JP2009507853A - Gm−csf発現ポックスウイルスを用いる転移性および/または全身播種性癌の全身処置 - Google Patents
Gm−csf発現ポックスウイルスを用いる転移性および/または全身播種性癌の全身処置 Download PDFInfo
- Publication number
- JP2009507853A JP2009507853A JP2008530209A JP2008530209A JP2009507853A JP 2009507853 A JP2009507853 A JP 2009507853A JP 2008530209 A JP2008530209 A JP 2008530209A JP 2008530209 A JP2008530209 A JP 2008530209A JP 2009507853 A JP2009507853 A JP 2009507853A
- Authority
- JP
- Japan
- Prior art keywords
- tumor
- virus
- cancer
- cells
- vaccinia virus
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 336
- 201000011510 cancer Diseases 0.000 title claims abstract description 110
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 title claims abstract description 68
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 title claims abstract description 68
- 238000011282 treatment Methods 0.000 title claims description 100
- 230000009885 systemic effect Effects 0.000 title description 22
- 206010061289 metastatic neoplasm Diseases 0.000 title description 4
- 230000001394 metastastic effect Effects 0.000 title 1
- 238000000034 method Methods 0.000 claims abstract description 121
- 241000700618 Vaccinia virus Species 0.000 claims abstract description 95
- 239000000203 mixture Substances 0.000 claims abstract description 56
- 230000003362 replicative effect Effects 0.000 claims abstract description 15
- 108090000623 proteins and genes Proteins 0.000 claims description 183
- 241000700605 Viruses Species 0.000 claims description 155
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 144
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 134
- 229920001184 polypeptide Polymers 0.000 claims description 124
- 150000007523 nucleic acids Chemical class 0.000 claims description 102
- 102000039446 nucleic acids Human genes 0.000 claims description 71
- 108020004707 nucleic acids Proteins 0.000 claims description 71
- 230000014509 gene expression Effects 0.000 claims description 70
- 230000035772 mutation Effects 0.000 claims description 61
- 238000012217 deletion Methods 0.000 claims description 43
- 230000037430 deletion Effects 0.000 claims description 43
- 206010046865 Vaccinia virus infection Diseases 0.000 claims description 39
- 208000007089 vaccinia Diseases 0.000 claims description 39
- 239000007924 injection Substances 0.000 claims description 38
- 238000002347 injection Methods 0.000 claims description 38
- 230000000295 complement effect Effects 0.000 claims description 32
- 238000001990 intravenous administration Methods 0.000 claims description 32
- 208000015181 infectious disease Diseases 0.000 claims description 27
- 108020004440 Thymidine kinase Proteins 0.000 claims description 23
- 230000001105 regulatory effect Effects 0.000 claims description 19
- 102000014150 Interferons Human genes 0.000 claims description 16
- 108010050904 Interferons Proteins 0.000 claims description 16
- 201000001441 melanoma Diseases 0.000 claims description 15
- 206010027476 Metastases Diseases 0.000 claims description 14
- 229940079322 interferon Drugs 0.000 claims description 14
- 238000013518 transcription Methods 0.000 claims description 14
- 230000035897 transcription Effects 0.000 claims description 14
- 108010012236 Chemokines Proteins 0.000 claims description 13
- 238000009472 formulation Methods 0.000 claims description 13
- 102000019034 Chemokines Human genes 0.000 claims description 12
- 206010025323 Lymphomas Diseases 0.000 claims description 9
- 230000002458 infectious effect Effects 0.000 claims description 9
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 8
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 8
- 208000032839 leukemia Diseases 0.000 claims description 8
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 7
- 101800003344 Vaccinia growth factor Proteins 0.000 claims description 7
- 201000005202 lung cancer Diseases 0.000 claims description 7
- 208000020816 lung neoplasm Diseases 0.000 claims description 7
- 239000003001 serine protease inhibitor Substances 0.000 claims description 7
- 101710172503 Chemokine-binding protein Proteins 0.000 claims description 6
- 101710197234 Inactive chemokine-binding protein Proteins 0.000 claims description 6
- 206010006187 Breast cancer Diseases 0.000 claims description 5
- 208000026310 Breast neoplasm Diseases 0.000 claims description 5
- 206010009944 Colon cancer Diseases 0.000 claims description 5
- 206010060862 Prostate cancer Diseases 0.000 claims description 5
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 5
- 238000001802 infusion Methods 0.000 claims description 5
- 101150045588 A38L gene Proteins 0.000 claims description 4
- 101150091700 A41L gene Proteins 0.000 claims description 4
- 101150020569 B3R gene Proteins 0.000 claims description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 4
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 4
- 229940122055 Serine protease inhibitor Drugs 0.000 claims description 4
- 101710102218 Serine protease inhibitor Proteins 0.000 claims description 4
- 101100540425 Vaccinia virus (strain Copenhagen) VGF gene Proteins 0.000 claims description 4
- 101100107503 Vaccinia virus (strain Western Reserve) VACWR166 gene Proteins 0.000 claims description 4
- 230000002147 killing effect Effects 0.000 claims description 4
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 4
- 201000002528 pancreatic cancer Diseases 0.000 claims description 4
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 4
- 101100118545 Holotrichia diomphalia EGF-like gene Proteins 0.000 claims description 3
- 101150035509 N1L gene Proteins 0.000 claims description 3
- 101150073111 NPH1 gene Proteins 0.000 claims description 3
- 101100459380 Vaccinia virus (strain Western Reserve) VACWR028 gene Proteins 0.000 claims description 3
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 3
- 101100272147 Vaccinia virus (strain Copenhagen) B7R gene Proteins 0.000 claims description 2
- 101100272148 Vaccinia virus (strain Western Reserve) VACWR189 gene Proteins 0.000 claims description 2
- 208000037819 metastatic cancer Diseases 0.000 claims description 2
- 208000011575 metastatic malignant neoplasm Diseases 0.000 claims description 2
- 210000004027 cell Anatomy 0.000 description 191
- 238000002604 ultrasonography Methods 0.000 description 139
- 102000004169 proteins and genes Human genes 0.000 description 96
- 235000018102 proteins Nutrition 0.000 description 90
- 230000003612 virological effect Effects 0.000 description 54
- 108020004414 DNA Proteins 0.000 description 52
- 230000000694 effects Effects 0.000 description 52
- 210000004881 tumor cell Anatomy 0.000 description 49
- 210000001519 tissue Anatomy 0.000 description 47
- 235000001014 amino acid Nutrition 0.000 description 43
- 239000013598 vector Substances 0.000 description 40
- 241001465754 Metazoa Species 0.000 description 39
- 230000006870 function Effects 0.000 description 39
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 35
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 35
- 229940024606 amino acid Drugs 0.000 description 34
- 150000001413 amino acids Chemical class 0.000 description 34
- 239000000047 product Substances 0.000 description 33
- 241000699670 Mus sp. Species 0.000 description 31
- 230000006907 apoptotic process Effects 0.000 description 30
- 150000002632 lipids Chemical class 0.000 description 30
- 239000000126 substance Substances 0.000 description 29
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 28
- 238000003199 nucleic acid amplification method Methods 0.000 description 28
- 230000003321 amplification Effects 0.000 description 27
- 108091028043 Nucleic acid sequence Proteins 0.000 description 26
- 230000007246 mechanism Effects 0.000 description 26
- 238000009169 immunotherapy Methods 0.000 description 25
- 102000004127 Cytokines Human genes 0.000 description 24
- 108090000695 Cytokines Proteins 0.000 description 24
- 238000003556 assay Methods 0.000 description 24
- 230000001965 increasing effect Effects 0.000 description 24
- 230000010076 replication Effects 0.000 description 24
- 241000283973 Oryctolagus cuniculus Species 0.000 description 23
- 239000000523 sample Substances 0.000 description 23
- 230000001225 therapeutic effect Effects 0.000 description 22
- 230000000259 anti-tumor effect Effects 0.000 description 21
- 238000009396 hybridization Methods 0.000 description 21
- 102000006601 Thymidine Kinase Human genes 0.000 description 19
- 125000003275 alpha amino acid group Chemical group 0.000 description 19
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 19
- 230000006698 induction Effects 0.000 description 19
- 238000006467 substitution reaction Methods 0.000 description 19
- 238000013459 approach Methods 0.000 description 18
- 239000003623 enhancer Substances 0.000 description 18
- 230000001404 mediated effect Effects 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- 102000000589 Interleukin-1 Human genes 0.000 description 17
- 108010002352 Interleukin-1 Proteins 0.000 description 17
- 238000002512 chemotherapy Methods 0.000 description 17
- 238000003780 insertion Methods 0.000 description 17
- 230000037431 insertion Effects 0.000 description 17
- 239000003550 marker Substances 0.000 description 17
- 102000004190 Enzymes Human genes 0.000 description 16
- 108090000790 Enzymes Proteins 0.000 description 16
- 101000746373 Homo sapiens Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 16
- 102100033254 Tumor suppressor ARF Human genes 0.000 description 16
- 230000002601 intratumoral effect Effects 0.000 description 16
- 230000003211 malignant effect Effects 0.000 description 16
- 238000004519 manufacturing process Methods 0.000 description 16
- 238000002703 mutagenesis Methods 0.000 description 16
- 231100000350 mutagenesis Toxicity 0.000 description 16
- 230000008685 targeting Effects 0.000 description 16
- 239000002246 antineoplastic agent Substances 0.000 description 15
- 102000046157 human CSF2 Human genes 0.000 description 15
- 210000004185 liver Anatomy 0.000 description 15
- 125000003729 nucleotide group Chemical group 0.000 description 15
- 238000001959 radiotherapy Methods 0.000 description 15
- 239000002773 nucleotide Substances 0.000 description 14
- 230000004083 survival effect Effects 0.000 description 14
- 238000012384 transportation and delivery Methods 0.000 description 14
- 210000003462 vein Anatomy 0.000 description 14
- 108091026890 Coding region Proteins 0.000 description 13
- 108010074328 Interferon-gamma Proteins 0.000 description 13
- 108700012411 TNFSF10 Proteins 0.000 description 13
- 102100024598 Tumor necrosis factor ligand superfamily member 10 Human genes 0.000 description 13
- 230000015572 biosynthetic process Effects 0.000 description 13
- 230000004663 cell proliferation Effects 0.000 description 13
- 238000001514 detection method Methods 0.000 description 13
- 241000701161 unidentified adenovirus Species 0.000 description 13
- 206010027458 Metastases to lung Diseases 0.000 description 12
- 108700020796 Oncogene Proteins 0.000 description 12
- 230000004913 activation Effects 0.000 description 12
- 239000000427 antigen Substances 0.000 description 12
- 108091007433 antigens Proteins 0.000 description 12
- 102000036639 antigens Human genes 0.000 description 12
- 238000001415 gene therapy Methods 0.000 description 12
- 210000000056 organ Anatomy 0.000 description 12
- 102000005962 receptors Human genes 0.000 description 12
- 108020003175 receptors Proteins 0.000 description 12
- 230000004044 response Effects 0.000 description 12
- -1 B8R Proteins 0.000 description 11
- 241000699666 Mus <mouse, genus> Species 0.000 description 11
- 230000002779 inactivation Effects 0.000 description 11
- 210000004072 lung Anatomy 0.000 description 11
- 230000008488 polyadenylation Effects 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 238000007920 subcutaneous administration Methods 0.000 description 11
- 238000001356 surgical procedure Methods 0.000 description 11
- 101150039990 B13R gene Proteins 0.000 description 10
- 102000053602 DNA Human genes 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 230000001472 cytotoxic effect Effects 0.000 description 10
- 230000001419 dependent effect Effects 0.000 description 10
- 230000029142 excretion Effects 0.000 description 10
- 230000028993 immune response Effects 0.000 description 10
- 230000001939 inductive effect Effects 0.000 description 10
- 102000040430 polynucleotide Human genes 0.000 description 10
- 108091033319 polynucleotide Proteins 0.000 description 10
- 239000002157 polynucleotide Substances 0.000 description 10
- 230000005855 radiation Effects 0.000 description 10
- 241000894007 species Species 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- 230000032258 transport Effects 0.000 description 10
- 230000001018 virulence Effects 0.000 description 10
- 108020004705 Codon Proteins 0.000 description 9
- 230000008901 benefit Effects 0.000 description 9
- 230000030833 cell death Effects 0.000 description 9
- 231100000433 cytotoxic Toxicity 0.000 description 9
- 230000006378 damage Effects 0.000 description 9
- 239000003102 growth factor Substances 0.000 description 9
- 239000003112 inhibitor Substances 0.000 description 9
- 230000008569 process Effects 0.000 description 9
- 230000002829 reductive effect Effects 0.000 description 9
- 230000035945 sensitivity Effects 0.000 description 9
- 108091007914 CDKs Proteins 0.000 description 8
- 108700026244 Open Reading Frames Proteins 0.000 description 8
- 101150071286 SPI-2 gene Proteins 0.000 description 8
- 108010067390 Viral Proteins Proteins 0.000 description 8
- 210000004204 blood vessel Anatomy 0.000 description 8
- 238000003776 cleavage reaction Methods 0.000 description 8
- 150000001875 compounds Chemical class 0.000 description 8
- 238000002591 computed tomography Methods 0.000 description 8
- 230000002950 deficient Effects 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- 230000012010 growth Effects 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 208000014018 liver neoplasm Diseases 0.000 description 8
- 210000004698 lymphocyte Anatomy 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 102000016914 ras Proteins Human genes 0.000 description 8
- 108010014186 ras Proteins Proteins 0.000 description 8
- 238000002271 resection Methods 0.000 description 8
- 150000003839 salts Chemical class 0.000 description 8
- 230000007017 scission Effects 0.000 description 8
- 238000012385 systemic delivery Methods 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- 238000013519 translation Methods 0.000 description 8
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 7
- 101150023320 B16R gene Proteins 0.000 description 7
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 7
- 206010019695 Hepatic neoplasm Diseases 0.000 description 7
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 7
- 102100037850 Interferon gamma Human genes 0.000 description 7
- 102000006992 Interferon-alpha Human genes 0.000 description 7
- 108010047761 Interferon-alpha Proteins 0.000 description 7
- 108010002350 Interleukin-2 Proteins 0.000 description 7
- 102000000588 Interleukin-2 Human genes 0.000 description 7
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 7
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 7
- 108091007178 TNFRSF10A Proteins 0.000 description 7
- 102100040113 Tumor necrosis factor receptor superfamily member 10A Human genes 0.000 description 7
- 208000009956 adenocarcinoma Diseases 0.000 description 7
- 230000004071 biological effect Effects 0.000 description 7
- 230000005540 biological transmission Effects 0.000 description 7
- 239000002299 complementary DNA Substances 0.000 description 7
- 238000010790 dilution Methods 0.000 description 7
- 239000012895 dilution Substances 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 239000013604 expression vector Substances 0.000 description 7
- 239000000411 inducer Substances 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 230000003472 neutralizing effect Effects 0.000 description 7
- 230000037361 pathway Effects 0.000 description 7
- 239000013612 plasmid Substances 0.000 description 7
- 230000000770 proinflammatory effect Effects 0.000 description 7
- 108091008146 restriction endonucleases Proteins 0.000 description 7
- 229960005486 vaccine Drugs 0.000 description 7
- 230000029812 viral genome replication Effects 0.000 description 7
- 201000009030 Carcinoma Diseases 0.000 description 6
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 6
- 101000980932 Homo sapiens Cyclin-dependent kinase inhibitor 2A Proteins 0.000 description 6
- 101000733249 Homo sapiens Tumor suppressor ARF Proteins 0.000 description 6
- 102000008070 Interferon-gamma Human genes 0.000 description 6
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 6
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 6
- 108060001084 Luciferase Proteins 0.000 description 6
- 239000005089 Luciferase Substances 0.000 description 6
- 102000043276 Oncogene Human genes 0.000 description 6
- 101100316831 Vaccinia virus (strain Copenhagen) B18R gene Proteins 0.000 description 6
- 101100004099 Vaccinia virus (strain Western Reserve) VACWR200 gene Proteins 0.000 description 6
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 6
- 241000700647 Variola virus Species 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 238000007792 addition Methods 0.000 description 6
- 230000001093 anti-cancer Effects 0.000 description 6
- 238000011394 anticancer treatment Methods 0.000 description 6
- 230000002238 attenuated effect Effects 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 210000001185 bone marrow Anatomy 0.000 description 6
- 230000022534 cell killing Effects 0.000 description 6
- 229940127089 cytotoxic agent Drugs 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 239000012634 fragment Substances 0.000 description 6
- 229940121354 immunomodulator Drugs 0.000 description 6
- 230000001976 improved effect Effects 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 230000003834 intracellular effect Effects 0.000 description 6
- 230000005865 ionizing radiation Effects 0.000 description 6
- 229960000310 isoleucine Drugs 0.000 description 6
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- 210000005075 mammary gland Anatomy 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 244000309459 oncolytic virus Species 0.000 description 6
- 238000002741 site-directed mutagenesis Methods 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 231100000331 toxic Toxicity 0.000 description 6
- 230000002588 toxic effect Effects 0.000 description 6
- 239000004474 valine Substances 0.000 description 6
- 239000004475 Arginine Substances 0.000 description 5
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 5
- 101710137943 Complement control protein C3 Proteins 0.000 description 5
- 108090000467 Interferon-beta Proteins 0.000 description 5
- 208000008839 Kidney Neoplasms Diseases 0.000 description 5
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 5
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 239000004472 Lysine Substances 0.000 description 5
- 108091034117 Oligonucleotide Proteins 0.000 description 5
- 108020002230 Pancreatic Ribonuclease Proteins 0.000 description 5
- 102000005891 Pancreatic ribonuclease Human genes 0.000 description 5
- 108700020978 Proto-Oncogene Proteins 0.000 description 5
- 102000052575 Proto-Oncogene Human genes 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- 206010038389 Renal cancer Diseases 0.000 description 5
- 206010039491 Sarcoma Diseases 0.000 description 5
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 5
- 208000036142 Viral infection Diseases 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 5
- 235000009582 asparagine Nutrition 0.000 description 5
- 229960001230 asparagine Drugs 0.000 description 5
- 210000004556 brain Anatomy 0.000 description 5
- 230000007910 cell fusion Effects 0.000 description 5
- 210000001072 colon Anatomy 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 239000012636 effector Substances 0.000 description 5
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 5
- 238000003384 imaging method Methods 0.000 description 5
- 238000010253 intravenous injection Methods 0.000 description 5
- 201000010982 kidney cancer Diseases 0.000 description 5
- 210000002540 macrophage Anatomy 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 210000000822 natural killer cell Anatomy 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 230000010412 perfusion Effects 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 230000002062 proliferating effect Effects 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 230000009466 transformation Effects 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- 230000009385 viral infection Effects 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 4
- FMMWHPNWAFZXNH-UHFFFAOYSA-N Benz[a]pyrene Chemical compound C1=C2C3=CC=CC=C3C=C(C=C3)C2=C2C3=CC=CC2=C1 FMMWHPNWAFZXNH-UHFFFAOYSA-N 0.000 description 4
- 208000003174 Brain Neoplasms Diseases 0.000 description 4
- 108090000565 Capsid Proteins Proteins 0.000 description 4
- 206010008342 Cervix carcinoma Diseases 0.000 description 4
- 241000700626 Cowpox virus Species 0.000 description 4
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 4
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 4
- 102000001398 Granzyme Human genes 0.000 description 4
- 108060005986 Granzyme Proteins 0.000 description 4
- 102000003996 Interferon-beta Human genes 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- ZKXDGKXYMTYWTB-UHFFFAOYSA-N N-nitrosomorpholine Chemical compound O=NN1CCOCC1 ZKXDGKXYMTYWTB-UHFFFAOYSA-N 0.000 description 4
- 206010061309 Neoplasm progression Diseases 0.000 description 4
- 241000700629 Orthopoxvirus Species 0.000 description 4
- 108020004511 Recombinant DNA Proteins 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- 102100040112 Tumor necrosis factor receptor superfamily member 10B Human genes 0.000 description 4
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 4
- 230000004075 alteration Effects 0.000 description 4
- 230000005809 anti-tumor immunity Effects 0.000 description 4
- 230000000840 anti-viral effect Effects 0.000 description 4
- 235000003704 aspartic acid Nutrition 0.000 description 4
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 4
- 230000029918 bioluminescence Effects 0.000 description 4
- 238000005415 bioluminescence Methods 0.000 description 4
- 230000003833 cell viability Effects 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 201000010881 cervical cancer Diseases 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 238000010367 cloning Methods 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 108020001507 fusion proteins Proteins 0.000 description 4
- 102000037865 fusion proteins Human genes 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 4
- 206010020718 hyperplasia Diseases 0.000 description 4
- 239000012642 immune effector Substances 0.000 description 4
- 230000003308 immunostimulating effect Effects 0.000 description 4
- 230000008595 infiltration Effects 0.000 description 4
- 238000001764 infiltration Methods 0.000 description 4
- 229960003130 interferon gamma Drugs 0.000 description 4
- 108010027775 interleukin-1beta-converting enzyme inhibitor Proteins 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 238000012423 maintenance Methods 0.000 description 4
- 230000009401 metastasis Effects 0.000 description 4
- 238000006386 neutralization reaction Methods 0.000 description 4
- 230000000174 oncolytic effect Effects 0.000 description 4
- 210000003463 organelle Anatomy 0.000 description 4
- 230000002611 ovarian Effects 0.000 description 4
- 210000001672 ovary Anatomy 0.000 description 4
- 230000002018 overexpression Effects 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 230000000861 pro-apoptotic effect Effects 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 210000002307 prostate Anatomy 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 210000003491 skin Anatomy 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 238000007910 systemic administration Methods 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 230000005751 tumor progression Effects 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- 230000006648 viral gene expression Effects 0.000 description 4
- 210000002845 virion Anatomy 0.000 description 4
- 101150115056 A31R gene Proteins 0.000 description 3
- 101150049392 A34R gene Proteins 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 206010003571 Astrocytoma Diseases 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 208000005623 Carcinogenesis Diseases 0.000 description 3
- 102000000844 Cell Surface Receptors Human genes 0.000 description 3
- 108010001857 Cell Surface Receptors Proteins 0.000 description 3
- 230000005778 DNA damage Effects 0.000 description 3
- 231100000277 DNA damage Toxicity 0.000 description 3
- 101100044298 Drosophila melanogaster fand gene Proteins 0.000 description 3
- 206010059866 Drug resistance Diseases 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- 241000588724 Escherichia coli Species 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 101150064015 FAS gene Proteins 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- 102220603448 Homeobox protein SIX3_A34R_mutation Human genes 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 102000013462 Interleukin-12 Human genes 0.000 description 3
- 108010065805 Interleukin-12 Proteins 0.000 description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 3
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 3
- 206010029260 Neuroblastoma Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 101100335198 Pneumocystis carinii fol1 gene Proteins 0.000 description 3
- 241000700625 Poxviridae Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108010090931 Proto-Oncogene Proteins c-bcl-2 Proteins 0.000 description 3
- 102000013535 Proto-Oncogene Proteins c-bcl-2 Human genes 0.000 description 3
- 108020004682 Single-Stranded DNA Proteins 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000002105 Southern blotting Methods 0.000 description 3
- 108091081024 Start codon Proteins 0.000 description 3
- 108091023040 Transcription factor Proteins 0.000 description 3
- 102000040945 Transcription factor Human genes 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 3
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 3
- 102100031988 Tumor necrosis factor ligand superfamily member 6 Human genes 0.000 description 3
- 108050002568 Tumor necrosis factor ligand superfamily member 6 Proteins 0.000 description 3
- 101150004676 VGF gene Proteins 0.000 description 3
- 101100000228 Vaccinia virus (strain Western Reserve) VACWR157 gene Proteins 0.000 description 3
- 108700005077 Viral Genes Proteins 0.000 description 3
- 241000021375 Xenogenes Species 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000003110 anti-inflammatory effect Effects 0.000 description 3
- 229940041181 antineoplastic drug Drugs 0.000 description 3
- 238000003782 apoptosis assay Methods 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 210000001367 artery Anatomy 0.000 description 3
- 210000003719 b-lymphocyte Anatomy 0.000 description 3
- 239000012620 biological material Substances 0.000 description 3
- 238000001815 biotherapy Methods 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 230000036952 cancer formation Effects 0.000 description 3
- 231100000504 carcinogenesis Toxicity 0.000 description 3
- 230000021164 cell adhesion Effects 0.000 description 3
- 210000003679 cervix uteri Anatomy 0.000 description 3
- 208000006990 cholangiocarcinoma Diseases 0.000 description 3
- 210000000349 chromosome Anatomy 0.000 description 3
- 238000011284 combination treatment Methods 0.000 description 3
- 238000011498 curative surgery Methods 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 231100000517 death Toxicity 0.000 description 3
- 239000002612 dispersion medium Substances 0.000 description 3
- 238000001962 electrophoresis Methods 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 210000003979 eosinophil Anatomy 0.000 description 3
- 210000000981 epithelium Anatomy 0.000 description 3
- 210000003527 eukaryotic cell Anatomy 0.000 description 3
- 230000001605 fetal effect Effects 0.000 description 3
- 210000002950 fibroblast Anatomy 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 238000012224 gene deletion Methods 0.000 description 3
- 235000013922 glutamic acid Nutrition 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- 210000004565 granule cell Anatomy 0.000 description 3
- 210000003714 granulocyte Anatomy 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 230000003463 hyperproliferative effect Effects 0.000 description 3
- 230000002519 immonomodulatory effect Effects 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 230000028709 inflammatory response Effects 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 230000000977 initiatory effect Effects 0.000 description 3
- 239000007972 injectable composition Substances 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 230000009545 invasion Effects 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 101150066555 lacZ gene Proteins 0.000 description 3
- 230000003902 lesion Effects 0.000 description 3
- 210000000265 leukocyte Anatomy 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 229930182817 methionine Natural products 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- 102000035118 modified proteins Human genes 0.000 description 3
- 108091005573 modified proteins Proteins 0.000 description 3
- 230000009456 molecular mechanism Effects 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 239000003471 mutagenic agent Substances 0.000 description 3
- 231100000707 mutagenic chemical Toxicity 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 238000011587 new zealand white rabbit Methods 0.000 description 3
- 201000008968 osteosarcoma Diseases 0.000 description 3
- 210000000496 pancreas Anatomy 0.000 description 3
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 3
- 230000002093 peripheral effect Effects 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 230000005522 programmed cell death Effects 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000011555 rabbit model Methods 0.000 description 3
- 230000006798 recombination Effects 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 230000028327 secretion Effects 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 150000003505 terpenes Chemical class 0.000 description 3
- 235000007586 terpenes Nutrition 0.000 description 3
- 239000003053 toxin Substances 0.000 description 3
- 231100000765 toxin Toxicity 0.000 description 3
- 108700012359 toxins Proteins 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 3
- 244000052613 viral pathogen Species 0.000 description 3
- 239000013603 viral vector Substances 0.000 description 3
- 230000003442 weekly effect Effects 0.000 description 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- 201000004384 Alopecia Diseases 0.000 description 2
- 101150019032 B29R gene Proteins 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- BPYKTIZUTYGOLE-IFADSCNNSA-N Bilirubin Chemical compound N1C(=O)C(C)=C(C=C)\C1=C\C1=C(C)C(CCC(O)=O)=C(CC2=C(C(C)=C(\C=C/3C(=C(C=C)C(=O)N\3)C)N2)CCC(O)=O)N1 BPYKTIZUTYGOLE-IFADSCNNSA-N 0.000 description 2
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 2
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 2
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 2
- 108090000426 Caspase-1 Proteins 0.000 description 2
- 108010055166 Chemokine CCL5 Proteins 0.000 description 2
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 2
- 208000005243 Chondrosarcoma Diseases 0.000 description 2
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 2
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 2
- 102400000739 Corticotropin Human genes 0.000 description 2
- 101800000414 Corticotropin Proteins 0.000 description 2
- 101100232885 Cowpox virus (strain Brighton Red) CPXV209 gene Proteins 0.000 description 2
- 101710169745 Cytokine response-modifying protein B Proteins 0.000 description 2
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 102000010170 Death domains Human genes 0.000 description 2
- 108050001718 Death domains Proteins 0.000 description 2
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 2
- 206010012735 Diarrhoea Diseases 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- VYZAHLCBVHPDDF-UHFFFAOYSA-N Dinitrochlorobenzene Chemical compound [O-][N+](=O)C1=CC=C(Cl)C([N+]([O-])=O)=C1 VYZAHLCBVHPDDF-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 206010058314 Dysplasia Diseases 0.000 description 2
- 201000008808 Fibrosarcoma Diseases 0.000 description 2
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 2
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 2
- 208000004463 Follicular Adenocarcinoma Diseases 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 108700004714 Gelonium multiflorum GEL Proteins 0.000 description 2
- 102000005720 Glutathione transferase Human genes 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- 102000009465 Growth Factor Receptors Human genes 0.000 description 2
- 108010009202 Growth Factor Receptors Proteins 0.000 description 2
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 2
- 108091027305 Heteroduplex Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 206010020843 Hyperthermia Diseases 0.000 description 2
- 102100040019 Interferon alpha-1/13 Human genes 0.000 description 2
- 108010002616 Interleukin-5 Proteins 0.000 description 2
- 108090001007 Interleukin-8 Proteins 0.000 description 2
- 108091092195 Intron Proteins 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- 102000003960 Ligases Human genes 0.000 description 2
- 108090000364 Ligases Proteins 0.000 description 2
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 2
- 206010027406 Mesothelioma Diseases 0.000 description 2
- 206010054949 Metaplasia Diseases 0.000 description 2
- 101710151805 Mitochondrial intermediate peptidase 1 Proteins 0.000 description 2
- 241000186366 Mycobacterium bovis Species 0.000 description 2
- 241000700562 Myxoma virus Species 0.000 description 2
- 206010028813 Nausea Diseases 0.000 description 2
- 102000048850 Neoplasm Genes Human genes 0.000 description 2
- 108700019961 Neoplasm Genes Proteins 0.000 description 2
- 102000007530 Neurofibromin 1 Human genes 0.000 description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 2
- 108091092724 Noncoding DNA Proteins 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 108010011536 PTEN Phosphohydrolase Proteins 0.000 description 2
- 102000014160 PTEN Phosphohydrolase Human genes 0.000 description 2
- 206010061332 Paraganglion neoplasm Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000223960 Plasmodium falciparum Species 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 208000005585 Poxviridae Infections Diseases 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 2
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 238000011579 SCID mouse model Methods 0.000 description 2
- 101710142113 Serine protease inhibitor A3K Proteins 0.000 description 2
- 230000024932 T cell mediated immunity Effects 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 101150003725 TK gene Proteins 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 description 2
- 206010057644 Testis cancer Diseases 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 108700019146 Transgenes Proteins 0.000 description 2
- 206010054094 Tumour necrosis Diseases 0.000 description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 description 2
- 101100004091 Vaccinia virus (strain Copenhagen) B15R gene Proteins 0.000 description 2
- 101100102446 Vaccinia virus (strain Copenhagen) C21L gene Proteins 0.000 description 2
- 101100219233 Vaccinia virus (strain Western Reserve) B28R gene Proteins 0.000 description 2
- 101100340726 Vaccinia virus (strain Western Reserve) VACWR197 gene Proteins 0.000 description 2
- 101900335902 Vaccinia virus Complement control protein C3 Proteins 0.000 description 2
- 108020005202 Viral DNA Proteins 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000002424 anti-apoptotic effect Effects 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 108010008730 anticomplement Proteins 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 230000001640 apoptogenic effect Effects 0.000 description 2
- 230000009925 apoptotic mechanism Effects 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 210000001772 blood platelet Anatomy 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 108010006025 bovine growth hormone Proteins 0.000 description 2
- 239000003183 carcinogenic agent Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 238000013170 computed tomography imaging Methods 0.000 description 2
- 230000030944 contact inhibition Effects 0.000 description 2
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 2
- 229960000258 corticotropin Drugs 0.000 description 2
- 201000003740 cowpox Diseases 0.000 description 2
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 210000000805 cytoplasm Anatomy 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000003935 denaturing gradient gel electrophoresis Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 230000001066 destructive effect Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 2
- 229960002986 dinoprostone Drugs 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- 238000006073 displacement reaction Methods 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000003651 drinking water Substances 0.000 description 2
- 235000020188 drinking water Nutrition 0.000 description 2
- 206010013781 dry mouth Diseases 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- 102000015694 estrogen receptors Human genes 0.000 description 2
- 108010038795 estrogen receptors Proteins 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 201000003444 follicular lymphoma Diseases 0.000 description 2
- 210000003976 gap junction Anatomy 0.000 description 2
- 102000034356 gene-regulatory proteins Human genes 0.000 description 2
- 108091006104 gene-regulatory proteins Proteins 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 210000004195 gingiva Anatomy 0.000 description 2
- 208000005017 glioblastoma Diseases 0.000 description 2
- 230000003676 hair loss Effects 0.000 description 2
- 208000024963 hair loss Diseases 0.000 description 2
- 201000010536 head and neck cancer Diseases 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 229940022353 herceptin Drugs 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 238000001794 hormone therapy Methods 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 230000002390 hyperplastic effect Effects 0.000 description 2
- 230000036031 hyperthermia Effects 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 239000000568 immunological adjuvant Substances 0.000 description 2
- 239000002955 immunomodulating agent Substances 0.000 description 2
- 230000004957 immunoregulator effect Effects 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 230000006882 induction of apoptosis Effects 0.000 description 2
- 210000004969 inflammatory cell Anatomy 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 230000014828 interferon-gamma production Effects 0.000 description 2
- 229940047124 interferons Drugs 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 230000031146 intracellular signal transduction Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 238000011901 isothermal amplification Methods 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- 231100001231 less toxic Toxicity 0.000 description 2
- 208000037841 lung tumor Diseases 0.000 description 2
- 201000009020 malignant peripheral nerve sheath tumor Diseases 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 210000001758 mesenteric vein Anatomy 0.000 description 2
- 230000015689 metaplastic ossification Effects 0.000 description 2
- 239000000693 micelle Substances 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 230000003990 molecular pathway Effects 0.000 description 2
- OHDXDNUPVVYWOV-UHFFFAOYSA-N n-methyl-1-(2-naphthalen-1-ylsulfanylphenyl)methanamine Chemical compound CNCC1=CC=CC=C1SC1=CC=CC2=CC=CC=C12 OHDXDNUPVVYWOV-UHFFFAOYSA-N 0.000 description 2
- 230000008693 nausea Effects 0.000 description 2
- 230000006654 negative regulation of apoptotic process Effects 0.000 description 2
- 208000029974 neurofibrosarcoma Diseases 0.000 description 2
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 2
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 2
- 208000007312 paraganglioma Diseases 0.000 description 2
- 238000003752 polymerase chain reaction Methods 0.000 description 2
- 230000004481 post-translational protein modification Effects 0.000 description 2
- 230000037452 priming Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 2
- 210000001938 protoplast Anatomy 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 238000002708 random mutagenesis Methods 0.000 description 2
- 108700015048 receptor decoy activity proteins Proteins 0.000 description 2
- 238000005215 recombination Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000026267 regulation of growth Effects 0.000 description 2
- 238000007894 restriction fragment length polymorphism technique Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 238000012868 site-directed mutagenesis technique Methods 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 238000011477 surgical intervention Methods 0.000 description 2
- 230000009747 swallowing Effects 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 201000003120 testicular cancer Diseases 0.000 description 2
- 210000001550 testis Anatomy 0.000 description 2
- 231100001274 therapeutic index Toxicity 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 208000030901 thyroid gland follicular carcinoma Diseases 0.000 description 2
- 210000002105 tongue Anatomy 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- 210000003932 urinary bladder Anatomy 0.000 description 2
- 206010046766 uterine cancer Diseases 0.000 description 2
- 238000002255 vaccination Methods 0.000 description 2
- 230000009278 visceral effect Effects 0.000 description 2
- JSPNNZKWADNWHI-PNANGNLXSA-N (2r)-2-hydroxy-n-[(2s,3r,4e,8e)-3-hydroxy-9-methyl-1-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoctadeca-4,8-dien-2-yl]heptadecanamide Chemical compound CCCCCCCCCCCCCCC[C@@H](O)C(=O)N[C@H]([C@H](O)\C=C\CC\C=C(/C)CCCCCCCCC)CO[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O JSPNNZKWADNWHI-PNANGNLXSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- XAXNKAGAUFXFDO-JVJDXIHNSA-N (e)-n-[(4r,4as,7ar,12br)-3-(cyclopropylmethyl)-9-hydroxy-7-oxo-2,4,5,6,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-4a-yl]-3-(4-chlorophenyl)prop-2-enamide;methanesulfonic acid Chemical compound CS(O)(=O)=O.N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)NC(=O)\C=C\C=2C=CC(Cl)=CC=2)CC1)O)CC1CC1 XAXNKAGAUFXFDO-JVJDXIHNSA-N 0.000 description 1
- VVJYUAYZJAKGRQ-BGZDPUMWSA-N 1-[(2r,4r,5s,6r)-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)C1 VVJYUAYZJAKGRQ-BGZDPUMWSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- MEOVPKDOYAIVHZ-UHFFFAOYSA-N 2-chloro-1-(1-methylpyrrol-2-yl)ethanol Chemical compound CN1C=CC=C1C(O)CCl MEOVPKDOYAIVHZ-UHFFFAOYSA-N 0.000 description 1
- OPIFSICVWOWJMJ-AEOCFKNESA-N 5-bromo-4-chloro-3-indolyl beta-D-galactoside Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OC1=CNC2=CC=C(Br)C(Cl)=C12 OPIFSICVWOWJMJ-AEOCFKNESA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- 208000016557 Acute basophilic leukemia Diseases 0.000 description 1
- 208000003200 Adenoma Diseases 0.000 description 1
- 206010001233 Adenoma benign Diseases 0.000 description 1
- 206010068873 Adenosquamous cell carcinoma Diseases 0.000 description 1
- 108010027410 Adenovirus E3 Proteins Proteins 0.000 description 1
- 208000006468 Adrenal Cortex Neoplasms Diseases 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 241000589158 Agrobacterium Species 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 244000144725 Amygdalus communis Species 0.000 description 1
- 241000207875 Antirrhinum Species 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 241000219194 Arabidopsis Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 241000700663 Avipoxvirus Species 0.000 description 1
- 101150006352 B18R gene Proteins 0.000 description 1
- 101150095960 B19R gene Proteins 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 206010004593 Bile duct cancer Diseases 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 206010006002 Bone pain Diseases 0.000 description 1
- 101100263837 Bovine ephemeral fever virus (strain BB7721) beta gene Proteins 0.000 description 1
- 208000007690 Brenner tumor Diseases 0.000 description 1
- 208000003170 Bronchiolo-Alveolar Adenocarcinoma Diseases 0.000 description 1
- 241001455646 Buffalopox virus Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 102100031092 C-C motif chemokine 3 Human genes 0.000 description 1
- 101710155856 C-C motif chemokine 3 Proteins 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- 102100040839 C-type lectin domain family 6 member A Human genes 0.000 description 1
- 101710125370 C-type lectin domain family 6 member A Proteins 0.000 description 1
- 102100040840 C-type lectin domain family 7 member A Human genes 0.000 description 1
- 101150018201 C11L gene Proteins 0.000 description 1
- 102000001902 CC Chemokines Human genes 0.000 description 1
- 108010040471 CC Chemokines Proteins 0.000 description 1
- 108010062802 CD66 antigens Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- 241000700664 Capripoxvirus Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 1
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 1
- 201000000274 Carcinosarcoma Diseases 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 102100035904 Caspase-1 Human genes 0.000 description 1
- 108010076667 Caspases Proteins 0.000 description 1
- 102000011727 Caspases Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 206010050337 Cerumen impaction Diseases 0.000 description 1
- 102000001326 Chemokine CCL4 Human genes 0.000 description 1
- 108010055165 Chemokine CCL4 Proteins 0.000 description 1
- 206010008583 Chloroma Diseases 0.000 description 1
- 108010049048 Cholera Toxin Proteins 0.000 description 1
- 102000009016 Cholera Toxin Human genes 0.000 description 1
- 241000251730 Chondrichthyes Species 0.000 description 1
- 201000009047 Chordoma Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 108010034753 Complement Membrane Attack Complex Proteins 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 1
- 101100233978 Cowpox virus (strain GRI-90 / Grishak) KBTB2 gene Proteins 0.000 description 1
- 108050006400 Cyclin Proteins 0.000 description 1
- 102000016736 Cyclin Human genes 0.000 description 1
- 102000013701 Cyclin-Dependent Kinase 4 Human genes 0.000 description 1
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 1
- 102000013698 Cyclin-Dependent Kinase 6 Human genes 0.000 description 1
- 108010025468 Cyclin-Dependent Kinase 6 Proteins 0.000 description 1
- 229940083347 Cyclin-dependent kinase 4 inhibitor Drugs 0.000 description 1
- 102100033270 Cyclin-dependent kinase inhibitor 1 Human genes 0.000 description 1
- 102100033233 Cyclin-dependent kinase inhibitor 1B Human genes 0.000 description 1
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 1
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 102000004594 DNA Polymerase I Human genes 0.000 description 1
- 108010017826 DNA Polymerase I Proteins 0.000 description 1
- 102000011724 DNA Repair Enzymes Human genes 0.000 description 1
- 108010076525 DNA Repair Enzymes Proteins 0.000 description 1
- 102000003915 DNA Topoisomerases Human genes 0.000 description 1
- 108090000323 DNA Topoisomerases Proteins 0.000 description 1
- 239000012623 DNA damaging agent Substances 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 108700010025 DRD1 Proteins 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- 241000275449 Diplectrum formosum Species 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 101710140859 E3 ubiquitin ligase TRAF3IP2 Proteins 0.000 description 1
- 102100026620 E3 ubiquitin ligase TRAF3IP2 Human genes 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000710188 Encephalomyocarditis virus Species 0.000 description 1
- 208000005431 Endometrioid Carcinoma Diseases 0.000 description 1
- 101100316840 Enterobacteria phage P4 Beta gene Proteins 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 102100023688 Eotaxin Human genes 0.000 description 1
- 101710139422 Eotaxin Proteins 0.000 description 1
- 206010014967 Ependymoma Diseases 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 208000031637 Erythroblastic Acute Leukemia Diseases 0.000 description 1
- 208000036566 Erythroleukaemia Diseases 0.000 description 1
- 241001524679 Escherichia virus M13 Species 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 208000014061 Extranodal Extension Diseases 0.000 description 1
- 101150021185 FGF gene Proteins 0.000 description 1
- 206010016059 Facial pain Diseases 0.000 description 1
- VTLYFUHAOXGGBS-UHFFFAOYSA-N Fe3+ Chemical compound [Fe+3] VTLYFUHAOXGGBS-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 238000000729 Fisher's exact test Methods 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 102000034348 GTPases Human genes 0.000 description 1
- 108091006109 GTPases Proteins 0.000 description 1
- 229940032072 GVAX vaccine Drugs 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 108700007698 Genetic Terminator Regions Proteins 0.000 description 1
- 208000021309 Germ cell tumor Diseases 0.000 description 1
- 208000002966 Giant Cell Tumor of Bone Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 206010018498 Goitre Diseases 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 208000006050 Hemangiopericytoma Diseases 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 208000009889 Herpes Simplex Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 208000002291 Histiocytic Sarcoma Diseases 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 101100005713 Homo sapiens CD4 gene Proteins 0.000 description 1
- 101000944380 Homo sapiens Cyclin-dependent kinase inhibitor 1 Proteins 0.000 description 1
- 101000944361 Homo sapiens Cyclin-dependent kinase inhibitor 1B Proteins 0.000 description 1
- 101001056180 Homo sapiens Induced myeloid leukemia cell differentiation protein Mcl-1 Proteins 0.000 description 1
- 101001033249 Homo sapiens Interleukin-1 beta Proteins 0.000 description 1
- 101100343328 Homo sapiens LIMK2 gene Proteins 0.000 description 1
- 101001034314 Homo sapiens Lactadherin Proteins 0.000 description 1
- 101001116302 Homo sapiens Platelet endothelial cell adhesion molecule Proteins 0.000 description 1
- 101000621344 Homo sapiens Protein Wnt-2 Proteins 0.000 description 1
- 101000651211 Homo sapiens Transcription factor PU.1 Proteins 0.000 description 1
- 101000610604 Homo sapiens Tumor necrosis factor receptor superfamily member 10B Proteins 0.000 description 1
- 101000610602 Homo sapiens Tumor necrosis factor receptor superfamily member 10C Proteins 0.000 description 1
- 101000610609 Homo sapiens Tumor necrosis factor receptor superfamily member 10D Proteins 0.000 description 1
- 241000598171 Human adenovirus sp. Species 0.000 description 1
- 101000829171 Hypocrea virens (strain Gv29-8 / FGSC 10586) Effector TSP1 Proteins 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 102100026539 Induced myeloid leukemia cell differentiation protein Mcl-1 Human genes 0.000 description 1
- 108020005350 Initiator Codon Proteins 0.000 description 1
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 1
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 102100026720 Interferon beta Human genes 0.000 description 1
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 1
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 102000004388 Interleukin-4 Human genes 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- ZQISRDCJNBUVMM-UHFFFAOYSA-N L-Histidinol Natural products OCC(N)CC1=CN=CN1 ZQISRDCJNBUVMM-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZQISRDCJNBUVMM-YFKPBYRVSA-N L-histidinol Chemical compound OC[C@@H](N)CC1=CNC=N1 ZQISRDCJNBUVMM-YFKPBYRVSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 102100039648 Lactadherin Human genes 0.000 description 1
- 108010000851 Laminin Receptors Proteins 0.000 description 1
- 102000002297 Laminin Receptors Human genes 0.000 description 1
- 208000018142 Leiomyosarcoma Diseases 0.000 description 1
- UPYKUZBSLRQECL-UKMVMLAPSA-N Lycopene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1C(=C)CCCC1(C)C)C=CC=C(/C)C=CC2C(=C)CCCC2(C)C UPYKUZBSLRQECL-UKMVMLAPSA-N 0.000 description 1
- JEVVKJMRZMXFBT-XWDZUXABSA-N Lycophyll Natural products OC/C(=C/CC/C(=C\C=C\C(=C/C=C/C(=C\C=C\C=C(/C=C/C=C(\C=C\C=C(/CC/C=C(/CO)\C)\C)/C)\C)/C)\C)/C)/C JEVVKJMRZMXFBT-XWDZUXABSA-N 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- 102000008072 Lymphokines Human genes 0.000 description 1
- 108010074338 Lymphokines Proteins 0.000 description 1
- 108090000542 Lymphotoxin-alpha Proteins 0.000 description 1
- 102000004083 Lymphotoxin-alpha Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 238000000719 MTS assay Methods 0.000 description 1
- 231100000070 MTS assay Toxicity 0.000 description 1
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 208000007369 Malignant Mixed Tumor Diseases 0.000 description 1
- 206010025566 Malignant haemangiopericytoma Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 206010027145 Melanocytic naevus Diseases 0.000 description 1
- 206010027193 Meningioma malignant Diseases 0.000 description 1
- 201000009574 Mesenchymal Chondrosarcoma Diseases 0.000 description 1
- 206010027457 Metastases to liver Diseases 0.000 description 1
- 206010027465 Metastases to skin Diseases 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 208000013811 Mueller mixed tumour Diseases 0.000 description 1
- 206010073148 Multiple endocrine neoplasia type 2A Diseases 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 101000746372 Mus musculus Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 1
- 101100020679 Mus musculus Lcn5 gene Proteins 0.000 description 1
- 102000010645 MutS Proteins Human genes 0.000 description 1
- 108010038272 MutS Proteins Proteins 0.000 description 1
- 208000000112 Myalgia Diseases 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- NFOMHWALMFWNAQ-UHFFFAOYSA-N N-acetoxy-2-acetamidofluorene Chemical compound C1=CC=C2C3=CC=C(N(C(C)=O)OC(=O)C)C=C3CC2=C1 NFOMHWALMFWNAQ-UHFFFAOYSA-N 0.000 description 1
- 150000004008 N-nitroso compounds Chemical class 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 208000003788 Neoplasm Micrometastasis Diseases 0.000 description 1
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 1
- 101100007739 Neosartorya fumigata (strain ATCC MYA-4609 / Af293 / CBS 101355 / FGSC A1100) crmA gene Proteins 0.000 description 1
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 1
- 102000008021 Nucleoside-Triphosphatase Human genes 0.000 description 1
- 108010075285 Nucleoside-Triphosphatase Proteins 0.000 description 1
- 208000007871 Odontogenic Tumors Diseases 0.000 description 1
- 201000010133 Oligodendroglioma Diseases 0.000 description 1
- 208000001388 Opportunistic Infections Diseases 0.000 description 1
- 206010068319 Oropharyngeal pain Diseases 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 101000911993 Ovis aries CD59 glycoprotein Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000027868 Paget disease Diseases 0.000 description 1
- 241000700639 Parapoxvirus Species 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000004503 Perforin Human genes 0.000 description 1
- 108010056995 Perforin Proteins 0.000 description 1
- KHGNFPUMBJSZSM-UHFFFAOYSA-N Perforine Natural products COC1=C2CCC(O)C(CCC(C)(C)O)(OC)C2=NC2=C1C=CO2 KHGNFPUMBJSZSM-UHFFFAOYSA-N 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 201000007100 Pharyngitis Diseases 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 208000009077 Pigmented Nevus Diseases 0.000 description 1
- 208000007641 Pinealoma Diseases 0.000 description 1
- 102100022427 Plasmalemma vesicle-associated protein Human genes 0.000 description 1
- 101710193105 Plasmalemma vesicle-associated protein Proteins 0.000 description 1
- 102000037602 Platelet Endothelial Cell Adhesion Molecule-1 Human genes 0.000 description 1
- 102000004211 Platelet factor 4 Human genes 0.000 description 1
- 108090000778 Platelet factor 4 Proteins 0.000 description 1
- 208000000474 Poliomyelitis Diseases 0.000 description 1
- 101710124239 Poly(A) polymerase Proteins 0.000 description 1
- 208000037062 Polyps Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 102100038277 Prostaglandin G/H synthase 1 Human genes 0.000 description 1
- 108050003243 Prostaglandin G/H synthase 1 Proteins 0.000 description 1
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 1
- 102000007066 Prostate-Specific Antigen Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102000055027 Protein Methyltransferases Human genes 0.000 description 1
- 108700040121 Protein Methyltransferases Proteins 0.000 description 1
- 102100022805 Protein Wnt-2 Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 108010066717 Q beta Replicase Proteins 0.000 description 1
- 108020004518 RNA Probes Proteins 0.000 description 1
- 239000003391 RNA probe Substances 0.000 description 1
- 239000013614 RNA sample Substances 0.000 description 1
- 230000007022 RNA scission Effects 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 101000836070 Rattus norvegicus Serine protease inhibitor A3L Proteins 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 101710100968 Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 206010070308 Refractory cancer Diseases 0.000 description 1
- 208000007660 Residual Neoplasm Diseases 0.000 description 1
- 108050002653 Retinoblastoma protein Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 206010070834 Sensitisation Diseases 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 108050000761 Serpin Proteins 0.000 description 1
- 102000008847 Serpin Human genes 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 206010040880 Skin irritation Diseases 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 241000700568 Suipoxvirus Species 0.000 description 1
- 101100064373 Swinepox virus (strain Kasza) DUT gene Proteins 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- 108060008245 Thrombospondin Proteins 0.000 description 1
- 102000002938 Thrombospondin Human genes 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical class IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- 102100030951 Tissue factor pathway inhibitor Human genes 0.000 description 1
- 102100040250 Transcription elongation factor A protein-like 1 Human genes 0.000 description 1
- 102100027654 Transcription factor PU.1 Human genes 0.000 description 1
- 108010091356 Tumor Protein p73 Proteins 0.000 description 1
- 102000018252 Tumor Protein p73 Human genes 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 102000015098 Tumor Suppressor Protein p53 Human genes 0.000 description 1
- 108010078814 Tumor Suppressor Protein p53 Proteins 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 102100040115 Tumor necrosis factor receptor superfamily member 10C Human genes 0.000 description 1
- 102100040110 Tumor necrosis factor receptor superfamily member 10D Human genes 0.000 description 1
- 102000003425 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 108010046334 Urease Proteins 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 101100534077 Vaccinia virus (strain Western Reserve) SPI-1 gene Proteins 0.000 description 1
- 101100004100 Vaccinia virus (strain Western Reserve) VACWR199 gene Proteins 0.000 description 1
- 101900209501 Vaccinia virus Thymidine kinase Proteins 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 206010058874 Viraemia Diseases 0.000 description 1
- 101710145727 Viral Fc-gamma receptor-like protein UL119 Proteins 0.000 description 1
- 230000010530 Virus Neutralization Effects 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 238000001793 Wilcoxon signed-rank test Methods 0.000 description 1
- 241000700574 Yatapoxvirus Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000016383 Zea mays subsp huehuetenangensis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 208000021841 acute erythroid leukemia Diseases 0.000 description 1
- 208000002517 adenoid cystic carcinoma Diseases 0.000 description 1
- 201000008395 adenosquamous carcinoma Diseases 0.000 description 1
- 201000002454 adrenal cortex cancer Diseases 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 238000011256 aggressive treatment Methods 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 238000012867 alanine scanning Methods 0.000 description 1
- 150000001295 alanines Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 206010065867 alveolar rhabdomyosarcoma Diseases 0.000 description 1
- 208000010029 ameloblastoma Diseases 0.000 description 1
- 150000003862 amino acid derivatives Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- BSJGASKRWFKGMV-UHFFFAOYSA-L ammonia dichloroplatinum(2+) Chemical compound N.N.Cl[Pt+2]Cl BSJGASKRWFKGMV-UHFFFAOYSA-L 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000002391 anti-complement effect Effects 0.000 description 1
- 230000003302 anti-idiotype Effects 0.000 description 1
- 230000002785 anti-thrombosis Effects 0.000 description 1
- 230000002137 anti-vascular effect Effects 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 230000007416 antiviral immune response Effects 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- 230000005775 apoptotic pathway Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 210000002565 arteriole Anatomy 0.000 description 1
- 210000004507 artificial chromosome Anatomy 0.000 description 1
- 210000001106 artificial yeast chromosome Anatomy 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 201000005476 astroblastoma Diseases 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000003305 autocrine Effects 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 239000013602 bacteriophage vector Substances 0.000 description 1
- 230000037429 base substitution Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 208000026900 bile duct neoplasm Diseases 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 201000000053 blastoma Diseases 0.000 description 1
- 208000034158 bleeding Diseases 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 238000009534 blood test Methods 0.000 description 1
- 208000007047 blue nevus Diseases 0.000 description 1
- 201000011143 bone giant cell tumor Diseases 0.000 description 1
- 210000002302 brachial artery Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 230000000981 bystander Effects 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 231100000357 carcinogen Toxicity 0.000 description 1
- 210000004004 carotid artery internal Anatomy 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 230000011748 cell maturation Effects 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 230000010307 cell transformation Effects 0.000 description 1
- 108091092328 cellular RNA Proteins 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 229930183167 cerebroside Natural products 0.000 description 1
- RIZIAUKTHDLMQX-UHFFFAOYSA-N cerebroside D Natural products CCCCCCCCCCCCCCCCC(O)C(=O)NC(C(O)C=CCCC=C(C)CCCCCCCCC)COC1OC(CO)C(O)C(O)C1O RIZIAUKTHDLMQX-UHFFFAOYSA-N 0.000 description 1
- 210000002939 cerumen Anatomy 0.000 description 1
- 235000015111 chews Nutrition 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 229930002875 chlorophyll Natural products 0.000 description 1
- 235000019804 chlorophyll Nutrition 0.000 description 1
- ATNHDLDRLWWWCB-AENOIHSZSA-M chlorophyll a Chemical compound C1([C@@H](C(=O)OC)C(=O)C2=C3C)=C2N2C3=CC(C(CC)=C3C)=[N+]4C3=CC3=C(C=C)C(C)=C5N3[Mg-2]42[N+]2=C1[C@@H](CCC(=O)OC\C=C(/C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)[C@H](C)C2=C5 ATNHDLDRLWWWCB-AENOIHSZSA-M 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 201000005217 chondroblastoma Diseases 0.000 description 1
- 210000003287 ciliary artery Anatomy 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 208000029664 classic familial adenomatous polyposis Diseases 0.000 description 1
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000000084 colloidal system Substances 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000005757 colony formation Effects 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 239000003283 colorimetric indicator Substances 0.000 description 1
- 238000009096 combination chemotherapy Methods 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 230000004154 complement system Effects 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 229940109239 creatinine Drugs 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 238000002681 cryosurgery Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 208000030381 cutaneous melanoma Diseases 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 208000002445 cystadenocarcinoma Diseases 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 230000021040 cytoplasmic transport Effects 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 108010025838 dectin 1 Proteins 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- 230000005860 defense response to virus Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 210000003298 dental enamel Anatomy 0.000 description 1
- CGMRCMMOCQYHAD-UHFFFAOYSA-J dicalcium hydroxide phosphate Chemical compound [OH-].[Ca++].[Ca++].[O-]P([O-])([O-])=O CGMRCMMOCQYHAD-UHFFFAOYSA-J 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 201000008184 embryoma Diseases 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 208000028730 endometrioid adenocarcinoma Diseases 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000001976 enzyme digestion Methods 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 201000010877 epithelioid cell melanoma Diseases 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 206010016256 fatigue Diseases 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000001105 femoral artery Anatomy 0.000 description 1
- 210000003191 femoral vein Anatomy 0.000 description 1
- 230000008175 fetal development Effects 0.000 description 1
- 201000008825 fibrosarcoma of bone Diseases 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 108700014844 flt3 ligand Proteins 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 238000001215 fluorescent labelling Methods 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 230000037433 frameshift Effects 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 150000002270 gangliosides Chemical class 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 208000015419 gastrin-producing neuroendocrine tumor Diseases 0.000 description 1
- 201000000052 gastrinoma Diseases 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 238000010448 genetic screening Methods 0.000 description 1
- 210000002165 glioblast Anatomy 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002327 glycerophospholipids Chemical class 0.000 description 1
- 150000002339 glycosphingolipids Chemical class 0.000 description 1
- 201000003872 goiter Diseases 0.000 description 1
- 208000030316 grade III meningioma Diseases 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 210000004209 hair Anatomy 0.000 description 1
- 210000003780 hair follicle Anatomy 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 210000002989 hepatic vein Anatomy 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 208000029824 high grade glioma Diseases 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 235000011167 hydrochloric acid Nutrition 0.000 description 1
- 230000006951 hyperphosphorylation Effects 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000001571 immunoadjuvant effect Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 230000028802 immunoglobulin-mediated neutralization Effects 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 238000011532 immunohistochemical staining Methods 0.000 description 1
- 229960001438 immunostimulant agent Drugs 0.000 description 1
- 230000001024 immunotherapeutic effect Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 108091008042 inhibitory receptors Proteins 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000010468 interferon response Effects 0.000 description 1
- 230000031261 interleukin-10 production Effects 0.000 description 1
- 108010070145 interleukin-18 binding protein Proteins 0.000 description 1
- 102000044166 interleukin-18 binding protein Human genes 0.000 description 1
- 108090000237 interleukin-24 Proteins 0.000 description 1
- 102000003898 interleukin-24 Human genes 0.000 description 1
- 210000002490 intestinal epithelial cell Anatomy 0.000 description 1
- 102000027411 intracellular receptors Human genes 0.000 description 1
- 108091008582 intracellular receptors Proteins 0.000 description 1
- 230000004068 intracellular signaling Effects 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 238000002430 laser surgery Methods 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 238000007834 ligase chain reaction Methods 0.000 description 1
- 125000003473 lipid group Chemical group 0.000 description 1
- 108010013555 lipoprotein-associated coagulation inhibitor Proteins 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 229960004999 lycopene Drugs 0.000 description 1
- OAIJSZIZWZSQBC-GYZMGTAESA-N lycopene Chemical compound CC(C)=CCC\C(C)=C\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C=C(/C)CCC=C(C)C OAIJSZIZWZSQBC-GYZMGTAESA-N 0.000 description 1
- 239000001751 lycopene Substances 0.000 description 1
- 235000012661 lycopene Nutrition 0.000 description 1
- 208000012804 lymphangiosarcoma Diseases 0.000 description 1
- 210000001365 lymphatic vessel Anatomy 0.000 description 1
- 230000000527 lymphocytic effect Effects 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 208000025036 lymphosarcoma Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 235000009973 maize Nutrition 0.000 description 1
- 201000007055 malignant Leydig cell tumor Diseases 0.000 description 1
- 201000011614 malignant glioma Diseases 0.000 description 1
- 201000006812 malignant histiocytosis Diseases 0.000 description 1
- 208000006178 malignant mesothelioma Diseases 0.000 description 1
- 208000021810 malignant mixed neoplasm Diseases 0.000 description 1
- 208000028676 malignant spindle cell neoplasm Diseases 0.000 description 1
- 201000000289 malignant teratoma Diseases 0.000 description 1
- 208000025854 malignant tumor of adrenal cortex Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 208000027202 mammary Paget disease Diseases 0.000 description 1
- 208000000516 mast-cell leukemia Diseases 0.000 description 1
- 201000008749 mast-cell sarcoma Diseases 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 210000003593 megakaryocyte Anatomy 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 206010027191 meningioma Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000000051 modifying effect Effects 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 208000010492 mucinous cystadenocarcinoma Diseases 0.000 description 1
- 230000036457 multidrug resistance Effects 0.000 description 1
- 206010051747 multiple endocrine neoplasia Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 208000013465 muscle pain Diseases 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 201000005962 mycosis fungoides Diseases 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 201000005987 myeloid sarcoma Diseases 0.000 description 1
- 208000009091 myxoma Diseases 0.000 description 1
- 208000001611 myxosarcoma Diseases 0.000 description 1
- 210000001989 nasopharynx Anatomy 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 210000005170 neoplastic cell Anatomy 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 208000027831 neuroepithelial neoplasm Diseases 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 231100000957 no side effect Toxicity 0.000 description 1
- 210000004967 non-hematopoietic stem cell Anatomy 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 230000008689 nuclear function Effects 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 239000002853 nucleic acid probe Substances 0.000 description 1
- 238000001668 nucleic acid synthesis Methods 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 208000027825 odontogenic neoplasm Diseases 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 210000004798 organs belonging to the digestive system Anatomy 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 238000011499 palliative surgery Methods 0.000 description 1
- 208000004019 papillary adenocarcinoma Diseases 0.000 description 1
- 201000010210 papillary cystadenocarcinoma Diseases 0.000 description 1
- 208000024641 papillary serous cystadenocarcinoma Diseases 0.000 description 1
- 201000001494 papillary transitional carcinoma Diseases 0.000 description 1
- 208000031101 papillary transitional cell carcinoma Diseases 0.000 description 1
- 230000003076 paracrine Effects 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 229930192851 perforin Natural products 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 102000013415 peroxidase activity proteins Human genes 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 208000024724 pineal body neoplasm Diseases 0.000 description 1
- BLFWHYXWBKKRHI-JYBILGDPSA-N plap Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(=O)[C@@H]1CCCN1C(=O)[C@H](CO)NC(=O)[C@@H](N)CCC(O)=O BLFWHYXWBKKRHI-JYBILGDPSA-N 0.000 description 1
- 208000031223 plasma cell leukemia Diseases 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 230000029279 positive regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000012342 propidium iodide staining Methods 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 description 1
- 230000012846 protein folding Effects 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 210000001147 pulmonary artery Anatomy 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 230000003439 radiotherapeutic effect Effects 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 208000016691 refractory malignant neoplasm Diseases 0.000 description 1
- 230000012121 regulation of immune response Effects 0.000 description 1
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000009711 regulatory function Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000003307 reticuloendothelial effect Effects 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 238000012340 reverse transcriptase PCR Methods 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 201000007416 salivary gland adenoid cystic carcinoma Diseases 0.000 description 1
- 230000002784 sclerotic effect Effects 0.000 description 1
- 201000008407 sebaceous adenocarcinoma Diseases 0.000 description 1
- 238000011519 second-line treatment Methods 0.000 description 1
- 230000008313 sensitization Effects 0.000 description 1
- 230000000405 serological effect Effects 0.000 description 1
- 210000000717 sertoli cell Anatomy 0.000 description 1
- 231100000004 severe toxicity Toxicity 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 125000005630 sialyl group Chemical group 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 1
- 229910010271 silicon carbide Inorganic materials 0.000 description 1
- 108700021652 sis Genes Proteins 0.000 description 1
- 230000036556 skin irritation Effects 0.000 description 1
- 231100000475 skin irritation Toxicity 0.000 description 1
- 201000003708 skin melanoma Diseases 0.000 description 1
- 208000000649 small cell carcinoma Diseases 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 208000021795 small intestine disease Diseases 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 108090000586 somatostatin receptor 2 Proteins 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 210000000955 splenic vein Anatomy 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000011301 standard therapy Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 208000028210 stromal sarcoma Diseases 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 238000000015 thermotherapy Methods 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 239000005495 thyroid hormone Substances 0.000 description 1
- 229940036555 thyroid hormone Drugs 0.000 description 1
- 102000004217 thyroid hormone receptors Human genes 0.000 description 1
- 108090000721 thyroid hormone receptors Proteins 0.000 description 1
- 230000030968 tissue homeostasis Effects 0.000 description 1
- 231100000155 toxicity by organ Toxicity 0.000 description 1
- 230000007675 toxicity by organ Effects 0.000 description 1
- 238000013417 toxicology model Methods 0.000 description 1
- 238000002627 tracheal intubation Methods 0.000 description 1
- ZCIHMQAPACOQHT-ZGMPDRQDSA-N trans-isorenieratene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/c1c(C)ccc(C)c1C)C=CC=C(/C)C=Cc2c(C)ccc(C)c2C ZCIHMQAPACOQHT-ZGMPDRQDSA-N 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 230000014599 transmission of virus Effects 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 230000005909 tumor killing Effects 0.000 description 1
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 238000012285 ultrasound imaging Methods 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000001631 vena cava inferior Anatomy 0.000 description 1
- 210000002620 vena cava superior Anatomy 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 238000007794 visualization technique Methods 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
Images












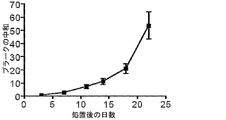
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
- A61K35/768—Oncolytic viruses not provided for in groups A61K35/761 - A61K35/766
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0271—Chimeric vertebrates, e.g. comprising exogenous cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/0004—Screening or testing of compounds for diagnosis of disorders, assessment of conditions, e.g. renal clearance, gastric emptying, testing for diabetes, allergy, rheuma, pancreas functions
- A61K49/0008—Screening agents using (non-human) animal models or transgenic animal models or chimeric hosts, e.g. Alzheimer disease animal model, transgenic model for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/53—Colony-stimulating factor [CSF]
- C07K14/535—Granulocyte CSF; Granulocyte-macrophage CSF
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2207/00—Modified animals
- A01K2207/20—Animals treated with compounds which are neither proteins nor nucleic acids
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/105—Murine
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/03—Animal model, e.g. for test or diseases
- A01K2267/0331—Animal model for proliferative diseases
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/03—Animal model, e.g. for test or diseases
- A01K2267/0393—Animal model comprising a reporter system for screening tests
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
- C12N2710/24132—Use of virus as therapeutic agent, other than vaccine, e.g. as cytolytic agent
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/24011—Poxviridae
- C12N2710/24111—Orthopoxvirus, e.g. vaccinia virus, variola
- C12N2710/24141—Use of virus, viral particle or viral elements as a vector
- C12N2710/24143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Virology (AREA)
- Genetics & Genomics (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biomedical Technology (AREA)
- Biochemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Toxicology (AREA)
- Immunology (AREA)
- Mycology (AREA)
- Oncology (AREA)
- Environmental Sciences (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Diabetes (AREA)
- Animal Husbandry (AREA)
- Rheumatology (AREA)
- Cell Biology (AREA)
- Pathology (AREA)
- Urology & Nephrology (AREA)
Abstract
Description
本発明は、一般的に腫瘍学およびウイルス学の分野に関する。より具体的には、本発明は、GM-CSFを発現するワクシニアウイルスおよび癌を処置するための全身投与におけるその利用に関する。なお、本出願はその全内容が参照により本明細書に組み入れられる、2005年9月7日に提出された米国特許仮出願第60/714,679号に対する優先権を主張する。
正常組織の恒常性は、細胞増殖および細胞死の高度に調節されたプロセスである。細胞増殖または細胞死のいずれかの不均衡は、癌様状態に発展しうる(Solyanikら、1995;Stokkeら、1997;Mumby and Walter、1991;Natoliら、1998;Magi-Galluzziら、1998)。例えば、子宮頚癌、腎臓癌、肺癌、膵臓癌、結腸直腸癌、および脳癌は、起こりうる多くの癌のごく少数の例である(Erlandsson、1998;Kolmel、1998;Mangray and King、1998;Gertig and Hunter、1997;Mouginら、1998)。実際に、癌の発生は非常に高く、米国だけでも毎年500,000人以上の死亡は癌が原因である。
このように、本発明に従って、投与が血管内である、顆粒球-マクロファージコロニー刺激因子(GM-CSF)をコードする核酸の発現を指示するプロモーターと共に発現領域を有する複製型ワクシニアウイルスの有効量を被験者に投与する段階を含む、被験者における癌細胞を殺す方法が提供される。核酸はヒトGM-CSFをコードすることが具体的に企図される。
GM-CSFを発現するポックスウイルスは、黒色腫を有する動物および患者における腫瘍内注射後の有効性および安全性が証明された(Mastrangelo et al, 1999; 2000)。腫瘍の退縮および安定化を含む、局所注射部位および遠隔効果が認められた(Mastrangelo et al, 1999)。有効性は哺乳動物における癌細胞に対する免疫応答の誘導によると提唱された。米国特許第6,093,700号および第6,475,999号は、黒色腫、頭頚部癌、前立腺癌、および膀胱癌に対してインサイチューで免疫するために、ヒトにおいてGM-CSFを腫瘍内注射によって送達するためにポックスウイルスを用いることを提唱している(Mastrangelo et al, 2002)。これらの癌は、腫瘍内注射を指示するためにその表在性の本質および近づきやすさのために選択された。これらの癌はまた、全身性の腫瘍特異的細胞障害性Tリンパ球を誘導する免疫治療アプローチに対しても感受性があることが報告されている。GM-CSFは腫瘍特異的細胞障害性Tリンパ球(CTL)の強力な誘導物質であることが証明された(Dranoff et al., 1993)。ウイルスは、免疫細胞の浸潤および前炎症性サイトカインを誘導することから、ウイルスは、腫瘍塊内でGM-CSFを送達および発現する理想的な方法となる可能性がある。
A.ワクシニアウイルス
ワクシニアウイルスはウイルス学にとって不可解である。ワクシニアウイルスが遺伝子組み換えの産物であるか否か、持続的な連続継代による牛痘ウイルスもしくは天然痘ウイルスに由来する種であるか、または現在は絶滅したウイルスの生存例であるか否かはわかっていない。ワクシニアウイルスは、上腕の皮膚の表層への接種による天然痘ワクチン接種のために用いられた。しかし、天然痘の絶滅により、ワクシニアウイルスによるルーチンのワクチン接種は終了した。ワクシニアに関する最近の関心は、他のウイルスに対する免疫および遺伝子治療のためのベクターとしての可能性がある用途に集中した。
オルトポックスウイルス属は、チョルドポックスウイルス亜科の他のメンバーより比較的均一であり、これには、ワクシニアウイルス、痘瘡ウイルス(天然痘の原因物質)、牛痘ウイルス、バッファロー痘ウイルス、サル痘ウイルス、マウス痘ウイルス、およびウマ痘ウイルス種と共にその他が含まれる、異なるが近縁の11種が含まれる(Moss、1996を参照されたい)。本明細書において記述されるように本発明の特定の態様は、オルトポックスウイルス属の他のメンバーのみならず、パラポックスウイルス、アビポックスウイルス、カプリポックスウイルス、レポリポックスウイルス、スイポックスウイルス、モルシポックスウイルス、およびヤタポックスウイルス属に拡大してもよい。ポックスウイルス科の属は、一般的に、実験動物における中和および交叉反応性を含む血清学的手段によって定義される。オルトポックスウイルス属の様々なメンバーと共にチョルドポックスウイルス亜科の他のメンバーは、宿主生物の免疫応答に拮抗するために免疫調節分子を利用し、その例を本明細書において提供する。このように、本明細書に記述の本発明は、ワクシニアウイルスに限定されないが、多くのウイルスに応用可能でありうる。
ウイルスは、しばしばインターフェロン(-α、-β、-γ)および腫瘍壊死因子-α(TNF)(Moss、1996)のような免疫調節分子によって不活化、阻害、または排泄される。宿主組織および炎症免疫細胞はしばしば、ウイルス感染に反応してこれらの分子を分泌する。これらの分子は、直接の抗ウイルス作用を有しうる、および/または炎症細胞およびリンパ球の動員および/または活性化を通して間接的な作用を有しうる。これらの免疫学的排泄メカニズムの重要性を考慮すると、ウイルスは、これらのサイトカイン/ケモカインおよびインターフェロンの誘導および/または機能を阻害する遺伝子産物の発現に関与する。例えば、ワクシニアウイルス(VV;およびいくつかの他のポックスウイルス)は、CCケモカイン(例えば、RANTES、エオタキシン、MIP-1-α)に結合して阻害する分泌型タンパク質vCKBP(B29R)をコードする(Alcamiら、1998)。いくつかのVV株も同様に、TNFに結合して不活化する分泌型ウイルスタンパク質(例えば、リスターA53R)を発現する(Alcamiら、1999)。ほとんどのポックスウイルス株は、インターフェロン-α/β(例えば、B18R)またはインターフェロン-γ(B8R)に結合してその機能を阻害する分泌型蛋白質をコードする遺伝子を有する。vC12Lは、IL-18によるIFN-γおよびNK細胞/細胞障害性T細胞活性化の誘導を妨害するIL-18結合タンパク質である。
1.インターフェロン調節ポリペプチド
インターフェロン-α/βは、いくつかのメカニズムを通してウイルス複製を遮断する。インターフェロン-γは、直接のウイルス阻害作用はより弱いが、いくつかのメカニズムを通して細胞媒介免疫の強力な誘導物質である。ウイルスは、インターフェロンの抗ウイルス作用に拮抗できる分泌型遺伝子産物を発現するように進化した。例えば、ワクシニアウイルス(および他のポックスウイルス)は、インターフェロン-γおよびα/βにそれぞれ結合する分泌型蛋白質B8RおよびB18Rをコードする(Smithら、1997;Symonsら、1995;Alcamiら、2000)。インターフェロン誘導を減少させるワクシニア遺伝子産物のさらなる例は、インターフェロン-γ誘導因子であるIL-18の活性化を阻害するカスパーゼ-1阻害剤B13Rである。インターフェロン調節ポリペプチドには、ワクシニアウイルスのコペンハーゲン株のような、他のウイルス株ではB19Rと呼ばれることもあるB18R;B8R;B13R;vC12L;A53R;E3Lおよび類似の活性または特性を有する他のウイルスポリペプチドが含まれるがこれらに限定されない。IFN調節ポリペプチドは、選択的にIFNαおよび/またはβ経路を調節するポリペプチド(B18R、B8R、B13R、またはvC12Lのような)、およびIFNγ経路を調節するポリペプチド(例えば、B8R、B13R、またはvC12L)の非排他的分類に分類してもよい。
ウイルス病原体の排泄に関する主なメカニズムは、補体依存的メカニズムによって宿主細胞内の感染細胞または生物内のビリオンを殺すことである。感染細胞は死滅するため、感染ウイルスを産生し続けることは不可能である。さらに、アポトーシスの際に、DNAを分解する細胞内酵素が放出される。これらの酵素によってウイルスDNAの分解およびウイルスの不活化が起こりうる。アポトーシスは、活性化された補体の結合および補体膜攻撃複合体を含む多数のメカニズムによって誘導することができる。ワクシニアのようなポックスウイルスは、ウイルスおよび/またはウイルス感染細胞の補体媒介排泄に拮抗することができる遺伝子産物を発現するように進化した。これらの遺伝子はそれによってアポトーシスを阻害して、補体依存的メカニズムによってウイルス排泄を阻害し、このように、ウイルス感染を進行させて、ウイルスのビルレンスを増加させることができる。例えば、ワクシニアウイルス補体制御タンパク質(VCP;例えばC21L)は、補体媒介殺細胞および/またはウイルス不活化の防止において役割を有する(Isaacsら、1992)。VCPはまた、その発現によってウイルス感染組織への白血球の浸潤が減少することから、抗炎症作用も有する。補体制御ポリペプチドには、C3LまたはC21Lとしても知られるVCPが含まれるがこれらに限定されない。
ウイルス病原体の排泄に関する様々なメカニズムの一つは、上記のようにアポトーシスの誘導によって宿主内で感染細胞を殺すことである。アポトーシスは、細胞内シグナル伝達カスケードの誘因となる細胞TNF受容体に対するTNFおよびリンフォトキシン-α(LTα)の結合を含む、多数のメカニズムによって誘導することができる。TNF受容体の活性化は、免疫および炎症反応の調節と共にアポトーシス細胞死の誘導において機能する(Wallachら、1999)。
ウイルス病原体の排泄に関する主なメカニズムは、宿主における感染細胞のアポトーシスの誘導である。感染細胞は死滅するため、感染ウイルスを産生し続けることは不可能である。さらに、アポトーシスの際に、DNAを分解する細胞内酵素が放出される。これらの酵素はウイルスDNAの分解およびウイルスの不活化を引き起こしうる。アポトーシスは、サイトカイン(例えば、腫瘍壊死因子)の結合、細胞障害性Tリンパ球によるグランザイム産生、またはfas-リガンド結合を含む多数のメカニズムによって誘導することができる;カスパーゼ活性化は、最終的に一般的なアポトーシス経路の重要な部分である。ウイルスは、fasリガンドまたは腫瘍壊死因子(TNF)/TNF関連分子(例えば、アデノウイルスのE3 10.4/14.5、14.7遺伝子(Woldら、1994);アデノウイルスのE1B-19 kD(Boydら、1994);牛痘ウイルスからのcrmA;ワクシニアウイルスからのB13R)を含むそのような分子によって誘導される細胞内シグナル伝達カスケードに拮抗することができる遺伝子産物を発現するように進化した(Dobbelsteinら、1996;Kettleら、1997)。これらの遺伝子産物は、アポトーシス誘導分子によるアポトーシスを阻害して、このように抗ウイルス性のアポトーシス誘導サイトカイン、fas、グランザイム、または他のアポトーシス刺激物質の存在にもかかわらず、ウイルス複製を進行させる。
IL-1βは、局所そして全身性に作用する生物活性因子である。IL-1βとIL-1αとのあいだの機能的な差はごくわずかであることが記述されている。IL-1βの多数の生物活性の例は、IL-1を記述する際の多くの異なる頭字語によって示されている。IL-1は、ブタ細胞において不活性であるヒトIL-1βを例外として種特異性を示さない。IL-1の生物活性のいくつかは、ACTH(コルチコトロピン)、PGE2(プロスタグランジンE2)、PF4(血小板因子-4)、CSF(コロニー刺激因子)、IL-6、およびIL-8を含む他のメディエータの合成の誘導によって間接的に媒介される。IL-1の合成は、TNF-α、IFN-α、IFN-βおよびIFN-γを含む他のサイトカインによって誘導される可能性があり、同様に細菌エンドトキシン、ウイルス、マイトゲンおよび抗原によって誘導される可能性がある。IL-1の主な生物活性は、T-ヘルパー細胞の刺激であり、これはIL-2を分泌して、IL-2受容体を発現するように誘導される。ウイルス感染マクロファージは、大量のIL-1阻害剤を産生し、これはT細胞成熟欠損患者における日和見感染および細胞のトランスフォーメーションを支持する可能性がある。IL-1は、B細胞に直接作用して、その増殖および免疫グロブリンの合成を促進する。IL-1はまた、B細胞をIL-5に対して反応性にするプライミング因子の一つとしても機能する。IL-1は、NK細胞および線維芽細胞、胸腺細胞、神経膠芽細胞の増殖および活性化を刺激する。
ウイルスは伝幡して腫瘍部位に転移し、さらに感染した固形腫瘍塊内でも伝幡するが、一般的に非効率的である(Heiseら、1999)。静脈内投与では典型的に、抗体(例えば、アデノウイルス)(Kayら、1997)および/または補体系(例えば、HSV)(Ikedaら、1999)によるウイルスの排泄または不活化が起こる。これらの免疫媒介メカニズムの他に、これらのウイルスの生体内分泌によって、腫瘍塊よりむしろ正常組織内で静脈内ウイルスの大多数が沈着する。例えば、静脈内のアデノウイルスは、主に肝臓および脾臓にたどり着く;免疫欠損マウスにおいても、腫瘍内に沈着するのは投与されたウイルスの0.1%未満である(Heiseら、1999)。したがって、免疫欠損マウスの腫瘍モデルにおいて、極端に高い相対量によって何らかの控えめな抗腫瘍有効性が証明されうるものの、静脈内輸送は極めて非効率的であり、有効性を有意に制限する。
他のウイルス免疫調節ポリペプチドには、免疫応答の他のメディエータに結合しておよび/または免疫応答に関連した分子経路を調節するポリペプチドが含まれてもよい。例えば、B29R(このタンパク質は存在するが、ワクシニアウイルスのコペンハーゲン株では不活性である可能性がある)、C23L、vCKBP、A41Lおよび類似の活性または特性を有するポリペプチドのようなケモカイン結合ポリペプチド。ウイルスEGF様増殖因子であるワクシニアウイルス増殖因子(例えば、C11L)のような他のワクシニアウイルスタンパク質も同様に、本発明のいくつかの態様において改変の標的となる可能性がある。ウイルス調節因子として分類されてもよい他のポリペプチドには、B7R、N1L、またはその活性もしくは特性がポックスウイルスのビルレンスを増加させる他のポリペプチドが含まれるがこれらに限定されない。
本発明の特定の態様において、A56RまたはK2Lコード核酸遺伝子の改変、欠失、または変異によって、VV感染症によって誘導される細胞細胞融合または融合体形成が起こる可能性がある。ワクシニアウイルス誘導細胞融合は典型的に、腫瘍内ウイルス伝幡によりVVの抗腫瘍効率を増加させるであろう。細胞融合による腫瘍内のウイルス伝幡によって、典型的に、ウイルスは中和抗体および免疫応答を回避することができるであろう。隣接する非感染細胞(すなわち、「傍観者作用」)の殺細胞および感染は、これらの遺伝子の一つまたは双方において変異を有するVVではより効率的となる可能性があり、それによって改善された局所抗腫瘍効果が得られる可能性がある。
ワクシニアウイルスは、参照として本明細書に組み入れられる、Ausbelら、「Current Protocols in Molecular Biology」、16.15.1〜16.18.10頁においてEarlおよびMossによって記述された方法を用いて増殖させてもよい。
本発明は、患者における癌細胞および癌の試験および治療において都合がよいポックスウイルスに関する。これは、ウイルスが癌細胞に対して用いるために所望の特性を有するが、非癌細胞に対しては毒性が低い、または非毒性となるように、野生型と比較して一つまたはそれ以上の変異を有するように構築されていてもよいワクシニアウイルスに関する。そのようなポックスウイルスは、参照により本明細書に組み入れられるPCT/US2003/025141に記載されている。下記の教示は、例として、本発明の方法および組成物を実行するための様々なプロトコールを提供する。それらは、組換えDNA技術を用いて変異ウイルスを作製するためのバックグラウンドを提供する。
特定の態様において、本発明は、一つまたはそれ以上の機能的ポリペプチドまたはタンパク質を欠損していてもよいワクシニアウイルスを作製すること、および/または感染性のEEV型のようなウイルスの特定の型をより多く放出する能力を有するポックスウイルスを産生することに関する。他の態様において、本発明は、ポックスウイルスに関し、および薬学的に許容される製剤の一部としての蛋白質様組成物と併用してそれらを利用することに関する。
本出願がウイルスタンパク質またはポリペプチドの機能または活性に言及する場合、特に明記していない限り、生理的条件でのウイルスタンパク質またはポリペプチドの活性または機能を指すと意味する。例えば、インターフェロン調節ポリペプチドは、少なくとも一つのインターフェロンおよびその活性に直接または間接的に影響を及ぼすポリペプチドを指す。ポリペプチドは、インターフェロンの活性を直接または間接的に誘導、増強、上昇、増加、減少、弱化、低下、阻害、または隠す可能性がある。インターフェロンに直接影響を及ぼす例は、いくつかの態様において、インターフェロンに特異的に結合するインターフェロン調節ポリペプチドを含む。どの分子がこの活性を有するかを決定することは、当業者に周知のアッセイを用いて行ってもよい。例えば、インターフェロンまたはその変種を調節する産物をコードする遺伝子を、そのような遺伝子の移入を有する細胞と比較してインターフェロン活性が誘導されている細胞に移入して、インターフェロン反応の異なるレベルによって、インターフェロン調節機能を有するそれらの分子を同定してもよい。
本発明のポリペプチドのアミノ酸配列変種は、置換、挿入、または欠失変種となりうる。ウイルスタンパク質をコードする遺伝子における変異は、野生型と比較して、ポリペプチドの1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、275、300、325、350、375、400、425、450、475、500個またはそれ以上の非隣接または隣接アミノ酸に影響を及ぼしてもよい。ワクシニアウイルスによってコードされる様々なポリペプチドは、そのそれぞれが参照として本明細書に組み入れられる、Roselら(1986)、Goebelら(1990)、およびGenBankアクセッション番号NC_001559号を参照して同定される可能性がある。

A.天然のタンパク質または改変タンパク質をコードするポリヌクレオチド
本発明は、タンパク質またはポリペプチドの全てまたは一部を発現することができる、細胞から単離可能なポリヌクレオチドに関する。本発明のいくつかの態様において、これは、特定の機能的ウイルスポリペプチドを欠損するウイルスを作製するように特異的に変異させたウイルスゲノムに関する。ポリヌクレオチドは、ウイルスアミノ酸配列の全てもしくは一部を含むペプチドもしくはポリペプチドをコードしてもよく、またはそれらがそのようなウイルスポリペプチドをコードしないように、もしくは少なくとも一つの機能もしくは活性が低下、減少もしくは存在しないウイルスポリペプチドをコードするように操作してもよい。組換え型蛋白質は、活性なタンパク質を生じるように発現細胞から精製することができる。ゲノムは、ワクシニアウイルスのコード領域に関する定義と同様に、そのそれぞれが参照として本明細書に組み入れられる、Roselら(1986)、Goebelら(1990)、および/またはGenBankアクセッション番号NC_00159号において認められる可能性がある。
様々な態様において、ポックスウイルスポリヌクレオチドを改変してもよくまたは変異誘発してもよい。改変または変異には、挿入、欠失、点突然変異、逆位等が含まれてもよく、それによって特定の経路または分子メカニズムの調節、活性化、および/または不活化と共に、特に遺伝子産物を非機能的にするような、遺伝子産物の機能、位置、または発現の変化が起こってもよい。用いられる場合、ポックスウイルスの全てまたは一部をコードするポリヌクレオチドの変異誘発は、多様な標準的な変異誘発技法によって行ってもよい(Sambrookら、1989)。変異は、それによって生物の量または構造に変化が起こるプロセスである。変異は、単一の遺伝子、遺伝子のブロックまたは染色体全体のヌクレオチド配列の改変を含みうる。単一の遺伝子における変化は、DNA配列内の一つのヌクレオチド塩基の除去、付加、もしくは置換を含む点突然変異の結果であってもよく、またはそれらは多数のヌクレオチドの挿入もしくは欠失を含む変化の結果であってもよい。
a.挿入変異誘発
挿入変異誘発は、既知のDNA断片の挿入による遺伝子の不活化に基づく。これはいくつかのタイプのDNA断片の挿入を含むことから、生成された変異は一般的に、機能獲得変異よりはむしろ機能喪失変異である。しかし、機能獲得変異を生じる挿入にはいくつかの例がある。挿入的変異は細菌およびショウジョウバエ(Cooleyら、1988)において非常に成功しており、近年トウモロコシ(シロイヌナズナ(Arabidopsis;(Marksら、1991;Konczら、1990);およびキンギョソウ(Sommerら、1990)において強力なツールとなった。挿入変異誘発は、標準的な分子生物学技術を用いて行ってもよい。
化学的変異誘発は、表現型の重症度が異なる広範な変異を発見する能力のような特定の長所を提供し、実施が容易で安価である。多数の化学的発癌物質がDNAにおいて変異を生じる。ベンゾ[a]ピレン、N-アセトキシ-2-アセチルアミノフルオレンおよびアフロトキシンB1は、細菌および哺乳類細胞においてGCからTAのトランスバージョンを引き起こす。ベンゾ[a]ピレンはまた、ATからTAのような塩基置換を生じうる。N-ニトロソ化合物は、GCからATの転移を生じる。n-ニトロソウレアへの暴露によるチミンのO4位置でのアルキル化によって、TAからCGの転移が起こる。
生体分子は電離放射線によって分解される。入射エネルギーの吸着によって、イオンおよびフリーラジカルの形成が起こり、いくつかの共有結合の切断が起こる。放射線障害に対する感受性は、分子間で、そして同じ分子であっても異なる結晶構造間で非常に多様であるように思われる。これは、総蓄積線量に依存し、同様に線量率にも依存する(フリーラジカルが存在する場合、それらが引き起こす分子の損傷はその本来の拡散速度およびこのようにリアルタイムに依存する)。損傷は、試料を可能な限り低温にすることによって減少および制御される。電離放射線は、一般的に線量率に比例してDNA損傷を引き起こす。
ランダム変異誘発はまた、誤りがちなPCRを用いて導入してもよい。変異誘発率は、鋳型の希釈液を含む多数の試験管においてPCRを行うことによって増加させてもよい。
構造に従う部位特異的変異誘発は、タンパク質-リガンド相互作用を解剖して操作するための強力なツールである(Wells、1996;Braistedら、1996)。この技術は、選択されたDNAに一つまたはそれ以上のヌクレオチド配列を導入することによって配列変種の調製および試験を提供する。
「ベクター」という用語は、外因性の核酸配列が、それが複製されうる細胞に導入するために挿入されうる担体核酸分子を指すために用いられる。核酸配列は、ベクターが導入される細胞に対して異物であること、または配列は細胞における配列と相同であるが、配列が本来認められない宿主細胞内での位置に存在することを意味する「外因性」となりうる。ベクターには、プラスミド、コスミド、ウイルス(バクテリオファージ、動物ウイルス、および植物ウイルス)、および人工染色体(例えば、YAC)が含まれる。当業者は、いずれも参照として本明細書に組み入れられる、Sambrookら(1989)およびAusubelら(1994)に記述される標準的な組換え技術によってベクターを構築するために必要なものを十分に有するであろう。改変ゲロニンのような改変されたポリペプチドをコードする他に、ベクターは、タグまたはターゲティング分子のような非改変ポリペプチド配列をコードしてもよい。そのような融合タンパク質をコードする有用なベクターには、pINベクター(Inouyeら、1985)、ヒスチジン鎖をコードするベクター、および後に精製および分離または切断するためのグルタチオンS-トランスフェラーゼ(GST)可溶性融合タンパク質を作製するために用いられるpGEXベクターが含まれる。ターゲティング分子は、改変ポリペプチドを被験体の体における特定の臓器、組織、細胞、または他の位置に向ける分子である。
「プロモーター」は、そこで転写の開始および速度が制御される核酸配列の領域である。これは、RNAポリメラーゼおよび他の転写因子のような、調節タンパク質および分子が結合する可能性がある遺伝子要素を含んでもよい。「機能的に配置された」、「機能的に結合した」、「制御下である」、および「転写制御下である」という句は、その配列の転写開始および/または発現を制御するために核酸配列に関してプロモーターが正確な機能的位置および/または方向に存在することを意味する。プロモーターは、核酸配列の転写活性化に関与するシス作用調節配列を指す、「エンハンサー」と共に用いてもよく用いなくてもよい。
特異的開始シグナルも同様に、コード配列の効率的な翻訳にとって必要であるかも知れない。これらのシグナルには、ATG開始コドンまたは隣接配列が含まれる。ATG開始コドンを含む外因性の翻訳制御シグナルを提供する必要があるかも知れない。当業者は、容易にこれを決定して、必要なシグナルを提供することができるであろう。開始コドンは、インサート全体の翻訳を確実にするために所望のコード配列の読み取り枠と「フレームが一致して」いなければならない。外因性の翻訳制御シグナルおよび開始コドンは天然または合成のいずれかとなりうる。発現効率は、適当な転写エンハンサー要素を含めることによって増強してもよい。
ベクターには、多数の制限酵素部位を含む核酸領域であって、そのいずれもがベクターを消化するために標準的な組換え技術と共に用いることができる多クローニング部位(MCS)が含まれうる。(参照として本明細書に組み入れられる、Carbonelliら、1999、Levensonら、1998、およびCocea、1997を参照されたい)。「制限酵素消化」は、核酸分子における特定の位置に限って機能する酵素によって核酸分子を触媒的に切断することを指す。これらの制限酵素の多くは市販されている。そのような酵素を用いることは当業者に広く理解されている。しばしば、ベクターは、外因性の配列をベクターにライゲーションできるように、MCS内で切断する制限酵素を用いて直線または断片にする。「ライゲーション」とは、互いに隣接してもしなくてもよい二つの核酸断片のあいだのホスホジエステル結合を形成するプロセスを指す。制限酵素およびライゲーション反応を含む技術は、組換え技術の当業者に周知である。
ほとんどの転写された真核細胞RNA分子は、一次転写物からイントロンを除去するためにRNAスプライシングを受けるであろう。ゲノム真核細胞配列を含むベクターは、タンパク質発現のために転写物の適切なプロセシングを確実にするためにドナーおよび/またはアクセプタースプライシング部位を必要とする可能性がある(参照として本明細書に組み入れられる、Chandlerら、1997を参照されたい)。
本発明のベクターまたは構築物は一般的に、少なくとも一つの終結シグナルを含むであろう。「終結シグナル」または「ターミネータ」は、RNAポリメラーゼによってRNA転写物の特異的終結に関係するDNA配列を含む。このように、特定の態様において、RNA転写物の産生で終結する終結シグナルが企図される。ターミネータは、所望のメッセージレベルを得るためにインビボで必要となる可能性がある。
発現において、特に真核細胞発現において、典型的に、転写物の適当なポリアデニル化を行うためにポリアデニル化シグナルを含めるであろう。ポリアデニル化シグナルの特性は、本発明の実践の成功にとって重要ではないと考えられ、および/またはそのような如何なる配列を用いてもよい。好ましい態様には、様々な標的細胞において都合がよく、および/または十分に機能することがわかっている、SV40ポリアデニル化シグナルおよび/またはウシ成長ホルモンポリアデニル化シグナルが含まれる。ポリアデニル化は、転写物の安定性を増加させる可能性があり、または細胞質輸送を促進する可能性がある。
宿主細胞においてベクターを増殖させるために、ベクターは、複製が開始される特異的核酸配列である一つまたはそれ以上の複製開始部位(しばしば「ori」と呼ばれる)を含んでもよい。または、宿主細胞が酵母である場合、自律的に複製する配列(ARS)を用いることができる。
本発明の特定の態様において、本発明の核酸構築物を含む細胞は、発現ベクターにマーカーを含めることによってインビトロまたはインビボで同定してもよい。そのようなマーカーは、細胞に対して同定可能な変更を付与して、発現ベクターを含む細胞の容易な同定を可能にするであろう。一般的に、選択マーカーは、選択を可能にする特性を付与するマーカーである。陽性選択マーカーは、マーカーが存在することによって選択が可能となるマーカーであり、陰性選択マーカーは、マーカーが存在することによってその選択が妨害されるマーカーである。陽性選択マーカーの例は、薬物耐性マーカーである。
ポックスウイルスタンパク質、ポリペプチド、および/またはペプチドの発現を指示するために用いられる他に、本明細書に開示の核酸配列は、多様な他の用途を有する。例えば、それらは、核酸ハイブリダイゼーションを含む態様に関してプローブまたはプライマーとして有用性を有する。それらは、本発明の診断またはスクリーニング法において用いてもよい。ポックスウイルスまたはポックスウイルスポリペプチド調節物質をコードする核酸の検出は本発明に含まれる。
長さが13〜100ヌクレオチド、好ましくは17〜100ヌクレオチド、または本発明のいくつかの局面において長さが1〜2キロベースもしくはそれ以上のプローブまたはプライマーを用いることによって、安定かつ選択的な二本鎖分子が形成される。長さが20塩基より大きい隣接する枝に対して相補的配列を有する分子は、得られたハイブリッド分子の安定性および/または選択性を増加させるために一般的に好ましい。一般的に20〜30ヌクレオチド、または望ましい場合にはそれより長い一つまたはそれ以上の相補的配列を有する核酸分子をハイブリダイゼーションのために設計することが好まれるであろう。そのような断片は、例えば化学的手段によって断片を直接合成することによって、または選択した配列を組換え産生のために組換えベクターに導入することによって、容易に調製されるであろう。
増幅の鋳型として用いられる核酸は、標準的な方法論に従って(Sambrookら、1989)細胞、組織、または他の試料から単離してもよい。特定の態様において、鋳型核酸を実質的に精製することなく、細胞全体もしくは組織ホモジネート、または生体液試料について分析を行う。核酸は、ゲノムDNAまたは分画もしくは全細胞RNAであってもよい。RNAを用いる場合、まずRNAを相補的DNAに変換することが望ましいかも知れない。
任意の増幅の後、鋳型および/または過剰のプライマーから増幅産物を分離することが望ましいかも知れない。一つの態様において、増幅産物は、標準的な方法(Sambrookら、1989)を用いてアガロース、アガロース-アクリルアミド、またはポリアクリルアミドゲル電気泳動によって分離される。分離された増幅産物を、さらに操作するためにゲルから切断して溶出させてもよい。低融点アガロースゲルを用いて、ゲルを加熱することによって、分離したバンドを採取してその後核酸の抽出を行う。
遺伝子スクリーニングのための他のアッセイは、例えばゲノムDNA、cDNAおよび/またはRNA試料における変異を検出するために、本発明の範囲において用いてもよい。点突然変異を検出するために用いられる方法には、変性勾配ゲル電気泳動(「DGGE」)、制限断片長多形分析(「RFLP」)、化学または酵素切断法、PCR(商標)によって増幅された標的領域の直接シークエンシング(上記を参照されたい)、一本鎖構造多形分析(「SSCP」)、および当技術分野で周知の他の方法が含まれる。
本発明の組成物の発現を行うために核酸輸送の適した方法は、本明細書に記述されるように、または当業者に既知であるように、それによって核酸(例えば、ウイルスおよび非ウイルスベクターを含むDNA)がオルガネラ、細胞、組織、または生物に導入されうる実質的に如何なる方法も含まれると考えられる。そのような方法には、マイクロインジェクション(Harlan and Weintraub、1985;参照として本明細書に組み入れられる、米国特許第5,789,215号)を含む注入(参照として本明細書に組み入れられる、米国特許第5,994,624号、第5,981,274号、第5,945,100号、第5,780,448号、第5,736,524号、第5,702,932号、第5,656,610号、第5,589,466号、および第5,580,859号);エレクトロポレーション(参照として本明細書に組み入れられる、米国特許第5,384,253号);燐酸カルシウム沈殿(Graham and Van Der Eb、1973;Chen and Okayama、1987;Rippeら、1990);DEAE-デキストランの後にポリエチレングリコールを用いることによって(Gopal、1985);直接超音波負荷(Fechheimerら、1987);リポソーム媒介トランスフェクション(Nicolau and Sene、1982;Fraleyら、1979;Nicolauら、1987;Wongら、1980;Kanedaら、1989;Katoら、1991);微小発射物の衝突(そのそれぞれが参照として本明細書に組み入れられる、PCT出願国際公開公報第94/09699号および第95/06128号;米国特許第5,610,042号、第5,322,783号、第5,563,055号、第5,550,318号、第5,538,877号、および第5,538,880号);炭化ケイ素繊維との攪拌(Kaepplerら、1990;それぞれが参照として本明細書に組み入れられる、米国特許第5,302,523号および第5,464,765号);アグロバクテリウム(Agrobacterium)媒介形質転換(そのそれぞれが参照として本明細書に組み入れられる、米国特許第5,591,616号および第5,563,055号);またはプロトプラストのPEG媒介形質転換(Omirullehら、1993;そのそれぞれが参照として本明細書に組み入れられる、米国特許第4,684,611号および第4,952,500号);乾燥/阻害媒介DNA取り込み(Potrykusら、1985)のような方法によるDNAの直接輸送が含まれるがこれらに限定されない。これらのような技術の応用によって、オルガネラ、細胞、組織、または生物は、安定または一過性に形質転換される可能性がある。
特定の態様において、本発明は、核酸、ペプチドのようなアミノ酸分子、またはもう一つの低分子化合物に関連した一つまたはそれ以上の脂質を含む組成物に関する。本明細書において考察した任意の態様において、分子は、ポックスウイルスポリペプチドまたはポックスウイルスポリペプチド調節物質、例えばポックスウイルスポリペプチドの全てもしくは一部をコードする核酸、またはポックスウイルスポリペプチド調節物質の全てもしくは一部をコードするアミノ酸分子であってもよい。脂質は、特徴的に水に不溶性で、有機溶媒によって抽出可能な物質である。本明細書において特に記述した化合物以外の化合物は、当業者によって脂質であると理解され、それらも本発明の組成物および方法に含まれる。脂質成分および非脂質は、共有結合または非共有結合によって互いに結合させてもよい。
本発明の特定の局面において、ワクシニアウイルスは、GM-CSFをコードする遺伝子を有するであろう。GM-CSFは、より多くの白血球、特に顆粒球、マクロファージ、および血小板になる細胞を作製するために役立つ物質である顆粒球-マクロファージコロニー刺激因子である。これは造血(血液形成)物質と呼ばれる薬物ファミリーに属するサイトカインであり、サルグラモスチムとしても知られる。GM-CSFは、Cantrell et al. (1985)によって1985年に初めてクローニングおよびシークエンシングされた。ヒトGM-CSFは、予想分子量16,293ダルトンの1つのオープンリーディングフレームによってコードされるアミノ酸144個の糖タンパク質である。ヒトGM-CSFは、マウスGM-CSFと69%のヌクレオチド相同性および54%のアミノ酸相同性を示し、1コピー遺伝子として存在する。
いくつかの態様において、本発明の方法において用いられるワクシニアウイルスは、GM-CSFをコードしないが、もう1つの異種配列をコードする異種配列を発現する核酸配列を含む。特定の態様において、異種配列はもう1つのサイトカインをコードする。またはもしくはさらに、ワクシニアウイルスは、IL-12、チミジンデアミナーゼ、TNF等をコードする核酸を含んでもよい。さらに、本明細書において考察されるいかなる遺伝子産物も、ワクシニアウイルス内に含まれる核酸によってコードされてもよく、本発明の方法において用いてもよい。
本発明の一つの態様において、ワクシニアウイルスの輸送によって、癌のような過増殖性疾患を治療するための方法が企図される。治療に企図される癌の例には、肺癌、頭頚部癌、乳癌、膵臓癌、前立腺癌、腎臓癌、子宮癌、骨癌、精巣癌、子宮頚癌、消化器癌、リンパ腫、肺の前新生物病変、結腸癌、黒色腫、膀胱癌、および処置しうる任意の他の癌または腫瘍が含まれる。
本発明の方法および組成物を用いて、細胞を殺すため、細胞増殖を阻害するため、転移を阻害するため、腫瘍または組織の大きさを減少させるため、およびそうでなければ腫瘍細胞の悪性の表現型を逆転または減少させるために、一般的に、ポリペプチドまたはポリペプチドをコードする発現構築物のような治療化合物を、過増殖細胞に接触させるであろう。投与経路は当然、病変の位置および特性によって変化して、これには例えば、皮内、経皮、非経口、静脈内、筋肉内、鼻腔内、皮下、局所、経皮、気管内、腹腔内、動脈内、小胞内、腫瘍内、吸入、還流、洗浄、直接注射、および経口投与ならびに製剤が含まれるであろう。
本発明において、ポックスウイルスゲノムの全てまたは一部をコードする発現構築物またはウイルスを癌または腫瘍細胞に輸送するための好ましい方法は、腫瘍内注射である。しかし、または本明細書に開示の薬学的組成物は、米国特許第5,543,158号、米国特許第5,641,515号、および米国特許第5,399,363号(そのそれぞれの全文が参照として本明細書に組み入れられる)に記述されるように、非経口、静脈内、皮内、筋肉内、経皮、または腹腔内投与してもよい。
本発明の化合物および方法は、癌およびアテローム性動脈硬化症を含む過増殖性疾患/病態の状況において用いてもよい。弱毒化ワクシニアウイルスのような本発明の組成物による治療の有効性を増加させるために、それらの疾患および病態の治療において有効な他の物質とこれらの組成物とを併用することが望ましいかも知れない。例えば、癌の治療は、本発明の治療化合物と、抗癌物質または手術のような他の抗癌治療とによって行ってもよい。
「抗癌」物質は、例えば、癌細胞を殺す、癌細胞においてアポトーシスを誘導する、癌細胞の増殖速度を減少させる、転移の発生または数を減少させる、腫瘍の大きさを減少させる、腫瘍の増殖を阻害する、腫瘍または癌細胞への血液供給を減少させる、癌細胞または腫瘍に対する免疫応答を促進させる、癌の進行を予防または阻害する、または癌を有する被験体の生命を延長させることによって被験体における癌に負の影響を及ぼすことができる。抗癌剤には、生体物質(生物治療)、化学療法剤、および放射線療法が含まれる。より一般的に、これらの他の組成物は、細胞を殺すまたは増殖を阻害するために有効な併用量で提供されるであろう。このプロセスは、発現構築物および物質または多数の因子を細胞に同時に接触させることを含んでもよい。これは、単一の組成物もしくは双方の物質を含む薬理学的製剤を細胞に接触させることによって、または一つの組成物に発現構築物が含まれ、他の組成物に第二の物質が含まれる、二つの異なる組成物もしくは製剤を細胞に同時に接触させることによって行ってもよい。
癌の療法にはまた、化学および放射線に基づく治療との多様な併用治療が含まれる。併用化学療法には、例えばシスプラチン(CDDP)、カルボプラチン、プロカルバジン、メクロレタミン、シクロホスファミド、カンプトテシン、イフォスファミド、メルファラン、クロラムブシル、ブスルファン、ニトロソウレア、ダクチノマイシン、ダウノルビシン、ドキソルビシン、ブレオマイシン、プリコマイシン、マイトマイシン、エトポシド(VP16)、タモキシフェン、ラロキシフェン、エストロゲン受容体結合剤、タキソール、ゲムシタビン、ナベルビン、ファルネシル-タンパク質トランスフェラーゼ阻害剤、トランスプラチン、5-フルオロウラシル、ビンクリスチン、ビンブラスチン、およびメトトレキセート、テマゾロミド(DTICの水溶型)、または前述の任意の誘導体もしくは類似体が含まれる。化学療法と生物療法との併用は生化学療法として知られている。
DNA損傷を引き起こし、広範に用いられている他の物質には、一般的にγ線、X線として一般的に知られている放射線、および/または放射性同位元素の腫瘍細胞への直接輸送が含まれる。マイクロ波およびUV照射のような他の型のDNA損傷剤も同様に企図される。これらの因子は全て、DNA、DNAの前駆体、DNAの複製および修復、ならびに染色体の構築および維持に対して広範囲の損傷を与える可能性が最も高い。X線の線量範囲は、1日量50〜200レントゲンを長期間(3〜4週間)から、2000〜6000レントゲンの1回線量に及ぶ。放射性同位元素の用量範囲は広く異なり、同位元素の半減期、放出される放射線の強度およびタイプ、ならびに新生物細胞による取り込みに依存する。
免疫療法は一般的に、癌細胞を標的として破壊するために免疫エフェクター細胞および分子を用いることに依存する。免疫エフェクターは、例えば腫瘍細胞の表面上のいくつかのマーカーに対して特異的な抗体であってもよい。抗体単独は、治療のエフェクターとして役立ってもよく、または殺細胞を実際に行うために他の細胞を動員してもよい。抗体はまた、薬物または毒素(化学療法剤、放射性核種、リシンA鎖、コレラ毒素、百日咳毒素等)に結合させてもよく、単にターゲティング剤として作用してもよい。または、エフェクターは、腫瘍細胞標的と直接または間接的に相互作用する表面分子を有するリンパ球であってもよい。様々なエフェクター細胞には、細胞障害性T細胞およびNK細胞が含まれる。治療様相の併用、すなわち直接細胞障害活性と特定のポックスウイルスポリペプチドの阻害または減少との併用は、癌の治療において治療利益を提供するであろう。
癌の受動免疫療法には多くの異なるアプローチが存在する。それらは、以下の分類に広く分類される可能性がある:抗体単独の注射、毒素または化学療法剤に共役させた抗体の注射、放射活性同位元素に共役させた抗体の注射、抗イディオタイプ抗体の注射、および最後に、骨髄における腫瘍細胞の追放。
能動的免疫治療において、抗原性ペプチド、ポリペプチド、またはタンパク質、または自己もしくは同種異系腫瘍細胞組成物または「ワクチン」は、一般的に異なる細菌アジュバント(Ravindranath and Morton、1991;Mortonら、1992;Mitchellら、1990;Mitchellら、1993)と共に投与される。黒色腫の免疫療法において、高いIgM反応を誘発する患者はしばしば、全くIgM抗体を誘発しないまたは低いIgM抗体を誘発する患者より長く生存する(Mortonら、1992)。IgM抗体はしばしば一過性の抗体であるが、この規則の例外は、抗ガングリオシドまたは抗炭化水素抗体であるように思われる。
養子免疫療法において、患者の循環中のリンパ球または腫瘍浸潤リンパ球をインビトロで単離して、IL-2のようなリンフォカインによって活性化して、腫瘍壊死に関する遺伝子を形質導入して再投与する(Rosenbergら、1988;1989)。これを行うために、動物またはヒト患者に、本明細書に記述のアジュバントを組み入れた抗原ペプチド組成物と併用して活性化リンパ球の免疫学的有効量が投与されるであろう。活性化リンパ球は最も好ましくは、血液または腫瘍試料から予め単離されてインビトロで活性化された(または「増殖させた」)患者自身の細胞であろう。この型の免疫治療によって、黒色腫および腎癌の数例の退縮が引き起こされたが、反応者の割合は反応しなかった人と比較すると少数であった。
さらにもう一つの態様において、二次治療は、弱毒化ポックスウイルスが投与される前、後、または同時に治療ポリヌクレオチドが投与される遺伝子治療である。以下の遺伝子産物の一つをコードするベクターと共にポックスウイルスを輸送すれば、標的組織に対して併用抗癌作用を有するであろう。または、ポックスウイルスは、ウイルスベクターとして治療ポリヌクレオチドを含めるように操作してもよい。多様なタンパク質が本発明に含まれ、そのいくつかを下記に記す。表7は、本発明と共にいくつかの型の遺伝子治療のために標的とされる可能性がある様々な遺伝子を一覧にする。
細胞増殖を誘導するタンパク質はさらに、機能に応じて様々な分類に分けられる。これらのタンパク質の全ての一般性は、細胞増殖の調節能である。例えば、PDGFの一つの型であるsis腫瘍遺伝子は、分泌型増殖因子である。腫瘍遺伝子はまれに増殖因子をコードする遺伝子から生じるが、現在のところ、sisは、既知の唯一の天然に存在する腫瘍遺伝子増殖因子である。本発明の一つの態様において、細胞増殖の特定の誘導物質に向けられるアンチセンスmRNAを用いて、細胞増殖の誘導物質の発現を阻害することが企図される。
腫瘍抑制癌遺伝子は、過度の細胞増殖を阻害するように機能する。これらの遺伝子の不活化は、その阻害活性を破壊して調節されない増殖を生じる。腫瘍抑制因子p53、p16およびC-CAMを下記に記述する。
アポトーシスまたはプログラムされた細胞死は、正常な胎児発達の本質的なプロセスであり、成体組織における恒常性を維持し、発癌を抑制する(Kerrら、1972)。Bcl-2ファミリータンパク質およびICE-様プロテアーゼは、他の系におけるアポトーシスの重要な調節物質およびエフェクターであることが証明されている。濾胞性リンパ腫に関連して発見されたBcl-2タンパク質は、アポトーシスの制御および多様なアポトーシス刺激に反応した細胞生存の増強に顕著な役割を有する(Bakhshiら、1985;Cleary and Sklar、1985;Clearyら、1986;Tsujimotoら、1985;Tsujimoto and Croce、1986)。進化的に保存されたBcl-2タンパク質は現在では、デスアゴニストまたはデスアンタゴニストとして分類することができる関連タンパク質ファミリーのメンバーであると認識されている。
癌患者の約60%が何らかのタイプの手術を受け、その中には、予防的、診断的または進行期決定、治癒的および待期的手術が含まれる。治癒的手術は、本発明の治療、化学療法、放射線療法、ホルモン療法、遺伝子治療、免疫療法および/またはもう一つの治療のような他の治療と共に用いてもよい癌治療である。
治療の治療的有効性を改善するために、他の物質を本発明と併用して用いてもよいと企図される。これらのさらなる物質には、免疫調節剤、細胞表面受容体のアップレギュレーションおよびGAP接合部に影響を及ぼす物質、細胞抑制および分化物質、細胞接着阻害剤、アポトーシス誘導物質に対する過増殖細胞の感受性を増加させる物質、または他の生体物質が含まれる。免疫調節剤には、腫瘍壊死因子;インターフェロンα、β、およびγ;IL-2および他のサイトカイン;F42Kおよび他のサイトカイン類似体;またはMIP-1、MIP-1β、MCP-1、RANTES、および他のケモカインが含まれる。Fas/Fasリガンド、DR4またはDR5/TRAIL(Apo-2リガンド)のような細胞表面受容体またはそのリガンドのアップレギュレーションは、過増殖細胞に対するオートクラインまたはパラクライン効果を確立することによって本発明のアポトーシス誘導能を増強するであろう。GAP接合部の数を増加させることによる細胞内シグナル伝達の増加は、隣接する過増殖細胞集団に対する抗過増殖効果を増加するであろう。他の態様において、細胞抑制または分化因子を、治療の抗過増殖有効性を改善するために本発明と併用して用いることができる。細胞接着阻害剤は、本発明の有効性を改善すると企図される。細胞接着阻害剤の例は、局所接着キナーゼ(FAK)阻害剤およびロバスタチンである。抗体c225のようなアポトーシスに対する過増殖細胞の感受性を増加させる他の物質を、治療の有効性を改善するために本発明と併用して用いることができるであろう。
以下の実施例は、本発明の好ましい態様を証明するために含まれる。下記の実施例に開示された技術は、本発明の実践にあたって十分に機能すると本発明者らによって発見された技術の代表的なものであり、このように、本発明の実践の好ましい様式を構成すると見なすことができると当業者によって認識されるべきである。しかし、当業者は、本開示に照らして、開示される特定の態様に多くの変更を行うことができ、それでも同様または類似の結果を得ることができるが、それらも本発明の趣旨および範囲に含まれることを認識しなければならない。
ウイルスおよび細胞株
野生型ポックスウイルスのパネル(Wyeth、Western Reserve (WR)、USSR、Tian Tan、Tash Kent、Patwadangar、Lister、King、IHD-W、IHD-J、およびEvans)は、Dr Geoff Smith, Imperial College, Londonの厚意により提供された。ヒトアデノウイルス血清型5型(Ad5)は、ATCCから得た。WRのウイルス増殖因子(VGF)欠失株(vSC20)は、Dr Bernie Moss, NIHの厚意により提供された。WRのチミジンキナーゼ欠失株(vJS6)およびWRのTK-、VGF-二重欠失株(vvDD)は、Puhlmann et al. (2000)およびMcCart et al. (2001)において記述されている。発光飛翔昆虫のルシフェラーゼを発現するWR株は、Dr Gary Luker (Uni Michigan)の厚意によって提供された。
細胞株を6ウェルプレートにおいて5×106個/ウェルで播種して、終夜放置した。次に、ウイルスを感染多重度(MOI)1.0プラーク形成単位(PFU)/細胞で加えて、2時間感染させた。感染の終了時、培地を交換して、プレートを48時間インキュベートした後、細胞を培地の中にこすり取って採取した。細胞を凍結融解3ラウンドの後に超音波によって溶解した後、粗ウイルス溶解物の連続希釈液をBSC-1細胞に加えて、ウイルス力価を測定した。プラークアッセイは既に記述されているように(Earl et al, 1998)行った。アデノウイルスはA549細胞(Earl et al, 1998)において力価を測定した。試験は典型的に1試料あたり3個ずつ行う。
免疫コンピテントマウスの皮下に同系腫瘍細胞(1×106個/マウス)を、JCおよびTIB-75細胞がBALB/cマウスに、ならびにMC38およびCMT 64細胞がC57/B6マウスに移植されるように移植した。特定のヒト異種移植片モデルは、SCIDマウス(マウスは全て8〜10週齢であり性別を一致させた)に皮下移植したHT29細胞1×107個を含んだ。腫瘍が50〜100 mm3に達した後、動物を再度群分けして、表記のように処置した。腫瘍の大きさをキャリパーによる測定によって追跡した。
VX2腫瘍のニュージーランドホワイトウサギの肝臓への移植ならびにCTおよび超音波スキャンによる腫瘍の進行および肺への転移の測定は、既に記述されている(Paeng et al, 2003)。
これはVX2腫瘍細胞に関して示されているように処置したウサギから得た標識末梢血リンパ球(PBL)を混合することによって行う。4時間後、細胞のアポトーシスをヨウ化プロピジウム染色およびフローサイトメトリーによって測定した。
抗ワクシニア中和抗体の産生は、処置後のウサギから得られた血漿において測定する。血漿の希釈液をワクシニア1000 PFUと終夜混合した後、A2780細胞を含む96ウェルプレートに加える。72時間後、細胞の生存率をMTSアッセイによって測定する。ウイルスの中和は、ウイルス不活化を防止するために必要な血漿の希釈液として測定される。
カプラン-マイヤー曲線を、一般化ウィルコキソン検定を用いて比較する。腫瘍の反応率および無転移率は、典型的にフィッシャーの正確確率検定によって比較する。
ラット(スプレージ-ドーリー、雄性)をその飲料水において発癌物質(N-ニトロソモルホリン、NNM)(175 mg/L)に8週間曝露して、その期間に肝硬変を発症した後、平均して16〜20週のあいだ(Oh et al, 2002において既に記述されたモデル)に肝臓内でインサイチューで腫瘍(肝細胞癌または胆管癌)が発生した。腫瘍の検出および評価は、超音波造影を用いて経験のある超音波技師が行った。腫瘍の大きさは、処置開始直前でのベースラインで直径約0.75〜1.5 cmであった;腫瘍の体積は、対照群と処置群のあいだでベースラインにおいて有意差がなかった(推定平均体積は400〜500 mm3)。対照動物(n=17)には、処置を与えず、処置動物(n=6)には、合成初期-後期プロモーター(Mastrangelo et al, 1999において記述されたウイルス構築物)からヒトGM-CSFを発現するポックスウイルス(Wyeth株、チミジンキナーぜ遺伝子欠失が存在する)の静脈内注射(尾静脈により)を行った。ウイルスを、108プラーク形成単位(Earl et al, 1998のように力価測定)の用量で全量0.75 ml(ウイルス浮遊液を10 mM Trisと混合して所望の容積とする)で尾静脈により60秒間かけて静脈内投与した。処置は、2週間毎に繰り返して全体で3回投与(1、15および29日目)した。
試験はVX2ウサギ癌モデルにおいて行った(Paeng et al, 2003において記述されるとおり)。ヒトGM-CSFはこれまでにウサギにおいて有意な生物活性を有することが証明されたことから、ウサギ(マウスに対して)を種として選択した。VX2腫瘍をニュージーランドホワイトウサギの筋肉において成長させて、腫瘍の1〜2 mm3断片からの細胞を解離させて、生理食塩液0.1 mlに浮遊させて、肝臓嚢の下に注射して(21ゲージ針;手術用小片を巾着縫合することによって注射部位を覆う)、原発腫瘍が確立される(平均直径、1.5〜2.0 cm;推定体積2〜4 cm3)まで14日間成長させた。VX2細胞は、標準的なバーストアッセイにおいてエクスビボでワクシニアポックスウイルスによって感染可能であることが証明された。腫瘍の大きさをCTスキャンおよび超音波によって経時的にモニターした。翌7週間にわたって、対照(無処置)動物(n=18)は、肝臓内で腫瘍の進行を示し、推定平均腫瘍体積は約100 cm3(S.E.約20)に達した。さらに、多数の腫瘍の転移が進行して、肺および肝臓において経時的に検出可能となった(図2A〜B)。7週目までに、対照動物は全て検出可能な転移を有し、肺転移の平均数は17(S.E. 2.3)であった。これらの対照動物の生存期間の中央値は55日(処置動物において処置開始後)であり、全てのマウスが80日以内に死亡した。
標的化治療は、癌の処置にとって非常に有望であるが、標的分子の変異および/または経路の重複による腫瘍の逃避を通してしばしば耐性が発達することから、新規物質がなおも必要である。腫瘍崩壊性のウイルスは、固有に、または遺伝子操作を通してその複製が悪性細胞タイプに限定されるウイルスである(Thorne et al, 2005)1。選択的な腫瘍内複製により、ウイルスの繁殖、独自のおよびアポトーシス非依存的メカニズム(腫瘍崩壊)による感染した癌細胞の殺細胞、ならびに他の腫瘍細胞へのウイルスの伝搬が起こる。したがって、ウイルス治療物質は、不応性の癌を有効に処置する能力を有し、いくつかの腫瘍崩壊性ウイルスの局所または限局的投与に関して、臨床での概念の証明が得られている(Parato et al, 2005)2。しかし、腫瘍崩壊性ウイルスが患者の生存に関して主要な影響を有するためには、全身での有効性および静脈内送達が必要であろう。
以下の参考文献は、本明細書に記載の内容に対して補助的な例示的技法または他の詳細をそれらが提供する程度に、参照により本明細書に具体的に組み入れられる:
米国特許第4,554,101号
米国特許第4,683,195号
米国特許第4,683,202号
米国特許第4,684,611号
米国特許第4,800,159号
米国特許第4,879,236号
米国特許第4,883,750号
米国特許第4,946,773号
米国特許第4,952,500号
米国特許第5,220,007号
米国特許第5,279,721号
米国特許第5,284,760号
米国特許第5,302,523号
米国特許第5,322,783号
米国特許第5,354,670号
米国特許第5,366,878号
米国特許第5,384,253号
米国特許第5,389,514号
米国特許第5,399,363号
米国特許第5,464,765号
米国特許第5,466,468号
米国特許第5,538,877号
米国特許第5,538,880号
米国特許第5,543,158号
米国特許第5,550,318号
米国特許第5,563,055号
米国特許第5,580,859号
米国特許第5,589,466号
米国特許第5,591,616号
米国特許第5,610,042号
米国特許第5,633,016号
米国特許第5,635,377号
米国特許第5,641,515号
米国特許第5,656,610号
米国特許第5,702,932号
米国特許第5,736,524号
米国特許第5,739,169号
米国特許第5,780,448号
米国特許第5,789,166号
米国特許第5,789,215号
米国特許第5,798,208号
米国特許第5,798,339号
米国特許第5,801,005号
米国特許第5,824,311号
米国特許第5,824,348号
米国特許第5,830,650号
米国特許第5,830,880号
米国特許第5,840,873号
米国特許第5,843,640号
米国特許第5,843,650号
米国特許第5,843,651号
米国特許第5,843,663号
米国特許第5,846,225号
米国特許第5,846,233号
米国特許第5,846,708号
米国特許第5,846,709号
米国特許第5,846,717号
米国特許第5,846,726号
米国特許第5,846,729号
米国特許第5,846,783号
米国特許第5,846,945号
米国特許第5,849,481号
米国特許第5,849,483号
米国特許第5,849,486号
米国特許第5,849,487号
米国特許第5,849,497号
米国特許第5,849,546号
米国特許第5,849,547号
米国特許第5,851,770号
米国特許第5,851,772号
米国特許第5,853,990号
米国特許第5,853,992号
米国特許第5,853,993号
米国特許第5,856,092号
米国特許第5,858,652号
米国特許第5,861,244号
米国特許第5,863,732号
米国特許第5,863,753号
米国特許第5,866,331号
米国特許第5,866,337号
米国特許第5,866,366号
米国特許第5,871,740号
米国特許第5,871,986号
米国特許第5,882,864号
米国特許第5,900,481号
米国特許第5,905,024号
米国特許第5,910,407号
米国特許第5,912,124号
米国特許第5,912,145号
米国特許第5,912,148号
米国特許第5,916,776号
米国特許第5,916,779号
米国特許第5,919,626号
米国特許第5,919,630号
米国特許第5,922,574号
米国特許第5,925,517号
米国特許第5,925,525号
米国特許第5,925,565号
米国特許第5,928,862号
米国特許第5,928,869号
米国特許第5,928,870号
米国特許第5,928,905号
米国特許第5,928,906号
米国特許第5,928,906号
米国特許第5,929,227号
米国特許第5,932,413号
米国特許第5,932,451号
米国特許第5,935,791号
米国特許第5,935,819号
米国特許第5,935,825号
米国特許第5,939,291号
米国特許第5,942,391号
米国特許第5,945,100号
米国特許第5,981,274号
米国特許第5,994,624号
Claims (29)
- 投与が血管内である、顆粒球-マクロファージコロニー刺激因子(GM-CSF)をコードする核酸の発現を指示するプロモーターと共に発現領域を有する複製型ワクシニアウイルスの有効量を被験者に投与する段階を含む、被験者における癌細胞を殺す方法。
- ワクシニアウイルスが静脈内投与される、請求項1記載の方法。
- ワクシニアウイルスが動脈内投与される、請求項1記載の方法。
- ワクシニアウイルスが薬学的に許容される製剤で存在する、請求項1記載の方法。
- ワクシニアウイルスがそのゲノムに欠失を有する、請求項1記載の方法。
- ワクシニアウイルスが1つまたは複数の遺伝子において変異を有する、請求項5記載の方法。
- 変異が欠失である、請求項6記載の方法。
- 少なくともチミジンキナーゼ遺伝子が欠失している、請求項6記載の方法。
- ワクシニアウイルスが、
(a)ワクシニアウイルス増殖因子;
(b)インターフェロンに直接結合する、機能的インターフェロン調節ポリペプチド;
(c)変異によって少なくとも1つの機能的補体制御ポリペプチドを欠損するウイルスが得られる、補体制御ポリペプチド;
(d)変異によって少なくとも1つの機能的TNF調節ポリペプチドを欠損するウイルスが得られる、TNF調節ポリペプチド;
(e)変異によって少なくとも1つの機能的セリンプロテアーゼ阻害剤を欠損するウイルスが得られる、セリンプロテアーゼ阻害剤;
(f)変異によって少なくとも1つの機能的IL-1β調節ポリペプチドを欠損するウイルスが得られる、IL-1β調節ポリペプチド;
(g)変異によってA41L、B7R、N1L、vCKBP、またはC11Rの少なくとも1つの機能を欠損するウイルスが得られる、機能的A41L、B7R、N1L、もしくはvCKBPケモカイン結合ポリペプチド、またはC11R EGF様ポリペプチド;または
(h)変異によってワクシニアウイルスの感染性EEV型の増加が起こる、ポリペプチド、
をコードする遺伝子において変異を有する、請求項6記載の方法。 - ワクシニアウイルス増殖因子において変異をさらに含む、請求項8記載の方法。
- ワクシニアウイルスがWyethまたはWestern Reserve(WR)株である、請求項1記載の方法。
- ワクシニアウイルスがWR株である、請求項11記載の方法。
- プロモーターがワクシニアウイルスプロモーターである、請求項1記載の方法。
- プロモーターが合成プロモーターである、請求項13記載の方法。
- プロモーターが感染の少なくとも初期相のあいだに転写を指示する、請求項13記載の方法。
- プロモーターが感染の少なくとも後期相のあいだに転写を指示する、請求項13記載の方法。
- 被験者がワクシニアウイルスを複数回投与される、請求項1記載の方法。
- 第二の処置が第一の処置の3週間以内に起こる、請求項17記載の方法。
- 第二の処置が第一の処置の2週間以内に起こる、請求項18記載の方法。
- 同じ用量が投与される、請求項17記載の方法。
- 被験者がウイルス約105〜約1013 pfuを投与される、請求項1記載の方法。
- 被験者がウイルス約107〜約1010 pfuを投与される、請求項1記載の方法。
- 投与が注射によって血管内で起こる、請求項1記載の方法。
- 投与が静脈内点滴またはボーラスを用いて血管内で起こる、請求項1記載の方法。
- 投与がポンプを用いて血管内で起こる、請求項1記載の方法。
- 癌細胞が転移癌細胞である、請求項1記載の方法。
- 被験者が、肺癌、結腸直腸癌、乳癌、前立腺癌、膵臓癌、肝細胞癌、白血病、リンパ腫、骨髄腫、または黒色腫を有する、請求項1記載の方法。
- 投与が血管内である、顆粒球-マクロファージコロニー刺激因子(GM-CSF)をコードする核酸の発現を指示するプロモーターと共に発現領域を有する複製型ワクシニアウイルスの有効量を被験者に投与する段階を含む、被験者における癌を処置するための方法。
- 投与が血管内である、顆粒球-マクロファージコロニー刺激因子(GM-CSF)をコードする核酸の発現を指示するプロモーターと共に発現領域を有する複製型ワクシニアの有効量を被験者に投与する段階を含む、被験者における1つまたは複数の転移を処置するための方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71467905P | 2005-09-07 | 2005-09-07 | |
| PCT/US2006/034945 WO2007030668A2 (en) | 2005-09-07 | 2006-09-07 | Systemic treatment of metastatic and/or systemically-disseminated cancers using gm-csf-expressing poxviruses |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013113888A Division JP5927143B2 (ja) | 2005-09-07 | 2013-05-30 | Gm−csf発現ポックスウイルスを用いる転移性および/または全身播種性癌の全身処置 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2009507853A true JP2009507853A (ja) | 2009-02-26 |
Family
ID=37758563
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008530209A Withdrawn JP2009507853A (ja) | 2005-09-07 | 2006-09-07 | Gm−csf発現ポックスウイルスを用いる転移性および/または全身播種性癌の全身処置 |
| JP2013113888A Active JP5927143B2 (ja) | 2005-09-07 | 2013-05-30 | Gm−csf発現ポックスウイルスを用いる転移性および/または全身播種性癌の全身処置 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013113888A Active JP5927143B2 (ja) | 2005-09-07 | 2013-05-30 | Gm−csf発現ポックスウイルスを用いる転移性および/または全身播種性癌の全身処置 |
Country Status (8)
| Country | Link |
|---|---|
| US (3) | US9180149B2 (ja) |
| EP (1) | EP1933857A2 (ja) |
| JP (2) | JP2009507853A (ja) |
| KR (2) | KR20090004839A (ja) |
| CN (1) | CN101351213B (ja) |
| AU (1) | AU2006287441B2 (ja) |
| CA (1) | CA2621982C (ja) |
| WO (1) | WO2007030668A2 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8392560B2 (en) | 2006-04-28 | 2013-03-05 | Microsoft Corporation | Offering and provisioning secured wireless virtual private network services |
| JP2014502970A (ja) * | 2011-01-04 | 2014-02-06 | ジェンネレックス インコーポレイティッド | 腫瘍溶解性ワクシニアウイルスの投与による腫瘍抗原に対する抗体の生成および腫瘍特異的補体依存性細胞傷害の生成 |
| JP2016527920A (ja) * | 2013-08-22 | 2016-09-15 | ユニヴァーシティ オヴ ピッツバーグ オヴ ザ コモンウェルス システム オヴ ハイアー エデュケーション | 免疫腫瘍溶解療法 |
| JP2020514324A (ja) * | 2017-02-03 | 2020-05-21 | ユニバーシティ オブ ピッツバーグ −オブ ザ コモンウェルス システム オブ ハイヤー エデュケイション | 腫瘍溶解性ウイルス療法 |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2818693C (en) | 2002-08-12 | 2016-05-17 | Jennerex, Inc. | Methods and compositions concerning poxviruses and cancer |
| US8980246B2 (en) * | 2005-09-07 | 2015-03-17 | Sillajen Biotherapeutics, Inc. | Oncolytic vaccinia virus cancer therapy |
| CN101351213B (zh) * | 2005-09-07 | 2013-10-02 | 詹纳里克斯公司 | 使用表达gm-csf的痘病毒系统性治疗转移性癌和/或全身散布性癌 |
| US20100086522A1 (en) * | 2006-07-18 | 2010-04-08 | Ottawa Health Research Institute | Disparate suicide carrier cells for tumor targeting of promiscuous oncolytic viruses |
| WO2008100292A2 (en) | 2006-10-16 | 2008-08-21 | Genelux Corporation | Modified vaccinia virus strains for use in diagnostic and therapeutic methods |
| KR20080084528A (ko) * | 2007-03-15 | 2008-09-19 | 제네렉스 바이오테라퓨틱스 인크. | 종양살상형 백시니아 바이러스 암 치료 |
| US20090117034A1 (en) * | 2007-06-15 | 2009-05-07 | Nanhai Chen | Microorganisms for imaging and/or treatment of tumors |
| AU2010291901C1 (en) | 2009-09-14 | 2016-03-03 | Sillajen Biotherapeutics, Inc. | Oncolytic vaccinia virus combination cancer therapy |
| US20120237481A1 (en) * | 2011-03-17 | 2012-09-20 | Xiaoliu Zhang | Incorporation of the B18R gene to enhance antitumor effect of virotherapy |
| US11149254B2 (en) | 2011-04-15 | 2021-10-19 | Genelux Corporation | Clonal strains of attenuated vaccinia viruses and methods of use thereof |
| DK2739293T3 (da) * | 2011-08-05 | 2020-08-24 | Sillajen Biotherapeutics Inc | Fremgangsmåder og sammensætninger til frembringelse af vaccinavirus |
| EP2764119A2 (en) | 2011-10-05 | 2014-08-13 | Genelux Corporation | Method for detecting replication or colonization of a biological therapeutic |
| TWI690322B (zh) | 2012-10-02 | 2020-04-11 | 法商傳斯堅公司 | 含病毒的調配物及其使用 |
| CN105121635B (zh) * | 2013-04-10 | 2018-10-16 | 东源生物医药科技(上海)有限公司 | 突变型痘苗病毒株、其用途及其制造方法 |
| GB201405834D0 (en) * | 2014-04-01 | 2014-05-14 | Univ London Queen Mary | Oncolytic virus |
| ES2753364T3 (es) | 2014-12-01 | 2020-04-08 | Transgene Sa | Formulaciones líquidas estables de virus vaccinia |
| CA2990133A1 (en) * | 2015-06-19 | 2017-03-09 | Sillajen, Inc. | Compositions and methods for viral embolization |
| EP3347460A4 (en) * | 2015-09-08 | 2019-04-10 | Sillajen, Inc. | MODIFIED ONCOLYTIC VACCINE VIRUSES EXPRESSING CYTOKINE AND CARBOXYESTERASE AND METHODS OF USE THEREOF |
| JP6757897B2 (ja) * | 2016-02-24 | 2020-09-23 | 国立大学法人大阪大学 | 試験方法 |
| BR112019024556A2 (pt) | 2017-05-24 | 2020-06-23 | Novartis Ag | Proteínas enxertadas com citocina de anticorpo e métodos para uso no tratamento de câncer |
| CN107164337B (zh) * | 2017-05-26 | 2020-08-04 | 云南省第一人民医院 | 含ccl5和sstr2基因的重组痘病毒及其制备方法 |
| CN109554353B (zh) * | 2017-09-26 | 2021-08-06 | 杭州康万达医药科技有限公司 | 分离的重组溶瘤痘病毒、药物组合物及其在治疗肿瘤和/或癌症的药物中的用途 |
| KR102661908B1 (ko) * | 2019-08-29 | 2024-04-29 | 주식회사 바이오녹스 | 백시니아 바이러스 및 과립백혈구 형성 억제제를 유효성분으로 포함하는 암 치료용 약학 조성물 |
| CN111150748A (zh) * | 2019-12-27 | 2020-05-15 | 杭州荣谷生物科技有限公司 | 重组溶瘤病毒在制备治疗消化道癌药物中的用途 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09502993A (ja) * | 1993-09-29 | 1997-03-25 | トランジェーヌ、ソシエテ、アノニム | 免疫または炎症応答の調節による抗がん遺伝子療法 |
| JPH09503902A (ja) * | 1993-01-21 | 1997-04-22 | ヴァイロジェネティクス コーポレイション | 組み換えウイルス免疫治療 |
| US20030206886A1 (en) * | 2002-05-03 | 2003-11-06 | University Of Medicine & Dentistry Of New Jersey | Neutralization of immune suppressive factors for the immunotherapy of cancer |
Family Cites Families (137)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US553880A (en) * | 1896-02-04 | Workman s time-recorder | ||
| US4554101A (en) * | 1981-01-09 | 1985-11-19 | New York Blood Center, Inc. | Identification and preparation of epitopes on antigens and allergens on the basis of hydrophilicity |
| NL8200523A (nl) * | 1982-02-11 | 1983-09-01 | Univ Leiden | Werkwijze voor het in vitro transformeren van planteprotoplasten met plasmide-dna. |
| US4879236A (en) * | 1984-05-16 | 1989-11-07 | The Texas A&M University System | Method for producing a recombinant baculovirus expression vector |
| US4957858A (en) | 1986-04-16 | 1990-09-18 | The Salk Instute For Biological Studies | Replicative RNA reporter systems |
| US4883750A (en) * | 1984-12-13 | 1989-11-28 | Applied Biosystems, Inc. | Detection of specific sequences in nucleic acids |
| US4683202A (en) * | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683195A (en) * | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4946773A (en) * | 1985-12-23 | 1990-08-07 | President And Fellows Of Harvard College | Detection of base pair mismatches using RNAase A |
| US4800159A (en) * | 1986-02-07 | 1989-01-24 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences |
| US5824311A (en) | 1987-11-30 | 1998-10-20 | Trustees Of The University Of Pennsylvania | Treatment of tumors with monoclonal antibodies against oncogene antigens |
| US4952500A (en) * | 1988-02-01 | 1990-08-28 | University Of Georgia Research Foundation, Inc. | Cloning systems for Rhodococcus and related bacteria |
| EP0365627B1 (en) | 1988-03-24 | 1993-12-22 | University Of Iowa Research Foundation | Catalytic hybridization systems for the detection of nucleic acid sequences based on their activity as cofactors in catalytic reactions in which a complementary labeled nucleic acid probe is cleaved |
| US5858652A (en) * | 1988-08-30 | 1999-01-12 | Abbott Laboratories | Detection and amplification of target nucleic acid sequences |
| US5151509A (en) * | 1988-12-16 | 1992-09-29 | United States Of America | Gene encoding serine protease inhibitor |
| US5856092A (en) * | 1989-02-13 | 1999-01-05 | Geneco Pty Ltd | Detection of a nucleic acid sequence or a change therein |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| US5284760A (en) * | 1989-04-03 | 1994-02-08 | Feinstone Stephen M | Techniques for producing site-directed mutagenesis of cloned DNA |
| US5302523A (en) * | 1989-06-21 | 1994-04-12 | Zeneca Limited | Transformation of plant cells |
| US7705215B1 (en) | 1990-04-17 | 2010-04-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US5550318A (en) * | 1990-04-17 | 1996-08-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US5073627A (en) | 1989-08-22 | 1991-12-17 | Immunex Corporation | Fusion proteins comprising GM-CSF and IL-3 |
| US5322783A (en) * | 1989-10-17 | 1994-06-21 | Pioneer Hi-Bred International, Inc. | Soybean transformation by microparticle bombardment |
| US5484956A (en) | 1990-01-22 | 1996-01-16 | Dekalb Genetics Corporation | Fertile transgenic Zea mays plant comprising heterologous DNA encoding Bacillus thuringiensis endotoxin |
| US5149797A (en) * | 1990-02-15 | 1992-09-22 | The Worcester Foundation For Experimental Biology | Method of site-specific alteration of rna and production of encoded polypeptides |
| US5466468A (en) * | 1990-04-03 | 1995-11-14 | Ciba-Geigy Corporation | Parenterally administrable liposome formulation comprising synthetic lipids |
| CA2079802C (en) * | 1990-04-05 | 2001-09-18 | Roberto Crea | Walk-through mutagenesis |
| US5849481A (en) | 1990-07-27 | 1998-12-15 | Chiron Corporation | Nucleic acid hybridization assays employing large comb-type branched polynucleotides |
| US5645987A (en) * | 1990-09-21 | 1997-07-08 | Amgen Inc. | Enzymatic synthesis of oligonucleotides |
| US5798339A (en) * | 1990-12-17 | 1998-08-25 | University Of Manitoba | Treatment method for cancer |
| US5384253A (en) * | 1990-12-28 | 1995-01-24 | Dekalb Genetics Corporation | Genetic transformation of maize cells by electroporation of cells pretreated with pectin degrading enzymes |
| US5399363A (en) * | 1991-01-25 | 1995-03-21 | Eastman Kodak Company | Surface modified anticancer nanoparticles |
| ATE247163T1 (de) * | 1991-03-07 | 2003-08-15 | Virogenetics Corp | Gentechnologisch hergestellter stamm für impfstoffe |
| CA2105277C (en) | 1991-03-07 | 2006-12-12 | William I. Cox | Genetically engineered vaccine strain |
| GB9105383D0 (en) * | 1991-03-14 | 1991-05-01 | Immunology Ltd | An immunotherapeutic for cervical cancer |
| AU2515992A (en) * | 1991-08-20 | 1993-03-16 | Genpharm International, Inc. | Gene targeting in animal cells using isogenic dna constructs |
| US5846717A (en) | 1996-01-24 | 1998-12-08 | Third Wave Technologies, Inc. | Detection of nucleic acid sequences by invader-directed cleavage |
| US5610042A (en) * | 1991-10-07 | 1997-03-11 | Ciba-Geigy Corporation | Methods for stable transformation of wheat |
| US5849486A (en) | 1993-11-01 | 1998-12-15 | Nanogen, Inc. | Methods for hybridization analysis utilizing electrically controlled hybridization |
| ATE247471T1 (de) * | 1991-11-15 | 2003-09-15 | Smithkline Beecham Corp | Cisplatin und topotecan enthaltende zusammensetzung als antitumor. |
| US5846708A (en) | 1991-11-19 | 1998-12-08 | Massachusetts Institiute Of Technology | Optical and electrical methods and apparatus for molecule detection |
| ATE239089T1 (de) * | 1991-12-24 | 2003-05-15 | Harvard College | Gezielte punkt-mutagenese von dna |
| US20020146702A1 (en) | 1992-01-31 | 2002-10-10 | Vielkind Juergen R. | Nucleic acid molecule associated with prostate cancer and melanoma immunodetection and immunotherapy |
| AT400033B (de) * | 1992-03-10 | 1995-09-25 | Biochemie Gmbh | Neues verfahren zur isolierung und reinigung von clavulansäure und zur herstellung von pharmakologisch verträglichen salzen derselben |
| JP3633932B2 (ja) * | 1992-04-01 | 2005-03-30 | ザ ジョーンズ ホプキンズ ユニバーシティー スクール オブ メディシン | 糞便試料から単離した哺乳類の核酸を検出する方法、およびその検出用試薬 |
| US5843640A (en) | 1992-06-19 | 1998-12-01 | Northwestern University | Method of simultaneously detecting amplified nucleic acid sequences and cellular antigens in cells |
| DK0604662T3 (da) * | 1992-07-07 | 2008-10-20 | Japan Tobacco Inc | Fremgangsmåde til transformering af monocotyledon |
| US5702932A (en) | 1992-07-20 | 1997-12-30 | University Of Florida | Microinjection methods to transform arthropods with exogenous DNA |
| HUT70467A (en) * | 1992-07-27 | 1995-10-30 | Pioneer Hi Bred Int | An improved method of agrobactenium-mediated transformation of cultvred soyhean cells |
| US5636377A (en) * | 1992-08-19 | 1997-06-10 | Hipco, Inc. | Hip protection device for the elderly |
| DE4228457A1 (de) * | 1992-08-27 | 1994-04-28 | Beiersdorf Ag | Herstellung von heterodimerem PDGF-AB mit Hilfe eines bicistronischen Vektorsystems in Säugerzellen |
| US5389514A (en) * | 1992-08-28 | 1995-02-14 | Fox Chase Cancer Center | Method for specifically altering the nucleotide sequence of RNA |
| US5861244A (en) * | 1992-10-29 | 1999-01-19 | Profile Diagnostic Sciences, Inc. | Genetic sequence assay using DNA triple strand formation |
| GB9222888D0 (en) | 1992-10-30 | 1992-12-16 | British Tech Group | Tomography |
| US5846945A (en) | 1993-02-16 | 1998-12-08 | Onyx Pharmaceuticals, Inc. | Cytopathic viruses for therapy and prophylaxis of neoplasia |
| US5801005A (en) * | 1993-03-17 | 1998-09-01 | University Of Washington | Immune reactivity to HER-2/neu protein for diagnosis of malignancies in which the HER-2/neu oncogene is associated |
| US5658751A (en) * | 1993-04-13 | 1997-08-19 | Molecular Probes, Inc. | Substituted unsymmetrical cyanine dyes with selected permeability |
| US5279721A (en) * | 1993-04-22 | 1994-01-18 | Peter Schmid | Apparatus and method for an automated electrophoresis system |
| GB9311386D0 (en) * | 1993-06-02 | 1993-07-21 | Pna Diagnostics As | Nucleic acid analogue assay procedures |
| US5846709A (en) | 1993-06-15 | 1998-12-08 | Imclone Systems Incorporated | Chemical process for amplifying and detecting nucleic acid sequences |
| US5543158A (en) * | 1993-07-23 | 1996-08-06 | Massachusetts Institute Of Technology | Biodegradable injectable nanoparticles |
| FR2708288B1 (fr) | 1993-07-26 | 1995-09-01 | Bio Merieux | Procédé d'amplification d'acides nucléiques par transcription utilisant le déplacement, réactifs et nécessaire pour la mise en Óoeuvre de ce procédé. |
| US5969094A (en) * | 1993-10-12 | 1999-10-19 | Emory University | Anti-paramyxovirus screening method and vaccine |
| US5925517A (en) * | 1993-11-12 | 1999-07-20 | The Public Health Research Institute Of The City Of New York, Inc. | Detectably labeled dual conformation oligonucleotide probes, assays and kits |
| GB2284208A (en) | 1993-11-25 | 1995-05-31 | Pna Diagnostics As | Nucleic acid analogues with a chelating functionality for metal ions |
| JP2935950B2 (ja) * | 1993-12-03 | 1999-08-16 | 株式会社山田製作所 | ステアリングシャフト及びその製造装置 |
| EP0663447B1 (en) | 1993-12-28 | 2003-07-09 | Eiken Chemical Co., Ltd. | Method of detecting a specific polynucleotide |
| US5851770A (en) | 1994-04-25 | 1998-12-22 | Variagenics, Inc. | Detection of mismatches by resolvase cleavage using a magnetic bead support |
| US5656465A (en) * | 1994-05-04 | 1997-08-12 | Therion Biologics Corporation | Methods of in vivo gene delivery |
| US6093700A (en) * | 1995-05-11 | 2000-07-25 | Thomas Jefferson University | Method of inducing an immune response using vaccinia virus recombinants encoding GM-CSF |
| DE69527355T2 (de) * | 1994-05-28 | 2003-03-06 | Tepnel Medical Ltd., Manchester | Herstellung von nukleinsaeurekopien |
| US5656610A (en) * | 1994-06-21 | 1997-08-12 | University Of Southern California | Producing a protein in a mammal by injection of a DNA-sequence into the tongue |
| US5942391A (en) * | 1994-06-22 | 1999-08-24 | Mount Sinai School Of Medicine | Nucleic acid amplification method: ramification-extension amplification method (RAM) |
| US5849483A (en) | 1994-07-28 | 1998-12-15 | Ig Laboratories, Inc. | High throughput screening method for sequences or genetic alterations in nucleic acids |
| CA2195562A1 (en) * | 1994-08-19 | 1996-02-29 | Pe Corporation (Ny) | Coupled amplification and ligation method |
| GB9506466D0 (en) * | 1994-08-26 | 1995-05-17 | Prolifix Ltd | Cell cycle regulated repressor and dna element |
| US5599668A (en) | 1994-09-22 | 1997-02-04 | Abbott Laboratories | Light scattering optical waveguide method for detecting specific binding events |
| US5871986A (en) * | 1994-09-23 | 1999-02-16 | The General Hospital Corporation | Use of a baculovirus to express and exogenous gene in a mammalian cell |
| ES2271944T3 (es) * | 1994-10-28 | 2007-04-16 | Gen-Probe Incorporated | Composiciones y metodos para la deteccion y cuantificacion simultaneas de multiples secuencias especificas de acidos nucleicos. |
| US5736524A (en) * | 1994-11-14 | 1998-04-07 | Merck & Co.,. Inc. | Polynucleotide tuberculosis vaccine |
| US5935825A (en) * | 1994-11-18 | 1999-08-10 | Shimadzu Corporation | Process and reagent for amplifying nucleic acid sequences |
| US5599302A (en) | 1995-01-09 | 1997-02-04 | Medi-Ject Corporation | Medical injection system and method, gas spring thereof and launching device using gas spring |
| US5866337A (en) * | 1995-03-24 | 1999-02-02 | The Trustees Of Columbia University In The City Of New York | Method to detect mutations in a nucleic acid using a hybridization-ligation procedure |
| IE80468B1 (en) * | 1995-04-04 | 1998-07-29 | Elan Corp Plc | Controlled release biodegradable nanoparticles containing insulin |
| US5843650A (en) | 1995-05-01 | 1998-12-01 | Segev; David | Nucleic acid detection and amplification by chemical linkage of oligonucleotides |
| US5916779A (en) * | 1995-09-21 | 1999-06-29 | Becton, Dickinson And Company | Strand displacement amplification of RNA targets |
| IL123653A0 (en) * | 1995-09-29 | 1998-10-30 | Immunex Corp | Chemokine inhibitor |
| US5866331A (en) * | 1995-10-20 | 1999-02-02 | University Of Massachusetts | Single molecule detection by in situ hybridization |
| DE19541450C2 (de) * | 1995-11-07 | 1997-10-02 | Gsf Forschungszentrum Umwelt | Genkonstrukt und dessen Verwendung |
| US5780448A (en) * | 1995-11-07 | 1998-07-14 | Ottawa Civic Hospital Loeb Research | DNA-based vaccination of fish |
| US5789166A (en) * | 1995-12-08 | 1998-08-04 | Stratagene | Circular site-directed mutagenesis |
| US5612473A (en) | 1996-01-16 | 1997-03-18 | Gull Laboratories | Methods, kits and solutions for preparing sample material for nucleic acid amplification |
| US5851772A (en) | 1996-01-29 | 1998-12-22 | University Of Chicago | Microchip method for the enrichment of specific DNA sequences |
| US5739169A (en) * | 1996-05-31 | 1998-04-14 | Procept, Incorporated | Aromatic compounds for inhibiting immune response |
| US5939291A (en) * | 1996-06-14 | 1999-08-17 | Sarnoff Corporation | Microfluidic method for nucleic acid amplification |
| US5912124A (en) * | 1996-06-14 | 1999-06-15 | Sarnoff Corporation | Padlock probe detection |
| US5853990A (en) | 1996-07-26 | 1998-12-29 | Edward E. Winger | Real time homogeneous nucleotide assay |
| CA2744096C (en) * | 1996-07-31 | 2013-07-30 | Laboratory Corporation Of America Holdings | Biomarkers and targets for diagnosis, prognosis and management of prostate disease |
| US5945100A (en) * | 1996-07-31 | 1999-08-31 | Fbp Corporation | Tumor delivery vehicles |
| US5928870A (en) * | 1997-06-16 | 1999-07-27 | Exact Laboratories, Inc. | Methods for the detection of loss of heterozygosity |
| US5849546A (en) | 1996-09-13 | 1998-12-15 | Epicentre Technologies Corporation | Methods for using mutant RNA polymerases with reduced discrimination between non-canonical and canonical nucleoside triphosphates |
| US5981274A (en) * | 1996-09-18 | 1999-11-09 | Tyrrell; D. Lorne J. | Recombinant hepatitis virus vectors |
| US5853992A (en) | 1996-10-04 | 1998-12-29 | The Regents Of The University Of California | Cyanine dyes with high-absorbance cross section as donor chromophores in energy transfer labels |
| US5853993A (en) | 1996-10-21 | 1998-12-29 | Hewlett-Packard Company | Signal enhancement method and kit |
| US5900481A (en) * | 1996-11-06 | 1999-05-04 | Sequenom, Inc. | Bead linkers for immobilizing nucleic acids to solid supports |
| US5905024A (en) * | 1996-12-17 | 1999-05-18 | University Of Chicago | Method for performing site-specific affinity fractionation for use in DNA sequencing |
| US5846225A (en) | 1997-02-19 | 1998-12-08 | Cornell Research Foundation, Inc. | Gene transfer therapy delivery device and method |
| EP0985046B1 (en) * | 1997-02-21 | 2006-04-12 | Oxxon Therapeutics Limited | Recombinant poxvirus incapable of expressing a41l gene, coding for a soluble protein that binds chemokines |
| US5846729A (en) | 1997-02-27 | 1998-12-08 | Lorne Park Research, Inc. | Assaying nucleotides in solution using a fluorescent intensity quenching effect |
| US5849497A (en) | 1997-04-03 | 1998-12-15 | The Research Foundation Of State University Of New York | Specific inhibition of the polymerase chain reaction using a non-extendable oligonucleotide blocker |
| US5827311A (en) * | 1997-05-08 | 1998-10-27 | Biomet Inc | Carpal tunnel tome |
| US5846726A (en) * | 1997-05-13 | 1998-12-08 | Becton, Dickinson And Company | Detection of nucleic acids by fluorescence quenching |
| US5919626A (en) * | 1997-06-06 | 1999-07-06 | Orchid Bio Computer, Inc. | Attachment of unmodified nucleic acids to silanized solid phase surfaces |
| US5866366A (en) * | 1997-07-01 | 1999-02-02 | Smithkline Beecham Corporation | gidB |
| US5916776A (en) * | 1997-08-27 | 1999-06-29 | Sarnoff Corporation | Amplification method for a polynucleotide |
| HUP0003911A3 (en) | 1997-10-09 | 2007-11-28 | Pro Virus | Treatment of neoplasms with viruses |
| US20030044384A1 (en) | 1997-10-09 | 2003-03-06 | Pro-Virus, Inc. | Treatment of neoplasms with viruses |
| US5994624A (en) * | 1997-10-20 | 1999-11-30 | Cotton Incorporated | In planta method for the production of transgenic plants |
| US6177076B1 (en) * | 1997-12-09 | 2001-01-23 | Thomas Jefferson University | Method of treating bladder cancer with wild type vaccinia virus |
| US6528054B1 (en) * | 1998-12-28 | 2003-03-04 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of breast cancer |
| HUP0302278A3 (en) | 1999-04-15 | 2011-01-28 | Pro Virus | Treatment of neoplasms with viruses |
| EP1180157B1 (en) * | 1999-05-28 | 2012-11-28 | THE GOVERNMENT OF THE UNITED STATES OF AMERICA, as represented by THE SECRETARY, DEPARTMENT OF HEALTH AND HUMAN SERVICES | A combined growth factor-deleted and thymidine kinase-deleted vaccinia virus vector |
| MXPA02004736A (es) | 1999-11-12 | 2003-01-28 | Oncolytics Biotech Inc | Virus para el tratamiento de trastornos proliferativos celulares. |
| US20020048777A1 (en) * | 1999-12-06 | 2002-04-25 | Shujath Ali | Method of diagnosing monitoring, staging, imaging and treating prostate cancer |
| AU2001236525A1 (en) | 2000-02-14 | 2001-08-27 | Gary R. Davis, M.D., L.L.C. | Neutralizing antibody and immunomodulatory enhancing compositions |
| US20040091995A1 (en) * | 2001-06-15 | 2004-05-13 | Jeffrey Schlom | Recombinant non-replicating virus expressing gm-csf and uses thereof to enhance immune responses |
| DE10134955C1 (de) * | 2001-07-23 | 2003-03-06 | Infineon Technologies Ag | Anordnung von Gräben in einem Halbleitersubstrat, insbesondere für Grabenkondensatoren |
| CA2818693C (en) | 2002-08-12 | 2016-05-17 | Jennerex, Inc. | Methods and compositions concerning poxviruses and cancer |
| CN1279056C (zh) | 2003-06-06 | 2006-10-11 | 马菁 | 肿瘤相关抗原sm5-1的特异性抗体及其应用 |
| EP1644492B1 (en) * | 2003-06-18 | 2009-01-07 | Genelux Corporation | Modified recombinant vaccinia viruses, uses thereof |
| US20050207974A1 (en) | 2004-03-17 | 2005-09-22 | Deng David X | Endothelial cell markers and related reagents and methods of use thereof |
| CN101351213B (zh) | 2005-09-07 | 2013-10-02 | 詹纳里克斯公司 | 使用表达gm-csf的痘病毒系统性治疗转移性癌和/或全身散布性癌 |
| GB0519303D0 (en) | 2005-09-21 | 2005-11-02 | Oxford Biomedica Ltd | Chemo-immunotherapy method |
| US20090317456A1 (en) | 2006-10-13 | 2009-12-24 | Medigene Ag | Use of oncolytic viruses and antiangiogenic agents in the treatment of cancer |
| WO2008100292A2 (en) | 2006-10-16 | 2008-08-21 | Genelux Corporation | Modified vaccinia virus strains for use in diagnostic and therapeutic methods |
| EP1914242A1 (en) | 2006-10-19 | 2008-04-23 | Sanofi-Aventis | Novel anti-CD38 antibodies for the treatment of cancer |
| KR20080084528A (ko) | 2007-03-15 | 2008-09-19 | 제네렉스 바이오테라퓨틱스 인크. | 종양살상형 백시니아 바이러스 암 치료 |
-
2006
- 2006-09-07 CN CN2006800413898A patent/CN101351213B/zh active Active
- 2006-09-07 KR KR1020087008408A patent/KR20090004839A/ko active Application Filing
- 2006-09-07 WO PCT/US2006/034945 patent/WO2007030668A2/en active Application Filing
- 2006-09-07 CA CA2621982A patent/CA2621982C/en active Active
- 2006-09-07 EP EP06814307A patent/EP1933857A2/en not_active Withdrawn
- 2006-09-07 KR KR1020147006129A patent/KR101772375B1/ko active IP Right Grant
- 2006-09-07 US US11/470,951 patent/US9180149B2/en active Active
- 2006-09-07 JP JP2008530209A patent/JP2009507853A/ja not_active Withdrawn
- 2006-09-07 AU AU2006287441A patent/AU2006287441B2/en active Active
-
2007
- 2007-08-14 US US11/838,774 patent/US20080286237A1/en not_active Abandoned
-
2013
- 2013-05-30 JP JP2013113888A patent/JP5927143B2/ja active Active
-
2015
- 2015-10-06 US US14/876,195 patent/US9827278B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09503902A (ja) * | 1993-01-21 | 1997-04-22 | ヴァイロジェネティクス コーポレイション | 組み換えウイルス免疫治療 |
| JPH09502993A (ja) * | 1993-09-29 | 1997-03-25 | トランジェーヌ、ソシエテ、アノニム | 免疫または炎症応答の調節による抗がん遺伝子療法 |
| US20030206886A1 (en) * | 2002-05-03 | 2003-11-06 | University Of Medicine & Dentistry Of New Jersey | Neutralization of immune suppressive factors for the immunotherapy of cancer |
Non-Patent Citations (4)
| Title |
|---|
| JPN5008013575; THORNE STEVE H: EXPERT OPINION ON BIOLOGICAL THERAPY V4 N8, 200408, P1307-1321 * |
| JPN6012022143; THORNE,S.H. et al: 'Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic' J Clin Invest Vol.117, No.11, 2007, p.3350-3358 * |
| JPN6012022144; MCCART,J.A. et al: 'Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and va' Cancer Res Vol.61, No.24, 2001, p.8751-8757 * |
| JPN7012001605; KIM, JH et al.: 'Both Oncolysis and Tumor Immunity Are Involved in an Antitumoral Efficacy by Intratumoral Injection' MOLECULAR THERAPY Vol.11, 20050815, p.S67, 167, ACADEMIC PRESS * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8392560B2 (en) | 2006-04-28 | 2013-03-05 | Microsoft Corporation | Offering and provisioning secured wireless virtual private network services |
| JP2014502970A (ja) * | 2011-01-04 | 2014-02-06 | ジェンネレックス インコーポレイティッド | 腫瘍溶解性ワクシニアウイルスの投与による腫瘍抗原に対する抗体の生成および腫瘍特異的補体依存性細胞傷害の生成 |
| JP2016527920A (ja) * | 2013-08-22 | 2016-09-15 | ユニヴァーシティ オヴ ピッツバーグ オヴ ザ コモンウェルス システム オヴ ハイアー エデュケーション | 免疫腫瘍溶解療法 |
| JP2019205451A (ja) * | 2013-08-22 | 2019-12-05 | ユニヴァーシティ オヴ ピッツバーグ オヴ ザ コモンウェルス システム オヴ ハイアー エデュケーション | 免疫腫瘍溶解療法 |
| JP7021154B2 (ja) | 2013-08-22 | 2022-02-16 | ユニヴァーシティ オヴ ピッツバーグ オヴ ザ コモンウェルス システム オヴ ハイアー エデュケーション | 免疫腫瘍溶解療法 |
| JP2022062171A (ja) * | 2013-08-22 | 2022-04-19 | ユニヴァーシティ オヴ ピッツバーグ オヴ ザ コモンウェルス システム オヴ ハイアー エデュケーション | 免疫腫瘍溶解療法 |
| US11478518B2 (en) | 2013-08-22 | 2022-10-25 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Immuno-oncolytic therapies |
| JP7333431B2 (ja) | 2013-08-22 | 2023-08-24 | ユニヴァーシティ オヴ ピッツバーグ オヴ ザ コモンウェルス システム オヴ ハイアー エデュケーション | 免疫腫瘍溶解療法 |
| JP2020514324A (ja) * | 2017-02-03 | 2020-05-21 | ユニバーシティ オブ ピッツバーグ −オブ ザ コモンウェルス システム オブ ハイヤー エデュケイション | 腫瘍溶解性ウイルス療法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013189456A (ja) | 2013-09-26 |
| EP1933857A2 (en) | 2008-06-25 |
| KR20090004839A (ko) | 2009-01-12 |
| KR20140036333A (ko) | 2014-03-25 |
| US9180149B2 (en) | 2015-11-10 |
| KR101772375B1 (ko) | 2017-08-29 |
| AU2006287441B2 (en) | 2012-09-06 |
| US20070065411A1 (en) | 2007-03-22 |
| JP5927143B2 (ja) | 2016-05-25 |
| WO2007030668A2 (en) | 2007-03-15 |
| WO2007030668A3 (en) | 2007-07-19 |
| US20160038548A1 (en) | 2016-02-11 |
| AU2006287441A1 (en) | 2007-03-15 |
| US9827278B2 (en) | 2017-11-28 |
| CA2621982A1 (en) | 2007-03-15 |
| US20080286237A1 (en) | 2008-11-20 |
| CN101351213B (zh) | 2013-10-02 |
| CA2621982C (en) | 2017-11-28 |
| CN101351213A (zh) | 2009-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5927143B2 (ja) | Gm−csf発現ポックスウイルスを用いる転移性および/または全身播種性癌の全身処置 | |
| JP4796299B2 (ja) | ポックスウイルスおよび癌に関する方法および組成物 | |
| US20210322578A1 (en) | Oncolytic vaccinia virus cancer therapy | |
| AU2012244210B2 (en) | Systemic treatment of metastatic and/or systemically-disseminated cancers using gm-csf-expressing poxviruses | |
| AU2012261681B2 (en) | Methods and compositions concerning poxviruses and cancer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090819 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100819 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120502 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120731 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120807 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121102 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20130130 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130530 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130712 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130712 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20130805 |
|
| A912 | Re-examination (zenchi) completed and case transferred to appeal board |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20130906 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20140507 |