WO2007131446A1 - Acide bêta-phényl-alpha-hydroxy propanoïque substitué, procédé de synthèse et son utilisation - Google Patents
Acide bêta-phényl-alpha-hydroxy propanoïque substitué, procédé de synthèse et son utilisation Download PDFInfo
- Publication number
- WO2007131446A1 WO2007131446A1 PCT/CN2007/001550 CN2007001550W WO2007131446A1 WO 2007131446 A1 WO2007131446 A1 WO 2007131446A1 CN 2007001550 W CN2007001550 W CN 2007001550W WO 2007131446 A1 WO2007131446 A1 WO 2007131446A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- formula
- compound according
- synthesis
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/732—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids of unsaturated hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/612—Esters of carboxylic acids having a carboxyl group bound to an acyclic carbon atom and having a six-membered aromatic ring in the acid moiety
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/46—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/48—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to an acyclic carbon atom of a saturated carbon skeleton containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/32—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
- C07C235/34—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/62—Halogen-containing esters
- C07C69/65—Halogen-containing esters of unsaturated acids
- C07C69/653—Acrylic acid esters; Methacrylic acid esters; Haloacrylic acid esters; Halomethacrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/80—Acids; Esters in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/60—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/36—Systems containing two condensed rings the rings having more than two atoms in common
- C07C2602/42—Systems containing two condensed rings the rings having more than two atoms in common the bicyclo ring system containing seven carbon atoms
Definitions
- the present invention relates to a substituted ?-phenyl-'-hydroxypropionic acid derivative, a synthetic method thereof and use thereof for the preparation of a medicament for preventing and treating cardiovascular and cerebrovascular diseases.
- Salvia miltiorrhiza is a traditional Chinese medicine for the treatment of cardiovascular and cerebrovascular diseases. It is generally believed that Danshensu (the chemical name is: ?-(3,4-dihydroxyphenyl)- «-hydroxypropionic acid) is a water-soluble group of Salvia miltiorrhiza. The main active ingredient of the fraction, pharmacological tests showed that the phenyl---hydroxypropyl danshenin drug ⁇ ⁇ group, but its efficacy is not significant. Therefore, the structure of the substituted ?-phenyl- «-hydroxypropionic acid may be modified, and the modified derivative may have or have a greater potency than the prototype compound, and may improve its efficacy in preventing and treating cardiovascular and cerebrovascular diseases. For example, water tablets can pass through the heart-brain barrier, while danshensu is not easy to pass through the heart-brain barrier. Therefore, in the structure of Danshensu, the chemical structure of borneol is added to structurally transform Danshensu.
- the object of the present invention is to provide a substituted /?-phenyl-"-hydroxypropionic acid derivative and a synthetic method thereof, and a substituted ?-phenylhydroxypropionic acid derivative for the preparation of a medicament for preventing and treating cardiovascular and cerebrovascular diseases. ⁇ Use of the object.
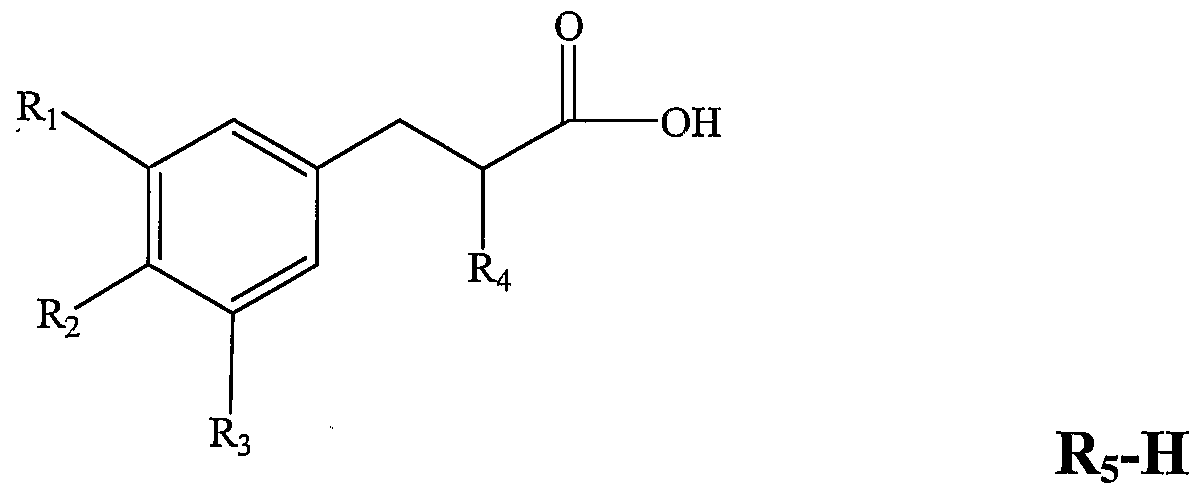
- a substituted ff-phenyl- «, hydroxypropionic acid derivative in particular a compound represented by the general formula (I):
- R 2 and R 3 are independently selected from the group consisting of H, OH, F Cl, Br, and methoxy And B or alternatively, R 2 together form -OCH 2 0-, R 3 is selected from the group consisting of H, OH, ⁇ #L ⁇ , B and halogen;
- R 5 is selected from a cycloalkoxy group and a substituted amino group, and in the case where R 5 is an amino group, Ri, R 2 and R 3 are not all H.
- 3 ⁇ 4 is OH.
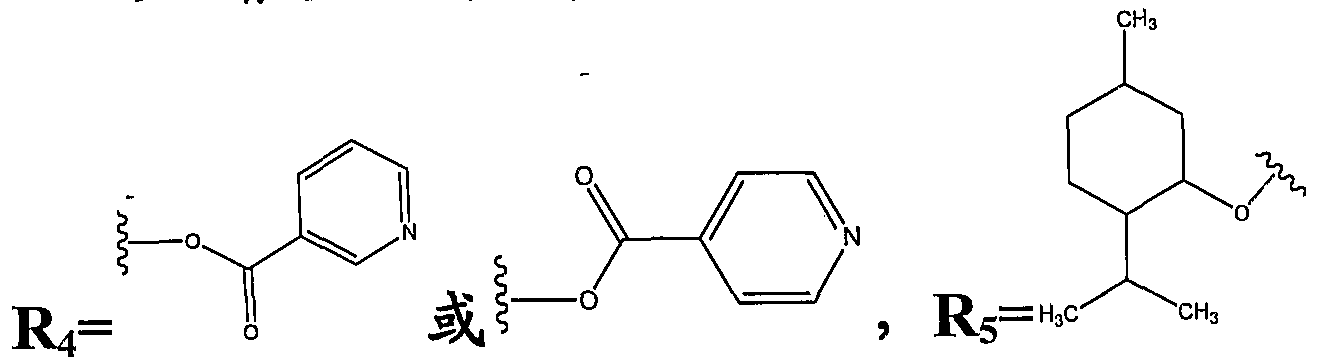
- R4 is an aroyloxy group or a heterocyclic substituted acyloxy group.
- 3 ⁇ 4 is o-acetoxybenzoyloxy, 3-pyridinebenzoyloxy or 4-pyridinebenzoyloxy.
- R 5 is:
- R 2 is OH, respectively.
- R 2 together form -OCH 2 0-.
- Ri and R 2 are respectively OH
- R 2 is formed
- the catalyst is concentrated H 2 S0 4 , silicotungstic acid, phosphomolybdic acid, p-nonyl acid, S 2 0—/Zr0 2 , aluminum trichloride, zinc chloride and/or magnesium chloride.
- the catalyst is p-toluenesulfonic acid, S 2 0-/Zr0 2 , aluminum trichloride and/or zinc chloride. It is especially advantageous to use p-toluenesulfonic acid and/or S 2 0 ⁇ -/Zr0 2 .
- the reaction molar ratio of the compound of the formula (m) to the compound of the formula (IV) is 1: 0.8-1: 1.5, preferably 1: 1 to 1: 1.5, more preferably 1: 1.25-1: 1.5, most preferably 1: 1.5.
- the reaction molar ratio of the compound of the formula (V) to the compound of the formula (VI) is 1: 0.8-1: 1.5, preferably 1: 1 to 1: 1.5, more preferably 1: 1.25-1: 1.5, most preferably 1: 1.5.
- the reaction is carried out in a solvent.
- the solvent used is selected from the group consisting of ethyl acetate, dichloromethane, tetrahydrofuran, acetone, toluene, 1,4-dioxane and ⁇ yv-dimethylformamide.
- the solvent used is selected from the group consisting of tetrahydrofuran, acetone, toluene, 1,4-dioxane, Ay-dimethylformamide.
- the solvent used is selected from the group consisting of tetrahydrofuran and acetone.
- the use of tetrahydrofuran as a solvent is most preferred.
- the solvents may be used singly or in combination.
- the reaction temperature varies depending on the solvent selected, and it is advantageous to control from 0 ° C to 150 ° C.
- the reaction temperature is from 25 ° C to 100 ° C. More preferably, the reaction temperature is 65 °C.
- the reaction time can be 21! ⁇ 24h, preferably 5h ⁇ 15h, more preferably 8h ⁇ 12h, most preferably 8h.
- the synthesis of a compound of formula (II) comprises: reacting ?-(3,4-dihydroxyphenyl)hydroxypropionic acid with borneol in the presence of a catalyst.
- the catalyst is a Lewis acid catalyst, such as methylbenzene sulfonic acid,
- the reaction is carried out in a solvent, and the solvent is selected from tetrahydrofuran, toluene, 1,4-dioxane or Ayv-dimethylformamide, preferably tetrahydrofuran.
- the degree of reactivity varies depending on the solvent used, and is usually controlled at 65. C ⁇ 150 ° C, preferably controlled at 65. C.
- the reaction time is from 8 h to 12 h, preferably 8 h.
- s 2 o [- /Zr0 2 can be optionally prepared by the following method: adding ammonia water to the ZrOCl 2 solution at 0 to 10 ° C Medium, to pH 9-12, aged, washed and precipitated to no Cr, dried, ground, placed in (NH 4 ) 2 S 2 0 8 solution, soaked, filtered, dried, ground, at 500 ⁇ 700. C roasting 2-5h prepared ⁇ s 2 o _ / Zr0 2. ⁇
- a compound of the invention for the manufacture of a medicament for the prophylaxis and treatment of cardiovascular and cerebrovascular diseases, in particular, ?-(3,4-dihydroxyphenyl)-hydroxypropionic acid borneol Esters (compounds of formula II) are useful in the preparation of medicaments for the prevention and treatment of cardiovascular and cerebrovascular diseases.
- FIG. 1 is a view showing the synthesis route of a compound of the formula (II) in Example 1, i.e., ?-(3,4-dihydroxyphenyl)-hydroxypropionic acid borneol.
- Figure 2 shows the mass spectrum of the final product obtained in Example 1.
- Fig. 3 shows an infrared spectrum of the final product obtained in Example 1.
- Fig. 4 shows the ?NMR spectrum of the final product obtained in Example 1.
- Figure 5 shows the 13 C NMR spectrum of the final product obtained in Example 1.
- the preparation method of the catalyst S 2 0 8 2 7Zr0 2 is as follows: 0.025 mol of ZrOCl 2 -8H 2 0 is prepared into a 1 mol.I/ 1 ZrOCl 2 solution, stirred in a water bath, and slowly dropped into 6 moH / 1 Ammonia water, until the pH was 10, aged for 12 h, suction filtered, and the precipitate was washed with distilled water until no CT (tested with 0.1 mol'l AgNOs).
- the precipitate was baked at 110 ° C for 10 h, finely ground, and then 0.5
- the solution was immersed for 12 h, suction filtered, dried, ground, and fired at 600 ° C for 3 h in a muffle furnace to obtain S 2 0 8 2 7Zr0 2 .
- Figure 2 is a mass spectrum of the obtained pale yellow oil. It can be seen that 351.7 is
- iHNMlU CD3COCD3, 500MHz ⁇ 6.57-7.64(m, 3H, Ar-H), 4.10-4.32(m, lH, -CH(OH)-), 4.83(t, 1H, -CH- ), 2.79-2.92 (m, 2H, -CH 2 -);
- Example 2 Synthesis of ?-(3,4-dihydroxyphenyl)-hydroxypropionic acid hydroflavonate The other two steps were the same as in Example 1, except that 0.12 mol of ⁇ -(3,4-di) was added to the three-necked flask. Hydroxyphenyl)- ⁇ -hydroxypropionic acid and 0.15 mol of borneol, then 0.86 g of p-toluenesulfonic acid catalyst, 500 mL of tetrahydrofuran, and reacted at 65 ° C for 12 h.
- Example 3 Synthesis of ?-(3,4-dihydroxyphenyl)--hydroxypropionic acid borneol ester Three additional steps were the same as in Example 1, except that 0.1 mol of ⁇ -(3,4-dihydroxyl was added to a three-necked flask Phenyl) hydroxypropionic acid and 0.12 mol of borneol, then 1.33 g of catalyst 8 2 0 / ⁇ 1:0 2 , 1,4-dioxane 400 ml were added and reacted at 100 ° C for 8 h. After the completion of the reaction, the catalyst S 2 0 ⁇ -/Zr0 2 was removed by suction filtration, and then the solvent was distilled off under reduced pressure.
- the obtained viscous material was removed from the borneol by an oil pump (1.3 ⁇ l (T 3 Pa )) in a boiling water bath to obtain a black-brown viscosity.
- the viscous material was separated into a pale yellow oil.
- the mass spectrum and the infrared spectrum of the obtained product were the same as in Example 1.
- Example 4 -(3,4-dihydroxyphenyl; synthesis of Hz-hydroxypropionic acid hydroflavonate four other steps are the same as in Example 1, except that 0.06 mol ?-(3,4-dihydroxyl) is added to the three-necked vial Phenyl group: hydroxypropionic acid and 0.09 mol of water, then 0.60 g of a catalyst aluminum trichloride, 200 ml of a solvent A V-dimethyl phthalamide, and reacted at 150 ° C for 10 h. After the reaction, the solvent was distilled off under reduced pressure.
- the obtained viscous material was taken out in a boiling water bath by an oil pump (1.3 ⁇ l (T 3 Pa ) to obtain a black ochre substance, and then separated by a column color to obtain a pale yellow oil.
- Mass spectrum of the obtained product and infrared light pan The same as in the first embodiment.
- Example 5 Synthesis of ?-(4-chlorophenyl)- «-hydroxypropionic acid hydroflavonate (1) Synthesis of 2-mercapto-4-(4-chlorobenzylidene)oxazolone similar to Example 1 (2) except that 4-chlorophenylhydrazine was used instead of 3,4-dihydroxybenzaldehyde. Brownish yellow crystals, yield 87.4%.
- Steps (1) to (4) are equivalent to Examples 7 (1) to (4).
- Steps (1) to (4) are equivalent to Examples 7 (1) to (4).
- Steps (1) to (4) are equivalent to those of Examples 7 (1) to (4).
- mice 60 SD rats, weighing 220 ⁇ 20g. Randomly divided into normal control group, model control group, Danshensu injection group (ip lmL/kg) and yff-(3,4-dihydroxyphenyl)-ct-hydroxypropionic acid hydroflavonate small, medium and large doses Group (ip 5, 15, 35 mg/kg).
- Normal control group and model control group ip equal volume of normal saline. Animals were intraperitoneally injected with 1% pentobarbital sodium 40 mg/kg anesthesia, supine position, fixed head, midline incision skin, tracheal intubation, spontaneous breathing. The right common carotid vein and the common carotid artery were separated and threaded for use.
- the threading speed should be slowed down, and the blood flow of the brain microcirculation displayed by the laser Doppler microcirculation blood flow analyzer should be observed at the same time.
- the middle cerebral artery is worn, There may be a sudden drop in the microcirculation perfusion flow, and after the microcirculation blood flow drops, the inner diameter is penetrated by about 1 mm, and then the distal end of the incision and the wire in the artery are firmly ligated to remove the excess thread.
- the J 12200 laser Doppler microcirculation blood flow probe was fixed at the cranial window, and the probe was maintained without displacement and rotation during the whole experiment. Recording before and after ligation in the area 5, 15 The microcirculation blood flow at 30, 45, and 60 min was the same for the medication group.
- the average microcirculation perfusion flow within the observation time lmin is the microcirculation perfusion flow of the observation time.
- the tracheal intubation was performed, and the ventilation frequency was 60 times/ Min, open the chest, with 6/0 line, a sputum from the root of the anterior descending coronary artery lmm ⁇ 2mm, through a plastic tube, tighten the sputum, observe the ECG changes, ST elevation or decrease are successful ligation
- the color of the myocardial tissue below the ligature is darkened.
- the plastic tube was pulled out, the coronary blood flow was recanalized, and the local tissue was congested.
- the myocardial infarct size was recorded before the experiment, ischemia lmin and 30 min, and reperfusion for 30 min.
- the heart tissue was taken and fixed with 10% formalin. , paraffin-embedded, 4 ⁇ thick serial sections, respectively, immunohistochemical detection; sham operation group only threaded, but did not ligature coronary artery.
- myocardial tissue is divided into: blue is normal myocardium, light red is ischemic myocardium, grayish white is necrotic myocardium.
- Infarct myocardium was calculated by computer image analysis software Percentage of area (nec/aar) of the myocardium in the risk zone (ie, ischemic myocardium, including ischemic infarction and ischemic infarcted myocardium, aar) and percentage of infarcted myocardium (nec/lv) to the extent of infarction At the same time, the percentage of left ventricular area (aar/lv) in the dangerous area was calculated.
- Bax anti-rabbit polyclonal antibody (Santa C zBio. Inc.), dilution 1:200; Bcl22: anti-rabbit polyclonal antibody (TBD Tianjin Biotechnology Center), dilution 1100; caspase-3: anti-rabbit Cloning antibody (Normarkers Fromont, CA), diluted; i at 1:200; MMP-2: anti-mouse monoclonal antibody (Normarkers Fromont, CA), dilution 1:200; PPARy: anti-sheep polyclonal antibody (Santa Cruz Bio. Inc.
- the specific steps are as described in the ABC and SP kit instructions, DAB color development, neutral resin sealing.
- the primary antibody was replaced with PBS as a negative control.
- the cells with positive expression were brown-yellow, MMP-2 protein was in the cytoplasm, Bcl-2 was expressed in the nuclear membrane and cytoplasm, Bax was mainly in the cytoplasm, part was expressed in the nucleus, caspase-3 was the main To be expressed in the nucleus, part is expressed in the cytosol.
- the CIAAS image analysis system was used to perform random selection and automatic point analysis on the sections, and the average optical density values or integrated optical densities of the obtained myocardial tissue sections were statistically processed.
- Table 3 shows the effects of Bax, Bcl-2, caspase-3, MMP-2, and PPARy protein expression ("10)
- Danshensu group lmL/kg 0.08 ⁇ 0.02 A 0.18 ⁇ 0.04 0.21 ⁇ 0.12** 0.12 ⁇ 0.05 0.34 ⁇ 0.08 AA test group 5 mg/kg 0.12 ⁇ 0.03 0.15 ⁇ 0.03 0.35 ⁇ 0.14 0.14 ⁇ 0.05 0.16 ⁇ 0.
- MMP-2 is involved in myocardial I/R injury and is achieved by cleavage of troponin I, whereas cleavage of muscle 4 protein I directly leads to apoptosis.
- Specific MMP-2 inhibitors can improve myocardial I/R cardiac function in rats. What results show? Reduction of ⁇ -2 protein by the action of monos(3,4-dihydroxyphenyl)- ⁇ -hydroxypropionic acid hydroflavonate, which may be ff-(3,4-dihydroxyphenyl)- ⁇ -hydroxypropionic acid Another mechanism by which borneol esters are resistant to myocardial I/R injury. 3.
- Rats were anesthetized by intraperitoneal injection of 20% urethane 5mL/kg, fixed; cut the neck skin of rats, separate the anterior cervical muscles, expose the trachea, insert Endotracheal intubation; separation of the common carotid artery, insertion into the left ventricle through the common carotid artery through the cardiac catheter, through the RM-6000 multi-channel physiological recorder pressure transducer (T-200) and RM-6000 multi-channel physiological recorder : Large (AP-601G) measures the left chamber pressure, and then inputs the left chamber pressure signal to the RM-6000 multi-channel physiological recorder differential amplifier (ED-601G) to record the left chamber pressure rise or fall maximum rate p/dt max - Dp/dt max ); The right femoral artery was isolated, the arterial blood pressure was measured by cannulation; the ECG recording electrode was connected, and the standard ECG was
- the catheter was duodenal and the observations were recorded at 5, 15, 30, 60, 90, and 120 minutes after the drug.
- the rate of change was calculated by the following formula, and statistical analysis of the comparison between groups was performed with the rate of change.
- Rate of change (%) xlOO
- Danshensu hydrolone ester 18mg/kg group can significantly reduce the mean arterial pressure, systolic blood pressure and diastolic blood pressure of anesthetized rats after administration. Compared with the blank control group, there are significant differences at 15, 60, 90, 120 minutes. (P ⁇ 0.05 or P ⁇ 0.01); Danshensu borneol The ester 9mg/kg group reduced the mean arterial pressure, systolic blood pressure and diastolic blood pressure in anesthetized rats.
- the Danshensu hydrolone ester 18mg/kg group can significantly reduce the left ventricular pressure of rats after administration. Compared with the blank control group, there is a significant difference at 15, 30, 60, 90, 120 minutes (PO.05). Or P ⁇ 0.01); Danshensu hydrol ester 9mg/kg group, the left ventricular pressure tendency of drunk rats after administration, compared with the blank control group, there was a significant difference at 60 minutes (PO.05 or P ⁇ 0.01); Danshensu hydroslip ester 4.5mg/kg group had no significant effect on left ventricular pressure of anesthetized rats after administration, no significant difference compared with blank control group; verapamil hydrochloride group, administered After the treatment, the left internal pressure of the anesthetized rats was significantly reduced, which was significantly different from the blank control group (PO.01) (Table 8).
- Danshensu borneol ester 18mg/kg group can significantly reduce dp/dt after administration, compared with the blank control group, there is significant difference at 15, 30, 60, 90, 120 minutes (P ⁇ 0.05 or P ⁇ 0.01); Danshensu borneol ester 9mg/kg group, after administration, decreased the trend of dp/dt in anesthetized rats, compared with the blank control group, there was significant difference at 60 and 120 minutes (P ⁇ 0.05 or PO.
- Danshensu hydrolone ester 9mg/kg and 18mg/kg group had a trend of -dp/dt in drunken rats after administration, compared with the blank control group, there was a significant difference at 60 and 120 minutes (P ⁇ 0.05). Or P ⁇ 0.01); Danshensu borneol 4.5mg/kg group had no significant effect on -dp/dt of anesthetized rats after administration, and there was no significant difference compared with the blank control group; verapamil hydrochloride group, Significantly reduce the anesthetized rats after administration -dp/dt, there was a significant difference compared with the blank control group ( ⁇ 0 ⁇ 01). (Tables 9, 10).
- the rats were anesthetized with ether, fixed on the back, and normal electrocardiograms were recorded (before modeling). Under sterile conditions, the left chest is lifted, the muscles are bluntly separated in the fourth intercostal space, the right chest is gently squeezed out of the heart, between the pulmonary artery cone and the left atrial appendage, and the coronary artery is ligated 2 to 3 mm from the origin of the left coronary artery. The left anterior descending artery is then immediately returned to the chest and the wound is sutured. Topical application of penicillin to prevent infection. Immediately after the operation, the electrocardiogram after ischemia (Omin after modeling) was recorded, and the height of the ST-T segment was measured.
- Omin after modeling the electrocardiogram after ischemia
- Rate of change (%) 140.08 ⁇ 161.56 22.34 ⁇ 21.51**
- Model control group 1 10 0.20 ⁇ 0.09 0.19 ⁇ 0.41 0.23 ⁇ 0.10
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cardiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Heart & Thoracic Surgery (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description









Claims









Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07721123A EP2019090B1 (en) | 2006-05-15 | 2007-05-14 | Substituted beta-phenyl-alpha-hydroxy propanoic acid, synthesis method and use thereof |
| NZ572958A NZ572958A (en) | 2006-05-15 | 2007-05-14 | Substituted beta-phenyl-alpha-hydroxy propanoic acid, synthesis method and use thereof |
| BRPI0712101A BRPI0712101B8 (pt) | 2006-05-15 | 2007-05-14 | ácido beta-fenil-alfa-hidróxi propanóico substituído, método de síntese e uso do mesmo |
| PL07721123T PL2019090T3 (pl) | 2006-05-15 | 2007-05-14 | Podstawiony kwas beta-fenylo-alfa-hydroksypropionowy, sposób syntezy i jego zastosowanie |
| JP2009510260A JP5094845B2 (ja) | 2006-05-15 | 2007-05-14 | 置換β−フェニル−α−ヒドロキシプロピオン酸、その合成方法及び使用 |
| DK07721123.3T DK2019090T3 (da) | 2006-05-15 | 2007-05-14 | Substitueret beta-phenyl-alfa-hydroxypropansyre, fremgangsmåde til syntese og anvendelse deraf |
| US12/301,069 US8017786B2 (en) | 2006-05-15 | 2007-05-14 | Substituted β-phenyl-α-hydroxy-propanoic acid, synthesis method and use thereof |
| AU2007250364A AU2007250364B8 (en) | 2006-05-15 | 2007-05-14 | Substituted beta-phenyl-alpha-hydroxy propanoic acid, synthesis method and use thereof |
| KR1020087030201A KR101059639B1 (ko) | 2006-05-15 | 2007-05-14 | 치환된 베타-페닐-알파-하이드록시 프로파노산, 그의 합성 방법 및 사용 |
| SI200731105T SI2019090T1 (sl) | 2006-05-15 | 2007-05-14 | Substituirana beta-fenil-alfa hidroksi propanojska kislina, postopek sinteze in njena uporaba |
| CA2652299A CA2652299C (en) | 2006-05-15 | 2007-05-14 | Substituted beta-phenyl-alpha-hydroxy propanoic acid, synthesis method and use thereof |
| IL195313A IL195313A (en) | 2006-05-15 | 2008-11-16 | Alternate beta-phenyl-alpha-hydroxyl acid, a method of synthesis and use |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB2006100427873A CN100415709C (zh) | 2006-05-15 | 2006-05-15 | β-(3,4-二羟基苯基)-α-羟基丙酸冰片酯、其合成方法和用途 |
| CN200610042787.3 | 2006-05-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007131446A1 true WO2007131446A1 (fr) | 2007-11-22 |
Family
ID=37442796
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2007/001550 WO2007131446A1 (fr) | 2006-05-15 | 2007-05-14 | Acide bêta-phényl-alpha-hydroxy propanoïque substitué, procédé de synthèse et son utilisation |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US8017786B2 (zh) |
| EP (2) | EP2514739B1 (zh) |
| JP (1) | JP5094845B2 (zh) |
| KR (1) | KR101059639B1 (zh) |
| CN (1) | CN100415709C (zh) |
| AU (1) | AU2007250364B8 (zh) |
| BR (1) | BRPI0712101B8 (zh) |
| CA (1) | CA2652299C (zh) |
| CY (2) | CY1113676T1 (zh) |
| DK (2) | DK2019090T3 (zh) |
| ES (1) | ES2618677T3 (zh) |
| HU (1) | HUE032505T2 (zh) |
| IL (1) | IL195313A (zh) |
| MY (1) | MY148134A (zh) |
| NZ (1) | NZ572958A (zh) |
| PL (2) | PL2019090T3 (zh) |
| PT (1) | PT2514739T (zh) |
| RU (1) | RU2421443C2 (zh) |
| SI (2) | SI2019090T1 (zh) |
| WO (1) | WO2007131446A1 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103570546A (zh) * | 2013-10-09 | 2014-02-12 | 西安石油大学 | 一种丹参素冰片酯的工业化合成方法 |
| WO2014091465A2 (en) * | 2012-12-16 | 2014-06-19 | Mahesh Kandula | Compositions and methods for the treatment of metabolic syndrome and lipid disorders |
| CN109608356A (zh) * | 2018-12-14 | 2019-04-12 | 南京医科大学 | 一类丙二酸单酯酰化的氨基酸(+)2-崁醇酯的衍生物及其应用 |
| WO2019127746A1 (zh) * | 2017-12-30 | 2019-07-04 | 苏州沪云肿瘤研究中心股份有限公司 | 一种苯丙酸酯类化合物及其制备方法和应用 |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100415709C (zh) | 2006-05-15 | 2008-09-03 | 西北大学 | β-(3,4-二羟基苯基)-α-羟基丙酸冰片酯、其合成方法和用途 |
| CN101607897B (zh) * | 2008-06-18 | 2012-10-24 | 复旦大学 | (z)-2-乙酰氧基-3-(3,4-二羟基苯基)丙烯酸及其合成方法和用途 |
| CA2795408C (en) | 2010-04-12 | 2018-03-13 | Supernus Pharmaceuticals Inc. | Methods for producing viloxazine salts and novel polymorphs thereof |
| CN102030648B (zh) * | 2010-12-07 | 2013-04-03 | 西北大学 | 3-(3,4-二羟基苯基)-2-羟基丙酸酯的不对称合成方法 |
| CN102432468B (zh) * | 2011-11-21 | 2014-03-26 | 中国人民解放军第四军医大学 | 丹参素冰片酯的不对称合成方法 |
| CN103232347B (zh) * | 2013-04-12 | 2015-09-16 | 合肥工业大学 | 一种cns药物的脑靶向前药及其制备方法以及冰片在cns药物脑靶向前药中的用途 |
| CN103570547B (zh) * | 2013-10-09 | 2016-04-20 | 西安石油大学 | 一种丹参素异丙酯的工业化合成方法 |
| CN105693817B (zh) | 2014-11-27 | 2020-06-05 | 西北大学 | 一类三肽化合物及其制备方法与应用 |
| CN106496897A (zh) * | 2016-10-14 | 2017-03-15 | 无锡三帝特种高分子材料有限公司 | 一种聚酯镀铝膜 |
| CN107619376A (zh) * | 2017-11-09 | 2018-01-23 | 宁波职业技术学院 | 一种缬氨酸冰片酯及其制备方法和应用 |
| CN109928959B (zh) * | 2017-12-18 | 2020-11-06 | 中国科学院上海营养与健康研究所 | 抗心肌肥厚的药物、制备方法和用途 |
| JP6768868B2 (ja) * | 2019-03-29 | 2020-10-14 | 健裕生技股▲分▼有限公司 | 心筋再生を促進させるための化合物、その調製方法及びこれらの使用 |
| JP2020164528A (ja) * | 2020-04-28 | 2020-10-08 | 健裕生技股▲分▼有限公司 | 心筋再生を促進させるための化合物、その調製方法、医薬組成物及びこれらの使用 |
| CN114394901A (zh) * | 2021-12-30 | 2022-04-26 | 广东医科大学附属第二医院 | 一种丹参素特戊酸甲酯衍生物及其制备方法 |
| US11891353B1 (en) | 2023-08-28 | 2024-02-06 | King Faisal University | 3,3′-(hydrazine-1,2-diyl)bis(1-(naphthalen-2-yloxy)propan-2-ol) as an ecofriendly insecticidal agent against Spodoptera littoralis (boisd.) |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60255774A (ja) * | 1984-06-01 | 1985-12-17 | Dai Ichi Seiyaku Co Ltd | テトラヒドロナフタレン誘導体 |
| RU2092169C1 (ru) | 1992-08-20 | 1997-10-10 | Раиса Павловна Свистунова | Местное противовоспалительное средство "сальвинол" |
| JP3050733B2 (ja) * | 1992-10-16 | 2000-06-12 | 中外製薬株式会社 | 4−アルコキシ−2,6−ジ−t−ブチルフェノール誘導体 |
| US5736537A (en) * | 1995-09-12 | 1998-04-07 | Estee Lauder, Inc. | Dehydroep:androsterone sailcylate useful against skin atrophy |
| WO1997015546A1 (fr) * | 1995-10-26 | 1997-05-01 | Nippon Shinyaku Co., Ltd. | Derives d'acide carboxylique et compositions pharmaceutiques |
| CN1161140C (zh) * | 2001-11-09 | 2004-08-11 | 天津天士力制药股份有限公司 | 一种预防和治疗冠心病心绞痛的药物及其制备方法和其它用途 |
| US20040248849A1 (en) * | 2001-06-28 | 2004-12-09 | Potlapally Rajender Kumar | 3-Aryl-A-oxy substituted propanoic acids and a process for their preparation |
| FR2842523A1 (fr) * | 2002-07-17 | 2004-01-23 | Sanofi Synthelabo | Derives d'acylaminothiazole, leur preparation et leur application en therapeutique |
| CN100441563C (zh) * | 2004-06-03 | 2008-12-10 | 西安交通大学 | β-(3,4-二羟基苯基)-α-羟基丙酸异丙酯及其合成方法 |
| CN100415709C (zh) | 2006-05-15 | 2008-09-03 | 西北大学 | β-(3,4-二羟基苯基)-α-羟基丙酸冰片酯、其合成方法和用途 |
-
2006
- 2006-05-15 CN CNB2006100427873A patent/CN100415709C/zh active Active
-
2007
- 2007-05-14 DK DK07721123.3T patent/DK2019090T3/da active
- 2007-05-14 EP EP12002538.2A patent/EP2514739B1/en active Active
- 2007-05-14 SI SI200731105T patent/SI2019090T1/sl unknown
- 2007-05-14 PL PL07721123T patent/PL2019090T3/pl unknown
- 2007-05-14 JP JP2009510260A patent/JP5094845B2/ja active Active
- 2007-05-14 SI SI200731907A patent/SI2514739T1/sl unknown
- 2007-05-14 NZ NZ572958A patent/NZ572958A/en unknown
- 2007-05-14 WO PCT/CN2007/001550 patent/WO2007131446A1/zh active Application Filing
- 2007-05-14 KR KR1020087030201A patent/KR101059639B1/ko active IP Right Grant
- 2007-05-14 MY MYPI20084629A patent/MY148134A/en unknown
- 2007-05-14 CA CA2652299A patent/CA2652299C/en active Active
- 2007-05-14 EP EP07721123A patent/EP2019090B1/en active Active
- 2007-05-14 BR BRPI0712101A patent/BRPI0712101B8/pt active IP Right Grant
- 2007-05-14 PL PL12002538T patent/PL2514739T3/pl unknown
- 2007-05-14 RU RU2008145090/04A patent/RU2421443C2/ru active
- 2007-05-14 DK DK12002538.2T patent/DK2514739T3/en active
- 2007-05-14 PT PT120025382T patent/PT2514739T/pt unknown
- 2007-05-14 US US12/301,069 patent/US8017786B2/en active Active
- 2007-05-14 AU AU2007250364A patent/AU2007250364B8/en active Active
- 2007-05-14 HU HUE12002538A patent/HUE032505T2/en unknown
- 2007-05-14 ES ES12002538.2T patent/ES2618677T3/es active Active
-
2008
- 2008-11-16 IL IL195313A patent/IL195313A/en active IP Right Grant
-
2013
- 2013-02-13 CY CY20131100125T patent/CY1113676T1/el unknown
-
2017
- 2017-03-15 CY CY20171100327T patent/CY1118722T1/el unknown
Non-Patent Citations (4)
| Title |
|---|
| ANDRUS M.B. ET AL.: "Asymmetric Phase-Transfer Catalyzed Glycolate Alkylation, Investigation of the Scope, and Application to the Synthesis of (-)-Ragaglitazar", JOURNAL OF ORGANIC CHEMISTRY, vol. 70, no. 23, 2005, pages 9470 - 9479, XP003024722 * |
| DALKO P.I. ET AL.: "Stereoselective hetero-Claisen rearrangement of camphor derived oxazoline-N-oxides", TETRAHEDRON LETTERS, vol. 39, no. 15, 1998, pages 2107 - 2110, XP003024723 * |
| GAMBONI R. ET AL.: "Structure and diastereoselectivity of the alpha-hydroxylation of chiral ester enolates by molybdenum peroxo complex", TETRAHEDRON LETTERS, vol. 27, no. 34, 1986, pages 3999 - 4002, XP003024724 * |
| See also references of EP2019090A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014091465A2 (en) * | 2012-12-16 | 2014-06-19 | Mahesh Kandula | Compositions and methods for the treatment of metabolic syndrome and lipid disorders |
| WO2014091465A3 (en) * | 2012-12-16 | 2014-12-24 | Mahesh Kandula | Compositions and methods for the treatment of metabolic syndrome and lipid disorders |
| CN103570546A (zh) * | 2013-10-09 | 2014-02-12 | 西安石油大学 | 一种丹参素冰片酯的工业化合成方法 |
| WO2019127746A1 (zh) * | 2017-12-30 | 2019-07-04 | 苏州沪云肿瘤研究中心股份有限公司 | 一种苯丙酸酯类化合物及其制备方法和应用 |
| US11767286B2 (en) | 2017-12-30 | 2023-09-26 | Suzhou Pharmavan Co., Ltd | Phenylpropionate compound, preparation method for same, and applications thereof |
| CN109608356A (zh) * | 2018-12-14 | 2019-04-12 | 南京医科大学 | 一类丙二酸单酯酰化的氨基酸(+)2-崁醇酯的衍生物及其应用 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007131446A1 (fr) | Acide bêta-phényl-alpha-hydroxy propanoïque substitué, procédé de synthèse et son utilisation | |
| RU2431634C2 (ru) | Соединения флавоноидов и их применение | |
| JP6076737B2 (ja) | 高血圧症の防止及び治療のための組成物並びに方法 | |
| TWI689489B (zh) | 用於治療肺纖維化、肝纖維化、皮膚纖維化及心臟纖維化之經取代之芳族化合物 | |
| WO2002089809A1 (en) | Medicinal uses of hydrazones and hydrazines | |
| BR112015022008A2 (pt) | compostos aromáticos substituídos e métodos relacionados para o tratamento da fibrose | |
| ES2380454T3 (es) | Composición farmacéutica para prevenir y tratar enfermedades óseas metabólicas que contiene derivados de alfa-arilmetoxiacrilato | |
| KR20110117257A (ko) | 트리에틸아세틸기-3-히드록실기페닐기아데노신 및 혈지조절에 대한 용도 | |
| WO2002089799A2 (en) | Use of dihydropyrazoles to increase erythropoietin and vasculari zation | |
| WO2017124949A1 (zh) | 二氢黄酮衍生物、其制备方法和用途 | |
| WO2006007794A1 (fr) | Derives de stilbene cis-1,2-substitues et utilisation de ceux-ci pour la preparation de medicaments servant au traitement ou a la prevention du diabete | |
| WO2021032218A1 (zh) | 一种并环THRβ受体激动剂化合物及其制备方法和用途 | |
| CN113166024A (zh) | 芳族化合物及其医药用途 | |
| WO2012145899A1 (zh) | 酯类化合物、其制备方法和应用 | |
| CN110835335A (zh) | 2,5-二酮哌嗪类化合物及其在制备抗癌药物中的应用 | |
| CN113387909B (zh) | 2,3-环氧丁二酰衍生物的医药用途 | |
| CN114853630A (zh) | 一种2,6-二苯亚甲基环己酮肟类化合物及其制备方法和应用 | |
| WO2020038279A1 (zh) | 取代吡唑类化合物、其制备方法、药物组合物及用途 | |
| JP2020083811A (ja) | 1,5−アンヒドロフルクトース誘導体を含むampk活性化剤 | |
| WO2018053587A1 (en) | Compositions for the treatment of hypertension and/or fibrosis | |
| KR20240094390A (ko) | 골질환의 예방 또는 치료용 약학 조성물 | |
| WO2018166504A1 (zh) | 苯乙酮类化合物、其制备方法及其在调血脂方面的应用 | |
| JP2021529782A (ja) | 新生物障害及び神経性障害の診断、治療及び予防のための組成物及び方法 | |
| JP2003034690A (ja) | イノシトール誘導体、その製造方法、およびシクロオキシゲナーゼ−2発現抑制剤 | |
| WO2006034604A1 (fr) | Composes s-heterocycliques a 5 elements et leur utilisation dans la preparation de medicaments pour le traitement ou la prevention de maladies associees a l’obesite |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07721123 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2652299 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009510260 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 572958 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007721123 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007250364 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10230/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087030201 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008145090 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2007250364 Country of ref document: AU Date of ref document: 20070514 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12301069 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0712101 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081117 |