WO2009047240A1 - Indole derivatives useful as ppar activators - Google Patents
Indole derivatives useful as ppar activators Download PDFInfo
- Publication number
- WO2009047240A1 WO2009047240A1 PCT/EP2008/063371 EP2008063371W WO2009047240A1 WO 2009047240 A1 WO2009047240 A1 WO 2009047240A1 EP 2008063371 W EP2008063371 W EP 2008063371W WO 2009047240 A1 WO2009047240 A1 WO 2009047240A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- indole
- amino
- methyl
- carboxylic acid
- Prior art date
Links
- 0 COC(c(cc1c(CCOCC(N*)=O)c2)ccc1[n]2-c1ccccc1)=O Chemical compound COC(c(cc1c(CCOCC(N*)=O)c2)ccc1[n]2-c1ccccc1)=O 0.000 description 2
- CPEDHEFNYKVGBP-UHFFFAOYSA-N CC(C)(C)OC(NCC(C)(C)NCc(c1cc(C(OC)=O)ccc11)c[n]1-c1ccccc1)=O Chemical compound CC(C)(C)OC(NCC(C)(C)NCc(c1cc(C(OC)=O)ccc11)c[n]1-c1ccccc1)=O CPEDHEFNYKVGBP-UHFFFAOYSA-N 0.000 description 1
- KKTLFTDYPQTOQU-UHFFFAOYSA-N CC(C)c(cc1)ccc1C(NC(CC1)CN1C(OC(C)(C)C)=O)=O Chemical compound CC(C)c(cc1)ccc1C(NC(CC1)CN1C(OC(C)(C)C)=O)=O KKTLFTDYPQTOQU-UHFFFAOYSA-N 0.000 description 1
- WUDGGENIYONOGD-UHFFFAOYSA-N CC(C)c(cc1)ccc1C(NCCCCc(c1cc(C(OC)=O)ccc11)c[n]1-c(cc1)ccc1F)=O Chemical compound CC(C)c(cc1)ccc1C(NCCCCc(c1cc(C(OC)=O)ccc11)c[n]1-c(cc1)ccc1F)=O WUDGGENIYONOGD-UHFFFAOYSA-N 0.000 description 1
- JNRWMLJQHJUGFS-UHFFFAOYSA-N CC(C)c(cc1)ccc1NC(CCCCc(c1cc(C(OC)=O)ccc11)c[n]1-c1ccccc1)=O Chemical compound CC(C)c(cc1)ccc1NC(CCCCc(c1cc(C(OC)=O)ccc11)c[n]1-c1ccccc1)=O JNRWMLJQHJUGFS-UHFFFAOYSA-N 0.000 description 1
- DALUCNUGMDNXET-UHFFFAOYSA-N CCc1c(C)nc(C)[s]1 Chemical compound CCc1c(C)nc(C)[s]1 DALUCNUGMDNXET-UHFFFAOYSA-N 0.000 description 1
- OUAHZNNXQDRDQF-UHFFFAOYSA-N COC(c(cc1c(CCN)c2)ccc1[n]2-c1ccccn1)=O Chemical compound COC(c(cc1c(CCN)c2)ccc1[n]2-c1ccccn1)=O OUAHZNNXQDRDQF-UHFFFAOYSA-N 0.000 description 1
- CSFZISMCWGJXSB-UHFFFAOYSA-N COC(c(cc1c(CCOCC(O)=O)c2)ccc1[n]2-c1ccccc1)=O Chemical compound COC(c(cc1c(CCOCC(O)=O)c2)ccc1[n]2-c1ccccc1)=O CSFZISMCWGJXSB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/12—Radicals substituted by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to indole derivatives, compositions and medicaments containing said indole derivatives and processes for their preparation.
- the invention also relates to the use of said indole derivatives, compositions and medicaments, for example as activators of the alpha subtype of the human peroxisome proliferator activated receptor (hPPAR) and/or for the treatment and/or prophylaxis of diseases or conditions mediated by hPPAR.
- hPPAR human peroxisome proliferator activated receptor
- HMG CoA reductase inhibitors are useful for treating conditions characterized by high LDL-c levels. It has been shown that lowering LDL-c is not sufficient for reducing the risk of cardiovascular disease in some patients, particularly those with normal LDL-c levels. This population pool is identified by the independent risk factor of low HDL-c.
- the increased risk of cardiovascular disease associated with low HDL-c levels has not yet been successfully addressed by drug therapy (i.e., currently there are no drugs on the market that are useful for raising HDL-c >40%). (Bisgaier, C. L.; Pape, M. E. Curr. Pharm. Des. 1998, 4, 53-70).
- Syndrome X (including metabolic syndrome) is loosely defined as a collection of abnormalities including hyperinsuinlemia, obesity, elevated levels of trigycerides, uric acid, fibrinogen, small dense LDL-c particles, and plasminogen activator inhibitor 1 (PAI-1 ), and decreased levels of HDL-c.
- abnormalities including hyperinsuinlemia, obesity, elevated levels of trigycerides, uric acid, fibrinogen, small dense LDL-c particles, and plasminogen activator inhibitor 1 (PAI-1 ), and decreased levels of HDL-c.
- NIDDM is described as insulin resistance which in turn causes anomalous glucose output and a decrease in glucose uptake by skeletal muscle. These factors eventually lead to impaired glucose tolerance (IGT) and hyperinsulinemia.
- ITT impaired glucose tolerance
- Peroxisome Proliferator Activated Receptors are orphan receptors belonging to the steroid/retinoid receptor superfamily of ligand-activated transcription factors. See, for example, Willson, T. M. and Wahli, W., Curr. Opin. Chem. Biol., (1997), Vol. 1 , pp 235- 241.
- Three mammalian Peroxisome Proliferator-Activated Receptors have been isolated and termed PPAR-alpha, PPAR-gamma, and PPAR-delta (also known as NUC1 or PPAR- beta). These PPARs regulate expression of target genes by binding to DNA sequence elements, termed PPAR response elements (PPRE).
- Certain compounds that activate or otherwise interact with one or more of the PPARs have been implicated in the regulation of triglyceride and cholesterol levels in animal models. See, for example, WO 01/40207, WO 01/00603, WO 97/31907, WO 02/46174 (Glaxo Group Ltd et al).
- Z is phenyl (optionally substituted by halogen), pyridinyl;
- Y is phenyl (optionally substituted by one or more groups each independently selected from -Ci-6 alkyl, -Ci -6 alkoxy, -Ci -6 haloalkyl, -Ci -6 haloalkoxy, -halogen, -OH, -C 3-7 cycloalkyl, -NR 1 R 2 ), or phenyl fused to a 5 or 6 membered heterocyclic or cycloalkyl ring;
- R 1 and R 2 independently represent H or d- 3 alkyl
- X is a linker group of 5-7 atoms in shortest length between the carbon of the indole group and Y;
- X-Y represents a group:
- R 3 is C 1-3 alkyl or C 1-3 haloalkyl.
- a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof for use in therapy, in particular in the treatment of hPPAR mediated diseases or conditions.
- a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof and one or more of pharmaceutically acceptable carriers, diluents and excipients.
- a method of treating hPPAR mediated diseases or conditions comprising administering to said mammal a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
- alkyl refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms.
- C 1-6 alkyl means a straight or branched alkyl chain containing at least 1 , and at most 6 carbon atoms.
- alkyl as used herein include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, t-butyl, n-pentyl.
- alkylene refers to a straight or branched chain divalent hydrocarbon radical having from 1-10 carbon atoms.
- alkylene include but are not limited to methylene, ethylene, n-propylene.
- alkenyl refers to a straight or branched hydrocarbon radical having from two to ten carbons and at least one carbon-carbon double bond.
- Examples of "C 2 -C 10 alkenyl” groups used in the present invention include, but are not limited to ethenyl, propenyl, 1-butenyl, 2-butenyl, and isobutenyl.
- cycloalkyl refers to a non-aromatic hydrocarbon ring containing the specified number of carbon atoms.
- C 3 - 7 cycloalkyl means a non-aromatic ring containing at least three, and at most seven, ring carbon atoms.
- Examples of "cycloalkyl” as used herein include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl.
- halogen refers to fluorine (F), chlorine (Cl), bromine (Br) or iodine (I), and the radicals, thereof, fluoro (-F), chloro (-Cl), bromo (-Br) and iodo (-I).
- CrC 6 haloalkyl refers to an alkyl group as defined above containing at least 1 , and at most 6, carbon atoms substituted with at least one halo group, halo being as defined herein.
- Examples of branched or straight chained "C 1 -C 6 haloalkyl” groups useful in the present invention include, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl and n-butyl substituted independently with one or more halos, e.g., fluoro, chloro, bromo and iodo.
- alkoxy refers to the group R 3 O-, where R 3 is alkyl as defined above and the term "C 1 -C 6 alkoxy” refers to an alkoxy group as defined herein wherein the alkyl moiety contains at least 1 , and at most 6, carbon atoms.
- Exemplary C 1 -C 6 alkoxy groups useful in the present invention include, but are not limited to, methoxy, ethoxy, n- propoxy, isopropoxy, n-butoxy, and t-butoxy.
- heterocyclic or the term “heterocyclyl” refers to a non-aromatic three to seven-membered heterocyclic ring being satured or having one or more degrees of unsaturation containing one or more heteroatoms selected from S, SO, SO 2 , O, or N.
- heterocyclic moieties include, but are not limited to, tetrahydrofuran, pyran, 1 ,4-dioxane, 1 ,3-dioxane, piperidine, pyrrolidine, morpholine, tetrahydrothiopyran, tetrahydrothiophene, di-oxo tetrahydrothiophene, and the like.
- haloalkoxy refers to the group R 3 O-, where R 3 is haloalkyl as defined above and the term "C 1 -C 6 haloalkoxy” refers to a haloalkoxy group as defined herein wherein the haloalkyl moiety contains at least 1 and at most 6, carbon atoms.
- Exemplary C 1 -C 6 haloalkoxy groups useful in the present invention include, but is not limited to, trifluoromethoxy.
- hPPAR alpha activator is used to mean a compound which binds to and activates hPPAR alpha, for example in the binding and transfection assays described below.
- hPPAR mediated diseases or conditions is used to mean any disease state mediated or modulated by hPPAR, including dyslipidemia including associated diabetic dyslipidemia and mixed dyslipidemia, obesity, syndrome X (as defined in this application this embraces metabolic syndrome), heart failure, hypercholesteremia, cardiovascular disease including atherosclerosis, arteriosclerosis, and hypertriglyceridemia, type Il diabetes mellitus, type I diabetes, insulin resistance, hyperlipidemia, inflammation, epithelial hyperproliferative diseases including eczema and psoriasis and conditions associated with the lining and gut, regulation of appetite and food intake in subjects suffering from disorders such as obesity, bulimia, and anorexia nervosa, Alzheimers disease, multiple sclerosis or other cognitive disorders.
- the compounds of the invention are potentially useful in the treatment and prevention of cardiovascular disorders including atherosclerosis, arteriosclerosis, hypertriglyceridemia, and dyslipidemia including mixed dyslipidaemia.
- a compound of the invention means a compound of formula (I) or a salt or solvate thereof.
- the term "effective amount” means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal or human that is being sought, for instance, by a researcher or clinician.
- therapeutically effective amount means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder.
- the term also includes within its scope amounts effective to enhance normal physiological function.
- solvate refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of formula (I), or a salt thereof) and a solvent.
- solvents for the purpose of the invention may not interfere with the biological activity of the solute.
- suitable solvents include, but are not limited to, water, acetone, methanol, ethanol and acetic acid.
- the solvent used is a pharmaceutically acceptable solvent.
- suitable pharmaceutically acceptable solvents include water, ethanol and acetic acid. Most preferably the solvent is water.
- the compounds of formula (I) may have the ability to crystallize in more than one form, a characteristic, which is known as polymorphism, and it is understood that such polymorphic forms (“polymorphs”) are within the scope of formula (I).
- Polymorphism generally can occur as a response to changes in temperature or pressure or both and can also result from variations in the crystallization process. Polymorphs can be distinguished by various physical characteristics known in the art such as x-ray diffraction patterns, solubility and melting point.
- the compounds described herein may contain one or more chiral atoms, or may otherwise be capable of existing as two enantiomers. Accordingly, the compounds of this invention include mixtures of enantiomers as well as purified enantiomers or enantiomerically enriched mixtures. Also included within the scope of the invention are the individual isomers of the compounds represented by formula (I) above as well as any wholly or partially equilibrated mixtures thereof. The present invention also covers the individual isomers of the compounds represented by the formulas above as mixtures with isomers thereof in which one or more chiral centres are inverted.
- Y is phenyl (substituted by one or more groups independently selected from -OH, -C 1-4 alkyl, -CF 3 -Cl, -OCr 3 alkyl, -cyclohexyl), or a fused structure as shown below:
- X is a linker selected from
- -CH CH-(CH 2 ), C(O)NH-;
- X is a linker 5 atoms in shortest length which is -(CH 2 ) 2 NHC(O)CH 2 - .
- X is a linker 7 atoms in shortest length which is
- X in formula (Ia) is -CH 2 CH 2 OCH 2 CH-.
- the compounds of the invention are modulators of human PPARs, particularly PPAR alpha and PPAR gamma. In one aspect they are agonists or partial agonists.
- the hPPAR agonists of formula (I) may be agonists of only one type ("selective agonists"), agonists for two PPAR subtypes ("dual agonists"), or agonists for all three subtypes ("pan agonists").
- agonist or “activating compound”, or “activator”, or the like, is meant those compounds which have a pKi of at least 6.0 preferably at least 7.0 to the relevant PPAR, for example hPPAR alpha in the binding assay described below, and which achieve at least 50% activation of the relevant PPAR relative to the appropriate indicated positive control in the transfection assay described below at concentrations of 10 "5 M or less.
- EC 50 is defined in the transfection assay described below and is the concentration at which a compound achieves 50% of its maximum activity.
- Partial agonists can be defined as compounds that transactivate the relevant PPAR, for example PPAR alpha in CV1 cells with less than 50% fold activation compared to the reference PPAR alpha full agonist in the transfection assays of the type described below.
- the compounds of formula (1 ) are hPPAR alpha agonists.
- the compounds of the present invention may be in the form of and/or may be administered as a pharmaceutically acceptable salt.
- suitable salts see Berge et al, J. Pharm. Sci. 1977, 66, 1-19.
- the salts of the present invention are pharmaceutically acceptable salts.
- Salts encompassed within the term “pharmaceutically acceptable salts” refer to non-toxic salts of the compounds of this invention.
- Suitable pharmaceutically acceptable salts can include acid addition salts.
- a pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic or organic acid (such as hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic), optionally in a suitable solvent such as an organic solvent, to give the salt which is usually isolated for example by crystallisation and filtration.
- a suitable inorganic or organic acid such as hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic
- a suitable solvent such as an organic solvent
- a pharmaceutically acceptable acid addition salt of a compound of formula (I) can comprise or be for example a hydrobromide, hydrochloride, sulfate, nitrate, phosphate, p-toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulfonate, naphthalenesulfonate (e.g. 2- naphthalenesulfonate) salt.
- non-pharmaceutically acceptable salts e.g. trifluoroacetates
- the invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the compounds of formula (I).
- the compounds of formula (1 ) are believed to have utility as a result of activation of hPPARs.
- the invention thus provides compounds of formula (I) and salts and solvates thereof for use in therapy, and particularly in the treatment of hPPAR mediated diseases or conditions.
- a method of treating hPPAR mediated diseases or conditions comprising administering a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
- hPPAR mediated diseases or conditions include dyslipidemia including associated diabetic dyslipidemia and mixed dyslipidemia, syndrome X (as defined in this application this embraces metabolic syndrome), heart failure, hyperchloresterolemia, cardiovascular disease including atherosclerosis, arteriosclerosis, and hypertriglyceridemia, type Il diabetes mellitus, type 1 diabetes, insulin resistance, hyperlipidemia, obesity, inflammation, epithelial hyperproliferative diseases including eczema and psoriasis and conditions associated with the lung and gut and regulation of appetite and food intake in subjects suffering from disorders such as obesity, anorexia bulimia, and anorexia nervosa, cancer, Alzheimers disease, multiple sclerosis or other cognitive disorders.
- the compounds of this invention are useful in the treatment and prevention of diabetes and cardiovascular diseases and conditions including atherosclerosis, arteriosclerosis, hypertriglyceridemia, and dyslipidaemia.
- the invention further provides pharmaceutical compositions comprising a compound of the formula (I) and pharmaceutically acceptable salts, or solvates thereof, and one or more pharmaceutically acceptable carriers, diluents, or excipients.
- the compounds of the formula (I) and pharmaceutically acceptable salts, or solvates thereof, are as described above.
- the carrier(s), diluent(s) or excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof.
- a process for the preparation of a pharmaceutical composition including admixing a compound of the formula (I), or pharmaceutically acceptable salts, or solvates thereof, with one or more pharmaceutically acceptable carriers, diluents or excipients.
- the pharmaceutical composition can be for use in the treatment and/or prophylaxis of any of the conditions described herein.
- the compounds of formula (I) are intended for use in pharmaceutical compositions it will be readily understood that they are each preferably provided in substantially pure form, for example, at least 60% pure, more suitably at least 75% pure and preferably at least 85% pure, especially at least 98% pure (% in a weight for weight basis).
- compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose.
- Preferred unit dosage compositions are those containing a daily dose or sub-dose, or an appropriate fraction thereof, of an active ingredient. Such unit doses may therefore be administered once or more than once a day.
- Preferred unit dosage compositions are those containing a daily dose or sub-dose (for administration more than once a day), as herein above recited, or an appropriate fraction thereof, of an active ingredient.
- such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
- compositions may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual), rectal, inhaled, intranasal, topical
- compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier(s) or excipient(s).
- compositions adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like.
- an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like.
- Powders are prepared by reducing the compound to a suitable fine size and mixing with a similarly prepared pharmaceutical carrier such as an edible carbohydrate, as, for example, starch or mannitol. Flavoring, preservative, dispersing and coloring agent can also be present.
- Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths.
- Glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation.
- a disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
- suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like.
- Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
- Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like. Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets.
- a powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate.
- the powder mixture can be granulated by wetting with a binder such as syrup, starch paste, acadia mucilage or solutions of cellulosic or polymeric materials and forcing through a screen.
- the powder mixture can be run through the tablet machine and the result is imperfectly formed slugs broken into granules.
- the granules can be lubricated to prevent sticking to the tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil.
- the lubricated mixture is then compressed into tablets.
- the compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps.
- a clear or opaque protective coating consisting of a sealing coat of shellac, a coating of sugar or polymeric material and a polish coating of wax can be provided. Dyestuffs can be added to these coatings to distinguish different unit dosages.
- Oral fluids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of the compound.
- Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle.
- Suspensions can be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
- dosage unit compositions for oral administration can be microencapsulated.
- the formulation can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
- the compounds of formula (I), and salts, solvates and physiological functional derivatives thereof, can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
- Pharmaceutical compositions adapted for transdermal administration may be presented as discrete patches intended to remain in intimate contact with the epidermis of the recipient for a prolonged period of time.
- compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- compositions are preferably applied as a topical ointment or cream.
- the active ingredient may be employed with either a paraffinic or a water- miscible ointment base.
- the active ingredient may be formulated in a cream with an oil-in-water cream base or a water-in-oil base.
- compositions adapted for topical administrations to the eye include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
- compositions adapted for topical administration in the mouth include lozenges, pastilles and mouth washes.
- compositions adapted for rectal administration may be presented as suppositories or as enemas.
- Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, suspensions drops, gels or dry powders.
- compositions suitable and/or adapted for inhaled administration it is preferred that the compound or salt of formula (I) is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation.
- the preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction).
- compositions adapted for administration by inhalation include the particle dusts or mists.
- compositions wherein the carrier is a liquid for administration as a nasal spray or drops include aqueous or oil solutions/suspensions of the active ingredient which may be generated by means of various types of metered dose pressurised aerosols, nebulizers or insufflators.
- Aerosol formulations can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or nonaqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
- a metering valve metered dose inhaler
- the dosage form comprises an aerosol dispenser
- it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC).
- suitable HFC propellants include
- the aerosol dosage forms can also take the form of a pump-atomiser.
- the pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol. Other excipient modifiers may also be incorporated to improve, for example, the stability and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
- the pharmaceutical composition may be a dry powder inhalable composition.
- a dry powder inhalable composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of formula (I) or salt or solvate thereof (preferably in particle-size- reduced form, e.g. in micronised form), and optionally a performance modifier such as L- leucine or another amino acid, cellobiose octaacetate and/or metals salts of stearic acid such as magnesium or calcium stearate.
- a powder base such as lactose, glucose, trehalose, mannitol or starch
- the compound of formula (I) or salt or solvate thereof preferably in particle-size- reduced form, e.g. in micronised form
- a performance modifier such as L- leucine or another amino acid, cellobiose octaacetate and/or metals salts of stearic acid such as magnesium or calcium
- Aerosol formulations are preferably arranged so that each metered dose or "puff" of aerosol contains a particular amount of a compound of the invention. Administration may be once daily or several times daily, for example 2, 3 4 or 8 times, giving for example 1 , 2 or 3 doses each time.
- the overall daily dose and the metered dose delivered by capsules and cartridges in an inhaler or insufflator will generally be double those with aerosol formulations.
- compositions adapted for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
- compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the composition isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the compositions may be presented in unit- dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- compositions may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
- a therapeutically effective amount of a compound of the present invention will depend upon a number of factors including, for example, the age and weight of the animal, the precise condition requiring treatment and its severity, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant physician or veterinarian.
- the pharmaceutically acceptable compounds or salts of the invention may be administered in a daily dose (for an adult patient) of, for example, an oral or parenteral dose of 0.01 mg to 100 mg/kg per day body weight. This amount may be given in a single dose per day or more usually in a number (such as two, three, four, five or six) of sub- doses per day such that the total daily dose is the same.
- An effective amount of a salt or solvate, thereof, may be determined as a proportion of the effective amount of the compound of formula (I) per se.
- the actual amount per day would usually be from 70 to 700 mg and this amount may be given in a single dose per day or more usually in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same.
- An effective amount of a salt or solvate, or physiologically functional derivative thereof, may be determined as a proportion of the effective amount of active ingredient.
- Combination therapies according to the present invention thus comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, and the use of at least one other pharmaceutically active agent.
- combination therapies according to the present invention comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, and at least one other pharmaceutically active agent.
- the compound(s) of formula (I) and the other pharmaceutically active agent(s) may be administered together in a single pharmaceutical composition or separately and, when administered separately this may occur simultaneously or sequentially in any order.
- the amounts of the compound(s) of formula (I) and the other pharmaceutically active agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
- the compound and pharmaceutical compositions according to the invention may be used in combination with or include one or more other therapeutic agents, for example selected from statins (HMG Co A reductase inhibitors) and/or other lipid lowering drugs for example MTP inhibitors and LDLR upregulators.
- the compounds of the invention may also be used in combination with antidiabetic agents, e.g. metformin, sulfonylureas and/or PPAR activators (for example thiazolidinediones such as e.g. Pioglitazone and Rosiglitazone).
- the compounds may also be used in combination with antihypertensive agents such as angiotensin antagonists (eg Telmisartan) calcium channel antagonists and ACE inhibitors.
- the invention thus provides in a further aspect the use of a combination comprising a compound of formula (I) with a further therapeutic agent in the treatment of a hPPAR mediated disease.
- the other therapeutic ingredient(s) may be used in the form of salts, for example as alkali metal or amine salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates, to optimise the activity and/or stability and/or physical characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
- the compounds When a compound of formula (I) is used in combination with other therapeutic agents, the compounds may be administered either sequentially or simultaneously by any convenient route.
- compositions comprising a combination as defined above optimally together with a pharmaceutically acceptable carrier or excipient comprise a further aspect of the invention.
- the individual components of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical compositions.
- the two compounds When combined in the same composition it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the composition and may be formulated for administration. When formulated separately they may be provided in any convenient composition, conveniently in such a manner as are known for such compounds in the art.
- the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
- the compounds of this invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention are prepared in the Working Examples.
- '880 Ammonia' or '0.880 ammonia' refers to concentrated aqueous ammonia (specific gravity 0.880).
- 'Hydrophobic Frit' This refers to a Whatman PTFE filter medium (frit), pore size 5.0 ⁇ m, housed in a polypropylene tube.
- Room temperature this is usually in the range of about 20 to about 25 0 C.
- Methyl 4-amino-3-iodobenzoate (27.7 g, 0.1 mol), LiCI (4.24 g, 0.1 mol), Cs 2 CO 3 (81.5 g, 0.25 mol), tri(o-tolyl)phosphine (1.52 g, 5 mmol) was added to DMF (500 ml). The mixture was degassed with nitrogen and 4-trimethylsilyl-3-butyn-1-ol (26.67 ml, 0.16 mol) and palladium (II) acetate (1.12 g, 5 mmol) was added, The mixture was degassed with nitrogen and stirred at 100 0 C for 3 hours. The reaction mixture was filtered on celite (5 cm) and evaporated.
- Methyl 3-formyl-1-phenyl-1 /-/-indole-5-carboxylate (Intermediate 40) 2.2 g, 7.9 mmol) and benzyl (triphenylphosphoranylidene)acetate (3.55 g, 8.7 mmol) in toluene were heated to reflux for 4 hours. The solvent was evaporated and the residue was purified on SiC> 2 eluting with dichloromethane/cyclohexane 50/50 to give the title compound (2.3 g, 70%).
- the reaction was stirred at 100 0 C for 24 hours and was re-added K 3 PO 4 (28.8 g, 136 mmol), CuI (616 mg, 3.24 mmol), N,N'-dimethylethylenediamine (2.1 ml, 19.4 mmol) and iodobenzene (8.7 ml, 77.6 mmol).
- K 3 PO 4 28.8 g, 136 mmol
- CuI 616 mg, 3.24 mmol
- N,N'-dimethylethylenediamine 2.1 ml, 19.4 mmol
- iodobenzene 8.7 ml, 77.6 mmol
- the white precipitate was filtrate and washed with water.
- the precipitate was dissolved in ethyl acetate, the solution was washed with water and the organic phase was dried over MgSO 4 , filtered and evaporated to dryness to give the title compound as a white solid (32.5 g, 93%).
- Methyl 3-formyl-1-phenyl-1 H-indole-5-carboxylate (Intermediate 38) (586 mg, 2.1 mmol), 1-(phenylmethyl)piperazine (470 ⁇ l, 2.62 mmol) and acetic acid (130 ⁇ l, 2.1 mmol) were dissolved in methanol. The mixture was stirred at room temperature for one hour. NaBH 3 CN was added and the mixture was stirred at room temperature for 18 hours. The solvent was evaporated and the residue was diluted with HCI 1 N and neutralised with NaOH 1 N. The mixture was extracted with diethyl ether.
- reaction mixture was stirred at room temperature for 1 H30. The reaction mixture was quenched with water and evaporated. A saturated solution of NaHCU3 was added and the mixture was extracted with ethyl acetate (3 times). The organic phases were washed with brine, dried over Na 2 SO 4 , filtered and evaporated. The residue was purified on SiO 2 eluting with dichloromethane/methanol 98/2 to 95/5 to give the title compound as colourless oil (170 mg, 57%).
- PPAR ligand binding domain (LBD) was expressed in E. coli as polyHis tagged fusion proteins and purified. The LBD was then labelled with biotin and immobilised on streptavidin-modified scintillation proximity beads. The beads were then incubated with a constant amount of the appropriate radioligand (5- ⁇ 4-[2-(Methyl-pyridin-2-yl-amino)-ethoxy]-benzyl ⁇ -thiazolidine- 2,4-dione (J.Med.Chem.
- Transfection assay Compounds were screened for functional potency in transient transfection assays in CV-1 cells for their ability to activate the PPAR subtypes (transactivation assay).
- a previously established chimeric receptor system was utilized to allow comparison of the relative transcriptional activity of the receptor subtypes on the same target gene and to prevent endogenous receptor activation from complicating the interpretation of results. See, for example, Lehmann, J. M.; Moore, L. B.; Smith-Oliver, T. A.; Wilkison, W. O.; Willson, T. M.; Kliewer, S.
- An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARgamma), J. Biol. Chem., 270, 12953-6 (1995).
- the ligand binding domains for murine and human PPAR alpha, PPAR gamma, and PPAR delta were each fused to the yeast transcription factor GAL4 DNA binding domain.
- CV-1 cells were transiently transfected with expression vectors for the respective PPAR chimera along with a reporter construct containing five copies of the GAL4 DNA binding site driving expression of secreted placental alkaline phosphatase (SPAP) and beta-galactosidase.
- SPAP secreted placental alkaline phosphatase
- the positive control in the hPPAR alpha assays was 2-4-[2-(3-[4-fluorophenyl]-1- heptylureido)ethyl]-phenoxy-(2-methyl propionic acid (WO 97/36579).
- the positive control for PPAR delta assays was 2- ⁇ 2-methyl-4-[( ⁇ 4-methyl-2- ⁇ trifluoromethyl)phenyl]-1 ,3- thiazol-5-yl ⁇ methyl)sulfanyl]phenoxy ⁇ acetic acid (WO 01/00603).
- the positive control was (5- ⁇ 4-[2-(Methyl-pyridin-2-yl-amino)-ethoxy]-benzyl ⁇ -thiazolidine-2,4-dione (J. Med. Chem. 1994, 37(23), 3977), for PPAR gamma.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
There is provided according to the invention novel compounds of Formula (I) or pharmaceutically acceptable salts or solvates thereof: (I) useful as PPAR activators.
Description
INDOLE DERIVATIVES USEFUL AS PPAR ACTIVATORS
Field of the Invention
The present invention relates to indole derivatives, compositions and medicaments containing said indole derivatives and processes for their preparation. The invention also relates to the use of said indole derivatives, compositions and medicaments, for example as activators of the alpha subtype of the human peroxisome proliferator activated receptor (hPPAR) and/or for the treatment and/or prophylaxis of diseases or conditions mediated by hPPAR.
Background to the Invention
Several independent risk factors have been associated with cardiovascular disease. These include hypertension, increased fibrinogen levels, high levels of triglycerides, elevated LDL cholesterol, elevated total cholesterol, and low levels of HDL cholesterol. HMG CoA reductase inhibitors ("statins") are useful for treating conditions characterized by high LDL-c levels. It has been shown that lowering LDL-c is not sufficient for reducing the risk of cardiovascular disease in some patients, particularly those with normal LDL-c levels. This population pool is identified by the independent risk factor of low HDL-c. The increased risk of cardiovascular disease associated with low HDL-c levels has not yet been successfully addressed by drug therapy (i.e., currently there are no drugs on the market that are useful for raising HDL-c >40%). (Bisgaier, C. L.; Pape, M. E. Curr. Pharm. Des. 1998, 4, 53-70).
Syndrome X (including metabolic syndrome) is loosely defined as a collection of abnormalities including hyperinsuinlemia, obesity, elevated levels of trigycerides, uric acid, fibrinogen, small dense LDL-c particles, and plasminogen activator inhibitor 1 (PAI-1 ), and decreased levels of HDL-c.
NIDDM is described as insulin resistance which in turn causes anomalous glucose output and a decrease in glucose uptake by skeletal muscle. These factors eventually lead to impaired glucose tolerance (IGT) and hyperinsulinemia.
Peroxisome Proliferator Activated Receptors (PPARs) are orphan receptors belonging to the steroid/retinoid receptor superfamily of ligand-activated transcription factors. See, for example, Willson, T. M. and Wahli, W., Curr. Opin. Chem. Biol., (1997), Vol. 1 , pp 235- 241.
Three mammalian Peroxisome Proliferator-Activated Receptors have been isolated and termed PPAR-alpha, PPAR-gamma, and PPAR-delta (also known as NUC1 or PPAR- beta). These PPARs regulate expression of target genes by binding to DNA sequence elements, termed PPAR response elements (PPRE). To date, PPRE's have been identified in the enhancers of a number of genes encoding proteins that regulate lipid metabolism suggesting that PPARs play a pivotal role in the adipogenic signaling cascade and lipid homeostasis (H. Keller and W. Wahli, Trends Endocrin. Met 291-296, 4 (1993)).
Certain compounds that activate or otherwise interact with one or more of the PPARs have been implicated in the regulation of triglyceride and cholesterol levels in animal models. See, for example, WO 01/40207, WO 01/00603, WO 97/31907, WO 02/46174 (Glaxo Group Ltd et al).
Summary of the Invention
In one aspect of the invention there is provided a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof:

Z is phenyl (optionally substituted by halogen), pyridinyl;
Y is phenyl (optionally substituted by one or more groups each independently selected from -Ci-6 alkyl, -Ci-6 alkoxy, -Ci-6 haloalkyl, -Ci-6 haloalkoxy, -halogen, -OH, -C3-7 cycloalkyl, -NR1R2), or phenyl fused to a 5 or 6 membered heterocyclic or cycloalkyl ring;
R1 and R2 independently represent H or d-3 alkyl;
X is a linker group of 5-7 atoms in shortest length between the carbon of the indole group and Y;
or X-Y represents a group:

R3 is C1-3 alkyl or C1-3 haloalkyl.
In a further aspect of the present invention, there is provided a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof for use in therapy, in particular in the treatment of hPPAR mediated diseases or conditions.
In a further aspect of the present invention, there is provided a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof and one or more of pharmaceutically acceptable carriers, diluents and excipients.
In a further aspect of the present invention, there is provided a method of treating hPPAR mediated diseases or conditions comprising administering to said mammal a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
In an further aspect of the present invention, there is provided the use of a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof in the preparation of a medicament for use in the treatment of hPPAR mediated diseases or conditions.
In an further aspect of the invention there is provided a compound of formula (I) or a pharmaceutically acceptable salt, or solvate thereof for use in the treatment of hPPAR mediated diseases or conditions.
Detailed Description of the Invention
As used herein, the term "alkyl" refers to straight or branched hydrocarbon chains containing the specified number of carbon atoms. For example, C1-6 alkyl means a
straight or branched alkyl chain containing at least 1 , and at most 6 carbon atoms. Examples of "alkyl" as used herein include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, t-butyl, n-pentyl.
As used herein the term "alkylene" refers to a straight or branched chain divalent hydrocarbon radical having from 1-10 carbon atoms. Examples of "alkylene" include but are not limited to methylene, ethylene, n-propylene.
As used herein, the term "alkenyl" refers to a straight or branched hydrocarbon radical having from two to ten carbons and at least one carbon-carbon double bond. Examples of "C2-C10 alkenyl" groups used in the present invention include, but are not limited to ethenyl, propenyl, 1-butenyl, 2-butenyl, and isobutenyl.
As used herein, the term "cycloalkyl" refers to a non-aromatic hydrocarbon ring containing the specified number of carbon atoms. For example C3-7 cycloalkyl means a non-aromatic ring containing at least three, and at most seven, ring carbon atoms. Examples of "cycloalkyl" as used herein include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl.
As used herein the term "halogen" refers to fluorine (F), chlorine (Cl), bromine (Br) or iodine (I), and the radicals, thereof, fluoro (-F), chloro (-Cl), bromo (-Br) and iodo (-I).
As used herein, the term "CrC6 haloalkyl" refers to an alkyl group as defined above containing at least 1 , and at most 6, carbon atoms substituted with at least one halo group, halo being as defined herein. Examples of branched or straight chained "C1-C6 haloalkyl" groups useful in the present invention include, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl and n-butyl substituted independently with one or more halos, e.g., fluoro, chloro, bromo and iodo.
As used herein, the term "alkoxy" refers to the group R3O-, where R3 is alkyl as defined above and the term "C1-C6 alkoxy" refers to an alkoxy group as defined herein wherein the alkyl moiety contains at least 1 , and at most 6, carbon atoms. Exemplary C1-C6 alkoxy groups useful in the present invention include, but are not limited to, methoxy, ethoxy, n- propoxy, isopropoxy, n-butoxy, and t-butoxy.
As used herein, the term "heterocyclic" or the term "heterocyclyl" refers to a non-aromatic three to seven-membered heterocyclic ring being satured or having one or more degrees of unsaturation containing one or more heteroatoms selected from S, SO, SO2, O, or N. Examples of "heterocyclic" moieties include, but are not limited to, tetrahydrofuran, pyran, 1 ,4-dioxane, 1 ,3-dioxane, piperidine, pyrrolidine, morpholine, tetrahydrothiopyran, tetrahydrothiophene, di-oxo tetrahydrothiophene, and the like.
As used herein, the term "haloalkoxy" refers to the group R3O-, where R3 is haloalkyl as defined above and the term "C1-C6 haloalkoxy" refers to a haloalkoxy group as defined herein wherein the haloalkyl moiety contains at least 1 and at most 6, carbon atoms. Exemplary C1-C6 haloalkoxy groups useful in the present invention include, but is not limited to, trifluoromethoxy.
The term hPPAR alpha activator is used to mean a compound which binds to and activates hPPAR alpha, for example in the binding and transfection assays described below.
The term "hPPAR mediated diseases or conditions" is used to mean any disease state mediated or modulated by hPPAR, including dyslipidemia including associated diabetic dyslipidemia and mixed dyslipidemia, obesity, syndrome X (as defined in this application this embraces metabolic syndrome), heart failure, hypercholesteremia, cardiovascular disease including atherosclerosis, arteriosclerosis, and hypertriglyceridemia, type Il diabetes mellitus, type I diabetes, insulin resistance, hyperlipidemia, inflammation, epithelial hyperproliferative diseases including eczema and psoriasis and conditions associated with the lining and gut, regulation of appetite and food intake in subjects suffering from disorders such as obesity, bulimia, and anorexia nervosa, Alzheimers disease, multiple sclerosis or other cognitive disorders. In particular, the compounds of the invention are potentially useful in the treatment and prevention of cardiovascular disorders including atherosclerosis, arteriosclerosis, hypertriglyceridemia, and dyslipidemia including mixed dyslipidaemia.
As used herein, "a compound of the invention" means a compound of formula (I) or a salt or solvate thereof.
As used herein, the term "effective amount" means that amount of a drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system,
animal or human that is being sought, for instance, by a researcher or clinician. Furthermore, the term "therapeutically effective amount" means any amount which, as compared to a corresponding subject who has not received such amount, results in improved treatment, healing, prevention, or amelioration of a disease, disorder, or side effect, or a decrease in the rate of advancement of a disease or disorder. The term also includes within its scope amounts effective to enhance normal physiological function.
As used herein, the term "solvate" refers to a complex of variable stoichiometry formed by a solute (in this invention, a compound of formula (I), or a salt thereof) and a solvent. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute. Examples of suitable solvents include, but are not limited to, water, acetone, methanol, ethanol and acetic acid. Preferably the solvent used is a pharmaceutically acceptable solvent. Examples of suitable pharmaceutically acceptable solvents include water, ethanol and acetic acid. Most preferably the solvent is water.
The compounds of formula (I) may have the ability to crystallize in more than one form, a characteristic, which is known as polymorphism, and it is understood that such polymorphic forms ("polymorphs") are within the scope of formula (I). Polymorphism generally can occur as a response to changes in temperature or pressure or both and can also result from variations in the crystallization process. Polymorphs can be distinguished by various physical characteristics known in the art such as x-ray diffraction patterns, solubility and melting point.
Certain of the compounds described herein may contain one or more chiral atoms, or may otherwise be capable of existing as two enantiomers. Accordingly, the compounds of this invention include mixtures of enantiomers as well as purified enantiomers or enantiomerically enriched mixtures. Also included within the scope of the invention are the individual isomers of the compounds represented by formula (I) above as well as any wholly or partially equilibrated mixtures thereof. The present invention also covers the individual isomers of the compounds represented by the formulas above as mixtures with isomers thereof in which one or more chiral centres are inverted.
It is also noted that the compounds of Formula (I) may form tautomers. It is understood that all tautomers and mixtures of tautomers of the compounds of the present invention are included within the scope of the compounds of the present invention.
In one aspect, Z is

In one aspect Y is phenyl (substituted by one or more groups independently selected from -OH, -C1-4 alkyl, -CF3 -Cl, -OCr3 alkyl, -cyclohexyl), or a fused structure as shown below:

In one aspect X is C1-10 alkylene linker or C2-io alkenylene linker, each of which may contain within the alkylene chain one or more C=O, NR, and/or O moieties (R=H or Ci-3 alkyl) forming a 6 atom linker in shortest length.
In a further aspect, X is a linker which includes a 5-6 membered heteroaryl or a 5-6 membered heterocyclic nitrogen containing ring, together with one or more C1-6 alkylene, NR1 (R1 = H or C1-3 alkyl), and/or C=O moieties to form a 6 atom linker in shortest length.
In one aspect X is a linker selected from
-(CH2J4 NHC(O)-;
-(CHz)4 CONH;
-(CHz)2 OCH2 C(O)NH-; -(CHz)2 NHC(O)CH2O-;
-(CH2)2 NH C(O) (CH2)2-
-(CHz)2 N(CH3) CH2 C(O)NH-
-CH2 N(CH3) CH2CH2 NHC(O)-
-CH2 N(CH3) C(CH3)2 CH2 NHCO-; -CH2 NH(CH2)2 NHC(O-);
-CH = CH-(CH2), C(O)NH-;
-C(O)NH CH2CH2 C(O)NH-;
-(CH2)2 C(O)NHCH2CH2-;





-CH-N N-CO- and

In a further aspect X is a linker 5 atoms in shortest length which is -(CH2)2NHC(O)CH2- .
In a further aspect, X is a linker 7 atoms in shortest length which is

In one aspect X-Y together represent

While aspects for each variable have generally been listed above separately for each variable this invention includes those compounds in which several or each aspect in formula (I) is selected from each of the aspects listed above. Therefore, this invention is intended to include all combinations of aspects for each variable.
In a particular aspect, there is provided a compound for formula (I) which is of formula (Ia)
wherein X is defined as above for formula (I).
In a further aspect, X in formula (Ia) is -CH2CH2OCH2CH-.
The compounds of the invention are modulators of human PPARs, particularly PPAR alpha and PPAR gamma. In one aspect they are agonists or partial agonists. The hPPAR agonists of formula (I) may be agonists of only one type ("selective agonists"), agonists for two PPAR subtypes ("dual agonists"), or agonists for all three subtypes ("pan agonists").
As used herein, by "agonist", or "activating compound", or "activator", or the like, is meant those compounds which have a pKi of at least 6.0 preferably at least 7.0 to the relevant PPAR, for example hPPAR alpha in the binding assay described below, and which achieve at least 50% activation of the relevant PPAR relative to the appropriate indicated positive control in the transfection assay described below at concentrations of 10"5 M or less. EC50 is defined in the transfection assay described below and is the concentration at which a compound achieves 50% of its maximum activity.
Partial agonists can be defined as compounds that transactivate the relevant PPAR, for example PPAR alpha in CV1 cells with less than 50% fold activation compared to the reference PPAR alpha full agonist in the transfection assays of the type described below. Suitably the compounds of formula (1 ) are hPPAR alpha agonists.
Specific examples of compounds of the present invention include:
3-{2-[(2-{[4-(1-methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 H-indole-5- carboxylic acid;
3-{2-[(2-{[4-(1 ,1-dimethylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 H-indole-5- carboxylic acid; 3-[2-({2-[(4-methylphenyl)amino]-2-oxoethyl}oxy)ethyl]-1-phenyl-1 H-indole-5-carboxylic acid;
3-[2-({2-[(4-ethylphenyl)amino]-2-oxoethyl}oxy)ethyl]-1-phenyl-1 H-indole-5-carboxylic acid;
3-(2-{[2-(2,3-dihydro-1 H-inden-5-ylamino)-2-oxoethyl]oxy}ethyl)-1-phenyl-1 H-indole-5- carboxylic acid; 3-(2-{[2-(2,3-dihydro-1 ,4-benzodioxin-6-ylamino)-2-oxoethyl]oxy}ethyl)-1-phenyl-1 H- indole-5-carboxylic acid;
3-{2-[(2-{[3-(1-methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 H-indole-5- carboxylic acid;
3-[2-({2-[(4-cyclohexylphenyl)amino]-2-oxoethyl}oxy)ethyl]-1-phenyl-1 H-indole-5- carboxylic acid;
3-{2-[(2-{[4-(dimethylamino)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 /-/-indole-5- carboxylic acid;
3-{2-[({[4-(methyloxy)phenyl]oxy}acetyl)amino]ethyl}-1-(2-pyridinyl)-1/-/-indole-5-carboxylic acid; 3-{2-[(2-{[4-(1-methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-(2-pyridinyl)-1 H-indole-5- carboxylic acid;
3-{2-[({[4-(1-methylethyl)phenyl]oxy}acetyl)amino]ethyl}-1-phenyl-1 H-indole-5-carboxylic acid;
3-[2-({[(3,4-dichlorophenyl)oxy]acetyl}amino)ethyl]-1-phenyl-1 H-indole-5-carboxylic acid; 3-{2-[({4-methyl-2-[4-(1-methylethyl)phenyl]-1 ,3-thiazol-5-yl}carbonyl)amino]ethyl}-1- phenyl-1 H-indole-5-carboxylic acid;
3-{2-[({[4-(1 ,1-dimethylethyl)phenyl]oxy}acetyl)amino]ethyl}-1-phenyl-1 H-indole-5- carboxylic acid;
3-{2-[({[3-(methyloxy)phenyl]oxy}acetyl)amino]ethyl}-1 -phenyl-1 H-indole-5-carboxylic acid; 3-{2-[({[4-(methyloxy)phenyl]oxy}acetyl)amino]ethyl}-1 -phenyl-1 H-indole-5-carboxylic acid;
3-[2-({[4-(1-methylethyl)phenyl]acetyl}amino)ethyl]-1 -phenyl-1 H-indole-5-carboxylic acid;
3-[2-({3-[4-(1-methylethyl)phenyl]propanoyl}amino)ethyl]-1 -phenyl-1 H-indole-5-carboxylic acid;
1 -phenyl-3-{4-[6-(trifluoromethyl)-3,4-dihydro-2(1 H)-isoquinolinyl]butyl}-1 H-indole-5- carboxylic acid;
3-[4-({[4-(1 -methylethyl)phenyl]carbonyl}amino)butyl]-1 -(2-pyridinyl)-1 H-indole-5- carboxylic acid;
3-[4-({[4-(1 -methylethyl)phenyl]carbonyl}amino)butyl]-1 -phenyl-1 H-indole-5-carboxylic acid; 1-(4-fluorophenyl)-3-[4-({[4-(1-methylethyl)phenyl]carbonyl}amino)butyl]-1H-indole-5- carboxylic acid;
3-[4-({[4-(1 ,1-dimethylethyl)phenyl]carbonyl}amino)butyl]-1 -phenyl-1 H-indole-5-carboxylic acid;
3-[4-({[2,4-bis(methyloxy)phenyl]carbonyl}amino)butyl]-1-(2-pyridinyl)-1 H-indole-5- carboxylic acid;
3-[4-({[2-hydroxy-3-(1-methylethyl)phenyl]carbonyl}amino)butyl]-1-(2-pyridinyl)-1 H-indole-
5-carboxylic acid;
3-{4-[6-(1 -methylethyl)-3,4-dihydro-2(1 H)-isoquinolinyl]butyl}-1 -phenyl-1 H-indole-5- carboxylic acid; 1 -phenyl-3-[4-({[4-(trifluoromethyl)phenyl]carbonyl}amino)butyl]-1 H-indole-5-carboxylic acid;
2-methyl-3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-piperidinyl]methyl}-1 -phenyl-
IH-indole-5-carboxylic acid;
3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-piperidinyl]methyl}-1-(2-pyridinyl)-1 /-/- indole- 5-carboxylic acid; 1-(4-fluorophenyl)-3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-piperidinyl]methyl}-
1H-indole- 5-carboxylic acid;
3-({methyl[2-({[4-(1-methylethyl)phenyl]carbonyl}amino)ethyl]amino}methyl)-1-phenyl-1 /-/- indole- 5-carboxylic acid;
3-{[[2-({[2-hydroxy-3-(1-methylethyl)phenyl]carbonyl}amino)ethyl](methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[methyl(1-{[4-(1-methylethyl)phenyl]carbonyl}-3-piperidinyl)amino]methyl}-1-phenyl-1H- indole- 5-carboxylic acid;
3-{[[1 ,1-dimethyl-2-({[4-(1-methylethyl)phenyl]carbonyl}amino)ethyl](methyl)amino]methyl}-
1-phenyl-1 H-indole- 5-carboxylic acid; 3-{[methyl({4-methyl-2-[4-(trifluoromethyl)phenyl]-1 ,3-thiazol-5-yl}methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[methyl({4-methyl-2-[4-(1-methylethyl)phenyl]-1 !3-thiazol-5-yl}methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[(1-{[4-(1 ,1-dimethylethyl)phenyl]carbonyl}-3-piperidinyl)(methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[({2-[4-(1 ,1-dimethylethyl)phenyl]-4-methyl-1 ,3-thiazol-5- yl}methyl)(methyl)amino]methyl}-1 -phenyl-1 H-indole-5-carboxylic acid;
3-({[2-({[4-(1-methylethyl)phenyl]carbonyl}amino)ethyl]amino}methyl)-1 -phenyl-1 H-indole-
5-carboxylic acid; 3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-pyrrolidinyl]methyl}-1 -phenyl-1 H-indole- 5-carboxylic acid;
3-((1 £)-5-{[4-(1 -methylethyl)phenyl]amino}-5-oxo-1 -penten-1 -yl)-1 -phenyl-1 H-indole- 5- carboxylic acid;
3-{[(3-{[4-(1-methylethyl)phenyl]amino}-3-oxopropyl)amino]carbonyl}-1 -phenyl-1 H-indole- 5-carboxylic acid;
3-(5-{[4-(1-methylethyl)phenyl]amino}-5-oxopentyl)-1 -phenyl-1 H-indole-5-carboxylic acid;
3-{2-[methyl(2-{[4-(1-methylethyl)phenyl]amino}-2-oxoethyl)amino]ethyl}-1-(2-pyridinyl)-
1 H-indole-5-carboxylic acid;
3-{2-[methyl(2-{[4-(1 -methylethyl)phenyl]amino}-2-oxoethyl)amino]ethyl}-1 -phenyl-1 H- indole- 5-carboxylic acid;
6-methyl-3-{[(1-{[4-(1-methylethyl)phenyl]carbonyl}-3-piperidinyl)amino]methyl}-1 -phenyl- IH-indole-5-carboxylic acid;
3-[3-({2-[4-(1-methylethyl)phenyl]ethyl}amino)-3-oxopropyl]-1-phenyl-1H-indole-5- carboxylic acid; 3-[(4-{[4-(1-methylethyl)phenyl]carbonyl}-1-piperazinyl)methyl]-1-phenyl-1 H-indole-5- carboxylic acid;
3-{[3-{[4-(1-methylethyl)phenyl]acetyl}tetrahydro-1 (2/-/)-pyrimidinyl]methyl}-1-phenyl-1H- indole- 5-carboxylic acid;
and salts and solvates thereof.
The compounds of the present invention may be in the form of and/or may be administered as a pharmaceutically acceptable salt. For a review on suitable salts see Berge et al, J. Pharm. Sci. 1977, 66, 1-19.
Typically, the salts of the present invention are pharmaceutically acceptable salts. Salts encompassed within the term "pharmaceutically acceptable salts" refer to non-toxic salts of the compounds of this invention.
Suitable pharmaceutically acceptable salts can include acid addition salts.
A pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of formula (I) with a suitable inorganic or organic acid (such as hydrobromic, hydrochloric, sulfuric, nitric, phosphoric, p-toluenesulfonic, benzenesulfonic, methanesulfonic, ethanesulfonic, naphthalenesulfonic such as 2-naphthalenesulfonic), optionally in a suitable solvent such as an organic solvent, to give the salt which is usually isolated for example by crystallisation and filtration. A pharmaceutically acceptable acid addition salt of a compound of formula (I) can comprise or be for example a hydrobromide, hydrochloride, sulfate, nitrate, phosphate, p-toluenesulfonate, benzenesulfonate, methanesulfonate, ethanesulfonate, naphthalenesulfonate (e.g. 2- naphthalenesulfonate) salt.
Other non-pharmaceutically acceptable salts, e.g. trifluoroacetates, may be used, for example in the isolation of compounds of the invention, and are included within the scope of this invention.
The invention includes within its scope all possible stoichiometric and non-stoichiometric forms of the compounds of formula (I).
The compounds of formula (1 ) are believed to have utility as a result of activation of hPPARs.
In particular the compounds of formula (I) and salts and solvates thereof are believed to be activators of hPPAR alpha, and thus be potentially useful in the treatment of hPPAR mediated diseases or conditions.
The invention thus provides compounds of formula (I) and salts and solvates thereof for use in therapy, and particularly in the treatment of hPPAR mediated diseases or conditions.
In a further aspect of the present invention, there is provided a method of treating hPPAR mediated diseases or conditions comprising administering a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect of the present invention, there is provided the use of a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof in the preparation of a medicament for use in the treatment of hPPAR mediated diseases or conditions.
In further aspect of the invention there is provided a compound of formula (I) or a pharmaceutically acceptable salt, or solvate thereof for use in the treatment of hPPAR mediated diseases or conditions.
hPPAR mediated diseases or conditions include dyslipidemia including associated diabetic dyslipidemia and mixed dyslipidemia, syndrome X (as defined in this application this embraces metabolic syndrome), heart failure, hyperchloresterolemia, cardiovascular disease including atherosclerosis, arteriosclerosis, and hypertriglyceridemia, type Il diabetes mellitus, type 1 diabetes, insulin resistance, hyperlipidemia, obesity, inflammation, epithelial hyperproliferative diseases including eczema and psoriasis and conditions associated with the lung and gut and regulation of appetite and food intake in subjects suffering from disorders such as obesity, anorexia bulimia, and anorexia nervosa, cancer, Alzheimers disease, multiple sclerosis or other cognitive disorders. In particular, the compounds of this invention are useful in the treatment and prevention of diabetes
and cardiovascular diseases and conditions including atherosclerosis, arteriosclerosis, hypertriglyceridemia, and dyslipidaemia.
While it is possible that, for use in therapy, a compound of formula (I), as well as pharmaceutically acceptable solvates thereof, may be administered as the raw chemical, it is possible to present the active ingredient as a pharmaceutical composition. Accordingly, the invention further provides pharmaceutical compositions comprising a compound of the formula (I) and pharmaceutically acceptable salts, or solvates thereof, and one or more pharmaceutically acceptable carriers, diluents, or excipients. The compounds of the formula (I) and pharmaceutically acceptable salts, or solvates thereof, are as described above. The carrier(s), diluent(s) or excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipient thereof. In accordance with another aspect of the invention there is also provided a process for the preparation of a pharmaceutical composition including admixing a compound of the formula (I), or pharmaceutically acceptable salts, or solvates thereof, with one or more pharmaceutically acceptable carriers, diluents or excipients. The pharmaceutical composition can be for use in the treatment and/or prophylaxis of any of the conditions described herein.
Since the compounds of formula (I) are intended for use in pharmaceutical compositions it will be readily understood that they are each preferably provided in substantially pure form, for example, at least 60% pure, more suitably at least 75% pure and preferably at least 85% pure, especially at least 98% pure (% in a weight for weight basis).
Pharmaceutical compositions may be presented in unit dose forms containing a predetermined amount of active ingredient per unit dose. Preferred unit dosage compositions are those containing a daily dose or sub-dose, or an appropriate fraction thereof, of an active ingredient. Such unit doses may therefore be administered once or more than once a day. Preferred unit dosage compositions are those containing a daily dose or sub-dose (for administration more than once a day), as herein above recited, or an appropriate fraction thereof, of an active ingredient. Furthermore, such pharmaceutical compositions may be prepared by any of the methods well known in the pharmacy art.
Pharmaceutical compositions may be adapted for administration by any appropriate route, for example by the oral (including buccal or sublingual), rectal, inhaled, intranasal, topical
(including buccal, sublingual or transdermal), vaginal or parenteral (including
subcutaneous, intramuscular, intravenous or intradermal) route. Such compositions may be prepared by any method known in the art of pharmacy, for example by bringing into association the active ingredient with the carrier(s) or excipient(s).
Pharmaceutical compositions adapted for oral administration may be presented as discrete units such as capsules or tablets; powders or granules; solutions or suspensions in aqueous or non-aqueous liquids; edible foams or whips; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
For instance, for oral administration in the form of a tablet or capsule, the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like. Powders are prepared by reducing the compound to a suitable fine size and mixing with a similarly prepared pharmaceutical carrier such as an edible carbohydrate, as, for example, starch or mannitol. Flavoring, preservative, dispersing and coloring agent can also be present.
Capsules are made by preparing a powder mixture, as described above, and filling formed gelatin sheaths. Glidants and lubricants such as colloidal silica, talc, magnesium stearate, calcium stearate or solid polyethylene glycol can be added to the powder mixture before the filling operation. A disintegrating or solubilizing agent such as agar-agar, calcium carbonate or sodium carbonate can also be added to improve the availability of the medicament when the capsule is ingested.
Moreover, when desired or necessary, suitable binders, glidants, lubricants, sweetening agents, flavours, disintegrating agents and coloring agents can also be incorporated into the mixture. Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes and the like.
Lubricants used in these dosage forms include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
Disintegrators include, without limitation, starch, methyl cellulose, agar, bentonite, xanthan gum and the like. Tablets are formulated, for example, by preparing a powder mixture, granulating or slugging, adding a lubricant and disintegrant and pressing into tablets. A powder mixture is prepared by mixing the compound, suitably comminuted, with a diluent or base as described above, and optionally, with a binder such as carboxymethylcellulose, an aliginate, gelatin, or polyvinyl pyrrolidone, a solution retardant such as paraffin, a
resorption accelerator such as a quaternary salt and/or an absorption agent such as bentonite, kaolin or dicalcium phosphate. The powder mixture can be granulated by wetting with a binder such as syrup, starch paste, acadia mucilage or solutions of cellulosic or polymeric materials and forcing through a screen. As an alternative to granulating, the powder mixture can be run through the tablet machine and the result is imperfectly formed slugs broken into granules. The granules can be lubricated to prevent sticking to the tablet forming dies by means of the addition of stearic acid, a stearate salt, talc or mineral oil. The lubricated mixture is then compressed into tablets. The compounds of the present invention can also be combined with a free flowing inert carrier and compressed into tablets directly without going through the granulating or slugging steps. A clear or opaque protective coating consisting of a sealing coat of shellac, a coating of sugar or polymeric material and a polish coating of wax can be provided. Dyestuffs can be added to these coatings to distinguish different unit dosages.
Oral fluids such as solution, syrups and elixirs can be prepared in dosage unit form so that a given quantity contains a predetermined amount of the compound. Syrups can be prepared by dissolving the compound in a suitably flavored aqueous solution, while elixirs are prepared through the use of a non-toxic alcoholic vehicle. Suspensions can be formulated by dispersing the compound in a non-toxic vehicle. Solubilizers and emulsifiers such as ethoxylated isostearyl alcohols and polyoxy ethylene sorbitol ethers, preservatives, flavor additive such as peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, and the like can also be added.
Where appropriate, dosage unit compositions for oral administration can be microencapsulated. The formulation can also be prepared to prolong or sustain the release as for example by coating or embedding particulate material in polymers, wax or the like.
The compounds of formula (I), and salts, solvates and physiological functional derivatives thereof, can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles. Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
Pharmaceutical compositions adapted for transdermal administration may be presented as discrete patches intended to remain in intimate contact with the epidermis of the recipient for a prolonged period of time.
Pharmaceutical compositions adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
For treatments of the eye or other external tissues, for example mouth and skin, the compositions are preferably applied as a topical ointment or cream. When formulated in an ointment, the active ingredient may be employed with either a paraffinic or a water- miscible ointment base. Alternatively, the active ingredient may be formulated in a cream with an oil-in-water cream base or a water-in-oil base.
Pharmaceutical compositions adapted for topical administrations to the eye include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
Pharmaceutical compositions adapted for topical administration in the mouth include lozenges, pastilles and mouth washes.
Pharmaceutical compositions adapted for rectal administration may be presented as suppositories or as enemas.
Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, suspensions drops, gels or dry powders.
For compositions suitable and/or adapted for inhaled administration, it is preferred that the compound or salt of formula (I) is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation. The preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction). Compositions adapted for administration by inhalation include the particle dusts or mists. Suitable compositions wherein the carrier is a liquid for administration as a nasal spray or drops include aqueous or oil solutions/suspensions of the active ingredient which may be
generated by means of various types of metered dose pressurised aerosols, nebulizers or insufflators.
Aerosol formulations, e.g. for inhaled administration, can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or nonaqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
Where the dosage form comprises an aerosol dispenser, it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC). Suitable HFC propellants include
1 ,1 ,1 ,2,3,3,3-heptafluoropropane and 1 ,1 ,1 ,2-tetrafluoroethane. The aerosol dosage forms can also take the form of a pump-atomiser. The pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol. Other excipient modifiers may also be incorporated to improve, for example, the stability and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
For pharmaceutical compositions suitable and/or adapted for inhaled administration, the pharmaceutical composition may be a dry powder inhalable composition. Such a composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of formula (I) or salt or solvate thereof (preferably in particle-size- reduced form, e.g. in micronised form), and optionally a performance modifier such as L- leucine or another amino acid, cellobiose octaacetate and/or metals salts of stearic acid such as magnesium or calcium stearate.
Aerosol formulations are preferably arranged so that each metered dose or "puff" of aerosol contains a particular amount of a compound of the invention. Administration may be once daily or several times daily, for example 2, 3 4 or 8 times, giving for example 1 , 2 or 3 doses each time. The overall daily dose and the metered dose delivered by capsules
and cartridges in an inhaler or insufflator will generally be double those with aerosol formulations.
Pharmaceutical compositions adapted for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations.
Pharmaceutical compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the composition isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. The compositions may be presented in unit- dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
It should be understood that in addition to the ingredients particularly mentioned above, the compositions may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
A therapeutically effective amount of a compound of the present invention will depend upon a number of factors including, for example, the age and weight of the animal, the precise condition requiring treatment and its severity, the nature of the formulation, and the route of administration, and will ultimately be at the discretion of the attendant physician or veterinarian.
The pharmaceutically acceptable compounds or salts of the invention may be administered in a daily dose (for an adult patient) of, for example, an oral or parenteral dose of 0.01 mg to 100 mg/kg per day body weight. This amount may be given in a single dose per day or more usually in a number (such as two, three, four, five or six) of sub- doses per day such that the total daily dose is the same. An effective amount of a salt or solvate, thereof, may be determined as a proportion of the effective amount of the compound of formula (I) per se. Thus, for a 70kg adult mammal, the actual amount per day would usually be from 70 to 700 mg and this amount may be given in a single dose
per day or more usually in a number (such as two, three, four, five or six) of sub-doses per day such that the total daily dose is the same. An effective amount of a salt or solvate, or physiologically functional derivative thereof, may be determined as a proportion of the effective amount of active ingredient.
The compounds of the present invention and their pharmaceutically acceptable salts and solvates thereof, may be employed alone or in combination with other therapeutic agents. Combination therapies according to the present invention thus comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, and the use of at least one other pharmaceutically active agent. Preferably, combination therapies according to the present invention comprise the administration of at least one compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, and at least one other pharmaceutically active agent. The compound(s) of formula (I) and the other pharmaceutically active agent(s) may be administered together in a single pharmaceutical composition or separately and, when administered separately this may occur simultaneously or sequentially in any order. The amounts of the compound(s) of formula (I) and the other pharmaceutically active agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
The compound and pharmaceutical compositions according to the invention may be used in combination with or include one or more other therapeutic agents, for example selected from statins (HMG Co A reductase inhibitors) and/or other lipid lowering drugs for example MTP inhibitors and LDLR upregulators. The compounds of the invention may also be used in combination with antidiabetic agents, e.g. metformin, sulfonylureas and/or PPAR activators (for example thiazolidinediones such as e.g. Pioglitazone and Rosiglitazone). The compounds may also be used in combination with antihypertensive agents such as angiotensin antagonists (eg Telmisartan) calcium channel antagonists and ACE inhibitors. The invention thus provides in a further aspect the use of a combination comprising a compound of formula (I) with a further therapeutic agent in the treatment of a hPPAR mediated disease.
It will be clear to a person skilled in the art that, where appropriate, the other therapeutic ingredient(s) may be used in the form of salts, for example as alkali metal or amine salts or as acid addition salts, or prodrugs, or as esters, for example lower alkyl esters, or as solvates, for example hydrates, to optimise the activity and/or stability and/or physical
characteristics, such as solubility, of the therapeutic ingredient. It will be clear also that, where appropriate, the therapeutic ingredients may be used in optically pure form.
When a compound of formula (I) is used in combination with other therapeutic agents, the compounds may be administered either sequentially or simultaneously by any convenient route.
The combinations referred to above may conveniently be presented for use in the form of a pharmaceutical composition and thus pharmaceutical compositions comprising a combination as defined above optimally together with a pharmaceutically acceptable carrier or excipient comprise a further aspect of the invention. The individual components of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical compositions.
When combined in the same composition it will be appreciated that the two compounds must be stable and compatible with each other and the other components of the composition and may be formulated for administration. When formulated separately they may be provided in any convenient composition, conveniently in such a manner as are known for such compounds in the art.
When the compound of formula (I) is used in combination with a second therapeutic agent active against the same hPPAR mediated disease, the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
The compounds of this invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention are prepared in the Working Examples.
Compounds of general formula (I) may be prepared by methods known in the art of organic synthesis as set forth in the schemes below and/or the specific Examples described below. In all of the methods, it is well understood that protecting groups for sensitive or reactive groups may be employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard
methods of organic synthesis (T. W. Green and P. G. M. Wuts (1999) Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection of processes as well as the reaction conditions and order of their execution shall be consistent with the preparation of compounds of Formula
(I)-
These schemes show preparation of compounds having a defined linker, but could be adapted to embrace other linkers as detailed in examples below.
Scheme 1 :
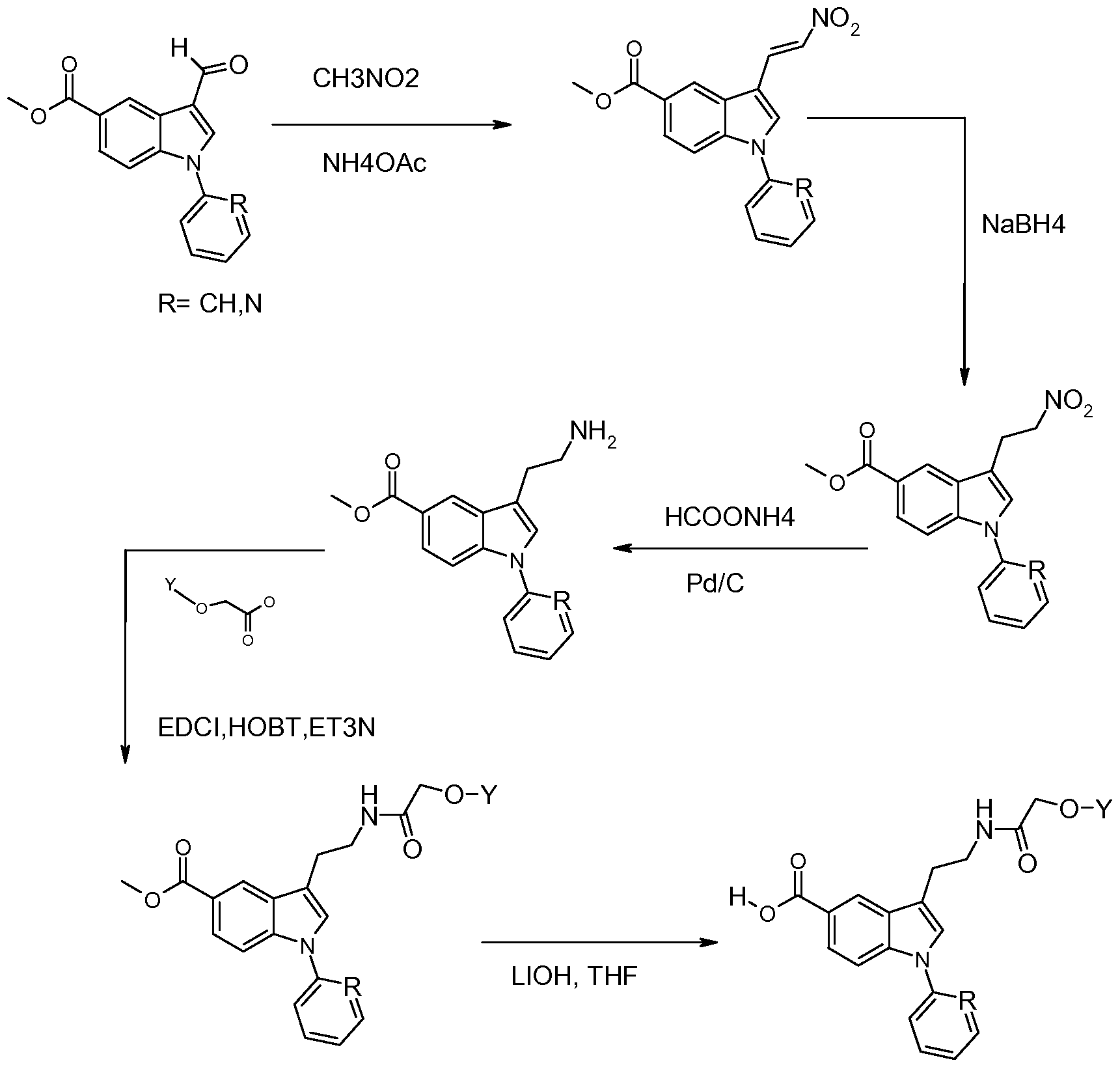

EXAMPLES
The various aspects of the invention will now be described by reference to the following examples. These examples are merely illustrative and are not to be construed as a limitation of the scope of the present invention.
Abbreviations used herein:
NBS Λ/-bromosucunamide DCM dichloromethane
DMF Λ/,Λ/-dimethyl formamide
DIPEA Λ/,Λ/-diisopropylethyl amine (1Pr2NEt)
EtOAc ethyl acetate
EtOH ethanol
HCI hydrogen chloride or hydrochloric acid
MeCN acetonitrile
MeOH methanol
NaHCO3 sodium bicarbonate
NaOH sodium hydroxide
Na2SO4 sodium sulphate
THF tetrahydrofuran
HPLC high pressure liquid chromatography
SPE solid phase extraction
NMR nuclear magnetic resonance (in which: s = singlet, d = doublet, t = triplet, q
= quartet, dd = doublet of doublets, m = multiplet, H = no. of protons)
LCMS liquid chromatography/mass spectroscopy
TLC thin layer chromatography h hours
TEA triethylamine
APTS Para-toluene sulfonic acid
TRET retention time
'880 Ammonia' or '0.880 ammonia' refers to concentrated aqueous ammonia (specific gravity 0.880). 'Hydrophobic Frit'. This refers to a Whatman PTFE filter medium (frit), pore size 5.0μm, housed in a polypropylene tube.
Room temperature this is usually in the range of about 20 to about 25 0C.
General Experimental Details
Intermediates and Examples
When the name of a commercial supplier is given after the name of a compound or a reagent, for instance "compound X (Aldrich)" or "compound X/AIdrich", this means that compound X is obtainable from a commercial supplier, such as the commercial supplier named.
Similarly, when a literature or a patent reference is given after the name of a compound, for instance compound Y (EP 0 123 456), this means that the preparation of the compound is described in the named reference.
Intermediates
Intermediate 1
Methyl 2-(trimethylsilyl)-3-f2-r(trimethylsilyl)oxy1ethyl)-1H-indole-5-carboxylate

Methyl 4-amino-3-iodobenzoate (27.7 g, 0.1 mol), LiCI (4.24 g, 0.1 mol), Cs2CO3 (81.5 g, 0.25 mol), tri(o-tolyl)phosphine (1.52 g, 5 mmol) was added to DMF (500 ml). The mixture was degassed with nitrogen and 4-trimethylsilyl-3-butyn-1-ol (26.67 ml, 0.16 mol) and palladium (II) acetate (1.12 g, 5 mmol) was added, The mixture was degassed with nitrogen and stirred at 100 0C for 3 hours. The reaction mixture was filtered on celite (5 cm) and evaporated. The crude extract was diluted with ethyl acetate, the organic phase was washed with brine and dried over Na2SO4, filtered and concentrated. The residue was purified on SiO2 eluting with dichloromethane/ethyl acetate 95/5 to give a first fraction of the title compound (2.1 g) and a second fraction after evaporation and addition of heptane, precipitation and filtration : (Intermediate 1) (19.3 g, 66 %). LC/MS : m/z 292 (M+H)+, Rt: 2.96
Intermediate 2 Methyl 3-(2-hydroxyethyl)-1H-indole-5-carboxylate FOPI/U1435/115/1

Concentrated HCI (1 1 ml) at 00C was added to a solution of methyl 2-(trimethylsilyl)-3-{2- [(trimethylsilyl)oxy]ethyl}-1H-indole-5-carboxylate (Intermediate 1) (19.13 g, 65.7 mmol) in methanol (250 ml). The reaction mixture was stirred at room temperature for 5 hours. The reaction was not complete, concentrated HCI (5 ml) was added and the mixture was stirred for 1 hour. Dichloromethane was evaporated and the aqueous phase was extracted with ethyl acetate. The organic phase was washed with brine (4 times), dried over Na2SO4 and filtered. The phase was concentrated to 30 ml and after addition of heptane, the precipitate formed was filtered to give, after drying, the title compound (13.35 mg, 92%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.41 (s, 1 H), 7.93 (dd, 1 H, J=1.49 Hz, 8.60 Hz), 7.39 (d, 1 H, J=8.81 Hz), 7.18 (s, 1 H), 3.96 (m, 5H), 3.08 (t, 2H, J=6.25 Hz)
Intermediate 3 1,1-dimethylethyl fr4-(trimethylsilyl)-3-butvn-1 -vπoxy)acetate FOPI/U1435/59/1

1 ,1-dimethylethyl bromoacetate (2.95 ml, 20 mmol) and NaOH dropwise (0.880 g, 22 mmol) at 00C was added to a solution of 4-(trimethylsilyl)-3-butyn-1-ol (3.33 ml, 20 mmol) in anhydrous DMF (70 ml) was added. The reaction mixture was stirred at room temperature overnight. Heptane and water was added. The mixture was extracted and the organic phase was washed with brine, dried over Na2SO4, filtered and evaporated to give a mixture of product as colourless oil (3.8 g, 74%).
NMR1H NMR (300 MHz), CDCI3 δ: 3.90 (m, 2H), 3.44 (m, 2H), 2.38 (m, 2H), 1.34 (s, 9H), 0 (m, 9H).
Intermediate 4
Methyl 3-r2-α2-r(1.1-dimethylethyl)oxy1-2-oxoethyl)oxy)ethvπ-2-(trimethylsilyl)-1H- indole-5-carboxylate FOPI/U1435/60/1

Pd(OAc)2 (106 mg, 0.47 mmol) was added to a solution of methyl 4-amino-3-iodobenzoate (2.58 g, 9.3 mmol), 1 ,1-dimethylethyl {[4-(trimethylsilyl)-3-butyn-1-yl]oxy}acetate (Intermediate 3) (3.8 g, 14.8 mmol), LiCI (394 mg, 9.3 mmol), Cs2CO3 (7.58 g, 23.25 mmol) and tri-o-tolylphosphine (143 mg, 0.47 mmol) in anhydrous DMF. The reaction mixture was stirred at 100 0C for 3 hours. The reaction mixture was filtered and rinse with dichloromethane. The filtrate was washed with brine, dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with dichloromethane to give the title compound (1.65 g, 44%). LC/MS : m/z 406 (M+H)+, Rt: 3.96.
Intermediate 5
Methyl 2-methyl-4-nitrobenzoate
Intermediate 6
Methyl 3-f (1 E)-3-oxo-3-r(phenylmethyl)oxy1-1 -propen-1 -yl)-1 -phenyl-1 H-indole-5- carboxylate

Intermediate 7
Methyl 4-amino-2-methylbenzoate

To a solution of methyl 2-methyl-4-nitrobenzoate (Intermediate 5) (3.25 g, 16.6 mmol) in ethanol was added Pd/C 10% (catalytic quantity) and ammonium formate (10.5 g, 0.17 mmol). The mixture was stirred at 400C for 2 hours. The mixture was filtered on celite, washed with diethyl ether. The filtrate was evaporated and the residue was diluted with diethyl ether, washed with water. The organic phase was dried over Na2SC>4, filtered and evaporated to give the title compound as brown oil (2.8 g, quantitative yield). NMR1H NMR (300 MHz), CDCI3 δ: 7.74 (d, 1 H, J=9.20 Hz), 6.41 (m, 2H), 3.75 (s, 3H), 2.47 (s, 3H).
The following Intermediates were prepared in an analogous manner to methyl 4-amino-2- methylbenzoate (Intermediate 7):


Intermediate 9 methyl 4-amino-5-iodo-2-methylbenzoate

To a solution of methyl 4-amino-2-methylbenzoate (Intermediate 7) (2.9 g, 16.6 mmol) in DMF (15 ml) was added sodium periodate (1.45 g, 6.6 mmol) and iodine (3.4 g, 13.3 mmol). The reaction mixture was stirred at 500C for 2 hours. The mixture was poured into cold water with NaHSO3 (0.5 g). After stirring, the mixture was left overnight. The precipitate was filtered, washed with water and diluted with dichloromethane. This solution was dried over Na2SO4, filtered and evaporated to give the title compound as orange solid (4.1 g, 85%). NMR1H NMR (300 MHz), CDCI3 δ: 8.20 (s, 1 H), 6.47 (s, 1 H), 3.75 (s, 3H), 2.42 (s, 3H).
Intermediate 10
Methyl 4-(acetylamino)-5-iodo-2-methylbenzoate
To a solution of methyl 4-amino-5-iodo-2-methylbenzoate (Intermediate 9) (4.1 g, 14 mmol) in pyridine was added DMAP (catalytic quantity) and acetyl chloride (3 ml, 42 mmol) dropwise. The reaction mixture was stirred at room temperature for 20 hours. The solvent was evaporated and the residue was diluted with water, filtered and washed with water. The obtained solid was purified on SiO2 eluting with cyclohexane/ethyl acetate 70/30, 50/50 and dichloromethane. The obtained product was recrystallised in cyclohexane/ethyl acetate 75/25 to give the title compound as light yellow solid (2 g, 43%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.36 (s, 1 H), 7.28 (s, 1 H), 3.89 (s, 3H), 2.60 (s, 3H), 2.28 (s, 3H).
Intermediate 11
Methyl 4-(acetylamino)-2-methyl-5-r(trimethylsilyl)ethynvnbenzoate

To a solution of methyl 4-(acetylamino)-5-iodo-2-methylbenzoate (Intermediate 10) in dichloromethane (40 ml) was added triethylamine (1.65 ml, 1 1.7 mmol), CuI (15 mg, 0.078 mmol) and Pd(PPh3)2CI2 (30 mg, 0.04 mmol). The reaction mixture was sonicated for divided CuI and ethynyl(trimethyl)silane (1.65 ml, 11.7 mmol) was added drop by drop. The reaction mixture was stirred at room temperature for 18 hours. The mixture was extracted with diethyl ether and HCI 1 N. The organic phase was dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with cyclohexane/ethyl acetate 80/20 to give the title compound as orange solid (2.3 g, 97%). NMR1H NMR (300 MHz), CDCI3 δ: 8.38 (s, 1 H), 8.07 (s, 1 H), 3.90 (s, 3H), 2.66 (s, 3H), 2.26(s, 3H), 0.34 (s, 9H).
Intermediate 12
Methyl 3-r2-α2-rn.1-dimethylethyl)oxyl-2-oxoethyl)oxy)ethyll-1H-indole-5- carboxylate

To a solution of methyl 3-[2-({2-[(1 ,1-dimethylethyl)oxy]-2-oxoethyl}oxy)ethyl]-2- (trimethylsilyl)-1 H-indole-5-carboxylate (Intermediate 4) (1.65 g, 4.07 mmol) in THF was added nBu4NF 1 M in THF (4.5 ml, 4.5 mmol). The reaction mixture was stirred at room temperature for 16 hours. The reaction wasn't complete, nBu4NF (4.9 ml, 4.9 mmol) was added. The reaction mixture was concentrated; ethyl acetate and water were added. The mixture was extracted, the organic phase was washed with brine, dried over Na2SO4,
filtered and concentrated. The residue was purified on SiC>2 eluting with dichloromethane to dichloromethane/ethyl acetate 95/5 to give the title compound (650 mg, 48%). LC/MS : m/z 334 (M+H)+, Rt: 3.24 Min.
The following Intermediate was prepared in an analogous manner to methyl 3-[2-({2-[(1 ,1- dimethylethyl)oxy]-2-oxoethyl}oxy)ethyl]-1 H-indole-5-carboxylate (Intermediate 12)

Intermediate 14
Methyl 3-(2-hydroxyethyl)-1 -phenyl-1 H-indole-5-carboxylate

To a solution of methyl 3-(2-hydroxyethyl)-1 H-indole-5-carboxylate (Intermediate 2) (14.16 g, 64.7 mmol) in toluene was added K3PO4 (28.8 g, 136 mmol) and CuI (616 mg, 3.24 mmol). The mixture was degassed with nitrogen and N,N'-dimethylethylenediamine (2.1 ml, 19.4 mmol) and iodobenzene (8.7 ml, 77.6 mmol) were added. The reaction was stirred at 1000C for 24 hours and was re-added K3PO4 (28.8 g, 136 mmol), CuI (616 mg, 3.24 mmol), N,N'-dimethylethylenediamine (2.1 ml, 19.4 mmol) and iodobenzene (8.7 ml, 77.6 mmol). The reaction was continued at 1100C for 48 hours. The reaction mixture was filtered on celite and the filtrate was concentrated. The residue was purified on SiO2 eluting with dichloromethane/ethyl acetate 96/4 to give the title compound (12.2 g, 63%). LC/MS : m/z 296 (M+H)+, Rt: 3.18
The following Intermediate was prepared in an analogous manner to methyl 3-(2- hydroxyethyl)-1-phenyl-1 H-indole-5-carboxylate (Intermediate 14)

Intermediate 16
Methyl 3-r2-(f2-r(1.1 -dimethylethyl)oxy1-2-oxoethyl)oxy)ethvπ-1 -phenyl-1H-indole-5- carboxylate

To a solution of methyl 3-(2-hydroxyethyl)-1-phenyl-1 H-indole-5-carboxylate (Intermediate 14) (11.2 g, 38 mmol) in dichloromethane (120 ml) was added tert-butyl bromoacetate (16.8 ml, 114 mmol), a concentrated solution of NaOH (120 ml) and tetrabutylammonium hydrogen sulfate (12.9 g, 38 mmol). The reaction mixture was stirred at room temperature for 24 hours. The reaction mixture was decanted and the aqueous phase was extracted with dichloromethane. The organic phases were washed with brine 2 times, dried over Na2SO4 and filtered. After concentration under vacuum the crude
material wass purified on SiC>2 eluting with dichloromethane to give a mix between the expected product and tert-butyl bromoacetate (22.9 g). LC/MS : m/z 410 (M+H)+, Rt: 4.04 min
Intermediate 17 r(2-{5-r(methyloxy)carbonvn-1 -phenyl-1H-indol-3-yl)ethyl)oxy1acetic acid

To a solution of methyl 3-[2-({2-[(1 ,1-dimethylethyl)oxy]-2-oxoethyl}oxy)ethyl]-1-phenyl- 1H-indole-5-carboxylate (Intermediate 16) (22.9 g, 37.9 mmol) in dichloromethane (200 ml) was added TFA (56 ml, 0.76 mol). The reaction mixture was stirred at room temperature for 24 hours. Brine was added and the aqueous phase was extracted 2 times with dichloromethane. The organic phase was washed with brine 2 times and dried over Na2SO4 and filtered. n-Hexane was added to the filtrate and dichloromethane was evaporated. The precipitate was filtered to remove n-hexane After drying, the title compound was obtained as a white solid (11.8 g, 88%). LC/MS : m/z 354 (M+H)+, Rt: 2.61 min.
The following Intermediate was prepared in an analogous manner to [(2-{5- [(methyloxy)carbonyl]-1-phenyl-1 H-indol-3-yl}ethyl)oxy]acetic acid (Intermediate 17 )


Intermediate 19
Methyl 3-(2-[(2-([4-(I -methylethyl)phenyllamino)-2-oxoethyl)oxylethyl)-1 -phenyl-1 H- indole-5-carboxylate

To a solution of [(2-{5-[(methyloxy)carbonyl]-1 -phenyl-1 H-indol-3-yl}ethyl)oxy]acetic acid (Intermediate 17) (11.8 g, 33 mmol) in anhydrous DMF (100 ml) was added, HOBT (5.4 g, 40 mmol), EDCI (7.68 g, 40 mmol), 4-(1-methylethyl)aniline (5.46 ml, 40 mmol) and triethylamine (11 m, 80 mmol). The reaction mixture was stirred at room temperature for 16 hours and was concentrated. The residue was diluted with ethyl acetate. The organic phase was washed with brine, HCI 1 N (2 times) and re-washed with brine, dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with dichloromethane to dichloromethane/ethyl acetate 95/5 to give after recrystallisation in a mixture of heptane/ethyl acetate (9/1 ) the title compound as a white solid (12.05 g, 78%). LC/MS : m/z 471 (M+H)+, Rt: 4.01 min
Example 1
3-{2-r(2-{ F4-(1 -methylethyl)phenvπamino)-2-oxoethyl)oxy1ethyl)-1 -phenyl-1 H-indole- 5-carboxylic acid

To a solution of methyl 3-{2-[(2-{[4-(1-methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1- phenyl-1 H-indole-5-carboxylate (Intermediate 19) (14.7 g, 31.3 mmol) in 150ml of THF was added 150ml of MeOH, and 156ml (156 mmol) of LiOH 1 M, the reaction mixture was
heated at 700C for 1 H20, then 200 ml of HCI 1 N was added immediately, after cooling at
RT, ethyl acetate was added and reaction mixture was washed with brine then dried with
Na2SC>4, and filtered. After concentration under vacuum the crude material obtained was purified on SiO2 and re-crystallized in acetonitrile to give the title compound as white solid
(yield of the re-crystallisation: 80%).
NMR1H NMR (300 MHz), DMSO δ: 8.61 (s, 1 H), 8.05 (m, 2H), 7.50 (m, 6H), 7.35 (s, 1 H),
7.12 (dd, 4H, J=8.3 Hz, 21 Hz), 4.15 (s, 2H), 3.98 (t, 2H, J=6.40 Hz), 3.26 (t, 2H, J=6 Hz),
2.84 (m, 1 H), 1.21 (d, 6H, J=6.36Hz).
TOF MS ES+ exact mass calculated for C28H28N2O4: 457.2127 (M+H)+ Found:
457.2162 (M+H)+ ; RT= 2.74 min.
The following Intermediates were prepared in an analogous manner to 3-{2-[(2-{[4-(1- methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1H-indole-5-carboxylate Intermediate 19


Methyl 3-[2-({2-[(4- [(2-{5- ethylphenyl)amino]-2- [(methyloxy)carbonyl]-1- LC/MS : m/z oxoethyl}oxy)ethyl]- 1 - phenyl-1 H-indol-3- 457 (M-H)+,
 phenyl-1 H-indole- yl}ethyl)oxy]acetic acid Rt: 3.89 min (Intermediate 22) (Intermediate 17)
phenyl-1 H-indole- yl}ethyl)oxy]acetic acid Rt: 3.89 min (Intermediate 22) (Intermediate 17)
Methyl 3-(2-{[2-(2,3- dihydro-1 H-inden-5- [(2-{5- ylamino)-2- [(methyloxy)carbonyl]-1- LC/MS : m/z oxoethyl]oxy}ethyl)-1- phenyl-1 H-indol-3- 469 (M-H)+,
 phenyl-1 H-indole-5- yl}ethyl)oxy]acetic acid Rt: 3.92 min carboxylate (Intermediate 17)
phenyl-1 H-indole-5- yl}ethyl)oxy]acetic acid Rt: 3.92 min carboxylate (Intermediate 17)
(Intermediate 23)
Methyl 3-(2-{[2-(2,3-
[(2-{5- dihydro-1 ,4-benzodioxin-6-
[(methyloxy)carbonyl]-1- LC/MS : m/z ylamino)-2- oxoethyl]oxy}ethyl)-1- } phenyl-1 H-indol-3- 487 (M-H)+, yl}ethyl)oxy]acetic acid Rt: 3.56min phenyl-1 H-indo (Intermediate 17) (Intermediate 24)
Methyl 3-(2-{[2-(2,3-
[(2-{5- dihydro-1 ,4-benzodioxin-6-
[(methyloxy)carbonyl]-1- LC/MS : m/z ylamino)-2- phenyl-1 H-indol-3- 471 (M-H)+, oxoethyl]oxy}ethyl)-1-
 yl}ethyl)oxy]acetic acid Rt: 4.00 phenyl-1 H-indo (Intermediate 17) (Intermediate 25)
yl}ethyl)oxy]acetic acid Rt: 4.00 phenyl-1 H-indo (Intermediate 17) (Intermediate 25)
Methyl 3-[2-({2-[(4- cyclohexylphenyl)amino]- [(2-{5- 2-oxoethyl}oxy)ethyl]-1 - [(methyloxy)carbonyl]-1- LC/MS : m/z phenyl-1 H-indole-5- phenyl-1 H-indol-3- 51 1 (M-H)+, carboxylate
 yl}ethyl)oxy]acetic acid Rt: 4.33 min (Intermediate 17)
yl}ethyl)oxy]acetic acid Rt: 4.33 min (Intermediate 17)
(Intermediate 26)



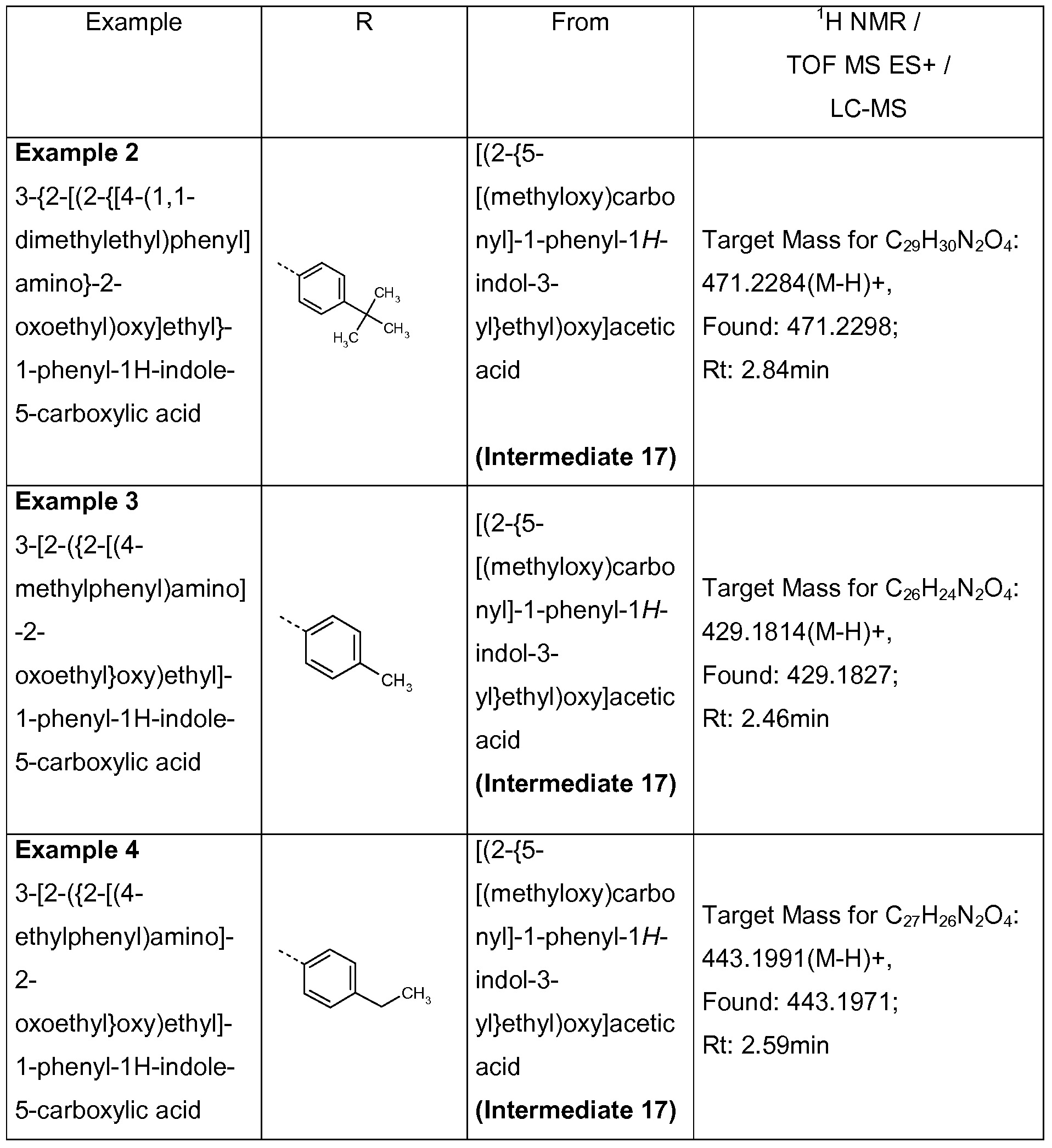

(dιmethylamιno)ph 2H, J=6.84 Hz), 2.84 (s, 6H). enyl]amιno}-2- oxoethyl)oxy]ethyl}
-1-phenyl-1 H- ιndole-5- carboxylate
(Intermediate 29)
The following examples were prepared in an analogous manner to 3-{2-[(2-{[4-(1- methylethyl)phenyl]amιno}-2-oxoethyl)oxy]ethyl}-1-phenyl-1H-ιndole-5-carboxylιc acid (Example 1 ) :


Intermediate 30
Ethyl 1H-indole-5-carboxylate

Intermediate 31
Methyl 3-r(E)-2-(dimethylamino)ethenyll-4-nitrobenzoate

To a solution of methyl 3-methyl-4-nitrobenzoate (25 g, 128 mmol) in DMF (100 ml) was added Λ/,Λ/-dimethyl-1 ,1-bis(methyloxy)methanamine (75 ml, in excess). The reaction mixture was stirred at reflux for 48 hours and concentrated. The red dark solid was diluted with toluene (150 ml), heptane (1.5 I) was added and the product was precipitated. This precipitate was filtered and washed with heptane to give the title compound as purple solid (20.5 g, 64%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.14 (dd, 1 H, J=1.80 Hz), 7.83 (d, 1 H, J=8.56 Hz), 7.08 (d, 1 H, J=13.50 Hz), 5.78 (d, 1 H, J=13.33Hz), 3.95 (s, 3H), 2.95 (s, 6H).
Intermediate 32
Methyl 4-nitro-3-(2-oxopropyl)benzoate

To solution of methyl 3-[(£)-2-(dimethylamino)ethenyl]-4-nitrobenzoate (Intermediate 31) 20.4 g, 81.6 mmol) in anhydrous dichloromethane (200 ml) was added pyridine (10.86 ml, 135 mmol) and acetyl chloride (8.75 ml, 122 mmol) in anhydrous dichloromethane (50 ml) drop by drop. The reaction mixture was stirred at room temperature for 72 hours. Water (250 ml) was added. The mixture was extracted and the organic phase was concentrated. The brown residue was diluted with dioxane (250 ml) and water (70 ml) and this mixture was heated at reflux for 24 hours. The reaction mixture was concentrated and the residue was extracted with ethyl acetate and washed with brine. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified on SiO2 eluting with hexane/ethyl acetate 2/1 to give the title compound as orange solid (12.3 g, 63%). NMR1H NMR (300 MHz), CDCI3 δ: 8.24 (m, 2H), 8.07 (s, 1 H), 4.30 (s, 2H), 4.08 (s, 3H), 2.45 (s, 3H).
Intermediate 33
Methyl 1 -phenyl-1 H-indole-5-carboxylate

To solution of methyl 1 /-/-indole- 5-carboxylate (15 g, 85.6 mmol) in DMF (410 ml) under argon was added K2CO3 (35.5 g, 257 mmol), CuI (1.63 g, 8.56 mmol) and diphenyliodonium chloride (27.10 g, 85.6 mmol). The reaction mixture was stirred at reflux for 3 hours. CuI (1.63 g, 8.56 mmol) and diphenyliodonium chloride (5.42 g, 17 mmol) was added and the reaction mixture was stirred at reflux for 3 hours. The mixture was cooled at room temperature and diluted with ethyl acetate, washed with a saturated aqueous solution of NH4CI until the aqueous phase became colourless. The aqueous phase was
re-extracted with ethyl acetate and the organic phase was washed with brine, dried over MgSO4 and filtered. After concentration under vacuum the crude material obtained was purified on SiC>2 eluting with cyclohexane/ethyl acetate 95/5 to 70/30 to give the title compound as yellow oil (31.4 g, %).
NMR1H NMR (300 MHz), CDCI3 δ: 8.45 (s, 1 H), 7.9 (dd, 1 H), 7.52 (m, 5H), 7.4 (m, 2H), 6.78 (d, 1 H), 3.95 (s, 3H).
The followings compounds were similarly prepared by analogous method to that described before :

Intermediate 36
Methyl 1 -(4-fluorophenyl)-1 H-indole-5-carboxylate

To a solution of methyl 1 H-indole-5-carboxylate (5.25 g, 30 mmol) in toluene (50 ml) was added K2CO3 (8 g, 42 mmol), 1-fluoro-4-iodobenzene (8.65 ml, 75 mmol), CuBr (260 mg,
1.8 mmol) and 1 ,2-ethanediamine (500 μl, 7.5 mmol). The mixture was stirred at 1200C for 6 hours and the same quantity of reagent was added (except for methyl 1 H-indole-5- carboxylate). The mixture was stirred at 1200C for 16 hours. After cooling, water was added, the mixture was extracted with ethyl acetate. The organic phase was washed with a saturated solution of NH4CI and brine, dried over Na2SO4, filtered and concentrated. The residue was purified on SiO2 eluting with cyclohexane/ethyl acetate 9/1 to give the title compound as white solid (5.88 g, 73%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.38 (d, 1 H, J=1.49 Hz), 7.86 (dd, 1 H, J=1.52 Hz, 8.63 Hz), 7.39 (m, 3H), 7.27 (d, 1 H, J=3.31 Hz), 7.17 (m, 3H), 6.70 (d, 1 H, J=3.31 Hz), 3.88 (s, 3H).
Intermediate 37
Methyl 1 -(2-pyridinyl)-1H-indole-5-carboxylate

To a solution of methyl 1 H-indole-5-carboxylate (1.75 g, 10 mmol) in DMF (10 ml) was added slowly NaH (0.4 g, 10 mmol). 2-fluoropyridine (2.92 ml, 34 mmol) was added and the reaction mixture was stirred at 100 °C for 18 hours. The mixture was not complete but was hydrolysed with concentrated HCI and extracted with a mix of ethyl acetate/water. The mixture was basified with NaOH 1 N, the organic phase was dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with dichloromethane to give a mix of the expected product and methyl 1 H-indole-5-carboxylate. This mix was diluted with DMF and, was added NaH (15 mmol) and 2-fluoropyridine (20 mmol). The reaction was stirred at reflux for 18 hours. The treatment was the same as before to give the title compound as colourless oil (1.2 g, 24%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.43 (bd, 1 H, J=4.86 Hz), 8.24 (sd, 1 H, J=1.53 Hz), 8.06 (d, 1 H, 8.75 Hz), 7.83 (dd, 1 H, J=1.67 Hz, 8.75 Hz), 7.70 (td, 1 H, J=1.86 Hz, 8.08Hz), 7.59 (d, 1 H, J=3.52 Hz), 7.33 (d, 1 H, J=8.29 Hz), 7.07 (m, 1 H), 6.63 (d, 1 H, J=3.52 Hz), 3.78 (s, 3H).
Intermediate 38
Methyl 3-formyl-1 -phenyl-1 H-indole-5-carboxylate

POCI3 (1 1.6 ml, 125 mmol) was added drop by drop to DMF (22.7 ml, 294 mmol) at 00C under argon. The mixture was stirred at 00C for 30 min, and then methyl 1 -phenyl-1 H- indole-5-carboxylate (Intermediate 33) (31.4 g, 125 mmol) in DMF (170 ml) was added, drop by drop at 00C. The reaction mixture was stirred at room temperature overnight. The reaction mixture was poured into a mix of water/ice, KOH (powder) diluted with a little of water was added to reach basic pH. The mixture was stirred at room temperature for 30 minutes. The white precipitate was filtrate and washed with water. The precipitate was dissolved in ethyl acetate, the solution was washed with water and the organic phase was dried over MgSO4, filtered and evaporated to dryness to give the title compound as a white solid (32.5 g, 93%).
NMR1H NMR (300 MHz), CDCI3 δ: 10.16 (s, 1 H), 9.09 (s, 1 H), 8.05 (d, 1 H, J=8.80 Hz), 8.00 (s, 1 H), 7.52 (m, 6H), 3.97 (s, 3H).
The following compounds were similarly prepared by analogous method to that described before :

Intermediate 43
5-r(methyloxy)carbonyl1-1 -phenyl-1 H-indole-3-carboxylic acid

To a solution of methyl 3-formyl-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 38) (1.6 g, 5.73 mmol) in a mix of THF/tBuOH (100 ml / 30 ml) was added 2-methyl-2-butene (3.3 ml, 6.6 mmol) and a mix of NaCIO2 (1.55 g, 17.2 mmol) / NaH2PO4 (3.1 g, 25.8 mmol) in water. The reaction mixture was stirred at room temperature for 24 hours. The solvents were evaporated and the precipitate was filtered with H2O, isopropyl ether and pentane to give the title compound as white solid (1.69 g, 100%). LC/MS : m/z 296 (M+H)+, Rt: 2.34
Intermediate 44
Methyl 3-r(-Ξ)-2-nitroethenyll-1 -phenyl-1 H-indole-5-carboxylate
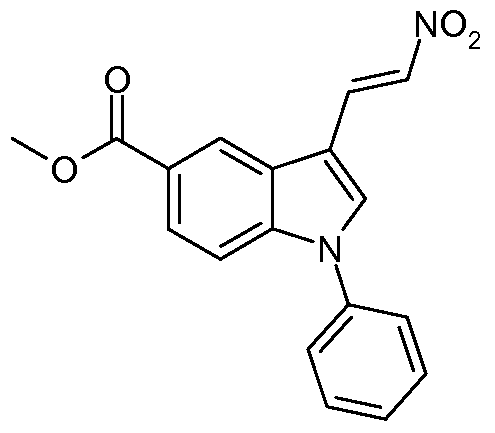
To a solution of methyl 3-formyl-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 38) (4 g, 14 mmol) in nitromethane (50 ml, 12 vol) was added ammonium acetate (1.3 g, 16.8 mmol). The mixture was stirred at 120°c during 3 hours. The mixture was cooled to room temperature and ethanol (5 ml) and water (40 ml) were added. The precipitate was filtered and washed with ethanol and pentane and dry to give the title compound as yellow powder (3.917 g, 85%). LC/MS : m/z 323 (M+H)+, Rt: 3.52
Intermediate 45
Methyl 3-(2-nitroethyl)-1 -phenyl-1 H-indole-5-carboxylate

To a solution of methyl 3-[(£)-2-nitroethenyl]-1-phenyl-1H-indole-5-carboxylate (Intermediate 44). (917 g, 12 mmol) in THF/methanol (100 ml/ 40 ml) was added at 00C sodium tetraborohydride (1.38 g, 36 mmol). The mixture was stirred at room temperature overnight. A saturated aqueous solution of NH4CI was added, the mixture was extracted with ethyl acetate (2 times). The organic phase was washed with brine and dried over Na2SO4, filtered and evaporated to give the title compound (4 g, quantitative yield). LC/MS : m/z 325 (M+H)+, Rt: 3.52
Intermediate 46
Methyl 3-(2-aminoethyl)-1 -phenyl-1 H-indole-5-carboxylate

To a solution of methyl 3-(2-n itroethy I )- 1 -phenyl-1 H-indole-5-carboxylate (Intermediate 45) (4 g, 12 mmol) in ethanol/THF (150 ml/150 ml) was added Pd/C 10% (600 mg, 15%) and ammonium formiate (4.6 g, 72 mmol). The reaction mixture was stirred at 75°C under argon for 4 hours. Ammonium formiate (5 g) was added, the reaction mixture was stirred at 75°C under argon for 1 hour and one night at 45°C. Ammonium formiate (4.6 g) and Pd/C 10% (300 mg) were added, the reaction mixture was stirred at 75°C under argon for 3 hours. The reaction was blocked at 64% of expected product. The reaction mixture was filtered and the filtrate was stirred with ammonium formiate (9 g) and Pd/C 10% (600 mg) over the week-end. The reaction mixture was filtered and evaporated. The residue was washed with a saturated aqueous solution of NaHCO3, extracted with dichloromethane, the organic phase was washed with brine, dried over Na2SO4, filtered and evaporated to give the title compound as a yellow gum (3.4 g, 97%). LC/MS : m/z 295 (M+H)+, Rt: 2.70
Intermediate 47
Methyl 3-f2-({f(1,1 -dimethylethyl)oxy1carbonyl)amino)ethvn-1-phenyl-1H-indole-5- carboxylate

To a solution of methyl 3-(2-aminoethyl)-1-phenyl-1 H-indole-5-carboxylate (Intermediate 46) (1.02 g, 3.46 mmol) in dichloromethane (20 ml) at 00C was added Di-te/t-butyl dicarbonate (1.13 g, 5.2 mmol) and triethylamine (960 μl, 6.9 mmol). The reaction mixture was stirred at 00C. When reaction was finished, water (25 ml) was added and the reaction mixture was washed with a solution of sodium hydrogenocarbonate and then washed with brine. The organic phase was dried over Na2SO4 and filtered. After concentration under vacuum, the crude material obtained was purified on SiO2 eluting with methanol/ethyl acetate 9/1 to give the title compound as orange oil (1.6 g, quantitative yield). LC/MS : m/z 395 (M+H)+, Rt: 3.64 min.
Intermediate 48
Methyl 3-(2-aminoethyl)-1 -phenyl-1 H-indole-5-carboxylate hydrochloride

To a solution of methyl 3-[2-({[(1 ,1-dimethylethyl)oxy]carbonyl}amino)ethyl]-1 -phenyl-1 H- indole-5-carboxylate (Intermediate 47) (1.368 g, 3.47 mmol) in ethanol (10 ml) was added concentrated solution of HCI (16 ml). The mixture was stirred at room temperature for 2 days and evaporated to give the title compound (1.6 g, quantitative yield). LC/MS : m/z 295 (M+H)+, Rt: 2.56 min.
Intermediate 49
Methyl 3-{2-r({f4-(1 -methylethyl)phenvπoxy)acetyl)amino1ethyl)-1 -phenyl-1 H-indole- 5-carboxylate

To a solution of methyl 3-(2-aminoethyl)-1 -phenyl-1 H-indole-5-carboxylate hydrochloride (Intermediate 48) (200 mg, 0.6 mmol) in 20 ml of DMF was added, HOBT (107 mg, 0.79 mmol), EDCI (152 mg, 0.79 mmol), {[4-(1-methylethyl)phenyl]oxy}acetic acid (153.4 mg, 0.79 mmol), and triethylamine (335 μl, 2.42 mmol), then the reaction mixture was stirred 16H at room temperature. Water (200 ml) and ethyl acetate (30 ml) were added. The aqueous phase was extracted with ethyl acetate and the organic phase was dried over Na2SO4, filtered and concentrated to give the title compound (305 mg, quantitative yield). LC/MS : m/z 471 (M+H)+, Rt: 4.09
Intermediate 50 4-(1-methylethyl)benzenecarbothioamide

HCI gas was bubbled in a solution of
4-isopropyl benzonitrile (10 g, 68.8 mmol) in DMF (50 ml) during 20 min. Then, thioacetamide (10.3 g, 137 mmol) was added and the reaction mixture was stirred at
1000C for 23 hours. The reaction mixture was poured into water (150 ml) and diethyl ether
(150 ml). The mixture was extracted with diethyl ether (2x150 ml). The organic phase was washed with water (2x150 ml) and dried over Na2SO4, filtered and evaporated. The residue was triturated in 2,2,4-trimethylpentane, filtered and dry to give the title compound
(10.31 g, 84%).
NMR1H NMR (300 MHz), CDCI3 δ: 7.75 (d, 2H, J=8.23 Hz), 7.18 (d, 2H, J=7.99 Hz), 2.88
(m, 1 H), 1.18 (d, 6H, J=6.78 Hz).
Intermediate 51
4-methyl-2-r4-(1 -methylethvDphenyli-i .S-thiazole-δ-carboxylic acid
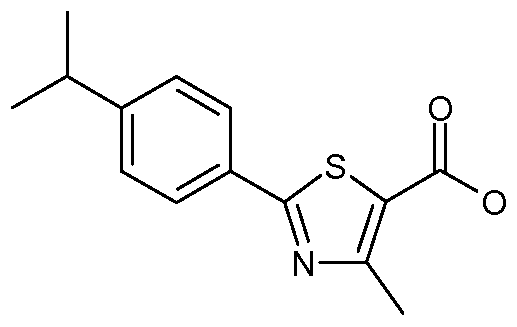
A solution of 4-(1-methylethyl)benzenecarbothioamide (Intermediate 50) (10 g, 56 mmol) and ethyl 2-chloro-3-oxobutanoate (7.76 ml, 56 mmol) in ethanol (50 ml) was stirred at reflux for 5 hours. The solvent was evaporated, the residue was dissolved in ethanol (80 ml); NaOH (4.49 g, 112 mmol) in water (80 ml) was added at room temperature. The mixture was heated at reflux during 2 hours. The solvent was evaporated, and the residue was extracted with ethyl acetate (2x100 ml), acidified with HCI 1 N (90 ml), the precipitate was filtered and washed with water. The solid was dissolved in dichloromethane, the solution was dried over Na2SO4, filtered and evaporated. The solid was re-crystallized in toluene to give the title compound as yellow powder (7.67 g, 52%)).
NMR1H NMR (300 MHz), CDCI3 δ: 8.16 (d, 2H, J=8.2 7Hz), 7.57 (d, 2H, J=8.2 7Hz), 3.21 (m, 1 H), 3.06 (s, 3H), 1.53 (d, 6H, J=6.89 Hz).
Example 12 3-{2-r({f4-(1 -methylethyl)phenvπoxy)acetyl)amino1ethyl)-1 -phenyl-1 H-indole-5- carboxylic acid

To a solution of 4-(1-methylethyl)benzenecarbothioamide (Intermediate 49) (282 mg, 0.6 mmol) in 2.4 ml of THF was added 1.2 ml of ethanol, and 1.2 ml (1.2 mmol) of LiOH 1 M. The reaction mixture was heated at 700C for 1 hour and 20 minutes, then 1.2 ml of HCI 1 N was added and the mixture was extracted with ethyl acetate. The organic phase was dried with Na2SO4 and filtered. After concentration under vacuum the crude material obtained was purified on SiO2 eluting with dichloromethane/methanol 97/3. The solid obtained was recrystallized with ethanol to give the title compound as white powder (80 mg, 30%).
PB62659C
NMR1H NMR (300 MHz), CDCI3 δ: 8.51 (s, 1 H), 8 (dd, 1 H, J=1.51 Hz, 8.91 Hz), 7.52 (m, 6H), 7.20 (s, 1 H), 7.09 (d, 2H, J=8.16 Hz), 6.82 (m, 1 H), 6.74 (d, 2H, J=8.67 Hz), 4.51 (s, 2H), 3.78 (m, 2H), 3.13 (t, 2H, J=5.95 Hz), 2.84 (m, 1 H), 1.21 (d, 6H, J=6.84 Hz). TOF MS ES+ exact mass calculated for C28H28N2O4: 457.2127 (M+H)+ Found: 457.2144 (M+H)+ ; RT= 2.68 min.
The following Intermediates were prepared in an analogous manner to Methyl 3-{2-[({[4- (1-methylethyl)phenyl]oxy}acetyl)amino]ethyl}-1-phenyl-1 /-/-indole-5-carboxylate intermediate 52:

Intermediate R From 1H NMR ( 300 MHz)/ LC- MS
Methyl 3-[2-({[(3,4-
Methyl 3-(2- dichlorophenyl)oxy]acetyl} aminoethyl)-1- amino)ethyl]-1-phenyl-1 H- LC/MS : m/z 497 (M-H)+, phenyl-1 H-indole- indole-5-carboxylate Rt: 3.98
 5-carboxylate (Intermediate 52) (Intermediate 46)
5-carboxylate (Intermediate 52) (Intermediate 46)
Methyl 3-(2-
Methyl 3-{2-[({4-methyl-2- aminoethyl)-1- [4-(1 -methylethyl)phenyl]- phenyl-1 /-/-indole- 1 ,3-thiazol-5- 5-carboxylate yl}carbonyl)amino]ethyl}-1 - (Intermediate 46) LC/MS : m/z 538 (M-H)+, phenyl-1 H-indole-5- and Rt: 4.33 carboxylate
 4-methyl-2-[4-(1 - (Intermediate 53) methylethyl)phenyl
4-methyl-2-[4-(1 - (Intermediate 53) methylethyl)phenyl
]-1 ,3-thiazole-5- carboxylic acid
PB62659C

The following examples were prepared in an analogous manner to 3-{2-[({[4-(1- methylethyl)phenyl]oxy}acetyl)amino]ethyl}-1 -phenyl-1 /-/-indole- 5-carboxylic acid Example 12 :




Intermediate 59
Methyl 3-r(-=)-2-nitroethenyll-1 -(2-pyridinyl)-1 W-indole-5-carboxylate
PB62659C

To a suspension of methyl 3-formyl-1 -(2-pyridinyl)-1 H-indole-5-carboxylate (Intermediate 40) (5.6 g, 20 mmol) in nitromethane (75 ml) was added ammonium acetate (1.85 g, 24 mmol). The mixture was stirred at 1200C for 2 hours. The mixture was cooled at room temperature, water (45 ml) and ethanol (10 ml) was added. The yellow precipitate was filtered and washed with heptane and ethanol to give the title compound (6.12 g, 94%). LC/MS : m/z 324 (M+H)+, Rt: 3.31 Min.
The following compound was similarly prepared by analogous method to that described before :

Intermediate 64 Methyl 3-(2-nitroethyl)-1 -(2-pyridinyl)-1 fY-indole-5-carboxylate

To a solution of methyl 3-[(E)-2-nitroethenyl]-1-(2-pyridinyl)-1 H-indole-5-carboxylate (Intermediate 59) in THF (200 ml) and methanol (45 ml) was added NaBH4 (1.13 g, 30
PB62659C
mmol). An important gas escape was observed and the reaction mixture was stirred at room temperature overnight. The solvents were evaporated and the residue was diluted with ethyl acetate and washed with a saturated aqueous solution of NH4CI. HCI 1 N was added and the organic phase was extracted, washed with brine, dried over Na2SO4, filtered and concentrated. The obtained product was recrystallised in a mix of heptane/ethyl acetate (9/1 ) to give the title compound as a yellow solid (1.2 g, 37%). LC/MS : m/z 326 (M+H)+, Rt: 3.22 Min.
The following compound was similarly prepared by analogous method to that described before :

Intermediate 63
Methyl 3-(2-aminoethyl)-1 -phenyl-1H-indole-5-carboxylate

To a solution of methyl 3-(2-nitroethyl)-1-phenyl-1 H-indole-5-carboxylate (Intermediate 62) (5.95 g, 17 mmol) in methanol was added, under nitrogen, Pd/C (680 mg) and NaBH4 (1.61 g, 42.5 mmol). The mixture was stirred at room temperature for 24 hours. The reaction wasn't finished, NaBH4 (1.61 g, 42.5 mmol) was added and the mixture was stirred 24 hours. The reaction was not complete so over one week, NaBH4 (1.61 g, 42.5 mmol) was re-added everyday. The reaction mixture was filtered on celite. The filtrate was diluted with ethyl acetate and washed with brine (3 times). The organic phase was dried over Na∑SCv, filtered and concentrated. The residue was purified on SiC>2 eluting with dichloromethane/methanol 8/2 to give the title compound (840 mg, 17%).
LC/MS : m/z 295 (M+H)+, Rt: 2.58 Min.
Intermediate 64
Methyl 1 -phenyl -3-{r4-(phenylmethyl)-1 -piperazinyllmethyll-1 H-indole-5-carboxylate

Methyl 3-formyl-1-phenyl-1 H-indole-5-carboxylate (Intermediate 38) (586 mg, 2.1 mmol), 1-(phenylmethyl)piperazine (470 μl, 2.62 mmol) and acetic acid (130 μl, 2.1 mmol) were dissolved in methanol. The mixture was stirred at room temperature for one hour. NaBH3CN was added and the mixture was stirred at room temperature for 18 hours. The solvent was evaporated and the residue was diluted with HCI 1 N and neutralised with NaOH 1 N. The mixture was extracted with diethyl ether. The organic phase was evaporated and the residue was purified on SiC>2 eluting with dichloromethane/cyclohexane (50/50) and dichloromethane 100% and dichloromethane /methanol 99.5/05 to 99/1 to give the title compound as oil (650 mg, 70%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.35 (sd, 1 H, J=1.12 Hz), 7.74 (dd, 1 H, J=1.47 Hz, 8.67 Hz), 7.32 (m, 5H), 7.15 (m, 7H), 3.77 (s, 3H), 3.63 (s, 2H), 3.33 (s, 2H), 2.39 (m, 8H).
Intermediate 65
Methyl 1 -phenyl -3-(1 -piperazinylmethylH H-indole-5-carboxylate

A solution of Methyl 1-phenyl-3-{[4-(phenylmethyl)-1-piperazinyl]methyl}-1 H-indole-5- carboxylate (Intermediate 64) 650 mg, 1.5 mmol) in ethanol in presence of Pd/C 10% was hydrogenated under 3 bars at 600C for 6 hours. The reaction mixture was filtered and the solvent was evaporated to give the title compound (500 mg, 98%). NMR1H NMR (300 MHz), CDCI3 δ: 8.44 (s, 1 H), 7.83 (dd, 1 H, J=1.41 Hz, 8.74 Hz), 7.42 (m, 5H), 7.25 (m, 2H), 3.86 (s, 3H), 3.70 (s, 2H), 2.88 (m, 4H), 2.50 (m, 4H).
Intermediate 66
Methyl 3-(2-aminoethyl)-1 -(2-pyridinyl)-1 H-indole-5-carboxylate

Intermediate 67
Methyl 2-(phenylmethyl)-1,2,3.4-tetrahvdro-6-isoquinolinecarboxylate

To a solution of methyl 1 ,2,3,4-tetrahydro-6-isoquinolinecarboxylate hydrochloride (230 mg, 1.01 mmol) in ethanol (10 ml) was added triethylamine (380 μl, 2.02 mmol) and (bromomethyl)benzene (120 μl, 1.01 mmol). The reaction mixture was stirred at room temperature overnight. The mixture was concentrated and the residue was diluted with a saturated solution of NaHCO3 and dichloromethane. The organic phase was extracted, dried over Na2SO4, filtered and concentrated to give the title compound as yellow oil (260 mg, 86%). LC/MS : m/z 282 (M+H)+, Rt: 3.47.
The followings compounds were similarly prepared by analogous method to that described before :

Intermediate 71
2-r2-(phenylmethyl)-1,2,3,4-tetrahydro-6-isoquinolinvn-2-propanol

To a solution of methyl 2-(phenylmethyl)-1 ,2,3,4-tetrahydro-6-isoquinolinecarboxylate (Intermediate 67) (260 mg, 0.93 mmol) in anhydrous THF (10 ml) was added at 00C, MeMgBr (1.65 ml, 2.32 mmol). The reaction mixture was stirred at room temperature overnight. The reaction was not complete, MeMgBr (1.4 mmol) was added and the reaction was stirred at room temperature for 24 hours. The reaction was stopped (there are still 7% of reagent) and quenched with a saturated solution of NH4CI. The organic phase was extracted with ethyl acetate, dried over Na2SC>4, filtered and evaporated to dryness to give the title compound as yellow oil (170 mg, 65%). LC/MS : m/z 282 (M+H)+, Rt: 2.99.
Intermediate 72 6-(1-methylethyl)-2-(phenylmethyl)-1 ,2,3,4-tetrahvdroisoquinoline

To a solution of 2-[2-(phenylmethyl)-1 ,2,3,4-tetrahydro-6-isoquinolinyl]-2-propanol (Intermediate 71) (170 mg, 0.6 mmol) in dichloromethane (10 ml) was added Et3SiH (1 10 μl, 0.66 mmol), and TFA (450 μl, 6 mmol). The reaction mixture was stirred at room temperature overnight and at reflux for 6 hours. A saturated solution of NaHCO3 was added and the organic phase was extracted, dried over Na2SO4, filtered and concentrated. The residue was purified on SiO2 eluting with dichloromethane to give the title compound as yellow oil (80 mg, 50%). LC/MS : m/z 266 (M+H)+, Rt: 3.96.
Intermediate 73
6-(1 -methylethvD-1 ,2,3,4-tetrahvdroisoquinoline

To a solution of 6-(1-methylethyl)-2-(phenylmethyl)-1 ,2,3,4-tetrahydroisoquinoline
(Intermediate 72) (80 mg, 0.3 mmol) in methanol (10 ml) was added Pd(OH)2 (8 mg, 30%w/w), HCOONH4 (190 mg, 3 mmol). The reaction mixture was stirred at reflux for 3
hours. The mixture was filtered after cooling and the filtrate was concentrated. The residue was triturating with dichloromethane and NaOH 1 N. The organic phase was extracted, dried over Na2SC>4, filtered and concentrated to dryness to give the title compound as brown oil (45 mg, 85%). NMR1H NMR (300 MHz), CDCI3 δ: 6.9 (m, 3H), 3.9 (bs, 2H), 3.05 (bs, 2H), 2.75 (m, 3H), 1.1 (d, 6H).
Intermediate 74
Methyl 3-((1 E)-5-( F4-(1 -methylethyl)phenyllamino)-5-oxo-1 -penten-1 -yl)-1 -phenyl-1 H- indole-5-carboxylate

To a solution of (4-{[4-(1-methylethyl)phenyl]amino}-4-oxobutyl)(triphenyl)phosphonium bromide (Intermediate 140) (1.02 g, 1.9 mmol) in anhydrous THF (20 ml) was added under nitrogen at -400C, BuLi (3.72 mmol) drop by drop. The reaction mixture was stirred at room temperature for 2 hours. Methyl 3-formyl-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 38) (400 mg, 1.43 mmol) was added at -400C, the reaction was stirred at room temperature for 5 hours. Brine and ethyl acetate was added and the organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified on SiO2 eluting with dichloromethane to dichloromethane /methanol 99/1 to give the title compound as a white solid (150 mg, 22%). LC/MS : m/z 467 (M+H)+, Rt: 3.95
Intermediate 75
Methyl 3-(5-U4-(1-methylethyl)phenyl1amino)-5-oxopentyl)-1-phenyl-1H-indole-5- carboxylate

The following compound was similarly prepared by analogous method to that described before :

Intermediate 77
Methyl 1 -phenyl -3-f (1 E)-4-r(phenylmethyl)oxy1-1 -buten-1 -yl)-1 H-indole-5-carboxylate

To a solution of Methyl 3-formyl-1 -phenyl-1 /-/-indole-5-carboxylate (Intermediate 38) (1.4 g, 5.02 mmol) in THF (60 ml) was added triphenyl{3- [(phenylmethyl)oxy]propyl}phosphonium bromide (3.45 g, 7.03 mmol) and tBuONa (675 mg, 7.03 mmol). The reaction mixture was stirred at room temperature for 2 hours. The reaction was quenched with methanol and brine and ethyl acetate were added. The organic phase was extracted, dried over Na2SO4, filtered and concentrated. The residue was purified on SiO2 eluting with dichloromethane/cyclohexane 5/5 to 8/2 to give the title compound as yellow oil (1.65 g, 80%). LC/MS : m/z 412 (M+H)+, Rt: 4.27.
The followings compounds were similarly prepared by analogous method to that described before :


Intermediate 81
Methyl 3-bromo-1 H-indole-5-carboxylate

To a solution of methyl -1 /-/-indole- 5-carboxylate (20 g, 0.1 14 mmol) in DMF (200 ml) was added at 00C, NBS in solution of DMF (100 ml), drop by drop. The reaction mixture was stirred for 4 hours. DMF was evaporated and the residue was diluted with ethyl acetate. This solution was washed with a saturated solution of NaHCO3 (2 times) and brine (2 times). The organic phase was dried over Na2SO4 and filtered. After 4 days, a precipitate was formed and filtered to give the title compound with 7% of reagent (30.86 g, quantitative yield). NMR1H NMR (300 MHz), DMSO δ: 8.13 (sd, 1 H, J=1.12 Hz), 7.84 (dd, 1 H, J=1.61 Hz, 8.47 Hz), 7.74 (s, 1 H), 7.56 (d, 1 H, J=8.69 Hz), 3.90 (s, 3H).
Intermediate 82
Methyl 3-(4-hydroxybutyl)-1 -phenyl-i/y-indole-5-carboxylate

To a solution of methyl 1-phenyl-3-{(1 E)-4-[(phenylmethyl)oxy]-1-buten-1-yl}-1 H-indole-5- carboxylate (Intermediate 77) (1.65 g, 4 mmol) in a mix of THF/methanol (50 ml/50 ml) was added Pd/C (165 mg). The reaction mixture was hydrogenated under 4 bars at 600C during 4 hours. Pd/C was filtered and the filtrate was concentrated to give the title compound as a yellow oil (800 mg, 62%). LC/MS : m/z 324 (M+H)+, Rt: 3.33.
The followings compounds were similarly prepared by analogous method to that described before :


Intermediate 86
Methyl 3-(4-aminobutyl)-1 -phenyl-1 H-indole-5-carboxylate

To a solution of methyl 3-[4-(1 ,3-dioxo-1 , 3-dihydro-2H-isoindol-2-yl)butyl]-1 -phenyl-1 H- indole-5-carboxylate (Intermediate 83) (2 g, 4.4 mmol) in THF (30 ml) was added ethanol (100 ml) and hydrazine hydrate (1.28 ml, 26.4 mmol). The mixture was stirred at 7O0C for
16 hours. The solvents were evaporated and the residue was diluted with dichloromethane (40 ml). The solid was filtered and the filtrate was evaporated to give the title compound as orange oil (1.55 g, quantitative yield).
NMR1H NMR (300 MHz), CDCI3 δ: 8.32 (sd, 1 H, J=1.49 Hz), 7.83 (dd, 1 H, 1.71 Hz, 8.83 Hz), 7.44 (m, 5H), 7.29 (m, 1 H), 7.12 (s, 1 H), 3.88 (s, 3H), 2.78 (t, 2H, J=7.54 Hz), 2.70 (t,
2H, J=6.93 Hz), 1.72 (m, 2H), 1 .53 (m, 2H).
The followings compounds were similarly prepared by analogous method to that described before :
 PB62659C
PB62659C
H, 7.36 H),
Hz), H,

Intermediate 89
Methyl 3-r4-(ir4-(1-methylethyl)phenyllcarbonyl}amino)butyll-1 -(2-pyridinyl)-1H- indole-5-carboxylate

To a solution of 4-(1-methylethyl)benzoic acid (76 mg, 0.46 mmol) in toluene (5 ml) was added thionyl chloride. The mixture was stirred at reflux for 1 hour. Then, thionyl chloride was eliminated by co-evaporation with toluene to give 4-(1 -methylethyl)benzoyl chloride, methyl 3-(4-aminobutyl)-1 -(2-pyridinyl)-1 H-indole-5-carboxylate (Intermediate 88) (100
PB62659C
mg, 0.31 mmol) was dissolved in anhydrous THF (10 ml), triethylamine (62 μl, 0.46 mmol) was added and 4-(1-methylethyl)benzoyl chloride was poured into the mixture. The reaction is stirred at room temperature for 2 days. The mixture was hydrolysed with water and extracted with ethyl acetate. The organic phase was washed with water and dried over Na2SO4, filtered and evaporated to dryness. The residue was purified on SiO2 eluting with dichloromethane to give the title compound as brown oil (1 10 mg, 76%). LC/MS : m/z 470 (M+H)+, Rt: 3.68.
The following compound was similarly prepared by analogous method to that described before :

Intermediate 91
Methyl 3-r4-(ir2-hvdroxy-3-(1 -methylethyl)phenyllcarponyl}amino)butyll-1 -(2- pyridinvh-1H-indole-5-carboxylate

To a solution of 2-hydroxy-3-(1 -methylethyl)benzoic acid (92 mg, 0.51 mmol) in THF (10 ml) was added DMAP (17 mg, 0.14 mmol), EDCI (98 mg, 0.51 mmol) and methyl 3-(4- aminobutyl)-1-(2-pyridinyl)-1 H-indole-5-carboxylate (Intermediate 88) (150 mg, 0.46 mmol). The reaction was stirred at room temperature overnight. The mixture was extracted with ethyl acetate and water. The organic phase was dried over Na2SO4, filtered
and evaporated to dryness. The residue was purified on SiC>2 eluting with dichloromethane to give the title compound as colorless oil (50 mg, 23%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.48 (m, 1 H), 8.27 (s, 1 H), 8.14 (d, 1 H, J=8.89 Hz), 7.90 (dd, 1 H, J=1.48 Hz, 8.64 Hz), 7.75 (td,1 H, J=1.97 Hz, 7.90 Hz), 7.48 (s, 1 H), 7.38 (d, 1 H, J=8.39 Hz), 7.23 (d, 1 H, J=7.65 Hz), 7.09 (m, 2H), 6.69 (t, 1 H, J=7.65 Hz), 6.33 (m, 1 H), 3.87 (s, 3H), 3.41 (q, 2H, J=6.91 Hz), 3.30 (m, 1 H), 2.79 (t, 2H, J=6.91 Hz), 1.71 (m, 4H), 1.15 (d, 6H, J=6.87 Hz).
Intermediate 92 Methyl 3-[4-(I [4-(1 ,1 -dimethylethyl)phenyllcarbonyl)amino)butyll-1 -phenyl-1 H- indole-5-carboxylate

To a solution of Methyl 3-(4-aminobutyl)-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 86) (250 mg, 0.77 mmol) in dichloromethane was added triethylamine (95 mg, 0.93 mmol) and 4-(1 ,1-dimethylethyl)benzoyl chloride (168 mg, 0.85 mmol). The reaction mixture was stirred at room temperature for 2 hours. The mixture was washed with brine (2 times), the organic phase was dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with dichloromethane/ethyl acetate 95/5 to give the title compound (166 mg, 45%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.33 (sd, 1 H, J=1.64 Hz), 7.84 (dd, 1 H, J=1.49 Hz), 8.65 Hz), 7.59 (d, 2H, J=8.50 Hz), 7.44 (m, 5H), 7.34 (d, 2H, J=8.65 Hz), 7.29 (m, 1 H), 7.94 (s, 2H), 3.87 (s, 3H), 3.45 (q, 2H, J=6.13 Hz), 2.83 (t, 2H, J=7.25 Hz), 1.74 (m, 4H), 1.25 (s, 9H). The following Intermediates were prepared in an analogous manner to Methyl 3-[4-({[4- (1 ,1-dimethylethyl)phenyl]carbonyl}amino)butyl]-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 92):
PB62659C

Intermediate 95
Methyl 3-U-r6-π -methylethvn-3.4-dihvdro-2πH)-isoαuinolinvnbutyl>-1 -phenyl-1H- indole-5-carboxylate

To a solution of methyl 3-{4-[(methylsulfonyl)oxy]butyl}-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 93) (1 15 mg, 0.28 mmol) in MIBK was added 6-(1-methylethyl)-1 , 2,3,4- tetrahydroisoquinoline (Intermediate 73) (45 mg, 0.26 mmol), K2CO3 (45 mg, 0.32 mmol), Kl (45 mg, 0.26 mmol). The reaction mixture was stirred at reflux overnight and concentrated. The residue was diluted with dichloromethane/water. The organic phase was extracted and washed with a saturated solution of NaHCO3. The organic phase was
dry over Na2SO4, filtered and concentrated. The residue was purified on SiC>2 eluting with dichloromethane and dichloromethane/MeOH 97/3 to give the title compound as yellow oil (45 mg, 37%). LC/MS : m/z 481 (M+H)+, Rt: 4.36 min.
Intermediate 96
Methyl 1 -(4-fluorophenyl)-3-r4-(( F4-(1 -methylethyl)phenyllcarbonyl)amino )butyl1-1 H- indole-5-carboxylate

To a solution of 4-(1-methylethyl)benzoic acid (197 mg, 1.2 mmol) in DMF (3 ml) was added HATU (456 mg, 1.2 mmol), methyl 3-(4-aminobutyl)-1-(4-fluorophenyl)-1 H-indole-5- carboxylate (Intermediate 87) (340 mg, 1 mmol) in dichloromethane (4 ml) and triethylamine (280 μl, 2 mmol). The reaction was stirred at room temperature for 16 hours. The mixture was extracted with ethyl acetate (150 ml), the organic phase was washed with brine and dried over Na2SO4, filtered and evaporated to dryness. The residue was purified on SiO2 eluting with dichloromethane/ethyl acetate 95/5 to give the title compound (400 mg, 86%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.31 (sd, 1 H, J=1.33 Hz), 7.82 (dd, 1 H, J=1.61 Hz, 8.70 Hz), 7.59 (d, 2H, J=8.22 Hz), 7.33 (m, 3H), 7.13 (m, 4H), 7.04 (s, 1 H), 6.21 (m, 1 H), 3.84 (s, 3H), 3.42 (q, 2H, J=6.59 Hz), 2.84 (m, 1 H), 2.78 (t, 2H, J=7.32 Hz), 1.71 (m, 4H), 1.15 (d, 6H, J=6.95 Hz).
The following compound was similarly prepared by analogous method to that described before :
 H),
H),

Intermediate 98
Methyl 3-U(1-U(1 J-dimethylethyl)oxylcarbonyl)-3-piperidinyl)aminolmethyl)-6- methyl-1 -phenyl-1 H-indole-5-carboxylate

To a solution of methyl 3-formyl-6-methyl-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 41) (300 mg, 1 mmol) in dichloromethane was added 1 ,1-dimethylethyl 3-amino-1- piperidinecarboxylate (370 mg, 1.5 mmol) and sodium acetate (130 mg, 1.5 mmol). The reaction mixture was stirred at room temperature for 10 min. NaHB(OAc)3 (650 mg, 3 mmol) was added. The reaction mixture was stirred for 18 hours. The mixture was diluted with diethyl ether and washed with water. The organic phase was dried over Na2SO4, filtered and evaporated to give the title compound as light oil (420 mg, 94%). LC/MS : m/z 498 (M+H)+, Rt: 3.74 min
Intermediate 99
4-(1,1-dimethylethyl )benzenecarbothioamide

A solution of 4-(1 ,1-dimethylethyl)benzonitrile in DMF (50 ml) was saturated with HCI gas, the temperature climbed to 800C. Ethanethioamide (9.5 g, 0.12 mol) was added at 700C and the mixture was stirred at 100°C for 26 hours. The solution was re-saturated with HCI gas and heated to 1000C for 6 hours. After cooling to room temperature, the mixture was poured into water (150 ml) and extracted with diethyl ether (3x150 ml). The organic phase was washed with water (2x100 ml), dried over MgSO4, filtered and evaporated. The obtained product was washed with pentane, filtered and dried to give the title compound (9.6 g, 79%).
NMR1H NMR (300 MHz), CDCI3 δ: 7.83 (d, 2H, J=8.65 Hz), 7.43 (d, 2H, J=8.87 Hz), 1.34 (s, 9H).
Intermediate 100 2-f4-(1,1-dimethylethyl)phenvn-4-methyl-1,3-thiazole-5-carboxylic acid

Ethyl 2-chloro-3-oxobutanoate (6.43 ml, 46.5 mmol) and 4-(1 ,1-dimethylethyl )benzenecarbothioamide (Intermediate 99) (9 g, 46.5 mmol) in ethanol (45 ml) were stirred at reflux for 5H30. The reaction mixture was evaporated and the residue was diluted with ethanol (70 ml). NaOH (3.72g) diluted in water (70 ml) was added at room temperature and the reaction mixture was stirred at reflux for 1 H30. Ethanol was evaporated and water (150 ml) was added. The mixture was extracted with ethyl acetate (150 ml + 100 ml) and acidified with HCL 1 N (100 ml) until pH=1. The precipitate was filtrate and washed with water. The precipitate was dissolved with dichloromethane (300 ml), this solution was dried over Na2SO4, filtered and evaporated. The obtained solid was recrystallised in toluene, filtrate and washed with pentane to give the title compound (7.3 g, 57%).
NMR1H NMR (300 MHz), CDCI3 δ: 7.84 (d, 2H, J=8.49 Hz), 7.42 (d, 2H, J=8.72 Hz), 2.75 (s, 3H), 1.29 (s, 9H).
Intermediate 101
Λ/,4-dimethyl-2-r4-(1-methylethyl)phenvn-1,3-thiazole-5-carboxamide

To a solution of 4-methyl-2-[4-(1-methylethyl)phenyl]-1 ,3-thiazole-5-carboxylic acid (0.5 g, 1.91 mmol) in toluene (5 ml) was added thionyl chloride (0.7 ml, 9.56 mmol). The mixture was stirred at 1100C for 2h30. The thionyl chloride was co-evaporated with toluene. The acyl chloride obtained diluted with dichloromethane was added drop by drop to a solution of triethylamine ( 0.8 ml, 5.73 mmol) and methyl amine (2 ml, 3.83 ml) in dichloromethane (5 ml). The mixture was stirred 15 min at room temperature. The solvent was evaporated and the residue was diluted with ethyl acetate, washed with HCI 0.5N, NaHCC>3 and brine. The organic phase was dried over Na2SO4, filtered and evaporated to give the title compound (0.453 g, 86%). LC/MS : m/z 275 (M+H)+, Rt: 2.85 min
The following compounds were prepared in an analogous manner to Intermediate 101 :
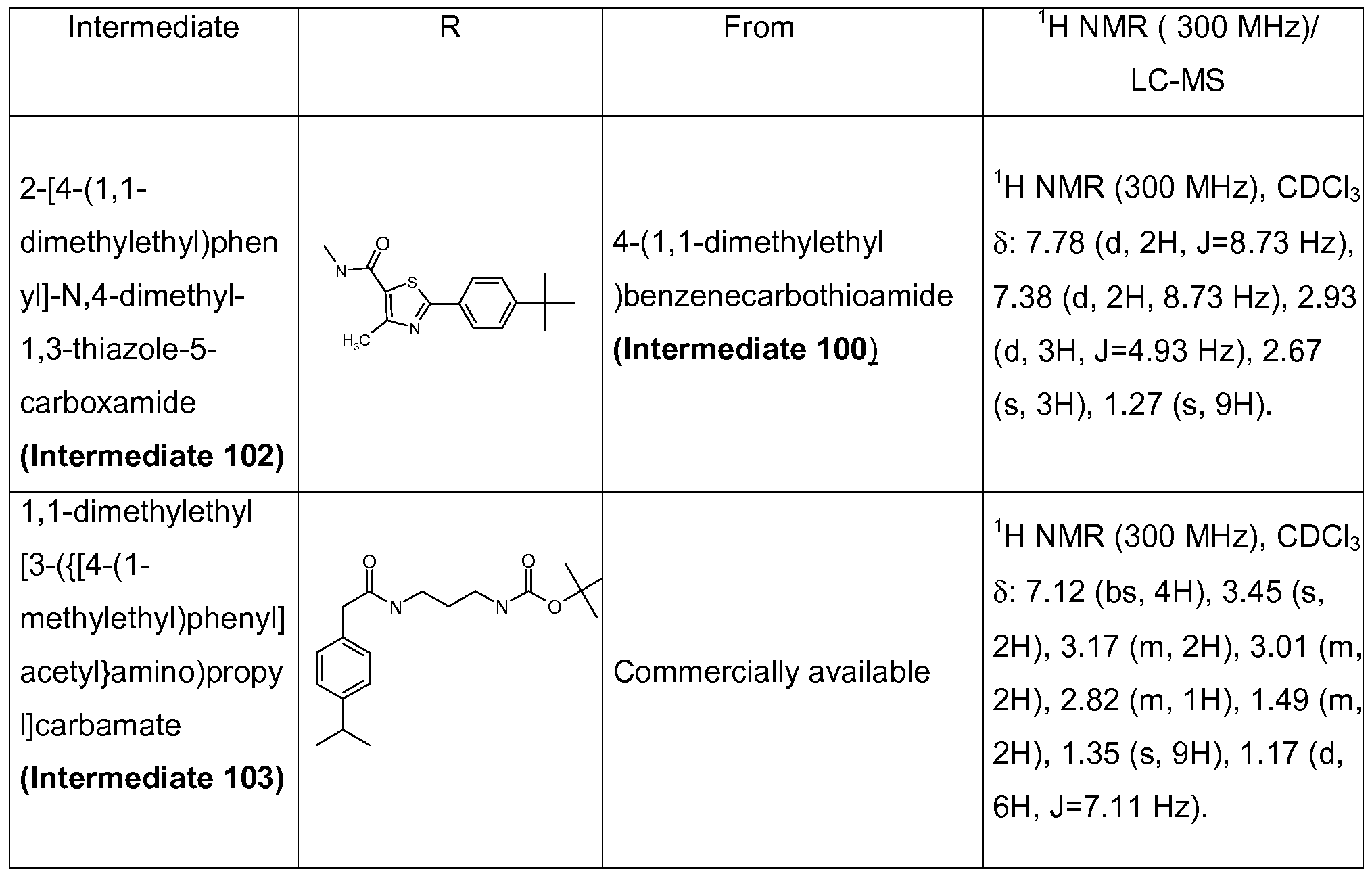

HCI gas was bubbled in a solution of 1 ,1-dimethylethyl [2-({[4-(1- methylethyl)phenyl]carbonyl}amino)ethyl]carbamate (Intermediate 149) (3 g, 8.6 mmol) in ethyl acetate up to saturation. The reaction mixture was stirred at room temperature for 18 hours. A precipitate was formed, diethylether was added and the precipitate was filtered under nitrogen flow. The solid was dried by co-evaporation to give the title compound as a white powder (hygroscopic, 2.3 g, 94%).
NMR1H NMR (300 MHz), DMSO δ: 8.50 (d, 1 H, J=7.65 Hz), 7.82 (d, 2H, J=8.08 Hz), 7.32 (d, 2H, J=8.08 Hz), 3.26 (m, 2H), 3.14 (m, 1 H), 2.93 (m, 1 H), 2.85 (m, 2H), 1.86 (m, 2H), 1.65 (m, 2H), 1.20 (d, 6H, J=6.80 Hz).
The following compounds were prepared in an analogous manner to Methyl 3-{[(1-{[(1 ,1- dimethylethyl)oxy]carbonyl}-3-piperidinyl)amino]methyl}-6-methyl-1-phenyl-1H-indole-5- carboxylate (Intermediate 104):
Intermediate R From 1H NMR ( 300 MHz)/
LC-MS
1 ,1-dimethylethyl
N-(3-aminopropyl)-2-[4- 1H NMR (300 MHz),
ClH [3-({[4-(1-
(1- methylethyl)phenyl CDCI3 δ:7.15 (m, 2H), methylethyl)phenyl]aceta ]acetyl}amino)prop 6.95 (m, 2H), 3.6 (m, 1 H), mide hydrochloride yl]carbamate 3.2 (m, 4H), 2.7 (m, 2H),
(Intermediate 105) (Intermediate 2. (m, 2H), 1.05 (d, 6H).
103)
Methyl 6-methyl-1- Methyl 3-{[(1- phenyl-3-[(3- {[(1 ,1-
LC/MS : m/z 378 (M+H)+, piperidinylamino)methyl]- dimethylethyl)oxy] Rt: 2.80 min 1 H-indole-5-carboxylate carbonyl}-3- hydrochloride piperidinyl)amino]

Intermediate 107
Λ/-methyl-1 -f4-methyl-2-r4-(1 -methylethyl)phenyl1-1 ,3-thiazol-5-yl)methanamine

Under nitrogen, introduce LiAIH4 1 M in THF (2.2 ml, 2.2 mmol) in THF (5 ml) at 00C. The mixture was stirred at 00C and Λ/,4-dimethyl-2-[4-(1-methylethyl)phenyl]-1 ,3-thiazole-5- carboxamide (Intermediate 101) (0.2 g, 0.73 mmol) in solution of THF (5 ml) was added drop by drop. When the gas escape was finished, the mixture was stirred at room temperature for 15 min and heated at reflux for 3 hours. The reaction was quenched at 00C with water over Na2SO4 slowly. When the gas escape was finished, the mixture was filtered and washed with THF. The solvent was evaporated and the residue was diluted with ethyl acetate, washed with brine and the organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified on SiO2 eluting with dichloromethane/methanol 100/0 to 90/10 to give the title compound (116 mg, 61%). LC/MS : m/z 261 (M+H)+, Rt: 2.67 min
The following compound was similarly prepared by analogous method to that described before :


Intermediate 109
Methyl 2 -methyl -3-{ f3-({f4-(1 -methylethyl)phenyl1carbonyl)amino)-1 -piperidinyl
1methyl)-1 -phenyl-1 H-indole-5-carboxylate

To a solution of 4-(1-methylethyl)-Λ/-3-piperidinylbenzamide hydrochloride (Intermediate 104) (270 mg, 0.96 mmol) in anhydrous THF was added NaHB(OAc)3 (509 mg, 2.4 mmol) and the mixture was stirred at room temperature for 30 min. Then, methyl 3-formyl-2- methyl-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 115a) (235 mg, 0.8 mmol) was added and the mixture was stirred at room temperature for 30 min and NaHB(OAc)3 (305 mg, 1.44 mmol) was added. The reaction mixture was stirred at room temperature for 16 hours. The mixture was quenched with brine and extracted with AcOEt. The organic phase was washed with brine, dried over Na2SO4 and concentrated. The residue was purified on SiO2 eluting with dichloromethane/methanol 96/4 to give the title compound as colourless oil which solidified (260 mg, 62%). LC/MS : m/z 524 (M+H)+, Rt: 4.06 min
Intermediate 115a was prepared in a manner analagous to Intermediate 38.
The following compounds were prepared in an analogous manner to Methyl 2-methyl-3- {[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1 -piperidinyl ]methyl}-1 -phenyl-1 H-indole-5- carboxylate (Intermediate 109)
PB62659C



Intermediate 118
Methyl 1 -(4-fluorophenyl)-3-{r3-({[4-(1 -methylethyl)phenvHcarbonyl)amino)-1 ■ piperidinyllmethyll-I H-indole-S-carboxylate

4-(1-methylethyl)-/V-3-piperidinylbenzamide hydrochloride (Intermediate 104) (190 mg, 0.67 mmol) and sodium acetate (55 mg, 0.67 mmol) were diluted with ethanol. A precipitate was formed so, ethanol was evaporated and the reagent were diluted with
dichloromethane. methyl 1-(4-fluorophenyl)-3-formyl-1 H-indole-5-carboxylate
(Intermediate 39) (200 mg, 0.67 mmol) was added. The mixture was stirred 2 hours and NaHB(OAc)3 (215 mg, 1 mmol) was added. The mixture was stirred at room temperature for 18 hours. The reaction mixture was washed with water and NaOH 1 N. The organic phase was evaporated and the residue was purified on SiC>2 eluting with dichloromethane, dichloromethane/MeOH 99/1 and 98/2 to give the title compound as light oil (230 mg, 65%).
NMR1H NMR (300 MHz), DMSO δ: 8.53 (s, 1 H), 7.89 (dd, 1 H, J=1.78 Hz, 8.88 Hz), 7.45 (d, 2H, J=8.07 Hz), 7.37 (m, 3H), 7.18 (m, 2H), 7.1 1 (t, 3H, J=8.56 Hz), 6.77 (m, 1 H), 4.20 (m, 1 H), 3.82 (s, 2H), 3.79 (d, 2H, J=13Hz), 3.58 (d, 2H, J=13.33 Hz), 2.84 (m, 2H), 2.71 (m, 1 H), 2.31 (m, 1 H), 2.20 (m, 1 H), 1.76 (m, 2H), 1.52 (m, 2H), 1.17 (d, 6H, J=6.75 Hz).
Intermediate 119 r4-(1-methylethyl)phenyllacetonitrile

4-lsopropyl benzyl bromide (1.65 ml, 9.38 mmol) and NaCN (920 mg, 18.77 mmol) were dissolved in a mix of ethanol (26 ml) and water (4 ml). The mixture was stirred at 800C for 1 H20. The mixture was cooled to room temperature and ethanol was evaporated. The residue was diluted with water and extracted with diethyl ether. The organic phase was dried over MgSO4, filtered and evaporated to give the title compound as yellow oil (1.56 g, quantitative yield). NMR1H NMR (300 MHz), CDCI3 δ : 7.25 (s, 4H), 3.71 (s, 2H), 2.90 (m, 1 H), 1.25 (d, 6H).
Intermediate 120 (2-[4-(I -methylethyl)phenyllethyl)amine hydrochloride

A solution of LiAIH4 (838 mg, 20.97 mmol) in diethyl ether (15 ml) was cooled at 00C under argon. [4-(1-methylethyl)phenyl]acetonitrile (Intermediate 119) (1.67 g, 10.48 mmol) in solution of diethyl ether (10 ml) was added drop by drop at 00C. The mixture was stirred at reflux for 1 hour. The mixture was cooled at 0°C, NH4CI (1.246 g) was added and 1.2 ml of water was added. Diethyl ether was added and the mixture was dried over
MgSO4 and filtered on celite and washed with diethyl ether. The filtrate was evaporated.
The residue was diluted with a solution of methanol saturated with HCI (25 ml) to formed chlorhydrate. The mixture was stirred at room temperature 35 min. The mixture was evaporated and co-evaporated with dichloromethane. The obtained solid was triturated with a mix of dichloromethane/diethyl ether to give the title compound as a pale yellow solid (734 mg, 35%).
NMR1H NMR (300 MHz), DMSO δ : 7.18 (m, 4H), 2.98 (m, 2H), 2.84 (m, 2H), 2.50 (m, 1 H), 1.17 (d, 6H).
Intermediate 121 Λ/-(2-aminoethyl)-4-(1 -methylethvDbenzamide hydrochloride

To a solution of 1 ,1-dimethylethyl [2-({[4-(1- methylethyl)phenyl]carbonyl}amino)ethyl]carbamate (Intermediate 149) (3.06 g, 10 mmol) in methanol (80 ml) was added HCI 6M (16.7 ml, 100 mmol). The reaction mixture was stirred at room temperature for 2 hours and HCI 12 N (10 ml) was added. The mixture was stirred at room temperature for 24 hours. The mixture was concentrated to dryness and the residue was diluted with ethanol (100 ml) and this mixture was re-concentrated to dryness to give the title compound as a white solid (2.39 g, 98%). NMR1H NMR (300 MHz), DMSO δ: 7.86 (d, 2H, J=7.86 Hz), 7.33 (d, 2H, J=8.61 Hz), 3.53 (m, 2H), 2.97 (m, 2H; m, 1 H), 1.21 (d, 6H, J=6.98 Hz).
The following compound was similarly prepared by analogous method to that described before :

Methyl 1-phenyl-3- {[(1 ,1-
1 H NMR (300 MHz), DMSO
[(3- dimethylethyl)oxy]
ClH δ: 8.6 (s, 1 H), 8.1 (s, 1 H), piperidinylamino)met carbonyl}-3- 7.9 (dd, 1 H), 7.65 (m, 5H), hyl]-1 H-indole-5- piperidinyl)amino] 7.5 (m, 1 H), 4.55 (m, 2H), carboxylate methyl}-1-phenyl-
 3.9 (s, 3H), 3.60 (m, 2H), dihydrochloride 1 H-indole-5- 3.15 (m, 2H), 2.35 (m, 1 H), (Intermediate 122) carboxylate 2.00 (m, 1 H), 1.75 (m, 2H).
3.9 (s, 3H), 3.60 (m, 2H), dihydrochloride 1 H-indole-5- 3.15 (m, 2H), 2.35 (m, 1 H), (Intermediate 122) carboxylate 2.00 (m, 1 H), 1.75 (m, 2H).
117)
Intermediate 123
Methyl 3-{[(1-{[4-(1 J -dimethylethyOphenyllcarbonyll-S-piperidinvOamino 1methyl)-1- phenyl-1 H-indole-5-carboxylate

To a mix of methyl 1-phenyl-3-[(3-piperidinylamino)methyl]-1 H-indole-5-carboxylate dihydrochloride (Intermediate 122) (150 mg, 0.35 mmol) and triethylamine (253 μl, 1.75 mmol) in dichloromethane (15 ml) was added 4-(1 ,1-dimethylethyl)benzoic acid (62.3 mg, 0.35 mmol), HOBT (57 mg, 0.42 mmol) and EDCI (81 mg, 0.42 mmol). The reaction mixture was stirred at room temperature for 3 hours. The solvent was evaporated and the residue was diluted with a mix of ethyl acetate and water (50/50). The organic phase was decanted and washed with brine, dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with dichloromethane/methanol 100/0 to 93/7 to give the title compound (105 mg, 58%).
LC/MS : m/z 524 (M+H)+, Rt: 3.80 min
Intermediate 124
Methyl 3-[3-((2-[4-(I -methylethyl)phenyl1ethyl)amino)-3-oxopropyl1-1 -phenyl-1 H- indole-5-carboxylate

To a solution of 3-{5-[(methyloxy)carbonyl]-1-phenyl-1H-indol-3-yl}propanoic acid (Intermediate 8) (318 mg, 0.983 mmol) and {2-[4-(1-methylethyl)phenyl]ethyl}amine hydrochloride (Intermediate 120) (196 mg, 0.983 mmol) in dichloromethane (7 ml) was added EDCI (21 1 mg, 1.082 mmol) and DMAP (156 mg, 1.278 mmol). The reaction mixture was stirred at reflux for 18 hours. The mixture was washed with water and the organic phase was dried over MgSO4, filtered and concentrated. The residue was purified on SiC>2 eluting with dichloromethane and dichloromethane/ ethylacetate (98/2 to 95/5) to give the title compound as pale yellow solid (348 mg, 75%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.40 (s, 1 H), 7.94 (d, 1 H), 7.40 (m, 6H), 7.04 (d, 2H), 6.91 (d, 2H), 5.39 (d, 2H), 3.92 (s, 3H), 3.48 (m, 2H), 3.19 (m, 2H), 2.82 (m, 1 H), 2.63 (m, 4H), 1.21 (d, 6H).
Intermediate 125
Methyl 3-U(1-U4-(1 J-dimethylethyl)phenyllcarbonyl)-3-piperidinyl)(methyl) amino1methyl)-1 -phenyl-1 H-indole-5-carboxylate

To a solution of Methyl 3-{[(1-{[4-(1 ,1-dimethylethyl)phenyl]carbonyl}-3-piperidinyl)amino ]methyl}-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 123) (105 mg, 0.2 mmol) in THF (15 ml) was added formaldehyde (75 μl, 1 mmol), NaHB(OAc)3 (212 mg, 1 mmol). The reaction mixture was stirred at room temperature for 4 hours. The mixture was diluted with brine and extracted with ethyl acetate (2 times). The organic phase was washed with a saturated solution of NaHCO3 and brine. It was dried over Na2SO4, filtered and evaporated to give the title compound as colourless oil (108 mg, quantitative yield) LC/MS : m/z 538 (M+H)+, Rt: 4.09 min
The following compounds were prepared in an analogous manner to Methyl 3-{[(1-{[4- (1 ,1-dimethylethyl)phenyl]carbonyl}-3-piperidinyl)(methyl) amino]methyl}-1-phenyl-1 H- indole-5-carboxylate (Intermediate 125):


Intermediate 130
1,1-dimethylethyl (2-amino-2-methylpropyl)carbamate

To a solution of 2-methyl-1 ,2-propanediamine (300 mg, 3.40 mmol) in ethanol (15 ml) was added phenyl-tBu-carbamate (1.32 g, 6.80 mmol). The reaction mixture was stirred at 800C overnight. The solvent was evaporated, water was added and HCI 1 N until pH=3. The mixture was washed with dichloromethane (2 times). The aqueous phase was basified with NaOH 1 N until pH=11-12 and was extracted with dichloromethane (4 times). The organic phase was dried over Na2SO4, filtered and concentrated to give the title compound as colourless liquid (422 mg, 66%). LC/MS : m/z 189 (M+23)+, Rt: 1.62 min.
Intermediate 131
Methyl 3-(U2-(U(1,1 -dimethylethyl)oxy1carbonyl)amino)-1,1-dimethylethvπ amino)methyl)-1 -phenyl-1 H-indole-5-carboxylate

A solution of 1 ,1-dimethylethyl (2-amino-2-methylpropyl)carbamate 1 ,1 -dimethylethyl (2- amino-2-methylpropyl)carbamate (Intermediate 130) (150 mg, 0.798 mmol) and Methyl 3- formyl-1-phenyl-1 H-indole-5-carboxylate (Intermediate 38) (185 mg, 0.66 mmol) in anhydrous THF (10 ml) was stirred at 8O0C overnight. Molecular sieves were added and the reaction was stirred at reflux for 2H30. The reaction mixture was filtered and evaporated. Methanol was added and NaBH4 (50 mg, 1.33 mmol). The reaction mixture was stirred at room temperature for 1 H30.The reaction mixture was quenched with water and evaporated. A saturated solution of NaHCU3 was added and the mixture was extracted with ethyl acetate (3 times). The organic phases were washed with brine, dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with dichloromethane/methanol 98/2 to 95/5 to give the title compound as colourless oil (170 mg, 57%).
NMR1H NMR (300 MHz), CDCI3 δ: 8.36 (s, 1 H), 7.84 (dd, 1 H, J=1.54 Hz, 8.95 Hz), 7.44 (m, 5H), 7.30 (m, 2H), 3.92 (s, 2H), 3.87 (s, 3H), 3.09 (bs, 2H), 1.35 (s, 9H), 1.15 (s, 6H).
The following compounds were prepared in an analogous manner to Methyl 3-({[2-({[(1 ,1- dimethylethyl)oxy]carbonyl}amino)-1 ,1-dimethylethyl] amino}methyl)-1-phenyl-1 /-/-indole- 5-carboxylate (Intermediate 131):


Intermediate 134
Methyl 3-fr[2-(ir(1 J -dimethylethyl)oxylcarbonyl)amino)-1 ,1 -dimethylethyll
(methyl)aminolmethyl>-1 -phenyl-1 H-indole-5-carboxylate

To a solution of methyl 3-({[2-({[(1 ,1-dimethylethyl)oxy]carbonyl}amino)-1 ,1-dimethylethyl] amino}methyl)-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 131) (165 mg, 0.366 mmol) in a mix of acetonitrile (8 ml)/ THF (1 ml) was added acetic acid (42 μl, 0.732 mmol), NaHB(OAc)3 (465 mg, 2.195 mmol) and formaldehyde (60 μl, 0.734 mmol). The reaction mixture was stirred at room temperature for one hour and 30 minutes. A saturated solution of NaHCO3 was added, the mixture was extracted with dichloromethane (3 times). The organic phase was dried over Na2SO4, filtered and concentrated to give the title compound (166 mg, 98%). LC/MS : m/z 466 (M+23)+, Rt: 3.80 min.
The following compounds were prepared in an analogous manner to Methyl 3-{[[2-({[(1 ,1- dimethylethyl)oxy]carbonyl}amino)-1 ,1-dimethylethyl] (methyl)amino]methyl}-1 -phenyl-1 H- indole-5-carboxylate (Intermediate 134):

Intermediate 138
1 ,1 -dimethylethyl 3-amino-1 -pyrrolidinecarboxylate

3-pyrrolidinamine (500 mg, 5.80 mmol), Tert-butyl phenyl carbonate (1.24 g, 6.38 mmol) was dissolved in DMF (5 ml) and stirred for 24 hours. Water and HCI 1 N was added until pH=3. The mixture was washed with dichloromethane (2 times). NaOH 1 N was added to the aqueous phase until pH=1 1-12 and was extracted with dichloromethane (4 times). The
organic phase was dried over Na2SC>4, filtered and evaporated to give the title compound as light orange liquid (1.063 g, 98%). LC/MS : m/z 187 (M+23)+, Rt: 1.54 min.
Intermediate 139
1,1-dimethylethyl 3-({f4-(1 -methylethyl)phenvncarbonyl)amino)-1- pyrrol i d i necarboxyl ate

To a solution of 1 ,1-dimethylethyl 3-amino-i-pyrrolidinecarboxylate (Intermediate 138) (400 mg, 2.15 mmol) in acetontrile (10 ml) was added 4-(1-methylethyl)benzoic acid (388 mg, 2.36 mmol), EDCI (495 mg, 2.58 mmol) and HOBT (349 mg, 2.58 mmol). The reaction mixture was stirred at room temperature overnight. The solvent was evaporated and a saturated solution of K2CO3 was added. The mixture was extracted with ethyl acetate (3 times). The organic phase was washed with brine, dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with cyclohexane/ethyl acetate 8/2 to 5/5 to give the title compound as colourless oil (605 mg, 85%). LC/MS : m/z 333 (M+23)+, Rt: 3.26 min.
Intermediate 140 (4-{r4-(1-methylethyl)phenvπamino)-4-oxobutyl)(triphenyl)phosphonium bromide

To a solution of HOBT (3.6 g, 26.6 mmol), EDAC (5.1 g, 26.6 mmol) and triethylamine (5.3 ml, 66.6 mmol) in DMF was added (3-carboxypropyl)(triphenyl)phosphonium bromide (10.5 g, 24.4 mmol). The reaction mixture was stirred at room temperature for 15 min and 4-(1-methylethyl)aniline (3 g, 22.2 mmol) was added. The mixture was stirred at room temperature overnight. DMF was evaporated and the residue was diluted with dichloromethane and a saturated solution of NaHCO3. The organic phase was extracted, dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with
PB62659C
dichloromethane to dichloromethane/methanol 9/1. The obtained product was triturated in isopropyl ether and sonicated to give the title compound as a white product (9 g, 74%). NMR1H NMR (300 MHz), CDCI3 δ: 7.86 (m, 9H), 7.76 (m, 8 H), 7.21 (d, 2H, J=8.45 HZ), 3.82 (m, 2H), 3.1 1 (t, 2H, J=6.34 Hz), 2.92 (m, 1 H), 2.14 (m, 2H), 1.28 (d, 6H, J=7.08 H z).
Intermediate 141
Methyl 3-fr(2-amino-1.1-dimethylethyl)(methvπamino1methyl'y-1-phenyl-1/y-indole-5- carboxylate

The following compounds were prepared in an analogous manner to Methyl 3-{[(2-amino- 1 ,1-dimethylethyl)(methyl)amino]methyl}-1-phenyl-1 H-indole-5-carboxylate (Intermediate 141):


Intermediate 145
Methyl 3-<Tmethyl(1 -fF4-(1 -methylethyl)phenyllcarbonyl'y-3-piperidinyl)aminol methyl)-1 -phenyl-1 H-indole-5-carboxylate

Methyl 3-{[methyl(3-piperidinyl)amino]methyl}-1 -phenyl-1 H-indole-5-carboxylate
(Intermediate 142) (130 mg, 0.345 mmol), 4-(1-methylethyl)benzoic acid (74 mg, 0.448 mmol), HOBT (70 mg, 0.517 mmol), triethylamine (96 μl, 0.69 mmol) and EDCI (100 mg, 0.517 mmol) was stirred in acetonitrile for a week-end. The solvent was evaporated and a saturated solution of K2CO3 was added. The mixture was extracted with ethyl acetate (3 times). The organic phase was washed with brine, dried over Na2SO4, filtered and evaporated. The residue was purified on SiO2 eluting with dichloromethane/methanol 98/2 to give the title compound (130 mg, 72%). LC/MS : m/z 546 (M+23)+, Rt: 3.80 min.
PB62659C
The following compounds were prepared in an analogous manner to Methyl 3-{[methyl(1- {[4-(1-methylethyl)phenyl]carbonyl}-3-piperidinyl)amino] methyl}-1 -phenyl-1 H-indole-5- carboxylate (Intermediate 145):



Example 20
1 -phenyl -S-U-re-ftrifluoromethvD-S^-dihvdro-Σd ffl-isoαuinolinvnbutvD-IH-indole-S- carboxylic acid

To a solution of methyl 3-{4-[(methylsulfonyl)oxy]butyl}-1 -phenyl-1 H-indole-5-carboxylate (Intermediate 93) (120 mg, 0.3 mmol) in MIBK (10 ml), was added K2CO3 (105 mg, 0.75 mmol) and 6-(trifluoromethyl)-1 ,2,3,4-tetrahydroisoquinoline hydrochloride (72 mg, 0.36 mmol). The reaction mixture was stirred at 12O0C for 2 days. The mixture was filtered and the filtrate was evaporated. The residue was purified on SiO2 eluting with dichloromethane to dichloromethane/ethyl acetate 95/5 to give the ester (15 mg). The ester was diluted with methanol, NaOH 1 N (3 ml) was added and the mixture was stirred at reflux for 24 hours. The mixture was cooled and HCI 1 N ( 3 ml) was added. The mixture was concentrated and, the residue was purified on SiO2 eluting with dichloromethane to dichloromethane/ethyl acetate 95/5. The product was triturating in cyclohexane and the solid obtained was filtered and washed with pentane to give after drying the title compound as a cream solid (9 mg, 6%).
MR1H (300 MHz), CDCI3 δ: 8.36 (s, 1 H), 7.83 (d, 1 H, J=9.63 Hz), 7.41 (m, 5H), 7.29 (m, 3H), 7.1 1 (s, 1 H), 7.06 (d, 1 H, J=8.61 Hz), 3.79 (m, 2H), 2.96 (m, 2H), 2.90 (m, 2H), 2.82 (m, 2H), 2.68 (m, 2H), 1.78 (m, 4H). TOF MS ES+ exact mass calculated for C29H27F3N2O2: 493.2103 (M+H)+ Found: 493.2105 (M+H)+ ; RT= 2.97 min.
PB62659C
Example 21
3-r4-(ff4-(1-methylethyl)phenvncarbonyl>amino)butvn-1-(2-pyridinyl)-1 H-indole-5- carboxylic acid

To a solution of methyl 3-[4-({[4-(1-methylethyl)phenyl]carbonyl}amino)butyl]-1-(2- pyridinyl)-1 H-indole-5-carboxylate (Intermediate 89) (100 mg, 0.21 mmol) in ethanol, was added NaOH 1 N (425 μl, 0.42 mmol). The reaction mixture is heated at 900C for 2 hours. The solvent was evaporated. The crude is acidified with HCI 1 N, the precipitate formed is filtrate and washed with isopropanol. The solid is recrystallized with acetontrilie to give the title compound as cream powder (35 mg, 37%).
NMR1H (300 MHz), DMSO δ: 12.78 (s, 1 H), 8.66 (s, 1 H), 8.50 (m, 2H), 8.35 (s, 1 H), 7.95 (m, 6H), 7.37 (m, 2H), 3.42 (m; 2H), 2.92 (m, 2H), 1.80 (m, 4H), 1.27 (d, 6H,J=7.96 Hz). TOF MS ES+ exact mass calculated for C28H29N3O3: 456.2287 (M+H)+ Found: 456.2294 (M+H)+ ; RT= 2.48 min.
The following examples were prepared in an analogous manner to 3-[4-({[4-(1- methylethyl)phenyl]carbonyl}amino)butyl]-1-(2-pyridinyl)-1 H-indole-5-carboxylic acid Example 21 from:
Examples Structures From NMR1H NMR
Intermediate ( 300MHz)/LC-MS
Example 22 Methyl 3-[4-({[4-
3-[4-({[4-(1 - (1- TOF MS ES+ exact mass methylethyl)phenyl en calculated for
]carbonyl}amino)b CX i methylethyl)ph yl]carbonyl}amino C29H30N2O3: 453.2178 utyl]-1-phenyl-1 H- y )butyl]-1-phenyl- (M+H)+ Found: 453.2216 indole-5-carboxylic 1 H-indole-5- (M+H)+ ; RT= 2.72 min acid carboxylate
PB62659C



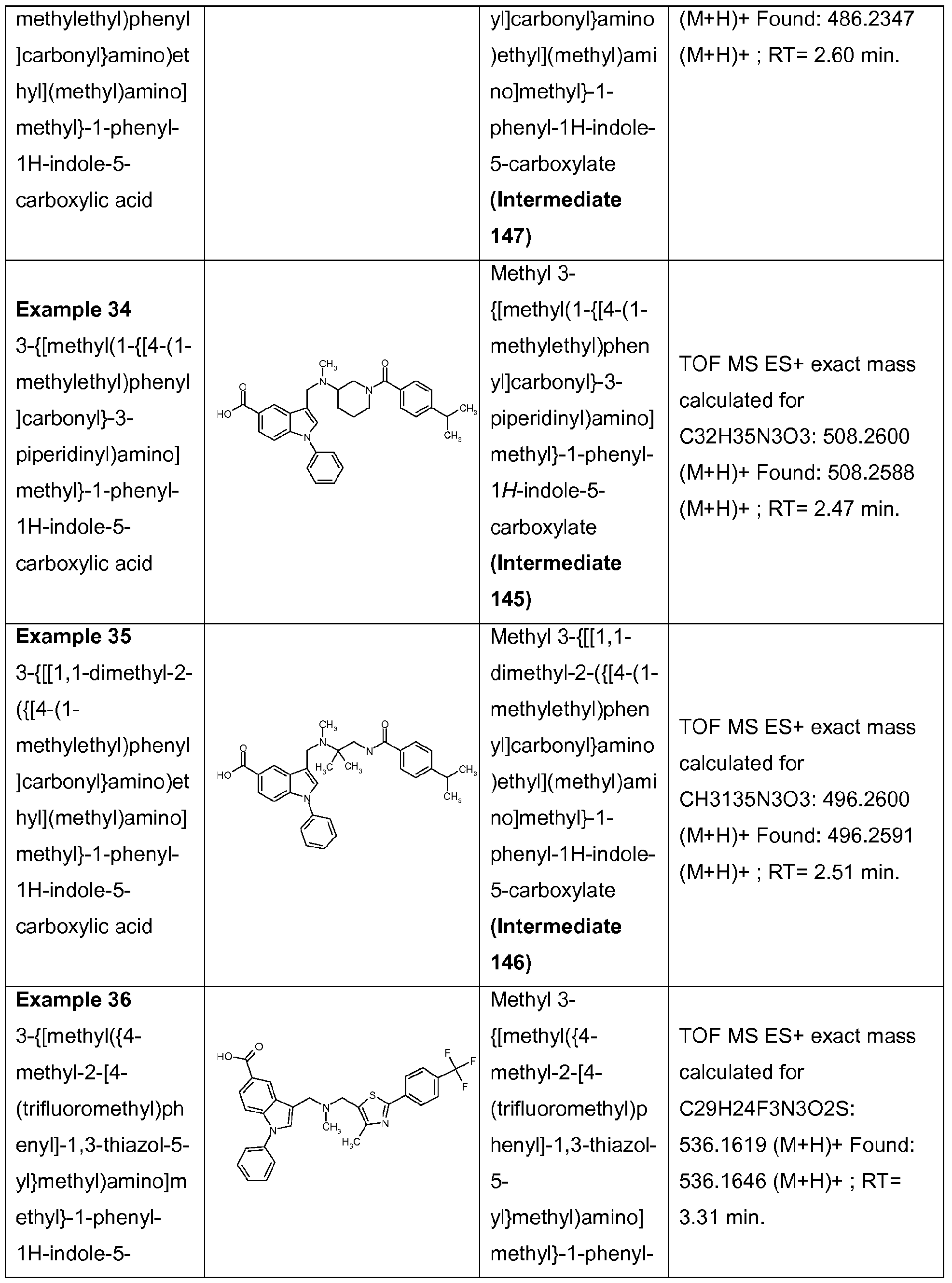




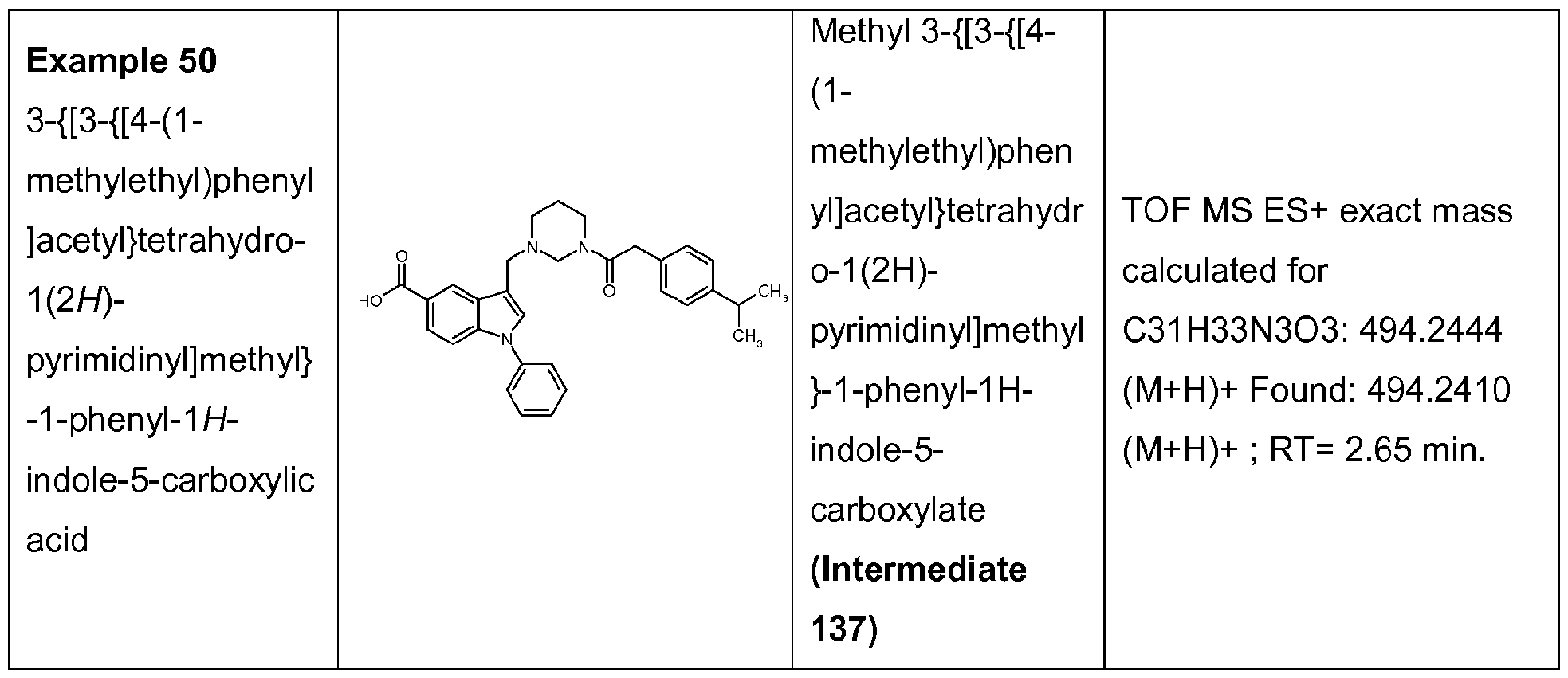
Biological Assays
Binding Assay:
Compounds were tested for their ability to bind to hPPAR gamma hPPARalpha or PPARdelta using a Scintillation Proximity Assay (SPA). The PPAR ligand binding domain (LBD) was expressed in E. coli as polyHis tagged fusion proteins and purified. The LBD was then labelled with biotin and immobilised on streptavidin-modified scintillation proximity beads. The beads were then incubated with a constant amount of the appropriate radioligand (5-{4-[2-(Methyl-pyridin-2-yl-amino)-ethoxy]-benzyl}-thiazolidine- 2,4-dione (J.Med.Chem. 1994, 37(23), 3977), for PPARgamma), and labelled GW 2433 (see Brown, P. J et al . Chem. Biol., 4, 909-918 (1997), for the structure and synthesis of this ligand) for PPAR alpha and PPAR delta) and variable concentrations of test compound, and after equilibration the radioactivity bound to the beads was measured by a scintillation counter. The amount of nonspecific binding, as assessed by control wells containing 50 μM of the corresponding unlabeled ligand, was subtracted from each data point. For each compound tested, plots of ligand concentration vs. CPM of radioligand bound were constructed and apparent Kl values were estimated from nonlinear least squares fit of the data assuming simple competitive binding. The details of this assay have been reported elsewhere (see, Blanchard, S. G. et. al. Development of a Scintillation Proximity Assay for Peroxisome Proliferator-Activated Receptor gamma Ligand Binding Domain. Anal. Biochem., 257, 112-119 (1998)).
Transfection assay:
Compounds were screened for functional potency in transient transfection assays in CV-1 cells for their ability to activate the PPAR subtypes (transactivation assay). A previously established chimeric receptor system was utilized to allow comparison of the relative transcriptional activity of the receptor subtypes on the same target gene and to prevent endogenous receptor activation from complicating the interpretation of results. See, for example, Lehmann, J. M.; Moore, L. B.; Smith-Oliver, T. A.; Wilkison, W. O.; Willson, T. M.; Kliewer, S. A., An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARgamma), J. Biol. Chem., 270, 12953-6 (1995). The ligand binding domains for murine and human PPAR alpha, PPAR gamma, and PPAR delta were each fused to the yeast transcription factor GAL4 DNA binding domain. CV-1 cells were transiently transfected with expression vectors for the respective PPAR chimera along with a reporter construct containing five copies of the GAL4 DNA binding site driving expression of secreted placental alkaline phosphatase (SPAP) and beta-galactosidase. After 16 h, the medium was exchanged to DME medium supplemented with 10% delipidated fetal calf serum and the test compound at the appropriate concentration. After an additional 24h, cell extracts were prepared and assayed for alkaline phosphatase and β-galactosidase activity. Alkaline phosphatase activity was corrected for transfection efficiency using the beta-galactosidase activity as an internal standard (see, for example, Kliewer, S. A., et. al. Cell 83, 813-819 {1995)). Rosiglitazone (BRL 49653) was used as a positive control in the hPPAR gamma assay. The positive control in the hPPAR alpha assays was 2-4-[2-(3-[4-fluorophenyl]-1- heptylureido)ethyl]-phenoxy-(2-methyl propionic acid (WO 97/36579). The positive control for PPAR delta assays was 2-{2-methyl-4-[({4-methyl-2-{trifluoromethyl)phenyl]-1 ,3- thiazol-5-yl}methyl)sulfanyl]phenoxy}acetic acid (WO 01/00603). The positive control was (5-{4-[2-(Methyl-pyridin-2-yl-amino)-ethoxy]-benzyl}-thiazolidine-2,4-dione (J. Med. Chem. 1994, 37(23), 3977), for PPAR gamma.
The examples above demonstrated activity of <1 μM in the above transfaction assay (or similar) on the PPAR alpha subunit.
Claims
1. A compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof:

Z is phenyl (optionally substituted by halogen), pyridinyl;
Y is phenyl (optionally substituted by one or more groups each independently selected from
-Ci-6 alkyl, -Ci-6 alkoxy, -Ci-6 haloalkyl, -Ci-6 haloalkoxy, -halogen, -OH, -C3-7 cycloalkyl, -
NR1R2), or phenyl fused to a 5 or 6 membered heterocyclic or cycloalkyl ring;
R1 and R2 independently represent H or d-3 alkyl;
X is a linker group of 5-7 atoms in shortest length between the carbon of the indole group and Y;
or X-Y represents a group:

R3 is Ci-3 alkyl or Ci-3 haloalkyl.
2. A compound according to claim 1 wherein Z is: 
3. A compound according to claims 1 to 2 wherein Y is phenyl (substituted by one or more groups independently selected from -OH, -C1-4 alkyl, -CF3 -Cl, -OCr3 alkyl, - cyclohexyl), or a fused structure as shown below:

4. A compound according to claims 1 to 3 wherein X is C1-1O alkylene linker or C2-10 alkenylene linker, each of which may contain within the alkylene chain one or more C=O, NR, and/or O moieties (R=H or Ci-3 alkyl) forming a 6 atom linker in shortest length.
5. A compound according to claims 1 to 3 wherein X is a linker which includes a 5-6 membered heteroaryl or a 5-6 membered heterocyclic nitrogen containing ring, together with one or more C1-6 alkylene, NR1 (R1 = H or C1-3 alkyl), and/or C=O moieties to form a 6 atom linker in shortest length.
6. A compound according to claims 1 to 3 wherein X is a linker selected from:
-(CH2)4 NHC(O)-;
-(CH2J4 CONH;
-(CH2)2 OCH2 C(O)NH-;
-(CH2)2 NHC(O)CH2O-; -(CHz)2 NH C(O) (CH2)2-
-(CHz)2 N(CH3) CH2 C(O)NH-
-CH2 N(CH3) CH2CH2 NHC(O)-
-CH2 N(CH3) C(CH3)2 CH2 NHCO-;
-CH2 NH(CH2), NHC(O)-; -CH = CH-(CH2)2 C(O)NH-;
-C(O)NH CH2CH2 C(O)NH-;
-(CHz)2 C(O)NHCH2CH2-; 




-CH2-N N-CO-

7. A compound according to claims 1 to 3 wherein X is a linker 5 atoms in shortest length which is:
-(CH2)2NHC(O)CH2- .
8. A compound according to claims 1 to 3 wherein X is a linker 7 atoms in shortest length which is: 
9. A compound according to claims 1 to 3 wherein X-Y together represent:

10. A compound according to claim 1 wherein there is provided a compound for formula (Ia):
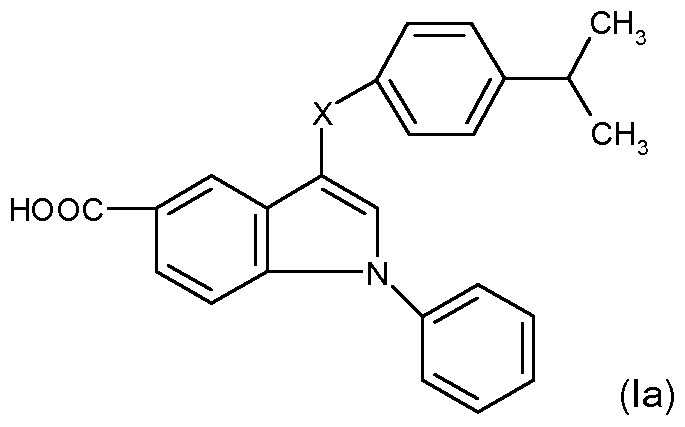
wherein X is as defined in claim 1.
11. A compound according to claim 10 wherein X in formula (Ia) is -CH2CH2OCH2CH-.
12. A compound of formula (I) selected from the group consisting of: 3-{2-[(2-{[4-(1-methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 H-indole-5- carboxylic acid; 3-{2-[(2-{[4-(1 ,1-dimethylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 H-indole-5- carboxylic acid;
3-[2-({2-[(4-methylphenyl)amino]-2-oxoethyl}oxy)ethyl]-1-phenyl-1 H-indole-5-carboxylic acid; 3-[2-({2-[(4-ethylphenyl)amino]-2-oxoethyl}oxy)ethyl]-1-phenyl-1H-indole-5-carboxylic acid;
3-(2-{[2-(2,3-dihydro-1 /-/-inden-5-ylamino)-2-oxoethyl]oxy}ethyl)-1-phenyl-1/-/-indole-5- carboxylic acid;
3-(2-{[2-(2>dihydro-1 ,4-benzodioxin-6-ylamino)-2-oxoethyl]oxy}ethyl)-1 -phenyl-1 H- indole- 5-carboxylic acid;
3-{2-[(2-{[3-(1-methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 H-indole-5- carboxylic acid;
3-[2-({2-[(4-cyclohexylphenyl)amino]-2-oxoethyl}oxy)ethyl]-1-phenyl-1 H-indole-5- carboxylic acid; 3-{2-[(2-{[4-(dimethylamino)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-phenyl-1 /-/-indole-5- carboxylic acid;
3-{2-[({[4-(methyloxy)phenyl]oxy}acetyl)amino]ethyl}-1-(2-pyridinyl)-1 /-/-indole-5-carboxylic acid;
3-{2-[(2-{[4-(1-methylethyl)phenyl]amino}-2-oxoethyl)oxy]ethyl}-1-(2-pyridinyl)-1 H-indole-5- carboxylic acid;
3-{2-[({[4-(1-methylethyl)phenyl]oxy}acetyl)amino]ethyl}-1-phenyl-1 H-indole- 5-carboxylic acid;
3-[2-({[(3,4-dichlorophenyl)oxy]acetyl}amino)ethyl]-1-phenyl-1 H-indole- 5-carboxylic acid;
3-{2-[({4-methyl-2-[4-(1-methylethyl)phenyl]-1 ,3-thiazol-5-yl}carbonyl)amino]ethyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{2-[({[4-(1 ,1-dimethylethyl)phenyl]oxy}acetyl)amino]ethyl}-1-phenyl-1 /-/-indole-5- carboxylic acid;
3-{2-[({[3-(methyloxy)phenyl]oxy}acetyl)amino]ethyl}-1-phenyl-1 H-indole- 5-carboxylic acid;
3-{2-[({[4-(methyloxy)phenyl]oxy}acetyl)amino]ethyl}-1 -phenyl-1 H-indole- 5-carboxylic acid; 3-[2-({[4-(1 -methylethyl)phenyl]acetyl}amino)ethyl]-1 -phenyl-1 H-indole-5-carboxylic acid;
3-[2-({3-[4-(1-methylethyl)phenyl]propanoyl}amino)ethyl]-1 -phenyl-1 H-indole- 5-carboxylic acid;
1-phenyl-3-{4-[6-(trifluoromethyl)-3,4-dihydro-2(1 H)-isoquinolinyl]butyl}-1 /-/-indole-5- carboxylic acid; 3-[4-({[4-(1 -methylethyl)phenyl]carbonyl}amino)butyl]-1 -(2-pyridinyl)-1 H-indole-5- carboxylic acid;
3-[4-({[4-(1 -methylethyl)phenyl]carbonyl}amino)butyl]-1 -phenyl-1 H-indole-5-carboxylic acid;
1-(4-fluorophenyl)-3-[4-({[4-(1-methylethyl)phenyl]carbonyl}amino)butyl]-1 H-indole- 5- carboxylic acid; 3-[4-({[4-(1 ,1-dimethylethyl)phenyl]carbonyl}amino)butyl]-1-phenyl-1 /-/-indole-5-carboxylic acid;
3-[4-({[2,4-bis(methyloxy)phenyl]carbonyl}amino)butyl]-1-(2-pyridinyl)-1 H-indole-5- carboxylic acid; 3-[4-({[2-hydroxy-3-(1-methylethyl)phenyl]carbonyl}amino)butyl]-1-(2-pyridinyl)-1 /-/-indole-
5-carboxylic acid;
3-{4-[6-(1 -methylethyl)-3,4-dihydro-2(1 H)-isoquinolinyl]butyl}-1 -phenyl-1 H-indole-5- carboxylic acid;
1-phenyl-3-[4-({[4-(trifluoromethyl)phenyl]carbonyl}amino)butyl]-1 /-/-indole-5-carboxylic acid;
2-methyl-3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-piperidinyl]methyl}-1 -phenyl-
1 H-indole- 5-carboxylic acid;
3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-piperidinyl]methyl}-1-(2-pyridinyl)-1 /-/- indole- 5-carboxylic acid; 1-(4-fluorophenyl)-3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-piperidinyl]methyl}-
1 H-indole- 5-carboxylic acid;
3-({methyl[2-({[4-(1-methylethyl)phenyl]carbonyl}amino)ethyl]amino}methyl)-1 -phenyl-1 H- indole- 5-carboxylic acid;
3-{[[2-({[2-hydroxy-3-(1-methylethyl)phenyl]carbonyl}amino)ethyl](methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[methyl(1-{[4-(1-methylethyl)phenyl]carbonyl}-3-piperidinyl)amino]methyl}-1 -phenyl-1 H- indole- 5-carboxylic acid;
3-{[[1 ,1-dimethyl-2-({[4-(1-methylethyl)phenyl]carbonyl}amino)ethyl](methyl)amino]methyl}-
1 -phenyl-1 H-indole- 5-carboxylic acid; 3-{[methyl({4-methyl-2-[4-(trifluoromethyl)phenyl]-1 ,3-thiazol-5-yl}methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[methyl({4-methyl-2-[4-(1-methylethyl)phenyl]-1 ,3-thiazol-5-yl}methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[(1-{[4-(1 ,1-dimethylethyl)phenyl]carbonyl}-3-piperidinyl)(methyl)amino]methyl}-1- phenyl-1 H-indole- 5-carboxylic acid;
3-{[({2-[4-(1 ,1-dimethylethyl)phenyl]-4-methyl-1 ,3-thiazol-5- yl}methyl)(methyl)amino]methyl}-1 -phenyl-1 H-indole-5-carboxylic acid;
3-({[2-({[4-(1-methylethyl)phenyl]carbonyl}amino)ethyl]amino}methyl)-1 -phenyl-1 H-indole-
5-carboxylic acid; 3-{[3-({[4-(1-methylethyl)phenyl]carbonyl}amino)-1-pyrrolidinyl]methyl}-1 -phenyl-1 H-indole- 5-carboxylic acid; 3-((1 £)-5-{[4-(1-methylethyl)phenyl]amino}-5-oxo-1-penten-1-yl)-1-phenyl-1 /-/-indole-5- carboxylic acid;
3-{[(3-{[4-(1-methylethyl)phenyl]amino}-3-oxopropyl)amino]carbonyl}-1-phenyl-1 /-/-indole-
5-carboxylic acid; 3-(5-{[4-(1-methylethyl)phenyl]amino}-5-oxopentyl)-1-phenyl-1 /-/-indole-5-carboxylic acid;
3-{2-[methyl(2-{[4-(1-methylethyl)phenyl]amino}-2-oxoethyl)amino]ethyl}-1-(2-pyridinyl)-
1H-indole- 5-carboxylic acid;
3-{2-[methyl(2-{[4-(1 -methylethyl)phenyl]amino}-2-oxoethyl)amino]ethyl}-1 -phenyl-1 H- indole- 5-carboxylic acid; 6-methyl-3-{[(1-{[4-(1-methylethyl)phenyl]carbonyl}-3-piperidinyl)amino]methyl}-1 -phenyl-
1H-indole- 5-carboxylic acid;
3-[3-({2-[4-(1-methylethyl)phenyl]ethyl}amino)-3-oxopropyl]-1 -phenyl-1 H-indole-5- carboxylic acid;
3-[(4-{[4-(1-methylethyl)phenyl]carbonyl}-1-piperazinyl)methyl]-1 -phenyl-1 H-indole-5- carboxylic acid;
3-{[3-{[4-(1-methylethyl)phenyl]acetyl}tetrahydro-1 (2H)-pyrimidinyl]methyl}-1 -phenyl-1 H- indole- 5-carboxylic acid;
and salts and solvates thereof.
13. A method of treatment and/or prophylaxis of a hPPAR mediated disease or a condition, which comprises administration of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof according to any of claims 1 to 12.
14. A compound or a pharmaceutically acceptable salt or solvate thereof, according to claims 1 to 12 for use in therapy.
15. A compound or a pharmaceutically acceptable salt or solvate thereof, according to claims 1 to 12 for use in the treatment or prophylaxis of a hPPAR mediated disease or condition.
16. The use of a compound according to claims 1 to 12, or a pharmaceutically acceptable salt or solvate thereof, in the manufacture of a medicament for the treatment or prophylaxis of a hPPAR mediated disease or condition.
17. A pharmaceutical composition which comprises a compound according to claims 1 to 12 or a pharmaceutically acceptable salt or solvate thereof optionally with a pharmaceutically acceptable carrier or excipient.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US97858407P | 2007-10-09 | 2007-10-09 | |
| US60/978,584 | 2007-10-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009047240A1 true WO2009047240A1 (en) | 2009-04-16 |
Family
ID=40227876
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/063371 WO2009047240A1 (en) | 2007-10-09 | 2008-10-07 | Indole derivatives useful as ppar activators |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009047240A1 (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009147121A1 (en) * | 2008-06-02 | 2009-12-10 | Smithkline Beecham Corporation | Carboxyl substituted indoles for use as ppar alpha modulators |
| WO2011037223A1 (en) | 2009-09-28 | 2011-03-31 | 興和株式会社 | Agent for reducing visceral fat weight |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| CN102690211A (en) * | 2012-06-05 | 2012-09-26 | 郑州明泽医药科技有限公司 | Method for preparing tolvaptan intermediate |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8507499B2 (en) | 2010-12-06 | 2013-08-13 | Confluence Life Sciences, Inc. | Substituted indole/indazole-pyrimidinyl compounds |
| WO2014050134A1 (en) | 2012-09-27 | 2014-04-03 | 興和株式会社 | Therapeutic agent for dyslipidemia |
| WO2015030033A1 (en) | 2013-08-28 | 2015-03-05 | 興和株式会社 | Therapeutic agent for dyslipidemia |
| WO2015104602A2 (en) | 2014-01-08 | 2015-07-16 | Wockhardt Limited | A process for the preparation of anagliptin and its intermediates thereof |
| WO2015150887A1 (en) | 2014-03-29 | 2015-10-08 | Wockhardt Limited | Process for the preparation of anagliptin or its salts |
| CN109053801A (en) * | 2018-08-17 | 2018-12-21 | 中国农业大学 | A kind of triphenyl phosphonium salt compound and the preparation method and application thereof |
| WO2020034181A1 (en) * | 2018-08-17 | 2020-02-20 | 中国农业大学 | Quaternary triphenylphosphonium salt compound, preparation method therefor, and uses thereof |
| EP3463362A4 (en) * | 2016-05-25 | 2020-04-22 | Merck Sharp & Dohme Corp. | Substituted tetrahydroisoquinoline compounds useful as gpr120 agonists |
| CN113692276A (en) * | 2019-02-19 | 2021-11-23 | 加利福尼亚大学董事会 | NURR1 receptor modulators |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002030895A1 (en) * | 2000-10-10 | 2002-04-18 | Smithkline Beecham Corporation | SUBSTITUTED INDOLES, PHARMACEUTICAL COMPOSITIONS CONTAINING SUCH INDOLES AND THEIR USE AS PPAR-η BINDING AGENTS |
-
2008
- 2008-10-07 WO PCT/EP2008/063371 patent/WO2009047240A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002030895A1 (en) * | 2000-10-10 | 2002-04-18 | Smithkline Beecham Corporation | SUBSTITUTED INDOLES, PHARMACEUTICAL COMPOSITIONS CONTAINING SUCH INDOLES AND THEIR USE AS PPAR-η BINDING AGENTS |
Non-Patent Citations (1)
| Title |
|---|
| SZEWCZYK, JASON W. ET AL: "SAR studies: Designing potent and selective LXR agonists", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 16(11), 3055-3060 CODEN: BMCLE8; ISSN: 0960-894X, 2006, XP002510895 * |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009147121A1 (en) * | 2008-06-02 | 2009-12-10 | Smithkline Beecham Corporation | Carboxyl substituted indoles for use as ppar alpha modulators |
| WO2011037223A1 (en) | 2009-09-28 | 2011-03-31 | 興和株式会社 | Agent for reducing visceral fat weight |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8507499B2 (en) | 2010-12-06 | 2013-08-13 | Confluence Life Sciences, Inc. | Substituted indole/indazole-pyrimidinyl compounds |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| CN102690211A (en) * | 2012-06-05 | 2012-09-26 | 郑州明泽医药科技有限公司 | Method for preparing tolvaptan intermediate |
| WO2014050134A1 (en) | 2012-09-27 | 2014-04-03 | 興和株式会社 | Therapeutic agent for dyslipidemia |
| KR20150063035A (en) | 2012-09-27 | 2015-06-08 | 교와 가부시키가이샤 | Therapeutic agent for dyslipidemia |
| US11013722B2 (en) | 2012-09-27 | 2021-05-25 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US9572798B2 (en) | 2012-09-27 | 2017-02-21 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US9931321B2 (en) | 2012-09-27 | 2018-04-03 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US10258609B2 (en) | 2012-09-27 | 2019-04-16 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| WO2015030033A1 (en) | 2013-08-28 | 2015-03-05 | 興和株式会社 | Therapeutic agent for dyslipidemia |
| KR20230050472A (en) | 2013-08-28 | 2023-04-14 | 교와 가부시키가이샤 | Therapeutic agent for dyslipidemia |
| EP3939586A1 (en) | 2013-08-28 | 2022-01-19 | Kowa Company, Ltd. | Combination of pemafibrate and omega-3 fatty acids for dyslipedmia |
| KR20210107168A (en) | 2013-08-28 | 2021-08-31 | 교와 가부시키가이샤 | Therapeutic agent for dyslipidemia |
| WO2015104602A2 (en) | 2014-01-08 | 2015-07-16 | Wockhardt Limited | A process for the preparation of anagliptin and its intermediates thereof |
| WO2015104602A3 (en) * | 2014-01-08 | 2015-12-10 | Wockhardt Limited | A process for the preparation of anagliptin and its intermediates thereof |
| WO2015150887A1 (en) | 2014-03-29 | 2015-10-08 | Wockhardt Limited | Process for the preparation of anagliptin or its salts |
| EP3463362A4 (en) * | 2016-05-25 | 2020-04-22 | Merck Sharp & Dohme Corp. | Substituted tetrahydroisoquinoline compounds useful as gpr120 agonists |
| US11161819B2 (en) | 2016-05-25 | 2021-11-02 | Merck Sharp & Dohme Corp. | Substituted tetrahydroisoquinoline compounds useful as GPR120 agonists |
| WO2020034181A1 (en) * | 2018-08-17 | 2020-02-20 | 中国农业大学 | Quaternary triphenylphosphonium salt compound, preparation method therefor, and uses thereof |
| CN109053801A (en) * | 2018-08-17 | 2018-12-21 | 中国农业大学 | A kind of triphenyl phosphonium salt compound and the preparation method and application thereof |
| AU2018436544B2 (en) * | 2018-08-17 | 2022-09-08 | China Agricultural University | Quaternary triphenylphosphonium salt compound, preparation method therefor, and uses thereof |
| CN113692276A (en) * | 2019-02-19 | 2021-11-23 | 加利福尼亚大学董事会 | NURR1 receptor modulators |
| EP3927330A4 (en) * | 2019-02-19 | 2022-11-30 | The Regents of the University of California | Nurr1 receptor modulators |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009047240A1 (en) | Indole derivatives useful as ppar activators | |
| AU758758B2 (en) | Substituted oxazoles and thiazoles derivatives as HPPAR alpha activators | |
| JP3507494B2 (en) | Tachykinin antagonist | |
| AU728812B2 (en) | Thiazole benzenesulfonamides as beta3 agonists for the treatment of diabetes and obesity | |
| TWI404712B (en) | Benzoazepin-oxy-acetic acid derivatives as ppar-delta agonists used for the increase of hdl-c, lower ldl-c and lower cholesterol | |
| ES2402805T3 (en) | Aromatic carboxamide and urea derivatives substituted as vanilloid receptor ligands | |
| US20040133008A1 (en) | Amide compounds | |
| JP3785397B2 (en) | 5-membered ring compounds | |
| JP2000516593A (en) | Substituted sulfonamides as selective β agonists used in the treatment of diabetes and obesity | |
| WO2009147121A1 (en) | Carboxyl substituted indoles for use as ppar alpha modulators | |
| JP2007511485A (en) | Substituted pyrazoles as PPAR agonists | |
| WO2001002349A1 (en) | Phenyl sulfamate derivatives | |
| US20040147571A1 (en) | Substituted oxazoles and thiazoles as hppar alpha agonists | |
| KR20140040104A (en) | Azetidine derivatives useful for the treatment of metabolic and inflammatory diseases | |
| WO2013085890A1 (en) | Therapeutic methods | |
| AU701560B2 (en) | 1-aryl-2-acylamino-ethane compounds and their use as neurokinin especially neurokinin 1 antagonists | |
| SK13292000A3 (en) | Vitronectin receptor antagonists | |
| WO2004000785A2 (en) | Phenyloxyalkanonic acid derivatives as hppar activators | |
| JP4986927B2 (en) | Medicine | |
| CN100491355C (en) | Cyclic diamine compound having five-membered cyclic group | |
| CA2320120C (en) | Benzisoxazole derivatives having d4-antagonistic activity | |
| ES2296965T3 (en) | DERIVATIVES OF OXAZOL / TIAZOL AS ACTIVATORS OF THE PPARH-ALFA RECEPTOR. | |
| KR100248643B1 (en) | Aryl and heteroaryl alkoxynaphthalene derivatives | |
| US7141591B2 (en) | 1,2,4-oxadiazole derivatives as hPPAR alpha agonists | |
| JP2003192591A (en) | Medicine composed of 5-membered ring compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08837247 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08837247 Country of ref document: EP Kind code of ref document: A1 |