WO2009026658A1 - Ppar agonists - Google Patents
Ppar agonists Download PDFInfo
- Publication number
- WO2009026658A1 WO2009026658A1 PCT/AU2008/001292 AU2008001292W WO2009026658A1 WO 2009026658 A1 WO2009026658 A1 WO 2009026658A1 AU 2008001292 W AU2008001292 W AU 2008001292W WO 2009026658 A1 WO2009026658 A1 WO 2009026658A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- hydrogen
- methyl
- compound
- ppar
- Prior art date
Links
- 0 CC(c(c(*)c1)ccc1O)=O Chemical compound CC(c(c(*)c1)ccc1O)=O 0.000 description 4
- YASAKCUCGLMORW-UHFFFAOYSA-N CN(CCOc1ccc(CC(C(N2)=O)SC2=O)cc1)c1ncccc1 Chemical compound CN(CCOc1ccc(CC(C(N2)=O)SC2=O)cc1)c1ncccc1 YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 1
- MQRZSVGUMJCOKS-PMERELPUSA-N Cc1c(CCOc2cc([C@@H](C(O)=O)Nc(cccc3)c3C(c3ccccc3)=O)ccc2)nc(-c2ccccc2)[o]1 Chemical compound Cc1c(CCOc2cc([C@@H](C(O)=O)Nc(cccc3)c3C(c3ccccc3)=O)ccc2)nc(-c2ccccc2)[o]1 MQRZSVGUMJCOKS-PMERELPUSA-N 0.000 description 1
- HUMNYLRZRPPJDN-UHFFFAOYSA-N O=Cc1ccccc1 Chemical compound O=Cc1ccccc1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 1
- GHMLBKRAJCXXBS-UHFFFAOYSA-N Oc1cc(O)ccc1 Chemical compound Oc1cc(O)ccc1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/76—Ketones containing a keto group bound to a six-membered aromatic ring
- C07C49/84—Ketones containing a keto group bound to a six-membered aromatic ring containing ether groups, groups, groups, or groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/02—Non-specific cardiovascular stimulants, e.g. drugs for syncope, antihypotensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/46—Friedel-Crafts reactions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/72—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction of compounds containing >C = O groups with the same or other compounds containing >C = O groups
- C07C45/74—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction of compounds containing >C = O groups with the same or other compounds containing >C = O groups combined with dehydration
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/76—Ketones containing a keto group bound to a six-membered aromatic ring
- C07C49/82—Ketones containing a keto group bound to a six-membered aromatic ring containing hydroxy groups
- C07C49/835—Ketones containing a keto group bound to a six-membered aromatic ring containing hydroxy groups having unsaturation outside an aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/22—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4
- C07D311/26—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3
- C07D311/34—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3 with aromatic rings attached in position 3 only
- C07D311/36—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3 with aromatic rings attached in position 3 only not hydrogenated in the hetero ring, e.g. isoflavones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/54—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to PPAR agonists and their use in therapy, including the treatment of disease.
- PPARs peroxisome proliferator-activated receptors
- PPAR- ⁇ The peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors that belong to the nuclear hormone superfamily.
- PPAR- ⁇ three isoforms of PPAR have been identified: PPAR- ⁇ , - ⁇ and - ⁇ .
- PP AR- ⁇ is the most abundant receptor expressed in adipocytes and macrophages, where, apart from its involvement in adipocyte differentiation and lipid storage, it serves as the primary receptor modulating insulin sensitization and maintaining lipid and glucose homeostasis.
- PPAR- ⁇ is the target of numerous drug discovery efforts because of its role in numerous disease states, including Type II diabetes.
- the thiazolidinediones (TZDs; or glitazones) and the L-tyrosine analogues are anti-diabetic synthetic agonists that selectively target PPAR- ⁇ .
- Their mode of action begins with sensitizing tissue to insulin, lowering glucose levels and reducing serum lipids in diabetic patients by potently binding, and subsequently activating, PPAR- ⁇ .
- Rosiglitazone (Avandia®), shown below, is a prototypical TZD and serves as a reference compound for this class, which also includes pioglitazone (Actos®) and troglitazone. Rosiglitazone is active in vivo as an anti-diabetic agent in the ob/ob mouse model and is presently being used as an oral hypoglycaemic agent for the treatment of Type II diabetes.
- the L-tyrosine analogue class of compounds such as Farglitazar (GI262570) shown below, represent the most potent and selective class of synthetic PPAR- ⁇ agonists currently in existence.
- PPAR- ⁇ receptor mainly expressed in the liver, play an important role in fatty acid oxidation and lipoprotein metabolism.
- Fibrates Feenofibrate, Clofibrate
- WY- 14643 shown below, show effects such as lowering triglycerides and elevating HDL levels through activation of PPAR- ⁇ .
- the majority of type II diabetes patients suffer from atherogenic lipid abnormalities in addition to insulin resistance, termed as metabolic syndrome.
- metabolic syndrome The importance of controlling both glucose and lipid levels in metabolic syndrome, gave rise to the concept of identifying dual agonists, which can activate both PPAR- ⁇ and PPAR- ⁇ .
- PPAR- ⁇ agonists reduce body weight gain which led to a hypothesis that activation of PPAR- ⁇ may mitigate the weight gain induced by PPAR- ⁇ activation in humans.
- PPAR- ⁇ and PPAR- ⁇ agonists have been implicated in the pathology of various disorders including atherosclerosis, coronary heart disease, obesity and inflammation.
- Compounds that are dual PPAR- ⁇ and PPAR- ⁇ agonists can have fewer therapeutic side- effects than those that act solely at the PPAR- ⁇ receptor or those that act solely at the
- the present invention relates to compounds having PPAR agonist activity, and the therapeutic use thereof.
- the PPAR agonist is a PPAR- ⁇ agonist.
- the PPAR agonist is a PPAR- ⁇ agonist.
- the PPAR agonist is a dual PPAR ⁇ / ⁇ agonist.
- the present invention provides compounds of general formula (3):
- A is a C 5-6 heteroaryl ring optionally substituted with one or more substituents independently selected from halogen, C 1-4 alkyl, O-C 1-4 alkyl, haloCi -4 alkyl, hydroxy- C M alkyl and CO 2 R;
- R a -R J are each independently selected from hydrogen, hydroxyl, halogen, C 1-4 alkyl,
- each R is independently selected from hydrogen and C 1-4 alkyl; and pharmaceutically acceptable salts thereof.
- L is selected from Ci -4 alkylene and C 2-4 alkenylene
- R Q -R U are each independently selected from hydrogen, hydroxyl, halogen, C M alkyl, haloC 1-4 alkyl, hydroxyC 1-4 alkyl, O-C M alkyl, O-C M alkyl-CO 2 R, O-C 3-6 cycloalkyl, O-C 3-6 heteroaryl, N(R) 2 C 1-4 alkyl, N(R) 3 C 1-4 alkyl, O-Ci -4 alkyl-N(R) 2 , O-C 1-4 alkyl-N(R) 3> C 6-10 aryl, O-C 6 -i 0 aryl, O-C M alkyl-C ⁇ oheterocycloalkyl, O-C M alkyl- C 6-10 heteroaryl, CO 2 R, O-Q-eheterocycloalkyl, O-C 1-4 alkyl-C 6- i 0 aryl, 0-C(O)-C M alkyl, 0-C(O)
- R Q and R R , R R and R s , R s and R ⁇ and R ⁇ and R u together form
- R v -R z are each independently selected from hydrogen, hydroxyl, halogen, C 1-4 alkyl, haloC 1-4 alkyl, hydroxyCi -4 alkyl, O-C 1-4 alkyl, O-C M alkyl-CO 2 R, O-C ⁇ cycloalkyl, O-C 3-6 heteroaryl, N(R) 2 C 1-4 alkyl, N(R) 3 Ci -4 alkyl, O-Ci -4 alky 1-N(R) 2 ,
- A is a heteroaryl ring selected from imidazolyl, pyrazolyl, oxazolyl, and isoxazolyl, wherein the heteroaryl ring may be substituted with one or more substituents independently selected from methyl, ethyl, CO 2 R and CF 3 ;
- R a is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi -2 alkyl, O-methyl and 2s CO 2 R;
- R b , R c , R d , R e , R f , R 1 , and R j are each independently selected from hydrogen, hydroxyl, C 1-2 alkyl, O-C 1-2 alkyl, CO 2 R, and O-benzyl;
- R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
- the invention provides compounds of general formula (4a):
- L is -C(O)Ci. 3 alky-ene-, -C(O)C 2-3 alkenylene-, -Ci -3 alkyleneC(O)- and s -C 2-3 alkenyleneC(O)-;
- R z is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi -2 alkyl, O-methyl and CO 2 R;
- R Q , R R , R u , R v , R w , R x , and R ⁇ are each independently selected from hydrogen, hydroxyl, C 1-2 alkyl, O-C 1-2 alkyl, CO 2 R and O-benzyl; o R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
- the present invention provides a pharmaceutical composition comprising one or more compounds of formula (3), (4), (3a) or (4a), or a prodrug thereof, together with a pharmaceutically acceptable adjuvant, diluent or carrier.
- the present invention provides a method of treating or preventing a disease in a vertebrate, the method comprising administering to a vertebrate in need thereof an effective amount of a compound of formula (3), (4), (3a), or (4a) as defined herein, or a prodrug thereof, or a composition according to the fifth aspect of the invention, wherein the disease is selected from Type II diabetes, obesity, hyperlipidemia,0 cardiovascular disease, anti-neoplastic diseases and tumours, inflammatory conditions and neurodegenerative diseases.
- the present invention relates to the use of a compound of formula (3), (4), (3 a), or (4a) as defined herein, or a prodrug thereof, in the manufacture of a medicament for treating a disease selected from Type II diabetes, obesity, 5 hyperlipidemia, cardiovascular disease, anti-neoplastic diseases and tumours, inflammatory conditions and neurodegenerative diseases.
- the invention relates to a method for identifying a PPAR agonist, the method comprising determining ligand-receptor interactions of a candidate compound with docking0 templates; comparing the ligand-receptor interactions of the candidate compound with the interactions of a known PPAR agonist; and thereby determining whether a candidate compound is a PPAR agonist.
- the PPAR agonist is a PPAR- ⁇ agonist.
- the PPAR agonist is a PPAR- ⁇ agonist.
- the PPAR agonist is a dual PPAR ⁇ / ⁇ agonist.
- the method of the invention may be used to identify PPAR agonists that share little similarity with known ligands.
- the method may further comprise testing a compound identified as a PPAR agonist I 0 in vitro for PPAR activation efficacy using either a transcriptional factor or a reporter gene luciferase assay.
- PPAR peroxisome proliferator-activated receptor
- LBD LBD
- C 1-4 alkyl group includes within its meaning monovalent
- alkyl and divalent (“alkylene”) straight chain or branched chain saturated aliphatic groups having from 1 to 4 carbon atoms.
- the alkyl group may be C 1-3 alkyl or Cj -2 alkyl.
- Ci -4 alkyl includes, but is not limited to, methyl, ethyl, 1- propyl, isopropyl, 1 -butyl, 2-butyl, isobutyl, tert-butyl, and the like.
- C 2-4 alkenyl group includes within its meaning monovalent (“alkenyl”) and divalent (“alkenylene”) straight or branched chain unsaturated aliphatic hydrocarbon groups having from 2 to 4 carbon atoms and at least one double bond anywhere in the chain.
- the alkenyl group may be C 2-3 alkenyl. Unless indicated otherwise, the stereochemistry about each double bond may independently be cis, trans, E or Z as
- C 2-4 alkenyl groups include but are not limited to ethenyl, vinyl, allyl, 1-methylvinyl, 1-propenyl, 2-propenyl, 2-methyl-l-propenyl, 2-methyl-l-propenyl, 1-butenyl, 2-butenyl, 3-butentyl, 1,3-butadienyl, and the like.
- amino refers to groups of the form -NR a R b wherein R a and R b are individually selected from hydrogen, optionally substituted (Ci -4 )alkyl,
- the amino group may be a primary, secondary or tertiary amino group.
- amino acid as used herein includes naturally and non-naturally occurring amino acids, as well as substituted variants thereof. Thus, (L) and (D) forms of amino acids are included in the scope of the term “amino acid”.
- amino acid includes within its scope glycine, alanine, valine, leucine, isoleucine, methionine, proline, phenylalanine, tryptophan, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartate, lysine, arginine, and histidine.
- the backbone of the amino acid residue may be substituted with one or more groups independently selected from (C ⁇ alkyl, halogen, hydroxy, hydroxy(C 1-6 )alkyl, aryl (e.g, phenyl), aryl(Ci- 3 )alkyl (e.g, benzyl), and (C 3-6 )cycloalkyl.
- arylalkyl or variants such as “arylalkyl” as used herein, includes within its meaning monovalent (“aryl”) and divalent (“arylene”), single, polynuclear, conjugated and fused aromatic hydrocarbon radicals attached to divalent, saturated, straight or branched chain alkylene radicals.
- C 6-10 aromatic group or variants such as “C 6- I 0 aryl” or “C 6- I 0 arylene” as used herein refers to monovalent (“aryl”) and divalent (“arylene”) single, polynuclear, conjugated and fused residues of aromatic hydrocarbons having from 6 to 10 carbon atoms.
- aromatic groups include phenyl, naphthyl, phenanthrenyl, and the like.
- C 3-6 cycloalkyl refers to cyclic saturated aliphatic groups and includes within its meaning monovalent (“cycloalkyl”), and divalent (“cycloalkylene”), saturated, monocyclic, bicyclic, polycyclic or fused polycyclic hydrocarbon radicals having from 3 to 6 carbon atoms.
- the cycloalkyl group may be C 3-5 cycloalkyl. Examples of cycloalkyl groups include but are not limited to cyclopropyl, 2- methylcyclopropyl, cyclobutyl, cyclopentyl, 2-methylcyclopentyl, 3-methylcyclopentyl, cyclohexyl, and the like.
- C 3-6 cycloalkenyl refers to cyclic unsaturated aliphatic groups and includes within its meaning monovalent (“cycloalkenyl”) and divalent (“cycloalkenylene”), monocyclic, bicyclic, polycyclic or fused polycyclic hydrocarbon radicals having from 3 to 6 carbon atoms and having at least one double bond anywhere in the alkyl chain.
- the cycloalkenyl group may be C 3-5 cycloalkenyl. Unless indicated otherwise, the stereochemistry about each double bond may be independently cis, trans, E or Z as appropriate.
- C 3-6 heterocycloalkyl includes within its meaning monovalent (“heterocycloalkyl”) and divalent (“heterocycloalkylene”), saturated, monocyclic, bicyclic, polycyclic or fused hydrocarbon radicals having from 3 to 6 ring atoms, wherein from 1 to 3, ring atoms are heteroatoms independently selected from O, N, NH, or S.
- the heterocycloalkyl group may be C 3-5 heterocycloalkyl.
- heterocycloalkyl groups include aziridinyl, pyrrolidinyl, piperidinyl, piperazinyl, quinuclidinyl, azetidinyl, morpholinyl, tetrahydrothiophenyl, tetrahydrofuranyl, tetrahydropyranyl, and the like.
- C 5-10 heteroaromatic group and variants such as “heteroaryl” or “heteroarylene” as used herein, includes within its meaning monovalent (“heteroaryl”) and divalent (“heteroarylene”), single, polynuclear, conjugated and fused aromatic radicals having from 5 to 10 atoms, wherein 1 to 4, or 1 to 2 ring atoms are heteroatoms independently selected from O, N, NH and S.
- the heteroaromatic group may be C 5-8 heteroaromatic.
- heteroaromatic groups include pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, 2,2'-bipyridyl, phenanthrolinyl, quinolinyl, isoquinolinyl, imidazolinyl, thiazolinyl, pyrrolyl, furanyl, thiophenyl, oxazolyl, isoxazolyl, isothiazolyl, triazolyl, and the like.
- halogen or variants such as “halide” or “halo” as used herein refers to fluorine, chlorine, bromine and iodine.
- heteroatom or variants such as “hetero-” as used herein refers to O, N, and S or the group NH.
- optionally substituted means the group to which this term refers may be unsubstituted, or may be substituted with one or more groups independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, halo, haloalkyl, haloalkynyl, hydroxyl, hydroxyalkyl, alkoxy, thioalkoxy, alkenyloxy, haloalkoxy, haloalkenyloxy, NO 2 , NR a R , nitroalkyl, nitroalkenyl, nitroalkynyl, nitroheterocyclyl, alkylamino, dialkylamino, alkenylamine, alkynylamino, acyl, alkenoyl, alkynoyl, acylamino, diacylamino, acyloxy, alkylsulfonyloxy,
- Preferred substituents include C 1-3 alkyl, Ci -3 alkoxy, -CH 2 -(C 1-3 )alkoxy, C 6-10 aryl, -CH 2 -phenyl, halo, hydroxyl, hydroxy-(C 1-3 )alkyl, and halo-(C 1-3 )alkyl, e.g, CF 3 , CH 2 CF 3 .
- administering and variations of that term including "administer” and “administration”, includes contacting, applying, delivering or providing a compound or composition of the invention to an organism, or a surface by any appropriate means.
- vertebrate includes humans and individuals of any species of social, economic or research importance including but not limited to members of the genus ovine, bovine, equine, porcine, feline, canine, primates (including human and non-human primates), rodents, murine, caprine, leporine, and avian.
- the vertebrate may be a human.
- treatment refers to any and all uses which remedy a disease state or symptoms, prevent the establishment of disease, or otherwise prevent, hinder, retard, or reverse the progression of disease or other undesirable symptoms in any way whatsoever.
- terapéuticaally effective amount and “diagnostically effective amount”, include within their meaning a sufficient but non-toxic amount of a compound or composition of the invention to provide the desired therapeutic or diagnostic effect.
- the exact amount required will vary from subject to subject depending on factors such as the species being treated, the age and general condition of the subject, the severity of the condition being treated, the particular agent being administered, the mode of administration, and so forth. Thus, it is not possible to specify an exact “effective amount”. However, for any given case, an appropriate "effective amount” may be determined by one of ordinary skill in the art using only routine experimentation.
- Figure 1 a) X-ray crystallographic pose of the known PPAR-alpha agonist AZ-242. b) Induced-fit docking pose of ( ⁇ indicating the similarity of the binding pose in this compound to the known structure AZ-242. Both ligands establish H-bond interactions to the PPAR-alpha receptor.
- Figure 2 PPAR- ⁇ reporter gene activity of compounds 4, 8 and 11 in HEK 293 cell line. The HEK 293 cells were transiently transfected with tK-PPREx3-Luc, pSG5-hPPAR- ⁇ and pSV- ⁇ -galactosidase control plasmid. Cells were treated with test compounds (5 ⁇ M and 25 ⁇ M).
- Rosiglitazone (25 ⁇ M) and GWl 929 (25 ⁇ M) were used as positive controls and DMSO (0.1%) as a negative control.
- DMSO 0.1%)
- the cells were lysed and assayed for luciferase and ⁇ -galactosidase activities. The results are expressed as relative luciferase activity (fold difference compared to negative control).
- the present invention is directed to compounds which are agonists of the PPAR receptors.
- the invention relates to compounds which are PPAR- ⁇ agonists.
- the present invention also relates to compounds that are agonists of the PPAR- ⁇ receptor.
- the present invention is further directed to compounds which are dual agonists of the PPAR- ⁇ and PPAR- ⁇ receptors.
- Compounds according to the present invention may be useful in therapy, including for example, the treatment of Type II diabetes, obesity, hyperlipidemia, cardiovascular disease (e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease), anti-neoplastic diseases and tumours (e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer), inflammatory conditions (e.g, inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma, rheumatoid arthritis), and neurodegenerative diseases (e.g, Parkinson's disease, Alzheimer's disease).
- cardiovascular disease e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease
- anti-neoplastic diseases and tumours e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer
- inflammatory conditions e.g, inflammatory bowel diseases, psoriasis,
- A is a C 5-6 heteroaryl ring optionally substituted with one or more substituents independently selected from halogen, d. 4 alkyl, O-Ci -4 alkyl, haloCi. 4 alkyl, hydroxy- Ci -4 alkyl and CO 2 R;
- R a -R j are each independently selected from hydrogen, hydroxyl, halogen, Ci. 4 alkyl, C 3-6 cycloalkyl, haloC 1-4 alkyl, hydroxyC]. 4 alkyl, O-C] -4 alkyl, O-C 3 .
- each R is independently selected from hydrogen and C 1-4 alkyl; and pharmaceutically acceptable salts thereof.
- A is selected from pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, pyrazolyl, imidazolyl, thiazolyl, pyrrolyl, furanyl, thiophenyl, oxazolyl, isoxazolyl, isothiazolyl, and triazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci -3 alkyl, 0-C 1-3 alkyl, haloC 1-3 alkyl, hydroxyC 1-3 alkyl and CO 2 R.
- A is selected from pyrazolyl, imidazolyl, thiazolyl, pyrrolyl, furanyl, thiophenyl, oxazolyl, isoxazolyl, isothiazolyl, and triazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci -3 alkyl, 0-C 1-3 alkyl, haloC 1-3 alkyl, hydroxyC 1-3 alkyl and CO 2 R.
- A is selected from pyrazolyl, imidazolyl, pyrrolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci -3 alkyl, 0-Ci -3 alkyl, haloC 1-3 alkyl, hydroxyCi- 3 alkyl and CO 2 R.
- A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci -4 alkyl, O-Ci -4 alkyl, haloCi -4 alkyl, hydroxyCi -4 alkyl and CO 2 R.
- A is selected from pyrazolyl and isoxazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from Ci -4 alykl, haloCi -4 alkyl and COOH.
- A is pyrazolyl optionally substituted with methyl or CF 3 .
- each optional substituent on the heteroaryl ring may be independently selected from methyl, ethyl, O-methyl, O-ethyl, fluorine, chlorine, halomethyl, hydroxymethyl, CO 2 H, CO 2 Me and CO 2 Et.
- each optional substituent is selected from methyl, ethyl, O-methyl, fluorine, hydroxymethyl, CH 2 F, CHF 2 , CF 3 and CO 2 H.
- each optional substituent is selected from methyl, fluorine, CF 3 and CO 2 H.
- each optional substituent is selected from methyl, CF 3 and CO 2 H.
- each R is independently selected from hydrogen, methyl and ethyl.
- R a -R j are each independently selected from hydrogen, hydroxyl, halogen, Ci -4 alkyl, haloC] -4 alkyl, hydroxyC 1-4 alkyl, and O-C 1-4 alkyl.
- R a -R j are each independently selected from hydrogen, hydroxyl, methyl, ethyl, CF 3 , CH 2 OH, O-methyl and O-ethyl.
- R a -R j are each independently selected from hydrogen, hydroxyl, methyl, ethyl, and CF 3 .
- R a -R j are each independently selected from hydrogen, hydroxyl, O-methyl, fluorine, methyl and ethyl. In a further embodiment, R a -R J are each independently selected from hydrogen, hydroxyl, O-methyl and fluorine.
- R 8 and R h together form ° * .
- A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with methyl, fluorine, CF 3 or CO 2 H; and R g and R h together form 0 ⁇ .
- A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with methyl, fluorine, CF 3 or CO 2 H; R a and R c are each hydroxyl; and R 8 and
- A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with methyl, fluorine, CF 3 or CO 2 H; R a and R c are each hydroxyl; R b R d and
- R e are each independently selected from hydrogen, C ⁇ -2 alkyl and CF 3 , and R 8 and R h
- A is pyrazolyl optionally substituted with methyl or CF 3 ;
- R a and R c are each hydroxyl;
- R h is fluorine or O-methyl
- L is selected from C 1-4 alkylene and C 2-4 alkenylene;
- R Q -R U are each independently selected from hydrogen, hydroxyl, halogen, Ci. 4 alkyl, haloC 1-4 alkyl, hydroxyCi -4 alkyl, O-C 1-4 alkyl, O-C M alkyl-CO 2 R, O-C ⁇ cycloalkyl, O-C 3-6 heteroaryl, N(R) 2 C 1-4 alkyl, N(R) 3 C 1-4 alkyl, O-C 1-4 alkyl-N(R) 2 , O-C 1-4 alkyl-N(R) 3> C 6- i 0 aryl, O-C 6- i 0 aryl, O-C M alkyl-Q-ioheterocycloalkyl, O-C M alkyl- C 6-10 heteroaryl, CO 2 R, O-C 3-6 heterocycloalkyl, O-C 1-4 alkyl-C 6-10 aryl,
- R v -R z are each independently selected from hydrogen, hydroxyl, halogen,
- C 6-10 heteroaryl CO 2 R, O-C 3-6 heterocycloalkyl, O-C 1-4 alkyl-C 6- i 0 aryl, O-C(O)-C t-4 alkyl,
- each R is independently selected from hydrogen and and pharmaceutically acceptable salts thereof.
- L is C 1-2 alkylene.
- R R and R s or R s
- R v -R z are each independently selected from hydrogen, hydroxyl, halogen, C ⁇ --,alkyl, haloCi -4 alkyl, hydroxyC 1-4 alkyl, O-Ci -4 alkyl, THP, O-benzyl, and CO 2 H.
- the sugar may be a monosaccharide or a disaccharide. Examples of suitable sugar moieties include but are not limited to glucose, rhamnose, arabinglucose, neohesperidose, and rutinose.
- R x and R z are each hydroxyl
- R ⁇ -R u are each independently selected from hydrogen, hydroxyl and O- methyl.
- R v is selected from hydrogen and hydroxyl and R R and R s together form ⁇ ° ° ⁇ .
- L is -CH 2 -, R" and R z are each hydrogen.
- R x and R z are each hydroxyl
- R v , R w , R Q , R ⁇ and R u are each hydrogen
- R R and R s together form
- R ⁇ is not methyl.
- R x and R z are each hydroxyl
- R v , R ⁇ , R Q , R ⁇ and R u are each hydrogen
- R R and R s together form
- R w is not ethyl.
- R x and R z are
- R x and R z are each
- R w , R ⁇ , R Q , R ⁇ and R u are each hydrogen, and R R and R s together form 0 ⁇ , then R v is not ethyl.
- R x and R z are each hydroxyl, and R u is O-methyl, then R v , R w , R ⁇ , R Q , R R and R s and R ⁇ are not each hydrogen.
- A is a heteroaryl ring selected from imidazolyl, pyrazolyl, oxazolyl, and isoxazolyl, wherein the heteroaryl ring may be substituted with one or more substituents independently selected from methyl, ethyl, CO 2 R and CF 3 ;
- R a is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi -2 alkyl, O-methyl and CO 2 R; o R b , R c , R d , R e , R f , R 1 , and R" are each independently selected from hydrogen, hydroxyl, C 1-2 alkyl, CO 2 R, O-benzyl and O-C 1-2 alkyl;
- R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
- R a and R c are hydroxyl. s
- the present invention relates to compounds of general formula
- L is -C(O)C 1-3 alkylene-, -C(O)C 2-3 alkenylene-, -C 1-3 alkyleneC(O)- and0 -C 2 . 3 alkenyleneC(O)-;
- R z is selected from hydrogen, hydroxyl, methyl, ethyl, haloC 1-2 alkyl, O-methyl and CO 2 R;
- R Q , R R , R u , R v , R w , R x , and R ⁇ are each independently selected from hydrogen, hydroxyl, C 1-2 alkyl, O-Ci -2 alkyl, CO 2 R and O-benzyl; s R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
- R x is selected from hydroxyl, methyl, ethyl, O- methyl and O-ethyl.
- the compound is selected from compounds 6, 7, 8, 11, 12, 14, 15, 16, 17, 18, 19, 20, 23, 24, 26, 27, 28, 30, 31, 32, 33 and 34 above.
- the compound is selected from compounds 1, 2, 3, 4, 5, 13, 14, 21, 22 and 35 above.
- the compound is selected from compounds 1, 2, 3, 4 and 5 above. In further embodiments the compound is compound 14 above.
- the compound is selected from compounds 6, 7, 8, 11, 12, 15, 16, 17, 18, 19, 20, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 and 34 above.
- the compound is selected from compounds 6, 7, 8, 11, 12, 15, 16, 17, 18, 19, 20, 23, 24, 26, 27, 28, 30, 31, 32, 33 and 34 above.
- the compound is selected from compounds 6, 7, 8, 11 and 12 above.
- the compound is selected from compounds 4, 8 and 11 above.
- R OMe (12) _ Reagents and Conditions: (a) BF 3 OEt 2 , 90 C for 90 min; (b) Ac 2 O, K 2 CO 3 , DMF 1
- Reagents and Conditions (a) BF 3 -OEt 2, 11O 0 C; (b) (CF 3 CO) 2 O, Pyridine; (c) i. CICOCOOEt, Pyridine; ii. NaOH (d) i. Ac 2 O, Pyridine, ii. NaOH; (e) NH 2 NH 2 -H 2 O, EtOH; (f) NH 2 OH-HCI, Pyridine.
- Compounds for use in accordance with the present invention may be PPAR agonists.
- the compound may be a PPAR- ⁇ agonist.
- the compound may be a PPAR- ⁇ agonist.
- compounds in accordance with the present invention may exhibit dual PPAR ⁇ / ⁇ agonist activity.
- 'pro-drugs' of the compounds of the invention are so-called 'pro-drugs' of the compounds of the invention.
- certain derivatives of compounds of formulae (3)-(4) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of the invention having the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as 'prodrugs'.
- Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association).
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of the invention with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
- prodrugs in accordance with the invention include: (i) where the compound contains a carboxylic acid functionality (COOH), an ester thereof, for example, a compound wherein the hydrogen of the carboxylic acid functionality of a compound of formulae (3)-(4) is replaced by (Ci-C 4 )alkyl; (ii) where the compound contains an hydroxyl functionality, an ether thereof, for example, a compound wherein the hydrogen of the alcohol functionality of a compound of formulae (3)-(4) is replaced by (Ci-C 4 )alkanoyloxymethyl, or a phosphonate ester thereof; and (iii) where the compound contains a primary or secondary amino functionality (-NH 2 or -NHR where R ⁇ H), an amide thereof, for example, a compound wherein, as the case may be, one or both hydrogens of the amino functionality of the compound is/are replaced by (Q-C ⁇ alkanoyl.
- COOH carboxylic acid functionality
- an ester thereof for example, a compound wherein the hydrogen
- Stereoisomers include all stereoisomers, geometric isomers and tautomeric forms of the compounds of formulae (3)-(4) [and (3a)-(4a)], including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof.
- the present disclosure encompasses all such compounds, including cis- isomers, trans-isomers, (E)-isomers, (Z)-isomers, (i?)-enantiomers, (S)-enantiomers and mixtures thereof including racemic mixtures.
- acid addition or base salts wherein the counterion is optically active, for example, an amino acid, e.g, J-lactate or /-lysine, etc, or racemic, for example, (//-tartrate or J/-arginine, and the like.
- Cisl trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
- Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (3) or (4) contains an acidic or basic moiety, a base or acid such as 1- phenylethylamine or tartaric acid.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound of formula (3) or (4) contains an acidic or basic moiety, a base or acid such as 1- phenylethylamine or tartaric acid.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
- Chiral compounds of the invention may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
- chromatography typically HPLC
- a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine.
- Stereoisomeric conglomerates may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by ⁇ . L. ⁇ liel and S. H. Wilen (Wiley, New York, 1994). Therapeutic applications
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising one or more compounds of formula (3), (4), (3a) or (4a), or a prodrug thereof, together with a pharmaceutically acceptable adjuvant, diluent or carrier.
- Pharmaceutical compositions comprising one or more compounds of formula (3),
- the pharmaceutical compositions of the present invention may contain one or more other active ingredients, in addition to a compound of formula (3), (4), (3a) or (4a).
- the optimal dosage of the drug/s to be administered in combination with the compound/s of the present invention can be readily determined by one of ordinary skill in the art.
- the additional drug/s may be administered simultaneously or sequentially with the compounds of the present invention. When administered simultaneously, it is preferable to use a pharmaceutical composition in unit dosage form containing the compound/s of the present invention and other drug/s. When administered in combination, either simultaneously or sequentially, the compound/s of the present invention and additional other drug/s may be used in lower doses than when each is used alone.
- Agents which improve a patient's lipid profile including PPAR alpha agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), PPAR alpha/gamma dual agonists such as KRP-297, muraglitazar, tesaglitazar, farglitazar, and JT-501, PPAR delta, nicotinyl alcohol, nicotinic acid or a salt thereof, bile acid sequestrants (cholestyramine, colestipol, and dialkylaminoalkyl derivatives of a cross- linked dextran), HMG-CoA reductase inhibitors (lovastatin, simvastatin, rosuva
- Further active ingredients that may be administered in combination with the compounds of the present invention include ileal bile acid transporter inhibitors, antiobesity compounds such as fenfluramine, dexfenfluramine, phentiramine, subitramine, orlistat, neuropeptide Y5 inhibitors, Mc4r agonists, cannabinoid receptor 1 (CB-I) antagonists/inverse agonists, and beta 3 adrenergic receptor agonists, biguanides including metformin and phenformin, protein tyrosine phosphatase-lB (PTP-IB) inhibitors, dipeptidyl peptidase IV (DP-IV) inhibitors, insulin or insulin mimetics, sulfonylureas including tolbutamide and glipizide or related materials, PPAR gamma agonists and partial agonists such as glitazones and non-glitazones (e.g.
- alpha-glucosidase inhibitors including acarbose, agonists disclosed in WO097/28149, agents for the treatment of inflammatory conditions such as non-steroidal anti-inflammatory drugs, aspirin, glucocorticoids, azulfidine, and cyclo- oxygenase 2 selective inhibitors, glucagon receptor antagonists, and GLP-I, GIP-I and GLP-I analogs such as exendins (for example exenitide).
- the compounds of the present invention may also be administered in combination with multiple active compounds, for example, biguanides PPAR agonists, PTP-IB inhibitors, anti-obesity compounds, sulfonylureas, HMG-CoA reductase inhibitors, and DP-IV inhibitors.
- active compounds for example, biguanides PPAR agonists, PTP-IB inhibitors, anti-obesity compounds, sulfonylureas, HMG-CoA reductase inhibitors, and DP-IV inhibitors.
- the present invention relates to a method of treating or preventing a disease in a vertebrate, the method comprising administering to a vertebrate in need thereof an effective amount of a compound of formula (3), (4), (3a), or (4a) as defined herein, or a prodrug thereof, or a composition according to the second aspect of the invention, wherein the disease is selected from Type II diabetes, obesity, hyperlipidemia, cardiovascular disease (e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease), anti-neoplastic diseases and tumours (e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer), inflammatory conditions (e.g, inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma, rheumatoid arthritis), and neurodegenerative diseases (e.g, Parkinson's disease, Alzheimer's disease).
- the disease is selected from Type II diabetes, obesity, hyperlipidemia, cardiovascular disease (e.
- Compounds for use in accordance with the present invention may be PPAR agonists.
- the compound may be a PPAR- ⁇ agonist.
- the compound may be a PPAR- ⁇ agonist.
- compounds in accordance with the present invention may exhibit dual PPAR ⁇ / ⁇ agonist activity.
- One aspect of the invention is the treatment in vertebrates of diseases that are amenable to amelioration through the activation of PPAR including for example type II diabetes, obesity, hyperlipidemia, cardiovascular disease, anti-neoplastic diseases and tumors, inflammatory conditions and neurogenerative diseases.
- the method comprises administering a compound of formula (3) as defined herein. In another embodiment the method comprises administering a compound of formula (4) as defined herein. In another embodiment the method comprises administering a compound of formula (3a) or (3) as defined herein. In another embodiment the method comprises administering a compound of formula (4a) or (4) as defined herein. In another aspect the present invention relates to the use of a compound of formula
- a disease selected from Type II diabetes, obesity, hyperlipidemia, cardiovascular disease (e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease), anti-neoplastic diseases and tumours (e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer), inflammatory conditions (e.g, inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma, rheumatoid arthritis), and neurodegenerative diseases (e.g, Parkinson's disease, Alzheimer's disease).
- cardiovascular disease e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease
- anti-neoplastic diseases and tumours e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer
- inflammatory conditions e.g, inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma, rheumatoi
- the invention provides for use of a compound of formula (3) as defined herein, or a prodrug thereof, in the manufacture of the medicament. In another embodiment the invention provides for use of a compound of formula (4) as defined herein, or a prodrug thereof, in the manufacture of the medicament. In an embodiment the invention provides for use of a compound of formula (3) or (3a) as defined herein, or a prodrug thereof, in the manufacture of the medicament. In another embodiment the invention provides for use of a compound of formula (4) or (4a) as defined herein, or a prodrug thereof, in the manufacture of the medicament.
- the disease or condition to be treated is Type II diabetes, obesity, or hyperlipidemia. In another embodiment the disease or condition to be treated is Type II diabetes.
- salts of the compounds of formulae (3)-(4) [and (3a)- (4a)] will be pharmaceutically acceptable salts; although other salts may be used in the preparation of the inventive compounds or of the pharmaceutically acceptable salt thereof.
- pharmaceutically acceptable salt it is meant those salts which, within the scope of sound medical judgement, are suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- Pharmaceutically acceptable salts are well known in the art.
- compositions of formulae (3)-(4) [and (3a)-(4a)] may be prepared by methods known to those skilled in the art, including for example, (i) by reacting a compound of formula (3) or (4) with the desired acid or base; (ii) by removing an acid- or base-labile protecting group from a suitable precursor of the compound or by ring-opening a suitable cyclic precursor, for example, a lactone or lactam, using the desired acid or base; or (iii) by converting one salt of the compound to another by reaction with an appropriate acid or base or by means of a suitable ion exchange column.
- the resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
- suitable pharmaceutically acceptable salts of compounds according to the present invention may be prepared by mixing a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, methanesulfonic acid, succinic acid, fumaric acid, maleic acid, benzoic acid, phosphoric acid, acetic acid, oxalic acid, carbonic acid, tartaric acid, or citric acid with the compounds of the invention.
- a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, methanesulfonic acid, succinic acid, fumaric acid, maleic acid, benzoic acid, phosphoric acid, acetic acid, oxalic acid, carbonic acid, tartaric acid, or citric acid.
- Suitable pharmaceutically acceptable salts of the compounds of the present invention therefore include acid addition salts.
- S. M. Berge et al. describe pharmaceutically acceptable salts in detail in J.
- the salts can be prepared in situ during the final isolation and purification of the compounds of the invention, or separately by reacting the free base function with a suitable organic acid.
- Representative acid addition salts include acetate, adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, digluconate, cyclopentanepropionate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptonate, glycerophosphate, hemisulfate, heptonate, hexanoate,' hydrobromide, hydrochloride, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanes
- alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like, as well as non-toxic ammonium, quaternary ammonium, and amine cations, including, but not limited to ammonium, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, triethanolamine and the like.
- Convenient modes of administration include injection (subcutaneous, intravenous, etc.), oral administration, inhalation, transdermal application, topical creams or gels or powders, or rectal administration.
- the mode of administration is parenteral.
- the mode of administration is oral.
- the formulation and/or compound may be coated with a material to protect the compound from the action of enzymes, acids and other natural conditions which may inactivate the therapeutic activity of the compound.
- the compound also may be administered parenterally or intraperitoneally.
- Dispersions of compounds according to the invention may also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof and in oils. Under ordinary conditions of storage and use, pharmaceutical preparations may contain a preservative to prevent the growth of microorganisms.
- Pharmaceutical compositions suitable for injection include sterile aqueous solutions
- the composition is stable under the conditions of manufacture and storage and may include a preservative to stabilise the composition against the contaminating action of microorganisms such as bacteria and fungi.
- Compounds of formulae (3)-(4) [and (3a)-(4a)] according to the present invention may be administered orally, for example, with an inert diluent or an assimilable edible carrier.
- the compound(s) and other ingredients may also be enclosed in a hard or soft shell gelatin capsule, compressed into tablets, or incorporated directly into an individual's diet.
- the compound(s) may be incorporated with excipients and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like.
- such compositions and preparations may contain at least 1% by weight of active compound.
- the percentage of the compound(s) in pharmaceutical compositions and preparations may, of course, be varied and, for example, may conveniently range from about 2% to about 90%, about 5% to about 80%, about 10% to about 75%, about 15% to about 65%; about 20% to about 60%, about 25% to about 50%, about 30% to about 45%, or about 35% to about 45%, of the weight of the dosage unit.
- the amount of compound in therapeutically useful compositions is such that a suitable dosage will be obtained.
- pharmaceutically acceptable carrier is intended to include solvents, dispersion media, coatings, anti-bacterial and anti-fungal agents, isotonic and absorption delaying agents, and the like. The use of such media and agents for pharmaceutically active substances is well known in the art.
- compositions according to the present invention may also be incorporated into the compositions according to the present invention. It is especially advantageous to formulate parenteral compositions in dosage unit form for ease of administration and uniformity of dosage.
- Dosage unit form refers to physically discrete units suited as unitary dosages for the individual to be treated; each unit containing a predetermined quantity of compound(s) is calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- the compound(s) may be formulated for convenient and effective administration in effective amounts with a suitable pharmaceutically acceptable carrier in an acceptable dosage unit. In the case of compositions containing supplementary active ingredients, the dosages are determined by reference to the usual dose and manner of administration of the said ingredients.
- the carrier is an orally administrable carrier.
- Another form of a pharmaceutical composition is a dosage form formulated as enterically coated granules, tablets or capsules suitable for oral administration.
- prodrug is an inactive form of a compound which is transformed in vivo to the active form.
- Suitable prodrugs include esters, phosphonate esters etc, of the active form of the compound.
- the compound of formulae (3)-(4) [and (3a)-(4a)] may be administered by injection.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyethylene glycol, and the like), suitable mixtures thereof, and vegetable oils.
- the proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- Prevention of the action of microorganisms can be achieved by including various anti-bacterial and/or anti-fungal agents.
- Suitable agents are well known to those skilled in the art and include, for example, parabens, chlorobutanol, phenol, benzyl alcohol, ascorbic acid, thimerosal, and the like.
- isotonic agents for example, sugars, polyalcohols such as mannitol, sorbitol, sodium chloride in the composition.
- Prolonged absorption of the injectable compositions can be brought about by including in the composition an agent which delays absorption, for example, aluminium monostearate and gelatin.
- Sterile injectable solutions can be prepared by incorporating the analogue in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by filtered sterilisation.
- dispersions are prepared by incorporating the analogue into a sterile vehicle which contains a basic dispersion medium and the required other ingredients from those enumerated above.
- Tablets, troches, pills, capsules and the like can also contain the following: a binder such as gum gragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid and the like; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin or a flavouring agent such as peppermint, oil of wintergreen, or cherry flavouring.
- a binder such as gum gragacanth, acacia, corn starch or gelatin
- excipients such as dicalcium phosphate
- a disintegrating agent such as corn starch, potato starch, alginic acid and the like
- a lubricant such as magnesium stearate
- a sweetening agent such as sucrose, lactose or saccharin or a flavouring agent such as peppermint, oil of wintergreen, or
- tablets, pills, or capsules can be coated with shellac, sugar or both.
- a syrup or elixir can contain the analogue, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavouring such as cherry or orange flavour.
- any material used in preparing any dosage unit form should be pharmaceutically pure and substantially non-toxic in the amounts employed.
- the analogue can be incorporated into sustained-release preparations and formulations.
- the pharmaceutical composition may further include a suitable buffer to minimise acid hydrolysis. Suitable buffer agent agents are well known to those skilled in the art and include, but are not limited to, phosphates, citrates, carbonates and mixtures thereof.
- Single or multiple administrations of the compounds and/or pharmaceutical compositions according to the invention may be carried out.
- One skilled in the art would be able, by routine experimentation, to determine effective, non-toxic dosage levels of the compound and/or composition of the invention and an administration pattern which would be suitable for treating the diseases and/or infections to which the compounds and compositions are applicable.
- the optimal course of treatment such as the number of doses of the compound or composition of the invention given per day for a defined number of days, can be ascertained using convention course of treatment determination tests.
- an effective dosage per 24 hours may be in the range of about 0.0001 mg to about 1000 mg per kg body weight; for example, about 0.001 mg to about 750 mg per kg body weight; about 0.01 mg to about 500 mg per kg body weight; about 0.1 mg to about 500 mg per kg body weight; about 0.1 mg to about 250 mg per kg body weight; or about 1.0 mg to about 250 mg per kg body weight. More suitably, an effective dosage per
- I 0 24 hours may be in the range of about 1.0 mg to about 200 mg per kg body weight; about 1.0 mg to about 100 mg per kg body weight; about 1.0 mg to about 50 mg per kg body weight; about 1.0 mg to about 25 mg per kg body weight; about 5.0 mg to about 50 mg per kg body weight; about 5.0 mg to about 20 mg per kg body weight; or about 5.0 mg to about 15 mg per kg body weight.
- an effective dosage may be up to about 500mg/m 2 .
- an effective dosage is expected to be in the range of about 25 to about 500mg/m 2 , about 25 to about 350mg/m 2 , about 25 to about 300mg/m 2 , about 25 to about 250mg/m 2 , about 50 to about 250mg/m 2 , and about 75 to about 150mg/m 2 .
- a compound of Formula (3) or (4) may be administered in another embodiment.
- 20 an amount in the range from about 100 to about 1000 mg per day, for example, about 200 mg to about 750 mg per day, about 250 to about 500 mg per day, about 250 to about 300 mg per day, or about 270 mg to about 280 mg per day.
- compound(s) according to the present invention may be administered in combination therapy with known antidiabetic or antilipidemic agents.
- compositions at least one of which contains a compound according to the present invention, may be combined in the form of a kit suitable for co-administration of the compositions.
- the respective agents may be administered simultaneously, or sequentially in any order.
- the invention relates to a method for identifying a PPAR agonist, comprising determining ligand-receptor interactions of a candidate compound with docking templates; comparing the ligand-receptor interactions of the candidate compound with the interactions of a known PPAR agonist; and thereby determining whether a candidate compound is a PPAR agonist.
- the agonist is a PPAR- ⁇ agonist.
- the agonist is a PPAR- ⁇ agonist.
- the agonist may be a dual PPAR ⁇ / ⁇ agonist.
- the method of the invention may be used to identify PPAR agonists that share little similarity with known ligands.
- the present invention utilises the approach of "Induced Fit Docking", or IFD.
- IFD protocol was run from the graphical user interface accessible within Maestro 8.5. It was carried out on PPAR- ⁇ [(figure 1), (PDB Code: 117G)] with claim 3 and 4 compounds, and AZ-242, the known PPAR- ⁇ agonist.
- the overall procedure has four stages: Briefly, during Stage 1 initial softened-potential Glide docking is performed on a vdW scaled-down rigid-receptor; a scaling of 0.5/ 0.5 was set for receptor / ligand vdW radii, respectively. The top 20 poses for each test ligand was retained. In Stage 2, receptor sampling and refinement was performed on residues within 5.0 of each ligand for each of the 20 ligand:protein complexes.
- Ligands showing high affinity towards PPAR- ⁇ and PPAR- ⁇ in silico are subsequently tested in vitro using a PPAR- ⁇ transcriptional factor assay, a PPAR- ⁇ reporter gene luciferase assay and a PPAR- ⁇ reporter gene luciferase assay.
- the activity of compounds identified by this process in inducing PPAR- ⁇ mRNA and protein expression in vitro may be also examined to see if the effect can be abolished in the presence of GW9662, a potent synthetic PPAR- ⁇ antagonist (Bendixen et al., 2001).
- Biological data from the transactivation assays may be used to characterize compounds as agonists of human PPAR.
- TLC Thin layer chromatography
- silica 0.2 mm, 60F 254
- UV fluorescence 254 nm
- Flash vacuum chromatography was performed on silica gel (Merck silica gel 6OH, particle size 5 ⁇ 0 ⁇ m). Chemicals were purchased from Aldrich, Boron Molecular and at the highest available grade.
- Example 3 is l-(2,4-Dihydroxyphenyl)-3-(3-methoxyphenyl)prop-2-en-l-one (3)
- a solution of trifluoroacetic anhydride (0.22 mL) is added to a solution of 52 (400 mg) in 2 mL of dry pyridine at O 0 C.
- the reaction mixture was shaken, with ice cooling, for 10-15 min and is left overnight. On the following day, it is heated to 40-50 0 C for 10- 15 min and again left at room temperature for 12 h. Then it is poured into 20-30 mL cold water, and the precipitate is filtered off and crystallized from ethanol to give 53 as a cream colour solid.
- Resorcinol (1 g, 1 mmol) and 4-fluorophenylacetic acid (1.4 g, 1 mmol) in boron trifluoride etherate (5.68 mL, 0.2 mol) were heated, under a nitrogen atmosphere, at 90 0 C for 90 mins to give a red solution.
- the solution was allowed to cool and poured into aqueous sodium acetate (10% aq.; 100 mL) and the mixture stirred to give a pale yellow precipitate.
- the solids were removed by filtration and washed with water (100 mL).
- the solids were taken up in ethyl acetate (150 mL) and washed with water (100 mL).
- the solution was dried over anhydrous sodium sulfate and concentrated to a yellow semi- solid. Trituration with diethyl ether (50 mL) gave 9b (626 mg, 28%) as a pale orange solid after drying in vacuo.
- Acetic anhydride (0.77 mL, 4 mmol) was added to a suspension of potassium carbonate (1.02 g, 4 mmol) and 2-(4-fluorophenyl)-l-(2,4-dihydroxyphenyl)ethenone (9b) (500 mg, 1 mmol) in DMF (5 mL), and the resulting suspension heated at 115 °C for 120 mins. The mixture was allowed to cool and poured into water (100 mL), to give an off-white precipitate.
- Resorcinol (1 g, 1 mmol) and 4-methoxyphenylacetic acid (1.50 g, 1 mmol) in boron trifluoride etherate (5.68 mL, 0.2 mol) were heated, under a nitrogen atmosphere, at 90 0 C for 90 mins to give a pale red solution.
- the solution was allowed to cool and poured into aqueous sodium acetate (10% aq.; 100 mL) and the mixture stirred to give a pale yellow precipitate.
- the solids were removed by filtration and washed with water (100 mL).
- the solids were taken up in ethyl acetate (150 mL) and washed with water (100 mL).
- the solution was dried over anhydrous sodium sulfate and concentrated to a yellow semi-solid.
- Trituration with diethyl ether (50 mL) gave 10b (469 mg, 20%) as a pale orange solid after drying in vacuo.
- Acetic anhydride (0.53 mL, 4 mmol) was added to a suspension of potassium carbonate (782 mg, 4 mmol) and l-(2,4-dihydroxyphenyl)-2-(4-methoxyphenyl)ethanone (10b) (400 mg, 1 mmol) in DMF (5 mL), and the resulting suspension heated at 115 0 C for 120 mins. The mixture was allowed to cool and poured into water (100 mL), to give an off-white precipitate.
- Example 22 Screening assay to identify PPAR- ⁇ and PPAR- ⁇ agonists (HEK cell lines)
- Human embryonic kidney (HEK) 293 cell line was obtained from American Type
- HEK 293 cells were grown in Dulbecco's modified
- DMEM/F-12 Eagle's medium/F-12
- penicillin 100 U/mL
- streptomycin 100 mg/mL
- 10% v/v heat- inactivated foetal bovine serum in a humidified atmosphere of 5% CO 2 and 95% O 2 at 37 0 C.
- the transfection and luciferase procedures were performed as described previously (Bramlett et al., 2003) with slight modification.
- the HEK 293 cell line was transfected with tK-PPREx3-Luc plasmid, pSG5-hPPAR- ⁇ plasmid and pSV- ⁇ -galactosidase (Promega, Australia) control plasmid.
- Cells were transfected with FuGENE 6 transfection reagent (Roche, Australia) in accordance with the manufacturer's instructions. After 24 h at 37°C, cells were harvested and plated into 96-well plates at 5 x 10 4 cells per well in complete transfection media and allowed to attach over night at 37 °C.
- the cells were then treated with rosiglitazone and GW1929 as positive controls, DMSO (0.1%) as a negative control and the test samples. After 48 hours, the cells were lysed and assayed for luciferase and ⁇ -galactosidase activities using the Bright-Glo Luciferase Assay system and Beta-Glo Assay system (Promega, Australia), respectively. The results were expressed as relative luciferase activity normalized to the ⁇ -galactosidase signal (fold difference compared to negative control).
- Human embryonic kidney cell line (HEK 293) is seeded overnight then treated with
- PPAR- ⁇ Transfection and Luciferase Assay
- the transfection and luciferase procedures will be performed as described previously (Bramlett et al., 2003) with slight modification.
- the HEK 293 cell line will be transfected with tK-PPREx3-Luc plasmid, pBI-G-hPPAR- ⁇ plasmid and pSV- ⁇ - galactosidase (Promega, Australia) control plasmid.
- Cells will be transfected with FuGENE 6 transfection reagent (Roche, Australia) in accordance with the manufacturer's instructions.
- HEK 293 cells will be seeded overnight then treated with various concentrations of WY-14643, Fenofibrate and test compounds (0 - 100 ⁇ M) and incubated for 48 hours at 37 0 C in a humidified atmosphere with 5% CO 2 .
- MTS (tetrazolium salt) reagent CellTiter96® Aqueous One Solution Cell Proliferation Assay, Promega, Sydney, Australia
- CellTiter96® Aqueous One Solution Cell Proliferation Assay Promega, Sydney, Australia
- Example 23 Screening assay to identify PPAR- ⁇ agonists (THP-I cell lines)
- Anti-actin primary antibody bovine serum albumin (BSA), dimethyl sulfoxide (DMSO), GW9662 and phorbol 12-myristate 13-acetate (PMA) will be purchased from Sigma-Aldrich (Sydney, Australia). Natural products, totaling 200 compounds, will be sourced from the Herbal Medicines Research and Education Center (Faculty of Pharmacy, University of Sydney). Cell Culture The THP-I monocytes and macrophages will be grown in RPMI 1640 in the presence of 50 (M ( ⁇ -mercaptoethanol.
- BSA bovine serum albumin
- DMSO dimethyl sulfoxide
- PMA phorbol 12-myristate 13-acetate
- All media will contain L-glutamine supplemented with penicillin (100 U/ml)/ streptomycin (100 (g/ml), and 10% (v/v) heat-inactivated fetal bovine serum (FBS) in a humidified atmosphere of 5% CO 2 and 95% O 2 at 37°C.
- FBS heat-inactivated fetal bovine serum
- the THP-I monocytes will be treated with PMA (400 ng/ml) for 72 h, before selective PPAR- ⁇ ( agonist rosiglitazone, test samples or vehicle (0.1% DMSO) will be added and incubated for a further 48 h in culture medium for the macrophage treatment experiments.
- the PPAR- ⁇ antagonist, GW9662 (5 ⁇ M) will be added 1 h prior to addition of positive control or test samples.
- Cell-Based Transcriptional Factor Assay The PPAR- ⁇ Transcription Factor Assay is a sensitive ELISA method for detecting
- PPAR- ⁇ transcription factor DNA binding activity in nuclear extracts of THP-I derived macrophage cell line The ELISA assay will be conducted according to the manufacturer's instructions (Cayman Chemical, Sydney, Australia). The purification of cellular nuclear extract from the cultured cells will be prepared with CelLyticTM NuCLEARTM Extraction Kit (Sigma, Sydney, Australia). Cell Proliferation Assay
- THP-I macrophage cells will be seeded overnight, then treated with various concentrations of rosiglitazone, GW9662 and test compounds (0.01 - 100 ⁇ M) before further incubation for 3 days at 37°C in a humidified atmosphere with 5% CO 2 .
- MTS (tetrazolium salt) reagents (the CellTiter96® Aqueous One Solution Cell Proliferation Assay, Promega, Sydney, Australia) will be added, incubated for 4 h and finally analyzed using a microtiter plate reader (model 3550, Bio-Rad) ( ⁇ : 490 nm).
- Total mRNA will be prepared separately from the THP-I macrophage cells using TRIzol (Invitrogen, Sydney, Australia). The relative levels of specific mRNAs will be assessed by RT-PCR as described previously (Abe et al., 2002). Single-stranded cDNA is synthesized from 1 ⁇ g of total RNA using Superscript II RNAse H Reverse Transcriptase, as per instructions of the manufacturer (Invitrogen, Sydney, Australia). PCR will be performed on a thermocycler, PTC-200 DNA engine (MJ Research Inc, USA). The required cDNA will be synthesized with the Platinum ® Pfx DNA Polymerase method (Invitrogen, Sydney, Australia). The genes examined will be PPAR- ⁇ (L40904; 382bp; sense: 5'-GAGCCCAAGTTTGAGTTTGC-S'; 5'-
- PCR samples will be electrophoresed on 5-12% acrylamide gel (29:1, acrylamide:N,N'- methylene-bis-acrylamide) in TBE buffer [89 mM Tris-base pH 7.6, 89 mM boric acid, 2 mM EDTA].
- TBE buffer 89 mM Tris-base pH 7.6, 89 mM boric acid, 2 mM EDTA.
- the gels will be stained with ethidium bromide (10 ⁇ g/ml) and photographed on top of a 280 nm UV light box (Biorad ® Gel Doc 1000, Australia).
- the gel images will be digitally captured with a CCD camera and analyzed with ImageJ 1.29x (NIH, USA). RT-PCR values will be presented as a ratio of the specified gene signal in the selected linear amplification cycle divided by the ⁇ -actin signal.
- THP-I cells will be seeded and treated with PMA (400 ng/ml) for 72 h to obtain THP-I macrophages.
- the macrophages will be treated with 0.1% DMSO, test compounds (30 ⁇ M) and rosiglitazone (30 ⁇ M) for 48 h. Then the cells will be washed with PBS and lysed with RIPA lysis buffer for protein extraction. The protein contents in the samples will be measured using BCA protein estimation kit and 20 ⁇ g of sample will be loaded onto 4-12% NuP AGE® Bis-Tris Gel (Invitrogen, Sydney, Australia).
- the protein After electrophoresis at 200 V for 1 h, the protein will be transferred to PVDF membrane and blocked overnight in skim milk (5% skim milk in tris buffered saline). On the subsequent day the PVDF membranes will be treated with antihuman PPAR- ⁇ mouse monoclonal primary antibody (1:500 dilution; Santa Cruz Biotechnology, USA) followed with horseradish peroxidase-conjugated anti-mouse secondary antibody (1 : 10,000 dilution; Promega, USA). The antibody treatment will be performed for Ih followed by 30 min wash with the washing buffer (Tris buffered saline with 0.1% Tween-20). Protein expression will be detected by chemiluminescence method (Roche).
- the PVDF membranes will be exposed to X-ray film (Kodak, USA) and developed using the SRX- 101 A X-ray developer (Konica, Taiwan). Quantitation of the results are performed by using the NIH Image J software. After stripping with stripping buffer (Glycine (15 g), SDS (1 g), Tween-20 (10 mL), pH 2.2) the membranes will be re-probed with anti-actin primary antibody (1:10,000 dilution; Sigma, Australia) will be re- incubated with the secondary horseradish peroxidase antibody, and protein bands are detected as described above.
- stripping buffer Glycine (15 g), SDS (1 g), Tween-20 (10 mL), pH 2.2
Abstract
The present invention relates to PPAR agonists and their use in therapy, including the treatment of disease. In particular, the invention relates to compounds which are PPAR-gamma agonists and/or PPAR alpha/gamma dual agonists.
Description
PPAR AGONISTS
Technical Field
The present invention relates to PPAR agonists and their use in therapy, including the treatment of disease. Background
The peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors that belong to the nuclear hormone superfamily. To date, three isoforms of PPAR have been identified: PPAR-α, -δ and -γ. PP AR-γ is the most abundant receptor expressed in adipocytes and macrophages, where, apart from its involvement in adipocyte differentiation and lipid storage, it serves as the primary receptor modulating insulin sensitization and maintaining lipid and glucose homeostasis.
PPAR-γ is the target of numerous drug discovery efforts because of its role in numerous disease states, including Type II diabetes. The thiazolidinediones (TZDs; or glitazones) and the L-tyrosine analogues are anti-diabetic synthetic agonists that selectively target PPAR-γ. Their mode of action begins with sensitizing tissue to insulin, lowering glucose levels and reducing serum lipids in diabetic patients by potently binding, and subsequently activating, PPAR-γ.
Rosiglitazone (Avandia®), shown below, is a prototypical TZD and serves as a reference compound for this class, which also includes pioglitazone (Actos®) and troglitazone. Rosiglitazone is active in vivo as an anti-diabetic agent in the ob/ob mouse model and is presently being used as an oral hypoglycaemic agent for the treatment of Type II diabetes. The L-tyrosine analogue class of compounds, such as Farglitazar (GI262570) shown below, represent the most potent and selective class of synthetic PPAR-γ agonists currently in existence.

Rosiglitazone Farglitazar
Unfortunately, despite their excellent potencies, several of the known PPAR-γ agonists have been found to present unwanted adverse therapeutic profiles, such as fluid retention, weight gain and cardiac hypertrophy. Troglitazone, for example, was withdrawn from therapeutic use due to liver toxicity, and Farglitazar failed to pass phase
III clinical trials due to the emergence of peripheral oedema.
The PPAR-α receptor, mainly expressed in the liver, play an important role in fatty acid oxidation and lipoprotein metabolism. Fibrates (Fenofibrate, Clofibrate) and WY- 14643, shown below, show effects such as lowering triglycerides and elevating HDL levels through activation of PPAR-α. The majority of type II diabetes patients suffer from atherogenic lipid abnormalities in addition to insulin resistance, termed as metabolic syndrome. The importance of controlling both glucose and lipid levels in metabolic syndrome, gave rise to the concept of identifying dual agonists, which can activate both PPAR-α and PPAR-γ. In addition to their hypolipidemic effects, PPAR-α agonists reduce body weight gain which led to a hypothesis that activation of PPAR-α may mitigate the weight gain induced by PPAR-γ activation in humans.

PPAR-γ and PPAR-α agonists have been implicated in the pathology of various disorders including atherosclerosis, coronary heart disease, obesity and inflammation.
Compounds that are dual PPAR-γ and PPAR-α agonists can have fewer therapeutic side- effects than those that act solely at the PPAR-γ receptor or those that act solely at the
PPAR-α receptor. Thus development of a safer and efficacious dual PPAR-γ and PPAR-α agonists are of considerable therapeutic value.
There is a need to discover new PPAR agonists. More particularly there is a need for PPAR-γ and PPAR-α agonists and/or dual PPAR-α/γ agonists that are suitable for use in therapy. A further need exists for PPAR-γ and PPAR-α agonists and/or dual PPAR-α/γ agonists that have a therapeutically acceptable side-effect profile.
Summary
The present invention relates to compounds having PPAR agonist activity, and the therapeutic use thereof. In one embodiment the PPAR agonist is a PPAR-γ agonist. In another embodiment the PPAR agonist is a PPAR-α agonist. In a further embodiment the PPAR agonist is a dual PPAR α/γ agonist.
In a first aspect the present invention provides compounds of general formula (3):

(3) wherein
A is a C5-6 heteroaryl ring optionally substituted with one or more substituents independently selected from halogen, C1-4alkyl, O-C1-4alkyl, haloCi-4alkyl, hydroxy- CMalkyl and CO2R;
Ra-RJ are each independently selected from hydrogen, hydroxyl, halogen, C1-4alkyl,
C3-6CyClOaIlCyI, haloC1-4alkyl, hydroxyCi-4alkyl, O-C1-4alkyl, O-C3-6cycloalkyl, Ci-4alkyl-
CO2R, O-C1-4alkyl-CO2R, O-C3-6heterocycloalkyl, O-C3-6heteroaryl, N(R)2C i-4alkyl, N(R)3C1-4alkyl, O-C1-4alkyl-N(R)2, O-Ci-4alkyl-N(R)3, C6-10aryl, O-Ci-4alkyl-C6. loheterocycloalkyl, 0-C1-4alkyl-C6-ioheteroaryl, CO2R, O-sugar, O-Cs^cycloalkyl, O-C6.i0 aryl, O-C1-4alkyl-C6-i0aryl, , O-benzyl, O-benzoyl, O-C(O)-CMalkyl, CO2Ci.4alkyl,
CON(R)2, OC(O)-N(R)2, C^alkanoyloxymethyl, and -P(O)(OH)(OCi-4alkyl), -
P(O)(OC1-4alkyl)2 and OS(O)(O)NH2; or one or more ofRa and Rb, Rb and Rc, Rc and Rd, Rd and Re, Rfand Rg, Rg and Rh, R'
and
 each R is independently selected from hydrogen and C1-4alkyl; and pharmaceutically acceptable salts thereof.
each R is independently selected from hydrogen and C1-4alkyl; and pharmaceutically acceptable salts thereof.
In a second aspect the present invention provides compounds of general formula (4):

(4) wherein L is selected from Ci-4alkylene and C2-4alkenylene;
RQ-RU are each independently selected from hydrogen, hydroxyl, halogen, CMalkyl, haloC1-4alkyl, hydroxyC1-4alkyl, O-CMalkyl, O-CMalkyl-CO2R, O-C3-6cycloalkyl, O-C3-6heteroaryl, N(R)2C 1-4alkyl, N(R)3C1-4alkyl, O-Ci-4alkyl-N(R)2,
O-C1-4alkyl-N(R)3> C6-10aryl, O-C6-i0aryl, O-CMalkyl-C^oheterocycloalkyl, O-CMalkyl- C6-10heteroaryl, CO2R, O-Q-eheterocycloalkyl, O-C1-4alkyl-C6-i0aryl, 0-C(O)-C Malkyl, 0-C(O)-N(R)2, CON(R)2, d^alkanoyloxymethyl, -OP(O)(OH)(OCMalkyl),. - OP(O)(OC1-4alkyl)2, OS(O)(O)NH2 and O-sugar; or one or more of RQ and RR, RR and Rs, Rs and Rτ and Rτ and Ru together form

Rv-Rz are each independently selected from hydrogen, hydroxyl, halogen, C1-4alkyl, haloC1-4alkyl, hydroxyCi-4alkyl, O-C1-4alkyl, O-CMalkyl-CO2R, O-C^cycloalkyl, O-C3-6heteroaryl, N(R)2C1-4alkyl, N(R)3Ci-4alkyl, O-Ci-4alky 1-N(R)2,
10 O-CMalkyl-N(R)3; C6.10aryl, O-C6-i0 aryl, O-C1-4alkyl-C6-10heterocycloalkyl, O-CMalkyl- C6-10heteroaryl, CO2R, O-C^heterocycloalkyl, 0-C1-4alkyl-C6-ioaryl, 0-C(0)-C1-4alkyl, 0-C(O)-N(R)2, CON(R)2, Ci-4alkanoyloxymethyl, -OP(O)(OH)(OCi-4alkyl), - OP(O)(OCi-4alkyl)2 and O-sugar; or one or more of Rv and Rw, Rw and Rx, Rx and Rγ and Rγ and Rz together form
 ; each R is independently selected from hydrogen and C1-4 alkyl; and pharmaceutically acceptable salts thereof. In a third aspect the invention provides compounds of general formula (3 a):
; each R is independently selected from hydrogen and C1-4 alkyl; and pharmaceutically acceptable salts thereof. In a third aspect the invention provides compounds of general formula (3 a):

20 (3a) wherein A is a heteroaryl ring selected from imidazolyl, pyrazolyl, oxazolyl, and isoxazolyl, wherein the heteroaryl ring may be substituted with one or more substituents independently selected from methyl, ethyl, CO2R and CF3;
Ra is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi-2alkyl, O-methyl and 2s CO2R;
Rb, Rc, Rd, Re, Rf, R1, and Rj are each independently selected from hydrogen, hydroxyl, C1-2alkyl, O-C1-2alkyl, CO2R, and O-benzyl;
R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
In a fourth aspect the invention provides compounds of general formula (4a):

(4a) wherein
L is -C(O)Ci.3alky-ene-, -C(O)C2-3alkenylene-, -Ci-3alkyleneC(O)- and s -C2-3alkenyleneC(O)-;
Rz is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi-2alkyl, O-methyl and CO2R;
RQ, RR, Ru, Rv, Rw, Rx, and Rγ are each independently selected from hydrogen, hydroxyl, C1-2alkyl, O-C1-2alkyl, CO2R and O-benzyl; o R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
In an fifth aspect the present invention provides a pharmaceutical composition comprising one or more compounds of formula (3), (4), (3a) or (4a), or a prodrug thereof, together with a pharmaceutically acceptable adjuvant, diluent or carrier. s In a sixth aspect the present invention provides a method of treating or preventing a disease in a vertebrate, the method comprising administering to a vertebrate in need thereof an effective amount of a compound of formula (3), (4), (3a), or (4a) as defined herein, or a prodrug thereof, or a composition according to the fifth aspect of the invention, wherein the disease is selected from Type II diabetes, obesity, hyperlipidemia,0 cardiovascular disease, anti-neoplastic diseases and tumours, inflammatory conditions and neurodegenerative diseases.
In a seventh aspect the present invention relates to the use of a compound of formula (3), (4), (3 a), or (4a) as defined herein, or a prodrug thereof, in the manufacture of a medicament for treating a disease selected from Type II diabetes, obesity,5 hyperlipidemia, cardiovascular disease, anti-neoplastic diseases and tumours, inflammatory conditions and neurodegenerative diseases.
In an eighth aspect the invention relates to a method for identifying a PPAR agonist, the method comprising determining ligand-receptor interactions of a candidate compound with docking0 templates;
comparing the ligand-receptor interactions of the candidate compound with the interactions of a known PPAR agonist; and thereby determining whether a candidate compound is a PPAR agonist. In one embodiment the PPAR agonist is a PPAR-γ agonist. In another embodiment s the PPAR agonist is a PPAR-α agonist. In a further embodiment the PPAR agonist is a dual PPAR α/γ agonist.
The method of the invention may be used to identify PPAR agonists that share little similarity with known ligands.
The method may further comprise testing a compound identified as a PPAR agonist I0 in vitro for PPAR activation efficacy using either a transcriptional factor or a reporter gene luciferase assay.
Abbreviations
PPAR, peroxisome proliferator-activated receptor is LBD, ligand binding domain
TZD, thiazolidinediones
THP Ether, Tetrahydropyranyl Ether
DHP, 3,4-Dihydro-2H-Pyran
PPTS, Pyridinium-p-toluenesulfonate 20 EC so, the half maximal effective concentration
HEK293, human embryonic kidney cell line
DMSO, N,N-Dimethylsulfoxide
PPRE, PPAR response element
IFD, induced-fit docking 25 DMF, N,N-Dimethylformamide
DMF-DMA, N,N-Dimethylformamide-dimethyl acetal
THF, tetrahydrofuran
TEA, triethylamine
DME, dimethoxyethane 30 vdW, van der Waals
Definitions
The following are some definitions that may be helpful in understanding the description of the present invention. These are intended as general definitions and should
in no way limit the scope of the present invention to those terms alone, but are put forth for a better understanding of the following description.
Unless the context requires otherwise or specifically stated to the contrary, integers, steps, or elements of the invention recited herein as singular integers, steps or elements
5 clearly encompass both singular and plural forms of the recited integers, steps or elements.
Throughout this specification, unless the context requires otherwise, the word "comprise", or variations such as "comprises" or "comprising", will be understood to imply the inclusion of a stated step or element or integer or group of steps or elements or
I0 integers, but not the exclusion of any other step or element or integer or group of elements or integers. Thus, in the context of this specification, the term "comprising" means "including principally, but not necessarily solely".
Those skilled in the art will appreciate that the invention described herein is susceptible to variations and modifications other than those specifically described. It is to is be understood that the invention includes all such variations and modifications. For the avoidance of any doubt, the invention also includes all of the steps, features, compositions and compounds referred to or indicated in this specification, individually or collectively, and any and all combinations or any two or more of said steps or features, in any appropriate order.
20 As used herein, the term "C1-4 alkyl group" includes within its meaning monovalent
("alkyl") and divalent ("alkylene") straight chain or branched chain saturated aliphatic groups having from 1 to 4 carbon atoms. The alkyl group may be C1-3 alkyl or Cj-2 alkyl. Thus, for example, the term Ci-4 alkyl includes, but is not limited to, methyl, ethyl, 1- propyl, isopropyl, 1 -butyl, 2-butyl, isobutyl, tert-butyl, and the like.
2s The term "C2-4 alkenyl group" includes within its meaning monovalent ("alkenyl") and divalent ("alkenylene") straight or branched chain unsaturated aliphatic hydrocarbon groups having from 2 to 4 carbon atoms and at least one double bond anywhere in the chain. The alkenyl group may be C2-3 alkenyl. Unless indicated otherwise, the stereochemistry about each double bond may independently be cis, trans, E or Z as
30 appropriate. Examples of C2-4 alkenyl groups include but are not limited to ethenyl, vinyl, allyl, 1-methylvinyl, 1-propenyl, 2-propenyl, 2-methyl-l-propenyl, 2-methyl-l-propenyl, 1-butenyl, 2-butenyl, 3-butentyl, 1,3-butadienyl, and the like.
The term "amino" as used herein refers to groups of the form -NRaRb wherein Ra and Rb are individually selected from hydrogen, optionally substituted (Ci-4)alkyl,
35 optionally substituted (C2-4)alkenyl, optionally substituted (C2-4)alkynyl, optionally
substituted (C6-10)aryl and optionally substituted aralkyl groups, such as benzyl. The amino group may be a primary, secondary or tertiary amino group.
The term "amino acid" as used herein includes naturally and non-naturally occurring amino acids, as well as substituted variants thereof. Thus, (L) and (D) forms of amino acids are included in the scope of the term "amino acid". The term "amino acid" includes within its scope glycine, alanine, valine, leucine, isoleucine, methionine, proline, phenylalanine, tryptophan, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartate, lysine, arginine, and histidine. The backbone of the amino acid residue may be substituted with one or more groups independently selected from (C^alkyl, halogen, hydroxy, hydroxy(C1-6)alkyl, aryl (e.g, phenyl), aryl(Ci-3)alkyl (e.g, benzyl), and (C3-6)cycloalkyl.
The term "aralkyl" or variants such as "arylalkyl" as used herein, includes within its meaning monovalent ("aryl") and divalent ("arylene"), single, polynuclear, conjugated and fused aromatic hydrocarbon radicals attached to divalent, saturated, straight or branched chain alkylene radicals.
The term "C6-10 aromatic group", or variants such as "C6-I0 aryl" or "C6-I0 arylene" as used herein refers to monovalent ("aryl") and divalent ("arylene") single, polynuclear, conjugated and fused residues of aromatic hydrocarbons having from 6 to 10 carbon atoms. Examples of aromatic groups include phenyl, naphthyl, phenanthrenyl, and the like.
The term "C3-6 cycloalkyl" as used herein refers to cyclic saturated aliphatic groups and includes within its meaning monovalent ("cycloalkyl"), and divalent ("cycloalkylene"), saturated, monocyclic, bicyclic, polycyclic or fused polycyclic hydrocarbon radicals having from 3 to 6 carbon atoms. The cycloalkyl group may be C3-5 cycloalkyl. Examples of cycloalkyl groups include but are not limited to cyclopropyl, 2- methylcyclopropyl, cyclobutyl, cyclopentyl, 2-methylcyclopentyl, 3-methylcyclopentyl, cyclohexyl, and the like.
The term "C3-6 cycloalkenyl" as used herein, refers to cyclic unsaturated aliphatic groups and includes within its meaning monovalent ("cycloalkenyl") and divalent ("cycloalkenylene"), monocyclic, bicyclic, polycyclic or fused polycyclic hydrocarbon radicals having from 3 to 6 carbon atoms and having at least one double bond anywhere in the alkyl chain. The cycloalkenyl group may be C3-5 cycloalkenyl. Unless indicated otherwise, the stereochemistry about each double bond may be independently cis, trans, E or Z as appropriate. Examples of cycloalkenyl groups include but are not limited to cyclopropenyl, cyclopentenyl, cyclohexenyl, and the like.
The term "C3-6 heterocycloalkyl" as used herein, includes within its meaning monovalent ("heterocycloalkyl") and divalent ("heterocycloalkylene"), saturated, monocyclic, bicyclic, polycyclic or fused hydrocarbon radicals having from 3 to 6 ring atoms, wherein from 1 to 3, ring atoms are heteroatoms independently selected from O, N, NH, or S. The heterocycloalkyl group may be C3-5 heterocycloalkyl. Examples of heterocycloalkyl groups include aziridinyl, pyrrolidinyl, piperidinyl, piperazinyl, quinuclidinyl, azetidinyl, morpholinyl, tetrahydrothiophenyl, tetrahydrofuranyl, tetrahydropyranyl, and the like.
The term "C5-10 heteroaromatic group" and variants such as "heteroaryl" or "heteroarylene" as used herein, includes within its meaning monovalent ("heteroaryl") and divalent ("heteroarylene"), single, polynuclear, conjugated and fused aromatic radicals having from 5 to 10 atoms, wherein 1 to 4, or 1 to 2 ring atoms are heteroatoms independently selected from O, N, NH and S. The heteroaromatic group may be C5-8 heteroaromatic. Examples of heteroaromatic groups include pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, 2,2'-bipyridyl, phenanthrolinyl, quinolinyl, isoquinolinyl, imidazolinyl, thiazolinyl, pyrrolyl, furanyl, thiophenyl, oxazolyl, isoxazolyl, isothiazolyl, triazolyl, and the like.
The term "halogen" or variants such as "halide" or "halo" as used herein refers to fluorine, chlorine, bromine and iodine. The term "heteroatom" or variants such as "hetero-" as used herein refers to O, N, and S or the group NH.
The term "optionally substituted" as used herein means the group to which this term refers may be unsubstituted, or may be substituted with one or more groups independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, halo, haloalkyl, haloalkynyl, hydroxyl, hydroxyalkyl, alkoxy, thioalkoxy, alkenyloxy, haloalkoxy, haloalkenyloxy, NO2, NRaR , nitroalkyl, nitroalkenyl, nitroalkynyl, nitroheterocyclyl, alkylamino, dialkylamino, alkenylamine, alkynylamino, acyl, alkenoyl, alkynoyl, acylamino, diacylamino, acyloxy, alkylsulfonyloxy, heterocycloxy, heterocycloamino, haloheterocycloalkyl, alkylsulfenyl, alkylcarbonyloxy, alkylthio, acylthio, phosphorus-containing groups such as phosphono and phosphinyl, aryl, heteroaryl, alkylaryl, aralkyl, alkylheteroaryl, cyano, cyanate, isocyanate, CO2H, CO2(alkyl), C(O)NH2, -C(O)NH(alkyl), and -C(O)N(alkyl)2. Preferred substituents include C1-3 alkyl, Ci-3 alkoxy, -CH2-(C1-3)alkoxy, C6-10 aryl, -CH2-phenyl, halo, hydroxyl, hydroxy-(C1-3)alkyl, and halo-(C1-3)alkyl, e.g, CF3, CH2CF3.
In the context of this specification the term "administering" and variations of that term including "administer" and "administration", includes contacting, applying, delivering or providing a compound or composition of the invention to an organism, or a surface by any appropriate means. In the context of this specification, the term "vertebrate" includes humans and individuals of any species of social, economic or research importance including but not limited to members of the genus ovine, bovine, equine, porcine, feline, canine, primates (including human and non-human primates), rodents, murine, caprine, leporine, and avian. The vertebrate may be a human. In the context of this specification, the term "treatment", refers to any and all uses which remedy a disease state or symptoms, prevent the establishment of disease, or otherwise prevent, hinder, retard, or reverse the progression of disease or other undesirable symptoms in any way whatsoever.
In the context of this specification the terms "therapeutically effective amount" and "diagnostically effective amount", include within their meaning a sufficient but non-toxic amount of a compound or composition of the invention to provide the desired therapeutic or diagnostic effect. The exact amount required will vary from subject to subject depending on factors such as the species being treated, the age and general condition of the subject, the severity of the condition being treated, the particular agent being administered, the mode of administration, and so forth. Thus, it is not possible to specify an exact "effective amount". However, for any given case, an appropriate "effective amount" may be determined by one of ordinary skill in the art using only routine experimentation.
Brief Description of the Figures
Figure 1. a) X-ray crystallographic pose of the known PPAR-alpha agonist AZ-242. b) Induced-fit docking pose of (^indicating the similarity of the binding pose in this compound to the known structure AZ-242. Both ligands establish H-bond interactions to the PPAR-alpha receptor. Figure 2: PPAR-γ reporter gene activity of compounds 4, 8 and 11 in HEK 293 cell line. The HEK 293 cells were transiently transfected with tK-PPREx3-Luc, pSG5-hPPAR-γ and pSV-β-galactosidase control plasmid. Cells were treated with test compounds (5 μM and 25 μM). Rosiglitazone (25 μM) and GWl 929 (25 μM) were used as positive controls and DMSO (0.1%) as a negative control. At the end of the incubation period the cells
were lysed and assayed for luciferase and β-galactosidase activities. The results are expressed as relative luciferase activity (fold difference compared to negative control).
Detailed Description
The present invention is directed to compounds which are agonists of the PPAR receptors. In one embodiment the invention relates to compounds which are PPAR-γ agonists. The present invention also relates to compounds that are agonists of the PPAR-α receptor. The present invention is further directed to compounds which are dual agonists of the PPAR-γ and PPAR-α receptors.
Compounds according to the present invention may be useful in therapy, including for example, the treatment of Type II diabetes, obesity, hyperlipidemia, cardiovascular disease (e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease), anti-neoplastic diseases and tumours (e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer), inflammatory conditions (e.g, inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma, rheumatoid arthritis), and neurodegenerative diseases (e.g, Parkinson's disease, Alzheimer's disease). In an embodiment, compounds in accordance with the invention may be useful for treating Type II diabetes.
In a first aspect the invention provides compounds of general formula (3):

(3) wherein
A is a C5-6 heteroaryl ring optionally substituted with one or more substituents independently selected from halogen, d.4alkyl, O-Ci-4alkyl, haloCi.4alkyl, hydroxy- Ci-4alkyl and CO2R;
Ra-Rj are each independently selected from hydrogen, hydroxyl, halogen, Ci.4alkyl, C3-6 cycloalkyl, haloC1-4alkyl, hydroxyC].4alkyl, O-C]-4alkyl, O-C3.6cycloalkyl, Ci-4alkyl- CO2R, O-CMalkyl-CO2R, O-C^heterocycloalkyl, O-C3-6heteroaryl, N(R)2C i-4alkyl, N(R)3Ci-4alkyl, O-C1-4alkyl-N(R)2, O-C1-4alkyl-N(R)3> C6-10aryl, O-Ci-4alkyl-C6. 10heterocycloalkyl, O-Ci-4alkyl-C6-i0heteroaryl, CO2R, O-sugar, O-Cs-όCycloalkyl, O-C6-i0 aryl, O-Ci-4alkyl-C6-10 aryl, O-benzyl, O-benzoyl, O-C(O)-C1-4alkyl, CO2Ci-4alkyl,
CON(R)2, OC(O)-N(R)2, Ci-4alkanoyloxymethyl, P(O)(OH)(OC1-4alkyl), -P(O)(OC1. 4alkyl)2 and OS(O)(O)NH2; or one or more of Ra and Rb, Rb and Rc, Rc and Rd, Rd and Re, Rf and Rg, Rg and Rh, R1
and Rj and R1 and Rh together form
 each R is independently selected from hydrogen and C1-4alkyl; and pharmaceutically acceptable salts thereof.
each R is independently selected from hydrogen and C1-4alkyl; and pharmaceutically acceptable salts thereof.
In an embodiment of compounds of formula (3), A is selected from pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, pyrazolyl, imidazolyl, thiazolyl, pyrrolyl, furanyl, thiophenyl, oxazolyl, isoxazolyl, isothiazolyl, and triazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci-3 alkyl, 0-C1-3 alkyl, haloC1-3 alkyl, hydroxyC1-3 alkyl and CO2R. In another embodiment, A is selected from pyrazolyl, imidazolyl, thiazolyl, pyrrolyl, furanyl, thiophenyl, oxazolyl, isoxazolyl, isothiazolyl, and triazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci-3 alkyl, 0-C1-3 alkyl, haloC1-3 alkyl, hydroxyC1-3 alkyl and CO2R. In another embodiment A is selected from pyrazolyl, imidazolyl, pyrrolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci-3 alkyl, 0-Ci-3 alkyl, haloC1-3 alkyl, hydroxyCi-3 alkyl and CO2R. In another embodiment A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from halogen, Ci-4alkyl, O-Ci-4alkyl, haloCi-4alkyl, hydroxyCi-4alkyl and CO2R. In another embodiment, A is selected from pyrazolyl and isoxazolyl, wherein each heteroaryl ring may be optionally substituted with one or more substituents independently selected from Ci-4alykl, haloCi-4alkyl and COOH. In another embodiment, A is pyrazolyl optionally substituted with methyl or CF3.
In embodiments of formula (3), each optional substituent on the heteroaryl ring may be independently selected from methyl, ethyl, O-methyl, O-ethyl, fluorine, chlorine, halomethyl, hydroxymethyl, CO2H, CO2Me and CO2Et. In another embodiment, each optional substituent is selected from methyl, ethyl, O-methyl, fluorine, hydroxymethyl, CH2F, CHF2, CF3 and CO2H. In another embodiment, each optional substituent is selected from methyl, fluorine, CF3 and CO2H. In another embodiment, each optional substituent is selected from methyl, CF3 and CO2H.
In embodiments of compounds of formula (3), each R is independently selected from hydrogen, methyl and ethyl.
In embodiments of compounds of formula (3), Ra-Rj are each independently selected from hydrogen, hydroxyl, halogen, Ci-4alkyl, haloC]-4alkyl, hydroxyC1-4alkyl, and O-C1-4alkyl. In another embodiment, Ra-Rj are each independently selected from hydrogen, hydroxyl, methyl, ethyl, CF3, CH2OH, O-methyl and O-ethyl. In a further embodiment, Ra-Rj are each independently selected from hydrogen, hydroxyl, methyl, ethyl, and CF3. In a further embodiment, Ra-Rj are each independently selected from hydrogen, hydroxyl, O-methyl, fluorine, methyl and ethyl. In a further embodiment, Ra-RJ are each independently selected from hydrogen, hydroxyl, O-methyl and fluorine.
In embodiments of compounds of formula (3), one or more of Ra and Rb, Rb and Rc,
<°1 Rc and Rd, Rd and Re, Rf and Rg, Rg and Rh, R1 and RJ and R1 and Rh together form ° * ,
or °"* . In another embodiment, R8 and Rh together form ° * .
In an embodiment of compounds of formula (3), A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally
 substituted with methyl, fluorine, CF3 or CO2H; and Rg and Rh together form 0 ^ . In another embodiment of compounds of formula (3), A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with methyl, fluorine, CF3 or CO2H; Ra and Rc are each hydroxyl; and R8 and
substituted with methyl, fluorine, CF3 or CO2H; and Rg and Rh together form 0 ^ . In another embodiment of compounds of formula (3), A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with methyl, fluorine, CF3 or CO2H; Ra and Rc are each hydroxyl; and R8 and
<°1
Rh together form ° * .
In another embodiment of compounds of formula (3), A is selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with methyl, fluorine, CF3 or CO2H; Ra and Rc are each hydroxyl; Rb Rd and
Re are each independently selected from hydrogen, Cι-2alkyl and CF3, and R8 and Rh
together form 0^ .
In another embodiment of compounds of formula (3), A is pyrazolyl optionally substituted with methyl or CF3; Ra and Rc are each hydroxyl; Rh is fluorine or O-methyl
<°1 or Rg and Rh together form 0 ^ .
In another embodiment of compounds of formula (3), when A is pyrazolyl substituted with methyl or CO2H, Rb, Re, Rf, R*, Rj are each hydrogen, Ra and Rc are each
<°1 hydroxyl, and R8 and Rh together form ° < , then Rd is not ethyl.
In another aspect the present invention relates to compounds of formula (4):

(4) wherein L is selected from C1-4alkylene and C2-4alkenylene; RQ-RU are each independently selected from hydrogen, hydroxyl, halogen, Ci.4alkyl, haloC1-4alkyl, hydroxyCi-4alkyl, O-C1-4alkyl, O-CMalkyl-CO2R, O-C^cycloalkyl, O-C3-6heteroaryl, N(R)2C 1-4alkyl, N(R)3C 1-4alkyl, O-C1-4alkyl-N(R)2, O-C1-4alkyl-N(R)3> C6-i0aryl, O-C6-i0aryl, O-CMalkyl-Q-ioheterocycloalkyl, O-CMalkyl- C6-10heteroaryl, CO2R, O-C3-6heterocycloalkyl, O-C1-4alkyl-C6-10aryl, 0-C(O)-C 1-4alkyl, 0-C(O)-N(R)2, CON(R)2, Ci-4alkanoyloxymethyl, -OP(O)(OH)(OC1-4alkyl), - OP(O)(OC1-4alkyl)2, OS(O)(O)NH2 and O-sugar; or one or more of RQ and RR, RR and Rs, Rs and Rτ and Rτ and Ru together form

Rv-Rz are each independently selected from hydrogen, hydroxyl, halogen,
CMalkyl, haloCi-4alkyl, hydroxyCi-4alkyl, O-C1-4alkyl, O-C1-4alkyl-CO2R, O-Cs-ecycloalkyl, O-C3-6heteroaryl, N(R)2C1-4alkyl, N(R)3C 1-4alkyl, O-C1-4alkyl-N(R)2,
O-CMalkyl-N(R)3, C6-i0aryl, O-C6-i0 aryl, O-C^alkyl-Ce-ioheterocycloalkyl, O-C1-4alkyl-
C6-10heteroaryl, CO2R, O-C3-6heterocycloalkyl, O-C1-4alkyl-C6-i0aryl, O-C(O)-Ct-4alkyl,
0-C(O)-N(R)2, CON(R)2, d^alkanoyloxymethyl, -OP(O)(OH)(OC1-4alkyl), -
OP(O)(OC i-4alkyl)2 and O-sugar; or one or more of Rv and Rw, Rw and Rx, Rx and Rγ and Rγ and Rz together form
 each R is independently selected from hydrogen and
each R is independently selected from hydrogen and
 and pharmaceutically acceptable salts thereof.
and pharmaceutically acceptable salts thereof.
In one embodiment L is C1-2alkylene.
In one embodiment, RR and Rs, or Rs

In one embodiment Rv-Rz are each independently selected from hydrogen, hydroxyl, halogen, Cι--,alkyl, haloCi-4alkyl, hydroxyC1-4alkyl, O-Ci-4alkyl, THP, O-benzyl, and CO2H.
The sugar may be a monosaccharide or a disaccharide. Examples of suitable sugar moieties include but are not limited to glucose, rhamnose, arabinglucose, neohesperidose, and rutinose.
In one embodiment of compounds of formula (4), L is -CH=CH-, Rx and Rz are each hydroxyl, R^-Ru are each independently selected from hydrogen, hydroxyl and O- methyl.
In one embodiment of compounds of formula (4), L is -CH=CH- or -CH2-, Rx and Rz are each hydroxyl; Ry and Rw are each independently selected from hydrogen, methyl
and ethyl; Rv is selected from hydrogen and hydroxyl and RR and Rs together form <° °ϊ< . In one embodiment of compounds of formula (4), L is -CH2-, R" and Rz are each
 hydrogen.
hydrogen.
In one embodiment of formula (4), when L is -CH=CH-, Rv is not hydroxyl.
In one embodiment of compounds of formula (4), when L is -CH2-, Rx and Rz are each hydroxyl, Rv, Rw, RQ, Rτ and Ru are each hydrogen, and RR and Rs together form
<° 01 < , then Rγ is not methyl. In another embodiment, when L is -CH2-, Rx and Rz are each hydroxyl, Rv, Rγ, RQ, Rτ and Ru are each hydrogen, and RR and Rs together form
<°1
° * , then Rw is not ethyl. In another embodiment, when L is -CH=CH-, Rx and Rz are
<°1 each hydroxyl, and RR and Rs together form 0^ , then Rv, Rw, Rγ, RQ, Rτ and Ru are not each hydrogen. In another embodiment, when L is -CH2-, Rx and Rz are each
<°1 hydroxyl, Rw, Rγ, RQ, Rτ and Ru are each hydrogen, and RR and Rs together form 0^ , then Rv is not ethyl. In another embodiment, when L is -CH=CH-, Rx and Rz are each hydroxyl, and Ru is O-methyl, then Rv, Rw, Rγ, RQ, RR and Rs and Rτ are not each hydrogen. In another embodiment, when L is -CH=CH-, Rx and Rz are each hydroxyl, and Rτ is O-methyl, then Rv, Rw, Rγ, RQ, RR and Rs and Ru are not each hydrogen. In another embodiment, when L is -CH=CH-, Rx and Rz are each hydroxyl, and Rs is O- methyl or hydroxyl, then Rv, Rw, Rγ, RQ, RR and Ru and Rτ are not each hydrogen.
In another aspect the present invention relates to compounds of general formula (3a):

(3a) s wherein A is a heteroaryl ring selected from imidazolyl, pyrazolyl, oxazolyl, and isoxazolyl, wherein the heteroaryl ring may be substituted with one or more substituents independently selected from methyl, ethyl, CO2R and CF3;
Ra is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi-2alkyl, O-methyl and CO2R; o Rb, Rc, Rd, Re, Rf, R1, and R" are each independently selected from hydrogen, hydroxyl, C1-2alkyl, CO2R, O-benzyl and O-C1-2alkyl;
R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof. In an embodiment of formula (3a), Ra and Rc are hydroxyl. s In another aspect the present invention relates to compounds of general formula

L is -C(O)C1-3alkylene-, -C(O)C2-3alkenylene-, -C1-3alkyleneC(O)- and0 -C2.3alkenyleneC(O)-;
Rz is selected from hydrogen, hydroxyl, methyl, ethyl, haloC1-2alkyl, O-methyl and CO2R;
RQ, RR, Ru, Rv, Rw, Rx, and Rγ are each independently selected from hydrogen, hydroxyl, C1-2alkyl, O-Ci-2alkyl, CO2R and O-benzyl; s R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
In an embodiment of formula (4a), Rx is selected from hydroxyl, methyl, ethyl, O- methyl and O-ethyl.
Compounds of formulae (3a)-(4a) are subsets of formulae (3)-(4), respectively.
Particular compounds in accordance with embodiments of the present invention include:

Other compounds in accordance with the invention include:



In embodiments of the invention the compound is selected from compounds 6, 7, 8, 11, 12, 14, 15, 16, 17, 18, 19, 20, 23, 24, 26, 27, 28, 30, 31, 32, 33 and 34 above.
In embodiments of the invention the compound is selected from compounds 1, 2, 3, 4, 5, 13, 14, 21, 22 and 35 above.
In other embodiments the compound is selected from compounds 1, 2, 3, 4 and 5 above. In further embodiments the compound is compound 14 above.
In other embodiments of the invention the compound is selected from compounds 6, 7, 8, 11, 12, 15, 16, 17, 18, 19, 20, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 and 34 above.
In further embodiments of the invention the compound is selected from compounds 6, 7, 8, 11, 12, 15, 16, 17, 18, 19, 20, 23, 24, 26, 27, 28, 30, 31, 32, 33 and 34 above.
In further embodiments of the invention the compound is selected from compounds 6, 7, 8, 11 and 12 above.
In further embodiments of the invention the compound is selected from compounds 4, 8 and 11 above.
Representative Synthetic Schemes
Compounds in accordance with the present invention can be prepared using methods known to those skilled in the art. Representative synthetic schemes are shown below.
Scheme 1:

X = OH, R = 2-OMe (1),
X = OH, R = 4-OH (2),
X = OH1 R = 3-OMe (3),
X = H, R = 3, 4-OMe (4),
X = OH, R = 4-OMe (5)
Reagents and Conditions: (a) KOH, MeOH, rt for 6-12 hrs.
10
I5
20
25
Scheme 2:

40c,e
Reagents and Conditions: (a) DHP, PPTS, DCM, rt for 4 hrs; (b) DMF-DMA, 950C for 3 hrs; (c) I2,
Pyridine, CHCI3, rt for 12 hrs; (d) ArB(OH)2, 10% Pd/C, Na2CO3, DME/H20, 450C for 1-3 hrs; (e) p-TsOH, MeOH, THF, 600C for 1-2 hrs.
Compd. R7 R8 Ry R 10

39e, 4Oe H H H a3τR>2^ and RJ together = -0-CH2-O-
Scheme 3:

Reagents and Conditions: (a) Anhydrous HCI, ZnCI2-Et2O, then aq. HCI, heat;
(b) (CF3CO)2O, pyridine, O0C to rt for 12 hrs.
Scheme 4:



Reagents and Conditions: (a) Hydrazine hydrate, EtOH, 700C, 8 hrs.
Scheme 5:

R = F (9b), R = OMe (IOa)

R = OMe (12) _ Reagents and Conditions: (a) BF3OEt2, 90 C for 90 min; (b) Ac2O, K2CO3, DMF1
1150C for 2 hrs; (c) Hydrazine hydrate, EtOH, 800C, 8 hrs.
Compounds in accordance with the present invention could also be prepared according to the following representative synthetic schemes.
Scheme 6:
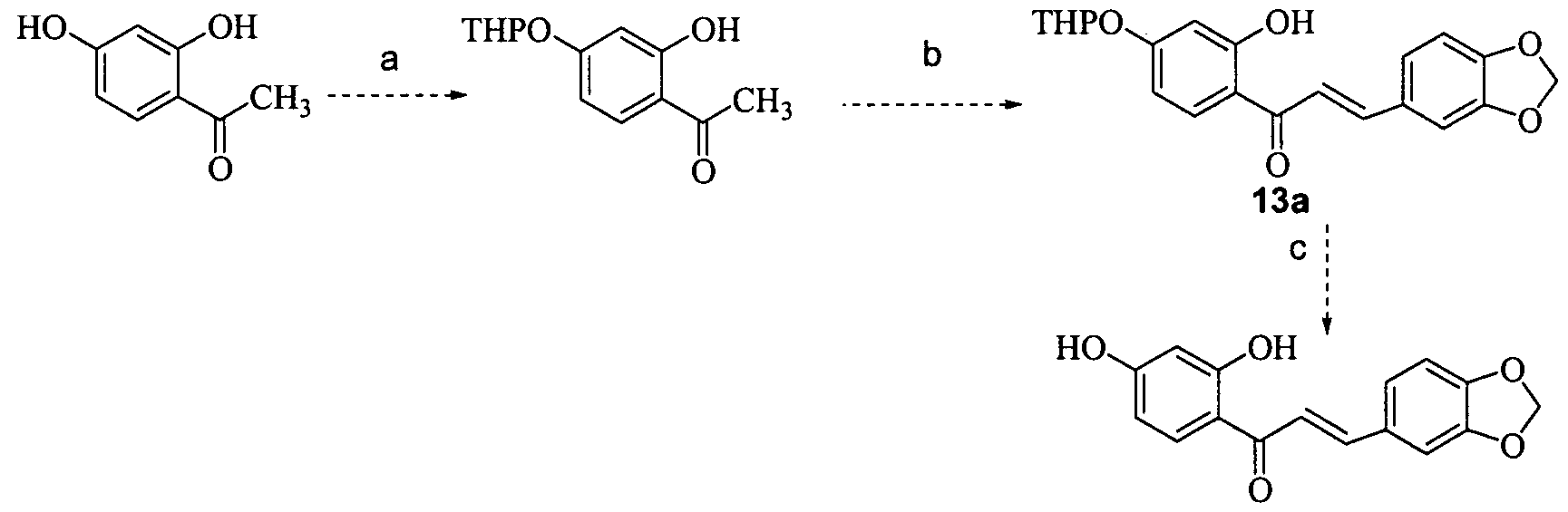
13
Reagents and Conditions: (a) DHP, PPTS, DCM, rt for 4 hrs; (b) Piperonal, KOH, MeOH; (c) P-TsOH, MeOH, THF.
Scheme 7:

17
Reagents and Conditions: (a) BF3-OEt2, 11O0C; (b) (CF3CO)2O, Pyridine; (c) i. CICOCOOEt, Pyridine; ii. NaOH (d) i. Ac2O, Pyridine, ii. NaOH; (e) NH2NH2-H2O, EtOH; (f) NH2OH-HCI, Pyridine.
Scheme 8:
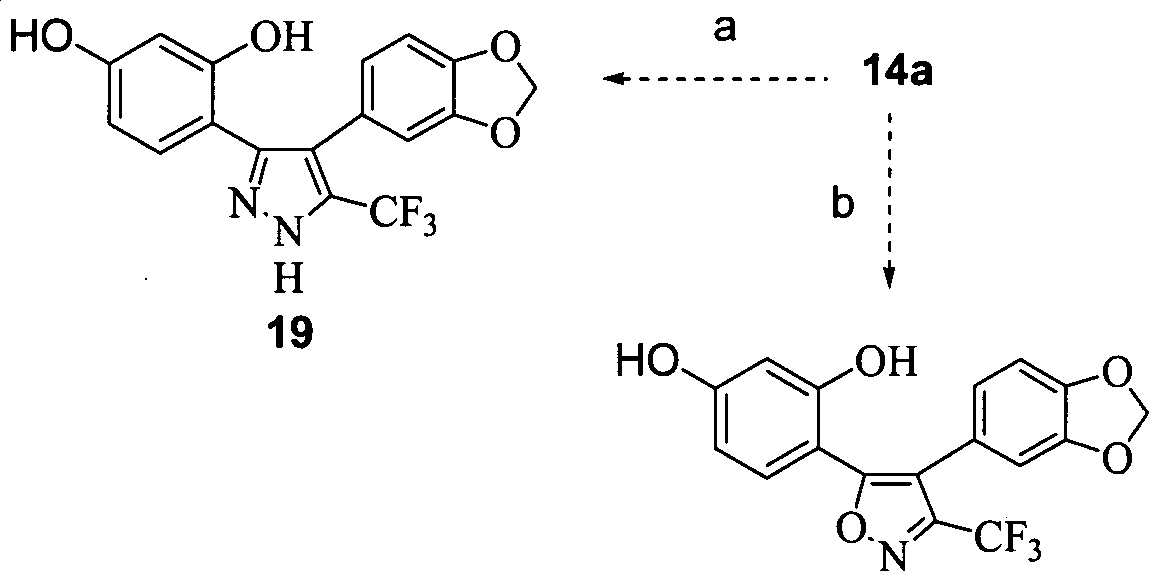
Reagents and Conditions: (a) NH2NH2-H2O, EtOH; (b) NH2OH. HCI, Pyridine.
Scheme 9:

Reagents and Conditions: (a) BF3OEt2, 1100C; (b) i. Ac2O, Pyridine, ii. NaOH; (c) NH2NH2-H2O, EtOH; (d) NH2OKHCI1 Pyridine.
Scheme 10:

Reagents and Conditions: (a) i. CICOCOOEt, Pyridine ii. NaOH; (b) NH2NH2.H2O, EtOH; (c) NH2OH-HCI, Pyridine.
Scheme 11:

Reagents and Conditions: (a) (CF3CO)2O, Pyridine; (b) NH2NH2-H2O, EtOH; (c) NH2OH.HCI, Pyridine.
Scheme 12:

Reagents and Conditions: (a)Anhy. HCI, ZnCI2, Et2O.
Compounds for use in accordance with the present invention may be PPAR agonists. In an embodiment of the invention the compound may be a PPAR-γ agonist. In another embodiment of the invention the compound may be a PPAR-α agonist. In other embodiments compounds in accordance with the present invention may exhibit dual PPAR α/γ agonist activity.
Also within the scope of the present invention are so-called 'pro-drugs' of the compounds of the invention. Thus, certain derivatives of compounds of formulae (3)-(4) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of the invention having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as 'prodrugs'. Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association).
Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of the invention with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
Some examples of prodrugs in accordance with the invention include: (i) where the compound contains a carboxylic acid functionality (COOH), an ester thereof, for example, a compound wherein the hydrogen of the carboxylic acid functionality of a compound of formulae (3)-(4) is replaced by (Ci-C4)alkyl; (ii) where the compound contains an hydroxyl functionality, an ether thereof, for example, a compound wherein the hydrogen of the alcohol functionality of a compound of formulae (3)-(4) is replaced by (Ci-C4)alkanoyloxymethyl, or a phosphonate ester thereof; and
(iii) where the compound contains a primary or secondary amino functionality (-NH2 or -NHR where R ≠ H), an amide thereof, for example, a compound wherein, as the case may be, one or both hydrogens of the amino functionality of the compound is/are replaced by (Q-C^alkanoyl. Included within the scope of the present invention are all stereoisomers, geometric isomers and tautomeric forms of the compounds of formulae (3)-(4) [and (3a)-(4a)], including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof. The present disclosure encompasses all such compounds, including cis- isomers, trans-isomers, (E)-isomers, (Z)-isomers, (i?)-enantiomers, (S)-enantiomers and mixtures thereof including racemic mixtures. Also included are acid addition or base salts wherein the counterion is optically active, for example, an amino acid, e.g, J-lactate or /-lysine, etc, or racemic, for example, (//-tartrate or J/-arginine, and the like.
Cisl trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation. Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (3) or (4) contains an acidic or basic moiety, a base or acid such as 1- phenylethylamine or tartaric acid. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
Chiral compounds of the invention (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
Stereoisomeric conglomerates may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by Ε. L. Εliel and S. H. Wilen (Wiley, New York, 1994).
Therapeutic applications
In a further aspect the present invention relates to a pharmaceutical composition comprising one or more compounds of formula (3), (4), (3a) or (4a), or a prodrug thereof, together with a pharmaceutically acceptable adjuvant, diluent or carrier. Pharmaceutical compositions comprising one or more compounds of formula (3),
(4), (3 a) or (4a) may be used in combination with drugs beneficial for treating a targeted condition or disease. Accordingly, the pharmaceutical compositions of the present invention may contain one or more other active ingredients, in addition to a compound of formula (3), (4), (3a) or (4a). The optimal dosage of the drug/s to be administered in combination with the compound/s of the present invention can be readily determined by one of ordinary skill in the art. The additional drug/s may be administered simultaneously or sequentially with the compounds of the present invention. When administered simultaneously, it is preferable to use a pharmaceutical composition in unit dosage form containing the compound/s of the present invention and other drug/s. When administered in combination, either simultaneously or sequentially, the compound/s of the present invention and additional other drug/s may be used in lower doses than when each is used alone.
Examples of other active ingredients that may be administered in combination with a compound of formula (3), (4), (3 a) or (4a), either separately or in the same pharmaceutical composition, include, but are not limited to the following examples. Agents which improve a patient's lipid profile, including PPAR alpha agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), PPAR alpha/gamma dual agonists such as KRP-297, muraglitazar, tesaglitazar, farglitazar, and JT-501, PPAR delta, nicotinyl alcohol, nicotinic acid or a salt thereof, bile acid sequestrants (cholestyramine, colestipol, and dialkylaminoalkyl derivatives of a cross- linked dextran), HMG-CoA reductase inhibitors (lovastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, atorvastatin, rivastatin, itavastatin, ZD-4522 and other statins), cholesterol absorption inhibitors such as ezetinibe, acyl CoA: cholesterol acyltransferase (ACAT) inhibitors, such as avasimibe, CETP inhibitors, and phenolic anti-oxidants, such as probucol.
Further active ingredients that may be administered in combination with the compounds of the present invention include ileal bile acid transporter inhibitors, antiobesity compounds such as fenfluramine, dexfenfluramine, phentiramine, subitramine, orlistat, neuropeptide Y5 inhibitors, Mc4r agonists, cannabinoid receptor 1 (CB-I) antagonists/inverse agonists, and beta 3 adrenergic receptor agonists, biguanides
including metformin and phenformin, protein tyrosine phosphatase-lB (PTP-IB) inhibitors, dipeptidyl peptidase IV (DP-IV) inhibitors, insulin or insulin mimetics, sulfonylureas including tolbutamide and glipizide or related materials, PPAR gamma agonists and partial agonists such as glitazones and non-glitazones (e.g. rosiglitazone, LY-818, englitazone, balaglitazone, netoglitazone, troglitazone, T-131, LY-300512, MCC-555, and pioglitazone), alpha-glucosidase inhibitors including acarbose, agonists disclosed in WO097/28149, agents for the treatment of inflammatory conditions such as non-steroidal anti-inflammatory drugs, aspirin, glucocorticoids, azulfidine, and cyclo- oxygenase 2 selective inhibitors, glucagon receptor antagonists, and GLP-I, GIP-I and GLP-I analogs such as exendins (for example exenitide).
The compounds of the present invention may also be administered in combination with multiple active compounds, for example, biguanides PPAR agonists, PTP-IB inhibitors, anti-obesity compounds, sulfonylureas, HMG-CoA reductase inhibitors, and DP-IV inhibitors. In a further aspect the present invention relates to a method of treating or preventing a disease in a vertebrate, the method comprising administering to a vertebrate in need thereof an effective amount of a compound of formula (3), (4), (3a), or (4a) as defined herein, or a prodrug thereof, or a composition according to the second aspect of the invention, wherein the disease is selected from Type II diabetes, obesity, hyperlipidemia, cardiovascular disease (e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease), anti-neoplastic diseases and tumours (e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer), inflammatory conditions (e.g, inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma, rheumatoid arthritis), and neurodegenerative diseases (e.g, Parkinson's disease, Alzheimer's disease).
Compounds for use in accordance with the present invention may be PPAR agonists. In an embodiment of the invention the compound may be a PPAR-γ agonist. In another embodiment of the invention the compound may be a PPAR-α agonist. In other embodiments compounds in accordance with the present invention may exhibit dual PPAR α/γ agonist activity. One aspect of the invention is the treatment in vertebrates of diseases that are amenable to amelioration through the activation of PPAR including for example type II diabetes, obesity, hyperlipidemia, cardiovascular disease, anti-neoplastic diseases and tumors, inflammatory conditions and neurogenerative diseases.
In an embodiment the method comprises administering a compound of formula (3) as defined herein. In another embodiment the method comprises administering a
compound of formula (4) as defined herein. In another embodiment the method comprises administering a compound of formula (3a) or (3) as defined herein. In another embodiment the method comprises administering a compound of formula (4a) or (4) as defined herein. In another aspect the present invention relates to the use of a compound of formula
(3), (4), (3a), or (4a) as defined herein, or a prodrug thereof, in the manufacture of a medicament for treating a disease selected from Type II diabetes, obesity, hyperlipidemia, cardiovascular disease (e.g, coronary and ischemic heart disease, atherosclerosis, peripheral vascular disease), anti-neoplastic diseases and tumours (e.g, control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma, breast cancer), inflammatory conditions (e.g, inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma, rheumatoid arthritis), and neurodegenerative diseases (e.g, Parkinson's disease, Alzheimer's disease).
In an embodiment the invention provides for use of a compound of formula (3) as defined herein, or a prodrug thereof, in the manufacture of the medicament. In another embodiment the invention provides for use of a compound of formula (4) as defined herein, or a prodrug thereof, in the manufacture of the medicament. In an embodiment the invention provides for use of a compound of formula (3) or (3a) as defined herein, or a prodrug thereof, in the manufacture of the medicament. In another embodiment the invention provides for use of a compound of formula (4) or (4a) as defined herein, or a prodrug thereof, in the manufacture of the medicament.
In a further embodiment the disease or condition to be treated is Type II diabetes, obesity, or hyperlipidemia. In another embodiment the disease or condition to be treated is Type II diabetes.
Pharmaceutical and/or Therapeutic Formulations
Typically, for medical use, salts of the compounds of formulae (3)-(4) [and (3a)- (4a)] will be pharmaceutically acceptable salts; although other salts may be used in the preparation of the inventive compounds or of the pharmaceutically acceptable salt thereof. By pharmaceutically acceptable salt it is meant those salts which, within the scope of sound medical judgement, are suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio. Pharmaceutically acceptable salts are well known in the art.
Pharmaceutically acceptable salts of compounds of formulae (3)-(4) [and (3a)-(4a)] may be prepared by methods known to those skilled in the art, including for example, (i) by reacting a compound of formula (3) or (4) with the desired acid or base; (ii) by removing an acid- or base-labile protecting group from a suitable precursor of the compound or by ring-opening a suitable cyclic precursor, for example, a lactone or lactam, using the desired acid or base; or (iii) by converting one salt of the compound to another by reaction with an appropriate acid or base or by means of a suitable ion exchange column.
All three reactions are typically carried out in solution. The resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent. The degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
Thus, for instance, suitable pharmaceutically acceptable salts of compounds according to the present invention may be prepared by mixing a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, methanesulfonic acid, succinic acid, fumaric acid, maleic acid, benzoic acid, phosphoric acid, acetic acid, oxalic acid, carbonic acid, tartaric acid, or citric acid with the compounds of the invention. Suitable pharmaceutically acceptable salts of the compounds of the present invention therefore include acid addition salts. S. M. Berge et al. describe pharmaceutically acceptable salts in detail in J.
Pharmaceutical Sciences, 1977, 66Λ-19. The salts can be prepared in situ during the final isolation and purification of the compounds of the invention, or separately by reacting the free base function with a suitable organic acid. Representative acid addition salts include acetate, adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, digluconate, cyclopentanepropionate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptonate, glycerophosphate, hemisulfate, heptonate, hexanoate,' hydrobromide, hydrochloride, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate, pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate, toluenesulfonate, undecanoate, valerate salts, and the like. Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like, as well as non-toxic ammonium, quaternary ammonium, and amine cations, including, but not limited to ammonium, tetramethylammonium, tetraethylammonium,
methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, triethanolamine and the like.
Convenient modes of administration include injection (subcutaneous, intravenous, etc.), oral administration, inhalation, transdermal application, topical creams or gels or powders, or rectal administration. In one embodiment, the mode of administration is parenteral. In another embodiment, the mode of administration is oral. Depending on the route of administration, the formulation and/or compound may be coated with a material to protect the compound from the action of enzymes, acids and other natural conditions which may inactivate the therapeutic activity of the compound. The compound also may be administered parenterally or intraperitoneally.
Dispersions of compounds according to the invention may also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof and in oils. Under ordinary conditions of storage and use, pharmaceutical preparations may contain a preservative to prevent the growth of microorganisms. Pharmaceutical compositions suitable for injection include sterile aqueous solutions
(where water soluble) or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. Ideally, the composition is stable under the conditions of manufacture and storage and may include a preservative to stabilise the composition against the contaminating action of microorganisms such as bacteria and fungi.
Compounds of formulae (3)-(4) [and (3a)-(4a)] according to the present invention may be administered orally, for example, with an inert diluent or an assimilable edible carrier. The compound(s) and other ingredients may also be enclosed in a hard or soft shell gelatin capsule, compressed into tablets, or incorporated directly into an individual's diet. For oral therapeutic administration, the compound(s) may be incorporated with excipients and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like. Suitably, such compositions and preparations may contain at least 1% by weight of active compound. The percentage of the compound(s) in pharmaceutical compositions and preparations may, of course, be varied and, for example, may conveniently range from about 2% to about 90%, about 5% to about 80%, about 10% to about 75%, about 15% to about 65%; about 20% to about 60%, about 25% to about 50%, about 30% to about 45%, or about 35% to about 45%, of the weight of the dosage unit. The amount of compound in therapeutically useful compositions is such that a suitable dosage will be obtained.
The language "pharmaceutically acceptable carrier" is intended to include solvents, dispersion media, coatings, anti-bacterial and anti-fungal agents, isotonic and absorption delaying agents, and the like. The use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the compound, use thereof in the therapeutic compositions and methods of treatment and prophylaxis is contemplated. Supplementary active compounds may also be incorporated into the compositions according to the present invention. It is especially advantageous to formulate parenteral compositions in dosage unit form for ease of administration and uniformity of dosage. "Dosage unit form" as used herein refers to physically discrete units suited as unitary dosages for the individual to be treated; each unit containing a predetermined quantity of compound(s) is calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier. The compound(s) may be formulated for convenient and effective administration in effective amounts with a suitable pharmaceutically acceptable carrier in an acceptable dosage unit. In the case of compositions containing supplementary active ingredients, the dosages are determined by reference to the usual dose and manner of administration of the said ingredients.
In one embodiment, the carrier is an orally administrable carrier.
Another form of a pharmaceutical composition is a dosage form formulated as enterically coated granules, tablets or capsules suitable for oral administration.
Also included in the scope of this invention are delayed release formulations. Compounds of formulae (3)-(4) [and (3a)-(4a)] according to the invention also may be administered in the form of a "prodrug". A prodrug is an inactive form of a compound which is transformed in vivo to the active form. Suitable prodrugs include esters, phosphonate esters etc, of the active form of the compound.
In one embodiment, the compound of formulae (3)-(4) [and (3a)-(4a)] may be administered by injection. In the case of injectable solutions, the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyethylene glycol, and the like), suitable mixtures thereof, and vegetable oils. The proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants. Prevention of the action of microorganisms can be achieved by including various anti-bacterial and/or anti-fungal agents. Suitable agents are well known to those skilled in the art and include, for example, parabens, chlorobutanol, phenol, benzyl alcohol, ascorbic acid, thimerosal, and
the like. In many cases, it may be preferable to include isotonic agents, for example, sugars, polyalcohols such as mannitol, sorbitol, sodium chloride in the composition. Prolonged absorption of the injectable compositions can be brought about by including in the composition an agent which delays absorption, for example, aluminium monostearate and gelatin.
Sterile injectable solutions can be prepared by incorporating the analogue in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by filtered sterilisation. Generally, dispersions are prepared by incorporating the analogue into a sterile vehicle which contains a basic dispersion medium and the required other ingredients from those enumerated above.
Tablets, troches, pills, capsules and the like can also contain the following: a binder such as gum gragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid and the like; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin or a flavouring agent such as peppermint, oil of wintergreen, or cherry flavouring. When the dosage unit form is a capsule, it can contain, in addition to materials of the above type, a liquid carrier. Various other materials can be present as coatings or to otherwise modify the physical form of the dosage unit. For instance, tablets, pills, or capsules can be coated with shellac, sugar or both. A syrup or elixir can contain the analogue, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavouring such as cherry or orange flavour. Of course, any material used in preparing any dosage unit form should be pharmaceutically pure and substantially non-toxic in the amounts employed. In addition, the analogue can be incorporated into sustained-release preparations and formulations. Preferably, the pharmaceutical composition may further include a suitable buffer to minimise acid hydrolysis. Suitable buffer agent agents are well known to those skilled in the art and include, but are not limited to, phosphates, citrates, carbonates and mixtures thereof.
Single or multiple administrations of the compounds and/or pharmaceutical compositions according to the invention may be carried out. One skilled in the art would be able, by routine experimentation, to determine effective, non-toxic dosage levels of the compound and/or composition of the invention and an administration pattern which would be suitable for treating the diseases and/or infections to which the compounds and compositions are applicable.
Further, it will be apparent to one of ordinary skill in the art that the optimal course of treatment, such as the number of doses of the compound or composition of the invention given per day for a defined number of days, can be ascertained using convention course of treatment determination tests.
5 Generally, an effective dosage per 24 hours may be in the range of about 0.0001 mg to about 1000 mg per kg body weight; for example, about 0.001 mg to about 750 mg per kg body weight; about 0.01 mg to about 500 mg per kg body weight; about 0.1 mg to about 500 mg per kg body weight; about 0.1 mg to about 250 mg per kg body weight; or about 1.0 mg to about 250 mg per kg body weight. More suitably, an effective dosage per
I0 24 hours may be in the range of about 1.0 mg to about 200 mg per kg body weight; about 1.0 mg to about 100 mg per kg body weight; about 1.0 mg to about 50 mg per kg body weight; about 1.0 mg to about 25 mg per kg body weight; about 5.0 mg to about 50 mg per kg body weight; about 5.0 mg to about 20 mg per kg body weight; or about 5.0 mg to about 15 mg per kg body weight. is Alternatively, an effective dosage may be up to about 500mg/m2. For example, generally, an effective dosage is expected to be in the range of about 25 to about 500mg/m2, about 25 to about 350mg/m2, about 25 to about 300mg/m2, about 25 to about 250mg/m2, about 50 to about 250mg/m2, and about 75 to about 150mg/m2.
In another embodiment, a compound of Formula (3) or (4) may be administered in
20 an amount in the range from about 100 to about 1000 mg per day, for example, about 200 mg to about 750 mg per day, about 250 to about 500 mg per day, about 250 to about 300 mg per day, or about 270 mg to about 280 mg per day.
Therapeutic advantages may be realised through combination regimens. Thus, compounds in accordance with the present invention may be administered as part of a
25 therapeutic regimen with other drugs. It may be desirable to administer a combination of active compounds, for example, for the purpose of treating a particular disease or condition. For example, compound(s) according to the present invention may be administered in combination therapy with known antidiabetic or antilipidemic agents.
Accordingly, it is within the scope of the present invention that two or more
30 pharmaceutical compositions, at least one of which contains a compound according to the present invention, may be combined in the form of a kit suitable for co-administration of the compositions. In combination therapy the respective agents may be administered simultaneously, or sequentially in any order.
35
Screening assay to identify PPAR agonists
Another aspect of the present invention relates to an assay for identifying PPAR agonists. Thus, the invention relates to a method for identifying a PPAR agonist, comprising determining ligand-receptor interactions of a candidate compound with docking templates; comparing the ligand-receptor interactions of the candidate compound with the interactions of a known PPAR agonist; and thereby determining whether a candidate compound is a PPAR agonist. In one embodiment the agonist is a PPAR-γ agonist. In another embodiment the agonist is a PPAR-α agonist. In a further embodiment the agonist may be a dual PPAR α/ γ agonist.
The method of the invention may be used to identify PPAR agonists that share little similarity with known ligands. The present invention utilises the approach of "Induced Fit Docking", or IFD. The
IFD protocol was run from the graphical user interface accessible within Maestro 8.5. It was carried out on PPAR-α [(figure 1), (PDB Code: 117G)] with claim 3 and 4 compounds, and AZ-242, the known PPAR-α agonist. The overall procedure has four stages: Briefly, during Stage 1 initial softened-potential Glide docking is performed on a vdW scaled-down rigid-receptor; a scaling of 0.5/ 0.5 was set for receptor / ligand vdW radii, respectively. The top 20 poses for each test ligand was retained. In Stage 2, receptor sampling and refinement was performed on residues within 5.0 of each ligand for each of the 20 ligand:protein complexes. Here, Prime side-chain sampling and prediction was carried out. The side-chains, as well as the backbone and ligand, underwent subsequent energy minimizations. A total of 20 induced fit receptor conformations were generated for each of the five test ligands. Stage 3 involved re-docking the test ligands into their respective 20 structures that are within 30.0 kcal / mol of their lowest energy structure. Finally, the ligand poses were scored in Stage 4 using a combination of Prime and GlideScore scoring functions in which the top ranked pose for each ligand was chosen as the final result. The XP scoring function was used in all docking stages.
Ligands showing high affinity towards PPAR-γ and PPAR-α in silico are subsequently tested in vitro using a PPAR-γ transcriptional factor assay, a PPAR-γ reporter gene luciferase assay and a PPAR-α reporter gene luciferase assay. The activity of compounds identified by this process in inducing PPAR-γ mRNA and protein
expression in vitro may be also examined to see if the effect can be abolished in the presence of GW9662, a potent synthetic PPAR-γ antagonist (Bendixen et al., 2001). Biological data from the transactivation assays may be used to characterize compounds as agonists of human PPAR. The invention will now be described in more detail, by way of illustration only, with respect to the following examples. The examples are intended to serve to illustrate this invention and should not be construed as limiting the generality of the disclosure of the description throughout this specification.
EXAMPLES
General
1H NMR spectra were recorded at 300 MHz using a Varian Gemini 300 spectrometer. Chemical shifts (δπ) are quoted in parts per million (ppm), referenced internally to tetramethylsilane (TMS) at 0 ppm in CDCl3 and Coupling constants (J) are reported in Hertz. Low resolution mass spectra were recorded on a Finnigan/MAT TSQ 7000 LCMS/MS spectrometer and high resolution mass spectra were recorded on a Micromass Qtof II spectrometer; only molecular ions (M+ 1). Thin layer chromatography (TLC) was performed on Merck aluminium backed plates pre-coated with silica (0.2 mm, 60F254) which were developed using UV fluorescence (254 nm). Flash vacuum chromatography was performed on silica gel (Merck silica gel 6OH, particle size 5^0 μm). Chemicals were purchased from Aldrich, Boron Molecular and at the highest available grade.
General synthetic procedure for compound 1 to 5: Dihydroxyacetophenone (1 mmol) in MeOH (3 mL) is treated with KOH (1 mmol) under magnetically stirred condition for 10 min at room temperature followed by addition of benzaldehyde (1 mmol) The mixture is stirred magnetically for 6-12 hrs at room temperature. After the completion of the reaction, a yellow precipitate was formed and this served as indicator for monitoring the reaction visually. MeOH was removed under reduced pressure. The residue was diluted with water (5 mL), neutralized with 2% aqueous HCl and extracted with EtOAc (3><20 mL). The combined EtOAc extracts were washed with brine solution (5 mL), dried (Na2SO4) and concentrated under reduced pressure to afford 1 to 5 (32-45 %).
Example 1 l-(2,4-Dihydroxyphenyl)-3-(2-methoxyphenyl)prop-2-en-l-one (l)
Yellow solid, yield: 32 %
1H NMR (300 MHz, acetone-dg): δ 12.90 (s, IH), 8.12 (d, IH, J= 8.3 Hz), 7.90 (d, IH, J 5 = 15.2 Hz), 7.72 (d, IH, J= 15.1 Hz), 7.56-6.60 (m, 6H), 3.97 (s, 3H). Mass (+ve ion mode m/z): 271 (M++l).
Example 2 l-(2,4-Dihydroxyphenyl)-3-(4-hydroxyphenyl)prop-2-en-l-one (2)
Pale yellow solid, yield: 41 % io 1H NMR (300 MHz, acetone-de): δ 12.40 (s, IH), 8.98 (s, IH), 8.03 (s, IH), 7.86 (d, IH, J = 15.1 Hz), 7.70 (d, IH, J= 15.1 Hz), 7.52 (d, IH, J = 3.0 Hz), 7.05 (dd, IH, J= 3.0, 8.6 Hz), 6.91 (d, 2H, J= 8.6 Hz), 6.79 (d, IH, J= 8.6 Hz). Mass (+ve ion mode m/z): 257 (M++l).
Example 3 is l-(2,4-Dihydroxyphenyl)-3-(3-methoxyphenyl)prop-2-en-l-one (3)
Pale Yellow solid, yield: 36 %
1H NMR (300 MHz, acetone-d6): δ 12.80 (s, IH), 8.08 (d, IH, J = 8.2 Hz), 7.88 (d, IH, J
= 15.1 Hz), 7.68 (d, IH, J = 15.1 Hz), 7.52-6.50 (m, 6H), 3.91 (s, 3H).
Mass (+ve ion mode m/z): 271 (M++l).
20 Example 4 l-(4-hydroxyphenyl)-3-(3,4-dimethoxyphenyl)prop-2-en- 1-one (4)
Yellow plate, yield: 38 %
1H NMR (300 MHz, acetone-de): δ 12.98 (s, IH), 8.03 (d, 2H, J = 7.3 Hz), 7.78 (d, IH, J = 15.5 Hz), 7.56 (d, IH, J = 15.3 Hz), 7.34 (d, IH, J= 8.4 Hz), 7.01 (s, IH), 7.26 (d, IH, J 25. = 2.1 Hz), 6.78 (d, 2H, J = 7.1 Hz), 3.87 (s, 3H), 3.85 (s, 3H). Mass (+ve ion mode m/z): 285 (M++l).
Example 5 l-(2,4-Dihydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-l-one (5)
Yellow needle, yield: 45 % 3o δ 13.22 (s, IH), 8.12 (d, IH, J= 8.7 Hz), 7.80 (d, IH, J= 15.3 Hz), 7.70 (d, IH, J= 15.3
Hz), 7.49 (d, 2H, J= 8.7 Hz), 7.01 (d, 2H, J= 8.7 Hz), 6.36 (IH, d, J= 2.1 Hz), 6.46 (dd, IH, J= 2.1, 8.7 Hz), 3.84 (s, 3H). Mass (+ve ion mode m/z): 271 (M++ 1).
Example 6
3-Iodo-7-(tetrahydro-2//-pyran-2-yloxy)-4iy-chromen-4-one (38)
A solution of DHP (3,4-dihydro-2H-pyran) (9 mL, 98.7 mmol) in CH2Cl2 (54 mL) was added dropwise to a solution of acetophenone (5 g, 32.8 mmol) and PPTS (Pyridinium-p-toluenesulfonate) (296 mg) at rt. The resulting mixture was stirred for 4 h at rt, then washed with saturated aqueous NaHCO3 solution, and extracted with CH2Cl2. The collected organic extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude was diluted with DMF/DMA (6.54 mL, 49.3 mmol) and the resulting mixture was stirred at 95 °C for 3 h. After evaporation of volatiles, the obtained solid was dissolved in CHCl3 (53 mL) and successively treated with pyridine (2.66 mL, 33 mmol) and I2 (16.7 g, 66 mmol). The resulting mixture was stirred at rt for 12 h. The reaction was hydrolyzed with saturated aqueous Na2S2O3 solution and stirred for 30 min at room temperature (rt). The aqueous phase was extracted with CH2Cl2. The collected organic extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography (20% EtOAc/hexane then 40% EtOAc/hexane) gave 38 (11 g, 90% yield) as a colorless solid.
1H NMR (300 MHz, CDCl3): δ 8.27 (s, IH), 8.18 (d, IH, J= 9.6 Hz), 7.16-7.10 (m, 2H),
5.57 (m, IH), 3.91-3.80 (m, IH), 3.70-3.62 (m, IH), 2.10-1.90 (m, 3H), 1.82-1.58 (m,
3H).
Mass (+ve ion mode m/z): 373 (M++l).
Example 7
3-(Benzo[</l[l,3]dioxol-5-yl)-7-(tetrahydro-2H-pyran-2-yloxy)-4Λ|r-chromen-4-one (39c)
To a solution of 3-iodo-7-(tetrahydro-2H-pyran-2-yloxy)-4//-chromen-4-one (38) (500 mg, 1 mmol) in DME (4 mL) and H2O (4 mL) were added Na2CO3 (427 mg, 3 mmol), 3,4-methylenedioxyphenylboronic acid (267 mg, 1.2 mmol), and Pd/C (71 mg, 5 mol %). The resulting mixture was stirred for 1 h at 45 °C and then filtered. The catalyst was washed with H2O (3 mL) and CH2Cl2 (5 mL). The aqueous phase was extracted twice with CH2Cl2. The collected organic extracts were dried (Na2SO4), filtered, and
concentrated under reduced pressure. The crude was purified by flash chromatography (50% EtOAc/hexane) to give 39c (472 mg, 96% yield) as a colourless solid. 1H NMR (300 MHz, CDCl3): δ 8.24 (d, IH, J= 9.3 Hz), 7.95 (s, IH), 7.15-7.11 (m, 3H), 7.0 (dd, IH, J= 1.5 Hz, 8.1 Hz), 6.90 (d, IH, J= 8.1Hz), 6.03 (s, 2H), 5.59 (m, IH), 3.94- s 3.84 (m, IH), 3.73-3.65 (m, IH), 2.10-1.89 (m, 3H), 1.80-1.60 (m, 3H). Mass (+ve ion mode m/z): 366 (M++ 1).
Example 8 3-(4-Fluorophenyl)-7-(tetrahydro-2//-pyran-2-yloxy)-4//-chromen-4-one (39e)o To a solution of 3-iodo-7-(tetrahydro-2H-pyran-2-yloxy)-4H-chromen-4-one (38)
(500 mg, 1 mmol) in DME (4 mL) and H2O (4 mL) were added Na2CO3 (427 mg, 3 mmol), 4-fluorophenylboronic acid (282 mg, 1.5 mmol), and Pd/C (71 mg, 5 mol %). The resulting mixture was stirred for 1.5 h at 45 0C and then filtered. The catalyst was washed with H2O (3 mL) and CH2Cl2 (5 mL). The aqueous phase was extracted twices with CH2Cl2. The collected organic extracts were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude was purified by flash chromatography (60% EtOAc/hexane) to give 39e (356 mg, 78% yield) as a colourless solid. 1H NMR (300 MHz, CDCl3): δ 8.42 (d, IH, J= 8.7 Hz), 8.01 (s, IH), 7.65 (m, 2H), 7.28 ( t, 2H, J = 8.7 Hz), 6.99 (dd, IH, J = 2.1 Hz, 8.4 Hz), 6.90 (d, IH, J = 2.4 Hz), 5.57 (m,0 IH), 3.94-3.70 (m, IH), 3.71-3.60 (m, IH), 2.11-1.90 (m, 3H), 1.81- 1.63 (m, 3H). Mass (+ve ion mode m/z): 341 (M++ 1).
Example 9 3-(Benzo[</] [l,3]dioxol-5-yl)-7-hydroxy-4//-chromen-4-one (40c)
To a solution of 3-(Benzo[d][l,3]dioxol-5-yl)-7-(tetrahydro-2H-pyran-2-yloxy)-4H-s chromen-4-one (39c) (450 mg) in MeOH (30 mL) and THF (30 mL) was added p-TsOH (35 mg) at rt. The resulting mixture was stirred at 60 °C for 1 h, then Et3N (0.3 mL) was added, and volatiles were removed under reduced pressure. Purification by flash chromatography (40% EtOAc/hexane then 5% MeOH/EtOAc) provided 40c (277 mg, 80% yield) as a colorless solid. 0 1H NMR (300 MHz, DMSO): δ 10.82 (br s, IH), 8.36 (s, IH), 7.99 (d, IH, J = 9 Hz), 7.17-6.88 (m, 5H), 6.06 (s, 2H). Mass (+ve ion mode m/z): 283 (M++ 1).
5
Example 10 3-(4-Fluorophenyl)-7-hydroxy-4//-chromen-4-one (40e)
To a solution of 3-(4-fluorophenyl)-7-(tetrahydro-2H-pyran-2-yloxy)-4H-chromen-
4-one (39e) (300 nig) in MeOH (30 mL) and THF (30 mL) was added /?-TsOH (30 mg) at rt. The resulting mixture was stirred at 60 °C for 1 h, then Et3N (0.3 mL) was added, and volatiles were removed under reduced pressure. Purification by flash chromatography
(40% EtOAc/hexane then 5% MeOH/EtOAc) provided 4Oe (187 mg, 83% yield) as a colorless solid.
1H NMR (300 MHz, DMSO): δ 10.84 (br s, IH), 8.42 (s, IH), 8.01 (d, IH, J = 8.7 Hz), 7.65 (m, 2H), 7.28 (t, 2H, J= 8.7 Hz), 6.99 (dd, IH5 J= 2.1 Hz, 8.4 Hz), 6.90 (d, IH, J =
2.4 Hz).
Mass (+ve ion mode m/z): 257 (M++l).
Example 11 2-(4-Fluorophenyl)-l-(2,4-dihydroxyphenyl)ethanone (52)
With stirring, a rapid current of dry hydrogen chloride is passed for 10 min into a solution of 4-fluorobenzyl cyanide (500 mg, 1 mmol) in dry toluene (10 mL) cooled to O0C. Then a solution of resorcinol (448 mg, 1.1 mmol) and fused zinc chloride (251 mg, 0.5 mmol) in dry ether (5 mL) is added. Saturation with hydrogen chloride is continued for 4 h and then kept overnight at room temperature. The solvent was decanted and triturated twice with dry toluene. The hot water (50 mL) added and the mixture is kept at 900C and pH 1 for 30 min. The product is separated from the hot solution and washed several times with water. The crude was purified by flash chromatography to give 52 (455 mg, 50% yield) as an oily product. 1H NMR (300 MHz, CDCl3): δ 12.11 (br s, IH), 7.55 (d, IH, J = 8.9 Hz), 7.05-6.95 (m, 4H), 6.20-6.10 (m, 2H), 3.95 (s, 2H). Mass (+ve ion mode m/z): 247 (M++ 1).
Example 12 2-(Trifluoromethyl)-3-(4-fluorophenyl)-7-hydroxy-4//-chromen-4-one (53)
A solution of trifluoroacetic anhydride (0.22 mL) is added to a solution of 52 (400 mg) in 2 mL of dry pyridine at O0C. The reaction mixture was shaken, with ice cooling, for 10-15 min and is left overnight. On the following day, it is heated to 40-500C for 10- 15 min and again left at room temperature for 12 h. Then it is poured into 20-30 mL cold
water, and the precipitate is filtered off and crystallized from ethanol to give 53 as a cream colour solid.
1H NMR (300 MHz, DMSO): δ 11.18 (s, IH), 7.93 (d, IH, J = 8.7 Hz), 7.36-7.25 (m, 4H), 7.03 (dd, IH, J= 2.4, 9 Hz), 6.95 (d, IH, J= 2.4 Hz). Mass (+ve ion mode m/z): 325 (M++l).
General synthetic procedure for compounds 6 to 8:
Hydrazine hydrate (-55% aq.; 2mL) was added to a suspension of flavonoids (1 mmol) in ethanol (10 mL) and the mixture heated under reflux for 8 h to give a brown solution. The solution was allowed to cool and concentrated to a pale yellow solid. The solids were washed with water, to give crude product. A sample was recrystallised from boiling toluene, to give the pyrazole, washed with hexane and dried in vacuo.
Example 13
4-(4-(Benzo[d][lv3]dioxol-6-yl)-li7-pyrazol-3-yl)benzene-l,3-diol (6) Colourless solid, yield: 77%
1H NMR (300 MHz, DMSO): δ 12.89 (s, IH), 9.57 (s, IH), 9.53 (s, IH), 7.78 (s, IH)5 6.80-6-45 (m, 6H), 6.02 (s, 2H). Mass (+ve ion mode m/z): 297 (M++ 1).
Example 14 4-(4-(4-Fluorophenyl)-l//-pyrazol-3-yl)benzene-l,3-diol (7)
Colourless solid, yield: 73%
1H NMR (300 MHz, DMSO): δ 13.31 (s, IH), 9.69 (s, IH), 9.54 (s, IH), 7.86 (s, IH),
7.27-7.11 (m, 4H), 6.72 (d, IH, J = 8.4 Hz), 6.32 (d, IH, J = 2.2 Hz), 6.13 (dd, IH, J =
2.3, 8.4 Hz). Mass (+ve ion mode m/z): 271 (M++l).
Example 15
4-(5-(Trifluoromethyl)-4-(441uorophenyl)-liϊ-pyrazol-3-yl)benzene-l,3-diol (8)
Colourless solid, yield: 70%
1H NMR (300 MHz, DMSO): δ 13.51 (s, IH), 9.76 (s, IH), 9.57 (s, IH), 7.22-7.12 (m, 4H), 6.74 (d, IH, J= 8.1 Hz), 6.35 (d, IH, J= 2.4 Hz), 6.15 (dd, IH, J= 2.1, 8.1 Hz). Mass (+ve ion mode m/z): 239 (M++l).
Example 16
2-(4-Fluorophenyl)-l-(2,4-dihydroxyphenyl)ethenone (9b)
Resorcinol (1 g, 1 mmol) and 4-fluorophenylacetic acid (1.4 g, 1 mmol) in boron trifluoride etherate (5.68 mL, 0.2 mol) were heated, under a nitrogen atmosphere, at 900C for 90 mins to give a red solution. The solution was allowed to cool and poured into aqueous sodium acetate (10% aq.; 100 mL) and the mixture stirred to give a pale yellow precipitate. The solids were removed by filtration and washed with water (100 mL). The solids were taken up in ethyl acetate (150 mL) and washed with water (100 mL). The solution was dried over anhydrous sodium sulfate and concentrated to a yellow semi- solid. Trituration with diethyl ether (50 mL) gave 9b (626 mg, 28%) as a pale orange solid after drying in vacuo.
1H NMR (300 MHz, CDCl3): 1H NMR (300 MHz, CDCl3): δ 12.11 (br s, IH), 7.55 (d,
IH5 J= 8.9 Hz), 7.05-6.95 (m, 4H), 6.20-6.10 (m, 2H), 3.95 (s, 2H). Mass (+ve ion mode m/z): 247 (M++l).
Example 17
3-(4-fluorophenyl)-7-hydroxy-2-methyl-4//-chromen-4-one (9c)
Acetic anhydride (0.77 mL, 4 mmol) was added to a suspension of potassium carbonate (1.02 g, 4 mmol) and 2-(4-fluorophenyl)-l-(2,4-dihydroxyphenyl)ethenone (9b) (500 mg, 1 mmol) in DMF (5 mL), and the resulting suspension heated at 115 °C for 120 mins. The mixture was allowed to cool and poured into water (100 mL), to give an off-white precipitate. The solids were removed by filtration and washed with water (50 mL) and diethyl ether (2 x 40 mL), to give 9c (466 mg, 85%) as an off-white powder after drying in vacuo 1H NMR (300 MHz, DMSO): 11.21 (s, IH), 7.94 (d, IH, J= 8.7 Hz), 7.42-7.25 (m, 4H), 7.10-6.90 (m, 2H), 2.20 (s, 3H). Mass (+ve ion mode m/z): 271 (M++l).
Example 18
4-(4-(4-Fluorophenyl)-5-methyl-l//-pyrazol-3-yl)benzene-l,3-diol (11)
Hydrazine hydrate (~55% aq.; 1.5 mL) was added to a suspension of 3-(4- fluorophenyl)-2-methyl-4-oxo-4H-chromen-7-yl acetate (9c) (400 mg) in ethanol (10 mL) and the mixture heated under reflux for 8 h to give a brown solution. The solution was allowed to cool and concentrated to a pale yellow solid. The solids were washed with water, to give 11 (294 mg, 70%) as an off-white solid after drying in vacuo. A sample
was recrystallised from boiling toluene, to give the pyrazole 11 as a white crystalline solid, washed with hexane and dried in vacuo.
1H NMR (300 MHz, DMSO): δ 12.94 (s, IH), 9.51 (s, IH), 9.40 (s, IH), 7.24-7.13 (m, 4H), 6.74 (d, IH, J= 8.4 Hz), 6.32-6.02 (m, 2H), 2.16 (s, 3H). Mass (+ve ion mode m/z): 285 (M++ 1).
Example 19 l-(2,4-Dihydroxyphenyl)-2-(4-methoxyphenyl)ethanone (10b)
Resorcinol (1 g, 1 mmol) and 4-methoxyphenylacetic acid (1.50 g, 1 mmol) in boron trifluoride etherate (5.68 mL, 0.2 mol) were heated, under a nitrogen atmosphere, at 900C for 90 mins to give a pale red solution. The solution was allowed to cool and poured into aqueous sodium acetate (10% aq.; 100 mL) and the mixture stirred to give a pale yellow precipitate. The solids were removed by filtration and washed with water (100 mL). The solids were taken up in ethyl acetate (150 mL) and washed with water (100 mL). The solution was dried over anhydrous sodium sulfate and concentrated to a yellow semi-solid. Trituration with diethyl ether (50 mL) gave 10b (469 mg, 20%) as a pale orange solid after drying in vacuo.
1H NMR (DMSO, 300 MHz): δ 7.95 (d, IH, J = 8.9 Hz), 7.22 (d, 2H, J = 8.7 Hz), 6.9 (d, 2H, J = 8.7 Hz), 6.42 (d, IH, J = 9.8 Hz), 6.25 (s, IH), 4.18 (s, 2H), 3.75 (s, 3H). Mass (+ve ion mode m/z): 259 (M++l).
Example 20
7-Hydroxy-3-(4-methoxyphenyl)-2-methyl-4//-chromen-4-one (10c)
Acetic anhydride (0.53 mL, 4 mmol) was added to a suspension of potassium carbonate (782 mg, 4 mmol) and l-(2,4-dihydroxyphenyl)-2-(4-methoxyphenyl)ethanone (10b) (400 mg, 1 mmol) in DMF (5 mL), and the resulting suspension heated at 115 0C for 120 mins. The mixture was allowed to cool and poured into water (100 mL), to give an off-white precipitate. The solids were removed by filtration and washed with water (50 mL) and diethyl ether (2 x 40 mL), to give 10c (332 mg, 76%) as an off-white powder after drying in vacuo 1H NMR (DMSO, 300 MHz): δ 7.84 (d, IH, J = 8.7 Hz), 7.12 (d, 2H, J = 8.8 Hz), 7.05 (d, 2H, J = 8.8 Hz), 6.93 (d, IH, J = 8.7 Hz), 6.81 (s, IH), 3.83 (s, 3H), 2.25 (s, 3H). Mass (+ve ion mode m/z): 283 (M++l).
Example 21
7-hydroxy-3-(4-methoxyphenyl)-2-methyl-4H-chromen-4-one (12)
Hydrazine hydrate (-55% aq.; 1 mL) was added to a suspension of 7-hydroxy-3-(4- methoxyphenyl)-2-methyl-4//-chromen-4-one (10c) (300 mg) in ethanol (10 mL) and the mixture heated under reflux for 8 h to give a pale brown solution. The solution was allowed to cool and concentrated to a pale yellow solid. The solids were washed with water, to give 12 (236 mg, 75%) as an off-white solid after drying in vacuo. A sample was recrystallised from boiling toluene, to give the pyrazole 12 as a colourless solid, washed with hexane and dried in vacuo. 1H NMR (DMSO, 300 MHz): δ 7.21 (d, 2H, J = 8.8 Hz), 7.03 (d, 2H, J = 8.8 Hz), 6.92 (d, IH, J = 8.6 Hz), 6.35 (s, IH), 6.14 (d, IH, J= 8.7 Hz), 3.85 (s, 3H), 2.25 (s, 3H).
Mass (+ve ion mode m/z): 297 (M++l).
Example 22 Screening assay to identify PPAR-γ and PPAR-α agonists (HEK cell lines)
EXPERIMENTAL PROCEDURE
Cell Culture
Human embryonic kidney (HEK) 293 cell line was obtained from American Type
Culture Collection (USA). All materials used for tissue culture were purchased from Invitrogen, Australia unless specified. HEK 293 cells were grown in Dulbecco's modified
Eagle's medium/F-12 (DMEM/F-12), containing L-glutamine supplemented with penicillin (100 U/mL), streptomycin (100 mg/mL) and 10% (v/v) heat- inactivated foetal bovine serum in a humidified atmosphere of 5% CO2 and 95% O2 at 37 0C. (Bramlett et al, 2003; Frederiksen et al, 2004).
Transfection and Luciferase Assay (PPAR-γ)
The transfection and luciferase procedures were performed as described previously (Bramlett et al., 2003) with slight modification. The HEK 293 cell line was transfected with tK-PPREx3-Luc plasmid, pSG5-hPPAR-γ plasmid and pSV-β-galactosidase (Promega, Australia) control plasmid. Cells were transfected with FuGENE 6 transfection reagent (Roche, Australia) in accordance with the manufacturer's instructions. After 24 h at 37°C, cells were harvested and plated into 96-well plates at 5 x 104 cells per well in complete transfection media and allowed to attach over night at 37 °C. The cells were
then treated with rosiglitazone and GW1929 as positive controls, DMSO (0.1%) as a negative control and the test samples. After 48 hours, the cells were lysed and assayed for luciferase and β-galactosidase activities using the Bright-Glo Luciferase Assay system and Beta-Glo Assay system (Promega, Australia), respectively. The results were expressed as relative luciferase activity normalized to the β-galactosidase signal (fold difference compared to negative control).
Cell Proliferation Assay (PPAR-γ)
Human embryonic kidney cell line (HEK 293) is seeded overnight then treated with
10 various concentrations of rosiglitazone, GWl 929 and test compounds (0 - 100 μM) and incubated for 48 hours at 37°C in a humidified atmosphere with 5% CO2. MTS (tetrazolium salt) reagent (CellTiter96® Aqueous One Solution Cell Proliferation Assay, Promega, Sydney, Australia) is added and samples are incubated for a further 1-4 hours before finally being analyzed using a BMG POLARstar Galaxy Microplate Reader (λ:
I5 490 nm). Results: Reporter Gene Luciferase Assay (PPAR-γ)
A series of compounds (Table 3) have been designed, synthesized and evaluated for PPAR-γ fold activation activity in the Human Embryonic Kidney cell line (HEK 293) at
20 various concentrations (5 and 25 μM). Known PPAR-γ agonist rosiglitazone was used as positive control, while DMSO (0.1%) acted as a negative control. As shown in Table 3 and figure 2, compounds 4, 8 and 11 demonstrated a comparable PPAR-γ fold activation activity (3.5, 3.9 and 5.3, respectively) compared to the known PPAR-γ agonist rosiglitazone (4) at 25 μM.
2S
Table 3 Structures and PPAR-γ fold activation using HEK 293 cells (Luciferase assay).
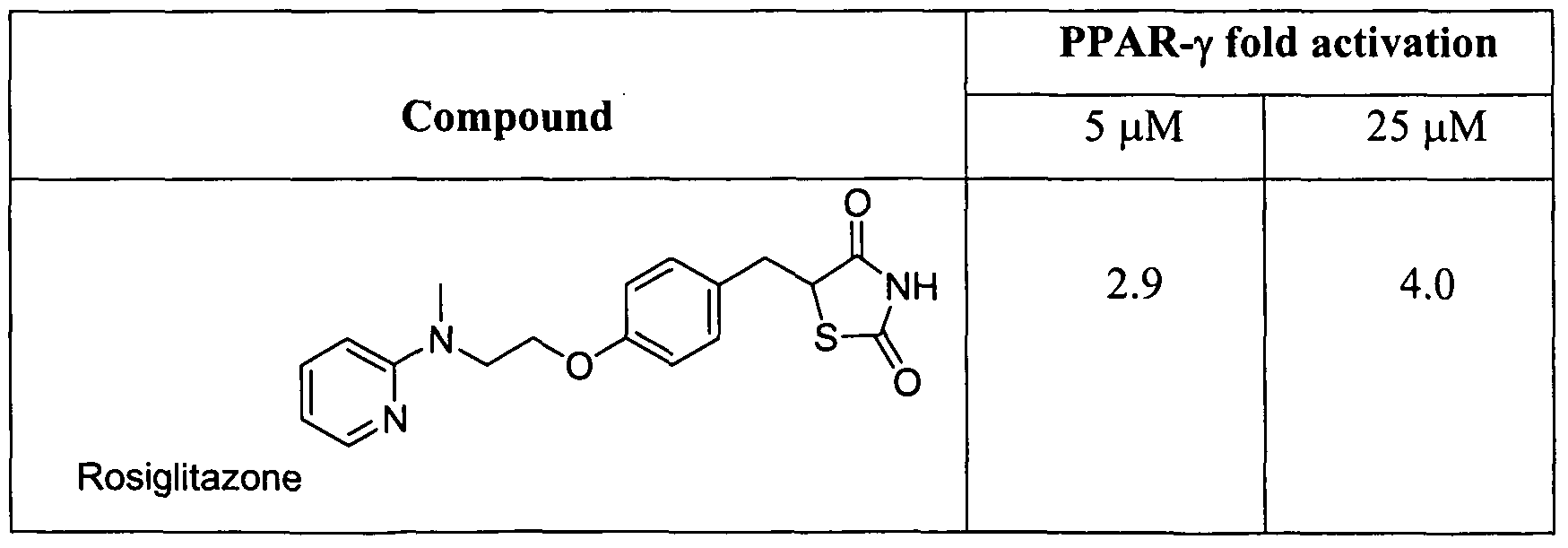


The equivalent experiments will be conducted for PPAR-α in accordance with the following procedures.
Transfection and Luciferase Assay (PPAR-α) The transfection and luciferase procedures will be performed as described previously (Bramlett et al., 2003) with slight modification. The HEK 293 cell line will be transfected with tK-PPREx3-Luc plasmid, pBI-G-hPPAR-α plasmid and pSV-β- galactosidase (Promega, Australia) control plasmid. Cells will be transfected with FuGENE 6 transfection reagent (Roche, Australia) in accordance with the manufacturer's instructions. After 24 h at 37°C, cells will be harvested and plated into 96-well plates at 5 x 104 cells per well in complete transfection media and allowed to attach over night at 37°C. The cells will then be treated with WY-14643, Fenofibrate as positive controls, DMSO (0.1%) as a negative control and the test samples. After 48 hours, the cells will be lysed and assayed for luciferase and β-galactosidase activities using the Bright-Glo Luciferase Assay system and Beta-Glo Assay system (Promega, Australia), respectively. Cell Proliferation Assay (PPAR-α)
HEK 293 cells will be seeded overnight then treated with various concentrations of WY-14643, Fenofibrate and test compounds (0 - 100 μM) and incubated for 48 hours at 37 0C in a humidified atmosphere with 5% CO2. MTS (tetrazolium salt) reagent (CellTiter96® Aqueous One Solution Cell Proliferation Assay, Promega, Sydney,
Australia) will be added and samples will be incubated for a further 1-4 hours before finally being analyzed using a BMG POLARstar Galaxy Microplate Reader (λ: 490 nm).
Example 23 Screening assay to identify PPAR-γ agonists (THP-I cell lines)
Future experiments will involve a screening assay to identify PPAR-γ agonists (THP-I cell lines). They will be conducted according to the following experimental procedures. General
Anti-actin primary antibody, bovine serum albumin (BSA), dimethyl sulfoxide (DMSO), GW9662 and phorbol 12-myristate 13-acetate (PMA) will be purchased from Sigma-Aldrich (Sydney, Australia). Natural products, totaling 200 compounds, will be sourced from the Herbal Medicines Research and Education Center (Faculty of Pharmacy, University of Sydney). Cell Culture The THP-I monocytes and macrophages will be grown in RPMI 1640 in the presence of 50 (M (β-mercaptoethanol. All media will contain L-glutamine supplemented with penicillin (100 U/ml)/ streptomycin (100 (g/ml), and 10% (v/v) heat-inactivated fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 and 95% O2 at 37°C. To induce monocyte differentiation into macrophages the THP-I monocytes will be treated with PMA (400 ng/ml) for 72 h, before selective PPAR-γ ( agonist rosiglitazone, test samples or vehicle (0.1% DMSO) will be added and incubated for a further 48 h in culture medium for the macrophage treatment experiments. The PPAR-γ antagonist, GW9662 (5 μM) will be added 1 h prior to addition of positive control or test samples. Cell-Based Transcriptional Factor Assay The PPAR-γ Transcription Factor Assay is a sensitive ELISA method for detecting
PPAR-γ transcription factor DNA binding activity in nuclear extracts of THP-I derived macrophage cell line. The ELISA assay will be conducted according to the manufacturer's instructions (Cayman Chemical, Sydney, Australia). The purification of cellular nuclear extract from the cultured cells will be prepared with CelLytic™ NuCLEAR™ Extraction Kit (Sigma, Sydney, Australia). Cell Proliferation Assay
THP-I macrophage cells will be seeded overnight, then treated with various concentrations of rosiglitazone, GW9662 and test compounds (0.01 - 100 μM) before further incubation for 3 days at 37°C in a humidified atmosphere with 5% CO2. MTS (tetrazolium salt) reagents (the CellTiter96® Aqueous One Solution Cell Proliferation
Assay, Promega, Sydney, Australia) will be added, incubated for 4 h and finally analyzed using a microtiter plate reader (model 3550, Bio-Rad) (λ: 490 nm). mRNA Analysis
Total mRNA will be prepared separately from the THP-I macrophage cells using TRIzol (Invitrogen, Sydney, Australia). The relative levels of specific mRNAs will be assessed by RT-PCR as described previously (Abe et al., 2002). Single-stranded cDNA is synthesized from 1 μg of total RNA using Superscript II RNAse H Reverse Transcriptase, as per instructions of the manufacturer (Invitrogen, Sydney, Australia). PCR will be performed on a thermocycler, PTC-200 DNA engine (MJ Research Inc, USA). The required cDNA will be synthesized with the Platinum® Pfx DNA Polymerase method (Invitrogen, Sydney, Australia). The genes examined will be PPAR-γ (L40904; 382bp; sense: 5'-GAGCCCAAGTTTGAGTTTGC-S'; 5'-
TGGAAGAAGGGAAATGTTGG-3') and β-actin (NMOOI lOl; 629bp; sense: 5'- GGAGTAACCAGGTCGTCCAA-3'; 5'-GAAGGTGCCCAGAATACCAA-S'). The PCR samples will be electrophoresed on 5-12% acrylamide gel (29:1, acrylamide:N,N'- methylene-bis-acrylamide) in TBE buffer [89 mM Tris-base pH 7.6, 89 mM boric acid, 2 mM EDTA]. The gels will be stained with ethidium bromide (10 μg/ml) and photographed on top of a 280 nm UV light box (Biorad® Gel Doc 1000, Australia). The gel images will be digitally captured with a CCD camera and analyzed with ImageJ 1.29x (NIH, USA). RT-PCR values will be presented as a ratio of the specified gene signal in the selected linear amplification cycle divided by the β-actin signal. Western Blot
THP-I cells will be seeded and treated with PMA (400 ng/ml) for 72 h to obtain THP-I macrophages. The macrophages will be treated with 0.1% DMSO, test compounds (30 μM) and rosiglitazone (30 μM) for 48 h. Then the cells will be washed with PBS and lysed with RIPA lysis buffer for protein extraction. The protein contents in the samples will be measured using BCA protein estimation kit and 20 μg of sample will be loaded onto 4-12% NuP AGE® Bis-Tris Gel (Invitrogen, Sydney, Australia). After electrophoresis at 200 V for 1 h, the protein will be transferred to PVDF membrane and blocked overnight in skim milk (5% skim milk in tris buffered saline). On the subsequent day the PVDF membranes will be treated with antihuman PPAR-γ mouse monoclonal primary antibody (1:500 dilution; Santa Cruz Biotechnology, USA) followed with horseradish peroxidase-conjugated anti-mouse secondary antibody (1 : 10,000 dilution; Promega, USA). The antibody treatment will be performed for Ih followed by 30 min wash with the washing buffer (Tris buffered saline with 0.1% Tween-20). Protein
expression will be detected by chemiluminescence method (Roche). The PVDF membranes will be exposed to X-ray film (Kodak, USA) and developed using the SRX- 101 A X-ray developer (Konica, Taiwan). Quantitation of the results are performed by using the NIH Image J software. After stripping with stripping buffer (Glycine (15 g), SDS (1 g), Tween-20 (10 mL), pH 2.2) the membranes will be re-probed with anti-actin primary antibody (1:10,000 dilution; Sigma, Australia) will be re- incubated with the secondary horseradish peroxidase antibody, and protein bands are detected as described above.
REFERENCES
Abe A, Kiriyama Y, Hirano M, Miura T, Kamiya H, Harashima H and Tokumitsu Y (2002) Troglitazone suppresses cell growth of KU812 cells independently of PPARgamma. Eur J Pharmacol 436(l-2):7-13.
Bendixen AC, Shevde NK, Dienger KM, Willson TM, Funk CD and Pike JW (2001) IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor gamma 1. Proc Natl Acad Sci U S A 98(5):2443-2448.
Bramlett KS, Houck KA, Borchert KM, Dowless MS, Kulanthaivel P, Zhang Y, Beyer TP, Schmidt R, Thomas JS, Michael LF, Barr R, Montrose C, Eacho PI, Cao G and Burris TP (2003) A natural product ligand of the oxysterol receptor, liver X receptor. J Pharmacol Exp Ther 307(1 ):291-296.
Davies GF, McFie PJ, Khandelwal RL and Roesler WJ (2002) Unique ability of troglitazone to up-regulate peroxisome proliferator-activated receptor-gamma expression in hepatocytes. J Pharmacol Exp Ther 300(l):72-77.
Frederiksen KS, Wulff EM, Sauerberg P, Mogensen JP, Jeppesen L and Fleckner J (2004) Prediction of PPAR-alpha ligand-mediated physiological changes using gene expression profiles. J Lipid Res 45(3):592-601.
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC and Mainz DT (2006) Extra precision glide: docking and scoring
incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49(21):6177-6196.
Gampe RT, Jr., Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM and Xu HE (2000) Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. MoI Cell 5(3):545-555.
Liang YC, Tsai SH, Tsai DC, Lin-Shiau SY and Lin JK (2001) Suppression of inducible cyclooxygenase and nitric oxide synthase through activation of peroxisome proliferator- activated receptor-gamma by flavonoids in mouse macrophages. FEBS Lett 496(1): 12-18.
Nestel P (2003) Isoflavones: their effects on cardiovascular risk and functions. Curr Opin Lipidol 14(l):3-8.
Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK and Milburn MV (1998) Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395(6698):137-143.
Sherman W, Day T, Jacobson MP, Friesner RA and Farid R (2006) Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49(2):534-553.
Penning TD, Kramer SW, Lee LF, Collins PW, Koboldt CM, Seibert K, Veenhuizen AW, Zhang YY and Isakson PC (1997) 3,4-Diarylpyrazoles: potent and selective inhibitors of cyclooxygenase-2. Biorg Med Chem Lett 7(16):2121-2124.
Khilya VP, Aitmambetov, Ismailov AM, Grishko LG (1994) Synthetic and modified isoflavonoids xv. Interaction of synthetic analogs of isoflavones with hydrazine hydrate and its derivatives. Chem Natural Comp 30(5):580-583.
Dymock BW, Barril X, Brough PA, Cansfield JE, Massey A, McDonald E, Hubbard RE, Surgenor A, Roughley SD, Webb P, Workman P, Wright L and Drysdale MJ (2005)
Novel, potent small-molecule inhibitors of the molecular chaperone Hsp90 discovered through structure-based design. J Med Chem 48:4212-4215.
Claims
CLAIMS:
1. A compound of general formula (3):

(3) wherein
5 A is a C5-6heteroaryl ring optionally substituted with one or more substituents independently selected from halogen, C1-4alkyl, O-Ci-4alkyl, haloCi-4alkyl, hydroxy- C1-4alkyl and CO2R;
Ra-RJ are each independently selected from hydrogen, hydroxyl, halogen, C1-4alkyl,
C3-6cycloalkyl, haloC1-4alkyl, hydroxyC1-4alkyl, O-Ci-4alkyl, Ci-4alkyl-CO2R, 0-Ci- io 4alkyl-CO2R, O-Q.eheterocycloalkyl, O-C3-6heteroaryl, N(R)2C ]-4alkyl, N(R)3C1-4alkyl,
O-C1-4alkyl-N(R)2, O-C1-4alkyl-N(R)3, C6-i0aryl, O-Ci^alkyl-Ce-ioheterocycloalkyl, 0-Ci- 4alkyl-C6-10heteroaryl, CO2R, O-sugar, O-C^cycloalkyl, O-C5-6cycloalkyl, O-C6-i0 aryl,
O-C1-4alkyl-C6-10 aryl, O-benzyl, O-benzoyl, O-C(O)-C].4alkyl, CHO, CO2CMalkyl,
CON(R)2, OC(O)-N(R)2, Ci-4alkanoyloxymethyl, and -P(O)(OH)(OCMalkyl), - i5 P(O)(OCi-4alkyl)2 and OS(O)(O)NH2; or
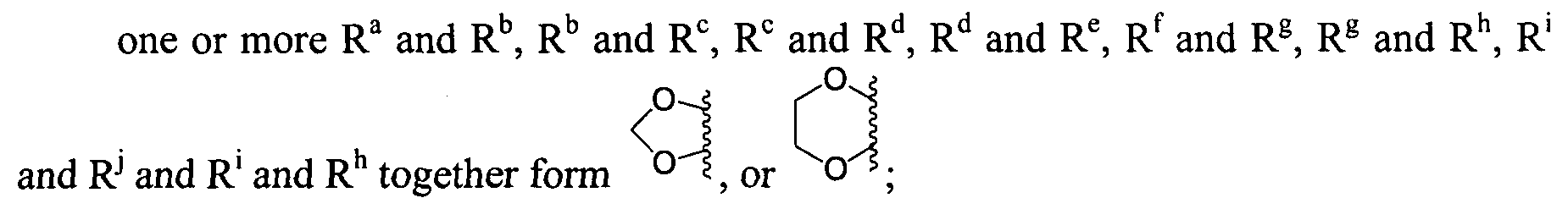
20 2. A compound according to claim 1, wherein A is a heteroaryl ring selected from pyrazolyl, imidazolyl, pyrrolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with one or more substituents independently selected from halogen, C1-3 alkyl, 0-C1-3 alkyl, haloC1-3 alkyl, hydroxyCi-3 alkyl and CO2R, wherein R is hydrogen or C1-4 alkyl.
25 3. A compound according to claim 1, wherein A is a heteroaryl ring selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with one or more substituents independently selected from methyl, ethyl, O-methyl, O-ethyl, fluorine, chlorine, halomethyl, hydroxymethyl, CO2H, CO2Me and CO2Et.
4. A compound according to any one of claims 1 to 3, wherein A is a heteroaryl ring selected from pyrazolyl, imidazolyl, oxazolyl, and isoxazolyl, wherein each heteroaryl ring is optionally substituted with methyl, fluorine, CF3 or CO2H; R laa aanndd R "cc are each hydroxyl;
R >b r R>d and j r R>ee are each independently selected from hydrogen, Ci-2alkyl and CF3, and R8
 and Rh together form 0 ^
and Rh together form 0 ^
5. A compound according to any one of claims 1 to 3, wherein A is pyrazolyl optionally substituted with methyl or CF3;
Ra and Rc are each hydroxyl; and
<°1
10 Rh is fluorine or O-methyl; or Rg and Rh together form 0 ^ .
6. A compound according to any one of claims 1 to 3, wherein when A is pyrazolyl substituted with methyl or CO2H, Rb, Re, Rf , R1, RJ are each hydrogen, and Ra
<°1 and Rc are each hydroxyl, and Rg and Rh together form 0 ^ , then Rd is not ethyl.
I5 7. A compound according to any one of claims 1 to 3 selected from:



8. A compound of general formula (3 a):

(3a) wherein A is a heteroaryl ring selected from imidazolyl, pyrazolyl, oxazolyl, and isoxazolyl, wherein the heteroaryl ring may be substituted with one or more substituents independently selected from methyl, ethyl, CO2R and CF3;
Ra is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi.2alkyl, O-methyl and CO2R;
10 Rb, Rc, Rd, Re, Rf, R1, and Rj are each independently selected from hydrogen, hydroxyl, Ci-2alkyl, CO2R, O-benzyl and O-C1-2alkyl;
R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
9. A compound of general formula (4):

I5 (4) wherein L is selected from Ci-4alkylene and C2-4alkenylene;
RQ-RU are each independently selected from hydrogen, hydroxyl, halogen, C1-4alkyl, haloC1-4alkyl, hydroxyCi-4alkyl, O-Ci-4alkyl, O-Ci-4alkyl-CO2R, O-C3-6cycloalkyl, O-C3-6heteroaryl, N(R)2C i^alkyl, N(R)3C 1-4alkyl, O-C1-4alkyl-N(R)2,
20 O-C1-4alkyl-N(R)3, C6-ioaryl, O-C6-i0aryl, O-CMalkyl-Cβ-ioheterocycloalkyl, O-Ci-4alkyl- C6-ioheteroaryl, CO2R, O-C3-6heterocycloalkyl, O-C1-4alkyl-C6-10aryl, O-C(O)-Ci-4alkyl, 0-C(O)-N(R)2, CON(R)2, C^alkanoyloxymethyl, -OP(O)(OH)(OC)-4alkyl), - OP(O)(OC1-4alkyl)2, OS(O)(O)NH2 and O-sugar; or
one or more of RQ and RR, RR and Rs, Rs and Rτ and Rτ and Ru together form

Rv-Rz are each independently selected from hydrogen, hydroxyl, halogen,
C1-4alkyl, haloC1-4alkyl, hydroxyC1-4alkyl, O-C1-4alkyl, O-Ci-4alkyl-CO2R, s O-C3-6cycloalkyl, O-C3-6heteroaryl, N(R)2C1-4alkyl, N(R)3C1-4alkyl, O-C1-4alkyl-N(R)2,
O-C1-4alkyl-N(R)3, C6-i0aryl, 0-C6-10 aryl, O-Ci-4alkyl-C6-i0heterocycloalkyl, O-CMalkyl-
C6.ioheteroaryl, CO2R,O-C3.6heterocycloalkyl, O-Ci-4alkyl-C6-10aryl, 0-C(O)-C 1-4alkyl,
0-C(O)-N(R)2, CON(R)2, C^alkanoyloxymethyl, -OP(O)(OH)(OCMalkyl), -
OP(O)(OC1-4alkyl)2 and O-sugar; or o one or more of Rv and Rw, Rw and Rx, Rx and Rγ and Rγ and Rz together form
M or V^ each R is independently selected from hydrogen and C1-4 alkyl; and pharmaceutically acceptable salts thereof. to claim 9, wherein RR and Rs, or Rs and Rτ together

Rv-Rz are each independently selected from hydrogen, hydroxyl, halogen, C1-4alkyl, haloC1-4alkyl, hydroxyC1-4alkyl, O-Ci-4alkyl, THP, O-benzyl, and CO2H.
11. A. compound according to claim 9, wherein when L is -CH=CH-, Rv is not hydroxyl. 0 12. A compound of general formula (4a):

(4a) wherein
L is -C(O)C1-3alkylene-, -C(O)C2-3alkenylene-, -Ci.3alkyleneC(O)- and -C2-3alkenyleneC(O)-; 5 Rz is selected from hydrogen, hydroxyl, methyl, ethyl, haloCi-2alkyl, O-methyl and
CO2R;
RQ, RR, Ru, Rv, Rw, Rx, and Rγ are each independently selected from hydrogen, hydroxyl, C1-2alkyl, O-C1-2alkyl, CO2R and O-benzyl;
R is selected from hydrogen, methyl and ethyl; and pharmaceutically acceptable salts thereof.
13. A pharmaceutical composition comprising one or more compounds of formula (3), (4), (3a) or (4a) according to any one of claims 1 to 12, or a prodrug thereof,
5 together with a pharmaceutically acceptable adjuvant, diluent or carrier.
14. A method of treating or preventing a disease in a vertebrate, the method comprising administering to a vertebrate in need thereof an effective amount of a compound of formula (3), (4), (3a) or (4a) according to any one of claims 1 to 12 or a prodrug thereof, or a composition according to claim 13, wherein the disease is selectedo from Type II diabetes, obesity, hyperlipidemia, cardiovascular disease, anti-neoplastic diseases and tumours, inflammatory conditions, and neurodegenerative diseases.
15. The method according to claim 14, wherein the disease or condition to be treated is Type II diabetes, obesity, hyperlipidemia or cardiovascular disease.
16. The method according to claim 14 or 15, wherein the cardiovascular disease iss selected from the group consisting of coronary and ischemic heart disease, atherosclerosis and peripheral vascular disease.
17. The method according to claim 14, wherein the anti-neoplastic diseases and tumours are selected from the group consisting of control of cell growth, cell differentiation, motility and apoptosis, neuroblastoma and breast cancer. o 18. The method according to claim 14, wherein the inflammatory condition is selected from the group consisting of inflammatory bowel diseases, psoriasis, chronic inflammatory airway disease, asthma and rheumatoid arthritis.
19. The method according to claim 14, wherein the neurodegenerative disease is selected from Parkinson's disease and Alzheimer's disease. 5 20. The method according to claim 14 or 15, wherein the disease or condition to be treated is Type II diabetes.
21. A method for identifying a PPAR agonist, the method comprising determining ligand-receptor interactions of a candidate compound with docking templates; 0 comparing the ligand-receptor interactions of the candidate compound with the interactions of a known PPAR agonist; and thereby determining whether a candidate compound is a PPAR agonist.
22. The method according to claim 20, wherein the PPAR agonist is a PPAR-γ agonist. 5
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007904674A AU2007904674A0 (en) | 2007-08-29 | PPAR - Gamma agonists | |
| AU2007904674 | 2007-08-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009026658A1 true WO2009026658A1 (en) | 2009-03-05 |
Family
ID=40386589
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2008/001292 WO2009026658A1 (en) | 2007-08-29 | 2008-08-29 | Ppar agonists |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009026658A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013022951A1 (en) * | 2011-08-10 | 2013-02-14 | Kaohsiung Medical University | Composition for treating diabetes and metabolic diseases and a preparation method thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US20140194474A1 (en) * | 2011-08-10 | 2014-07-10 | Kaohsiung Medical University | Composition for treating atherosclerosis and a preparation method thereof |
| US8865754B2 (en) | 2011-03-03 | 2014-10-21 | Proteotech Inc. | Compounds for the treatment of neurodegenerative diseases |
| CN108904481A (en) * | 2018-10-11 | 2018-11-30 | 温州医科大学 | O-hydroxylate chalcone analog is preparing the application in anti-oxidation medicine |
| US11034669B2 (en) | 2018-11-30 | 2021-06-15 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| CN115671097A (en) * | 2022-08-23 | 2023-02-03 | 上海交通大学医学院附属第九人民医院 | Compound for preventing and treating atherosclerosis and plaque instability and application thereof |
| CN115894422A (en) * | 2022-12-26 | 2023-04-04 | 上海中医药大学 | PPAR gamma agonist and combination and application thereof |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003055860A1 (en) * | 2001-12-21 | 2003-07-10 | Vernalis (Cambridge) Limited | 3,4-diarylpyrazoles and their use in the therapy of cancer |
| WO2003101927A1 (en) * | 2002-05-31 | 2003-12-11 | Proteotech, Inc. | Compounds, compositions and methods for the treatment of amyloid diseases and synucleinopathies such as alzheimer's disease, type 2 diabetes, and parkinson's disease |
| WO2004000817A2 (en) * | 2002-06-24 | 2003-12-31 | Pfizer Products Inc. | Benzimidazole compounds and their use as estrogen agonists/antagonists |
| WO2004065374A1 (en) * | 2003-01-17 | 2004-08-05 | Astellas Pharma Inc. | Oxazole derivatives as inhibitors of cyclooxygenase |
| WO2004072051A1 (en) * | 2003-02-11 | 2004-08-26 | Vernalis (Cambridge) Limited | Isoxazole compounds as inhibitors of heat shock proteins |
| US20050131036A1 (en) * | 2003-12-11 | 2005-06-16 | Byoung-Mog Kwon | Pharmaceutical compositions of diaryl-isoxazole derivatives for the prevention and treatment of cancers |
| WO2008041610A1 (en) * | 2006-10-03 | 2008-04-10 | Nippon Kayaku Kabushiki Kaisha | Compound of resorcinol derivative with polymer |
-
2008
- 2008-08-29 WO PCT/AU2008/001292 patent/WO2009026658A1/en active Application Filing
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003055860A1 (en) * | 2001-12-21 | 2003-07-10 | Vernalis (Cambridge) Limited | 3,4-diarylpyrazoles and their use in the therapy of cancer |
| WO2003101927A1 (en) * | 2002-05-31 | 2003-12-11 | Proteotech, Inc. | Compounds, compositions and methods for the treatment of amyloid diseases and synucleinopathies such as alzheimer's disease, type 2 diabetes, and parkinson's disease |
| WO2004000817A2 (en) * | 2002-06-24 | 2003-12-31 | Pfizer Products Inc. | Benzimidazole compounds and their use as estrogen agonists/antagonists |
| WO2004065374A1 (en) * | 2003-01-17 | 2004-08-05 | Astellas Pharma Inc. | Oxazole derivatives as inhibitors of cyclooxygenase |
| WO2004072051A1 (en) * | 2003-02-11 | 2004-08-26 | Vernalis (Cambridge) Limited | Isoxazole compounds as inhibitors of heat shock proteins |
| US20050131036A1 (en) * | 2003-12-11 | 2005-06-16 | Byoung-Mog Kwon | Pharmaceutical compositions of diaryl-isoxazole derivatives for the prevention and treatment of cancers |
| WO2008041610A1 (en) * | 2006-10-03 | 2008-04-10 | Nippon Kayaku Kabushiki Kaisha | Compound of resorcinol derivative with polymer |
Non-Patent Citations (4)
| Title |
|---|
| DYMOCK, B. W. ET AL.: "Novel, Potent Small-Molecule Inhibitors of the Molecular Chaperone Hsp90 Discovered through Structure-Based Design", J. MED. CHEM., vol. 48, 2005, pages 4212 - 4215, XP055354595 * |
| SMITH, N. F. ET AL.: "Preclinical pharmacokinetics and metabolism of a novel diaryl pyrazole resorcinol series of heat shock protein 90 inhibitors", MOL. CANCER. THER., vol. 5, no. 6, 2006, pages 1628 - 1637, XP055149018 * |
| SUMANASEKERA, W. K. ET AL.: "Heat Shock Protein-90 (Hsp90) Acts as a Repressor of Peroxisome Proliferator-Activated Receptor-alpha (PPARalpha) and PPARbeta Activity", BIOCHEMISTRY, vol. 42, 2003, pages 10726 - 10735, XP055354600 * |
| TANAKA, K. ET AL.: "Diaryl Pyrroles: A New Series of Anti-Inflammatory Agents", EXPERIENTIA, vol. 28, no. 8, 1972, pages 937 - 938 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8865754B2 (en) | 2011-03-03 | 2014-10-21 | Proteotech Inc. | Compounds for the treatment of neurodegenerative diseases |
| TWI417088B (en) * | 2011-08-10 | 2013-12-01 | Univ Kaohsiung Medical | Composition for treating diabetes and metabolic diseases and a preparation method thereof |
| US20140194474A1 (en) * | 2011-08-10 | 2014-07-10 | Kaohsiung Medical University | Composition for treating atherosclerosis and a preparation method thereof |
| WO2013022951A1 (en) * | 2011-08-10 | 2013-02-14 | Kaohsiung Medical University | Composition for treating diabetes and metabolic diseases and a preparation method thereof |
| US9085520B2 (en) | 2011-08-10 | 2015-07-21 | Kaohsiung Medical University | Composition for treating diabetes and metabolic diseases and a preparation method thereof |
| US10098853B2 (en) * | 2011-08-10 | 2018-10-16 | Kaohsiung Medical University | Composition for treating atherosclerosis and a preparation method thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| CN108904481A (en) * | 2018-10-11 | 2018-11-30 | 温州医科大学 | O-hydroxylate chalcone analog is preparing the application in anti-oxidation medicine |
| US11034669B2 (en) | 2018-11-30 | 2021-06-15 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| CN115671097A (en) * | 2022-08-23 | 2023-02-03 | 上海交通大学医学院附属第九人民医院 | Compound for preventing and treating atherosclerosis and plaque instability and application thereof |
| CN115671097B (en) * | 2022-08-23 | 2024-02-13 | 上海交通大学医学院附属第九人民医院 | Compound for preventing and treating atherosclerosis and plaque instability and application thereof |
| CN115894422A (en) * | 2022-12-26 | 2023-04-04 | 上海中医药大学 | PPAR gamma agonist and combination and application thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009026658A1 (en) | Ppar agonists | |
| WO2009026657A1 (en) | Flavonoid ppar agonists | |
| CN110770226B (en) | Sulfoximine glycosidase inhibitors | |
| US7939545B2 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP3349761B1 (en) | Hepatitis b core protein modulators | |
| TWI691496B (en) | Substituted chromanes and method of use | |
| KR20210089195A (en) | Biphenyl-based compounds, intermediates thereof, preparation methods, pharmaceutical compositions and uses | |
| KR20190026827A (en) | Aromatic acetylene or aromatic ethylenic compounds, intermediates thereof, process for their preparation, pharmaceutical compositions and uses | |
| KR20100108337A (en) | Inhibitors of human immunodeficiency virus replication | |
| UA121379C2 (en) | Prodrugs of pyridone amides useful as modulators of sodium channels | |
| JP7398605B2 (en) | FXR small molecule agonists and their preparation methods and uses | |
| US8829196B2 (en) | TRPA1 antagonists | |
| US9725463B2 (en) | Substituted heterocyclic compounds as CRAC modulators | |
| KR20080073337A (en) | Oxazole compound and pharmaceutical composition | |
| KR20140021655A (en) | Inhibitors of viral replication, their process of preparation and their therapeutical uses | |
| US7998993B2 (en) | TRPV1 antagonists | |
| JP2006508930A (en) | Novel bioactive diphenylethene compounds and their therapeutic applications | |
| TW201805278A (en) | Pyridinyl derivatives, pharmaceutical compositions and uses thereof | |
| JP2023545784A (en) | Small molecule compounds and their uses as JAK kinase inhibitors | |
| JP2010504322A (en) | 3-Amino-pyridine derivatives for the treatment of metabolic disorders | |
| CN111801314B (en) | ASK1 inhibitor compounds and uses thereof | |
| JP7041129B2 (en) | Anti-infective agent | |
| WO2023085392A1 (en) | Anti-sars-cov-2 drug | |
| WO2023244946A1 (en) | Prodrugs of stat3 inhibitors | |
| KR20230074404A (en) | Novel flavonoid derivatives and pharmaceutical composition for prevention or treatment of metabolic diseases comprising the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08783036 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08783036 Country of ref document: EP Kind code of ref document: A1 |