JP2014237669A - 最終分化を誘導する方法 - Google Patents
最終分化を誘導する方法 Download PDFInfo
- Publication number
- JP2014237669A JP2014237669A JP2014146512A JP2014146512A JP2014237669A JP 2014237669 A JP2014237669 A JP 2014237669A JP 2014146512 A JP2014146512 A JP 2014146512A JP 2014146512 A JP2014146512 A JP 2014146512A JP 2014237669 A JP2014237669 A JP 2014237669A
- Authority
- JP
- Japan
- Prior art keywords
- saha
- group
- acid
- methanol
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 *NC(N*1*=C1)=O Chemical compound *NC(N*1*=C1)=O 0.000 description 2
- UKIMVQKYXFBPCC-UHFFFAOYSA-N COC(CCCCCCC(Nc1ccccc1)=O)=O Chemical compound COC(CCCCCCC(Nc1ccccc1)=O)=O UKIMVQKYXFBPCC-UHFFFAOYSA-N 0.000 description 1
Images














Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/164—Amides, e.g. hydroxamic acids of a carboxylic acid with an aminoalcohol, e.g. ceramides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/04—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
Abstract
【課題】新生物細胞の最終分化、細胞増殖停止及び/又はアポトーシスを選択的に誘導し、癌に対して有効なヒストンデアセチラーゼインヒビターである、スベロイルアニリドヒドロキサム酸(SAHA)に関し、バイオアベイラビリティーの高い結晶を製造する方法の提供。【解決手段】下記式に表される結晶形態のSAHAの製造方法。(a)スベリン酸をアニリンと反応させ、スベルアニリン酸又はその塩を生成させる工程、(b)スベルアニリン酸又はその塩をメタノールと反応させ、スベルアニリン酸メチルを生成させる工程、(c)スベルアニリン酸メチルをヒドロキシアミン塩酸塩と反応させ、粗SAHAを生成させる工程、(d)メタノール及び水の混合物からSAHAの粗生成物を再結晶する工程、からなる結晶形態のSAHAの製造方法。【選択図】なし
Description
(関連出願への交互参照)
本発明は、2002年3月4日に出願された合衆国仮出願第60/361,759号の利益を主張する。この仮出願の全体の教示は、本明細書に参考として援用される。
本発明は、2002年3月4日に出願された合衆国仮出願第60/361,759号の利益を主張する。この仮出願の全体の教示は、本明細書に参考として援用される。
(政府の利益の陳述)
本発明は、国立癌研究所によって授与される認可番号1R21 CA 096228−01の下、全部または一部が政府の支持でなされた。政府は、本発明において特定の権利を有し得る。
本発明は、国立癌研究所によって授与される認可番号1R21 CA 096228−01の下、全部または一部が政府の支持でなされた。政府は、本発明において特定の権利を有し得る。
(発明の分野)
本発明は、新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導すること、および/またはヒストンデアセチラーゼ(HDAC)インヒビターを含む薬学的組成物のHDAC阻害投与の方法を提供する。この薬学的組成物の経口処方物は、高いバイオアベイラビリティーのような好ましい薬物動態学プロフィールを有し、そして驚くべきことに、延長された時間の期間に亘り、活性化合物の高い血中レベルを生じる。
本発明は、新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導すること、および/またはヒストンデアセチラーゼ(HDAC)インヒビターを含む薬学的組成物のHDAC阻害投与の方法を提供する。この薬学的組成物の経口処方物は、高いバイオアベイラビリティーのような好ましい薬物動態学プロフィールを有し、そして驚くべきことに、延長された時間の期間に亘り、活性化合物の高い血中レベルを生じる。
(発明の背景)
本出願を通じて、種々の刊行物が括弧内のアラビア数字によって参照される。これら刊行物の完全な引用が、特許請求の範囲のすぐ前にある明細書の末端に見出され得る。これら刊行物のそれら全部の開示は、本発明が関係する当該技術分野の状態をより詳細に記載するために、本出願中に参考として本明細書によって援用される。
本出願を通じて、種々の刊行物が括弧内のアラビア数字によって参照される。これら刊行物の完全な引用が、特許請求の範囲のすぐ前にある明細書の末端に見出され得る。これら刊行物のそれら全部の開示は、本発明が関係する当該技術分野の状態をより詳細に記載するために、本出願中に参考として本明細書によって援用される。
癌は、細胞の集団が、変化する程度で、通常は、増殖および分化を支配する制御機構に非応答性になった疾患である。多年の間、癌の化学療法処置には、2つの原則的戦略:a)性ホルモンの産性または末梢作用での妨害によるホルモン依存性腫瘍細胞増殖をブロックすること;およびb)新生物細胞集団および正常細胞集団の両方を損傷する細胞傷害性物質に癌細胞を直接曝すことによって癌細胞を殺傷すること、が存在している。
癌治療はまた、新生物細胞の最終分化の誘導によって試みられている(1)。細胞培養モデルでは、分化は、サイクリックAMPおよびレチノイン酸(2、3)、アクラルビシンおよびその他のアンスラサイクリン(4)を含む種々の刺激剤への細胞の曝露によって報告されている。
腫瘍学の分野における多くの進展に拘らず、大部分の固形腫瘍は、進行したステージでは不治のままである。細胞傷害性治療が大部分の事例で用いられるが、しかし、それは、しばしば、有意な臨床的利点なくして、患者に有意な死亡率を引き起こす。進行した悪性腫瘍を処置および制御するために、より毒性が少なく、そしてより特異的な薬剤が開発されている。
新生物形質転換は、癌細胞が分化する能力を必ずしも破壊しないという多くの証拠が存在している(1、5、6)。通常の増殖の調節剤に反応せず、そしてそれらの分化プログラムの発現がブロックされているように見え、そしてなお、分化が誘導されかつ複製を止め得る腫瘍細胞の多くの例が存在している。いくつかの比較的単純な極性化合物(5、7〜9)、ビタミンDおよびレチノイン酸の誘導体(10〜12)、ステロイドホルモン(13)、成長因子(6、14)、プロテアーゼ(15、16)、腫瘍プロモーター(17、18)、およびDNAまたはRNA合成のインヒビター(4、19〜24)を含む種々の薬剤が、種々の形質転換細胞株および原発性ヒト腫瘍外植片を誘導し得、より分化した特徴を発現する。
初期の研究は、多くの形質転換細胞株において分化の有効な誘導剤であった一連の極性化合物を同定した(8、9)。これらのうち、最も有効な誘導剤は、極性/非極性ハイブリッド化合物であるN,N’−ヘキサメチレンビスアセタミド(HMBA)であった(9)。この極性/非極性化合物の、マウスの赤白血病細胞(MELC)が発癌性を抑制して赤血球分化を起こすように誘導するための使用は、形質転換細胞の誘導剤媒介分化を研究するための有用なモデルであることが証明されている(5、7〜9)。HMBA誘導MELC終末赤血球分化は、複数ステップのプロセスである。培養中のMELC(745A−DS19)へのHMBAの添加に際し、最終分化への拘束(commitment)が検出される前に、10〜12時間の潜伏期間がある。拘束は、細胞が、誘導剤の除去に拘らず、最終分化を発現する能力として規定される(25)。HMBAへの連続した曝露に際し、分化する細胞の進行する召集がある。本発明者らは、比較的低レベルのビンクリスチンに耐性にしたMELC細胞株が、HMBAの誘導作用に著しくより感受性になり、そして潜伏期間がほとんどないか、または全くなく誘導されて分化され得ることを報告した(26)。
HMBAは、広範な種類の細胞株において分化に一致する表現型変化を誘導し得る(5)。薬物誘導効果の特徴は、マウス赤白血病細胞系(MELC)で最も広範に研究されている(5、25、27、28)。分化のMELC誘導は、時間および濃度の両方に依存する。大部分の株においてインビトロで効果を示すために必要な最小濃度は2〜3mMであり;継続する薬物曝露なくして集団の実質的部分(>20%)における分化を誘導するために一般に必要な連続曝露の最小持続時間は、約36時間である。
HMBAの作用の主要な標的は未知である。プロテインキナーゼCが誘導剤媒介分化の経路に関与しているという証拠がある(29)。このインビトロ研究は、HMBAの能力を、ヒト癌の処置における細胞分化剤として評価するための基礎を提供した(30)。HMBAを用いるいくつかのフェーズI臨床試験が終了している(31〜36)。臨床試験は、この化合物が、癌をもつ患者における治療応答を誘導し得ることを示した(35、36)。しかし、これらのフェーズI臨床試験はまた、HMBAの潜在的な効き目が、一部、最適血中レベルの達成を防ぐ用量関連細胞傷害性によって、および延長された期間に亘る、この薬剤の大量の静脈内投与の必要性によって制限されることを示した。
モル濃度ベースで、非極性結合によって分離される極性基をもつ、HMBAに関連する多くの化合物が、活性であるか(37)、またはHMBAより100倍より活性であること(38)が報告されている。しかし、あるクラスとして、HMBAのような対称ダイマーおよび関連する化合物は、最良の細胞分化剤ではないことが見出されている。
最良の化合物は、メチレン基の柔軟性の鎖によって分離された2つの極性末端基を含むことが予期せぬことに見出されており、ここで、この極性末端基の1つまたは両方は、大きな疎水性基である。好ましくは、これら極性末端基は異なり、そして1つだけが大きな疎水性基である。これら化合物は、予期せぬことに、HMBAより1000倍より活性であり、そしてHMBA関連化合物より10倍より活性である。
スベロイルアニリドヒドロシアミド酸(SAHA)のようなヒストンデアセチラーゼのインヒビターは、腫瘍細胞成長停止、分化および/またはアポトーシスを誘導する能力を有するこのクラスの薬剤に属する(39)。これら化合物は、新生物細胞が悪性になる能力に固有の機構の方に標的される。なぜなら、それらは、動物における腫瘍成長の阻害のために有効な用量で細胞傷害性を有するようには見えないからである(40)。ヒストンアセチル化および脱アセチル化が、細胞における転写調節が達成される機構であるといういくつかの系統の証拠がある(41)。これらの効果は、ヌクレオソーム中のコイル状DNAに対するヒストンタンパク質の親和性を改変することによるクロマチンの構造中の変化を通じて起こると考えられている。ヌクレオソーム中で同定されている5つのタイプのヒストンが存在する(H1、H2A、H2B、H3およびH4と称される)。各ヌクレオソームは、ヌクレオソーム構造の外側の部分中で単一で存在しているH1を除き、そのコア内に2つの各ヒストンタイプを含む。ヒストンタンパク質が、低アセチル化されるとき、DNAリン酸骨格に対するヒストンのより大きな親和性が存在すると考えられている。この親和性は、DNAをヒストンに堅く結合させ、そしてDNAを、転写調節エレメントおよび転写調節機械に接近不能にする。アセチル化状態の調節は、2つの酵素複合体、ヒストンアセチルトランスフェラーゼ(HAT)およびヒストンデアセチラーゼ(HDAC)間の活性のバランスを通じて起こる。この低アセチル化状態は、関連するDNAの転写を阻害すると考えられている。この低アセチル化状態は、HDAC酵素を含む大きな複数タンパク質複合体によって触媒される。特に、HDACは、クロマチンコアヒストンからアセチル基の除去を触媒することが示されている。
HDACのSAHAによる阻害は、X線結晶学研究によって示されるように、酵素の触媒部位との直接相互作用によって起こると考えられている(42)。HDAC阻害の結果は、ゲノムに対して一般化された効果を有するとは考えられておらず、むしろ、小サブセットのゲノムに影響するのみである(43)。HDACインヒビターとともに培養した悪性細胞株を用いるDNAマイクロアレイによって提供された証拠は、その産物が改変される有限(1〜2%)数の遺伝子が存在することを示す。例えば、HDACインヒビターとの培養で処理された細胞は、サイクリン依存性キナーゼインヒビターp21の一貫した誘導を示す(44)。このタンパク質は、細胞周期停止で重要な役割を演じている。HDACインヒビターは、p21遺伝子の領域中のヒストンの高アセチル化状態を進行することによってp21の転写速度を増加し、それによって、この遺伝子を転写機構に接近可能にすると考えられている。HDACインヒビターによってその発現が影響されない遺伝子は、領域関連ヒストンのアセチル化で変化を示さない(45)。
いくつかの事例で、HATまたはHDAC活性の破壊が悪性表現型の発症に関与するということが示されている。例えば、急性前骨髄球白血病(promyelocytic)では、PMLおよびRARαの融合によって産生される腫瘍性タンパク質が、HDACの召集によって特異的遺伝子転写を抑制するようである(46)。このようにして、新生物細胞は、分化を終了することができず、そして白血病細胞株の過剰な増殖に至る。
本発明者の幾人かに発行された、米国特許第5,369,108号、同第5,932,616号、同5,700,811号、同第6,087,367号および同第6,511,990号は、新生物細胞の最終分化を選択的に誘導するために有用な化合物を開示し、これら化合物は、メチレン基の柔軟な鎖によるか、または剛直なフェニル基によって分離された2つの極性末端基を有し、ここで、この極性末端基の1つまたは両方は、大きな疎水性基である。これら化合物のいくつかは、第1の疎水性基と分子の同じ末端にさらなる大きな疎水性基を有し、これは、酵素アッセイにおいて約100倍、そして細胞分化アッセイにおいて約50倍分化活性をさらに増加する。本発明の方法および薬学的組成物で用いられる化合物を合成する方法は、前述の特許に完全に記載されており、それらの全体の内容は、本明細書に参考として援用される。
前述の特許は、HDACインヒビターの詳細な経口処方物、または記載された化合物の詳細な投薬量および投与スケジュールを記載していない。重要なことには、前述の特許は、延長された時間の期間に亘り、活性化合物の高い血中レベルを生じる高いバイオアベイラビリティーのような好ましい薬物動態学プロフィールをもつ経口処方物を開示していない。
本発明のクラスの化合物は、新生物細胞の最終分化を選択的に誘導するために有用であり得、そしてそれ故、患者における腫瘍の処置を支援する。従って、これら化合物の適切な投薬量および投与スケジュールを発見し、そして処方物、好ましくは、延長された期間に亘り、活性化合物の安定な、治療的に有効な血中レベルを生じる経口処方物を開発する緊急の必要性がある。
(発明の要旨)
本発明は、投与の後少なくとも2時間の期間に亘り、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のヒストンデアセチラーゼ(HDAC)インヒビターを生成する方法を提供し、これは、上記被験体に、HDACインヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を投与する工程を包含する。
本発明は、投与の後少なくとも2時間の期間に亘り、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のヒストンデアセチラーゼ(HDAC)インヒビターを生成する方法を提供し、これは、上記被験体に、HDACインヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を投与する工程を包含する。
本発明はまた、新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導し、それによって、このような細胞の増殖を阻害する方法、および投与後少なくとも2時間の期間に亘って被験体中でインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のヒストンデアセチラーゼ(HDAC)インヒビターを生成することにより、上記被験体に、HDACインヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を投与することにより、腫瘍細胞の分化を誘導する方法を提供する。
本発明は、投与後少なくとも2時間の期間に亘って被験体中でインビボで少なくとも約10nMの平均血漿濃度のスベロイルアニリドヒドロキサム酸(SAHA)を生成する方法をさらに提供し、これは、上記被験体に、SAHAまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を投与する工程を包含する。
本発明はまた、新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導し、それによって、このような細胞の増殖を阻害する方法、および投与後少なくとも2時間の期間に亘って被験体中でインビボで少なくとも約10nMの平均血漿濃度のSAHAを生成することにより、上記被験体に、SAHAまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を投与することにより、腫瘍細胞の分化を誘導する方法を提供する。
本発明は、経口投与に適切な薬学的組成物を提供し、これは、新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導するために有用であり、および/またはヒストンデアセチラーゼ(HDAC)の潜在的なインヒビターである化合物を含む。この薬学的組成物は、さらに微結晶セルロース、クロスカルメロースナトリウム(croscarmellose sodium)およびステアリン酸マグネシウムから構成される。本発明はまた、SAHA、微結晶セルロース、クロスカルメロースナトリウムおよびステアリン酸マグネシウムを含む、経口投与のための薬学的組成物を提供する。本発明の処方物中の活性化合物の経口バイオアベイラビリティーは、驚くべきことに高い。さらに、これら処方物は、予期せぬことに、延長された時間の期間に亘り、活性化合物の高い、治療的に有効な血中レベルを生じる。本発明は、これら処方物の、従うのが容易であり、かつ守るのが容易である安全な、毎日の投与レジメンをさらに提供する。
本明細書で示されるように、驚くべきことに、かつ予期せぬことに、HDACインヒビター、特に、スベロイルアニリドヒドロキサム酸(SAHA)を含む経口処方物が、インビボで、活性化合物の非常に高い全体の経口バイオアベイラビリティーを有することが見出された。さらにこれら処方物は、活性化合物の高い血中レベルを生じ、これは、予期せぬことに、例えば、10〜12時間までの延長された時間の期間に亘り、高く維持される。本発明の経口処方物は、特に非経口処方物と比較したとき、多くの利点を有している。なぜなら、一方で、それらは、HDACインヒビターの高い安定なかつ延長された治療的に有効な血中レベルを提供し、他方で、経口投与の任意の従来様式によって患者に投与するのが容易であるからである。
従って、本発明は、ヒストンデアセチラーゼ(HDAC)インヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む経口投与のための薬学的組成物を提供し;ここで、この組成物は、投与後少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するに有効な平均血漿濃度のHDACインヒビターを提供する。好ましい実施形態では、このHDACインヒビターの濃度は、投与後、少なくとも10時間の期間、HDACを阻害するために有効である。
好ましい実施形態では、本発明は、SAHAまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む経口投与のための薬学的組成物を提供し;ここで、この組成物は、投与後少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するに有効なSAHAの平均血漿濃度を提供する。好ましい実施形態では、このSAHAの濃度は、投与後、少なくとも10時間の期間、HDACを阻害するために有効である。
本発明の処方物は、新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導し、そしてそれ故、患者において腫瘍の処置を支援するために有用である。
従って、本発明はまた、被験体における新生物細胞の最終分化を選択的に誘導し、そしてそれによって、被験体においてこのような細胞の増殖を阻害する方法を提供し、この被験体に、ヒストンデアセチラーゼ(HDAC)インヒビターまたはその薬学的に受容可能な塩または水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を経口的に投与する工程を包含し;ここで、この組成物は、投与の後、少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するために有効な平均血漿濃度のHDACインヒビターを提供する。
さらに、本発明はまた、被験体において新生物細胞の細胞増殖停止を選択的に誘導し、そしてそれによって、被験体においてそのような細胞の増殖を阻害する方法を提供し、この被験体に、ヒストンデアセチラーゼ(HDAC)インヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を経口的に投与する工程を包含し;ここで、この組成物は、投与の後、少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するために有効な平均血漿濃度のHDACインヒビターを提供する。
さらに、本発明はまた、被験体において新生物細胞のアポトーシスを選択的に誘導し、そしてそれによって、被験体においてそのような細胞の増殖を阻害する方法を提供し、この被験体に、ヒストンデアセチラーゼ(HDAC)インヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を経口的に投与する工程を包含し;ここで、この組成物は、投与の後、少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するために有効な平均血漿濃度のHDACインヒビターを提供する。
さらに、本発明はまた、腫瘍を有する被験体において腫瘍細胞の分化を誘導する方法を提供し、この被験体に、ヒストンデアセチラーゼ(HDAC)インヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を経口的に投与する工程を包含し;ここで、この組成物は、投与の後、少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するために有効な平均血漿濃度のHDACインヒビターを提供する。
さらに、本発明は、被験体においてヒストンデアセチラーゼの活性を阻害する方法を提供し、この被験体に、ヒストンデアセチラーゼ(HDAC)インヒビターまたは薬学的に受容可能なその塩またはその水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む薬学的組成物の有効量を経口的に投与する工程を包含し;ここで、この組成物は、投与の後、少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するために有効な平均血漿濃度のHDACインヒビターを提供する。
さらに、本発明はまた、被験体において新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導し、そしてそれによって、被験体においてこのような細胞の増殖を阻害する方法を提供し、この被験体に、SAHAまたは薬学的に受容可能なその塩またはその水和物を含む薬学的組成物の有効量を投与する工程を包含し;ここで、この組成物は、投与の後、少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するために有効な平均血漿濃度のSAHAを提供する。
さらに、本発明はまた、腫瘍を有する被験体において腫瘍細胞の分化を誘導する方法を提供し、この被験体に、SAHAまたは薬学的に受容可能なその塩またはその水和物を含む薬学的組成物の有効量を投与する工程を包含し;ここで、この組成物は、投与の後、少なくとも2時間の期間、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するために有効な平均血漿濃度のSAHAを提供する。
さらに、本発明は、被験体のヒストンデアセチラーゼの活性を阻害する方法を提供し、この方法は、被験体に、有効量の薬学的組成物(SAHAまたはその薬学的に受容可能な塩または水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む)を経口投与する工程を包含し、ここで、この組成物は、投与後少なくとも2時間の期間にわたって、インビボで、ヒストンデアセチラーゼ(HDAC)を阻害するのに有効な平均血漿濃度のSAHAを提供する。
好ましい実施形態において、SAHAまたは任意のHDACインヒビターは、25〜4000mg/m2の間の合計の毎日の投薬量で患者に投与される。別の好ましい実施形態において、SAHAまたは任意のHDACインヒビターは、200mgの合計の毎日の投薬量で患者に投与される。SAHAまたは任意のHDACインヒビターは、400mgの合計の毎日の投薬量で患者に投与される。
1つの好ましい実施形態において、組成物は、投与に続く少なくとも2時間の期間にわたって、ヒストンデアセチラーゼを阻害し得る平均血漿濃度のHDACインヒビター(例えば、SAHA)を提供し、これは、好ましくは、少なくとも約10nMの濃度である。なお別の好ましい実施形態において、この組成物は、投与に続く少なくとも10時間の期間にわたって、少なくとも約10nMの平均血漿濃度のHDACインヒビターを提供する。
1つの好ましい実施形態において、組成物は、最終分化、細胞増殖停止および/または新生物細胞のアポトーシスを選択的に誘導し得るか、あるいは腫瘍中の腫瘍細胞の分化を誘導し得る平均血漿濃度のHDACインヒビター(例えば、SAHA)を提供し、ここで、この濃度は、投与後少なくとも2時間の期間にわたって維持され、これは、好ましくは、約2.5μMの濃度である。なお別の好ましい実施形態において、この組成物は、投与後少なくとも10時間の期間にわたって、約2.5μMの平均血漿濃度のHDACインヒビターを提供する。
本発明の組成物は、経口投与に適切な任意の単位投薬形態(液体または固体)(例えば、ペレット、錠剤、コーティングされた錠剤、カプセル、ゼラチンカプセル、溶液、懸濁液、または分散剤の形態)で処方され得る。好ましい実施形態において、この組成物は、ゼラチンカプセルの形態である。
キャリアまたは希釈剤として共通に使用される任意の不活性賦形剤は、本発明の処方物(例えば、ガム、デンプン、糖、セルロース材料、アクリレート、またはその混合物)で使用され得る。好ましい希釈剤は、微結晶セルロースである。この組成物は、さらに、崩壊剤(例えば、ナトリウムクロスカルメロース)および潤滑剤(例えば、ステアリン酸マグネシウム)を含み得、さらに、結合剤、緩衝液、プロテアーゼインヒビター、界面活性剤、可溶化剤、可塑剤、乳化剤、安定化剤、粘性増加剤、甘味料、フィルム形成剤、またはこれらの任意の組合せから選択される1つ以上の添加剤を含み得る。さらに、本発明の組成物は、制御放出処方物または迅速放出処方物の形態であり得る。
幅広い種々のHDACインヒビターが、本発明の組成物においての使用に適切である。好ましい実施形態において、HDACインヒビターは、スベロイルアニリド(suberoylanilide)ヒドロキサム酸(SAHA)である。
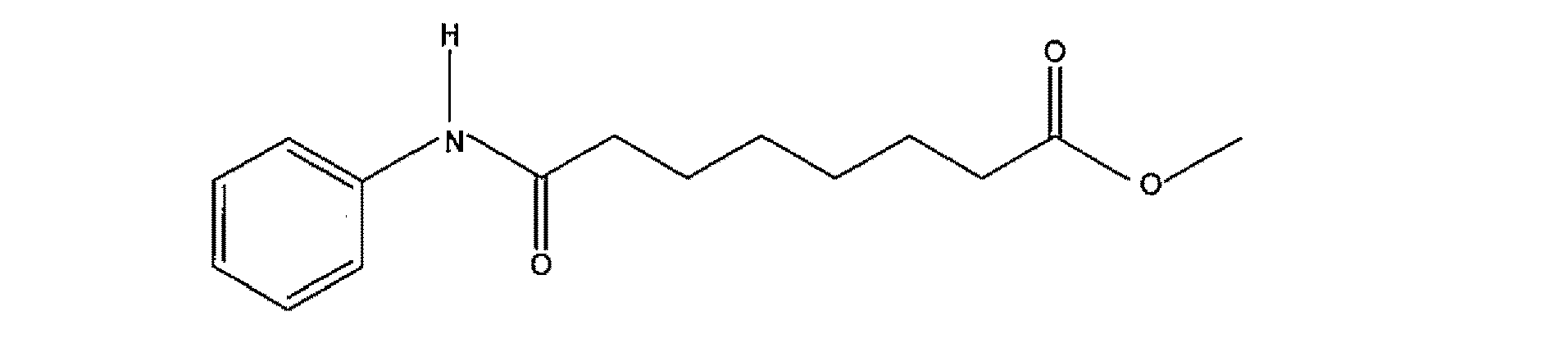
本発明の組成物においての使用に適切なHDACインヒビターの他の非限定的な例は、以下である:
以下の構造によって表されるピロキサミド:
以下の構造によって表されるピロキサミド:
以下の構造式によって表される化合物:
ここで、Rは、置換または非置換の、フェニル、ピペリジン、チアゾール、2−ピリジン、3−ピリジンまたは4−ピリジンであり、そしてnは、約4〜約8の整数である。
以下の構造式によって表される化合物:
さらに、本発明の特定の実施形態に従って、ヒストンデアセチラーゼ(HDAC)インヒビターまたはその薬学的に受容可能な塩または水和物;キャリアまたは希釈剤としての微結晶セルロース;崩壊剤としてのクロスカルメロース;および潤滑剤としてのステアリン酸マグネシウムを含む経口投与のための薬学的組成物が提供され;ここで、この組成物は、投与後少なくとも2時間の期間にわたって、インビボでヒストンデアセチラーゼを阻害するのに有効な平均血漿濃度のHDACインヒビターを提供する。好ましい実施形態において、HDACインヒビターは、スベロイルアニリドヒドロキサム酸(SAHA)である。
さらに、本発明の特定の実施形態に従って、スベロイルアニリドヒドロキサム酸(SAHA)またはその薬学的に受容可能な塩もしくは水和物;キャリアまたは希釈剤としての微結晶セルロース;崩壊剤としてのクロスカルメロース;および潤滑剤としてのステアリン酸マグネシウムを含む経口投与のための薬学的組成物が提供され;ここで、この組成物は、投与後少なくとも2時間の期間にわたって、インビボでヒストンデアセチラーゼを阻害するのに有効な平均血漿濃度のHDACインヒビターを提供する。別の好ましい実施形態において、この組成物は、50〜70重量%のSAHAまたはその薬学的に受容可能な塩もしくは水和物;キャリアまたは希釈剤としての20〜40重量%の微結晶セルロース;5〜15重量%の崩壊剤としてのクロスカルメロース;および0.1〜5重量%の潤滑剤としてのステアリン酸マグネシウムを含む。別の好ましい実施形態において、この組成物は、約50〜200mgのSAHAを含む。特に好ましい実施形態において、この組成物は、ゼラチンカプセルの形態である。
本発明は、さらに、これらの処方物の安全な毎日の投薬レジメンを提供し、これは、従いそして固守するのが容易である。本発明の処方物は、最終分化、細胞増殖停止および/または新生物細胞のアポトーシスを選択的に誘導するために有用であり、従って、患者における腫瘍の処置を補助する。
本発明の上記および他の目的、特徴および利点は、添付の図面(ここで、類似の参照文字は、異なる図面全体にわたって同じ部分をいう)に示されるように、本発明の好ましい実施形態の以下のより詳細な説明から明らかである。図面は、必ずしも一定の比率ではなく、その代わり、本発明の原理を説明する際に強調がなされる。
(発明の詳細な説明)
本発明は、投与後の少なくとも2時間の期間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のヒストンデアセチラーゼ(HDAC)を生成する方法を提供し、この方法は、この被験体に、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む有効量の薬学的組成物を投与する工程を包含する。
本発明は、投与後の少なくとも2時間の期間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のヒストンデアセチラーゼ(HDAC)を生成する方法を提供し、この方法は、この被験体に、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む有効量の薬学的組成物を投与する工程を包含する。
本発明はまた、最終分化、細胞増殖停止および/または新生物細胞のアポトーシスを選択的に誘導し、それによって、このような細胞の増殖を阻害するための方法、ならびに被験体に、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む有効量の薬学的組成物を投与することによって、投与後の少なくとも2時間の期間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のヒストンデアセチラーゼ(HDAC)を生成することより腫瘍細胞の分化を誘導するための方法を提供する。
本発明は、さらに、投与後の少なくとも2時間の期間にわたって、被験体においてインビボで、少なくとも約10nMの平均血漿濃度のスベロイルアニリドヒドロキサム酸(SAHA)を生成する方法を提供し、この方法は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む有効量の薬学的組成物を被験体に投与する工程を包含する。
本発明はまた、最終分化、細胞増殖停止および/または新生物細胞のアポトーシスを選択的に誘導し、それによって、このような細胞の増殖を阻害するための方法、ならびに被験体に、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む有効量の薬学的組成物を投与することによって、投与後の少なくとも2時間の期間にわたって、被験体においてインビボで少なくとも10nMの平均血漿濃度のSAHAを生成することより腫瘍細胞の分化を誘導するための方法を提供する。
本発明は、経口投与に適切な薬学的組成物を提供し、この組成物は、最終分化、細胞増殖停止および/または新生物細胞のアポトーシスを選択的に誘導するのに有用な化合物、ならびに/あるいはヒストンデアセチラーゼ(HDAC)の強力なインヒビターである化合物を含む。薬学的組成物は、さらに、微結晶セルロース、クロスカルメロースナトリウムおよびステアリン酸マグネシウムから構成される。本発明はまた、SAHA、微結晶セルロース、クロスカルメロースナトリウムおよびステアリン酸マグネシウムを含む、経口投与のための薬学的組成物を提供する。本発明の処方物の活性化合物の経口バイオアベイラビリティーは、驚くほど高い。さらに、この処方物は、長期間にわたって、活性化合物の高い治療的に有効な血液レベルに予期せず上昇する。本発明はさらに、これらの処方物の安全な毎日の投薬レジメンを提供し、これは、従い、そして固守しやすい。
本発明の処方物中の活性化合物の経口バイオアベイラビリティーは、驚くほど高い。さらに、この処方物は、長期間にわたって、活性化合物の高い治療的に有効な血液レベルに予期せず上昇する。本発明はさらに、これらの処方物の安全な毎日の投薬レジメンを提供し、これは、従い、そして固守しやすく、インビボで、記載された化合物の治療的有効量に上昇する。本発明の処方物は、最終分化、細胞増殖停止および/または新生物細胞のアポトーシスを選択的に誘導するのに有用であり、従って、患者において腫瘍の処置を補助する。
本明細書中で実証されるように、本発明で提供される薬学的組成物は、最初の平均血漿濃度(すなわち、処方物の投与直後に得られる濃度)に上昇し、これは、長期間にわたって、予期せず高いままである。同じ投薬量を有する親処方物(例えば、IV処方物)(ここで、この活性化合物は、ほとんどすぐに、排除される)と比較した場合、経口組成物は、長期間(少なくとも2時間、より代表的には、少なくとも10時間または12時間)にわたって、高い平均血漿濃度の活性化合物を保持する。代表的に、経口投薬処方物の平均血漿濃度は、12時間またはそれより長い時間の間、初期の平均血漿濃度の50%未満に低下しない。
本発明の発見まで、本明細書中に記載されるHDACインヒビターの静脈内投与は、最も効果的であることが証明されている。この化合物の静脈内投与は、連続的に(すなわち、毎日)、長期の期間(例えば、少なくとも3日間、好ましくは5日より長く)実施されなければならない。これは、明らかに、この処置を受ける患者に対して、重い負荷を与える。本発明の予期しない驚くべき発見によって、IV注入により、この薬物を連続的に投与する必要性無しに、インビボで、活性化合物の高い安定したレベルに上昇する経口投薬形態を処方することが可能であり、これは、この処置を受ける患者に莫大な利点を提供する。
従って、本発明は、ヒストンデアセチラーゼ(HDAC)インヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能な塩もしくは水和物を含む、経口投与のための薬学的組成物を提供し、ここで、この組成物は、投与後、少なくとも2時間の間、インビボでヒストンデアセチラーゼ(HDAC)を阻害するのに有効な平均血漿濃度のHDACインヒビターを提供する。好ましい実施形態において、HDACインヒビターの濃度は、投与後、少なくとも8時間の間、HDACを阻害するのに有効である。別の好ましい実施形態において、HDACインヒビターの濃度は、投与後、少なくとも10時間の間、HDACを阻害するのに有効である。別の実施形態において、HDACインヒビターの濃度は、投与後、少なくとも12時間の間、HDACを阻害するのに有効である。
好ましい実施形態において、本発明は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、経口投与のための薬学的組成物を提供し、ここで、この組成物は、投与後、少なくとも2時間の間、インビボでヒストンデアセチラーゼ(HDAC)を阻害するのに有効な平均血漿濃度のSAHAを提供する。好ましい実施形態において、SAHAの濃度は、投与後、少なくとも8時間の間、HDACを阻害するのに有効である。別の好ましい実施形態において、SAHAの濃度は、投与後、少なくとも10時間の間、HDACを阻害するのに有効である。別の好ましい実施形態において、SAHAの濃度は、投与後、少なくとも12時間の間、HDACを阻害するのに有効である。
本発明の処方物は、最終分化、細胞増殖停止および/または新生物細胞のアポトーシスを選択的に誘導するために有用であり、従って、患者において腫瘍の処置を補助する。
従って、本発明はまた、被験体において、新生物細胞の最終分化を選択的に誘導し、そしてそれによって、被験体において、このような細胞の増殖を阻害する方法を提供し、この方法は、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアもしくは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによって、投与後、少なくとも2時間の期間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のHDACインヒビターを生成する工程を包含する。
さらに、本発明はまた、被験体において新生物細胞の細胞増殖停止を選択的に誘導し、それによって、被験体においてこのような細胞の増殖を阻害する方法を提供し、この方法は、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアもしくは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによって、投与後、少なくとも2時間の期間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のHDACインヒビターを生成する工程を包含する。
さらに、本発明はまた、被験体において新生物細胞のアポトーシスを選択的に誘導し、それによって、被験体においてこのような細胞の増殖を阻害する方法を提供し、この方法は、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアもしくは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによって、投与後、少なくとも2時間の期間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のHDACインヒビターを生成する工程を包含する。
さらに、本発明は、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することにより、投与の後少なくとも2時間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る、平均血漿濃度のHDACインヒビターを生成することによる、腫瘍を有する被験体中の腫瘍細胞の分化を誘導する方法もまた、提供する。
さらに、本発明は、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによる投与の後少なくとも2時間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のHDACインヒビターを生成する工程を包含する、被験体においてヒストンデアセチラーゼの活性を阻害する方法を提供する。
さらに、本発明は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによる投与の後少なくとも2時間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のSAHAを生成する工程を包含する、被験体における新生物細胞の最終的分化、増殖停止および/またはアポトーシスを選択的に誘導し、それによって、被験体中のこのような細胞の増殖を阻害する方法もまた、提供する。
さらに、本発明は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによる投与の後少なくとも2時間にわたって、被験体においてインビボで少なくとも10nMのSAHA平均血漿濃度を生成する工程を包含する、被験体における新生物細胞の最終的分化、増殖停止および/またはアポトーシスを選択的に誘導し、それによって、被験体中のこのような細胞の増殖を阻害する方法もまた、提供する。
さらに、本発明は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによる投与の後少なくとも2時間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得る平均血漿濃度のSAHAを生成する工程を包含する、腫瘍を有する被験体中の腫瘍細胞の分化を誘導する方法もまた提供する。
さらに、本発明は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによる投与の後少なくとも2時間にわたって、被験体においてインビボで少なくとも10nMのSAHA平均血漿濃度を生成する工程を包含する、腫瘍を有する被験体中の腫瘍細胞の分化を誘導する方法もまた提供する。
さらに、本発明は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによる投与の後少なくとも2時間にわたって、被験体においてインビボでヒストンデアセチラーゼを阻害し得るSAHA平均血漿濃度を生成する工程を包含する、被験体においてヒストンデアセチラーゼの活性を阻害する方法を提供する。
さらに、本発明は、SAHAまたはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは希釈剤を含む、有効量の薬学的組成物をこの被験体に投与することによる投与の後少なくとも2時間にわたって、被験体においてインビボで少なくとも10nMのSAHA平均血漿濃度を生成する工程を包含する、被験体においてヒストンデアセチラーゼの活性を阻害する方法を提供する。
別の好ましい実施形態において、この組成物は、投与後少なくとも2時間にわたってヒストンデアセチラーゼを阻害し得るHDACインヒビター(例えば、SAHA)の平均血漿濃度を提供し、これは、好ましくは、少なくとも約10nMの濃度である。別の実施形態において、この組成物は、投与後少なくとも8時間にわたって少なくとも約10nMの平均血漿濃度のHDACインヒビターを提供する。なお別の好ましい実施形態において、この組成物は、投与後少なくとも10時間にわたって少なくとも約10nMの平均血漿濃度のHDACインヒビターを提供する。なお別の好ましい実施形態において、この組成物は、投与後少なくとも12時間にわたって少なくとも約10nMの平均血漿濃度のHDACインヒビターを提供する。平均血漿濃度の非限定的な例は、約10nM、25nM、40nM、45nM、50nM、100nM、1μM、2μM、2.5μM、5μM、10μM、25μM、50μM、100μMなどである。これらの用量は、本発明の範囲を如何様にも限定せず、そしてヒストンデアセチラーゼを阻害し得る任意の平均血漿濃度が適切であることは、当業者に明らかであるはずである。
1つの好ましい実施形態において、この組成物は、新生物細胞の最終分化、細胞増殖停止および/もしくはアポトーシスを選択的に誘導し得るか、または腫瘍中の腫瘍細胞の分化を誘導し得る、平均血漿濃度のHDACインヒビター(例えば、SAHA)を提供し、ここで、この濃度は、投与後少なくとも2時間にわたって維持され、これは、好ましくは少なくとも約2.5μMの濃度である。別の実施形態において、この組成物は、投与後少なくとも8時間にわたって少なくとも約2.5μMの平均血漿濃度のHDACインヒビターを提供する。なお別の好ましい実施形態において、この組成物は、投与後少なくとも10時間にわたって少なくとも約2.5μMの平均血漿濃度のHDACインヒビターを提供する。なお別の好ましい実施形態において、この組成物は、投与後少なくとも12時間にわたって少なくとも約2.5μMの平均血漿濃度のHDACインヒビターを提供する。平均血漿濃度の非限定的な例は、約10nM、25nM、40nM、45nM、50nM、100nM、1μM、2μM、2.5μM、5μM、10μM、25μM、50μM、100μMなどである。これらの用量は、本発明の範囲を如何様にも限定せず、そして新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを誘導し得る任意の平均血漿濃度が適切であることは、当業者に明らかであるはずである。
別の好ましい実施形態において、この組成物は、腫瘍を有する被験体において腫瘍細胞の分化を誘導するのに有効な、平均血漿濃度のHDACインヒビター(例えば、SAHA)を提供し、ここで、この量は、被験体に投与された後に少なくとも2時間にわたって維持される。別の好ましい実施形態において、この組成物は、腫瘍を有する被験体において腫瘍細胞の分化を誘導するのに有効な、HDACインヒビターの平均血漿濃度を提供し、ここで、この量は、被験体に投与された後に少なくとも8時間にわたって維持される。別の好ましい実施形態において、この組成物は、腫瘍を有する被験体において腫瘍細胞の分化を誘導するのに有効な、平均血漿濃度のHDACインヒビターを提供し、ここで、この量は、被験体に投与された後に少なくとも10時間にわたって維持される。平均血漿濃度の非限定的な例は、約10nM、25nM、40nM、45nM、50nM、100nM、1μM、2μM、2.5μM、5μM、10μM、25μM、50μM、100μMなどである。これらの用量は、本発明の範囲を如何様にも限定せず、そして腫瘍中の腫瘍細胞の分化を誘導し得る任意の平均血漿濃度が適切であることは、当業者に明らかであるはずである。
本発明の方法は、インビトロおよびインビボでの実施に適切である。これらの方法がインビトロで実施される場合、細胞をこの化合物と共にインキュベートすることによって、接触がもたらされ得る。細胞と接触した化合物の濃度は、約1nM〜約25mM、例えば、約10nM〜約1mM、約40nM〜約0.5mMであるべきである。特定の用量の非限定的な例は、10nM、25nM、40nM、45nM、50nM、100nM、1μM、2μM、2.5μM、5μM、10μM、25μM、50μM、100μMなどである。この濃度は、個々の化合物および新生物細胞の状態に依存する。
本発明の方法はインビトロで実施され得るが、新生物細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導する方法についての好ましい実施形態は、インビボで細胞を接触させる工程(すなわち、処置を必要とする、新生物細胞または腫瘍細胞を有する被験体に、この化合物を投与することによる)を包含することが、企図される。
本発明の方法はまた、被験体中の新生物細胞を、抗腫瘍剤に対して耐性にするように、抗腫瘍剤を被験体に最初に投与する工程、ならびに、このような細胞の最終分化、細胞増殖停止および/またはアポトーシスを選択的に誘導するのに有効な、本発明の任意の組成物の有効量を引き続いて投与する工程を包含し得る。
抗腫瘍剤は、多数の化学療法剤(例えば、アルキル化剤、代謝拮抗物質、ホルモン剤、抗生物質、コルヒチン、ビンカアルカロイド、L−アスパラギナーゼ、プロカルバジン、ヒドロキシウレア、ミトーテン、ニトロソウレアまたはイミダゾールカルボキサミド)の1つであり得る。適切な薬剤は、チューブリンの脱分極を促進する薬剤である。好ましくは、抗腫瘍剤は、コルヒチンまたはビンカアルカロイドであり;ビンブラスチンおよびビンクリスチンが特に好ましい。抗腫瘍剤がビンリスチンである実施形態において、これらの細胞は、好ましくは、約5mg/mlの濃度のビンクリスチンに対して耐性であるように、処理される。これらの細胞を抗腫瘍剤に対して耐性にする細胞処理は、少なくとも3〜5日間にわたって、細胞をこの薬剤と接触させることによってもたらされ得る。得られた細胞を、上記の任意の化合物と接触させることは、上記のように実施される。上記の化学療法剤に加えて、これらの化合物は、放射線療法と一緒にも投与され得る。
本発明はまた、新生物細胞の増殖によって特徴づけられる腫瘍を有する患者を処置する方法を提供し、この方法は、このような新生物細胞の最終分化を選択的に誘導し、それによって、これらの細胞の増殖を阻害するのに有効な、上記の本発明の任意の組成物の有効量を、この患者に投与する工程を包含する。
本発明の方法は、腫瘍を有するヒト患者の処置のために意図される。しかし、この方法は、他の哺乳動物における腫瘍の処置において有効である可能性もある。用語腫瘍は、新生物細胞の増殖によって引き起こされる任意の癌(例えば、肺癌、急性リンパ性骨髄腫、ホジキンリンパ腫、非ホジキンリンパ腫、膀胱黒色腫、腎臓癌腫、乳房癌腫、前立腺癌腫、卵巣癌種または結腸直腸癌腫)を含むことが意図される。
薬学的組成物の投与は、1日1回、1日2回、1日3回などで経口投与され得る、単位投薬で実施され得る。現在好ましい実施形態は、1日1回の投与、1日2回の投与および1日3回の投与である。
(ヒストンデアセチラーゼおよびヒストンデアセチラーゼインヒビター)
ヒストンデアセチラーゼ(HDAC)は、この用語が本明細書中で使用される場合、ヌクレオソームコアヒストンのアミノ末端テイル中のリジン残基からのアセチル基の除去を触媒する酵素である。それ自体、HDACは、ヒストンアセチルトランスフェラーゼ(HAT)と共に、ヒストンのアセチル化状態を調節する。ヒストンアセチル化は、HDACの遺伝子発現およびインヒビターに影響を与える(例えば、ヒドロキサム酸ベースのハイブリッド極性化合物であるスベロイルアニリドヒドロキサム酸(suberoylanilide hydroxamic acid)(SAHA)は、形質転換した細胞の増殖停止、分化および/またはアポトーシスをインビトロで誘導し、そして腫瘍増殖をインビボで阻害する)。HDACは、構造的相同性に基づいて3つのクラスに分割され得る。クラスI HDAC(HDAC1、2、3および8)は、酵母RPD3タンパク質に対する類似性を保有し、核に位置し、そして転写共リプレッサーと関連して複合体中に見出される。クラスII HDAC(HDAC4、5、6、7および9)は、酵母HDA1タンパク質と類似であり、核および細胞質の両方の細胞内局在を有する。クラスIおよびクラスIIの両方のHDACは、ヒドロキサム酸ベースのHDACインヒビター(例えば、SAHA)によって阻害される。クラスIII HDACは、酵母SIR2タンパク質に関連するNAD依存性酵素の構造的に遠いクラスを形成し、そしてヒドロキサム酸ベースのHDACインヒビターによって阻害されない。
ヒストンデアセチラーゼ(HDAC)は、この用語が本明細書中で使用される場合、ヌクレオソームコアヒストンのアミノ末端テイル中のリジン残基からのアセチル基の除去を触媒する酵素である。それ自体、HDACは、ヒストンアセチルトランスフェラーゼ(HAT)と共に、ヒストンのアセチル化状態を調節する。ヒストンアセチル化は、HDACの遺伝子発現およびインヒビターに影響を与える(例えば、ヒドロキサム酸ベースのハイブリッド極性化合物であるスベロイルアニリドヒドロキサム酸(suberoylanilide hydroxamic acid)(SAHA)は、形質転換した細胞の増殖停止、分化および/またはアポトーシスをインビトロで誘導し、そして腫瘍増殖をインビボで阻害する)。HDACは、構造的相同性に基づいて3つのクラスに分割され得る。クラスI HDAC(HDAC1、2、3および8)は、酵母RPD3タンパク質に対する類似性を保有し、核に位置し、そして転写共リプレッサーと関連して複合体中に見出される。クラスII HDAC(HDAC4、5、6、7および9)は、酵母HDA1タンパク質と類似であり、核および細胞質の両方の細胞内局在を有する。クラスIおよびクラスIIの両方のHDACは、ヒドロキサム酸ベースのHDACインヒビター(例えば、SAHA)によって阻害される。クラスIII HDACは、酵母SIR2タンパク質に関連するNAD依存性酵素の構造的に遠いクラスを形成し、そしてヒドロキサム酸ベースのHDACインヒビターによって阻害されない。
ヒストンデアセチラーゼインヒビターすなわちHDACインヒビターは、この用語が本明細書中で使用される場合、インビボ、インビトロまたはその両方で、ヒストンの脱アセチル化を阻害し得る化合物である。それ自体、HDACインヒビターは、少なくとも1つのヒストンデアセチラーゼの活性を阻害する。少なくとも1つのヒストンの脱アシル化を阻害することの結果として、アセチル化ヒストンの増大が生じ、そしてアセチル化ヒストンの蓄積は、HDACインヒビターの活性を評価するのに適切な生物学的マーカーである。従って、アセチル化ヒストンの蓄積について評価し得る手順は、目的の化合物のHDAC阻害活性を決定するために使用され得る。ヒストンデアセチラーゼ活性を阻害し得る化合物はまた、他の基質に結合し得、それ自体、酵素のような他の生物学的に活性な分子を阻害し得ることが、理解される。本発明の化合物が上記のヒストンデアセチラーゼのいずれか、または任意の他のヒストンデアセチラーゼを阻害し得ることもまた、理解されるべきである。
例えば、HDACインヒビターを受ける患者において、末梢血単核球細胞になあらびにHDACインヒビターで処置された組織におけるアセチル化ヒストンの蓄積は、適切なコントロールに対して決定され得る。
特定の化合物のHDAC阻害活性は、例えば、少なくとも1つのヒストンデアセチラーゼの阻害を示す酵素アッセイを使用して、インビトロで決定され得る。さらに、特定の組成物で処理した細胞中のアセチル化ヒストンの蓄積の決定は、化合物のHDAC阻害活性の決定であり得る。
アセチル化ヒストンの蓄積についてのアッセイは、文献中で周知である。例えば、Marks,P.A.ら、J.Natl.Cancer Inst.,92:1210−1215,2000,Butler,L.M.ら、Cancer Res.60:5165−5170(2000),Richon,V.M.ら、Proc.Natl.Acad.Sci.,USA,95:3003−3007,1998,およびYoshida,M.ら、J.Biol.Chem265:17174−17179,1990を参照のこと。
例えば、ヒストンデアセチラーゼインヒビター化合物の活性を決定するための酵素アッセイは、以下のように実施され得る。簡潔には、アフィニティ精製したヒトエピトープタグ化(Flag)HDAC1に対するHDACインヒビター化合物の効果は、示された量のインヒビター化合物と共に、基質の非存在下で氷上で20分間酵素調製物をインキュベートすることによって、アッセイされ得る。基質([3H]アセチル標識化マウス赤白血病細胞由来ヒストン)が添加され得、そしてこのサンプルは、30μlの総容量で、37℃で20分間インキュベートされ得る。次いで、この反応が停止され得、そして放出されたアセテートが抽出され得、そして放射能放出量がシンチレーション計数によって決定される。ヒストンデアセチラーゼインヒビター化合物の活性を決定するために有用な代替的アッセイは、BIOMOL(登録商標)Research Laboratories,Inc.,Plymouth Meeting,PAから入手可能な、「HDAC Fluorescent Activity Assay;Drug Discovery Kit−AK−500」である。
インビボ研究は、以下のようにして実施され得る。動物(例えば、マウス)に、HDACインヒビター化合物を腹腔内注射し得る。選択された組織(例えば、脳、脾臓、肝臓など)は、投与後の所定の時間で単離され得る。ヒストンは、Yoshidaら、J.Biol.Chem265:17174−17179,1990に本質的に記載されるように、これらの組織から単離され得る。等量のヒストン(約1μg)は、15%のSDS−ポリアクリルアミドゲルで電気泳動され得、そしてHybond−Pフィルタ(Amershamから入手可能)に移され得る。フィルタを、3%ミルクでブロッキングし得、そして精製ウサギポリクローナル抗アセチル化ヒストンH4抗体(αAc−H4)および抗アセチル化ヒストンH3抗体(αAc−H3)(Upstate Biotechnology,Inc.)を用いて探索し得る。アセチル化ヒストンのレベルは、西洋ワサビペルオキシダーゼ結合体化ヤギ抗ウサギ抗体(1:5000)およびSuperSignal化学発光基質(Pierce)を使用して、可視化され得る。ヒストンタンパク質についての充填コントロールとして、平行ゲルを泳動させ得、そしてクマシーブルー(CB)で染色し得る。
さらに、ヒドロキサム酸ベースのHDACインヒビターは、p21WAF1遺伝子の発現をアップレギュレートすることが示されている。p21WAF1タンパク質は、標準的な方法を使用して、種々の形質転換細胞中においてHDACインヒビターとの培養の2時間以内に、誘導される。p21WAF1遺伝子の誘導は、この遺伝子のクロマチン領域中のアセチル化ヒストンの蓄積に関連する。従って、p21WAF1の誘導は、形質転換細胞においてHDACインヒビターによって引き起こされるG1細胞周期停止に関与すると認識され得る。
代表的に、HDACインヒビターは、5つの一般的クラスに入る:1)ヒドロキサム酸誘導体;2)短鎖脂肪酸(SCFA);3)環状テトラペプチド;4)ベンズアミド;および5)求電子性ケトン。
従って、本発明は、その広範な範囲内に、ヒストンデアセチラーゼを阻害し、新生物細胞における最終分化を誘導し、そして/または腫瘍中の腫瘍細胞の分化を誘導する際に使用するための、1)ヒドロキサム酸誘導体;2)短鎖脂肪酸(SCFA);3)環状テトラペプチド;4)ベンズアミド;5)求電子性ケトン;および/またはヒストンデアセチラーゼを阻害し得る任意の他のクラスの化合物である、HDACインヒビターを含む組成物を包含する。
このようなHDACインヒビターの例としては、以下が挙げられるが、これらに限定されない:
(A.ヒドロキサム誘導体)
例えば、スベロイルアニリドヒドロキサム酸(SAHA)(Richonら、Proc.Natl.Acad.Sci.USA 95,3003−3007(1998));m−カルボキシ桂皮酸ビスヒドロキサミド(CBHA)(Richonら、前出);ピロキサミド;トリコスタチン(trichostatin)アナログ(例えば、トリコスタチンA(TSA)およびトリコスタチンC(Kogheら、1998.Biochem.Pharmacol.56:1359−1364));サリチルヒドロキサム酸(SBHA)(Andrewsら、International J.Parasitology 30,761−768(2000));スベロイルビスヒドロキサム酸(SBHA)(米国特許第5,608,108号);アゼライック(azelaic)ビスヒドロキサム酸(ABHA)(Andrewsら、前出);アゼライック−1−ヒドロキサメート−9−アニリド(AAHA)(Qiuら、Mol.Biol.Cell 11,2069−2083(2000));6−(3−クロロフェニルウレイド)カプロニックヒドロキサム酸(3C1−UCHA);オキサムフラチン[(2E)−5−[3−[(フェニルスルホニル)アミノールフェニル]−ペンタ−2−エン−4−イノヒドロキサム酸](oxamflatin[(2E)−5−[3−[(phenylsufonyl)aminol phenyl]−pent−2−en−4−ynohydroxamic acid])(Kimら、Oncogene,18:2461−2470(1999));A−161906,Scriptaid(Suら、2000 Cancer Research,60:3137−3142);PXD−101(Prolifix);LAQ−824;CHAP;MW2796(Andrewsら、前出);MW2996(Andrewsら、前出);または米国特許第5,369,108号、同第5,932,616号、同第5,700,811号、同第6,087,367号および同第6,511,990号に開示されるようなヒドロキサム酸のいずれか。
(A.ヒドロキサム誘導体)
例えば、スベロイルアニリドヒドロキサム酸(SAHA)(Richonら、Proc.Natl.Acad.Sci.USA 95,3003−3007(1998));m−カルボキシ桂皮酸ビスヒドロキサミド(CBHA)(Richonら、前出);ピロキサミド;トリコスタチン(trichostatin)アナログ(例えば、トリコスタチンA(TSA)およびトリコスタチンC(Kogheら、1998.Biochem.Pharmacol.56:1359−1364));サリチルヒドロキサム酸(SBHA)(Andrewsら、International J.Parasitology 30,761−768(2000));スベロイルビスヒドロキサム酸(SBHA)(米国特許第5,608,108号);アゼライック(azelaic)ビスヒドロキサム酸(ABHA)(Andrewsら、前出);アゼライック−1−ヒドロキサメート−9−アニリド(AAHA)(Qiuら、Mol.Biol.Cell 11,2069−2083(2000));6−(3−クロロフェニルウレイド)カプロニックヒドロキサム酸(3C1−UCHA);オキサムフラチン[(2E)−5−[3−[(フェニルスルホニル)アミノールフェニル]−ペンタ−2−エン−4−イノヒドロキサム酸](oxamflatin[(2E)−5−[3−[(phenylsufonyl)aminol phenyl]−pent−2−en−4−ynohydroxamic acid])(Kimら、Oncogene,18:2461−2470(1999));A−161906,Scriptaid(Suら、2000 Cancer Research,60:3137−3142);PXD−101(Prolifix);LAQ−824;CHAP;MW2796(Andrewsら、前出);MW2996(Andrewsら、前出);または米国特許第5,369,108号、同第5,932,616号、同第5,700,811号、同第6,087,367号および同第6,511,990号に開示されるようなヒドロキサム酸のいずれか。
(B.環状テトラペプチド)
例えば、トラポキシン(trapoxin)A(TPX)−環状テトラペプチド(シクロ−(L−フェニルアラニル−L−フェニルアラニル−D−ピペコリニル−L−2−アミノ−8−オキソ−9,10−エポキシデカノイル))(Kijimaら、J Biol.Chem268,22429−22435(1993));FR901228(FK 228,デプシペプチド)(Nakajimaら、Ex.Cell Res.241,126−133(1998));FR225497環状テトラペプチド(H.Moriら、PCT出願WO00/08048(2000年2月17日));アピシジン(apicidin)環状テトラペプチド[シクロ(N−O−メチル−L−トリプトファニル−L−イソロイシニル−D−ピペコリニル−L−2−アミノ−8−オキソデカノイル)](Darkin−Rattrayら、Proc.Natl.Acad.Sci.USA 93,13143−13147(1996));アピシジンIa、アピシジンIb、アピシジンIc、アピシジンIIaおよびアピシジンIIb(P.Dulskiら、PCT出願WO97/11366);CHAP、HC−毒素環状テトラペプチド(Boschら、Plant Cell 7,1941−1950(1995));WF27082環状テトラペプチド(PCT出願WO98/48825);およびクラミドシン(chlamydocin)(Boschら、前出)。
例えば、トラポキシン(trapoxin)A(TPX)−環状テトラペプチド(シクロ−(L−フェニルアラニル−L−フェニルアラニル−D−ピペコリニル−L−2−アミノ−8−オキソ−9,10−エポキシデカノイル))(Kijimaら、J Biol.Chem268,22429−22435(1993));FR901228(FK 228,デプシペプチド)(Nakajimaら、Ex.Cell Res.241,126−133(1998));FR225497環状テトラペプチド(H.Moriら、PCT出願WO00/08048(2000年2月17日));アピシジン(apicidin)環状テトラペプチド[シクロ(N−O−メチル−L−トリプトファニル−L−イソロイシニル−D−ピペコリニル−L−2−アミノ−8−オキソデカノイル)](Darkin−Rattrayら、Proc.Natl.Acad.Sci.USA 93,13143−13147(1996));アピシジンIa、アピシジンIb、アピシジンIc、アピシジンIIaおよびアピシジンIIb(P.Dulskiら、PCT出願WO97/11366);CHAP、HC−毒素環状テトラペプチド(Boschら、Plant Cell 7,1941−1950(1995));WF27082環状テトラペプチド(PCT出願WO98/48825);およびクラミドシン(chlamydocin)(Boschら、前出)。
(C.短鎖脂肪酸(SCFA)誘導体)
例えば、酪酸ナトリウム(Cousensら、J.Biol.Chem254,1716−1723(1979));イソバレレート(McBainら、Biochem.Pharm.53:1357−1368(1997));バレレート(McBainら、前出);4−フェニルブチレート(4−PBA)(LeaおよびTulsyan,Anticancer Research,15,879−873(1995));フェニルブチレート(PB)(Wangら、Cancer Research,59,2766−2799(1999));プロピオネート(McBainら、前出);ブチルアミド(LeaおよびTulsyan、前出);イソブチルアミド(LeaおよびTulsyan、前出);フェニルアセテート(LeaおよびTulsyan、前出);3−ブロモプロピオネート(LeaおよびTulsyan、前出);トリブチリン(Guanら、Cancer Research,60,749−755(2000));バルプロ酸およびバルプロエート。
例えば、酪酸ナトリウム(Cousensら、J.Biol.Chem254,1716−1723(1979));イソバレレート(McBainら、Biochem.Pharm.53:1357−1368(1997));バレレート(McBainら、前出);4−フェニルブチレート(4−PBA)(LeaおよびTulsyan,Anticancer Research,15,879−873(1995));フェニルブチレート(PB)(Wangら、Cancer Research,59,2766−2799(1999));プロピオネート(McBainら、前出);ブチルアミド(LeaおよびTulsyan、前出);イソブチルアミド(LeaおよびTulsyan、前出);フェニルアセテート(LeaおよびTulsyan、前出);3−ブロモプロピオネート(LeaおよびTulsyan、前出);トリブチリン(Guanら、Cancer Research,60,749−755(2000));バルプロ酸およびバルプロエート。
(D.ベンズアミド誘導体)
例えば、CI−994;MS−27−275[N−(2−アミノフェニル)−4−[N−(ピリジン−3−イルメトキシカルボニル)アミノメチル]ベンズアミド](Saitoら、Proc.Natl.Acad.Sci.USA 96,4592−4597(1999));およびMS−27−275の3’−アミノ誘導体(Saitoら、前出)。
例えば、CI−994;MS−27−275[N−(2−アミノフェニル)−4−[N−(ピリジン−3−イルメトキシカルボニル)アミノメチル]ベンズアミド](Saitoら、Proc.Natl.Acad.Sci.USA 96,4592−4597(1999));およびMS−27−275の3’−アミノ誘導体(Saitoら、前出)。
(E.求電子性ケトン誘導体)
例えば、トリフルオロメチルケトン(Freyら、Bioorganic&Med.Chem.Lett.(2002),12,3443−3447;米国特許第6,511,990号)およびα−ケトアミド(例えば、N−メチル−α−ケトアミド)。
例えば、トリフルオロメチルケトン(Freyら、Bioorganic&Med.Chem.Lett.(2002),12,3443−3447;米国特許第6,511,990号)およびα−ケトアミド(例えば、N−メチル−α−ケトアミド)。
(F.他のHDACインヒビター)
例えば、デプデシン(depudecin)(Kwonら、1998.PNAS 95:3356−3361)。
例えば、デプデシン(depudecin)(Kwonら、1998.PNAS 95:3356−3361)。
好ましいヒドロキサム酸ベースのHDACインヒビターは、スベロイルアニリドヒドロキサム酸(SAHA)、m−カルボキシ桂皮酸ビスヒドロキサメート(CBHA)およびピロキサミドである。SAHAは、ヒストンデアセチラーゼ酵素の触媒ポケット中に直接結合することが示されている。SAHAは、培養物中の形質転換細胞の細胞周期停止、分化および/またはアポトーシスを誘導し、そしてげっ歯類において腫瘍増殖を阻害する。SAHAは、固形腫瘍および血液学的癌の両方においてこれらの影響を誘導する際に有効である。SAHAは、動物に対する毒性なしに、動物において腫瘍増殖を阻害する際に有効であることが示されている。腫瘍増殖のSAHA誘導性阻害は、腫瘍におけるアセチル化ヒストンの蓄積に関連する。SAHAは、ラットにおける発癌物質(N−メチルニトロソウレア)誘導性の乳腺腫瘍の発達および継続的な増殖を阻害する際に有効である。SAHAは、130日間の研究期間にわたって、ラットの食餌中で、ラットに投与された。従って、SAHAは、非毒性の経口的に活性な抗腫瘍剤であり、その作用機構は、ヒストンデアセチラーゼ活性の阻害を含む。
好ましいHDACインヒビターは、本発明者らが開示した化合物のいくつかに対して発行された米国特許第5,369,108号、同第5,932,616号、同第5,700,811号、同第6,087,367号および同第6,511,990号に開示される化合物であり、これらの米国特許の全内容は、本明細書中で参考として援用され、その非限定的な例は、以下に示される:
従って、1実施形態において、本発明は、式1の構造:
従って、1実施形態において、本発明は、式1の構造:
式1の特定の実施形態において、R1およびR2は同じであり、そして置換または非置換のチアゾールアミノ基であり;そしてnは、約4〜約8の整数である。
別の実施形態において、本発明は、式2の構造:
式2の特定の実施形態において、R3およびR4の各々は、独立して、互いに同じかまたは異なり、そして水素原子、ヒドロキシル基、置換または非置換の、分枝または非分枝の、アルキル基、アルケニル基、シクロアルキル基、アリール基、アルキルオキシ基、アリールオキシ基、アリールアルキルオキシ基またはピリジン基であるか、あるいは、R3およびR4は、一緒に結合して、ピペリジン基を形成し;R2は、ヒドロキシルアミノ基、ヒドロキシル基、アミノ基、アルキルアミノ基またはアルキルオキシ基であり;nは、5〜7の整数であり;そしてR3−N−R4およびR2は異なる。
式2の別の特定の実施形態において、nは6である。式IIのなお別の実施形態において、R4は水素原子であり、R3は置換または非置換のフェニルであり、そしてnは6である。式IIのなお別の実施形態において、R4は水素原子であり、R3は、置換されたフェニルであり、そしてnは6であり、ここで、このフェニル置換基は、以下からなる群より選択される:メチル基、シアノ基、ニトロ基、トリフルオロメチル基、アミノ基、アミノカルボニル基、メチルシアノ基、クロロ基、フルオロ基、ブロモ基、ヨード基、2,3−ジフルオロ基、2,4−ジフルオロ基、2,5−ジフルオロ基、3,4−ジフルオロ基、3,5−ジフルオロ基、2,6−ジフルオロ基、1,2,3−トリフルオロ基、2,3,6−トリフルオロ基、2,4,6−トリフルオロ基、3,4,5−トリフルオロ基、2,3,5、6−テトラフルオロ基,2,3,4,5,6−ペンタフルオロ基、アジド基、ヘキシル基、t−ブチル基、フェニル基、カルボキシル基、ヒドロキシル基、メトキシ基、フェニルオキシ基、ベンジルオキシ基、フェニルアミノオキシ基、フェニルアミノカルボニル基、メトキシカルボニル基、メチルアミノカルボニル基、ジメチルアミノ基、ジメチルアミノカルボニル基またはヒドロキシルアミノカルボニル基。
式2の別の実施形態において、nは6であり、R4は水素原子であり、そしてR3はシクロヘキシル基である。式2の別の実施形態において、nは6であり、R4は水素原子であり、そしてR3はメトキシ基である。式2の別の実施形態において、nは6であり、R3およびR4は、一緒に結合して、ピペリジン基を形成する。式2の別の実施形態において、nは6であり、R4は水素原子であり、そしてR3はベンジルオキシ基である。式2の別の実施形態において、R4は水素原子であり、そしてR3はγ−ピリジン基である。式2の別の実施形態において、R4は水素原子であり、そしてR3はβ−ピリジン基である。式2の別の実施形態において、R4は水素原子であり、そしてR3はα−ピリジン基である。式2の別の実施形態において、nは6であり、そしてR3およびR4は、両方ともメチル基である。式IIの別の実施形態において、nは6であり、R4はメチル基であり、そしてR3はフェニル基である。
別の実施形態において、本発明は、式3
式3の好ましい実施形態において、nは6である。この実施形態によれば、本発明は、SAHA(4)、またはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは賦形剤を含む、薬学的組成物を提供する。SAHAは、以下の構造式によって示され得る。
別の実施形態において、本発明は、式6(ピロキサミド)
別の実施形態において、本発明は、式7
別の実施形態において、本発明は、式8
別の実施形態において、本発明は、式9
別の実施形態において、本発明は、式10
別の実施形態において、本発明は、式11
別の実施形態において、本発明は、式12
特定の実施形態において、HDACインヒビターは、式XIの化合物であり、ここで、X、YおよびRは、各々ヒドロキシルであり、そしてmおよびnは共に5である。
別の実施形態において、本発明は、式13
式13の1つの特定の実施形態において、XおよびYの各々はヒドロキシル基であり、R1およびR2の各々は、メチル基である。式13の別の特定の実施形態において、XおよびYの各々はヒドロキシル基であり、R1およびR2の各々は、メチル基であり、nおよびoの各々は6であり、そしてmは2である。
別の実施形態において、本発明は、式14
別の実施形態において、本発明は、式15
式1の1つの特定の実施形態において、XおよびYの各々は、ヒドロキシル基であり、そしてmおよびnの各々は5である。
別の実施形態において、本発明は、式16
別の実施形態において、本発明は、式17
式17の1つの特定の実施形態において、XおよびYは各々ヒドロキシルアミノ基であり;R1はメチル基であり、R2は水素原子であり;そしてmおよびnの各々は2である。式17の別の特定の実施形態において、XおよびYは各々ヒドロキシルアミノ基であり;R1はカルボニルヒドロキシルアミノ基であり、R2は水素原子であり;そしてmおよびnの各々は5である。式17の別の特定の実施形態において、XおよびYは各々ヒドロキシルアミノ基であり;R1およびR2の各々はフルオロ基であり、そしてmおよびnは各々2である。
別の実施形態において、本発明は、式18
別の実施形態において、本発明は、式19
式19の1つの特定の実施形態において、R1はフェニルアミノ基であり、そしてR2はヒドロキシルアミノ基である。
別の実施形態において、本発明は、式20
式XVIIIの1つの特定の実施形態において、R1はヒドロキシルアミノ基である。式21の別の特定の実施形態において、R2はヒドロキシルアミノ基である。
別の実施形態において、本発明は、式22
式23の1つの特定の実施形態において、R1はフェニルアミノ基であり、そしてR2はヒドロキシルアミノ基である。
別の実施形態において、本発明は、式24
別の実施形態において、本発明は、式25(CBHA)
別の実施形態において、本発明は、式26
別の実施形態において、本発明は、式27の構造によって表される化合物またはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは賦形剤を含む薬学的組成物を提供する:
式27の1つの特定の実施形態において、Rは、置換フェニル基である。式27の別の特定の実施形態において、Rは、置換フェニル基であり、ここで、この置換基は、メチル、シアノ、ニトロ、チオ、トリフルオロメチル、アミノ、アミノカルボニル、メチルシアノ、クロロ、フルオロ、ブロモ、ヨード、2,3−ジフルオロ、2,4−ジフルオロ、2,5−ジフルオロ、3,4−ジフルオロ、3,5−ジフルオロ、2,6−ジフルオロ、1,2,3−トリフルオロ、2,3,6−トリフルオロ、2,4,6−トリフルオロ、3,4,5−トリフルオロ、2,3,5,6−テトラフルオロ、2,3,4,5,6−ペンタフルオロ、アジド、ヘキシル、t−ブチル、フェニル、カルボキシル、ヒドロキシル、メチルオキシ、フェニルオキシ、ベンジルオキシ、フェニルアミノオキシ、フェニルアミノカルボニル、メチルオキシカルボニル、メチルアミノカルボニル、ジメチルアミノ、ジメチルアミノカルボニルまたはヒドロキシルアミノカルボニル基からなる群から選択される。
式27の別の特定の実施形態において、Rは、置換または非置換の2−ピリジン、3−ピリジン、または4−ピリジンであり、nは、約4〜約8の整数である。
別の実施形態において、本発明は、式28の構造によって表される化合物またはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは賦形剤を含む薬学的組成物を提供する:
式28の特定の実施形態において、Rは、置換フェニル基である。式28の別の特定の実施形態において、Rは、置換フェニル基であり、ここで、この置換基は、メチル、シアノ、ニトロ、チオ、トリフルオロメチル、アミノ、アミノカルボニル、メチルシアノ、クロロ、フルオロ、ブロモ、ヨード、2,3−ジフルオロ、2,4−ジフルオロ、2,5−ジフルオロ、3,4−ジフルオロ、3,5−ジフルオロ、2,6−ジフルオロ、1,2,3−トリフルオロ、2,3,6−トリフルオロ、2,4,6−トリフルオロ、3,4,5−トリフルオロ、2,3,5,6−テトラフルオロ、2,3,4,5,6−ペンタフルオロ、アジド、ヘキシル、t−ブチル、フェニル、カルボキシル、ヒドロキシル、メチルオキシ、フェニルオキシ、ベンジルオキシ、フェニルアミノオキシ、フェニルアミノカルボニル、メチルオキシカルボニル、メチルアミノカルボニル、ジメチルアミノ、ジメチルアミノカルボニルまたはヒドロキシルアミノカルボニル基からなる群から選択される。
式28の別の特定の実施形態において、Rは、フェニルであり、nは、5である。別の実施形態において、nは、5であり、Rは、3−クロロフェニルである。
別の実施形態において、本発明は、式29の構造によって表される化合物またはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは賦形剤を含む薬学的組成物を提供する:
式29の特定の実施形態において、R1は、−NH−R4であり、ここで、R4は、置換または非置換のアリール(例えば、フェニル)、アリールアルキル(例えば、ベンジル)、ナフチル、シクロアルキル、シクロアルキルアミノ、ピリジンアミノ、ピペリジノ、9−プリン−6−アミノ、チアゾールアミノ、ヒドロキシル、分枝もしくは非分枝のアルキル、アルケニル、アルキルオキシ、アリールオキシ、アリールアルキルオキシ、ピリジル、またはキノリニルもしくはイソキノリニルである。
別の実施形態において、本発明は、式30の構造によって表される化合物またはその薬学的に受容可能な塩もしくは水和物、および薬学的に受容可能なキャリアまたは賦形剤を含む薬学的組成物を提供する:
さらなる特定の実施形態において、化合物29または30の範囲内のより特定された構造を有する化合物は、以下である:
式31の構造によって表される化合物:
式31の構造によって表される化合物:
例えば、式30の化合物は、構造31または32を有し得る:
式33の構造によって表される化合物:
例えば、式36の化合物は、式37または38を有し得る:
式39の構造によって表される化合物:
例えば、式39の化合物は、以下であり得る:
本発明で使用するのに適切な他のHDACインヒビターとしては、以下のより特定された式で示されるインヒビターが挙げられる:
このような化合物の他の例および他のHDACインヒビターは、以下に見出され得る:米国特許第5,369,108号(1994年11月29日発行)、米国特許第5,700,811号(1997年12月23日発行)、米国特許第5,773,474号(1998年6月30日発行)、米国特許第5,932,616号(1999年8月3日発行)、および米国特許第6,511,990号(2003年1月28日発行)(これら全てはBreslowらによる);米国特許第5,055,608号(1991年10月8日発行)、米国特許第5,175,191号(1992年12月29日発行)、および米国特許第5,608,108号(1997年3月4日発行)(これら全ては、Marksらによる);ならびにYoshida,M.ら,Bioassays 17,423−430(1995);Saito,A.ら,PNAS USA 96,4592−4597,(1999);Furamai R.ら,PNAS USA 98(1),87−92(2001);Komatsu,Y.ら,Cancer Res.61(11),4459−4466(2001);Su,G.H.ら,Cancer Res.60,3137−3142(2000);Lee.B.I.ら,Cancer Res.61(3),931−934;Suzuki,T.ら,J.Med.Chem.42(15),3001−3003(1999);公開PCT出願WO01/18171(2110年3月15日公開、Sloan−Kettering Institute for Cancer ResearchおよびThe Trustees of Columbia University);公開PCT出願WO02/246144(Hoffmann−La Roche);公開PCT出願WO02/22577(Novartis);公開PCT出願WO02/30879(Prolifix);公開PCT出願WO01/38322(2001年5月31日公開)、WO01/70675(2001年9月27日公開)およびWO00/71703(2000年11月30日公開)(全てMethylgene、Inc.);公開PCT出願WO00/21979(1999年10月8日公開、Fujisawa Pharmaceutical Co.,Ltd.);公開PCT出願WO98/40080(1998年3月11日公開、Beacon Laboratories,L.L.C.);およびCurtin M.(ヒストンデアセチラーゼインヒビターの現在の特許状態 Expert Opin.Ther.Patents(2002)12(9):1375−1384およびその中で引用された参考文献)。
SAHAまたは他のHDACのいずれかは、詳細な実験の章で概説される方法に従って、または米国特許第5,369,108号、同第5,700,811号、同第5,932,616号および同第6,511,990号(これらの内容は、その全体が本明細書中で参考として援用される)に記載の方法に従って、または当業者に公知の任意の他の方法に従って、合成され得る。
本発明は、上記の化合物に加えて、このような化合物のホモログおよびアナログの使用を包含することを意図する。これに関して、ホモログは、上記の化合物と実質的な構造類似性を有する分子であり、そしてアナログは、構造類似性に関係無く、実質的な生物学的類似性を有する分子である。
本発明はまた、以下の有機酸および無機酸とHDACインヒビターとの薬学的に受容可能な塩(例えば、酸付加塩)を含む薬学的組成物を包含する:酸付加塩(例えば、塩酸、硫酸、メタンスルホン酸、フマル酸、マレイン酸、コハク酸、酢酸、安息香酸、シュウ酸、クエン酸、酒石酸、炭酸、リン酸などであり得る)。薬学的に受容可能な塩はまた、無機塩基(例えば、水酸化ナトリウム、水酸化カリウム、水酸化アンモニウム、水酸化カルシウムまたは水酸化鉄)および有機塩基(例えば、イソプロピルアミン、トリメチルアミン、2−エチルアミノエタノール、ヒスチジン、プロカインなど)で処理することによって調製され得る。
本発明はまた、HDACインヒビターの水和物を含む薬学的組成物を包含する。用語「水和物」は、半水和物、一水和物、二水和物、三水和物などを含むが、これらに限定されない。
本発明はまた、SAHAの任意の固体または液体の物理形態、あるいは他のHDACインヒビターの任意のものを含む薬学的組成物を包含する。例えば、HDACインヒビターは、結晶形態、アモルファス形態であり得、任意の粒子サイズを有する。HDACインヒビター粒子は、微粉化され得るか、または凝集された粒子状顆粒、粉末、油、油状懸濁液または固体もしくは液体の物理形態の任意の他の形態であり得る。
(薬学的組成物)
本発明の化合物、その誘導体、フラグメント、アナログ、ホモログ、薬学的に受容可能な塩または水和物は、経口投与するのに適切な薬学的組成物に薬学的に受容可能なキャリアまたは賦形剤と共に組み込まれ得る。このような組成物は、代表的に、治療有効量の上記の化合物のいずれか、および薬学的に受容可能なキャリアを含む。好ましくは、有効量とは、適切な新生物細胞の末端分化を選択的に誘導するのに有効であり、かつ患者に毒性を引き起こす量よりも小さい量である。
本発明の化合物、その誘導体、フラグメント、アナログ、ホモログ、薬学的に受容可能な塩または水和物は、経口投与するのに適切な薬学的組成物に薬学的に受容可能なキャリアまたは賦形剤と共に組み込まれ得る。このような組成物は、代表的に、治療有効量の上記の化合物のいずれか、および薬学的に受容可能なキャリアを含む。好ましくは、有効量とは、適切な新生物細胞の末端分化を選択的に誘導するのに有効であり、かつ患者に毒性を引き起こす量よりも小さい量である。
キャリアまたは希釈剤として一般的に使用される任意の不活性賦形剤(例えば、ゴム、デンプン、糖、セルロース材料、アクリレートまたはそれらの混合物)が、本発明の処方物で使用され得る。好ましい希釈剤は、微結晶性セルロースである。この組成物は、崩壊剤(例えば、クロスカルメロースナトリウム)、および滑沢剤(例えば、ステアリン酸マグネシウム)をさらに含み得、さらに、結合剤、緩衝剤、プロテアーゼインヒビター、界面活性剤、可溶化剤、可塑剤、乳化剤、安定化剤、粘度向上剤、甘味剤、フィルム形成剤から選択される1つ以上の添加剤、または任意のそれらの組合せを含み得る。さらに、本発明の組成物は、制御放出処方物または徐放性処方物の形態であり得る。
一実施形態は、HDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、微結晶性セルロール、クロスカルメロースナトリウムおよびステアリン酸マグネシウムを含む、経口投与のための薬学的組成物である。別の実施形態は、HDACインヒビターとしてSAHAを有する。別の実施形態は、50〜70重量%のHDACインヒビターまたはその薬学的に受容可能な塩もしくは水和物、20〜40重量%の微結晶性セルロース、5〜15重量%のクロスカルメロースナトリウム、および0.1〜5重量%のステアリン酸マグネシウムを含む。別の実施形態は、約50〜200gのHDACインヒビターを含む。
一実施形態において、この薬学的組成物は、経口投与され、従って、経口投与に適切な形態、すなわち、固体調製物または液体調製物として処方される。適切な固体経口処方物としては、錠剤、カプセル剤、丸剤、顆粒剤、ペレットなどが挙げられる。適切な液体経口処方物としては、溶液、懸濁液、分散液、エマルジョン、油などが挙げられる。本発明の一実施形態において、この組成物は、カプセル剤に処方される。この実施形態によると、本発明の組成物は、HDACインヒビター活性化合物および不活性キャリアまたは希釈剤に加えて、硬質ゼラチンカプセルを含む。
本明細書中で使用される場合、「薬学的に受容可能なキャリア」とは、薬剤の投与に適合性の、任意および全ての溶媒、分散媒体、コーティング、殺菌剤および抗真菌剤、等張剤、吸収遅延剤など(例えば、滅菌発熱物質を含まない水)を含むことを意図する。適切なキャリアは、この分野の基本的な参考テキストであるRemington’s Pharmaceutical Sciencesの最新版(これは本明細書中に参考として援用される)に記載される。このようなキャリアまたは希釈剤の好ましい例としては、水、生理食塩水、フィンガー溶液、デキストロース溶液、および5%ヒト血清アルブミンが挙げられるが、これらに限定されない。リポソームおよび非水性ビヒクル(例えば、不揮発性油)もまた使用され得る。薬学的に活性な物質のためのこのような媒体および薬剤の使用は、当該分野で周知である。任意の従来の媒体または薬剤が活性化合物と適合性である限り、組成物におけるこれらの使用が企図される。補助活性化合物もまた、この組成物に組み込まれ得る。
固体キャリア/希釈剤としては、ゴム、デンプン(例えば、コーンスターチ、アルファ化デンプン)、糖(例えば、ラクトース、マンニトール、スクロース、デキストロース)、セルロース材料(例えば、微結晶性セルロール)、アクリレート(例えば、ポリメチルアクリレート)、炭酸カルシウム、酸化マグネシウム、タルク、またはこれらの混合物が挙げられるが、これらに限定されない。
液体処方物について、薬学的に受容可能なキャリアは、水性または非水性の溶液、懸濁液、エマルジョンまたは油であり得る。非水性溶媒の例は、プロピレングリコール、ポリエチレングリコールおよび注射用有機エステル(例えば、オレイン酸エチル)である。水性キャリアとしては、水、アルコール性/水性の溶液、エマルジョンまたは懸濁液(生理食塩水および緩衝化媒体を含む)が挙げられる。油の例は、石油、動物性油、植物性油、または合成系油(例えば、落花生油、大豆油、鉱油、オリーブ油、ヒマワリ油および魚肝油)である。溶液または懸濁液はまた、以下の成分を含み得る:滅菌希釈剤(例えば、注射用の水、生理食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒);抗菌剤(例えば、ベンジルアルコールまたはメチルパラベン);酸化防止剤(例えば、アスコルビン酸または重亜硫酸ナトリウム);キレート剤(例えば、エチレンジアミン四酢酸(EDTA));緩衝剤(例えば、酢酸塩、クエン酸塩またはリン酸塩);および張度を調節するための薬剤(例えば、塩化ナトリウムまたはデキストロース)。pHは、酸または塩基(例えば、塩酸または水酸化ナトリウム)を用いて調節され得る。
さらに、この組成物は、以下をさらに含み得る:結合剤(例えば、アカシア、コーンスターチ、ゼラチン、カルボマー(carbomer)、エチルセルロース、ガーゴム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポビドン)、崩壊剤(例えば、コーンスターチ、ポテトスターチ、アルギン酸、二酸化ケイ素、クロスカルメロースナトリウム、クロスポビドン、ガーゴム、ナトリウムスターチグリコレート、Primogel)、種々のpHおよびイオン強度の緩衝剤(例えば、tris−塩酸、酢酸塩、リン酸塩)、添加剤(例えば、アルブミンまたは表面への吸収を防止するためのゼラチン)、洗剤(例えば、Tween 20、Tween 80、Pluronic F68、胆汁酸塩)、プロテアーゼインヒビター、界面活性剤(例えば、ラウリル硫酸ナトリウム)、浸透促進剤、可溶化剤(例えば、グリセロール、ポリエチレングリコール)、グリダント(例えば、コロイド状二酸化ケイ素)、抗酸化剤(例えば、アスコルビン酸、メタ重亜硫酸ナトリウム、ブチルヒドロキシアニソール)、安定剤(例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース)、粘度向上剤(例えば、カルボマー、コロイド状二酸化ケイ素、エチルセルロース、ガーゴム)、甘味剤(例えば、スクロース、アスパルテーム、クエン酸)、香味剤(例えば、ペパーミント、サリチル酸メチル、またはオレンジフレーバー)、防腐剤(例えば、Thimerosal、ベンジルアルコール、パラベン)、滑沢剤(例えば、ステアリン酸、ステアリン酸マグネシウム、ポリエチレングリコール、ラウリル硫酸ナトリウム)、流動補助剤(例えば、コロイド状二酸化ケイ素)、可塑剤(例えば、フタル酸ジエチル、クエン酸トリエチル)、乳化剤(例えば、カルボマー、ヒドロキシプロピルセルロース、ラウリル硫酸ナトリウム)、ポリマーコーティング(例えば、ポロキサマーまたはポロキサミン)、コーティングおよびフィルム形成剤(例えば、エチルセルロール、アクリレート、ポリメタクリレート)、および/またはアジュバント。
1つの実施形態において、活性化合物は、身体からの迅速な排除に対して化合物を保護するキャリア(例えば、制御放出処方物(移植片およびマイクロカプセル化送達系を含む))を用いて調製される。生分解性、生体適合性ポリマー(例えば、エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸)が使用され得る。このような処方物の調製方法は、当業者に明らかである。これらの材料はまた、Alza CorporationおよびNova Pharmaceuticals,Inc.から市販される。リポソーム懸濁液(ウイルス抗原に対するモノクローナル抗体を用いて感染される細胞に標的化されるリポソームを含む)はまた、薬学的に受容可能なキャリアとして使用され得る。これらは、当業者に公知の方法(例えば、米国特許第4,522,811号に記載される)に従って調製され得る。
投与の容易さおよび投薬量の均一性のために投薬量単位形態で経口処方物を処方することが特に有利である。本明細書中で使用される投薬量単位形態とは、処置される被験体に対して単位投薬量として適切な物理的別個の単位をいい;各々の単位が、必要とされる薬学的キャリアとともに所望の治療効果を生じるように計算された活性化合物の所定量を含む。本発明の投薬量単位形態のための詳細は、活性化合物の独特の特性および達成されるべき特定の治療効果、および個体の処置のための活性化合物のような調剤の分野に固有の制限によって決められ、そして直接的に依存する。
薬学的組成物は、投与のための指示書とともに、容器、パック、またはディスペンサーに含まれ得る。
次いで、毎日の投与は、数日〜数年の期間の間、連続的に繰り返される。経口処置は、1週間〜患者の寿命の間、続けられ得る。好ましくは、投与は、さらなる投与が必要とされるか否かを決定するために患者が評価され得るときより後に、連続した5日の間、行われる。投与は、連続的または断続的(すなわち、多くの連続した日の間の処置、続く、休止期間)であり得る。
本発明の化合物は、処置の第1日目に静脈内投与され得、第2日目およびその後の全ての連続した日において、経口投与される。
本発明の化合物は、疾患の進行を妨げるためまたは腫瘍増殖を安定化させるために投与され得る。
活性成分を含む薬学的組成物の調製は、例えば、混合、粒状化、または錠剤形成プロセスによって、当該分野において十分に理解される。活性治療成分は、しばしば、薬学的に受容可能であり、活性成分と適合性である賦形剤と混合される。経口投与のために、活性成分は、この目的のために習慣的な添加剤(例えば、ビヒクル、安定化剤、または不活性希釈剤)と混合され、そして投与のための適切な形態(例えば、上記に詳細に記載されるように、錠剤、コーティングされた錠剤、硬質ゼラチンカプセルまたは軟質ゼラチンカプセル、水性、アルコール性または油性溶液など)に習慣的な方法で変換される。
本発明の化合物は、25〜4000mg/m2の間、例えば、約25〜1000mg、50〜1000mg、100mg、200mg、300mg、400mg、600mg、800mg、1000mgなどの毎日の合計の用量で経口的に投与され得る。代表的に、化合物は、患者に対して、400mgまでの投与の場合、単一用量として投与される。より高い合計の投薬量(すなわち、400mgより上)について、全体は、複数用量(例えば、毎日2回、毎日3回など)に分割され、好ましくは、その日の間で等しい時間の間隔に分散される。例えば、2つの用量(例えば、それぞれ、500mg)は、一日に、1000mgの合計の投薬量を達成するために、12時間空けて投与され得る。
1つの現在好ましい実施形態において、SAHAまたはHDACインヒビターのいずれかは、200mgの毎日の合計の投薬量で患者に投与される。別の現在好ましい実施形態において、SAHAまたはHDACインヒビターのいずれかは、400mgの毎日の合計の投薬量で患者に投与される。別の現在好ましい実施形態において、SAHAまたはHDACインヒビターのいずれかは、600mgの毎日の合計の投薬量で患者に投与される。
患者に投与される化合物の量は、患者において毒性を引き起こす量より少ない。特定の実施形態において、患者に投与される化合物の量は、化合物の毒性レベルに等しいかまたはそれを超えるレベルの、患者の血漿中の化合物の濃度をもたらす量より少ない。好ましくは、患者の血漿中の化合物の濃度は、約10nMに維持される。別の実施形態において、患者の血漿中の化合物の濃度は、約25nMに維持される。別の実施形態において、患者の血漿中の化合物の濃度は、約50nMに維持される。別の実施形態において、患者の血漿中の化合物の濃度は、約100nMに維持される。別の実施形態において、患者の血漿中の化合物の濃度は、約500nMに維持される。別の実施形態において、患者の血漿中の化合物の濃度は、約1000nMに維持される。別の実施形態において、患者の血漿中の化合物の濃度は、約2500nMに維持される。別の実施形態において、患者の血漿中の化合物の濃度は、約5000nMに維持される。HMBAを用いて、約5gm/m2/日〜約30gm/m2/日、特に、約20gm/m2/日の量での化合物の投与が、患者において毒性を生じることなく有効であることが見出された。本発明の実施において患者に投与されるべき化合物の最適量は、使用される特定の化合物および処置される癌の型に依存する。
本発明の現在好ましい実施形態において、薬学的組成物は、ヒストンデアセチラーゼ(HDAC)インヒビター;キャリアまたは希釈剤としての微結晶セルロース;崩壊剤としてのクロスカルメロースナトリウム;および潤滑剤としてのステアリン酸マグネシウムを含む。特に好ましい実施形態において、HDACインヒビターは、スベロイルアニリドヒドロキサム酸(SAHA)である。
処方物中の活性成分および種々の賦形剤の割合は、変化し得る。例えば、この組成物は、20%重量と90重量%との間、好ましくは、50〜70重量%のヒストンデアセチラーゼ(HDAC)を含み得る。さらに、この組成物は、10重量%と70重量%との間、好ましくは、20〜40重量%の微結晶セルロースをキャリアまたは希釈剤として含み得る。さらに、この組成物は、1重量%と30重量%との間、好ましくは、5〜15重量%のクロスカルメロースナトリウムを崩壊剤として含み得る。さらに、この組成物は、潤滑剤として0.1〜5重量%のステアリン酸マグネシウムを含み得る。別の好ましい実施形態において、この組成物は、約50〜200mgのHDACインヒビター(例えば、HDACインヒビター(例えば、SAHA)について、50mg、100mg、および200mg)を含む。特に好ましい実施形態において、この組成物は、ゼラチンカプセルの形態である。
本発明の現在好ましい実施形態は、ゼラチンカプセル中に含まれる、微結晶セルロース、NF(Avicel Ph 101)、クロスカルメロースナトリウム、NF(AC−Di−Sol)およびステアリン酸マグネシウム、NFを伴うSAHAの固体処方物である。さらに好ましい実施形態は、ゼラチンカプセル中に含まれる、89.5mgの微結晶セルロース、9mgのクロスカルメロースナトリウムおよび1.5mgのステアリン酸マグネシウムを伴う200mgの固形SAHAである。
本発明の薬学的組成物が、新生物細胞誘導の増殖の阻害および癌の処置に有用であるだけではないこと、ならびにこれらの化合物が、HDACインヒビターが有用であることが見出されている幅広い範囲の疾患を処置する際に有用であることが当業者に明らかである。
例えば、HDACインヒビター、および特にSAHAは、種々の急性および慢性の炎症性疾患、自己免疫疾患、アレルギー疾患、酸化ストレスに関連する疾患、および細胞性過剰増側によって特徴付けられる疾患の処置に有用であることが見出されている。非限定的な例は、以下である:関節の炎症性状態(慢性関節リウマチ(RA)および乾癬性関節炎を含む);炎症性腸疾患(例えば、クローン病および潰瘍性大腸炎);破壊性脊椎症;強皮症;乾癬(T細胞媒介乾癬を含む)および炎症性皮膚症(dermatose)(例えば、皮膚炎、湿疹、アトピー性皮膚炎、アレルギー性接触皮膚炎、じんましん);脈管炎(例えば、壊死性脈管炎、皮膚性脈管炎、および過敏性脈管炎);好酸球性筋炎(eosinphilic myositis)、好酸球性筋膜炎;皮膚または器官の白血球浸潤を伴う癌、虚血性傷害(大脳虚血(例えば、外傷、てんかん、出血または発作の結果のような脳損傷(これらの各々が、神経変性を導き得る)));HIV、心不全、慢性、急性または悪性の肝臓疾患、自己免疫甲状腺炎;全身性エリテマトーデス、シェーグレン症候群、肺疾患(例えば、ARDS);急性膵炎;筋萎縮性側索硬化症(ALS);アルツハイマー病;悪液質/食欲不振;喘息;アテローム性動脈硬化症;慢性疲労症候群、発熱;糖尿病(例えば、インシュリン糖尿病または若年型糖尿病);糸球体腎炎;対宿主性移植片拒絶(例えば、移植);出血性ショック;痛覚過敏;炎症性腸疾患;多発性硬化症;筋障害(例えば、筋肉タンパク質代謝、特に、敗血症);骨粗鬆症;パーキンソン病;疼痛;早期出産;乾癬;再潅流障害;サイトカイン誘導毒性(例えば、敗血症ショック、内毒素ショック);放射線療法由来の副作用、時間的顎関節疾患、腫瘍転移;あるいは挫傷、捻挫、軟骨損傷、外傷(例えば、火傷、整形外科手術、感染または他の疾患プロセス)から生じる炎症状態。アレルギー性疾患および状態としては、以下が挙げられるがこれらに限定されない:呼吸性アレルギー性疾患(例えば、喘息、アレルギー性鼻炎、過敏性肺疾患、過敏性肺炎、好酸球性肺炎(例えば、レフレル症候群、慢性好酸球性肺炎)、遅延型過敏症、間質性肺疾患(ILD)(例えば、特発性肺線維症、または慢性関節リウマチに関連するILD、全身性エリテマトーデス、強直性脊椎炎、全身性硬化症、シェーグレン症候群、多発性筋炎または皮膚筋炎);全身アナフィラキシーまたは過敏性応答、薬物アレルギー(例えば、ペニシリン、セファロスポリンに対する)、虫刺し傷アレルギーなど。
例えば、HDACインヒビター、および特にSAHAは、以下の非徹底的なリストの種々の神経変性疾患の処置において有用であることが見出されている:
I.他の顕著な神経学的徴候(例えば、アルツハイマー病;アルツハイマー型の老人性痴呆症;およびピック病(脳葉萎縮))の非存在下での進行性痴呆によって特徴付けられる障害。
I.他の顕著な神経学的徴候(例えば、アルツハイマー病;アルツハイマー型の老人性痴呆症;およびピック病(脳葉萎縮))の非存在下での進行性痴呆によって特徴付けられる障害。
II.進行性痴呆と他の顕著な神経学的異常、例えば、A)主に成体において見られる症候群(例えば、ハンチントン病、痴呆とパーキンソン病の運動失調症および/または発現とを組み合わせる複数の系の萎縮、進行性核上性麻痺(Steel−Richardson−Olszewski)、びまん性レヴィー小体病、および大脳基質(corticodentatonigral)変性;ならびにB)主に子供または若い成体において見られる症候群(例えば、Hallervorden−Spatz疾患および進行性家族性ミオクローヌスてんかん)とを組み合わせた症候群。
III.姿勢および運動の異常(例えば、振戦麻痺(パーキンソン病)、線条体黒質系変性、進行性核上麻痺、ねじれ失調症(捻転痙攣;変形性筋失調症)、痙性斜頸および他の運動異常症、家族性振戦、ならびにGilles de la Tourette症候群)を徐々に発生する症候群。
IV.小脳変性(例えば、小脳皮質変性およびオリーブ橋小脳萎縮症(OPCA));ならびに脊髄小脳変性症(フリートライヒ運動失調(atazia)および関連障害)のような進行性失調症の症状。
V.中枢自律神経系不全の症候群(Shy−Drager症候群)。
VI.感覚変化のない筋肉の弱化および消耗の症候群(運動ニューロン疾患(例えば、筋萎縮性側索硬化症、脊髄筋肉萎縮症(例えば、乳児脊髄筋萎縮(Werdnig−Hoffman)、若年性脊髄筋萎縮(Wohlfart−Kugelberg−Welander)および他の形態の家族性脊髄筋萎縮)、原発性側索硬化症、および遺伝性痙性対麻痺。
VII.筋肉の弱化および消耗と感覚の変化を組み合わせた症候群(進行性神経筋肉萎縮;慢性家族性多発性神経障害)(例えば、腓骨筋萎縮(Charcot−Marie−Tooth)、B.Hypertrophic間質性多発神経障害(Dejerine−Sottas)、および慢性進行性神経炎のC.Miscellaneous形態)。
VIII.網膜の色素変性(色素性網膜炎)、および遺伝性視神経萎縮(Leber病)のような進行性の視角喪失の症候群。
本発明は、以下に続く実験の詳細の節において実施例で示される。この節は、本発明の理解を補助するために記載されるが、その後に続く特許請求の範囲に記載される本発明をいかなるようにも制限することを意図せず、解釈されるべきではない。
(実験の詳細の節)
(SAHAの合成)
SAHAは、以下に概説される方法、または米国特許第5,369,108号(この内容は、その全体が参考として援用される)に記載される方法もしくは任意の他の方法に従って、合成され得る。
SAHAは、以下に概説される方法、または米国特許第5,369,108号(この内容は、その全体が参考として援用される)に記載される方法もしくは任意の他の方法に従って、合成され得る。
(SAHAの合成)
(工程1−スベルアニリン酸(suberanilic acid)の合成)
(工程1−スベルアニリン酸(suberanilic acid)の合成)
22Lのフラスコにおいて、3,500g(20.09モル)のスベリン酸を入れ、そしてこの酸を加熱しながら融解させた。温度を175℃に上昇させ、次いで、2,040g(21.92モル)のアニリンを添加した。温度を190℃に上昇させ、そして20分間その温度で維持した。50Lの水中に溶解させた融解物を、4,017gの水酸化カリウムを含むNalgeneタンク中に注いだ。この混合物を、融解物の添加後20分間攪拌した。反応を同じスケールで繰り返し、そして第2の融解物を同じ水酸化カリウムの溶液中に注いだ。この混合物を徹底的に攪拌後、スターラーを止め、そして混合物を静止させた。次いで、この混合物を、セライト(4,200g)パッドを通して濾過した(生成物を、中性の副生成物(スベリン酸の両側に対するアニリンによる攻撃由来)を除去するために濾過した。濾液は、生成物の塩を含み、そしてまた、未反応スベリン酸の塩を含んだ。この混合物を静止させた。なぜなら、濾過は、非常にゆっくりであり、数日かかるからである)。濾液を、5Lの濃塩酸を使用して酸性化した;この混合物を1時間攪拌し;次いで、一晩静止させた。生成物を濾過によって集め、そして漏斗において脱イオン水(4×5L)を用いて洗浄した。湿潤したフィルターケーキを、44Lの脱イオン水を用いて72Lのフラスコ中に配置し、この混合物を50℃に加熱し、そして固体を熱濾過によって単離した(所望の生成物は、熱水中においてかなり高い溶解度を有するスベリン酸を混入していた。数回の熱粉砕を行い、スベリン酸を除去した。その生成物を、スベリン酸の除去をモニターするために、NMR[D6DMSO]によってチェックした)。熱粉砕を44Lの水を用いて50℃で繰り返した。生成物を再び、濾過によって単離し、そして4Lの熱水でリンスした。それを減圧供給源としてNashポンプを使用して、65℃で減圧オーブンにおいて週末にわたって乾燥させた(Nashポンプは、液体リングポンプ(水)であり、約29インチ水銀の減圧に引っ張る。断続的なアルゴンパージを使用して水を運び去ることを助けた);4,182.8gのスベルアニリン酸を得た。
生成物は、なお、少量のスベリン酸を含んだ;従って、一度に約300gの生成物を使用して、熱粉砕を、65℃で一部ずつ行った。各部分を濾過し、そしてさらなる熱水で徹底的にリンスした(合計約6L)。これを、バッチ全体を精製するために繰り返した。これは、生成物からスベリン酸を完全に除去した。固体生成物をフラスコ中で組合せ、そして6Lのメタノール/水(1:2)とともに攪拌し、次いで、濾過によって単離し、そして週末にわたってフィルターで空気乾燥した。これをトレーに置き、そしてNashポンプおよびアルゴンブリード(bleed)を使用して、65℃で45時間減圧中で乾燥させた。最終生成物は、3,278.4g(32.7%収率)を有した。
(工程2−スベルアニリン酸メチルの合成)
メカニカルスターラーおよび凝縮器を取り付けた50Lのフラスコに、先の工程からの3,229gのスベルアニリン酸、20Lのメタノールおよび398.7gのDowex 50WX2−400樹脂を配置した。この混合物を加熱還流し、18時間還流を維持した。この混合物を濾過して樹脂ビーズを除き、そして濾液をロータリーエバポレーターの残渣にとった。
ロータリーエバポレーターからの残渣を、凝縮器およびメカニカルスターラーを取り付けた50Lフラスコ中に移した。フラスコに6Lのメタノールを加え、そしてこの混合物を、溶液を与えるように加熱した。次いで、2Lの脱イオン水を添加し、そして熱を遮断した。攪拌される混合物を冷却させ、次いで、フラスコを氷浴中に配置し、そして混合物を冷却した。固体生成物を濾過によって単離し、そしてフィルターケーキを4Lの冷メタノール/水(1:1)でリンスした。生成物を、合計64時間、Nashポンプを使用して、減圧オーブン中で、45℃で乾燥させ、2,850.2g(84%収率)のスベルアニリン酸メチル(CSL Lot#98−794−92−3 1を得た。
(工程3−粗SAHAの合成)
メカニカルスターラー、熱電対、および不活性雰囲気のための入口を備えた50Lフラスコに、1,451.9gのヒドロキシアミン塩酸塩、19Lの無水メタノール、およびメタノール中の3.93Lの30%ナトリウムメトキシド溶液を加えた。次いで、フラスコに、2,748.0gのスベルアニリン酸メチルを入れ、続いて、1.9Lの30%ナトリウムメトキシドメタノール溶液を入れた。この混合物を、16時間10分間攪拌させた。反応混合物の約半分を、反応フラスコ(フラスコ1)からメカニカルスターラーを取り付けた50Lのフラスコ(フラスコ2)に移した。次いで、27Lの脱イオン水をフラスコ1に加え、そしてこの混合物を10分間攪拌した。pHメーターを使用してpHを測定した;pHは、11.56であった。この混合物のpHを、100mlの30%ナトリウムメトキシドメタノール溶液の添加によって、12.02に調節した;これは、透明な溶液を与えた(このとき、この反応混合物は、少量の固体を含んだ。透明な溶液を与えるようにpHを調製し、この溶液から、生成物の沈殿が、沈殿する)。フラスコ2中の反応混合物を、同じ方法で希釈した;27Lの脱イオン水を添加し、そしてpHを、この混合物への100mlの30%ナトリウムメトキシド溶液の添加によって調節して、12.01のpHを得た(透明溶液)。
各フラスコ中の反応混合物を、氷酢酸の添加によって酸性化し、生成物を沈殿させた。フラスコ1は、8.98の最終pHを有し、そしてフラスコ2は、8.70の最終pHを有した。両方のフラスコ由来の生成物を、Buchner漏斗および濾布を用いる濾過によって単離した。フィルターケーキを15Lの脱イオン水で洗浄し、そして漏斗をカバーし、そして生成物を、15.5時間、減圧下で漏斗上で部分的に乾燥させた。生成物を取り出し、そして5個のガラストレー内に配置した。トレーを減圧オーブンに配置し、そしてこの生成物を一定の重量まで乾燥させた。第1の乾燥期間は、アルゴンブリードを伴う減圧供給源としてのNashポンプを使用して、60℃で22時間であった。トレーを減圧オーブンから取り出し、そして秤量した。トレーをオーブンに戻し、そしてアルゴンブリード無しで、減圧供給源としてオイルポンプを使用して、さらに4時間10分間生成物を乾燥した。この物質を、二重4−ミルポリエチレンバッグに包装し、そしてプラスチック外側容器内に配置した。サンプリング後の最終重量は、2633.4g(95.6%)であった。
(工程4−粗SAHAの再結晶)
粗SAHAを、メタノール/水から再結晶化した。メカニカルスターラー、熱電対、凝縮器および不活性雰囲気のための入口を備えた50Lフラスコに、結晶化させる粗SAHA(2,525.7g)、続いて、2,625mlの脱イオン水および15,755mlのメタノールを入れた。この物質を加熱還流させて、溶液を得た。次いで、5,250mlの脱イオン水をこの反応混合物に添加した。熱を遮断し、そしてこの混合物を、冷却させた。フラスコを安全に操作し得るようにこの混合物を十分に冷却した(28℃)とき、フラスコを熱マントルから除き、そして冷却浴として使用するためにタブに配置した。氷/水をタブに添加して、この混合物を−5℃に冷却した。この混合物を2時間その温度未満に維持した。この生成物を濾過によって単離し、そしてフィルターケーキを、1.5Lの冷メタノール/水(2:1)で洗浄した。漏斗をカバーし、そして生成物を1.75時間、減圧下で部分的に乾燥させた。生成物を漏斗から取り出し、そして6個のガラストレーに配置した。トレーを減圧オーブンに配置し、そして生成物を、減圧供給源としてNashポンプを使用し、そしてアルゴンブリードを使用して、60℃で64.75時間乾燥させた。トレーを秤量のために取り出し、次いで、オーブンに戻し、そしてさらに4時間60℃で乾燥して、一定の重量を得た。第2の乾燥期間の間、減圧供給源は、オイルポンプであり、アルゴンブリードを使用しなかった。この物質を、二重4−ミルポリエチレンバッグに包装し、そしてプラスチック外側容器内に配置した。サンプリング後の最終重量は、2,540.9g(92.5%)であった。
粗SAHAを、メタノール/水から再結晶化した。メカニカルスターラー、熱電対、凝縮器および不活性雰囲気のための入口を備えた50Lフラスコに、結晶化させる粗SAHA(2,525.7g)、続いて、2,625mlの脱イオン水および15,755mlのメタノールを入れた。この物質を加熱還流させて、溶液を得た。次いで、5,250mlの脱イオン水をこの反応混合物に添加した。熱を遮断し、そしてこの混合物を、冷却させた。フラスコを安全に操作し得るようにこの混合物を十分に冷却した(28℃)とき、フラスコを熱マントルから除き、そして冷却浴として使用するためにタブに配置した。氷/水をタブに添加して、この混合物を−5℃に冷却した。この混合物を2時間その温度未満に維持した。この生成物を濾過によって単離し、そしてフィルターケーキを、1.5Lの冷メタノール/水(2:1)で洗浄した。漏斗をカバーし、そして生成物を1.75時間、減圧下で部分的に乾燥させた。生成物を漏斗から取り出し、そして6個のガラストレーに配置した。トレーを減圧オーブンに配置し、そして生成物を、減圧供給源としてNashポンプを使用し、そしてアルゴンブリードを使用して、60℃で64.75時間乾燥させた。トレーを秤量のために取り出し、次いで、オーブンに戻し、そしてさらに4時間60℃で乾燥して、一定の重量を得た。第2の乾燥期間の間、減圧供給源は、オイルポンプであり、アルゴンブリードを使用しなかった。この物質を、二重4−ミルポリエチレンバッグに包装し、そしてプラスチック外側容器内に配置した。サンプリング後の最終重量は、2,540.9g(92.5%)であった。
(スベロイルアニリドヒドロキサム酸(SAHA)の経口投薬)
(背景):ハイブリッド極性細胞性分化剤での処置は、ヒト固形腫瘍由来細胞株および移植片の増殖を阻害した。この効果は、一部、ヒストンデアセチラーゼの阻害によって媒介される。SAHAは、実験室および前臨床研究における腫瘍細胞増殖停止、分化およびアポトーシスを誘導する能力を有することが示された強力なヒストンデアセチラーゼインヒビターである。
(背景):ハイブリッド極性細胞性分化剤での処置は、ヒト固形腫瘍由来細胞株および移植片の増殖を阻害した。この効果は、一部、ヒストンデアセチラーゼの阻害によって媒介される。SAHAは、実験室および前臨床研究における腫瘍細胞増殖停止、分化およびアポトーシスを誘導する能力を有することが示された強力なヒストンデアセチラーゼインヒビターである。
(目的):フェーズII研究において使用され得るSAHAの安全な毎日の経口レジメンを規定すること。さらに、SAHAの経口処方物の薬物動態的なプロフィールを評価した。絶食状態 対 非絶食状態のヒトにおけるSAHAの経口バイオアベイラビリティー、および処置の抗腫瘍効果もまたモニターした。さらに、正常組織および腫瘍細胞に対するSAHAの生物学的効果を評価し、そしてヒストンアセチル化のレベルに関する応答を記録した。
(患者):組織学的に記録された進行した段階、標準的な治療法に対して不応答性であるかまたは治療的に標準的な治療法が存在しない原発性または転移性成体固形腫瘍を有する患者。患者は、70%以上のKarnofsky動作状態、ならびに適切な血液学的機能、肝臓機能および腎臓機能を有しなければならない。患者は、任意の先の化学療法、放射線療法または他の研究用の抗癌薬物から少なくとも4週間なければならない。
(投薬スケジュール):第1日目に、患者を、最初に、200mgの静脈内投与されるSAHAで処置した。第2日目に開始して、患者を、表1に従って、毎日の用量の経口SAHAで処置した。各コホートは、異なる用量のSAHAを受容した。「QD」は、1日に1回の投薬を示し:「Q12時間」は、1日に2回の投薬を示す。例えば、コホートIVの患者は、1日当たり2回の800mgの用量のSAHAを受容した。用量を患者に毎日連続的に投与した。血液サンプルを経口処置の1日目および21日目に採取した。患者は、疾患進行、腫瘍後退、受容しがたい副作用、または他の治療を用いる処置に起因して、経口SAHAを止めた。
(結果):血清血漿レベルの比較は、患者が絶食している場合および患者が絶食していない場合の両方において、静脈内投与されたSAHA(IV SAHA)と比較して、経口投与されるSAHAの高いバイオアベイラビリティーを示す。「AUC」は、SAHA(ng/ml)分のバイオアベイラビリティーの推定であり、ここで、660ng/mlは、2.5μM SAHAに等しい。半減期(t1/2)とともにAUCは、経口SAHAの全体的なバイオアベイラビリティーが、IV SAHAのバイオアベイラビリティーよりも良いことを示す。Cmaxは、投与後に観察されるSAHAの最大濃度である。IV SAHAは、2時間にわたって注入される200mgで投与される。経口SAHAを、単一カプセルで200mgで投与した。表2および表3は、HPLCアッセイ(重水素化標準を使用するLCMS)の結果を要約し、これは、マーカーとしてアセチル化ヒストン−4を使用して、患者の血液血漿中のSAHAの量 対 時間を定量化する。
図1〜8は、コホートIおよびIIの患者におけるα−AcH4の量を示すHPLCスライドであり、経口投薬後10時間まで測定され、SAHAが静脈内投与された場合のα−AcH4レベルと比較した。図9は、投与後の示された時点での平均血漿濃度のSAHA(ng/ml)を示す。図9A:8日目に絶食下の経口用量(200mgおよび400mg)。図9B:9日目の食事を伴う経口用量。図9C:1日目のIV用量。図10は、8日目、9日目および22日目のSAHA200mgおよび400mgの経口用量の見かけの半減期を示す。図11は、8日目、9日目および22日目のSAHA200mgおよび400mgのAUC(ng/ml/時間)を示す。図12は、8日目、9日目および22日目のSAHA200mgおよび400mgの経口用量後のSAHAのバイオアベイラビリティーを示す。
(スベロイルアニリドヒドロキサム酸(SAHA)の経口投薬−用量増加)
別の実験において、表4に示されるように、固形腫瘍を有する25人の患者は、アームAに登録され、ホジキンリンパ腫または非ホジキンリンパ腫を有する13人の患者は、アームBに登録され、そして急性白血病を有する1人の患者および骨髄形成異常を有する1人の患者は、アームCに登録された。
別の実験において、表4に示されるように、固形腫瘍を有する25人の患者は、アームAに登録され、ホジキンリンパ腫または非ホジキンリンパ腫を有する13人の患者は、アームBに登録され、そして急性白血病を有する1人の患者および骨髄形成異常を有する1人の患者は、アームCに登録された。
(結果):
コホートIIで処置された11人の患者のうち、1人の患者が、最初の処置サイクルの間、グレード3の下痢およびグレード3の脱水のDLTを経験した。9人の患者が、コホートIIIに入力された。2人の患者は、迅速な疾患の進行に起因して、初期に研究を停止したので、28日毒性評価について評価不可能であった。7人の残りの患者のうち、5人が、最初の処置サイクルの間、DLTを経験した:下痢/脱水(n=1)、疲労/脱水(n=1)、食欲不振(n=1)、脱水(n=1)および食欲不振/脱水(n=1)。これらの5人の患者は、研究薬物を持続した後約1週間で回復した。これらの患者は、後に、400mgQDに減少した用量であり、これは十分に耐えられるようであった。コホートIIIにおける全ての患者について400mgのBIDで中央日は、21日であった。これらの知見に基づいて、400mg q12時間投薬スケジュールは、最大寛容用量を超えていると判断した。プロトコル訂正後、1日1回の600mgの投薬でコホートIVで自然増加(accrual)を続けた。コホートIVに登録された7人の患者のうち、2人が、迅速な疾患の進行に起因して、初期に研究を停止したので、28日毒性評価について評価不可能であった。3人の患者は、最初の処置サイクルの間、DLTを経験した:食欲不振/脱水/疲労(n=1)、および下痢/脱水(n=2)。従って、600mgの用量は、最大の許容用量を超えていると判断し、1日当たり1回400mgの用量は、毎日1回の経口投与について最大の許容用量として規定された。連続的に投与される200mgのBIDおよび300mgのBIDで、1日2回の投薬スケジュールのさらなる用量レベルを評価するために、プロトコルを訂正した。
コホートIIで処置された11人の患者のうち、1人の患者が、最初の処置サイクルの間、グレード3の下痢およびグレード3の脱水のDLTを経験した。9人の患者が、コホートIIIに入力された。2人の患者は、迅速な疾患の進行に起因して、初期に研究を停止したので、28日毒性評価について評価不可能であった。7人の残りの患者のうち、5人が、最初の処置サイクルの間、DLTを経験した:下痢/脱水(n=1)、疲労/脱水(n=1)、食欲不振(n=1)、脱水(n=1)および食欲不振/脱水(n=1)。これらの5人の患者は、研究薬物を持続した後約1週間で回復した。これらの患者は、後に、400mgQDに減少した用量であり、これは十分に耐えられるようであった。コホートIIIにおける全ての患者について400mgのBIDで中央日は、21日であった。これらの知見に基づいて、400mg q12時間投薬スケジュールは、最大寛容用量を超えていると判断した。プロトコル訂正後、1日1回の600mgの投薬でコホートIVで自然増加(accrual)を続けた。コホートIVに登録された7人の患者のうち、2人が、迅速な疾患の進行に起因して、初期に研究を停止したので、28日毒性評価について評価不可能であった。3人の患者は、最初の処置サイクルの間、DLTを経験した:食欲不振/脱水/疲労(n=1)、および下痢/脱水(n=2)。従って、600mgの用量は、最大の許容用量を超えていると判断し、1日当たり1回400mgの用量は、毎日1回の経口投与について最大の許容用量として規定された。連続的に投与される200mgのBIDおよび300mgのBIDで、1日2回の投薬スケジュールのさらなる用量レベルを評価するために、プロトコルを訂正した。
暫定的な薬物動態学的な分析は、200mg QD、400mg QD、および400mg BIDの用量レベルで処置された18人の患者に基づく。一般的に、絶食条件下または食物条件下で経口投与されるSAHAのCmaxおよびAUCinfの平均推定値は、200mg〜400mgの用量範囲で、用量に比例して増加した。全体的に、外挿に起因したAUCinfの割合は、1%以下であった。見かけの半減期についての平均推定値は、絶食条件下または食物条件下で用量群にわたって、種々であり、61〜114分の範囲であった。Cmaxの平均推定値は、233ng/ml(0.88μM)から570ng/ml(2.3μM)まで変化する。SAHAのバイオアベイラビリティーの割合(IV注入および経口経路後のAUCinf値から計算される)は、約0.48であることが見出された。
末梢血単核細胞を、処置前、注入直後およびSAHAカプセルの経口摂取後2〜10時間で収集して、正常な宿主細胞におけるヒストンアセチル化の程度に対するSAHAの効果を評価した。ヒストンを単離し、そして抗アセチル化ヒストン(H3)抗体、続いて、HRP−二次抗体でプローブした。予備的分析は、1日当たり400mgの用量レベルでのSAHAカプセルの消化後10時間までに検出され得る末梢単核細胞のアセチル化ヒストンの蓄積の増加を実証した。
応答性の疾患または安定な疾患を有する13人の患者で、3〜12ヶ月間処置を続けた:甲状腺(n=3)、汗腺(n=1)、腎臓(n=2)、喉頭(n=1)、前立腺(n=1)、ホジキンリンパ腫(n=2)、非ホジキンリンパ腫(n=2)、および白血病(n=1)。
6人の患者は、CTスキャンにおいて腫瘍収縮を有した。これら6人のうち3人の患者は、部分的応答の基準に適合する(転移性咽頭癌を有する1人の患者および非ホジキンリンパ腫を有する2人の患者)。これらの部分的応答は、400mg BID(n=2)および600mg QD(n=1)の用量レベルで生じた。
(SAHAの静脈内投薬)
表5は、静脈内でSAHAを受容する患者についての投薬スケジュールを示す。患者は、コホートIで開始し、300mg/m2のSAHAを、1週間週に5日の連続した日で、合計1500mg/m2の用量を受容する。次いで、患者は、処置が、疾患の進行、腫瘍後退、受容不可能な副作用または他の処置を受けた患者に起因して終了しない限り、2週間の間、観察され、そしてコホートIIに続けられ、次いで、コホートを通じて進めた。
表5は、静脈内でSAHAを受容する患者についての投薬スケジュールを示す。患者は、コホートIで開始し、300mg/m2のSAHAを、1週間週に5日の連続した日で、合計1500mg/m2の用量を受容する。次いで、患者は、処置が、疾患の進行、腫瘍後退、受容不可能な副作用または他の処置を受けた患者に起因して終了しない限り、2週間の間、観察され、そしてコホートIIに続けられ、次いで、コホートを通じて進めた。
本発明は、その好ましい実施形態を参照して詳細に示され、そして記載されるものの、形態および詳細において種々の変更が、記載される発明の意義から逸脱することなく、なされ得ることが当業者に理解される。むしろ、本発明の範囲は、添付の特許請求の範囲によって規定される。
(参考文献)
Claims (14)
- 結晶形態のスベロイルアニリドヒドロキサム酸(SAHA)を製造する方法であって、粗生成SAHAをメタノール及び水の混合物から再結晶する工程を含む方法。
- メタノールと水の混合比が2:1である請求項1に記載の方法。
- 粗生成SAHAをメタノール及び水の混合物から再結晶する工程を含む方法によって得られる結晶形態のSAHAの物理的特性を有するSAHAの結晶形態からなる活性成分。
- メタノールと水の混合比が2:1である請求項3に記載の活性成分。
- 結晶形態のスベロイルアニリドヒドロキサム酸(SAHA)を製造する方法であって、
(a)スベリン酸をアニリンと反応させ、下記構造を有するスベルアニリン酸又はその塩を生成させる工程、
(d)メタノール及び水の混合物からSAHAの粗生成物を再結晶する工程、
を含む方法。 - メタノールと水の混合比が2:1である請求項5に記載の方法。
- 工程(c)が、さらに
(1)反応混合物にナトリウムメトキシドを加え透明な溶液を得る工程、及び
(2)氷酢酸を該透明な溶液に加え粗スベロイルアニリドヒドロキサム酸を含む沈殿を生成させる工程を含む請求項5に記載の方法。 - 以下の工程を含む方法によって得られる結晶形態のSAHAの物理的特性を有するスベロイルアニリドヒドロキサム酸(SAHA)の結晶形態からなる活性成分、
(a)スベリン酸をアニリンと反応させ、下記構造を有するスベルアニリン酸又はその塩を生成させる工程、
(d)SAHAの粗生成物をメタノールと水の混合物で再結晶する工程。 - メタノールと水の混合比が2:1である請求項8に記載の活性成分。
- 工程(c)がさらに、
(1)ナトリウムメトキシドを反応混合物に加え、透明な溶液を得る工程、及び
(2)氷酢酸を該透明な溶液に加えて、粗スベロイルアニリドヒドロキサム酸を含む沈殿を得る工程、
を含む請求項8に記載の活性成分。 - スベロイルアニリドヒドロキサム酸(SAHA)を含む経口投与の薬学的組成物であって、該SAHAは、メタノールと水の混合物でSAHAの粗生成物を再結晶する工程を含む方法によって得られる結晶性のSAHAの物理的性質を有する結晶性のSAHAを含む薬学的組成物。
- 結晶形態のSAHAが以下の工程によって得られる請求項11に記載の薬学的組成物、
(a)スベリン酸をアニリンと反応させ、下記の構造で表されるスベルアニリン酸またはその塩を生成させる工程、
(d)メタノールと水の混合物からSAHAの粗生成物を再結晶する工程。 - 結晶形態のSAHAが以下の工程によって得られる請求項11に記載の薬学的組成物、
(a)スベリン酸をアニリンと反応させ下記の構造を有するスベルアニリン酸を生成する工程、
(d)ナトリウムメトキシドを反応混合物に加え透明な溶液を得る工程、
(e)該透明な溶液に氷酢酸を加え、粗スベロイルアニリドヒドロキサム酸を含む沈殿を生成させる工程、及び
(f)メタノールと水の混合物からSAHAの粗生成物を再結晶する工程。 - メタノールと水の混合比が2:1である請求項11〜13のいずれかに記載の薬学的組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US36175902P | 2002-03-04 | 2002-03-04 | |
| US60/361,759 | 2002-03-04 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009188712A Division JP5675071B2 (ja) | 2002-03-04 | 2009-08-17 | 最終分化を誘導する方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2014237669A true JP2014237669A (ja) | 2014-12-18 |
Family
ID=27805074
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003574115A Expired - Lifetime JP4732693B2 (ja) | 2002-03-04 | 2003-03-04 | 最終分化を誘導する方法 |
| JP2009188712A Expired - Lifetime JP5675071B2 (ja) | 2002-03-04 | 2009-08-17 | 最終分化を誘導する方法 |
| JP2009188710A Expired - Lifetime JP5586896B2 (ja) | 2002-03-04 | 2009-08-17 | 最終分化を誘導する方法 |
| JP2014146512A Pending JP2014237669A (ja) | 2002-03-04 | 2014-07-17 | 最終分化を誘導する方法 |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003574115A Expired - Lifetime JP4732693B2 (ja) | 2002-03-04 | 2003-03-04 | 最終分化を誘導する方法 |
| JP2009188712A Expired - Lifetime JP5675071B2 (ja) | 2002-03-04 | 2009-08-17 | 最終分化を誘導する方法 |
| JP2009188710A Expired - Lifetime JP5586896B2 (ja) | 2002-03-04 | 2009-08-17 | 最終分化を誘導する方法 |
Country Status (18)
| Country | Link |
|---|---|
| US (7) | US20040072735A1 (ja) |
| EP (4) | EP2082737B1 (ja) |
| JP (4) | JP4732693B2 (ja) |
| KR (2) | KR100868813B1 (ja) |
| CN (3) | CN103393630A (ja) |
| AU (1) | AU2003213684C1 (ja) |
| BR (1) | BR0308250A (ja) |
| CA (3) | CA2671976C (ja) |
| ES (1) | ES2532607T3 (ja) |
| HK (1) | HK1072362A1 (ja) |
| IL (3) | IL163909A0 (ja) |
| MX (1) | MXPA04008577A (ja) |
| NO (2) | NO329984B1 (ja) |
| NZ (3) | NZ567758A (ja) |
| PL (1) | PL372239A1 (ja) |
| RU (3) | RU2394022C2 (ja) |
| WO (1) | WO2003075839A2 (ja) |
| ZA (2) | ZA200407942B (ja) |
Families Citing this family (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6777217B1 (en) | 1996-03-26 | 2004-08-17 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| US20030129724A1 (en) | 2000-03-03 | 2003-07-10 | Grozinger Christina M. | Class II human histone deacetylases, and uses related thereto |
| US7244853B2 (en) | 2001-05-09 | 2007-07-17 | President And Fellows Of Harvard College | Dioxanes and uses thereof |
| AU2002340253C1 (en) * | 2001-10-16 | 2011-03-31 | Sloan-Kettering Institute For Cancer Research | Treatment of neurodegenerative diseases and cancer of the brain |
| EP1482962A4 (en) * | 2002-02-15 | 2009-12-23 | Sloan Kettering Inst Cancer | METHOD OF TREATING THIOREDOXIN-MEDIATED DISEASES (TRX) |
| US20060276547A1 (en) * | 2002-03-04 | 2006-12-07 | Bacopoulos Nicholas G | Methods of treating cancer with HDAC inhibitors |
| US20040132825A1 (en) * | 2002-03-04 | 2004-07-08 | Bacopoulos Nicholas G. | Methods of treating cancer with HDAC inhibitors |
| US20070060614A1 (en) * | 2002-03-04 | 2007-03-15 | Bacopoulos Nicholas G | Methods of treating cancer with hdac inhibitors |
| EP2082737B1 (en) * | 2002-03-04 | 2014-12-31 | Merck HDAC Research, LLC | Methods of inducing terminal differentiation |
| US7456219B2 (en) | 2002-03-04 | 2008-11-25 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7148257B2 (en) | 2002-03-04 | 2006-12-12 | Merck Hdac Research, Llc | Methods of treating mesothelioma with suberoylanilide hydroxamic acid |
| TWI319387B (en) | 2002-04-05 | 2010-01-11 | Astrazeneca Ab | Benzamide derivatives |
| WO2003088954A1 (en) * | 2002-04-15 | 2003-10-30 | Sloan-Kettering Institute For Cancer Research | Combination therapy for the treatment of cancer |
| TW559390U (en) * | 2002-08-27 | 2003-10-21 | Molex Inc | Electrical connector |
| FR2847817B1 (fr) * | 2002-11-28 | 2006-11-10 | Centre Nat Rech Scient | Utilisation d'un inhibiteur d'histone deacetylase pour le traitement des dystrophies musculaires |
| KR20050122210A (ko) * | 2003-03-17 | 2005-12-28 | 다케다 샌디에고, 인코포레이티드 | 히스톤 탈아세틸화 효소 억제제 |
| CN1839121A (zh) * | 2003-04-01 | 2006-09-27 | 斯隆-凯特林癌症研究所 | 异羟肟酸化合物及其使用方法 |
| PL1651612T3 (pl) | 2003-07-22 | 2012-09-28 | Astex Therapeutics Ltd | Związki 3,4-pochodne 1h-pirazolu i ich zastosowanie jako kinazy zależne od cyklin (cdk) i modulatory kinazy syntazy glikogenu-3 (gsk-3) |
| KR20050022216A (ko) * | 2003-08-25 | 2005-03-07 | 윤덕구 | 동결건조 즉석소면 및 그 제조방법 |
| ATE462426T1 (de) * | 2003-08-26 | 2010-04-15 | Merck Hdac Res Llc | Verwendung von saha zur behandlung von mesotheliom |
| US20050137234A1 (en) * | 2003-12-19 | 2005-06-23 | Syrrx, Inc. | Histone deacetylase inhibitors |
| WO2005065681A1 (en) * | 2003-12-19 | 2005-07-21 | Takeda San Diego, Inc. | N- hydroxy-3-(3-(1h-imidazol-2-yl)-phenyl)-acrylamide derivatives and related compounds as histone deacetylase (hdac) inhibitors for the treatment of cancer |
| MX2007001550A (es) * | 2004-08-09 | 2007-04-10 | Astellas Pharma Inc | Compuestos de hidroxiamida que tienen actividad como inhibidores de histona desacetilasa (hdac). |
| CN101115495B (zh) * | 2004-12-14 | 2011-06-22 | 生物如恩克斯株式会社 | 通过runx2乙酰化作用增强bmp的骨形成活性的方法 |
| US7642275B2 (en) * | 2004-12-16 | 2010-01-05 | Takeda San Diego, Inc. | Histone deacetylase inhibitors |
| AU2006207321B2 (en) * | 2005-01-21 | 2012-09-06 | Astex Therapeutics Limited | Pharmaceutical compounds |
| US8404718B2 (en) | 2005-01-21 | 2013-03-26 | Astex Therapeutics Limited | Combinations of pyrazole kinase inhibitors |
| AR054425A1 (es) | 2005-01-21 | 2007-06-27 | Astex Therapeutics Ltd | Sales de adicion de piperidin 4-il- amida de acido 4-(2,6-dicloro-benzoilamino) 1h-pirazol-3-carboxilico. |
| AU2006207322A1 (en) * | 2005-01-21 | 2006-07-27 | Astex Therapeutics Limited | Combinations of pyrazole kinase inhibitors and further antitumor agents |
| EP1845973B1 (en) * | 2005-01-21 | 2015-08-12 | Astex Therapeutics Limited | Pharmaceutical compounds |
| WO2006082428A2 (en) | 2005-02-03 | 2006-08-10 | Topotarget Uk Limited | Combination therapies using hdac inhibitors |
| EP2491926B1 (en) | 2005-03-22 | 2018-05-09 | President and Fellows of Harvard College | Treatment of protein degradation disorders |
| WO2006113606A2 (en) * | 2005-04-18 | 2006-10-26 | Johns Hopkins University | Histone deacetylase inhibitors |
| EP1715334A1 (fr) * | 2005-04-22 | 2006-10-25 | Adamant Technologies SA | Procédé utilisant un capteur électrochimique et électrodes formant ce capteur |
| US20090215800A1 (en) * | 2005-05-05 | 2009-08-27 | Chroma Therapeutics Ltd | Enzyme and Receptor Modulation |
| US7642253B2 (en) * | 2005-05-11 | 2010-01-05 | Takeda San Diego, Inc. | Histone deacetylase inhibitors |
| PT1901729E (pt) * | 2005-05-13 | 2012-04-30 | Topotarget Uk Ltd | Formulações farmacêuticas de inibidores de hdac |
| TWI415603B (zh) * | 2005-05-20 | 2013-11-21 | Merck Sharp & Dohme | 1,8-辛二醯基苯胺羥胺酸(suberoylanilide hydroxamic acid)之調配物及其製配方法 |
| EP2229941A1 (en) * | 2005-05-20 | 2010-09-22 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| JP2009501236A (ja) * | 2005-07-14 | 2009-01-15 | タケダ サン ディエゴ インコーポレイテッド | ヒストンデアセチラーゼ阻害剤 |
| CA2621560A1 (en) * | 2005-09-07 | 2007-03-15 | Braincells, Inc. | Modulation of neurogenesis by hdac inhibition |
| GB0518720D0 (en) * | 2005-09-14 | 2005-10-19 | Hamlet Pharma Ab | Therapeutic combination |
| WO2007117272A2 (en) * | 2005-09-30 | 2007-10-18 | The Henry M. Jackson Foundation For The Advancement Of Military Medicine | Methods for treatment of hemorrhagic shock and related disorders |
| CA2626679C (en) * | 2005-11-04 | 2011-08-16 | Merck & Co., Inc. | Methods of treating cancers with saha, carboplatin, and paclitaxel and other combination therapies |
| WO2007056244A2 (en) * | 2005-11-04 | 2007-05-18 | Merck & Co., Inc. | Methods of using saha and erlotinib for treating cancer |
| EP1947936A4 (en) * | 2005-11-04 | 2010-02-10 | Merck & Co Inc | METHOD FOR THE USE OF SAHA AND BORTEZOMIB FOR THE TREATMENT OF CANCER |
| AU2006313517B2 (en) | 2005-11-10 | 2013-06-27 | Topotarget Uk Limited | Histone deacetylase (HDAC) inhibitors (PXD101) for the treatment of cancer alone or in combination with chemotherapeutic agent |
| CA2630216A1 (en) * | 2005-11-18 | 2007-05-31 | Gloucester Pharmaceuticals, Inc. | Metabolite derivatives of the hdac inhibitor fk228 |
| EP1960384A4 (en) * | 2005-12-05 | 2010-03-24 | Astrazeneca Ab | NOVEL PROCESS FOR THE PRODUCTION OF NON-SALINE ESOMEPRAZOLE |
| US20070207950A1 (en) * | 2005-12-21 | 2007-09-06 | Duke University | Methods and compositions for regulating HDAC6 activity |
| JP2009525955A (ja) * | 2006-01-13 | 2009-07-16 | タケダ サン ディエゴ インコーポレイテッド | ヒストンデアセチラーゼ阻害剤 |
| CN101400362B (zh) | 2006-02-14 | 2016-10-12 | 哈佛大学校长及研究员协会 | 双官能组蛋白去乙酰化酶抑制剂 |
| MX2008010462A (es) | 2006-02-14 | 2009-04-17 | Harvard College | Inhibidores de histona desacetilasa. |
| US20100137239A1 (en) * | 2006-04-24 | 2010-06-03 | Gloucester Pharmaceuticals | Gemcitabine combination therapy |
| CA2654540C (en) * | 2006-05-03 | 2017-01-17 | President And Fellows Of Harvard College | Histone deacetylase and tubulin deacetylase inhibitors |
| WO2007129062A1 (en) * | 2006-05-08 | 2007-11-15 | Astex Therapeutics Limited | Pharmaceutical combinations of diazole derivatives for cancer treatment |
| EP2018366A4 (en) * | 2006-05-16 | 2010-08-04 | Univ Mcgill | HYBRID MOLECULES HAVING MIXED PROPERTIES OF VITAMIN D RECEPTOR AGONISM AND HISTONE DEACETYLASE INHIBITOR |
| US8957027B2 (en) * | 2006-06-08 | 2015-02-17 | Celgene Corporation | Deacetylase inhibitor therapy |
| CA2663569A1 (en) * | 2006-09-28 | 2008-04-03 | Merck & Co., Inc. | Pharmaceutical compositions of hdac inhibitors and chelatable metal compounds, and metal-hdac inhibitor chelate complexes |
| CA2668070A1 (en) * | 2006-10-30 | 2008-05-08 | Chroma Therapeutics Ltd. | Hydroxamates as inhibitors of histone deacetylase |
| EP2086323A4 (en) * | 2006-11-03 | 2010-01-06 | Univ Maryland | METHOD OF USE OF SAHA AND BORTEZOMIB FOR THE TREATMENT OF MULTIPLE MYELOMA |
| WO2008083288A2 (en) * | 2006-12-29 | 2008-07-10 | Gloucester Pharmaceuticals | Purifiction of romidepsin |
| EP2815761A1 (en) * | 2006-12-29 | 2014-12-24 | Celgene Corporation | Romidepsin-based treatments for cancer |
| EP2124916A1 (en) * | 2007-02-27 | 2009-12-02 | Government of the United States of America, as represented by the Secretary, Department of Health and Human Services | Use of histone deacetylase inhibitors for the treatment of central nervous system metastases |
| CA2700173C (en) * | 2007-09-25 | 2016-10-11 | Topotarget Uk Limited | Methods of synthesis of certain hydroxamic acid compounds |
| RS52343B (en) * | 2007-12-20 | 2012-12-31 | Pharma Mar S.A. | ANTITUMOR UNITS |
| PT2262493E (pt) * | 2008-03-07 | 2015-06-02 | Topotarget As | Métodos de tratamento empregando infusão contínua prolongada de belinostat |
| RU2515611C2 (ru) | 2008-07-23 | 2014-05-20 | Президент Энд Феллоуз Оф Гарвард Колледж | Ингибиторы деацетилазы и их применение |
| NZ592686A (en) | 2008-10-15 | 2012-12-21 | Generics Uk Ltd | Process for the preparation of vorinostat from aniline, hydroxylamine and suberic acid starting materials |
| CN102282126A (zh) | 2008-11-26 | 2011-12-14 | 基因里克斯(英国)有限公司 | 多晶型物 |
| GB0900555D0 (en) * | 2009-01-14 | 2009-02-11 | Topotarget As | New methods |
| EP2236503B1 (en) | 2009-04-03 | 2014-02-26 | NatureWise Biotech & Medicals Corporation | Cinamic compounds and derivatives therefrom for the inhibition of histone deacetylase |
| US7994357B2 (en) * | 2009-04-03 | 2011-08-09 | Naturewise Biotech & Medicals Corporation | Cinamic compounds and derivatives therefrom for the inhibition of histone deacetylase |
| EP2277387B1 (en) | 2009-07-22 | 2016-10-19 | NatureWise Biotech & Medicals Corporation | New use of histone deacetylase inhibitors in changing mrjp3 protein in royal jelly |
| WO2011019393A2 (en) | 2009-08-11 | 2011-02-17 | President And Fellows Of Harvard College | Class- and isoform-specific hdac inhibitors and uses thereof |
| JP2013508458A (ja) | 2009-10-26 | 2013-03-07 | ラモット・アット・テル−アヴィヴ・ユニヴァーシティ・リミテッド | Ftsとhdac阻害剤との組合せを用いたがん治療 |
| WO2011084623A1 (en) | 2009-12-16 | 2011-07-14 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Method of sensitizing cancer cells to the cytotoxic effects of death receptor ligands in cancer treatment |
| US20110195400A1 (en) * | 2010-02-10 | 2011-08-11 | Selinfreund Richard H | Systems and methods for diagnosing cancer |
| WO2011127467A1 (en) * | 2010-04-09 | 2011-10-13 | Companion Diagnostics, Inc. | Devices, systems, and methods for biomarker stabilization |
| MX2012012695A (es) | 2010-05-05 | 2013-02-26 | Teva Womens Health Inc | Metodos para reducir sintomas en sujetos utilizando formas de dosificacion individuales con velocidades de liberacion que disminuyen gradualmente. |
| ES2755909T3 (es) | 2010-07-12 | 2020-04-24 | Celgene Corp | Formas sólidas de la romidepsina y sus usos |
| US8859502B2 (en) | 2010-09-13 | 2014-10-14 | Celgene Corporation | Therapy for MLL-rearranged leukemia |
| DK2624832T3 (en) * | 2010-10-08 | 2018-01-08 | Vib Vzw | HDAC INHIBITORS FOR TREATMENT OF CHARCOT-MARIE-TOOTHS DISEASE |
| US20120164674A1 (en) * | 2010-10-28 | 2012-06-28 | Selinfreund Richard H | Devices and washes for biomarker stabilization |
| EP3750544A3 (en) | 2011-11-30 | 2021-03-24 | Emory University | Jak inhibitors for use in the prevention or treatment of viral infection |
| CN102793693A (zh) * | 2012-09-07 | 2012-11-28 | 天津医科大学 | 伏立诺他在制备治疗自身免疫及炎症性疾病药物方面的应用 |
| AU2013202506B2 (en) | 2012-09-07 | 2015-06-18 | Celgene Corporation | Resistance biomarkers for hdac inhibitors |
| AU2013202507B9 (en) | 2012-11-14 | 2015-08-13 | Celgene Corporation | Inhibition of drug resistant cancer cells |
| EP2968565A2 (en) | 2013-03-14 | 2016-01-20 | Genentech, Inc. | Methods of treating cancer and preventing cancer drug resistance |
| NZ630311A (en) | 2013-12-27 | 2016-03-31 | Celgene Corp | Romidepsin formulations and uses thereof |
| CN103819364B (zh) * | 2014-01-24 | 2015-12-02 | 中南大学 | N-烃酰基环内酰胺衍生物的一锅合成方法 |
| CN107405321A (zh) | 2015-01-23 | 2017-11-28 | 坦普尔大学 | 短链脂肪酸在癌症预防中的应用 |
| WO2016164392A1 (en) | 2015-04-06 | 2016-10-13 | The Johns Hopkins University | A h3t3a mutant protein efficiently reduces h3t3p and causes increased cell death of rapidly dividing cells |
| CN105055386A (zh) * | 2015-08-12 | 2015-11-18 | 天津医科大学总医院 | 伏立诺他用于制备免疫调节药物的应用 |
| CN106187818B (zh) * | 2016-06-27 | 2017-12-08 | 刘美新 | 一种制备抗癌药物伏立诺他的方法 |
| US11065217B2 (en) | 2017-01-27 | 2021-07-20 | Temple University—Of the Commonwealth System of Higher Education | Use of short chain fatty acids for the treatment and prevention of diseases and disorders |
| CN110891982B (zh) | 2017-04-17 | 2023-12-22 | 芝加哥大学 | 向肠道递送短链脂肪酸以用于人体保健和疾病治疗的聚合物材料 |
| US11453661B2 (en) | 2019-09-27 | 2022-09-27 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| US11583521B2 (en) * | 2020-07-01 | 2023-02-21 | Jubilant Pharma Holdings Inc. | Long-acting injection dosage form of beta 3 adrenoreceptor agonists |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS51125238A (en) * | 1974-12-20 | 1976-11-01 | Klinge Co Chem Pharm Fab | Process for preparing novel substituted propanol*2**derivative and its nicotic acid ester |
| JPS61251649A (ja) * | 1985-04-05 | 1986-11-08 | Mitsubishi Petrochem Co Ltd | ジカルボン酸ジアニリドの製造法 |
| JPH07502494A (ja) * | 1991-10-04 | 1995-03-16 | スローン−ケタリング・インスティテュート・フォー・キャンサー・リサーチ | 新規の有効な末端分化誘発剤およびその使用方法 |
| WO1995031977A1 (en) * | 1994-05-19 | 1995-11-30 | Sloan-Kettering Institute For Cancer Research | Novel potent inducers of terminal differentiation and methods of use thereof |
| JP2003509343A (ja) * | 1999-09-08 | 2003-03-11 | スローン−ケターリング インスティチュート フォー キャンサー リサーチ | 新規クラスの細胞分化剤およびヒストンデアセチラーゼならびにそれらの使用方法 |
| JP5675071B2 (ja) * | 2002-03-04 | 2015-02-25 | メルク・エイチ・デイ・エイ・シー・リサーチ,エル・エル・シー | 最終分化を誘導する方法 |
Family Cites Families (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA622801A (en) | 1961-06-27 | L. Clark Robert | Substituted benzimidazolones | |
| FR901228A (fr) | 1943-01-16 | 1945-07-20 | Deutsche Edelstahlwerke Ag | Système d'aimant à entrefer annulaire |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| JPS61176523A (ja) * | 1985-01-30 | 1986-08-08 | Teruhiko Beppu | 制癌剤 |
| US5055608A (en) | 1988-11-14 | 1991-10-08 | Sloan-Kettering Institute For Cancer Research | Novel potent inducers of thermal differentiation and method of use thereof |
| US5175191A (en) | 1988-11-14 | 1992-12-29 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| US5608108A (en) | 1988-11-14 | 1997-03-04 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and method of use thereof |
| USRE38506E1 (en) * | 1991-10-04 | 2004-04-20 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| US5635532A (en) * | 1991-10-21 | 1997-06-03 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Compositions and methods for therapy and prevention of pathologies including cancer, AIDS and anemia |
| HU217629B (hu) | 1991-12-12 | 2000-03-28 | Novartis Ag. | Eljárás fluvasztatint tartalmazó stabilizált gyógyszerkészítmények előállítására |
| CA2231251A1 (en) | 1995-09-20 | 1997-03-27 | Merck & Co., Inc. | Histone deacetylase as target for antiprotozoal agents |
| US6239178B1 (en) * | 1996-05-17 | 2001-05-29 | George Dimelow Alexander Lord | Method of treating mammals |
| US6030961A (en) | 1997-03-11 | 2000-02-29 | Bar-Ilan Research & Development Co., Ltd. | Oxyalkylene phosphate compounds and uses thereof |
| US6043389A (en) * | 1997-03-11 | 2000-03-28 | Mor Research Applications, Ltd. | Hydroxy and ether-containing oxyalkylene esters and uses thereof |
| US6124495A (en) | 1997-03-11 | 2000-09-26 | Beacon Laboratories, Inc. | Unsaturated oxyalkylene esters and uses thereof |
| JPH10262694A (ja) | 1997-03-28 | 1998-10-06 | Sumitomo Forestry Co Ltd | B細胞性リンパ腫の骨髄転移検査法 |
| US6387673B1 (en) | 1997-05-01 | 2002-05-14 | The Salk Institute For Biological Studies | Compounds useful for the modulation of processes mediated by nuclear hormone receptors, methods for the identification and use of such compounds |
| US6231880B1 (en) * | 1997-05-30 | 2001-05-15 | Susan P. Perrine | Compositions and administration of compositions for the treatment of blood disorders |
| AUPO721997A0 (en) | 1997-06-06 | 1997-07-03 | Queensland Institute Of Medical Research, The | Anticancer compounds |
| US6262116B1 (en) * | 1998-01-23 | 2001-07-17 | Sloan-Kettering Institute For Cancer Research | Transcription therapy for cancers |
| DK1051187T3 (da) | 1998-01-30 | 2004-03-08 | Univ New York | Behandling af follikulære lymfomer ved anvendelse af inhibitorer af lymfotoxin(LT)-syntesevejen |
| AUPP505798A0 (en) | 1998-08-04 | 1998-08-27 | Fujisawa Pharmaceutical Co., Ltd. | Novel compound fr225497 substance |
| NZ510504A (en) * | 1998-09-25 | 2003-09-26 | Warner Lambert Co | Chemotherapy of cancer with acetyldinaline in combination with gemcitabine, capecitabine or cisplatin |
| CZ20011342A3 (cs) | 1998-10-13 | 2001-09-12 | Fujisawa Pharmaceutical Co., Ltd. | Cyklické tetrapeptidy |
| AU6718200A (en) | 1999-05-03 | 2000-12-12 | Methylgene, Inc. | Inhibition of histone deacetylase |
| US6450992B1 (en) | 1999-07-02 | 2002-09-17 | Smith & Nephew, Inc. | Cannula interface |
| JP2001081031A (ja) | 1999-08-30 | 2001-03-27 | Schering Ag | 溶解性および経口吸収性を改善したベンズアミド誘導体含有製剤 |
| US6673564B2 (en) | 1999-10-18 | 2004-01-06 | Millennium Pharmaceuticals, Inc. | Methods for using 22045, a human cyclic nucleotide phosphodiesterase |
| KR20020070285A (ko) | 1999-11-23 | 2002-09-05 | 메틸진, 인크. | 히스톤 디아세틸라제의 억제제 |
| AU2001248701A1 (en) | 2000-03-24 | 2001-10-03 | Methylgene, Inc. | Inhibitors of histone deacetylase |
| AU2001285042A1 (en) | 2000-08-18 | 2002-03-04 | The Governement Of The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Methods of treating cutaneous and peripheral t-cell lymphoma by a histone deacetylase inhibitor |
| PE20020354A1 (es) | 2000-09-01 | 2002-06-12 | Novartis Ag | Compuestos de hidroxamato como inhibidores de histona-desacetilasa (hda) |
| AU2001287157A1 (en) * | 2000-09-12 | 2002-03-26 | Virginia Commonwealth University | Promotion of adoptosis in cancer cells by co-administration of cyclin dependent kinase inhibitors and cellular differentiation agents |
| GB0023983D0 (en) | 2000-09-29 | 2000-11-15 | Prolifix Ltd | Therapeutic compounds |
| AU9013101A (en) | 2000-09-29 | 2002-04-22 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| AU2002243231A1 (en) * | 2000-11-21 | 2002-07-24 | Wake Forest University | Method of treating autoimmune diseases |
| AR035659A1 (es) | 2000-12-07 | 2004-06-23 | Hoffmann La Roche | Hidroxiamidas de acido (1-oxo-1,2,3,4-tetrahidro-naftalen-2-il)-alcanoico, proceso para la manufactura de estos compuestos, composiciones farmaceuticas que contienen dichos compuestos y los usos de los mismos |
| US6533803B2 (en) * | 2000-12-22 | 2003-03-18 | Advanced Medical Applications, Inc. | Wound treatment method and device with combination of ultrasound and laser energy |
| WO2002060430A1 (en) * | 2001-02-01 | 2002-08-08 | Cornell Research Foundation, Inc. | Use of retinoids plus histone deacetylase inhibitors to inhibit the growth of solid tumors |
| US7314953B2 (en) * | 2001-03-27 | 2008-01-01 | Errant Gene Therapeutics, Llc | Treatment of lung cells with histone deacetylase inhibitors |
| US6495719B2 (en) * | 2001-03-27 | 2002-12-17 | Circagen Pharmaceutical | Histone deacetylase inhibitors |
| US6905669B2 (en) * | 2001-04-24 | 2005-06-14 | Supergen, Inc. | Compositions and methods for reestablishing gene transcription through inhibition of DNA methylation and histone deacetylase |
| CA2450129A1 (en) * | 2001-06-14 | 2002-12-27 | Donald G. Jackson | Novel human histone deacetylases |
| ITMI20011733A1 (it) | 2001-08-07 | 2003-02-07 | Italfarmaco Spa | Derivati dell'acido idrossamico inibitori degli enzimi istone deacetilasi, quali nuovi farmaci antiinfiammatori inibenti la sintesi di citoc |
| AU2002340253C1 (en) * | 2001-10-16 | 2011-03-31 | Sloan-Kettering Institute For Cancer Research | Treatment of neurodegenerative diseases and cancer of the brain |
| US20040132643A1 (en) * | 2002-01-09 | 2004-07-08 | Fojo Antonio Tito | Histone deacelylase inhibitors in diagnosis and treatment of thyroid neoplasms |
| EP1482962A4 (en) * | 2002-02-15 | 2009-12-23 | Sloan Kettering Inst Cancer | METHOD OF TREATING THIOREDOXIN-MEDIATED DISEASES (TRX) |
| US20060276547A1 (en) * | 2002-03-04 | 2006-12-07 | Bacopoulos Nicholas G | Methods of treating cancer with HDAC inhibitors |
| US20040132825A1 (en) * | 2002-03-04 | 2004-07-08 | Bacopoulos Nicholas G. | Methods of treating cancer with HDAC inhibitors |
| US20070060614A1 (en) * | 2002-03-04 | 2007-03-15 | Bacopoulos Nicholas G | Methods of treating cancer with hdac inhibitors |
| US7456219B2 (en) * | 2002-03-04 | 2008-11-25 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7148257B2 (en) * | 2002-03-04 | 2006-12-12 | Merck Hdac Research, Llc | Methods of treating mesothelioma with suberoylanilide hydroxamic acid |
| WO2003088954A1 (en) * | 2002-04-15 | 2003-10-30 | Sloan-Kettering Institute For Cancer Research | Combination therapy for the treatment of cancer |
| CN1839121A (zh) * | 2003-04-01 | 2006-09-27 | 斯隆-凯特林癌症研究所 | 异羟肟酸化合物及其使用方法 |
| ATE462426T1 (de) * | 2003-08-26 | 2010-04-15 | Merck Hdac Res Llc | Verwendung von saha zur behandlung von mesotheliom |
| CN102349927A (zh) * | 2003-08-29 | 2012-02-15 | Hdac默克研究有限责任公司 | 联合治疗癌症的方法 |
| US20090227674A1 (en) | 2005-08-18 | 2009-09-10 | Richon Victoria M | Combination methods fo saha and targretin for treating cancer |
| CA2626679C (en) | 2005-11-04 | 2011-08-16 | Merck & Co., Inc. | Methods of treating cancers with saha, carboplatin, and paclitaxel and other combination therapies |
| WO2007056244A2 (en) * | 2005-11-04 | 2007-05-18 | Merck & Co., Inc. | Methods of using saha and erlotinib for treating cancer |
| EP1947936A4 (en) * | 2005-11-04 | 2010-02-10 | Merck & Co Inc | METHOD FOR THE USE OF SAHA AND BORTEZOMIB FOR THE TREATMENT OF CANCER |
-
2003
- 2003-03-04 EP EP20090005103 patent/EP2082737B1/en not_active Expired - Lifetime
- 2003-03-04 MX MXPA04008577A patent/MXPA04008577A/es active IP Right Grant
- 2003-03-04 CA CA2671976A patent/CA2671976C/en not_active Expired - Lifetime
- 2003-03-04 EP EP03711372A patent/EP1487426B1/en not_active Revoked
- 2003-03-04 NZ NZ567758A patent/NZ567758A/en not_active IP Right Cessation
- 2003-03-04 KR KR1020077003262A patent/KR100868813B1/ko active IP Right Grant
- 2003-03-04 RU RU2007112879A patent/RU2394022C2/ru active
- 2003-03-04 US US10/379,149 patent/US20040072735A1/en not_active Abandoned
- 2003-03-04 PL PL37223903A patent/PL372239A1/xx unknown
- 2003-03-04 ES ES09005103.8T patent/ES2532607T3/es not_active Expired - Lifetime
- 2003-03-04 JP JP2003574115A patent/JP4732693B2/ja not_active Expired - Lifetime
- 2003-03-04 CN CN2013102683183A patent/CN103393630A/zh active Pending
- 2003-03-04 CA CA 2478094 patent/CA2478094C/en not_active Expired - Lifetime
- 2003-03-04 WO PCT/US2003/006451 patent/WO2003075839A2/en active IP Right Grant
- 2003-03-04 NZ NZ55018503A patent/NZ550185A/xx not_active IP Right Cessation
- 2003-03-04 CA CA 2632078 patent/CA2632078C/en not_active Expired - Lifetime
- 2003-03-04 EP EP20100183586 patent/EP2322160A1/en not_active Withdrawn
- 2003-03-04 CN CNA038095890A patent/CN1720034A/zh active Pending
- 2003-03-04 IL IL16390903A patent/IL163909A0/xx unknown
- 2003-03-04 BR BR0308250A patent/BR0308250A/pt not_active Application Discontinuation
- 2003-03-04 AU AU2003213684A patent/AU2003213684C1/en active Active
- 2003-03-04 RU RU2004133675A patent/RU2320331C2/ru active Protection Beyond IP Right Term
- 2003-03-04 EP EP20100184834 patent/EP2266552A3/en not_active Withdrawn
- 2003-03-04 NZ NZ535578A patent/NZ535578A/en not_active IP Right Cessation
- 2003-03-04 KR KR1020087010088A patent/KR100892016B1/ko active IP Right Grant
- 2003-03-04 CN CN2008100838935A patent/CN101259120B/zh not_active Expired - Lifetime
- 2003-07-09 US US10/616,649 patent/US7399787B2/en active Active
- 2003-09-16 US US10/665,079 patent/US20040127523A1/en not_active Abandoned
-
2004
- 2004-03-01 ZA ZA200407942A patent/ZA200407942B/xx unknown
- 2004-09-05 IL IL16390904A patent/IL163909A/en not_active IP Right Cessation
- 2004-09-28 NO NO20044112A patent/NO329984B1/no not_active IP Right Cessation
-
2005
- 2005-05-25 HK HK05104380A patent/HK1072362A1/xx not_active IP Right Cessation
-
2006
- 2006-02-23 ZA ZA200601757A patent/ZA200601757B/en unknown
-
2007
- 2007-09-11 US US11/853,700 patent/US7732490B2/en active Active
- 2007-11-09 US US11/983,469 patent/US20080114069A1/en not_active Abandoned
-
2009
- 2009-03-12 IL IL197582A patent/IL197582A/en active IP Right Grant
- 2009-07-20 NO NO20092714A patent/NO332749B1/no not_active IP Right Cessation
- 2009-08-17 JP JP2009188712A patent/JP5675071B2/ja not_active Expired - Lifetime
- 2009-08-17 JP JP2009188710A patent/JP5586896B2/ja not_active Expired - Lifetime
-
2010
- 2010-03-09 RU RU2010108448/15A patent/RU2530648C2/ru active
- 2010-04-23 US US12/799,368 patent/US8067472B2/en not_active Expired - Fee Related
-
2011
- 2011-10-19 US US13/276,710 patent/US20120041067A1/en not_active Abandoned
-
2014
- 2014-07-17 JP JP2014146512A patent/JP2014237669A/ja active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS51125238A (en) * | 1974-12-20 | 1976-11-01 | Klinge Co Chem Pharm Fab | Process for preparing novel substituted propanol*2**derivative and its nicotic acid ester |
| JPS61251649A (ja) * | 1985-04-05 | 1986-11-08 | Mitsubishi Petrochem Co Ltd | ジカルボン酸ジアニリドの製造法 |
| JPH07502494A (ja) * | 1991-10-04 | 1995-03-16 | スローン−ケタリング・インスティテュート・フォー・キャンサー・リサーチ | 新規の有効な末端分化誘発剤およびその使用方法 |
| WO1995031977A1 (en) * | 1994-05-19 | 1995-11-30 | Sloan-Kettering Institute For Cancer Research | Novel potent inducers of terminal differentiation and methods of use thereof |
| JP2003509343A (ja) * | 1999-09-08 | 2003-03-11 | スローン−ケターリング インスティチュート フォー キャンサー リサーチ | 新規クラスの細胞分化剤およびヒストンデアセチラーゼならびにそれらの使用方法 |
| JP5675071B2 (ja) * | 2002-03-04 | 2015-02-25 | メルク・エイチ・デイ・エイ・シー・リサーチ,エル・エル・シー | 最終分化を誘導する方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5675071B2 (ja) | 最終分化を誘導する方法 | |
| US7148257B2 (en) | Methods of treating mesothelioma with suberoylanilide hydroxamic acid | |
| JP4338734B2 (ja) | Hdac阻害剤による癌処置法 | |
| US7456219B2 (en) | Polymorphs of suberoylanilide hydroxamic acid | |
| JP2007509171A (ja) | Hdac阻害剤による癌治療法 | |
| AU2007203525C1 (en) | Methods of Inducing Terminal Differentiation | |
| KR20050020760A (ko) | 말단 분화의 유도 방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A132 Effective date: 20150217 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150511 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20151027 |