RU2679309C2 - Способ получения метионина - Google Patents
Способ получения метионина Download PDFInfo
- Publication number
- RU2679309C2 RU2679309C2 RU2016114077A RU2016114077A RU2679309C2 RU 2679309 C2 RU2679309 C2 RU 2679309C2 RU 2016114077 A RU2016114077 A RU 2016114077A RU 2016114077 A RU2016114077 A RU 2016114077A RU 2679309 C2 RU2679309 C2 RU 2679309C2
- Authority
- RU
- Russia
- Prior art keywords
- methionine
- suspension
- solution
- crystallization
- ammonium salt
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 59
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 title claims description 40
- 229930182817 methionine Natural products 0.000 title claims description 40
- FFEARJCKVFRZRR-UHFFFAOYSA-N methionine Chemical compound CSCCC(N)C(O)=O FFEARJCKVFRZRR-UHFFFAOYSA-N 0.000 claims abstract description 149
- 239000000243 solution Substances 0.000 claims abstract description 74
- 239000000725 suspension Substances 0.000 claims abstract description 65
- 239000000654 additive Substances 0.000 claims abstract description 53
- 150000003863 ammonium salts Chemical class 0.000 claims abstract description 38
- 230000000996 additive effect Effects 0.000 claims abstract description 37
- 239000007864 aqueous solution Substances 0.000 claims abstract description 31
- 239000000203 mixture Substances 0.000 claims abstract description 26
- 239000007900 aqueous suspension Substances 0.000 claims abstract description 16
- 239000011734 sodium Substances 0.000 claims abstract description 14
- 235000014113 dietary fatty acids Nutrition 0.000 claims abstract description 13
- 239000000194 fatty acid Substances 0.000 claims abstract description 13
- 229930195729 fatty acid Natural products 0.000 claims abstract description 13
- 239000003945 anionic surfactant Substances 0.000 claims abstract description 12
- 150000004665 fatty acids Chemical class 0.000 claims abstract description 12
- 239000002736 nonionic surfactant Substances 0.000 claims abstract description 9
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 claims abstract description 8
- 150000002148 esters Chemical class 0.000 claims abstract description 8
- 238000004519 manufacturing process Methods 0.000 claims abstract description 8
- 229920006395 saturated elastomer Polymers 0.000 claims abstract description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical group [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims abstract description 4
- 229910052700 potassium Chemical group 0.000 claims abstract description 4
- 239000011591 potassium Chemical group 0.000 claims abstract description 4
- 239000007787 solid Substances 0.000 claims abstract description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical group C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims abstract description 3
- 125000003118 aryl group Chemical group 0.000 claims abstract description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 3
- 125000004122 cyclic group Chemical group 0.000 claims abstract description 3
- 229910052708 sodium Inorganic materials 0.000 claims abstract description 3
- 238000002425 crystallisation Methods 0.000 claims description 46
- 230000008025 crystallization Effects 0.000 claims description 45
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 40
- 229960004452 methionine Drugs 0.000 claims description 39
- 229910021529 ammonia Inorganic materials 0.000 claims description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 20
- 239000002518 antifoaming agent Substances 0.000 claims description 19
- 239000004480 active ingredient Substances 0.000 claims description 10
- 239000012452 mother liquor Substances 0.000 claims description 9
- 239000003795 chemical substances by application Substances 0.000 claims description 7
- 229920002545 silicone oil Polymers 0.000 claims description 7
- 238000004821 distillation Methods 0.000 claims description 6
- 238000001704 evaporation Methods 0.000 claims description 5
- 230000008020 evaporation Effects 0.000 claims description 5
- 238000000926 separation method Methods 0.000 claims description 5
- 239000004094 surface-active agent Substances 0.000 claims description 5
- 150000001408 amides Chemical class 0.000 claims description 4
- 150000002825 nitriles Chemical class 0.000 claims description 4
- 230000007062 hydrolysis Effects 0.000 claims description 3
- 238000006460 hydrolysis reaction Methods 0.000 claims description 3
- 238000004090 dissolution Methods 0.000 claims description 2
- 125000000129 anionic group Chemical group 0.000 claims 1
- 239000002002 slurry Substances 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 4
- 239000002244 precipitate Substances 0.000 abstract description 4
- 239000000126 substance Substances 0.000 abstract description 2
- 230000015572 biosynthetic process Effects 0.000 abstract 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 238000001035 drying Methods 0.000 description 6
- 239000006260 foam Substances 0.000 description 6
- 239000000047 product Substances 0.000 description 5
- 238000007127 saponification reaction Methods 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 4
- GKBFFPJHOLVCHW-WCCKRBBISA-N azanium;(2s)-2-amino-4-methylsulfanylbutanoate Chemical compound [NH4+].CSCC[C@H](N)C([O-])=O GKBFFPJHOLVCHW-WCCKRBBISA-N 0.000 description 4
- 238000005086 pumping Methods 0.000 description 4
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 4
- 235000012239 silicon dioxide Nutrition 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000011942 biocatalyst Substances 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- RYFIVUXDWBPMNT-UHFFFAOYSA-N diazanium;methanedisulfonate Chemical compound [NH4+].[NH4+].[O-]S(=O)(=O)CS([O-])(=O)=O RYFIVUXDWBPMNT-UHFFFAOYSA-N 0.000 description 3
- 150000002191 fatty alcohols Chemical class 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- ASFAZLDBJLQTSG-WCCKRBBISA-N (2s)-2-amino-4-methylsulfanylbutanoic acid;imidazolidine-2,4-dione Chemical compound O=C1CNC(=O)N1.CSCC[C@H](N)C(O)=O ASFAZLDBJLQTSG-WCCKRBBISA-N 0.000 description 2
- MWLKEJXYXYRWIH-UHFFFAOYSA-N 2-amino-4-methylsulfanylbutanenitrile Chemical compound CSCCC(N)C#N MWLKEJXYXYRWIH-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- -1 Sorbitan fatty acid esters Chemical class 0.000 description 2
- 238000004833 X-ray photoelectron spectroscopy Methods 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 239000013530 defoamer Substances 0.000 description 2
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- SBKRXUMXMKBCLD-SCSAIBSYSA-N (R)-5-[2-(methylthio)ethyl]hydantoin Chemical compound CSCC[C@H]1NC(=O)NC1=O SBKRXUMXMKBCLD-SCSAIBSYSA-N 0.000 description 1
- IJCWFDPJFXGQBN-UHFFFAOYSA-N 2-[4-Hydroxy-3-(octadecanoyloxy)oxolan-2-yl]-2-(octadecanoyloxy)ethyl octadecanoate Polymers CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)C1OCC(O)C1OC(=O)CCCCCCCCCCCCCCCCC IJCWFDPJFXGQBN-UHFFFAOYSA-N 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- GSYTVXOARWSQSV-BYPYZUCNSA-N L-methioninamide Chemical compound CSCC[C@H](N)C(N)=O GSYTVXOARWSQSV-BYPYZUCNSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229910010413 TiO 2 Inorganic materials 0.000 description 1
- 229920004482 WACKER® Polymers 0.000 description 1
- IJCWFDPJFXGQBN-RYNSOKOISA-N [(2R)-2-[(2R,3R,4S)-4-hydroxy-3-octadecanoyloxyoxolan-2-yl]-2-octadecanoyloxyethyl] octadecanoate Polymers CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCCCCCCCCCCCC IJCWFDPJFXGQBN-RYNSOKOISA-N 0.000 description 1
- IJCWFDPJFXGQBN-JYVCTSCWSA-N [2-[(2R,3R)-4-hydroxy-3-octadecanoyloxyoxolan-2-yl]-2-octadecanoyloxyethyl] octadecanoate Polymers CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)[C@H]1OCC(O)[C@H]1OC(=O)CCCCCCCCCCCCCCCCC IJCWFDPJFXGQBN-JYVCTSCWSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000011552 falling film Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- WJRBRSLFGCUECM-UHFFFAOYSA-N hydantoin Chemical compound O=C1CNC(=O)N1 WJRBRSLFGCUECM-UHFFFAOYSA-N 0.000 description 1
- 229940091173 hydantoin Drugs 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- CDWZRHFHYAOGGK-WCCKRBBISA-M potassium;(2s)-2-amino-4-methylsulfanylbutanoate Chemical compound [K+].CSCC[C@H](N)C([O-])=O CDWZRHFHYAOGGK-WCCKRBBISA-M 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- IJCWFDPJFXGQBN-BIFNRIDTSA-N sorbitan tristearate Polymers CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)[C@H]1OC[C@@H](O)[C@@H]1OC(=O)CCCCCCCCCCCCCCCCC IJCWFDPJFXGQBN-BIFNRIDTSA-N 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C321/00—Thiols, sulfides, hydropolysulfides or polysulfides
- C07C321/12—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to acyclic carbon atoms
- C07C321/14—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/14—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides
- C07C319/20—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides by reactions not involving the formation of sulfide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/26—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/26—Separation; Purification; Stabilisation; Use of additives
- C07C319/28—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Изобретение относится к способу кристаллизации D,L-метионина из водных растворов и/или суспензий, которые содержат D,L-метионин и аммониевую соль D,L-метионина и имеют содержание Met 70-180 г/кг раствора и/или суспензии, предпочтительно 90-150 г/кг раствора и/или суспензии, и содержание NH1-5 г/кг раствора и/или суспензии. Способ осуществляют в присутствии кристаллизующей добавки, которая содержит неионогенное или анионное поверхностно-активное средство или смесь различных неионогенных или анионных поверхностно-активных средств. При этом температуру раствора и/или суспензии снижают сразу или постепенно от T=85-110°C до Т=30-50°С таким образом, что D,L-метионин оседает в виде твердого вещества. Способ характеризуется тем, что анионное поверхностно-активное средство, применяемое для образования кристаллизующей добавки, является одним из соединений, отображенных в формулах 1-3, или их смесью. В формулах 1-3 n является целым числом от 1 до 10, М является натрием или калием, a R, Rи Rявляются линейной, разветвленной или циклической, насыщенной или ненасыщенной С-С-алкильной группой или арильной группой. При этом неионогенная кристаллизующая добавка является сложным эфиром сорбитана и жирной кислоты или смесью различных сложных эфиров сорбитана и жирной кислоты. Предлагаемый способ позволяет получать D,L-метионин с высокими объемными плотностями. Изобретение относится также к способу получения D,L-метионина с использованием указанного способа кристаллизации D,L-метионина. 2 н. и 20 з.п. ф-лы, 1 ил., 3 табл., 4 пр.
Description
В настоящем изобретении предусматривают способ выделения D,L-метионина с объемными плотностями > 550 г/л из растворов, содержащих аммониевую соль D,L-метионина.
Уровень техники
Известны способы получения D,L-метионина, в которых D,L-метионин вначале получают в качестве метионината аммония.
В соответствии с US 20050176115 водный, аммоний-содержащий раствор D,L-метионина получают посредством реагирования водных растворов 2-амино-4-метилтиобутиронитрила уже в присутствии NH3 с использованием биокатализаторов с получением D,L-метионина. D,L-метионин затем осаждают путем удаления аммиака при пониженном давлении. Полученный таким образом D,L-метионин имеет чистоту 99%. Объемная плотность полученного D,L-метионина не указывается.
Схожим образом в JP 2004-254690, D,L-метионин и NH3 образуются приведением в реакцию 2-амино-4-метилтиобутиронитрила с биокатализатором. Растворимость метионина повышается в присутствии аммиака, что облегчает удаление биокатализатора. Точные условия выделения и характеристики продукта D,L-метионина в JP 2004-254690 не указаны.
В DE 60127538 описывают способ, при котором D,L-метионин получают из амида метионина путем реакции омыления в присутствии катализатора. В данном случае аммиак полностью удаляют из получаемого раствора метионината аммония путем отгонки и метионин кристаллизуют, при этом объемные плотности полученного метионина не указаны.
В WO 2008006977, D,L-метионин получают из гидантоина метионина путем омыления с NH3 и D,L-метионин получают путем выпаривания NH3 и CO2 при пониженном давлении. Относительно характеристик продукта ничего не указано.
В WO 2007034065 также описывают аммонийное омыление гидантоина метионина. Аммиак удаляют из метионината аммония в отгоночной колонне и метионин затем осаждают путем охлаждения раствора. Характеристики образованного метионина не упоминаются в указанном документе.
В DE 10238212 описывают способ, при котором гидантоин метионина омыляют в воде при высоких температурах в присутствии или без катализатора. NH3 и CO2 частично удаляют перед кристаллизацией получаемого метионина. Нет упоминания об остаточных количествах CO2 и аммиака в растворе, из которого осуществляют кристаллизацию, также не проясняется ничего относительно объемной плотности получаемого метионина.
В WO 2003050071 описывают водные смеси сложных эфиров полиэтиленгликоля и жирных кислот с модифицированными целлюлозами, которые применяют в качестве вспомогательных средств при кристаллизации метионина из растворов метионината калия, нейтрализованных диоксидом углерода из щелочного омыления гидантоина метионина. В этих конкретных способах в присутствии больших количеств калия получают объемные плотности до 586 г/л, в одном случае 620 г/л (с гидроксиэтилцеллюлозной добавкой). В то же время не приводится подробной информации о кристаллизации D,L-метионина из растворов, содержащих D,L-метионинат аммония.
Растворимость D,L-метионина повышается в присутствии NH3 (смотри кривые растворимости из JP 2004-254690).
Для экономичного способа выделения D,L-метионина, следовательно, является преимуществом как можно более полное удаление аммиака с целью максимизировать количество способного кристаллизоваться D,L-метионина. С этой целью аммиак можно удалять рядом способов, известных из литературы, таких как отгонка, выпаривание при пониженном давлении и т. д.
Цель настоящего изобретения
Целью настоящего изобретения является создание способа выделения D,L-метионина, исходя из растворов, содержащих метионинат аммония, в которых D,L-метионин получают с высокими объемными плотностями > 550 г/л.
Описание
Настоящие исследования показали, что D,L-метионин при кристаллизации из водных растворов обычно получают с низкими объемными плотностями < 550 г/л. Заметного увеличения объемной плотности нельзя достигнуть исключительно добавлением вспомогательных средств для кристаллизации. Неожиданно было показано, что остаточное количество NH3 в комбинации с добавленными добавками приводит к получению D,L-метионина с более высокой объемной плотностью.
Поскольку в водных растворах, содержащих и NH3 и метионин, устанавливается равновесие между метионинатом аммония с одной стороны и аммиаком с другой стороны, ссылка ниже для простоты относится только к концентрациям NH4 +, независимо от того, относится это к NH4 + или к NH3.
Цель, упомянутая выше, а также другие, связанные, но не указанные в явной форме цели, достигаются обеспечением
способа кристаллизации D,L-метионина из водных растворов и/или суспензий, содержащих D,L-метионин и аммонийную соль D,L-метионина, характеризующихся содержанием Met (включающим в себя общее количество метионина в форме D,L-метионина и аммонийной соли D,L-метионина) 70–180 г/кг раствора и/или суспензии (7–18% по весу), предпочтительно 90–150 г/кг (9–15% по весу),
содержания NH4 + 1–5 г/кг раствора и/или суспензии (0,1–0,5% по весу), предпочтительно от 1,5 до 3,0 г/кг (0,15–0,3% по весу), в присутствии кристаллизующей добавки,
которая содержит неионогенное или анионное поверхностно-активное средство или смесь различных неионогенных или анионных поверхностно-активных средств,
при котором температуру раствора и/или суспензии снижают сразу или постепенно от диапазона температур T1 = 85–110°C до диапазона температур T2 = 30–50°C таким образом, что метионин оседает в виде твердого вещества из раствора и/или суспензии.
Водный раствор, содержащий D,L-метионин и аммониевую соль D,L-метионина, в данном документе нужно понимать как раствор, в котором преобладающая доля общего метионина (Мет) находится в растворенном виде и лишь небольшие доли Met макс. до 5% находятся в нерастворенном виде, т. е. суспендированы.
Водную суспензию, содержащую D,L-метионин и аммониевую соль D,L-метионина, в данном документе нужно понимать как суспензию, в которой значительная доля, а именно > 5%, общего присутствующего метионина суспендирована, в то время как оставшаяся доля находится в растворенной форме.
Соответственно, оптимальная концентрация NH4 +, исходя из метионина, составляет по меньшей мере приблизительно 5 г NH4 + / кг Met и не более чем приблизительно 60 г NH4 + / кг Met, в то время как концентрация метионина в растворе находится в диапазоне от 90 г/кг до 150 г/кг.
Концентрацию NH4 + в этом случае можно определять, например, путем использования NH4 +-чувствительного электрода, в соответствии с известными способами. Концентрацию NH4 + в этом случае обычно определяют путем измерения образца раствора, приведенного к pH 11 и сравнением с измерением растворов NH4Cl известной концентрации, также приведенных к pH 11.
Концентрацию метионина в растворе и/или в суспензии проще всего определять с помощью HPLC.
Путем описанной комбинации присутствия ионов NH4 +, добавления кристаллизующих добавок, регуляции температуры кристаллизации и применения способа по настоящему изобретению получают крупнозернистые кристаллы метионина, которые можно легко отфильтровать и которые после высушивания имеют объемную плотность > 550 г/л.
В качестве анионных поверхностно-активных средств пригодными являются, в частности, поверхностно-активные средства, соответствующие одному из соединений, отображенных в формулах 1-3, или их смесям:
R1–O–SO3M (формула 1);
R2–O–(CH2)n–SO3M (формула 2);
R3-(O-C2H4)n-O-SO3M (формула 3);
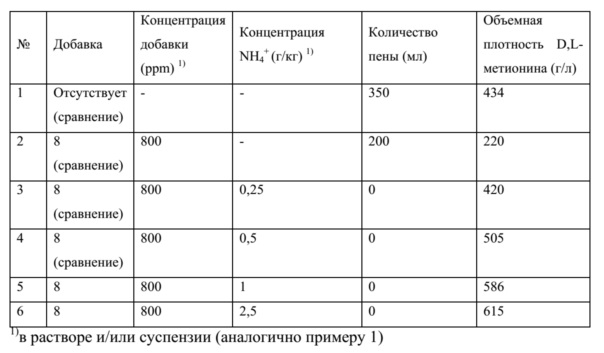
где n является целым числом от 1 до 10, M является натрием или калием, а R1, R2 и R3 являются линейной, разветвленной или циклической насыщенной или ненасыщенной от C8- до C20-алкильной группой или арильной группой. Относительно высокие объемные плотности от 564 до 588 кг/л получали с этими поверхностно-активными средствами, как показано в таблице 1, в примере 1, с добавками 2, 3 и 4.
Предпочтение отдают применению поверхностно-активных средств, в которых n равен 2, а R1, R2 и R3 являются линейными насыщенными C8-C18-алкильными группами, поскольку они являются коммерчески легкодоступными и эффективными.
Особое предпочтение отдается применению анионных поверхностно-активных средств формул
CnH2n+1-O-SO3Na, где n = 12-18 (Sulfopon® 1218G, Oleochemicals);
CnH2n+1-O-C2H4-SO3Na, где n = 8-18 (Hostapon® SCI 85, Clariant);
CnH2n+1-(OC2H4)2-O-SO3Na, где n = 12 (Disponil® FES 27, Cognis).
В предпочтительном варианте осуществления данного способа применяемое неионогенное поверхностно-активное средство является сложным эфиром сорбитана и жирной кислоты или смесью различных сложных эфиров сорбитана и жирной кислоты, особенно предпочтительно полиэтоксилированными сложными эфирами сорбитана и жирной кислоты. Сложные эфиры сорбитана и жирной кислоты имеют преимущество, поскольку они являются эффективными и коммерчески легкодоступными. В наиболее предпочтительном варианте осуществления неионогенным поверхностно-активным средством является полиэтоксилированный сорбитан тристеарат, согласно формуле 4:

где w+x+y+z = 20.
С помощью данных поверхностно-активных средств получали относительно высокую объемную плотность 578 кг/л, как показано в примере 3.
Концентрация кристаллизующей добавки (на основе активного ингредиента) в растворе и/или суспензии, из которых осуществляют кристаллизацию, составляет предпочтительно по меньшей мере от 700 ppm до не более 4000 ppm, исходя из общей массы раствора и/или суспензии, особенно предпочтительно по меньшей мере от 750 ppm до не более 2000 ppm, наиболее особенно предпочтительно по меньшей мере от 800 ppm до не более 1000 ppm. Это обеспечивает хорошее развитие эффекта добавки без вовлечения слишком многих посторонних материалов в раствор продукта.
Для того, чтобы достигнуть оптимального дозирования и распределения кристаллизующей добавки последнюю обычно применяют в форме водного раствора или эмульсии, где концентрация кристаллизующей добавки в растворе или эмульсии составляет предпочтительно от 2 до 15% по весу.
В предпочтительном варианте осуществления способа по данному изобретению раствор, из которого осуществляют кристаллизацию, дополнительно содержит противовспениватель. Назначение противовспенивателя – подавлять пену, возникающую во время обработки раствора и/или суспензии метионина и вызванную или интенсифицированную некоторыми из кристаллизующих добавок, упомянутых выше. Более того, неожиданно при одновременном применении противовспенивателя и кристаллизующих добавок в случае достижения объемных плотностей метионина возникает синергический эффект, в силу чего достигают объемных плотностей даже более 600 г/л и одновременно избегают негативных эффектов процессов насыщения и, таким образом, способ по настоящему изобретению можно также осуществлять в непрерывном режиме. Это особенно очевидно при сравнении экспериментов 2, 3 и 4 (без добавления противовспенивателя -> объемные плотности 564-588 кг/л) с экспериментами 8, 9 и 10 (с добавлением противовспенивателя -> объемные плотности 630-634 кг/л) соответственно в таблице 1 примера 1. Кроме того, этот вариант осуществления позволяет применять меньшее количество кристаллизующей добавки.
Предпочтение отдают применению противовспенивателей, содержащих силиконовое масло, поскольку было доказано, что оно особенно эффективно, при этом предпочтительно используют силиконовое масло, имеющее коэффициент кинематической вязкости от 0,65 до 10000 мм2/с (измеренный при 25°C в соответствии с DIN 53018), особенно предпочтительно от 90 до 1500 мм2/с. Противовспениватель может дополнительно содержать компоненты, которые эффективны в качестве эмульгаторов, например, смеси полиэтоксилированных жирных кислот и полиэтоксилированных жирных спиртов. Противовспениватель может подобным образом содержать кремниевую кислоту. В предпочтительном варианте осуществления противовспениватель является водным раствором, содержащим от 5 до 10% по весу силиконового масла, от 0,05 до 1% по весу кремниевой кислоты, от 0,5 до 5% по весу смеси полиэтоксилированных жирных кислот и от 2 до 7% по весу смеси полиэтоксилированных жирных спиртов.
Предпочтение отдают применению противовспенивателя в смеси с кристаллизующей добавкой. Для того, чтобы достичь непрерывного стабильного дозирования противовспенивателя, его предпочтительно дополнительно разбавляют водой перед применением.
Применение противовспенивателей на основе силиконового масла означает, что кремний можно обнаружить в метионине, полученном способом согласно настоящему изобретению при применении приемлемого аналитического способа (например, рентгеновская фотоэлектронная спектроскопия, сокращенно XPS).
Предпочтение отдают применению противовспенивателя в способе согласно настоящему изобретению так, чтобы весовое отношение противовспениватель:кристаллизующая добавка (на основе активного ингредиента) в растворе или суспензии, из которых осуществляют кристаллизацию, находится в диапазоне от 4:1 до 1:1, предпочтительно в диапазоне от 3:1 до 2:1 и концентрация кристаллизующей добавки (на основе активного ингредиента) в этом случае составляет по меньшей мере от 50 ppm до не более 1200 ppm, исходя из общей массы раствора и/или суспензии, предпочтительно от 100 ppm до 600 ppm, особенно предпочтительно от 200 ppm до 400 ppm. Высокие объемные плотности D,L-метионина значительно выше 600 г/л, достигаемые в этом случае, можно видеть особенно эффективно в примере 4/таблице 3.
Способ согласно настоящему изобретению в этом случае предпочтительно осуществлять таким образом, чтобы кристаллизация проходила путем введения водного раствора и/или суспензии, нагретых до 85-110°C, содержащих D,L-метионин и аммониевую соль D,L-метионина, в водный раствор и/или суспензию, нагретые до 30-50°C, содержащие D,L-метионин и аммониевую соль D,L-метионина, при этом температуру получаемой смеси постоянно поддерживают на уровне от 30 до 50°C.
Особенное предпочтение отдают в этом случае введению водного раствора, нагретого до 85-110°C, содержащего D,L-метионин и аммониевую соль D,L-метионина, в водную суспензию, нагретую до 30-50°C, содержащую D,L-метионин и аммониевую соль D,L-метионина. Преимущество этого заключается в том, что таким образом получают кристаллы, которые можно особенно эффективно отфильтровывать.
Дополнительный предпочтительный вариант осуществления данного способа характеризуется тем, что кристаллизацию осуществляют в две стадии, при этом на первой стадии кристаллизации раствор и/или суспензию, нагретые до 85-110°C, содержащие D,L-метионин и аммониевую соль D,L-метионина, вводят в суспензию, нагретую до 60-80°C, содержащую D,L-метионин и аммониевую соль D,L-метионина, и температуру получаемой смеси постоянно поддерживают на уровне от 60 до 80°C, и при этом суспензию, нагретую до 60-80°C, содержащую D,L-метионин и аммониевую соль D,L-метионина, полученную на первой стадии кристаллизации, вводят на второй стадии кристаллизации в суспензию, нагретую до 30-50°C, содержащую D,L-метионин и аммониевую соль D,L-метионина, при этом температуру получаемой смеси постоянно поддерживают на уровне от 30 до 50°C. В этом случае долю примесей в кристаллизационной среде можно особенно эффективно контролировать или можно уменьшать выделением на приемлемом этапе без ослабления положительного эффекта объемной плотности выше 550 г/л.
Дополнительный предпочтительный вариант осуществления способа согласно настоящему изобретению характеризуется тем, что кристаллизацию осуществляют путем вакуумной кристаллизации, при этом давление на первой стадии кристаллизации составляет от 60 до 1000 мбар и, если кристаллизацию осуществляют в две стадии, давление на второй стадии кристаллизации составляет от 35 до 200 мбар. В этом случае преимуществом является то, что задействованные поверхности являются менее холодными. Холодные поверхности могут привести к нежелательному локальному комкованию.
Водные растворы и/или суспензии, содержащие D,L-метионин и аммониевые соли D,L-метионина, применяемые для способа кристаллизации согласно настоящему изобретению, могут быть получены предварительно растворением и/или суспендированием D,L-метионина в воде в присутствии соответствующих количеств аммиака. Метионин может происходить из любого производственного процесса, что предполагает повсеместно применимый способ.
Как доказано, в этом случае будет предпочтительным то, что D,L-метионин, характеризующийся содержанием метионина по меньшей мере 90% по весу, предпочтительно по меньшей мере 95% по весу, применяют для растворения.
Особенно приемлемым для этой цели является D,L-метионин в форме чистого метионина и/или необработанного метионина из любого производственного процесса, характеризующийся остаточной влажностью от 0,1 до 9,5% по весу, предпочтительно от 0,2 до 4,5% по весу. Применение чистого или необработанного метионина такого качества, что он еще содержит влагу после фильтрации в промышленном способе получения D,L-метионина, является предпочтительным, поскольку таким образом непосредственно в конце процесса после высушивания можно получать D,L-метионин с требуемыми свойствами, в частности объемной плотностью 550 кг/л.
Способ согласно настоящему изобретению является также особенно приемлемым для выделения D,L-метионина из водных растворов и/или суспензий, содержащих D,L-метионин и аммониевые соли D,L-метионина, которые были получены путем гидролиза нитрила D,L-метионина и/или амида D,L-метионина, а именно без применения солеобразующих кислотных или основных омыляющих средств (таких как HCl, H2SO4 или гидроксиды щелочных металлов, такие как NaOH или KOH), отличных от аммиака. Такие несолеобразующие омыляющие средства или омыляющие катализаторы, такие, как TiO2 или MnO2, известны в соответствующей патентной литературе и, подобно аммиаку в качестве омыляющего средства при омылении нитрила D,L-метионина и/или амида D,L-метионина, непосредственно приводят к получению водных растворов и/или суспензий, содержащих D,L-метионин и аммониевые соли D,L-метионина, из которых можно получать D,L-метионин путем кристаллизации способом согласно настоящему изобретению. Такие водные растворы и/или суспензии, таким образом, практически не содержат ионов щелочных металлов, таких как ионы Na+ или K+.
Следовательно, дополнительным объектом настоящего изобретения является
способ получения D,L-метионина, при котором водный раствор аммониевой соли D,L-метионина изначально образуют путем гидролиза нитрила D,L-метионина и/или амида D,L-метионина, и водный раствор и/или суспензию, содержащие D,L-метионин и аммониевую соль D,L-метионина, получают из него путем частичного удаления аммиака, который присутствует в связанном виде в качестве иона аммония, из аммониевой соли D,L-метионина, из которых затем получают D,L-метионин способом кристаллизации согласно настоящему изобретению.
В способе согласно настоящему изобретению первично образованный раствор метионината аммония, следовательно, вначале подвергают обеднению в отношении аммиака. Способы для этой цели известны: этого можно достигнуть, например, нагреванием при пониженном давлении или отгонкой с паром.
Соответственно, способ согласно настоящему изобретению для получения D,L-метионина предпочтительно осуществляют таким образом, что водный раствор аммониевой соли D,L-метионина доводят до содержания NH4 + 1-5 г/кг раствора и/или суспензии, предпочтительно 1,5-3,0 г/кг, путем выпаривания и/или отгонки аммиака до кристаллизации, например, путем нагревания при пониженном давлении или отгонки с паром.
Тем не менее, при этом концентрация аммония в соответствии с настоящим изобретением уменьшается только до значений, находящихся в диапазоне от приблизительно 5 до приблизительно 60 г NH3/кг метионина. В этом случае будет преимуществом выбрать достаточно высокую температуру, чтобы D,L-метионин не выпадал в осадок уже во время удаления аммиака, а оставался в растворе. Горячий раствор метионина предпочтительно быстро охлаждают путем подачи в предварительно загруженную, более холодную суспензию метионина, таким образом создают избыточную концентрацию растворенного D,L-метионина и D,L-метионин выпадает в осадок из раствора.
Процедуру также можно проводить таким образом, что водный раствор и/или суспензию D,L-метионина и/или аммониевой соли D,L-метионина доводят до соответствующего содержания Met 70-180 г/кг раствора, предпочтительно 90-150 г/кг раствора, путем добавления воды и/или D,L-метионина, что предполагает изменяемый и гибкий в применении способ.
Выпавший в осадок D,L-метионин предпочтительно отделяют от получаемого маточного раствора и высушивают или изначально перекристаллизовывают и высушивают после отделения от маточного раствора, полученного в этом случае, вследствие этого после высушивания наконец получают D,L-метионин со степенью чистоты по меньшей мере 99% по весу и объемной плотностью по меньшей мере 550 г/л.
Полученный при этом маточный раствор предпочтительно подают обратно на стадию кристаллизации, что приводит к минимизации потерь метионина.
Способ можно осуществлять как непрерывно, так и периодически.
На фиг. 1 показано в виде примера и схемы непрерывную процедуру способа согласно настоящему изобретению. Раствор метионината аммония вначале подают в приемлемое устройство A для уменьшения концентрации аммиака. Обычно это выпарная система, например, содержащая выпарной аппарат с падающей пленкой или циркуляционный выпарной аппарат. При этом условия выбирают так, что количество NH3 от 1 до 5 г/кг раствора и/или суспензии присутствует в потоке продукта, в то время как концентрация Met находится в диапазоне 70-180 г/кг раствора и/или суспензии, предпочтительно 90-150 г/кг. Кристаллизирущую добавку согласно настоящему изобретению, необязательно содержащую противовспениватель, непрерывно добавляют к этому потоку продукта. Температура раствора и/или суспензии, содержащей D,L-метионин и/или аммониевую соль D,L-метионина, составляет предпочтительно от 90 до 100°C. Данный раствор Met и/или суспензию можно нагревать до 100-110°C, если требуется, посредством одного или более теплообменников B, и затем можно предпочтительно быстро охлаждать в течение одной или более стадий до температур 30-50°C в приемлемом кристаллизационном устройстве C, где кристаллизуется D,L-метионин. Если требуется, суспензию D,L-метионина можно подавать в промежуточный сосуд D, для того чтобы обеспечить последующее осаждение D,L-метионина. Наконец, D,L-метионин выделяют на приемлемой стадии E отделения твердого вещества и жидкости, как, например, фильтрация или центрифугирование, где полученный фильтрат можно подавать обратно, если требуется, с загрузочным материалом в устройство A. Это может вести к обогащению добавками согласно настоящему изобретению.
Следующие примеры предназначены для иллюстрирования изобретения в подробностях, но не его ограничения.
Примеры
Пример 1. Подбор анионной поверхностно-активной добавки
40 г D,L-метионина и 360 г воды загружали в колбу и обеспечивали циркуляцию через теплообменник путем прокачивания при температуре 40°C. К данной суспензии добавляли со скоростью 18 мл/мин. раствор 125 г D,L-метионина в 1125 г воды, нагретый до 90°C, при этом поддерживали температуру загруженной суспензии 40°C. После добавления 650 мл горячего раствора, удаляли 500 мл суспензии и затем дополнительно добавляли 500 мл горячего раствора со скоростью 18 мл/мин. Получаемую суспензию сливали, количество пены определяли в мл и D,L-метионин отфильтровывали и промывали с помощью 300 мл ацетона. После высушивания D,L-метионина определяли объемную плотность.
Для экспериментов с NH3 требуемую концентрацию рассчитывали как концентрацию NH4 + и корректировали как в исходных растворах, так и в исходных суспензиях. Дополнительно, количество метионина увеличивали количеством, эквимолярным количеству добавленного NH3.
Эксперименты по кристаллизации осуществляли в присутствии следующих добавок, где указанную концентрацию корректировали добавлением добавки как в исходные растворы, так и в исходные суспензии.
Добавка 1
Водную смесь, содержащую 6,9% по весу силиконового масла с коэффициентом кинематической вязкости 1000 мм2/с (AK 1000, Wacker-Chemie GmbH), 0,27% по весу гидрофобизированной кремниевой кислоты (Sipernat D10, Evonik Degussa GmbH) и 17,9% по весу смеси полиэтоксилированных жирных кислот (Intrasol® FS 18/90/7, Ashland Deutschland GmbH), применяли в качестве чистого противовспенивателя 1 (добавка 1, сравнительный пример).
Применяемые чистые кристаллизующие добавки представляли собой следующие анионные поверхностно-активные средства:
добавка 2) CnH2n+1-O-SO3Na, где n = 12-18 (Sulfopon® 1218G, Oleochemicals);
добавка 3) CnH2n+1-O-C2H4-SO3Na, где n = 8-18 (Hostapon® SCI 85, Clariant);
добавка 4) CnH2n+1-(OC2H4)2-O-SO3Na, где n = 12 (Disponil® FES 27, Cognis);
сравнительный пример, добавка 5) CnH2n+1-(OC2H4)12-O-SO3Na, где n = 12 (Disponil® FES 993, Cognis);
сравнительный пример, добавка 6) CnH2n+1-(OC2H4)30-O-SO3Na, где n = 12 (Disponil® FES 77, Cognis).
Для комбинирования кристаллизующих добавок с противовспенивателем применяли водную смесь 7, содержащую 6,1% по весу силиконового масла с коэффициентом кинематической вязкости 1000 мм2/с (AK 1000, Wacker Chemie GmbH), 0,25% по весу гидрофобизированной кремниевой кислоты (Sipernat D10, Evonik Degussa GmbH), 2,6% по весу смеси полиэтоксилированных жирных кислот (Intrasol® FS 18/90/7, Ashland Deutschland GmbH), 3,7% по весу смеси полиэтоксилированных жирных спиртов (2,35% по весу Marlipal®, Sasol Germany GmbH, 1,35% по весу Brij C2, Croda Chemicals Europe), в воде (соответствует 12,65% по весу активного ингредиента).
В каждом случае смесь применяли с 5,1% по весу соответствующей кристаллизующей добавки (2, 3 или 4) в воде (соответствует 17,75% по весу общего активного ингредиента, доведенного до 100% по весу с водой). Применяли следующие добавки:
добавка 8) = (7) + (2);
добавка 9) = (7) + (3);
добавка 10)= (7) + (4).
Соотношение противовспениватель 7:кристаллизующая добавка (2, 3, 4) в каждом случае составляло 2,5:1 (на основе активного игредиента). Данные по концентрации в таблице 1 показывают общее содержание активного ингредиента добавки без воды, исходя из общей массы раствора или суспензии.
Таблица 1

Пример 2. Влияние концентрации NH4 + на объемную плотность метионина
40 г метионина и 360 г воды загружали в колбу и обеспечивали циркуляцию через теплообменник путем прокачивания при температуре 40°C. К данной суспензии добавляли со скоростью 18 мл/мин. раствор 125 г метионина в 1125 г воды, нагретый до 90°C, при этом поддерживали температуру загруженной суспензии 40°C. После добавления 650 мл горячего раствора, удаляли 500 мл суспензии и затем дополнительно добавляли 500 мл горячего раствора со скоростью 18 мл/мин. Получаемую суспензию сливали, определяли количество пены и метионин отфильтровывали и промывали с помощью 300 мл ацетона. После высушивания метионина определяли объемную плотность.
Для экспериментов с NH3 требуемую концентрацию рассчитывали как концентрацию NH4 + и корректировали как в исходных растворах, так и в исходных суспензиях путем добавления водного раствора NH3.
Эксперименты по кристаллизации осуществляли в присутствии добавки 8 (согласно примеру 1), где указанную концентрацию корректировали добавлением добавки таким же образом как в исходные растворы, так и в исходные суспензии согласно таблице 2.
Данные по концентрации в таблице 2 показывают общее содержание активного ингредиента добавки без воды, исходя из общей массы раствора или суспензии.
Таблица 2
Пример 3. Неионогенное поверхностно-активное средство
48 г D,L-метионина, 348 г воды, 3,8 г водного раствора NH3 (25%) и 0,32 г Tween 65 загружали в колбу и обеспечивали циркуляцию через теплообменник путем прокачивания при температуре 40°C. К этой суспензии добавляли со скоростью 18 мл/мин. раствор 151 г D,L-метионина, 1087 г воды, 11,8 г водного раствора NH3 (25%) и 1,0 г Tween 65, нагретый до 90°C, при этом поддерживали температуру загруженной суспензии 40°C. После добавления 650 мл горячего раствора удаляли 500 мл суспензии и затем дополнительно добавляли 500 мл горячего раствора со скоростью 18 мл/мин. Получаемую суспензию сливали, определяли количество пены и D,L-метионин отфильтровывали и промывали с помощью 300 мл ацетона. После высушивания D,L-метионина определяли объемную плотность.
Наблюдали 0 мл пены, и объемная плотность выделенного D,L-метионина составляла 578 г/л.
Пример 4. Эксперименты, зависящие от концентрации
48 г D,L-метионина, 348 г воды, 3,8 г водного раствора NH3 (25%) загружали в колбу и обеспечивали циркуляцию через теплообменник путем прокачивания при температуре 40°C. Добавляли к этой суспензии со скоростью 18 мл/мин. раствор 151 г D,L-метионина, 1087 г воды и 11,8 г водного раствора NH3 (25%), нагретый до 90°C, при этом поддерживали температуру загруженной суспензии 40°C. После добавления 650 мл горячего раствора удаляли 500 мл суспензии и затем дополнительно добавляли 500 мл горячего раствора со скоростью 18 мл/мин. Получаемую суспензию сливали, определяли количество пены и D,L-метионин отфильтровывали и промывали с помощью 300 мл ацетона. После высушивания D,L-метионина определяли объемную плотность.
Эксперименты по кристаллизации осуществляли в присутствии добавок 8, 9 или 10 ниже (согласно примеру 1), где указанную концентрацию корректировали добавлением добавки как в исходные растворы, так и в исходные суспензии согласно таблице 3.
Добавка 8) (7) + (2)
Добавка 9) (7) + (3)
Добавка 10) (7) + (4)
Данные по концентрации в таблице 3 показывают общее содержание активного ингредиента добавки без воды, исходя из общей массы раствора или суспензии.
Таблица 3

Можно видеть, что добавки согласно настоящему изобретению (с противовспенивателем) с диапазоном концентрации от 400 до 4000 ppm улучшают объемную плотность D,L-метионина до значений > 600 г/л.
Claims (32)
1. Способ кристаллизации D,L-метионина из водных растворов и/или суспензий, содержащих D,L-метионин и аммониевую соль D,L-метионина, имеющих содержание Met 70-180 г/кг раствора и/или суспензии, предпочтительно 90-150 г/кг раствора и/или суспензии, содержание NH4 + 1-5 г/кг раствора и/или суспензии, в присутствии кристаллизующей добавки, которая содержит неионогенное или анионное поверхностно-активное средство или смесь различных неионогенных или анионных поверхностно-активных средств, при котором температуру раствора и/или суспензии снижают сразу или постепенно от T1=85-110°C до Т2=30-50°С таким образом, что D,L-метионин оседает в виде твердого вещества, отличающийся тем, что анионное поверхностно-активное средство, применяемое для образования кристаллизующей добавки, является одним из соединений, отображенных в формулах 1-3, или их смесью:
где n является целым числом от 1 до 10, М является натрием или калием, a R1, R2 и R3 являются линейной, разветвленной или циклической, насыщенной или ненасыщенной С8-С20-алкильной группой или арильной группой, при этом неионогенная кристаллизующая добавка является сложным эфиром сорбитана и жирной кислоты или смесью различных сложных эфиров сорбитана и жирной кислоты.
2. Способ по п.1, отличающийся тем, что n равен 2, a R1, R2 и R3 являются линейными насыщенными C8-C18-алкильными группами.
3. Способ по п.1 или 2, отличающийся тем, что
CnH2n+1-O-SO3Na, где n=12-18,
CnH2n+1-O-C2H4-SO3Na, где n=8-18, или
CnH2n+1-(OC2H4)2-O-SO3Na, где n=12,
применяют в качестве анионного поверхностно-активного средства.
4. Способ по п.1, отличающийся тем, что применяют сложный эфир сорбитана и жирной кислоты, соответствующий формуле 4:

где w+x+y+z=20 и где каждый из w, х, у и z может принимать значения от 1 до 17.
5. Способ по п.1 или 2, отличающийся тем, что концентрация кристаллизующей добавки в растворе и/или суспензии, из которых осуществляют кристаллизацию, составляет по меньшей мере от 700 ppm до не более 4000 ppm исходя из общей массы раствора и/или суспензии, предпочтительно по меньшей мере от 750 ppm до не более 2000 ppm, особенно предпочтительно по меньшей мере от 800 ppm до не более 1000 ppm.
6. Способ по п.1 или 2, отличающийся тем, что раствор, из которого осуществляют кристаллизацию, дополнительно содержит противовспениватель.
7. Способ по п.6, отличающийся тем, что противовспениватель содержит силиконовое масло.
8. Способ по п.6, отличающийся тем, что весовое соотношение противовспениватель : кристаллизующая добавка на основе активного ингредиента находится в диапазоне от 4:1 до 1:1, предпочтительно в диапазоне от 3:1 до 2:1, и в этом случае концентрация кристаллизующей добавки составляет по меньшей мере от 50 ppm до не более 1200 ppm исходя из общей массы раствора и/или суспензии, предпочтительно от 100 ppm до 600 ppm, особенно предпочтительно от 200 ppm до 400 ppm.
9. Способ по п.1 или 2, отличающийся тем, что кристаллизацию осуществляют путем введения водного раствора и/или суспензии, нагретых до 85-110°С, содержащих D,L-метионин и аммониевую соль D,L-метионина, в водный раствор и/или суспензию, нагретые до 30-50°С, содержащие D,L-метионин и аммониевую соль D,L-метионина, где температуру получаемой смеси постоянно поддерживают на уровне от 30 до 50°С.
10. Способ по п.9, отличающийся тем, что кристаллизацию осуществляют путем введения раствора, нагретого до 85-110°С, содержащего D,L-метионин и аммониевую соль D,L-метионина, в суспензию, нагретую до 30-50°С, содержащую D,L-метионин и аммониевую соль D,L-метионина.
11. Способ по п.1 или 2, отличающийся тем, что кристаллизацию осуществляют в две стадии, где на первой стадии кристаллизации раствор и/или суспензию, нагретые до 85-110°С, содержащие D,L-метионин и аммониевую соль D,L-метионина, вводят в суспензию, нагретую до 60-80°С, содержащую D,L-метионин и аммониевую соль D,L-метионина, и температуру получаемой смеси постоянно поддерживают на уровне от 60 до 80°С, и где суспензию, нагретую до 60-80°С, содержащую D,L-метионин и аммониевую соль D,L-метионина, полученную на первой стадии кристаллизации, вводят на второй стадии кристаллизации в суспензию, нагретую до 30-50°С, содержащую D,L-метионин и аммониевую соль D,L-метионина, где температуру получаемой смеси постоянно поддерживают на уровне от 30 до 50°С.
12. Способ по п.1 или 2, отличающийся тем, что кристаллизацию осуществляют путем вакуумной кристаллизации, где давление на стадии кристаллизации составляет от 60 до 1000 мбар, и если кристаллизацию осуществляют в две стадии, давление на второй стадии кристаллизации составляет от 35 до 200 мбар.
13. Способ по любому из пп.1 или 2, отличающийся тем, что водные растворы и/или суспензии, содержащие D,L-метионин и аммониевые соли D,L-метионина, предварительно получают путем растворения и/или суспендирования D,L-метионина в воде в присутствии соответствующих количеств аммиака.
14. Способ по п.13, отличающийся тем, что для растворения применяют D,L-метионин, характеризующийся содержанием метионина по меньшей мере 90% по весу, предпочтительно по меньшей мере 95% по весу.
15. Способ по п.14, отличающийся тем, что применяют D,L-метионин в форме чистого метионина и/или необработанного метионина из любого производственного процесса, характеризующийся остаточной влажностью от 0,1 до 9,5% по весу, предпочтительно от 0,2 до 4,5% по весу.
16. Способ по п.1 или 2, отличающийся тем, что выпавший в осадок D,L-метионин отделяют от маточного раствора и высушивают или сначала перекристаллизовывают и высушивают после отделения от маточного раствора, полученного в этом случае.
17. Способ по п.16, отличающийся тем, что маточный раствор подают обратно на стадию кристаллизации.
18. Способ получения D,L-метионина, в котором водный раствор аммониевой соли D,L-метионина изначально образуют путем гидролиза нитрила D,L-метионина и/или амида D,L-метионина, и водный раствор и/или суспензию, содержащие D,L-метионин и аммониевую соль D,L-метионина, получают из него путем частичного удаления аммиака из аммониевой соли D,L-метионина, из которых затем получают метионин путем кристаллизации по п.1 или 2.
19. Способ по п.18, отличающийся тем, что водный раствор аммониевой соли D,L-метионина доводят до содержания NH4 + 1-5 г/кг раствора и/или суспензии, предпочтительно 1,5-3,0 г/кг, путем выпаривания и/или отгонки аммиака до кристаллизации.
20. Способ по п.18 или 19, отличающийся тем, что водный раствор и/или суспензию D,L-метионина и аммониевой соли D,L-метионина доводят до содержания Met 70-180 г/кг раствора и/или суспензии, предпочтительно 90-150 г/кг, путем добавления воды и/или D,L-метионина.
21. Способ по п.18 или 19, отличающийся тем, что выпавший в осадок D,L-метионин отделяют от маточного раствора и высушивают или сначала перекристаллизовывают и высушивают после отделения от маточного раствора, полученного в этом случае.
22. Способ по п.21, отличающийся тем, что маточный раствор подают обратно на стадию кристаллизации.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13184831.9A EP2848607A1 (de) | 2013-09-17 | 2013-09-17 | Verfahren zur Gewinnung von Methionin |
| EP13184831.9 | 2013-09-17 | ||
| PCT/EP2014/069247 WO2015039935A1 (de) | 2013-09-17 | 2014-09-10 | Verfahren zur gewinnung von methionin |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| RU2016114077A RU2016114077A (ru) | 2017-10-23 |
| RU2016114077A3 RU2016114077A3 (ru) | 2018-06-08 |
| RU2679309C2 true RU2679309C2 (ru) | 2019-02-07 |
Family
ID=49165674
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2016114077A RU2679309C2 (ru) | 2013-09-17 | 2014-09-10 | Способ получения метионина |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US9617209B2 (ru) |
| EP (2) | EP2848607A1 (ru) |
| JP (1) | JP6490079B2 (ru) |
| KR (1) | KR20160058149A (ru) |
| CN (1) | CN105764886B (ru) |
| BR (1) | BR112016005751B1 (ru) |
| MX (1) | MX2016003134A (ru) |
| MY (1) | MY173792A (ru) |
| RU (1) | RU2679309C2 (ru) |
| SG (1) | SG11201601432WA (ru) |
| WO (1) | WO2015039935A1 (ru) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109384696A (zh) * | 2017-08-03 | 2019-02-26 | 宁夏紫光天化蛋氨酸有限责任公司 | 一种获得高纯度高堆积密度蛋氨酸的方法 |
| EP3689851A1 (en) | 2019-02-04 | 2020-08-05 | Evonik Operations GmbH | Salt-free production of methionine from methionine nitrile |
| CN111100051B (zh) * | 2019-12-31 | 2022-01-28 | 山东新和成氨基酸有限公司 | 在甲硫氨酸制备过程中使用的添加剂及甲硫氨酸的制备方法 |
| CN113416159A (zh) * | 2021-04-21 | 2021-09-21 | 重庆紫光化工股份有限公司 | 一种蛋氨酸铵盐的脱色结晶制备dl-蛋氨酸的方法 |
| CN114920675B (zh) * | 2022-04-20 | 2024-02-06 | 天津大学 | 一种蛋氨酸晶体及其制备方法和应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11158140A (ja) * | 1997-11-26 | 1999-06-15 | Sumitomo Chem Co Ltd | メチオニンの製造方法 |
| US20050176115A1 (en) * | 2002-07-23 | 2005-08-11 | Nippon Soda Co., Ltd. | Process for the production of methionine |
| RU2294922C2 (ru) * | 2001-12-08 | 2007-03-10 | Дегусса Аг | Способ получения метионина |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4619610B1 (ru) * | 1966-08-25 | 1971-06-01 | ||
| JPS5715756B2 (ru) * | 1974-06-05 | 1982-04-01 | ||
| JPS5720947B2 (ru) * | 1974-06-05 | 1982-05-04 | ||
| DE19547236A1 (de) * | 1995-12-18 | 1997-07-03 | Degussa | Verfahren zur Herstellung von D,L-Methionin oder dessen Salz |
| DE19707380A1 (de) | 1997-02-25 | 1998-08-27 | Degussa | Verfahren zur Herstellung eines rieselfähigen Tierfuttermittelsupplements auf Methioninsalzbasis und das so erhältliche Granulat |
| US6545179B2 (en) | 2000-02-15 | 2003-04-08 | Aventis Animal Nutrition, Sa | Process for the production of methionine |
| DE10238212A1 (de) | 2002-08-21 | 2004-03-04 | Degussa Ag | Verfahren zur Herstellung von α-Aminosäuren durch Hydrolyse von Hydantoinen bei erhöhtem Druck und erhöhter Temperatur |
| JP2004254690A (ja) | 2003-02-03 | 2004-09-16 | Nippon Soda Co Ltd | Dl−メチオニンの製造法 |
| EP1564208B1 (en) | 2004-02-14 | 2011-05-18 | Evonik Degussa GmbH | Process for producing methionine |
| FR2890965A1 (fr) | 2005-09-21 | 2007-03-23 | Adisseo France Sas Soc Par Act | Hydrolyse ammoniacale de l'hydantoine de methionine sans catalyseur |
| WO2008006977A1 (fr) | 2006-07-11 | 2008-01-17 | Adisseo France S.A.S. | Procédé de préparation du 2-hydroxy-4-(méthylthio)butyronitrile et de la méthionine |
| JP5307512B2 (ja) * | 2008-11-07 | 2013-10-02 | 住友化学株式会社 | メチオニンの製造方法 |
| RU2618042C2 (ru) | 2011-08-30 | 2017-05-02 | Эвоник Дегусса Гмбх | Способ получения соли метионина |
| DE102011081828A1 (de) | 2011-08-30 | 2013-02-28 | Evonik Degussa Gmbh | Verfahren zur Umsetzung von Methylmercaptopropionaldehyd aus Roh-Acrolein und Roh-Methylmercaptan |
| EP2641898A1 (de) | 2012-03-20 | 2013-09-25 | Evonik Industries AG | Verfahren zur Herstellung von Methionin |
-
2013
- 2013-09-17 EP EP13184831.9A patent/EP2848607A1/de not_active Withdrawn
-
2014
- 2014-09-10 RU RU2016114077A patent/RU2679309C2/ru active
- 2014-09-10 SG SG11201601432WA patent/SG11201601432WA/en unknown
- 2014-09-10 BR BR112016005751-1A patent/BR112016005751B1/pt not_active IP Right Cessation
- 2014-09-10 EP EP14771817.5A patent/EP3046903B1/de active Active
- 2014-09-10 WO PCT/EP2014/069247 patent/WO2015039935A1/de active Application Filing
- 2014-09-10 KR KR1020167009927A patent/KR20160058149A/ko not_active Application Discontinuation
- 2014-09-10 US US15/021,541 patent/US9617209B2/en active Active
- 2014-09-10 MX MX2016003134A patent/MX2016003134A/es active IP Right Grant
- 2014-09-10 CN CN201480051391.8A patent/CN105764886B/zh active Active
- 2014-09-10 MY MYPI2016700871A patent/MY173792A/en unknown
- 2014-09-10 JP JP2016543363A patent/JP6490079B2/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11158140A (ja) * | 1997-11-26 | 1999-06-15 | Sumitomo Chem Co Ltd | メチオニンの製造方法 |
| RU2294922C2 (ru) * | 2001-12-08 | 2007-03-10 | Дегусса Аг | Способ получения метионина |
| US20050176115A1 (en) * | 2002-07-23 | 2005-08-11 | Nippon Soda Co., Ltd. | Process for the production of methionine |
Non-Patent Citations (1)
| Title |
|---|
| База данных DWPI, запись AN 1971-36844S, документ JP 46-019610 B. * |
Also Published As
| Publication number | Publication date |
|---|---|
| SG11201601432WA (en) | 2016-04-28 |
| KR20160058149A (ko) | 2016-05-24 |
| US9617209B2 (en) | 2017-04-11 |
| JP2016534144A (ja) | 2016-11-04 |
| MX2016003134A (es) | 2016-06-24 |
| US20160229799A1 (en) | 2016-08-11 |
| RU2016114077A3 (ru) | 2018-06-08 |
| BR112016005751B1 (pt) | 2021-02-02 |
| WO2015039935A1 (de) | 2015-03-26 |
| EP2848607A1 (de) | 2015-03-18 |
| EP3046903B1 (de) | 2021-03-24 |
| CN105764886B (zh) | 2018-04-13 |
| CN105764886A (zh) | 2016-07-13 |
| JP6490079B2 (ja) | 2019-03-27 |
| EP3046903A1 (de) | 2016-07-27 |
| MY173792A (en) | 2020-02-24 |
| RU2016114077A (ru) | 2017-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2640656C2 (ru) | Способ получения метионина | |
| RU2679309C2 (ru) | Способ получения метионина | |
| RU2678585C2 (ru) | Способ непрерывной подготовки кристаллов метионина высокой насыпной плотности | |
| JP4213038B2 (ja) | メチオニンの製造方法 | |
| JP2921097B2 (ja) | メチオニンの製造方法 | |
| JP7388647B2 (ja) | メチオニンの製造プロセスで用いられる添加剤及びメチオニンの製造方法 | |
| JP5125908B2 (ja) | シトルリンの晶析方法 | |
| JPH04244056A (ja) | メチオニンの製造方法 | |
| CN106928146A (zh) | 一种柠檬黄的纯化方法 | |
| US20220306574A1 (en) | Process for the preparation of d,l-methionine | |
| JP2018039755A (ja) | ナトリウム2α−メチル−2β−(1,2,3−トリアゾール−1−イル)−メチルペナム−3α−カルボン酸1,1−ジオキシド一水和物結晶の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PD4A | Correction of name of patent owner |