JP5777953B2 - 電子写真用トナーバインダー及びトナー組成物 - Google Patents
電子写真用トナーバインダー及びトナー組成物 Download PDFInfo
- Publication number
- JP5777953B2 JP5777953B2 JP2011146559A JP2011146559A JP5777953B2 JP 5777953 B2 JP5777953 B2 JP 5777953B2 JP 2011146559 A JP2011146559 A JP 2011146559A JP 2011146559 A JP2011146559 A JP 2011146559A JP 5777953 B2 JP5777953 B2 JP 5777953B2
- Authority
- JP
- Japan
- Prior art keywords
- polyester resin
- acid
- toner
- parts
- mol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000000203 mixture Substances 0.000 title claims description 40
- 239000011230 binding agent Substances 0.000 title claims description 28
- 229920001225 polyester resin Polymers 0.000 claims description 162
- 239000004645 polyester resin Substances 0.000 claims description 162
- 125000004432 carbon atom Chemical group C* 0.000 claims description 37
- 229920005862 polyol Polymers 0.000 claims description 37
- 150000003077 polyols Chemical class 0.000 claims description 36
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 claims description 32
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 23
- OYQYHJRSHHYEIG-UHFFFAOYSA-N ethyl carbamate;urea Chemical compound NC(N)=O.CCOC(N)=O OYQYHJRSHHYEIG-UHFFFAOYSA-N 0.000 claims description 23
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 21
- 239000003795 chemical substances by application Substances 0.000 claims description 17
- 239000002253 acid Substances 0.000 claims description 16
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 claims description 16
- 238000003860 storage Methods 0.000 claims description 16
- ARCGXLSVLAOJQL-UHFFFAOYSA-N trimellitic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1 ARCGXLSVLAOJQL-UHFFFAOYSA-N 0.000 claims description 16
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 claims description 12
- 125000003118 aryl group Chemical group 0.000 claims description 9
- 239000003086 colorant Substances 0.000 claims description 8
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 claims description 8
- 150000001991 dicarboxylic acids Chemical class 0.000 claims description 7
- 150000002148 esters Chemical class 0.000 claims description 5
- 239000000654 additive Substances 0.000 claims description 4
- 238000005227 gel permeation chromatography Methods 0.000 claims description 4
- 230000009477 glass transition Effects 0.000 claims description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 110
- 238000006243 chemical reaction Methods 0.000 description 52
- -1 aromatic dicarboxylic acids Chemical class 0.000 description 46
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 39
- 229920005989 resin Polymers 0.000 description 36
- 239000011347 resin Substances 0.000 description 36
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 35
- 238000004519 manufacturing process Methods 0.000 description 34
- 230000015572 biosynthetic process Effects 0.000 description 31
- 239000002245 particle Substances 0.000 description 27
- 239000005056 polyisocyanate Substances 0.000 description 27
- 229920001228 polyisocyanate Polymers 0.000 description 27
- 238000000034 method Methods 0.000 description 25
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 21
- 229920000768 polyamine Polymers 0.000 description 21
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 19
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 18
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 229910052757 nitrogen Inorganic materials 0.000 description 17
- 238000003786 synthesis reaction Methods 0.000 description 17
- 239000003054 catalyst Substances 0.000 description 15
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 14
- 238000002156 mixing Methods 0.000 description 13
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 12
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 12
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 12
- 230000000052 comparative effect Effects 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- SRPWOOOHEPICQU-UHFFFAOYSA-N trimellitic anhydride Chemical compound OC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 SRPWOOOHEPICQU-UHFFFAOYSA-N 0.000 description 10
- UPMLOUAZCHDJJD-UHFFFAOYSA-N 4,4'-Diphenylmethane Diisocyanate Chemical compound C1=CC(N=C=O)=CC=C1CC1=CC=C(N=C=O)C=C1 UPMLOUAZCHDJJD-UHFFFAOYSA-N 0.000 description 9
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical class C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 9
- 239000007795 chemical reaction product Substances 0.000 description 9
- 230000000903 blocking effect Effects 0.000 description 8
- 238000010298 pulverizing process Methods 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 7
- 125000001931 aliphatic group Chemical group 0.000 description 7
- 235000010233 benzoic acid Nutrition 0.000 description 7
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 7
- 238000009833 condensation Methods 0.000 description 7
- 230000005494 condensation Effects 0.000 description 7
- 239000003960 organic solvent Substances 0.000 description 7
- 238000006068 polycondensation reaction Methods 0.000 description 7
- 239000002685 polymerization catalyst Substances 0.000 description 7
- 230000035484 reaction time Effects 0.000 description 7
- 239000005711 Benzoic acid Substances 0.000 description 6
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 6
- 150000004984 aromatic diamines Chemical class 0.000 description 6
- YPFDHNVEDLHUCE-UHFFFAOYSA-N propane-1,3-diol Chemical compound OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 6
- 239000010936 titanium Substances 0.000 description 6
- 229910052719 titanium Inorganic materials 0.000 description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 6
- 239000001993 wax Substances 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 239000011521 glass Substances 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 238000004898 kneading Methods 0.000 description 5
- 239000011976 maleic acid Substances 0.000 description 5
- 229920003986 novolac Polymers 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- VPWNQTHUCYMVMZ-UHFFFAOYSA-N 4,4'-sulfonyldiphenol Chemical class C1=CC(O)=CC=C1S(=O)(=O)C1=CC=C(O)C=C1 VPWNQTHUCYMVMZ-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Natural products CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 4
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 4
- 101710148027 Ribulose bisphosphate carboxylase/oxygenase activase 1, chloroplastic Proteins 0.000 description 4
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 4
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 150000002009 diols Chemical class 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 239000000049 pigment Substances 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- CYIDZMCFTVVTJO-UHFFFAOYSA-N pyromellitic acid Chemical compound OC(=O)C1=CC(C(O)=O)=C(C(O)=O)C=C1C(O)=O CYIDZMCFTVVTJO-UHFFFAOYSA-N 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 230000001629 suppression Effects 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 229930185605 Bisphenol Natural products 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 239000005057 Hexamethylene diisocyanate Substances 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 125000002723 alicyclic group Chemical group 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- KORSJDCBLAPZEQ-UHFFFAOYSA-N dicyclohexylmethane-4,4'-diisocyanate Chemical compound C1CC(N=C=O)CCC1CC1CCC(N=C=O)CC1 KORSJDCBLAPZEQ-UHFFFAOYSA-N 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 230000032050 esterification Effects 0.000 description 3
- 238000005886 esterification reaction Methods 0.000 description 3
- 150000002170 ethers Chemical class 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- RRAMGCGOFNQTLD-UHFFFAOYSA-N hexamethylene diisocyanate Chemical compound O=C=NCCCCCCN=C=O RRAMGCGOFNQTLD-UHFFFAOYSA-N 0.000 description 3
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 238000006116 polymerization reaction Methods 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 150000005846 sugar alcohols Polymers 0.000 description 3
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 3
- 229910000859 α-Fe Inorganic materials 0.000 description 3
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 2
- OHLKMGYGBHFODF-UHFFFAOYSA-N 1,4-bis(isocyanatomethyl)benzene Chemical compound O=C=NCC1=CC=C(CN=C=O)C=C1 OHLKMGYGBHFODF-UHFFFAOYSA-N 0.000 description 2
- ALVZNPYWJMLXKV-UHFFFAOYSA-N 1,9-Nonanediol Chemical compound OCCCCCCCCCO ALVZNPYWJMLXKV-UHFFFAOYSA-N 0.000 description 2
- CRSBERNSMYQZNG-UHFFFAOYSA-N 1-dodecene Chemical compound CCCCCCCCCCC=C CRSBERNSMYQZNG-UHFFFAOYSA-N 0.000 description 2
- LIKMAJRDDDTEIG-UHFFFAOYSA-N 1-hexene Chemical compound CCCCC=C LIKMAJRDDDTEIG-UHFFFAOYSA-N 0.000 description 2
- RNLHGQLZWXBQNY-UHFFFAOYSA-N 3-(aminomethyl)-3,5,5-trimethylcyclohexan-1-amine Chemical compound CC1(C)CC(N)CC(C)(CN)C1 RNLHGQLZWXBQNY-UHFFFAOYSA-N 0.000 description 2
- WECDUOXQLAIPQW-UHFFFAOYSA-N 4,4'-Methylene bis(2-methylaniline) Chemical compound C1=C(N)C(C)=CC(CC=2C=C(C)C(N)=CC=2)=C1 WECDUOXQLAIPQW-UHFFFAOYSA-N 0.000 description 2
- 229910002012 Aerosil® Inorganic materials 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229920000089 Cyclic olefin copolymer Polymers 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 239000004952 Polyamide Substances 0.000 description 2
- 239000004721 Polyphenylene oxide Substances 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 125000005263 alkylenediamine group Chemical group 0.000 description 2
- ADCOVFLJGNWWNZ-UHFFFAOYSA-N antimony trioxide Chemical compound O=[Sb]O[Sb]=O ADCOVFLJGNWWNZ-UHFFFAOYSA-N 0.000 description 2
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000006229 carbon black Substances 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000004203 carnauba wax Substances 0.000 description 2
- 235000013869 carnauba wax Nutrition 0.000 description 2
- 239000008119 colloidal silica Substances 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- FOTKYAAJKYLFFN-UHFFFAOYSA-N decane-1,10-diol Chemical compound OCCCCCCCCCCO FOTKYAAJKYLFFN-UHFFFAOYSA-N 0.000 description 2
- 150000004985 diamines Chemical class 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- GHLKSLMMWAKNBM-UHFFFAOYSA-N dodecane-1,12-diol Chemical compound OCCCCCCCCCCCCO GHLKSLMMWAKNBM-UHFFFAOYSA-N 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- NAQMVNRVTILPCV-UHFFFAOYSA-N hexane-1,6-diamine Chemical compound NCCCCCCN NAQMVNRVTILPCV-UHFFFAOYSA-N 0.000 description 2
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- SZVJSHCCFOBDDC-UHFFFAOYSA-N iron(II,III) oxide Inorganic materials O=[Fe]O[Fe]O[Fe]=O SZVJSHCCFOBDDC-UHFFFAOYSA-N 0.000 description 2
- NIMLQBUJDJZYEJ-UHFFFAOYSA-N isophorone diisocyanate Chemical compound CC1(C)CC(N=C=O)CC(C)(CN=C=O)C1 NIMLQBUJDJZYEJ-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000006247 magnetic powder Substances 0.000 description 2
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 2
- RNVCVTLRINQCPJ-UHFFFAOYSA-N o-toluidine Chemical compound CC1=CC=CC=C1N RNVCVTLRINQCPJ-UHFFFAOYSA-N 0.000 description 2
- CCCMONHAUSKTEQ-UHFFFAOYSA-N octadec-1-ene Chemical compound CCCCCCCCCCCCCCCCC=C CCCMONHAUSKTEQ-UHFFFAOYSA-N 0.000 description 2
- 125000005702 oxyalkylene group Chemical group 0.000 description 2
- 238000000614 phase inversion technique Methods 0.000 description 2
- LCPDWSOZIOUXRV-UHFFFAOYSA-N phenoxyacetic acid Chemical compound OC(=O)COC1=CC=CC=C1 LCPDWSOZIOUXRV-UHFFFAOYSA-N 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229920001281 polyalkylene Polymers 0.000 description 2
- 229920002647 polyamide Polymers 0.000 description 2
- 229920000570 polyether Polymers 0.000 description 2
- 229920000098 polyolefin Polymers 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 2
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- REZQBEBOWJAQKS-UHFFFAOYSA-N triacontan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCO REZQBEBOWJAQKS-UHFFFAOYSA-N 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- HOVAGTYPODGVJG-UVSYOFPXSA-N (3s,5r)-2-(hydroxymethyl)-6-methoxyoxane-3,4,5-triol Chemical compound COC1OC(CO)[C@@H](O)C(O)[C@H]1O HOVAGTYPODGVJG-UVSYOFPXSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- GFNDFCFPJQPVQL-UHFFFAOYSA-N 1,12-diisocyanatododecane Chemical compound O=C=NCCCCCCCCCCCCN=C=O GFNDFCFPJQPVQL-UHFFFAOYSA-N 0.000 description 1
- ZTNJGMFHJYGMDR-UHFFFAOYSA-N 1,2-diisocyanatoethane Chemical compound O=C=NCCN=C=O ZTNJGMFHJYGMDR-UHFFFAOYSA-N 0.000 description 1
- AZYRZNIYJDKRHO-UHFFFAOYSA-N 1,3-bis(2-isocyanatopropan-2-yl)benzene Chemical compound O=C=NC(C)(C)C1=CC=CC(C(C)(C)N=C=O)=C1 AZYRZNIYJDKRHO-UHFFFAOYSA-N 0.000 description 1
- RTTZISZSHSCFRH-UHFFFAOYSA-N 1,3-bis(isocyanatomethyl)benzene Chemical compound O=C=NCC1=CC=CC(CN=C=O)=C1 RTTZISZSHSCFRH-UHFFFAOYSA-N 0.000 description 1
- PCHXZXKMYCGVFA-UHFFFAOYSA-N 1,3-diazetidine-2,4-dione Chemical group O=C1NC(=O)N1 PCHXZXKMYCGVFA-UHFFFAOYSA-N 0.000 description 1
- WZCQRUWWHSTZEM-UHFFFAOYSA-N 1,3-phenylenediamine Chemical compound NC1=CC=CC(N)=C1 WZCQRUWWHSTZEM-UHFFFAOYSA-N 0.000 description 1
- ALQLPWJFHRMHIU-UHFFFAOYSA-N 1,4-diisocyanatobenzene Chemical compound O=C=NC1=CC=C(N=C=O)C=C1 ALQLPWJFHRMHIU-UHFFFAOYSA-N 0.000 description 1
- OVBFMUAFNIIQAL-UHFFFAOYSA-N 1,4-diisocyanatobutane Chemical compound O=C=NCCCCN=C=O OVBFMUAFNIIQAL-UHFFFAOYSA-N 0.000 description 1
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical compound NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 1
- SBJCUZQNHOLYMD-UHFFFAOYSA-N 1,5-Naphthalene diisocyanate Chemical compound C1=CC=C2C(N=C=O)=CC=CC2=C1N=C=O SBJCUZQNHOLYMD-UHFFFAOYSA-N 0.000 description 1
- OSNILPMOSNGHLC-UHFFFAOYSA-N 1-[4-methoxy-3-(piperidin-1-ylmethyl)phenyl]ethanone Chemical compound COC1=CC=C(C(C)=O)C=C1CN1CCCCC1 OSNILPMOSNGHLC-UHFFFAOYSA-N 0.000 description 1
- AEYNYHSOGNVQRY-UHFFFAOYSA-N 1-n,1-n-diethyl-4-methylbenzene-1,3-diamine Chemical compound CCN(CC)C1=CC=C(C)C(N)=C1 AEYNYHSOGNVQRY-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical compound NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 description 1
- OLYCWGBQORTQQX-UHFFFAOYSA-N 2,3-dimethylnaphthalene-1,4-diamine Chemical compound C1=CC=CC2=C(N)C(C)=C(C)C(N)=C21 OLYCWGBQORTQQX-UHFFFAOYSA-N 0.000 description 1
- XVBLEUZLLURXTF-UHFFFAOYSA-N 2,4-dimethylbenzene-1,3-diamine Chemical compound CC1=CC=C(N)C(C)=C1N XVBLEUZLLURXTF-UHFFFAOYSA-N 0.000 description 1
- QAYVHDDEMLNVMO-UHFFFAOYSA-N 2,5-dichlorobenzene-1,4-diamine Chemical compound NC1=CC(Cl)=C(N)C=C1Cl QAYVHDDEMLNVMO-UHFFFAOYSA-N 0.000 description 1
- RLYCRLGLCUXUPO-UHFFFAOYSA-N 2,6-diaminotoluene Chemical compound CC1=C(N)C=CC=C1N RLYCRLGLCUXUPO-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- SXERGJJQSKIUIC-UHFFFAOYSA-N 2-Phenoxypropionic acid Chemical compound OC(=O)C(C)OC1=CC=CC=C1 SXERGJJQSKIUIC-UHFFFAOYSA-N 0.000 description 1
- QDCPNGVVOWVKJG-VAWYXSNFSA-N 2-[(e)-dodec-1-enyl]butanedioic acid Chemical compound CCCCCCCCCC\C=C\C(C(O)=O)CC(O)=O QDCPNGVVOWVKJG-VAWYXSNFSA-N 0.000 description 1
- TXBCBTDQIULDIA-UHFFFAOYSA-N 2-[[3-hydroxy-2,2-bis(hydroxymethyl)propoxy]methyl]-2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(CO)(CO)COCC(CO)(CO)CO TXBCBTDQIULDIA-UHFFFAOYSA-N 0.000 description 1
- FGTYTUFKXYPTML-UHFFFAOYSA-N 2-benzoylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1C(=O)C1=CC=CC=C1 FGTYTUFKXYPTML-UHFFFAOYSA-N 0.000 description 1
- MGLZGLAFFOMWPB-UHFFFAOYSA-N 2-chloro-1,4-phenylenediamine Chemical compound NC1=CC=C(N)C(Cl)=C1 MGLZGLAFFOMWPB-UHFFFAOYSA-N 0.000 description 1
- FZZMTSNZRBFGGU-UHFFFAOYSA-N 2-chloro-7-fluoroquinazolin-4-amine Chemical compound FC1=CC=C2C(N)=NC(Cl)=NC2=C1 FZZMTSNZRBFGGU-UHFFFAOYSA-N 0.000 description 1
- WBJWXIQDBDZMAW-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carbonyl chloride Chemical compound C1=CC=CC2=C(C(Cl)=O)C(O)=CC=C21 WBJWXIQDBDZMAW-UHFFFAOYSA-N 0.000 description 1
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 1
- WDGCBNTXZHJTHJ-UHFFFAOYSA-N 2h-1,3-oxazol-2-id-4-one Chemical group O=C1CO[C-]=N1 WDGCBNTXZHJTHJ-UHFFFAOYSA-N 0.000 description 1
- JRBJSXQPQWSCCF-UHFFFAOYSA-N 3,3'-Dimethoxybenzidine Chemical compound C1=C(N)C(OC)=CC(C=2C=C(OC)C(N)=CC=2)=C1 JRBJSXQPQWSCCF-UHFFFAOYSA-N 0.000 description 1
- ZDBWYUOUYNQZBM-UHFFFAOYSA-N 3-(aminomethyl)aniline Chemical compound NCC1=CC=CC(N)=C1 ZDBWYUOUYNQZBM-UHFFFAOYSA-N 0.000 description 1
- ANOPCGQVRXJHHD-UHFFFAOYSA-N 3-[3-(3-aminopropyl)-2,4,8,10-tetraoxaspiro[5.5]undecan-9-yl]propan-1-amine Chemical compound C1OC(CCCN)OCC21COC(CCCN)OC2 ANOPCGQVRXJHHD-UHFFFAOYSA-N 0.000 description 1
- XYUINKARGUCCQJ-UHFFFAOYSA-N 3-imino-n-propylpropan-1-amine Chemical compound CCCNCCC=N XYUINKARGUCCQJ-UHFFFAOYSA-N 0.000 description 1
- RGBBCHBCGNDCRL-UHFFFAOYSA-N 3-n,4-dimethylbenzene-1,3-diamine Chemical compound CNC1=CC(N)=CC=C1C RGBBCHBCGNDCRL-UHFFFAOYSA-N 0.000 description 1
- YBRVSVVVWCFQMG-UHFFFAOYSA-N 4,4'-diaminodiphenylmethane Chemical compound C1=CC(N)=CC=C1CC1=CC=C(N)C=C1 YBRVSVVVWCFQMG-UHFFFAOYSA-N 0.000 description 1
- ZDTYWWRZDUKNNY-UHFFFAOYSA-N 4-(1-aminoethyl)piperazin-1-amine Chemical compound CC(N)N1CCN(N)CC1 ZDTYWWRZDUKNNY-UHFFFAOYSA-N 0.000 description 1
- ZWUBBMDHSZDNTA-UHFFFAOYSA-N 4-Chloro-meta-phenylenediamine Chemical compound NC1=CC=C(Cl)C(N)=C1 ZWUBBMDHSZDNTA-UHFFFAOYSA-N 0.000 description 1
- HQDCQNCMUSAKQU-UHFFFAOYSA-N 4-bromobenzene-1,3-diamine Chemical compound NC1=CC=C(Br)C(N)=C1 HQDCQNCMUSAKQU-UHFFFAOYSA-N 0.000 description 1
- TYMLOMAKGOJONV-UHFFFAOYSA-N 4-nitroaniline Chemical compound NC1=CC=C([N+]([O-])=O)C=C1 TYMLOMAKGOJONV-UHFFFAOYSA-N 0.000 description 1
- NNJMFJSKMRYHSR-UHFFFAOYSA-N 4-phenylbenzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=CC=C1 NNJMFJSKMRYHSR-UHFFFAOYSA-N 0.000 description 1
- DFWXYHZQNLIBLY-UHFFFAOYSA-N 5-nitrobenzene-1,3-diamine Chemical compound NC1=CC(N)=CC([N+]([O-])=O)=C1 DFWXYHZQNLIBLY-UHFFFAOYSA-N 0.000 description 1
- 229920000178 Acrylic resin Polymers 0.000 description 1
- 239000004925 Acrylic resin Substances 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- RLSFZHQORSPXDW-UHFFFAOYSA-J C(C1=CC=C(C(=O)[O-])C=C1)(=O)OOC(C)C.C(C1=CC=C(C(=O)[O-])C=C1)(=O)OOC(C)C.[Ti+4].C(C)(C)OOC(C1=CC=C(C(=O)[O-])C=C1)=O.C(C)(C)OOC(C1=CC=C(C(=O)[O-])C=C1)=O Chemical compound C(C1=CC=C(C(=O)[O-])C=C1)(=O)OOC(C)C.C(C1=CC=C(C(=O)[O-])C=C1)(=O)OOC(C)C.[Ti+4].C(C)(C)OOC(C1=CC=C(C(=O)[O-])C=C1)=O.C(C)(C)OOC(C1=CC=C(C(=O)[O-])C=C1)=O RLSFZHQORSPXDW-UHFFFAOYSA-J 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- PMVSDNDAUGGCCE-TYYBGVCCSA-L Ferrous fumarate Chemical compound [Fe+2].[O-]C(=O)\C=C\C([O-])=O PMVSDNDAUGGCCE-TYYBGVCCSA-L 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- 239000005058 Isophorone diisocyanate Substances 0.000 description 1
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- GKXVJHDEWHKBFH-UHFFFAOYSA-N [2-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=CC=C1CN GKXVJHDEWHKBFH-UHFFFAOYSA-N 0.000 description 1
- YIMQCDZDWXUDCA-UHFFFAOYSA-N [4-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1CCC(CO)CC1 YIMQCDZDWXUDCA-UHFFFAOYSA-N 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- ZOIORXHNWRGPMV-UHFFFAOYSA-N acetic acid;zinc Chemical compound [Zn].CC(O)=O.CC(O)=O ZOIORXHNWRGPMV-UHFFFAOYSA-N 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000007824 aliphatic compounds Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000005250 alkyl acrylate group Chemical group 0.000 description 1
- PYHXGXCGESYPCW-UHFFFAOYSA-N alpha-phenylbenzeneacetic acid Natural products C=1C=CC=CC=1C(C(=O)O)C1=CC=CC=C1 PYHXGXCGESYPCW-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- LHIJANUOQQMGNT-UHFFFAOYSA-N aminoethylethanolamine Chemical compound NCCNCCO LHIJANUOQQMGNT-UHFFFAOYSA-N 0.000 description 1
- IMUDHTPIFIBORV-UHFFFAOYSA-N aminoethylpiperazine Chemical compound NCCN1CCNCC1 IMUDHTPIFIBORV-UHFFFAOYSA-N 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 239000000987 azo dye Substances 0.000 description 1
- WXLFIFHRGFOVCD-UHFFFAOYSA-L azophloxine Chemical compound [Na+].[Na+].OC1=C2C(NC(=O)C)=CC(S([O-])(=O)=O)=CC2=CC(S([O-])(=O)=O)=C1N=NC1=CC=CC=C1 WXLFIFHRGFOVCD-UHFFFAOYSA-L 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- HFACYLZERDEVSX-UHFFFAOYSA-N benzidine Chemical compound C1=CC(N)=CC=C1C1=CC=C(N)C=C1 HFACYLZERDEVSX-UHFFFAOYSA-N 0.000 description 1
- UKXSKSHDVLQNKG-UHFFFAOYSA-N benzilic acid Chemical compound C=1C=CC=CC=1C(O)(C(=O)O)C1=CC=CC=C1 UKXSKSHDVLQNKG-UHFFFAOYSA-N 0.000 description 1
- 150000001559 benzoic acids Chemical class 0.000 description 1
- VBIAXKVXACZQFW-OWOJBTEDSA-N bis(2-isocyanatoethyl) (e)-but-2-enedioate Chemical compound O=C=NCCOC(=O)\C=C\C(=O)OCCN=C=O VBIAXKVXACZQFW-OWOJBTEDSA-N 0.000 description 1
- MRNZSTMRDWRNNR-UHFFFAOYSA-N bis(hexamethylene)triamine Chemical compound NCCCCCCNCCCCCCN MRNZSTMRDWRNNR-UHFFFAOYSA-N 0.000 description 1
- 150000001638 boron Chemical class 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- UTOVMEACOLCUCK-PLNGDYQASA-N butyl maleate Chemical compound CCCCOC(=O)\C=C/C(O)=O UTOVMEACOLCUCK-PLNGDYQASA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- VPKDCDLSJZCGKE-UHFFFAOYSA-N carbodiimide group Chemical group N=C=N VPKDCDLSJZCGKE-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- ZLFVRXUOSPRRKQ-UHFFFAOYSA-N chembl2138372 Chemical compound [O-][N+](=O)C1=CC(C)=CC=C1N=NC1=C(O)C=CC2=CC=CC=C12 ZLFVRXUOSPRRKQ-UHFFFAOYSA-N 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- HNEGQIOMVPPMNR-IHWYPQMZSA-N citraconic acid Chemical compound OC(=O)C(/C)=C\C(O)=O HNEGQIOMVPPMNR-IHWYPQMZSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000008094 contradictory effect Effects 0.000 description 1
- XCJYREBRNVKWGJ-UHFFFAOYSA-N copper(II) phthalocyanine Chemical compound [Cu+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 XCJYREBRNVKWGJ-UHFFFAOYSA-N 0.000 description 1
- 229930003836 cresol Natural products 0.000 description 1
- ZXJXZNDDNMQXFV-UHFFFAOYSA-M crystal violet Chemical compound [Cl-].C1=CC(N(C)C)=CC=C1[C+](C=1C=CC(=CC=1)N(C)C)C1=CC=C(N(C)C)C=C1 ZXJXZNDDNMQXFV-UHFFFAOYSA-M 0.000 description 1
- GEQHKFFSPGPGLN-UHFFFAOYSA-N cyclohexane-1,3-diamine Chemical compound NC1CCCC(N)C1 GEQHKFFSPGPGLN-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- JGFBRKRYDCGYKD-UHFFFAOYSA-N dibutyl(oxo)tin Chemical compound CCCC[Sn](=O)CCCC JGFBRKRYDCGYKD-UHFFFAOYSA-N 0.000 description 1
- 125000005442 diisocyanate group Chemical group 0.000 description 1
- LDCRTTXIJACKKU-ARJAWSKDSA-N dimethyl maleate Chemical compound COC(=O)\C=C/C(=O)OC LDCRTTXIJACKKU-ARJAWSKDSA-N 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- ZZTCPWRAHWXWCH-UHFFFAOYSA-N diphenylmethanediamine Chemical compound C=1C=CC=CC=1C(N)(N)C1=CC=CC=C1 ZZTCPWRAHWXWCH-UHFFFAOYSA-N 0.000 description 1
- IRXRGVFLQOSHOH-UHFFFAOYSA-L dipotassium;oxalate Chemical compound [K+].[K+].[O-]C(=O)C([O-])=O IRXRGVFLQOSHOH-UHFFFAOYSA-L 0.000 description 1
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229940069096 dodecene Drugs 0.000 description 1
- 125000006575 electron-withdrawing group Chemical class 0.000 description 1
- 239000003822 epoxy resin Substances 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 230000005294 ferromagnetic effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- NKHAVTQWNUWKEO-UHFFFAOYSA-N fumaric acid monomethyl ester Natural products COC(=O)C=CC(O)=O NKHAVTQWNUWKEO-UHFFFAOYSA-N 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- RBTKNAXYKSUFRK-UHFFFAOYSA-N heliogen blue Chemical compound [Cu].[N-]1C2=C(C=CC=C3)C3=C1N=C([N-]1)C3=CC=CC=C3C1=NC([N-]1)=C(C=CC=C3)C3=C1N=C([N-]1)C3=CC=CC=C3C1=N2 RBTKNAXYKSUFRK-UHFFFAOYSA-N 0.000 description 1
- 229910052595 hematite Inorganic materials 0.000 description 1
- 239000011019 hematite Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- SEIUOYFQDIJJEO-UHFFFAOYSA-N hexane-1,1,1-tricarboxylic acid Chemical compound CCCCCC(C(O)=O)(C(O)=O)C(O)=O SEIUOYFQDIJJEO-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- UCNNJGDEJXIUCC-UHFFFAOYSA-L hydroxy(oxo)iron;iron Chemical compound [Fe].O[Fe]=O.O[Fe]=O UCNNJGDEJXIUCC-UHFFFAOYSA-L 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229940079865 intestinal antiinfectives imidazole derivative Drugs 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- LIKBJVNGSGBSGK-UHFFFAOYSA-N iron(3+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Fe+3].[Fe+3] LIKBJVNGSGBSGK-UHFFFAOYSA-N 0.000 description 1
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical group OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 235000020778 linoleic acid Nutrition 0.000 description 1
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- HNEGQIOMVPPMNR-NSCUHMNNSA-N mesaconic acid Chemical compound OC(=O)C(/C)=C/C(O)=O HNEGQIOMVPPMNR-NSCUHMNNSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- AYLRODJJLADBOB-QMMMGPOBSA-N methyl (2s)-2,6-diisocyanatohexanoate Chemical compound COC(=O)[C@@H](N=C=O)CCCCN=C=O AYLRODJJLADBOB-QMMMGPOBSA-N 0.000 description 1
- HOVAGTYPODGVJG-UHFFFAOYSA-N methyl beta-galactoside Natural products COC1OC(CO)C(O)C(O)C1O HOVAGTYPODGVJG-UHFFFAOYSA-N 0.000 description 1
- NKHAVTQWNUWKEO-IHWYPQMZSA-N methyl hydrogen fumarate Chemical compound COC(=O)\C=C/C(O)=O NKHAVTQWNUWKEO-IHWYPQMZSA-N 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- HNEGQIOMVPPMNR-UHFFFAOYSA-N methylfumaric acid Natural products OC(=O)C(C)=CC(O)=O HNEGQIOMVPPMNR-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000006082 mold release agent Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 239000012170 montan wax Substances 0.000 description 1
- KMBPCQSCMCEPMU-UHFFFAOYSA-N n'-(3-aminopropyl)-n'-methylpropane-1,3-diamine Chemical compound NCCCN(C)CCCN KMBPCQSCMCEPMU-UHFFFAOYSA-N 0.000 description 1
- LSHROXHEILXKHM-UHFFFAOYSA-N n'-[2-[2-[2-(2-aminoethylamino)ethylamino]ethylamino]ethyl]ethane-1,2-diamine Chemical compound NCCNCCNCCNCCNCCN LSHROXHEILXKHM-UHFFFAOYSA-N 0.000 description 1
- ZETYUTMSJWMKNQ-UHFFFAOYSA-N n,n',n'-trimethylhexane-1,6-diamine Chemical compound CNCCCCCCN(C)C ZETYUTMSJWMKNQ-UHFFFAOYSA-N 0.000 description 1
- YYHPPOGFPXBRRX-UHFFFAOYSA-N n,n-dichloro-1-[4-[(dichloroamino)methyl]phenyl]methanamine Chemical compound ClN(Cl)CC1=CC=C(CN(Cl)Cl)C=C1 YYHPPOGFPXBRRX-UHFFFAOYSA-N 0.000 description 1
- YDLYQMBWCWFRAI-UHFFFAOYSA-N n-Hexatriacontane Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC YDLYQMBWCWFRAI-UHFFFAOYSA-N 0.000 description 1
- JXTPJDDICSTXJX-UHFFFAOYSA-N n-Triacontane Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC JXTPJDDICSTXJX-UHFFFAOYSA-N 0.000 description 1
- OLAKSHDLGIUUET-UHFFFAOYSA-N n-anilinosulfanylaniline Chemical compound C=1C=CC=CC=1NSNC1=CC=CC=C1 OLAKSHDLGIUUET-UHFFFAOYSA-N 0.000 description 1
- SHDMMLFAFLZUEV-UHFFFAOYSA-N n-methyl-1,1-diphenylmethanamine Chemical compound C=1C=CC=CC=1C(NC)C1=CC=CC=C1 SHDMMLFAFLZUEV-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KYTZHLUVELPASH-UHFFFAOYSA-N naphthalene-1,2-dicarboxylic acid Chemical compound C1=CC=CC2=C(C(O)=O)C(C(=O)O)=CC=C21 KYTZHLUVELPASH-UHFFFAOYSA-N 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- ZWLPBLYKEWSWPD-UHFFFAOYSA-N o-toluic acid Chemical compound CC1=CC=CC=C1C(O)=O ZWLPBLYKEWSWPD-UHFFFAOYSA-N 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- LYTNHSCLZRMKON-UHFFFAOYSA-L oxygen(2-);zirconium(4+);diacetate Chemical compound [O-2].[Zr+4].CC([O-])=O.CC([O-])=O LYTNHSCLZRMKON-UHFFFAOYSA-L 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 1
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical compound N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 1
- 239000001007 phthalocyanine dye Substances 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920000647 polyepoxide Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920006267 polyester film Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920005672 polyolefin resin Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920000909 polytetrahydrofuran Polymers 0.000 description 1
- 229920005749 polyurethane resin Polymers 0.000 description 1
- AOHJOMMDDJHIJH-UHFFFAOYSA-N propylenediamine Chemical compound CC(N)CN AOHJOMMDDJHIJH-UHFFFAOYSA-N 0.000 description 1
- VHNQIURBCCNWDN-UHFFFAOYSA-N pyridine-2,6-diamine Chemical compound NC1=CC=CC(N)=N1 VHNQIURBCCNWDN-UHFFFAOYSA-N 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 235000012739 red 2G Nutrition 0.000 description 1
- 239000004180 red 2G Substances 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229940043267 rhodamine b Drugs 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 229920002050 silicone resin Polymers 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- KKEYFWRCBNTPAC-UHFFFAOYSA-L terephthalate(2-) Chemical compound [O-]C(=O)C1=CC=C(C([O-])=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-L 0.000 description 1
- CXJNRRJXWSODHK-UHFFFAOYSA-J terephthalate;titanium(4+) Chemical compound [Ti+4].[O-]C(=O)C1=CC=C(C([O-])=O)C=C1.[O-]C(=O)C1=CC=C(C([O-])=O)C=C1 CXJNRRJXWSODHK-UHFFFAOYSA-J 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- FAGUFWYHJQFNRV-UHFFFAOYSA-N tetraethylenepentamine Chemical compound NCCNCCNCCNCCN FAGUFWYHJQFNRV-UHFFFAOYSA-N 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- WJJKZTNEIIXZDN-UHFFFAOYSA-J titanium(4+);2,3,5-tributoxyterephthalate Chemical compound [Ti+4].CCCCOC1=CC(C([O-])=O)=C(OCCCC)C(OCCCC)=C1C([O-])=O.CCCCOC1=CC(C([O-])=O)=C(OCCCC)C(OCCCC)=C1C([O-])=O WJJKZTNEIIXZDN-UHFFFAOYSA-J 0.000 description 1
- RUELTTOHQODFPA-UHFFFAOYSA-N toluene 2,6-diisocyanate Chemical compound CC1=C(N=C=O)C=CC=C1N=C=O RUELTTOHQODFPA-UHFFFAOYSA-N 0.000 description 1
- OLTHARGIAFTREU-UHFFFAOYSA-N triacontane Natural products CCCCCCCCCCCCCCCCCCCCC(C)CCCCCCCC OLTHARGIAFTREU-UHFFFAOYSA-N 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- QXJQHYBHAIHNGG-UHFFFAOYSA-N trimethylolethane Chemical compound OCC(C)(CO)CO QXJQHYBHAIHNGG-UHFFFAOYSA-N 0.000 description 1
- AAAQKTZKLRYKHR-UHFFFAOYSA-N triphenylmethane Chemical compound C1=CC=CC=C1C(C=1C=CC=CC=1)C1=CC=CC=C1 AAAQKTZKLRYKHR-UHFFFAOYSA-N 0.000 description 1
- AVWRKZWQTYIKIY-UHFFFAOYSA-N urea-1-carboxylic acid Chemical group NC(=O)NC(O)=O AVWRKZWQTYIKIY-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 239000004246 zinc acetate Substances 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Developing Agents For Electrophotography (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description
すなわち本発明は、下記2発明である。
(I) カルボン酸成分(x)とポリオール成分(y)とを構成単位として有し、カルボン酸成分(x)が1,2,4−ベンゼントリカルボン酸(x1)と、芳香族ジカルボン酸およびそのエステル形成性誘導体から選ばれる2種以上のジカルボン酸(x2)を合計で80〜99.5モル%含有し、ポリオール成分(y)が、炭素数が2〜10の脂肪族ジオール(y1)を80モル%以上含有する、酸価が2(mgKOH/g)以下のポリエステル樹脂(A)と、ウレタン(ウレア)変性ポリエステル樹脂(B)とで構成されるポリエステル樹脂組成物(P)を含有し、(A)、(B)、および(P)が次の式(1)〜(3)を満たす電子写真用トナーバインダー。
{(P)の〔G’180〕}/{(A)の〔G’180〕と(B)の〔G’180〕の加重平均}≧1.5 ・・・式(1)
(A)の〔G’150〕≦10000 ・・・式(2)
(B)の〔G’150〕≦10000 ・・・式(3)
[上記式中、〔G’180〕は180℃における貯蔵弾性率(dyn/cm2)、〔G’
150〕は150℃における貯蔵弾性率(dyn/cm2)を意味する。]
(II) 上記の電子写真用トナーバインダーと着色剤、並びに必要により、離型剤、荷電制御剤、および流動化剤から選ばれる1種類以上の添加剤を含有するトナー組成物。
本発明の電子写真用トナーバインダーは、カルボン酸成分(x)とポリオール成分(y)とを構成単位として有するポリエステル樹脂(A)と、ウレタン(ウレア)変性ポリエステル樹脂(B)とで構成されるポリエステル樹脂組成物(P)を含有する。
上記「ウレタン(ウレア)変性ポリエステル樹脂」とは、ウレア変性されていてもよいウレタン変性ポリエステル樹脂を意味する。
本発明におけるポリエステル樹脂(A)は、カルボン酸成分(x)中に、定着性と粉砕性および耐久性の観点から、1,2,4−ベンゼントリカルボン酸(x1)を必須構成単位として含有する。(x)中の(x1)量は、同様の観点から0.5〜20モル%が好ましく、より好ましくは0.8〜17モル%、さらに好ましくは1〜15モル%である。
(A)は、低温定着性と耐ホットオフセット性を両立させる(定着温度幅の拡大)観点から、カルボン酸成分(x)が、さらに芳香族ジカルボン酸およびそのエステル形成性誘導体から選ばれる2種以上のジカルボン酸(x2)を合計で80〜99.5モル%含有し、ポリオール成分(y)が炭素数が2〜10の脂肪族ジオール(y1)を80モル%以上含有するのが好ましい。
上記エステル形成性誘導体としては、酸無水物、アルキル(炭素数1〜24:メチル、エチル、ブチル、ステアリル等、好ましくは炭素数1〜4)エステル、および部分アルキル(上記と同様)エステル等が挙げられる。以下のエステル形成性誘導体についても同様である。
なお、本発明においては、芳香族ジカルボン酸およびそのエステル形成性誘導体から選ばれる2種以上のジカルボン酸(x2)において、芳香族ジカルボン酸とその同一ジカルボン酸のエステル形成性誘導体とは、1種として数える。
これら(x2)のうち、低温定着性と耐ホットオフセット性の両立の観点から好ましくは、以下に挙げた(1)〜(3)から選ばれる2種以上である。
(1)テレフタル酸および/またはそのエステル形成性誘導体
(2)イソフタル酸および/またはそのエステル形成性誘導体
(3)フタル酸および/またはそのエステル形成性誘導体
好ましい組合せとしては(1)と(2)、および(1)と(3)であり、さらに好ましくは、(1)と(2)の重量比が(1)/(2)=4/6〜8/2であり、(1)と(3)の重量比が(1)/(3)=4/6〜8/2である。
ポリエステル樹脂(A)のカルボン酸成分(x)中のジカルボン酸(x2)の量は、好ましくは80〜99.5モル%であり、さらに好ましくは83〜99.2モル%、とくに好ましくは85〜99モル%である。
カルボン酸成分(x)のうち、(x2)以外のジカルボン酸としては、炭素数4〜36のアルカンジカルボン酸(例えばコハク、アジピン、およびセバシン酸);炭素数6〜40の脂環式ジカルボン酸〔例えばダイマー酸(2量化リノール酸)〕;炭素数4〜36のアルケンジカルボン酸(例えば、ドデセニルコハク酸等のアルケニルコハク酸、マレイン、フマル、シトラコン、およびメサコン酸)およびこれらのエステル形成性誘導体;等が挙げられる。
これらのうち好ましいものは、炭素数4〜20のアルカンジカルボン酸;炭素数4〜36のアルケンジカルボン酸、およびこれらのエステル形成性誘導体であり、さらに好ましくは、コハク酸、アジピン酸、マレイン酸、フマル酸、および/またはそれらのエステル形成性誘導体である。
これらのうち好ましいものは、ピロメリット酸、およびそのエステル形成性誘導体である。
(x)中の(x1)以外の3価以上のポリカルボン酸の量は、10モル%以下が好ましく、さらに好ましくは7モル%以下、とくに好ましくは5モル%以下である。
また、(x)中の芳香族モノカルボン酸の量は、10モル%以下が好ましく、さらに好ましくは9モル%以下、とくに好ましくは8モル%以下である。
これらのうち、1,2,4−ベンゼントリカルボン酸(x1)との反応性の観点から、分子末端に1級水酸基を有する脂肪族ジオール(エチレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、ネオペンチルグリコール、1,6−ヘキサンジオール、1,9−ノナンジオールおよび1,10−デカンジオール等)が好ましく、エチレングリコール、1,3−プロピレングリコールおよびネオペンチルグリコールがさらに好ましく、エチレングリコール、およびエチレングリコールとネオペンチルグリコールの併用(重量比が、好ましくは100/0〜50/50、さらに好ましくは99/1〜60/40)がとくに好ましい。
ポリオール成分(y)のうち、(y1)以外のジオールとしては、炭素数11〜36のアルキレングリコール(1,12−ドデカンジオール等);炭素数11〜36のアルキレンエーテルグリコール(ポリエチレングリコール、ポリプロピレングリコール、およびポリテトラメチレンエーテルグリコール等);炭素数6〜36の脂環式ジオール(1,4−シクロヘキサンジメタノール、および水素添加ビスフェノールA等);上記脂環式ジオールの(ポリ)オキシアルキレン〔アルキレン基の炭素数2〜4(オキシエチレン、オキシプロピレン等)。以下のポリオキシアルキレン基も同じ〕エーテル〔オキシアルキレン単位(以下AO単位と略記)の数1〜30〕;および2価フェノール〔単環2価フェノール(例えばハイドロキノン)、およびビスフェノール類(ビスフェノールA、ビスフェノールFおよびビスフェノールS等)〕のポリオキシアルキレンエーテル(AO単位の数2〜30);等が挙げられる。
これらのうち好ましいものは、ビスフェノール類のポリオキシアルキレンエーテル(AO単位の数2〜30)である。
これらのうち好ましいものは、3〜8価もしくはそれ以上の脂肪族多価アルコール、およびノボラック樹脂のポリオキシアルキレンエーテル(AO単位の数2〜30)であり、さらに好ましいものはノボラック樹脂のポリオキシアルキレンエーテル(AO単位の数2〜30)である。
ポリオール成分(y)とカルボン酸成分(x)との反応比率は、水酸基とカルボキシル基の当量比[OH]/[COOH]として、好ましくは2/1〜1.01/1、さらに好ましくは1.5/1〜1.02/1、とくに好ましくは1.3/1〜1.03/1である。
このとき必要に応じてエステル化触媒を使用することができる。エステル化触媒の例には、スズ含有触媒(例えばジブチルスズオキシド)、三酸化アンチモン、チタン含有触媒[例えばチタンアルコキシド、シュウ酸チタン酸カリウム、テレフタル酸チタン、特開2006−243715号公報に記載の触媒〔チタニウムジヒドロキシビス(トリエタノールアミネート)、チタニウムモノヒドロキシトリス(トリエタノールアミネート)、およびそれらの分子内重縮合物等〕、および特開2007−11307号公報に記載の触媒(チタントリブトキシテレフタレート、チタントリイソプロポキシテレフタレート、およびチタンジイソプロポキシジテレフタレート等)]、ジルコニウム含有触媒(例えば酢酸ジルコニル)、および酢酸亜鉛等が挙げられる。これらの中で好ましくはチタン含有触媒である。
また、(A)の水酸基価〔OHV〕(mgKOH/g、以下同じ。)は、好ましくは0〜100、さらに好ましくは0〜80、とくに好ましくは0〜70である。水酸基価が100以下であるとトナー化時の耐ホットオフセット性がより良好となる。
なお、試料中に架橋にともなう溶剤不溶解分がある場合は、以下の方法で溶融混練後のものを試料として用いる。
混練装置 : 東洋精機(株)製ラボプラストミルMODEL4M150
混練条件 : 130℃、70rpmにて30分
装置(一例) : 東ソー(株)製 HLC−8120
カラム(一例): TSK GEL GMH6 2本 〔東ソー(株)製〕
測定温度 : 40℃
試料溶液 : 0.25重量%のTHF(テトラヒドロフラン)溶液
溶液注入量 : 100μl
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000)
得られたクロマトグラム上で最大のピーク高さを示す分子量をピークトップ分子量(Mp)と称する。また、分子量の測定は、ポリエステル樹脂をTHFに溶解し、不溶解分をグラスフィルターでろ別したものを試料溶液とする。
なお、上記および以下において、Tgはセイコー電子工業(株)製DSC20、SSC/580を用いて、ASTM D3418−82に規定の方法(DSC法)で測定される。
この範囲であると、耐ホットオフセット性と低温定着性の両立が良好となる。本発明において、T1/2は以下の方法で測定される。
<軟化点〔T1/2〕>
高化式フローテスター{たとえば、(株)島津製作所製、CFT−500D}を用いて、1gの測定試料を昇温速度6℃/分で加熱しながら、プランジャーにより1.96MPaの荷重を与え、直径1mm、長さ1mmのノズルから押し出して、「プランジャー降下量(流れ値)」と「温度」とのグラフを描き、プランジャーの降下量の最大値の1/2に対応する温度をグラフから読み取り、この値(測定試料の半分が流出したときの温度)を軟化点〔T1/2〕とする。
本発明におけるTHF不溶解分は、以下の方法で求めたものである。
試料0.5gに50mlのTHFを加え、3時間撹拌還流させる。冷却後、グラスフィルターにて不溶解分をろ別し、グラスフィルター上の樹脂分を80℃で3時間減圧乾燥する。グラスフィルター上の乾燥した樹脂分の重量と試料の重量比から、不溶解分を算出する。
〔1〕脂肪族ジアミン{C2〜C6アルキレンジアミン(エチレンジアミン、プロピレンジアミン、トリメチレンジアミン、テトラメチレンジアミン、ヘキサメチレンジアミンなど)、ポリアルキレン(C2〜C6)ジアミン〔ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン,トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミンなど〕};
〔2〕これらのアルキル(C1〜C4)またはヒドロキシアルキル(C2〜C4)置換体〔ジアルキル(C1〜C3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン、メチルイミノビスプロピルアミンなど〕;
〔3〕脂環または複素環含有脂肪族ジアミン{脂環式ジアミン(C4〜C15)〔1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)など〕、複素環式ジアミン(C4〜C15)〔ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなど〕;
〔4〕芳香環含有脂肪族アミン類(C8〜C15)(キシリレンジアミン、テトラクロル−p−キシリレンジアミンなど);等が挙げられる。
〔1〕非置換芳香族ジアミン〔1,2−、1,3−および1,4−フェニレンジアミン、2,4´−および4,4´−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4´,4′′−トリアミン、ナフチレンジアミンなど;
〔2〕核置換アルキル基〔メチル,エチル,n−およびi−プロピル、ブチルなどのC1〜C4アルキル基〕を有する芳香族ジアミン、たとえば2,4−および2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、4,4´−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタンなど〕、およびこれらの異性体の種々の割合の混合物;
〔3〕核置換電子吸引基(Cl,Br,I,Fなどのハロゲン;メトキシ、エトキシなどのアルコキシ基;ニトロ基など)を有する芳香族ジアミン〔メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロロ−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロロ−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン、3−ジメトキシ−4−アミノアニリンなど〕;
〔4〕2級アミノ基を有する芳香族ジアミン〔上記〔1〕〜〔3〕の芳香族ジアミンの−NH2の一部または全部が−NH−R´(R´はアルキル基たとえばメチル,エチルなどの低級アルキル基)で置き換ったもの〕〔4,4´−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼンなど〕が挙げられる。
上記モル比率は、ウレタン(ウレア)変性ポリエステル樹脂(B)を製造する際に使用した、ポリイソシアネート(i)と、ポリアミン(j)および(i)と反応する水の重量から、(B)中に含有されるウレタン基(―NHCOO―)のモル数とウレア基(―NHCONH―)のモル数の比を、計算により求めたものである。
製造法〔1〕;カルボン酸成分(x)とポリオール成分(y)とを重縮合させて得られる、水酸基を有するポリエステル樹脂(a)の有機溶剤(S)溶液を、ポリイソシアネート(i)と反応させてウレタン変性ポリエステル樹脂(B1)を製造する、または、さらに上記の未反応のイソシアネート基を有する反応生成物をポリアミン(j)と反応させてウレタンウレア変性ポリエステル樹脂(B2)を製造する方法。
製造法〔2〕;カルボン酸成分(x)とポリオール成分(y)とを重縮合させて得られる、水酸基を有するポリエステル樹脂(a)を、液体状態で、ポリイソシアネート(i)と反応させてウレタン変性ポリエステル樹脂(B1)を製造する、または、さらに上記の未反応のイソシアネート基を有する反応生成物をポリアミン(j)と反応させてウレタンウレア変性ポリエステル樹脂(B2)を製造する方法。
製造法〔3〕;ポリイソシアネート(i)とポリアミン(j)を、[(i)中のイソシアネート基]/[(j)中のアミノ基]=1.5/1〜3/1の当量比で反応させ、次いで未反応のイソシアネート基を有する反応生成物とポリオール成分(y)とを反応させて得られる変性ポリオール(y*)を含むポリオール成分(y)と、カルボン酸成分(x)とを重縮合させて、ウレタンウレア変性ポリエステル樹脂(B2)を製造する方法。
ポリオール成分(y)、カルボン酸成分(x)は前記の成分を特に限定なく用いることができる。必要により、前記のエステル化触媒を用いてもよい。
ポリエステル樹脂(a)の水酸基とポリイソシアネート(i)のイソシアネート基の当量比、および、(a)と(i)の反応生成物中の未反応イソシアネート基とポリアミン(j)のアミノ基の当量比は、製造法〔1〕と同様でよい。反応温度はアロファネート化及びビューレット化の開裂の観点から150〜250℃で反応させることが好ましく、より好ましくは170〜230℃、最も好ましくは180〜220℃である。
(B2)を製造する場合、ポリエステル樹脂(a)とポリイソシアネート(i)の反応が完了した後、反応生成物の未反応イソシアネート基とポリアミン(j)のアミノ基とを反応させることが好ましい。(a)と(i)の反応時間は1時間以下が好ましく、より好ましくは30分以下、最も好ましくは20分以下である。(a)と(i)との反応生成物と(j)との反応時間は30分以下が好ましく、より好ましくは20分以下、最も好ましくは15分以下である。
反応の際、ウレタン基とウレア基の導入率、及び貯蔵弾性率の観点から、ポリイソシアネート(i)中のイソシアネート基とポリアミン(j)中のアミノ基の当量比[NCO]/[NH2]が、1.5/1〜3/1であることが好ましく、より好ましくは1.7/1〜2.8/1、さらに好ましくは1.8/1〜2.5/1である。
なお、(y)を過剰に用いると、変性ポリオール(y*)と(y*)以外のポリオールを含むポリオール成分(y)が得られる。
ポリオール成分(y)中の変性ポリオール(y*)の含有量は、好ましくは0.5モル%以上、さらに好ましくは1〜80モル%である。
この範囲であると、耐ホットオフセット性と低温定着性の両立が良好となる。
[〔T1/2〕−〔Tf〕]は、より好ましくは46〜63℃、とくに好ましくは47〜60℃である。
[〔T1/2〕−〔Tf〕]の値を大きくする場合、架橋点の数を増やす、分子量分布を広くする、またはウレタン基濃度、ウレア基濃度を上げる等で達成できる。
<流出開始温度〔Tf〕>
高化式フローテスター{たとえば、(株)島津製作所製、CFT−500D}を用いて、1gの測定試料を昇温速度6℃/分で加熱しながら、プランジャーにより1.96MPaの荷重を与え、直径1mm、長さ1mmのノズルから押し出して、「プランジャー降下量(流れ値)」と「温度」とのグラフを描き、プランジャーの降下が始まり、樹脂の流出が始まる温度を流出開始温度〔Tf〕とする。
この範囲であると、定着性と粉砕性および耐久性の両立が良好となる。
{(P)の〔G’180〕}/{(A)の〔G’180〕と(B)の〔G’180〕の加重平均}≧1.5 ・・・式(1)
{(P)の〔G’180〕}/{(A)の〔G’180〕と(B)の〔G’180〕の加重平均}≧1.7 ・・・式(1’)
{(P)の〔G’180〕}/{(A)の〔G’180〕と(B)の〔G’180〕の加重平均}≧2.0 ・・・式(1”)
(A)の〔G’150〕≦10000 ・・・式(2)
(B)の〔G’150〕≦10000 ・・・式(3)
(A)および(B)の樹脂物性を全て前記の好ましい範囲にすると、式(1)の関係を達成するのが容易である。式(1)を満たすと、高温領域でも実用範囲において粘度が低くなりすぎないと考えられ、トナーとして使用したときの耐ホットオフセット性が良好となる。
ポリエステル樹脂(A)の貯蔵弾性率(G’)を増加させるには、(A)のT1/2を上げる、3価以上の構成成分の比率を上げ架橋点の数を増やす、分子量を大きくする、および/またはTgを高くする、等で達成できる。
ウレタン(ウレア)変性ポリエステル樹脂(B)の貯蔵弾性率(G’)を増加させるには、(B)のT1/2を上げる、ウレタン基(およびウレア基)含有量を増やす、分子量を大きくする、および/またはTgを高くする、等で達成できる。
装置 :ARES−24A(レオメトリック社製)
治具 :25mmパラレルプレート
周波数 :1Hz
歪み率 :5%
昇温速度:5℃/min
ポリオール成分(y)とカルボン酸成分(x)との反応比率は、水酸基とカルボキシル基の当量比[OH]/[COOH]が、好ましくは3/1〜1/3、さらに好ましくは2.5/1〜1/2.5、とくに好ましくは2/1〜1/2である。
また、本発明の電子写真用トナーバインダーのフローテスターで測定した軟化点〔T1/2〕は、耐ホットオフセット性と低温定着性の両立の観点から、好ましくは120〜170℃、さらに好ましくは125〜160℃である。
溶融混合する場合の混合装置としては、反応槽等のバッチ式混合装置、および連続式混合装置が挙げられる。適正な温度で短時間で均一に混合するためには、連続式混合装置が好ましい。連続混合装置としては、エクストルーダー、コンティニアスニーダー、3本ロール等が挙げられる。
粉体混合する場合の混合装置としては、ヘンシェルミキサー、ナウターミキサー、およびバンバリーミキサー等が挙げられる。好ましくはヘンシェルミキサーである。
着色剤の含有量は、本発明の電子写真用トナーバインダー100部に対して、好ましくは1〜40部、さらに好ましくは3〜10部である。なお、磁性粉を用いる場合は、好ましくは20〜150部、さらに好ましくは40〜120部である。上記および以下において、部は重量部を意味する。
ポリオレフィンワックスとしては、オレフィン(例えばエチレン、プロピレン、1−ブテン、イソブチレン、1−ヘキセン、1−ドデセン、1−オクタデセンおよびこれらの混合物等)の(共)重合体[(共)重合により得られるものおよび熱減成型ポリオレフィンを含む]、オレフィンの(共)重合体の酸素および/またはオゾンによる酸化物、オレフィンの(共)重合体のマレイン酸変性物[例えばマレイン酸およびその誘導体(無水マレイン酸、マレイン酸モノメチル、マレイン酸モノブチルおよびマレイン酸ジメチル等)変性物]、オレフィンと不飽和カルボン酸[(メタ)アクリル酸、イタコン酸および無水マレイン酸等]および/または不飽和カルボン酸アルキルエステル[(メタ)アクリル酸アルキル(アルキルの炭素数1〜18)エステルおよびマレイン酸アルキル(アルキルの炭素数1〜18)エステル等]等との共重合体、およびサゾールワックス等が挙げられる。
また、乳化転相法によりトナーを得る場合、流動化剤を除くトナーを構成する成分を有機溶剤に溶解または分散後、水を添加する等によりエマルジョン化し、次いで分離、分級して製造することができる。トナーの体積平均粒径は、3〜15μmが好ましい。
[ポリエステル樹脂(A−1)の合成]
反応槽中に、テレフタル酸561部(3.4モル)、無水フタル酸214部(1.4モル)、エチレングリコール550部(8.9モル)、ネオペンチルグリコール81部(0.8モル)、重合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水を留去しながら5時間反応させた後、無水1,2,4−ベンゼントリカルボン酸(以下無水トリメリット酸と記載)16部(0.1モル)を入れ、5〜20mmHgの減圧下で反応させ所定の粘度で取り出した。回収されたエチレングリコールは275部(4.4モル)であった。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(A−1)とする。
ポリエステル樹脂(A−1)のTgは58℃、T1/2は125℃、Mpは7800、OHVは30、AVは1、THF不溶解分は2%であった。
[ポリエステル樹脂(A−2)の合成]
反応槽中に、テレフタル酸549部(3.3モル)、無水フタル酸210部(1.4モル)、エチレングリコール537部(8.7モル)、ネオペンチルグリコール82部(0.8モル)、重合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水を留去しながら5時間反応させた後、無水トリメリット酸32部(0.2モル)を入れ、5〜20mmHgの減圧下で反応させ所定の粘度で取り出した。回収されたエチレングリコールは261部(4.2モル)であった。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(A−2)とする。
ポリエステル樹脂(A−2)のTgは60℃、T1/2は131℃、Mpは6200、OHVは35、AVは1、THF不溶解分は4%であった。
[ポリエステル樹脂(A−3)の合成]
反応槽中に、テレフタル酸477部(2.9モル)、イソフタル酸318部(1.9モル)、エチレングリコール544部(8.8モル)、ネオペンチルグリコール83部(0.8モル)、重合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水を留去しながら5時間反応させた後、無水トリメリット酸16部(0.1モル)を入れ、5〜20mmHgの減圧下で反応させ所定の粘度で取り出した。回収されたエチレングリコールは264部(4.3モル)であった。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(A−3)とする。
ポリエステル樹脂(A−3)のTgは58℃、T1/2は121℃、Mpは5100、OHVは41、AVは1、THF不溶解分は1%であった。
[ポリエステル樹脂(A−4)の合成]
反応槽中に、テレフタル酸411部(2.5モル)、イソフタル酸411部(2.5モル)、エチレングリコール613部(9.9モル)、重合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水を留去しながら5時間反応させた後、無水トリメリット酸22部(0.1モル)を入れ、5〜20mmHgの減圧下で反応させ所定の粘度で取り出した。回収されたエチレングリコールは277部(4.5モル)であった。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(A−4)とする。
ポリエステル樹脂(A−4)のTgは60℃、T1/2は119℃、Mpは6100、OHVは36、AVは1、THF不溶解分は2%であった。
[ポリエステル樹脂(a−1)の合成]
反応槽中に、テレフタル酸558部(3.4モル)、無水フタル酸217部(1.5モル)、エチレングリコール558部(9.0モル)、ネオペンチルグリコール82部(0.8モル)および縮合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水とエチレングリコールを留去しながら5時間反応させた後、5〜20mmHgの減圧下に2時間反応させた後取出した。回収されたエチレングリコールは279部(4.5モル)であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(a−1)とする。
ポリエステル樹脂(a−1)のTgは53℃、Mnは2500、OHVは45、AVは1であった。
[ウレタン変性ポリエステル樹脂(B−1)の合成]
反応槽中に、ポリエステル樹脂(a−1)484部(0.17モル)を入れ、120℃まで加熱し熔融した。窒素気流下で4,4’−ジフェニルメタンジイソシアネート(以下、MDIと記載)を16部(0.06モル)加え、3時間反応させ取り出した。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(B−1)とする。
ウレタン変性ポリエステル樹脂(B−1)のTgは60℃、T1/2は135℃、Mpは8500、Tfは85℃、AVは1、OHVは25、THF不溶解分は5%であった。
[ポリエステル樹脂(a−2)の合成]
反応槽中に、テレフタル酸570部(3.4モル)、イソフタル酸244部(1.5モル)、エチレングリコール560部(9.0モル)、ネオペンチルグリコール82部(0.8モル)および縮合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水とエチレングリコールを留去しながら5時間反応させた後、5〜20mmHgの減圧下に2時間反応させた後取出した。回収されたエチレングリコールは282部(4.6モル)であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(a−2)とする。
ポリエステル樹脂(a−2)のTgは55℃、Mnは2800、OHVは40、AVは1であった。
[ウレタン変性ポリエステル樹脂(B−2)の合成]
反応槽中に、ポリエステル樹脂(a−2)480部(0.17モル)、テトラヒドロフラン800部を入れ80℃まで加熱し、(a−2)を溶解した。窒素気流下でMDIを20部(0.08モル)加え、12時間反応させた後、200℃まで加熱しながら5〜20mmHgの減圧下でテトラヒドロフランを10時間かけて留去し、取出した。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(B−2)とする。
ウレタン変性ポリエステル樹脂(B−2)のTgは61℃、T1/2は137℃、Mpは8100、Tfは84℃、AVは1、OHVは21、THF不溶解分は4%であった。
[ポリエステル樹脂(a−3)の合成]
反応槽中に、テレフタル酸592部(3.6モル)、イソフタル酸254部(1.5モル)、エチレングリコール632部(10.2モル)および重合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水を留去しながら5時間反応させた後、5〜20mmHgの減圧下で反応させ所定の粘度で取り出した。回収されたエチレングリコールは297部(4.8モル)であった。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(a−3)とする。
ポリエステル樹脂(a−3)のTgは56℃、Mnは3200、OHVは35、AVは1であった。
[ウレタンウレア変性ポリエステル樹脂(B−3)の合成]
反応槽中に、ポリエステル樹脂(a−3)486部(0.15モル)を入れ、120℃まで加熱し熔融した。窒素気流下でMDIを14部(0.06モル)加え、3時間反応させた後、さらにIPDAを11部(0.06モル)加え、3時間攪拌した後取出した。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(B−3)とする。
ウレタンウレア変性ポリエステル樹脂(B−3)のTgは60℃、T1/2は135℃、Mpは8500、Tfは85℃、AVは1、OHVは25、THF不溶解分は5%であった。
[線形ポリエステル樹脂(C−1)の合成]
反応槽中に、テレフタル酸525部(3.2モル)、無水フタル酸201部(1.4モル)、エチレングリコール515部(8.3モル)、ネオペンチルグリコール76部(0.7モル)、および縮合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水とエチレングリコールを留去しながら5時間反応させた後、5〜20mmHgの減圧下に1時間反応させた。安息香酸を43部(0.3モル)入れ、さらに5〜20mmHgの減圧下に反応させた。AVが2以下になった時点で、180℃まで冷却後無水トリメリット酸を43部(0.2モル)仕込み、180℃で1時間保持した後取出した。回収されたエチレングリコールは260部(4.2モル)であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化した。これを線形ポリエステル樹脂(C−1)とする。
線形ポリエステル樹脂(C−1)のTgは61℃、Mpは6500、OHVは5、AVは25であった。
[線形ポリエステル樹脂(C−2)の合成]
反応槽中に、テレフタル酸548部(3.3モル)、イソフタル酸235部(1.4モル)、エチレングリコール538部(8.7モル)、ネオペンチルグリコール78部(0.8モル)、および縮合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水とエチレングリコールを留去しながら5時間反応させた後、5〜20mmHgの減圧下に2時間反応させた。AVが2以下になった時点で、180℃まで冷却後無水トリメリット酸を43部(0.2モル)仕込み、180℃で1時間保持した後取出した。回収されたエチレングリコールは274部(4.4モル)であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化した。これを線形ポリエステル樹脂(C−2)とする。
線形ポリエステル樹脂(C−2)のTgは62℃、Mpは7100、OHVは22、AVは24であった。
[線形ポリエステル樹脂(C−3)の合成]
反応槽中に、テレフタル酸510部(3.1モル)、無水フタル酸245部(1.7モル)、エチレングリコール586部(9.5モル)、および縮合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水とエチレングリコールを留去しながら5時間反応させた後、5〜20mmHgの減圧下に1時間反応させた。安息香酸を45部(0.4モル)入れ、さらに5〜20mmHgの減圧下に反応させた。AVが2以下になった時点で、180℃まで冷却後無水トリメリット酸を34部(0.2モル)仕込み、180℃で1時間保持した後取出した。回収されたエチレングリコールは275部(4.4モル)であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化した。これを線形ポリエステル樹脂(C−3)とする。
線形ポリエステル樹脂(C−3)のTgは60℃、Mpは6900、OHVは2、AVは20であった。
[線形ポリエステル樹脂(C−4)の合成]
反応槽中に、テレフタル酸245部(1.5モル)、ビスフェノールA・プロピレンオキサイド2モル付加物720部(2.1モル)、および縮合触媒としてテトラブトキシチタネート3部を入れ、230℃で窒素気流下に、生成する水を留去しながら5時間反応させた。次いで5〜20mmHgの減圧下に反応させ、AVが2以下になった時点で、180℃まで冷却後無水トリメリット酸を85部(0.4モル)仕込み、180℃で1時間保持した後取出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化した。これを線形ポリエステル樹脂(C−4)とする。
線形ポリエステル樹脂(C−4)のTgは61℃、Mpは3500、AVは50、OHVは48であった。
[比較用ポリエステル樹脂(RA−1)〔(C−5)〕の合成]
反応槽中に、テレフタル酸540部(3.3モル)、無水フタル酸206部(1.4モル)、エチレングリコール529部(8.5モル)、ネオペンチルグリコール79部(0.8モル)、重合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水とエチレングリコールを留去しながら5時間反応させた後、5〜20mmHgの減圧下に1時間反応させた。次いで、無水トリメリット酸52部(0.1モル)を加え、常圧下で1時間反応させた後、20〜40mmHgの減圧下で反応させ所定の粘度で取り出した。回収されたエチレングリコールは264部(4.3モル)であった。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(RA−1)〔(C−5)〕とする。
ポリエステル樹脂(RA−1)のTgは62℃、T1/2は148℃、Mpは10500、AVは20、OHVは21、THF不溶解分は5%であった。
[比較用ポリエステル樹脂(RA−2)〔(C−6)〕の合成]
反応槽中に、テレフタル酸574部(3.5モル)、無水フタル酸219部(1.5モル)、エチレングリコール564部(9.1モル)、ネオペンチルグリコール81部(0.8モル)、重合触媒としてテトラブトキシチタネート3部を入れ、210℃で窒素気流下に生成する水を留去しながら5時間反応させた後、5〜20mmHgの減圧下で反応させ所定の粘度で取り出した。回収されたエチレングリコールは290部(4.7モル)であった。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(RA−2)〔(C−6)〕とする。
ポリエステル樹脂(RA−2)のTgは56℃、T1/2は110℃、Mpは8100、OHVは30、AVは1、THF不溶解分は1%であった。
[比較用ポリエステル樹脂(RB−1)の合成]
反応槽中に、ポリエステル樹脂(a−1)469部(0.17モル)を入れ、120℃まで加熱し熔融した。窒素気流下でMDIを31部(0.13モル)加え、3時間反応させ取り出した。得られた樹脂を室温まで冷却後、粉砕し粒子化した。これをポリエステル樹脂(RB−1)とする。
ウレタン変性ポリエステル樹脂(RB−1)のTgは63℃、T1/2は155℃、Mpは12500、Tfは98℃、AVは1、OHVは18、THF不溶解分は6%であった。
上記製造例で得られたポリエステル樹脂(A−1)〜(A−4)、(B−1)〜(B−3)、(C−1)〜(C−4)、および比較製造例で得られた比較用ポリエステル樹脂(RA−1)、(RA−2)、(RB−1)を表1の配合比(部)に従い配合し、本発明の電子写真用トナーバインダー、および比較の電子写真用トナーバインダーを得て、下記の方法でトナー化した。(カーボンブラックMA−100[三菱化学(株)製]、カルナバワックス、荷電制御剤T−77[保土谷化学(製)])
まず、ヘンシェルミキサー[三井三池化工機(株)製 FM10B]を用いて予備混合した後、二軸混練機[(株)池貝製 PCM−30]で140℃で混練した。ついで超音速ジェット粉砕機ラボジェット[日本ニューマチック工業(株)製]を用いて微粉砕した後、気流分級機[日本ニューマチック工業(株)製 MDS−I]で分級し、粒径D50が8μmのトナー粒子を得た。ついで、トナー粒子100部にコロイダルシリカ(アエロジルR972:日本アエロジル製)0.5部をサンプルミルにて混合して、本発明のトナー組成物(T−1)〜(T−9)、および比較用のトナー組成物(RT−1)〜(RT−3)を得た。
前記の方法で測定した、ポリエステル樹脂(A)、ウレタン(ウレア)変性ポリエステル樹脂(B)、ポリエステル樹脂組成物(P)、線形ポリエステル樹脂(C)、およびトナーバインダーの物性値、並びに下記評価方法で評価したトナー組成物の評価結果を表2に示す。
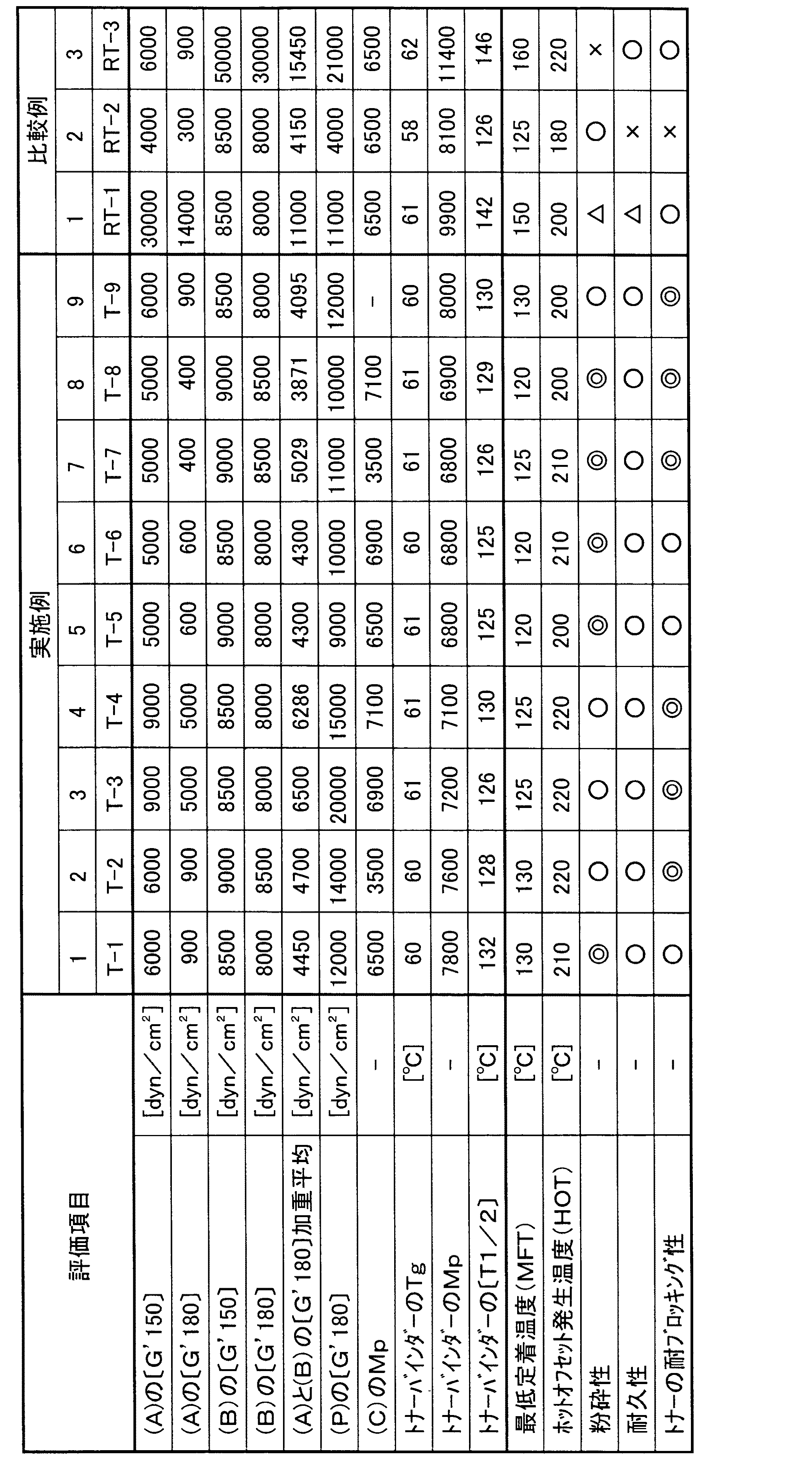
〔1〕最低定着温度(MFT)
上記トナー組成物を使用し、市販複写機(AR5030;シャープ製)を用いて現像した未定着画像を、市販複写機(AR5030;シャープ製)の定着機を用いて評価した。定着画像をパットで擦った後の画像濃度の残存率が70%以上となる下限温度を最低定着温度とした。
〔2〕ホットオフセット発生温度(HOT)
上記MFTと同様に定着評価し、定着画像へのホットオフセットの有無を目視評価した。定着ロール通過後ホットオフセットが発生しない上限温度をホットオフセット発生温度とした。
〔3〕粉砕性
二軸混練機で混練、冷却したトナー粗粉砕物(8.6メッシュパス〜30メッシュオンのもの)を、超音速ジェット粉砕機ラボジェット[日本ニューマチック工業(株)製]により下記条件で微粉砕した。
粉砕圧:0.5MPa
アジャスターリング:15mm
ルーバーの大きさ:中
粉砕時間:10分
これを分級せずに、体積平均粒径をコールターカウンター−TAII(米国コールター・エレクトロニクス社製)により測定し、粉砕性のテストとした。評価基準を下記の通りとした。(単位:μm)
◎ : 10未満
○ : 10以上11未満
△ : 11以上12未満
× : 12以上
〔4〕耐久性
上記トナー組成物を市販のプリンタLP−1400(エプソン製)用のカートリッジに充填し、同機を用いてべた画像を3000枚連続印刷し、3000枚後の画像を目視で判定した。
判定基準
○ : スジ・ムラなし。
△ : わずかにスジ・ムラがある。
× : スジ・ムラがある。
〔5〕トナーの耐ブロッキング性試験
上記トナー組成物を、40℃・95%R.H.の高温高湿環境下で、48時間調湿した。同環境下において該現像剤のブロッキング状態を目視判定し、さらに市販複写機(AR5030:シャープ製)でコピーした時の画質を観察した。
判定基準
◎ : トナーのブロッキングがなく、3000枚複写後の画質も良好。
○ : トナーのブロッキングはないが、3000枚複写後の画質に僅かに乱れが観察される。
× : トナーのブロッキングが目視でき、3000枚までに画像が出なくなる
Claims (9)
- カルボン酸成分(x)とポリオール成分(y)とを構成単位として有し、カルボン酸成分(x)が1,2,4−ベンゼントリカルボン酸(x1)と、芳香族ジカルボン酸およびそのエステル形成性誘導体から選ばれる2種以上のジカルボン酸(x2)を合計で80〜99.5モル%含有し、ポリオール成分(y)が、炭素数が2〜10の脂肪族ジオール(y1)を80モル%以上含有する、酸価が2(mgKOH/g)以下のポリエステル樹脂(A)と、ウレタン(ウレア)変性ポリエステル樹脂(B)とで構成されるポリエステル樹脂組成物(P)を含有し、(A)、(B)、および(P)が次の式(1)〜(3)を満たす電子写真用トナーバインダー。
{(P)の〔G’180〕}/{(A)の〔G’180〕と(B)の〔G’180〕の加重平均}≧1.5 ・・・式(1)
(A)の〔G’150〕≦10000 ・・・式(2)
(B)の〔G’150〕≦10000 ・・・式(3)
[上記式中、〔G’180〕は180℃における貯蔵弾性率(dyn/cm2)、〔G’
150〕は150℃における貯蔵弾性率(dyn/cm2)を意味する。] - ポリエステル樹脂(A)を構成するジカルボン酸(x2)が、下記(1)〜(3)から選ばれる2種以上である請求項1記載の電子写真用トナーバインダー:
(1)テレフタル酸および/またはそのエステル形成性誘導体
(2)イソフタル酸および/またはそのエステル形成性誘導体
(3)フタル酸および/またはそのエステル形成性誘導体。 - ポリエステル樹脂組成物(P)中のポリエステル樹脂(A)の含有量が、(P)の重量に対して30〜70重量%である請求項1または2記載の電子写真用トナーバインダー。
- さらに、ポリエステル樹脂(A)以外の線形ポリエステル樹脂(C)を含有する請求項1〜3いずれか記載の電子写真用トナーバインダー。
- 線形ポリエステル樹脂(C)のテトラヒドロフラン可溶分のゲルパーミエーションクロマトグラフィーのピークトップ分子量が1000〜10000である請求項4記載の電子写真用トナーバインダー。
- 線形ポリエステル樹脂(C)の含有量が、ポリエステル樹脂組成物(P)と線形ポリエステル樹脂(C)の合計重量に対して70重量%以下である請求項4または5記載の電子写真用トナーバインダー。
- テトラヒドロフラン可溶分のゲルパーミエーションクロマトグラフィーのピークトップ分子量が2000〜20000である請求項1〜6いずれか記載の電子写真用トナーバインダー。
- ガラス転移温度が30〜75℃であり、フローテスターによる軟化点が120〜132℃である請求項1〜7いずれか記載の電子写真用トナーバインダー。
- 請求項1〜8いずれか記載の電子写真用トナーバインダーと着色剤、並びに必要により、離型剤、荷電制御剤、および流動化剤から選ばれる1種類以上の添加剤を含有するトナー組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011146559A JP5777953B2 (ja) | 2011-06-30 | 2011-06-30 | 電子写真用トナーバインダー及びトナー組成物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011146559A JP5777953B2 (ja) | 2011-06-30 | 2011-06-30 | 電子写真用トナーバインダー及びトナー組成物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013015584A JP2013015584A (ja) | 2013-01-24 |
| JP5777953B2 true JP5777953B2 (ja) | 2015-09-16 |
Family
ID=47688340
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011146559A Active JP5777953B2 (ja) | 2011-06-30 | 2011-06-30 | 電子写真用トナーバインダー及びトナー組成物 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5777953B2 (ja) |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06128367A (ja) * | 1992-10-14 | 1994-05-10 | Toray Ind Inc | トナーバインダー用ポリエステル樹脂 |
| DE60109433T2 (de) * | 2000-09-07 | 2005-12-29 | Mitsui Chemicals, Inc. | Tonerzusammensetzung und verfahren zu deren herstellung |
| JP2005010368A (ja) * | 2003-06-18 | 2005-01-13 | Ricoh Co Ltd | 静電荷像現像用トナー及び製造方法 |
| JP4518479B2 (ja) * | 2003-06-24 | 2010-08-04 | 株式会社リコー | 静電荷像現像用トナー |
| JP4101165B2 (ja) * | 2003-12-09 | 2008-06-18 | 株式会社リコー | 静電荷像現像用トナーおよびその製造方法 |
| JP2006154686A (ja) * | 2003-12-10 | 2006-06-15 | Sanyo Chem Ind Ltd | トナー用ポリエステル樹脂、トナー組成物及び樹脂粒子 |
| JP2006106707A (ja) * | 2004-09-07 | 2006-04-20 | Ricoh Co Ltd | 静電荷像現像用トナー及び製造方法、現像剤、プロセスカートリッジ、画像形成装置 |
| JP4707087B2 (ja) * | 2005-01-13 | 2011-06-22 | 株式会社リコー | 画像形成方法 |
| JP4662475B2 (ja) * | 2006-03-10 | 2011-03-30 | 株式会社リコー | 静電荷像現像用トナー |
| JP2010072199A (ja) * | 2008-09-17 | 2010-04-02 | Ricoh Co Ltd | 静電荷像現像用トナー及び製造方法 |
| EP2416219B1 (en) * | 2009-03-31 | 2016-03-30 | Sanyo Chemical Industries, Ltd. | Toner binder and toner composition |
| JP5511315B2 (ja) * | 2009-10-30 | 2014-06-04 | 株式会社リコー | 静電荷像現像用トナー、その製造方法、現像剤、画像形成方法及び画像形成装置 |
| JP5448247B2 (ja) * | 2009-11-30 | 2014-03-19 | 株式会社リコー | トナーとその製造方法、現像剤、現像剤収容容器および画像形成方法 |
-
2011
- 2011-06-30 JP JP2011146559A patent/JP5777953B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013015584A (ja) | 2013-01-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5301722B2 (ja) | トナーバインダー及びトナー組成物 | |
| JP5763497B2 (ja) | トナーバインダーおよびトナー組成物 | |
| JP6081259B2 (ja) | トナーバインダーおよびトナー組成物 | |
| WO2013128872A1 (ja) | トナーバインダーおよびトナー | |
| JP2011028170A (ja) | トナーバインダー及びトナー組成物 | |
| JP5698026B2 (ja) | トナーバインダーおよびトナー組成物 | |
| JP5497511B2 (ja) | トナー用樹脂およびトナー組成物 | |
| JP6348361B2 (ja) | トナーバインダーおよびトナー組成物 | |
| JP5723549B2 (ja) | トナーバインダーおよびトナー組成物 | |
| JP2013178504A (ja) | 粉砕トナー用ポリエステル樹脂及びトナー組成物 | |
| JP2017215587A (ja) | トナーバインダー及びトナー | |
| JP5554125B2 (ja) | トナー用バインダー樹脂およびトナー組成物 | |
| JP5985922B2 (ja) | 電子写真用トナーバインダー及びトナー組成物 | |
| JP5415143B2 (ja) | トナー用樹脂組成物およびトナー組成物 | |
| JP5777953B2 (ja) | 電子写真用トナーバインダー及びトナー組成物 | |
| JP5642566B2 (ja) | トナーバインダーおよびトナー組成物 | |
| JP6829102B2 (ja) | トナーバインダーの製造方法及びトナーの製造方法 | |
| JP6316863B2 (ja) | トナーバインダーの製造方法 | |
| JP6227571B2 (ja) | トナーバインダーおよびトナー組成物 | |
| JP2017223944A (ja) | トナー用樹脂及びトナー | |
| JP2018169613A (ja) | トナーバインダー及びトナー | |
| JP6401199B2 (ja) | トナーバインダーの製造方法 | |
| JP6316862B2 (ja) | トナーバインダーの製造方法 | |
| JP2012013772A (ja) | トナーバインダー及びトナー組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140401 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20150128 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150210 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150408 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150707 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150708 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5777953 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |