WO2008152178A1 - Inhibidores de cisteína-proteasas - Google Patents
Inhibidores de cisteína-proteasas Download PDFInfo
- Publication number
- WO2008152178A1 WO2008152178A1 PCT/ES2008/070116 ES2008070116W WO2008152178A1 WO 2008152178 A1 WO2008152178 A1 WO 2008152178A1 ES 2008070116 W ES2008070116 W ES 2008070116W WO 2008152178 A1 WO2008152178 A1 WO 2008152178A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- radical
- alkyl
- pharmaceutically acceptable
- formula
- group
- Prior art date
Links
- 108010005843 Cysteine Proteases Proteins 0.000 title claims description 7
- 102000005927 Cysteine Proteases Human genes 0.000 title claims description 7
- 239000003112 inhibitor Substances 0.000 title abstract description 9
- 150000001875 compounds Chemical class 0.000 claims abstract description 88
- -1 4-hydroxy-phenylmethyl Chemical group 0.000 claims abstract description 54
- 150000003254 radicals Chemical class 0.000 claims abstract description 31
- 150000003839 salts Chemical class 0.000 claims abstract description 27
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 15
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims abstract description 11
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 11
- 201000004792 malaria Diseases 0.000 claims abstract description 11
- 201000010099 disease Diseases 0.000 claims abstract description 10
- 208000000230 African Trypanosomiasis Diseases 0.000 claims abstract description 9
- 208000029080 human African trypanosomiasis Diseases 0.000 claims abstract description 9
- 125000006239 protecting group Chemical group 0.000 claims abstract description 9
- 201000002612 sleeping sickness Diseases 0.000 claims abstract description 9
- 241000223105 Trypanosoma brucei Species 0.000 claims abstract description 8
- 230000002265 prevention Effects 0.000 claims abstract description 5
- 125000006650 (C2-C4) alkynyl group Chemical group 0.000 claims abstract description 4
- 125000004399 C1-C4 alkenyl group Chemical group 0.000 claims abstract description 4
- 108010007459 falcipain Proteins 0.000 claims abstract description 4
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 4
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract 2
- 125000000217 alkyl group Chemical group 0.000 claims description 26
- 238000000034 method Methods 0.000 claims description 16
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 claims description 14
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 claims description 13
- 239000012453 solvate Substances 0.000 claims description 13
- 239000003814 drug Substances 0.000 claims description 11
- 101000831241 Trypanosoma cruzi Cruzipain Proteins 0.000 claims description 10
- 206010001935 American trypanosomiasis Diseases 0.000 claims description 9
- 241000223109 Trypanosoma cruzi Species 0.000 claims description 9
- 238000002360 preparation method Methods 0.000 claims description 9
- 239000003795 chemical substances by application Substances 0.000 claims description 8
- 230000005764 inhibitory process Effects 0.000 claims description 8
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 8
- 208000024699 Chagas disease Diseases 0.000 claims description 7
- 125000005842 heteroatom Chemical group 0.000 claims description 5
- 125000006518 morpholino carbonyl group Chemical group [H]C1([H])OC([H])([H])C([H])([H])N(C(*)=O)C1([H])[H] 0.000 claims description 5
- 229910052760 oxygen Inorganic materials 0.000 claims description 5
- 229910052717 sulfur Inorganic materials 0.000 claims description 5
- VOLGAXAGEUPBDM-UHFFFAOYSA-N $l^{1}-oxidanylethane Chemical group CC[O] VOLGAXAGEUPBDM-UHFFFAOYSA-N 0.000 claims description 4
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 3
- 125000002619 bicyclic group Chemical group 0.000 claims description 3
- 229910052799 carbon Inorganic materials 0.000 claims description 3
- 238000006735 epoxidation reaction Methods 0.000 claims description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen(.) Chemical compound [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 3
- 239000007800 oxidant agent Substances 0.000 claims description 3
- 230000003647 oxidation Effects 0.000 claims description 3
- 238000007254 oxidation reaction Methods 0.000 claims description 3
- 230000006806 disease prevention Effects 0.000 claims description 2
- 230000001404 mediated effect Effects 0.000 claims description 2
- 150000004677 hydrates Chemical class 0.000 abstract description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 abstract description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 abstract 1
- 108090000711 cruzipain Proteins 0.000 abstract 1
- 230000007170 pathology Effects 0.000 abstract 1
- 239000000543 intermediate Substances 0.000 description 50
- 239000000203 mixture Substances 0.000 description 27
- 238000006243 chemical reaction Methods 0.000 description 20
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 16
- 239000000243 solution Substances 0.000 description 16
- 239000011734 sodium Substances 0.000 description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000002904 solvent Substances 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 11
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 9
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 102000004190 Enzymes Human genes 0.000 description 9
- 108090000790 Enzymes Proteins 0.000 description 9
- 238000004587 chromatography analysis Methods 0.000 description 7
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 6
- HSJKGGMUJITCBW-UHFFFAOYSA-N 3-hydroxybutanal Chemical compound CC(O)CC=O HSJKGGMUJITCBW-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 108010085937 benzyloxycarbonyl-phenylalanylarginine-4-methylcoumaryl-7-amide Proteins 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 4
- 108090000712 Cathepsin B Proteins 0.000 description 4
- 102000004225 Cathepsin B Human genes 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- WLLIXJBWWFGEHT-UHFFFAOYSA-N [tert-butyl(dimethyl)silyl] trifluoromethanesulfonate Chemical compound CC(C)(C)[Si](C)(C)OS(=O)(=O)C(F)(F)F WLLIXJBWWFGEHT-UHFFFAOYSA-N 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 239000012948 isocyanate Substances 0.000 description 4
- 150000002513 isocyanates Chemical class 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 0 *[C@@]([C@@](C=C*)O)NC([C@](N*)I)=O Chemical compound *[C@@]([C@@](C=C*)O)NC([C@](N*)I)=O 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 244000089409 Erythrina poeppigiana Species 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000030852 Parasitic disease Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000223960 Plasmodium falciparum Species 0.000 description 2
- 235000009776 Rathbunia alamosensis Nutrition 0.000 description 2
- 229910004161 SiNa Inorganic materials 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 241000224557 Trypanosoma brucei brucei Species 0.000 description 2
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 150000001723 carbon free-radicals Chemical group 0.000 description 2
- GLNDAGDHSLMOKX-UHFFFAOYSA-N coumarin 120 Chemical compound C1=C(N)C=CC2=C1OC(=O)C=C2C GLNDAGDHSLMOKX-UHFFFAOYSA-N 0.000 description 2
- 239000002852 cysteine proteinase inhibitor Substances 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 229920002113 octoxynol Polymers 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- OTGHWLKHGCENJV-UHFFFAOYSA-M oxirane-2-carboxylate Chemical compound [O-]C(=O)C1CO1 OTGHWLKHGCENJV-UHFFFAOYSA-M 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- JSPLKZUTYZBBKA-UHFFFAOYSA-N trioxidane Chemical compound OOO JSPLKZUTYZBBKA-UHFFFAOYSA-N 0.000 description 2
- 201000002311 trypanosomiasis Diseases 0.000 description 2
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 2
- XEEQGYMUWCZPDN-DOMZBBRYSA-N (-)-(11S,2'R)-erythro-mefloquine Chemical compound C([C@@H]1[C@@H](O)C=2C3=CC=CC(=C3N=C(C=2)C(F)(F)F)C(F)(F)F)CCCN1 XEEQGYMUWCZPDN-DOMZBBRYSA-N 0.000 description 1
- SLDUGQISGRPGAW-VIFPVBQESA-N (4s)-4-benzyl-1,3-thiazolidine-2-thione Chemical compound C1SC(=S)N[C@H]1CC1=CC=CC=C1 SLDUGQISGRPGAW-VIFPVBQESA-N 0.000 description 1
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- SUNMBRGCANLOEG-UHFFFAOYSA-N 1,3-dichloroacetone Chemical class ClCC(=O)CCl SUNMBRGCANLOEG-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- PAFVKDRJXASKEG-SFHVURJKSA-N 1-[(4s)-4-benzyl-2-sulfanylidene-1,3-thiazolidin-3-yl]-4-phenylbutan-1-one Chemical compound C([C@@H]1CC=2C=CC=CC=2)SC(=S)N1C(=O)CCCC1=CC=CC=C1 PAFVKDRJXASKEG-SFHVURJKSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- WSNDAYQNZRJGMJ-UHFFFAOYSA-N 2,2,2-trifluoroethanone Chemical compound FC(F)(F)[C]=O WSNDAYQNZRJGMJ-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- VQDQISMDUHBUFF-UHFFFAOYSA-N 4-phenylbutanoyl chloride Chemical compound ClC(=O)CCCC1=CC=CC=C1 VQDQISMDUHBUFF-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 241000256186 Anopheles <genus> Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000008316 Arsenic Poisoning Diseases 0.000 description 1
- CULUWZNBISUWAS-UHFFFAOYSA-N Benznidazole Chemical compound [O-][N+](=O)C1=NC=CN1CC(=O)NCC1=CC=CC=C1 CULUWZNBISUWAS-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 101000749287 Clitocybe nebularis Clitocypin Proteins 0.000 description 1
- 101000767029 Clitocybe nebularis Clitocypin-1 Proteins 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 229940094664 Cysteine protease inhibitor Drugs 0.000 description 1
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical class C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- LLQPHQFNMLZJMP-UHFFFAOYSA-N Fentrazamide Chemical compound N1=NN(C=2C(=CC=CC=2)Cl)C(=O)N1C(=O)N(CC)C1CCCCC1 LLQPHQFNMLZJMP-UHFFFAOYSA-N 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 241001502121 Glossina brevipalpis Species 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- JTTHKOPSMAVJFE-VIFPVBQESA-N L-homophenylalanine Chemical compound OC(=O)[C@@H](N)CCC1=CC=CC=C1 JTTHKOPSMAVJFE-VIFPVBQESA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- JCYZMTMYPZHVBF-UHFFFAOYSA-N Melarsoprol Chemical compound NC1=NC(N)=NC(NC=2C=CC(=CC=2)[As]2SC(CO)CS2)=N1 JCYZMTMYPZHVBF-UHFFFAOYSA-N 0.000 description 1
- VZSXPUDQSLKVIR-JDXGNMNLSA-N N-[(2S)-1-[[(3S)-1-(benzenesulfonyl)-5-phenylpentan-3-yl]amino]-1-oxo-3-phenylpropan-2-yl]-4-methylpiperazine-1-carboxamide Chemical compound C1CN(C)CCN1C(=O)N[C@H](C(=O)N[C@@H](CCC=1C=CC=CC=1)CCS(=O)(=O)C=1C=CC=CC=1)CC1=CC=CC=C1 VZSXPUDQSLKVIR-JDXGNMNLSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- ARFHIAQFJWUCFH-IZZDOVSWSA-N Nifurtimox Chemical compound CC1CS(=O)(=O)CCN1\N=C\C1=CC=C([N+]([O-])=O)O1 ARFHIAQFJWUCFH-IZZDOVSWSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000224016 Plasmodium Species 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 241001442399 Trypanosoma brucei gambiense Species 0.000 description 1
- 241001442397 Trypanosoma brucei rhodesiense Species 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- JUCMRTZQCZRJDC-UHFFFAOYSA-N acetyl fluoride Chemical class CC(F)=O JUCMRTZQCZRJDC-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 238000005575 aldol reaction Methods 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000000729 antidote Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000009697 arginine Nutrition 0.000 description 1
- 150000001495 arsenic compounds Chemical class 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 229960004001 benznidazole Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000005415 bioluminescence Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 150000001767 cationic compounds Chemical class 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229960003677 chloroquine Drugs 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- AFOSIXZFDONLBT-UHFFFAOYSA-N divinyl sulfone Chemical class C=CS(=O)(=O)C=C AFOSIXZFDONLBT-UHFFFAOYSA-N 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000007908 dry granulation Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 1
- 229960002759 eflornithine Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 229940125532 enzyme inhibitor Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- BLHLJVCOVBYQQS-UHFFFAOYSA-N ethyllithium Chemical compound [Li]CC BLHLJVCOVBYQQS-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 150000002367 halogens Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 229910001411 inorganic cation Inorganic materials 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 239000013038 irreversible inhibitor Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 229960001962 mefloquine Drugs 0.000 description 1
- 229960001728 melarsoprol Drugs 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229960002644 nifurtimox Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- BSCHIACBONPEOB-UHFFFAOYSA-N oxolane;hydrate Chemical compound O.C1CCOC1 BSCHIACBONPEOB-UHFFFAOYSA-N 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229960005190 phenylalanine Drugs 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 230000000391 smoking effect Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 208000037972 tropical disease Diseases 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/336—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having three-membered rings, e.g. oxirane, fumagillin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/48—Compounds containing oxirane rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/05—Dipeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D301/00—Preparation of oxiranes
- C07D301/02—Synthesis of the oxirane ring
- C07D301/03—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds
- C07D301/14—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds with organic peracids, or salts, anhydrides or esters thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to new cysteine protease inhibitors, as well as intermediates and methods useful for their preparation, and to pharmaceutical compositions containing them.
- Chagas disease or American trypanosomiasis is a tropical disease caused by the Trypanosoma cruzi protozoan that affects between 16 and 18 million people in Latin America where it causes 50,000 deaths per year. It is estimated that 25% of the population of Latin America (90 million) is at risk of contracting the disease (Weekly Epidemiological Record, World Health Organization, 2000, 2: 10-12).
- African trypanosomiasis or sleeping sickness affects about half a million people in sub-Saharan Africa, an estimated 66 million people in 36 countries are at risk of contracting it.
- the eastern variant of the disease is caused by the Trypanosoma brucei rhodesiense protozoan and the western one by Trypanosoma brucei gambiense, the transmitting agent is in both cases Ia tse-tse fly, widespread throughout Africa.
- malaria is caused by protozoa of the genus Plasmodium, mainly Plasmodium falciparum and is transmitted to people through the Anopheles mosquito. Malaria affects almost 40% of the world's population where more than three billion are at risk, occurs in about one hundred countries in very depressed tropical areas of Africa, Asia and Latin America. More than 90% of cases and 80% of deaths occur in tropical Africa (World Malaria Report 2005).
- Chagas disease African trypanosomiasis and malaria have a similar infectious cycle in which a transmitting agent (insect) transmits the pathogen to man. All the pathogens of these diseases have a common essential enzymatic activity for their life cycle, the cysteine-protease activity: cruzaine in Trypanosoma cruzi, rhodesa ⁇ na in T. Brucei and falcipaine in Plasmodium falciparum (CR. Caffrey et al. Mol. Biochem Parasitol, 2001, vol. 1. pp. 61-73)
- the inventors have found new cysteine protease inhibitor compounds cruzaine, rhodesaine and falcipain; which are also selective against cathepsin B.
- one aspect of the present invention refers to a substantially pure diastereoisomeric compound of formula Ia, alternatively Ib, or a pharmaceutically acceptable salt thereof or a pharmaceutically acceptable solvate thereof, including hydrates,
- GP is a protective group
- Ri is a radical selected from the group consisting of phenylmethyl, A-hydroxyphenylmethyl, (1 / - / - indole-3-yl) methyl and (1 / - / - imidazol-4-yl) methyl;
- R 2 is a radical selected from the group consisting of -H, -CH 3 , -CH 2 SH, -CH 2 OH, -CH 2 Ph, -CH 2 CO 2 H, -CH 2 CONH 2 , -CH (OH ) CH 3 , -CH (CH 3 ) CH 2 CH 3 , -CH (CH 3 ) 2 , -CH 2 CH (CH 3 ) 2 , - (CH 2 J 2 SCH 3 , - (CH 2 J 2 CO 2 H, - (CH 2 ) CONH 2 , - (CH 2 ) 3 NHC (NH) NH 2 , - (CH 2 ) 4 NH 2 , imidazol-4-ylmethyl, 4-hydroxyphenylmethyl, (1 / - / - indole-3-yl) methyl, (1 / - / -imidazol-4-yl) methyl and - (CH 2 ) n -Ar; Y
- R 3 is a radical selected from the group consisting of -O (dC 4 ) alkyl, -O (C 2 -C 4 ) alkenyl, -O (C 2 -C 4 ) alkynyl, -O (C r C 4 ) alkyl -Ar, -OAr, -NR 3 Ar, -N (R a ) [(dC 4 ) alkyl-Ar], -NR 3 OAr and -N (R a ) [O (C r C 4 ) alkyl-Ar] ;
- n represents a value selected between 2 and 3;
- Ar is a carbon or nitrogen radical of a known 5 to 6 membered monocyclic aromatic carbocyclic ring or 8 to 10 membered bicyclic ring, optionally containing 1 to 3 heteroatoms selected from N 1 S and O, and which may be optionally substituted by one or more radicals independently selected from the group consisting of -OH, -CHO, -SH, -NO 2 , -CN, -F, -Cl, -Br, - (dC 4 ) alkyl optionally substituted by one or more radicals independently selected from the group consisting of -F, -Cl, -Br and -OH; -O (dC 4 ) alkyl optionally substituted by one or more radicals independently selected from the group consisting of -F, -Cl, -Br and -OH; -CO (C r C 4 ) alkyl, -OCO (C r C 4 ) alkyl, -S (C r C 4 )
- R a and R b independently represent a radical -H or - (dC 4 ) alkyl.
- the object of the present invention is each individual stereoisomer as well as mixtures thereof.
- some of the compounds of the present invention may have cis / trans isomerism.
- the object of the present invention is each of the geometric isomers as well as their mixtures.
- the compounds of the invention of formula Ia are inhibitors of a cysteine protease selected from the group consisting of cruzaine, rhodesaine and falcipain. Therefore, another aspect of the present invention relates to a pharmaceutical composition comprising a therapeutically effective amount of a substantially pure diastereoisomeric compound of formula Ia, alternatively Ib, or a pharmaceutically acceptable salt thereof or a pharmaceutically acceptable solvate thereof. , including a hydrate, together with appropriate amounts of pharmaceutically acceptable excipients.
- Another aspect of the present invention relates to the use of the compounds of the invention of formula Ia, alternatively Ib, for the preparation of a medicament for the treatment and / or prevention of diseases mediated by the inhibition of a cysteine protease selected from the group consisting of cruzaine, rhodesa ⁇ na and falcipa ⁇ na.
- a cysteine protease selected from the group consisting of cruzaine, rhodesa ⁇ na and falcipa ⁇ na.
- Another aspect of the present invention relates to the use of the compounds of the invention of formula Ia, alternatively Ib, for the preparation of a medicament for the treatment and / or prevention of Chagas disease or African trypanosomiasis.
- Another aspect of the present invention relates to the use of the compounds of the invention of formula Ia, alternatively Ib, for the preparation of a medicament for the treatment and / or prevention of malaria.
- the invention is also related to a method for the treatment and / or prevention of a mammal, including a human, who suffers or is susceptible to suffering from a disease selected from the group consisting of Chagas disease, African trypanosomiasis and malaria.
- Said method comprises administering to said patient a therapeutically effective amount of a compound of the invention of formula Ia, alternatively Ib, together with pharmaceutically acceptable excipients or carriers.
- Another aspect of the invention relates to a process for preparing the compounds of the invention of formula Ia, alternatively Ib, which comprises the oxidation of a compound of formula Ma, alternatively Mb, with a suitable oxidizing agent:
- Another aspect of the present invention relates to an intermediate diastereoisomeric compound of formula Ma, alternatively Mb, or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate thereof, including hydrates,
- Another aspect of the present invention relates to a process for preparing a compound of the invention of formula Ma, alternatively Mb, which comprises the epoxidation of a compound of formula MIa, alternatively MIb, with a suitable epoxidant agent:
- (Ci-C 4 ) alkyl means a radical of a linear or branched saturated hydrocarbon chain containing 1 to 4 carbon atoms. Examples include among others the methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and tere-butyl groups.
- (C 2 -C 4 ) alkenyl means a radical of a linear or branched unsaturated hydrocarbon chain containing 1 to 4 carbon atoms and at least one double bond. Examples include among others the groups ethenyl, propenyl, isopropenyl, 1-butenyl, 2-butenyl, 3-butenyl and 1,3-butadienyl.
- (C2-C 4 ) alkynyl means a radical of a linear or branched unsaturated hydrocarbon chain containing 1 to 4 carbon atoms and at least one triple bond. Examples include among others the ethynyl, propynyl, isopropyl, 1-butynyl, 2-butynyl and 3-butynyl groups.
- Ar is a carbon or nitrogen radical of a known 5 to 6 membered monocyclic aromatic carbocyclic ring or 8 to 10 membered bicyclic ring, optionally containing 1 to 3 heteroatoms selected from N 1 S and O, and which may be optionally substituted as indicated previously.
- Ar radicals include, without limitation, thiophenyl, furanyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, triazolyl, thiadiazolyl, oxadiazolyl, phenyl, pyridinyl, pyrazinyl, pyridazinyl, pirimidininilo, triazinyl, tienofuranilo, tienopirrolilo, pirrolopirazolilo , benzothiophenyl, benzofuranyl, indolyl, isoindolyl, benzimidazolyl, benzothiazolyl, imidazopyridinyl, pyrazolopyridinyl, isoquinolinyl, quinolinyl, quinolizinyl, naphthyridinyl, quinazolinyl,
- substantially pure diastereoisomeric compound means a compound with sufficient diastereoisomeric excess for its preparation on an industrial scale, which depends on each specific case as the person skilled in the art will decide. In most cases, a diastereoisomeric excess greater than 95% is sufficient.
- a particular embodiment of the invention refers to a substantially pure diastereoisomeric compound of formula Ia, or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate thereof, including a hydrate.
- Ri represents phenylmethyl
- R 2 is a radical - (CH 2 ) n -Ar
- R 3 is a radical selected from the group consisting of -O (dC 4 ) alkyl, -O (dC 4 ) alkyl-Ar, -OAr, n represents 2
- Ar is a carbon radical of a known 5- to 6-membered monocyclic aromatic carbocyclic ring, optionally containing 1 to 3 heteroatoms selected from N 1 S and O.
- GP is a radical selected from the group consisting of benzyloxycarbonyl, morpholinocarbonyl and ⁇ / -methylcarbonyl; Ri is a phenylmethyl radical, R 2 is a 2-phenylethyl radical and R3 is an -O (dC 4 ) alkyl radical.
- GP is a benzyloxycarbonyl radical
- Ri is a phenylmethyl radical
- R 2 is a 2-phenylethyl radical
- R3 is an ethoxy radical.
- the invention also relates to intermediates of formula Ma, alternatively Mb, to its pharmaceutically acceptable salts and pharmaceutically acceptable solvates, including hydrates.
- Another particular embodiment relates to a substantially pure diastereoisomeric intermediate of formula Ma, or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate thereof, including a hydrate.
- Ri represents phenylmethyl
- R 2 is a radical - (CH 2 ) n -Ar
- R 3 is a radical selected from the group consisting of -O (dC 4 ) alkyl, -O (dC 4 ) alkyl-Ar, -OAr, n represents 2
- Ar is a carbon radical of a known 5- to 6-membered monocyclic aromatic carbocyclic ring, optionally containing 1 to 3 heteroatoms selected from N, S and O.
- Ma alternatively Mb, GP is a radical selected from the group consisting of benzyloxycarbonyl, morpholinocarbonyl and ⁇ / -methylcarbonyl; Ri is a phenylmethyl radical, R 2 is a 2-phenylethyl radical and R3 is an -O (d-C 4 ) alkyl radical.
- GP is a benzyloxycarbonyl radical
- Ri is a phenylmethyl radical
- R 2 is a 2-phenylethyl radical
- R 3 is an ethoxy radical
- the compounds of the present invention may contain one or more basic nitrogen and therefore may form salts with acids, both organic and inorganic, which are also part of the present invention.
- salts include: salts with inorganic acids such as hydrochloric acid, hydrobromic acid, iodhydric acid, nitric acid, perchloric acid, sulfuric acid or phosphoric acid; and salts with organic acids, such as methanesulfonic acid, trifluoromethanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, fumaric acid, oxalic acid, acetic acid or maleic acid, among others.
- the compounds of the present invention may contain one or more acidic protons and therefore could form salts with bases, which are also part of the present invention.
- salts include: salts with inorganic cations such as sodium, potassium, calcium, magnesium, lithium, aluminum, zinc, etc .; salts formed with pharmaceutically acceptable amines such as ammonia, alkylamines, hydroxyalkylamines, lysine, arginine, N-methylglucamine, procaine and the like.
- salts with inorganic cations such as sodium, potassium, calcium, magnesium, lithium, aluminum, zinc, etc .
- salts formed with pharmaceutically acceptable amines such as ammonia, alkylamines, hydroxyalkylamines, lysine, arginine, N-methylglucamine, procaine and the like.
- the salts can be prepared by treating the compound of formula Ia, Ib, Ma or Mb with a sufficient amount of the desired acid or base to give the salt in a conventional manner.
- the salts of the compounds of formula Ia, Ib, Ma or Mb can in turn be transformed into other salts of compounds of formula Ia, Ib, Ma or Mb respectively, by ion exchange using an ion exchange resin.
- the present invention also includes pharmaceutical compositions.
- the formulation chosen depends on the nature of the compound administered and the route of administration. Different routes of administration can be used, for example oral or topical administration.
- Solid compositions for oral administration include tablets, granules and capsules.
- the preparation method can be based on simple mixing, dry granulation or wet granulation of the active ingredient with excipients.
- excipients can be, for example, diluents, binding agents, disintegrants, and lubricating agents.
- the tablets may be coated with suitable excipients to delay their disintegration and absorption in the gastrointestinal tract or to improve their organoleptic properties or their stability.
- the active ingredient can also be incorporated by coating on inert pellets by using natural or synthetic smoking polymers.
- the active substance is mixed with water or oily medium.
- Oral suspensions of powders and granules can be prepared by adding water, mixing the active ingredient with dispersing or wetting agents; suspending and preservative.
- Other excipients can also be added, for example sweeteners, flavorings and dyes.
- compositions for oral administration, emulsions, solutions, suspensions, syrups and elixirs containing commonly used inert diluents can be included.
- Such compositions may also contain adjuvants such as wetting, suspending, sweetening, flavoring, preservative and pH regulating agents.
- Topical formulations include creams, lotions, gels, ointments, powders, solutions and patches in which the compound is dispersed or dissolved in suitable excipients.
- the substantially pure diastereoisomeric compounds of formula Ia can be prepared by the procedures described below. As will be apparent to one skilled in the art, the precise method used for the preparation of a given compound may vary depending on its chemical structure. If the opposite is not specified, the meanings of the GP, Ri, R 2 and R3 variables are as indicated above.
- protecting groups of an amino function are the 9-fluorenylmethoxycarbonyl (Fmoc), tert-butoxycarbonyl (Boc), benzyl (Bn), benzyloxycarbonyl (Cbz), trifluoroacetyl (TFA), morpholinocarbonyl and ⁇ / groups -methylcarbonyl.
- the hydroxyl groups can be protected, for example, with trimethylsilyl groups (TMS), tere- butyldimethylsilyl (TBS) or tetrahydropyranyl (THP).
- TMS trimethylsilyl groups
- TBS tere- butyldimethylsilyl
- THP tetrahydropyranyl
- a substantially pure diastereoisomeric compound of formula Ia can be obtained by oxidation of a compound of formula Ma. This reaction is carried out in the presence of an oxidizing agent, such as D-Martin Periodine, in a suitable solvent, such as example dichloromethane, at a suitable temperature, preferably room temperature.
- an oxidizing agent such as D-Martin Periodine
- a suitable solvent such as example dichloromethane
- a compound of formula Ma can be obtained by an epoxidation reaction of an allyl alcohol of formula MIa. This reaction is carried out in the presence of an epoxidant agent, such as lithium ⁇ erc-butylhydroperoxide, in a suitable solvent, such as tetrahydrofuran, at a suitable temperature, preferably room temperature. Under these conditions, the epoxyalcohol is obtained mostly without respect to the hydroxyl group (S. Rodr ⁇ guez et al, Tetrahedron 2006, pp. 1112-11123).
- an epoxidant agent such as lithium ⁇ erc-butylhydroperoxide
- a compound of formula Illa can be prepared from a carboxylic acid of formula Va, where GP 'is a hydroxyl protecting group, for example tert-butyl dimethylsilyl, by a sequence of several steps.
- a first step the carboxylic acid group of the compound of formula Va is transformed into an amino group by a Curtius transposition.
- This reaction can be carried out with diphenylphosphorylazide, in the presence of a base, such as triethylamine, in a suitable solvent, such as ⁇ /, ⁇ / -dimethylformamide, at a suitable temperature, preferably room temperature, forming an intermediate isocyanate.
- the intermediate isocyanate can be formed by reacting the carboxylic acid of formula Va with methyl chloroformate and subsequently sodium azide.
- the amine obtained in the first stage is converted into an amide of formula IVa by reaction with an amino acid of formula VIa with the amino group conveniently protected if necessary, for example protected with the benzyloxycarbonyl group.
- This reaction can be carried out in the presence of a suitable condensing agent such as for example N- (3-dimethylaminopropyl)-) / '-ethylcarbodiimide or dicyclohexylcarbodiimide optionally in the presence of 1-hydroxybenzotriazole, or in the presence of a suitable base, such as 4-dimethylaminopriridine or pyridine, within a suitable solvent, such as dichloromethane or dimethylformamide.
- a suitable condensing agent such as for example N- (3-dimethylaminopropyl)-) / '-ethylcarbodiimide or dicyclohexylcarbodiimide optionally in the presence of 1-hydroxybenzotriazole, or in the presence of a suitable base, such as 4-dimethylaminopriridine or pyridine, within a suitable solvent, such as dichloromethane or dimethylformamide.
- the isocyanate obtained in the first stage is converted into an amide of formula IVa by reaction with an amino acid of formula VIa with the amino group conveniently protected if necessary, for example, protected with the benzyloxycarbonyl group.
- This reaction can be carried out in the presence of a suitable base, such as 4-dimethylaminopriridine, within a suitable solvent, such as dichloromethane.
- a compound of formula Va can be obtained from a compound of formula XIII following the synthetic route indicated in the following scheme: introduction of
- the carboxylic acid of formula XIII is converted into an acylated derivative of formula Xl where X represents halogen, preferably chlorine, by reaction with a halogenating agent, for example thionyl chloride, in a suitable solvent, such as benzene, at a suitable temperature. , preferably heating.
- a halogenating agent for example thionyl chloride
- the compound of formula Xl is converted into a compound of formula IX by reaction with a compound of formula XII, where X c represents a chiral auxiliary, such as (S) -4-benzyl-2-thioxothiazolidin-3-yl.
- This reaction is carried out in the presence of a base, such as triethylamine or 2,6-lutidine and optionally in the presence of ferc-butyldimethylsilyl triflate (TBSOTf), in a suitable solvent, such as dichloromethane or without solvent and suitable temperature, for example room temperature.
- a base such as triethylamine or 2,6-lutidine
- TBSOTf ferc-butyldimethylsilyl triflate
- Hydrolysis of a compound of formula VIIa results in a compound of formula Va.
- This reaction can be carried out in the presence of hydrogen peroxide and a base, such as lithium hydroxide, in a suitable solvent, such as a tetrahydrofuran-water mixture and at a suitable temperature, preferably room temperature.
- a base such as lithium hydroxide
- a substantially pure diastereoisomeric compound of formula Ib may Obtaining analogously to obtaining a compound of formula Ia from the corresponding aldol of formula VIIIb:
- a compound of formula Ia, alternatively Ib can be transformed into another compound of formula Ia, alternatively Ib.
- a compound of formula Ia, alternatively Ib, where R3 represents -O (dC 4 ) alkyl, -O (dC 4 ) alkyl-Ar or -OAr can be transformed into a compound of formula Ia, alternatively Ib, where R3 represents -NHAr or -NH-OAr, through reactions well known to the person skilled in the art.
- the interconversion reactions of a radical R 3 into another radical R 3 can also be carried out on any of the intermediates of the compounds of the invention of formula Ia, alternatively Ib.
- IR spectra were recorded in oily films on NaCI pellets on a Perkin-Elmer 2000 FT-IR spectrometer.
- EM Science Silica GeI 60 was used for column chromatography while TLC was performed with aluminum sheets loaded with E. Merck silica (Kieselgel 60, F 2 5 4 , 0.25 mm). Except in the indicated cases, all reactions were carried out under a nitrogen atmosphere with magnetic stirring.
- TX-100 Triton-X (trimethylbenzylammonium hydroxide) Z: benzyloxycarbonyl
- Intermediate Vlla. 1 ⁇ sin IR ⁇ 3063, 3027, 2954, 2930, 2857, 1718, 1659, 1604, 1496, 1471, 1455, 1365, 1341, 1262, 1193, 1161, 1135, 1083, 1083, 1039, 983, 939, 883, 838, 779, 747, 701, 667 cm "1 .
- the inhibition of cysteine proteases cruzaine, rhodesaine, cathepsin B (Tb CatB) and falcipaine (FP2) was evaluated by quantification of the fluorescence emission resulting from the proteolytic breakdown of the synthetic compound Z-Phe-Arg-AMC (Bachem).
- the Ia or Ib inhibitors were used in concentrations ranging from 10 nM to 10,000 nM (from 500 nM to 10.00OnM for Tb CatB); thus, serial dilutions were prepared in DMSO with the minimum concentration of inhibitor Ia or Ib used in the calculations and at least 10 times greater than that of the enzyme.
- the IC50s were determined by a direct reading of the semi log plot of the reaction rate versus the inhibitor concentration.
- Table 1 shows the values of percent inhibition corresponding to Trypanosoma brucei brucei (Tbb), cruzaine, rhodesaine and cathepsin B.
- Table 2 shows the IC 50 values of the enzymes falcipaine, cruzaine, rhodesaine and TbCat B.
- the effectiveness of the compounds of the invention can also be verified by cellular assays or animal models in vivo.
- the animal models of trypanosomiasis, Chagas disease and malaria are known in the state of the art.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Animal Behavior & Ethology (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Tropical Medicine & Parasitology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Epoxy Compounds (AREA)
Abstract
Compuestos diastereoisoméricos sustancialmente puros de fórmula Ia, alternativamente Ib, o una sal farmacéuticamente aceptable de los mismos o un solvato farmacéuticamente aceptable de los mismos, incluyendo hidratos, donde GP es un grupo protector; R1es un radical seleccionado del grupo que consiste en fenilmetilo, 4-hidroxifenilmetilo, (1H-indol-3-il)metilo y (1H- imidazol-4-il)metilo; R2es un radical seleccionado del grupo que consiste en -H, -CH3, -CH2SH, -CH2OH, -CH2Ph, -CH2CO 2H, -CH2CONH2, -CH(OH)CH3, -CH(CH3)CH2CH3, -CH(CH3)2, -CH2CH(CH3)2,-(CH2)2SCH3, -(CH2)2CO2H, -(CH2)CONH2, -(CH2)3NHC(NH)NH2, -(CH2)4NH2, imidazol-4-ilmetilo, 4- hidroxifenilmetilo, (1H-indol-3-il)metilo, (1H-imidazol-4-il)metilo y -(CH 2) n-Ar; y R3 es un radical seleccionado del grupo que consiste en -O(C1-C4)alquilo, -O(C2-C4)alquenilo,-O(C2-C4)alquinilo,-O(C1-C4)alquilo-Ar, -OAr, -NRa Ar, -N(Ra )[(C1-C4)alquilo-Ar], y-NRa OAry -N(Ra )[O(C1-C4)alquilo-Ar]. Dichos compuestos son inhibidores de las cisteína-proteasas cruzaína, rhodesaína y falcipaína, y por tanto, útiles en el tratamiento y/o prevención de patologías tales como la enfermedad de Chagas, la tripanosomiasis africana o la malaria.
Description
Inhibidores de cisteína-proteasas
La presente invención se refiere a nuevos inhibidores de cisteína-proteasas, así como intermedios y procedimientos útiles para su preparación, y a composiciones farmacéuticas que los contienen.
ESTADO DE LA TÉCNICA ANTERIOR
La tripanosomiasis y Ia malaria son enfermedades parasitarias mayoritarias en países en vías de desarrollo.
La enfermedad de Chagas o tripanosomiasis americana es una enfermedad tropical causada por el protozoo Trypanosoma cruzi que afecta entre 16 y 18 millones de personas en Latinoamérica donde causa 50.000 muertes por año. Se estima que el 25% de Ia población de Latinoamérica (90 millones) está en riesgo de contraer Ia enfermedad (Weekly Epidemiological Record, World Health Organization, 2000, 2:10-12).
La tripanosomiasis africana o enfermedad del sueño afecta a alrededor de medio millón de personas del África subsahariana, se calcula que 66 millones de personas en 36 países están en riesgo de contraerla. La variante oriental de Ia enfermedad está causada por el protozoo Trypanosoma brucei rhodesiense y Ia occidental por Trypanosoma brucei gambiense, el agente transmisor es en ambos casos Ia mosca tse-tse, muy extendida por todo África.
Por otra parte, Ia malaria está causada por protozoos del género Plasmodium, principalmente Plasmodium falciparum y se transmite a las personas a través del mosquito anofeles. La malaria afecta a casi el 40% de Ia población mundial donde más de tres mil millones están en riesgo, se da en unos cien países de áreas tropicales muy deprimidas de África, Asia y Latinoamérica. Más del 90% de los casos y el 80% de las muertes se dan en el África tropical (World Malaria Report 2005).
No existe a día de hoy ninguna vacuna para Ia tripanosomiasis americana o enfermedad de Chagas, los fármacos empleados (benznidazole y nifurtimox) sólo sirven en las etapas tempranas, muestran baja eficacia y efectos
secundarios indeseados. Por otro lado, el único fármaco disponible para Ia tripanosomiasis africana o enfermedad del sueño es el melarsoprol que consiste en un compuesto de arsénico tóxico que se suministra con su antídoto y que expone a un 10% de riesgo de encefalopatía arsénica, por ello unos mil pacientes mueren cada año a causa de Ia toxicidad del fármaco. Recientemente, se ha desarrollado Ia eflornithina para el tratamiento de Ia enfermedad que, a pesar de que ha mostrado ser efectiva y ser bien tolerada, presenta muchas desventajas.
Finalmente, aunque existen algunos fármacos para prevenir Ia enfermedad de Ia malaria como Ia cloroquina y Ia mefloquina, éstos muestran efectos secundarios y no existe ninguno que garantice Ia protección total.
La enfermedad de Chagas, Ia tripanosomiasis africana y Ia malaria tienen un ciclo infeccioso parecido en el que un agente transmisor (insecto) transmite el agente patógeno al hombre. Todos los agentes patógenos de estas enfermedades poseen una actividad enzimática esencial común para su ciclo de vida, Ia actividad cisteína-proteasa: cruzaína en Tripanosoma cruzi, rhodesaína en T. Brucei y falcipaína en Plasmodium falciparum (CR. Caffrey et al. Mol. Biochem. Parasitol. 2001 , vol. 1. pp. 61-73)
Una línea de investigación reciente dirigida a Ia búsqueda de fármacos para estas enfermedades se basa en Ia inhibición de las cisteína-proteasas mencionadas, de manera que Ia síntesis de inhibidores de estos enzimas podría dar lugar a nuevos fármacos con los cuales combatir las enfermedades, especialmente como alternativa a Ia terapia tradicional en organismos resistentes (J. H. McKerrow et al, Bioorq. Med. Chem. 1999, vol. 7, pp. 639-644).
En este sentido, se han desarrollado varios inhibidores irreversibles de cisteína-proteasas basados en péptidos, como por ejemplo halometil cetonas, diazometanos, derivados de epoxisuccinilo, y derivados de sulfonas vinílicas (H.Otto et al, Chem. Rev. 1997, vol. 97, pp.133-171 ). Una desventaja de las clorometil cetonas es su elevada reactividad (reaccionan con serina- proteasas y otras moléculas que contienen grupos SH) y consecuente falta de selectividad de modo que resultan tóxicas in vivo. Para incrementar Ia selectividad y reducir Ia reactividad de estos compuestos, se han
desarrollado otras moléculas menos reactivas, como por ejemplo las monofluoro metil cetonas (D.Rasnick, Anal. Biochem. 1985, vol. 149, pp. 461- 465) y epoxiderivados (W. R. Roush et al, Bioorg. Med. Chem. Lett. 1998, pp. 2809-2812). Sin embargo, estos compuestos no son efectivos in vivo debido a su baja biodisponibilidad.
Así, a pesar de los esfuerzos invertidos en investigación en el pasado, el tratamiento de estas enfermedades parasitarias está lejos de ser satisfactorio. Por tanto, el desarrollo de nuevos compuestos para el tratamiento de estas enfermedades es todavía de gran interés.
EXPLICACIÓN DE LA INVENCIÓN
Los inventores han encontrado nuevos compuestos inhibidores de las cisteína-proteasas cruzaína, rhodesaína y falcipaína; que son además selectivos frente a Ia catepsina B.
Así, un aspecto de Ia presente invención se refiere a un compuesto diastereoisomérico sustancialmente puro de fórmula Ia, alternativamente Ib, o una sal farmacéuticamente aceptable de los mismos o un solvato farmacéuticamente aceptable de los mismos, incluyendo hidratos,

donde GP es un grupo protector;
Ri es un radical seleccionado del grupo que consiste en fenilmetilo, A- hidroxifenilmetilo, (1/-/-indol-3-il)metilo y (1/-/-imidazol-4-il)metilo;
R2 es un radical seleccionado del grupo que consiste en -H, -CH3, -CH2SH, -CH2OH, -CH2Ph, -CH2CO2H, -CH2CONH2, -CH(OH)CH3, -CH(CH3)CH2CH3, -CH(CH3)2, -CH2CH(CH3)2, -(CH2J2SCH3, -(CH2J2CO2H, -(CH2)CONH2,
-(CH2)3NHC(NH)NH2, -(CH2)4NH2, imidazol-4-ilmetilo, 4-hidroxifenilmetilo, (1/-/-indol-3-il)metilo, (1/-/-imidazol-4-il)metilo y -(CH2)n-Ar; y
R3 es un radical seleccionado del grupo que consiste en -O(d-C4)alquilo, -O(C2-C4)alquenilo, -O(C2-C4)alquinilo, -O(CrC4)alquilo-Ar, -OAr, -NR3Ar, -N(Ra)[(d-C4)alquilo-Ar], -NR3OAr y -N(Ra)[O(CrC4)alquilo-Ar];
donde n representa un valor seleccionado entre 2 y 3;
Ar es un radical de carbono o nitrógeno de un anillo conocido carbocíclico aromático monocíclico de 5 a 6 miembros o bicíclico de 8 a 10 miembros, que contiene opcionalmente de 1 a 3 heteroátomos seleccionados entre N1 S y O, y que puede estar opcionalmente sustituido por uno o más radicales seleccionados independientemente del grupo que consiste en -OH, -CHO, -SH, -NO2, -CN, -F, -Cl, -Br, -(d-C4)alquilo opcionalmente sustituido por uno o más radicales seleccionados independientemente del grupo que consiste en -F, -Cl, -Br y -OH; -O(d-C4)alquilo opcionalmente sustituido por uno o más radicales seleccionados independientemente del grupo que consiste en -F, -Cl, -Br y -OH; -CO(CrC4)alquilo, -OCO(CrC4)alquilo, -S(CrC4)alquilo, -SO(d-C4)alquilo, -SO2(d-C4)alquilo, -SO2O(d-C4)alquilo, -OSO2(CrC4)alquilo, -NRaRb, -CONRaRb; y
Ra y Rb representan independientemente un radical -H o -(d-C4)alquilo.
Algunos compuestos de Ia invención de fórmula Ia, alternativamente Ib, además de los centros quirales indicados, pueden poseer otros centros quirales que pueden dar lugar a diversos estereoisómeros. Son objeto de Ia presente invención cada uno de los estereoisómeros individuales así como sus mezclas.
Asimismo, algunos de los compuestos de Ia presente invención pueden presentar isomería cis/trans. Son objeto de Ia presente invención cada uno de los isómeros geométricos así como sus mezclas.
Los compuestos de Ia invención de fórmula Ia, alternativamente Ib, son inhibidores de una cisteína-proteasa seleccionada del grupo que consiste en cruzaína, rhodesaína y falcipaína.
Por ello, otro aspecto de Ia presente invención se refiere a una composición farmacéutica que comprende una cantidad terapéuticamente efectiva de un compuesto diastereoisómerico sustancialmente puro de fórmula Ia, alternativamente Ib, o una sal farmacéuticamente aceptable de los mismos o un solvato farmacéuticamente aceptable de los mismos, incluyendo un hidrato, junto con cantidades apropiadas de excipientes farmacéuticamente aceptables.
Otro aspecto de Ia presente invención se refiere al uso de los compuestos de Ia invención de fórmula Ia, alternativamente Ib, para Ia preparación de un medicamento para el tratamiento y/o prevención de enfermedades mediadas por Ia inhibición de una cisteína-proteasa seleccionada del grupo que consiste en cruzaína, rhodesaína y falcipaína.
Otro aspecto de Ia presente invención se refiere al uso de los compuestos de Ia invención de fórmula Ia, alternativamente Ib, para Ia preparación de un medicamento para el tratamiento y/o prevención de Ia enfermedad de Chagas o Ia tripanosomiasis africana. Otro aspecto de Ia presente invención se refiere al uso de los compuestos de Ia invención de fórmula Ia, alternativamente Ib, para Ia preparación de un medicamento para el tratamiento y/o prevención de Ia malaria.
La invención también está relacionada con un método para el tratamiento y/o prevención de un mamífero, incluyendo un humano, que padece o es susceptible de padecer una enfermedad seleccionada del grupo que consiste en Ia enfermedad de Chagas, Ia tripanosomiasis africana y Ia malaria. Dicho método comprende Ia administración a dicho paciente de una cantidad terapéuticamente efectiva de un compuesto de Ia invención de fórmula Ia, alternativamente Ib, junto con excipientes o portadores farmacéuticamente aceptables.
Otro aspecto de Ia invención se refiere a un procedimiento de preparación de los compuestos de Ia invención de fórmula Ia, alternativamente Ib, que comprende Ia oxidación de un compuesto de fórmula Ma, alternativamente Mb, con un agente oxidante adecuado:


Mb Ib
donde GP, R-i, R2 y R3 tienen el significado indicado anteriormente.
Otro aspecto de Ia presente invención se refiere a un compuesto diastereoisomérico intermedio de fórmula Ma, alternativamente Mb, o una sal farmacéuticamente aceptable de los mismos, o un solvato farmacéuticamente aceptable de los mismos, incluyendo hidratos,

Ma Mb donde GP, R-i, R2 y R3 tienen el significado indicado anteriormente.
Otro aspecto de Ia presente invención se refiere a un procedimiento de preparación de un compuesto de Ia invención de fórmula Ma, alternativamente Mb, que comprende Ia epoxidación de un compuesto de fórmula MIa, alternativamente MIb, con un agente epoxidante adecuado:


En las definiciones anteriores, el término (Ci-C4)alquilo significa un radical de una cadena hidrocarbonada saturada lineal o ramificada que contiene de 1 a 4 átomos de carbono. Ejemplos incluyen entre otros los grupos metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo y tere-butilo.
El término (C2-C4)alquenilo significa un radical de una cadena hidrocarbonada insaturada lineal o ramificada que contiene de 1 a 4 átomos de carbono y al menos un doble enlace. Ejemplos incluyen entre otros los grupos etenilo, propenilo, isopropenilo, 1-butenilo, 2-butenilo, 3-butenilo y 1 ,3-butadienilo.
El término (C2-C4)alquinilo significa un radical de una cadena hidrocarbonada insaturada lineal o ramificada que contiene de 1 a 4 átomos de carbono y al menos un triple enlace. Ejemplos incluyen entre otros los grupos etinilo, propinilo, isopropinilo, 1 -butinilo, 2-butinilo y 3-butinilo.
La expresión "opcionalmente sustituido por uno o más sustituyentes" significa
Ia posibilidad de un grupo de estar sustituido por uno o más, preferiblemente por 1 , 2, 3 ó 4 sustituyentes, siempre que dicho grupo disponga de 1 , 2, 3 ó 4 posiciones susceptibles de ser sustituidas.
Ar es un radical de carbono o nitrógeno de un anillo conocido carbocíclico aromático monocíclico de 5 a 6 miembros o bicíclico de 8 a 10 miembros, que contiene opcionalmente de 1 a 3 heteroátomos seleccionados entre N1 S y O, y que puede estar opcionalmente sustituido según se ha indicado
anteriormente. Ejemplos de radicales Ar incluyen, sin limitación, tiofenilo, furanilo, pirrolilo, pirazolilo, imidazolilo, tiazolilo, isotiazolilo, oxazolilo, isoxazolilo, triazolilo, tiadiazolilo, oxadiazolilo, fenilo, piridinilo, pirazinilo, piridazinilo, pirimidininilo, triazinilo, tienofuranilo, tienopirrolilo, pirrolopirazolilo, benzotiofenilo, benzofuranilo, indolilo, isoindolilo, benzimidazolilo, benzotiazolilo, imidazopiridinilo, pirazolopiridinilo, isoquinolinilo, quinolinilo, quinolizinilo, naftiridinilo, quinazolinilo, quinoxalinilo y naftilo.
En el contexto de esta invención, el término "compuesto diastereoisomérico sustancialmente puro" significa un compuesto con suficiente exceso diastereoisomérico para su preparación a escala industrial, Io cual depende de cada caso concreto como decidirá el experto en Ia materia. En Ia mayoría de los casos, un exceso diastereoisomérico superior al 95% es suficiente.
Aunque Ia presente invención incluye todos los compuestos arriba mencionados, una realización particular de Ia invención se refiere a un compuesto diastereoisomérico sustancialmente puro de fórmula Ia, o una sal farmacéuticamente aceptable del mismo, o un solvato farmacéuticamente aceptable del mismo, incluyendo un hidrato.
En otra realización particular, en un compuesto diastereoisomérico sustancialmente puro de fórmula Ia, alternativamente Ib, Ri representa fenilmetilo; R2 es un radical -(CH2)n-Ar; R3 es un radical seleccionado del grupo que consiste en -O(d-C4)alquilo, -O(d-C4)alquilo-Ar, -OAr, n representa 2; y Ar es un radical de carbono de un anillo conocido carbocíclico aromático monocíclico de 5 a 6 miembros, que contiene opcionalmente de 1 a 3 heteroátomos seleccionados entre N1 S y O.
En otra realización particular, en un compuesto de Ia invención de fórmula Ia, alternativamente Ib, GP es un radical seleccionado del grupo que consiste en benciloxicarbonilo, morfolinocarbonilo y Λ/-metilcarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2-feniletilo y R3 es un radical -O(d-C4)alquilo.
En otra realización particular, en un compuesto de Ia invención de fórmula Ia, alternativamente Ib, GP es un radical benciloxicarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2-feniletilo y R3 es un radical etoxilo.
Como se ha mencionado anteriormente, Ia invención también se refiere a los intermedios de fórmula Ma, alternativamente Mb, a sus sales farmacéuticamente aceptables y a sus solvatos farmacéuticamente aceptables, incluyendo hidratos.
Así, otra realización particular se refiere a un intermedio diastereoisomérico sustancialmente puro de fórmula Ma, o una sal farmacéuticamente aceptable del mismo, o un solvato farmacéuticamente aceptable del mismo, incluyendo un hidrato.
En otra realización particular, en un compuesto de Ia invención de fórmula Ma, alternativamente Mb, Ri representa fenilmetilo; R2 es un radical -(CH2)n- Ar; R3 es un radical seleccionado del grupo que consiste en -O(d-C4)alquilo, -O(d-C4)alquilo-Ar, -OAr, n representa 2; y Ar es un radical de carbono de un anillo conocido carbocíclico aromático monocíclico de 5 a 6 miembros, que contiene opcionalmente de 1 a 3 heteroátomos seleccionados entre N, S y O.
En otra realización particular, en un compuesto de Ia invención de fórmula
Ma, alternativamente Mb, GP es un radical seleccionado del grupo que consiste en benciloxicarbonilo, morfolinocarbonilo y Λ/-metilcarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2-feniletilo y R3 es un radical -O(d- C4)alquilo.
En otra realización particular, en un compuesto de Ia invención de fórmula Ma, alternativamente Mb, GP es un radical benciloxicarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2-feniletilo y R3 es un radical etoxilo.
Además, todas las posibles combinaciones de las realizaciones mencionadas arriba forman también parte de Ia invención.
Los compuestos de Ia presente invención pueden contener uno o más nitrógenos básicos y por tanto pueden formar sales con ácidos, tanto orgánicos como inorgánicos, que forman también parte de Ia presente invención. Ejemplos de dichas sales incluyen: sales con ácidos inorgánicos como ácido clorhídrico, ácido bromhídrico, ácido iodhídrico, ácido nítrico,
ácido perclórico, ácido sulfúrico o ácido fosfórico; y sales con ácidos orgánicos, como ácido metanosulfónico, ácido trifluorometanosulfónico, ácido etanosulfónico, ácido bencenosulfónico, ácido p-toluenosulfónico, ácido fumárico, ácido oxálico, ácido acético o ácido maleico, entre otros. Los compuestos de Ia presente invención pueden contener uno o más protones ácidos y por tanto podrían formar sales con bases, que forman también parte de Ia presente invención. Ejemplos de dichas sales incluyen: sales con cationes inorgánicos como sodio, potasio, calcio, magnesio, litio, aluminio, zinc, etc.; sales formadas con aminas farmacéuticamente aceptables como amoníaco, alquilaminas, hidroxialquilaminas, lisina, arginina, N- metilglucamina, procaína y similares. No hay limitación en Ia naturaleza de dichas sales, en el supuesto de que cuando se usen con fines terapéuticos sean farmacéuticamente aceptables. Las sales se pueden preparar por tratamiento del compuesto de fórmula Ia, Ib, Ma o Mb con una cantidad suficiente del ácido o Ia base deseados para dar Ia sal de una forma convencional. Las sales de los compuestos de fórmula Ia, Ib, Ma o Mb se pueden transformar a su vez en otras sales de compuestos de fórmula Ia, Ib, Ma o Mb respectivamente, por intercambio de iones mediante una resina de intercambio iónico.
La presente invención también incluye composiciones farmacéuticas. La formulación escogida depende de Ia naturaleza del compuesto administrado y de Ia vía de administración. Se pueden utilizar distintas vías de administración, por ejemplo administración oral o tópica.
Las composiciones sólidas para Ia administración oral incluyen comprimidos, granulados y cápsulas. El método de preparación puede basarse en mezcla simple, granulación seca o granulación húmeda del principio activo con excipientes. Estos excipientes pueden ser, por ejemplo, diluyentes, agentes aglutinantes, disgregantes, y agentes lubricantes. Los comprimidos pueden estar recubiertos con excipientes adecuados para retrasar su disgregación y absorción en el tracto gastrointestinal o para mejorar sus propiedades organolépticas o su estabilidad. El principio activo puede también ser incorporado por recubrimiento sobre pellets inertes mediante el uso de polímeros fumógenos naturales o sintéticos.
En el caso de utilizar cápsulas de gelatina blanda, el principio activo se mezcla con agua o con medio oleoso. Las suspensiones orales de polvos y granulados se pueden preparar mediante Ia adición de agua, mezclando el principio activo con agentes dispersantes o humectantes; suspensantes y conservantes. También pueden añadirse otros excipientes, por ejemplo edulcorantes, aromatizantes y colorantes.
Como formas líquidas para Ia administración oral se pueden incluir emulsiones, soluciones, suspensiones, jarabes y elixires que contienen diluyentes inertes comúnmente utilizados. Dichas composiciones pueden también contener coadyuvantes como agentes humectantes, suspensantes, edulcorantes, aromatizantes, conservantes y reguladores de pH.
El compuesto puede también ser formulado para su aplicación tópica. Formulaciones tópicas incluyen cremas, lociones, geles, ungüentos, polvos, soluciones y parches en las que el compuesto se encuentra dispersado o disuelto en excipientes adecuados.
Los compuestos diastereoisoméricos sustancialmente puros de fórmula Ia, alternativamente Ib, se pueden preparar mediante los procedimientos descritos a continuación. Como será evidente para un experto en Ia materia, el método preciso utilizado para Ia preparación de un compuesto dado puede variar en función de su estructura química. Si no se especifica Io contrario, los significados de las variables GP, Ri, R2 y R3 son los indicados anteriormente.
Asimismo, en alguno de los procedimientos indicados puede ser necesario o conveniente proteger los grupos reactivos o lábiles mediante grupos protectores convencionales. Tanto Ia naturaleza de dichos grupos protectores como los procedimientos para su introducción y eliminación son bien conocidos y forman parte del estado de Ia técnica (Greene T. W. y Wuts P. G. M, "Protective Groups in Organic Synthesis", John Wiley & Sons, 3rd edition, 1999). Algunos ejemplos de grupos protectores de una función amino que pueden emplearse son los grupos 9-fluorenilmetoxi-carbonilo (Fmoc), terc-butoxicarbonilo (Boc), bencilo (Bn), benciloxicarbonilo (Cbz), trifluoroacetilo (TFA), morfolinocarbonilo y Λ/-metilcarbonilo. Los grupos hidroxilo pueden protegerse por ejemplo con grupos trimetilisililo (TMS), tere-
butildimetilsililo (TBS) o tetrahidropiranilo (THP). La reacción de desprotección de los grupos anteriores se realiza en condiciones habituales en síntesis orgánica, como las descritas en Ia referencia arriba mencionada.
Un compuesto diastereoisomérico sustancialmente puro de fórmula Ia puede obtenerse por oxidación de un compuesto de fórmula Ma. Esta reacción se lleva a cabo en presencia de un agente oxidante, como por ejemplo el peryodinano de D ess- Martin, en un disolvente adecuado, como por ejemplo diclorometano, a una temperatura adecuada, preferiblemente temperatura ambiente.

Illa βpoxidacion

Ia Ma
Un compuesto de fórmula Ma puede obtenerse mediante una reacción de epoxidación de un alcohol alílico de fórmula MIa. Esta reacción se lleva a cabo en presencia de un agente epoxidante, como íerc-butilhidroperóxido de litio, en un disolvente adecuado, como por ejemplo tetrahidrofurano, a una temperatura adecuada, preferiblemente temperatura ambiente. En estas condiciones se obtiene mayoritariamente el epoxialcohol sin con respecto el grupo hidroxilo (S. Rodríguez et al, Tetrahedron 2006, pp.11112-11123).
Un compuesto de fórmula Illa puede prepararse a partir de un ácido carboxílico de fórmula Va, donde GP' es un grupo protector de hidroxilo, por ejemplo terc-butilidimetilsililo, mediante una secuencia de varias etapas. En una primera etapa, el grupo ácido carboxílico del compuesto de fórmula Va se transforma en un grupo amino mediante una transposición de Curtius. Esta reacción puede llevarse a cabo con difenifosforilazida, en presencia de una base, como por ejemplo trietilamina, en un disolvente adecuado, como por ejemplo Λ/,Λ/-dimetilformamida, a una temperatura adecuada, preferiblemente temperatura ambiente, formándose un isocianato intermedio. Alternativamente, el isocianato intermedio puede formarse haciendo reaccionar el ácido carboxílico de fórmula Va con cloroformiato de metilo y posteriormente azida sódica.
El tratamiento acuoso del isocianato obtenido intermedio a temperatura adecuada, preferiblemente a 100 0C, da lugar a una amina primaria. En una segunda etapa, se convierte Ia amina obtenida en Ia primera etapa en una amida de fórmula IVa por reacción con un aminoácido de fórmula VIa con el grupo amino convenientemente protegido en caso necesario, por ejemplo protegido con el grupo benciloxicarbonilo. Esta reacción puede llevarse a cabo en presencia de un agente condensante adecuado como por ejemplo N- (3-dimetilaminopropil)-Λ/'-etilcarbodiimida o diciclohexilcarbodiimida opcionalmente en presencia de 1-hidroxibenzotriazol, o bien en presencia de una base adecuada, tal como 4-dimetilaminopriridina o piridina, en el seno de un disolvente adecuado, tal como diclorometano o dimetilformamida.
Alternativamente, se convierte el isocianato obtenido en Ia primera etapa en una amida de fórmula IVa por reacción con un aminoácido de fórmula VIa con el grupo amino convenientemente protegido en caso necesario, por ejemplo, protegido con el grupo benciloxicarbonilo. Esta reacción puede llevarse a cabo en presencia de una base adecuada, tal como 4-dimetilaminopriridina, en el seno de un disolvente adecuado, tal como diclorometano.
Un compuesto de fórmula Va puede obtenerse a partir de un compuesto de fórmula XIII siguiendo Ia ruta sintética que se indica en el siguiente esquema:
 introducción del
introducción del
XIII Xl auxiliar quiral |χ

Villa VIIIb
Prtotección del alcohol
O OGP1
^COR3 R2
VIIa
hidrólisis

Va
El ácido carboxílico de fórmula XIII se convierte en un derivado acilado de fórmula Xl donde X representa halógeno, preferiblemente cloro, mediante reacción con un agente halogenante, por ejemplo cloruro de tionilo, en un disolvente adecuado, como por ejemplo benceno, a una temperatura adecuada, preferiblemente calentando.
Posteriormente, el compuesto de fórmula Xl se convierte en un compuesto de
fórmula IX por reacción con un compuesto de fórmula XII, donde Xc representa un auxiliar quiral, como por ejemplo (S)-4-bencil-2-tioxotiazolidin- 3-ilo. Esta reacción se lleva a cabo en presencia de una base, como por ejemplo trietilamina o 2,6-lutidina y opcionalmente en presencia de triflato de ferc-butildimetilsililo (TBSOTf), en un disolvente adecuado, como por ejemplo diclorometano o sin disolvente y a una temperatura adecuada, por ejemplo temperatura ambiente.
La reacción aldólica asimétrica entre Ia cetona de fórmula IX y un aldehido de fórmula X da lugar a los aldoles sin y anti de fórmulas Villa y VIIIb respectivamente, de estereoquímica absoluta definida y separables mediante cromatografía. Esta reacción se puede llevar a cabo, por ejemplo, según se indica en D.A. Evans et al, Orq. Lett. 2002, vol. 4, pp. 1127-1130; D.A. Evans et al, J. Am. Chem. Soc. 2002, vol. 124, pp. 392-393 y D.A. Evans et al, ¿h Am. Chem. Soc. 1981 , vol. 103, pp. 2127-2129. Cuando Xc es un radical (S)- 4-bencil-2-tioxotiazolidin-3-ilo y Ia reacción se lleva a cabo según se describe en D.A. Evans et al, Org. Lett. 2002, 4, 1127, se obtiene mayoritariamente el compuesto de fórmula VIIIb.
Tras separar los compuestos de fórmula Villa y VIIIb, el grupo hidroxilo del compuesto de fórmula Villa se puede proteger para dar un compuesto de fórmula VIIa, donde GP' representa un grupo protector, por ejemplo terc- butilidimetilsililo. Esta reacción se lleva a cabo en condiciones bien conocidas para el experto, como por ejemplo las descritas en Greene T.W. y Wuts P. G. M, "Protective Groups in Organic Synthesis", John Wiley & Sons,
3rd edition, 1999.
La hidrólisis de un compuesto de fórmula VIIa da lugar a un compuesto de fórmula Va. Esta reacción se puede llevar a cabo en presencia de peróxido de hidrógeno y de una base, como por ejemplo hidróxido de litio, en un disolvente adecuado, como por ejemplo una mezcla de tetrahidrofurano-agua y a una temperatura adecuada, preferiblemente temperatura ambiente.
Los compuestos XIII, XII, X y VIa son comerciales o pueden obtenerse fácilmente mediante métodos convencionales.
Un compuesto diastereoisomérico sustancialmente puro de fórmula Ib puede
obtenerse de manera análoga a Ia obtención de un compuesto de fórmula Ia a partir del correspondiente aldol de fórmula VIIIb:

Además, un compuesto de fórmula Ia, alternativamente Ib, puede transformarse en otro compuesto de fórmula Ia, alternativamente Ib. Por ejemplo, un compuesto de fórmula Ia, alternativamente Ib, donde R3 representa -O(d-C4)alquilo, -O(d-C4)alquilo-Ar o -OAr puede transformarse en un compuesto de fórmula Ia, alternativamente Ib, donde R3 representa -NHAr o -NH-OAr, mediante reacciones bien conocidas por el experto en Ia materia. Asimismo las reacciones de interconversión de un radical R3 en otro radical R3 pueden también llevarse a cabo sobre cualquiera de los intermedios de los compuestos de Ia invención de fórmula Ia, alternativamente Ib.
A Io largo de Ia descripción y las reivindicaciones Ia palabra "comprende" y sus variantes no pretenden excluir otras características técnicas, aditivos, componentes o pasos.
Para los expertos en Ia materia, otros objetos, ventajas y características de Ia invención se desprenderán en parte de Ia descripción y en parte de Ia práctica de Ia invención. Los siguientes ejemplos se proporcionan a modo de ilustración, y no se pretende que sean limitativos de Ia presente invención.
EJEMPLOS
Todos los disolventes usados en las reacciones se destilaron en el momento de ser utilizados mediante agentes desecantes adecuados. Los espectros de 1H-RMN y de 13C-RMN se registraron en disolución de CDCI3 (1H, 7.24 ppm; 13C 77.0 ppm) a 30 0C en un espectrómetro de 300 MHz Mercury Varían o un 500 MHz Innova Varían RMN en los Servéis Centráis d'lnstrumentació
Científica de Ia Universitat Jaume I. Los espectros de masas se registraron en un espectrómetro de masas QTOF I (quadrupole-hexapole-TOF) con interface ortogonal Z-spray-electrospray (Micromass, Manchester, UK). Los espectros de IR se registraron en películas aceitosas sobre pastillas de NaCI en un espectrómetro Perkin-Elmer 2000 FT-IR. Para las cromatografías de columna se empleó EM Science Silica GeI 60 mientras que las TLC se realizaron con folios de aluminio cargados con silica E. Merck (Kieselgel 60, F254, 0.25 mm). Excepto en los casos indicados, todas las reacciones se llevaron a cabo bajo atmósfera de nitrógeno con agitación magnética.
En los ejemplos descritos a continuación se han utilizado las siguientes abreviaciones:
AcOEt: acetato de etilo AMC: 7-Amino-4-metil coumarina
Arg: L-Arginina
4-DMAP: 4-Λ/,Λ/-dimetilaminopiridina
DMSO: dimetilsulfóxido
DTT: ditiotreitol Et3N: trietilamina
Et2O: dietil éter
Hex: hexano
HPhe: L-Homofenilalanina
Phe: L-Fenilalanina TBHP: terc-butilhidroperóxido
TBSOTf: triflato de terc-butildimetilsililo
THF: tetrahidrofurano
TMSCI: cloruro de trimetilsililo
TX-100: tritón-X (hidróxido de trimetilbencilamonio) Z: benciloxicarbonil
Intermedio XI.1 (R2 = 2-feniletilo; X = Cl): Cloruro de 4-fenilbutanoílo.
Una disolución de ácido 4-fenilbutírico (18.76g, 114.23mmol) en benceno (147mL) se trató con cloruro de tionilo (25.27mL, 348mmol) y Ia mezcla resultante se reflujo durante 2h, entonces el solvente se eliminó bajo vacío y
el aceite marrón resultante se sometió al siguiente paso sin ninguna purificación.
Intermedio IX.1 (R2 = 2-feniletilo, Xc: (S)-4-bencil-2-tioxotiazolidin-3-ilo): 1- ((S)-4-bencil-2-tioxotiazolidin-3-il)-4-fenilbutan-1 -ona.
Una disolución enfriada en baño de hielo de (S)-4-benciltiazolidin-2-tiona (18.37g, 87.87mmol) en CH2CI2 (266mL) se trató con Et3N (2 eq) y después con cloruro de 4-fenilbutanoilo (Intermedio Xl.1 , 114.23mmol). La mezcla resultante se agitó a temperatura ambiente durante 12 días y luego se paró con salmuera y se extrajo con CH2CI2, se secó con Na2SO4, se filtró y se concentró. La purificación por cromatografía sobre gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (95:5, 9:1 , 8:2, 7:3 y 6:4) dio lugar a 24g (70%) de un aceite amarillo.
13C-RMN (500MHz, CDCI3) δ 201.06, 173.73, 141.53, 136.56, 129.44, 128.88, 128.53, 128.36, 127.19, 125.96, 68.53, 37.93, 36.79, 35.11 , 31.90, 26.43.
Intermedios Vllla.1 (sin) v Vlllb.1 (anti) (R2 = 2-feniletilo, R3 = etoxilo, Xc: (S)- 4-bencil-2-tioxotiazolidin-3-ilo): (2E,4R,5S)-5-((4S)-4-Benzil-2- tioxotiazolidin-3-carbonil)-7-fenil-4-hidroxihept-2-enoato de etilo y (2£,4S,5S)-5-((4S)-4-Benzil-2-tioxotiazolidin-3-carbonil)-7-fenil-4- hidroxihept-2-enoato de etilo.
MgBr EtO2 (440.47mg, 1.71 mol) se introdujo en un matraz de dos bocas con un agitador magnético usando Ia caja seca. Luego se añadieron secuencialmente una disolución de 1-((S)-4-bencil-2-tioxotiazolidin-3-il)-4- fenilbutan-1-ona (Intermedio IX.1 , 4.79g, 13.12mmol) en AcOEt (33mL), (E)- 3-formilacrilato de etilo (Intermedio X.1 (R3 = etoxilo), 5.04g, 39.35mmol), Et3N (4.02mL, 28.86mmol) y TMSCI (2.83mL, 22.30mmol) y Ia mezcla resultante se agitó a temperatura ambiente durante 12días. Pasado este tiempo, el crudo se filtró a través de gel de sílice usando Et2O como eluyente y se concentró bajo vacío. El crudo resultante se disolvió en THF (20OmL) y se trató con 1 M HCI acuoso (46mL). La mezcla resultante se agitó a temperature ambiente durante 1.5h, luego se paró con NaHCO3 sólido, se filtró y se concentró. La purificación por cromatografía sobre gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (8:2, 1 :1 , 1 :2, 1 :4 y 1 :7)
dio lugar a los dos compuestos deseados en forma de aceites amarillos (peso de los dos compuestos conjuntamente: 3.80 g (60%)).
Intermedio Vllla.1 {sin): 1H-RMN (500MHz, CDCI3) δ 7.20-7.35 (m, 10H), 6.94 (1 H, dd, J= 15.5 and 3.5Hz), 6.14 (1 H, dd, J= 15.5 y 1.0Hz), 5.14 (1 H, m), 4.73 (1 H, m), 4.46 (1 H, m), 4.08 (3H, m), 3.31 (1 H, dd, J= 12.0 y 7.5Hz), 3.08 (1 H, dd, J= 13.0 y 3.5Hz), 2.94 (1 H, dd, J= 13.5 y 10.5Hz), 2.80 (1 H, m), 2.60 (m, 2H), 2.45 (1 H, m), 2.15 (m, 1 H), 1.85 (m, 1 H), 1.22 (3H, t, J= 7.0Hz).
Intermedio Vlllb.1 {anti): 1H-RMN (500MHz, CDCI3) δ 7.10-7.25 (m, 10H), 6.84 (1 H, dd, J= 15.5 and 3.5Hz), 6.03 (1 H, dd, J= 15.5 and 1.0Hz), 5.24 (1 H, m), 4.83 (1 H, m), 4.55 (1 H, m), 4.17 (3H, m), 3.24 (1 H, dd, J= 11.0 y 6.5Hz), 3.03 (1 H, dd, J= 13.5 y 4.0Hz), 2.91 (1 H, dd, J= 13.0 y 10.0Hz), 2.76 (2H, m), 2.57 (m, 1 H), 2.12 (m, 1 H), 2.03 (m, 1 H), 1.19 (3H, t, J= 7.0Hz).
Intermedios Vlla.1 (s/n) v Vllb.1 (anti) (R2 = 2-feniletilo, R3 = etoxilo, Xc: (S)-4- bencil-2-tioxotiazolidin-3-ilo, GP': terc-butildimetilsililo): (2£,4R,5S)-5-((4S)-4- Benzil-2-tioxotiazolidin-3-carbonil)-4-íerc-butildimetilsililoxi-7-fenilhept- 2-enoato de etilo y (2E,4S,5S)-5-((4S)-4-Benzil-2-tioxotiazolidin-3- carbonil)-4-íerc-butildimetilsililoxi-7-fenilhept-2-enoato de etilo.
Una disolución del aldol (intermedio Vlllb.1 , 3.63g, 8.05mmol) enfriada a -7O0C en CH2CI2 (8OmL) se trató con 2,6-lutidina (3.75ml_, 32.19) y TBSOTf (5.53ml_, 24.15mmol). La mezcla resultante se agitó a dicha temperatura durante 5.5h. Luego se paró con una disolución acuosa saturada de NaHCO3 y se extrajo con CH2CI2, se secó (Na2SO4), se filtró y se concentró. La purificación por cromatografía sobre gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (7:3 y 1 :1 ) dio lugar a 4.47g (93%) de un aceite amarillo, correspondiente al intermedio VIIb.1.
Siguiendo un procedimiento análogo al descrito anteriormente pero utilizando como producto de partida el intermedio Vllla.1 , se obtuvo el producto Vlla.1.
Intermedio Vlla.1 {sin): IR δ 3063, 3027, 2954, 2930, 2857, 1718, 1659, 1604, 1496, 1471 , 1455, 1365, 1341 , 1262, 1193, 1161 , 1135, 1083, 1083, 1039, 983, 939, 883, 838, 779, 747, 701 , 667 cm"1.
Intermedio Vllb.1 (anti): IR 5 3063, 3027, 2930, 2857, 1716, 1659, 1604, 1496, 1455, 1367, 1341 , 1159, 1038, 984, 837, 779, 746, 701 , 667 crrϊ1.
Intermedios Va.1 (s/n) y Vb.1 (anti) (R2 = 2-feniletilo, R3 = etoxilo, GP': terc- butildimetilsililo): Ácido (E,2S,3R)-5-(etoxicarbonil)-3-ferc- butildimetilsililoxi-2-fenetilpent-4-enoico y Ácido (£,2S,3S)-5- (etoxicarbonil)-3-íerc-butildimetilsililoxi-2-fenetilpent-4-enoico.
A una disolución del aldol protegido (intermedio Vllb.1 , 2.02g, 4.98mmol) en THF-H2O (3:1 ) (5OmL) enfriada en baño de hielo se Ie añadió gota a gota H2O2 (30%) (3.08ml_, 29.90mmol) y luego una disolución de LiOH (238mg, 9.96mmol) en agua (2mL). La mezcla resultante se agitó a temperatura ambiente durante 2 h. Pasado este tiempo Ia reacción se paró con Na2SOs (1 ,6M solución acuosa) (8mL) y se agitó durante 20min, luego se concentró bajo vacío y Ia disolución acuosa resultante se extrajo con CH2CI2, se secó (Na2SO4), se filtró y se concentró. La purificación por cromatografía sobre gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (7:3, 6:4 y 1 :1 ) dio lugar a 1.86 g (83%) de un aceite amarillo-pálido, correspondiente al intermedio Vb.1.
Siguiendo un procedimiento análogo al descrito anteriormente pero utilizando como producto de partida el intermedio VIIa.1 , se obtuvo el producto Va.1.
Intermedio Va.1 (sin): —
Intermedio Vb.1 (anti): 1HRMS m/z caled, for C22H34O5SiNa [IvRNa+]: 429.2073, found: 429.2099.
Intermedios IVa.1 (sin) y IVb.1 (anti) (GP = benciloxicarbonilo, Ri = bencilo, R2 = 2-feniletilo, R3 = etoxilo, GP': terc-butildimetilsililo): (2£,4R,5S)-5-((2S)- 2-Benciloxicarbonilamino-3-fenilpropionilamino)-4-ferc- butildimetilsililoxi-7-fenilhept-2-enoato de etilo y (2E,4S,5S)-5-((2S)-2- Benciloxicarbonilamino-3-fenilpropionilamino)-4-íerc-butildimetilsililoxi- 7-fenilhept-2-enoato de etilo.
Una disolución del ácido carboxílico (intermedio Vb.1 , 969mg, 2.38mmol) en acetona-H2O (12mL) enfriada en baño de hielo se trató con Et3N (389μL,
2.79mmol) y luego con cloroformiato de metilo (247μl_, 3.19mmol). La mezcla resultante se agitó a esta temperatura durante 1.5h. Luego se añadió una disolución de NaN3 (309mg, 4.76mmol) en agua (1.5mL) y Ia mezcla se agitó enfriada en baño de hielo durante 4h, luego se vertió en un embudo de decantación y se extrajo con Et2O, se secó (Na2SO4), se filtró y se concentró. El aceite amarillo resultante se disolvió en tolueno (12mL) y se reflujo durante 35min, luego se concentró bajo vacío y el crudo se sometió al siguiente paso sin ninguna purificación. Después una disolución del crudo anterior y (S)-benciloxicarbonilfenilalanina (911 mg, 3.05mmol) en CH2CI2 (35mL) enfriada en baño de hielo se trató con 4-DMAP (71 mg, 0.58mmol). La mezcla resultante se agitó a dicha temperatura durante 1.5h y luego a temperatura ambiente durante 6h. Luego Ia mezcla se concentró bajo vacío. La purificación por cromatografía sobre gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (6:4) dio lugar a 752mg (48%) de un aceite, correspondiente al intermedio IVb.1.
Siguiendo un procedimiento análogo al descrito anteriormente pero utilizando como producto de partida el intermedio Va.1 , se obtuvo el producto IVa.1.
Intermedio IVa.1 {sin): 13C-RMN (500MHz, CDCI3) δ 173.91 , 165.43, 147.09, 141.25, 136.03, 129.00, 128.45, 127.92, 126.77, 125.50, 122.10, 73.98, 68.60, 60.13, 49.00, 36.43, 32.65, 31.17, 30.34, 25.23, 13.73, -4.63, -5.44.
Intermedio IVb.1 {anti): IR 5 3321 , 3027, 2929, 2858, 1719, 1659, 1535, 1497, 1455, 1366, 1260, 1174, 1130, 1031 , 837, 778, 746, 698 crrϊ1.
Intermedios llla.1 (sin) y lllb.1 (anti) (GP = benciloxicarbonilo, Ri = bencilo, R2 = 2-feniletilo, R3 = etoxilo): (2£,4/?,5S)-5-((2S)-2-Benciloxicarbonilamino-3- fenil-propionilamino)-7-fenil-4-hidroxihept-2-enoato de etilo y (2£,4S,5S)- 5-((2S)-2-Benciloxicarbonilamino-3-fenilpropionilamino)-7-fenil-4- hidroxi-hept-2-enoato de etilo.
A una disolución de compuesto protegido (intermedio IVb.1 , 725mg, 1.10mmol) in THF (11 mL) enfriado en baño de hielo se añadió fluoruro de tetra-n-butilamonio (1 M en THF) (5.5mL, 5.5mmol). La mezcla resultante se agitó a temperatura ambiente durante 7.5 h. Pasado este tiempo, el solvente se eliminó bajo vacío y el crudo resultante se purificó por cromatografía sobre
gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (1 :1 ) dando lugar a 467mg (78%) de un sólido blanco, correspondiente al intermedio IMb.1
Siguiendo un procedimiento análogo al descrito anteriormente pero utilizando como producto de partida el intermedio IVa.1 , se obtuvo el producto llla.1.
Intermedio llla.1 {sin): 13C-RMN (500MHz, CDCI3) δ 171.98, 166.44, 156.39, 145.97, 141.32, 136.69, 136.27, 129.00, 128.45, 127.92, 126.77, 125.60, 73.36, 67.50, 60.76, 54.07, 38.73, 32.64, 30.67, 14.44.
Intermedio lllb.1 {anti): IR 5 3438, 3309, 3029, 2927, 1690, 1642, 1532, 1495, 1455, 1385, 1369, 1302, 1258, 1219, 1190, 1135, 1041 , 971 , 911 , 873, 750, 700, 673 cm"1.
Intermedios lla.1 (sin-sin) y llb.1 (anti-sin): (GP = benciloxicarbonilo, Ri = bencilo, R2 = 2-feniletilo, R3 = etoxilo): (2S,3R)-3-[(1S,2S)-2-((2S)-2- Benciloxicarbonilamino-3-fenilpropionilamino)-4-fenil-1- hidroxibutil]oxirano-2-carboxilato de etilo y (2f?,3S)-3-[(1f?,2S)-2-((2S)-2- Benciloxicarbonilamino-3-fenilpropionilamino)-4-fenil-1 - hidroxibutil]oxirano-2-carboxilato de etilo.
A una disolución de TBHP (3.3M en tolueno, preparado según HiII, J. G.; Rossiter, B. E.; Sharpless, B.; J. Org. Chem. 1983, 48, 3607-3608; 616μl_, 2.34 mmol) en THF (5 ml_) enfriada a -780C se añadió etil litio (0.5M in benceno/ciclohexano (9/1 )) (3.43ml_, 1.72 mmol). La mezcla resultante se agitó a -780C durante 15 min y luego una disolución del ester insaturado (intermedio lllb.1 , 425mg, 0.78 mmol) en THF (3 ml_) se añadió gota a gota y Ia mezcla resultante se agitó a temperatura ambiente durante 3 días. Luego se añadió Na2SO3 (120mg) de una vez y se agitó durante 15 min.
Posteriormente se diluyó con una disolución acuosa saturada de NH4CI y se extrajo con Et2O (3 x 30 mL), las fases orgánicas se lavaron con salmuera, se secaron y se concentraron. El aceite crudo resultante se purificó por cromatografía sobre gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (7:3), (6:4), (1 :1 ), (1 :2) y AcOEt) dando lugar a 175mg de un sólido blanco, correspondiente al intermedio Mb.1.
Siguiendo un procedimiento análogo al descrito anteriormente pero utilizando como producto de partida el intermedio llla.1 , se obtuvo el producto Ma.1.
Intermedio lla.1 {sin-sin): HRMS m/z caled, for C22H34O5SiNa [IVRNa+]: 429.2073, found: 429.2099.
Intermedio llb.1 (anti-sin): 13C-RMN (500MHz, CDCI3) δ 171.73, 168.60, 155.81 , 141.11 , 136.46, 136.06, 129.31 , 129.27, 128.87, 128.83, 128.57, 128.52, 128.36, 128.27, 127.14, 126.14, 70.71 , 67.26, 61.80, 58.49, 56.63, 53.05, 51.29, 50.05, 38.06, 33.07, 32.40, 32.30, 32.24, 29.72, 14.14.
Compuestos la.1 y lb.1 (GP = benciloxicarbonilo, Ri = bencilo, R2 = 2- feniletilo, R3 = etoxilo): (2S,3S)-3-[(2S)-2-((2S)-2-Benciloxicarbonilamino-3- fenilpropionilamino)-4-fenilbutiril]oxirano-2-carboxilato de etilo y (2R,3/?)-3-[(2S)-2-((2S)-2-Benciloxicarbonilamino-3-fenilpropionilamino)- 4-fenilbutiril]oxirano-2-carboxilato de etilo.
Una disolución del epoxialcohol (intermedio Mb, 50mg, 0.09mmol) en CH2CI2 (3ml_) enfriada en baño de hielo se trató con piridina (36μl_, 0.45mmol) y el peryodinano de Dess-Martin (169mg, 0.45mmol).
La mezcla resultante se agitó a temperatura ambiente durante 4 h. Luego se paró con una disolución saturada acuosa de NaHCO3/Na2S2O3, se diluyó con Et2O, luego se extrajo con Et2O (3x20mL), las fases orgánicas se lavaron (salmuera), se secaron (Na2SO4) y se concentraron. El crudo se purificó a través de cromatografía sobre gel de sílice usando mezclas Hex:AcOEt de polaridad creciente (8:2, 7:3, 6:4, 1 :1 y AcOEt) para dar 34mg (68 %) de un sólido blanco, correspondiente al intermedio Mb.1.
Siguiendo un procedimiento análogo al descrito anteriormente pero utilizando como producto de partida el intermedio MIa.1 , se obtuvo el producto Ma.1.
Compuesto la.1 : HRMS m/z caled, for C32H34O5N2O7Na [M+Na+]: 581.2264, found: 581.2251.
Compuesto lb.1 : HRMS m/z caled, for C32H34O5N2O7Na [M+Na+]: 581.2264, found: 581.2258.
Estudios de inhibición enzimática y determinación de los valores ICsn
La inhibición de las cisteína proteasas cruzaína, rhodesaína, catepsina B (Tb CatB) y falcipaína (FP2) se evaluó por cuantificación de Ia emisión de fluorescencia resultante de Ia ruptura proteolítica del compuesto sintético Z- Phe-Arg-AMC (Bachem). Los inhibidores Ia o Ib se usaron en concentraciones que iban desde 10 nM a 10.000 nM (de 500 nM a 10.00OnM para Ia Tb CatB); así se prepararon diluciones en serie en DMSO con Ia concentración mínima de inhibidor Ia o Ib empleada en los cálculos y al menos 10 veces mayor que Ia del enzima. Estos ensayos se realizaron repartiendo el inhibidor Ia o Ib en los 96 pocilios, Ia fluorescencia se registró con un fluorímetro de Ia marca Flex Station de Molecular Devices, excitando a 355 nm y emisión de 460 nm a 250C, repartiendo 100 μl del substrato con pipetas multicanal a un volumen igual de inhibidor enzima preincubado a 250C durante 5 minutos. En cada serie de ensayos se emplearon como controles el enzima con DMSO y el enzima en presencia del conocido inhibidor irreversible K11777 (N methyl piperazine-Phe- HPhe-(CH=CHSO2 Ph)), Arris Pharmaceuticals Inc., South San Francisco, CA).
Los datos de inhibición para todos los enzimas se determinaron de forma similar: cruzaína recombinante a 4 nM con 5μM de Z-Phe-Arg-AMC (Km=1 μM) en el tampón A (100 mM de acetato de sodio pH 5.5 con 5 mM de DTT y 0.001 % de Triton-X 100 (Sigma)); rhodesaína recombinante a 4 nM de enzima y 5 μM Z-Phe-Arg-AMC (Km = 1 μM) en tampón A; Tb CatB recombinante a 40 nM de enzima y 5 μM de Z-Phe-Arg-AMC (Km = 60 μM) en tampón A y FP2 recombinante a 4 nM con 5μM de Z-Phe-Arg-AMC (Km=1 μM) en el tampón A . Los IC50 se determinaron mediante una lectura directa del semi log plot de Ia velocidad de reacción versus Ia concentración de inhibidor.
Los ensayos de inhibición sobre Trypanosoma brucei brucei se realizaron a través de ATP-bioluminiscencia (Z. B. Mackey et al, Chem. Biol. Druq Des. 2006, 67, pp. 355-363).
En Ia tabla 1 se muestran los valores de porcentajes de inhibición correspondientes a Tripanosoma brucei brucei (Tbb), cruzaína, rhodesaína y cathepsina B.

En Ia tabla 2 se muestran los valores de IC50 de las enzimas falcipaína, cruzaína, rhodesaína y TbCat B.
Tabla 2

Los compuestos Ia.1 y lb.1 son muy activos frente a rhodesaína y cruzaína pero no para Ia cathepsina B Io que denota su selectividad.
La efectividad de los compuestos de Ia invención también se puede comprobar mediante ensayos celulares o modelos animales in vivo. Los modelos animales de trypanosomiasis, enfermedad de Chagas y malaria son conocidos en el estado de Ia técnica.
Claims
1. Compuesto diastereoisomérico sustancialmente puro de fórmula Ia, alternativamente Ib, o una sal farmacéuticamente aceptable de los mismos o 5 un solvato farmacéuticamente aceptable de los mismos, incluyendo un hidrato,

R2 es un radical seleccionado del grupo que consiste en -H, -CH3, -CH2SH, -CH2OH, -CH2Ph, -CH2CO2H, -CH2CONH2, -CH(OH)CH3, -CH(CH3)CH2CH3, O -CH(CH3)2, -CH2CH(CH3)2, -(CH2J2SCH3, -(CH2J2CO2H, -(CH2)CONH2,
-(CH2)3NHC(NH)NH2, -(CH2J4NH2, imidazol-4-ilmetilo, 4-hidroxifenilmetilo, (1/-/-indol-3-il)metilo, (1/-/-imidazol-4-il)metilo y -(CH2)n-Ar; y
R3 es un radical seleccionado del grupo que consiste en -O(d-C4)alquilo, 5 -O(C2-C4)alquenilo, -O(C2-C4)alquinilo, -O(d-C4)alquilo-Ar, -OAr, -NR3Ar,
-N(Ra)[(d-C4)alquilo-Ar], -NR3OAr y -N(Ra)[O(CrC4)alquilo-Ar];
donde n representa un valor seleccionado entre 2 y 3;
0 Ar es un radical de carbono o nitrógeno de un anillo conocido carbocíclico aromático monocíclico de 5 a 6 miembros o bicíclico de 8 a 10 miembros, que contiene opcionalmente de 1 a 3 heteroátomos seleccionados entre N1 S y O, y que puede estar opcionalmente sustituido por uno o más radicales seleccionados independientemente del grupo que consiste en -OH, -CHO, 5 -SH, -NO2, -CN, -F, -Cl, -Br, -(d-C4)alquilo opcionalmente sustituido por uno o más radicales seleccionados independientemente del grupo que consiste en -F, -Cl, -Br y -OH; -O(d-C4)alquilo opcionalmente sustituido por uno o más radicales seleccionados independientemente del grupo que consiste en -F, -Cl, -Br y -OH; -CO(d-C4)alquilo, -OCO(d-C4)alquilo, -S(d-C4)alquilo, -SO(Ci-C4)alquilo, -SO2(Ci-C4)alquilo, -SO2O(Ci-C4)alquilo, -OSO2(C1- C4)alquilo, -NRaRb, -CONRaRb; y
Ra y Rb representan independientemente un radical -H o -(d-C4)alquilo.
2. Compuesto según Ia reivindicación 1 de fórmula Ia, o una sal farmacéuticamente aceptable del mismo, o un solvato farmacéuticamente aceptable del mismo, incluyendo un hidrato.
3. Compuesto según cualquiera de las reivindicaciones 1 a 2 donde GP es un radical seleccionado del grupo que consiste en benciloxicarbonilo, morfolinocarbonilo y Λ/-metilcarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2-feniletilo y R3 es un radical -O(d-C4)alquilo.
4. Compuesto según Ia reivindicación 3, donde GP es un radical benciloxicarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2-feniletilo y R3 es un radical etoxilo.
5. Composición farmacéutica que comprende una cantidad terapéuticamente efectiva de un compuesto definido en cualquiera de las reivindicaciones 1 a 4, o una sal farmacéuticamente aceptable del mismo, o un solvato farmacéuticamente aceptable del mismo, incluyendo un hidrato junto con cantidades apropiadas de excipientes farmacéuticamente aceptables.
6. Uso de un compuesto definido en cualquiera de las reivindicaciones 1 a 4 para Ia preparación de un medicamento para el tratamiento y/o prevención de enfermedades mediadas por Ia inhibición de una cisteína-proteasa seleccionada del grupo que consiste en cruzaína, rhodesaína y falcipaína.
7. Uso de un compuesto definido en cualquiera de las reivindicaciones 1 a 4 para Ia preparación de un medicamento para el tratamiento y/o prevención de una enfermedad seleccionada del grupo que consiste en Ia enfermedad de Chagas, Ia tripanosomiasis africana y Ia malaria.
8. Procedimiento de preparación de un compuesto diastereoisomérico sustancialmente puro de fórmula Ia, alternativamente Ib, o una sal farmacéuticamente aceptable de los mismos, o un solvato farmacéuticamente aceptable de los mismos, incluyendo un hidrato, que comprende Ia oxidación de un compuesto de fórmula Ma, alternativamente Mb con un agente oxidante adecuado:

Ma Mb
donde GP, R-i, R2 y R3 tienen el significado indicado en Ia reivindicación 1.
9. Compuesto diastereoisomérico de fórmula Ma, alternativamente Mb, o una sal farmacéuticamente aceptable de los mismos o un solvato farmacéuticamente aceptable de los mismos, incluyendo un hidrato,
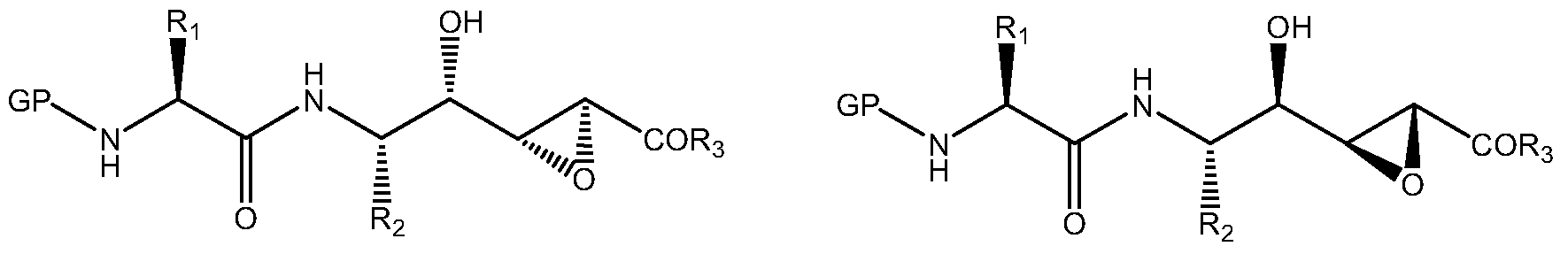
Ha Mb donde GP, R-i, R2 y R3 tienen el significado indicado en Ia reivindicación 1.
10. Compuesto según Ia reivindicación 9 de fórmula Ma, o una sal farmacéuticamente aceptable del mismo, o un solvato farmacéuticamente aceptable del mismo, incluyendo un hidrato.
11. Compuesto según cualquiera de las reivindicaciones 9 a 10 donde GP es un radical seleccionado del grupo que consiste en benciloxicarbonilo, morfolinocarbonilo y Λ/-metilcarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2-feniletilo y R3 es un radical -O(d-C4)alquilo.
12. Compuesto según cualquiera de las reivindicaciones 9 a 11 donde GP es un radical benciloxicarbonilo; Ri es un radical fenilmetilo, R2 es un radical 2- feniletilo y R3 es un radical etoxilo.
13. Procedimiento de preparación de un compuesto diastereoisomérico de fórmula Ma, alternativamente Mb, o una sal farmacéuticamente aceptable de los mismos, o un solvato farmacéuticamente aceptable de los mismos, incluyendo un hidrato, que comprende Ia epoxidación de un compuesto de fórmula MIa o MIb respectivamente con un agente epoxidante adecuado:

Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010511668A JP2010529979A (ja) | 2007-06-15 | 2008-06-12 | システイン−プロテアーゼ阻害剤 |
| EP08787650A EP2168954A4 (en) | 2007-06-15 | 2008-06-12 | CYSTEINE PROTEASES INHIBITORS |
| US12/664,445 US20100190848A1 (en) | 2007-06-15 | 2008-06-16 | Cysteine-protease inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ESP200701717 | 2007-06-15 | ||
| ES200701717A ES2310143B1 (es) | 2007-06-15 | 2007-06-15 | Inhibidores de cisteina proteasas. |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008152178A1 true WO2008152178A1 (es) | 2008-12-18 |
Family
ID=40084603
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/ES2008/070116 WO2008152178A1 (es) | 2007-06-15 | 2008-06-12 | Inhibidores de cisteína-proteasas |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20100190848A1 (es) |
| EP (1) | EP2168954A4 (es) |
| JP (1) | JP2010529979A (es) |
| ES (1) | ES2310143B1 (es) |
| WO (1) | WO2008152178A1 (es) |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11263783A (ja) * | 1998-01-09 | 1999-09-28 | Taisho Pharmaceut Co Ltd | エポキシスクシナム酸誘導体 |
| CA2306741A1 (en) * | 1998-08-25 | 2000-03-02 | Kazumi Okuro | Process for the preparation of (2r, 3s)-3-amino-1,2-oxirane |
| HU229338B1 (en) * | 2000-08-16 | 2013-11-28 | Bristol Myers Squibb Holdings Ireland | Stereoselective reduction of substituted oxo-butanes |
-
2007
- 2007-06-15 ES ES200701717A patent/ES2310143B1/es active Active
-
2008
- 2008-06-12 JP JP2010511668A patent/JP2010529979A/ja active Pending
- 2008-06-12 EP EP08787650A patent/EP2168954A4/en not_active Withdrawn
- 2008-06-12 WO PCT/ES2008/070116 patent/WO2008152178A1/es active Application Filing
- 2008-06-16 US US12/664,445 patent/US20100190848A1/en not_active Abandoned
Non-Patent Citations (6)
| Title |
|---|
| DRAHL C. ET AL.: "Protein-Reactive Natural Products", ANGEWANDTE CHEMIE INTERNATIONAL EDITION, vol. 44, 2005, pages 5788 - 5809, XP008134899 * |
| GONZALEZ F.V. ET AL.: "Dipeptidyl-alpha,beta-epoxyesters as potent irreversible inhibitors of the cysteine proteases cruzain and rhodesain", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 17, no. 24, 2007, pages 6697 - 6700, XP022339557 * |
| GROLL M. ET AL.: "Crystal Structure of Epoxomicin: 20S Proteasome Reveals a Molecular Basis of Selectivity of alpha',beta'-Epoxyketone Proteasome Inhibitors", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 122, 2000, pages 1237 - 1238, XP008128036 * |
| ROUSH W.R. ET AL.: "Design and synthesis of dipeptidyl alpha',beta'-Epoxy ketones, potent irreversible inhibitors of the cysteine protease cruzain", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 8, no. 19, 1998, pages 2809 - 2812, XP004139625 * |
| See also references of EP2168954A4 * |
| SPALTENSTEIN A. ET AL.: "Design and Synthesis of Novel Protease Inhibitors, Tripeptide alpha',beta'-Epoxyketones as Nanomolar Inactivators of the Proteasome", TETRAHEDRON LETTERS, vol. 37, no. 9, 1996, pages 1343 - 1346, XP004705209 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20100190848A1 (en) | 2010-07-29 |
| JP2010529979A (ja) | 2010-09-02 |
| EP2168954A1 (en) | 2010-03-31 |
| ES2310143A1 (es) | 2008-12-16 |
| ES2310143B1 (es) | 2010-02-08 |
| EP2168954A4 (en) | 2012-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3034043B2 (ja) | ピロリジンおよびチアゾリジン誘導体、それらの製造およびそれらを含んでいる薬剤 | |
| US7491748B2 (en) | Carboxylic acid derivative compounds and drugs comprising these compounds as the active ingredient | |
| ES2353077T3 (es) | Derivados de urea con sustitución en posición 3 y uso medicinal de los mismos. | |
| JP4443037B2 (ja) | ペプチド含有α−ケトアミドのシステインおよびセリンプロテアーゼ阻害剤 | |
| ES2226870T3 (es) | Derivados de indol y su uso para el tratamiento de la osteoporosis entre otras aplicaciones. | |
| JPH0789935A (ja) | ファルネシル蛋白質トランスフェラーゼの複素環式抑制剤 | |
| JPH08504815A (ja) | 環状スルホン含有レトロウイルスプロテアーゼ阻害物質 | |
| JP2005539089A (ja) | ニトロソ化非ステロイド性抗炎症化合物、組成物および使用方法 | |
| US20080300191A1 (en) | Protease inhibitors for coronaviruses and sars-cov and the use thereof | |
| EP1135151A4 (en) | ALPHA-KETO OXADIAZOLE CONTAINING PEPTOIDS AND NON-PEPTOIDS AS SERINE PROTEASE INHIBITORS | |
| BRPI0620736A2 (pt) | compostos orgánicos | |
| ES2235050T3 (es) | Derivados de pirrolidina como inhibifdores del factor xa. | |
| JPH09512528A (ja) | バソプレッシン拮抗物質としてのベンズアミド誘導体 | |
| KR940003297B1 (ko) | 1,3,2-디옥사티오렌옥시드 유도체 | |
| ES2619844T3 (es) | Derivados de biarilo-metanosulfinilo tio-sustituidos | |
| FI110938B (fi) | Menetelmä virusperäisten proteaasien inhiboimiseksi käyttökelpoisten karboksiamidijohdannaisten valmistamiseksi ja menetelmässä käytettäviä välituotteita | |
| ES2213563T3 (es) | Compuestos de tiopirano como inhibidores de mmp. | |
| WO2008152178A1 (es) | Inhibidores de cisteína-proteasas | |
| JP7113815B2 (ja) | リシンジンジパインのケトン阻害剤 | |
| JPH07504417A (ja) | レトロウイルスプロテアーゼ拮抗薬 | |
| JP2003523337A (ja) | マトリックスメタロプロタアーゼ阻害剤としてのチアゼピニルヒドロキサム酸誘導体 | |
| ES2301793T3 (es) | Cianamidas utiles como inhibidores reversibles de cisteina proteasas. | |
| JPH07157459A (ja) | ビフェニルメチル置換バレリルアミド誘導体 | |
| ES2346964T3 (es) | Derivados del acido hidroxamico. | |
| ES2254679T3 (es) | Inhibidores de la peptido desformilasa. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08787650 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12664445 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010511668 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008787650 Country of ref document: EP |