JP2013527567A - 充電式金属空気電池のための可溶性酸素発生触媒 - Google Patents
充電式金属空気電池のための可溶性酸素発生触媒 Download PDFInfo
- Publication number
- JP2013527567A JP2013527567A JP2013506357A JP2013506357A JP2013527567A JP 2013527567 A JP2013527567 A JP 2013527567A JP 2013506357 A JP2013506357 A JP 2013506357A JP 2013506357 A JP2013506357 A JP 2013506357A JP 2013527567 A JP2013527567 A JP 2013527567A
- Authority
- JP
- Japan
- Prior art keywords
- battery
- group
- oxygen
- battery according
- groups
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 229910052760 oxygen Inorganic materials 0.000 title claims abstract description 192
- 239000001301 oxygen Substances 0.000 title claims abstract description 153
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 title claims abstract description 131
- 239000003054 catalyst Substances 0.000 title claims abstract description 68
- 239000003792 electrolyte Substances 0.000 claims abstract description 70
- 229910044991 metal oxide Inorganic materials 0.000 claims abstract description 57
- 150000004706 metal oxides Chemical class 0.000 claims abstract description 57
- 239000007788 liquid Substances 0.000 claims abstract description 28
- 238000000034 method Methods 0.000 claims abstract description 27
- 230000001590 oxidative effect Effects 0.000 claims abstract description 22
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 claims abstract description 16
- 229910001882 dioxygen Inorganic materials 0.000 claims abstract description 16
- 229910021645 metal ion Inorganic materials 0.000 claims abstract description 14
- 239000011148 porous material Substances 0.000 claims abstract description 13
- 239000011263 electroactive material Substances 0.000 claims abstract description 5
- 150000002500 ions Chemical class 0.000 claims abstract description 5
- -1 or Te Inorganic materials 0.000 claims description 72
- 229910018071 Li 2 O 2 Inorganic materials 0.000 claims description 68
- 229910052717 sulfur Inorganic materials 0.000 claims description 50
- XTFIVUDBNACUBN-UHFFFAOYSA-N 1,3,5-trinitro-1,3,5-triazinane Chemical compound [O-][N+](=O)N1CN([N+]([O-])=O)CN([N+]([O-])=O)C1 XTFIVUDBNACUBN-UHFFFAOYSA-N 0.000 claims description 45
- 125000005842 heteroatom Chemical group 0.000 claims description 45
- 125000003118 aryl group Chemical group 0.000 claims description 37
- 229910052711 selenium Inorganic materials 0.000 claims description 35
- 239000011669 selenium Substances 0.000 claims description 35
- 229910052736 halogen Inorganic materials 0.000 claims description 31
- 150000002367 halogens Chemical class 0.000 claims description 31
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims description 30
- 125000000623 heterocyclic group Chemical group 0.000 claims description 30
- 125000002837 carbocyclic group Chemical group 0.000 claims description 29
- 239000002904 solvent Substances 0.000 claims description 29
- 239000000463 material Substances 0.000 claims description 28
- 229910052714 tellurium Inorganic materials 0.000 claims description 23
- 230000007306 turnover Effects 0.000 claims description 23
- 229910052723 transition metal Inorganic materials 0.000 claims description 20
- 229910001416 lithium ion Inorganic materials 0.000 claims description 18
- 150000003624 transition metals Chemical class 0.000 claims description 18
- 229910052751 metal Inorganic materials 0.000 claims description 17
- 229920000642 polymer Polymers 0.000 claims description 17
- 150000001491 aromatic compounds Chemical class 0.000 claims description 13
- 239000002184 metal Substances 0.000 claims description 13
- 229910052744 lithium Inorganic materials 0.000 claims description 10
- 150000001449 anionic compounds Chemical class 0.000 claims description 9
- 229910001412 inorganic anion Inorganic materials 0.000 claims description 9
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 claims description 8
- 229910018068 Li 2 O Inorganic materials 0.000 claims description 8
- 239000003880 polar aprotic solvent Substances 0.000 claims description 8
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 239000011593 sulfur Substances 0.000 claims description 7
- 150000004820 halides Chemical class 0.000 claims description 6
- 229910052782 aluminium Inorganic materials 0.000 claims description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 5
- 125000002577 pseudohalo group Chemical group 0.000 claims description 5
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 claims description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 claims description 4
- 239000000654 additive Substances 0.000 claims description 4
- 239000000956 alloy Substances 0.000 claims description 4
- 150000001408 amides Chemical class 0.000 claims description 4
- 150000002825 nitriles Chemical class 0.000 claims description 4
- 229910052698 phosphorus Inorganic materials 0.000 claims description 4
- 229910052697 platinum Inorganic materials 0.000 claims description 4
- 229910052708 sodium Inorganic materials 0.000 claims description 4
- 239000007784 solid electrolyte Substances 0.000 claims description 4
- 125000005259 triarylamine group Chemical group 0.000 claims description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 3
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 claims description 3
- 150000001412 amines Chemical class 0.000 claims description 3
- 229910052787 antimony Inorganic materials 0.000 claims description 3
- 229910052797 bismuth Inorganic materials 0.000 claims description 3
- 229910001424 calcium ion Inorganic materials 0.000 claims description 3
- 229910052804 chromium Inorganic materials 0.000 claims description 3
- 229910052802 copper Inorganic materials 0.000 claims description 3
- 239000002241 glass-ceramic Substances 0.000 claims description 3
- 239000002608 ionic liquid Substances 0.000 claims description 3
- 229910052741 iridium Inorganic materials 0.000 claims description 3
- 229910052742 iron Inorganic materials 0.000 claims description 3
- 229910052749 magnesium Inorganic materials 0.000 claims description 3
- 229910001425 magnesium ion Inorganic materials 0.000 claims description 3
- 229910052748 manganese Inorganic materials 0.000 claims description 3
- 229910052750 molybdenum Inorganic materials 0.000 claims description 3
- 229910052759 nickel Inorganic materials 0.000 claims description 3
- 229910052758 niobium Inorganic materials 0.000 claims description 3
- 125000001741 organic sulfur group Chemical group 0.000 claims description 3
- 229910052762 osmium Inorganic materials 0.000 claims description 3
- 229910052763 palladium Inorganic materials 0.000 claims description 3
- 229950000688 phenothiazine Drugs 0.000 claims description 3
- 239000011574 phosphorus Substances 0.000 claims description 3
- 229910052707 ruthenium Inorganic materials 0.000 claims description 3
- 229910052710 silicon Inorganic materials 0.000 claims description 3
- 229910001415 sodium ion Inorganic materials 0.000 claims description 3
- 229910052718 tin Inorganic materials 0.000 claims description 3
- 229910052719 titanium Inorganic materials 0.000 claims description 3
- 229910052720 vanadium Inorganic materials 0.000 claims description 3
- 229910052725 zinc Inorganic materials 0.000 claims description 3
- UJOBWOGCFQCDNV-UHFFFAOYSA-N Carbazole Natural products C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 claims description 2
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 claims description 2
- 125000000609 carbazolyl group Chemical class C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 claims description 2
- 125000004122 cyclic group Chemical group 0.000 claims description 2
- 229910052732 germanium Inorganic materials 0.000 claims description 2
- 229910001512 metal fluoride Inorganic materials 0.000 claims description 2
- 229910052987 metal hydride Inorganic materials 0.000 claims description 2
- 150000004681 metal hydrides Chemical class 0.000 claims description 2
- 150000004767 nitrides Chemical class 0.000 claims description 2
- 239000013460 polyoxometalate Substances 0.000 claims description 2
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 claims description 2
- 150000005029 thianthrenes Chemical class 0.000 claims description 2
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 claims 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims 1
- 239000003795 chemical substances by application Substances 0.000 claims 1
- 239000003245 coal Substances 0.000 claims 1
- 229910052737 gold Inorganic materials 0.000 claims 1
- 239000003446 ligand Substances 0.000 claims 1
- 229910052976 metal sulfide Inorganic materials 0.000 claims 1
- 125000001484 phenothiazinyl group Chemical class C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 claims 1
- 239000010703 silicon Substances 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 abstract description 13
- 239000003570 air Substances 0.000 description 167
- 210000004027 cell Anatomy 0.000 description 84
- 239000000047 product Substances 0.000 description 81
- 230000003647 oxidation Effects 0.000 description 62
- 238000007254 oxidation reaction Methods 0.000 description 62
- 238000012360 testing method Methods 0.000 description 52
- 150000001875 compounds Chemical class 0.000 description 43
- 241000894007 species Species 0.000 description 37
- 238000002474 experimental method Methods 0.000 description 36
- 239000000203 mixture Substances 0.000 description 34
- QXBUYALKJGBACG-UHFFFAOYSA-N 10-methylphenothiazine Chemical compound C1=CC=C2N(C)C3=CC=CC=C3SC2=C1 QXBUYALKJGBACG-UHFFFAOYSA-N 0.000 description 32
- 239000012071 phase Substances 0.000 description 31
- 229910052739 hydrogen Inorganic materials 0.000 description 30
- 230000000670 limiting effect Effects 0.000 description 29
- 238000006243 chemical reaction Methods 0.000 description 28
- 238000009792 diffusion process Methods 0.000 description 26
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 25
- 230000009467 reduction Effects 0.000 description 24
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 20
- 229910020366 ClO 4 Inorganic materials 0.000 description 17
- 229910052740 iodine Inorganic materials 0.000 description 17
- 239000000243 solution Substances 0.000 description 17
- 239000000126 substance Substances 0.000 description 17
- 229910052801 chlorine Inorganic materials 0.000 description 16
- 229910052731 fluorine Inorganic materials 0.000 description 16
- 101710178035 Chorismate synthase 2 Proteins 0.000 description 15
- 101710152694 Cysteine synthase 2 Proteins 0.000 description 15
- 229910052789 astatine Inorganic materials 0.000 description 15
- 229910052794 bromium Inorganic materials 0.000 description 15
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 14
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000011734 sodium Substances 0.000 description 12
- 239000000843 powder Substances 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- 230000000052 comparative effect Effects 0.000 description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 229910003473 lithium bis(trifluoromethanesulfonyl)imide Inorganic materials 0.000 description 9
- QSZMZKBZAYQGRS-UHFFFAOYSA-N lithium;bis(trifluoromethylsulfonyl)azanide Chemical compound [Li+].FC(F)(F)S(=O)(=O)[N-]S(=O)(=O)C(F)(F)F QSZMZKBZAYQGRS-UHFFFAOYSA-N 0.000 description 9
- 230000002829 reductive effect Effects 0.000 description 9
- 235000019439 ethyl acetate Nutrition 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 238000004502 linear sweep voltammetry Methods 0.000 description 8
- CJAOGUFAAWZWNI-UHFFFAOYSA-N 1-n,1-n,4-n,4-n-tetramethylbenzene-1,4-diamine Chemical compound CN(C)C1=CC=C(N(C)C)C=C1 CJAOGUFAAWZWNI-UHFFFAOYSA-N 0.000 description 7
- 230000000875 corresponding effect Effects 0.000 description 7
- 238000002484 cyclic voltammetry Methods 0.000 description 7
- 239000004810 polytetrafluoroethylene Substances 0.000 description 7
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- FHCPAXDKURNIOZ-UHFFFAOYSA-N tetrathiafulvalene Chemical class S1C=CSC1=C1SC=CS1 FHCPAXDKURNIOZ-UHFFFAOYSA-N 0.000 description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 230000003247 decreasing effect Effects 0.000 description 6
- 239000008151 electrolyte solution Substances 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 239000002638 heterogeneous catalyst Substances 0.000 description 6
- 239000007773 negative electrode material Substances 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 150000001450 anions Chemical class 0.000 description 5
- 239000010411 electrocatalyst Substances 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 239000007772 electrode material Substances 0.000 description 4
- 238000006056 electrooxidation reaction Methods 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 230000002427 irreversible effect Effects 0.000 description 4
- 150000004053 quinones Chemical class 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- HMPWFXQSDUNCMF-UHFFFAOYSA-N 1-(2-ethylphenyl)-n-[[(2-ethylphenyl)methylamino]disulfanyl]methanamine Chemical compound CCC1=CC=CC=C1CNSSNCC1=CC=CC=C1CC HMPWFXQSDUNCMF-UHFFFAOYSA-N 0.000 description 3
- HNQNFZRNOJJPFM-UHFFFAOYSA-N 3-ethyl-1,3-benzothiazole-2-thione Chemical compound C1=CC=C2SC(=S)N(CC)C2=C1 HNQNFZRNOJJPFM-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 3
- 239000007791 liquid phase Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- REHUVSDWCKMIAO-UHFFFAOYSA-N 3,6-difluoro-1-n,1-n,2-n,2-n,4-n,4-n,5-n,5-n-octamethylbenzene-1,2,4,5-tetramine Chemical compound CN(C)C1=C(F)C(N(C)C)=C(N(C)C)C(F)=C1N(C)C REHUVSDWCKMIAO-UHFFFAOYSA-N 0.000 description 2
- PVXFCOPBOYHONF-UHFFFAOYSA-N 3-ethyl-1,3-benzothiazol-2-one Chemical compound C1=CC=C2SC(=O)N(CC)C2=C1 PVXFCOPBOYHONF-UHFFFAOYSA-N 0.000 description 2
- JLHIDOFPSLZSHM-UHFFFAOYSA-N 3-ethyl-2-methylsulfanyl-2h-1,3-benzothiazole Chemical compound C1=CC=C2N(CC)C(SC)SC2=C1 JLHIDOFPSLZSHM-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 101100317222 Borrelia hermsii vsp3 gene Proteins 0.000 description 2
- 229910003771 Gold(I) chloride Inorganic materials 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 229910000733 Li alloy Inorganic materials 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 230000001351 cycling effect Effects 0.000 description 2
- BOXSCYUXSBYGRD-UHFFFAOYSA-N cyclopenta-1,3-diene;iron(3+) Chemical compound [Fe+3].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 BOXSCYUXSBYGRD-UHFFFAOYSA-N 0.000 description 2
- 210000001787 dendrite Anatomy 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 229910021397 glassy carbon Inorganic materials 0.000 description 2
- FDWREHZXQUYJFJ-UHFFFAOYSA-M gold monochloride Chemical compound [Cl-].[Au+] FDWREHZXQUYJFJ-UHFFFAOYSA-M 0.000 description 2
- 229910002804 graphite Inorganic materials 0.000 description 2
- 239000010439 graphite Substances 0.000 description 2
- ZQBFAOFFOQMSGJ-UHFFFAOYSA-N hexafluorobenzene Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1F ZQBFAOFFOQMSGJ-UHFFFAOYSA-N 0.000 description 2
- 238000012613 in situ experiment Methods 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 239000001989 lithium alloy Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 239000013110 organic ligand Substances 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- XSCHRSMBECNVNS-UHFFFAOYSA-N quinoxaline Chemical compound N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 2
- 230000036647 reaction Effects 0.000 description 2
- 230000027756 respiratory electron transport chain Effects 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 239000010935 stainless steel Substances 0.000 description 2
- 229910001220 stainless steel Inorganic materials 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- VIPKQVRVQSOXGZ-UHFFFAOYSA-N 1,4-diethyl-2,3-dihydroquinoxaline Chemical compound C1=CC=C2N(CC)CCN(CC)C2=C1 VIPKQVRVQSOXGZ-UHFFFAOYSA-N 0.000 description 1
- NAMDIHYPBYVYAP-UHFFFAOYSA-N 1-methoxy-2-(2-methoxyethoxy)ethane Chemical compound COCCOCCOC.COCCOCCOC NAMDIHYPBYVYAP-UHFFFAOYSA-N 0.000 description 1
- YPUWDDMFYRNEFX-UHFFFAOYSA-N 1-methoxy-2-[2-(2-methoxyethoxy)ethoxy]ethane Chemical compound COCCOCCOCCOC.COCCOCCOCCOC YPUWDDMFYRNEFX-UHFFFAOYSA-N 0.000 description 1
- RTYCZCFQHXCMGC-UHFFFAOYSA-N 1-methoxy-2-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]ethane Chemical compound COCCOCCOCCOCCOC.COCCOCCOCCOCCOC RTYCZCFQHXCMGC-UHFFFAOYSA-N 0.000 description 1
- XABNHQJAAXYGOW-UHFFFAOYSA-N 10-(4-methoxyphenyl)phenothiazine Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2SC2=CC=CC=C21 XABNHQJAAXYGOW-UHFFFAOYSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- YRNWIFYIFSBPAU-UHFFFAOYSA-N 4-[4-(dimethylamino)phenyl]-n,n-dimethylaniline Chemical compound C1=CC(N(C)C)=CC=C1C1=CC=C(N(C)C)C=C1 YRNWIFYIFSBPAU-UHFFFAOYSA-N 0.000 description 1
- QJPJQTDYNZXKQF-UHFFFAOYSA-N 4-bromoanisole Chemical compound COC1=CC=C(Br)C=C1 QJPJQTDYNZXKQF-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Divinylene sulfide Natural products C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 1
- 239000002000 Electrolyte additive Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910013188 LiBOB Inorganic materials 0.000 description 1
- 229910013684 LiClO 4 Inorganic materials 0.000 description 1
- 229910010707 LiFePO 4 Inorganic materials 0.000 description 1
- 229910013870 LiPF 6 Inorganic materials 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Natural products P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 1
- 235000008331 Pinus X rigitaeda Nutrition 0.000 description 1
- 235000011613 Pinus brutia Nutrition 0.000 description 1
- 241000018646 Pinus brutia Species 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910045601 alloy Inorganic materials 0.000 description 1
- 239000012080 ambient air Substances 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000002485 combustion reaction Methods 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 238000007596 consolidation process Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 150000001913 cyanates Chemical class 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000004042 decolorization Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 125000005331 diazinyl group Chemical class N1=NC(=CC=C1)* 0.000 description 1
- SQKZZFWTOOPCDQ-UHFFFAOYSA-N dichloromethane;ethyl acetate;hexane Chemical compound ClCCl.CCCCCC.CCOC(C)=O SQKZZFWTOOPCDQ-UHFFFAOYSA-N 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- CNXMDTWQWLGCPE-UHFFFAOYSA-N ditert-butyl-(2-phenylphenyl)phosphane Chemical compound CC(C)(C)P(C(C)(C)C)C1=CC=CC=C1C1=CC=CC=C1 CNXMDTWQWLGCPE-UHFFFAOYSA-N 0.000 description 1
- 238000000840 electrochemical analysis Methods 0.000 description 1
- 238000003487 electrochemical reaction Methods 0.000 description 1
- 238000009713 electroplating Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000004146 energy storage Methods 0.000 description 1
- 150000002170 ethers Chemical group 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 238000011066 ex-situ storage Methods 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000011245 gel electrolyte Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 238000004770 highest occupied molecular orbital Methods 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 238000006713 insertion reaction Methods 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 150000003951 lactams Chemical group 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- YDGSUPBDGKOGQT-UHFFFAOYSA-N lithium;dimethylazanide Chemical compound [Li+].C[N-]C YDGSUPBDGKOGQT-UHFFFAOYSA-N 0.000 description 1
- MCVFFRWZNYZUIJ-UHFFFAOYSA-M lithium;trifluoromethanesulfonate Chemical compound [Li+].[O-]S(=O)(=O)C(F)(F)F MCVFFRWZNYZUIJ-UHFFFAOYSA-M 0.000 description 1
- 238000004768 lowest unoccupied molecular orbital Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000004660 morphological change Effects 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 150000002990 phenothiazines Chemical class 0.000 description 1
- GJSGGHOYGKMUPT-UHFFFAOYSA-N phenoxathiine Chemical class C1=CC=C2OC3=CC=CC=C3SC2=C1 GJSGGHOYGKMUPT-UHFFFAOYSA-N 0.000 description 1
- 150000004986 phenylenediamines Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 239000005518 polymer electrolyte Substances 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 238000010248 power generation Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004151 quinonyl group Chemical group 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000010183 spectrum analysis Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- ZUHZGEOKBKGPSW-UHFFFAOYSA-N tetraglyme Chemical compound COCCOCCOCCOCCOC ZUHZGEOKBKGPSW-UHFFFAOYSA-N 0.000 description 1
- 150000003567 thiocyanates Chemical class 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 150000003577 thiophenes Chemical class 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 229910021561 transition metal fluoride Inorganic materials 0.000 description 1
- 229910000045 transition metal hydride Inorganic materials 0.000 description 1
- 229910000314 transition metal oxide Inorganic materials 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
- 238000001075 voltammogram Methods 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images

























Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/02—Details
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M12/00—Hybrid cells; Manufacture thereof
- H01M12/08—Hybrid cells; Manufacture thereof composed of a half-cell of a fuel-cell type and a half-cell of the secondary-cell type
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M12/00—Hybrid cells; Manufacture thereof
- H01M12/04—Hybrid cells; Manufacture thereof composed of a half-cell of the fuel-cell type and of a half-cell of the primary-cell type
- H01M12/06—Hybrid cells; Manufacture thereof composed of a half-cell of the fuel-cell type and of a half-cell of the primary-cell type with one metallic and one gaseous electrode
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/38—Selection of substances as active materials, active masses, active liquids of elements or alloys
- H01M4/381—Alkaline or alkaline earth metals elements
- H01M4/382—Lithium
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/8605—Porous electrodes
- H01M4/8626—Porous electrodes characterised by the form
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/90—Selection of catalytic material
- H01M4/9008—Organic or organo-metallic compounds
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/90—Selection of catalytic material
- H01M4/92—Metals of platinum group
- H01M4/923—Compounds thereof with non-metallic elements
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
Abstract
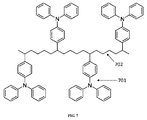
【選択図】図4
Description
本明細書は、2010年4月23日に出願の米国特許出願第61/327,304号、および2010年10月11日に出願の米国特許出願第61/392,014号の先の出願日の利益を主張し、その内容は参照によりその全体が本明細書に組み込まれる。

図1は、特定の実施形態によるOECの作用原理の一般的な例証を提供する。電池充電中に、セルは、概して、平衡セル電圧よりも高い電圧で動作する。本明細書で使用される場合、「平衡セル電圧」という用語は、セル反応全体と関連付けられる熱力学的参照値から計算することができる量を指す(表2を参照されたい)。この電位範囲内で、OECは、電気化学的に酸化された時に活性化した状態になる。酸化型(OEC+)は、溶液中に拡散し、金属酸化物放電生成物を酸化させて、分子酸素および金属イオンを放出する。金属酸化物の酸化に従って、還元したOECが溶液中に拡散し、再度、電気化学的に酸化させるために利用可能である。理論に拘束されることを望むものではないが、OECは、空気電極表面上で電気化学的に酸化および再酸化されてもよい。結果的に、OECの電気化学的酸化は、OEC+を発生または再生し、金属酸化物放電生成物から空気電極まで電子を移動させる役目をすることができる。図1で例証されるように、主な利益は、金属酸化物の間接的な酸化およびOECの電気化学的再生といった過程に関連し得る。特定の実施形態では、間接的な酸化の機構は、空気電極に直接接触していない、または不十分な電子伝導性を有する金属酸化物放電生成物の効率的な充電を可能にすることができる。対照的に、従来の不均質触媒(図3の303dを参照されたい。以下で詳細に説明する)は、電極表面上の固定場所での電荷移動だけにしか影響を与え得ず、これは、充電過程中に酸素発生反応の効率を改善する触媒の能力を制限する。
1)Cl-、Br-、I-、を含むハロゲン化物。
2)シアン化物、シアネート、イソシアネート、ロダン化物(すなわち、チオシアネートおよびイソチオシアネート)、セレノロダン化物(selenorhodanide)、テルロロダン化物(tellurorhodanide)、およびアジド等の、対応する疑ハロゲン基のアニオン(または官能基)を含む擬ハロゲン化物。
3)Keggin型アニオンと、Dawson型アニオンとを含むポリオキソメタレート。
1)置換されたトリアリールアミン:
2)置換されたフェニレンジアミン:
3)置換された芳香族ポリアリールアミン:
4)置換されたフェノチアジン:
5)置換された1,2−ビス(3−アルキル−2,3−ジヒドロ−1,3−ベンゾチアゾール−2−イリデン)ヒドラジン:
式中、R1〜R10は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基の任意の組み合わせから選択され、ヘテロ原子は、N、O、S、Se、Te、任意のハロゲン(例えば、F、Cl、Br、I、At)、または任意の短分子(例えば、CR2、CR、CR3、OR、Ph、O−Ph、CHO、CN、COR、CO2R、COSH、CS2H、SR、CSSH、NR、NR2、NO2、OH、OPO3H2、OSO3H、PO3H2、SO2、SO3H、式中、Rは、R1〜R10について定義されたものである)のうちの1つ以上である。
6)置換されたカルバゾール:
式中、R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基の任意の組み合わせから選択され、ヘテロ原子は、N、O、S、Se、Te、任意のハロゲン(例えば、F、Cl、Br、I、At)、または任意の短分子(例えば、CR2、CR、CR3、OR、Ph、O−Ph、CHO、CN、COR、CO2R、COSH、CS2H、SR、CSSH、NR、NR2、NO2、OH、OPO3H2、OSO3H、PO3H2、SO2、SO3H、式中、Rは、R1〜R8について定義されたものである)のうちの1つ以上である。
7)置換されたテトラチアフルバレン:
式中、R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基の任意の組み合わせから選択され、ヘテロ原子は、N、O、S、Se、Te、任意のハロゲン(例えば、F、Cl、Br、I、At)、または任意の短分子(例えば、CR2、CR、CR3、OR、Ph、O−Ph、CHO、CN、COR、CO2R、COSH、CS2H、SR、CSSH、NR、NR2、NO2、OH、OPO3H2、OSO3H、PO3H2、SO2、SO3H、式中、Rは、R1〜R8について定義されたものである)のうちの1つ以上である。
8)置換されたチオフェン:
式中、R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基の任意の組み合わせから選択され、ヘテロ原子は、N、O、S、Se、Te、任意のハロゲン(例えば、F、Cl、Br、I、At)、または任意の短分子(例えば、CR2、CR、CR3、OR、Ph、O−Ph、CHO、CN、COR、CO2R、COSH、CS2H、SR、CSSH、NR、NR2、NO2、OH、OPO3H2、OSO3H、PO3H2、SO2、SO3H、式中、Rは、R1〜R8について定義されたものである)のうちの1つ以上である。nは、0〜5の範囲であってもよい。
9)置換されたチアントレンおよびフェノキサチイン:
式中、R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基の任意の組み合わせから選択され、ヘテロ原子は、N、O、S、Se、Te、任意のハロゲン(例えば、F、Cl、Br、I、At)、または任意の短分子(例えば、CR2、CR、CR3、OR、Ph、O−Ph、CHO、CN、COR、CO2R、COSH、CS2H、SR、CSSH、NR、NR2、NO2、OH、OPO3H2、OSO3H、PO3H2、SO2、SO3H、式中、Rは、R1〜R8について定義されたものである)のうちの1つ以上である。
10)置換されたジおよびポリアルコキシベンゼン:
11)置換されたホスフィンイミド:
12)置換された多環芳香族化合物:
式中、R1〜R10は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基の任意の組み合わせから選択され、ヘテロ原子は、N、O、S、Se、Te、任意のハロゲン(例えば、F、Cl、Br、I、At)、または任意の短分子(例えば、CR2、CR、CR3、OR、Ph、O−Ph、CHO、CN、COR、CO2R、COSH、CS2H、SR、CSSH、NR、NR2、NO2、OH、OPO3H2、OSO3H、PO3H2、SO2、SO3H、式中、Rは、R1〜R10について定義されたものである)のうちの1つ以上である。nは、1〜10の範囲であってもよい。
13)置換されたジアジン:
式中、R1〜R16は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基の任意の組み合わせから選択され、ヘテロ原子は、N、O、S、Se、Te、任意のハロゲン(例えば、F、Cl、Br、I、At)、または任意の短分子(例えば、CR2、CR、CR3、OR、Ph、O−Ph、CHO、CN、COR、CO2R、COSH、CS2H、SR、CSSH、NR、NR2、NO2、OH、OPO3H2、OSO3H、PO3H2、SO2、SO3H、式中、Rは、R1〜R16について定義されたものである)のうちの1つ以上である。
充電式金属空気電池は、種々の負電極材料によって調製することができる。Liは、比較的に高い電気陽性および低い分子量を有するので、Li−空気電池は、高容量を必要とする応用のための有望な技術である。非プロトン電解質を含有するLi−空気電池は、とりわけ高い理論セル電圧および容量を有する。以下のセルの反応によれば、この種類のLi−空気電池は、3,459Wh/kgの理論的比エネルギー、および7,955Wh/Lのエネルギー密度を有する。
リチオ化された負電極材料は、酸素および/または水と反応し、したがって、費用のかかる、または煩雑な取り扱い方法が必要になる可能性があるので、特定の種類の負不電極材料を脱リチオ化された状態で電池の中に組み立てることができる。例えば、これは、Liイオン電池に一般的に用いられるグラファイト陽極の場合であり得るが、Li合金、Li転換反応電極、およびリチウム金属自体等の数多くの高容量材料にも当てはまる。
比較の目的で、この実施例は、Li2O2および適切な電解質を含有する、事前に製造した「放電済みの」空気電極とともに組み立てた、Li−空気電池の充電を例証する。この比較実施例のLi−空気電池は、OECを含有しない。60重量%の乳濁液を、200mLのイソプロパノール/H2O(1:2、v/v)中に懸濁したSuper Pカーボンブラックと機械式回転装置で5分間混合することによって、Super P/PTFE粉末を調製した。溶媒は、最初にロータリ蒸発器によって、次に2日間80℃で真空乾燥することによって、2ステップで除去した。乾燥ペーストは、90重量%のSuper Pおよび10重量%のPTFEから成る微粉末を形成するために、混合機で粉末化した。
1) 1,2,4,5−テトラキス(ジメチルアミノ)−3,6−ジフルオロベンゼン。Ar中で火炎乾燥した丸底フラスコにおいて、リチウムジメチルアミド(40mLのヘキサン中5%の懸濁液、26.70mmol)、および無水THF(20mL以下、または塩を溶解するのに十分な量)を化合させて、−20℃に冷却した。次に、ヘキサフルオロベンゼン(0.62g、3.30mmol)を滴下し、1時間攪拌を継続した。次いで、反応物を、20%のKOH溶液中に注入し、酢酸エチルで抽出し、水および塩水で洗浄し、そして、硫酸ナトリウム上で乾燥した。この反応の生成物を、ごく少量のメタノールで洗浄することによって精製して、0.65gを得た。
2) Arグローブボックス中で、ジメトキシエタン(40mL)、ナトリウム(0.35g、15.28mmol)、およびビフェニル(1.62g、10.50mmol)を、丸底フラスコの中で化合させて、2時間撹拌した。次に、1,2,4,5−テトラキス(ジメチルアミノ)−3,6−ジフルオロベンゼン(0.41g、1.43mmol)を加え、一晩反応を進行させた。溶液の脱色が見られるまで希釈したHClを数滴加え、その後に、反応混合物を20mLの20%のHCl溶液中に注入し、ヘキサンでビフェニルを抽出し、アルカリ性になるまでアンモニア溶液を水層に加えた。次いで、水層を酢酸エチルで抽出し、水および塩水で洗浄し、MgSO4上で乾燥し、そして、結果として生じた白色の固形物をジクロロメタン/メタノールから再結晶させて、0.32gの生成物を得た。化合物の特徴付けは、文献で報告されている値に一致した。
1) 3−エチル−ベンゾチアゾール−2−one。室温で、11.6g(208.31mmol)のNaOHペレットを、250mLの丸底フラスコの中のDMF(30mL)中7.0g(46.29mmol)のベンゾチアゾロンの溶液に加えた。混合物を、油浴中で5分間60℃に加熱し、4.15mL(55.56mmol)の臭化エチルを混合物に滴下した。茶色のppt.が直ちに形成された。反応混合物を、60℃で1時間加熱し、次いで、加熱を止めて、室温に達するまでしばらくの間そのままにした。50mLのEtOAcを反応混合物に加え、次いで、蒸留したH2Oを加えた。次いで、生成物をEtOAcで抽出し、1MのHClで洗浄した。EtOAc抽出物を塩水で洗浄し、MgSO4上で乾燥した。化合した抽出物をロータリ蒸発器を使用して真空下で濃縮し、次いで、溶出液としてEtOAc−ヘキサンを使用して、シリカゲル上でカラムクロマトグラフィによって精製した。無色油としての生成物が、99%の収率で得られた。合成した化合物は、1H−NMR(400MHz)、13C−NMR(100MHz)、DEPT−135(100MHz)、CoSy、およびGC−MSスペクトルデータ分析によって特徴付けた。
2) 2−エチル−ベンジルアミノ−ジスルフィド。2.6g(14.5mmol)の3−エチル−ベンゾチアゾール−2−oneを、還流凝縮器をその上に伴う250mLの丸底フラスコに加えた。200mLのMeOH:H2O(1:1)をこのフラスコに加えて、混合物を15分間撹拌した。次いで、反応混合物を、大気に開口して13時間加熱還流し、次いで、6時間室温で放置して、生成物がジスルフィドに完全に酸化することを確実にした。生成物を、EtOAcで抽出し、1MのHClおよび塩水で洗浄し、次いで、MgSO4上で乾燥した。粗生成物を、溶出液として3%のEtOAc−ヘキサンを使用して、シリカゲル上でカラムクロマトグラフィによって精製した。2−エチル−ベンジルアミノ−ジスルフィドの黄色油が、74%の収率(2つのステップ全体)で得られ、次いでこれを、1H−NMR(400MHz)、13C−NMR(400MHz)、DEPT−135およびGCM分析によって完全に特徴付けた。
3) 3−エチル−ベンゾチアゾール−2−チオン。室温で、H2O中10MのNaOHを、1.8g(5.91mmol)の2−エチル−ベンジルアミノ−ジスルフィドの溶液に加えた。混合物を、5分間撹拌し、次いで、3.6mL(59.10mmol)の二硫化炭素を加えた。反応混合物を、Ar中で還流した。室温まで冷却した後に、混合物を次いでさらに2時間撹拌した。粗生成物を、EtOAcで抽出し、1MのHClで洗浄し、塩水で洗浄し、MgSO4上で乾燥した。化合した抽出物を、ロータリ蒸発器を使用して真空下で濃縮し、次いで、溶出液として12%のEtOAc−ヘキサンを使用して、シリカゲル上でカラムクロマトグラフィによって精製した。淡黄色の結晶性生成物が、96%の収率で得られた。3−エチル−ベンゾチアゾール−2−チオンの構造は、1H−NMR(400MHz)、13C−NMR(100MHz)、DEPT−135、およびGCM分析によって確認した。
4) 2−メチルスルファニル−3−エチル−ベンゾチアゾール。1.7mL(17.66mmol)のジメチル硫酸を、250mL丸底フラスコの中のアセトニトリル(60mL)中2.3g(11.77mmol)の3−エチル−ベンゾチアゾール−2−チオンに加えた。反応混合物を、4時間Ar中で還流した。反応物を、室温まで冷却し、次いで、ロータリ蒸発器を使用して濃縮した。室温で、200mLのEt2Oを、濃縮したアセトニトリル溶液に加えた。オフホワイトのppt.が形成され、次いでそれを濾過し、Et2Oで洗浄した。塩を、一晩高真空中で乾燥した。白色粉体が、100%の収率で得られた。2−メチルサルファニル−3−エチル−ベンゾチアゾールの構造は、1H−NMR(400MHz)、13C−NMR(100MHz)、DEPT−135分析によって確認した。
5) ABT−DE。Ar雰囲気中で、Et3N(3.12mL、22.4mmol)およびピリジン(0.05mL、0.56mmol)を、無水EtOH(10mL)中2−メチルサルファニル−3−エチル−ベンゾチアゾール(3.6g、11.20mmol)の溶液に加えた。混合物を、室温で15分間撹拌した。EtOH中に希釈した無水ヒドラジン(0.16mL、5.0mmol)を、反応混合物に滴下した。反応混合物を、室温で20時間撹拌した。20時間後、ABT−DEの白色ppt.が反応フラスコの中に観察された。生成物の完全な沈殿のために、ヘキサン(30mL)を、反応混合物に加えた。白色の沈殿物を、ブフナー漏斗を使用して濾過し、ヘキサンで洗浄した(100mL×2)。最後に、生成物を、溶媒系としてヘキサン−ジクロロメタン−酢酸エチルを使用して、シリカゲル上でカラムクロマトグラフィによって精製した。ABT−DEの白色の結晶性生成物が、90%の収率で得られた。生成物の構造は、1H−NMR(400MHz)、13C−NMR(100MHz)、DEPT−135、CoSy、およびGC−MSスペクトル分析によって完全に特徴付けた。
Claims (77)
- a)活性金属イオンを取り込み、放出することが可能な負電極と、
b)電気活性材料として酸素を使用する、多孔質正電極と、
c)前記負電極と正電極との間でイオンを伝導するように構成され、かつ1つ以上の相を含む、電解質と、
を備え、少なくとも1つの相は、液体を含み、少なくとも部分的に前記正電極の孔を満たし、
前記液体は、酸素発生触媒を備える、
充電式金属空気電池。 - 前記酸素発生触媒は、無機アニオンを含む、請求項1に記載の電池。
- 前記酸素発生触媒は、ハロゲン化物を含む、請求項1に記載の電池。
- 前記ハロゲン化物は、I-である、請求項3に記載の電池。
- 前記酸素発生触媒は、擬ハロゲン化物を含む、請求項1に記載の電池。
- 前記酸素発生触媒は、ポリオキソメタレートを含む、請求項1に記載の電池。
- 前記酸素発生触媒は、1つ以上のリガンドに結合される1つ以上の遷移金属中心を含む、遷移金属錯体を備える、請求項1に記載の電池。
- 前記1つ以上の遷移金属中心は、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn、Nb、Mo、Ru、Pd、Ag、W、Os、Ir、Pt、Au、およびそれらの組み合わせから成る群から選択される、請求項7に記載の電池。
- 前記酸素発生触媒は、
式中、Mは独立して、Li、Na、Al、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn、Nb、Mo、Ru、Pd、Ag、W、Os、Ir、Pt、またはAuから選択され、
R1〜R16は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項1に記載の電池。 - 前記酸素発生触媒は、キノンまたはキノイドを含む、請求項1に記載の電池。
- 前記酸素発生触媒は、
式中、R1〜R4は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項10に記載の電池。 - 前記酸素発生触媒は、芳香族化合物を含む、請求項1に記載の電池。
- 前記酸素発生触媒は、窒素含有芳香族化合物を含む、請求項1に記載の電池。
- 前記酸素発生触媒は、以下の構造を有する置換されたトリアリールアミンを含み、
- 前記酸素発生触媒は、
式中、R1〜R12は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項13に記載の電池。 - 前記酸素発生触媒は、
式中、R1〜R12は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項13に記載の電池。 - 前記酸素発生触媒は、以下の構造を有する置換されたフェノチアジンを含み、
- 前記酸素発生触媒は、以下の構造を有する置換された1,2−ビス(3−アルキル−2,3−ジヒドロ−1,3−ベンゾチアゾール−2−イリデン)ヒドラジンを含み、
R1〜R10は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項1に記載の電池。 - 前記酸素発生触媒は、以下の構造を有する置換されたカルバゾールを含み、
R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項1に記載の電池。 - 前記酸素発生触媒は、硫黄、セレニウム、およびテルリウムのうちの1つ以上を含有する芳香族化合物を含む、請求項1に記載の電池。
- 前記酸素発生触媒は、
式中、X1〜X4は独立して、S、Se、O、またはTeから選択され、
R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項1に記載の電池。 - 前記酸素発生触媒は、
式中、X1〜X3は独立して、S、Se、O、C=CR2、C=O、またはTeから選択され、
R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択され、
nは、0〜100の範囲である、請求項1に記載の電池。 - 前記酸素発生触媒は、以下の構造を有する置換されたチアントレンを含み、
R1〜R8は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項1に記載の電池。 - 前記酸素発生触媒は、酸素含有芳香族化合物を含む、請求項1に記載の電池。
- 前記酸素発生触媒は、
式中、R1〜R6は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項1に記載の電池。 - 前記酸素発生触媒は、リン含有芳香族化合物を含む、請求項1に記載の電池。
- 前記酸素発生触媒は、
式中、R1〜R14は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、前記ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択され、
nは、1〜100の範囲である、請求項1に記載の電池。 - 前記酸素発生触媒は、
式中、X1〜X3は、S、Se、O、C=CR2、C=O、NR、またはTeから独立して選択され、
R1〜R10は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、前記ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択され、
nは、1〜100の範囲である、請求項1に記載の電池。 - 前記酸素発生触媒は、
式中、R1〜R4は独立して、置換された又は非置換の、C1−C10アルキル基、アリール基、C4−C8炭素環式基、C4−C8複素環式基から選択され、ヘテロ原子は独立して、N、O、S、Se、もしくはTe、ハロゲン、または短分子から選択される、請求項1に記載の電池。 - 前記酸素発生触媒は、ポリマー構造に取り付けられる、請求項1に記載の電池。
- 前記酸素発生触媒は、平衡セル電圧より1.5V未満高い平衡電位を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、前記平衡セル電圧より1V未満高い平衡電位を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、前記平衡セル電圧より0.5V未満高い平衡電位を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、前記平衡セル電圧より0.4V未満高い平衡電位を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、前記平衡セル電圧より0.3V未満高い平衡電位を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、前記平衡セル電圧より0.2V未満高い平衡電位を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、前記平衡セル電圧より0.1V未満高い平衡電位を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、100以上のターンオーバー数を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、500以上のターンオーバー数を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、1000以上のターンオーバー数を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、5000以上のターンオーバー数を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、10,000以上のターンオーバー数を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、0.05M以上の前記液体への溶解性を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、0.1M以上の前記液体への溶解性を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、0.5M以上の前記液体への溶解性を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、1.0M以上の前記液体への溶解性を有する、請求項1に記載の電池。
- 前記酸素発生触媒は、2.0M以上の前記液体への溶解性を有する、請求項1に記載の電池。
- 前記液体は、極性非プロトン溶媒である、請求項1に記載の電池。
- 前記極性非プロトン溶媒は、エーテル、グリム、炭酸塩、ニトリル、アミド、アミン、有機硫黄溶媒、有機リン溶媒、有機ケイ素溶媒、フッ素化溶媒、およびイオン液体から成る群から選択される、1つ以上の溶媒を含む、請求項48に記載の電池。
- 前記電解質は、前記正電極と負電極との間に間置される第2の相を含み、前記酸素発生触媒に対して半透過性および実質的に不透過性である、請求項1に記載の電池。
- 前記第2の電解質相は、ポリマーを含む、請求項50に記載の電池。
- 前記第2の電解質相は、ガラスセラミックを含む、請求項50に記載の電池。
- 前記第2の電解質相は、固体電解質中間相を含む、請求項50に記載の電池。
- 前記電解質は、アニオン受容体、カチオン受容体、および固体電解質中間相形成剤から成る群から選択される、1つ以上の添加剤を含有する、請求項1に記載の電池。
- 前記負電極は、活性Liイオンを取り込み、放出することが可能である、請求項1に記載の電池。
- 前記正電極は、Li2O2またはLi2Oをさらに含む、請求項55に記載の電池。
- 前記負電極は、活性Naイオンを取り込み、放出することが可能である、請求項1に記載の電池。
- 前記正電極は、Na2O2またはNa2Oをさらに含む、請求項57に記載の電池。
- 前記負電極は、活性Mgイオンを取り込み、放出することが可能である、請求項1に記載の電池。
- 前記正電極は、MgOまたはMgO2をさらに含む、請求項59に記載の電池。
- 前記負電極は、活性Caイオンを取り込み、放出することが可能である、請求項1に記載の電池。
- 前記正電極は、CaOまたはCaO2をさらに含む、請求項61に記載の電池。
- 前記負電極は、Si、Ge、Sn、Sb、Al、Mg、およびBiから成る群から選択される、1つ以上の合金材料をさらに含む、請求項1に記載の電池。
- 前記負電極は、金属酸化物、金属水素化物、金属窒化物、金属フッ化物、金属硫化物、金属アンチモン化物、および金属リン化物から成る群から選択される、転換反応材料をさらに含む、請求項1に記載の電池。
- 酸素発生触媒を備える、第1の構成要素を提供することと、
金属酸化物放電生成物を含む、第2の構成要素を提供することと、
前記第1の構成要素および前記第2の構成要素を備える、空気電極を形成することと、
を含む、方法。 - 活性金属イオンを取り込み、放出することが可能な負電極を提供することと、
電解質を使用して前記負電極と前記空気電極の間の接続を形成することと、
をさらに含む、請求項65に記載の方法。 - 金属空気電池で使用するための空気電極であって、
a) 電子伝導性成分と、
b) 金属酸化物と、
c) 酸素発生触媒と、
を備える、空気電極。 - 前記金属酸化物は、20質量%を超える量で前記空気電極中に含有される、請求項67に記載の空気電極。
- 前記金属酸化物は、40質量%を超える量で前記空気電極中に含有される、請求項67に記載の空気電極。
- 前記金属酸化物は、60質量%を超える量で前記空気電極中に含有される、請求項67に記載の空気電極。
- 前記金属酸化物は、80質量%を超える量で前記空気電極中に含有される、請求項67に記載の空気電極。
- 前記金属酸化物は、Na2O2またはNa2Oである、請求項67に記載の空気電極。
- 前記金属酸化物は、MgOまたはMgO2である、請求項67に記載の空気電極。
- 前記金属酸化物は、CaOまたはCaO2である、請求項67に記載の空気電極。
- 前記金属酸化物は、Li2O2またはLi2Oである、請求項67に記載の空気電極。
- 前記空気電極は、90%を超える前記金属酸化物が酸化されるように、0.2mA/cm2を超える電流密度で、電池のOCVより1V以下高い電圧まで前記電池に充電することが可能である、請求項75に記載の空気電極。
- 充電式金属空気電池で使用するための材料であって、
a) 前記電池に用いられる液体に可溶であり、
b) 前記平衡セル電圧を超える電位で電気化学的に活性化され、
c) 前記充電式金属空気電池の放電中に生成される金属酸化物放電生成物を酸化させることによって、酸素ガスを発生させることが可能である、
材料。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US32730410P | 2010-04-23 | 2010-04-23 | |
| US61/327,304 | 2010-04-23 | ||
| US39201410P | 2010-10-11 | 2010-10-11 | |
| US61/392,014 | 2010-10-11 | ||
| PCT/US2011/033821 WO2011133982A1 (en) | 2010-04-23 | 2011-04-25 | Soluble oxygen evolving catalysts for rechargeable metal-air batteries |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013527567A true JP2013527567A (ja) | 2013-06-27 |
| JP2013527567A5 JP2013527567A5 (ja) | 2014-10-02 |
Family
ID=44834547
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013506357A Pending JP2013527567A (ja) | 2010-04-23 | 2011-04-25 | 充電式金属空気電池のための可溶性酸素発生触媒 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US20120028137A1 (ja) |
| EP (1) | EP2561573B1 (ja) |
| JP (1) | JP2013527567A (ja) |
| KR (1) | KR20130103333A (ja) |
| WO (1) | WO2011133982A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015022858A1 (ja) * | 2013-08-15 | 2015-02-19 | 国立大学法人 東京大学 | リチウム-空気電池用電解液 |
| WO2015133139A1 (en) * | 2014-03-06 | 2015-09-11 | Sharp Kabushiki Kaisha | Battery anode with preloaded metals |
| US9178254B2 (en) | 2010-12-07 | 2015-11-03 | Samsung Electronics Co., Ltd. | Lithium air battery |
| JP2016110699A (ja) * | 2014-12-02 | 2016-06-20 | 日本電信電話株式会社 | リチウム空気二次電池 |
| JP2017507472A (ja) * | 2012-03-28 | 2017-03-16 | シャープ株式会社 | 予め充填された金属を有する電池の負極 |
| JP2017054760A (ja) * | 2015-09-11 | 2017-03-16 | 日本電信電話株式会社 | リチウム空気二次電池 |
| CN106732766A (zh) * | 2015-11-20 | 2017-05-31 | 现代自动车株式会社 | 用于锂‑空气电池的可溶性催化剂 |
| JP2017103215A (ja) * | 2015-12-01 | 2017-06-08 | 株式会社デンソー | 非水型マグネシウム酸素電池 |
| JP2017107786A (ja) * | 2015-12-11 | 2017-06-15 | 日本電信電話株式会社 | リチウム空気二次電池およびリチウム空気二次電池用電解質 |
| JP2019003940A (ja) * | 2017-06-12 | 2019-01-10 | パナソニックIpマネジメント株式会社 | 空気電池 |
| JP2019003935A (ja) * | 2017-06-12 | 2019-01-10 | パナソニックIpマネジメント株式会社 | リチウム空気電池 |
| JP2019046784A (ja) * | 2017-08-31 | 2019-03-22 | パナソニックIpマネジメント株式会社 | リチウム空気電池 |
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9431660B2 (en) | 2010-09-23 | 2016-08-30 | Robert Bosch Gmbh | Lithium battery with charging redox couple |
| US9761878B2 (en) | 2010-09-23 | 2017-09-12 | Robert Bosch Gmbh | Metal/oxygen battery with a clean surface for oxidizing redox additives |
| WO2013077863A1 (en) * | 2011-11-22 | 2013-05-30 | Robert Bosch Gmbh | Lithium battery with charging redox couple |
| KR101899483B1 (ko) * | 2011-11-30 | 2018-09-18 | 삼성전자주식회사 | 리튬 공기 전지 |
| KR101887534B1 (ko) * | 2012-04-03 | 2018-08-13 | 삼성전자주식회사 | 리튬 공기 전지의 충전 방법 |
| WO2013183655A1 (ja) * | 2012-06-05 | 2013-12-12 | 日本電気株式会社 | リチウム二次電池 |
| JP5660086B2 (ja) * | 2012-08-08 | 2015-01-28 | 株式会社デンソー | マグネシウム二次電池 |
| EP2883269B1 (en) * | 2012-08-10 | 2019-05-08 | Robert Bosch GmbH | Controlling the location of product distribution and removal in a metal/oxygen cell |
| KR101571588B1 (ko) | 2012-09-11 | 2015-11-24 | 주식회사 두산 | 퀴녹살린계 유기발광 화합물 및 이를 이용한 유기 전계 발광 소자 |
| WO2014080871A1 (ja) | 2012-11-20 | 2014-05-30 | 日本電気株式会社 | リチウムイオン二次電池 |
| US9450278B2 (en) | 2012-12-20 | 2016-09-20 | International Business Machines Corporation | Cathode material for lithium—oxygen battery |
| JP5854009B2 (ja) * | 2012-12-26 | 2016-02-09 | 株式会社デンソー | マグネシウム二次電池用負極の表面処理方法 |
| DE102013200585A1 (de) * | 2013-01-16 | 2014-07-31 | Siemens Aktiengesellschaft | Wiederaufladbarer elektrischer Energiespeicher |
| CN103474671B (zh) * | 2013-09-13 | 2015-10-28 | 深圳大学 | 一种锂空气电池用碳-过氧化锂正极及其制备方法 |
| KR101600141B1 (ko) | 2013-10-11 | 2016-03-04 | 서울대학교산학협력단 | 레독스 플로우 전지용 전해액 및 이를 포함하는 레독스 플로우 전지 |
| KR20150057260A (ko) * | 2013-11-19 | 2015-05-28 | 한양대학교 산학협력단 | 리튬 공기 전지용 양극 및 이를 포함하는 리튬 공기 전지 |
| CN103633308A (zh) * | 2013-11-28 | 2014-03-12 | 宁波金和新材料股份有限公司 | 一种富锂镍钴铝氧正极材料及其制备方法 |
| US10079401B2 (en) | 2014-03-24 | 2018-09-18 | Cornell University | Symmetric redox flow battery containing organic redox active molecule |
| WO2015148358A1 (en) | 2014-03-24 | 2015-10-01 | Cornell University | Solar flow battery |
| US9343787B2 (en) | 2014-07-30 | 2016-05-17 | Toyota Motor Engineering & Manufacturing North America, Inc. | Lithium-air battery with sodium salt as mediator |
| WO2016024919A1 (en) * | 2014-08-15 | 2016-02-18 | National University Of Singapore | A battery system |
| US20160172676A1 (en) * | 2014-12-10 | 2016-06-16 | Basf Corporation | Metal Hydride Compositions and Lithium Ion Batteries |
| CN106328964B (zh) * | 2015-06-25 | 2019-04-23 | 清华大学 | 金属空气电池正极及金属空气电池 |
| EP3116052A1 (en) | 2015-07-08 | 2017-01-11 | Basf Se | Rechargeable metal-oxygen cells |
| US10103402B2 (en) | 2015-12-01 | 2018-10-16 | University Of Kentucky Research Foundation | Liquid phenothiazine catholytes for non-aqueous redox flow batteries |
| JP2017022096A (ja) * | 2016-06-28 | 2017-01-26 | ロベルト・ボッシュ・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツングRobert Bosch Gmbh | 電気化学セル |
| EP3267514B1 (en) * | 2016-07-06 | 2021-01-27 | LiCAP Technologies, Inc. | Lithium attached electrodes and method of making same |
| US10177427B2 (en) * | 2017-02-10 | 2019-01-08 | General Electric Company | Electrochemical cell for use in high temperature metal-air battery |
| US10826145B2 (en) | 2017-02-10 | 2020-11-03 | General Electric Company | Electrochemical cell for use in high temperature metal-air battery |
| US11515566B2 (en) | 2020-04-26 | 2022-11-29 | International Business Machines Corporation | Liquid cathode formulation for rechargeable metal halide battery |
| CN113976099B (zh) * | 2021-11-23 | 2023-06-27 | 山东交通学院 | 一种负载氧化钙的磁性多孔碱性碳材料、其制备方法及应用 |
| CN116207313B (zh) * | 2023-05-06 | 2023-07-11 | 苏州擎动动力科技有限公司 | 自增湿膜电极及其制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080096061A1 (en) * | 2006-06-12 | 2008-04-24 | Revolt Technology Ltd | Metal-Air Battery or Fuel Cell |
| JP2010033890A (ja) * | 2008-07-29 | 2010-02-12 | Toyota Central R&D Labs Inc | リチウム空気電池 |
| JP2010167390A (ja) * | 2009-01-26 | 2010-08-05 | Nec Corp | 酸素還元触媒、それを用いた燃料電池および空気電池、酸素還元触媒の製造方法 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3316126A (en) * | 1962-03-01 | 1967-04-25 | Pullman Inc | Fuel cell |
| US4255498A (en) * | 1979-10-26 | 1981-03-10 | Toshiba Ray-O-Vac Co., Ltd. | Button-type air cell |
| US6908710B2 (en) * | 2001-10-09 | 2005-06-21 | Valence Technology, Inc. | Lithiated molybdenum oxide active materials |
| US7645543B2 (en) * | 2002-10-15 | 2010-01-12 | Polyplus Battery Company | Active metal/aqueous electrochemical cells and systems |
| US6855453B2 (en) * | 2002-12-30 | 2005-02-15 | Utc Fuel Cells, Llc | Fuel cell having a corrosion resistant and protected cathode catalyst layer |
| JP2004319324A (ja) * | 2003-04-17 | 2004-11-11 | Matsushita Electric Ind Co Ltd | 電気化学デバイス |
| US7238440B2 (en) * | 2003-10-03 | 2007-07-03 | E. I. Du Pont De Nemours And Company | Membrane free fuel cell |
| US7282295B2 (en) * | 2004-02-06 | 2007-10-16 | Polyplus Battery Company | Protected active metal electrode and battery cell structures with non-aqueous interlayer architecture |
| KR100570359B1 (ko) * | 2004-12-23 | 2006-04-12 | 비나텍주식회사 | 하이브리드 전지 |
| GB0505087D0 (en) * | 2005-03-12 | 2005-04-20 | Acal Energy Ltd | Fuel cells |
| CN101147286A (zh) * | 2005-08-25 | 2008-03-19 | 松下电器产业株式会社 | 氧还原用电极 |
| EP1977475B1 (en) * | 2005-12-06 | 2012-02-29 | ReVolt Technology Ltd | Bifunctional air electrode |
| US20110123877A1 (en) * | 2007-08-23 | 2011-05-26 | Nec Corporation | Catalyst for oxygen reduction electrode and oxygen reduction electrode |
| US20090053594A1 (en) * | 2007-08-23 | 2009-02-26 | Johnson Lonnie G | Rechargeable air battery and manufacturing method |
| JP5315831B2 (ja) * | 2008-03-24 | 2013-10-16 | 株式会社豊田中央研究所 | リチウム空気電池 |
| US20090288946A1 (en) * | 2008-05-23 | 2009-11-26 | Lumimove, Inc. Dba Crosslink | Electroactivated film with layered structure |
| US20100266907A1 (en) * | 2008-11-04 | 2010-10-21 | Rachid Yazami | Metal air battery system |
| CN101960664B (zh) * | 2008-12-25 | 2014-06-04 | 丰田自动车株式会社 | 锂空气电池 |
| KR20120063163A (ko) * | 2010-12-07 | 2012-06-15 | 삼성전자주식회사 | 리튬 공기 전지 |
-
2011
- 2011-04-25 EP EP11772854.3A patent/EP2561573B1/en active Active
- 2011-04-25 JP JP2013506357A patent/JP2013527567A/ja active Pending
- 2011-04-25 WO PCT/US2011/033821 patent/WO2011133982A1/en active Application Filing
- 2011-04-25 US US13/093,759 patent/US20120028137A1/en not_active Abandoned
- 2011-04-25 KR KR1020127030602A patent/KR20130103333A/ko not_active Application Discontinuation
-
2020
- 2020-09-11 US US17/018,965 patent/US20200411933A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080096061A1 (en) * | 2006-06-12 | 2008-04-24 | Revolt Technology Ltd | Metal-Air Battery or Fuel Cell |
| JP2010033890A (ja) * | 2008-07-29 | 2010-02-12 | Toyota Central R&D Labs Inc | リチウム空気電池 |
| JP2010167390A (ja) * | 2009-01-26 | 2010-08-05 | Nec Corp | 酸素還元触媒、それを用いた燃料電池および空気電池、酸素還元触媒の製造方法 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9178254B2 (en) | 2010-12-07 | 2015-11-03 | Samsung Electronics Co., Ltd. | Lithium air battery |
| JP2017507472A (ja) * | 2012-03-28 | 2017-03-16 | シャープ株式会社 | 予め充填された金属を有する電池の負極 |
| WO2015022858A1 (ja) * | 2013-08-15 | 2015-02-19 | 国立大学法人 東京大学 | リチウム-空気電池用電解液 |
| WO2015133139A1 (en) * | 2014-03-06 | 2015-09-11 | Sharp Kabushiki Kaisha | Battery anode with preloaded metals |
| JP2016110699A (ja) * | 2014-12-02 | 2016-06-20 | 日本電信電話株式会社 | リチウム空気二次電池 |
| JP2017054760A (ja) * | 2015-09-11 | 2017-03-16 | 日本電信電話株式会社 | リチウム空気二次電池 |
| CN106732766B (zh) * | 2015-11-20 | 2021-04-27 | 现代自动车株式会社 | 用于锂-空气电池的可溶性催化剂 |
| CN106732766A (zh) * | 2015-11-20 | 2017-05-31 | 现代自动车株式会社 | 用于锂‑空气电池的可溶性催化剂 |
| JP2017098215A (ja) * | 2015-11-20 | 2017-06-01 | 現代自動車株式会社Hyundai Motor Company | リチウム−空気電池用液相触媒 |
| JP2017103215A (ja) * | 2015-12-01 | 2017-06-08 | 株式会社デンソー | 非水型マグネシウム酸素電池 |
| JP2017107786A (ja) * | 2015-12-11 | 2017-06-15 | 日本電信電話株式会社 | リチウム空気二次電池およびリチウム空気二次電池用電解質 |
| JP2019003940A (ja) * | 2017-06-12 | 2019-01-10 | パナソニックIpマネジメント株式会社 | 空気電池 |
| JP2019003935A (ja) * | 2017-06-12 | 2019-01-10 | パナソニックIpマネジメント株式会社 | リチウム空気電池 |
| US10629970B2 (en) | 2017-06-12 | 2020-04-21 | Panasonic Intellectual Property Management Co., Ltd. | Lithium air battery including negative electrode, positive electrode, nonaqueous lithium ion conductor, and copper ion |
| US10665867B2 (en) | 2017-06-12 | 2020-05-26 | Panasonic Intellectual Property Management Co., Ltd. | Air battery including negative electrode, positive electrode, nonaqueous metal ion conductor, and oxygen evolving catalyst |
| JP2019046784A (ja) * | 2017-08-31 | 2019-03-22 | パナソニックIpマネジメント株式会社 | リチウム空気電池 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200411933A1 (en) | 2020-12-31 |
| KR20130103333A (ko) | 2013-09-23 |
| WO2011133982A1 (en) | 2011-10-27 |
| US20120028137A1 (en) | 2012-02-02 |
| EP2561573B1 (en) | 2020-03-25 |
| EP2561573A4 (en) | 2016-09-14 |
| EP2561573A1 (en) | 2013-02-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20200411933A1 (en) | Soluble oxygen evolving catalysts for rechargeable metal-air batteries | |
| Shu et al. | Understanding the reaction chemistry during charging in aprotic lithium–oxygen batteries: existing problems and solutions | |
| Yadegari et al. | Sodium‐oxygen batteries: a comparative review from chemical and electrochemical fundamentals to future perspective | |
| Bergner et al. | How to improve capacity and cycling stability for next generation Li–O2 batteries: approach with a solid electrolyte and elevated redox mediator concentrations | |
| Qin et al. | Superoxide-based K–O2 batteries: highly reversible oxygen redox solves challenges in air electrodes | |
| Lin et al. | Controlling reversible expansion of Li2O2 formation and decomposition by modifying electrolyte in Li-O2 batteries | |
| JP3555097B2 (ja) | 電極材料及び二次電池 | |
| JP3525403B2 (ja) | 電極材料及び二次電池 | |
| Kwon et al. | Stability of Glyme Solvate Ionic Liquid as an Electrolyte for Rechargeable Li− O2 Batteries | |
| Trahan et al. | Solvent-coupled catalysis of the oxygen electrode reactions in lithium-air batteries | |
| Lee et al. | Polyoxometalate as a Nature-Inspired Bifunctional Catalyst for Lithium–Oxygen Batteries | |
| JP6716364B2 (ja) | リチウム−空気電池用液相触媒 | |
| Lyu et al. | A dicyanobenzoquinone based cathode material for rechargeable lithium and sodium ion batteries | |
| JP5589821B2 (ja) | 蓄電デバイス及び電極活物質の製造方法 | |
| US10727488B2 (en) | Redox mediators for metal-sulfur batteries | |
| Liu et al. | Prevention of na corrosion and dendrite growth for Long-Life Flexible Na–Air Batteries | |
| JP2017027868A (ja) | レドックスフロー電池 | |
| JP2008112630A (ja) | 二次電池 | |
| US9431678B2 (en) | Functionalized carboranyl magnesium electrolyte for magnesium battery | |
| Wang et al. | Benzene-bridged anthraquinones as a high-rate and long-lifespan organic cathode for advanced Na-ion batteries | |
| Chen et al. | Charging processes in lithium-oxygen batteries unraveled through the lens of the distribution of relaxation times | |
| Zhang et al. | Real-Time Monitoring of Surface Effects on the Oxygen Reduction Reaction Mechanism for Aprotic Na–O2 Batteries | |
| Wang et al. | Bio-derived 4-electron-accepting carbonyl-N-methylpyridinium species for high-performance lithium-organic batteries | |
| KR20130008830A (ko) | 신규한 이온성 액체, 이온성 액체를 포함하는 리튬 공기 전지용 겔 고분자 전해질 및 리튬 공기 전지 | |
| He et al. | The Key Role of Magnesium Polysulfides in the Development of Mg-S Batteries |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140425 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140425 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20140606 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140704 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140626 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20150128 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150303 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150602 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150703 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150801 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20151203 |