T” (“Caisson” is a registered trademark), 2-n-octyl-4-isothiazolin-3-one, molecular weight 213, melting point: less than 20 ° C., solubility in water: 300 ppm, solubility parameter ⁇ Hydrogen term of dipole force term ⁇ p, compound : 5.47 [(J / cm 3
- flusilazole Bis (4-fluorophenyl) methyl (1H-1,2,4-triazol-1-ylmethylsilane, molecular weight 315, melting point: 54 ° C., solubility in water: 45 ppm, dipole force term with solubility parameter ⁇ ⁇ p , compound : 5.95 [(J / cm 3 ) 1/2 ], hydrogen bond term of solubility parameter ⁇ ⁇ , compound : 6.85 [(J / cm 3 ) 1/2 ], Air Brown MBACT manufactured by KK: trade name “Irgalol 1071” (“Irgalol” is a registered trademark), 2-methylthio-4-tert-butylamino-6-cyclopropylamino-s-triazine, The amount 253, melting point: 133 ° C., solubility in water: 7 ppm, solubility parameter polar term [delta] p, Compound of ⁇ : 7.18 [(J / cm 3) 1/2], hydrogen bonding solub
- TCP-10U trade name, tribasic calcium phosphate (3 [Ca 3 ( PO 4 ) 2 ] ⁇ Ca (OH) 2 ) 10 wt% aqueous suspension, DBN manufactured by Matsuo Pharmaceutical Co., Ltd .: Trade name “Neopelex No.
- Example 1 (Formulation of suspension containing IPBC-containing sustained release particles) A 200 mL beaker (1) was charged with 40 g of IPBC, 54 g of methyl methacrylate, 6 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion exchange water, 20 g of a 10% aqueous solution of PVA-217, and 200 mg of a 5% aqueous solution of DBN, and stirred at room temperature to obtain a uniform aqueous solution.
- hydrophobic solution was added to a 500 mL beaker (2).
- K A suspension was prepared by dispersing the hydrophobic solution in water by stirring with a homomixer MARK 2.5 type (manufactured by Primix) for 10 minutes at a rotational speed of 5000 rpm.
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 1 hour, 70 ⁇ 2 ° C. for 3 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or less to obtain a suspension (suspension) of sustained release particles containing IPBC having a median diameter of 7.4 ⁇ m.
- the median diameter of the sustained release particles was measured with a laser diffraction / scattering particle size distribution analyzer LA-920 (manufactured by Horiba, Ltd.). The measurement of the median diameter is the same for each of the following examples and comparative examples.
- Example 2 (Formulation of suspension containing IPBC-containing sustained release particles) A 200 mL beaker (1) was charged with 40 g of IPBC, 42 g of methyl methacrylate, 18 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion-exchanged water, 20 g of TCP-10U and 200 mg of 5% aqueous solution of Perex SS-L, and stirred at room temperature to obtain a uniform aqueous solution.
- hydrophobic solution was added to a 500 mL beaker (2).
- K A suspension was prepared by dispersing the hydrophobic solution in water by stirring with a homomixer MARK 2.5 type (manufactured by Primix) for 10 minutes at a rotational speed of 5000 rpm.
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 1 hour, 70 ⁇ 2 ° C. for 3 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or lower to obtain a suspension (suspension) of sustained release particles containing IPBC having a median diameter of 8.2 ⁇ m.
- Example 3 (Formulation of suspension containing IPBC-containing sustained release particles) Except that the amounts of methyl methacrylate and ethylene glycol dimethacrylate were both changed to 30 g, in the same manner as in Example 1, the hydrophobic solution was dispersed in water, followed by suspension polymerization to obtain IPBC. A suspension (suspension) of sustained release particles having a median diameter of 9.6 ⁇ m was obtained.
- Example 4 (Formulation of suspension containing OIT-containing sustained release particles) A 200 mL beaker (1) was charged with 40 g of OIT, 54 g of methyl methacrylate, 6 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring with a homomixer MARK 2.5 type (manufactured by Primix) for 10 minutes at a rotational speed of 2000 rpm.
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 2 hours, 70 ⁇ 2 ° C. for 2 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or lower to obtain a suspension (suspension) of sustained release particles containing OIT and having a median diameter of 14 ⁇ m.
- Example 5 (Formulation of suspension containing OIT-containing sustained release particles) The hydrophobic solution was dispersed in water, followed by suspension polymerization in the same manner as in Example 4 except that both the amounts of methyl methacrylate and ethylene glycol dimethacrylate were changed to 30 g. A suspension (suspension) of sustained release particles having a median diameter of 14 ⁇ m was obtained.
- Example 6 (Formulation of suspension containing OIT-containing sustained release particles)
- the hydrophobic solution was dispersed in water in the same manner as in Example 4 except that the amounts of OIT, methyl methacrylate and ethylene glycol dimethacrylate were changed to 50 g, 45 g and 5 g, respectively, followed by suspension polymerization.
- a suspension (suspension) of sustained-release particles containing OIT and having a median diameter of 9.8 ⁇ m was obtained.
- Example 7 (Formulation of suspension containing OIT-containing sustained release particles) T. at the time of water dispersion. K. By dispersing the hydrophobic solution in water and then carrying out suspension polymerization in the same manner as in Example 4 except that the stirring condition of the homomixer MARK 2.5 type (manufactured by Primix) was changed to 1000 rpm. As a result, a suspension (suspension) of sustained-release particles containing IPBC and having a median diameter of 29 ⁇ m was obtained.
- Example 8 (Formulation of suspension containing fine particles containing cyfluthrin) A 200 mL beaker (1) was charged with 40 g of cyfluthrin, 54 g of methyl methacrylate, 6 g of trimethylolpropane trimethacrylate, and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion-exchanged water, 20 g of a 10% aqueous solution of PVA-217, and 200 mg of a 5% aqueous solution of DBN, and stirred at room temperature to obtain a uniform aqueous solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring with a homomixer MARK 2.5 type (manufactured by Primix) for 10 minutes at a rotational speed of 3000 rpm.
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 2 hours, 70 ⁇ 2 ° C. for 2 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or lower to obtain a suspension (suspension) of sustained release particles containing cyfluthrin having a median diameter of 15 ⁇ m.
- Example 9 (Formulation of suspension containing OIT-containing sustained release particles) A 200 mL beaker (1) was charged with 40 g of OIT, 36 g of methyl methacrylate, 6 g of methacrylic acid, 18 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion-exchanged water, 20 g of TCP-10U and 200 mg of 5% aqueous solution of Perex SS-L, and stirred at room temperature to obtain a uniform aqueous solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring for 10 minutes at 2000 rpm with a homomixer MARK 2.5 (manufactured by Primix).
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 2 hours, 70 ⁇ 2 ° C. for 2 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or lower to obtain a suspension (suspension) of sustained release particles containing OIT and having a median diameter of 36 ⁇ m.
- Example 10 (Formulation of suspension containing cyfluthrin-containing sustained release particles) A 200 mL beaker (1) was charged with 40 g of cyfluthrin, 42 g of methyl methacrylate, 18 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion-exchanged water, 20 g of TCP-10U and 200 mg of 5% aqueous solution of Perex SS-L, and stirred at room temperature to obtain a uniform aqueous solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring for 10 minutes at 3000 rpm with a homomixer MARK 2.5 (manufactured by Primix).
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer and a reflux cooler, and heated with stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 2 hours, 70 ⁇ 2 ° C. for 2 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension After the reaction is cooled to 30 ° C. or lower, whereby a suspension of sustained-release particles having a median diameter of 22 ⁇ m and containing cyfluthrin (mixture of isomer I, isomer II and isomer III) ( Suspension) was obtained.
- Example 11 (Formulation of suspension containing propiconazole-containing sustained-release particles) A median containing propiconazole was obtained by dispersing the hydrophobic solution in water and then performing suspension polymerization in the same manner as in Example 10 except that 40 g of propiconazole was used instead of 40 g of cyfluthrin. A suspension (suspension) of sustained-release particles having a diameter of 19 ⁇ m was obtained.
- Example 12 (Formulation of suspension containing prochloraz-containing sustained-release particles) Except that 40 g of prochloraz was used instead of 40 g of sifluthrin, a hydrophobic solution was dispersed in water in the same manner as in Example 10 and then suspension polymerization was carried out to gradually adjust the median diameter of 30 ⁇ m containing prochloraz. A suspension of suspension particles was obtained.
- Example 13 (Formulation of suspension containing flusilazole-containing sustained-release particles) A hydrophobic solution was dispersed in water in the same manner as in Example 10 except that 40 g of flusilazole was used instead of 40 g of cyfluthrin. Subsequently, suspension polymerization was performed to obtain a gradil having a median diameter of 32 ⁇ m containing flusilazole. A suspension of suspension particles was obtained.
- Example 14 (Formulation of suspension containing IPBC-containing sustained release particles) Into a 200 mL beaker (1), 40 g of IPBC, 24 g of methyl methacrylate, 18 g of n-butyl methacrylate, 18 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide are prepared and stirred at room temperature to prepare a uniform hydrophobic solution. did.
- a 500 mL beaker (2) was charged with 280 g of ion exchange water, 20 g of a 10% aqueous solution of PVA-217, and 200 mg of a 5% aqueous solution of DBN, and stirred at room temperature to obtain a uniform aqueous solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring for 10 minutes at 3000 rpm with a homomixer MARK 2.5 (manufactured by Primix).
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 1 hour, 70 ⁇ 2 ° C. for 3 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or lower to obtain a suspension (suspension) of sustained release particles containing IPBC having a median diameter of 24 ⁇ m.
- Example 15 (Formulation of suspension containing IPBC-containing sustained release particles) A hydrophobic solution was dispersed in water as in Example 14 except that 18 g of ethyl acrylate was used instead of 18 g of n-butyl methacrylate, and subsequently contained IPBC by suspension polymerization. A suspension (suspension) of sustained release particles having a median diameter of 22 ⁇ m was obtained.
- Example 16 (Formulation of suspension containing IPBC-containing sustained release particles) An IPBC was prepared by dispersing the hydrophobic solution in water and then performing suspension polymerization in the same manner as in Example 14 except that 18 g of n-butyl acrylate was used instead of 18 g of n-butyl methacrylate. A suspension (suspension agent) of sustained-release particles having a median diameter of 22 ⁇ m was obtained.
- Comparative Example 1 (Formulation of suspension containing MBACT-containing sustained release particles) A 200 mL beaker (1) was charged with 10 g of MBACT, 81 g of methyl methacrylate, 9 g of ethylene glycol dimethacrylate, and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion exchange water, 20 g of a 10% aqueous solution of PVA-217, and 200 mg of a 5% aqueous solution of DBN, and stirred at room temperature to obtain a uniform aqueous solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring with a homomixer MARK 2.5 type (manufactured by Primix) for 10 minutes at a rotational speed of 3000 rpm.
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 2 hours, 70 ⁇ 2 ° C. for 3 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or lower to obtain a suspension (suspension) of sustained-release particles containing MBACT and having a median diameter of 20 ⁇ m.
- Comparative Example 2 (Formulation of suspension containing MBACT coexisting particles) A 200 mL beaker (1) was charged with 40 g of MBACT, 54 g of methyl methacrylate, and 6 g of ethylene glycol dimethacrylate, and heated with stirring to prepare a uniform hydrophobic solution at 70 ° C.
- a 500 mL beaker (2) was charged with 280 g of ion-exchanged water, 20 g of a 10% aqueous solution of PVA-217 and 200 mg of a 5% aqueous solution of DBN, and heated with stirring to obtain a uniform aqueous solution at 70 ° C.
- this monomer solution was quickly added to the beaker (2), while maintaining the liquid temperature at 70 ° C. K.
- the suspension was prepared by dispersing the hydrophobic solution in water by stirring with a homomixer MARK 2.5 type (manufactured by Primix) for 10 minutes at a rotational speed of 3000 rpm.
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was carried out continuously at 70 ⁇ 2 ° C. for 5 hours and at 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or less to obtain a suspension (suspension) of particles having a median diameter of 26 ⁇ m in which MBACT coexists.
- Comparative Example 3 (Formulation of suspension containing capric acid-containing sustained-release particles) A 200 mL beaker (1) was charged with 40 g of capric acid, 42 g of methyl methacrylate, 18 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion-exchanged water, 20 g of TCP-10U and 200 mg of 5% aqueous solution of Perex SS-L, and stirred at room temperature to obtain a uniform aqueous solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring for 10 minutes at 2000 rpm with a homomixer MARK 2.5 (manufactured by Primix).
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 1 hour, 70 ⁇ 2 ° C. for 2 hours, and 80 ⁇ 2 ° C. for 2 hours.
- Comparative Example 4 (Formulation of suspension containing IPBC coexisting particles) A 200 mL beaker (1) was charged with 40 g of IPBC, 42 g of n-butyl methacrylate, 18 g of ethylene glycol dimethacrylate and 300 mg of dilauroyl peroxide, and stirred at room temperature to prepare a uniform hydrophobic solution.
- a 500 mL beaker (2) was charged with 280 g of ion exchange water, 20 g of a 10% aqueous solution of PVA-217, and 200 mg of a 5% aqueous solution of DBN, and stirred at room temperature to obtain a uniform aqueous solution.
- the hydrophobic solution was added to a 500 mL beaker (2).
- K The suspension was prepared by dispersing the hydrophobic solution in water by stirring with a homomixer MARK 2.5 type (manufactured by Primix) for 10 minutes at a rotational speed of 3000 rpm.
- the suspension was transferred to a 500 mL 4-neck Kolben equipped with a stirrer, a reflux condenser, a thermometer, and a nitrogen introduction tube, and the temperature was increased while stirring in a nitrogen stream to perform suspension polymerization.
- Suspension polymerization was started at the time when the temperature reached 55 ° C., and then continuously performed at 60 ⁇ 2 ° C. for 1 hour, 70 ⁇ 2 ° C. for 3 hours, and 80 ⁇ 2 ° C. for 2 hours.
- suspension after the reaction was cooled to 30 ° C. or lower to obtain a suspension (suspension) of particles having a median diameter of 28 ⁇ m in which IPBC coexists.
- Comparative Example 5 (Formulation of suspension containing IPBC coexisting particles) By performing suspension polymerization in the same manner as in Example 2 except that 42 g of styrene was charged instead of 42 g of methyl methacrylate, and the rotation speed of the homomixer in the aqueous dispersion of the hydrophobic solution was changed to 3000 rpm. A suspension (suspension) of particles having a median diameter of 30 ⁇ m in which IPBC coexists was obtained.
- Comparative Example 6 (Formulation of suspension containing IPBC coexisting particles) IPBC coexisted by dispersing the hydrophobic solution in water and then performing suspension polymerization in the same manner as in Comparative Example 4 except that 42 g of methyl acrylate was used instead of 42 g of n-butyl methacrylate. A suspension (suspension) of particles having a median diameter of 11 ⁇ m was obtained.
- Tables 2 to 4 show the formulation of each component in each Example and each Comparative Example.
- surface shows g number (solid content. However, ion-exchange water is remove
- Example 17 (Formulation of powder and re-formulation of suspension) The suspension of Example 2 was transferred to a stainless steel vat and air-dried at room temperature to formulate a powder having excellent fluidity.
- deionized water was added to 50 g of the formulated powder so that the solid concentration was 25%. K.
- the suspension was re-formulated (regenerated) by stirring with a disper (manufactured by Primix) and again dispersing (suspending) the powder in water.
- the re-formulated sustained release particles of the suspension of Example 17 had the same median diameter and particle size distribution as the sustained release particles of the suspension of Example 2.
- Examples 18-22 (Formulation of powder and re-formulation of suspension) Examples 9 to 13 were also treated in the same manner as Example 17 to formulate powders, followed by re-formulation of suspensions, which were respectively used as powders and suspensions of Examples 18-22. It was.
- the re-formulated suspensions of Examples 18 to 22 had the same median diameter and particle size distribution as the sustained-release particles of the suspensions of Examples 9 to 13, respectively.
- Table 5 shows the correspondence between the powders and suspensions of Examples 17 to 22 (after resuspension) and the suspensions of Examples 2 and 9 to 13.
- FIGS. 1 to 10 Image processing diagrams of SEM photographs of Examples 1, 5, 8, 14 to 16 and Comparative Examples 2, 4 to 6 are shown in FIGS. 1 to 10, respectively.
- Example 6 due to the effect of OIT as a plasticizer, when water was distilled off, a transparent film was formed, so SEM observation was not possible. Therefore, it can be seen from this fact that Example 6 is a compatible system. 2. Observation by TEM (Transmission Electron Microscope) Each of the suspensions (suspensions) of Examples 1 to 16 was freeze-dried, then dispersed in a bisphenol-type liquid epoxy resin containing an amine, and then cured. I let you.
- the cured product is cut with an ultramicrotome to obtain a cross section, and the cross section is stained with osmium tetroxide, and further, if necessary, further stained with ruthenium tetroxide, and this is cut into ultrathin sections with an ultramicrotome.
- Samples were prepared. The prepared sample was observed with a transmission electron microscope (model number “H-7100”, manufactured by Hitachi, Ltd.) by TEM.
- Fig. 11 to 16 show image processing diagrams of TEM photographs of Examples 1 and 9 to 13, respectively.
- the cross-section of the sustained-release particles is uniform, so that the antibiotic compound is uniformly incorporated into the sustained-release particles. It was confirmed that it was dissolved and contained.
- the antibiotic compound of the present invention that is, isomer I (melting point: 57 ° C.), isomer II (melting point: 74 ° C.) and isomer III of cyfluthrin ( A mixture having a melting point of 66 ° C. is uniformly mixed and contained in the polymer, while isomer IV (melting point: 102 ° C.) contained as an impurity in an amount of about 20% by weight is precipitated inside the polymer. It was observed that the sustained release particles were formed in the form of microphase separation.
- the sustained release rate of IPBC was calculated using HPLC from the amount of IPBC in the obtained filtrate and the amount of IPBC remaining on the filter paper.
- the sustained release rate in each water flow amount was calculated as an integrated value (that is, total sustained release rate).
- Example 11 The results of Examples 1 to 3 are shown in FIG. 17, and the results of Examples 14 to 16 are shown in FIG. (2) Sustained release test of propiconazole-containing sustained release particles (Example 11) Based on the operation of “(1) Sustained release test of IPBC-containing sustained release particles” described above, the sustained release test of propiconazole-containing sustained release particles of Example 11 was performed.
- the suspension of the sustained release particles obtained in Examples 4 to 7 and 9 and the OIT as a blank were added to a commercially available white acrylic silicone emulsion paint with an OIT concentration of 0.2.
- the coating material added to a weight percentage and then the suspension of sustained-release particles was diluted 1.5 times with ion-exchanged water.
- filter paper (corresponding to two types of Toyo filter paper No. 2 and JIS P 3801) was cut into 3.5 cm ⁇ 3.5 cm, precisely weighed, and immersed in the above-described paint.
- the filter paper was put into a glass bottle, and 15 mL of ion exchange water was added and shaken for 18 hours. Subsequently, ion-exchanged water was collected, and 15 mL of ion-exchanged water was newly added and shaken for 18 hours. Thereafter, the above-described ion exchange water exchange operation was repeated twice.
- the sustained release rate of OIT was calculated from the amount of OIT in each ion-exchanged water collected as described above using HPLC. In addition, the sustained release rate in each number of times was calculated as an integrated value (that is, total sustained release rate).
- suspensions (suspension agents) of sustained release particles of Examples 8 and 10 (cyfluthrin concentration 10%) and cyfluthrin were dissolved to prepare a 10% acetonitrile solution as a blank.
- the sustained release amount of cyfluthrin was calculated from the ion-exchanged water / methanol mixture collected each time using TOF-MS. In addition, the sustained release amount in each number of times was calculated as an integrated value (that is, total sustained release amount).
- the sustained-release particles of the present invention can be applied to various industrial products, such as indoor and outdoor paints, rubber, fibers, resins, plastics, adhesives, joint agents, sealing agents, building materials, caulking agents, soils. It can be blended into a processing agent, wood, white water in a papermaking process, pigment, printing plate processing liquid, cooling water, ink, cutting oil, cosmetics, nonwoven fabric, spinning oil, leather, and the like.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Inorganic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Manufacturing Of Micro-Capsules (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
Description
(1)融点が100℃以下であり、Hansenで定義され、van Klevelen and Hoftyzer法で算出される溶解度パラメータδの双極子間力項δp,compoundが2~8[(J/cm3)1/2]、前記溶解度パラメータδの水素結合力項δh,compoundが5.5~9.5[(J/cm3)1/2]である疎水性の抗生物活性化合物を、溶剤の不存在下、疎水性の重合性ビニルモノマーで溶解することにより、疎水性溶液を調製し、その疎水性溶液を水分散させ、前記重合性ビニルモノマーを、油溶性開始剤の存在下、ラジカル重合して、前記溶解度パラメータδの双極子間力項δp,polymerが5~7[(J/cm3)1/2]であり、前記溶解度パラメータδの水素結合力項δh,polymerが8~10[(J/cm3)1/2]である重合体を生成することにより得られることを特徴とする、徐放性粒子、
(2)前記重合体の双極子間力項δp,polymerから前記抗生物活性化合物の双極子間力項δp,compoundを差し引いた値Δδpが-1.1~2.7[(J/cm3)1/2]であり、前記重合体の水素結合力項δh,polymerから前記抗生物活性化合物の水素結合力項δh,compoundを差し引いた値Δδhが、0~4.2[(J/cm3)1/2]であることを特徴とする、前記(1)に記載の徐放性粒子、
(3)前記抗生物活性化合物の前記重合性ビニルモノマーに対する配合割合が、重量基準で、0.11~1.5であることを特徴とする、前記(1)に記載の徐放性粒子、
(4)融点が100℃以下であり、Hansenで定義され、van Klevelen and Hoftyzer法で算出される溶解度パラメータδの双極子間力項δp,compoundが2~8[(J/cm3)1/2]、前記溶解度パラメータδの水素結合力項δh,compoundが5.5~9.5[(J/cm3)1/2]である疎水性の抗生物活性化合物を、溶剤の不存在下、疎水性の重合性ビニルモノマーで溶解することにより、疎水性溶液を調製する工程、前記疎水性溶液を水分散させる工程、および、水分散された前記疎水性溶液の前記重合性ビニルモノマーを、油溶性開始剤の存在下、ラジカル重合して、前記溶解度パラメータδの双極子間力項δp,polymerが5~7[(J/cm3)1/2]であり、前記溶解度パラメータδの水素結合力項δh,polymerが8~10[(J/cm3)1/2]である重合体を生成する工程を備えることを特徴とする、徐放性粒子の製造方法、
(5)前記重合体の双極子間力項δp,polymerから前記抗生物活性化合物の双極子間力項δp,compoundを差し引いた値Δδpが-1.1~2.7[(J/cm3)1/2]であり、前記重合体の水素結合力項δh,polymerから前記抗生物活性化合物の水素結合力項δh,compoundを差し引いた値Δδhが、0~4.2[(J/cm3)1/2]であることを特徴とする、前記(4)に記載の徐放性粒子の製造方法
である。


上記したFp、EhおよびVの数値は、「Properties of Polymers」(3rd Edition、第7章、第189~225頁、van Klevelen著、ELSEVIER、2003年発行)に、原子団毎に記載されている。
-I Fp:0(J1/2・cm3/2・mol-1)
Eh:0(J・mol-1)
>Si< Fp:0(J1/2・cm3/2・mol-1)
Eh:0(J・mol-1)
=N- Fp:800(J1/2・cm3/2・mol-1)
Eh:3000(J・mol-1)
≡C- Fp:0(J1/2・cm3/2・mol-1)
Eh:0(J・mol-1)
次に、重合体の一例として、メタクリル酸メチルの重合体であるポリメタクリル酸メチル(PMMA)を例示し、かかるポリメタクリル酸メチルの溶解度パラメータδの双極子間力項δp,PMMAおよび水素結合力項δh,PMMAを算出する。
1.ホモポリマーの双極子間力項δpおよび水素結合力項δh
(1)ポリメタクリル酸メチルの構造式
ポリメタクリル酸メチルは、下記式(3)で表される。

(2)双極子間力項δp,PMMMA
上記式(3)のモノマー単位(-CH2-C(CH3)COOCH3-)において、各原子団に対応するFpおよびVを以下に記載する。
-CH3 Fp:0(J1/2・cm3/2・mol-1)
V:33.5(cm3・mol)
-CH2- Fp:0(J1/2・cm3/2・mol-1)
V:16.1(cm3・mol)
>C< Fp:0(J1/2・cm3/2・mol-1)
V:-19.2(cm3・mol)
-COO- Fp:490(J1/2・cm3/2・mol-1)
V:18(cm3・mol)
従って、モノマー単位の双極子間力項δp,monomer unitは、下記式(4)に示すように、5.98[(J/cm3)1/2]と算出される。

(3)水素結合力項δh,PMMA
上記式(3)のモノマー単位(-CH2-C(CH3)COOCH3-)において、各原子団に対応するEhを以下に記載する。
-CH3 Eh:0(J・mol-1)
-CH2- Eh:0(J・mol-1)
>C< Eh:0(J・mol-1)
-COO- Eh:7000(J・mol-1)
従って、モノマー単位の水素結合力項δh,monomer unitは、下記式(5)に示すように、9.25[(J/cm3)1/2]と算出される。

2.共重合体の双極子間力項δpおよび水素結合力項δh
次に、共重合体の双極子間力項δpおよび水素結合力項δhを算出する。
(1)双極子間力項δp,PMMA-EGDMA
メタクリル酸メチルのモノマー単位の双極子間力項δp,MMA unitは、上記で算出したように、5.98[(J/cm3)1/2]である。
δp,PMMA-EGDMA=(90/100)δp、MMA unit+(10/100)δp、EGDMA unit
=(90/100)×5.98+(10/100)×5.37
=5.92[(J/cm3)1/2] (6)
(2)水素結合力項δh,PMMA-EGDMA
メタクリル酸メチルのモノマー単位の水素結合力項δh,MMA unitは、9.25[(J/cm3)1/2]である。
δh,PMMA-EGDMA=(90/100)δh,MMA unit+(10/100)δh,EGDMA unit
=(90/100)×9.25+(10/100)×10.42
=9.36[(J/cm3)1/2] (7)
そして、重合体の溶解度パラメータδの双極子間力項δp,polymerは、好ましくは、5~6.5[(J/cm3)1/2]であり、重合体の溶解度パラメータδの水素結合力項δh,polymerは、好ましくは、9~10[(J/cm3)1/2]である。
3.抗生物活性化合物の溶解度δの双極子間力項δp,compoundおよび水素結合力項δh,compound
抗生物活性化合物の溶解度δの双極子間力項δp,compoundおよび水素結合力項δh,compoundについても、上記したモノマー単位のそれと同様にして算出される。

4.溶解度パラメータの双極子間力項δpの差(Δδp)および水素結合力項δhの差(Δδh)
本発明では、溶解度パラメータδにおいて、双極子間力項δp,polymerから抗生物活性化合物の双極子間力項δp,compoundを差し引いた値Δδp(=δp,polymer-δp,compound)は、例えば、-1.1~2.7[(J/cm3)1/2]である。
IPBC:商品名「ファンギトロール400」、3-ヨード-2-プロピニルブチルカルバメート、分子量281、融点:60℃、水への溶解度:150ppm、溶解度パラメータδの双極子間力項δp,compound:3.23[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,compound:7.83[(J/cm3)1/2]、インターナショナル・スペシャリティ・プロダクツ社製
OIT:商品名「ケーソン893T」(「ケーソン」は登録商標)、2-n-オクチル-4-イソチアゾリン-3-オン、分子量213、融点:20℃未満、水への溶解度:300ppm、溶解度パラメータδの双極子間力項δp,compound:5.47[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,compound:5.87[(J/cm3)1/2]、ローム・アンド・ハース社製
シフルトリン:商品名「プリベントールHS12」(「プリベントール」は登録商標)、(RS)-α-シアノ-4-フルオロ-3-フェノキシベンジル=(1RS,3RS)-(1RS,3RS)-3-(2,2-ジクロロビニル)-2,2-メチルシクロプロパンカルボキシラート、分子量434、水への溶解度:1~2ppb、異性体I(融点57℃)と異性体II(融点74℃)と異性体III(融点66℃)と異性体IV(融点102℃)との混合物、溶解度パラメータδの双極子間力項δp,compound:3.46[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,compound:6.09[(J/cm3)1/2]、ランクセス社製
プロピコナゾール:1-[2-(2,4-ジクロロフェニル)-4-n-プロピル-1,3-ジオキソラン-2-イルメチル]-1H-1,2,4-トリアゾール、分子量342、融点:20℃未満、水への溶解度:110ppm、溶解度パラメータδの双極子間力項δp,compound:6.55[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,compound:9.44[(J/cm3)1/2]、八幸通商社製
プロクロラズ:N-プロピル-N-[2-(2,4,6-トリクロロ-フェノキシ)エチル]イミダゾール-1-カルボキサミド、分子量375、融点45~52℃、水への溶解度:55ppm、溶解度パラメータδの双極子間力項δp,compound:6.87[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,compound:8.85[(J/cm3)1/2]、丸善薬品社製
フルシラゾール:ビス(4-フルオロフェニル)メチル(1H-1,2,4-トリアゾール-1-イルメチルシラン、分子量315、融点:54℃、水への溶解度:45ppm、溶解度パラメータδの双極子間力項δp,compound:5.95[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,compound:6.85[(J/cm3)1/2]、エアブラウン社製
MBACT:商品名「イルガロール1071」(「イルガロール」は登録商標)、2-メチルチオ-4-t-ブチルアミノ-6-シクロプロピルアミノ-s-トリアジン、分子量253、融点:133℃、水への溶解度:7ppm、溶解度パラメータδの双極子間力項δp,compound:7.18[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,compound:8.77[(J/cm3)1/2]、チバ・スペシャリティ・ケミカルズ社製
カプリン酸:分子量172、融点:29~32℃、水への溶解度:1.5重量%、溶解度パラメータδの双極子間力項δp,monomer unit:2.20[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,monomer unit:7.24[(J/cm3)1/2]
メタクリル酸メチル:商品名「アクリルエステルM」、水への溶解度:1.6重量%、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:5.98[(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer unit:9.25[(J/cm3)1/2]、三菱レイヨン社製
メタクリル酸n-ブチル:水への溶解度:0.08重量%、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:3.76(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer unit:7.33[(J/cm3)1/2]、三菱レイヨン社製
アクリル酸メチル:水への溶解度:5.7重量%、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:7.36[(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer unit:10.25[(J/cm3)1/2]、日本触媒社製
アクリル酸エチル:水への溶解度:1.5重量%、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:5.93[(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer unit:9.20[(J/cm3)1/2]、日本触媒社製
アクリル酸n-ブチル:水への溶解度:0.2重量%、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:4.26[(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer unit:7.81[(J/cm3)1/2]、日本触媒社製
メタクリル酸:水への溶解度:8.9重量%、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:7.13[(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer unit:13.03[(J/cm3)1/2]、三菱レイヨン製
スチレン:水に不溶、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:1.27[(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer unit:0.00[(J/cm3)1/2]
エチレングリコールジメタクリレート:商品名「ライトエステルEG」、水に不溶、モノマー単位としての溶解度パラメータδの双極子間力項δp,monomer unit:5.37[(J/cm3)1/2]、モノマー単位としての溶解度パラメータδの水素結合力項δh,monomer:10.42[(J/cm3)1/2]、共栄社化学社製
トリメチロールプロパントリメタクリレート:商品名「ライトエステルTMP」、水に不溶、溶解度パラメータδの双極子間力項δp,monomer unit:3.79[(J/cm3)1/2]、溶解度パラメータδの水素結合力項δh,monomer unit:9.68[(J/cm3)1/2]、共栄社化学社製
ジラウロイルパーオキシド:商品名「パーロイルL」(「パーロイル」は登録商標)、日油社製
PVA-217:商品名「クラレポバール217」、部分鹸化ポリビニルアルコール、クラレ社製
TCP-10U:商品名、第三燐酸カルシウム(3[Ca3(PO4)2]・Ca(OH)2)の10重量%水懸濁液、松尾薬品産業社製
DBN:商品名「ネオペレックスNo.6パウダー」(「ネオペレックス」は登録商標)、ドデシルベンゼンスルホン酸ナトリウム、花王社製
ペレックスSS-L:商品名(「ペレックス」は登録商標)、ドデシルジフェニルエーテルジスルホン酸ナトリウム、花王社製
実施例1
(IPBC含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、IPBC40g、メタクリル酸メチル54g、エチレングリコールジメタクリレート6gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(IPBC含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、IPBC40g、メタクリル酸メチル42g、エチレングリコールジメタクリレート18gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(IPBC含有徐放性粒子を含む懸濁剤の製剤化)
メタクリル酸メチルおよびエチレングリコールジメタクリレートの仕込み量を、ともに30gに変更した以外は、実施例1と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、IPBCを含有するメジアン径9.6μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(OIT含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、OIT40g、メタクリル酸メチル54g、エチレングリコールジメタクリレート6gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(OIT含有徐放性粒子を含む懸濁剤の製剤化)
メタクリル酸メチルおよびエチレングリコールジメタクリレートの仕込み量を、ともに30gに変更した以外は、実施例4と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、OITを含有するメジアン径14μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(OIT含有徐放性粒子を含む懸濁剤の製剤化)
OIT、メタクリル酸メチルおよびエチレングリコールジメタクリレートの仕込み量を、それぞれ、50g、45gおよび5gに変更した以外は、実施例4と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、OITを含有するメジアン径9.8μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(OIT含有徐放性粒子を含む懸濁剤の製剤化)
水分散時におけるT.K.ホモミクサーMARK2.5型(プライミクス社製)の攪拌条件を、回転数1000rpmに変更した以外は、実施例4と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、IPBCを含有するメジアン径29μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(シフルトリン含有微粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、シフルトリン40g、メタクリル酸メチル54g、トリメチロールプロパントリメタクリレート6g、ジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(OIT含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、OIT40g、メタクリル酸メチル36g、メタクリル酸6g、エチレングリコールジメタクリレート18gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(シフルトリン含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、シフルトリン40g、メタクリル酸メチル42g、エチレングリコールジメタクリレート18gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(プロピコナゾール含有徐放性粒子を含む懸濁剤の製剤化)
シフルトリン40gに代えて、プロピコナゾール40gを仕込んだ以外は、実施例10と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、プロピコナゾールを含有するメジアン径19μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(プロクロラズ含有徐放性粒子を含む懸濁剤の製剤化)
シフルトリン40gに代えて、プロクロラズ40gを仕込んだ以外は、実施例10と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、プロクロラズを含有するメジアン径30μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(フルシラゾール含有徐放性粒子を含む懸濁剤の製剤化)
シフルトリン40gに代えて、フルシラゾール40gを仕込んだ以外は、実施例10と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、フルシラゾールを含有するメジアン径32μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(IPBC含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、IPBC40g、メタクリル酸メチル24g、メタクリル酸n-ブチル18g、エチレングリコールジメタクリレート18gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(IPBC含有徐放性粒子を含む懸濁剤の製剤化)
メタクリル酸n-ブチル18gに代えて、アクリル酸エチル18gを仕込んだ以外は、実施例14と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、IPBCを含有するメジアン径22μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(IPBC含有徐放性粒子を含む懸濁剤の製剤化)
メタクリル酸n-ブチル18gに代えて、アクリル酸n-ブチル18gを仕込んだ以外は、実施例14と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、IPBCを含有するメジアン径22μmの徐放性粒子の懸濁液(懸濁剤)を得た。
(MBACT含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、MBACT10g、メタクリル酸メチル81g、エチレングリコールジメタクリレート9g、ジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(MBACT共存粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、MBACT40g、メタクリル酸メチル54g、エチレングリコールジメタクリレート6gを仕込み、攪拌下、昇温して、70℃の均一な疎水性溶液を調製した。
(カプリン酸含有徐放性粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、カプリン酸40g、メタクリル酸メチル42g、エチレングリコールジメタクリレート18gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(IPBC共存粒子を含む懸濁剤の製剤化)
200mLのビーカー(1)に、IPBC40g、メタクリル酸n-ブチル42g、エチレングリコールジメタクリレート18gおよびジラウロイルパーオキシド300mgを仕込み、室温で攪拌することにより、均一な疎水性溶液を調製した。
(IPBC共存粒子を含む懸濁剤の製剤化)
メタクリル酸メチル42gに代えて、スチレン42gを仕込み、さらに、疎水性溶液の水分散におけるホモミクサーの回転数を3000rpmに変更した以外は、実施例2と同様にして、懸濁重合を行なうことにより、IPBCが共存するメジアン径30μmの粒子の懸濁液(懸濁剤)を得た。
(IPBC共存粒子を含む懸濁剤の製剤化)
メタクリル酸n-ブチル42gに代えて、アクリル酸メチル42gを仕込んだ以外は、比較例4と同様にして、疎水性溶液を水分散させ、続いて、懸濁重合を行なうことにより、IPBCが共存するメジアン径11μmの粒子の懸濁液(懸濁剤)を得た。



(粉剤の製剤化および懸濁剤の再製剤化)
実施例2の懸濁液をステンレス製バットに移し、室温で風乾することにより、流動性に優れた粉剤を製剤化した。
(粉剤の製剤化および懸濁剤の再製剤化)
実施例9~13についても、実施例17と同様に処理して、粉剤を製剤化し、続いて、懸濁剤を再製剤化し、それらを、それぞれ、実施例18~22の粉剤および懸濁剤とした。

1. 重合体の溶解度パラメータδの双極子間力項δp,polymerおよび水素結合力項δh,polymerを、上記に準拠して、算出した。
2. Δδp(=δp,polymer-δp,compound)およびΔδh(=δh,polymer-δh,compound)をそれぞれ算出した。
1.SEM(走査型電子顕微鏡、Scanning Electron Microscope)観察
実施例1~16および比較例2、4~6の懸濁液(懸濁剤)を、試料台に滴下し、その後、水を留去した後、得られた徐放性粒子(比較例2および4~6では、粒子)を、走査型電子顕微鏡日立TM-100(日立ハイテクノロジーズ社製)で、SEM観察した。
2.TEM(透過型電子顕微鏡、Transmission Electron Microscope)観察
実施例1~16の懸濁液(懸濁剤)をそれぞれ凍結乾燥し、次いで、アミンを含むビスフェノール型液状エポキシ樹脂に分散して、その後、硬化させた。次いで、硬化物をウルトラミクロトームで切断して断面を出し、かかる断面を四酸化オスミウムによって染色し、必要に応じて、さらに四酸化ルテニウムで染色し、これをウルトラミクロトームで超薄切片に切り出すことにより、サンプルを調製した。調製したサンプルを、透過型電子顕微鏡(型番「H-7100」、日立製作所社製)で、TEM観察した。
3.徐放性試験
(1)IPBC含有徐放性粒子の徐放性試験(実施例1~3および14~16)
以下の操作に従って、実施例1~3および14~16のIPBC含有徐放性粒子について、徐放性試験を実施した。
(2)プロピコナゾール含有徐放性粒子の徐放性試験(実施例11)
上記した「(1)IPBC含有徐放性粒子の徐放性試験」の操作に準拠して、実施例11のプロピコナゾール含有徐放性粒子の徐放性試験を実施した。
(3)プロクロラズ含有徐放性粒子の徐放性試験(実施例12)
上記した「(1)IPBC含有徐放性粒子の徐放性試験」の操作に準拠して、実施例12のプロクロラズ含有徐放性粒子の徐放性試験を実施した。
(4)フルシラゾール含有徐放性粒子の徐放性試験(実施例13)
上記した「(1)IPBC含有徐放性粒子の徐放性試験」の操作に準拠して、実施例13のフルシラゾール含有徐放性粒子の徐放性試験を実施した。
(5)OIT含有徐放性粒子の徐放性試験(実施例4~7および9)
以下の操作に従って、実施例4~7および9のOIT含有徐放性粒子について、徐放性試験を実施した。
(6)シフルトリン含有徐放性粒子の徐放性試験(実施例8および10)
以下の操作に従って、実施例8および10のシフルトリン含有徐放性粒子について、徐放性試験を実施した。
(7)MBACT含有徐放性粒子(比較例1)およびMBACT共存徐放性粒子(比較例2)の徐放性試験
OIT含有徐放性粒子に代えて、MBACT含有徐放性粒子(比較例1)およびMBACT共存徐放性粒子(比較例2)を用いた以外は、上記した(5)OIT含有徐放性粒子の徐放性試験と同様の方法により、MBACTの徐放性試験を実施した。
Claims (5)
- 融点が100℃以下であり、Hansenで定義され、van Klevelen and Hoftyzer法で算出される溶解度パラメータδの双極子間力項δp,compoundが2~8[(J/cm3)1/2]、前記溶解度パラメータδの水素結合力項δh,compoundが5.5~9.5[(J/cm3)1/2]である疎水性の抗生物活性化合物を、溶剤の不存在下、疎水性の重合性ビニルモノマーで溶解することにより、疎水性溶液を調製し、その疎水性溶液を水分散させ、前記重合性ビニルモノマーを、油溶性開始剤の存在下、ラジカル重合して、前記溶解度パラメータδの双極子間力項δp,polymerが5~7[(J/cm3)1/2]であり、前記溶解度パラメータδの水素結合力項δh,polymerが8~10[(J/cm3)1/2]である重合体を生成することにより得られることを特徴とする、徐放性粒子。
- 前記重合体の双極子間力項δp,polymerから前記抗生物活性化合物の双極子間力項δp,compoundを差し引いた値Δδpが-1.1~2.7[(J/cm3)1/2]であり、
前記重合体の水素結合力項δh,polymerから前記抗生物活性化合物の水素結合力項δh,compoundを差し引いた値Δδhが、0~4.2[(J/cm3)1/2]であることを特徴とする、請求項1に記載の徐放性粒子。 - 前記抗生物活性化合物の前記重合性ビニルモノマーに対する配合割合が、重量基準で、0.11~1.5であることを特徴とする、請求項1に記載の徐放性粒子。
- 融点が100℃以下であり、Hansenで定義され、van Klevelen and Hoftyzer法で算出される溶解度パラメータδの双極子間力項δp,compoundが2~8[(J/cm3)1/2]、前記溶解度パラメータδの水素結合力項δh,compoundが5.5~9.5[(J/cm3)1/2]である疎水性の抗生物活性化合物を、溶剤の不存在下、疎水性の重合性ビニルモノマーで溶解することにより、疎水性溶液を調製する工程、
前記疎水性溶液を水分散させる工程、および、
水分散された前記疎水性溶液の前記重合性ビニルモノマーを、油溶性開始剤の存在下、ラジカル重合して、前記溶解度パラメータδの双極子間力項δp,polymerが5~7[(J/cm3)1/2]であり、前記溶解度パラメータδの水素結合力項δh,polymerが8~10[(J/cm3)1/2]である重合体を生成する工程
を備えることを特徴とする、徐放性粒子の製造方法。 - 前記重合体の双極子間力項δp,polymerから前記抗生物活性化合物の双極子間力項δp,compoundを差し引いた値Δδpが-1.1~2.7[(J/cm3)1/2]であり、
前記重合体の水素結合力項δh,polymerから前記抗生物活性化合物の水素結合力項δh,compoundを差し引いた値Δδhが、0~4.2[(J/cm3)1/2]であることを特徴とする、請求項4に記載の徐放性粒子の製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/394,960 US8808752B2 (en) | 2009-09-11 | 2010-09-09 | Controlled release particles and method for producing the same |
| CN201080040294.0A CN102480939B (zh) | 2009-09-11 | 2010-09-09 | 缓释性颗粒及其制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009211143 | 2009-09-11 | ||
| JP2009-211143 | 2009-09-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011030824A1 true WO2011030824A1 (ja) | 2011-03-17 |
Family
ID=43732493
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/065521 WO2011030824A1 (ja) | 2009-09-11 | 2010-09-09 | 徐放性粒子およびその製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8808752B2 (ja) |
| JP (1) | JP5547589B2 (ja) |
| CN (1) | CN102480939B (ja) |
| WO (1) | WO2011030824A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150141549A1 (en) * | 2012-07-13 | 2015-05-21 | Japan Envirochemicals, Ltd. | Antibiotic particles and production method thereof |
| JP2015528519A (ja) * | 2012-08-09 | 2015-09-28 | ローム アンド ハース カンパニーRohm And Haas Company | 殺生物剤を有するコーティング組成物 |
| US20180116539A1 (en) * | 2016-10-28 | 2018-05-03 | St. Jude Medical, Cardiology Division, Inc. | Flexible high-density mapping catheter |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6083936B2 (ja) * | 2011-03-11 | 2017-02-22 | 大阪ガスケミカル株式会社 | 徐放性粒子の製造方法 |
| JP5763570B2 (ja) * | 2011-03-11 | 2015-08-12 | 大阪ガスケミカル株式会社 | 徐放性粒子およびその製造方法 |
| JP5873790B2 (ja) * | 2011-12-28 | 2016-03-01 | 大阪ガスケミカル株式会社 | 徐放性粒子およびその製造方法 |
| JP5873714B2 (ja) * | 2011-12-28 | 2016-03-01 | 大阪ガスケミカル株式会社 | 徐放性粒子の製造方法 |
| JP6109502B2 (ja) * | 2012-07-13 | 2017-04-05 | 大阪ガスケミカル株式会社 | 抗生物活性粒子およびその製造方法 |
| JP5873843B2 (ja) * | 2012-10-12 | 2016-03-01 | 大阪ガスケミカル株式会社 | 乳濁液の製造方法 |
| JP5873842B2 (ja) * | 2012-11-21 | 2016-03-01 | 大阪ガスケミカル株式会社 | 徐放性粒子、その製造方法およびこれを用いた木部処理剤 |
| WO2015030213A1 (ja) * | 2013-08-30 | 2015-03-05 | 日本エンバイロケミカルズ株式会社 | 徐放性粒子、その製造方法、成形材料および成形品 |
| JP7228393B2 (ja) * | 2018-02-02 | 2023-02-24 | 大阪ガスケミカル株式会社 | 防藻性粒子、その製造方法および防藻性塗料 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001502733A (ja) * | 1996-10-24 | 2001-02-27 | バイエル・アクチエンゲゼルシヤフト | 汚止めペイント |
| JP2002513039A (ja) * | 1998-05-01 | 2002-05-08 | スリーエム イノベイティブ プロパティズ カンパニー | 抗微生物剤送達系 |
| JP2007246527A (ja) * | 2006-03-16 | 2007-09-27 | Rohm & Haas Co | カプセル化殺生物剤のブレンド |
| JP2008239561A (ja) * | 2007-03-28 | 2008-10-09 | Sumitomo Chemical Co Ltd | 常温で固体の生理活性物質のマイクロカプセル組成物の製造方法 |
| JP2008239562A (ja) * | 2007-03-28 | 2008-10-09 | Sumitomo Chemical Co Ltd | 常温で固体の生理活性物質のマイクロカプセル組成物の製造方法 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59145265A (ja) * | 1983-02-04 | 1984-08-20 | Sumitomo Naugatuck Co Ltd | 接着剤組成物 |
| EP0143608B1 (en) | 1983-11-25 | 1992-07-22 | Ciba Specialty Chemicals Water Treatments Limited | Manufacture and use of polymeric beads |
| GB8331546D0 (en) | 1983-11-25 | 1984-01-04 | Exxon Research Engineering Co | Polymeric compositions |
| JPS6186941A (ja) | 1984-10-03 | 1986-05-02 | Japan Synthetic Rubber Co Ltd | 含油マイクロカプセルの製造方法 |
| DE4137619A1 (de) | 1991-11-15 | 1993-05-19 | Basf Ag | Mikrokapseln mit feststoff-kern |
| DE19807118A1 (de) | 1998-02-20 | 1999-08-26 | Bayer Ag | Perlpolymerisat-Formulierungen |
| US6471975B1 (en) | 1998-05-01 | 2002-10-29 | 3M Innovative Properties Company | Microspheres as a delivery vehicle for bio-active agents useful in agricultural applications |
| JP4514077B2 (ja) | 1999-12-27 | 2010-07-28 | 日本エンバイロケミカルズ株式会社 | 微生物増殖抑制剤含有マイクロカプセルおよび微生物増殖抑制剤含有マイクロカプセルの製造方法 |
| JP2004331625A (ja) * | 2003-05-12 | 2004-11-25 | Nof Corp | 水分散型のフェロモン徐放製剤およびその製造方法 |
| JP4716435B2 (ja) | 2007-03-02 | 2011-07-06 | シチズン電子株式会社 | 光源装置及び光源装置を備えた表示装置 |
| JP5212271B2 (ja) | 2009-06-24 | 2013-06-19 | トヨタ自動車株式会社 | 車両用制御装置 |
-
2010
- 2010-09-09 US US13/394,960 patent/US8808752B2/en not_active Expired - Fee Related
- 2010-09-09 WO PCT/JP2010/065521 patent/WO2011030824A1/ja active Application Filing
- 2010-09-09 CN CN201080040294.0A patent/CN102480939B/zh not_active Expired - Fee Related
- 2010-09-09 JP JP2010201604A patent/JP5547589B2/ja active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001502733A (ja) * | 1996-10-24 | 2001-02-27 | バイエル・アクチエンゲゼルシヤフト | 汚止めペイント |
| JP2002513039A (ja) * | 1998-05-01 | 2002-05-08 | スリーエム イノベイティブ プロパティズ カンパニー | 抗微生物剤送達系 |
| JP2007246527A (ja) * | 2006-03-16 | 2007-09-27 | Rohm & Haas Co | カプセル化殺生物剤のブレンド |
| JP2008239561A (ja) * | 2007-03-28 | 2008-10-09 | Sumitomo Chemical Co Ltd | 常温で固体の生理活性物質のマイクロカプセル組成物の製造方法 |
| JP2008239562A (ja) * | 2007-03-28 | 2008-10-09 | Sumitomo Chemical Co Ltd | 常温で固体の生理活性物質のマイクロカプセル組成物の製造方法 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150141549A1 (en) * | 2012-07-13 | 2015-05-21 | Japan Envirochemicals, Ltd. | Antibiotic particles and production method thereof |
| JP2015528519A (ja) * | 2012-08-09 | 2015-09-28 | ローム アンド ハース カンパニーRohm And Haas Company | 殺生物剤を有するコーティング組成物 |
| US20180116539A1 (en) * | 2016-10-28 | 2018-05-03 | St. Jude Medical, Cardiology Division, Inc. | Flexible high-density mapping catheter |
| US11172858B2 (en) * | 2016-10-28 | 2021-11-16 | St. Jude Medical, Cardiology Division, Inc. | Flexible high-density mapping catheter |
Also Published As
| Publication number | Publication date |
|---|---|
| US20120172334A1 (en) | 2012-07-05 |
| CN102480939B (zh) | 2014-11-05 |
| CN102480939A (zh) | 2012-05-30 |
| JP5547589B2 (ja) | 2014-07-16 |
| US8808752B2 (en) | 2014-08-19 |
| JP2011079816A (ja) | 2011-04-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5547589B2 (ja) | 徐放性粒子およびその製造方法 | |
| JP6083936B2 (ja) | 徐放性粒子の製造方法 | |
| JP5763570B2 (ja) | 徐放性粒子およびその製造方法 | |
| JP6147115B2 (ja) | 抗生物活性粒子およびその製造方法 | |
| JP2011079816A5 (ja) | ||
| JP5873790B2 (ja) | 徐放性粒子およびその製造方法 | |
| EP2729002A1 (en) | Micelle-coated crystalline particles | |
| JP2012207012A5 (ja) | ||
| RU2363493C2 (ru) | Содержащие действующие вещества полимерные частицы | |
| JP6646950B2 (ja) | 木材保存剤および木材保護塗料 | |
| WO2013100102A1 (ja) | 徐放性粒子、木材処理剤およびその製造方法 | |
| JP6355485B2 (ja) | 徐放性粒子、その製造方法、成形材料および成形品 | |
| JP5873842B2 (ja) | 徐放性粒子、その製造方法およびこれを用いた木部処理剤 | |
| JP6622981B2 (ja) | 粒子の製造方法 | |
| JP2015221892A (ja) | 粒子 | |
| JP6114879B2 (ja) | 徐放性粒子およびその製造方法 | |
| US20150010635A1 (en) | Controlled release particles, wood treatment agent, and producing method thereof | |
| JP6109502B2 (ja) | 抗生物活性粒子およびその製造方法 | |
| JP5873843B2 (ja) | 乳濁液の製造方法 | |
| WO2015030213A1 (ja) | 徐放性粒子、その製造方法、成形材料および成形品 | |
| JP2016194185A (ja) | 繊維処理剤および繊維処理方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080040294.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10815419 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13394960 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10815419 Country of ref document: EP Kind code of ref document: A1 |