WO2004100875A2 - Benzimidazoles, compositions containing such compounds and methods of use - Google Patents
Benzimidazoles, compositions containing such compounds and methods of use Download PDFInfo
- Publication number
- WO2004100875A2 WO2004100875A2 PCT/US2004/013874 US2004013874W WO2004100875A2 WO 2004100875 A2 WO2004100875 A2 WO 2004100875A2 US 2004013874 W US2004013874 W US 2004013874W WO 2004100875 A2 WO2004100875 A2 WO 2004100875A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- groups
- optionally substituted
- alkyl
- halo
- group
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 154
- 238000000034 method Methods 0.000 title claims abstract description 43
- 239000000203 mixture Substances 0.000 title abstract description 30
- 150000001556 benzimidazoles Chemical class 0.000 title abstract description 4
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims abstract description 30
- 125000005843 halogen group Chemical group 0.000 claims description 297
- 125000000217 alkyl group Chemical group 0.000 claims description 110
- 125000003118 aryl group Chemical group 0.000 claims description 91
- 125000003545 alkoxy group Chemical group 0.000 claims description 86
- -1 1-2 OH Chemical group 0.000 claims description 53
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 53
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 53
- 229910052717 sulfur Inorganic materials 0.000 claims description 40
- 150000003839 salts Chemical class 0.000 claims description 35
- 229910052739 hydrogen Inorganic materials 0.000 claims description 32
- 229910052757 nitrogen Inorganic materials 0.000 claims description 28
- 229910052799 carbon Inorganic materials 0.000 claims description 25
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 24
- 125000004043 oxo group Chemical group O=* 0.000 claims description 23
- 239000012453 solvate Substances 0.000 claims description 21
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 19
- 229910052731 fluorine Inorganic materials 0.000 claims description 10
- 125000000623 heterocyclic group Chemical group 0.000 claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- 125000003342 alkenyl group Chemical group 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 5
- 125000001743 benzylic group Chemical group 0.000 claims description 4
- 229940122904 Glucagon receptor antagonist Drugs 0.000 abstract description 4
- 229910002092 carbon dioxide Inorganic materials 0.000 description 105
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 102
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 88
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 80
- 238000004128 high performance liquid chromatography Methods 0.000 description 56
- 239000000047 product Substances 0.000 description 47
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 45
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 42
- 230000002829 reductive effect Effects 0.000 description 42
- 239000000243 solution Substances 0.000 description 42
- 235000019439 ethyl acetate Nutrition 0.000 description 40
- 238000006243 chemical reaction Methods 0.000 description 37
- 239000007787 solid Substances 0.000 description 37
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 35
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 32
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 26
- ULWOJODHECIZAU-UHFFFAOYSA-N n,n-diethylpropan-2-amine Chemical compound CCN(CC)C(C)C ULWOJODHECIZAU-UHFFFAOYSA-N 0.000 description 26
- 229910001868 water Inorganic materials 0.000 description 26
- 239000011541 reaction mixture Substances 0.000 description 25
- 239000012074 organic phase Substances 0.000 description 23
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 22
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 19
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- WMFOQBRAJBCJND-UHFFFAOYSA-M lithium hydroxide Inorganic materials [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 18
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 17
- 239000008103 glucose Substances 0.000 description 17
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 16
- 102000004877 Insulin Human genes 0.000 description 16
- 108090001061 Insulin Proteins 0.000 description 16
- 239000000556 agonist Substances 0.000 description 16
- 229940125396 insulin Drugs 0.000 description 16
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 15
- 150000001412 amines Chemical class 0.000 description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 15
- 238000003818 flash chromatography Methods 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N dimethyl sulfoxide Natural products CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- 239000000543 intermediate Substances 0.000 description 14
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 14
- 239000002904 solvent Substances 0.000 description 14
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 13
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 12
- 239000004480 active ingredient Substances 0.000 description 12
- 239000000377 silicon dioxide Substances 0.000 description 12
- 201000001320 Atherosclerosis Diseases 0.000 description 11
- 239000008346 aqueous phase Substances 0.000 description 11
- 239000012267 brine Substances 0.000 description 11
- 239000003112 inhibitor Substances 0.000 description 11
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 11
- 206010022489 Insulin Resistance Diseases 0.000 description 10
- 108010007622 LDL Lipoproteins Proteins 0.000 description 10
- 102000007330 LDL Lipoproteins Human genes 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 10
- 229960004666 glucagon Drugs 0.000 description 10
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 10
- 239000012044 organic layer Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 10
- 238000005160 1H NMR spectroscopy Methods 0.000 description 9
- 102000051325 Glucagon Human genes 0.000 description 9
- 108060003199 Glucagon Proteins 0.000 description 9
- 108010010234 HDL Lipoproteins Proteins 0.000 description 9
- 102000015779 HDL Lipoproteins Human genes 0.000 description 9
- 238000005859 coupling reaction Methods 0.000 description 9
- 239000011734 sodium Substances 0.000 description 9
- 239000003826 tablet Substances 0.000 description 9
- 208000032928 Dyslipidaemia Diseases 0.000 description 8
- 208000035150 Hypercholesterolemia Diseases 0.000 description 8
- 208000031226 Hyperlipidaemia Diseases 0.000 description 8
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 8
- 208000035475 disorder Diseases 0.000 description 8
- 208000006575 hypertriglyceridemia Diseases 0.000 description 8
- 239000004615 ingredient Substances 0.000 description 8
- 208000017170 Lipid metabolism disease Diseases 0.000 description 7
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 201000001421 hyperglycemia Diseases 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 6
- ULRPISSMEBPJLN-UHFFFAOYSA-N 2h-tetrazol-5-amine Chemical compound NC1=NN=NN1 ULRPISSMEBPJLN-UHFFFAOYSA-N 0.000 description 6
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 description 6
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 6
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 6
- 102000023984 PPAR alpha Human genes 0.000 description 6
- 102000002808 Pituitary adenylate cyclase-activating polypeptide Human genes 0.000 description 6
- 108010004684 Pituitary adenylate cyclase-activating polypeptide Proteins 0.000 description 6
- 102100040918 Pro-glucagon Human genes 0.000 description 6
- 208000017442 Retinal disease Diseases 0.000 description 6
- 206010038923 Retinopathy Diseases 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical class NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- 206010012601 diabetes mellitus Diseases 0.000 description 6
- 239000003814 drug Substances 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 239000012065 filter cake Substances 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- 208000017169 kidney disease Diseases 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 150000002632 lipids Chemical class 0.000 description 6
- 201000001119 neuropathy Diseases 0.000 description 6
- 230000007823 neuropathy Effects 0.000 description 6
- 229910052763 palladium Inorganic materials 0.000 description 6
- 208000033808 peripheral neuropathy Diseases 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- 239000002002 slurry Substances 0.000 description 6
- ZWZVWGITAAIFPS-UHFFFAOYSA-N thiophosgene Chemical compound ClC(Cl)=S ZWZVWGITAAIFPS-UHFFFAOYSA-N 0.000 description 6
- JVSMPWHQUPKRNV-UHFFFAOYSA-N 2h-tetrazol-5-amine;hydrate Chemical compound O.NC=1N=NNN=1 JVSMPWHQUPKRNV-UHFFFAOYSA-N 0.000 description 5
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 5
- 102100039994 Gastric inhibitory polypeptide Human genes 0.000 description 5
- 108010028924 PPAR alpha Proteins 0.000 description 5
- 230000029936 alkylation Effects 0.000 description 5
- 238000005804 alkylation reaction Methods 0.000 description 5
- 239000000010 aprotic solvent Substances 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 5
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 4
- VJPPLCNBDLZIFG-ZDUSSCGKSA-N 4-[(3S)-3-(but-2-ynoylamino)piperidin-1-yl]-5-fluoro-2,3-dimethyl-1H-indole-7-carboxamide Chemical compound C(C#CC)(=O)N[C@@H]1CN(CCC1)C1=C2C(=C(NC2=C(C=C1F)C(=O)N)C)C VJPPLCNBDLZIFG-ZDUSSCGKSA-N 0.000 description 4
- 208000004611 Abdominal Obesity Diseases 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 4
- 206010065941 Central obesity Diseases 0.000 description 4
- 229940122502 Cholesterol absorption inhibitor Drugs 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- 229940122199 Insulin secretagogue Drugs 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- 208000008589 Obesity Diseases 0.000 description 4
- 206010033645 Pancreatitis Diseases 0.000 description 4
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 description 4
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 238000013019 agitation Methods 0.000 description 4
- WMHOESUUCMEQMS-UHFFFAOYSA-L bis[(2,2,2-trifluoroacetyl)oxy]mercury Chemical compound [Hg+2].[O-]C(=O)C(F)(F)F.[O-]C(=O)C(F)(F)F WMHOESUUCMEQMS-UHFFFAOYSA-L 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 230000004110 gluconeogenesis Effects 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- UZCXPYDBYUEZCV-UHFFFAOYSA-N methyl 3-aminopropanoate Chemical compound COC(=O)CCN UZCXPYDBYUEZCV-UHFFFAOYSA-N 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000004770 neurodegeneration Effects 0.000 description 4
- 208000015122 neurodegenerative disease Diseases 0.000 description 4
- 235000020824 obesity Nutrition 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 208000037803 restenosis Diseases 0.000 description 4
- 208000011580 syndromic disease Diseases 0.000 description 4
- 230000002792 vascular Effects 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- DPJCXCZTLWNFOH-UHFFFAOYSA-N 2-nitroaniline Chemical compound NC1=CC=CC=C1[N+]([O-])=O DPJCXCZTLWNFOH-UHFFFAOYSA-N 0.000 description 3
- MVQVNTPHUGQQHK-UHFFFAOYSA-N 3-pyridinemethanol Chemical compound OCC1=CC=CN=C1 MVQVNTPHUGQQHK-UHFFFAOYSA-N 0.000 description 3
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 229940123208 Biguanide Drugs 0.000 description 3
- 229940089838 Glucagon-like peptide 1 receptor agonist Drugs 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 206010020772 Hypertension Diseases 0.000 description 3
- 229940122355 Insulin sensitizer Drugs 0.000 description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- 108010015181 PPAR delta Proteins 0.000 description 3
- 102000014743 Pituitary Adenylate Cyclase-Activating Polypeptide Receptors Human genes 0.000 description 3
- 108010064032 Pituitary Adenylate Cyclase-Activating Polypeptide Receptors Proteins 0.000 description 3
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- 229940100389 Sulfonylurea Drugs 0.000 description 3
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000002404 acyltransferase inhibitor Substances 0.000 description 3
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 3
- 229940121363 anti-inflammatory agent Drugs 0.000 description 3
- 239000002260 anti-inflammatory agent Substances 0.000 description 3
- 230000003579 anti-obesity Effects 0.000 description 3
- 239000003529 anticholesteremic agent Substances 0.000 description 3
- 229940127226 anticholesterol agent Drugs 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 235000006708 antioxidants Nutrition 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 150000004283 biguanides Chemical class 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000001906 cholesterol absorption Effects 0.000 description 3
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000005595 deprotonation Effects 0.000 description 3
- 238000010537 deprotonation reaction Methods 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 230000009977 dual effect Effects 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- 108010036598 gastric inhibitory polypeptide receptor Proteins 0.000 description 3
- 239000003862 glucocorticoid Substances 0.000 description 3
- 230000009229 glucose formation Effects 0.000 description 3
- 230000002440 hepatic effect Effects 0.000 description 3
- 125000001072 heteroaryl group Chemical group 0.000 description 3
- 229940121380 ileal bile acid transporter inhibitor Drugs 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 235000010446 mineral oil Nutrition 0.000 description 3
- 229960003512 nicotinic acid Drugs 0.000 description 3
- 235000001968 nicotinic acid Nutrition 0.000 description 3
- 239000011664 nicotinic acid Substances 0.000 description 3
- 229960004738 nicotinyl alcohol Drugs 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 3
- 239000003880 polar aprotic solvent Substances 0.000 description 3
- 238000012746 preparative thin layer chromatography Methods 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 229940044601 receptor agonist Drugs 0.000 description 3
- 239000000018 receptor agonist Substances 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 238000006268 reductive amination reaction Methods 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 239000003352 sequestering agent Substances 0.000 description 3
- 229960002855 simvastatin Drugs 0.000 description 3
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 229940037128 systemic glucocorticoids Drugs 0.000 description 3
- ZJXHVYSDMUKUCA-UHFFFAOYSA-N tert-butyl 3-aminopropanoate Chemical compound CC(C)(C)OC(=O)CCN ZJXHVYSDMUKUCA-UHFFFAOYSA-N 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 238000001665 trituration Methods 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- 229920002554 vinyl polymer Polymers 0.000 description 3
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 2
- 0 *C1=C(*)C=IC=C1 Chemical compound *C1=C(*)C=IC=C1 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 2
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 101001040075 Homo sapiens Glucagon receptor Proteins 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 229940126033 PPAR agonist Drugs 0.000 description 2
- 108010016731 PPAR gamma Proteins 0.000 description 2
- 102000000536 PPAR gamma Human genes 0.000 description 2
- 208000018262 Peripheral vascular disease Diseases 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 102000001494 Sterol O-Acyltransferase Human genes 0.000 description 2
- 108010054082 Sterol O-acyltransferase Proteins 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 150000001351 alkyl iodides Chemical class 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 230000003042 antagnostic effect Effects 0.000 description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 2
- 229960005370 atorvastatin Drugs 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- GPUADMRJQVPIAS-QCVDVZFFSA-M cerivastatin sodium Chemical compound [Na+].COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 GPUADMRJQVPIAS-QCVDVZFFSA-M 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 208000029078 coronary artery disease Diseases 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- VDQVEACBQKUUSU-UHFFFAOYSA-M disodium;sulfanide Chemical compound [Na+].[Na+].[SH-] VDQVEACBQKUUSU-UHFFFAOYSA-M 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 239000012039 electrophile Substances 0.000 description 2
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 2
- 229960000815 ezetimibe Drugs 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229960003765 fluvastatin Drugs 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 230000004116 glycogenolysis Effects 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 229960004844 lovastatin Drugs 0.000 description 2
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 2
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- LFVPHXOOLUCFFB-UHFFFAOYSA-N methyl 3-amino-2-hydroxypropanoate Chemical compound COC(=O)C(O)CN LFVPHXOOLUCFFB-UHFFFAOYSA-N 0.000 description 2
- NLWBJPPMPLPZIE-UHFFFAOYSA-N methyl 4-(bromomethyl)benzoate Chemical compound COC(=O)C1=CC=C(CBr)C=C1 NLWBJPPMPLPZIE-UHFFFAOYSA-N 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 238000005897 peptide coupling reaction Methods 0.000 description 2
- 239000002307 peroxisome proliferator activated receptor agonist Substances 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 229960002797 pitavastatin Drugs 0.000 description 2
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229960002965 pravastatin Drugs 0.000 description 2
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- LALFOYNTGMUKGG-BGRFNVSISA-L rosuvastatin calcium Chemical compound [Ca+2].CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O.CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O LALFOYNTGMUKGG-BGRFNVSISA-L 0.000 description 2
- 238000007127 saponification reaction Methods 0.000 description 2
- 229910052979 sodium sulfide Inorganic materials 0.000 description 2
- 229910000080 stannane Inorganic materials 0.000 description 2
- 239000001119 stannous chloride Substances 0.000 description 2
- 235000011150 stannous chloride Nutrition 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 150000003536 tetrazoles Chemical class 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- BOVGTQGAOIONJV-BETUJISGSA-N 1-[(3ar,6as)-3,3a,4,5,6,6a-hexahydro-1h-cyclopenta[c]pyrrol-2-yl]-3-(4-methylphenyl)sulfonylurea Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1C[C@H]2CCC[C@H]2C1 BOVGTQGAOIONJV-BETUJISGSA-N 0.000 description 1
- PWKNBLFSJAVFAB-UHFFFAOYSA-N 1-fluoro-2-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1F PWKNBLFSJAVFAB-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- WTTWSMJHJFNCQB-UHFFFAOYSA-N 2-(dibenzylamino)ethanol Chemical compound C=1C=CC=CC=1CN(CCO)CC1=CC=CC=C1 WTTWSMJHJFNCQB-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000006029 2-methyl-2-butenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- ZHCLIFKUVIFYBY-UHFFFAOYSA-N 2h-tetrazol-5-ylmethanamine Chemical compound NCC1=NN=NN1 ZHCLIFKUVIFYBY-UHFFFAOYSA-N 0.000 description 1
- UQRLKWGPEVNVHT-UHFFFAOYSA-N 3,5-dichloroaniline Chemical compound NC1=CC(Cl)=CC(Cl)=C1 UQRLKWGPEVNVHT-UHFFFAOYSA-N 0.000 description 1
- MCSXGCZMEPXKIW-UHFFFAOYSA-N 3-hydroxy-4-[(4-methyl-2-nitrophenyl)diazenyl]-N-(3-nitrophenyl)naphthalene-2-carboxamide Chemical compound Cc1ccc(N=Nc2c(O)c(cc3ccccc23)C(=O)Nc2cccc(c2)[N+]([O-])=O)c(c1)[N+]([O-])=O MCSXGCZMEPXKIW-UHFFFAOYSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- MPOYBFYHRQBZPM-UHFFFAOYSA-N 3h-pyridin-4-one Chemical class O=C1CC=NC=C1 MPOYBFYHRQBZPM-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- LJGJDFAHHABYEF-UHFFFAOYSA-N 4-methoxy-1-n-methylbenzene-1,2-diamine Chemical compound CNC1=CC=C(OC)C=C1N LJGJDFAHHABYEF-UHFFFAOYSA-N 0.000 description 1
- QFMJFXFXQAFGBO-UHFFFAOYSA-N 4-methoxy-2-nitroaniline Chemical compound COC1=CC=C(N)C([N+]([O-])=O)=C1 QFMJFXFXQAFGBO-UHFFFAOYSA-N 0.000 description 1
- LAOZSCRCYVBSJA-UHFFFAOYSA-N 5,5-dimethyl-1,3-diazinane-2,4,6-trione Chemical compound CC1(C)C(=O)NC(=O)NC1=O LAOZSCRCYVBSJA-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 1
- 102000007592 Apolipoproteins Human genes 0.000 description 1
- 108010071619 Apolipoproteins Proteins 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- OABPVPIQBUOWRM-UHFFFAOYSA-N CC(C)(C)C(CC1)CCC1N(Cc(cc1)ccc1C(Nc1nnn[nH]1)=O)c([n](C)c1c2)nc1ccc2OCC=C Chemical compound CC(C)(C)C(CC1)CCC1N(Cc(cc1)ccc1C(Nc1nnn[nH]1)=O)c([n](C)c1c2)nc1ccc2OCC=C OABPVPIQBUOWRM-UHFFFAOYSA-N 0.000 description 1
- 101150041968 CDC13 gene Proteins 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RKWGIWYCVPQPMF-UHFFFAOYSA-N Chloropropamide Chemical compound CCCNC(=O)NS(=O)(=O)C1=CC=C(Cl)C=C1 RKWGIWYCVPQPMF-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 108010063919 Glucagon Receptors Proteins 0.000 description 1
- 102100040890 Glucagon receptor Human genes 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 108010023302 HDL Cholesterol Proteins 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 108010028554 LDL Cholesterol Proteins 0.000 description 1
- 238000008214 LDL Cholesterol Methods 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- IBAQFPQHRJAVAV-ULAWRXDQSA-N Miglitol Chemical compound OCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO IBAQFPQHRJAVAV-ULAWRXDQSA-N 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-O Piperidinium(1+) Chemical compound C1CC[NH2+]CC1 NQRYJNQNLNOLGT-UHFFFAOYSA-O 0.000 description 1
- 102000005157 Somatostatin Human genes 0.000 description 1
- 108010056088 Somatostatin Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric Acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 229960001466 acetohexamide Drugs 0.000 description 1
- VGZSUPCWNCWDAN-UHFFFAOYSA-N acetohexamide Chemical compound C1=CC(C(=O)C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 VGZSUPCWNCWDAN-UHFFFAOYSA-N 0.000 description 1
- 108060000200 adenylate cyclase Proteins 0.000 description 1
- 102000030621 adenylate cyclase Human genes 0.000 description 1
- OQIQSTLJSLGHID-WNWIJWBNSA-N aflatoxin B1 Chemical compound C=1([C@@H]2C=CO[C@@H]2OC=1C=C(C1=2)OC)C=2OC(=O)C2=C1CCC2=O OQIQSTLJSLGHID-WNWIJWBNSA-N 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000003178 anti-diabetic effect Effects 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 229940058303 antinematodal benzimidazole derivative Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229940000635 beta-alanine Drugs 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229960004111 buformin Drugs 0.000 description 1
- XSEUMFJMFFMCIU-UHFFFAOYSA-N buformin Chemical compound CCCC\N=C(/N)N=C(N)N XSEUMFJMFFMCIU-UHFFFAOYSA-N 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 229960003362 carbutamide Drugs 0.000 description 1
- VDTNNGKXZGSZIP-UHFFFAOYSA-N carbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(N)C=C1 VDTNNGKXZGSZIP-UHFFFAOYSA-N 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 229960001761 chlorpropamide Drugs 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 239000012024 dehydrating agents Substances 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 238000006266 etherification reaction Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- 229960001764 glibornuride Drugs 0.000 description 1
- RMTYNAPTNBJHQI-LLDVTBCESA-N glibornuride Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)N[C@H]1[C@H](C2(C)C)CC[C@@]2(C)[C@H]1O RMTYNAPTNBJHQI-LLDVTBCESA-N 0.000 description 1
- 229960000346 gliclazide Drugs 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960003236 glisoxepide Drugs 0.000 description 1
- ZKUDBRCEOBOWLF-UHFFFAOYSA-N glisoxepide Chemical compound O1C(C)=CC(C(=O)NCCC=2C=CC(=CC=2)S(=O)(=O)NC(=O)NN2CCCCCC2)=N1 ZKUDBRCEOBOWLF-UHFFFAOYSA-N 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- RIGBPMDIGYBTBJ-UHFFFAOYSA-N glycyclamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 RIGBPMDIGYBTBJ-UHFFFAOYSA-N 0.000 description 1
- 229950005514 glycyclamide Drugs 0.000 description 1
- NFRPNQDSKJJQGV-UHFFFAOYSA-N glyhexamide Chemical compound C=1C=C2CCCC2=CC=1S(=O)(=O)NC(=O)NC1CCCCC1 NFRPNQDSKJJQGV-UHFFFAOYSA-N 0.000 description 1
- 229950008290 glyhexamide Drugs 0.000 description 1
- 229950009188 glypinamide Drugs 0.000 description 1
- RHQSNARBXHRBNP-UHFFFAOYSA-N glypinamide Chemical compound C1=CC(Cl)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 RHQSNARBXHRBNP-UHFFFAOYSA-N 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 1
- 201000008980 hyperinsulinism Diseases 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 230000004130 lipolysis Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- OFXSXYCSPVKZPF-UHFFFAOYSA-N methoxyperoxymethane Chemical compound COOOC OFXSXYCSPVKZPF-UHFFFAOYSA-N 0.000 description 1
- NHFBYYMNJUMVOT-UHFFFAOYSA-N methyl 2-bromo-2-phenylacetate Chemical compound COC(=O)C(Br)C1=CC=CC=C1 NHFBYYMNJUMVOT-UHFFFAOYSA-N 0.000 description 1
- YRMODRRGEUGHTF-UHFFFAOYSA-N methyl 2-formylbenzoate Chemical compound COC(=O)C1=CC=CC=C1C=O YRMODRRGEUGHTF-UHFFFAOYSA-N 0.000 description 1
- 229960001110 miglitol Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000007410 oral glucose tolerance test Methods 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- AFOGBLYPWJJVAL-UHFFFAOYSA-N phenbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=CC=C1 AFOGBLYPWJJVAL-UHFFFAOYSA-N 0.000 description 1
- 229950008557 phenbutamide Drugs 0.000 description 1
- 229960003243 phenformin Drugs 0.000 description 1
- ICFJFFQQTFMIBG-UHFFFAOYSA-N phenformin Chemical compound NC(=N)NC(=N)NCCC1=CC=CC=C1 ICFJFFQQTFMIBG-UHFFFAOYSA-N 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 238000001525 receptor binding assay Methods 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 206010038464 renal hypertension Diseases 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 1
- 229960000553 somatostatin Drugs 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 229960002277 tolazamide Drugs 0.000 description 1
- OUDSBRTVNLOZBN-UHFFFAOYSA-N tolazamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 OUDSBRTVNLOZBN-UHFFFAOYSA-N 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 1
- 229960001641 troglitazone Drugs 0.000 description 1
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229960001729 voglibose Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D235/30—Nitrogen atoms not forming part of a nitro radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to substituted benzimidazole derivatives, compositions containing such compounds and methods of treating type 2 diabetes mellitus.
- Diabetes refers to a disease process derived from multiple causative factors and is characterized by elevated levels of plasma glucose (hyperglycemia) in the fasting state or following glucose administration during an oral glucose tolerance test.
- Frank diabetes mellitus e.g., a blood glucose level >126 mg/dL in a fasting state
- Type 2 diabetes mellitus Patients with non-insulin dependent diabetes mellitus (type 2 diabetes mellitus), approximately 95% of patients with diabetes mellitus, frequently display elevated levels of serum lipids, such as cholesterol and triglycerides, and have poor blood-lipid profiles, with high levels of LDL- cholesterol and low levels of HDL-cholesterol.
- Those suffering from Type 2 diabetes mellitus are thus at an increased risk of developing macrovascular and microvascular complications, including coronary heart disease, stroke, peripheral vascular disease, hypertension (for example, blood pressure > 130/80 mmHg in a resting state), nephropathy, neuropathy and retinopathy.
- Type 2 diabetes at least early in the natural progression of the disease is characterized primarily by insulin resistance rather than by a decrease in insulin production, resulting in insufficient uptake, oxidation and storage of glucose in muscle, inadequate repression of lipolysis in adipose tissue, and excess glucose production and secretion by the liver.
- the net effect of decreased sensitivity to insulin is high levels of insulin circulating in the blood without appropriate reduction in plasma glucose (hyperglycemia). Hyperinsulinemia is a risk factor for developing hypertension and may also contribute to vascular disease.
- Glucagon serves as the major regulatory hormone attenuating the effect of insulin in its inhibition of liver gluconeogenesis and is normally secreted by pancreatic islet cells in response to falling blood glucose levels.
- the hormone binds to specific receptors in liver cells that triggers glycogenolysis and an increase in gluconeogenesis through cAMP-mediated events. These responses generate glucose (e.g. hepatic glucose production) to help maintain euglyce ia by preventing blood glucose levels from falling significantly.
- type II diabetics In addition to elevated levels of circulating insulin, type II diabetics have elevated levels of plasma glucagon and increased rates of hepatic glucose production. Antagonists of glucagon are useful in improving insulin responsiveness in the liver, decreasing the rate of gluconeogenesis and lowering the rate of hepatic glucose output resulting in a decrease in the levels of plasma glucose.
- the present invention is directed to a compound represented by formula I:
- R 1 represents H or is independently selected from the group consisting of: a) OH, halo, C0 2 R ⁇ C ⁇ NR'R 0 , NR b R c , CN or S(0) p R d ; b) .ioalkyl, C 2 - ⁇ 0 alkenyl, C 2 . ⁇ oalkynyl, OC ⁇ _ ⁇ 0 alkyl, OC 3 .
- alkenyl and OC 3 _ ⁇ 0 alkynyl said groups being optionally substituted with: (1) 1-5 halo groups up to a perhaloalkyl group; (2) 1 oxo group; (3) 1-2 OH groups; (4) 1-2 C ⁇ - ⁇ 0 alkoxy groups, each optionally substituted with: up to five halo or a perhaloalkoxy, 1 OH or C0 2 R a group; (5) 1 C0 2 R a or S(0) p R d ; (6)1-2 Aryl, Hetcy or HAR groups, each optionally substituted as follows: (a) 1-5 halo groups, (b) 1 OH, C0 2 R a , CN, S(0) p R d , N0 2 or C(0)NR b R c group, (c) 1-2 Q.ioalkyl or alkoxy groups, each optionally substituted with: 1-5 halo, up to perhaloalkyl, and 1-2 OH or C
- HAR group each optionally substituted as follows: (a) 1-5 halo groups, (b) 1 OH, C0 2 R a , CN, S(0) p R d , N0 2 or C(0)NR b R c group, (c) 1-2 C ⁇ _ ⁇ 0 alkyl or alkoxy groups, each optionally substituted with: 1-5 halo, up to perhaloalkyl, and 1-2 OH or C0 2 R a groups; and (d) 1-2 phenyl rings, each of which is optionally substituted as follows: 1-5 halo groups up to perhalo; 1-3 Ci.ioalkyl or alkoxy groups, each being further optionally substituted with 1-5 halo up to perhalo; and 1-2 hydroxy or C0 2 R a groups; c) Aryl, HAR, Hetcy, -O-Aryl, -O-HAR and -O-Hetcy, each optionally substituted as set forth below: (1) 1-3 C ⁇ - ⁇ -
- R 3 represents H or is selected from the group consisting of: a) C ⁇ _ ⁇ 0 alkyl or C 2 - ⁇ o lkenyl, each optionally substituted with 1-5 halo groups up to perhalo; 1-2 OH, C ⁇ _ 3 alkoxy or haloC ⁇ _ 3 alkoxy groups; 1-2 NR c R d groups; and 1-2 Aryl, HAR or Hetcy groups, each optionally substituted with 1-3 halo groups and 1-2 groups selected from CN, N0 2 , C ⁇ _ 3 alkyl, haloC ⁇ _ 3 alkyl, C ⁇ - 3 alkoxy and haloC ⁇ - 3 alkoxy groups; and b) Aryl, HAR or Hetcy, each optionally substituted with 1-3 halo groups and 1-2 groups selected from CN, N0 2 , C ⁇ _ 3 alkyl, haloC ⁇ _ 3 alkyl, C ⁇ _ 3 alkoxy and haloC ⁇ _ 3 alkoxy groups; R
- R 5 represents H or C ⁇ - 6 alkyl
- R 6 is selected from the group consisting of H, OH, F or C ⁇ _ 3 alkyl;
- R 7 is H or F, or R 6 and R 7 are taken in combination and represent oxo;
- R 8 represents H or C ⁇ _ ⁇ alkyl, optionally substituted with OH and 1-5 halo groups up to perhalo;
- R 9 represents H, halo, OH, C ⁇ _ 6 alkyl, optionally substituted with 1-5 halo groups up to perhalo, or C ⁇ _
- R 8 and R 9 can be taken together and represent a -(CH 2 ) 24 - or a -0-(CH 2 ) ⁇ _ 3 - group;
- R a is H or C ⁇ - ⁇ 0 alkyl, optionally substituted with phenyl, OH, OC ⁇ - 6 alkyl, C0 2 H, C0 2 C ⁇ _ 6 alkyl and 1-3 halo groups;
- R b is H or C 0 alkyl
- R c is H or is independently selected from: (a) Ci.ioalkyl, optionally substituted with OH, OC ⁇ - 6 alkyl, C0 2 H, C0 2 C ⁇ . 6 alkyl, and 1-3 halo groups; (b) Aryl or Ar-C ⁇ _ 6 alkyl, each optionally substituted with 1-5 halos and 1-3 members selected from the group consisting of: CN, OH, C ⁇ _ ⁇ 0 alkyl and OCi.jo alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo; (c) Hetcy or Hetcy-C ⁇ - 6 alkyl, optionally substituted with 1-5 halo groups and 1-3 groups selected from: oxo, C ⁇ - ⁇ 0 alkyl and OC MO alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo; and (d) HAR or HAR-C ⁇ .
- alkyl optionally substituted with 1-5 halo groups and 1-3 groups selected from: C ⁇ - ⁇ 0 alkyl and OC MO alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo;
- R d is Ci_i 0 alkyl, Aryl or Ax-C]_i 0 alkyl; m is an integer selected from 0, 1 and 2; n is an integer selected from 0 to 6; p is an integer selected from 0, 1 and 2, and when at least one of m and n is other than 0, Z is selected from C0 2 R a , 5-tetrazolyl and 5-(2-oxo-l,3,4-oxadiazolyl), and when both m and n are 0, Z is selected from 5-tetrazolyl and 5-(2-oxo- 1,3,4-oxadiazolyl).
- Alkyl as well as other groups having the prefix "alk”, such as alkoxy, alkanoyl and the like, means carbon chains which may be linear, branched or cyclic, or combinations thereof, containing the indicated number of carbon atoms. If no number is specified, 1-10 carbon atoms are intended for linear or branched alkyl groups. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl and the like.
- Cycloalkyl is a subset of alkyl; if no number of atoms is specified, 3-10 carbon atoms are intended, forming 1-3 carbocyclic rings that are fused. "Cycloalkyl” also includes monocyclic rings fused to an aryl group in which the point of attachment is on the non-aromatic portion. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, tetrahydronaphthyl, decahydronaphthyl, indanyl and the like.
- alkenyl means carbon chains which contain at least one carbon-carbon double bond, and which may be linear or branched or combinations thereof. Examples of alkenyl include vinyl, allyl, isopropenyl, pentenyl, hexenyl, heptenyl, 1-propenyl, 2-butenyl, 2-methyl-2-butenyl, and the like.
- alkynyl means carbon chains which contain at least one carbon-carbon triple bond, and which may be linear or branched or combinations thereof. Examples of alkynyl include ethynyl, propargyl, 3-methyl-l-pentynyl, 2-heptynyl and the like.
- Aryl (Ar) means mono- and bicyclic aromatic rings containing 6-12 carbon atoms. Examples of aryl include phenyl, naphthyl, indenyl and the like.
- Heteroaryl means a mono- or bicyclic aromatic ring or ring system containing at least one heteroatom selected from O, S and N, with each ring containing 5 to 6 atoms. Examples include pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl, furo(2,3-b)pyridyl, quinolyl, indolyl, isoquinolyl and the like.
- Heteroaryl also includes aromatic heterocyclic groups fused to heterocycles that are non-aromatic or partially aromatic, and aromatic heterocyclic groups fused to cycloalkyl rings. Heteroaryl also includes such groups in charged form, e.g., pyridinium.
- Heterocyclyl (Hetcy) means mono- and bicyclic saturated rings and ring systems containing at least one heteroatom selected from N, S and O, each of said ring having from 3 to 10 atoms in which the point of attachment may be carbon or nitrogen.
- heterocyclyl include pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, 2,3-dihydrofuro(2,3-b)pyridyl, benzoxazinyl, tetrahydrohydroquinolinyl, tetrahydroisoquinolinyl, dihydroindolyl, and the like.
- the term also includes partially unsaturated monocyclic rings that are not aromatic, such as 2- or 4-pyridones attached through the nitrogen or N-substituted-(lH,3H)-pyrimidine-2,4-diones (N-substituted uracils).
- Heterocyclyl moreover includes such moieties in charged form, e.g., piperidinium.
- Halogen includes fluorine, chlorine, bromine and iodine, preferably F and Cl, more preferably F.
- the invention is directed to a compound represented by formula I:
- R 1 represents H or is independently selected from the group consisting of: a) OH, halo, C0 2 R a , C(0)NR b R c , NR 1 ⁇ 0 , CN or S(0) p R d ; b) C ⁇ _ ⁇ 0 alkyl, C 2 _ ⁇ oalkenyl, C 2 - ⁇ oalkynyl, OC ⁇ _ ⁇ 0 alkyl, OC 3 _ ⁇ 0 alkenyl and OC 3 _ ⁇ 0 alkynyl, said groups being optionally substituted with: (1) 1-5 halo groups up to a perhaloalkyl group; (2) 1 oxo group; (3) 1-2 OH groups; (4) 1-2 C ⁇ _ ⁇ 0 alkoxy groups, each optionally substituted with: up to five halo or a perhaloalkoxy, 1 OH or C0 2 R a group; (5) 1 C0 2 R a or S(0) p R d ; (6)1-2 A
- alkyl or C ⁇ - 6 alkoxy groups the alkyl and alkoxy groups being further optionally substituted with 1-3 halo groups; C0 2 R a ; CN or S(0) p R d groups; and (2) 1-3 C ⁇ . ⁇ 0 alkoxy groups, the alkyl portion of which is optionally substituted with 1-5 halo groups, 1-2 OH; phenyl optionally substituted with 1-3 halo, C ⁇ .
- R 3 represents H or is selected from the group consisting of: a) C]. ⁇ 0 alkyl or C 2 - ⁇ oalkenyl, each optionally substituted with 1-5 halo groups up to perhalo; 1-2 OH, C ⁇ . 3 alkoxy or haloC ⁇ . 3 alkoxy groups; 1-2 NR c R d groups; and 1-2 Aryl, HAR or Hetcy groups, each optionally substituted with 1-3 halo groups and 1-2 groups selected from CN, N0 2 , C ⁇ - 3 alkyl, haloC ⁇ - 3 alkyl, C ⁇ .
- Aryl, HAR or Hetcy each optionally substituted with 1-3 halo groups and 1-2 groups selected from CN, N0 2 , C ⁇ - 3 alkyl, haloC ⁇ - 3 alkyl, C ⁇ - 3 alkoxy and haloC ⁇ - 3 alkoxy groups;
- R 4 is independently selected from the group consisting of: a) C ⁇ - ⁇ 4 alkyl, C 2 - ⁇ oalkenyl and C 2 . ⁇ 0 alkynyl, said groups being optionally substituted with: (1) 1-5 halo groups up to perhaloalkyl; (2) 1 oxo group; (3) 1-2 OH groups; (4) 1-2 C ⁇ - ⁇ 0 alkoxy groups, each optionally substituted with up to five halo or a perhaloalkoxy, 1 OH or C0 2 R a group; (5) 1 C0 2 R a or S(0) p R d ; (6) 1-2 Aryl, Hetcy or HAR groups, each optionally substituted as follows: (i) 1-5 halo groups, (ii) 1 OH, C0 2 R a , CN, S(0) p R d , N0 2 or C(0)NR b R c group, (iii) 1-2 C ⁇ . ⁇ 0 alkyl or alkoxy groups,
- R 5 represents H or C ⁇ . 6 alkyl
- R 6 is selected from the group consisting of H, OH, F or C ⁇ - 3 alkyl
- R 7 is H or F, or R 6 and R 7 are taken in combination and represent oxo;
- R 8 represents H or - ⁇ alkyl, optionally substituted with OH and 1-5 halo groups up to perhalo;
- R 9 represents H, halo, OH, C ⁇ - 6 alkyl, optionally substituted with 1-5 halo groups up to perhalo, or Q. 6 alkoxy, optionally substituted with 1-3 halo groups up to perhalo, or when R 9 is ortho to the benzylic group, R 8 and R 9 can be taken together and represent a -(CH 2 ) 2 . 4 - or a -0-(CH 2 ) ⁇ - 3 - group;
- R a is H or C ⁇ - ⁇ 0 alkyl, optionally substituted with phenyl, OH, OC ⁇ alkyl, C0 2 H, C0 2 C ⁇ . ⁇ alkyl and 1-3 halo groups;
- R b is H or Ci-ioalkyl
- R c is H or is independently selected from: (a) Ci.ioalkyl, optionally substituted with OH, OC ⁇ - 6 alkyl, C0 2 H, C0 2 C ⁇ . 6 alkyl, and 1-3 halo groups; (b) Aryl or Ar-C ⁇ . 6 alkyl, each optionally substituted with 1-5 halos and 1-3 members selected from the group consisting of: CN, OH, C ⁇ - ⁇ 0 alkyl and OC MO alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo; (c) Hetcy or Hetcy-C ⁇ .
- 6 alkyl optionally substituted with 1-5 halo groups and 1-3 groups selected from: oxo, C ⁇ - ⁇ 0 alkyl and OC MO alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo; and (d) HAR or HAR-C ⁇ . 6 alkyl, optionally substituted with 1-5 halo groups and 1-3 groups selected from: C ⁇ - ⁇ 0 alkyl and OC O alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo;
- R d is Ci.ioalkyl, Aryl or Ar-C ⁇ - ⁇ 0 alkyl; m is an integer selected from 0, 1 and 2; n is an integer selected from 0 to 6; p is an integer selected from 0, 1 and 2, and when at least one of m and n is other than 0, Z is selected from C0 2 R a , 5-tetrazolyl and
- One aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 1 represents H. Within this aspect of the invention, all other variables are as originally defined with respect to formula I.
- Another aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein one R 2 represents H, halo or d- 6 alkyl, and the other is selected from the group consisting of: H, halo, OH, optionally substituted with 1-3 halo groups, C ⁇ alkoxy optionally substituted with 1-3 halo groups or 1 phenyl or heterocyclic ring, C . 4 alkenyl or OC - alkenyl. Within this aspect of the invention, all other variables are as originally defined with respect to formula I.
- R 3 is selected from the group consisting of: H, C 2 . 4 alkenyl and C ⁇ . 6 alkyl optionally substituted as follows: a) up to 3 halo groups; b) NR c R d wherein R c and R d are H or C ⁇ . 4 alkyl; c) OH; and d) Aryl optionally substituted with 1-3 halo groups, C M alkyl, OC ⁇ . 3 alkyl, CN, N0 2 , haloC ⁇ - 3 alkyl or 0-haloC ⁇ _ 3 alkyl.
- all other variables are as originally defined with respect to formula I.
- R 4 is independently selected from the group consisting of:
- (a) C]- ⁇ 4 alkyl optionally substituted with: (1) 1-5 halo groups up to perhaloalkyl; (2) 1-2 C ⁇ - ⁇ 0 alkoxy groups, each optionally substituted with 1-5 halo groups up to perhaloalkoxy; (3) 1-2 Aryl groups, each optionally substituted as follows: (i) 1-5 halo groups, (ii) CN or N0 2 , (iii) 1-2 C ⁇ .
- ⁇ oalkyl or alkoxy groups each optionally substituted with: 1-5 halo, up to perhaloalkyl; and (b) Aryl, HAR or Hetcy, each optionally substituted as follows: (1) 1-2 C ⁇ - ⁇ 0 alkyl or C 2 - ⁇ 0 alkenyl groups, optionally substituted with 1-5 halo groups, phenyl or C0 2 R a groups; (2) 1-2 .
- ⁇ 0 alkoxy groups the alkyl portion of which is optionally substituted with 1-5 halo groups; (3) 1-2 Aryl, HAR or Hetcy, OAryl, OHAR or OHetcy groups, each optionally substituted as follows: (i) 1-3 halo groups; (ii) 1-2 Ci.ioalkyl or C 2 . ⁇ 0 alkenyl, each optionally substituted with 1-3 halo groups; (iii) 1-2 .
- Another aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 9 represents H or halo.
- R 9 represents H or halo.
- Yet another aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 8 and R 9 are taken in combination and represent -(CH 2 ) 2 . 4 -- More particularly, an aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein R 8 and R 9 are taken in combination and represent ethylene.
- all other variables are as originally defined with respect to formula I.
- an aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein
- R 1 represents H
- one R 2 represents H, halo or Ci- ⁇ alkyl, and the other is selected from the group consisting of: H, halo, OH, optionally substituted with 1-3 halo groups, C ⁇ - 6 alkoxy optionally substituted with 1-3 halo groups or 1 phenyl or heterocyclic ring, C 2 . 4 alkenyl or OC 2 . 4 alkenyl;
- R 3 is selected from the group consisting of: H, C 2 . 4 alkenyl and C ⁇ . 6 alkyl optionally substituted as follows: a) up to 3 halo groups; b) NR c R d wherein R c and R d are H or C ⁇ - 4 alkyl; c) OH; and d) Aryl optionally substituted with 1-3 halo groups, C ⁇ - 3 alkyl, OC ⁇ - 3 alkyl, CN, N0 2 , haloC ⁇ - 3 alkyl or 0-haloC ⁇ - 3 alkyl; ⁇
- R 4 is independently selected from the group consisting of:
- Aryl, HAR or Hetcy each optionally substituted as follows: (1) 1-2 Ci.ioalkyl or C 2 . l oalkenyl, optionally substituted with 1-5 halo groups, phenyl or C0 2 R a groups; (2) 1-2 Ci-ioalkoxy groups, the alkyl portion of which is optionally substituted with 1-5 halo groups; (3) 1-2 Aryl, HAR or Hetcy, OAryl, OHAR or OHetcy groups, each optionally substituted as follows: (i) 1-3 halo groups; (ii) 1-2 C ⁇ - ⁇ 0 alkyl or C 2 - ⁇ oalkenyl, each optionally substituted with 1-3 halo groups; (iii) 1-2 C ⁇ . ⁇ 0 alkoxy groups the alkyl portion of which being optionally substituted with 1-3 halo groups, and (iv) 1-2 C0 2 R a , S(0) p R d , CN, NR
- R 8 represents H or C ⁇ _ 6 alkyl
- R 9 represents H or halo
- R 5 represents H or C]- 6 alkyl
- R 6 is selected from the group consisting of H, OH, F or C ⁇ - 3 alkyl;
- R 7 is H or F, or R 6 and R 7 are taken in combination and represent oxo;
- R a is H or Ci.ioalkyl, optionally substituted with phenyl, OH, OC ⁇ - 6 alkyl, C0 2 H, C0 2 C ⁇ - 6 alkyl and 1-3 halo groups;
- R is H or Ci-ioalkyl
- R c is H or is independently selected from: (a) Ci.ioalkyl, optionally substituted with OH, OC ⁇ - 6 alkyl, C0 2 H, C0 2 C ⁇ . 6 alkyl, and 1-3 halo groups; (b) Aryl or Ar-C ⁇ - 6 alkyl, each optionally substituted with 1-5 halos and 1-3 members selected from the group consisting of: CN, OH, C ⁇ . ⁇ 0 alkyl and OC MO alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo; (c) Hetcy or Hetcy-Ci- ⁇ alkyl, optionally substituted with 1-5 halo groups and 1-3 groups selected from: oxo, C ⁇ - ⁇ 0 alkyl and OC O alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo; and (d) HAR or HAR-C ⁇ .
- alkyl optionally substituted with 1-5 halo groups and 1-3 groups selected from: C ⁇ - ⁇ 0 alkyl and OC MO alkyl, said alkyl and alkoxy being further optionally substituted with 1-5 halo groups up to perhalo;
- R d is Ci.ioalkyl, Aryl or Ar-C ⁇ - ⁇ 0 alkyl; m is an integer selected from 0, 1 and 2; n is an integer selected from 0 to 6; p is an integer selected from 0, 1 and 2, and when at least one of m and n is other than 0, Z is selected from C0 2 R a , 5-tetrazolyl and 5-(2-oxo-l,3,4-oxadiazolyl), and when both m and n are 0, Z is selected from 5-tetrazolyl and 5-(2-oxo- 1,3,4-oxadiazolyl).
- all other variables are as originally defined with respect to formula I.
- Another more particular aspect of the invention that is of interest relates to a compound of formula I or a pharmaceutically acceptable salt or solvate thereof wherein:
- alkenyl and Cj- ⁇ alkyl optionally substituted as follows: a) up to 3 halo groups; b) NR c R d wherein R c and R d are H or C M alkyl; c) OH; and d) Aryl optionally substituted with 1-3 halo groups, C ⁇ - 3 alkyl, OC ⁇ - 3 alkyl, CN, N0 2 , haloC ⁇ .
- R 4 is independently selected from the group consisting of: a) C].
- ⁇ 4 a ⁇ kyl optionally substituted with: (1) 1-5 halo groups up to perhaloalkyl; (2) 1- 2 Ci-ioalkoxy groups, each optionally substituted with 1-5 halo groups up to perhaloalkoxy; (3) 1-2 Aryl groups, each optionally substituted as follows: (i) 1-5 halo groups, (ii) CN or N0 2 , and (iii) 1-2 Ci- ⁇ 0 alkyl or alkoxy groups, each optionally substituted with: 1-5 halo, up to perhaloalkyl; and b) Aryl, HAR or Hetcy, each optionally substituted as follows: (1) 1-2 C ⁇ - ⁇ 0 alkyl or C 2 - i 0 alkenyl, optionally substituted with 1-5 halo groups, phenyl or C0 2 R a groups; (2)
- R 8 and R 9 are taken in combination and represent -(CH 2 ) 2 . 4 -;
- R 5 represents H or C ⁇ . ⁇ alkyl;
- R 6 is selected from the group consisting of H, OH, F or C ⁇ . 3 alkyl
- R 7 is H or F, or R 6 and R 7 are taken in combination and represent oxo;
- R a is H or Ci.ioalkyl, optionally substituted with phenyl, OH, OC ⁇ alkyl, C0 2 H, C0 2 C ⁇ . 6 alkyl and 1-3 halo groups;
- R D is H or C,., 0 alkyl;
- R c is H or is independently selected from: (a) C ⁇ - ⁇ 0 alkyl, optionally substituted with OH, OC ⁇ . 6 alkyl, C0 2 H, C0 2 C ⁇ - 6 alkyl, and 1-3 halo groups; (b) Aryl or Ar-C ⁇ .
- R d is Ci-ioalkyl, Aryl or Ar-C ⁇ . ⁇ 0 alkyl; m is an integer selected from 0, 1 and 2; n is an integer selected from 0 to 6; p is an integer selected from 0, 1 and 2, and when at least one of m and n is other than 0, Z is selected from C0 2 R a , 5-tetrazolyl and 5-(2-oxo-l,3,4-oxadiazolyl), and when both m and n are 0, Z is selected from 5-tetrazolyl and 5-(2-oxo- 1,3,4-oxadiazolyl).
- all other variables are as originally defined with respect to formula I
- the invention further includes a pharmaceutical composition which is comprised of a compound of formula I in combination with a pharmaceutically acceptable carrier.
- disorders include diseases and conditions selected from the group consisting of: dyslipidemias, (e.g., hyperlipidemia), such as elevated levels of cholesterol (hypercholesterolemia), triglycerides (hypertriglyceridemia) or low density lipoproteins (LDL) (high LDL levels), low levels of high density lipoprotein (HDL), microvascular or macrovascular changes and the sequellae of such conditions, such as coronary heart disease, stroke, peripheral vascular disease, hypertension, renal hypertension, nephropathy, neuropathy and retinopathy.
- dyslipidemias e.g., hyperlipidemia
- hyperlipidemia such as elevated levels of cholesterol (hypercholesterolemia), triglycerides (hypertriglyceridemia) or low density lipoproteins (LDL) (high LDL levels), low levels of high density lipoprotein (HDL)
- LDL low density lipoproteins
- HDL high density lipoprotein
- microvascular or macrovascular changes and the sequellae of such conditions such as coronar
- the method entails administering to a type 2 diabetic patient, e.g., a human patient, an amount of a compound of formula I that is effective for treating, preventing or delaying the onset of such diseases or conditions. Also included is a method of treating atherosclerosis in a mammalian patient in need of such treatment, comprising administering to said patient a compound of formula I in an amount effective to treat atherosclerosis.
- a condition selected from the group consisting of: (1) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) obesity, (5) lipid disorders, (6) dyslipidemia,
- a condition selected from the group consisting of (1) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) obesity, (5) lipid disorders, (6) dyslipidemia,
- tautomers Some of the compounds described herein may exist with different points of attachment of hydrogen, referred to as tautomers. Such an example may be a ketone and its enol form known as keto-enol tautomers. The individual tautomers as well as mixture thereof are encompassed with compounds of Formula I.
- salts refers to salts prepared from pharmaceutically acceptable substantially non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids, as well as salts that can be converted into pharmaceutically acceptable salts.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- basic ion exchange resins such as ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, pipe
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- Solvates as used herein refers to the compound of formula I or a salt thereof, in association with a solvent, such as water. Representative examples include hydrates, hemihydrates, trihydrates and the like.
- references to the compounds of Formula I include the pharmaceutically acceptable salts and solvates.
- This invention relates to method of antagonizing or inhibiting the production or activity of glucagon, thereby reducing the rate of gluconeogenesis and glycogenolysis, and the concentration of glucose in plasma.
- the compounds of formula I can be used in the manufacture of a medicament for the prophylactic or therapeutic treatment of disease states in mammals caused by elevated levels of glucose, comprised of combining the compound of formula I with the carrier materials to provide the medicament.
- the prophylactic or therapeutic dose of the compound of formula I will, of course, vary with the nature of the condition to be treated, the particular compound selected and its route of administration. It will also vary according to the age, weight and response of the individual patient. In general, the daily dose range lie within the range of from about 0.001 mg to about 100 mg per kg body weight, preferably about 0.01 mg to about 50 mg per kg, and more preferably 0.1 to 10 mg per kg, in single or divided doses. It may be necessary to use dosages outside of these limits in some cases.
- the terms "effective amount” "anti-diabetic effective amount” and the other terms appearing throughout the application addressing the amount of the compound to be used refer to the dosage ranges provided, taking into account any necessary variation outside of these ranges, as determined by the skilled physician.
- Representative dosages for adults range from about 0.1 mg to about 1.0 g per day, preferably about 1 mg to about 200 mg, in single or divided doses.
- a representative dosage range is from about 0.001 mg to about 100 mg (preferably from 0.01 mg to about 10 mg) of a compound of Formula I per kg of body weight per day, and more preferably, about 0.1 mg to about 10 mg of a compound of Formula I per kg of body weight per day.
- the pharmaceutical composition comprises a compound of Formula I or a pharmaceutically acceptable salt or solvate thereof and a pharmaceutically acceptable carrier.
- composition encompasses a product comprising the active and inert ingredient(s), (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from the combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions between ingredients.
- the composition is comprised of a compound of formula I in an amount that is effective to treat, prevent or delay the onset of type 2 diabetes mellitus, in combination with the pharmaceutically acceptable carrier.
- Any suitable route of administration may be employed for providing a mammal, especially a human with an effective dosage of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols and the like, with oral tablets being preferred.
- a compound of formula I for preparing a pharmaceutical composition which is comprised of combining the compound of formula I with the carrier.
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquids, e.g., suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solids, e.g., powders, capsules and tablets, with the solid oral preparations being preferred. Because of their ease of administration, tablets and capsules represent the most advantageous oral dosage unit forms. If desired, tablets may be coated by standard aqueous or nonaqueous techniques.
- the compounds of Formula I may also be administered by controlled release means and/or delivery devices such as those described in U.S. Patent Nos. 3,845,770; 3,916,899; 3,536,809; 3,598,123; 3,630,200 and 4,008,719.
- compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient, as a powder or granules or as a solution or a suspension in an aqueous liquid, a non-aqueous liquid, an oil-in-water emulsion or a water-in-oil liquid emulsion.
- Such compositions may be prepared by any of the methods of pharmacy but all methods include the step of bringing into association the active ingredient with the carrier which constitutes one or more necessary ingredients.
- the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired presentation.
- a tablet may be prepared by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent.
- each tablet contains from about 1 mg to about lg of the active ingredient and each cachet or capsule contains from about 1 to about 500 mg of the active ingredient.
- Combination Therapy Compounds of Formula I may be used in combination with other drugs that are used in the treatment/prevention/delaying the onset of type 2 diabetes mellitus, as well as the diseases and conditions associated with type 2 diabetes mellitus, for which compounds of Formula I are useful.
- Other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I.
- a pharmaceutical composition containing such other drugs in addition to the compound of Formula I is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of Formula I.
- Examples of other active ingredients that may be combined with a compound of Formula I, either administered separately or in the same pharmaceutical compositions include, but are not limited to: (a) bis-guanides (e.g., buformin, metformin, phenformin), (b) PPAR agonists (e.g., troglitazone, pioglitazone, rosiglitazone), (c) insulin, (d) somatostatin, (e) D-glucosidase inhibitors (e.g., voglibose, miglitol, acarbose), (f) DP-1V inhibitors, (g) LXR modulators and (h) insulin secretagogues (e.g., acetohexamide, carbutamide, chlorpropamide, glibornuride, gliclazide, glimerpiride, glipizide, gliquidine, glisoxepid, glyburide, glyhexamide, glypin
- the weight ratio of the compound of the Formula I to the second active ingredient may be varied within wide limits and depends upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the Formula I is combined with a PPAR agonist the weight ratio of the compound of the Formula I to the PPAR agonist will generally range from about 1000: 1 to about 1 : 1000, preferably about 200: 1 to about 1 :200. Combinations of a compound of the Formula I and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. For combination products, the compound of formula I may be combined with any other active ingredients and then added to the carrier ingredients; alternatively the order of mixing may be varied.
- Examples of pharmaceutical combination compositions include: 1) a compound according to formula I, 2) a compound selected from the group consisting of: a) DP-IV inhibitors; b) insulin sensitizers selected from the group consisting of (i) PPAR agonists and (ii) biguanides; c) insulin and insulin mimetics; d) sulfonylureas and other insulin secretagogues; e) alpha glucosidase inhibitors; f) glucagon receptor antagonists; g) GLP-1, GLP-1 mimetics, and GLP-1 receptor agonists; h) GIP, G1P mimetics, and GIP receptor agonists; i) PACAP, PACAP mimetics, and PACAP receptor 3 agonists; j) cholesterol lowering agents selected from the group consisting of (i) HMG-CoA reductase inhibitors, (ii) sequestrants, (iii) nicotinyl alcohol, nicotinic
- a method that is of particular interest relates to a method of treating, preventing or delaying the onset of diabetes, and in particular, type 2 diabetes, in a mammalian patient in need thereof, comprising administering to the patient 1) a compound according to formula I, and 2) a compound selected from the group consisting of: a) DP-1N inhibitors; b) insulin sensitizers selected from the group consisting of (i) PPAR agonists and (ii) biguanides; c) insulin and insulin mimetics; d) sulfonylureas and other insulin secretagogues; e) alpha glucosidase inhibitors; f) glucagon receptor antagonists; g) GLP-1, GLP-1 mimetics, and GLP-1 receptor agonists; h) GIP, GIP mimetics, and GIP receptor agonists; i) PACAP, PACAP mimetics, and PACAP receptor 3 agonists; j) cholesterol lowering agents selected from the group consisting
- PPAR alpha/gamma dual agonists (vi) inhibitors of cholesterol absorption, (vii) acyl CoA: cholesterol acyltransferase inhibitors, (viii) anti-oxidants and (ix) LXR modulators; (k) PPAR delta agonists; (1) antiobesity compounds; (m) an ileal bile acid transporter inhibitor; (n) anti-inflammatory agents other than glucocorticoids; and (o) protein tyrosine phosphatase-lB (PTP-1B) inhibitors; said compounds being administered in an amount that is effective to treat, prevent or delay the onset of type 2 diabetes.
- PTP-1B protein tyrosine phosphatase-lB
- one method that is of interest relates to a method of treating a condition selected from the group consisting of (1) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) obesity, (5) lipid disorders, (6) dyslipidemia, (7) hyperlipidemia, (8) hypertriglyceridemia, (9) hypercholesterolemia, (10) low HDL levels, (11) high LDL levels, (12) atherosclerosis and its sequelae, (13) vascular restenosis, (14) pancreatitis, (15) abdominal obesity, (16) neurodegenerative disease, (17) retinopathy, (18) nephropathy, (19) neuropathy, (20) Syndrome X, and other conditions and disorders where insulin resistance is a component, in a mammalian patient in need of such treatment, comprising administering to the patient an effective amount of a compound of formula I and a compound selected from the group consisting of: (a) DP-IV inhibitors; (b) insulin sensitizers selected from the group consisting of (i) PP
- a method that is of interest relates to a method of treating a condition selected from the group consisting of hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperlipidemia, hypertriglyceridemia and dyslipidemia, in a mammalina patient in need of such treatment, comprising administering to the patient a therapeutically effective amount of a compound of formula I and an HMG-CoA reductase inhibitor.
- the method that is of interest comprises administering to the patient a therapeutically effective amount of a compound of formula I and an HMG-CoA reductase inhibitor wherein the HMG-CoA reductase inhibitor is a statin, and even more particularly, the statin is selected from the group consisting of lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, itavastatin, ZD-4522 and rivastatin.
- a different aspect of the invention relates to a method of reducing the risk of developing a condition selected from the group consisting of hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperlipidemia, hypertriglyceridemia and dyslipidemia, and the sequelae of such conditions comprising administering to a mammalian patient in need of such treatment a therapeutically effective amount of a compound of formula I and an HMG-CoA reductase inhibitor.
- Another aspect of the invention relates to a method for delaying the onset or reducing the risk of developing atherosclerosis in a human patient in need of such treatment comprising administering to said patient an effective amount of a compound of formula I and an HMG-CoA reductase inhibitor. More particularly, the method comprises administering an effective amount of a compound of formula I and an HMG-CoA reductase inhibitor wherein the HMG-CoA reductase inhibitor is a statin.
- the method comprises administering a compound of formula I and a statin selected from the group consisting of: lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, itavastatin, ZD-4522 and rivastatin. Still more particularly, the method comprises administering a compound of formula I and the statin known as simvastatin.
- Another aspect of the invention relates to a method of reducing the risk of developing a condition selected from the group consisting of hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperlipidemia, hypertriglyceridemia and dyslipidemia, and the sequelae of such conditions comprising administering to a mammalian patient in need of such treatment a therapeutically effective amount of a compound of formula I and a cholesterol absorption inhibitor.
- the method comprises administering an effective amount of a compound of formula I and the cholesterol absorption inhibitor known as ezetimibe.
- a method for delaying the onset or reducing the risk of developing atherosclerosis in a human patient in need of such treatment comprises administering to said patient an effective amount of a compound of formula I and a cholesterol absorption inhibitor. More particularly, the method comprises administering a compound of formula I and the cholesterol absorption inhibitor known as ezetimibe.
- the compounds (la) where R 3 is hydrogen may be prepared from ester Ha (vide infra),
- R l , R 2 , R 4 , R 8 , and R 9 are as defined above and R 10 represents an alkyl or aryl group.
- Amine 1 may be commercially available or readily prepared via a reductive amination sequence by treating, for example, carbomethoxy benzaldehyde 2 (if R 8 and R 9 are hydrogen) and an amine 3 with a reducing agent such as sodium triacetoxyborohydride or cyanoborohydride in a solvent such as dichloroethane at ambient temperature.
- a reducing agent such as sodium triacetoxyborohydride or cyanoborohydride in a solvent such as dichloroethane at ambient temperature.
- the benzylamine 4 can be reacted with the appropriate R 4 carbonyl containing substituent under the same conditions to give amine I.
- Amine 1 is then treated with thiophosgene in the presence of a base such as diethylisopropylamine (DIEA) in a nonpolar aprotic solvent such as dichloromethane at temperatures of zero to 25° C followed by direct addition of a 1,2-diaminobenzene and either mercury (II) trifluoroacetate or methyl iodide (for example /. Med. Chem., 1985, 28, 1925 and Synthesis, 1974, 41). The reaction is stirred a further 30 min to 6h before isolation of benzimidazole 5 with an aqueous work-up.
- a base such as diethylisopropylamine (DIEA) in a nonpolar aprotic solvent such as dichloromethane
- a nonpolar aprotic solvent such as dichloromethane
- 1,2-Diaminobenzene analogs are commercially available, or readily prepared by those skilled in the art by reduction of the corresponding 2-nitroaniline with, for example hydrogen and a palladium catalyst or stannous chloride. Either reaction is effected in an alcoholic solvent such as methanol or ethanol.
- An alternative approach to synthesizing benzimidazole Ha involves reaction of amine 1 with triphosgene in the presence of a base, such as triethylamine, in a nonpolar aprotic solvent such as dichloromethane at temperatures of zero to 25 °C, as shown in Scheme 2.
- a base such as triethylamine
- a nonpolar aprotic solvent such as dichloromethane
- the carbamoyl chloride 6 formed in the reaction can be readily isolated and treated with a 1,2-diaminobenzene to give the urea which is treated directly with a dehydrating agent, usually phosphorus oxychloride, at elevated temperatures for 6 - 24h, followed by an aqueous work-up to yield the benzimidazole 5.
- Coupling of the acid with an amine is then achieved using l-ethyl-3-(3- dimethylaminopropyl)-carbodiimide (EDC), 1-hydroxybenzotriazole (HOBt), and a base, generally diisopropylethylamine, in a solvent such as N,N-dimethylformamide (DMF) or methylene chloride for 3 to 48 hours at ambient temperature to yield the compounds Ia-7 and Ia-8.
- EDC l-ethyl-3-(3- dimethylaminopropyl)-carbodiimide
- HOBt 1-hydroxybenzotriazole
- a base generally diisopropylethylamine
- the product is purified from unwanted side products by recrystallization, trituration, preparative thin layer chromatography, flash chromatography on silica gel as described by W. C. Still et al, J. Org. Chem., 43, 2923, (1978), or HPLC.
- Compounds purified by HPLC may be isolated as the corresponding salt. Purification of intermediates is achieved in the same manner.
- enantiomerically pure compounds enantiomerically pure starting materials should be used for the preparation of enantiomerically pure compounds.
- intermediates such as 5 can be undertaken in one of several different ways. These manipulations may include, but are not limited to substitution, reduction, oxidation, alkylation, acylation, and hydrolysis reactions, which are commonly known to those skilled in the art.
- the compounds (lb) (which are defined as compounds of formula I wherein R 3 is not hydrogen) may be prepared from ester lib (vide infra),
- R 1 , R 2 , R 3 , R 4 , R 8 , and R 9 are as defined above and R 10 represents an alkyl or aryl group.
- N-alkylated 1 ,2-diaminobenzene K are commercially available or readily prepared by those skilled in art.
- One such method involves alkylation of a 2-nitro aniline. This is effected by deprotonation with a base such as sodium hydride in a polar aprotic solvent such as dimethylformamide (DMF) at 0 - 25 °C for 15min to 2h, followed by addition of an electrophile such as an alkyl iodide, Scheme 5. The reaction is stirred for an additional 1 - 24 h to give intermediate J_l , which can be reduced with, for example hydrogen and a palladium catalyst or stannous chloride in an alcoholic solvent.
- the alkylated 2-nitro aniline JJ. can also be prepared by nucleophilic displacement of fluorine from a 2-fluoronitrobenzene 12 with an amine as described in J. Org. Chem., 1999, 64, 3060. This is achieved in a solvent such as methylene chloride or DMF with a base such as DIEA, at temperatures of 25 - 80 °C for l-6h, Scheme 5.
- the diaminobenzene H) can then be converted to the benzimidazole 9 using amine 1 or carbamoyl chloride 6 in an identical fashion to that described above and illustrated in Schemes 6 and 7.
- a third route to intermediates Hb involves alkylation of the 2-aminobenzimdazole 3 with a benzylic bromide, for example carbomethoxy benzyl-bromide, as illustrated in Scheme 8.
- Amine 3 is converted to the isothiocyanate by reaction with thiophosgene in the presence of a base such as DIEA in a nonpolar aprotic solvent such as dichloromethane at temperatures of zero to 25° C followed by addition of diamine 10 and cyclization with an agent such as methyl iodide. This reaction is effected at 25 - 50 °C for 1 - 24 h to give amine 13.
- Deprotonation is achieved with a base such as sodium hydride or potassium carbonate in a nonpolar aprotic solvent such as DMF to give a mixture of the desired compound 9 and its isomer 14.
- a base such as sodium hydride or potassium carbonate
- a nonpolar aprotic solvent such as DMF
- ester 9 Conversion of ester 9 to the final products is achieved by saponification of the ester using a base such as aqueous lithium or sodium hydroxide in a polar solvent such as tetrahydrofuran, methanol, ethanol or a mixture of similar solvents, Scheme 9.
- a base such as aqueous lithium or sodium hydroxide in a polar solvent such as tetrahydrofuran, methanol, ethanol or a mixture of similar solvents, Scheme 9.
- Coupling of the acid with an amine is then achieved using l-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), 1-hydroxybenzotriazole (HOBt), and a base, generally diisopropylethylamine, in a solvent such as N,N-dimethylformamide (DMF) or methylene chloride for 3 to 48 hours at ambient temperature to yield the compounds Ib-7 and Ib-8.
- EDC l-ethyl-3-(3-dimethylaminopropyl)-carbodiimide
- HOBt 1-hydroxybenzotriazole
- a base generally diisopropylethylamine
- DMF N,N-dimethylformamide
- methylene chloride methylene chloride
- the product is purified from unwanted side products by recrystallization, trituration, preparative thin layer chromatography, flash chromatography on silica gel as described by W. C. Still et al, /. Org. Chem., 43, 2923, (1978), or HPLC.
- Compounds purified by HPLC may be isolated as the corresponding salt. Purification of intermediates is achieved in the same manner.
- Deprotection of a methoxy ether is routinely effected by treatment of the compound with boron tribromide in a solvent such as methylene chloride for a period of 1 - 16h at ambient temperatures. Finally, if the alcohol is protected as an allyl ether, this is removed by treatment with dimethylbarbituric acid and a palladium catalyst, routinely tris(dibenzylideneacetone)dipalladium(0), with a ligand such as l,4-bis-(diphenylphospino)butane in an aprotic solvent such as methylene chloride for 15min to 2h. See “Protective Groups in Organic Synthesis", Greene, published by Wiley and Sons.
- the free hydroxyl group may then be further modified to prepare ethers using an alcohol and coupling agent, such as diisopropylazodicarboxylate, and triphenylphosphine in a non polar solvent such as methylene chloride at temperatures of 0 to 40°C for 1 to 16h, Scheme 10.
- an alcohol and coupling agent such as diisopropylazodicarboxylate, and triphenylphosphine in a non polar solvent such as methylene chloride at temperatures of 0 to 40°C for 1 to 16h, Scheme 10.
- Intermediates J_6 and 17 can then be converted to the desired products as previously described, vide supra.
- R 4 contains an aromatic halide as in 18, Scheme 11
- R 4 contains an aromatic halide as in 18, Scheme 11
- the halide is coupled with a boronic acid, exemplified here with phenyl boronic acid, using a palladium catalyst such as palladium acetate and tris-o-tolylphosphine or triphenyl phosphine.
- the solvent is generally DMF or ethanol, and cesium carbonate or aqueous sodium carbonate is also added to the reaction, which is performed at elevated temperatures for 12-24 h (see Helv. Chim.
- R and R 9 form a 5-membered ring alternate conditions were used for the synthesis of the amine intermediate 21, Scheme 12.
- commercially available ketone 22 was converted to amine 23 by a reductive amination sequence using a Lewis acid such as titanium isopropoxide in ethanol at ambient temperature for 6 - 24 h, followed by further reduction with a hydride reducing agent such as sodium borohydride ( J. C. S., Perkin Trans 1, 1998, 2527-2531).
- a hydride reducing agent such as sodium borohydride
- decaborane in methanol at ambient temperature can be used for the reductive amination (J.C.S. Perkin Trans 1, 2000 145-146).
- ester linkage is then installed by treatment of the bromide with a base such as butyl lithium at -78 °C in a polar aprotic solvent such as THF, followed by quenching the reaction with solid carbon dioxide to give the acid.
- a base such as butyl lithium at -78 °C in a polar aprotic solvent such as THF
- THF polar aprotic solvent
- Intermediate 21 can then be converted to the desired products as previously described, vide supra.
- Condition B 20 to 60% acetonitrile in water (each containing 0.1% trifluoroacetic acid).
- Condition C 20 to 80% acetonitrile in water (each containing 0.1% trifluoroacetic acid).
- Condition D 20 to 100% acetonitrile in water (each containing 0.1% trifluoroacetic acid).
- Step A l-Isothiocyanato-2-nitrobenzene.
- 2-nitroaniline 10 mmol, 1.38 g
- DIEA 15 mmol, 2.6 mL
- thiophosgene 15 mmol, 1.14 mL
- HPLC A 2.24 min.
- Step B Methyl 4- (r(4-tert-butylcyclohexyl)aminol methyl Ibenzoate
- the reaction mixture was stirred at ambient temperature for 1.5 h, then concentrated under reduced pressure to ca. 25 % of the initial volume. 400 mL of EtOAc was added to the solution and the mixture was washed with 3 x 200 mL of 5 % NaHC0 3 followed by brine. The organic phase was dried over MgS0 and concentrated under reduced pressure. The residue was purified by flash chromatography on silica eluting with 1:1 EtOAc/hexanes to afford the trans isomer as a white solid.
- Step C Methyl 4- ⁇ riH-benzimidazol-2-yl(4-tert-butylcvclohexyl)amino1methyl)-benzoate
- DIEA 0.8 mL
- the reaction mixture was concentrated under reduced pressure.
- the residue was taken up in 5 mL of DMF containing 0.4 mL of H 2 0, and SnCl 2 (2 g) was added (exothermic).
- the crude thiourea was concentrated under reduced pressure and the residue was taken up in 10 mL of EtOH.
- Step D 4- ⁇ riH-Benzimidazol-2-yl(4-tgrt-butylcvclohexyl)aminolmethyl)benzoic acid
- Step E 4- ⁇ riH-Benzimidazol-2-yl(4-tgrt-butylcvclohexyl)aminolmethyIl-N-(lH-tetra-azol-5- vDbenzamide
- DIEA 0.5 mmol, 90 ⁇ L
- Step A 4- ⁇ r(4-tgrt-Butylcvclohexyl)(l-methyl-lH-benzimidazol-2-v ⁇ aminolmethv ⁇ -benzoic acid
- Step B N-(4- (4-tgrt-ButylcvclohexylKl-methyl-lH-benzimidazol-2-yl)aminolmethyll-benzoylVD- alanine
- Step A Methyl 4-(r(4-fgrt-butylcvclohexyl ' )(l-(2-r(trimethylsilyl ' )oxylethyll-lH-benz-imidazol-2- yDaminol methyl Ibenzoate
- Step B 4- ⁇ r(4-tgrt-Butylcvclohexyl)(l-l2-hydroxyethyU-lH-benzimidazol-2-yl ' )-aminolmethyllbenzoic acid
- Step C 4-( ⁇ (4-tgrt-Butylc vclohexyl) I " 1 -(2-hvdroxyethvD- lH-benzimidazol-2-y 11 amino ) -methyl)-N-( 1H- tetraazol-5 -vDbenzamide
- Example 1 Step A To the product of Example 1 Step A (3 mmol, 910 mg) and DIEA (3.6 mmol, 626 ⁇ L) in 12 mL of DCM was added thiophosgene (3 mmol, 229 ⁇ L). After 1 h additional DIEA (3.6 mmol) and the title compound of Example 11 Step C (2.75 mmol, 489 mg) were added. After 1.5 h Hg(0 2 CCF 3 ) 2 (3 mmol, 1.27 g) was added (exothermic), and the slurry was allowed to stand overnight. HPLC analysis revealed the cyclization was incomplete, so additional Hg(0 2 CCF 3 ) 2 (1.5 mmol, 650 mg) was added to the reaction.
- Step E 4-f rf6-(Allyloxy)-l-methyl-lH-benzimidazol-2-yll(4-tgrt-butylcvclohexyl)aminolmethyl Ibenzoic acid
- a solution of LiOH 0.4 mmol, 10 mg
- H 2 400 ⁇ L
- the resulting solution was stirred at ambient temperature overnight.
- the reaction mixture was taken up in a pH 7 buffer solution and EtOAc.
- Step F 4-1 rr6-(AlIyloxy)-l-methyl-lH-benzimidazol-2-yll(4-tgrt-butylcvclohexyl ' )-aminolmethvU-N- ( lH-tetraazol-5-yl)benzamide
- 1H- tetraazol-5-amine monohydrate (0.12 mmol, 12 mg)
- ⁇ OBt (0.08 mmol, 12 mg
- EDC 0.08 mmol, 15 mg
- Step A Methyl 4- ( r(4-tgrt-butylcvclohexyl )(6-hvdroxy- 1 -methyl- 1 H-benzimidazol-2- yl , a ⁇ -inol methyl 1 benzoate
- Step B 4-(r(4-tgrt-Butylcyclohexyl)(6-hydroxy-l-methyl-lH-benzimidazol-2-yl)aminol-methyl)benzoic acid
- Step C 4- ⁇ r(4-tgrt-Butylcyclohe ⁇ yl)(6-hvdroxy-l-methyl-lH-benzimidazol-2-yl)amino1-methyll-N-(lH- tetraazol-5-yl)benzamide
- lH-tetraazol-5 -amine monohydrate (0.12 mmol, 12 mg)
- HOBt (0.08 mmol, 12 mg
- EDC 0.08 mmol, 15 mg
- Step A Methyl 4-1 f (4-tgrt-butylcvclohexyl)(l-methyl-6-propoxy-lH-benzimidazol-2- yDaminol methyl Ibenzoate
- n- propanol 0.25 mmol, 19 ⁇ L
- diisopropyl azodicarboxylate 0.2 mmol, 37 ⁇ L
- Step B 4- ⁇ r(4-tert-Butylcvclohexyl)(l-methyl-6-propoxy-lH-benzimidazol-2-yl)aminol- methyl ibenzoate
- Step A ⁇ -Methyl-4-methoxy-2-nitroaniline.
- Step B N-Methyl-4-methoxy-1.2-phenylenediamine.
- Step D 4-1 f (4-tgrt-Butylcvclohexyl)( 5-methoxy-l-methyl-lH-benzimidazol-2-yl)aminolmethyl)benzoic acid
- Step E 4-( f(4-tgrt-Butylcvclohexyl)(5-methoxy-l-methyl-lH-benzimidazol-2-yl)-aminolmethyl ⁇ -N-(lH- tetraazol-5-yl)benzamide
- Step A Methyl 4-( [(4-tgrt-butylcvclohexyl)( 5-hvdroxy-l-methyl-lH-benzimidazol-2- yl.aminolmethyl Ibenzoate
- BBr 3 5 mmol, 5 mL of a IM solution in DCM
- Step C 4-(r(4-tgrt-Butylcvclohexyl)(5-hydroxy-l-methyl-lH-benzimidazol-2-yl)-amino-lmethy ⁇ -N-(lH- tetraazol-5-yl .benzamide
- EDC 0.2 mmol, 38 mg
- Step A Methyl 4-1 r(4-tgrt-butylcvclohexyl)( 5-benzyloxy-l-methyl-lH-benzimidazol-2- yl .aminolmethyl Ibenzoate
- Step B 4-(r(4-tert-Butylcvclohexyl)( 5 -benzyloxy-1 -methyl- 1 H-benzimidazol-2-yl.- aminol methyl I benzoic acid
- Step C 4-(r(4-tgrt-Butylcyclohexyl)(5-benzyloxy-l-methyl-lH-benzimidazol-2-yl)-aminolmethyll-N- ( lH-tetraazol-5-yl)benzamide
- Step B l-
- Step C 4-Cvclohex-l-en-l-ylphenylamine The title compound from Example 221 Step B was taken up in a solution of 150 mL of
- Step D N-(4-Cvclohex- 1 -en- 1 -ylpheny 1)- 1 -methyl- lH-benzimidazol-2-amine
- Step E Methyl 4-(r(4-cvclohex-l-en-l-ylphenyl)(l-methyl-li t - ' -benzi idazol-2-yl)- aminolmethyl 1 benzoate
- To the title compound from Example 221 Step D (0.53 mmol, 161 mg) and NaH (0.80 mmol, 32 mg of a 60% dispersion in mineral oil) was added 1 mL of DMF (gas evolution). The mixture was stirred for 30 min at ambient temperature, then methyl 4-(bromomethyl)benzoate (0.80 mmol, 182 mg) was added.
- Step F 4- ⁇ f(4-Cvclohex-l-en-l-ylphenyl (l-methyl-l//-benzimidazol-2-yl)amino1-methyl
- a solution of LiOH 2.6 mmol, 62 mg
- the reaction was stirred at 50°C for 30 min.
- the dioxane was removed under reduced pressure and the remaining aqueous solution was acidified with 2 N HCl.
- the resulting precipitate was filtered, washed with H 2 0 and dried under reduced pressure, affording the product as a white solid.
- HPLC A 2.11 min.
- Step G 4-( r(4-Cvclohex-l-en-l-ylphenyl)(l-methyl-lH-benzimidazol-2-yl)aminol-methyll-N-(lH- tetraazol-5 -vDbenzamide
- Step B Methyl 4-( r(3.5-dichlorophenyl)(l-methyl-lH-benzimidazol-2-yl)aminol-methyl Ibenzoate
- ⁇ aH 0.24 mmol, 6 mg of a 60% slurry in mineral oil
- DMF gas evolution
- methyl-4-(bromomethyl)benzoate 0.24 mmol, 55 mg was added and the reaction mixture was allowed to stand at ambient temperature overnight.
- the mixture was partitioned between DCM and ⁇ aHC0 3 .
- the organic phase was collected and the aqueous phase was extracted 2 x with DCM.
- Step C 4-f r(3,5-Dichlorophenyl)(l -methyl- lH-benzimidazol-2-yl)aminolmethyl I -benzoic acid
- a solution of LiOH 0.8 mmol, 19 mg
- the reaction was allowed to stir at ambient temperature overnight.
- the crude reaction mixture was poured into pH 7 buffer/EtOAc, which was acidified with 2 N HCl until two clear layers formed after agitation.
- the organic phase was collected and the aqueous phase was extracted twice with EtOAc.
- the combined organic phase was dried over MgS0 , then concentrated under reduced pressure to afford the product as a white foam.
- HPLC A 1.79 min.
- Step P 4- ⁇ [(3,5-Dichlorophenyl)(l-methyl-lH-benzimidazol-2-yl)aminolmethyl)-N-(l, I - f -tetraazol-5- vDbenzamide
- DIEA 0.48 mmol, 83 ⁇ L
- Step P Methyl 3-bromo-4- ⁇ r(4-t g rt-butylcvclohexyl)(5-methoxy-l-methyl-lH-benz-imidazol-2- vDaminol methyl Ibenzoate
- Step F 3-Bromo-4- ⁇ f(4-tgrt-butylcvcIohexyl)(5-methoxy-l-methyl-lH-benzimidazol-2- yl .aminolmethyl 1 -N-( lH-tetraazol-5-yl)benzamide
- Step A Methyl 3-bromo-4- ⁇ r(4-tgrt-butylcvclohexyl)(5-hvdroxy-l-methyl-lH-benz-imidazol-2- vDaminolmethyl 1 benzoate
- Step B Methyl 3-bromo-4- ⁇ r(4-tgrt-butylcvclohexyl)(5-cvclopentyloxy-l-methyl-lH-benz-imidazol-2- yDaminol methyl Ibenzoate
- Step C 3-Bromo-4- ⁇ r(4-tgrt-butylcyclohexyl)(5-cyclopentyloxy-l -methyl- lH-benz-imidazol -2- y Daminol methyl Ibenzoic acid
- Step D 3-Bromo-4-l r(4-tgrt-butylcvclohexyl)(5-cvclopentyloxy-l-methyl-lH-benzimidazol-2- y 1. aminol methyl ) -N-( 1 H-tetraazol-5 -vDbenzamide
- Step A Ethyl 4-1 l-f(tr ⁇ fi-.-4-tgrt-butylcvclohexyl)aminolethyl Ibenzoate
- Step B Ethyl 4-1 H(tr ⁇ n,y-4-tgrt-butylcvclohexyl)(l-methyl-lH-benzimidazol-2- vDaminolethyl Ibenzoate
- ethyl 4- ⁇ l-[(tran-;-4-tgrt-butylcyclohexyl)amino]ethyl ⁇ benzoate (0.55 g, 1.66 mmol) and DIEA (0.35 mL, 1.99 mmol) in dry dichloromethane (15 mL) was slowly added thiophosgene (0.13 mL, 1.66 mmol).
- Step C 4-1 l-r(tr ⁇ w-.-4-tgrt-ButylcvcIohexyl)(l-methyl-lH-benzimidazol-2-yl)amino1ethyll-N-lH- tetrazol-5-ylbenzamide. isomer A and B.
- Step A (5-Bromo-2,3-dihydro- lH-inden- 1 -ylXtrans -4-tgrt-butylcvclohexyl)amine
- 5-bromoindan-l-one (6.33 g, 30.0 mmol)
- titanium (IV) isopropoxide (17.8 mL, 60.0 mmol)
- 4-tgrt-butylcyclohexyl amine (9.32 g, 60.0 mmol) in absolute ethanol (200 mL) was stirred under nitrogen at room temperature for 12 h.
- Sodium borohydride (1.70 g, 45.0 mmol) was then added and the resulting mixture was stirred for an additional 8 h at room temperature.
- Step B Methyl l-r(tr ⁇ n-.-4-tgrt-butylcvclohexyl)aminolindane-5-carboxylate
- Step C Methyl l-r(tr ⁇ » -4-tgrt-butylcvclohexyl)(l-methyl-lH-benzimidazol-2-yl)aminolindane-5- carboxylate
- Step P l-[(tr ⁇ n-.-4-tgrt-Butylcvclohexyl)(l-methyl-lH-benzimidazol-2-yl)aminol-N-lH-tetrazol-5- ylindane-5-carboxamide.
- Step B Butyl l-[(4-cvclohexylphenyl)(l-methyl-lH-benzimidazol-2-yl)aminolindane-5-carboxyIate To a 0°C solution of butyl l-[(4-cyclohexylphenyl)amino]indane-5-carboxylate (0.50 g,
- Step C l-f(4-Cvclohexylphenyl " )(l-methyl-lH-benzimidazol-2-yl)aminol-N-lH-tetrazol-5-ylindane-5- carboxamide. isomer B.
- Butyl l-[(4-cyclohexylphenyl)(l-methyl-lH-benzimidazol-2-yl)amino]indane-5- carboxylate (0.10 g, 0.18 mmol) was dissolved in EtO ⁇ /n- ⁇ eptane (1: 1, 4 mL) and eluted with 10% isopropanol in n- ⁇ eptane on ChiralPak AD column. The fast moving component was collected as isomer A and the slow moving component as isomer B.
- Step A Butyl l-r(tr ⁇ n-.-4-tgrt-butylcyclohexyl)(5-methoxy-l-methyl-lH-benzimidazol-2- yl)aminolindane-5-carboxylate
- Step B Butyl l-l(tr ⁇ n .-4-t g rt-butylcvclohexyl)(5-hydroxy-l-methyl-lH-benzimidazol-2- yl)aminolindane-5-carboxylate
- Butyl l-[(tr n-.-4-tgrt-butylcyclohexyl)(l-methyl-5-propoxy-lH-benzimidazol-2- yl)amino]indane-5-carboxylate (36.0 mg, 0.06 mmol) was dissolved in T ⁇ F/MeO ⁇ (1:1, 6 mL) and aq. LiO ⁇ (1.0 M, 3 mL) was added. After stirred at room temperature for 16 h, the reaction was neutralized with aqueous ⁇ C1 (IN, 3.5 mL) until white precipitate started to appear. The resulting mixture was poured into brine (10 mL) and extracted with EtOAc (3x10 mL).
- BIOLOGICAL ASSAYS The ability of the compounds of the present invention to inhibit the binding of glucagon and their utility in treating or preventing type 2 diabetes mellitus and the related conditions can be demonstrated by the following in vitro assays.
- Glucagon Receptor Binding Assay A stable CHO (Chinese hamster ovary) cell line expressing cloned human glucagon receptor was maintained as described (Chicchi et al._J Biol Chem 272, 7765-9(1997); Cascieri et al. J Biol Chem 274, 8694-7(1999)).
- Glucagon-stimulated Intracellular cAMP Formation Exponentially growing CHO cells expressing human glucagon receptor were harvested with the aid of enzyme-free dissociation media (Specialty Media), pelleted at low speed, and re- suspended in the Cell Stimulation Buffer included in the Flash Plate cAMP kit (New England Nuclear, SMP0004A). The adenylate cyclase assay was setup as per manufacturer instructions. Briefly, compounds were diluted from stocks in DMSO and added to cells at a final DMSO concentration of 5%.
- Cells prepared as above were preincubated in flash plates coated with anti-cAMP antibodies (NEN) in presence of compounds or DMSO controls for 30 minutes, and then stimulated with glucagon (250 pM) for an additional 30 minutes.
- the cell stimulation was stopped by addition of equal amount of a detection buffer containing lysis buffer as well as 1 5 I-labeled cAMP tracer (NEN). After 3 hours of incubation at room temperature the bound radioactivity was determined in a liquid scintillation counter (TopCount-Packard Instruments). Basal activity (100% inhibition) was determined using the DMSO control while 0% inhibition was defined at the amount of pmol cAMP produced by 250pM glucagon.
Abstract
Description














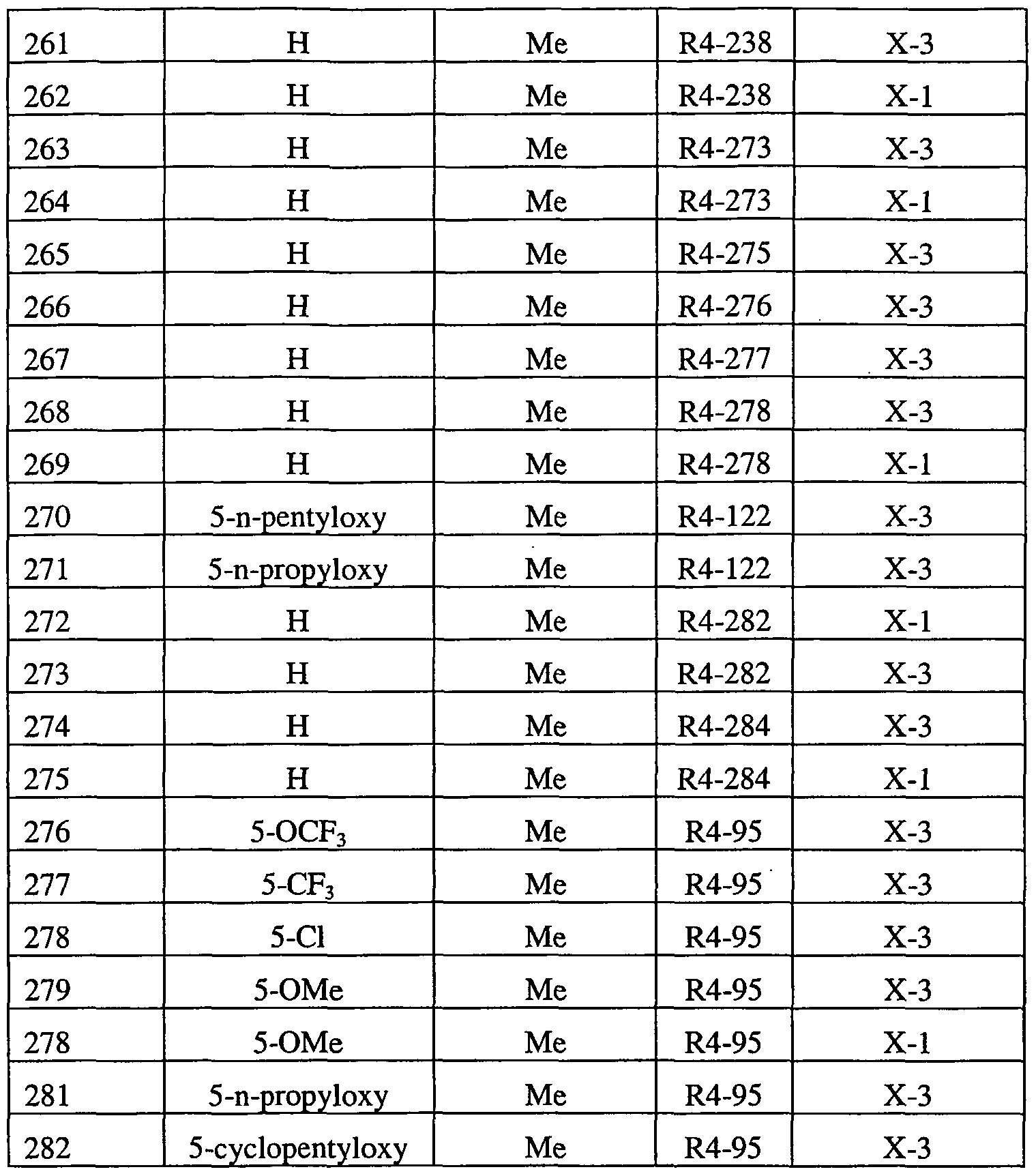


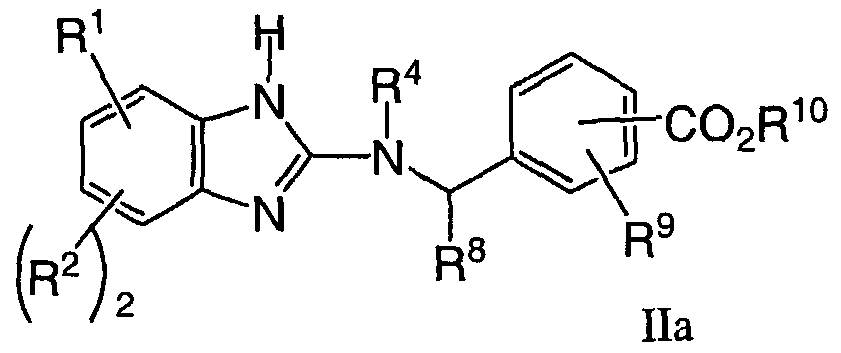



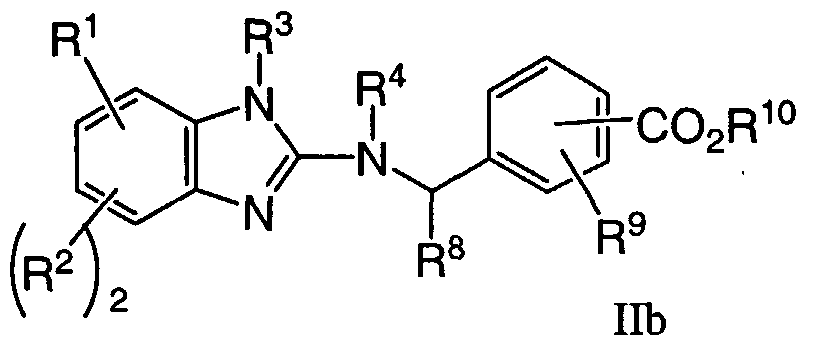



















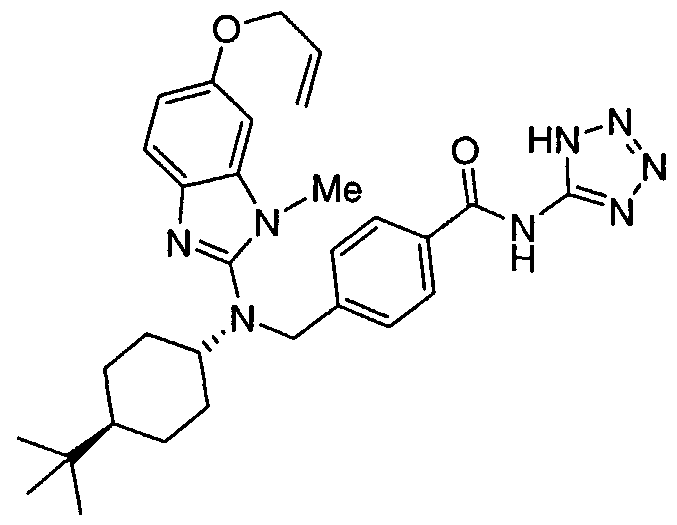
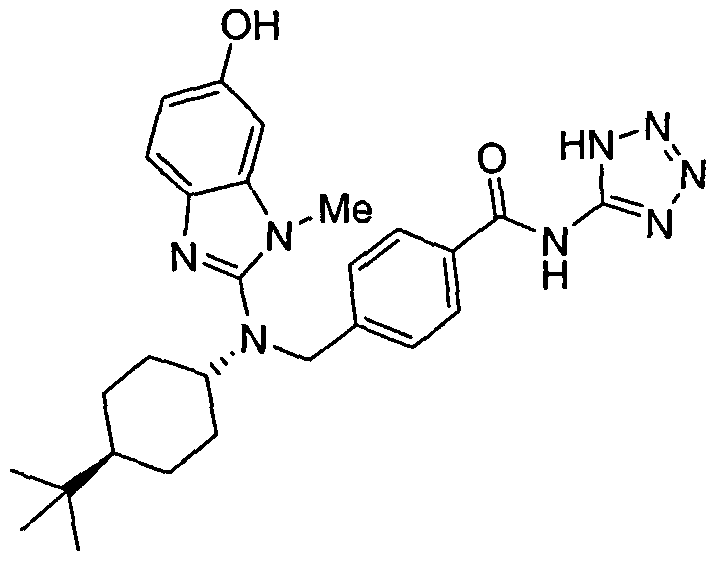

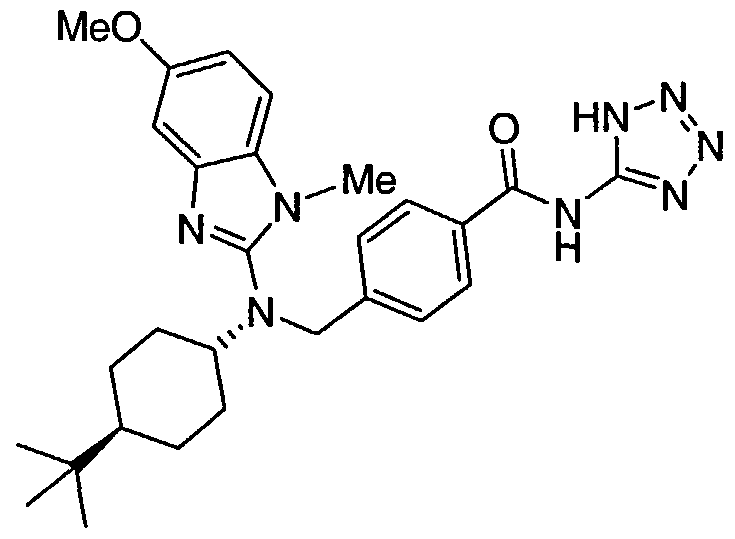





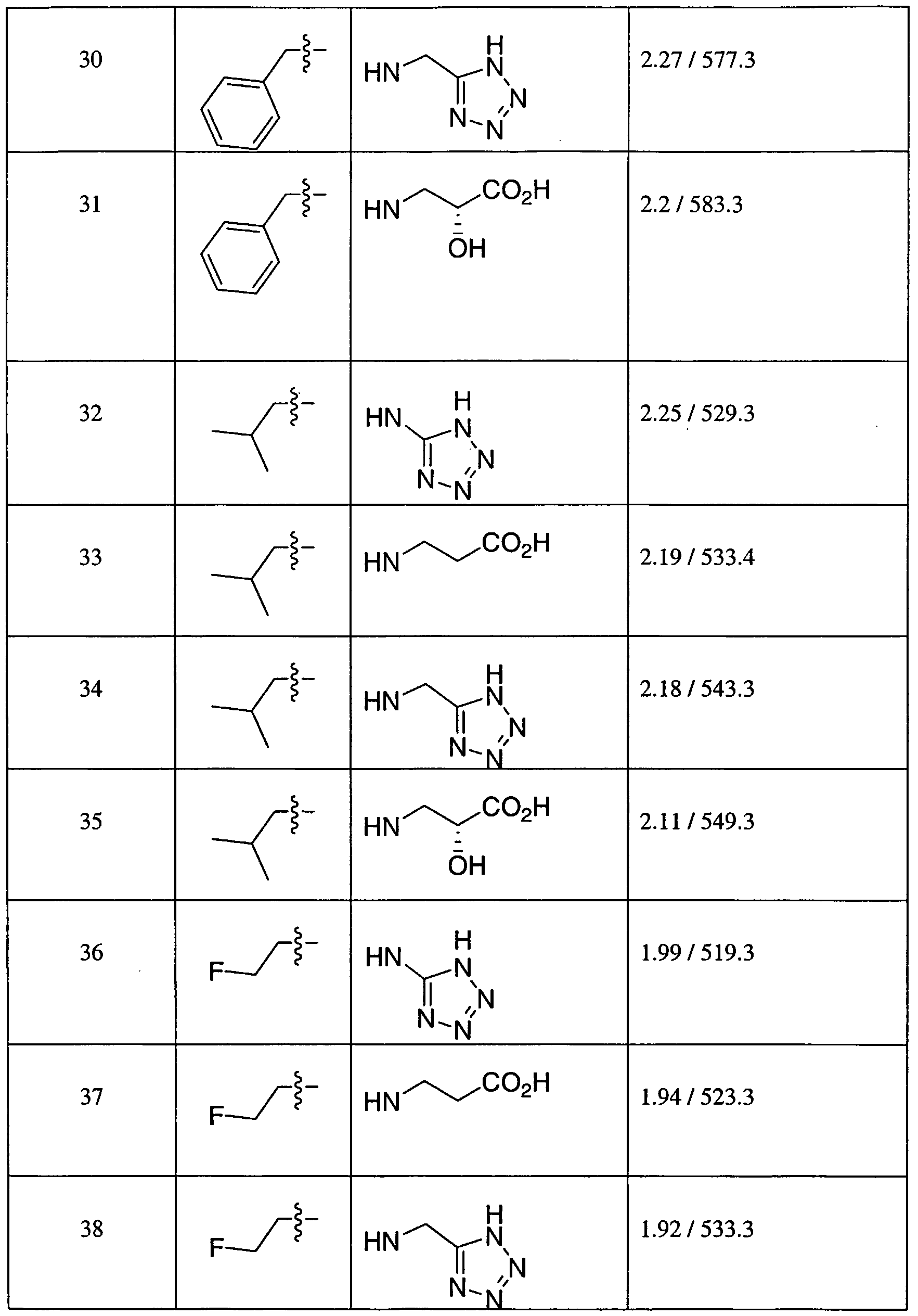




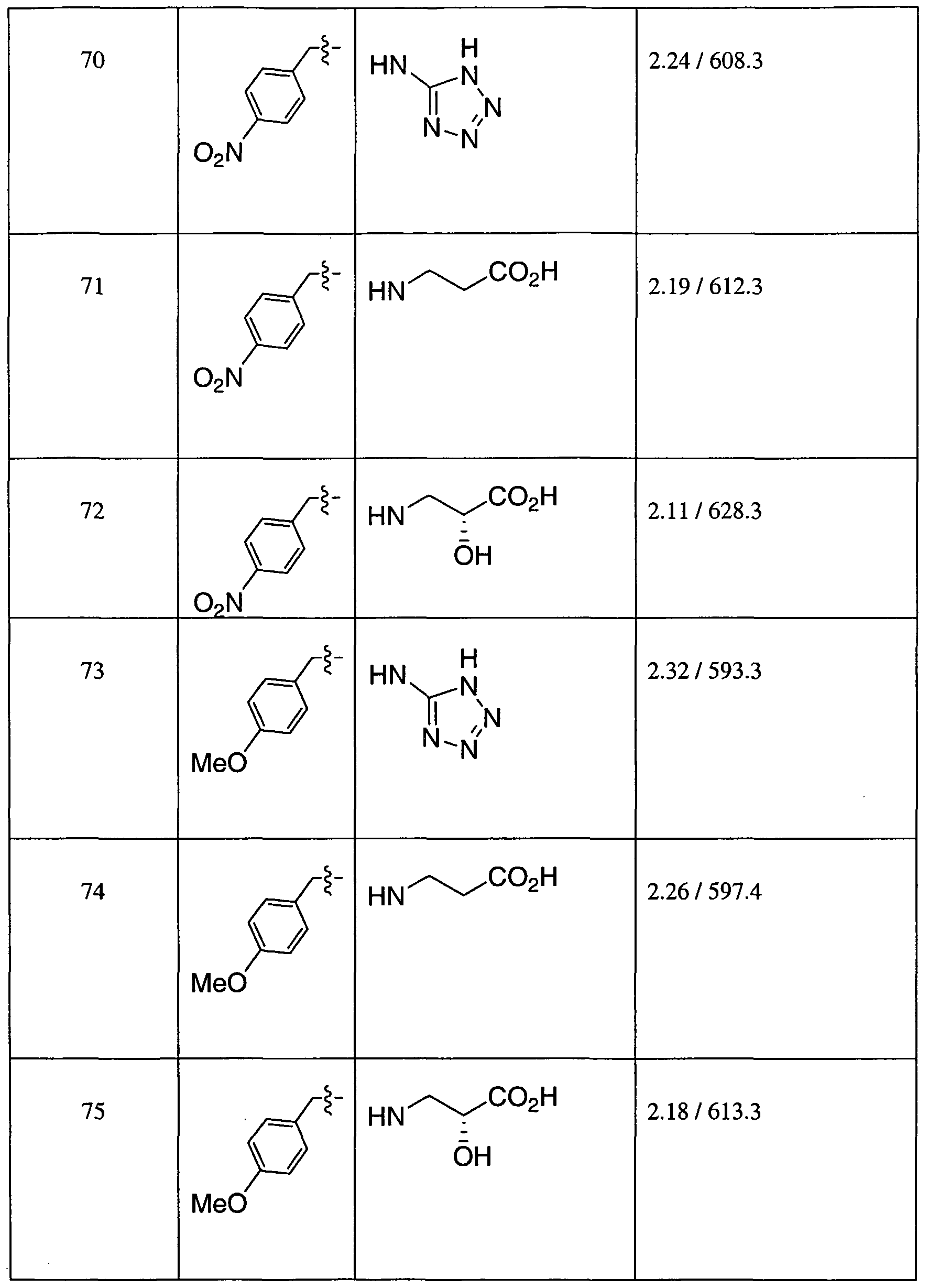

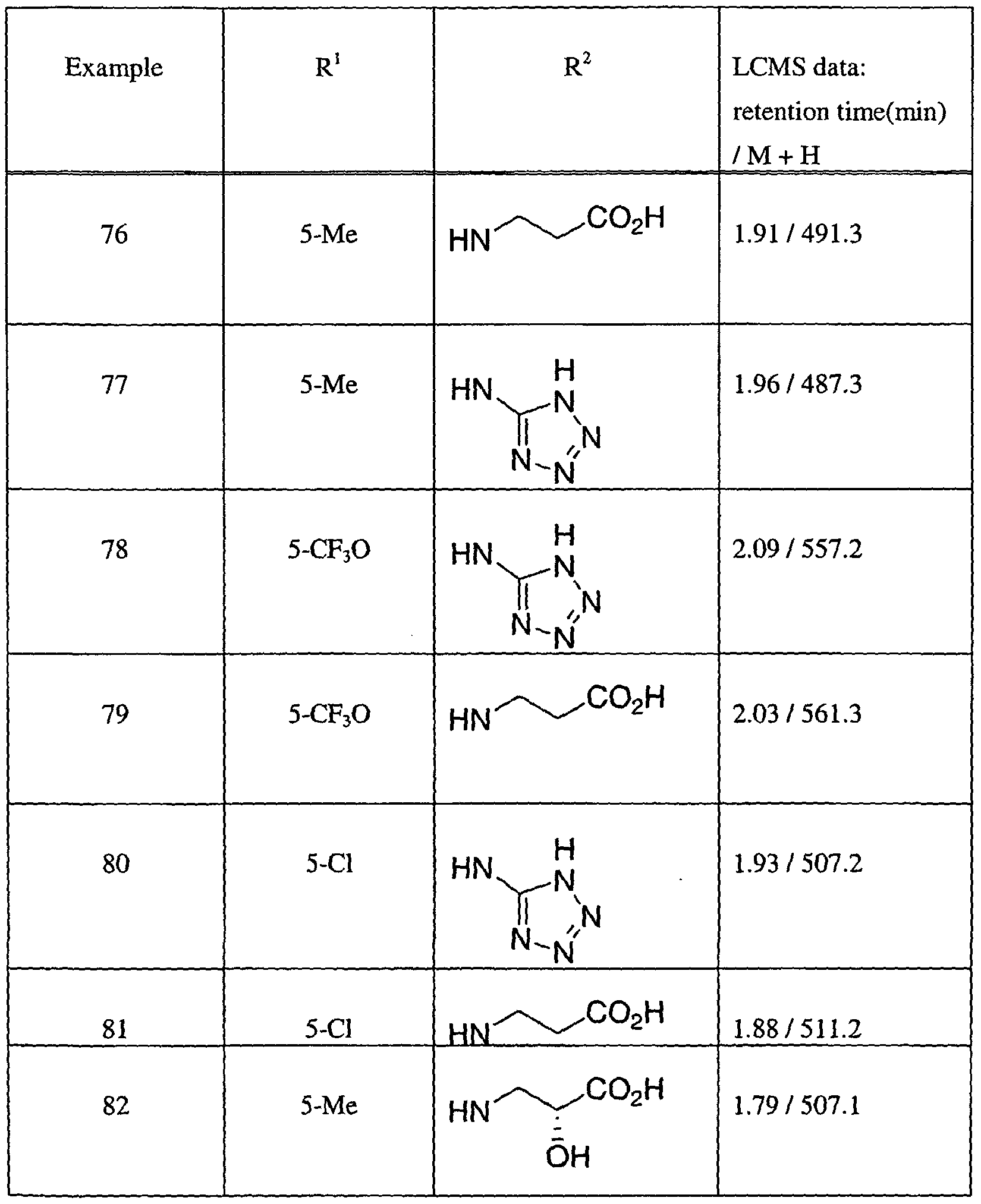









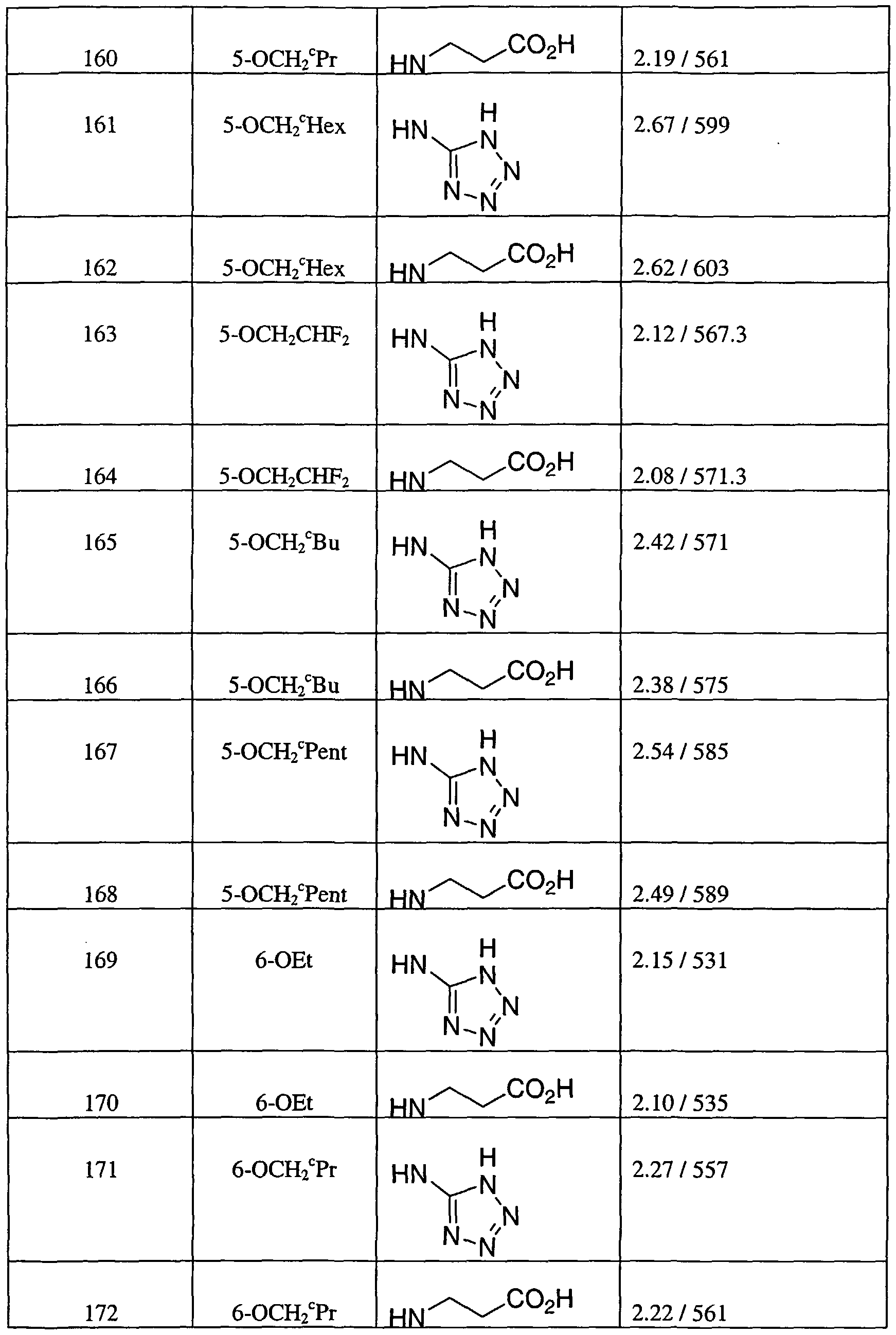




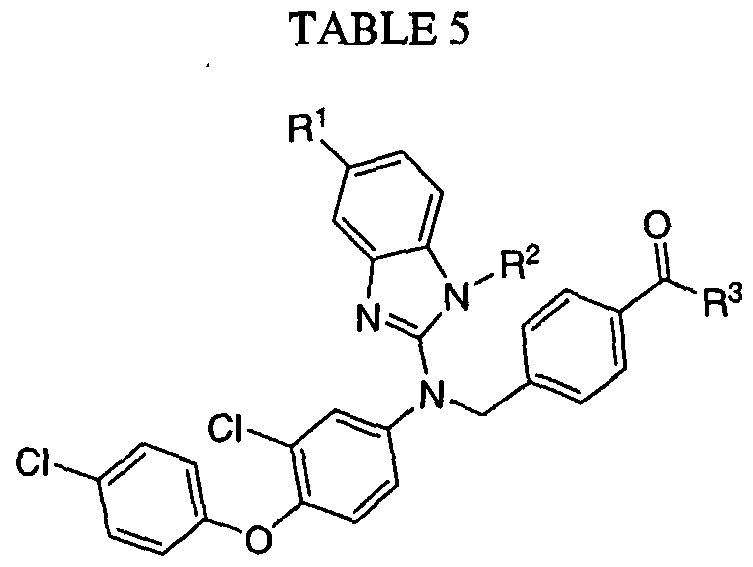



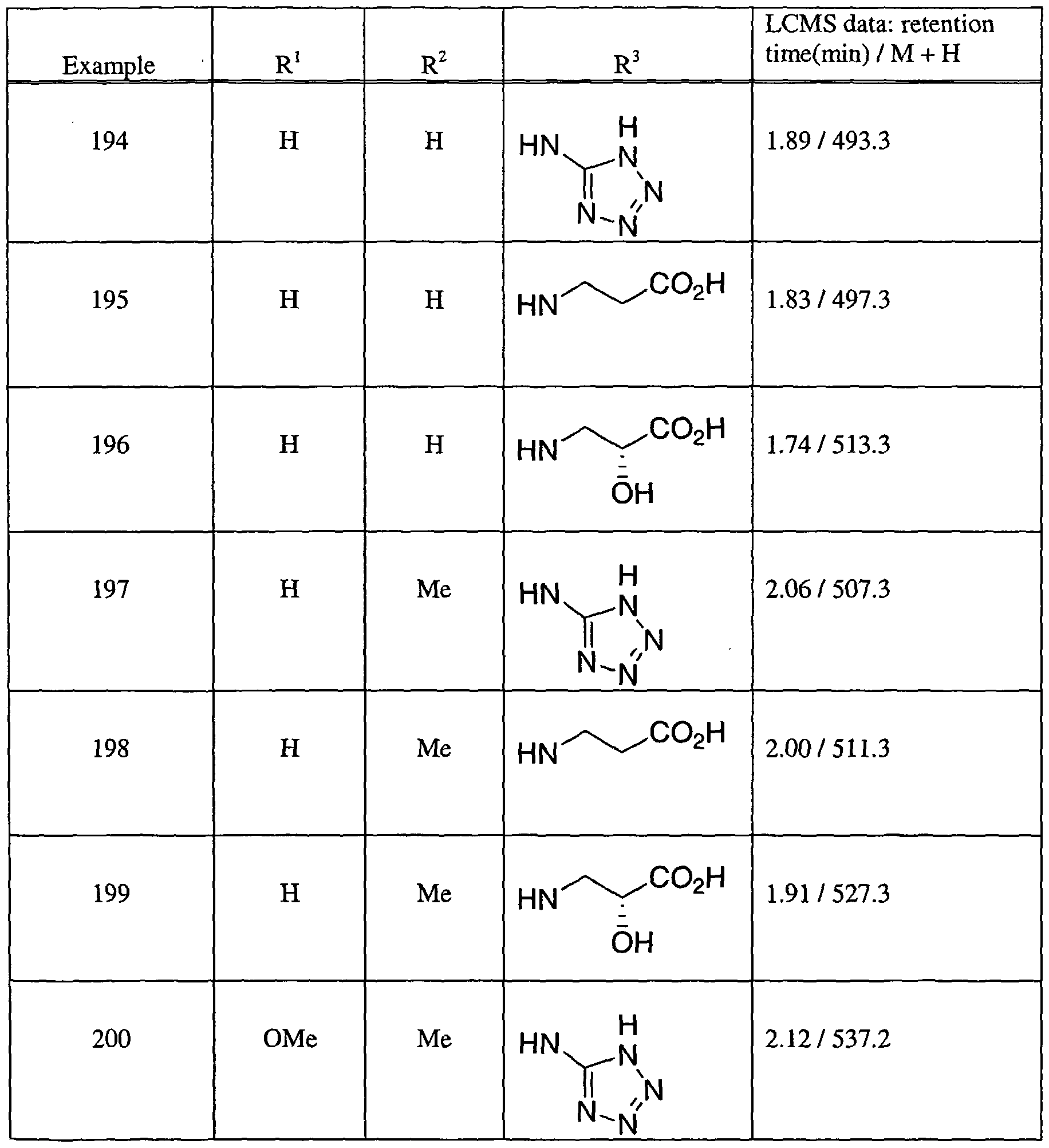





















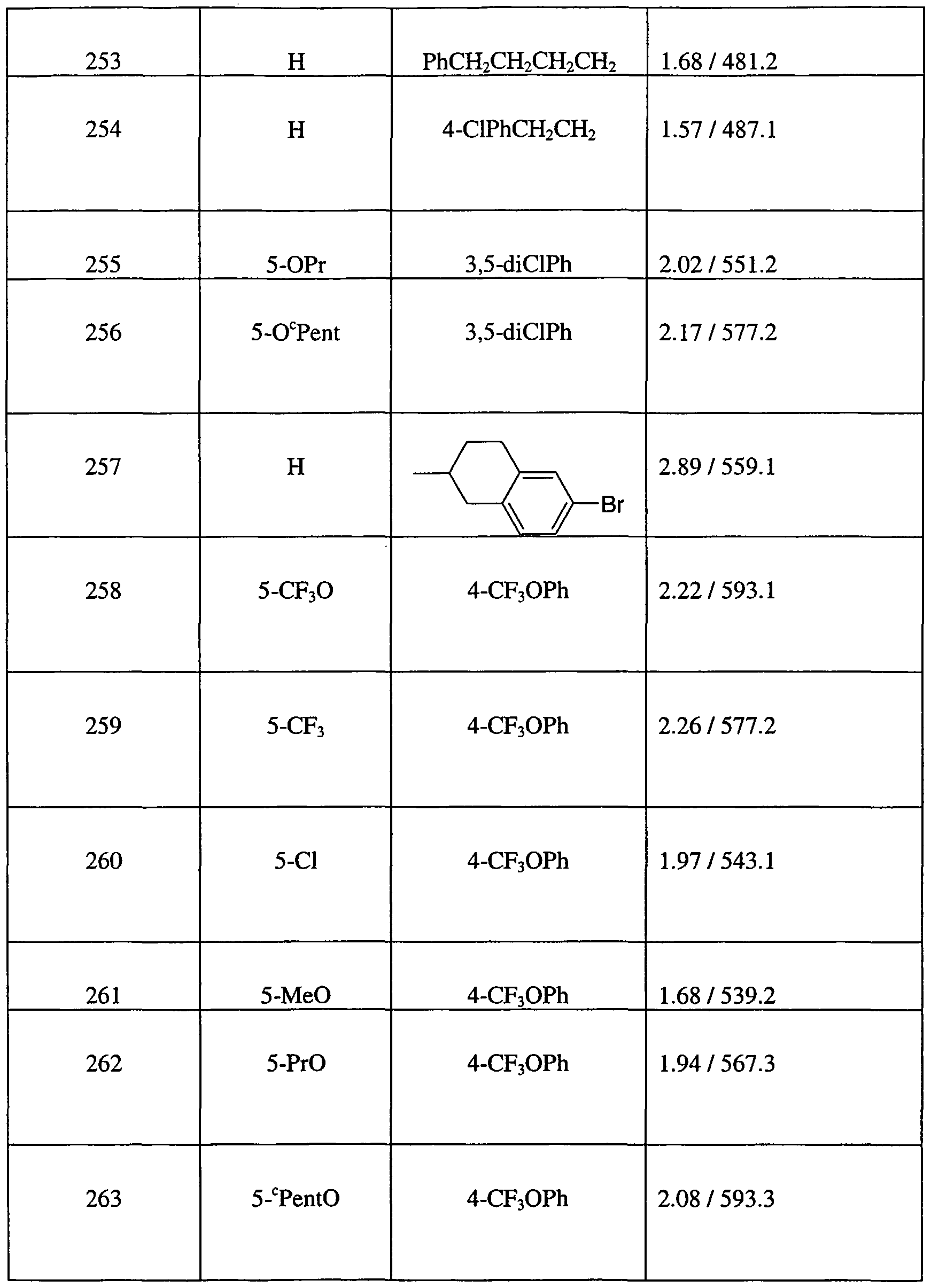












Claims



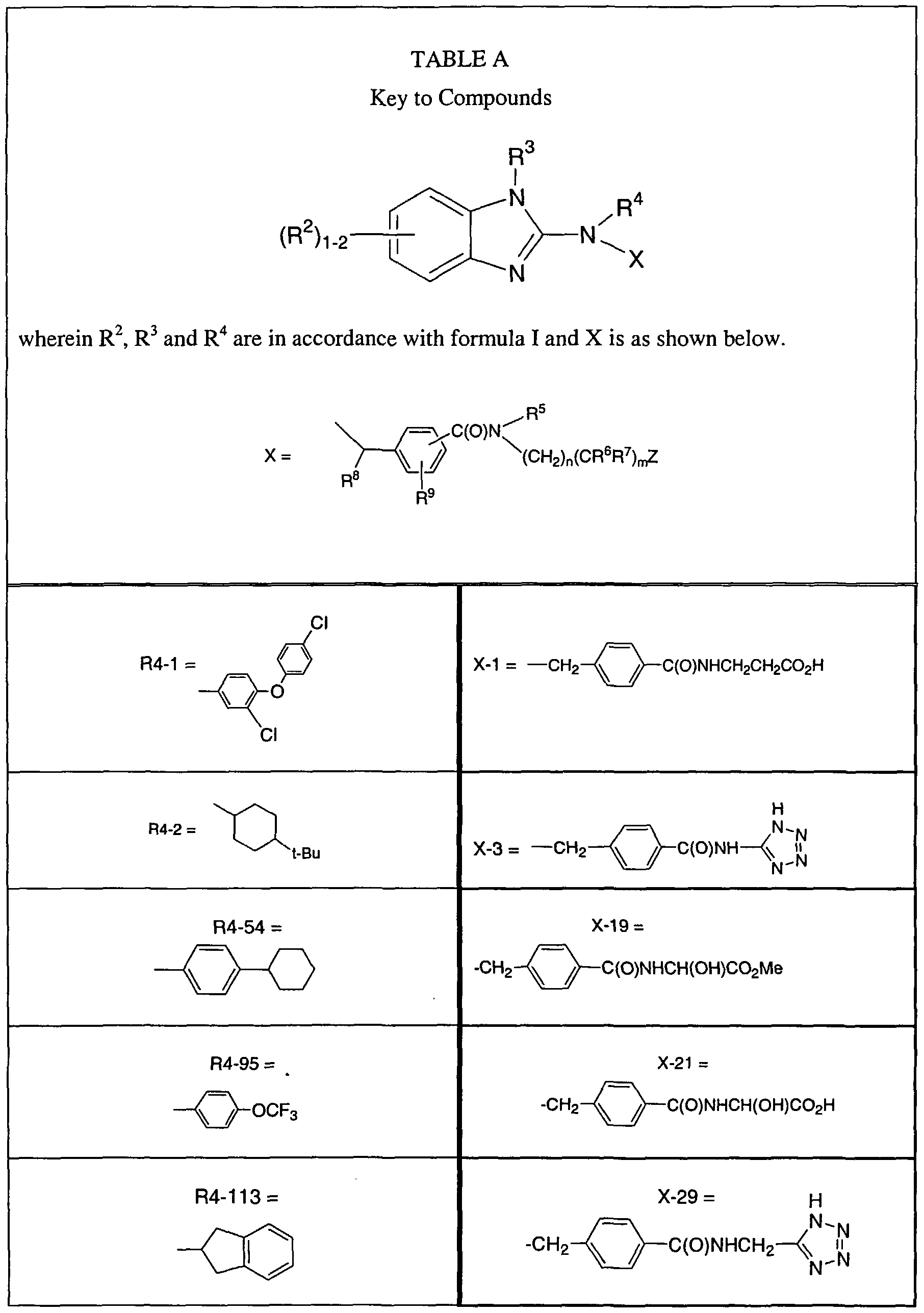












Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2004238240A AU2004238240A1 (en) | 2003-05-09 | 2004-05-05 | Benzimidazoles, compositions containing such compounds and methods of use |
| JP2006532564A JP2006528687A (en) | 2003-05-09 | 2004-05-05 | Benzimidazoles, compositions containing such compounds and methods of use |
| CA002524436A CA2524436A1 (en) | 2003-05-09 | 2004-05-05 | Benzimidazoles, compositions containing such compounds and methods of use |
| US10/556,230 US7563815B2 (en) | 2003-05-05 | 2004-05-05 | Benzamidazoles, compositions containing such compounds and methods of use |
| EP04751318A EP1626717A4 (en) | 2003-05-09 | 2004-05-05 | Benzimidazoles, compositions containing such compounds and methods of use |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US46933203P | 2003-05-09 | 2003-05-09 | |
| US60/469,332 | 2003-05-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004100875A2 true WO2004100875A2 (en) | 2004-11-25 |
| WO2004100875A3 WO2004100875A3 (en) | 2005-03-17 |
Family
ID=33452275
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/013874 WO2004100875A2 (en) | 2003-05-05 | 2004-05-05 | Benzimidazoles, compositions containing such compounds and methods of use |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1626717A4 (en) |
| JP (1) | JP2006528687A (en) |
| CN (1) | CN1784226A (en) |
| AU (1) | AU2004238240A1 (en) |
| CA (1) | CA2524436A1 (en) |
| WO (1) | WO2004100875A2 (en) |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005121097A2 (en) * | 2004-06-04 | 2005-12-22 | Merck & Co., Inc. | Pyrazole derivatives, compositions containing such compounds and methods of use |
| US7030113B2 (en) * | 2003-10-02 | 2006-04-18 | Schering Corporation | Aminobenzimidazoles as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| WO2007047177A1 (en) * | 2005-10-13 | 2007-04-26 | Merck & Co., Inc. | Acyl indoles, compositions containing such compounds and methods of use |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| DE102007005045A1 (en) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | New phenothiazine derivative for use in preparing medicine for blood sugar lowering and for treatment of diabetes, nicotine dependence, alcohol dependence, central nervous system disorders, schizophrenia, and Alzheimer's disease |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US7572922B2 (en) | 2003-01-27 | 2009-08-11 | Merck & Co., Inc. | Substituted pyrazoles, compositions containing such compounds and methods of use |
| US7625938B2 (en) | 2004-07-22 | 2009-12-01 | Merck & Co., Inc. | Substituted pyrazoles, compositions containing such compounds and methods of use |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US7709658B2 (en) | 2005-07-26 | 2010-05-04 | Merck Sharp & Dohme Corp. | Process for synthesizing a substituted pyrazole |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| CN102015641A (en) * | 2008-03-05 | 2011-04-13 | 武田药品工业株式会社 | Heterocyclic compound |
| US7994331B2 (en) | 2005-07-13 | 2011-08-09 | Msd K.K. | Heterocycle-substituted benzimidazole derivative |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| JP2012503661A (en) * | 2008-09-26 | 2012-02-09 | メルク・シャープ・エンド・ドーム・コーポレイション | Novel cyclic benzimidazole derivatives useful as antidiabetic agents |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8507533B2 (en) | 2011-02-08 | 2013-08-13 | Pfizer Inc. | Glucagon receptor modulators |
| US8710236B2 (en) | 2007-02-09 | 2014-04-29 | Metabasis Therapeutics, Inc. | Antagonists of the glucagon receptor |
| US8809342B2 (en) | 2010-12-23 | 2014-08-19 | Pfizer Inc. | Glucagon receptor modulators |
| EP2799428A2 (en) | 2008-08-13 | 2014-11-05 | Metabasis Therapeutics, Inc. | Glucagon antagonists |
| US8927577B2 (en) | 2011-07-22 | 2015-01-06 | Pfizer Inc. | Quinolinyl glucagon receptor modulators |
| TWI475018B (en) * | 2009-02-06 | 2015-03-01 | Takeda Pharmaceutical | Heterocyclic compound |
| US10076504B2 (en) | 2014-06-12 | 2018-09-18 | Ligand Pharmaceuticals, Inc. | Glucagon antagonists |
| WO2019160940A1 (en) | 2018-02-13 | 2019-08-22 | Ligand Pharmaceuticals Incorporated | Glucagon receptor antagonists |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011027849A1 (en) * | 2009-09-04 | 2011-03-10 | 武田薬品工業株式会社 | Heterocyclic compound |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5705515A (en) * | 1994-04-26 | 1998-01-06 | Merck & Co., Inc. | Substituted sulfonamides as selective β-3 agonists for the treatment of diabetes and obesity |
| EP1183229B1 (en) * | 1999-05-17 | 2005-10-26 | Novo Nordisk A/S | Glucagon antagonists/inverse agonists |
| US6340681B1 (en) * | 1999-07-16 | 2002-01-22 | Pfizer Inc | 2-benzimidazolylamine compounds as ORL-1-receptor agonists |
-
2004
- 2004-05-05 JP JP2006532564A patent/JP2006528687A/en not_active Withdrawn
- 2004-05-05 CN CNA2004800124899A patent/CN1784226A/en active Pending
- 2004-05-05 AU AU2004238240A patent/AU2004238240A1/en not_active Abandoned
- 2004-05-05 CA CA002524436A patent/CA2524436A1/en not_active Abandoned
- 2004-05-05 WO PCT/US2004/013874 patent/WO2004100875A2/en active Application Filing
- 2004-05-05 EP EP04751318A patent/EP1626717A4/en not_active Withdrawn
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1626717A4 * |
Cited By (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7572922B2 (en) | 2003-01-27 | 2009-08-11 | Merck & Co., Inc. | Substituted pyrazoles, compositions containing such compounds and methods of use |
| US7989475B2 (en) | 2003-01-27 | 2011-08-02 | Merck Sharp & Dohme Corp. | Substituted pyrazoles, compositions containing such compounds and methods of use |
| US7030113B2 (en) * | 2003-10-02 | 2006-04-18 | Schering Corporation | Aminobenzimidazoles as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders |
| US7799818B2 (en) | 2004-06-04 | 2010-09-21 | Merck Sharp & Dohme Corp. | Pyrazole derivatives, compositions containing such compounds and methods of use |
| AU2008229701B2 (en) * | 2004-06-04 | 2010-03-04 | Merck Sharp & Dohme Corp. | Pyrazole derivatives, compositions containing such compounds and methods of use |
| AU2005252183B2 (en) * | 2004-06-04 | 2008-07-17 | Merck Sharp & Dohme Corp. | Pyrazole derivatives, compositions containing such compounds and methods of use |
| WO2005121097A3 (en) * | 2004-06-04 | 2006-02-16 | Merck & Co Inc | Pyrazole derivatives, compositions containing such compounds and methods of use |
| AU2008229701C1 (en) * | 2004-06-04 | 2010-07-29 | Merck Sharp & Dohme Corp. | Pyrazole derivatives, compositions containing such compounds and methods of use |
| US7598285B2 (en) | 2004-06-04 | 2009-10-06 | Merck & Co., Inc | Pyrazole derivatives, compositions containing such compounds and methods of use |
| EA012431B1 (en) * | 2004-06-04 | 2009-10-30 | Мерк Энд Ко., Инк. | Pyrazole derivatives, pharmaceutical compositions and use thereof |
| WO2005121097A2 (en) * | 2004-06-04 | 2005-12-22 | Merck & Co., Inc. | Pyrazole derivatives, compositions containing such compounds and methods of use |
| US7625938B2 (en) | 2004-07-22 | 2009-12-01 | Merck & Co., Inc. | Substituted pyrazoles, compositions containing such compounds and methods of use |
| US7994331B2 (en) | 2005-07-13 | 2011-08-09 | Msd K.K. | Heterocycle-substituted benzimidazole derivative |
| US7709658B2 (en) | 2005-07-26 | 2010-05-04 | Merck Sharp & Dohme Corp. | Process for synthesizing a substituted pyrazole |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| WO2007047177A1 (en) * | 2005-10-13 | 2007-04-26 | Merck & Co., Inc. | Acyl indoles, compositions containing such compounds and methods of use |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| DE102007005045A1 (en) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | New phenothiazine derivative for use in preparing medicine for blood sugar lowering and for treatment of diabetes, nicotine dependence, alcohol dependence, central nervous system disorders, schizophrenia, and Alzheimer's disease |
| US9169201B2 (en) | 2007-02-09 | 2015-10-27 | Metabasis Therapeutics, Inc. | Antagonists of the glucagon receptor |
| US10807946B2 (en) | 2007-02-09 | 2020-10-20 | Metabasis Therapeutics, Inc. | Antagonists of the glucagon receptor |
| US9701626B2 (en) | 2007-02-09 | 2017-07-11 | Metabasis Therapeutics, Inc. | Antagonists of the glucagon receptor |
| US8710236B2 (en) | 2007-02-09 | 2014-04-29 | Metabasis Therapeutics, Inc. | Antagonists of the glucagon receptor |
| US10239829B2 (en) | 2007-02-09 | 2019-03-26 | Metabasis Therapeutics, Inc. | Antagonists of the glucagon receptor |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| CN102015641A (en) * | 2008-03-05 | 2011-04-13 | 武田药品工业株式会社 | Heterocyclic compound |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US10221130B2 (en) | 2008-08-13 | 2019-03-05 | Metabasis Therapeutics, Inc. | Glucagon antagonists |
| EP2799428A2 (en) | 2008-08-13 | 2014-11-05 | Metabasis Therapeutics, Inc. | Glucagon antagonists |
| US8907103B2 (en) | 2008-08-13 | 2014-12-09 | Metabasis Therapeutics, Inc. | Glucagon antagonists |
| US9783494B2 (en) | 2008-08-13 | 2017-10-10 | Metabasis Therapeutics, Inc. | Glucagon antagonists |
| US11352321B2 (en) | 2008-08-13 | 2022-06-07 | Metabasis Therapeutics, Inc. | Glucagon antagonists |
| JP2012503661A (en) * | 2008-09-26 | 2012-02-09 | メルク・シャープ・エンド・ドーム・コーポレイション | Novel cyclic benzimidazole derivatives useful as antidiabetic agents |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| TWI475018B (en) * | 2009-02-06 | 2015-03-01 | Takeda Pharmaceutical | Heterocyclic compound |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| US8933104B2 (en) | 2010-12-23 | 2015-01-13 | Pfizer Inc. | Glucagon receptor modulators |
| US9056834B2 (en) | 2010-12-23 | 2015-06-16 | Pfizer Inc. | Glucagon receptor modulators |
| US8809342B2 (en) | 2010-12-23 | 2014-08-19 | Pfizer Inc. | Glucagon receptor modulators |
| US8507533B2 (en) | 2011-02-08 | 2013-08-13 | Pfizer Inc. | Glucagon receptor modulators |
| US8859591B2 (en) | 2011-02-08 | 2014-10-14 | Pfizer Inc. | Glucagon receptor modulators |
| US9452999B2 (en) | 2011-02-08 | 2016-09-27 | Pfizer Inc. | Glucagon receptor modulators |
| US9073871B2 (en) | 2011-02-08 | 2015-07-07 | Pfizer Inc. | Glucagon receptor modulators |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| US8927577B2 (en) | 2011-07-22 | 2015-01-06 | Pfizer Inc. | Quinolinyl glucagon receptor modulators |
| US9139538B2 (en) | 2011-07-22 | 2015-09-22 | Pfizer Inc. | Quinolinyl glucagon receptor modulators |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US10076504B2 (en) | 2014-06-12 | 2018-09-18 | Ligand Pharmaceuticals, Inc. | Glucagon antagonists |
| WO2019160940A1 (en) | 2018-02-13 | 2019-08-22 | Ligand Pharmaceuticals Incorporated | Glucagon receptor antagonists |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1626717A2 (en) | 2006-02-22 |
| JP2006528687A (en) | 2006-12-21 |
| AU2004238240A1 (en) | 2004-11-25 |
| CN1784226A (en) | 2006-06-07 |
| EP1626717A4 (en) | 2009-09-09 |
| WO2004100875A3 (en) | 2005-03-17 |
| CA2524436A1 (en) | 2004-11-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004100875A2 (en) | Benzimidazoles, compositions containing such compounds and methods of use | |
| US7301036B2 (en) | Cyclic guanidines, compositions containing such compounds and methods of use | |
| EP1590336B1 (en) | Substituted pyrazoles, compositions containing such compounds and methods of use | |
| EP1773330B1 (en) | Substituted pyrazoles, compositions containing such compounds and methods of use | |
| EP1765335B1 (en) | Pyrazole amide derivatives, compositions containing such compounds and methods of use | |
| US7803951B2 (en) | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use | |
| EP1569915A2 (en) | Spirocyclic ureas, compositions containing such compounds and methods of use | |
| WO2007047177A1 (en) | Acyl indoles, compositions containing such compounds and methods of use | |
| AU2007229850A1 (en) | Glucagon receptor antagonist compounds, compositions containing such compounds and methods of use | |
| US7563815B2 (en) | Benzamidazoles, compositions containing such compounds and methods of use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004238240 Country of ref document: AU Ref document number: 4615/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004751318 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2524436 Country of ref document: CA |
|
| ENP | Entry into the national phase in: |
Ref document number: 2004238240 Country of ref document: AU Date of ref document: 20040505 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004238240 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006532564 Country of ref document: JP Ref document number: 20048124899 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007093544 Country of ref document: US Ref document number: 10556230 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004751318 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10556230 Country of ref document: US |