WO2008108602A1 - Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same - Google Patents
Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same Download PDFInfo
- Publication number
- WO2008108602A1 WO2008108602A1 PCT/KR2008/001322 KR2008001322W WO2008108602A1 WO 2008108602 A1 WO2008108602 A1 WO 2008108602A1 KR 2008001322 W KR2008001322 W KR 2008001322W WO 2008108602 A1 WO2008108602 A1 WO 2008108602A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- methoxy
- compound
- ethoxy
- methyl
- Prior art date
Links
- 0 *c1ccc(C=O)cc1 Chemical compound *c1ccc(C=O)cc1 0.000 description 7
- HAXMZFDSJLRIEM-UHFFFAOYSA-N CC1(C)OB(c(cc2)ccc2C(CC2)=NN(C)C2=O)OC1(C)C Chemical compound CC1(C)OB(c(cc2)ccc2C(CC2)=NN(C)C2=O)OC1(C)C HAXMZFDSJLRIEM-UHFFFAOYSA-N 0.000 description 1
- DXQFYZLTLNJNQH-INIZCTEOSA-N CCO[C@@H](Cc(cc1)ccc1OCc([o]1)ccc1Br)C(OCC)=O Chemical compound CCO[C@@H](Cc(cc1)ccc1OCc([o]1)ccc1Br)C(OCC)=O DXQFYZLTLNJNQH-INIZCTEOSA-N 0.000 description 1
- LLSHFVSTBZJGDZ-UHFFFAOYSA-N CN(C(CC1)=O)N=C1c(cc1)ccc1Br Chemical compound CN(C(CC1)=O)N=C1c(cc1)ccc1Br LLSHFVSTBZJGDZ-UHFFFAOYSA-N 0.000 description 1
- IPORKSQKGSJZRN-UHFFFAOYSA-N CNC(c(cc1)ccc1Br)NO Chemical compound CNC(c(cc1)ccc1Br)NO IPORKSQKGSJZRN-UHFFFAOYSA-N 0.000 description 1
- UAQJEIWAMCLVTE-UHFFFAOYSA-N Cc1c(CO)[s]cc1 Chemical compound Cc1c(CO)[s]cc1 UAQJEIWAMCLVTE-UHFFFAOYSA-N 0.000 description 1
- XLVPVZKGRKXLIA-UHFFFAOYSA-O Cc1c(C[OH2+])[s]c(Br)n1 Chemical compound Cc1c(C[OH2+])[s]c(Br)n1 XLVPVZKGRKXLIA-UHFFFAOYSA-O 0.000 description 1
- YWBIOYGLRGIJRT-UHFFFAOYSA-N Cc1nc(-c(cc2)ccc2Br)n[o]1 Chemical compound Cc1nc(-c(cc2)ccc2Br)n[o]1 YWBIOYGLRGIJRT-UHFFFAOYSA-N 0.000 description 1
- HQSCPPCMBMFJJN-UHFFFAOYSA-N N#Cc(cc1)ccc1Br Chemical compound N#Cc(cc1)ccc1Br HQSCPPCMBMFJJN-UHFFFAOYSA-N 0.000 description 1
- CDDNLMGOLYMTSF-UHFFFAOYSA-N O=C(CC1)NN=C1c(cc1)ccc1Br Chemical compound O=C(CC1)NN=C1c(cc1)ccc1Br CDDNLMGOLYMTSF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/14—Radicals substituted by singly bound hetero atoms other than halogen
- C07D333/16—Radicals substituted by singly bound hetero atoms other than halogen by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
Definitions
- the present invention relates to novel compounds represented by formula 1, and preparation and use thereof:
- the compound of formula 1 has modulatory effects on peroxisome proliferator-activated receptor gamma (hereinafter, referred to as "PPAR- ⁇ ") and therefore can be effective for hypoglycemic (blood glucose-lowering) effects, hypolipidemic (blood lipid-lowering) effects, and alleviation of insulin resistance.
- PPAR- ⁇ peroxisome proliferator-activated receptor gamma
- Diabetes mellitus is a chronic metabolic disease which has a prevalence rate of nearly 5% among populations of industrialized countries.
- An incidence rate of Type 2 diabetes mellitus (formerly called non-insulin-dependent diabetes mellitus, NIDDM), which accounts for 90% or higher of diabetic conditions, is gradually increasing with generalization of high- calorie diet and advanced country-type lifestyle habits (Rondinone et al, Exp Opin Ther Targets (2005) 9:415-419).
- Type 2 diabetic patients frequently suffer from attendant diseases such as hyperglycemia, hyperlipidemia, atherosclerosis and obesity.
- a primary etiological factor of Type 2 diabetes mellitus is insulin resistance. That is, the incidence of Type 2 diabetes mellitus is initiated with manifestation of insulin resistance at the early stage, followed by hypoinsulinaemia due to dysfunction of pancreatic beta cells.
- PPAR- ⁇ is a transcriptional activator or transactivator that mediates adipogenic differentiation.
- Rosiglitazone and pioglitazone drugs which are synthetic ligands for PPAR- ⁇ , have been clinically proven to be excellent therapeutic agents that are capable of regulating an elevated blood glucose level by enhancing insulin sensitivity of Type 2 diabetic patients to thereby alleviate insulin resistance.
- conventional glitazone drugs entail adverse side effects such as potential risks of edema and weight gain in practical clinical applications and development of cardiac hypertrophy in preclinical animal models, even though these drugs exhibit excellent drug efficacy.
- a selective PPAR- ⁇ modulator is a drug that elicits a relatively low PPAR- ⁇ transcriptional activity, as compared to a ligand species which theoretically exhibits 100% transcriptional activity, such as rosiglitazone, and that has hypoglycemic effects simultaneously with reduction of the above-mentioned adverse side effects. Further, improvement of insulin sensitivity does not necessarily require 100% activation of PPAR- ⁇ .
- the selective PPAR- ⁇ modulator nTZDpa shows different adipocyte-specific gene expression patterns than those of a ligand that exhibits 100% transcriptional activity, such as rosiglitazone.
- a ligand that exhibits 100% transcriptional activity such as rosiglitazone.
- comparable drug efficacy was achieved with significantly low weight gain of adipose tissues while not causing significant differences in blood glucose and insulin levels, as compared to a control group fed with high-fat diet and a group treated with a ligand exhibiting 100% transcriptional activity.
- metaglidasen and INT-131 were demonstrated in animal models. Specifically, administration of these drugs resulted in amelioration in development of edema and weight gain (Abstract 44-OR, 65 th ADA, 2005; and Abstract 659-P, 64 th ADA, 2004). 12- week clinical results of metaglidasen showed that co-administration of metaglidasen with insulin exhibits excellent drug efficacy with a 0.7% decrease of glycosylated hemoglobin as compared to a non-treated control group, in conjunction with a 21% reduction in the blood triglyceride level. However, there was no significant difference in the body weight (+0.6 kg vs. +1.3 kg in control group; P>0.05) and an edema incidence rate (7.2% vs.
- the inventors of the present invention succeeded in synthesis of a novel compound which is capable of achieving improved insulin sensitivity and efficient control of the blood glucose level through the selective modulation of PPAR- ⁇ while simultaneously reducing adverse side effects that were shown by conventional drugs.
- the present invention has been completed based on this finding.
- PPAR- ⁇ modulator comprising a phenylpropionic acid derivative represented by formula 1 or a pharmaceutically acceptable salt thereof, as an active ingredient.
- a novel compound having a structure of formula 1 and a pharmaceutically acceptable salt thereof.
- racemates, optical isomers and pharmaceutically acceptable salts of a compound of formula 1 fall within the scope of the present invention.
- Ri is hydrogen, ethyl, or an alkali metal
- R 2 is hydrogen or methyl
- X is S or O; Y is N or C;
- R 3 is hydrogen, lower alkyl or lower alkoxy
- R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-/f-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, Ci -6 alkoxy, hydroxy, amino, trifluoromethyl, phenyl, benzyl, benzoyl, furan, thiophene, piperidine and morpholine; and n is
- lower alkyl is selected from methyl, ethyl and isopropyl; lower alkoxy is selected from methoxy and ethoxy; and halide is selected from Cl, F and Br.
- alkylcarbamoyl is selected from:
- oxadiazole is selected from:
- isoxazole is selected from:
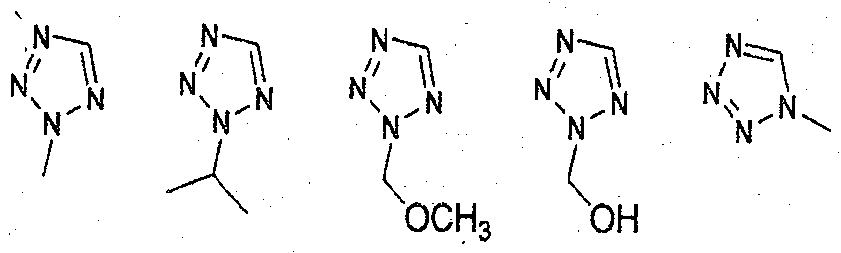
- tetrazole is selected from:
- Representative examples of compounds in accordance with the present invention may include the following compounds:
- the phenylpropionic acid derivatives in accordance with the present invention have an asymmetric carbon center, and may be present in the form of racemates and corresponding optical isomers. All kinds of these isomers fall within the scope of the present invention.
- the optical isomers were given optical selectivity via enzymatic reactions of racemic intermediates.
- the enzyme used in synthesis of the compounds in accordance with the present invention was Viscozyme-L (Novozyme) as disclosed in Korean Patent Application No. 2006-66440.
- Racemic resolution for producing optically active isomers of a compound represented by formula 1 may be carried out by a conventional resolution method known in the art. For example, a base of the compound of formula 1 is reacted with an optically active acid to form a salt of the compound of formula 1 , and then dextro (right) and levo (left) forms of optical isomers are then separated by fractional crystallization.
- acids suitable for resolution of the compound of formula 1 may include optically active forms of tartaric acid, ditolyltartaric acid, dibenzoyltartaric acid, malic acid, mandelic acid and camphorsulfonic acid and any optically active acid known in the related art. In this case, more biologically and optically active stereoisomeric forms of the compound of formula 1 are preferably separated.
- the compound of formula 1 in accordance with the present invention include pharmaceutically acceptable salts thereof, for example salts with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, and sulfuric acid; salts with organic carboxylic acids such as acetic acid, trifluoroacetic acid, citric acid, maleic acid, oxalic acid, succinic acid, benzoic acid, tartaric acid, fumaric acid, mandelic acid, ascorbic acid, and malic acid; salts with sulfonic acids such as methanesulfonic acid, and p-toluenesulfonic acid; salts with alkali metals such as sodium, potassium, and lithium; salts with organic amines such as ethanolamine; and salts with any acid known in the art. Further, the present invention provides a method for preparing a compound represented by formula 1 or a pharmaceutically acceptable salt thereof.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, and
- the preparation method of the present invention comprises (1) reacting a compound of formula 2 with a compound of formula 3, 4, 5 or 6 to form a compound of formula 7, 8, 9 or 10; and (2) reacting the compound of formula 7, 8, 9 or 10 with a boron compound of formula 11 to form a compound of formula 1 wherein R 1 is ethyl.
- R 1 is hydrogen
- the method may further comprise hydrolysis of the ethyl ester compound of Step 2 by the reaction with a base.
- R 1 is an alkali metal
- the method may further comprise reacting the hydrolysate, obtained from reaction of the ester compound of Step 2 with the base, with an alkali metal salt to prepare a desired compound of formula 1 :
- R 1 is hydrogen, ethyl, or an alkali metal
- R 2 is hydrogen or methyl
- X is S or O
- Y is N or C
- R 3 is hydrogen, lower alkyl or lower alkoxy
- R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-/f-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2//)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Ci
- the compound of formula 1 in accordance with the present invention may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 3 through the Mitsunobu reaction to form an ether bond, followed by bromination of the reaction product with N-bromosuccinimide to form Compound 7; and Step 2: Suzuki coupling of Compound 7 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond.
- the resulting compound of formula 1 may be a compound of formula 1-1.
- Rj is ethyl; X is S; Y is C; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-i/-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, Ci -6 alkoxy, hydroxy, amino, trifluor
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 4 through the Mitsunobu reaction to form Compound 8 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 8 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond.
- the resulting compound of formula 1 may be a compound of formula 1-2.
- R 1 is ethyl; X is O; Y is C; R 2 is hydrogen; R 3 and R 4 are as defined in formula 1, and n is 1.
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 5 through the Mitsunobu reaction to form Compound 9 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 9 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond.
- the resulting compound of formula 1 may be a compound of formula 1-3.
- R 1 is ethyl; X is O; Y is C; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-//-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2/f)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Cj -6 alkyl, Ci -6 alkoxy, hydroxy, amino, tri
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 6 through the Mitsunobu reaction to form Compound 10 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 10 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond.
- the resulting compound of formula 1 may be a compound of formula 1-4.
- R 1 is ethyl; X is S; Y is N; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-H-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Ci -6 alkyl, Ci -6 alkoxy, hydroxy, amino, trifluor
- the method may further comprise hydrolysis of the reaction product of Step 2 after reaction with a boron compound to thereby form a compound of formula 1 wherein R 1 is hydrogen. From the compound of formula 1 wherein Ri is hydrogen, a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate.
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 3 through the Mitsunobu reaction to form an ether bond, followed by bromination of the reaction product with N-bromosuccinimide to form
- Compound 7, and Step 2 Suzuki coupling of Compound 7 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon-carbon bond, followed by hydrolysis.
- the resulting compound of formula 1 may be a compound of formula 1-5.
- a carbon- carbon bond of Step 2 is formed in the preparation process of Compound 1-5, a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate.
- Ri is hydrogen; X is S; Y is C; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-H-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Ci -6 alkyl, C] -6 alkoxy, hydroxy, amino, trifluoromethyl,
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 4 through the Mitsunobu reaction to form Compound 8 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 8 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond, followed by hydrolysis.
- the resulting compound of formula 1 may be a compound of formula 1-6.
- a carbon-carbon bond of Step 2 is formed in the preparation process of Compound 1-6, a compound of formula 1 wherein Ri is ethyl may be obtained as an intermediate.
- Ri is hydrogen; X is O; Y is C; R 2 is hydrogen; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-/f-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl., morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Ci -6 alkyl, Ci -6 alkoxy, hydroxy, amino, trifluoromethyl,
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 5 through the Mitsunobu reaction to form Compound 9 via formation of an ether bond, and Step 2 Suzuki coupling of Compound 9 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond, followed by hydrolysis.
- the resulting compound of formula 1 may be a compound of formula 1-7.
- a carbon-carbon bond of Step 2 is formed in the preparation process of Compound 1-7, a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate.
- Ri is hydrogen; X is O; Y is C; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-H-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Cj -6 alkyl, C 1-6 alkoxy, hydroxy, amino, trifluoromethyl,
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 6 through the Mitsunobu reaction to form Compound 10 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 10 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond, followed by hydrolysis.
- the resulting compound of formula 1 may be a compound of formula 1-8.
- a carbon-carbon bond of Step 2 is formed in the preparation process of Compound 1-8, a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate..
- R 1 is hydrogen; X is S; Y is N; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-i/-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Ci -6 alkyl, Ci -6 alkoxy, hydroxy, amino, trifluoromethyl,
- the method may further comprise reacting the hydrolysate of Step 2, obtained from hydrolysis of the reaction product after reaction with a boron compound, with sodium, lithium or potassium ethyl-2 hexanoate to prepare a compound of formula 1 wherein R 1 is an alkali metal.
- a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate.
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 3 through the Mitsunobu reaction to form an ether bond, followed by bromination of the reaction product with N-bromosuccinimide to form Compound 7, and Step 2: Suzuki coupling of Compound 7 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon-carbon bond, followed by hydrolysis.
- the resulting compound of formula 1 may be a compound of formula 1 -9.
- a carbon- carbon bond of Step 2 is formed in the preparation process of Compound 1-9, a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate.
- R 1 is an alkali metal
- X is S
- Y is C
- R 2 is methyl
- Rs is hydrogen, lower alkyl or lower alkoxy
- R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-//-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, Ci -6 alkoxy, hydroxy, amino
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 4 through the Mitsunobu reaction to form Compound 8 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 8 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond, followed by hydrolysis.
- the resulting compound of formula 1 may be a compound of formula 1-10.
- a carbon-carbon bond of Step 2 is formed in the preparation process of Compound 1-10, a compound of formula 1 wherein Ri is ethyl may be obtained as an intermediate.
- R 1 is an alkali metal; X is O; Y is C; R 2 is hydrogen; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-/f-benzo[b][l,4]oxazine, morpholine, thiazole, mo ⁇ holinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)- one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Cj -6 alkyl, C 1-6 alkoxy, hydroxy, amino, trifluor
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 5 through the Mitsunobu reaction to form Compound 9 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 9 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond, followed by hydrolysis.
- the resulting compound of formula 1 may be a compound of formula 1-11.
- a carbon-carbon bond of Step 2 is formed in the preparation process of Compound 1-11, a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate.
- R 1 is an alkali metal; X is O; Y is C; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-H-benzo[b][l,4]oxazine, morpholine, thiazole, mo ⁇ holinosulfonyl, mo ⁇ holinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, Ci -6 alkyl, C i- 6 alkoxy, hydroxy, amino,
- the compound of formula 1 may be prepared by Step 1 : nucleophilic substitution of Compound 2 with Compound 6 through the Mitsunobu reaction to form Compound 10 via formation of an ether bond, and Step 2: Suzuki coupling of Compound 10 using boronic acid or dioxaborolan of formula 11 and a palladium catalyst to form a carbon- carbon bond, followed by hydrolysis.
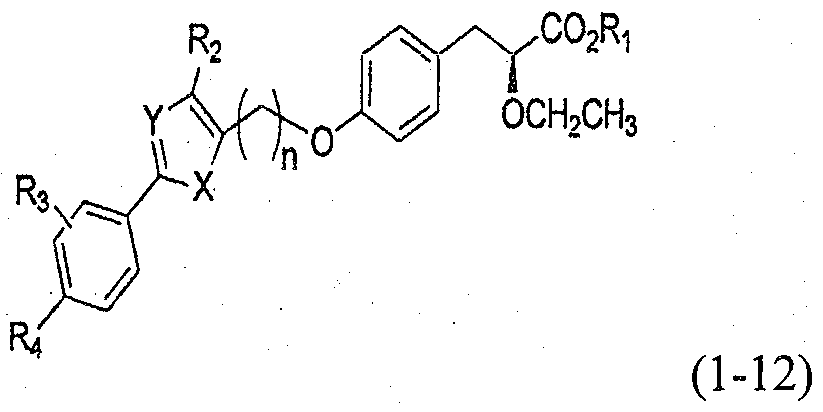
- the resulting compound of formula 1 may be a compound of formula 1-12.
- a carbon-carbon bond of Step 2 is formed in the preparation process of Compound 1-12, a compound of formula 1 wherein R 1 is ethyl may be obtained as an intermediate.
- R 1 is an alkali metal; X is S; Y is N; R 2 is methyl; R 3 is hydrogen, lower alkyl or lower alkoxy; R 4 is hydrogen, lower alkyl, lower alkoxy, halide, cyano, acetyl, acetamino, benzoyl, carbamoyl, alkylcarbamoyl, aminosulfonyl, 2-H-benzo[b][l,4]oxazine, morpholine, thiazole, morpholinosulfonyl, morpholinocarbonyl, 4,5-dihydropyridazin-3(2H)-one, thiadiazole, oxadiazole, tetrazole, oxazole, or isoxazole, each of which being optionally substituted by at least one selected from the group consisting of hydrogen, halogen, C] -6 alkyl, Ci -6 alkoxy, hydroxy, amino, trifluor
- reaction solvent there is no particular limit to the reaction solvent, so long as it facilitates nucleophilic substitution and hydrolysis while not having adverse effects on the reaction of interest.
- the reaction solvent may include alcohols such as methanol, ethers such as dioxane and tetrahydrofuran (THF), aromatic solvents such as benzene and toluene, chlorinated hydrocarbons such as methylene chloride and dichloroethane, and organic solvents such as acetonitrile and N,N-dimethylformamide (DMF). These materials may be used alone or in any combination thereof.
- the reaction may be carried out at a temperature of 0 to 150 ° C .
- the compound of formula 2 encompasses an optical isomeric form thereof.
- Compound 2 may be prepared by condensation of commercially available 4-benzyloxybenzaldehyde as a starting material with triethyl 2-phosphonobutyrate, followed by hydrogenation. The condensation step may be carried out as a Wittig-type reaction (cf. Comprehensive Organic Synthesis vol. 1 p. 755-781, Pergamon Press) or as described in the Preparation Examples hereinafter.
- an olefin intermediate is synthesized by reaction of reaction materials in the presence of hydrogenation products of alkali metals such as sodium hydride (NaH) and potassium hydride (KH), organolithium such as methyl lithium (CH 3 Li) and butyl lithium (BuLi), alkoxides such as sodium methoxide (NaOMe), sodium ethoxide (NaOEt) and potassium t-butoxide (t-BuOK), or bases such as lithium hydroxide (LiOH) and sodium hydroxide (NaOH), which is followed by reduction of the intermediate using hydrogen gas and a Pd/C, Rh/C or Pt/C catalyst or a mixture thereof.
- alkali metals such as sodium hydride (NaH) and potassium hydride (KH)
- organolithium such as methyl lithium (CH 3 Li) and butyl lithium (BuLi)
- alkoxides such as sodium methoxide (NaOMe), sodium ethoxide (
- reaction solvent may include dioxane, acetic acid, ethyl acetate, and ethanol. Properties of the solvent are not particularly important.
- the reaction may be carried out under pressure of 80 psi.
- the catalyst is preferably 5 to 10% Pd/C, and may be used in a range of 1 to 100% w/w.
- Synthesis of Compound 2 is illustrated in the following Reaction Scheme.
- Z represents a linear or branched saturated hydrocarbon, preferably ethyl.
- the resulting hydrogenation product is obtained in the form of a racemic mixture. Therefore, for synthesis of an optical isomeric form, hydrolysis of Compound 2 is carried out via the selective enzymatic reaction to prepare a preferred (s)-form of carboxylic acid, followed by esterif ⁇ cation (Mats T. Liderberg et al., Organic Process Research & Development 2004, 8, 838-845).
- the enzyme used in synthesis of the desired compound was Viscozyme-L (Novozyme) as disclosed in Korean Patent Application No. 2006-66440.
- Compound 7 may be prepared by nucleophilic substitution of Compound 2 with
- N-bromosuccinimide hereinafter, referred to as "NBS"
- Compound 8 was prepared from Compounds 2 and 4 through the Mitsunobu reaction.
- Compound 9 was prepared from Compounds 2 and 5 through the Mitsunobu reaction.
- Compound 10 was prepared from Compounds 2 and 6 through the Mitsunobu reaction.
- Compound 12 was prepared by formation of a carbon-carbon bond using Compounds 7 and 11 in the presence of a palladium catalyst, followed by hydrolysis of the reaction product. Further, the alkali metal salt may be prepared by reacting the hydrolyzed compound with a certain reagent.
- R 1 , R 3 , and R 4 are as defined in formula 1.
- Reaction Scheme 1 illustrates a general method for preparation of a compound represented by formula 1.
- Reagents and reaction conditions a) Diisopropyl azodicarboxylate, triphenylphosphine, room temperature, 2 hours. b) Boronic acid or 4,4,5,5-tetramethyl-l,3,2-dioxaborolan derivative, tetrakis(triphenylphosphine)palladium, cesium carbonate, dioxane, 90 "C, 2 hours. Alternatively, tetrakis(triphenylphosphine)palladium, aq. potassium carbonate (K 2 CO 3 ), toluene, ethanol, 90 0 C , 1 hour.
- K 2 CO 3 potassium carbonate
- the compound of formula 1 was prepared by formation of an ether bond by nucleophilic substitution through the Mitsunobu reaction, formation of a carbon- carbon bond through Suzuki coupling reaction (Suzuki A. et al, Synth. Commun. (1981), 11, 513) using boronic acid or dioxaborolan as defined in formula 11 and a palladium catalyst, and then hydrolysis of the reaction product under basic conditions to synthesize a desired form of propionic acid.
- R 1 , R 2 , R 3 , R 4 , X, and Y are as defined in formula 1.
- Reaction Scheme 2 illustrates a method for preparation of Compound 2.
- Reagents and reaction conditions a) Triethyl 2-ethoxyphosphonoacetate, potassium, t-butoxide (t-BuOH), toluene, room temperature, 3 hours. b) H 2 A 0% Pd-C, ethanol (EtOH), 12 hours. c) Viscozyme L (Viscozyme L), phosphate buffer, room temperature, 48 hours. d) Thionyl chloride (SOCl 2 ), ethanol (EtOH), reflux, 3 hours.
- the optical activity of Compound 2 was assayed by determining the optical purity (ee: enantiomeric excess) of the compound in the form of carboxylic acid using the following column.
- the optical purity of the compound was 99.54%.
- Reagents and reaction conditions a) Sodium borohydride, ethanol, room temperature, 1 hour.
- Reaction Scheme 4 illustrates synthesis of Compound 4.
- Compound 4 was prepared in the same manner as in Reaction Scheme 3.
- Reagents and reaction conditions a) Sodium borohydride, ethanol, room temperature, 1 hour.
- Reagents and reaction conditions a) Lithium aluminum hydride (LAH), tetrahydrofuran (THF), room temperature, 1 hour.
- Reaction Scheme 6 below illustrates synthesis of Compound 6.
- Reagents and reaction conditions a) Lithium aluminum hydride (LAH), tetrahydrofuran (THF), room temperature, 1 hour.
- LAH Lithium aluminum hydride
- THF tetrahydrofuran
- Reagents and reaction conditions a) Diisopropyl azodicarboxylate, triphenylphosphine, room temperature, 2 hours. b) N-bromosuccinimide (NBS), N,N-dimethylformamide (hereinafter, referred to as "DMF”), room temperature, 3 hours.
- NBS N-bromosuccinimide
- DMF N,N-dimethylformamide
- Reaction Scheme 8 illustrates synthesis of Compound 8.
- Reaction Scheme 11 illustrates synthesis of 5-substituted-3-(4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)isoxazole among compounds of formula 11.
- the isoxazole compound was prepared by reacting 4-bromobenzaldehyde (as a starting material) with hydroxylamine to form an oxime compound, introducing chloride into the oxime compound via use of N-chlorosuccinimide (hereinafter, referred to as "NCS"), and reacting the resulting compound with a certain butyne compound to obtain isoxazole. Thereafter, a desired compound was prepared by replacement of bromine into dioxaborolan using bis(pinacolato)diboron.
- NCS N-chlorosuccinimide
- Reagents and reaction conditions a) Hydroxylamine hydrogen chloride (NH?OH ⁇ C1), pyridine, room temperature, 2 hours. b) N-chlorosuccinimide (NCS), N,N-dimethylformamide (DMF), room temperature, 1 hour. c) 3,3-dimethyl-l-butyne, triethylamine (hereinafter, referred to as "Et 3 N"), methylene chloride (CH 2 Cl 2 ) , room temperature, 5 hours. d) 2-propyn-l-ol, triethylamine (Et 3 N), methylene chloride (CH 2 Cl 2 ), room temperature, 5 hours.
- e Sodium hydride (NaH), methyl iodide (MeI), N,N-dimethylformamide (DMF), room temperature, 1 hour.
- f Bis(pinacolato)diboron, [l,r-bis(diphenylphosphino)ferrocene]dichloropalladium (II) complex with dichloromethane (1:1), potassium acetate, dioxane, 90 ° C, 2 hours.
- Reaction Scheme 12 illustrates synthesis of 3-substituted-5-(4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)isoxazole among compounds of formula 11.
- the isoxazole compound was prepared by reacting 4-bromoacetophenone (as a starting material) with N,N-dimethylacetamide dimethyl acetal or diethyl oxalate to synthesize Compounds 12a and 12c, and reacting Compounds 12a or 12c with hydroxylamine to synthesize isoxazole compounds. Thereafter, a compound for formation of a carbon-carbon bond was synthesized by replacement of bromine of isoxazole compounds (12b, 12e, 12f, and 12i) into dioxaborolan via use of bis(pinacolato)diboron.
- Reagents and reaction conditions a) N,N-dimethylacetamide dimethyl acetal, 1,4-dioxane, reflux, 12 hours. b) Hydroxylamine, ethanol, reflux, 2 hours. c) Diethyl oxalate (CO 2 Et) 2 ), 60% sodium hydride, toluene, reflux, 1 hour. d) Lithium aluminum hydride, tetrahydrofuran (THF), 0 °C , 2 hours. e) Sodium hydride, MeI (methyl iodide), N,N-dimethylformamide (DMF), room temperature, 1 hour.
- Reaction Scheme 13 illustrates synthesis of a tetrazole compound among compounds of formula 11.
- 4-bromophenyl cyanide as a starting material was reacted with sodium azide and ammonium chloride to form tetrazole.
- nucleophilic substitution was made at position 1 or 2 of the tetrazole compound to synthesize a 1- or 2- substituted tetrazole compound.
- a compound for formation of a carbon-carbon bond was prepared by replacement of bromine of the tetrazole compound into dioxaborolan via use of bis(pinacolato)diboron.
- Reagents and reaction conditions a) Methyl iodide (CH 3 I), 60% sodium hydride, N,N-dimethylformamide (DMF), room temperature, 4 hours. b) Isopropyl bromide, 60% sodium hydride, N,N-dimethylfomiamide (DMF), room temperature, 4 hours. c) Bromomethyl methyl ether, sodium hydroxide (NaOH), N,N-dimethylformamide (DMF), 0 ° C, 4 hours.
- Reaction Scheme 14 illustrates synthesis of N-methyl-N-(4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)phenyl)acetamide among compounds of formula 11.
- Reagents and reaction conditions a) Methyl iodide, 60% sodium hydride, N,N-dimethylformamide (DMF), room temperature, 1 hour.
- Reaction Scheme 15 illustrates synthesis of 2-methyl-6-(4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)phenyl)-4,5-dihydropyridazin-3(2H)-one among compounds of formula 11.
- Reagents and reaction conditions a) Methyl iodide, triethylamine (Et 3 N), tetrahydrofuran (THF), room temperature, 3 hours. b) Bis(pinacolato)diboron, [l,r-bis(diphenylphosphino)ferrocene]dichloropalladium (II) complex with dichloromethane (1:1), potassium acetate, dioxane, 90 ° C, 2 hours.
- Reaction Scheme 16 illustrates synthesis of 5-methyl-3-(4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)phenyl)l,2,4-oxadiazole among compounds of formula 11.
- Reagents and reaction conditions a) Hydroxylamine, sodium bicarbonate, ethanol, 90 ° C , 3 hours. b) N,N-dimethylacetamide dimethyl acetal, 1,4-dioxane, reflux, 12 hours. c) Bis(pinacolato)diboron, [l,r-bis(diphenylphosphino)ferrocene]dichloropalladium (II) complex with dichloromethane (1:1), potassium acetate, dioxane, 90 ° C, 2 hours.
- Reaction Scheme 17 illustrates synthesis of 2-methyl-5-(4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl) phenyl)- 1, 3, 4-oxadiazole among compounds of formula 11.
- Reagents and reaction conditions a) Acetic anhydride, pyridine, reflux, 2 hours. b) Bis(pinacolato)diboron, [l,r-bis(diphenylphosphino)ferrocene]dichloropalladium (II) complex with dichloromethane (1:1), potassium acetate, dioxane, 90 ° C, 2 hours.
- Reaction Scheme 18 illustrates synthesis of 2-(4-(4,4,5,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)phenyl)-5-(trifluoromethyl)-l,3,4-oxadiazole among compounds of formula 11.
- Reagents and reaction conditions a) Trifluoroacetic anhydride, pyridine, reflux. b) Bis(pinacolato)diboron, [l,l'-bis(diphenylphosphino)ferrocene]dichloropalladium (II) complex with dichloromethane (1 :1), potassium acetate (KOAc), N,N-dimethylformamide (DMF), 120 ° C, 2 hours.
- Reaction Scheme 19 illustrates synthesis of 2-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)-l,3,4-oxadiazole among compounds of formula 11.
- 19c ISd Reagents and reaction conditions: a) Sulfuric acid, ethanol, 100 ° C, 6 hours. b) Hydrazine, ethanol, reflux, 12 hours. c) Acetic anhydride (AC 2 O), 1-4 dioxane, reflux, 4 hours. d) Bis(pinacolato)diboron), [1,1 '-bis(diphenylphosphino)ferrocene]dichloropalladium (II) complex with dichloromethane (1:1), potassium acetate, dioxane, 90 0 C, 2 hours.
- Reaction Scheme 20 illustrates synthesis of 4,5-dimethyl-2-(4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)oxazole among compounds of formula 11.
- Reagents and reaction conditions a) Triethylamine (Et 3 N), alanine methyl ester, ethyl chloroformate, tetrahydrofuran (THF), methanol, room temperature, 4 hours. b) 2N-NaOH, methanol, reflux, 2 hours. c) Acetic anhydride (AC 2 O), pyridine, 90 ° C, 3 hours. d) Sulfuric acid, acetic anhydride (AC 2 O), 90 ° C, 1.5 hours.
- Reaction Scheme 21 illustrates synthesis of 1, 3-dimethyl-5-(4-(4 ,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl-lH-pyrazole among compounds of formula 11. ⁇ Reaction Scheme 21>
- Reagents and reaction conditions a) Hydrazine, ethanol, 90 ° C , 6 hours. b) Methyl iodide, N,N-dimethylformamide (DMF), 60% sodium hydride, room temperature, 1 hour. c) Bis(pinacolato)diboron, [l,r-bis(diphenylphosphino)ferrocene]dichloropalladium (II) complex with dichloromethane (1 :1), potassium acetate, dioxane, 90 °C , 2 hours.
- Reaction Scheme 22 illustrates synthesis of compounds shown in the following Examples.
- R 1 , R 4 , and n are as defined in formula 1.
- each of brominated compounds (7b, 8a, 9a, and 10a) as a starting material was reacted with a boron compound as defined in formula 11 in the presence of a palladium catalyst to thereby form a carbon-carbon bond, and the resulting reaction product was hydrolyzed under basic conditions to afford a desired compound.
- Reagents and reaction conditions a) Tetrakis(triphenylphosphine)palladium, cesium carbonate, dioxane, 90 °C, 2 hours. Alternatively, tetrakis(triphenylphosphine)palladium, aq. potassium carbonate, toluene, ethanol, 90 ° C, 1 hour. b) IN-NaOH, ethanol, tetrahydrofuran (THF), 50 ° C, 1 hour.
- Reaction Scheme 23 illustrates a method for preparation of an alkali metal salt from the acid compound of Reaction Scheme 21.
- Reagents and reaction conditions a) 2-ethylhexanoic acid lithium salt, or 2-ethylhexanoic acid sodium salt, or 2- ethylhexanoic acid potassium salt, ethyl acetate/acetone, room temperature, 1 hour.
- Rj is an alkali metal, specifically lithium, sodium, or potassium.
- the present invention provides a pharmaceutical composition for modulation of peroxisome proliferator-activated receptor gamma (PPAR- ⁇ ), comprising a compound represented by formula 1, an optical isomer thereof or a pharmaceutically acceptable salt thereof, as an active ingredient.
- PPAR- ⁇ peroxisome proliferator-activated receptor gamma
- the present invention provides a use of the aforesaid pharmaceutical composition for modulation of peroxisome proliferator-activated receptor gamma (PPAR- ⁇ ), and a method for modulation of peroxisome proliferator-activated receptor gamma (PPAR- ⁇ ), comprising administering the aforesaid pharmaceutical composition to a subject.
- PPAR- ⁇ peroxisome proliferator-activated receptor gamma
- PPAR- ⁇ peroxisome proliferator-activated receptor gamma
- a pharmaceutical composition comprising a compound of formula 1 in accordance with the present invention will be effective as a PPAR agonist that exhibits hypoglycemic, hypolipidemic and insulin resistance-reducing effects while alleviating adverse side effects.
- a compound of formula 1 has hypoglycemic, hypolipidemic and insulin resistance-reducing effects on PPAR- mediated diseases or disorders, so it can be prophylactically or therapeutically effective for symptoms of PPAR-related diseases and conditions, such as obesity, diabetes, hypertension, and hyperlipidemia. Therefore, the present invention provides a use of the aforesaid composition for prevention or treatment of PPAR-mediated diseases (including obesity, diabetes, hypertension and hyperlipidemia), and a method for prevention or treatment of PPAR-mediated diseases (including obesity, diabetes, hypertension and hyperlipidemia), comprising administering the aforesaid composition to a subject.
- Dosage forms of the composition of the present invention may include oral formulations such as powders, granules, tablets, capsules, suspensions, emulsions, syrups and aerosols, and parenteral formulations such as external preparations, suppositories, and sterile injections. That is, the composition may be formulated into a desired dosage form, depending upon diseases to be treated and ingredients, using any appropriate method known in the art, as disclosed in "Remington's Pharmaceutical Sciences,” Mack Publishing Co., Easton, PA.
- the pharmaceutical composition of the present invention can be administered via a conventional route, for example orally, intradermally, subcutaneously, intravenously, intramuscularly, rectally, intraorally, intranasally, intraocularly, etc.
- the pharmaceutical composition may further comprise one or more pharmaceutically acceptable additives such as excipients, disintegrating agents, sweeteners, binders, coating agents, blowing agents, lubricants, glidants, solubilizers, etc, depending upon dosage forms of the composition.
- the compound of formula 1 may be administered at a dose of 0.1 mg to 1000 mg/kg BW once or several times a day.
- the effective dose of the active compound may vary depending upon various factors such as particular factors of patients, co-administered drugs, and severity of diseases.
- the present invention provides a novel phenylpropionic acid derivative of formula 1 and a method for preparing the same.
- the compound of the present invention has modulatory activity on peroxisome proliferator-activated receptor gamma (PPAR- ⁇ ) and therefore exhibits hypoglycemic, hypolipidemic and insulin resistance-reducing effects on PPAR-mediated diseases or disorders.
- PPAR- ⁇ peroxisome proliferator-activated receptor gamma
- the compound of formula 1 can be effective for prevention or treatment of PPAR- related diseases such as obesity, diabetes, hypertension, hypertriglyceridemia, etc.
- Fig. 1 shows test results for binding capacity of inventive compounds with Trap220 which is a main cofactor implicated in adipogenic differentiation.
- Step 1 Preparation of 3-(4-(benzyloxy)phenyI)-2-ethoxy acrylic acid ethyl ester (2a)
- Step 2 Preparation of 2-ethoxy-3-(4-hydroxyphenyI)-propionic acid ethyl ester (2b) 3-(4-(benzyloxy)phenyl)-2-ethoxy acrylic acid ethyl ester (2a, 8.0 g, 24.53 mmol) obtained in Step 1 was subjected to hydrogenation using 10% Pd/C to give 2-ethoxy-3-(4- hydroxyphenyl)-butyric acid ethyl ester (2b) as a colorless oil. Yield: 91%.
- Step 3 was dissolved in ethanol (20 mL), to which thionyl chloride (SOCl 2 , 1.2 niL) was then added, followed by reflux for 3 hours. After completion of the reaction was confirmed by thin layer chromatography (TLC), the solvent was removed under reduced pressure, followed by extraction with water (100 mL) and ethyl acetate (100 mL). An organic layer was washed with water (50 mL X 2) and brine (30 mL). The organic layer was separated, dried over anhydrous magnesium sulfate, and filtered under reduced pressure to remove ethyl acetate.
- TLC thin layer chromatography
- Step 3 Preparation of (S)-3-(4-((5-bromo-3-methylthiophen-2-yl)methoxy)phenyI)- 2-ethoxypropionic acid ethyl ester (7b)
- Step 2 Preparation of (S)-ethyl 3-(4-((5-bromofuran-2-yl)methoxy)phenyl)-2- ethoxypropanoate (8a)
- Step 1 Preparation of (5-bromo-3-methyIfuran-2-yl)methanol (5a) Ethyl 5-bromo-3-methylfuran-2-carboxylate (4.7 g, 20.16 mmol) was dissolved in tetrahydrofuran (THF, 20 mL). 2 equivalents of lithium aluminum hydride (LAH) were gradually added to the solution while being maintained at 0 ° C , followed by reaction for 1 hour.
- THF tetrahydrofuran
- Step 2 Preparation of rS)-3-(4-((5-bromo-3-methyIfuran-2-yl)methoxy)phenyl)-2- ethoxypropionic acid ethyl ester (9a) Analogously to Step 2 of Preparation Example 1, the title compound (S)-3-(4-((5-bromo-
- Step 1 Preparation of (2-bromo-4-methyIthiazol-5-yI)methanoI (6a) Analogously to Step 1 of Preparation Example 4, the title compound (2-bromo-4- methylthiazol-5-yl)methanol (6a) was synthesized from ethyl 2-bromo-4-methylthiazole-5- carboxylate (5.0 g, 20.00 mmol).
- Step 2 Preparation of (S)-ethyl 3-(4-((2-bromo-4-methylthiazoI-2- y ⁇ )methoxy)phenyl)-2-ethoxypropanoate (10a)
- the title compound (S)-ethyl 3-(4-((2- bromo-4-methylthiazol-2-yl)methoxy)phenyl)-2-ethoxypropanoate (10a) was synthesized from Compound 6a and Compound 2d of Preparation Example 1 through the Mitsunobu reaction. Yield: 35%.
- Step 4 Preparation of 5-tert-butyI-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaboroIan-2- vDphenvDisoxazole (lid)
- Step 2 Preparation of (3-(4-(4,4,5,5-tetramethvI-l,3,2-dioxaboroIan-2- vI)phenvI)isoxazoI-5-yl)methanoI (llh)
- Step 1 Preparation of l-(4-bromophen ⁇ I)-3-(dimethylamino)but-2-en-l-one (12a)
- Step 2 Preparation of 5-(4-bromophenvI)-3-meth ⁇ Iisoxazole (12b) l-(4-bromophenyl)-3-(dimethylamino)but-2-en-l-one (2.68 g, 10 mmol) synthesized in Step 1 of Preparation Example 9, and ammonium hydroxide (3 eq.) were dissolved in ethanol (50 mL) and the solution was warmed to 90 ° C, followed by reaction for 3 hours. After completion of the reaction was confirmed by TLC, extraction was carried out with water (100 mL) and ethyl acetate (250 mL).
- N,N-dimethylforniamide (DMF, 30 mL) were added 5-(4-bromophenyl)-3- methylisoxazole (2.5 g, 10.45 mmol), bis(pinacolato)diboron (5.0 g, 19.69 mmol), bis(diphenylphosphino)ferrocene dichloropalladium (900 mg, 1.1 mmol), and potassium acetate (3 g, 30.56 mmol), followed by reaction at 90 ° C for 2 hours. After completion of the reaction was confirmed by TLC, reactants were filtered through celite. The filtrate was extracted with water (10 mL) and ethyl acetate (10 mL).
- Step 1 Preparation of ethyl 4-(4-bromophenyl)-4-hvdroxy-2-oxobut-3-enoate (12d)
- Step 3 Preparation of ethyl 2-oxo-2-(5-(4-(4,4,5,,5-tetramethyl-l,3,2-dioxaboroIan- 2-vDphenyl)isoxazoI-3-yl)acetate (12h)
- Step 2 Preparation of 5-(4-(4,4,5,5-tetramethyI-l,3,2-dioxaboroIan-2- yl)phenvI)isoxazoI-3-vI)methanoI (12i)
- Step 1 Preparation of 5-(4-bromophenyI)-3-(methoxymeth ⁇ I)isoxazoIe (12g) 5-(4-bromophenyl)isoxazol-3-yl)methanol (1.0 g, 3.94 mmol) synthesized in Step 1 of
- Preparation Example 11 and 60% sodium hydride (200 mg) were added to N,N- dimethylformamide (50 mL) and the mixture was stirred for 11 min. Methyl iodide was added to the mixture, followed by reaction for 1 hour. After completion of the reaction was confirmed by TLC, extraction was carried with water (20 mL) and ethyl acetate (100 mL). The organic layer was washed with water (50 mL X 2) and brine (20 mL). The organic layer was separated, dried over anhydrous magnesium sulfate, and filtered under reduced pressure to remove methylene chloride.
- Step 3 Preparation of N-methyI-5-(4-(4,4,5,5-tetramethyl-l,3i2-dioxaborolan-2- yl)phenyI)isoxazole-3-earboxamide (12m) Analogously to Step 4 of Preparation Example 6, N-methyl-5-(4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)phenyl)isoxazole-3-carboxamide (12m) was prepared from 5-(4- bromophenyl)-N-methylisoxazole-3-carboxamide (105 mg, 0.37 mmol). Yield: 82%.
- Step 1 Preparation of 5-(4-bromophenyI)-2-methyl-2H-tetrazoIe (13a) To N,N-dimethylformamide (10 niL) were added 5-(4-bromophenyl)-2H-tetrazole (5 g,
- Step 2 Preparation of l-methyl-5-(4-(4,4,5,5-tetramethyI-l,3i2-dioxaborolan-2- yI)phenyl)-lH-tetrazole (13b-l)
- Step 2 Preparation of 2-(methoxymethyl)-5-(4-(4,4,5,5-tetramethyI-l,3i2- dioxaborolan-2-vDphenvD-2H-tetrazoIe (13f)
- Preparation Example 18 Preparation of (5-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)-2H-tetrazol-2-yl)methanol (13g) 2-(methoxymethyl)-5-(4-(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl)-2H- tetrazole (500 mg, 1.58 mmol) synthesized in Step 2 of Preparation Example 17 was dissolved in methylene chloride (20 mL) to which tribromoborane (BBr 3 , 2 eq.) was then added, followed by reaction for 5 hours.
- BBr 3 tribromoborane
- N-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)acetamide (1.0 g, 3.83 mmol), methyl iodide (1.2 eq.) and triethylamine were dissolved in tetrahydrofuran (10 mL), and the reactants were stirred at room temperature for 4 hours. After completion of the reaction was confirmed by TLC, water (50 mL) and ethyl acetate (500 mL) were added to the reactants, followed by extraction. The organic layer was washed with brine and distilled under reduced pressure.
- Step 2 Preparation of 3-(4-bromophenyD-5-methyl-l,,2,4-oxadiazole (16b) 4-bromo-N'-hydroxybenzimidamide (1.56 g, 7.25 mmol) and N,N-dimethylacetamide dimethyl acetal (DMA acetal, 2.9 mL, 21.8 mmol) were dissolved in dioxane (30 mL), followed by reaction under reflux for 12 hours. After completion of the reaction was confirmed by TLC, water (50 mL) and ethyl acetate (100 mL) were added to the reactants, followed by extraction. The organic layer was washed with brine and distilled under reduced pressure. The resulting solids were dried and recrystallized from hexane to afford the title compound 3-(4-bromophenyl)-5-methyl-l,2,4-oxadiazole (16b). Yield: 85%.
- Step 3 Preparation of 5-meth> f I-3-(4-(4,4 ⁇ 5 ⁇ 5-tetramethyl-l,3 ⁇ 2-dioxaboroIan-2- vDphenvD-l,2,4-oxadiazole (16c)
- Step 2 Preparation of 2-methyl-5-(4-(4,4,5,5-tetramethyi-l,3 ⁇ 2-dioxaboroIan-2- yI)phenvD-l,3 ⁇ -oxadiazole (17b)
- Step 1 Preparation of 2-(4-bromophenyI)-5-(trifluoromethyI)-l,3 ⁇ 4-o ⁇ adiazoIe (18a) 5-(4-bromophenyl)-lH-tetrazole (3 g, 13.3 mmol) and (CF 3 CO) 2 O (9 mL, 40 mmol)were dissolved in pyridine (10 mL), followed by reflux for 12 hours. After completion of the reaction was confirmed by TLC, extraction was carried out with water (20 mL) and ethyl acetate (50 mL). The organic layer was washed with water (20 mL X 2) and brine (20 mL).
- Step 2 Preparation of 2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaboroIan-2-yl)phen ⁇ l)-5- (trifluoromethyl)-l,3,4-oxadiazole (18b)
- the title compound 2-(4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)-5-(trifluoromethyl)-l,3,4-oxadiazole (18b) was prepared from 2-(4-bromophenyl)-5-(trifluoromethyl)-l,3,4-oxadiazole (2.22 g, 7.58 mmol). Yield: 85%.
- Step 5 Preparation of 4,5-dimethyl-2-(4-(4,4,5,5-tetramethyl-l,3 ⁇ 2-dioxaborolan-2- yDphenyPoxazoIe (2Oe)
- Step 1 Preparation of 5-(4-bromophenyl)-3-methyI-lH-pyrazoIe (21a) l-(4-bromophenyl)-3-(dimethylamino)but-2-en-l-one (2.68 g, 10 mmol) synthesized in
- Step 1 of Preparation Example 9 was dissolved in ethanol (10 mL) to which hydrazine (2eq.) was then added, followed by reaction at 90 " C for 6 hours. After completion of the reaction was confirmed by TLC, reactants were filtered through celite. The filtrate was extracted with water (100 mL) and ethyl acetate (100 mL). The organic layer was washed with water (100 mL X 2) and brine (50 mL). The organic layer was separated, dried over anhydrous magnesium sulfate, and filtered under reduced pressure to remove ethyl acetate.
- Step 2 Preparation of 5-(4-bromophenvI)-l,3-dimethyI-lH-pyrazoIe
- N,N- dimethylformamide 10 niL
- 60% sodium hydride NaH, 1.3 eq.
- methyl iodide 1.5 eq.
- Step 3 Preparation of l,3-dimethyl-5-(4-(4.,4,5,5-tetramethyl-l,3.,2-dioxaborolan-2- yDphenvD-lH-pyrazole (21c)
- Step 1 Preparation of (S)-2-ethoxy-3-(4-((5-(3-methoxyphenyI)-3-methylthiophen- 2-yl)methoxy)phen ⁇ l)propionic acid ethyl ester
- Step 2 Preparation of (S)-2-ethoxy-3-(4-(Y5-(3-methoxyphenvI)-3-methylthiophen- 2-vI)methoxy)phenvI)propionic acid
- an ester compound was prepared from (S)-2- ethoxy-3 -(4-((5 -(4-fluorophenyl)-3 -methylthiophen-2-yl)methoxy)phenyl)propionic acid ethyl ester (600 mg) and 4-fluorophenylboronic acid (1.2 eq.).
- the ester compound was then hydro lyzed to afford the title compound (S)-2-ethoxy-3-(4-((5-
- Example 3 Preparation of (S)-3-(4-((5-(3,4-dimethoxyphenyQ-3-methylthiophen-2- vI)methoxy)phenvD-2-ethoxypropionic acid Analogously to Step 1 of Example 1, an ester compound was prepared from (S)-3-(4-((5-
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-p-phenylthiophen-2-yl)methoxy)phenyl)propionic acid ethyl ester (400 mg) and 4-methylphenylboronic acid (1.2 eq.).
- the ester compound was then hydrolyzed to afford the title compound (S)-2-ethoxy-3-(4-((3- methyl-5-p-phenylthiophen-2-yl)methoxy)phenyl)propionic acid (Example 7).
- an ester compound was prepared from (S)-2- ethoxy-3 -(4-((3 -methyl-5 -(4-(trifluoromethoxy)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (600 mg) and 4- (trifluoromethoxy)phenylboronic acid (1.2 eq.).
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-isopropylphenyl)thiophen-2-yl)methoxy)phenyl)propionic acid ethyl ester (600 mg) and 4-isopropylphenylboronic acid (1.2 eq.).
- the ester compound was then hydrolyzed to afford the title compound (S)-2- ethoxy-3-(4-((5-(4-isopropylphenyl)-3-methylthiophen-2-yl)methoxy)phenyl)propionic acid (Example 9).
- Example 10 Preparation of (S)-2-ethoxy-3-(4-((3-methyl-5-phenylthiophen-2- yl)methoxy)phenyl)propionic acid Analogously to Step 1 of Example 1, an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-phenylthiophen-2-yl)methoxy)phenyl)propionic acid ethyl ester
- Example 13 Preparation of (S)-3-(4-((5-(4-acetamidophenyI)-3-methyIthiophen-2- yl)methoxy)phenyl)-2-ethoxypropionic acid
- an ester compound was prepared from (S)-2- ethoxy-3 -(4-((3 -methyl-5 -(4-(N-methylacetamido)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (520 mg) and N-(4-(4,4,5,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)phenyl)acetamide (1.2 eq.) as defined in formula 11.
- Example 14 Preparation of (S)-2-ethoxy-3-(4-((3-methyl-5-(4-(N- methyIacetamido)phenyI)thiophen-2-yl)methoxy)phenyl)propionic acid Analogously to Step 1 of Example 1, an ester compound was prepared from (S)-3-(4-((5-)
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((5-(4-(furan-2-yl-methylcarbamoyl)phenyl)-3-methylthiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (400 mg) and 4-(furan-2-yl- methylcarbamoyl)phenylboronic acid (1.2 eq.).
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-(morpholine-4-carbonyl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (400 mg) and 4-(morpholino-4- carbonyl)phenylboronic acid (1.2 eq.).
- Example 18 Preparation of (S)-2-ethoxy-3-(4-((3-methyI-5-(4- (morpholinosulfonyl)phenyl)thiophen-2-yl)methoxy)phenyl)propionie acid
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-(mo ⁇ holinosulfonyl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (330 mg) and 4- (morpholinosulfonyl)phenylboronic acid (1.2 eq.).
- Example 19 Preparation of (S)-3-f4-(Y5-(4-(5,6-dihydro-4H-l,3-oxazin-2- v ⁇ phenyl)-3-methylthiophen-2-yl)methoxy)phenyl)-2-ethoxypropionic acid Analogously to Step 1 of Example 1, an ester compound was prepared from (S)-3-(4-((5-
- Example 1 the ester compound was then hydrolyzed to afford the title compound (S)-2- ethoxy-3-(4-((3-methyl-5-(4-morpholinophenyl)thiophen-2-yl)methoxy)phenyl)propionic acid (Example 20).
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-(l -methyl-6-oxo- 1 ,4,5,6-tetrahydropyridazin-3- yl)phenyl)thiophen-2-yl)methoxy)phenyl)propionic acid ethyl ester (560 mg) and Compound 15b (1.2 eq.) synthesized in Preparation Example 20.
- an ester compound was prepared from (S)-3-(4-((5- (4-(2H-benzo[b] [1 ,4]oxazin-3-yl)phenyl)-3-methylthiophen-2-yl)methoxy)phenyl)-2- ethoxypropionic acid ethyl ester (320 mg) and 4-(2H-benzo[b][l,4]oxazine)phenylboronic acid (1.2 eq.).
- an ester compound was prepared from (S)-3-(4-((5- (4-( 1 ,2,3 -thiadiazol-4-yl)phenyl)-3 -methylthiophen-2-yl)methoxy)phenyl)-2-ethoxypropionic acid ethyl ester (220 mg) and 4-(l,2,3-thiadiazole)phenylboronic acid (1.2 eq.).
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-(5-methyl- 1 ,2,4-oxadiazol-3-yl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (430 mg) and Compound 16c (1.2 eq.) synthesized in Preparation Example 21.
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (510 mg) and Compound 17b (1.2 eq.) synthesized in Preparation Example 22.
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-(5-(trifluoromethyl)-l,3,4-oxadiazol-2-yl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (460 mg) and Compound 18b (1.2 eq.) synthesized in Preparation Example 23.
- Example 29 Preparation of (S)-2-ethoxy-3-(4-((3-methyl-5-(4-(2-methyI-2H- tetrazol-5-yI)phenyl)thiophen-2-yI)methoxy)phenyl)propionic acid
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((3-methyl-5-(4-(2-methyl-2H-tetrazol-5-yl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (390 mg) and Compound 13b (1.2 eq.) synthesized in Preparation Example 14.
- an ester compound was prepared from (S)-3-(4-((5- (4-(4,5-dimethyloxazol-2-yl)phenyl)-3-methylthiophen-2-yl)methoxy)phenyl)-2- ethoxypropionic acid ethyl ester (430 mg) and Compound 2Oe (1.2 eq.) synthesized in Preparation Example 25.
- Example 35 Preparation of fS)-2-ethoxy-3-(4-((5-(4-(5-(hydroxymethyI)isoxazol-3- yl)phenyI)-3-methylthiophen-2-yl)methoxy)phenyl)propionie acid
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((5-(4-(5-(hydroxymethyl)isoxazol-3-yl)phenyl)-3-methylthiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (600 mg) and Compound Hh (1.2 eq.) synthesized in Preparation Example 7.
- an ester compound was prepared from (S)-2- ethoxy-3 -(4-((3 -methyl-5 -(4-(3 -(methylcarbamoyl)isoxazol-5-yl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (320 mg) and Compound 12m (1.2 eq.) synthesized in Preparation Example 13.
- an ester compound was prepared from (S)-2- ethoxy-3-(4-((5-(4-(3-(methoxymethyl)isoxazol-5-yl)phenyl)-3-methylthiophen-2- yl)methoxy)phenyl)propionic acid ethyl ester (540 mg) and Compound 12j (1.2 eq.) synthesized in Preparation Example 12.
- Example 43 Preparation of sodium (S)-2-ethoxy-3-(4-((3-methyl-5-(4-(3- methyIisoxazoI-5-yl)phenyI)thiophen-2-yI)methoxy)phenyI)propanoate (S)-2-ethoxy-3-(4-((3-methyl-5-(4-(3-methylisoxazol-5-yl)phenyl)thio ⁇ hen-2- yl)methoxy)phenyl)propionic acid (2.0 g) synthesized in Example 41 was dissolved in a mixture of ethyl acetate (7 mL) and acetone (1 mL), which was followed by addition of 2- ethylhexanoic acid sodium salt (1.2 eq.) and stirring at room temperature for 1 hour, as disclosed in Reaction Scheme 23.
- Example 44 Preparation of potassium (S)-2-ethoxy-3-(4-(Y3-methyI-5-(4-(3- methylisoxazol-5-vI)phenyl)thiophen-2-vI)methoxy)phenyl)propanoate (S)-2-ethoxy-3-(4-((3-methyl-5-(4-(3-methylisoxazol-5-yl)phenyl)thiophen-2- yl)methoxy)phenyl)propionic acid (4.0 g) synthesized in Example 41 was dissolved in a mixture of ethyl acetate (15 niL) and acetone (2 mL), which was followed by addition of 2- ethylhexanoic acid potassium salt (1.2 eq.) and stirring at room temperature for 1 hour, as disclosed in Reaction Scheme 23.
- Example 46 Preparation of (S)-2-ethoxy-3-(4-((3-methyl-5-(4-(3-methylisoxazol-5- yl)phenyl)fiiran-2-yl)methoxy)phepyl)propionic acid
- (S)-2-ethoxy-3-(4-((3-methyl-5-(4-(3- methylisoxazol-5-yl)phenyl)furan-2-yl)methoxy)phenyl)propionic acid ethyl ester 300 mg
- Compound 9a 1.0 g, 2.43 mmol
- Example 48 Preparation of (S)-3-(4-((5-(4-(5-tert-butyIisoxazol-3-yDphenyD-3- methvIthiophen-2-yl)methox ⁇ )phen ⁇ D-2-ethoxypropionic acid Analogously to Step 1 of Example 1, an ester compound was prepared from (S)-3-(4-((5-)
- Example 52 Preparation of (S)-3-(4-((5-(4-(5-tert-butyIisoxazol-3-yI)phenyI)furan- 2-yl)methoxy)phenyl)-2-ethoxypropionie aeid
- (S)-3-(4-((5-(4-(5-tert-butylisoxazol-3- yl)phenyl)furan-2-yl)methoxy)phenyl)-2-ethoxypropionic acid ethyl ester 400 mg
- Example 53 Preparation of (S)-3-(4-((5-(4-(5-tert-butvIisoxazoI-3-yI)phenvI)-3- methylfuran-2-yl)methoxy)phenyI)-2-ethoxypropionic acid Analogously to Step 1 of Example 1, (S)-3-(4-((5-(4-(5-tert-butylisoxazol-3-yl)phenyl)-
- the African green monkey kidney cell line CV-I (CCL-70, ATCC) was used as a test cell line, and PPAR- ⁇ and - ⁇ were murine- and human-derived PPARs. Samples used were compounds prepared in Examples 19, 21, 23, 25, 26, 27, 31, 33, 34, 36, 38, 40, 42, 43 and 50.
- 3-4-[2-(2-phenyl-4-methyl-l,3- oxazole)ethyloxy]phenyl-(2S)-[(l -methyl-3-oxo-3-phenyl)propenyl]aminopropionic acid was used that is a PPAR- ⁇ or - ⁇ agonist which was once under development and whose clinical trials and studies were suspended at phase III.
- a chimeric receptor was adopted to circumvent the probable interference due to endogenous receptor activation (Jian-Shen Q. et al., MoI Cell Biol (1995) 15(3):1817-1825).
- the chimeric receptor was constructed as a fusion of a PPAR- ⁇ or - ⁇ ligand-binding domain with a DNA-binding domain of GAL4 which is a yeast transactivator.
- the CV-I cells were transiently transfected with each of chimeric receptor-expressing
- DNA constructs and each of DNA constructs comprising 5 copies of the GAL4 DNA-binding domain and capable of inducing expression of firefly luciferase or Renilla luciferase using a Lipofectamine Plus reagent (Invitrogen, USA).
- the culture media were replaced with DMEM containing the above samples and 10% fetal bovine serum.
- the firefly luciferase activity and Renilla luciferase activity were continuously assayed while adding an equal amount of a dual luciferase assay reagent (Promega, USA) to the cell-containing media.
- the transfection efficiency was normalized against Renilla luciferase activity (Motomura W.
- the PPAR- ⁇ and - ⁇ activity was determined by calculating Relative Response % to maximum effects of the positive control drug, and conducting multiple dose evaluation of the inventive compounds to calculate EC 50 , which is the concentration of a drug which produces 50% activation relative to maximum effects of the inventive compounds, by nonlinear regression analysis.
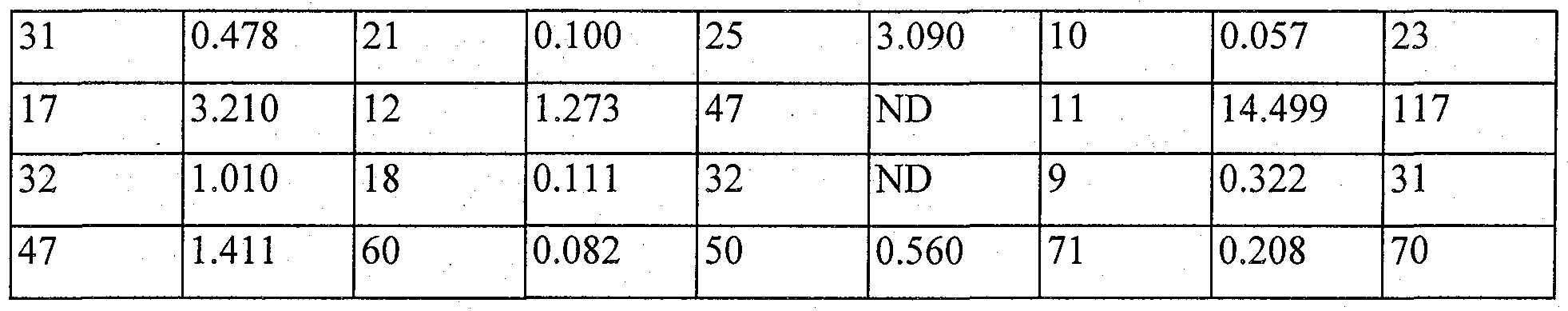
- Representative compounds of the present invention exhibited EC 50 of 400 to 6000 nM for human PPAR- ⁇ and EC 50 of 7 to 100OnM for human PPAR- ⁇ (see Table 1).
- the maximum response of the inventive compounds for human PPAR- ⁇ was found to be a 15 to 80% level of the positive control drug that causes 100% activation of PPAR- ⁇ . That is, the compounds of formula 1 in accordance with the present invention were identified as drugs which activate PPAR- ⁇ even at a low concentration, but exhibit a relatively low responsiveness as compared to the positive control drug inducing 100% activation and have higher activity for PPAR- ⁇ than for PPAR- ⁇ . Therefore, a pharmaceutical composition comprising the compound of the present invention can be effectively used as a PPAR agonist that is expected to exhibit hypoglycemic, hypolipidemic and insulin resistance-reducing effects simultaneously with decreased adverse side effects of the drug.
- mice were orally administered with 5 PPAR compounds that exhibit partial agonism on PPAR- ⁇ and - ⁇ in the in vitro reporter assay, and the PPAR- ⁇ modulator INT-131, respectively.
- INT-131 as a control drug exhibited ED 30 of 4 mg/kg.
- the compound of formula 1 in accordance with the present invention was shown to have excellent hypoglycemic activity comparable to or higher than INT-131.
- a monkey ovary cell line CHO-Kl (CCL-61, ATCC) was used as a test cell line.
- DNA constructs used in this assay were an expression vector pVP16 (Clontech) constructed to express a fusion of a human PPAR- ⁇ 2 ligand-binding domain with an activation domain of the yeast transactivator GAL4, and an expression vector pM (Clontech) constructed to express a fusion of human Trap220 with the GAL4 DNA-binding domain.
- Rosiglitazone maleate (Alcon Biosciences Private Limited), which is clinically used as a PPAR- ⁇ agonist, was employed as a control drug.
- the CHO-Kl cells were transiently transfected with two DNA constructs expressing the chimeric receptors and DNA constructs comprising 5 copies of the GAL4 DNA-binding domain and capable of inducing expression of firefly luciferase or Renilla luciferase using a Lipofectamine Plus reagent (Invitrogen, USA). Subsequent processes were carried out in the same manner as in the transactivation assay. The experimental results were expressed as an increase of the responsiveness vs. the negative control group with no addition of the drug. The results thus obtained are shown in Fig. 1.
- the compound of the present invention has modulatory activity on peroxisome proliferator-activated gamma receptor (PPAR- ⁇ ). That is, the compound shows hypoglycemic, hypolipidemic and insulin resistance-reducing effects on PPAR-mediated diseases or disorders, so it can be prophylactically or therapeutically effective for PPAR-related diseases and conditions, such as obesity, diabetes, hypertension, and hyperlipidemia.
- PPAR- ⁇ peroxisome proliferator-activated gamma receptor
Abstract
Description





































































Claims






























Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/449,979 US20100063041A1 (en) | 2007-03-07 | 2008-03-07 | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same |
| AU2008221718A AU2008221718A1 (en) | 2007-03-07 | 2008-03-07 | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same |
| JP2009552595A JP2010520873A (en) | 2007-03-07 | 2008-03-07 | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, methods thereof and pharmaceutical compositions containing the same |
| GB0917415A GB2460784A (en) | 2007-03-07 | 2008-03-07 | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators,method of the same,and pharmaceutical composition |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2007-0022681 | 2007-03-07 | ||
| KR20070022681 | 2007-03-07 | ||
| KR1020080021695A KR20080082541A (en) | 2007-03-07 | 2008-03-07 | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same |
| KR10-2008-0021695 | 2008-03-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008108602A1 true WO2008108602A1 (en) | 2008-09-12 |
Family
ID=39738429
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2008/001322 WO2008108602A1 (en) | 2007-03-07 | 2008-03-07 | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008108602A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| JP2012506386A (en) * | 2008-10-21 | 2012-03-15 | メタボレックス, インコーポレイテッド | Aryl GPR120 receptor agonist and uses thereof |
| WO2012175513A1 (en) | 2011-06-20 | 2012-12-27 | Bayer Intellectual Property Gmbh | Thienylpyri(mi)dinylpyrazole |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013041621A1 (en) | 2011-09-20 | 2013-03-28 | Basf Se | Low molecular weight modulators of the cold-menthol receptor trpm8 and use thereof |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013068486A1 (en) | 2011-11-08 | 2013-05-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of male infertility |
| WO2019025554A1 (en) * | 2017-08-04 | 2019-02-07 | Bayer Aktiengesellschaft | 6-phenyl-4,5-dihydropyridazin-3(2h)-one derivatives as pde3a and pde3b inhibitors for treating cancer |
| CN112851604A (en) * | 2021-01-11 | 2021-05-28 | 河南中医药大学 | Compound D extracted from Cornus officinalis and having hypoglycemic effect, and preparation method and application thereof |
| US11427553B2 (en) | 2017-08-04 | 2022-08-30 | Bayer Aktiengesellschaft | Dihydrooxadiazinones |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004020420A1 (en) * | 2002-08-30 | 2004-03-11 | F.Hoffmann-La Roche Ag | Novel 2-arylthiazole compounds as pparalpha and ppargama agonists |
-
2008
- 2008-03-07 WO PCT/KR2008/001322 patent/WO2008108602A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004020420A1 (en) * | 2002-08-30 | 2004-03-11 | F.Hoffmann-La Roche Ag | Novel 2-arylthiazole compounds as pparalpha and ppargama agonists |
Non-Patent Citations (3)
| Title |
|---|
| ELTE J.W. ET AL.: "Thiazolidinediones for the treatment of type 2 diabetes", EUR. J. INTERN. MED., vol. 18, no. 1, January 2007 (2007-01-01), pages 18 - 25, XP005827282 * |
| LEHMANN J.M. ET AL.: "An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma)", J. BIOL. CHEM., vol. 270, no. 22, 2 June 1995 (1995-06-02), pages 12953 - 12956, XP000577082 * |
| ZHANG F. ET AL.: "Selective Modulators of PPAR-gamma Activity: Molecular Aspects Related to Obesity and Side-Effects", PPAR RES., vol. 32696, 2007, pages 7 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012506386A (en) * | 2008-10-21 | 2012-03-15 | メタボレックス, インコーポレイテッド | Aryl GPR120 receptor agonist and uses thereof |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012175513A1 (en) | 2011-06-20 | 2012-12-27 | Bayer Intellectual Property Gmbh | Thienylpyri(mi)dinylpyrazole |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013041621A1 (en) | 2011-09-20 | 2013-03-28 | Basf Se | Low molecular weight modulators of the cold-menthol receptor trpm8 and use thereof |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013068486A1 (en) | 2011-11-08 | 2013-05-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of male infertility |
| WO2019025554A1 (en) * | 2017-08-04 | 2019-02-07 | Bayer Aktiengesellschaft | 6-phenyl-4,5-dihydropyridazin-3(2h)-one derivatives as pde3a and pde3b inhibitors for treating cancer |
| US11427553B2 (en) | 2017-08-04 | 2022-08-30 | Bayer Aktiengesellschaft | Dihydrooxadiazinones |
| US11773070B2 (en) | 2017-08-04 | 2023-10-03 | Bayer Aktiengesellschaft | Dihydrooxadiazinones |
| US11897867B2 (en) | 2017-08-04 | 2024-02-13 | Bayer Aktiengesellschaft | 6-phenyl-4,5-dihydropyridazin-3(2H)-one derivatives as PDE3A and PDE3B inhibitors for treating cancer |
| CN112851604A (en) * | 2021-01-11 | 2021-05-28 | 河南中医药大学 | Compound D extracted from Cornus officinalis and having hypoglycemic effect, and preparation method and application thereof |
| CN112851604B (en) * | 2021-01-11 | 2022-06-17 | 河南中医药大学 | Compound D extracted from Cornus officinalis and having hypoglycemic effect, and preparation method and application thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008108602A1 (en) | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same | |
| JP4931893B2 (en) | PPAR activating compound and pharmaceutical composition containing the same | |
| US8003648B2 (en) | Heterocyclic GPR40 modulators | |
| US20100063041A1 (en) | Novel phenylpropionic acid derivatives as peroxisome proliferator-activated gamma receptor modulators, method of the same, and pharmaceutical composition comprising the same | |
| KR100654516B1 (en) | Carboxylic acid substituted oxazole derivatives for use as ppar-alpha and -gamma activators in the treatment of diabetes | |
| JP3997491B2 (en) | Carbazole derivative, solvate thereof, or pharmaceutically acceptable salt thereof | |
| US7282501B2 (en) | Modulators of peroxisome proliferator activated receptors (PPAR) | |
| TWI440633B (en) | Activator of Activated Receptors for Peroxisome Producers | |
| US20100137323A1 (en) | Benzo-fused compounds for use in treating metabolic disorders | |
| WO2002014291A1 (en) | PPARδ ACTIVATORS | |
| JP2005529077A (en) | Peroxisome proliferator-activated receptor modulator | |
| WO2006127503A2 (en) | Compounds, pharmaceutical compositions and methods for their use in treating metabolic disorders | |
| JPWO2002076957A1 (en) | Activator of peroxisome proliferator-activated receptor | |
| JP2006516254A (en) | Fused heterocyclic derivatives as PPAR modulators | |
| NO330171B1 (en) | Chiral oxazole-arylpropionic acid derivatives, processes for the preparation thereof, pharmaceutical compositions containing them, such compounds for use as therapeutically active substances, and such compounds for the treatment and / or prevention of disease | |
| JP2005504778A (en) | Oral antidiabetic agent | |
| CA2418104A1 (en) | Oxazolyl-arylpropionic acid derivatives and their use as ppar agonists | |
| JP2004506722A (en) | Methods of modulating peroxisome proliferator-activated receptors | |
| US7504433B2 (en) | Thiophene derivative PPAR modulators | |
| US20080207685A1 (en) | Heterocyclic Compounds As Modulators Of Peroxisome Proliferator Activated Receptors, Useful For The Treatment And/Or Prevention Of Disorders Modulated By A Ppar | |
| JPWO2006041197A1 (en) | Activator of peroxisome proliferator activated receptor δ | |
| TWI328585B (en) | Oxazol derivatives | |
| KR100958831B1 (en) | Heteroarylalkoxy-phenyl derivatives, processes for the preparation thereof, and compositions comprising the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08723359 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 12449979 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009552595 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008221718 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 0917415 Country of ref document: GB Kind code of ref document: A Free format text: PCT FILING DATE = 20080307 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2008221718 Country of ref document: AU Date of ref document: 20080307 Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08723359 Country of ref document: EP Kind code of ref document: A1 |