WO2007105753A1 - トリアゾール誘導体またはその塩 - Google Patents
トリアゾール誘導体またはその塩 Download PDFInfo
- Publication number
- WO2007105753A1 WO2007105753A1 PCT/JP2007/055048 JP2007055048W WO2007105753A1 WO 2007105753 A1 WO2007105753 A1 WO 2007105753A1 JP 2007055048 W JP2007055048 W JP 2007055048W WO 2007105753 A1 WO2007105753 A1 WO 2007105753A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lower alkyl
- triazole
- substituted
- methyl
- cycloalkyl
- Prior art date
Links
- 150000003839 salts Chemical class 0.000 title claims abstract description 33
- 150000003852 triazoles Chemical class 0.000 title claims abstract description 25
- 150000001875 compounds Chemical class 0.000 claims abstract description 130
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 79
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 33
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 18
- 206010012601 diabetes mellitus Diseases 0.000 claims abstract description 16
- 238000011282 treatment Methods 0.000 claims abstract description 8
- 230000003389 potentiating effect Effects 0.000 claims abstract 2
- -1 5-methyl-4-phenyl-4H- 1,2,4-triazol-3-yl Chemical group 0.000 claims description 99
- 238000004519 manufacturing process Methods 0.000 claims description 76
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 35
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 24
- 125000000623 heterocyclic group Chemical group 0.000 claims description 23
- 239000000126 substance Substances 0.000 claims description 22
- 125000003118 aryl group Chemical group 0.000 claims description 20
- 229910052736 halogen Inorganic materials 0.000 claims description 19
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 19
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 18
- 238000000034 method Methods 0.000 claims description 18
- 125000005843 halogen group Chemical group 0.000 claims description 17
- 239000003814 drug Substances 0.000 claims description 14
- 150000002367 halogens Chemical class 0.000 claims description 14
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 12
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims description 11
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 239000003112 inhibitor Substances 0.000 claims description 10
- 150000001721 carbon Chemical group 0.000 claims description 9
- 125000004076 pyridyl group Chemical group 0.000 claims description 8
- 229940124597 therapeutic agent Drugs 0.000 claims description 8
- 230000003449 preventive effect Effects 0.000 claims description 6
- 125000005275 alkylenearyl group Chemical group 0.000 claims description 5
- 229940122355 Insulin sensitizer Drugs 0.000 claims description 4
- 108090000874 11-beta-hydroxysteroid dehydrogenases Proteins 0.000 claims description 3
- 102000004277 11-beta-hydroxysteroid dehydrogenases Human genes 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 claims description 2
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 claims description 2
- 238000011321 prophylaxis Methods 0.000 claims description 2
- 230000002401 inhibitory effect Effects 0.000 abstract description 18
- 206010022489 Insulin Resistance Diseases 0.000 abstract description 15
- 208000001072 type 2 diabetes mellitus Diseases 0.000 abstract description 15
- 108010088011 11-beta-Hydroxysteroid Dehydrogenase Type 1 Proteins 0.000 abstract description 6
- 102000008645 11-beta-Hydroxysteroid Dehydrogenase Type 1 Human genes 0.000 abstract description 6
- 201000010099 disease Diseases 0.000 abstract description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 5
- 230000002058 anti-hyperglycaemic effect Effects 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 105
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 80
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 75
- 238000006243 chemical reaction Methods 0.000 description 72
- 239000000203 mixture Substances 0.000 description 72
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 68
- 230000002829 reductive effect Effects 0.000 description 63
- 239000007787 solid Substances 0.000 description 58
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 54
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 51
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 45
- 239000012044 organic layer Substances 0.000 description 42
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 41
- 238000010898 silica gel chromatography Methods 0.000 description 41
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 40
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 39
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 38
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 37
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 36
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 34
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 33
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 33
- 229920006395 saturated elastomer Polymers 0.000 description 32
- 239000002904 solvent Substances 0.000 description 31
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 29
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 27
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 25
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 23
- 238000010438 heat treatment Methods 0.000 description 23
- 239000000047 product Substances 0.000 description 18
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 17
- 235000017557 sodium bicarbonate Nutrition 0.000 description 17
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 17
- 238000000926 separation method Methods 0.000 description 16
- 235000011054 acetic acid Nutrition 0.000 description 15
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 15
- 239000007788 liquid Substances 0.000 description 15
- 239000011780 sodium chloride Substances 0.000 description 15
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 14
- 239000002253 acid Substances 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 125000001424 substituent group Chemical group 0.000 description 12
- 239000008280 blood Substances 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 230000000875 corresponding effect Effects 0.000 description 11
- 238000001914 filtration Methods 0.000 description 11
- 239000002994 raw material Substances 0.000 description 11
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 10
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 10
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 10
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 10
- 239000007864 aqueous solution Substances 0.000 description 10
- 125000001246 bromo group Chemical group Br* 0.000 description 10
- 238000001816 cooling Methods 0.000 description 10
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- 239000008103 glucose Substances 0.000 description 9
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 8
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 8
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 8
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 7
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000011541 reaction mixture Substances 0.000 description 7
- 239000012312 sodium hydride Substances 0.000 description 7
- 229910000104 sodium hydride Inorganic materials 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 6
- 208000008589 Obesity Diseases 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 125000002947 alkylene group Chemical group 0.000 description 6
- KTHXBEHDVMTNOH-UHFFFAOYSA-N cyclobutanol Chemical compound OC1CCC1 KTHXBEHDVMTNOH-UHFFFAOYSA-N 0.000 description 6
- 229910052739 hydrogen Inorganic materials 0.000 description 6
- 235000020824 obesity Nutrition 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 229910052717 sulfur Inorganic materials 0.000 description 6
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 6
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 5
- OLFDFWKBAMKMQO-UHFFFAOYSA-N 3,4-dicyclopropyl-5-(2-pyrrol-1-ylpropan-2-yl)-1,2,4-triazole Chemical compound C1=CC=CN1C(C)(C)C(N1C2CC2)=NN=C1C1CC1 OLFDFWKBAMKMQO-UHFFFAOYSA-N 0.000 description 5
- 101710088194 Dehydrogenase Proteins 0.000 description 5
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 208000031226 Hyperlipidaemia Diseases 0.000 description 5
- 206010020772 Hypertension Diseases 0.000 description 5
- 150000001298 alcohols Chemical class 0.000 description 5
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- MVEAAGBEUOMFRX-UHFFFAOYSA-N ethyl acetate;hydrochloride Chemical compound Cl.CCOC(C)=O MVEAAGBEUOMFRX-UHFFFAOYSA-N 0.000 description 5
- 125000001153 fluoro group Chemical group F* 0.000 description 5
- 125000000524 functional group Chemical group 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 201000001421 hyperglycemia Diseases 0.000 description 5
- OIRDBPQYVWXNSJ-UHFFFAOYSA-N methyl trifluoromethansulfonate Chemical compound COS(=O)(=O)C(F)(F)F OIRDBPQYVWXNSJ-UHFFFAOYSA-N 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 239000002798 polar solvent Substances 0.000 description 5
- 239000001294 propane Substances 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 239000012279 sodium borohydride Substances 0.000 description 5
- 229910000033 sodium borohydride Inorganic materials 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- 208000010412 Glaucoma Diseases 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 125000001118 alkylidene group Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 4
- 238000006911 enzymatic reaction Methods 0.000 description 4
- 229960000890 hydrocortisone Drugs 0.000 description 4
- 239000003701 inert diluent Substances 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000011259 mixed solution Substances 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 125000000714 pyrimidinyl group Chemical group 0.000 description 4
- 125000000168 pyrrolyl group Chemical group 0.000 description 4
- 239000008279 sol Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 4
- 125000000335 thiazolyl group Chemical group 0.000 description 4
- VWHRMADLSUBXLK-UHFFFAOYSA-N 1,3-dibromo-2-(methoxymethoxy)propane Chemical compound COCOC(CBr)CBr VWHRMADLSUBXLK-UHFFFAOYSA-N 0.000 description 3
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 208000001132 Osteoporosis Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 210000000577 adipose tissue Anatomy 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical group 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 150000002170 ethers Chemical class 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 229960002897 heparin Drugs 0.000 description 3
- 229920000669 heparin Polymers 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 229910052500 inorganic mineral Inorganic materials 0.000 description 3
- 235000010755 mineral Nutrition 0.000 description 3
- 239000011707 mineral Substances 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 125000005493 quinolyl group Chemical group 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 238000007363 ring formation reaction Methods 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- ORACYDGVNJGDMI-VAWYXSNFSA-N (e)-1,3-diphenylprop-2-en-1-ol Chemical compound C=1C=CC=CC=1C(O)\C=C\C1=CC=CC=C1 ORACYDGVNJGDMI-VAWYXSNFSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- 102000006739 11-beta-Hydroxysteroid Dehydrogenase Type 2 Human genes 0.000 description 2
- 108010086356 11-beta-Hydroxysteroid Dehydrogenase Type 2 Proteins 0.000 description 2
- 229940126558 11β-HSD1 inhibitor Drugs 0.000 description 2
- SNTWKPAKVQFCCF-UHFFFAOYSA-N 2,3-dihydro-1h-triazole Chemical compound N1NC=CN1 SNTWKPAKVQFCCF-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- KTFDYVNEGTXQCV-UHFFFAOYSA-N 2-Thiophenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CS1 KTFDYVNEGTXQCV-UHFFFAOYSA-N 0.000 description 2
- AVHBGFUJNUPHSU-UHFFFAOYSA-N 2-[2-(4,5-dicyclopropyl-1,2,4-triazol-3-yl)propan-2-yl]pyridine Chemical compound C=1C=CC=NC=1C(C)(C)C(N1C2CC2)=NN=C1C1CC1 AVHBGFUJNUPHSU-UHFFFAOYSA-N 0.000 description 2
- XLIQGIRXGKJQIR-UHFFFAOYSA-N 2-cyclopropyl-5-(2-pyridin-2-ylpropan-2-yl)-1,3,4-oxadiazole Chemical compound C=1C=CC=NC=1C(C)(C)C(O1)=NN=C1C1CC1 XLIQGIRXGKJQIR-UHFFFAOYSA-N 0.000 description 2
- LQLLQLCAIKJGJP-UHFFFAOYSA-N 2-methyl-2-pyridin-2-ylpropanehydrazide Chemical compound NNC(=O)C(C)(C)C1=CC=CC=N1 LQLLQLCAIKJGJP-UHFFFAOYSA-N 0.000 description 2
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical compound CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 2
- MJQMXMPFQBZROO-UHFFFAOYSA-N 3,5-dicyclopropyl-1h-1,2,4-triazole Chemical compound C1CC1C1=NNC(C2CC2)=N1 MJQMXMPFQBZROO-UHFFFAOYSA-N 0.000 description 2
- CNPURSDMOWDNOQ-UHFFFAOYSA-N 4-methoxy-7h-pyrrolo[2,3-d]pyrimidin-2-amine Chemical compound COC1=NC(N)=NC2=C1C=CN2 CNPURSDMOWDNOQ-UHFFFAOYSA-N 0.000 description 2
- CVICEEPAFUYBJG-UHFFFAOYSA-N 5-chloro-2,2-difluoro-1,3-benzodioxole Chemical group C1=C(Cl)C=C2OC(F)(F)OC2=C1 CVICEEPAFUYBJG-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- RMEUGDGEMREPAT-UHFFFAOYSA-N C(C)(=O)OCCCl.[Na] Chemical compound C(C)(=O)OCCCl.[Na] RMEUGDGEMREPAT-UHFFFAOYSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 2
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 2
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229960000530 carbenoxolone Drugs 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 229920001429 chelating resin Polymers 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- HXUAVKFUQQQZRL-UHFFFAOYSA-N chloroform formaldehyde Chemical compound C(Cl)(Cl)Cl.C=O HXUAVKFUQQQZRL-UHFFFAOYSA-N 0.000 description 2
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000006999 cognitive decline Effects 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- TXWOGHSRPAYOML-UHFFFAOYSA-M cyclobutanecarboxylate Chemical compound [O-]C(=O)C1CCC1 TXWOGHSRPAYOML-UHFFFAOYSA-M 0.000 description 2
- TXWOGHSRPAYOML-UHFFFAOYSA-N cyclobutanecarboxylic acid Chemical compound OC(=O)C1CCC1 TXWOGHSRPAYOML-UHFFFAOYSA-N 0.000 description 2
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 2
- XURKNVQJECSGJP-UHFFFAOYSA-N cyclobutyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OC1CCC1 XURKNVQJECSGJP-UHFFFAOYSA-N 0.000 description 2
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000012024 dehydrating agents Substances 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- 125000004611 dihydroisoindolyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 230000010030 glucose lowering effect Effects 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 230000002218 hypoglycaemic effect Effects 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 150000002466 imines Chemical class 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000003914 insulin secretion Effects 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000006371 metabolic abnormality Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 2
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 2
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 2
- 235000019799 monosodium phosphate Nutrition 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- HCCHMKIFYLZSOQ-UHFFFAOYSA-N n-cyclopropylcyclopropanecarboxamide Chemical compound C1CC1C(=O)NC1CC1 HCCHMKIFYLZSOQ-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 239000012457 nonaqueous media Substances 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 125000001715 oxadiazolyl group Chemical group 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 125000003566 oxetanyl group Chemical group 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- DXGIRFAFSFKYCF-UHFFFAOYSA-N propanehydrazide Chemical compound CCC(=O)NN DXGIRFAFSFKYCF-UHFFFAOYSA-N 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 2
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000005932 reductive alkylation reaction Methods 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 2
- 229960002218 sodium chlorite Drugs 0.000 description 2
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 125000005750 substituted cyclic group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 229940042055 systemic antimycotics triazole derivative Drugs 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- QVESIVAXVFLNLD-UHFFFAOYSA-N tert-butyl n-[1-(hydrazinecarbonyl)cyclobutyl]carbamate Chemical compound CC(C)(C)OC(=O)NC1(C(=O)NN)CCC1 QVESIVAXVFLNLD-UHFFFAOYSA-N 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- UKSZBOKPHAQOMP-SVLSSHOZSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 UKSZBOKPHAQOMP-SVLSSHOZSA-N 0.000 description 1
- GHPYJLCQYMAXGG-WCCKRBBISA-N (2R)-2-amino-3-(2-boronoethylsulfanyl)propanoic acid hydrochloride Chemical compound Cl.N[C@@H](CSCCB(O)O)C(O)=O GHPYJLCQYMAXGG-WCCKRBBISA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- ZYZCALPXKGUGJI-DDVDASKDSA-M (e,3r,5s)-7-[3-(4-fluorophenyl)-2-phenyl-5-propan-2-ylimidazol-4-yl]-3,5-dihydroxyhept-6-enoate Chemical compound C=1C=C(F)C=CC=1N1C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C(C)C)N=C1C1=CC=CC=C1 ZYZCALPXKGUGJI-DDVDASKDSA-M 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- KGKAYWMGPDWLQZ-UHFFFAOYSA-N 1,2-bis(bromomethyl)benzene Chemical compound BrCC1=CC=CC=C1CBr KGKAYWMGPDWLQZ-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- DNUTZBZXLPWRJG-UHFFFAOYSA-N 1-Piperidine carboxylic acid Chemical compound OC(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-N 0.000 description 1
- BNYLUVXSPAIYMI-UHFFFAOYSA-N 1-hydroxycyclopropane-1-carbohydrazide Chemical compound NNC(=O)C1(O)CC1 BNYLUVXSPAIYMI-UHFFFAOYSA-N 0.000 description 1
- 229940124681 11 beta HSD inhibitor Drugs 0.000 description 1
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 1
- GFISDBXSWQMOND-UHFFFAOYSA-N 2,5-dimethoxyoxolane Chemical compound COC1CCC(OC)O1 GFISDBXSWQMOND-UHFFFAOYSA-N 0.000 description 1
- WJMGOQFPVZDUMV-UHFFFAOYSA-N 2-(6-chloropyridin-2-yl)acetonitrile Chemical compound ClC1=CC=CC(CC#N)=N1 WJMGOQFPVZDUMV-UHFFFAOYSA-N 0.000 description 1
- MOMFXATYAINJML-UHFFFAOYSA-N 2-Acetylthiazole Chemical group CC(=O)C1=NC=CS1 MOMFXATYAINJML-UHFFFAOYSA-N 0.000 description 1
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 1
- JDMBMJTYPCGDDN-UHFFFAOYSA-N 2-[2-(4,5-dicyclopropyl-1,2,4-triazol-3-yl)propan-2-yl]-3h-isoindol-1-one Chemical compound C1C2=CC=CC=C2C(=O)N1C(C)(C)C(N1C2CC2)=NN=C1C1CC1 JDMBMJTYPCGDDN-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- IQHSSYROJYPFDV-UHFFFAOYSA-N 2-bromo-1,3-dichloro-5-(trifluoromethyl)benzene Chemical group FC(F)(F)C1=CC(Cl)=C(Br)C(Cl)=C1 IQHSSYROJYPFDV-UHFFFAOYSA-N 0.000 description 1
- IMRWILPUOVGIMU-UHFFFAOYSA-N 2-bromopyridine Chemical compound BrC1=CC=CC=N1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 1
- FPYUJUBAXZAQNL-UHFFFAOYSA-N 2-chlorobenzaldehyde Chemical compound ClC1=CC=CC=C1C=O FPYUJUBAXZAQNL-UHFFFAOYSA-N 0.000 description 1
- KVVDRQDTODKIJD-UHFFFAOYSA-N 2-cyclopropylacetic acid Chemical compound OC(=O)CC1CC1 KVVDRQDTODKIJD-UHFFFAOYSA-N 0.000 description 1
- OMLXJKPVHUWPQX-UHFFFAOYSA-N 2-methyl-1h-pyrrole Chemical compound CC1=CC=[C]N1 OMLXJKPVHUWPQX-UHFFFAOYSA-N 0.000 description 1
- CVTKARRWMZVHLJ-UHFFFAOYSA-N 2-methylbut-2-ene;2-methylpropan-2-ol Chemical compound CC=C(C)C.CC(C)(C)O CVTKARRWMZVHLJ-UHFFFAOYSA-N 0.000 description 1
- PLNNJQXIITYYTN-UHFFFAOYSA-N 2-methylpropanehydrazide Chemical compound CC(C)C(=O)NN PLNNJQXIITYYTN-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- CLSHQIDDCJTHAJ-UHFFFAOYSA-N 2-thienylacetonitrile Chemical compound N#CCC1=CC=CS1 CLSHQIDDCJTHAJ-UHFFFAOYSA-N 0.000 description 1
- RUBKAILYDADALU-UHFFFAOYSA-N 3,4-dicyclopropyl-1,2,4-triazole Chemical compound C1CC1C1=NN=CN1C1CC1 RUBKAILYDADALU-UHFFFAOYSA-N 0.000 description 1
- GBPJFTBEFZCMJO-UHFFFAOYSA-N 3-(methoxymethyl)-1h-pyrrole Chemical compound COCC=1C=CNC=1 GBPJFTBEFZCMJO-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZZHFDFIWLDELCX-UHFFFAOYSA-N 3-bromo-1h-pyrrole Chemical compound BrC=1C=CNC=1 ZZHFDFIWLDELCX-UHFFFAOYSA-N 0.000 description 1
- VLRGXXKFHVJQOL-UHFFFAOYSA-N 3-chloropentane-2,4-dione Chemical compound CC(=O)C(Cl)C(C)=O VLRGXXKFHVJQOL-UHFFFAOYSA-N 0.000 description 1
- OKCOIPJALQSOSJ-UHFFFAOYSA-N 3-cyclopropyl-2,2-dimethylpropanamide Chemical compound NC(=O)C(C)(C)CC1CC1 OKCOIPJALQSOSJ-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- AQBFKBMMIDHCFS-UHFFFAOYSA-N 4-bromo-2-benzofuran-1,3-dione Chemical compound BrC1=CC=CC2=C1C(=O)OC2=O AQBFKBMMIDHCFS-UHFFFAOYSA-N 0.000 description 1
- XQURBTZGKJENJS-UHFFFAOYSA-N 5-fluorothiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(F)S1 XQURBTZGKJENJS-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 229910015900 BF3 Inorganic materials 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- RNJRMTBMJBNYRD-UHFFFAOYSA-N C=O.CO.C(Cl)(Cl)Cl Chemical compound C=O.CO.C(Cl)(Cl)Cl RNJRMTBMJBNYRD-UHFFFAOYSA-N 0.000 description 1
- AQBPZOCARNHSIQ-UHFFFAOYSA-N CN(C)[S](N(C)C)N(C)C Chemical compound CN(C)[S](N(C)C)N(C)C AQBPZOCARNHSIQ-UHFFFAOYSA-N 0.000 description 1
- 101100451537 Caenorhabditis elegans hsd-1 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- DQFBYFPFKXHELB-UHFFFAOYSA-N Chalcone Natural products C=1C=CC=CC=1C(=O)C=CC1=CC=CC=C1 DQFBYFPFKXHELB-UHFFFAOYSA-N 0.000 description 1
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010023302 HDL Cholesterol Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000928753 Homo sapiens 11-beta-hydroxysteroid dehydrogenase 1 Proteins 0.000 description 1
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 1
- 235000003332 Ilex aquifolium Nutrition 0.000 description 1
- 241000209027 Ilex aquifolium Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- OHLUUHNLEMFGTQ-UHFFFAOYSA-N N-methylacetamide Chemical compound CNC(C)=O OHLUUHNLEMFGTQ-UHFFFAOYSA-N 0.000 description 1
- BAWFJGJZGIEFAR-NNYOXOHSSA-N NAD zwitterion Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-N 0.000 description 1
- XJLXINKUBYWONI-NNYOXOHSSA-N NADP zwitterion Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-NNYOXOHSSA-N 0.000 description 1
- ACFIXJIJDZMPPO-NNYOXOHSSA-N NADPH Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](OP(O)(O)=O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-NNYOXOHSSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 241000709400 Ruba Species 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- AVRWEULSKHQETA-UHFFFAOYSA-N Thiophene-2 Chemical compound S1C=2CCCCCC=2C(C(=O)OC)=C1NC(=O)C1=C(F)C(F)=C(F)C(F)=C1F AVRWEULSKHQETA-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- MOQOOKGPCBQMCY-UHFFFAOYSA-N acetic acid;hexane Chemical compound CC(O)=O.CCCCCC MOQOOKGPCBQMCY-UHFFFAOYSA-N 0.000 description 1
- WJGAPUXHSQQWQF-UHFFFAOYSA-N acetic acid;hydrochloride Chemical compound Cl.CC(O)=O WJGAPUXHSQQWQF-UHFFFAOYSA-N 0.000 description 1
- PHENPABYBHPABM-UHFFFAOYSA-N acetic acid;octane Chemical compound CC(O)=O.CCCCCCCC PHENPABYBHPABM-UHFFFAOYSA-N 0.000 description 1
- AVMNFQHJOOYCAP-UHFFFAOYSA-N acetic acid;propanoic acid Chemical compound CC(O)=O.CCC(O)=O AVMNFQHJOOYCAP-UHFFFAOYSA-N 0.000 description 1
- DQFAAIWCCHDCLO-UHFFFAOYSA-N acetonitrile;formic acid;hydrate Chemical compound O.CC#N.OC=O DQFAAIWCCHDCLO-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical group C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- BIVUUOPIAYRCAP-UHFFFAOYSA-N aminoazanium;chloride Chemical compound Cl.NN BIVUUOPIAYRCAP-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000000561 anti-psychotic effect Effects 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 230000000949 anxiolytic effect Effects 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 101150117004 atg18 gene Proteins 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 239000003899 bactericide agent Substances 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- MOOAHMCRPCTRLV-UHFFFAOYSA-N boron sodium Chemical compound [B].[Na] MOOAHMCRPCTRLV-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- DLIJPAHLBJIQHE-UHFFFAOYSA-N butylphosphane Chemical compound CCCCP DLIJPAHLBJIQHE-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- JHRWWRDRBPCWTF-OLQVQODUSA-N captafol Chemical compound C1C=CC[C@H]2C(=O)N(SC(Cl)(Cl)C(Cl)Cl)C(=O)[C@H]21 JHRWWRDRBPCWTF-OLQVQODUSA-N 0.000 description 1
- OBZHEBDUNPOCJG-SZTGPWMUSA-N carbenoxolone Chemical compound C([C@H]1C2=CC(=O)[C@@H]34)[C@](C)(C(O)=O)CC[C@@]1(C)CC[C@@]2(C)[C@]4(C)CC[C@H]1[C@@]3(C)CC[C@@H](OC(=O)CCC(O)=O)C1(C)C OBZHEBDUNPOCJG-SZTGPWMUSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 235000005513 chalcones Nutrition 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 238000000262 chemical ionisation mass spectrometry Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- SKCNIGRBPJIUBQ-UHFFFAOYSA-N chloroform;ethyl acetate Chemical compound ClC(Cl)Cl.CCOC(C)=O SKCNIGRBPJIUBQ-UHFFFAOYSA-N 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 229960004544 cortisone Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- KZZKOVLJUKWSKX-UHFFFAOYSA-N cyclobutanamine Chemical compound NC1CCC1 KZZKOVLJUKWSKX-UHFFFAOYSA-N 0.000 description 1
- KYUUPPKLZSVGMG-UHFFFAOYSA-N cyclobutanecarbohydrazide Chemical compound NNC(=O)C1CCC1 KYUUPPKLZSVGMG-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- ACYMGUSQXQEHGA-UHFFFAOYSA-N cyclohex-2-en-1-amine Chemical compound NC1CCCC=C1 ACYMGUSQXQEHGA-UHFFFAOYSA-N 0.000 description 1
- JFYKIEHOOZWARC-UHFFFAOYSA-N cyclopropanecarbohydrazide Chemical compound NNC(=O)C1CC1 JFYKIEHOOZWARC-UHFFFAOYSA-N 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 1
- MIOUTPXNKVBIFR-UHFFFAOYSA-J dicalcium 2-hydroxyacetate Chemical compound C(CO)(=O)[O-].[Ca+2].[Ca+2].C(CO)(=O)[O-].C(CO)(=O)[O-].C(CO)(=O)[O-] MIOUTPXNKVBIFR-UHFFFAOYSA-J 0.000 description 1
- CVKBMWWNKUWISK-UHFFFAOYSA-L dichloromethane;dichloropalladium Chemical compound ClCCl.Cl[Pd]Cl CVKBMWWNKUWISK-UHFFFAOYSA-L 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- IPZJQDSFZGZEOY-UHFFFAOYSA-N dimethylmethylene Chemical group C[C]C IPZJQDSFZGZEOY-UHFFFAOYSA-N 0.000 description 1
- FODIHFPARWJLJA-UHFFFAOYSA-N disodium;dicyanide Chemical compound [Na+].[Na+].N#[C-].N#[C-] FODIHFPARWJLJA-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- IOLQWGVDEFWYNP-UHFFFAOYSA-N ethyl 2-bromo-2-methylpropanoate Chemical compound CCOC(=O)C(C)(C)Br IOLQWGVDEFWYNP-UHFFFAOYSA-N 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- RQIOMXQOMYOGKD-UHFFFAOYSA-N fluorocyclohexane Chemical compound FC1[CH]CCCC1 RQIOMXQOMYOGKD-UHFFFAOYSA-N 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- XZBIXDPGRMLSTC-UHFFFAOYSA-N formohydrazide Chemical compound NNC=O XZBIXDPGRMLSTC-UHFFFAOYSA-N 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- JISVIRFOSOKJIU-UHFFFAOYSA-N hexylidene Chemical group [CH2+]CCCC[CH-] JISVIRFOSOKJIU-UHFFFAOYSA-N 0.000 description 1
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 208000013403 hyperactivity Diseases 0.000 description 1
- 125000005946 imidazo[1,2-a]pyridyl group Chemical group 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Substances C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000003933 intellectual function Effects 0.000 description 1
- 210000001596 intra-abdominal fat Anatomy 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- PKAUVIXBZJUYRV-UHFFFAOYSA-N methane;hydroiodide Chemical compound C.I PKAUVIXBZJUYRV-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- MDBZEGAGINSHQW-UHFFFAOYSA-N methyl 2-methyl-2-pyridin-2-ylpropanoate Chemical compound COC(=O)C(C)(C)C1=CC=CC=N1 MDBZEGAGINSHQW-UHFFFAOYSA-N 0.000 description 1
- BGTMJCYYKAHCFQ-UHFFFAOYSA-N methylsulfanyloxymethane Chemical compound COSC BGTMJCYYKAHCFQ-UHFFFAOYSA-N 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- TULYMWRNUWOELC-UHFFFAOYSA-N n,2-dicyclopropylacetamide Chemical compound C1CC1NC(=O)CC1CC1 TULYMWRNUWOELC-UHFFFAOYSA-N 0.000 description 1
- IXHFNEAFAWRVCF-UHFFFAOYSA-N n,2-dimethylpropanamide Chemical compound CNC(=O)C(C)C IXHFNEAFAWRVCF-UHFFFAOYSA-N 0.000 description 1
- JTARSJRCZDCRHE-UHFFFAOYSA-N n-(2-methyl-2-pyridin-2-ylpropanoyl)cyclopropanecarbohydrazide Chemical compound C=1C=CC=NC=1C(C)(C)C(=O)N(N)C(=O)C1CC1 JTARSJRCZDCRHE-UHFFFAOYSA-N 0.000 description 1
- LPOIGVZLNWEGJG-UHFFFAOYSA-N n-benzyl-5-(4-methylpiperazin-1-yl)-2-nitroaniline Chemical compound C1CN(C)CCN1C1=CC=C([N+]([O-])=O)C(NCC=2C=CC=CC=2)=C1 LPOIGVZLNWEGJG-UHFFFAOYSA-N 0.000 description 1
- 229940101270 nicotinamide adenine dinucleotide (nad) Drugs 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 235000020925 non fasting Nutrition 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 125000005498 phthalate group Chemical class 0.000 description 1
- 125000005542 phthalazyl group Chemical group 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- OHEDMAIVEXQCOZ-UHFFFAOYSA-N piperidine;dihydrochloride Chemical compound Cl.Cl.C1CCNCC1 OHEDMAIVEXQCOZ-UHFFFAOYSA-N 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 239000000932 sedative agent Substances 0.000 description 1
- 230000001624 sedative effect Effects 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- UYCAUPASBSROMS-UHFFFAOYSA-M sodium;2,2,2-trifluoroacetate Chemical compound [Na+].[O-]C(=O)C(F)(F)F UYCAUPASBSROMS-UHFFFAOYSA-M 0.000 description 1
- XHFLOLLMZOTPSM-UHFFFAOYSA-M sodium;hydrogen carbonate;hydrate Chemical class [OH-].[Na+].OC(O)=O XHFLOLLMZOTPSM-UHFFFAOYSA-M 0.000 description 1
- SUBJHSREKVAVAR-UHFFFAOYSA-N sodium;methanol;methanolate Chemical compound [Na+].OC.[O-]C SUBJHSREKVAVAR-UHFFFAOYSA-N 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000009469 supplementation Effects 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- BMWZMDDFCPGUSM-UHFFFAOYSA-N thiophene-2-carboxylic acid;hydrochloride Chemical compound Cl.OC(=O)C1=CC=CS1 BMWZMDDFCPGUSM-UHFFFAOYSA-N 0.000 description 1
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 1
- 229940125725 tranquilizer Drugs 0.000 description 1
- 239000003204 tranquilizing agent Substances 0.000 description 1
- 230000002936 tranquilizing effect Effects 0.000 description 1
- DQFBYFPFKXHELB-VAWYXSNFSA-N trans-chalcone Chemical compound C=1C=CC=CC=1C(=O)\C=C\C1=CC=CC=C1 DQFBYFPFKXHELB-VAWYXSNFSA-N 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- BZVJOYBTLHNRDW-UHFFFAOYSA-N triphenylmethanamine Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(N)C1=CC=CC=C1 BZVJOYBTLHNRDW-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000000052 vinegar Substances 0.000 description 1
- 235000021419 vinegar Nutrition 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/08—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/08—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- the present invention relates to a novel triazole derivative useful as a medicament, particularly as a therapeutic or prophylactic agent for a disease involving 11 ⁇ -hydroxysteroid dehydrogenase type 1, such as diabetes, insulin resistance, etc. Relates to a salt that is pharmaceutically acceptable.
- Darcocorticoids are hormones that cause metabolic abnormalities such as hyperglycemia, insulin resistance, obesity, hyperlipidemia, and hypertension, and are not only produced by the adrenal gland but also inactive at the tissue level. It is converted to the active form and acts through its receptor.
- 11 ⁇ -hydroxysteroid dehydrogenase (11 ⁇ -HSD) is an enzyme that catalyzes this conversion, and it is known that there are two subtypes.
- 11 j8 -Hydroxysteroid dehydrogenase type 1 (11 -HSDl) is an enzyme that converts an inactive form into an active form and is highly expressed in the liver
- 11 ⁇ -hydroxysteroid dehydrogenase type 2 (11 beta -HSD2) is an active form It is an enzyme that converts to an inactive form and is highly expressed in the kidney.
- Non-patent Documents 3 and 4 Transgenic mice overexpressed with 11 iS-HSDl selectively in adipose tissue show elevated darcocorticoids in adipose tissue, insulin resistance, visceral fat obesity, hyperlipidemia, and hypertension
- 8 -HSD1 knockout mice have been reported to exhibit improved glucose tolerance, decreased blood triglycerides, and increased HDL-cholesterol (Non-patent Document 5).
- 11-HSD1-selective inhibitors suppress the conversion of darcocorticoids into tissues by inhibiting the conversion to active darcocorticoids, resulting in hyperglycemia and insulin resistance induced by darcocorticoids. It is expected to correct metabolic abnormalities such as sex, obesity, hyperlipidemia, and hypertension.
- non-selective 11 ⁇ -HSD inhibitor carbenoxolone has been reported to improve insulin secretion decrease by inactive glucocorticoid supplementation in mouse spleen ⁇ -cells (Non-patent document 6).
- 11 -HSD1 inhibitors may correct hyperglycemia by promoting insulin secretion as well as improving insulin resistance.
- HSD1 osteoporosis
- glaucoma Non-patent document 8
- cognitive decline Non-patent document 9
- Patent Documents 1 to 9 are known as triazole derivatives having 11 ⁇ -HSD1 inhibitory action.
- Patent Document 1 reports a triazole derivative represented by the formula ( ⁇ ). However, it differs from the compound of the present invention in that there is no part corresponding to ⁇ and ⁇ of the compound of the present invention.
- R 1 represents an optionally substituted adamantyl
- X represents CH or a single bond
- Z represents S.
- Patent Document 2 reports a triazole derivative represented by the formula (B). However, it differs in that there is no structure corresponding to A and B of the compound of the present invention.
- R 1 is an aryl reel,-(CH) -aryl and-(CH) -hetero reel
- Patent Documents 3 and 4 report triazole derivatives represented by the formula (C). Shi However, it is substituted !, but is different from the compound of the present invention in that the phenyl ring is bonded to the triazo ring via one carbon atom.
- each R 3 is optionally substituted C alkyl.
- A is a C, optionally substituted C alkyl, OC alkyl or phenyl
- B is H, C, respectively.
- Patent Document 5 a triazole derivative represented by the formula (D) is reported. However, the difference is that the 3- or 5-position of the triazole ring is bonded by an oxygen atom or a sulfur atom.
- Patent Document 6 reports a triazole derivative represented by the formula (E). However, it differs from the compound of the present invention in that the triazole ring is condensed.
- Patent Document 7 reports a triazole derivative represented by the formula (F).
- Ar 1 corresponding to R 1 of the compound of the present invention is heteroaryl, only compounds in which Z-Ar 2 is a phenol are disclosed as examples.
- Patent Document 8 reports a compound represented by the formula (G) including a wide range of compounds!
- R 1 represents a hydrogen atom or an optionally substituted cyclic group
- R 2 represents an optionally substituted cyclic group
- Ar represents an optionally substituted 5- or 6-membered aromatic heterocyclic ring
- L 1 and L 2 may be the same or different (1) a bond (2) substituted !, which may be a divalent hydrocarbon group o
- Patent Document 9 filed by the present applicant and published after the priority date of the present application, a triazole derivative represented by the formula (H) is reported. Specific compounds which are groups are disclosed, it, according to R 3 in R 3 is the invention onset Akira compound of formula (H) with and force.
- Patent Document 10 reports that a triazole derivative represented by the formula ⁇ is useful as an antipsychotic drug and an analgesic drug.
- ⁇ - [2- (4-Chroosteal) ethyl]- ⁇ - Methyl-1- (5-methyl-4-phenol-4 ⁇ -1,2,4-triazol-3-yl) cyclohex-2-en-1-amine is described as an example Yes.
- Ij8-HSD1 inhibitory action and effectiveness for diabetes treatment is not mention of Ij8-HSD1 inhibitory action and effectiveness for diabetes treatment.
- Patent Document 11 reports that the triazole derivative represented by the formula (K) is useful as a sedative, a tranquilizer, and an anxiolytic, (Dimethylamino) cyclopropyl] -5-methyl-4H-1,2,4-triazol-4-yl ⁇ phenyl) (2-chlorophenyl) methanone is described as an example. However, I lj8-HSD1 inhibitory action and effectiveness for diabetes treatment are not described.
- Non-Patent Document 1 Rask E. et al., “The Journal of Clinical Endocrinology & Metabolism” (USA), 2001, 86th, p.1418-1421
- Non-Patent Document 2 Lindsay RS et al., “The Journal of Clinical Endocrinology & Metabolism” J, 2003, 88th, p.2738-2744
- Non-Patent Document 3 Masuzaki H. et al., “Science” (USA), 2000, 294, pp.2166-2170
- Non-Patent Document 4 Masaki H. et al., “The Journal of Clinical Investigation” (USA), 2003, 112th ⁇ , p.83-90
- Non-Patent Document 5 Morton NM et al., "The Journal of Biological Chemistry” (USA), 2001, 27th , P.41293- 41300
- Non-Patent Document 6 Davani B. et al., “The Journal of Biological Chemistry” (USA), 2000, 275th p. 3484 1-34844
- Non-Patent Document 7 Cooper M.S., et al., “Bone” (USA), 2000, No. 27, p.375-381
- Non-Patent Document 8 Rauz S. et al., “Investigative Opthalmology & Visual Science” (USA), 2001, No. 42 , P.2037-2042
- Non-Patent Document 9 Sandeep TC, et al., “Proceedings of the National Academy of Sciences” (USA) 2004, No. 101, p.6734-6739
- Patent Document 1 International Publication No. 03/65983 Pamphlet
- Patent Document 2 US Patent Application Publication No. 2004/133011
- Patent Document 3 International Publication No. 03/104207 Pamphlet
- Patent Document 4 International Publication No. 03/104208 Pamphlet
- Patent Document 5 Pamphlet of International Publication No. 04/089367
- Patent Document 6 International Publication No. 04/089380 Pamphlet
- Patent Document 7 International Publication No. 05/044192 Pamphlet
- Patent Document 8 JP-A-2005-170939
- Patent Document 9 Pamphlet of International Publication No. 06/030805
- Patent Document 10 US Patent No. 4577020
- Patent Document 11 US Pat. No. 3,090,781 specification
- the 11 ⁇ -HSD1 inhibitor described in the above document is not satisfactory in terms of efficacy, selectivity, safety, and economy, and provides an excellent selective 11 j8-HSD1 inhibitor Is anxious.
- the present inventors have conducted extensive research on a compound having 11 jS-HSD1 inhibitory activity that can be expected to improve diabetes and insulin resistance.
- the novel triazole derivative of the present invention or its The present inventors have found that a salt has an excellent selective inhibitory action on 11 ⁇ -HSD1 and completed the present invention.
- these compounds have, for example, in vivo drug efficacy (such as hypoglycemic action and / or tridallylide lowering action), oral absorbability or metabolic stability compared to the conventionally known 11 j8-HSD1 inhibitors. It is excellent in any of efficacy, selectivity, safety, and economy, such as selectivity with cytochrome p450 (Cytochrome p450; CYP) inhibitory action that may cause drug interaction. This is useful.
- the present invention relates to a triazole derivative represented by the following formula (I) or a pharmaceutically acceptable salt thereof that is useful as an 11 ⁇ -HSD1 inhibitor.
- R 1 heterocyclic group, or —N (R °) -R 4 .
- the heterocyclic group in R 1 may be substituted!
- R ° -H or lower alkyl.
- R 4 C alkyl, halogeno lower alkyl, lower alkyl substituted with cycloalkyl
- a and B the same or different from each other, lower alkyl.
- a and B may be combined to form a cycloalkyl ring, which may be bonded together and substituted with a carbon atom.
- R 2 lower alkyl, halogeno lower alkyl, cycloalkyl, aryl, lower alkylene-CO R °, lower alkylene-cycloalkyl, lower alkylene-aryl or lower alkyl
- cycloalkyl, aryl and aromatic heterocyclic group in R 2 may be substituted respectively.
- R 3 -H, halogen, lower alkyl, halogeno lower alkyl, -OR 0 , -CO R °, cycloal
- cycloalkyl and saturated heterocyclic group in R 3 may be substituted.
- the present application also relates to a pharmaceutical composition
- a pharmaceutical composition comprising a triazole derivative represented by the general formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, in particular, 11 18 -hydroxy steroid dehydrogenase. It also relates to a pharmaceutical composition that is a type 1 inhibitor, an insulin sensitizer, or a preventive or therapeutic agent for diabetes. Further, the present application relates to a compound represented by formula (I) or a pharmaceutical product thereof for the manufacture of 11 j8 -hydroxysteroid dehydrogenase 'type 1 inhibitor, insulin resistance ameliorating agent, diabetes preventive or therapeutic agent. And a method for preventing or treating diabetes, comprising administering to a patient an effective amount of a compound represented by formula (I) or a pharmaceutically acceptable salt thereof. .
- a pharmaceutical composition comprising a compound according to formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
- composition according to (1) which is a preventive or therapeutic agent for diabetes.
- a method for the prophylaxis or treatment of diabetes comprising administering an effective amount of the compound according to formula (I) or a salt thereof to a patient.
- the procedure for measuring 11 18 -HSD1 inhibitory activity is as follows. The enzyme reaction and measurement were performed using a 384 well plate. The enzyme was prepared according to the literature (Walker EA et al., Journal of Biologi cal Chemistry, 2001, 276, p. 21343-21350). Reactions consisted of 10 mM phosphate buffer (PH6.6), 200 nM cortisone, 40 M reduced nicotinamide adenine dinucleotide phosphate (NADPH), and human recombinant 11 j8-HSD1 in various concentrations. The test compound was added and then incubated at room temperature for 1 hour (10 ⁇ l / well).
- the test compound was dissolved in dimethyl sulfoxide (DMSO) to prepare a DMS 0 concentration of 1% in the reaction solution.
- DMSO dimethyl sulfoxide
- cortisol is analyzed by time-resolved fluorescence (Ho).
- HTRF time-resolved fluorescence
- XL-665 labeled cortisol containing 400 ⁇ - carbenoxolone and Cryptate labeled cortisol antibody (CIS bio international) 5 ⁇ 1 / Welka ⁇ e After incubating at room temperature for 2 hours, measure the fluorescence intensity using a fluorometer (trade name: Discovery, PerkinElmer). From the fluorescence intensity ratio of two wavelengths (665nm / 620nm) Enzyme inhibitory activity was calculated.
- the 11 ⁇ -HSD2 inhibitory activity was measured in the same manner as the 11 ⁇ -HSD1 inhibitory activity except for the enzyme reaction conditions.
- Enzymatic reactions can be performed in various reaction solutions consisting of 40 mM Tris-HCl buffer (Tris-HC1) (pH 8.0), 200 nM cortisol, 200 nicotinamide adenine dinucleotide (NAD), and human recombinant 11 i8 -HSD2. After incubating the test substance at a concentration of 37 ° C, incubation was performed at 37 ° C for 2 hours (10 1 / well).
- the measurement results were calculated by averaging the values of 3 wells under the same conditions.
- the ratio when DMSO is added instead of the test compound is 0%, the ratio when 11 ⁇ -HSD1 or 11 ⁇ -HSD2 is not added is 100%, and the concentration at which the test compound is inhibited by 50%.
- IC for compound inhibitory activity is a ratio when DMSO is added instead of the test compound.
- Example Compound 3 As a result, 27% of Example Compound 3, 21% of Example Compound 154, 24% of Example Compound 167, 27% of Example Compound 172, and 35% of Example Compound 280 showed a blood glucose lowering effect, It was confirmed that the compound of the present invention has an excellent hypoglycemic effect.
- Example Compound 3 showed a triglyceride lowering action of 50% and Example Compound 280 of 42%, confirming that the compound of the present invention has an excellent triglyceride lowering action.
- the compound of the present invention has an 11 ⁇ -HSD1 inhibitory action. Therefore, prevention / treatment of hyperglycemia, insulin resistance, obesity, hyperlipidemia, hypertension, osteoporosis, glaucoma, cognitive decline, etc., particularly diabetes, insulin resistance, etc. involving 11 j8-HSD1 It is clear that it is useful as a therapeutic agent for diseases such as drugs.
- lower alkyl lower alkenyl
- lower alkylidene lower alkylene
- “Lower alkenyl” refers to a C alkaryl having a plurality of double bonds.
- “Lower alkylidene” means a group in which a free valence formed by removing one carbon nuclear hydrogen having a lower alkyl bond is a double bond. Specific examples include methylidene, ethylidene, propylidene, butylidene, pentylidene, and hexylidene. Preferably C 1 alkylidene, more preferably methylidene.
- Alkylene means a divalent group formed by removing one hydrogen at any position of alkyl.
- Lower alkylene means C alkylene. Specifically, methylene,
- Tylene methylmethylene, dimethylmethylene, propylene, butylene, pentylene, hexylene and the like.
- Preferred is C alkylene, and more preferred is methylene, ethylene,
- Tinolemethylene dimethylmethylene and propylene.
- Cycloalkyl means a non-aromatic hydrocarbon ring of C 1, and the bridged ring is a spiro ring.
- cyclopropyl cyclobutyl, cyclopentyl, cyclohexyl, cyclohexyl, cyclohexyl, cyclobutyr, adamantyl, norbornyl, etc., preferably C cycloalkyl, more preferably.
- Cycloalkyl ring formed by combining A and B together with the carbon atom to which they are bonded is a non-aromatic group of C that is bonded to A and B at the same carbon atom.
- carbonized water Of carbonized water
- a bridged ring may form a spiro ring. Moreover, it may have a partially unsaturated bond.
- cyclopropyl ring cyclopropane-1,1-diyl
- cyclobutyl ring cyclobutane-1,1-diyl
- cyclopentyl ring cyclopentane-1,1-diyl
- cyclohexane ring Cyclohexane-1,1-diyl
- cyclobutyr ring cyclobut-2-ene-1,1-diyl
- a cyclopropyl ring, a cyclobutyl ring or a cyclobutenyl ring More preferably a cyclopropyl ring, a cyclobutyl ring or a cyclobutenyl ring.
- Halogen means a halogen atom, and specific examples include fluoro, black mouth, bromine and iodine, and preferred are fluoro and black mouth.
- halogeno lower alkyl means one or more arbitrary hydrogen atom forces of the “lower alkyl” which are the same or different from each other and substituted with the “halogen”. Specific examples include trifluoromethyl, pentafluoroethyl and the like. Preferred is trifluoromethyl.
- aryl means a monocyclic to tricyclic C aromatic hydrocarbon ring, specifically
- a "heterocyclic” group is i) a monocyclic 3 to 8 membered (preferably 5 to 7 membered) heterocyclic ring containing 1 to 4 selected heteroatoms, 0, S and N forces, ii )
- the monocyclic heterocycle is condensed with one or two rings selected from the group consisting of a monocyclic heterocycle, a benzene ring, and a C cycloalkyl group.
- Bicyclic 8- to 14-membered (preferably 9-: L 1-membered) heterocycles and tricyclic 11- to 20-membered (containing 1 to 5 heteroatoms also selected for 0, S and N forces) (Preferably 12 to 15 membered) means a heterocyclic group or a cyclic group composed of the heterocyclic group.
- the ring atom S or N may be oxidized to form an oxide or dioxide.
- heterocyclic Preferred as the “heterocyclic” group are aziridyl-, azetidyl, pyrrolidyl-, piberidinyl, piperazinyl, homopiperazinyl, oxylanyl, oxetanyl, tetrahydrofuranyl, tetrahydrobiranyl, morpholinyl, homomorpholinyl, tetrahydrothiobilanyl, pyrrolyl, imidazolyl, triazolyl, Tetrazolyl, pyridyl, pyrimidyl, pyrajyl, furyl, chenyl, oxazolyl, oxadiazolyl, thiazolyl, thiadiazolyl, indolyl, dihydroisoindolyl, indolizyl, benzimidazolyl, imidazo [1,2-a] pyridyl, quinoxalinyl , Quino
- the "saturated heterocycle” group is a saturated heterocycle group among the above-mentioned “heterocycle” groups, for example, aziridinyl, azetidyl, pyrrolidinyl, piperidinyl, piperazinyl, homopiperazinyl, oxylanyl, Examples include oxetanyl, tetrahydrofuranyl, tetrahydrobilanyl, morpholinyl, homomorpholinyl, and tetrahydrothiobilal.
- Aromatic heterocycle means a monocyclic 3 to 8 member, preferably 1 to 4 heteroatoms selected from 0, S and N forces among the above “heterocyclic” groups, preferably Means a 5- to 7-membered monocyclic aromatic hetero ring and a bi- or tri-cyclic hetero ring in which the aromatic hetero rings or the aromatic hetero rings are condensed with a benzene ring.
- the ring atom S or N may be oxidized to form an oxide.
- Pyridyl, pyrimidyl, pyradyl, chenyl, pyrrolyl, thiazolyl and quinolyl are preferable, and pyridyl and chelyl are more preferable.
- Substituted in R 1 may preferably be a group selected from the following G 1 group, more preferably as a substituent allowed in the "heterocyclic group". , Halogen, lower alkyl, halogeno lower alkyl, cyan-containing -0-lower alkyl and -0-halogeno lower alkyl, more preferably, halogen, lower alkyl and halogeno lower alkyl. Groups selected from the group consisting of strong groups, and even more preferably groups selected from the group consisting of halogen and halogeno lower alkyl groups.
- G Group 1 neurogen, lower alkyl, lower alkyl, halogeno-lower alkyl, siaoyl -0 R °, -0-norgeno lower alkyl, lower alkylene-OR °, lower alkylene-0-lower alkylene-cyclo Alkyl, -C (0) R °, -CO R. And oxo.
- R 4 is preferably substituted !, may! /, “Cycloalkyl”, “aryl”, and “aromatic heterocyclic group”, preferably as halogenated and lower alkyl. And a group selected from the group consisting of a strong group, and a halogen is more preferable.
- a and B are converted into a single body, and the carbon atoms to which they are bonded are replaced with each other! /, May! /
- Preferred examples of the permissible substituent include a group selected from the group consisting of a group consisting of no, a rogen, and an -OH force.
- aryl and aromatic heterocyclic group which may be substituted in R 2
- substituents include groups selected from the group consisting of halogen, lower alkyl, —OR Q, and —0-norogeno lower alkyl force, and more preferably halogen.
- Cycloalkyl is preferably a group selected from the group consisting of halogen, lower alkyl, and arylalkyl. And more preferably halogen.
- R 1 is preferably an optionally substituted aromatic heterocyclic group, more preferably each optionally substituted pyridyl, chael, pyrrolyl, quinolyl, thiazolyl, pyrimidyl or bilazyl, Even more preferably, they are each substituted with a group selected from the group consisting of lower alkyl, halogen and neurogen lower alkyl, and may be pyridyl or chenyl, particularly preferably lower alkyl, halogen and neurogen. Substituted with a group selected from the group consisting of lower alkyl !, may! /, Chaels.
- a and B are preferably methyl.
- the ring formed by combining A and B together with the carbon atom to which they are bonded is preferably a cyclobutyl ring or a cyclobutyl ring, and more preferably a nonogen.
- it may be substituted with —OH, but is a cyclobutyl ring or a cyclobutyr ring, and more preferably substituted with halogen or —OH! It may be a cyclobutynole ring.
- R 2 is preferably lower alkyl, cycloalkyl, lower alkylene- (optionally substituted or aryl) or lower alkylene-aromatic heterocyclic group, more preferably lower alkyl, cycloalkyl or lower alkylene. -(Substituted, optionally, aryl) and even more preferably methyl, cyclopropyl,-(CH)-(no, substituted with rogen)
- (CH 3) 2 -pyridyl more preferably cyclopropyl or-( CH 2)-(no, a phenyl optionally substituted with a rogen), and even more preferably cyclo
- R 3 is preferably lower alkyl or substituted or cycloalkyl, more preferably cycloalkyl optionally substituted with lower alkyl, and more preferably substituted with lower alkyl. It may be cyclopropyl or cyclobutyl, even more preferably cyclopropyl or cyclobutyl optionally substituted with methyl, particularly preferably cyclopropyl.
- a and B forces are both methyl; or A and B are isomers, and the ring formed together with the carbon atom to which they are bonded may be substituted with a cyclobutyl ring or a cyclobutenyl ring, respectively.
- R 2 is cyclopropyl or — (CH 2) ⁇ (a phenyl optionally substituted with halogen)
- the triazole derivative represented by the formula (I) may form a salt, and is included in the compound of the present invention as long as the salt to be obtained is a pharmaceutically acceptable salt.
- inorganic salts such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, Acid addition salts with organic acids such as maleic acid, lactic acid, malic acid, tartaric acid, citrate, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, asnoginic acid, glutamic acid, sodium, potassium, calcium, Examples thereof include inorganic bases containing metals such as magnesium, addition salts with organic bases such as methylamine, ethylamine, ethanolamine, lysine and ortho, ammonium salt
- the compound of the present invention may contain an asymmetric carbon atom depending on the type of substituent, and optical isomers based on this may exist.
- the present invention encompasses all of these optical isomer mixtures and isolated ones.
- the compounds of the present invention may have tautomers, but the present invention includes a mixture of these isomers or a mixture thereof.
- a label that is, a compound obtained by substituting one or more atoms of the compound of the present invention with a radioactive isotope or a non-radioactive isotope is also included in the present invention.
- the present invention includes various hydrates and solvates of the compounds of the present invention and substances having crystal polymorphs.
- the compounds of the present invention include all of the derivatives represented by the formula (I) and pharmaceutically acceptable salts thereof, which are not limited to the compounds described in the Examples below. Is.
- the compounds of the present invention include all compounds that are metabolized in vivo and converted into the compounds of the present invention, so-called prodrugs.
- Groups that form prodrugs of the compounds of the present invention include “Progress in Medicine”, Lifecycle The group described in Ens' Medica, 1985, May 5, p.2157-2161, and the group described in Yodogawa Shoten 1999, “Development of Drugs”, 7th Molecular Design, pages 163-198 Are listed.
- the compound of the present invention and a pharmaceutically acceptable salt thereof can be produced by applying various known synthetic methods using characteristics based on the basic skeleton or the type of substituent.
- a typical production method is illustrated below.
- it is effective in terms of production technology to replace the functional group with a suitable protecting group at the raw material or intermediate stage, that is, a group that can be easily converted to the functional group. There are cases. Thereafter, the protecting group can be removed as necessary to obtain the desired compound.
- functional groups include a hydroxyl group, a carboxyl group, and an amino group.
- protective groups for these functional groups include “Protective”, “Groups”, “In” and “Organic” by Greene and Wuts.
- L 1 represents a leaving group
- This production method is a method for producing the compound (I) of the present invention by a cyclization reaction between the compound (II) and the compound (III).
- the leaving group for L 1 include black mouth, promo, methoxy, methylsulfur and the like.
- Reactions include ethers such as tetrahydrofuran (THF), 1,4 dioxane, diglyme, alcohols such as methanol, ethanol, propanol, butanol or ⁇ , ⁇ dimethylformamide (DMF), dimethylimidazolidinone, di
- THF tetrahydrofuran
- 1,4 dioxane diglyme
- alcohols such as methanol, ethanol, propanol, butanol or ⁇ , ⁇ dimethylformamide (DMF), dimethylimidazolidinone, di
- the reaction can be carried out in a solvent such as methylacetamide or DMSO or an aprotic polar solvent at room temperature or under heating conditions.
- This production method is a method for producing the compound (I) of the present invention by a compound (IV) force alkylation reaction.
- a compound (IV) force alkylation reaction In the alkylation reaction in this step, sodium hydride, potassium hydride, butyllithium, lithium diisopropylamide or the like can be used as the base, and the corresponding alkyl halide, dihalogen-alkane or the like can be used as the electrophilic reagent.
- the reaction can be carried out in a solvent such as an ether or an aprotic polar solvent under cooling, at room temperature or under heating conditions.
- phase transfer catalyst such as tetra-n-butylammodium.
- This production method is a method for producing the compound (I) of the present invention by cyclization reaction between the activated carboxylic acid derivative compound (V) and the compound (VI).
- the leaving group for L 2 include black mouth, bromo, fluoro, and acyloxy.
- Reaction is ether Can be carried out in a solvent such as alcohols, alcohols or aprotic polar solvents at room temperature or under heating conditions.
- an acid such as an organic acid such as acetic acid or P-toluenesulfonic acid, or a mineral acid such as sulfuric acid or hydrochloric acid.
- This production method is a method for obtaining the compound (I) of the present invention by reacting the compound ( ⁇ ) with the compound (vm).
- the reaction is carried out using an equivalent amount of compound ( ⁇ ) and compound (vm), or an excess of one of them, in an alcohol, aromatic hydrocarbons such as benzene, toluene, xylene, acetic acid, etc. in a solvent inert to the reaction.
- the reaction can be performed without solvent, at room temperature or under heating, preferably under heating.
- an acid such as acetic acid, organic acids such as P-toluenesulfonic acid, and mineral acids such as sulfuric acid and hydrochloric acid. It may also be advantageous to perform the reaction using microwaves.
- R 4 ° is C alkyl, halogeno lower alkyl, lower substituted with cycloalkyl.
- Alkyl, cycloalkyl, aryl, lower alkylene-aryl or lower alkylene-aromatic heterocyclic group is shown. The same applies below. )
- This production method is a method for obtaining the compound (Ia) of the present invention by reductive alkylation of the compound (IX).
- an equivalent amount of compound (IX) and aldehyde or ketone corresponding to R 4Q is used in an excess amount of H or one, and inactive in the reaction with alcohols, ethers, etc. in the presence of a reducing agent.
- the cooling power can be carried out with heating under reflux.
- 3 examples are sodium cyanide sodium borohydride, sodium triacetoxyborohydride, sodium borohydride and the like. It may be preferable to carry out the reaction in the presence of a dehydrating agent such as molecular sieves, or an acid such as acetic acid, hydrochloric acid, titanium (IV) isopropoxide complex. Depending on the reaction, when an imine formed in the reaction system as an intermediate can be stably isolated, a separate reduction reaction may be performed after obtaining the imine.
- a dehydrating agent such as molecular sieves
- an acid such as acetic acid, hydrochloric acid, titanium (IV) isopropoxide complex.
- L 3 represents a leaving group
- R 41 represents a lower alkyl, aryl or aromatic heterocyclic group. The same shall apply hereinafter.
- This production method is a method in which the compound (IX) and the compound (X) are reacted to obtain the compound (Ib) of the present invention.
- examples of the leaving group for L 3 include black mouth, bromo, and fluoro.
- compound (IX) and compound (X) are used in the same amount or in an excess amount, and ethers, halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane or chloroform, and aprotic polar solvents.
- the reaction can be carried out under cooling to heating in a solvent inert to the reaction.
- the reaction proceeds smoothly in the presence of an organic base such as triethylamine, ⁇ , ⁇ -diisopropylethylamine or ⁇ ⁇ ⁇ -methylmorpholine, or an inorganic base such as potassium carbonate or sodium carbonate.
- the raw materials used in the production of the compound of the present invention are, for example, by applying the following method, the method described in Reference Examples described later, a known method, a method obvious to those skilled in the art, or a modification thereof. Can be manufactured.
- Compound (VII) can be produced by a cyclization reaction between compound (II) and compound (XI).
- examples of the leaving group for L 4 include black mouth, bromo, fluoro, and hydroxy.
- the reaction is carried out by condensing (II) and (XI) at room temperature or under heating conditions in a solvent such as an aprotic polar solvent such as a halogenated hydrocarbon, and the resulting diacyl is converted to phosphorus oxychloride or trifluoromethanesulfone. It can be produced by the action of a dehydrating agent such as an acid anhydride.
- a dehydrating agent such as an acid anhydride.
- reacting in the presence of an organic base such as triethylamine, ⁇ , ⁇ -diisopropylethylamine or pyridine, or an inorganic base such as potassium carbonate or sodium carbonate may facilitate the reaction. May be advantageous.
- Boc means a tert-ptoxycarbon group. The same applies hereinafter.
- Compound (IX) can be produced by deprotecting compound (XIII).
- Deprotection of the Boc group can be carried out by a method commonly used by those skilled in the art. For example, it can be carried out by the method described in “Protective ⁇ Groups ⁇ Organic ⁇ Synthesis”.
- Compound (XIII) can be produced from compound (XII) and compound (III) in the same manner as in the first production process.
- the compound of the present invention produced as described above is isolated or purified as a salt by leaving it as it is or subjecting it to a conventional salt formation treatment. Isolation and purification are performed by applying ordinary chemical operations such as extraction, concentration, distillation, distillation, crystallization, filtration, recrystallization, and various chromatography.
- Various isomers can be isolated by conventional methods utilizing the difference in physicochemical properties between isomers.
- a racemic mixture can be converted to an optically pure heteroisomer by a general racemic resolution method, such as a method of optical resolution by diastereomeric salt with a general optically active acid such as tartaric acid.
- the diastereomeric mixture can be separated by, for example, fractional crystallization or various types of chromatography.
- An optically active compound can also be produced by using an appropriate optically active raw material.
- a pharmaceutical composition containing the compound of the present invention or one or more of pharmaceutically acceptable salts thereof as an active ingredient comprises a carrier carrier, excipient, and other additives for commonly used formulations. It is prepared into tablets, powders, fine granules, granules, capsules, pills, solutions, injections, suppositories, ointments, patches, etc. and administered orally or parenterally.
- the clinical dose of the compound of the present invention for humans is appropriately determined in consideration of the symptoms, body weight, age, sex, etc. of the patient to which it is applied.
- the daily dose is usually about 0.0001 per body weight. ⁇ 50 mg / kg, preferably about 0.001 to 10 mg / kg, more preferably 0.01 to 1 mg / kg, which is administered once or divided into 2 to 4 times.
- the daily dose is about 0.0001 to 1 mg / kg, preferably about 0.0001 to 0.1 mg / kg per body weight, and is administered once to several times a day. Since the dose varies depending on various conditions, a sufficient effect may be obtained if the dose is less than the above dose range.
- the solid composition for oral administration tablets, powders, granules and the like are used.
- one or more active substances are present in at least one inert diluent such as lactose, mannitol, glucose, hydroxypropyl cellulose, microcrystalline cellulose, starch, poly Mixed with bull pyrrolidone, magnesium metasilicate aluminate, etc.
- the composition contains additives other than inert diluents, for example, lubricants such as magnesium stearate, disintegrating agents such as calcium calcium glycolate, stabilizers, solubilizing agents and the like according to conventional methods. It may be. If necessary, tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate, or a gastric or enteric film.
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, elixirs, and the like, commonly used inert diluents, For example, purified water and ethanol (EtOH) are included.
- the composition may contain adjuvants such as wetting agents and suspending agents, sweeteners, flavors, fragrances and preservatives.
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions.
- aqueous solutions and suspensions include distilled water for injection and physiological saline.
- non-aqueous solutions and suspensions include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, alcohols such as EtOH, and polysorbate 80.
- Such a composition may further contain auxiliary agents such as preservatives, wetting agents, emulsifiers, dispersants, stabilizers, and solubilizing agents. These are sterilized, for example, by filtration through a bacteria-retaining filter, blending with a bactericide, or irradiation. These can also be used by producing a sterile solid composition and dissolving it in sterile water or a sterile solvent for injection before use.
- the number in front of the substituent indicates the substitution position, and thus 4-C ⁇ 5-F, for example, indicates 4-chloro-5-fluoro.
- Syn production method (number indicates production using the corresponding raw material in the same manner as the example compound having the number as an example number), RSyn: production method (the number is the production example number) This indicates that the product was produced using the corresponding raw material in the same manner as in the production example compound.
- Production Example 4 2- (3 Chloro-2 chalcone) 2-methylpropanecarboxylic acid is obtained by reacting 2-methylpropane-tolyl and potassium hydroxide in ethylene glycol under heating. It was.
- Ethyl (1-oxide pyridine-4-inole) acetate was obtained by reacting ethyl pyridine-4-yl acetate and methacro-benzoic acid peracid benzoate in di-cyclo methan and saturated aqueous sodium bicarbonate at room temperature.
- Acetyl 2-anilino 2-methylpropanoate was obtained by reacting a phosphorus, 2-ethyl bromoisobutyrate and potassium carbonate in DMF at 90 ° C.
- Lithium amide obtained from THF solution of dicyclohexylamine and 1.6M n-butyllithium / hexane solution, toluene solution of cyclobutanecarboxylate ethyl, bis (dibenzylideneacetone) palladium, 2-bromopyridine and 10% tert- Ethyl 1-pyridine-2-yl-cyclobutanecarboxylate was obtained by reacting a mixture of butylphosphine / hexane solution at room temperature.
- Ethyl 2- (5-iodo-2-cell) -2-methylpropanoate was heated and stirred at 95 ° C with methyl difluoro (fluorosulfol) acetate and copper iodide in DMF.
- Ethyl 2-methyl-2- [5- (trifluoromethyl) -2-chel] propanoate was obtained.
- N 2-dicyclopropylacetamide was obtained by reacting cyclopropylacetic acid with cyclopropylamine, HOBt 'monohydrate and WSC' 1 hydrochloride in dichloromethane at room temperature.
- Methyl trifluoromethanesulfonate (1.61 ml) was added to N-cyclopropylcyclopropanecarboxamide (1.67 g) and heated at 60 ° C. for 30 minutes.
- Toluene 25 ml
- triethylamine (3.97 ml)
- 2-methyl 2- (2 chalc) propanohydrazide (1.64 g) were added to the resulting mixture, and the mixture was stirred at 60 ° C for 10 hours and at 100 ° C for 12 hours. did.
- the reaction solution is black mouth form (100m
- the mixture was diluted with 1), washed successively with saturated aqueous sodium hydrogen carbonate (100 ml) and saturated brine (50 ml), the organic layer was dried and concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (black mouth form: methanol).
- the resulting solid was washed with jetyl ether, and 3, 4-dicyclopropyl-5- [1-methyl-1- (2-chel) ethyl] -4H- 1, 2, 4-triazole (colorless solid) 813 mg was obtained.
- the mixture was stirred at ° C for 2 hours. After distilling off the reaction solution under reduced pressure, it was diluted with chloroform and saturated sodium bicarbonate water.
- the obtained residue was dissolved in dichloromethane (20 ml), tris (dimethylamino) sulfur (trimethylsilyl) difluoride (861 mg) was added, and the mixture was stirred at room temperature for 15 hours.
- the reaction mixture was diluted with saturated aqueous sodium hydrogen carbonate (40 ml) and extracted with black mouth form (20 mk2).
- the organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure, and the residue was purified by silica gel column chromatography. The obtained solids were washed with jetyl ether, respectively.
- Sulfur trifluoride dimethoxyethylamino complex (175 ⁇ 1) is dissolved in dichloromethane (10 ml) and cooled under ice-cooling.
- the reaction solution was diluted with saturated aqueous sodium hydrogen carbonate, and extracted with chloroform.
- Example 33 4-dicyclopropyl-5— ⁇ 1 [5— (difluoromethyl) -2-ethyl] -1-methylethyl ⁇ - 4H— 1, 2, 4-triazole (87 mg) (Example 31) was obtained from the highly polar product ⁇ 5— [1 (4,5-dicyclopropyl-4H— 1, 2, 4 triazole -3- L) -1-methylethyl] -2-chel ⁇ methanol (23 mg) (Example 32) was obtained. [0141] Example 33
- the obtained residue was dissolved in dichloromethane (3 ml), triethylamine (62 ⁇ 1) and trifluoromethanesulfonic anhydride (361) were added at -78 ° C, and the mixture was naturally warmed to room temperature. Stir for hours.
- the reaction solution was diluted in saturated aqueous sodium hydrogen carbonate and extracted with black mouth form.
- the obtained solid was diluted with ethyl acetate, tetraethyl acetate hydrogen acetate (0.2 ml) was added, and the mixture was concentrated under reduced pressure.
- reaction solution was cooled to room temperature, diluted with ethyl acetate, washed successively with water, 1M aqueous sodium hydroxide solution, aqueous citrate solution and saturated brine, and then dried over anhydrous magnesium sulfate.
- the solvent was concentrated under reduced pressure, and the resulting residue was washed with diisopropyl ether to give 1- ⁇ 5- [1- (5-chloro-2-chloro) -1-methylethyl] -4-cyclopropyl-4H. — 1, 2, 4-Triazol-3-yl ⁇ cyclopropanol (532 mg) was obtained as a white solid.
- Examples 48 to 284 shown in Tables 22 to 59 described later were produced using the corresponding raw materials.
- Tables 22 to 74 show the structures and physicochemical data of the example compounds.
- Tables 75 to 82 below show structures of other compounds of the present invention. These can be easily produced by using the production methods described above, the methods described in the examples, methods obvious to those skilled in the art, or variations thereof.
- the compound of the present invention has an excellent 11-HSD1 inhibitory action, hyperglycemia, insulin resistance, obesity, hyperlipidemia, hypertension, osteoporosis, glaucoma, and glaucoma associated with 11 ⁇ -HSD1 are recognized. It is useful as a prophylactic / therapeutic agent for diseases such as decreased intellectual function, especially diabetes and insulin resistance.
Abstract
Description





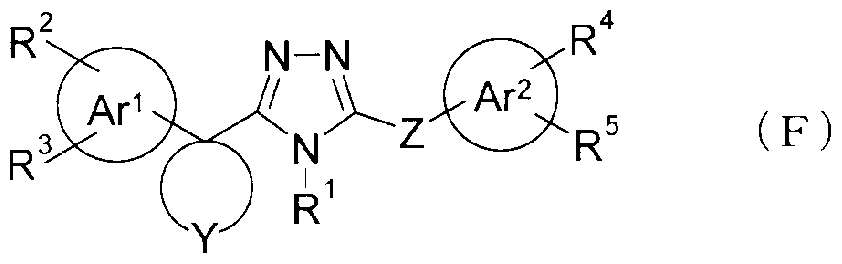
















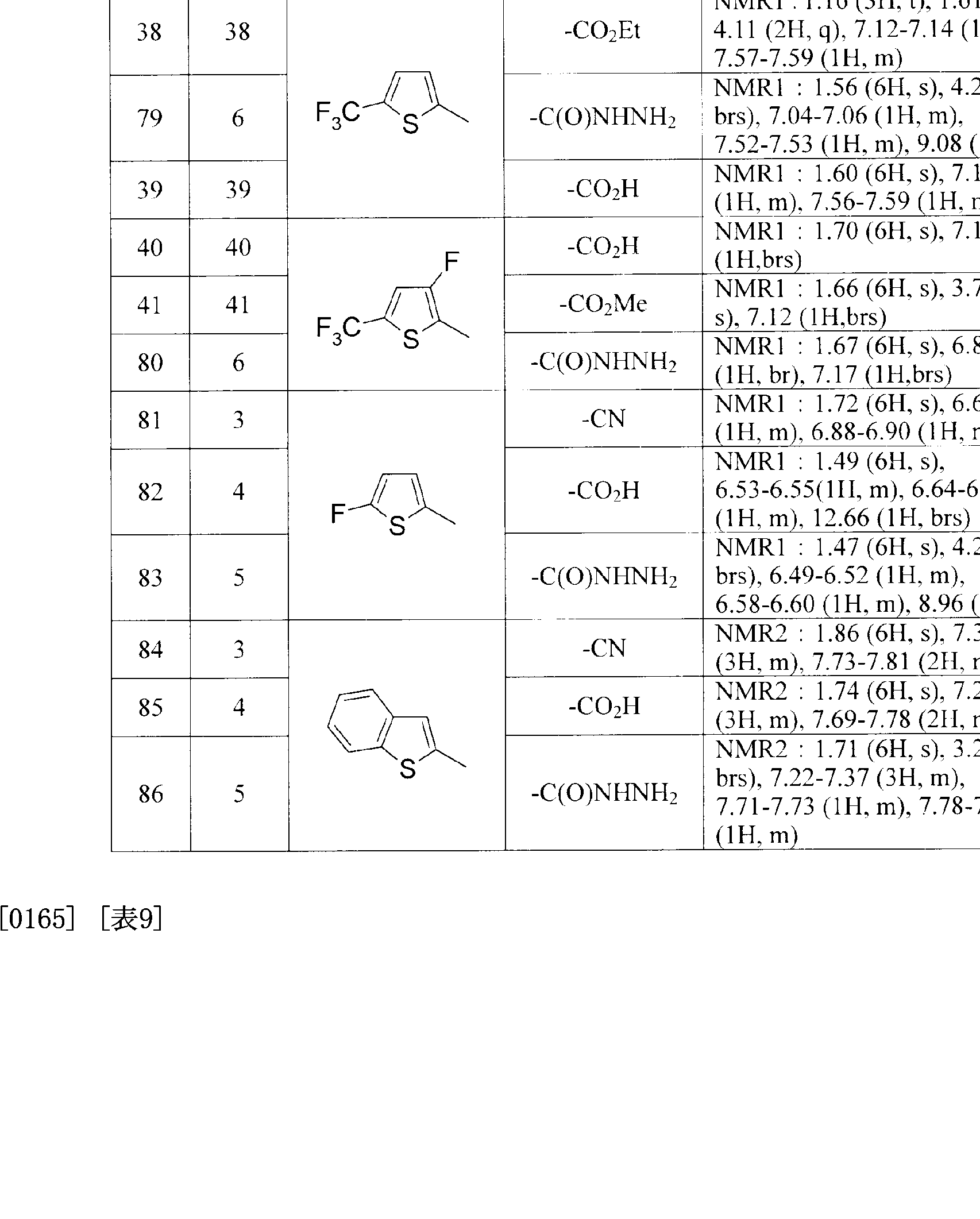









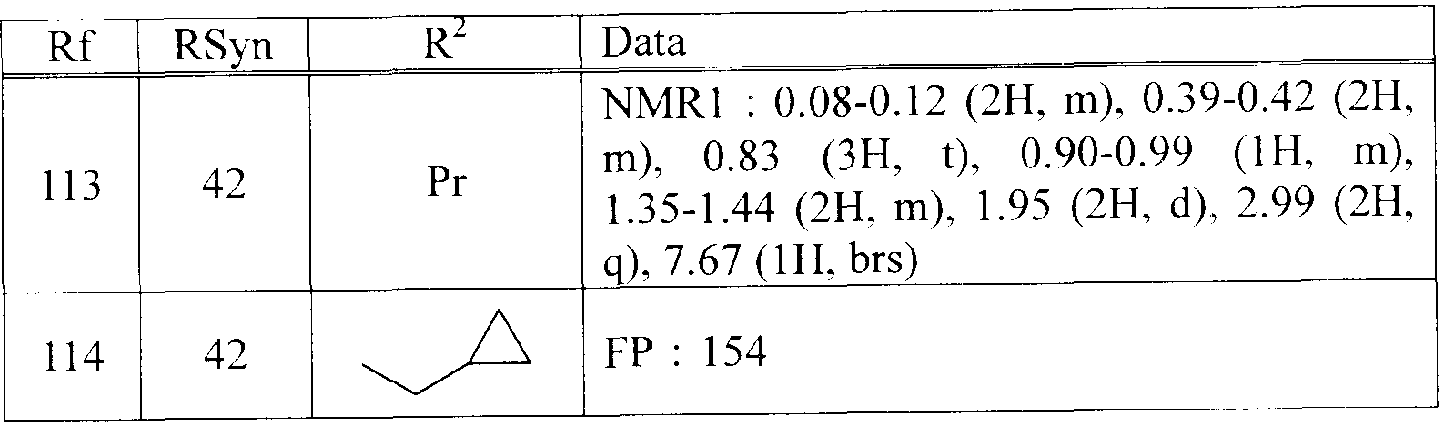














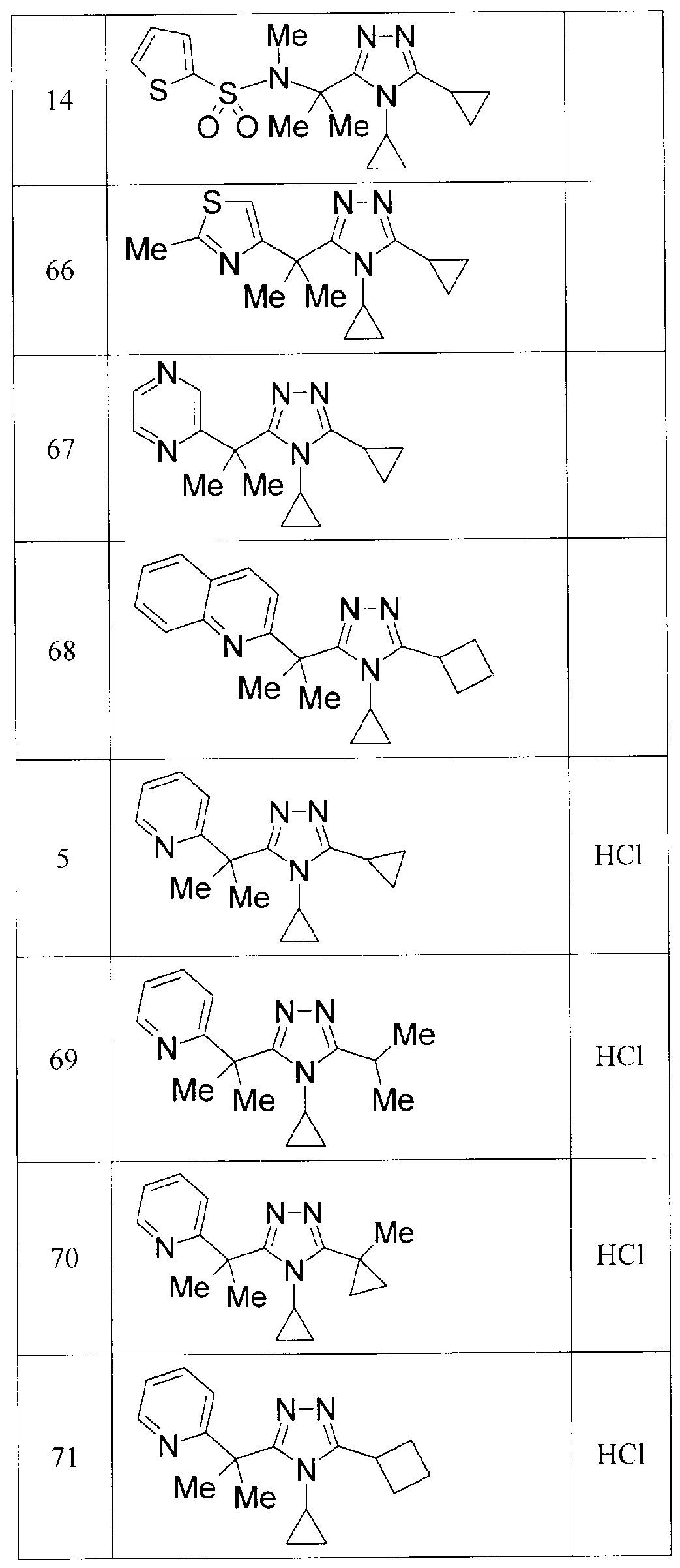














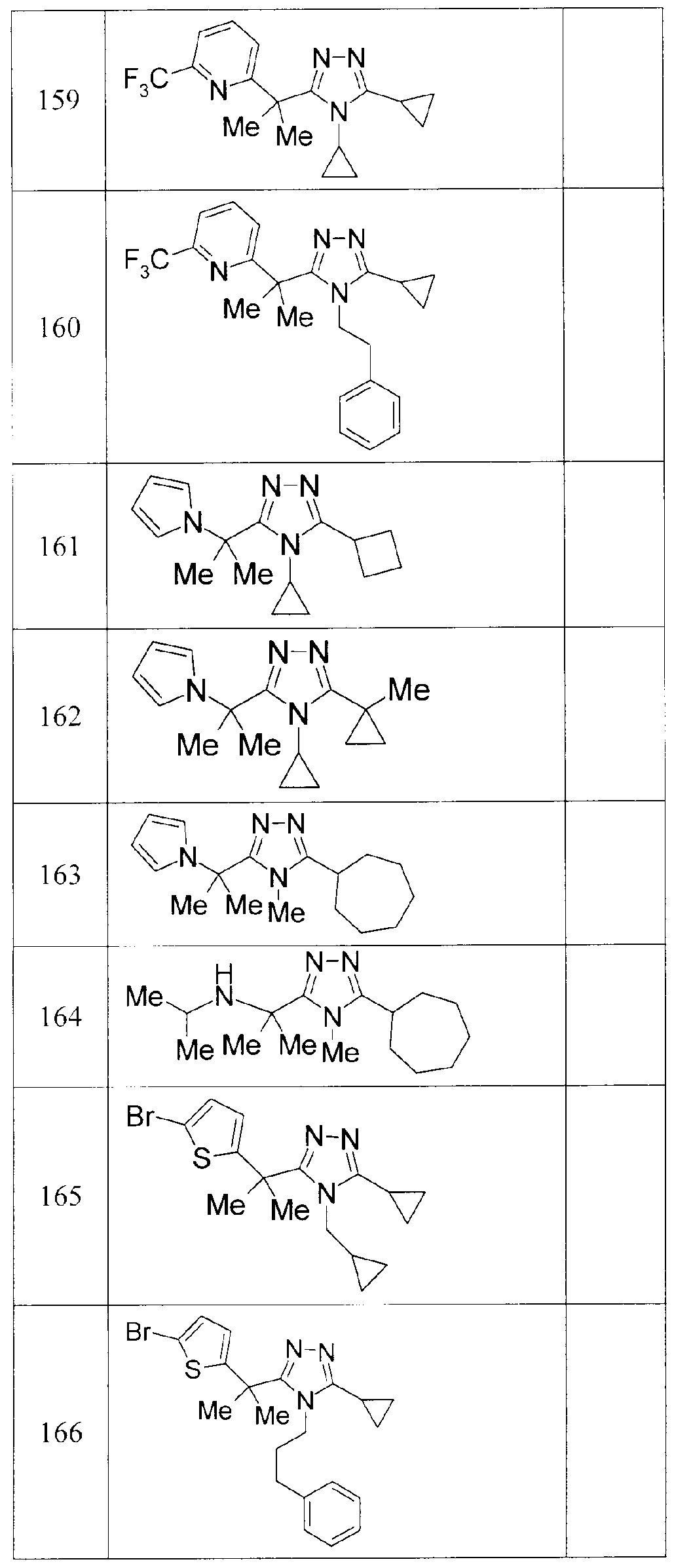








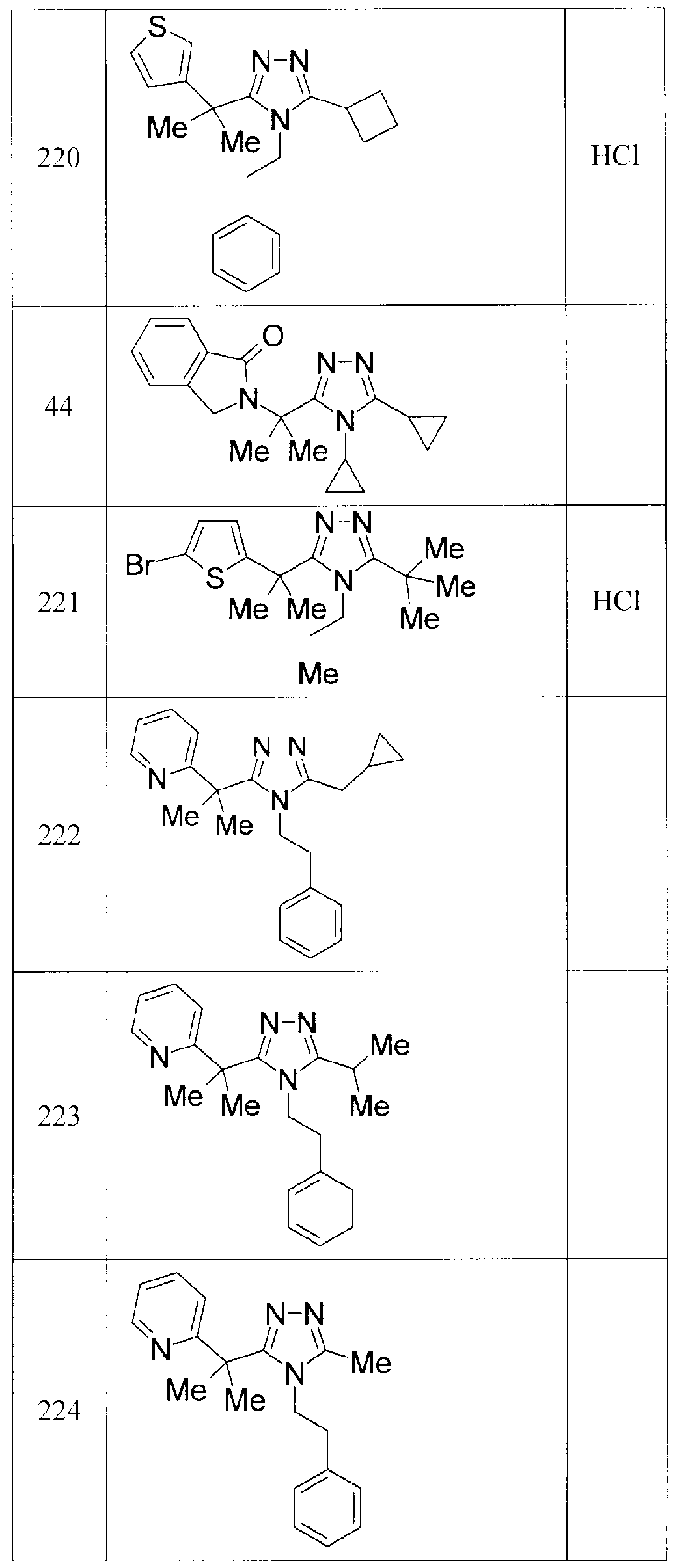





































Claims

Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/293,214 US20090082367A1 (en) | 2006-03-16 | 2007-03-14 | Triazole derivative or a salt thereof |
| EP07738519A EP1995243A4 (en) | 2006-03-16 | 2007-03-14 | DERIVATIVE OF TRIAZOLE OR SALT |
| BRPI0708782-9A BRPI0708782A2 (pt) | 2006-03-16 | 2007-03-14 | derivado de triazol ou um sal deste |
| AU2007225680A AU2007225680A1 (en) | 2006-03-16 | 2007-03-14 | Triazole derivative or salt thereof |
| MX2008011723A MX2008011723A (es) | 2006-03-16 | 2007-03-14 | Derivado de triazol o su sal. |
| JP2008505180A JPWO2007105753A1 (ja) | 2006-03-16 | 2007-03-14 | トリアゾール誘導体またはその塩 |
| CA002645712A CA2645712A1 (en) | 2006-03-16 | 2007-03-14 | Triazole derivative or salt thereof |
| IL193741A IL193741A0 (en) | 2006-03-16 | 2008-08-28 | A triazole derivative or a salt thereof |
| NO20084321A NO20084321L (no) | 2006-03-16 | 2008-10-15 | Triazolderivat eller salt derav |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006072146 | 2006-03-16 | ||
| JP2006-072146 | 2006-03-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007105753A1 true WO2007105753A1 (ja) | 2007-09-20 |
Family
ID=38509569
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/055048 WO2007105753A1 (ja) | 2006-03-16 | 2007-03-14 | トリアゾール誘導体またはその塩 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20090082367A1 (ja) |
| EP (1) | EP1995243A4 (ja) |
| JP (1) | JPWO2007105753A1 (ja) |
| KR (1) | KR20080112299A (ja) |
| CN (1) | CN101400660A (ja) |
| AU (1) | AU2007225680A1 (ja) |
| BR (1) | BRPI0708782A2 (ja) |
| CA (1) | CA2645712A1 (ja) |
| IL (1) | IL193741A0 (ja) |
| MX (1) | MX2008011723A (ja) |
| NO (1) | NO20084321L (ja) |
| RU (1) | RU2008140940A (ja) |
| WO (1) | WO2007105753A1 (ja) |
| ZA (1) | ZA200807451B (ja) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| JP2011503110A (ja) * | 2007-11-09 | 2011-01-27 | ユニバーシティ オブ テネシー リサーチ ファウンデーション | 経口投与用の抗炎症性キナ酸誘導体 |
| KR20110025688A (ko) | 2008-07-03 | 2011-03-10 | 아스텔라스세이야쿠 가부시키가이샤 | 트리아졸 유도체 또는 그의 염 |
| WO2011074560A1 (ja) * | 2009-12-15 | 2011-06-23 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有するオキサジアゾール誘導体 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9765040B2 (en) | 2010-09-07 | 2017-09-19 | Astellas Pharma Inc. | Therapeutic agent for pain |
| JP2020519644A (ja) * | 2017-05-12 | 2020-07-02 | ボード オブ トラスティーズ オブ ザ サザン イリノイ ユニバーシティ オン ビハーフ オブ サザン イリノイ ユニバーシティ エドワーズビル | 3,4,5−三置換−1,2,4−トリアゾールおよび3,4,5−三置換−3−チオ−1,2,4−トリアゾール、ならびにそれらの使用 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006030805A1 (ja) * | 2004-09-16 | 2006-03-23 | Astellas Pharma Inc. | トリアゾール誘導体またはその塩 |
| KR102087918B1 (ko) | 2015-03-24 | 2020-03-12 | 상하이 잉리 파마슈티컬 컴퍼니 리미티드 | 융합 고리 유도체, 이의 제조방법, 중간체, 약물 조성물 및 응용 |
| CN107759477A (zh) * | 2017-11-20 | 2018-03-06 | 阿里化学(常州)有限公司 | 一种对硝基苯乙胺盐酸盐的生产制备方法 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3907821A (en) | 1974-11-20 | 1975-09-23 | Upjohn Co | 2-{8 3-{8 1-(DIMETHYLAMINO)CYCLOPROPYL{8 -4H-1,2,4-triazol-4-yl{8 {0 benzophenone |
| US4577020A (en) | 1983-01-25 | 1986-03-18 | The Upjohn Company | Aminoalkyl and aminoalkenyl triazoles as anti-psychotic agents |
| WO2003065983A2 (en) | 2002-02-01 | 2003-08-14 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003104207A2 (en) | 2002-06-10 | 2003-12-18 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| US20040133011A1 (en) | 2002-12-20 | 2004-07-08 | Waddell Sherman T. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2004089367A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of substituted 1,2,4-triazoles |
| WO2004089380A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of fused 1,2,4-triazoles |
| WO2005044192A2 (en) | 2003-10-28 | 2005-05-19 | Amgen Inc. | Triazole compounds and uses related thereto |
| JP2005170939A (ja) | 2003-11-20 | 2005-06-30 | Takeda Chem Ind Ltd | 糖尿病の予防・治療剤 |
| WO2006030805A1 (ja) | 2004-09-16 | 2006-03-23 | Astellas Pharma Inc. | トリアゾール誘導体またはその塩 |
| WO2006080533A1 (ja) * | 2005-01-31 | 2006-08-03 | Mochida Pharmaceutical Co., Ltd. | 3-アミノ-1,2,4-トリアゾール誘導体 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2002361861A1 (en) * | 2001-12-21 | 2003-07-30 | Rhode Island Hospital | SELECTIVE 11Beta-HSD INHIBITORS AND METHODS FOR USE THEREOF |
| EP1798226A4 (en) * | 2004-08-04 | 2009-06-17 | Taisho Pharmaceutical Co Ltd | TRAIZOL DERIVATIVE |
-
2007
- 2007-03-14 EP EP07738519A patent/EP1995243A4/en not_active Withdrawn
- 2007-03-14 WO PCT/JP2007/055048 patent/WO2007105753A1/ja active Application Filing
- 2007-03-14 CA CA002645712A patent/CA2645712A1/en not_active Abandoned
- 2007-03-14 AU AU2007225680A patent/AU2007225680A1/en not_active Abandoned
- 2007-03-14 BR BRPI0708782-9A patent/BRPI0708782A2/pt not_active Application Discontinuation
- 2007-03-14 ZA ZA200807451A patent/ZA200807451B/xx unknown
- 2007-03-14 MX MX2008011723A patent/MX2008011723A/es not_active Application Discontinuation
- 2007-03-14 CN CNA2007800091367A patent/CN101400660A/zh active Pending
- 2007-03-14 JP JP2008505180A patent/JPWO2007105753A1/ja not_active Withdrawn
- 2007-03-14 KR KR1020087025140A patent/KR20080112299A/ko not_active Application Discontinuation
- 2007-03-14 US US12/293,214 patent/US20090082367A1/en not_active Abandoned
- 2007-03-14 RU RU2008140940/04A patent/RU2008140940A/ru not_active Application Discontinuation
-
2008
- 2008-08-28 IL IL193741A patent/IL193741A0/en unknown
- 2008-10-15 NO NO20084321A patent/NO20084321L/no not_active Application Discontinuation
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3907821A (en) | 1974-11-20 | 1975-09-23 | Upjohn Co | 2-{8 3-{8 1-(DIMETHYLAMINO)CYCLOPROPYL{8 -4H-1,2,4-triazol-4-yl{8 {0 benzophenone |
| US4577020A (en) | 1983-01-25 | 1986-03-18 | The Upjohn Company | Aminoalkyl and aminoalkenyl triazoles as anti-psychotic agents |
| WO2003065983A2 (en) | 2002-02-01 | 2003-08-14 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003104207A2 (en) | 2002-06-10 | 2003-12-18 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003104208A1 (en) | 2002-06-10 | 2003-12-18 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| US20040133011A1 (en) | 2002-12-20 | 2004-07-08 | Waddell Sherman T. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2004089367A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of substituted 1,2,4-triazoles |
| WO2004089380A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of fused 1,2,4-triazoles |
| WO2005044192A2 (en) | 2003-10-28 | 2005-05-19 | Amgen Inc. | Triazole compounds and uses related thereto |
| JP2005170939A (ja) | 2003-11-20 | 2005-06-30 | Takeda Chem Ind Ltd | 糖尿病の予防・治療剤 |
| WO2006030805A1 (ja) | 2004-09-16 | 2006-03-23 | Astellas Pharma Inc. | トリアゾール誘導体またはその塩 |
| WO2006080533A1 (ja) * | 2005-01-31 | 2006-08-03 | Mochida Pharmaceutical Co., Ltd. | 3-アミノ-1,2,4-トリアゾール誘導体 |
Non-Patent Citations (14)
| Title |
|---|
| "Bunshi Sekkei", vol. 7, 1990, HIROKAWA SHOTEN, article "Iyakuhin no Kaihatsu", pages: 163 - 198 |
| "Progress in Medicines", LIFE SCIENCE MEDICA, vol. 5, 1985, pages 2157 - 2161 |
| "Protective Groups in Organic Synthesis", 1999, JOHN WILLEY & SONS |
| COOPER M.S. ET AL., BONE, vol. 27, 2000, pages 375 - 381 |
| DAVANI B. ET AL., THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 275, 2000, pages 34841 - 34844 |
| LINDSAY R.S. ET AL., THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM, vol. 88, 2003, pages 2738 - 2744 |
| MASUZAKI H. ET AL., SCIENCE, vol. 294, 2001, pages 2166 - 2170 |
| MASUZAKI H. ET AL., THE JOURNAL OF CLINICAL INVESTIGATION, vol. 112, 2003, pages 83 - 90 |
| MORTON N.M. ET AL., THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 276, 2001, pages 41293 - 41300 |
| RASK E. ET AL., THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM, vol. 86, 2001, pages 1418 - 1421 |
| RAUZ S. ET AL., INVESTIGATIVE OPHTHALMOLOGY & VISUAL SCIENCE, vol. 42, 2001, pages 2037 - 2042 |
| SANDEEP T.C. ET AL., PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, vol. 101, 2004, pages 6734 - 6739 |
| See also references of EP1995243A4 |
| WALKER E.A. ET AL., JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 276, 2001, pages 21343 - 21350 |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| JP2011503110A (ja) * | 2007-11-09 | 2011-01-27 | ユニバーシティ オブ テネシー リサーチ ファウンデーション | 経口投与用の抗炎症性キナ酸誘導体 |
| AU2008323683B2 (en) * | 2007-11-09 | 2014-10-02 | University Of Tennessee Research Foundation | Anti-inflammatory quinic acid derivatives for oral administration |
| US8377923B2 (en) | 2008-07-03 | 2013-02-19 | Astellas Pharma Inc. | Triazole derivative or salt thereof |
| KR20110025688A (ko) | 2008-07-03 | 2011-03-10 | 아스텔라스세이야쿠 가부시키가이샤 | 트리아졸 유도체 또는 그의 염 |
| WO2011074560A1 (ja) * | 2009-12-15 | 2011-06-23 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有するオキサジアゾール誘導体 |
| JP5791150B2 (ja) * | 2009-12-15 | 2015-10-07 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有するオキサジアゾール誘導体 |
| US8754113B2 (en) | 2009-12-15 | 2014-06-17 | Shionogi & Co., Ltd. | Oxadiazole derivative having endothelial lipase inhibitory activity |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US9765040B2 (en) | 2010-09-07 | 2017-09-19 | Astellas Pharma Inc. | Therapeutic agent for pain |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| JP2020519644A (ja) * | 2017-05-12 | 2020-07-02 | ボード オブ トラスティーズ オブ ザ サザン イリノイ ユニバーシティ オン ビハーフ オブ サザン イリノイ ユニバーシティ エドワーズビル | 3,4,5−三置換−1,2,4−トリアゾールおよび3,4,5−三置換−3−チオ−1,2,4−トリアゾール、ならびにそれらの使用 |
| JP7343170B2 (ja) | 2017-05-12 | 2023-09-12 | ボード オブ トラスティーズ オブ ザ サザン イリノイ ユニバーシティ オン ビハーフ オブ サザン イリノイ ユニバーシティ エドワーズビル | 3,4,5-三置換-1,2,4-トリアゾールおよび3,4,5-三置換-3-チオ-1,2,4-トリアゾール、ならびにそれらの使用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2007105753A1 (ja) | 2009-07-30 |
| RU2008140940A (ru) | 2010-04-27 |
| NO20084321L (no) | 2008-11-20 |
| US20090082367A1 (en) | 2009-03-26 |
| ZA200807451B (en) | 2010-01-27 |
| MX2008011723A (es) | 2008-09-26 |
| CN101400660A (zh) | 2009-04-01 |
| IL193741A0 (en) | 2009-05-04 |
| BRPI0708782A2 (pt) | 2011-06-14 |
| AU2007225680A1 (en) | 2007-09-20 |
| KR20080112299A (ko) | 2008-12-24 |
| EP1995243A1 (en) | 2008-11-26 |
| EP1995243A4 (en) | 2009-07-22 |
| CA2645712A1 (en) | 2007-09-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007105753A1 (ja) | トリアゾール誘導体またはその塩 | |
| JP4882748B2 (ja) | トリアゾール誘導体またはその塩 | |
| JP6391076B2 (ja) | アンドロゲン受容体モジュレーターおよびその使用 | |
| JP5487214B2 (ja) | キナーゼ阻害剤として有用なカルバゾールカルボキシアミド化合物 | |
| TWI353834B (en) | Aryl- and heteroarylpiperidinecarboxylate derivati | |
| AU2018360577A1 (en) | Bridged bicyclic compounds as farnesoid X receptor modulators | |
| WO2014075575A1 (zh) | 吡咯磺酰类衍生物、其制备方法及其在医药上的应用 | |
| WO2008059854A1 (fr) | Dérivés de pipéridine ou sels de ceux-ci | |
| JP5494482B2 (ja) | トリアゾール誘導体またはその塩 | |
| AU2012327076B2 (en) | Bicyclic heterocyclic compound | |
| JP5049134B2 (ja) | ドーパミンd3受容体の調節に応答する障害の治療に好適なトリアゾール化合物 | |
| EP1988075A1 (en) | Pyrrole derivative or salt thereof | |
| TW200914439A (en) | Novel pyrazolone-derivatives | |
| WO2019062657A1 (zh) | 氮杂环类衍生物、其制备方法及其医药用途 | |
| EA010154B1 (ru) | Пиридиновые производные алкилоксиндолов в качестве агентов, активных в отношении рецептора 5-нт7 | |
| WO2015162452A1 (en) | Substituted pyrazole compounds as cb1 receptor antagonists and uses thereof | |
| AU2020221371A1 (en) | Substituted amide compounds useful as farnesoid x receptor modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07738519 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2008505180 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 193741 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008501950 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007225680 Country of ref document: AU Ref document number: 570948 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2645712 Country of ref document: CA Ref document number: 2007738519 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/011723 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780009136.7 Country of ref document: CN Ref document number: 12293214 Country of ref document: US Ref document number: 4914/CHENP/2008 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007225680 Country of ref document: AU Date of ref document: 20070314 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087025140 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2008140940 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0708782 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080911 |