RU2631243C2 - СОЛЬ (СОЛИ) ДИМЕТИЛАМИДА 7-ЦИКЛОПЕНТИЛ-2-(5-ПИПЕРАЗИН-1-ИЛ-ПИРИДИН-2-ИЛАМИНО)-7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-6-КАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ - Google Patents
СОЛЬ (СОЛИ) ДИМЕТИЛАМИДА 7-ЦИКЛОПЕНТИЛ-2-(5-ПИПЕРАЗИН-1-ИЛ-ПИРИДИН-2-ИЛАМИНО)-7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-6-КАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ Download PDFInfo
- Publication number
- RU2631243C2 RU2631243C2 RU2013126112A RU2013126112A RU2631243C2 RU 2631243 C2 RU2631243 C2 RU 2631243C2 RU 2013126112 A RU2013126112 A RU 2013126112A RU 2013126112 A RU2013126112 A RU 2013126112A RU 2631243 C2 RU2631243 C2 RU 2631243C2
- Authority
- RU
- Russia
- Prior art keywords
- compound
- formula
- pyrrolo
- cancer
- ylamino
- Prior art date
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C55/00—Saturated compounds having more than one carboxyl group bound to acyclic carbon atoms
- C07C55/02—Dicarboxylic acids
- C07C55/10—Succinic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/73—Unsubstituted amino or imino radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/74—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Изобретение относится к новой сукцинатной соли диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты, представленной формулой (II), в виде гидратной и негидратной формы. Соединение может найти применение для лечения заболевания, которое ослабляется ингибированием активности циклинзависимых киназ, в частности киназ CDK1, CDK2, CDK3, CDK4 CDK5, CDK6 и CDK9. Таким заболеванием может быть рак молочной железы, рак мочеполовых органов, рак легких, рак поджелудочной железы и т.д. Сукцинатная соль соответствует структурной формуле, указанной ниже.

3 н. и 2 з.п. ф-лы, 6 ил., 2 табл., 7 пр.
Description
Область техники, к которой относится изобретение
Настоящее изобретение предлагает (1) способ получения диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты и его соли (солей); (2) новые соли (соль) диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты; (3) содержащие их фармацевтические композиции и (4) использующие их способы лечения.
Уровень техники, к которой относится изобретение
Соединение диметиламид 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты формулы (I):

и его синтез конкретно описан в примере 74 международной патентной заявки WO 2010/020675 A1.
Международная патентная заявка WO 2010/020675 описывает, что соединение формулы (I) имеет ценные фармакологические свойства и может быть использовано, например, (1) в качестве ингибиторов циклинзависимых киназ (в частности, циклинзависимых киназ, выбранных из CDK1, CDK2, CDK3, CDK4, CDK5, CDK6 и CDK9); и (2) в качестве модуляторов и/или ингибиторов киназы-3 гликогенсинтазы (GSK-3).
Международная патентная заявка WO 2010/020675 не описывает и не предлагает сукцинатную соль диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты.
Сущность изобретения
Настоящее изобретение относится к сукцинатной соли диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты.
Сукцинатная соль представлена формулой (II):

Настоящее изобретение также относится к способу получения соединения формулы (II).
Настоящее изобретение также относится к способу получения соединения формулы (I).
Настоящее изобретение также относится к способу получения соединения формулы (III):

Настоящее изобретение также относится к способу получения соединения формулы (IV):

Настоящее изобретение также относится к фармацевтическим композициям, содержащим соль формулы (II) и по меньшей мере один фармацевтически приемлемый носитель, разбавитель, растворитель или эксципиент.
Настоящее изобретение также относится к способу лечения заболевания, которое ослабляется ингибированием циклинзависимых киназ (в частности, циклинзависимых киназ, выбранных из CDK1, CDK2, CDK3, CDK4, CDK5, CDK6 и CDK9), включающему стадию введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (II).
Краткое описание чертежей
Фиг.1 представляет график изотермы динамической сорбции пара (DVS) соединения формулы (II) (цикл относительной влажности (RH) 0-90-0%).
Фиг.2 представляет рентгеновскую дифрактограмму порошка (XRPD) соединения формулы (II) после DVS (цикл RH 0-90-0%).
Фиг.3 представляет данные дифференциальной сканирующей калориметрии (DSC) соединения формулы (II) после DVS (цикл RH 0-90-0%).
Фиг.4 представляет данные термогравиметрического анализа (TGA) соединения формулы (II) после DVS (цикл RH 0-90-0%).
Фиг.5 представляет график изотермы DVS соединения формулы (II) (цикл RH 0-80-0%).
Фиг.6 представляет XRPD соединения формулы (II) после DVS (цикл RH 0-80-0%).
Подробное описание изобретения
Настоящее изобретение относится к сукцинатной соли диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты.
Сукцинатная соль представлена формулой (II):

Сукцинатная соль диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты может находиться в негидратной, гидратной форме или представлять собой их смеси.
В одном варианте осуществления сукцинатная соль содержит более чем 99,9% негидратной формы.
В одном варианте осуществления сукцинатная соль содержит более чем 99% негидратной формы.
В одном варианте осуществления сукцинатная соль содержит более чем 97% негидратной формы.
В одном варианте осуществления сукцинатная соль содержит более чем 95% негидратной формы.
В одном варианте осуществления сукцинатная соль содержит более чем 90% негидратной формы.
Негидратная форма сукцинатной соли диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты проявляет хорошую устойчивость, отсутствие гигроскопичности, а также хорошую растворимость.
Настоящее изобретение также относится к способу получения соединения формулы (I) и соединения формулы (II):
Технологическая схема получения соединения формулы (I) и соединения формулы (II)

Настоящее изобретение также относится к способу получения соединения формулы (III):
Технологическая схема получения соединения формулы (III)

По сравнению с предшествующим способом получения соединения формулы (III) в настоящем способе увеличивается общий выход соединения формулы (III) (т.е. A1) от 4% до 30%. Кроме того, для данного усовершенствованного способа не требуется пяти стадий колоночной очистки, требуемой для предшествующего способа.
Настоящее изобретение также относится к способу получения соединения формулы (IV):
Технологическая схема получения соединения формулы (IV)

В настоящем синтезе соединения формулы (IV) (т.е. A2) разработан простой способ для замещения хлорида в A2d на A2c, используя н-бутанол в качестве растворителя. Данный способ увеличивает выход и не требует хроматографической очистки соединения формулы (IV) для следующих процессов.
Таким образом, разработан масштабируемый, более безопасный, упрощенный, дающий повышенный выход и более экономичный эффективный способ получения исходных соединений A1 (соединение формулы (III)), A2 (соединение формулы (IV)), свободного основания A4 (соединение формулы (I)) и сукцинатной соли A6 (соединение формулы (II)). По сравнению с предшествующими способами синтеза в общей методике сокращено число стадий синтеза и увеличен общий выход от 0,9% до 12%.
Настоящее изобретение относится также к фармацевтическим композициям, содержащим соль формулы (II) и по меньшей мере один фармацевтически приемлемый носитель, разбавитель, растворитель или эксципиент.
Настоящее изобретение относится также к способу лечения заболевания, которое ослабляется ингибированием циклинзависимых киназ (в частности, циклинзависимых киназ, выбранных из CDK1, CDK2, CDK3, CDK4, CDK5, CDK6 и CDK9), включающему стадию введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (II).
Такие заболевания, которые ослабляются ингибированием циклинзависимых киназ, включают, но не ограничиваются ими, рак молочной железы, рак мочеполовых органов, рак легких, рак желудочно-кишечного тракта, эпидермоидный рак, меланому, рак яичника, рак поджелудочной железы, нейробластому, рак головы и/или шеи или рак мочевого пузыря, или, в более широком смысле, рак почки, головного мозга или желудка; лейкемии, гиперплазии, рак желудка, рак толстой кишки, рак гортани, рак лимфатической системы, рак мочеполовой системы, рак кости, рак простаты, мелкоклеточный рак легких, глиомный рак, рак толстой и прямой кишки, рак почки, рак эпидермиса, рак печени, рак пищевода, рак кроветворной системы, лимфому, миелому, рак щитовидной железы; опухоль мезенхимального происхождения, например, фибросаркому или хабдомиосаркому; опухоль центральной или периферической нервной системы, например, астроцитому, нейробластому, глиому или шванному; меланому; семиному; тератокарциному; остеосаркому; пигментную ксеродермию; кератоакантому; рак щитовидной железы; саркому Капоши (Kaposi), хроническую лимфоцитарную лейкемию, лимфому клеток мантии, B-крупноклеточную лимфому.
«Терапевтически эффективное количество» означает количество соли по изобретению, которое при введении нуждающемуся в этом пациенту является достаточным, чтобы осуществлять лечение в состоянии заболевания, которое ослабляется ингибированием активности циклинзависимых киназ. Количество данного соединения по настоящему изобретению, которое является терапевтически эффективным, будет изменяться в зависимости от факторов, таких как состояние заболевания и его тяжесть, личность нуждающегося в этом пациента и т.д., причем данное количество может быть определено стандартным образом обычным специалистом в данной области.
«По меньшей мере один фармацевтически приемлемый носитель, разбавитель, растворитель или эксципиент» может быть легко выбран обычным специалистом в данной области, причем выбор будет определяться желательным путем введения. Иллюстративные примеры подходящих путей введения включают пероральный, назальный, парентеральный, местный, трансдермальный и ректальный. Фармацевтические композиции по настоящему изобретению могут иметь любую фармацевтическую форму, признаваемую специалистом в данной области как подходящую. Подходящие фармацевтические формы включают твердые, полутвердые, жидкие или лиофилизированные готовые формы, такие как таблетки, порошки, капсулы, суппозитории, суспензии, липосомы и аэрозоли.
Конкретные варианты осуществления настоящего изобретения далее будут продемонстрированы со ссылкой на следующие примеры. Следует понимать, что данные примеры представлены исключительно для цели иллюстрации настоящего изобретения, и их не следует рассматривать в качестве ограничивающих каким-либо образом объем настоящего изобретения.
Пример 1. Получение соединения A1 (т.е. соединения формулы (III))
Соединение A1  (т.е. соединение формулы (III), 2-хлор-7-циклопентил-N,N-диметил-7H-пирроло[2,3-d]пиримидин-6-карбоксамид) получали согласно представленной ниже схеме синтеза.
(т.е. соединение формулы (III), 2-хлор-7-циклопентил-N,N-диметил-7H-пирроло[2,3-d]пиримидин-6-карбоксамид) получали согласно представленной ниже схеме синтеза.
Схема синтеза

Подробные описания каждой из стадий представлены ниже как стадии 1.1-1.4. Стадия 1.5 является необязательной стадией очистки.
1.1. 5-Бром-2-хлор-N-циклопентилпиримидин-4-амин (A1f)

В продуваемую азотом, оборудованную надлежащим образом 5-литровую четырехгорлую круглодонную колбу загружали 250 г (1,097 моль, 140,4 мл, 1,0 экв.) 5-бром-2,4-дихлорпиримидина (A1h) и 1127 г (1250 мл) этилацетата. Содержимое перемешивали при 20°C и добавляли 283,5 г (2,194 моль, 382,0 мл, 2,0 экв.) N,N-диизопропилэтиламина. Раствор 102,8 г (1,207 моль, 119 мл, 1,1 экв.) циклопентиламина (A1g), растворенного в 1127 г (1250 мл) этилацетата, добавляли в течение 60 минут. Наблюдали экзотерму от 18°C до 36°C. Раствор нагревали до 40°C. Данную температуру поддерживали в течение по меньшей мере 6 часов или до тех пор, пока не израсходуется все исходное соединение A1h, что определяли путем анализа методом ВЭЖХ. Полученную в результате суспензию охлаждали до 25°C и добавляли 500 г (500 мл) воды. Содержимое перемешивали в течение 15 минут и оставляли для разделения фаз. Нижний (водный) слой отделяли, а органический слой промывали еще раз, используя 500 г (500 мл) воды. Образец перемешивали в течение 15 минут и оставляли для разделения фаз. Нижний (водный) слой отделяли. Органическую фазу концентрировали (при атмосферном давлении) до объема 1500 мл (температура смеси составляла 82°C). Добавляли 684 г (1 л) гептана и повторяли концентрирование до объема 1500 мл (температура смеси составляла 85°C). Снова добавляли 684 г (1 л) гептана и повторяли концентрирование до объема 1500 мл (температура смеси составляла 96°C). Образец охлаждали до 50°C и вносили затравочные кристаллы. Охлаждение продолжали до 4°C и температуру поддерживали на уровне 4°C в течение 1 часа. Твердое вещество отфильтровывали и осадок на фильтре промывали однократно, используя 137 г (200 мл) холодного (4°C) гептана. Твердое вещество сушили при 50°C в течение 16 часов, получая 259,0 г (выход с поправкой 88,0%) соединения A1f в виде кристаллического твердого вещества белого цвета, температура плавления которого составляла 95-96°C.
1.2. 3-[2-Хлор-4-(циклопентиламино)пиримидин-5-ил]проп-2-ин-1-ол (A1d)

В продуваемую азотом, оборудованную надлежащим образом 5-литровую четырехгорлую круглодонную колбу загружали 200 г (0,723 моль, 1,0 экв.) 5-бром-2-хлор-N-циклопентилпиримидин-4-амина (A1f) и 2303 г (2600 мл) тетрагидрофурана. Смесь перемешивали, нагревали с обратным холодильником (67°C) и собирали 200 мл дистиллята. Образец охлаждали до 25°C и добавляли 52,7 г (0,940 моль, 55,6 мл, 1,3 экв.) пропаргилового спирта (A1e), 570,3 г (1,808 моль, 2,5 экв.) тригидрата фторида тетрабутиламмония и 25,4 г (0,036 моль, 0,05 экв.) дихлорида бис(трифенилфосфин)палладия(II). Образец перемешивали, нагревали с обратным холодильником (67°C) и выдерживали при данной температуре в течение 2 часов или до тех пор, пока не останется 5-7% исходного соединения A1f, что определяли путем анализа методом ВЭЖХ. Образец охлаждали до 25°C и концентрировали при пониженном давлении (100 мбар (10 кПа), максимальная внутренняя температура 30°C) до объема 1150 мл, чтобы удалить тетрагидрофуран. Добавляли 541 г (600 мл) этилацетата. Образец снова концентрировали при пониженном давлении (100 мбар (10 кПа), максимальная внутренняя температура 30°C) до объема 1150 мл, чтобы удалить остаточный тетрагидрофуран. Добавляли 2706 г (3000 мл) этилацетата и раствор 63 г бикарбоната натрия в 1500 г (1500 мл) воды. Образец перемешивали при 25°C в течение 10 минут и разделяли фазы. Органическую (верхнюю) фазу промывали однократно, используя 1500 г (1500 мл) воды. Образец перемешивали в течение 10 минут и разделяли фазы. Органическую (верхнюю) фазу концентрировали при пониженном давлении (100 мбар (10 кПа), максимальная внутренняя температура 30°C) до объема 625 мл, чтобы удалить этилацетат. В концентрат добавляли 1582 г (2000 мл) ацетона. Образец перемешивали, нагревали с обратным холодильником (58°C) и выдерживали при данной температуре в течение 30 минут. Затем его охлаждали до 40°C и очищали фильтрованием через слой фильтровальной целлюлозы. Колбу и осадок на фильтре промывали дважды, используя 158 г (200 мл, 2×100 мл на каждое промывание) ацетона. Образец концентрировали при пониженном давлении (100 мбар (10 кПа), максимальная внутренняя температура 30°C) до объема 460 мл. Затем его охлаждали до 4°C и выдерживали при данной температуре в течение 1 часа. Твердое вещество отфильтровывали и осадок на фильтре промывали дважды, используя 158 г (2×100 мл) холодного (4°C) ацетона. Твердое вещество сушили при 50°C в течение 16 часов, получая 85,6 г (выход с поправкой 47,4%) соединения A1d в виде кристаллического твердого вещества желто-коричневого цвета, температура плавления которого составляла 162-163°C.
1.3. (2-Хлор-7-циклопентил-7H-пирроло[2,3-d]пиримидин-6-ил)метанол (A1c)

В продуваемую сухим азотом 5-литровую четырехгорлую круглодонную колбу загружали 100 г (чистота 98%, 0,389 моль, 1,0 экв.) 3-(2-хлор-4-(циклопентиламино)пиримидин-5-ил)проп-2-ин-1-ола (A1d), 880 г (1000 мл) не содержащего пероксида тетрагидрофурана и 753 г (856 мл) фторида тетрабутиламмония, в виде 1,0М раствора в ТГФ. Содержимое перемешивали при 25°C в течение 10 минут и затем раствор нагревали до 60°C. Данную температуру поддерживали в течение 1,5 часов до тех пор, пока исходное соединение A1d не составит ≤2,5±0,5%, что определяли путем анализа методом ВЭЖХ. Полученный в результате раствор охлаждали до температуры ниже 30±3°C и подвергали дистилляции при пониженном давлении, чтобы удалить ТГФ. Добавляли 79 г (100 мл) 2-пропанола. Образец перемешивали в течение 15 минут и затем 1000 г (1000 мл) воды медленно добавляли в течение 30 минут. Образец перемешивали при 20±3°C в течение 30 минут и затем фильтровали. Осадок на фильтре промывали дважды, используя 200 г (2×100 мл) воды. Твердое вещество сушили при 50°C в течение 16 часов, получая соединение A1c в виде кристаллического твердого вещества желто-коричневого цвета, температура плавления которого составляла 174-176°C.
1.4. 2-Хлор-7-циклопентил-N,N-диметил-7H-пирроло[2,3-d]пиримидин-6-карбоксамид (A1)

В продуваемый сухим азотом 100-литровый реакционный сосуд ACE-100L загружали 97,3 г цианида натрия, 2500 г (2-хлор-7-циклопентил-7H-пирроло[2,3-d]пиримидин-6-ил)метанола A1c, 16680 г (19,5 л) диметиламина A1a (2,0М раствор в ТГФ) и 28320 г (30,0 л) безводного N,N-диметилформамида. Смесь перемешивали при 20±3°C в течение 15 минут. Затем добавляли 2,06 кг оксида марганца(IV). Темную суспензию перемешивали в течение 30 минут и добавляли 12,36 кг оксида марганца(IV) тремя порциями (первая порция: 2,06 кг, вторая порция: 4,12 кг и третья порция: 6,18 кг) с интервалами по 30 минут. После добавления последней порции образец выдерживали в течение 1 часа и затем добавляли 6,18 кг оксида марганца(IV). Образец выдерживали в течение 1 часа. Затем из реакционной смеси отбирали образцы. Реакцию считали завершенной, когда исходное соединение A1c составило ≤1,0±0,5%, что определяли путем анализа методом ВЭЖХ. Реакционную смесь затем фильтровали через слой целита, чтобы удалить оксид марганца(IV). Реактор и осадок на фильтре промывали, используя 23 л этилацетата. Фильтрат подвергали дистилляции при пониженном давлении (20 мбар (2 кПа), температура 45±3°C), чтобы удалить ТГФ, диметиламин и этилацетат. Образец затем подвергали дистилляции при пониженном давлении (5 мбар (500 Па), температура 70±5°C), чтобы удалить ДМФА. Концентрат разбавляли 35 л этилацетата. Полученный в результате темный раствор промывали водным раствором сульфата железа(II) (1 кг FeSO4·7H2O в 14 л воды), 15 л воды и, наконец, 15 л 10% водного раствора NaCl. Фазы разделяли после каждого промывания. Органическую фазу подвергали дистилляции (45°C, 50 мбар (5 кПа)), чтобы азеотропно отделить воду. Полученный в результате неочищенный A1 (2,788 г темного густого полутвердого остатка) можно использовать непосредственно на следующей стадии.
1.5. Методика получения чистого A1 из неочищенного A1
Неочищенный A1 после стадии 1.4 может быть необязательно очищен либо способом 1, либо способом 2, приведенными ниже.
Способ 1
Слабо нагревали 10 г неочищенного A1 и 9 мл 1-пропанола до тех пор, пока не получится однородный темный раствор. Раствор охлаждали до 25±3°C и медленно добавляли 30-40 мл гексана. В образец помещали затравочные кристаллы и перемешивали его до тех пор, пока не начнется кристаллизация. Дополнительно добавляли медленно 50-60 мл гексана. Общий объем добавленного гексана составлял приблизительно 90 мл. Суспензию выдерживали при 22±3°C в течение 2 часов, затем охлаждали до 4°C и выдерживали дополнительно в течение 2 часов. Твердое вещество отфильтровывали. Колбу и осадок на фильтре промывали гексаном по мере необходимости. Осадок на фильтре сушили при 50°C и 50 мбар (5 кПа), получая 6,35 г очищенного A1 в виде кристаллического твердого вещества светло-коричневого цвета. Выход: 63,5%.
Способ 2
Готовили раствор 10 г неочищенного A1 в 10 мл EtOAc и наносили на 100 г слоя силикагеля в колонке. Колонку элюировали 300 мл смеси EtOAc/гексан (2/8) и элюат отбрасывали. Затем колонку элюировали 800 мл смеси EtOAC/гексан (5/5) и второй элюат собирали для выделения продукта. Второй элюат концентрировали, получая жидкое масло. Медленно добавляли 100 мл гексана и образец перемешивали при 22±3°C в течение 2 часов. Образец охлаждали до 4°C и выдерживали дополнительно в течение 2 часов. Твердое вещество отфильтровывали. Колбу и осадок на фильтре промывали гексаном при необходимости. Осадок на фильтре сушили при 50°C и 50 мбар (5 кПа), получая 6,05 г очищенного A1 в виде кристаллического твердого вещества светло-коричневого цвета. Выход: 60,5%.
Пример 2. Получение соединения A2 (т.е. соединения формулы (IV), трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилата)
Соединение A2  (т.е. соединение формулы (IV), трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилат) получали согласно представленной ниже схеме синтеза.
(т.е. соединение формулы (IV), трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилат) получали согласно представленной ниже схеме синтеза.
Схема синтеза

Подробное описание каждой из стадий представлено ниже как стадии 2.1-2.4.
2.1. Гидрохлорид 1-(6-нитропиридин-3-ил)пиперазина (A2b)

В продуваемую азотом, оборудованную надлежащим образом 22-литровую четырехгорлую круглодонную колбу загружали 1392 г (8,78 моль, 1,0 экв.) 5-хлор-2-нитропиридина (A2d), 1512 г (17,56 моль, 2,0 экв.) пиперазина (A2c) и 11340 г (14000 мл) н-бутанола. Полученную в результате суспензию перемешивали и нагревали до 95°C. Данную температуру поддерживали в течение по меньшей мере 24 часов или до тех пор, пока оставшееся исходное соединение A2d не составит ≥2% (по нормированной площади), что определяли путем анализа методом ВЭЖХ. Полученную в результате суспензию охлаждали до 25°C в течение 1 часа. Твердое вещество отфильтровывали через слой полипропиленового фильтра. Осадок на фильтре промывали дважды, используя в сумме 2267 г (2×1300 мл) изопропилацетата. Твердое вещество сушили при 60°C в течение 16 часов, получая 1769 г (выход без поправки 82,3%) A2b в виде кристаллического твердого вещества желтого цвета, температура плавления которого составляла >230°C (с разложением).
2.2. трет-Бутил 4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилат (A2a)

В продуваемую азотом, оборудованную надлежащим образом 22-литровую четырехгорлую круглодонную колбу загружали 589 г (2,41 моль, 1,0 экв.) гидрохлорида 1-(6-нитропиридин-3-ил)пиперазина (A2b). Готовили раствор 630,5 г (2,89 моль, 1,2 экв.) ди-трет-бутилдикарбоната в 10223 г (11500 мл) тетрагидрофурана и выливали в колбу. Полученную в результате суспензию перемешивали и охлаждали до 8±3°C. В продуваемую азотом, оборудованную надлежащим образом 5-литровую четырехгорлую круглодонную колбу загружали 499 г (3,61 моль, 1,5 экв.) карбоната калия. В 5-литровую колбу добавляли 3600 г (3600 мл) воды. После перемешивания получали раствор. Данный раствор охлаждали до 25±3°C и переносили в реакционную смесь в течение 30 минут. В процессе добавления поддерживали температуру смеси ≤12±3°C. Смесь нагревали до 22±3°C и выдерживали при этой температуре в течение дополнительного 1 часа или до тех пор, пока не перестанет обнаруживаться исходное соединение, A2b, определяемое аналитическим методом тонкослойной хроматографии (TLC). Двухфазную смесь фильтровали через слой целита массой 250 г. Осадок на фильтре промывали дважды, используя в сумме 800 г (2×450 мл) тетрагидрофурана. Растворитель после промывания объединяли с фильтратом. Фазы разделяли и водную (нижнюю) фазу отбрасывали. Фильтрат концентрировали при пониженном давлении (100 мбар (10 кПа), максимальная внутренняя температура 40°C), получая густую пасту.
Данную процедуру полностью повторяли еще два раза. Концентраты из всех трех экспериментов объединяли и загружали в продуваемую азотом, оборудованную надлежащим образом 22-литровую четырехгорлую круглодонную колбу. К указанным концентратам добавляли 4719 г (6900 мл) гептана. Образец перемешивали и концентрировали при пониженном давлении (100 мбар (10 кПа), максимальная внутренняя температура 40°C), получая густую пасту. К концентрированному образцу добавляли еще 3146 г (4600 мл) гептана. Полученную в результате суспензию перемешивали при 37±3°C в течение 1 часа, охлаждали до 22±3°C и выдерживали в течение 15 минут. Твердое вещество отфильтровывали через слой полипропиленового фильтра и осадок дважды промывали, используя 615 г (2×450 мл) гептана. Твердое вещество сушили при 55°C в токе азота в течение 16 часов, получая 2088 г (93,8%) соединения A2a в виде кристаллического твердого вещества желтого цвета, температура плавления которого составляла 173-174°C.
2.3. трет-Бутил 4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилат (A2)

В продуваемый азотом, 2,5-литровый толстостенный реакционный сосуд Парра, который выдерживает давление 60 фунтов на кв. дюйм (413,7 кПа), загружали 68 г (0,22 моль) трет-бутил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилата (A2a), 6,8 г 10% палладия на угле, содержащего 50% воды, в качестве влажного катализатора и 807 г (1020 мл) метанола. Реакционный сосуд трижды заполняли инертным газом (азотом) при давлении, составляющем приблизительно 30 фунтов на кв. дюйм (206,8 кПа), каждый раз откачивая атмосферу над реакционной смесью. В сосуд дважды нагнетали водород при давлении, составляющем (приблизительно 30 фунтов на кв. дюйм (206,8 кПа), каждый раз откачивая атмосферу над реакционной смесью. В реакционный сосуд нагнетали водород при давлении 45 фунтов на кв. дюйм (310,3 кПа). Включали мотор встряхивателя. Протекала экзотермическая реакция. Наблюдали повышение температуры от 19 до 54°C в течение 15 минут, после чего впуск водорода прекращали. Смеси давали охладиться до 30°C в течение 1 часа, после чего встряхиватель останавливали. Водород в атмосфере реакционного сосуда заменяли инертным азотом, как описано выше. Катализатор отделяли фильтрованием через слой фильтровальной целлюлозы массой 10 г. Данную процедуру полностью повторяли еще один раз, оба фильтрата объединяли и загружали в чистую 3-литровую четырехгорлую круглодонную колбу.
2.4. Выделение продукта
Фильтраты после стадии 2.3 перемешивали и концентрировали при пониженном давлении (50 мбар (5 кПа), максимальная внутренняя температура 40°C), получая густую пасту. К остатку добавляли 190 г (250 мл) трет-бутилметилового эфира. Образец снова перемешивали и концентрировали при пониженном давлении (50 мбар (5 кПа), максимальная внутренняя температура 30°C), получая густую пасту. К остатку добавляли 342 г (500 мл) гептана и полученную в результате суспензию перемешивали в течение 15 минут при 22±3°C. Твердое вещество отфильтровывали и осадок на фильтре промывали, используя 68 г (100 мл) гептана. Твердое вещество сушили при 50°C в течение 16 часов, получая 112,3 г (93,4%) соединения A2 в виде пластинок желто-коричневого цвета, температура плавления которого составляла 124-126°C.
Пример 3. Получение диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (т.е. соединения формулы (I))
Соединение A4  (т.е. соединение формулы (I), диметиламид 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты) получали согласно представленной ниже схеме синтеза.
(т.е. соединение формулы (I), диметиламид 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты) получали согласно представленной ниже схеме синтеза.
Схема синтеза
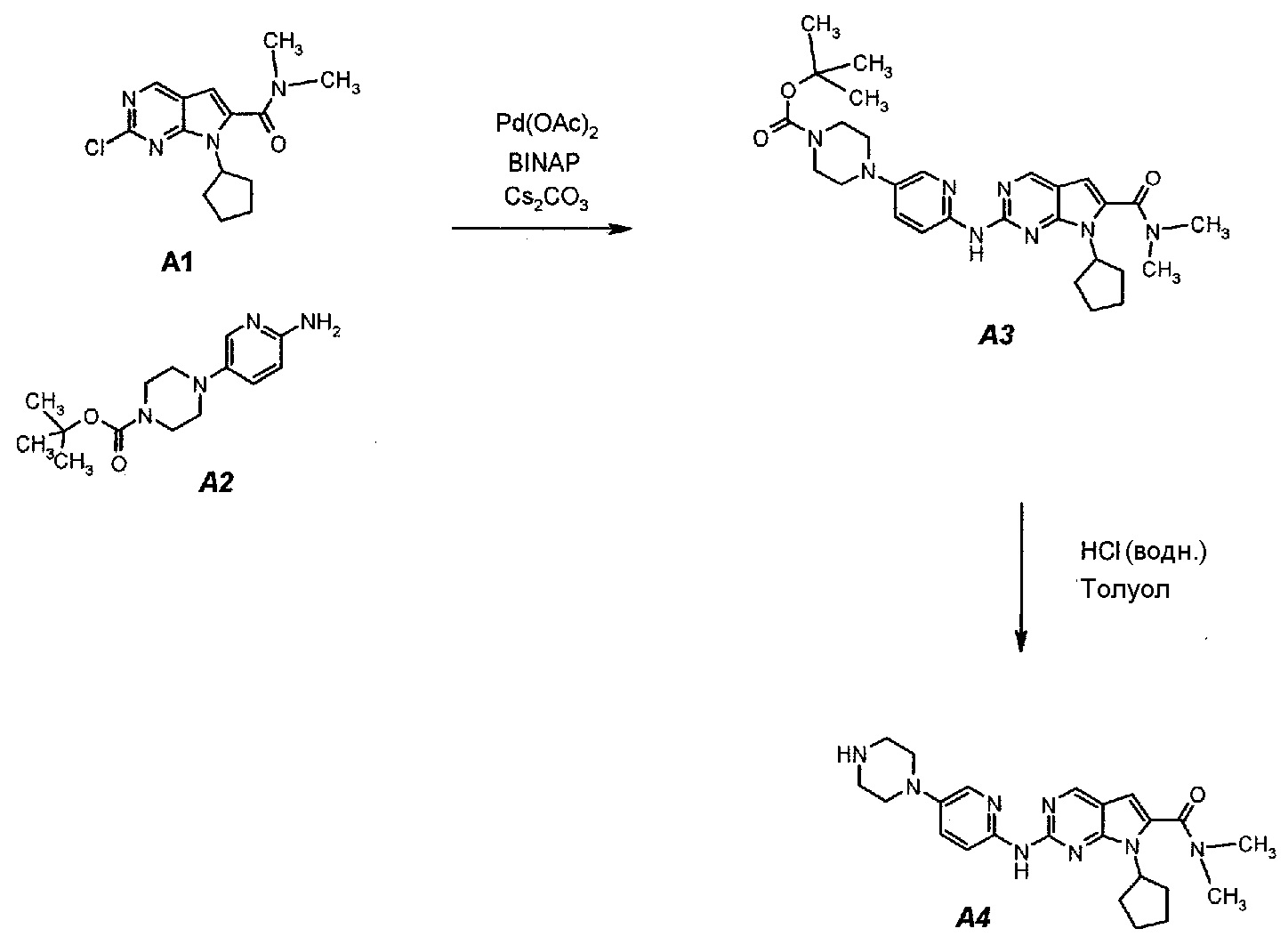
Подробное описание каждой из стадий представлено ниже как стадии 3.1 и 3.2.
3.1. трет-Бутил 4-(6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2-иламино)пиридин-3-ил)пиперазин-1-карбоксилат (A3)

В продуваемый азотом 3-литровый реактор системы Argonaut загружали 43,9 г (0,15 моль, 1,0 экв.) 2-хлор-7-циклопентил-N,N-диметил-7H-пирроло[2,3-d]пиримидин-6-карбоксамида (неочищенный A1 со стадии 1.4, приведенной выше), 45,9 г (0,165 моль, 1,1 экв.) трет-бутил 4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилата (A2), 0,67 г (3,0 ммоль, 0,02 экв.) ацетата палладия(II), 3,73 г (6,0 ммоль, 0,04 экв.) (±)2,2’-бис(дифенилфосфино)-1,1’-бинафталина, ±BINAP (2,2’-бис(дифенилфосфино)-1,1’-бинафтил) и 275 г (344 мл) 4-метил-2-пентанона. Полученную в результате суспензию перемешивали и нагревали до 40±3°C. В течение 15 минут добавляли 73,3 г (0,225 моль, 1,5 экв.) карбоната цезия порциями от 5 до 10 г. Полученную в результате суспензию перемешивали и нагревали до 100±3°C. Данную температуру поддерживали в течение 3 часов или до тех пор, пока остаток исходного соединения A1 не составит ≤2% (по нормированной площади), что определяли путем анализа методом ВЭЖХ. Протекание реакции контролировали, используя систему управления процессом. Образец охлаждали до 70±3°C и добавляли 344 г (344 мл) воды в течение 5 минут. Образец охлаждали до 50±3°C и выдерживали при этой температуре в течение 30 минут. В течение 30 минут добавляли 353 г (516 мл) гептана и образец перемешивали в течение 2 часов. После этого смесь охлаждали до 22±3°C и выдерживали по меньшей мере в течение 4 часов (точка контроля). Твердое вещество отфильтровывали через слой полипропиленового фильтра. Осадок на фильтре промывали холодной (4°C) смесью 24 г (30 мл) 4-метил-2-пентанона и 41 г (60 мл) гептана. Твердое вещество сушили при 60°C до тех пор, пока анализ методом HSGC PSC не покажет, что содержание органического растворителя составляет ≤1%, получая 72,6 г A3 в виде твердого вещества желто-коричневого цвета, температура плавления которого составляла 215-217°C (с разложением).
3.2. 7-Циклопентил-N,N-диметил-2-(5-(пиперазин-1-ил)пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоксамид (A4)

В продуваемый азотом 3-литровый реактор системы Argonaut загружали 67,4 г (0,126 моль, 1,0 экв.) трет-бутил 4-(6-(7-циклопентил-6-(диметилкарбамоил)-7H-пирроло[2,3-d]пиримидин-2-иламинопиридин-3-ил)пиперазин-1-карбоксилата (A3) и 329 г (380 мл) толуола. Суспензию перемешивали и охлаждали до 12±3°C. В течение 30 минут добавляли 138 г (126 мл, 6,0 экв.) 6 н. водного раствора хлористоводородной кислоты, поддерживая температуру смеси ≤15±3°C. Полученный в результате двухфазный раствор нагревали до 25±3°C и выдерживали при этой температуре в течение 30 минут или до тех пор, пока остаток исходного соединения A3 не составит ≤2% (по нормированной площади), что определяли путем анализа методом ВЭЖХ. Протекание реакции контролировали, используя систему управления процессом. Добавляли 250 г (250 мл) 1 н. водного раствора хлористоводородной кислоты и смесь перемешивали в течение 5 минут. Двухфазную реакционную смесь фильтровали через 25 г фильтровальной целлюлозы. Фазы разделяли. Водную фазу (содержащую продукт) загружали в 2-литровую четырехгорлую круглодонную колбу (оборудованную согласно описанию устройства 4) и охлаждали до 15±3°C. Значение pH доводили до 3,2±0,3, медленно добавляя 62 г (41 мл) 50% водного раствора гидроксида натрия, в процессе его добавления температуру смеси поддерживали на уровне ≤27±3°C. Добавляли 16,4 г функционализированного силикагеля Si-Thiol. Суспензию перемешивали в течение 3 часов при 50±3°C. Полимер отфильтровывали, колбу и осадок на фильтре промывали, используя 50 мл воды. Промывную воду объединяли с фильтратом. Фильтрат возвращали в колбу и добавляли 16,4 г функционализированного силикагеля Si-Thiol. Суспензию перемешивали в течение 3 часов при 50±3°C. Силикагель отфильтровывали. Колбу и осадок на фильтре промывали, используя 50 мл воды. Промывную воду объединяли с фильтратом. Фильтрат возвращали в колбу и снова добавляли 16,4 г функционализированного силикагеля Si-Thiol. Суспензию перемешивали в течение 3 часов при 50±3°C. Силикагель отфильтровывали. Колбу и осадок на фильтре промывали, используя 50 мл воды. Промывную воду объединяли с фильтратом. В продуваемый азотом 3-литровый реактор системы Argonaut загружали фильтрат и охлаждали его до 15±3°C. Значение pH доводили до 12,5±0,5, медленно добавляя 17 г (18 мл) 50% водного раствора гидроксида натрия, чтобы осадить продукт (максимальный объем смеси составлял 900 мл). Образец перемешивали в течение по меньшей мере 6 часов при 22±3°C. Твердое вещество отфильтровывали через слой полипропиленового фильтра. Осадок на фильтре четырехкратно промывали, используя 340 г (4×85 мл) воды, до тех пор, пока значение pH промывной воды не составит ≤9. Твердое вещество сушили при 60°C в течение по меньшей мере 16 часов или до тех пор, пока потеря массы при высушивании (LOD) не составит ≤1%, получая 45,7 г (выход с поправкой 84,9%) соединения A4 в виде твердого вещества желто-коричневого цвета, температура плавления которого составляла 194-195°C.
Пример 4. Получение сукцинатной соли диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоновой кислоты (т.е. соединения A6, соединения формулы (II))
Соединение A6  (т.е. соединение формулы (II), сукцинат 7-циклопентил-N,N-диметил-2-(5-(пиперазин-1-ил)пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоксамида) получали согласно представленной ниже схеме синтеза.
(т.е. соединение формулы (II), сукцинат 7-циклопентил-N,N-диметил-2-(5-(пиперазин-1-ил)пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоксамида) получали согласно представленной ниже схеме синтеза.

В продуваемую азотом 1-литровую четырехгорлую круглодонную колбу загружали 11,16 г (0,0945 моль, 1,05 экв.) янтарной кислоты (A5) и 245 г (312 мл) 2-пропанола. Суспензию перемешивали и нагревали до 65±3°C, получая прозрачный раствор. Раствор фильтровали при нагревании через бумажно-стекловолоконный фильтр. Фильтрат выдерживали при 30±3°C в процессе добавления к A4. В продуваемую азотом 2-литровую четырехгорлую круглодонную колбу загружали 39,11 г (0,09 моль, 1,0 экв.) 7-циклопентил-N,N-диметил-2-(5-(пиперазин-1-ил)пиридин-2-иламино)-7H-пирроло[2,3-d]пиримидин-6-карбоксамида (A4) и 1115 г (1420 мл) 2-пропанола. Полученную в результате суспензию перемешивали и нагревали до 80±3°C, получая мутный раствор желтого цвета. Раствор охлаждали до 70±3°C и фильтровали через слой целита массой 25 г. Теплый профильтрованный раствор A4 переносили в продуваемый азотом 3-литровый реактор системы Argonaut и повторно нагревали до 80±3°C. Раствор янтарной кислоты в 2-пропаноле добавляли в течение 1 часа, поддерживая в процессе добавления температуру на уровне 80±3°C. В смесь помещали затравочные кристаллы после добавления 80% раствора янтарной кислоты. Образец перемешивали при 80±3°C в течение 1 часа после завершения добавления, охлаждали до 20±3°C в течение 1 часа, выдерживали в течение 30 минут и твердое вещество отфильтровывали. Осадок на фильтре промывали, используя 78 г (100 мл) 2-пропанола. Твердое вещество сушили при 60°C в течение по меньшей мере 16 часов или до тех пор, пока потеря массы при высушивании (LOD) не составит ≤1%, получая 47,16 г (выход с поправкой 94,9%) соединения A6 в виде кристаллического твердого вещества желтого цвета, температура плавления которого составляла 202-203°C.
Пример 5. Исследование физической формы соединения формулы (II) при относительной влажности 90%
Чтобы понять его гигроскопичную природу, полученное в примере 4 соединение A6 исследовали в двух циклах изменения относительной влажности (RH) от 0 до 90 и обратно до 0%. Как показано в таблице 1 и на фиг.1, соединение A6 абсорбирует до 2% влаги при 90% RH в каждом цикле, демонстрируя слегка гигроскопичное поведение в условиях высокой влажности. Кроме того, в условиях 90% RH в каждом цикле наблюдали резкое повышение поглощения влаги, и различное поведение при сорбции и десорбции отражает образование гидратной формы, которое происходит в условиях 90% RH. На Фиг.2, 3 и 4 показано изменение физической формы соединения A6 при воздействии вплоть до 90% RH, поскольку наблюдали другую кристаллическую форму с эндотермическим переходом, который показывал потерю массы 3% около 100°C, с одновременным превращением в гидратную форму после воздействия 90% RH.
При 90% RH приблизительно 7,35% полученного в примере 4 соединения A6 превращалось из негидратной формы в гидратную форму.
Пример 6. Исследование физической формы соединения формулы (II) при относительной влажности 80%
Для подтверждения того, что изменение формы, наблюдаемое при 90% RH, не происходит при 80% RH, полученное в примере 4 соединение A6 исследовали в цикле изменения RH от 0 до 80 и обратно до 0%. Как показано в таблице 2 и на фиг.5, соединение A6 абсорбирует вплоть до 0,5% влаги при 80% RH, отражая почти негигроскопичное поведение при 80°C. На фиг.5 и 6 показана устойчивость физической формы соединения формулы (II) при воздействии 80% RH, что является признаком негидратной формы. Поскольку для устойчивости входящего в лекарственное средство вещества и полученного лекарственного средства желательной является устойчивость физической формы при 75% RH, соединение формулы (II) (негидратная форма) подходит для разработки.
При 80% RH только приблизительно 0,52% полученного в примере 4 соединения A6 превращается из негидратной формы в гидратную форму.
Пример 7. Растворимость
Растворимость негидратной формы в воде составляет приблизительно 30 мг/мл. Напротив, растворимость гидратной формы значительно ниже и составляет менее чем 0,5 мг/мл.


Claims (9)
1. Сукцинатная соль диметиламида 7-циклопентил-2-(5-пиперазин-1-ил-пиридин-2-иламино)-7Н-пирроло[2,3-d]пиримидин-6-карбоновой кислоты, представленная формулой (II)

или его гидратная форма.
2. Соль по п. 1 в негидратной форме.
3. Соль по п. 1 в гидратной форме.
4. Фармацевтическая композиция, обладающая свойствами циклинзависимой киназы, содержащая
(a) терапевтически эффективное количество соли по любому из пп. 1-3; и
(b) по меньшей мере один фармацевтически приемлемый носитель, разбавитель, растворитель или эксципиент.
5. Способ лечения заболевания, которое ослабляется ингибированием активности циклинзависимых киназ, включающий стадию введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соли по любому из пп. 1-3.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US41206410P | 2010-11-10 | 2010-11-10 | |
| US61/412,064 | 2010-11-10 | ||
| PCT/US2011/059890 WO2012064805A1 (en) | 2010-11-10 | 2011-11-09 | Salt(s) of 7-cyclopentyl-2 -(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2013126112A RU2013126112A (ru) | 2014-12-20 |
| RU2631243C2 true RU2631243C2 (ru) | 2017-09-20 |
Family
ID=45094234
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2013126112A RU2631243C2 (ru) | 2010-11-10 | 2011-11-09 | СОЛЬ (СОЛИ) ДИМЕТИЛАМИДА 7-ЦИКЛОПЕНТИЛ-2-(5-ПИПЕРАЗИН-1-ИЛ-ПИРИДИН-2-ИЛАМИНО)-7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-6-КАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ |
Country Status (24)
| Country | Link |
|---|---|
| US (4) | US20120115878A1 (ru) |
| EP (2) | EP2638030B1 (ru) |
| JP (4) | JP2013542257A (ru) |
| KR (4) | KR20190099090A (ru) |
| CN (4) | CN113956257A (ru) |
| AR (2) | AR083797A1 (ru) |
| AU (4) | AU2011326620A1 (ru) |
| BR (2) | BR112013010167B1 (ru) |
| CA (1) | CA2815839C (ru) |
| CL (1) | CL2013001282A1 (ru) |
| CO (1) | CO6781544A2 (ru) |
| EC (1) | ECSP13012619A (ru) |
| ES (1) | ES2803432T3 (ru) |
| GT (1) | GT201300123A (ru) |
| IL (1) | IL225580B (ru) |
| JO (1) | JO3670B1 (ru) |
| MA (1) | MA34653B1 (ru) |
| MX (1) | MX370852B (ru) |
| MY (1) | MY164274A (ru) |
| PE (1) | PE20140065A1 (ru) |
| RU (1) | RU2631243C2 (ru) |
| SG (1) | SG189229A1 (ru) |
| TW (1) | TWI549953B (ru) |
| WO (1) | WO2012064805A1 (ru) |
Families Citing this family (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UY33227A (es) | 2010-02-19 | 2011-09-30 | Novartis Ag | Compuestos de pirrolopirimidina como inhibidores de la cdk4/6 |
| US20120115878A1 (en) | 2010-11-10 | 2012-05-10 | John Vincent Calienni | Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof |
| ES2676180T3 (es) | 2011-07-01 | 2018-07-17 | Novartis Ag | Terapia de combinación que comprende un inhibidor de CDK4/6 y un inhibidor de PI3K para el uso en el tratamiento de cáncer |
| US9867825B2 (en) | 2012-12-20 | 2018-01-16 | Novartis Ag | Pharmaceutical combination comprising binimetinib |
| JP2017516855A (ja) * | 2014-05-28 | 2017-06-22 | シャンハイ フォチョン ファーマシューティカル カンパニー リミテッド | 特定のタンパク質キナーゼ阻害剤 |
| CN105294737B (zh) * | 2014-07-26 | 2019-02-12 | 广东东阳光药业有限公司 | Cdk类小分子抑制剂的化合物及其用途 |
| EP3218005B1 (en) | 2014-11-12 | 2023-01-04 | Seagen Inc. | Glycan-interacting compounds and methods of use |
| CN105111215B (zh) * | 2014-12-12 | 2019-06-18 | 苏州晶云药物科技股份有限公司 | 一种周期蛋白依赖性激酶抑制剂的晶型及其制备方法 |
| EP3231805B1 (en) * | 2014-12-12 | 2020-03-04 | Crystal Pharmatech Co. Ltd. | Salt of pyrrolo[2,3-d]pyrimidine compound and novel polymorph of salt |
| US10138250B2 (en) | 2014-12-12 | 2018-11-27 | Crystal Pharmatech Co., Ltd. | Salt of pyrrolo[2,3-D]pyrimidine compound and novel polymorph of salt |
| CN107787226A (zh) * | 2015-03-25 | 2018-03-09 | 诺华股份有限公司 | 药物组合 |
| US10323035B2 (en) | 2015-04-01 | 2019-06-18 | Crystal Pharmatech Co., Ltd. | Co-crystal of a CDK inhibitor and an MEK inhibitor and process of preparation thereof |
| PT3283058T (pt) | 2015-04-16 | 2023-02-03 | Novartis Ag | Comprimido de ribociclib |
| CN105037236B (zh) | 2015-06-04 | 2017-07-28 | 苏州明锐医药科技有限公司 | 瑞博西尼中间体及其制备方法 |
| JP2018526376A (ja) * | 2015-08-28 | 2018-09-13 | ノバルティス アーゲー | がんの治療または予防のための(a)サイクリン依存性キナーゼ4/6(cdk4/6)阻害剤のlee011(=リボシクリブ)および(b)上皮成長因子受容体(egfr)阻害剤のエルロチニブを含む医薬組み合わせ物 |
| EP3156406A1 (en) | 2015-10-14 | 2017-04-19 | ratiopharm GmbH | Crystalline forms of ribociclib free base |
| EP3373969A4 (en) | 2015-11-12 | 2019-08-14 | Siamab Therapeutics, Inc. | GLYCANINTERAGING COMPOUNDS AND METHOD OF USE |
| CN106749259B (zh) * | 2015-11-19 | 2019-02-01 | 华东师范大学 | 一种环戊基嘧啶并吡咯类化合物的合成方法 |
| CN107266451B (zh) * | 2016-04-07 | 2021-12-31 | 上海医药工业研究院 | 瑞布昔利布中间体的制备方法 |
| DK3452484T3 (da) | 2016-05-07 | 2023-09-25 | Fochon Pharmaceuticals Ltd | Visse protein-kinase-inhibitorer |
| KR102466192B1 (ko) | 2016-08-23 | 2022-11-14 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 간세포성 암종의 치료를 위한 조합 요법 |
| WO2018051280A1 (en) * | 2016-09-15 | 2018-03-22 | Dr. Reddy’S Laboratories Limited | Process for preparation of ribociclib, its acid addition salts |
| MX2019003095A (es) | 2016-09-19 | 2019-07-04 | Novartis Ag | Combinaciones terapeuticas que comprenden un inhibidor de raf y un inhibidor de erk. |
| CN106478641B (zh) * | 2016-10-09 | 2018-07-24 | 杭州科巢生物科技有限公司 | 瑞博西尼中间体的合成方法 |
| EP3541847A4 (en) | 2016-11-17 | 2020-07-08 | Seattle Genetics, Inc. | COMPOUNDS INTERACTING WITH GLYCANE AND METHODS OF USE |
| CN108314686A (zh) * | 2017-01-18 | 2018-07-24 | 西南民族大学 | 一种新颖的Ribociclib制备方法 |
| EP3589319A4 (en) | 2017-03-03 | 2021-07-14 | Seagen Inc. | COMPOUNDS INTERACTING WITH GLYCAN AND METHODS OF USE |
| CN108586356B (zh) * | 2017-03-16 | 2021-02-19 | 杭州科巢生物科技有限公司 | 瑞博西尼新中间体及其制备瑞博西尼的合成方法 |
| WO2018170447A1 (en) | 2017-03-16 | 2018-09-20 | Eisai R&D Management Co., Ltd. | Combination therapies for the treatment of breast cancer |
| CN108623599A (zh) * | 2017-03-17 | 2018-10-09 | 西南民族大学 | 一种快速合成Ribociclib方法 |
| PT3618875T (pt) | 2017-05-02 | 2023-08-07 | Novartis Ag | Terapia de combinação que compreende um inibidor de raf e trametinib |
| CN106928236B (zh) * | 2017-05-06 | 2019-05-31 | 山东君瑞医药科技有限公司 | 一种瑞博西尼的合成工艺 |
| CN109206373B (zh) * | 2017-07-07 | 2022-02-15 | 上海医药工业研究院 | 一种帕博昔布中间体5-溴-2-氯-4-环戊基氨基嘧啶的制备工艺 |
| WO2019019959A1 (zh) * | 2017-07-27 | 2019-01-31 | 苏州晶云药物科技股份有限公司 | 瑞博西尼的单琥珀酸盐晶型及其制备方法和用途 |
| WO2019040567A1 (en) | 2017-08-25 | 2019-02-28 | Teva Pharmaceuticals Usa, Inc. | RIBOCICLIB SALTS AND SOLID STATE FORMS THEREOF |
| WO2019043504A1 (en) | 2017-08-31 | 2019-03-07 | Novartis Ag | METHODS OF SELECTING TREATMENT FOR PATIENTS WITH CANCER |
| WO2019062854A1 (zh) | 2017-09-29 | 2019-04-04 | 杭州领业医药科技有限公司 | 瑞博西林的共晶和瑞博西林单琥珀酸盐的共晶、其制备方法、组合物和用途 |
| AU2018354972B2 (en) * | 2017-10-27 | 2021-07-08 | Fresenius Kabi Oncology Ltd. | An improved process for the preparation of ribociclib and its salts |
| WO2019111160A1 (en) * | 2017-12-04 | 2019-06-13 | Sun Pharmaceutical Industries Limited | Crystalline forms of ribociclib succinate |
| WO2019123364A1 (en) * | 2017-12-22 | 2019-06-27 | Shilpa Medicare Limited | Novel polymorphs of ribociclib mono succinate |
| IN201741000072A (ru) * | 2017-12-29 | 2018-07-06 | ||
| CN107936029B (zh) * | 2018-01-08 | 2020-06-30 | 南京奇可药业有限公司 | 一种合成瑞博西尼的方法 |
| CN110016024B (zh) * | 2018-01-09 | 2021-09-03 | 南京药石科技股份有限公司 | 一种合成cdk4/6双重抑制剂的关键中间体及其制备方法和应用 |
| US11414421B2 (en) | 2018-01-20 | 2022-08-16 | Natco Pharma Ltd | Process for the preparation of ribociclib succinate and its novel crystalline forms thereof |
| WO2019150181A1 (en) * | 2018-02-05 | 2019-08-08 | Biophore India Pharmaceuticals Pvt Ltd | Improved process for the preparation of 7-cyclopentyl-n, n-dimethyl-2-(5-(piperazin-1-yl) pyridin-2-ylaminuteso)-7h-pyrrolo[2,3-d] pyrimidine-6-carboxamide succinate (ribociclib succinate) and its crystalline forms thereof |
| WO2019166987A1 (en) * | 2018-02-28 | 2019-09-06 | Sun Pharmaceutical Industries Limited | A process for the preparation of ribociclib and its intermediates |
| WO2019167068A1 (en) | 2018-03-01 | 2019-09-06 | Cipla Limited | Novel polymorphs of ribociclib succinate |
| US10723739B2 (en) | 2018-05-14 | 2020-07-28 | Apotex Inc. | Processes for the preparation of Ribociclib and intermediates thereof |
| TWI675662B (zh) | 2018-05-17 | 2019-11-01 | 中化合成生技股份有限公司 | 瑞博西尼琥珀酸鹽(Ribociclib succinate salt)晶型B、C及D與其衍生物,以及其等之製造方法與藥物組合物 |
| CA3048036A1 (en) | 2018-07-02 | 2020-01-02 | Apotex Inc | Novel crystalline form of ribociclib succinate |
| CN110156793B (zh) * | 2018-07-13 | 2022-01-14 | 安礼特(上海)医药科技有限公司 | 瑞博西尼单琥珀酸盐新晶型及制备方法 |
| WO2020084389A1 (en) | 2018-10-23 | 2020-04-30 | Lupin Limited | Ribociclib intermediate and process for preparation thereof |
| CN111100128B (zh) * | 2018-10-26 | 2022-09-06 | 广安凯特制药有限公司 | 一种瑞博西尼中间产品的合成方法及其中间体化合物 |
| CN114269345A (zh) | 2018-10-26 | 2022-04-01 | 科乐斯疗法公司 | Alk2抑制剂的晶体形式 |
| CN113195000A (zh) | 2018-12-21 | 2021-07-30 | 第一三共株式会社 | 抗体-药物缀合物和激酶抑制剂的组合 |
| CN109400612A (zh) * | 2018-12-24 | 2019-03-01 | 重庆三圣实业股份有限公司 | 一种瑞博西尼的制备方法及其产品和用途 |
| WO2020152629A1 (en) | 2019-01-23 | 2020-07-30 | Novartis Ag | New crystalline forms of a succinate salt of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine -6-carboxylic acid dimethylamide |
| WO2020225827A1 (en) * | 2019-05-08 | 2020-11-12 | Mylan Laboratories Limited | Novel polymorphs of ribociclib succinate |
| CN112010857B (zh) * | 2019-05-30 | 2021-11-05 | 常州制药厂有限公司 | 一种瑞博西尼单丁二酸盐的晶型 |
| WO2021038590A1 (en) | 2019-08-30 | 2021-03-04 | Mylan Laboratories Limited | Novel polymorph of ribociclib succinate |
| CN115551509A (zh) | 2020-05-12 | 2022-12-30 | 诺华股份有限公司 | 包含craf抑制剂的治疗组合 |
| WO2022207788A2 (en) | 2021-04-01 | 2022-10-06 | Krka, D.D., Novo Mesto | Process for the preparation of ribociclib and pharmaceutically acceptable salts thereof |
| CN113636973B (zh) * | 2021-09-07 | 2023-04-07 | 山东铂源药业股份有限公司 | 一种4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的工业化制备方法 |
| IL313422A (en) | 2021-12-10 | 2024-08-01 | Lilly Co Eli | CDK4 and 6 inhibitor in combination with FULVESTRANT for the treatment of advanced or metastatic breast cancer in patients with advanced or metastatic breast cancer before CDK 4 and 4 |
| WO2023114264A1 (en) | 2021-12-15 | 2023-06-22 | Eli Lilly And Company | Combination for treatment of high-risk metastatic hormone-sensitive prostate cancer |
| IL314046A (en) | 2022-01-25 | 2024-09-01 | Novartis Ag | RIBOCICLIB medicinal preparations |
| WO2024049926A1 (en) | 2022-08-31 | 2024-03-07 | Arvinas Operations, Inc. | Dosage regimens of estrogen receptor degraders |
| WO2024097206A1 (en) | 2022-11-02 | 2024-05-10 | Petra Pharma Corporation | Allosteric chromenone inhibitors of phosphoinositide 3-kinase (pi3k) for the treatment of disease |
| WO2024115680A1 (en) | 2022-12-01 | 2024-06-06 | Krka, D.D., Novo Mesto | Ribociclib salts and formulations thereof |
| CN117069663B (zh) * | 2023-08-31 | 2023-12-26 | 四川维亚本苑生物科技有限公司 | 一种瑞博西尼中间体v的合成方法及瑞博西尼的合成方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006008545A2 (en) * | 2004-07-22 | 2006-01-26 | Astex Therapeutics Limited | Thiazole and isothiazole derivatives as protein kinase inhibitors |
| WO2007075783A2 (en) * | 2005-12-22 | 2007-07-05 | Wyeth | Substituted isoquinoline-1,3(2h,4h)-diones, 1-thioxo-1,4-dihydro-2h-isoquinoline-3-ones and 1,4-dihydro-3(2h)-isoquinolones and use thereof as kinase inhibitor |
| WO2010020675A1 (en) * | 2008-08-22 | 2010-02-25 | Novartis Ag | Pyrrolopyrimidine compounds as cdk inhibitors |
| EA012926B1 (ru) * | 2001-12-20 | 2010-02-26 | Оси Фармасьютикалз, Инк. | Пирролопиримидиновые соединения, относящиеся к a-селективным антагонистам, их синтез и применение |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITMI992711A1 (it) | 1999-12-27 | 2001-06-27 | Novartis Ag | Composti organici |
| AU2004270733B2 (en) | 2003-09-11 | 2011-05-19 | Itherx Pharma, Inc. | Cytokine inhibitors |
| ES2424642T3 (es) * | 2004-02-14 | 2013-10-07 | Irm Llc | Compuestos y composiciones como inhibidores de la proteína quinasa |
| WO2006091737A1 (en) * | 2005-02-24 | 2006-08-31 | Kemia, Inc. | Modulators of gsk-3 activity |
| BRPI0612997A2 (pt) | 2005-07-22 | 2010-12-14 | Qualcomm Inc | dispositivos mems e respectivos mÉtodos de fabrico |
| JO3235B1 (ar) * | 2006-05-26 | 2018-03-08 | Astex Therapeutics Ltd | مركبات بيررولوبيريميدين و استعمالاتها |
| AR081331A1 (es) | 2010-04-23 | 2012-08-08 | Cytokinetics Inc | Amino- pirimidinas composiciones de las mismas y metodos para el uso de los mismos |
| US20120115878A1 (en) * | 2010-11-10 | 2012-05-10 | John Vincent Calienni | Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof |
-
2011
- 2011-11-08 US US13/291,187 patent/US20120115878A1/en not_active Abandoned
- 2011-11-08 AR ARP110104165A patent/AR083797A1/es active IP Right Grant
- 2011-11-09 SG SG2013024534A patent/SG189229A1/en unknown
- 2011-11-09 CN CN202110832802.9A patent/CN113956257A/zh active Pending
- 2011-11-09 KR KR1020197023852A patent/KR20190099090A/ko not_active Application Discontinuation
- 2011-11-09 EP EP11791110.7A patent/EP2638030B1/en active Active
- 2011-11-09 KR KR1020187027991A patent/KR20180112079A/ko active Search and Examination
- 2011-11-09 RU RU2013126112A patent/RU2631243C2/ru active
- 2011-11-09 WO PCT/US2011/059890 patent/WO2012064805A1/en active Application Filing
- 2011-11-09 ES ES11791110T patent/ES2803432T3/es active Active
- 2011-11-09 KR KR1020137011989A patent/KR20130132433A/ko active Application Filing
- 2011-11-09 BR BR112013010167-9A patent/BR112013010167B1/pt active IP Right Grant
- 2011-11-09 TW TW100140959A patent/TWI549953B/zh active
- 2011-11-09 BR BR122021022703-9A patent/BR122021022703B1/pt active IP Right Grant
- 2011-11-09 CN CN201510746385.0A patent/CN105384741B/zh active Active
- 2011-11-09 CN CN2011800541083A patent/CN103201275A/zh not_active Withdrawn
- 2011-11-09 MX MX2013005292A patent/MX370852B/es active IP Right Grant
- 2011-11-09 MY MYPI2013001156A patent/MY164274A/en unknown
- 2011-11-09 JP JP2013538838A patent/JP2013542257A/ja active Pending
- 2011-11-09 AU AU2011326620A patent/AU2011326620A1/en not_active Abandoned
- 2011-11-09 CA CA2815839A patent/CA2815839C/en active Active
- 2011-11-09 KR KR1020207033901A patent/KR20200136493A/ko not_active Application Discontinuation
- 2011-11-09 US US13/882,353 patent/US9193732B2/en active Active
- 2011-11-09 CN CN201510746374.2A patent/CN105399743A/zh active Pending
- 2011-11-09 EP EP20168336.4A patent/EP3753933A1/en not_active Withdrawn
- 2011-11-09 PE PE2013000978A patent/PE20140065A1/es not_active Application Discontinuation
- 2011-11-12 JO JOP/2011/0332A patent/JO3670B1/ar active
-
2013
- 2013-04-04 IL IL225580A patent/IL225580B/en active IP Right Grant
- 2013-05-07 MA MA35884A patent/MA34653B1/fr unknown
- 2013-05-08 CO CO13115442A patent/CO6781544A2/es not_active Application Discontinuation
- 2013-05-09 CL CL2013001282A patent/CL2013001282A1/es unknown
- 2013-05-10 GT GT201300123A patent/GT201300123A/es unknown
- 2013-05-10 EC ECSP13012619 patent/ECSP13012619A/es unknown
-
2014
- 2014-06-18 US US14/307,901 patent/US20150099760A1/en not_active Abandoned
-
2015
- 2015-10-20 US US14/887,790 patent/US9868739B2/en active Active
-
2016
- 2016-05-27 AU AU2016203534A patent/AU2016203534B2/en active Active
- 2016-10-14 JP JP2016202167A patent/JP6534375B2/ja active Active
- 2016-10-14 JP JP2016202168A patent/JP2017039757A/ja not_active Withdrawn
-
2017
- 2017-08-30 AU AU2017221805A patent/AU2017221805A1/en not_active Abandoned
-
2018
- 2018-09-28 JP JP2018184166A patent/JP2019038809A/ja active Pending
- 2018-12-04 AU AU2018274883A patent/AU2018274883B2/en active Active
-
2020
- 2020-01-10 AR ARP200100071A patent/AR117799A2/es unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA012926B1 (ru) * | 2001-12-20 | 2010-02-26 | Оси Фармасьютикалз, Инк. | Пирролопиримидиновые соединения, относящиеся к a-селективным антагонистам, их синтез и применение |
| WO2006008545A2 (en) * | 2004-07-22 | 2006-01-26 | Astex Therapeutics Limited | Thiazole and isothiazole derivatives as protein kinase inhibitors |
| WO2007075783A2 (en) * | 2005-12-22 | 2007-07-05 | Wyeth | Substituted isoquinoline-1,3(2h,4h)-diones, 1-thioxo-1,4-dihydro-2h-isoquinoline-3-ones and 1,4-dihydro-3(2h)-isoquinolones and use thereof as kinase inhibitor |
| WO2010020675A1 (en) * | 2008-08-22 | 2010-02-25 | Novartis Ag | Pyrrolopyrimidine compounds as cdk inhibitors |
| EA019094B1 (ru) * | 2008-08-22 | 2014-01-30 | Новартис Аг | Пирролопиримидины и их применение |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2631243C2 (ru) | СОЛЬ (СОЛИ) ДИМЕТИЛАМИДА 7-ЦИКЛОПЕНТИЛ-2-(5-ПИПЕРАЗИН-1-ИЛ-ПИРИДИН-2-ИЛАМИНО)-7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-6-КАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | |
| AU2019340569B2 (en) | Improved method for the manufacture of 3-[(1S)-1-imidazo[1,2-a]pyridin-6-ylethyl]-5-(1-methylpyrazol-4-yl)triazolo[4,5-b]pyrazine and polymorphic forms thereof | |
| CN112125908B (zh) | Cdk激酶抑制剂、其制备方法、药物组合物和应用 | |
| CN105924431B (zh) | 化合物克唑替尼的合成工艺 | |
| JP7369798B2 (ja) | Cdkキナーゼ阻害剤 | |
| CN112424187B (zh) | 抑制jak的化合物的昔萘酸盐 | |
| CN115028633A (zh) | 吡咯并嘧啶类化合物的制备及其应用 | |
| WO2023212147A1 (en) | Heterocyclic compounds as modulators of bcl6 as ligand directed degraders | |
| CN117209538A (zh) | 一种降解egfr的氘代物及其在医药上的应用 | |
| CN115403624A (zh) | 一种二芳基硫代乙内酰脲化合物及其制备方法与应用 |