WO2009083553A1 - Azine compounds as glucokinase activators - Google Patents
Azine compounds as glucokinase activators Download PDFInfo
- Publication number
- WO2009083553A1 WO2009083553A1 PCT/EP2008/068232 EP2008068232W WO2009083553A1 WO 2009083553 A1 WO2009083553 A1 WO 2009083553A1 EP 2008068232 W EP2008068232 W EP 2008068232W WO 2009083553 A1 WO2009083553 A1 WO 2009083553A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- substituted
- unsubstituted
- halogen
- independently selected
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- Type 1 is usually due to autoimmune destruction of the pancreatic beta cells which produce insulin.
- Type 2 is characterized by tissue -wide insulin resistance and varies widely; it sometimes progresses to loss of beta cell function.
- Gestational diabetes is similar to type 2 diabetes, in that it involves insulin resistance; the hormones of pregnancy cause insulin resistance in those women genetically predisposed to developing this condition.
- G5 is selected from the group consisting of:
- compounds of the invention meet at least one of the following criteria.
- Recombinant GK protein is expressed in Escherichia coli as a GST-tagged fusion protein and is purified by glutathione-agarose affinity chromatography.
- the recombinant GK protein after the removal of GST- tag is further purified by size exclusion chromatography using Superdex S-200 (GE health Sciences) column on an FPLC using 2OmM Tris, pH 7.5, 50 mM NacCl, 5 mM TCEP and 20 mM Glucose.
- the recombinant GK is incubated with the compound of the invention at a concentration of 150 to 250 ⁇ M for 30 min to 1 Hr at room temperature and concentrated to 10 mg/ml using 50 KDa cut-off
- Human recombinant glucokinase (-200 ng) is added of to the assay reaction except the blank.
- the increase in A 34o nm is monitored in Spectramax at 30° C for 15 min .
- the A 34o nm is obtained for both test and positive control after substracting the blank.
- G 3 , G 4 , G 5 , G 6 , G 7 , X, Y and Z are as defined above.
- Azines signifies the member of the azabenzene series, having 1, 2 or 3 of the methylidyne groups of the benzene molecule replaced with nitrogen atoms, such as pyridine, pyrimidine and 1,3,5-triazine.
- alkyl as well as other groups having the prefix “alk”, such as alkoxy means carbon chains which may be linear or branched, and combinations thereof, unless the carbon chain is defined otherwise.
- alkyl groups include but are not limited to methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, and the like.
- Aryl means a mono- or poly cyclic aromatic ring system containing carbon ring atoms.
- the preferred aryl is monocyclic or bicyclic 6-10 membered aromatic ring systems. Phenyl and naphthyl are preferred aryls. The most preferred aryl is phenyl.
- Heterocycle and “heterocyclyl” refer to saturated or unsaturated non-aromatic rings or ring systems containing at least one heteroatom selected from O, S and N, further including the oxidized forms of sulfur, namely SO and SO 2 .
- heterocycles include tetrahydrofuran (THF), dihydrofuran, 1,4-dioxane, morpholine, 1 ,4-dithiane, piperazine, piperidine, 1,3-dioxolane, imidazolidine, imidazoline, pyrroline, pyrrolidine, tetrahydropyran, dihydropyran, oxathiolane, dithiolane, 1,3-dioxane, 1,3-dithiane, oxathiane, thiomorpholine, and the like.
- THF tetrahydrofuran
- dihydrofuran 1,4-dioxane
- morpholine 1 ,4-dithiane
- piperazine piperidine
- 1,3-dioxolane imidazolidine
- imidazoline imidazoline
- pyrroline pyrrolidine
- tetrahydropyran dihydropyran
- Heteroaryl means an aromatic or partially aromatic heterocycle that contains at least one ring heteroatom selected from O, S and N. Heteroaryls also include heteroaryls fused to other kinds of rings, such as aryls, cycloalkyls and heterocycles that are not aromatic.
- group G4 Especially preferred for group G4 are:
- Z in ZG5 is preferably O.
- G3 groups examples include:
- Preferred G5 groups are:
- R be chloro or -O-isopropyl or -0-2-methylpropyl and that G4 be one of the preferred G4 groups described above.
- the compounds of the invention may be administered in the form of a pro-drug.
- a pro-drug is a bioprecursor or pharmaceutically acceptable compound being degradable in the body to produce a compound of the invention (such as an ester or amide of a compound of the invention, particularly an in- vivo hydro lysable ester).
- a compound of the invention such as an ester or amide of a compound of the invention, particularly an in- vivo hydro lysable ester.
- Various forms of prodrugs are known in the art.
- the compounds of the present invention may be administered in the form of a pro-drug.
- a pro-drug is a bioprecursor or pharmaceutically acceptable compound being degradable in the body to produce a compound of the invention such as esters, particularly an in- vivo hydrolysable ester, amides, N-oxides, phosphate esters, glycoamide esters, glyceride conjugates or the pharmaceutically-acceptable salts thereof.
- the prodrugs may add to the value of the present compound's advantages in adsorption, pharmacokinetics or in distribution. (Ref : US6624176) Various forms of prodrugs are known and have been described in the art, for example:
- pro-drugs are as follows- an in vivo hydrolysable ester of a compound of formula (I) containing a carboxy or hydroxy group; a pharmaceutically acceptable ester which is cleaved in the human or animal body to produce the parent acid or alcohol.
- Suitable pharmaceutically acceptable esters for carboxy include Ci_6alkoxymethyl esters for example methoxymethyl, C 1-6 alkanoyloxymethyl esters for example pivaloyloxymethyl, phthalidyl esters, C3-8 cycloalkoxycarbonyloxy, C 1-6 alkyl esters for example & 1- eye lohexylcarbonyloxy ethyl; l,3-dioxolen-2- onylmethyl esters for example 5-methyl-l,3-dioxolen-2-onylmethyl; and Ci_ 6 alkoxycarbonyloxyethyl esters for example 1 -methoxycarbonyloxyethyl and may be formed at any carboxy group in the compounds of this invention.
- Suitable pharmaceutically-acceptable esters for hydroxy include inorganic esters such as phosphate esters (including phosphorarnidic cyclic esters) and ⁇ -acyloxyalkyl ethers and related compounds which as a result of the in -vivo hydrolysis of the ester breakdown to give the parent hydroxy group/s.
- inorganic esters such as phosphate esters (including phosphorarnidic cyclic esters) and ⁇ -acyloxyalkyl ethers and related compounds which as a result of the in -vivo hydrolysis of the ester breakdown to give the parent hydroxy group/s.
- ⁇ -acyloxyalkyl ethers include acetoxymethoxy and 2,2-dimethylpropionyloxymethoxy.
- a selection of in- vivo hydro lysable ester forming groups for hydroxy include C 1-10 alkanoyl, for example acetyl; benzoyl; phenylacetyl; substituted benzoyl and phenylacetyl, Ci-io alkoxycarbonyl (to give alkyl carbonate esters), for example ethoxycarbonyl; di-(Ci_4)alkylcarbamoyl and N- (di-(Ci_4) alkylaminoethyl)-N- 1 (C 1-4 ) alkylcarbamoyl (to give carbamates); di-( C 1-4 ) alkylaminoacetyl and carboxyacetyl.
- Other interesting in- vivo hyrolysable esters include, for example, R a C(O) 0(C
- Pharmaceutically acceptable salts also include compounds in which the main compound functions as an acid and is reacted with an appropriate base to form a salt e.g. sodium, potassium, magnesium, ammonium and chlorine salts.
- a salt e.g. sodium, potassium, magnesium, ammonium and chlorine salts.
- acid addition salts of the claimed compounds may be prepared by reaction of the compounds with the appropriate inorganic or organic acid via any of a number of known methods.
- alkaline earth metals salts can be prepared by reacting the compounds of the invention with an appropriate base via a variety of known methods.
- the invention includes a pharmaceutical or veterinary composition comprising one or more compounds of the invention together with a pharmaceutically or veterinarily acceptable carrier and further includes a compound of the invention formulated for pharmaceutical or veterinary administration.
- the invention further includes any such compound for use in medicine, and especially for use in the treatment of any metabolic disorder, as described herein, particularly for the treatment of diabetes.
- compositions of the invention may be in a form suitable for oral use (for example as tablets, lozenges, hard or soft capsules, aqueous or oily suspensions, emulsions, dispersible powders or granules, syrups or elixirs), for topical use (for example as creams, ointments, gels, or aqueous or oily solutions or suspensions), for administration by inhalation (for example as a finely divided powder or a liquid aerosol), for administration by insufflation (for example as a finely divided powder) or for parenteral administration (for example as a sterile aqueous or oily solution for intravenous, subcutaneous, intramuscular or intramuscular dosing or as a suppository for rectal dosing).
- a therapeutically effective amount of a compound in accordance with this invention means an amount of compound that is effective to treat obesity and/or type II diabetes.
- the therapeutically effective amount or dosage of a compound according to this invention can vary within wide limits and may be determined in a manner known in the art. Such dosage will be adjusted to the individual requirements in each particular case including the specific compound(s) being administered, the route of administration, the condition being treated, as well as the patient being treated.
- the disclosure also relates to identifying these compounds described in Formula (I), (Ia), (Ib), and (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, which are beneficial for the prophylaxis, management, treatment, control of progression, or adjunct treatment of diseases and/or medical conditions where the activation of glucokinase would be beneficial, such as diabetes, metabolic syndrome X and/or diabetes-related complications and as therapeutic and/or prophylactic agents for obesity.
- the disclosure further relates to compounds of Formula (I), (Ia), (Ib), and (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, for use in the manufacture of medicament for the treatment of diabetes, obesity, metabolic syndrome X, insulin resistance, impaired glucose tolerance and dyslipidemia.
- the pharmaceutical preparations can also contain preserving agents, solubilizing agents, stabilizing agents, wetting agents, emulsifying agents, sweetening agents, coloring agents, flavoring agents, salts for varying the osmotic pressure, buffers, coating agents or antioxidants. They can also contain other therapeutically valuable substances, including additional active ingredients other than those of formula (I), (Ia), (Ib) or (Ic).
- R 3 is selected from the group consisting of aryl, wherein aryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from C1-C12 alkyl and preferably C 1-6 alkyl, cycloalkyl and preferably C 3 -Cs cycloalkyl; heteroaryl, wherein heteroaryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from hydroxy, halogen, CO 2 H, C 1-6 alkyloxycarbonyl, C 1-6 alkyl, C 3-6 cycloalkyl, and C 1-6 alkoxy and the like, wherein the alkyl and alkoxy are unsubstituted or substituted with one or more groups selected from groups such as halogens, hydroxyl, oxo, CO 2 H.
- R 3 is selected from the group consisting of aryl, wherein aryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from C 1 -C 12 alkyl and preferably C 1-6 alkyl, cycloalkyl and preferably C 3 -Cs cycloalkyl; heteroaryl, wherein heteroaryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from hydroxy, halogen, CO 2 H, C 1-6 alkyloxycarbonyl, C 1-6 alkyl, C 3 _ 6 cycloalkyl, and C j _ 6 alkoxy and the like, wherein the alkyl and alkoxy are unsubstituted or substituted with one or more groups selected from groups such as halogens, hydroxyl, oxo, CO 2 H.
- Example 2 1. Preparation of 2-hydroxy-6-isopropoxy-isonicotinicacid isopropyl ester.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- General Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Azine derivatives, of the general structural Formula (I) wherein G1 and G2 are selected independently from N and CH; R is halogen, or -OG3, or NHG3 where G3 is selected from the group consisting of hydrogen, C1-6, alkyl or alkylene, wherein the said alkyl or alkylene is straight chain or branched unsubstituted or substituted wherein; the substituents are selected from halogen, hydroxyl, amino, C1-2alkylamino, C1-4alkoxy, oxo, and CO2H, Z is O, NH, S or SO2 G5 is selected from the group consisting of: optionally substituted -C1 -12 alkyl, -C2-12 alkenyl, -(CH2)n-aryl, -(CH2)n- heteroaryl, -(CH2)n-heterocyclyl, or -(CH2)n-C3-8 cycloalkyl; Y is either O or S; and G4 represents optionally substituted mono or fused cycles selected from heteroaryl moieties; or salts, solvates, hydrates, or prodrugs thereof are useful in treating hyperglycemia and diabetes.
Description
AZINE COMPOUNDS AS GLUCOKINASE ACTIVATORS
Field of the invention
This disclosure relates to derivatives of azines, their pharmaceutically acceptable salts, the polymorphs of these derivatives and the salts, and compositions thereof useful as glucokinase activators that are useful in the prophylactic or therapeutic treatment of hyperglycemia and diabetes, particularly type II diabetes. Further, methods of treatment of diseases mediated by Glucokinase using said compounds are also disclosed.
Background of the invention
Diabetes is a metabolic disorder due to multiple causative factors, characterized by hyperglycemia (high blood sugar) in the fasting state or after administration of glucose during an oral glucose tolerance test, and other signs, as distinct from a single illness or condition. Acute complications of diabetes as hypoglycemia, ketoacidosis or nonketotic hyperosmolar coma may occur if the disease is not adequately controlled. Serious long-term complications include cardiovascular disease, chronic renal failure (diabetic nephropathy), retinal damage (which can lead to blindness and is the most significant cause of adult blindness in the non-elderly in the developed world), nerve damage of several kinds, and microvascular damage, which may cause erectile dysfunction (impotence) and poor healing. Poor healing of wounds, particularly of the feet, can lead to gangrene which can require amputation-the leading cause of non-traumatic amputation in adults in the developed world.
The World Health Organization recognizes three main forms of diabetes: type 1, type 2, and gestational diabetes (occurring during pregnancy), which have similar signs, symptoms, and consequences, but different causes and population distributions. Ultimately, all forms are due to the beta cells of the pancreas being unable to produce sufficient insulin to prevent hyperglycemia. Type 1 is usually due to autoimmune destruction of the pancreatic beta cells which produce insulin. Type 2 is characterized by tissue -wide insulin resistance and varies widely; it sometimes progresses to loss of beta cell function. Gestational diabetes
is similar to type 2 diabetes, in that it involves insulin resistance; the hormones of pregnancy cause insulin resistance in those women genetically predisposed to developing this condition.
Glucokinase (GK), also known as hexokinase IV or D, is a key regulatory enzyme in the liver and in the pancreatic β-cell that contributes to the maintenance of blood glucose homeostasis. Glucokinase, like other hexokinases, facilitates the phosphorylation of glucose to glucose-6-phosphate, however, glucokinase has a 100-fold lower substrate affinity, K1n, for glucose compared to hexokinases I, II, and III, and is not subjected to feed-back inhibition by glucose-6-phosphate.
Glucokinase occurs in cells in the liver, pancreas, gut, and brain of humans and most other vertebrates. In each of these organs, especially in the liver and in the pancreatic β-cell it plays an important role in the regulation of carbohydrate metabolism by acting as a glucose sensor, triggering shifts in metabolism or cell function in response to a rising or falling glucose level.
Molecular biological studies of the glucokinase gene and its products have advanced our understanding of how this gene is differentially regulated in the liver and beta cell. The production of an active glucokinase isoform is determined by both transcriptional and post- transcriptional events. Two different promoter regions that are widely separated in a single glucokinase gene are used to produce glucokinase mRNAs in the liver, pancreatic beta cell, and pituitary. The different transcription control regions are tissue-specific in their expression and are differentially regulated.
In liver, glucokinase gene expression is regulated by insulin and cAMP, whereas in the beta cell it is regulated by glucose. The upstream glucokinase promoter region, which gives rise to the glucokinase mRNA in pituitary and pancreas, is structurally and functionally different from the downstream promoter region, which gives rise to the glucokinase mRNA in liver. The use of distinct promoter regions in a single glucokinase gene enables a different set of transcription factors to be utilized in the liver and islet, thus allowing a functionally similar gene product to be regulated in a manner consistent with the different functions of these two tissues. In addition, the splicing of the glucokinase pre-mRNA is regulated in a tissue-specific manner and can affect the activity of the gene product. It plays a crucial role
in the regulation of insulin secretion and has been termed the pancreatic β-cell sensor (Matschinsky FM, 2002).
A peculiar property of the monomeric GK is its cooperative kinetics with its substrate glucose [Hill coefficient of 1.7 where values of > 1 indicate positive cooperativity — that is, the binding of one ligand facilitates binding of subsequent ligands at other sites on a multimeric receptor complex; the Hill coefficient was originally worked out for the binding of oxygen to hemoglobin (Hill coefficient of 2.8)]. Most importantly, single -point mutations
(V62M, D158A, Y214A, V455M, F456V) in a region distinct from the substrate binding sites of the enzyme lead to allosteric activation of GK — this unexpected finding has fueled the hunt for GK activators .
Glucokinase activators (GKAs) may represent a new approach to diabetes therapy as these compounds attenuate hyperglycemia by increasing hepatic glucose metabolism and/or glucose-stimulated pancreatic insulin release. Several activators of glucokinase have been described in the literature (Grimsby J, Sarabu et.al. 2003, Brocklehurst KJ et al, 2004, Kamata K, et al 2004, Castelhano AL, et al 2005, Efanov AM, et al, 2005, McKerrecher D, et al, 2005, McKerrecher D, et al 2006, Futamura M, et al, 2006, Coope GJ, et al, 2006). The potential use of these agents, GKAs, in the therapeutic management of type 2 diabetes has also been reviewed (Leighton B, et al, 2005, Sarabu R,et al, 2005, Matschinsky FM, et al, 2006).
US 2007/0078168 discloses GK activators of for instance the formula:

Summary of the invention
The present disclosure is directed to azine derivatives which are glucokinase activators and are useful in the treatment or prevention of diseases in which glucokinase activator is involved, such as type I and type II diabetes and obesity. The invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which the glucokinase activator is involved.
Detailed description of the invention
The present invention provides azine derivatives, of the general structural Formula 1

Gi and G2 are selected independently from N and CH;
R is halogen, or -OG3, or NHG3 where
G3 is selected from the group consisting of hydrogen, C1-6, alkyl or alkylene, wherein the said alkyl or alkylene is straight chain or branched unsubstituted or substituted wherein; the substituents are selected from halogen, hydroxyl, amino, Ci-2alkylamino, Ci_4alkoxy, oxo, and CO2H,
Z is O, NH, S or SO2
G5 is selected from the group consisting of: optionally substituted -Cl -12 alkyl, -C2-12 alkenyl, -(CH2)n-aryl, -(CH2)n- heteroaryl, -(CH2)n-heterocyclyl, or -(CH2)n-C3-8 cycloalkyl;
Y is either O or S; and
G4 represents optionally substituted mono or fused cycles selected from heteroaryl moieties;
or a pharmaceutically or veterinarily acceptable salt, solvate, hydrate, or prodrug thereof.
Preferably, G5 is selected from the group consisting of:
-Cl -12 alkyl, wherein the alkyl is straight chain or branched and is unsubstituted or substituted with one or more substituents independently selected from halogen, hydroxyl, Ci- 4alkoxy, Ci-4alkylamino, oxo, carbamimidoyl, amino, amido, or CO2H,
-C2-12 alkenyl, wherein the alkenyl is unsubstituted or substituted with one or more groups, independently selected from halogen, hydroxyl, Ci-4alkoxy, Ci-4alkylamino, oxo, carbamimidoyl, amino, amido, or CO2H,
-(CH2)n-aryl, wherein aryl is unsubstituted or substituted with one or more substituents which are independently selected from hydroxy, halogen, CO2H, C 1-6 alkyloxycarbonyl, Cl- 6 alkyl, C 1-6 alkoxy, -NH2; -NHV, -NVV, -NHCO-V, -SO2-W or -CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more substituents
independently selected from halogens, hydroxyl, oxo, or CO2H, wherein V is Ci _i 2 alkyl, Ci _i 2 alkenyl, -(CH2)n-aryl, -(CH2)n-heteroaryl, -(CH2)n- heterocyclyl, -(CH2)n-cycloalkyl, wherein W is Ci_i2alkyl, Ci .gcycloalkyl, or -NR R2 wherein each of R and R independently is Ci .3 alkyl or together form an non-aromatic or aromatic heterocycle, and wherein V and W are each optionally substituted with one or more substituents independently selected from halogens, hydroxyl, oxo, and CO2H,
-(CH2)n-heteroaryl, wherein heteroaryl is unsubstituted or substituted with one or more substituents independently selected from hydroxy, halogen, CO2H, Cl -6 alkyloxycarbonyl,
Cl-6 alkyl, Cl-6 alkoxy, -NH2; -NHV, -NW, -NHCO-V, -SO2-W or -CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more substituents independently selected from halogens, hydroxyl, oxo, or CO2H, and V and W are as defined above,
-(CH2)n-heterocyclyl, wherein heterocyclyl is unsubstituted or substituted with one or more substituents independently selected from oxo, hydroxy, halogen, CO2H, Cl-6 alkyloxycarbonyl, Cl-6 alkyl, Cl- 6 alkoxy, -NH2, -NHV, -NW, -NHCO-V, -SO2-W or -
CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more substituents independently selected from halogens, hydroxyl, oxo, or CO2H, and V and W are as defined above,
or -(CH2)n-C3-8 cycloalkyl; and preferably (CH2)n-C3-6 cycloalkyl, wherein cycloalkyl is unsubstituted or substituted with one to three substituents independently selected from halogen, hydroxy, CO2H, Cl-6 alkyloxycarbonyl, alkyl, alkoxy, -NH2 -NHV, -NVV, - NHCO-V, -SO2-W or -CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more halogens, hydroxyl, oxo, or CO2H, and V and W are as defined above; and wherein each "n" is an integer of any value ranging from 0-10 and is more preferably 1-6, more preferably 1, 2, 3, or 4.
Y is preferably O.
Compounds in which R is OG3 where G3 is isopropyl are particularly preferred.
Preferably, compounds of the invention meet at least one of the following criteria.
Criterion 1: Co-crystallisability
Preferably, compounds of the invention are co-crystallisable with recombinantly expressed glucokinase (GK). Preferably, such compounds are co-crystallisable therewith according to the following protocol.
The co-crystallization of Glucokinase (GK) with activators is set up using purified recombinant human enzyme (11-465 aa of liver isoform).
Recombinant GK protein is expressed in Escherichia coli as a GST-tagged fusion protein and is purified by glutathione-agarose affinity chromatography. The recombinant GK protein after the removal of GST- tag is further purified by size exclusion chromatography using Superdex S-200 (GE health Sciences) column on an FPLC using 2OmM Tris, pH 7.5, 50 mM NacCl, 5 mM TCEP and 20 mM Glucose. The recombinant GK is incubated with the compound of the invention at a concentration of 150 to 250 μM for 30 min to 1 Hr at room temperature and concentrated to 10 mg/ml using 50 KDa cut-off
Centricon (Millipore). Crystallization is set up by the hanging drop vapor diffusion method at room temperature. 1 - 2.5μl of protein is mixed with equal volumes of well solution (0.1 M Hepes, pH 6.6 and 25-31 % PEG 1500) and crystallization is set up. Bi-pyramidal crystals appear within 3 days and are allowed to grow for 7 - 15 days.
Criterion 2: Ability to increase the V-max of GK by a factor of 1.2 or more
Criterion 3: An EC^n value of < 10 uM
Criterion 4: A Kn value of < 5mM at a test concentration of a compound of the invention of 5O uM
Criterion 5 a) an activation (see below) at a 50 μM concentration of test compound of at least 100% at 2mM glucose; or b) an activation at a 50 μM concentration of test compound of at least 50% at 7mM glucose; or c) an activation at a 50 μM concentration of test compound of at least 25% at 12mM glucose.
Each of the activation, V-max, EC50 and Km may be measured in an enzymatic assay as follows. An enzymatic Glucokinase (GK) assay is set up using purified recombinant human enzyme (pancreatic full length).
In this coupled enzymatic assay, GK calatyzes glucose phosphorylation in the presence of Mg-ATP. The product formed is Glucose-6-phosphate which is oxidized to 6-phospho D-gluconate by an excess of glucose-6-phosphate dehydrogenase in the presence of nicotinamide adenine dinucleotide phosphate (NADP). The production of reduced nicotinamide adenine dinucleotide phosphate (NADPH) shows as an increase in absorbance used to monitor GK activity. Recombinant GK is expressed in Escherichia coli as a GST-tagged fusion protein and is purified by glutathione-agarose affinity chromatography. The recombinant GK after the removal of GST- tag is used for the enzymatic assay. The assay is performed in a final incubation volume of 100 μl in a 96- well transparent flat bottom microplates (Greiner Bio-one 655101).
The assay mixture consisting of a selected concentration of a compound according to the invention, 25 mM HEPES pH 7.1 , 25 mM KCl, 5 mM β Mercaptoethanol, 1 mM ATP, 2.5 mM MgCl2 ,D-Glucose- 2 mM/7 mM /12 mM, 2 units/ ml yeast glucose 6- phosphate dehydrogenase (Roche Applied Sciences, Catalog no. 10127655001) and 5 % DMSO is pre-incubated at 300C for 5 minutes. (Ref : Brocklehurst K.J., et al (2004) Diabetes 53: 535-541). Human recombinant glucokinase (-200 ng) is added of to the assay reaction except the blank. The increase in A 34onm is monitored in Spectramax at
30° C for 15 min . The A 34onm is obtained for both test and positive control after substracting the blank.
The percentage activation is calculated by the equation:
% Activation = (V3 /V0 -1) x 100 where:
V3 = Activity in mmoles product/min in the presence of tested compound concentration (50 μM).
Vo = Activity in the absence of compound.
Km and vmax are related by the standard Michaelis-Menton equation:
V = K + M ' ^ being the concentration of the substrate glucose.
For Km for Glucose and vmax determination, glucose is used in 12 increasing concentrations. Suitable concentrations where the compound of the invention is very effective as an activator are from 0.05 mM -100 mM. Suitable concentrations where the compound under test is active, but less effective, as an activator are 0.2 mM - 100 mM. A dose response curve is generated using GraphPad Prism software Version 4 (San Diego, California, USA) using non linear regression curve fit for sigmoidal dose response (variable slope). Typically the control K mwithout the compound of the invention should give values in the range of 8 to 9 mM with a Hill slope of 1.6. The procedure is repeated over a suitable range of concentrations of the compound of the invention under test.
For EC50 determination at 10 mM Glucose, the effective compound concentration that produces 50 % of maximal increase in GK activity observed using a saturating activator concentration is calculated. Two fold activation (saturating concentration) is observed at 1OmM glucose in general. A dose response curve is generated using GraphPad Prism software Version 4 (San Diego, California, USA) using non linear regression curve fit for sigmoidal dose response ( variable slope).
More preferred compounds of the invention satisfy any two, more preferably any three, more preferably any four, more preferably any five, more preferably any six, and most preferably all seven of the above criteria. The required increase in V-max is the
single most preferred criterion to satisfy and where compounds do not satisfy all the criteria, compounds satisfying combinations of criteria including the V-max criterion are preferred.
Preferred values to satisfy in each of criteria 2-5(c) are as follows: an increase in Vmax by a factor of 1.3, more preferably 1.4, most preferably 1.5;
an EC50 value of 5< μM, more preferably <1 μM, more preferably <500 nM;
a Km value of < 4mM, more preferably < 3 mM, at a test concentration of a compound of the invention of 50 μM;
activations of higher than 200% at 2mM glucose, or of higher than 100% at 7 mM glucose, or of higher than 50% at 12 mM glucose using 50 μM of test compound.
Exemplary G4 rings are illustrated by way of representatives only, and should in no way be construed to be limited to those listed below but include:




wherein
* is the point of attachment to Formula 1 ,
G6 is selected from group of H, Ci_io alkyl, wherein the alkyl is an optionally substituted straight or branched chain moiety
G7 is independently selected from any of the groups such as CX3 wherein X is a halogen and preferably fluorine; CMO alkyl wherein the alkyl is unsubstituted or substituted, straight or branched chain; or an aryl which is optionally substituted.
One embodiment of the present disclosure encompasses the compounds represented by formula Ia below;

wherein R, G2 , G3, G4, G5, G6, G7, X, Y and Zare as defined above.
In another embodiment of the present disclosure, as depicted in the formula Ib below

wherein; R, Gi, G3, G4, G5, G6, G7, X, Y and Z are as defined above.
In yet another embodiment of the present disclosure both Gi and G2 represent N , as depicted in formula 1 c as below,

and R, G3, G4, G5, G6, G7, X, Y and Z are as defined above.
As used herein the following definitions are applicable:
"Azines" signifies the member of the azabenzene series, having 1, 2 or 3 of the methylidyne groups of the benzene molecule replaced with nitrogen atoms, such as pyridine, pyrimidine and 1,3,5-triazine.
"Alkyl", as well as other groups having the prefix "alk", such as alkoxy means carbon chains which may be linear or branched, and combinations thereof, unless the carbon chain is defined otherwise. Examples of alkyl groups include but are not limited to methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, and the like.
"Cycloalkyl" means a saturated carbocyclic ring having a specified number of carbon atoms. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, and the like. A cycloalkyl group generally is monocyclic unless stated otherwise. Cycloalkyl groups are saturated unless otherwise defined.
The term "alkoxy" refers to straight or branched chain alkoxides, wherein the number of carbon atoms specified (e.g., C 1-6 alkoxy), or any number within this range [i.e., methoxy (MeO-), ethoxy, isopropoxy, etc.]
"Aryl" means a mono- or poly cyclic aromatic ring system containing carbon ring atoms. The preferred aryl is monocyclic or bicyclic 6-10 membered aromatic ring systems. Phenyl and naphthyl are preferred aryls. The most preferred aryl is phenyl.
"Heterocycle" and "heterocyclyl" refer to saturated or unsaturated non-aromatic rings or ring systems containing at least one heteroatom selected from O, S and N, further including the oxidized forms of sulfur, namely SO and SO2. Examples of heterocycles include tetrahydrofuran (THF), dihydrofuran, 1,4-dioxane, morpholine, 1 ,4-dithiane, piperazine, piperidine, 1,3-dioxolane, imidazolidine, imidazoline, pyrroline, pyrrolidine, tetrahydropyran, dihydropyran, oxathiolane, dithiolane, 1,3-dioxane, 1,3-dithiane, oxathiane, thiomorpholine, and the like.
"Heteroaryl" means an aromatic or partially aromatic heterocycle that contains at least one ring heteroatom selected from O, S and N. Heteroaryls also include heteroaryls fused to other kinds of rings, such as aryls, cycloalkyls and heterocycles that are not aromatic. Examples of heteroaryl groups include pyrrolyl, isothiazolyl, pyrazolyl, pyridinyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, pyrimidinyl, pyrazinyl, benzothiazolyl, benzothiadiazolyl, indolinyl, pyridazinyl, isoindolyl, cinnolinyl, phthalazinyl,
quinazolinyl, naphthyridinyl, purinyl, benzimidazolyl, quinolyl, indolyl, isoquinolyl, and the like.
"Halogen" refers to fluorine, chlorine, bromine and iodine. Chlorine and fluorine are generally preferred. Fluorine is most preferred when the halogens are substituted on an alkyl or alkoxy group (e.g. CF3O and CF3CH2O).
Especially preferred for group G4 are:


Z in ZG5 is preferably O.
Examples of suitable G3 groups to be used in any combination include:




Especially, but not only, in connection with each of these, it is preferred that R be chloro or -O-isopropyl or -0-2-methylpropyl and that G4 be one of the preferred G4 groups described above.
The disclosure relates to compounds described in formula (I), (Ia), (Ib), and (Ic), prodrugs thereof, pharmaceutically acceptable salts, stereoisomers, tautomers, solvates, hydrates, prodrugs thereof or mixtures thereof.
The disclosure also relates to compounds of interest in formula (I), (Ia), (Ib), and (Ic) also includes its polymorphs, and suitable formulations for oral dosage.
The compounds of the invention may be administered in the form of a pro-drug. A pro-drug is a bioprecursor or pharmaceutically acceptable compound being degradable in the body to produce a compound of the invention (such as an ester or amide of a compound of the invention, particularly an in- vivo hydro lysable ester). Various forms of prodrugs are known in the art.
The compounds of the present invention may be administered in the form of a pro-drug. A pro-drug is a bioprecursor or pharmaceutically acceptable compound being degradable in the body to produce a compound of the invention such as esters, particularly an in- vivo hydrolysable ester, amides, N-oxides, phosphate esters, glycoamide esters, glyceride conjugates or the pharmaceutically-acceptable salts thereof.
The prodrugs may add to the value of the present compound's advantages in adsorption, pharmacokinetics or in distribution. (Ref : US6624176) Various forms of prodrugs are known and have been described in the art, for example:
a) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985) and Methods in 5 Enzymology, Vol. 42, p. 309- 396, edited by K. Widder, et al. (Academic Press, 1985); b) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H. Bundgaard, Chapter 5 "Design and Application of Prodrugs", by H. Bundgaard p. 113-
191 (1991); c) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992); 10 d) H. Bundgaard, et al., Journal of Pharmaceutical Sciences, 77, 285 (1988); and e) N. Kakeya, et al., Chem Pharm Bull, 32, 692 (1984). f) Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry, and
Enzymology By Bernard Testa and Joachim M. Mayer. Volkmar Wehner, Aventis
Pharma Deutschland GmbH, Frankfurt am Main, Germany
[WO03074485A2]
Examples of pro-drugs are as follows- an in vivo hydrolysable ester of a compound of formula (I) containing a carboxy or hydroxy group; a pharmaceutically
acceptable ester which is cleaved in the human or animal body to produce the parent acid or alcohol.
Suitable pharmaceutically acceptable esters for carboxy include Ci_6alkoxymethyl esters for example methoxymethyl, C1-6 alkanoyloxymethyl esters for example pivaloyloxymethyl, phthalidyl esters, C3-8 cycloalkoxycarbonyloxy, C 1-6 alkyl esters for example & 1- eye lohexylcarbonyloxy ethyl; l,3-dioxolen-2- onylmethyl esters for example 5-methyl-l,3-dioxolen-2-onylmethyl; and Ci_6 alkoxycarbonyloxyethyl esters for example 1 -methoxycarbonyloxyethyl and may be formed at any carboxy group in the compounds of this invention. Suitable pharmaceutically-acceptable esters for hydroxy include inorganic esters such as phosphate esters (including phosphorarnidic cyclic esters) and α-acyloxyalkyl ethers and related compounds which as a result of the in -vivo hydrolysis of the ester breakdown to give the parent hydroxy group/s. Examples of α -acyloxyalkyl ethers include acetoxymethoxy and 2,2-dimethylpropionyloxymethoxy. A selection of in- vivo hydro lysable ester forming groups for hydroxy include C1-10 alkanoyl, for example acetyl; benzoyl; phenylacetyl; substituted benzoyl and phenylacetyl, Ci-io alkoxycarbonyl (to give alkyl carbonate esters), for example ethoxycarbonyl; di-(Ci_4)alkylcarbamoyl and N- (di-(Ci_4) alkylaminoethyl)-N- 1 (C1-4) alkylcarbamoyl (to give carbamates); di-( C1-4) alkylaminoacetyl and carboxyacetyl. Other interesting in- vivo hyrolysable esters include, for example, RaC(O) 0(C
6)alkyl CO-, wherein Ra is for example, benzyloxy-( C1-4 )alkyl, or phenyl). Suitable substituents on a phenyl group in such esters include, for example, 4- (C1-4 ) piperazino-( C1-4 )alkyl, piperazino (C-4) alkyl and morpholino-( C1-4 ) alkyl. Ref :[ WO03074485A2] The term "pharmaceutically acceptable salts" and the term 'veterinarily acceptable salts' as used herein include any salt with both inorganic or organic pharmaceutically or veterinarily acceptable acids such as hydrochloric acid, hydrobromic acid, trifluoroacetic acid, nitric acid, sulfuric acid, phosphoric acid, citric acid, formic acid, maleic acid, acetic acid, succinic acid, tartaric acid, methanesulfonic acid, para-toluene sulfonic acid and the like. In addition, a suitable pharmaceutically-acceptable salt or veterinarily acceptable salt also includes any pharmaceutically or veterinarily acceptable base salt such as amine salts,
trialkyl amine salts and the like. Such salts can be formed quite readily by those skilled in the art using standard techniques.
The term "pharmaceutically acceptable salts" and the term 'veterinarily acceptable salts' as used herein include those obtained by reacting the main compound functioning as a base with an inorganic or organic acid to form a salt for example, salts of hydrochloric acid, hydrobromic acid, sulfuric acid , phosphoric, and methane sulfonic acid, camphor sulfonic acid oxalic acid, maleic acid, acetic acid, succinic acid, tartaric acid, para-toluene sulfonic acid, citric acid, benzoic acid, salicylic acid, madelic acid, pamoic acid, and carbonic acid.
Pharmaceutically acceptable salts also include compounds in which the main compound functions as an acid and is reacted with an appropriate base to form a salt e.g. sodium, potassium, magnesium, ammonium and chlorine salts. It is obvious to those skilled in the art that acid addition salts of the claimed compounds may be prepared by reaction of the compounds with the appropriate inorganic or organic acid via any of a number of known methods. Alternatively alkaline earth metals salts can be prepared by reacting the compounds of the invention with an appropriate base via a variety of known methods.
The following are further examples of acid salts that can be obtained by reaction with inorganic or organic acids: acetates, aDIPEAtes, alginates, citrates, aspartates, benzoates, benzenesulfonates, bisulfates, butyrates, camphorates, digluconates, cyclopentanepropionates, dodecylsulfates, ethanesulfonates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, fumarates, hydrobromides, hydroiodides, 2-hydroxy-ethanesulfonates, lactates, maleates, methanesulfonates, nicotinates, 2-naphthalenesulfonates, oxalates, pamoates, pectinates, persulfates, 3- phenylpropionates, picrates, pivalates, propionates, succinates, tartrates, thiocyanates, tosylates, mesylates and undecanoates.
For example, the pharmaceutically acceptable salt can be a hydrochloride, a hydrobromide, a hydroformate, or a maleate.
Preferably, the pharmaceutically acceptable salts formed are 'veterinarily acceptable salts' and they are particularly pharmaceutically acceptable for administration for mammals. However, pharmaceutically unacceptable salts of the compounds are
suitable as intermediates, for example, for isolating the compound as a salt and then converting the salt back to the free base compound by treatment with an alkaline reagent. The free base can then, if desired, be converted to a pharmaceutically acceptable acid addition salt.
The invention includes a pharmaceutical or veterinary composition comprising one or more compounds of the invention together with a pharmaceutically or veterinarily acceptable carrier and further includes a compound of the invention formulated for pharmaceutical or veterinary administration. The invention further includes any such compound for use in medicine, and especially for use in the treatment of any metabolic disorder, as described herein, particularly for the treatment of diabetes.
The compositions of the invention may be in a form suitable for oral use (for example as tablets, lozenges, hard or soft capsules, aqueous or oily suspensions, emulsions, dispersible powders or granules, syrups or elixirs), for topical use (for example as creams, ointments, gels, or aqueous or oily solutions or suspensions), for administration by inhalation (for example as a finely divided powder or a liquid aerosol), for administration by insufflation (for example as a finely divided powder) or for parenteral administration (for example as a sterile aqueous or oily solution for intravenous, subcutaneous, intramuscular or intramuscular dosing or as a suppository for rectal dosing). A therapeutically effective amount of a compound in accordance with this invention means an amount of compound that is effective to treat obesity and/or type II diabetes.
Determination of a therapeutically effective amount is within the skill in the art.
The therapeutically effective amount or dosage of a compound according to this invention can vary within wide limits and may be determined in a manner known in the art. Such dosage will be adjusted to the individual requirements in each particular case including the specific compound(s) being administered, the route of administration, the condition being treated, as well as the patient being treated.
In using a compound of the Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, for therapeutic or prophylactic purposes it will
generally be administered so that a daily dose in the range, for example, 0.05 mg to 75 mg per kg body weight is received, given if required in divided doses. In general lower doses will be administered when a parenteral route is employed. Thus, for example, for intravenous administration, a dose in the range, for example, 0.05 mg to 30 mg per kg body weight will generally be used. Similarly, for administration by inhalation, a dose in the range, for example, 0.05 mg to 25 mg per kg body weight will be used.
The disclosure also relates to identifying these compounds described in Formula (I), (Ia), (Ib), and (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, which are beneficial for the prophylaxis, management, treatment, control of progression, or adjunct treatment of diseases and/or medical conditions where the activation of glucokinase would be beneficial, such as diabetes, metabolic syndrome X and/or diabetes-related complications and as therapeutic and/or prophylactic agents for obesity. The disclosure further relates to compounds of Formula (I), (Ia), (Ib), and (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, for use in the manufacture of medicament for the treatment of diabetes, obesity, metabolic syndrome X, insulin resistance, impaired glucose tolerance and dyslipidemia.
The disclosure also relates to a method of prophylactic or therapeutic treatment of hyperglycemia or diabetes, particularly type II diabetes, comprising a step of administering an effective amount of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof.
The disclosure also relates to a method for the prevention of diabetes, particularly type II diabetes, in a human demonstrating pre-diabetic hyperglycemia or impaired glucose tolerance comprising a step of administering an effective prophylactic amount of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof.
Such methods may include a step of selecting a patient on the basis of diagnosis of such a metabolic disorder.
The disclosure also relates to the use of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, as a Glucokinase activator.
The disclosure also relates to the use of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, for the prophylactic or therapeutic treatment of hyperglycemia or diabetes, particularly type II diabetes.
The disclosure also relates to the use of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, for the prevention of diabetes, particularly type II diabetes, in a human demonstrating pre-diabetic hyperglycemia or impaired glucose tolerance.
The disclosure also relates to the use of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, in the manufacture of a medicament for the activation of Glucokinase.
The disclosure also relates to the use of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, in the manufacture of a medicament for the prophylactic or therapeutic treatment of hyperglycemia or diabetes, particularly type II diabetes. The disclosure also relates to the use of a compound of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, in the manufacture of a medicament for the prevention of diabetes, particularly type II diabetes, in a human demonstrating pre-diabetic hyperglycemia or impaired glucose tolerance. The compounds and compositions of the present invention may be optionally employed in combination with one or more other anti-diabetic agents or anti-hyperglycemic agents, which include, for example, sulfonylureas (e.g. glyburide, glimepiride, glipyride, glipizide, chlorpropamide, gliclazide, glisoxepid, acetohexamide, glibornuride, tolbutamide, tolazamide, carbutamide, gliquidone, glyhexamide, phenbutamide, tolcyclamide, etc.), biguanides (e.g. metformin, phenformin, buformin, etc.), glucagon antagonists (e.g. a peptide or non-peptide glucagon antagonist), glucosidase inhibitors (e.g. acarbose, miglitol, etc.), insulin secetagogues, insulin sensitizers (e.g. troglitazone, rosiglitazone, pioglitazone, etc.) and the like; or anti-obesity agents (e.g. sibutramine, orlistat, etc.) and the like. The compounds and compositions of the present invention and the other anti-diabetic agents or anti-hyperglycemic agents may be administered simultaneously, sequentially or separately.
The pharmaceutical compositions of the present invention comprise a compound of Formula (I), (Ia), (Ib), or (Ic) or a pharmaceutically acceptable salt thereof, as an active ingredient, a pharmaceutically acceptable carrier and optionally other therapeutic agent
The pharmaceutical compositions of the present invention comprising compounds of Formula (I), (Ia), (Ib), or (Ic), or pharmaceutically acceptable salt, solvate or pro-drug thereof, may be manufactured in a manner that is known in the art, e.g. by means of conventional mixing, encapsulating, dissolving, granulating, emulsifying, entrapping, dragee- making, or lyophilizing processes. These pharmaceutical preparations can be formulated with therapeutically inert, inorganic or organic carriers. Lactose, corn starch or derivatives thereof, talc, steric acid or its salts can be used as such carriers for tablets, coated tablets, dragees and hard gelatin capsules. Suitable carriers for soft gelatin capsules include vegetable oils, waxes and fats. Depending on the nature of the active substance, no carriers are generally required in the case of soft gelatin capsules. In such cases, pharmaceutically acceptable carriers are considered to include soft gelatin capsules. Suitable carriers for the manufacture of solutions and syrups are water, polyols, saccharose, invert sugar and glucose. Suitable carriers for injection are water, alcohols, polyols, glycerine, vegetable oils, phospholipids and surfactants. Suitable carriers for suppositories are natural or hardened oils, waxes, fats and semi-liquid polyols.
The pharmaceutical preparations can also contain preserving agents, solubilizing agents, stabilizing agents, wetting agents, emulsifying agents, sweetening agents, coloring agents, flavoring agents, salts for varying the osmotic pressure, buffers, coating agents or antioxidants. They can also contain other therapeutically valuable substances, including additional active ingredients other than those of formula (I), (Ia), (Ib) or (Ic).
The present disclosure also relates to the process of preparation of compounds covered by formula (I) as described below under Schemes 1 -5.
Scheme 1:

wherein Ri=G3 and R2=Gs, provided Ri ≠ R2,
R3 is selected from the group consisting of aryl, wherein aryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from C1-C12 alkyl and preferably C1-6 alkyl, cycloalkyl and preferably C3-Cs cycloalkyl; heteroaryl, wherein heteroaryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from hydroxy, halogen, CO2H, C1-6 alkyloxycarbonyl, C1-6 alkyl, C3-6 cycloalkyl, and C1-6 alkoxy and the like, wherein the alkyl and alkoxy are unsubstituted or
substituted with one or more groups selected from groups such as halogens, hydroxyl, oxo, CO2H.
Example 1: Preparation of 6-[(2-isopropoxy-6-methoxy-pyridine-4-carbonyl)- a mi no I -nicotinic acid. Step-1
Preparation of 2-hydroxy-6-methoxy-isonicotinic acid methyl ester.

To a solution of 2, 6-dihydroxy-isonicotinicacid (1Og, 64.5 mmole) in methanol (60 mL) was added concentrated sulfuric acid (10 mL) slowly at 10 0C. After complete addition, reaction mixture was refluxed for 72 hrs. Methanol was removed at reduced pressure and the resultant was basified with saturated sodium bicarbonate solution (pH = 8) and extracted with ethyl acetate (200 ml x 3). The organic layer was washed with saturated sodium bicarbonate solution (2 x 100 mL), brine (2 x 100 mL), dried over sodium sulfate and concentrated at reduced pressure. Crude product was purified by column chromatography over silica gel (60-120 mesh), with 5% ethyl acetate in hexane to give 2- hydroxy-6-methoxy-isonicotinic acid methyl ester as a white solid (4.3 g, 39.4%); MS m/z 184 (M+H+); 1H NMR δ (300 MHz, CDCl3): 3.8 (s, 3H), 3.9 (s, 3H), 6.6 (s, 2H), 11.2 (bs, IH).
Step-2
Preparation of 2-isopropoxy-6-methoxy-isonicotinic acid methyl ester.

To a stirred solution of 2-hydroxy-6-methoxy-isonicotinic acid methyl ester (0.2g, 0.95 mmole) in N,N'-dimethylformamide (10 mL), activated potassium carbonate (0.32g, 2.6 mmole) was added and the resultant reaction mass was stirred at room temperature for 15 min. To the above reaction mixture 2-bromopropane (0.49g, 3.5 mmole) was added at room temperature and stirred at room temperature for 12 hrs. N,N'-dimethylformamide was evaporated at reduced pressure. Distilled water (50 mL) was added to the residue and the resultant was extracted with ethyl acetate (3 x 50 mL). The combined organic layer was washed with brine solution (2 x 50 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. Crude product was purified by column chromatography using 5% ethyl acetate in hexane as the eluant to give 2-isopropoxy-6- methoxy-isonicotinic acid methyl ester as thick oil (0.185g, 86%). MS m/z 226 (M+H+); 1H NMR δ (300 MHz, DMSOd6): 1.1 (d, 6H), 3.8 (d, 6H), 5.2 (m, IH), 6.7 (d, 2H).
Step-3
Preparation of 2-isopropoxy-6-methoxy-isonicotinic acid.

To a stirred solution of 2-isopropoxy-6-methoxy-isonicotinic acid methyl ester (2.3Og, 1.00 mmole) in methanol (10 mL) and water (1 mL), potassium carbonate (2.11g, 1.50 mmole) was added to the reaction mixture and the resultant was stirred at room temperature for 12 hrs. Methanol was evaporated under reduced pressure, the residue obtained was diluted with water (100 mL) and acidified with 10% aqueous solution of
hydrochloric acid (pH = 3). The solid that precipitated out was filtered, washed with water and dried to give 2-isopropoxy-6-methoxy-isonicotinic acid as a white solid (1.85g, 88.1%). MS m/z 210 (M-H+); 1H NMR δ (300 MHz, DMSO-d6): 1.3 (d, 6H), 3.9 (s, 3H), 5.2 (m, IH), 6.8 (s, 2H).
Step-4
Preparation of 6-[(2-isopropoxy-6-methoxy-pyridine-4-carbonyl)-amino]-nicotinic acid methyl ester.
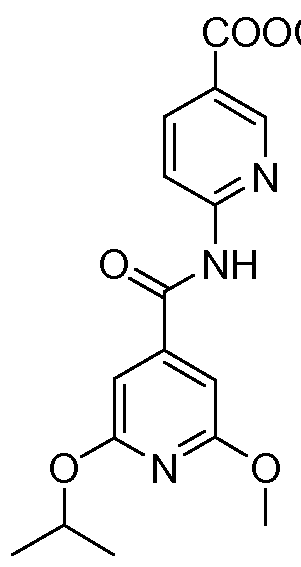
To a stirred solution of 2-isopropoxy-6-methoxy-isonicotinic acid (0.2g, 0.94 mmole) in N,N'-dimethylformamide (10 mL), l-hydroxybenzotriazole (0.15g, 1.13 mmole) followed by N,N'-diisopropylcarbadiimide (0.14g, 1.13 mmole) were added and the reaction mixture stirred at room temperature for 15 min. To the above reaction mixture 6-amino- nicotinic acid methyl ester (0.22g, 1.42 mmole) was added at room temperature and the resulting reaction mixture was heated at 100 0C for 1 h. N,N'-dimethylformamide was evaporated at reduced pressure. To the resultant, distilled water (50 mL) was added followed by extraction with diethyl ether (2 x 100 mL). The combined organic layer was washed with 10% aqueous hydrochloric acid solution (2 x 50 mL), saturated aqueous sodium bicarbonate solution (2 x 50 mL) and brine solution (2 x 50 mL). Organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude product so obtained was purified by crystallization using hexane and chloroform to give 6-[(2-isopropoxy-6-methoxy-pyridine-4-carbonyl)-amino]-nicotinic acid methyl
ester as a white solid (0.28g, 90.62%). MS m/z 346 (M+H+); 1H NMR δ (300 MHz, DMSO-d6): 1.38 (d, 6H), 3.94 (s, 6H), 5.3 (m, IH), 6.7 (d, 2H), 8.4 (m, 2H), 8.6 (bs, IH), 8.9 (d, IH).
Step-5
Preparation of 6-[(2-isopropoxy-6-methoxy-pyridine-4-carbonyl)-amino]-nicotinic acid.

To a stirred solution of 6-[(2-ispropoxy-6-methoxy-pyridine-4-carbonyl)-amino]- nicotinic acid methyl ester (0.2g, 0.58 mmole) in tetrahydrofuran (20 mL) and water (20 mL), sodium hydroxide (0.12g, 2.89 mmole) was added and stirred at room temperature for 1.5 hrs. After reducing the volume to half, the reaction mixture was acidified with 10% hydrochloric acid (pH = 3) to give a precipitate which was filtered off, washed with water and dried at reduced pressure to give a white solid. This was stirred in methanol (15 mL), refluxed, cooled, filtered and dried at reduced pressure to give 6-[(2-isopropoxy-6- methoxy-pyridine-4-carbonyl)-amino]-nicotinic acid as a white solid (0.17Og, 88.5%); MS m/z 330 (M-H+); 1H NMR δ (300 MHz, DMSO-d6): 1.3 (d, 6H), 3.9 (s, 3H), 5.2 (m, IH), 6.8 (d, 2H), 8.3 (m, 2H), 8.9 (m, IH), 11.3 (s, IH), 13.3 (bs, IH).
Scheme 2:


wherein Ri=G3 and R2=Gs,
R3 is selected from the group consisting of aryl, wherein aryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from C1-C12 alkyl and preferably C1-6 alkyl, cycloalkyl and preferably C3-Cs cycloalkyl; heteroaryl, wherein heteroaryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from hydroxy, halogen, CO2H, C1-6 alkyloxycarbonyl, C1-6 alkyl, C3-6 cycloalkyl, and C1-6 alkoxy and the like, wherein the alkyl and alkoxy are unsubstituted or
substituted with one or more groups selected from groups such as halogens, hydroxyl, oxo, CO2H.
Scheme 3:

wherein Ri=G3 and R2=Gs, R3 is selected from the group consisting of aryl, wherein aryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from C1-C12 alkyl and preferably C1-6 alkyl, cycloalkyl and preferably C3-Cs cycloalkyl;
heteroaryl, wherein heteroaryl is unsubstituted or substituted with one or more substituents wherein, the substituents are independently selected from hydroxy, halogen, CO2H, C1-6 alkyloxycarbonyl, C1-6 alkyl, C3_6 cycloalkyl, and Cj_6 alkoxy and the like, wherein the alkyl and alkoxy are unsubstituted or substituted with one or more groups selected from groups such as halogens, hydroxyl, oxo, CO2H.
Scheme 4

I Q R= H, CH3, CH2COOH
Conditions: i. Diethyl Oxalate, Ethanol reflux; ii. K2CO3, MeOH, H2O, RT iii. HOBT, DIC, amines, DMF, 1000C 15 iv. 2,5-dimethoxytetrahydrofuran, AcOH, 10O 0C
References:
1) Chem Pharm Bull. 1968, 16(3), 474-479
2) Molecules 2003, 8, 678-686 0

Example 2: 1. Preparation of 2-hydroxy-6-isopropoxy-isonicotinicacid isopropyl ester.

2. Preparation of 2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-isonicotinic acid Isopropyl ester

To a stirred solution of 2-hydroxy-6-isopropoxy-isonicotinic acid isopropyl ester (0.5g, 2.092mmole) in N,N'-dimethylacetamide (10 mL), cesium carbonate (1.36g, 4.18 mmol) was added and stirred the reaction mass at room temperature for 15 min. To the above reaction mixture 4-fiuoro phenyl methyl sulfone (0.62g, 3.53 mmole) was added at room temperature and raised the temperature to 115°C and stirred at 115°C for 24 hrs. NJST- dimethylacetamide was evaporated using high vacuum and distilled water (150 mL) was added to the residue and extracted with ethyl acetate (3 x 50 mL). The combined organic layer was washed with brine solution (2 x 50 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. Crude product was purified by column
chromatography using eluent 10% ethyl acetate in hexane to afford 2-isopropoxy-6-(4- methanesulfonyl-phenoxy)-isonicotinic acid isopropyl ester as solid. Yield: (0.125g, 15%); MS m/z 394.1 (M+H+); 1HNMR δ (300 MHz, CDCl3): 1.04 (d, 6H), 1.1 (d, 6H), 3.05 (s, 3H), 4.95 (m, IH), 5.01 (m, IH), 7.05 (d, 2H), 7.4 (d, 2H), 8.0 (d, 2H).
3. Preparation of 2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-iso nicotinic acid

To a stirred solution of 2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-iso nicotinic acid isopropyl ester (0.125g, 0.315 mmole) in methanol (9 mL) and water (1 mL), potassium carbonate (0.075g, 0.543 mmole) was added to the reaction and stirred at room temperature for 6 hrs. Methanol was evaporated under reduced pressure, residue obtained was diluted with water (100 mL) and acidified with 10% aqueous solution of hydrochloric acid (pH = 2), solid was precipitated out. Solid was filtered and washed with water, dried to afford. 2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-iso nicotinic acid as a white solid. Yield (O.lOg, 88.52%); MS m/z 352 (M+H+); 1HNMR δ (300 MHz, CDCl3): 1.25 (d, 6H), 3.05 (s, 3H), 4.95 (m, IH), 5.01 (m, IH), 7.10 (d, 2H), 7.35 (d, 2H), 8.0 (d, 2H).
4. Preparation of 2-((2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-pyridine-4- carbonyl)-amino)-thiazol-4-yl)-acetic acid ethyl ester

To a stirred solution of 2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-iso nicotinic acid (0.15g, 0.427mmole) in N,N'-dimethylformamide (5 mL) and 1-hydroxybenzotriazole (0.074g ,0.548 mmol) followed by diisopropylcarbadiimide (0.09mL, 0.634 mmol) was added and stirred the reaction mixture at room temperature for 15 min. To the above reaction mixture(2-amino-thiazol-4-yl)acetic acid ethyl ester (0.087g, 0.467 mmole) was added at room temperature and reaction mixture was heated to 100 0C for 1 h. NJST- dimethylformamide was evaporated using high vacuum, distilled water (50 mL) was added to the residue and extracted with diethyl ether (2 x 100 mL). The combined organic layer was washed with 10% aqueous hydrochloric acid solution (2 x 50 mL), saturated aqueous sodium bicarbonate solution (2 x 50 mL) and brine solution (2 x 50 mL). Organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. Crude product was purified by column chromatography over silica gel (60-120 mesh), with 12% ethyl acetate in hexane to give as a white solid. Yield: (0.08g, 40.75%); MS m/z 519.9 (M+H+); 1H NMR δ (DMSO, 300 MHz): 1.2 (m, 9H), 3.2 (s, 3H), 3.8 (s, 2H), 4.04 (m, 2H), 4.9 (s, IH), 7.1 (s, IH), 7.2 (d, 2H) ,7.49 (d, 2H), 8.0 (d, 2H).
5. Preparation of 2-((2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-pyridine-4- carbonyl)-amino)-thiazol-4-yl)-acetic acid.

To a stirred solution of 2-((2-isopropoxy-6-(4-methanesulfonyl-phenoxy)-pyridine-4- carbonyl)-amino)-thiazol-4-yl)-acetic acid ethyl ester(0.05g, 0.096 mmole) in tetrahydrofuran (10 mL) and water (10 mL), sodium hydroxide (0.02g, 0.5 mmole) was added to the reaction and stirred at room temperature for 1 hrs. After evaporating to half volume the reaction mixture was acidified with 10% hydrochloric acid (pH = 2) to give precipitate. The precipitate was filtered off, washed with water and dried under vacuum to give a white solid. This product was stirred in methanol (10 mL) at reflux, cooled, filtered and dried under vacuum to give the 2-(2-isopropoxy-6-(4-methanesulfonyl- phenoxy)-pyridine-4-carbonyl)-amino)-thiazol-4-yl)-acetic acid as a white solid (0.03Og, 63.48%); MS m/z 492.0 (M+H+); 1H NMR δ (300 MHz, DMSO-d6): 1.2 (m, 6H), 3.2 (s, 3H), 3.8 (s, 2H), 4.9 (s, IH), 7.1 (s, IH), 7.2 (d, 2H), 7.49 (d, 2H), 8.0 (d, 2H).
6. Preparation of 2-isopropoxy-6-(4-methane sulfonyl-phenoxy)-Λ^-thiazol-2-yl- isonicotinamide.

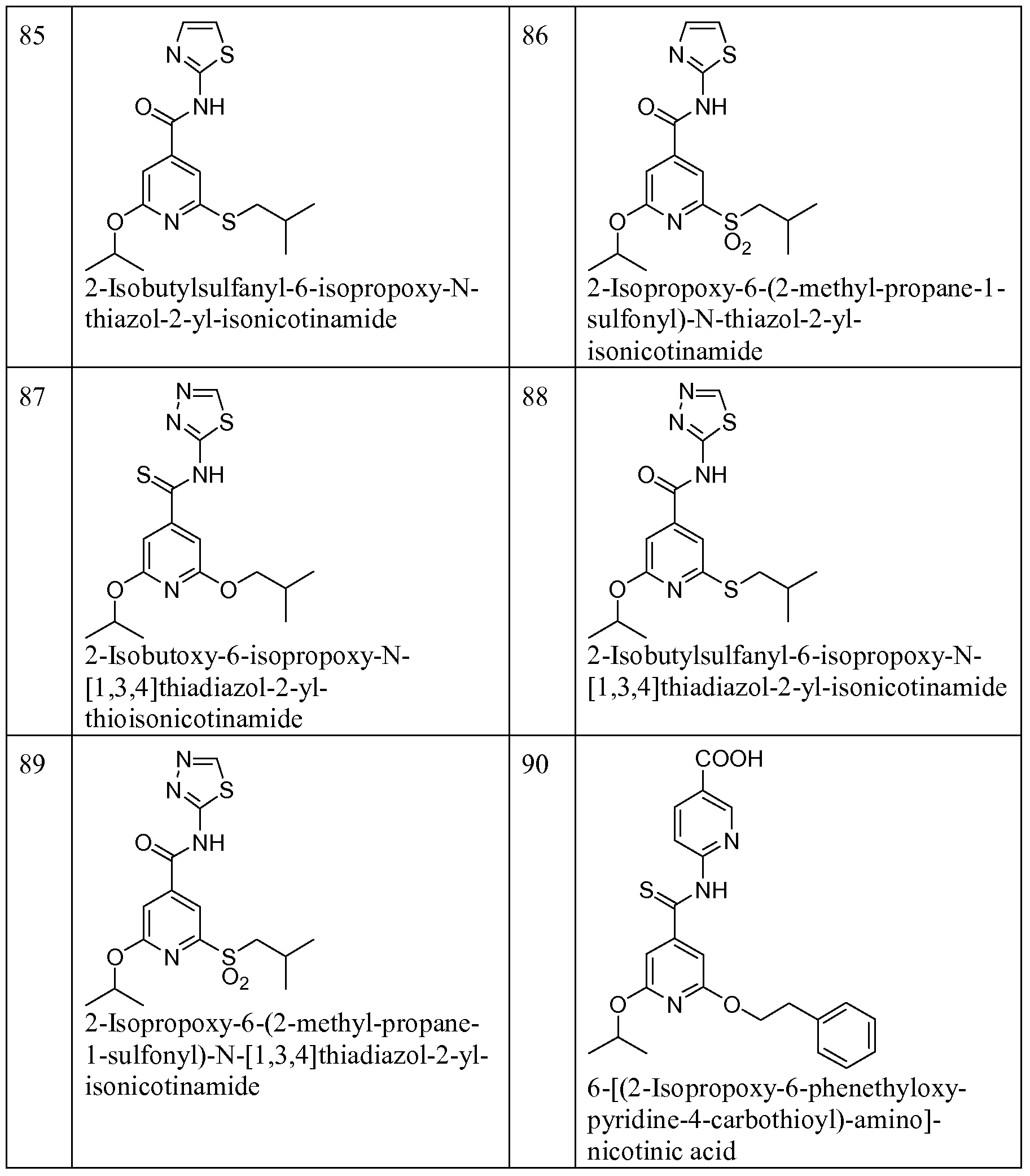
Illustrative, but non-limiting examples, of compounds of the present invention that are useful as glucokinase activator are given in table 1.
Table Ia:


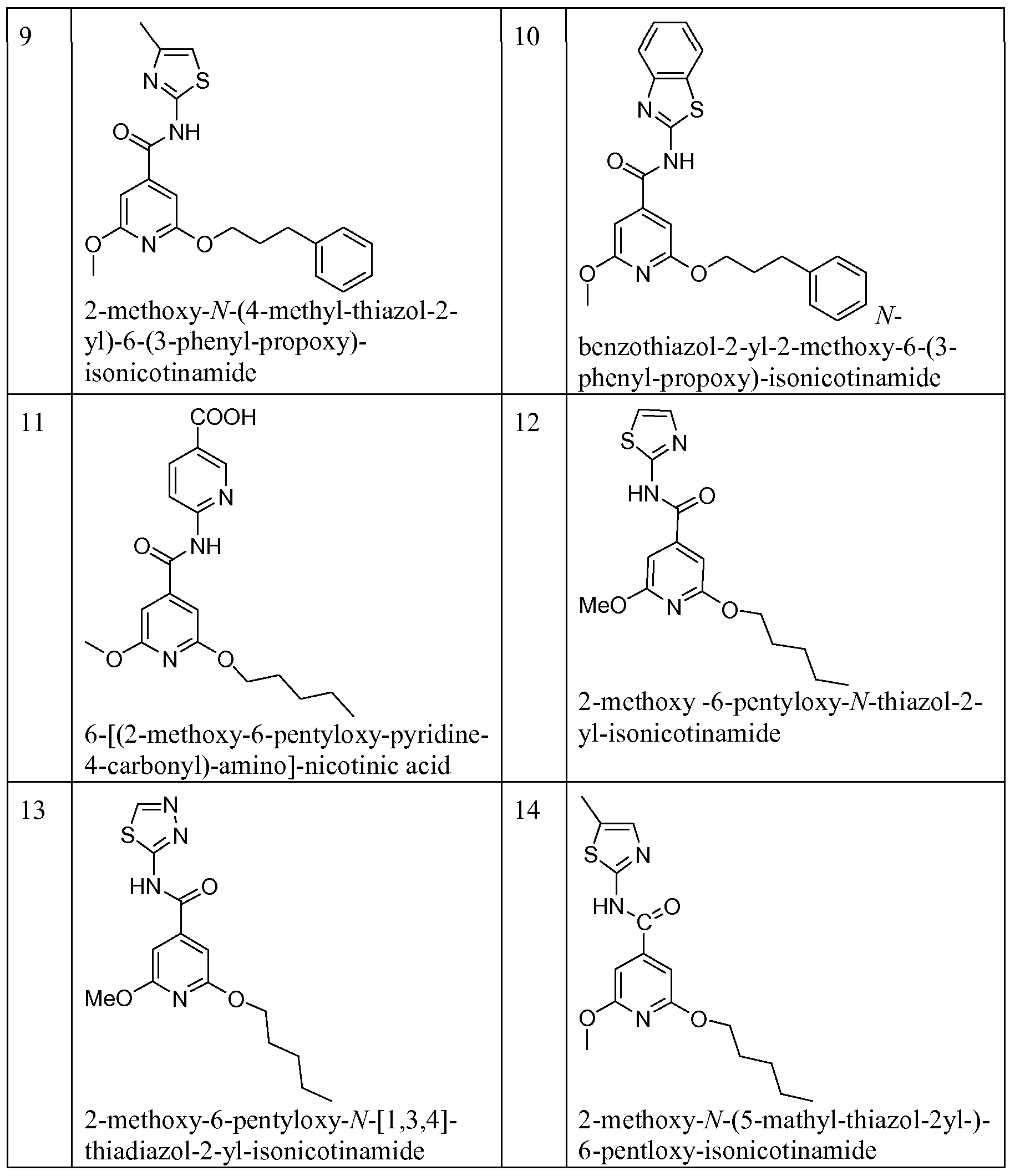























1 -


 - thiazol-2-yl-isonicotinamide
- thiazol-2-yl-isonicotinamide
 [l,3,4]thiadiazol-2-yl-isonicotinamide
[l,3,4]thiadiazol-2-yl-isonicotinamide

















Table Ib: 1-3 -























 -
-









































The biological effects of selected compounds of the invention have been tested in the following way:
Enzymatic assay of Glucokinase:
An enzymatic Glucokinase (GK) assay was set up according to the protocol given above using purified recombinant human enzyme (pancreatic full length). This was used to evaluate the effects of potential small molecule activators.
In this coupled enzymatic assay, GK calatyzes glucose phosphorylation in the presence of Mg-ATP. The product formed is Glucose-6-phosphate which is oxidized to 6- phospho D-gluconate by an excess of glucose-6-phosphate dehydrogenase in the presence of nicotinamide adenine dinucleotide phosphate (NADP). The production of reduced nicotinamide adenine dinucleotide phosphate (NADPH) shows increase in absorbance that was used to monitor GK activity. Recombinant GK was expressed in Escherichia coli as a GST-tagged fusion protein and was purified by glutathione-agarose affinity chromatography. The recombinant GK after the removal of GST- tag was used for enzymatic assay. The assay was performed in a final incubation volume of 100 μl in a 96- well transparent flat bottom microplates (Greiner Bio-one 655101).
The assay mixture consisting of 25 mM HEPES pH 7.1, 25 mM KCl, 5 mM β Mercaptoethanol, 1 mM ATP, 2.5 mM MgCl2 ,D-Glucose- 2 mM/7 mM /12 mM, 2 units/ ml yeast glucose 6-phosphate dehydrogenase (Roche Applied Sciences, Catalog no. 10127655001) and 5 % DMSO was pre-incubated at 300C for 5 minutes. (Ref : Brocklehurst K.J., et al (2004) Diabetes 53: 535-541). Human recombinant glucokinase (-200 ng) is added of to the assay reaction except the blank. The increase in A 34onm was monitored in Spectramax at 30° C for 15 min .The A 34onm was obtained for both test and positive control after substracting the blank.
For Km for Glucose and vmax determination, Glucose is used in 12 increasing concentrations from 0.05 mM -100 mM ( very active> 500 % activation at 2 mM) and 0.2 mM - 100 mM ( active). Dose response curve was generated using GraphPad Prism software Version 4 (San Diego, California, USA) using non linear regression curve fit for sigmoidal dose response ( variable slope). Typically the control K m without Activator should give values in the range of 8 to 9 mM with a Hill slope of 1.6.
For EC50 determination at 10 mM Glucose, the effective compound concentration that produces 50 % of maximal increase in GK activity observed using saturating activator concentration was calculated. Two fold activation (saturating concentration) was observed at 1OmM Glucose in general. Dose response curve was generated using
GraphPad Prism software Version 4 (San Diego, California, USA) using non linear regression curve fit for sigmoidal dose response ( variable slope).
The results obtained are summarized in the following tables:
Table Id: Compounds of the formula


*The 'Activitation' figures given represent the increase in GK activity obtainable using a concentration of the test compound of 50 μM when GK is used with each of three glucose concentrations 2mM, 7mM and 12mM as described above, the three results given being for these concentrations of glucose in this order.
ı52
Table 2a









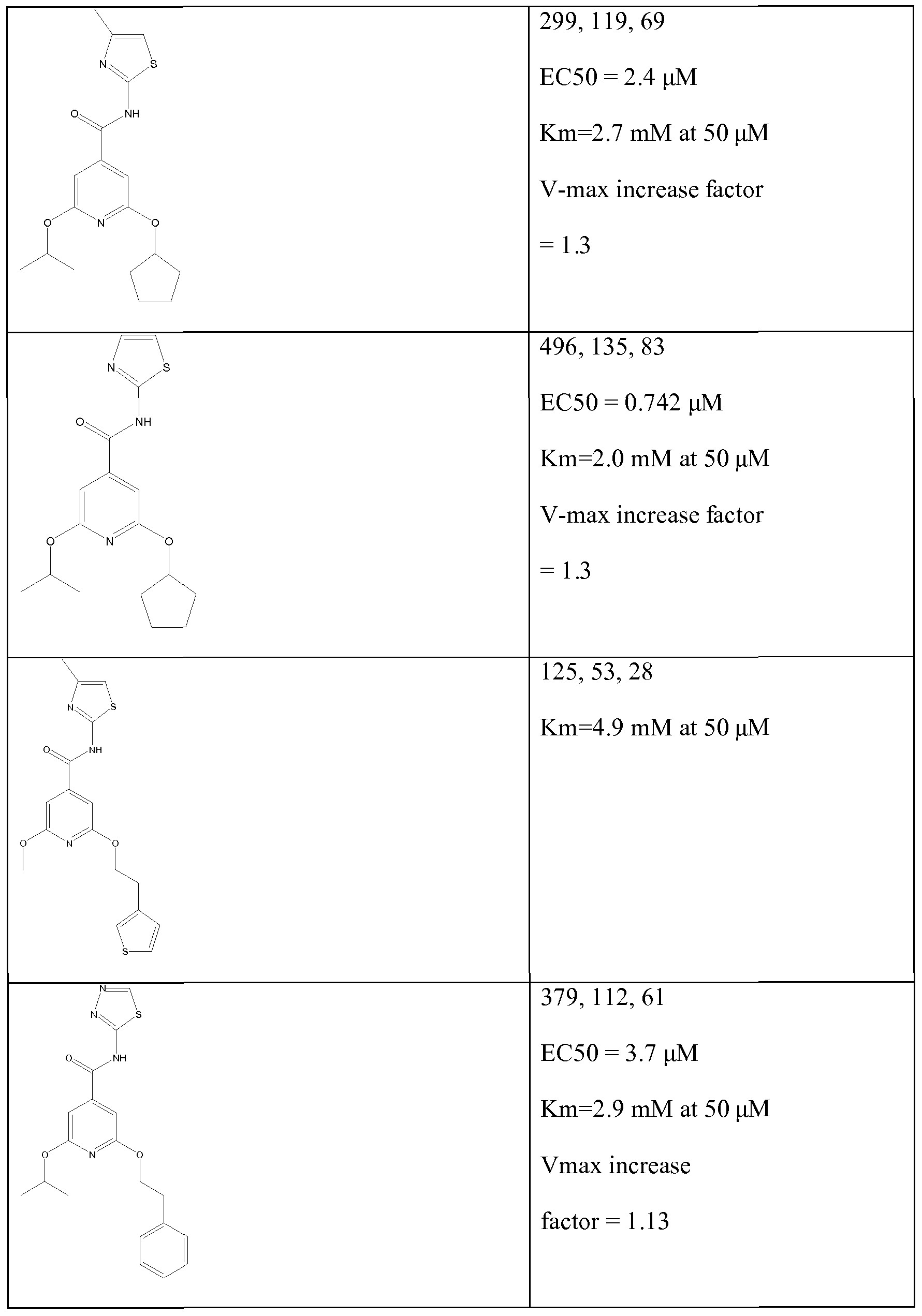



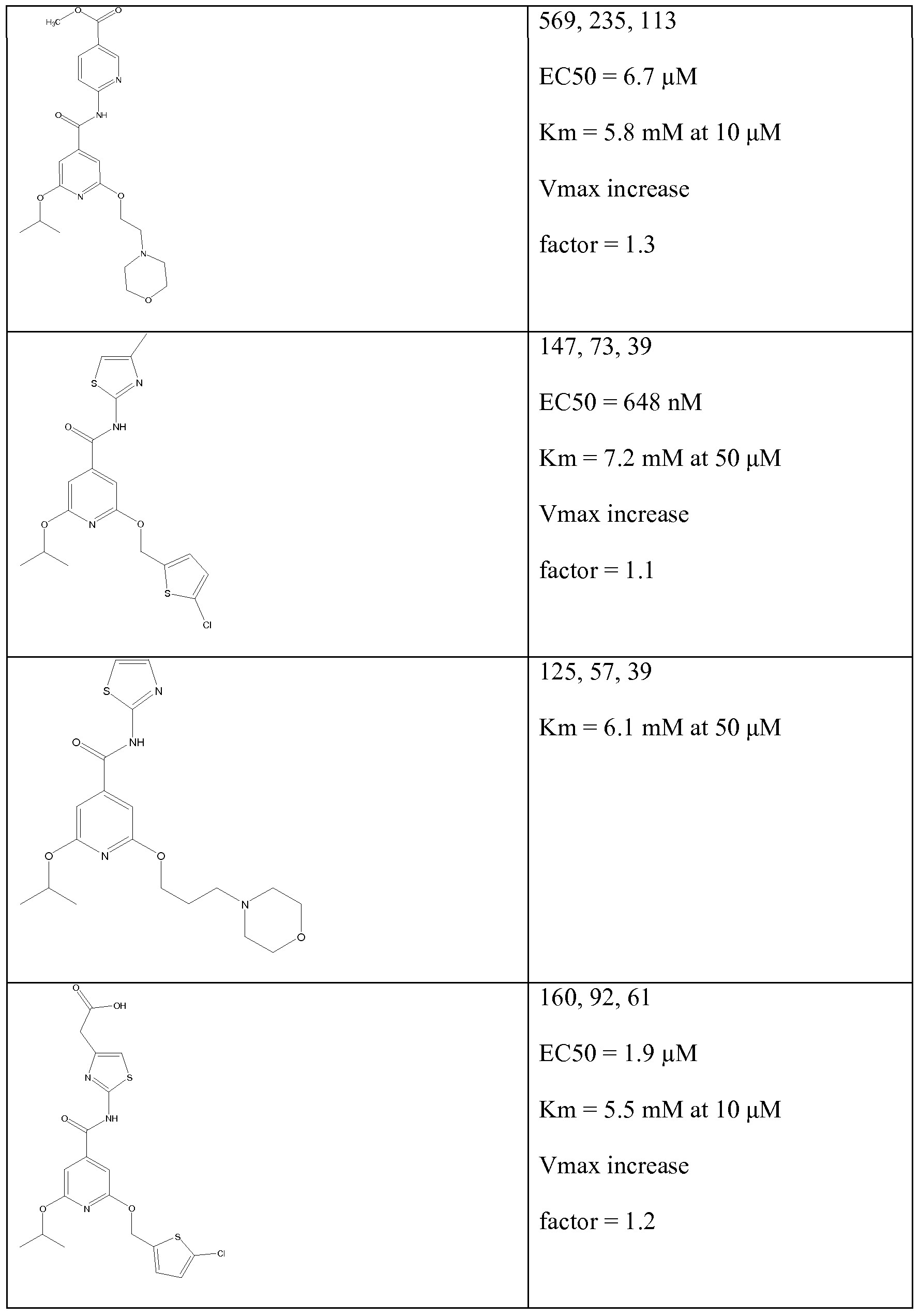




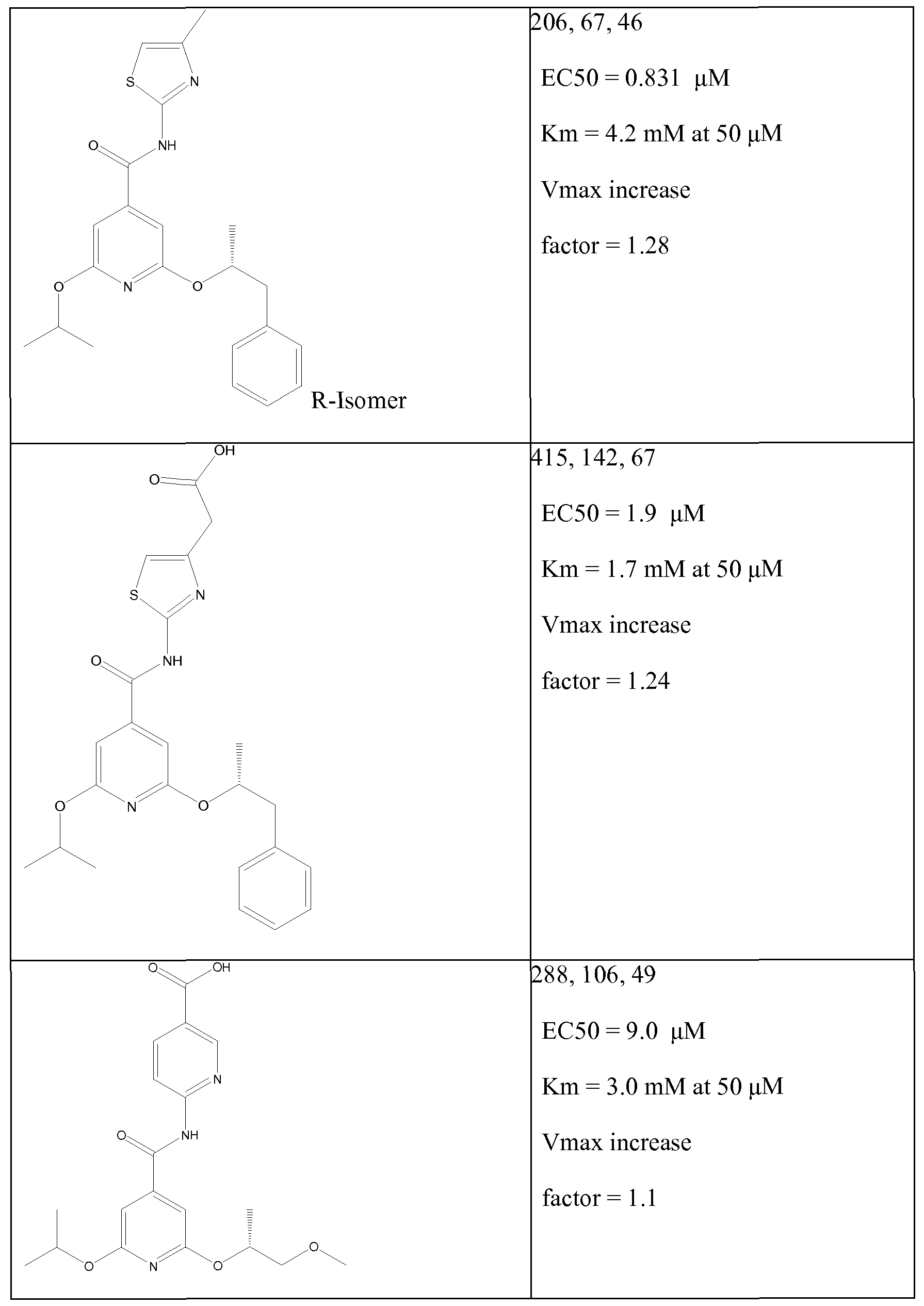








Table 2b:


In this specification, unless expressly otherwise indicated, the word 'or' is used in the sense of an operator that returns a true value when either or both of the stated conditions is met, as opposed to the operator 'exclusive or' which requires that only one of the conditions is met. The word 'comprising' is used in the sense of 'including' rather than in to mean
'consisting of. All prior teachings acknowledged above are hereby incorporated by reference. No acknowledgement of any prior published document herein should be taken to be an admission or representation that the teaching thereof was common general knowledge in Australia or elsewhere at the date hereof.
Claims
1. An azine derivative, of the general structural Formula 1

wherein
Gi and G2 are selected independently from N and CH;
R is halogen, or -OG3, or NHG3 where
G3 is selected from the group consisting of hydrogen, C1-6, alkyl or alkylene, wherein the said alkyl or alkylene is straight chain or branched unsubstituted or substituted wherein; the substituents are selected from halogen, hydroxyl, amino, Ci_2alkylamino, Ci_4alkoxy, oxo, and CO2H,
Z is O, NH, S or SO2
G5 is selected from the group consisting of: optionally substituted -Cl -12 alkyl, -C2-12 alkenyl, -(CH2)n-aryl, -(CH2)n- heteroaryl, -(CH2)n-heterocyclyl, or -(CH2)n-C3-8 cycloalkyl;
Y is either O or S; and
G4 represents optionally substituted mono or fused cycles selected from heteroaryl moieties;
or a pharmaceutically or veterinarily acceptable salt, solvate, hydrate, or prodrug thereof.
2. An azine derivative as claimed in claim 1 , wherein G5 is selected from the group consisting of:
-Cl -12 alkyl, wherein the alkyl is straight chain or branched and is unsubstituted or substituted with one or more substituents independently selected from halogen, hydro xyl, Ci_4alkoxy, Ci_4alkylamino, oxo, carbamimidoyl, amino, amido, or
CO2H,
-C2-12 alkenyl, wherein the alkenyl is unsubstituted or substituted with one or more groups, independently selected from halogen, hydroxyl, Ci_4alkoxy, Ci . 4alkylamino, oxo, carbamimidoyl, amino, amido,or CO2H,
-(CH2)n-aryl, wherein aryl is unsubstituted or substituted with one or more substituents which are independently selected from hydroxy, halogen, CO2H, Cl- 6 alkyloxycarbonyl, C 1-6 alkyl, C 1-6 alkoxy, -NH2j -NHV, -NW, -NHCO-V, - SO2-W or-CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more substituents independently selected from halogens, hydroxyl, oxo, or CO2H, wherein V is Cj_j2 alkyl, £\-\2 alkeriyl -(CH2)n-aryl, - (CH2 )n-hetero aryl, - (CH2)n-heterocyclyl, -(CH2)n-cycloalkyl, wherein W is C^.^2alkyl, C^.gcycloalkyl, or -NR R2 wherein each of R and R independently is C 1.3 alkyl or together form an non-aromatic or aromatic heterocycle, and wherein V and W are each optionally substituted with one or more substituents independently selected from halogens, hydroxyl, oxo, and CO2H,
-(CH2)n-heteroaryl, wherein heteroaryl is unsubstituted or substituted with one or more substituents independently selected from hydroxy, halogen, CO2H, C 1-6 alkyloxycarbonyl, C 1-6 alkyl, C 1-6 alkoxy, -NH2; -NHV, -NVV, -NHCO-V, - SO2-W or -CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more substituents independently selected from halogens, hydroxyl, oxo, or CO2H, and V and W are as defined above,
-(CH2)n-heterocyclyl, wherein heterocyclyl is unsubstituted or substituted with one or more substituents independently selected from oxo, hydroxy, halogen,
CO2H, C 1-6 alkyloxycarbonyl, C 1-6 alkyl, Cl- 6 alkoxy, -NH2; -NHV, -NW, -
NHCO-V, -SO2-W or -CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more substituents independently selected from halogens, hydroxyl, oxo, or CO2H, and V and W are as defined above,
or -(CH2)n-C3-8 cycloalkyl; and preferably (CH2VC3-6 cycloalkyl, wherein cycloalkyl is unsubstituted or substituted with one to three substituents independently selected from halogen, hydroxy, CO2H, C 1-6 alkyloxycarbonyl, alkyl, alkoxy, -NH2; -NHV, -NW, -NHCO-V, -SO2-W or -CO-W, wherein alkyl and alkoxy are unsubstituted or substituted with one or more halogens, hydroxyl, oxo, or CO2H, and V and W are as defined above; and wherein each "n" is an integer of any value ranging from 0-10.
3. An azine derivative as claimed in claim 1, wherein R is Cl or is -OG3 wherein G3 is Cj.4 alkyl, optionally substituted by halogen, hydroxyl, or C\_
2alkoxy.
4. An azine derivative as claimed in claim 2, wherein R is -OG3 and is any one

5. An azine derivative as claimed in claim 2, wherein R is OG3 wherein G3 is isopropyl, or 2-methylpropyl, optionally substituted with methoxy.
6. An azine derivative as claimed in any preceding claim, wherein -ZG5 is any one of:





7. An azine derivative as claimed in any preceding claim, wherein G4 is optionally substituted thiazole, thiadiazole, pyridine, purine, benzothiazole, pyrimidine, isothiazole, pyrazole, pyridothiazole, pyridoimidazole, benzimidazole, or pyrazine.
8. An azine derivative as claimed in claim 7, wherein said G4 group is substituted with C 1-4 alkyl, haloalkyl, carbonyl, carbonyl ester, C 1.4 alkylcarbonyl, or C 1.4 alkylcarbonylester.
9. An azine as claimed in claim 7, wherein said G4 group is any one of:




wherein * is the point of attachment to Formula 1;
G6 is selected from group of H, and CMO alkyl,
G7 is independently selected from CX3 wherein X is halogen, Ci_io alkyl, or aryl.
10. An azine derivative as claimed in any preceding claim, wherein both G^ and G2 are C.
11. An azine derivative as claimed in any preceding claim, having the ability to increase the V-max of human glucokinase by a factor of at least 1.2.
12. An azine derivative as claimed in any preceding claim that is co-crystallisable with isolated human glucokinase.
13. An azine derivative as claimed in any preceding claim, for use in a method of therapy.
14. An azine derivative as claimed in any preceding claim for use in the preventative or curative treatment of hyperglycaemia, diabetes, or the sequelae thereof.
15. An azine derivative as claimed in any one of claims 1 to 12, formulated for pharmaceutical or veterinary use.
16. A method of preventative or curative treatment of hyperglycaemia or diabetes or any sequelae thereof comprising the administration to a patient in need thereof of an azine derivative as claimed in any one of claims 1-12 or formulated in accordance with claim 15.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2780DE2007 | 2007-12-31 | ||
| IN2780/DEL/2007 | 2007-12-31 | ||
| IN889/CHE/2008 | 2008-04-09 | ||
| IN889CH2008 | 2008-04-09 | ||
| US5662708P | 2008-05-28 | 2008-05-28 | |
| US61/056,627 | 2008-05-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009083553A1 true WO2009083553A1 (en) | 2009-07-09 |
Family
ID=40394544
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/068232 WO2009083553A1 (en) | 2007-12-31 | 2008-12-23 | Azine compounds as glucokinase activators |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009083553A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011123572A1 (en) | 2010-03-31 | 2011-10-06 | The Scripps Research Institute | Reprogramming cells |
| WO2011122458A1 (en) * | 2010-03-29 | 2011-10-06 | 第一三共株式会社 | Aromatic ring compound containing nitrogen |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| CN102558149A (en) * | 2010-12-29 | 2012-07-11 | 中国医学科学院药物研究所 | Pyrimidine derivative, preparation method thereof, medicinal composition and application thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015022546A1 (en) * | 2013-08-14 | 2015-02-19 | Kalvista Pharmaceuticals Limited | Inhibitors of plasma kallikrein |
| WO2016187384A1 (en) * | 2015-05-20 | 2016-11-24 | Eli Lilly And Company | Novel dgat2 inhibitors |
| KR20170034902A (en) * | 2014-07-16 | 2017-03-29 | 라이프에스씨아이 파마슈티컬스, 인크. | Therapeutic inhibitory compounds |
| EP3089746A4 (en) * | 2013-12-30 | 2017-08-30 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| WO2017155909A1 (en) | 2016-03-07 | 2017-09-14 | The Global Alliance For Tb Drug Development, Inc. | Antibacterial compounds and uses thereof |
| US10023557B2 (en) | 2015-07-01 | 2018-07-17 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| US10301284B2 (en) | 2016-07-11 | 2019-05-28 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| WO2019229250A1 (en) * | 2018-05-31 | 2019-12-05 | Universiteit Leiden | Inhibitors of n-acylphosphatidylethanolamine phospholipase d (nape-pld) |
| US10611758B2 (en) | 2014-11-27 | 2020-04-07 | Kalvista Pharmaceuticals Limited | N-((het)arylmethyl)-heteroaryl-carboxamides compounds as kallikrein inhibitors |
| US10752607B2 (en) | 2016-06-01 | 2020-08-25 | Kalvista Pharmaceuticals Limited | Polymorphs of N-[(6-cyano-2-fluoro)-3-methoxyphenyl)methyl]-3-(methoxymethyl)-1-({4-[(2-oxopyridin-1-yl)methyl]phenyl}methyl)pyrazole-4-carboxamide as kallikrein inhibitors |
| US10781181B2 (en) | 2014-11-27 | 2020-09-22 | Kalvista Pharmaceuticals Limited | N-((het) arylmethyl)-heteroaryl-carboxamides compounds as plasma kallikrein inhibitors |
| US11180484B2 (en) | 2016-05-31 | 2021-11-23 | Kalvista Pharmaceuticals Limited | Pyrazole derivatives as plasma kallikrein inhibitors |
| US11230537B2 (en) | 2016-06-01 | 2022-01-25 | Kalvista Pharmaceuticals Limited | Polymorphs of n-[(3-fluoro-4-methoxypyridin-2-yl)methyl]-3-(methoxymethyl)-1-({4-[2-oxopyridin-1-yl)methyl]phenyl} methyl)pyrazole-4-carboxamide as iallikrein inhibitors |
| US11234939B2 (en) | 2017-11-29 | 2022-02-01 | Kalvista Pharmaceuticals Limited | Dosage forms comprising a plasma kallikrein inhibitor |
| WO2022005976A3 (en) * | 2020-06-29 | 2022-02-03 | University Of Notre Dame Du Lac | Antibacterial picolinamide compounds |
| US11242333B2 (en) | 2013-08-14 | 2022-02-08 | Kalvista Pharmaceuticals Limited | Inhibitors of plasma kallikrein |
| US11584735B2 (en) | 2017-11-29 | 2023-02-21 | Kalvista Pharmaceuticals Limited | Solid forms of a plasma kallikrein inhibitor and salts thereof |
| US11613527B2 (en) | 2019-08-09 | 2023-03-28 | Kalvista Pharmaceuticals Limited | Enzyme inhibitors |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08208613A (en) * | 1995-02-06 | 1996-08-13 | Dainippon Ink & Chem Inc | 2-chloro-6-hydroxyisonicotinic acid derivative and plant disease injury-controlling agent |
| WO1999024404A1 (en) * | 1997-11-07 | 1999-05-20 | Amgen Inc. | Substituted pyridine compounds as anti-inflammatory agents |
| WO2005044801A1 (en) * | 2003-10-31 | 2005-05-19 | Astrazeneca Ab | Pyridine carboxylic acid derivatives as glucokinase modulators |
| EP1598349A1 (en) * | 2003-02-13 | 2005-11-23 | Banyu Pharmaceutical Co., Ltd. | Novel 2-pyridinecarboxamide derivatives |
| WO2006099379A2 (en) * | 2005-03-14 | 2006-09-21 | Transtech Pharma, Inc. | Benzazole derivatives, compositions, and methods of use as b-secretase inhibitors |
-
2008
- 2008-12-23 WO PCT/EP2008/068232 patent/WO2009083553A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08208613A (en) * | 1995-02-06 | 1996-08-13 | Dainippon Ink & Chem Inc | 2-chloro-6-hydroxyisonicotinic acid derivative and plant disease injury-controlling agent |
| WO1999024404A1 (en) * | 1997-11-07 | 1999-05-20 | Amgen Inc. | Substituted pyridine compounds as anti-inflammatory agents |
| EP1598349A1 (en) * | 2003-02-13 | 2005-11-23 | Banyu Pharmaceutical Co., Ltd. | Novel 2-pyridinecarboxamide derivatives |
| WO2005044801A1 (en) * | 2003-10-31 | 2005-05-19 | Astrazeneca Ab | Pyridine carboxylic acid derivatives as glucokinase modulators |
| WO2006099379A2 (en) * | 2005-03-14 | 2006-09-21 | Transtech Pharma, Inc. | Benzazole derivatives, compositions, and methods of use as b-secretase inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| F. E. GODA ET AL: "Synthesis and Antiviral Activity of 1,3,5-triazine Derivatives", MANSOURA JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 20, 2004, pages 1 - 10, XP009113493 * |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011122458A1 (en) * | 2010-03-29 | 2011-10-06 | 第一三共株式会社 | Aromatic ring compound containing nitrogen |
| EP3199623A1 (en) | 2010-03-31 | 2017-08-02 | The Scripps Research Institute | Reprogramming cells |
| WO2011123572A1 (en) | 2010-03-31 | 2011-10-06 | The Scripps Research Institute | Reprogramming cells |
| EP3936608A1 (en) | 2010-03-31 | 2022-01-12 | The Scripps Research Institute | Reprogramming cells |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| CN102558149A (en) * | 2010-12-29 | 2012-07-11 | 中国医学科学院药物研究所 | Pyrimidine derivative, preparation method thereof, medicinal composition and application thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015022546A1 (en) * | 2013-08-14 | 2015-02-19 | Kalvista Pharmaceuticals Limited | Inhibitors of plasma kallikrein |
| US11242333B2 (en) | 2013-08-14 | 2022-02-08 | Kalvista Pharmaceuticals Limited | Inhibitors of plasma kallikrein |
| CN105683179A (en) * | 2013-08-14 | 2016-06-15 | 卡尔维斯塔制药有限公司 | Inhibitors of plasma kallikrein |
| CN105683179B (en) * | 2013-08-14 | 2018-10-26 | 卡尔维斯塔制药有限公司 | The inhibitor of plasma kallikrein |
| US10221161B2 (en) | 2013-08-14 | 2019-03-05 | Kalvista Pharmaceuticals Limited | Inhibitors of plasma kallikrein |
| EP3089746A4 (en) * | 2013-12-30 | 2017-08-30 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| US10259803B2 (en) | 2013-12-30 | 2019-04-16 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| US11021463B2 (en) | 2013-12-30 | 2021-06-01 | Attune Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| US10266515B2 (en) | 2013-12-30 | 2019-04-23 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| KR20170034902A (en) * | 2014-07-16 | 2017-03-29 | 라이프에스씨아이 파마슈티컬스, 인크. | Therapeutic inhibitory compounds |
| KR102286305B1 (en) | 2014-07-16 | 2021-08-09 | 라이프에스씨아이 파마슈티컬스, 인크. | Therapeutic inhibitory compounds |
| US11084809B2 (en) | 2014-11-27 | 2021-08-10 | Kalvista Pharmaceuticals Limited | N-((HET)arylmethyl)-heteroaryl-carboxamides compounds as kallikrein inhibitors |
| US10611758B2 (en) | 2014-11-27 | 2020-04-07 | Kalvista Pharmaceuticals Limited | N-((het)arylmethyl)-heteroaryl-carboxamides compounds as kallikrein inhibitors |
| US11001578B2 (en) | 2014-11-27 | 2021-05-11 | Kalvista Pharmaceuticals Limited | N-((HET)arylmethyl)-heteroaryl-carboxamides compounds as plasma kallikrein inhibitors |
| US10781181B2 (en) | 2014-11-27 | 2020-09-22 | Kalvista Pharmaceuticals Limited | N-((het) arylmethyl)-heteroaryl-carboxamides compounds as plasma kallikrein inhibitors |
| US11198691B2 (en) | 2014-11-27 | 2021-12-14 | Kalvista Pharmaceuticals Limited | N-((het)arylmethyl)-heteroaryl-carboxamides compounds as kallikrein inhibitors |
| WO2016187384A1 (en) * | 2015-05-20 | 2016-11-24 | Eli Lilly And Company | Novel dgat2 inhibitors |
| CN107635975B (en) * | 2015-05-20 | 2020-07-24 | 伊莱利利公司 | DGAT2 inhibitors |
| US10053429B2 (en) | 2015-05-20 | 2018-08-21 | Eli Lilly And Company | DGAT2 inhibitors |
| JP2018515554A (en) * | 2015-05-20 | 2018-06-14 | イーライ リリー アンド カンパニー | Novel DGAT2 inhibitor |
| CN107635975A (en) * | 2015-05-20 | 2018-01-26 | 伊莱利利公司 | New dgat2 inhibitor |
| US10308637B2 (en) | 2015-07-01 | 2019-06-04 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| US10023557B2 (en) | 2015-07-01 | 2018-07-17 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| WO2017155909A1 (en) | 2016-03-07 | 2017-09-14 | The Global Alliance For Tb Drug Development, Inc. | Antibacterial compounds and uses thereof |
| EP4148053A1 (en) | 2016-03-07 | 2023-03-15 | The Global Alliance for TB Drug Development, Inc. | Antibacterial compounds and uses thereof |
| US11180484B2 (en) | 2016-05-31 | 2021-11-23 | Kalvista Pharmaceuticals Limited | Pyrazole derivatives as plasma kallikrein inhibitors |
| US10752607B2 (en) | 2016-06-01 | 2020-08-25 | Kalvista Pharmaceuticals Limited | Polymorphs of N-[(6-cyano-2-fluoro)-3-methoxyphenyl)methyl]-3-(methoxymethyl)-1-({4-[(2-oxopyridin-1-yl)methyl]phenyl}methyl)pyrazole-4-carboxamide as kallikrein inhibitors |
| US11230537B2 (en) | 2016-06-01 | 2022-01-25 | Kalvista Pharmaceuticals Limited | Polymorphs of n-[(3-fluoro-4-methoxypyridin-2-yl)methyl]-3-(methoxymethyl)-1-({4-[2-oxopyridin-1-yl)methyl]phenyl} methyl)pyrazole-4-carboxamide as iallikrein inhibitors |
| US11739068B2 (en) | 2016-06-01 | 2023-08-29 | Kalvista Pharmaceuticals Limited | Polymorphs of N-[(3-fluoro-4-methoxypyridin-2-yl)methyl]-3-(methoxymethyl)-1-({4-[(2-oxopyridin-1-yl)methyl]phenyl}methyl)pyrazole-4-carboxamide and salts thereof |
| US10781200B2 (en) | 2016-07-11 | 2020-09-22 | Attune Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| US10301284B2 (en) | 2016-07-11 | 2019-05-28 | Lifesci Pharmaceuticals, Inc. | Therapeutic inhibitory compounds |
| US11234939B2 (en) | 2017-11-29 | 2022-02-01 | Kalvista Pharmaceuticals Limited | Dosage forms comprising a plasma kallikrein inhibitor |
| US11584735B2 (en) | 2017-11-29 | 2023-02-21 | Kalvista Pharmaceuticals Limited | Solid forms of a plasma kallikrein inhibitor and salts thereof |
| WO2019229250A1 (en) * | 2018-05-31 | 2019-12-05 | Universiteit Leiden | Inhibitors of n-acylphosphatidylethanolamine phospholipase d (nape-pld) |
| US11613527B2 (en) | 2019-08-09 | 2023-03-28 | Kalvista Pharmaceuticals Limited | Enzyme inhibitors |
| WO2022005976A3 (en) * | 2020-06-29 | 2022-02-03 | University Of Notre Dame Du Lac | Antibacterial picolinamide compounds |
| EP4171221A4 (en) * | 2020-06-29 | 2024-07-24 | Univ Notre Dame Du Lac | Antibacterial picolinamide compounds |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009083553A1 (en) | Azine compounds as glucokinase activators | |
| US20210221814A1 (en) | Inhibitors of cyclin-dependent kinase 7 (cdk7) | |
| US20100234341A1 (en) | Substituted pyrimidines as adenosine receptor antagonists | |
| AU2001265914B2 (en) | Para-amine substituted phenylamide glucokinase activators | |
| DE602004011603T2 (en) | SULFONAMIDE-SUBSTITUTED XANTHINE DERIVATIVES FOR USE AS PEPCK INHIBITORS | |
| US8268999B2 (en) | Tri-cyclic pyrazolopyridine kinase inhibitors | |
| AU2021269397B2 (en) | Compounds and compositions for the treatment of cancer | |
| WO2009010871A2 (en) | Pyrazole derivatives as antagonists of adenosine a3 receptor | |
| IL163286A (en) | 1-(substituted glycyl)pyrrolidine-2-carbonitriles, their preparation and pharmaceutical compositions containing them | |
| KR20070084035A (en) | Pyrimidine derivative fused with nonaromatic ring | |
| ZA200504617B (en) | Novel compounds having selective inhibiting effect at GSK3 | |
| AU2001265914A1 (en) | Para-amine substituted phenylamide glucokinase activators | |
| CA2669708A1 (en) | Benzamide glucokinase activators | |
| NZ566862A (en) | Diarylamine-containing compounds and compositions, and their use as modulators of C-kit receptors | |
| CA2649577A1 (en) | Benzamide glucokinase activators | |
| US8524906B2 (en) | Tetrahydrothiazolopyridine inhibitors of phosphatidylinositol 3-kinase | |
| US7329678B2 (en) | Chemical compounds | |
| WO2000021934A1 (en) | Compounds | |
| JP2005516961A (en) | Use of oxindole derivatives in the treatment of dementia related diseases, Alzheimer's disease and glycogen synthase kinase-3 related symptoms | |
| DE602004010529T2 (en) | 2-AMINO-5-BENZOYLTHIAZOL NPY ANTAGONISTS | |
| US7507834B2 (en) | Substituted anilinopyrazoles | |
| NO330392B1 (en) | Imidazole derivatives with affinity for alpha 2 receptor activity and uses thereof, as well as pharmaceutical preparations comprising imidazole derivatives | |
| EP1656374B1 (en) | Thiazole derivatives as npy antagonists | |
| WO2021070957A1 (en) | Benzene condensed ring compound and medical composition containing same | |
| EP4377300A1 (en) | 6-substituted naphthalene-1,3-disulfonic acid derivatives as modulators of the extracellular nicotinamide phosphoribosyl transferase (enampt) for the treatment of e.g. diabetes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08866391 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08866391 Country of ref document: EP Kind code of ref document: A1 |