JP6554792B2 - 炭素繊維強化樹脂組成物、ペレット、成形品および電子機器筐体 - Google Patents
炭素繊維強化樹脂組成物、ペレット、成形品および電子機器筐体 Download PDFInfo
- Publication number
- JP6554792B2 JP6554792B2 JP2014549236A JP2014549236A JP6554792B2 JP 6554792 B2 JP6554792 B2 JP 6554792B2 JP 2014549236 A JP2014549236 A JP 2014549236A JP 2014549236 A JP2014549236 A JP 2014549236A JP 6554792 B2 JP6554792 B2 JP 6554792B2
- Authority
- JP
- Japan
- Prior art keywords
- acid
- carbon fiber
- resin composition
- weight
- fiber reinforced
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
- C08G69/265—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids from at least two different diamines or at least two different dicarboxylic acids
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/80—Component parts, details or accessories; Auxiliary operations
- B29B7/88—Adding charges, i.e. additives
- B29B7/90—Fillers or reinforcements, e.g. fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/12—Making granules characterised by structure or composition
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/12—Making granules characterised by structure or composition
- B29B9/14—Making granules characterised by structure or composition fibre-reinforced
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/0405—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres
- C08J5/042—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres with carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/30—Sulfur-, selenium- or tellurium-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/02—Fibres or whiskers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/02—Fibres or whiskers
- C08K7/04—Fibres or whiskers inorganic
- C08K7/06—Elements
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/06—Polyamides derived from polyamines and polycarboxylic acids
Landscapes
- Chemical & Material Sciences (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Inorganic Chemistry (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polyamides (AREA)
- Reinforced Plastic Materials (AREA)
- Polyesters Or Polycarbonates (AREA)
Description
すなわち、
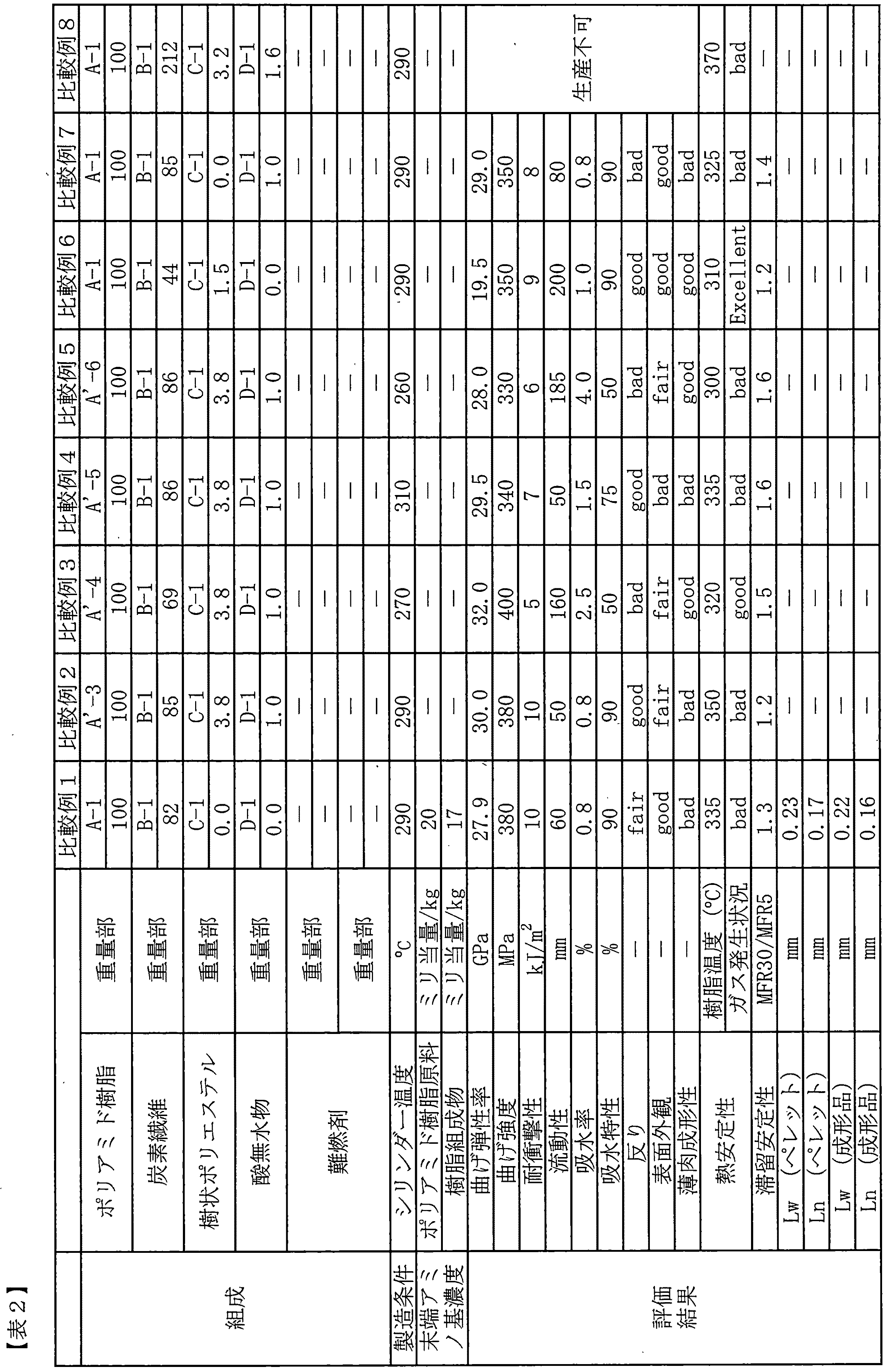
(A)ジカルボン酸総量中テレフタル酸を60〜100モル%含有するジカルボン酸と、ジアミン総量中1,9−ノナンジアミンおよび/または2−メチル−1,8−オクタンジアミンを合計60〜100モル%含有するジアミンとを重縮合して得られる、融点が220〜300℃である半芳香族ポリアミド樹脂100重量部に対して、(B)炭素繊維60〜200重量部、(C)樹状ポリエステル0.01〜10重量部および(D)酸無水物0.01〜5重量部を配合してなる炭素繊維強化樹脂組成物、である。
上記炭素繊維強化樹脂組成物を成形してなるペレットであって、ペレット中の炭素繊維の重量平均繊維長が0.1〜0.5mmであるペレット、である。
ペレットを射出成形して得られる成形品、である。
ペレットを射出成形して得られる電子機器筐体、である。
なお、ペレット中の炭素繊維の重量平均繊維長は、ペレットを500℃で1時間焼成し、得られた灰分を水分散させた後、濾過を行い、その残渣を光学顕微鏡にて観察し、1,000本の長さを測定した結果を重量平均繊維長に換算することにより求めることができる。具体的には、樹脂組成物のペレットを10g程度ルツボに入れ、電気コンロにて可燃性ガスが発生しなくなるまで蒸し焼きにした後、500℃に設定した電気炉内でさらに1時間焼成することにより炭素繊維の残渣のみを得る。その残渣を光学顕微鏡にて50〜100倍に拡大した画像を観察し、無作為に選んだ1,000本の長さを測定し、その測定値(mm)(小数点2桁が有効数字)を用いて、次の式(1)または式(2)に基づき算出することができる。
ここで、Liは炭素繊維の繊維長、niは繊維長Liの炭素繊維の本数、Wiは繊維長Liの炭素繊維の重量、riは繊維長Liの炭素繊維の繊維径、ρは炭素繊維の密度、πは円周率を示し、炭素繊維の断面形状を繊維径riの真円と近似している。繊維径ri、および密度ρが一定である場合、上記式(1)は次の通りに近似され、式(2)により重量平均繊維長を求めることができる。
(B)炭素繊維の重量平均繊維長を前記範囲に調整する手段としては、例えば、目的の繊維長に合わせて任意の繊維長分布を有する炭素繊維を原料とする方法、使用する熱可塑性樹脂の溶融粘度を調整することにより炭素繊維への剪断付与を調整する方法、後述する樹脂組成物の溶融混練時のスクリュー回転数、シリンダー温度、吐出量を調整する方法などを挙げることができる。
ポリアミド樹脂0.1gを98%硫酸溶液50mLに溶解し、ウベローデ粘度計を使用し、30℃±0.05℃の条件下で試料溶液の流下秒数を測定し、以下の式に基づき極限粘度を算出した。
上記の式中、[η]は極限粘度(dL/g)、ηSPは比粘度、Cは試料濃度(g/dL)、tは試料溶液の流下秒数(秒)、t0は硫酸の流下秒数(秒)を示す。
セイコーインスツルメンツ(株)製示差走査熱量計EXSTAR DSC6000を用い、ポリアミド樹脂を一旦330℃で5分間保持し、次いで10℃/分の速度で23℃まで降温せしめた後、10℃/分で昇温したときの融解吸熱ピークを測定し、これを融点とした。
各実施例および比較例により得られた樹脂組成物ペレットを、住友重機械工業(株)製の75トン射出成形機を使用して、シリンダー温度300℃、金型温度80℃の条件で曲げ試験片を射出成形し、ISO178に従い23℃で曲げ強度および曲げ弾性率を評価した。
各実施例および比較例により得られた樹脂組成物ペレットを、住友重機械工業(株)製の75トン射出成形機を使用して、シリンダー温度300℃、金型温度80℃の条件でシャルピー衝撃試験片を射出成形し、ISO179に従い23℃でシャルピー衝撃強さ(ノッチ付き)を評価した。
各実施例および比較例により得られた樹脂組成物ペレットを、住友重機械工業(株)製の75トン射出成形機を使用して、シリンダー温度320℃、金型温度130℃、射出圧力55MPa、射出時間5秒、成形品厚み0.7mmの条件で射出成形した。最初の20ショットを成形した後、続けて成形した10ショットの成形品の流動長を平均し、その値をバーフロー流動長とした。この値が大きい程、流動性に優れている。
各実施例および比較例により得られた樹脂組成物ペレットを、住友重機械工業(株)製の75トン射出成形機を使用して、シリンダー温度300℃、金型温度80℃の条件でダンベル試験片を射出成形した。得られたダンベル試験片を用いて、80℃、95%RH環境下にて静置して重量の経時変化を測定する吸水試験を行い、乾燥時(吸水試験前)および1,000時間経過後の重量を測定し、以下の式より吸水率を求めた。なお、以下の式において、乾燥時重量とは、吸水試験に供する前のダンベル試験片の初期の重量のことである。
(7)成形品の吸水特性
前記(6)の吸水試験後のダンベル試験片を用いて、前記(3)と同様の方法で曲げ強度を評価し、以下の式より強度保持率を求めた。
(8)成形品の反り
各実施例および比較例により得られた樹脂組成物ペレットを、住友重機械工業(株)製の75トン射出成形機を使用して、シリンダー温度320℃、金型温度130℃、射出時間10秒、冷却時間20秒の条件で、80mm×80mm×厚み1mmの金型内に樹脂を充填して射出成形を行った後、冷却し、ゲート部分をカットしないまま取り出し、反り評価用の試験片とした。この試験片を、25℃、湿度65%の条件で24時間静置した後、ゲート側を基準として、これと反対側の端面の浮き上がった高さ(反り量)を測定し、以下の4段階で評価した。
前記(8)で作製した80mm×80mm×厚み1mmの試験片の表面光沢、表面の凹凸を目視観察し、以下の基準により評価を行った。
薄肉電子機器筐体への適用性を実証するため、各実施例および比較例により得られた樹脂組成物ペレットに対して、750トン射出成形機を使用し、シリンダー温度320℃、金型温度100〜120℃の条件で、ホットランナーを使用せず、220mm×300mm×0.8mm厚の金型(11点ゲート)での射出成形を行った。その結果を以下の3段階で評価した。
各実施例および比較例における二軸押出機での溶融混練時の樹脂温度を測定し、併せてガスの発生状況を以下の3段階で評価した。
(12)ポリアミド樹脂および樹脂組成物の末端アミノ基濃度
ポリアミド樹脂または樹脂組成物ペレット0.2gをヘキサフルオロイソプロパノール10mLに溶解して試料溶液とし、0.02N塩酸水溶液を使用して電位差滴定を行うことにより測定した。
(13)滞留安定性
作製した樹脂組成物ペレットを用いて、ASTM D−1238−82に準拠し、荷重2.16kgおよび温度300℃の条件で、滞留時間5分および30分におけるMFRを測定し、これらをMFR5、MFR30とした時の比:MFR30/MFR5を算出した。
(14)ペレットおよび成形品の炭素繊維の平均繊維長
ペレットおよび引張試験片からサンプル10gを切り出し、500℃に設定した電気炉中で1時間焼成した後、イオン交換水に分散、濾過を行い、その残渣を光学顕微鏡にて50〜100倍の倍率で観察しながら、1,000本の長さを測定し、ペレット、成形品の炭素繊維の重量平均繊維長(Lw)および数平均繊維長(Ln)をそれぞれ求めた。
(製造例1)ポリアミド樹脂(A−1)の製造
テレフタル酸4,539.3g(27.3モル)、(a)1,9−ノナンジアミンと(b)2−メチル−1,8−オクタンジアミンの混合物〔(a)/(b)=50/50(モル比)〕4,478.8g(28.3モル)、安息香酸101.6g(0.83モル)、次亜リン酸ナトリウム一水和物9.12g(原料の総質量に対して0.1質量%)および蒸留水2.5リットルを、内容積20リットルのオートクレーブに入れ、窒素置換した。この混合物を、100℃で30分間撹拌し、2時間かけてオートクレーブ内部の温度を220℃に昇温した。この時、オートクレーブ内部の圧力は2MPaまで昇圧した。そのまま2時間反応を続けた後230℃に昇温し、その後2時間、230℃に温度を保ち、水蒸気を徐々に抜いて圧力を2MPaに保ちながら反応させた。次に、30分間かけて圧力を1MPaまで下げ、さらに1時間反応させて、極限粘度[η]が0.18dL/gのプレポリマーを得た。
(製造例2)ポリアミド樹脂(A−2)の製造
製造例1において、固相重合時間のみを2時間に変更し、融点が262℃、極限粘度が0.52dL/gのポリアミド樹脂(A−2)を製造した。
(製造例3)ポリアミド樹脂(A−3)の製造
製造例1において、安息香酸の添加量を1.02g(0.008モル)に変更した以外は、同様の方法により、ポリアミド樹脂(A−3)を製造した。(A−3)の融点は262℃、極限粘度は0.9dL/gであった。
(製造例4)ポリアミド樹脂(A’−3)の製造
(a)1,9−ノナンジアミンと(b)2−メチル−1,8−オクタンジアミンの混合物のモル比を(a)/(b)=80/20に変更した以外は、製造例1と同様の方法でポリアミド樹脂(A’−3)を製造した。(A’−3)の融点は302℃、極限粘度は0.91dL/gであった。
(製造例5)樹状ポリエステル(C−1)の製造
撹拌翼および留出管を備えた500mLの反応容器に、p−ヒドロキシ安息香酸66.3g(0.48モル)、4,4’−ジヒドロキシビフェニル8.38g(0.045モル)、テレフタル酸7.48g(0.045モル)、固有粘度が約0.6dL/gのポリエチレンテレフタレ−ト14.41g(0.075モル)および無水酢酸62.48g(フェノール性水酸基合計の1.00当量)を仕込み、窒素ガス雰囲気下で撹拌しながら145℃で2時間反応させた。その後、トリメシン酸31.52g(0.15モル)を加えて260℃まで昇温し、3時間撹拌し、理論留出量の91%の酢酸が留出したところで加熱および撹拌を停止し、内容物を冷水中に吐出し、樹状ポリエステル(C−1)を得た。
(A’−4)ポリアミドMXD6樹脂“レニー”(登録商標)#6002(三菱エンジニアリングプラスチックス(株)製)(融点:238℃)
(A’−5)ポリアミド10T樹脂“Vestamid”(登録商標)HTPlus M3000(ダイセル・エボニック(株)製)(融点:285℃)
(A’−6)ポリアミド6樹脂“アミラン”(登録商標)CM1001(東レ(株)製)(融点:222℃)
(B−1)PAN系炭素繊維“トレカ”(登録商標)カットファイバーTV14−006(東レ(株)製、原糸T700SC−12K:ストランド強度4.9GPa、ストランド弾性率230GPa)
(D−1)無水コハク酸(シグマアルドリッチジャパン(株)製)、SAJ1級
(E−1)ホスフィン酸塩化合物“EXOLIT”(登録商標)OP1230(クラリアントジャパン(株)製)
(E−2)アクリル変性テトラフルオロエチレン“メタブレン”(登録商標)A3800(三菱レイヨン(株)製)。
[実施例1〜5、参考例1〜3、比較例1〜8]
シリンダー温度を表1に示す温度に設定し、スクリュー回転数を200rpmに設定した二軸押出機((株)日本製鋼所製TEX30α)を用いた。主ホッパーからポリアミド樹脂、樹状ポリエステル、酸無水物および難燃剤を表1〜2に示す配合で供給し、サイドフィーダーから炭素繊維を溶融樹脂中に供給して溶融混練した。ダイから吐出されたストランドを水中にて冷却し、ストランドカッターにより長さ3.0mm長にカットしてペレット化し、炭素繊維強化樹脂組成物ペレットを得た。作製したペレットを用いて、上述した方法により、各種特性評価を行った。結果を表1〜2に示す。
Claims (9)
- (A)ジカルボン酸総量中テレフタル酸を60〜100モル%含有するジカルボン酸と、ジアミン総量中1,9−ノナンジアミンおよび/または2−メチル−1,8−オクタンジアミンを合計60〜100モル%含有するジアミンとを重縮合して得られる、融点が220〜300℃である半芳香族ポリアミド樹脂100重量部に対して、(B)炭素繊維60〜200重量部、(C)樹状ポリエステル0.01〜10重量部および(D)酸無水物0.01〜5重量部を配合してなる炭素繊維強化樹脂組成物。
- (A)半芳香族ポリアミド樹脂の0.2g/dL濃硫酸中30℃で測定した極限粘度が0.5〜1.3dL/gの範囲である請求項1に記載の炭素繊維強化樹脂組成物。
- 炭素繊維強化樹脂組成物中における(A)半芳香族ポリアミド樹脂1kgあたりの末端アミノ基濃度が0.1〜30ミリ当量/kgである請求項1または2に記載の炭素繊維強化樹脂組成物。
- 請求項1〜3のいずれかに記載の炭素繊維強化熱可塑性樹脂組成物を成形してなる、炭素繊維の重量平均繊維長が0.1〜0.5mmであるペレット。
- 請求項4に記載のペレットを射出成形して得られる成形品。
- 成形品中の炭素繊維の重量平均繊維長が0.01〜0.5mmである請求項5に記載の成形品。
- 成形品中の炭素繊維の重量平均繊維長/数平均繊維長の比(Lw/Ln)が1.0以上1.3未満である請求項5または6に記載の成形品。
- 請求項4に記載のペレットを射出成形して得られる電子機器筐体。
- 平均肉厚が0.5〜1.0mmである請求項8記載の電子機器筐体。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013213347 | 2013-10-11 | ||
| JP2013213347 | 2013-10-11 | ||
| PCT/JP2014/076517 WO2015053181A1 (ja) | 2013-10-11 | 2014-10-03 | 炭素繊維強化樹脂組成物、ペレット、成形品および電子機器筐体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2015053181A1 JPWO2015053181A1 (ja) | 2017-03-09 |
| JP6554792B2 true JP6554792B2 (ja) | 2019-08-07 |
Family
ID=52812997
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014549236A Expired - Fee Related JP6554792B2 (ja) | 2013-10-11 | 2014-10-03 | 炭素繊維強化樹脂組成物、ペレット、成形品および電子機器筐体 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP6554792B2 (ja) |
| KR (1) | KR20160070062A (ja) |
| CN (1) | CN105492537B (ja) |
| TW (1) | TWI641653B (ja) |
| WO (1) | WO2015053181A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4052894A4 (en) * | 2019-12-17 | 2024-02-14 | Fukuvi Chemical Industry Co., Ltd. | FIBER REINFORCED RESIN COMPOSITE SHEET, FIBER REINFORCED RESIN COMPOSITE MATERIAL AND MOLDED RESIN ARTICLE THEREFROM |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016118607A (ja) * | 2014-12-19 | 2016-06-30 | オリンパス株式会社 | 鏡枠、鏡筒、および光学機器 |
| JP6703389B2 (ja) * | 2015-10-20 | 2020-06-03 | ダイセルポリマー株式会社 | 成形品の製造方法 |
| CN109691248B (zh) * | 2016-09-26 | 2021-07-30 | 东丽株式会社 | 电子设备壳体及其制造方法 |
| EP3587472A4 (en) | 2017-02-21 | 2020-04-01 | Mitsubishi Gas Chemical Company, Inc. | AMORPHOUS POLYAMIDE RESIN AND MOLDED ARTICLE |
| EP3378884A1 (en) * | 2017-03-21 | 2018-09-26 | Solvay Specialty Polymers USA, LLC. | Thermoplastic composites and corresponding fabrication methods and articles |
| EP3960809A4 (en) * | 2019-04-26 | 2022-06-22 | Unitika Ltd. | POLYAMIDE RESIN COMPOSITION AND MOLDED ARTICLE OBTAINED BY MOLDING THEREOF |
| EP3974469B1 (en) * | 2019-05-20 | 2024-07-03 | TOYOBO MC Corporation | Polyamide-based resin composition for injection molding and sliding component formed therefrom |
| JP6741834B1 (ja) * | 2019-08-09 | 2020-08-19 | 住友化学株式会社 | 液晶ポリエステル樹脂ペレット、及びその製造方法、並びに成形体の製造方法 |
| EP4261250A4 (en) * | 2020-12-09 | 2024-04-24 | Mitsubishi Chemical Corporation | RESIN COMPOSITION, PELLET, MOLDED BODY AND METHOD FOR PRODUCING THE RESIN COMPOSITION |
| CN113651956B (zh) * | 2021-08-23 | 2022-09-16 | 安徽农业大学 | 超高韧性支化聚酰胺共聚物的制备方法、制得的聚酰胺共聚物 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5014610B2 (ja) * | 2005-10-14 | 2012-08-29 | 株式会社クラレ | ポリアミド樹脂組成物およびその成形品 |
| KR101351165B1 (ko) * | 2006-03-30 | 2014-01-14 | 도레이 카부시키가이샤 | 수지상 폴리에스테르, 그의 제조 방법 및 열가소성 수지 조성물 |
| JP2011080092A (ja) * | 2006-03-30 | 2011-04-21 | Toray Ind Inc | 樹状ポリエステル、その製造方法および熱可塑性樹脂組成物 |
| JP5182914B2 (ja) * | 2006-03-30 | 2013-04-17 | 東レ株式会社 | 樹状ポリエステル、その製造方法および熱可塑性樹脂組成物 |
| JP5386870B2 (ja) * | 2007-07-18 | 2014-01-15 | 東レ株式会社 | 樹状ポリエステル、その製造方法および熱可塑性樹脂組成物 |
| JP2009298853A (ja) * | 2008-06-10 | 2009-12-24 | Ube Ind Ltd | ポリアミド樹脂組成物 |
| JP5790005B2 (ja) * | 2010-02-26 | 2015-10-07 | 東レ株式会社 | ポリアミド樹脂組成物およびその製造方法 |
| JP2012116917A (ja) * | 2010-11-30 | 2012-06-21 | Toray Ind Inc | 繊維強化樹脂ペレット |
| CN102532840B (zh) * | 2010-12-29 | 2014-04-02 | 合肥杰事杰新材料股份有限公司 | 用于笔记本电脑外壳的热塑树脂复合材料及其制造方法 |
| CN103958612B (zh) * | 2011-11-29 | 2016-08-24 | 东丽株式会社 | 碳纤维增强热塑性树脂组合物、该组合物的粒料和成型品 |
-
2014
- 2014-10-03 WO PCT/JP2014/076517 patent/WO2015053181A1/ja not_active Ceased
- 2014-10-03 KR KR1020167008530A patent/KR20160070062A/ko not_active Withdrawn
- 2014-10-03 CN CN201480046695.5A patent/CN105492537B/zh not_active Expired - Fee Related
- 2014-10-03 JP JP2014549236A patent/JP6554792B2/ja not_active Expired - Fee Related
- 2014-10-07 TW TW103134858A patent/TWI641653B/zh not_active IP Right Cessation
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4052894A4 (en) * | 2019-12-17 | 2024-02-14 | Fukuvi Chemical Industry Co., Ltd. | FIBER REINFORCED RESIN COMPOSITE SHEET, FIBER REINFORCED RESIN COMPOSITE MATERIAL AND MOLDED RESIN ARTICLE THEREFROM |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20160070062A (ko) | 2016-06-17 |
| CN105492537A (zh) | 2016-04-13 |
| JPWO2015053181A1 (ja) | 2017-03-09 |
| WO2015053181A1 (ja) | 2015-04-16 |
| TWI641653B (zh) | 2018-11-21 |
| TW201522502A (zh) | 2015-06-16 |
| CN105492537B (zh) | 2018-01-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6554792B2 (ja) | 炭素繊維強化樹脂組成物、ペレット、成形品および電子機器筐体 | |
| JP5857742B2 (ja) | 難燃性ポリアミド樹脂組成物 | |
| EP2436717B1 (en) | Polyamide resin | |
| TWI549989B (zh) | Liquid crystal polyester amide, liquid crystal polyesteramide resin composition and molded body | |
| CN108137907B (zh) | 照相机组件用液晶性聚酯树脂组合物及由其形成的照相机组件用成型品 | |
| JP2015129271A (ja) | 炭素繊維強化ポリアミド樹脂組成物およびそれを成形してなる成形品 | |
| JP2013166848A (ja) | 液晶ポリエステル組成物及び成形体 | |
| JP2020186323A (ja) | ポリアミド樹脂組成物 | |
| CN103003330B (zh) | 芳香族液晶聚酯树脂的制造方法及芳香族液晶聚酯树脂复合物的制造方法 | |
| CN113825804A (zh) | 聚酰胺树脂组合物 | |
| CN103282404B (zh) | 液晶性聚酯及其制造方法 | |
| JP2015021063A (ja) | 液晶ポリエステル樹脂組成物 | |
| JP5572922B2 (ja) | エンジン冷却水系部品用ポリアミド樹脂組成物、及び当該組成物から成形させたエンジン冷却水系部品 | |
| JP6985828B2 (ja) | 振動溶着用ポリアミド樹脂組成物、成形体及び溶着成形体 | |
| JP5321434B2 (ja) | Smtコネクタ用ポリアミド樹脂組成物 | |
| JP5532533B2 (ja) | ポリアミド樹脂組成物およびその成形品 | |
| JP2010163552A (ja) | 繊維強化複合材料およびその製造方法 | |
| JP3549624B2 (ja) | 熱可塑性樹脂組成物 | |
| JP5621220B2 (ja) | 導電性ポリアミド樹脂組成物及びケーブルハウジング | |
| KR101582808B1 (ko) | 우수한 물성을 갖는 전방향족 액정 폴리에스테르 수지의 제조방법 및 전방향족 액정 폴리에스테르 수지 컴파운드의 제조방법 | |
| JP7824126B2 (ja) | ポリアミド樹脂組成物、及び成形体 | |
| JP5584966B2 (ja) | 耐熱剤含有樹脂組成物及び該耐熱剤含有樹脂組成物から形成された成形物 | |
| JP2009298856A (ja) | 耐熱剤含有樹脂組成物及び該耐熱剤含有樹脂組成物から形成された成形物 | |
| JP5446795B2 (ja) | Icトレイ用ポリアミド樹脂組成物及びicトレイ | |
| JP7017969B2 (ja) | ポリアミド樹脂組成物、およびその成形品 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170914 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20170914 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20181016 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20181205 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190611 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190624 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 6554792 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| LAPS | Cancellation because of no payment of annual fees |