JP2012521197A - 担体免疫グロブリンおよびその使用 - Google Patents
担体免疫グロブリンおよびその使用 Download PDFInfo
- Publication number
- JP2012521197A JP2012521197A JP2012501019A JP2012501019A JP2012521197A JP 2012521197 A JP2012521197 A JP 2012521197A JP 2012501019 A JP2012501019 A JP 2012501019A JP 2012501019 A JP2012501019 A JP 2012501019A JP 2012521197 A JP2012521197 A JP 2012521197A
- Authority
- JP
- Japan
- Prior art keywords
- seq
- binding protein
- antigen binding
- amino acid
- peptide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 108060003951 Immunoglobulin Proteins 0.000 title description 61
- 102000018358 immunoglobulin Human genes 0.000 title description 60
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 331
- 102000025171 antigen binding proteins Human genes 0.000 claims abstract description 277
- 108091000831 antigen binding proteins Proteins 0.000 claims abstract description 277
- 210000004027 cell Anatomy 0.000 claims abstract description 164
- 102000004196 processed proteins & peptides Human genes 0.000 claims abstract description 153
- 229920001184 polypeptide Polymers 0.000 claims abstract description 99
- 150000007523 nucleic acids Chemical class 0.000 claims abstract description 93
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 88
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 88
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims abstract description 59
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims abstract description 59
- 239000000126 substance Substances 0.000 claims abstract description 46
- 210000004408 hybridoma Anatomy 0.000 claims abstract description 39
- 239000013598 vector Substances 0.000 claims abstract description 20
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 8
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 139
- 230000027455 binding Effects 0.000 claims description 134
- 238000009739 binding Methods 0.000 claims description 130
- 238000000034 method Methods 0.000 claims description 127
- 239000000427 antigen Substances 0.000 claims description 104
- 108091007433 antigens Proteins 0.000 claims description 102
- 102000036639 antigens Human genes 0.000 claims description 102
- 108010047041 Complementarity Determining Regions Proteins 0.000 claims description 81
- 231100000765 toxin Toxicity 0.000 claims description 80
- 239000003053 toxin Substances 0.000 claims description 78
- 108700012359 toxins Proteins 0.000 claims description 78
- 125000000539 amino acid group Chemical group 0.000 claims description 75
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 claims description 31
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 claims description 31
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 claims description 24
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 claims description 24
- 210000004899 c-terminal region Anatomy 0.000 claims description 22
- 239000001963 growth medium Substances 0.000 claims description 22
- 239000005557 antagonist Substances 0.000 claims description 21
- 239000013604 expression vector Substances 0.000 claims description 20
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 claims description 12
- 108090000932 Calcitonin Gene-Related Peptide Proteins 0.000 claims description 10
- 101001008255 Homo sapiens Immunoglobulin kappa variable 1D-8 Proteins 0.000 claims description 10
- 101001047628 Homo sapiens Immunoglobulin kappa variable 2-29 Proteins 0.000 claims description 10
- 101001008321 Homo sapiens Immunoglobulin kappa variable 2D-26 Proteins 0.000 claims description 10
- 101001047619 Homo sapiens Immunoglobulin kappa variable 3-20 Proteins 0.000 claims description 10
- 101001008263 Homo sapiens Immunoglobulin kappa variable 3D-15 Proteins 0.000 claims description 10
- 102100022964 Immunoglobulin kappa variable 3-20 Human genes 0.000 claims description 10
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 10
- 238000012258 culturing Methods 0.000 claims description 9
- 239000000556 agonist Substances 0.000 claims description 8
- 102100023995 Beta-nerve growth factor Human genes 0.000 claims description 7
- 108010025020 Nerve Growth Factor Proteins 0.000 claims description 7
- 229940083963 Peptide antagonist Drugs 0.000 claims description 7
- TWSALRJGPBVBQU-PKQQPRCHSA-N glucagon-like peptide 2 Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(O)=O)[C@@H](C)CC)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=CC=C1 TWSALRJGPBVBQU-PKQQPRCHSA-N 0.000 claims description 7
- 239000000816 peptidomimetic Substances 0.000 claims description 7
- 102000017916 BDKRB1 Human genes 0.000 claims description 6
- 101800000221 Glucagon-like peptide 2 Proteins 0.000 claims description 6
- 101000998953 Homo sapiens Immunoglobulin heavy variable 1-2 Proteins 0.000 claims description 6
- 102100036887 Immunoglobulin heavy variable 1-2 Human genes 0.000 claims description 6
- 108010044231 Bradykinin B1 Receptor Proteins 0.000 claims description 5
- 102100025588 Calcitonin gene-related peptide 1 Human genes 0.000 claims description 5
- 102100039939 Growth/differentiation factor 8 Human genes 0.000 claims description 4
- 108090001005 Interleukin-6 Proteins 0.000 claims description 4
- 108010056852 Myostatin Proteins 0.000 claims description 4
- 102000004889 Interleukin-6 Human genes 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 claims 2
- 102400000322 Glucagon-like peptide 1 Human genes 0.000 claims 2
- 102400000326 Glucagon-like peptide 2 Human genes 0.000 claims 2
- 238000004519 manufacturing process Methods 0.000 abstract description 31
- 239000008177 pharmaceutical agent Substances 0.000 abstract 1
- 108090000623 proteins and genes Proteins 0.000 description 199
- 102000004169 proteins and genes Human genes 0.000 description 170
- 235000018102 proteins Nutrition 0.000 description 159
- 239000000047 product Substances 0.000 description 118
- 235000001014 amino acid Nutrition 0.000 description 112
- 229940024606 amino acid Drugs 0.000 description 92
- 230000002829 reductive effect Effects 0.000 description 90
- 150000001413 amino acids Chemical class 0.000 description 87
- 150000001720 carbohydrates Chemical class 0.000 description 81
- 238000006467 substitution reaction Methods 0.000 description 53
- 239000012634 fragment Substances 0.000 description 52
- 125000005647 linker group Chemical group 0.000 description 44
- 102000053602 DNA Human genes 0.000 description 39
- 238000004458 analytical method Methods 0.000 description 36
- 108020004414 DNA Proteins 0.000 description 35
- 230000004048 modification Effects 0.000 description 35
- 238000012986 modification Methods 0.000 description 35
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 34
- 102220531375 Uncharacterized protein KIAA2012_Q16K_mutation Human genes 0.000 description 34
- 230000000694 effects Effects 0.000 description 34
- 238000006722 reduction reaction Methods 0.000 description 32
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 31
- 241001465754 Metazoa Species 0.000 description 30
- 230000004927 fusion Effects 0.000 description 29
- 230000035772 mutation Effects 0.000 description 29
- 239000000203 mixture Substances 0.000 description 28
- UFBJCMHMOXMLKC-UHFFFAOYSA-N 2,4-dinitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O UFBJCMHMOXMLKC-UHFFFAOYSA-N 0.000 description 25
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 24
- 230000009467 reduction Effects 0.000 description 24
- 239000003814 drug Substances 0.000 description 23
- 230000014509 gene expression Effects 0.000 description 23
- 239000000178 monomer Substances 0.000 description 23
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 23
- -1 dithiophosphate Chemical compound 0.000 description 22
- 125000003729 nucleotide group Chemical group 0.000 description 22
- AXAVXPMQTGXXJZ-UHFFFAOYSA-N 2-aminoacetic acid;2-amino-2-(hydroxymethyl)propane-1,3-diol Chemical compound NCC(O)=O.OCC(N)(CO)CO AXAVXPMQTGXXJZ-UHFFFAOYSA-N 0.000 description 21
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 20
- 239000000523 sample Substances 0.000 description 20
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 19
- 238000012217 deletion Methods 0.000 description 18
- 230000037430 deletion Effects 0.000 description 18
- 230000001965 increasing effect Effects 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- 108091026890 Coding region Proteins 0.000 description 17
- 238000002835 absorbance Methods 0.000 description 17
- 230000004071 biological effect Effects 0.000 description 17
- 238000003780 insertion Methods 0.000 description 17
- 230000037431 insertion Effects 0.000 description 17
- 239000002773 nucleotide Substances 0.000 description 17
- 102000005962 receptors Human genes 0.000 description 17
- 239000011780 sodium chloride Substances 0.000 description 17
- 230000001225 therapeutic effect Effects 0.000 description 17
- 239000003643 water by type Substances 0.000 description 17
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 16
- 239000012636 effector Substances 0.000 description 16
- 230000006870 function Effects 0.000 description 16
- 230000036039 immunity Effects 0.000 description 16
- 108020003175 receptors Proteins 0.000 description 16
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 15
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 15
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 15
- 102000003982 Parathyroid hormone Human genes 0.000 description 15
- 108090000445 Parathyroid hormone Proteins 0.000 description 15
- 239000002202 Polyethylene glycol Substances 0.000 description 15
- 108010076504 Protein Sorting Signals Proteins 0.000 description 15
- 235000004279 alanine Nutrition 0.000 description 15
- 125000000217 alkyl group Chemical group 0.000 description 15
- 238000000338 in vitro Methods 0.000 description 15
- 239000000199 parathyroid hormone Substances 0.000 description 15
- 229960001319 parathyroid hormone Drugs 0.000 description 15
- 229920001223 polyethylene glycol Polymers 0.000 description 15
- 102000040430 polynucleotide Human genes 0.000 description 15
- 108091033319 polynucleotide Proteins 0.000 description 15
- 239000002157 polynucleotide Substances 0.000 description 15
- 102000006395 Globulins Human genes 0.000 description 14
- 108010044091 Globulins Proteins 0.000 description 14
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 14
- 108091028043 Nucleic acid sequence Proteins 0.000 description 14
- 238000010586 diagram Methods 0.000 description 14
- 238000010186 staining Methods 0.000 description 14
- 102000004190 Enzymes Human genes 0.000 description 13
- 108090000790 Enzymes Proteins 0.000 description 13
- 108091034117 Oligonucleotide Proteins 0.000 description 13
- 229960000074 biopharmaceutical Drugs 0.000 description 13
- 239000003795 chemical substances by application Substances 0.000 description 13
- 229940088598 enzyme Drugs 0.000 description 13
- 239000012160 loading buffer Substances 0.000 description 13
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 12
- 241000700159 Rattus Species 0.000 description 12
- 238000003556 assay Methods 0.000 description 12
- 235000018417 cysteine Nutrition 0.000 description 12
- 238000000816 matrix-assisted laser desorption--ionisation Methods 0.000 description 12
- 238000002823 phage display Methods 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 238000010183 spectrum analysis Methods 0.000 description 12
- 229940124597 therapeutic agent Drugs 0.000 description 12
- 108020004705 Codon Proteins 0.000 description 11
- 239000004472 Lysine Substances 0.000 description 11
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 11
- CKLJMWTZIZZHCS-REOHCLBHSA-N aspartic acid group Chemical group N[C@@H](CC(=O)O)C(=O)O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 11
- 238000004113 cell culture Methods 0.000 description 11
- 239000000562 conjugate Substances 0.000 description 11
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 11
- 229960002433 cysteine Drugs 0.000 description 11
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- 238000001727 in vivo Methods 0.000 description 11
- 235000018977 lysine Nutrition 0.000 description 11
- 210000002966 serum Anatomy 0.000 description 11
- 238000001542 size-exclusion chromatography Methods 0.000 description 11
- 239000007790 solid phase Substances 0.000 description 11
- 208000024891 symptom Diseases 0.000 description 11
- 238000013518 transcription Methods 0.000 description 11
- 230000035897 transcription Effects 0.000 description 11
- 239000004475 Arginine Substances 0.000 description 10
- 102100040918 Pro-glucagon Human genes 0.000 description 10
- 238000007792 addition Methods 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 238000010494 dissociation reaction Methods 0.000 description 10
- 230000005593 dissociations Effects 0.000 description 10
- 238000005516 engineering process Methods 0.000 description 10
- 230000002209 hydrophobic effect Effects 0.000 description 10
- 229940072221 immunoglobulins Drugs 0.000 description 10
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- 238000001228 spectrum Methods 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- 238000001890 transfection Methods 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 241000588724 Escherichia coli Species 0.000 description 9
- 239000004471 Glycine Substances 0.000 description 9
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 9
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 9
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 9
- 206010035226 Plasma cell myeloma Diseases 0.000 description 9
- 108020004511 Recombinant DNA Proteins 0.000 description 9
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 9
- 235000009697 arginine Nutrition 0.000 description 9
- 125000003118 aryl group Chemical group 0.000 description 9
- 230000021615 conjugation Effects 0.000 description 9
- NKLPQNGYXWVELD-UHFFFAOYSA-M coomassie brilliant blue Chemical compound [Na+].C1=CC(OCC)=CC=C1NC1=CC=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C=CC(=CC=2)N(CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=C1 NKLPQNGYXWVELD-UHFFFAOYSA-M 0.000 description 9
- 108020001507 fusion proteins Proteins 0.000 description 9
- 102000037865 fusion proteins Human genes 0.000 description 9
- 230000013595 glycosylation Effects 0.000 description 9
- 238000006206 glycosylation reaction Methods 0.000 description 9
- 201000000050 myeloid neoplasm Diseases 0.000 description 9
- 241000894006 Bacteria Species 0.000 description 8
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 8
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 8
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 8
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 8
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 8
- 101100242312 Oryza sativa subsp. japonica OSK1 gene Proteins 0.000 description 8
- 101100533605 Oryza sativa subsp. japonica SKP1 gene Proteins 0.000 description 8
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 8
- 239000004473 Threonine Substances 0.000 description 8
- 230000009824 affinity maturation Effects 0.000 description 8
- 125000000151 cysteine group Chemical class N[C@@H](CS)C(=O)* 0.000 description 8
- 201000010099 disease Diseases 0.000 description 8
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 8
- 229960002743 glutamine Drugs 0.000 description 8
- 235000004554 glutamine Nutrition 0.000 description 8
- 239000011159 matrix material Substances 0.000 description 8
- 238000001906 matrix-assisted laser desorption--ionisation mass spectrometry Methods 0.000 description 8
- 239000002609 medium Substances 0.000 description 8
- 238000003752 polymerase chain reaction Methods 0.000 description 8
- 235000013930 proline Nutrition 0.000 description 8
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 7
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 7
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 7
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 7
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 7
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 7
- 238000012867 alanine scanning Methods 0.000 description 7
- 235000009582 asparagine Nutrition 0.000 description 7
- 229960001230 asparagine Drugs 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 238000011033 desalting Methods 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 239000000539 dimer Substances 0.000 description 7
- 239000003623 enhancer Substances 0.000 description 7
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 7
- 229960002885 histidine Drugs 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 238000001819 mass spectrum Methods 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 229930182817 methionine Natural products 0.000 description 7
- 235000006109 methionine Nutrition 0.000 description 7
- 239000002245 particle Substances 0.000 description 7
- 238000010647 peptide synthesis reaction Methods 0.000 description 7
- COLNVLDHVKWLRT-QMMMGPOBSA-N phenylalanine group Chemical group N[C@@H](CC1=CC=CC=C1)C(=O)O COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 7
- 238000003127 radioimmunoassay Methods 0.000 description 7
- 238000000926 separation method Methods 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 7
- 229920002307 Dextran Polymers 0.000 description 6
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 6
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 6
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 6
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 6
- 102000035554 Proglucagon Human genes 0.000 description 6
- 108010058003 Proglucagon Proteins 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 210000001744 T-lymphocyte Anatomy 0.000 description 6
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 6
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 6
- 230000002378 acidificating effect Effects 0.000 description 6
- 230000004913 activation Effects 0.000 description 6
- 238000001994 activation Methods 0.000 description 6
- 210000003719 b-lymphocyte Anatomy 0.000 description 6
- 239000011230 binding agent Substances 0.000 description 6
- 238000004422 calculation algorithm Methods 0.000 description 6
- 238000013368 capillary electrophoresis sodium dodecyl sulfate analysis Methods 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- 238000010367 cloning Methods 0.000 description 6
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 6
- 239000002299 complementary DNA Substances 0.000 description 6
- 238000001212 derivatisation Methods 0.000 description 6
- 239000005350 fused silica glass Substances 0.000 description 6
- 230000001900 immune effect Effects 0.000 description 6
- 230000001976 improved effect Effects 0.000 description 6
- 239000003112 inhibitor Substances 0.000 description 6
- 229960000310 isoleucine Drugs 0.000 description 6
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 6
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 229920000642 polymer Polymers 0.000 description 6
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 238000012216 screening Methods 0.000 description 6
- 150000003384 small molecules Chemical class 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 230000009870 specific binding Effects 0.000 description 6
- 238000010561 standard procedure Methods 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- 230000009261 transgenic effect Effects 0.000 description 6
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 6
- 239000004474 valine Substances 0.000 description 6
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 5
- 101000994669 Homo sapiens Potassium voltage-gated channel subfamily A member 3 Proteins 0.000 description 5
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 5
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 5
- 208000019695 Migraine disease Diseases 0.000 description 5
- 108010021466 Mutant Proteins Proteins 0.000 description 5
- 102000008300 Mutant Proteins Human genes 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 108010067902 Peptide Library Proteins 0.000 description 5
- 108010041111 Thrombopoietin Proteins 0.000 description 5
- 102000036693 Thrombopoietin Human genes 0.000 description 5
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 5
- LNTHITQWFMADLM-UHFFFAOYSA-N anhydrous gallic acid Natural products OC(=O)C1=CC(O)=C(O)C(O)=C1 LNTHITQWFMADLM-UHFFFAOYSA-N 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 235000014633 carbohydrates Nutrition 0.000 description 5
- 230000000295 complement effect Effects 0.000 description 5
- 239000000306 component Substances 0.000 description 5
- MWRBNPKJOOWZPW-CLFAGFIQSA-N dioleoyl phosphatidylethanolamine Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(COP(O)(=O)OCCN)OC(=O)CCCCCCC\C=C/CCCCCCCC MWRBNPKJOOWZPW-CLFAGFIQSA-N 0.000 description 5
- 210000001723 extracellular space Anatomy 0.000 description 5
- 229940049906 glutamate Drugs 0.000 description 5
- 229930195712 glutamate Natural products 0.000 description 5
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 5
- XKUKSGPZAADMRA-UHFFFAOYSA-N glycyl-glycyl-glycine Chemical compound NCC(=O)NCC(=O)NCC(O)=O XKUKSGPZAADMRA-UHFFFAOYSA-N 0.000 description 5
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 5
- 230000028993 immune response Effects 0.000 description 5
- 230000003053 immunization Effects 0.000 description 5
- 230000002163 immunogen Effects 0.000 description 5
- 230000016784 immunoglobulin production Effects 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 238000002372 labelling Methods 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 230000000670 limiting effect Effects 0.000 description 5
- 150000002632 lipids Chemical class 0.000 description 5
- 210000004962 mammalian cell Anatomy 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- 206010027599 migraine Diseases 0.000 description 5
- 201000006417 multiple sclerosis Diseases 0.000 description 5
- 229940053128 nerve growth factor Drugs 0.000 description 5
- 238000005457 optimization Methods 0.000 description 5
- 238000004091 panning Methods 0.000 description 5
- 125000001151 peptidyl group Chemical group 0.000 description 5
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 230000028327 secretion Effects 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- 230000009466 transformation Effects 0.000 description 5
- 230000001052 transient effect Effects 0.000 description 5
- 238000013519 translation Methods 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- QJYNZEYHSMRWBK-NIKIMHBISA-N 1,2,3,4,6-pentakis-O-galloyl-beta-D-glucose Chemical compound OC1=C(O)C(O)=CC(C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(O)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(O)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(O)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(O)C(O)=C(O)C=2)=C1 QJYNZEYHSMRWBK-NIKIMHBISA-N 0.000 description 4
- 102000014914 Carrier Proteins Human genes 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- 108010087819 Fc receptors Proteins 0.000 description 4
- 102000009109 Fc receptors Human genes 0.000 description 4
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 4
- 101800004266 Glucagon-like peptide 1(7-37) Proteins 0.000 description 4
- 108010051041 HC toxin Proteins 0.000 description 4
- 241000238631 Hexapoda Species 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 4
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 4
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 4
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 4
- 241000239299 Orthochirus scrobiculosus Species 0.000 description 4
- 241000239226 Scorpiones Species 0.000 description 4
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- 239000002671 adjuvant Substances 0.000 description 4
- 238000011091 antibody purification Methods 0.000 description 4
- 229940009098 aspartate Drugs 0.000 description 4
- 230000001580 bacterial effect Effects 0.000 description 4
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical compound NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 description 4
- 108091008324 binding proteins Proteins 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 238000004590 computer program Methods 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 230000007717 exclusion Effects 0.000 description 4
- 238000000855 fermentation Methods 0.000 description 4
- 230000004151 fermentation Effects 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 125000001475 halogen functional group Chemical group 0.000 description 4
- 238000002649 immunization Methods 0.000 description 4
- 230000005847 immunogenicity Effects 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 238000010369 molecular cloning Methods 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 4
- 229960005190 phenylalanine Drugs 0.000 description 4
- 230000004481 post-translational protein modification Effects 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- 230000008707 rearrangement Effects 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 239000002795 scorpion venom Substances 0.000 description 4
- 230000003248 secreting effect Effects 0.000 description 4
- 239000001488 sodium phosphate Substances 0.000 description 4
- 229910000162 sodium phosphate Inorganic materials 0.000 description 4
- 235000011008 sodium phosphates Nutrition 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000004611 spectroscopical analysis Methods 0.000 description 4
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 4
- 210000004989 spleen cell Anatomy 0.000 description 4
- 238000012453 sprague-dawley rat model Methods 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 150000003573 thiols Chemical class 0.000 description 4
- 239000013638 trimer Substances 0.000 description 4
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 4
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 description 3
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- 241000242759 Actiniaria Species 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 3
- 241000282693 Cercopithecidae Species 0.000 description 3
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 3
- 108091006146 Channels Proteins 0.000 description 3
- 241000251730 Chondrichthyes Species 0.000 description 3
- 108091035707 Consensus sequence Proteins 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- 108090000394 Erythropoietin Proteins 0.000 description 3
- 102000003951 Erythropoietin Human genes 0.000 description 3
- 102100031939 Erythropoietin Human genes 0.000 description 3
- 241000206602 Eukaryota Species 0.000 description 3
- 108091029865 Exogenous DNA Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 102100029098 Hypoxanthine-guanine phosphoribosyltransferase Human genes 0.000 description 3
- 108010073807 IgG Receptors Proteins 0.000 description 3
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 3
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 3
- 108090000862 Ion Channels Proteins 0.000 description 3
- 102000004310 Ion Channels Human genes 0.000 description 3
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 3
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 3
- 108010052285 Membrane Proteins Proteins 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 3
- 230000004989 O-glycosylation Effects 0.000 description 3
- 101001046988 Orthochirus scrobiculosus Potassium channel toxin alpha-KTx 3.7 Proteins 0.000 description 3
- 101001121312 Oryza sativa subsp. japonica Serine/threonine protein kinase OSK1 Proteins 0.000 description 3
- 229920002873 Polyethylenimine Polymers 0.000 description 3
- 102100034355 Potassium voltage-gated channel subfamily A member 3 Human genes 0.000 description 3
- 108010071690 Prealbumin Proteins 0.000 description 3
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 3
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- 102000009190 Transthyretin Human genes 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 3
- 229910052789 astatine Inorganic materials 0.000 description 3
- 230000000975 bioactive effect Effects 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 238000001311 chemical methods and process Methods 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 210000000349 chromosome Anatomy 0.000 description 3
- 239000002131 composite material Substances 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 3
- 239000005547 deoxyribonucleotide Substances 0.000 description 3
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 3
- 229940105423 erythropoietin Drugs 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 230000033581 fucosylation Effects 0.000 description 3
- 235000013922 glutamic acid Nutrition 0.000 description 3
- 229960002989 glutamic acid Drugs 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 208000010726 hind limb paralysis Diseases 0.000 description 3
- 229940088597 hormone Drugs 0.000 description 3
- 239000005556 hormone Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 210000003292 kidney cell Anatomy 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- UNAFTICPPXVTTN-UHFFFAOYSA-N n-dodecyldodecan-1-amine;hydrobromide Chemical compound [Br-].CCCCCCCCCCCC[NH2+]CCCCCCCCCCCC UNAFTICPPXVTTN-UHFFFAOYSA-N 0.000 description 3
- 230000000269 nucleophilic effect Effects 0.000 description 3
- 230000007170 pathology Effects 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 230000002797 proteolythic effect Effects 0.000 description 3
- 230000006798 recombination Effects 0.000 description 3
- 238000005215 recombination Methods 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000001632 sodium acetate Substances 0.000 description 3
- 235000017281 sodium acetate Nutrition 0.000 description 3
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000005030 transcription termination Effects 0.000 description 3
- 238000010361 transduction Methods 0.000 description 3
- 230000026683 transduction Effects 0.000 description 3
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical group N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- WMBWREPUVVBILR-WIYYLYMNSA-N (-)-Epigallocatechin-3-o-gallate Chemical compound O([C@@H]1CC2=C(O)C=C(C=C2O[C@@H]1C=1C=C(O)C(O)=C(O)C=1)O)C(=O)C1=CC(O)=C(O)C(O)=C1 WMBWREPUVVBILR-WIYYLYMNSA-N 0.000 description 2
- NMDDZEVVQDPECF-LURJTMIESA-N (2s)-2,7-diaminoheptanoic acid Chemical compound NCCCCC[C@H](N)C(O)=O NMDDZEVVQDPECF-LURJTMIESA-N 0.000 description 2
- DDYAPMZTJAYBOF-ZMYDTDHYSA-N (3S)-4-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-amino-1-[[(2S,3R)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-4-amino-1-[[(2S)-1-[[(2S)-4-amino-1-[[(2S)-4-amino-1-[[(2S,3S)-1-[[(1S)-1-carboxyethyl]amino]-3-methyl-1-oxopentan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-4-methylsulfanyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S,3R)-2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]-3-hydroxybutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]amino]-5-oxopentanoyl]amino]-4-oxobutanoic acid Chemical class [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O DDYAPMZTJAYBOF-ZMYDTDHYSA-N 0.000 description 2
- KWVJHCQQUFDPLU-YEUCEMRASA-N 2,3-bis[[(z)-octadec-9-enoyl]oxy]propyl-trimethylazanium Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC KWVJHCQQUFDPLU-YEUCEMRASA-N 0.000 description 2
- BRMWTNUJHUMWMS-UHFFFAOYSA-N 3-Methylhistidine Natural products CN1C=NC(CC(N)C(O)=O)=C1 BRMWTNUJHUMWMS-UHFFFAOYSA-N 0.000 description 2
- 229940117976 5-hydroxylysine Drugs 0.000 description 2
- GANZODCWZFAEGN-UHFFFAOYSA-N 5-mercapto-2-nitro-benzoic acid Chemical compound OC(=O)C1=CC(S)=CC=C1[N+]([O-])=O GANZODCWZFAEGN-UHFFFAOYSA-N 0.000 description 2
- 229920000936 Agarose Polymers 0.000 description 2
- 206010003445 Ascites Diseases 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 229940104754 Bradykinin B1 receptor antagonist Drugs 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 241000557626 Corvus corax Species 0.000 description 2
- 241000699800 Cricetinae Species 0.000 description 2
- 102000036364 Cullin Ring E3 Ligases Human genes 0.000 description 2
- 108091007045 Cullin Ring E3 Ligases Proteins 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- QSJXEFYPDANLFS-UHFFFAOYSA-N Diacetyl Chemical compound CC(=O)C(C)=O QSJXEFYPDANLFS-UHFFFAOYSA-N 0.000 description 2
- 241000255925 Diptera Species 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 108010008177 Fd immunoglobulins Proteins 0.000 description 2
- 241000724791 Filamentous phage Species 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 2
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 2
- WMBWREPUVVBILR-UHFFFAOYSA-N GCG Natural products C=1C(O)=C(O)C(O)=CC=1C1OC2=CC(O)=CC(O)=C2CC1OC(=O)C1=CC(O)=C(O)C(O)=C1 WMBWREPUVVBILR-UHFFFAOYSA-N 0.000 description 2
- 102000051325 Glucagon Human genes 0.000 description 2
- 108060003199 Glucagon Proteins 0.000 description 2
- 108010088406 Glucagon-Like Peptides Proteins 0.000 description 2
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 2
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 2
- 101001135770 Homo sapiens Parathyroid hormone Proteins 0.000 description 2
- 101001135995 Homo sapiens Probable peptidyl-tRNA hydrolase Proteins 0.000 description 2
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 2
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 2
- QUOGESRFPZDMMT-YFKPBYRVSA-N L-homoarginine Chemical compound OC(=O)[C@@H](N)CCCCNC(N)=N QUOGESRFPZDMMT-YFKPBYRVSA-N 0.000 description 2
- FFFHZYDWPBMWHY-VKHMYHEASA-N L-homocysteine Chemical group OC(=O)[C@@H](N)CCS FFFHZYDWPBMWHY-VKHMYHEASA-N 0.000 description 2
- 241000282567 Macaca fascicularis Species 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 102000047918 Myelin Basic Human genes 0.000 description 2
- 101710107068 Myelin basic protein Proteins 0.000 description 2
- 241000237536 Mytilus edulis Species 0.000 description 2
- JDHILDINMRGULE-LURJTMIESA-N N(pros)-methyl-L-histidine Chemical compound CN1C=NC=C1C[C@H](N)C(O)=O JDHILDINMRGULE-LURJTMIESA-N 0.000 description 2
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical compound CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 description 2
- 230000004988 N-glycosylation Effects 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 108010038807 Oligopeptides Proteins 0.000 description 2
- 102000015636 Oligopeptides Human genes 0.000 description 2
- 208000002193 Pain Diseases 0.000 description 2
- 206010033799 Paralysis Diseases 0.000 description 2
- 102100030869 Parathyroid hormone 2 receptor Human genes 0.000 description 2
- 101710149426 Parathyroid hormone 2 receptor Proteins 0.000 description 2
- 102100032256 Parathyroid hormone/parathyroid hormone-related peptide receptor Human genes 0.000 description 2
- 101710180613 Parathyroid hormone/parathyroid hormone-related peptide receptor Proteins 0.000 description 2
- 206010057249 Phagocytosis Diseases 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 108010039918 Polylysine Proteins 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- RADKZDMFGJYCBB-UHFFFAOYSA-N Pyridoxal Chemical compound CC1=NC=C(CO)C(C=O)=C1O RADKZDMFGJYCBB-UHFFFAOYSA-N 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 108091028664 Ribonucleotide Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 241000235088 Saccharomyces sp. Species 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- 241000607720 Serratia Species 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 108091008874 T cell receptors Proteins 0.000 description 2
- 230000005867 T cell response Effects 0.000 description 2
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 2
- NYTOUQBROMCLBJ-UHFFFAOYSA-N Tetranitromethane Chemical compound [O-][N+](=O)C([N+]([O-])=O)([N+]([O-])=O)[N+]([O-])=O NYTOUQBROMCLBJ-UHFFFAOYSA-N 0.000 description 2
- 101710120037 Toxin CcdB Proteins 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 101000980463 Treponema pallidum (strain Nichols) Chaperonin GroEL Proteins 0.000 description 2
- 101710181056 Tumor necrosis factor ligand superfamily member 13B Proteins 0.000 description 2
- 102100036922 Tumor necrosis factor ligand superfamily member 13B Human genes 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- HIHOWBSBBDRPDW-PTHRTHQKSA-N [(3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-yl] n-[2-(dimethylamino)ethyl]carbamate Chemical compound C1C=C2C[C@@H](OC(=O)NCCN(C)C)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HIHOWBSBBDRPDW-PTHRTHQKSA-N 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 230000006229 amino acid addition Effects 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- 230000008827 biological function Effects 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 239000003144 bradykinin B1 receptor antagonist Substances 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 150000001718 carbodiimides Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000007910 cell fusion Effects 0.000 description 2
- 238000012875 competitive assay Methods 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- UFULAYFCSOUIOV-UHFFFAOYSA-N cysteamine Chemical compound NCCS UFULAYFCSOUIOV-UHFFFAOYSA-N 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 238000005034 decoration Methods 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 230000022811 deglycosylation Effects 0.000 description 2
- VEVRNHHLCPGNDU-MUGJNUQGSA-O desmosine Chemical compound OC(=O)[C@@H](N)CCCC[N+]1=CC(CC[C@H](N)C(O)=O)=C(CCC[C@H](N)C(O)=O)C(CC[C@H](N)C(O)=O)=C1 VEVRNHHLCPGNDU-MUGJNUQGSA-O 0.000 description 2
- 239000000032 diagnostic agent Substances 0.000 description 2
- 229940039227 diagnostic agent Drugs 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 229940030275 epigallocatechin gallate Drugs 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 210000003414 extremity Anatomy 0.000 description 2
- 230000005714 functional activity Effects 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 229940074391 gallic acid Drugs 0.000 description 2
- 235000004515 gallic acid Nutrition 0.000 description 2
- 238000001415 gene therapy Methods 0.000 description 2
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 2
- 150000004676 glycans Chemical class 0.000 description 2
- 239000000710 homodimer Substances 0.000 description 2
- 102000046611 human KCNA3 Human genes 0.000 description 2
- 102000058004 human PTH Human genes 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000012872 hydroxylapatite chromatography Methods 0.000 description 2
- 229960002591 hydroxyproline Drugs 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 210000003000 inclusion body Anatomy 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 238000005304 joining Methods 0.000 description 2
- 238000012004 kinetic exclusion assay Methods 0.000 description 2
- 238000011694 lewis rat Methods 0.000 description 2
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 108091005601 modified peptides Proteins 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 235000020638 mussel Nutrition 0.000 description 2
- AHLPHDHHMVZTML-BYPYZUCNSA-N ornithyl group Chemical group N[C@@H](CCCN)C(=O)O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 230000036407 pain Effects 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000006320 pegylation Effects 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 210000001322 periplasm Anatomy 0.000 description 2
- 230000008782 phagocytosis Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- OJUGVDODNPJEEC-UHFFFAOYSA-N phenylglyoxal Chemical compound O=CC(=O)C1=CC=CC=C1 OJUGVDODNPJEEC-UHFFFAOYSA-N 0.000 description 2
- 230000036470 plasma concentration Effects 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 230000008488 polyadenylation Effects 0.000 description 2
- 108010054442 polyalanine Proteins 0.000 description 2
- 229920000656 polylysine Polymers 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 230000012743 protein tagging Effects 0.000 description 2
- 230000017854 proteolysis Effects 0.000 description 2
- 230000005180 public health Effects 0.000 description 2
- NGVDGCNFYWLIFO-UHFFFAOYSA-N pyridoxal 5'-phosphate Chemical compound CC1=NC=C(COP(O)(O)=O)C(C=O)=C1O NGVDGCNFYWLIFO-UHFFFAOYSA-N 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 238000010188 recombinant method Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 239000002336 ribonucleotide Substances 0.000 description 2
- 125000002652 ribonucleotide group Chemical group 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 2
- 238000010532 solid phase synthesis reaction Methods 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- 229940063675 spermine Drugs 0.000 description 2
- 238000010254 subcutaneous injection Methods 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 238000011830 transgenic mouse model Methods 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 239000002435 venom Substances 0.000 description 2
- 231100000611 venom Toxicity 0.000 description 2
- 210000001048 venom Anatomy 0.000 description 2
- PFWOJTLVNOZSGQ-RIMJLNMDSA-N (1R,2aS,4S,7S,10S,13S,19S,22S,25S,28S,31R,36R,39S,45R,48S,51S,54S,57R,60S,63S,66S,69S,72S,75S,78S,81S,84S,87S,90R,93S,96S,99S)-10,51,87-tris(4-aminobutyl)-31-[[(2S)-2-[[(2S)-2-amino-5-carbamimidamidopentanoyl]amino]-3-hydroxypropanoyl]amino]-75-(aminomethyl)-93-(3-amino-3-oxopropyl)-60,96-dibenzyl-19,28-bis[(2S)-butan-2-yl]-4,54,69-tris(3-carbamimidamidopropyl)-25-(carboxymethyl)-2a,22,39,48-tetrakis[(1R)-1-hydroxyethyl]-7,63,81-tris(hydroxymethyl)-72-[(4-hydroxyphenyl)methyl]-84-(1H-imidazol-5-ylmethyl)-99-methyl-66-(2-methylpropyl)-78-(2-methylsulfanylethyl)-1a,3,4a,6,9,12,18,21,24,27,30,38,41,44,47,50,53,56,59,62,65,68,71,74,77,80,83,86,89,92,95,98-dotriacontaoxo-6a,7a,10a,11a,33,34-hexathia-a,2,3a,5,8,11,17,20,23,26,29,37,40,43,46,49,52,55,58,61,64,67,70,73,76,79,82,85,88,91,94,97-dotriacontazatetracyclo[55.47.4.445,90.013,17]dodecahectane-36-carboxylic acid Chemical compound CC[C@H](C)[C@@H]1NC(=O)[C@H](CSSC[C@H](NC(=O)[C@@H](NC(=O)CNC(=O)[C@@H]2CSSC[C@@H]3NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](Cc4ccccc4)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CSSC[C@H](NC(=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc4ccc(O)cc4)NC(=O)[C@H](CN)NC(=O)[C@H](CCSC)NC(=O)[C@H](CO)NC(=O)[C@H](Cc4cnc[nH]4)NC(=O)[C@H](CCCCN)NC3=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(=O)N2)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@@H]2CCCN2C(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC1=O)[C@@H](C)O)[C@@H](C)CC)[C@@H](C)O)[C@@H](C)O)C(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CCCNC(N)=N PFWOJTLVNOZSGQ-RIMJLNMDSA-N 0.000 description 1
- VUNNYEFBYZVJCK-RXMQYKEDSA-N (2R)-2-amino-2-(hydroxymethyl)-3-oxobutanoic acid Chemical compound CC(=O)[C@](N)(CO)C(O)=O VUNNYEFBYZVJCK-RXMQYKEDSA-N 0.000 description 1
- AMHXPZLIADYCSB-ZCFIWIBFSA-N (2R)-2-amino-2-formyl-4-methylsulfanylbutanoic acid Chemical compound CSCC[C@@](N)(C=O)C(O)=O AMHXPZLIADYCSB-ZCFIWIBFSA-N 0.000 description 1
- GMKMEZVLHJARHF-UHFFFAOYSA-N (2R,6R)-form-2.6-Diaminoheptanedioic acid Natural products OC(=O)C(N)CCCC(N)C(O)=O GMKMEZVLHJARHF-UHFFFAOYSA-N 0.000 description 1
- OPCHFPHZPIURNA-MFERNQICSA-N (2s)-2,5-bis(3-aminopropylamino)-n-[2-(dioctadecylamino)acetyl]pentanamide Chemical compound CCCCCCCCCCCCCCCCCCN(CC(=O)NC(=O)[C@H](CCCNCCCN)NCCCN)CCCCCCCCCCCCCCCCCC OPCHFPHZPIURNA-MFERNQICSA-N 0.000 description 1
- GPYTYOMSQHBYTK-LURJTMIESA-N (2s)-2-azaniumyl-2,3-dimethylbutanoate Chemical compound CC(C)[C@](C)([NH3+])C([O-])=O GPYTYOMSQHBYTK-LURJTMIESA-N 0.000 description 1
- KYBXNPIASYUWLN-WUCPZUCCSA-N (2s)-5-hydroxypyrrolidine-2-carboxylic acid Chemical compound OC1CC[C@@H](C(O)=O)N1 KYBXNPIASYUWLN-WUCPZUCCSA-N 0.000 description 1
- RSPOGBIHKNKRFJ-FSPLSTOPSA-N (2s,3s)-2-amino-2,3-dimethylpentanoic acid Chemical compound CC[C@H](C)[C@](C)(N)C(O)=O RSPOGBIHKNKRFJ-FSPLSTOPSA-N 0.000 description 1
- NGJOFQZEYQGZMB-KTKZVXAJSA-N (4S)-5-[[2-[[(2S,3R)-1-[[(2S)-1-[[(2S,3R)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[2-[[(1S)-4-carbamimidamido-1-carboxybutyl]amino]-2-oxoethyl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-2-oxoethyl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-2-oxoethyl]amino]-4-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]propanoyl]amino]-5-oxopentanoic acid Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NGJOFQZEYQGZMB-KTKZVXAJSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- HXRVTZVVSGPFEC-BTJKTKAUSA-N (z)-but-2-enedioic acid;2,3-dihydroxybutanedioic acid Chemical compound OC(=O)\C=C/C(O)=O.OC(=O)C(O)C(O)C(O)=O HXRVTZVVSGPFEC-BTJKTKAUSA-N 0.000 description 1
- LUTLAXLNPLZCOF-UHFFFAOYSA-N 1-Methylhistidine Chemical group OC(=O)C(N)(C)CC1=NC=CN1 LUTLAXLNPLZCOF-UHFFFAOYSA-N 0.000 description 1
- YAMUFBLWGFFICM-PTGWMXDISA-N 1-O-oleoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)COP([O-])(=O)OCC[N+](C)(C)C YAMUFBLWGFFICM-PTGWMXDISA-N 0.000 description 1
- RYCNUMLMNKHWPZ-SNVBAGLBSA-N 1-acetyl-sn-glycero-3-phosphocholine Chemical compound CC(=O)OC[C@@H](O)COP([O-])(=O)OCC[N+](C)(C)C RYCNUMLMNKHWPZ-SNVBAGLBSA-N 0.000 description 1
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical group COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 1
- 125000003287 1H-imidazol-4-ylmethyl group Chemical group [H]N1C([H])=NC(C([H])([H])[*])=C1[H] 0.000 description 1
- LDGWQMRUWMSZIU-LQDDAWAPSA-M 2,3-bis[(z)-octadec-9-enoxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCCOCC(C[N+](C)(C)C)OCCCCCCCC\C=C/CCCCCCCC LDGWQMRUWMSZIU-LQDDAWAPSA-M 0.000 description 1
- NHJVRSWLHSJWIN-UHFFFAOYSA-N 2,4,6-trinitrobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O NHJVRSWLHSJWIN-UHFFFAOYSA-N 0.000 description 1
- OGNSCSPNOLGXSM-UHFFFAOYSA-N 2,4-diaminobutyric acid Chemical group NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- BHANCCMWYDZQOR-UHFFFAOYSA-N 2-(methyldisulfanyl)pyridine Chemical compound CSSC1=CC=CC=N1 BHANCCMWYDZQOR-UHFFFAOYSA-N 0.000 description 1
- GSCHIGXDTVYEEM-UHFFFAOYSA-N 2-[2-[[3-[6-[[4,5-dihydroxy-3-[3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxymethyl]-3,4-dihydroxy-5-[3,4,5-trihydroxy-6-[(3,4,5-trihydroxyoxan-2-yl)oxymethyl]oxan-2-yl]oxyoxan-2-yl]oxy-4,5-dihydroxy-6-[4,5,6-trihydroxy-2-(hydroxymethyl)oxan Chemical compound OC1C(O)C(O)C(CO)OC1OC1C(OCC2C(C(O)C(O)C(OC3C(OC(O)C(O)C3O)CO)O2)OC2C(C(O)C(OC3C(C(O)C(O)C(COC4C(C(O)C(O)CO4)O)O3)O)C(COC3C(C(O)C(O)CO3)OC3C(C(O)C(O)C(CO)O3)O)O2)O)OCC(O)C1O GSCHIGXDTVYEEM-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 125000000979 2-amino-2-oxoethyl group Chemical group [H]C([*])([H])C(=O)N([H])[H] 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- BVVPXQPZVXOEKA-UHFFFAOYSA-N 2-amino-N-[3-[4-(3-aminopropylamino)butylamino]propyl]acetamide N-octadecyloctadecan-1-amine Chemical compound NCCCNCCCCNCCCNC(=O)CN.CCCCCCCCCCCCCCCCCCNCCCCCCCCCCCCCCCCCC BVVPXQPZVXOEKA-UHFFFAOYSA-N 0.000 description 1
- RDFMDVXONNIGBC-UHFFFAOYSA-N 2-aminoheptanoic acid Chemical compound CCCCCC(N)C(O)=O RDFMDVXONNIGBC-UHFFFAOYSA-N 0.000 description 1
- JUQLUIFNNFIIKC-UHFFFAOYSA-N 2-aminopimelic acid Chemical compound OC(=O)C(N)CCCCC(O)=O JUQLUIFNNFIIKC-UHFFFAOYSA-N 0.000 description 1
- OFYAYGJCPXRNBL-UHFFFAOYSA-N 2-azaniumyl-3-naphthalen-1-ylpropanoate Chemical compound C1=CC=C2C(CC(N)C(O)=O)=CC=CC2=C1 OFYAYGJCPXRNBL-UHFFFAOYSA-N 0.000 description 1
- FKJSFKCZZIXQIP-UHFFFAOYSA-N 2-bromo-1-(4-bromophenyl)ethanone Chemical compound BrCC(=O)C1=CC=C(Br)C=C1 FKJSFKCZZIXQIP-UHFFFAOYSA-N 0.000 description 1
- CRTWOYOCILXSSX-UHFFFAOYSA-N 2-bromo-3-(1h-imidazol-5-yl)propanoic acid Chemical compound OC(=O)C(Br)CC1=CN=CN1 CRTWOYOCILXSSX-UHFFFAOYSA-N 0.000 description 1
- JQPFYXFVUKHERX-UHFFFAOYSA-N 2-hydroxy-2-cyclohexen-1-one Natural products OC1=CCCCC1=O JQPFYXFVUKHERX-UHFFFAOYSA-N 0.000 description 1
- NFJMWGFHZFBGJQ-LQDDAWAPSA-M 3,3-bis[[(z)-octadec-9-enoyl]oxy]propyl-trimethylazanium;bromide Chemical compound [Br-].CCCCCCCC\C=C/CCCCCCCC(=O)OC(CC[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC NFJMWGFHZFBGJQ-LQDDAWAPSA-M 0.000 description 1
- VJINKBZUJYGZGP-UHFFFAOYSA-N 3-(1-aminopropylideneamino)propyl-trimethylazanium Chemical compound CCC(N)=NCCC[N+](C)(C)C VJINKBZUJYGZGP-UHFFFAOYSA-N 0.000 description 1
- PECYZEOJVXMISF-UHFFFAOYSA-N 3-aminoalanine Chemical group [NH3+]CC(N)C([O-])=O PECYZEOJVXMISF-UHFFFAOYSA-N 0.000 description 1
- ONZQYZKCUHFORE-UHFFFAOYSA-N 3-bromo-1,1,1-trifluoropropan-2-one Chemical compound FC(F)(F)C(=O)CBr ONZQYZKCUHFORE-UHFFFAOYSA-N 0.000 description 1
- QHSXWDVVFHXHHB-UHFFFAOYSA-N 3-nitro-2-[(3-nitropyridin-2-yl)disulfanyl]pyridine Chemical compound [O-][N+](=O)C1=CC=CN=C1SSC1=NC=CC=C1[N+]([O-])=O QHSXWDVVFHXHHB-UHFFFAOYSA-N 0.000 description 1
- QFVHZQCOUORWEI-UHFFFAOYSA-N 4-[(4-anilino-5-sulfonaphthalen-1-yl)diazenyl]-5-hydroxynaphthalene-2,7-disulfonic acid Chemical compound C=12C(O)=CC(S(O)(=O)=O)=CC2=CC(S(O)(=O)=O)=CC=1N=NC(C1=CC=CC(=C11)S(O)(=O)=O)=CC=C1NC1=CC=CC=C1 QFVHZQCOUORWEI-UHFFFAOYSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- AGFIRQJZCNVMCW-UAKXSSHOSA-N 5-bromouridine Chemical class O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 AGFIRQJZCNVMCW-UAKXSSHOSA-N 0.000 description 1
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 241000256118 Aedes aegypti Species 0.000 description 1
- 241000256173 Aedes albopictus Species 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 241000024188 Andala Species 0.000 description 1
- 101001023095 Anemonia sulcata Delta-actitoxin-Avd1a Proteins 0.000 description 1
- 235000002198 Annona diversifolia Nutrition 0.000 description 1
- 108010032595 Antibody Binding Sites Proteins 0.000 description 1
- 101710170231 Antimicrobial peptide 2 Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 101100107610 Arabidopsis thaliana ABCF4 gene Proteins 0.000 description 1
- 241000239290 Araneae Species 0.000 description 1
- 101000641989 Araneus ventricosus Kunitz-type U1-aranetoxin-Av1a Proteins 0.000 description 1
- 208000002109 Argyria Diseases 0.000 description 1
- 206010003178 Arterial thrombosis Diseases 0.000 description 1
- FTCGGKNCJZOPNB-WHFBIAKZSA-N Asn-Gly-Ser Chemical compound NC(=O)C[C@H](N)C(=O)NCC(=O)N[C@@H](CO)C(O)=O FTCGGKNCJZOPNB-WHFBIAKZSA-N 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 241000351920 Aspergillus nidulans Species 0.000 description 1
- 241000228245 Aspergillus niger Species 0.000 description 1
- 206010003591 Ataxia Diseases 0.000 description 1
- 102100022717 Atypical chemokine receptor 1 Human genes 0.000 description 1
- 241001203868 Autographa californica Species 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- 241000194108 Bacillus licheniformis Species 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 241000409811 Bombyx mori nucleopolyhedrovirus Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000282832 Camelidae Species 0.000 description 1
- 241000282836 Camelus dromedarius Species 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 101001028691 Carybdea rastonii Toxin CrTX-A Proteins 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 101000685083 Centruroides infamatus Beta-toxin Cii1 Proteins 0.000 description 1
- 241000239328 Centruroides noxius Species 0.000 description 1
- 101000685085 Centruroides noxius Toxin Cn1 Proteins 0.000 description 1
- 241000239282 Centruroides suffusus Species 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 101001028688 Chironex fleckeri Toxin CfTX-1 Proteins 0.000 description 1
- 241000867607 Chlorocebus sabaeus Species 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- 101000644407 Cyriopagopus schmidti U6-theraphotoxin-Hs1a Proteins 0.000 description 1
- 101150097493 D gene Proteins 0.000 description 1
- 150000008574 D-amino acids Chemical class 0.000 description 1
- ODKSFYDXXFIFQN-SCSAIBSYSA-N D-arginine Chemical compound OC(=O)[C@H](N)CCCNC(N)=N ODKSFYDXXFIFQN-SCSAIBSYSA-N 0.000 description 1
- 229930028154 D-arginine Natural products 0.000 description 1
- LEVWYRKDKASIDU-QWWZWVQMSA-N D-cystine Chemical compound OC(=O)[C@H](N)CSSC[C@@H](N)C(O)=O LEVWYRKDKASIDU-QWWZWVQMSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Polymers OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical class OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- 101000761020 Dinoponera quadriceps Poneritoxin Proteins 0.000 description 1
- 241000255601 Drosophila melanogaster Species 0.000 description 1
- 241000588914 Enterobacter Species 0.000 description 1
- 241000588921 Enterobacteriaceae Species 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000588698 Erwinia Species 0.000 description 1
- 241000588722 Escherichia Species 0.000 description 1
- 108010011459 Exenatide Proteins 0.000 description 1
- 108091006020 Fc-tagged proteins Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 241000237858 Gastropoda Species 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- 101800004295 Glucagon-like peptide 1(7-36) Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 102000002254 Glycogen Synthase Kinase 3 Human genes 0.000 description 1
- 108010014905 Glycogen Synthase Kinase 3 Proteins 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical class OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- HVLSXIKZNLPZJJ-TXZCQADKSA-N HA peptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 HVLSXIKZNLPZJJ-TXZCQADKSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 208000009889 Herpes Simplex Diseases 0.000 description 1
- 229920000209 Hexadimethrine bromide Polymers 0.000 description 1
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 1
- 101000678879 Homo sapiens Atypical chemokine receptor 1 Proteins 0.000 description 1
- 101500026352 Homo sapiens Bradykinin Proteins 0.000 description 1
- 101500028771 Homo sapiens Glucagon-like peptide 2 Proteins 0.000 description 1
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 description 1
- 101001055314 Homo sapiens Immunoglobulin heavy constant alpha 2 Proteins 0.000 description 1
- 101001055308 Homo sapiens Immunoglobulin heavy constant epsilon Proteins 0.000 description 1
- 101000961156 Homo sapiens Immunoglobulin heavy constant gamma 1 Proteins 0.000 description 1
- 101000961146 Homo sapiens Immunoglobulin heavy constant gamma 2 Proteins 0.000 description 1
- 101000961145 Homo sapiens Immunoglobulin heavy constant gamma 3 Proteins 0.000 description 1
- 101000917826 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-a Proteins 0.000 description 1
- 101000917824 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-b Proteins 0.000 description 1
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 1
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- LCWXJXMHJVIJFK-UHFFFAOYSA-N Hydroxylysine Natural products NCC(O)CC(N)CC(O)=O LCWXJXMHJVIJFK-UHFFFAOYSA-N 0.000 description 1
- 241000257303 Hymenoptera Species 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 1
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 1
- 102000012745 Immunoglobulin Subunits Human genes 0.000 description 1
- 108010079585 Immunoglobulin Subunits Proteins 0.000 description 1
- 102100026216 Immunoglobulin heavy constant alpha 2 Human genes 0.000 description 1
- 102100039345 Immunoglobulin heavy constant gamma 1 Human genes 0.000 description 1
- 102100039346 Immunoglobulin heavy constant gamma 2 Human genes 0.000 description 1
- 102100039348 Immunoglobulin heavy constant gamma 3 Human genes 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 229930010555 Inosine Chemical class 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical class O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- 241000235649 Kluyveromyces Species 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- QUOGESRFPZDMMT-UHFFFAOYSA-N L-Homoarginine Natural products OC(=O)C(N)CCCCNC(N)=N QUOGESRFPZDMMT-UHFFFAOYSA-N 0.000 description 1
- AGPKZVBTJJNPAG-UHNVWZDZSA-N L-allo-Isoleucine Chemical compound CC[C@@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-UHNVWZDZSA-N 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical group C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 125000000510 L-tryptophano group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C(C([H])([H])[C@@]([H])(C(O[H])=O)N([H])[*])C2=C1[H] 0.000 description 1
- 241000282838 Lama Species 0.000 description 1
- 102100029204 Low affinity immunoglobulin gamma Fc region receptor II-a Human genes 0.000 description 1
- NPBGTPKLVJEOBE-IUCAKERBSA-N Lys-Arg Chemical compound NCCCC[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=N NPBGTPKLVJEOBE-IUCAKERBSA-N 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000542857 Megathura Species 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 101001050257 Mesobuthus martensii Potassium channel toxin alpha-KTx 16.3 Proteins 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- DTERQYGMUDWYAZ-ZETCQYMHSA-N N(6)-acetyl-L-lysine Chemical compound CC(=O)NCCCC[C@H]([NH3+])C([O-])=O DTERQYGMUDWYAZ-ZETCQYMHSA-N 0.000 description 1
- NTNWOCRCBQPEKQ-YFKPBYRVSA-N N(omega)-methyl-L-arginine Chemical group CN=C(N)NCCC[C@H](N)C(O)=O NTNWOCRCBQPEKQ-YFKPBYRVSA-N 0.000 description 1
- BRMWTNUJHUMWMS-LURJTMIESA-N N(tele)-methyl-L-histidine Chemical group CN1C=NC(C[C@H](N)C(O)=O)=C1 BRMWTNUJHUMWMS-LURJTMIESA-N 0.000 description 1
- NTWVQPHTOUKMDI-YFKPBYRVSA-N N-Methyl-arginine Chemical group CN[C@H](C(O)=O)CCCN=C(N)N NTWVQPHTOUKMDI-YFKPBYRVSA-N 0.000 description 1
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 description 1
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 1
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 241000221960 Neurospora Species 0.000 description 1
- 241000221961 Neurospora crassa Species 0.000 description 1
- 101100395023 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) his-7 gene Proteins 0.000 description 1
- NCHSYZVVWKVWFQ-BYPYZUCNSA-N Ng-amino-l-arginine Chemical group NNC(N)=NCCC[C@H](N)C(O)=O NCHSYZVVWKVWFQ-BYPYZUCNSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Chemical group NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Chemical group OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 108010058846 Ovalbumin Proteins 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 206010033885 Paraparesis Diseases 0.000 description 1
- 206010033892 Paraplegia Diseases 0.000 description 1
- 102000043299 Parathyroid hormone-related Human genes 0.000 description 1
- 101710123753 Parathyroid hormone-related protein Proteins 0.000 description 1
- 241000237988 Patellidae Species 0.000 description 1
- 241000228143 Penicillium Species 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091093037 Peptide nucleic acid Proteins 0.000 description 1
- 101000679608 Phaeosphaeria nodorum (strain SN15 / ATCC MYA-4574 / FGSC 10173) Cysteine rich necrotrophic effector Tox1 Proteins 0.000 description 1
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 1
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 1
- 101710124951 Phospholipase C Proteins 0.000 description 1
- 241000235648 Pichia Species 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 241000588769 Proteus <enterobacteria> Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 208000010378 Pulmonary Embolism Diseases 0.000 description 1
- 230000026279 RNA modification Effects 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 101100068078 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) GCN4 gene Proteins 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 1
- 108010084592 Saporins Proteins 0.000 description 1
- 108010077895 Sarcosine Proteins 0.000 description 1
- 241000235347 Schizosaccharomyces pombe Species 0.000 description 1
- 241000311088 Schwanniomyces Species 0.000 description 1
- 241001123650 Schwanniomyces occidentalis Species 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 241000270295 Serpentes Species 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 241000607768 Shigella Species 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- FBPFZTCFMRRESA-NQAPHZHOSA-N Sorbitol Polymers OCC(O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-NQAPHZHOSA-N 0.000 description 1
- 241000256251 Spodoptera frugiperda Species 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 102000002248 Thyroxine-Binding Globulin Human genes 0.000 description 1
- 108010000259 Thyroxine-Binding Globulin Proteins 0.000 description 1
- 101000994422 Tityus discrepans Potassium channel toxin alpha-KTx 15.6 Proteins 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 241000223259 Trichoderma Species 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 101710162629 Trypsin inhibitor Proteins 0.000 description 1
- 229940122618 Trypsin inhibitor Drugs 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 101150117115 V gene Proteins 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 241001416177 Vicugna pacos Species 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 241000235013 Yarrowia Species 0.000 description 1
- CWRILEGKIAOYKP-SSDOTTSWSA-M [(2r)-3-acetyloxy-2-hydroxypropyl] 2-aminoethyl phosphate Chemical compound CC(=O)OC[C@@H](O)COP([O-])(=O)OCCN CWRILEGKIAOYKP-SSDOTTSWSA-M 0.000 description 1
- HMNZFMSWFCAGGW-XPWSMXQVSA-N [3-[hydroxy(2-hydroxyethoxy)phosphoryl]oxy-2-[(e)-octadec-9-enoyl]oxypropyl] (e)-octadec-9-enoate Chemical compound CCCCCCCC\C=C\CCCCCCCC(=O)OCC(COP(O)(=O)OCCO)OC(=O)CCCCCCC\C=C\CCCCCCCC HMNZFMSWFCAGGW-XPWSMXQVSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229940037003 alum Drugs 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- 125000002344 aminooxy group Chemical group [H]N([H])O[*] 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 238000012870 ammonium sulfate precipitation Methods 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 238000005571 anion exchange chromatography Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001064 anti-interferon Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000009830 antibody antigen interaction Effects 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical class [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- 229940000635 beta-alanine Drugs 0.000 description 1
- 150000001576 beta-amino acids Chemical class 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 239000012503 blood component Substances 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 238000005277 cation exchange chromatography Methods 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 230000009920 chelation Effects 0.000 description 1
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- VXIVSQZSERGHQP-UHFFFAOYSA-N chloroacetamide Chemical compound NC(=O)CCl VXIVSQZSERGHQP-UHFFFAOYSA-N 0.000 description 1
- FOCAUTSVDIKZOP-UHFFFAOYSA-N chloroacetic acid Chemical compound OC(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-N 0.000 description 1
- 229940106681 chloroacetic acid Drugs 0.000 description 1
- 210000003763 chloroplast Anatomy 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- BHYOQNUELFTYRT-DPAQBDIFSA-N cholesterol sulfate Chemical compound C1C=C2C[C@@H](OS(O)(=O)=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 BHYOQNUELFTYRT-DPAQBDIFSA-N 0.000 description 1
- 229940080277 cholesteryl sulfate Drugs 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 108050003126 conotoxin Proteins 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 239000005289 controlled pore glass Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- OILAIQUEIWYQPH-UHFFFAOYSA-N cyclohexane-1,2-dione Chemical compound O=C1CCCCC1=O OILAIQUEIWYQPH-UHFFFAOYSA-N 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 210000004395 cytoplasmic granule Anatomy 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000003412 degenerative effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 150000005690 diesters Chemical class 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- FFYPMLJYZAEMQB-UHFFFAOYSA-N diethyl pyrocarbonate Chemical compound CCOC(=O)OC(=O)OCC FFYPMLJYZAEMQB-UHFFFAOYSA-N 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- OGQYPPBGSLZBEG-UHFFFAOYSA-N dimethyl(dioctadecyl)azanium Chemical compound CCCCCCCCCCCCCCCCCC[N+](C)(C)CCCCCCCCCCCCCCCCCC OGQYPPBGSLZBEG-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical group 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- 150000004662 dithiols Chemical class 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 208000033068 episodic angioedema with eosinophilia Diseases 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 229960001519 exenatide Drugs 0.000 description 1
- HTQBXNHDCUEHJF-URRANESESA-N exendin-4 Chemical compound C([C@@H](C(=O)N[C@@H](C(C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1N=CNC=1)C(C)O)C(C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-URRANESESA-N 0.000 description 1
- 230000028023 exocytosis Effects 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 230000006529 extracellular process Effects 0.000 description 1
- 238000009313 farming Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- UHBYWPGGCSDKFX-VKHMYHEASA-N gamma-carboxy-L-glutamic acid Chemical class OC(=O)[C@@H](N)CC(C(O)=O)C(O)=O UHBYWPGGCSDKFX-VKHMYHEASA-N 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- UKVFVQPAANCXIL-FJVFSOETSA-N glp-1 (1-37) amide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 UKVFVQPAANCXIL-FJVFSOETSA-N 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 229940121355 glucagon like peptide 2 (glp-2) analogues Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002303 glucose derivatives Polymers 0.000 description 1
- 150000002304 glucoses Polymers 0.000 description 1
- 125000002791 glucosyl group Polymers C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 150000002314 glycerols Polymers 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 150000002338 glycosides Chemical class 0.000 description 1
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N guanidine group Chemical group NC(=N)N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 239000012433 hydrogen halide Substances 0.000 description 1
- 229910000039 hydrogen halide Inorganic materials 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- QJHBJHUKURJDLG-UHFFFAOYSA-N hydroxy-L-lysine Natural products NCCCCC(NO)C(O)=O QJHBJHUKURJDLG-UHFFFAOYSA-N 0.000 description 1
- 125000002349 hydroxyamino group Chemical group [H]ON([H])[*] 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Substances C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 1
- 239000012642 immune effector Substances 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 150000002484 inorganic compounds Chemical class 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- RGXCTRIQQODGIZ-UHFFFAOYSA-O isodesmosine Chemical compound OC(=O)C(N)CCCC[N+]1=CC(CCC(N)C(O)=O)=CC(CCC(N)C(O)=O)=C1CCCC(N)C(O)=O RGXCTRIQQODGIZ-UHFFFAOYSA-O 0.000 description 1
- 238000001155 isoelectric focusing Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 231100000225 lethality Toxicity 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 108020001756 ligand binding domains Proteins 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 230000002132 lysosomal effect Effects 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000013178 mathematical model Methods 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 230000004630 mental health Effects 0.000 description 1
- 229960003151 mercaptamine Drugs 0.000 description 1
- GMKMEZVLHJARHF-SYDPRGILSA-N meso-2,6-diaminopimelic acid Chemical compound [O-]C(=O)[C@@H]([NH3+])CCC[C@@H]([NH3+])C([O-])=O GMKMEZVLHJARHF-SYDPRGILSA-N 0.000 description 1
- 229960004452 methionine Drugs 0.000 description 1
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- NEGQCMNHXHSFGU-UHFFFAOYSA-N methyl pyridine-2-carboximidate Chemical compound COC(=N)C1=CC=CC=N1 NEGQCMNHXHSFGU-UHFFFAOYSA-N 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- ZFRKLDBGVWUUEE-UHFFFAOYSA-N n,n-dimethyl-1-phenylmethanamine;hydrate Chemical compound [OH-].C[NH+](C)CC1=CC=CC=C1 ZFRKLDBGVWUUEE-UHFFFAOYSA-N 0.000 description 1
- 229950006780 n-acetylglucosamine Drugs 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- FEMOMIGRRWSMCU-UHFFFAOYSA-N ninhydrin Chemical compound C1=CC=C2C(=O)C(O)(O)C(=O)C2=C1 FEMOMIGRRWSMCU-UHFFFAOYSA-N 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 229940092253 ovalbumin Drugs 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 230000037324 pain perception Effects 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 210000002571 pancreatic alpha cell Anatomy 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 230000007030 peptide scission Effects 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 1
- HMFAQQIORZDPJG-UHFFFAOYSA-N phosphono 2-chloroacetate Chemical compound OP(O)(=O)OC(=O)CCl HMFAQQIORZDPJG-UHFFFAOYSA-N 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical compound OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 1
- 230000037081 physical activity Effects 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 108010087782 poly(glycyl-alanyl) Proteins 0.000 description 1
- 229920001583 poly(oxyethylated polyols) Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 108010094020 polyglycine Proteins 0.000 description 1
- 229920000232 polyglycine polymer Polymers 0.000 description 1
- 210000004896 polypeptide structure Anatomy 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 108010000222 polyserine Proteins 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 108020001213 potassium channel Proteins 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 238000000734 protein sequencing Methods 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 229960003581 pyridoxal Drugs 0.000 description 1
- 235000008164 pyridoxal Nutrition 0.000 description 1
- 239000011674 pyridoxal Substances 0.000 description 1
- 235000007682 pyridoxal 5'-phosphate Nutrition 0.000 description 1
- 239000011589 pyridoxal 5'-phosphate Substances 0.000 description 1
- 229960001327 pyridoxal phosphate Drugs 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000005932 reductive alkylation reaction Methods 0.000 description 1
- 102000037983 regulatory factors Human genes 0.000 description 1
- 108091008025 regulatory factors Proteins 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000002702 ribosome display Methods 0.000 description 1
- XMVJITFPVVRMHC-UHFFFAOYSA-N roxarsone Chemical group OC1=CC=C([As](O)(O)=O)C=C1[N+]([O-])=O XMVJITFPVVRMHC-UHFFFAOYSA-N 0.000 description 1
- 238000013391 scatchard analysis Methods 0.000 description 1
- 210000004739 secretory vesicle Anatomy 0.000 description 1
- 239000006152 selective media Substances 0.000 description 1
- 125000001554 selenocysteine group Chemical group [H][Se]C([H])([H])C(N([H])[H])C(=O)O* 0.000 description 1
- JRPHGDYSKGJTKZ-UHFFFAOYSA-K selenophosphate Chemical compound [O-]P([O-])([O-])=[Se] JRPHGDYSKGJTKZ-UHFFFAOYSA-K 0.000 description 1
- 210000000717 sertoli cell Anatomy 0.000 description 1
- 230000009450 sialylation Effects 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- PTLRDCMBXHILCL-UHFFFAOYSA-M sodium arsenite Chemical compound [Na+].[O-][As]=O PTLRDCMBXHILCL-UHFFFAOYSA-M 0.000 description 1
- IHQKEDIOMGYHEB-UHFFFAOYSA-M sodium dimethylarsinate Chemical compound [Na+].C[As](C)([O-])=O IHQKEDIOMGYHEB-UHFFFAOYSA-M 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 238000004114 suspension culture Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- YSMODUONRAFBET-WHFBIAKZSA-N threo-5-hydroxy-L-lysine Chemical compound NC[C@@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-WHFBIAKZSA-N 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- 125000000430 tryptophan group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
Images















































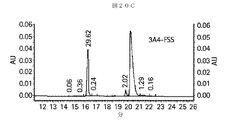




































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/44—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material not provided for elsewhere, e.g. haptens, metals, DNA, RNA, amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39591—Stabilisation, fragmentation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
- A61K47/6817—Toxins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6843—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a material from animals or humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/43504—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from invertebrates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/605—Glucagons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/66—Thymopoietins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/244—Interleukins [IL]
- C07K16/248—IL-6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/55—Fusion polypeptide containing a fusion with a toxin, e.g. diphteria toxin
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Toxicology (AREA)
- Zoology (AREA)
- Epidemiology (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Diabetes (AREA)
- Biomedical Technology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Urology & Nephrology (AREA)
Abstract
Description
本発明は、薬物動態特性を改善するために1つまたは複数の薬理学的に活性な化学部分に結合できる担体抗体に関する。
「担体」部分とは、非ペプチド有機部分(すなわち、「低分子」)またはポリペプチド薬剤など、薬理学的に活性な化学部分が共有結合または融合できる薬理学的に不活性な分子を指す。薬理学的に活性な化学部分のタンパク質分解もしくは他のインビボ活性を低下させる化学修飾によって薬理学的に活性な部分のインビボ減成を防止もしくは軽減するため、または腎臓クリアランスを低下させるため、インビボ半減期もしくは他の治療的な薬物動態特性を高める(薬理学的に活性な部分の非結合形態と比較して、吸収率を高める、毒性もしくは免疫原性を低減する、溶解度を改善する、ならびに/または製造可能性もしくは保存安定性を増加するなどの)ため、効果的な担体が探究されている。
本明細書において別段の定めがない限り、本出願に関連して使用する科学用語および専門用語は、当業者によって一般的に理解されている意味を有する。さらに、文脈において別段の必要がない限り、単数形の用語は複数形を含み、複数形の用語は単数形を含む。したがって、本明細書および添付の特許請求の範囲で使用する単数形「a」、「an」、および「the」は、文脈上はっきりと別段の指摘がされない限り、複数形の指示対象を含む。例えば、「タンパク質(a protein)」という言及は複数のタンパク質を含む;「細胞(a cell)」という言及は複数の細胞集団を含む。
アルゴリズム:Needleman et al., 1970, J. Mol. Biol. 48:443−453;
比較行列:上記Henikoff et al., 1992のBLOSUM62;
ギャップペナルティ:12(ただし、末端ギャップペナルティなし)
ギャップ長ペナルティ:4
類似性の閾値:0
完全長の免疫グロブリン軽鎖および重鎖では、可変および定常領域は、アミノ酸が約10個多い「D」領域も含む重鎖とアミノ酸約12個以上の「J」領域により結合している。例えば、Fundamental Immunology, 2nd ed., Ch. 7 (Paul, W., ed.) 1989, New York: Raven Pressを参照されたい(すべての目的のために参照によりその全体が本明細書に組み込まれる)。各軽鎖/重鎖ペアの可変領域は典型的には抗原結合部位を形成する。
ASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//配列番号86を有する。
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//配列番号87を有する。
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//配列番号88を有する。
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//配列番号89を有する。
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//配列番号90を有する。
GQPKANPTVTLFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASSYLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS//配列番号91である(「CL−1」と命名)。
GQPKAAPSVTLFPPSSEELQANKATLVCLISDFYPGAVTVAWKADSSPVKAGVETTTPSKQSNNKYAASSYLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS//配列番号92を有する。
GQPKAAPSVTLFPPSSEELQANKATLVCLISDFYPGAVTVAWKADSSPVKAGVETTTPSKQSNNKYAASSYLSLTPEQWKSHKSYSCQVTHEGSTVEKTVAPTECS//配列番号93を有する。
GQPKAAPSVTLFPPSSEELQANKATLVCLVSDFYPGAVTVAWKADGSPVKVGVETTKPSKQSNNKYAASSYLSLTPEQWKSHRSYSCRVTHEGSTVEKTVAPAECS//配列番号94を有する。

(a)配列番号77、配列番号107、配列番号111、配列番号113、配列番号115、配列番号117、配列番号119、配列番号123、配列番号129、配列番号144、配列番号145、配列番号181、配列番号182、配列番号183、配列番号184、もしくは配列番号185のアミノ酸配列を含むか、またはN末端かC末端、もしくは両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン重鎖;
(b)配列番号105、配列番号109、配列番号121;配列番号125、もしくは配列番号127のアミノ酸配列を含むか、またはN末端もしくはC末端、または両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン軽鎖;または
(c)(a)の免疫グロブリン重鎖および(b)の免疫グロブリン軽鎖。
(a)配列番号46、配列番号133、配列番号139、配列番号143、配列番号186、もしくは配列番号187、配列番号366、もしくは配列番号367のアミノ酸配列を含むか、またはN末端かC末端、もしくは両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン重鎖;
(b)配列番号28、配列番号131、配列番号135、配列番号137;もしくは配列番号141のアミノ酸配列を含むか、またはN末端もしくはC末端、または両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン軽鎖;または
(c)(a)の免疫グロブリン重鎖および(b)の免疫グロブリン軽鎖。
本明細書に提供する各種重鎖および軽鎖可変領域を表2A〜Bに示す。これらの可変領域はそれぞれ上記の重鎖および軽鎖定常領域に結合して、それぞれ完全な抗体重鎖および軽鎖を形成し得る。さらに、このように産生された重鎖および軽鎖配列はそれぞれ結合して完全な抗体構造を形成し得る。本明細書に提供する重鎖および軽鎖可変領域は、上記に列挙した配列例と異なる配列を有する他の定常ドメインに結合することもできることが理解されるべきである。
(a)重鎖可変領域が配列番号250、配列番号252、配列番号254、配列番号256、配列番号258、もしくは配列番号260の配列と少なくとも95%、96%、97%、98%、99%、もしくは100%同一であるアミノ酸配列を含む;または
(b)軽鎖可変領域が配列番号232、配列番号234、配列番号236、配列番号238、もしくは配列番号240の配列と少なくとも95%、96%、97%、98%、99%、もしくは100%同一であるアミノ酸配列を含む;または
(c)(a)の重鎖可変領域および(b)の軽鎖可変領域。
(a)重鎖可変領域が配列番号262、配列番号264、もしくは配列番号266の配列と少なくとも95%、96%、97%、98%、99%、もしくは100%同一であるアミノ酸配列を含む;または
(b)軽鎖可変領域が配列番号242、配列番号244、配列番号246、もしくは配列番号248の配列と少なくとも95%、96%、97%、98%、99%、もしくは100%同一であるアミノ酸配列を含む;または
(c)(a)の重鎖可変領域および(b)の軽鎖可変領域。
本明細書で開示する抗原結合タンパク質は、1つまたは複数のCDR内にグラフト化、挿入/または結合されるポリペプチドである。抗原結合タンパク質は、1、2、3、4、5または6個のCDRを有することができる。したがって抗原結合タンパク質は、例えば、1つの重鎖CDR1(「CDRH1」)、および/または1つの重鎖CDR2(「CDRH2」)、および/または1つの重鎖CDR3(「CDRH3」)、および/または1つの軽鎖CDR1(「CDRL1」)、および/または1つの軽鎖CDR2(「CDRL2」)、および/または1つの軽鎖CDR3(「CDRL3」)を有することができる。一部の抗原結合タンパク質は、CDRH3とCDRL3の両方を含む。具体的な重鎖および軽鎖CDRを、それぞれ表3A〜B(抗DNP)および表3C〜D(抗KLH)に同定する。
(a)CDRH1は配列番号188、配列番号189、配列番号190、もしくは配列番号191のアミノ酸配列を有する;ならびに/または
(b)CDRH2は配列番号192、配列番号193、配列番号194、もしくは配列番号195のアミノ酸配列を有する;ならびに/または
(c)CDRH3は配列番号196、配列番号197、配列番号198、配列番号199、配列番号200、もしくは配列番号201のアミノ酸配列を有する;ならびに/または
(d)CDRL1は配列番号202、配列番号203、配列番号204、もしくは配列番号205のアミノ酸配列を有する;ならびに/または
(e)CDRL2は配列番号206または配列番号207のアミノ酸配列を有する;ならびに/または
(f)CDRL3は配列番号208、配列番号209、配列番号210、配列番号211、もしくは配列番号212のアミノ酸配列を有する。
(a)CDRH1は配列番号213、配列番号214、もしくは配列番号215のアミノ酸配列を有する;ならびに/または
(b)CDRH2は配列番号216、配列番号217、もしくは配列番号218のアミノ酸配列を有する;ならびに/または
(c)CDRH3は配列番号219、配列番号220、もしくは配列番号221のアミノ酸配列を有する;ならびに/または
(d)CDRL1は配列番号204、配列番号222、配列番号223、もしくは配列番号224のアミノ酸配列を有する;ならびに/または
(e)CDRL2は配列番号206、配列番号225、もしくは配列番号226のアミノ酸配列を有する;ならびに/または
(f)CDRL3は配列番号227、配列番号228、配列番号229、もしくは配列番号230のアミノ酸配列を有する。
HGEGTFTSDQSSYLEGQAAKEFIAWLVKGRG//(配列番号290);
HGEGTFTSDQSSYLEGQAAKEFIAWLQKGRG//(配列番号291);
HGEGTFTSDVSSYQEGQAAKEFIAWLVKGRG//(配列番号292);
HGEGTFTSDVSSYLEGQAAKEFIAQLVKGRG//(配列番号293);
HGEGTFTSDVSSYLEGQAAKEFIAQLQKGRG//(配列番号294);
HGEGTFTSDVSSYLEGQAAKEFIAWLQKGRG//(配列番号295);
HNETTFTSDVSSYLEGQAAKEFIAWLVKGRG//(配列番号296)
HGEGTFTSDVSSYLENQTAKEFIAWLVKGRG//(配列番号297);
HGEGTFTSDVSSYLEGNATKEFIAWLVKGRG//(配列番号298);
HGEGTFTSDVSSYLEGQAAKEFIAWLVNGTG//(配列番号299);
HGEGTFTSDVSSYLEGQAAKEFIAWLVKNRT//(配列番号300);
HGEGTFTSDVSSYLEGQAAKEFIAWLVKGRNGT//(配列番号301);
HGEGTFTSDVSSYLEGQAAKEFIAWLVKGRGGTGNGT//(配列番号302);ならびに
HGEGTFTSDVSSYLEGQAAKEFIAWLVKGRGGSGNGT//(配列番号303)。
ポリクローナル抗体。ポリクローナル抗体は、好ましくは関連抗原およびアジュバントの複数回皮下(sc)または腹腔内(ip)注射した動物において増加する。あるいは、抗原は動物のリンパ節内に直接注射し得る(Kilpatrick et al., Hybridoma, 16:381−389, 1997を参照されたい)。二機能性薬剤または誘導化剤、例えば、マレイミドベンジルスルホコハク酸イミドエステル(システイン残基を介した接合)、N−ヒドロキシコハク酸イミド(リジン残基を介して)、グルタルアルデヒド、コハク酸無水物または当技術分野において知られている他の薬剤を用いて、免疫化対象種において免疫原性であるタンパク質(例えば、キーホールリンペットヘモシアニン、血清アルブミン、ウシチログロブリン、または大豆トリプシンインヒビター)に関連抗原を接合することにより、改善された抗体反応が得られ得る。
(a)ハイブリドーマにより抗原結合タンパク質を発現させる条件下の培養基でハイブリドーマを培養すること;ならびに
(b)培養基から抗原結合タンパク質を回収することであり、これは既知の抗体精製技術(これに限定されないが、本明細書の実施例1に開示のモノクローナル抗体精製技術など)により成し遂げることができる。
ATGGACATGAGGGTGCCCGCTCAGCTCCTGGGGCTCCTGCTGCTGTGGCTGAGAGGTGCGCGCTGT//配列番号102、これはVK−1シグナルペプチド配列MDMRVPAQLLGLLLLWLRGARC//配列番号103をコードする)、複製起点、1つもしくは複数の選択的マーカー遺伝子(例えば、抗生剤もしくは他の薬剤耐性、相補性栄養要求欠陥を付与し得るか、または培地中で入手不能な重要栄養剤を補給し得る)、エンハンサーエレメント、プロモーター、および転写終止配列であり、これらはすべて当技術分野において周知である。
組換えヒト抗体遺伝子のレパートリー作製のための技術開発および線維状バクテリオファージ表面上のコードされた抗体断片のディスプレイにより、ヒト由来抗体を生成する別の手順が提供されている。ファージディスプレイについては、例えば、Dower et al.、WO91/17271号、McCafferty et al.、WO92/01047号、およびCaton and Koprowski, Proc. Natl. Acad. Sci. USA, 87:6450−6454 (1990)(これらはそれぞれ参照によりそれらの全体が本明細書に組み込まれる)に記載されている。ファージ技術により産生された抗体は、通常、抗原結合断片(例えばFvまたはFab断片)として細菌内に産生されるため、エフェクター機能を欠く。エフェクター機能は、2つの戦略の1つにより導入できる:断片は、哺乳類細胞内における発現のために完全な抗体内か、またはエフェクター機能を惹起できる第二結合部位を有する二重特異性抗体断片内のいずれかに加工できる。
上記のとおり、抗体断片はインタクトな完全長抗体の一部、好ましくはインタクト抗体の抗原結合領域もしくは可変領域を含み、抗体断片から形成される線状抗体および多重特異性抗体を含む。限定されない抗体断片の例としては、抗体が所望の生体活性を保持する限り、Fab、Fab’、F(ab’)2、Fv、Fd、ドメイン抗体(dAb)、相補性決定領域(CDR)断片、単鎖抗体(scFv)、単鎖抗体断片、マキシボディ、ダイアボディ、トリアボディ、テトラボディ、ミニボディ、線状抗体、キレート化組換え抗体、トリボディまたはバイボディ、細胞内抗体、ナノボディ、小モジュール免疫薬剤(SMIP)、抗原結合ドメイン免疫グロブリン融合タンパク質、ラクダ化抗体、VHHを含む抗体、またはそれらの変異タンパク質もしくは誘導体、ならびにポリペプチドへ結合する特異的抗原を付与する上で十分な免疫グロブリンを少なくとも一部含むポリペプチド(CDR配列など)が挙げられる。かかる抗原断片は、完全抗体の修飾により産生してもよいし、組換えDNA技術またはペプチド合成を用いてde novo合成してもよい。
いくつかの実施形態では、多価またはさらには多重特異性(例えば二重特異性、三重特異性など)モノクローナル抗体を産生することが望ましい場合がある。かかる抗体は、標的抗原の少なくとも2つの異なるエピトープに対して特異的に結合し得るか、あるいは2つの異なる分子、例えば標的抗原と細胞表面タンパク質または受容体に結合し得る。例えば、二重特異性抗体は、細胞防御機序を標的発現細胞に集中させるために、標的に結合する腕と、(T細胞受容体分子(例えば、CD2またはCD3)などの白血球上の引き金分子、またはIgGのためのFc受容体(FcγR)、例えばFcγRI(CD64)、FcγRII(CD32)およびFcγRIII(CD16)に結合する別の腕を含み得る。別の例では、二重特異性抗体を、標的抗原を発現する細胞に細胞障害性薬を局在化させるために用いてよい。これらの抗体は、標的結合腕と細胞障害性薬(例えば、サポリン、抗インターフェロン60、ビンカアルカロイド、リシンA鎖、メトトレキサートまたは放射性同位体ハプテン)に結合する腕を有する。多重特異性抗体は、完全長抗体または抗体断片として調製できる。
発明の抗原結合タンパク質は、ペプチボディも含む。「ペプチボディ」という用語は、少なくとも1つのペプチドに結合した抗体Fcドメインを含む分子を指す。ペプチボディの産生については、PCT公開WO00/24782号(2000年5月4日公開)に一般的に記載されている。これらのペプチドはいずれも、リンカーを伴いまたは伴わず、直列で(すなわち、連続的に)結合し得る。システイニル残基を含むペプチドは、別のCys含有ペプチドと架橋し得、この片方または両方がビヒクルに結合し得る。同様に、複数のCys残基を有するペプチドはいずれも、ペプチド間でジスルフィド結合を形成し得る。これらのペプチドはいずれも誘導体化し得、例えばカルボキシル末端はアミノ基とキャッピングし得、システインはキャッピングし得、またはアミノ酸残基はアミノ酸残基以外の部分により置換し得る(例えば、Bhatnagar et al., J. Med. Chem. 39: 3814−9 (1996), and Cuthbertson et al., J. Med. Chem. 40: 2876−82 (1997)を参照されたく、これらは参照によりそれらの全体が本明細書に組み込まれる)。ペプチド配列は至適化し得、抗体の親和性成熟に類似しているか、あるいはアラニンスキャニングまたは無作為もしくは部位指向変異により改変され、続くスクリーニングにより最高の結合剤が同定される。Lowman, Ann. Rev. Biophys. Biomol. Struct. 26: 401−24 (1997)。抗原結合タンパク質の所望の活性を維持しつつ、各種分子を抗原結合タンパク質構造、例えば、抗原結合タンパク質のペプチド部分自体内またはペプチドとビヒクル部分の間に挿入できる。例えば、Fcドメインもしくはその断片、ポリエチレングリコールなどの分子、またはデキストラン、脂肪酸、脂質、コレステロール基、小さな炭水化物、ペプチド、本明細書に記載の検出可能な部分(蛍光剤、放射同位体などの放射標識を含む)、オリゴ糖、オリゴヌクレオチド、ポリヌクレオチド、干渉(または他の)RNA、酵素、ホルモンなどの他の関連分子を容易に挿入できる。この方法での挿入に適した他の分子は当業者に理解されており、本発明の範囲内に包含される。これは、例えば、任意選択的に適切なリンカーにより結合した2つの連続したアミノ酸間へ所望の分子を挿入することを含む。
(Gly)3Lys(Gly)4(配列番号159);
(Gly)3AsnGlySer(Gly)2(配列番号156);
(Gly)3Cys(Gly)4(配列番号157);ならびに
GlyProAsnGlyGly(配列番号158)。
GGEGGG(配列番号160);
GGEEEGGG(配列番号161);
GEEEG(配列番号162);
GEEE(配列番号163);
GGDGGG(配列番号164);
GGDDDGG(配列番号165);
GDDDG(配列番号166);
GDDD(配列番号167);
GGGGSDDSDEGSDGEDGGGGS(配列番号168);
WEWEW(配列番号169);
FEFEF(配列番号170);
EEEWWW(配列番号171);
EEEFFF(配列番号172);
WWEEEWW(配列番号173);または
FFEEEFF(配列番号174)。
1)疎水性:ノルロイシン(NorまたはNle)、Met、Ala、Val、Leu、Ile;
2)中性で親水性:Cys、Ser、Thr、Asn、Gln;
3)酸性:Asp、Glu;
4)塩基性:His、Lys、Arg;
5)鎖の方向に影響を与える残基:Gly、Pro;ならびに
6)芳香族:Trp、Tyr、Phe。
例えば、抗原結合タンパク質へ結合した1つもしくは複数の炭水化物部分の添加もしくは欠失、ならびに/または抗原結合タンパク質における1つもしくは複数のグリコシル化部位の添加もしくは欠失により、親ポリペプチドと比較して修飾グリコシル化パターンを有する抗原結合タンパク質変異体も産生できる。
システイン残基(1つまたは複数)は、抗体またはFc含有ポリペプチドのFc領域で除去または誘発され得、その結果、この領域で鎖間ジスルフィド結合形態を除去または増加する。そのように産生されたホモ二量体抗原結合タンパク質は内部移行能力を改善しならびに/または相補性媒介性細胞殺傷および抗体依存性細胞傷害(ADCC)を増大し得る。Caron et al., J. Exp Med. 176: 1191−1195 (1992) and Shopes, B. J. Immunol. 148: 2918−2922 (1992)を参照されたい。ホモ二量体抗原結合タンパク質または抗体は、Wolff et al., Cancer Research 53: 2560−2565 (1993)に記載のヘテロ二機能性交差リンカーを用いることによっても調製し得る。あるいは、二重Fc領域を有する抗原結合タンパク質を加工でき、その結果、高まった相補性溶解およびADCC能力を有し得る。Stevenson et al., Anti−CancerDrug Design 3: 219−230 (1989)を参照されたい。
抗DNPまたは抗KLH抗原結合タンパク質の他の特定の共有結合修飾も本発明の範囲内に含まれる。それらは、妥当な場合、抗原結合タンパク質または抗体の化学合成によりまたは酵素もしくは化学切断により作製し得る。他のタイプの共有結合修飾は、選択された側鎖またはN末端残基もしくはC末端残基と反応できる有機誘導化剤と標的アミノ酸残基を反応させることにより導入できる。
本発明の別の態様は、本発明の抗原結合タンパク質をコードする単離核酸、例えばこれらに限定されないが、本発明の抗体または抗体断片をコードする単離核酸などである。かかる核酸は、当技術分野において知られているおよび/または本明細書に開示する組換え技術により作製される。
(a)CDRH1は配列番号188、配列番号189、配列番号190、もしくは配列番号191のアミノ酸配列を含む;
(b)CDRH2は配列番号192、配列番号193、配列番号194、もしくは配列番号195のアミノ酸配列を含む;ならびに
(c)CDRH3は配列番号196、配列番号197、配列番号198、配列番号199、配列番号200、もしくは配列番号201のアミノ酸配列を含む。
(a)CDRL1は配列番号202、配列番号203、配列番号204、もしくは配列番号205のアミノ酸配列を含む;
(b)CDRL2は配列番号206もしくは配列番号207のアミノ酸配列を含む;ならびに
(c)CDRL3は配列番号208、配列番号209、配列番号210、配列番号211、もしくは配列番号212のアミノ酸配列を含む。
(a)CDRH1は配列番号213、配列番号214、もしくは配列番号215のアミノ酸配列を含む;
(b)CDRH2は配列番号216、配列番号217、もしくは配列番号218のアミノ酸配列を含む;ならびに
(c)CDRH3は配列番号219、配列番号220、もしくは配列番号221のアミノ酸配列を含む。
(a)CDRL1は配列番号204、配列番号222、配列番号223、もしくは配列番号224のアミノ酸配列を含む;
(b)CDRL2は配列番号206、配列番号225、もしくは配列番号226のアミノ酸配列を含む;ならびに
(c)CDRL3は配列番号227、配列番号228、配列番号229、もしくは配列番号230のアミノ酸配列を含む。
(a)発現ベクターによりコードされた抗原結合タンパク質を発現させる条件下の培養基で宿主細胞を培養すること;ならびに
(b)培養基から抗原結合タンパク質を回収すること。抗原結合タンパク質の回収は、抗体の既知の精製方法(これらに限定されないが、本明細書の実施例1および他の箇所に開示の抗体精製技術など)により成し遂げられる。
適切な細胞への治療抗原結合タンパク質の送達は、当技術分野において知られている任意の適切なアプローチを用いてエクスビボ、原位置、またはインビボ遺伝子療法を介してもたらすことができる。例えば、インビボ療法において、所望の抗原結合タンパク質もしくは抗体をコードする核酸を、単独またはベクター、リポソーム、もしくは沈殿物と連結するかのいずれかで対象に直接注射し得、いくつかの実施形態では、抗原結合タンパク質化合物の発現が望ましい部位で注射し得る。エクスビボ処置において、対象の細胞を除去し、これらの細胞内に核酸を導入し、直接または、例えば、患者に移植する多孔膜に被包するかのいずれかで修飾細胞を対象に戻す。例えば米国特許第4,892,538号および同第5,283,187号を参照されたい。
本発明の方法の実践に用いられる抗DNPまたは抗KLH抗原結合タンパク質または抗体は所望の送達方法に適した担体を含む医薬組成物および薬剤に製剤化し得る。適切な担体は、抗DNPまたは抗KLH抗原結合タンパク質または抗体と組み合わせた場合、それぞれDNPまたはKLHの高親和性結合を保持し、対象の免疫系と反応しない任意の物質を含む。例としては、滅菌リン酸緩衝生理食塩水、静菌水などの任意のいくつかの標準的な医薬担体が挙げられるが、これらに限定されない。例えば、水、緩衝水、0.4%生理食塩水、0.3%グリシンなどの各種水性担体を用いてよく、軽度化学修飾などに供するアルブミン、リポタンパク質、グロブリンなどの安定性向上のための他のタンパク質を含み得る。
免疫化。XenoMouse(商標)マウスをDNP−KLHにより4週間免疫化して抗DNP抗体を生成し、DNP−リジンに結合する抗体をスクリーニングした。より詳細には、国際特許第WO98/24893号、およびWO00/76310号(これらの開示はすべて、参照により本明細書に組み込まれる)に開示の方法に従い、XenoMouse(登録商標)のXMG2系統の初回免疫化のため、既に記載されている(Mendez et al., Nat. Genet. 15:146−156 (1997);国際特許第WO98/24893号、およびWO00/76310号公開、これらの開示は参照により本明細書に組み込まれる)ようにマウスのXenoMouse(商標)XMG2系統を一般に生成し、TiterMax Goldアジュバント(Sigma−Aldrich, Oakville, Ontario)中で乳化した免疫原(マウス1匹当たり10〜30μgの範囲)を用いて、2,4−ジニトロフェニル−キーホールリンペットヘモシアニン(DNP−KLH conjugate; BioSearch Technologies, Novato, CA)で免疫化した。初回免疫化後、続く免疫原(マウス1匹当たり5〜20μg)ブーストをマウスにおいて適切な抗DNP価を導入するために必要なスケジュールおよび期間で投与した。固定化したDNP−BSA(BioSearch Technologies, Novato, CA)を用いた酵素免疫アッセイにより力価を決定し、最終DNP:BSAモル比が30:1であるようにこの結合体を調製した。
5’RACEとして知られているポリメラーゼ連鎖反応(PCR)増幅技術(cDNA末端の急速な増幅)により、XenoMouse配列由来のヒト抗KLH抗体を得た。TRIzol試薬(Invitrogen)を用いて、KLH結合モノクローナル抗体を発現する3つのハイブリドーマ;16.3.1、108.1.2および120.6から総RNAを単離してから、RNeasy Miniキット(Qiagen)を用いてさらに精製した。混合した無作為およびオリゴ−dTプライマー第一鎖、RACE ready cDNAをGeneRacerキット(Invitrogen)を用いて調製した。順行プライマー、GeneRacer(登録商標)ネステッドプライマーを用いたAdvantage HF2 DNAポリメラーゼ(Clontech)でcDNAのPCR増幅を実施した:
5’−GGA CAC TGA CAT GGA CTG AAG GAG TA−3’//(配列番号271);ならびに逆行プライマー:
軽鎖において5’−CTC CTG GGA GTT ACC CGA TTG−3’//(配列番号272)、および重鎖において5’−GAT GGG CCC TTG GTG GAG GCT GAG GAG ACG GTG ACC GTG G−3’//(配列番号273)。PCR反応サイクルは94℃で30秒のcDNA変性からなり、94℃で20秒間;55℃で30秒間;ならびに72℃で90秒間からなる各サイクルの増幅の3サイクル+94℃で20秒間;65℃で30秒間;ならびに72℃で90秒間からなる追加の27サイクルが続く。次いで反応物を72℃で7分間インキュベートして、最終PCRサイクルが続いて完全な延長部を保証する。RACE PCR生成物をpCR4−TOPO(Invitrogen)内にクローン化して、ABI DNA配列決定機器(Perkin Elmer)を用いてそれらの配列を決定した。ベクターNTI 8.0ソフトウェア(Invitrogen)を用いてコンセンサス配列を決定し、完全長抗体鎖PCR増幅プライマーを設計するために用いられる。
5’−AAG CTC GAG GTC GAC TAG ACC ACC ATG GAC ATG AGG−3’//(配列番号274)であり、およびカルボキシル末端をコードした3’プライマーおよび終止コドン、ならびにNotI制限部位は:
5’−AAC CGT TTA AAC GCG GCC GCT CAA CAC TCT CCC CTG TTG AA−3’//(配列番号275)であった。
5’−AAG CTC GAG GTC GAC TAG ACC ACC ATG GAA TTG GGA CTG AG−3’//(配列番号276)であり、カルボキシル末端をコードした3’プライマーおよび終止コドン、ならびにNotI制限部位は:
5’−AAC CGT TTA AAC GCG GCC GCT CAT TTA CCC GGA GAC AGG GA−3’//(配列番号277)であった。
5’ TTT TTT TTG CGC GCT GTG ACA TCC AGA TGA CCC AGT C 3’//(配列番号278)、
および3’プライマー 5’ AAA AAA CGT ACG TTT GAT ATC CAC TTT GGT CC 3’//(配列番号279)。
5’ CTG TGT ATT ACT GTG CGA GGT ATA ACT TCA ACT ACG GTA TGG ACG TCT GG 3’//(配列番号280)および(−)鎖プライマー:
5’ CCA GAC GTC CAT ACC GTA GTT GAA GTT ATA CCT CGC ACA GTA ATA CAC AG 3’//(配列番号281)を用いたPCRによりフェニルアラニンに変異し、
重鎖5末端プライマー
5’ AAG CTC GAG GTC GAC TAG ACC ACC ATG GAC ATG AGG GTG CCC GCT CAG CTC CTG GGG CT 3’//(配列番号284)および重鎖3’プライマー:
5’ AAC CGT TTA AAC GCG GCC GCT CAT TTA CCC GGA GAC AGG GA 3’//(配列番号285)と連結している(+)鎖プライマー:
5’ CTG TGT ATT ACT GTG CGA GGT ATA ACT ACA ACT ACG GTA TGG ACG TCT GG 3’//(配列番号282)および(−)鎖プライマー:
5’ CCA GAC GTC CAT ACC GTA GTT GTA GTT ATA CCT CGC ACA GTA ATA CAC AG 3’//(配列番号283)を用いたPCRによりチロシンに変異した。また、3A4 IgG2重鎖のヒンジ領域のジスルフィドスクランブリングを減らすため、例として、ヒンジシステイン219および220(EU番号付け)を
重鎖5末端プライマー:
5’ AAG CTC GAG GTC GAC TAG ACC ACC ATG GAC ATG AGG GTG CCC GCT CAG CTC CTG GGG CT 3’//(配列番号288)および重鎖3’プライマー:
5’ AAC CGT TTA AAC GCG GCC GCT CAT TTA CCC GGA GAC AGG GA 3’//(配列番号289)と連結している(+)鎖プライマー:
5’ GGA CAA GAC AGT TGA GCG CAA ATC TTC TGT CGA GTG CCC ACC GTG CCC AG 3’//(配列番号286)および(−)鎖プライマー:
5’ CTG GGC ACG GTG GGC ACT CGA CAG AAG ATT TGC GCT CAA CTG TCT TGT CC 3’//(配列番号287)を用いたPCRにより変異させた。
3B1、3H4、3C2、3A1および3A4抗体の生成物品質を、還元負荷緩衝液を用いて1.0mmトリス−グリシン4〜20%SDS−PAGE(Novex)上で分析した(図11)。aDNP3H4抗体が安定した細胞系から不均一な生成物を産生したというこれらのデータから、aDNP3H4抗体は担体抗体として良好な候補ではなかったことが示された(均一生成物が望ましいため)。aDNP3A1、3A4、3C2、および3B1ならびにaKLH120.6抗体の生成物品質を、非還元負荷緩衝液を用いて1.0mmトリス−グリシン4〜20%SDS−PAGE(Novex)上で分析した(図12A〜B)。aDNP3C2抗体は例外的な高分子質量物質を伴う不均一生成物を産生し、これは担体抗体として理想的な候補ではないことが示された(高分子質量物質が含まれる生成物は望ましくないため)。加えて、aDNP3B1抗体は、これらの条件下で二重線を示した。次いで、トリス−グリシンSDS−PAGEならびにビス−トリスNuPAGE系の両方を用いて、非還元条件下でaDNP3B1抗体とaDNP3A1抗体を比較した(図13A〜B)。aDNP3B1抗体では、トリス−グリシンSDS−PAGE上のaDNP3A1では観察されなかった二重線がはっきりと生じることが見出された;しかしながら、ビス−トリスNuPAGEにより分析した場合、aDNP3B1抗体はaDNP3A1抗体よりも均一であると考えられ、二重線は分析法のアーチファクトであり得ることが示された。aDNP3B1抗体を室温、85℃、または100℃で非還元試料緩衝液で処置後にトリス−グリシンSDS−PAGEにより分析した場合、二重線は除去されなかった(図14A)。しかしながら、aDNP3B1抗体を通常の0.1%ではなく0.4%SDSゲルの流れる緩衝液を用いてトリス−グリシンSDS−PAGEにより評価した際、二重線は激減し(図14B)、二重線は分析系のアーチファクトであったというさらなる証拠が得られた。
aDNP3A4−F、aDNP3A4−FSSおよびaDNP3B1抗体の薬物動態プロファイルについて、成体Sprague−Dawleyラット(8〜12週齢)に5mg/kgを皮下注射し、投与から0、0.25、1、4、24、48、72、96、168、336、504、672、840および1008時間後に側面尾静脈から血液約250μLをMicrotainer(商標)血清分離管に採取して決定した(図22)。各試料を採取後に室温で維持し、30〜40分凝固させた後に、目盛り付きEppendorf 5417R遠心分離系(Brinkmann Instruments, Inc., Westbury, NY)を用いて試料を2〜8℃、11,500rpmで約10分間遠心分離した。次いで採取した血清を分析のために(各ラットにおいて)前標識した低温貯蔵管内に移して、−60℃〜−80℃で保存した。PK試験試料から血清試料濃度を測定するため、以下の方法を用いた:2分の1部分黒プレート(Corning3694)を2μg/mLの抗hu FC、Ab1.35.1の1×PBS溶液でコーティングしてから4℃で一晩インキュベートした。プレートを洗浄し、I−Block(登録商標)(Applied Biosystems)で4℃で一晩ブロックした。試料を希釈する必要があった場合は、ラットSD血清で希釈した。標準および試料をI−Block(登録商標)+5%BSAで1:20希釈し380μLの希釈緩衝液とした。プレートを洗浄し、前処置した標準の50μL試料および各試料をAb1.35.1コーティングプレートに移して室温で1.5時間インキュベートした。プレートを洗浄してから、抗hu FC Ab21.1−HRP結合体のI−Block(登録商標)+5%BSA 100ng/mLを50μL添加し、1.5時間インキュベートした。プレートを洗浄してから、ピコ基質50μLを添加後、プレートを照度計で直ちに分析した。すべての抗体の薬物動態プロファイルが良好であったが、aDNP3B1が最高の全体プロファイルを示した。
概して、ヒト使用のためのモノクローナル抗体生成物の製造および検査における考慮点(Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use)(U.S. Department of Health and Human Services, Food and Drug Administraton, Center for Biologics Evaluation and Research (1997))におけるガイダンスに従い、発明の抗体の各種ヒト組織との交差反応を決定する非GLP予備試験を実施した。抗体による薬剤発現が意図される場合、GLP条件下のより広範な検査が必要である。抗体DNP 3A4−FおよびaKLH120.6の組織交差反応を、試験物質のAlexa Fluor 488標識形態を用いて選択されたヒト組織の凍結切片で検討した(Charles River Laboratories, Preclinical Services, Reno, NV)。(別段の指定のない限り)互いに依存しない2人の正常ヒト組織をSpecial Pathology Services Human Tissue Bank collected by the National Disease Research Interchange (NDRI, Philadelphia, PA)、Cureline, Inc. (Burlingame, CA)、Cybrdi (Rockville, MD)、またはRocky Mountain Lions Eye Bank (Aurora, CO)から得た。試験組織は、ヒト小脳、肺、大脳皮質、卵巣(成熟女性由来)、眼部、胎座、消化管(小腸)、皮膚(1人)、心臓、脾臓、腎臓(1人)、甲状腺、肝臓、精巣を含む。新しい凍結ヒト組織切片および対照ビーズブロック(DNP[31]−ウシ血清アルブミン[BSA]ビーズ[陽性]、およびヒト血清アルブミン[HSA]ビーズ[陰性])を冷却器上で切断し、キャピラリーギャップスライド上に乗せて解凍した。組織および対照ビーズのスライドを冷却アセトン中で−10℃〜−25℃で約10分間固定した。固定スライドを少なくとも1時間(〜一晩)乾燥させた。凍結保存の場合、実験の前日に固定スライドを凍結器から除去して使用前に一晩解凍させた。以降の工程はすべて別段の定めがない限り室温で実施した。スライドを1×Morphosave(登録商標)で約15分間インキュベートして組織形態を保持してから、1×リン酸緩衝生理食塩水(PBS)で2回、それぞれ約5分間洗浄した。内因性ペルオキシダーゼをブロックするため、スライドをグルコースオキシダーゼ溶液中で約37℃で約1時間インキュベートした。スライドを1×PBSで2回、それぞれ約5分間洗浄した。内因性ビオチンをアビジンおよびビオチン溶液中の逐次インキュベーション(それぞれ約15分間)によりブロックした。ビオチン内でインキュベーション後、組織切片をブロック抗体溶液で約25分間ブロックした。Alexa Fluor 488−Ab3A4 W101F(抗DNP)、およびAlexa Fluor 488抗KLH(抗KLH Ab)を至適濃度(2.0μg/mL)または至適濃度の5倍(10.0μg/mL)で切片に約25分間適用した。スライドを洗浄緩衝液で3回洗浄してから、二次抗体(ウサギ抗Alexa Fluor 488)で約25分間インキュベートした。二次抗体でインキュベーション後、スライドを洗浄緩衝液で4回洗浄してから、三次抗体(セイヨウワサビペルオキシダーゼ結合ヤギ抗ウサギIgG抗体)で約25分間インキュベートし、ジアミノ酪酸(DAB)色素原基質で結合を視覚化した。すべての実験においてDNP(31)−BSAビーズを陽性対照として用いた。HSAビーズを陰性対照として用いた。CD31に対する抗体(抗CD31)、すなわち、血小板内皮細胞接着分子(PECAM−1)染色を介して免疫組織化学に妥当な組織を適格とした。任意の試験抗体の2.0または10.0μg/mL濃度のいずれかで評価したいずれのヒト組織においても特定の染色はなかった。
本発明の例示的な実施形態を含み、一種別の1価、2価および3価構造を比較のために発現させて精製した。それらには、aKLH IgG2/Fc−ShK変異体(図1E:「ヘミボディ」配置の図解を参照されたい)、および抗KLH IgG2−ShK変異体(図1F〜Lを参照されたい)が含まれる。例えば、2価Fc−L10−ShK[1−35]、1価抗キーホールリンペットヘモシアニン(KLH)免疫グロブリン重鎖−[Lys16]ShK融合抗体(「aKLH HC−[Lys16]ShK Ab」と命名;図1Fを参照されたい)、および1価抗KLH免疫グロブリン軽鎖−[Lys16]ShK抗体(「aKLH LC−[Lys16]ShK Ab」と命名;図1Jを参照されたい)。IgG2 Fc/Fc−ShK変異体(図1Aを参照されたい)、2価Fc−L10−ShK[2−35]、1価Fc/Fc−L10−ShK[2−35]を比較のため、Sullivan et al.、WO2008/088422A2号、特に実施例1、2、および56(参照によりその全体が本明細書に組み込まれる)に記載の、または本明細書に修正されるように組換え方法により作製した。
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号1)は、標準的なPCR技術を用いて、Kv1.3阻害剤ペプチドShK[2−35]の単量体にインフレーム融合させたかまたは変異ShK[2−35、Q16K]を構築した。PCR反応においてpcDNA3.1(+)CMVi内の元のFc−2×L−ShK[2−35]を鋳型として用いて、ShK[2−35]もしくはShK[2−35、Q16K]および分子の10個のアミノ酸リンカー部分を生成した(Sullivan et al.、WO2008/088422A2号の実施例2、図15A〜Bを参照されたい)。PCR反応においてpcDNA3.1(+)CMVi内の元のFc−2×L−ShK[1−35]を鋳型として用いてShK[1−35]を生成した(Sullivan et al.、WO2008/088422A2号の実施例1、図14A〜B)。これらのShK構築物は、
以下のオリゴ:
5’−CAT GAA TTC CCC ACC ATG GAA TGG AGC TGG−3’(配列番号3);および
5’−CA CGG TGG GCA CTC GAC TTT GCG CTC GGA GTG GAC ACC−3’(配列番号4)
を有するpSelexis−Vh21−hIgG2−Fc鋳型から産生した修飾VH21シグナルペプチドのアミノ酸配列MEWSWVFLFFLSVTTGVHSERKVECPPCP//配列番号2を有する。
GGGGSGGGGSSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC//配列番号6)を有する野生型ShK[2−35]を:
GGAGGAGGAGGATCCGGAGGAGGAGGAAGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCCAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGC//(配列番号5)のDNA配列によりコードした。このコード配列(配列番号5)を含む断片を、以下のオリゴ(配列番号7および配列番号8)を用いて、ならびにpcDNA3.1(+)CMVi中の元のFc−L10−ShK[2−35]を鋳型として用いて生成した(参照により組み込まれるSullivan et al.、WO2008/088422A2号の実施例2、図15A〜B):
5’−GTC CAC TCC GAG CGC AAA GTC GAG TGC CCA CCG TGC C−3’(配列番号7);および
5’−TCC TCC TCC TTT ACC CGG AGA CAG GGA GAG−3’//(配列番号8)。
5’−GCT GCA CCG CCT TCA AGT GCA AGC ACA GC 3’(配列番号9);および
5’−GCT GTG CTT GCA CTT GAA GGC GGT GCA GC−3’(配列番号10);であり、pcDNA3.1(+)CMVi中の元のFc−L10−ShK[2−35]を鋳型として用いて、DNAコード配列
GGAGGAGGAGGATCCGGAGGAGGAGGAAGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCAAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGC//(配列番号11)を生成し、これはN末端リンカー伸張部分:GGGGSGGGGSSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//配列番号12)を有するShk(2−35、K16)アミノ酸配列をコードする(Sullivan et al.、WO2008/088422A2号の実施例2、図15A〜B)。
5’−GTC CAC TCC GAG CGC AAA GTC GAG TGC CCA CCG TGC C−3’(配列番号7);および
5’−TCC TCC TCC TTT ACC CGG AGA CAG GGA GAG−3’(配列番号8)
を用いてShK[1−35]WT断片を生成した(Sullivan et al.、WO2008/088422A2号の実施例1、図14A〜B)。
5’−CAT GCG GCC GCT CAT TAG CAG GTG−3’(配列番号14)のオリゴを用いて、ならびにpSelexis Vh21−hIgG2−Fc鋳型を用いてIgG2Fc領域を生成し、アミノ酸配列
APPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK配列番号16)をコードするコード配列:
GCACCACCTGTGGCAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGACACCCTCATGATCTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCCACGGGAGGAGCAGTTCAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTTGTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGGCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGGCAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAAGACCACACCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACGTCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCACTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTAAA//配列番号15を含むDNA断片を得た。
5’−CAT GAA TTC CCC ACC ATG GAA TGG AGC TGG−3’(配列番号3);および
5’−CAT GCG GCC GCT CAT TAG CAG GTG−3’(配列番号14)
と一緒に縫い合わせた。
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGGGSGGGGSSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC//(配列番号17)をコードした。
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGGGSGGGGSSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//配列番号18をコードした;
また、pTT14−VH21SP−IgG2−Fc ShK1−35構築物には、以下の配列:
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGGGSGGGGSRSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC//(配列番号19)
を有するIgG2 Fc−L10−ShK(1−35)融合ポリペプチドのためのコード配列が含まれた。
5’−CAT AAG CTT CCC ACC ATG GAA TGG AGC TGG−3’(配列番号20);および
5’−CA CGG TGG GCA CTC GAC TTT GCG CTC GGA GTG GAC ACC−3’(配列番号4)を用いて、ならびに上記pSelexis鋳型を用いてVH21シグナルペプチドを生成した。
5’−GTC CAC TCC GAG CGC AAA GTC GAG TGC CCA CCG TGC C−3’(配列番号7);および
5’−CAT GGA TCC TCA TTT ACC CGG AGA CAG GGA G−3’(配列番号21)
を用いてFc領域を生成した。
GGATCCGGAGGAGGAGGAAGCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCAAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGCTAATGAGCGGCCGCTCGAGGCCGGCAAGGCCGGATCC//(配列番号22)
を含む断片をBamHI/BamHIを用いて切断した。このコード配列(配列番号23)はN末端リンカー配列:
GSGGGGSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号23)を有するShK(1−35、Q16K)をコードする。
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGG//(配列番号25)をコードするコード配列
ATGGAATGGAGCTGGGTCTTTCTCTTCTTCCTGTCAGTAACGACTGGTGTCCACTCCGAGCGCAAAGTCGAGTGCCCACCGTGCCCAGCACCACCTGTGGCAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGACACCCTCATGATCTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCCACGGGAGGAGCAGTTCAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTTGTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGGCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGGCAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAAGACCACACCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACGTCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCACTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTAAAGGAGGAGGA//(配列番号24)を得た。
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGGGSGGGGSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号26)をコードした。
(a)N末端VK−1SPシグナルペプチド配列(配列番号103)を組み入れるaKLH120.6κLC(以下の配列番号28):
MDMRVPAQLLGLLLLWLRGARCDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC//(配列番号28);
(b)N末端VK−1SPシグナルペプチド配列(配列番号103)を組み入れるaKLH120.6IgG2 HC(以下の配列番号29):
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号29);
および、
(c)IgG2 Fc−L10−ShK(1−35):
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGGGSGGGGSRSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC//(配列番号30)。
aKLH120.6IgG2−ShK融合抗体の成分(図1Fに図解)は以下の単量体を含有した。
(a)aKLH120.6κLC(上記の配列番号28);
(b)aKLH120.6IgG2 HC(上記の配列番号29);および
(c)以下のHC配列を有するaKLH120.6IgG2−ShK融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSRSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC//(配列番号31)。
(a)aKLH120.6κLC(上記の配列番号28);
(b)aKLH120.6IgG2 HC(上記の配列番号29);および
(c)以下の配列を有するaKLH120.6IgG2−ShK[1−35、Q16K]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号32)。
(a)aKLH120.6κLC(配列番号28);
(b)aKLH120.6IgG2 HC(配列番号29);および
(c)以下のアミノ酸配列を有するaKLH120.6IgG2 HC−ShK[1−35、R1A、I4A、Q16K]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSASCADTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号304)。
所望の1価aKLH120.6IgG2 HC−ShKアナログ生成物は1本の重鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖:重鎖−ShKの比率は1:2:1であった。予期した発現生成物はaKLH120.6IgG2抗体、1価aKLH120.6IgG2 HC−ShKペプチドアナログ、および2価aKLH120.6IgG2 HC−ShKペプチドアナログである。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 HC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6κLC(配列番号28);
(b)aKLH120.6IgG2 HC(配列番号29);および
(c)以下の配列を有するaKLH120.6IgG2−ShK[1−35、R1A、Q16K、K30E]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSASCIDTIPKSRCTAFKCKHSMKYRLSFCRETCGTC//(配列番号305)。
所望の1価aKLH120.6IgG2 HC−ShKアナログ生成物は1本の重鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖:重鎖−ShKの比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2抗体、1価aKLH120.6IgG2 HC−ShKペプチドアナログ、および2価aKLH120.6IgG2 HC−ShKペプチドアナログである。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 HC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6κLC(配列番号28);
(b)aKLH120.6IgG2 HC(配列番号29);および
(c)以下のアミノ酸配列を有するaKLH120.6HC IgG2−ShK[1−35、R1H、I4A、Q16K]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSHSCADTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号306)。
所望の1価aKLH120.6IgG2 HC−ShKアナログ生成物は1本の重鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖:重鎖−ShKの比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2抗体、1価aKLH120.6IgG2 HC−ShKペプチドアナログ、および2価aKLH120.6IgG2 HC−ShKペプチドアナログである。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 HC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6κLC(配列番号28);
(b)aKLH120.6IgG2 HC(配列番号29);および
(c)以下の配列を有するaKLH120.6IgG2−ShK[1−35、R1H、Q16K、K30E]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSHSCIDTIPKSRCTAFKCKHSMKYRLSFCRETCGTC//(配列番号307)。
所望の1価aKLH120.6IgG2 HC−ShKアナログ生成物は1本の重鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖:重鎖−ShKの比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2抗体、1価aKLH120.6IgG2 HC−ShKペプチドアナログ、および2価aKLH120.6IgG2 HC−ShKペプチドアナログである。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 HC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6κLC(配列番号28);
(b)aKLH120.6IgG2 HC(配列番号29);および
(c)以下の配列を有するaKLH120.6HC(IgG2)−ShK[1−35、R1K、I4A、Q16K]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSKSCADTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号308)。
所望の1価aKLH120.6IgG2 HC−ShKアナログ生成物は1本の重鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖:重鎖−ShKの比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2抗体、1価aKLH120.6IgG2 HC−ShKペプチドアナログ、および2価aKLH120.6IgG2 HC−ShKペプチドアナログである。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 HC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6κLC(配列番号28);
(b)aKLH120.6IgG2 HC(配列番号29);および
(c)以下のアミノ酸配列を有するaKLH120.6IgG2−ShK[1−35、R1K、Q16K、K30E]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSKSCIDTIPKSRCTAFKCKHSMKYRLSFCRETCGTC//(配列番号309)。
所望の1価aKLH120.6IgG2 HC−ShKアナログ生成物は1本の重鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖:重鎖−ShKの比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2抗体、1価aKLH120.6IgG2 HC−ShKペプチドアナログ、および2価aKLH120.6IgG2 HC−ShKペプチドアナログである。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 HC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6κLC(上記の配列番号28);
(b)aKLH120.6IgG2 HC(上記の配列番号29);および
(c)以下のHC配列を有するaKLH120.6IgG2−ShK[2−35、Q16K]融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号33)。
所望のaKLH120.6IgG2−ShKアナログ生成物は、図1Fに企図する1本の重鎖のC末端に融合したShKペプチドを有する完全抗体である。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖:重鎖−ShKの比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2、1価aKLH120.6IgG2−ShK、および2価aKLH120.6IgG2−ShKである。本明細書に記載のとおり、1価aKLH120.6IgG2毒素ペプチド(または毒素ペプチドアナログ)融合抗体を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6κLC(上記の配列番号28);
(b)aKLH120.6IgG1 HC:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号34);
および、
(c)aKLH120.6IgG1−ループ−ShK:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELGGRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTCGGTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号35)。
(a)aKLH120.6IgG2 HC(配列番号29);
(b)aKLH120.6κLC(配列番号28);および
(c)以下の配列を有するaKLH120.6κLC−ShK[1−35、Q16K]融合物:
MDMRVPAQLLGLLLLWLRGARCDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGECGGGGSGGGGSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号267)。
1価aKLH120.6IgG2 LC−ShK[1−35、Q16K]生成物のこの実施形態は、図1Jに示すように、1本の軽鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の重鎖を共有する2つの異なる軽鎖を含み、軽鎖:重鎖:軽鎖−ShK[1−35、Q16K]の比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2、1価aKLH120.6IgG2 LC−ShK[1−35、Q16K]、および2価aKLH120.6IgG2 LC−ShK[1−35、Q16K]である。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 LC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6IgG2 HC(配列番号29);
(b)aKLH120.6κLC(配列番号28);および
(c)以下の配列を有するaKLH120.6κLC−ShK[2−35、Q16K]融合物:
MDMRVPAQLLGLLLLWLRGARCDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGECGGGGSGGGGSSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号268)。
1価aKLH120.6IgG2 LC−ShK[2−35、Q16K]生成物のこの実施形態は、図1Jに示すように、1本の軽鎖のC末端に融合したShKペプチドを有する完全抗体であった。1種類の重鎖を共有する2つの異なる軽鎖を含み、軽鎖:重鎖:軽鎖−ShK[2−35、Q16K]の比率は1:2:1であった。予期した発現生成物は、aKLH120.6IgG2、1価aKLH120.6IgG2 LC−ShK[2−35、Q16K]、および2価aKLH120.6IgG2 LC−ShK[2−35、Q16K]である。本明細書に記載のとおり、抗体を含む1価aKLH120.6IgG2 LC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6IgG2 HC(配列番号29);および
(b)上記のaKLH120.6κLC−ShK[1−35、Q16K]融合物(配列番号267)。
2価aKLH120.6IgG2 LC−ShK[1−35、Q16K]抗体生成物のこの実施形態は、図1Kに示すように、両軽鎖のC末端に融合したShKペプチドを有する完全抗体であった。重鎖:軽鎖−ShK[1−35、Q16K]の比率は1:1であった。予期した発現生成物は、2価aKLH120.6IgG2 LC−ShK[1−35、Q16K]である。本明細書に記載のとおり、抗体分子を含む2価aKLH120.6IgG2 LC−ShK[1−35、Q16K]ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6IgG2 HC(配列番号29);および
(b)上記のaKLH120.6κLC−ShK[2−35、Q16K]融合物(配列番号268)。
2価aKLH120.6IgG2 LC−ShK[2−35、Q16K]生成物のこの実施形態は、図1Kに示すように、両軽鎖のC末端に融合したShKペプチドを有する完全抗体であった。重鎖:軽鎖−ShK[2−35、Q16K]の比率は1:1であった。予期した発現生成物は、2価aKLH120.6IgG2 LC−ShK[2−35、Q16K]抗体である。本明細書に記載のとおり、抗体を含む2価aKLH120.6IgG2 LC−ShK[2−35、Q16K]毒素融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
(a)aKLH120.6IgG2 HC(上記の配列番号29);
(b)上記の配列番号32のアミノ酸を有するaKLH120.6IgG2 HC−Shk[1−35、Q16K]融合物;および
(c)上記の配列番号267のアミノ酸配列を有するaKLH120.6κLC−ShK[1−35、Q16K]融合物。
3価aKLH120.6IgG2 LC−ShK生成物のこの実施形態は、図1Lに示すように、両軽鎖のC末端に融合したShK[1−35、Q16K]ペプチドおよび1本の重鎖を有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖−ShK[1−35、Q16K]:重鎖−ShK[1−35、Q16K]の比率は1:2:1であった。予期した発現生成物は2価aKLH120.6IgG2 LC−ShK[1−35、Q16K]抗体、3価aKLH120.6IgG2 LC−ShK[1−35、Q16K]抗体、および4価aKLH120.6IgG2 LC−ShK[1−35、Q16K]抗体であった。本明細書に記載のとおり、抗体分子を含む3価aKLH120.6IgG2 LC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
a)aKLH120.6IgG2 HC(配列番号29);
(b)上記のaKLH120.6IgG2 HC−Shk[2−35、Q16K]融合物(配列番号33);および
(c)上記のaKLH120.6κLC−ShK[2−35、Q16K]融合物(配列番号268)。
3価aKLH120.6IgG2 LC−ShK[2−35、Q16K]抗体生成物のこの実施形態は、図1Lに示すように、両軽鎖のC末端に融合したShK[2−35、Q16K]ペプチドおよび1本の重鎖を有する完全抗体であった。1種類の軽鎖を共有する2つの異なる重鎖を含み、重鎖:軽鎖−ShK[2−35、Q16K]:重鎖−ShK[2−35、Q16K]の比率は1:2:1であった。予期した発現生成物は2価aKLH120.6IgG2 LC−ShK[2−35、Q16K]抗体、3価aKLH120.6IgG2 LC−ShK[2−35、Q16K]抗体、および4価aKLH120.6IgG2 LC−ShK[2−35、Q16K]抗体であった。本明細書に記載のとおり、抗体分子を含む3価aKLH120.6IgG2 LC毒素ペプチド融合物を陽イオン交換クロマトグラフィーを用いて混合物から単離した。
MDMRVPAQLLGLLLLWLRGARCDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC//(配列番号28)をコードする。
5’−AAC AGG GGA GAG TGT GGA GGA GGA GGA TCC GGA G−3’(配列番号269);および
5’−CAT GCG GCC GCT CAT TAG CAG G−3’(配列番号270)
を用いてPCRによりShk[1−35、Q16K]断片を生成した。
以下のオリゴ
5’−CAT TCT AGA CCC ACC ATG GAC ATG AGG GTG−3’(配列番号43);および
5’−GGA TCC TCC TCC TCC ACC CGG AGA CAG GGA GAG G−3’(配列番号44)
を用いて、
aKLH120.6−VK1SP−IgG2重鎖(配列番号46;以下)をコードするコード配列(配列番号45;以下)を含むpTT5−aKLH120.6−VK1SP−IgG2重鎖(HC)構築物からaKLH−IgG2−重鎖領域をPCRにより増幅した:
ATGGACATGAGGGTGCCCGCTCAGCTCCTGGGGCTCCTGCTGCTGTGGCTGAGAGGTGCCAGATGTCAGGTGCAGCTGGTGCAGTCTGGGGCTGAGGTGAAGAAGCCTGGGGCCTCAGTGAAGGTCTCCTGCAAGGCTTCTGGATACACCTTCACCGGCTACCACATGCACTGGGTGCGACAGGCCCCTGGACAAGGGCTTGAGTGGATGGGATGGATCAACCCTAACAGTGGTGGCACAAACTATGCACAGAAGTTTCAGGGCAGGGTCACCATGACCAGGGACACGTCCATCAGCACAGCCTACATGGAGCTGAGCAGGCTGAGATCTGACGACACGGCCGTGTATTACTGTGCGAGAGATCGTGGGAGCTACTACTGGTTCGACCCCTGGGGCCAGGGAACCCTGGTCACCGTCTCCTCAGCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGGCGCCCTGCTCCAGGAGCACCTCCGAGAGCACAGCGGCCCTGGGCTGCCTGGTCAAGGACTACTTCCCCGAACCGGTGACGGTGTCGTGGAACTCAGGCGCTCTGACCAGCGGCGTGCACACCTTCCCAGCTGTCCTACAGTCCTCAGGACTCTACTCCCTCAGCAGCGTGGTGACCGTGCCCTCCAGCAACTTCGGCACCCAGACCTACACCTGCAACGTAGATCACAAGCCCAGCAACACCAAGGTGGACAAGACAGTTGAGCGCAAATGTTGTGTCGAGTGCCCACCGTGCCCAGCACCACCTGTGGCAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGACACCCTCATGATCTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCCACGGGAGGAGCAGTTCAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTTGTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGGCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGGCAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAAGACCACACCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACGTCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCACTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGT//(配列番号45)、
これはアミノ酸配列
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG//(配列番号46)をコードする。
5’−TCC CTG TCT CCG GGT GGA GGA GGA GGA TCC GGA G−3’(配列番号47);および5’−CAT GCG GCC GCT CAT TAG CAG GTG−3’(配列番号14)を用いて生成した。PCR生成物を1%アガロースゲル上に流した。バンドにアガロースプラグ用の穴を開けてプラグを新しいPCR反応管に入れた。次いで外プライマー配列番号357および配列番号330を用いてPCRによりアガロースプラグを増幅した。次いでPCR生成物をXbaIおよびNotIにより消化して、PCRクリーンアップキット(Qiagen)で精製した。同時に、pTT5ベクター(CMVプロモーターおよびPoly A tailを含むAmgenベクター)をXbaIおよびNotIにより切断した。pTT5ベクターを1%アガロースゲル上に流して、大型断片を切断し、QiagenのGel Purificationキットによりゲル精製した。精製したPCR生成物を大型ベクター断片と結紮して、OneShot Top10細菌に形質転換した。形質転換細菌コロニーからDNAを単離し、XbaIおよびNotI制限酵素による消化に供して、1%アガロースゲル上で分解した。予期したパターンに至るDNAを配列決定用に提出した。いくつかのクローン配列分析において上記配列と100%適合したが、1クローンのみを大規模プラスミド精製用に選択した。最終pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35]構築物はアミノ酸配列:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSRSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC//(配列番号48)
とのIgG2−HC−L10−ShK[1−35]融合ポリペプチドをコードした。
5’−GCT GCA CCG CCT TCA AGT GCA AGC ACA GC 3’(配列番号9);および
5’−GCT GTG CTT GCA CTT GAA GGC GGT GCA GC−3’(配列番号10)
を用いて部位指向変異を実施した。最終構築物pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35、Q16K]はアミノ酸配列:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号49)
とのIgG2−HC−L10−ShK[1−35、Q16K]融合ポリペプチドをコードした。
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGG//(配列番号51)をコードするコード配列:
ATGGACATGAGGGTGCCCGCTCAGCTCCTGGGGCTCCTGCTGCTGTGGCTGAGAGGTGCCAGATGTCAGGTGCAGCTGGTGCAGTCTGGGGCTGAGGTGAAGAAGCCTGGGGCCTCAGTGAAGGTCTCCTGCAAGGCTTCTGGATACACCTTCACCGGCTACCACATGCACTGGGTGCGACAGGCCCCTGGACAAGGGCTTGAGTGGATGGGATGGATCAACCCTAACAGTGGTGGCACAAACTATGCACAGAAGTTTCAGGGCAGGGTCACCATGACCAGGGACACGTCCATCAGCACAGCCTACATGGAGCTGAGCAGGCTGAGATCTGACGACACGGCCGTGTATTACTGTGCGAGAGATCGTGGGAGCTACTACTGGTTCGACCCCTGGGGCCAGGGAACCCTGGTCACCGTCTCCTCAGCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGGCGCCCTGCTCCAGGAGCACCTCCGAGAGCACAGCGGCCCTGGGCTGCCTGGTCAAGGACTACTTCCCCGAACCGGTGACGGTGTCGTGGAACTCAGGCGCTCTGACCAGCGGCGTGCACACCTTCCCAGCTGTCCTACAGTCCTCAGGACTCTACTCCCTCAGCAGCGTGGTGACCGTGCCCTCCAGCAACTTCGGCACCCAGACCTACACCTGCAACGTAGATCACAAGCCCAGCAACACCAAGGTGGACAAGACAGTTGAGCGCAAATGTTGTGTCGAGTGCCCACCGTGCCCAGCACCACCTGTGGCAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGACACCCTCATGATCTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCCACGGGAGGAGCAGTTCAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTTGTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGGCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGGCAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAAGACCACACCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACGTCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCACTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTGGAGGAGGA//(配列番号50)を含有した。次いでベクター断片をCalf Intestine Phosphatase(CIP)で処理して5’リン酸基を除去し、フェノール/クロロホルムを抽出してベクター自体の再結紮を防止した。挿入物はIgG2 Fc−L10−ShK(2−35、Q16K)をコードするpTT14−VH21SP−IgG2−Fc−ShK[2−35、Q16K]に由来した:
MEWSWVFLFFLSVTTGVHSERKVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGGGSGGGGSSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号18)、
また、挿入物はBamHI/BamHIを用いて消化もさせた。挿入ShK[2−35、Q16K]断片をそのベクターからゲル精製して、QiagenのGel Purificationキットでクリーンアップした。精製したDNA挿入物は、アミノ酸配列GSGGGGSSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC(配列番号53)をコードするコード配列GGA TCC GGA GGA GGA GGA AGC AGC TGC ATC GAC ACC ATC CCC AAG AGC CGC TGC ACC GCC TTC AAG TGC AAG CAC AGC ATG AAG TAC CGC CTG AGC TTC TGC CGC AAG ACC TGC GGC ACC TGC TAA TGA//(配列番号52)を含み、大型ベクター断片と結紮して、OneShot Top10細菌に形質転換した。形質転換細菌コロニーからDNAを単離し、BamHI制限酵素による消化に供して、1%アガロースゲル上で分解した。予期したパターンに至るDNAを配列決定用に提出した。いくつかのクローン配列分析において上記配列と100%適合したが、1クローンのみを大規模プラスミド精製用に選択した。最終構築物pTT5−aKLH−IgG2 HC−L10−ShK[2−35、Q16K]はIgG2 HC−L10−ShK[2−35、Q16K]融合ポリペプチド:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号54)をコードした。
5’−AGG AGG AGG AAG CGC CAG CTG CGC CGA CAC CAT CCC C−3’//(配列番号310);および
5’−GGG GAT GGT GTC GGC GCA GCT GGC GCT TCC TCC TCC T−3’//(配列番号311)
を用いて生成した。
Stratagene QuikChange Multi site Directed Mutagenesisキットを製造業者の説明書に従いつつ用いて部位指向変異を実施した。最終pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35 R1A、I4A、Q16K]構築物はIgG2−HC−L10−ShK[1−35、R1A、I4A、Q16K]融合ポリペプチド(配列番号304)をコードした。
5’−GAG GAG GAG GAA GCG CCA GCT GCA TCG ACA−3’//(配列番号312);
5’−GAG CTT CTG CCG CGA GAC CTG CGG CAC−3’//(配列番号313);
5’−CGA TGC AGC TGG CGC TTC CTC CTC CTC−3’//(配列番号314);および
5’−GTG CCG CAG GTC TCG CGG CAG AAG CTC−3’//(配列番号315)
を用いて上記のとおりShk[1−35、R1A、Q16K、K30E]断片を生成した。
最終pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35 R1A、Q16K、K30E]構築物はIgG2−HC−L10−ShK[1−35、R1A、Q16K、K30E]融合ポリペプチド(配列番号305)をコードした。
5’−GGA GGA GGA AGC CAC AGC TGC GCC GAC ACC ATC CCC−3’//(配列番号316);および
5’−GGG GAT GGT GTC GGC GCA GCT GTG GCT TCC TCC TCC−3’//(配列番号317)
を用いてShK[1−35、R1H、I4A、Q16K]断片を生成した。
Stratagene QuikChange Multi site Directed Mutagenesisキット(Cat#200531)を製造業者の説明書に従いつつ用いて部位指向変異を実施した。最終pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35 R1H、I4A、Q16K]構築物はIgG2−HC−L10−ShK[1−35、R1H、I4A、Q16K]融合ポリペプチド(配列番号306)をコードした。
5’−GGA GGA GGA AGC CAC AGC TGC ATC GAC−3’//(配列番号318)および配列番号313;
5’−GTC GAT GCA GCT GTG GCT TCC TCC TCC−3’//(配列番号319)および配列番号315を用いて上記のとおりShk[1−35、R1H、Q16K、K30E]断片を生成した。
最終pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35 R1H、Q16K、K30E]構築物はIgG2−HC−L10−ShK[1−35、R1H、Q16K、K30E]融合ポリペプチド(配列番号307)をコードした。
5’−CCG GAG GAG GAG GAA GCA AGA GCT GCG CCG ACA CCA TCC CCA AGA−3’//(配列番号320);および
5’−TCT TGG GGA TGG TGT CGG CGC AGC TCT TGC TTC CTC CTC CTC CGG−3’//(配列番号321)
を用いてShk[1−35、R1K、I4A、Q16K]断片を生成した。
Stratagene QuikChange Multi site Directed Mutagenesisキット(Cat#200531)を製造業者の説明書に従いつつ用いて部位指向変異を実施した。最終pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35 R1K、I4A、Q16K]構築物はIgG2−HC−L10−ShK[1−35、R1K、I4A、Q16K]融合ポリペプチド(配列番号308)をコードした。
5’−CGG AGG AGG AGG AAG CAA GAG CTG CAT CGA CAC CA −3’//(配列番号322)および配列番号313;
5’−TGG TGT CGA TGC AGC TCT TGC TTC CTC CTC CTC CG −3’//(配列番号323)および配列番号315を用いてShk[1−35、R1K、Q16K、K30E]断片を上記のとおり生成した。
最終pTT5−aKLH120.6−VK1SP−IgG2−HC−L10−ShK[1−35 R1H、Q16K、K30E]構築物はIgG2−HC−L10−ShK[1−35、R1K、Q16K、K30E]融合ポリペプチド(配列番号309)をコードした。
順行プライマー:
TGCAGAAGCTTCTAGACCACCATGGAATGGAGCTGGGTCTTTCTCTTCTTCCTGTCAGTAACGACTGGTGTCCACTCCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCCAGT//(配列番号55);および
逆行プライマー:
CTCCGGATCCTCCTCCTCCGCAGGTGCCGCAGGTCTTGCGGCAGAAGCTCAGGCGGTACTTCATGCTGTGCTTGCACTGGAAGGCGGTGCAGCGGCTCTTGGGGATGGTGTCGAT//(配列番号56)。
順行プライマー:
GTAGGATCCGGAGGAGGAGGAAGCGACAAAACTCACAC//(配列番号57);および
逆行プライマー:
CGAGCGGCCGCTTACTATTTACCCGGAGACAGGGA//(配列番号58)。
MEWSWVFLFFLSVTTGVHSRSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTCGGGGSGGGGSDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号60)をコードするコード配列
ATGGAATGGAGCTGGGTCTTTCTCTTCTTCCTGTCAGTAACGACTGGTGTCCACTCCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCCAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGCGGAGGAGGAGGATCCGGAGGAGGAGGAAGCGACAAAACTCACACATGCCCACCGTGCCCAGCACCTGAACTCCTGGGGGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGACACCCTCATGATCTCCCGGACCCCTGAGGTCACATGCGTGGTGGTGGACGTGAGCCACGAAGACCCTGAGGTCAAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCCGCGGGAGGAGCAGTACAACAGCACGTACCGTGTGGTCAGCGTCCTCACCGTCCTGCACCAGGACTGGCTGAATGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGCCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAGCCAAAGGGCAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGATGAGCTGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTATCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAAGACCACGCCTCCCGTGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACGTCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCACTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTAAATAGTAA//(配列番号59)を含有した。
5’−GCT GCA CCG CCT TCA AGT GCA AGC ACA GC−3’//(配列番号9);および
5’−GCT GTG CTT GCA CTT GAA GGC GGT GCA GC−3’(配列番号10)
を用いる部位指向変異を実施してShK領域にQ16K変異を産生した。
Stratagene QuikChange Multi site Directed Mutagenesisキットを製造業者の説明書に従いつつ用いた。pCMVi−N末端−ShK[1−35Q16K]−L10−IgG1−Fcのための最終構築物は、以下のシグナルペプチド(VH21SP)−ShK[1−35、Q16K]−L10−IgG1−Fc融合ポリペプチド:
MEWSWVFLFFLSVTTGVHSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTCGGGGSGGGGSDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号61)をコードした。
5’−CAT TCT AGA CCA CCA TGG AAT GG−3’(配列番号62);
5’−CAG CTG CAC CTG GCT TCC TCC TCC TCC GG−3’(配列番号63);
およびpCMVi−N末端−ShK[1−35、Q16K]−L10−IgG1−Fc鋳型を用いて産生し、VH21SP−ShK(1−35、Q16K)−L10アミノ酸配列
MEWSWVFLFFLSVTTGVHSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTCGGGGSGGGGS//(配列番号65)をコードするコード配列
ATGGAATGGAGCTGGGTCTTTCTCTTCTTCCTGTCAGTAACGACTGGTGTCCACTCCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCAAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGCGGAGGAGGAGGATCCGGAGGAGGAGGAAGC//(配列番号64)を含む断片を得た。
5’−GGA GGA GGA AGC CAG GTG CAG CTG GTG CAG−3’(配列番号66);
5’−CAT GCG GCC GCT CAT TTA CCC−3’(配列番号67);
およびpTT5−aKLH120.6−HC鋳型を用いてPCR生成物を作製し、アミノ酸配列
QVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号69)をコードするコード配列
CAGGTGCAGCTGGTGCAGTCTGGGGCTGAGGTGAAGAAGCCTGGGGCCTCAGTGAAGGTCTCCTGCAAGGCTTCTGGATACACCTTCACCGGCTACCACATGCACTGGGTGCGACAGGCCCCTGGACAAGGGCTTGAGTGGATGGGATGGATCAACCCTAACAGTGGTGGCACAAACTATGCACAGAAGTTTCAGGGCAGGGTCACCATGACCAGGGACACGTCCATCAGCACAGCCTACATGGAGCTGAGCAGGCTGAGATCTGACGACACGGCCGTGTATTACTGTGCGAGAGATCGTGGGAGCTACTACTGGTTCGACCCCTGGGGCCAGGGAACCCTGGTCACCGTCTCCTCAGCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGGCGCCCTGCTCCAGGAGCACCTCCGAGAGCACAGCGGCCCTGGGCTGCCTGGTCAAGGACTACTTCCCCGAACCGGTGACGGTGTCGTGGAACTCAGGCGCTCTGACCAGCGGCGTGCACACCTTCCCAGCTGTCCTACAGTCCTCAGGACTCTACTCCCTCAGCAGCGTGGTGACCGTGCCCTCCAGCAACTTCGGCACCCAGACCTACACCTGCAACGTAGATCACAAGCCCAGCAACACCAAGGTGGACAAGACAGTTGAGCGCAAATGTTGTGTCGAGTGCCCACCGTGCCCAGCACCACCTGTGGCAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGACACCCTCATGATCTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCCACGGGAGGAGCAGTTCAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTTGTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGGCCTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGGCAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAAGACCACACCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACGTCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCACTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTAAATGA//(配列番号68)を含むDNA断片を得た。
MEWSWVFLFFLSVTTGVHSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTCGGGGSGGGGSQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK//(配列番号70)をコードした。
5’−CAT TCT AGA CCA CCA TGG AAT GG−3’(配列番号62);および
5’−CAT CTG GAT GTC GCT TCC TCC TCC TCC GG−3’(配列番号71);
ならびにpCMVi−N末端−ShK[1−35Q16K]−L10−IgG1−Fc鋳型を用いて作製し、シグナルペプチド(VH21SP)−ShK(1−35、Q16K)−L10リンカーのためのアミノ酸配列:
MEWSWVFLFFLSVTTGVHSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTCGGGGSGGGGS//(配列番号65)をコードするコード配列
ATGGAATGGAGCTGGGTCTTTCTCTTCTTCCTGTCAGTAACGACTGGTGTCCACTCCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCAAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGCGGAGGAGGAGGATCCGGAGGAGGAGGAAGC//(配列番号64)を含むDNA断片を得た。
鋳型ならびに以下のオリゴ:
5’−GGA GGA GGA AGC GAC ATC CAG ATG ACC CAG TC−3’(配列番号72);および
5’−CAT CTC GAG CGG CCG CTC AAC−3’(配列番号73)を用いた。
N末端シグナルペプチド:
MEWSWVFLFFLSVTTGVHSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTCGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC//(配列番号75)
を有するN末端VH21SP−ShK[1−35、Q16K]−L10−aKLH120.6軽鎖(LC)のためのアミノ酸配列をコードするコード配列
ATGGAATGGAGCTGGGTCTTTCTCTTCTTCCTGTCAGTAACGACTGGTGTCCACTCCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCAAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGCGGAGGAGGAGGATCCGGAGGAGGAGGAAGCGACATCCAGATGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGAAATGATTTAGGCTGGTATCAGCAGAAACCAGGGAAAGCCCCTAAACGCCTGATCTATGCTGCATCCAGTTTGCAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGAATTCACTCTCACAATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCTACAGCATAATAGTTACCCGCTCACTTTCGGCGGAGGGACCAAGGTGGAGATCAAACGAACTGTGGCTGCACCATCTGTCTTCATCTTCCCGCCATCTGATGAGCAGTTGAAATCTGGAACTGCCTCTGTTGTGTGCCTGCTGAATAACTTCTATCCCAGAGAGGCCAAAGTACAGTGGAAGGTGGATAACGCCCTCCAATCGGGTAACTCCCAGGAGAGTGTCACAGAGCAGGACAGCAAGGACAGCACCTACAGCCTCAGCAGCACCCTGACGCTGAGCAAAGCAGACTACGAGAAACACAAAGTCTACGCCTGCGAAGTCACCCATCAGGGCCTGAGCTCGCCCGTCACAAAGAGCTTCAACAGGGGAGAGTGTTGA//(配列番号74)を含むクローン化PCR断片を生成した。
プラスミドpTT5−aDNP3A4(W101F)IgG2−Shk[1−35Q16K]の作製:
Kv1.3阻害剤毒素ペプチドアナログShK[1−35、Q16K](配列番号76)の単量体にインフレーム融合させたヒト抗2,4−ジニトロフェニル(aDNP)抗体の重鎖をコードするDNA配列を標準的なクローニング技術を用いて構築した。以下のアミノ酸配列:
MDMRVPAQLLGLLLLWLRGARCQVQLVESGGGVVQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAVIWYDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARYNFNYGMDVWGQGTTVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG//(配列番号77)を有する抗DNP 3A4(W101F)IgG2重鎖の一部(pDC324:aDNP3A4 HC(W101F)およびIgG2Fc−Shk[1−35、Q16K]の一部にpTT5ベクターを結紮する3つの方法によりプラスミドpTT5−aDNP3A4(W101F)IgG2−Shk[1−35、Q16K]を生成した。複数のクローニング部位を放出するSalI/NotIによりpTT5ベクターを切断した。次いでベクターをCalf Intestine Phosphatase(CIP)で処理して背景を減らした。第一挿入物はpDC324由来であった:aDNP3A4 HC(W101F)をSalI/StuIで切断することにより、アミノ酸配列
MDMRVPAQLLGLLLLWLRGARCQVQLVESGGGVVQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAVIWYDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARYNFNYGMDVWGQGTTVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKG//(配列番号79)をコードするコード配列
ATGGACATGAGGGTGCCCGCTCAGCTCCTGGGGCTCCTGCTGCTGTGGCTGAGAGGTGCGCGCTGTCAGGTGCAGCTGGTGGAGTCTGGGGGAGGCGTGGTCCAGCCTGGGAGGTCCCTGAGACTCTCCTGTGCAGCGTCTGGATTCACCTTCAGTAGCTATGGCATGCACTGGGTCCGCCAGGCTCCAGGCAAGGGGCTGGAGTGGGTGGCAGTTATATGGTATGATGGAAGTAATAAATACTATGCAGACTCCGTGAAGGGCCGATTCACTATCTCCAGAGACAATTCCAAGAACACGCTGTATCTGCAAATGAACAGCCTGAGAGCCGAGGACACGGCTGTGTATTACTGTGCGAGGTATAACTTCAACTACGGTATGGACGTCTGGGGCCAAGGGACCACGGTCACCGTCTCTAGTGCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGGCGCCCTGCTCCAGGAGCACCTCCGAGAGCACAGCGGCCCTGGGCTGCCTGGTCAAGGACTACTTCCCCGAACCGGTGACGGTGTCGTGGAACTCAGGCGCTCTGACCAGCGGCGTGCACACCTTCCCAGCTGTCCTACAGTCCTCAGGACTCTACTCCCTCAGCAGCGTGGTGACCGTGCCCTCCAGCAACTTCGGCACCCAGACCTACACCTGCAACGTAGATCACAAGCCCAGCAACACCAAGGTGGACAAGACAGTTGAGCGCAAATGTTGTGTCGAGTGCCCACCGTGCCCAGCACCACCTGTGGCAGGACCGTCAGTCTTCCTCTTCCCCCCAAAACCCAAGGACACCCTCATGATCTCCCGGACCCCTGAGGTCACGTGCGTGGTGGTGGACGTGAGCCACGAAGACCCCGAGGTCCAGTTCAACTGGTACGTGGACGGCGTGGAGGTGCATAATGCCAAGACAAAGCCACGGGAGGAGCAGTTCAACAGCACGTTCCGTGTGGTCAGCGTCCTCACCGTTGTGCACCAGGACTGGCTGAACGGCAAGGAGTACAAGTGCAAGGTCTCCAACAAAGGC//(配列番号78)を含むDNA断片を得た。
第二挿入物をStuI/NotIを用いて消化し、以下の切断されたIgG2 Fc−L10−ShK(1−35、Q16K)アミノ酸配列
LPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKGGGGSGGGGSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号81)をコードするコード配列
CTCCCAGCCCCCATCGAGAAAACCATCTCCAAAACCAAAGGGCAGCCCCGAGAACCACAGGTGTACACCCTGCCCCCATCCCGGGAGGAGATGACCAAGAACCAGGTCAGCCTGACCTGCCTGGTCAAAGGCTTCTACCCCAGCGACATCGCCGTGGAGTGGGAGAGCAATGGGCAGCCGGAGAACAACTACAAGACCACACCTCCCATGCTGGACTCCGACGGCTCCTTCTTCCTCTACAGCAAGCTCACCGTGGACAAGAGCAGGTGGCAGCAGGGGAACGTCTTCTCATGCTCCGTGATGCATGAGGCTCTGCACAACCACTACACGCAGAAGAGCCTCTCCCTGTCTCCGGGTAAAGGAGGAGGAGGATCCGGAGGAGGAGGAAGCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCAAGTGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGCTAATGA//(配列番号80)を含有した。
MDMRVPAQLLGLLLLWLRGARCQVQLVESGGGVVQPGRSLRLSCAASGFTFSSYGMHWVRQAPGKGLEWVAVIWYDGSNKYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARYNFNYGMDVWGQGTTVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGSGGGGSRSCIDTIPKSRCTAFKCKHSMKYRLSFCRKTCGTC//(配列番号82)を有するaDNP3A4(W101F)IgG2 HC−L10−Shk[1−35、Q16K]をコードした。
MDMRVPAQLLGLLLLWLRGARCDIQMTQSPSSVSASVGDRVTITCRASQGISRRLAWYQQKPGKAPKLLIYAASSLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQANSFPFTFGPGTKVDIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC//(配列番号109)。
薬理学的に活性なポリペプチドのための免疫グロブリン担体として用いられる本発明の抗原結合タンパク質の実施形態は、良好な薬物動態および薬力学特性を提供することが示された。1価もしくは2価Fc−L10−ShK[2−35]、1価もしくは2価Fc−L10−ShK[1−35]、1価もしくは2価Fc−L10−ShK(1−35、Q16K)、1価もしくは2価抗KLH HC−ShK(1−35、Q16K)Ab、1価もしくは2価抗KLH Abループ−[Lys16]ShK融合タンパク質、1価Fc−ShK(1−35 Q16K)/aKLH Abヘテロ三量体、ならびに表7Hに列挙する他の例示的な実施形態を、実施例4に記載の手段により発現させ、単離して精製した。表7HのPEG化および非PEG化毒素ペプチド比較物を以下のとおり合成的に調製した。
ShKに対する抗体。ShK(配列番号361)に対するウサギポリクローナルおよびマウスモノクローナル抗体を動物のFc−ShKペプチボディ結合体による免疫化により生成した。抗ShK特異的ポリクローナル抗体を抗血清から親和性精製して結合体のShK部分に特異的な抗体のみを単離した。融合およびスクリーニング後、ShKに特異的なハイブリドーマを選択し、単離した。マウス抗ShK特異的モノクローナル抗体をクローン条件培地から精製した。ELISA分析により、精製した抗ShKポリクローナルおよびモノクローナル抗体はShKペプチド単独のみに反応し、Fcとは交差反応しなかった。
(a)ストレプトアビジンマイクロタイタープレートを250ng/mLビオチン化−抗ShKマウスモノクローナル抗体(mAb2.10、Amgen)のIブロック緩衝液[1リットルにつき:1000mL1×PBS、CaCl2、MgCl2非含有、5mLのTween 20(Thermo Scientific)、2gのIブロック試薬(Tropix)]で4℃でコーティングし、一晩撹拌せずにインキュベートした。
(b)プレートをKPL洗浄緩衝液(Kirkegaard & Perry Laboratories)で3回洗浄した。
(c)プールした血清100%を有する標準(STD)、品質管理(QC)および試料希釈を調製してから、Iブロック緩衝液で5分の1に希釈した(前処置)。前処置したSTD、QCおよび試料を洗浄したプレートに添加し、室温で2時間インキュベートした。(STD、QCの連続希釈をプールした血清100%内で調製した。希釈する必要のある試料はプールした血清100%でも調製した。std、QCおよび試料の両方に対して前処置を行い、基質効果を最小化した。)
(d)プレートをKPL洗浄緩衝液で3回洗浄した。
(e)HRP標識ウサギ抗ShKポリクローナルAb、250ng/mLのIブロック緩衝液を添加し、プレートを室温で1時間撹拌しながらインキュベートした。
(f)プレートをKPL洗浄緩衝液で再び3回洗浄し、Femto[Thermo Scientific]基質を添加した。
(g)プレートをLmax II 384(Molecular Devices)照度計で読み込んだ。
(a)ストレプトアビジンマイクロタイタープレートを250ng/mLビオチン化−抗ShKマウスモノクローナル抗体(mab2.10、Amgen)のIブロック緩衝液[1リットルにつき:1000mLの1×PBS、CaCl2、MgCl2非含有、5mLのTween 20(Thermo Scientific)で、2gのIブロック試薬(Tropix)]で4℃で、一晩撹拌せずにコーティングした;
(b)プレートをKPL洗浄緩衝液(Kirkegaard & Perry Laboratories)で3回洗浄した
(c)プールした血清100%を有する標準(STD)、品質管理(QC)および試料希釈を調製してから、Iブロック緩衝液で5分の1に希釈した(前処置)。前処置したSTD、QCおよび試料を洗浄したプレートに添加した。インキュベーションは室温で2時間行った。(STD、QCの連続希釈をプールした血清100%内で調製した。希釈する必要のある試料はプールした血清100%でも調製した。std、QCおよび試料の両方に対して前処置を行い、基質効果を最小化した。);
(d)プレートをKPL洗浄緩衝液で3回洗浄した;
(e)HRP標識Ab35(ヒトIgG Fcに対して)、150ng/mLのIブロック緩衝液を添加し、プレートを室温で1時間撹拌しながらインキュベートした。
(f)プレートをKPL洗浄緩衝液で3回洗浄し、Femto[Thermo Scientific]基質を添加した;
(g)プレートをLmax II 384[Molecular Devices]照度計で読み込んだ。
(a)1×コーティング緩衝液で希釈したCostar3590 96ウェルEIA/RIAプレートを0.1mL/ウェルの2μg/mLヤギ抗HuFc、Fab2、(Sigma I−3391)(10×コーティング緩衝液:1.59g Na2CO3、2.93g NaHCO3のH2O溶液100mL)でコーティングした。プレートを密封して4℃で一晩インキュベートした;
(b)プレートをPBST(PBS+0.1%Tween−20)で3回洗浄し、0.3mLのblotto(PBS、0.1%Tween−20、5%脱脂粉乳)を各ウェルへ添加してブロックし、室温(RT)で撹拌しながら1時間インキュベートした;
(c)プレートをKP洗浄溶液(Cat#50−63−00, KPL, Gaithersburg, MD)で洗浄した;
(d)希釈した血清試料および対照/標準の希釈緩衝液(PBS、0.1%BSA、0.1%Tween−20)+ラット血清で、必要な場合、最終ラット血清10%とし、0.1mL試料を各ウェルへ添加した。プレートを室温で撹拌しながら、1時間インキュベートした;
(e)プレートをKP洗浄溶液(Cat#50−63−00, KPL, Gaithersburg, MD)で洗浄した;
(f)HRP標識二次抗体(Pierce#31416−HRPヤギα−Hu IgG Fc)をPBSTで1:5000希釈してから100μL/ウェルを追加し、RTで撹拌しながら、1時間インキュベートした;
(g)プレートをKP洗浄溶液(Cat#50−63−00, KPL, Gaithersburg, MD)で洗浄してABTS基質(ABTS Microwell Substrate 1−Component, Cat#50−66−018, KPL)100μL/ウェルを添加した;
(h)基質を添加して撹拌した後、適切な時点で、SpectraMax340[Molecular Devices]プレート読取り器でプレートを読み込んだ。
Kv1.7を介したKv1.1発現細胞系。CHO−K1細胞をヒトKv1.3で、またはカウンタースクリーンのため(本明細書の実施例8を参照されたい)、hKv1.4、hKv1.6、もしくはhKv1.7で安定的にトランスフェクトした;HEK293細胞はヒトKV1.3もしくはヒトKv1.1を安定的に発現していた。細胞系はAmgenまたはBioFocus DPI(A Galapagos Company)から得た。hKv1.2を安定的に発現しているCHO K1細胞は、カウンタースクリーンのため、Millipore(Cat#.CYL3015)から購入した。
1.ベースライン電流は安定していなければならない
2.初回ピーク電流は300pA超でなければならない
3.初回Rmおよび最終Rmは300Ohm超でなければならない
4.ピーク電流は第一化合物の添加前に定常状態に達さなければならない。
hKv1.3を安定的に発現しているCHO細胞およびhKv1.1を安定的に発現しているHEK293細胞上で電気生理学的手法を行った。IWQ電気生理学的手法のためのShKアナログおよび結合体を含む「アッセイプレート」の調製手順は以下のとおりであった:0.3%BSAを含む細胞外緩衝液(0.9mM Ca2+および0.5mM Mg2+含有PBS)ですべてのアナログを溶解し、96ウェルポリプロピレンプレートのH列にカラム1〜カラム10に濃度100nMで分配した。カラム11および12を陰性および陽性対照のために保留してから、1:3比で連続希釈してA列で分配した。IonWorks Quattro(IWQ)電気生理学的手法およびデータ分析を以下のとおり成し遂げた:再懸濁した細胞(細胞外緩衝液中)、アッセイプレート、Population Patch Clamp(PPC)パッチプレートならびに適切な細胞内(90mMグルコン酸カリウム、20mM KF、2mM NaCl、1mM MgCl2、10mM EGTA、10mM HEPES、pH7.35)および細胞外緩衝液をIonWorks Quattro上に位置した。アナログをパッチプレートに添加した場合、アッセイプレートから0.1%BSAでさらに3倍希釈して33.3nM〜15pMの範囲の最終試験濃度に達した。アンホテリシンベースの穿孔パッチクランプ手順を用いてCHO−Kv1.3およびHEK−Kv1.1細胞の電気生理記録を取った。IonWorks Quattroの電圧クランプ回路構成を用いて、細胞を膜電位−80mVで保持し、膜電位を400ミリ秒間+30mVに進めることにより電圧活性化K+電流を引き起こした。対照条件下すなわち実験開始時に阻害剤の非存在下、10分インキュベーション後にアナログおよび対照の存在下でK+電流を引き起こした。430〜440ミリ秒の平均K+電流振幅を測定し、データをMicrosoft社のエクセルシートにエクスポートした。各濃度のアナログおよび対照の存在下におけるK+電流振幅を同じウェル中の前化合物の電流振幅のK+電流率として表した。これらの対照値%を濃度関数としてプロット時、各化合物のIC50値は以下の方程式を用いるエクセル適合プログラムにおいて用量反応適合モデル201を用いて算出し得る:
IL−2およびIFN−g分泌に与える毒素ペプチドアナログKv1.3阻害剤の影響を評価するためのEx vivoアッセイ。既に記載されているエクスビボアッセイ(その全体が参照により本明細書に組み込まれるWO2008/088422A2号の実施例46を参照されたい)を用いて、ShKアナログおよび結合体がヒト全血中のT細胞炎症をブロックする効能を評価した。簡単に述べると、50%ヒト全血液をタプシガルジンで刺激してストア枯渇、カルシウム可動化およびサイトカイン分泌を誘発する。T細胞サイトカイン分泌のブロックにおける分子の効能を評価するため、タプシガルジン刺激を加える30〜60分前に各種濃度のKv1.3ブロックペプチドおよびペプチド結合体をヒト全血液試料でプレインキュベートした。48時間後、37℃、5%CO2で、条件培地を収集し、MesoScale Discoveryから4点電気化学発光免疫アッセイを用いてサイトカイン分泌レベルを決定した。タプシガルジン刺激を用いて、複数ドナーから単離した血液からロバスト的にサイトカインIL−2およびIFN−gを分泌した。タプシガルジン刺激後にヒト全血中に産生されたIL−2およびIFN−gを、細胞内サイトカイン染色および蛍光標示式細胞分取器(FACS)分析により明らかにしたようにT細胞から産生した。
Kv1.1、Kv1.2、Kv1.3、Kv1.4、Kv1.6およびKv1.7、PatchXpress(商標)、平面パッチクランプ電気生理学的手法。上記の実施例6に記載の方法および細胞を用いるMDCのPatchXpress(商標)7000A電気生理系を用いて、イオンチャネル電流を室温で記録できる。各チャネルの電圧プロトコールを下表7Mに示す。
AMP5 TPO模倣ペプチドを全4つの可能な末端融合配置(図1F〜1K;図45に図解)で本発明の抗KLH抗体に遺伝的に融合した、すなわち、両免疫グロブリン軽鎖単量体および両免疫グロブリン重鎖単量体にN末端融合およびC末端融合し、哺乳類(CHO)細胞内で発現させた。次いで融合物をタンパク質Aクロマトグラフィー(GE Life Sciences)により2価陽イオンを含まないダルベッコのPBS 10カラム容量を洗浄緩衝液として、および100mM酢酸を溶出緩衝液として7℃で用いて精製した。クロマトグラムに基づき溶出ピークをプールして、2Mトリス塩基を用いてpHを約5.0に増大した。次いでプールを少なくとも4容量の水で希釈してからSP−HPセファロースカラム(GE Life Sciences)上に負荷し、10カラム容量のS緩衝液A(20mM酢酸、pH5.0)で洗浄した後、60%S緩衝液B勾配(20mM酢酸、1M NaCl、pH5.0)の20カラム容量を用いて7℃で溶出した。クロマトグラムに基づきプールを作製し、10kDa Slide−A−Lyzers(Pierce)を4℃で用いて物質を20容量超の10mM酢酸、9%ショ糖(pH5.0)に対して透析した。次いで透析物質を0.22μm酢酸セルロースフィルターを介してろ過し、吸光度280nmで濃度を決定した。各抗体50μgを融合していない対照と共にPhenomenex SEC 3000カラム(7.8×300mm)の50mM NaH2PO4、pH6.5、250mM NaCl溶液に注入し、1mL/分で展開して吸光度280nmで観察した(図39)。各抗体の分析を、220Vで展開した1.0mmトリス−グリシン4〜20%SDS−PAGE(Novex)を用いて、還元および非還元負荷緩衝液を用いて、QuickBlue(Boston Biologicals)により染色して行い(図40A〜E)、LC−MSにより質量を決定した(図41A〜D)。
(a)aKLH120.6κLC(上記の配列番号28);
(b)aKLH120.6IgG2 HC(上記の配列番号29);
(c)aKLH120.6IgG1 HC(上記の配列番号34);
(c)以下のアミノ酸配列を有するaKLH120.6IgG2 HC−Amp5:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGGQGCSSGGPTLREWQQCRRAQHS//(配列番号324);
(d)以下のアミノ酸配列を有するAmp5−aKLH120.6IgG2 HC(配列番号332):
MDMRVPAQLLGLLLLWLRGARCQGCSSGGPTLREWQQCRRAQHSGGGGGQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYQSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG//(配列番号332)。
(e)以下のアミノ酸配列を有するaKLH120.6hIgG1 N297Q−Amp5 Fc(CH3)ループ:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYQSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMGGQGCSSGGPTLREWQQCRRAQHSGGTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG//(配列番号341);
(f)以下のアミノ酸配列を有するAmp5−aKLH120.6κLCポリペプチド融合物:
MDMRVPAQLLGLLLLWLRGARCQGCSSGGPTLREWQQCRRAQHSGGGGGDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC//(配列番号342)。
AAG CTC GAG GTC GAC TAG ACC ACC ATG GAC ATG AGG GTG CCC GCT CAG CTC CTG GGG CT//(配列番号325)であり;および
逆行プライマー配列は:
GCC GCT GCT GCA GCC CTG ACC ACC ACC TCC ACC ACC CGG AGA CAG GGA GAG//(配列番号326)であった。
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGGQGC//(配列番号327)であった。
CTC TCC CTG TCT CCG GGT GGT GGA GGT GGT GGT CAG GGC TGC AGC AGC GGC//(配列番号328)であり;および
逆行プライマー配列は:
CTA CTA GCG GCC GCT CAG CTA TGC TGA GCG CGG CG//(配列番号329)であった。PCR2から産生した断片によりコードされたアミノ酸配列は:
LSLSPGGGGGGQGCSSGGPTLREWQQCRRAQHS//(配列番号330)であった。
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGGQGCSSGGPTLREWQQCRRAQHS//(配列番号331)。
単量体を含む所望のAMP5−aKLH120.6IgG2 DesK HC生成物(上記の配列番号332)は、1本の重鎖のN末端に融合するAMP5ペプチドを有する完全抗体であり、図1Iの図解を企図し、PFU High Fidelity Ultra(Stratagene製)を用いてPCRの2つの別々のラウンドにより会合した。第一ラウンドのPCRにより3つの断片:VK1sp−AMP5、AMP5−G5、およびG5−aKLH120.6IgG2 DesK HC断片が産生された。これらの断片を産生するために用いたオリゴおよびPCR鋳型を以下に列挙する。ポリメラーゼ連鎖反応1(PCR1)によりVK1sp−AMP5が産生され、VK1spをコードする既存のDNAを鋳型として用いた。この断片も、VK1sp−AMP5−G5−aKLH120.6κ LC構築において用いた点に留意されたい。
AAG CTC GAG GTC GAC TAG ACC ACC ATG GAC ATG AGG GTG CCC GCT CAG CTC CTG GGG CT//(配列番号325)であり;および
逆行プライマー配列は:
GCC GCT GCT GCA GCC CTG ACA TCT GGC ACC TCT CAA CC//(配列番号333)であった。PCR1から産生した断片によりコードされたアミノ酸配列は:
MDMRVPAQLLGLLLLWLRGARCQGCSSG//(配列番号334)であった。
GGT TGA GAG GTG CCA GAT GTC AGG GCT GCA GCA GCG GC//(配列番号335)であり;および
逆行プライマー配列は:
CAG CTG CAC CTG ACC ACC ACC TCC ACC GCT ATG CTG AGC GCG//(配列番号336)であった。
WLRGARCQGCSSGGPTLREWQQCRRAQHSGGGGGQVQLV//(配列番号337)であった。
CGC GCT CAG CAT AGC GGT GGA GGT GGT GGT CAG GTG CAG CTG//(配列番号338)であり;および
逆行プライマー配列は:
CTA CTA GCG GCC GCT CAA CCC GGA GAC AGG GAG A//(配列番号339)であった。
RAQHSGGGGGQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYQSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG//(配列番号340)であった。
WLRGARCQGCSSGGPTLREWQQCRRAQHSGGGGGDIQMTQ//(配列番号345)であった。
CGC GCT CAG CAT AGC GGT GGA GGT GGT GGT GAC ATC CAG ATG ACC CAG//配列番号346)であり;および
逆行プライマー配列は:
AAC CGT TTA AAC GCG GCC GCT CAA CAC TCT CCC CTG TTG AA//(配列番号347)であった。PCR3から産生した断片のペプチド配列は:
RAQHSGGGGGDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC//(配列番号348)であった。
エキセンディン−4ペプチド(HGEGTFTSDL SKQMEEEAVR LFIEWLKNGG PSSGAPPPS//配列番号349)を1kGリンカーを介して抗KLH120.6抗体の軽鎖のN末端に遺伝的に融合し(「Ex−4−1kG−aKLH120.6−Ab」と命名)、哺乳類細胞内で発現させた。図42は、エキセンディン−4(「Ex4」)−1kG−aKLH120.6LC融合構築物の図式マップである。
(a)以下のアミノ酸配列を有するEx−4−1kG−aKLH120.6κLC:
MDMRVPAQLLGLLLLWLRGARCHGEGTFTSDL SKQMEEEAVR LFIEWLKNGG PSSGAPPPSG SGSATGGSGSGASSGSGSAT GSDIQMTQSP SSLSASVGDR VTITCRASQG IRNDLGWYQQKPGKAPKRLI YAASSLQSGV PSRFSGSGSG TEFTLTISSL QPEDFATYYCLQHNSYPLTF GGGTKVEIKR TVAAPSVFIF PPSDEQLKSG TASVVCLLNNFYPREAKVQW KVDNALQSGN SQESVTEQDS KDSTYSLSST LTLSKADYEKHKVYACEVTH QGLSSPVTKS FNRGEC//(配列番号355);および
(b)aKLH120.6IgG2 HC(上記の配列番号29)。
SalI
〜〜〜〜〜〜
GTCGACTAGACCACCATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTCCTGCTATTGTGGTTGAGAGGTGCCAGATGTCATGGGGAGGGAACATTTACAAGCGATCTGAGCAAACAAATGGAGGAAGAGGCAGTTAGACTGTTCATTGAATGGCTCAAGAACGGCGGACCGAGTAGTGGTGCTCCGCCTCCCAGCGGATCTGGCAGCGCTACTGGTG
GATCTGGATCGGGTGCATCCTCTGGATCTGGAAGCGCTACCGGATCC//(配列番号351)
〜〜〜〜〜〜
BamHI
AAT GGA TCC GAC ATC CAG ATG ACC CAG TC/(配列番号352);およびAAT GCG GCC GCT CAA CAC TCT CC//(配列番号353)と上記(pTT5−aKLH120.6−VK1SP−κ軽鎖(LC)構築物)のaKLH120.6−Ab LC DNA鋳型から増幅した。
BamHI
〜〜〜〜〜〜
GGATCCGACATCCAGATGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGAAATGATTTAGGCTGGTATCAGCAGAAACCAGGGAAAGCCCCTAAACGCCTGATCTATGCTGCATCCAGTTTGCAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGAATTCACTCTCACAATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCTACAGCATAATAGTTACCCGCTCACTTTCGGCGGAGGGACCAAGGTGGAGATCAAACGAACTGTGGCTGCACCATCTGTCTTCATCTTCCCGCCATCTGATGAGCAGTTGAAATCTGGAACTGCCTCTGTTGTGTGCCTGCTGAATAACTTCTATCCCAGAGAGGCCAAAGTACAGTGGAAGGTGGATAACGCCCTCCAATCGGGTAACTCCCAGGAGAGTGTCACAGAGCAGGACAGCAAGGACAGCACCTACAGCCTCAGCAGCACCCTGACGCTGAGCAAAGCAGACTACGAGAAACACAAAGTCTACGCCTGCGAAGTCACCCATCAGGGCCTGAGCTCGCCCGTCACAAAGAGCTTCAACAGGGGAGAGTGTTGAGCGGCCGC//(配列番号368)
〜〜〜〜〜〜
NotI
SalI
〜〜〜〜〜〜
GTCGACTAGACCACCATGGACATGAGGGTCCCCGCTCAGCTCCTGGGGCTCCTGCTATTGTGGTTGAGAGGTGCCAGATGTCATGGGGAGGGAACATTTACAAGCGATCTGAGCAAACAAATGGAGGAAGAGGCAGTTAGACTGTTCATTGAATGGCTCAAGAACGGCGGACCGAGTAGTGGTGCTCCGCCTCCCAGCGGATCTGGCAGCGCTACTGGTG GATCTGGATCGGGTGCATCCTCTGGATCTGGAAGCGCTACCGGATCCGACATCCAGATGACCCAGTCTCCATCCTCCCTGTCTGCATCTGTAGGAGACAGAGTCACCATCACTTGCCGGGCAAGTCAGGGCATTAGAAATGATTTAGGCTGGTATCAGCAGAAACCAGGGAAAGCCCCTAAACGCCTGATCTATGCTGCATCCAGTTTGCAAAGTGGGGTCCCATCAAGGTTCAGCGGCAGTGGATCTGGGACAGAATTCACTCTCACAATCAGCAGCCTGCAGCCTGAAGATTTTGCAACTTATTACTGTCTACAGCATAATAGTTACCCGCTCACTTTCGGCGGAGGGACCAAGGTGGAGATCAAACGAACTGTGGCTGCACCATCTGTCTTCATCTTCCCGCCATCTGATGAGCAGTTGAAATCTGGAACTGCCTCTGTTGTGTGCCTGCTGAATAACTTCTATCCCAGAGAGGCCAAAGTACAGTGGAAGGTGGATAACGCCCTCCAATCGGGTAACTCCCAGGAGAGTGTCACAGAGCAGGACAGCAAGGACAGCACCTACAGCCTCAGCAGCACCCTGACGCTGAGCAAAGCAGACTACGAGAAACACAAAGTCTACGCCTGCGAAGTCACCCATCAGGGCCTGAGCTCGCCCGTCACAAAGAGCTTCAACAGGGGAGAGTGTTGAGCGGCCGC//(配列番号354)
〜〜〜〜〜〜
NotI
C681ポリペプチドは、いわゆるアビマー構造のIL−6結合ポリペプチドである。(例えば、Kolkman et al., Novel Proteins with Targeted Binding、米国特許第2005/0089932号;Baker et al., IL−6 Binding Proteins、米国特許第2008/0281076号;Stemmer et al., Protein Scaffolds and Uses Thereof、米国特許第2006/0223114号および米国特許第2006/0234299号を参照されたい)。
(a)aKLH120.6κLC(配列番号28);および
(b)以下のアミノ酸配列を有する(VK−1SP)−C681−(G5)−aKLH120.6IgG2 HC融合物:
MDMRVPAQLLGLLLLWLRGARCSGGSCLPDQFRCGNGQCIPLDWVCDGVNDCPDDSDEEGCPPRTCAPSQFQCGSGYCISQRWVCDGENDCEDGSDEANCAGSVPTCPSDEFRCRNGRCIPRAWRCDGVNDCADNSDEEDCTEHTGGGGGQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG//(配列番号356)。
MDMRVPAQLLGLLLLWLRGARCSGGSCLPDQFRCGNGQCIPLDWVCDGVNDCPDDSDEEGCPPRTCAPSQFQCGSGYCISQRWVCDGENDCEDGSDEANCAGSVPTCPSDEFRCRNGRCIPRAWRCDGVNDCADNSDEEDCTEHTGGGGGQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTK//(配列番号350)。
(a)aKLH120.6κLC(配列番号28);および
(b)以下のアミノ酸配列を有するaKLH120.6IgG2 HC−C681融合物:
MDMRVPAQLLGLLLLWLRGARCQVQLVQSGAEVKKPGASVKVSCKASGYTFTGYHMHWVRQAPGQGLEWMGWINPNSGGTNYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARDRGSYYWFDPWGQGTLVTVSSASTKGPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGGSGGSCLPDQFRCGNGQCIPLDWVCDGVNDCPDDSDEEGCPPRTCAPSQFQCGSGYCISQRWVCDGENDCEDGSDEANCAGSVPTCPSDEFRCRNGRCIPRAWRCDGVNDCADNSDEEDCTEHT//(配列番号357)。
MTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGGGGGGSGGSCLPDQFRCGNGQCIPLDWVCDGVNDCPDDSDEEGCPPRTCAPSQFQCGSGYCISQRWVCDGENDCEDGSDEANCAGSVPTCPSDEFRCRNGRCIPRAWRCDGVNDCADNSDEEDCTEHT//(配列番号358)。
(a)aKLH120.6IgG2 HC(配列番号29);および
(b)以下のアミノ酸配列を有するC681−aKLH120.6κLC融合物:
MDMRVPAQLLGLLLLWLRGARCSGGSCLPDQFRCGNGQCIPLDWVCDGVNDCPDDSDEEGCPPRTCAPSQFQCGSGYCISQRWVCDGENDCEDGSDEANCAGSVPTCPSDEFRCRNGRCIPRAWRCDGVNDCADNSDEEDCTEHTGGGGGDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC//(配列番号359)。
MDMRVPAQLLGLLLLWLRGARCSGGSCLPDQFRCGNGQCIPLDWVCDGVNDCPDDSDEEGCPPRTCAPSQFQCGSGYCISQRWVCDGENDCEDGSDEANCAGSVPTCPSDEFRCRNGRCIPRAWRCDGVNDCADNSDEEDCTEHTGGGGGDIQMTQSPSSLSASVGDRVTITCRASQGIRNDLGWYQQKPGKAPKRLIYAASSLQSGVPSRFSGSGSGTEFTLTISSLQPEDFATYYCLQHNSYPLTFGGGTKVEIKRTVA//(配列番号360)
タンパク質Aセファロース(GE Healthcare)を用いたFc領域の親和性高速タンパク質液体クロマトグラフィー(FPLC)捕獲、続いてトリス緩衝生理食塩水、1mM CaCl2(Teknova)によるカラム洗浄および100mM酢酸、1mM CaCl2(pH3.5)を2.5cm/分で流した溶出工程により条件培地の初回精製を行った。タンパク質を含む留分をプールし、10N NaOHを用いてpHを8.0に調整し、5容量の水でさらに希釈した。物質を0.45μm酢酸セルロースフィルター(Corning)を介してろ過し、陽イオン交換FPLC(SP Sepharose High Performance; GE Healthcare)によりさらに精製した。試料を100%緩衝液A(20mMトリス、1mM、pH8.0)で平衡させたカラム上に負荷し、勾配0〜80%緩衝液B(20mMトリス、1M NaCl、1mM CaCl2、pH8.0)30カラム容量、流速1.5cm/分で溶出した。ピークを含む標的種をプールして、10mMトリス、150mM NaCl、1mM CaCl2(pH8.0)中に製剤化した。N末端HCおよびN末端LCならびにC末端HC融合タンパク質の精製例を図36〜38に示す。非還元SDS−PAGE分析(図36)により、完全に組み立てられた抗体が形成され得ることが示され、還元SDS−PAGE分析により所望の成分が存在することが示される。サイズ排除クロマトグラム(図37)により、精製した生成物の大部分が所望の非凝集状態にあることが示される。最終的に、質量スペクトル分析(図38)により、所望の融合生成物が存在することが示される。まとめると、これらの例により、aKLH120.6抗体は、所望の生成物を形成するアビマーとの融合物を受容できることが示される。
物質。ハイブリドーマ(3A1、3C2、3A4および3B1)または組換えCHO(3A4−F−G2および3B1−G2)のいずれか由来の精製した抗DNP抗体の発現を試験した。抗ヒトIgG、Fcγ−特異性抗体はJackson ImmunoResearch Laboratories, Inc. (West Grove, PA)から得た。DNP−BSA(ウシ血清アルブミンに結合される2,4−ジニトロフェノール)はBiosearch Technologies, Inc.(Novato, CA)から得た。BIACore 2000, research gradeセンサーチップCM5、界面活性剤P−20(ポリオキシエチレンソルビタン)、HBS−EP(10mM HEPES、0.15M NaCl、3.4mM EDTA、0.005%P−20、pH7.4)、アミンカップリング試薬、10mM酢酸塩(pH4.0)および10mMグリシン(pH1.5)はBIACore, Inc.(Piscataway, NJ)から得た。リン酸緩衝生理食塩水(PBS、1×、塩化カルシウム非含有、塩化マグネシウム非含有)はInvitrogen(Carlsbad, CA)から得た。ウシ血清アルブミン(BSA、分留V、IgG非含有)はSigma (St. Louis, MO)から得た。
本明細書を通して使用する略語は、特定の状況で別段の指定のない限り、以下のとおり定義する。
Ac アセチル(以前はアセチル化残基を指していた)
AcBpa アセチル化p−ベンジル−L−フェニルアラニン
ACN アセトニトリル
AcOH 酢酸
ADCC 抗体依存性細胞傷害
Aib アミノイソ酪酸
bA β−アラニン
Bpa p−ベンゾイル−L−フェニルアラニン
BrAc ブロモアセチル(BrCH2C(O)
BSA ウシ血清アルブミン
Bzl ベンジル
Cap カプロン酸
CBC 全血球計算値
COPD 慢性閉塞性肺疾患
CTL 細胞傷害性Tリンパ球
DCC ジシクロヘキシルカルボジイミド
Dde 1−(4,4−ジメチル−2,6−ジオキソ−シクロヘキシリデン)エチル
DNP 2,4−ジニトロフェノール
DOPC 1,2−ジオレオイル−sn−グリセロ−3−ホスホコリン
DOPE 1,2−ジオレオイル−sn−グリセロ−3−ホスホエタノールアミン
DPPC 1,2−ジパルミトイル−sn−グリセロ−3−ホスホコリン
DSPC 1,2−ジステアロイル−sn−グリセロ−3−ホスホコリン
DTT ジチオスレイトール
EAE 実験的自己免疫性脳脊髄炎
ECL 電気化学発光
ESI−MS エレクトロスプレーイオン化質量分析
FACS 蛍光標示式細胞分取器
Fmoc フルオレニルメトキシカルボニル
HOBt 1−ヒドロキシベンゾトリアゾール
HPLC 高速液体クロマトグラフィー
HSL ホモセリンラクトン
IB 封入体
KCa カルシウム活性化カリウムチャネル(IKCa、BKCa、SKCa含有)
KLH キーホールリンペットヘモシアニン
Kv 電位依存性カリウムチャネル
Lau ラウリン酸
LPS リポ多糖類
LYMPH リンパ液
MALDI−MS マトリックス支援レーザー脱離イオン化法質量分析
Me メチル
MeO メトキシ
MeOH メタノール
MHC 主要組織適合複合体
MMP マトリックスメタロプロテイナーゼ
MW 分子量
MWCO 分子量カットオフ
1−Nap 1−ナフチルアラニン
NEUT 好中球
Nle ノルロイシン
NMP N−メチル−2−ピロリジノン
OAc 酢酸塩
PAGE ポリアクリルアミドゲル電気泳動
PBMC 末梢血液単核細胞細胞
PBS リン酸緩衝生理食塩水
Pbf 2,2,4,6,7−ペンダメチルジヒドロベンゾフラン−5−スルホニル
PCR ポリメラーゼ連鎖反応
PD 薬力学
Pec ピペコリン酸
PEG ポリ(エチレングリコール)
pGlu ピログルタミン酸
Pic ピコリン酸
PK 薬物動態
pY ホスホチロシン
RBS リボソーム結合部位
RT 室温(約25℃)
Sar サルコシン
SDS ドデシル硫酸ナトリウム
STK セリン−トレオニンキナーゼ
t−Boc tert−ブトキシカルボニル
tBu tert−ブチル
TCR T細胞受容体
TFA トリフルオロ酢酸
THF 胸腺液性因子
Trt トリチル
Claims (60)
- 免疫グロブリン重鎖可変領域および免疫グロブリン軽鎖可変領域を含み、前記重鎖可変領域が配列番号250、配列番号252、配列番号254、配列番号256、配列番号258、もしくは配列番号260の配列と少なくとも95%同一であるアミノ酸配列を含む、単離抗原結合タンパク質。
- 軽鎖可変領域が配列番号232、配列番号234、配列番号236、配列番号238、もしくは配列番号240のアミノ酸配列を含む、請求項1の単離抗原結合タンパク質。
- 重鎖可変領域がCDRH1、CDRH2およびCDRH3と命名された3つの相補性決定領域を含み:
(a)CDRH1は配列番号188、配列番号189、配列番号190、もしくは配列番号191のアミノ酸配列を含む;
(b)CDRH2は配列番号192、配列番号193、配列番号194、もしくは配列番号195のアミノ酸配列を含む;ならびに
(c)CDRH3は配列番号196、配列番号197、配列番号198、配列番号199、配列番号200、もしくは配列番号201のアミノ酸配列を含む、請求項1の単離抗原結合タンパク質。 - 免疫グロブリン軽鎖可変領域を含み、前記軽鎖可変領域がCDRLl、CDRL2およびCDRL3と命名された3つのCDRを含み:
(a)CDRL1は配列番号202、配列番号203、配列番号204、もしくは配列番号205のアミノ酸配列を含む;
(b)CDRL2は配列番号206もしくは配列番号207のアミノ酸配列を含む;ならびに
(c)CDRL3は配列番号208、配列番号209、配列番号210、配列番号211、もしくは配列番号212のアミノ酸配列を含む、請求項1の単離抗原結合タンパク質。 - 重鎖可変領域がCDRH1、CDRH2およびCDRH3と命名された3つの相補性決定領域を含み、ならびに軽鎖可変領域がCDRLl、CDRL2およびCDRL3と命名された3つのCDRを含み:
(a)CDRH1は配列番号188、配列番号189、配列番号190、もしくは配列番号191のアミノ酸配列を含む;
(b)CDRH2は配列番号192、配列番号193、配列番号194、もしくは配列番号195のアミノ酸配列を含む;
(c)CDRH3は配列番号196、配列番号197、配列番号198、配列番号199、配列番号200、もしくは配列番号201のアミノ酸配列を含む;
(d)CDRL1は配列番号202、配列番号203、配列番号204、もしくは配列番号205のアミノ酸配列を含む;
(e)CDRL2は配列番号206もしくは配列番号207のアミノ酸配列を含む;ならびに
(f)CDRL3は配列番号208、配列番号209、配列番号210、配列番号211、もしくは配列番号212のアミノ酸配列を含む、請求項1の単離抗原結合タンパク質。 - 抗体または抗体断片を含む、請求項1の単離抗原結合タンパク質。
- IgG1、IgG2、IgG3またはIgG4を含む請求項6の単離抗原結合タンパク質。
- モノクローナル抗体を含む請求項6の単離抗原結合タンパク質。
- キメラまたはヒト化抗体を含む請求項8の単離抗原結合タンパク質。
- ヒト抗体を含む請求項8の単離抗原結合タンパク質。
- (a)配列番号77、配列番号107、配列番号111、配列番号113、配列番号115、配列番号117、配列番号119、配列番号123、配列番号129、配列番号144、配列番号145、配列番号181、配列番号182、配列番号183、配列番号184、もしくは配列番号185のアミノ酸配列を含むか、またはN末端かC末端、もしくは両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン重鎖;
(b)配列番号105、配列番号109、配列番号121;配列番号125、もしくは配列番号127のアミノ酸配列を含むか、またはN末端かC末端、もしくは両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン軽鎖;または
(c)(a)の免疫グロブリン重鎖および(b)の免疫グロブリン軽鎖
を含む請求項6の単離抗原結合タンパク質。 - 1から24の、結合された薬理学的に活性な化学部分をさらに含む、請求項1乃至請求項11のうちのいずれかの単離抗原結合タンパク質。
- 薬理学的に活性な化学部分が薬理学的に活性なポリペプチドである、請求項12の単離抗原結合タンパク質。
- 抗原結合ペプチドが組換え産生される、請求項13の単離抗原結合タンパク質。
- 抗原結合タンパク質が少なくとも1本の免疫グロブリン重鎖および少なくとも1本の免疫グロブリン軽鎖を含み、ならびに前記免疫グロブリン重鎖の一次アミノ酸配列において免疫グロブリン重鎖Fcドメイン内部ループ内に薬理学的に活性なポリペプチドが挿入されている、請求項14の単離抗原結合タンパク質。
- 抗原結合タンパク質が少なくとも1本の免疫グロブリン重鎖および少なくとも1本の免疫グロブリン軽鎖を含み、ならびに前記免疫グロブリン重鎖のN末端またはC末端に薬理学的に活性なポリペプチドが結合されている、請求項13の単離抗原結合タンパク質。
- 抗原結合タンパク質が少なくとも1本の免疫グロブリン重鎖および少なくとも1本の免疫グロブリン軽鎖を含み、ならびに前記免疫グロブリン軽鎖のN末端またはC末端に薬理学的に活性なポリペプチドが結合されている、請求項13の単離抗原結合タンパク質。
- 薬理学的に活性なポリペプチドが毒素ペプチド、IL−6結合ペプチド、CGRPペプチドアンタゴニスト、ブラジキニンB1受容体ペプチドアンタゴニスト、PTHアゴニストペプチド、PTHアンタゴニストペプチド、ang−1結合ペプチド、ang−2結合ペプチド、ミオスタチン結合ペプチド、EPO模倣ペプチド、TPO模倣ペプチド、NGF結合ペプチド、BAFFアンタゴニストペプチド、GLP−1もしくはそのペプチド模倣物、またはGLP−2もしくはそのペプチド模倣物である、請求項13の単離抗原結合タンパク質。
- 毒素ペプチドがShKまたはShKペプチドアナログである、請求項18の単離抗原結合タンパク質。
- 請求項1乃至請求項19のうちのいずれかの抗原結合タンパク質、ならびに医薬上許容可能な希釈液、賦形剤もしくは担体、を含む医薬組成物。
- 請求項1乃至請求項4のうちのいずれかの抗原結合タンパク質をコードする単離核酸。
- 請求項5の抗原結合タンパク質をコードする単離核酸。
- 請求項11の抗原結合タンパク質をコードする単離核酸。
- 請求項14乃至請求項19のうちのいずれかの抗原結合タンパク質をコードする単離核酸。
- 請求項21乃至請求項24のうちのいずれかの単離核酸を含むベクター。
- 発現ベクターを含む請求項25のベクター。
- 請求項26の発現ベクターを含む単離宿主細胞。
- (a)発現ベクターによりコードされた抗原結合タンパク質を発現させる条件下の培養基で請求項27の宿主細胞を培養すること;および
(b)培養基から抗原結合タンパク質を回収すること、
を含む方法。 - 請求項11の抗原結合タンパク質を産生するハイブリドーマ。
- (a)ハイブリドーマにより抗原結合タンパク質を発現させる条件下の培養基で請求項29のハイブリドーマを培養すること;および
(b)培養基から抗原結合タンパク質を回収すること、
を含む方法。 - 免疫グロブリン重鎖可変領域および免疫グロブリン軽鎖可変領域を含み、前記重鎖可変領域が配列番号262、配列番号264、もしくは配列番号266の配列と少なくとも95%同一であるアミノ酸配列を含む、単離抗原結合タンパク質。
- 軽鎖可変領域が配列番号242、配列番号244、配列番号246、もしくは配列番号248のアミノ酸配列を含む、請求項31の単離抗原結合タンパク質。
- 重鎖可変領域がCDRH1、CDRH2およびCDRH3と命名された3つの相補性決定領域を含み:
(a)CDRH1は配列番号213、配列番号214、もしくは配列番号215のアミノ酸配列を含む;
(b)CDRH2は配列番号216、配列番号217、もしくは配列番号218のアミノ酸配列を含む;ならびに
(c)CDRH3は配列番号219、配列番号220、もしくは配列番号221のアミノ酸配列を含む、請求項31の単離抗原結合タンパク質。 - 免疫グロブリン軽鎖可変領域を含み、前記軽鎖可変領域がCDRLl、CDRL2およびCDRL3と命名された3つのCDRを含み:
(a)CDRL1は配列番号204、配列番号222、配列番号223、もしくは配列番号224のアミノ酸配列を含む;
(b)CDRL2は配列番号206、配列番号225、もしくは配列番号226のアミノ酸配列を含む;ならびに
(c)CDRL3は配列番号227、配列番号228、配列番号229、もしくは配列番号230のアミノ酸配列を含む、請求項31の単離抗原結合タンパク質。 - 重鎖可変領域がCDRH1、CDRH2およびCDRH3と命名された3つの相補性決定領域を含み、ならびに軽鎖可変領域がCDRLl、CDRL2およびCDRL3と命名された3つのCDRを含み:
(a)CDRH1は配列番号213、配列番号214、もしくは配列番号215のアミノ酸配列を含む;
(b)CDRH2は配列番号216、配列番号217、もしくは配列番号218のアミノ酸配列を含む;ならびに
(c)CDRH3は配列番号219、配列番号220、もしくは配列番号221のアミノ酸配列を含む;
(d)CDRL1は配列番号204、配列番号222、配列番号223、もしくは配列番号224のアミノ酸配列を含む;
(e)CDRL2は配列番号206、配列番号225、もしくは配列番号226のアミノ酸配列を含む;ならびに
(f)CDRL3は配列番号227、配列番号228、配列番号229、もしくは配列番号230のアミノ酸配列を含む、請求項31の単離抗原結合タンパク質。 - 抗体または抗体断片を含む、請求項31の単離抗原結合タンパク質。
- IgG1、IgG2、IgG3またはIgG4を含む請求項36の単離抗原結合タンパク質。
- モノクローナル抗体を含む請求項36の単離抗原結合タンパク質。
- キメラまたはヒト化抗体を含む請求項38の単離抗原結合タンパク質。
- ヒト抗体を含む請求項38の単離抗原結合タンパク質。
- (a)配列番号46、配列番号133、配列番号139、配列番号143、配列番号186、もしくは配列番号187、配列番号366、もしくは配列番号367のアミノ酸配列を含むか、またはN末端かC末端、もしくは両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン重鎖;
(b)配列番号28、配列番号131、配列番号135、配列番号137;もしくは配列番号141のアミノ酸配列を含むか、またはN末端もしくはC末端、または両端のアミノ酸残基が1個、2個、3個、4個もしくは5個欠けている前述の任意の一配列を含む免疫グロブリン軽鎖;または
(c)(a)の免疫グロブリン重鎖および(b)の免疫グロブリン軽鎖、
を含む請求項36の単離抗原結合タンパク質。 - 1から24の、結合された薬理学的に活性な化学部分をさらに含む、請求項31乃至請求項41のうちのいずれかの単離抗原結合タンパク質。
- 薬理学的に活性な化学部分が薬理学的に活性なポリペプチドである、請求項42の単離抗原結合タンパク質。
- 抗原結合ペプチドが組換え産生される、請求項43の単離抗原結合タンパク質。
- 抗原結合タンパク質が少なくとも1本の免疫グロブリン重鎖および少なくとも1本の免疫グロブリン軽鎖を含み、ならびに前記免疫グロブリン重鎖の一次アミノ酸配列において免疫グロブリン重鎖Fcドメイン内部ループ内に薬理学的に活性なポリペプチドが挿入されている、請求項44の単離抗原結合タンパク質。
- 抗原結合タンパク質が少なくとも1本の免疫グロブリン重鎖および少なくとも1本の免疫グロブリン軽鎖を含み、ならびに前記免疫グロブリン重鎖のN末端またはC末端に薬理学的に活性なポリペプチドが結合されている、請求項43の単離抗原結合タンパク質。
- 抗原結合タンパク質が少なくとも1本の免疫グロブリン重鎖および少なくとも1本の免疫グロブリン軽鎖を含み、ならびに前記免疫グロブリン軽鎖のN末端またはC末端に薬理学的に活性なポリペプチドが結合されている、請求項43の単離抗原結合タンパク質。
- 薬理学的に活性なポリペプチドが毒素ペプチド、IL−6結合ペプチド、CGRPペプチドアンタゴニスト、ブラジキニンB1受容体ペプチドアンタゴニスト、PTHアゴニストペプチド、PTHアンタゴニストペプチド、ang−1結合ペプチド、ang−2結合ペプチド、ミオスタチン結合ペプチド、EPO模倣ペプチド、TPO模倣ペプチド、NGF結合ペプチド、BAFFアンタゴニストペプチド、GLP−1もしくはそのペプチド模倣物、またはGLP−2もしくはそのペプチド模倣物である、請求項43の単離抗原結合タンパク質。
- 毒素ペプチドがShKまたはShKペプチドアナログである、請求項48の単離抗原結合タンパク質。
- 請求項31乃至請求項49のうちのいずれかの抗原結合タンパク質、ならびに医薬上許容可能な希釈液、賦形剤もしくは担体、を含む医薬組成物。
- 請求項31乃至請求項34のうちのいずれかの抗原結合タンパク質をコードする単離核酸。
- 請求項35の抗原結合タンパク質をコードする単離核酸。
- 請求項41の抗原結合タンパク質をコードする単離核酸。
- 請求項44乃至請求項49のうちのいずれかの抗原結合タンパク質をコードする単離核酸。
- 請求項51乃至請求項54のうちのいずれかの単離核酸を含むベクター。
- 発現ベクターを含む請求項55のベクター。
- 請求項56の発現ベクターを含む、単離宿主細胞。
- (a)発現ベクターによりコードされた抗原結合タンパク質を発現させる条件下の培養基で請求項57の宿主細胞を培養すること;および
(b)培養基から抗原結合タンパク質を回収すること、
を含む方法。 - 請求項41の抗原結合タンパク質を産生するハイブリドーマ。
- (a)ハイブリドーマにより抗原結合タンパク質を発現させる条件下の培養基で請求項59のハイブリドーマを培養すること;および
(b)培養基から抗原結合タンパク質を回収すること、
を含む方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21059409P | 2009-03-20 | 2009-03-20 | |
| US61/210,594 | 2009-03-20 | ||
| PCT/US2010/028060 WO2010108153A2 (en) | 2009-03-20 | 2010-03-19 | Carrier immunoglobulins and uses thereof |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015001336A Division JP6173359B2 (ja) | 2009-03-20 | 2015-01-07 | 担体免疫グロブリンおよびその使用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012521197A true JP2012521197A (ja) | 2012-09-13 |
Family
ID=42740266
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012501019A Pending JP2012521197A (ja) | 2009-03-20 | 2010-03-19 | 担体免疫グロブリンおよびその使用 |
| JP2012501020A Pending JP2012521360A (ja) | 2009-03-20 | 2010-03-19 | Kv1.3の選択的かつ強力なペプチド阻害剤 |
| JP2015001336A Active JP6173359B2 (ja) | 2009-03-20 | 2015-01-07 | 担体免疫グロブリンおよびその使用 |
| JP2017077334A Active JP6345832B2 (ja) | 2009-03-20 | 2017-04-10 | 担体免疫グロブリンおよびその使用 |
| JP2018097620A Active JP6609347B2 (ja) | 2009-03-20 | 2018-05-22 | 担体免疫グロブリンおよびその使用 |
Family Applications After (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012501020A Pending JP2012521360A (ja) | 2009-03-20 | 2010-03-19 | Kv1.3の選択的かつ強力なペプチド阻害剤 |
| JP2015001336A Active JP6173359B2 (ja) | 2009-03-20 | 2015-01-07 | 担体免疫グロブリンおよびその使用 |
| JP2017077334A Active JP6345832B2 (ja) | 2009-03-20 | 2017-04-10 | 担体免疫グロブリンおよびその使用 |
| JP2018097620A Active JP6609347B2 (ja) | 2009-03-20 | 2018-05-22 | 担体免疫グロブリンおよびその使用 |
Country Status (7)
| Country | Link |
|---|---|
| US (5) | US8734796B2 (ja) |
| EP (3) | EP3385279B1 (ja) |
| JP (5) | JP2012521197A (ja) |
| AU (2) | AU2010226392B9 (ja) |
| CA (4) | CA2889453C (ja) |
| MX (4) | MX347291B (ja) |
| WO (2) | WO2010108154A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017501728A (ja) * | 2013-12-23 | 2017-01-19 | ザイムワークス インコーポレイティド | C末端軽鎖ポリペプチド伸長を含有する抗体、ならびにその複合体及びその使用方法 |
Families Citing this family (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7833979B2 (en) | 2005-04-22 | 2010-11-16 | Amgen Inc. | Toxin peptide therapeutic agents |
| MX2009012609A (es) | 2007-05-22 | 2009-12-07 | Amgen Inc | Composiciones y metodos para producir proteinas de fusion bioactivas. |
| US8557243B2 (en) | 2008-01-03 | 2013-10-15 | The Scripps Research Institute | EFGR antibodies comprising modular recognition domains |
| US8557242B2 (en) | 2008-01-03 | 2013-10-15 | The Scripps Research Institute | ERBB2 antibodies comprising modular recognition domains |
| GEP20156390B (en) | 2008-01-03 | 2015-11-10 | Scripps Research Inst | Antibody targeting through a modular recognition domain |
| US8574577B2 (en) | 2008-01-03 | 2013-11-05 | The Scripps Research Institute | VEGF antibodies comprising modular recognition domains |
| US8454960B2 (en) | 2008-01-03 | 2013-06-04 | The Scripps Research Institute | Multispecific antibody targeting and multivalency through modular recognition domains |
| CA2889453C (en) | 2009-03-20 | 2018-11-06 | Amgen Inc. | Carrier immunoglobulins and uses thereof |
| AU2010321832B2 (en) | 2009-11-20 | 2014-08-14 | Amgen Inc. | Anti-Orai1 antigen binding proteins and uses thereof |
| US20120100166A1 (en) | 2010-07-15 | 2012-04-26 | Zyngenia, Inc. | Ang-2 Binding Complexes and Uses Thereof |
| KR20130110169A (ko) | 2010-09-22 | 2013-10-08 | 암젠 인크 | 담체 면역글로뷸린 및 이것의 용도 |
| KR20140145947A (ko) | 2011-03-16 | 2014-12-24 | 암젠 인크 | Nav1.3과 nav1.7의 유효한 선별적 저해제 |
| CN102718858B (zh) * | 2011-03-29 | 2014-07-02 | 天津药物研究院 | 胰高血糖素样肽-1类似物单体、二聚体及其制备方法与应用 |
| CA2837169C (en) | 2011-05-24 | 2021-11-09 | Zyngenia, Inc. | Multispecific complexes comprising angiopoietin-2-binding peptide and their uses |
| BR112013031370A2 (pt) * | 2011-06-06 | 2016-11-29 | Kineta One Llc | composições farmacêuticas à base de shk e métodos de fabricação e uso destas |
| WO2012170807A2 (en) | 2011-06-10 | 2012-12-13 | Medimmune, Llc | Anti-pseudomonas psl binding molecules and uses thereof |
| ES2692187T3 (es) | 2011-06-10 | 2018-11-30 | Hanmi Science Co., Ltd. | Nuevos derivados de oxintomodulina y composición farmacéutica para el tratamiento de obesidad que lo comprende |
| KR101577734B1 (ko) | 2011-06-17 | 2015-12-29 | 한미사이언스 주식회사 | 옥신토모듈린과 면역글로불린 단편을 포함하는 결합체 및 그의 용도 |
| SI2776065T1 (sl) | 2011-11-07 | 2020-10-30 | Medimmune Limited | Kombinacijske terapije z uporabo molekul, ki vežejo anti-Pseudomonas Psl in PcrV |
| KR101895047B1 (ko) * | 2011-12-30 | 2018-09-06 | 한미사이언스 주식회사 | 면역글로불린 단편을 이용한 위치 특이적 글루카곤 유사 펩타이드-2 약물 결합체 |
| EP2855520B1 (en) * | 2012-06-04 | 2018-09-26 | Novartis AG | Site-specific labeling methods and molecules produced thereby |
| US9283241B2 (en) * | 2012-07-10 | 2016-03-15 | Clemson University | Treatment to render implants resistant to diabetes |
| KR101968344B1 (ko) | 2012-07-25 | 2019-04-12 | 한미약품 주식회사 | 옥신토모듈린 유도체를 포함하는 고지혈증 치료용 조성물 |
| US10066019B2 (en) | 2012-10-12 | 2018-09-04 | Agency For Science, Technology And Research | Optimised heavy chain and light chain signal peptides for the production of recombinant antibody therapeutics |
| KR101993393B1 (ko) | 2012-11-06 | 2019-10-01 | 한미약품 주식회사 | 옥신토모듈린 유도체를 포함하는 당뇨병 또는 비만성 당뇨병 치료용 조성물 |
| WO2014073842A1 (en) * | 2012-11-06 | 2014-05-15 | Hanmi Pharm. Co., Ltd. | Liquid formulation of protein conjugate comprising the oxyntomodulin and an immunoglobulin fragment |
| HUE039465T2 (hu) * | 2013-01-25 | 2019-01-28 | Janssen Biotech Inc | KV1.3 antagonisták és alkalmazási eljárások |
| AR096927A1 (es) | 2013-03-12 | 2016-02-10 | Amgen Inc | INHIBIDORES POTENTES Y SELECTIVOS DE NaV1.7 |
| TW201446792A (zh) * | 2013-03-12 | 2014-12-16 | Amgen Inc | Nav1.7之強效及選擇性抑制劑 |
| BR112015023752B1 (pt) | 2013-03-15 | 2023-11-14 | Zyngenia, Inc. | Domínio de reconhecimento modular (mrd), complexo compreendendo mrd e cetuximabe, usos do complexo para inibir a angiogênese e tratar câncer e composição farmacêutica compreendendo o dito complexo |
| WO2015061826A1 (en) | 2013-10-28 | 2015-05-07 | Monash University | Novel scorpion toxin analogue and method for treating autoimmune diseases |
| EP3152238A4 (en) * | 2014-06-06 | 2018-01-03 | The California Institute for Biomedical Research | Methods of constructing amino terminal immunoglobulin fusion proteins and compositions thereof |
| WO2015191781A2 (en) | 2014-06-10 | 2015-12-17 | Amgen Inc. | Apelin polypeptides |
| US9710451B2 (en) * | 2014-06-30 | 2017-07-18 | International Business Machines Corporation | Natural-language processing based on DNA computing |
| WO2016023072A1 (en) * | 2014-08-15 | 2016-02-18 | Monash University | Novel potassium channel blockers and use thereof in the treatment of autoimmune diseases |
| TWI802396B (zh) | 2014-09-16 | 2023-05-11 | 南韓商韓美藥品股份有限公司 | 長效glp-1/高血糖素受體雙促效劑治療非酒精性脂肝疾病之用途 |
| US10035823B2 (en) | 2014-09-29 | 2018-07-31 | The Regents Of The University Of California | Compositions for expanding regulatory T cells (TREG), and treating autoimmune and inflammatory diseases and conditions |
| KR102418477B1 (ko) | 2014-12-30 | 2022-07-08 | 한미약품 주식회사 | 글루카곤 유도체 |
| WO2016112208A2 (en) * | 2015-01-09 | 2016-07-14 | Kineta One, Llp | Topical applications of kv1.3 channel blocking peptides to treat skin inflammation |
| WO2016187356A1 (en) | 2015-05-18 | 2016-11-24 | Agensys, Inc. | Antibodies that bind to axl proteins |
| US10787516B2 (en) | 2015-05-18 | 2020-09-29 | Agensys, Inc. | Antibodies that bind to AXL proteins |
| WO2017000059A1 (en) * | 2015-06-29 | 2017-01-05 | The University Of British Columbia | B1sp fusion protein therapeutics, methods, and uses |
| PE20181322A1 (es) * | 2015-09-01 | 2018-08-14 | Agenus Inc | Anticuerpo anti-pd1 y sus metodos de uso |
| TW201718629A (zh) * | 2015-09-25 | 2017-06-01 | 韓美藥品股份有限公司 | 包含多個生理多肽及免疫球蛋白Fc區之蛋白質接合物 |
| US10388404B2 (en) | 2015-10-27 | 2019-08-20 | International Business Machines Corporation | Using machine-learning to perform linear regression on a DNA-computing platform |
| WO2017083604A1 (en) | 2015-11-12 | 2017-05-18 | Amgen Inc. | Triazine mediated pharmacokinetic enhancement of therapeutics |
| US11161891B2 (en) | 2015-12-09 | 2021-11-02 | The Scripps Research Institute | Relaxin immunoglobulin fusion proteins and methods of use |
| JOP20170013B1 (ar) | 2016-01-22 | 2021-08-17 | Merck Sharp & Dohme | أجسام xi مضادة لعامل مضاد للتجلط |
| WO2017184619A2 (en) | 2016-04-18 | 2017-10-26 | Celldex Therapeutics, Inc. | Agonistic antibodies that bind human cd40 and uses thereof |
| IL315266A (en) | 2016-06-14 | 2024-10-01 | Merck Sharp ַ& Dohme Llc | Antibodies to coagulation factor XI |
| WO2018013483A1 (en) * | 2016-07-11 | 2018-01-18 | The California Institute For Biomedical Research | Kv1.3 channel blocking peptides and uses thereof |
| BR112019011582A2 (pt) | 2016-12-07 | 2019-10-22 | Agenus Inc. | anticorpos e métodos de utilização dos mesmos |
| WO2018135598A1 (ja) * | 2017-01-19 | 2018-07-26 | 国立大学法人 東京大学 | 新規蛍光標識方法 |
| JP2022527812A (ja) | 2019-04-01 | 2022-06-06 | ノヴォ ノルディスク アー/エス | リラグルチドに対する抗体およびその使用 |
| US11028132B1 (en) | 2020-04-07 | 2021-06-08 | Yitzhak Rosen | Half-life optimized linker composition |
| JP7458085B2 (ja) * | 2021-07-28 | 2024-03-29 | 株式会社三協 | 錠剤用崩壊剤及びこれを利用した錠剤 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002522090A (ja) * | 1998-08-17 | 2002-07-23 | トーマス ジェファーソン ユニヴァーシティー | 抗体エンベロープ融合タンパク質および野生型エンベロープタンパク質を含有するレトロウイルスベクターを用いる細胞型特異的遺伝子移入 |
| JP2008501302A (ja) * | 2003-10-16 | 2008-01-24 | イムクローン システムズ インコーポレイティド | 繊維芽細胞増殖因子レセプター−1阻害物質及びその治療方法 |
| WO2008088422A2 (en) * | 2006-10-25 | 2008-07-24 | Amgen Inc. | Toxin peptide therapeutic agents |
Family Cites Families (135)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US30985A (en) | 1860-12-18 | Thomas l | ||
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| US3941763A (en) | 1975-03-28 | 1976-03-02 | American Home Products Corporation | PGlu-D-Met-Trp-Ser-Tyr-D-Ala-Leu-Arg-Pro-Gly-NH2 and intermediates |
| JPS6023084B2 (ja) | 1979-07-11 | 1985-06-05 | 味の素株式会社 | 代用血液 |
| US4640835A (en) | 1981-10-30 | 1987-02-03 | Nippon Chemiphar Company, Ltd. | Plasminogen activator derivatives |
| JPS5896026A (ja) | 1981-10-30 | 1983-06-07 | Nippon Chemiphar Co Ltd | 新規ウロキナ−ゼ誘導体およびその製造法ならびにそれを含有する血栓溶解剤 |
| DE3380726D1 (en) | 1982-06-24 | 1989-11-23 | Japan Chem Res | Long-acting composition |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4496689A (en) | 1983-12-27 | 1985-01-29 | Miles Laboratories, Inc. | Covalently attached complex of alpha-1-proteinase inhibitor with a water soluble polymer |
| DE3675588D1 (de) | 1985-06-19 | 1990-12-20 | Ajinomoto Kk | Haemoglobin, das an ein poly(alkenylenoxid) gebunden ist. |
| US4766106A (en) | 1985-06-26 | 1988-08-23 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| GB8516415D0 (en) | 1985-06-28 | 1985-07-31 | Celltech Ltd | Culture of animal cells |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| AU597574B2 (en) | 1986-03-07 | 1990-06-07 | Massachusetts Institute Of Technology | Method for enhancing glycoprotein stability |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US4791192A (en) | 1986-06-26 | 1988-12-13 | Takeda Chemical Industries, Ltd. | Chemically modified protein with polyethyleneglycol |
| US5260203A (en) | 1986-09-02 | 1993-11-09 | Enzon, Inc. | Single polypeptide chain binding molecules |
| ATE87659T1 (de) | 1986-09-02 | 1993-04-15 | Enzon Lab Inc | Bindungsmolekuele mit einzelpolypeptidkette. |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| US5567610A (en) | 1986-09-04 | 1996-10-22 | Bioinvent International Ab | Method of producing human monoclonal antibodies and kit therefor |
| DE3889853D1 (de) | 1987-11-05 | 1994-07-07 | Hybritech Inc | Polysaccharidmodifizierte Immunglobuline mit reduziertem immunogenem Potential oder verbesserter Pharmakokinetik. |
| US4892538A (en) | 1987-11-17 | 1990-01-09 | Brown University Research Foundation | In vivo delivery of neurotransmitters by implanted, encapsulated cells |
| US5283187A (en) | 1987-11-17 | 1994-02-01 | Brown University Research Foundation | Cell culture-containing tubular capsule produced by co-extrusion |
| EP0387319B1 (en) | 1988-07-23 | 1996-03-06 | Delta Biotechnology Limited | Secretory leader sequences |
| ATE135397T1 (de) | 1988-09-23 | 1996-03-15 | Cetus Oncology Corp | Zellenzuchtmedium für erhöhtes zellenwachstum, zur erhöhung der langlebigkeit und expression der produkte |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US5175384A (en) | 1988-12-05 | 1992-12-29 | Genpharm International | Transgenic mice depleted in mature t-cells and methods for making transgenic mice |
| DE3920358A1 (de) | 1989-06-22 | 1991-01-17 | Behringwerke Ag | Bispezifische und oligospezifische, mono- und oligovalente antikoerperkonstrukte, ihre herstellung und verwendung |
| DE69120146T2 (de) | 1990-01-12 | 1996-12-12 | Cell Genesys Inc | Erzeugung xenogener antikörper |
| US5229275A (en) | 1990-04-26 | 1993-07-20 | Akzo N.V. | In-vitro method for producing antigen-specific human monoclonal antibodies |
| US5427908A (en) | 1990-05-01 | 1995-06-27 | Affymax Technologies N.V. | Recombinant library screening methods |
| GB9014932D0 (en) | 1990-07-05 | 1990-08-22 | Celltech Ltd | Recombinant dna product and method |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| GB9022543D0 (en) | 1990-10-17 | 1990-11-28 | Wellcome Found | Antibody production |
| ES2136092T3 (es) | 1991-09-23 | 1999-11-16 | Medical Res Council | Procedimientos para la produccion de anticuerpos humanizados. |
| ES2241710T3 (es) | 1991-11-25 | 2005-11-01 | Enzon, Inc. | Procedimiento para producir proteinas multivalentes de union a antigeno. |
| FI92507C (fi) | 1991-12-19 | 1994-11-25 | Valto Ilomaeki | Menetelmä ja laite maaperään pakotettavien putkien ohjaamiseksi |
| ATE244763T1 (de) | 1992-02-11 | 2003-07-15 | Cell Genesys Inc | Erzielen von homozygotem durch zielgerichtete genetische ereignisse |
| WO1993019172A1 (en) | 1992-03-24 | 1993-09-30 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5573905A (en) | 1992-03-30 | 1996-11-12 | The Scripps Research Institute | Encoded combinatorial chemical libraries |
| EP0646178A1 (en) | 1992-06-04 | 1995-04-05 | The Regents Of The University Of California | expression cassette with regularoty regions functional in the mammmlian host |
| CA2140638C (en) | 1992-07-24 | 2010-05-04 | Raju Kucherlapati | Generation of xenogeneic antibodies |
| DE69327229T2 (de) | 1992-12-11 | 2000-03-30 | The Dow Chemical Co., Midland | Multivalente einkettige Antikörper |
| US6342225B1 (en) | 1993-08-13 | 2002-01-29 | Deutshces Wollforschungsinstitut | Pharmaceutical active conjugates |
| EP0731842A1 (en) | 1993-12-03 | 1996-09-18 | Medical Research Council | Recombinant binding proteins and peptides |
| JPH09509428A (ja) | 1994-02-23 | 1997-09-22 | カイロン コーポレイション | 薬理学的に活性な薬剤の血清半減期を増加させるための方法および組成物 |
| US6054287A (en) | 1994-05-27 | 2000-04-25 | Methodist Hospital Of Indiana, Inc. | Cell-type-specific methods and devices for the low temperature preservation of the cells of an animal species |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| CA2215394C (en) | 1995-03-23 | 2011-04-26 | Immunex Corporation | Il-17 receptor |
| US6130364A (en) | 1995-03-29 | 2000-10-10 | Abgenix, Inc. | Production of antibodies using Cre-mediated site-specific recombination |
| US6096871A (en) | 1995-04-14 | 2000-08-01 | Genentech, Inc. | Polypeptides altered to contain an epitope from the Fc region of an IgG molecule for increased half-life |
| EP1709970A1 (en) | 1995-04-27 | 2006-10-11 | Abgenix, Inc. | Human antibodies against EGFR, derived from immunized xenomice |
| US5869451A (en) | 1995-06-07 | 1999-02-09 | Glaxo Group Limited | Peptides and compounds that bind to a receptor |
| US5746516A (en) | 1995-08-11 | 1998-05-05 | Hitachi Powdered Metals Co., Ltd. | Porous bearing system having internal grooves and electric motor provided with the same |
| US5849863A (en) | 1995-09-08 | 1998-12-15 | University Of Colorado | Cytolytic bradykinin antagonists |
| US5834431A (en) | 1995-09-08 | 1998-11-10 | Cortech, Inc. | Des-Arg9 -BK antagonists |
| US5989830A (en) | 1995-10-16 | 1999-11-23 | Unilever Patent Holdings Bv | Bifunctional or bivalent antibody fragment analogue |
| AU728657B2 (en) | 1996-03-18 | 2001-01-18 | Board Of Regents, The University Of Texas System | Immunoglobulin-like domains with increased half-lives |
| GB9712818D0 (en) | 1996-07-08 | 1997-08-20 | Cambridge Antibody Tech | Labelling and selection of specific binding molecules |
| WO1998023639A2 (en) * | 1996-11-27 | 1998-06-04 | University Of Florida | ShK TOXIN COMPOSITIONS AND METHODS OF USE |
| CA2273194C (en) | 1996-12-03 | 2011-02-01 | Abgenix, Inc. | Transgenic mammals having human ig loci including plural vh and vk regions and antibodies produced therefrom |
| US6689749B1 (en) | 1996-12-19 | 2004-02-10 | Suntory Limited | Neuropeptides originating in scorpion |
| US20020032315A1 (en) | 1997-08-06 | 2002-03-14 | Manuel Baca | Anti-vegf antibodies |
| US6171586B1 (en) | 1997-06-13 | 2001-01-09 | Genentech, Inc. | Antibody formulation |
| US20030044772A1 (en) | 1997-08-04 | 2003-03-06 | Applied Molecular Evolution [Formerly Ixsys] | Methods for identifying ligand specific binding molecules |
| US6022952A (en) | 1998-04-01 | 2000-02-08 | University Of Alberta | Compositions and methods for protein secretion |
| US20020029391A1 (en) | 1998-04-15 | 2002-03-07 | Claude Geoffrey Davis | Epitope-driven human antibody production and gene expression profiling |
| JP3820105B2 (ja) | 1998-10-23 | 2006-09-13 | キリン−アムジエン・インコーポレーテツド | Mp1受容体に結合し血小板生成活性を有する二量体トロンボポエチンペプチド模倣体 |
| US6660843B1 (en) | 1998-10-23 | 2003-12-09 | Amgen Inc. | Modified peptides as therapeutic agents |
| ATE429504T1 (de) | 1998-11-20 | 2009-05-15 | Fuso Pharmaceutical Ind | Proteinexpressionsvektor und benutzung desselbigen |
| CA2368068A1 (en) | 1999-03-18 | 2000-09-21 | Steven M. Ruben | 27 human secreted proteins |
| US6887470B1 (en) | 1999-09-10 | 2005-05-03 | Conjuchem, Inc. | Protection of endogenous therapeutic peptides from peptidase activity through conjugation to blood components |
| FR2794461B1 (fr) * | 1999-06-07 | 2004-01-23 | Lab Francais Du Fractionnement | Procede de preparation de nouvelles fractions d'ig humaines possedant une activite immunomodulatrice |
| US6833268B1 (en) | 1999-06-10 | 2004-12-21 | Abgenix, Inc. | Transgenic animals for producing specific isotypes of human antibodies via non-cognate switch regions |
| JO2291B1 (en) | 1999-07-02 | 2005-09-12 | اف . هوفمان لاروش ايه جي | Erythropoietin derivatives |
| US6475220B1 (en) | 1999-10-15 | 2002-11-05 | Whiteside Biomechanics, Inc. | Spinal cable system |
| RU2317999C2 (ru) | 2000-02-25 | 2008-02-27 | Те Гавернмент Оф Те Юнайтед Стейтс, Эз Репризентед Бай Те Секретари Оф Те Департмент Оф Хелт Энд Хьюман Сервисез | SCFV ПРОТИВ EGFRvIII С УЛУЧШЕННОЙ ЦИТОТОКСИЧНОСТЬЮ И ВЫХОДОМ, ИММУНОТОКСИНЫ НА ИХ ОСНОВЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ |
| CA2405557C (en) | 2000-04-12 | 2013-09-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US6756480B2 (en) | 2000-04-27 | 2004-06-29 | Amgen Inc. | Modulators of receptors for parathyroid hormone and parathyroid hormone-related protein |
| EP1916303B1 (en) | 2000-11-30 | 2013-02-27 | Medarex, Inc. | Nucleic acids encoding rearranged human immunoglobulin sequences from transgenic transchromosomal mice |
| US20030133939A1 (en) | 2001-01-17 | 2003-07-17 | Genecraft, Inc. | Binding domain-immunoglobulin fusion proteins |
| US7754208B2 (en) | 2001-01-17 | 2010-07-13 | Trubion Pharmaceuticals, Inc. | Binding domain-immunoglobulin fusion proteins |
| US7829084B2 (en) | 2001-01-17 | 2010-11-09 | Trubion Pharmaceuticals, Inc. | Binding constructs and methods for use thereof |
| US7211395B2 (en) | 2001-03-09 | 2007-05-01 | Dyax Corp. | Serum albumin binding moieties |
| US20050054051A1 (en) | 2001-04-12 | 2005-03-10 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US20050089932A1 (en) | 2001-04-26 | 2005-04-28 | Avidia Research Institute | Novel proteins with targeted binding |
| US20060223114A1 (en) | 2001-04-26 | 2006-10-05 | Avidia Research Institute | Protein scaffolds and uses thereof |
| EE05294B1 (et) | 2001-05-11 | 2010-04-15 | Amgen Inc. | TALL-1-ga seostuv ainekompositsioon |
| US20030113316A1 (en) | 2001-07-25 | 2003-06-19 | Kaisheva Elizabet A. | Stable lyophilized pharmaceutical formulation of IgG antibodies |
| US7371849B2 (en) | 2001-09-13 | 2008-05-13 | Institute For Antibodies Co., Ltd. | Methods of constructing camel antibody libraries |
| US7332474B2 (en) | 2001-10-11 | 2008-02-19 | Amgen Inc. | Peptides and related compounds having thrombopoietic activity |
| US7205275B2 (en) | 2001-10-11 | 2007-04-17 | Amgen Inc. | Methods of treatment using specific binding agents of human angiopoietin-2 |
| US7138370B2 (en) | 2001-10-11 | 2006-11-21 | Amgen Inc. | Specific binding agents of human angiopoietin-2 |
| JP2005516965A (ja) | 2001-12-28 | 2005-06-09 | アブジェニックス・インコーポレーテッド | 抗muc18抗体を使用する方法 |
| KR100708398B1 (ko) | 2002-03-22 | 2007-04-18 | (주) 에이프로젠 | 인간화 항체 및 이의 제조방법 |
| US20030191056A1 (en) | 2002-04-04 | 2003-10-09 | Kenneth Walker | Use of transthyretin peptide/protein fusions to increase the serum half-life of pharmacologically active peptides/proteins |
| US6919426B2 (en) | 2002-09-19 | 2005-07-19 | Amgen Inc. | Peptides and related molecules that modulate nerve growth factor activity |
| ES2639301T3 (es) | 2003-04-30 | 2017-10-26 | Universität Zürich | Procedimientos de tratamiento de cáncer usando una inmunotoxina |
| AR045563A1 (es) | 2003-09-10 | 2005-11-02 | Warner Lambert Co | Anticuerpos dirigidos a m-csf |
| US7605120B2 (en) | 2003-10-22 | 2009-10-20 | Amgen Inc. | Antagonists of the brandykinin B1 receptor |
| AU2004284090A1 (en) | 2003-10-24 | 2005-05-06 | Avidia, Inc. | LDL receptor class A and EGF domain monomers and multimers |
| PE20051053A1 (es) * | 2004-01-09 | 2005-12-12 | Pfizer | ANTICUERPOS CONTRA MAdCAM |
| GB0414272D0 (en) | 2004-06-25 | 2004-07-28 | Cellpep Sa | OsK1 derivatives |
| CA2580796C (en) | 2004-09-24 | 2013-03-26 | Amgen Inc. | Modified fc molecules having peptides inserted in internal loop regions |
| WO2006042151A2 (en) * | 2004-10-07 | 2006-04-20 | The Regents Of The University Of California | Analogs of shk toxin and their uses in selective inhibition of kv1.3 potassium channels |
| AU2005307789A1 (en) | 2004-11-16 | 2006-05-26 | Avidia Research Institute | Protein scaffolds and uses thereof |
| WO2006127040A2 (en) | 2004-11-16 | 2006-11-30 | Avidia Research Institute | Protein scaffolds and uses therof |
| WO2006055638A2 (en) | 2004-11-17 | 2006-05-26 | Abgenix, Inc. | Fully human monoclonal antibodies to il-13 |
| CN105085678B (zh) * | 2004-12-21 | 2019-05-07 | 阿斯利康公司 | 血管生成素-2的抗体及其应用 |
| US7833979B2 (en) | 2005-04-22 | 2010-11-16 | Amgen Inc. | Toxin peptide therapeutic agents |
| BRPI0614222A2 (pt) | 2005-07-13 | 2011-03-15 | Amgen Mountain View Inc | proteìnas ligantes de il-6 |
| US8008453B2 (en) | 2005-08-12 | 2011-08-30 | Amgen Inc. | Modified Fc molecules |
| PE20071101A1 (es) | 2005-08-31 | 2007-12-21 | Amgen Inc | Polipeptidos y anticuerpos |
| US8168592B2 (en) | 2005-10-21 | 2012-05-01 | Amgen Inc. | CGRP peptide antagonists and conjugates |
| TW200722436A (en) | 2005-10-21 | 2007-06-16 | Hoffmann La Roche | A peptide-immunoglobulin-conjugate |
| EA015992B1 (ru) | 2006-03-17 | 2012-01-30 | Байоджен Айдек Эмэй Инк. | Стабилизированное антитело и многовалентная антигенсвязывающая молекула на его основе, способы получения и использования вышеназванного стабилизированного антитела |
| TW200813091A (en) | 2006-04-10 | 2008-03-16 | Amgen Fremont Inc | Targeted binding agents directed to uPAR and uses thereof |
| US7833527B2 (en) | 2006-10-02 | 2010-11-16 | Amgen Inc. | Methods of treating psoriasis using IL-17 Receptor A antibodies |
| CA3069091C (en) | 2006-11-01 | 2021-09-14 | Ventana Medical Systems, Inc. | Haptens, hapten conjugates, compositions thereof and method for their preparation and use |
| EP2118138A1 (en) | 2007-03-12 | 2009-11-18 | Esbatech AG | Sequence based engineering and optimization of single chain antibodies |
| US8793074B2 (en) | 2007-06-21 | 2014-07-29 | Saint Louis University | Sequence covariance networks, methods and uses therefor |
| MX2009013328A (es) | 2007-06-25 | 2010-06-02 | Esbatech An Alcon Biomedical R | Metodos para modificar anticuerpos, y anticuerpos modificados con propiedades funcionales mejoradas. |
| EP2069401A4 (en) | 2007-07-31 | 2011-02-23 | Medimmune Llc | MULTISPECIENT EPITOP BINDING PROTEINS AND THEIR USE |
| US20090203038A1 (en) | 2007-12-27 | 2009-08-13 | Abbott Laboratories | Negative Mimic Antibody For Use as a Blocking Reagent In BNP Immunoassays |
| KR20100129319A (ko) | 2008-03-20 | 2010-12-08 | 체에스엘 베링 게엠베하 | 병리학적 혈전 형성에 중요한 칼슘 센서 STIM1 및 혈소판 SOC 채널 Orai1 (CRACM1) |
| EP2356270B1 (en) | 2008-11-07 | 2016-08-24 | Fabrus Llc | Combinatorial antibody libraries and uses thereof |
| US8530629B2 (en) | 2009-01-30 | 2013-09-10 | Ab Biosciences, Inc. | Lowered affinity antibodies and uses therefor |
| CA2889453C (en) | 2009-03-20 | 2018-11-06 | Amgen Inc. | Carrier immunoglobulins and uses thereof |
| CN102971342B (zh) | 2010-01-28 | 2016-08-03 | Ab生物科学公司 | 亲和力降低的新抗体和制备所述抗体的方法 |
| KR20130110169A (ko) | 2010-09-22 | 2013-10-08 | 암젠 인크 | 담체 면역글로뷸린 및 이것의 용도 |
-
2010
- 2010-03-19 CA CA2889453A patent/CA2889453C/en active Active
- 2010-03-19 AU AU2010226392A patent/AU2010226392B9/en not_active Ceased
- 2010-03-19 CA CA3018235A patent/CA3018235C/en active Active
- 2010-03-19 JP JP2012501019A patent/JP2012521197A/ja active Pending
- 2010-03-19 EP EP18173194.4A patent/EP3385279B1/en active Active
- 2010-03-19 US US13/258,668 patent/US8734796B2/en active Active
- 2010-03-19 CA CA2755336A patent/CA2755336C/en active Active
- 2010-03-19 CA CA2755133A patent/CA2755133A1/en not_active Abandoned
- 2010-03-19 MX MX2014010300A patent/MX347291B/es unknown
- 2010-03-19 MX MX2014010302A patent/MX347295B/es unknown
- 2010-03-19 EP EP10754210.2A patent/EP2408814B1/en active Active
- 2010-03-19 AU AU2010226391A patent/AU2010226391C1/en active Active
- 2010-03-19 US US13/258,454 patent/US20120121591A1/en not_active Abandoned
- 2010-03-19 MX MX2011009797A patent/MX2011009797A/es not_active Application Discontinuation
- 2010-03-19 WO PCT/US2010/028061 patent/WO2010108154A2/en active Application Filing
- 2010-03-19 MX MX2011009798A patent/MX2011009798A/es active IP Right Grant
- 2010-03-19 WO PCT/US2010/028060 patent/WO2010108153A2/en active Application Filing
- 2010-03-19 EP EP10754211.0A patent/EP2432488A4/en not_active Withdrawn
- 2010-03-19 JP JP2012501020A patent/JP2012521360A/ja active Pending
-
2014
- 2014-05-22 US US14/285,579 patent/US9562108B2/en active Active
- 2014-05-22 US US14/285,576 patent/US9562107B2/en active Active
-
2015
- 2015-01-07 JP JP2015001336A patent/JP6173359B2/ja active Active
-
2017
- 2017-01-12 US US15/405,233 patent/US10189912B2/en active Active
- 2017-04-10 JP JP2017077334A patent/JP6345832B2/ja active Active
-
2018
- 2018-05-22 JP JP2018097620A patent/JP6609347B2/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002522090A (ja) * | 1998-08-17 | 2002-07-23 | トーマス ジェファーソン ユニヴァーシティー | 抗体エンベロープ融合タンパク質および野生型エンベロープタンパク質を含有するレトロウイルスベクターを用いる細胞型特異的遺伝子移入 |
| JP2008501302A (ja) * | 2003-10-16 | 2008-01-24 | イムクローン システムズ インコーポレイティド | 繊維芽細胞増殖因子レセプター−1阻害物質及びその治療方法 |
| WO2008088422A2 (en) * | 2006-10-25 | 2008-07-24 | Amgen Inc. | Toxin peptide therapeutic agents |
Non-Patent Citations (1)
| Title |
|---|
| JPN6014028796; Journal of Immunological Methods Vol.313, 2006, p.20-28 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017501728A (ja) * | 2013-12-23 | 2017-01-19 | ザイムワークス インコーポレイティド | C末端軽鎖ポリペプチド伸長を含有する抗体、ならびにその複合体及びその使用方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6609347B2 (ja) | 担体免疫グロブリンおよびその使用 | |
| JP6284566B2 (ja) | 担体としての免疫グロブリンおよびその使用 | |
| AU2014200459B2 (en) | Carrier immunoglobulins and uses thereof | |
| AU2015201449B2 (en) | Carrier Immunoglobulins And Uses Thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130225 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140708 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20141006 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20141014 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20150310 |