WO2006022272A1 - 炭酸塩の製造方法 - Google Patents
炭酸塩の製造方法 Download PDFInfo
- Publication number
- WO2006022272A1 WO2006022272A1 PCT/JP2005/015294 JP2005015294W WO2006022272A1 WO 2006022272 A1 WO2006022272 A1 WO 2006022272A1 JP 2005015294 W JP2005015294 W JP 2005015294W WO 2006022272 A1 WO2006022272 A1 WO 2006022272A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbonate
- producing
- carbon dioxide
- source
- dioxide gas
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F11/00—Compounds of calcium, strontium, or barium
- C01F11/18—Carbonates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/60—Preparation of carbonates or bicarbonates in general
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F11/00—Compounds of calcium, strontium, or barium
- C01F11/18—Carbonates
- C01F11/182—Preparation of calcium carbonate by carbonation of aqueous solutions and characterised by an additive other than CaCO3-seeds
- C01F11/183—Preparation of calcium carbonate by carbonation of aqueous solutions and characterised by an additive other than CaCO3-seeds the additive being an organic compound
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F11/00—Compounds of calcium, strontium, or barium
- C01F11/18—Carbonates
- C01F11/186—Strontium or barium carbonate
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G21/00—Compounds of lead
- C01G21/14—Carbonates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G9/00—Compounds of zinc
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/02—Compounds of alkaline earth metals or magnesium
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/02—Compounds of alkaline earth metals or magnesium
- C09C1/021—Calcium carbonates
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/14—Compounds of lead
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/01—Particle morphology depicted by an image
- C01P2004/03—Particle morphology depicted by an image obtained by SEM
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/10—Particle morphology extending in one dimension, e.g. needle-like
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/54—Particles characterised by their aspect ratio, i.e. the ratio of sizes in the longest to the shortest dimension
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/62—Submicrometer sized, i.e. from 0.1-1 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/60—Optical properties, e.g. expressed in CIELAB-values
Definitions
- the present invention relates to a carbonate having orientation birefringence, in particular, a carbonate having any one of a needle shape and a rod shape at an efficiency near room temperature without heating control at 50 ° C or higher.
- Background art related to a method for producing a carbonate that can be formed easily and easily and can control the particle size
- carbonates for example, calcium carbonate
- carbonates with high functionality have been developed one after another. Depending on the shape and particle size, etc., it is used for many purposes and for many purposes.
- crystal form of the carbonate include calacite, aragonite, and patelite.
- aragonite is acicular and is useful for various applications because of its excellent strength and elastic modulus.
- a method for producing carbonate there are a method of producing carbonate by reacting a solution containing carbonate ions and a solution of chloride, and a method of producing carbonate by reacting chloride and carbon dioxide. Such methods are generally known.
- a method for producing a needle-like carbonate having an aragonite structure for example, in the former method, a reaction between a solution containing carbonate ions and a salt solution is performed under ultrasonic irradiation (Patent Literature). 1) and Ca (OH) water
- Patent Document 2 A method in which acicular aragonite crystals to be seed crystals are added and the seed crystals are grown only in a certain direction (see Patent Document 2), sodium aluminate is added to the calcium hydroxide slurry, and 50 ° A method of heating to C or higher and blowing carbon dioxide gas (see Patent Document 3) has been proposed.
- a deflection beam is used as a reading beam or a writing beam, and when a birefringent optical element (for example, the disk itself, a lens, etc.) exists in the optical path, reading is performed. Or it will adversely affect writing accuracy.
- a birefringent optical element for example, the disk itself, a lens, etc.
- Patent Document 4 a non-birefringent optical resin material using a polymer resin and inorganic fine particles having different birefringence signs.
- the optical resin material is obtained by a technique called a crystal doping method. Specifically, a large number of inorganic fine particles are dispersed in a polymer resin, and a molding force is applied from the outside by stretching or the like. The polymer resin binding chains and a large number of inorganic fine particles are oriented almost in parallel, and the birefringence caused by the orientation of the polymer resin binding chains is reduced by the birefringence of the inorganic particles with different signs. It is a thing.
- inorganic fine particles that can be used for the crystal dope method are indispensable. It has been recognized that fine needle-like or rod-like carbonates can be used particularly suitably.
- Patent Document 1 Japanese Patent Application Laid-Open No. 59-203728
- Patent Document 2 U.S. Pat.No. 5,164,172
- Patent Document 3 Japanese Patent Laid-Open No. 8-2914
- Patent Document 4 Pamphlet of International Publication No. 01Z25364
- the present invention relates to a carbonate having orientation birefringence, in particular, a carbonate having either a needle shape or a rod shape, in an efficient and simple manner near room temperature without performing heating control. It is an object of the present invention to provide a method for producing a carbonate which can be formed and can control the particle size.
- the present invention is based on the above knowledge obtained by the present inventor, and means for solving the above problems are as follows. That is,
- Carbon dioxide gas derived from a carbon dioxide source is released into the gas phase, and the carbon dioxide gas released into the gas phase is converted into Sr 2+ ions, Ca 2+ ions, Ba 2+ ions, Zn 2+ ions.
- a carbonate having an aspect ratio greater than 1 by being dissolved in a liquid containing a metal ion source containing at least one metal ion selected from Pb 2+ This is a method for producing a salt.
- the carbon dioxide gas derived from the carbonic acid source is released into the gas phase.
- the carbon dioxide gas released into the gas phase dissolves in the liquid containing the metal ion source. At this time, the carbon dioxide gas slowly diffuses and dissolves in the liquid. The result is a carbonate with an aspect ratio greater than 1.
- ⁇ 2> The method for producing a carbonate according to ⁇ 1>, wherein a carbonate having a needle-like shape or a rod-like shape is produced.
- the carbonate Since the carbonate having any one of the needle shape and the rod shape is produced, the carbonate can be used for multipurpose and versatile purposes. It can also be applied to non-birefringent optical resin materials.
- ⁇ 3> The carbonic acid according to any one of ⁇ 1> to ⁇ 2>, wherein the metal ion source is a hydroxide of at least one metal selected from Sr, Ca, Ba, Zn, and Pb.
- the metal ion source is a metal hydroxide
- the carbonate is synthesized in a high alkali region. As a result, a fine carbonate having a high aspect ratio can be obtained.
- ⁇ 4> The method for producing carbonate according to any one of ⁇ 1> to ⁇ 3>, wherein the carbonic acid source is solid or gaseous.
- ⁇ 6> The method for producing carbonate according to any one of ⁇ 1> to ⁇ 5>, wherein the carbon dioxide gas is released in a closed container.
- ⁇ 9> The method for producing a carbonate according to any one of ⁇ 1> to ⁇ 8>, wherein the solution contains a solvent.
- the solvent since the solvent is contained, the solubility of the obtained carbonate can be reduced.
- ⁇ 10> The method for producing a carbonate according to ⁇ 9>, wherein the solvent is at least one selected from methanol, ethanol, and isopropyl alcohol.
- a conventional problem can be solved, and carbonates having orientation birefringence, particularly carbonates having either a needle shape or a rod shape, are brought to the vicinity of room temperature without performing heating control. Therefore, it is possible to provide a method for producing a carbonate that can be formed efficiently and simply and whose particle size can be controlled.
- FIG. 1 is a conceptual diagram illustrating an example of a method for producing a carbonate of the present invention.
- FIG. 2A is a graph showing the relationship between pH and elapsed time of a strontium hydroxide suspension in Example 1.
- FIG. 2B is an SEM photograph of the strontium carbonate crystal produced in Example 1.
- FIG. 3A is an SEM photograph (magnification: 1,020 times) of the strontium carbonate crystal produced in Comparative Example 1.
- FIG. 3B is an SEM photograph (magnification: 5, 0 to 30 times) of the strontium carbonate crystal produced in Comparative Example 1.
- carbon dioxide gas derived from a carbon dioxide source is released into a gas phase, and the carbon dioxide gas released into the gas phase is dissolved in a liquid containing a metal ion source.
- the carbonic acid source is a force that can be appropriately selected depending on the purpose without particular limitation as long as it generates carbon dioxide gas.
- the state is preferably either solid or gas.
- the solid carbonic acid source can be appropriately selected according to the purpose without any particular limitation.
- ammonium carbonate [(NH 3) 2 CO 3] is preferable.
- gaseous carbon dioxide source examples include carbon dioxide gas.
- the metal ion source can be appropriately selected depending on the purpose without particular limitation as long as it contains metal ions, but it reacts with the carbon dioxide gas to be any one of calacite, aragonite, battery, and amorphous. Those forming a carbonate having a morphology are preferred, and those forming a carbonate having an aragonite-type crystal structure are particularly preferred.
- the crystal structure of the aragonite type is represented by a metal ion and CO 2 — unit, and the CO 2 "
- 3 3 Units are stacked to form a carbonate with either a needle or bar shape. For this reason, when the carbonate is added to a polymer film or the like, Thus, when the film is stretched in any one direction, the crystals are arranged in a state where the major axis direction of the particles coincides with the stretching direction.
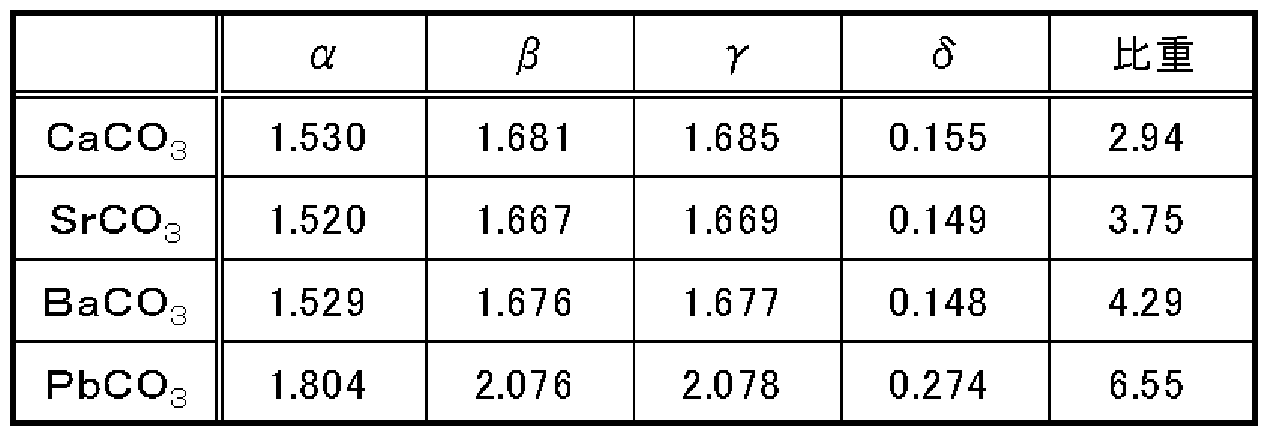
- Table 1 shows the refractive index of aragonite-type minerals. As shown in Table 1, the carbonate having an aragonite-type crystal structure has a large birefringence ⁇ , and therefore can be suitably used for doping a polymer having orientation birefringence.
- the metal ion source is not particularly limited as long as it contains at least one metal ion selected from Sr ion, Ca ion, Ba ion, Zn ion, and Pb2 + ion force. Examples thereof include nitrates, chlorides, and hydroxides of at least one metal selected from Sr, Ca, Ba, Zn, and Pb. Among these, the metal hydroxide is particularly preferable.
- the metal ion source is a hydroxide of the metal, carbonate is synthesized in a high alkali region, so that a fine carbonate with a high aspect ratio and strength can be obtained.
- the liquid containing the metal ion source contains water. Therefore, the liquid containing the metal ion source is preferably an aqueous solution or a suspension.
- the solubility of the carbonate crystals to be synthesized it is preferable to include a solvent in the liquid.
- the solvent is not particularly limited and can be appropriately selected according to the purpose, but preferably includes methanol, ethanol, isopropyl alcohol and the like. These may be used alone or in combination of two or more.
- the amount of the solvent added is not particularly limited and can be appropriately selected depending on the purpose, but 1 to 80% by volume of the solvent amount after the carbonate production is preferable, and 10 to 80% by volume is more preferable. Good.
- the method for releasing the carbon dioxide gas into the gas phase can be appropriately selected according to the purpose without any particular limitation, but it is preferable that the carbon dioxide gas is released in a sealed container.
- the method for dissolving the carbon dioxide gas in the liquid containing the metal ion source can be appropriately selected according to the purpose without any particular limitation.
- the carbon dioxide gas can be slowly diffused and dissolved. preferable.
- liquid containing the metal ion source and the carbonic acid source are accommodated in one sealed container.
- the manner in which the liquid containing the metal ion source and the carbonic acid source are accommodated in the sealed container can be appropriately selected according to the purpose without any particular limitation.
- the carbonic acid source is a solid
- the solid carbonic acid source may be attached to a side surface disposed on the bottom surface of the sealed container, or may be housed by being suspended from the top surface.
- the closed container is provided so that the discharge pressure of the carbon dioxide gas can be controlled.
- the control mode of the discharge pressure of the carbon dioxide gas can be appropriately selected according to the purpose for which there is no particular limitation.
- the carbon dioxide gas is contained in the liquid containing the metal ion source. It is preferable to set the discharge pressure of the carbon dioxide gas so that a desired mass melts.
- the carbonate source is a solid
- the liquid 2 containing the metal ion source is placed in a (sealed) container 1 and placed together with the carbonic acid source 3 on the bottom surface of the one (sealed) container 1 for storage.
- carbon dioxide gas 4 is generated from the carbon dioxide source 3, and the carbon dioxide source 3 releases the carbon dioxide gas 4 into the gas phase in the (closed) container 1.
- the carbon dioxide gas 4 released into the gas phase diffuses slowly and dissolves in the liquid 2 containing the metal ion source.
- the metal ions in the liquid 2 react with carbonate ions generated by the carbon dioxide gas 4 dissolved in the liquid 2 to produce a carbonate.
- strontium carbonate (SrCO 3) as the carbonate is synthesized.
- the reaction temperature in the synthesis reaction is not particularly limited, and can be appropriately selected according to the purpose.
- the ability to synthesize carbonates from about room temperature to less than 50 ° C Power 25 ° C A temperature around room temperature is preferable.
- the reaction time is not particularly limited and may be appropriately selected according to the purpose. However, it is preferably 15 to 360 minutes, more preferably 30 to 240 minutes.
- the synthesis reaction is preferably performed while stirring the liquid containing the metal ion source.
- the stirring speed is preferably 500 to 1,500 rpm.
- the carbonate produced by the method for producing a carbonate of the present invention needs to have an aspect ratio of more than 1, and preferably has a needle-like shape or a rod-like shape.
- the aspect ratio represents the ratio between the length and the diameter of the carbonate, and the larger the value, the better.
- the aspect ratio is preferably 1.5 to 20, and more preferably 1.5 to 8.
- the average particle length of the carbonate is 0.05-30111, more preferably 0.05-5111. If the average particle length exceeds 30 m, it may be greatly affected by scattering, and the adaptability to optical applications may be reduced.
- the proportion of carbonates having a length of [average particle length] is 60% or more, more preferably 70% or more, and more preferably 75% or more. % Or more is particularly preferable. When the proportion is 60% or more, it is recognized that the control of the particle size is highly accurate.
- 0.05 to 2 ⁇ m force is preferable, 0.05 to: L 0 ⁇ m force ⁇ is more preferable, 0.05 to 0.8 m force is more preferable, and 0.05 to 0. 1 m power ⁇ Especially preferred!
- the carbonate produced by the carbonate production method of the present invention has little orientation inside the molded article. It is useful as a plastic reinforcement, friction material, thermal insulation, filter, etc. In particular, in a composite material subjected to deformation such as a stretched material, the strength and optical properties can be improved by orienting the particles.
- a carbonate (crystal) produced by the carbonate production method of the present invention is dispersed in an optical polymer having birefringence, and subjected to a stretching treatment to form a binding chain of the optical polymer and the carbonate. And the birefringence caused by the orientation of the bond chain of the optical polymer can be canceled by the birefringence of the carbonate.
- the stretching treatment can be appropriately selected depending on the purpose without particular limitation, and examples thereof include uniaxial stretching.
- An example of the method of the axial stretching is stretching with a stretching machine to a desired stretching ratio while heating as necessary.
- a carbonate having orientation birefringence particularly a carbonate having an acicular or rod-like shape, without any heating control at 50 ° C. or higher. It can be formed efficiently and simply near room temperature. Further, it is possible to obtain a carbonate having a constant particle size at a high rate by controlling the particle size.
- Ammonium carbonate [(NH 3) 2 CO 3] was placed in a closed container. Then (NH) CO
- Figure 2A shows the relationship between pH and elapsed time of strontium hydroxide [Sr (OH)] suspension
- strontium carbonate crystals were removed by filtration and dried.
- the dried strontium carbonate crystals were observed with a scanning electron microscope (SEM) (Hitachi, S-900).
- Example 1 a 0.01 M strontium hydroxide [Sr (OH)] suspension was added to a 0.01 M Except for replacing with calcium hydroxide [Ca (OH)] suspension, the same method as in Example 1 was used.
- Example 1 isopropyl alcohol (IPA) as the solvent in the strontium hydroxide [Sr (OH)] suspension was used for the purpose of reducing the solubility of the crystals obtained.
- IPA isopropyl alcohol
- Sr (OH) strontium hydroxide
- a strontium carbonate crystal as the carbonate was produced in the same manner as in Example 1 except that was added. The obtained strontium carbonate crystals were observed by SEM photographs. Table 3 shows the results of various measurements.
- Example 1 a 0.01 M strontium hydroxide [Sr (OH)] suspension was added to a 0.01 M
- Barium carbonate crystals as the acid salt were prepared. The obtained barium carbonate crystals were observed by SEM. Table 4 shows the various measurement results.
- Example 1 a 0.01 M strontium hydroxide [Sr (OH)] suspension was added to a 0.01 M
- Carbonic acid was prepared in the same manner as in Example 1 except that it was replaced with a zinc hydroxide [Zn (OH)] suspension.
- Zinc carbonate crystals as a salt were produced.
- the obtained zinc carbonate crystals were observed by SEM.
- Table 4 shows the various measurement results.
- Example 1 a 0.01 M strontium hydroxide [Sr (OH)] suspension was added to a 0.01 M
- the reaction was carried out with stirring to produce strontium carbonate crystals.
- the stirring speed is
- the obtained strontium carbonate crystals were removed by filtration and dried.
- the dried strontium carbonate crystals were observed by SEM.
- the SEM photograph at this time is shown in Fig. 3A and Fig. 3B.
- the method produced strontium carbonate crystals.
- the obtained strontium carbonate crystals were observed by SEM photographs.
- Table 5 shows the various measurement results.
- a strontium carbonate crystal as the carbonate was produced in the same manner as in Comparative Example 1 except that it was added.
- the obtained strontium carbonate crystals were observed by SEM photographs. Table 5 shows the measurement results.
- Example 1 Example 2
- Example 3 Metal ion source Sr (OH) 2 Ca (OH) 2 Sr (OH) 2 Carbonate source (NH 4 ) 2 C0 3 (NH 4 ) 2 C0 3 (NH 4 ) 2 C0 3 Isopropyl solvent ⁇ ⁇
- the method for producing carbonate of the present invention can control the particle size, and can efficiently and easily produce a carbonate having a certain particle size at a high ratio.
- the carbonate produced by the method for producing a carbonate of the present invention has a high crystallinity and an aspect ratio that is difficult to aggregate and is larger than 1 (particularly, needle-like, rod-like, etc.). It exhibits isotropic properties with little orientation, and can be suitably used for plastic reinforcing materials, friction materials, heat insulating materials, filters, and the like. In particular, in a composite material subjected to deformation such as stretching, the strength and optical properties can be improved by orienting the particles.
- the carbonate (crystal) produced by the carbonate production method of the present invention is dispersed in an optical polymer having birefringence, and subjected to a stretching treatment, whereby the binding chain of the optical polymer and the carbonate are substantially reduced.
- the birefringence caused by the orientation of the bond chain of the optical polymer can be canceled by the birefringence of the carbonate.
- it can be suitably used for an optical component, in particular, an optical element in which deflection characteristics are important and high accuracy is required.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Compounds Of Alkaline-Earth Elements, Aluminum Or Rare-Earth Metals (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
Abstract
Description


Claims
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006531924A JPWO2006022272A1 (ja) | 2004-08-25 | 2005-08-23 | 炭酸塩の製造方法 |
| US11/660,948 US20080260614A1 (en) | 2004-08-25 | 2005-08-23 | Process for Producing Carbonate Particles |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004-245116 | 2004-08-25 | ||
| JP2004245116 | 2004-08-25 | ||
| JP2005222081 | 2005-07-29 | ||
| JP2005-222081 | 2005-07-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006022272A1 true WO2006022272A1 (ja) | 2006-03-02 |
Family
ID=35967483
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2005/015294 WO2006022272A1 (ja) | 2004-08-25 | 2005-08-23 | 炭酸塩の製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20080260614A1 (ja) |
| JP (1) | JPWO2006022272A1 (ja) |
| KR (1) | KR20070040418A (ja) |
| WO (1) | WO2006022272A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006117487A (ja) * | 2004-10-22 | 2006-05-11 | Ube Material Industries Ltd | 平板状炭酸ストロンチウム粒子 |
| JP2006193411A (ja) * | 2004-12-15 | 2006-07-27 | Fuji Photo Film Co Ltd | 炭酸塩の製造方法 |
| JP2007076934A (ja) * | 2005-09-12 | 2007-03-29 | Ube Material Industries Ltd | 炭酸ストロンチウム微粉末及びその製造方法 |
| JP2008247692A (ja) * | 2007-03-30 | 2008-10-16 | Fujifilm Corp | 炭酸塩及びその製造方法 |
| JP2009078970A (ja) * | 2008-12-02 | 2009-04-16 | Ube Material Industries Ltd | 針状炭酸ストロンチウム粒子の製造方法 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4800671B2 (ja) * | 2004-12-15 | 2011-10-26 | 富士フイルム株式会社 | 炭酸塩結晶及びその製造方法、並びに透明光学用樹脂組成物 |
| US20100163199A1 (en) * | 2008-12-31 | 2010-07-01 | Weyerhaeuser Company | Readily defibered pulp product |
| US20100163200A1 (en) * | 2008-12-31 | 2010-07-01 | Weyerhaeuser Company | Method for making readily defibered pulp product |
| JP6058250B2 (ja) * | 2010-04-12 | 2017-01-11 | 日東電工株式会社 | 粒子分散樹脂組成物、粒子分散樹脂成形体およびそれらの製造方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63100011A (ja) * | 1986-10-14 | 1988-05-02 | Sumitomo Chem Co Ltd | 球形微粒子の製造方法 |
| JPH01212209A (ja) * | 1988-02-17 | 1989-08-25 | Mitsui Mining & Smelting Co Ltd | 金属炭酸塩の製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4421729A (en) * | 1982-01-11 | 1983-12-20 | Fmc Corporation | Preparation of strontium carbonate |
| DE3587060T2 (de) * | 1984-10-18 | 1993-05-19 | Pfizer | Kugelfoermiges ausgefaelltes calciumcarbonat, seine herstellung und seine verwendung. |
| JPS6350316A (ja) * | 1986-08-21 | 1988-03-03 | Yahashi Kogyo Kk | 六角板状粒子炭酸カルシウムの生成方法 |
| JP2684112B2 (ja) * | 1989-06-29 | 1997-12-03 | 丸尾カルシウム株式会社 | 針状形状をしたアラゴナイト結晶形炭酸カルシウムの製造方法 |
| US5059407A (en) * | 1990-03-28 | 1991-10-22 | Liquid Carbonic Corporation | Liquid carbon dioxide injection in exothermic chemical reactions |
| ATE182559T1 (de) * | 1995-10-26 | 1999-08-15 | Solvay Barium Strontium Gmbh | Mikronisiertes erdalkalimetallcarbonat |
| US6159436A (en) * | 1996-11-18 | 2000-12-12 | Centro De Investigacion Y De Estudios Avanzados Del I.P.N. | Conversion process for strontium sulfate in carbonate rich celestite ores to strontium carbonate using sodium carbonate in an air/vapor-lift loop reactor |
| CN1257209C (zh) * | 1999-10-05 | 2006-05-24 | 小池康博 | 非双折射性的光学树脂材料 |
-
2005
- 2005-08-23 JP JP2006531924A patent/JPWO2006022272A1/ja active Pending
- 2005-08-23 WO PCT/JP2005/015294 patent/WO2006022272A1/ja active Application Filing
- 2005-08-23 KR KR1020077006652A patent/KR20070040418A/ko not_active IP Right Cessation
- 2005-08-23 US US11/660,948 patent/US20080260614A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63100011A (ja) * | 1986-10-14 | 1988-05-02 | Sumitomo Chem Co Ltd | 球形微粒子の製造方法 |
| JPH01212209A (ja) * | 1988-02-17 | 1989-08-25 | Mitsui Mining & Smelting Co Ltd | 金属炭酸塩の製造方法 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006117487A (ja) * | 2004-10-22 | 2006-05-11 | Ube Material Industries Ltd | 平板状炭酸ストロンチウム粒子 |
| JP2006193411A (ja) * | 2004-12-15 | 2006-07-27 | Fuji Photo Film Co Ltd | 炭酸塩の製造方法 |
| JP2007076934A (ja) * | 2005-09-12 | 2007-03-29 | Ube Material Industries Ltd | 炭酸ストロンチウム微粉末及びその製造方法 |
| JP2008247692A (ja) * | 2007-03-30 | 2008-10-16 | Fujifilm Corp | 炭酸塩及びその製造方法 |
| JP2009078970A (ja) * | 2008-12-02 | 2009-04-16 | Ube Material Industries Ltd | 針状炭酸ストロンチウム粒子の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20070040418A (ko) | 2007-04-16 |
| JPWO2006022272A1 (ja) | 2008-05-08 |
| US20080260614A1 (en) | 2008-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006022272A1 (ja) | 炭酸塩の製造方法 | |
| JP4140884B2 (ja) | 炭酸ストロンチウムの製造方法、非複屈折性光学樹脂材料並びに光学素子 | |
| JP2006021988A (ja) | 炭酸塩の製造方法 | |
| KR101207827B1 (ko) | 카보네이트 결정과 그 제조 방법 및 투명 광학용 수지조성물 | |
| TWI373453B (en) | Method for producing carbonate | |
| JP4884702B2 (ja) | 炭酸塩の製造方法 | |
| JP2009001475A (ja) | 炭酸ストロンチウム微粒子の製造方法 | |
| JP5081372B2 (ja) | 炭酸塩の製造方法 | |
| JP5242934B2 (ja) | 炭酸塩及びその製造方法 | |
| KR101232520B1 (ko) | 탄산염 제조 방법 | |
| JP5123546B2 (ja) | 炭酸塩及びその製造方法 | |
| JP2006176367A (ja) | 炭酸塩結晶の製造方法 | |
| JP5144012B2 (ja) | 炭酸塩の製造方法 | |
| WO2006137344A1 (ja) | 光学用樹脂組成物及びその製造方法 | |
| JP4273066B2 (ja) | 針状炭酸ストロンチウム粒子 | |
| JP2007055866A (ja) | 炭酸ストロンチウム結晶及びその製造方法、並びに樹脂組成物 | |
| JP4920666B2 (ja) | 針状炭酸ストロンチウム粒子の製造方法 | |
| JP2008222514A (ja) | 炭酸ストロンチウム及びその製造方法 | |
| TWI392701B (zh) | 光學用樹脂組成物及其製法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006531924 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077006652 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11660948 Country of ref document: US |
|
| 122 | Ep: pct application non-entry in european phase |