WO2008054674A2 - Antidiabetic bicyclic compounds - Google Patents
Antidiabetic bicyclic compounds Download PDFInfo
- Publication number
- WO2008054674A2 WO2008054674A2 PCT/US2007/022643 US2007022643W WO2008054674A2 WO 2008054674 A2 WO2008054674 A2 WO 2008054674A2 US 2007022643 W US2007022643 W US 2007022643W WO 2008054674 A2 WO2008054674 A2 WO 2008054674A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- c3alkyl
- optionally substituted
- independently selected
- group
- pharmaceutically acceptable
- Prior art date
Links
- 0 *c(c(*)nc(*)c1*)c1Oc1cc(**C2*(BC(N3)=O)C3=O)c2cc1 Chemical compound *c(c(*)nc(*)c1*)c1Oc1cc(**C2*(BC(N3)=O)C3=O)c2cc1 0.000 description 1
- VUCZAYDFIHRKSR-UHFFFAOYSA-N CCOC(C(C1c(ccc(OC)c2)c2SCC1)Br)=O Chemical compound CCOC(C(C1c(ccc(OC)c2)c2SCC1)Br)=O VUCZAYDFIHRKSR-UHFFFAOYSA-N 0.000 description 1
- XSGCUQLKTRSCDY-SNVBAGLBSA-N COc1ccc([C@@H](CC(O)=O)CCC2)c2c1 Chemical compound COc1ccc([C@@H](CC(O)=O)CCC2)c2c1 XSGCUQLKTRSCDY-SNVBAGLBSA-N 0.000 description 1
- BJHOIWIKUDSVQJ-UHFFFAOYSA-N O=C1c(ccc(OCc2ccccc2)c2)c2OC1 Chemical compound O=C1c(ccc(OCc2ccccc2)c2)c2OC1 BJHOIWIKUDSVQJ-UHFFFAOYSA-N 0.000 description 1
- XEUXURCJOSWFGB-VUWPPUDQSA-N Oc1ccc([C@H](CCC2)C(C(N3)=O)SC3=O)c2c1 Chemical compound Oc1ccc([C@H](CCC2)C(C(N3)=O)SC3=O)c2c1 XEUXURCJOSWFGB-VUWPPUDQSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the instant invention is concerned with biaryl ethers in which one of the aromatic rings is fused to a cycloalkyl or heterocyclic ring, which is attached to a thiazohdinedione or oxazohdinedione ring.
- the compounds are agonists of G-protem coupled receptor 40 (GPR40) and are useful as therapeutic compounds, particularly in the treatment of Type 2 diabetes mellitus, and of conditions that are often associated with this disease, including insulin resistance, obesity, and lipid disorders.
- GPR40 G-protem coupled receptor 40
- Diabetes is a disease derived from multiple causative factors and characterized by elevated levels of plasma glucose (hyperglycemia) in the fasting state or after administration of glucose during an oral glucose tolerance test.
- hyperglycemia plasma glucose
- type 1 diabetes or insulin-dependent diabetes mellitus (DDDM)
- DDDM insulin-dependent diabetes mellitus
- NIDDM noninsuhn-dependent diabetes mellitus
- Patients having type 2 diabetes have a resistance to the effects of insulin in stimulating glucose and lipid metabolism in the main msulm-sensitive tissues, which are muscle, liver and adipose tissues These patients often have normal levels of insulin, and may have hype ⁇ nsulinemia (elevated plasma insulin levels), as they compensate for the reduced effectiveness of insulin by secreting increased amounts of insulin Insulin resistance is not primarily caused by a diminished number of insulin receptors but rather by a post-insulin receptor binding defect that is not yet completely understood. This lack of responsiveness to insulin results in insufficient insulin-mediated activation of uptake, oxidation and storage of glucose in muscle, and inadequate insulin-mediated repression of hpolysis in adipose tissue and of glucose production and secretion in the liver.
- Persistent or uncontrolled hyperglycemia that occurs with diabetes is associated with increased and premature morbidity and mortality. Often abnormal glucose homeostasis is associated both directly and indirectly with obesity, hypertension, and alterations of the lipid, lipoprotein and apohpoprotem metabolism, as well as other metabolic and hemodynamic disease Patients with type 2 diabetes mellitus have a significantly increased risk of macrovascular and microvascular complications, including atherosclerosis, coronary heart disease, stroke, peripheral vascular disease, hypertension, nephropathy, neuropathy, and retinopathy.
- a patient having metabolic syndrome is characterized as having three or more symptoms selected from the following group of five symptoms: (1) abdominal obesity; (2) hypertriglyceridemia; (3) low high-density lipoprotein cholesterol (HDL); (4) high blood pressure; and (5) elevated fasting glucose, which may be in the range characteristic of Type 2 diabetes if the patient is also diabetic.
- the biguanides are a class of drugs that are widely used to treat type 2 diabetes.
- the two best known biguanides, phenformin and metformin cause some correction of hyperglycemia.
- the biguanides act primarily by inhibiting hepatic glucose production, and they also are believed to modestly improve insulin sensitivity
- the biguanides can be used as monotherapy or m combination with other anti-diabetic drugs, such as insulin or an insulin secretagogues, without increasing the risk of hypoglycemia.
- phenformin and metformin can induce lactic acidosis and nausea/diarrhea Metformin has a lower risk of side effects than phenformin and is widely prescribed for the treatment of Type 2 diabetes.
- the ghtazones are a newer class of compounds that can ameliorate hyperglycemia and other symptoms of type 2 diabetes.
- the ghtazones that are currently marketed are agonists of the peroxisome prohferator activated receptor (PPAR) gamma subtype.
- PPAR peroxisome prohferator activated receptor
- the PPAR-gamma agonists substantially increase insulin sensitivity in muscle, liver and adipose tissue in several animal models of type 2 diabetes, resulting in partial or complete correction of elevated plasma glucose levels without the occurrence of hypoglycemia.
- PPAR-gamma agonism is believed to be responsible for the improved insulin sensitization that is observed in human patients who are treated with the ghtazones.
- New PPAR agonists are currently being developed. Many of the newer PPAR compounds are agonists of one or more of the PPAR alpha, gamma and delta subtypes. Compounds that are agonists of both the PPAR alpha and PPAR gamma subtypes (PPAR alpha/gamma dual agonists) are promising because they reduce hyperglycemia and also improve lipid metabolism.
- the currently marketed PPAR gamma agonists are modestly effective in reducing plasma glucose and HemoglobmAlC. The currently marketed compounds do not greatly improve lipid metabolism and may actually have a negative effect on the lipid profile. Thus, the PPAR compounds represent an important advance in diabetic therapy, but further improvements are still needed.
- insulin secretagogues such as the sulfonylureas (e g. tolbutamide and glipizide).
- sulfonylureas e g. tolbutamide and glipizide
- These drugs increase the plasma level of insulin by stimulating the pancreatic ⁇ -cells to secrete more insulin.
- Insulin secretion m the pancreatic ⁇ -cell is under strict regulation by glucose and an array of metabolic, neural and hormonal signals. Glucose stimulates insulin production and secretion through its metabolism to generate ATP and other signaling molecules, whereas other extracellular signals act as potentiators or inhibitors of insulin secretion through GPCR's present on the plasma membrane.
- Sulfonylureas and related insulin secretagogues act by blocking the ATP-dependent K+ channel in /3-cells, which causes depolarization of the cell and the opening of the voltage-dependent Ca2+ channels with stimulation of insulin release.
- This mechanism is non-glucose dependent, and hence insulin secretion can occur regardless of the ambient glucose levels. This can cause insulin secretion even if the glucose level is low, resulting in hypoglycemia, which can be fatal in severe cases.
- the administration of insulin secretagogues must therefore be carefully controlled.
- the insulin secretagogues are often used as a first-line drug treatment for Type 2 diabetes.
- GPCR G-protem coupled receptors
- GPR40 is a cell-surface GPCR that is highly expressed in human (and rodent) islets as well as in insulin-secreting cell lines
- F's medium to long- chain fatty acids
- synthetic compounds including several members of the thiazohdinedione class of PPAR ⁇ agonists, have recently been identified as ligands for GPR40 (Itoh, Y. et al., Nature 422: 173 [2003], B ⁇ scoe, CP. et al., J. Biol. Chem. 278: 11303 [2003], Kotarsky, K. et al., Biochem. Biophys. Res Comm 301: 406 [2003].
- GPR40 agonists are capable of augmenting the release of insulin from islet cells.
- the specificity of this response is suggested by results showing that the inhibition of GPR40 activity by siRNA attenuates FA-induced amplification of GSIS
- GPR40-mediated insulin secretion is glucose dependent, there is little or no ⁇ sk of hypoglycemia.
- the limited tissue distribution of GPR40 suggests that there would be less chance for side effects associated with GPR40 activity in other tissues.
- GPR40 agonists that are active m the islets may have the potential to restore or preserve islet function. This would be highly advantageous, because long term diabetes therapy often leads to the gradual diminution of islet activity, so that after extended periods of treatment, it is often necessary to treat type 2 diabetic patients with daily insulin injections. By restoring or preserving islet function, GPR40 agonists may delay or prevent the diminution and loss of islet function in a type 2 diabetic patient.
- the class of compounds described herein is a new class of GPR40 agonists.
- the compounds are useful in the treatment of diseases that are modulated by GPR40 agonists, including type 2 diabetes, hyperglycemia that may be associated with type 2 diabetes or pre-diabetic insulin resistance, and obesity.
- the present invention is directed to a compound of formula I, or a pharmaceutically acceptable salt thereof, including individual diastereomers and enantiomers thereof, and mixtures of diastereomers and/or enantiomers thereof:
- A is selected from the group consisting of -CH- and -N-;
- B is selected from the group consisting of -S-, -O-, -NH-, and -CH2-,
- Z is selected from the group consisting of -CH 2 - and -CH 2 CH 2 -;
- Heterocycle is a 5-6 membered saturated or partly saturated monocyclic heterocyclic ⁇ ng having 1-3 heteroatoms independently selected from O, N and S;
- Heteroaryl is a 5-6 membered monocyclic heteroaromatic ring having 1-3 heteroatoms independently selected from O, N and S;
- Each R 6 is independently selected from the group consisting of H and -Cj-C ⁇ alkyl.
- alkyl, alkenyl, and alkynyl groups can each be linear or branched, unless otherwise defined.
- the invention has numerous embodiments, which are summarized below.
- the invention includes the specific compounds as shown, and also includes individual diastereomers, enantiomers, and epimers of the compounds, and mixtures of diastereomers and/or enantiomers thereof.
- the invention also includes pharmaceutically acceptable salts of the compounds, and pharmaceutical compositions composing the compounds and a pharmaceutically acceptable carrier.
- the compounds are useful m treating insulin resistance, type 2 diabetes, and dyshpidemia that is associated with type 2 diabetes and insulin resistance.
- each R" is independently selected from H and CH 3
- R 1 , R 2 , R 3 , and R 4 are each independently selected from H, halogen, Ci-C 3 alkyl, CF 3 ,
- the fused phenyl ring at the Rl and R 2 positions is optionally substituted with 1-2 substituents independently selected from halogen, -NO2, -CH3, -CF3, -OCH3, and -OCF3.
- the fused furan or thiophene ring at the Rl and R2 positions is optionally substituted with 1-2 substituents independently selected from halogen, -NO2, -CH3, -CF3, -OCH3, and -OCF3.
- A is CH- or -N-; and B is selected from -O- and -S-.
- Z is selected from the group consisting of -CH2- and -CH2CH2-.
- W is selected from the group consisting of -CH2-, -0-, and -S-; and Z is selected from the group consisting of -CH2- and -CH2CH2-.
- Each R6 IS independently selected from H and CH3;
- W is selected from the group consisting of -CH2-, -O-, and -S-;
- Z is selected from the group consisting of-CH2- and -CH2CH2-;
- A is -CH- or N
- B is selected from -S- and -O-.
- R ⁇ is selected from the group consisting of H, CH3, and halogen.
- R ⁇ is selected from the group consisting of H, CH3, and halogen.
- R ⁇ is selected from the group consisting of H, halogen, CH3, CF3, and -S(O)2NH2.
- W is selected from the group consisting of -CH2-, -O-, and -S-.
- Z is selected from the group consisting of -CH2- and -CH2CH2-;
- A is -CH- or N; and B is selected from -S- and -O-.
- stereoisomers including diastereomers, enantiomers, epimers, and mixtures of these may also have utility in treating GPR40 mediated diseases.
- Inactive or less active diastereoisomers and enantiomers are useful for scientific studies relating to the receptor and the mechanism of activation.
- the compounds of this invention may be used in pharmaceutical compositions comprising (a) the compound(s) or pharmaceutically acceptable salts thereof, and (b) a pharmaceutically acceptable carrier.
- the compounds of this invention may be used in pharmaceutical compositions that include one or more other active pharmaceutical ingredients
- the compounds of this invention may also be used in pharmaceutical compositions in which the compound of Formula I or a pharmaceutically acceptable salt thereof is the only active ingredient.
- a compound of Formula I, or a pharmaceutically acceptable salt thereof may be used in the manufacture of a medicament for the treatment of type 2 diabetes melhtus in a human or other mammalian patient.
- a method of treating type 2 diabetes comprises the administration of a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising the compound, to a patient in need of treatment.
- Other medical uses of the compounds of Formula I are described hereinafter.
- Alkyl means saturated carbon chains which may be linear or branched or combinations thereof, unless the carbon chain is defined otherwise.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, and the like.
- Alkenyl means carbon chains which contain at least one carbon-carbon double bond, and which may be linear or branched or combinations thereof.
- alkenyl examples include vinyl, allyl, isopropenyl, pentenyl, hexenyl, heptenyl, 1-propenyl, 2-butenyl, 2-methyl-2-butenyl, and the like.
- Alkynyl means carbon chains which contain at least one carbon-carbon triple bond, and which may be linear or branched or combinations thereof. Examples of alkynyl include ethynyl, propargyl, 3-methyl-l-pentynyl, 2-heptynyl and the like.
- Cycloalkyl means a saturated carbocyclic ring, having a specified number of carbon atoms. The term may also be used to describe a carbocyclic ring fused to an aryl group. Examples of cycloalkyl include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like. Cycloalkenyl rings comprise a double bond in the ring.
- Aryl is commonly used to refer to carbocyclic aromatic structures. The most common aryl groups are phenyl and naphthyl. Phenyl is generally the most preferred aryl group.
- Heterocycle means a fully or partially saturated ring or ⁇ ng system containing at least one heteroatom selected from N, S and O, wherein the number of heteroatoms and the ⁇ ng size are defined herein.
- heterocycles include tetrahydrofuran, piperazme, pipe ⁇ dme, and morphohne.
- Heteroaryl means an aromatic ring or two fused aromatic rings containing at least one ring heteroatom selected from N, O and S (including SO and SO2), as defined more specifically herein.
- heteroaryl examples include pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, lmidazolyl, t ⁇ azolyl, tetrazolyl, furanyl, triazinyl, thienyl, py ⁇ midyl, pyridazinyl, pyrazmyl, benzisoxazolyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl (including S-oxide and dioxide), furo(2,3-b)pyridyl, quinolyl, indolyl, isoquinolyl, quinazohnyl, dibenzofuranyl, and the like.
- Halogen includes fluorine, chlorine, bromine and iodine
- Me represents methyl
- composition as in pharmaceutical composition, is intended to encompass a product comp ⁇ sing the active mgredient(s), and the inert ⁇ ngredient(s) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable earner.
- the substituent "tetrazole” means a 2H-tetrazol-5-yl substituent group and tautomers thereof.
- Optical Isomers - Diastereomers - Geometric Isomers - Tautomers Compounds of Formula I may contain one or more asymmetric centers and can thus occur as racemates, racemic mixtures, single enantiomers, individual diastereomers, and mixtures of diastereomers and/or enantiomers.
- the present invention is meant to comprehend all such isomeric forms of the compounds of Formula I.
- the compounds of the instant invention have at least one asymmetric center, which is on the ⁇ ng that is fused to the phenyl ring at the point where the heterocyclic ring is attached. There may also a second asymmetric center in the heterocyclic ring.
- asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers, and it is intended that all of the possible optical isomers, stereoisomers, and diastereomers in mixtures and as pure or partially purified compounds are included within the scope of this invention (i.e. all possible combinations of the asymmetric centers as pure compounds or in mixtures).
- Some of the compounds described herein may contain olef ⁇ nic double bonds, and unless specified otherwise, are meant to include both E and Z geometric isomers.
- keto-enol tautomers Some of the compounds described herein may exist with different points of attachment of hydrogen, referred to as tautomers.
- An example is a ketone and its enol form, known as keto-enol tautomers.
- keto-enol tautomers The individual tautomers as well as mixtures thereof are encompassed with compounds of Formula I.
- enantiomers and other compounds with chiral centers may be synthesized by stereospecific synthesis using optically pure starting materials and/or reagents of known configuration.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts denved from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'- dibenzylethylenediamine, diethylamme, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morphohne, N-ethylpipe ⁇ dme, glucamme, glucosamine, histidine, hydrabamine, lsopropylamme, lysine, methylglucamme, morphohne, piperazme, pipe ⁇ dme, polyamine resms, procaine, purines, theobromine, triethylamine, trimethylamine, t ⁇ propylamine, tromethamme, and the like.
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfomc, benzoic, camphorsulfonic, citric, ethanesulfonic, fuma ⁇ c, gluconic, glutamic, hydrobromic, hydrochloric, lsethionic, lactic, maleic, malic, mandehc, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfu ⁇ c, tartaric, p-toluenesulfonic acid, and the like.
- Particularly preferred are citric, hydrobromic, hydrochlo ⁇ c, maleic, phosphoric, sulfuric, and tartaric acids.
- Therapeutically active metabolites where the metabolites themselves fall within the scope of the claimed invention, are also compounds of the current invention.
- Prodrugs which are compounds that are converted to the claimed compounds as they are being administered to a patient or after they have been administered to a patient, are compounds of this invention
- the claimed chemical structures of this application in some cases may themselves be prodrugs.
- Compounds of the present invention are potent agonists of the GPR40 receptor.
- the compounds of the invention, and pharmaceutically acceptable salts thereof, may be efficacious m the treatment of diseases that are modulated by GPR40 hgands and agonists. Many of these diseases are summarized below.
- One or more of the following diseases may be treated by the administration of a therapeutically effective amount of a compound of this invention, or a pharmaceutically acceptable salt thereof, to a patient m need of treatment
- the compounds of the invention may be used for the manufacture of a medicament for treating one or more of these diseases:
- non-insulin dependent diabetes melhtus type 2 diabetes
- hypertriglyceridemia (elevated levels of t ⁇ glycende- ⁇ ch-hpoproteins); (7) mixed or diabetic dyshpidemia;
- Preferred uses of the compounds are for the treatment of one or more of the following diseases by administering a therapeutically effective amount to a patient m need of treatment.
- the compounds may be used for manufacturing a medicament for the treatment of one or more of these diseases: (1) Type 2 diabetes, and specifically hyperglycemia;
- the compounds are expected to be effective in lowering glucose and lipids in diabetic patients and m non-diabetic patients who have impaired glucose tolerance and/or are m a pre -diabetic condition.
- the compounds may ameliorate hype ⁇ nsulinemia, which often occurs in diabetic or pre- diabetic patients, by modulating the swings in the level of serum glucose that often occurs in these patients.
- the compounds may also be effective m treating or reducing insulin resistance.
- the compounds may be effective in treating or preventing gestational diabetes.
- the compounds, compositions, and medicaments as described herein may also be effective in reducing the risks of adverse sequelae associated with metabolic syndrome, and in reducing the risk of developing atherosclerosis, delaying the onset of atherosclerosis, and/or reducing the risk of sequelae of atherosclerosis.
- Sequelae of atherosclerosis include angina, claudication, heart attack, stroke, and others
- the compounds may also be effective in delaying or preventing vascular restenosis and diabetic retinopathy
- the compounds of this invention may also have utility in improving or restoring /S-cell function, so that they may be useful in treating type 1 diabetes or in delaying or preventing a patient with type 2 diabetes from needing insulin therapy
- the compounds generally will be efficacious in treating one or more of the following diseases: (1) type 2 diabetes (also known as non-insulin dependent diabetes mellitus, or NIDDM), (2) hyperglycemia, (3) impaired glucose tolerance, (4) insulin resistance, (5) obesity, (6) lipid disorders, (7) dyshpidemia, (8) hyperlipidemia, (9) hypertriglyceridemia, (10) hypercholesterolemia, (11) low HDL levels, (12) high LDL levels, (13) atherosclerosis and its sequelae, (14) vascular restenosis, (15) abdominal obesity, (16) retinopathy, (17) metabolic syndrome, (18) high blood pressure, and (19) insulin resistance.
- type 2 diabetes also known as non-insulin dependent diabetes mellitus, or NIDDM
- hyperglycemia also known as non-insulin dependent
- One aspect of the invention provides a method for the treatment and control of mixed or diabetic dyslipidemia, hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperhpidemia, and/or hypertriglyceridemia, which comprises administering to a patient in need of such treatment a therapeutically effective amount of a compound having formula I.
- the compound may be used alone or advantageously may be administered with a cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor such as lovastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, atorvastatin, ⁇ vastatm, itavastatin, or ZD-4522.
- the compound may also be used advantageously m combination with other lipid lowering drugs such as cholesterol absorption inhibitors (for example stanol esters, sterol glycosides such as tiqueside, and azetidinones such as ezetimibe), ACAT inhibitors (such as avasimibe), CETP inhibitors (for example torcetrapib), niacin and niacin receptor agonists, bile acid sequestrants, microsomal triglyceride transport inhibitors, and bile acid reuptake inhibitors.
- cholesterol absorption inhibitors for example stanol esters, sterol glycosides such as tiqueside, and azetidinones such as ezetimibe
- ACAT inhibitors such as avasimibe
- CETP inhibitors for example torcetrapib
- niacin and niacin receptor agonists for example torcetrapib
- bile acid sequestrants for example torcetrapi
- Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dose of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compounds of Formula I are administered orally.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the seventy of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art.
- a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 milligrams to about 1000 milligrams.
- the total daily dose will generally be from about 1 milligram to about 500 milligrams.
- the dosage for an adult human may be as low as 0.1 mg.
- the dosage regimen may be adjusted within this range or even outside of this range to provide the optimal therapeutic response.
- Oral administration will usually be carried out using tablets or capsules. Examples of doses in tablets and capsules are 0.1 mg, 0.25 mg, 0.5 mg, 1 mg, 2 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, and 500 mg. Other oral forms may also have the same or similar dosages.
- compositions which comprise a compound of Formula I and a pharmaceutically acceptable carrier.
- the pharmaceutical compositions of the present invention comprise a compound of Formula I or a pharmaceutically acceptable salt as an active ingredient, as well as a pharmaceutically acceptable earner and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- a pharmaceutical composition may also comprise a prodrug, or a pharmaceutically acceptable salt thereof, if a prodrug is administered.
- compositions include compositions suitable for oral, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ocular (ophthalmic), pulmonary (nasal or buccal inhalation), or nasal administration, although the most suitable route in any given case will depend on the nature and seventy of the conditions being treated and on the nature of the active ingredient They may be conveniently presented m unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
- the compounds of Formula I can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the earner may take a wide variety of forms depending on the form of preparation desired for administration, e g., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions, or carriers such as starches, sugars, microcrystallme cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations.
- oral liquid preparations such as, for example, suspensions, elixirs and solutions, or carriers such as starches, sugars, microcrystallme cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations
- tablets and capsules represent the most advantageous oral dosage unit form m which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques. Such compositions and preparations should contain at least 0.1 percent of active compound. The percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2 percent to about 60 percent of the weight of the unit. The amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained.
- the active compounds can also be administered intranasally as, for example, liquid drops or spray.
- the tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, algmic acid; a lubncant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- the compound or salt may be advantageous to formulate the compound or salt as a solution in an oil such as a triglyceride of one or more medium chain fatty acids, a lipophilic solvent such as t ⁇ acetm, a hydrophilic solvent (e.g. propylene glycol), or a mixture of two or more of these, also optionally including one or more ionic or nomonic surfactants, such as sodium lauryl sulfate, polysorbate 80, polyethoxylated triglycerides, and mono and/or diglyce ⁇ des of one or more medium chain fatty acids.
- an oil such as a triglyceride of one or more medium chain fatty acids, a lipophilic solvent such as t ⁇ acetm, a hydrophilic solvent (e.g. propylene glycol), or a mixture of two or more of these, also optionally including one or more ionic or nomonic surfactants, such as sodium lauryl sulfate, polysorbate 80, polyeth
- Solutions containing surfactants will form emulsions or microemulsions on contact with water.
- the compound may also be formulated in a water soluble polymer in which it has been dispersed as an amorphous phase by such methods as hot melt extrusion and spray drying, such polymers including HPMCAS, HPMCS, and polyvmylpyrrolidinones.
- Va ⁇ ous other mate ⁇ als may be present as coatings or to modify the physical form of the dosage unit.
- tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor
- Compounds of formula I may also be administered parenterally. Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxypropylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy sy ⁇ ngabihty exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- Compounds of Formula I may be used m combination with other drugs that may also be useful m the treatment or amelioration of the diseases or conditions for which compounds of Formula I are useful.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I.
- more than one drug is commonly administered.
- the compounds of this invention may generally be administered to a patient who is already taking one or more other drugs for these conditions.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of Formula I is preferred.
- the combination therapy also includes therapies in which the compound of Formula I and one or more other drugs are administered on different overlapping schedules. It is also contemplated that when used in combination with one or more other active ingredients, the compound of the present invention and the other active ingredients may be used in lower doses than when each is used singly. Accordingly, the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of Formula I.
- Examples of other active ingredients that may be administered in combination with a compound of Formula I, and either administered separately or in the same pharmaceutical composition include, but are not limited to:
- PPAR gamma agonists and partial agonists including both ghtazones and non- ghtazones (e.g. troghtazone, pioghtazone, enghtazone, MCC-555, rosiglitazone, balaghtazone, netoghtazone, T-131, LY-300512, and LY-818;
- biguanides such as metformin and phenformin;
- PTP-IB protein tyrosine phosphatase- IB
- D dipeptidyl peptidase IV (DP-FV) inhibitors, such as sitaghptin, saxaghptin, and vildaghptin;
- D-FV dipeptidyl peptidase IV
- sulfonylureas such as tolbutamide, glimepi ⁇ de, glipizide, and related materials
- ⁇ -glucosidase inhibitors such as acarbose
- agents which improve a patient's lipid profile such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, atorvastatin, ⁇ vastatin, itavastatin, ZD-4522 and other statins), ( ⁇ ) bile acid sequestrants (cholestyramine, colestipol, and dialkylammoalkyl derivatives of a cross-lmked dextran), (in) niacin receptor agonists, nicotmyl alcohol, nicotinic acid, or a salt thereof, (iv) PP ARa agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (v) cholesterol absorption inhibitors, such as for example ezetimibe, (vi) acyl CoA:cholesterol acy
- antiobesity compounds such as fenfluramine, dexfenfluramine, phentiramine, subitramine, orhstat, neuropeptide Y5 inhibitors, Mc4r agonists, cannabmoid receptor 1 (CB-I) antagonists/inverse agonists, and ⁇ 3 adrenergic receptor agonists; (1) ileal bile acid transporter inhibitors;
- agents intended for use in inflammatory conditions such as aspinn, non-steroidal anti-inflammatory drugs, glucocorticoids, azulfidine, and cyclo-oxygenase 2 selective inhibitors;
- agents intended for use in inflammatory conditions such as aspinn, non-steroidal anti-inflammatory drugs, glucocorticoids, azulfidine, and cyclo-oxygenase 2 selective inhibitors;
- glucagon receptor antagonists such as aspinn, non-steroidal anti-inflammatory drugs, glucocorticoids, azulfidine, and cyclo-oxygenase 2 selective inhibitors;
- GLP-I analogs such as exendms, for example exenatide (Byetta)
- HSD-I Hydroxysterol dehydrogenase- 1
- Non- hmiting examples include combinations of compounds having Formula I with two or more active compounds selected from biguanides, sulfonylureas, HMG-CoA reductase inhibitors, other PPAR agonists, PTP-IB inhibitors, DP-IV inhibitors, and anti-obesity compounds.
- Human and mouse GPR40 stable cell-lmes were generated in CHO cells stably expressing NFAT BLA (Beta-lactamase).
- a human GPR40 stable cell-line was generated in HEK cells stably expressing the aequorm expressing reporter.
- the expression plasmids were transfected using lipofectamine (Life Technologies) following manufacturer's instructions. Stable cell-lines were generated following drug selection.
- FLIPR Fluorescence output was measured using FLIPR.
- Compounds were dissolved in DMSO and diluted to desired concentrations with assay buffer 13.3 ⁇ l/well of compound solution was added.
- EC50 activities were measured for the compounds using both the human and mouse GPR40 cell lines.
- the activities for the compounds herein are as follows: human FLEPR EC50: 0.033 to 40 ⁇ M; mouse FLIPR EC50: 0.051 to 40 ⁇ M.
- the preferred compounds have an EC50 ⁇ 10 ⁇ M.
- the assay is performed in 96-well format.
- HEK cells stably expressing human GPR40 are plated to be 60-80% confluent within 72 hours. After 72 hours, the plates are aspirated and the cells washed with lnositol-free DMEM (ICN).
- the wash media is replaced with 15OuL of 3H-inositol labeling media (lnositol-free media containing 0.4% human albumin or 0.4% mouse albumin, IX pen/strep antibiotics, glutamme, 25mM HEPES to which is added 3H-myo-inositol NEN #NET114A lmCi/mL, 25Ci/mmol diluted 1 :150 in loading media with a final specific radioactivity of 1UCI/150UL).
- the human and mouse albumin can be added after the overnight labeling step before the addition of LiCl.
- the assay is typically run the next day after 18 hours labeling. On the day of the assay,
- step B The product from step B was mixed with 50 mL of 2N aq. HCl and 50 mL of ethanol. The mixture was refluxed overnight (monitored by LC-MS until a complete conversion was observed). After removal of ethanol under vacuum, the residue was extracted with ethyl acetate, d ⁇ ed over anhydrous sodium sulfate, evaporated and dried under high vacuum to afford a yellow solid.
- the (S,R) salt from the above step B (23.2 g) was stirred for one hour with 200 mL of 3N HCl and 200 mL of ethyl acetate. The organic phase was separated and washed with 3N aq. HCl (2 x 100 mL), dried over sodium sulfate, filtered and evaporated to give the desired (S)-acid as a light brown solid.
- the (S)-acid from the above step D (14 g) was dissolved in 150 mL of ethanol, and 19 mL of trimethylsilyl chloride was added. The mixture was stirred at RT overnight, and was then evaporated and mixed with ethyl acetate (100 mL). The organic phase was washed with water and saturated aq. sodium hydrogen carbonate, dried over sodium sulfate, and purified on FC (Silica gel, 20% ethyl acetate/hexane) to give the desired (S)-ester as a colorless oil.
- FC Silica gel, 20% ethyl acetate/hexane
- step F The product from the above step F was mixed with 50 mL of 4N aq. HCl and 50 mL of ethanol. The mixture was refluxed overnight (monitored by LC-MS until a complete conversion was observed). After removal of ethanol under vacuum, the residue was extracted with ethyl acetate, dried over anhydrous sodium sulfate, evaporated, and dried under high vacuum to afford a yellow solid.
- step D The product from the above step D was mixed with 50 mL of 4N aq. HCl and 50 mL of ethanol. The mixture was refluxed overnight (monitored by LC-MS until a complete conversion was observed). After removal of ethanol under vacuum, the residue was extracted with ethyl acetate, dried over anhydrous sodium sulfate, and evaporated and dried under high vacuum to afford a yellow solid.
- LC-MS calc. for C14H15NO3S: 277 Found: 278 (M+H).
- reaction was stirred for an additional 30 min., then the reaction vessel was gradually warmed to room temperature. Solvent was then removed in vacuo (roto-evaporation) and then ca. 75 mL of pentane was added to the residue. Rapid filtration and removal of solvent in vacuo yielded crude alkyl trimethyl ketene acetal.
- step E To the crude material from step E ( ⁇ 20 mmol) in 100 mL of EtOH was added thiourea (M. W. 76.12, 1.979 g, 26 mmol) and sodium acetate (M.W. 82.03, 3.281 g, 40 mmol). The reaction was refluxed overnight. The organic solvent was removed in vacuo and the residue was partitioned between 50 mL of 6 N HCl (aq) and 50 mL of EtOAc. The organic layer was further extracted with 6 N HCl (aq) (2 x 25 mL). The aqueous layers were combined and further washed with EtOAc (1 x 10 mL).
- the INTERMEDIATE 7 was prepared according to the same procedures as in the preparation of INTERMEDIATE 5 by replacing ethyl [(lS)-5-methoxy-2,3-dihydro-lH-inden-l-yl]acetate with the product of Step D in INTERMEDIATE 2.
- LC-MS for C 13 H 13 NO 4 calculated 247.1 , found 246.1 [M-H].
- This material was prepared according to the procedure used in the Steps A - E in the preparation of INTERMEDIATE 6, using 7-methoxythiochroman-4-one (prepared according to the procedure used in Tetrahedron, 1970, 26(10), 2353-2363) as the starting material. This crude material was used without further purification.
- INTERMEDIATE 8 was prepared from material obtained in the above Step A using a similar procedure to that used in Steps B-D in the preparation of INTERMEDIATE 1.
- LC-MS for C 12 HnNO 3 S 2 calculated 281.02, found 280 [M-H].
- Example 47 was prepared following a procedure similar to the one used in the preparation of Example 46, replacing 4-chloro-7-(trifluoromethyl)quinoline with 4,7-dichloroquinoline.
- LC-MS calc. for C20H14C1N3O4: 395; Found: 396 (M+H).
Abstract
Biaryl ethers in which one of the aromatic πngs is fused to a cycloalkyl or heterocyclic πng, which is attached to a thiazohdinedione or oxazohdmedione πng, including pharmaceutically acceptable salts and prodrugs thereof, are agonists of G-protein coupled receptor 40 (GPR40) and are useful as therapeutic compounds, particularly in the treatment of Type 2 diabetes melhtus, and of conditions that often occur with type 2 diabetes, including insulin resistance, obesity, and lipid disorders.
Description
TITLE OF THE INVENTION ANTIDIABETIC BICYCLIC COMPOUNDS
FIELD OF THE INVENTION The instant invention is concerned with biaryl ethers in which one of the aromatic rings is fused to a cycloalkyl or heterocyclic ring, which is attached to a thiazohdinedione or oxazohdinedione ring. The compounds are agonists of G-protem coupled receptor 40 (GPR40) and are useful as therapeutic compounds, particularly in the treatment of Type 2 diabetes mellitus, and of conditions that are often associated with this disease, including insulin resistance, obesity, and lipid disorders.
BACKGROUND OF THE INVENTION
Diabetes is a disease derived from multiple causative factors and characterized by elevated levels of plasma glucose (hyperglycemia) in the fasting state or after administration of glucose during an oral glucose tolerance test. There are two generally recognized forms of diabetes. In type 1 diabetes, or insulin-dependent diabetes mellitus (DDDM), patients produce little or no insulin, the hormone which regulates glucose utilization. In type 2 diabetes, or noninsuhn-dependent diabetes mellitus (NIDDM), insulin is still produced in the body. Patients having type 2 diabetes have a resistance to the effects of insulin in stimulating glucose and lipid metabolism in the main msulm-sensitive tissues, which are muscle, liver and adipose tissues These patients often have normal levels of insulin, and may have hypeπnsulinemia (elevated plasma insulin levels), as they compensate for the reduced effectiveness of insulin by secreting increased amounts of insulin Insulin resistance is not primarily caused by a diminished number of insulin receptors but rather by a post-insulin receptor binding defect that is not yet completely understood. This lack of responsiveness to insulin results in insufficient insulin-mediated activation of uptake, oxidation and storage of glucose in muscle, and inadequate insulin-mediated repression of hpolysis in adipose tissue and of glucose production and secretion in the liver.
Persistent or uncontrolled hyperglycemia that occurs with diabetes is associated with increased and premature morbidity and mortality. Often abnormal glucose homeostasis is associated both directly and indirectly with obesity, hypertension, and alterations of the lipid, lipoprotein and apohpoprotem metabolism, as well as other metabolic and hemodynamic disease Patients with type 2 diabetes mellitus have a significantly increased risk of macrovascular and microvascular complications, including atherosclerosis, coronary heart disease, stroke, peripheral vascular disease, hypertension, nephropathy, neuropathy, and retinopathy. Therefore, therapeutic control of glucose homeostasis, lipid metabolism, obesity, and hypertension are critically important m the clinical management and treatment of diabetes mellitus Patients who have insulin resistance often have several symptoms that together are referred to as syndrome X, or the metabolic syndrome. According to one widely used definition, a patient having metabolic syndrome is characterized as having three or more symptoms selected from the
following group of five symptoms: (1) abdominal obesity; (2) hypertriglyceridemia; (3) low high-density lipoprotein cholesterol (HDL); (4) high blood pressure; and (5) elevated fasting glucose, which may be in the range characteristic of Type 2 diabetes if the patient is also diabetic. Each of these symptoms is defined clinically m the Third Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III, or ATP III), National Institutes of Health, 2001, NIH Publication No. 01-3670. Patients with metabolic syndrome, whether or not they have or develop overt diabetes mellitus, have an increased risk of developing the macrovascular and microvascular complications that occur with type 2 diabetes, such as atherosclerosis and coronary heart disease. There are several available treatments for type 2 diabetes, each of which has its own limitations and potential risks. Physical exercise and a reduction in dietary intake of calories often dramatically improve the diabetic condition and are the usual recommended first-line treatment of type 2 diabetes and of pre-diabetic conditions associated with insulin resistance. Compliance with this treatment is very poor because of well-entrenched sedentary lifestyles and excess food consumption, especially of foods containing high amounts of fat and carbohydrates. Pharmacologic treatments have focused on three areas of pathophysiology: (1) Hepatic glucose production (biguanides), (2) insulin resistance (PPAR agonists), and (3) insulin secretion.
The biguanides are a class of drugs that are widely used to treat type 2 diabetes. The two best known biguanides, phenformin and metformin, cause some correction of hyperglycemia. The biguanides act primarily by inhibiting hepatic glucose production, and they also are believed to modestly improve insulin sensitivity The biguanides can be used as monotherapy or m combination with other anti-diabetic drugs, such as insulin or an insulin secretagogues, without increasing the risk of hypoglycemia. However, phenformin and metformin can induce lactic acidosis and nausea/diarrhea Metformin has a lower risk of side effects than phenformin and is widely prescribed for the treatment of Type 2 diabetes.
The ghtazones (i e 5-benzylthiazohdine-2,4-diones) are a newer class of compounds that can ameliorate hyperglycemia and other symptoms of type 2 diabetes. The ghtazones that are currently marketed (rosightazone and pioghtazone) are agonists of the peroxisome prohferator activated receptor (PPAR) gamma subtype. The PPAR-gamma agonists substantially increase insulin sensitivity in muscle, liver and adipose tissue in several animal models of type 2 diabetes, resulting in partial or complete correction of elevated plasma glucose levels without the occurrence of hypoglycemia. PPAR-gamma agonism is believed to be responsible for the improved insulin sensitization that is observed in human patients who are treated with the ghtazones. New PPAR agonists are currently being developed. Many of the newer PPAR compounds are agonists of one or more of the PPAR alpha, gamma and delta subtypes. Compounds that are agonists of both the PPAR alpha and PPAR gamma subtypes (PPAR alpha/gamma dual agonists) are promising because they reduce hyperglycemia and also improve lipid metabolism. The currently marketed PPAR gamma agonists are modestly effective in reducing plasma
glucose and HemoglobmAlC. The currently marketed compounds do not greatly improve lipid metabolism and may actually have a negative effect on the lipid profile. Thus, the PPAR compounds represent an important advance in diabetic therapy, but further improvements are still needed.
Another widely used drug treatment involves the administration of insulin secretagogues, such as the sulfonylureas (e g. tolbutamide and glipizide). These drugs increase the plasma level of insulin by stimulating the pancreatic β-cells to secrete more insulin. Insulin secretion m the pancreatic β-cell is under strict regulation by glucose and an array of metabolic, neural and hormonal signals. Glucose stimulates insulin production and secretion through its metabolism to generate ATP and other signaling molecules, whereas other extracellular signals act as potentiators or inhibitors of insulin secretion through GPCR's present on the plasma membrane. Sulfonylureas and related insulin secretagogues act by blocking the ATP-dependent K+ channel in /3-cells, which causes depolarization of the cell and the opening of the voltage-dependent Ca2+ channels with stimulation of insulin release. This mechanism is non-glucose dependent, and hence insulin secretion can occur regardless of the ambient glucose levels. This can cause insulin secretion even if the glucose level is low, resulting in hypoglycemia, which can be fatal in severe cases. The administration of insulin secretagogues must therefore be carefully controlled. The insulin secretagogues are often used as a first-line drug treatment for Type 2 diabetes.
There has been a renewed focus on pancreatic islet-based insulin secretion that is controlled by glucose-dependent insulin secretion. This approach has the potential for stabilization and restoration of /3-cell function. In this regard, several orphan G-protem coupled receptors (GPCR's) have recently been identified that are preferentially expressed in the /3-cell and that are implicated m glucose stimulated insulin secretion (GSIS). GPR40 is a cell-surface GPCR that is highly expressed in human (and rodent) islets as well as in insulin-secreting cell lines Several naturally-occurring medium to long- chain fatty acids (FA's) as well as synthetic compounds, including several members of the thiazohdinedione class of PPARγ agonists, have recently been identified as ligands for GPR40 (Itoh, Y. et al., Nature 422: 173 [2003], Bπscoe, CP. et al., J. Biol. Chem. 278: 11303 [2003], Kotarsky, K. et al., Biochem. Biophys. Res Comm 301: 406 [2003]. Under hyperglycemic conditions, GPR40 agonists are capable of augmenting the release of insulin from islet cells. The specificity of this response is suggested by results showing that the inhibition of GPR40 activity by siRNA attenuates FA-induced amplification of GSIS These findings indicate that, in addition to the intracellular generation of hpid-deπvatives of FA's that are thought to promote insulin release, FA's (and other synthetic GPR40 agonists) may also act as extracellular ligands that bind to GPR40 in mediating FA-induced insulin secretion. There are several potential advantages of GPR40 as a potential target for the treatment of type 2 diabetes. First, since GPR40-mediated insulin secretion is glucose dependent, there is little or no πsk of hypoglycemia. Second, the limited tissue distribution of GPR40 (mainly in islets) suggests that there would be less chance for side effects associated with GPR40 activity in other tissues. Third, GPR40 agonists that are active m the islets may have the potential to restore or preserve islet function. This would be highly
advantageous, because long term diabetes therapy often leads to the gradual diminution of islet activity, so that after extended periods of treatment, it is often necessary to treat type 2 diabetic patients with daily insulin injections. By restoring or preserving islet function, GPR40 agonists may delay or prevent the diminution and loss of islet function in a type 2 diabetic patient.
SUMMARY OF THE INVENTION
The class of compounds described herein is a new class of GPR40 agonists. The compounds are useful in the treatment of diseases that are modulated by GPR40 agonists, including type 2 diabetes, hyperglycemia that may be associated with type 2 diabetes or pre-diabetic insulin resistance, and obesity.
The present invention is directed to a compound of formula I, or a pharmaceutically acceptable salt thereof, including individual diastereomers and enantiomers thereof, and mixtures of diastereomers and/or enantiomers thereof:

I In the compound of formula I, A is selected from the group consisting of -CH- and -N-;
B is selected from the group consisting of -S-, -O-, -NH-, and -CH2-,
W is selected from the group consisting of -CH2-, -CF2-, -O-, -C(=O)-, -NR.6-, -S-, -S(O)-, and -S(O)2-;
Z is selected from the group consisting of -CH2- and -CH2CH2-;
or alternatively -W-Z- is selected from the group consisting of -N(R6)C(=O)- and -C(=O)N(R6)-, or -W-Z- represents two atoms that are connected to form one side of a 5-6-membered heteroaromatic πng having 1-3 heteroatoms independently selected from O, N and S, where the 5- membered heteroaromatic πng is optionally substituted with 1-3 groups independently selected from halogen, CH3, CF3, -OCH3, and -OCF3;
Heterocycle is a 5-6 membered saturated or partly saturated monocyclic heterocyclic πng having 1-3 heteroatoms independently selected from O, N and S;
Heteroaryl is a 5-6 membered monocyclic heteroaromatic ring having 1-3 heteroatoms independently selected from O, N and S;
R.1, R^, R3 and R^ are each independently selected from the group consisting of H, halogen, -CN, -NO2, -Q-Coalkyl, -OCi-Cβalkyl, -SQ-Coalkyl, -S(O)2Ci-C6alkyl, -N(R6)(R6), -N(R6)C(=O)Ci-C6alkyl, -N(R6)S(O)2Ci-C6alkyl, -S(O)2N(R6)(R6), -C(O)H, -C(O)OH, -C(O)OC i-Cβalkyl, -C(O)C i-Cβalkyl, -C(=O)N(R6)(R6), -C(O)Phenyl, -C(=O)Naphthyl, -C(=O)Heterocyle, Heterocycle, Heteroaryl, C3-C7-Cycloalkyl, Phenyl and Naphthyl; wherein -C]-C6alkyl and the alkyl groups of -0Ci-C6alkyl, -SQ-Cόalkyl,
-S(O)2Ci-C6alkyl, -N(R6)C(=O)Ci-C6alkyl, -N(R6)S(O)2Ci-C6alkyl, -C(=0)0Ci-C6alkyl, and -C(=0)Ci-C6alkyl are optionally substituted with 1-5 halogens and optionally substituted with 1-2 groups independently selected from -OH, -OCi-Cβalkyl which is optionally substituted with 1-5 halogens, -S(O)2C]-C3alkyl which is optionally substituted with 1-5 halogens, -C(=0)Ci-C3alkyl which is optionally substituted with 1-5 halogens, -0C(=0)Ci-C6alkyl which is optionally substituted with 1-5 halogens, -NHC(=0)CH3, -NHC(=0)0Ci-C6alkyl which is optionally substituted with 1-5 halogens, -NHS(O)2CH3, -N(R6XR6), Heterocycle, Heteroaryl, C3-C7-Cycloalkyl, Phenyl, and Naphthyl; wherein -C(=O)Phenyl, -C(=O)Naphthyl, -C(=O)Heterocyle, Heterocycle, Heteroaryl, C3-C7-Cycloalkyl, Phenyl and Naphthyl either as Rl, R2, R3, Or R4, or as substituents on Rl, R2, R3 and R4 are optionally substituted with 1-4 substituents independently selected from halogen, -CF3, -OCF3, -CN, -NO2, -OH, -Ci-C3alkyl, -C(O)Ci-C3alkyl, -S(O)2Ci-C3alkyl, and -0Ci-C3alkyl, wherein said -Ci-C3alkyl, -0Ci-C3alkyl, -S(O)2Ci-C3alkyl, and -C(=0)Ci-C3alkyl substituents are optionally substituted with 1-3 halogens; wherein the pair of substituents R^ and R2 may together represent a bridging divalent 4- carbon chain -CH=CH-CH=CH-, forming a fused phenyl ring at the Rl and R2 positions, wherein said fused phenyl ring is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Ci-C3alkyl, -0Ci-C3alkyl, -SCi-C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3; or alternatively the pair of substituents R^ and R2 may together represent a bridging divalent 3 -atom chain selected from -CH=CHO-, -OCH=CH-, -CH=CH-S-, and -SCH=CH-, forming a fused furan or thiophene ring at the Rl and R2 positions, wherein said fused furan or thiophene ring is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Q- C3alkyl, -OCi-Csalkyl, -SCi-C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3; and
Each R6 is independently selected from the group consisting of H and -Cj-Cβalkyl.
In the above description, the optional bπdging groups as drawn that connect pairs of groups R land R2 are attached to the ring with the left-hand side of the structure attached where Rl is attached and the right-hand side of the structure attached where R2 IS attached.
In the description above and in subsequent description below, alkyl, alkenyl, and alkynyl groups can each be linear or branched, unless otherwise defined.
DETAILED DESCRIPTION OF THE INVENTION
The invention has numerous embodiments, which are summarized below. The invention includes the specific compounds as shown, and also includes individual diastereomers, enantiomers, and epimers of the compounds, and mixtures of diastereomers and/or enantiomers thereof. The invention also includes pharmaceutically acceptable salts of the compounds, and pharmaceutical compositions composing the compounds and a pharmaceutically acceptable carrier. The compounds are useful m treating insulin resistance, type 2 diabetes, and dyshpidemia that is associated with type 2 diabetes and insulin resistance.
Subgroups of the compounds of Formula I compπse compounds, including pharmaceutically acceptable salts thereof, in which R^, R2, R3, and R4 are independently selected from (I) H; (2) halogen; (3) -NO2; (4) -CN; (5) Ci -C4alkyl, which is optionally substituted with 1-5 halogens and optionally with 1-2 substituents independently selected from -OH, -C(=0)Ci-C3alkyl, and - OCi-C3alkyl, where the Cl-C3alkyl groups are optionally substituted with 1-3 halogens; (6) -OCi- C4alkyl, which is optionally substituted with 1-5 halogens and optionally with one -C(=O)Ci-C3 alkyl group; (7) -C(O)C i-C3alkyl, which is optionally substituted with 1-5 halogens; (8) -C(=0)H; (9) -C(=O)N(R6)(R6); (10) -S(O)2N(R6)(R6); (l l) -S(O)2Ci-C3alkyl; (12) -N(R6)(R6); (I3) C3- C7Cycloalkyl; (14) Phenyl; and (15) Heterocycle; wherein C3.C7Cycloalkyl, Phenyl, and Heterocycle are each optionally substituted with 1-3 substituents independently selected from halogen, -OH, -OCi- C3alkyl, CF3, and -C(=O)Ci-C3alkyl.
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof, each R" is independently selected from H and CH3
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof, R1, R2, R3, and R4 are each independently selected from H, halogen, Ci-C3alkyl, CF3,
-OCi-C3alkyl, -OCF3, -C(=O)H, -C(=O)N(R6)(R6), -S(O)2N(R6)(R6), -C(=O)Ci -C3 alkyl, -CN, -NO2, -S(O)2C i-C3alkyl, and -N(R6)(R6).
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof, R1 , R2, R3, and R4 are each independently selected from H, F, Br, Cl, Ci-C3alkyl, CF3, -C(=0)NH2, -S(O)2NH2, -C(=O)CH3, -CN, -OCH3, -OCF3, and -NO2.
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof, the pair of ortho substituents R^ and R2 together represent a bridging divalent 4-carbon chain -CH=CH-CH=CH-, forming a fused phenyl ring at the Rl and R2 positions, wherein the fused phenyl ring is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -Nθ2, -Ci-C3alkyl, -OCi-Csalkyl, -SCi-C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3. In subgroups of the compounds of Formula I described immediately above, including pharmaceutically acceptable salts thereof, the fused phenyl ring at the Rl and R2 positions is optionally substituted with 1-2 substituents independently selected from halogen, -NO2, -CH3, -CF3, -OCH3, and -OCF3.
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof, R^ and R2 together represent a bridging divalent group selected from -CH=CHS-, -SCH=CH-, -CH=CHO-, and -OCH=CH-, forming a fused furan or thiophene at the Rl and R2 positions, wherein said fused furan or thiophene πng is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Ci-C3alkyl, -0Ci-C3alkyl, -SCi-C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3. In subgroups of the compounds of Formula I described immediately above, including pharmaceutically acceptable salts thereof, the fused furan or thiophene ring at the Rl and R2 positions is optionally substituted with 1-2 substituents independently selected from halogen, -NO2, -CH3, -CF3, -OCH3, and -OCF3.
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof, A is CH- or -N-; and B is selected from -O- and -S-.
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof, W is selected from the group consisting of -CH2-, -CF2-, -0-, -C(=0)-, -NR6-, -S-,
-S(O)-, and -S(0)2-; and
Z is selected from the group consisting of -CH2- and -CH2CH2-.
In subgroups of the compounds of Formula I, including pharmaceutically acceptable salts thereof,
W is selected from the group consisting of -CH2-, -0-, and -S-; and Z is selected from the group consisting of -CH2- and -CH2CH2-.
In a subgroup of the compound of Formula I, including pharmaceutically acceptable salts thereof, R^ R2, R3, and R4 are independently selected from (I) H; (2) halogen; (3) -NO2; (4) -CN; (5) -Ci-C4alkyl, which is optionally substituted with 1-5 halogens and optionally with 1-2 substituents independently selected from -OH, -C(=O)Ci-C3alkyl, and -0Ci-C3alkyl, where the Ci-C3alkyl groups
are optionally substituted with 1-3 halogens; (6) -OCi-Ojalkyl, which is optionally substituted with 1-5 halogens and optionally with one -C(=O)Ci-C3alkyl group; (7) -C(=O)Ci-C3alkyl, which is optionally substituted with 1-5 halogens; (8) -C(=O)H; (9) -C(=O)N(R6)(R6); (10) -S(O)2N(R6)(R6); (11) -S(O)2Ci-C3alkyl; (12) -N(R6)(R6); (13) C3.C7Cycloa.kyl; (14) Phenyl; and (15) Heterocycle, wherein C3-C7Cycloalkyl, Phenyl, and Heterocycle are each optionally substituted with 1-3 substituents independently selected from halogen, -OH, -OCi-Cβalkyl, CF3, and -C(=O)Ci-C3alkyl; wherein the pair of substituents R^ and R^ may together represent a bridging divalent 4- carbon chain -CH=CH-CH=CH- or a bπdging divalent 3-atom chain selected from -CH=CHO-, -OCH=CH-, -CH=CH-S-, and -SCH=CH-, forming a fused phenyl, furan or thiophene πng at the Rl and R2 positions, wherein said fused phenyl, furan or thiophene πng is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Ci-C3alkyl, -OCi-C3alkyl, -SCi- C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3;
Each R6 IS independently selected from H and CH3;
W is selected from the group consisting of -CH2-, -O-, and -S-; Z is selected from the group consisting of-CH2- and -CH2CH2-;
A is -CH- or N; and
B is selected from -S- and -O-.
In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, R^ is selected from the group consisting of H, CH3, and halogen.
In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, R^ is selected from the group consisting of H, CH3, and halogen.
In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, Rl and R2 together represent a bridging group selected from -CH=CH-CH=CH-, -CH=CHS-, -SCH=CH-, and -OCH=CH-, forming a fused phenyl, thiophene, or furan ring at the Rl and R2 positions, wherein said fused ring is optionally substituted with 1 -2 substituents independently selected from halogen, CF3, -OCF3, -OCH3, -CN, -NO2, and Ci.3alkyl.
In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, R^ is selected from the group consisting of H, halogen, Ci_3alkyl, -OCH3, -OCF3, CF3, -C(=O)NH2, -CN, and -NO2
In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, R^ is selected from the group consisting of H, halogen, CH3, CF3, and -S(O)2NH2.
In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, W is selected from the group consisting of -CH2-, -O-, and -S-. In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, Z is selected from the group consisting of -CH2- and -CH2CH2-;
In subgroups of the compound of Formula I, including pharmaceutically acceptable salts thereof, A is -CH- or N; and B is selected from -S- and -O-.
Although the specific stereochemistries described above are preferred, all other stereoisomers, including diastereomers, enantiomers, epimers, and mixtures of these may also have utility in treating GPR40 mediated diseases. Inactive or less active diastereoisomers and enantiomers are useful for scientific studies relating to the receptor and the mechanism of activation.
Structures of specific compounds and synthetic methods for making the compounds are disclosed in the Examples. Some of the Examples are disclosed in tables in the specification, along with analytical information. These examples are readily made by one of ordinary skill in the art using the information disclosed herein. Where a stereochemical center is not defined (as for example, A in figure I, where A is -CH-), the compound is a mixture of stereoisomers at that center. For such compounds, the individual stereoisomers, including enantiomers, diastereomers, and mixtures of these are also compounds of the invention. The compounds of the invention also include pharmaceutically acceptable salts.
The compounds of this invention may be used in pharmaceutical compositions comprising (a) the compound(s) or pharmaceutically acceptable salts thereof, and (b) a pharmaceutically acceptable carrier. The compounds of this invention may be used in pharmaceutical compositions that include one or more other active pharmaceutical ingredients The compounds of this invention may also be used in pharmaceutical compositions in which the compound of Formula I or a pharmaceutically acceptable salt thereof is the only active ingredient.
A compound of Formula I, or a pharmaceutically acceptable salt thereof, may be used in the manufacture of a medicament for the treatment of type 2 diabetes melhtus in a human or other mammalian patient. A method of treating type 2 diabetes comprises the administration of a therapeutically effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising the compound, to a patient in need of treatment. Other medical uses of the compounds of Formula I are described hereinafter.
Definitions
"Ac" is acetyl, which is CH3C(=O)-.
"Alkyl" means saturated carbon chains which may be linear or branched or combinations thereof, unless the carbon chain is defined otherwise. Other groups having the prefix "alk", such as alkoxy and alkanoyl, also may be linear or branched or combinations thereof, unless the carbon chain is defined otherwise. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, and the like.
"Alkenyl" means carbon chains which contain at least one carbon-carbon double bond, and which may be linear or branched or combinations thereof. Examples of alkenyl include vinyl, allyl, isopropenyl, pentenyl, hexenyl, heptenyl, 1-propenyl, 2-butenyl, 2-methyl-2-butenyl, and the like.
"Alkynyl" means carbon chains which contain at least one carbon-carbon triple bond, and which may be linear or branched or combinations thereof. Examples of alkynyl include ethynyl, propargyl, 3-methyl-l-pentynyl, 2-heptynyl and the like.
"Cycloalkyl" means a saturated carbocyclic ring, having a specified number of carbon atoms. The term may also be used to describe a carbocyclic ring fused to an aryl group. Examples of cycloalkyl include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like. Cycloalkenyl rings comprise a double bond in the ring.
"Aryl" is commonly used to refer to carbocyclic aromatic structures. The most common aryl groups are phenyl and naphthyl. Phenyl is generally the most preferred aryl group.
"Heterocycle" means a fully or partially saturated ring or πng system containing at least one heteroatom selected from N, S and O, wherein the number of heteroatoms and the πng size are defined herein. Examples of heterocycles include tetrahydrofuran, piperazme, pipeπdme, and morphohne.
"Heteroaryl" means an aromatic ring or two fused aromatic rings containing at least one ring heteroatom selected from N, O and S (including SO and SO2), as defined more specifically herein.
Examples of heteroaryl include pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, lmidazolyl, tπazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyπmidyl, pyridazinyl, pyrazmyl, benzisoxazolyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl (including S-oxide and dioxide), furo(2,3-b)pyridyl, quinolyl, indolyl, isoquinolyl, quinazohnyl, dibenzofuranyl, and the like.
"Halogen" includes fluorine, chlorine, bromine and iodine "Me" represents methyl.
The phrase "pharmaceutically acceptable" is employed herein to refer to those compounds, materials, compositions, salts and/or dosage forms which are, using sound medical judgment, and following all applicable government regulations, safe and suitable for administration to a human being or an animal. The term "composition," as in pharmaceutical composition, is intended to encompass a product compπsing the active mgredient(s), and the inert ιngredient(s) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable earner.
The substituent "tetrazole" means a 2H-tetrazol-5-yl substituent group and tautomers thereof.
Optical Isomers - Diastereomers - Geometric Isomers - Tautomers Compounds of Formula I may contain one or more asymmetric centers and can thus occur as racemates, racemic mixtures, single enantiomers, individual diastereomers, and mixtures of diastereomers and/or enantiomers. The present invention is meant to comprehend all such isomeric forms of the compounds of Formula I. Specifically, the compounds of the instant invention have at least one asymmetric center, which is on the πng that is fused to the phenyl ring at the point where the heterocyclic ring is attached. There may also a second asymmetric center in the heterocyclic ring.
Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers, and it is intended that all of the possible optical isomers, stereoisomers, and diastereomers in mixtures and as pure or partially purified compounds are included within the scope of this invention (i.e. all possible combinations of the asymmetric centers as pure compounds or in mixtures).
Some of the compounds described herein may contain olefϊnic double bonds, and unless specified otherwise, are meant to include both E and Z geometric isomers.
Some of the compounds described herein may exist with different points of attachment of hydrogen, referred to as tautomers. An example is a ketone and its enol form, known as keto-enol tautomers. The individual tautomers as well as mixtures thereof are encompassed with compounds of Formula I.
Compounds of Formula I having one or more asymmetric centers may be separated into diastereoisomers, enantiomers, and the like by methods well known in the art
Alternatively, enantiomers and other compounds with chiral centers may be synthesized by stereospecific synthesis using optically pure starting materials and/or reagents of known configuration.
Salts
The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates. Salts denved from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-
dibenzylethylenediamine, diethylamme, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morphohne, N-ethylpipeπdme, glucamme, glucosamine, histidine, hydrabamine, lsopropylamme, lysine, methylglucamme, morphohne, piperazme, pipeπdme, polyamine resms, procaine, purines, theobromine, triethylamine, trimethylamine, tπpropylamine, tromethamme, and the like.
When the compound of the present invention is basic, or when it has a basic substituent group in its structure, salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids. Such acids include acetic, benzenesulfomc, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaπc, gluconic, glutamic, hydrobromic, hydrochloric, lsethionic, lactic, maleic, malic, mandehc, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuπc, tartaric, p-toluenesulfonic acid, and the like. Particularly preferred are citric, hydrobromic, hydrochloπc, maleic, phosphoric, sulfuric, and tartaric acids.
It will be understood that, as used herein, references to the compounds of Formula I are meant to also include the pharmaceutically acceptable salts.
Metabolites - Prodrugs
Therapeutically active metabolites, where the metabolites themselves fall within the scope of the claimed invention, are also compounds of the current invention. Prodrugs, which are compounds that are converted to the claimed compounds as they are being administered to a patient or after they have been administered to a patient, are compounds of this invention The claimed chemical structures of this application in some cases may themselves be prodrugs.
Utilities
Compounds of the present invention are potent agonists of the GPR40 receptor. The compounds of the invention, and pharmaceutically acceptable salts thereof, may be efficacious m the treatment of diseases that are modulated by GPR40 hgands and agonists. Many of these diseases are summarized below.
One or more of the following diseases may be treated by the administration of a therapeutically effective amount of a compound of this invention, or a pharmaceutically acceptable salt thereof, to a patient m need of treatment Also, the compounds of the invention may be used for the manufacture of a medicament for treating one or more of these diseases:
(1) non-insulin dependent diabetes melhtus (type 2 diabetes);
(2) hyperglycemia;
(3) the metabolic syndrome; (4) obesity;
(5) hypercholesterolemia;
(6) hypertriglyceridemia (elevated levels of tπglycende-πch-hpoproteins);
(7) mixed or diabetic dyshpidemia;
(8) low HDL cholesterol;
(9) high LDL cholesterol;
(10) hyperapoBhproteinemia; and (11) atherosclerosis.
Preferred uses of the compounds are for the treatment of one or more of the following diseases by administering a therapeutically effective amount to a patient m need of treatment. The compounds may be used for manufacturing a medicament for the treatment of one or more of these diseases: (1) Type 2 diabetes, and specifically hyperglycemia;
(2) Metabolic syndrome;
(3) Obesity; and
(4) Hypercholesterolemia.
The compounds are expected to be effective in lowering glucose and lipids in diabetic patients and m non-diabetic patients who have impaired glucose tolerance and/or are m a pre -diabetic condition. The compounds may ameliorate hypeπnsulinemia, which often occurs in diabetic or pre- diabetic patients, by modulating the swings in the level of serum glucose that often occurs in these patients. The compounds may also be effective m treating or reducing insulin resistance. The compounds may be effective in treating or preventing gestational diabetes. The compounds, compositions, and medicaments as described herein may also be effective in reducing the risks of adverse sequelae associated with metabolic syndrome, and in reducing the risk of developing atherosclerosis, delaying the onset of atherosclerosis, and/or reducing the risk of sequelae of atherosclerosis. Sequelae of atherosclerosis include angina, claudication, heart attack, stroke, and others By keeping hyperglycemia under control, the compounds may also be effective in delaying or preventing vascular restenosis and diabetic retinopathy
The compounds of this invention may also have utility in improving or restoring /S-cell function, so that they may be useful in treating type 1 diabetes or in delaying or preventing a patient with type 2 diabetes from needing insulin therapy The compounds generally will be efficacious in treating one or more of the following diseases: (1) type 2 diabetes (also known as non-insulin dependent diabetes mellitus, or NIDDM), (2) hyperglycemia, (3) impaired glucose tolerance, (4) insulin resistance, (5) obesity, (6) lipid disorders, (7) dyshpidemia, (8) hyperlipidemia, (9) hypertriglyceridemia, (10) hypercholesterolemia, (11) low HDL levels, (12) high LDL levels, (13) atherosclerosis and its sequelae, (14) vascular restenosis, (15) abdominal obesity, (16) retinopathy, (17) metabolic syndrome, (18) high blood pressure, and (19) insulin resistance.
One aspect of the invention provides a method for the treatment and control of mixed or diabetic dyslipidemia, hypercholesterolemia, atherosclerosis, low HDL levels, high LDL levels, hyperhpidemia, and/or hypertriglyceridemia, which comprises administering to a patient in need of such treatment a therapeutically effective amount of a compound having formula I. The compound may be used alone or advantageously may be administered with a cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor such as lovastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, atorvastatin, πvastatm, itavastatin, or ZD-4522. The compound may also be used advantageously m combination with other lipid lowering drugs such as cholesterol absorption inhibitors (for example stanol esters, sterol glycosides such as tiqueside, and azetidinones such as ezetimibe), ACAT inhibitors (such as avasimibe), CETP inhibitors (for example torcetrapib), niacin and niacin receptor agonists, bile acid sequestrants, microsomal triglyceride transport inhibitors, and bile acid reuptake inhibitors. These combination treatments may be effective for the treatment or control of one or more related conditions selected from the group consisting of hypercholesterolemia, atherosclerosis, hyperhpidemia, hypertriglyceridemia, dyslipidemia, high LDL, and low HDL.
Administration and Dose Ranges
Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dose of a compound of the present invention. For example, oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed. Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like. Preferably compounds of Formula I are administered orally.
The effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the seventy of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art. When treating or controlling diabetes melhtus and/or hyperglycemia or hypertriglyceridemia or other diseases for which compounds of Formula I are indicated, generally satisfactory results are obtained when the compounds of the present invention are administered at a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form. For most large mammals, the total daily dosage is from about 1.0 milligrams to about 1000 milligrams. In the case of a 70 kg adult human, the total daily dose will generally be from about 1 milligram to about 500 milligrams. For a particularly potent compound, the dosage for an adult human may be as low as 0.1 mg. The dosage regimen may be adjusted within this range or even outside of this range to provide the optimal therapeutic response. Oral administration will usually be carried out using tablets or capsules. Examples of doses in tablets and capsules are 0.1 mg, 0.25 mg, 0.5 mg, 1 mg, 2 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, and 500 mg. Other oral forms may also have the same or similar dosages.
Pharmaceutical Compositions
Another aspect of the present invention provides pharmaceutical compositions which comprise a compound of Formula I and a pharmaceutically acceptable carrier. The pharmaceutical compositions of the present invention comprise a compound of Formula I or a pharmaceutically acceptable salt as an active ingredient, as well as a pharmaceutically acceptable earner and optionally other therapeutic ingredients. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids. A pharmaceutical composition may also comprise a prodrug, or a pharmaceutically acceptable salt thereof, if a prodrug is administered.
The compositions include compositions suitable for oral, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ocular (ophthalmic), pulmonary (nasal or buccal inhalation), or nasal administration, although the most suitable route in any given case will depend on the nature and seventy of the conditions being treated and on the nature of the active ingredient They may be conveniently presented m unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
In practical use, the compounds of Formula I can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques. The earner may take a wide variety of forms depending on the form of preparation desired for administration, e g., oral or parenteral (including intravenous). In preparing the compositions for oral dosage form, any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions, or carriers such as starches, sugars, microcrystallme cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations.
Because of their ease of administration, tablets and capsules represent the most advantageous oral dosage unit form m which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques. Such compositions and preparations should contain at least 0.1 percent of active compound. The percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2 percent to about 60 percent of the weight of the unit. The amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained. The active compounds can also be administered intranasally as, for example, liquid drops or spray. The tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, algmic acid; a lubncant such as magnesium stearate; and a sweetening
agent such as sucrose, lactose or saccharin. When a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
In some instances, depending on the solubility of the compound or salt being administered, it may be advantageous to formulate the compound or salt as a solution in an oil such as a triglyceride of one or more medium chain fatty acids, a lipophilic solvent such as tπacetm, a hydrophilic solvent (e.g. propylene glycol), or a mixture of two or more of these, also optionally including one or more ionic or nomonic surfactants, such as sodium lauryl sulfate, polysorbate 80, polyethoxylated triglycerides, and mono and/or diglyceπdes of one or more medium chain fatty acids. Solutions containing surfactants (especially 2 or more surfactants) will form emulsions or microemulsions on contact with water. The compound may also be formulated in a water soluble polymer in which it has been dispersed as an amorphous phase by such methods as hot melt extrusion and spray drying, such polymers including HPMCAS, HPMCS, and polyvmylpyrrolidinones.
Vaπous other mateπals may be present as coatings or to modify the physical form of the dosage unit. For instance, tablets may be coated with shellac, sugar or both. A syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor
Compounds of formula I may also be administered parenterally. Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxypropylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
The pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases, the form must be sterile and must be fluid to the extent that easy syπngabihty exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
Combination Therapy
Compounds of Formula I may be used m combination with other drugs that may also be useful m the treatment or amelioration of the diseases or conditions for which compounds of Formula I are useful. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I. In the treatment of patients who have type 2 diabetes, insulin resistance, obesity, metabolic syndrome, and co-morbidities that accompany these diseases, more than one drug is commonly administered. The compounds of this invention may generally be administered to a patient who is already taking one or more other drugs for these conditions.
When a compound of Formula I is used contemporaneously with one or more other drugs, a pharmaceutical composition in unit dosage form containing such other drugs and the compound of Formula I is preferred. However, the combination therapy also includes therapies in which the compound of Formula I and one or more other drugs are administered on different overlapping schedules. It is also contemplated that when used in combination with one or more other active ingredients, the compound of the present invention and the other active ingredients may be used in lower doses than when each is used singly. Accordingly, the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of Formula I.
Examples of other active ingredients that may be administered in combination with a compound of Formula I, and either administered separately or in the same pharmaceutical composition, include, but are not limited to:
(a) PPAR gamma agonists and partial agonists, including both ghtazones and non- ghtazones (e.g. troghtazone, pioghtazone, enghtazone, MCC-555, rosiglitazone, balaghtazone, netoghtazone, T-131, LY-300512, and LY-818; (b) biguanides such as metformin and phenformin;
(c) protein tyrosine phosphatase- IB (PTP-IB) inhibitors;
(d) dipeptidyl peptidase IV (DP-FV) inhibitors, such as sitaghptin, saxaghptin, and vildaghptin;
(e) insulin or insulin mimetics; (f) sulfonylureas such as tolbutamide, glimepiπde, glipizide, and related materials;
(g) α-glucosidase inhibitors (such as acarbose);
(h) agents which improve a patient's lipid profile, such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, rosuvastatin, pravastatin, fluvastatin, atorvastatin, πvastatin, itavastatin, ZD-4522 and other statins), (π) bile acid sequestrants (cholestyramine, colestipol, and dialkylammoalkyl derivatives of a cross-lmked dextran), (in) niacin receptor agonists, nicotmyl alcohol, nicotinic acid, or a salt thereof, (iv) PP ARa agonists such as fenofibric acid derivatives (gemfibrozil, clofibrate, fenofibrate and bezafibrate), (v) cholesterol absorption inhibitors, such as for example ezetimibe, (vi) acyl CoA:cholesterol acyltransferase (ACAT) inhibitors, such as avasimibe, (vπ) CETP inhibitors, such as torcetrapib, and (vin) phenolic anti -oxidants, such as probucol; (i) PPARα/γ dual agonists, such as muraghtazar, tesaghtazar, farghtazar, and JT-501;
(]) PPARδ agonists such as those disclosed in WO97/28149;
(k) antiobesity compounds such as fenfluramine, dexfenfluramine, phentiramine, subitramine, orhstat, neuropeptide Y5 inhibitors, Mc4r agonists, cannabmoid receptor 1 (CB-I) antagonists/inverse agonists, and β3 adrenergic receptor agonists; (1) ileal bile acid transporter inhibitors;
(m) agents intended for use in inflammatory conditions such as aspinn, non-steroidal anti-inflammatory drugs, glucocorticoids, azulfidine, and cyclo-oxygenase 2 selective inhibitors;
(n) glucagon receptor antagonists;
(o) GLP-I,
(P) GIP-I,
(q) GLP-I analogs, such as exendms, for example exenatide (Byetta), and (r) Hydroxysterol dehydrogenase- 1 (HSD-I) inhibitors.
The above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds. Non- hmiting examples include combinations of compounds having Formula I with two or more active compounds selected from biguanides, sulfonylureas, HMG-CoA reductase inhibitors, other PPAR agonists, PTP-IB inhibitors, DP-IV inhibitors, and anti-obesity compounds.
BIOLOGICAL ASSAYS Generation of GPR40-Expressmg Cells
Human and mouse GPR40 stable cell-lmes were generated in CHO cells stably expressing NFAT BLA (Beta-lactamase). A human GPR40 stable cell-line was generated in HEK cells stably expressing the aequorm expressing reporter. The expression plasmids were transfected using lipofectamine (Life Technologies) following manufacturer's instructions. Stable cell-lines were generated following drug selection.
FLIPR Assays FLIPR (Fluoπmetπc Imaging Plate Reader, Molecular Devices) assays were performed to measure agonist-induced calcium mobilization of the stable clones. For the FLIPR assay, one day before assay, GPR40/CHO NFAT BLA cells were seeded into black- wall-clear-bottom 384-well plates (Costar) at 1.4 x 10e4 cells / 20 μl medium / well. The cells were incubated with 20 μl / well of the assay buffer (HBSS, 0.1 % BSA, 20 mM HEPES, 2.5 mM probenecid, pH 7.4) containing 8μM fluo-4,AM, 0.08 % pluronic acid at room temperature for 100 minutes. Fluorescence output was measured using FLIPR. Compounds were dissolved in DMSO and diluted to desired concentrations with assay buffer 13.3 μl/well of compound solution was added.
EC50 activities were measured for the compounds using both the human and mouse GPR40 cell lines. The activities for the compounds herein are as follows: human FLEPR EC50: 0.033 to 40 μM; mouse FLIPR EC50: 0.051 to 40 μM. The preferred compounds have an EC50 <10 μM.
Inositol Phosphate Turnover Assay
The assay is performed in 96-well format. HEK cells stably expressing human GPR40 are plated to be 60-80% confluent within 72 hours. After 72 hours, the plates are aspirated and the cells washed with lnositol-free DMEM (ICN). The wash media is replaced with 15OuL of 3H-inositol labeling media (lnositol-free media containing 0.4% human albumin or 0.4% mouse albumin, IX pen/strep
antibiotics, glutamme, 25mM HEPES to which is added 3H-myo-inositol NEN #NET114A lmCi/mL, 25Ci/mmol diluted 1 :150 in loading media with a final specific radioactivity of 1UCI/150UL). Alternatively, the human and mouse albumin can be added after the overnight labeling step before the addition of LiCl. The assay is typically run the next day after 18 hours labeling. On the day of the assay,
5uL of 30OmM LiCl is added to all wells and incubated at 37 degrees for 20 mms. 0.75uL of 200X compounds are added and incubated with the cells for 60 minutes at 37 degrees. The media is then aspirated off and the assay terminated with the addition of 6OuL 1OmM formic acid. The cells are lysed for 60 mms at room temperature. 15-3OuL of lysate is mixed with 70uL/lmg YSi SPA beads (Amersham) in clear bottom Isoplates. The plates are shaken for 2 hours at room temperature. Beads are allowed to settle and the plates are counted in the Wallac Microbeta.
In Vivo Studies
Male C57BL/6N mice (7-12 weeks of age) are housed 10 per cage and given access to normal diet rodent chow and water ad libitum. Mice are randomly assigned to treatment groups and fasted 4 to 6 hours. Baseline blood glucose concentrations are determined by glucometer from tail nick blood Animals are then treated orally with vehicle (0.25% methylcellulose) or test compound. Blood glucose concentration is measured at a set time point after treatment (t = 0 mm) and mice are then intrapeπtoneally-challenged with dextrose (2 g/kg). One group of vehicle-treated mice is challenged with salme as a negative control. Blood glucose levels are determined from tail bleeds taken at 20, 40, 60 minutes after dextrose challenge. The blood glucose excursion profile from t = 0 to t = 60 mm is used to integrate an area under the curve (AUC) for each treatment. Percent inhibition values for each treatment are generated from the AUC data normalized to the saline-challenged controls.
EXAMPLES
The following Examples are provided to illustrate the invention and are not to be construed as limiting the invention in any manner The scope of the invention is defined by the appended claims.
Methods for preparing the compounds of this invention are illustrated in the following Examples. Syntheses of several Intermediates that are used for making the exemplified compounds are also provided. Starting materials are either commercially available or made by known procedures in the literature or as illustrated. The present invention further provides processes for the preparation of compounds of formula I as defined above.
INTERMEDIATE 1

To a cooled (-78 0C) solution of ethyl [(lS)-5-methoxy-2,3-dihydro-lH-mden-l-yl]acetate (2.34 g, 10 mmol), prepared according to a published procedure (WO 20040011446), in 20 mL of anhydrous THF was added a solution of sodium bis(trimethylsilyl)amide (1.0 M, 12 mL, 12 mmol) dropwise. The mixture was stirred at -78 0C for 30 mm, then a neat solution of tπmethylsilyl chloπde (1.4 mL, 11 mmol) was added dropwise. The reaction was stirred for an additional 10 min., solid NBS (2.0 g, 11 mmol) was added in one portion, the reaction was warmed to RT for one hour, quenched with water, and extracted with ethyl acetate. The organic phase was washed with water and brine, dried over anhydrous sodium sulfate, evaporated to afford a crude oil which was used in the next step without further purification.

The crude product (3.70 g) from step A was treated with thiourea (0.76 g, 10 mmol) and sodium acetate (0.82 g, 10 mmol) in 50 mL of ethanol. The mixture was refluxed for 13 h, cooled at RT. After addition of 20 mL of ether and 20 mL of hexane, the resulting solid was collected by filtration and washing with hexane. The desired product was obtained as off white solid. LC-MS: calc. for C13H14N2O2S: 262; Found: 263 (M+H). IH NMR (400 MHz, CD3OD) δ 7.11, 6.90 (dd, J = 8.1, 8.3 Hz, ratio = 2: 1, IH),
6.58-6.76 (m, 2H), 5.03, 4.66 (dd, J = 3.0, 2.8 Hz, ratio = 2:1, IH), 3.95 (m, IH), 3.70 (s, 3H), 2.92 (m, 2 H), 2.42, 2.05, 1.82, 1.70 (m, 2H).


To a stirred, cool (-78 0C) solution of the product (1.40 g, 5.3 mmol) from step C m IO mL of dichloromethane was added a solution of boron tribromide in dichloromethane (1.0 M, 15 mL, 15 mmol). The reaction was then warmed to RT for 30 mm., then quenched with ice-water. The product was extracted with ethyl acetate twice. The organic phase was washed with water twice, dried over anhydrous sodium sulfate, and evaporated. The residue was dried under high vacuum to afford a light brown solid which could be used in next step without further purification. LC-MS: calc. for C12H11NO3S: 249 Found: 250 (M+H). IH NMR (400 MHz, CD3OD) δ 7.0, 6.9 (dd, J = 8.2, 8.2 Hz, ratio = 3:1, IH), 6.50-6.62 (m, 2H), 5.08, 4.71 (dd, J = 3.8, 4.2 Hz ratio = 3: 1, IH), 3.90 (m, IH), 3.72 (s, 3H), 2.92-2.70 (m, 2 H), 2.38, 2.06, 1.86 (mmm, 2H).
INTERMEDIATE 2

Step A

To a stirred solution of the racemic [6-methoxy-l,2,3,4-tetrahydronaphthalen-l-yl]acetic acid (69.4 g, prepared according to a published procedure (WO 20040011446) in 1500 mL of acetone was added 38.7
mL of (S)-alpha-methylbenzylamine in one portion. The mixture was stirred at RT for 30 min, then 1500 mL of hexane was added. The mixture was stirred for one hour. The resulting solid was removed by filtration and washing with hexane/acetone (4:1 v/v), and was then dried m air to give the first batch of solid. The combined mother liquids were stored at 0-5 0C overnight, the resulting solid was collected by filtration to give a second batch of solid. The two batches of the salt were combined, dissolved in a warm acetone (500 mL). 750 mL of hexane was added, and the mixture was stirred at RT for one hour. The resulting solid was collected by filtration, washed with hexane/acetone (4:1), and dried in air to give off-white crystals of (R,S)-salt.
Step B

All mother liquids from the above step A were combined and condensed to give a light brown solid. 3N aq. HCl was added to adjust pH <3, stirred with ethyl acetate (500 mL), and separated. The organic phase was washed with 3N aq. HCl, dried over sodium sulfate, filtered and evaporated to afford a light brown solid (32 g, 145 mmol, S-enπched acid). This solid was dissolved in 500 mL of acetone, (R)-(+)- alpha-methylbenzylamine (16.6 mL, 145 mmol) was added, the mixture was refluxed until all the solid dissolved, and was then cooled to RT. The resulting precipitate was collected by filtration and washing with acetone to afford a white solid salt (S,R). The (S)-absolute configuration of the acid was confirmed by x-ray crystallography of the amide formed by treatment of the above salt with EDAC.
Step C

The (S,R) salt from the above step B (23.2 g) was stirred for one hour with 200 mL of 3N HCl and 200 mL of ethyl acetate. The organic phase was separated and washed with 3N aq. HCl (2 x 100 mL), dried over sodium sulfate, filtered and evaporated to give the desired (S)-acid as a light brown solid.
Step D

Step E

To a cooled (-78 0C) solution of ethyl [(lS)-6-methoxy-l,2,3,4-tetrahydronaphthalen-l-yl] acetate (7.45 g, 30 mmol), from the above step D, in 50 mL of anhydrous THF was added a solution of sodium bis(tπmethylsilyl)amide (1.0 M, 36 mL, 36 mmol) dropwise. The mixture was stirred at -78 0C for 30 mm, then a neat solution of trimethylsilyl chloride (4.22 mL, 33 mmol) was added dropwise. The reaction was stirred for additional 10 mm. Solid NBS (5 87 g, 33 mmol) was added in one portion. The reaction was warmed to RT during one hour, quenched with water, and extracted with ethyl acetate The organic phase was washed with water and brme, dried over anhydrous sodium sulfate, and evaporated to afford a crude oil which was used in the next step without further purification.
Step F

The crude product from the above step E was treated with thiourea (2.28 g, 30 mmol) and sodium acetate (2.46 g, 30 mmol) in 50 mL of ethanol. The mixture was refluxed for 13 h, cooled at RT. After addition of 20 mL of ether and 20 mL of hexane, the resulting solid was collected by filtration and washing with hexane. The desired product was obtained as an off white solid. LC-MS: calc. for C14H16N2O2S: 276; Found: 277 (M+H).
Step G

The product from the above step F was mixed with 50 mL of 4N aq. HCl and 50 mL of ethanol. The mixture was refluxed overnight (monitored by LC-MS until a complete conversion was observed). After removal of ethanol under vacuum, the residue was extracted with ethyl acetate, dried over anhydrous sodium sulfate, evaporated, and dried under high vacuum to afford a yellow solid. LC-MS: calc. for C14H15NO3S: 277 Found: 278 (M+H).
Step H
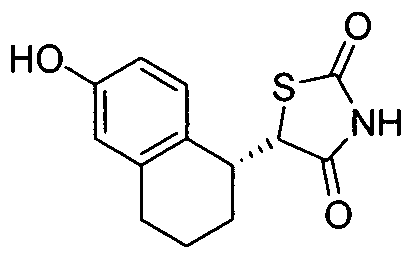
To a stirred, cool (-78 0C) solution of the product from the above step G (5.2 g, 18.7 mmol) in 50 mL of dichloromethane was added a solution of boron tπbromide in dichloromethane (1.0 M, 57 mL, 57 mmol). The reaction was then warmed to RT for 30 min. and quenched with ice-water. The product was extracted with ethyl acetate twice. The organic phase was washed with water twice, dried with anhydrous sodium sulfate, and evaporated. The residue was dried under high vacuum to afford a light brown solid which could be used in next step without further purification. LC-MS: calc. for C13H13NO3S: 263 Found: 264 (M+H).
INTERMEDIATE 3

Step A

The (R,S) salt (24.5 g) from step A of the synthesis of Intermediate 2 was stirred for one hour with 200 mL of 3N HCl and 200 mL of ethyl acetate. The organic phase was separated and washed with 3N aq. HCl (2 x 100 mL), dried over sodium sulfate, filtered, and evaporated to give the desired (R)-acid as a light brown solid.
Step B

The (R)-acid from the above step A (15.6 g) was dissolved in 150 mL of ethanol followed by addition of 19 mL of trimethylsilyl chloπde. The mixture was stirred at RT overnight, evaporated and mixed with ethyl acetate (100 mL). The organic phase was washed with water and saturated aq. sodium hydrogen carbonate, dried over sodium sulfate, and purified on FC (Silica gel, 5% ethyl acetate/hexane) to give the desired (R)-ester as a colorless oil.

To a cooled (-78 0C) solution of ethyl [(lR)-6-methoxy-l,2,3,4-tetrahydronaphthalen-l-yl] acetate (7 45 g, 30 mmol), from the above step B, in 50 mL of anhydrous THF was added a solution of sodium bis(tπmethylsilyl)amide (1 0 M, 36 mL, 36 mmol) dropwise. The mixture was stirred at -78 0C for 30 mm. A neat solution of trimethylsilyl chloride (4.22 mL, 33 mmol) was added dropwise. The reaction was stirred for an additional 10 mm., and solid NBS (5 87 g, 33 mmol) was added in one portion. The reaction was warmed to RT during one hour, quenched with water, and extracted with ethyl acetate. The organic phase was washed with water and brine, dried over anhydrous sodium sulfate, and evaporated to afford a crude oil which was used in the next step without further purification.
Step D

The crude product from the above step C was treated with thiourea (2.28 g, 30 mmol) and sodium acetate (2.46 g, 30 mmol) in 50 mL of ethanol. The mixture was refluxed for 13 h, and cooled to RT. After addition of 20 mL of ether and 20 mL of hexane, the resulting solid was collected by filtration and washed with hexane. The desired product was obtained as an off white solid. LC-MS: calc. for C14H16N2O2S: 276; Found: 277 (M+H).
Step E
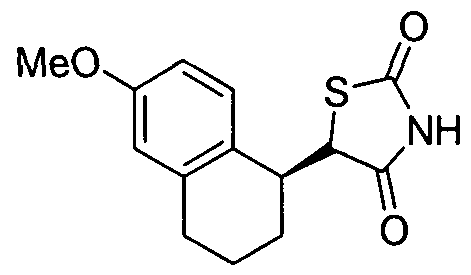
The product from the above step D was mixed with 50 mL of 4N aq. HCl and 50 mL of ethanol. The mixture was refluxed overnight (monitored by LC-MS until a complete conversion was observed). After removal of ethanol under vacuum, the residue was extracted with ethyl acetate, dried over anhydrous sodium sulfate, and evaporated and dried under high vacuum to afford a yellow solid. LC-MS: calc. for C14H15NO3S: 277 Found: 278 (M+H).
Step F

To a stirred, cool (-78 0C) solution of the product from the above step E (4.02 g, 14.5 mmol) in 50 mL of dichloromethane was added a solution of boron tribromide in dichloromethane (1.0 M, 30 mL, 30 mmol). The reaction was then warmed to RT for 30 min., and quenched with ice-water. The product was extracted with ethyl acetate twice. The organic phase was washed with water twice, dried with anhydrous sodium sulfate, and evaporated. The residue was dried under high vacuum to afford a light brown solid which could be used in the next step without further purification. LC-MS: calc. for C13H13NO3S: 263 Found: 264 (M+H).
INTEMEDIATE 4

Step A

To 6-hydroxyl-2, 3-dihydrobenzofuran-3-one (30 g, 200 mmol) in DMF (600 mL) was added K2CO3 (220 mmol, 30.4 g) followed by BnBr (200 mmol, 24 mL). After stirring at room temperature for 3
hours, the reaction mixture was partitioned between methyl t-butyl ether (MTBE, 500 mL) and water (IL). The aqueous layer was separated and further extracted with MTBE (2 x 500 mL). The organic layers were combined, washed with water (500 mL), Brine (500 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give 6 as the product yellow solid. LC-MS for C 15Hl 303 [M+H]: calculated 241, found 241.
Step B

To a suspension of NaH (60% in mineral oil, 381 mmol, 15.2 g) in anhydrous THF (900 mL) was added triethyl phosphonoacetate (381 mmol, 76 mL) dropwisely in an ice bath. After addition, the reaction was stirred at room temperature for 20 minutes until a clear solution was obtained. A solution of the ketone (45.7 g, 190 mmol) from Step A in THF (100 mL) was then added to the reaction. The reaction was stirred overnight at room temperature and then quenched with 0.1N HCl (1 L). The aqueous layer was separated and extracted with EtOAc (2 x 500 mL). The organic layers were combined, washed with water (500 mL), then Brine (500 mL), then dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography (10% to 30% EtOAc/hexanes) to give the product as a yellow solid. LC-MS for C 19Hl 904 [M+H]: calculated 311.1, found 311.3.
Step C
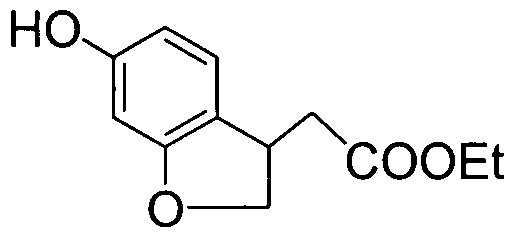
To a solution of the unsaturated ester (6.6 g, 21.3 mmol) from Step B in ethanol (75 mL) and EtOAc (75 mL) was added 10% Pd/C (2 g). The mixture was hydrogenated in a Parr-shaker at 50 psi for 2 hours. The mixture was then filtered through celite. The filtrate was concentrated in vacuo to give the product as a red oil. LC-MS for C12H15O4 [M+H]: calculated 223, found 223.
Step D

To a solution of the acid (2 g, 9 mmol) from Step C in DMF (15 mL) and acetone (60 mL) was added K2CO3 (11 mmol, 1.5 g) followed by BnBr (11 mmol, 1.3 mL). The reaction was stirred overnight at
room temperature and then concentrated in vacuo. The residue was partitioned between EtOAc (100 mL) and water (200 mL). The aqueous layer was separated and further extracted with EtOAc (2 x 100 mL). The organic layers were combined, washed with water (100 mL), then Brine (100 mL), then dried with anhydrous Na2SO4, filtered and concentrated m vacuo. The residue was purified by silica gel flash chromatography (0% to 15% EtOAc/hexanes) to give the product as an oil. IH NMR (500 MHz, CDC13) δ 7.50-7.30 (m, 5H), 7.06 (d, J = 8.0 Hz,lH), 6.52 (d, J = 8.2 Hz, IH), 6.50 (bs, IH), 5.0 (s, 2H), 4.8 (t, J = 8.9 Hz, 1 H), 4.30 (dd, J = 6.1, 8.9 Hz, 1 H), 4.2 (q, J = 7.1 Hz, 2 H), 4.85 (m, 1 H), 2.75 (dd, J = 5.5, 16.5 Hz, I H), 2.58 (dd, J = 9.1, 16.2 Hz, 1 H), 1.30 (t, J = 7.1 Hz, 3 H). LC-MS for Cl 9H2104 [M+H]: calculated 313.4, found 313.2.
Step E

To a flame-dried flask was added anhydrous THF (30 mL) followed by NaHMDS (7.5 mmol, 7.5 mL of 1 M THF solution). After cooling to -78 °C, a solution of the ester (2.0 g, 6.3 mmol) from Step D m THF (10 mL) was added to the reaction slowly. After addition, the reaction was stirred at -78 °C for 15 minutes before TMSCl (7.2 mL of 1 M solution m THF, 7.2 mmol) was added. After another 30 minutes at -78 °C, NBS (6.9 mmol, 1.2 g) was added in one portion. The reaction was allowed to warm up to 0 °C over 2 hours before being quenched with 0.1 N HCl (200 mL). The aqueous layer was separated and further extracted with EtOAc (2 x 100 mL). The organic layers were combined, washed with water (100 mL), then Brine (100 mL), then dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel flash chromatography (0% to 18% EtOAc/hexanes) to give 2.3 g of oil. This oil was then dissolved in ethanol (40 mL), and thiourea (7.0 mmol, 0.54 g) and NaOAc (12 mmol, 0.96 g) were added. The reaction was refluxed for 24 hours and then cooled back to room temperature. The suspension was then filtered. The solid was further washed with cold EtOH (4 mL) and dried in air to give the product as a white solid. LC-MS for C 18H 17N2O3 S [M+H] : calculated 341.1, found 341.1.
Step F
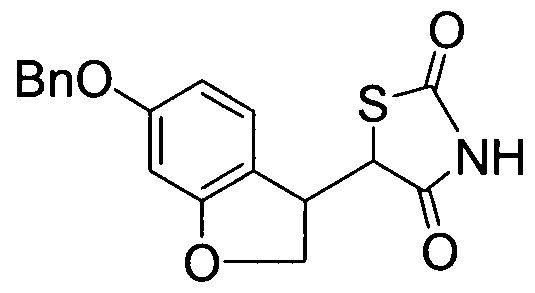
A suspension of the cyclic product (1.5 g, 4.4 mmol) from Step E in EtOH (20 mL) and 6 N HCl (4 mL) was refluxed overnight. The reaction was then concentrated in vacuo. The residue was purified by silica
gel flash chromatography (0% to 50% EtOAc/hexanes) to give the desired TZD as a mixture of diastereomers. LC-MS for C18H16NO4S [M+H]: calculated 342.1, found 342.1.
Step G

To a suspension of the methoxy TZD (300 mg, 0.88 mmol) from Step F in EtOH (20 mL) was added 4 N HCl in dioxane (500 uL and 10% Pc/C (500 mg). The reaction was hydrogenated at 1 arm for 2 hours to give a completed reaction. The mixture was then filtered through cehte. The filtrate was concentrated in vacuo to give INTERMEDIATE 4 as a yellow solid. LC-MS for Cl 1H10NO4S [M+H]: calculated 252.0, found 252.1.
INTERMEDIATE 5

Step A

To a cooled (-78 0C) solution of ethyl [(lS)-5-methoxy-2,3-dihydro-lH-inden-l-yl]acetate (2.34 g, 10 mmol), prepared according to a published procedure (WO 20040011446), in 20 mL of anhydrous THF was added a solution of sodium bis(tπmethylsilyl)amide (1.0 M, 12 mL, 12 mmol) dropwise. The mixture was stirred at -78 0C for 30 mm, then a neat solution of tπmethylsilyl chloride (1.4 mL, 11 mmol) was added dropwise. The reaction was stirred for an additional 30 min., then the reaction vessel was gradually warmed to room temperature. Solvent was then removed in vacuo (roto-evaporation) and then ca. 75 mL of pentane was added to the residue. Rapid filtration and removal of solvent in vacuo yielded crude alkyl trimethyl ketene acetal.
Step B


The hydroxyl ester obtained from Step B was mixed with 4N ammonia-methanol (50 mL) and stirred overnight, then was evaporated, and the residue was mixed with 5 mL of ethyl acetate and 20 mL of hexane. The resulting white powder was filtered and washed with hexane, dried under high vacuum to give the pure product as single isomer. LC-MS for C12H15NO3: calculated 221, found 222 [M+H]. IH NMR (400 MHz, CDC13) δ 7.12 (d, J = 8.1 Hz, IH), 6.77 (m, 2H), 6.55 (bs, IH), 5.53 (bs, IH), 4.54 (s, IH), 3.76 (s, 3 H), 2.82 (m, 2H), 2.12 (m, IH), 2.00 (m, IH), 1.98 (m, IH).
Step D

The hydroxy amide from Step C (280 mg, 1.267 mmol) and diethyl carbonate (747 mg, 6.335 mmol) were mixed with sodium methoxide (345 mg, 6.335 mmol) and ethanol (10 mL). The mixture was refluxed for 1.5 h, then evaporated. The residue was acidified with 3N aq. HCl, extracted with ethyl acetate, dried over sodium sulfate, evaporated and purified on Comb-Flash (5-30% ethyl acetate/hexane) to give the product. LC-MS calculated for C13H13NO4 [M+H] calculated: 247; Found: [M+H]. IH NMR (400 MHz, CDC13) (major isomer) δ 7.17 (d, J = 8.0 Hz, IH), 6.75 (m, 2H), 5.10 (s, IH), 3.75 (s, 3H), 3.00 (m, IH), 2.84 (m, IH), 2.22 (m, 2H), 2.04 (m, IH). Major/minor - 6:1.
Step E

To a stirred, cool (-78 0C) solution of the product from the above step D (100 mg, 0.4 mmol) in 5 mL of dichloromethane was added a solution of boron tribromide in dichloromethane (1.0 M, 1.0 mL, 1.0 mmol). The reaction was warmed to RT for 50 min., then quenched with ice-water. The product was extracted with ethyl acetate twice. The organic phase was washed with water twice, dried with anhydrous sodium sulfate, and evaporated. The residue was dried under high vacuum to afford INTERMEDIATE 5 which could be used in next step without further purification. LC-MS: calc. for C12H11NO4: 233 Found: 234 (M+H).
INTERMEDIATE 6

Step A
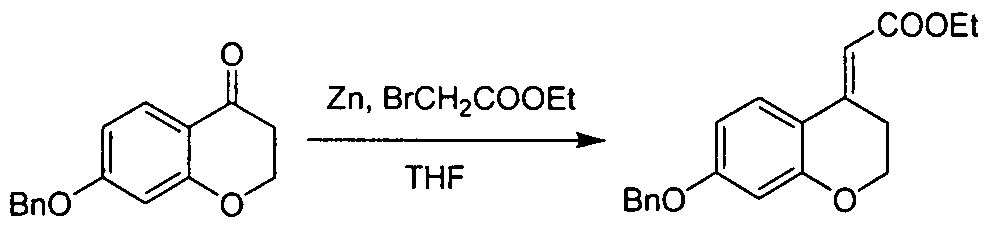
To a dried 3-neck 2 L round bottom was added freshly azeotroped 7-(benzyloxy)chroman-4-one (287 g, 1.13 mol, synthesized according to J. Med. Chem. 1998, 41, 1 172-1184) and 2 L of anhydrous THF (no inhibitor). Zinc (124.9 g, 1.92 mol) and CuI (10.7 g, 56.5 mmol) were then quickly added to the reaction solution. After refluxing for 30 minutes under N2 atmosphere, 81 mL of ethyl bromoacetate (1/2 of total needed, F.W. 167.01, d 1.506, 0.7 mol) was added dropwise to the refluxing mixture. Heat was then turned off and the reaction was stirred at ambient temperature for 4-5 h. Another 81 ml of ethyl bromoaceate (F.W. 167.01, d 1.506, 0.7 mol) was then added dropwise and the reaction was stirred without heating until the reaction temperature returned to ambient temperature. Solids were removed by vacuum filtration through celite and the filtrate was concentrated to ~800 mL by rotary evaporation, which was poured into 1 L of IN HCl (aq) with 1000 g of ice, and stirred vigorously for 30 min. The mixture was extracted with EtOAc (1 x 2 L, 2 x 1 L). The combined organic layers were washed with
H2O (1 x 3 L), Brine (1 x 2 L), dried over Na2SO4, and concentrated in vacuo. The crude compound was used without further purification.
Step B

To a solution of crude product (-356 g, 1.1 mol) from step A in THF/MeOH/ H2O (2:2:1, 2.5 L) was added LiOKH2O (92.4 g, M.W. 41.96, 2.2 mol). The reaction was stirred at ambient temperature overnight. The organic solvents were removed in vacuo and the residue was diluted with water to 3 L in volume. This aqueous solution was washed with diethyl ether (2 x 500 mL) and the aqueous layer was then acidified to pH=l with 10 N HCl (aq). The solid was isolated by vacuum filtration, washed with EtOAc and dried under vacuum. The filtrate was extracted with EtOAc (2 x 500 mL). The combined organics were washed with brine (400 mL) and concentrated in vacuo. All solids were combined, triturated with minimal EtOAc, and dried under high vacuum to give a mixture of two isomers.

A solution of product from step B (20 g, 67.6 mmol) in anhydrous methanol (800 mL) was degassed by bubbling through N2 for 1 hour. (R)-BINAP RuCl2 (1.11 g, F.W. 794.65, 1.4 mmol) and 950 μL of freshly degassed tπethylamine (F.W. 101.19, d 0.72, 6.76 mmol) were quickly added under N2 atmosphere. The mixture was hydrogenated under H2 (50 psi) for 4 days. The mixture was then filtered and the filtrate was concentrated in vacuo. The residue was purified by column chromatography to give the desired product (70% ee) and recovered starting material. The product was dissolved in minimal EtOAc (~ 20 mL) and petroleum ether (~20 mL) and re-crystallized to give chiral acid (~95%ee).


To a solution of chiral ester from step D (7.3 g, 22.4 mmol) in anhydrous THF (100 mL) was added
NaHMDS (2.0M m THF, 14.6 mL, 29.2 mmol) at -78 0C. After addition, the reaction was stirred at -78 0C for 30 minutes before TMSCl (2.0M m THF, 13.5 mL, 26.9 mmol) was added. After the addition of TMSCl, the reaction was stirred for another 30 minuted and NBS (4.4 g, 24.7 mmol) was added in one portion. The reaction was allowed to warm up to 0 0C over 2-3 hours. The reaction was partitioned between 0.1 N HCl aq (200 mL) and ethyl acetate (200 mL). The organic layer was washed with 0.1 N HCl aq (1 x 200 mL). The aqueous layers were combined and back-extracted with EtOAc (1 x 100 mL). The organic layers were combined and washed with Brine (2 x 100 mL), dried over Na2SO4 and concentrated in vacuo to give the desired product. This crude material was used without further purification.
Step F

To the crude material from step E (~20 mmol) in 100 mL of EtOH was added thiourea (M. W. 76.12, 1.979 g, 26 mmol) and sodium acetate (M.W. 82.03, 3.281 g, 40 mmol). The reaction was refluxed overnight. The organic solvent was removed in vacuo and the residue was partitioned between 50 mL of 6 N HCl (aq) and 50 mL of EtOAc. The organic layer was further extracted with 6 N HCl (aq) (2 x 25 mL). The aqueous layers were combined and further washed with EtOAc (1 x 10 mL). The aqueous layer was separated and EtOH (50 mL) was added to the aqueous solution. This solution was refluxed for 24 hours and then cooled to room temperature. The reaction was diluted with water (400 mL) and extracted with EtOAc (1 x 400 mL, 2 x 200 mL). The organic layers were combined and washed with
Bπne (1 x 100 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (silica, 0-20% EtOAc/CH2Cl2) to afford the desired INTERMEDIATE 6. LC-MS negative[M-H]: calc. for C12H10NO4S: 264 Found: 264.
INTERMEDIATE 7

The INTERMEDIATE 7 was prepared according to the same procedures as in the preparation of INTERMEDIATE 5 by replacing ethyl [(lS)-5-methoxy-2,3-dihydro-lH-inden-l-yl]acetate with the product of Step D in INTERMEDIATE 2. LC-MS for C13H13NO4: calculated 247.1 , found 246.1 [M-H].
INTERMEDIATE 8

Step A

This material was prepared according to the procedure used in the Steps A - E in the preparation of INTERMEDIATE 6, using 7-methoxythiochroman-4-one (prepared according to the procedure used in Tetrahedron, 1970, 26(10), 2353-2363) as the starting material. This crude material was used without further purification.
Step B

EXAMPLE l

INTERMEDIATE 2 (52.6 mg, 0.2 mmol) was combined with 4-chloroquinoline (39 mg, 0.24 mmol) and Cs2CO3 (156 mg, 0.48 mmol) in 1 rtiL of N,N-dimethylformamide. The reaction mixture was stirred at 110 0C for 6 hours, then was poured into water and acidified with 2N aq. HCl to pH <2. The resulting solid precipitate was extracted with ethyl acetate and the combined organic layers were washed with water followed by brine, dried over anhydrous Na2SO4, filtered, and concentrated. Purification by preparative thin layer chromatography (silica, 6% methanol in dichloromethane) gave the product. LC- MS calc. for C22H18N2O3S: 390; Found- 391 (M+H). EXAMPLES 2-45
The following examples in Table 1 were prepared following a procedure similar to the one used in the preparation of EXAMPLE 1. These are readily prepared by one of skill in the art of synthetic organic chemistry or medicinal chemistry using the information provided herein.
Table 1







EXAMPLE 46

Step A

To a stirred solution of 5 -hydroxy- 1-indanone (296.3 mg, 2 mmol) in 3 mL of DMF was added 4-chloro- 7-(trifluoromethyl)quinoline (473.2 mg, 2 mmol) and cesium carbonate (977.4 mg, 3 mmol). The reaction mixture was heated at 80 0C for 16 hours. After cooling to room temperature, it was diluted with ethyl acetate, washed with water and brine, dried over magnesium sulfate, filtered and concentrated. The crude product was purified by silica gel column chromatography using ethyl acetate/hexanes as the eluant (10 - 70%). LC-MS LC-MS calc. for C19H12F3NO2: 343; Found: 344 (M+H).
Step B

To a stirred solution of (R)-2-methyl-CBS-oxazaborohdine (1 M in toluene, 57 uL, 5.68 mmol) m 3 mL of dichloromethane at -20 0C was added slowly borane-methyl sulfide complex (1 M in dichloromethane, 1.14 mL, 1.136 mmol). Then the ketone obtained in Step A (195 mg, 0.568 mmol) in 3 mL of dichloromethane was added at -20 0C over 3 h. The reaction mixture was stored in the freezer (-15 0C) over night. The reaction was quenched with methanol, and heated at 70 0C for 1 hour before concentration under reduced pressure. The residue was purified with preparative TLC (40% EtOAC/hexane) to give the desired product. LC-MS calc. for C19H14F3NO2: 345; Found: 346 (M+H).
Step C

To a stirred solution of the product obtained in Step B (90.0 mg, 0.261 mmol) in 1 mL of THF was added l,2,4-oxadiazohdme-3,5-dione (26 6 mg, 0 261 mmol), triphenylphosphine (102.7 mg, 0.392 mmol) and dπsopropyl azo-dicarboxylate (50 6 uL, 0 261 mmol). The reaction mixture was treated in a microwave reactor at 80 0C for 2 mm before being concentrated under reduced pressure. The residue was purified with preparative TLC (8% MeOH/CH2Cl2 plus 0.5% HOAc) to give the desired product. LC-MS calc. for C21H14F3N3O4: 429; Found- 430 (M+H).
EXAMPLE 47

Claims
1. A compound of formula I, or a pharmaceutically acceptable salt thereof:

I wherein A is selected from the group consisting of -CH- and -N-;
B is selected from the group consisting of -S-, -O-, -NH-, and -CH2-;
W is selected from the group consisting of -CH2-, -CF2-, -O-, -C(=O)-, -NR.6-, -S-, -S(O)-, and -S(O)2-;
Z is selected from the group consisting of -CH2- and -CH2CH2-;
or alternatively -W-Z- is selected from the group consisting of -N(R6)C(=O)- and -C(=O)N(R6)-, or -W-Z- represents two atoms that are connected to form one side of a 5-6-membered heteroaromatic ring having 1-3 heteroatoms independently selected from O, N and S, where the 5- membered heteroaromatic ring is optionally substituted with 1 -3 groups independently selected from halogen, CH3 , CF3 , -OCH3 , and -OCF3 ;
Heterocycle is a 5-6 membered saturated or partly saturated monocyclic heterocyclic ring having 1-3 heteroatoms independently selected from O, N and S;
Heteroaryl is a 5-6 membered monocyclic heteroaromatic ring having 1-3 heteroatoms independently selected from O, N and S;
R', R% R^ and R^ are each independently selected from the group consisting of H, halogen, -CN, -NO2, -Q-Coalkyl, -0Ci-C6alkyl, -SCi-C6alkyl, -S(O)2Ci-C6alkyl, -N(R6)(R6), -N(R6)C(=O)C 1 -Cόalkyl, -N(R6)S(O)2C 1 -Cόalkyl, -S(O)2N(R6)(R6), -C(=O)H, -C(O)OH, -C(=O)OCi-C6alkyl, -C(O)Ci -Cόalkyl, -C(=O)N(R6)(R6), -C(=O)Phenyl, -C(=O)Naphthyl, -C(0)Heterocyle, Heterocycle, Heteroaryl, C3-C7-Cycloalkyl, Phenyl and Naphthyl; wherein -Ci-C6alkyl and the alkyl groups of -OCi-Cβalkyl, -SCi-Cόalkyl, -S(O)2Ci-C6alkyl, -N(R6)C(=O)Ci-C6alkyl, -N(R6)S(O)2Ci-C6alkyl, -C(O)OC i-C6alkyl, and -C(O)C i-Cgalkyl are optionally substituted with 1-5 halogens and optionally substituted with 1-2 groups independently selected from -OH, -0C]-C3alkyl which is optionally substituted with 1-5 halogens, -S(O)2Ci-C3alkyl which is optionally substituted with 1-5 halogens, -C(=0)Ci-C3alkyl which is optionally substituted with 1-5 halogens, -0C(=0)Ci-C6alkyl which is optionally substituted with 1-5 halogens, -NHC(=0)CH3, -NHC(=0)0Ci-C6alkyl which is optionally substituted with 1-5 halogens, -NHS(O)2CH3, -N(R6XR6), Heterocycle, Heteroaryl, C3-C7-Cycloalkyl, Phenyl, and Naphthyl; wherein -C(=O)Phenyl, -C(=O)Naphthyl, -C(=O)Heterocyle, Heterocycle, Heteroaryl, C3-C7-Cycloalkyl, Phenyl and Naphthyl either as Rl, R2, R3, or R4, or as substituents on Rl, R2, R3 and R4 are optionally substituted with 1-4 substituents independently selected from halogen, -CF3, -OCF3, -CN, -NO2, -OH, -Ci-C3alkyl, -C(=0)Ci-C3alkyl, -S(O)2Ci-C3alkyl, and -0Ci-C3alkyl, wherein said -Ci-C3alkyl, -0Ci-C3alkyl, -S(O)2Ci-C3alkyl, and -C(=0)Ci-C3alkyl substituents are optionally substituted with 1 -3 halogens; wherein the pair of substituents R^ and R2 may together represent a bridging divalent 4- carbon chain -CH=CH-CH=CH-, forming a fused phenyl ring at the Rl and R2 positions, wherein said fused phenyl ring is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Ci-C3alkyl, -0Ci-C3alkyl, -SCi-C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3; or alternatively the pair of substituents R^ and R2 may together represent a bridging divalent 3-atom chain selected from -CH=CHO-, -OCH=CH-, -CH=CH-S-, and -SCH=CH-, forming a fused furan or thiophene ring at the Rl and R2 positions, wherein said fused furan or thiophene ring is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Q- C3alkyl, -0Ci-C3alkyl, -SCi-C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3, and
Each R6 is independently selected from the group consisting of H and -Ci-Cβalkyl.
2. The compound of Claim 1, or a pharmaceutically acceptable salt thereof, wherein R1, R2, R3, and R4 are independently selected from (I) H; (2) halogen; (3) -NO2; (4) -CN; (5) Ci-C4alkyl, which is optionally substituted with 1-5 halogens and optionally with 1-2 substituents independently selected from -OH, -C(=O)C 1 -C3alkyl, and -OC 1 -C3alkyl, where the C 1 -C3alkyl groups are optionally substituted with 1-3 halogens; (6) -0Ci_C4alkyl, which is optionally substituted with 1-5 halogens and optionally with one -C(=0)Ci-C3 alkyl group; (7) -C(=0)Ci-C3alkyl, which is optionally substituted with 1-5 halogens; (8) -C(O)H; (9) -C(O)N(R6)(R6); (10) -S(O)2N(R6)(R6); (11) -S(O)2Ci-C3alkyl; (12) -N(R6)(R6); (13) C3_C7Cycloalkyl; (14) Phenyl; and (15) Heterocycle; wherein C3-C7Cycloalkyl, Phenyl, and Heterocycle are each optionally substituted with 1-3 substituents independently selected from halogen, -OH, -0Ci-C3alkyl, CF3, and -C(O)C i-C3alkyl; and Each R" is independently selected from H and CH3.
3. The compound of Claim 1, or a pharmaceutically acceptable salt thereof, wherein R^, R2, R3 ; and R4 are each independently selected from H, halogen, Ci-C3alkyl, CF3,
-OCi-C3alkyl, -OCF3, -C(=O)H, -C(=O)N(R6)(R6), -S(O)2N(R6)(R6), -C(=O)Ci-C3alkyl, -CN, -NO2, -S(O)2Ci-C3alkyl, and -N(R6)(R6); and
Each R^ IS independently selected from H and CH3.
4. The compound of Claim 3, or a pharmaceutically acceptable salt thereof, wherein R1, R2, R3, and R4 are each independently selected from H, F, Br, Cl, Ci-C3alkyl, CF3, -C(O)NH2, -S(O)2NH2, -C(=O)CH3, -CN, -OCH3, -OCF3, and -NO2.
5. The compound of Claim 1, or a pharmaceutically acceptable salt thereof, wherein the pair of ortho substituents R^ and R2 together represent a bridging divalent 4-carbon chain -CH=CH-CH=CH-, forming a fused phenyl ring at the Rl and R2 positions, wherein the fused phenyl ring is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Ci-C3alkyl, -OCi-C3alkyl, -SCi-C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3.
6. The compound of Claim 5, or a pharmaceutically acceptable salt thereof, wherein the fused phenyl ring at the Rl and R2 positions is optionally substituted with 1-2 substituents independently selected from halogen, -NO2, -CH3, -CF3, -OCH3, and -OCF3.
7. The compound of Claim 1, or a pharmaceutically acceptable salt thereof, wherein R* and R2 together represent a bridging divalent group selected from -CH=CHS-, -SCH=CH-, -CH=CHO-, and -OCH=CH-, forming a fused furan or thiophene at the Rl and R2 positions, wherein said fused furan or thiophene ring is optionally substituted with 1 -3 substituents independently selected from halogen, -OH, -CN, -NO2, -Ci-C3alkyl, -OQ^alkyl, -SCi-Csalkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3.
8. The compound of Claim 7, or a pharmaceutically acceptable salt thereof, wherein the fused furan or thiophene ring at the Rl and R2 positions is optionally substituted with 1 -2 substituents independently selected from halogen, -NO2, -CH3, -CF3, -OCH3, and -OCF3.
9. The compound of Claim 1, or a pharmaceutically acceptable salt thereof, wherein A is CH- or -N-; and B is selected from -O- and -S-.
10. The compound of Claim 1, or a pharmaceutically acceptable salt thereof, wherein W is selected from the group consisting of -CH2-, -CF2-, -O-, -C(=O)-, -NR6-, - S-, -S(O)-, and -S(0)2S and
Z is selected from the group consisting of -CH2- and -CH2CH2-.
11. The compound of Claim 1 , or a pharmaceutically acceptable salt thereof, wherein W is selected from the group consisting of -CH2-, -O-, and -S-; and Z is selected from the group consisting of -CH2- and -CH2CH2-.
12. The compound of Claim 1, or a pharmaceutically acceptable salt thereof, wherein R^, R^, R3, and R^ are independently selected from (1) H; (2) halogen; (3) -NO2; (4) -CN; (5) -C i-C4alkyl, which is optionally substituted with 1-5 halogens and optionally with 1-2 substituents independently selected from -OH, -C(=O)Ci-C3alkyl, and -OCi X^alkyl, where the Q- C3alkyl groups are optionally substituted with 1-3 halogens; (6) -OCi-C4alkyl, which is optionally substituted with 1-5 halogens and optionally with one -C(=O)Ci-C3alkyl group; (7) -C(=O)Ci-
Cβalkyl, which is optionally substituted with 1-5 halogens; (8) -C(=O)H; (9) -C(=O)N(R6)(R6); (10) -S(O)2N(R6)(R6); (11) -S(O)2Ci-C3alkyl, (12) -N(R6)(R6); (13) C3.C7Cycloalkyl; (14) Phenyl; and (15) Heterocycle, wherein C3_C7Cycloalkyl, Phenyl, and Heterocycle are each optionally substituted with 1-3 substituents independently selected from halogen, -OH, -OCi-C3alkyl, CF3, and -C(=O)Ci- C3alkyl; wherein the pair of substituents R* and R^ may together represent a bridging divalent 4- carbon chain -CH=CH-CH=CH- or a bridging divalent 3-atom chain selected from -CH=CHO-, -OCH=CH-, -CH=CH-S-, and -SCH=CH-, forming a fused phenyl, furan or thiophene ring at the Rl and R2 positions, wherein said fused phenyl, furan or thiophene ring is optionally substituted with 1-3 substituents independently selected from halogen, -OH, -CN, -NO2, -Ci-C3alkyl, -0Ci-C3alkyl, -SC 1- C3alkyl, -S(O)2Ci-C3alkyl, -CF3, and -OCF3;
Each R^ IS independently selected from H and CH3;
W is selected from the group consisting of -CH2-, -0-, and -S-;
Z is selected from the group consisting of-CH2- and -CH2CH2-; A is -CH- or N; and
B is selected from -S- and -O-.
13. The compound of Claim 12, or a pharmaceutically acceptable salt thereof, wherein R^, R^, R^, and R^ are each independently selected from H, F, Br, Cl, Ci-C3alkyl, CF3, -C(=0)NH2, -S(O)2NH2, -C(=O)CH3, -CN, -OCH3, -CN, -OCF3, and -NO2.
14. The compound of Claim 13, or a pharmaceutically acceptable salt thereof, wherein
RI IS selected from the group consisting of H, CH3, and halogen;
R.2 IS selected from the group consisting of H, CH3, and halogen; or alternatively Rl and R2 together represent a bridging group selected from
-CH-CH-CH=CH-, -CH=CHS-, -SCH=CH-, and -OCH=CH-, forming a fused phenyl, thiophene, or furan ring at the Rl and R2 positions, wherein said fused ring is optionally substituted with 1-2 substituents independently selected from halogen, CF3, -OCF3, -OCH3, -CN, -NO2, and Ci-3alkyl;
R^ IS selected from the group consisting of H, halogen, Ci_3alkyl, -OCH3, -OCF3, CF3, -C(=O)NH2, -CN, and -NO2;
R^ IS selected from the group consisting of H, halogen, CH3, CF3, and -S(O)2NH2;
W is selected from the group consisting of -CH2-, -O-, and -S-;
Z is selected from the group consisting of -CH2- and -CH2CH2-;
A is -CH- or N; and
B is selected from -S- and -O-.
15 The compound of Claim 14, which is selected from the group consisting of the compounds below, or a pharmaceutically acceptable salt thereof:





16. A pharmaceutical composition comprising the compound of Claim 1 , or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
17. The use of the compound of Claim 1 or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for the treatment of Type 2 diabetes mellitus.
18. A method of treating type 2 diabetes melhtus in a patient m need of treatment comprising the administration to the patient of a therapeutically effective amount of the compound of formula I, or a pharmaceutically acceptable salt thereof.
19. A pharmaceutical composition comprising (1) the compound of Claim 1 or a pharmaceutically acceptable salt thereof;
(2) one or more compounds selected from the group consisting of :
(a) PPAR gamma agonists and partial agonists;
(b) biguanides;
(c) protein tyrosine phosphatase- IB (PTP-IB) inhibitors; (d) dφeptidyl peptidase IV (DP-IV) inhibitors;
(e) insulin or an insulin mimetic;
(f) sulfonylureas;
(g) α-glucosidase inhibitors;
(h) agents which improve a patient's lipid profile, said agents being selected from the group consisting of (i) HMG-CoA reductase inhibitors, (n) bile acid sequestrants, (in) nicotinyl alcohol, nicotinic acid or a salt thereof, (iv) PP ARa agonists, (v) cholesterol absorption inhibitors, (vi) acyl CoAxholesterol acyltransferase (ACAT) inhibitors, (vii) CETP inhibitors, and (via) phenolic anti -oxidants;
(i) PPARα/γ dual agonists, 0) PP ARδ agonists,
(k) antiobesity compounds,
(1) ileal bile acid transporter inhibitors;
(m) anti-inflammatory agents;
(n) glucagon receptor antagonists; (o) GLP-I,
(p) GIP-I ;
(q) GLP-I analogs; and
(r) HSD-I inhibitors; and
(3) a pharmaceutically acceptable carrier.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US85566506P | 2006-10-31 | 2006-10-31 | |
| US60/855,665 | 2006-10-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008054674A2 true WO2008054674A2 (en) | 2008-05-08 |
| WO2008054674A3 WO2008054674A3 (en) | 2009-04-16 |
Family
ID=39344854
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/022643 WO2008054674A2 (en) | 2006-10-31 | 2007-10-26 | Antidiabetic bicyclic compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008054674A2 (en) |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008054674A2 (en) * | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
| WO2008063202A2 (en) * | 2006-01-30 | 2008-05-29 | Array Biopharma Inc. | Heterobicyclic thiophene compounds for the treatment of cancer |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010085525A1 (en) | 2009-01-23 | 2010-07-29 | Schering Corporation | Bridged and fused heterocyclic antidiabetic compounds |
| WO2010085528A1 (en) | 2009-01-23 | 2010-07-29 | Schering Corporation | Bridged and fused antidiabetic compounds |
| WO2010091176A1 (en) | 2009-02-05 | 2010-08-12 | Schering Corporation | Phthalazine-containing antidiabetic compounds |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| CN102307860A (en) * | 2008-12-18 | 2012-01-04 | 麦它波莱克斯股份有限公司 | GPR120 receptor agonists and uses thereof |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8476308B2 (en) | 2010-06-16 | 2013-07-02 | Metabolex, Inc. | GPR120 receptor agonists and uses thereof |
| WO2014011926A1 (en) | 2012-07-11 | 2014-01-16 | Elcelyx Therapeutics, Inc. | Compositions comprising statins, biguanides and further agents for reducing cardiometabolic risk |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2015095256A1 (en) | 2013-12-19 | 2015-06-25 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| WO2015097713A1 (en) | 2013-11-14 | 2015-07-02 | Cadila Healthcare Limited | Novel heterocyclic compounds |
| WO2016022742A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016022448A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016019863A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [7,6]-fused bicyclic antidiabetic compounds |
| WO2016022446A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [5,6]-fused bicyclic antidiabetic compounds |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| US9957219B2 (en) | 2013-12-04 | 2018-05-01 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| US10000454B2 (en) | 2014-05-22 | 2018-06-19 | Merck Sharp & Dohme | Antidiabetic tricyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US10059667B2 (en) | 2014-02-06 | 2018-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic compounds |
| US10519115B2 (en) | 2013-11-15 | 2019-12-31 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US10676458B2 (en) | 2016-03-29 | 2020-06-09 | Merch Sharp & Dohne Corp. Rahway | Antidiabetic bicyclic compounds |
| US10710986B2 (en) | 2018-02-13 | 2020-07-14 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10774071B2 (en) | 2018-07-13 | 2020-09-15 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10899735B2 (en) | 2018-04-19 | 2021-01-26 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11225471B2 (en) | 2017-11-16 | 2022-01-18 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US11236085B2 (en) | 2018-10-24 | 2022-02-01 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
| US11512065B2 (en) | 2019-10-07 | 2022-11-29 | Kallyope, Inc. | GPR119 agonists |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008054674A2 (en) * | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
-
2007
- 2007-10-26 WO PCT/US2007/022643 patent/WO2008054674A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008054674A2 (en) * | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8003662B2 (en) | 2006-01-30 | 2011-08-23 | Array Biopharma, Inc. | Heterobicyclic thiophene compounds and methods of use |
| WO2008063202A2 (en) * | 2006-01-30 | 2008-05-29 | Array Biopharma Inc. | Heterobicyclic thiophene compounds for the treatment of cancer |
| WO2008063202A3 (en) * | 2006-01-30 | 2009-01-22 | Array Biopharma Inc | Heterobicyclic thiophene compounds for the treatment of cancer |
| WO2008054674A3 (en) * | 2006-10-31 | 2009-04-16 | Merck & Co Inc | Antidiabetic bicyclic compounds |
| WO2008054674A2 (en) * | 2006-10-31 | 2008-05-08 | Merck & Co., Inc. | Antidiabetic bicyclic compounds |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| JP2012512893A (en) * | 2008-12-18 | 2012-06-07 | メタボレックス, インコーポレイテッド | GPR120 receptor agonist and use thereof |
| EP2514745A1 (en) * | 2008-12-18 | 2012-10-24 | Metabolex Inc. | GPR120 receptor agonists and uses thereof |
| US8598374B2 (en) | 2008-12-18 | 2013-12-03 | Metabolex, Inc. | GPR120 receptor agonists and uses thereof |
| CN102307860A (en) * | 2008-12-18 | 2012-01-04 | 麦它波莱克斯股份有限公司 | GPR120 receptor agonists and uses thereof |
| WO2010085528A1 (en) | 2009-01-23 | 2010-07-29 | Schering Corporation | Bridged and fused antidiabetic compounds |
| US9278965B2 (en) | 2009-01-23 | 2016-03-08 | Merck Sharp & Dohme Corp. | Bridged and fused antidiabetic compounds |
| WO2010085525A1 (en) | 2009-01-23 | 2010-07-29 | Schering Corporation | Bridged and fused heterocyclic antidiabetic compounds |
| US8575166B2 (en) | 2009-02-05 | 2013-11-05 | Merck Sharp & Dohme Corp. | Phthalazine-containing antidiabetic compounds |
| WO2010091176A1 (en) | 2009-02-05 | 2010-08-12 | Schering Corporation | Phthalazine-containing antidiabetic compounds |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| US8476308B2 (en) | 2010-06-16 | 2013-07-02 | Metabolex, Inc. | GPR120 receptor agonists and uses thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014011926A1 (en) | 2012-07-11 | 2014-01-16 | Elcelyx Therapeutics, Inc. | Compositions comprising statins, biguanides and further agents for reducing cardiometabolic risk |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US9527875B2 (en) | 2012-08-02 | 2016-12-27 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US9840512B2 (en) | 2013-02-22 | 2017-12-12 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US9932311B2 (en) | 2013-10-08 | 2018-04-03 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2015097713A1 (en) | 2013-11-14 | 2015-07-02 | Cadila Healthcare Limited | Novel heterocyclic compounds |
| US10246470B2 (en) | 2013-11-14 | 2019-04-02 | Cadila Healthcare Limited | Heterocyclic compounds |
| US10011609B2 (en) | 2013-11-14 | 2018-07-03 | Cadila Healthcare Limited | Heterocyclic compounds |
| US10519115B2 (en) | 2013-11-15 | 2019-12-31 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US9957219B2 (en) | 2013-12-04 | 2018-05-01 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US9834563B2 (en) | 2013-12-19 | 2017-12-05 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| WO2015095256A1 (en) | 2013-12-19 | 2015-06-25 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| EP3974413A1 (en) | 2013-12-19 | 2022-03-30 | Merck Sharp & Dohme Corp. | Antidiabetic substituted heteroaryl compounds |
| US10059667B2 (en) | 2014-02-06 | 2018-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic compounds |
| US10000454B2 (en) | 2014-05-22 | 2018-06-19 | Merck Sharp & Dohme | Antidiabetic tricyclic compounds |
| WO2016022448A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US10968193B2 (en) | 2014-08-08 | 2021-04-06 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016019863A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [7,6]-fused bicyclic antidiabetic compounds |
| US10100042B2 (en) | 2014-08-08 | 2018-10-16 | Merck Sharp & Dohme Corp. | [5,6]—fused bicyclic antidiabetic compounds |
| US10131651B2 (en) | 2014-08-08 | 2018-11-20 | Merck Sharp & Dohme Corp. | [7,6]-fused bicyclic antidiabetic compounds |
| WO2016022742A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016022446A1 (en) | 2014-08-08 | 2016-02-11 | Merck Sharp & Dohme Corp. | [5,6]-fused bicyclic antidiabetic compounds |
| US10662171B2 (en) | 2014-08-08 | 2020-05-26 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| US10676458B2 (en) | 2016-03-29 | 2020-06-09 | Merch Sharp & Dohne Corp. Rahway | Antidiabetic bicyclic compounds |
| US11072602B2 (en) | 2016-12-06 | 2021-07-27 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| US10968232B2 (en) | 2016-12-20 | 2021-04-06 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
| US11225471B2 (en) | 2017-11-16 | 2022-01-18 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| US10710986B2 (en) | 2018-02-13 | 2020-07-14 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11555029B2 (en) | 2018-02-13 | 2023-01-17 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10899735B2 (en) | 2018-04-19 | 2021-01-26 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US10774071B2 (en) | 2018-07-13 | 2020-09-15 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11236085B2 (en) | 2018-10-24 | 2022-02-01 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| US11512065B2 (en) | 2019-10-07 | 2022-11-29 | Kallyope, Inc. | GPR119 agonists |
| US11279702B2 (en) | 2020-05-19 | 2022-03-22 | Kallyope, Inc. | AMPK activators |
| US11851429B2 (en) | 2020-05-19 | 2023-12-26 | Kallyope, Inc. | AMPK activators |
| US11407768B2 (en) | 2020-06-26 | 2022-08-09 | Kallyope, Inc. | AMPK activators |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008054674A3 (en) | 2009-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008054674A2 (en) | Antidiabetic bicyclic compounds | |
| EP2079467B1 (en) | Antidiabetic bicyclic compounds | |
| EP1846379B1 (en) | Antidiabetic bicyclic compounds | |
| EP2021327B1 (en) | Antidiabetic bicyclic compounds | |
| US20080090865A1 (en) | Antidiabetic Bicyclic Compounds | |
| JP4759517B2 (en) | Production and use of arylalkyl acid derivatives for the treatment of obesity | |
| CA2749930A1 (en) | Bridged and fused heterocyclic antidiabetic compounds | |
| AU3821401A (en) | Aryloxyacetic acids for diabetes and lipid disorders | |
| WO2003065983A2 (en) | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia | |
| AU2010206783A1 (en) | Pentafluorosulpholane-containing antidiabetic compounds | |
| CA2571789A1 (en) | Indoles having anti-diabetic activity | |
| WO2013056679A1 (en) | Novel heteroaryl-amino derivatives | |
| WO2010129729A1 (en) | Substituted spirocyclic amines useful as antidiabetic compounds | |
| WO2007018956A2 (en) | Antidiabetic oxazolidinediones and thiazolidinediones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07839786 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07839786 Country of ref document: EP Kind code of ref document: A2 |