WO2010084872A1 - (ポリ)カーボネートポリオールおよび該(ポリ)カーボネートポリオールを原料とするカルボキシル基含有ポリウレタン - Google Patents
(ポリ)カーボネートポリオールおよび該(ポリ)カーボネートポリオールを原料とするカルボキシル基含有ポリウレタン Download PDFInfo
- Publication number
- WO2010084872A1 WO2010084872A1 PCT/JP2010/050611 JP2010050611W WO2010084872A1 WO 2010084872 A1 WO2010084872 A1 WO 2010084872A1 JP 2010050611 W JP2010050611 W JP 2010050611W WO 2010084872 A1 WO2010084872 A1 WO 2010084872A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- component
- carboxyl group
- containing polyurethane
- polyol
- poly
- Prior art date
Links
- 0 CC(C)(C)c1cc(*C(C)(*)*)cc(C(C)(C)C)c1* Chemical compound CC(C)(C)c1cc(*C(C)(*)*)cc(C(C)(C)C)c1* 0.000 description 1
Images
















Classifications
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K3/00—Apparatus or processes for manufacturing printed circuits
- H05K3/22—Secondary treatment of printed circuits
- H05K3/28—Applying non-metallic protective coatings
- H05K3/285—Permanent coating compositions
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/02—Aliphatic polycarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/10—Prepolymer processes involving reaction of isocyanates or isothiocyanates with compounds having active hydrogen in a first reaction step
- C08G18/12—Prepolymer processes involving reaction of isocyanates or isothiocyanates with compounds having active hydrogen in a first reaction step using two or more compounds having active hydrogen in the first polymerisation step
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/30—Low-molecular-weight compounds
- C08G18/34—Carboxylic acids; Esters thereof with monohydroxyl compounds
- C08G18/341—Dicarboxylic acids, esters of polycarboxylic acids containing two carboxylic acid groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
- C08G18/44—Polycarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/65—Low-molecular-weight compounds having active hydrogen with high-molecular-weight compounds having active hydrogen
- C08G18/66—Compounds of groups C08G18/42, C08G18/48, or C08G18/52
- C08G18/6633—Compounds of group C08G18/42
- C08G18/6659—Compounds of group C08G18/42 with compounds of group C08G18/34
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/75—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic
- C08G18/758—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing two or more cycloaliphatic rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L75/00—Compositions of polyureas or polyurethanes; Compositions of derivatives of such polymers
- C08L75/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D175/00—Coating compositions based on polyureas or polyurethanes; Coating compositions based on derivatives of such polymers
- C09D175/04—Polyurethanes
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K3/00—Apparatus or processes for manufacturing printed circuits
- H05K3/22—Secondary treatment of printed circuits
- H05K3/28—Applying non-metallic protective coatings
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/03—Use of materials for the substrate
- H05K1/0393—Flexible materials
Definitions
- the present invention relates to a (poly) carbonate polyol, a carboxyl group-containing polyurethane using the (poly) carbonate polyol as a raw material, a carboxyl group-containing polyurethane solution, a method for producing a carboxyl group-containing polyurethane, a curable composition containing a carboxyl group-containing polyurethane,
- the present invention relates to a cured product obtained from the composition, a flexible wiring board coated with the cured product, and a method for producing a flexible wiring board.
- surface protection films for flexible wiring circuits are made by punching a die that matches the pattern of a polyimide film called a coverlay film, and then pasting it with an adhesive, or UV curing with flexibility.
- a type or a thermosetting type overcoat agent is applied by a screen printing method, and the latter is particularly useful in terms of workability.
- curable overcoat agents resin compositions composed mainly of epoxy resin, acrylic resin, or composites thereof are known. These are often mainly composed of resins that have been modified, such as the introduction of a butadiene skeleton, a siloxane skeleton, a polycarbonate diol skeleton, a long-chain aliphatic skeleton, and the like.
- the improvement of flexibility and the occurrence of warpage due to curing shrinkage have been suppressed.
- Patent Document 1 JP-A-11-61038 discloses a resin composition using a polybutadiene blocked isocyanate and a polyol, but the cured product is excellent in terms of flexibility and shrinkage. However, heat resistance is not enough.
- Patent Document 2 discloses a polyamide-imide resin obtained by reacting both ends of a diisocyanate polyurethane obtained by reacting a polycarbonate diol and a diisocyanate compound with trimellitic acid. There is a drawback that the long-term reliability of the electrical properties of the cured product is not sufficient.
- Patent Document 3 discloses a polyamideimide resin having an organosiloxane skeleton, but the adhesion between the cured product and the substrate is not good. It is necessary to use a special solvent such as methyl-2-pyrrolidone, which may cause a problem since the emulsion may be dissolved particularly during screen printing.
- Patent Document 4 discloses a carboxyl group-containing polyurethane having a polyol unit selected from the group consisting of polybutadiene polyol, polyisoprene polyol, and hydrogenated polybutadiene polyol.
- the wiring that is currently widely used in the COF mounting method is produced by the subtractive method.
- the carboxyl group-containing polyurethane disclosed in Patent Document 4 exhibits sufficient insulating performance.
- the distance between wirings of the flexible wiring board will be further narrowed (for example, 20 ⁇ m pitch or less).
- polycarbonate diols using dimer diol as a raw material that is, having a structural unit derived from dimer diol
- dimer diol for example, JP-A-10-231360 (Patent Document 5) or JP-A-10-251398). No. (Patent Document 6)).
- a polycarbonate diol using a polyol having an alicyclic structure having 10 to 20 carbon atoms as a raw material (that is, having a structural unit derived from a polyol having an alicyclic structure having 10 to 20 carbon atoms) is also known (for example, JP-A-2006-31729 (Patent Document 7)).
- a carboxyl group-containing polyurethane using dimer diol as a raw material is known (for example, JP 2000-7909 (Patent Document 8) or JP 2007-100037 (Patent Document 9)).
- Polyurethanes using polycarbonate diol having a structural unit derived from dimer diol as a raw material are also known (for example, JP-A-10-273514 (Patent Document 10), JP-A-10-251369 (Patent Document). 11)).
- the present invention has been made in view of the above-described problems of the prior art, and includes a compound having a low warpage during curing, an excellent electrical insulating property of the cured product, and excellent flexibility of the cured product, and the compound.
- An object of the present invention is to provide a curable composition capable of obtaining a cured product having good electrical insulation characteristics, a cured product obtained from the composition, a flexible wiring board coated with the cured product, and a method for producing a flexible wiring board.
- the present invention (I) relates to a (poly) carbonate polyol containing an organic residue derived from a dimer diol and an organic residue derived from a polyol having an alicyclic structure having 10 to 20 carbon atoms.
- the present invention (II) relates to a carboxyl group-containing polyurethane using at least the following components (a), (b) and (c) as raw materials.
- the present invention (III) comprises the carboxyl group-containing polyurethane of the present invention (II).
- the present invention relates to a carboxyl group-containing polyurethane solution comprising a solvent having a boiling point of 120 to 300 ° C.
- the present invention (IV) includes diethylene glycol dimethyl ether, diethylene glycol diethyl ether, diethylene glycol ethyl methyl ether, diethylene glycol dibutyl ether, diethylene glycol butyl methyl ether, diethylene glycol isopropyl methyl ether, triethylene glycol dimethyl ether, triethylene glycol butyl methyl ether, tetraethylene glycol dimethyl ether, Dipropylene glycol dimethyl ether, tripropylene glycol dimethyl ether, anisole, ethylene glycol monomethyl ether acetate, ethylene glycol monoethyl ether acetate, propylene glycol monomethyl ether acetate, propylene glycol monoethyl ether Acetate, dipropylene glycol monomethyl ether acetate, dipropylene glycol monoethyl ether acetate, diethylene glycol monoethyl ether acetate, diethylene glycol monomethyl ether acetate, methyl methoxy
- the present invention (V) is a carboxyl group-containing polyurethane of the present invention (II)
- the present invention relates to a curable composition comprising a solvent having a boiling point of 120 to 300 ° C. and a curing agent.
- the present invention (VI) relates to a cured product obtained by curing the curable resin composition of the present invention (V).
- the present invention (VII) is characterized in that a part of or the entire surface of the flexible wiring board in which the wiring is formed on the flexible substrate is covered with the cured product of the present invention (VI). And a flexible wiring board coated with a cured product.
- a printed film is formed on the pattern by printing the curable composition of the present invention (V) on a tin-plated wiring pattern portion of a flexible wiring board.
- the present invention relates to a method for producing a flexible wiring board covered with a protective film, wherein the protective film is formed by heat curing at ⁇ 130 ° C.
- the present invention relates to the following [1] to [14].
- each R 6 independently represents an alkylene group of the following groups (A), (B) or (C), t is an integer of 1 or more, and (t + 1) Among R 6 , at least one R 6 is an alkylene group represented by the following group (A), and among (t + 1) R 6 , at least one R 6 is represented by the following group (B): An alkylene group.) (A) group: alkylene group represented by the following formula (35) or (36) (B) group: alkylene group represented by the following formula (37) or (38) (C) group: chain having 9 to 12 carbon atoms Aliphatic alkylene group
- R 1 and R 2 are both alkyl groups, and the total number of carbon atoms contained in R 1 and R 2 and p and q is 30.
- R 3 and R 4 are both alkyl groups, and the total number of carbon atoms contained in R 3 and R 4 and r and s is 34.
- a carboxyl group-containing polyurethane using at least the following component (a), component (b) and component (c) as raw materials.
- Component (c): Carboxyl group-containing polyol [5] At least the following component (a), A carboxyl group-containing polyurethane using the component (b), the component (c) and the component (d) as raw materials.
- Component (a) (Poly) carbonate polyol according to any one of [1] to [3]
- a carboxyl group-containing polyurethane solution comprising the carboxyl group-containing polyurethane according to any of [4] to [4] to [7] and a solvent having a boiling point of 120 to 300 ° C.
- a curable composition comprising the carboxyl group-containing polyurethane according to any one of the above, a solvent having a boiling point of 120 to 300 ° C., and a curing agent.
- a curing characterized in that a part of or all of the surface of the flexible wiring board in which the wiring is formed on the flexible substrate is covered with the cured product described in [12]. Flexible wiring board covered with objects.
- a printed film is formed on the pattern by printing the curable composition according to [10] or [11] on a tin-plated wiring pattern portion of a flexible wiring board.
- the cured product of the present invention has no tack, good handling properties, good flexibility and moisture resistance, long-term electrical insulation reliability at a high level, and low warpage. Good adhesion to the underfill material and good solvent resistance. For this reason, when applying the curable composition of the present invention to a flexible substrate such as a flexible wiring board or a polyimide film, and then creating a cured product (protective film) by a curing reaction, a flexible wiring board with a protective film or The warp of the flexible base material with the protective film is small, and the alignment of the IC chip mounting process is facilitated thereafter.
- the cured product of the present invention has flexibility, it is possible to provide a flexible wiring board with an electrically insulating protective film (for example, a flexible printed wiring board such as COF) that is not easily cracked.
- an electrically insulating protective film for example, a flexible printed wiring board such as COF
- FIG. 1 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of product A1 in the examples.
- FIG. 2 is an IR spectrum of the product A1 in the example.
- FIG. 3 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of product A2 in the examples.
- FIG. 4 is an IR spectrum of the product A2 in the example.
- FIG. 5 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of the product A3 in the example.
- FIG. 6 is an IR spectrum of the product A3 in the example.
- FIG. 7 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of the product A4 in the examples.
- FIG. 8 is an IR spectrum of the product A4 in the example.
- FIG. 1 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of product A1 in the examples.
- FIG. 2 is an IR spectrum of the product A1 in the example
- FIG. 9 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of the carboxyl group-containing polyurethane BU1 in the examples.
- FIG. 10 is an IR spectrum of carboxyl group-containing polyurethane BU1 in Examples.
- FIG. 11 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of carboxyl group-containing polyurethane BU2 in the examples.
- FIG. 12 is an IR spectrum of carboxyl group-containing polyurethane BU2 in Examples.
- FIG. 13 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of carboxyl group-containing polyurethane BU3 in the examples.
- FIG. 14 is an IR spectrum of carboxyl group-containing polyurethane BU3 in Examples.
- FIG. 15 is a 1 H-NMR spectrum (solvent: CDCl 3 ) of carboxyl group-containing polyurethane BU4 in the examples.
- FIG. 16 is an IR spectrum of carboxyl group-containing polyurethane BU4 in the examples.
- the “dimer diol” means that a dimer acid and / or a lower alcohol ester thereof is reduced in the presence of a catalyst, and a diol having 36 carbon atoms in which the carboxylic acid portion of the dimer acid is an alcohol is a main component.
- the dimer acid is a known dibasic acid obtained by an intermolecular polymerization reaction of an unsaturated fatty acid, and is obtained, for example, by dimerizing an unsaturated fatty acid having 11 to 22 carbon atoms with a clay catalyst or the like.
- dimer acids that are industrially obtained include arbitrary amounts of trimer acid and monomer acid depending on the degree of purification.
- the main component means that it is present in an amount of 50% by mass or more.
- the dimer diol in the present invention a hydrogenated dimer diol obtained by hydrogenating a carbon-carbon double bond derived from dimer acid is particularly preferable.
- Examples of commercially available dimer diols include PRIPOL 2033 (manufactured by Croda) and Sovermol 908 (manufactured by Cognis).
- PRIPOL 2033 is mainly composed of a mixture of compounds represented by the following formulas (1) and (2).
- Examples of commercially available dimer acids include PRIPOL 1006, 1009, 1015, 1025, etc. (manufactured by Croda), and EMPOL1062 (Cognis).
- Examples of the structure of a typical compound of dimer diol include the following formulas (1) and (2).
- R 1 and R 2 are both alkyl groups, and the total number of carbon atoms contained in R 1 and R 2 and p and q is 30.
- the “organic residue derived from dimer diol” in the present invention means a structure obtained by removing hydrogen from at least one alcoholic hydroxyl group of the dimer diol.
- an organic residue derived from a polyol having an alicyclic structure having 10 to 20 carbon atoms means hydrogen of at least one alcoholic hydroxyl group of the polyol having an alicyclic structure having 10 to 20 carbon atoms. It means the structure excluding.
- the present invention (I) is a (poly) carbonate polyol containing an organic residue derived from a dimer diol and an organic residue derived from a polyol having an alicyclic structure having 10 to 20 carbon atoms.
- the (poly) carbonate polyol of the present invention (I) contains an organic residue derived from a dimer diol and an organic residue derived from a polyol having an alicyclic structure having 10 to 20 carbon atoms. If so, there is no particular limitation.
- (poly) carbonate in the (poly) carbonate polyol described in this specification means that the molecule has one or more carbonate bonds. Therefore, “(poly) carbonate polyol” described in the present specification means a compound having one or more carbonate bonds and two or more alcoholic hydroxyl groups in the molecule. Examples of the (poly) carbonate polyol include (poly) carbonate diol having two hydroxyl groups in one molecule, (poly) carbonate triol having three hydroxyl groups in one molecule, and one hydroxyl group in one molecule. (Poly) carbonate tetraol having 4 in the above. (Poly) carbonate polyol is generally produced by polymerizing a raw material polyol via a carbonate bond.
- a polyhydric alcohol containing dimer diol and a polyol having an alicyclic structure having 10 to 20 carbon atoms is used as the raw material polyol.
- these polyhydric alcohols and carbonate esters such as dialkyl carbonates, diaryl carbonates or alkylene carbonates are used in the presence of a catalyst. It can be synthesized by transesterification.
- the obtained (poly) carbonate polyol may contain a raw material polyol component, but in this specification, the remaining polyol component is It is defined as not included in “(poly) carbonate polyol”.
- Examples of the polyol having an alicyclic structure having 10 to 20 carbon atoms include, for example, tricyclo [5.2.1.0 2,6 ] decandimethanol, tricyclo [3.3.1.1 3,7 ] decane.
- Tricyclodecane dimethanol such as 1,3-dimethanol, tricyclo [5.2.1.0 2,6 ] decanediol, tricyclo [3.3.1.1 3,7 ] decane-1,3- Examples include tricyclodecanediol such as diol, decahydronaphthalenediol, decahydronaphthalenedimethanol, 2,2-bis (4-hydroxycyclohexyl) propane, bis (4-hydroxycyclohexyl) methane, and bis (4-hydroxycyclohexyl). be able to.
- the primary hydroxyl group is more reactive than the secondary hydroxyl group, so among the plurality of alcoholic hydroxyl groups in the polyol having an alicyclic structure having 10 to 20 carbon atoms,
- the at least one alcoholic hydroxyl group is preferably a primary hydroxyl group.
- tricyclo [5.2.1.0 2,6 ] decanedimethanol tricyclo [3.3.1.1 3,7 ] decane.
- Tricyclodecane dimethanol such as 1,3-dimethanol and decahydronaphthalene dimethanol are preferred, and tricyclo [5.2.1.0 2,6 ] decandimethanol and tricyclo [3.3.1.1 3 , 7 ] decane-1,3-dimimethanol and the like are more preferable, and tricyclo [5.2.1.0 2,6 ] decane dimethanol is most preferable in view of availability.
- the method for producing the (poly) carbonate polyol of the present invention (I) includes a polyvalent carbonate containing a dimer diol and a polyol having an alicyclic structure having 10 to 20 carbon atoms in the presence of a transesterification reaction catalyst.
- a method for producing alcohol by transesterification with a carbonate such as dialkyl carbonate, diaryl carbonate or alkylene carbonate.
- a polyhydric alcohol containing a dimer diol and a polyol having an alicyclic structure having 10 to 20 carbon atoms and the carbonate ester in the presence of a transesterification catalyst at a temperature of 110 to 280 ° C. under normal pressure By transesterifying while distilling off alcohols or phenols by-produced, and further by transesterifying with distilling off by-produced alcohols or phenols at a temperature of 110 to 280 ° C. Can be manufactured.
- the (poly) carbonate polyol of the present invention (I) contains an organic residue derived from a dimer diol and an organic residue derived from a polyol having an alicyclic structure having 10 to 20 carbon atoms as described above. Furthermore, it may contain an organic residue derived from other polyols.
- polystyrene resin examples include ethylene glycol, diethylene glycol, propylene glycol, 1,3-propanediol, 1,4-butanediol, 1,3-butanediol, 1,5-pentanediol, and neopentyl glycol.
- 2 is preferable.
- -Methyl-1,8-octanediol, 1,9-nonanediol, 2-ethyl-2-butyl-1,3-propanediol, 2,4-diethyl-1,5-pentanediol, 1,10-decane A chain aliphatic polyol having 9 to 12 carbon atoms such as diol and 1,12-dodecanediol.
- the (poly) carbonate polyol of the present invention (I) is preferably a (poly) carbonate polyol represented by the following formula (34).
- each R 6 independently represents an alkylene group of the following groups (A), (B) or (C), t is an integer of 1 or more, and (t + 1) Among R 6 , at least one R 6 is an alkylene group represented by the following group (A), and among (t + 1) R 6 , at least one R 6 is represented by the following group (B): An alkylene group.) (A) group: alkylene group represented by the following formula (35) or (36) (B) group: alkylene group represented by the following formula (37) or (38) (C) group: chain having 9 to 12 carbon atoms Aliphatic alkylene group
- R 1 and R 2 are both alkyl groups, and the total number of carbon atoms contained in R 1 and R 2 and p and q is 30.
- R 3 and R 4 are both alkyl groups, and the total number of carbon atoms contained in R 3 and R 4 and r and s is 34.
- the “dimer diol” with respect to the sum of the total amount of organic residues derived from the polyol component in the (poly) carbonate polyol and the total amount of the component (d) is preferably 10 to 70% by mass, more preferably 15 to 65% by mass, and most preferably 20 to 60% by mass. It is.
- the “carbon number” relative to the sum of the total amount of organic residues derived from the polyol component and the total amount of component (d) in the (poly) carbonate polyol is preferably 10 to 50% by mass, and more preferably Is 13 to 45% by mass, and most preferably 15 to 40% by mass.
- the hydroxyl value of the (poly) carbonate polyol of the present invention (I) is not particularly limited. However, when the carboxyl group-containing polyurethane of the present invention (II) described later is produced, the hydroxyl value of the mixture of the (poly) carbonate polyol of the present invention (I) and the component (d) described later is 30 to 200. It is preferably from 40 to 180, more preferably from 45 to 160.
- the hydroxyl value of the mixture of the (poly) carbonate polyol of the present invention (I) and the component (d) refers to the (poly) carbonate polyol of the present invention (I) and the component (d). It means the value of the hydroxyl value determined by the neutralization titration method of JIS K0070 using a mixture mixed at the ratio used when producing the carboxyl group-containing polyurethane of the present invention (II) described later.
- the component (d) when the component (d) is not used in the production of the carboxyl group-containing polyurethane of the present invention (II), neutralization titration according to JIS K0070 using the (poly) carbonate polyol of the present invention (I).
- the value of the hydroxyl value determined by the method is defined as “the hydroxyl value of the mixture of the (poly) carbonate polyol of the present invention (I) and the component (d)”.
- the present invention (II) is a carboxyl group-containing polyurethane using at least the following components (a), (b) and (c) as raw materials.
- the (poly) carbonate polyol of the present invention (I) is as described above.
- the (poly) carbonate polyols may be used alone or in combination of two or more.
- the hydroxyl value of component (a), which is a raw material of the present invention (II), is not particularly limited, but the hydroxyl value of a mixture of component (a) and component (d) described later is 30 to It is preferably 200, more preferably 40 to 180, and particularly preferably 45 to 160.
- hydroxyl value of the mixture of component (a) and component (d) is the ratio used when producing the carboxyl group-containing polyurethane of the present invention (II), and component (a) It means the value of the hydroxyl value determined by the neutralization titration method of JIS K0070 using a mixture obtained by mixing the component and component (d).
- the component (d) is not used in the carboxyl group-containing polyurethane of the present invention (II)
- the hydroxyl value obtained by the neutralization titration method of JIS K0070 is used as the component (a).
- Component (b), which is a raw material of the present invention (II), is a polyisocyanate and is not particularly limited as long as it is a compound having two or more isocyanate groups.
- Specific examples of the component (b) as a raw material of the present invention (II) include, for example, 1,4-cyclohexane diisocyanate, isophorone diisocyanate, methylene bis (4-cyclohexyl isocyanate), 1,3-bis (isocyanatomethyl) cyclohexane.
- component (b) includes 1,4-cyclohexane diisocyanate, isophorone diisocyanate, methylene bis (4-cyclohexyl isocyanate), 1,3-bis (isocyanatomethyl) cyclohexane, 1, 4-bis (isocyanatomethyl) cyclohexane, diphenylmethane-4,4′-diisocyanate, 1,3-xylylene diisocyanate, 1,4-xylylene diisocyanate, 2,4,4-trimethylhexamethylene diisocyanate, 2,2, 4-trimethylhexanemethylene diisocyanate and norbornane diisocyanate are preferred, more preferably methylenebis (4-cyclohexylisocyanate), diphenylmethane-4,4′-diisocyanate and norbornene. Renan is a diisocyanate.
- Component (c) which is a raw material of the present invention (II) is a carboxyl group-containing polyol, such as dimethylolpropionic acid, 2,2-dimethylolbutanoic acid, N, N-bis (hydroxyethyl) glycine, N , N-bis (hydroxyethyl) glycine and the like.
- dimethylolpropionic acid and 2,2-dimethylolbutanoic acid are particularly preferred from the viewpoint of solubility in a solvent.
- These carboxyl group-containing polyols may be used alone or in combination of two or more.
- the raw material of the carboxyl group-containing polyurethane of the present invention (II) may be only the above three components of component (a), component (b) and component (c), but for the purpose of balancing the physical properties of the carboxyl group-containing polyurethane. It is preferable to use a polyol (also referred to as component (d)) other than component (a) and component (c) as a raw material. That is, the carboxyl group-containing polyurethane of the present invention (II) preferably uses at least component (a), component (b), component (c) and component (d) as raw materials.
- component (d) examples include dimer diol, trimer triol, ethylene glycol, diethylene glycol, propylene glycol, 1,3-propanediol, 1,4-butanediol, 1,3-butanediol, 1,5-pentanediol.
- Neopentyl glycol 3-methyl-1,5-pentanediol, 1,6-hexanediol, 2-methyl-1,8-octanediol, 1,9-nonanediol, 2-ethyl-2-butyl-1 , 3-propanediol, chain aliphatic polyols such as 2,4-diethyl-1,5-pentanediol, 1,4-cyclohexanedimethanol, 1,3-cyclohexanedimethanol, tricyclo [5.2.1.
- R 5 s each independently represent a divalent aliphatic or aromatic hydrocarbon group
- Xs each independently represent a residue obtained by removing a carboxyl group of a tetracarboxylic acid.
- m Y's each independently represent a residue obtained by removing the amino group of diamine, and m represents an integer of 0 to 20.
- a polyol component for example, dimer diol, having 10 to 20 carbon atoms
- the remaining polyol component is not included in the “(poly) carbonate polyol” but is included in the component (d).
- the carboxyl group-containing polyurethane of the present invention (II) can be synthesized using the above-mentioned component (a), component (b), component (c), and, if necessary, three or four components of component (d) as raw materials.
- a monohydroxy compound (component (e)) can be further reacted for the purpose of eliminating the influence of the terminal isocyanate group. That is, the carboxyl group-containing polyurethane of the present invention (II) may further use a monohydroxy compound (component (e)) as a raw material.
- a monoisocyanate compound (component (f)) is further added for the purpose of eliminating the influence of the terminal hydroxyl group. Can be reacted. That is, the carboxyl group-containing polyurethane of the present invention (II) may further use a monoisocyanate compound (component (f)) as a raw material.
- Component (e) includes methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, sec-butanol, t-butanol, ethylene glycol monoethyl ether, diethylene glycol monoethyl ether, diethylene glycol monoisopropyl ether, diethylene glycol mono Examples thereof include isobutyl ether and dipropylene glycol monopropyl ether.
- ethanol preferred among these are ethanol, n-propanol, isopropanol, n-butanol, isobutanol, ethylene glycol monoethyl ether, diethylene glycol monoethyl ether, diethylene glycol.
- ethanol preferred among these are ethanol, n-propanol, isopropanol, n-butanol, isobutanol, ethylene glycol monoethyl ether, diethylene glycol monoethyl ether, diethylene glycol.
- Monoisopropyl ether, diethylene glycol monoisobutyl ether and dipropylene glycol monopropyl ether preferred among these are ethanol, n-propanol, isopropanol, n-butanol, isobutanol, ethylene glycol monoethyl ether, diethylene glycol monoethyl ether, diethylene glycol.
- Monoisopropyl ether diethylene glycol
- These monohydroxy compounds may be used alone or in combination of two or more.
- cyclohexyl isocyanate, octadecyl isocyanate, phenyl isocyanate, toluyl isocyanate and the like can be used. Considering heat resistance, cyclohexyl isocyanate and octadecyl isocyanate are preferable.
- These monoisocyanate compounds may be used alone or in combination of two or more.
- the number average molecular weight of the carboxyl group-containing polyurethane of the present invention (II) is preferably 1,000 to 100,000, more preferably 3,000 to 50,000, particularly preferably 5,000. ⁇ 30,000.
- the “number average molecular weight of the carboxyl group-containing polyurethane” referred to here is a value in terms of polystyrene measured by gel permeation chromatography (hereinafter referred to as GPC). If the molecular weight is less than 1,000, the elongation, flexibility, and strength of the cured film, which will be described later, may be impaired. If the molecular weight exceeds 100,000, the solubility in a solvent becomes low, and the viscosity even if dissolved. Since it becomes too high, restrictions may be increased in terms of use.
- GPC measurement conditions are as follows.
- the acid value of the carboxyl group-containing polyurethane of the present invention is preferably 5 to 120 mgKOH / g, more preferably 10 to 50 mgKOH. / G.
- the acid value is less than 5 mgKOH / g
- the reactivity with other curable resins such as a curing agent described later may be reduced, and heat resistance may be impaired. If it exceeds 120 mgKOH / g, the cured film described later may be too hard and brittle.
- the carboxyl group-containing polyurethane of the present invention (II) is preferably a carboxyl group-containing polyurethane having a number average molecular weight of 1,000 to 100,000 and an acid value of 5 to 120 mgKOH / g, more preferably The average molecular weight is 3,000 to 50,000, and the acid value is 10 to 50 mgKOH / g.
- the acid value of the carboxyl group-containing polyurethane of the present invention is a value of an acid value measured by a potentiometric titration method of JIS K0070.
- the present invention (III) is a carboxyl group-containing polyurethane solution comprising the carboxyl group-containing polyurethane of the present invention (II) and a solvent having a boiling point of 120 to 300 ° C.
- the carboxyl group-containing polyurethane of the present invention (II), which is a constituent component of the carboxyl group-containing polyurethane solution of the present invention (III), is as described above.
- the solvent which is a constituent component of the carboxyl group-containing polyurethane solution of the present invention (III) has a boiling point of 120 to 300 ° C., and any solvent can be used as long as it can dissolve the carboxyl group-containing polyurethane of the present invention (II).
- any solvent can be used as long as it can dissolve the carboxyl group-containing polyurethane of the present invention (II).
- the solvent that is a constituent component of the carboxyl group-containing polyurethane solution of the present invention (III) is preferably a solvent having a boiling point of 150 to 250 ° C., more preferably a solvent having a boiling point of 170 to 230 ° C.
- the boiling point of the solvent means the boiling point at 1 atm unless otherwise specified.
- Examples of the solvent that is a constituent of the carboxyl group-containing polyurethane solution of the present invention (III) include anisole, diethylene glycol dimethyl ether, diethylene glycol diethyl ether, diethylene glycol ethyl methyl ether, diethylene glycol dibutyl ether, diethylene glycol butyl methyl ether, diethylene glycol isopropyl methyl ether, Ether solvents such as triethylene glycol dimethyl ether, triethylene glycol butyl methyl ether, tetraethylene glycol dimethyl ether, dipropylene glycol dimethyl ether, tripropylene glycol dimethyl ether, ethylene glycol monomethyl ether acetate, ethylene glycol monoethyl ether acetate , Propylene glycol monomethyl ether acetate, propylene glycol monoethyl ether acetate, dipropylene glycol monomethyl ether acetate, dipropylene glycol monoethyl ether acetate, di
- the carboxyl group-containing polyurethane of the present invention (II) When producing the carboxyl group-containing polyurethane of the present invention (II), the solubility of the carboxyl group-containing polyurethane of the present invention (II), the effect of suppressing bleeding during printing, the drying property of the solvent (that is, the boiling point of the solvent), and the carboxyl group-containing polyurethane of the present invention (II)
- preferred among these solvents is diethylene glycol diethyl Ether, diethylene glycol ethyl methyl ether, diethylene glycol butyl methyl ether, diethylene glycol isopropyl methyl ether, triethylene glycol dimethyl ether, dipropylene glycol dimethyl ether, tripropylene glycol dimethyl ether, Propylene glycol monomethyl ether acetate, dipropylene glycol monoethyl ether acetate
- a particularly preferable solvent is a solvent containing at least one selected from the group consisting of diethylene glycol diethyl ether and ⁇ -butyrolactone.
- the preferred solid content concentration of the carboxyl group-containing polyurethane solution of the present invention (III) is 10 to 90% by mass. More preferably, it is 15 to 70% by mass, and particularly preferably 20 to 60% by mass.
- the solution viscosity of the carboxyl group-containing polyurethane solution is, for example, 5,000 under the measurement conditions described in the examples. From 1 to 1 million mPa ⁇ s is preferable from the viewpoint of uniform dispersion.
- the present invention (IV) includes diethylene glycol dimethyl ether, diethylene glycol diethyl ether, diethylene glycol ethyl methyl ether, diethylene glycol dibutyl ether, diethylene glycol butyl methyl ether, diethylene glycol isopropyl methyl ether, triethylene glycol dimethyl ether, triethylene glycol butyl methyl ether, tetraethylene glycol dimethyl ether, Dipropylene glycol dimethyl ether, tripropylene glycol dimethyl ether, anisole, ethylene glycol monomethyl ether acetate, ethylene glycol monoethyl ether acetate, propylene glycol monomethyl ether acetate, propylene glycol monoethyl ether Acetate, dipropylene glycol monomethyl ether acetate, dipropylene glycol monoethyl ether acetate, diethylene glycol monoethyl ether acetate, diethylene glycol monomethyl ether acetate, methyl methoxy
- the component (d), the component (e), and the component (f) selected according to the above may be reacted together with the aforementioned component (a), component (b), and component (c).
- Component (d) polyol other than component (a) and component (c), Component (e): monohydroxyl compound, Component (f): monoisocyanate compound.
- the solvent used in the method for producing a carboxyl group-containing polyurethane of the present invention (IV) is diethylene glycol dimethyl ether, diethylene glycol diethyl ether, diethylene glycol ethyl methyl ether, diethylene glycol dibutyl ether, diethylene glycol butyl methyl ether, diethylene glycol isopropyl methyl ether, triethylene glycol dimethyl ether.
- diethylene glycol diethyl is preferable.
- Ether diethylene glycol ethyl methyl ether, diethylene glycol butyl methyl ether, diethylene glycol isopropyl methyl ether, triethylene glycol dimethyl ether, dipropylene glycol dimethyl ether, tripropylene glycol dimethyl ether, dipropylene glycol monomethyl ether acetate, dipropylene glycol monoethyl ether acetate, diethylene glycol Monoethyl ether acetate, diethylene glycol monomethyl ether acetate, Is a solvent containing at least one selected from the group consisting of pre- ⁇ - butyrolactone.
- a particularly preferable solvent is a solvent containing at least one selected from the group consisting of diethylene glycol diethyl ether and ⁇ -butyrolactone.
- the component (a) and the component (b) are prepared using the above solvent in the presence or absence of a known urethanization catalyst such as dibutyltin dilaurate. ), Component (c) and optionally selected component (d), component (e), and component (f) can be synthesized. Since the physical property value at the time of actual use as is improved, it is preferable.
- the order in which the raw materials are charged is not particularly limited. Usually, however, the component (a), the component (c) and, if necessary, the component (d) are charged first and dissolved in a solvent, and then 30 to 140 ° C. More preferably, component (b) is added dropwise at 60 to 120 ° C., and then reacted at 50 to 160 ° C., more preferably 60 to 150 ° C.
- the raw material charge molar ratio is adjusted according to the molecular weight and acid value of the target carboxyl group-containing polyurethane.
- the component (e) is used as a raw material for the carboxyl group-containing polyurethane, if the carboxyl group-containing polyurethane being produced by the above method reaches the target number average molecular weight (or if the target number average molecular weight is approached)
- the component (e) is added for the purpose of blocking the isocyanate group at the terminal of the carboxyl group-containing polyurethane being produced and suppressing the increase in the number average molecular weight.
- the number of isocyanate groups in component (b) may be less than, equal to or greater than the total number of hydroxyl groups in component (a), component (c) and component (d). But no problem.
- the component (e) when used in excess, the result is that the unreacted component (e) remains.
- the excess component (e) is used as part of the solvent. Alternatively, it may be removed by distillation or the like.
- the reason why the component (e) is used as a raw material for the carboxyl group-containing polyurethane is to suppress an increase in the molecular weight of the carboxyl group-containing polyurethane (that is, to stop the reaction), and the component (e) is added to the solution at 30 to 150 ° C. More preferably, the solution is added dropwise at 70 to 140 ° C., and then maintained at the same temperature to complete the reaction.
- the component (f) in order to use the component (f) as a raw material for the carboxyl group-containing polyurethane, the component (a), so that the terminal of the carboxyl group-containing polyurethane during production before using the component (f) in the reaction is a hydroxyl group. It is necessary to use it so that the number of isocyanate groups in component (b) is smaller than the number of total hydroxyl groups in component (c) and component (d). When the reaction between the total hydroxyl groups of component (a), component (c) and component (d) and the isocyanate group of component (b) is almost complete, it remains at the end of the carboxyl group-containing polyurethane being produced.
- the component (f) is dropped into the solution of the carboxyl group-containing polyurethane being produced at 30 to 150 ° C., more preferably 70 to 140 ° C., and then maintained at the same temperature. To complete the reaction.
- the blending amount of each component in the method for producing a carboxyl group-containing polyurethane is as follows.
- the amount of component (c) is preferably 1 to 32% by weight, more preferably 2 to 2% by weight based on the total amount of component (a) + component (b) + component (c) + component (d). 15% by mass.
- the ratio of the total number of hydroxyl groups in component (a) + component (b) + component (c) to the number of isocyanate groups in component (b) is 1: 0.9 to 0.9: 1. More preferably, it is 1: 0.92 to 0.95: 1.
- the amount of component (a) relative to the total amount of component (a) + component (d) is preferably 50% by mass or more, and more preferably 60% by mass or more.
- the present invention (V) is a curable composition comprising the carboxyl group-containing polyurethane of the present invention (II), a solvent having a boiling point of 120 to 300 ° C., and a curing agent.
- the carboxyl group-containing polyurethane of the present invention (II), which is a constituent component of the curable composition of the present invention (V), is as described above.
- the solvent having a boiling point of 120 to 300 ° C. which is a constituent component of the curable composition of the present invention (V)
- the concentration of the solvent in the curable resin composition of the present invention (V) is preferably 10 to 90% by mass, more preferably 20 to 70% by mass.
- a compound having two or more epoxy groups in one molecule is usually used.
- the compound having two or more epoxy groups in one molecule include phenol novolac type epoxy resin, orthocresol novolac type epoxy resin, phenol, cresol, xylenol, resorcin, catechol, phenols and / or ⁇ .
- -Epoxy novolak resins obtained by condensation or cocondensation of naphthols such as naphthol, ⁇ -naphthol, dihydroxynaphthalene and the like with compounds having an aldehyde group such as formaldehyde, acetaldehyde, propionaldehyde, benzaldehyde, salicylaldehyde in the presence of an acidic catalyst.
- naphthols such as naphthol, ⁇ -naphthol, dihydroxynaphthalene and the like
- aldehyde group such as formaldehyde, acetaldehyde, propionaldehyde, benzaldehyde, salicylaldehyde in the presence of an acidic catalyst.
- Novolac epoxy resin bisphenol A, bisphenol F, bisphenol S, alkyl-substituted or unsubstituted biphenol, stilbene phenols and other diglycidyl Ethers (bisphenol A type epoxy compounds, bisphenol F type epoxy compounds, bisphenol S type epoxy compounds, biphenyl type epoxy compounds, stilbene type epoxy compounds), glycidyl ethers of alcohols such as butanediol, polyethylene glycol, polypropylene glycol, phthalic acid, Glycidyl type such as glycidyl ester type epoxy resin of carboxylic acids such as isophthalic acid, tetrahydrophthalic acid, etc., substituted with active hydrogen bonded to nitrogen atom such as aniline, bis (4-aminophenyl) methane, isocyanuric acid, etc.
- bisphenol A type epoxy compounds bisphenol F type epoxy compounds, bisphenol S type epoxy compounds, biphenyl type epoxy compounds, stilbene type epoxy compounds
- the active hydrogen bonded to the nitrogen atom of aminophenols such as methylglycidyl type epoxy resin and p-aminophenol and the active hydrogen of the phenolic hydroxyl group are replaced with glycidyl groups.
- Glycidyl type or methyl glycidyl type epoxy resins vinylcyclohexene diepoxide obtained by epoxidizing olefin bonds in the molecule, 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate, 2- (3 , 4-epoxy) cyclohexyl-5,5-spiro (3,4-epoxy) cyclohexane-m-dioxane and other alicyclic epoxy resins, glycidyl ether of paraxylylene and / or metaxylylene modified phenolic resin, glycidyl of terpene modified phenolic resin Glycidyl ether of ether, di
- a compound having two or more epoxy groups in one molecule a compound having two or more epoxy groups in one molecule and having an aromatic ring structure and / or an alicyclic structure is preferable.
- dicyclopentadiene-modified phenolic resin of glycidyl ether i.e., tricyclo [5,2,1,0 2,6] have decane structure and aromatic ring structure and a compound having two or more epoxy groups
- 1, 3-bis (1-adamantyl) -4,6-bis (glycidylyl) benzene 1, 3-bis (1-adamantyl) -4,6-bis (glycidylyl) benzene, 1- [2 ′, 4′-bis (glycidylyl) phenyl] adamantane, 1,3-bis (4′-glycidylylphenyl) Epoxy resins having an adamantane structure such as adamantane and 1,3-bis [2 ′, 4′-bis (glycidylyl) phenyl] adamantane (ie, tricycl
- Amino group and aromatic ring structure such as glycidyl type or methyl glycidyl type epoxy resin, and two or more epoxy groups
- the compound which has this is preferable, Especially preferably, it is a compound as described in following formula (5).
- a compound having two or more epoxy groups in one molecule may be used alone or in combination of two or more.
- the blending amount of the curing agent with respect to 100 parts by mass of the carboxyl group-containing polyurethane of the present invention (II) cannot be generally described because it differs depending on the acid value of the carboxyl group-containing polyurethane of the present invention (II).
- the ratio of the number of carboxyl groups contained in the carboxyl group-containing polyurethane of the present invention (II) to the number of epoxy groups in the compound having two or more epoxy groups in one molecule is 1/3 to 2 / It is preferably in the range of 1, more preferably in the range of 1 / 2.5 to 1.5 / 1.
- the ratio of the number of carboxyl groups contained in the carboxyl group-containing polyurethane of the present invention (II) to the number of epoxy groups in the compound having two or more epoxy groups in one molecule is less than 1/3, there is no reaction. It is not preferable that a large number of compounds having two or more epoxy groups remain in one molecule. On the other hand, if this ratio is larger than 2/1, many unreacted carboxyl groups remain in the carboxyl group-containing polyurethane, which is not preferable in terms of electrical insulation performance.
- the curable composition of the present invention (V) can further contain a curing accelerator.
- the curing accelerator is not particularly limited as long as it is a compound that accelerates the reaction between an epoxy group and a carboxyl group.
- melamine, acetoguanamine, benzoguanamine, 2,4-diamino-6-methacryloyloxyethyl-S Triazines such as triazine, 2,4-methacryloyloxyethyl-s-triazine, 2,4-diamino-6-vinyl-s-triazine, 2,4-diamino-6-vinyl-s-triazine and isocyanuric acid adducts
- imidazole 2-methylimidazole, 2-ethyl-4-methylimidazole, 2-phenylimidazole, 2-undecylimidazole, 2-heptadecylimidazole, 1-benzyl-2-methyl
- These curing accelerators may be used alone or in combination of two or more.
- melamine in view of making the curing acceleration action and electrical insulation performance compatible, melamine, imidazole compound, cycloamidine compound, derivatives of cycloamidine compound, phosphine compound and amine system are preferable. More preferred are melamine, 1,5-diazabicyclo (4.3.0) nonene-5 and salts thereof, and 1,8-diazabicyclo (5.4.0) undecene-7 and salts thereof.
- the blending amount of these curing accelerators is not particularly limited as long as the curing acceleration effect can be achieved.
- the present invention (V ) In the range of 0.05 to 5 parts by mass, more preferably 0.1 to 3.0 parts, relative to 100 parts by mass of the total amount of the carboxyl group-containing polyurethane and the curing agent in the curable composition. Part by mass.
- the blending amount is less than 0.05 parts by mass, it is difficult to cure in a short time, and when it exceeds 5 parts by mass, the electrical insulation characteristics and water resistance of the cured product obtained by curing the composition are deteriorated. There is.
- inorganic fine particles and / or organic fine particles can be blended and preferably blended for the purpose of adjusting fluidity.
- inorganic fine particles and / or organic fine particles means not only inorganic fine particles and organic fine particles, but also a powdery inorganic compound physically coated with an organic compound or chemically surfaced with an organic compound. It is defined to include organic / inorganic composite-based fine particles that have been treated.
- Inorganic fine particles and / or organic fine particles that may be used for the purpose of blending with the curable composition of the present invention (V) include the carboxyl group-containing polyurethane of the present invention (II) and the carboxyl group-containing composition of the present invention (III).
- the paste is formed by dispersing in a polyurethane solution, a curing agent or a curing agent solution.
- inorganic fine particles examples include silica (SiO 2 ), alumina (Al 2 O 3 ), titania (TiO 2 ), tantalum oxide (Ta 2 O 5 ), zirconia (ZrO 2 ), silicon nitride (Si 3 ).
- Such organic fine particles are preferably heat-resistant resin fine particles having an amide bond, an imide bond, an ester bond or an ether bond.
- the resin is preferably a polyimide resin or a precursor thereof, a polyamideimide resin or a precursor thereof, or a polyamide resin from the viewpoint of heat resistance and mechanical properties.
- the average particle diameter of these inorganic fine particles and / or organic fine particles is preferably 0.01 to 10 ⁇ m, more preferably 0.1 to 5 ⁇ m.
- the blending amount of the inorganic fine particles and / or organic fine particles is 1 to 150 parts by weight, preferably 1 with respect to 100 parts by weight of the total amount of the carboxyl group-containing polyurethane, solvent and curing agent contained in the curable resin composition. Is 120 parts by mass, more preferably 1-60 parts by mass.
- the curable composition of this invention (V) is a curable composition from which the hardened
- the curable composition of the present invention (V) is used as a composition for a solder resist (that is, a solder resist ink composition), for the purpose of eliminating or suppressing the generation of bubbles during printing, Agents can and are preferably used.
- the above defoaming agent is not particularly limited as long as it literally has an action of eliminating or suppressing bubbles generated when the solder resist ink composition is printed.
- antifoaming agent used in the curable composition of the present invention (V) include, for example, BYK-077 (manufactured by Big Chemie Japan), SN deformer 470 (manufactured by San Nopco), and TSA750S (momentive performance). ⁇ Materials Co., Ltd.), Silicone Antifoaming Agents such as Silicone Oil SH-203 (Toray Dow Corning Co.), Dappo SN-348 (San Nopco), Dappo SN-354 (San Nopco), Dappo SN -368 (manufactured by Sannopco), acrylic polymer antifoaming agents such as Disparon 230HF (manufactured by Enomoto Kasei), Surfynol DF-110D (manufactured by Nisshin Chemical Industry), Surfynol DF-37 (Nisshin Chemical Industries, Ltd.) Acetylene diol-based antifoaming agents such as FA-630 and
- curable composition of the present invention (V)
- surfactants such as a leveling agent, phthalocyanine blue, phthalocyanine green, iodin green, disazo yellow, crystal violet, carbon black, A known colorant such as naphthalene black can be added.
- the curable composition of the present invention (V) is added with a phenolic antioxidant, a phosphite antioxidant, a thioether oxidation.
- Antioxidants such as inhibitors can be added and are preferably added.
- phenolic antioxidants include compounds represented by the following formulas (6) to (16).
- n is an integer of 1 to 5.
- Examples of the phosphite antioxidant include compounds represented by the following formulas (17) to (27).
- Examples of the thioether-based antioxidant include compounds represented by the following formulas (28) to (33).
- flame retardants and lubricants can be added as necessary.
- the curable composition of the present invention (V) can be obtained by uniformly kneading and mixing part or all of the blending components with a roll mill, a bead mill or the like. When a part of the blending components is mixed, the remaining components can be mixed when actually used.
- the curable composition of this invention (V) when using the curable composition of this invention (V) as a soldering resist ink composition, in order to improve the printability of the curable composition of this invention (V), it has a thixotropy index of a certain range. It is desirable.
- the “thixotropy index” described in this specification is a cone / plate viscometer (Brookfield model: DV-II + Pro spindle model number: CPE-52) measured at 1 rpm at 25 ° C. It is defined as the ratio of the viscosity at the time of 10 to the viscosity at a rotation speed of 10 rpm at 25 ° C.
- the thixotropy index of the composition at 25 ° C. is preferably in the range of 1.1 to 3.0, more preferably in the range of 1.1 to 2.5.
- the curable composition of the present invention (V) is used as a solder resist ink composition, if the thixotropy index at 25 ° C. of the curable composition is less than 1.1, after printing the curable composition, The composition may flow and may not have a constant film thickness or may not maintain a printed pattern.
- the thixotropy index at 25 ° C. of the curable composition is larger than 3.0, the defoaming property of the coating film of the printed composition may be deteriorated.
- the present invention (VI) relates to a cured product obtained by curing the curable resin composition of the present invention (V).
- the cured product of the present invention (VI) is generally obtained by removing a part or all of the solvent in the curable composition of the present invention (V), and then proceeding with a curing reaction by heating to obtain a cured product. is there.
- a coating film comprising the cured product can be obtained through the following first to third steps.
- First step A step of printing the curable composition of the present invention (V) to obtain a coating film.
- Second step A step of obtaining a coating film from which a part or all of the solvent has been removed by evaporating the solvent from the coating film obtained in the first step in an atmosphere of 20 ° C. to 100 ° C.
- Third step A step of thermally curing the coating film obtained in the second step in an atmosphere of 100 ° C. to 250 ° C. to obtain a thermally cured coating film (that is, a coating film made of a cured product).
- the first step is a step of printing the curable composition of the present invention (V) to obtain a coating film, but there is no particular limitation on the printing method of the curable composition of the present invention (V), for example, a screen A coating film can be obtained by coating by a printing method, a roll coater method, a spray method, a curtain coater method, or the like.
- the second step is a step of obtaining a coating film from which a part or all of the solvent has been removed by evaporating the solvent from the coating film obtained in the first step in an atmosphere of 20 ° C. to 100 ° C.
- the time for removing the solvent is preferably 4 hours or less, more preferably 2 hours or less.
- the coating film obtained in the second step is thermally cured in an atmosphere of 100 ° C. to 250 ° C. to obtain a thermally cured coating film (that is, a cured coating film). It is.
- the heat curing time is preferably in the range of 20 minutes to 4 hours, more preferably in the range of 30 minutes to 2 hours.
- the present invention (VII) is characterized in that a part of or the entire surface of the flexible wiring board in which the wiring is formed on the flexible substrate is covered with the cured product of the present invention (VI). And a flexible wiring board covered with a cured product.
- a printed film is formed on the pattern by printing the curable composition of the present invention (V) on a tin-plated wiring pattern portion of a flexible wiring board.
- the curable composition of the present invention (V) can be used, for example, as a solder resist ink, and the cured product of the present invention (VI) can be used as an insulating protective film.
- the cured product of the present invention (VI) can be used as a solder resist by covering the whole or part of the wiring of a flexible wiring board such as a chip-on film.
- a protective film for a flexible wiring board can be formed through the following steps A to C.
- Step A A step of obtaining a coating film by screen-printing the curable composition of the present invention (V) on the wiring pattern portion of the flexible wiring board that has been previously tin-plated.
- the coating film obtained in this step is called a printed film.
- Step B A step of obtaining a coating film from which a part or all of the solvent has been removed by evaporating the solvent from the coating film obtained in Step A in an atmosphere of 20 to 100 ° C.
- Process C A process of obtaining a protective film for a flexible printed circuit board by thermally curing the coating film obtained in Process B in an atmosphere of 80 to 130 ° C.
- the temperature for evaporating the solvent in Step B is 20 to 100 ° C., preferably 60 to 100 ° C., more preferably, considering the evaporation rate of the solvent and the rapid transition to the next step (Step C). 70 to 90 ° C.
- the time for evaporating the solvent in Step B is not particularly limited, but is preferably 10 to 120 minutes, and more preferably 20 to 100 minutes.
- the operation of the step B is an operation performed as necessary.
- the operation of the step C may be performed immediately after the operation of the step A, and the curing reaction and the removal of the solvent may be performed together.
- the conditions of thermosetting performed in the step C are in the range of 80 to 130 ° C. from the viewpoint of preventing the plating layer from being diffused and obtaining warpage and flexibility suitable as a protective film.
- the temperature is preferably 90 to 130 ° C, and 110 to 130 ° C.
- the time for thermosetting performed in Step C is not particularly limited, but is preferably 20 to 150 minutes, and more preferably 30 to 120 minutes.
- the acid value was measured according to the potentiometric titration method of JIS K0070.
- thixotropy index of the curable composition was measured by the following method.
- the thixotropy index was rotated at a temperature of 25.0 ° C. using a cone / plate viscometer (Brookfield model: DV-II + Pro spindle model number: CPE-52). The viscosity after 7 minutes from the start of measurement under the condition of several tens rpm was measured. Thereafter, the viscosity after 7 minutes from the start of measurement was measured under the conditions of a temperature of 25.0 ° C. and a rotation speed of 1 rpm.
- the thixotropy index was calculated by the following method.
- Thixotropic index [viscosity at 1 rpm] ⁇ [viscosity at 10 rpm] ⁇ Synthesis of (poly) carbonate polyol containing an organic residue derived from dimer diol and an organic residue derived from a polyol having an alicyclic structure having 10 to 20 carbon atoms>
- Example 1 In a 1000 ml four-necked round bottom flask equipped with a stirrer, thermometer and rectifying column, a mixture of 2,4-diethyl-1,5-pentanediol and 2-ethyl-2-butyl-1,3-propanediol ( Kyowa Hakko Chemical Co., Ltd.
- the hydroxyl value of product A1 was 123 mgKOH / g.
- Example 2 To a 500 ml four-necked round bottom flask equipped with a stirrer, a thermometer and a rectifying column, 117.8 g (0.600 mol) of tricyclo [5.2.1.0 2,6 ] decandimethanol (manufactured by Tokyo Chemical Industry Co., Ltd.) ), PRIPOL 2033 (98.2% by weight of dimer diol, 0.6% by weight of monool, 1.2% by weight of trimer triol, hydroxyl value: 205 mg KOH / g), 162.6 g, diethyl carbonate (manufactured by Wako Pure Chemical Industries, Ltd.) 70 .878 g (0.600 mol) and 1.405 g (4.1 mmol) of tetra n-butyl titanate (Mitsubishi Gas Chemical Co., Ltd.) were added, the nitrogen stream was maintained for 30 minutes, and then the supply of nitrogen was stopped and an oil bath at 150 ° C.
- PRIPOL 2033
- the hydroxyl value of product A2 was 123 mgKOH / g.
- Example 3 In a 500 ml four-necked round bottom flask equipped with a stirrer, a thermometer and a rectifying column, a mixture of 2,4-diethyl-1,5-pentanediol and 2-ethyl-2-butyl-1,3-propanediol ( Kyowa Hakko Chemical Co., Ltd. product name: PD-9) 68.6 g (0.426 mol), tricyclo [5.2.1.0 2,6 ] decandimethanol (Tokyo Chemical Industry Co., Ltd.) 68.6 g (0.
- PRIPOL 2033 (98.2% by weight of dimer diol, 0.6% by weight of monool, 1.2% by weight of trimer, hydroxyl value; 205 mg KOH / g), 68.6 g, diethyl carbonate (manufactured by Wako Pure Chemical Industries, Ltd.) 81.8 g (0.693 mol) and tetra n-butyl titanate (Mitsubishi Gas Chemical Co., Ltd.) 0.823 g (2.42 mmol) were added, and nitrogen was added for 30 minutes. Maintaining the flow, then nitrogen feed was stopped and heated set in an oil bath 0.99 ° C..
- the product A3 was analyzed by gas chromatography. As a result, 2,4-diethyl-1,5-pentanediol, 2-ethyl-2-butyl-1,3-propanediol and tricyclo [5.2.1.0 2 , 6 ] decanedimethanol was confirmed to remain in 4.2% by mass, 0.3% by mass, and 4.5% by mass, respectively, in the product A3. As a result of analyzing the product A3 by liquid chromatography, it was confirmed that 5.0% by mass of dimer diol remained in the product A3, and that the monool and trimer triol were below the detection limit.
- the hydroxyl value of product A3 was 110 mgKOH / g.
- Example 4 A mixture of 2-methyl-1,8-octanediol and 1,9-nonanediol (trade name: ND-15, manufactured by Kuraray Co., Ltd.) was added to a 1000 ml four-necked round bottom flask equipped with a stirrer, thermometer and rectifying column.
- the hydroxyl value of product A5 was 116 mgKOH / g.
- the temperature of the reaction solution was lowered to 90 ° C., and 139.0 g of methylenebis (4-cyclohexylisocyanate) (trade name: Desmodur-W, manufactured by Sumika Bayer Urethane Co., Ltd.) was added dropwise over 30 minutes with a dropping funnel. After reacting at 120 ° C. for 6 hours and confirming that the isocyanate almost disappeared, 4.0 g of isobutanol (manufactured by Wako Pure Chemical Industries, Ltd.) was added dropwise, and further reacted at 120 ° C. for 3 hours to contain a carboxyl group.
- methylenebis (4-cyclohexylisocyanate) trade name: Desmodur-W, manufactured by Sumika Bayer Urethane Co., Ltd.
- a polyurethane solution (hereinafter referred to as “carboxyl group-containing polyurethane solution B1”) was obtained.
- the viscosity of the obtained carboxyl group-containing polyurethane solution B1 was 11500 mPa ⁇ s.
- the number average molecular weight of the carboxyl group-containing polyurethane contained in the carboxyl group-containing polyurethane solution B1 (hereinafter referred to as “carboxyl group-containing polyurethane BU1”) is 14,000, and the acid value of the carboxyl group-containing polyurethane BU1 is 30. It was 0 mgKOH / g.
- the solid content concentration in the carboxyl group-containing polyurethane solution B1 was 45.0% by mass.
- Example 6 In a reaction vessel equipped with a stirrer, a thermometer and a condenser, 204.5 g of the product A2 obtained in Example 2, 2,4-diethyl-1,5-pentanediol and 2-ethyl-2-butyl- Mixture of 1,3-propanediol (trade name: PD-9, manufactured by Kyowa Hakko Chemical Co., Ltd.) 33.1 g, 2,2-dimethylolbutanoic acid (manufactured by Nippon Kasei Co., Ltd.) as the carboxyl group-containing diol, 35.7 g as the solvent 250.0 g of ⁇ -butyrolactone (manufactured by Mitsubishi Chemical Corporation) and 250.0 g of diethylene glycol diethyl ether (manufactured by Nippon Emulsifier Co., Ltd.) were charged and heated to 100 ° C.
- PD-9 manufactured by Kyowa Hakko Chemical Co., Ltd.
- the viscosity of the obtained carboxyl group-containing polyurethane solution B2 was 301000 mPa ⁇ s.
- the number average molecular weight of the carboxyl group-containing polyurethane (hereinafter referred to as “carboxyl group-containing polyurethane BU2”) contained in the carboxyl group-containing polyurethane solution B2 is 14,000, and the acid value of the carboxyl group-containing polyurethane BU2 is 30. It was 0 mgKOH / g.
- the solid content concentration in the carboxyl group-containing polyurethane solution B2 was 45.0% by mass.
- Example 7 In a reaction vessel equipped with a stirrer, a thermometer and a condenser, 277.7 g of the product A3 obtained in Example 3 and 2,2-dimethylolbutanoic acid (manufactured by Nippon Kasei Co., Ltd.) as a carboxyl group-containing diol 35. 7 g, 330.0 g of ⁇ -butyrolactone (manufactured by Mitsubishi Chemical Corporation) and 22.0 g of diethylene glycol diethyl ether (manufactured by Nippon Emulsifier Co., Ltd.) were charged as a solvent, and heated to 100 ° C. to dissolve all raw materials.
- 2,2-dimethylolbutanoic acid manufactured by Nippon Kasei Co., Ltd.
- ⁇ -butyrolactone manufactured by Mitsubishi Chemical Corporation
- diethylene glycol diethyl ether manufactured by Nippon Emulsifier Co., Ltd.
- the viscosity of the obtained carboxyl group-containing polyurethane solution B3 was 12000 mPa ⁇ s.
- the number average molecular weight of the carboxyl group-containing polyurethane contained in the carboxyl group-containing polyurethane solution B3 (hereinafter referred to as “carboxyl group-containing polyurethane BU3”) is 14,000, and the acid value of the carboxyl group-containing polyurethane BU3 is 30. It was 0 mgKOH / g.
- the solid content concentration in the carboxyl group-containing polyurethane solution B3 was 45.0% by mass.
- Example 8 In a reaction vessel equipped with a stirrer, a thermometer and a condenser, 274.6 g of the product A5 obtained in Example 4 and 2,2-dimethylolbutanoic acid (manufactured by Nippon Kasei Co., Ltd.) as a carboxyl group-containing diol 35. 7 g, 330.0 g of ⁇ -butyrolactone (manufactured by Mitsubishi Chemical Corporation) and 22.0 g of diethylene glycol diethyl ether (manufactured by Nippon Emulsifier Co., Ltd.) were charged as a solvent and heated to 100 ° C. to dissolve all the raw materials.
- 2,2-dimethylolbutanoic acid manufactured by Nippon Kasei Co., Ltd.
- reaction solution B4 The temperature of the reaction solution was lowered to 90 ° C., and 135.7 g of methylene bis (4-cyclohexylisocyanate) (trade name; Desmodur-W, manufactured by Sumika Bayer Urethane Co., Ltd.) was added dropwise over 30 minutes with a dropping funnel. After reacting at 120 ° C. for 6 hours and confirming that the isocyanate almost disappeared, 4.0 g of isobutanol (manufactured by Wako Pure Chemical Industries, Ltd.) was added dropwise, and further reacted at 120 ° C. for 3 hours to contain a carboxyl group. A polyurethane solution (hereinafter referred to as “carboxyl group-containing polyurethane solution B4”) was obtained.
- the viscosity of the obtained carboxyl group-containing polyurethane solution B4 was 12000 mPa ⁇ s.
- the number average molecular weight of the carboxyl group-containing polyurethane contained in the carboxyl group-containing polyurethane solution B4 (hereinafter referred to as “carboxyl group-containing polyurethane BU4”) is 14,000, and the acid value of the carboxyl group-containing polyurethane BU4 is 30. It was 0 mgKOH / g.
- the solid content concentration in the carboxyl group-containing polyurethane solution B4 was 45.0% by mass.
- the number average molecular weight of the carboxyl group-containing polyurethane (hereinafter referred to as “carboxyl group-containing polyurethane CU1”) contained in the obtained carboxyl group-containing polyurethane solution C1 is 6800, and the acid value of the carboxyl group-containing polyurethane CU1 is 39. 0.9 mg KOH / g.
- the solid content concentration in the carboxyl group-containing polyurethane solution C1 was 49.5% by mass.
- the temperature of the reaction solution was lowered to 70 ° C., and 437.3 g of methylene bis (4-cyclohexylisocyanate) (trade name; Desmodur-W, manufactured by Sumika Bayer Urethane Co., Ltd.) was dropped as a polyisocyanate over 30 minutes with a dropping funnel. After completion of dropping, the reaction was carried out at 80 ° C. for 1 hour, then at 100 ° C. for 1 hour, and then at 120 ° C. for 2 hours. After confirming that the isocyanate almost disappeared, isobutanol (manufactured by Wako Pure Chemical Industries, Ltd.) 5 g was added dropwise, and the reaction was further carried out at 120 ° C. for 1.5 hours to obtain a carboxyl group-containing polyurethane solution (hereinafter referred to as “carboxyl group-containing polyurethane solution C2”).
- carboxyl group-containing polyurethane solution C2 hereinafter referred to as “carboxyl group-
- the number average molecular weight of the carboxyl group-containing polyurethane (hereinafter referred to as “carboxyl group-containing polyurethane CU2”) contained in the obtained carboxyl group-containing polyurethane solution C2 is 13800, and the acid value of the carboxyl group-containing polyurethane CU2 is 40. 0.2 mg KOH / g.
- the solid content concentration in the carboxyl group-containing polyurethane solution C2 was 50.1% by mass.
- Example formulation examples 2 to 4 and comparative formulation examples 1 to 2 According to the same method as in Example 1 of blending, blending was performed according to the blending composition shown in Table 1.
- the formulations prepared in Examples 2 to 4 were referred to as main agent formulations D2 to D4, respectively, and the formulations prepared in comparative formulation example 1 and comparative formulation example 2 were respectively referred to as main agent formulation E1 and main agent formulation E2. did.
- an epoxy resin having a structure of the following formula (5) (Japan Epoxy Resin grade name; jER604, epoxy equivalent 120 g / eq) 300 g, ⁇ -butyrolactone (Mitsubishi Chemical Corp.) 180 g and 120 g of diethylene glycol diethyl ether (manufactured by Toho Chemical Industry Co., Ltd.) was added and stirring was started. Stirring was continued for 1 hour. Thereafter, it was confirmed that jER604 was completely dissolved, and a jER604-containing solution having a concentration of 50% by mass was obtained. This solution is used as a curing agent solution F2.
- a solution obtained by mixing F1 and F2 at a ratio of 1: 1 (mass ratio) is referred to as a curing agent solution F3.
- Curable composition H1 a curable composition
- curable composition G1 is applied to a film [Kapton (registered trademark) 300H, manufactured by Toray DuPont Co., Ltd.] by screen printing so that the thickness of the curable composition is 15 ⁇ m (thickness after drying). It was cured by placing it in a hot air circulation dryer at 80 ° C. for 30 minutes and then placing it in a 120 ° C. hot air circulation dryer for 120 minutes.
- the peeling tape was manufactured by Nitto Denko Corporation and evaluated according to the following criteria.
- ⁇ When 80 or more grids remain, ⁇ : When the number of grids is 50 or more and less than 80, ⁇ : When the number of grids remains less than 50.
- Warpage Curing Composition G1 was applied to a substrate by screen printing, placed in a hot air circulating dryer at 80 ° C. for 30 minutes, and then cured by placing in a hot air circulating dryer at 120 ° C. for 60 minutes. It was. A 38 ⁇ m-thick polyimide film (Kapton (registered trademark) 150EN, manufactured by Toray DuPont) was used as the substrate.
- the curable composition was applied, and the coating film cured using a dryer was cut to 50 mm ⁇ with a circle cutter. What is cut into a circle exhibits a deformation in which the vicinity of the center warps in a convex or concave shape.
- the test piece After leaving the test piece at a temperature of 23 ⁇ 0.5 ° C. and a humidity of 60 ⁇ 5% RH for 12 hours or more, the test piece is left in a convex state, and the portion that is most warped from the plane, The height of the warp from the plane was measured using a length meter and averaged at two locations that were symmetric with respect to the center of.
- the sign represents the direction of warping, and when left in a downwardly convex state, “+” indicates that the cured film is on the upper side of the polyimide film, and “ ⁇ ” indicates that the cured film is on the lower side.
- the curable composition of the present invention (V) containing the carboxyl group-containing polyurethane, solvent and curing agent of the present invention (II) using the (poly) carbonate diol of the present invention (I) as a raw material Is a composition that can exhibit long-term electrical insulation characteristics at a high level by being cured, and can provide a cured product obtained by curing the composition.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Materials Engineering (AREA)
- Wood Science & Technology (AREA)
- Polyurethanes Or Polyureas (AREA)
- Non-Metallic Protective Coatings For Printed Circuits (AREA)
- Polyesters Or Polycarbonates (AREA)
Abstract
Description
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
本発明(III)は、本発明(II)のカルボキシル基含有ポリウレタンと沸点が120~300℃である溶媒とを含有することを特徴とするカルボキシル基含有ポリウレタン溶液に関する。
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
本発明(V)は、本発明(II)のカルボキシル基含有ポリウレタン、沸点が120~300℃である溶媒および硬化剤を含む硬化性組成物に関する。
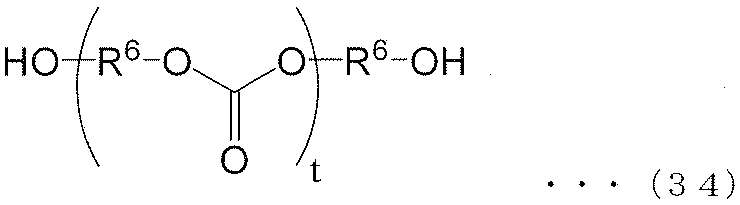
(A)群:下記式(35)または(36)で表わされるアルキレン基
(B)群:下記式(37)または(38)で表わされるアルキレン基
(C)群:炭素数9~12の鎖状脂肪族アルキレン基

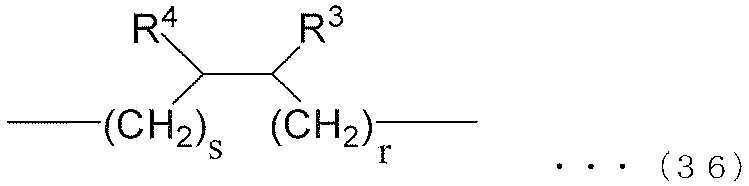

成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
[5] 少なくとも以下の成分(a)、成分(b)、成分(c)および成分(d)を原料とするカルボキシル基含有ポリウレタン。
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
成分(d):成分(a)および成分(c)以外のポリオ-ル。
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
[10] [4]~[7]のいずれかに記載のカルボキシル基含有ポリウレタン、沸点が120~300℃である溶媒および硬化剤を含む硬化性組成物。
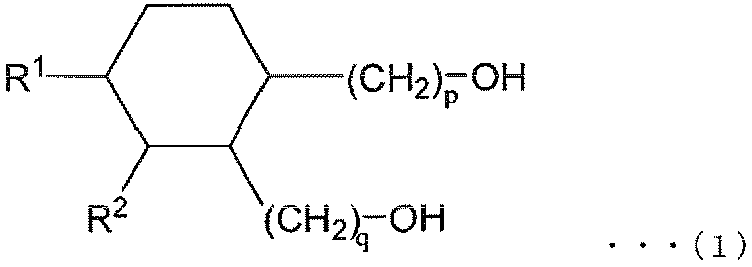

なお、本発明における「ダイマージオールから誘導された有機残基」とは、前記ダイマージオ-ルの少なくとも1つのアルコール性水酸基の水素を除いた構造を意味する。
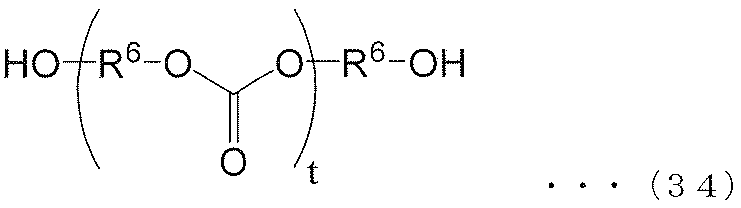
(A)群:下記式(35)または(36)で表わされるアルキレン基
(B)群:下記式(37)または(38)で表わされるアルキレン基
(C)群:炭素数9~12の鎖状脂肪族アルキレン基


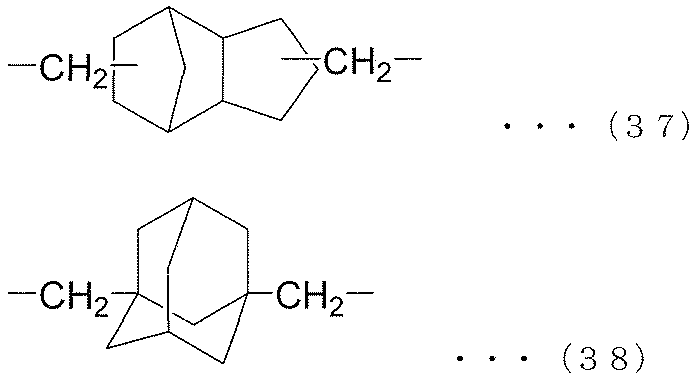
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
本発明(II)の原料の1つである成分(a)は、本発明(I)の(ポリ)カーボネートポリオールであり、前述の通りである。前記(ポリ)カーボネートポリオールは単独で用いても、2種以上を組み合わせて用いても良い。

これらの中で好ましいものとしては、2-メチル-1,8-オクタンジオール、1,9-ノナンジオール、2-エチル-2-ブチル-1,3-プロパンジオール、2,4-ジエチル-1,5-ペンタンジオール等の炭素数9以上の鎖状脂肪族ポリオール、1,4-シクロヘキサンジメタノール、1,3-シクロヘキサンジメタノール、トリシクロ[5,2,1,02,6]デカンジメタノール、2-メチルシクロヘキサン-1,1-ジメタノール等の脂環構造を有するポリオールおよびダイマージオールである。これらのポリオ-ルを単独で用いても、2種以上を併用しても良い。
カラム:ShodexカラムLF-804を3本連結(直列)
移動相:テトラヒドロフラン
流速:1.0mL/min
検出器:日本分光(株)製 RI-2031Plus
温度:40.0℃
試料量:サンプルループ 100μリットル
試料濃度:0.1質量%前後に調製
本発明(II)のカルボキシル基含有ポリウレタンの酸価は、5~120mgKOH/gであると好ましく、さらに好ましくは、10~50mgKOH/gである。酸価が5mgKOH/g未満では、後述の硬化剤等の他の硬化性樹脂との反応性が低下し耐熱性を損ねることがある。120mgKOH/gを超えると後述する硬化膜が硬く脆くなりすぎることがある。
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
本発明(IV)のカルボキシル基含有ポリウレタンの製造方法においては、さらに必要に応じて選ばれる成分(d)、成分(e)、成分(f)を、前述の成分(a)、成分(b)および成分(c)と共に反応させてもよい。
成分(e):モノヒドロキシル化合物、
成分(f):モノイソシアネート化合物。

一方、本発明(II)のカルボキシル基含有ポリウレタンとの反応性を重視する場合には、1分子中に2個以上のエポキシ基を有しかつ芳香環構造および/または脂環構造を有する化合物の中で、アニリン、ビス(4-アミノフェニル)メタンの窒素原子に結合した活性水素をグリシジル基で置換したもの等のグリシジル型またはメチルグリシジル型のエポキシ樹脂、p-アミノフェノール等のアミノフェノール類の窒素原子に結合した活性水素およびフェノール性水酸基の活性水素をグリシジル基で置換したもの等のグリシジル型またはメチルグリシジル型のエポキシ樹脂等のアミノ基および芳香環構造を有しかつ2個以上のエポキシ基を有する化合物が好ましく、特に好ましくは、下記式(5)に記載の化合物である。


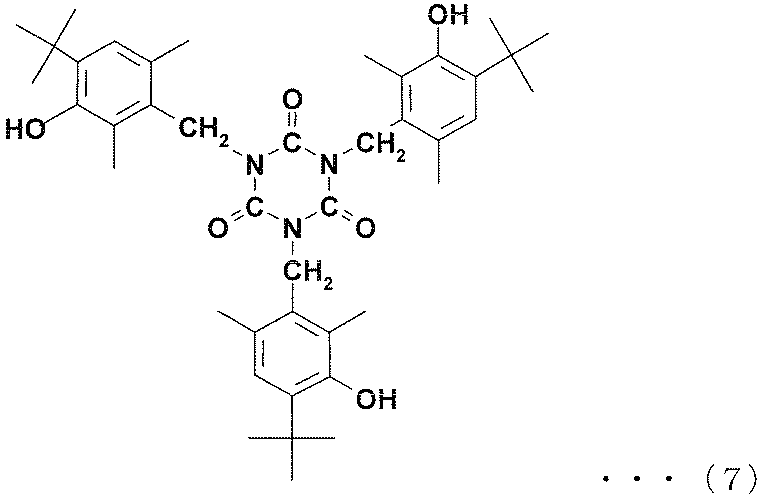




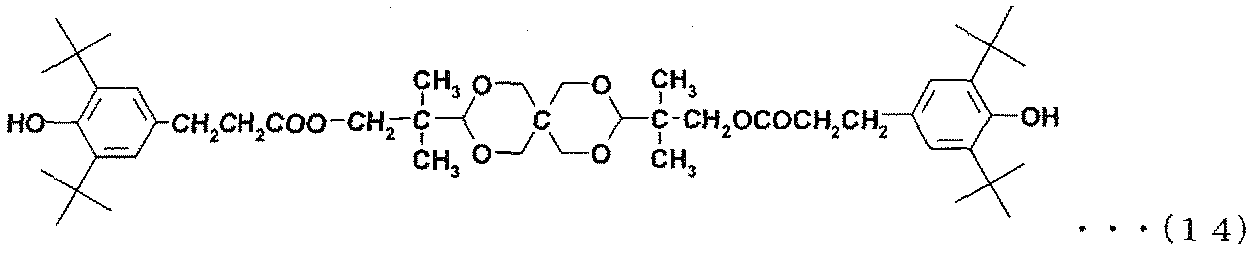


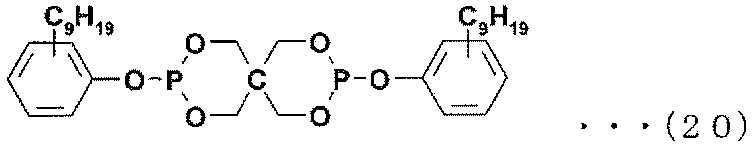
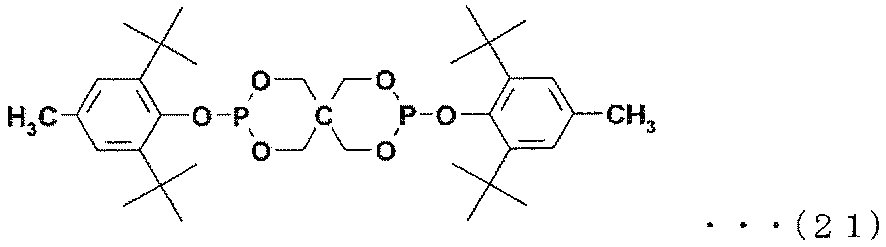




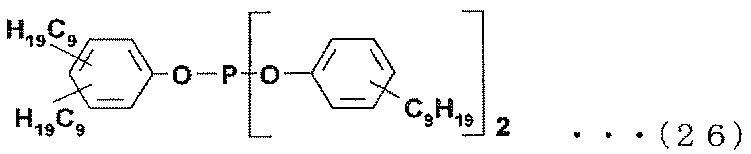



本発明(III)のカルボキシル基含有ポリウレタン溶液中の溶媒を加熱下で、減圧留去して本発明(II)のカルボキシル基含有ポリウレタンを得た。
電極:京都電子工業社製 複合ガラス電極C-173
<成分(a)と成分(d)の混合物の水酸基価の測定>
JIS K0070の中和滴定法に従い、成分(a)と成分(d)の混合物の水酸基価を測定した。
GPCで測定したポリスチレン換算の値であり、GPCの測定条件は以下のとおりである。
カラム:ShodexカラムLF-804を3本連結(直列)
移動相:テトラヒドロフラン
流速:1.0mL/min
検出器:日本分光(株)製 RI-2031Plus
温度:40.0℃
試料量:サンプルループ 100μリットル
試料濃度:0.1質量%に調製
<カルボキシル基含有ポリウレタン溶液の粘度の測定>
カルボキシル基含有ポリウレタン溶液の粘度を以下の方法により測定した。
硬化性組成物のチクソトロピー指数を以下の方法により測定した。
チクソトロピー指数=[1rpmの粘度]÷[10rpmの粘度]
<ダイマージオールから誘導された有機残基および炭素数10~20の脂環構造を有するポリオールから誘導された有機残基を含有する(ポリ)カーボネートポリオールの合成>
[実施例1]
撹拌機、温度計および精留塔のついた1000ml四口丸底フラスコに、2,4-ジエチル-1,5-ペンタンジオールと2-エチル-2-ブチル-1,3-プロパンジオールの混合物(協和発酵ケミカル社製 商品名:PD-9)150.2g(0.937mol)、トリシクロ[5.2.1.02,6]デカンジメタノール(東京化成工業社製)128.7g(0.656mol)、PRIPOL2033(ダイマージオール98.2質量%、モノオール0.6質量%、トリマートリオール1.2質量%、水酸基価;205mgKOH/g)150.2g、ジエチルカーボネート(和光純薬工業社製)166.3g(1.408mol)およびテトラn-ブチルチタネート(三菱ガス化学社製)1.717g(5.04mmol)を入れ、30分間窒素気流を保ち、その後、窒素の供給停止し、オイルバス150℃に設定し加熱した。反応の進行とともに、生成してくる少量のジエチルカーボネートを含むエタノールを精留塔から留出させて300mlナス型フラスコで取得した。エタノールの留出速度が小さくなるのを観察し、徐々にオイルバスの設定温度を上げて、最終的には200℃まで昇温した。その後、反応の進行に伴い、徐々に1000ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、計8時間反応を行った。その後、取得した少量のジエチルカーボネートを含むエタノール中のジエチルカーボネートの質量をガスクロマトグラフ法により分析した。その後、留出した質量のジエチルカーボネートを新たに添加し、オイルバスの温度を190℃に設定し反応を再開し、常圧で2時間反応を継続した。その間徐々にオイルバスの温度を200℃まで昇温した。その後、再び、徐々に1000ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、反応開始から計12時間反応を行った。反応終了後、1000ml四口丸底フラスコ内に微黄色粘稠液状物(以下、生成物A1と記す。)を得た。
撹拌機、温度計および精留塔のついた500ml四口丸底フラスコに、トリシクロ[5.2.1.02,6]デカンジメタノール(東京化成工業社製)117.8g(0.600mol)、PRIPOL2033(ダイマージオール98.2質量%、モノオール0.6質量%、トリマートリオール1.2質量%、水酸基価;205mgKOH/g)162.6g、ジエチルカーボネート(和光純薬工業社製)70.878g(0.600mol)およびテトラn-ブチルチタネート(三菱ガス化学社製)1.405g(4.1mmol)を入れ、30分間窒素気流を保ち、その後、窒素の供給停止し、オイルバス150℃に設定し加熱した。反応の進行とともに、生成してくる少量のジエチルカーボネートを含むエタノールを精留塔から留出させて300mlナス型フラスコで取得した。エタノールの留出速度が小さくなるのを観察し、徐々にオイルバスの設定温度を上げて、最終的には200℃まで昇温した。その後、反応の進行に伴い、徐々に500ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、計8時間反応を行った。その後、取得した少量のジエチルカーボネートを含むエタノール中のジエチルカーボネートの質量をガスクロマトグラフ法により分析した。その後、留出した質量のジエチルカーボネートを新たに添加し、オイルバスの温度を190℃に設定し反応を再開し、常圧で2時間反応を継続した。その間徐々にオイルバスの温度を200℃まで昇温した。その後、再び、徐々に500ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、反応開始から計12時間反応を行った。反応終了後、500ml四口丸底フラスコ内に微黄色粘稠液状物(以下、生成物A2と記す。)を得た。
撹拌機、温度計および精留塔のついた500ml四口丸底フラスコに、2,4-ジエチル-1,5-ペンタンジオールと2-エチル-2-ブチル-1,3-プロパンジオールの混合物(協和発酵ケミカル社製 商品名:PD-9)68.6g(0.426mol)、トリシクロ[5.2.1.02,6]デカンジメタノール(東京化成工業社製)68.6g(0.349mol)、PRIPOL2033(ダイマージオール98.2質量%、モノオール0.6質量%、トリマートリオール1.2質量%、水酸基価;205mgKOH/g)68.6g、ジエチルカーボネート(和光純薬工業社製)81.8g(0.693mol)およびテトラn-ブチルチタネート(三菱ガス化学社製)0.823g(2.42mmol)を入れ、30分間窒素気流を保ち、その後、窒素の供給停止し、オイルバス150℃に設定し加熱した。反応の進行とともに、生成してくる少量のジエチルカーボネートを含むエタノールを精留塔から留出させて300mlナス型フラスコで取得した。エタノールの留出速度が小さくなるのを観察し、徐々にオイルバスの設定温度を上げて、最終的には200℃まで昇温した。その後、反応の進行に伴い、徐々に500ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、計8時間反応を行った。その後、取得した少量のジエチルカーボネートを含むエタノール中のジエチルカーボネートの質量をガスクロマトグラフ法により分析した。その後、留出した質量のジエチルカーボネートを新たに添加し、オイルバスの温度を190℃に設定し反応を再開し、常圧で2時間反応を継続した。その間徐々にオイルバスの温度を200℃まで昇温した。その後、再び、徐々に500ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、反応開始から計12時間反応を行った。反応終了後、500ml四口丸底フラスコ内に微黄色粘稠液状物(以下、生成物A3と記す。)を得た。
撹拌機、温度計および精留塔のついた1000ml四口丸底フラスコに、2-メチル-1,8-オクタンジオールと1,9-ノナンジオールの混合物(クラレ社製 商品名:ND-15)150.2g(0.937mol)、トリシクロ[5.2.1.02,6]デカンジメタノール(東京化成工業社製)150.2g(0.765mol)、PRIPOL2033(ダイマージオール98.2質量%、モノオール0.6質量%、トリマートリオール1.2質量%、水酸基価;205mgKOH/g)150.2g、ジエチルカーボネート(和光純薬工業社製)179.2g(1.517mol)およびテトラn-ブチルチタネート(三菱ガス化学社製)1.802g(5.30mmol)を入れ、30分間窒素気流を保ち、その後、窒素の供給停止し、オイルバス150℃に設定し加熱した。反応の進行とともに、生成してくる少量のジエチルカーボネートを含むエタノールを精留塔から留出させて300mlナス型フラスコで取得した。エタノールの留出速度が小さくなるのを観察し、徐々にオイルバスの設定温度を上げて、最終的には200℃まで昇温した。その後、反応の進行に伴い、徐々に1000ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、計8時間反応を行った。その後、取得した少量のジエチルカーボネートを含むエタノール中のジエチルカーボネートの質量をガスクロマトグラフ法により分析した。その後、留出した質量のジエチルカーボネートを新たに添加し、オイルバスの温度を190℃に設定し反応を再開し、常圧で2時間反応を継続した。その間徐々にオイルバスの温度を200℃まで昇温した。その後、再び、徐々に1000ml四口丸底フラスコ内を減圧にし、最終的には5333Paまで減圧し、反応開始から計12時間反応を行った。反応終了後、1000ml四口丸底フラスコ内に微黄色粘稠液状物(以下、生成物A5と記す。)を得た。
[実施例5]
攪拌装置、温度計およびコンデンサーを備えた反応容器に、実施例1で得られた生成物A1を271.4g、カルボキシル基含有ジオールとして2,2-ジメチロールブタン酸(日本化成社製)35.7g、溶媒としてγ―ブチロラクトン(三菱化学社製)330.0gとジエチレングリコールジエチルエーテル(日本乳化剤社製)220.0gを仕込み、100℃に加熱してすべての原料を溶解した。反応液の温度を90℃まで下げ、滴下ロートにより、ポリイソシアネートとしてメチレンビス(4-シクロヘキシルイソシアネート)(住化バイエルウレタン社製 商品名:デスモジュール-W)139.0gを30分かけて滴下した。120℃で6時間反応を行い、ほぼイソシアネートが消失したことを確認した後、イソブタノール(和光純薬工業社製)4.0gを滴下し、さらに120℃で3時間反応を行い、カルボキシル基含有ポリウレタン溶液(以下、「カルボキシル基含有ポリウレタン溶液B1」と記す。)を得た。
得られたカルボキシル基含有ポリウレタン溶液B1の粘度は11500mPa・sであった。また、カルボキシル基含有ポリウレタン溶液B1中に含まれるカルボキシル基含有ポリウレタン(以下、「カルボキシル基含有ポリウレタンBU1」と記す。)の数平均分子量は14000であり、カルボキシル基含有ポリウレタンBU1の酸価は30.0mgKOH/gであった。
攪拌装置、温度計およびコンデンサーを備えた反応容器に、実施例2で得られた生成物A2を204.5gと2,4-ジエチル-1,5-ペンタンジオールと2-エチル-2-ブチル-1,3-プロパンジオールの混合物(協和発酵ケミカル社製 商品名:PD-9)33.1g、カルボキシル基含有ジオールとして2,2-ジメチロールブタン酸(日本化成社製)35.7g、溶媒としてγ―ブチロラクトン(三菱化学社製)250.0gとジエチレングリコールジエチルエーテル(日本乳化剤(株)製)250.0gを仕込み、100℃に加熱してすべての原料を溶解した。反応液の温度を90℃まで下げ、滴下ロートにより、ポリイソシアネートとしてメチレンビス(4-シクロヘキシルイソシアネート)(住化バイエルウレタン社製 商品名:デスモジュール-W)176.5gを30分かけて滴下した。120℃で6時間反応を行い、ほぼイソシアネートが消失したことを確認した後、イソブタノール(和光純薬工業社製)4.0gを滴下し、さらに120℃で3時間反応を行い、カルボキシル基含有ポリウレタン溶液(以下、「カルボキシル基含有ポリウレタン溶液B2」と記す。)を得た。
攪拌装置、温度計およびコンデンサーを備えた反応容器に、実施例3で得られた生成物A3を277.7g、カルボキシル基含有ジオールとして2,2-ジメチロールブタン酸(日本化成社製)35.7g、溶媒としてγ―ブチロラクトン(三菱化学社製)330.0gとジエチレングリコールジエチルエーテル(日本乳化剤(株)製)220.0gを仕込み、100℃に加熱してすべての原料を溶解した。反応液の温度を90℃まで下げ、滴下ロートにより、ポリイソシアネートとしてメチレンビス(4-シクロヘキシルイソシアネート)(住化バイエルウレタン社製 商品名:デスモジュール-W)132.6gを30分かけて滴下した。120℃で6時間反応を行い、ほぼイソシアネートが消失したことを確認した後、イソブタノール(和光純薬工業社製)4.0gを滴下し、さらに120℃で3時間反応を行い、カルボキシル基含有ポリウレタン溶液(以下、「カルボキシル基含有ポリウレタン溶液B3」と記す。)を得た。
攪拌装置、温度計およびコンデンサーを備えた反応容器に、実施例4で得られた生成物A5を274.6g、カルボキシル基含有ジオールとして2,2-ジメチロールブタン酸(日本化成社製)35.7g、溶媒としてγ―ブチロラクトン(三菱化学社製)330.0gとジエチレングリコールジエチルエーテル(日本乳化剤社製)220.0gを仕込み、100℃に加熱してすべての原料を溶解した。反応液の温度を90℃まで下げ、滴下ロートにより、ポリイソシアネートとしてメチレンビス(4-シクロヘキシルイソシアネート)(住化バイエルウレタン社製 商品名;デスモジュール-W)135.7gを30分かけて滴下した。120℃で6時間反応を行い、ほぼイソシアネートが消失したことを確認した後、イソブタノール(和光純薬(株)製)4.0gを滴下し、さらに120℃で3時間反応を行いカルボキシル基含有ポリウレタン溶液(以下、「カルボキシル基含有ポリウレタン溶液B4」と記す。)を得た。
得られたカルボキシル基含有ポリウレタン溶液B4の粘度は12000mPa・sであった。また、カルボキシル基含有ポリウレタン溶液B4中に含まれるカルボキシル基含有ポリウレタン(以下、「カルボキシル基含有ポリウレタンBU4」と記す。)の数平均分子量は14000であり、カルボキシル基含有ポリウレタンBU4の酸価は30.0mgKOH/gであった。
攪拌機、温度計、コンデンサーを備えた反応容器に、C-1065N(クラレ社製 (ポリ)カーボネートジオールと原料ジオール(即ち、1,9-ノナンジオールおよび2-メチル-1,8-オクタンジオール)の混合物、原料ジオールの仕込みモル比;1,9-ノナンジオール:2-メチル-1,8-オクタンジオール=65:35、水酸基価113.2mgKOH/g、1,9-ノナンジオールの残存濃度7.5質量%、2-メチル-1,8-オクタンジオールの残存濃度4.4質量%)70.7g、カルボキシル基含有ジオールとして2,2-ジメチロールブタン酸(日本化成社製)13.5g、ジエチレングリコールモノエチルエーテルアセテート(ダイセル化学工業社製)128.9gを仕込み、90℃ですべての原料を溶解した。反応液の温度を70℃まで下げ、滴下ロートにより、ポリイソシアネートとしてメチレンビス(4-シクロヘキシルイソシアネート)(住化バイエルウレタン社製 商品名;デスモジュール-W)42.4gを30分かけて滴下した。滴下終了後、80℃で1時間、90℃で1時間、100℃で2時間反応を行い、ほぼイソシアネートが消失したことを確認した後、イソブタノール(和光純薬工業社製)1.46gを滴下し、更に105℃にて1.5時間反応を行い、カルボキシル基含有ポリウレタン溶液(以下、「カルボキシル基含有ポリウレタン溶液C1」と記す。)を得た。
撹拌機、温度計およびコンデンサーを備えた反応容器に、C-1015N(クラレ社製(ポリ)カーボネートジオールと原料ジオール(即ち、1,9-ノナンジオールおよび2-メチル-1,8-オクタンジオール)の混合物、原料ジオールの仕込みモル比;1,9-ノナンジオール:2-メチル-1,8-オクタンジオール=15:85、水酸基価116.4mgKOH/g、1,9-ノナンジオールの残存濃度2.1質量%、2-メチル-1,8-オクタンジオールの残存濃度9.3質量%)660.6g、G-1000(日本曹達社製、両末端水酸基化1,2-ポリブタジエン、数平均分子量1548)73.39g、カルボキシル基含有ジオールとして2,2-ジメチロールブタン酸(日本化成社製)138.4g、およびジエチレングリコールモノエチルエーテルアセテート(ダイセル化学工業社製)1303gを仕込み、90℃で前記2,2-ジメチロールブタン酸を溶解させた。
(実施配合例1)
カルボキシル基含有ポリウレタン溶液B1 111.1g、シリカ粉(日本アエロジル社製 商品名;アエロジルR-974)5.0g、硬化促進剤としてメラミン(日産化学工業社製)0.36gおよび消泡剤(モメンティブ・パフォーマンス・マテリアルズ社製 商品名;TSA750S)0.70gを混合し、三本ロールミル(井上製作所社製 型式:S-4セ×11)を用いて、カルボキシル基含有ポリウレタン溶液B1へのシリカ粉、硬化促進剤および消泡剤の混合を行った。この配合物を主剤配合物D1とした。
実施配合例1の同様の方法によって、表1に示す配合組成に従って配合した。実施配合例2~4で調整した配合物を、それぞれ主剤配合物D2~D4とし、比較配合例1および比較配合例2で調整した配合物を、それぞれ、主剤配合物E1および主剤配合物E2とした。
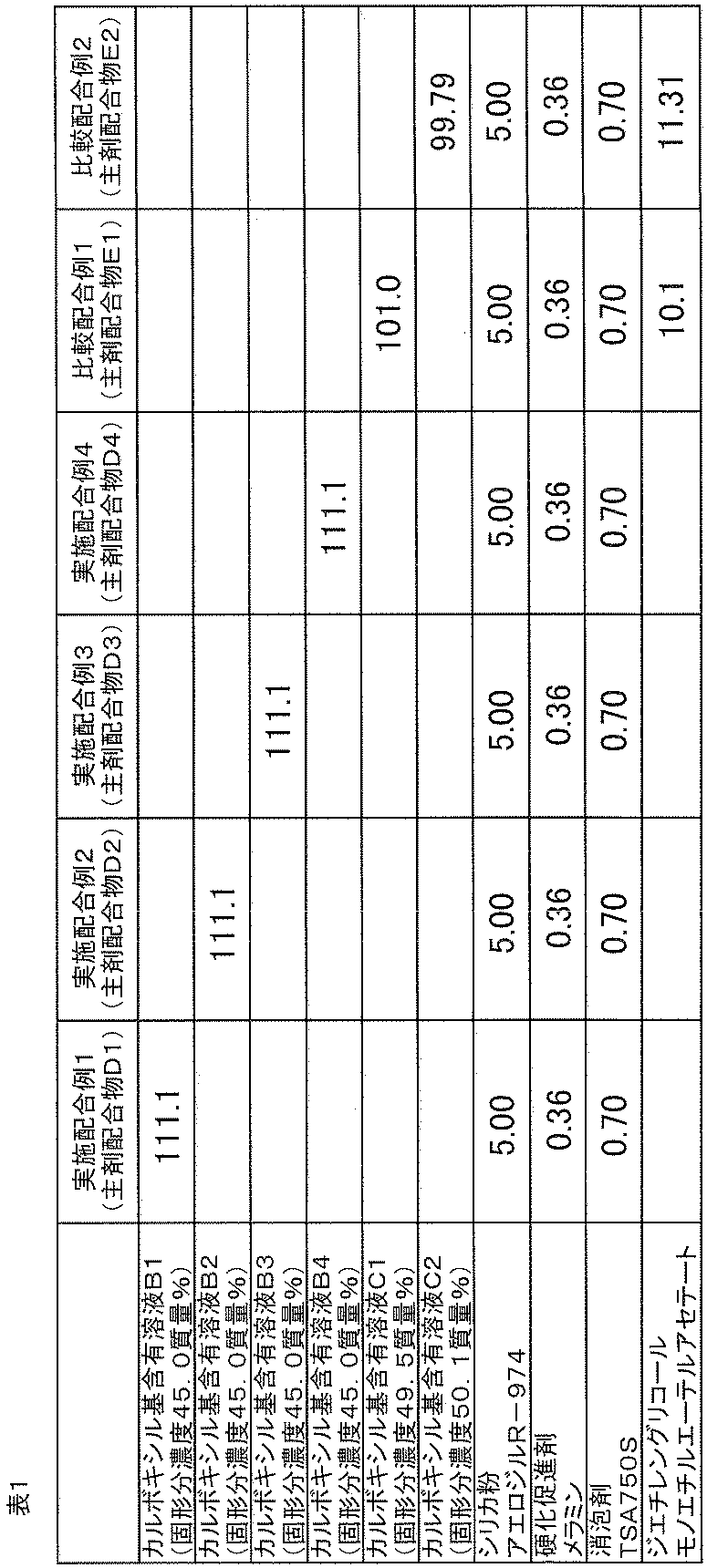
撹拌機、温度計およびコンデンサーを備えた容器に、下記式(4)の構造を有するエポキシ樹脂(DIC社製 グレード名;HP-7200H エポキシ当量278g/eq)300g、γ-ブチロラクトン(三菱化学社製)180gおよびジエチレングリコールジエチルエーテル(東邦化学工業社製)120gを添加し、撹拌を開始した。撹拌を継続しながら、オイルバスを用いて、容器内の温度を70℃に昇温した。内温を70℃に昇温後、30分間撹拌を継続した。その後、HP-7200Hが完全に溶解したのを確認して、室温まで冷却し、濃度50質量%のHP-7200H含有溶液を取得した。この溶液を硬化剤溶液F1する。
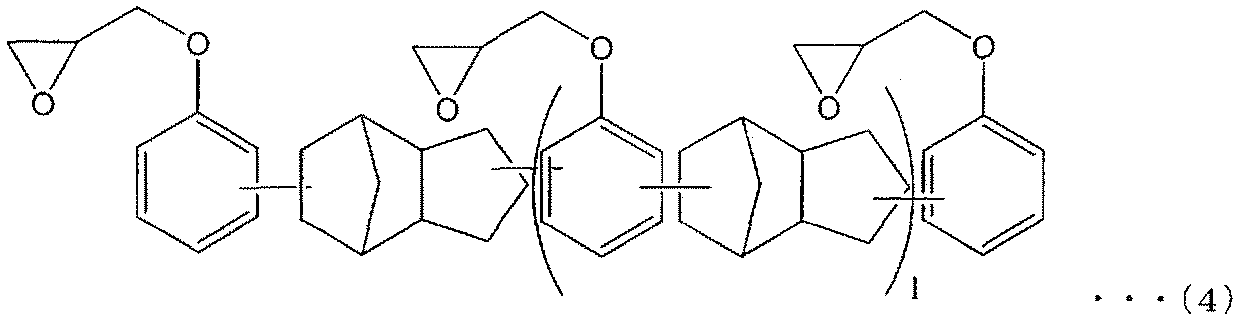
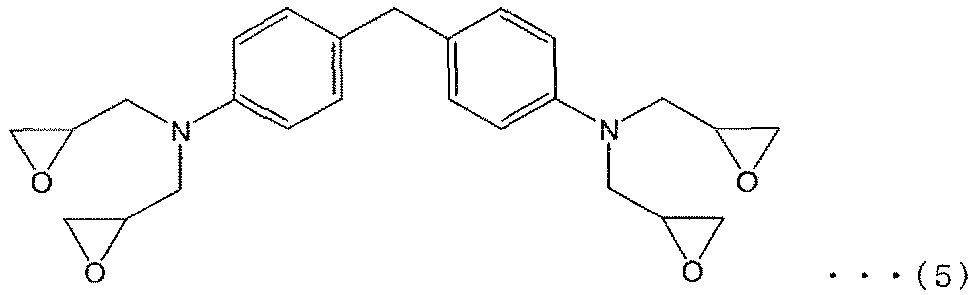
[実施例9]
200.0gの主剤配合物D1および25.4gの硬化剤溶液F1を混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G1」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G2」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G3」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G4」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G5」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G6」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G7」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G8」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のγ-ブチロラクトン:ジエチレングリコールジエチルエーテル=3:2(質量比)の混合溶媒を添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物G9」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のジエチレングリコールモノエチルエーテルアセテートを添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物H1」と記す。)を得た。
表2に記載の配合比で混合し、スパチュラを用いて、充分に撹拌混合した。その後、チクソトロピー指数を1.3に合わせるのに必要量のジエチレングリコールモノエチルエーテルアセテートを添加して、粘度調整を行い、硬化性組成物(以下、「硬化性組成物H2」と記す。)を得た。

表3に記したように、それぞれ硬化性組成物G1~硬化性組成物G9、硬化性組成物H1および硬化性組成物H2を用いて、後述の方法により、ポリイミドおよび錫メッキ処理を施した銅への密着性の評価、反り性の評価および長期電気絶縁信頼性の評価を行った。その結果を表3に記す。
フレキシブル銅張り積層板(住友金属鉱山社製 グレード名;エスパーフレックス 銅厚;8μm、ポリイミド厚;38μm)に錫メッキを施した基板およびポリイミドフィルム〔カプトン(登録商標)300H、東レ・デュポン社製〕に、硬化性組成物G1を、スクリーン印刷法により、硬化性組成物の厚みが15μmの厚さ(乾燥後の厚さ)になるように塗布し、80℃の熱風循環式乾燥機に30分間入れ、その後、120℃の熱風循環式乾燥機に120分間入れることにより硬化させた。この硬化塗膜に、1mm間隔で100個の格子状パターンを切込み、約75mmの長さに切ったテープを格子の部分に接着し、剥離用テープ(JIS Z 1522に規定する物)を60°に近い角度で0.5~1.0秒の時間で引きはがした。
△:碁盤目の数が50個以上80個未満残る場合、
×:碁盤目の数が50個未満しか残らない場合。
硬化性組成物G1を、基板にスクリーン印刷により塗布し、80℃の熱風循環式乾燥機に30分間入れ、その後、120℃の熱風循環式乾燥機に60分間入れることにより硬化させた。基板は38μm厚ポリイミドフィルム〔カプトン(登録商標)150EN、東レ・デュポン社製〕を用いた。
フレキシブル銅張り積層板(住友金属鉱山社製 グレード名;エスパーフレックス 銅厚;8μm、ポリイミド厚;38μm)の銅上に、幅75mm、長さ110mmの大きさ、硬化後の塗膜の厚み15μmになるように硬化性組成物G1を、クリーン印刷により塗布し、10分間室温で保持し、120℃の熱風循環式乾燥機に60分間入れることにより硬化させた。作製した試験片の裏打ちのPETフィルムを剥離し、幅10mmの短冊状にカッターナイフで切り出した後、塗膜面が外側になる様に約180度折り曲げ、圧縮機を用いて0.5±0.2MPaで3秒間圧縮した。屈曲部を曲げた状態で30倍の顕微鏡で観察し、クラックの発生の有無を確認した。
フレキシブル銅張り積層板(住友金属鉱山社製 グレード名;エスパーフレックス 銅厚;8μm、ポリイミド厚:38μm)をエッチングして製造した、JPCA-ET01に記載の微細くし形パターン形状の基板(銅配線幅/銅配線間幅=15μm/15μm)に錫メッキ処理を施したフレキシブル配線板に、硬化性組成物G1を、スクリーン印刷法により、ポリイミド面からの厚みが15μmの厚さ(乾燥後)になるように塗布し、80℃の熱風循環式乾燥機に30分間入れ、その後、120℃の熱風循環式乾燥機に120分間入れることにより硬化させた。
この試験片を用いて、バイアス電圧60Vを印加し、温度120℃、湿度95%RHの条件での温湿度定常試験を、MIGRATION TESTER MODEL MIG-8600(IMV社製)を用いて行った。上記温湿度定常試験をスタート初期およびスタートしてから30時間後、50時間後、100時間後の抵抗値を表3に記す。

Claims (14)
- ダイマージオールから誘導された有機残基および炭素数10~20の脂環構造を有するポリオールから誘導された有機残基を含有する(ポリ)カーボネートポリオール。
- 前記炭素数10~20の脂環構造を有するポリオールが、トリシクロデカンジメタノールであることを特徴とする請求項1に記載の(ポリ)カーボネートポリオール。
- 下記式(34)で表される(ポリ)カーボネートポリオール。

(A)群:下記式(35)または(36)で表わされるアルキレン基
(B)群:下記式(37)または(38)で表わされるアルキレン基
(C)群:炭素数9~12の鎖状脂肪族アルキレン基
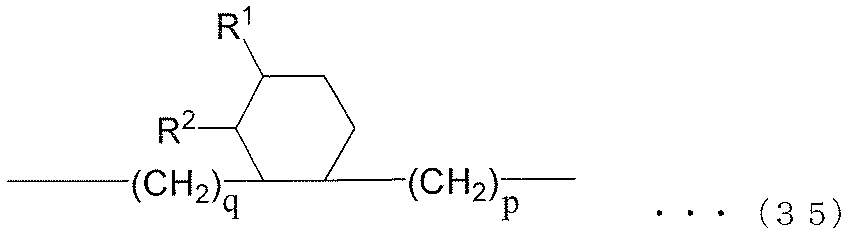
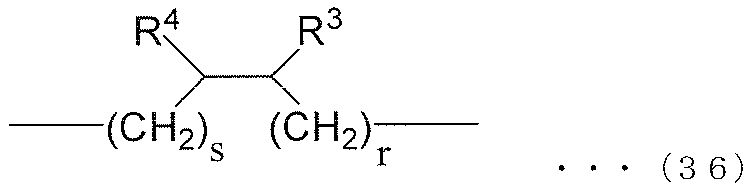
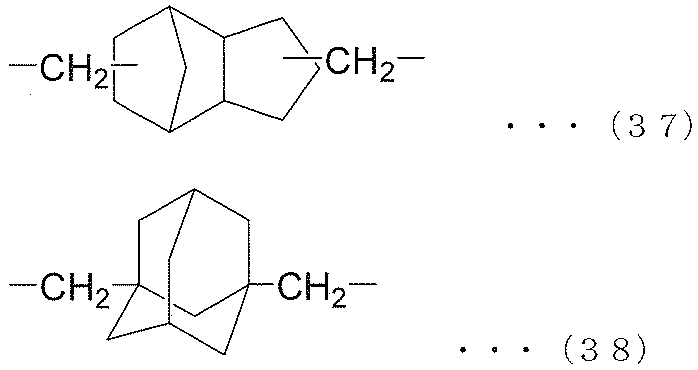
- 少なくとも以下の成分(a)、成分(b)および成分(c)を原料とするカルボキシル基含有ポリウレタン。
成分(a):請求項1~3のいずれか一項に記載の(ポリ)カーボネートポリオール
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール - 少なくとも以下の成分(a)、成分(b)、成分(c)および成分(d)を原料とするカルボキシル基含有ポリウレタン。
成分(a):請求項1~3のいずれか一項に記載の(ポリ)カーボネートポリオール
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール
成分(d):成分(a)および成分(c)以外のポリオ-ル。 - さらに、モノヒドロキシル化合物(成分(e))を原料とする請求項4または5に記載のカルボキシル基含有ポリウレタン。
- さらに、モノイソシアネート化合物(成分(f))を原料とする請求項4~6のいずれか1項に記載のカルボキシル基含有ポリウレタン。
- 請求項4~7のいずれか1項に記載のカルボキシル基含有ポリウレタンと沸点が120~300℃である溶媒とを含有することを特徴とするカルボキシル基含有ポリウレタン溶液。
- ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジエチレングリコールエチルメチルエーテル、ジエチレングリコールジブチルエーテル、ジエチレングリコールブチルメチルエーテル、ジエチレングリコールイソプロピルメチルエーテル、トリエチレングリコールジメチルエーテル、トリエチレングリコールブチルメチルエーテル、テトラエチレングリコールジメチルエーテル、ジプロピレングリコ-ルジメチルエーテル、トリプロピレングリコールジメチルエーテル、アニソール、エチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテート、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、ジプロピレングリコールモノメチルエーテルアセテート、ジプロピレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノメチルエーテルアセテート、メトキシプロピオン酸メチル、メトキシプロピオン酸エチル、エトキシプロピオン酸メチル、エトキシプロピオン酸エチル、デカヒドロナフタリン、シクロヘキサノンおよびγ-ブチロラクトンからなる群より選ばれる少なくとも1種を含む溶媒中で、少なくとも以下の成分(a)、成分(b)および成分(c)を、30℃~160℃の温度範囲で反応させることを特徴とするカルボキシル基含有ポリウレタンの製造方法。
成分(a):請求項1~3のいずれか一項に記載の(ポリ)カーボネートポリオール
成分(b):ポリイソシアネート
成分(c):カルボキシル基含有ポリオール - 請求項4~7のいずれか1項に記載のカルボキシル基含有ポリウレタン、沸点が120~300℃である溶媒および硬化剤を含む硬化性組成物。
- 硬化剤が、1分子中に2個以上のエポキシ基を有する化合物であることを特徴とする請求項10に記載の硬化性組成物。
- 請求項10または11に記載の硬化性樹脂組成物を硬化して得られる硬化物。
- フレキシブル基板上に配線が形成されてなるフレキシブル配線板の、配線が形成されている表面の一部または全部が請求項12に記載の硬化物によって被覆されたことを特徴とする、硬化物によって被覆されたフレキシブル配線板。
- 請求項10または11に記載の硬化性組成物をフレキシブル配線板の錫メッキ処理された配線パターン部に印刷することで該パターン上に印刷膜を形成し、該印刷膜を80~130℃で加熱硬化させることで保護膜を形成することを特徴とする、保護膜によって被覆されたフレキシブル配線板の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020117016222A KR101276986B1 (ko) | 2009-01-20 | 2010-01-20 | (폴리)카르보네이트 폴리올 및 상기 (폴리)카르보네이트 폴리올을 원료로 하는 카르복실기 함유 폴리우레탄 |
| US13/145,241 US8674017B2 (en) | 2009-01-20 | 2010-01-20 | (Poly)carbonate polyol and carboxyl group-containing polyurethane obtained from the (poly)carbonate polyol |
| JP2010547493A JP5717448B2 (ja) | 2009-01-20 | 2010-01-20 | (ポリ)カーボネートポリオールおよび該(ポリ)カーボネートポリオールを原料とするカルボキシル基含有ポリウレタン |
| CN201080004295XA CN102272186B (zh) | 2009-01-20 | 2010-01-20 | (聚)碳酸酯多元醇和以该(聚)碳酸酯多元醇为原料而成的含羧基的聚氨酯 |
| EP10733479.9A EP2390277B1 (en) | 2009-01-20 | 2010-01-20 | (poly)carbonate polyol and carboxyl group-containing polyurethane using the (poly)carbonate polyol as starting material |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009010238 | 2009-01-20 | ||
| JP2009-010238 | 2009-01-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010084872A1 true WO2010084872A1 (ja) | 2010-07-29 |
Family
ID=42355924
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/050611 WO2010084872A1 (ja) | 2009-01-20 | 2010-01-20 | (ポリ)カーボネートポリオールおよび該(ポリ)カーボネートポリオールを原料とするカルボキシル基含有ポリウレタン |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8674017B2 (ja) |
| EP (1) | EP2390277B1 (ja) |
| JP (1) | JP5717448B2 (ja) |
| KR (1) | KR101276986B1 (ja) |
| CN (1) | CN102272186B (ja) |
| TW (1) | TWI482797B (ja) |
| WO (1) | WO2010084872A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012043775A1 (ja) * | 2010-09-30 | 2012-04-05 | 宇部興産株式会社 | テープキャリアパッケージの製造方法、及びテープキャリアパッケージ用柔軟性配線板の製造方法 |
| WO2015041158A1 (ja) | 2013-09-20 | 2015-03-26 | 三菱瓦斯化学株式会社 | 油吸着材として使用できるポリカーボネート樹脂 |
| JP2015064922A (ja) * | 2013-08-30 | 2015-04-09 | 富士フイルム株式会社 | 磁気記録媒体用組成物および磁気記録媒体 |
| JP2015147940A (ja) * | 2009-10-07 | 2015-08-20 | 日立化成株式会社 | 熱硬化性樹脂組成物及び硬化膜 |
| WO2016052370A1 (ja) * | 2014-09-30 | 2016-04-07 | 三菱瓦斯化学株式会社 | ポリカーボネート樹脂および光学レンズ |
| DE102015014864A1 (de) | 2015-11-17 | 2017-05-18 | Gt Elektrotechnische Produkte Gmbh | Verfahren zur Herstellung von zelligen, elastischen Polycarbonaturethan-Materialien sowie die Polycarbonaturethan-Materialien |
| JP2018522076A (ja) * | 2015-05-21 | 2018-08-09 | クローダ インターナショナル パブリック リミティド カンパニー | ポリウレタン |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5265854B2 (ja) * | 2005-12-08 | 2013-08-14 | 昭和電工株式会社 | 熱硬化性樹脂組成物、熱可塑性樹脂溶液および皮膜形成材料ならびにこれらの硬化物 |
| US20120305295A1 (en) * | 2010-02-03 | 2012-12-06 | Showa Denko K.K. | Thermosetting composition |
| KR101136951B1 (ko) * | 2011-11-16 | 2012-04-26 | (주)삼일물산 | 불소화 폴리카보네이트 디올, 이의 제조방법 및 이를 이용한 불소화 우레탄 고분자의 제조방법 |
| JP7192353B2 (ja) * | 2017-10-30 | 2022-12-20 | Dic株式会社 | ウレタン樹脂組成物、皮膜、及び合成皮革 |
| CN111307888B (zh) * | 2018-12-11 | 2022-04-12 | 辽宁奥克化学股份有限公司 | 一种嵌段聚醚多元醇中伯羟基含量的检测方法 |
| JP2023531880A (ja) * | 2020-06-25 | 2023-07-26 | ダウ グローバル テクノロジーズ エルエルシー | ポリオール及びそれから作製されたフォーム |
| CN111995725B (zh) * | 2020-09-07 | 2022-02-11 | 江苏湘园化工有限公司 | 一种环保型液态聚氨酯固化剂及其应用方法 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08283399A (ja) * | 1995-04-07 | 1996-10-29 | Bayer Ag | ポリカーボネートジオール、その製造方法及びポリウレタンプラスチックの出発材料としての使用 |
| JPH10231360A (ja) | 1997-02-18 | 1998-09-02 | Ube Ind Ltd | ポリカーボネートジオールの製造方法 |
| JPH10251398A (ja) | 1997-03-14 | 1998-09-22 | Ube Ind Ltd | ポリカーボネートジオールの製造法 |
| JPH10251369A (ja) | 1997-03-14 | 1998-09-22 | Ube Ind Ltd | ポリウレタン樹脂の製造方法 |
| JPH10273514A (ja) | 1997-03-31 | 1998-10-13 | Ube Ind Ltd | ポリウレタン樹脂の製造法 |
| JPH11508636A (ja) * | 1995-07-12 | 1999-07-27 | ヘンケル・コマンディットゲゼルシャフト・アウフ・アクチエン | 二量体ジオールからの液体オリゴカーボネート誘導体の製造および使用 |
| JPH11236443A (ja) * | 1998-02-23 | 1999-08-31 | Lion Corp | ポリカーボネートポリオール |
| JP2000007909A (ja) | 1998-06-19 | 2000-01-11 | Sekisui Chem Co Ltd | ウレタン系水性組成物 |
| JP2004137370A (ja) | 2002-10-17 | 2004-05-13 | Hitachi Chem Co Ltd | ポリアミドイミド樹脂ペースト及びそれを含む被膜形成材料 |
| JP2004182792A (ja) | 2002-11-29 | 2004-07-02 | Hitachi Chem Co Ltd | ポリアミドイミド樹脂及びそれを用いた接着剤組成物 |
| JP2006312729A (ja) | 2005-04-06 | 2006-11-16 | Showa Denko Kk | 脂環骨格を有するポリカーボネートジオール重合体及びその製造方法 |
| JP2006348278A (ja) | 2005-05-16 | 2006-12-28 | Showa Denko Kk | カルボキシル基含有ポリウレタン、熱硬化性樹脂組成物およびそれらの用途 |
| JP2007100037A (ja) | 2005-10-07 | 2007-04-19 | Showa Denko Kk | カルボキシル基含有ポリウレタンおよび熱硬化性ポリウレタン樹脂組成物 |
| WO2008029760A1 (fr) * | 2006-09-06 | 2008-03-13 | Showa Denko K.K. | Nouvelle résine, son procédé de production, nouvelle composition, et produit durci obtenu par durcissement de la composition |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU669567B2 (en) * | 1992-10-30 | 1996-06-13 | Basf Corporation | Water dispersible ionic and nonionic polyamide modified polyurethane resins for use in coating compositions |
| JP3844025B2 (ja) | 1997-08-14 | 2006-11-08 | 味の素株式会社 | オーバーコート用樹脂組成物 |
| TW393494B (en) | 1997-08-14 | 2000-06-11 | Ajinomoto Kk | Curable resin composition for overcoat of flexible circuit |
| JP4237944B2 (ja) | 1999-03-19 | 2009-03-11 | 日本化薬株式会社 | ウレタンオリゴマー、その樹脂組成物、その硬化物 |
| TW200643059A (en) | 2005-04-06 | 2006-12-16 | Showa Denko Kk | Polymer of polycarbonate diol having an alicyclic structure and production process thereof |
| US20090082518A1 (en) | 2005-05-16 | 2009-03-26 | Showa Denko K.K. | Carboxyl group-containing polyurethane, heat-curable resin composition and uses thereof |
| DE602006019092D1 (de) * | 2005-07-04 | 2011-02-03 | Showa Denko Kk | Polyurethan mit einer carboxylgruppe und seine verwendungen |
| JP5265854B2 (ja) | 2005-12-08 | 2013-08-14 | 昭和電工株式会社 | 熱硬化性樹脂組成物、熱可塑性樹脂溶液および皮膜形成材料ならびにこれらの硬化物 |
| US7666972B2 (en) * | 2007-10-18 | 2010-02-23 | SABIC Innovative Plastics IP B., V. | Isosorbide-based polycarbonates, method of making, and articles formed therefrom |
| US20110190471A1 (en) * | 2008-08-01 | 2011-08-04 | Masahiko Watanabe | Polycarbonate diol and polycarbonate diol copolymer |
-
2010
- 2010-01-20 TW TW099101481A patent/TWI482797B/zh not_active IP Right Cessation
- 2010-01-20 EP EP10733479.9A patent/EP2390277B1/en not_active Not-in-force
- 2010-01-20 KR KR1020117016222A patent/KR101276986B1/ko active IP Right Grant
- 2010-01-20 JP JP2010547493A patent/JP5717448B2/ja active Active
- 2010-01-20 US US13/145,241 patent/US8674017B2/en not_active Expired - Fee Related
- 2010-01-20 WO PCT/JP2010/050611 patent/WO2010084872A1/ja active Application Filing
- 2010-01-20 CN CN201080004295XA patent/CN102272186B/zh not_active Expired - Fee Related
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08283399A (ja) * | 1995-04-07 | 1996-10-29 | Bayer Ag | ポリカーボネートジオール、その製造方法及びポリウレタンプラスチックの出発材料としての使用 |
| JPH11508636A (ja) * | 1995-07-12 | 1999-07-27 | ヘンケル・コマンディットゲゼルシャフト・アウフ・アクチエン | 二量体ジオールからの液体オリゴカーボネート誘導体の製造および使用 |
| JPH10231360A (ja) | 1997-02-18 | 1998-09-02 | Ube Ind Ltd | ポリカーボネートジオールの製造方法 |
| JPH10251398A (ja) | 1997-03-14 | 1998-09-22 | Ube Ind Ltd | ポリカーボネートジオールの製造法 |
| JPH10251369A (ja) | 1997-03-14 | 1998-09-22 | Ube Ind Ltd | ポリウレタン樹脂の製造方法 |
| JPH10273514A (ja) | 1997-03-31 | 1998-10-13 | Ube Ind Ltd | ポリウレタン樹脂の製造法 |
| JPH11236443A (ja) * | 1998-02-23 | 1999-08-31 | Lion Corp | ポリカーボネートポリオール |
| JP2000007909A (ja) | 1998-06-19 | 2000-01-11 | Sekisui Chem Co Ltd | ウレタン系水性組成物 |
| JP2004137370A (ja) | 2002-10-17 | 2004-05-13 | Hitachi Chem Co Ltd | ポリアミドイミド樹脂ペースト及びそれを含む被膜形成材料 |
| JP2004182792A (ja) | 2002-11-29 | 2004-07-02 | Hitachi Chem Co Ltd | ポリアミドイミド樹脂及びそれを用いた接着剤組成物 |
| JP2006312729A (ja) | 2005-04-06 | 2006-11-16 | Showa Denko Kk | 脂環骨格を有するポリカーボネートジオール重合体及びその製造方法 |
| JP2006348278A (ja) | 2005-05-16 | 2006-12-28 | Showa Denko Kk | カルボキシル基含有ポリウレタン、熱硬化性樹脂組成物およびそれらの用途 |
| JP2007100037A (ja) | 2005-10-07 | 2007-04-19 | Showa Denko Kk | カルボキシル基含有ポリウレタンおよび熱硬化性ポリウレタン樹脂組成物 |
| WO2008029760A1 (fr) * | 2006-09-06 | 2008-03-13 | Showa Denko K.K. | Nouvelle résine, son procédé de production, nouvelle composition, et produit durci obtenu par durcissement de la composition |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2390277A4 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015147940A (ja) * | 2009-10-07 | 2015-08-20 | 日立化成株式会社 | 熱硬化性樹脂組成物及び硬化膜 |
| WO2012043775A1 (ja) * | 2010-09-30 | 2012-04-05 | 宇部興産株式会社 | テープキャリアパッケージの製造方法、及びテープキャリアパッケージ用柔軟性配線板の製造方法 |
| US10081701B2 (en) | 2013-08-30 | 2018-09-25 | Fujifilm Corporation | Composition for magnetic recording medium and magnetic recording medium |
| JP2015064922A (ja) * | 2013-08-30 | 2015-04-09 | 富士フイルム株式会社 | 磁気記録媒体用組成物および磁気記録媒体 |
| US9708445B2 (en) | 2013-09-20 | 2017-07-18 | Mitsubishi Gas Chemical Company, Inc. | Polycarbonate resin usable as oil-adsorbing material |
| KR20160058820A (ko) | 2013-09-20 | 2016-05-25 | 미츠비시 가스 가가쿠 가부시키가이샤 | 기름 흡착재로서 사용할 수 있는 폴리카보네이트 수지 |
| WO2015041158A1 (ja) | 2013-09-20 | 2015-03-26 | 三菱瓦斯化学株式会社 | 油吸着材として使用できるポリカーボネート樹脂 |
| CN106661216A (zh) * | 2014-09-30 | 2017-05-10 | 三菱瓦斯化学株式会社 | 聚碳酸酯树脂和光学透镜 |
| WO2016052370A1 (ja) * | 2014-09-30 | 2016-04-07 | 三菱瓦斯化学株式会社 | ポリカーボネート樹脂および光学レンズ |
| JPWO2016052370A1 (ja) * | 2014-09-30 | 2017-07-27 | 三菱瓦斯化学株式会社 | ポリカーボネート樹脂および光学レンズ |
| US10048404B2 (en) | 2014-09-30 | 2018-08-14 | Mitsubishi Gas Chemical Company, Inc. | Polycarbonate resin and optical lens |
| US10605956B2 (en) | 2014-09-30 | 2020-03-31 | Mitsubishi Gas Chemical Company, Inc. | Polycarbonate resin and optical lens |
| JP2018522076A (ja) * | 2015-05-21 | 2018-08-09 | クローダ インターナショナル パブリック リミティド カンパニー | ポリウレタン |
| US10882943B2 (en) | 2015-05-21 | 2021-01-05 | Croda International Plc | Polyurethane |
| JP6993232B2 (ja) | 2015-05-21 | 2022-01-13 | クローダ インターナショナル パブリック リミティド カンパニー | ポリウレタン |
| DE102015014864A1 (de) | 2015-11-17 | 2017-05-18 | Gt Elektrotechnische Produkte Gmbh | Verfahren zur Herstellung von zelligen, elastischen Polycarbonaturethan-Materialien sowie die Polycarbonaturethan-Materialien |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20110094339A (ko) | 2011-08-23 |
| US8674017B2 (en) | 2014-03-18 |
| EP2390277A4 (en) | 2013-04-03 |
| JPWO2010084872A1 (ja) | 2012-07-19 |
| CN102272186B (zh) | 2013-06-05 |
| TW201041933A (en) | 2010-12-01 |
| US20110272183A1 (en) | 2011-11-10 |
| EP2390277B1 (en) | 2014-04-16 |
| KR101276986B1 (ko) | 2013-06-24 |
| JP5717448B2 (ja) | 2015-05-13 |
| TWI482797B (zh) | 2015-05-01 |
| EP2390277A1 (en) | 2011-11-30 |
| CN102272186A (zh) | 2011-12-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5717448B2 (ja) | (ポリ)カーボネートポリオールおよび該(ポリ)カーボネートポリオールを原料とするカルボキシル基含有ポリウレタン | |
| JP6912385B2 (ja) | 硬化性組成物、硬化物、オーバーコート膜、被覆フレキシブル配線板およびその製造方法 | |
| US11044807B2 (en) | Polyurethane, curable composition, overcoat film, and flexible wiring board and production method therefor | |
| JP5579081B2 (ja) | カルボキシル基含有ポリウレタン | |
| JP5726094B2 (ja) | 熱硬化性組成物 | |
| JP5796961B2 (ja) | 硬化性組成物 | |
| TWI696639B (zh) | 硬化性組成物及其用途 | |
| WO2020137347A1 (ja) | ポリウレタンの製造方法、硬化性組成物の製造方法、硬化物の製造方法、オーバーコート膜の製造方法、及びフレキシブル配線板の製造方法 | |
| WO2021005913A1 (ja) | 硬化性組成物、硬化物、オーバーコート膜、並びにフレキシブル配線板及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080004295.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10733479 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010547493 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 20117016222 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010733479 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13145241 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |