WO2013037390A1 - 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors - Google Patents
6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors Download PDFInfo
- Publication number
- WO2013037390A1 WO2013037390A1 PCT/EP2011/065719 EP2011065719W WO2013037390A1 WO 2013037390 A1 WO2013037390 A1 WO 2013037390A1 EP 2011065719 W EP2011065719 W EP 2011065719W WO 2013037390 A1 WO2013037390 A1 WO 2013037390A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- hydroxy
- pyrazolo
- pyridin
- piperazin
- Prior art date
Links
- 0 CC(C)C(*1CC[C@](C[C@](CC(OCCCCO*(O)=O)=O)OC)OC)=C(C(*(C)(C)c2ccccc2)=O)C(c2ccccc2)=C1c(cc1)ccc1N Chemical compound CC(C)C(*1CC[C@](C[C@](CC(OCCCCO*(O)=O)=O)OC)OC)=C(C(*(C)(C)c2ccccc2)=O)C(c2ccccc2)=C1c(cc1)ccc1N 0.000 description 2
- HJLDTPJKEPKCLW-YCRREMRBSA-N CN(Cc1ccc(/C=C/c2n[nH]c3c2c(C(N2CCNCC2)=O)cc(-c(cc2)ccc2O)n3)cc11)C1=O Chemical compound CN(Cc1ccc(/C=C/c2n[nH]c3c2c(C(N2CCNCC2)=O)cc(-c(cc2)ccc2O)n3)cc11)C1=O HJLDTPJKEPKCLW-YCRREMRBSA-N 0.000 description 2
- BHPGHONWCCZVCE-LPSZOUNZSA-N CC(C)(C)OC(N(CC1)C[C@H](CO)N1C(c1c(c(/C=C/c2ccccc2)n[n]2C3OCCCC3)c2nc(-c(cc2)ccc2O)c1)=O)=O Chemical compound CC(C)(C)OC(N(CC1)C[C@H](CO)N1C(c1c(c(/C=C/c2ccccc2)n[n]2C3OCCCC3)c2nc(-c(cc2)ccc2O)c1)=O)=O BHPGHONWCCZVCE-LPSZOUNZSA-N 0.000 description 1
- CMLUGNQVANVZHY-POURPWNDSA-N CC(C)(CN(c(cc1)c([C@@H](c2cccc(OC)c2OC)O[C@@H]2CC(N3CCC(CC(O)=O)CC3)=O)cc1Cl)C2=O)COC(C)=O Chemical compound CC(C)(CN(c(cc1)c([C@@H](c2cccc(OC)c2OC)O[C@@H]2CC(N3CCC(CC(O)=O)CC3)=O)cc1Cl)C2=O)COC(C)=O CMLUGNQVANVZHY-POURPWNDSA-N 0.000 description 1
- QNAZTOHXCZPOSA-UHFFFAOYSA-N CC(C)c(cc(Cc(c(C)c1)c(C)cc1OCC(O)=O)cc1)c1O Chemical compound CC(C)c(cc(Cc(c(C)c1)c(C)cc1OCC(O)=O)cc1)c1O QNAZTOHXCZPOSA-UHFFFAOYSA-N 0.000 description 1
- ZYJMDFSUXXSPLI-UHFFFAOYSA-O CCCCC[NH+]=C Chemical compound CCCCC[NH+]=C ZYJMDFSUXXSPLI-UHFFFAOYSA-O 0.000 description 1
- KECQKNGBRNTZCJ-UHFFFAOYSA-N CCOC(c1c(cn[nH]2)c2nc(O)c1)=O Chemical compound CCOC(c1c(cn[nH]2)c2nc(O)c1)=O KECQKNGBRNTZCJ-UHFFFAOYSA-N 0.000 description 1
- DPUVAXLWVRZASN-UHFFFAOYSA-N CN(Cc(c1c2)ccc2Br)C1=O Chemical compound CN(Cc(c1c2)ccc2Br)C1=O DPUVAXLWVRZASN-UHFFFAOYSA-N 0.000 description 1
- IQQBRKLVEALROM-UHFFFAOYSA-N CS(N(C(C1)CN1C(c(cc1)ccc1Cl)c(cc1)ccc1Cl)c1cc(F)cc(F)c1)(=O)=O Chemical compound CS(N(C(C1)CN1C(c(cc1)ccc1Cl)c(cc1)ccc1Cl)c1cc(F)cc(F)c1)(=O)=O IQQBRKLVEALROM-UHFFFAOYSA-N 0.000 description 1
- CTESJDQKVOEUOY-UHFFFAOYSA-N N#CC(C(Nc1c2c(-c(cc3)ccc3-c3ccccc3O)c[s]1)=O)=C2O Chemical compound N#CC(C(Nc1c2c(-c(cc3)ccc3-c3ccccc3O)c[s]1)=O)=C2O CTESJDQKVOEUOY-UHFFFAOYSA-N 0.000 description 1
- SGOYQRYHNPHJHX-TZHYSIJRSA-N OC(C1(CCN(CC2)CC(C3)[C@H]2Cc2c3c3cc(C(F)(F)F)ccc3[nH]2)CCCCC1)=O Chemical compound OC(C1(CCN(CC2)CC(C3)[C@H]2Cc2c3c3cc(C(F)(F)F)ccc3[nH]2)CCCCC1)=O SGOYQRYHNPHJHX-TZHYSIJRSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N OS(c1ccccc1)(=O)=O Chemical compound OS(c1ccccc1)(=O)=O SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- QCYOJXCKWQJIAU-IZZDOVSWSA-N Oc(cc1)ccc1-c1nc([nH]nc2/C=C/c3cc(N(CCC4)C4=O)ccc3)c2c(C(N2CCNCC2)=O)c1 Chemical compound Oc(cc1)ccc1-c1nc([nH]nc2/C=C/c3cc(N(CCC4)C4=O)ccc3)c2c(C(N2CCNCC2)=O)c1 QCYOJXCKWQJIAU-IZZDOVSWSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- PKC-beta II Protein kinase C-beta II expression in patients with colorectal cancer.
- angiogenesis Nakamura S, Chikaraishi Y, Tsuruma K,
- R 4 is piperidin-4-yl, piperidin-3-yl or pyrrolidin-3-yl, which are unsubstituted or
- R 5 is H
- R 17 is H, or CH 3 ;
- R 1 is a) a residue of the formula la wherein
- R b is H, F, CI, (Ci-C 3 )-alkyl or 0-(Ci-C 3 )-alkyl, wherein (Ci-C 3 )-alkyl is unsubstituted or substituted by one to five F; or R 6 is H, F, CI, CH 3 , CF 3 , or 0-CH 3 ; or R 6 is H;
- R 9 is H, F, CH 3 , O-CH 3 ; or R 9 is H;
- R is H
- R 17 is H
- R 2 is H, F, CI, or CH 3 ; or
- R 12 is H, or CH 3 ;
- R 5 is H
- R 9 is H, F, CH 3 , or O-CH 3 ;
- R 3 is H
- R and R 12 together with the C-atoms carrying them are 1 '-spiro- cyclopentyl
- the terms “including” and “comprising” are used in their open, non- limiting sense.
- the terms “(C-i-Ce)” or “(Cs-Ce)” and so forth refer to moieties having 1 to 8 or 5 to 8 carbon atoms, respectively.
- Within terms like "(Co-Ce)- alkyl” or “(C 0 -C 6 )-alkylen” "C 0 -alkyl” or “(Co)-alkylen” refer to a bond, or in case of an unsubstituted "(Co)-alkyl” it refers to a hydrogen.
- Substituted alkyl groups, alkenyl groups and alkynyl groups can be substituted in any positions, provided that the respective compound is sufficiently stable and is suitable for the desired purpose such as use as a drug substance.
- the prerequisite that a specific group and a compound of the formula I are sufficiently stable and suitable for the desired purpose such as use as a drug substance applies in general with respect to the definitions of all groups in the compounds of the formula I.
- bicyclic or tricyclic fused ring cycloalkyls are derived from, but not limited to, the following ring systems: bicyclo[3.1.0]hexane, bicyclo[4.1 .0]heptane, bicycle- [5.1 .0]octane, bicyclo[3.2.0]heptane, bicyclo[4.2.0]octane, octahydro-pentalene, octa- hydro-indene, decahydro-azulene, decahydro-naphthalene, decahydro-benzo- cycloheptene, dodecahydro-heptalene, 1 ,2,3,3a,4,6a-hexahydro-pentalene, 1 ,2,3,4- tetrahydro-pentalene, 2,3,3a,4,5,7a-hexahydro-1 H-indene, 2,3,3a,4,7,7a-hexahydro- 1
- Exemplary bicyclic or tricyclic spiro ring cycloalkyls are derived from, but not limited to, the following ring systems: spiro[2.4]heptane, spiro[2.5]octane, spiro[2.6]nonane, spiro[3.3]heptane, spiro[3.4]octane, spiro[3.5]nonane, spiro[3.6]decane, spiro[4.4]no- nane, spiro[4.5]decane, spiro[4.6]undecane, spiro[5.5]undecane, spiro[5.6]dodecane, spiro[6.6]tridecane, dispiro[2.2.4.2]dodecane, dispiro[2.2.3.2]undecane, dispiro- [2.1 .4.2]undecane and spiro[5.5]undec-2-ene
- Exemplary non-fused or bridged bicyclic or tricyclic ring cycloalkyls are derived from, but not limited to, the following ring systems: bicyclo[2.2.1 ]heptane, bicyclo[2.2.2]oc- tane, bicyclo[3.2.1 ]octane, bicyclo[3.2.2]nonane and adamantane.
- the number of ring heteroatoms which can be present in a heterocyclic group is 1 , 2, 3 or 4, in another embodiment 1 , 2 or 3, in another embodiment 1 or 2, in another embodiment 2, in another embodiment 1 , wherein the ring heteroatoms can be identical or different.
- the heterocycloalkyl group can be attached by any ring carbon atom or saturated ring nitrogen atom, with the exeption of spiro- or bridgehead atoms.
- monocyclic heterocycloalkyls are derived from azetidine, pyrrolidine, piperidine, piperazine, morpholine or 1 ,4-diazepane:
- bicyclic fused ring heterocycloalkyls are derived from 3-aza- bicyclo[3.1 .0]hexane, octahydro-pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4-b]pyrrole, octahydro-thieno[3,4-b]pyrazine, octahydro-pyrrolo[1 ,2-a]pyrazine, decahydro- quinoxaline, octahydro-pyrido[1 ,2-a]pyrazine or decahydro-[1 ,6]naphthyridine:
- heterocycloalkyls in which a ring is fused to one ring of a bicyclic spiro system, are derived from, but not limited to, the following ring systems: octahydro- spiro[cyclopentane-1 ,2'(3'H)-quinoxalin], 1 ',4'-dihydro-spiro[cyclopentane-1 ,2'(3'H)- quinoxalin], 1 ',2',4,5-tetrahydro-spiro[furan-3(2H),3'-[3H]indol], 1 ,3-dihydro-spiro[in- dene-2,2'-piperazine], 2,3-dihydro-spiro[1 H-indene-1 ,4'-piperidin] and 1 ,2-dihydro-5- spiro[3H-indole-3,4'-piperidin]:
- Exemplary non-fused or bridged bicyclic or tricyclic ring heterocycloalkyls are derived from, but not limited to, the following ring systems: 2-aza-bicyclo[2.2.1 ]heptane, 1 -aza- bicyclo[2.2.2]octane, 8-aza-bicyclo[3.2.1 ]octane, 3-aza-bicyclo[3.2.1 ]octane, 9-aza- bicyclo[3.3.1 ]nonane, 2,5-diaza-bicyclo[2.2.1 ]heptane, 2,5-diaza-bicyclo[2.2.2]octane, 3,8-diaza-bicyclo[3.2.1 ]octaneand 3,7-diaza-bicyclo[3.3.1 ]nonane:
- heteroaryl refers to a radical derived from an aromatic mono- or bicyclic ring system, in which 1 , 2, 3, 4 or 5 carbon atoms are replaced by heteroatoms.
- the ring heteroatoms are generally chosen from N, 0 and S, wherein N includes ring nitrogen atoms which carry a hydrogen atom or a substituent as well as ring nitrogen atoms which do not carry a hydrogen atom or a substituent.
- Ring heteroatoms can be located in any position, provided that the heterocyclic system is stable and suitable as a subgroup for the desired purpose of the compound of the formula I such as use as a drug substance.
- Heteroaryl radicals are derived from 5- membered or 6-membered monocyclic rings or 8-membered, 9-membered or 10- membered bicyclic rings, in another embodiment 5-membered or 6-membered monocyclic rings or 9-membered or 10-membered bicyclic rings, in another
- Suitable ring nitrogen atoms in aromatic heterocycles in the compounds of the formula I can in general also be present as N-oxide or as quaternary salt, for example as N-(Ci-C 4 )-alkyl salt such as N-methyl salt, wherein in one embodiment of the invention the counter anion in such quaternary salt is a physiologically acceptable anion which is derived from an acid that forms a physiologically acceptable salt.
- the substituent can be located in the 2-position, the 3-position or the 4-position.
- the substituents can be located in 2, 3-position, 2, 4-position, 2,5-position, 2,6-position, 3, 4-position or 3,5- position.
- the substituents can be located in 2,3,4- position, 2,3,5-position, 2,3,6-position, 2,4,5-position, 2,4,6-position or 3,4,5-position.
- Ring heteroatoms can be located in any positions, provided that the heterocyclic system is known in the art and is stable and suitable as a subgroup for the desired purpose of the compound of the formula I such as use as a drug substance.
- two ring oxygen atoms cannot be present in adjacent ring positions of any heterocycle
- two ring heteroatoms chosen from oxygen and sulfur cannot be present in adjacent ring positions of any heterocycle.
- Substituents on heterocyclic groups can be located in any positions. For example, in a pyridin-2-yl group substituents can be located in the 3-position and/or 4-position and/or
- the compounds of the present invention can be widely combined with other pharmacologically active compounds, e.g., with all antihypertensives and nephroprotectives, mentioned in the Rote Liste 201 1 , antidiabetics mentioned in the Rote Liste 201 1 , chapter 12; all weight-reducing agents/appetite suppressants mentioned in the Rote Liste 201 1 , chapter 1 ; all diuretics mentioned in the Rote Liste 201 1 , chapter 36; all lipid-lowering agents mentioned in the Rote Liste 201 1 , chapter 58. They can be combined with the inventive compound of the formula I, especially for a synergistic improvement in action.
- the active ingredient combination can be administered either by separate administration of the active ingredients to the patient or in the form of combination products in which a plurality of active ingredients are present in one pharmaceutical preparation. When the active ingredients are
- WO2009133099 or insulins which can be administered transdermally; additionally included are also those insulin derivatives which are bonded to albumin by a
- WO2009125424, WO2009129696, WO2009149148 peptides, for example obinepitide (TM-30338), orally active GLP-1 analogs (e.g. NN9924 from Novo Nordisk), amylin receptor agonists, as described, for example, in WO2007104789, WO20090341 19, analogs of the human GLP-1 , as described in WO2007120899, WO2008022015, WO2008056726, chimeric pegylated peptides containing both GLP-1 and glucagon residues, as described, for example, in WO2008101017, WO2009155257,
- Antidiabetics additionally include poly- or monoclonal antibodies directed, for example, against interleukin 1 beta (IL-1 ⁇ ), for example XOMA-052.
- Antidiabetics additionally include peptides which can bind to the human pro-islet peptide (HIP) receptor, as described, for example, in WO2009049222.
- HIP human pro-islet peptide
- Antidiabetics additionally include encapsulated insulin-producing porcine cells, for example DiabeCell(R).
- the orally active hypoglycemic ingredients preferably include sulfonylureas, biguanidines,
- glucosidase inhibitors inhibitors of glycogen phosphorylase
- potassium channel openers for example pinacidil, cromakalim, diazoxide, diazoxide choline salt, or those as described in R. D. Carr et al., Diabetes 52, 2003, 2513.2518, in J. B. Hansen et al., Current Medicinal Chemistry 11 , 2004, 1595-1615, in T. M.
- DPP-IV dipeptidyl peptidase-IV
- liver enzymes involved in stimulating gluconeogenesis and/or
- estrogen receptor gamma agonists (ERR ⁇ agonists)
- the compound of the formula I is administered with a combination of mitiglinide with an alpha-glucosidase inhibitor.
- the compound of the formula I is administered in combination with a PPAR alpha agonist or mixed PPAR alpha/PPAR delta agonist, for example GW9578, GW-590735, K-1 1 1 , LY-674, KRP-101 , DRF-10945, LY-518674, CP-900691 , BMS-687453, BMS-71 1939, or those as described in WO2001040207, WO2002096894, WO2005097076, WO2007056771 , WO2007087448,
- the compound of the formula I is administered in combination with an inhibitor of the interaction of liver glycogen phosphorylase with the protein PPP1 R3 (GL subunit of glycogen-associated protein phosphatase 1 (PP1 )), as described, for example, in WO2009030715.
- PPP1 R3 GL subunit of glycogen-associated protein phosphatase 1 (PP1 )
- the compound of the formula I is administered in combination with inhibitors of fructose 1 ,6-bisphosphatase (FBPase), for example MB-07729, CS-917 (MB-06322) or MB-07803, or those as described in WO2006023515, WO2006104030, WO2007014619, WO2007137962, WO2008019309, WO2008037628,
- FBPase fructose 1 ,6-bisphosphatase
- the compound of the formula I is administered in combination with a combination of a DPP-IV inhibitor with metformin hydrochloride, as described, for example, in WO2009121945.
- the compound of the formula I is administered in combination with a combination of a DPP-IV inhibitor with a GPR-1 19 agonist, as described, for example, in WO2009123992.
- the compound of the formula I is administered in combination with a combination of a DPP-IV inhibitor with miglitol, as described, for example, in
- the compound of the formula I is administered in combination with a substance which enhances insulin secretion, for example KCP-265
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound of the formula I is administered in combination with a stimulator of glucose transport, as described, for example, in WO2008136392, WO2008136393.
- the compound of the formula I is administered in combination with inhibitors of 1 1 -beta-hydroxysteroid dehydrogenase 1 (1 ⁇ ⁇ -HSDI ), for example BVT- 2733, JNJ-25918646, INCB-13739, INCB-20817, DIO-92 ((-)-ketoconazole) or those as described, for example, in WO200190090-94, WO200343999, WO20041 12782, WO200344000, WO200344009, WO20041 12779, WO20041 13310, WO2004103980, WO20041 12784, WO2003065983, WO2003104207, WO2003104208,
- the compound of the formula I is administered in combination with inhibitors of protein tyrosine phosphatase-1 B (PTP-1 B), as described, for example, in WO2001 19830-31 , WO2001 17516, WO2004506446, WO2005012295,
- PTP-1 B protein tyrosine phosphatase-1 B
- the compound of the formula I is administered in combination with stimulators of tyrosine kinase B (Trk-B), as described, for example, in
- the compound of the formula I is administered in combination with beta 3 agonists (also called beta-3 adrenoceptor agonists), as described, for example, in Physiol. Behav. 2004 Sep 15;82(2-3):489-96, J Clin Invest (1998) 101 : 2387-93, Curr. Pharma. Des.2001 Sep;7(14): 1433-49., Bioorganic & Medicinal Chemistry Letters volume 14, number 13, July 5, 2004, pages 3525-3529 (BMS- 201620).
- beta 3 agonists also called beta-3 adrenoceptor agonists
- the compound of the formula I is administered in combination with an agonist of GPR109A (HM74A receptor agonists; NAR agonists (nicotinic acid receptor agonists)), for example nicotinic acid or extended release niacin in conjunction with MK-0524A (laropiprant) or MK-0524, or those compounds as described in WO2004041274, WO2006045565, WO2006045564, WO2006069242, WO2006085108, WO20060851 12, WO20060851 13, WO2006124490,
- GPR109A HM74A receptor agonists
- NAR agonists nicotinic acid receptor agonists
- WO2007120575 WO2007134986, WO2007150025, WO2007150026,
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound of the formula I is administered in combination with nicotinic acid or another nicotinic acid receptor agonist and a prostaglandin DP receptor antagonist, for example those as described in
- the compound of the formula I is administered in combination with a solid combination of niacin with meloxicam, as described, for example, in WO2009149056.
- the compound of the formula I is administered in combination with modulators of GPR40, as described, for example, in WO2007013689,
- the compound of the formula I is administered in combination with modulators of GPR1 19 (G-protein-coupled glucose-dependent insulinotropic receptor), for example PSN-1 19-1 , PSN-821 , PSN-1 19-2, MBX-2982 or those as described, for example, in WO2004065380, WO2005061489 (PSN-632408), WO2006083491 , WO2007003960-62 and WO2007003964, WO2007035355, WO20071 16229,
- GPR1 19 G-protein-coupled glucose-dependent insulinotropic receptor
- the compound of the formula I is administered in combination with modulators of GPR120, as described, for example, in EP1688138,
- the compound of the formula I is administered in combination with antagonists of GPR105, as described, for example, in WO2009000087,
- the compound of the formula I is administered in combination with a phospholipase A2 inhibitor, for example darapladib or A-002, or those as described in WO2008048866, WO20080488867, US2009062369.
- a phospholipase A2 inhibitor for example darapladib or A-002, or those as described in WO2008048866, WO20080488867, US2009062369.
- the compound of the formula I is administered in combination with myricitrin, a lipase inhibitor (WO20071 19827).
- the compound of the formula I is administered in combination with an inhibitor of glycogen synthase kinase-3 beta (GSK-3 beta), as described, for example, in US2005222220, WO2005085230, WO20051 1 1018, WO2003078403, WO2004022544, WO2003106410, WO2005058908, US2005038023,
- GSK-3 beta glycogen synthase kinase-3 beta
- WO20081 13469 WO2008121063, WO2008121064, EP-1992620, EP-1992621 , EP1992624, EP-1992625, WO2008130312, WO2009007029, EP2020232,
- the compound of the formula I is administered in combination with an inhibitor of phosphoinositide kinase-3 (PI3K), for example those as described in WO2008027584, WO2008070150, WO2008125833, WO2008125835,
- PI3K phosphoinositide kinase-3
- the compound of the formula I is administered in combination with an inhibitor of serum/glucocorticoid-regulated kinase (SGK), as described, for example, in WO2006072354, WO2007093264, WO2008009335, WO2008086854,
- SGK serum/glucocorticoid-regulated kinase
- the compound of the formula I is administered in combination with a modulator of the glucocorticoid receptor, as described, for example, in
- the compound of the formula I is administered in combination with a modulator of the mineralocorticoid receptor (MR), for example drospirenone, or those as described in WO2008104306, WO20081 19918.
- MR mineralocorticoid receptor
- the compound of the formula I is administered in combination with an inhibitor of protein kinase C beta (PKC beta), for example ruboxistaurin, or those as described in WO2008096260, WO2008125945.
- PDC beta protein kinase C beta
- the compound of the formula I is administered in combination with an inhibitor of protein kinase D, for example doxazosin (WO2008088006).
- an inhibitor of protein kinase D for example doxazosin (WO2008088006).
- the compound of the formula I is administered in combination with an activator/modulator of the AMP-activated protein kinase (AMPK), as described, for example, in WO2007062568, WO2008006432, WO2008016278, WO2008016730, WO2008020607, WO2008083124, WO2008136642, WO2009019445,
- AMPK AMP-activated protein kinase
- the compound of the formula I is administered in combination with an inhibitor of ceramide kinase, as described, for example, in WO20071 12914, WO2007149865.
- the compound of the formula I is administered in combination with an inhibitor of MAPK-interacting kinase 1 or 2 (MNK1 or 2), as described, for example, in WO2007104053, WO20071 15822, WO2008008547, WO2008075741 .
- MNK1 or 2 MAPK-interacting kinase 1 or 2
- the compound of the formula I is administered in combination with inhibitors of "l-kappaB kinase" (IKK inhibitors), as described, for example, in
- the compound of the formula I is administered in combination with inhibitors of NF-kappaB (NFKB) activation, for example salsalate.
- NFKB NF-kappaB
- the compound of the formula I is administered in combination with inhibitors of ASK-1 (apoptosis signal-regulating kinase 1 ), as described, for example, in WO2008016131 , WO2009123986.
- ASK-1 apoptosis signal-regulating kinase 1
- the compound of the formula I is administered in combination with an HMG-CoA reductase inhibitor such as simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin, pitavastatin, L-659699, BMS-644950, NCX-6560, or those as described in US2007249583, WO2008083551 , WO2009054682.
- an HMG-CoA reductase inhibitor such as simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin, pitavastatin, L-659699, BMS-644950, NCX-6560, or those as described in US2007249583, WO2008083551 , WO2009054682.
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound of the formula I is administered in combination with a ligand of the liver X receptor (LXR), as described, for example, in WO2007092965, WO2008041003, WO2008049047, WO2008065754,
- LXR liver X receptor
- the compound of the formula I is administered in combination with a fibrate, for example fenofibrate, clofibrate, bezafibrate, or those as described in WO2008093655.
- the compound of the formula I is administered in combination with fibrates, for example the choline salt of fenofibrate (SLV-348;
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- the compound of the formula I is administered in combination with a solid combination of metformin with an MTP inhibitor, as described in WO2009090210.
- the compound of the formula I is administered in combination with a cholesterol reabsorption inhibitor, for example ezetimibe, tiqueside, pamaqueside, FM-VP4 (sitostanol/campesterol ascorbyl phosphate; Forbes Medi- Tech, WO2005042692, WO2005005453), MD-0727 (Microbia Inc., WO2005021497, WO2005021495) or with compounds as described in WO2002066464, WO2005000353 (Kotobuki Pharmaceutical Co. Ltd.) or WO2005044256 or
- a cholesterol reabsorption inhibitor for example ezetimibe, tiqueside, pamaqueside, FM-VP4 (sitostanol/campesterol ascorbyl phosphate; Forbes Medi- Tech, WO2005042692, WO2005005453), MD-0727 (Microbia Inc., WO2005021497, WO2005021495) or with compounds as described in WO2002066464, WO
- WO2005062824 Merck & Co.
- WO2005061451 and WO2005061452 Alignment AB
- WO2006017257 Phenomix
- WO2005033100 Lipideon Biotechnology AG
- the compound of the formula I is administered in combination with an NPC1 L1 antagonist, for example those as described in
- the compound of the formula I is administered in combination with a solid combination of ezetimibe with atorvastatin. In one embodiment of the invention, the compound of the formula I is administered in combination with a solid combination of ezetimibe with fenofibrate.
- the further active ingredient is a
- the further active ingredient is a diphenylazetidinone derivative, as described, for example, in US 6,992,067 or US 7,205,290, combined with a statin, for example simvastatin, fluvastatin, pravastatin, lovastatin, cerivastatin, atorvastatin, pitavastatin or rosuvastatin.
- a statin for example simvastatin, fluvastatin, pravastatin, lovastatin, cerivastatin, atorvastatin, pitavastatin or rosuvastatin.
- the compound of the formula I is administered in combination with a solid combination of lapaquistat, a squalene synthase inhibitor, with atorvastatin.
- the compound of the formula I is administered in combination with a solid combination of lapaquistat, a squalene synthase inhibitor, with atorvastatin.
- the compound of the formula I is administered in combination with a CETP inhibitor, for example torcetrapib, anacetrapib or JTT-705 (dalcetrapib), or those as described in WO2006002342, WO2006010422,
- a CETP inhibitor for example torcetrapib, anacetrapib or JTT-705 (dalcetrapib), or those as described in WO2006002342, WO2006010422,
- the compound of the formula I is administered in combination with agonists of GPBAR1 (G-protein-coupled bile acid receptor 1 ; TGR5), for example INT- 777 or those as described, for example, in US20060199795, WO20071 10237,
- GPBAR1 G-protein-coupled bile acid receptor 1 ; TGR5
- the compound of the formula I is administered in combination with modulators of histone deacetylase, for example ursodeoxycholic acid, as described in WO200901 1420.
- modulators of histone deacetylase for example ursodeoxycholic acid, as described in WO200901 1420.
- the compound of the formula I is administered in combination with inhibitors/modulators of the TRPA1 channel (TRP cation channel A1 ), as described, for example, in US2009176883, WO2009089083, WO2009144548. In one embodiment, the compound of the formula I is administered in combination with inhibitors/modulators of the TRPV3 channel (TRP cation channel V3), as described, for example, in WO2009084034, WO2009130560.
- the compound of the formula I is administered in combination with colesevelam hydrochloride and metformin or a sulfonylurea or insulin.
- the compound of the formula I is administered in combination with tocotrienol and insulin or an insulin derivative. In one embodiment of the invention, the compound of the formula I is administered in combination with a chewing gum comprising phytosterols (ReductolTM).
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- a cholesterol absorption inhibitor for example ezetimibe
- MTP inhibitor the triglyceride transfer protein
- the compound of the formula I is administered in combination with an antagonist of the somatostatin 5 receptor (SST5 receptor), for example those as described in WO2006094682.
- SST5 receptor somatostatin 5 receptor
- the compound of the formula I is administered in combination with an ACAT inhibitor, for example avasimibe, SMP-797 or KY-382, or those as described in WO2008087029, WO2008087030, WO2008095189,
- an ACAT inhibitor for example avasimibe, SMP-797 or KY-382, or those as described in WO2008087029, WO2008087030, WO2008095189,
- the compound of the formula I is administered in combination with an inhibitor of liver carnitine palmitoyltransferase-1 (L-CPT1 ), as described, for example, in WO2007063012, WO2007096251 (ST-3473), WO2008015081 , US2008103182, WO2008074692, WO2008145596,
- the compound of the formula I is administered in combination with an inhibitor of carnitin O-palmitoyltransferase II (CPT2), as described, for example, in US2009270500, US2009270505, WO2009132978,
- CPT2 carnitin O-palmitoyltransferase II
- the compound of the formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- SPT serine palmitoyltransferase
- the compound of the formula I is administered in combination with a squalene synthetase inhibitor, for example BMS-188494, TAK-475 (lapaquistat acetate), or as described in WO2005077907, JP2007022943,
- the compound of the formula I is administered in combination with ISIS-301012 (mipomersen), an antisense oligonucleotide which is capable of regulating the apolipoprotein B gene.
- the compound of the formula I is administered in combination with apolipoprotein (ApoB) SNALP, a therapeutic product which comprises an siRNA (directed against the ApoB gene).
- the compound of the formula I is administered in combination with a stimulator of the ApoA-1 gene, as described, for example, in WO2008092231 .
- the compound of the formula I is administered in combination with a modulator of the synthesis of apolipoprotein —l 11 , for example ISIS- APOCIIIRx.
- the compound of the formula I is administered in combination with an LDL receptor inducer (see US 6,342,512), for example HMR1 171 , HMR1586, or those as described in WO2005097738, WO2008020607.
- an LDL receptor inducer see US 6,342,512
- the compound of the formula I is administered in combination with modulators of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase 3 (GPAT3, described in WO2007100789) or with modulators of microsomal acyl- CoA:glycerol-3-phosphate acyltransferase 4 (GPAT4, described in WO2007100833) or with modulators of mitochondrial glycerol-3-phosphate O-acyltransferase, described in WO2010005922.
- modulators of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase 3 GPAT3, described in WO2007100789
- modulators of microsomal acyl- CoA:glycerol-3-phosphate acyltransferase 4 GPAT4, described in WO2007100833
- modulators of mitochondrial glycerol-3-phosphate O-acyltransferase described in WO2010005922.
Abstract
The present invention relates to pyrazolo[3,4-b]pyridine compounds of the formula I, in which R1, R2, R3, R4 and R5 are defined as indicated below. The compounds of the formula I are kinase inhibitors, and are useful for the treatment of diseases associated with diabetes and diabetic complications, such as, diabetic nephropathy, diabetic neuropathy and diabetic retinopathy, for example. The invention furthermore relates to the use of compounds of the formula I, in particular as active ingredients in pharmaceuticals, and pharmaceutical compositions comprising them.
Description
6-(4-Hydroxy-phenyl)-3-styryl-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors
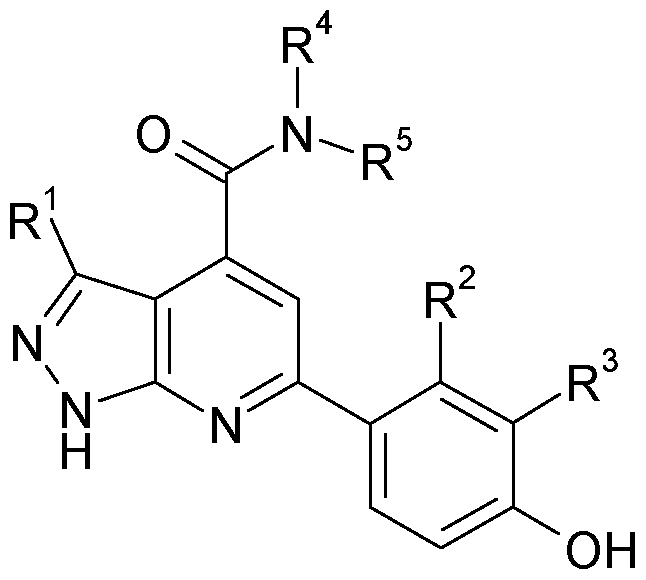
The present invention relates to pyrazolo[3,4-b]pyridine compounds of the formula I,
PKC is involved in the regulation of smooth muscle contractility. Upon stimulation PKC phosphorylates the regulatory myosin light chain (MLC20) and inhibits the myosin associated phosphatase (MYPT). Phosphorylation of MLC20 and inhibition of MYPT leads to an increased activity of the acto-myosin complex and to vasoconstriction in different vascular beds, e.g. resistance-sized, retinal, cerebral, coronary, conduit arteries and veins (Merkel LA, Rivera LM, Colussi DJ, Perrone MH. Protein kinase C
and vascular smooth muscle contractility: effects of inhibitors and down-regulation. J Pharmacol Exp Ther 1991 ; 257(1 ): 134-140; Sehic E, Malik KU. Influence of protein kinase C activators on vascular tone and adrenergic neuroeffector events in the isolated rat kidney. J Pharmacol Exp Ther 1989; 251 (2):634-639.).
Overexpressed or overactivated PKC detrimentally affects heart function. Upon activation PKC affects the intracellular calcium homeostasis which results in reduced myocardial contractility and relaxation of the myocardium. Overall this effect leads to myocardial contractile insufficiency (Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, Thai K, Krum H, Gilbert RE. Inhibition of protein kinase C-beta by ruboxi- staurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail 2009; 2(2): 129-137). Moreover, activated PKC mediates organ damage during end-organ injuries, e.g. during ischemia in heart (Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, Thai K, Krum H, Gilbert RE. Inhibition of protein kinase C-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail 2009; 2(2): 129-137; Hambleton M, Hahn H, Pleger ST, Kuhn MC, Klevitsky R, Carr AN, Kimball TF, Hewett TE, Dorn GW, Koch WJ, Molkentin JD. Pharmacological- and gene therapy-based inhibition of protein kinase Calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation 2006; 1 14(6): 574-582) or kidney (Tuttle KR. Protein kinase C-beta inhibition for diabetic kidney disease. Diabetes Res Clin Pract 2008; 82 SuppI 1 :S70-S74; Anderson PW, McGill JB, Tuttle KR. Protein kinase C beta inhibition: the promise for treatment of diabetic nephropathy. Curr Opin Nephrol Hypertens 2007; 16(5):397-402). PKC and especially the PKC-beta II isoform is overexpressed or overactivated in diabetes in various different types of tissue and exerts its deleterious effect to the cells, tissues and end-organs, e.g. kidney (Tuttle KR. Protein kinase C-beta inhibition for diabetic kidney disease. Diabetes Res Clin Pract 2008; 82 SuppI 1 :S70-S74; Anderson PW, McGill JB, Tuttle KR. Protein kinase C beta inhibition: the promise for treatment of diabetic nephropathy. Curr Opin Nephrol Hypertens 2007; 16(5):397-402; Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care 2005; 28(1 1 ):2686-2690; Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, Wilkinson-Berka JL, Gilbert RE. Protein kinase C beta inhibition attenuates
the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes 2003; 52(2):512-518), heart (Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, Thai K, Krum H, Gilbert RE. Inhibition of protein kinase C- beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail 2009; 2(2): 129-137; Guo M, Wu MH, Korompai F, Yuan SY. Upregulation of PKC genes and isozymes in
cardiovascular tissues during early stages of experimental diabetes. Physiol Genomics 2003; 12(2): 139-146), or in tissues like the retina (Aiello LP, Clermont A, Arora V, Davis MD, Sheetz MJ, Bursell SE. Inhibition of PKC beta by oral administration of ruboxistaurin is well tolerated and ameliorates diabetes-induced retinal hemodynamic abnormalities in patients. Invest Ophthalmol Vis Sci 2006; 47(1 ):86-92; Aiello LP. The potential role of PKC beta in diabetic retinopathy and macular edema. Surv
Ophthalmol 2002; 47 Suppl 2:S263-S269; Kimura M, Ishizawa M, Miura A, Itaya S, Kanoh Y, Yasuda K, Uno Y, Morita H, Ishizuka T. Platelet protein kinase C isoform content in type 2 diabetes complicated with retinopathy and nephropathy. Platelets 2001 ; 12(3): 138-143) or neuronal tissue (Krishnan ST, Rayman G. New treatments for diabetic neuropathy: symptomatic treatments. Curr Diab Rep 2003; 3(6):459-467; Kim H, Sasaki T, Maeda K, Koya D, Kashiwagi A, Yasuda H. Protein kinase Cbeta selective inhibitor LY333531 attenuates diabetic hyperalgesia through ameliorating cGMP level of dorsal root ganglion neurons. Diabetes 2003; 52(8):2102-2109; Cotter MA, Jack AM, Cameron NE. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci (Lond) 2002; 103(3):31 1 -321 ; Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, Naruse K, Kasuya Y, Mizubayashi R, Miwa K, Yasuda Y, Kamiya H, lenaga K, Sakakibara F, Koh N, Hotta N. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes 1999; 48(10):2090-2095) or in platelets (Assert R, Scherk G, Bumbure A, Pirags V, Schatz H, Pfeiffer AF. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia 2001 ; 44(2): 188-195; Bynagari-Settipalli YS, Chari R, Kilpatrick L, Kunapuli SP. Protein kinase C - possible therapeutic target to treat cardiovascular diseases. Cardiovasc Hematol Disord Drug Targets 2010;
10(4):292-308; Kimura M, Ishizawa M, Miura A, Itaya S, Kanoh Y, Yasuda K, Uno Y,
Morita H, Ishizuka T. Platelet protein kinase C isoform content in type 2 diabetes complicated with retinopathy and nephropathy. Platelets 2001 ; 12(3): 138-143;
Oskarsson HJ, Hofmeyer TG, Coppey L, Yorek MA. Effect of protein kinase C and phospholipase A2 inhibitors on the impaired ability of human diabetic platelets to cause vasodilation. Br J Pharmacol 1999; 127(4):903-908) or induces endothelial dysfunction (Chiasson VL, Quinn MA, Young KJ, Mitchell BM. Protein kinase Cbetall-mediated phosphorylation of endothelial nitric oxide synthase threonine 495 mediates the endothelial dysfunction induced by FK506 (tacrolimus). J Pharmacol Exp Ther 201 1 ; 337(3):718-723; Xu Y, Wang S, Feng L, Zhu Q, Xiang P, He B. Blockade of PKC-beta protects HUVEC from advanced glycation end products induced inflammation. Int
Immunopharmacol 2010; 10(12): 1552-1559; Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 2010;
106(8): 1319-1331 ; Nacci C, Tarquinio M, Montagnani M. Molecular and clinical aspects of endothelial dysfunction in diabetes. Intern Emerg Med 2009; 4(2): 107-1 16). Furthermore, it has been suggested that PKC signalling is involved in tumour formation (Gonelli A, Mischiati C, Guerrini R, Voltan R, Salvadori S, Zauli G. Perspectives of protein kinase C (PKC) inhibitors as anti-cancer agents. Mini Rev Med Chem 2009; 9(4):498-509; AN AS, AN S, El-Rayes BF, Philip PA, Sarkar FH. Exploitation of protein kinase C: a useful target for cancer therapy. Cancer Treat Rev 2009; 35(1 ): 1 -8), e.g. in hematological tumours (Mischiati C, Melloni E, Corallini F, Milani D, Bergamini C, Vaccarezza M. Potential role of PKC inhibitors in the treatment of hematological malignancies. Curr Pharm Des 2008; 14(21 ):2075-2084; Cheson BD, Zwiebel JA, Dancey J, Murgo A. Novel therapeutic agents for the treatment of myelodysplastic syndromes. Semin Oncol 2000; 27(5):560-577; Deng X, Kornblau SM, Ruvolo PP, May WS, Jr. Regulation of Bcl2 phosphorylation and potential significance for leukemic cell chemoresistance. J Natl Cancer Inst Monogr 2001 ;(28):30-37), in glioma formation (Baltuch GH, Dooley NP, Villemure JG, Yong VW. Protein kinase C and growth regulation of malignant gliomas. Can J Neurol Sci 1995; 22(4):264-271 ; Blobe GC, Obeid LM, Hannun YA. Regulation of protein kinase C and role in cancer biology. Cancer Metastasis Rev 1994; 13(3-4):41 1 -431 ; Bredel M, Pollack IF. The role of protein kinase C (PKC) in the evolution and proliferation of malignant gliomas, and the application of PKC inhibition as a novel approach to anti-glioma therapy. Acta
Neurochir (Wien ) 1997; 139(1 1 ): 1000-1013), in gastric and intestinal cancer (Atten MJ, Godoy-Romero E, Attar BM, Milson T, Zopel M, Holian O. Resveratrol regulates cellular PKC alpha and delta to inhibit growth and induce apoptosis in gastric cancer cells. Invest New Drugs 2005; 23(2): 1 1 1 -1 19; Fahrmann M. Targeting protein kinase C (PKC) in physiology and cancer of the gastric cell system. Curr Med Chem 2008;
15(12): 1 175-1 191 ), in skin cancer (Birt DF, Yaktine A, Duysen E. Glucocorticoid mediation of dietary energy restriction inhibition of mouse skin carcinogenesis. J Nutr 1999; 129 (2S Suppl):571 S-574S; Birt DF, Przybyszewski J, Wang W, Stewart J, Liu Y. Identification of molecular targets for dietary energy restriction prevention of skin carcinogenesis: an idea cultivated by Edward Bresnick. J Cell Biochem 2004;
91 (2):258-264), lung cancer (Herbst RS, Oh Y, Wagle A, Lahn M. Enzastaurin, a protein kinase Cbeta- selective inhibitor, and its potential application as an anticancer agent in lung cancer. Clin Cancer Res 2007; 13(15 Pt 2):s4641 -s4646; Herbst RS. Targeted therapy in non-small-cell lung cancer. Oncology (Williston Park) 2002; 16(9 Suppl 9): 19-24) and others. PKC is an important signal transducer of events in autoimmune responses, e.g. in T-cell (Birchall AM, Bishop J, Bradshaw D, Cline A, Coffey J, Elliott LH, Gibson VM, Greenham A, Hallam TJ, Harris W, . Ro 32-0432, a selective and orally active inhibitor of protein kinase C prevents T-cell activation. J Pharmacol Exp Ther 1994; 268(2):922-929; Isakov N, Altman A. Protein kinase C(theta) in T cell activation. Annu Rev Immunol 2002; 20:761 -794) or B-cell (Shinohara H, Kurosaki T. Comprehending the complex connection between PKCbeta, TAK1 , and IKK in BCR signaling. Immunol Rev 2009; 232(1 ):300-318; Venkataraman C, Chen XC, Na S, Lee L, Neote K, Tan SL. Selective role of PKCbeta enzymatic function in regulating cell survival mediated by B cell antigen receptor cross-linking. Immunol Lett 2006; 105(1 ):83-89) linked autoimmune signalling, and in inflammatory processes.
The above mentioned effects of the PKC-mediated signalling leads to induction or promotion of the progression of asthma (Boschelli DH. Small molecule inhibitors of PKCTheta as potential antiinflammatory therapeutics. Curr Top Med Chem 2009; 9(7):640-654), chronic obstructive pulmonary disease (Mercer BA, D'Armiento JM. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int J Chron Obstruct Pulmon Dis 2006; 1 (2): 137-150; Adcock IM, Chung KF, Caramori G, Ito K. Kinase inhibitors and airway inflammation. Eur J Pharmacol 2006; 533(1 -3): 1 18-132;
Dempsey EC, Cool CD, Littler CM. Lung disease and PKCs. Pharmacol Res 2007; 55(6):545-559; Ishii M, Kurachi Y. Muscarinic acetylcholine receptors. Curr Pharm Des 2006; 12(28):3573-3581 ; Medina-Tato DA, Watson ML, Ward SG. Leukocyte navigation mechanisms as targets in airway diseases. Drug Discov Today 2006; 1 1 (19- 20):866-879), pulmonary hypertension (Agbani EO, Coats P, Mills A, Wadsworth RM. Peroxynitrite stimulates pulmonary artery endothelial and smooth muscle cell proliferation: involvement of ERK and PKC. Pulm Pharmacol Ther 201 1 ; 24(1 ): 100- 109; Littler CM, Wehling CA, Wick MJ, Fagan KA, Cool CD, Messing RO, Dempsey EC. Divergent contractile and structural responses of the murine PKC-epsilon null pulmonary circulation to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 2005; 289(6): L1083-L1093), retinopathy, like retinal ischemia and neovascularization (Galvez Ml. Protein kinase C inhibitors in the treatment of diabetic retinopathy. Review. Curr Pharm Biotechnol 201 1 ; 12(3):386-391 ; Schwartz SG, Flynn HW, Jr., Aiello LP.
Ruboxistaurin mesilate hydrate for diabetic retinopathy. Drugs Today (Bare) 2009; 45(4):269-274), nephropathy, including hypertension-induced (Kelly DJ, Edgley AJ, Zhang Y, Thai K, Tan SM, Cox AJ, Advani A, Connelly KA, Whiteside CI, Gilbert RE. Protein kinase C-beta inhibition attenuates the progression of nephropathy in non- diabetic kidney disease. Nephrol Dial Transplant 2009; 24(6): 1782-1790; Hayashi K, Wakino S, Ozawa Y, Homma K, Kanda T, Okubo K, Takamatsu I, Tatematsu S, Kumagai H, Saruta T. Role of protein kinase C in Ca channel blocker-induced renal arteriolar dilation in spontaneously hypertensive rats-studies in the isolated perfused hydronephrotic kidney. Keio J Med 2005; 54(2): 102-108; Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, Wilkinson-Berka JL, Gilbert RE. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes 2003; 52(2):512-518), non- hypertension-induced, and diabetic nephropathies (Danis RP, Sheetz MJ.
Ruboxistaurin: PKC-beta inhibition for complications of diabetes. Expert Opin Pharma- cother 2009; 10(17):2913-2925; Tuttle KR. Protein kinase C-beta inhibition for diabetic kidney disease. Diabetes Res Clin Pract 2008; 82 Suppl 1 :S70-S74), renal failure (Danis RP, Sheetz MJ. Ruboxistaurin: PKC-beta inhibition for complications of diabetes. Expert Opin Pharmacother 2009; 10(17):2913-2925; Yamagishi S, Fukami K, Ueda S, Okuda S. Molecular mechanisms of diabetic nephropathy and its therapeutic
intervention. Curr Drug Targets 2007; 8(8):952-959) and myocardial infarction (Byna- gari-Settipalli YS, Chari R, Kilpatrick L, Kunapuli SP. Protein kinase C - possible therapeutic target to treat cardiovascular diseases. Cardiovasc Hematol Disord Drug Targets 2010; 10(4):292-308; Rohilla A, Singh G, Singh M, Bala kP. Possible involvement of PKC-delta in the abrogated cardioprotective potential of ischemic preconditioning in hyperhomocysteinemic rat hearts. Biomed Pharmacother 2010; 64(3): 195-202; Liu Q, Chen X, Macdonnell SM, Kranias EG, Lorenz JN, Leitges M, Houser SR, Molkentin JD. Protein kinase C{alpha}, but not PKC{beta} or PKC
{gamma}, regulates contractility and heart failure susceptibility: implications for ruboxi- staurin as a novel therapeutic approach. Circ Res 2009; 105(2): 194-200; Yonezawa T, Kurata R, Kimura M, Inoko H. PKC delta and epsilon in drug targeting and therapeutics. Recent Pat DNA Gene Seq 2009; 3(2):96-101 ) cardiac hypertrophy and failure (Ferreira JC, Brum PC, Mochly-Rosen D. betallPKC and epsilonPKC isozymes as potential pharmacological targets in cardiac hypertrophy and heart failure. J Mol Cell Cardiol 2010; Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D. Protein kinase C in heart failure: a therapeutic target? Cardiovasc Res 2009; 82(2):229-239), coronary heart disease, artherosclerosis, restenosis (Ding RQ, Tsao J, Chai H, Mochly-Rosen D, Zhou W. Therapeutic potential for protein kinase C inhibitor in vascular restenosis. J Cardiovasc Pharmacol Ther 201 1 ; 16(2): 160-167; Schleicher E, Friess U. Oxidative stress, AGE, and atherosclerosis. Kidney Int Suppl 2007;(106):S17-S26), diabetes, diabetic complications, glucose utilization and metabolic syndrome (Bynagari-Settipalli YS, Chari R, Kilpatrick L, Kunapuli SP. Protein kinase C - possible therapeutic target to treat cardiovascular diseases. Cardiovasc Hematol Disord Drug Targets 2010;
10(4):292-308; Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 2010; 106(8): 1319-1331 ; Danis RP, Sheetz MJ. Ruboxistaurin: PKC-beta inhibition for complications of diabetes. Expert Opin Pharmacother 2009; 10(17):2913-2925), immune diseases (Baier G, Wagner J. PKC inhibitors: potential in T cell-dependent immune diseases. Curr Opin Cell Biol 2009; 21 (2):262-267; Mecklenbrauker I, Saijo K, Zheng NY, Leitges M, Tarakhovsky A.
Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature 2002; 416(6883):860-865; Wilkinson SE, Hallam TJ. Protein kinase C: is its pivotal role in cellular activation over-stated? Trends Pharmacol Sci 1994; 15(2):53-57; Costello R,
Mawas C, Olive D. Differential immuno-suppressive effects of metabolic inhibitors on T-lymphocyte activation. Eur Cytokine Netw 1993; 4(2): 139-146), like psoriasis
(Sommerer C, Zeier M. AEB071 -a promising immunosuppressive agent. Clin
Transplant 2009; 23 Suppl 21 : 15-18; Rasmussen HH, Celis JE. Evidence for an altered protein kinase C (PKC) signaling pathway in psoriasis. J Invest Dermatol 1993; 101 (4):560-566; Fisher GJ, Tavakkol A, Leach K, Burns D, Basta P, Loomis C, Griffiths CE, Cooper KD, Reynolds NJ, Elder JT, . Differential expression of protein kinase C isoenzymes in normal and psoriatic adult human skin: reduced expression of protein kinase C-beta II in psoriasis. J Invest Dermatol 1993; 101 (4):553-559), rheumatoid athritis (Healy AM, Izmailova E, Fitzgerald M, Walker R, Hattersley M, Silva M, Siebert E, Terkelsen J, Picarella D, Pickard MD, LeClair B, Chandra S, Jaffee B. PKC-theta- deficient mice are protected from Th1 -dependent antigen-induced arthritis. J Immunol 2006; 177(3): 1886-1893; Ji JD, Tassiulas I, Park-Min KH, Aydin A, Mecklenbrauker I, Tarakhovsky A, Pricop L, Salmon JE, Ivashkiv LB. Inhibition of interleukin 10 signaling after Fc receptor ligation and during rheumatoid arthritis. J Exp Med 2003;
197(1 1 ): 1573-1583; Kehlen A, Thiele K, Riemann D, Langner J. Expression, modulation and signalling of IL-17 receptor in fibroblast-like synoviocytes of patients with rheumatoid arthritis. Clin Exp Immunol 2002; 127(3):539-546), or other
autoimmune disorders (Zanin-Zhorov A, Dustin ML, Blazar BR. PKC-theta function at the immunological synapse: prospects for therapeutic targeting. Trends Immunol 201 1 ; 32(8):358-363), central nervous system disorders (Liang J, Takeuchi H, Jin S, Noda M, Li H, Doi Y, Kawanokuchi J, Sonobe Y, Mizuno T, Suzumura A. Glutamate induces neurotrophic factor production from microglia via protein kinase C pathway. Brain Res 2010; 1322:8-23; Bastianetto S, Zheng WH, Quirion R. Neuroprotective abilities of resveratrol and other red wine constituents against nitric oxide-related toxicity in cultured hippocampal neurons. Br J Pharmacol 2000; 131 (4):71 1 -720), cerebral ischemia or cerebral vasospasm (Bu X, Zhang N, Yang X, Liu Y, Du J, Liang J, Xu Q, Li J. Proteomic analysis of cPKCbetall-interacting proteins involved in HPC-induced neuroprotection against cerebral ischemia of mice. J Neurochem 201 1 ; 1 17(2):346- 356), neuropathies and pain, e.g. neuropathic pain (Nakajima A, Tsuboi Y, Suzuki I, Honda K, Shinoda M, Kondo M, Matsuura S, Shibuta K, Yasuda M, Shimizu N, Iwata K. PKCgamma in Vc and C1/C2 is involved in trigeminal neuropathic pain. J Dent Res
201 1 ; 90(6):777-781 ; Malmberg AB, Chen C, Tonegawa S, Basbaum Al. Preserved acute pain and reduced neuropathic pain in mice lacking PKCgamma. Science 1997; 278(5336):279-283), cancer development and progression, neoplasia where inhibition of protein kinase C has been shown to inhibit tumor cell growth and metastasis (Kim J, Thorne SH, Sun L, Huang B, Mochly-Rosen D. Sustained inhibition of PKCalpha reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene 201 1 ; 30(3):323-333; Spindler KL, Lindebjerg J, Lahn M, Kjaer-Frifeldt S, Jakobsen A. Protein kinase C-beta II (PKC-beta II) expression in patients with colorectal cancer. Int J Colorectal Dis 2009; 24(6):641 -645; Guo K, Li Y, Kang X, Sun L, Cui J, Gao D, Liu Y. Role of PKCbeta in hepatocellular carcinoma cells migration and invasion in vitro: a potential therapeutic target. Clin Exp Metastasis 2009; 26(3): 189-195), angiogenesis (Nakamura S, Chikaraishi Y, Tsuruma K,
Shimazawa M, Hara H. Ruboxistaurin, a PKCbeta inhibitor, inhibits retinal
neovascularization via suppression of phosphorylation of ERK1/2 and Akt. Exp Eye Res 2010; 90(1 ): 137-145; AN AS, AN S, El-Rayes BF, Philip PA, Sarkar FH.
Exploitation of protein kinase C: a useful target for cancer therapy. Cancer Treat Rev 2009; 35(1 ): 1 -8; Tekle C, Giovannetti E, Sigmond J, Graff JR, Smid K, Peters GJ.
Molecular pathways involved in the synergistic interaction of the PKC beta inhibitor enzastaurin with the antifolate pemetrexed in non-small cell lung cancer cells. Br J Cancer 2008; 99(5):750-759; Mischiati C, Melloni E, Corallini F, Milani D, Bergamini C, Vaccarezza M. Potential role of PKC inhibitors in the treatment of hematological malignancies. Curr Pharm Des 2008; 14(21 ):2075-2084), platelet disorders leading to thrombosis (Gilio K, Harper MT, Cosemans JM, Konopatskaya O, Munnix IC, Prinzen L, Leitges M, Liu Q, Molkentin JD, Heemskerk JW, Poole AW. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J Biol Chem 2010; 285(30):23410-23419; Chari R, Getz T, Nagy B, Jr., Bhavaraju K, Mao Y, Bynagari YS, Murugappan S, Nakayama K, Kunapuli SP. Protein kinase C[delta] differentially regulates platelet functional responses. Arterioscler Thromb Vase Biol 2009; 29(5):699-705; Nagy B, Jr., Bhavaraju K, Getz T, Bynagari YS, Kim S, Kunapuli SP. Impaired activation of platelets lacking protein kinase C-theta isoform. Blood 2009; 1 13(1 1 ):2557-2567; Harper MT, Poole AW. Isoform-specific functions of protein kinase C: the platelet paradigm. Biochem Soc Trans 2007; 35(Pt 5): 1005-1008; Strehl A,
Munnix IC, Kuijpers MJ, van der Meijden PE, Cosemans JM, Feijge MA, Nieswandt B, Heemskerk JW. Dual role of platelet protein kinase C in thrombus formation:
stimulation of pro-aggregatory and suppression of procoagulant activity in platelets. J Biol Chem 2007; 282(10):7046-7055; London FS. The protein kinase C inhibitor RO318220 potentiates thrombin-stimulated platelet-supported prothrombinase activity. Blood 2003; 102(7):2472-2481 ; Wheeler-Jones CP, Patel Y, Kakkar W, Krishnamurthi S. Translocation of protein kinase C (PKC) in stimulated platelets: a role for
aggregation in PKC degradation. Br J Pharmacol 1989; 98 Suppl:845P), and leukocyte aggregation (Hu H, Zhang W, Li N. Glycoprotein llb/llla inhibition attenuates platelet- activating factor-induced platelet activation by reducing protein kinase C activity. J
Thromb Haemost 2003; 1 (8): 1805-1812; Kotovuori A, Pessa-Morikawa T, Kotovuori P, Nortamo P, Gahmberg CG. ICAM-2 and a peptide from its binding domain are efficient activators of leukocyte adhesion and integrin affinity. J Immunol 1999; 162(1 1 ):6613- 6620; Lorenz HM, Lagoo AS, Hardy KJ. The cell and molecular basis of leukocyte common antigen (CD45)-triggered, lymphocyte function-associated antigen-1 - /intercellular adhesion molecule-1 -dependent, leukocyte adhesion. Blood 1994;
83(7): 1862-1870).
Until now, mainly staurosporine derivatives have been described as PKC inhibitors in the prior art, for example Ruboxistaurin (e.g. EP 657458), Enzastaurin (e.g. WO
9517182), Midostaurin (e.g. EP 2961 10) or Sotrastaurin (e.g. WO 2002038561 ). Only very few PKCB inhibitors, which not derived from staurosporine have been described, such as 3-amido-pyrrolo[3,4-c]pyrazole-5(1 H, 4H,6H)carbaldehydes in WO
2008125945.
However, there continues to be a need for further effective low molecular weight PKCB inhibitors, in particular in view of safety and selectivity. The present invention satisfies this need by providing the pyrazolo[3,4-b]pyridine compounds of the formula I.
Pyrazolo[3,4-b]pyridine derivatives which are useful for pharmaceutical applications, have already been disclosed, for example in WO 2005028480 (Neurogen Corp. and Aventis Pharmaceuticals Inc.), in WO 2005009389 (Exelixis Inc.) or in WO
2000058307 (Neurogen Corp.).
Accordingly, a subject of the present invention is a compound of the formula I,

R1 is a) a residue of the formula la

R6 is H, F, CI, (Ci-C3)-alkyl or O-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F;
R7 is H, F, CI, (Ci-C3)-alkyl or O-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is CN, (C0-C2)- alkylene-SO2(Ci-C3)-alkyl, (Co-C2)-alkylene-N((Ci-C4)-alkyl)2, CO- N((Ci-C4)-alkyl)2, 1 -pyrrolidinyl, 1 -piperidinyl, 1 -pyrrolidinyl-2-on, 1 - piperidinyl-2-on, or 4-morpholinyl;
R8 is H, F, CI, (Ci-C5)-alkyl or O-(Ci-C5)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is CN, OH, O- phenyl, SO2(Ci-C3)-alkyl, CO-(Ci-C )-alkyl, CO-N((Ci-C )-alkyl)2, CO-1 -piperidinyl, CO-1 -pyrrolidinyl, or CO-4-morpholinyl;
R9 is H, F, CH3, or O-CH3;
R10 is H; or
R6 and R7 form, together with the two carbon atoms, to which they are attached, a five to six membered heterocycloalkyl ring, which contains one or two identical or different heteroatoms selected from nitrogen and oxygen, and wherein said heterocycloalkyl is unsubstituted or mono-substituted by (Ci-C4)-alkyl or oxo (=0); or
R7 and R8 form, together with the two carbon atoms, to which they are attached, a five to six membered heterocycloalkyl ring, which contains one or two identical or different heteroatoms selected from nitrogen and oxygen, and wherein said heterocycloalkyl is unsubstituted or mono-substituted by (Ci-C4)-alkyl or oxo (=0); b) ethylene-phenyl, wherein phenyl is unsubstituted or substituted by one or two identical or different substituents selected from F, CI, CH3, 0-CH3, CF3, O-CFs, CN, SO2CH3, CO-CH3, and CO-N((CH3)2;
c) 2-phenylcyclopropyl, wherein phenyl is unsubstituted or substituted by one or two identical or different substituents selected from F, CI, CH3, 0- CH3, CF3, 0-CF3, CN, S02CH3, CO-CH3, and CO-N((CH3)2; d) ethylene-(C5-C7)cycloalkyl;
R2 is H, F, CI, or CH3;
R3 is H, F, or 0-CH3;
R4 is piperidin-4-yl, piperidin-3-yl or pyrrolidin-3-yl, which are unsubstituted or
monosubstituted by (Ci-C3)-alkyl, which is unsubstituted or substituted by one to five F;
R5 is H; or
R4 and R5 together with the nitrogen carrying them denote
a) a residue of the formula lb
 wherein
wherein
R11 is H, (C3-C5)-cycloalkyl, (Ci-C4)-alkylene-OH or (Ci-C4)-alkyl, which is unsubstituted or substituted by one to five F;
R12 is H, or (Ci-C )-alkyl,
R13 is H, (Ci-C )-alkyl, or (C3-C5)-cycloalkyl;
R14 is H, or (Ci-C )-alkyl; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclo-(C3-C6)-alkyl; and
R15 is H, CH2-CO-N((Ci-C )-alkyl)2, CO-(Ci-C6)-alkyl,
CO-(Ci-C )-alkylene-OH, CO-(Ci-C )-alkylene-0-CH3, CO-CH2-4- morpholinyl, CO-0-(Ci-C )-alkyl, CO-N((Ci-C )-alkyl)2, (C3-C6)- cycloalkyl, or pyridyl;
R16 is H, or (Ci-C )-alkyl;
R17 is H, or CH3;
R18 is H, or CH3;
R19 is H; b) a 9 to 1 1 membered spiro-heterocycloalkyl containing 2 nitrogen atoms, which is attached via a nitrogen atom; in any of its stereoisomeric forms, or a mixture of stereoisomeric forms in any ratio, or a physiologically acceptable salt thereof, or a physiologically acceptable solvate of any of them.
Another group of embodiments are compounds of the formula I wherein
R1 is a) a residue of the formula la
 wherein
wherein
Rb is H, F, CI, (Ci-C3)-alkyl or 0-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F; or R6 is H, F, CI, CH3, CF3, or 0-CH3; or R6 is H;
R7 is H, F, CI, (Ci-C3)-alkyl or 0-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is CN, SO2(Ci-C3)- alkyl, N((Ci-C4)-alkyl)2, 1 -piperidinyl, or 4-morpholinyl; or R7 is H, F, CI, CH3, CF3, CN, 0-CH3, S02CH3, N(CH3)2, 1 -piperidinyl, or 4- morpholinyl; or R7 is H;
R8 is H, F, CI, (Ci-C5)-alkyl or 0-(Ci-C5)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is OH, O-phenyl, SO2(Ci-C3)-alkyl, CO-(Ci-C )-alkyl, CO-N((Ci-C )-alkyl)2; or R8 is H, F, CI, CH3, CH2CH3, CH(CH3)2, C(CH3)3, CF3, OH, O-CH3, O- C2H5, O-CHF2, O-CF3, O-phenyl, SO2CH3, CO-CH3, or CO- N(CH3)2; or R8 is H;
R9 is H, F, CH3, O-CH3; or R9 is H;
R10 is H; or
R7 and R8 together are
 b) ethylene-phenyl, wherein phenyl is unsubstituted or substituted by one to two identical or different substituents selected from F, CI, CH3, O-CH3, CF3, O-CF3, CN, SO2CH3, CO-CH3, and CO-N((CH3)2; or R1 is phenethyl.
b) ethylene-phenyl, wherein phenyl is unsubstituted or substituted by one to two identical or different substituents selected from F, CI, CH3, O-CH3, CF3, O-CF3, CN, SO2CH3, CO-CH3, and CO-N((CH3)2; or R1 is phenethyl.
Another group of embodiments are compounds of the formula I wherein
R2 is H, F, CH3. or R2 is H, CH3 or R2 is H, F, or R2 is H.
Another group of embodiments are compounds of the formula I wherein
Another group of embodiments are compounds of the formula I wherein
R4 is piperidin-4-yl, piperidin-3-yl or pyrrolidin-3-yl, which are unsubstituted or monosubstituted by (Ci-C3)-alkyl, which is unsubstituted or substituted by one to five F; or R4 is 4-methyl-piperidin-4-yl and
R5 is H.
Another group of embodiments are compounds of the formula I wherein
R4 and R5 together with the nitrogen carrying them denote
a) a residue of the formula lb

R is H, CH3, (C3-C4)-alkyl, or (C3-C )-cycloalkyl; or R11 is H, CH3, or (CH2)3-CH3; or R11 is H;
R is H;
R13 is H, (Ci-C )-alkyl, or (C3-C )-cycloalkyl; or R13 is H, CH3, or CH2-
CH(CH3)2; or R1 J is H or CH3; or R1 J is H;
R1 is H, or CH3; or R14 is H; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclo-(C3-C6)-alkyl; preferably 1 '-spiro-cyclopentyl; and
R15 is H, CH2-CO-N((Ci-C )-alkyl)2, CO-(Ci-C6)-alkyl,
CO-(Ci-C )-alkylene-OH, CO-(Ci-C )-alkylene-0-CH3, CO-CH2-4- morpholinyl, CO-0-(Ci-C )-alkyl, CO-N(-(Ci-C )-alkyl)2, -(C3-C6)-
cycloalkyl, or pyridyl; or R1 S is H, CH2-CO-N(CH3)2, CO-CH(C2H5)2, CO-C(CH3)2-OH, CO-C(CH3)2-CH2-OH, CO-(CH2)2-0-CH3, CO- CH2-4-morpholinyl, CO-OCH3, CO-N(CH3)2, cyclopropyl, or 3-pyridyl; or R15 is H;
R16 is H, or (Ci-C4)-alkyl; or R16 is H, or CH3; or R16 is H;
R17 is H;
b) a 9 to 1 1 membered spiro-heterocycloalkyl containing two nitrogen
atoms, which is attached via a nitrogen atom, or R1 is (2,7-Diaza- spiro[4.4]non-2-yl) .
Another group of embodiments are compounds of the formula I
wherein
R1 is a) a residue of the formula la

R6 is H, F, CI, CH3, CF3, or 0-CH3;
R7 is H, F, CI, CH3, CH2-S02-CH3, CF3, CN, CH2-N(CH3)2, CO- N(CH3)2, 0-CH3, S02CH3, N(CH3)2, 1 -pyrrolidinyl, 1 -piperidinyl, 4- morpholinyl, or 1 -pyrrolidinyl-2-on;
R8 is H, F, CI, CH3, CH2CH3, CH(CH3)2, C(CH3)3, CF3, CN, OH, O- CH3, O-C2H5, O-CHF2, O-CF3, O-phenyl, SO2CH3, CO-CH3, CO- N(CH3)2, or CO-1 -piperidinyl;
R9 is H, CI, F, CH3, or O-CH3;
R10 is H; or
R6 and R7 togeth

R7 and R8 together are

c) 2-phenylcyclopropyl;
d) 2-cyclohexylethyl; or
R2 is H, F, CI, or CH3; or
R3 is H, F, or 0-CH3; or
R4 is 4-methyl-piperidin-4-yl;
R5 is H; or
R4 and R5 together with the nitrogen carrying them denote a) a residue of the formula lb

R is H, CH3, C2H5, (CH2)3-CH3, or CH2-OH;
R12 is H, or CH3;
R13 is H, CH3, or CH2-CH(CH3)2;
R14 is H, or CH3; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclopentyl; and
R15 is H, CH2-CO-N(CH3)2, CO-CH(C2H5)2, CO-C(CH3)2-OH,
CO-C(CH3)2-CH2-OH, CO-(CH2)2-0-CH3, CO-CH2-4-morpholinyl, CO-OCH3, CO-N(CH3)2, cyclopropyl, or CO-3-pyridyl;
R16 is H, or CH3;
R17 is H;
R18 is H;
R19 is H; b) (2,7-Diaza-spiro[4.4]non-2-yl) (= Bsp 60).
Another group of embodiments are compounds of the formula I
wherein
R1 is a) a residue of the formula la
wherein
R6 is H, F, CI, CH3, CF3, or 0-CH3;
R7 is H, F, CI, CH3, CF3, CN, 0-CH3, S02CH3, N(CH3)2, 1 -piperidinyl, or 4-morpholinyl;
R8 is H, F, CI, CH3, CH2CH3, CH(CH3)2, C(CH3)3, CF3, OH, O- CH3, O-C2H5, O-CHF2, O-CF3, O-phenyl, SO2CH3, CO-CH3, or CO- N(CH3)2;
R9 is H, F, CH3, or O-CH3;
R10 is H; or
R7 and R8 together are
 b) phenethyl; R2 is H, or CH3;
b) phenethyl; R2 is H, or CH3;
R3 is H, or 0-CH3;
R4 is 4-methyl-piperidin-4-yl;
R5 is H; or
R4 and R5 together with the nitrogen carrying them denote
a) a residue of the formula lb

R11 is H, CH3, or (CH2)3-CH3;
R12 is H;
R13 is H, CH3, or CH2-CH(CH3)2;
R14 is H, or CH3; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclopentyl; and
R15 is H, CH2-CO-N(CH3)2, CO-CH(C2H5)2, CO-C(CH3)2-OH, CO-
C(CH3)2-CH2-OH, CO-(CH2)2-0-CH3, CO-CH2-4-morpholinyl, CO-
OCH3, CO-N(CH3)2, cyclopropyl, or 3-pyridyl;
R16 is H, or CH3;
R17 is H;
R11 and R12 together with the C-atoms carrying them are 1 '-spiro- cyclopentyl;
R18 is H;
R19 is H; b) (2,7-Diaza-spiro[4.4]non-2-yl) .
Another group of embodiments are compounds of the formula I
wherein
R1 is a residue of the formula la

R6 is H, F, CI, CH3, CF3, or 0-CH3;
R7 is H, F, CI, CH3, CF3, CN, 0-CH3, S02CH3, N(CH3)2, 1 -piperidinyl, or 4-morpholinyl;
R8 is H, F, CI, CH3, CH2CH3, CH(CH3)2, C(CH3)3, CF3, OH, O- CH3, O-C2H5, O-CHF2, O-CF3, O-phenyl, SO2CH3, CO-CH3, or CO- N(CH3)2;
R9 is H, F, CH3, or O-CH3;
R10 is H;
R2 is H;
R3 is H;
R4 and R5 together with the nitrogen carrying them denote
a residue of the formula lb
 wherein
wherein
R11 is H, CH3, or (CH2)3-CH3;
R12 is H;
R13 is H, CH3, or CH2-CH(CH3)2;
R14 is H, or CH3; or
R 3 and R14 together with the C-atom carrying them is
1 '-spiro-cyclopentyl; and
R15 is H;
R16 is H, or CH3;
R17 is H;
R and R12 together with the C-atoms carrying them are 1 '-spiro- cyclopentyl;
R18 is H;
Another group of embodiments are compounds selected from the group consisting of 1
[3-[(E)-2-(4-Chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin- -yl-methanone
2
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
3
(2,2-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
4
(3,3-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
5
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-3-isobutyl- piperazin-1 -yl)-methanone
6
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-2-methyl- piperazin-1 -yl)-methanone
7
[6-(4-Hydroxy-2-methyl-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin- 1 -yl-methanone
8
((2S,5R)-2,5-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone
9
[6-(4-Hydroxy-3-methoxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
10
((S)-2-Ethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
1 1
[6-(2-Fluoro-4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-2- methyl-piperazin-1 -yl)-methanone
12
[6-(3-Fluoro-4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
13
[6-(2-Chloro-4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
14
[3-[2-(3-Dimethylaminomethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
15
{6-(4-Hydroxy-phenyl)-3-[2-(3-methanesulfonylmethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
16
((R)-2-Hydroxymethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone
17
[6-(4-Hydroxy-phenyl)-3-phenethyl-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
18
[6-(4-Hydroxy-phenyl)-3-((1 R,2R)-2-phenyl-cyclopropyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
19
(6-(4-Hydroxy-phenyl)-3-{2-[4-(piperidine-1 -carbonyl)-phenyl]-vinyl}-1 H-pyrazolo[3,4- b]pyridin-4-yl)-piperazin-1 -yl-methanone
20
1 -(3-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-phenyl)-pyrrolidin-2-one
21
[6-(4-Hydroxy-phenyl)-3-((E)-2-p-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 - yl-methanone
22
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methanesulfonyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
23
[3-[(E)-2-(3-Dimethylamino-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-methanesulfonyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
25
3- {(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-benzonitrile
26
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-piperidin-1 -yl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
27
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-morpholin-4-yl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
28
{6-(4-Hydroxy-phenyl)-3-[2-(3-pyrrolidin-1 -yl-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
29
4- {2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-benzonitrile
30
[3-[2-(3-Chloro-5-piperidin-1 -yl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
31
3- {2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-N,N-dimethyl-benzamide
32
5-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one
33
[3-[2-(2,3-Dihydro-1 H-indol-7-yl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-
4- yl]-piperazin-1 -yl-methanone
34
3-{2-[4-(2,2-Dimethyl-piperazine-1 -carbonyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-3-yl]-vinyl}-N,N-dimethyl-benzamide
35
6-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one
36
[3-(2-Cyclohexyl-ethyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin- 1 -yl-methanone
37
[3-[(E)-2-(4-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
38
[6-(4-Hydroxy-phenyl)-3-((E)-2-m-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-
1 -yl-methanone
39
[3-[(E)-2-(2,4-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
40
[3-[(E)-2-(3,5-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
41
[3-[(E)-2-(4-Difluoromethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
42
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
43
[3-[(E)-2-(2,4-Dimethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-
4-yl]-piperazin-1 -yl-methanone
44
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-phenoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
45
[3-[(E)-2-(3,4-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
46
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
47
[3-[(E)-2-(4-Hydroxy-3-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
48
[3-[(E)-2-(3-Chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
49
[3-[(E)-2-(2,4-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
50
[3-[(E)-2-(2,5-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
51
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-isopropyl-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
52
[3-[(E)-2-(2-Chloro-4-fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
53
[3-[(E)-2-(4-Ethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
54
[3-[(E)-2-(4-tert-Butyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
55
[3-[(E)-2-(3-Fluoro-4-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
56
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
57
1 -(4-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin- 3-yl]-vinyl}-phenyl)-ethanone
58
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-trifluoromethoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
59
[3-[(E)-2-(3,4-Difluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
60
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-methoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
61
[3-[(E)-2-(4-Fluoro-2-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
62
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3,4,5-tnmethoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
63
[3-[(E)-2-(2-Fluoro-4-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
64
[3-[(E)-2-(2,3-Dihydro-benzofuran-5-yl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
65
[3-((E)-2-Benzo[1 ,3]dioxol-5-yl-vinyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
66
4-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-N,N-dimethyl-benzamide
67
[3-[(E)-2-(4-Ethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
68
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
69
[6-(4-Hydroxy-phenyl)-3-((E)-2-o-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 - yl-methanone
70
[3-[2-(3,5-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
71
[3-[(E)-2-(3-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
72
[3-[(E)-2-(2-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
73
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-methoxy-3-methyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
74
4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazine-1 -carboxylic acid dimethylamide
75
6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (4- methyl-piperidin-4-yl)-amide
76
((S)-2-Butyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
77
((R)-2-Butyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
78
(2,7-Diaza-spiro[4.4]non-2-yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
79
(6,9-Diaza-spiro[4.5]dec-9-yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
80
2-Ethyl-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-butan-1 -one
81
1 - {4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-2-morpholin-4-yl-ethanone
82
4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazine-1 -carboxylic acid methyl ester
83
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-[4-(pyridine-3- carbonyl)-piperazin-1 -yl]-methanone
84
2- {4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-N,N-dimethyl-acetamide
85
(4-Cyclopropyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
86
2-Hydroxy-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-2-methyl-propan-1 -one
87
3-Hydroxy-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-2,2-dimethyl-propan-1 -one
88
1 -{4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-3-methoxy-propan-1 -one
Another group of embodiments are compounds selected from the group consisting of
[6-(4-hydroxyphenyl)-3-(2-phenylethenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl](4-methyl-1 - piperazinyl)-methanone
(4,7-Diaza-spiro[2.5]oct-4-yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone. Another group of embodiments are compounds selected from the group consisting of 1
[3-[(E)-2-(4-Chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
2
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
4
(3,3-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
5
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-3-isobutyl- piperazin-1 -yl)-methanone
6
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-2-methyl- piperazin-1 -yl)-methanone
7
[6-(4-Hydroxy-2-methyl-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-
1 -yl-methanone
8
((2S,5R)-2,5-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone
9
[6-(4-Hydroxy-3-methoxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
17
[6-(4-Hydroxy-phenyl)-3-phenethyl-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
21
[6-(4-Hydroxy-phenyl)-3-((E)-2-p-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 - yl-methanone
22
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methanesulfonyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
23
[3-[(E)-2-(3-Dimethylamino-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
24
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-methanesulfonyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
25
3-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-benzonitrile
26
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-piperidin-1 -yl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
27
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-morpholin-4-yl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
37
[3-[(E)-2-(4-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
38
[6-(4-Hydroxy-phenyl)-3-((E)-2-m-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin- 1 -yl-methanone
39
[3-[(E)-2-(2,4-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
40
[3-[(E)-2-(3,5-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
41
[3-[(E)-2-(4-Difluoromethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
42
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
43
[3-[(E)-2-(2,4-Dimethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin- 4-yl]-piperazin-1 -yl-methanone
44
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-phenoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
45
[3-[(E)-2-(3,4-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
46
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
47
[3-[(E)-2-(4-Hydroxy-3-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
48
[3-[(E)-2-(3-Chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
49
[3-[(E)-2-(2,4-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
50
[3-[(E)-2-(2,5-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
51
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-isopropyl-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
52
[3-[(E)-2-(2-Chloro-4-fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
53
[3-[(E)-2-(4-Ethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
54
[3-[(E)-2-(4-tert-Butyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
55
[3-[(E)-2-(3-Fluoro-4-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
56
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
57
1 -(4-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin- 3-yl]-vinyl}-phenyl)-ethanone
58
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-trifluoromethoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
59
[3-[(E)-2-(3,4-Difluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
60
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-methoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
61
[3-[(E)-2-(4-Fluoro-2-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
62
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3,4,5-tnmethoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
63
[3-[(E)-2-(2-Fluoro-4-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
64
[3-[(E)-2-(2,3-Dihydro-benzofuran-5-yl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
65
[3-((E)-2-Benzo[1 ,3]dioxol-5-yl-vinyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
66
4-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-N,N-dimethyl-benzamide
67
[3-[(E)-2-(4-Ethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
68
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
69
[6-(4-Hydroxy-phenyl)-3-((E)-2-o-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 - yl-methanone
71
[3-[(E)-2-(3-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
72
[3-[(E)-2-(2-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
73
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-methoxy-3-methyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
74
4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazine-1 -carboxylic acid dimethylamide
75
6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (4- methyl-piperidin-4-yl)-amide
76
((S)-2-Butyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
77
((R)-2-Butyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
78
(2,7-Diaza-spiro[4.4]non-2-yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
79
(6,9-Diaza-spiro[4.5]dec-9-yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
80
2-Ethyl-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-butan-1 -one
81
1 -{4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-2-morpholin-4-yl-ethanone
82
4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazine-1 -carboxylic acid methyl ester
83
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-[4-(pyridine-3- carbonyl)-piperazin-1 -yl]-methanone
84
2- {4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-N,N-dimethyl-acetamide
85
(4-Cyclopropyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
86
2-Hydroxy-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-2-methyl-propan-1 -one
87
3- Hydroxy-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-2,2-dimethyl-propan-1 -one
88
1 -{4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-3-methoxy-propan-1 -one
Another group of embodiments are compounds selected from the series consisting of 3
(2,2-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
10
((S)-2-Ethyl-piperazin-1 -yl)-[6-(4-hydroxy-ph
b]pyridin-4-yl]-methanone
1 1
[6-(2-Fluoro-4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-2- methyl-piperazin-1 -yl)-methanone
12
[6-(3-Fluoro-4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
13
[6-(2-Chloro-4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
14
[3-[2-(3-Dimethylaminomethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
15
{6-(4-Hydroxy-phenyl)-3-[2-(3-methanesulfonylmethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
16
((R)-2-Hydroxymethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone
18
[6-(4-Hydroxy-phenyl)-3-((1 R,2R)-2-phenyl-cyclopropyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
19
(6-(4-Hydroxy-phenyl)-3-{2-[4-(piperidine-1 -carbonyl)-phenyl]-vinyl}-1 H-pyrazolo[3,4- b]pyridin-4-yl)-piperazin-1 -yl-methanone
20
1 -(3-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-phenyl)-pyrrolidin-2-one
28
{6-(4-Hydroxy-phenyl)-3-[2-(3-pyrrolidin-1 -yl-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
29
4- {2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-benzonitrile
30
[3-[2-(3-Chloro-5-piperidin-1 -yl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
31
3-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-N,N-dimethyl-benzamide
32
5- {2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one
33
[3-[2-(2,3-Dihydro-1 H-indol-7-yl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-
4-yl]-piperazin-1 -yl-methanone
34
3-{2-[4-(2,2-Dimethyl-piperazine-1 -carbonyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-3-yl]-vinyl}-N,N-dimethyl-benzamide
35
6- {2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one
36
[3-(2-Cyclohexyl-ethyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin- 1 -yl-methanone
70
[3-[2-(3,5-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
Structural elements such as groups, substituents, hetero ring members, numbers or other features, for example alkyl groups, groups like R1, R2, R3 etc., which can occur
several times in the compounds of the formula I, can all independently of one another have any of the indicated meanings and can in each case be identical to or different from one another. For example, the alkyl groups in a dialkylamino group can be identical or different.
As used here, the terms "including" and "comprising" are used in their open, non- limiting sense. As used herein, the terms "(C-i-Ce)" or "(Cs-Ce)" and so forth, refer to moieties having 1 to 8 or 5 to 8 carbon atoms, respectively. Within terms like "(Co-Ce)- alkyl" or "(C0-C6)-alkylen" "C0-alkyl" or "(Co)-alkylen" refer to a bond, or in case of an unsubstituted "(Co)-alkyl" it refers to a hydrogen.
The term "alkyl", as used herein, refers to saturated, monovalent hydrocarbon radicals. The term "alkenyl", as used herein, refers to monovalent hydrocarbon radicals, which contain at least one carbon-carbon double bond, wherein each double bond can have E- or Z-configuration. The term "alkinyl", as used herein, refers to monovalent hydrocarbon radicals, which contain at least one carbon-carbon triple bond. The alkyl, alkenyl and alkynyl groups can be linear, i.e. straight-chain, or branched. This also applies when they are part of other groups, for example alkyloxy groups (= alkoxy groups, O-alkylgroups), alkyloxycarbonyl groups or alkyl-substituted amino groups, or when they are substituted. Depending on the respective definition, the number of carbon atoms in an alkyl group can be 1 , 2, 3, 4, 5, 6, 7 or 8, or 1 , 2, 3, 4, 5 or 6, or 1 , 2, 3 or 4, or 1 , 2 or 3. Examples of alkyl are methyl, ethyl, propyl including n-propyl and isopropyl, butyl including n-butyl, sec-butyl, isobutyl and tert-butyl, pentyl including n-pentyl, 1 -methylbutyl, isopentyl, neopentyl and tert-pentyl, hexyl including n-hexyl, 3,3-dimethylbutyl and isohexyl, heptyl and octyl. Double bonds and triple bonds in alkenyl groups and alkynyl groups can be present in any positions. In one embodiment of the invention, alkenyl groups contain one double bond and alkynyl groups contain one triple bond. In one embodiment of the invention, an alkenyl group or alkynyl group contains at least three carbon atoms and is bonded to the remainder of the molecule via a carbon atom which is not part of a double bond or triple bond. Examples of alkenyl and alkynyl are ethenyl, prop-1 -enyl, prop-2-enyl (= allyl), but-2-enyl, 2- methylprop-2-enyl, 3-methylbut-2-enyl, hex-3-enyl, hex-4-enyl, prop-2-ynyl (=
propargyl), but-2-ynyl, but-3-ynyl, hex-4-ynyl or hex-5-ynyl. Substituted alkyl groups, alkenyl groups and alkynyl groups can be substituted in any positions, provided that the respective compound is sufficiently stable and is suitable for the desired purpose such as use as a drug substance. The prerequisite that a specific group and a compound of the formula I are sufficiently stable and suitable for the desired purpose such as use as a drug substance, applies in general with respect to the definitions of all groups in the compounds of the formula I.
Independently of one another and independently of any other substituents, alkyl groups, divalent alkyl groups, alkenyl groups, alkynyl groups, cycloalkyl groups and heterocycloalkyl groups are optionally substituted by one or more fluorine substituents which can be located in any positions, i.e., the said groups can be unsubstituted by fluorine substituents or substituted by fluorine substituents, for example by 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 1 1 , 12 or 13, or by 1 , 2, 3, 4, 5, 6, 7, 8 or 9, or by 1 , 2, 3, 4, 5, 6 or 7, or by 1 , 2, 3, 4 or 5, or by 1 , 2 or 3, or by 1 or 2, fluorine substituents. Examples of fluorine-substituted said groups are trifluoromethyl, 2-fluoroethyl, 2,2,2-trifluoroethyl, pentafluoroethyl, 3,3,3-trifluoropropyl, 2,2,3,3,3-pentafluoropropyl, 4,4,4-trifluorobutyl, heptafluoroisopropyl, -CHF-, -CF2-, -CF2-CH2-,
-CH2-CF2-, -CF2-CF2-, -CF(CH3)-, -C(CF3)2-, -C(CH3)2-CF2-, -CF2-C(CH3)2-, 1 -fluoro- cyclopropyl, 2,2-difluorocyclopropyl, 3,3-difluorocyclobutyl, 1 -fluorocyclohexyl, 4,4- difluorocyclohexyl, 3,3,4,4,5,5-hexafluorocyclohexyl. Examples of alkyloxy groups in which the alkyl moiety is fluorine-substituted, are trifluoromethoxy, 2,2,2-trifluoro- ethoxy, pentafluoroethoxy and 3,3,3-trifluoropropoxy. The term "alkanediyl" or "alkylene" ", as used herein, refers to saturated, divalent hydrocarbon radicals. The term "alkenediyl", as used herein, refers to divalent hydrocarbon radicals, which contain at least one carbon-carbon double bond, wherein each double bond can have E- or Z-configuration. The term "alkindiyl", as used herein, refers to divalent hydrocarbon radicals, which contain at least one carbon-carbon triple bond. As far as applicable, the preceding explanations regarding alkyl, alkenyl and alkinyl groups apply correspondingly to alkanediyl, alkendiyl and alkindiyl groups, which thus can likewise be linear and branched. Examples of divalent alkyl groups are
-CH2- (= methylene), -CH2-CH2-, -CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-, -CH(CH3)-, - C(CH3)2-, -CH(CH3)-CH2-, -CH2-CH(CH3)-, -C(CH3)2-CH2-, -CH2-C(CH3)2-.
The term "cycloalkyl", as used herein, unless otherwise indicated, refers to a mono- valent radical of a saturated or partially saturated hydrocarbon ring system, which can be monocyclic, bicyclic or tricyclic, i.e. which can contain one, two or three rings. The bicyclic or tricyclic ring system can be a fused ring system, in which two adjacent rings share two adjacent carbon atoms. The bicyclic or tricyclic ring system can be a spiro ring system or a di-spiro-ringsystem, in which two adjacent rings share a single carbon atom. The tricyclic ring system can also be a bicyclic spiro ring system, to which another ring is fused, that means that the latter ring and the ring in the spiro ring system, to which it is attached, share two adjacent carbon atoms; herein the latter ring can be an aromatic, saturated or partially saturated ring. The bicyclic or tricyclic system can also be a non-fused or bridged ring system, in which two adjacent rings share two non-adjacent carbon atoms. The bicyclic or tricyclic ring can be attached by any ring atom except a spiro- or a bridgehead atom.
In a monocyclic cycloalkyl group the number of ring carbon atoms can be 3, 4, 5, 6, 7 or 8. In one embodiment of the invention, the number of ring carbon atoms in a cyclo- alkyl group, independently of the number of ring carbon atoms in any other cycloalkyl group, is 3, 4, 5 or 6, in another embodiment 3, 4 or 5, in another embodiment 3 or 4, in another embodiment 3, in another embodiment 5, 6 or 7, in another embodiment 5 or 6, in another embodiment 6 or 7, in another embodiment 5, in another embodiment 6. Examples of cycloalkyl groups are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
In a bicyclic cycloalkyl group the number of ring carbon atoms can be 6, 7, 8, 9, 10, 1 1 or 12. In one embodiment of the invention, the number of ring carbon atoms in a bicyclic cycloalkyl group can be 7, 8, 9, 10 or 1 1 , in another embodiment 8, 9 or 10. In a tricyclic cycloalkyl group the number of ring carbon atoms can be 7, 8, 9, 10, 1 1 , 12, 13, 14 or 15. In one embodiment of the invention, the number of ring carbon atoms in a tricyclic cycloalkyl group can be 10, 1 1 or 12.
Exemplary bicyclic or tricyclic fused ring cycloalkyls are derived from, but not limited to, the following ring systems: bicyclo[3.1.0]hexane, bicyclo[4.1 .0]heptane, bicycle- [5.1 .0]octane, bicyclo[3.2.0]heptane, bicyclo[4.2.0]octane, octahydro-pentalene, octa- hydro-indene, decahydro-azulene, decahydro-naphthalene, decahydro-benzo- cycloheptene, dodecahydro-heptalene, 1 ,2,3,3a,4,6a-hexahydro-pentalene, 1 ,2,3,4- tetrahydro-pentalene, 2,3,3a,4,5,7a-hexahydro-1 H-indene, 2,3,3a,4,7,7a-hexahydro- 1 H-indene, 3a,4,5,6,7,7a-hexahydro-1 H-indene, 4, 5,6, 7-tetrahydro-1 H-indene, indane, 1 ,2,3,4,4a,5,6,8a-octahydro-naphthalene, 1 ,2,3,4,4a,5,8,8a-octahydro-naphthalene, 1 ,2,4a,5,8,8a-hexahydro-naphthalene, 1 ,4,4a,5,8,8a-hexahydro-naphthalene, 1 ,2,3,4- tetrahydro-naphthalene, 2,3,4,4a,5,6,9,9a-octahydro-1 H-benzocycloheptene,
2,3,4,4a,5,9a-hexahydro-1 H-benzocycloheptene, 4,4a,5,6,7,8,9,9a-octahydro-1 H- benzocycloheptene, 6,7,8,9-tetrahydro-5H-benzocycloheptene, 1 , 2, 3, 4, 5,5a, 6,7, 8, 10a- decahydro-heptalene, dodecahydro-as-indacene and 2,3,3a,4,9,9a-hexahydro-1 H- cyclopenta[b]naphthalene:

Exemplary bicyclic or tricyclic spiro ring cycloalkyls are derived from, but not limited to, the following ring systems: spiro[2.4]heptane, spiro[2.5]octane, spiro[2.6]nonane, spiro[3.3]heptane, spiro[3.4]octane, spiro[3.5]nonane, spiro[3.6]decane, spiro[4.4]no- nane, spiro[4.5]decane, spiro[4.6]undecane, spiro[5.5]undecane, spiro[5.6]dodecane, spiro[6.6]tridecane, dispiro[2.2.4.2]dodecane, dispiro[2.2.3.2]undecane, dispiro- [2.1 .4.2]undecane and spiro[5.5]undec-2-ene:


Exemplary non-fused or bridged bicyclic or tricyclic ring cycloalkyls are derived from, but not limited to, the following ring systems: bicyclo[2.2.1 ]heptane, bicyclo[2.2.2]oc- tane, bicyclo[3.2.1 ]octane, bicyclo[3.2.2]nonane and adamantane.

The term "heterocycloalkyl" or "heterocyclyl", as used herein, unless otherwise indicated, refers to a cycloalkyi as defined above, in which 1 , 2, 3 or 4 carbon atoms
are replaced by nitrogen, oxygen or sulfur atoms, provided that a spiro atom is always a carbon atom and a bridgehead atom is either a carbon or a nitrogen atom and provided that the heterocycloalkyl system is stable and suitable as a subgroup for the desired purpose of the compound of the formula I such as use as a drug substance. Depending on the definition of the respective heterocyclic group, in one embodiment of the invention the number of ring heteroatoms which can be present in a heterocyclic group, independently of the number of ring heteroatoms in any other heterocyclic group, is 1 , 2, 3 or 4, in another embodiment 1 , 2 or 3, in another embodiment 1 or 2, in another embodiment 2, in another embodiment 1 , wherein the ring heteroatoms can be identical or different. The heterocycloalkyl group can be attached by any ring carbon atom or saturated ring nitrogen atom, with the exeption of spiro- or bridgehead atoms. A ring sulfur atom in a heterocycloalkyl group can carry zero, one or two oxo groups, it is a non-oxidized sulfur atom S in case it does not carry any oxo group, or it is an S(O) group (= sulfoxide group, S-oxide group) in case it carries one oxo group, or it is an S(0)2 group (= sulfone group, S,S-dioxide group) in case it carries two oxo groups.
Exemplary monocyclic heterocycloalkyls are derived from, but not limited to, the following ring systems: aziridine, oxirane, azetidine, oxetane, pyrrolidine, tetrahydrofu- rane, tetrahydrothiophene, 4,5-dihydrothiazole, piperidine, piperazine, morpholine, thiomorpholine, tetrahydropyran, 1 ,4-dioxane, 1 ,4-oxathiane, 1 ,2,3,6-tetrahydro- pyridine, azepane, 2,3,4,7-tetrahydro-1 H-azepine, 2,7-dihydro-1 H-azepine, 1 ,4-diaze- pane, 1 ,4-oxazepane, 1 ,4-thiazepane and 1 ,4-dioxepane:

In one embodiment monocyclic heterocycloalkyls are derived from azetidine, pyrrolidine, piperidine, piperazine, morpholine or 1 ,4-diazepane:

Exemplary bicyclic fused ring heterocycloalkyls are derived from, but not limited to, the following ring systems: 3-aza-bicyclo[3.1 .0]hexane, 2-aza-bicyclo[4.1 .0]heptane, 2- oxa-5-aza-bicyclo[5.1 .0]octane, 3-aza-bicyclo[3.2.0]heptane, 2-aza-bicyclo[4.2.0]oc- tane, octahydro-pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4-b]pyrrole, octahydro- pyrrolo[3,4-b]pyridine, octahydro-thieno[3,4-b]pyrazine, octahydro-furo[3,4-b]pyridine, octahydro-cyclopenta[1 ,4]oxazine, octahydro-pyrrolo[1 ,2-a]pyrimidine, octahydro- pyrrolo[1 ,2-a]pyrazine, octahydro-cyclopenta[e][1 ,4]oxazepine, decahydro-quinoxaline, decahydro-[1 ,6]naphthyridine, octahydro-benzo[1 ,4]oxazine, octahydro-benzo- [1 ,4]thiazine, octahydro-pyrido[1 ,2-a]pyrazine, octahydro-pyrano[3,2-b]pyridine, deca- hydro-1 -oxa-9-aza-benzocycloheptene, 1 ,2,3,3a,6,6a-hexahydro-cyclopenta[b]pyrrole, 5,6-dihydro-4H-cyclopenta[b]thiophene, 2,3,4,4a,7,7a-hexahydro-1 H-[2]pyrindine, 2,4a,5,6,7,7a-hexahydro-1 H-[1 ]pyrindine, 2,3,3a,4,7,7a-hexahydro-1 H-indole, 1 ,2,3,4- tetrahydro-quinoxaline, 4,5,6,7-tetrahydro-benzofuran, benzo[1 ,3]dioxole,
3,4,4a,7,8,8a-hexahydro-2H-benzo[1 ,4]oxazine, 1 ,2,3,4,4a,5,8,8a-octahydro-
quinoxaline, 4a,5,8,8a-tetrahydro-2H-thiopyrano[3,2-b]pyridine and 1 ,2,3,4-tetrahydro- [1 ,5]naphthyridine:

In one embodiment bicyclic fused ring heterocycloalkyls are derived from 3-aza- bicyclo[3.1 .0]hexane, octahydro-pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4-b]pyrrole,
octahydro-thieno[3,4-b]pyrazine, octahydro-pyrrolo[1 ,2-a]pyrazine, decahydro- quinoxaline, octahydro-pyrido[1 ,2-a]pyrazine or decahydro-[1 ,6]naphthyridine:

Exemplary bicyclic or tricyclic spiro ring heterocycloalkyls are derived from, but not limited to, the following ring systems:4-aza-spiro[2.4]heptane, 5-aza-spiro[3.4]octane, 1 -aza-spiro[4.4]nonane, 7-oxa-1 -aza-spiro[4.4]nonane, 7-thia-1 -aza-spiro[4.4]nonane, 4-aza-spiro[2.5]octane, 5-aza-spiro[2.5]octane, 6-aza-spiro[2.5]octane, 5-aza- spiro[3.5]nonane, 6-aza-spiro[3.5]nonane, 7-aza-spiro[3.5]nonane, 4,7-diaza- spiro[2.5]octane, 5,8-diaza-spiro[3.5]nonane, 6,9-diaza-spiro[4.5]decane, 1 ,4-diaza- spiro[5.5]undecane, 2-oxa-6,9-diaza-spiro[4.5]decane, 2-oxa-6-aza-spiro[4.5]decane, 2,7-diaza-spiro[3.5]nonane, 3,9-diaza-spiro[5.5]undecane, 1 -oxa-4,9-diaza-spiro- [5.5]undecane, 1 -oxa-4,8-diaza-spiro[5.5]undecane, 1 -thia-4,9-diaza-spiro[5.5]un- decane, 1 -thia-4,8-diaza-spiro[5.5]undecane, 4,8-diaza-spiro[2.6]nonane, 5,8-diaza- spiro[3.6]decane, 2-aza-spiro[5.5]undec-7-ene, 6,9-diaza-spiro[4.6]undecane, 4-aza- dispiro[2.1.4.2]undecane, 4, 10-diaza-dispiro[2.2.3.2]undecane and 4,1 1 -diaza- dispiro[2.2.4.2]dodecane:


Exemplary heterocycloalkyls, in which a ring is fused to one ring of a bicyclic spiro system, are derived from, but not limited to, the following ring systems: octahydro- spiro[cyclopentane-1 ,2'(3'H)-quinoxalin], 1 ',4'-dihydro-spiro[cyclopentane-1 ,2'(3'H)- quinoxalin], 1 ',2',4,5-tetrahydro-spiro[furan-3(2H),3'-[3H]indol], 1 ,3-dihydro-spiro[in- dene-2,2'-piperazine], 2,3-dihydro-spiro[1 H-indene-1 ,4'-piperidin] and 1 ,2-dihydro-5- spiro[3H-indole-3,4'-piperidin]:
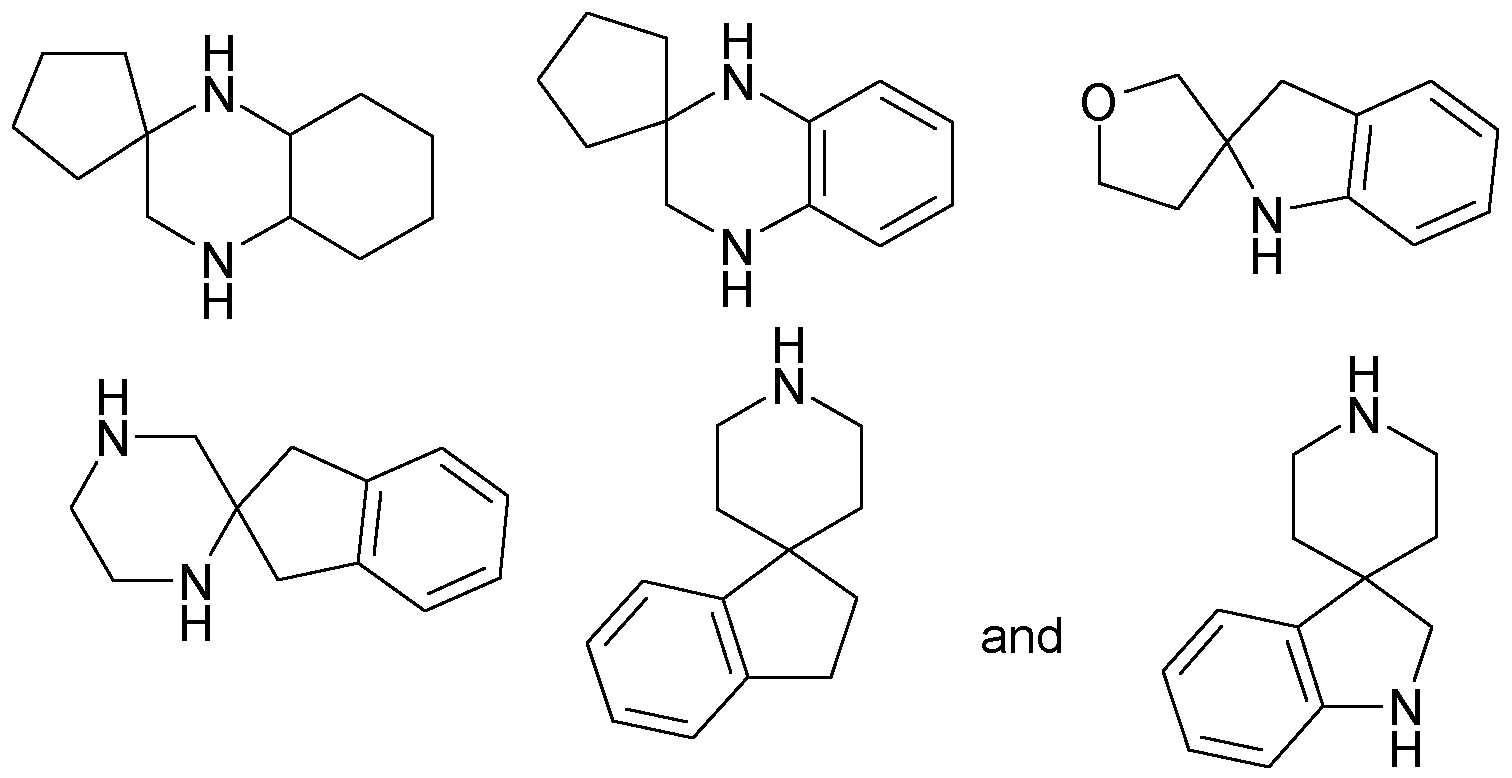
In one embodiment heterocycloalkyls, in which a ring is fused to one ring of a bicyclic spiro system, are derived from 3-dihydro-spiro[indene-2,2'-piperazine], 2,3-dihydro- -indene-1 ,4'-piperidin] or 1 ,2-dihydro-5-spiro[3H-indole-3,4'-piperidin]:

Exemplary non-fused or bridged bicyclic or tricyclic ring heterocycloalkyls are derived from, but not limited to, the following ring systems: 2-aza-bicyclo[2.2.1 ]heptane, 1 -aza- bicyclo[2.2.2]octane, 8-aza-bicyclo[3.2.1 ]octane, 3-aza-bicyclo[3.2.1 ]octane, 9-aza- bicyclo[3.3.1 ]nonane, 2,5-diaza-bicyclo[2.2.1 ]heptane, 2,5-diaza-bicyclo[2.2.2]octane, 3,8-diaza-bicyclo[3.2.1 ]octaneand 3,7-diaza-bicyclo[3.3.1 ]nonane:

The term "aryl", as used herein, refers to a radical derived from an aromatic hydrocarbon by removal of one hydrogen, such as phenyl or naphthyl (= naphthalenyl).
The term "heteroaryl" or "hetaryl" as used herein, refers to a radical derived from an aromatic mono- or bicyclic ring system, in which 1 , 2, 3, 4 or 5 carbon atoms are replaced by heteroatoms. The ring heteroatoms are generally chosen from N, 0 and S, wherein N includes ring nitrogen atoms which carry a hydrogen atom or a substituent as well as ring nitrogen atoms which do not carry a hydrogen atom or a substituent. Ring heteroatoms can be located in any position, provided that the heterocyclic system is stable and suitable as a subgroup for the desired purpose of the compound of the formula I such as use as a drug substance. Heteroaryl radicals are derived from 5- membered or 6-membered monocyclic rings or 8-membered, 9-membered or 10- membered bicyclic rings, in another embodiment 5-membered or 6-membered monocyclic rings or 9-membered or 10-membered bicyclic rings, in another
embodiment 5-membered or 6-membered monocyclic rings.
Exemplary heteroaryl systems are derived from, but not limited to, the following ring systems: pyrrole, furan, thiophene, imidazole, pyrazole, oxazole (= [1 ,3]oxazole), isoxazole (= [1 ,2]oxazole), thiazole (= [1 ,3]thiazole), isothiazole (= [1 ,2]thiazole),
[1 ,2,3]triazole, [1 ,2,4]triazole, [1 ,2,4]oxadiazole, [1 ,3,4]oxadiazole, [1 ,2,4]thiadiazole, [1 ,3,4]thiadiazole, tetrazole, pyridine, pyridazine, pyrimidine, pyrazine, [1 ,2,3]triazine, [1 ,2,4]triazine, [1 ,3,5]triazine, indole, isoindole, benzofuran, benzothiophene
[1 ,3]benzoxazole, [1 ,3]benzothiazole, benzoimidazolejndazole, quinoline, isoquinoline,
cinnoline, quinazoline, quinoxaline, phthalazine, different naphthyridines, e.g.
[1 ,8]naphthyridine, different thienopyridines, e.g. thieno[2,3-b]pyridine and purine:

Groups like phenyl, naphthyl (= naphthalenyl) and residues of aromatic heterocycles which are optionally substituted by one or more substituents, can be unsubstituted or substituted, for example by 1 , 2, 3, 4 or 5, or by 1 , 2, 3 or 4, or by 1 , 2 or 3, or by 1 or 2, or by 1 , identical or different substituents which can be located in any positions. Aromatic nitrogen heterocycles which in the parent ring system carry a hydrogen atom
on a ring nitrogen atom in a 5-membered ring, such as a pyrrole, imidazole, indole or benzoimidazole ring, for example, can be substituted on ring carbon atoms and/or on such ring nitrogen atoms. In one embodiment of the invention, substituents on such ring nitrogen atoms are chosen from (Ci-C4)-alkyl groups, i.e. such ring nitrogen atoms in aromatic heterocycles carry a hydrogen atom or a (Ci-C4)-alkyl substituent. When it is stated with respect to ring nitrogen atoms in aromatic heterocycles and any other heterocycles that they can carry a hydrogen atom or a substituent, such ring nitrogen atoms either carry a hydrogen atom or a substituent or they do not carry a hydrogen atom or substituent. Ring nitrogen atoms which carry a hydrogen atom or a
substituent, occur in a nitrogen-containing aromatic 5-membered ring as is present in pyrrole, imidazole, indole or benzoimidazole, for example, and in a non-aromatic ring including a saturated ring. Ring nitrogen atoms which do not carry a hydrogen atom or a substituent unless they are present in positively charged form, including any further ring nitrogen atoms in addition to ring nitrogen atoms which carry a hydrogen atom or a substituent, occur in an aromatic ring as is present in thiazole, imidazole, pyridine or benzoimidazole, for example, and in a non-aromatic ring in which they are part of a double bond, and they occur as ring nitrogen atoms via which a ring is bonded.
Suitable ring nitrogen atoms in aromatic heterocycles in the compounds of the formula I, such as the ring nitrogen atom in a pyridine ring or a quinoline ring, can in general also be present as N-oxide or as quaternary salt, for example as N-(Ci-C4)-alkyl salt such as N-methyl salt, wherein in one embodiment of the invention the counter anion in such quaternary salt is a physiologically acceptable anion which is derived from an acid that forms a physiologically acceptable salt. In monosubstituted phenyl groups, the substituent can be located in the 2-position, the 3-position or the 4-position. In disubstituted phenyl groups, the substituents can be located in 2, 3-position, 2, 4-position, 2,5-position, 2,6-position, 3, 4-position or 3,5- position. In trisubstituted phenyl groups, the substituents can be located in 2,3,4- position, 2,3,5-position, 2,3,6-position, 2,4,5-position, 2,4,6-position or 3,4,5-position. Naphthyl can be 1 -naphthyl (= naphthalen-1 -yl) or 2-naphthyl (= naphthalen-2-yl). In monosubstituted 1 -naphthyl groups, the substituent can be located in the 2-, 3-, 4-, 5-, 6-, 7- or 8-position. In monosubstituted 2-naphthyl groups, the substituent can be
located in the 1 -, 3-, 4-, 5-, 6-, 7- or 8-position. In disubstituted naphthyl groups, the substituents can likewise be located in any positions both in the ring via which the naphthyl group is bonded and/or in the other ring. Ring heteroatoms can be located in any positions, provided that the heterocyclic system is known in the art and is stable and suitable as a subgroup for the desired purpose of the compound of the formula I such as use as a drug substance. In one embodiment of the invention, two ring oxygen atoms cannot be present in adjacent ring positions of any heterocycle, in another embodiment two ring heteroatoms chosen from oxygen and sulfur cannot be present in adjacent ring positions of any heterocycle. Substituents on heterocyclic groups can be located in any positions. For example, in a pyridin-2-yl group substituents can be located in the 3-position and/or 4-position and/or
5- position and/or 6-position, in a pyridin-3-yl group substituent can be located in the 2- position and/or 4-position and/or 5-position and/or 6-position, in a pyridin-4-yl group substituents can be located in the 2-position and/or 3-position and/or 5-position and/or
6- position.
Halogen is fluorine, chlorine, bromine or iodine. In one embodiment of the invention, any halogen in a compound of the formula I is independently of any other halogen chosen from fluorine, chlorine and bromine, in another embodiment from fluorine and chlorine, and in yet another embodiment it is fluorine.
When an oxo group is bonded to a carbon atom, it replaces two hydrogen atoms on a carbon atom of the parent system. Thus, if a CH2 group in a chain or a ring is substituted by oxo, i.e. by a doubly bonded oxygen atom, it becomes a CO group.
Evidently, an oxo group cannot occur as a substituent on a carbon atom in an aromatic ring such as in a phenyl group, for example. When a ring sulfur atom in a heterocyclic group can carry one or two oxo groups, it is a non-oxidized sulfur atom S in case it does not carry any oxo group, or it is an S(O) group (= sulfoxide group, S-oxide group) in case it carries one oxo group, or it is an S(O)2 group (= sulfone group, S,S-dioxide group) in case it carries two oxo groups.
The present invention includes all stereoisomeric forms of the compounds of the formula I and their salts and solvates. With respect to each chiral center, independently of any other chiral center, the compounds of the formula I can be present in S configuration or substantially S configuration, or in R configuration or substantially R configuration, or as a mixture of the S isomer and the R isomer in any ratio. The invention includes all possible enantiomers and diastereomers and mixtures of two or more stereoisomers, for example mixtures of enantiomers and/or diastereomers, in all ratios. Thus, compounds according to the invention which can exist as enantiomers can be present in enantiomerically pure form, both as levorotatory and as
dextrorotatory antipodes, and in the form of mixtures of the two enantiomers in all ratios including racemates. In the case of a E/Z isomerism, or cis/trans isomerism, for example on double bonds or rings such as cycloalkyl rings, the invention includes both the E form and Z form, or the cis form and the trans form, as well as mixtures of these forms in all ratios. In one embodiment of the invention, a compound which can occur in two or more stereoisomeric forms is a pure, or substantially pure, individual
stereoisomer. The preparation of individual stereoisomers can be carried out, for example, by separation of a mixture of isomers by customary methods, for example by chromatography or crystallization, by the use of stereochemical^ uniform starting materials in the synthesis, or by stereoselective synthesis. Optionally, a derivatization can be carried out before a separation of stereoisomers. The separation of a mixture of stereoisomers can be carried out at the stage of the compound of the formula I or at the stage of a starting material or an intermediate during the synthesis. The present invention also includes all tautomeric forms of the compounds of the formula I and their salts and solvates.
In case the compounds of the formula I contain one or more acidic and/or basic groups, i.e. salt-forming groups, the invention also includes their corresponding physiologically or toxicologically acceptable salts, i.e. non-toxic salts, in particular their pharmaceutically acceptable salts.
The present invention furthermore includes all solvates of compounds of the formula I, for example hydrates or adducts with alcohols such as (Ci-C4)-alkanols, active
metabolites of the compounds of the formula I, and also prodrugs and derivatives of the compounds of the formula I which in vitro may not necessarily exhibit pharmacological activity but which in vivo are converted into pharmacologically active compounds, for example esters or amides of carboxylic acid groups.
The compounds of the present invention, PKC inhibitors, can be widely combined with other pharmacologically active compounds, e.g., with all antihypertensives and nephroprotectives, mentioned in the Rote Liste 201 1 , antidiabetics mentioned in the Rote Liste 201 1 , chapter 12; all weight-reducing agents/appetite suppressants mentioned in the Rote Liste 201 1 , chapter 1 ; all diuretics mentioned in the Rote Liste 201 1 , chapter 36; all lipid-lowering agents mentioned in the Rote Liste 201 1 , chapter 58. They can be combined with the inventive compound of the formula I, especially for a synergistic improvement in action. The active ingredient combination can be administered either by separate administration of the active ingredients to the patient or in the form of combination products in which a plurality of active ingredients are present in one pharmaceutical preparation. When the active ingredients are
administered by separate administration of the active ingredients, this can be done simultaneously or successively. Most of the active ingredients mentioned hereinafter are disclosed in the USP Dictionary of USAN and International Drug Names, US Pharmacopeia, Rockville 2006.
Antidiabetics include insulin and insulin derivatives, for example Lantus® (see www.lantus.com) or HMR 1964 or Levemir® (insulin detemir), Humalog(R) (Insulin Lispro), insulin degludec, insulin aspart, polyethylene glycosidized (PEGylated) Insulin Lispro as described in WO2009152128, Humulin(R), VIAject™, SuliXen(R), VIAject™ or those as described in WO2005005477 (Novo Nordisk), fast-acting insulins (see US 6,221 ,633), inhalable insulins, for example Exubera®, Nasulin™, or oral insulins, for example IN-105 (Nobex) or Oral-lyn™ (Generex Biotechnology), or Technosphere(R) insulin (MannKind) or Cobalamin™ oral insulin or ORMD-0801 or insulins or insulin precursors as described in WO2007128815, WO2007128817, WO2008034881 , WO200804971 1 , WO2008145721 , WO20090341 17, WO2009060071 ,
WO2009133099 or insulins which can be administered transdermally; additionally
included are also those insulin derivatives which are bonded to albumin by a
bifunctional linker, as described, for example, in WO2009121884;
GLP-1 derivatives and GLP-1 agonists, for example exenatide or specific formulations thereof, as described, for example, in WO2008061355, WO2009080024,
WO2009080032, liraglutide, taspoglutide (R-1583), albiglutide, lixisenatide or those which have been disclosed in WO 98/08871 , WO2005027978, WO200603781 1 , WO2006037810 by Novo Nordisk A/S, in WO 01/04156 by Zealand or in WO 00/34331 by Beaufour-lpsen, pramlintide acetate (Symlin; Amylin Pharmaceuticals), inhalable GLP-1 (MKC-253 from MannKind) AVE-0010, BIM-51077 (R-1583, ITM-077), PC- DAC:exendin-4 (an exendin-4 analog which is bonded covalently to recombinant human albumin), biotinylated exendin (WO2009107900), a specific formulation of exendin-4 as described in US2009238879, CVX-73, CVX-98 and CVx-96 (GLP-1 analogs which are bonded covalently to a monoclonal antibody which has specific binding sites for the GLP-1 peptide), CNTO-736 (a GLP-1 analog which is bonded to a domain which includes the Fc portion of an antibody), PGC-GLP-1 (GLP-1 bonded to a nanocarrier), agonists or modulators, as described, for example, in D. Chen et al., Proc. Natl. Acad. Sci. USA 104 (2007) 943, those as described in WO2006124529, WO2007124461 , WO2008062457, WO2008082274, WO2008101017,
WO2008081418, WO20081 12939, WO20081 12941 , WO20081 13601 ,
WO20081 16294, WO20081 16648, WO20081 19238, WO2008148839,
US2008299096, WO2008152403, WO2009030738, WO2009030771 ,
WO2009030774, WO2009035540, WO2009058734, WO20091 1 1700,
WO2009125424, WO2009129696, WO2009149148, peptides, for example obinepitide (TM-30338), orally active GLP-1 analogs (e.g. NN9924 from Novo Nordisk), amylin receptor agonists, as described, for example, in WO2007104789, WO20090341 19, analogs of the human GLP-1 , as described in WO2007120899, WO2008022015, WO2008056726, chimeric pegylated peptides containing both GLP-1 and glucagon residues, as described, for example, in WO2008101017, WO2009155257,
WO2009155258, glycosylated GLP-1 derivatives as described in WO2009153960, and orally active hypoglycemic ingredients.
Antidiabetics also include gastrin analogs, for example TT-223.
Antidiabetics additionally include poly- or monoclonal antibodies directed, for example, against interleukin 1 beta (IL-1 β), for example XOMA-052. Antidiabetics additionally include peptides which can bind to the human pro-islet peptide (HIP) receptor, as described, for example, in WO2009049222.
Antidiabetics also include agonists of the glucose-dependent insulinotropic polypeptide (GIP) receptor, as described, for example, in WO2006121860.
Antidiabetics also include the glucose-dependent insulinotropic polypeptide (GIP), and also analogous compounds, as described, for example, in WO2008021560,
WO2010016935, WO2010016936, WO2010016938, WO2010016940,
WO2010016944.
Additionally included are analogs and theivatives of human pancreatic polypeptide, as described, for example, in WO2009007714. Antidiabetics additionally include encapsulated insulin-producing porcine cells, for example DiabeCell(R).
Antidiabetics also include analogs and theivatives of fibroblast growth factor 21 (FGF- 21 ), as described, for example, in WO2009149171 , WO2010006214.
The orally active hypoglycemic ingredients preferably include sulfonylureas, biguanidines,
meglitinides,
oxadiazolidinediones,
thiazolidinediones,
PPAR and RXR modulators,
glucosidase inhibitors,
inhibitors of glycogen phosphorylase,
glucagon receptor antagonists,
glucokinase activators,
inhibitors of fructose 1 ,6-bisphosphatase,
modulators of glucose transporter 4 (GLUT4),
inhibitors of glutamine:fructose-6-phosphate amidotransferase (GFAT),
GLP-1 agonists,
potassium channel openers, for example pinacidil, cromakalim, diazoxide, diazoxide choline salt, or those as described in R. D. Carr et al., Diabetes 52, 2003, 2513.2518, in J. B. Hansen et al., Current Medicinal Chemistry 11 , 2004, 1595-1615, in T. M.
Tagmose et al., J. Med. Chem. 47, 2004, 3202-321 1 or in M. J. Coghlan et al., J. Med. Chem. 44, 2001, 1627-1653, or those which have been disclosed in WO 97/26265 and WO 99/03861 by Novo Nordisk A/S,
active ingredients which act on the ATP-dependent potassium channel of the beta cells,
inhibitors of dipeptidyl peptidase-IV (DPP-IV),
insulin sensitizers,
inhibitors of liver enzymes involved in stimulating gluconeogenesis and/or
glycogenolysis,
modulators of glucose uptake, of glucose transport and of glucose reabsorption, modulators of sodium-dependent glucose transporter 1 or 2 (SGLT1 , SGLT2), inhibitors of 1 1 -beta-hydroxysteroid dehydrogenase-1 (1 Ι β-HSDI ),
inhibitors of protein tyrosine phosphatase-1 B (PTP-1 B),
nicotinic acid receptor agonists,
inhibitors of hormone-sensitive or endothelial lipases,
inhibitors of acetyl-CoA carboxylase (ACC1 and/or ACC2) or
inhibitors of GSK-3 beta.
Also included are compounds which modify the lipid metabolism, such as active antihyperlipidemic ingredients and active antilipidemic ingredients,
HMG-CoA reductase inhibitors,
farnesoid X receptor (FXR) modulators,
fibrates,
cholesterol reabsorption inhibitors,
CETP inhibitors,
bile acid absorption inhibitors,
MTP inhibitors,
estrogen receptor gamma agonists (ERR γ agonists),
sigma-1 receptor antagonists,
antagonists of the somatostatin 5 receptor (SST5 receptor);
compounds which reduce food intake, and
compounds which increase thermogenesis.
In one embodiment of the invention, the compound of the formula I is administered in combination with insulin.
In another embodiment of the invention, the compound of the formula I is administered in combination with an insulin sensitizer, for example PN-2034 or ISIS-1 13715.
In one embodiment, the compound of the formula I is administered in combination with an active ingredient which acts on the ATP-dependent potassium channel of the beta cells, for example sulfonylureas, for example tolbutamide, glibenclamide, glipizide, gliclazide or glimepiride, or those formulations as described, for example, in
EP2103302.
In one embodiment, the compound of the formula I is administered in combination with a tablet which comprises both glimepiride, which is released rapidly, and metformin, which is released over a longer period (as described, for example, in US2007264331 , WO2008050987, WO2008062273).
In one embodiment, the compound of the formula I is administered in combination with a biguanide, for example metformin or one of its salts.
In a further embodiment, the compound of the formula I is administered in combination with a guanidine, for example benzylguanidine or one of its salts, or those guanidines as described in WO2009087395.
In another embodiment, the compound of the formula I is administered in combination with a meglitinide, for example repaglinide, nateglinide or mitiglinide.
In a further embodiment, the compound of the formula I is administered with a combination of mitiglinide with a glitazone, e.g. pioglitazone hydrochloride.
In a further embodiment, the compound of the formula I is administered with a combination of mitiglinide with an alpha-glucosidase inhibitor.
In a further embodiment, the compound of the formula I is administered in combination with antidiabetic compounds, as described in WO2007095462, WO2007101060, WO2007105650.
In a further embodiment, the compound of the formula I is administered in combination with antihypoglycemic compounds, as described in WO2007137008, WO2008020607. In one embodiment, the compound of the formula I is administered in combination with a thiazolidinedione, for example troglitazone, ciglitazone, pioglitazone, rosiglitazone or the compounds disclosed in WO 97/41097 by Dr. Reddy's Research Foundation, especially 5-[[4-[(3,4-dihydro-3-methyl-4-oxo-2-quinazolinylmethoxy]phenyl]methyl]- 2,4-thiazolidinedione. In one embodiment of the invention, the compound of the formula I is administered in combination with a PPAR gamma agonist, for example rosiglitazone, pioglitazone, JTT-501 , Gl 262570, R-483, CS-01 1 (rivoglitazone), DRL-17564, DRF-2593
(balaglitazone), INT-131 , T-2384, or those as described in WO2005086904,
WO2007060992, WO2007100027, WO2007103252, WO2007122970,
WO2007138485, WO2008006319, WO2008006969, WO2008010238,
WO2008017398, WO2008028188, WO2008066356, WO2008084303,
WO2008089461 -WO2008089464, WO2008093639, WO2008096769,
WO2008096820, WO2008096829, US2008194617, WO2008099944,
WO2008108602, WO2008109334, WO20081 10062, WO2008126731 ,
WO2008126732, WO2008137105, WO2009005672, WO2009038681 , WO2009046606, WO2009080821 , WO2009083526, WO2009102226,
WO2009128558, WO2009139340. In one embodiment of the invention, the compound of the formula I is administered in combination with Competact™, a solid combination of pioglitazone hydrochloride with metformin hydrochloride.
In one embodiment of the invention, the compound of the formula I is administered in combination with Tandemact™, a solid combination of pioglitazone with glimepiride.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a solid combination of pioglitazone hydrochloride with an angiotensin II agonist, for example TAK-536.
In one embodiment of the invention, the compound of the formula I is administered in combination with a PPAR alpha agonist or mixed PPAR alpha/PPAR delta agonist, for example GW9578, GW-590735, K-1 1 1 , LY-674, KRP-101 , DRF-10945, LY-518674, CP-900691 , BMS-687453, BMS-71 1939, or those as described in WO2001040207, WO2002096894, WO2005097076, WO2007056771 , WO2007087448,
WO2007089667, WO2007089557, WO2007102515, WO2007103252, JP2007246474, WO20071 18963, WO20071 18964, WO2007126043, WO2008006043,
WO2008006044, WO2008012470, WO2008035359, WO2008087365,
WO2008087366, WO2008087367, WO20081 17982, JP2009023975, WO2009033561 , WO2009047240, WO2009072581 , WO2009080248, WO2009080242,
WO2009149819, WO2009149820, WO2009147121 , WO2009153496,
WO2010008299, WO2010014771 .
In one embodiment of the invention, the compound of the formula I is administered in combination with a mixed PPAR alpha/gamma agonist, for example naveglitazar, aleglitazar, LY-510929, ONO-5129, E-3030, AVE 8042, AVE 8134, AVE 0847, AVE 0897, CKD-501 (lobeglitazone sulfate), MBX-213, KY-201 , BMS-759509, or as
described in WO 00/64888, WO 00/64876, WO03/020269, WO2004024726,
WO2007099553, US2007276041 , WO2007085135, WO2007085136,
WO2007141423, WO2008016175, WO2008053331 , WO2008109697,
WO2008109700, WO2008108735, WO2009026657, WO2009026658,
WO2009149819, WO2009149820 or in J.P.Berger et al., TRENDS in Pharmacological Sciences 28(5), 244-251 , 2005.
In one embodiment of the invention, the compound of the formula I is administered in combination with a PPAR delta agonist, for example GW-501516, or as described in WO2006059744, WO2006084176, WO2006029699, WO2007039172- WO2007039178, WO2007071766, WO2007101864, US2007244094,
WO20071 19887, WO2007141423, US2008004281 , WO2008016175,
WO2008066356, WO200807131 1 , WO2008084962, US2008176861 ,
WO2009012650, US2009137671 , WO2009080223, WO2009149819,
WO2009149820, WO2010000353.
In one embodiment of the invention, the compound of the formula I is administered in combination with a pan-SPPARM (selective PPAR modulator alpha, gamma, delta), for example GFT-505, indeglitazar, or those as described in WO2008035359,
WO2009072581 .
In one embodiment, the compound of the formula I is administered in combination with metaglidasen or with MBX-2044 or other partial PPAR gamma agonists/antagonists. In one embodiment, the compound of the formula I is administered in combination with an a-glucosidase inhibitor, for example miglitol or acarbose, or those as described, for example, in WO20071 14532, WO2007140230, US2007287674, US2008103201 , WO2008065796, WO2008082017, US2009076129.
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of glycogen phosphorylase, for example PSN-357 or FR-258900, or those as described in WO2003084922, WO2004007455, WO2005073229-31 ,
WO2005067932, WO2008062739, WO2008099000, WO20081 13760, WO20090161 18, WO20090161 19, WO2009030715, WO2009045830,
WO2009045831 , WO2009127723.
In another embodiment, the compound of the formula I is administered in combination with an inhibitor of the interaction of liver glycogen phosphorylase with the protein PPP1 R3 (GL subunit of glycogen-associated protein phosphatase 1 (PP1 )), as described, for example, in WO2009030715.
In one embodiment, the compound of the formula I is administered in combination with glucagon receptor antagonists, for example A-770077 or NNC-25-2504 or as described in WO2004100875, WO2005065680, WO2006086488, WO2007047177, WO2007106181 , WO20071 1 1864, WO2007120270, WO2007120284,
WO2007123581 , WO2007136577, WO2008042223, WO2008098244,
WO2009057784, WO2009058662, WO2009058734, WO20091 10520,
WO2009120530, WO2009140342, WO2010019828.
In a further embodiment, the compound of the formula I is administered in combination with an antisense compound, e.g. ISIS-325568, which inhibits the production of the glucagon receptor.
In one embodiment, the compound of the formula I is administered in combination with activators of glucokinase, for example LY-2121260 (WO2004063179), PSN-105, PSN- 1 10, GKA-50, or those as described, for example, in WO2004072031 ,
WO2004072066, WO2005080360, WO2005044801 , WO2006016194,
WO2006058923, WO20061 12549, WO2006125972, WO2007017549,
WO2007017649, WO2007007910, WO2007007040-42, WO2007006760-61 ,
WO2007006814, WO2007007886, WO2007028135, WO2007031739,
WO2007041365, WO2007041366, WO2007037534, WO2007043638,
WO2007053345, WO2007051846, WO2007051845, WO2007053765,
WO2007051847, WO2007061923, WO2007075847, WO2007089512,
WO2007104034, WO20071 17381 , WO2007122482, WO2007125103,
WO2007125105 US2007281942, WO2008005914, WO2008005964,
WO2008043701 WO2008044777 , WO2008047821 , US2008096877,
WO20080501 17 WO2008050101 , WO2008059625, US2008146625,
WO2008078674 WO2008079787 , WO2008084043, WO2008084044,
WO2008084872 WO2008089892 , WO2008091770, WO2008075073,
WO2008084043 WO2008084044 , WO2008084872, WO2008084873,
WO2008089892 WO2008091770 , JP2008189659, WO2008104994, WO20081 1 1473, WO20081 16107 WO20081 18718 , WO2008120754, US2008280875,
WO2008136428 WO2008136444 , WO2008149382, WO2008154563,
WO2008156174 WO2008156757 , US2009030046, WO2009018065,
WO2009023718 WO2009039944 , WO2009042435, WO2009046784,
WO2009046802 WO2009047798 , WO2009063821 , WO2009081782,
WO2009082152 WO2009083553 , WO2009091014, US2009181981 ,
WO2009092432 WO2009099080 , WO2009106203, WO2009106209,
WO2009109270 WO2009125873 , WO2009127544, WO2009127546,
WO2009128481 WO2009133687 , WO2009140624, WO2010013161 ,
WO2010015849 WO2010018800
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of gluconeogenesis, as described, for example, in FR-225654,
WO2008053446.
In one embodiment, the compound of the formula I is administered in combination with inhibitors of fructose 1 ,6-bisphosphatase (FBPase), for example MB-07729, CS-917 (MB-06322) or MB-07803, or those as described in WO2006023515, WO2006104030, WO2007014619, WO2007137962, WO2008019309, WO2008037628,
WO2009012039, EP2058308, WO2009068467, WO2009068468.
In one embodiment, the compound of the formula I is administered in combination with modulators of glucose transporter 4 (GLUT4), for example KST-48 (D.-O. Lee et al.: Arzneim.-Forsch. Drug Res. 54 (12), 835 (2004)).
In one embodiment, the compound of the formula I is administered in combination with inhibitors of glutamine:fructose-6-phosphate amidotransferase (GFAT), as described, for example, in WO2004101528. In one embodiment, the compound of the formula I is administered in combination with inhibitors of dipeptidyl peptidase-IV (DPP-IV), for example vildagliptin (LAF-237), sitagliptin (MK-0431 ), sitagliptin phosphate, saxagliptin (BMS-4771 18), GSK-823093, PSN-9301 , SYR-322, SYR-619, TA-6666, TS-021 , GRC-8200 (melogliptin), GW- 825964X, KRP-104, DP-893, ABT-341 , ABT-279 or another salt thereof, S-40010, S- 40755, PF-00734200, BI-1356, PHX-1 149, DSP-7238, alogliptin benzoate, linagliptin, melogliptin, carmegliptin, or those compounds as described in WO2003074500, WO2003106456, WO2004037169, WO200450658, WO2005037828, WO2005058901 , WO2005012312, WO2005/012308, WO2006039325, WO2006058064,
WO2006015691 , WO2006015701 , WO2006015699, WO2006015700,
WO20060181 17, WO2006099943, WO2006099941 , JP2006160733, WO2006071752, WO2006065826, WO2006078676, WO2006073167, WO2006068163,
WO2006085685, WO2006090915, WO2006104356, WO2006127530,
WO20061 1 1261 , US2006890898, US2006803357, US2006303661 , WO2007015767 (LY-2463665), WO2007024993, WO2007029086, WO2007063928, WO2007070434, WO2007071738, WO2007071576, WO2007077508, WO2007087231 ,
WO2007097931 , WO2007099385, WO2007100374, WO20071 12347,
WO20071 12669, WO20071 13226, WO20071 13634, WO20071 15821 ,
WO20071 16092, US2007259900, EP1852108, US2007270492, WO2007126745, WO2007136603, WO2007142253, WO2007148185, WO2008017670,
US2008051452, WO2008027273, WO2008028662, WO2008029217, JP2008031064, JP2008063256, WO2008033851 , WO2008040974, WO2008040995, WO2008060488, WO2008064107, WO2008066070, WO2008077597, JP2008156318, WO2008087560, WO2008089636, WO2008093960, WO2008096841 , WO2008101953,
WO20081 18848, WO20081 19005, WO20081 19208, WO2008120813,
WO2008121506, WO2008130151 , WO2008131 149, WO2009003681 ,
WO2009014676, WO2009025784, WO2009027276, WO2009037719,
WO2009068531 , WO2009070314, WO2009065298, WO2009082134,
WO2009082881 , WO2009084497, WO2009093269, WO2009099171 , WO2009099172, WO20091 1 1239, WO20091 13423, WO20091 16067,
US2009247532, WO2010000469, WO2010015664. In one embodiment, the compound of the formula I is administered in combination with Janumet™, a solid combination of sitagliptin phosphate with metformin hydrochloride.
In one embodiment, the compound of the formula I is administered in combination with Eucreas(R), a solid combination of vildagliptin with metformin hydrochloride.
In a further embodiment, the compound of the formula I is administered in combination with a solid combination of alogliptin benzoate with pioglitazone.
In one embodiment, the compound of the formula I is administered in combination with a solid combination of a salt of sitagliptin with metformin hydrochloride.
In one embodiment, the compound of the formula I is administered in combination with a combination of a DPP-IV inhibitor with omega-3 fatty acids or omega-3 fatty acid esters, as described, for example, in WO2007128801 .
In one embodiment, the compound of the formula I is administered in combination with a combination of a DPP-IV inhibitor with metformin hydrochloride, as described, for example, in WO2009121945.
In one embodiment, the compound of the formula I is administered in combination with a combination of a DPP-IV inhibitor with a GPR-1 19 agonist, as described, for example, in WO2009123992.
In one embodiment, the compound of the formula I is administered in combination with a combination of a DPP-IV inhibitor with miglitol, as described, for example, in
WO2009139362.
In one embodiment, the compound of the formula I is administered in combination with a solid combination of a salt of sitagliptin with metformin hydrochloride.
In one embodiment, the compound of the formula I is administered in combination with a solid combination of alopliptin benzoate with pioglitazone hydrochloride.
In one embodiment, the compound of the formula I is administered in combination with a substance which enhances insulin secretion, for example KCP-265
(WO2003097064), or those as described in WO2007026761 , WO2008045484, US2008194617, WO2009109259, WO2009109341 .
In one embodiment, the compound of the formula I is administered in combination with agonists of the glucose-dependent insulinotropic receptor (GDIR), for example APD- 668.
In one embodiment of the invention, the compound of the formula I is administered in combination with an ATP citrate lyase inhibitor, for example SB-204990.
In one embodiment, the compound of the formula I is administered in combination with modulators of the sodium-dependent glucose transporter 1 and/or 2 (SGLT1 , SGLT2), for example KGA-2727, T-1095, SGL-0010, AVE 2268, SAR 7226, SGL-5083, SGL- 5085, SGL-5094, ISIS-388626, sergliflozin, dapagliflozin or remogliflozin etabonate, canagliflozin, or as described, for example, in WO2004007517, WO200452903, WO200452902, PCT/EP2005/005959, WO2005085237, JP2004359630,
WO2005121 161 , WO2006018150, WO2006035796, WO2006062224,
WO2006058597, WO2006073197, WO2006080577, WO2006087997,
WO2006108842, WO2007000445, WO2007014895, WO2007080170,
WO2007093610, WO20071261 17, WO2007128480, WO2007129668,
US2007275907, WO20071361 16, WO2007143316, WO2007147478,
WO2008001864, WO2008002824, WO2008013277, WO2008013280,
WO2008013321 , WO2008013322, WO2008016132, WO200802001 1 , JP2008031 161 , WO2008034859, WO2008042688, WO2008044762, WO2008046497,
WO2008049923, WO2008055870, WO2008055940, WO2008069327,
WO2008070609, WO2008071288, WO2008072726, WO2008083200, WO2008090209, WO2008090210, WO2008101586, WO2008101939,
WO20081 16179, WO20081 16195, US2008242596, US2008287529, WO2009026537, WO2009049731 , WO2009076550, WO2009084531 , WO2009096503,
WO2009100936, WO2009121939, WO2009124638, WO2009128421 ,
WO2009135673, WO2010009197, WO2010018435, WO2010018438 or by A. L.
Handlon in Expert Opin. Ther. Patents (2005) 15(1 1 ), 1531 -1540.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a solid combination of an SGLT inhibitor with a DPP-IV inhibitor, as described in WO2009091082.
In one embodiment, the compound of the formula I is administered in combination with a stimulator of glucose transport, as described, for example, in WO2008136392, WO2008136393.
In one embodiment, the compound of the formula I is administered in combination with inhibitors of 1 1 -beta-hydroxysteroid dehydrogenase 1 (1 Ι β-HSDI ), for example BVT- 2733, JNJ-25918646, INCB-13739, INCB-20817, DIO-92 ((-)-ketoconazole) or those as described, for example, in WO200190090-94, WO200343999, WO20041 12782, WO200344000, WO200344009, WO20041 12779, WO20041 13310, WO2004103980, WO20041 12784, WO2003065983, WO2003104207, WO2003104208,
WO2004106294, WO200401 1410, WO2004033427, WO2004041264,
WO2004037251 , WO2004056744, WO2004058730, WO2004065351 ,
WO2004089367, WO2004089380, WO2004089470-71 , WO2004089896,
WO2005016877, WO2005063247, WO2005097759, WO2006010546,
WO2006012227, WO2006012173, WO2006017542, WO2006034804,
WO2006040329, WO2006051662, WO2006048750, WO2006049952,
WO2006048331 , WO2006050908, WO2006024627, WO2006040329,
WO2006066109, WO2006074244, WO2006078006, WO2006106423,
WO2006132436, WO2006134481 , WO2006134467, WO2006135795,
WO2006136502, WO2006138508, WO2006138695, WO2006133926,
WO2007003521 WO2007007688 US2007066584, WO2007029021 ,
WO2007047625 WO200705181 1 WO2007051810 WO2007057768,
WO2007058346 WO2007061661 WO2007068330 WO2007070506,
WO2007087150 WO2007092435 WO2007089683 WO2007101270,
WO2007105753 WO2007107470 WO2007107550 WO20071 1 1921 ,
US2007207985, US2007208001 , WO20071 15935, WO20071 18185, WO200712241 1 , WO2007124329 WO2007124337 WO2007124254 WO2007127688,
WO2007127693 WO2007127704 WO2007127726 WO2007127763,
WO2007127765 WO2007127901 US2007270424, JP2007291075, WO2007130898, WO2007135427 WO2007139992 WO2007144394 WO2007145834.
WO2007145835 WO2007146761 WO2008000950 WO2008000951 ,
WO200800361 1 WO2008005910 WO2008006702 WO2008006703,
WO200801 1453 WO2008012532 WO2008024497 WO2008024892,
WO2008032164 WO2008034032 WO2008043544 WO2008044656,
WO2008046758 WO2008052638 WO2008053194 WO2008071 169,
WO2008074384 WO2008076336 WO2008076862 WO2008078725,
WO2008087654 WO2008088540 WO2008099145 WO2008101885,
WO2008101886 WO2008101907 WO2008101914 WO2008106128,
WO20081 10196 WO20081 19017 WO2008120655 WO2008127924,
WO2008130951 WO2008134221 WO2008142859 WO2008142986,
WO2008157752 WO2009001817 WO2009010416 WO2009017664,
WO2009020140 WO2009023180 WO2009023181 WO2009023664,
WO2009026422 WO2009038064 WO2009045753 WO2009056881 ,
WO2009059666 WO2009061498 WO2009063061 WO2009070497,
WO2009074789 WO2009075835 WO2009088997 WO2009090239,
WO2009094169 WO2009098501 WO2009100872 WO2009102428,
WO2009102460 WO2009102761 WO2009106817 WO2009108332,
WO20091 12691 WO20091 12845 WO20091 14173 WO20091 17109,
US2009264401 , WO20091 18473, WO2009131669, WO2009132986,
WO2009134384 WO2009134387 WO2009134392 WO2009134400,
WO2009135581 WO2009138386 WO2010006940 WO2010010157,
WO2010010174 WO201001 1917
In one embodiment, the compound of the formula I is administered in combination with inhibitors of protein tyrosine phosphatase-1 B (PTP-1 B), as described, for example, in WO2001 19830-31 , WO2001 17516, WO2004506446, WO2005012295,
WO20051 16003, WO20051 16003, WO2006007959, DE 10 2004 060542.4,
WO200700991 1 , WO2007028145, WO2007067612-615, WO2007081755,
WO20071 15058, US2008004325, WO2008033455, WO2008033931 ,
WO2008033932, WO2008033934, WO2008089581 , WO2008148744,
WO2009032321 , WO2009109999, WO2009109998.
In a further embodiment, the compound of the formula I is administered in combination with stimulators of tyrosine kinase B (Trk-B), as described, for example, in
WO2010014613. In a further embodiment, the compound of the formula I is administered in combination with beta 3 agonists (also called beta-3 adrenoceptor agonists), as described, for example, in Physiol. Behav. 2004 Sep 15;82(2-3):489-96, J Clin Invest (1998) 101 : 2387-93, Curr. Pharma. Des.2001 Sep;7(14): 1433-49., Bioorganic & Medicinal Chemistry Letters volume 14, number 13, July 5, 2004, pages 3525-3529 (BMS- 201620).
In one embodiment of the invention, the compound of the formula I is administered in combination with an agonist of GPR109A (HM74A receptor agonists; NAR agonists (nicotinic acid receptor agonists)), for example nicotinic acid or extended release niacin in conjunction with MK-0524A (laropiprant) or MK-0524, or those compounds as described in WO2004041274, WO2006045565, WO2006045564, WO2006069242, WO2006085108, WO20060851 12, WO20060851 13, WO2006124490,
WO20061 13150, WO2007002557, WO2007017261 , WO2007017262,
WO2007017265, WO2007015744, WO2007027532, WO2007092364,
WO2007120575, WO2007134986, WO2007150025, WO2007150026,
WO2008016968, WO2008051403, WO2008086949, WO2008091338,
WO2008097535, WO2008099448, US2008234277, WO2008127591.
In another embodiment of the invention, the compound of the formula I is administered in combination with a solid combination of niacin with simvastatin. In another embodiment of the invention, the compound of the formula I is administered in combination with nicotinic acid or "extended release niacin" in conjunction with MK- 0524A (laropiprant).
In a further embodiment of the invention, the compound of the formula I is
administered in combination with nicotinic acid or "extended release niacin" in conjunction with MK-0524A (laropiprant) and with simvastatin.
In one embodiment of the invention, the compound of the formula I is administered in combination with nicotinic acid or another nicotinic acid receptor agonist and a prostaglandin DP receptor antagonist, for example those as described in
WO2008039882.
In another embodiment of the invention, the compound of the formula I is administered in combination with a solid combination of niacin with meloxicam, as described, for example, in WO2009149056.
In another embodiment of the invention, the compound of the formula I is administered in combination with an agonist of GPR1 16, as described, for example, in
WO2006067531 , WO2006067532.
In one embodiment, the compound of the formula I is administered in combination with modulators of GPR40, as described, for example, in WO2007013689,
WO2007033002, WO2007106469, US2007265332, WO2007123225,
WO2007131619, WO2007131620, WO2007131621 , US2007265332,
WO2007131622, WO2007136572, WO2008001931 , WO2008030520,
WO2008030618, WO2008054674, WO2008054675, WO2008066097,
US2008176912, WO2008130514, WO2009038204, WO2009039942,
WO2009039943, WO2009048527, WO2009054479, WO2009058237, WO20091 1 1056, WO2010012650.
In one embodiment, the compound of the formula I is administered in combination with modulators of GPR1 19 (G-protein-coupled glucose-dependent insulinotropic receptor), for example PSN-1 19-1 , PSN-821 , PSN-1 19-2, MBX-2982 or those as described, for example, in WO2004065380, WO2005061489 (PSN-632408), WO2006083491 , WO2007003960-62 and WO2007003964, WO2007035355, WO20071 16229,
WO20071 16230, WO2008005569, WO2008005576, WO2008008887,
WO2008008895, WO2008025798, WO2008025799, WO2008025800,
WO2008070692, WO2008076243, WO200807692, WO2008081204, WO2008081205, WO2008081206, WO2008081207, WO2008081208, WO2008083238,
WO2008085316, WO2008109702, WO2008130581 , WO2008130584,
WO2008130615, WO2008137435, WO2008137436, WO2009012275,
WO2009012277, WO2009014910, WO2009034388, WO2009038974,
WO2009050522, WO2009050523, WO2009055331 , WO2009105715,
WO2009105717, WO2009105722, WO2009106561 , WO2009106565,
WO20091 17421 , WO2009125434, WO2009126535, WO2009129036,
US2009286812, WO2009143049, WO2009150144, WO2010001 166,
WO2010004343, WO2010004344, WO2010004345, WO2010004346,
WO2010004347, WO2010004348, WO2010008739, WO2010006191 ,
WO2010009183, WO2010009195, WO2010009207, WO2010009208,
WO2010014593. In a further embodiment, the compound of the formula I is administered in combination with modulators of GPR120, as described, for example, in EP1688138,
WO2008066131 , WO2008066131 , WO2008103500, WO2008103501 ,
WO2008139879, WO2009038204, WO2009147990, WO2010008831. In another embodiment, the compound of the formula I is administered in combination with antagonists of GPR105, as described, for example, in WO2009000087,
WO2009070873.
In a further embodiment, the compound of the formula I is administered in combination with agonists of GPR43, for example ESN-282. In one embodiment, the compound of the formula I is administered in combination with inhibitors of hormone-sensitive lipase (HSL) and/or phospholipases, as described, for example, in WO2005073199, WO2006074957, WO2006087309, WO20061 1 1321 , WO2007042178, WO20071 19837, WO2008122352, WO2008122357,
WO2009009287.
In one embodiment, the compound of the formula I is administered in combination with inhibitors of endothelial lipase, as described, for example, in WO20071 10216.
In one embodiment, the compound of the formula I is administered in combination with a phospholipase A2 inhibitor, for example darapladib or A-002, or those as described in WO2008048866, WO20080488867, US2009062369.
In one embodiment, the compound of the formula I is administered in combination with myricitrin, a lipase inhibitor (WO20071 19827).
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of glycogen synthase kinase-3 beta (GSK-3 beta), as described, for example, in US2005222220, WO2005085230, WO20051 1 1018, WO2003078403, WO2004022544, WO2003106410, WO2005058908, US2005038023,
WO2005009997, US2005026984, WO2005000836, WO2004106343, EP1460075, WO2004014910, WO2003076442, WO2005087727, WO20040461 17,
WO20070731 17, WO2007083978, WO2007120102, WO2007122634,
WO2007125109, WO20071251 10, US2007281949, WO2008002244,
WO2008002245, WO2008016123, WO2008023239, WO2008044700,
WO2008056266, WO2008057940, WO2008077138, EP1939191 , EP1939192, WO2008078196, WO2008094992, WO20081 12642, WO20081 12651 ,
WO20081 13469, WO2008121063, WO2008121064, EP-1992620, EP-1992621 ,
EP1992624, EP-1992625, WO2008130312, WO2009007029, EP2020232,
WO2009017452, WO2009035634, WO2009035684, WO2009038385,
WO2009095787, WO2009095788, WO2009095789, WO2009095792,
WO2009145814, US2009291982, WO2009154697, WO2009156857,
WO2009156859, WO2009156860, WO2009156861 , WO2009156863,
WO2009156864, WO2009156865, WO2010013168, WO2010014794.
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of phosphoenolpyruvate carboxykinase (PEPCK), for example those as described in WO2004074288.
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of phosphoinositide kinase-3 (PI3K), for example those as described in WO2008027584, WO2008070150, WO2008125833, WO2008125835,
WO2008125839, WO2009010530, WO2009026345, WO2009071888,
WO2009071890, WO2009071895.
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of serum/glucocorticoid-regulated kinase (SGK), as described, for example, in WO2006072354, WO2007093264, WO2008009335, WO2008086854,
WO2008138448.
In one embodiment, the compound of the formula I is administered in combination with a modulator of the glucocorticoid receptor, as described, for example, in
WO2008057855, WO2008057856, WO2008057857, WO2008057859,
WO2008057862, WO2008059867, WO2008059866, WO2008059865,
WO2008070507, WO2008124665, WO2008124745, WO2008146871 ,
WO2009015067, WO2009040288, WO2009069736, WO2009149139. In one embodiment, the compound of the formula I is administered in combination with a modulator of the mineralocorticoid receptor (MR), for example drospirenone, or those as described in WO2008104306, WO20081 19918.
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of protein kinase C beta (PKC beta), for example ruboxistaurin, or those as described in WO2008096260, WO2008125945.
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of protein kinase D, for example doxazosin (WO2008088006).
In a further embodiment, the compound of the formula I is administered in combination with an activator/modulator of the AMP-activated protein kinase (AMPK), as described, for example, in WO2007062568, WO2008006432, WO2008016278, WO2008016730, WO2008020607, WO2008083124, WO2008136642, WO2009019445,
WO2009019446, WO2009019600, WO2009028891 , WO2009065131 ,
WO2009076631 , WO2009079921 , WO2009100130, WO2009124636,
WO2009135580, WO2009152909.
In one embodiment, the compound of the formula I is administered in combination with an inhibitor of ceramide kinase, as described, for example, in WO20071 12914, WO2007149865.
In a further embodiment, the compound of the formula I is administered in combination with an inhibitor of MAPK-interacting kinase 1 or 2 (MNK1 or 2), as described, for example, in WO2007104053, WO20071 15822, WO2008008547, WO2008075741 . In one embodiment, the compound of the formula I is administered in combination with inhibitors of "l-kappaB kinase" (IKK inhibitors), as described, for example, in
WO2001000610, WO2001030774, WO2004022057, WO2004022553,
WO2005097129, WO20051 13544, US2007244140, WO2008099072,
WO2008099073, WO2008099073, WO2008099074, WO2008099075,
WO2009056693, WO2009075277, WO2009089042, WO2009120801.
In another embodiment, the compound of the formula I is administered in combination with inhibitors of NF-kappaB (NFKB) activation, for example salsalate.
In a further embodiment, the compound of the formula I is administered in combination with inhibitors of ASK-1 (apoptosis signal-regulating kinase 1 ), as described, for example, in WO2008016131 , WO2009123986.
In one embodiment of the invention, the compound of the formula I is administered in combination with an HMG-CoA reductase inhibitor such as simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin, pitavastatin, L-659699, BMS-644950, NCX-6560, or those as described in US2007249583, WO2008083551 , WO2009054682.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a farnesoid X receptor (FXR) modulator, for example WAY-362450 or those as described in WO2003099821 , WO2005056554,
WO2007052843, WO2007070796, WO2007092751 , JP2007230909, WO2007095174, WO2007140174, WO2007140183, WO2008000643, WO2008002573,
WO2008025539, WO2008025540, JP2008214222, JP2008273847, WO2008157270, US20082991 18, US2008300235, WO2009005998, WO2009012125, WO2009027264, WO2009062874, US2009131409, US2009137554, US2009163552, WO2009127321 , EP2128158.
In another embodiment of the invention, the compound of the formula I is administered in combination with a ligand of the liver X receptor (LXR), as described, for example, in WO2007092965, WO2008041003, WO2008049047, WO2008065754,
WO2008073825, US2008242677, WO2009020683, US2009030082, WO2009021868, US2009069373, WO2009024550, WO2009040289, WO2009086123,
WO2009086129, WO2009086130, WO2009086138, WO2009107387,
US2009247587, WO2009133692, WO2008138438, WO2009144961 ,
WO2009150109.
In one embodiment of the invention, the compound of the formula I is administered in combination with a fibrate, for example fenofibrate, clofibrate, bezafibrate, or those as described in WO2008093655. In one embodiment of the invention, the compound of the formula I is administered in combination with fibrates, for example the choline salt of fenofibrate (SLV-348;
Trilipix™).
In one embodiment of the invention, the compound of the formula I is administered in combination with fibrates, for example the choline salt of fenofibrate (Trilipix™) and an HMG-CoA reductase inhibitor, for example rosuvastatin.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with bezafibrate and diflunisal.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a solid combination of fenofibrate or a salt thereof with simvastatin, rosuvastatin, fluvastatin, lovastatin, cerivastatin, pravastatin, pitavastatin or atorvastatin.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with Synordia (R), a solid combination of fenofibrate with metformin. In another embodiment of the invention, the compound of the formula I is administered in combination with a solid combination of metformin with an MTP inhibitor, as described in WO2009090210.
In one embodiment of the invention, the compound of the formula I is administered in combination with a cholesterol reabsorption inhibitor, for example ezetimibe, tiqueside, pamaqueside, FM-VP4 (sitostanol/campesterol ascorbyl phosphate; Forbes Medi- Tech, WO2005042692, WO2005005453), MD-0727 (Microbia Inc., WO2005021497,
WO2005021495) or with compounds as described in WO2002066464, WO2005000353 (Kotobuki Pharmaceutical Co. Ltd.) or WO2005044256 or
WO2005062824 (Merck & Co.) or WO2005061451 and WO2005061452 (AstraZeneca AB) and WO2006017257 (Phenomix) or WO2005033100 (Lipideon Biotechnology AG), or as described in WO2002050060, WO2002050068, WO2004000803,
WO2004000804, WO2004000805, WO2004087655, WO2004097655,
WO2005047248, WO2006086562, WO2006102674, WO20061 16499,
WO2006121861 , WO2006122186, WO2006122216, WO2006127893,
WO2006137794, WO2006137796, WO2006137782, WO2006137793,
WO2006137797, WO2006137795, WO2006137792, WO2006138163,
WO2007059871 , US2007232688, WO2007126358, WO2008033431 ,
WO2008033465, WO2008052658, WO2008057336, WO2008085300,
WO2008104875, US2008280836, WO2008108486. In one embodiment of the invention, the compound of the formula I is administered in combination with an NPC1 L1 antagonist, for example those as described in
WO2008033464, WO2008033465.
In one embodiment of the invention, the compound of the formula I is administered in combination with Vytorin™, a solid combination of ezetimibe with simvastatin.
In one embodiment of the invention, the compound of the formula I is administered in combination with a solid combination of ezetimibe with atorvastatin. In one embodiment of the invention, the compound of the formula I is administered in combination with a solid combination of ezetimibe with fenofibrate.
In one embodiment of the invention, the further active ingredient is a
diphenylazetidinone derivative, as described, for example, in US 6,992,067 or
US 7,205,290.
In a further embodiment of the invention, the further active ingredient is a diphenylazetidinone derivative, as described, for example, in US 6,992,067 or US 7,205,290, combined with a statin, for example simvastatin, fluvastatin, pravastatin, lovastatin, cerivastatin, atorvastatin, pitavastatin or rosuvastatin.
In one embodiment of the invention, the compound of the formula I is administered in combination with a solid combination of lapaquistat, a squalene synthase inhibitor, with atorvastatin. In a further embodiment of the invention, the compound of the formula I is
administered in combination with a conjugate consisting of the HMG-CoA reductase inhibitor atorvastatin with the renin inhibitor aliskiren (WO2009090158).
In one embodiment of the invention, the compound of the formula I is administered in combination with a CETP inhibitor, for example torcetrapib, anacetrapib or JTT-705 (dalcetrapib), or those as described in WO2006002342, WO2006010422,
WO2006012093, WO2006073973, WO2006072362, WO2007088996,
WO2007088999, US2007185058, US20071851 13, US2007185154, US2007185182, WO2006097169, WO2007041494, WO2007090752, WO2007107243,
WO2007120621 , US2007265252, US2007265304, WO2007128568, WO2007132906, WO2008006257, WO2008009435, WO2008018529, WO2008058961 ,
WO2008058967, WO2008059513, WO2008070496, WO20081 15442,
WO20081 1 1604, WO2008129951 , WO2008141077, US20091 18287,
WO2009062371 , WO2009071509.
In one embodiment of the invention, the compound of the formula I is administered in combination with bile acid reabsorption inhibitors (inhibitors of the intestinal bile acid transporter (IB AT)) (see, for example, US 6,245,744, US 6,221 ,897 or WO 00/61568), for example HMR 1741 , or those as described in DE 10 2005 033099.1 and DE 10 2005 033100.9, DE 10 2006 053635, DE 10 2006 053637, WO2007009655-56, WO2008058628, WO2008058629, WO2008058630, WO2008058631.
In one embodiment, the compound of the formula I is administered in combination with agonists of GPBAR1 (G-protein-coupled bile acid receptor 1 ; TGR5), for example INT- 777 or those as described, for example, in US20060199795, WO20071 10237,
WO2007127505, WO2008009407, WO2008067219, WO2008067222, FR2908310, WO2008091540, WO2008097976, US2009054304, WO2009026241 ,
WO2009146772, WO2010014739, WO2010014836.
In one embodiment, the compound of the formula I is administered in combination with modulators of histone deacetylase, for example ursodeoxycholic acid, as described in WO200901 1420.
In one embodiment, the compound of the formula I is administered in combination with inhibitors/modulators of the TRPM5 channel (TRP cation channel M5), as described, for example, in WO2008097504, WO2009038722.
In one embodiment, the compound of the formula I is administered in combination with inhibitors/modulators of the TRPA1 channel (TRP cation channel A1 ), as described, for example, in US2009176883, WO2009089083, WO2009144548. In one embodiment, the compound of the formula I is administered in combination with inhibitors/modulators of the TRPV3 channel (TRP cation channel V3), as described, for example, in WO2009084034, WO2009130560.
In one embodiment of the invention, the compound of the formula I is administered in combination with a polymeric bile acid adsorber, for example cholestyramine, colesevelam hydrochloride.
In one embodiment of the invention, the compound of the formula I is administered in combination with colesevelam hydrochloride and metformin or a sulfonylurea or insulin.
In one embodiment of the invention, the compound of the formula I is administered in combination with tocotrienol and insulin or an insulin derivative.
In one embodiment of the invention, the compound of the formula I is administered in combination with a chewing gum comprising phytosterols (Reductol™).
In one embodiment of the invention, the compound of the formula I is administered in combination with an inhibitor of the microsomal triglyceride transfer protein (MTP inhibitor), for example implitapide, BMS-201038, R-103757, AS-1552133, SLx-4090, AEGR-733, JTT-130, or those as described in WO2005085226, WO2005121091 , WO2006010423, WO20061 13910, WO2007143164, WO2008049806,
WO2008049808, WO2008090198, WO2008100423, WO2009014674.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a combination of a cholesterol absorption inhibitor, for example ezetimibe, and an inhibitor of the triglyceride transfer protein (MTP inhibitor), for example implitapide, as described in WO2008030382 or in
WO2008079398.
In one embodiment of the invention, the compound of the formula I is administered in combination with an active antihypertriglyceridemic ingredient, for example those as described in WO2008032980.
In another embodiment of the invention, the compound of the formula I is administered in combination with an antagonist of the somatostatin 5 receptor (SST5 receptor), for example those as described in WO2006094682.
In one embodiment of the invention, the compound of the formula I is administered in combination with an ACAT inhibitor, for example avasimibe, SMP-797 or KY-382, or those as described in WO2008087029, WO2008087030, WO2008095189,
WO2009030746, WO2009030747, WO2009030750, WO2009030752,
WO2009070130, WO2009081957, WO2009081957.
In a further embodiment of the invention, the compound of the formula I is administered in combination with an inhibitor of liver carnitine palmitoyltransferase-1 (L-CPT1 ), as described, for example, in WO2007063012, WO2007096251 (ST-3473), WO2008015081 , US2008103182, WO2008074692, WO2008145596,
WO2009019199, WO2009156479, WO2010008473.
In another embodiment of the invention, the compound of the formula I is administered in combination with an inhibitor of carnitin O-palmitoyltransferase II (CPT2), as described, for example, in US2009270500, US2009270505, WO2009132978,
WO2009132979.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a modulator of serine palmitoyltransferase (SPT), as described, for example, in WO2008031032, WO2008046071 , WO2008083280, WO2008084300.
In one embodiment of the invention, the compound of the formula I is administered in combination with a squalene synthetase inhibitor, for example BMS-188494, TAK-475 (lapaquistat acetate), or as described in WO2005077907, JP2007022943,
WO2008003424, WO2008132846, WO2008133288, WO2009136396.
In one embodiment of the invention, the compound of the formula I is administered in combination with ISIS-301012 (mipomersen), an antisense oligonucleotide which is capable of regulating the apolipoprotein B gene.
In one embodiment of the invention, the compound of the formula I is administered in combination with apolipoprotein (ApoB) SNALP, a therapeutic product which comprises an siRNA (directed against the ApoB gene). In one embodiment of the invention, the compound of the formula I is administered in combination with a stimulator of the ApoA-1 gene, as described, for example, in WO2008092231 .
In one embodiment of the invention, the compound of the formula I is administered in combination with a modulator of the synthesis of apolipoprotein —l 11 , for example ISIS- APOCIIIRx.
In one embodiment of the invention, the compound of the formula I is administered in combination with an LDL receptor inducer (see US 6,342,512), for example HMR1 171 , HMR1586, or those as described in WO2005097738, WO2008020607.
In another embodiment of the invention, the compound of the formula I is administered in combination with an HDL cholesterol-elevating agent, for example those as described in WO2008040651 , WO2008099278, WO2009071099, WO2009086096, US2009247550.
In one embodiment of the invention, the compound of the formula I is administered in combination with an ABCA1 expression enhancer, as described, for example, in WO2006072393, WO2008062830, WO2009100326.
In one embodiment of the invention, the compound of the formula I is administered in combination with a lipoprotein lipase modulator, for example ibrolipim (NO-1886).
In one embodiment of the invention, the compound of the formula I is administered in combination with a lipoprotein(a) antagonist, for example gemcabene (CI-1027).
In one embodiment of the invention, the compound of the formula I is administered in combination with a lipase inhibitor, for example orlistat or cetilistat (ATL-962).
In one embodiment of the invention, the compound of the formula I is administered in combination with an adenosine A1 receptor agonist (adenosine A1 R), for example CVT-3619 or those as described, for example, in EP1258247, EP1375508,
WO2008028590, WO2008077050, WO2009050199, WO2009080197, WO2009100827, WO20091 12155.
In one embodiment of the invention, the compound of the formula I is administered in combination with an adenosine A2B receptor agonist (adenosine A2B R), for example ATL-801 .
In another embodiment of the invention, the compound of the formula I is administered in combination with a modulator of adenosine A2A and/or adenosine A3 receptors, as described, for example, in WO20071 1 1954, WO2007121918, WO2007121921 , WO2007121923, WO2008070661 , WO2009010871 .
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a ligand of the adenosine A1/A2B receptors, as described, for example, in WO2008064788, WO2008064789, WO2009080198, WO2009100827, WO2009143992.
In one embodiment of the invention, the compound of the formula I is administered in combination with an adenosine A2B receptor antagonist (adenosine A2B R), as described in US2007270433, WO2008027585, WO2008080461 , WO2009037463, WO2009037467, WO2009037468, WO20091 18759.
In one embodiment, the compound of the formula I is administered in combination with inhibitors of acetyl-CoA carboxylase (ACC1 and/or ACC2), for example those as described in W0199946262, WO200372197, WO2003072197, WO2005044814,
WO2005108370, JP2006131559, WO200701 1809, WO200701 181 1 , WO2007013691 , WO2007095601 -603, WO20071 19833, WO2008065508, WO2008069500,
WO2008070609, WO2008072850, WO2008079610, WO2008088688,
WO2008088689, WO2008088692, US2008171761 , WO2008090944, JP2008179621 , US2008200461 , WO2008102749, WO2008103382, WO2008121592,
WO2009082346, US2009253725, JP2009196966, WO2009144554, WO2009144555, WO2010003624, WO2010002010.
In another embodiment, the compound of the formula I is administered in combination with modulators of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase 3 (GPAT3, described in WO2007100789) or with modulators of microsomal acyl- CoA:glycerol-3-phosphate acyltransferase 4 (GPAT4, described in WO2007100833) or with modulators of mitochondrial glycerol-3-phosphate O-acyltransferase, described in WO2010005922.
In a further embodiment, the compound of the formula I is administered in combination with modulators of xanthine oxidoreductase (XOR).
In another embodiment, the compound of the formula I is administered in combination with inhibitors of soluble epoxide hydrolase (sEH), as described, for example, in WO2008051873, WO2008051875, WO2008073623, WO2008094869,
WO20081 12022, WO200901 1872, WO2009049154, WO2009049157,
WO2009049165, WO2009073772, WO2009097476, WO20091 1 1207,
WO2009129508, WO2009151800.
In a further embodiment, the compound of the formula I is administered in combination with CART modulators (see "Cocaine-amphetamine-regulated transcript influences energy metabolism, anxiety and gastric emptying in mice" Asakawa, A. et al.: Hormone and Metabolic Research (2001 ), 33(9), 554-558); NPY antagonists, for example 4-[(4-aminoquinazolin-2-ylamino)methyl]- cyclohexylmethylnaphthalene-1 -sulfonamide hydrochloride (CGP 71683A) or velneperit or those as described in WO20091 10510;
NPY-5 receptor antagonists/receptor modulators, such as L-152804 or the compound "NPY-5-BY" from Banyu, or as described, for example, in WO2006001318,
WO2007103295, WO2007125952, WO2008026563, WO2008026564,
WO2008052769, WO2008092887, WO2008092888, WO2008092891 ,
WO2008129007, WO2008134228, WO2009054434, WO2009095377, WO2009131096;
NPY-4 receptor antagonists, as described, for example, in WO2007038942;
NPY-2 receptor antagonists/modulators, as described, for example, in
WO2007038943, WO2009006185, US2009099199, US2009099243, US2009099244, WO2009079593, WO2009079597; peptide YY 3-36 (PYY3-36) or analogous compounds, for example CJC-1682 (PYY3- 36 conjugated with human serum albumin via Cys34) or CJC-1643 (derivative of PYY3-36, which is conjugated in vivo to serum albumin), or those as described in WO2005080424, WO2006095166, WO2008003947, WO2009080608; NPY-2 receptor agonists, as described, for example, in WO2009080608; derivatives of the peptide obestatin, as described by WO2006096847;
CB1 R (cannabinoid receptor 1 ) antagonists/inverse agonists, for example rimonabant, surinabant (SR147778), SLV-319 (ibipinabant), AVE-1625, taranabant (MK-0364) or salts thereof, otenabant (CP-945,598), rosonabant, V-24343 or those compounds as described in, for example, EP 0656354, WO 00/15609, WO2001/64632-64634, WO 02/076949, WO2005080345, WO2005080328, WO2005080343, WO2005075450, WO2005080357, WO200170700, WO2003026647-48, WO200302776,
WO2003040107, WO2003007887, WO2003027069, US6,509,367, WO200132663, WO2003086288, WO2003087037, WO2004048317, WO2004058145,
WO2003084930, WO2003084943, WO2004058744, WO2004013120,
WO2004029204, WO2004035566, WO2004058249, WO2004058255,
WO2004058727, WO2004069838, US20040214837, US20040214855,
US20040214856, WO2004096209, WO2004096763, WO2004096794,
WO2005000809, WO2004099157, US20040266845, WO20041 10453,
WO2004108728, WO2004000817, WO2005000820, US20050009870,
WO200500974, WO20041 1 1033-34, WO20041 1038-39, WO2005016286, WO20050071 1 1 , WO2005007628, US20050054679, WO2005027837,
WO2005028456, WO2005063761 -62, WO2005061509, WO2005077897,
WO2006018662, WO2006047516, WO2006060461 , WO2006067428,
WO2006067443, WO2006087480, WO2006087476, WO2006100208,
WO2006106054, WO20061 1 1849, WO20061 13704, WO2007009705,
WO2007017124, WO2007017126, WO2007018459, WO2007018460,
WO2007016460, WO2007020502, WO2007026215, WO2007028849,
WO2007031720, WO2007031721 , WO2007036945, WO2007038045,
WO2007039740, US20070015810, WO2007046548, WO2007047737,
WO2007057687, WO2007062193, WO2007064272, WO2007079681 ,
WO2007084319, WO2007084450, WO2007086080, ΕΡ1816125, US2007213302, WO2007095513, WO2007096764, US2007254863, WO20071 19001 ,
WO2007120454, WO2007121687, WO2007123949, US2007259934,
WO2007131219, WO2007133820, WO2007136571 , WO2007136607,
WO2007136571 , US7297710, WO2007138050, WO2007139464, WO2007140385, WO2007140439, WO2007146761 , WO2007148061 , WO2007148062,
US2007293509, WO2008004698, WO2008017381 , US2008021031 , WO2008024284, WO2008031734, WO2008032164, WO2008034032, WO2008035356,
WO2008036021 , WO2008036022, WO2008039023, WO2998043544,
WO20080441 1 1 , WO2008048648, ΕΡ1921072-Α1 , WO2008053341 , WO2008056377, WO2008059207, WO2008059335, WO2008062424, WO2008068423,
WO2008068424, WO2008070305, WO2008070306, WO2008074816,
WO2008074982, WO2008075012, WO2008075013, WO2008075019,
WO20080751 18, WO2008076754, WO2008081009, WO2008084057, ΕΡ1944295,
US2008090809, US2008090810, WO2008092816, WO2008094473, WO2008094476, WO2008099076, WO2008099139, WO2008101995, US2008207704,
WO2008107179, WO2008109027, WO20081 12674, WO20081 15705,
WO20081 18414, WO20081 19999, WO200812000, WO2008121257, WO2008127585, WO2008129157, WO2008130616, WO2008134300, US2008262066, US2008287505, WO2009005645, WO2009005646, WO2009005671 , WO2009023292,
WO2009023653, WO2009024819, WO2009033125, ΕΡ2042175, WO2009053548-
WO2009053553, WO2009054923, WO2009054929, WO2009059264, WO2009073138, WO2009074782, WO2009075691 , WO2009078498,
WO2009087285, WO2009074782, WO2009097590, WO2009097995,
WO2009097996, WO2009097998, WO2009097999, WO2009098000,
WO2009106708, US2009239909, WO20091 18473, US2009264436, US2009264476, WO2009130234, WO2009131814, WO2009131815, US2009286758,
WO2009141532, WO2009141533, WO2009153569, WO2010003760,
WO2010012437, WO2010019762; cannabinoid receptor 1/cannabinoid receptor 2 (CB1 ,/CB2) modulating compounds, for example delta-9-tetrahydrocannabivarin, or those as described, for example, in
WO2007001939, WO2007044215, WO2007047737, WO2007095513,
WO2007096764, WO20071 12399, WO20071 12402, WO2008122618,
WO2009007697, WO2009012227, WO2009087564, WO2009093018,
WO2009095752, WO2009120660, WO2010012964; cannabinoid receptor 2 (CB2) modulating compounds, for example those as described, for example, in WO2008063625, WO2008157500, WO2009004171 , WO2009032754, WO2009055357, WO2009061652, WO2009063495, WO2009067613,
WO20091 14566; modulators of FAAH (fatty acid amide hydrolase), as described, for example,
WO2007140005, WO2008019357, WO2008021625, WO2008023720,
WO2008030532, WO2008129129, WO2008145839, WO2008145843,
WO2008147553, WO2008153752, WO200901 1904, WO2009048101 ,
WO2009084970, WO2009105220, WO2009109504, WO2009109743,
WO20091 17444, WO2009127944, WO2009138416, WO2009151991 ,
WO2009152025, WO2009154785, WO2010005572, WO2010017079; inhibitors of fatty acid synthase (FAS), as described, for example, in WO2008057585, WO2008059214, WO2008075064, WO2008075070, WO2008075077,
WO2009079860;
inhibitors of LCE (long chain fatty acid elongase)/long chain fatty acid CoA ligase, as described, for example, in WO2008120653, WO2009038021 , WO2009044788, WO2009081789, WO2009099086; vanilloid-1 receptor modulators (modulators of TRPV1 ), as described, for example, in WO2007091948, WO2007129188, WO2007133637, WO2008007780,
WO2008010061 WO200800721 1 WO2008010061 WO2008015335,
WO2008018827 WO2008024433 WO2008024438 WO2008032204,
WO2008050199 WO2008059339 WO2008059370 WO2008066664,
WO2008075150 WO2008090382 WO2008090434 WO2008093024,
WO2008107543 WO2008107544 WO20081 10863 WO2008125295,
WO2008125296 WO2008125337 WO2008125342 WO2008132600,
WO2008133973 WO2009010529 WO2009010824 WO2009016241 ,
WO2009023539 WO2009038812 WO2009050348 WO2009055629,
WO2009055749 WO2009064449 WO2009081222, WO2009089057,
WO2009109710WO20091 12677, WO20091 12678, WO20091 12679, WO2009121036, WO2009124551 , WO2009136625, WO2010002209; modulators, ligands, antagonists or inverse agonists of the opioid receptors, for example GSK-982 or those as described, for example, in WO2007047397,
WO2008021849, WO2008021851 , WO2008032156, WO2008059335,
WO2008125348, WO2008125349, WO2008142454, WO2009030962,
WO2009103552, WO20091 15257; modulators of the "orphan opioid (ORL-1 ) receptor", as described, for example, in US2008249122, WO2008089201 ; agonists of the prostaglandin receptor, for example bimatoprost or those compounds as described in WO20071 1 1806;
MC4 receptor agonists (melanocortin-4 receptor agonists, MC4R agonists, for example N-[2-(3a-benzyl-2-methyl-3-oxo-2, 3,3a, 4,6, 7-hexahydropyrazolo[4,3-c]pyridin-5-yl)-1 - (4-chlorophenyl)-2-oxoethyl]-1 -amino-1 ,2,3,4-tetrahydronaphthalene-2-carboxamide; (WO 01/91752)) or LB53280, LB53279, LB53278 or THIQ, MB243, RY764, CHIR-785, PT-141 , MK-0493, or those as described in WO2005060985, WO2005009950, WO2004087159, WO2004078717, WO2004078716, WO2004024720,
US20050124652, WO2005051391 , WO20041 12793, WOUS20050222014,
US20050176728, US20050164914, US20050124636, US20050130988,
US20040167201 , WO2004005324, WO2004037797, WO2004089307,
WO2005042516, WO2005040109, WO2005030797, US20040224901 ,
WO200501921 , WO200509184, WO2005000339, EP1460069, WO2005047253, WO2005047251 , WO20051 18573, EP1538159, WO2004072076, WO2004072077, WO2006021655-57, WO2007009894, WO2007015162, WO2007041061 ,
WO2007041052, JP2007131570, EP-1842846, WO2007096186, WO2007096763, WO2007141343, WO2008007930, WO2008017852, WO2008039418,
WO2008087186, WO2008087187, WO2008087189, WO2008087186- WO2008087190, WO2008090357, WO2008142319, WO2009015867,
WO200906141 1 , US2009076029, US2009131465, WO2009071 101 , US2009305960, WO2009144432, WO2009151383, WO2010015972;
MC4 receptor modulators (melanocortin-4 receptor modulators), as described, for example, in WO2009010299, WO2009074157; orexin receptor 1 antagonists (OX1 R antagonists), orexin receptor 2 antagonists (OX2R antagonists) or mixed OX1 R/OX2R antagonists (e.g. 1 -(2-methylbenzoxazol-6- yl)-3-[1 ,5]naphthyridin-4-ylurea hydrochloride (SB-334867-A), or those as described, for example, in WO200196302, WO200185693, WO2004085403, WO2005075458, WO2006067224, WO2007085718, WO2007088276, WO20071 16374,
WO2007122591 , WO2007126934, WO2007126935, WO2008008517,
WO2008008518, WO2008008551 , WO2008020405, WO2008026149,
WO2008038251 , US2008132490, WO2008065626, WO2008078291 ,
WO200808761 1 , WO2008081399, WO2008108991 , WO2008107335,
US2008249125, WO2008147518, WO2008150364, WO2009003993,
WO2009003997, WO200901 1775, WO2009016087, WO2009020642,
WO2009058238, US2009186920, US2009203736, WO2009092642, WO2009100994, WO2009104155, WO2009124956, WO2009133522, WO2009156951 ,
WO2010017260); histamine H3 receptor antagonists/inverse agonists (e.g. 3-cyclohexyl-1 -(4,4-dimethyl- 1 ,4,6,7-tetrahydroimidazo[4,5-c]pyridin-5-yl)propan-1 -one oxalic acid salt
(WO 00/63208), or those as described in WO200064884, WO2005082893,
WO2005123716, US2005171 181 (e.g. PF-00389027), WO2006107661 ,
WO2007003804, WO2007016496, WO2007020213, WO2007049798,
WO2007055418, WO2007057329, WO2007062999, WO2007065820,
WO2007068620, WO2007068641 , WO2007075629, WO2007080140,
WO2007082840, WO2007088450, WO2007088462, WO2007094962,
WO2007099423, WO2007100990, WO2007105053, WO2007106349,
WO20071 10364, WO20071 15938, WO2007131907, WO2007133561 ,
US2007270440, WO20071351 1 1 , WO2007137955, US2007281923, WO2007137968, WO2007138431 , WO2007146122, WO2008005338, WO2008012010,
WO2008015125, WO2008045371 , EP1757594, WO2008068173, WO2008068174, US20080171753, WO2008072703, WO2008072724, US2008188484, US2008188486, US2008188487, WO2008109333, WO2008109336, WO2008126886,
WO2008154126, WO2008151957, US2008318952, WO2009003003,
WO2009013195, WO2009036132, WO2009039431 , WO2009045313,
WO2009058300, WO2009063953, WO2009067401 , WO2009067405,
WO2009067406, US2009163464, WO2009100120, WO2009105206,
WO2009121812, WO2009126782, WO201001 1653, WO201001 1657); histamine H1/histamine H3 modulators, for example betahistine or its dihydrochloride; modulators of the histamine H3 transporter or of the histamine H3/serotonin
transporter, as described, for example, in WO2008002816, WO2008002817,
WO2008002818, WO2008002820;
modulators of vesicular monoamine transporter 2 (VMAT2), as described, for example, in WO2009126305; histamine H4 modulators, as described, for example, in WO20071 17399,
US2009156613;
CRF antagonists (e.g. [2-methyl-9-(2,4,6-trimethylphenyl)-9H-1 ,3,9-triazafluoren-4- yl]dipropylamine (WO 00/66585) or those CRF1 antagonists as described in
WO20071051 13, WO2007133756, WO2008036541 , WO2008036579,
WO2008083070, WO2010015628, WO2010015655);
CRF BP antagonists (e.g. urocortin); urocortin agonists; modulators of the beta-3 adrenoceptor, for example 1 -(4-chloro-3- methanesulfonylmethylphenyl)-2-[2-(2,3-dimethyl-1 H-indol-6-yloxy)ethylamino]ethanol hydrochloride (WO 01/83451 ) or solabegron (GW-427353) or N-5984 (KRP-204), or those as described in JP20061 1 1553, WO2002038543, WO2002038544,
WO2007048840-843, WO2008015558, EP1947103, WO2008132162;
MSH (melanocyte-stimulating hormone) agonists; MCH (melanine-concentrating hormone) receptor antagonists (for example NBI-845, A-761 , A-665798, A-798, ATC-0175, T-226296, T-71 (AMG-071 , AMG-076), GW- 856464, NGD-4715, ATC-0453, ATC-0759, GW-803430, or those compounds as described in WO2005085200, WO2005019240, WO200401 1438, WO2004012648, WO2003015769, WO2004072025, WO2005070898, WO2005070925,
WO2004039780, WO2004092181 , WO2003033476, WO2002006245,
WO2002089729, WO2002002744, WO2003004027, FR2868780, WO2006010446, WO2006038680, WO2006044293, WO2006044174, JP2006176443, WO2006018280,
WO2006018279 , WO20061 18320, WO2006130075, WO2007018248, WO2007012661 , WO2007029847, WO2007024004, WO2007039462,
WO2007042660 , WO2007042668, WO2007042669, US2007093508, US2007093509, WO2007048802 , JP2007091649, WO2007092416; WO2007093363-366,
WO20071 14902 , WO20071 14916, WO2007141200, WO2007142217,
US2007299062, WO2007146758, WO2007146759, WO2008001 160,
WO200801681 1 , WO2008020799, WO2008022979, WO2008038692,
WO2008041090 , WO2008044632, WO2008047544, WO2008061 109,
WO2008065021 , WO2008068265, WO2008071646, WO2008076562, JP2008088120, WO2008086404 , WO2008086409, US20082691 10, WO2008140239,
WO2009021740 , US200901 1994, US2009082359, WO2009041567, WO2009076387, WO2009089482 , WO2009103478, WO20091 19726, WO2009120655,
WO2009123194 , WO2009137270, WO2009146365, WO2009154132); CCK-A (CCK-1 ) modulators (for example {2-[4-(4-chloro-2,5-dimethoxyphenyl)-5-(2- cyclohexylethyl)-thiazol-2-ylcarbamoyl]-5,7-dimethylindol-1 -yl}acetic acid trifluoroacetic acid salt (WO 99/15525) or SR-146131 (WO 0244150) or SSR-125180) or those as described in WO20051 16034, WO2007120655, WO2007120688, WO2007120718, WO2008091631 ; serotonin reuptake inhibitors (e.g. dexfenfluramine), or those as described in
WO2007148341 , WO2008034142, WO2008081477, WO2008120761 ,
WO2008141081 , WO2008141082, WO2008145135, WO2008150848,
WO2009043834, WO2009077858; mixed serotonin/dopamine reuptake inhibitors (e.g. bupropion), or those as described in WO2008063673, or solid combinations of bupropion with naltrexone or bupropion with zonisamide; mixed reuptake inhibitors, for example DOV-21947 or those as described in
WO2009016214, WO2009016215, WO2009077584, WO2009098208,
WO2009098209, WO2009106769, WO2009109517, WO2009109518, WO2009109519, WO2009109608, WO2009145357, WO2009149258; mixed serotoninergic and noradrenergic compounds (e.g. WO 00/71549);
5-HT receptor agonists, for example 1 -(3-ethylbenzofuran-7-yl)piperazine oxalic acid salt (WO 01/091 1 1 ); mixed dopamine/norepinephrine/acetylcholine reuptake inhibitors (e.g. tesofensine), or those as described, for example, in WO20060851 18, WO2008150480; dopamine antagonists, as described, for example, in WO2008079838,
WO2008079839, WO2008079847, WO2008079848; norepinephrine reuptake inhibitors, as described, for example, in US2008076724, WO2009062318;
5-HT1 A receptor modulators, as described, for example, in WO2009006227,
WO2009137679, WO2009137732;
5-HT2A receptor antagonists, as described, for example, in WO2007138343;
5-HT2C receptor agonists (for example lorcaserine hydrochloride (APD-356) or BVT- 933, or those as described in WO200077010, WO200077001 -02, WO2005019180, WO2003064423, WO200242304, WO2005035533, WO2005082859, WO2006004937, US2006025601 , WO2006028961 , WO2006077025, WO200610351 1 ,
WO2007028132, WO2007084622, US2007249709; WO2007132841 ,
WO2007140213, WO2008007661 , WO2008007664, WO2008009125,
WO2008010073, WO2008108445, WO2009063991 , WO2009063992,
WO2009063993, WO2009079765);
5-HT6 receptor modulators, for example E-6837, BVT-74316, PF-3246799 or PRX- 07034, or those as described, for example, in WO2005058858, WO2007054257, WO2007107373, WO2007108569, WO2007108742-744, WO2008003703,
WO2008027073, WO2008034815, WO2008054288, EP1947085, WO2008084491 , WO2008084492, WO2008092665, WO2008092666, WO2008101247,
WO20081 10598, WO20081 16831 , WO20081 16833, WO20081 17169,
WO2008136017, WO2008147812, EP2036888, WO2009013010, WO2009034581 , WO2009053997, WO2009056632, WO20090731 18, WO20091 15515,
WO2009135925, WO2009135927, WO2010000456, WO2010012806, EP2145887; agonists of estrogen receptor gamma (ERRy agonists), as described, for example, in WO2007131005, WO2008052709; agonists of estrogen receptor alpha (ERRa / ERR1 agonists), as described, for example, in WO2008109727; agonists of estrogen receptor beta (ERRp agonists), as described, for example, in WO2009055734, WO2009100335, WO2009127686; sigma-1 receptor antagonists, as described, for example, in WO2007098953,
WO2007098961 , WO2008015266, WO2008055932, WO2008055933,
WO2009071657; muscarin 3 receptor (M3R) antagonists, as described, for example, in WO20071 10782, WO2008041 184; bombesin receptor agonists (BRS-3 agonists), as described, for example, in
WO2008051404, WO2008051405, WO2008051406, WO200807331 1 ; galanin receptor antagonists; growth hormone (e.g. human growth hormone or AOD-9604);
growth hormone releasing compounds (tert-butyl 6-benzyloxy-1 -(2- diisopropylaminoethylcarbamoyl)-3,4-dihydro-1 H-isoquinoline-2-carboxylate (WO 01/85695)); growth hormone secretagogue receptor antagonists (ghrelin antagonists), for example A-778193, or those as described in WO2005030734, WO2007127457,
WO2008008286, WO2009056707; growth hormone secretagogue receptor modulators (ghrelin modulators), for example JMV-2959, JMV-3002, JMV-2810, JMV-2951 , or those as described in
WO2006012577 (e.g. YIL-781 or YIL-870), WO2007079239, WO2008092681 , WO2008145749, WO2008148853, WO2008148854, WO2008148856,
WO2009047558, WO2009071283, WO20091 15503;
TRH agonists (see, for example, EP 0 462 884); decoupling protein 2 or 3 modulators (as described, for example, in WO2009128583); chemical decouplers (e.g. WO2008059023, WO2008059024, WO2008059025, WO2008059026); leptin receptor agonists (see, for example, Lee, Daniel W.; Leinung, Matthew C;
Rozhavskaya-Arena, Marina; Grasso, Patricia. Leptin agonists as a potential approach to the treatment of obesity. Drugs of the Future (2001 ), 26(9), 873-881 ); leptin receptor modulators, as described, for example, in WO2009019427,
WO2009071658, WO2009071668, WO2009071677, WO2009071678,
WO200914721 1 , WO2009147216, WO2009147219, WO2009147221 ;
DA agonists (bromocriptin, bromocriptin mesylate, doprexin) or those as described in US2009143390;
lipase/amylase inhibitors (e.g. WO 00/40569, WO2008107184, WO2009049428, WO2009125819); inhibitors of diacylglycerol O-acyltransferases (DGATs), for example BAY-74-41 13, or as described, for example, in US2004/0224997, WO2004094618, WO200058491 , WO2005044250, WO2005072740, JP2005206492, WO2005013907, WO2006004200, WO2006019020, WO2006064189, WO2006082952, WO2006120125,
WO20061 13919, WO2006134317, WO2007016538, WO2007060140, JP2007131584, WO2007071966, WO2007126957, WO2007137103, WO2007137107,
WO2007138304, WO200713831 1 , WO2007141502, WO2007141517,
WO2007141538, WO2007141545, WO2007144571 , WO200801 1 130,
WO200801 1 131 , WO2008039007, WO2008048991 , WO2008067257,
WO2008099221 , WO2008129319, WO2008141976, WO2008148840,
WO2008148849, WO2008148851 , WO2008148868, WO200901 1285,
WO2009016462, WO2009024821 , US2009076275, WO2009040410,
WO2009071483, WO2009081 195, WO20091 19534, WO2009126624,
WO2009126861 , WO2010007046, WO2010017040; inhibitors of monoacylglycerol acyltransferase (2-acylglycerol O-acyltransferase;
MGAT), as described, for example, in WO2008038768; inhibitors of fatty acid synthase (FAS), for example C75, or those as described in WO2004005277, WO20080061 13; inhibitors of stearoyl-CoA delta9 desaturase (SCD1 ), as described, for example, in WO2007009236, WO2007044085, WO2007046867, WO2007046868,
WO20070501 124, WO2007056846, WO2007071023, WO2007130075,
WO2007134457, WO2007136746, WO2007143597, WO2007143823,
WO2007143824, WO2008003753, WO2008017161 , WO2008024390,
WO2008029266, WO2008036715, WO2008043087, WO2008044767,
WO2008046226, WO2008056687, WO2008062276, WO2008064474,
WO2008074824 WO2008074832, WO2008074833, WO2008074834, WO2008074835 WO2008089580, WO2008096746, WO2008104524,
WO20081 16898 US2008249100, WO2008120744, WO2008120759,
WO2008123469 WO2008127349, WO2008128335, WO2008135141 ,
WO2008139845 WO2008141455, US20080255130, US2008255161 ,
WO2008141455 WO2009010560, WO2009016216, WO2009012573,
WO2009024287 JP2009019013, WO2009037542, WO2009056556, WO2009060053, WO2009060054 WO2009070533, WO2009073973, WO2009103739,
WO20091 17659 WO20091 17676, US2009253693, US2009253738, WO2009124259, WO2009126123 WO2009126527, WO2009129625, WO2009137201 ,
WO2009150196 WO2009156484, WO2010006962, WO2010007482; inhibitors of fatty acid desaturase 1 (delta5 desaturase), as described, for example, WO2008089310; inhibitors of monoglyceride lipase (MGL), as described in WO2008145842; hypoglycemic/hypertriglyceridemic indoline compounds, as described in
WO2008039087, WO2009051 1 19; inhibitors of "adipocyte fatty acid-binding protein aP2", for example BMS-309403 or those as described in WO2009028248; activators of adiponectin secretion, as described, for example, in WO2006082978, WO2008105533, WO2008136173;
promoters of adiponectin production, as described, for example, in WO2007125946, WO2008038712;
modified adiponectins, as described, for example, in WO2008121009; oxyntomodulin or analogs thereof (for example, TKS-1225); oleoyl-estrone
or agonists or partial agonists of the thyroid hormone receptor (thyroid hormone receptor agonists), for example: KB-21 15 (eprotirome), QRX-431 (sobetirome) or DITPA, or those as described in WO20058279, WO200172692, WO200194293, WO2003084915, WO2004018421 , WO2005092316, WO2007003419,
WO2007009913, WO2007039125, WO20071 10225, WO20071 10226,
WO2007128492, WO2007132475, WO2007134864, WO2008001959,
WO2008106213, JP2009155261 ; or agonists of the thyroid hormone receptor beta (TR-beta), for example MB-0781 1 or MB-07344, or those as described in WO2008062469.
In one embodiment of the invention, the compound of the formula I is administered in combination with a combination of eprotirome with ezetimibe.
In one embodiment of the invention, the compound of the formula I is administered in combination with an inhibitor of site-1 protease (S1 P), for example PF-429242.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with a modulator of the "trace amine associated receptor 1 " (TAAR1 ), as described, for example, in US2008146523, WO2008092785.
In one embodiment of the invention, the compound of the formula I is administered in combination with an inhibitor of growth factor receptor bound protein 2 (GRB2), as described, for example, in WO2008067270.
In a further embodiment of the invention, the compound of the formula I is
administered in combination with an RNAi (siRNA) therapeutic agent directed against PCSK9 (proprotein convertase subtilisin/kexin type 9).
In one embodiment, the compound of the formula I is administered in combination with Omacor® or Lovaza™ (omega-3 fatty acid ester; highly concentrated ethyl ester of eicosapentaenoic acid and of docosahexaenoic acid). In one embodiment, the compound of the formula I is administered in combination with lycopene.
In one embodiment of the invention, the compound of the formula I is administered in combination with an antioxidant, for example OPC-141 17, AGI-1067 (succinobucol), probucol, tocopherol, ascorbic acid, β-carotene or selenium, or those as described in WO2009135918.
In one embodiment of the invention, the compound of the formula I is administered in combination with a vitamin, for example vitamin B6 or vitamin B12.
In one embodiment, the compound of the formula I is administered in combination with more than one of the aforementioned compounds, for example in combination with a sulfonylurea and metformin, a sulfonylurea and acarbose, repaglinide and metformin (PrandiMet (TM)), insulin and a sulfonylurea, insulin and metformin, insulin and troglitazone, insulin and lovastatin, etc.
In a further embodiment, the compound of the formula I is administered in combination with an activator of soluble guanylate cyclase (sGC), as described, for example, in WO2009032249.
In another embodiment, the compound of the formula I is administered in combination with an inhibitor of carboanhydrase type 2 (carbonic anhydrase type 2), for example those as described in WO2007065948, WO2009050252.
In another embodiment, the compound of the formula I is administered in combination with topiramat or a derivative thereof, as described in WO2008027557,
US2009304789.
In a further embodiment, the compound of the formula I is administered in combination with a solid combination of topiramat with phentermin (Qnexa™).
In a further embodiment, the compound of the formula I is administered in combination with an antisense compound, e.g. ISIS-377131 , which inhibits the production of the glucocorticoid receptor.
In another embodiment, the compound of the formula I is administered in combination with an aldosterone synthase inhibitor and an antagonist of the glucocorticoid receptor, a Cortisol synthesis inhibitor and/or an antagonist of the corticotropin releasing factor, as described, for example, in EP1886695, WO20081 19744.
In one embodiment, the compound of the formula I is administered in combination with an agonist of the RUP3 receptor, as described, for example, in WO2007035355, WO2008005576.
In another embodiment, the compound of the formula I is administered in combination with an activator of the gene which codes for ataxia telangiectasia mutated (ATM) protein kinase, for example chloroquine.
In one embodiment, the compound of the formula I is administered in combination with a tau protein kinase 1 inhibitor (TPK1 inhibitor), as described, for example, in
WO20071 19463, WO2009035159, WO2009035162. In one embodiment, the compound of the formula I is administered in combination with a "c-Jun N-terminal kinase" inhibitor (JNK inhibitor), for example B1 -78D3 or those as described, for example, in WO2007125405, WO2008028860, WO20081 18626.
In one embodiment, the compound of the formula I is administered in combination with an endothelin A receptor antagonist, for example avosentan (SPP-301 ).
In one embodiment, the compound of the formula I is administered in combination with inhibitors of neutral endopeptidase (NEP inhibitors), as described, for example, in WO2009138122, WO2009135526. In one embodiment, the compound of the formula I is administered in combination with modulators of the glucocorticoid receptor (GR), for example KB-3305 or those compounds as described, for example, in WO2005090336, WO2006071609,
WO2006135826, WO2007105766, WO2008120661 , WO2009040288,
WO2009058944, WO2009108525, WO20091 1 1214.
In one embodiment, the further active ingredient is varenicline tartrate, a partial agonist of the alpha 4-beta 2 nicotinic acetylcholine receptor.
In one embodiment, the further active ingredient is an agonist of the alpha 7-nicotinic acetylcholine receptor, as described, for example, in WO2009018551 ,
WO2009071519, WO2009071576, WO2009071577.
In one embodiment, the further active ingredient is trodusquemine. In one embodiment, the further active ingredient is a modulator of the enzyme SIRT1 and/or SIRT3 (an NAD+-dependent protein deacetylase); this active ingredient may, for example, be resveratrol in suitable formulations, or those compounds as specified in WO2007019416 (e.g. SRT-1720), WO2008073451 , WO2008156866,
WO2008156869, WO2009026701 , WO2009049018, WO2009058348,
WO2009061453, WO2009134973, WO2009146358, WO2010003048.
In one embodiment of the invention, the further active ingredient is DM-71 (N-acetyl-L- cysteine with bethanechol). In one embodiment, the compound of the formula I is administered in combination with antihypercholesterolemic compounds, as described, for example, in WO2004000803, WO2006000804, WO2004000805, WO2004087655, WO20051 13496,
WO2007059871 , WO2007107587, WO20071 1 1994, WO2008052658,
WO2008106600, WO20081 13796, US2008280836, WO20091 13952, US2009312302
In a further embodiment, the compound of the formula I is administered in combination with inhibitors of SREBP (sterol regulatory element-binding protein), for example fatostatin, or those as described, for example, in WO2008097835.
In another embodiment, the compound of the formula I is administered in combination with a cyclic peptide agonist of the VPAC2 receptor, as described, for example, in WO2007101 146, WO2007133828.
In a further embodiment, the compound of the formula I is administered in combination with an agonist of the endothelin receptor, as described, for example, in
WO20071 12069.
In a further embodiment, the compound of the formula I is administered in combination with AKP-020 (bis(ethylmaltolato)oxovanadium(IV)). In another embodiment, the compound of the formula I is administered in combination with tissue-selective androgen receptor modulators (SARM), as described, for example, in WO2007099200, WO2007137874.
In a further embodiment, the compound of the formula I is administered in combination with an AGE (advanced glycation endproduct) inhibitor, as described, for example, in JP2008024673.
In one embodiment of the invention, the further active ingredient is leptin;
see, for example, "Perspectives in the therapeutic use of leptin", Salvador, Javier; Gomez-Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001 ), 2(10), 1615-1622.
In another embodiment of the invention, the further active ingredient is metreleptin (recombinant methionyl-leptin) combined with pramlintide.
In a further embodiment of the invention, the further active ingredient is the
tetrapeptide ISF-402.
In one embodiment, the further active ingredient is dexamphetamine or amphetamine.
In one embodiment, the further active ingredient is fenfluramine or dexfenfluramine. In another embodiment, the further active ingredient is sibutramine or those derivatives as described in WO2008034142.
In one embodiment, the further active ingredient is mazindol or phentermin. In a further embodiment, the further active ingredient is geniposidic acid
(WO2007100104) or derivatives thereof (JP2008106008).
In another embodiment, the further active ingredient is a neuropeptide FF2 agonist, as described, for example, in WO2009038012.
In one embodiment, the further active ingredient is a nasal calcium channel blocker, for example diltiazem, or those as described in US 7, 138, 107.
In one embodiment, the further active ingredient is an inhibitor of sodium-calcium ion exchange, for example those as described in WO2008028958, WO200808571 1 .
In a further embodiment, the further active ingredient is a blocker of calcium channels, for example of CaV3.2 or CaV2.2, as described in WO2008033431 , WO2008033447, WO2008033356, WO2008033460, WO2008033464, WO2008033465,
WO2008033468, WO2008073461 .
In one embodiment, the further active ingredient is a modulator of a calcium channel, for example those as described in WO2008073934, WO2008073936, WO2009107660.
In one embodiment, the further active ingredient is an inhibitor of the calcium
metabolism, for example those as described in US2009124680.
In one embodiment, the further active ingredient is a blocker of the "T-type calcium channel", as described, for example, in WO2008033431 , WO20081 10008,
US2008280900, WO2008141446, US2009270338, WO2009146540, US2009325979, WO2009146539.
In one embodiment, the further active ingredient is an inhibitor of KCNQ potassium channel 2 or 3, for example those as described in US2008027049, US2008027090. In one embodiment, the further active ingredient is a modulator of KCNN potassium channel 1 , 2 or 3 (modulators of the SK1 , SK2 and/or SK3 channel), for example those as described in US2009036475.
In one embodiment, the further active ingredient is an inhibitor of the potassium Kv1 .3 ion channel, for example those as described in WO2008040057, WO2008040058, WO2008046065, WO20090431 17.
In one embodiment, the further active ingredient is a potassium channel modulator, for example those as described in WO2008135447, WO2008135448, WO2008135591 , WO2009099820.
In a further embodiment, the further active ingredient is a hyperpolarization-activated cyclic nucleotide-gated (HCN) potassium-sodium channel inhibitor, for example those as described in US2009069296.
In another embodiment, the further active ingredient is an inhibitor of the sodium- potassium-2 chloride (NKCCI) cotransporter, for example those as described in
WO2009130735. In another embodiment, the further active ingredient is a voltage-gated sodium channel inhibitor, for example those as described in WO2009049180, WO2009049181 .
In another embodiment, the further active ingredient is a modulator of the MCP-1 receptor (monocyte chemoattractant protein-1 (MCP-1 )), for example those as described in WO2008014360, WO2008014381 .
In one embodiment, the further active ingredient is a modulator of somatostatin receptor 3 (SSTR3), for example those as described in WO200901 1836. In one embodiment, the further active ingredient is a modulator of somatostatin receptor 5 (SSTR5), for example those as described in WO2008019967,
US2008064697, US2008249101 , WO2008000692, US2008293756, WO2008148710.
In one embodiment, the further active ingredient is a modulator of somatostatin receptor 2 (SSTR2), for example those as described in WO2008051272.
In one embodiment, the further active ingredient is a compound which is capable of reducing the amount of retinol-binding protein 4 (RBP4), for example those as described in WO2009051244, WO2009145286.
In one embodiment, the further active ingredient is an erythropoietin-mimetic peptide which acts as an erythropoietin (EPO) receptor agonist. Such molecules are described, for example, in WO2008042800. In a further embodiment, the further active ingredient is an anorectic/a hypoglycemic compound, for example those as described in WO2008035305, WO2008035306, WO2008035686.
In one embodiment, the further active ingredient is an inductor of lipoic acid synthetase, for example those as described in WO2008036966, WO2008036967. In one embodiment, the further active ingredient is a stimulator of endothelial nitric oxide synthase (eNOS), for example those as described in WO2008058641 ,
WO2008074413.
In one embodiment, the further active ingredient is a modulator of carbohydrate and/or lipid metabolism, for example those as described in WO2008059023, WO2008059024, WO2008059025, WO2008059026.
In a further embodiment, the further active ingredient is an angiotensin II receptor antagonist, for example those as described in WO2008062905, WO2008067378, WO2008062905.
In one embodiment, the further active ingredient is an agonist of the sphingosine 1 - phosphate receptor (S1 P), for example those as described in WO2008064315, WO2008074820, WO2008074821 , WO2008135522, WO2009019167,
WO2009043013, WO2009080663, WO2009085847, WO2009151529,
WO2009151621 , WO2009151626, WO2009154737.
In one embodiment, the further active ingredient is an agent which retards gastric emptying, for example 4-hydroxyisoleucine (WO2008044770).
In one embodiment, the further active ingredient is a trytophan-5-hydroxylase inhibitor- 1 (TPH1 inhibitor), which modulates gastrointestinal motility, as described, for example, in WO2009014972. In one embodiment, the further active ingredient is a muscle-relaxing substance, as described, for example, in WO2008090200.
In a further embodiment, the further active ingredient is an inhibitor of monoamine oxidase B (MAO-B), for example those as described in WO2008092091 ,
WO2009066152. In a further embodiment, the further active ingredient is an inhibitor of monoamine oxidase A (MAO-A), for example those as described in WO2009030968.
In another embodiment, the further active ingredient is an inhibitor of the binding of cholesterol and/or triglycerides to the SCP-2 protein (sterol carrier protein-2), for example those as described in US2008194658.
In a further embodiment, the further active ingredient is a compound which binds to the β-subunit of the trimeric GTP-binding protein, for example those as described in WO2008126920.
In one embodiment, the further active ingredient is a urate anion exchanger inhibitor 1 , as described, for example, in WO2009070740.
In one embodiment, the further active ingredient is a modulator of the ATP transporter, as described, for example, in WO2009108657.
In another embodiment, the further active ingredient is lisofylline, which prevents autoimmune damage to insulin-producing cells. In yet another embodiment, the further active ingredient is an extract from Bidens pilosa with the ingredient cytopiloyne as described in EP1955701 .
In one embodiment, the further active ingredient is an inhibitor of glucosylceramide synthase, as described, for example, in WO2008150486.
In a further embodiment of the invention, the further active ingredient is a glycosidase inhibitor, as described, for example, in WO20091 17829, WO2009155753.
In another embodiment, the further active ingredient is an ingredient from the plant Hoodia Gordonii, as described in US2009042813, EP2044852. In one embodiment, the further active ingredient is an antidiabetic, for example D-tagatose.
In one embodiment, the further active ingredient is a zinc complex of curcumin, as described in WO2009079902.
In one embodiment, the further active ingredient is an inhibitor of the "cAMP response element binding protein" (CREB), as described in WO2009143391 .
In another embodiment, the further active ingredient is an antagonist of the bradykinin B1 receptor, as described in WO2009124746.
In a further embodiment, the further active ingredient is a compound which is capable of modulating diabetic peripheral neuropathy (DPN). Such modulators are, for example, FK-1706 or SB-509, or those as described in W01989005304,
WO2009092129, WO2010002956.
In one embodiment, the further active ingredient is a compound which is capable of modulating diabetic nephropathy. Such compounds are described, for example, in WO2009089545, WO2009153261 .
In one embodiment, the further active ingredient is an inhibitor (e.g. an anti-CD38 antibody) of CD38, as described in US2009196825.
In one embodiment, the further active ingredient is an inhibitor of human fibroblast growth factor receptor 4 (FGFR4), as described, for example, in WO2009046141 .
In a further embodiment of the invention, the further active ingredient is a compound which protects the beta cell, for example 14-alpha-lipolyl-andrographolide (AL-1 ).
In yet another embodiment of the invention, the further active ingredient is the INGAP (islet neogenesis associated protein) peptide, a peptide which reestablishes insulin production in patients with diabetes mellitus.
In one embodiment of the invention, the further active ingredient is a modulator of the CFTR (cystic fibrosis transmembrane conductance regulator), as described, for example, in US2009246137, US2009264433, US2009264441 , US2009264471 , US2009264481 , US2009264486, WO2010019239.
In one embodiment of the invention, the further active ingredient is a compound which stimulates/modulates insulin release, for example those as described in
WO2009109258, WO2009132739, US2009281057, WO2009157418 .
In one embodiment of the invention, the further active ingredient is an extract from Hippophae rhamnoides, as described, for example, in WO2009125071 . In one embodiment of the invention, the further active ingredient is an from Huangiian and Ku Ding Cha, as described, for example, in WO2009133458.
In another embodiment, the further active ingredient is a root extract from Cipadessa baccifera, as described in US2009238900.
In one embodiment of the invention, the further active ingredients are borapetoside A and/or borapetoside C, which can be isolated from the plant SDH-V, a species of Tinospora crispa, as described, for example, in US2010016213.
In one embodiment, the compound of the formula I is administered in combination with bulking agents, preferably insoluble bulking agents (see, for example,
Carob/Caromax® (Zunft H J; et al., Carob pulp preparation for treatment of hypercholesterolemia, ADVANCES IN THERAPY (2001 Sep-Oct), 18(5), 230-6).
Caromax is a carob-containing product from Nutrinova, Nutrition Specialties & Food Ingredients GmbH, Industriepark Hochst, 65926 Frankfurt/Main)). Combination with Caromax® is possible in one preparation or by separate administration of compounds of the formula I and Caromax®. Caromax® can in this connection also be administered in the form of food products such as, for example, in bakery products or muesli bars.
It will be appreciated that every suitable combination of the compounds of the invention with one or more of the aforementioned compounds and optionally one or more other pharmacologically active substances is considered to be covered within the scope of protection conferred by the present invention.


H-CI




PSN-632408 SYR-322

arenc ne arra

Lorcaserin Hydrochlorid



A-769662


BVT-74316 ABT-341





CKD-501 (lobeglitazone sulfate) MB-0781 1

JMV-2959 JMV-3002

JMV-2810 JMV-2951

BMS-309403 PSN-1 19-1

S-40755 LY-2463665

ST-3473 DOV-21947





DITPA DGAT-1 inhibitor from WO2007137103




MB-07229 MB-07803


alogliptin benzoate nicotinic acid / laropiprant


BI-78D3 FK-1706

-alpha-lipolyl-andrographolide (AL-1 ) fatostatin

PF-3246799
Also suitable are the following active ingredients for combination preparations:
all antiepileptics specified in the Rote Liste 201 1 , chapter 15;
all antihypertensives specified in the Rote Liste 201 1 , chapter 17;
all hypotonics specified in the Rote Liste 201 1 , chapter 19;
all anticoagulants specified in the Rote Liste 201 1 , chapter 20;
all arteriosclerosis drugs specified in the Rote Liste 201 1 , chapter 25;
all beta receptors, calcium channel blockers and inhibitors of the renin angiotensin system specified in the Rote Liste 201 1 , chapter 27;
all diuretics and perfusion-promoting drugs specified in the Rote Liste 201 1 , chapter 36 and 37;
all withdrawal drugs/drugs for the treatment of addictive disorders specified in the Rote Liste 201 1 , chapter 39;
all coronary drugs and gastrointestinal drugs specified in the Rote Liste 201 1 , chapter 55 and 60;
all migraine drugs, neuropathy preparations and Parkinson's drugs specified in the Rote Liste 201 1 , chapter 61 , 66 and 70.
It will be appreciated that every suitable combination of the compounds of the invention with one or more of the aforementioned compounds and optionally one or more other pharmacologically active substances is considered to be covered within the scope of protection conferred by the present invention.
PKC-inhibitors can be administered to animals, in particular to mammals including humans, as pharmaceuticals by themselves, in mixtures with one another, or in the form of pharmaceutical compositions. The administration can be carried out orally, for example in the form of tablets, film-coated tablets, sugar-coated tablets, granules, hard and soft gelatin capsules, solutions including aqueous, alcoholic and oily solutions, juices, drops, syrups, emulsions or suspensions, rectally, for example in the form of suppositories, or parenterally, for example in the form of solutions for subcutaneous, intramuscular or intravenous injection or infusion, in particular aqueous solutions.
Suitable pharmaceutical compounds for oral administration may be in the form of separate units, for example capsules, cachets, lozenges or tablets, each of which contains a defined amount of the compound of formula I; as powders or granules; as solution or suspension in an aqueous or nonaqueous liquid; or as an oil-in-water or water-in-oil emulsion. These compositions may, as already mentioned, be prepared by any suitable pharmaceutical method which includes a step in which the active ingredient and the carrier (which may consist of one or more additional ingredients) are brought into contact. The compositions are generally produced by uniform and homogeneous mixing of the active ingredient with a liquid and/or finely divided solid carrier, after which the product is shaped if necessary. Thus, for example, a tablet can be produced by compressing or molding a powder or granules of the compound, where appropriate with one or more additional ingredients. Compressed tablets can be produced by tableting the compound in free-flowing form such as, for example, a powder or granules, where appropriate mixed with a binder, glidant, inert diluent and/or one (or more) surfactant(s)/dispersant(s) in a suitable machine. Molded tablets can be
produced by molding the compound, which is in powder form and has been moistened with an inert liquid diluent, in a suitable machine.
Pharmaceutical compositions which are suitable for peroral (sublingual) administration comprise lozenges which contain a compound of formula I with a flavoring, typically sucrose, and gum arabic or tragacanth, and pastilles which comprise the compound in an inert base such as gelatin and glycerol or sucrose and gum arabic.
Coated formulations and coated slow-release formulations, especially acid- and gastric juice-resistant formulations, also belong within the framework of the invention. Suitable coatings resistant to gastric juice comprise cellulose acetate phthalate, polyvinyl acetate phthalate, hydroxypropylmethylcellulose phthalate and anionic polymers of methacrylic acid and methyl methacrylate. Pharmaceutical compositions suitable for rectal administration are preferably in the form of single-dose suppositories. These can be produced by mixing a compound of formula I with one or more conventional solid carriers, for example cocoa butter, and shaping resulting mixture. Pharmaceutical compositions suitable for parenteral administration comprise preferably sterile aqueous preparations of a compound of formula I, which are preferably isotonic with the blood of the intended recipient. These preparations are preferably administered intravenously, although administration may also take place by subcutaneous, intramuscular or intradermal injection. These preparations can preferably be produced by mixing the compound with water and making the resulting solution sterile and isotonic with blood. Injectable compositions of the invention generally contain 0.1 to 5% by weight of the active compound.
Other suitable administration forms are, for example, percutaneous or topical administration, for example in the form of ointments, creams, tinctures, sprays, powders or transdermal therapeutic systems, or inhalative administration, for example
in the form of nasal sprays or aerosol mixtures, or forms such as microcapsules, implants or rods.
Pharmaceutical compositions suitable for topical use on the skin are preferably in the form of ointment, cream, lotion, paste, spray, aerosol or oil. The carriers used may be petrolatum, lanolin, polyethylene glycols, alcohols and combinations of two or more of these substances. The active ingredient is generally present in a concentration of 0.1 to 15% by weight of the composition, for example 0.5 to 2%. Transdermal administration is also possible. Pharmaceutical compositions suitable for transdermal uses may be in the form of single patches which are suitable for long-term close contact with the patient's epidermis. Such patches suitably contain the active ingredient in an aqueous solution which is buffered where appropriate, dissolved and/or dispersed in an adhesive or dispersed in a polymer. A suitable active ingredient concentration is about 1 % to 35%, preferably about 3% to 15%. A particular option is for the active ingredient to be released by electrotransport or iontophoresis as described, for example, in Pharmaceutical Research, 2(6): 318 (1986).
PKC inhibitors can additionally be used in systems for local drug delivery, for example in coated stents for preventing or reducing in-stent restenosis or by applying them locally by means of a catheter. The appropriate administration form depends, among others, on the disease to be treated and on its severity.
The dosing of PKC inhibitors to achieve the desireabie therapeutic effect depends on a number of factors, for example the specific compound chosen, the intended use, the mode of administration and the clinical condition of the patient. The daily dose is generally in the range from 0.3 mg to 100 mg (typically from 3 mg to 50 mg) per day and per kilogram of body weight, for example 3-10 mg/kg/day. An intravenous dose may be, for example, in the range from 0.3 mg to 1 .0 mg/kg, which can suitably be administered as infusion of 10 ng to 100 ng per kilogram and per minute. Suitable infusion solutions for these purposes may contain, for example, 0.1 ng to 100 mg, typically 1 ng to 100 mg, per milliliter. Single doses may contain, for example, 1 mg to
10 g of the active ingredient. Thus, ampoules for injections may contain, for example, from 1 mg to 100 mg, and orally administrable single-dose formulations, for example tablets or capsules, may contain, for example, from 1 .0 to 1000 mg, typically from 10 to 600 mg. For treatment of the abovementioned conditions, the compounds of the formula I themselves may be used as the compound, but they are preferably present with a compatible carrier in the form of a pharmaceutical composition. The carrier must, of course, be acceptable in the sense that it is compatible with the other ingredients of the composition and is not harmful for the patient's health. The carrier may be a solid or a liquid or both and is preferably formulated with the compound as a single dose, for example as a tablet, which may contain 0.05% to 95% by weight of the active ingredient. Other pharmaceutically active substances may likewise be present, including other compounds of formula I. The pharmaceutical compositions of the invention can be produced by one of the known pharmaceutical methods, which essentially consist of mixing the ingredients with pharmacologically acceptable carriers and/or excipients.
The present invention provides novel and potent PKC inhibitors. The compounds of the present invention are selective to PKC over other kinases and are isozyme-selective. Selective PKC inhibitors are useful in preventing and treating diseases associated with diabetes and diabetic complications (e.g. diabetic cardiomyopathy, diabetic
nephropathy, diabetic micro- and macrovascular complications, diabetic neuropathy and diabetic retinopathy, preferably diabetic nephropathy, diabetic neuropathy and diabetic retinopathy), cardiovascular diseases, diseases associated with hypertension- and non-hypertension-related and ischemic and non-ischemic end-organ damage (e.g. mycardial infarction, coronary heart disease, atherosclerosis, cardiac and renal hypertrophy, stroke), diseases associated with inflammation and fibrosis, central nervous system disorders (e.g. neuropathic pain), dermatological diseases,
autoimmune diseases (e.g. psoriasis, type 1 diabetes) and cancer (e.g. hematological tumours, glioma, gastric and intestinal cancer, skin cancer and lung cancer). Another subject of the present invention are processes for the preparation of the compounds of the formula I and their salts and solvates, by which the compounds are obtainable and which are outlined in the following.
Scheme 1 shows a first process of preparing compounds of the formula I


wherein R1 is a residue of the formula la


The group G4 in the compounds of formulae XI and XII is defined as a hydrogen or a protecting group for a phenolic hydroxyl group, such as, for example, a trialkylsilyl- group such as tert-butyldimethylsilyl-group, a 2-tetrahydropyranyl-group, a benzyl group, a 4-methoxybenzyl or a 2,5-dimethoxybenzyl group. The group G5 in the compounds of formula XI is a boronic acid or a boronic ester or cyclic boronic ester as described above.
As it is known to the skilled person the employed protection groups in the invention should be chosen in a manner to be compatible with the desired reaction conditions for all subsequent steps. All protection and deprotection reactions used in all above- and
hereinafter-described transformations are per se well known to the skilled person and can be carried out under standard conditions according to, or analogously to, procedures described in the literature, for example in P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, Stuttgart, 1994 or T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley, New York, 1999).
Compounds of the formula II can be obtained by heating 3-aminopyrazole with a 2-oxo succinic acid derivative of formula lla (or a tautomer and/or salt thereof) in the presence of an acid, such as aqueous hydrochloric acid or acetic acid or trifluoro acetic acid at temperatures from about 20 °C to about 120 °C. The reaction can be carried out in neat conditions or in a suitable inert solvent. The reaction time is generally from 30 min to 48 h, preferably from 30 min to 16 h, depending on the composition of the mixture and the chosen temperature range. Depending on the solvents and conditions applied the preferred regioisomer of formula II can be easily isolated by precipitation from the reaction mixture.
Compounds of the formula III are obtained by reacting compounds of the formula II with a chlorinating agent such as phosphorous oxychloride. This reaction can be carried out in neat conditions or in a suitable inert solvent, for example a hydrocarbon, such as benzene or toluene, or a chlorinated hydrocarbon, such as 1 ,2-dichloroethane by heating at a temperature between 50°C to 1 10°C.
Subsequent iodination of compounds of the formula III to obtain compounds of the formula IV can be conducted by treatment with iodine in presence of a base such as Na2CO3, NaHCO3, tBuOK in water or an inert organic solvent such as acetonitrile, dimethylformamide, tetrahydrofurane and dioxane or a mixture of solvents. Preferably this iodination reaction is performed with iodine in a saturated sodium hydrogencar- bonate aqueous solution at room tempearature for 1 to 3 days to lead to compounds of the formula IV which can be subjected to protection on pyrazole ring nitrogen with an appropriate G2 using conventional chemistry well known to those skilled in the art to give compounds of the formula V. If G2 is a cyclic ether protecting group (e.g., a tetrahdropyran group), such groups may be applied and subsequently removed under known conditions. Such cyclic ethers may be attached to nitrogen moieties and hydroxyl groups via their enol ethers (e.g., 3,4-dihydro-2H-pyran) in the presence of a
suitable acid (e.g., p-toluenesulfonic acid monohydrate or methanesulfonic acid), and solvent (e.g., dichloromethane). After functionalization of the C-3 position with iodine, and protection of the pyrazole ring nitrogen with a suitable protecting group G2, the C-3 position of the pyrazole ring can be elaborated to a desired vinylic R1 group through a Suzuki-type or Heck-type reaction using the appropriate catalyst, ligand, and vinyl aryl species to obtain a compound of formula VII.
A Suzuki-type reaction between compounds of the formula V with compounds of the formula VI wherein G3 is a boronic acid or a boronic ester or cyclic boronic ester, is carried out in the presence of catalytic palladium compound, for example a palla- dium(ll) salt such as palladium(ll) acetate or palladium(ll) chloride, which can be employed in the presence of a phosphine such as tricyclohexylphosphine or triphenyl- phosphine, or a palladium complex such as tetrakis(triphenylphosphine)palladium(0), palladium(O) bis(tri-tert-butylphosphine) or bis(triphenylphosphine)palladium(ll) chloride, and favourably in the presence of a base, for example an alkaline metal car- bonate or phosphate such as sodium carbonate or tripotassium phosphate, in an inert solvent, for example a hydrocarbon, such as benzene, toluene or xylene, or an ether, such as THF, dioxane or DME, or water, or a mixture of solvents, at temperatures from about 20 °C to about 200 °C, for example at temperatures from about 80 °C to about 1 10 °C. The reaction time is generally from 30 min to 24 h, preferably from 30 min to 16 h, depending on the composition of the mixture and the chosen temperature range. Depending on the chosen reaction conditions to carry out the Suzuki-type coupling reaction, an in situ deprotection of the carboxylic group of compounds of the formula VII can occur to give directly compounds of the formula VIII. If the G1 protecting group is preserved in the chosen Suzuki conditions, a subsequent deprotection of the car- boxylic group, preferably in basic media (e.g., aqueous sodium hydroxide) allow the transformation of compounds of the formula VII into compounds of the formula VIII. Alternatively the vinylic linkage can be installed through a Heck- type reaction by heating compounds of the formula V with compounds of the formula VI wherein G3 is a hydrogen in the presence of catalyst such as palladium(ll) acetate, a ligand such as tri- o- tolylphosphine, a suitable base such as triethylamine or N,N-diisopropylethyl-amine, and a solvent such as acetonitrile or DMF to provide compounds of the formula VII.
Subsequent removal of G1 protecting group as described above affords compounds of the formula VIII.
The reaction of the compounds of the formula VIII with an amine of the formula IX to form an amide of the formula X is generally performed in the presence of activating agents, such as CDI, DCC, EDC, HO At, HOBt, HATU, TOTU, TBTU, BEP or combinations thereof, and optionally an additional base, such as TEA, DIPEA or N-methyl- morpholine in an appropriate inert solvent, for example a hydrocarbon or a chlorinated hydrocarbon such as benzene, toluene, chlorobenzene, dichloromethane, dichloro- ethane, chloroform, or an ether such as tetrahydrofurane, 1 ,4-dioxane, dibutylether, diisopropylether, methyl-tert-butylether, dimethoxyethane, or an ester such as ethyl acetate or ethyl butanoate or an amide such as Ν,Ν-dimethylformamide or N,N-di- methylacetamide or N-methyl-pyrrolidinone or a in mixture of solvents. The reaction temperature in this case is generally from 30°C to 200°C, preferably from -20°C to 80°C, more preferably from 0°C to 20 °C. The reaction time is generally from 15 min to 6 days, preferably from 15 min to 16 h, depending on the composition of the mixture and the chosen temperature range. The acids of formula VIII can be subjected to the reaction in form of their salts, for example their sodium salts. They can also be transformed into an activated derivative prior to the coupling with the amine, for example into an acid chloride or an acid anhydride by standard transformations. The amines of formula IX can be subjected to the reaction in form of their salts, for example as hydrochloride or triflate salts, in which case usually an additional equivalent of the base is added to the reaction.
The reaction of the compound of the formula X with compounds of the formula XI to a compound of formula XII is a Suzuki-type reaction and is generally carried out as already described above in the presence of catalytic palladium compound, for example a palladium(ll) salt such as palladium(ll) acetate or palladium(ll) chloride, which can be employed in the presence of a phosphine such as tricyclehexylphosphine or triphenylphosphine, or a palladium complex such as tetrakis(triphenylphos- phine)palladium(O), palladium(O) bis(tri-tert-butylphosphin) or bis(triphenylphosphine)- palladium(ll) chloride, and favourably in the presence of a base, for example an alkaline metal carbonate or phosphate such as sodium carbonate or tripotassium phosphate, in an inert solvent, for example a hydrocarbon, such as benzene, toluene
or xylene, or an ether, such as THF, dioxane or DME, or water, or a mixture of solvents, at temperatures from about 20 °C to about 200 °C, for example at temperatures from about 80 °C to about 120 °C. The reaction time is generally from 30 min to 48 h, preferably from 30 min to 16 h, depending on the composition of the mixture and the chosen temperature range.
A compound of formula XII can be transformed into a compound of formula I in one step or in several steps depending of the meaning of the groups G2, G4, R a and R5a. If the groups R a and/or R5a contain or if the groups G2 and/or G4 consist of protecting groups that can be cleaved by hydrogenation, e.g. a benzyl group or a 4-methoxy- benzyl group, one step in the transformation of a compound of formula la to a compound of formula I can be a catalytic hydrogenation or a transfer hydrogenation. If the groups R a and/or R5a contain or if the groups G2 and/or G4 consist of protecting groups that can be cleaved by treatment with acid, e.g. a 2-tetrahydropyranyl group, tri- methylsilylethoxymethyl-group, a trialkylsilyl-group such as tert-butyldimethylsilyl- group, a 4-methoxybenzyl group, a 2,4-dimethoxybenzyl group or a tert-butoxycar- bonyl group, one step in the transformation of a compound of formula XII to a compound of formula I can be an acidic deprotection. If the groups R a and/or R5a contain or if the groups G2 and/or G4 consist of protecting groups that can be cleaved by treatment with a suitable fluoride source, for example tetrabutylammomium fluoride, e.g. a 2- trimethylsilylethoxymethyl-group, a trialkylsilyl-group such as tert-butyldimethylsilyl- group, one step in the transformation of a compound of formula XII to a compound of formula I can be an deprotection with a suitable fluoride source. All deprotection reactions used in the above-described transformation of compounds of the formula XII, in which the groups R a and/or R5a contain or in which the groups G2 and/or G4 consist of protecting groups, to compounds of formula I are per se well known to the skilled person and can be carried out under standard conditions according to, or analogously to, procedures described in the literature, for example in P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, Stuttgart, 1994 or T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley, New York, 1999).
A transformation of a compound of formula XII, in which the groups R a and/or R5a are precursors to the groups R4 and/or R5 into a compound of formula I can also include a functionalization or modification of contained functional groups according to standard
procedures, for example by esterification, amidation, hydrolysis, etherification, alkylation, acylation, sulfonylation, reduction, oxidation, conversion into salts, and others. For example, a hydroxy group, which may be obtained from a protected hydroxy group by deprotection, can be esterified to give a carboxylic acid ester or a sulfonic acid ester, or etherified. Etherifications of hydroxy groups can favorably be performed by alkylation with the respective halogen compound, for example a bromide or iodide, in the presence of a base, for example an alkaline metal carbonate such potassium carbonate or cesium carbonate in an inert solvent, for example an amide like dimethylformamide or N-methylpyrrolidinone or a ketone like acetone or butan-2- one, or with the respective alcohol under the conditions of the Mitsunobu reaction referred to above. A hydroxy group can be converted into a halide by treatment with a halogenating agent. A halogen atom can be replaced with a variety of groups in a substitution reaction which may also be a transition-metal catalyzed reaction. A nitro group can be reduced to an amino group, for example by catalytic hydrogenation. An amino group, which may be obtained from a protected hydroxy group by deprotection, can be modified under standard conditions for alkylation, for example by reaction with a halogen compound or by reductive amination of a carbonyl compound, or for acylation or sulfonylation, for example by reaction with a reactive carboxylic acid derivative, like an acid chloride or anhydride or a sulfonic acid chloride, or with a carboxylic acid in the presence of activating agents, such as CDI, DCC, EDC, HOAt, HOBt, HATU, TOTU, TBTU, BEP or combinations thereof, or for carbamoylation, for example by reaction with an isocyanate. A carboxylic ester group can be hydrolyzed under acidic or basic conditions to give a carboxylic acid. A carboxylic acid group can be activated or converted into a reactive derivative as mentioned afore and reacted with an alcohol or an amine or ammonia to give an ester or amide. A primary amide can be dehydrated to give a nitrile. A sulfur atom, for example in an alkyl-S- group or in a heterocyclic ring, can be oxidized with a peroxide like hydrogen peroxide or a peracid to give a sulfoxide moiety S(O) or a sulfone moiety S(0)2. A carboxylic acid group, carboxylic acid ester group and a ketone group can be reduced to an alcohol, for example by means of a complex hydride such as lithium aluminium hydride, lithium borohydride or sodium borohydride. All reactions used in the above-described syntheses of the compounds of the formula I are per se well known to the skilled
person and can be carried out under standard conditions according to, or analogously to, procedures described in the literature, for example in Houben-Weyl, Methoden der Organischen Chemie (Methods of Organic Chemistry), Thieme-Verlag, Stuttgart, or Organic Reactions, John Wiley & Sons, New York.
Scheme 2 shows a second process of preparing compounds of the formula I starting from a compound of the formula V
Scheme 2

XII wherein R1 is a residue of the formula la

Compounds of the formula XIII are obtained by submitting compounds of the formula V to carboxylic acid deprotection for example in aqueous sodium hydroxide. Subsequent reaction of the compounds of the formula XIII with an amine of the formula IX to form an amide of the formula XIV is generally performed in the presence of activating agents, such as CDI, DCC, EDC, HOAt, HOBt, HATU, TOTU, TBTU, BEP or combinations thereof, and optionally an additional base, such as TEA, DIPEA or N- methylmorpholine in an appropriate inert solvent, for example a hydrocarbon or a
chlorinated hydrocarbon such as benzene, toluene, chlorobenzene, dichloromethane, dichloroethane, chloroform, or an ether such as tetrahydrofurane, 1 ,4-dioxane, dibutyl- ether, diisopropylether, methyl-tert-butylether, dimethoxyethane, or an ester such as ethyl acetate or ethyl butanoate or an amide such as Ν,Ν-dimethylformamide or N,N- dimethylacetamide or N-methyl-pyrrolidinone or a in mixture of solvents. The reaction temperature in this case is generally from 30°C to 200°C, preferably from -20°C to 80°C, more preferably from 0°C to 20 °C. The reaction time is generally from 15 min to 6 days, preferably from 15 min to 16 h, depending on the composition of the mixture and the chosen temperature range. The acids of formula XIII can be subjected to the reaction in form of their salts, for example their sodium salts. They can also be transformed into an activated derivative prior to the coupling with the amine, for example into an acid chloride or an acid anhydride by standard transformations. The amines of formula IX can be subjected to the reaction in form of their salts, for example as hydrochloride or triflate salts, in which case usually an additional equivalent of the base is added to the reaction.
Subsequently, the C-3 position of the pyrazoie ring can be elaborated to a desired vinyl group to lead to compounds of the formula XV by treatment of compounds of the formula XIV with a suitable CH2=CH-organometallic reagent such as a CH2=CH-tin reagent, for example tributyl(vinyl)stannane, or a CH2=CH-boron reagent, for example potassium vinyltrifluoroborate, and a suitable catalyst, for example a palladium complex such as tetrakis(triphenylphosphine)palladium(0), or bis(triphenylphos- phine)palladium(ll) chloride, or (diphenyphosphinoferrocene)palladium(ll) chloride in a suitable organic solvent such as tetrahydrofurane, dioxane, N,N-dimethylformamide, N-methylpyrrolidinone or a mixture of solvents, at temperatures from about 20 °C to about 120 °C, preferably in presence of a base when using a CH2=CH-boron reagent such as potassium vinyltrifluoroborate, for example a tertiary amine, such as tri- ethylamine, or Ν,Ν-ethyldiisopropylamine. A Heck- type reaction is conducted by heating compounds of the formula XV with compounds of the formula XVI in the presence of catalyst such as palladium(ll) acetate, a ligand such as tri-o- tolylphosphine, a suitable base such as triethylamine or Ν,Ν-diisopropylethyl-amine, and a solvent such as acetonitrile or dimethylformamide to provide compounds of the formula XII. The reaction of the compound of the formula XVII with compounds of the formula XI to a
compound of formula XII is a Suzuki-type reaction and is generally carried out as already described above in the presence of catalytic palladium compound, for example a palladium(ll) salt such as palladium(ll) acetate or palladium(ll) chloride, which can be employed in the presence of a phosphine such as tricyclohexylphosphine or triphenylphosphine, or a palladium complex such as tetrakis(triphenylphosphine)- palladium(O), palladium(O) bis(tri-tert-butylphosphin) or bis(triphenylphosphine)- palladium(ll) chloride, and favourably in the presence of a base, for example an alkaline metal carbonate or phosphate such as sodium carbonate or tripotassium phosphate, in an inert solvent, for example a hydrocarbon, such as benzene, toluene or xylene, or an ether, such as THF, dioxane or DME, or water, or a mixture of solvents, at temperatures from about 20 °C to about 200 °C, for example at temperatures from about 80 °C to about 120 °C. The reaction time is generally from 30 min to 48 h, preferably from 30 min to 16 h, depending on the composition of the mixture and the chosen temperature range. Compounds of the formula XII can be transformed into compounds of the formula I in one or several steps as already described above in the first process of the present invention (see Scheme 1 ).
Additionally, Schemes 3 and 4 show two examples of alternative processes that are also part of the invention where the order of the various synthetic steps shown in Scheme 2 is modified to prepare compounds of the formula I

Scheme 4

and the groups R2, R3, R4, R5, R6, R7,R8 R9 and R10 in the compounds of formulae XI, XII, XVI, XIX, XX, XXI, XXII, XXIII and XXIV are defined as above in the compounds of the formula I. The groups R a and R5a in the compounds of formulae IX, XII and XXIII are, independently of each other, either defined as the groups R4 and R5 in formula I, or they are precursors of the groups R4 and R5 in formula I, they can for example contain functional groups in protected form or functional groups which can be
converted to obtain the final groups R4 and R5. The group G1 in the compounds of formulae V, XVIII ,ΧΙΧ, XX and XXII is a protecting group for a carboxylic acid, such as, for example methyl, ethyl, propyl, tert-butyl or benzyl, preferably methyl or ethyl. The group G2 in the compounds of formulae V, XII, XVIII, XIX, XX, XXI, XXII, XXIII and XXIV is defined as a protecting group for a pyrazole nitrogen, such as, for example, a 2-tetrahydropyranyl-group, a trimethylsilylethoxymethyl-group, a benzyl group, a 4- methoxybenzyl , a 2,5-dimethoxybenzyl group, a trialkylsilyl-group such as tert- butyldimethylsilyl-group. The group G4 in the compounds of formulae XI, XII, XX, XXI, XXII, XXIII and XIV is defined as a hydrogen or a protecting group for a phenolic hydroxyl group, such as, for example, a trialkylsilyl-group such as tert-butyldimethyl- silyl-group, a 2-tetrahydropyranyl-group, a benzyl group, a 4-methoxybenzyl or a 2,5-dimethoxybenzyl group. The group G5 in the compounds of the formula XI is a boronic acid or a boronic ester or cyclic boronic ester. The group G6 in the compound of formula XVI is a leaving group, that can be replaced in a Heck-type reaction, such as a halide, e.g. iodide, bromide or chloride or as a sulfonate, e.g. a trifluoromethane- sulfonate.
As depicted in Scheme 3 and using procedures similar to those described in the previous process, compounds of the formula V can undergo a direct vinylation at C-3 position to give compounds of the formula XVIII followed by a Heck-type reaction with a suitable compound of the formula XVI lead to compounds of the formula XIX. Using procedures previously presented in the invention, a Suzuki-type reaction with a suitable compound of the formula XI affords compounds of the formula XX which undergo deprotection of the carboxylic acid group to give compounds of the formula XXI which are transformed to amides of formula XII by coupling with amines of the formula IX. Compounds of the formula XII can be transformed into compounds of the formula I in one or several steps as already described above in the first process of the present invention (see Scheme 1 ).
As depicted in Scheme 4 and using procedures described previously in the invention, compounds of the formula XVIII obtained through vinylation of V can be submitted to a Suzuki-type reaction with a compound of the formula XI to give compounds of the formula XXII. Subsequent cleavage of G2 protective group to obtain compounds of for-
mula XXIII is followed by coupling with an amine of the formula IX to afford amides of the formula XXIV. Subsequent Heck-type reaction with a compound of the formula XVI give compounds of the formula XII which are subsequently transformed to the desired compounds of the formula I in one or several steps as described in the first process of the present invention (see Scheme 1 ).
As another part of the invention, compounds of the formula I wherein R1 is ethylene- phenyl, wherein phenyl is unsubstituted or substituted by one or two identical or differrent substituents selected from F, CI, CH3, 0-CH3, CF3, 0-CF3, CN, S02CH3, CO- CH3, and CO-N((CH3)2 can be obtained in one step by reduction, for example by catalytic hydrogenation, of the corresponding vinylic compounds of the formula I which can be prepared following the synthetic sequences outlined in Schemes 1 , 2, 3 and 4.
As another part of the invention, Scheme 5 shows a process of preparing compounds of the formula I
Scheme 5

XXVIII I
wherein R1 is 2-phenylcyclopropyl, wherein phenyl is unsubstituted or substituted by one or two identical or different substituents selected from F, CI, CH3, 0-CH3, CF3, 0-CF3, CN, S02CH3, CO-CH3, and CO-N((CH3)2,
and the groups R2, R3 in the compounds of formulae XXVI, XXVII and XXVIII are defined as in the compounds of the formula I. The groups R a and R5a in the compounds of formulae IX and XXVIII are, independently of each other, either defined as the groups R4 and R5 in formula I, or they are precursors of the groups R4 and R5 in formula I, they can for example contain functional groups in protected form or functional groups which can be converted to obtain the final groups R4 and R5.The group G4 in the compounds of formulae XXVI, XXVII and XXVIII are defined as a hydrogen or a protecting group for a phenolic hydroxyl group, such as, for example, a trialkylsilyl-group such as tert-butyldimethylsilyl-group, a 2-tetrahydropyranyl-group, a benzyl group, a 4-methoxybenzyl or a 2,5-dimethoxybenzyl group.
The reaction of an aminopyrazole compound of the formula XXV (prepared as described in WO2008021924) with a benzaldehyde of the formula XXVI and pyruvic acid is conducted in the presence of acids, preferably at reflux in glacial acetic acid.
Depending on the solvents and conditions applied the preferred regioisomer of formula XXVII can be easily isolated by precipitation from the reaction mixture. A compound of formula XXVII is transformed to amide of the formula XXVIII by coupling with an amine of formula IX using a coupling procedure as described previously in Scheme 1 to transform compounds of the formula VIII into compounds of the formula XII.
A compound of the formula XXVIII can already be a compound of formula I, if G4 is H and if R a is R4 and R5a is R5. If a compound of formula XXVIII is not already a compound of formula I, it can be transformed into a compound of formula I in one step or in several steps depending of the meaning of the groups G4, R a and R5a. If the groups R a and/or R5a contain or if the group G4 consist of a protecting group that can be cleaved by hydrogenation, e.g. a benzyl group or a 4-methoxybenzyl group, one step in the transformation of a compound of formula XXVIII to a compound of formula I can be a catalytic hydrogenation or a transfer hydrogenation. If the groups R a and/or R5a contain or if the group G4 consist of a protecting group that can be cleaved by treatment with acid, e.g. a 2-tetrahydropyranyl group, a trialkylsilyl-group such as tert- butyldimethylsilyl-group, a 4-methoxybenzyl group, a 2,4-dimethoxybenzyl group or a tert-butoxycarbonyl group, one step in the transformation of a compound of formula XXVII to a compound of formula I can be an acidic deprotection. If the groups R a and/or R5a contain or if the group G4 consist of protecting group that can be cleaved by
treatment with a suitable fluoride source, for example tetrabutylammonium fluoride, e.g. a trialkylsilyl-group such as tert-butyldimethylsilyl-group, one step in the
transformation of a compound of formula XXVII to a compound of formula I can be an deprotection with a suitable fluoride source. All deprotection reactions used in the above-described transformation of compounds of the formula XII, in which the groups R a and/or R5a contain or in which the group G4 consist of protecting groups, to compounds of formula I are per se well known to the skilled person and can be carried out under standard conditions according to, or analogously to, procedures described in the literature, for example in P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, Stuttgart, 1994 or T. W. Greene, P. G. M. Wuts, Protective Groups in Organic
Synthesis, John Wiley, New York, 1999).
As another part of the invention, Scheme 6 shows a process of preparing compounds of the formula I
Scheme 6

XXXIV wherein R1 is ethylene-(C5-C7)cycloalkyl,
and the groups R2 and R3 in the compounds of formulae XI, XXXIII and XXXIV are de- fined as above in the compounds of the formula I. The groups R a and R5a in the compounds of formulae IX, XXXII, XXXIII and XXXIV are, independently of each other, either defined as the groups R4 and R5 in formula I, or they are precursors of the groups R4 and R5 in formula I, they can for example contain functional groups in protected form or functional groups which can be converted to obtain the final groups R4 and R5. The group G1 in the compounds of formulae V and XXX is a protecting group for a carboxylic acid, such as, for example methyl, ethyl, propyl, tert-butyl or benzyl,
preferably methyl or ethyl. The group G2 in the compounds of formulae V, XXX, XXXI, XXXII, XXXIII and XXXIV is defined as a protecting group for a pyrazole nitrogen, such as, for example, a 2-tetrahydropyranyl-group, a trimethylsilylethoxymethyl-group, a benzyl group, a 4-methoxybenzyl , a 2,5-dimethoxybenzyl group, a trialkylsilyl-group such as tert-butyldimethylsilyl-group. The group G4 in the compounds of formulae XI, XXXIII and XXXIV is defined as a hydrogen or a protecting group for a phenolic hydroxyl group, such as, for example, a trialkylsilyl-group such as tert- butyldimethylsilyl-group, a 2-tetrahydropyranyl-group, a benzyl group, a 4-methoxybenzyl or a 2,5-dimethoxybenzyl group. The group G5 in the compounds of the formula XI is a boronic acid or a boronic ester or cyclic boronic ester.
The alkyne linkage of compounds of the formula XXX can be installed under typical Sonogashira reaction conditions by heating a compound of the formula V with an ω- alkyne derivative of the formula XXIX in the presence of catalytic palladium compound, for example a palladium complex such as tetrakis(triphenylphosphine)palladium(0), and favourably in the presence of a base, for example a tertiary amine, such as triethylamine, and favourably in the presence of a copper (I) halide, for example copper(l) iodide, in an inert solvent, for example an ether, such as THF, dioxane or an amide, such as Ν,Ν-dimethylformamide or a mixture of solvents, at temperatures from about 20 °C to about 200 °C, for example at temperatures from about 20°C to about 1 10 °C. The reaction time is generally from 30 min to 24 h, preferably from 30 min to 16 h, depending on the composition of the mixture and the chosen temperature range. Subsequent removal of G1 protecting group preferably under basic conditions (e.g., aqueous sodium hydroxyde) affords compounds of the formula XXXI which are converted to amides of the formula XXXII by coupling with amines of the formula IX followed by a typical Suzuki-type reaction with compounds of the formula XI to afford compounds of the formula XXXIII by using conditions described above to transform compounds of the formula X to compounds of the formula XII in the first process of the present invention (see Scheme 1 ). Compounds of the formula XXXIII are transformed to compounds of the formula XXXIV through reduction , for example catalytic hydrogenation in presence of a palladium compound. A compound of formula XXXIV can be transformed into a compound of formula I in one step or in several steps depending of the meaning of the groups G2, G4, R a and R5a. If the groups R a and/or
R5a contain or if the groups G2 and/or G4 consist of protecting groups that can be cleaved by hydrogenation, e.g. a benzyl group or a 4-methoxybenzyl group, one step in the transformation of a compound of formula la to a compound of formula I can be a catalytic hydrogenation or a transfer hydrogenation. If the groups R a and/or R5a contain or if the groups G2 and/or G4 consist of protecting groups that can be cleaved by treatment with acid, e.g. a 2-tetrahydropyranyl group, trimethylsilylethoxymethyl- group, a trialkylsilyl-group such as tert-butyldimethylsilyl-group, a 4-methoxybenzyl group, a 2,4-dimethoxybenzyl group or a tert-butoxycarbonyl group, one step in the transformation of a compound of formula XII to a compound of formula I can be an acidic deprotection. If the groups R a and/or R5a contain or if the groups G2 and/or G4 consist of protecting groups that can be cleaved by treatment with a suitable fluoride source, for example tetrabutylammomium fluoride, e.g. a 2- trimethylsilylethoxymethyl- group, a trialkylsilyl-group such as tert-butyldimethylsilyl-group, one step in the transformation of a compound of formula XII to a compound of formula I can be an deprotection with a suitable fluoride source. All deprotection reactions used in the above-described transformation of compounds of the formula XXXIV, in which the groups R a and/or R5a contain or in which the groups G2 and/or G4 consist of protecting groups, to compounds of formula I are per se well known to the skilled person and can be carried out under standard conditions according to, or analogously to, procedures described in the literature, for example in P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, Stuttgart, 1994 or T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley, New York, 1999).
A another part of the invention, the compounds of the formula I, II, III, IV, XXVII and XXVIII may also be present in another tautomeric form as 2H-pyrazolo[3,4-b]pyridine derivatives in which the mobile hydrogen atom, which in formula I, II, III, IV, XXVII and XXVIII is bonded to the ring nitrogen atom in the 1 -position of the pyrazolo[3,4-b]- pyridine ring system, is bonded to the ring nitrogen atom in the 2-position of the pyrazolo[3,4-b]pyridine ring system. As far as applicable, it applies to all compounds occurring in the preparation of the compounds of the formula I that they can be present in any other tautomeric form than the one represented in their formulae.
As far as applicable and unless otherwise indicated, it applies to all acidic or basic compounds occurring in the preparation of the compounds of the formula I that they can be present in form of their salts. All reactions used in the above-described syntheses of the compounds of the formula I are per se well known to the skilled person and can be carried out under standard conditions according to, or analogously to, procedures described in the literature, for example in Houben-Weyl, Methoden der Organischen Chemie (Methods of Organic Chemistry), Thieme-Verlag, Stuttgart, or Organic Reactions, John Wiley & Sons, New York. If desired, the obtained compounds of the formula I, as well as any intermediate compounds, can be purified by customary purification procedures, for example by recrystallization or chromatography. As already mentioned, all starting compounds and intermediates employed into the above-described syntheses which contain an acidic or basic group, can also be employed in the form of salts, and all intermediates and final target compounds can also be obtained in the form of salts. As likewise mentioned above, depending on the circumstances of the specific case, in order to avoid an unwanted course of a reaction or side reactions during the synthesis of a compound it can generally be necessary or advantageous to temporarily block functional groups by introducing protective groups and deprotect them at a later stage of the synthesis, or to introduce functional groups in the form of precursor groups which later are converted into the desired functional groups. As examples of protecting groups amino-protecting groups may be mentioned which can be acyl groups or alkyloxycarbonyl groups, for example a tert-butyloxycarbonyl group (= Boc) which can be removed by treatment with trifluoroacetic acid (= TFA), a benzyloxycarbonyl group which can be removed by catalytic hydrogenation, or a fluoren-9-ylmethoxycarbonyl group which can be removed by treatment with piperidine, and protecting groups of carboxylic acid groups which can be protected as ester groups, such as tert-butyl esters which can be deprotected by treatment with trifluoroacetic acid, or benzyl esters which can be deprotected by catalytic hydrogenation. As an example of a precursor group the nitro group may be mentioned which can be converted into an amino group by reduction, for example by catalytic hydrogenation. Such synthesis strategies, and protective groups and precursor groups which are suitable in a specific case, are known to the skilled person.
Another subject of the present invention are the novel starting compounds and intermediates occurring in the synthesis of the compounds of the formula I, including the compounds of the formulae II, Ma, III, IV, V, VI, VII, VIII, X, XI, XII, XIII, XIV, XV, XVI, XVII, XVIII, XIX, XX, XXI, XXII, XXIII, XXIV, XXV, XXVI, XXVII, XXVIII, XXIX, XXX, XXXI , XXXII, XXXIII and XXXIV wherein R1, R2 R3, R4, R5 R a, R5a, G1, G2 G3, G4 G5 and G6 are defined as above, in any of their stereoisomeric forms or a mixture of stereoisomeric forms in any ratio, and their salts, and solvates of any of them, and their use as intermediates. The invention also includes all tautomeric forms of the said intermediates and starting compounds. All explanations given above and embodiments specified above with respect to the compounds of the formula I apply correspondingly to the said intermediates and starting compounds. Another subject of the invention are in particular the novel specific starting compounds and intermediates disclosed herein. Independently thereof whether they are disclosed as a free compound and/or as a specific salt, they are a subject of the invention both in the form of the free compounds and in the form of their salts, and if a specific salt is disclosed, additionally in the form of this specific salt, and in the form of solvates of any of them.
The following examples illustrate the invention.
When example compounds containing a basic group were purified by preparative high pressure liquid chromatography (HPLC) on reversed phase (RP) column material and, as customary, the eluent was a gradient mixture of water and acetonitrile containing trifluoroacetic acid (TFA), they were in part obtained in the form of their acid addition salt with trifluoroacetic acid, depending on the details of the workup such as evaporation or lyophilization conditions. In the names of the example compounds and their structural formulae any such contained trifluoroacetic acid is not specified. When example compounds containing a basic group were obtained after an acidic deprotec- tion step, they were in part obtained in the form of their acid addition salt with hydro- chloric acid, depending on the details of the workup such as washing steps with bases or evaporation or lyophilization conditions. In the names of the example compounds
and their structural formulae any such contained hydrochloric acid may or may not be specified.
The prepared compounds were in general characterized by spectroscopic data and chromatographic data, in particular mass spectra (MS) and HPLC retention times (Rt; in min) which were obtained by combined analytical HPLC/MS characterization
(LC/MS), and/or nuclear magnetic resonance (NMR) spectra. In the NMR characterization, the chemical shift δ (in ppm), the number of hydrogen atoms and the multiplicity (s = singlet, d = doublet, dd = double doublet, t = triplet, dt = double triplet, q = quartet, m = multiplet; br = broad) of the peaks is given. In the MS characterization, in general the mass number (m/z) of the peak of the molecular ion M, e.g. M+, or of a related ion such as the ion M+1 , e.g. [M+1 ]+, i.e. the protonated molecular ion [M+H]+, which was formed depending on the ionization method used, is given. Generally, the ionization method was electrospray ionization (ESI). The LC/MS conditions used were as follows.
Method LC1 :
Column: Waters Xbridge C18 4.6 mm x 50 mm, 2.5 μΜ; flow: 1 .7 ml/min; solvent A: water + 0.05% trifluoroacetic acid; solvent B: acetonitrile + 0.05% trifluoroacetic acid; gradient 95% A + 5% B for 0.2 min, then from 95% A + 5% B to 5% A + 95% B in 2.2 min, then 5% A + 95% B for 1 .1 min; MS ionisation method: EST.
Method LC2:
Column Waters Xbridge C18 4.6 mm x 50 mm, 2.5 μΜ; flow: 1 .3 ml/min; solvent A: water + 0.1 % formic acid; solvent B: acetonitrile + 0.1 % formic acid; gradient from 97% A + 3% B to 40% A + 60% B in 3.5min, then to 2% A + 98% B in 0.5min; then 2% A + 98% B fori .0 min; then to 97% A + 3% B in 0.2 min, then 97% A + 3% B for 1 .3 min; MS ionisation method: EST.
Method LC3:
Column: BEH C18 2.1 x 50 mm; 1.7μηη; flow: 0.9 ml; solvent A: water + 0.1 % formic acid; solvent B: acetonitrile + 0.08 % formic acid; gradient from 95% A + 5% B to 5% A + 95% B in 1 .1 min; then 5% A + 95 % B for 0.6 min; MS ionisation method: EST.
Method LC8:
Instrument: Waters Acquity UPLC/SQD; column: BEH C18 (2.1 *50mm, 1 .7μηι); 50°C; 0.8ml_/min; solvent: A: H20 + 0.1 % HC02H; solvent B: ACN +0.1 % HC02H: gradient: t=0: 5%B; t=0.05 min : 6%B; t=2.5min: 100%B; MS ionisation method: ESI+ or ESI-.
Method LC9:
UPLC / TOF - Column Acquity BEH C18 (50x2.1 mm ; 1 .7μηι); 40°C; gradient 3min; ; 1 .Oml/min; solvent A : H20 + 0.05% TFA ; solvent B : ACN + 0.035% TFA; gradient: TO : 98%A - T1.6 to T2.1 min : 100%B - T2.5 to T3min : 98%A; MS ionisation method: ESI+ or ESI-.
Method LC10:
Instrument: HPLC/ZQ; column Kromasil C18 (50*2, 1 mm ; 3,5μηπ); 40°C; 0.8ml_/min; solvent A : CH3COONH4 5mM + 3%ACN; solvent B : ACN; gradient: TO : 100%A - T5, 5 a T7min : 100%B - T7, 1 a T10min : 100%A; MS ionisation method: ESI+ or ESI-.
Method LC1 1
Instrument: HPLC/ZMD; column XBridge 4.6x30mm, 3pm, 1 ml/min; gradient 10 min; 1 . Oml/min; gradient A(0.1 % HC02H dans H20)/B (0.1 % HC02H dans ACN):t=0: 5% B; t=5.5min : 100% B; t=7min: 100% B; MS ionisation method: ESI+ or ESI-.
List of abbreviations
BEP 2-bromo-1 -ethyl-pyridinium tetrafluoroborate
BOC tert.butoxy carbonyl
CDI N,N'-carbonyldiimidazole
DCC 1 ,3-dicyclohexylcarbodiimide
DCM dichloromethane
DEA diethylamine
DMF N,N-dimethylformamide
EDC 1 -(3-dimethylaminopropyl)-3-ethylcarbodiimide
hydrochloride
EtOAc ethyl acetate
EtOH ethanol
Exp. No. Example number
h hour/s
HOAt 1 -hydroxy-aza-benzotriazole
HOBt 1 -hydroxy-benzotirazole
HATU 0-(7-azabenzotnazol-1 -yl)-N,N,N',N'-tetramethyl- uronium hexafluorophosphate
HPLC high pressure liquid chromatography
LC liquid chromatography
MeOH methanol
Min minutes
MS mass spectroscopy
Rt retention time
r.t. room temperature
sep. separation
TBTU [(benzotriazol-1 -yloxy)-dimethylamino-methylene]- dimethyl-ammonium tetrafluoroborate
TEA triethylamine
TFA trifluoroacetic acid
THF tetrahydrofurane
TOTU 0-(cyano(ethoxycarbonyl)methyleneam ino)-
Ν,Ν,Ν',Ν'-tetramethyluronium tetrafluoroborate
Example 1 :
[3-[(E)-2-(4-Chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridi piperazin-1 -yl-methanone

1 .1 : 6-Hydroxy-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester

To a solution of 3-aminopyrazole (224 g, 2.7 mol) in water (1 L) were added a solution of diethyl oxalacetate sodium salt (567 g, 2.7 mol) in water (1 .3 L) and then acetic acid in 4 portions (0.8 L). The reaction mixture was heated at 85°C for 15 hours then cooled to room temperature and filtered. The precipitate was washed with water and then dried to afford the title compound as an orange solid (159 g, 28%).
1 H NMR (250 MHz, DMSO): δ 1 .35 (t, 3H), 4.36 (q, 2H,), 6.69 (s, 1 H), 8.16 (s, 1 H).
1 .2: 6-chloro-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester

A mixture of 6-hydroxy-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester (1 16.4 g, 0.562 mol) and phosphorus oxychloride (500 ml_) was heated at 1 10°C for 6 hours then cooled to room temperature before being evaporated under reduced pressure. The residue was taken up in EtOAc and the precipitate filtered. The filtrate was
neutralized using triethylamine (500 ml_) and the resulting salt filtered. The filtrate was concentrated under vacuum and the residue subjected to silica gel column
chromatography using a 9/1 mixture of toluene/EtOAc to afford the title compound as a yellow solid (26.3 g, 21 %).
1 H NMR (200 MHz, DMSO): δ 1 .4 (t, 3H), 4.43 (q, 2H,), 7.60 (s, 1 H), 8.37 (s, 1 H).
1 .3: 6-chloro-3-iodo-1 H-pyrazolo[ -b]pyridine-4-carboxylic acid ethyl ester

In a 1 L single necked flask were charged 6-chloro-1 H-pyrazolo[3,4-b]pyridine-4-car- boxylic acid ethyl ester (10g, 44.3 mmol) and dioxane (400ml). To this resulting suspension were added a saturated solution of sodium hydrogen carbonate (0.5M, 260 ml) and iodine (1 1 .2g, 44.3 mmol). The reaction mixture was stirred at room temperature for 24 hours, iodine was then added (25g, 97.5 mmol) and stirring was pursued for 48 hours. Next an aqueous sodium thiosulfate solution (0.9M) was added until com- plete decolouration and was poured into a solution of ice and HCI at pH=2. The mixture was filtered to afford the title compound as a yellow solid (14.7g, 95%).
LC/MS (Method LC8): Rt = 1 .53 min; m/z = 351 [M+H]+.
1 .4: 6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxyli acid ethyl ester

To a solution of 6-chloro-3-iodo-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester (14g, 39.8 mmol) and p-toluenesulfonic acid monohydrate (0.75g, 3.9 mmol) in
anhydrous chloroform (200ml) at 0°C was added dropwise 3,4-dihydro-2H-pyran (5.45ml, 59.7 mmol). The reaction mixture was allowed to warm to room temperature, stirred overnight and concentrated under reduced pressure. The residue was subjected to silica gel column chromatography using a stepwise gradient of ethyl acetate (0-20%) in cyclohexane to afford the title compound as a yellow solid (9.4 g, 54%). LC/MS (Method LC8): Rt = 2.08 min; m/z = 351 [M+H-THP]+.
1 .5: 6-chloro-3-[(E)-2-(4-chloro-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo- [3,4-b]pyridine-4-carboxylic acid

To a solution of 6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4- carboxylic acid ethyl ester (2g, 4.59 mmol) in dioxane (51 ml) were added trans-2-(4- chlorophenyl)vinylboronic acid (0.83g, 4.59 mmol) and an aqueous solution of cesium carbonate (0.3M, 8.26 mmol). The resulting solution was degased using argon, tetra- kis(triphenylphosphine)palladium (0.26g, 0.23 mmol) was then added and the reaction mixture was refluxed overnight. Dioxane was removed under vacuum, methylene chloride and water were added, and the mixture was acidified until pH=2. The organic layer was washed with water and the aqueous phase was extracted using methylene chloride. The resulting organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the title compound (2g) as a yellow foam which was used without any further purification.
LC/MS (Method LC8): Rt = 2.10 min; m/z = 334 [M+H-THP]+.
1 .6: 4-[6-chloro-3-[(E)-2-(4-chloro-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 6-chloro-3-[(E)-2-(4-chloro-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carboxylic acid (2g, 4.79 mmol) in a mixture of anhydrous tetrahydrofuran (42ml) and N,N-dimethylacetamide (10 ml) were added 1 -boc-pipera- zine (0.98g, 5.27 mmol), 1 -hydroxybenzotriazole (0.06g, 0.48 mmol), and N-(3-di- methylaminopropyl)-N'-ethylcarbodiimide hydrochloride (0.91 g, 4.79 mmol). The reaction mixture was stirred at room temperature for 4 hours, tetrahydrofuran was eva- porated under reduced pressure, water and dichloromethane were then added. The organic phase was separated, dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The crude residue was purified on a silica gel column chromatography using a gradient of ethyl acetate (0-45%) in cyclohexane to yield the title compound as a yellow foam (1 .51 g, 54%).
LC/MS (Method LC8): Rt = 2.34 min; m/z = 586 [M+H]+.
1 .7: 4-[3-[(E)-2-(4-chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)- 1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 4-[6-chloro-3-[(E)-2-(4-chloro-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)- 1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester (1.5g, 2.56 mmol) in dioxane (38ml) were added 4-hydroxyphenylboronic acid (0.39g, 2.81 mmol) and an aqueous solution of cesium carbonate (0.3M, 4.6 mmol). The resulting solution was degased using argon, tetrakis(triphenylphosphine)palladium (0.14g, 0.13 mmol) was then added and the reaction mixture was refluxed for 3 hours. Solvents were removed under vacuum. The resulting crude mixture was submitted to a silica gel column chromatography using a gradient of ethyl acetate (0-45%) in cyclo- hexane to give the title compound (1.32g, 80%) as a white solid.
LC/MS (Method LC8): Rt = 2.25 min; m/z = 644 [M+H]+.
1 .8: [3-[(E)-2-(4-chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone

LC/MS (Method LC10): Rt = 3.47 min; m/z = 458 [M-H]+.
The following examples have been prepared in a similar fashion as the synthesis for [3-[(E)-2-(4-chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone described immediately above. In the fifth step trans-stryryl- boronic acid instead of trans-2-(4-chlorophenyl)vinylboronic acid was employed to yield 6-chloro-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (LC/MS (Method LC8): Rt = 1.88 min; m/z = 300.1 [M+H-THP]+). This was reacted in the sixth step with the respective pipirazines and the respective phenylboronic acids in the seventh step and deproteceted in the eigth step:



Example 14:
[3-[(E)-2-(3-Dimethylaminomethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone

14.1 : 6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid

To a solution of 6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4- carboxylic acid ethyl ester (25 g, 57 mmol) in 1 ,4-dioxane (200 ml_) was added an aqueous sodium hyroxide solution (1 M, 180 ml_). The reaction mixture was stirred at room temperature for 7 hours and then an aqueous HCI solution (1 M, 200 ml_) was carefully added. Water (140 ml_) was added, and after 5 minutes of stirring the precipitate was filtered, washed with water, cyclohexane and then dried to afford the title compound as a yellow solid (17.1 g, 73 %).
LC/MS (Method LC8): Rt = 1 .48 min; m/z = 324 [M+H-THP]+.
14.2: 4-[6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester
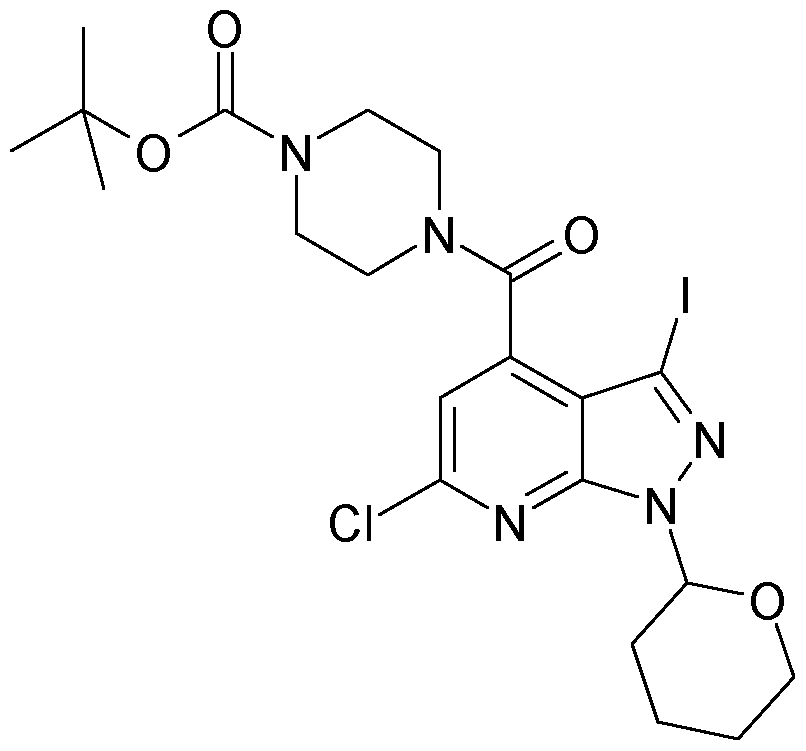
To a solution of 6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4- carboxylic acid (17 g, 42 mmol) in a mixture of tetrahydrofuran (250 ml_) and N,N-di- methylacetamide (40 ml_) were added 1 -boc-piperazine (8.6 g, 46 mmol), 1 -hydroxy- benzotriazole (0.56 g, 4.2 mmol), and N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (8.03 g, 42 mmol). The reaction mixture was stirred at room temperature for 20 hours and then the THF was evaporated under vacuum. An aqueous solution of bicarbonate (200 ml_) was then added and the mixture stirred for 1 hour. The precipitate was filtered, washed with water, and then dried to afford the title compound as a yellow solid (24 g, 100 %).
LC/MS (Method LC8): Rt = 1 .75 min; m/z = 436 [M+H-THP-tBu]+.
14.3: 4-[6-chloro-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carbo- nyl]-piperazine-1 -carboxylic acid tert-butyl ester

LC/MS (Method LC8): Rt = 1 .74 min; m/z = 475 [M+H]+. 14.4: (3-iodo-benzyl)-dimethyl-amine

To a cold (0°C) solution of 3-iodo-benzaldehyde (3g, 12.9 mmol) in anhydrous di- chloromethane (130 ml) were added acetic acid (1 .9ml) and N,N-dimethylacetamide. The yellow solution is stirred 10 minutes at 0°C and sodium triacetoxyborohydride (6.85g, 32.33 mmoles) is added by portions. The resulting mixture is stirred at room temperature for 90 minutes and evaporated to dryness under reduced pressure. Ethyl acetate and a saturated aqueous hydrogen carbonate solution were added to the residue and the two layers were separated The organic phase was washed with water, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum to yield (3-iodo-benzyl)-dimethyl-amine (3.05g, 90%) as a brown oil.
LC/MS (Method LC8): Rt = 0.86 min; m/z = 262 [M+H]+.
14.5: 4-[6-chloro-3-[(E)-2-(3-dimethylaminomethyl-phenyl)-vinyl]-1 -(tetrahydro-pyran-2- yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

In a 250 ml round bottom flask were charged 4-[6-chloro-1 -(tetrahydro-pyran-2-yl)-3- vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester (1.2g, 2.52 mmol), (3-iodo-benzyl)-dimethyl-amine (1.64g, 6.3 mmol), tetrabutyl- ammonium bromide (0.32g, 1 mmol), sodium hydrogen carbonate (0.53g, 6.3 mmol) and anhydrous dimethylformamide (42ml). The mixture was degased during 10 minutes using argon, palladium acetate (0.05g, 0.25 mmol) was then added. The resulting solution was heated at 100°C under argon overnight. Dimethylformamide was eliminated under vacuum, dichloromethane and water were added to the residue, after extraction the organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the crude product. Further purification on a silica gel column chromatography using a gradient of methanol (0-5%) in dichloromethane afforded the title compound as brown solid (0.44g, 29%).
LC/MS (Method LC8): Rt = 1 .17 min; m/z = 609 [M+H]+.
14.6: 4-[3-[(E)-2-(3-dimethylaminomethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 -(tetra- hydropyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester
O
N
/
Utilizing the procedure described in example 1 .7 except substituting 4-[6-chloro-3-[(E)- 2-(4-chloro-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-car- bonyl]-piperazine-1 -carboxylic acid tert-butyl ester with 4-[6-chloro-3-[(E)-2-(3-di- methylaminomethyl-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine- 4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester, 4-[3-[(E)-2-(3-dimethylamino- methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo- [3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester was obtained (0.25g, 52%) as a yellow solid.
LC/MS (Method LC8): Rt = 1 .22 min; m/z = 667 [M+H]+.
14.7: [3-[(E)-2-(3-dimethylaminomethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazo- lo[3,4-b]pyridin-4-yl]-piperazin-1 -yl-methanone

To a cold solution (0°C) of 4-[3-[(E)-2-(3-dimethylaminomethyl-phenyl)-vinyl]-6-(4- hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-pipe- razine-1 -carboxylic acid tert-butyl ester (0.25g, 0.37 mmol) in dry methanol (5 ml) was
added dropwise a solution of aqueous 6N HCI (0.7ml, 3.8 mmol). The reaction mixture was allowed to warm to room temperature and stirring was pursued for 2 hours before being concentrated under reduced pressure. Aqueous 6N HCI (0.7ml, 3.8 mmol) was added to the crude mixture and stirring was pursued for another hour. Solvents were evaporated under vacuum, methanol and diethyl ether were added, the resulting precipitate was filtered and dried to give a chlorhydrate salt of the title compound as an orange solid (0.21 g; 95%).
LC/MS (Method LC9): Rt = 0.68 min; m/z = 483 [M+H]+. Example 15:
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methanesulfonylmethyl-phenyl)-vinyl]-1 H- pyrazolo[3,4-b]pyridin-4-yl}-piperazin-1 -yl-methanone

Using the same synthetic sequence described in example 14 except substituting (3- iodo-benzyl)-dimethyl-amine with 1 -iodo-3-[(methylsulfonyl)methyl]-benzene (preparation described in WO0015609), {6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methanesulfonyl- methyl-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4-yl}-piperazin-1 -yl-methanone was obtained as a chlorhydrate salt.
LC/MS (Method LC9): Rt = 0.8 min; m/z = 518 [M+H]+.
Example 16:
((R)-2-Hydroxymethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone

16.1 : 6-chloro-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester
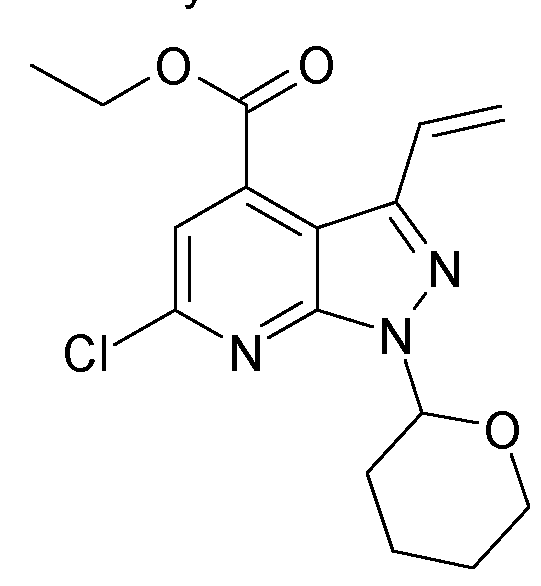
To a solution of 6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4- carboxylic acid ethyl ester (5.00 g, 1 1 .48 mmol) in anhydrous tetrahydrofuran (1 15ml) was added tributyl(vinyl)stannane (3.52 ml, 12.05 mmol). The mixture was degased for 15 minutes using argon, dichlorobis(triphenylphosphine)palladium (0.40 g, 0.57 mmol) was then added. The resulting solution was heated at 80°C overnight under argon, then evaporated under reduced pressure. Ethyl acetate and a 5% aqueous potassium fluoride solution were added to the residue, the resulting precipitate was filtered. The filtrate was recovered and the two layers separated, the organic phase was washed with brine, dried (Na2SO4), filtered and concentrated under vacuum. The crude product was purified on a silica gel column chromatography using a gradient of ethyl acetate (0-10%) in cyclohexane to afford the title compound as a yellow solid (3.24 g, 84%). LC/MS (Method LC8): Rt = 1 .97 min; m/z = 251 [M+H-THP]+.
16.2: 6-chloro-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-car- boxylic acid ethyl ester

Utilizing the procedure described in example 14.5 except substituting 4-[6-chloro-1 - (tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -car- boxylic acid tert-butyl ester with 6-chloro-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyra- zolo[3,4-b]pyridine-4-carboxylic acid ethyl ester and substituting (3-iodo-benzyl)-di- methyl-amine for iodobenzene, 6-chloro-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester was obtained (2.8g, 65%) as a yellow solid.
LC/MS (Method LC8): Rt = 2.22 min; m/z = 412 [M+H]+.
16.3: 6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]- pyridine-4-carboxylic acid

To a solution of 6-chloro-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyri dine-4-carboxylic acid ethyl ester (2.8 g, 6.80 mmol) in dioxane (60ml) were added 4- hydroxyphenylboronic acid (1 .03 g, 7.48 mmol) and an aqueous solution of cesium
carbonate (0.4M, 30 ml_). The resulting solution was degased using argon, tetrakis(tri- phenylphosphine)palladium (0.71 g, 0.61 mmol) was then added and the reaction mixture was refluxed overnight. Dioxane was removed under vacuum, methylene chloride and water were added, and the mixture was acidified until pH=2. The organic layer was washed with water and the aqueous phase was extracted using methylene chloride. The resulting organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the title compound (2.37g, 79%) as a yellow solid which was used without any further purification.
LC/MS (Method LC8): Rt = 1 .92 min; m/z = 442 [M+H]+.
16.4: (R)-3-hydroxymethyl-4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2- -1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

Utilizing the procedure described in example 1 .6 except substituting 6-chloro-3-[(E)-2- (4-chloro-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-car- boxylic acid with 6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyra- zolo[3,4-b]pyridine-4-carboxylic acid and 1 -boc-piperazine with (R)-3-hydroxymethyl- piperazine-1 -carboxylic acid tert-butyl ester, (R)-3-hydroxymethyl-4-[6-(4-hydroxy-phe- nyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-pi- perazine-1 -carboxylic acid tert-butyl ester (0.16g, 28%) was obtained as a yellow solid. LC/MS (Method LC8): Rt = 1 .86 min; m/z = 640 [M+H]+.
16.5: ((R)-2-hydroxymethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone

Utilizing the procedure described in example 1 .8 except substituting 4-[3-[(E)-2-(4- chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo- [3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester with (R)-3- hydroxymethyl-4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4 b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester, hydrochloride salt of ((R)-2-hydroxymethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)- styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-methanone was obtained (0.12g, 91 %) as a red solid.
LC/MS (Method LC9): Rt = 0.8 min; m/z = 456 [M+H]+. Example 17:
[6-(4-Hydroxy-phenyl)-3-phenethyl-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone

17.1 : 4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4- b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

Utilizing the procedure described in example 1 until the seventh step except substituting trans-2-(4-chlorophenyl)vinylboronic acid with trans-2-phenylvinylboronic acid, (4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyri- dine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester was obtained as a yellow solid.
LC/MS (Method LC8): Rt = 1 .99 min; m/z = 610 [M+H]+.
17.2: 4-[6-(4-hydroxy-phenyl)-3-phenethyl-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4- b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester (0.74g, 1 .22 mmol) in dry methanol (40 ml) was added Pd/C (150 mg). The reaction mixture was stirred under a hydrogen atmosphere for 1 hour, filtered and concentrated under reduced pressure. The crude residue was taken in dry methanol (30ml) and Pd/C (150
mg) was added. The mixture was submitted to a hydrogen pressure of 1 bar and was stirred for 1 hour, filtered and concentrated under vacuum to yield the title compound (0.72g, 96%).
LC/MS (Method LC8): Rt = 2.10 min; m/z = 612 [M+H]+.
17.3: [6-(4-hydroxy-phenyl)-3-phenethyl-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone

Utilizing the procedure described in example 1 .8 except substituting 4-[3-[(E)-2-(4-chlo- ro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyri- dine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester with 4-[6-(4-hydroxy-phe- nyl)-3-phenethyl-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-pipe- razine-1 -carboxylic acid tert-butyl ester, a chlorhydrate salt of [6-(4-hydroxy-phenyl)-3- phenethyl-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl-methanone was obtained (0.5g, quantitative) as a yellow solid.
LC/MS (Method LC9): Rt = 0.73 min; m/z = 428 [M+H]+.
Example 18:
(±)-[6-(4-Hydroxy-phenyl)-3 trans-(2-phenyl-cyclopropyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone

1 -3-oxo-3 trans-(2-phenyl-cyclopropyl)-propionitrile

To a suspension of sodium hydride (60% in oil, 2.03 g, 50.78 mmol) in dry dioxane (60 ml_) was added dropwise acetonitrile (2.65 ml_, 50.78 mmol). After 30 min, a solution of (±)-ethyl trans-2-phenylcyclopropanecarboxylate (8.05 g, 42.31 mmol) in dry dioxane (30 ml) was added dropwise to the reaction mixture. The suspension was heated at 100°C for 3 hours, cooled down to 0°C prior to the addition of water and di- chloromethane. Aqueous 1 M HCI solution was added to acidified the mixture to pH=5. The organic layer was separated and the aqueous layer extracted with dichloro- methane. The combined organic extracts were dried (MgSO4), filtered, concentrated under vacuum. The crude residue was purified on a silica gel column chromatography using a gradient of EtOAc (0-30%) in cyclohexane to yield the title compound (3.17 g, 40%) as a colourless oil.
LC/MS (Method LC8): Rt = 1 .28 min; m/z = 186 [M+H]+.
18.2: (±)-5 trans-(2-phenyl-cyclopropyl)-2H-pyrazol-3-ylamine

LC/MS (Method LC8): Rt = 0.71 min; m/z = 200 [M+H]+.
18.3: (±)-6-(4-Hydroxy-phenyl)-3-(trans-2-phenyl-cyclopropyl)-1 H-pyrazolo[3,4-b]pyri- dine-4-carboxylic acid

(±)-5-trans-(2-Phenyl-cyclopropyl)-2H-pyrazol-3-ylamine (3.24 g, 16.26 mmol), 4-hy- droxybenzaldehyde (1 .98 g, 16.26 mmol), pyruvic acid (1 .13 ml, 16.26 mmol) and glacial acetic acid (25 ml) were heated to reflux under nitrogen for 1.5 hours and then evaporated under reduced pressure. The residue was taken up in diethyl ether, the precipitate was filtered and washed with diethyl ether to afford the crude title compound (4 g, 67 %) as a brown solid.
LC/MS (Method LC8): Rt = 1 .53 min; m/z = 372 [M+H]+. 18.4: (±)- 4-[6-(4-Hydroxy-phenyl)-3 trans-(2-phenyl-cyclopropyl)-1 H-pyrazolo[3,4-b]py- ridin-4-yl]- piperazine-1 -carboxylic acid tert-butyl ester

To a solution of (±)-6-(4-hydroxy-phenyl)-3-(trans-2-phenyl-cyclopropyl)-1 H-pyrazolo- [3,4-b]pyridine-4-carboxylic acid (1 .2 g, 3.23 mmol) in a mixture of tetrahydrofuran (29 ml_) and N,N-dimethylacetamide (3 ml_) were added 1 -boc-piperazine (0.72 g, 3.88 mmol), 1 -hydroxybenzotriazole (52 mg, 0.39 mmol), and N-(3-dimethylamino- propyl)-N'-ethylcarbodiimide hydrochloride (0.74 g, 3.88 mmol). The reaction mixture was stirred at room temperature for 18 hours, EtOAc and an aqueous solution of citric acid were added. The organic layer was separated and the aqueous layer extracted with EtOAc. The combined organic extracts were washed with brine, dried (MgSO4), fil- tered, concentrated under vacuum. The crude residue was purified on a silica gel column chromatography using a gradient of EtOAc (20-60%) in cyclohexane to yield the title compound as a yellow wax (0.09 g, 9%).
LC/MS (Method LC8): Rt = 1 .68 min; m/z = 540 [M+H]+. 18.5: (±)-[6-(4-Hydroxy-phenyl)-3 trans-(2-phenyl-cyclopropyl)-1 H-pyrazolo[3,4-b]- pyridin-4-yl]-piperazin-1 -yl-methanone (SAR240501 A)

To a solution of (±)- 4-[6-(4-hydroxy-phenyl)-3 trans-(2-phenyl-cyclopropyl)-1 H-pyrazo- lo[3,4-b]pyridin-4-yl]- piperazine-1 -carboxylic acid tert-butyl ester (0.12 g, 0.23 mmol) in
methanol (3.3 ml) was added 4N HCI in dioxane (0.3 ml). The reaction mixture was stirred at room temperature for 18 hours, diethyl ether was then added, the resulting precipitated was filtered and washed once with diethyl ether to afford the hydrochloride salt of the title compound (0.09 g, 80%) as an orange solid.
LC/MS (Method LC9): Rt = 0.82 min; m/z = 440 [M+H]+.
Example 19:
(6-(4-Hydroxy-phenyl)-3-{(E)-2-[4-(piperidine-1 -carbonyl)-phenyl]-vinyl}-1 H- pyrazolo[3,4-b]pyridin-4-yl)-piperazin-1 -yl-methanone

19.1 : (4-iodo-phenyl)-piperidin-1 -yl-methanone

To a suspension of 4-iodo-benzoyl chloride (2.00 g, 7.51 mmol) in dichloromethane (25 ml_) was added piperidine (1 .85 ml_, 18.76 mmol). The reaction mixture was heated at reflux for 1 .5 hour and then cooled to room temperature. The organic layer was washed with an aqueous 1 M HCI solution, brine, dried (Na2SO4), filtered, concentrated under vacuum to give the title compound (2.37 g, 100%) as a white solid.
1 H NMR (400 MHz, CDCI3): δ 1 .40-1 .65 (m, 6H), 3.18-3.68 (m, 4H), 7.06-7.10 (m, 2H), 7.66-7.70 (m, 2H).
19.2: 6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4- b]pyridine-4-carboxylic acid

To a solution of 6-chloro-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine- 4-carboxylic acid ethyl ester (2.00 g, 5.96 mmol), described in example 16.1 , in dioxane (54 mL) were added 4-hydroxyphenylboronic acid (0.90 g, 6.55 mmol) and an aqueous solution of cesium carbonate (0.4 M, 27 mL, 10.8 mmol). The resulting solution was degased using argon, tetrakis(triphenylphosphine)palladium (0.62 g, 0.54 mmol) was then added and the reaction mixture was refluxed overnight. Dioxane was removed under vacuum, water was added and then the mixture was acidified to pH=2. The aqueous phase was extracted with a mixture of tetrahydrofuran and EtOAc. The combined organic extracts were washed with brine, dried (Na2SO4), filtered, concentrated under vacuum to give the title compound (3.13 g) as a brown foam which was used without further purification.
LC/MS (Method LC8): Rt = 1 .57 min; m/z = 366 [M+H]+.
19.3: 4-[6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyri- dine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4- b]pyridine-4-carboxylic acid (3.12 g, 6.62 mmol) in tetrahydrofuran (44 mL) were added 1 -boc-piperazine (1.35 g, 7.28 mmol), 1 -hydroxybenzotriazole (0.09 g, 0.66 mmol), and N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (1.27 g, 6.62 mmol). After 2 hours, N,N-dimethylacetamide (3 ml), 1 -boc-piperazine (0.24 g, 1 .32 mmol) and N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (0.25 g, 1.32 mmol) were added to the reaction mixture. After 1 hour, tetrahydrofuran was evaporated under reduced pressure, water and dichloromethane were then added. The organic layer was washed with an aqueous 1 M HCI solution, water, brine, dried (Na2S04), filtered, concentrated under vacuum. The crude residue was purified on a silica gel column chromatography using a gradient of methanol (0-5%) in dichloromethane to yield the title compound as a yellow foam (3.17 g, 90%).
LC/MS (Method LC8): Rt = 1.77 min; m/z = 534 [M+H]+.
19.4: 4-[6-[4-(tert-butyl-dimethyl-silanyloxy)-phenyl]-1 -(tetrahydro-pyran-2-yl)-3-vinyl- 1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 4-[6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo- [3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester (3.15 g,
5.90 mmol) in dichloromethane (22 ml) were added imidazole (0.96 g, 14.17 mmol) and tert-butyl(chloro)dimethylsilane (1 .07 g, 7.08 mmol). The reaction mixture was stirred at room temperature for 5 hours, water and dichloromethane were then added. The aqueous layer was separated and the organic layer was washed with water, brine, dried (Na2S04), filtered, concentrated under vacuum. The crude residue was purified on a silica gel column chromatography using a gradient of EtOAc (0-30%) in cyclo- hexane to yield the title compound as a yellow foam (2.05 g, 53%).
LC/MS (Method LC8): Rt = 2.41 min; m/z = 648 [M+H]+.
19.5: 4-[6-(4-Hydroxy-phenyl)-3-{(E)-2-[4-(piperidine-1 -carbonyl)-phenyl]-vinyl}-1 -(tetra- hydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 4-[6-[4-(tert-butyl-dimethyl-silanyloxy)-phenyl]-1 -(tetrahydro-pyran-2- yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert- butyl ester (0.3 g, 0.46 mmol) in DMF (2.1 mL) were added (4-iodo-phenyl)-piperidin-1 - yl-methanone (0.175 g, 0.56 mmol), tri(o-tolyl)phosphine (28 mg, 0.09 mmol) and tri- ethylamine (2.1 mL). The resulting solution was degased using argon, palladium acetate (5 mg, 0.02 mmol) was then added and the reaction mixture was heated at 125°C overnight. The reaction mixture was concentrated under vacuum and the residue taken up in water and then acidified to pH=1 . The precipitate was filtered, washed with water and then dried under vacuum. The crude residue was purified on a silica gel column chromatography using a gradient of methanol (0-3%) in dichloro- methane to yield the title compound as a light brown solid (0.25 g, 65%).
LC/MS (Method LC8): Rt = 1 .92 min; m/z = 721 [M+H]+. 19.6: (6-(4-Hydroxy-phenyl)-3-{(E)-2-[4-(piperidine-1 -carbonyl)-phenyl]-vinyl}-1 H-pyra- zolo[3,4-b]pyridin-4-yl)-piperazin-1 -yl-methanone

To a solution of 4-[4-[6-(4-Hydroxy-phenyl)-3-{(E)-2-[4-(piperidine-1 -carbonyl)-phenyl]- vinyl}-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 - carboxylic acid tert-butyl ester (0.25 g, 0.35 mmol) in methanol (5 ml) was added 4N HCI in dioxane (0.9 ml). The reaction mixture was stirred at room temperature for 18 hours, diethyl ether was then added, the resulting precipitated was filtered and washed once with diethyl ether to afford the hydrochloride salt of the title compound (0.175 g, 82%) as an orange solid.
LC/MS (Method LC9): Rt = 0.89 min; m/z = 537 [M+H]+.
Example 20:
1 -(3-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin- 3-yl]-vinyl}-phenyl)-pyrrolidin-2-one

20.1 : 4-Bromo-N-(3-iodo-phenyl)-butyramide

To a solution of 3-iodoaniline (1.5 g, 6.85 mmol) and triethylamine (1 .34 ml_,
9.59 mmol) in dichloromethane (28 ml_) at 0°C was added dropwise 4-bromobutyryl chloride (0.99 ml_, 8.56 mmol). The reaction mixture was then stirred at room tem- perature overnight. The organic extract was washed with an aqueous 1 M HCI solution, aqueous 1 M NaOH solution and brine, dried (Na2S04), filtered, concentrated under vacuum. The crude residue was purified by silica gel column chromatography using a gradient of EtOAc (0-15%) in cyclohexane to yield a sticky solid. Trituration in a mixture of cyclohexane and ether and then filtration give the title compound as a solid (1 .61 g, 64%).
1 H N MR (400 MHz, CDCI3): 5 2.17-2.33 (m, 2H), 2.58 (t, 2H), 3.54 (t, 1 H), 3.68 (t, 1 H), 7.03-7.08 (m, 1 H), 7.32-7.51 (m, 3H), 7.96 (s, 1 H).
20.2: 1 -(3-lodo-phenyl)-pyrrolidin-2-one

To a solution of 4-bromo-N-(3-iodo-phenyl)-butyramide (1.60 g, 4.36 mmol) in dry DMF (20 ml_) at 0°C was added portionwise sodium hydride (60% in oil, 0.20 g, 5.01 mmol). After 15 min at 0°C, the reaction mixture was then stirred at room temperature overnight. As the reaction was incomplete as judged by TLC, sodium hydride (60% in oil, 45 mg, 1 .09 mmol) was added. After two hours, water (40 ml_) and an aqueous solution of saturated ammonium chloride (40 ml_) were added to the reaction mixture. The aqueous phase was extractred with EtOAc and then the organic extracts were washed with brine, dried (Na2SO4), filtered, concentrated under vacuum. The crude residue was purified on a silica gel column chromatography using a gradient of EtOAc (0-30%) in cyclohexane to yield the title compound as a yellow solid (0.94 g, 75%).
1 H NMR (400 MHz, CDCI3): δ 2.14-2.23 (m, 2H), 2.63 (t, 2H), 3.85 (t, 2H), 7.1 (t, 1 H), 7.48-7.51 (m, 1 H), 7.66-7.70 (m, 1 H), 7.96 (t, 1 H).
20.3: 4-[6-(4-Hydroxy-phenyl)-3-{(E)-2-[3-(2-oxo-pyrrolidin-1 -yl)-phenyl]-vinyl}-1 -(tetra- hydropyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 4-[6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo- [3, 4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester (0.30 g, 0.56 mmol) in DMF (2.55 ml_) were added 1 -(3-iodo-phenyl)-pyrrolidin-2-one (0.19 g, 0.67 mmol), tri(o-tolyl)phosphine (34 mg, 0.1 1 mmol) and triethylamine (2.55 ml_). The resulting solution was degased using argon, palladium acetate (6 mg, 0.03 mmol) was then added and the reaction mixture was heated at 125°C overnight. The reaction mixture was concentrated under vacuum and the residue taken up in water and then acidified to pH=1 . The precipitate was filtered, washed with water and then dried under vacuum. The crude residue was purified by silica gel column chromatography using a gradient of EtOAc (0-100%) in cyclohexane to yield the title compound as a beige solid (0.26 g, 66%).
LC/MS (Method LC8): Rt = 1 .89 min; m/z = 693 [M+H]+.
20.4: 1 -(3-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]- pyridin-3-yl]-vinyl}-phenyl)-pyrrolidin-2-one

To a solution of 4-[6-(4-Hydroxy-phenyl)-3-{(E)-2-[3-(2-oxo-pyrrolidin-1 -yl)-phenyl]- vinyl}-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 - carboxylic acid tert-butyl ester (0.246 g, 0.36 mmol) in methanol (5 ml) was added 4N HCI in dioxane (0.9 ml). The reaction mixture was stirred at room temperature for 18 hours, diethyl ether was then added, the resulting precipitated was filtered and washed once with diethyl ether to afford the hydrochloride salt of the title compound (0.19 g, 93%) as an orange solid.
LC/MS (Method LC10): Rt = 2.98 min; m/z = 509 [M+H]+.
The following examples have been prepared in a similar fashion as the synthesis for 1 -(3-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin- 3-yl]-vinyl}-phenyl)-pyrrolidin-2-one described immediately above. In the third step 4-[6- (4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-car- bonyl]-piperazine-1 -carboxylic acid tert-butyl ester was reacted with the respective aryl halides, and deproteceted in the forth step:




Example 34:
3-{(E)-2-[4-(2,2-Dimethyl-piperazine-1 -carbonyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-3-yl]-vinyl}-N,N-dimethyl-benzamide

34.1 : 4-[6-(4-Hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]- pyridine-4-carbonyl]-3,3-dimethyl-piperazine-1 -carboxylic acid tert-butyl ester

4-[6-(4-Hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-3,3-dimethyl-piperazine-1 -carboxylic acid tert-butyl ester was obtained in a similar fashion as 4-[6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazo- lo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester employing 3,3-Dimethyl-piperazine-1 -carboxylic acid tert-butyl ester instead of piperazine-1 -car- boxylic acid tert-butyl ester.
LC/MS (Method LC8): Rt = 1 .86 min; m/z = 562.3 [M+H]+.
34.2: 3-{(E)-2-[4-(2,2-Dimethyl-piperazine-1 -carbonyl)-6-(4-hydroxy-phenyl)-1 H-pyra- zolo[3,4-b]pyridin-3-yl]-vinyl}-N,N-dimethyl-benzamide

3- {(E)-2-[4-(2,2-Dimethyl-piperazine-1 -carbonyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-3-yl]-vinyl}-N,N-dimethyl-benzamide has been prepared in a similar fashion as the synthesis for 1 -(3-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H- pyrazolo[3,4-b]pyridin-3-yl]-vinyl}-phenyl)-pyrrolidin-2-one. In the third step instead of
4- [6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester 4-[6-(4-Hydroxy-phenyl)-1 - (tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-3,3-dimethyl- piperazine-1 -carboxylic acid tert-butyl ester was reacted with 3-lodo-N,N-dimethyl- benzamide, and deproteceted in the forth step.
LC/MS (Method LC9): Rt = 0.78 min; m/z = 525 [M+H]+.
Example 35:
6-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one

35.1 : 6-Bromo-2-methylisoindolin-1 -one

To a solution of 5-bromo-2-bromomethyl-benzoic acid methyl ester (1.00 g, 3.25 mmol) in THF (6.5 ml_) was a solution of methylamine in THF (2M, 8.12 ml_, 16.24 mmol). The reaction mixture was heated at 50°C for 4 hours and then concentrated under vacuum. The residue was taken up in EtOAc and the organic layer was then washed with an aqueous 1 M HCI solution, brine, dried (Na2SO4), filtered, concentrated under vacuum to give the title compound (0.73 g, 100%) as a yellow solid.
1 H NMR (400 MHz, CDCI3): 5 3.19 (m, 3H), 4.32 (s, 2H), 7.31 (d, 1 H), 7.63 (dd, 1 H), 7.96 (d, 1 H).
35.2: 4-[6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-methyl-3-oxo-2,3-dihydro-1 H-isoindol-5-yl)- vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 - carboxylic acid tert-butyl ester

Utilizing the procedure described in example 20.3 except substituting 1 -(3-iodo- phenyl)-pyrrolidin-2-one with 6-bromo-2-methylisoindolin-1 -one lead to the title compound as a yellow solid.
LC/MS (Method LC8): Rt = 2.62 min; m/z = 679 [M+H]+.
35.3: 6-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyri- din-3- l]-vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one

To a solution of 4-[6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-methyl-3-oxo-2,3-dihydro-1 H-iso- indol-5-yl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-pipe- razine-1 -carboxylic acid tert-butyl ester (0.19 g, 0.28 mmol) in methanol (4 ml) was added 4N HCI in dioxane (0.7 ml). The reaction mixture was stirred at room tempera-
ture for 3 days, diethyl ether was then added, the resulting precipitated was filtered and washed once with diethyl ether to afford the hydrochloride salt of the title compound (0.15 g, 96%) as an orange solid.
LC/MS (Method LC10): Rt = 2.82 min; m/z = 495 [M+H]+.
Example 36:
[3-(2-cyclohexyl-ethyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin- 1 -yl-methanone

36.1 : 6-chloro-3-cyclohexylethynyl-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4- b]pyridine-4-carboxylic acid ethyl ester

A solution of 6-chloro-3-iodo-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4- carboxylic acid ethyl ester (1.0 g, 2.30 mmol) and triethylamine (1.92 mL, 13.77 mmol) in THF (7.65 mL) was degased using argon during 3 min. Cyclohexylacetylene (0.45 mL, 3.44 mmol), copper(l) iodide (0.08 g, 0.46 mmol) and tetrakis(triphenylphosphine)-
palladium (0.13 g, 0.1 1 mmol) were then added and the reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated under vacuum and the residue purified on a silica gel column chromatography using a gradient of ethyl acetate (0-5%) in cyclohexane to afford the title compound as a beige solid (0.93 g, 97%).
LC/MS (Method LC8): Rt = 2.27 min; m/z = 416 [M+H]+.
36.2: 3-cyclohexylethynyl-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carboxylic acid

Utilizing the procedure described in example 16.3 except substituting 6-chloro-3-((E)- styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid ethyl ester with 6-chloro-3-cyclohexylethynyl-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]py- ridine-4-carboxylic acid ethyl ester, 3-cyclohexylethynyl-6-(4-hydroxy-phenyl)-1 -(tetra- hydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (0.93 g, 97%) was obtained as a beige solid.
LC/MS (Method LC8): Rt = 1 .91 min; m/z = 446 [M+H]+.
36.3: 4-[3-cyclohexylethynyl-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

Utilizing the procedure described in example 1 .6 except substituting 6-chloro-3-[(E)-2- (4-chloro-phenyl)-vinyl]-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxy- lic acid with 3-cyclohexylethynyl-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H-py- razolo[3,4-b]pyridine-4-carboxylic acid, 4-[3-cyclohexylethynyl-6-(4-hydroxy-phenyl)-1 - (tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester (0.72 g, 96%) was obtained as a yellow solid.
LC/MS (Method LC8): Rt = 2.06 min; m/z = 614 [M+H]+. 36.4: 4-[3-(2-cyclohexyl-ethyl)-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester

To a solution of 4-[3-cyclohexylethynyl-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)- 1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester (0.34 g, 0.56 mmol) in methanol (15 ml) was added Pd/C (10%, 12 mg, 0.01 mmol).
The reaction mixture was hydrogenated at 4 bar for 18h, Pd/C (10%, 12 mg, 0.01 mmol) was added and hydrogenation pursued for an additional 8 hours. The reaction mixture was then filtered and concentrated under reduced pressure to yield the title compound (0.33 g, 95%) as a grey solid.
LC/MS (Method LC8): Rt = 2.17 min; m/z = 618 [M+H]+.
36.5: [3-(2-cyclohexyl-ethyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone

Utilizing the procedure described in example 1 .8 except substituting 4-[3-[(E)-2-(4-chlo- ro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyri- dine-4-carbonyl]-piperazine-1 -carboxylic acid tert-butyl ester with 4-[3-(2-cyclohexyl- ethyl)-6-(4-hydroxy-phenyl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-car- bonyl]-piperazine-1 -carboxylic acid tert-butyl ester, a chlorhydrate salt of [3-(2-cyclo- hexyl-ethyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl-methanone was obtained (0.15 g, 58%) as a yellow solid.
LC/MS (Method LC10): Rt = 3.47 min; m/z = 434 [M+H]+.
Example 37:
[3-[(E)-2-(4-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone

0.25 mmol of 1 -fluoro-4-iodo-benzene were weighted into a reaction tube. 2.5 mmol sodium hadrogencarbonate was added as solid, followed by 0.25 mmol tetrabutyl- ammonium fluoride in 0.5 ml DMF and 0.1 mmol of 4-[6-(4-hydroxy-phenyl)-1 -(tetra- hydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxy- lic acid tert-butyl ester in 1 ml DMF. The tube was purged with Argon. 0.01 mmol palla- dium(ll) acetate in 0.25 mmol DMF was added, the tube was closed with a screw cap and stirred for 8 h at 80 °C. New palladium(ll) acetate (0.01 mmol) was added and heating continued for 1 h at 100 °C. The reaction mixture was diluted with 20 ml ethyl acetate and washed with 20 ml water, dried and evaporated. The residue was dissolved in 5 ml THF and 1 ml methanol and shaken with 100 mg silica-bound propane thiol scavenger for 4h at RT. The mixture was filtered, the filter washed with THF, the filtrate evaporated and the residue submitted to SFC purification. The purified product was dissolved in 3 ml methanol and 0.5 ml 4N HCI in dioxane. The mixture was stirred for 30 min at 65 °C. The cooled solution was diluted slowly with 10 ml diethyl ether to precipitate the product, which was filtered off, washed with 1 ml diethyl ether, and dried in vacuo to afford [3-[(E)-2-(4-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo- [3,4-b]pyridin-4-yl]-piperazin-1 -yl-methanone (18 mg, 37%).
LC/MS (Method LC1 ): Rt = 1 ,74min; m/z = 444.17 [M+H]+.
1H-NMR (500MHz, d6-DMSO): 2,72-3,27 (bm, 4H), 3,4-3,5 (bm, 3H), 4,22-4,44 (bm, 1 H) 6,92 (d, 2H), 7, 1 1 (d, 1 H), 7,23 (m, 2H), 7,32 (d, 1 H), 7,61 (m, 2H), 7.79 (s, 1 H), 8.09 (d, 2H), 9.2 (bs, 1 H), 9.5 (bs, 1 H)
The following examples have been prepared in a similar fashion as the synthesis for [3-[(E)-2-(4-fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone described immediately above employing 4-[6-(4-hydroxy- phenyl)-1 -(tetrahydro-pyran-2-yl)-3-vinyl-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-pipera- zine-1 -carboxylic acid tert-butyl ester and the respective aryl iodides or bromides:

met anone


methanone

methanone


methanone
m/z
Exp. LC/MS Rt
Name Structure [M+H] No. method [min]
+
[3-[(E)-2-(2,3-
H
Dihydro- benzofuran-5-yl)- vinyl]-6-(4-
64 hydroxy-phenyl)- LC1 468.27 1 .74
1 H-pyrazolo[3,4- b]pyridin-4-yl]- piperazin-1 -yl- H
methanone
[3-((E)-2- H
Benzo[1 ,3]dioxol-
5-yl-vinyl)-6-(4- hydroxy-phenyl)-
65 LC1 470.24 1 .76
1 H-pyrazolo[3,4- b]pyridin-4-yl]- piperazin-1 -yl- methanone H
4-{(E)-2-[6-(4-
Hydroxy-phenyl)- H
4-(piperazine-1 - carbonyl)-1 H- O
66 pyrazolo[3,4- LC1 497.29 1 .63 b]pyridin-3-yl]- vinyl}-N,N- dimethyl- H
benzamide
[3-[(E)-2-(4- H
Ethoxy-phenyl)- vinyl]-6-(4- hydroxy-phenyl)-
67 LC1 470.26 1 .84
1 H-pyrazolo[3,4- b]pyridin-4-yl]- piperazin-1 -yl-
H
methanone
 m/z
m/z
Exp. LC/MS Rt
Name Structure [M+H]
No. method [min]
+
{6-(4-Hydroxy- phenyl)-3-[(E)-2-
(4-methoxy-3- methyl-phenyl)-
73 vinyl]-1 H- LC1 470.21 1 .84 pyrazolo[3,4- b]pyridin-4-yl}- piperazin-1 -yl-

methanone
Example 74:
4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazine-1 -carboxylic acid dimethylamide

0.24 mmol of Piperazine-1 -carboxylic acid dimethylamide were weighted into a reaction tube and dissolved/suspended in 1 ml THF. 1 ml of a DMF stock solution containing 0.24 mmol of 6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H- pyrazolo[3,4-b]pyridine-4-carboxylic acid, 1 mmol N-methyl morpholine, 0.01 mmol 4- dimethylaminopyridine, and 0.3 mmol 1 -hydroxybenzotnazole were added, followed by 0.3 mmol EDC. The tube was closed and shaken at 50 °C overnight. The THF was left to evaporate, 0.1 ml TFA was added, the volume was adjusted to 2ml with DMF. The solution was filtered and submitted to prep. RP HPLC purification to give 4-[6-(4-Hy- droxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-car- bonyl]-piperazine-1 -carboxylic acid dimethylamide. In order to transform the trifluoro
acetic acid salt into a hydrochloric acid salt and - for other examples to cleave acid labile protecting groups - the product obtained above was shaken with 3 ml methanol and 0.5 ml 4M HCI in dioxane at 65 °C for 30 minutes. Then 10 ml diethyl ether was slowly added to the cooled solution, and the formed precipitate was filtered off, washed with 1 ml diethyl ether, and dried in vacuo to afford 0.085 mmol of 4-[6-(4-Hydroxy- phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazine-1 -carboxylic acid dimethylamide (35%).
LC/MS (Method LC2): Rt = 3,82min; m/z = 497.26 [M+H]+.
1 H-NMR (400MHz, d6-DMSO): 2,70 (s, 6H), 2, 14-2,34 (m, 1 H), 2,90-3,40 (bm, 4H), 3,30-4,0 (bm, 4H), 6,91 (d, 2H), 7,20 (d, 1 H), 7,29-7,42 (m, 4H), 7,56 (d, 2H), 7,70 (s, 1 H), 8.09 (d, 2H), 10 (bs, 1 H).
The following examples have been prepared in a similar fashion as the synthesis for 4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]-piperazi- ne-1 -carboxylic acid dimethylamide described immediately above employing 6-(4-hy- droxy-phenyl)-3-((E)-styryl)-1 -(tetrahydro-pyran-2-yl)-1 H-pyrazolo[3,4-b]pyridine-4-car- boxylic acid and the respective amines:






Determination of PKC βΙΙ inhibition
PKC βΙΙ inhibition was determined according to the following protocol:
Active human full-length recombinant PKC βΙΙ and the peptide substrate, Fluorescein- RFARKGSLRQKNV, were purchased from Invitrogen GmbH, Darmstadt, Germany. Adenosine-5' -triphosphate (ATP), bovine serum albumine (BSA), dimethylsulphoxide (DMSO), 4-(2-hydroxyethyl)piperazine-1 -ethanesulfonic acid (Hepes), Triton X-100, 1 ,2-Dioleoyl-sn-glycerol (DAG), L-a-Phosphatidyl-L-serine (PS), calcium chloride (CaC ), and Pluronic F-68 were purchased from Sigma-Aldrich, Munich, Germany. Magnesium chloride, 1 M sodium hydroxide solution, 1 M hydrochloric acid solution and EDTA were obtained from Merck Biosciences, Darmstadt, Germany.
Test compounds were diluted to three times the test concentration in buffer 1 (30 mM Hepes-NaOH, pH 7.4, 0.01 % Pluronic F-68 and 3 % (v/v) DMSO). The PKC βΙΙ enzyme was diluted to a concentration of 30 ng/ml in buffer 2 (30 mM Hepes-NaOH, pH 7.4, 15 mM MgCI2, 150 μΜ CaCI2, 150 pg/ml PS, 60 pg/ml DAG, and 0.045 % (w/v) Triton X-100). The peptide substrate and ATP were diluted to concentrations of 3 μΜ and 120 μΜ, respectively, in buffer 2. Two μΙ of the compound solution were mixed with 2 μΙ of the diluted enzyme in a 384-well small volume microtiter plate (Greiner, Bio-One, Frickenhausen, Germany), and the kinase reaction was initiated by addition of 2 μΙ of the solution containing peptide substrate and ATP. After 60 min incubation at 32 °C, the reaction was stopped by addition of 20 μΙ of a solution containing 130 mM Hepes-NaOH, pH 7.4, 0.0195 % (v/v) Brij-35, 6.5 mM EDTA, 0.13 % chip coating reagent 3 (Caliper Lifescience Inc, Hopkinton, MA) and 6.5 % (v/v) DMSO. Phosphorylation of the substrate peptide was then detected on a Caliper 3000 instrument essentially as described by Pommereau et al (J. Biomol. Screening 2004, 9(5), 409- 416). Separation conditions were as follows: Pressure -0.8 psi, upstream voltage -3000 V, downstream voltage -800 V. Positive controls (buffer 1 instead of compound) and negative controls (buffer 1 instead of compound and buffer 2 instead of kinase solution) were run in parallel on each plate. Fractional turnover (R) was determined as the height of the peak of the phosphorylated peptide product divided by the sum of the unphosphorylated substrate and phosphorylated product peak heights. Percent-inhibition values for the test compounds were determined using the formula % Inhibition
— 100 (1 "(Rcompound - Rnegative control)/(Rpositive control " Rnegative control)-
In the following table % inhibition values observed with example compounds at a final concentration of 1 .14 μΜ (±0.15 μΜ) are listed:
Exp. No. % Inhibition
1 86
2 97
3 88
4 99
5 85
6 98
7 96
8 97
9 55
10 87
1 1 99
12 97
13 1 10
14 100
15 99
16 96
17 94
18 94
19 99
20 1 15
21 98
22 100
23 99
24 99
25 99
26 96
27 98
28 94
Exp. No. % Inhibition
29 96
30 90
31 99
32 99
33 99
34 98
35 98
36 81
37 97
38 98
39 39
40 99
41 97
42 66
43 87
44 97
45 61
46 97
47 98
48 97
49 88
50 92
51 94
52 91
53 95
54 95
55 76
56 100
57 98
58 89
59 97
60 98
Exp. No. % Inhibition
61 86
62 98
63 97
64 99
65 97
66 99
67 99
68 60
69 94
70 98
71 96
72 99
73 98
74 27
75 93
76 57
77 79
78 94
79 97
80 26
81 45
82 56
83 90
84 54
85 19
86 93
87 94
88 89
Claims
Claims
1 . A compound of the formula I

wherein
R1 is a) a residue of the formula la

R6 is H, F, CI, (Ci-C3)-alkyl or O-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F;
R7 is H, F, CI, (Ci-C3)-alkyl or O-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is CN, (C0-C2)- alkylene-SO2(Ci-C3)-alkyl, (Co-C2)-alkylene-N((Ci-C4)-alkyl)2, CO- N((Ci-C4)-alkyl)2, 1 -pyrrolidinyl, 1 -piperidinyl, 1 -pyrrolidinyl-2-on, 1 - piperidinyl-2-on, or 4-morpholinyl;
R8 is H, F, CI, (Ci-C5)-alkyl or O-(Ci-C5)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is CN, OH, O- phenyl, SO2(Ci-C3)-alkyl, CO-(Ci-C )-alkyl, CO-N((Ci-C )-alkyl)2, CO-1 -piperidinyl, CO-1 -pyrrolidinyl, or CO-4-morpholinyl;
R9 is H, F, CH3, or O-CH3;
R10 is H; or
R6 and R7 form, together with the two carbon atoms, to which they are attached, a five to six membered heterocyloalkyl ring, which contains one or two identical or different heteroatoms selected from nitrogen and oxygen, and wherein said heterocycloalkyl is unsubstituted or mono-substituted by (Ci-C4)-alkyl or oxo (=0); or
R7 and R8 form, together with the two carbon atoms, to which they are attached, a five to six membered heterocyloalkyl ring, which contains one or two identical or different heteroatoms selected from nitrogen and oxygen, and wherein said heterocycloalkyl is unsubstituted or mono-substituted by (Ci-C4)-alkyl or oxo (=0); b) ethylene-phenyl, wherein phenyl is unsubstituted or substituted by one or two identical or different substituents selected from F, CI, CH3, 0-CH3, CF3, O-CFs, CN, SO2CH3, CO-CH3, and CO-N((CH3)2;
c) 2-phenylcyclopropyl, wherein phenyl is unsubstituted or substituted by one or two identical or different substituents selected from F, CI, CH3, 0- CH3, CF3, 0-CF3, CN, S02CH3, CO-CH3, and CO-N((CH3)2; d) ethylene-(C5-C7)cylcoalkyl; R2 H, F, CI, or CH3;
R3 H, F, or 0-CH3;
R4 piperidin-4-yl, piperidin-3-yl or pyrrolidin-3-yl, which are unsubstituted or
monosubstituted by (Ci-C3)-alkyl, which is unsubstituted or substituted by one to five F;
R5 is H; or
J R5 together with the nitrogen carrying them denote
a) a residue of the formula lb
lb

R11 is H, (C3-C5)-cycloalkyl, (Ci-C4)-alkylene-OH or (Ci-C4)-alkyl, which is unsubstituted or substituted by one to five F;
R12 is H, or (Ci-C )-alkyl,
R13 is H, (Ci-C )-alkyl, or (C3-C5)-cycloalkyl;
R14 is H, or (Ci-C )-alkyl; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclo-(C3-C6)-alkyl; and
R15 is H, CH2-CO-N((Ci-C )-alkyl)2, CO-(Ci-C6)-alkyl,
CO-(Ci-C )-alkylene-OH, CO-(Ci-C )-alkylene-0-CH3, CO-CH2-4- morpholinyl, CO-0-(Ci-C )-alkyl, CO-N((Ci-C )-alkyl)2, (C3-C6)- cycloalkyl, or pyridyl;
R16 is H, or (Ci-C )-alkyl;
R17 is H, or CH3;
R18 is H, or CH3;
R19 is H; b) a 9 to 1 1 membered spiro-heterocycloalkyl containing 2 nitrogen atoms, which is attached via a nitrogen atom; in any of its stereoisomeric forms, or a mixture of stereoisomeric forms in any ratio, or a physiologically acceptable salt thereof, or a physiologically acceptable solvate of any of them.
2. A compound of the formula I as claimed in claim 1 , wherein
R1 is a) a residue of the formula la

R6 is H, F, CI, (Ci-C3)-alkyl or 0-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F;
R7 is H, F, CI, (Ci-C3)-alkyl or 0-(Ci-C3)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is CN, SO2(Ci-C3)- alkyl, N((Ci-C4)-alkyl)2, 1 -piperidinyl, or 4-morpholinyl;
R8 is H, F, CI, (Ci-C5)-alkyl or 0-(Ci-C5)-alkyl, wherein (Ci-C3)-alkyl is unsubstituted or substituted by one to five F, or is OH, O-phenyl, SO2(Ci-C3)-alkyl, CO-(Ci-C )-alkyl, CO-N((Ci-C )-alkyl)2;
R9 is H, F, CH3, O-CH3;
R10 is H; or
R7 and R8 together are
 b) ethylene-phenyl, wherein phenyl is unsubstituted or substituted by one to two identical or different substituents selected from F, CI, CH3, O-CH3, CF3, O-CF3, CN, SO2CH3, CO-CH3, and CO-N((CH3)2; R2 is H, F, CH3;
b) ethylene-phenyl, wherein phenyl is unsubstituted or substituted by one to two identical or different substituents selected from F, CI, CH3, O-CH3, CF3, O-CF3, CN, SO2CH3, CO-CH3, and CO-N((CH3)2; R2 is H, F, CH3;
R3 is H, O-CH3.
R4 is piperidin-4-yl, piperidin-3-yl or pyrrolidin-3-yl, which are unsubstituted or monosubstituted by (Ci-C3)-alkyl, which is unsubstituted or substituted by one to five F; preferably 4-methyl-piperidin-4-yl and
R5 is H; or
R4 and R5 together with the nitrogen carrying them denote
a) a residue of the formula lb
 wherein
wherein
R is H, CH3, (C3-C4)-alkyl, or (C3-C )-cycloalkyl;
R12 is H;
R13 is H, (Ci-C )-alkyl, or (C3-C )-cycloalkyl;
R14 is H, or CH3; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclo-(C3-C6)-alkyl; preferably 1 '-spiro-cyclopentyl; and
R15 is H, CH2-CO-N((Ci-C )-alkyl)2, CO-(Ci-C6)-alkyl,
CO-(Ci-C )-alkylene-OH, CO-(Ci-C )-alkylene-0-CH3, CO-CH2-4- morpholinyl, CO-0-(Ci-C )-alkyl, CO-N(-(Ci-C )-alkyl)2, -(C3-C6)- cycloalkyl, or pyridyl;
R16 is H, or (Ci-C )-alkyl;
b) a 9 to 1 1 membered spiro-heterocycloalkyl containing two nitrogen
atoms, which is attached via a nitrogen atom.
A compound of the formula I as claimed in claim 1 or 2, wherein a) a residue of the formula la
la

R6 is H, F, CI, CH3, CF3, or 0-CH3;
R7 is H, F, CI, CH3, CH2-S02-CH3, CF3, CN, CH2-N(CH3)2, CO-
N(CH3)2, 0-CH3, S02CH3, N(CH3)2, 1 -pyrrolidinyl, 1 -piperidinyl, 4 morpholinyl, or 1 - pyrrolidinyl-2-on;
R8 is H, F, CI, CH3, CH2CH3, CH(CH3)2, C(CH3)3, CF3, CN, OH, 0- CH3, O-C2H5, O-CHF2, O-CF3, O-phenyl, SO2CH3, CO-CH3, CO- N(CH3)2, or CO-1 -piperidinyl;
R9 is H, CI, F, CH3, or O-CH3;
R10 is H; or
R6 and R7 together are

R7 and R8 together are

c) 2-phenylcyclopropyl;
d) 2-cyclohexylethyl;
R2 is H, F, CI, or CH3;
R3 is H, F, or O-CH3;
R4 is 4-methyl-piperidin-4-yl;
R5 is H; or
R4 and R5 together with the nitrogen carrying them denote
a) a residue of the formula lb

R11 is H, CH3, C2H5, (CH2)3-CH3, or CH2-OH;
R 2 is H, or CH3;
R13 is H, CH3, or CH2-CH(CH3)2;
R 4 is H, or CH3; or
R 3 and R14 together with the C-atom carrying them is
1 '-spiro-cyclopentyl; and
R15 is H, CH2-CO-N(CH3)2, CO-CH(C2H5)2, CO-C(CH3)2-OH,
CO-C(CH3)2-CH2-OH, CO-(CH2)2-0-CH3, CO-CH2-4-morpholinyl, CO-OCH3, CO-N(CH3)2, cyclopropyl, or CO-3-pyridyl;
R 6 is H, or CH3;
R 7 is H;
R18 is H;
R19 is H; b) (2,7-Diaza-spiro[4.4]non-2-yl). 4. A compound of the formula I as claimed in claims 1 to 3, wherein
R1 is a) a residue of the formula la

R6 is H, F, CI, CH3, CF3, or 0-CH3;
R7 is H, F, CI, CH3, CF3, CN, 0-CH3, S02CH3, N(CH3)2, 1 -piperidinyl, or 4-morpholinyl;
R8 is H, F, CI, CH3, CH2CH3, CH(CH3)2, C(CH3)3, CF3, OH, O- CH3, O-C2H5, O-CHF2, O-CF3, O-phenyl, SO2CH3, CO-CH3, or CO- N(CH3)2;
R9 is H, F, CH3, or O-CH3;
R10 is H; or
R7 and R8 together are
 b) phenethyl;
b) phenethyl;
R2 is H, or CH3;
R3 is H, or O-CH3;
R4 is 4-methyl-piperidin-4-yl;
R5 is H; or
R4 and R5 together with the nitrogen carrying them denote
a) a residue of the formula lb

R is H, CH3, or (CH2)3-CH3;
R12 is H;
R13 is H, CH3, or CH2-CH(CH3)2;
R14 is H, or CH3; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclopentyl; and
R15 is H, CH2-CO-N(CH3)2, CO-CH(C2H5)2, CO-C(CH3)2-OH, CO-
C(CH3)2-CH2-OH, CO-(CH2)2-0-CH3, CO-CH2-4-morpholinyl, CO- OCH3, CO-N(CH3)2, cyclopropyl, or 3-pyridyl;
R16 is H, or CH3;
R17 is H;
R11 and R12 together with the C-atoms carrying them are 1 '-spiro- cyclopentyl;
R18 is H;
R19 is H;
(2,7-Diaza-spiro[4.4]non-2-yl)
5. A compound of the formula I as claimed in claims 1 to 4, wherein
R1 is a residue of the formula la

R6 is H, F, CI, CH3, CF3, or 0-CH3;
R7 is H, F, CI, CH3, CF3, CN, 0-CH3, S02CH3, N(CH3)2, 1 -piperidinyl, or 4-morpholinyl;
R8 is H, F, CI, CH3, CH2CH3, CH(CH3)2, C(CH3)3, CF3, OH, O- CH3, O-C2H5, O-CHF2, O-CF3, O-phenyl, SO2CH3, CO-CH3, or CO- N(CH3)2;
R9 is H, F, CH3, or O-CH3;
R2 is
R3 is H;
R4 and R5 together with the nitrogen carrying them denote
a residue of the formula lb
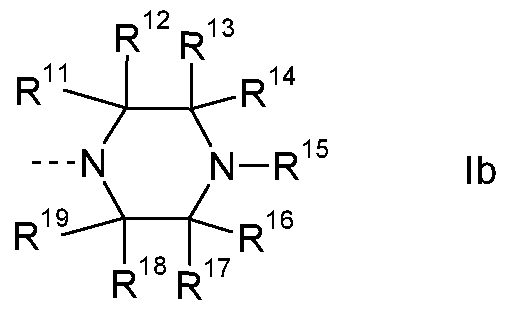
R11 is H, CH3, or (CH2)3-CH3;
R12 is H;
R13 is H, CH3, or CH2-CH(CH3)2;
R14 is H, or CH3; or
R13 and R14 together with the C-atom carrying them is
1 '-spiro-cyclopentyl; and
R15 is H;
R16 is H, or CH3;
R17 is H;
R11 and R 2 together with the C-atoms carrying them are 1 '-spiro- cyclopentyl;
6. A compound as claimed in claim 1 selected from the group consisting of 1
[3-[(E)-2-(4-Chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
2
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
3
(2,2-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
4
(3,3-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
5
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-3-isobutyl- piperazin-1 -yl)-methanone
6
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-2-methyl- piperazin-1 -yl)-methanone
7
[6-(4-Hydroxy-2-methyl-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-
1 -yl-methanone
8
((2S,5R)-2,5-Dimethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone
9
[6-(4-Hydroxy-3-methoxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
10
((S)-2-Ethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
1 1
[6-(2-Fluoro-4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-((S)-2- methyl-piperazin-1 -yl)-methanone
12
[6-(3-Fluoro-4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
13
[6-(2-Chloro-4-hydroxy-phenyl)-3-(styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
14
[3-[2-(3-Dimethylaminomethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
15
{6-(4-Hydroxy-phenyl)-3-[2-(3-methanesulfonylmethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
16
((R)-2-Hydroxymethyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H- pyrazolo[3,4-b]pyridin-4-yl]-methanone
17
[6-(4-Hydroxy-phenyl)-3-phenethyl-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 -yl- methanone
18
[6-(4-Hydroxy-phenyl)-3-((1 R,2R)-2-phenyl-cyclopropyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
19
(6-(4-Hydroxy-phenyl)-3-{2-[4-(piperidine-1 -carbonyl)-phenyl]-vinyl}-1 H-pyrazolo[3,4- b]pyridin-4-yl)-piperazin-1 -yl-methanone
20
1 -(3-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-phenyl)-pyrrolidin-2-one
21
[6-(4-Hydroxy-phenyl)-3-((E)-2-p-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 - yl-methanone
22
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methanesulfonyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
23
[3-[(E)-2-(3-Dimethylamino-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
24
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-methanesulfonyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
25
3- {(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-benzonitrile
26
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-piperidin-1 -yl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
27
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-morpholin-4-yl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
28
{6-(4-Hydroxy-phenyl)-3-[2-(3-pyrrolidin-1 -yl-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
29
4- {2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-benzonitrile
30
[3-[2-(3-Chloro-5-piperidin-1 -yl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
31
3-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-N,N-dimethyl-benzamide
32
5- {2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one
33
[3-[2-(2,3-Dihydro-1 H-indol-7-yl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin- 4-yl]-piperazin-1 -yl-methanone
34
3-{2-[4-(2,2-Dimethyl-piperazine-1 -carbonyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-3-yl]-vinyl}-N,N-dimethyl-benzamide
35
6-{2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]- vinyl}-2-methyl-2,3-dihydro-isoindol-1 -one
36
[3-(2-Cyclohexyl-ethyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin- 1 -yl-methanone
37
[3-[(E)-2-(4-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
38
[6-(4-Hydroxy-phenyl)-3-((E)-2-m-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin- 1 -yl-methanone
39
[3-[(E)-2-(2,4-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
40
[3-[(E)-2-(3,5-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
41
[3-[(E)-2-(4-Difluoromethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
42
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
43
[3-[(E)-2-(2,4-Dimethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin- 4-yl]-piperazin-1 -yl-methanone
44
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-phenoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
45
[3-[(E)-2-(3,4-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
46
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
47
[3-[(E)-2-(4-Hydroxy-3-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
48
[3-[(E)-2-(3-Chloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
49
[3-[(E)-2-(2,4-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
50
[3-[(E)-2-(2,5-Dimethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
51
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-isopropyl-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
52
[3-[(E)-2-(2-Chloro-4-fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
53
[3-[(E)-2-(4-Ethyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
54
[3-[(E)-2-(4-tert-Butyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
55
[3-[(E)-2-(3-Fluoro-4-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
56
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3-methoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
57
1 -(4-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin- 3-yl]-vinyl}-phenyl)-ethanone
58
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-trifluoromethoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
59
[3-[(E)-2-(3,4-Difluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
60
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-methoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4-b]pyridin-4- yl}-piperazin-1 -yl-methanone
61
[3-[(E)-2-(4-Fluoro-2-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
62
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(3,4,5-tnmethoxy-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
63
[3-[(E)-2-(2-Fluoro-4-methyl-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
64
[3-[(E)-2-(2,3-Dihydro-benzofuran-5-yl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-piperazin-1 -yl-methanone
65
[3-((E)-2-Benzo[1 ,3]dioxol-5-yl-vinyl)-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4- yl]-piperazin-1 -yl-methanone
66
4-{(E)-2-[6-(4-Hydroxy-phenyl)-4-(piperazine-1 -carbonyl)-1 H-pyrazolo[3,4-b]pyridin-3- yl]-vinyl}-N,N-dimethyl-benzamide
67
[3-[(E)-2-(4-Ethoxy-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
68
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(2-trifluoromethyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
69
[6-(4-Hydroxy-phenyl)-3-((E)-2-o-tolyl-vinyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-piperazin-1 - yl-methanone
70
[3-[2-(3,5-Dichloro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
71
[3-[(E)-2-(3-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
72
[3-[(E)-2-(2-Fluoro-phenyl)-vinyl]-6-(4-hydroxy-phenyl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]- piperazin-1 -yl-methanone
73
{6-(4-Hydroxy-phenyl)-3-[(E)-2-(4-methoxy-3-methyl-phenyl)-vinyl]-1 H-pyrazolo[3,4- b]pyridin-4-yl}-piperazin-1 -yl-methanone
74
4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazine-1 -carboxylic acid dimethylamide
75
6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carboxylic acid (4- methyl-piperidin-4-yl)-amide
76
((S)-2-Butyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
77
((R)-2-Butyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
78
(2,7-Diaza-spiro[4.4]non-2-yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
79
(6,9-Diaza-spiro[4.5]dec-9-yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
80
2-Ethyl-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-butan-1 -one
81
1 - {4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-2-morpholin-4-yl-ethanone
82
4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazine-1 -carboxylic acid methyl ester
83
[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridin-4-yl]-[4-(pyridine-3- carbonyl)-piperazin-1 -yl]-methanone
84
2- {4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-N,N-dimethyl-acetamide
85
(4-Cyclopropyl-piperazin-1 -yl)-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4- b]pyridin-4-yl]-methanone
86
2- Hydroxy-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-2-methyl-propan-1 -one
87
3- Hydroxy-1 -{4-[6-(4-hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4- carbonyl]-piperazin-1 -yl}-2,2-dimethyl-propan-1 -one
88
1 -{4-[6-(4-Hydroxy-phenyl)-3-((E)-styryl)-1 H-pyrazolo[3,4-b]pyridine-4-carbonyl]- piperazin-1 -yl}-3-methoxy-propan-1 -one. 7. A pharmaceutical composition, comprising at least one compound of the formula I as claimed in any of claims 1 to 6 or a physiologically acceptable salt thereof, or a physiologically acceptable solvate of any of them, and a pharmaceutically acceptable carrier. 8. A compound of the formula I as claimed in any of claims 1 to 6 or a
physiologically acceptable salt thereof, or a physiologically acceptable solvate of any of them, for use as a pharmaceutical.
9. The use of a compound of the formula I as claimed in any of claims 1 to 6 or a physiologically acceptable salt thereof, or a physiologically acceptable solvate of any of them, for the manufacture of a medicament for the treatment of diseases associated with diabetes and diabetic complications.
10. The use of a compound of the formula I as claimed in any of claims 1 to 6 or a physiologically acceptable salt thereof, or a physiologically acceptable solvate of any of them, for the manufacture of a medicament for the prevention and treatment of nephropathy, neuropathy, retinopathy, ischemia, inflammation, central nervous system disorders, cardiovascular diseases, dermatological diseases, autoimmune diseases and cancer.
1 1 . The use of a compound of the formula I as claimed in any of claims 1 to 6 or a physiologically acceptable salt thereof, or a physiologically acceptable solvate of any of
them, for the manufacture of a medicament for the treatment of a disease associated with the PKC receptor.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2011/065719 WO2013037390A1 (en) | 2011-09-12 | 2011-09-12 | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2011/065719 WO2013037390A1 (en) | 2011-09-12 | 2011-09-12 | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013037390A1 true WO2013037390A1 (en) | 2013-03-21 |
Family
ID=44645713
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/065719 WO2013037390A1 (en) | 2011-09-12 | 2011-09-12 | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2013037390A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150148352A1 (en) * | 2012-08-14 | 2015-05-28 | Cellixbio Private Limited | Compositions and methods for the treatment angina and cardiovascular conditions |
| WO2016081311A1 (en) * | 2014-11-21 | 2016-05-26 | Eli Lilly And Company | 1,2-benzothiazole compounds for the treatment of kidney disorders |
| US10392368B2 (en) | 2017-08-01 | 2019-08-27 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| KR20190133215A (en) * | 2017-03-28 | 2019-12-02 | 바스프 에스이 | Insecticide compound |
| US10604541B2 (en) | 2016-07-22 | 2020-03-31 | Bristol-Myers Squibb Company | Glucokinase activators and methods of using same |
| US10851102B2 (en) | 2019-01-23 | 2020-12-01 | Theravance Biopharma R&D Ip, Llc | Imidazole and triazole containing bicyclic compounds as JAK inhibitors |
Citations (981)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0296110A2 (en) | 1987-06-15 | 1988-12-21 | Ciba-Geigy Ag | Staurosporine derivatives substituted for the nitrogen atom of the methylamino group |
| EP0657458A1 (en) | 1993-12-07 | 1995-06-14 | Eli Lilly And Company | Protein kinase C inhibitors |
| WO1995017182A1 (en) | 1993-12-23 | 1995-06-29 | Eli Lilly And Company | Protein kinase c inhibitors |
| WO1997026265A1 (en) | 1996-01-17 | 1997-07-24 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| WO1997041097A2 (en) | 1996-12-31 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1999003861A1 (en) | 1997-07-16 | 1999-01-28 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO2000034331A2 (en) | 1998-12-07 | 2000-06-15 | Societe De Conseils De Recherches Et D'applications Scientifiques Sas | Analogues of glp-1 |
| WO2000058307A2 (en) | 1999-03-11 | 2000-10-05 | Neurogen Corporation | Aryl fused 2,4-disubstituted pyridines: nk3 receptor ligands |
| WO2000064876A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Tri-aryl acid derivatives as ppar receptor ligands |
| WO2000064888A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Di-aryl acid derivatives as ppar receptor ligands |
| WO2001000610A1 (en) | 1999-06-23 | 2001-01-04 | Aventis Pharma Deutschland Gmbh | Substituted benzimidazole |
| WO2001004156A1 (en) | 1999-07-12 | 2001-01-18 | Zealand Pharmaceuticals A/S | Peptides that lower blood glucose levels |
| WO2001017516A2 (en) | 1999-09-10 | 2001-03-15 | Novo Nordisk A/S | Method of inhibiting protein tyrosine phosphatase 1b and/or t-cell protein tyrosine phosphatase and/or other ptpases with an asp residue at position 48 |
| WO2001019831A1 (en) | 1999-09-10 | 2001-03-22 | Novo Nordisk A/S | MODULATORS OF PROTEIN TYROSINE PHOSPHATASES (PTPases) |
| WO2001019830A1 (en) | 1999-09-10 | 2001-03-22 | Novo Nordisk A/S | MODULATORS OF PROTEIN TYROSINE PHOSPHATASES (PTPases) |
| US6221633B1 (en) | 1997-06-20 | 2001-04-24 | Aventis Pharma Deutschland Gmbh | Insulin derivatives having a rapid onset of action |
| WO2001030774A1 (en) | 1999-10-26 | 2001-05-03 | Aventis Pharma Deutschland Gmbh | Substituted indoles for modulating nfkb activity |
| WO2001040207A1 (en) | 1999-12-02 | 2001-06-07 | Glaxo Group Limited | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
| WO2001090094A1 (en) | 2000-05-22 | 2001-11-29 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2002038561A1 (en) | 2000-11-07 | 2002-05-16 | Novartis Ag | Indolylmaleimide derivatives as protein kinase c inhibitors |
| WO2002096894A1 (en) | 2001-05-31 | 2002-12-05 | Glaxo Group Limited | Thiazole or oxazole derivatives which are useful in the treatment of cardiovascular and related diseases |
| WO2003020269A1 (en) | 2001-08-31 | 2003-03-13 | Aventis Pharma Deutschland Gmbh | Diaryl cycloalkyl derivatives, method for producing the same and the use thereof as ppar activators |
| WO2003044009A1 (en) | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003044000A1 (en) | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003043999A1 (en) | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003065983A2 (en) | 2002-02-01 | 2003-08-14 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003074500A2 (en) | 2002-03-06 | 2003-09-12 | Sanofi-Aventis | N-aminoacetyl-pyrrolidine-2-carbonitriles and their use as ddp-iv inhibitors |
| WO2003076442A1 (en) | 2002-03-05 | 2003-09-18 | Eli Lilly And Company | Purine derivatives as kinase inhibitors |
| WO2003078403A2 (en) | 2002-03-11 | 2003-09-25 | Aventis Pharma S.A. | Derives d’aminoindazoles comme inhibiteurs de proteine-kinase |
| WO2003084922A1 (en) | 2002-04-11 | 2003-10-16 | Aventis Pharma Deutschland Gmbh | Acyl-4-carboxyphenylurea derivatives, method for production and use thereof |
| WO2003097064A1 (en) | 2002-05-17 | 2003-11-27 | Kyowa Hakko Kogyo Co., Ltd. | Therapeutic agent for diabetes |
| WO2003104207A2 (en) | 2002-06-10 | 2003-12-18 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003106410A1 (en) | 2002-06-13 | 2003-12-24 | Aventis Pharma Deutschland Gmbh | Fluorinated cycloalkyl-derivatised benzoylguanidines and their use as a medicament |
| WO2003106456A2 (en) | 2002-06-14 | 2003-12-24 | Sanofi-Synthelabo | New compounds |
| WO2004007517A1 (en) | 2002-07-11 | 2004-01-22 | Aventis Pharma Deutschland Gmbh | Novel thiophenylglycoside derivatives, methods for production thereof, medicaments comprising said compounds and use thereof |
| WO2004007455A1 (en) | 2002-07-12 | 2004-01-22 | Aventis Pharma Deutschland Gmbh | Heterocyclically substituted benzoylureas, method for their production and their use as medicaments |
| WO2004011410A1 (en) | 2002-07-27 | 2004-02-05 | Astrazeneca Ab | Chemical compounds |
| WO2004014910A1 (en) | 2002-08-07 | 2004-02-19 | Mitsubishi Pharma Corporation | Dihydropyrazolopyridine compounds |
| WO2004022544A1 (en) | 2002-09-05 | 2004-03-18 | Aventis Pharma S.A. | Novel aminoindazole derivatives as medicines and pharmaceutical compositions containing same |
| WO2004024726A1 (en) | 2002-09-12 | 2004-03-25 | F. Hoffmann-La Roche Ag | N-substituted-1h-indol-5-propionic acid compounds as ppar agonists useful for the treatment of diabetes |
| WO2004033427A1 (en) | 2002-10-11 | 2004-04-22 | Astrazeneca Ab | 1,4-disubstituted piperidine derivatives and their use as 11-betahsd1 inhibitors |
| WO2004037251A1 (en) | 2002-10-24 | 2004-05-06 | Sterix Limited | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 and type 2 |
| WO2004037169A2 (en) | 2002-10-18 | 2004-05-06 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2004041264A1 (en) | 2002-11-07 | 2004-05-21 | Astrazeneca Ab | 2-oxo-ethanesulfonamide derivates |
| WO2004041274A1 (en) | 2002-11-05 | 2004-05-21 | Arena Pharmaceuticals, Inc. | Benzotriazoles and methods of prophylaxis or treatment of metabolic-related disorders thereof |
| WO2004046117A1 (en) | 2002-11-19 | 2004-06-03 | Aventis Pharma Deutschland Gmbh | Pyridazinone derivatives as gsk-3beta inhibitors |
| WO2004050646A1 (en) | 2002-11-29 | 2004-06-17 | Astrazeneca Ab | 1, 2, 5-thiadiazolidin-3-one 1, 1 dioxide derivatives as inhibitors of protein tyrosine phosphatase ptp1b |
| WO2004050658A1 (en) | 2002-12-03 | 2004-06-17 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel substituted imidazo-pyridinones and imidazo-pyridazeiones, the production and use thereof as medicaments |
| WO2004052903A1 (en) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Novel fluoroglycoside heterocyclic derivatives, pharmaceutical products containing said compounds and the use thereof |
| WO2004052902A1 (en) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Novel aromatic fluoroglycoside derivatives, pharmaceutical products containing said compounds and the use thereof |
| WO2004056744A1 (en) | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| WO2004058730A2 (en) | 2002-12-20 | 2004-07-15 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2004063179A1 (en) | 2003-01-06 | 2004-07-29 | Eli Lilly And Company | Substituted arylcyclopropylacetamides as glucokinase activators |
| WO2004065380A1 (en) | 2003-01-14 | 2004-08-05 | Arena Pharmaceuticals Inc. | 1,2,3-trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prpphylaxis and treatment of disorders related thereto such as diabetes and hyperglycemia |
| WO2004065351A1 (en) | 2003-01-24 | 2004-08-05 | Novartis Ag | Amide derivatives and their use as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2004072066A1 (en) | 2003-02-11 | 2004-08-26 | Prosidion Limited | Tri(cyclo) substituted amide glucokinase activator compounds |
| WO2004072031A2 (en) | 2003-02-11 | 2004-08-26 | Prosidion Limited | Phenylacetamides and their use as glucokinase modulators |
| WO2004074288A1 (en) | 2003-02-19 | 2004-09-02 | F. Hoffmann-La Roche Ag | Sulfonamide substituted xanthine derivatives for use as pepck inhibitors |
| EP1460075A1 (en) | 2003-03-21 | 2004-09-22 | Sanofi-Synthelabo | Substituted 8-Pyridinyl-6,7,8,9-Tetrahydropyrimido[1,2-a]Pyrimidin-4-one and 8-Phenyl-6-7,8,9-Tetrahydropyrimido[1,2-a]Pyrimidin-4-one derivatives |
| WO2004089896A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 ACTIVE COMPOUNDS |
| WO2004089367A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of substituted 1,2,4-triazoles |
| WO2004089471A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | NEW PYRAZOLO[1,5-a] PYRIMIDINES DERIVATIVES AND PHARMACEUTICAL USE THEREOF |
| WO2004089380A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of fused 1,2,4-triazoles |
| WO2004101528A2 (en) | 2003-05-19 | 2004-11-25 | F. Hoffmann-La Roche Ag | Isoquinoline derivatives and their use as gfat inhibitors |
| WO2004100875A2 (en) | 2003-05-09 | 2004-11-25 | Merck & Co., Inc. | Benzimidazoles, compositions containing such compounds and methods of use |
| WO2004103980A1 (en) | 2003-05-21 | 2004-12-02 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type i |
| WO2004106294A2 (en) | 2003-05-29 | 2004-12-09 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta hydroxysteroid dehydrogenase-1 |
| WO2004106343A2 (en) | 2003-05-30 | 2004-12-09 | Ufc Limited | Agelastatin derivatives of antitumour and gsk-3beta-inhibiting alkaloids |
| JP2004359630A (en) | 2003-06-06 | 2004-12-24 | Yamanouchi Pharmaceut Co Ltd | Difluorodiphenylmethane derivative and its salt |
| WO2004112782A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | New use v |
| WO2004112779A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | New use vii |
| WO2004112784A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | New use i |
| WO2004113310A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | Use of an inhibitor of 11-b-hydroxysteroid dehydrogenase type 1 compounds for promoting wound healing |
| WO2005000836A1 (en) | 2003-06-13 | 2005-01-06 | Janssen Pharmaceutica N.V. | Substituted indazolyl(indolyl)maleimide derivatives as kinase inhibitors |
| WO2005005477A2 (en) | 2003-07-11 | 2005-01-20 | Novo Nordisk A/S | Stabilised insulin compositions |
| WO2005009997A1 (en) | 2003-07-30 | 2005-02-03 | Pfizer Inc. | 3,5 disubstituted indazole compounds, pharmaceutical compositions, and methods for mediating or inhibiting cell proliferation |
| WO2005009389A2 (en) | 2003-07-23 | 2005-02-03 | Exelixis, Inc. | Anaplastic lymphoma kinase modulators and methods of use |
| US20050026984A1 (en) | 2003-07-29 | 2005-02-03 | Aventis Pharma S.A. | Substituted thieno [2,3-c] pyrazoles and their use as medicinal products |
| WO2005012295A1 (en) | 2003-07-28 | 2005-02-10 | Sanofi-Aventis Deutschland Gmbh | Substituted thiazole-benzoisothiazole dioxide derivatives, method for the production thereof and use of the same |
| WO2005012312A1 (en) | 2003-07-25 | 2005-02-10 | Sanofi-Aventis Deutschland Gmbh | Novel cyano thiazolides, methods for the production thereof, and use thereof as a medicament |
| WO2005012308A1 (en) | 2003-07-25 | 2005-02-10 | Sanofi-Aventis Deutschland Gmbh | Novel cyanopyrrolidides, methods for the production thereof, and use of the same as medicaments |
| US20050038023A1 (en) | 2000-12-21 | 2005-02-17 | David Bebbington | Pyrazole compounds useful as protein kinase inhibitors |
| WO2005016877A2 (en) | 2003-08-07 | 2005-02-24 | Merck & Co., Inc. | Pyrazole carboxamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2005027978A2 (en) | 2003-09-19 | 2005-03-31 | Novo Nordisk A/S | Albumin-binding derivatives of therapeutic peptides |
| WO2005028480A2 (en) | 2003-09-03 | 2005-03-31 | Neurogen Corporation | 5-aryl-pyrazolo[4,3-d]pyrimidines, pyridines, and pyrazines and related compounds |
| WO2005037828A1 (en) | 2003-10-20 | 2005-04-28 | Lg Life Sciences Ltd. | Novel inhibitors of dpp-iv, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| WO2005044801A1 (en) | 2003-10-31 | 2005-05-19 | Astrazeneca Ab | Pyridine carboxylic acid derivatives as glucokinase modulators |
| WO2005058901A1 (en) | 2003-12-17 | 2005-06-30 | Boehringer Ingelheim International Gmbh | Novel 2-(piperazin-1-yl)- and 2-([1,4]diazepan-1-yl)- imidazo[4,5-d]pyridazin-4-one, production and use thereof as medicament for the treatment of diabetes mellitus |
| WO2005058908A1 (en) | 2003-12-19 | 2005-06-30 | Sanofi-Aventis | SUBSTITUTED 8’-PYRI(MI)DINYL-DIHYDROSPIRO-[CYCLOALKYLAMINE]-PYRIMIDO[1,2-a]PYRIMIDIN-6-ONE DERIVATIVES |
| WO2005061489A1 (en) | 2003-12-24 | 2005-07-07 | Prosidion Limited | Heterocyclic derivatives as gpcr receptor agonists |
| WO2005063247A1 (en) | 2003-12-22 | 2005-07-14 | Amgen Inc. | Aryl sulfonamide compounds and uses related thereto |
| WO2005065680A1 (en) | 2003-12-19 | 2005-07-21 | Merck & Co., Inc. | Cyclic guanidines, compositions containing such compounds and methods of use |
| WO2005067931A2 (en) | 2004-01-14 | 2005-07-28 | Daniolabs Limited | Dopamine uptake inhibitors for the treatment of neurological disease |
| WO2005067932A1 (en) | 2004-01-06 | 2005-07-28 | Janssen Pharmaceutica, N.V. | (3-oxo-3, 4-dihydro-quinoxalin-2-yl-amino) -benzamide derivatives and related compound as glycogen phosphorylase inhibitors for the treatment of diabetes and obesity |
| WO2005073229A1 (en) | 2004-01-31 | 2005-08-11 | Sanofi-Aventis Deutschland Gmbh | 7-phenylamino-4-quinolone-3-carboxylic acid derivatives, methods for production and use thereof as medicaments |
| WO2005073199A1 (en) | 2004-02-02 | 2005-08-11 | Sanofi-Aventis Deutschland Gmbh | Indazole derivatives as inhibitors of hormone-sensitive lipases |
| WO2005080360A1 (en) | 2004-02-18 | 2005-09-01 | Astrazeneca Ab | Compounds |
| WO2005085237A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085230A1 (en) | 2004-03-02 | 2005-09-15 | Sanofi-Aventis Deutschland Gmbh | 4-benzimidazol-2-yl-pyridazine-3-one-derivatives, production and use thereof in medicaments |
| WO2005087727A1 (en) | 2004-03-12 | 2005-09-22 | Boehringer Ingelheim International Gmbh | Novel alkyl-containing 5-acylindolinones, their preparation and their use as pharmaceutical products |
| WO2005086904A2 (en) | 2004-03-08 | 2005-09-22 | Amgen Inc. | Therapeutic modulation of ppar (gamma) activity |
| US20050222220A1 (en) | 2004-04-05 | 2005-10-06 | Neuropharma, S.A. | GSK-3 inhibitors |
| WO2005097759A1 (en) | 2004-03-29 | 2005-10-20 | Merck & Co., Inc. | Diaryltriazoles as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2005097076A2 (en) | 2004-04-09 | 2005-10-20 | Smithkline Beecham Corporation | Low dose pharmaceutical products |
| WO2005111018A1 (en) | 2004-05-18 | 2005-11-24 | Sanofi-Aventis Deutschland Gmbh | Pyridazinone derivatives, methods for producing them and their use as pharmaceuticals |
| WO2005116003A2 (en) | 2004-05-29 | 2005-12-08 | Sanofi-Aventis Deutschland Gmbh | Substituted oxazolobenzoisothiazole dioxide derivatives method for production and use thereof |
| WO2005121161A1 (en) | 2004-06-11 | 2005-12-22 | Sanofi-Aventis Deutschland Gmbh | Novel fluoroglycoside derivatives of pyrazoles, medicaments containing these compounds, and the use thereof |
| WO2006007959A1 (en) | 2004-07-17 | 2006-01-26 | Sanofi-Aventis Deutschland Gmbh | Diphenylamine-substituted salicylthiazole derivatives and related compounds as phosphotyrosine phosphatase 1b (ptp1b) inhibitors for using as blood-sugar decreasing active ingredients for treating diabetes |
| WO2006012173A1 (en) | 2004-06-24 | 2006-02-02 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2006012227A2 (en) | 2004-06-24 | 2006-02-02 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2006010546A2 (en) | 2004-07-28 | 2006-02-02 | F. Hoffman-La Roche Ag | Aryl-pyridine derivatives as 11-beta-hsd1 inhibitors |
| WO2006017542A1 (en) | 2004-08-06 | 2006-02-16 | Merck & Co., Inc. | Sulfonyl compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2006015701A1 (en) | 2004-08-06 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted, bicyclic 8-pyrrolidinoxanthines, method for the production thereof and their use as medicaments |
| WO2006015700A1 (en) | 2004-08-06 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted, bicyclic 8-piperidinoxanthines, method for the production thereof and their use as medicaments |
| WO2006015699A1 (en) | 2004-08-06 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted bicyclic 8-pyrrolidino-xanthines and use thereof as inhibitors of the dipeptidyl peptidase iv |
| WO2006016194A1 (en) | 2004-08-12 | 2006-02-16 | Prosidion Limited | Substituted phenylacetamides and their use as glucokinase activators |
| WO2006015691A1 (en) | 2004-08-03 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted 8-aminoalkylthio-xanthines, and the use thereof as inhibitors of the dipeptidyl peptidase iv |
| WO2006018117A1 (en) | 2004-08-14 | 2006-02-23 | Sanofi-Aventis Deutschland Gmbh | Substituted 8-aminoalkoxi-xanthines, method for the production thereof and use thereof as medicaments |
| WO2006018150A1 (en) | 2004-08-11 | 2006-02-23 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituited cyclene, medicaments containing said compounds, use thereof and method for the production thereof |
| WO2006023515A2 (en) | 2004-08-18 | 2006-03-02 | Metabasis Therapeutics, Inc. | Novel thiazole inhibitors of fructose 1,6-bisphosphatase |
| WO2006024627A2 (en) | 2004-08-30 | 2006-03-09 | Janssen Pharmaceutica N.V. | N-2 adamantanyl-2-phenoxy-acetamide derivatives as 11-beta hydroxysteroid dehydrogenase inhibitors |
| WO2006029699A1 (en) | 2004-09-11 | 2006-03-23 | Sanofi-Aventis Deutschland Gmbh | 7-azaindoles and their use as ppar agonists |
| WO2006035796A1 (en) | 2004-09-29 | 2006-04-06 | Kissei Pharmaceutical Co., Ltd. | 1-(β-D-GLYCOPYRANOSYL)-3-SUBSTITUTED NITROGENOUS HETEROCYCLIC COMPOUND, MEDICINAL COMPOSITION CONTAINING THE SAME, AND MEDICINAL USE THEREOF |
| WO2006034804A1 (en) | 2004-09-29 | 2006-04-06 | F.Hoffmann-La Roche Ag | Indozolone derivatives as 11b-hsd1 inhibitors |
| WO2006037810A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted glp-1 compounds |
| WO2006037811A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted exendin-4 compounds |
| WO2006039325A2 (en) | 2004-10-01 | 2006-04-13 | Merck & Co., Inc. | Aminopiperidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006040329A1 (en) | 2004-10-12 | 2006-04-20 | Novo Nordisk A/S | 1 ibeta- hydroxysteroid dehydrogenase type 1 active spiro compounds |
| WO2006045564A1 (en) | 2004-10-22 | 2006-05-04 | Smithkline Beecham Corporation | Xanthine derivatives with hm74a receptor activity |
| WO2006045565A1 (en) | 2004-10-22 | 2006-05-04 | Smithkline Beecham Corporation | Xanthine derivatives with hm74a receptor activity |
| WO2006048331A1 (en) | 2004-11-08 | 2006-05-11 | Evotec Ag | 11β-HSD1 INHIBITORS |
| WO2006049952A1 (en) | 2004-10-29 | 2006-05-11 | Eli Lilly And Company | Cycloalkyl lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2006048750A2 (en) | 2004-11-02 | 2006-05-11 | Pfizer Inc. | Novel compounds of substituted and unsubstituted adamantyl amides |
| WO2006050908A1 (en) | 2004-11-08 | 2006-05-18 | Evotec Ag | Inhibitors of 11βετα-hydroxy steroid dehydrogenase type 1 (11beta-hsd1) |
| WO2006051662A1 (en) | 2004-11-09 | 2006-05-18 | Taisho Pharmaceutical Co., Ltd. | Thiazole derivative |
| WO2006058064A2 (en) | 2004-11-29 | 2006-06-01 | Merck & Co., Inc. | Fused aminopiperidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006058923A1 (en) | 2004-12-03 | 2006-06-08 | Novo Nordisk A/S | Heteroaromatic glucokinase activators |
| WO2006059744A1 (en) | 2004-11-30 | 2006-06-08 | Nippon Chemiphar Co., Ltd. | ACTIVATOR OF PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR δ |
| WO2006058597A1 (en) | 2004-12-03 | 2006-06-08 | Merck Patent Gmbh | Tetrahydropyrane derivatives for use as antidiabetics |
| WO2006062224A1 (en) | 2004-12-07 | 2006-06-15 | Takeda Pharmaceutical Company Limited | Carboxamide derivative |
| WO2006065826A2 (en) | 2004-12-15 | 2006-06-22 | Merck & Co., Inc. | Process to chiral beta amino acid derivatives by asymmetric hydrogenation |
| JP2006160733A (en) | 2004-11-15 | 2006-06-22 | Taisho Pharmaceut Co Ltd | Medicine containing cyanofluoropyrrolidine derivative as active ingredient |
| WO2006066109A2 (en) | 2004-12-17 | 2006-06-22 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
| WO2006069242A2 (en) | 2004-12-23 | 2006-06-29 | Arena Pharmaceuticals, Inc. | Fused pyrazole derivatives and uses thereof in methods of treatment of metabolic-related?disorders |
| WO2006067531A1 (en) | 2004-12-24 | 2006-06-29 | Prosidion Ltd | G-protein coupled receptor (gpr116) agonists and use thereof for treating obesity and diabetes |
| WO2006068163A1 (en) | 2004-12-24 | 2006-06-29 | Dainippon Sumitomo Pharma Co., Ltd. | Bicyclic pyrrole derivatives |
| WO2006067532A1 (en) | 2004-12-24 | 2006-06-29 | Prosidion Ltd | G-protein coupled receptor agonists |
| WO2006071752A1 (en) | 2004-12-29 | 2006-07-06 | Bristol-Myers Squibb Company | Azolopyrimidine-based inhibitors of dipeptidyl peptidase iv and methods |
| DE102004060542A1 (en) | 2004-12-16 | 2006-07-06 | Sanofi-Aventis Deutschland Gmbh | Hydroxybiphenyl carboxylic acids and derivatives, process for their preparation and their use |
| WO2006073197A1 (en) | 2005-01-07 | 2006-07-13 | Taisho Pharmaceutical Co., Ltd. | 1-thio-d-glucitol derivatives |
| WO2006073167A1 (en) | 2005-01-07 | 2006-07-13 | Ono Pharmaceutical Co., Ltd. | Pyrrolidine derivatives |
| WO2006072354A1 (en) | 2005-01-07 | 2006-07-13 | Merck Patent Gmbh | Squaric acid derivatives |
| WO2006074244A2 (en) | 2005-01-05 | 2006-07-13 | Abbott Laboratories | Adamantyl derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| WO2006074957A1 (en) | 2005-01-17 | 2006-07-20 | Sanofi-Aventis | Substituted aminomethylene sulphonamides, production and use thereof as medicaments |
| WO2006078006A1 (en) | 2005-01-24 | 2006-07-27 | Dainippon Sumitomo Pharma Co., Ltd. | Indole compound and pharmaceutical composition containing the same |
| WO2006078676A2 (en) | 2005-01-19 | 2006-07-27 | Merck & Co., Inc. | Bicyclic pyrimidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006080577A1 (en) | 2005-01-31 | 2006-08-03 | Tanabe Seiyaku Co., Ltd. | Indole derivatives |
| EP1688138A1 (en) | 2003-11-26 | 2006-08-09 | Takeda Pharmaceutical Company Limited | Receptor function regulating agent |
| WO2006083491A2 (en) | 2005-01-10 | 2006-08-10 | Arena Pharmaceuticals, Inc. | Substituted pyridinyl and pyrimidinyl derivatives as modulators of metabolism and the treatment of disorders related thereto |
| WO2006084176A2 (en) | 2005-02-03 | 2006-08-10 | Irm Llc | Compounds and compositions as ppar modulators |
| WO2006085108A1 (en) | 2005-02-14 | 2006-08-17 | Smithkline Beecham Corporation | Awthranilic acid derivatives and their use in treatment of diseases of lipid metabolism, in particular dyslipidaemia |
| WO2006085112A1 (en) | 2005-02-14 | 2006-08-17 | Smithkline Beecham Corporation | Anthranilic acid derivatives as hm74a receptor agonists |
| WO2006085113A2 (en) | 2005-02-14 | 2006-08-17 | Smithkline Beecham Corporation | 2-substituted 5-membered heteroaryl carboxylates as hm74a receptor agonists |
| WO2006086488A2 (en) | 2005-02-11 | 2006-08-17 | Eli Lilly And Company | Substituted thiophene derivatives as glucagon receptor antagonists, preparation and therapeutic uses |
| WO2006085685A1 (en) | 2005-02-09 | 2006-08-17 | Takeda Pharmaceutical Company Limited | Pyrazole compound |
| WO2006087309A1 (en) | 2005-02-15 | 2006-08-24 | Novo Nordisk A/S | 3,4-dihydro-1h-isoquinoline-2-carboxylic acid 5-aminopyridin-2-yl esters |
| WO2006087997A1 (en) | 2005-02-15 | 2006-08-24 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-7-(β-D-GLYCOPYRANOSYLOXY)(AZA)INDOLE COMPOUND AND PHARMACEUTICAL CONTAINING THE SAME |
| WO2006090915A1 (en) | 2005-02-25 | 2006-08-31 | Takeda Pharmaceutical Company Limited | Pyridyl acetic acid compounds |
| WO2006099943A1 (en) | 2005-03-19 | 2006-09-28 | Sanofi-Aventis | Amide-substituted 8-n-benzimidazoles, method for the production thereof, and use of the same as medicaments |
| WO2006099941A1 (en) | 2005-03-19 | 2006-09-28 | Sanofi-Aventis | Aminocarbonyl-substituted 8-n-benzimidazoles, method for their production and their use as medicaments |
| WO2006104030A1 (en) | 2005-03-25 | 2006-10-05 | Daiichi Sankyo Company, Limited | Thiazole compound |
| WO2006104356A1 (en) | 2005-04-01 | 2006-10-05 | Lg Life Sciences, Ltd. | Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| WO2006106423A2 (en) | 2005-04-07 | 2006-10-12 | Pfizer Inc. | Amino sulfonyl derivatives as inhibitors of human 11-.beta.-hydrosysteroid dehydrogenase |
| WO2006108842A1 (en) | 2005-04-15 | 2006-10-19 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted (heteroaryloxy-benzyl)-benzene derivatives as sglt inhibitors |
| WO2006112549A1 (en) | 2005-04-20 | 2006-10-26 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| WO2006111321A1 (en) | 2005-04-20 | 2006-10-26 | Sanofi-Aventis | Azole derivatives in the form of lipase and phospholipase inhibitors |
| WO2006111261A1 (en) | 2005-04-16 | 2006-10-26 | Sanofi-Aventis | Substituted 2-amin0alkylthi0-benzimidaz0les and use thereof for reducing blood sugar levels |
| WO2006113150A1 (en) | 2005-04-13 | 2006-10-26 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2006121860A2 (en) | 2005-05-06 | 2006-11-16 | Bayer Pharmaceuticals Corporation | Glucagon-like peptide 1 (glp-1) receptor agonists and their pharmacological methods of use |
| WO2006124490A2 (en) | 2005-05-17 | 2006-11-23 | Schering Corporation | Heterocycles as nicotinic acid receptor agonists for the treatment of dyslipidemia |
| WO2006124529A1 (en) | 2005-05-13 | 2006-11-23 | Eli Lilly And Company | Glp-1 pegylated compounds |
| WO2006127530A2 (en) | 2005-05-25 | 2006-11-30 | Merck & Co., Inc. | Aminocyclohexanes as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006125972A1 (en) | 2005-05-27 | 2006-11-30 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2006132436A1 (en) | 2005-06-08 | 2006-12-14 | Japan Tobacco Inc. | Heterocyclic compound |
| WO2006134467A1 (en) | 2005-06-16 | 2006-12-21 | Pfizer Inc. | N-(pyridin-2-yl)-sulfonamide derivatives |
| WO2006135795A1 (en) | 2005-06-09 | 2006-12-21 | Bristol-Myers Squibb Company | Imidazo- and triazolopyridines as inhibitors of 11-beta hydroxysteroid dehydrogenase type i |
| WO2006134481A1 (en) | 2005-06-16 | 2006-12-21 | Pfizer Inc. | Inhibitors of 11-beta hydroxysteroid dehydrogenase type 1 |
| WO2006133926A1 (en) | 2005-06-17 | 2006-12-21 | Carex Sa | Pyrazole derivates as cannabinoid receptor modulators |
| WO2006138508A2 (en) | 2005-06-17 | 2006-12-28 | Amgen Inc. | Benzamide derivatives and uses related thereto |
| WO2006136502A1 (en) | 2005-06-22 | 2006-12-28 | F. Hoffmann-La Roche Ag | ( 6-FLU0R0-BENZ0[l, 3] DIOXOLYL) -MORPHOLIN-4-YL-METHANONES AND THEIR USE AS CBl LIGANDS |
| WO2006138695A1 (en) | 2005-06-17 | 2006-12-28 | Bristol-Myers Squibb Company | Triazolopyridine derivatives as cannabinoid receptor 1 antagonists |
| WO2007002557A1 (en) | 2005-06-28 | 2007-01-04 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007000445A1 (en) | 2005-06-29 | 2007-01-04 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2007000006A2 (en) | 2005-06-29 | 2007-01-04 | Plansee Se | Halogen filament lamp comprising a dimming cover consisting of an mo alloy |
| WO2007003964A1 (en) | 2005-06-30 | 2007-01-11 | Prosidion Limited | G-protein coupled receptor agonists |
| WO2007003962A2 (en) | 2005-06-30 | 2007-01-11 | Prosidion Limited | Gpcr agonists |
| WO2007003960A1 (en) | 2005-06-30 | 2007-01-11 | Prosidion Limited | Gpcr agonists |
| WO2007003521A2 (en) | 2005-07-05 | 2007-01-11 | F. Hoffmann-La Roche Ag | Pyridazine derivatives as 11beta-hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2007007040A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | 2 -heterocyclyloxybenzoyl amino heterocyclyl compounds as modulators of glucokinase for the treatment of type 2 diabetes |
| WO2007006760A1 (en) | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkyl urea glucokinase activators |
| WO2007006761A1 (en) | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkylcarbamoyl ureas as glucokinase activators |
| WO2007006814A1 (en) | 2005-07-14 | 2007-01-18 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2007007910A1 (en) | 2005-07-13 | 2007-01-18 | Banyu Pharmaceutical Co., Ltd. | Heterocycle-substituted benzimidazole derivative |
| WO2007007688A1 (en) | 2005-07-08 | 2007-01-18 | Mochida Pharmaceutical Co., Ltd. | 3,5-diamino-1,2,4-triazole derivative |
| WO2007007886A1 (en) | 2005-07-11 | 2007-01-18 | Mitsubishi Tanabe Pharma Corporation | An oxime derivative and preparations thereof |
| WO2007007042A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2007009911A1 (en) | 2005-07-21 | 2007-01-25 | F. Hoffmann-La Roche Ag | PYRIDO [2 , 3-D] PYRIMIDINE-2 , 4-DIAMINE COMPOUNDS AS PTPlB INHIBITORS |
| WO2007013689A1 (en) | 2005-07-29 | 2007-02-01 | Takeda Pharmaceutical Company Limited | Cyclopropanecarboxylic acid compound |
| WO2007014895A2 (en) | 2005-07-28 | 2007-02-08 | Boehringer Ingelheim International Gmbh | Methods for preventing and treating metabolic disorders and new pyrazole-o-glycoside derivatives |
| WO2007014619A1 (en) | 2005-08-01 | 2007-02-08 | Merck Patent Gmbh | Novelimidazolecarboxamide derivatives as fructose-1,6-bisphosphatase inhibitors, and pharmaceutical compositions comprising same |
| WO2007015744A1 (en) | 2005-07-21 | 2007-02-08 | Incyte Corporation | Disubstituted thienyl compounds and their use as pharmaceuticals |
| WO2007015767A1 (en) | 2005-07-20 | 2007-02-08 | Eli Lilly And Company | Pyridine derivatives as dipeptedyl peptidase inhibitors |
| WO2007017649A1 (en) | 2005-08-09 | 2007-02-15 | Astrazeneca Ab | Heteroarylcarbamoylbenzene derivatives for the treatment of diabetes |
| WO2007017549A1 (en) | 2005-08-04 | 2007-02-15 | Universitat De Valencia | Aqueous base pigment compositions for the multicolour marking of inorganic materials with laser |
| WO2007017261A1 (en) | 2005-08-10 | 2007-02-15 | Smithkline Beecham Corporation | Xanthine derivatives as selective hm74a agonists |
| WO2007017265A1 (en) | 2005-08-10 | 2007-02-15 | Smithkline Beecham Corporation | Xanthine derivatives as selective hm74a agonists |
| WO2007024993A2 (en) | 2005-08-26 | 2007-03-01 | Merck & Co., Inc. | Fused aminopiperidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007028145A2 (en) | 2005-09-02 | 2007-03-08 | Dara Biosciences, Inc. | Agents and methods for reducing protein tyrosine phosphatase 1b activity in the central nervous system |
| WO2007028135A2 (en) | 2005-09-01 | 2007-03-08 | Takeda Pharmaceutical Company Limited | Imidazopyridine compounds |
| WO2007026761A1 (en) | 2005-08-31 | 2007-03-08 | Astellas Pharma Inc. | Thiazole derivative |
| WO2007027532A2 (en) | 2005-08-29 | 2007-03-08 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007029086A2 (en) | 2005-09-05 | 2007-03-15 | Ranbaxy Laboratories Limited | Derivatives of 3-azabicyclo[3.1.0]hexane as dipeptidyl peptidase-iv inhibitors |
| WO2007029021A1 (en) | 2005-09-08 | 2007-03-15 | The University Of Edinburgh | 1,5-substituted tetrazoles as therapeutic compounds |
| WO2007031739A1 (en) | 2005-09-16 | 2007-03-22 | Astrazeneca Ab | Heterobicyclic compounds as glucokinase activators |
| US20070066584A1 (en) | 2005-09-21 | 2007-03-22 | Wenqing Yao | Amido compounds and their use as pharmaceuticals |
| WO2007033002A1 (en) | 2005-09-14 | 2007-03-22 | Amgen Inc. | Conformationally constrained 3- (4-hydroxy-phenyl) - substituted-propanoic acids useful for treating metabolic disorders |
| WO2007035355A2 (en) | 2005-09-16 | 2007-03-29 | Arena Pharmaceuticals, Inc. | Modulators of metabolism and the treatment of disorders related thereto |
| WO2007037534A1 (en) | 2005-09-30 | 2007-04-05 | Banyu Pharmaceutical Co., Ltd. | 2-heteroaryl-substituted indole derivative |
| WO2007041365A2 (en) | 2005-09-30 | 2007-04-12 | Novartis Ag | 3-cyclyl-2- (4-sulfamo yl-phenyl) -n-cyclyl-propionamide derivatives useful in the treatment of impaired glucose tolerance and diabetes |
| WO2007039178A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl-[1,2,4]-oxadiazol-5-one derivatives with phenyl group, processes for their preparation and their use as pharmaceuticals |
| WO2007041366A1 (en) | 2005-09-30 | 2007-04-12 | Novartis Ag | Sulfonamide derivatives as glycokinase activators useful in the treatment of type 2 diabetes |
| WO2007042178A1 (en) | 2005-10-12 | 2007-04-19 | Sanofi-Aventis | Diacyl indazol derivatives as lipase and phospholipase inhibitors |
| WO2007043638A1 (en) | 2005-10-14 | 2007-04-19 | Astellas Pharma Inc. | Condensed heterocyclic compound |
| WO2007047625A2 (en) | 2005-10-20 | 2007-04-26 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2007047177A1 (en) | 2005-10-13 | 2007-04-26 | Merck & Co., Inc. | Acyl indoles, compositions containing such compounds and methods of use |
| WO2007053345A1 (en) | 2005-11-01 | 2007-05-10 | Array Biopharma Inc. | Glucokinase activators |
| WO2007051847A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides as glucokinase modulators |
| WO2007051810A2 (en) | 2005-11-01 | 2007-05-10 | Transtech Pharma | Pharmaceutical use of substituted amides |
| WO2007051845A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides |
| WO2007051811A2 (en) | 2005-11-01 | 2007-05-10 | Transtech Pharma | Pharmaceutical use of substituted amides |
| WO2007051846A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides |
| WO2007053765A2 (en) | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Substituted cycloalkylpyrrolones as allosteric modulators of glucokinase |
| WO2007056771A2 (en) | 2005-11-09 | 2007-05-18 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007058346A1 (en) | 2005-11-21 | 2007-05-24 | Shionogi & Co., Ltd. | HETEROCYCLIC COMPOUND HAVING INHIBITORY ACTIVITY ON 11-β-HYDROXYSTEROID DEHYDROGENASE TYPE I |
| WO2007057768A2 (en) | 2005-11-18 | 2007-05-24 | Pfizer Products Inc. | Sulfonyl derivatives |
| WO2007060992A1 (en) | 2005-11-25 | 2007-05-31 | Kaneka Corporation | Agent for preventing or improving metabolic syndrome or insulin-resistance syndrome |
| WO2007061923A2 (en) | 2005-11-18 | 2007-05-31 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2007061661A2 (en) | 2005-11-22 | 2007-05-31 | Amgen Inc. | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2007062568A1 (en) | 2005-12-02 | 2007-06-07 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Compounds used as mammalian cell amp-activated protein kinase(ampk) activators and their preparation methods and usages |
| WO2007063928A1 (en) | 2005-11-30 | 2007-06-07 | Toray Industries, Inc. | Novel noncyclic amine carboxamide derivative and salt thereof |
| WO2007068330A1 (en) | 2005-12-16 | 2007-06-21 | Merck Patent Gmbh | 2-ADAMANTYLUREA DERIVATIVES AS SELECTIVE 11β-HSD1 INHIBITORS |
| WO2007070506A2 (en) | 2005-12-14 | 2007-06-21 | Amgen Inc. | Diaza heterocyclic sulfonamide derivatives and their uses |
| WO2007070434A2 (en) | 2005-12-14 | 2007-06-21 | Merck & Co., Inc. | Fused aminopiperidines as dipeptidyl peptidase-4 inhibitors for the treatment or prevention of diabetes |
| WO2007071576A1 (en) | 2005-12-21 | 2007-06-28 | F. Hoffmann-La Roche Ag | New salt and polymorph of dpp-iv inhibitor |
| WO2007073117A1 (en) | 2005-12-22 | 2007-06-28 | Crystalgenomics, Inc. | Aminopyrimidine derivatives inhibiting protein kinase activity, method for the preparation thereof and pharmaceutical composition containing same |
| WO2007071766A2 (en) | 2005-12-22 | 2007-06-28 | Transtech Pharma | Phenoxy acetic acids as ppar delta activators |
| WO2007071738A1 (en) | 2005-12-23 | 2007-06-28 | Novartis Ag | Condensed heterocyclic compounds useful as dpp-iv inhibitors |
| WO2007075847A2 (en) | 2005-12-20 | 2007-07-05 | Takeda Pharmaceutical Company Limited | Glucokinase activators |
| WO2007077508A2 (en) | 2005-12-30 | 2007-07-12 | Ranbaxy Laboratories Limited | Derivatives of beta-amino acid as dipeptidyl peptidase-iv inhibitors |
| WO2007081755A2 (en) | 2006-01-09 | 2007-07-19 | Metabasis Therapeutics, Inc. | Indole-benzimidazole and indazole inhibitors of tyrosine phosphatases |
| WO2007080170A1 (en) | 2006-01-11 | 2007-07-19 | Boehringer Ingelheim International Gmbh | CRYSTALLINE FORM OF 1´-(1-METHYLETHYL)- 4´-[(2-FLUORO-4-METHOXYPHENYL)METHYL]-5´-METHYL-1H-PYRAZOL-3´-O-β-D-GLUCOPYRANOSIDE, A METHOD FOR ITS PREPARATION AND THE USE THEREOF FOR PREPARING MEDICAMENTS |
| WO2007083978A1 (en) | 2006-01-23 | 2007-07-26 | Crystalgenomics, Inc. | Imidazopyridine derivatives inhibiting protein kinase activity, method for the preparation thereof and pharmaceutical composition containing same |
| WO2007087231A2 (en) | 2006-01-25 | 2007-08-02 | Merck & Co., Inc. | Aminocyclohexanes as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007085135A1 (en) | 2006-01-25 | 2007-08-02 | Institute Of Pharmacology And Toxicology Academy Of Military Medical Sciences P.L.A. China | 1,3-benzodioxolecyclopentene-2,2-dicarboxylic acid derivates, preparation process and medical uses thereof |
| WO2007087150A2 (en) | 2006-01-13 | 2007-08-02 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2007085136A1 (en) | 2006-01-24 | 2007-08-02 | Institute Of Pharmacology And Toxicology Academy Of Military Medical Sciences P.L.A. China | 1,3-benzodioxolecyclopentene derivates, preparation process and medical uses thereof |
| WO2007087448A1 (en) | 2006-01-30 | 2007-08-02 | Irm Llc | Spiro imidazole derivatives as ppar modulators |
| WO2007089557A2 (en) | 2006-01-30 | 2007-08-09 | Irm Llc | Polycyclic 1, 2, 3, 4 -tetrahydro- isoquinoline derivatives and compositions comprising them as ppar modulators |
| WO2007089683A1 (en) | 2006-01-31 | 2007-08-09 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2007089512A1 (en) | 2006-01-27 | 2007-08-09 | Array Biopharma Inc. | Glucokinase activators |
| WO2007089667A1 (en) | 2006-01-30 | 2007-08-09 | Irm Llc | Compounds and compositions as ppar modulators |
| WO2007092435A2 (en) | 2006-02-07 | 2007-08-16 | Wyeth | 11-beta hsd1 inhibitors |
| WO2007092364A2 (en) | 2006-02-07 | 2007-08-16 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007095462A2 (en) | 2006-02-13 | 2007-08-23 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007093610A1 (en) | 2006-02-15 | 2007-08-23 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzonitrile derivatives, pharmaceutical compositions containing such compounds, their use and process for their manufacture |
| WO2007093264A1 (en) | 2006-02-14 | 2007-08-23 | Merck Patent Gmbh | Mandelic hydrazides |
| WO2007097931A2 (en) | 2006-02-15 | 2007-08-30 | Merck & Co., Inc. | Aminotetrahydropyrans as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| US20070208001A1 (en) | 2006-03-03 | 2007-09-06 | Jincong Zhuo | Modulators of 11- beta hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| US20070207985A1 (en) | 2006-03-01 | 2007-09-06 | Bristol-Myers Squibb Company | Triazine 11-beta hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2007101060A2 (en) | 2006-02-28 | 2007-09-07 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007101270A1 (en) | 2006-03-02 | 2007-09-07 | Incyte Corporation | MODULATORS OF 11-β HYDROXYL STEROID DEHYDROGENASE TYPE 1, PHARMACEUTICAL COMPOSITIONS THEREOF, AND METHODS OF USING THE SAME |
| WO2007099385A1 (en) | 2006-03-01 | 2007-09-07 | Glenmark Pharmaceuticals S.A. | Dipeptidyl peptidase iv inhibitor compounds and compositions |
| WO2007099553A2 (en) | 2006-02-27 | 2007-09-07 | Cadila Healthcare Limited | 1,3-dioxane carboxylic acids |
| WO2007100374A2 (en) | 2005-12-19 | 2007-09-07 | Trustees Of Tufts College | Soft protease inhibitors and pro-soft forms thereof |
| WO2007100027A1 (en) | 2006-03-02 | 2007-09-07 | Daiichi Sankyo Company, Limited | Optically active thiazolidinedione derivative |
| WO2007101864A2 (en) | 2006-03-09 | 2007-09-13 | High Point Pharmaceuticals, Llc | Compounds that modulate ppar activity, their preparation and use |
| WO2007104034A2 (en) | 2006-03-08 | 2007-09-13 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2007103252A2 (en) | 2006-03-03 | 2007-09-13 | Merck & Co., Inc. | Novel crystalline forms of antidiabetic compounds |
| WO2007104053A2 (en) | 2006-03-09 | 2007-09-13 | Pharmacopeia, Inc. | 8-heteroarylpurine mnk2 inhibitors for treating metabolic disorders |
| WO2007102515A1 (en) | 2006-03-07 | 2007-09-13 | Astellas Pharma Inc. | Phenylthiazole derivative |
| WO2007105650A1 (en) | 2006-03-10 | 2007-09-20 | Ajinomoto Co., Inc. | 4-hydroxyisoleucine derivative and process for producing the derivative |
| WO2007104789A2 (en) | 2006-03-15 | 2007-09-20 | Novo Nordisk A/S | Amylin derivatives |
| WO2007105753A1 (en) | 2006-03-16 | 2007-09-20 | Astellas Pharma Inc. | Triazole derivative or salt thereof |
| WO2007106181A2 (en) | 2005-11-17 | 2007-09-20 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007106469A2 (en) | 2006-03-14 | 2007-09-20 | Amgen Inc. | Bicyclic carboxylic acid derivatives useful for treating metabolic disorders |
| WO2007107550A1 (en) | 2006-03-21 | 2007-09-27 | High Point Pharmaceuticals, Llc | Adamantane derivatives for the treatment of the metabolic syndrome |
| WO2007107470A2 (en) | 2006-03-22 | 2007-09-27 | F. Hoffmann-La Roche Ag | Pyrazoles as 11-beta-hsd-1 |
| JP2007246474A (en) | 2006-03-17 | 2007-09-27 | Mitsubishi Pharma Corp | Benzene derivative |
| WO2007112347A1 (en) | 2006-03-28 | 2007-10-04 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2007111864A2 (en) | 2006-03-23 | 2007-10-04 | Merck & Co., Inc. | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use |
| WO2007110216A1 (en) | 2006-03-28 | 2007-10-04 | Sanofi-Aventis | Azolopyridin-3-one derivatives as inhibitors of endothelial lipase |
| WO2007111921A1 (en) | 2006-03-23 | 2007-10-04 | Amgen Inc. | 1-phenylsulfonyl-diaza heterocyclic amide compounds and their uses as modulators of hydroxsteroid dehydrogenases |
| WO2007114532A1 (en) | 2006-04-03 | 2007-10-11 | Industry-Academic Cooperation Foundation Gyeongsang National University | Novel chalcone derivatives, pharmaceutically acceptable salt, method for preparation and uses thereof |
| WO2007112914A2 (en) | 2006-03-30 | 2007-10-11 | Novartis Ag | Ceramide kinase modulation |
| WO2007113226A1 (en) | 2006-03-31 | 2007-10-11 | Novartis Ag | Organic compounds |
| WO2007113634A1 (en) | 2006-04-03 | 2007-10-11 | Matrix Laboratories Ltd. | Novel dipeptidyl peptidase iv inhibitors and processes for their preparation and pharmaceutical compositions containing them |
| WO2007115058A2 (en) | 2006-03-31 | 2007-10-11 | Novartis Ag | Thiadiazolidinone inhibitors of ptpase |
| WO2007112669A1 (en) | 2006-04-05 | 2007-10-11 | Shanghai Hengrui Pharmaceutical Co.Ltd. | Dicyclooctane derivates, preparation processes and medical uses thereof |
| WO2007115935A1 (en) | 2006-04-07 | 2007-10-18 | High Point Pharmaceuticals, Llc | 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 ACTIVE COMPOUNDS |
| WO2007117381A2 (en) | 2006-03-24 | 2007-10-18 | Array Biopharma Inc. | 2 -aminopyridine analogs as glucokinase activators |
| US20070244094A1 (en) | 2006-04-18 | 2007-10-18 | Gee-Hong Kuo | Benzoazepin-oxy-acetic acid derivatives as ppar-delta agonists used for the increase of hdl-c, lower ldl-c and lower cholesterol |
| WO2007116229A1 (en) | 2006-04-06 | 2007-10-18 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2007118185A2 (en) | 2006-04-07 | 2007-10-18 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| WO2007116092A1 (en) | 2006-04-12 | 2007-10-18 | Probiodrug Ag | Enzyme inhibitors |
| WO2007116230A1 (en) | 2006-04-11 | 2007-10-18 | Prosidion Ltd | Azetidine derivatives as g-protein coupled receptor (gpr119 ) agonists |
| WO2007115821A2 (en) | 2006-04-11 | 2007-10-18 | Novartis Ag | Organic compounds |
| WO2007115822A1 (en) | 2006-04-07 | 2007-10-18 | Develogen Aktiengesellschaft | Thienopyrimidines having mnkl /mnk2 inhibiting activity for pharmaceutical compositions |
| WO2007119827A1 (en) | 2006-04-14 | 2007-10-25 | Dic Plastics, Inc. | Locking structure of cover body and container |
| WO2007118964A1 (en) | 2006-03-24 | 2007-10-25 | Genfit | Substituted n-(phenethyl)benzamide derivatives, preparation and uses thereof |
| WO2007120284A2 (en) | 2005-11-23 | 2007-10-25 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007119887A1 (en) | 2006-04-18 | 2007-10-25 | Nippon Chemiphar Co., Ltd. | ACTIVATING AGENT FOR PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR δ |
| WO2007120102A1 (en) | 2006-04-19 | 2007-10-25 | Astrazeneca Ab | New substituted oxindole derivatives |
| WO2007119837A1 (en) | 2006-04-14 | 2007-10-25 | Ajinomoto Co., Inc. | Lipase inhibitor |
| WO2007118963A2 (en) | 2006-03-24 | 2007-10-25 | Genfit | Substituted n-(phenethyl)benzamide derivatives, preparation and uses thereof |
| WO2007120899A2 (en) | 2006-04-13 | 2007-10-25 | Societe De Conseils De Recherches Et D'applications Scientifiques, S.A.S. | Pharmaceutical compositions of hglp-1, exendin-4 and analogs thereof |
| WO2007120575A2 (en) | 2006-04-11 | 2007-10-25 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007120270A2 (en) | 2005-11-22 | 2007-10-25 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007122482A1 (en) | 2006-04-20 | 2007-11-01 | Pfizer Products Inc. | Fused phenyl amido heterocyclic compounds for the prevention and treatment of glucokinase-mediated diseases |
| WO2007122970A1 (en) | 2006-04-20 | 2007-11-01 | Osaka University | Ligand capable of binding to nuclear receptor |
| WO2007123581A1 (en) | 2005-11-17 | 2007-11-01 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007123225A1 (en) | 2006-04-24 | 2007-11-01 | Astellas Pharma Inc | Oxadiazolidinedione compound |
| WO2007124329A1 (en) | 2006-04-21 | 2007-11-01 | Eli Lilly And Company | Cyclohexylimidazole lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007122634A2 (en) | 2006-04-24 | 2007-11-01 | Jubilant Biosys Limited | Pyrimidinediones as tyrosine kinase inhibitors |
| WO2007124337A1 (en) | 2006-04-21 | 2007-11-01 | Eli Lilly And Company | Biphenyl amide lactam derivatives as inhibitors of 11- beta-hydroxysteroid dehydrogenase 1 |
| WO2007124461A2 (en) | 2006-04-20 | 2007-11-01 | Amgen Inc. | Glp-1 compounds |
| WO2007124254A2 (en) | 2006-04-21 | 2007-11-01 | Eli Lilly And Company | Cyclohexylpyrazole-lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007122411A1 (en) | 2006-04-26 | 2007-11-01 | Astrazeneca Ab | Diazepan-1-yl-sulfonyl derivatives for the treatment of metabolic syndrome |
| EP1852108A1 (en) | 2006-05-04 | 2007-11-07 | Boehringer Ingelheim Pharma GmbH & Co.KG | DPP IV inhibitor formulations |
| WO2007127693A1 (en) | 2006-04-24 | 2007-11-08 | Eli Lilly And Company | Substituted pyrrolidinones as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127726A2 (en) | 2006-04-25 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127688A2 (en) | 2006-04-24 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| US20070259900A1 (en) | 2006-05-04 | 2007-11-08 | Peter Sieger | Polymorphs |
| WO2007126745A2 (en) | 2006-03-28 | 2007-11-08 | Merck & Co., Inc. | Aminotetrahydropyrans as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007126043A1 (en) | 2006-04-27 | 2007-11-08 | Mitsubishi Tanabe Pharma Corporation | Use as drugs of carboxylic acid derivatives having thiazole rings |
| WO2007127704A1 (en) | 2006-04-24 | 2007-11-08 | Eli Lilly And Company | Cyclohexyl substituted pyrrolidinones as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| JP2007291075A (en) | 2006-03-27 | 2007-11-08 | Sankyo Co Ltd | New compound sterenin and method for producing the same |
| WO2007125105A2 (en) | 2006-04-28 | 2007-11-08 | Transtech Pharma, Inc. | Benzamide glucokinase activators |
| WO2007125103A2 (en) | 2006-04-28 | 2007-11-08 | Transtech Pharma, Inc. | Benzamide glucokinase activators |
| WO2007127763A2 (en) | 2006-04-25 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007125110A1 (en) | 2006-04-28 | 2007-11-08 | Noscira, S.A. | N- ( 2-thiazolyl) -amide derivatives as gsk-3 inhibitors |
| WO2007127901A1 (en) | 2006-04-28 | 2007-11-08 | Eli Lilly And Company | Pieridinyl substituted pyrrolidinones as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127765A1 (en) | 2006-04-25 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007126117A1 (en) | 2006-05-02 | 2007-11-08 | Taisho Pharmaceutical Co., Ltd. | Phenyl 5-thioglucoside compound |
| WO2007130898A1 (en) | 2006-05-01 | 2007-11-15 | Incyte Corporation | TETRASUBSTITUTED UREAS AS MODULATORS OF 11-β HYDROXYL STEROID DEHYDROGENASE TYPE 1 |
| WO2007128801A1 (en) | 2006-05-08 | 2007-11-15 | Novartis Ag | Combination of organic compounds |
| WO2007128815A1 (en) | 2006-05-09 | 2007-11-15 | Novo Nordisk A/S | Insulin derivative |
| US20070264331A1 (en) | 2005-09-08 | 2007-11-15 | Laboratorios Silanes, S.A De C.V. | Stable pharmaceutical composition of immediate-release glimepiride and extended-release metformin |
| WO2007128817A2 (en) | 2006-05-09 | 2007-11-15 | Novo Nordisk A/S | Insulin derivative |
| WO2007129668A1 (en) | 2006-05-02 | 2007-11-15 | Taisho Pharmaceutical Co., Ltd. | Pyrazolyl 5-thioglucoside compound |
| US20070265332A1 (en) | 2006-05-15 | 2007-11-15 | Min Ge | Antidiabetic bicyclic compounds |
| WO2007128480A2 (en) | 2006-05-02 | 2007-11-15 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Thioglycosides as pharmaceutically active agents |
| WO2007131620A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | 4,5-diphenyl-pyrimidinyl-amino substituted carboxylic acids, method for the production and use thereof as medicaments |
| US20070270424A1 (en) | 2006-05-17 | 2007-11-22 | Yun-Long Li | Heterocyclic inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 and methods of using the same |
| WO2007131622A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | Phenylamino-benzoxazole substituted carboxylic acids, method for their production and use thereof as medicaments |
| US20070270492A1 (en) | 2006-03-06 | 2007-11-22 | Avestha Gengraine Technologies Pvt. Ltd. | Nananoic acid derivatives as dipeptidyl peptidase inhibitors |
| WO2007131619A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | 4,5-diphenyl-pyrimidinyl-oxy or -mercapto substituted carboxylic acids, method for the production and use thereof as medicaments |
| WO2007131621A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | 4,5-diphenyl-pyrimidinyl substituted carboxylic acids, method for the production and use thereof as medicaments |
| WO2007137008A2 (en) | 2006-05-18 | 2007-11-29 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| US20070276041A1 (en) | 2006-05-25 | 2007-11-29 | Ajinomoto, Co., Inc. | Ppar activity regulators |
| WO2007136577A2 (en) | 2006-05-16 | 2007-11-29 | Merck & Co., Inc. | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use |
| WO2007135427A1 (en) | 2006-05-23 | 2007-11-29 | Astrazeneca Ab | 1,4-disubstituted piperazine and 1,4-disubstituted azepane as 11 -beta-hydroxysteroid dehydrogenase 1 inhibitors |
| WO2007136603A2 (en) | 2006-05-16 | 2007-11-29 | Merck & Co., Inc. | Aminotetrahydropyrans as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007134986A1 (en) | 2006-05-23 | 2007-11-29 | F. Hoffmann-La Roche Ag | Pyridopyrimidinone derivatives |
| US20070275907A1 (en) | 2006-05-23 | 2007-11-29 | Yuanwei Chen | Glucose transport inhibitors and methods of use |
| WO2007136116A2 (en) | 2006-05-19 | 2007-11-29 | Taisho Pharmaceutical Co., Ltd. | C-phenyl glycitol compound for the treatment of diabetes |
| US20070281942A1 (en) | 2006-05-31 | 2007-12-06 | Cao Sheldon X | Glucokinase activators |
| WO2007140230A2 (en) | 2006-05-26 | 2007-12-06 | Nestec S.A. | Methods of use and nutritional compositions of touchi extract |
| WO2007139992A2 (en) | 2006-05-26 | 2007-12-06 | Novartis Ag | ALDOSTERONE SYNTHASE AND/OR 11β-HYDROXYLASE INHIBITORS |
| WO2007137962A1 (en) | 2006-06-01 | 2007-12-06 | F. Hoffmann-La Roche Ag | Thiazole derivatives |
| US20070281949A1 (en) | 2006-05-15 | 2007-12-06 | Cephalon, Inc. | Substituted pyrazolopyrimidines |
| WO2007138485A2 (en) | 2006-01-18 | 2007-12-06 | Evolva Sa | Ppar modulators |
| WO2007141423A1 (en) | 2006-06-06 | 2007-12-13 | Negma-Lerads | Ppar enhancing derivatives, method for preparing same and therapeutic use thereof |
| WO2007143316A2 (en) | 2006-05-05 | 2007-12-13 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of sglt2 |
| WO2007142253A1 (en) | 2006-06-06 | 2007-12-13 | Mitsubishi Tanabe Pharma Corporation | 2-cyanopyrrolidine derivative |
| US20070287674A1 (en) | 2006-06-08 | 2007-12-13 | Hej Research Institute Of Chemistry | New treatment of diabetes mellitus |
| WO2007145834A2 (en) | 2006-06-08 | 2007-12-21 | Amgen Inc. | Benzamide derivatives and uses related thereto |
| WO2007146761A2 (en) | 2006-06-12 | 2007-12-21 | Neurogen Corporation | Diaryl pyrimidinones and related compounds |
| WO2007145835A2 (en) | 2006-06-08 | 2007-12-21 | Amgen Inc. | Benzamide derivatives as modulators of 11beta-hsd1 for treating diabetes and obesity |
| WO2007144394A2 (en) | 2006-06-16 | 2007-12-21 | High Point Pharmaceuticals, Llc. | Pharmaceutical use of substituted piperidine carboxamides |
| WO2007150026A2 (en) | 2006-06-23 | 2007-12-27 | Incyte Corporation | Purinone derivatives as hm74a agonists |
| WO2007150025A2 (en) | 2006-06-23 | 2007-12-27 | Incyte Corporation | Purinone derivatives as hm74a agonists |
| WO2007147478A1 (en) | 2006-06-23 | 2007-12-27 | Merck Patent Gmbh | 3 -amino-imidazo[1, 2-a]pyridine derivatives as sglt inhibitors |
| WO2007148185A2 (en) | 2006-06-21 | 2007-12-27 | Pfizer Products Inc. | Substituted 3 -amino- pyrrolidino-4 -lactams as dpp inhibitors |
| WO2007149865A2 (en) | 2006-06-19 | 2007-12-27 | University Of Utah Research Foundation | Methods and compositions related to inhibition of ceramide synthesis |
| US20080004281A1 (en) | 2006-06-28 | 2008-01-03 | Kalypsys, Inc. | Methods for the modulation of crp by the selective modulation of ppar delta |
| WO2008002824A1 (en) | 2006-06-28 | 2008-01-03 | Bristol-Myers Squibb Company | Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| WO2008001931A2 (en) | 2006-06-27 | 2008-01-03 | Takeda Pharmaceutical Company Limited | Fused cyclic compounds |
| US20080004325A1 (en) | 2006-06-29 | 2008-01-03 | Wyeth | PTP1B inhibitors |
| WO2008002245A2 (en) | 2006-06-27 | 2008-01-03 | Astrazeneca Ab | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| WO2008001864A1 (en) | 2006-06-29 | 2008-01-03 | Taisho Pharmaceutical Co., Ltd. | C-phenyl 1-thioglucitol compound |
| WO2008002244A2 (en) | 2006-06-27 | 2008-01-03 | Astrazeneca Ab | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| WO2008000950A2 (en) | 2006-06-27 | 2008-01-03 | Sanofi-Aventis | Derivatives of ureas of piperidine or pyrrolidine, their preparation and their therapeutical use |
| WO2008000951A2 (en) | 2006-06-27 | 2008-01-03 | Sanofi-Aventis | Urea derivatives of tropane, their preparation and their therapeutic application |
| WO2008005964A2 (en) | 2006-07-06 | 2008-01-10 | Bristol-Myers Squibb Company | Phosphonate and phosphinate compounds as glucokinase activators |
| WO2008005569A2 (en) | 2006-07-06 | 2008-01-10 | Arena Pharmaceuticals, Inc. | Modulators of metabolism and the treatment of disorders related thereto |
| WO2008006043A2 (en) | 2006-07-07 | 2008-01-10 | Bristol-Myers Squibb Company | 1,3-oxazole derivatives useful as anti-atherosclerotic, anti-dyslipidemic, anti-diabetic and anti-obesity agents |
| WO2008005910A2 (en) | 2006-07-06 | 2008-01-10 | Bristol-Myers Squibb Company | Pyridone/hydroxypyridine 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2008005914A2 (en) | 2006-07-06 | 2008-01-10 | Bristol-Myers Squibb Company | Novel glucokinase activators and methods of using same |
| WO2008005576A1 (en) | 2006-07-06 | 2008-01-10 | Arena Pharmaceuticals, Inc. | Modulators of metabolism and the treatment of disorders related thereto |
| WO2008003611A1 (en) | 2006-07-05 | 2008-01-10 | F. Hoffmann-La Roche Ag | Alkyl-pyridazine derivatives as inhibitors of 11 beta hydroxysteroid dehydrogenase type 1(11b-hsd 1) |
| WO2008006044A2 (en) | 2006-07-07 | 2008-01-10 | Bristol-Myers Squibb Company | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method |
| WO2008008887A2 (en) | 2006-07-13 | 2008-01-17 | Smithkline Beecham Corporation | Gpr119 agonists for treating metabolic disorders |
| WO2008008547A2 (en) | 2006-07-14 | 2008-01-17 | Ams Research Corporation | Balloon dilation for implantable prosthesis |
| WO2008006703A1 (en) | 2006-07-13 | 2008-01-17 | High Point Pharmaceuticals, Llc | 4-piperidylbenzamides as 11-beta-hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2008006702A1 (en) | 2006-07-13 | 2008-01-17 | High Point Pharmaceuticals, Llc. | 11beta-hydroxysteroid dehydrogenase type 1 active compounds |
| WO2008006969A2 (en) | 2006-07-10 | 2008-01-17 | Les Laboratoires Servier | Novel tetracyclic derivatives, process for the preparation thereof and pharmaceutical compositions which contain them |
| WO2008006319A1 (en) | 2006-07-07 | 2008-01-17 | Shanghai Allist Pharmaceutical., Inc. | BIBENZIMIDAZOLE DERIVATIVES HAVING PPARγ AGONIST ACTIVITY AND THEIR USES |
| WO2008006432A1 (en) | 2006-07-13 | 2008-01-17 | Merck Patent Gmbh | Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
| WO2008007692A1 (en) | 2006-07-14 | 2008-01-17 | Panasonic Corporation | Method for pretreating electrochemical capacitor negative electrode, method for manufacturing the electrochemical capacitor negative electrode, and method for manufacturing electrochemical capacitor using the method for manufacturing the electrochemical capacitor negative electrode |
| WO2008011453A2 (en) | 2006-07-20 | 2008-01-24 | Amgen Inc. | SUBSTITUTED AZOLE AROMATIC HETEROCYCLES AS INHIBITORS OF LLβ-HSD-1 |
| WO2008010238A2 (en) | 2006-07-21 | 2008-01-24 | Lupin Limited | Antidiabetic azabicyclo [3. 1. 0] hexan compounds |
| WO2008009335A2 (en) | 2006-07-18 | 2008-01-24 | Merck Patent Gmbh | Aminoindazole urea derivatives |
| WO2008013277A1 (en) | 2006-07-27 | 2008-01-31 | Chugai Seiyaku Kabushiki Kaisha | Fused ring spiroketal derivative and use thereof as drug for treating diabetes |
| WO2008013322A1 (en) | 2006-07-27 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | 1- (-d-glycopyranosyl) - 3 - (4-cyclopropylphenylmethyl) - 4 - halogeno indole derivatives and use thereof as sglt inhibitors. |
| WO2008012532A2 (en) | 2006-07-27 | 2008-01-31 | Astrazeneca Ab | : pyridine-3-carboxamide compounds and their use for inhibiting 11-beta-hydroxysteroid dehydrogenase |
| WO2008013321A1 (en) | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
| WO2008013280A1 (en) | 2006-07-27 | 2008-01-31 | Chugai Seiyaku Kabushiki Kaisha | Substituted spiroketal derivative and use thereof as drug for treating diabetes |
| WO2008012470A2 (en) | 2006-07-24 | 2008-01-31 | Genfit | Substituted imidazolone derivatives, preparation and uses |
| WO2008016730A2 (en) | 2006-08-02 | 2008-02-07 | Targeted Molecular Diagnostics, Llc | Compositions and methods for reducing cellular fat |
| WO2008016278A1 (en) | 2006-08-04 | 2008-02-07 | Korea Research Institute Of Chemical Technology | Furan-2-carboxylic acid derivatives and process for the preparation thereof |
| WO2008016968A2 (en) | 2006-08-03 | 2008-02-07 | Trustees Of Tufts College | Non-flushing niacin analogues, and methods of use thereof |
| WO2008016123A1 (en) | 2006-08-03 | 2008-02-07 | Takeda Pharmaceutical Company Limited | GSK-3β INHIBITOR |
| WO2008016132A1 (en) | 2006-08-04 | 2008-02-07 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
| WO2008016175A1 (en) | 2006-08-03 | 2008-02-07 | Nippon Chemiphar Co., Ltd. | Activator for peroxisome proliferator activated receptor |
| JP2008031161A (en) | 2006-07-06 | 2008-02-14 | Taisho Pharmaceut Co Ltd | Prophylactic or curative agent for diabetes containing 1-thio-d-glucitol derivative as active ingredient |
| JP2008031064A (en) | 2006-07-27 | 2008-02-14 | Astellas Pharma Inc | Diacylpiperazine derivative |
| WO2008017398A2 (en) | 2006-08-07 | 2008-02-14 | Krka | Polymorphic forms of rosiglitazone base |
| WO2008019309A1 (en) | 2006-08-04 | 2008-02-14 | Metabasis Therapeutics, Inc. | Novel inhibitors of fructose 1,6-bisphosphatase |
| WO2008017670A1 (en) | 2006-08-08 | 2008-02-14 | Boehringer Ingelheim International Gmbh | Pyrrolo [3, 2 -d] pyrimidines as dpp-iv inhibitors for the treatment of diabetes mellitus |
| WO2008020607A1 (en) | 2006-08-17 | 2008-02-21 | Dainippon Sumitomo Pharma Co., Ltd. | Phthalide derivative and salt thereof |
| WO2008020011A1 (en) | 2006-08-15 | 2008-02-21 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted cyclopropylbenzene derivatives, pharmaceutical compositions containing such compounds, their use as sglt inhibitors and process for their manufacture |
| WO2008021560A2 (en) | 2006-08-17 | 2008-02-21 | Amylin Pharmaceuticals, Inc. | Dpp-iv resistant gip hybrid polypeptides with selectable properties |
| WO2008022015A2 (en) | 2006-08-11 | 2008-02-21 | Trustees Of Tufts College | Retro-inverso incretin analogues, and methods of use thereof |
| US20080051452A1 (en) | 2006-03-06 | 2008-02-28 | Avestha Gengraine Technologies Pvt. Ltd. | Hexanoic acid derivatives as dipeptidyl peptidase inhibitors |
| WO2008024497A2 (en) | 2006-08-25 | 2008-02-28 | Vitae Pharmaceuticals, Inc. | INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE TYPE 1 |
| WO2008023239A1 (en) | 2006-08-23 | 2008-02-28 | Pfizer Products Inc. | Pyrimidone compounds as gsk-3 inhibitors |
| WO2008024892A2 (en) | 2006-08-24 | 2008-02-28 | Bristol-Myers Squibb Company | Cyclic 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2008027584A2 (en) | 2006-09-01 | 2008-03-06 | Vertex Pharmaceuticals Incorporated | 5- (2-furyl)-1, 3-thiazole derivatives useful as inhibitors of phosphatidylinositol 3-kinase |
| WO2008025799A1 (en) | 2006-08-30 | 2008-03-06 | Biovitrum Ab (Publ) | Pyridazine compounds for treating gpr119 related disorders |
| WO2008028188A2 (en) | 2006-09-01 | 2008-03-06 | The Ticket Reserve | Demand aggregation for future items contingent upon threshold demand |
| WO2008027273A2 (en) | 2006-08-30 | 2008-03-06 | Phenomix Corporation | Solid citrate and tartrate salts of dpp-iv inhibitors |
| WO2008029217A2 (en) | 2006-08-29 | 2008-03-13 | Orchid Research Laboratories Limited | Dipeptidyl peptidase iv inhibitors |
| WO2008030520A1 (en) | 2006-09-07 | 2008-03-13 | Amgen Inc. | Heterocyclic gpr40 modulators |
| WO2008028662A1 (en) | 2006-09-07 | 2008-03-13 | Santhera Pharmaceuticals (Schweiz) Ag | N-[1-(3-amino-4-phenyl-butyryl)-4-hydroxy-pyrrolidin-2-ylmethyl}-propionamide and related compounds as dpp-iv inhibitors for the treatment of type 2 diabetes mellitus |
| WO2008030618A1 (en) | 2006-09-07 | 2008-03-13 | Amgen Inc. | Benzo-fused compounds for use in treating metabolic disorders |
| WO2008033851A2 (en) | 2006-09-13 | 2008-03-20 | Takeda Pharmaceutical Company Limited | Use of 2-6- (3-amino-piperidin-l-yl) -3-methyl-2, 4-dioxo-3, 4-dihydr0-2h-pyrimidin-1-ylmet hyl-4-fluoro-benzonitrile |
| WO2008034032A2 (en) | 2006-09-14 | 2008-03-20 | Azevan Pharmaceuticals, Inc. | Beta-lactam cannabinoid receptor modulators |
| WO2008033455A2 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Biphenyl and heteroaryl phenyl derivatives as protein tyrosine phosphatases inhibitors |
| WO2008032164A2 (en) | 2006-09-12 | 2008-03-20 | Pfizer Products Inc. | Benzimidazolone derivatives |
| WO2008033931A1 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Para-xylylene carboxylic acids and isothiazolones useful as protein tyrosine phosphatases (ptps) in particular ptp-ib |
| WO2008033932A2 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Biarylthiazole carboxylic acid derivatives as protein tyrosine phosphatase-ib inhibitors |
| WO2008033934A1 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Substituted heteroaryl carboxylic acid derivatives as ptb-1b inhibitors |
| JP2008063256A (en) | 2006-09-06 | 2008-03-21 | Astellas Pharma Inc | beta-AMINO ACID DERIVATIVE |
| WO2008035359A2 (en) | 2006-06-12 | 2008-03-27 | Cadila Healthcare Limited | Oximinophenoxyalkanoic acid and phenylalkanoic acid derivatives |
| WO2008034859A1 (en) | 2006-09-21 | 2008-03-27 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted difluorobenzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008034881A1 (en) | 2006-09-22 | 2008-03-27 | Novo Nordisk A/S | Protease resistant insulin analogues |
| WO2008037628A1 (en) | 2006-09-29 | 2008-04-03 | F. Hoffmann-La Roche Ag | Sulfonamide derivatives |
| WO2008039882A1 (en) | 2006-09-30 | 2008-04-03 | Sanofi-Aventis U.S. Llc | A combination of niacin and a prostaglandin d2 receptor antagonist |
| WO2008042223A1 (en) | 2006-10-03 | 2008-04-10 | Merck & Co., Inc. | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use |
| WO2008042688A2 (en) | 2006-09-29 | 2008-04-10 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| WO2008040974A1 (en) | 2006-10-07 | 2008-04-10 | Peakdale Molecular Limited | Indoles for use as dpp-iv inhibitors |
| WO2008044777A1 (en) | 2006-10-06 | 2008-04-17 | Banyu Pharmaceutical Co., Ltd. | 2-pyridinecarboxamide derivative having gk-activating activity |
| WO2008044700A1 (en) | 2006-10-11 | 2008-04-17 | Takeda Pharmaceutical Company Limited | GSK-3β INHIBITOR |
| WO2008045484A1 (en) | 2006-10-10 | 2008-04-17 | Amgen Inc. | N-aryl pyrazole compounds for use against diabetes |
| WO2008044656A1 (en) | 2006-10-06 | 2008-04-17 | Taisho Pharmaceutical Co., Ltd. | Imidazolidinone derivative |
| WO2008043701A1 (en) | 2006-10-13 | 2008-04-17 | F. Hoffmann-La Roche Ag | Pharmaceutical solid dosage forms comprising compounds micro-embedded in ionic water-insoluble polymers |
| WO2008043544A1 (en) | 2006-10-11 | 2008-04-17 | Laboratorios Del Dr. Esteve, S.A. | Sulfonamide substituted pyrazoline compounds, their preparation and use as cb1 modulators |
| WO2008044762A1 (en) | 2006-10-13 | 2008-04-17 | Chugai Seiyaku Kabushiki Kaisha | Thioglucose spiroketal derivative and use thereof as therapeutic agent for diabetes |
| WO2008048866A2 (en) | 2006-10-13 | 2008-04-24 | Glaxo Group Limited | Bicyclic heteroaromatic compounds |
| WO2008047821A1 (en) | 2006-10-18 | 2008-04-24 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| US20080096877A1 (en) | 2006-10-19 | 2008-04-24 | Tsuneo Yasuma | Indole compound |
| WO2008048886A2 (en) | 2006-10-16 | 2008-04-24 | Vidyo, Inc. | Systems and methods for signaling and performing temporal level switching in scalable video coding |
| WO2008046758A2 (en) | 2006-10-19 | 2008-04-24 | F. Hoffmann-La Roche Ag | Imidazolone and imidazolidinone derivatives as 11b-hsd1 inhibitors for diabetes |
| WO2008046497A1 (en) | 2006-10-16 | 2008-04-24 | Merck Patent Gmbh | 3 -amino-imidazo [1,2 -a] pyridine derivatives having an sglt1 and sglt2 inhibiting action, for treating type 1 and type 2 diabetes |
| US20080103201A1 (en) | 2006-10-26 | 2008-05-01 | Wijayabandara Mirihanage Don J | Novel alpha-Glucosidase inhibitor from Tabernaemontana dichotoma |
| WO2008050117A1 (en) | 2006-10-26 | 2008-05-02 | Astrazeneca Ab | Benzoyl amino heterocyclyl compounds as glucokinase (glk) activators |
| WO2008049923A1 (en) | 2006-10-27 | 2008-05-02 | Boehringer Ingelheim International Gmbh | CRYSTALLINE FORM OF 4-(ß-D-GLUCOPYRANOS-1-YL)-1-METHYL-2-[4-((S)-TETRAHYDROFURAN-3-YLOXY)-BENZYL]-BENZENE, A METHOD FOR ITS PREPARATION AND THE USE THEREOF FOR PREPARING MEDICAMENTS |
| WO2008051403A2 (en) | 2006-10-20 | 2008-05-02 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2008049711A1 (en) | 2006-10-27 | 2008-05-02 | Novo Nordisk A/S | Peptide extended insulins |
| WO2008050101A2 (en) | 2006-10-23 | 2008-05-02 | Astrazeneca Ab | Benzoyl amino heterocyclyl compounds useful in the treatment of a disease mediated through glk |
| WO2008050987A1 (en) | 2006-10-23 | 2008-05-02 | Hanall Pharmaceutical Co., Ltd. | Combination formulation with controlled release comprising metformin and glimepiride |
| WO2008053446A2 (en) | 2006-11-02 | 2008-05-08 | Piramal Life Sciences Limited | Benzoxazepine compounds, their preparation and use |
| WO2008053194A2 (en) | 2006-11-03 | 2008-05-08 | Astrazeneca Ab | Pyridine carboxamides as 11-beta-hsd1 inhibitors |
| WO2008052638A1 (en) | 2006-11-03 | 2008-05-08 | Merk Patent Gmbh | DIAZEPANE-ACETAMIDE DERIVATIVES AS SELECTIVE 11β-HSD1 INHIBITORS |
| WO2008053331A1 (en) | 2006-11-01 | 2008-05-08 | Pronova Biopharma Norge A/S | Alpha-substituted omega-3 lipids that are activators or modulators of the peroxisome proliferators-activated receptor (ppar). |
| WO2008054675A2 (en) | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
| WO2008054674A2 (en) | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
| WO2008057859A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | Modulators of glucocorticoid receptor, ap-i and/or nf-kappab activity and use thereof |
| WO2008056266A2 (en) | 2006-11-07 | 2008-05-15 | Sanofi-Aventis | Substituted 8-piperidinyl-2-pyridinyl-pyrimido [1,2-a]pyrimidin-6-one and 8-piperidinyl-2-pyrimidinyl-pyrimido[1,2-a]pyrimidin-6-one derivatives |
| WO2008057856A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | Modulators of glucocorticoid receptor, ap-1 and/or nf-kappab activity and use thereof |
| WO2008057857A1 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | MODULATORS OF GLUCOCORTICOID RECEPTOR, AP-1, AND/OR NF-ϰB ACTIVITY AND USE THEREOF |
| WO2008057855A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | Heterocyclic compounds as modulators of glucocorticoid receptor, ap-i, and/or np-kappa-b activity |
| WO2008056726A1 (en) | 2006-11-09 | 2008-05-15 | Sanwa Kagaku Kenkyusho Co., Ltd. | Glp-1 derivative and use thereof |
| WO2008057862A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | MODULATORS OF GLUCOCORTICOID RECEPTOR, AP-1, AND/OR NF-ϰB ACTIVITY AND USE THEREOF |
| WO2008055940A2 (en) | 2006-11-09 | 2008-05-15 | Boehringer Ingelheim International Gmbh | Combination therapy with sglt-2 inhibitors and their pharmaceutical compositions |
| WO2008055870A1 (en) | 2006-11-06 | 2008-05-15 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzonitrile derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008057940A1 (en) | 2006-11-02 | 2008-05-15 | Vertex Pharmaceuticals Incorporated | Aminopyridines and aminopyrimidines useful as inhibitors of protein kinases |
| WO2008059625A1 (en) | 2006-11-17 | 2008-05-22 | National University Corporation Kagawa University | Utilization of the function of rare sugar as promoter for the migration of glucokinase from nucleus to cytoplasm |
| WO2008059866A1 (en) | 2006-11-14 | 2008-05-22 | Santen Pharmaceutical Co., Ltd. | Novel 1,2-dihydroquinoline derivative having substituted phenylamino lower alkyl group and ester-introduced phenyl group as substituents |
| WO2008060488A1 (en) | 2006-11-14 | 2008-05-22 | Merck & Co., Inc. | Tricyclic heteroaromatic compounds as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2008062273A2 (en) | 2006-11-20 | 2008-05-29 | Cadila Pharmaceuticals Limited | Solid oral dosage form having antidiabetic drug combination |
| WO2008062457A2 (en) | 2006-10-03 | 2008-05-29 | Cadila Healthcare Limited | Antidiabetic compounds |
| WO2008062739A1 (en) | 2006-11-20 | 2008-05-29 | Japan Tobacco Inc. | Pyrazoles and use thereof as drugs |
| WO2008064107A2 (en) | 2006-11-20 | 2008-05-29 | Bristol-Myers Squibb Company | 7,8-dihydro-1,6-naphthyridin-5(6h)-ones and related bicyclic compounds as inhibitors of dipeptidyl peptidase iv and methods |
| WO2008061355A1 (en) | 2006-11-24 | 2008-05-29 | Matregen Corp. | Glp-1 depot systems, and methods of manufacture and uses thereof |
| WO2008066070A1 (en) | 2006-11-29 | 2008-06-05 | Uha Mikakuto Co., Ltd. | Dipeptidyl peptidase-iv inhibitor |
| WO2008065796A1 (en) | 2006-11-30 | 2008-06-05 | Fuji-Sangyo Co., Ltd. | α-GLUCOSIDASE INHIBITOR |
| WO2008066097A1 (en) | 2006-12-01 | 2008-06-05 | Astellas Pharma Inc. | Carboxylic acid derivative |
| WO2008066131A1 (en) | 2006-12-01 | 2008-06-05 | Banyu Pharmaceutical Co., Ltd. | Novel phenyl-isoxazol-3-ol derivative |
| WO2008066356A1 (en) | 2006-12-02 | 2008-06-05 | Seoul National University Industry Foundation | Aryl compounds as ppar ligands and their use |
| WO2008070150A1 (en) | 2006-12-05 | 2008-06-12 | Bayer Schering Pharma Aktiengesellschaft | Substituted 2,3-dihydroimidazo[1,2-c]quinazoline derivatives useful for treating hyper-proliferative disorders and diseases associated with angiogenesis |
| WO2008070507A2 (en) | 2006-12-06 | 2008-06-12 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2008069327A1 (en) | 2006-12-04 | 2008-06-12 | Mitsubishi Tanabe Pharma Corporation | CRYSTALLINE FORM OF 1- (β-D-GLUCOPYRANOSYL) -4 -METHYL- 3- [5- (4 -FLUOROPHENYL) -2-THIENYLMETHYL] BENZENE HEMIHYDRATE |
| WO2008070609A1 (en) | 2006-12-04 | 2008-06-12 | Janssen Pharmaceutica N.V. | Thienyl-containing glycopyranosyl derivatives as antidiabetics |
| WO2008070692A2 (en) | 2006-12-06 | 2008-06-12 | Smithkline Beecham Corporation | Bicyclic compounds and use as antidiabetics |
| WO2008071311A1 (en) | 2006-12-14 | 2008-06-19 | Sanofi-Aventis | Sulfonyl-phenyl-2h-[1,2,4]oxodiazol-5-one derivatives, processes for their preparation and their use as pharmaceuticals |
| WO2008071288A1 (en) | 2006-12-11 | 2008-06-19 | Merck Patent Gmbh | Indolizine derivatives and their use as antidiabetics |
| WO2008071169A2 (en) | 2006-12-11 | 2008-06-19 | Universitätsklinikum Schleswig-Holstein | Method for the production of specific inhibitors of 11-beta-hydroxysteroid dehydrogenase, in particular type 1 with basic nor-oleanan or nor-ursan frameworks |
| US20080146625A1 (en) | 2006-12-14 | 2008-06-19 | Steven Joseph Berthel | Oxime glucokinase activators |
| WO2008072726A1 (en) | 2006-12-14 | 2008-06-19 | Taisho Pharmaceutical Co., Ltd. | 1-phenyl 1-thio-d-glucitol derivative |
| WO2008075741A1 (en) | 2006-12-20 | 2008-06-26 | Keio University | Therapeutic agent and prophylactic agent for diabetes |
| WO2008075073A1 (en) | 2006-12-21 | 2008-06-26 | Astrazeneca Ab | Novel crystalline compound useful as glk activator |
| WO2008076243A2 (en) | 2006-12-14 | 2008-06-26 | Merck & Co., Inc. | Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| WO2008077138A1 (en) | 2006-12-19 | 2008-06-26 | The Board Of Trustees Of The University Of Illinois | 3-benzofuranyl-4-indolyl maleimides as potent gsk3 inhibitors for neurogenerative disorders |
| WO2008076336A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | Imidazoles as aldosterone synthase inhibitors |
| WO2008076862A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | 1-substituted imidazole derivatives and their use as aldosterone synthase inhibitors |
| WO2008074384A1 (en) | 2006-12-21 | 2008-06-26 | Merck Patent Gmbh | 2-ADAMANTYL-BUTYRAMIDE DERIVATIVES AS SELECTIVE 11βETA-HSD1 INHIBITORS |
| EP1939192A1 (en) | 2006-12-28 | 2008-07-02 | Neuropharma S.A. | Cyclopentanone derivatives, method of synthesis and uses thereof |
| EP1939191A1 (en) | 2006-12-28 | 2008-07-02 | Neuropharma S.A. | Furan derivatives, method of synthesis and uses thereof |
| WO2008078196A2 (en) | 2006-12-20 | 2008-07-03 | Sanofi-Aventis | Substituted heteroaryl pyridopyrimidone derivatives |
| WO2008078725A1 (en) | 2006-12-26 | 2008-07-03 | Daiichi Sankyo Company, Limited | Thiazepine derivative |
| WO2008079787A2 (en) | 2006-12-20 | 2008-07-03 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2008078674A1 (en) | 2006-12-25 | 2008-07-03 | Kyorin Pharmaceutical Co., Ltd. | Glucokinase-activating substance |
| WO2008077597A1 (en) | 2006-12-22 | 2008-07-03 | Novartis Ag | 1-aminomethyl- l- phenyl- cyclohexane derivatives as ddp-iv inhibitors |
| WO2008082274A1 (en) | 2007-01-05 | 2008-07-10 | Hanmi Pharmaceutical Co., Ltd. | An insulinotropic complex using an immunoglobulin fragment |
| WO2008081205A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008082017A1 (en) | 2007-01-03 | 2008-07-10 | Takano Co., Ltd. | Cyclic sulfonium salt, method for production of cyclic sulfonium salt, and glycosidase inhibitor |
| WO2008081208A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081206A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| JP2008156318A (en) | 2006-12-26 | 2008-07-10 | Dainippon Sumitomo Pharma Co Ltd | 1,3,4-oxadiazol-2-one derivative |
| WO2008081204A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081207A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081418A1 (en) | 2007-01-05 | 2008-07-10 | Covx Technologies Ireland Limited | Glucagon-like protein-1 receptor (glp-1r) agonist compounds |
| WO2008083124A1 (en) | 2006-12-28 | 2008-07-10 | Rigel Pharmaceuticals, Inc. | N-substituted-heterocycloalkyloxybenzamide compounds and methods of use |
| WO2008083200A2 (en) | 2006-12-28 | 2008-07-10 | Theracos, Inc. | Spiroheterocyclic glycosides and methods of use |
| WO2008083238A2 (en) | 2006-12-28 | 2008-07-10 | Metabolex Inc. | Heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| WO2008084044A1 (en) | 2007-01-11 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008084043A1 (en) | 2007-01-09 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008084873A1 (en) | 2007-01-10 | 2008-07-17 | Mitsubishi Tanabe Pharma Corporation | Oxime derivative |
| WO2008084303A1 (en) | 2006-12-21 | 2008-07-17 | Pfizer Products Inc. | Compounds having both angiotensin ii receptor antagonism and ppary activating activities |
| WO2008084872A1 (en) | 2007-01-10 | 2008-07-17 | Mitsubishi Tanabe Pharma Corporation | Hydrazone derivative |
| WO2008084962A1 (en) | 2007-01-08 | 2008-07-17 | Seoul National University Industry Foundation | THIAZOLE COMPOUND (AS PPARδ) LIGAND AND PHARMACEUTICAL, COSMETIC AND HEALTH FOOD COMPRISED THEREOF |
| WO2008085316A1 (en) | 2006-12-20 | 2008-07-17 | Merck & Co., Inc. | Bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| WO2008086949A1 (en) | 2007-01-16 | 2008-07-24 | Sanofi-Aventis | Use of substituted pyranone acid derivatives for the treatment of metabolic syndrome |
| WO2008088006A1 (en) | 2007-01-19 | 2008-07-24 | Shinji Yokoyama | Ap2 inhibitor |
| WO2008089461A1 (en) | 2007-01-18 | 2008-07-24 | Evolva Sa | Substituted 1,3-dioxanes useful as ppar modulators |
| WO2008087367A2 (en) | 2006-12-29 | 2008-07-24 | Genfit | Substituted (phenylthiazolyl)-phenyl-propan-1-one and (phenyloxazodyl)-phenyl-propan-1-one derivatives, preparations and uses of same |
| WO2008088540A2 (en) | 2006-12-26 | 2008-07-24 | Amgen Inc. | N-cyclohexyl benzamides and benzeneacetamides as inhibitors of 11-beta-hydroxysteroid dehydrogenases |
| US20080176912A1 (en) | 2006-11-15 | 2008-07-24 | Gee-Hong Kuo | Gpr40 agonists |
| WO2008087560A2 (en) | 2007-01-16 | 2008-07-24 | Kainos Medicine, Inc., | Thiazolidine derivatives and methods for the preparation thereof |
| WO2008087366A2 (en) | 2006-12-29 | 2008-07-24 | Genfit | Substituted 3-phenyl-1-(phenylthienyl)propan-1-one and 3-phenyl-1-(phenylfuranyl)propan-1-one derivatives, and preparation and use of same |
| WO2008089464A1 (en) | 2007-01-18 | 2008-07-24 | Evolva Sa | Prodrugs of substituted 1,3-dioxanes and their uses |
| WO2008086854A1 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | 5-([1,3,4] oxadiazol-2-yl)-1h-indazol and 5-([1,3,4] thiadiazol-2-yl)-1h-indazol derivatives as sgk inhibitors for the treatment of diabetes |
| WO2008087365A2 (en) | 2006-12-29 | 2008-07-24 | Genfit | Substituted 1, 3-diphenylpropane derivatives, preparations and uses of same |
| WO2008087654A2 (en) | 2007-01-16 | 2008-07-24 | Cadila Healthcare Limited | PIPERIDINES AS INHIBITORS OF 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 |
| US20080176861A1 (en) | 2007-01-23 | 2008-07-24 | Kalypsys, Inc. | Sulfonyl-substituted bicyclic compounds as ppar modulators for the treatment of non-alcoholic steatohepatitis |
| WO2008089636A1 (en) | 2007-01-23 | 2008-07-31 | Shanghai Hengrui Pharmaceutical Co.Ltd. | Derivatives of azabicyclo octane, the method of making them and the uses thereof as inhibitors of dipeptidyl peptidase iv |
| WO2008089581A1 (en) | 2007-01-26 | 2008-07-31 | Merck Frosst Canada Ltd. | Fused aromatic ptp-1b inhibitors |
| WO2008090210A1 (en) | 2007-01-26 | 2008-07-31 | Boehringer Ingelheim International Gmbh | Methods for preventing and treating neurodegenerative disorders |
| WO2008091338A1 (en) | 2007-01-23 | 2008-07-31 | Reddy Us Therapeutics, Inc. | Methods and compositions for the treatment of insulin resistance, diabetes, and diabetes-associated dyslipidemia |
| WO2008090209A2 (en) | 2007-01-26 | 2008-07-31 | Boehringer Ingelheim International Gmbh | Use of glucopyranosyloxy- pyrazoles for preventing and treating neurodegenerative disorders |
| WO2008089892A1 (en) | 2007-01-26 | 2008-07-31 | Sanofi-Aventis | Phenothiazin derivates, method for the production thereof and use thereof as pharmaceuticals |
| WO2008091770A1 (en) | 2007-01-24 | 2008-07-31 | Array Biopharma Inc. | 2-aminopyridine derivatives as glucokinase activators |
| WO2008093960A1 (en) | 2007-01-30 | 2008-08-07 | Lg Life Sciences, Ltd. | Novel dipeptidyl peptidase-iv inhibitors |
| WO2008093639A1 (en) | 2007-01-29 | 2008-08-07 | Takeda Pharmaceutical Company Limited | Pyrazole compound |
| WO2008094992A2 (en) | 2007-01-31 | 2008-08-07 | Vertex Pharmaceuticals Incorporated | 2-aminopyridine derivatives useful as kinase inhibitors |
| WO2008096820A1 (en) | 2007-02-07 | 2008-08-14 | Kyowa Hakko Kirin Co., Ltd. | Biphenyl derivative |
| WO2008098244A1 (en) | 2007-02-09 | 2008-08-14 | Metabasis Therapeutics, Inc. | Novel antagonists of the glucagon receptor |
| US20080194617A1 (en) | 2007-02-09 | 2008-08-14 | Taisuke Tawaraishi | Fused ring compound |
| WO2008097535A2 (en) | 2007-02-08 | 2008-08-14 | Merck & Co., Inc. | Method of treating atherosclerosis, dyslipidemias and related conditions |
| WO2008096769A1 (en) | 2007-02-08 | 2008-08-14 | Daiichi Sankyo Company, Limited | Pharmaceutical compositions containing substituted cercosporamide derivatives |
| WO2008096260A1 (en) | 2007-02-07 | 2008-08-14 | Pfizer Inc. | 3-amino-pyrrolo[3,4-c] pyrazole- 5 (1h, 4h, 6h) carbaldehyde derivatives as pkc inhibitors |
| WO2008096829A1 (en) | 2007-02-07 | 2008-08-14 | Kyowa Hakko Kirin Co., Ltd. | Tricyclic compounds |
| WO2008096841A1 (en) | 2007-02-09 | 2008-08-14 | Kyorin Pharmaceutical Co., Ltd. | Dimerized cyclo derivative |
| WO2008099145A1 (en) | 2007-02-12 | 2008-08-21 | Astrazeneca Ab | Pyrazole derivatives as 11-beta-hsd1 inhibitors |
| WO2008099944A1 (en) | 2007-02-08 | 2008-08-21 | Daiichi Sankyo Company, Limited | Crystalline forms of thiazolidinedione derivative and its manufacturing method |
| WO2008099448A1 (en) | 2007-02-09 | 2008-08-21 | Masayoshi Yamaguchi | Preventive/therapeutic agent for diabetic diseases |
| WO2008099000A2 (en) | 2007-02-16 | 2008-08-21 | Boehringer Ingelheim International Gmbh | Substituted arylsulphonylglycines, the preparation thereof and the use thereof as pharmaceutical compositions |
| WO2008101017A2 (en) | 2007-02-15 | 2008-08-21 | Indiana Unversity Research And Technology Corporation | Glucagon/glp-1 receptor co-agonists |
| JP2008189659A (en) | 2007-01-10 | 2008-08-21 | Mitsubishi Tanabe Pharma Corp | Medical composition |
| WO2008101953A1 (en) | 2007-02-22 | 2008-08-28 | Novartis Ag | Compounds of formula (i) as serine protease inhibitors |
| WO2008101939A1 (en) | 2007-02-21 | 2008-08-28 | Boehringer Ingelheim International Gmbh | Tetrasubstituted glucopyranosylated benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008101914A2 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008101586A1 (en) | 2007-02-21 | 2008-08-28 | Merck Patent Gmbh | Benzimidazole derivatives |
| WO2008101907A2 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008101886A1 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008103501A1 (en) | 2007-02-22 | 2008-08-28 | Irm Llc | Compounds and methods for modulating g protein-coupled receptors |
| WO2008101885A1 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008103500A1 (en) | 2007-02-22 | 2008-08-28 | Irm Llc | Thiazole derivatives as modulators of g protein-coupled receptors |
| WO2008104994A2 (en) | 2007-02-28 | 2008-09-04 | Advinus Therapeutics Private Limited | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2008104306A2 (en) | 2007-02-27 | 2008-09-04 | Bayer Schering Pharma Aktiengesellschaft | Substituted 4-aryl-1,4-dihydro-1,6-naphthyridinamides and use thereof |
| WO2008106128A2 (en) | 2007-02-26 | 2008-09-04 | Vitae Pharmaceuticals, Inc. | CYCLIC UREA AND CARBAMATE INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2008109700A1 (en) | 2007-03-08 | 2008-09-12 | Plexxikon, Inc. | Ppar active compounds |
| WO2008108735A1 (en) | 2007-03-08 | 2008-09-12 | Albireo Ab | 2 -substituted- 3 -phenylpropionic acid derivatives and their use in the treatment of inflammatory bowel disease |
| WO2008108602A1 (en) | 2007-03-07 | 2008-09-12 | Dong-A Pharm. Co., Ltd. | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same |
| WO2008109697A2 (en) | 2007-03-08 | 2008-09-12 | Plexxikon, Inc. | Ppar active compounds |
| WO2008109334A1 (en) | 2007-03-02 | 2008-09-12 | Merck & Co., Inc. | Novel crystalline salt form of an antidiabetic compound |
| WO2008109702A1 (en) | 2007-03-08 | 2008-09-12 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2008110062A1 (en) | 2007-03-09 | 2008-09-18 | Shanghai Allist Pharmaceuticals, Inc. | Compounds with partial agonist activity of pparϝ and application thereof |
| WO2008110196A1 (en) | 2007-03-09 | 2008-09-18 | High Point Pharmaceuticals, Llc | Indole- and benzimidazole amides as hydroxysteroid dehydrogenase inhibitors |
| WO2008112939A2 (en) | 2007-03-13 | 2008-09-18 | Board Of Regents, The University Of Texas System | Composition and method for making oligo-benzamide compounds |
| WO2008112651A2 (en) | 2007-03-09 | 2008-09-18 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as inhibitors of protein kinases |
| WO2008111473A1 (en) | 2007-03-07 | 2008-09-18 | Kyorin Pharmaceutical Co., Ltd. | Glucokinase activator |
| WO2008112642A1 (en) | 2007-03-09 | 2008-09-18 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as inhibitors of protein kinases |
| WO2008116195A2 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Compositions comprising an sglt2 ingibitor for treating obesity |
| WO2008113760A2 (en) | 2007-03-16 | 2008-09-25 | Boehringer Ingelheim International Gmbh | Arylsulphonyglycine derivatives as suppressors of the interaction of glycogen phosphorylase a with the gl subunit of glycogen-associated protein phosphatase 1 (ppl) for the treatment of metabolic disorders, particulary diabetes |
| WO2008116179A1 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Pharmaceutical formulations containing dapagliflozin propylene glycol hydrate |
| WO2008113469A2 (en) | 2007-03-16 | 2008-09-25 | Bayer Schering Pharma Aktiengesellschaft | Substituted imidazopyrimidines and triazolopyrimidines |
| US20080234277A1 (en) | 2007-03-23 | 2008-09-25 | Aurelia Conte | Novel aza-pyridopyrimidinone derivatives |
| WO2008113601A1 (en) | 2007-03-21 | 2008-09-25 | Biocompatibles Uk Ltd. | Glp-1 fusion peptides conjugated to polymer(s), their production and use |
| WO2008116107A2 (en) | 2007-03-21 | 2008-09-25 | Takeda San Diego, Inc. | Piperazine derivatives as glucokinase activators |
| WO2008119005A1 (en) | 2007-03-27 | 2008-10-02 | Trustees Of Tufts College | 3,4-dehydro-proline-containing inhibitors of dipeptidylpeptidase iv |
| WO2008118848A1 (en) | 2007-03-23 | 2008-10-02 | Trustees Of Tufts College | N-substituted peptidomimetic inhibitors of dipeptidylpeptidase iv |
| WO2008116648A2 (en) | 2007-03-27 | 2008-10-02 | Biocompatibles Uk Ltd. | Novel glp-1 fusion peptides, their production and use |
| WO2008118718A2 (en) | 2007-03-23 | 2008-10-02 | Array Biopharma Inc. | 2-aminopyridine analogs as glucokinase activators |
| US20080242596A1 (en) | 2007-04-02 | 2008-10-02 | Theracos, Inc. | Benzylic glycoside derivatives and methods of use |
| WO2008119017A1 (en) | 2007-03-28 | 2008-10-02 | High Point Pharmaceuticals, Llc | 11beta-hsd1 active compounds |
| WO2008117982A1 (en) | 2007-03-28 | 2008-10-02 | Crystal Genomics, Inc. | Heterocyclic carboxylic acid derivatives and pharmaceutical composition for inhibiting lipid accumulation containing same |
| WO2008116294A1 (en) | 2007-03-23 | 2008-10-02 | Matregen Corp. | Exendin analogs |
| WO2008120754A1 (en) | 2007-03-30 | 2008-10-09 | Taisho Pharmaceutical Co., Ltd. | Picolinamide compound |
| WO2008121064A1 (en) | 2007-03-30 | 2008-10-09 | Astrazeneca Ab | New imidazo[4,5-b]pyridine-6-halo-7-aryl/heteroaryl compounds 705 |
| WO2008121063A1 (en) | 2007-03-30 | 2008-10-09 | Astrazeneca Ab | New imidazo[ 4,5-b]pyridine-7-carboxamides 704 |
| WO2008121506A2 (en) | 2007-03-30 | 2008-10-09 | Takeda Pharmaceutical Company Limited | Renin inhibitors |
| WO2008120655A1 (en) | 2007-03-30 | 2008-10-09 | Institute Of Medicinal Molecular Design, Inc. | Oxazolidinone derivative having inhibitory activity on 11β-hydroxysteroid dehydrogenase type i |
| WO2008119918A1 (en) | 2007-03-29 | 2008-10-09 | N.V. Organon | Mineralocorticoid receptor antagonists |
| WO2008120813A1 (en) | 2007-04-03 | 2008-10-09 | Mitsubishi Tanabe Pharma Corporation | Combined use of dipeptidyl peptidase iv inhibitor compound and sweetener |
| WO2008119238A1 (en) | 2007-03-30 | 2008-10-09 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Substituted five membered heterocycle compounds, preparation method and medical use thereof |
| WO2008119208A1 (en) | 2007-04-03 | 2008-10-09 | Beijing Molecule Science And Technology Co., Ltd. | N-substituted thiomorpholine derivatives as the inhibitors of dipeptidyl peptidase iv and the pharmaceutical uses thereof |
| WO2008124745A1 (en) | 2007-04-10 | 2008-10-16 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2008122352A1 (en) | 2007-04-05 | 2008-10-16 | Sanofi-Aventis | Imidazolidine carboxamide derivatives as lipase and phospholipase inhibitors |
| WO2008124665A1 (en) | 2007-04-10 | 2008-10-16 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2008122357A1 (en) | 2007-04-05 | 2008-10-16 | Sanofi-Aventis | 5-oxo-isoxazoles as inhibitors of lipases and phospholipases |
| WO2008127924A1 (en) | 2007-04-11 | 2008-10-23 | High Point Pharmaceuticals, Llc. | Novel compounds |
| WO2008127591A2 (en) | 2007-04-13 | 2008-10-23 | Schering Corporation | Pyrimidinedione derivatives and use thereof |
| WO2008126732A1 (en) | 2007-04-05 | 2008-10-23 | Daiichi Sankyo Company, Limited | Fused bicyclic heteroaryl derivatives |
| WO2008125833A1 (en) | 2007-04-12 | 2008-10-23 | Piramed Limited | Pharmaceutical compounds |
| WO2008125945A2 (en) | 2007-04-12 | 2008-10-23 | Pfizer Inc. | 3-amido-pyrrolo [3, 4-c] pyrazole-5 (1h, 4h, 6h) carbaldehyde derivatives as inhibitors of protein kinase c |
| WO2008125835A1 (en) | 2007-04-12 | 2008-10-23 | Piramed Limited | 2-morpholin-4-yl-pyrimidines as pi3k inhibitors |
| WO2008126731A1 (en) | 2007-04-05 | 2008-10-23 | Daiichi Sankyo Company, Limited | Aryl derivatives |
| WO2008125839A2 (en) | 2007-04-12 | 2008-10-23 | Piramed Limited | Pyrimidine derivatives as inhibitors of phosphatidylinositol-3-kinase |
| WO2008130514A1 (en) | 2007-04-16 | 2008-10-30 | Amgen Inc. | Substituted biphenyl phenoxy-, thiophenyl- and aminophenylpropanoic acid gpr40 modulators |
| WO2008130151A1 (en) | 2007-04-19 | 2008-10-30 | Dong-A Pharm. Co., Ltd. | Dpp-iv inhibitor including beta-amino group, preparation method thereof and pharmaceutical composition containing the same for preventing and treating a diabetes or an obesity |
| WO2008130951A1 (en) | 2007-04-17 | 2008-10-30 | Bristol-Myers Squibb Company | Fused heterocyclic 11-beta-hydroxysteroid dehydrogenase type i inhibitors |
| WO2008130581A1 (en) | 2007-04-20 | 2008-10-30 | Schering Corporation | Pyrimidinone derivatives and methods of use thereof |
| WO2008130615A1 (en) | 2007-04-20 | 2008-10-30 | Schering Corporation | Tetrahydropyrido[4,3-d]pyrimidinone derivatives and methods of use thereof |
| WO2008130584A1 (en) | 2007-04-20 | 2008-10-30 | Schering Corporation | Pyrimidinone derivatives and methods of use thereof |
| WO2008130312A1 (en) | 2007-04-18 | 2008-10-30 | Astrazeneca Ab | A new process for the manufacturing of the compound 2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1h-indole-5-carbonitrile 701 |
| WO2008131149A2 (en) | 2007-04-20 | 2008-10-30 | Bristol-Myers Squibb Company | Crystal forms of saxagliptin and processes for preparing same |
| WO2008134221A1 (en) | 2007-04-24 | 2008-11-06 | High Point Pharmaceuticals, Llc | Pharmaceutical use of substituted amides |
| WO2008137435A1 (en) | 2007-05-04 | 2008-11-13 | Bristol-Myers Squibb Company | [6,6] and [6,7]-bicyclic gpr119 g protein-coupled receptor agonists |
| WO2008136392A1 (en) | 2007-04-27 | 2008-11-13 | Ajinomoto Co., Inc. | Preparation for oral administration |
| WO2008137436A1 (en) | 2007-05-04 | 2008-11-13 | Bristol-Myers Squibb Company | [6,5]-bicyclic gpr119 g protein-coupled receptor agonists |
| WO2008137105A1 (en) | 2007-05-07 | 2008-11-13 | Merck & Co., Inc. | Method of treatment using fused aromatic compounds having anti-diabetic activity |
| WO2008136444A1 (en) | 2007-04-26 | 2008-11-13 | Dainippon Sumitomo Pharma Co., Ltd. | Fused heterocyclic derivative |
| WO2008136642A1 (en) | 2007-05-07 | 2008-11-13 | Mazence Inc. | Naphthoquinone-based pharmaceutical composition for treatment or prevention of diseases involving obesity, diabetes, metabolic syndrome, neuro-degenerative diseases and mitochondria dysfunction diseases |
| US20080280875A1 (en) | 2006-04-20 | 2008-11-13 | Pfizer Inc. | Fused phenyl amido heterocyclic compounds |
| WO2008136428A1 (en) | 2007-04-27 | 2008-11-13 | Takeda Pharmaceutical Company Limited | Nitrogen-containing five-membered heterocyclic compound |
| EP1992624A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Heteroarylamide pyrimidone compounds |
| EP1992625A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Arylamide pyrimidone compounds |
| EP1992621A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Heteroarylamide-substituted pyrimidone derivatives for the treatment of neurodegenerative diseases |
| EP1992620A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Arylamide pyrimidone derivatives for the treatment of neurodegenerative diseases |
| WO2008139879A1 (en) | 2007-04-26 | 2008-11-20 | Pharmafrontier, Co., Ltd. | G protein-coupled receptor inhibitor and pharmaceutical product |
| WO2008138448A2 (en) | 2007-05-14 | 2008-11-20 | Merck Patent Gmbh | Heterocyclic indazole derivatives |
| US20080287529A1 (en) | 2007-05-18 | 2008-11-20 | Bristol-Myers Squibb Company | Crystal structures of sglt2 inhibitors and processes for preparing same |
| WO2008144253A1 (en) * | 2007-05-14 | 2008-11-27 | Irm Llc | Protein kinase inhibitors and methods for using thereof |
| WO2008142986A1 (en) | 2007-05-18 | 2008-11-27 | Shionogi & Co., Ltd. | NITROGEN-CONTAINING HETEROCYCLIC DERIVATIVE HAVING 11 β-HYDROXYSTEROID DEHYDROGENASE TYPE I INHIBITORY ACTIVITY |
| WO2008142859A1 (en) | 2007-05-18 | 2008-11-27 | Kowa Company, Ltd. | Novel spiro-oxindole compound and pharmaceutical containing the same |
| US20080299096A1 (en) | 2007-06-01 | 2008-12-04 | Tatake Revati J | Modified nucleotide sequence encoding glucagon-like peptide-1 (glp-1), nucleic acid construct comprising same for production of glucagon-like peptide-1 (glp-1), human cells comprising said construct and insulin-producing constructs, and methods of use thereof |
| WO2008145721A2 (en) | 2007-06-01 | 2008-12-04 | Novo Nordisk A/S | N-terminal modification of polypeptides for protection against degradation by aminopeptidases |
| WO2008146871A1 (en) | 2007-05-29 | 2008-12-04 | Santen Pharmaceutical Co., Ltd. | Novel 1,2,3,4-tetrahydroquinoxaline derivative which has, as substituent, phenyl group having sulfonic acid ester structure or sulfonic acid amide structure introduced therein and has glucocorticoid receptor-binding activity |
| WO2008149382A1 (en) | 2007-06-08 | 2008-12-11 | Advinus Therapeutics Private Limited | Pyrrole-2-carboxamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2008148744A1 (en) | 2007-06-04 | 2008-12-11 | Novartis Ag | Thiadiazole derivatives as antidiabetic agents |
| WO2008148839A2 (en) | 2007-06-08 | 2008-12-11 | Ascendis Pharma As | Long-acting polymeric prodrugs of exendin |
| WO2008154563A1 (en) | 2007-06-11 | 2008-12-18 | Bristol-Myers Squibb Company | 1, 3 - dihydroxy substituted phenylamide glucokinase activators |
| WO2008152403A1 (en) | 2007-06-15 | 2008-12-18 | Zealand Pharma A/S | Glucagon analogues |
| WO2008156174A1 (en) | 2007-06-21 | 2008-12-24 | Taisho Pharmaceutical Co., Ltd. | Pyrazinamide compound |
| WO2008157752A1 (en) | 2007-06-21 | 2008-12-24 | Incyte Corporation | Spirocycles as inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 |
| WO2008156757A1 (en) | 2007-06-19 | 2008-12-24 | Takeda Pharmaceutical Company Limited | Indazole compounds for activating glucokinase |
| WO2009000087A1 (en) | 2007-06-28 | 2008-12-31 | Merck Frosst Canada Ltd. | Substituted fused pyrimidines as antagonists of gpr105 activity |
| WO2009001817A1 (en) | 2007-06-27 | 2008-12-31 | Taisho Pharmaceutical Co., Ltd. | COMPOUND HAVING 11β-HSD1 INHIBITORY ACTIVITY |
| WO2009005672A1 (en) | 2007-06-29 | 2009-01-08 | Merck & Co., Inc. | Antidiabetic azaindoles and diazaindoles |
| WO2009003681A1 (en) | 2007-07-02 | 2009-01-08 | Santhera Pharmaceuticals (Schweiz) Ag | Dpp-iv inhibitors |
| WO2009009287A2 (en) | 2007-07-12 | 2009-01-15 | Deviris Inc. | Hormone sensitive lipase modulators and methods of use |
| WO2009007714A2 (en) | 2007-07-09 | 2009-01-15 | Imperial Innovations Limited | Human pancreatic polypeptide (hpp) analogues and their effects on feeding behaviour |
| WO2009007029A1 (en) | 2007-07-11 | 2009-01-15 | Bayer Schering Pharma Aktiengesellschaft | Imidazo-, pyrazolopyrazines and imidazotriazines and their use |
| WO2009010416A2 (en) | 2007-07-17 | 2009-01-22 | F. Hoffmann-La Roche Ag | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| WO2009010530A1 (en) | 2007-07-18 | 2009-01-22 | Novartis Ag | Bicyclic heteroaryl compounds and their use as kinase inhibitors |
| WO2009012275A1 (en) | 2007-07-17 | 2009-01-22 | Bristol-Myers Squibb Company | Pyridone gpr119 g protein-coupled receptor agonists |
| WO2009012039A2 (en) | 2007-07-13 | 2009-01-22 | Metabasis Therapeutics Inc. | Crystalline polymorphs |
| WO2009015067A2 (en) | 2007-07-25 | 2009-01-29 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2009014910A2 (en) | 2007-07-19 | 2009-01-29 | Metabolex, Inc. | N-azacyclic substituted pyrrole, pyrazole, imidazole, triazole and tetrazole derivatives as agonists of the rup3 or gpr119 receptor for the treatment of diabetes and metabolic disorders |
| US20090030046A1 (en) | 2007-07-27 | 2009-01-29 | Bristol-Myers Squibb Company | Novel glucokinase activators and methods of using same |
| WO2009014676A1 (en) | 2007-07-23 | 2009-01-29 | Merck & Co., Inc. | Novel crystalline form of a dihydrochloride salt of a dipeptidyl peptidase-iv inhibitor |
| WO2009012650A1 (en) | 2007-07-25 | 2009-01-29 | Institute Of Pharmacology And Toxicology Academy Of Military Medical Sciences P.L.A. | Aryl pyrimidine derivatives, preparation methods and pharmaceutical uses thereof |
| EP2020232A1 (en) | 2007-08-03 | 2009-02-04 | Zeltia, S.A. | N-(1-thiazolyl)-amide derivatives for the treatment of obesity, diabetes and cardiovascular diseases |
| WO2009017452A1 (en) | 2007-07-30 | 2009-02-05 | Astrazeneca Ab | New crystalline forms of 2 -hydroxy- 3- [5- (morpholin- 4- ylmethyl) pyridin-2-yl] ih- indole- 5 -carbonitrile citrate |
| WO2009016118A1 (en) | 2007-07-27 | 2009-02-05 | Boehringer Ingelheim International Gmbh | Substituted arylsulfonylaminomethylphosphonic acid derivatives, their preparation and their use in the treatment of type i and ii diabetes mellitus |
| WO2009017664A1 (en) | 2007-07-26 | 2009-02-05 | Vitae Pharmaceuticals, Inc. | CYCLIC INHIBITORS OF 11β-HYDROXYSTERIOD DEHYDROGENASE 1 |
| JP2009023975A (en) | 2007-07-23 | 2009-02-05 | Nagoya Industrial Science Research Inst | PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR(PPAR)-alpha LIGAND AGENT |
| WO2009016119A1 (en) | 2007-07-27 | 2009-02-05 | Boehringer Ingelheim International Gmbh | Substituted arylsulfonylaminomethylphosphonic acid derivatives, their preparation and their use in the treatment of type i and ii diabetes mellitus |
| WO2009020140A1 (en) | 2007-08-06 | 2009-02-12 | Dainippon Sumitomo Pharma Co., Ltd. | Adamantylurea derivative |
| WO2009019446A1 (en) | 2007-08-03 | 2009-02-12 | Betagenon Ab | Compounds useful as medicaments |
| WO2009019600A2 (en) | 2007-08-03 | 2009-02-12 | Dr. Reddy's Laboratories Ltd. | Modulation of endogenous ampk levels for the treatment of obesity |
| WO2009023718A2 (en) | 2007-08-13 | 2009-02-19 | Metabasis Therapeutics, Inc. | Novel activators of glucokinase |
| WO2009023181A1 (en) | 2007-08-15 | 2009-02-19 | Schering Corporation | 6-SUBSTITUTED SULFONYL AZABICYCLO[3.2.1]OCTANES USEFUL TO INHIBIT 11β-HYDROXYSTEROID DEHYDROGENASE TYPE-1 |
| WO2009023664A2 (en) | 2007-08-15 | 2009-02-19 | Schering Corporation | Substituted azepine- and diazepine-sulfonamides useful to inhibit 11beta-hydroxysteroid dehydrogenase type-1 |
| WO2009023180A1 (en) | 2007-08-15 | 2009-02-19 | Schering Corporation | SUBSTITUTED BICYCLIC PIPERIDINYL-AND PIPERAZINYL- SULFONAMIDES USEFUL TO INHIBIT 11β-HYDROXYSTEROID DEHYDROGENASE TYPE-1 |
| WO2009025784A1 (en) | 2007-08-21 | 2009-02-26 | Merck & Co., Inc. | Heterocyclic compounds as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2009026345A1 (en) | 2007-08-20 | 2009-02-26 | Targegen Inc. | Thiazolidinone compounds, and methods of making and using same |
| WO2009026422A2 (en) | 2007-08-21 | 2009-02-26 | Abbott Laboratories | Treatment of central nervous system disorders |
| US20090062369A1 (en) | 2007-08-31 | 2009-03-05 | Joaquim Trias | Use of secretory phospholipase a2 (spla2) inhibitors to decrease spla2 levels |
| WO2009026657A1 (en) | 2007-08-29 | 2009-03-05 | The University Of Sydney | Flavonoid ppar agonists |
| WO2009026658A1 (en) | 2007-08-29 | 2009-03-05 | The University Of Sydney | Ppar agonists |
| WO2009027276A1 (en) | 2007-08-30 | 2009-03-05 | F. Hoffmann-La Roche Ag | Process for the preparation of pyrido [2, 1-a] isoquinoline derivatives and polymorphic froms thereof |
| WO2009028891A2 (en) | 2007-08-31 | 2009-03-05 | Hanall Pharmaceutical Company. Ltd | 1,3,5-triazine-2,4,6-triamine compound or pharmaceutical acceptable salt thereof, and pharmaceutical composition comprising the same |
| WO2009030771A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Peptides derivatized with a-b-c-d- and their therapeutical use |
| WO2009032321A2 (en) | 2007-09-06 | 2009-03-12 | Genaera Corporation | A method for treating diabetes |
| WO2009030715A1 (en) | 2007-09-05 | 2009-03-12 | Boehringer Ingelheim International Gmbh | Arylsulfonylaminomethylphosphonic acid derivatives, the preparation thereof and the use thereof as pharmaceutical compositions |
| WO2009030738A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Glucagon-like peptide-1 derivatives and their pharmaceutical use |
| WO2009030774A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Truncated glp-1 derivatives and their therapeutical use |
| WO2009034117A1 (en) | 2007-09-11 | 2009-03-19 | Novo Nordisk A/S | Mixture comprising an amylin peptide and a protracted insulin |
| WO2009034388A1 (en) | 2007-09-10 | 2009-03-19 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2009034119A1 (en) | 2007-09-11 | 2009-03-19 | Novo Nordisk A/S | Improved derivatives of amylin |
| WO2009033561A1 (en) | 2007-09-07 | 2009-03-19 | Bayer Schering Pharma Aktiengesellschaft | Substituted 6-pheylnicotinic acids and the use thereof |
| WO2009035684A1 (en) | 2007-09-12 | 2009-03-19 | Activx Biosciences, Inc. | Spirocyclic aminoquinolones as gsk-3 inhibitors |
| WO2009035634A2 (en) | 2007-09-11 | 2009-03-19 | Activx Biosciences, Inc. | Cyanoaminoquinolones and tetrazoloaminoquinolones as gsk-3 inhibitors |
| WO2009035540A2 (en) | 2007-09-07 | 2009-03-19 | Ipsen Pharma S.A.S. | Analogues of exendin-4 and exendin-3 |
| US20090076129A1 (en) | 2007-09-13 | 2009-03-19 | Gateway Health Alliances, Inc. | Methods and related compositions using specific flavonoids and indanes to reduce weight and inhibit lipase, alpha-amylase and alpha-glucosidase activity in mammals |
| WO2009038681A1 (en) | 2007-09-14 | 2009-03-26 | Metabolic Solutions Development Company | Thiazolidinedione analogues for the treatment of hypertension |
| WO2009038064A1 (en) | 2007-09-19 | 2009-03-26 | Institute Of Medicinal Molecular Design, Inc. | Heterocyclic derivative having inhibitory activity on type-i 11β-hydroxysteroid dehydrogenase |
| WO2009038204A1 (en) | 2007-09-17 | 2009-03-26 | Pharma Frontier Co., Ltd. | Novel long-chain fatty acid derivative compound and g-protein-coupled receptor agonist containing the compound as active ingredient |
| WO2009038974A1 (en) | 2007-09-20 | 2009-03-26 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009038385A2 (en) | 2007-09-21 | 2009-03-26 | Choongwae Pharma Corporation | Novel compounds having indazole frameworks, methods for preparing the same and pharmaceutical composition comprising the same |
| WO2009037719A1 (en) | 2007-09-21 | 2009-03-26 | Lupin Limited | Novel compounds as dipeptidyl peptidase iv (dpp iv) inhibitors |
| WO2009039944A1 (en) | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | Phenothiazine derivative having a double bond, method for the production thereof, and use thereof as a pharmaceutical |
| WO2009042435A1 (en) | 2007-09-21 | 2009-04-02 | Array Biopharma Inc. | Pyridin-2 -yl-amino-i, 2, 4 -thiadiazole derivatives as glucokinase activators for the treatment of diabetes mellitus |
| WO2009039943A1 (en) | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | (carboxylalkylene-phenyl)-phenyl-oxalamides, method for the production thereof, and use of same as a medicament |
| WO2009039942A1 (en) | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | (cyclopropyl-phenyl)-phenyl-oxalamides, method for the production thereof, and use of same as a medicament |
| WO2009040288A1 (en) | 2007-09-27 | 2009-04-02 | F. Hoffmann-La Roche Ag | 1,1,1-trifluoro-2-hydroxy-3-phenylpropane derivatives |
| WO2009045830A1 (en) | 2007-09-28 | 2009-04-09 | Smithkline Beecham Corporation | Glycogen phosphorylase inhibitor compound and pharmaceutical composition thereof |
| WO2009045831A1 (en) | 2007-09-28 | 2009-04-09 | Smithkline Beecham Corporation | Glycogen phosphorylase inhibitor compound and pharmaceutical composition thereof |
| WO2009045753A1 (en) | 2007-10-01 | 2009-04-09 | Bristol-Myers Squibb Company | Triazolopyridine 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2009047798A2 (en) | 2007-10-08 | 2009-04-16 | Advinus Therapeutics Private Limited | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| WO2009046784A1 (en) | 2007-10-09 | 2009-04-16 | Merck Patent Gmbh | Pyridine derivatives useful as glucokinase activators |
| WO2009047240A1 (en) | 2007-10-09 | 2009-04-16 | Smithkline Beecham Corporation | Indole derivatives useful as ppar activators |
| WO2009046802A1 (en) | 2007-10-09 | 2009-04-16 | Merck Patent Gmbh | N- ( pyrazole- 3 -yl) -benzamide derivatives as glucokinase activators |
| WO2009046606A1 (en) | 2007-10-11 | 2009-04-16 | Shanghai Institute Of Materia Medica, Cas | Pyrimidinyl-propionic acid derivatives and their use as ppar agonists |
| WO2009048527A1 (en) | 2007-10-10 | 2009-04-16 | Amgen Inc. | Substituted biphenyl gpr40 modulators |
| WO2009049222A1 (en) | 2007-10-12 | 2009-04-16 | Curedm, Inc. | Compositions and methods of using the human proislet peptide receptor |
| WO2009049731A1 (en) | 2007-10-11 | 2009-04-23 | Merck Patent Gmbh | Imidazo[1,2-a]pyrimidine derivatives for treating diseases such as diabetes |
| WO2009050523A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| WO2009050522A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| WO2009055331A2 (en) | 2007-10-22 | 2009-04-30 | Schering Corporation | Bicyclic heterocycle derivatives and their use as modulators of the activity of gpr119 |
| WO2009054479A1 (en) | 2007-10-26 | 2009-04-30 | Japan Tobacco Inc. | Spiro-ring compound and use thereof for medical purposes |
| WO2009058237A1 (en) | 2007-10-29 | 2009-05-07 | Merck & Co., Inc. | Antidiabetic tricyclic compounds |
| WO2009056881A1 (en) | 2007-10-29 | 2009-05-07 | Astrazeneca Ab | Chemical compounds 313 |
| WO2009058662A2 (en) | 2007-10-30 | 2009-05-07 | Indiana University Research And Technology Corporation | Glucagon antagonists |
| WO2009057784A1 (en) | 2007-11-01 | 2009-05-07 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| WO2009058734A1 (en) | 2007-10-30 | 2009-05-07 | Indiana University Research And Technology Corporation | Compounds exhibiting glucagon antagonist and glp-1 agonist activity |
| EP2058308A1 (en) | 2007-11-12 | 2009-05-13 | Merck Sante | Benzimidazoledihydrothiadiazinone derivatives used as fructose-1,6-biphosphatase inhibitors and pharmaceutical compositions containing same. |
| WO2009059666A1 (en) | 2007-11-05 | 2009-05-14 | Merck Patent Gmbh | 7-azaindole derivatives as selective 11-beta-hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2009060071A1 (en) | 2007-11-08 | 2009-05-14 | Novo Nordisk A/S | Insulin derivative |
| WO2009061498A1 (en) | 2007-11-07 | 2009-05-14 | Vitae Pharmaceuticals, Inc. | CYCLIC UREA INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2009063821A1 (en) | 2007-11-12 | 2009-05-22 | Banyu Pharmaceutical Co., Ltd. | Heteroaryloxy quinazoline derivative |
| WO2009063061A2 (en) | 2007-11-16 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of benzomorphanes and related scaffolds, medicaments containing such compounds and their use |
| WO2009065131A1 (en) | 2007-11-16 | 2009-05-22 | Rigel Pharmaceuticals, Inc. | Carboxamide, sulfonamide and amine compounds for metabolic disorders |
| WO2009065298A1 (en) | 2007-10-25 | 2009-05-28 | Shanghai Hengrui Pharmaceutical Co., Ltd. | Piperazine derivatives, preparation process and pharmaceutical use thereof |
| US20090137671A1 (en) | 2007-05-17 | 2009-05-28 | Noy Noa | Methods of treating metabolic disorders |
| WO2009068468A2 (en) | 2007-11-30 | 2009-06-04 | F. Hoffmann-La Roche Ag | Pyridine compounds |
| WO2009069736A1 (en) | 2007-11-28 | 2009-06-04 | Kyowa Hakko Kirin Co., Ltd. | Nitrogenated compound |
| WO2009070497A1 (en) | 2007-11-28 | 2009-06-04 | Smithkline Beecham Corporation | SEH AND 11 β-HSD1 INHIBITORS AND THEIR USE |
| WO2009068467A1 (en) | 2007-11-30 | 2009-06-04 | F. Hoffmann-La Roche Ag | Aminothiazole derivatives |
| WO2009070314A2 (en) | 2007-11-26 | 2009-06-04 | Teva Pharmaceutical Industries Ltd. | Crystalline form of sitagliptin |
| WO2009068531A2 (en) | 2007-11-30 | 2009-06-04 | Novartis Ag | Organic compounds |
| WO2009072581A1 (en) | 2007-12-05 | 2009-06-11 | Aska Pharmaceutical Co., Ltd. | Lactam compound or salt thereof, and ppar activator |
| WO2009071888A1 (en) | 2007-12-04 | 2009-06-11 | Ucb Pharma S.A. | Pyrrolothiazoles as pi3-kinase inhibitors |
| WO2009071895A1 (en) | 2007-12-04 | 2009-06-11 | Ucb Pharma S.A. | Fused thiazole and thiophene derivatives as kinase inhibitors |
| WO2009071890A1 (en) | 2007-12-04 | 2009-06-11 | Ucb Pharma S.A. | Tricyclic kinase inhibitors |
| WO2009070873A1 (en) | 2007-12-04 | 2009-06-11 | Merck Frosst Canada Ltd. | Substituted 2-naphthoic acids as antagonists of gpr105 activity |
| WO2009074789A1 (en) | 2007-12-12 | 2009-06-18 | The University Of Edinburgh | Amido-thiophene compounds and their use as 11-beta-hsd1 inhibitors |
| WO2009075835A1 (en) | 2007-12-11 | 2009-06-18 | Vitae Pharmaceutical, Inc | CYCLIC UREA INHIBITORS OF 11β-HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2009076550A1 (en) | 2007-12-13 | 2009-06-18 | Theracos, Inc. | Benzylphenyl cyclohexane derivatives and methods of use |
| WO2009076631A1 (en) | 2007-12-12 | 2009-06-18 | Rigel Pharmaceuticals, Inc. | Carboxamide, sulfonamide and amine compounds for metabolic disorders |
| WO2009080242A1 (en) | 2007-12-20 | 2009-07-02 | Bayer Schering Pharma Aktiengesellschaft | Substituted 4-aminopyrimidine-5-carboxylic acid and use thereof |
| WO2009080032A1 (en) | 2007-12-20 | 2009-07-02 | Fertin Pharma A/S | Compressed chewing gum comprising a systemically active small peptide |
| WO2009079921A1 (en) | 2007-11-30 | 2009-07-02 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Cucurbitane triterpenoid saponins, pharmaceutical composition thereof, preparation and use thereof |
| WO2009080024A1 (en) | 2007-12-20 | 2009-07-02 | Fertin Pharma A/S | Compressed chewing gum comprising an incretin mimetic |
| WO2009080223A1 (en) | 2007-12-26 | 2009-07-02 | Sanofi-Aventis | Cyclic pyridyl-n-(1,3,4)-thiadiazol-2-yl-benzene sulfonamides, processes for their preparation and their use as pharmaceuticals |
| WO2009080821A2 (en) | 2007-12-21 | 2009-07-02 | Giuliani International Limited | Receptor targeting ligands |
| WO2009081782A1 (en) | 2007-12-25 | 2009-07-02 | Banyu Pharmaceutical Co., Ltd. | N-pyrazole-2-pyridinecarboxamide derivative |
| WO2009080248A1 (en) | 2007-12-20 | 2009-07-02 | Bayer Schering Pharma Aktiengesellschaft | Substituted 2-phenylpyrimidine-5-carboxylic acids and the use thereof |
| WO2009082152A2 (en) | 2007-12-20 | 2009-07-02 | Lg Life Sciences Ltd. | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| WO2009082134A2 (en) | 2007-12-21 | 2009-07-02 | Lg Life Sciences, Ltd. | Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as active agent |
| WO2009084497A1 (en) | 2007-12-28 | 2009-07-09 | Dainippon Sumitomo Pharma Co., Ltd. | Methyl-substituted piperidine derivative |
| WO2009083526A1 (en) | 2008-01-02 | 2009-07-09 | Smithkline Beecham Corporation | Novel compounds |
| WO2009082881A1 (en) | 2007-12-26 | 2009-07-09 | Shanghai Hengrui Pharmaceutical Co., Ltd. | Tetrahydro-imidazo[1,5-a]pyrazine derivatives, preparation methods and medical uses thereof |
| WO2009083553A1 (en) | 2007-12-31 | 2009-07-09 | Rheoscience A/S | Azine compounds as glucokinase activators |
| WO2009084531A1 (en) | 2007-12-27 | 2009-07-09 | Kissei Pharmaceutical Co., Ltd. | Monosebacate of pyrazole derivative |
| US20090181981A1 (en) | 2008-01-15 | 2009-07-16 | Jeanette Tower Dunlap | Crystalline (r)-2-(4-cyclopropanesulphonyl-phenyl)-n-pyrazin-2-yl-3-(tetrahydropyran-4-yl)-propionamide |
| WO2009088997A1 (en) | 2008-01-07 | 2009-07-16 | Vitae Pharmaceuticals, Inc. | Lactam inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2009087395A1 (en) | 2008-01-10 | 2009-07-16 | University Of Strathclyde | Weight reducing compounds |
| WO2009091014A1 (en) | 2008-01-18 | 2009-07-23 | Astellas Pharma Inc. | Phenyl acetamide derivative |
| WO2009090239A1 (en) | 2008-01-17 | 2009-07-23 | Biovitrum Ab (Publ) | Isoxazole derivatives as modulators of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2009091082A1 (en) | 2008-01-17 | 2009-07-23 | Mitsubishi Tanabe Pharma Corporation | Combination therapy comprising sglt inhibitors and dpp4 inhibitors |
| WO2009093269A1 (en) | 2008-01-24 | 2009-07-30 | Panacea Biotec Limited | Novel heterocyclic compounds |
| WO2009092432A1 (en) | 2008-01-24 | 2009-07-30 | Merck Patent Gmbh | Beta-amino acid derivatives for treatment of diabetes |
| WO2009094169A1 (en) | 2008-01-24 | 2009-07-30 | Vitae Pharmaceuticals, Inc. | Cyclic carbazate and semicarbazide inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009095789A1 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted arylamide diazepinopyrimidone derivatives for the treatment of neurodegenerative diseases caused by abnormal activity of gsk3-beta |
| WO2009096503A1 (en) | 2008-01-31 | 2009-08-06 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
| WO2009095787A1 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted heteroarylamide oxazepinopyrimidone derivatives |
| WO2009095788A1 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted arylamide oxazepinopyrimidone derivatives |
| WO2009095792A2 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted heteroarylamide diazepinopyrimidone derivatives |
| WO2009099171A1 (en) | 2008-02-07 | 2009-08-13 | Takeda Pharmaceutical Company Limited | Pharmaceutical product |
| WO2009100130A1 (en) | 2008-02-04 | 2009-08-13 | Mercury Therapeutics, Inc. | Ampk modulators |
| WO2009099172A1 (en) | 2008-02-07 | 2009-08-13 | Takeda Pharmaceutical Company Limited | Pharmaceutical product |
| WO2009099080A1 (en) | 2008-02-06 | 2009-08-13 | Daiichi Sankyo Company, Limited | Novel phenylpyrrole derivative |
| WO2009098501A1 (en) | 2008-02-04 | 2009-08-13 | Astrazeneca Ab | Novel crystalline forms of 4- [4- (2-adamantylcarbam0yl) -5-tert-butyl-pyrazol-1-yl] benzoic acid |
| WO2009100872A1 (en) | 2008-02-12 | 2009-08-20 | Boehringer Ingelheim International Gmbh | Urea derivatives of benzomorphanes and related scaffolds, medicaments containing such compounds and their use |
| WO2009100936A2 (en) | 2008-02-13 | 2009-08-20 | Sanofi-Aventis | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds, and the use thereof |
| WO2009102761A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | 1,2,3-triazoles as 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2009102460A2 (en) | 2008-02-15 | 2009-08-20 | Vitae Pharmaceuticals, Inc. | Inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009102428A2 (en) | 2008-02-11 | 2009-08-20 | Vitae Pharmaceuticals, Inc. | 1,3-OXAZEPAN-2-ONE AND 1,3-DIAZEPAN-2-ONE INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2009102226A1 (en) | 2008-02-12 | 2009-08-20 | Adamed Sp. Z O.O. | Malonic acid salt of 5-[[4-[2-(methyl-2-pyridinylamino)ethoxy]phenyl]methyl]- 2,4-thiazolidinedione |
| WO2009105717A1 (en) | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009105722A1 (en) | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009105715A1 (en) | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009106565A1 (en) | 2008-02-27 | 2009-09-03 | Biovitrum Ab (Publ) | Agonists of gpr119 |
| WO2009106209A1 (en) | 2008-02-27 | 2009-09-03 | Merck Patent Gmbh | Carboxamide-heteroaryl derivatives for the treatment of diabetes |
| WO2009108332A1 (en) | 2008-02-27 | 2009-09-03 | Vitae Pharmaceuticals, Inc. | INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE TYPE 1 |
| WO2009106561A1 (en) | 2008-02-27 | 2009-09-03 | Biovitrum Ab (Publ) | Pyrazine compounds for treating gpr119 related disorders |
| WO2009106817A2 (en) | 2008-02-26 | 2009-09-03 | Sterix Limited | Compound |
| WO2009107900A1 (en) | 2008-02-25 | 2009-09-03 | Sungkyunkwan University Foundation For Corporate Collaboration | Exendin derivative linked biotin, method for the preparation thereof and pharmaceutical composition comprising the same |
| WO2009106203A1 (en) | 2008-02-25 | 2009-09-03 | Merck Patent Gmbh | Glucokinase activators |
| WO2009109259A1 (en) | 2008-03-05 | 2009-09-11 | Merck Patent Gmbh | Pyrazinone derivatives as insulin secretion stimulators, methods for obtaining them and use thereof for the treatment of diabetes |
| WO2009111239A2 (en) | 2008-03-05 | 2009-09-11 | National Health Research Institutes | Pyrrolidine derivatives |
| WO2009109341A1 (en) | 2008-03-05 | 2009-09-11 | Merck Patent Gmbh | Pyridopyrazinones derivatives insulin secretion stimulators, methods for obtaining them and use thereof for the treatment of diabetes |
| WO2009110520A1 (en) | 2008-03-05 | 2009-09-11 | 武田薬品工業株式会社 | Heterocyclic compound |
| WO2009109998A1 (en) | 2008-03-03 | 2009-09-11 | Lupin Limited | Novel protein tyrosine phosphatase - ib inhibitors |
| WO2009109270A1 (en) | 2008-03-01 | 2009-09-11 | Merck Patent Gmbh | 5-oxo-2, 3,4,5-tetrahydro-benzo[b]oxepine-4-carboxylic acid amides and 2,3-dihydro-benzo[b]oxepine-4-carboxylic acid amides for treatment and prevention of diabetes typ 1 and 2 |
| WO2009111056A1 (en) | 2008-03-06 | 2009-09-11 | Amgen Inc. | Conformationally constrained carboxylic acid derivatives useful for treating metabolic disorders |
| WO2009109999A1 (en) | 2008-03-03 | 2009-09-11 | Lupin Limited | Novel protein tyrosine phosphatase - ib inhibitors |
| WO2009111700A2 (en) | 2008-03-07 | 2009-09-11 | Transtech Pharma, Inc. | Oxadiazoanthracene compounds for the treatment of diabetes |
| WO2009112691A2 (en) | 2008-01-28 | 2009-09-17 | Sanofi-Aventis | Derivatives of tetrahydroquinoxaline urea, preparation thereof and therapeutic application thereof |
| WO2009114173A1 (en) | 2008-03-14 | 2009-09-17 | Exelixis, Inc. | Azabicyclo [3. 2. i] octyl derivatives as 11 beta-hsdl modulators |
| WO2009112845A1 (en) | 2008-03-13 | 2009-09-17 | The University Of Edinburgh | Amido-thiophene compounds and their use |
| WO2009113423A1 (en) | 2008-03-10 | 2009-09-17 | 大日本住友製薬株式会社 | Bicyclic pyrrole compound |
| EP2103302A1 (en) | 2008-03-21 | 2009-09-23 | Les Laboratoires Servier | Divisible dosage form allowing modified release of the active principle |
| WO2009116067A2 (en) | 2008-01-10 | 2009-09-24 | Sun Pharma Advanced Research Company Limited | Novel derivatives of acyl cyanopyrrolidines |
| WO2009117421A2 (en) | 2008-03-17 | 2009-09-24 | Kalypsys, Inc. | Heterocyclic modulators of gpr119 for treatment of disease |
| WO2009117109A1 (en) | 2008-03-18 | 2009-09-24 | Vitae Pharmaceuticals, Inc. | Inhibitors of 11beta-hydroxysteroid dehydrogenase type 1 |
| US20090238879A1 (en) | 2008-01-24 | 2009-09-24 | Northwestern University | Delivery scaffolds and related methods of use |
| US20090247532A1 (en) | 2008-03-28 | 2009-10-01 | Mae De Ltd. | Crystalline polymorph of sitagliptin phosphate and its preparation |
| WO2009118473A2 (en) | 2008-02-29 | 2009-10-01 | Sanofi-Aventis | Azetidine-derived compounds, preparation method therefor and therapeutic use of same |
| WO2009120530A1 (en) | 2008-03-27 | 2009-10-01 | Eli Lilly And Company | Glucagon receptor antagonists |
| WO2009121939A2 (en) | 2008-04-02 | 2009-10-08 | Tfchem | C-aryl glycoside compounds for the treatment of diabetes and obesity |
| WO2009121945A2 (en) | 2008-04-03 | 2009-10-08 | Boehringer Ingelheim International Gmbh | New formulations, tablets comprising such formulations, their use and process for their preparation |
| WO2009123992A1 (en) | 2008-03-31 | 2009-10-08 | Metabolex, Inc. | Oxymethylene aryl compounds and uses thereof |
| WO2009121884A1 (en) | 2008-04-01 | 2009-10-08 | Novo Nordisk A/S | Insulin albumin conjugates |
| WO2009124636A1 (en) | 2008-04-11 | 2009-10-15 | Merck Patent Gmbh | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| WO2009124638A1 (en) | 2008-04-07 | 2009-10-15 | Merck Patent Gmbh | Glucopyranoside derivatives |
| WO2009125424A2 (en) | 2007-12-11 | 2009-10-15 | Cadila Healthcare Limited | Peptidomimetics with glucagon antagonistic and glp-1 agonistic activities |
| WO2009126535A1 (en) | 2008-04-07 | 2009-10-15 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009125873A1 (en) | 2008-04-10 | 2009-10-15 | Takeda Pharmaceutical Company Limited | Fused ring compounds and use thereof |
| WO2009125434A2 (en) | 2008-04-07 | 2009-10-15 | Cadila Healthcare Limited | Oxime derivatives |
| WO2009127546A1 (en) | 2008-04-16 | 2009-10-22 | F. Hoffmann-La Roche Ag | Pyrrolidinone glucokinase activators |
| WO2009129036A1 (en) | 2008-04-14 | 2009-10-22 | Merck & Co., Inc. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| WO2009128421A1 (en) | 2008-04-16 | 2009-10-22 | キッセイ薬品工業株式会社 | Hemifumarate of a pyrazole derivative |
| WO2009128558A1 (en) | 2008-04-15 | 2009-10-22 | 日本ケミファ株式会社 | Activator for peroxisome proliferator-activated receptor |
| WO2009127723A1 (en) | 2008-04-19 | 2009-10-22 | Boehringer Ingelheim International Gmbh | New arylsulphonylglycine derivatives, the preparation thereof and their use as medicaments |
| US20090264401A1 (en) | 2008-04-22 | 2009-10-22 | Astrazeneca Ab | Substituted pyrimidin-5-carboxamides 281 |
| WO2009127544A1 (en) | 2008-04-16 | 2009-10-22 | F. Hoffmann-La Roche Ag | Pyridazinone glucokinase activators |
| WO2009128481A1 (en) | 2008-04-16 | 2009-10-22 | 武田薬品工業株式会社 | Nitrogenated 5-membered heterocyclic compound |
| WO2009129696A1 (en) | 2008-04-25 | 2009-10-29 | 国家新药筛选中心 | A kind of receptor signaling transoluction positive modulators, preparation methods and uses thereof |
| WO2009131669A2 (en) | 2008-04-22 | 2009-10-29 | Vitae Pharmaceuticals, Inc. | Carbamate and urea inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009133099A2 (en) | 2008-04-28 | 2009-11-05 | Novo Nordisk A/S | Insulin precursors for diabetes treatment |
| WO2009134400A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009134387A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009133687A1 (en) | 2008-04-28 | 2009-11-05 | 杏林製薬株式会社 | Cyclopentylacrylic acid amide derivative |
| WO2009134384A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009132986A1 (en) | 2008-04-30 | 2009-11-05 | F. Hoffmann-La Roche Ag | Imidazolidinone derivatives as 11b-hsd1 inhibitors |
| WO2009134392A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009135581A2 (en) | 2008-05-05 | 2009-11-12 | Merck Patent Gmbh | Novel nip thiazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2009135580A1 (en) | 2008-05-05 | 2009-11-12 | Merck Patent Gmbh | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| WO2009135673A2 (en) | 2008-05-09 | 2009-11-12 | Barbara La Ferla | Compounds with glycidic structure active in the therapy of systemic |
| WO2009139340A1 (en) | 2008-05-12 | 2009-11-19 | 武田薬品工業株式会社 | Pyrazole compound |
| US20090286812A1 (en) | 2008-05-19 | 2009-11-19 | Shawn David Erickson | GPR119 Receptor Agonists |
| WO2009139362A1 (en) | 2008-05-14 | 2009-11-19 | 株式会社 三和化学研究所 | Pharmaceutical preparation comprising dpp-iv inhibitor and other diabetes therapeutic agent in concomitant or combined form |
| WO2009138386A2 (en) | 2008-05-13 | 2009-11-19 | Boehringer Ingelheim International Gmbh | Alicyclic carboxylic acid derivatives of benzomorphans and related scaffolds, medicaments containing such compounds and their use |
| WO2009140624A2 (en) | 2008-05-16 | 2009-11-19 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2009140342A1 (en) | 2008-05-16 | 2009-11-19 | Schering Corporation | Glucagon receptor antagonists, compositions, and methods for their use |
| US20090291982A1 (en) | 2008-05-22 | 2009-11-26 | Astrazeneca Ab | New Substituted Oxindole Derivative 352 |
| WO2009143049A1 (en) | 2008-05-19 | 2009-11-26 | Schering Corporation | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| WO2009145814A2 (en) | 2008-03-10 | 2009-12-03 | Vertex Pharmaceuticals Incorporated | Pyrimidines and pyridines useful as inhibitors of protein kinases |
| WO2009147990A1 (en) | 2008-06-02 | 2009-12-10 | 萬有製薬株式会社 | Novel isoxazole derivative |
| WO2009149171A2 (en) | 2008-06-04 | 2009-12-10 | Amgen Inc. | Fgf21 mutants and uses thereof |
| WO2009149056A2 (en) | 2008-06-02 | 2009-12-10 | Dr. Reddy's Laboratories Ltd. | Combinations of niacin and an oxicam |
| WO2009149139A1 (en) | 2008-06-06 | 2009-12-10 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2009147121A1 (en) | 2008-06-02 | 2009-12-10 | Smithkline Beecham Corporation | Carboxyl substituted indoles for use as ppar alpha modulators |
| WO2009149148A2 (en) | 2008-06-03 | 2009-12-10 | Trustees Of Tufts College | Long-acting glp-1 derivatives, and methods of treating cardiac dysfunction |
| WO2009150144A1 (en) | 2008-06-10 | 2009-12-17 | Inovacia Ab | New gpr119modulators |
| WO2009149819A1 (en) | 2008-06-09 | 2009-12-17 | Sanofi-Aventis | Annelated pyrrolidin sulfonamides with oxadiazolone headgroup, processes for their preparation and their use as pharmaceuticals |
| WO2009149820A1 (en) | 2008-06-09 | 2009-12-17 | Sanofi-Aventis | Annelated n-heterocyclic sulfonamides with oxadiazolone headgroup, processes for their preparation and their use as pharmaceuticals |
| WO2009152128A1 (en) | 2008-06-13 | 2009-12-17 | Eli Lilly And Company | Pegylated insulin lispro compounds |
| WO2009152909A1 (en) | 2008-06-16 | 2009-12-23 | Merck Patent Gmbh | Quinoxalinedione derivatives |
| WO2009155257A1 (en) | 2008-06-17 | 2009-12-23 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting enhanced solubility and stability physiological ph buffers |
| WO2009153496A2 (en) | 2008-05-26 | 2009-12-23 | Genfit | Ppar agonist compounds, preparation and uses |
| WO2009153960A1 (en) | 2008-06-17 | 2009-12-23 | 大塚化学株式会社 | Glycosylated glp-1 peptide |
| WO2009154697A2 (en) | 2008-05-28 | 2009-12-23 | Massachusetts Institute Of Technology | Disc-1 pathway activators in the control of neurogenesis |
| WO2009155258A2 (en) | 2008-06-17 | 2009-12-23 | Indiana University Research And Technology Corporation | Glucagon/glp-1 receptor co-agonists |
| WO2009156857A1 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted n-oxide pyrazine derivatives |
| WO2009156863A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted tricyclic derivatives |
| WO2009156865A1 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted pyrimidin-4-one derivatives |
| WO2009156864A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted alkyl pyrimidin-4-one derivatives |
| WO2009156860A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted triazinone derivatives |
| WO2009156859A1 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted pyrimido [2, 1-a] isoquinolin-4-one derivatives |
| WO2009156861A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted pyrimidone derivatives |
| WO2010000353A1 (en) | 2008-06-09 | 2010-01-07 | Sanofi-Aventis | Sulfonamides with heterocycle and oxadiazolone headgroup, processes for their preparation and their use as pharmaceuticals |
| WO2010000469A2 (en) | 2008-07-03 | 2010-01-07 | Ratiopharm Gmbh | Crystalline salts of sitagliptin |
| WO2010001166A1 (en) | 2008-07-01 | 2010-01-07 | Prosidion Limited | Thiazole derivatives as gpr 119 modulators |
| WO2010004343A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidinyl gpcr agonists |
| WO2010004348A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2010006191A1 (en) | 2008-07-11 | 2010-01-14 | Irm Llc | 4-phenoxymethylpiperidines as modulators of gpr119 activity |
| WO2010004347A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2010004345A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidinyl gpcr agonists |
| WO2010004346A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidinyl gpcr agonists |
| WO2010004344A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidine gpcr agonists |
| WO2010006214A1 (en) | 2008-07-09 | 2010-01-14 | Ambrx, Inc. | Fgf-21 neutralizing antibodies and their uses |
| WO2010009197A1 (en) | 2008-07-17 | 2010-01-21 | Lexicon Pharmaceuticals, Inc. | Solid forms of (2s,3r,4r,5s,6r)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2h-pyran-3,4,5-triol and methods of their use |
| WO2010009207A1 (en) | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and their use as gpcr modulators |
| WO2010009183A1 (en) | 2008-07-16 | 2010-01-21 | Bristol-Myers Squibb Company | Pyridone and pyridazone analogues as gpr119 modulators |
| WO2010008831A2 (en) | 2008-06-24 | 2010-01-21 | Irm Llc | Compounds and methods for modulating g protein-coupled receptors |
| WO2010009195A1 (en) | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| WO2010009208A1 (en) | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and methods of use thereof |
| WO2010008739A2 (en) | 2008-06-20 | 2010-01-21 | Metabolex, Inc. | Aryl gpr119 agonists and uses thereof |
| WO2010008299A1 (en) | 2008-07-15 | 2010-01-21 | Pronova Biopharma Norge As | Novel sulphur containing lipids for use as food supplement or as medicament |
| WO2010006940A1 (en) | 2008-07-15 | 2010-01-21 | F. Hoffmann-La Roche Ag | Tetrahydrocinnolines as 11 beta hsd1 inhibitors for diabetes |
| WO2010011917A1 (en) | 2008-07-25 | 2010-01-28 | Glaxosmithkline Llc | SEH AND 11β-HSD1 DUAL INHIBITORS |
| WO2010010157A2 (en) | 2008-07-25 | 2010-01-28 | Boehringer Ingelheim International Gmbh | INHIBITORS OF 11beta-HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2010010174A1 (en) | 2008-07-25 | 2010-01-28 | Boehringer Ingelheim International Gmbh | 1,1'-diadamantyl carboxylic acids, medicaments containing such compounds and their use |
| WO2010011772A2 (en) * | 2008-07-23 | 2010-01-28 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010012650A1 (en) | 2008-07-28 | 2010-02-04 | Syddansk Universitet | Compounds for the treatment of metabolic diseases |
| WO2010014771A1 (en) | 2008-08-01 | 2010-02-04 | Ore Pharmaceuticals Inc. | Romazarit for treating metabolic diseases |
| WO2010014794A1 (en) | 2008-07-30 | 2010-02-04 | Oncotherapy Science, Inc. | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| WO2010013161A1 (en) | 2008-07-29 | 2010-02-04 | Pfizer Inc. | Fluorinated heteroaryls |
| WO2010013168A1 (en) | 2008-08-01 | 2010-02-04 | Centre National De La Recherche Scientifique (Cnrs) | 3',6-substituted indirubins and their biological applications |
| WO2010014613A2 (en) | 2008-07-28 | 2010-02-04 | Emory University | Treating various disorders using trkb agonists |
| WO2010014593A1 (en) | 2008-07-30 | 2010-02-04 | Smithkline Beecham Corporation | Chemical compounds and uses |
| WO2010016944A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Analogues of glucose-dependent insulinotropic polypeptide (gip) modified at n-terminal |
| WO2010015849A2 (en) | 2008-08-04 | 2010-02-11 | Astrazeneca Ab | Therapeutic agents 414 |
| WO2010016936A1 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Pharmaceutical compositions of analogues of glucose-dependent insulinotropic polypeptide |
| WO2010016935A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Truncated analogues of glucose-dependent insulinotropic polypeptide |
| WO2010016938A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Glucose-dependent insulinotropic polypeptide analogues |
| WO2010015664A1 (en) | 2008-08-06 | 2010-02-11 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients inappropriate for metformin therapy |
| WO2010016940A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Analogues of glucose-dependent insulinotropic polypeptide |
| WO2010018438A2 (en) | 2008-08-11 | 2010-02-18 | Hetero Research Foundation | Tetrazole glycosides |
| WO2010018435A1 (en) | 2008-08-11 | 2010-02-18 | Hetero Research Foundation | Amide glycosides |
| WO2010018800A1 (en) | 2008-08-15 | 2010-02-18 | Banyu Pharmaceutical Co.,Ltd. | Acetyl pyrrolidinyl indole derivative |
| WO2010019828A1 (en) | 2008-08-13 | 2010-02-18 | Metabasis Therapeutics, Inc. | Glucagon receptor antagonists |
| WO2010129668A1 (en) * | 2009-05-06 | 2010-11-11 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridines |
-
2011
- 2011-09-12 WO PCT/EP2011/065719 patent/WO2013037390A1/en active Application Filing
Patent Citations (999)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0296110A2 (en) | 1987-06-15 | 1988-12-21 | Ciba-Geigy Ag | Staurosporine derivatives substituted for the nitrogen atom of the methylamino group |
| EP0657458A1 (en) | 1993-12-07 | 1995-06-14 | Eli Lilly And Company | Protein kinase C inhibitors |
| WO1995017182A1 (en) | 1993-12-23 | 1995-06-29 | Eli Lilly And Company | Protein kinase c inhibitors |
| WO1997026265A1 (en) | 1996-01-17 | 1997-07-24 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1997041097A2 (en) | 1996-12-31 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| US6221633B1 (en) | 1997-06-20 | 2001-04-24 | Aventis Pharma Deutschland Gmbh | Insulin derivatives having a rapid onset of action |
| WO1999003861A1 (en) | 1997-07-16 | 1999-01-28 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO2000034331A2 (en) | 1998-12-07 | 2000-06-15 | Societe De Conseils De Recherches Et D'applications Scientifiques Sas | Analogues of glp-1 |
| WO2000058307A2 (en) | 1999-03-11 | 2000-10-05 | Neurogen Corporation | Aryl fused 2,4-disubstituted pyridines: nk3 receptor ligands |
| WO2000064876A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Tri-aryl acid derivatives as ppar receptor ligands |
| WO2000064888A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Di-aryl acid derivatives as ppar receptor ligands |
| WO2001000610A1 (en) | 1999-06-23 | 2001-01-04 | Aventis Pharma Deutschland Gmbh | Substituted benzimidazole |
| WO2001004156A1 (en) | 1999-07-12 | 2001-01-18 | Zealand Pharmaceuticals A/S | Peptides that lower blood glucose levels |
| WO2001019831A1 (en) | 1999-09-10 | 2001-03-22 | Novo Nordisk A/S | MODULATORS OF PROTEIN TYROSINE PHOSPHATASES (PTPases) |
| WO2001017516A2 (en) | 1999-09-10 | 2001-03-15 | Novo Nordisk A/S | Method of inhibiting protein tyrosine phosphatase 1b and/or t-cell protein tyrosine phosphatase and/or other ptpases with an asp residue at position 48 |
| WO2001019830A1 (en) | 1999-09-10 | 2001-03-22 | Novo Nordisk A/S | MODULATORS OF PROTEIN TYROSINE PHOSPHATASES (PTPases) |
| WO2001030774A1 (en) | 1999-10-26 | 2001-05-03 | Aventis Pharma Deutschland Gmbh | Substituted indoles for modulating nfkb activity |
| WO2001040207A1 (en) | 1999-12-02 | 2001-06-07 | Glaxo Group Limited | Substituted oxazoles and thiazoles derivatives as hppar alpha activators |
| WO2001090094A1 (en) | 2000-05-22 | 2001-11-29 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2001090090A1 (en) | 2000-05-22 | 2001-11-29 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2002038561A1 (en) | 2000-11-07 | 2002-05-16 | Novartis Ag | Indolylmaleimide derivatives as protein kinase c inhibitors |
| US20050038023A1 (en) | 2000-12-21 | 2005-02-17 | David Bebbington | Pyrazole compounds useful as protein kinase inhibitors |
| WO2002096894A1 (en) | 2001-05-31 | 2002-12-05 | Glaxo Group Limited | Thiazole or oxazole derivatives which are useful in the treatment of cardiovascular and related diseases |
| WO2003020269A1 (en) | 2001-08-31 | 2003-03-13 | Aventis Pharma Deutschland Gmbh | Diaryl cycloalkyl derivatives, method for producing the same and the use thereof as ppar activators |
| WO2003044000A1 (en) | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003043999A1 (en) | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003044009A1 (en) | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003065983A2 (en) | 2002-02-01 | 2003-08-14 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003076442A1 (en) | 2002-03-05 | 2003-09-18 | Eli Lilly And Company | Purine derivatives as kinase inhibitors |
| WO2003074500A2 (en) | 2002-03-06 | 2003-09-12 | Sanofi-Aventis | N-aminoacetyl-pyrrolidine-2-carbonitriles and their use as ddp-iv inhibitors |
| WO2003078403A2 (en) | 2002-03-11 | 2003-09-25 | Aventis Pharma S.A. | Derives d’aminoindazoles comme inhibiteurs de proteine-kinase |
| WO2003084922A1 (en) | 2002-04-11 | 2003-10-16 | Aventis Pharma Deutschland Gmbh | Acyl-4-carboxyphenylurea derivatives, method for production and use thereof |
| WO2003097064A1 (en) | 2002-05-17 | 2003-11-27 | Kyowa Hakko Kogyo Co., Ltd. | Therapeutic agent for diabetes |
| WO2003104207A2 (en) | 2002-06-10 | 2003-12-18 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003104208A1 (en) | 2002-06-10 | 2003-12-18 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003106410A1 (en) | 2002-06-13 | 2003-12-24 | Aventis Pharma Deutschland Gmbh | Fluorinated cycloalkyl-derivatised benzoylguanidines and their use as a medicament |
| WO2003106456A2 (en) | 2002-06-14 | 2003-12-24 | Sanofi-Synthelabo | New compounds |
| WO2004007517A1 (en) | 2002-07-11 | 2004-01-22 | Aventis Pharma Deutschland Gmbh | Novel thiophenylglycoside derivatives, methods for production thereof, medicaments comprising said compounds and use thereof |
| WO2004007455A1 (en) | 2002-07-12 | 2004-01-22 | Aventis Pharma Deutschland Gmbh | Heterocyclically substituted benzoylureas, method for their production and their use as medicaments |
| WO2004011410A1 (en) | 2002-07-27 | 2004-02-05 | Astrazeneca Ab | Chemical compounds |
| WO2004014910A1 (en) | 2002-08-07 | 2004-02-19 | Mitsubishi Pharma Corporation | Dihydropyrazolopyridine compounds |
| WO2004022544A1 (en) | 2002-09-05 | 2004-03-18 | Aventis Pharma S.A. | Novel aminoindazole derivatives as medicines and pharmaceutical compositions containing same |
| WO2004024726A1 (en) | 2002-09-12 | 2004-03-25 | F. Hoffmann-La Roche Ag | N-substituted-1h-indol-5-propionic acid compounds as ppar agonists useful for the treatment of diabetes |
| WO2004033427A1 (en) | 2002-10-11 | 2004-04-22 | Astrazeneca Ab | 1,4-disubstituted piperidine derivatives and their use as 11-betahsd1 inhibitors |
| WO2004037169A2 (en) | 2002-10-18 | 2004-05-06 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2004037251A1 (en) | 2002-10-24 | 2004-05-06 | Sterix Limited | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 and type 2 |
| WO2004041274A1 (en) | 2002-11-05 | 2004-05-21 | Arena Pharmaceuticals, Inc. | Benzotriazoles and methods of prophylaxis or treatment of metabolic-related disorders thereof |
| WO2004041264A1 (en) | 2002-11-07 | 2004-05-21 | Astrazeneca Ab | 2-oxo-ethanesulfonamide derivates |
| WO2004046117A1 (en) | 2002-11-19 | 2004-06-03 | Aventis Pharma Deutschland Gmbh | Pyridazinone derivatives as gsk-3beta inhibitors |
| WO2004050646A1 (en) | 2002-11-29 | 2004-06-17 | Astrazeneca Ab | 1, 2, 5-thiadiazolidin-3-one 1, 1 dioxide derivatives as inhibitors of protein tyrosine phosphatase ptp1b |
| WO2004050658A1 (en) | 2002-12-03 | 2004-06-17 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel substituted imidazo-pyridinones and imidazo-pyridazeiones, the production and use thereof as medicaments |
| WO2004052903A1 (en) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Novel fluoroglycoside heterocyclic derivatives, pharmaceutical products containing said compounds and the use thereof |
| WO2004052902A1 (en) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Novel aromatic fluoroglycoside derivatives, pharmaceutical products containing said compounds and the use thereof |
| WO2004058730A2 (en) | 2002-12-20 | 2004-07-15 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2004056744A1 (en) | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| WO2004063179A1 (en) | 2003-01-06 | 2004-07-29 | Eli Lilly And Company | Substituted arylcyclopropylacetamides as glucokinase activators |
| WO2004065380A1 (en) | 2003-01-14 | 2004-08-05 | Arena Pharmaceuticals Inc. | 1,2,3-trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prpphylaxis and treatment of disorders related thereto such as diabetes and hyperglycemia |
| WO2004065351A1 (en) | 2003-01-24 | 2004-08-05 | Novartis Ag | Amide derivatives and their use as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2004072066A1 (en) | 2003-02-11 | 2004-08-26 | Prosidion Limited | Tri(cyclo) substituted amide glucokinase activator compounds |
| WO2004072031A2 (en) | 2003-02-11 | 2004-08-26 | Prosidion Limited | Phenylacetamides and their use as glucokinase modulators |
| WO2004074288A1 (en) | 2003-02-19 | 2004-09-02 | F. Hoffmann-La Roche Ag | Sulfonamide substituted xanthine derivatives for use as pepck inhibitors |
| EP1460075A1 (en) | 2003-03-21 | 2004-09-22 | Sanofi-Synthelabo | Substituted 8-Pyridinyl-6,7,8,9-Tetrahydropyrimido[1,2-a]Pyrimidin-4-one and 8-Phenyl-6-7,8,9-Tetrahydropyrimido[1,2-a]Pyrimidin-4-one derivatives |
| WO2004089896A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 ACTIVE COMPOUNDS |
| WO2004089367A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of substituted 1,2,4-triazoles |
| WO2004089380A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Pharmaceutical use of fused 1,2,4-triazoles |
| WO2004089470A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | New amide derivatives and pharmaceutical use thereof |
| WO2004089471A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | NEW PYRAZOLO[1,5-a] PYRIMIDINES DERIVATIVES AND PHARMACEUTICAL USE THEREOF |
| WO2004100875A2 (en) | 2003-05-09 | 2004-11-25 | Merck & Co., Inc. | Benzimidazoles, compositions containing such compounds and methods of use |
| WO2004101528A2 (en) | 2003-05-19 | 2004-11-25 | F. Hoffmann-La Roche Ag | Isoquinoline derivatives and their use as gfat inhibitors |
| WO2004103980A1 (en) | 2003-05-21 | 2004-12-02 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type i |
| WO2004106294A2 (en) | 2003-05-29 | 2004-12-09 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta hydroxysteroid dehydrogenase-1 |
| WO2004106343A2 (en) | 2003-05-30 | 2004-12-09 | Ufc Limited | Agelastatin derivatives of antitumour and gsk-3beta-inhibiting alkaloids |
| JP2004359630A (en) | 2003-06-06 | 2004-12-24 | Yamanouchi Pharmaceut Co Ltd | Difluorodiphenylmethane derivative and its salt |
| WO2005000836A1 (en) | 2003-06-13 | 2005-01-06 | Janssen Pharmaceutica N.V. | Substituted indazolyl(indolyl)maleimide derivatives as kinase inhibitors |
| WO2004112782A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | New use v |
| WO2004112779A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | New use vii |
| WO2004112784A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | New use i |
| WO2004113310A1 (en) | 2003-06-25 | 2004-12-29 | Biovitrum Ab | Use of an inhibitor of 11-b-hydroxysteroid dehydrogenase type 1 compounds for promoting wound healing |
| WO2005005477A2 (en) | 2003-07-11 | 2005-01-20 | Novo Nordisk A/S | Stabilised insulin compositions |
| WO2005009389A2 (en) | 2003-07-23 | 2005-02-03 | Exelixis, Inc. | Anaplastic lymphoma kinase modulators and methods of use |
| WO2005012312A1 (en) | 2003-07-25 | 2005-02-10 | Sanofi-Aventis Deutschland Gmbh | Novel cyano thiazolides, methods for the production thereof, and use thereof as a medicament |
| WO2005012308A1 (en) | 2003-07-25 | 2005-02-10 | Sanofi-Aventis Deutschland Gmbh | Novel cyanopyrrolidides, methods for the production thereof, and use of the same as medicaments |
| WO2005012295A1 (en) | 2003-07-28 | 2005-02-10 | Sanofi-Aventis Deutschland Gmbh | Substituted thiazole-benzoisothiazole dioxide derivatives, method for the production thereof and use of the same |
| US20050026984A1 (en) | 2003-07-29 | 2005-02-03 | Aventis Pharma S.A. | Substituted thieno [2,3-c] pyrazoles and their use as medicinal products |
| WO2005009997A1 (en) | 2003-07-30 | 2005-02-03 | Pfizer Inc. | 3,5 disubstituted indazole compounds, pharmaceutical compositions, and methods for mediating or inhibiting cell proliferation |
| WO2005016877A2 (en) | 2003-08-07 | 2005-02-24 | Merck & Co., Inc. | Pyrazole carboxamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2005028480A2 (en) | 2003-09-03 | 2005-03-31 | Neurogen Corporation | 5-aryl-pyrazolo[4,3-d]pyrimidines, pyridines, and pyrazines and related compounds |
| WO2005027978A2 (en) | 2003-09-19 | 2005-03-31 | Novo Nordisk A/S | Albumin-binding derivatives of therapeutic peptides |
| WO2005037828A1 (en) | 2003-10-20 | 2005-04-28 | Lg Life Sciences Ltd. | Novel inhibitors of dpp-iv, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| WO2005044801A1 (en) | 2003-10-31 | 2005-05-19 | Astrazeneca Ab | Pyridine carboxylic acid derivatives as glucokinase modulators |
| EP1688138A1 (en) | 2003-11-26 | 2006-08-09 | Takeda Pharmaceutical Company Limited | Receptor function regulating agent |
| WO2005058901A1 (en) | 2003-12-17 | 2005-06-30 | Boehringer Ingelheim International Gmbh | Novel 2-(piperazin-1-yl)- and 2-([1,4]diazepan-1-yl)- imidazo[4,5-d]pyridazin-4-one, production and use thereof as medicament for the treatment of diabetes mellitus |
| WO2005058908A1 (en) | 2003-12-19 | 2005-06-30 | Sanofi-Aventis | SUBSTITUTED 8’-PYRI(MI)DINYL-DIHYDROSPIRO-[CYCLOALKYLAMINE]-PYRIMIDO[1,2-a]PYRIMIDIN-6-ONE DERIVATIVES |
| WO2005065680A1 (en) | 2003-12-19 | 2005-07-21 | Merck & Co., Inc. | Cyclic guanidines, compositions containing such compounds and methods of use |
| WO2005063247A1 (en) | 2003-12-22 | 2005-07-14 | Amgen Inc. | Aryl sulfonamide compounds and uses related thereto |
| WO2005061489A1 (en) | 2003-12-24 | 2005-07-07 | Prosidion Limited | Heterocyclic derivatives as gpcr receptor agonists |
| WO2005067932A1 (en) | 2004-01-06 | 2005-07-28 | Janssen Pharmaceutica, N.V. | (3-oxo-3, 4-dihydro-quinoxalin-2-yl-amino) -benzamide derivatives and related compound as glycogen phosphorylase inhibitors for the treatment of diabetes and obesity |
| WO2005067931A2 (en) | 2004-01-14 | 2005-07-28 | Daniolabs Limited | Dopamine uptake inhibitors for the treatment of neurological disease |
| WO2005073229A1 (en) | 2004-01-31 | 2005-08-11 | Sanofi-Aventis Deutschland Gmbh | 7-phenylamino-4-quinolone-3-carboxylic acid derivatives, methods for production and use thereof as medicaments |
| WO2005073199A1 (en) | 2004-02-02 | 2005-08-11 | Sanofi-Aventis Deutschland Gmbh | Indazole derivatives as inhibitors of hormone-sensitive lipases |
| WO2005080360A1 (en) | 2004-02-18 | 2005-09-01 | Astrazeneca Ab | Compounds |
| WO2005085230A1 (en) | 2004-03-02 | 2005-09-15 | Sanofi-Aventis Deutschland Gmbh | 4-benzimidazol-2-yl-pyridazine-3-one-derivatives, production and use thereof in medicaments |
| WO2005085237A1 (en) | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005086904A2 (en) | 2004-03-08 | 2005-09-22 | Amgen Inc. | Therapeutic modulation of ppar (gamma) activity |
| WO2005087727A1 (en) | 2004-03-12 | 2005-09-22 | Boehringer Ingelheim International Gmbh | Novel alkyl-containing 5-acylindolinones, their preparation and their use as pharmaceutical products |
| WO2005097759A1 (en) | 2004-03-29 | 2005-10-20 | Merck & Co., Inc. | Diaryltriazoles as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| US20050222220A1 (en) | 2004-04-05 | 2005-10-06 | Neuropharma, S.A. | GSK-3 inhibitors |
| WO2005097076A2 (en) | 2004-04-09 | 2005-10-20 | Smithkline Beecham Corporation | Low dose pharmaceutical products |
| WO2005111018A1 (en) | 2004-05-18 | 2005-11-24 | Sanofi-Aventis Deutschland Gmbh | Pyridazinone derivatives, methods for producing them and their use as pharmaceuticals |
| WO2005116003A2 (en) | 2004-05-29 | 2005-12-08 | Sanofi-Aventis Deutschland Gmbh | Substituted oxazolobenzoisothiazole dioxide derivatives method for production and use thereof |
| WO2005121161A1 (en) | 2004-06-11 | 2005-12-22 | Sanofi-Aventis Deutschland Gmbh | Novel fluoroglycoside derivatives of pyrazoles, medicaments containing these compounds, and the use thereof |
| WO2006012173A1 (en) | 2004-06-24 | 2006-02-02 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2006012227A2 (en) | 2004-06-24 | 2006-02-02 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2006007959A1 (en) | 2004-07-17 | 2006-01-26 | Sanofi-Aventis Deutschland Gmbh | Diphenylamine-substituted salicylthiazole derivatives and related compounds as phosphotyrosine phosphatase 1b (ptp1b) inhibitors for using as blood-sugar decreasing active ingredients for treating diabetes |
| WO2006010546A2 (en) | 2004-07-28 | 2006-02-02 | F. Hoffman-La Roche Ag | Aryl-pyridine derivatives as 11-beta-hsd1 inhibitors |
| WO2006015691A1 (en) | 2004-08-03 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted 8-aminoalkylthio-xanthines, and the use thereof as inhibitors of the dipeptidyl peptidase iv |
| WO2006015699A1 (en) | 2004-08-06 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted bicyclic 8-pyrrolidino-xanthines and use thereof as inhibitors of the dipeptidyl peptidase iv |
| WO2006015700A1 (en) | 2004-08-06 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted, bicyclic 8-piperidinoxanthines, method for the production thereof and their use as medicaments |
| WO2006015701A1 (en) | 2004-08-06 | 2006-02-16 | Sanofi-Aventis Deutschland Gmbh | Substituted, bicyclic 8-pyrrolidinoxanthines, method for the production thereof and their use as medicaments |
| WO2006017542A1 (en) | 2004-08-06 | 2006-02-16 | Merck & Co., Inc. | Sulfonyl compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2006018150A1 (en) | 2004-08-11 | 2006-02-23 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituited cyclene, medicaments containing said compounds, use thereof and method for the production thereof |
| WO2006016194A1 (en) | 2004-08-12 | 2006-02-16 | Prosidion Limited | Substituted phenylacetamides and their use as glucokinase activators |
| WO2006018117A1 (en) | 2004-08-14 | 2006-02-23 | Sanofi-Aventis Deutschland Gmbh | Substituted 8-aminoalkoxi-xanthines, method for the production thereof and use thereof as medicaments |
| WO2006023515A2 (en) | 2004-08-18 | 2006-03-02 | Metabasis Therapeutics, Inc. | Novel thiazole inhibitors of fructose 1,6-bisphosphatase |
| WO2006024627A2 (en) | 2004-08-30 | 2006-03-09 | Janssen Pharmaceutica N.V. | N-2 adamantanyl-2-phenoxy-acetamide derivatives as 11-beta hydroxysteroid dehydrogenase inhibitors |
| WO2006029699A1 (en) | 2004-09-11 | 2006-03-23 | Sanofi-Aventis Deutschland Gmbh | 7-azaindoles and their use as ppar agonists |
| WO2006035796A1 (en) | 2004-09-29 | 2006-04-06 | Kissei Pharmaceutical Co., Ltd. | 1-(β-D-GLYCOPYRANOSYL)-3-SUBSTITUTED NITROGENOUS HETEROCYCLIC COMPOUND, MEDICINAL COMPOSITION CONTAINING THE SAME, AND MEDICINAL USE THEREOF |
| WO2006034804A1 (en) | 2004-09-29 | 2006-04-06 | F.Hoffmann-La Roche Ag | Indozolone derivatives as 11b-hsd1 inhibitors |
| WO2006039325A2 (en) | 2004-10-01 | 2006-04-13 | Merck & Co., Inc. | Aminopiperidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006037810A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted glp-1 compounds |
| WO2006037811A2 (en) | 2004-10-07 | 2006-04-13 | Novo Nordisk A/S | Protracted exendin-4 compounds |
| WO2006040329A1 (en) | 2004-10-12 | 2006-04-20 | Novo Nordisk A/S | 1 ibeta- hydroxysteroid dehydrogenase type 1 active spiro compounds |
| WO2006045564A1 (en) | 2004-10-22 | 2006-05-04 | Smithkline Beecham Corporation | Xanthine derivatives with hm74a receptor activity |
| WO2006045565A1 (en) | 2004-10-22 | 2006-05-04 | Smithkline Beecham Corporation | Xanthine derivatives with hm74a receptor activity |
| WO2006049952A1 (en) | 2004-10-29 | 2006-05-11 | Eli Lilly And Company | Cycloalkyl lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2006048750A2 (en) | 2004-11-02 | 2006-05-11 | Pfizer Inc. | Novel compounds of substituted and unsubstituted adamantyl amides |
| WO2006050908A1 (en) | 2004-11-08 | 2006-05-18 | Evotec Ag | Inhibitors of 11βετα-hydroxy steroid dehydrogenase type 1 (11beta-hsd1) |
| WO2006048331A1 (en) | 2004-11-08 | 2006-05-11 | Evotec Ag | 11β-HSD1 INHIBITORS |
| WO2006051662A1 (en) | 2004-11-09 | 2006-05-18 | Taisho Pharmaceutical Co., Ltd. | Thiazole derivative |
| JP2006160733A (en) | 2004-11-15 | 2006-06-22 | Taisho Pharmaceut Co Ltd | Medicine containing cyanofluoropyrrolidine derivative as active ingredient |
| WO2006058064A2 (en) | 2004-11-29 | 2006-06-01 | Merck & Co., Inc. | Fused aminopiperidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006059744A1 (en) | 2004-11-30 | 2006-06-08 | Nippon Chemiphar Co., Ltd. | ACTIVATOR OF PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR δ |
| WO2006058923A1 (en) | 2004-12-03 | 2006-06-08 | Novo Nordisk A/S | Heteroaromatic glucokinase activators |
| WO2006058597A1 (en) | 2004-12-03 | 2006-06-08 | Merck Patent Gmbh | Tetrahydropyrane derivatives for use as antidiabetics |
| WO2006062224A1 (en) | 2004-12-07 | 2006-06-15 | Takeda Pharmaceutical Company Limited | Carboxamide derivative |
| WO2006065826A2 (en) | 2004-12-15 | 2006-06-22 | Merck & Co., Inc. | Process to chiral beta amino acid derivatives by asymmetric hydrogenation |
| DE102004060542A1 (en) | 2004-12-16 | 2006-07-06 | Sanofi-Aventis Deutschland Gmbh | Hydroxybiphenyl carboxylic acids and derivatives, process for their preparation and their use |
| WO2006066109A2 (en) | 2004-12-17 | 2006-06-22 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
| WO2006069242A2 (en) | 2004-12-23 | 2006-06-29 | Arena Pharmaceuticals, Inc. | Fused pyrazole derivatives and uses thereof in methods of treatment of metabolic-related?disorders |
| WO2006067531A1 (en) | 2004-12-24 | 2006-06-29 | Prosidion Ltd | G-protein coupled receptor (gpr116) agonists and use thereof for treating obesity and diabetes |
| WO2006068163A1 (en) | 2004-12-24 | 2006-06-29 | Dainippon Sumitomo Pharma Co., Ltd. | Bicyclic pyrrole derivatives |
| WO2006067532A1 (en) | 2004-12-24 | 2006-06-29 | Prosidion Ltd | G-protein coupled receptor agonists |
| WO2006071752A1 (en) | 2004-12-29 | 2006-07-06 | Bristol-Myers Squibb Company | Azolopyrimidine-based inhibitors of dipeptidyl peptidase iv and methods |
| WO2006074244A2 (en) | 2005-01-05 | 2006-07-13 | Abbott Laboratories | Adamantyl derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| WO2006072354A1 (en) | 2005-01-07 | 2006-07-13 | Merck Patent Gmbh | Squaric acid derivatives |
| WO2006073167A1 (en) | 2005-01-07 | 2006-07-13 | Ono Pharmaceutical Co., Ltd. | Pyrrolidine derivatives |
| WO2006073197A1 (en) | 2005-01-07 | 2006-07-13 | Taisho Pharmaceutical Co., Ltd. | 1-thio-d-glucitol derivatives |
| WO2006083491A2 (en) | 2005-01-10 | 2006-08-10 | Arena Pharmaceuticals, Inc. | Substituted pyridinyl and pyrimidinyl derivatives as modulators of metabolism and the treatment of disorders related thereto |
| WO2006074957A1 (en) | 2005-01-17 | 2006-07-20 | Sanofi-Aventis | Substituted aminomethylene sulphonamides, production and use thereof as medicaments |
| WO2006078676A2 (en) | 2005-01-19 | 2006-07-27 | Merck & Co., Inc. | Bicyclic pyrimidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006078006A1 (en) | 2005-01-24 | 2006-07-27 | Dainippon Sumitomo Pharma Co., Ltd. | Indole compound and pharmaceutical composition containing the same |
| WO2006080577A1 (en) | 2005-01-31 | 2006-08-03 | Tanabe Seiyaku Co., Ltd. | Indole derivatives |
| WO2006084176A2 (en) | 2005-02-03 | 2006-08-10 | Irm Llc | Compounds and compositions as ppar modulators |
| WO2006085685A1 (en) | 2005-02-09 | 2006-08-17 | Takeda Pharmaceutical Company Limited | Pyrazole compound |
| WO2006086488A2 (en) | 2005-02-11 | 2006-08-17 | Eli Lilly And Company | Substituted thiophene derivatives as glucagon receptor antagonists, preparation and therapeutic uses |
| WO2006085108A1 (en) | 2005-02-14 | 2006-08-17 | Smithkline Beecham Corporation | Awthranilic acid derivatives and their use in treatment of diseases of lipid metabolism, in particular dyslipidaemia |
| WO2006085113A2 (en) | 2005-02-14 | 2006-08-17 | Smithkline Beecham Corporation | 2-substituted 5-membered heteroaryl carboxylates as hm74a receptor agonists |
| WO2006085112A1 (en) | 2005-02-14 | 2006-08-17 | Smithkline Beecham Corporation | Anthranilic acid derivatives as hm74a receptor agonists |
| WO2006087309A1 (en) | 2005-02-15 | 2006-08-24 | Novo Nordisk A/S | 3,4-dihydro-1h-isoquinoline-2-carboxylic acid 5-aminopyridin-2-yl esters |
| WO2006087997A1 (en) | 2005-02-15 | 2006-08-24 | Kissei Pharmaceutical Co., Ltd. | 1-SUBSTITUTED-7-(β-D-GLYCOPYRANOSYLOXY)(AZA)INDOLE COMPOUND AND PHARMACEUTICAL CONTAINING THE SAME |
| WO2006090915A1 (en) | 2005-02-25 | 2006-08-31 | Takeda Pharmaceutical Company Limited | Pyridyl acetic acid compounds |
| WO2006099943A1 (en) | 2005-03-19 | 2006-09-28 | Sanofi-Aventis | Amide-substituted 8-n-benzimidazoles, method for the production thereof, and use of the same as medicaments |
| WO2006099941A1 (en) | 2005-03-19 | 2006-09-28 | Sanofi-Aventis | Aminocarbonyl-substituted 8-n-benzimidazoles, method for their production and their use as medicaments |
| WO2006104030A1 (en) | 2005-03-25 | 2006-10-05 | Daiichi Sankyo Company, Limited | Thiazole compound |
| WO2006104356A1 (en) | 2005-04-01 | 2006-10-05 | Lg Life Sciences, Ltd. | Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| WO2006106423A2 (en) | 2005-04-07 | 2006-10-12 | Pfizer Inc. | Amino sulfonyl derivatives as inhibitors of human 11-.beta.-hydrosysteroid dehydrogenase |
| WO2006113150A1 (en) | 2005-04-13 | 2006-10-26 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2006108842A1 (en) | 2005-04-15 | 2006-10-19 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted (heteroaryloxy-benzyl)-benzene derivatives as sglt inhibitors |
| WO2006111261A1 (en) | 2005-04-16 | 2006-10-26 | Sanofi-Aventis | Substituted 2-amin0alkylthi0-benzimidaz0les and use thereof for reducing blood sugar levels |
| WO2006112549A1 (en) | 2005-04-20 | 2006-10-26 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| WO2006111321A1 (en) | 2005-04-20 | 2006-10-26 | Sanofi-Aventis | Azole derivatives in the form of lipase and phospholipase inhibitors |
| WO2006121860A2 (en) | 2005-05-06 | 2006-11-16 | Bayer Pharmaceuticals Corporation | Glucagon-like peptide 1 (glp-1) receptor agonists and their pharmacological methods of use |
| WO2006124529A1 (en) | 2005-05-13 | 2006-11-23 | Eli Lilly And Company | Glp-1 pegylated compounds |
| WO2006124490A2 (en) | 2005-05-17 | 2006-11-23 | Schering Corporation | Heterocycles as nicotinic acid receptor agonists for the treatment of dyslipidemia |
| WO2006127530A2 (en) | 2005-05-25 | 2006-11-30 | Merck & Co., Inc. | Aminocyclohexanes as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006125972A1 (en) | 2005-05-27 | 2006-11-30 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2006132436A1 (en) | 2005-06-08 | 2006-12-14 | Japan Tobacco Inc. | Heterocyclic compound |
| WO2006135795A1 (en) | 2005-06-09 | 2006-12-21 | Bristol-Myers Squibb Company | Imidazo- and triazolopyridines as inhibitors of 11-beta hydroxysteroid dehydrogenase type i |
| WO2006134481A1 (en) | 2005-06-16 | 2006-12-21 | Pfizer Inc. | Inhibitors of 11-beta hydroxysteroid dehydrogenase type 1 |
| WO2006134467A1 (en) | 2005-06-16 | 2006-12-21 | Pfizer Inc. | N-(pyridin-2-yl)-sulfonamide derivatives |
| WO2006133926A1 (en) | 2005-06-17 | 2006-12-21 | Carex Sa | Pyrazole derivates as cannabinoid receptor modulators |
| WO2006138508A2 (en) | 2005-06-17 | 2006-12-28 | Amgen Inc. | Benzamide derivatives and uses related thereto |
| WO2006138695A1 (en) | 2005-06-17 | 2006-12-28 | Bristol-Myers Squibb Company | Triazolopyridine derivatives as cannabinoid receptor 1 antagonists |
| WO2006136502A1 (en) | 2005-06-22 | 2006-12-28 | F. Hoffmann-La Roche Ag | ( 6-FLU0R0-BENZ0[l, 3] DIOXOLYL) -MORPHOLIN-4-YL-METHANONES AND THEIR USE AS CBl LIGANDS |
| WO2007002557A1 (en) | 2005-06-28 | 2007-01-04 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007000445A1 (en) | 2005-06-29 | 2007-01-04 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2007000006A2 (en) | 2005-06-29 | 2007-01-04 | Plansee Se | Halogen filament lamp comprising a dimming cover consisting of an mo alloy |
| WO2007003964A1 (en) | 2005-06-30 | 2007-01-11 | Prosidion Limited | G-protein coupled receptor agonists |
| WO2007003962A2 (en) | 2005-06-30 | 2007-01-11 | Prosidion Limited | Gpcr agonists |
| WO2007003960A1 (en) | 2005-06-30 | 2007-01-11 | Prosidion Limited | Gpcr agonists |
| WO2007003521A2 (en) | 2005-07-05 | 2007-01-11 | F. Hoffmann-La Roche Ag | Pyridazine derivatives as 11beta-hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2007006760A1 (en) | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkyl urea glucokinase activators |
| WO2007006761A1 (en) | 2005-07-08 | 2007-01-18 | Novo Nordisk A/S | Dicycloalkylcarbamoyl ureas as glucokinase activators |
| WO2007007688A1 (en) | 2005-07-08 | 2007-01-18 | Mochida Pharmaceutical Co., Ltd. | 3,5-diamino-1,2,4-triazole derivative |
| WO2007007040A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | 2 -heterocyclyloxybenzoyl amino heterocyclyl compounds as modulators of glucokinase for the treatment of type 2 diabetes |
| WO2007007042A1 (en) | 2005-07-09 | 2007-01-18 | Astrazeneca Ab | Heteroaryl benzamide derivatives for use as glk activators in the treatment of diabetes |
| WO2007007886A1 (en) | 2005-07-11 | 2007-01-18 | Mitsubishi Tanabe Pharma Corporation | An oxime derivative and preparations thereof |
| WO2007007910A1 (en) | 2005-07-13 | 2007-01-18 | Banyu Pharmaceutical Co., Ltd. | Heterocycle-substituted benzimidazole derivative |
| WO2007006814A1 (en) | 2005-07-14 | 2007-01-18 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2007015767A1 (en) | 2005-07-20 | 2007-02-08 | Eli Lilly And Company | Pyridine derivatives as dipeptedyl peptidase inhibitors |
| WO2007015744A1 (en) | 2005-07-21 | 2007-02-08 | Incyte Corporation | Disubstituted thienyl compounds and their use as pharmaceuticals |
| WO2007009911A1 (en) | 2005-07-21 | 2007-01-25 | F. Hoffmann-La Roche Ag | PYRIDO [2 , 3-D] PYRIMIDINE-2 , 4-DIAMINE COMPOUNDS AS PTPlB INHIBITORS |
| WO2007014895A2 (en) | 2005-07-28 | 2007-02-08 | Boehringer Ingelheim International Gmbh | Methods for preventing and treating metabolic disorders and new pyrazole-o-glycoside derivatives |
| WO2007013689A1 (en) | 2005-07-29 | 2007-02-01 | Takeda Pharmaceutical Company Limited | Cyclopropanecarboxylic acid compound |
| WO2007014619A1 (en) | 2005-08-01 | 2007-02-08 | Merck Patent Gmbh | Novelimidazolecarboxamide derivatives as fructose-1,6-bisphosphatase inhibitors, and pharmaceutical compositions comprising same |
| WO2007017549A1 (en) | 2005-08-04 | 2007-02-15 | Universitat De Valencia | Aqueous base pigment compositions for the multicolour marking of inorganic materials with laser |
| WO2007017649A1 (en) | 2005-08-09 | 2007-02-15 | Astrazeneca Ab | Heteroarylcarbamoylbenzene derivatives for the treatment of diabetes |
| WO2007017261A1 (en) | 2005-08-10 | 2007-02-15 | Smithkline Beecham Corporation | Xanthine derivatives as selective hm74a agonists |
| WO2007017262A1 (en) | 2005-08-10 | 2007-02-15 | Smithkline Beecham Corporation | Xanthine derivatives as selective hm74a agonists |
| WO2007017265A1 (en) | 2005-08-10 | 2007-02-15 | Smithkline Beecham Corporation | Xanthine derivatives as selective hm74a agonists |
| WO2007024993A2 (en) | 2005-08-26 | 2007-03-01 | Merck & Co., Inc. | Fused aminopiperidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007027532A2 (en) | 2005-08-29 | 2007-03-08 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007026761A1 (en) | 2005-08-31 | 2007-03-08 | Astellas Pharma Inc. | Thiazole derivative |
| WO2007028135A2 (en) | 2005-09-01 | 2007-03-08 | Takeda Pharmaceutical Company Limited | Imidazopyridine compounds |
| WO2007028145A2 (en) | 2005-09-02 | 2007-03-08 | Dara Biosciences, Inc. | Agents and methods for reducing protein tyrosine phosphatase 1b activity in the central nervous system |
| WO2007029086A2 (en) | 2005-09-05 | 2007-03-15 | Ranbaxy Laboratories Limited | Derivatives of 3-azabicyclo[3.1.0]hexane as dipeptidyl peptidase-iv inhibitors |
| WO2007029021A1 (en) | 2005-09-08 | 2007-03-15 | The University Of Edinburgh | 1,5-substituted tetrazoles as therapeutic compounds |
| US20070264331A1 (en) | 2005-09-08 | 2007-11-15 | Laboratorios Silanes, S.A De C.V. | Stable pharmaceutical composition of immediate-release glimepiride and extended-release metformin |
| WO2007033002A1 (en) | 2005-09-14 | 2007-03-22 | Amgen Inc. | Conformationally constrained 3- (4-hydroxy-phenyl) - substituted-propanoic acids useful for treating metabolic disorders |
| WO2007031739A1 (en) | 2005-09-16 | 2007-03-22 | Astrazeneca Ab | Heterobicyclic compounds as glucokinase activators |
| WO2007035355A2 (en) | 2005-09-16 | 2007-03-29 | Arena Pharmaceuticals, Inc. | Modulators of metabolism and the treatment of disorders related thereto |
| US20070066584A1 (en) | 2005-09-21 | 2007-03-22 | Wenqing Yao | Amido compounds and their use as pharmaceuticals |
| WO2007039172A1 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridyl-i, 2 , 4 -oxadiazolone derivatives with phenyl group, processes for their preparation and their use as pharmaceuticals |
| WO2007039178A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl-[1,2,4]-oxadiazol-5-one derivatives with phenyl group, processes for their preparation and their use as pharmaceuticals |
| WO2007041365A2 (en) | 2005-09-30 | 2007-04-12 | Novartis Ag | 3-cyclyl-2- (4-sulfamo yl-phenyl) -n-cyclyl-propionamide derivatives useful in the treatment of impaired glucose tolerance and diabetes |
| WO2007041366A1 (en) | 2005-09-30 | 2007-04-12 | Novartis Ag | Sulfonamide derivatives as glycokinase activators useful in the treatment of type 2 diabetes |
| WO2007037534A1 (en) | 2005-09-30 | 2007-04-05 | Banyu Pharmaceutical Co., Ltd. | 2-heteroaryl-substituted indole derivative |
| WO2007042178A1 (en) | 2005-10-12 | 2007-04-19 | Sanofi-Aventis | Diacyl indazol derivatives as lipase and phospholipase inhibitors |
| WO2007047177A1 (en) | 2005-10-13 | 2007-04-26 | Merck & Co., Inc. | Acyl indoles, compositions containing such compounds and methods of use |
| WO2007043638A1 (en) | 2005-10-14 | 2007-04-19 | Astellas Pharma Inc. | Condensed heterocyclic compound |
| WO2007047625A2 (en) | 2005-10-20 | 2007-04-26 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2007051810A2 (en) | 2005-11-01 | 2007-05-10 | Transtech Pharma | Pharmaceutical use of substituted amides |
| WO2007051811A2 (en) | 2005-11-01 | 2007-05-10 | Transtech Pharma | Pharmaceutical use of substituted amides |
| WO2007053765A2 (en) | 2005-11-01 | 2007-05-10 | Janssen Pharmaceutica N.V. | Substituted cycloalkylpyrrolones as allosteric modulators of glucokinase |
| WO2007053345A1 (en) | 2005-11-01 | 2007-05-10 | Array Biopharma Inc. | Glucokinase activators |
| WO2007051847A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides as glucokinase modulators |
| WO2007051845A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides |
| WO2007051846A1 (en) | 2005-11-03 | 2007-05-10 | Prosidion Ltd | Tricyclo substituted amides |
| WO2007056771A2 (en) | 2005-11-09 | 2007-05-18 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007106181A2 (en) | 2005-11-17 | 2007-09-20 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007123581A1 (en) | 2005-11-17 | 2007-11-01 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007057768A2 (en) | 2005-11-18 | 2007-05-24 | Pfizer Products Inc. | Sulfonyl derivatives |
| WO2007061923A2 (en) | 2005-11-18 | 2007-05-31 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2007058346A1 (en) | 2005-11-21 | 2007-05-24 | Shionogi & Co., Ltd. | HETEROCYCLIC COMPOUND HAVING INHIBITORY ACTIVITY ON 11-β-HYDROXYSTEROID DEHYDROGENASE TYPE I |
| WO2007061661A2 (en) | 2005-11-22 | 2007-05-31 | Amgen Inc. | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2007120270A2 (en) | 2005-11-22 | 2007-10-25 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007120284A2 (en) | 2005-11-23 | 2007-10-25 | Eli Lilly And Company | Glucagon receptor antagonists, preparation and therapeutic uses |
| WO2007060992A1 (en) | 2005-11-25 | 2007-05-31 | Kaneka Corporation | Agent for preventing or improving metabolic syndrome or insulin-resistance syndrome |
| WO2007063928A1 (en) | 2005-11-30 | 2007-06-07 | Toray Industries, Inc. | Novel noncyclic amine carboxamide derivative and salt thereof |
| WO2007062568A1 (en) | 2005-12-02 | 2007-06-07 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Compounds used as mammalian cell amp-activated protein kinase(ampk) activators and their preparation methods and usages |
| WO2007070506A2 (en) | 2005-12-14 | 2007-06-21 | Amgen Inc. | Diaza heterocyclic sulfonamide derivatives and their uses |
| WO2007070434A2 (en) | 2005-12-14 | 2007-06-21 | Merck & Co., Inc. | Fused aminopiperidines as dipeptidyl peptidase-4 inhibitors for the treatment or prevention of diabetes |
| WO2007068330A1 (en) | 2005-12-16 | 2007-06-21 | Merck Patent Gmbh | 2-ADAMANTYLUREA DERIVATIVES AS SELECTIVE 11β-HSD1 INHIBITORS |
| WO2007100374A2 (en) | 2005-12-19 | 2007-09-07 | Trustees Of Tufts College | Soft protease inhibitors and pro-soft forms thereof |
| WO2007075847A2 (en) | 2005-12-20 | 2007-07-05 | Takeda Pharmaceutical Company Limited | Glucokinase activators |
| WO2007071576A1 (en) | 2005-12-21 | 2007-06-28 | F. Hoffmann-La Roche Ag | New salt and polymorph of dpp-iv inhibitor |
| WO2007071766A2 (en) | 2005-12-22 | 2007-06-28 | Transtech Pharma | Phenoxy acetic acids as ppar delta activators |
| WO2007073117A1 (en) | 2005-12-22 | 2007-06-28 | Crystalgenomics, Inc. | Aminopyrimidine derivatives inhibiting protein kinase activity, method for the preparation thereof and pharmaceutical composition containing same |
| WO2007071738A1 (en) | 2005-12-23 | 2007-06-28 | Novartis Ag | Condensed heterocyclic compounds useful as dpp-iv inhibitors |
| WO2007077508A2 (en) | 2005-12-30 | 2007-07-12 | Ranbaxy Laboratories Limited | Derivatives of beta-amino acid as dipeptidyl peptidase-iv inhibitors |
| WO2007081755A2 (en) | 2006-01-09 | 2007-07-19 | Metabasis Therapeutics, Inc. | Indole-benzimidazole and indazole inhibitors of tyrosine phosphatases |
| WO2007080170A1 (en) | 2006-01-11 | 2007-07-19 | Boehringer Ingelheim International Gmbh | CRYSTALLINE FORM OF 1´-(1-METHYLETHYL)- 4´-[(2-FLUORO-4-METHOXYPHENYL)METHYL]-5´-METHYL-1H-PYRAZOL-3´-O-β-D-GLUCOPYRANOSIDE, A METHOD FOR ITS PREPARATION AND THE USE THEREOF FOR PREPARING MEDICAMENTS |
| WO2007087150A2 (en) | 2006-01-13 | 2007-08-02 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2007138485A2 (en) | 2006-01-18 | 2007-12-06 | Evolva Sa | Ppar modulators |
| WO2007083978A1 (en) | 2006-01-23 | 2007-07-26 | Crystalgenomics, Inc. | Imidazopyridine derivatives inhibiting protein kinase activity, method for the preparation thereof and pharmaceutical composition containing same |
| WO2007085136A1 (en) | 2006-01-24 | 2007-08-02 | Institute Of Pharmacology And Toxicology Academy Of Military Medical Sciences P.L.A. China | 1,3-benzodioxolecyclopentene derivates, preparation process and medical uses thereof |
| WO2007085135A1 (en) | 2006-01-25 | 2007-08-02 | Institute Of Pharmacology And Toxicology Academy Of Military Medical Sciences P.L.A. China | 1,3-benzodioxolecyclopentene-2,2-dicarboxylic acid derivates, preparation process and medical uses thereof |
| WO2007087231A2 (en) | 2006-01-25 | 2007-08-02 | Merck & Co., Inc. | Aminocyclohexanes as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007089512A1 (en) | 2006-01-27 | 2007-08-09 | Array Biopharma Inc. | Glucokinase activators |
| WO2007089667A1 (en) | 2006-01-30 | 2007-08-09 | Irm Llc | Compounds and compositions as ppar modulators |
| WO2007089557A2 (en) | 2006-01-30 | 2007-08-09 | Irm Llc | Polycyclic 1, 2, 3, 4 -tetrahydro- isoquinoline derivatives and compositions comprising them as ppar modulators |
| WO2007087448A1 (en) | 2006-01-30 | 2007-08-02 | Irm Llc | Spiro imidazole derivatives as ppar modulators |
| WO2007089683A1 (en) | 2006-01-31 | 2007-08-09 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2007092435A2 (en) | 2006-02-07 | 2007-08-16 | Wyeth | 11-beta hsd1 inhibitors |
| WO2007092364A2 (en) | 2006-02-07 | 2007-08-16 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007095462A2 (en) | 2006-02-13 | 2007-08-23 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007093264A1 (en) | 2006-02-14 | 2007-08-23 | Merck Patent Gmbh | Mandelic hydrazides |
| WO2007093610A1 (en) | 2006-02-15 | 2007-08-23 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzonitrile derivatives, pharmaceutical compositions containing such compounds, their use and process for their manufacture |
| WO2007097931A2 (en) | 2006-02-15 | 2007-08-30 | Merck & Co., Inc. | Aminotetrahydropyrans as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007099553A2 (en) | 2006-02-27 | 2007-09-07 | Cadila Healthcare Limited | 1,3-dioxane carboxylic acids |
| WO2007101060A2 (en) | 2006-02-28 | 2007-09-07 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007099385A1 (en) | 2006-03-01 | 2007-09-07 | Glenmark Pharmaceuticals S.A. | Dipeptidyl peptidase iv inhibitor compounds and compositions |
| US20070207985A1 (en) | 2006-03-01 | 2007-09-06 | Bristol-Myers Squibb Company | Triazine 11-beta hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2007101270A1 (en) | 2006-03-02 | 2007-09-07 | Incyte Corporation | MODULATORS OF 11-β HYDROXYL STEROID DEHYDROGENASE TYPE 1, PHARMACEUTICAL COMPOSITIONS THEREOF, AND METHODS OF USING THE SAME |
| WO2007100027A1 (en) | 2006-03-02 | 2007-09-07 | Daiichi Sankyo Company, Limited | Optically active thiazolidinedione derivative |
| WO2007103252A2 (en) | 2006-03-03 | 2007-09-13 | Merck & Co., Inc. | Novel crystalline forms of antidiabetic compounds |
| US20070208001A1 (en) | 2006-03-03 | 2007-09-06 | Jincong Zhuo | Modulators of 11- beta hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| US20070270492A1 (en) | 2006-03-06 | 2007-11-22 | Avestha Gengraine Technologies Pvt. Ltd. | Nananoic acid derivatives as dipeptidyl peptidase inhibitors |
| US20080051452A1 (en) | 2006-03-06 | 2008-02-28 | Avestha Gengraine Technologies Pvt. Ltd. | Hexanoic acid derivatives as dipeptidyl peptidase inhibitors |
| WO2007102515A1 (en) | 2006-03-07 | 2007-09-13 | Astellas Pharma Inc. | Phenylthiazole derivative |
| WO2007104034A2 (en) | 2006-03-08 | 2007-09-13 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2007101864A2 (en) | 2006-03-09 | 2007-09-13 | High Point Pharmaceuticals, Llc | Compounds that modulate ppar activity, their preparation and use |
| WO2007104053A2 (en) | 2006-03-09 | 2007-09-13 | Pharmacopeia, Inc. | 8-heteroarylpurine mnk2 inhibitors for treating metabolic disorders |
| WO2007105650A1 (en) | 2006-03-10 | 2007-09-20 | Ajinomoto Co., Inc. | 4-hydroxyisoleucine derivative and process for producing the derivative |
| WO2007106469A2 (en) | 2006-03-14 | 2007-09-20 | Amgen Inc. | Bicyclic carboxylic acid derivatives useful for treating metabolic disorders |
| WO2007104789A2 (en) | 2006-03-15 | 2007-09-20 | Novo Nordisk A/S | Amylin derivatives |
| WO2007105753A1 (en) | 2006-03-16 | 2007-09-20 | Astellas Pharma Inc. | Triazole derivative or salt thereof |
| JP2007246474A (en) | 2006-03-17 | 2007-09-27 | Mitsubishi Pharma Corp | Benzene derivative |
| WO2007107550A1 (en) | 2006-03-21 | 2007-09-27 | High Point Pharmaceuticals, Llc | Adamantane derivatives for the treatment of the metabolic syndrome |
| WO2007107470A2 (en) | 2006-03-22 | 2007-09-27 | F. Hoffmann-La Roche Ag | Pyrazoles as 11-beta-hsd-1 |
| WO2007111921A1 (en) | 2006-03-23 | 2007-10-04 | Amgen Inc. | 1-phenylsulfonyl-diaza heterocyclic amide compounds and their uses as modulators of hydroxsteroid dehydrogenases |
| WO2007111864A2 (en) | 2006-03-23 | 2007-10-04 | Merck & Co., Inc. | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use |
| WO2007118963A2 (en) | 2006-03-24 | 2007-10-25 | Genfit | Substituted n-(phenethyl)benzamide derivatives, preparation and uses thereof |
| WO2007118964A1 (en) | 2006-03-24 | 2007-10-25 | Genfit | Substituted n-(phenethyl)benzamide derivatives, preparation and uses thereof |
| WO2007117381A2 (en) | 2006-03-24 | 2007-10-18 | Array Biopharma Inc. | 2 -aminopyridine analogs as glucokinase activators |
| JP2007291075A (en) | 2006-03-27 | 2007-11-08 | Sankyo Co Ltd | New compound sterenin and method for producing the same |
| WO2007126745A2 (en) | 2006-03-28 | 2007-11-08 | Merck & Co., Inc. | Aminotetrahydropyrans as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2007110216A1 (en) | 2006-03-28 | 2007-10-04 | Sanofi-Aventis | Azolopyridin-3-one derivatives as inhibitors of endothelial lipase |
| WO2007112347A1 (en) | 2006-03-28 | 2007-10-04 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2007112914A2 (en) | 2006-03-30 | 2007-10-11 | Novartis Ag | Ceramide kinase modulation |
| WO2007113226A1 (en) | 2006-03-31 | 2007-10-11 | Novartis Ag | Organic compounds |
| WO2007115058A2 (en) | 2006-03-31 | 2007-10-11 | Novartis Ag | Thiadiazolidinone inhibitors of ptpase |
| WO2007114532A1 (en) | 2006-04-03 | 2007-10-11 | Industry-Academic Cooperation Foundation Gyeongsang National University | Novel chalcone derivatives, pharmaceutically acceptable salt, method for preparation and uses thereof |
| WO2007113634A1 (en) | 2006-04-03 | 2007-10-11 | Matrix Laboratories Ltd. | Novel dipeptidyl peptidase iv inhibitors and processes for their preparation and pharmaceutical compositions containing them |
| WO2007112669A1 (en) | 2006-04-05 | 2007-10-11 | Shanghai Hengrui Pharmaceutical Co.Ltd. | Dicyclooctane derivates, preparation processes and medical uses thereof |
| WO2007116229A1 (en) | 2006-04-06 | 2007-10-18 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2007118185A2 (en) | 2006-04-07 | 2007-10-18 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| WO2007115822A1 (en) | 2006-04-07 | 2007-10-18 | Develogen Aktiengesellschaft | Thienopyrimidines having mnkl /mnk2 inhibiting activity for pharmaceutical compositions |
| WO2007115935A1 (en) | 2006-04-07 | 2007-10-18 | High Point Pharmaceuticals, Llc | 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 ACTIVE COMPOUNDS |
| WO2007120575A2 (en) | 2006-04-11 | 2007-10-25 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2007115821A2 (en) | 2006-04-11 | 2007-10-18 | Novartis Ag | Organic compounds |
| WO2007116230A1 (en) | 2006-04-11 | 2007-10-18 | Prosidion Ltd | Azetidine derivatives as g-protein coupled receptor (gpr119 ) agonists |
| WO2007116092A1 (en) | 2006-04-12 | 2007-10-18 | Probiodrug Ag | Enzyme inhibitors |
| WO2007120899A2 (en) | 2006-04-13 | 2007-10-25 | Societe De Conseils De Recherches Et D'applications Scientifiques, S.A.S. | Pharmaceutical compositions of hglp-1, exendin-4 and analogs thereof |
| WO2007119837A1 (en) | 2006-04-14 | 2007-10-25 | Ajinomoto Co., Inc. | Lipase inhibitor |
| WO2007119827A1 (en) | 2006-04-14 | 2007-10-25 | Dic Plastics, Inc. | Locking structure of cover body and container |
| WO2007119887A1 (en) | 2006-04-18 | 2007-10-25 | Nippon Chemiphar Co., Ltd. | ACTIVATING AGENT FOR PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR δ |
| US20070244094A1 (en) | 2006-04-18 | 2007-10-18 | Gee-Hong Kuo | Benzoazepin-oxy-acetic acid derivatives as ppar-delta agonists used for the increase of hdl-c, lower ldl-c and lower cholesterol |
| WO2007120102A1 (en) | 2006-04-19 | 2007-10-25 | Astrazeneca Ab | New substituted oxindole derivatives |
| WO2007122970A1 (en) | 2006-04-20 | 2007-11-01 | Osaka University | Ligand capable of binding to nuclear receptor |
| WO2007122482A1 (en) | 2006-04-20 | 2007-11-01 | Pfizer Products Inc. | Fused phenyl amido heterocyclic compounds for the prevention and treatment of glucokinase-mediated diseases |
| WO2007124461A2 (en) | 2006-04-20 | 2007-11-01 | Amgen Inc. | Glp-1 compounds |
| US20080280875A1 (en) | 2006-04-20 | 2008-11-13 | Pfizer Inc. | Fused phenyl amido heterocyclic compounds |
| WO2007124329A1 (en) | 2006-04-21 | 2007-11-01 | Eli Lilly And Company | Cyclohexylimidazole lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007124337A1 (en) | 2006-04-21 | 2007-11-01 | Eli Lilly And Company | Biphenyl amide lactam derivatives as inhibitors of 11- beta-hydroxysteroid dehydrogenase 1 |
| WO2007124254A2 (en) | 2006-04-21 | 2007-11-01 | Eli Lilly And Company | Cyclohexylpyrazole-lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007122634A2 (en) | 2006-04-24 | 2007-11-01 | Jubilant Biosys Limited | Pyrimidinediones as tyrosine kinase inhibitors |
| WO2007123225A1 (en) | 2006-04-24 | 2007-11-01 | Astellas Pharma Inc | Oxadiazolidinedione compound |
| WO2007127704A1 (en) | 2006-04-24 | 2007-11-08 | Eli Lilly And Company | Cyclohexyl substituted pyrrolidinones as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127688A2 (en) | 2006-04-24 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127693A1 (en) | 2006-04-24 | 2007-11-08 | Eli Lilly And Company | Substituted pyrrolidinones as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127726A2 (en) | 2006-04-25 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127763A2 (en) | 2006-04-25 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007127765A1 (en) | 2006-04-25 | 2007-11-08 | Eli Lilly And Company | Inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007122411A1 (en) | 2006-04-26 | 2007-11-01 | Astrazeneca Ab | Diazepan-1-yl-sulfonyl derivatives for the treatment of metabolic syndrome |
| WO2007126043A1 (en) | 2006-04-27 | 2007-11-08 | Mitsubishi Tanabe Pharma Corporation | Use as drugs of carboxylic acid derivatives having thiazole rings |
| WO2007125105A2 (en) | 2006-04-28 | 2007-11-08 | Transtech Pharma, Inc. | Benzamide glucokinase activators |
| WO2007125110A1 (en) | 2006-04-28 | 2007-11-08 | Noscira, S.A. | N- ( 2-thiazolyl) -amide derivatives as gsk-3 inhibitors |
| WO2007125103A2 (en) | 2006-04-28 | 2007-11-08 | Transtech Pharma, Inc. | Benzamide glucokinase activators |
| WO2007125109A1 (en) | 2006-04-28 | 2007-11-08 | Noscira, S.A. | N- (2-thiazolyl) -amide derivatives as gsk-3 inhibitors |
| WO2007127901A1 (en) | 2006-04-28 | 2007-11-08 | Eli Lilly And Company | Pieridinyl substituted pyrrolidinones as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2007130898A1 (en) | 2006-05-01 | 2007-11-15 | Incyte Corporation | TETRASUBSTITUTED UREAS AS MODULATORS OF 11-β HYDROXYL STEROID DEHYDROGENASE TYPE 1 |
| WO2007126117A1 (en) | 2006-05-02 | 2007-11-08 | Taisho Pharmaceutical Co., Ltd. | Phenyl 5-thioglucoside compound |
| WO2007129668A1 (en) | 2006-05-02 | 2007-11-15 | Taisho Pharmaceutical Co., Ltd. | Pyrazolyl 5-thioglucoside compound |
| WO2007128480A2 (en) | 2006-05-02 | 2007-11-15 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Thioglycosides as pharmaceutically active agents |
| US20070259900A1 (en) | 2006-05-04 | 2007-11-08 | Peter Sieger | Polymorphs |
| EP1852108A1 (en) | 2006-05-04 | 2007-11-07 | Boehringer Ingelheim Pharma GmbH & Co.KG | DPP IV inhibitor formulations |
| WO2007143316A2 (en) | 2006-05-05 | 2007-12-13 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of sglt2 |
| WO2007128801A1 (en) | 2006-05-08 | 2007-11-15 | Novartis Ag | Combination of organic compounds |
| WO2007128817A2 (en) | 2006-05-09 | 2007-11-15 | Novo Nordisk A/S | Insulin derivative |
| WO2007128815A1 (en) | 2006-05-09 | 2007-11-15 | Novo Nordisk A/S | Insulin derivative |
| WO2007131620A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | 4,5-diphenyl-pyrimidinyl-amino substituted carboxylic acids, method for the production and use thereof as medicaments |
| WO2007131621A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | 4,5-diphenyl-pyrimidinyl substituted carboxylic acids, method for the production and use thereof as medicaments |
| WO2007131619A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | 4,5-diphenyl-pyrimidinyl-oxy or -mercapto substituted carboxylic acids, method for the production and use thereof as medicaments |
| WO2007131622A1 (en) | 2006-05-11 | 2007-11-22 | Sanofi-Aventis | Phenylamino-benzoxazole substituted carboxylic acids, method for their production and use thereof as medicaments |
| US20070281949A1 (en) | 2006-05-15 | 2007-12-06 | Cephalon, Inc. | Substituted pyrazolopyrimidines |
| WO2007136572A2 (en) | 2006-05-15 | 2007-11-29 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
| US20070265332A1 (en) | 2006-05-15 | 2007-11-15 | Min Ge | Antidiabetic bicyclic compounds |
| WO2007136577A2 (en) | 2006-05-16 | 2007-11-29 | Merck & Co., Inc. | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use |
| WO2007136603A2 (en) | 2006-05-16 | 2007-11-29 | Merck & Co., Inc. | Aminotetrahydropyrans as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| US20070270424A1 (en) | 2006-05-17 | 2007-11-22 | Yun-Long Li | Heterocyclic inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 and methods of using the same |
| WO2007137008A2 (en) | 2006-05-18 | 2007-11-29 | Wellstat Therapeutics Corporation | Compounds for the treatment of metabolic disorders |
| WO2007136116A2 (en) | 2006-05-19 | 2007-11-29 | Taisho Pharmaceutical Co., Ltd. | C-phenyl glycitol compound for the treatment of diabetes |
| WO2007134986A1 (en) | 2006-05-23 | 2007-11-29 | F. Hoffmann-La Roche Ag | Pyridopyrimidinone derivatives |
| US20070275907A1 (en) | 2006-05-23 | 2007-11-29 | Yuanwei Chen | Glucose transport inhibitors and methods of use |
| WO2007135427A1 (en) | 2006-05-23 | 2007-11-29 | Astrazeneca Ab | 1,4-disubstituted piperazine and 1,4-disubstituted azepane as 11 -beta-hydroxysteroid dehydrogenase 1 inhibitors |
| US20070276041A1 (en) | 2006-05-25 | 2007-11-29 | Ajinomoto, Co., Inc. | Ppar activity regulators |
| WO2007140230A2 (en) | 2006-05-26 | 2007-12-06 | Nestec S.A. | Methods of use and nutritional compositions of touchi extract |
| WO2007139992A2 (en) | 2006-05-26 | 2007-12-06 | Novartis Ag | ALDOSTERONE SYNTHASE AND/OR 11β-HYDROXYLASE INHIBITORS |
| US20070281942A1 (en) | 2006-05-31 | 2007-12-06 | Cao Sheldon X | Glucokinase activators |
| WO2007137962A1 (en) | 2006-06-01 | 2007-12-06 | F. Hoffmann-La Roche Ag | Thiazole derivatives |
| WO2007141423A1 (en) | 2006-06-06 | 2007-12-13 | Negma-Lerads | Ppar enhancing derivatives, method for preparing same and therapeutic use thereof |
| WO2007142253A1 (en) | 2006-06-06 | 2007-12-13 | Mitsubishi Tanabe Pharma Corporation | 2-cyanopyrrolidine derivative |
| US20070287674A1 (en) | 2006-06-08 | 2007-12-13 | Hej Research Institute Of Chemistry | New treatment of diabetes mellitus |
| WO2007145835A2 (en) | 2006-06-08 | 2007-12-21 | Amgen Inc. | Benzamide derivatives as modulators of 11beta-hsd1 for treating diabetes and obesity |
| WO2007145834A2 (en) | 2006-06-08 | 2007-12-21 | Amgen Inc. | Benzamide derivatives and uses related thereto |
| WO2007146761A2 (en) | 2006-06-12 | 2007-12-21 | Neurogen Corporation | Diaryl pyrimidinones and related compounds |
| WO2008035359A2 (en) | 2006-06-12 | 2008-03-27 | Cadila Healthcare Limited | Oximinophenoxyalkanoic acid and phenylalkanoic acid derivatives |
| WO2007144394A2 (en) | 2006-06-16 | 2007-12-21 | High Point Pharmaceuticals, Llc. | Pharmaceutical use of substituted piperidine carboxamides |
| WO2007149865A2 (en) | 2006-06-19 | 2007-12-27 | University Of Utah Research Foundation | Methods and compositions related to inhibition of ceramide synthesis |
| WO2007148185A2 (en) | 2006-06-21 | 2007-12-27 | Pfizer Products Inc. | Substituted 3 -amino- pyrrolidino-4 -lactams as dpp inhibitors |
| WO2007150026A2 (en) | 2006-06-23 | 2007-12-27 | Incyte Corporation | Purinone derivatives as hm74a agonists |
| WO2007150025A2 (en) | 2006-06-23 | 2007-12-27 | Incyte Corporation | Purinone derivatives as hm74a agonists |
| WO2007147478A1 (en) | 2006-06-23 | 2007-12-27 | Merck Patent Gmbh | 3 -amino-imidazo[1, 2-a]pyridine derivatives as sglt inhibitors |
| WO2008000951A2 (en) | 2006-06-27 | 2008-01-03 | Sanofi-Aventis | Urea derivatives of tropane, their preparation and their therapeutic application |
| WO2008002244A2 (en) | 2006-06-27 | 2008-01-03 | Astrazeneca Ab | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| WO2008000950A2 (en) | 2006-06-27 | 2008-01-03 | Sanofi-Aventis | Derivatives of ureas of piperidine or pyrrolidine, their preparation and their therapeutical use |
| WO2008001931A2 (en) | 2006-06-27 | 2008-01-03 | Takeda Pharmaceutical Company Limited | Fused cyclic compounds |
| WO2008002245A2 (en) | 2006-06-27 | 2008-01-03 | Astrazeneca Ab | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| US20080004281A1 (en) | 2006-06-28 | 2008-01-03 | Kalypsys, Inc. | Methods for the modulation of crp by the selective modulation of ppar delta |
| WO2008002824A1 (en) | 2006-06-28 | 2008-01-03 | Bristol-Myers Squibb Company | Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| WO2008001864A1 (en) | 2006-06-29 | 2008-01-03 | Taisho Pharmaceutical Co., Ltd. | C-phenyl 1-thioglucitol compound |
| US20080004325A1 (en) | 2006-06-29 | 2008-01-03 | Wyeth | PTP1B inhibitors |
| WO2008003611A1 (en) | 2006-07-05 | 2008-01-10 | F. Hoffmann-La Roche Ag | Alkyl-pyridazine derivatives as inhibitors of 11 beta hydroxysteroid dehydrogenase type 1(11b-hsd 1) |
| WO2008005569A2 (en) | 2006-07-06 | 2008-01-10 | Arena Pharmaceuticals, Inc. | Modulators of metabolism and the treatment of disorders related thereto |
| WO2008005576A1 (en) | 2006-07-06 | 2008-01-10 | Arena Pharmaceuticals, Inc. | Modulators of metabolism and the treatment of disorders related thereto |
| WO2008005914A2 (en) | 2006-07-06 | 2008-01-10 | Bristol-Myers Squibb Company | Novel glucokinase activators and methods of using same |
| WO2008005910A2 (en) | 2006-07-06 | 2008-01-10 | Bristol-Myers Squibb Company | Pyridone/hydroxypyridine 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2008005964A2 (en) | 2006-07-06 | 2008-01-10 | Bristol-Myers Squibb Company | Phosphonate and phosphinate compounds as glucokinase activators |
| JP2008031161A (en) | 2006-07-06 | 2008-02-14 | Taisho Pharmaceut Co Ltd | Prophylactic or curative agent for diabetes containing 1-thio-d-glucitol derivative as active ingredient |
| WO2008006319A1 (en) | 2006-07-07 | 2008-01-17 | Shanghai Allist Pharmaceutical., Inc. | BIBENZIMIDAZOLE DERIVATIVES HAVING PPARγ AGONIST ACTIVITY AND THEIR USES |
| WO2008006043A2 (en) | 2006-07-07 | 2008-01-10 | Bristol-Myers Squibb Company | 1,3-oxazole derivatives useful as anti-atherosclerotic, anti-dyslipidemic, anti-diabetic and anti-obesity agents |
| WO2008006044A2 (en) | 2006-07-07 | 2008-01-10 | Bristol-Myers Squibb Company | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method |
| WO2008006969A2 (en) | 2006-07-10 | 2008-01-17 | Les Laboratoires Servier | Novel tetracyclic derivatives, process for the preparation thereof and pharmaceutical compositions which contain them |
| WO2008008895A1 (en) | 2006-07-13 | 2008-01-17 | Smithkline Beecham Corporation | Gpr119 agonists for the treatment of diabetes and related disorders |
| WO2008006432A1 (en) | 2006-07-13 | 2008-01-17 | Merck Patent Gmbh | Use of ampk-activating imidazole derivatives, preparation process therefor and pharmaceutical compositions comprising them |
| WO2008006702A1 (en) | 2006-07-13 | 2008-01-17 | High Point Pharmaceuticals, Llc. | 11beta-hydroxysteroid dehydrogenase type 1 active compounds |
| WO2008006703A1 (en) | 2006-07-13 | 2008-01-17 | High Point Pharmaceuticals, Llc | 4-piperidylbenzamides as 11-beta-hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2008008887A2 (en) | 2006-07-13 | 2008-01-17 | Smithkline Beecham Corporation | Gpr119 agonists for treating metabolic disorders |
| WO2008007692A1 (en) | 2006-07-14 | 2008-01-17 | Panasonic Corporation | Method for pretreating electrochemical capacitor negative electrode, method for manufacturing the electrochemical capacitor negative electrode, and method for manufacturing electrochemical capacitor using the method for manufacturing the electrochemical capacitor negative electrode |
| WO2008008547A2 (en) | 2006-07-14 | 2008-01-17 | Ams Research Corporation | Balloon dilation for implantable prosthesis |
| WO2008009335A2 (en) | 2006-07-18 | 2008-01-24 | Merck Patent Gmbh | Aminoindazole urea derivatives |
| WO2008011453A2 (en) | 2006-07-20 | 2008-01-24 | Amgen Inc. | SUBSTITUTED AZOLE AROMATIC HETEROCYCLES AS INHIBITORS OF LLβ-HSD-1 |
| WO2008010238A2 (en) | 2006-07-21 | 2008-01-24 | Lupin Limited | Antidiabetic azabicyclo [3. 1. 0] hexan compounds |
| WO2008012470A2 (en) | 2006-07-24 | 2008-01-31 | Genfit | Substituted imidazolone derivatives, preparation and uses |
| WO2008013322A1 (en) | 2006-07-27 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | 1- (-d-glycopyranosyl) - 3 - (4-cyclopropylphenylmethyl) - 4 - halogeno indole derivatives and use thereof as sglt inhibitors. |
| WO2008013280A1 (en) | 2006-07-27 | 2008-01-31 | Chugai Seiyaku Kabushiki Kaisha | Substituted spiroketal derivative and use thereof as drug for treating diabetes |
| WO2008012532A2 (en) | 2006-07-27 | 2008-01-31 | Astrazeneca Ab | : pyridine-3-carboxamide compounds and their use for inhibiting 11-beta-hydroxysteroid dehydrogenase |
| WO2008013277A1 (en) | 2006-07-27 | 2008-01-31 | Chugai Seiyaku Kabushiki Kaisha | Fused ring spiroketal derivative and use thereof as drug for treating diabetes |
| JP2008031064A (en) | 2006-07-27 | 2008-02-14 | Astellas Pharma Inc | Diacylpiperazine derivative |
| WO2008013321A1 (en) | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
| WO2008016730A2 (en) | 2006-08-02 | 2008-02-07 | Targeted Molecular Diagnostics, Llc | Compositions and methods for reducing cellular fat |
| WO2008016123A1 (en) | 2006-08-03 | 2008-02-07 | Takeda Pharmaceutical Company Limited | GSK-3β INHIBITOR |
| WO2008016968A2 (en) | 2006-08-03 | 2008-02-07 | Trustees Of Tufts College | Non-flushing niacin analogues, and methods of use thereof |
| WO2008016175A1 (en) | 2006-08-03 | 2008-02-07 | Nippon Chemiphar Co., Ltd. | Activator for peroxisome proliferator activated receptor |
| WO2008019309A1 (en) | 2006-08-04 | 2008-02-14 | Metabasis Therapeutics, Inc. | Novel inhibitors of fructose 1,6-bisphosphatase |
| WO2008016278A1 (en) | 2006-08-04 | 2008-02-07 | Korea Research Institute Of Chemical Technology | Furan-2-carboxylic acid derivatives and process for the preparation thereof |
| WO2008016132A1 (en) | 2006-08-04 | 2008-02-07 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
| WO2008017398A2 (en) | 2006-08-07 | 2008-02-14 | Krka | Polymorphic forms of rosiglitazone base |
| WO2008017670A1 (en) | 2006-08-08 | 2008-02-14 | Boehringer Ingelheim International Gmbh | Pyrrolo [3, 2 -d] pyrimidines as dpp-iv inhibitors for the treatment of diabetes mellitus |
| WO2008022015A2 (en) | 2006-08-11 | 2008-02-21 | Trustees Of Tufts College | Retro-inverso incretin analogues, and methods of use thereof |
| WO2008020011A1 (en) | 2006-08-15 | 2008-02-21 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted cyclopropylbenzene derivatives, pharmaceutical compositions containing such compounds, their use as sglt inhibitors and process for their manufacture |
| WO2008021560A2 (en) | 2006-08-17 | 2008-02-21 | Amylin Pharmaceuticals, Inc. | Dpp-iv resistant gip hybrid polypeptides with selectable properties |
| WO2008020607A1 (en) | 2006-08-17 | 2008-02-21 | Dainippon Sumitomo Pharma Co., Ltd. | Phthalide derivative and salt thereof |
| WO2008023239A1 (en) | 2006-08-23 | 2008-02-28 | Pfizer Products Inc. | Pyrimidone compounds as gsk-3 inhibitors |
| WO2008024892A2 (en) | 2006-08-24 | 2008-02-28 | Bristol-Myers Squibb Company | Cyclic 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2008024497A2 (en) | 2006-08-25 | 2008-02-28 | Vitae Pharmaceuticals, Inc. | INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE TYPE 1 |
| WO2008029217A2 (en) | 2006-08-29 | 2008-03-13 | Orchid Research Laboratories Limited | Dipeptidyl peptidase iv inhibitors |
| WO2008025800A1 (en) | 2006-08-30 | 2008-03-06 | Biovitrum Ab (Publ) | Pyrimidine compounds for treating gpr119 related disorders |
| WO2008027273A2 (en) | 2006-08-30 | 2008-03-06 | Phenomix Corporation | Solid citrate and tartrate salts of dpp-iv inhibitors |
| WO2008025798A1 (en) | 2006-08-30 | 2008-03-06 | Biovitrum Ab (Publ) | Pyridine compounds for treating gpr119 related disorders |
| WO2008025799A1 (en) | 2006-08-30 | 2008-03-06 | Biovitrum Ab (Publ) | Pyridazine compounds for treating gpr119 related disorders |
| WO2008028188A2 (en) | 2006-09-01 | 2008-03-06 | The Ticket Reserve | Demand aggregation for future items contingent upon threshold demand |
| WO2008027584A2 (en) | 2006-09-01 | 2008-03-06 | Vertex Pharmaceuticals Incorporated | 5- (2-furyl)-1, 3-thiazole derivatives useful as inhibitors of phosphatidylinositol 3-kinase |
| JP2008063256A (en) | 2006-09-06 | 2008-03-21 | Astellas Pharma Inc | beta-AMINO ACID DERIVATIVE |
| WO2008030520A1 (en) | 2006-09-07 | 2008-03-13 | Amgen Inc. | Heterocyclic gpr40 modulators |
| WO2008028662A1 (en) | 2006-09-07 | 2008-03-13 | Santhera Pharmaceuticals (Schweiz) Ag | N-[1-(3-amino-4-phenyl-butyryl)-4-hydroxy-pyrrolidin-2-ylmethyl}-propionamide and related compounds as dpp-iv inhibitors for the treatment of type 2 diabetes mellitus |
| WO2008030618A1 (en) | 2006-09-07 | 2008-03-13 | Amgen Inc. | Benzo-fused compounds for use in treating metabolic disorders |
| WO2008032164A2 (en) | 2006-09-12 | 2008-03-20 | Pfizer Products Inc. | Benzimidazolone derivatives |
| WO2008033932A2 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Biarylthiazole carboxylic acid derivatives as protein tyrosine phosphatase-ib inhibitors |
| WO2008033931A1 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Para-xylylene carboxylic acids and isothiazolones useful as protein tyrosine phosphatases (ptps) in particular ptp-ib |
| WO2008033934A1 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Substituted heteroaryl carboxylic acid derivatives as ptb-1b inhibitors |
| WO2008033455A2 (en) | 2006-09-13 | 2008-03-20 | The Institutes For Pharmaceutical Discovery, Llc | Biphenyl and heteroaryl phenyl derivatives as protein tyrosine phosphatases inhibitors |
| WO2008033851A2 (en) | 2006-09-13 | 2008-03-20 | Takeda Pharmaceutical Company Limited | Use of 2-6- (3-amino-piperidin-l-yl) -3-methyl-2, 4-dioxo-3, 4-dihydr0-2h-pyrimidin-1-ylmet hyl-4-fluoro-benzonitrile |
| WO2008034032A2 (en) | 2006-09-14 | 2008-03-20 | Azevan Pharmaceuticals, Inc. | Beta-lactam cannabinoid receptor modulators |
| WO2008034859A1 (en) | 2006-09-21 | 2008-03-27 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted difluorobenzyl-benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008034881A1 (en) | 2006-09-22 | 2008-03-27 | Novo Nordisk A/S | Protease resistant insulin analogues |
| WO2008037628A1 (en) | 2006-09-29 | 2008-04-03 | F. Hoffmann-La Roche Ag | Sulfonamide derivatives |
| WO2008042688A2 (en) | 2006-09-29 | 2008-04-10 | Lexicon Pharmaceuticals, Inc. | Phlorizin analogs as inhibitors of sodium glucose co-transporter 2 |
| WO2008039882A1 (en) | 2006-09-30 | 2008-04-03 | Sanofi-Aventis U.S. Llc | A combination of niacin and a prostaglandin d2 receptor antagonist |
| WO2008042223A1 (en) | 2006-10-03 | 2008-04-10 | Merck & Co., Inc. | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use |
| WO2008062457A2 (en) | 2006-10-03 | 2008-05-29 | Cadila Healthcare Limited | Antidiabetic compounds |
| WO2008044777A1 (en) | 2006-10-06 | 2008-04-17 | Banyu Pharmaceutical Co., Ltd. | 2-pyridinecarboxamide derivative having gk-activating activity |
| WO2008044656A1 (en) | 2006-10-06 | 2008-04-17 | Taisho Pharmaceutical Co., Ltd. | Imidazolidinone derivative |
| WO2008040995A1 (en) | 2006-10-07 | 2008-04-10 | Peakdale Molecular Limited | Indazoles for use as dpp-iv inhibitors |
| WO2008040974A1 (en) | 2006-10-07 | 2008-04-10 | Peakdale Molecular Limited | Indoles for use as dpp-iv inhibitors |
| WO2008045484A1 (en) | 2006-10-10 | 2008-04-17 | Amgen Inc. | N-aryl pyrazole compounds for use against diabetes |
| WO2008044700A1 (en) | 2006-10-11 | 2008-04-17 | Takeda Pharmaceutical Company Limited | GSK-3β INHIBITOR |
| WO2008043544A1 (en) | 2006-10-11 | 2008-04-17 | Laboratorios Del Dr. Esteve, S.A. | Sulfonamide substituted pyrazoline compounds, their preparation and use as cb1 modulators |
| WO2008043701A1 (en) | 2006-10-13 | 2008-04-17 | F. Hoffmann-La Roche Ag | Pharmaceutical solid dosage forms comprising compounds micro-embedded in ionic water-insoluble polymers |
| WO2008044762A1 (en) | 2006-10-13 | 2008-04-17 | Chugai Seiyaku Kabushiki Kaisha | Thioglucose spiroketal derivative and use thereof as therapeutic agent for diabetes |
| WO2008048866A2 (en) | 2006-10-13 | 2008-04-24 | Glaxo Group Limited | Bicyclic heteroaromatic compounds |
| WO2008048886A2 (en) | 2006-10-16 | 2008-04-24 | Vidyo, Inc. | Systems and methods for signaling and performing temporal level switching in scalable video coding |
| WO2008046497A1 (en) | 2006-10-16 | 2008-04-24 | Merck Patent Gmbh | 3 -amino-imidazo [1,2 -a] pyridine derivatives having an sglt1 and sglt2 inhibiting action, for treating type 1 and type 2 diabetes |
| WO2008047821A1 (en) | 2006-10-18 | 2008-04-24 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| WO2008046758A2 (en) | 2006-10-19 | 2008-04-24 | F. Hoffmann-La Roche Ag | Imidazolone and imidazolidinone derivatives as 11b-hsd1 inhibitors for diabetes |
| US20080096877A1 (en) | 2006-10-19 | 2008-04-24 | Tsuneo Yasuma | Indole compound |
| WO2008051403A2 (en) | 2006-10-20 | 2008-05-02 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2008050101A2 (en) | 2006-10-23 | 2008-05-02 | Astrazeneca Ab | Benzoyl amino heterocyclyl compounds useful in the treatment of a disease mediated through glk |
| WO2008050987A1 (en) | 2006-10-23 | 2008-05-02 | Hanall Pharmaceutical Co., Ltd. | Combination formulation with controlled release comprising metformin and glimepiride |
| WO2008050117A1 (en) | 2006-10-26 | 2008-05-02 | Astrazeneca Ab | Benzoyl amino heterocyclyl compounds as glucokinase (glk) activators |
| US20080103201A1 (en) | 2006-10-26 | 2008-05-01 | Wijayabandara Mirihanage Don J | Novel alpha-Glucosidase inhibitor from Tabernaemontana dichotoma |
| WO2008049711A1 (en) | 2006-10-27 | 2008-05-02 | Novo Nordisk A/S | Peptide extended insulins |
| WO2008049923A1 (en) | 2006-10-27 | 2008-05-02 | Boehringer Ingelheim International Gmbh | CRYSTALLINE FORM OF 4-(ß-D-GLUCOPYRANOS-1-YL)-1-METHYL-2-[4-((S)-TETRAHYDROFURAN-3-YLOXY)-BENZYL]-BENZENE, A METHOD FOR ITS PREPARATION AND THE USE THEREOF FOR PREPARING MEDICAMENTS |
| WO2008054675A2 (en) | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
| WO2008054674A2 (en) | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
| WO2008057856A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | Modulators of glucocorticoid receptor, ap-1 and/or nf-kappab activity and use thereof |
| WO2008053331A1 (en) | 2006-11-01 | 2008-05-08 | Pronova Biopharma Norge A/S | Alpha-substituted omega-3 lipids that are activators or modulators of the peroxisome proliferators-activated receptor (ppar). |
| WO2008057859A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | Modulators of glucocorticoid receptor, ap-i and/or nf-kappab activity and use thereof |
| WO2008057857A1 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | MODULATORS OF GLUCOCORTICOID RECEPTOR, AP-1, AND/OR NF-ϰB ACTIVITY AND USE THEREOF |
| WO2008057855A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | Heterocyclic compounds as modulators of glucocorticoid receptor, ap-i, and/or np-kappa-b activity |
| WO2008057862A2 (en) | 2006-11-01 | 2008-05-15 | Bristol-Myers Squibb Company | MODULATORS OF GLUCOCORTICOID RECEPTOR, AP-1, AND/OR NF-ϰB ACTIVITY AND USE THEREOF |
| WO2008053446A2 (en) | 2006-11-02 | 2008-05-08 | Piramal Life Sciences Limited | Benzoxazepine compounds, their preparation and use |
| WO2008057940A1 (en) | 2006-11-02 | 2008-05-15 | Vertex Pharmaceuticals Incorporated | Aminopyridines and aminopyrimidines useful as inhibitors of protein kinases |
| WO2008053194A2 (en) | 2006-11-03 | 2008-05-08 | Astrazeneca Ab | Pyridine carboxamides as 11-beta-hsd1 inhibitors |
| WO2008052638A1 (en) | 2006-11-03 | 2008-05-08 | Merk Patent Gmbh | DIAZEPANE-ACETAMIDE DERIVATIVES AS SELECTIVE 11β-HSD1 INHIBITORS |
| WO2008055870A1 (en) | 2006-11-06 | 2008-05-15 | Boehringer Ingelheim International Gmbh | Glucopyranosyl-substituted benzyl-benzonitrile derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008056266A2 (en) | 2006-11-07 | 2008-05-15 | Sanofi-Aventis | Substituted 8-piperidinyl-2-pyridinyl-pyrimido [1,2-a]pyrimidin-6-one and 8-piperidinyl-2-pyrimidinyl-pyrimido[1,2-a]pyrimidin-6-one derivatives |
| WO2008056726A1 (en) | 2006-11-09 | 2008-05-15 | Sanwa Kagaku Kenkyusho Co., Ltd. | Glp-1 derivative and use thereof |
| WO2008055940A2 (en) | 2006-11-09 | 2008-05-15 | Boehringer Ingelheim International Gmbh | Combination therapy with sglt-2 inhibitors and their pharmaceutical compositions |
| WO2008059866A1 (en) | 2006-11-14 | 2008-05-22 | Santen Pharmaceutical Co., Ltd. | Novel 1,2-dihydroquinoline derivative having substituted phenylamino lower alkyl group and ester-introduced phenyl group as substituents |
| WO2008059867A1 (en) | 2006-11-14 | 2008-05-22 | Santen Pharmaceutical Co., Ltd. | Novel 1,2-dihydroquinoline derivative having (substituted phenyl or substituted heterocyclic) carbonyloxy lower alkyl group and ester-introduced phenyl group as substituents |
| WO2008060488A1 (en) | 2006-11-14 | 2008-05-22 | Merck & Co., Inc. | Tricyclic heteroaromatic compounds as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2008059865A1 (en) | 2006-11-14 | 2008-05-22 | Santen Pharmaceutical Co., Ltd. | Novel 1,2-dihydroquinoline derivative having substituted phenylchalcogeno lower alkyl group and ester-introduced phenyl group as substituents |
| US20080176912A1 (en) | 2006-11-15 | 2008-07-24 | Gee-Hong Kuo | Gpr40 agonists |
| WO2008059625A1 (en) | 2006-11-17 | 2008-05-22 | National University Corporation Kagawa University | Utilization of the function of rare sugar as promoter for the migration of glucokinase from nucleus to cytoplasm |
| WO2008064107A2 (en) | 2006-11-20 | 2008-05-29 | Bristol-Myers Squibb Company | 7,8-dihydro-1,6-naphthyridin-5(6h)-ones and related bicyclic compounds as inhibitors of dipeptidyl peptidase iv and methods |
| WO2008062739A1 (en) | 2006-11-20 | 2008-05-29 | Japan Tobacco Inc. | Pyrazoles and use thereof as drugs |
| WO2008062273A2 (en) | 2006-11-20 | 2008-05-29 | Cadila Pharmaceuticals Limited | Solid oral dosage form having antidiabetic drug combination |
| WO2008061355A1 (en) | 2006-11-24 | 2008-05-29 | Matregen Corp. | Glp-1 depot systems, and methods of manufacture and uses thereof |
| WO2008066070A1 (en) | 2006-11-29 | 2008-06-05 | Uha Mikakuto Co., Ltd. | Dipeptidyl peptidase-iv inhibitor |
| WO2008065796A1 (en) | 2006-11-30 | 2008-06-05 | Fuji-Sangyo Co., Ltd. | α-GLUCOSIDASE INHIBITOR |
| WO2008066097A1 (en) | 2006-12-01 | 2008-06-05 | Astellas Pharma Inc. | Carboxylic acid derivative |
| WO2008066131A1 (en) | 2006-12-01 | 2008-06-05 | Banyu Pharmaceutical Co., Ltd. | Novel phenyl-isoxazol-3-ol derivative |
| WO2008066356A1 (en) | 2006-12-02 | 2008-06-05 | Seoul National University Industry Foundation | Aryl compounds as ppar ligands and their use |
| WO2008070609A1 (en) | 2006-12-04 | 2008-06-12 | Janssen Pharmaceutica N.V. | Thienyl-containing glycopyranosyl derivatives as antidiabetics |
| WO2008069327A1 (en) | 2006-12-04 | 2008-06-12 | Mitsubishi Tanabe Pharma Corporation | CRYSTALLINE FORM OF 1- (β-D-GLUCOPYRANOSYL) -4 -METHYL- 3- [5- (4 -FLUOROPHENYL) -2-THIENYLMETHYL] BENZENE HEMIHYDRATE |
| WO2008070150A1 (en) | 2006-12-05 | 2008-06-12 | Bayer Schering Pharma Aktiengesellschaft | Substituted 2,3-dihydroimidazo[1,2-c]quinazoline derivatives useful for treating hyper-proliferative disorders and diseases associated with angiogenesis |
| WO2008070692A2 (en) | 2006-12-06 | 2008-06-12 | Smithkline Beecham Corporation | Bicyclic compounds and use as antidiabetics |
| WO2008070507A2 (en) | 2006-12-06 | 2008-06-12 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2008071288A1 (en) | 2006-12-11 | 2008-06-19 | Merck Patent Gmbh | Indolizine derivatives and their use as antidiabetics |
| WO2008071169A2 (en) | 2006-12-11 | 2008-06-19 | Universitätsklinikum Schleswig-Holstein | Method for the production of specific inhibitors of 11-beta-hydroxysteroid dehydrogenase, in particular type 1 with basic nor-oleanan or nor-ursan frameworks |
| WO2008071311A1 (en) | 2006-12-14 | 2008-06-19 | Sanofi-Aventis | Sulfonyl-phenyl-2h-[1,2,4]oxodiazol-5-one derivatives, processes for their preparation and their use as pharmaceuticals |
| US20080146625A1 (en) | 2006-12-14 | 2008-06-19 | Steven Joseph Berthel | Oxime glucokinase activators |
| WO2008072726A1 (en) | 2006-12-14 | 2008-06-19 | Taisho Pharmaceutical Co., Ltd. | 1-phenyl 1-thio-d-glucitol derivative |
| WO2008076243A2 (en) | 2006-12-14 | 2008-06-26 | Merck & Co., Inc. | Acyl bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| WO2008076862A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | 1-substituted imidazole derivatives and their use as aldosterone synthase inhibitors |
| WO2008076336A2 (en) | 2006-12-18 | 2008-06-26 | Novartis Ag | Imidazoles as aldosterone synthase inhibitors |
| WO2008077138A1 (en) | 2006-12-19 | 2008-06-26 | The Board Of Trustees Of The University Of Illinois | 3-benzofuranyl-4-indolyl maleimides as potent gsk3 inhibitors for neurogenerative disorders |
| WO2008079787A2 (en) | 2006-12-20 | 2008-07-03 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2008085316A1 (en) | 2006-12-20 | 2008-07-17 | Merck & Co., Inc. | Bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| WO2008075741A1 (en) | 2006-12-20 | 2008-06-26 | Keio University | Therapeutic agent and prophylactic agent for diabetes |
| WO2008078196A2 (en) | 2006-12-20 | 2008-07-03 | Sanofi-Aventis | Substituted heteroaryl pyridopyrimidone derivatives |
| WO2008075073A1 (en) | 2006-12-21 | 2008-06-26 | Astrazeneca Ab | Novel crystalline compound useful as glk activator |
| WO2008084303A1 (en) | 2006-12-21 | 2008-07-17 | Pfizer Products Inc. | Compounds having both angiotensin ii receptor antagonism and ppary activating activities |
| WO2008074384A1 (en) | 2006-12-21 | 2008-06-26 | Merck Patent Gmbh | 2-ADAMANTYL-BUTYRAMIDE DERIVATIVES AS SELECTIVE 11βETA-HSD1 INHIBITORS |
| WO2008077597A1 (en) | 2006-12-22 | 2008-07-03 | Novartis Ag | 1-aminomethyl- l- phenyl- cyclohexane derivatives as ddp-iv inhibitors |
| WO2008078674A1 (en) | 2006-12-25 | 2008-07-03 | Kyorin Pharmaceutical Co., Ltd. | Glucokinase-activating substance |
| JP2008156318A (en) | 2006-12-26 | 2008-07-10 | Dainippon Sumitomo Pharma Co Ltd | 1,3,4-oxadiazol-2-one derivative |
| WO2008078725A1 (en) | 2006-12-26 | 2008-07-03 | Daiichi Sankyo Company, Limited | Thiazepine derivative |
| WO2008088540A2 (en) | 2006-12-26 | 2008-07-24 | Amgen Inc. | N-cyclohexyl benzamides and benzeneacetamides as inhibitors of 11-beta-hydroxysteroid dehydrogenases |
| EP1939192A1 (en) | 2006-12-28 | 2008-07-02 | Neuropharma S.A. | Cyclopentanone derivatives, method of synthesis and uses thereof |
| WO2008083124A1 (en) | 2006-12-28 | 2008-07-10 | Rigel Pharmaceuticals, Inc. | N-substituted-heterocycloalkyloxybenzamide compounds and methods of use |
| WO2008083200A2 (en) | 2006-12-28 | 2008-07-10 | Theracos, Inc. | Spiroheterocyclic glycosides and methods of use |
| WO2008083238A2 (en) | 2006-12-28 | 2008-07-10 | Metabolex Inc. | Heterocyclic receptor agonists for the treatment of diabetes and metabolic disorders |
| EP1939191A1 (en) | 2006-12-28 | 2008-07-02 | Neuropharma S.A. | Furan derivatives, method of synthesis and uses thereof |
| WO2008087366A2 (en) | 2006-12-29 | 2008-07-24 | Genfit | Substituted 3-phenyl-1-(phenylthienyl)propan-1-one and 3-phenyl-1-(phenylfuranyl)propan-1-one derivatives, and preparation and use of same |
| WO2008087365A2 (en) | 2006-12-29 | 2008-07-24 | Genfit | Substituted 1, 3-diphenylpropane derivatives, preparations and uses of same |
| WO2008087367A2 (en) | 2006-12-29 | 2008-07-24 | Genfit | Substituted (phenylthiazolyl)-phenyl-propan-1-one and (phenyloxazodyl)-phenyl-propan-1-one derivatives, preparations and uses of same |
| WO2008082017A1 (en) | 2007-01-03 | 2008-07-10 | Takano Co., Ltd. | Cyclic sulfonium salt, method for production of cyclic sulfonium salt, and glycosidase inhibitor |
| WO2008081207A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081205A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081206A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081204A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081208A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008082274A1 (en) | 2007-01-05 | 2008-07-10 | Hanmi Pharmaceutical Co., Ltd. | An insulinotropic complex using an immunoglobulin fragment |
| WO2008081418A1 (en) | 2007-01-05 | 2008-07-10 | Covx Technologies Ireland Limited | Glucagon-like protein-1 receptor (glp-1r) agonist compounds |
| WO2008084962A1 (en) | 2007-01-08 | 2008-07-17 | Seoul National University Industry Foundation | THIAZOLE COMPOUND (AS PPARδ) LIGAND AND PHARMACEUTICAL, COSMETIC AND HEALTH FOOD COMPRISED THEREOF |
| WO2008084043A1 (en) | 2007-01-09 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008084873A1 (en) | 2007-01-10 | 2008-07-17 | Mitsubishi Tanabe Pharma Corporation | Oxime derivative |
| WO2008084872A1 (en) | 2007-01-10 | 2008-07-17 | Mitsubishi Tanabe Pharma Corporation | Hydrazone derivative |
| JP2008189659A (en) | 2007-01-10 | 2008-08-21 | Mitsubishi Tanabe Pharma Corp | Medical composition |
| WO2008084044A1 (en) | 2007-01-11 | 2008-07-17 | Novo Nordisk A/S | Urea glucokinase activators |
| WO2008086949A1 (en) | 2007-01-16 | 2008-07-24 | Sanofi-Aventis | Use of substituted pyranone acid derivatives for the treatment of metabolic syndrome |
| WO2008087560A2 (en) | 2007-01-16 | 2008-07-24 | Kainos Medicine, Inc., | Thiazolidine derivatives and methods for the preparation thereof |
| WO2008087654A2 (en) | 2007-01-16 | 2008-07-24 | Cadila Healthcare Limited | PIPERIDINES AS INHIBITORS OF 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 |
| WO2008086854A1 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | 5-([1,3,4] oxadiazol-2-yl)-1h-indazol and 5-([1,3,4] thiadiazol-2-yl)-1h-indazol derivatives as sgk inhibitors for the treatment of diabetes |
| WO2008089461A1 (en) | 2007-01-18 | 2008-07-24 | Evolva Sa | Substituted 1,3-dioxanes useful as ppar modulators |
| WO2008089464A1 (en) | 2007-01-18 | 2008-07-24 | Evolva Sa | Prodrugs of substituted 1,3-dioxanes and their uses |
| WO2008088006A1 (en) | 2007-01-19 | 2008-07-24 | Shinji Yokoyama | Ap2 inhibitor |
| US20080176861A1 (en) | 2007-01-23 | 2008-07-24 | Kalypsys, Inc. | Sulfonyl-substituted bicyclic compounds as ppar modulators for the treatment of non-alcoholic steatohepatitis |
| WO2008089636A1 (en) | 2007-01-23 | 2008-07-31 | Shanghai Hengrui Pharmaceutical Co.Ltd. | Derivatives of azabicyclo octane, the method of making them and the uses thereof as inhibitors of dipeptidyl peptidase iv |
| WO2008091338A1 (en) | 2007-01-23 | 2008-07-31 | Reddy Us Therapeutics, Inc. | Methods and compositions for the treatment of insulin resistance, diabetes, and diabetes-associated dyslipidemia |
| WO2008091770A1 (en) | 2007-01-24 | 2008-07-31 | Array Biopharma Inc. | 2-aminopyridine derivatives as glucokinase activators |
| WO2008090209A2 (en) | 2007-01-26 | 2008-07-31 | Boehringer Ingelheim International Gmbh | Use of glucopyranosyloxy- pyrazoles for preventing and treating neurodegenerative disorders |
| WO2008089892A1 (en) | 2007-01-26 | 2008-07-31 | Sanofi-Aventis | Phenothiazin derivates, method for the production thereof and use thereof as pharmaceuticals |
| WO2008090210A1 (en) | 2007-01-26 | 2008-07-31 | Boehringer Ingelheim International Gmbh | Methods for preventing and treating neurodegenerative disorders |
| WO2008089581A1 (en) | 2007-01-26 | 2008-07-31 | Merck Frosst Canada Ltd. | Fused aromatic ptp-1b inhibitors |
| WO2008093639A1 (en) | 2007-01-29 | 2008-08-07 | Takeda Pharmaceutical Company Limited | Pyrazole compound |
| WO2008093960A1 (en) | 2007-01-30 | 2008-08-07 | Lg Life Sciences, Ltd. | Novel dipeptidyl peptidase-iv inhibitors |
| WO2008094992A2 (en) | 2007-01-31 | 2008-08-07 | Vertex Pharmaceuticals Incorporated | 2-aminopyridine derivatives useful as kinase inhibitors |
| WO2008096820A1 (en) | 2007-02-07 | 2008-08-14 | Kyowa Hakko Kirin Co., Ltd. | Biphenyl derivative |
| WO2008096260A1 (en) | 2007-02-07 | 2008-08-14 | Pfizer Inc. | 3-amino-pyrrolo[3,4-c] pyrazole- 5 (1h, 4h, 6h) carbaldehyde derivatives as pkc inhibitors |
| WO2008096829A1 (en) | 2007-02-07 | 2008-08-14 | Kyowa Hakko Kirin Co., Ltd. | Tricyclic compounds |
| WO2008097535A2 (en) | 2007-02-08 | 2008-08-14 | Merck & Co., Inc. | Method of treating atherosclerosis, dyslipidemias and related conditions |
| WO2008096769A1 (en) | 2007-02-08 | 2008-08-14 | Daiichi Sankyo Company, Limited | Pharmaceutical compositions containing substituted cercosporamide derivatives |
| WO2008099944A1 (en) | 2007-02-08 | 2008-08-21 | Daiichi Sankyo Company, Limited | Crystalline forms of thiazolidinedione derivative and its manufacturing method |
| WO2008096841A1 (en) | 2007-02-09 | 2008-08-14 | Kyorin Pharmaceutical Co., Ltd. | Dimerized cyclo derivative |
| WO2008099448A1 (en) | 2007-02-09 | 2008-08-21 | Masayoshi Yamaguchi | Preventive/therapeutic agent for diabetic diseases |
| US20080194617A1 (en) | 2007-02-09 | 2008-08-14 | Taisuke Tawaraishi | Fused ring compound |
| WO2008098244A1 (en) | 2007-02-09 | 2008-08-14 | Metabasis Therapeutics, Inc. | Novel antagonists of the glucagon receptor |
| WO2008099145A1 (en) | 2007-02-12 | 2008-08-21 | Astrazeneca Ab | Pyrazole derivatives as 11-beta-hsd1 inhibitors |
| WO2008101017A2 (en) | 2007-02-15 | 2008-08-21 | Indiana Unversity Research And Technology Corporation | Glucagon/glp-1 receptor co-agonists |
| WO2008099000A2 (en) | 2007-02-16 | 2008-08-21 | Boehringer Ingelheim International Gmbh | Substituted arylsulphonylglycines, the preparation thereof and the use thereof as pharmaceutical compositions |
| WO2008101939A1 (en) | 2007-02-21 | 2008-08-28 | Boehringer Ingelheim International Gmbh | Tetrasubstituted glucopyranosylated benzene derivatives, medicaments containing such compounds, their use and process for their manufacture |
| WO2008101586A1 (en) | 2007-02-21 | 2008-08-28 | Merck Patent Gmbh | Benzimidazole derivatives |
| WO2008103501A1 (en) | 2007-02-22 | 2008-08-28 | Irm Llc | Compounds and methods for modulating g protein-coupled receptors |
| WO2008101953A1 (en) | 2007-02-22 | 2008-08-28 | Novartis Ag | Compounds of formula (i) as serine protease inhibitors |
| WO2008103500A1 (en) | 2007-02-22 | 2008-08-28 | Irm Llc | Thiazole derivatives as modulators of g protein-coupled receptors |
| WO2008101886A1 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008101885A1 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008101914A2 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008101907A2 (en) | 2007-02-23 | 2008-08-28 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| WO2008106128A2 (en) | 2007-02-26 | 2008-09-04 | Vitae Pharmaceuticals, Inc. | CYCLIC UREA AND CARBAMATE INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2008104306A2 (en) | 2007-02-27 | 2008-09-04 | Bayer Schering Pharma Aktiengesellschaft | Substituted 4-aryl-1,4-dihydro-1,6-naphthyridinamides and use thereof |
| WO2008104994A2 (en) | 2007-02-28 | 2008-09-04 | Advinus Therapeutics Private Limited | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2008109334A1 (en) | 2007-03-02 | 2008-09-12 | Merck & Co., Inc. | Novel crystalline salt form of an antidiabetic compound |
| WO2008108602A1 (en) | 2007-03-07 | 2008-09-12 | Dong-A Pharm. Co., Ltd. | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same |
| WO2008111473A1 (en) | 2007-03-07 | 2008-09-18 | Kyorin Pharmaceutical Co., Ltd. | Glucokinase activator |
| WO2008109700A1 (en) | 2007-03-08 | 2008-09-12 | Plexxikon, Inc. | Ppar active compounds |
| WO2008108735A1 (en) | 2007-03-08 | 2008-09-12 | Albireo Ab | 2 -substituted- 3 -phenylpropionic acid derivatives and their use in the treatment of inflammatory bowel disease |
| WO2008109702A1 (en) | 2007-03-08 | 2008-09-12 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2008109697A2 (en) | 2007-03-08 | 2008-09-12 | Plexxikon, Inc. | Ppar active compounds |
| WO2008112642A1 (en) | 2007-03-09 | 2008-09-18 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as inhibitors of protein kinases |
| WO2008110062A1 (en) | 2007-03-09 | 2008-09-18 | Shanghai Allist Pharmaceuticals, Inc. | Compounds with partial agonist activity of pparϝ and application thereof |
| WO2008110196A1 (en) | 2007-03-09 | 2008-09-18 | High Point Pharmaceuticals, Llc | Indole- and benzimidazole amides as hydroxysteroid dehydrogenase inhibitors |
| WO2008112651A2 (en) | 2007-03-09 | 2008-09-18 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as inhibitors of protein kinases |
| WO2008112939A2 (en) | 2007-03-13 | 2008-09-18 | Board Of Regents, The University Of Texas System | Composition and method for making oligo-benzamide compounds |
| WO2008112941A1 (en) | 2007-03-13 | 2008-09-18 | Board Of Regents The University Of Texas System | Composition and method for the treatment of diseases affected by a peptide receptor |
| WO2008113760A2 (en) | 2007-03-16 | 2008-09-25 | Boehringer Ingelheim International Gmbh | Arylsulphonyglycine derivatives as suppressors of the interaction of glycogen phosphorylase a with the gl subunit of glycogen-associated protein phosphatase 1 (ppl) for the treatment of metabolic disorders, particulary diabetes |
| WO2008113469A2 (en) | 2007-03-16 | 2008-09-25 | Bayer Schering Pharma Aktiengesellschaft | Substituted imidazopyrimidines and triazolopyrimidines |
| WO2008113601A1 (en) | 2007-03-21 | 2008-09-25 | Biocompatibles Uk Ltd. | Glp-1 fusion peptides conjugated to polymer(s), their production and use |
| WO2008116107A2 (en) | 2007-03-21 | 2008-09-25 | Takeda San Diego, Inc. | Piperazine derivatives as glucokinase activators |
| WO2008116195A2 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Compositions comprising an sglt2 ingibitor for treating obesity |
| WO2008116179A1 (en) | 2007-03-22 | 2008-09-25 | Bristol-Myers Squibb | Pharmaceutical formulations containing dapagliflozin propylene glycol hydrate |
| US20080234277A1 (en) | 2007-03-23 | 2008-09-25 | Aurelia Conte | Novel aza-pyridopyrimidinone derivatives |
| WO2008118848A1 (en) | 2007-03-23 | 2008-10-02 | Trustees Of Tufts College | N-substituted peptidomimetic inhibitors of dipeptidylpeptidase iv |
| WO2008118718A2 (en) | 2007-03-23 | 2008-10-02 | Array Biopharma Inc. | 2-aminopyridine analogs as glucokinase activators |
| WO2008116294A1 (en) | 2007-03-23 | 2008-10-02 | Matregen Corp. | Exendin analogs |
| WO2008116648A2 (en) | 2007-03-27 | 2008-10-02 | Biocompatibles Uk Ltd. | Novel glp-1 fusion peptides, their production and use |
| WO2008119005A1 (en) | 2007-03-27 | 2008-10-02 | Trustees Of Tufts College | 3,4-dehydro-proline-containing inhibitors of dipeptidylpeptidase iv |
| WO2008119017A1 (en) | 2007-03-28 | 2008-10-02 | High Point Pharmaceuticals, Llc | 11beta-hsd1 active compounds |
| WO2008117982A1 (en) | 2007-03-28 | 2008-10-02 | Crystal Genomics, Inc. | Heterocyclic carboxylic acid derivatives and pharmaceutical composition for inhibiting lipid accumulation containing same |
| WO2008119918A1 (en) | 2007-03-29 | 2008-10-09 | N.V. Organon | Mineralocorticoid receptor antagonists |
| WO2008120655A1 (en) | 2007-03-30 | 2008-10-09 | Institute Of Medicinal Molecular Design, Inc. | Oxazolidinone derivative having inhibitory activity on 11β-hydroxysteroid dehydrogenase type i |
| WO2008120754A1 (en) | 2007-03-30 | 2008-10-09 | Taisho Pharmaceutical Co., Ltd. | Picolinamide compound |
| WO2008121506A2 (en) | 2007-03-30 | 2008-10-09 | Takeda Pharmaceutical Company Limited | Renin inhibitors |
| WO2008121064A1 (en) | 2007-03-30 | 2008-10-09 | Astrazeneca Ab | New imidazo[4,5-b]pyridine-6-halo-7-aryl/heteroaryl compounds 705 |
| WO2008121063A1 (en) | 2007-03-30 | 2008-10-09 | Astrazeneca Ab | New imidazo[ 4,5-b]pyridine-7-carboxamides 704 |
| WO2008119238A1 (en) | 2007-03-30 | 2008-10-09 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Substituted five membered heterocycle compounds, preparation method and medical use thereof |
| US20080242596A1 (en) | 2007-04-02 | 2008-10-02 | Theracos, Inc. | Benzylic glycoside derivatives and methods of use |
| WO2008119208A1 (en) | 2007-04-03 | 2008-10-09 | Beijing Molecule Science And Technology Co., Ltd. | N-substituted thiomorpholine derivatives as the inhibitors of dipeptidyl peptidase iv and the pharmaceutical uses thereof |
| WO2008120813A1 (en) | 2007-04-03 | 2008-10-09 | Mitsubishi Tanabe Pharma Corporation | Combined use of dipeptidyl peptidase iv inhibitor compound and sweetener |
| WO2008122352A1 (en) | 2007-04-05 | 2008-10-16 | Sanofi-Aventis | Imidazolidine carboxamide derivatives as lipase and phospholipase inhibitors |
| WO2008122357A1 (en) | 2007-04-05 | 2008-10-16 | Sanofi-Aventis | 5-oxo-isoxazoles as inhibitors of lipases and phospholipases |
| WO2008126731A1 (en) | 2007-04-05 | 2008-10-23 | Daiichi Sankyo Company, Limited | Aryl derivatives |
| WO2008126732A1 (en) | 2007-04-05 | 2008-10-23 | Daiichi Sankyo Company, Limited | Fused bicyclic heteroaryl derivatives |
| WO2008124745A1 (en) | 2007-04-10 | 2008-10-16 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2008124665A1 (en) | 2007-04-10 | 2008-10-16 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2008127924A1 (en) | 2007-04-11 | 2008-10-23 | High Point Pharmaceuticals, Llc. | Novel compounds |
| WO2008125833A1 (en) | 2007-04-12 | 2008-10-23 | Piramed Limited | Pharmaceutical compounds |
| WO2008125835A1 (en) | 2007-04-12 | 2008-10-23 | Piramed Limited | 2-morpholin-4-yl-pyrimidines as pi3k inhibitors |
| WO2008125945A2 (en) | 2007-04-12 | 2008-10-23 | Pfizer Inc. | 3-amido-pyrrolo [3, 4-c] pyrazole-5 (1h, 4h, 6h) carbaldehyde derivatives as inhibitors of protein kinase c |
| WO2008125839A2 (en) | 2007-04-12 | 2008-10-23 | Piramed Limited | Pyrimidine derivatives as inhibitors of phosphatidylinositol-3-kinase |
| WO2008127591A2 (en) | 2007-04-13 | 2008-10-23 | Schering Corporation | Pyrimidinedione derivatives and use thereof |
| WO2008130514A1 (en) | 2007-04-16 | 2008-10-30 | Amgen Inc. | Substituted biphenyl phenoxy-, thiophenyl- and aminophenylpropanoic acid gpr40 modulators |
| WO2008130951A1 (en) | 2007-04-17 | 2008-10-30 | Bristol-Myers Squibb Company | Fused heterocyclic 11-beta-hydroxysteroid dehydrogenase type i inhibitors |
| WO2008130312A1 (en) | 2007-04-18 | 2008-10-30 | Astrazeneca Ab | A new process for the manufacturing of the compound 2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1h-indole-5-carbonitrile 701 |
| WO2008130151A1 (en) | 2007-04-19 | 2008-10-30 | Dong-A Pharm. Co., Ltd. | Dpp-iv inhibitor including beta-amino group, preparation method thereof and pharmaceutical composition containing the same for preventing and treating a diabetes or an obesity |
| WO2008130615A1 (en) | 2007-04-20 | 2008-10-30 | Schering Corporation | Tetrahydropyrido[4,3-d]pyrimidinone derivatives and methods of use thereof |
| WO2008130584A1 (en) | 2007-04-20 | 2008-10-30 | Schering Corporation | Pyrimidinone derivatives and methods of use thereof |
| WO2008130581A1 (en) | 2007-04-20 | 2008-10-30 | Schering Corporation | Pyrimidinone derivatives and methods of use thereof |
| WO2008131149A2 (en) | 2007-04-20 | 2008-10-30 | Bristol-Myers Squibb Company | Crystal forms of saxagliptin and processes for preparing same |
| WO2008134221A1 (en) | 2007-04-24 | 2008-11-06 | High Point Pharmaceuticals, Llc | Pharmaceutical use of substituted amides |
| WO2008139879A1 (en) | 2007-04-26 | 2008-11-20 | Pharmafrontier, Co., Ltd. | G protein-coupled receptor inhibitor and pharmaceutical product |
| WO2008136444A1 (en) | 2007-04-26 | 2008-11-13 | Dainippon Sumitomo Pharma Co., Ltd. | Fused heterocyclic derivative |
| WO2008136428A1 (en) | 2007-04-27 | 2008-11-13 | Takeda Pharmaceutical Company Limited | Nitrogen-containing five-membered heterocyclic compound |
| WO2008136392A1 (en) | 2007-04-27 | 2008-11-13 | Ajinomoto Co., Inc. | Preparation for oral administration |
| WO2008136393A1 (en) | 2007-04-27 | 2008-11-13 | Ajinomoto Co., Inc. | Crystal of lactam compound, and method for production thereof |
| WO2008137435A1 (en) | 2007-05-04 | 2008-11-13 | Bristol-Myers Squibb Company | [6,6] and [6,7]-bicyclic gpr119 g protein-coupled receptor agonists |
| WO2008137436A1 (en) | 2007-05-04 | 2008-11-13 | Bristol-Myers Squibb Company | [6,5]-bicyclic gpr119 g protein-coupled receptor agonists |
| WO2008136642A1 (en) | 2007-05-07 | 2008-11-13 | Mazence Inc. | Naphthoquinone-based pharmaceutical composition for treatment or prevention of diseases involving obesity, diabetes, metabolic syndrome, neuro-degenerative diseases and mitochondria dysfunction diseases |
| WO2008137105A1 (en) | 2007-05-07 | 2008-11-13 | Merck & Co., Inc. | Method of treatment using fused aromatic compounds having anti-diabetic activity |
| WO2008138448A2 (en) | 2007-05-14 | 2008-11-20 | Merck Patent Gmbh | Heterocyclic indazole derivatives |
| WO2008144253A1 (en) * | 2007-05-14 | 2008-11-27 | Irm Llc | Protein kinase inhibitors and methods for using thereof |
| EP1992624A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Heteroarylamide pyrimidone compounds |
| EP1992625A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Arylamide pyrimidone compounds |
| EP1992621A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Heteroarylamide-substituted pyrimidone derivatives for the treatment of neurodegenerative diseases |
| EP1992620A1 (en) | 2007-05-16 | 2008-11-19 | Sanofi-Aventis | Arylamide pyrimidone derivatives for the treatment of neurodegenerative diseases |
| US20090137671A1 (en) | 2007-05-17 | 2009-05-28 | Noy Noa | Methods of treating metabolic disorders |
| WO2008142859A1 (en) | 2007-05-18 | 2008-11-27 | Kowa Company, Ltd. | Novel spiro-oxindole compound and pharmaceutical containing the same |
| US20080287529A1 (en) | 2007-05-18 | 2008-11-20 | Bristol-Myers Squibb Company | Crystal structures of sglt2 inhibitors and processes for preparing same |
| WO2008142986A1 (en) | 2007-05-18 | 2008-11-27 | Shionogi & Co., Ltd. | NITROGEN-CONTAINING HETEROCYCLIC DERIVATIVE HAVING 11 β-HYDROXYSTEROID DEHYDROGENASE TYPE I INHIBITORY ACTIVITY |
| WO2008146871A1 (en) | 2007-05-29 | 2008-12-04 | Santen Pharmaceutical Co., Ltd. | Novel 1,2,3,4-tetrahydroquinoxaline derivative which has, as substituent, phenyl group having sulfonic acid ester structure or sulfonic acid amide structure introduced therein and has glucocorticoid receptor-binding activity |
| US20080299096A1 (en) | 2007-06-01 | 2008-12-04 | Tatake Revati J | Modified nucleotide sequence encoding glucagon-like peptide-1 (glp-1), nucleic acid construct comprising same for production of glucagon-like peptide-1 (glp-1), human cells comprising said construct and insulin-producing constructs, and methods of use thereof |
| WO2008145721A2 (en) | 2007-06-01 | 2008-12-04 | Novo Nordisk A/S | N-terminal modification of polypeptides for protection against degradation by aminopeptidases |
| WO2008148744A1 (en) | 2007-06-04 | 2008-12-11 | Novartis Ag | Thiadiazole derivatives as antidiabetic agents |
| WO2008149382A1 (en) | 2007-06-08 | 2008-12-11 | Advinus Therapeutics Private Limited | Pyrrole-2-carboxamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2008148839A2 (en) | 2007-06-08 | 2008-12-11 | Ascendis Pharma As | Long-acting polymeric prodrugs of exendin |
| WO2008154563A1 (en) | 2007-06-11 | 2008-12-18 | Bristol-Myers Squibb Company | 1, 3 - dihydroxy substituted phenylamide glucokinase activators |
| WO2008152403A1 (en) | 2007-06-15 | 2008-12-18 | Zealand Pharma A/S | Glucagon analogues |
| WO2008156757A1 (en) | 2007-06-19 | 2008-12-24 | Takeda Pharmaceutical Company Limited | Indazole compounds for activating glucokinase |
| WO2008157752A1 (en) | 2007-06-21 | 2008-12-24 | Incyte Corporation | Spirocycles as inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 |
| WO2008156174A1 (en) | 2007-06-21 | 2008-12-24 | Taisho Pharmaceutical Co., Ltd. | Pyrazinamide compound |
| WO2009001817A1 (en) | 2007-06-27 | 2008-12-31 | Taisho Pharmaceutical Co., Ltd. | COMPOUND HAVING 11β-HSD1 INHIBITORY ACTIVITY |
| WO2009000087A1 (en) | 2007-06-28 | 2008-12-31 | Merck Frosst Canada Ltd. | Substituted fused pyrimidines as antagonists of gpr105 activity |
| WO2009005672A1 (en) | 2007-06-29 | 2009-01-08 | Merck & Co., Inc. | Antidiabetic azaindoles and diazaindoles |
| WO2009003681A1 (en) | 2007-07-02 | 2009-01-08 | Santhera Pharmaceuticals (Schweiz) Ag | Dpp-iv inhibitors |
| WO2009007714A2 (en) | 2007-07-09 | 2009-01-15 | Imperial Innovations Limited | Human pancreatic polypeptide (hpp) analogues and their effects on feeding behaviour |
| WO2009007029A1 (en) | 2007-07-11 | 2009-01-15 | Bayer Schering Pharma Aktiengesellschaft | Imidazo-, pyrazolopyrazines and imidazotriazines and their use |
| WO2009009287A2 (en) | 2007-07-12 | 2009-01-15 | Deviris Inc. | Hormone sensitive lipase modulators and methods of use |
| WO2009012039A2 (en) | 2007-07-13 | 2009-01-22 | Metabasis Therapeutics Inc. | Crystalline polymorphs |
| WO2009010416A2 (en) | 2007-07-17 | 2009-01-22 | F. Hoffmann-La Roche Ag | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| WO2009012275A1 (en) | 2007-07-17 | 2009-01-22 | Bristol-Myers Squibb Company | Pyridone gpr119 g protein-coupled receptor agonists |
| WO2009012277A1 (en) | 2007-07-17 | 2009-01-22 | Bristol-Myers Squibb Company | Method for modulating gpr119 g protein-coupled receptor and selected compounds |
| WO2009010530A1 (en) | 2007-07-18 | 2009-01-22 | Novartis Ag | Bicyclic heteroaryl compounds and their use as kinase inhibitors |
| WO2009014910A2 (en) | 2007-07-19 | 2009-01-29 | Metabolex, Inc. | N-azacyclic substituted pyrrole, pyrazole, imidazole, triazole and tetrazole derivatives as agonists of the rup3 or gpr119 receptor for the treatment of diabetes and metabolic disorders |
| WO2009014676A1 (en) | 2007-07-23 | 2009-01-29 | Merck & Co., Inc. | Novel crystalline form of a dihydrochloride salt of a dipeptidyl peptidase-iv inhibitor |
| JP2009023975A (en) | 2007-07-23 | 2009-02-05 | Nagoya Industrial Science Research Inst | PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR(PPAR)-alpha LIGAND AGENT |
| WO2009015067A2 (en) | 2007-07-25 | 2009-01-29 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2009012650A1 (en) | 2007-07-25 | 2009-01-29 | Institute Of Pharmacology And Toxicology Academy Of Military Medical Sciences P.L.A. | Aryl pyrimidine derivatives, preparation methods and pharmaceutical uses thereof |
| WO2009017664A1 (en) | 2007-07-26 | 2009-02-05 | Vitae Pharmaceuticals, Inc. | CYCLIC INHIBITORS OF 11β-HYDROXYSTERIOD DEHYDROGENASE 1 |
| WO2009016118A1 (en) | 2007-07-27 | 2009-02-05 | Boehringer Ingelheim International Gmbh | Substituted arylsulfonylaminomethylphosphonic acid derivatives, their preparation and their use in the treatment of type i and ii diabetes mellitus |
| WO2009018065A2 (en) | 2007-07-27 | 2009-02-05 | Bristol-Myers Squibb Company | Novel glucokinase activators and methods of using same |
| US20090030046A1 (en) | 2007-07-27 | 2009-01-29 | Bristol-Myers Squibb Company | Novel glucokinase activators and methods of using same |
| WO2009016119A1 (en) | 2007-07-27 | 2009-02-05 | Boehringer Ingelheim International Gmbh | Substituted arylsulfonylaminomethylphosphonic acid derivatives, their preparation and their use in the treatment of type i and ii diabetes mellitus |
| WO2009017452A1 (en) | 2007-07-30 | 2009-02-05 | Astrazeneca Ab | New crystalline forms of 2 -hydroxy- 3- [5- (morpholin- 4- ylmethyl) pyridin-2-yl] ih- indole- 5 -carbonitrile citrate |
| WO2009019600A2 (en) | 2007-08-03 | 2009-02-12 | Dr. Reddy's Laboratories Ltd. | Modulation of endogenous ampk levels for the treatment of obesity |
| WO2009019446A1 (en) | 2007-08-03 | 2009-02-12 | Betagenon Ab | Compounds useful as medicaments |
| WO2009019445A1 (en) | 2007-08-03 | 2009-02-12 | Betagenon Ab | Dithiazolidine and thiazolidine derivatives as anticancer agents |
| EP2020232A1 (en) | 2007-08-03 | 2009-02-04 | Zeltia, S.A. | N-(1-thiazolyl)-amide derivatives for the treatment of obesity, diabetes and cardiovascular diseases |
| WO2009020140A1 (en) | 2007-08-06 | 2009-02-12 | Dainippon Sumitomo Pharma Co., Ltd. | Adamantylurea derivative |
| WO2009023718A2 (en) | 2007-08-13 | 2009-02-19 | Metabasis Therapeutics, Inc. | Novel activators of glucokinase |
| WO2009023181A1 (en) | 2007-08-15 | 2009-02-19 | Schering Corporation | 6-SUBSTITUTED SULFONYL AZABICYCLO[3.2.1]OCTANES USEFUL TO INHIBIT 11β-HYDROXYSTEROID DEHYDROGENASE TYPE-1 |
| WO2009023664A2 (en) | 2007-08-15 | 2009-02-19 | Schering Corporation | Substituted azepine- and diazepine-sulfonamides useful to inhibit 11beta-hydroxysteroid dehydrogenase type-1 |
| WO2009023180A1 (en) | 2007-08-15 | 2009-02-19 | Schering Corporation | SUBSTITUTED BICYCLIC PIPERIDINYL-AND PIPERAZINYL- SULFONAMIDES USEFUL TO INHIBIT 11β-HYDROXYSTEROID DEHYDROGENASE TYPE-1 |
| WO2009026345A1 (en) | 2007-08-20 | 2009-02-26 | Targegen Inc. | Thiazolidinone compounds, and methods of making and using same |
| WO2009025784A1 (en) | 2007-08-21 | 2009-02-26 | Merck & Co., Inc. | Heterocyclic compounds as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2009026422A2 (en) | 2007-08-21 | 2009-02-26 | Abbott Laboratories | Treatment of central nervous system disorders |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2009026657A1 (en) | 2007-08-29 | 2009-03-05 | The University Of Sydney | Flavonoid ppar agonists |
| WO2009026658A1 (en) | 2007-08-29 | 2009-03-05 | The University Of Sydney | Ppar agonists |
| WO2009027276A1 (en) | 2007-08-30 | 2009-03-05 | F. Hoffmann-La Roche Ag | Process for the preparation of pyrido [2, 1-a] isoquinoline derivatives and polymorphic froms thereof |
| US20090062369A1 (en) | 2007-08-31 | 2009-03-05 | Joaquim Trias | Use of secretory phospholipase a2 (spla2) inhibitors to decrease spla2 levels |
| WO2009028891A2 (en) | 2007-08-31 | 2009-03-05 | Hanall Pharmaceutical Company. Ltd | 1,3,5-triazine-2,4,6-triamine compound or pharmaceutical acceptable salt thereof, and pharmaceutical composition comprising the same |
| WO2009030771A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Peptides derivatized with a-b-c-d- and their therapeutical use |
| WO2009030715A1 (en) | 2007-09-05 | 2009-03-12 | Boehringer Ingelheim International Gmbh | Arylsulfonylaminomethylphosphonic acid derivatives, the preparation thereof and the use thereof as pharmaceutical compositions |
| WO2009030738A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Glucagon-like peptide-1 derivatives and their pharmaceutical use |
| WO2009030774A1 (en) | 2007-09-05 | 2009-03-12 | Novo Nordisk A/S | Truncated glp-1 derivatives and their therapeutical use |
| WO2009032321A2 (en) | 2007-09-06 | 2009-03-12 | Genaera Corporation | A method for treating diabetes |
| WO2009035540A2 (en) | 2007-09-07 | 2009-03-19 | Ipsen Pharma S.A.S. | Analogues of exendin-4 and exendin-3 |
| WO2009033561A1 (en) | 2007-09-07 | 2009-03-19 | Bayer Schering Pharma Aktiengesellschaft | Substituted 6-pheylnicotinic acids and the use thereof |
| WO2009034388A1 (en) | 2007-09-10 | 2009-03-19 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2009034117A1 (en) | 2007-09-11 | 2009-03-19 | Novo Nordisk A/S | Mixture comprising an amylin peptide and a protracted insulin |
| WO2009035634A2 (en) | 2007-09-11 | 2009-03-19 | Activx Biosciences, Inc. | Cyanoaminoquinolones and tetrazoloaminoquinolones as gsk-3 inhibitors |
| WO2009034119A1 (en) | 2007-09-11 | 2009-03-19 | Novo Nordisk A/S | Improved derivatives of amylin |
| WO2009035684A1 (en) | 2007-09-12 | 2009-03-19 | Activx Biosciences, Inc. | Spirocyclic aminoquinolones as gsk-3 inhibitors |
| US20090076129A1 (en) | 2007-09-13 | 2009-03-19 | Gateway Health Alliances, Inc. | Methods and related compositions using specific flavonoids and indanes to reduce weight and inhibit lipase, alpha-amylase and alpha-glucosidase activity in mammals |
| WO2009038681A1 (en) | 2007-09-14 | 2009-03-26 | Metabolic Solutions Development Company | Thiazolidinedione analogues for the treatment of hypertension |
| WO2009038204A1 (en) | 2007-09-17 | 2009-03-26 | Pharma Frontier Co., Ltd. | Novel long-chain fatty acid derivative compound and g-protein-coupled receptor agonist containing the compound as active ingredient |
| WO2009038064A1 (en) | 2007-09-19 | 2009-03-26 | Institute Of Medicinal Molecular Design, Inc. | Heterocyclic derivative having inhibitory activity on type-i 11β-hydroxysteroid dehydrogenase |
| WO2009038974A1 (en) | 2007-09-20 | 2009-03-26 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009038385A2 (en) | 2007-09-21 | 2009-03-26 | Choongwae Pharma Corporation | Novel compounds having indazole frameworks, methods for preparing the same and pharmaceutical composition comprising the same |
| WO2009039944A1 (en) | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | Phenothiazine derivative having a double bond, method for the production thereof, and use thereof as a pharmaceutical |
| WO2009042435A1 (en) | 2007-09-21 | 2009-04-02 | Array Biopharma Inc. | Pyridin-2 -yl-amino-i, 2, 4 -thiadiazole derivatives as glucokinase activators for the treatment of diabetes mellitus |
| WO2009039943A1 (en) | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | (carboxylalkylene-phenyl)-phenyl-oxalamides, method for the production thereof, and use of same as a medicament |
| WO2009039942A1 (en) | 2007-09-21 | 2009-04-02 | Sanofi-Aventis | (cyclopropyl-phenyl)-phenyl-oxalamides, method for the production thereof, and use of same as a medicament |
| WO2009037719A1 (en) | 2007-09-21 | 2009-03-26 | Lupin Limited | Novel compounds as dipeptidyl peptidase iv (dpp iv) inhibitors |
| WO2009040288A1 (en) | 2007-09-27 | 2009-04-02 | F. Hoffmann-La Roche Ag | 1,1,1-trifluoro-2-hydroxy-3-phenylpropane derivatives |
| WO2009045830A1 (en) | 2007-09-28 | 2009-04-09 | Smithkline Beecham Corporation | Glycogen phosphorylase inhibitor compound and pharmaceutical composition thereof |
| WO2009045831A1 (en) | 2007-09-28 | 2009-04-09 | Smithkline Beecham Corporation | Glycogen phosphorylase inhibitor compound and pharmaceutical composition thereof |
| WO2009045753A1 (en) | 2007-10-01 | 2009-04-09 | Bristol-Myers Squibb Company | Triazolopyridine 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2009047798A2 (en) | 2007-10-08 | 2009-04-16 | Advinus Therapeutics Private Limited | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
| WO2009046784A1 (en) | 2007-10-09 | 2009-04-16 | Merck Patent Gmbh | Pyridine derivatives useful as glucokinase activators |
| WO2009046802A1 (en) | 2007-10-09 | 2009-04-16 | Merck Patent Gmbh | N- ( pyrazole- 3 -yl) -benzamide derivatives as glucokinase activators |
| WO2009047240A1 (en) | 2007-10-09 | 2009-04-16 | Smithkline Beecham Corporation | Indole derivatives useful as ppar activators |
| WO2009048527A1 (en) | 2007-10-10 | 2009-04-16 | Amgen Inc. | Substituted biphenyl gpr40 modulators |
| WO2009046606A1 (en) | 2007-10-11 | 2009-04-16 | Shanghai Institute Of Materia Medica, Cas | Pyrimidinyl-propionic acid derivatives and their use as ppar agonists |
| WO2009049731A1 (en) | 2007-10-11 | 2009-04-23 | Merck Patent Gmbh | Imidazo[1,2-a]pyrimidine derivatives for treating diseases such as diabetes |
| WO2009049222A1 (en) | 2007-10-12 | 2009-04-16 | Curedm, Inc. | Compositions and methods of using the human proislet peptide receptor |
| WO2009050523A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| WO2009050522A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| WO2009055331A2 (en) | 2007-10-22 | 2009-04-30 | Schering Corporation | Bicyclic heterocycle derivatives and their use as modulators of the activity of gpr119 |
| WO2009065298A1 (en) | 2007-10-25 | 2009-05-28 | Shanghai Hengrui Pharmaceutical Co., Ltd. | Piperazine derivatives, preparation process and pharmaceutical use thereof |
| WO2009054479A1 (en) | 2007-10-26 | 2009-04-30 | Japan Tobacco Inc. | Spiro-ring compound and use thereof for medical purposes |
| WO2009058237A1 (en) | 2007-10-29 | 2009-05-07 | Merck & Co., Inc. | Antidiabetic tricyclic compounds |
| WO2009056881A1 (en) | 2007-10-29 | 2009-05-07 | Astrazeneca Ab | Chemical compounds 313 |
| WO2009058734A1 (en) | 2007-10-30 | 2009-05-07 | Indiana University Research And Technology Corporation | Compounds exhibiting glucagon antagonist and glp-1 agonist activity |
| WO2009058662A2 (en) | 2007-10-30 | 2009-05-07 | Indiana University Research And Technology Corporation | Glucagon antagonists |
| WO2009057784A1 (en) | 2007-11-01 | 2009-05-07 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| WO2009059666A1 (en) | 2007-11-05 | 2009-05-14 | Merck Patent Gmbh | 7-azaindole derivatives as selective 11-beta-hydroxysteroid dehydrogenase type 1 inhibitors |
| WO2009061498A1 (en) | 2007-11-07 | 2009-05-14 | Vitae Pharmaceuticals, Inc. | CYCLIC UREA INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2009060071A1 (en) | 2007-11-08 | 2009-05-14 | Novo Nordisk A/S | Insulin derivative |
| WO2009063821A1 (en) | 2007-11-12 | 2009-05-22 | Banyu Pharmaceutical Co., Ltd. | Heteroaryloxy quinazoline derivative |
| EP2058308A1 (en) | 2007-11-12 | 2009-05-13 | Merck Sante | Benzimidazoledihydrothiadiazinone derivatives used as fructose-1,6-biphosphatase inhibitors and pharmaceutical compositions containing same. |
| WO2009065131A1 (en) | 2007-11-16 | 2009-05-22 | Rigel Pharmaceuticals, Inc. | Carboxamide, sulfonamide and amine compounds for metabolic disorders |
| WO2009063061A2 (en) | 2007-11-16 | 2009-05-22 | Boehringer Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of benzomorphanes and related scaffolds, medicaments containing such compounds and their use |
| WO2009070314A2 (en) | 2007-11-26 | 2009-06-04 | Teva Pharmaceutical Industries Ltd. | Crystalline form of sitagliptin |
| WO2009069736A1 (en) | 2007-11-28 | 2009-06-04 | Kyowa Hakko Kirin Co., Ltd. | Nitrogenated compound |
| WO2009070497A1 (en) | 2007-11-28 | 2009-06-04 | Smithkline Beecham Corporation | SEH AND 11 β-HSD1 INHIBITORS AND THEIR USE |
| WO2009068468A2 (en) | 2007-11-30 | 2009-06-04 | F. Hoffmann-La Roche Ag | Pyridine compounds |
| WO2009068467A1 (en) | 2007-11-30 | 2009-06-04 | F. Hoffmann-La Roche Ag | Aminothiazole derivatives |
| WO2009068531A2 (en) | 2007-11-30 | 2009-06-04 | Novartis Ag | Organic compounds |
| WO2009079921A1 (en) | 2007-11-30 | 2009-07-02 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Cucurbitane triterpenoid saponins, pharmaceutical composition thereof, preparation and use thereof |
| WO2009071895A1 (en) | 2007-12-04 | 2009-06-11 | Ucb Pharma S.A. | Fused thiazole and thiophene derivatives as kinase inhibitors |
| WO2009071888A1 (en) | 2007-12-04 | 2009-06-11 | Ucb Pharma S.A. | Pyrrolothiazoles as pi3-kinase inhibitors |
| WO2009071890A1 (en) | 2007-12-04 | 2009-06-11 | Ucb Pharma S.A. | Tricyclic kinase inhibitors |
| WO2009070873A1 (en) | 2007-12-04 | 2009-06-11 | Merck Frosst Canada Ltd. | Substituted 2-naphthoic acids as antagonists of gpr105 activity |
| WO2009072581A1 (en) | 2007-12-05 | 2009-06-11 | Aska Pharmaceutical Co., Ltd. | Lactam compound or salt thereof, and ppar activator |
| WO2009125424A2 (en) | 2007-12-11 | 2009-10-15 | Cadila Healthcare Limited | Peptidomimetics with glucagon antagonistic and glp-1 agonistic activities |
| WO2009075835A1 (en) | 2007-12-11 | 2009-06-18 | Vitae Pharmaceutical, Inc | CYCLIC UREA INHIBITORS OF 11β-HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2009074789A1 (en) | 2007-12-12 | 2009-06-18 | The University Of Edinburgh | Amido-thiophene compounds and their use as 11-beta-hsd1 inhibitors |
| WO2009076631A1 (en) | 2007-12-12 | 2009-06-18 | Rigel Pharmaceuticals, Inc. | Carboxamide, sulfonamide and amine compounds for metabolic disorders |
| WO2009076550A1 (en) | 2007-12-13 | 2009-06-18 | Theracos, Inc. | Benzylphenyl cyclohexane derivatives and methods of use |
| WO2009080242A1 (en) | 2007-12-20 | 2009-07-02 | Bayer Schering Pharma Aktiengesellschaft | Substituted 4-aminopyrimidine-5-carboxylic acid and use thereof |
| WO2009080032A1 (en) | 2007-12-20 | 2009-07-02 | Fertin Pharma A/S | Compressed chewing gum comprising a systemically active small peptide |
| WO2009080024A1 (en) | 2007-12-20 | 2009-07-02 | Fertin Pharma A/S | Compressed chewing gum comprising an incretin mimetic |
| WO2009080248A1 (en) | 2007-12-20 | 2009-07-02 | Bayer Schering Pharma Aktiengesellschaft | Substituted 2-phenylpyrimidine-5-carboxylic acids and the use thereof |
| WO2009082152A2 (en) | 2007-12-20 | 2009-07-02 | Lg Life Sciences Ltd. | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| WO2009082134A2 (en) | 2007-12-21 | 2009-07-02 | Lg Life Sciences, Ltd. | Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as active agent |
| WO2009080821A2 (en) | 2007-12-21 | 2009-07-02 | Giuliani International Limited | Receptor targeting ligands |
| WO2009081782A1 (en) | 2007-12-25 | 2009-07-02 | Banyu Pharmaceutical Co., Ltd. | N-pyrazole-2-pyridinecarboxamide derivative |
| WO2009080223A1 (en) | 2007-12-26 | 2009-07-02 | Sanofi-Aventis | Cyclic pyridyl-n-(1,3,4)-thiadiazol-2-yl-benzene sulfonamides, processes for their preparation and their use as pharmaceuticals |
| WO2009082881A1 (en) | 2007-12-26 | 2009-07-09 | Shanghai Hengrui Pharmaceutical Co., Ltd. | Tetrahydro-imidazo[1,5-a]pyrazine derivatives, preparation methods and medical uses thereof |
| WO2009084531A1 (en) | 2007-12-27 | 2009-07-09 | Kissei Pharmaceutical Co., Ltd. | Monosebacate of pyrazole derivative |
| WO2009084497A1 (en) | 2007-12-28 | 2009-07-09 | Dainippon Sumitomo Pharma Co., Ltd. | Methyl-substituted piperidine derivative |
| WO2009083553A1 (en) | 2007-12-31 | 2009-07-09 | Rheoscience A/S | Azine compounds as glucokinase activators |
| WO2009083526A1 (en) | 2008-01-02 | 2009-07-09 | Smithkline Beecham Corporation | Novel compounds |
| WO2009088997A1 (en) | 2008-01-07 | 2009-07-16 | Vitae Pharmaceuticals, Inc. | Lactam inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| WO2009087395A1 (en) | 2008-01-10 | 2009-07-16 | University Of Strathclyde | Weight reducing compounds |
| WO2009116067A2 (en) | 2008-01-10 | 2009-09-24 | Sun Pharma Advanced Research Company Limited | Novel derivatives of acyl cyanopyrrolidines |
| US20090181981A1 (en) | 2008-01-15 | 2009-07-16 | Jeanette Tower Dunlap | Crystalline (r)-2-(4-cyclopropanesulphonyl-phenyl)-n-pyrazin-2-yl-3-(tetrahydropyran-4-yl)-propionamide |
| WO2009090239A1 (en) | 2008-01-17 | 2009-07-23 | Biovitrum Ab (Publ) | Isoxazole derivatives as modulators of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2009091082A1 (en) | 2008-01-17 | 2009-07-23 | Mitsubishi Tanabe Pharma Corporation | Combination therapy comprising sglt inhibitors and dpp4 inhibitors |
| WO2009091014A1 (en) | 2008-01-18 | 2009-07-23 | Astellas Pharma Inc. | Phenyl acetamide derivative |
| WO2009094169A1 (en) | 2008-01-24 | 2009-07-30 | Vitae Pharmaceuticals, Inc. | Cyclic carbazate and semicarbazide inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009092432A1 (en) | 2008-01-24 | 2009-07-30 | Merck Patent Gmbh | Beta-amino acid derivatives for treatment of diabetes |
| US20090238879A1 (en) | 2008-01-24 | 2009-09-24 | Northwestern University | Delivery scaffolds and related methods of use |
| WO2009093269A1 (en) | 2008-01-24 | 2009-07-30 | Panacea Biotec Limited | Novel heterocyclic compounds |
| WO2009112691A2 (en) | 2008-01-28 | 2009-09-17 | Sanofi-Aventis | Derivatives of tetrahydroquinoxaline urea, preparation thereof and therapeutic application thereof |
| WO2009095787A1 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted heteroarylamide oxazepinopyrimidone derivatives |
| WO2009095792A2 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted heteroarylamide diazepinopyrimidone derivatives |
| WO2009095789A1 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted arylamide diazepinopyrimidone derivatives for the treatment of neurodegenerative diseases caused by abnormal activity of gsk3-beta |
| WO2009095788A1 (en) | 2008-01-29 | 2009-08-06 | Sanofi-Aventis | Substituted arylamide oxazepinopyrimidone derivatives |
| WO2009096503A1 (en) | 2008-01-31 | 2009-08-06 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
| WO2009100130A1 (en) | 2008-02-04 | 2009-08-13 | Mercury Therapeutics, Inc. | Ampk modulators |
| WO2009098501A1 (en) | 2008-02-04 | 2009-08-13 | Astrazeneca Ab | Novel crystalline forms of 4- [4- (2-adamantylcarbam0yl) -5-tert-butyl-pyrazol-1-yl] benzoic acid |
| WO2009099080A1 (en) | 2008-02-06 | 2009-08-13 | Daiichi Sankyo Company, Limited | Novel phenylpyrrole derivative |
| WO2009099172A1 (en) | 2008-02-07 | 2009-08-13 | Takeda Pharmaceutical Company Limited | Pharmaceutical product |
| WO2009099171A1 (en) | 2008-02-07 | 2009-08-13 | Takeda Pharmaceutical Company Limited | Pharmaceutical product |
| WO2009102428A2 (en) | 2008-02-11 | 2009-08-20 | Vitae Pharmaceuticals, Inc. | 1,3-OXAZEPAN-2-ONE AND 1,3-DIAZEPAN-2-ONE INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2009102761A1 (en) | 2008-02-12 | 2009-08-20 | Bristol-Myers Squibb Company | 1,2,3-triazoles as 11-beta hydroxysteroid dehydrogenase type i inhibitors |
| WO2009102226A1 (en) | 2008-02-12 | 2009-08-20 | Adamed Sp. Z O.O. | Malonic acid salt of 5-[[4-[2-(methyl-2-pyridinylamino)ethoxy]phenyl]methyl]- 2,4-thiazolidinedione |
| WO2009100872A1 (en) | 2008-02-12 | 2009-08-20 | Boehringer Ingelheim International Gmbh | Urea derivatives of benzomorphanes and related scaffolds, medicaments containing such compounds and their use |
| WO2009100936A2 (en) | 2008-02-13 | 2009-08-20 | Sanofi-Aventis | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds, and the use thereof |
| WO2009102460A2 (en) | 2008-02-15 | 2009-08-20 | Vitae Pharmaceuticals, Inc. | Inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009105722A1 (en) | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009105715A1 (en) | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009105717A1 (en) | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009107900A1 (en) | 2008-02-25 | 2009-09-03 | Sungkyunkwan University Foundation For Corporate Collaboration | Exendin derivative linked biotin, method for the preparation thereof and pharmaceutical composition comprising the same |
| WO2009106203A1 (en) | 2008-02-25 | 2009-09-03 | Merck Patent Gmbh | Glucokinase activators |
| WO2009106817A2 (en) | 2008-02-26 | 2009-09-03 | Sterix Limited | Compound |
| WO2009106209A1 (en) | 2008-02-27 | 2009-09-03 | Merck Patent Gmbh | Carboxamide-heteroaryl derivatives for the treatment of diabetes |
| WO2009106561A1 (en) | 2008-02-27 | 2009-09-03 | Biovitrum Ab (Publ) | Pyrazine compounds for treating gpr119 related disorders |
| WO2009108332A1 (en) | 2008-02-27 | 2009-09-03 | Vitae Pharmaceuticals, Inc. | INHIBITORS OF 11β -HYDROXYSTEROID DEHYDROGENASE TYPE 1 |
| WO2009106565A1 (en) | 2008-02-27 | 2009-09-03 | Biovitrum Ab (Publ) | Agonists of gpr119 |
| WO2009118473A2 (en) | 2008-02-29 | 2009-10-01 | Sanofi-Aventis | Azetidine-derived compounds, preparation method therefor and therapeutic use of same |
| WO2009109270A1 (en) | 2008-03-01 | 2009-09-11 | Merck Patent Gmbh | 5-oxo-2, 3,4,5-tetrahydro-benzo[b]oxepine-4-carboxylic acid amides and 2,3-dihydro-benzo[b]oxepine-4-carboxylic acid amides for treatment and prevention of diabetes typ 1 and 2 |
| WO2009109998A1 (en) | 2008-03-03 | 2009-09-11 | Lupin Limited | Novel protein tyrosine phosphatase - ib inhibitors |
| WO2009109999A1 (en) | 2008-03-03 | 2009-09-11 | Lupin Limited | Novel protein tyrosine phosphatase - ib inhibitors |
| WO2009111239A2 (en) | 2008-03-05 | 2009-09-11 | National Health Research Institutes | Pyrrolidine derivatives |
| WO2009109341A1 (en) | 2008-03-05 | 2009-09-11 | Merck Patent Gmbh | Pyridopyrazinones derivatives insulin secretion stimulators, methods for obtaining them and use thereof for the treatment of diabetes |
| WO2009109259A1 (en) | 2008-03-05 | 2009-09-11 | Merck Patent Gmbh | Pyrazinone derivatives as insulin secretion stimulators, methods for obtaining them and use thereof for the treatment of diabetes |
| WO2009110520A1 (en) | 2008-03-05 | 2009-09-11 | 武田薬品工業株式会社 | Heterocyclic compound |
| WO2009111056A1 (en) | 2008-03-06 | 2009-09-11 | Amgen Inc. | Conformationally constrained carboxylic acid derivatives useful for treating metabolic disorders |
| WO2009111700A2 (en) | 2008-03-07 | 2009-09-11 | Transtech Pharma, Inc. | Oxadiazoanthracene compounds for the treatment of diabetes |
| WO2009113423A1 (en) | 2008-03-10 | 2009-09-17 | 大日本住友製薬株式会社 | Bicyclic pyrrole compound |
| WO2009145814A2 (en) | 2008-03-10 | 2009-12-03 | Vertex Pharmaceuticals Incorporated | Pyrimidines and pyridines useful as inhibitors of protein kinases |
| WO2009112845A1 (en) | 2008-03-13 | 2009-09-17 | The University Of Edinburgh | Amido-thiophene compounds and their use |
| WO2009114173A1 (en) | 2008-03-14 | 2009-09-17 | Exelixis, Inc. | Azabicyclo [3. 2. i] octyl derivatives as 11 beta-hsdl modulators |
| WO2009117421A2 (en) | 2008-03-17 | 2009-09-24 | Kalypsys, Inc. | Heterocyclic modulators of gpr119 for treatment of disease |
| WO2009117109A1 (en) | 2008-03-18 | 2009-09-24 | Vitae Pharmaceuticals, Inc. | Inhibitors of 11beta-hydroxysteroid dehydrogenase type 1 |
| EP2103302A1 (en) | 2008-03-21 | 2009-09-23 | Les Laboratoires Servier | Divisible dosage form allowing modified release of the active principle |
| WO2009120530A1 (en) | 2008-03-27 | 2009-10-01 | Eli Lilly And Company | Glucagon receptor antagonists |
| US20090247532A1 (en) | 2008-03-28 | 2009-10-01 | Mae De Ltd. | Crystalline polymorph of sitagliptin phosphate and its preparation |
| WO2009123992A1 (en) | 2008-03-31 | 2009-10-08 | Metabolex, Inc. | Oxymethylene aryl compounds and uses thereof |
| WO2009121884A1 (en) | 2008-04-01 | 2009-10-08 | Novo Nordisk A/S | Insulin albumin conjugates |
| WO2009121939A2 (en) | 2008-04-02 | 2009-10-08 | Tfchem | C-aryl glycoside compounds for the treatment of diabetes and obesity |
| WO2009121945A2 (en) | 2008-04-03 | 2009-10-08 | Boehringer Ingelheim International Gmbh | New formulations, tablets comprising such formulations, their use and process for their preparation |
| WO2009126535A1 (en) | 2008-04-07 | 2009-10-15 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009124638A1 (en) | 2008-04-07 | 2009-10-15 | Merck Patent Gmbh | Glucopyranoside derivatives |
| WO2009125434A2 (en) | 2008-04-07 | 2009-10-15 | Cadila Healthcare Limited | Oxime derivatives |
| WO2009125873A1 (en) | 2008-04-10 | 2009-10-15 | Takeda Pharmaceutical Company Limited | Fused ring compounds and use thereof |
| WO2009124636A1 (en) | 2008-04-11 | 2009-10-15 | Merck Patent Gmbh | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| WO2009129036A1 (en) | 2008-04-14 | 2009-10-22 | Merck & Co., Inc. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| WO2009128558A1 (en) | 2008-04-15 | 2009-10-22 | 日本ケミファ株式会社 | Activator for peroxisome proliferator-activated receptor |
| WO2009128481A1 (en) | 2008-04-16 | 2009-10-22 | 武田薬品工業株式会社 | Nitrogenated 5-membered heterocyclic compound |
| WO2009127544A1 (en) | 2008-04-16 | 2009-10-22 | F. Hoffmann-La Roche Ag | Pyridazinone glucokinase activators |
| WO2009128421A1 (en) | 2008-04-16 | 2009-10-22 | キッセイ薬品工業株式会社 | Hemifumarate of a pyrazole derivative |
| WO2009127546A1 (en) | 2008-04-16 | 2009-10-22 | F. Hoffmann-La Roche Ag | Pyrrolidinone glucokinase activators |
| WO2009127723A1 (en) | 2008-04-19 | 2009-10-22 | Boehringer Ingelheim International Gmbh | New arylsulphonylglycine derivatives, the preparation thereof and their use as medicaments |
| US20090264401A1 (en) | 2008-04-22 | 2009-10-22 | Astrazeneca Ab | Substituted pyrimidin-5-carboxamides 281 |
| WO2009131669A2 (en) | 2008-04-22 | 2009-10-29 | Vitae Pharmaceuticals, Inc. | Carbamate and urea inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009129696A1 (en) | 2008-04-25 | 2009-10-29 | 国家新药筛选中心 | A kind of receptor signaling transoluction positive modulators, preparation methods and uses thereof |
| WO2009133099A2 (en) | 2008-04-28 | 2009-11-05 | Novo Nordisk A/S | Insulin precursors for diabetes treatment |
| WO2009133687A1 (en) | 2008-04-28 | 2009-11-05 | 杏林製薬株式会社 | Cyclopentylacrylic acid amide derivative |
| WO2009132986A1 (en) | 2008-04-30 | 2009-11-05 | F. Hoffmann-La Roche Ag | Imidazolidinone derivatives as 11b-hsd1 inhibitors |
| WO2009134387A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009134384A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009134392A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009134400A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009135581A2 (en) | 2008-05-05 | 2009-11-12 | Merck Patent Gmbh | Novel nip thiazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2009135580A1 (en) | 2008-05-05 | 2009-11-12 | Merck Patent Gmbh | Thienopyridone derivatives as amp-activated protein kinase (ampk) activators |
| WO2009135673A2 (en) | 2008-05-09 | 2009-11-12 | Barbara La Ferla | Compounds with glycidic structure active in the therapy of systemic |
| WO2009139340A1 (en) | 2008-05-12 | 2009-11-19 | 武田薬品工業株式会社 | Pyrazole compound |
| WO2009138386A2 (en) | 2008-05-13 | 2009-11-19 | Boehringer Ingelheim International Gmbh | Alicyclic carboxylic acid derivatives of benzomorphans and related scaffolds, medicaments containing such compounds and their use |
| WO2009139362A1 (en) | 2008-05-14 | 2009-11-19 | 株式会社 三和化学研究所 | Pharmaceutical preparation comprising dpp-iv inhibitor and other diabetes therapeutic agent in concomitant or combined form |
| WO2009140342A1 (en) | 2008-05-16 | 2009-11-19 | Schering Corporation | Glucagon receptor antagonists, compositions, and methods for their use |
| WO2009140624A2 (en) | 2008-05-16 | 2009-11-19 | Takeda San Diego, Inc. | Glucokinase activators |
| WO2009143049A1 (en) | 2008-05-19 | 2009-11-26 | Schering Corporation | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| US20090286812A1 (en) | 2008-05-19 | 2009-11-19 | Shawn David Erickson | GPR119 Receptor Agonists |
| US20090291982A1 (en) | 2008-05-22 | 2009-11-26 | Astrazeneca Ab | New Substituted Oxindole Derivative 352 |
| WO2009153496A2 (en) | 2008-05-26 | 2009-12-23 | Genfit | Ppar agonist compounds, preparation and uses |
| WO2009154697A2 (en) | 2008-05-28 | 2009-12-23 | Massachusetts Institute Of Technology | Disc-1 pathway activators in the control of neurogenesis |
| WO2009147990A1 (en) | 2008-06-02 | 2009-12-10 | 萬有製薬株式会社 | Novel isoxazole derivative |
| WO2009149056A2 (en) | 2008-06-02 | 2009-12-10 | Dr. Reddy's Laboratories Ltd. | Combinations of niacin and an oxicam |
| WO2009147121A1 (en) | 2008-06-02 | 2009-12-10 | Smithkline Beecham Corporation | Carboxyl substituted indoles for use as ppar alpha modulators |
| WO2009149148A2 (en) | 2008-06-03 | 2009-12-10 | Trustees Of Tufts College | Long-acting glp-1 derivatives, and methods of treating cardiac dysfunction |
| WO2009149171A2 (en) | 2008-06-04 | 2009-12-10 | Amgen Inc. | Fgf21 mutants and uses thereof |
| WO2009149139A1 (en) | 2008-06-06 | 2009-12-10 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| WO2009149820A1 (en) | 2008-06-09 | 2009-12-17 | Sanofi-Aventis | Annelated n-heterocyclic sulfonamides with oxadiazolone headgroup, processes for their preparation and their use as pharmaceuticals |
| WO2010000353A1 (en) | 2008-06-09 | 2010-01-07 | Sanofi-Aventis | Sulfonamides with heterocycle and oxadiazolone headgroup, processes for their preparation and their use as pharmaceuticals |
| WO2009149819A1 (en) | 2008-06-09 | 2009-12-17 | Sanofi-Aventis | Annelated pyrrolidin sulfonamides with oxadiazolone headgroup, processes for their preparation and their use as pharmaceuticals |
| WO2009150144A1 (en) | 2008-06-10 | 2009-12-17 | Inovacia Ab | New gpr119modulators |
| WO2009152128A1 (en) | 2008-06-13 | 2009-12-17 | Eli Lilly And Company | Pegylated insulin lispro compounds |
| WO2009152909A1 (en) | 2008-06-16 | 2009-12-23 | Merck Patent Gmbh | Quinoxalinedione derivatives |
| WO2009155257A1 (en) | 2008-06-17 | 2009-12-23 | Indiana University Research And Technology Corporation | Glucagon analogs exhibiting enhanced solubility and stability physiological ph buffers |
| WO2009153960A1 (en) | 2008-06-17 | 2009-12-23 | 大塚化学株式会社 | Glycosylated glp-1 peptide |
| WO2009155258A2 (en) | 2008-06-17 | 2009-12-23 | Indiana University Research And Technology Corporation | Glucagon/glp-1 receptor co-agonists |
| WO2010008739A2 (en) | 2008-06-20 | 2010-01-21 | Metabolex, Inc. | Aryl gpr119 agonists and uses thereof |
| WO2010008831A2 (en) | 2008-06-24 | 2010-01-21 | Irm Llc | Compounds and methods for modulating g protein-coupled receptors |
| WO2009156857A1 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted n-oxide pyrazine derivatives |
| WO2009156860A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted triazinone derivatives |
| WO2009156859A1 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted pyrimido [2, 1-a] isoquinolin-4-one derivatives |
| WO2009156861A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted pyrimidone derivatives |
| WO2009156864A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted alkyl pyrimidin-4-one derivatives |
| WO2009156863A2 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted tricyclic derivatives |
| WO2009156865A1 (en) | 2008-06-26 | 2009-12-30 | Mitsubishi Tanabe Pharma Corporation | Substituted pyrimidin-4-one derivatives |
| WO2010001166A1 (en) | 2008-07-01 | 2010-01-07 | Prosidion Limited | Thiazole derivatives as gpr 119 modulators |
| WO2010000469A2 (en) | 2008-07-03 | 2010-01-07 | Ratiopharm Gmbh | Crystalline salts of sitagliptin |
| WO2010006214A1 (en) | 2008-07-09 | 2010-01-14 | Ambrx, Inc. | Fgf-21 neutralizing antibodies and their uses |
| WO2010004344A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidine gpcr agonists |
| WO2010004345A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidinyl gpcr agonists |
| WO2010004346A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidinyl gpcr agonists |
| WO2010004347A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2010004348A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Heterocyclic gpcr agonists |
| WO2010004343A1 (en) | 2008-07-10 | 2010-01-14 | Prosidion Limited | Piperidinyl gpcr agonists |
| WO2010006191A1 (en) | 2008-07-11 | 2010-01-14 | Irm Llc | 4-phenoxymethylpiperidines as modulators of gpr119 activity |
| WO2010006940A1 (en) | 2008-07-15 | 2010-01-21 | F. Hoffmann-La Roche Ag | Tetrahydrocinnolines as 11 beta hsd1 inhibitors for diabetes |
| WO2010008299A1 (en) | 2008-07-15 | 2010-01-21 | Pronova Biopharma Norge As | Novel sulphur containing lipids for use as food supplement or as medicament |
| WO2010009183A1 (en) | 2008-07-16 | 2010-01-21 | Bristol-Myers Squibb Company | Pyridone and pyridazone analogues as gpr119 modulators |
| WO2010009195A1 (en) | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| WO2010009207A1 (en) | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and their use as gpcr modulators |
| WO2010009208A1 (en) | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and methods of use thereof |
| WO2010009197A1 (en) | 2008-07-17 | 2010-01-21 | Lexicon Pharmaceuticals, Inc. | Solid forms of (2s,3r,4r,5s,6r)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2h-pyran-3,4,5-triol and methods of their use |
| WO2010011772A2 (en) * | 2008-07-23 | 2010-01-28 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010010174A1 (en) | 2008-07-25 | 2010-01-28 | Boehringer Ingelheim International Gmbh | 1,1'-diadamantyl carboxylic acids, medicaments containing such compounds and their use |
| WO2010010157A2 (en) | 2008-07-25 | 2010-01-28 | Boehringer Ingelheim International Gmbh | INHIBITORS OF 11beta-HYDROXYSTEROID DEHYDROGENASE 1 |
| WO2010011917A1 (en) | 2008-07-25 | 2010-01-28 | Glaxosmithkline Llc | SEH AND 11β-HSD1 DUAL INHIBITORS |
| WO2010014613A2 (en) | 2008-07-28 | 2010-02-04 | Emory University | Treating various disorders using trkb agonists |
| WO2010012650A1 (en) | 2008-07-28 | 2010-02-04 | Syddansk Universitet | Compounds for the treatment of metabolic diseases |
| WO2010013161A1 (en) | 2008-07-29 | 2010-02-04 | Pfizer Inc. | Fluorinated heteroaryls |
| WO2010014593A1 (en) | 2008-07-30 | 2010-02-04 | Smithkline Beecham Corporation | Chemical compounds and uses |
| WO2010014794A1 (en) | 2008-07-30 | 2010-02-04 | Oncotherapy Science, Inc. | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| WO2010013168A1 (en) | 2008-08-01 | 2010-02-04 | Centre National De La Recherche Scientifique (Cnrs) | 3',6-substituted indirubins and their biological applications |
| WO2010014771A1 (en) | 2008-08-01 | 2010-02-04 | Ore Pharmaceuticals Inc. | Romazarit for treating metabolic diseases |
| WO2010015849A2 (en) | 2008-08-04 | 2010-02-11 | Astrazeneca Ab | Therapeutic agents 414 |
| WO2010015664A1 (en) | 2008-08-06 | 2010-02-11 | Boehringer Ingelheim International Gmbh | Treatment for diabetes in patients inappropriate for metformin therapy |
| WO2010016936A1 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Pharmaceutical compositions of analogues of glucose-dependent insulinotropic polypeptide |
| WO2010016935A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Truncated analogues of glucose-dependent insulinotropic polypeptide |
| WO2010016938A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Glucose-dependent insulinotropic polypeptide analogues |
| WO2010016944A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Analogues of glucose-dependent insulinotropic polypeptide (gip) modified at n-terminal |
| WO2010016940A2 (en) | 2008-08-07 | 2010-02-11 | Ipsen Pharma S.A.S. | Analogues of glucose-dependent insulinotropic polypeptide |
| WO2010018438A2 (en) | 2008-08-11 | 2010-02-18 | Hetero Research Foundation | Tetrazole glycosides |
| WO2010018435A1 (en) | 2008-08-11 | 2010-02-18 | Hetero Research Foundation | Amide glycosides |
| WO2010019828A1 (en) | 2008-08-13 | 2010-02-18 | Metabasis Therapeutics, Inc. | Glucagon receptor antagonists |
| WO2010018800A1 (en) | 2008-08-15 | 2010-02-18 | Banyu Pharmaceutical Co.,Ltd. | Acetyl pyrrolidinyl indole derivative |
| WO2010129668A1 (en) * | 2009-05-06 | 2010-11-11 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridines |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150148352A1 (en) * | 2012-08-14 | 2015-05-28 | Cellixbio Private Limited | Compositions and methods for the treatment angina and cardiovascular conditions |
| WO2016081311A1 (en) * | 2014-11-21 | 2016-05-26 | Eli Lilly And Company | 1,2-benzothiazole compounds for the treatment of kidney disorders |
| US10604541B2 (en) | 2016-07-22 | 2020-03-31 | Bristol-Myers Squibb Company | Glucokinase activators and methods of using same |
| KR20190133215A (en) * | 2017-03-28 | 2019-12-02 | 바스프 에스이 | Insecticide compound |
| KR102596592B1 (en) | 2017-03-28 | 2023-10-31 | 바스프 에스이 | insecticidal compounds |
| US10392368B2 (en) | 2017-08-01 | 2019-08-27 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| US10538513B2 (en) | 2017-08-01 | 2020-01-21 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| US10968205B2 (en) | 2017-08-01 | 2021-04-06 | Theravance Biopharma R&D Ip, Llc | Pyrazolo and triazolo bicyclic compounds as JAK kinase inhibitors |
| US10851102B2 (en) | 2019-01-23 | 2020-12-01 | Theravance Biopharma R&D Ip, Llc | Imidazole and triazole containing bicyclic compounds as JAK inhibitors |
| US11339160B2 (en) | 2019-01-23 | 2022-05-24 | Theravance Biopharma R&D Ip, Llc | Imidazole and triazole containing bicyclic compounds as JAK inhibitors |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2771340B1 (en) | 6-(4-hydroxy-phenyl)-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors | |
| EP2760862B1 (en) | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors | |
| EP2897939B1 (en) | Benzoimidazole-carboxylic acid amide derivatives for treating metabolic or cardiovascular diseases | |
| US8673917B2 (en) | 2-heteroaryl-pyrrolo [3,4-C]pyrrole derivatives, and use thereof as SCD inhibitors | |
| US8530413B2 (en) | Heterocyclically substituted methoxyphenyl derivatives with an oxo group, processes for preparation thereof and use thereof as medicaments | |
| US8003677B2 (en) | 2-heteroaryl-pyrrolo[3,4-c]pyrrole derivatives and their use as SCD inhibitors | |
| US8648038B2 (en) | (2-aryloxyacetylamino)phenylpropionic acid derivatives, processes for preparation thereof and use thereof as medicaments | |
| US8828991B2 (en) | Azacyclyl-substituted arylthienopyrimidinones, process for their preparation and their use as medicaments | |
| EP1987006B1 (en) | Novel aminoalcohol-substituted aryldihydroisoquinolinones, process for their preparation and their use as medicaments | |
| EP1987042B1 (en) | Novel amino alcohol-substituted arylthienopyrimidinones, process for their preparation and their use as medicaments | |
| US8846712B2 (en) | 6-(4-hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors | |
| EP2310372A2 (en) | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof | |
| WO2013037390A1 (en) | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors | |
| US9133181B2 (en) | Substituted 6-(4-hydroxy-phenyl)-1H-pyrazolo[3,4-b]pyridine derivatives as kinase inhibitors | |
| EP2567959B1 (en) | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors | |
| CN102993201A (en) | 6-(4-hydroxyl-phenyl)-3-styryl-1H-pyrazole[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitor | |
| JP2013060370A (en) | 6-(4-HYDROXYPHENYL)-3-STYRYL-1H-PYRAZOLO[3,4-b]PYRIDINE-4-CARBOXYLIC ACID AMIDE DERIVATIVE AS KINASE INHIBITOR | |
| MX2008010121A (en) | Novel aminoalcohol-substituted aryldihydroisoquinolinones, process for their preparation and their use as medicaments |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11755335 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11755335 Country of ref document: EP Kind code of ref document: A1 |