JP6571497B2 - ミルタザピンの製造方法 - Google Patents
ミルタザピンの製造方法 Download PDFInfo
- Publication number
- JP6571497B2 JP6571497B2 JP2015222831A JP2015222831A JP6571497B2 JP 6571497 B2 JP6571497 B2 JP 6571497B2 JP 2015222831 A JP2015222831 A JP 2015222831A JP 2015222831 A JP2015222831 A JP 2015222831A JP 6571497 B2 JP6571497 B2 JP 6571497B2
- Authority
- JP
- Japan
- Prior art keywords
- mirtazapine
- solvent
- methyl
- reaction
- crude
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 C*Cc1cccnc1N(CCN(C)C1)C1c1ccccc1 Chemical compound C*Cc1cccnc1N(CCN(C)C1)C1c1ccccc1 0.000 description 1
- RONZAEMNMFQXRA-UHFFFAOYSA-N CN(CC1)CC(c2c(C3)cccc2)N1c1c3cccn1 Chemical compound CN(CC1)CC(c2c(C3)cccc2)N1c1c3cccn1 RONZAEMNMFQXRA-UHFFFAOYSA-N 0.000 description 1
- PYZPABZGIRHQTA-UHFFFAOYSA-N CN(CC1)CC(c2ccccc2)N1c1ncccc1CO Chemical compound CN(CC1)CC(c2ccccc2)N1c1ncccc1CO PYZPABZGIRHQTA-UHFFFAOYSA-N 0.000 description 1
Description
装置:液体クロマトグラフ装置及び質量分析計(Waters Corporation
製)
検出器:紫外吸光光度計(Waters Corporation製)
測定波長:240nm
カラム:内径4.6mm、長さ25cmのステンレス管に5μmの液体クロマトグラ
フィー用オクタデシルシリル化シリカゲルが充填されたもの。
移動相a:酢酸アンモニウム0.39gを水1000mLに添加し溶解させた混合液。
移動相b:アセトニトリル。
移動相の送液:移動相A及びBの混合比を表1のように変えて濃度勾配制御する。
カラム温度:40℃付近の一定温度。
イオン化法:エレクトロスプレーイオン化法(ESI)。
検出モード:正イオンモード。
本発明に使用するピリジンメタノール化合物は、特に制限されることなく、公知の方法により製造することができる。公知の方法の一例として、特公昭59−042678号公報等に記載されているように、2−(4−メチル−2−フェニル−1−ピペラジニル)−3−ピリジンカルボン酸を有機溶媒中、金属水素化物と反応させて還元する方法が挙げられる。具体的には、2−(4−メチル−2−フェニル−1−ピペラジニル)−3−ピリジンカルボン酸とテトラヒドロフランとの溶液を得る。次いで、窒素雰囲気下、水素化リチウムアルミニウムとテトラヒドロフランとの懸濁液に、当該溶液を徐々に加える。還流温度まで加温し、HPLC等により2−(4−メチル−2−フェニル−1−ピペラジニル)−3−ピリジンカルボン酸の消失が確認されるまで反応させる。反応終了後、水を加え、析出した固体を濾別し、ピリジンメタノール化合物を含む溶液を得る。当該溶液を濃縮する、或いは、濃縮残渣をエーテル等の溶媒による再結晶で精製することでピリジンメタノール化合物を単離することができる。単離後、乾燥することにより、残留する有機溶媒や水等を除去でき、高純度なピリジンメタノール化合物を製造できる。
ピリジンメタノール化合物を硫酸中で環化してミルタザピンとする方法としては、特許文献1、特許文献2又は特許文献3等に記載されている公知の方法を用いることができる。具体的にはピリジンメタノール化合物と閉環試薬である濃硫酸とを混合した後、得られた混合物を所定の温度範囲で反応させる。当該混合操作は、ガラス製容器、ステンレス製容器、テフロン(登録商標)製容器、グラスライニング容器等の容器にて実施し、さらに、メカニカルスターラー、マグネティックスターラー等を用いて撹拌下で実施することが、操作性や均一性の点から好ましい。
本発明において、上記のようにして得られた反応終了後の混合物から、公知の方法による後処理操作を行うことにより、粗体のミルタザピンを得ることができる。例えば、特許文献1のように、反応終了後の反応混合物を冷却し、水を加え希釈した後、水酸化ナトリウムを加え中和する。さらに、トルエンを加え、析出したミルタザピンをトルエン中に溶解させ抽出する。当該トルエン層を水層と分離し、ヘプタンを加えて結晶化させ、結晶を固液分離により単離し、粗体のミルタザピンを得ることができる。
上記のようにして得られた粗体のミルタザピンを、本発明による精製操作を行うことにより、高純度のミルタザピンを得ることができる。具体的には、ミルタザピンを、良溶媒としてエタノール等のアルコール系溶媒、アセトン等のケトン系溶媒、酢酸エチル等のエステル系溶媒、及びテトラヒドロフラン等のエーテル系溶媒から選択される良溶媒に加熱溶解後、ヘプタン及びヘキサンから選択される貧溶媒を添加し、良溶媒と貧溶媒との混合溶媒の溶液とする、または良溶媒としてアセトニトリルを用いて粗体のミルタザピンを加熱溶解後、貧溶媒として水を添加し、良溶媒と貧溶媒との混合溶媒の溶液とし、その後混合溶媒の溶液を冷却し、スラリー状態とする。該スラリーをろ別し、ろ別したスラリーを必要に応じて上記良溶媒と貧溶媒の混合溶媒で洗浄することで、ダイマー1及び2が効果的に低減されたミルタザピンを取得することができる。
HPLCによるミルタザピンの純度及びダイマー不純物の含有量は、下記の装置、条件により測定した。当該条件によるHPLC分析において、ミルタザピンの保持時間は14.0分付近、ダイマー1は20.3分付近、ダイマー2は26.6分付近である。なお、ミルタザピンの純度とは、得られたクロマトグラムにおけるミルタザピンのピーク面積値の、全てのピークの面積値の合計に対する百分率で示した値である。また、ダイマー1及び2の含有量は、各不純物のピーク面積値の、全てのピークの面積値の合計に対する百分率で示した値である。
検出器:紫外吸光光度計(ウォーターズ2489)
検出波長:240nm
カラム:内径4.6mm、長さ25cmのステンレス管に5μmの液体クロマトグラフィー用オクタデシルシリカゲルが充填されたもの。
移動相及び送液方法:以下に示す移動相A及びBを用い、試料注入後の経過時間に従い、両者の混合比を下記表2に示す様に制御し、送液した。
移動相A:リン酸水素二ナトリウム12水和物7.2gを水1000mLに溶解し、リン酸を加えてpH7.4とした。
移動相B:アセトニトリル
流量:毎分1.0mL
カラム温度:40℃付近の一定温度
撹拌翼、温度計を取り付けた1Lの四口フラスコに、2−(4−メチル−2−フェニル−1−ピペラジニル)−3−ピリジンカルボン酸480g(1.61mol)、テトラヒドロフラン2160mLを加え懸濁させ、10℃に冷却した。窒素雰囲気下にて、70%ソジウムジヒドロビス(2−メトキシエトキシ)アルミネートのトルエン溶液1920g(5.65mol)を滴下した後、40℃で5時間反応させた。反応終了後、水2200mLを加え、生じた沈殿をろ過により除去し、有機層と水層を分液した。有機層に25wt%食塩水960mLを加えて洗浄後、分液を行い、得られた有機層を減圧下、濃縮した。得られた残査にトルエン1920mLを加えた後、60〜65℃に加熱し、不溶物を濾別した。トルエン層を減圧濃縮した後にピリジンメタノール化合物の粗体を得た。
撹拌翼、温度計を取り付けた100mLの四口フラスコに、窒素雰囲気下、濃硫酸17.6g(176.4mmol)を入れ、15℃付近に冷却した。次いで、製造例1で得られたピリジンメタノール化合物5.0g(17.64mmol)を、35℃以下で20分間かけて少量ずつ加えた。得られた混合物を35℃に加温し、9時間反応させた(反応転化率:99.6%、ダイマー1:0.20%、ダイマー2:0.19%)。
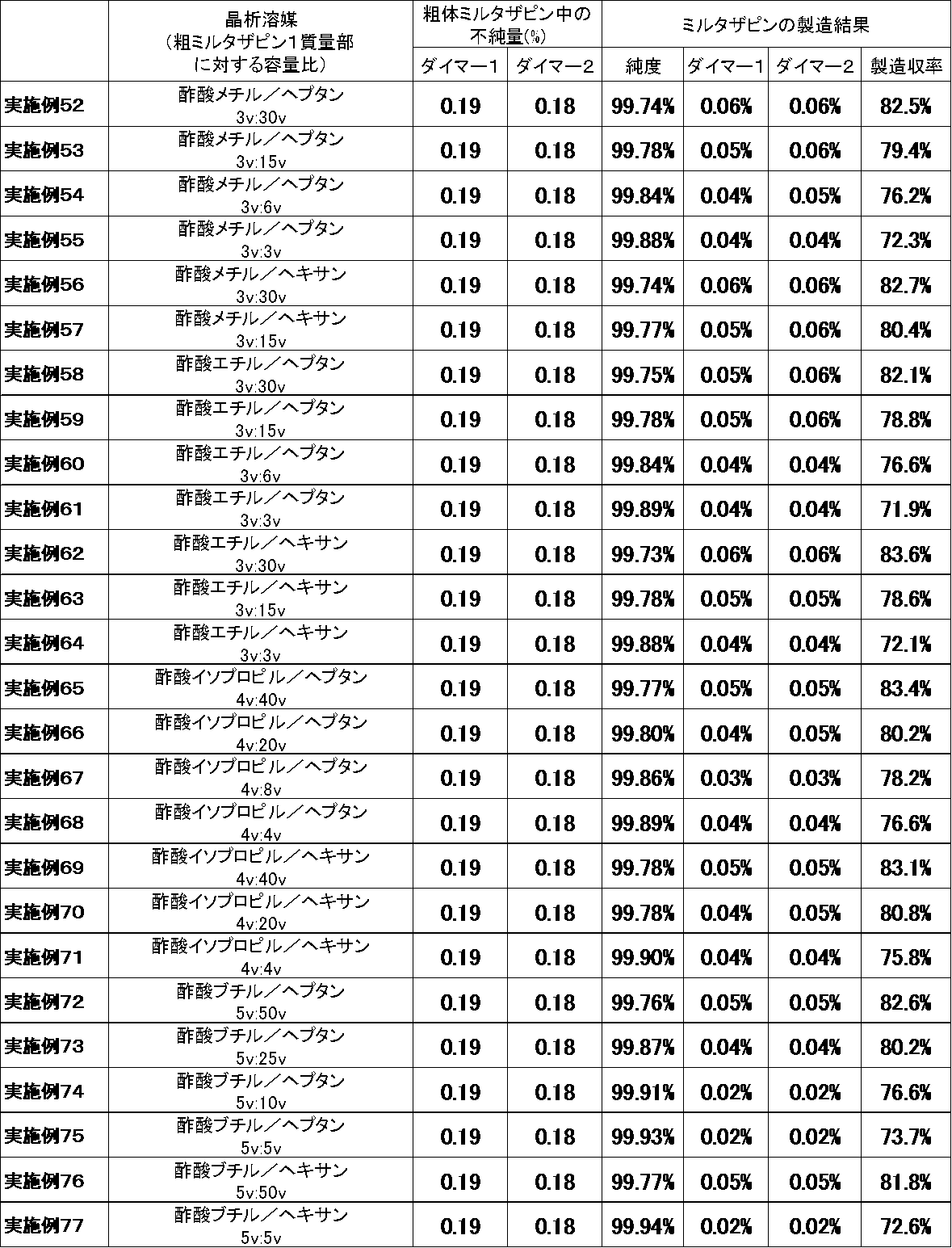
良溶媒の種類又は貧溶媒の種類を変えた以外は参考例1と同様の方法でミルタザピンを得た。製造結果を表3〜6に示した。なお、表3中、実施例1〜24は参考例である。
良溶媒としてアセトニトリル、貧溶媒として水を用いたこと以外は実施例1と同様の方法でミルタザピンを得た。製造結果を表7に示した。
良溶媒をメタノールに、貧溶媒を水に変えたこと以外は実施例1と同様の方法でミルタザピンを得た。製造結果を表8に示した。
Claims (6)
- 2−(4−メチル−2−フェニル−1−ピペラジニル)−3−ピリジンメタノールと硫酸とを反応させてミルタザピンを製造する方法において、ケトン系溶媒、エーテル系溶媒及びエステル系溶媒から選択される良溶媒と、ヘプタン及びヘキサンから選択される貧溶媒との混合溶媒を用いて、ミルタザピンを結晶化することを特徴とするミルタザピンの製造方法。
- 前記良溶媒がアセトン、メチルエチルケトン、メチルプロピルケトン、ジエチルケトン及びジイソプロピルケトンから選択されるケトン系溶媒である請求項1に記載のミルタザピンの製造方法。
- 前記良溶媒が酢酸メチル、酢酸エチル、酢酸イソプロピル及び酢酸ブチルから選択されるエステル系溶媒である請求項1に記載のミルタザピンの製造方法。
- 前記良溶媒がジイソプロピルエーテル、t-ブチルメチルエーテル、テトラヒドロフラン及びジオキサンから選択されるエーテル系溶媒である請求項1に記載のミルタザピンの製造方法。
- 2−(4−メチル−2−フェニル−1−ピペラジニル)−3−ピリジンメタノールと硫酸とを反応させてミルタザピンを製造する方法において、良溶媒がアセトニトリル、貧溶媒が水である、良溶媒と貧溶媒との混合溶媒を用いて、ミルタザピンを結晶化することを特徴とするミルタザピンの製造方法。
- 良溶媒と貧溶媒との混合比率が容量比1:1〜1:10である、請求項1又は請求項5に記載のミルタザピンの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015222831A JP6571497B2 (ja) | 2015-11-13 | 2015-11-13 | ミルタザピンの製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015222831A JP6571497B2 (ja) | 2015-11-13 | 2015-11-13 | ミルタザピンの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017088565A JP2017088565A (ja) | 2017-05-25 |
| JP6571497B2 true JP6571497B2 (ja) | 2019-09-04 |
Family
ID=58771571
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015222831A Expired - Fee Related JP6571497B2 (ja) | 2015-11-13 | 2015-11-13 | ミルタザピンの製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6571497B2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017088564A (ja) * | 2015-11-13 | 2017-05-25 | 株式会社トクヤマ | ミルタザピンの製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SK14672001A3 (sk) * | 1999-04-19 | 2002-11-06 | Teva Pharmaceutical Industries Ltd. | Spôsob syntézy a kryštalizácie zlúčenín obsahujúcich piperazínový kruh |
| WO2001038329A1 (fr) * | 1999-11-24 | 2001-05-31 | Sumika Fine Chemicals Co., Ltd. | Cristaux de mirtazapine anhydre et leur procede d'obtention |
| AU6474200A (en) * | 1999-12-13 | 2001-06-18 | Sumika Fine Chemicals Co., Ltd. | Process for the preparation of a pyridinemethanol compound |
| JP4321983B2 (ja) * | 2000-11-27 | 2009-08-26 | 住友化学株式会社 | 無水ミルタザピンおよびその製法 |
| JP5192707B2 (ja) * | 2007-03-22 | 2013-05-08 | 住友化学株式会社 | ミルタザピンの製造方法 |
-
2015
- 2015-11-13 JP JP2015222831A patent/JP6571497B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017088565A (ja) | 2017-05-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102240865B1 (ko) | Pde4 억제제의 제조 방법 | |
| EP3988545A1 (en) | Methods for preparing cdk4/6 inhibitor and salt and intermediate thereof | |
| CN111646922B (zh) | 一种2-(4-溴-2-氰基-6-氟苯基)乙酸的合成方法 | |
| CN108699068B (zh) | 一种三氟甲基取代的吡喃衍生物制备方法 | |
| WO2009006861A2 (en) | A method for isolation and purification of montelukast | |
| JP6571497B2 (ja) | ミルタザピンの製造方法 | |
| JP6452575B2 (ja) | ミルタザピンの製造方法 | |
| JP2017088564A (ja) | ミルタザピンの製造方法 | |
| CN107814757B (zh) | 一种合成多取代吡咯衍生物的方法 | |
| CN105753733A (zh) | Ahu377的晶型及其制备方法与用途 | |
| TW202304931A (zh) | 生物素之製造方法,以及生物素之l-離胺酸鹽及其製造方法 | |
| JP2013527239A (ja) | イクサベピロンの固体形 | |
| SI21850A (sl) | Soli olanzapina in njihova pretvorba v prosto bazo olanzapina | |
| JP2012020970A (ja) | {2−アミノ−1,4−ジヒドロ−6−メチル−4−(3−ニトロフェニル)−3,5−ピリジンジカルボン酸3−(1−ジフェニルメチルアゼチジン−3−イル)エステル5−イソプロピルエステル}の製造方法 | |
| JP6622634B2 (ja) | ミルタザピンの製造方法 | |
| CN109265407B (zh) | 一种双利奈唑胺的合成方法 | |
| EP1730153B1 (en) | Isopropanol water solvate of olanzapine | |
| JP6008734B2 (ja) | オランザピンii型結晶の製造方法 | |
| JP6433809B2 (ja) | 1−(3−ヒドロキシメチルピリジル−2−)−2−フェニル−4−メチルピペラジンの製造方法 | |
| KR100911720B1 (ko) | 결정형 염산 사포그릴레이트의 제조방법 | |
| JP2017222608A (ja) | ミルタザピンの製造方法 | |
| CN115368317A (zh) | 一种用于制备阿立哌唑中间体的改善方法 | |
| JP6516638B2 (ja) | ピリジンメタノール化合物の製造方法及びミルタザピンの製造方法 | |
| CN115850154A (zh) | 一种吲哚布芬的制备方法 | |
| CN106938981A (zh) | 一种奈韦拉平中间体球形晶体的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180802 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20190418 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190423 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190620 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190723 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190808 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6571497 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |