ES2955037T3 - Nuevos compuestos heterocíclicos - Google Patents
Nuevos compuestos heterocíclicos Download PDFInfo
- Publication number
- ES2955037T3 ES2955037T3 ES19809026T ES19809026T ES2955037T3 ES 2955037 T3 ES2955037 T3 ES 2955037T3 ES 19809026 T ES19809026 T ES 19809026T ES 19809026 T ES19809026 T ES 19809026T ES 2955037 T3 ES2955037 T3 ES 2955037T3
- Authority
- ES
- Spain
- Prior art keywords
- carbonyl
- pyrido
- hexahydro
- oxacin
- heptane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000002391 heterocyclic compounds Chemical class 0.000 title abstract description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 231
- 238000000034 method Methods 0.000 claims abstract description 100
- 238000004519 manufacturing process Methods 0.000 claims abstract description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 107
- 239000001257 hydrogen Substances 0.000 claims description 107
- 150000003839 salts Chemical class 0.000 claims description 103
- -1 spiro[4.5]decan-3-yl Chemical group 0.000 claims description 77
- 125000000217 alkyl group Chemical group 0.000 claims description 66
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 60
- 125000002619 bicyclic group Chemical group 0.000 claims description 52
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 49
- 150000002431 hydrogen Chemical class 0.000 claims description 48
- 229910052736 halogen Inorganic materials 0.000 claims description 36
- 150000002367 halogens Chemical class 0.000 claims description 36
- 238000011282 treatment Methods 0.000 claims description 31
- 241000124008 Mammalia Species 0.000 claims description 29
- 208000002193 Pain Diseases 0.000 claims description 27
- 239000000460 chlorine Substances 0.000 claims description 23
- 238000011321 prophylaxis Methods 0.000 claims description 21
- 125000003118 aryl group Chemical group 0.000 claims description 20
- 239000003153 chemical reaction reagent Substances 0.000 claims description 20
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims description 19
- 229910052801 chlorine Inorganic materials 0.000 claims description 18
- 230000036407 pain Effects 0.000 claims description 18
- 239000011737 fluorine Substances 0.000 claims description 17
- 229910052731 fluorine Inorganic materials 0.000 claims description 17
- 230000004770 neurodegeneration Effects 0.000 claims description 17
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 claims description 16
- 208000036110 Neuroinflammatory disease Diseases 0.000 claims description 16
- 230000003959 neuroinflammation Effects 0.000 claims description 16
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 16
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 15
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 15
- 125000006708 (C5-C14) heteroaryl group Chemical group 0.000 claims description 14
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 14
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 14
- 201000006417 multiple sclerosis Diseases 0.000 claims description 14
- 206010028980 Neoplasm Diseases 0.000 claims description 13
- 150000001412 amines Chemical class 0.000 claims description 13
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 12
- 208000024827 Alzheimer disease Diseases 0.000 claims description 12
- 125000001072 heteroaryl group Chemical group 0.000 claims description 12
- 125000005915 C6-C14 aryl group Chemical group 0.000 claims description 11
- 208000018737 Parkinson disease Diseases 0.000 claims description 11
- 201000011510 cancer Diseases 0.000 claims description 11
- 208000020016 psychiatric disease Diseases 0.000 claims description 11
- 125000000623 heterocyclic group Chemical group 0.000 claims description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- 206010029350 Neurotoxicity Diseases 0.000 claims description 9
- 206010044221 Toxic encephalopathy Diseases 0.000 claims description 9
- 231100000228 neurotoxicity Toxicity 0.000 claims description 9
- 230000007135 neurotoxicity Effects 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 230000008569 process Effects 0.000 claims description 8
- 208000019901 Anxiety disease Diseases 0.000 claims description 7
- 230000036506 anxiety Effects 0.000 claims description 7
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 7
- 238000002512 chemotherapy Methods 0.000 claims description 7
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 7
- 208000004296 neuralgia Diseases 0.000 claims description 7
- 208000021722 neuropathic pain Diseases 0.000 claims description 7
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 7
- 208000019695 Migraine disease Diseases 0.000 claims description 6
- 208000006011 Stroke Diseases 0.000 claims description 6
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 6
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 6
- 206010015037 epilepsy Diseases 0.000 claims description 6
- 206010027599 migraine Diseases 0.000 claims description 6
- 201000001119 neuropathy Diseases 0.000 claims description 6
- 230000007823 neuropathy Effects 0.000 claims description 6
- 208000033808 peripheral neuropathy Diseases 0.000 claims description 6
- HCMJWOGOISXSDL-UHFFFAOYSA-N (2-isothiocyanato-1-phenylethyl)benzene Chemical compound C=1C=CC=CC=1C(CN=C=S)C1=CC=CC=C1 HCMJWOGOISXSDL-UHFFFAOYSA-N 0.000 claims description 5
- 208000000094 Chronic Pain Diseases 0.000 claims description 5
- 206010033128 Ovarian cancer Diseases 0.000 claims description 5
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 5
- 230000004736 colon carcinogenesis Effects 0.000 claims description 5
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 5
- 208000008238 Muscle Spasticity Diseases 0.000 claims description 4
- 208000005298 acute pain Diseases 0.000 claims description 4
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 claims description 4
- 125000005226 heteroaryloxycarbonyl group Chemical group 0.000 claims description 4
- 125000004076 pyridyl group Chemical group 0.000 claims description 4
- 208000018198 spasticity Diseases 0.000 claims description 4
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims description 3
- IWSMFTHUILISSF-UHOSZYNNSA-N O=C(N(CC1)CCC1(C1)OC[C@H]1NS(C1=CC(C(F)(F)F)=CC=C1)(=O)=O)N(CC[C@@H]1OC2)C[C@H]1NC2=O Chemical compound O=C(N(CC1)CCC1(C1)OC[C@H]1NS(C1=CC(C(F)(F)F)=CC=C1)(=O)=O)N(CC[C@@H]1OC2)C[C@H]1NC2=O IWSMFTHUILISSF-UHOSZYNNSA-N 0.000 claims description 3
- YPJLRSCTNWRNMP-UHOSZYNNSA-N O=C(N(CC1)CCC1(C1)OC[C@H]1NS(C1=CC=C(C(F)(F)F)C=C1)(=O)=O)N(CC[C@@H]1OC2)C[C@H]1NC2=O Chemical compound O=C(N(CC1)CCC1(C1)OC[C@H]1NS(C1=CC=C(C(F)(F)F)C=C1)(=O)=O)N(CC[C@@H]1OC2)C[C@H]1NC2=O YPJLRSCTNWRNMP-UHOSZYNNSA-N 0.000 claims description 3
- 239000013543 active substance Substances 0.000 claims description 3
- 230000001131 transforming effect Effects 0.000 claims description 3
- VABYZYZVRBFITR-ZBFHGGJFSA-N (4aR,8aS)-6-[6-[2,5-bis(trifluoromethyl)phenyl]-2,6-diazaspiro[3.3]heptane-2-carbonyl]-4,4a,5,7,8,8a-hexahydropyrido[4,3-b][1,4]oxazin-3-one Chemical compound C1CN(C[C@@H]2[C@H]1OCC(=O)N2)C(=O)N3CC4(C3)CN(C4)C5=C(C=CC(=C5)C(F)(F)F)C(F)(F)F VABYZYZVRBFITR-ZBFHGGJFSA-N 0.000 claims description 2
- 206010019196 Head injury Diseases 0.000 claims description 2
- 125000004043 oxo group Chemical group O=* 0.000 claims description 2
- 239000000203 mixture Substances 0.000 abstract description 50
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 137
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 129
- 239000000543 intermediate Substances 0.000 description 129
- 238000006243 chemical reaction Methods 0.000 description 84
- 239000000243 solution Substances 0.000 description 80
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 77
- 235000019439 ethyl acetate Nutrition 0.000 description 63
- 239000011541 reaction mixture Substances 0.000 description 56
- 239000002904 solvent Substances 0.000 description 55
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 53
- 125000006239 protecting group Chemical group 0.000 description 52
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 49
- 102000005398 Monoacylglycerol Lipase Human genes 0.000 description 46
- 108020002334 Monoacylglycerol lipase Proteins 0.000 description 46
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 46
- 239000000047 product Substances 0.000 description 45
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 42
- 239000007787 solid Substances 0.000 description 38
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 38
- 238000000746 purification Methods 0.000 description 37
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 36
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 31
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 31
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 29
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 28
- 239000000725 suspension Substances 0.000 description 28
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 27
- 239000003921 oil Substances 0.000 description 27
- 235000019198 oils Nutrition 0.000 description 27
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 26
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 26
- 239000012044 organic layer Substances 0.000 description 26
- RCRCTBLIHCHWDZ-UHFFFAOYSA-N 2-Arachidonoyl Glycerol Chemical compound CCCCCC=CCC=CCC=CCC=CCCCC(=O)OC(CO)CO RCRCTBLIHCHWDZ-UHFFFAOYSA-N 0.000 description 25
- 238000003786 synthesis reaction Methods 0.000 description 24
- 230000015572 biosynthetic process Effects 0.000 description 23
- 125000003545 alkoxy group Chemical group 0.000 description 22
- 229910052786 argon Inorganic materials 0.000 description 21
- 239000012267 brine Substances 0.000 description 20
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 20
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 239000002585 base Substances 0.000 description 17
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 17
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 16
- 238000003556 assay Methods 0.000 description 16
- 230000000670 limiting effect Effects 0.000 description 16
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 15
- 238000002953 preparative HPLC Methods 0.000 description 15
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 14
- 229940114079 arachidonic acid Drugs 0.000 description 14
- 235000021342 arachidonic acid Nutrition 0.000 description 14
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 14
- 239000012071 phase Substances 0.000 description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 14
- 239000000741 silica gel Substances 0.000 description 14
- 229910002027 silica gel Inorganic materials 0.000 description 14
- 229910052938 sodium sulfate Inorganic materials 0.000 description 14
- 235000011152 sodium sulphate Nutrition 0.000 description 14
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 13
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 13
- 210000004556 brain Anatomy 0.000 description 13
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 13
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 13
- 239000010410 layer Substances 0.000 description 13
- 239000011734 sodium Substances 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- 229910000104 sodium hydride Inorganic materials 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- 238000003818 flash chromatography Methods 0.000 description 11
- 239000012074 organic phase Substances 0.000 description 11
- 229910052763 palladium Inorganic materials 0.000 description 11
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 10
- 239000008346 aqueous phase Substances 0.000 description 10
- 239000012230 colorless oil Substances 0.000 description 10
- 239000013058 crude material Substances 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 229920006395 saturated elastomer Polymers 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 10
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 208000030886 Traumatic Brain injury Diseases 0.000 description 9
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 9
- 150000001805 chlorine compounds Chemical class 0.000 description 9
- 125000006413 ring segment Chemical group 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 8
- KFEHNXLFIGPWNB-UHFFFAOYSA-N 2-fluoro-4-(trifluoromethyl)benzaldehyde Chemical compound FC1=CC(C(F)(F)F)=CC=C1C=O KFEHNXLFIGPWNB-UHFFFAOYSA-N 0.000 description 8
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- 229910052794 bromium Inorganic materials 0.000 description 8
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 8
- 239000003054 catalyst Substances 0.000 description 8
- 125000000753 cycloalkyl group Chemical group 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 125000005843 halogen group Chemical group 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 230000009529 traumatic brain injury Effects 0.000 description 8
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 7
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 7
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000012043 crude product Substances 0.000 description 7
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 description 7
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 7
- 239000007858 starting material Substances 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 238000002835 absorbance Methods 0.000 description 6
- 239000002671 adjuvant Substances 0.000 description 6
- 230000029936 alkylation Effects 0.000 description 6
- 238000005804 alkylation reaction Methods 0.000 description 6
- 239000012131 assay buffer Substances 0.000 description 6
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 6
- CODNYICXDISAEA-UHFFFAOYSA-N bromine monochloride Chemical compound BrCl CODNYICXDISAEA-UHFFFAOYSA-N 0.000 description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 description 6
- 150000001735 carboxylic acids Chemical class 0.000 description 6
- 238000004296 chiral HPLC Methods 0.000 description 6
- 125000004093 cyano group Chemical group *C#N 0.000 description 6
- 238000010790 dilution Methods 0.000 description 6
- 239000012895 dilution Substances 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 6
- 210000004248 oligodendroglia Anatomy 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 6
- 238000004808 supercritical fluid chromatography Methods 0.000 description 6
- PYRAMVPOMDHTNX-UHFFFAOYSA-N tert-butyl 2,6-diazaspiro[3.3]heptane-2-carboxylate;oxalic acid Chemical compound OC(=O)C(O)=O.C1N(C(=O)OC(C)(C)C)CC11CNC1.C1N(C(=O)OC(C)(C)C)CC11CNC1 PYRAMVPOMDHTNX-UHFFFAOYSA-N 0.000 description 6
- UMXXHZDEAZUQKZ-UHFFFAOYSA-N tert-butyl 6-hydroxy-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC11CC(O)C1 UMXXHZDEAZUQKZ-UHFFFAOYSA-N 0.000 description 6
- PSSWASGEGXCINO-RFZPGFLSSA-N (3R,4R)-3-aminopiperidin-4-ol Chemical class N[C@@H]1CNCC[C@H]1O PSSWASGEGXCINO-RFZPGFLSSA-N 0.000 description 5
- YBADLXQNJCMBKR-UHFFFAOYSA-N (4-nitrophenyl)acetic acid Chemical compound OC(=O)CC1=CC=C([N+]([O-])=O)C=C1 YBADLXQNJCMBKR-UHFFFAOYSA-N 0.000 description 5
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 5
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 description 5
- VKMLQSOYCDSKFO-UHFFFAOYSA-N ClC1=C(CNC2COC3(C2)CCN(CC3)C(=O)OC(C)(C)C)C=CC(=C1)F Chemical compound ClC1=C(CNC2COC3(C2)CCN(CC3)C(=O)OC(C)(C)C)C=CC(=C1)F VKMLQSOYCDSKFO-UHFFFAOYSA-N 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 5
- 238000006751 Mitsunobu reaction Methods 0.000 description 5
- 150000001200 N-acyl ethanolamides Chemical class 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 238000009835 boiling Methods 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 210000003169 central nervous system Anatomy 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 239000002621 endocannabinoid Substances 0.000 description 5
- 239000002158 endotoxin Substances 0.000 description 5
- 235000019253 formic acid Nutrition 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 229920006008 lipopolysaccharide Polymers 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 210000000653 nervous system Anatomy 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- NRADOPGBTAJXKB-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[3.5]nonane-7-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC11CNC1 NRADOPGBTAJXKB-UHFFFAOYSA-N 0.000 description 5
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 5
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 4
- LOVPHSMOAVXQIH-UHFFFAOYSA-N (4-nitrophenyl) hydrogen carbonate Chemical compound OC(=O)OC1=CC=C([N+]([O-])=O)C=C1 LOVPHSMOAVXQIH-UHFFFAOYSA-N 0.000 description 4
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 4
- FCFPJKHVCHKCMP-UHFFFAOYSA-N 2-chloro-4-fluorobenzenesulfonyl chloride Chemical compound FC1=CC=C(S(Cl)(=O)=O)C(Cl)=C1 FCFPJKHVCHKCMP-UHFFFAOYSA-N 0.000 description 4
- IGYXYGDEYHNFFT-UHFFFAOYSA-N 2-chloro-4-fluorophenol Chemical compound OC1=CC=C(F)C=C1Cl IGYXYGDEYHNFFT-UHFFFAOYSA-N 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 235000019270 ammonium chloride Nutrition 0.000 description 4
- 239000012300 argon atmosphere Substances 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 4
- HCUYBXPSSCRKRF-UHFFFAOYSA-N diphosgene Chemical compound ClC(=O)OC(Cl)(Cl)Cl HCUYBXPSSCRKRF-UHFFFAOYSA-N 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 150000007529 inorganic bases Chemical class 0.000 description 4
- 150000002576 ketones Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 4
- 210000000274 microglia Anatomy 0.000 description 4
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 150000007530 organic bases Chemical class 0.000 description 4
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 4
- 229920005862 polyol Polymers 0.000 description 4
- 150000003077 polyols Chemical class 0.000 description 4
- 235000015320 potassium carbonate Nutrition 0.000 description 4
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Substances [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 4
- 238000010898 silica gel chromatography Methods 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- BGUYAMZPJMTFRU-UHFFFAOYSA-N tert-butyl 2,6-diazaspiro[3.4]octane-6-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC11CNC1 BGUYAMZPJMTFRU-UHFFFAOYSA-N 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 229940099259 vaseline Drugs 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- YZXBAPSDXZZRGB-PNXWREOESA-N (5z,8z,11z,14z)-2,2,3,3,4,4,5,6-octadeuterioicosa-5,8,11,14-tetraenoic acid Chemical compound OC(=O)C([2H])([2H])C([2H])([2H])C([2H])([2H])C(/[2H])=C(/[2H])C\C=C/C\C=C/C\C=C/CCCCC YZXBAPSDXZZRGB-PNXWREOESA-N 0.000 description 3
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 description 3
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 3
- KMQWNQKESAHDKD-UHFFFAOYSA-N 2-chloro-4-fluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C(Cl)=C1 KMQWNQKESAHDKD-UHFFFAOYSA-N 0.000 description 3
- WEUIJSDNBZVSNY-UHFFFAOYSA-N 2-fluoro-4-(trifluoromethyl)phenol Chemical compound OC1=CC=C(C(F)(F)F)C=C1F WEUIJSDNBZVSNY-UHFFFAOYSA-N 0.000 description 3
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- 229910015845 BBr3 Inorganic materials 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 102100036214 Cannabinoid receptor 2 Human genes 0.000 description 3
- 101710187022 Cannabinoid receptor 2 Proteins 0.000 description 3
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical class C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 102100037611 Lysophospholipase Human genes 0.000 description 3
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 239000004743 Polypropylene Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 238000006880 cross-coupling reaction Methods 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 3
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 3
- 239000008298 dragée Substances 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 125000004438 haloalkoxy group Chemical group 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 150000002617 leukotrienes Chemical class 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 210000002569 neuron Anatomy 0.000 description 3
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- VBQCHPIMZGQLAZ-UHFFFAOYSA-N phosphorane Chemical class [PH5] VBQCHPIMZGQLAZ-UHFFFAOYSA-N 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- 150000003180 prostaglandins Chemical class 0.000 description 3
- 230000009257 reactivity Effects 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 238000006268 reductive amination reaction Methods 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- GBLUNQGZHFQTPW-SNVBAGLBSA-N tert-butyl (3r)-3-amino-1-oxa-8-azaspiro[4.5]decane-8-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC11OC[C@H](N)C1 GBLUNQGZHFQTPW-SNVBAGLBSA-N 0.000 description 3
- QTKGBUXGQNHIIM-UHFFFAOYSA-N tert-butyl 2-hydroxy-7-azaspiro[3.5]nonane-7-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC11CC(O)C1 QTKGBUXGQNHIIM-UHFFFAOYSA-N 0.000 description 3
- HQHRAGXKFOTSQE-UHFFFAOYSA-N tert-butyl 6-oxo-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC21CC(=O)C2 HQHRAGXKFOTSQE-UHFFFAOYSA-N 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 150000003672 ureas Chemical class 0.000 description 3
- BARRCLLWVNPCJS-NEPJUHHUSA-N (4-nitrophenyl) (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahydropyrido[4,3-b][1,4]oxazine-6-carboxylate Chemical compound [O-][N+](=O)C1=CC=C(OC(=O)N2CC[C@@H]3OCC(=O)N[C@@H]3C2)C=C1 BARRCLLWVNPCJS-NEPJUHHUSA-N 0.000 description 2
- GBOWGKOVMBDPJF-UHFFFAOYSA-N 1-fluoro-3-(trifluoromethyl)benzene Chemical compound FC1=CC=CC(C(F)(F)F)=C1 GBOWGKOVMBDPJF-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- ZNOVTXRBGFNYRX-UHFFFAOYSA-N 2-[[4-[(2-amino-5-methyl-4-oxo-1,6,7,8-tetrahydropteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 ZNOVTXRBGFNYRX-UHFFFAOYSA-N 0.000 description 2
- FNYJQRYWXHLCSX-UHFFFAOYSA-N 2-fluoro-4-(trifluoromethyl)benzenesulfonyl chloride Chemical compound FC1=CC(C(F)(F)F)=CC=C1S(Cl)(=O)=O FNYJQRYWXHLCSX-UHFFFAOYSA-N 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- OZDCZHDOIBUGAJ-UHFFFAOYSA-N 4-(trifluoromethyl)benzenesulfonyl chloride Chemical compound FC(F)(F)C1=CC=C(S(Cl)(=O)=O)C=C1 OZDCZHDOIBUGAJ-UHFFFAOYSA-N 0.000 description 2
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 2
- PTPTZLXZHPPVKG-UHFFFAOYSA-N 4-bromo-2-fluoropyridine Chemical compound FC1=CC(Br)=CC=N1 PTPTZLXZHPPVKG-UHFFFAOYSA-N 0.000 description 2
- TXNLQUKVUJITMX-UHFFFAOYSA-N 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine Chemical group CC(C)(C)C1=CC=NC(C=2N=CC=C(C=2)C(C)(C)C)=C1 TXNLQUKVUJITMX-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- 238000006443 Buchwald-Hartwig cross coupling reaction Methods 0.000 description 2
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 description 2
- 102000018208 Cannabinoid Receptor Human genes 0.000 description 2
- 108050007331 Cannabinoid receptor Proteins 0.000 description 2
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 2
- YURLRIFFSBUNMI-CQSZACIVSA-N ClC1=C(C=CC(=C1)F)S(=O)(=O)N[C@H]1COC2(C1)CCN(CC2)C(=O)OC(C)(C)C Chemical compound ClC1=C(C=CC(=C1)F)S(=O)(=O)N[C@H]1COC2(C1)CCN(CC2)C(=O)OC(C)(C)C YURLRIFFSBUNMI-CQSZACIVSA-N 0.000 description 2
- XAMIZLHXUMYUFQ-UHFFFAOYSA-N ClC1=C(CN(C2COC3(C2)CCN(CC3)C(=O)OC(C)(C)C)C)C=CC(=C1)F Chemical compound ClC1=C(CN(C2COC3(C2)CCN(CC3)C(=O)OC(C)(C)C)C)C=CC(=C1)F XAMIZLHXUMYUFQ-UHFFFAOYSA-N 0.000 description 2
- SBUKLPSBNFWJCU-UHFFFAOYSA-N ClIBr Chemical compound ClIBr SBUKLPSBNFWJCU-UHFFFAOYSA-N 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 238000006130 Horner-Wadsworth-Emmons olefination reaction Methods 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 102000017055 Lipoprotein Lipase Human genes 0.000 description 2
- 108010013563 Lipoprotein Lipase Proteins 0.000 description 2
- DAKFKMOSSGVLIN-UHFFFAOYSA-N OC(C(F)(F)F)=O.BrC1=CC=C(C(C2)CC22CNC2)C=C1 Chemical compound OC(C(F)(F)F)=O.BrC1=CC=C(C(C2)CC22CNC2)C=C1 DAKFKMOSSGVLIN-UHFFFAOYSA-N 0.000 description 2
- GRYQYKWVTGLYOQ-UHFFFAOYSA-N OC(CCN(C1)C(OCC2=CC=CC=C2)=O)C1NC(CCl)=O Chemical compound OC(CCN(C1)C(OCC2=CC=CC=C2)=O)C1NC(CCl)=O GRYQYKWVTGLYOQ-UHFFFAOYSA-N 0.000 description 2
- 229910002666 PdCl2 Inorganic materials 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 108010058864 Phospholipases A2 Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical class [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 102000004005 Prostaglandin-endoperoxide synthases Human genes 0.000 description 2
- 108090000459 Prostaglandin-endoperoxide synthases Proteins 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- XNRNNGPBEPRNAR-UHFFFAOYSA-N Thromboxane B2 Natural products CCCCCC(O)C=CC1OC(O)CC(O)C1CC=CCCCC(O)=O XNRNNGPBEPRNAR-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- LGEQQWMQCRIYKG-DOFZRALJSA-N anandamide Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(=O)NCCO LGEQQWMQCRIYKG-DOFZRALJSA-N 0.000 description 2
- 210000001130 astrocyte Anatomy 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- VUQWJABSPMOXEK-UHFFFAOYSA-N benzyl 3-oxo-4,4a,5,7,8,8a-hexahydropyrido[4,3-b][1,4]oxazine-6-carboxylate Chemical compound C1CN(CC2C1OCC(=O)N2)C(=O)OCC3=CC=CC=C3 VUQWJABSPMOXEK-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000003821 enantio-separation Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000003925 fat Substances 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- HQVFCQRVQFYGRJ-UHFFFAOYSA-N formic acid;hydrate Chemical compound O.OC=O HQVFCQRVQFYGRJ-UHFFFAOYSA-N 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 125000000814 indol-3-yl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C([*])C2=C1[H] 0.000 description 2
- 239000013067 intermediate product Substances 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 230000037356 lipid metabolism Effects 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000003340 mental effect Effects 0.000 description 2
- KVKFRMCSXWQSNT-UHFFFAOYSA-N n,n'-dimethylethane-1,2-diamine Chemical compound CNCCNC KVKFRMCSXWQSNT-UHFFFAOYSA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- 210000001428 peripheral nervous system Anatomy 0.000 description 2
- 230000009038 pharmacological inhibition Effects 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- 150000004714 phosphonium salts Chemical class 0.000 description 2
- 239000011941 photocatalyst Substances 0.000 description 2
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 238000012877 positron emission topography Methods 0.000 description 2
- 239000011591 potassium Chemical class 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011736 potassium bicarbonate Substances 0.000 description 2
- 235000015497 potassium bicarbonate Nutrition 0.000 description 2
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- 239000012041 precatalyst Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical class ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- HPPARSNAMZJAPZ-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.4]nonane-2-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC11CNCC1 HPPARSNAMZJAPZ-UHFFFAOYSA-N 0.000 description 2
- UTNKFENGOAIODD-UHFFFAOYSA-N tert-butyl 2-(2-chloro-4-fluorophenyl)sulfonyl-2,6-diazaspiro[3.3]heptane-6-carboxylate Chemical compound ClC1=C(C=CC(=C1)F)S(=O)(=O)N1CC2(CN(C2)C(=O)OC(C)(C)C)C1 UTNKFENGOAIODD-UHFFFAOYSA-N 0.000 description 2
- SHNYGXMIHRGFBJ-UHFFFAOYSA-N tert-butyl 2-(2-chloro-4-fluorophenyl)sulfonyl-2,7-diazaspiro[3.5]nonane-7-carboxylate Chemical compound ClC1=C(C=CC(=C1)F)S(=O)(=O)N1CC2(C1)CCN(CC2)C(=O)OC(C)(C)C SHNYGXMIHRGFBJ-UHFFFAOYSA-N 0.000 description 2
- UFUWDJDYOGGGIP-UHFFFAOYSA-N tert-butyl 2-[2-fluoro-4-(trifluoromethyl)phenoxy]-7-azaspiro[3.5]nonane-7-carboxylate Chemical compound FC1=C(OC2CC3(C2)CCN(CC3)C(=O)OC(C)(C)C)C=CC(=C1)C(F)(F)F UFUWDJDYOGGGIP-UHFFFAOYSA-N 0.000 description 2
- OTNFAVFLKOLOQE-UHFFFAOYSA-N tert-butyl 2-[2-fluoro-4-(trifluoromethyl)phenyl]sulfonyl-2,6-diazaspiro[3.3]heptane-6-carboxylate Chemical compound FC1=C(C=CC(=C1)C(F)(F)F)S(=O)(=O)N1CC2(CN(C2)C(=O)OC(C)(C)C)C1 OTNFAVFLKOLOQE-UHFFFAOYSA-N 0.000 description 2
- IDLIXSQPUIZUFM-UHFFFAOYSA-N tert-butyl 2-[4-(trifluoromethyl)phenyl]sulfonyl-2,7-diazaspiro[3.5]nonane-7-carboxylate Chemical compound FC(C1=CC=C(C=C1)S(=O)(=O)N1CC2(C1)CCN(CC2)C(=O)OC(C)(C)C)(F)F IDLIXSQPUIZUFM-UHFFFAOYSA-N 0.000 description 2
- CQGHICWYAUAEMI-UHFFFAOYSA-N tert-butyl 2-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,7-diazaspiro[3.4]octane-7-carboxylate Chemical compound FC1=C(CN2CC3(C2)CN(CC3)C(=O)OC(C)(C)C)C=CC(=C1)C(F)(F)F CQGHICWYAUAEMI-UHFFFAOYSA-N 0.000 description 2
- KDROZGRZOZCBKT-UHFFFAOYSA-N tert-butyl 2-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,7-diazaspiro[3.5]nonane-7-carboxylate Chemical compound FC1=C(CN2CC3(C2)CCN(CC3)C(=O)OC(C)(C)C)C=CC(=C1)C(F)(F)F KDROZGRZOZCBKT-UHFFFAOYSA-N 0.000 description 2
- PGDPKZJGCWOSAZ-UHFFFAOYSA-N tert-butyl 6-(2-chloro-4-fluorophenoxy)-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound ClC1=C(OC2CC3(CN(C3)C(=O)OC(C)(C)C)C2)C=CC(=C1)F PGDPKZJGCWOSAZ-UHFFFAOYSA-N 0.000 description 2
- DDYDRGNYVNDPNR-UHFFFAOYSA-N tert-butyl 6-(hydroxymethyl)-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC11CC(CO)C1 DDYDRGNYVNDPNR-UHFFFAOYSA-N 0.000 description 2
- SYVORJBNYHZGIO-UHFFFAOYSA-N tert-butyl 6-[(2-chloro-4-fluorophenyl)methyl]-2,6-diazaspiro[3.3]heptane-2-carboxylate Chemical compound ClC1=C(CN2CC3(CN(C3)C(=O)OC(C)(C)C)C2)C=CC(=C1)F SYVORJBNYHZGIO-UHFFFAOYSA-N 0.000 description 2
- BFEKATJLENXJSK-UHFFFAOYSA-N tert-butyl 6-[2-fluoro-4-(trifluoromethyl)phenyl]-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound FC1=C(C=CC(=C1)C(F)(F)F)C1CC2(CN(C2)C(=O)OC(C)(C)C)C1 BFEKATJLENXJSK-UHFFFAOYSA-N 0.000 description 2
- XLPMQQVVWMVIEG-UHFFFAOYSA-N tert-butyl 6-[4,5-bis(trifluoromethyl)pyridin-2-yl]oxy-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound FC(C1=CC(=NC=C1C(F)(F)F)OC1CC2(CN(C2)C(=O)OC(C)(C)C)C1)(F)F XLPMQQVVWMVIEG-UHFFFAOYSA-N 0.000 description 2
- TXRWUZYKAJWQJM-UHFFFAOYSA-N tert-butyl 6-[5-(trifluoromethyl)pyridin-2-yl]oxy-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound FC(C=1C=CC(=NC=1)OC1CC2(CN(C2)C(=O)OC(C)(C)C)C1)(F)F TXRWUZYKAJWQJM-UHFFFAOYSA-N 0.000 description 2
- UKZZSAKGUUYQHG-UHFFFAOYSA-N tert-butyl 6-[[2-fluoro-4-(trifluoromethyl)phenoxy]methyl]-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound FC1=C(OCC2CC3(CN(C3)C(=O)OC(C)(C)C)C2)C=CC(=C1)C(F)(F)F UKZZSAKGUUYQHG-UHFFFAOYSA-N 0.000 description 2
- FBSUEALECACMCG-UHFFFAOYSA-N tert-butyl 6-[[2-fluoro-4-(trifluoromethyl)phenyl]methoxy]-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound FC1=C(COC2CC3(CN(C3)C(=O)OC(C)(C)C)C2)C=CC(=C1)C(F)(F)F FBSUEALECACMCG-UHFFFAOYSA-N 0.000 description 2
- CJPHJWNFVKLHMJ-UHFFFAOYSA-N tert-butyl 6-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC2(C1)CC(C2)CC3=C(C=C(C=C3)C(F)(F)F)F CJPHJWNFVKLHMJ-UHFFFAOYSA-N 0.000 description 2
- BBZFGJIVFWJPPD-UHFFFAOYSA-N tert-butyl 6-[[4-methyl-3-(trifluoromethyl)phenyl]methoxy]-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound CC1=C(C=C(COC2CC3(CN(C3)C(=O)OC(C)(C)C)C2)C=C1)C(F)(F)F BBZFGJIVFWJPPD-UHFFFAOYSA-N 0.000 description 2
- HQEIXMTWVJWYMA-UHFFFAOYSA-N tert-butyl 6-bromo-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound BrC1CC2(CN(C2)C(=O)OC(C)(C)C)C1 HQEIXMTWVJWYMA-UHFFFAOYSA-N 0.000 description 2
- FDWWZBWSXCUBRR-UHFFFAOYSA-N tert-butyl 7-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,7-diazaspiro[4.4]nonane-2-carboxylate Chemical compound FC1=C(CN2CC3(CCN(C3)C(=O)OC(C)(C)C)CC2)C=CC(=C1)C(F)(F)F FDWWZBWSXCUBRR-UHFFFAOYSA-N 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- XNRNNGPBEPRNAR-JQBLCGNGSA-N thromboxane B2 Chemical compound CCCCC[C@H](O)\C=C\[C@H]1OC(O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O XNRNNGPBEPRNAR-JQBLCGNGSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- SCHZCUMIENIQMY-UHFFFAOYSA-N tris(trimethylsilyl)silicon Chemical compound C[Si](C)(C)[Si]([Si](C)(C)C)[Si](C)(C)C SCHZCUMIENIQMY-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- MGNBKNBEZGLHNF-UHFFFAOYSA-N (2-methylpyrazol-3-yl)boronic acid Chemical compound CN1N=CC=C1B(O)O MGNBKNBEZGLHNF-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- BARRCLLWVNPCJS-NWDGAFQWSA-N (4-nitrophenyl) (4aS,8aR)-3-oxo-4,4a,5,7,8,8a-hexahydropyrido[4,3-b][1,4]oxazine-6-carboxylate Chemical compound O=C1N[C@@H]2[C@H](OC1)CCN(C2)C(=O)OC1=CC=C(C=C1)[N+](=O)[O-] BARRCLLWVNPCJS-NWDGAFQWSA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- ASVDYYHIICJWOE-SJORKVTESA-N (4aR,8aS)-6-[6-[2-fluoro-4-(trifluoromethyl)phenyl]-2-azaspiro[3.3]heptane-2-carbonyl]-4,4a,5,7,8,8a-hexahydropyrido[4,3-b][1,4]oxazin-3-one Chemical compound C1CN(C[C@@H]2[C@H]1OCC(=O)N2)C(=O)N3CC4(C3)CC(C4)C5=C(C=C(C=C5)C(F)(F)F)F ASVDYYHIICJWOE-SJORKVTESA-N 0.000 description 1
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- CBPHUQIHJKGXHY-UHFFFAOYSA-N 1,2-dimethoxyethane;hydrochloride Chemical compound Cl.COCCOC CBPHUQIHJKGXHY-UHFFFAOYSA-N 0.000 description 1
- SWJPEBQEEAHIGZ-UHFFFAOYSA-N 1,4-dibromobenzene Chemical compound BrC1=CC=C(Br)C=C1 SWJPEBQEEAHIGZ-UHFFFAOYSA-N 0.000 description 1
- YINFEFUSAQRZGG-UHFFFAOYSA-N 1-(4-bromophenyl)pyrrolidin-2-one Chemical compound C1=CC(Br)=CC=C1N1C(=O)CCC1 YINFEFUSAQRZGG-UHFFFAOYSA-N 0.000 description 1
- IBLMYGXJKQIGSN-UHFFFAOYSA-N 1-(bromomethyl)-2,4-difluorobenzene Chemical compound FC1=CC=C(CBr)C(F)=C1 IBLMYGXJKQIGSN-UHFFFAOYSA-N 0.000 description 1
- GAUUDQVOPUKGJD-UHFFFAOYSA-N 1-(bromomethyl)-2-chloro-4-fluorobenzene Chemical compound FC1=CC=C(CBr)C(Cl)=C1 GAUUDQVOPUKGJD-UHFFFAOYSA-N 0.000 description 1
- MQUCRCQNRDHRPL-UHFFFAOYSA-N 1-(bromomethyl)-2-fluoro-4-(trifluoromethyl)benzene Chemical compound FC1=CC(C(F)(F)F)=CC=C1CBr MQUCRCQNRDHRPL-UHFFFAOYSA-N 0.000 description 1
- WWAGWLZAZFSJRH-UHFFFAOYSA-N 1-(bromomethyl)-2-methoxy-4-(trifluoromethyl)benzene Chemical compound COC1=CC(C(F)(F)F)=CC=C1CBr WWAGWLZAZFSJRH-UHFFFAOYSA-N 0.000 description 1
- WAUNFWSVXRPCDQ-UHFFFAOYSA-N 1-(bromomethyl)-2-methoxybenzene Chemical compound COC1=CC=CC=C1CBr WAUNFWSVXRPCDQ-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical group C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- NNMBNYHMJRJUBC-UHFFFAOYSA-N 1-bromo-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC(Br)=C1 NNMBNYHMJRJUBC-UHFFFAOYSA-N 0.000 description 1
- MPAOOLLBWUEXOM-UHFFFAOYSA-N 1-bromo-4-propan-2-yloxybenzene Chemical compound CC(C)OC1=CC=C(Br)C=C1 MPAOOLLBWUEXOM-UHFFFAOYSA-N 0.000 description 1
- KVYOWXUQJLOCIA-UHFFFAOYSA-N 1-oxa-8-azaspiro[4.5]decane Chemical compound C1CCOC21CCNCC2 KVYOWXUQJLOCIA-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- KSOZFWPHXQNOEB-UHFFFAOYSA-N 2,4-bis(trifluoromethyl)benzenesulfonyl chloride Chemical compound FC(F)(F)C1=CC=C(S(Cl)(=O)=O)C(C(F)(F)F)=C1 KSOZFWPHXQNOEB-UHFFFAOYSA-N 0.000 description 1
- NVWVWEWVLBKPSM-UHFFFAOYSA-N 2,4-difluorophenol Chemical compound OC1=CC=C(F)C=C1F NVWVWEWVLBKPSM-UHFFFAOYSA-N 0.000 description 1
- UDSAJFSYJMHNFI-UHFFFAOYSA-N 2,6-diazaspiro[3.3]heptane Chemical compound C1NCC11CNC1 UDSAJFSYJMHNFI-UHFFFAOYSA-N 0.000 description 1
- OXPIFHNJGOJJQZ-UHFFFAOYSA-N 2,7-diazaspiro[3.4]octane Chemical compound C1NCC11CNCC1 OXPIFHNJGOJJQZ-UHFFFAOYSA-N 0.000 description 1
- DROZYMFJWSYDRY-UHFFFAOYSA-N 2,7-diazaspiro[3.5]nonane Chemical compound C1NCC11CCNCC1 DROZYMFJWSYDRY-UHFFFAOYSA-N 0.000 description 1
- DDVRNOMZDQTUNS-UHFFFAOYSA-N 2,7-diazaspiro[4.4]nonane Chemical compound C1NCCC11CNCC1 DDVRNOMZDQTUNS-UHFFFAOYSA-N 0.000 description 1
- ODHKGRIXVOBNMW-UHFFFAOYSA-N 2-(2-azaspiro[3.3]heptan-6-ylmethyl)-3-fluorophenol Chemical compound C1NCC11CC(C1)CC1=C(C=CC=C1F)O ODHKGRIXVOBNMW-UHFFFAOYSA-N 0.000 description 1
- XSOHPEHRLNXOPK-UHFFFAOYSA-N 2-(2-azaspiro[3.3]heptan-6-ylmethyl)phenol Chemical compound C1NCC11CC(C1)CC1=C(C=CC=C1)O XSOHPEHRLNXOPK-UHFFFAOYSA-N 0.000 description 1
- KIEBJACUQXGWKN-UHFFFAOYSA-N 2-(2-chloro-4-fluorophenyl)sulfonyl-2,7-diazaspiro[3.5]nonane hydrochloride Chemical compound Cl.ClC1=C(C=CC(=C1)F)S(=O)(=O)N1CC2(C1)CCNCC2 KIEBJACUQXGWKN-UHFFFAOYSA-N 0.000 description 1
- XAQJPSCBRKBFFK-UHFFFAOYSA-N 2-(benzenesulfonyl)-2,7-diazaspiro[3.5]nonane hydrochloride Chemical compound Cl.C1(=CC=CC=C1)S(=O)(=O)N1CC2(C1)CCNCC2 XAQJPSCBRKBFFK-UHFFFAOYSA-N 0.000 description 1
- LSXJPJGBWSZHTM-UHFFFAOYSA-N 2-(bromomethyl)-1,3-difluorobenzene Chemical compound FC1=CC=CC(F)=C1CBr LSXJPJGBWSZHTM-UHFFFAOYSA-N 0.000 description 1
- RINUERVPFANASB-UHFFFAOYSA-N 2-(bromomethyl)-1-fluoro-3-(trifluoromethyl)benzene Chemical compound FC1=CC=CC(C(F)(F)F)=C1CBr RINUERVPFANASB-UHFFFAOYSA-N 0.000 description 1
- PVKFBCBTHHTDEX-UHFFFAOYSA-N 2-(bromomethyl)-1-fluoro-3-methoxybenzene Chemical compound COC1=CC=CC(F)=C1CBr PVKFBCBTHHTDEX-UHFFFAOYSA-N 0.000 description 1
- ZOEWNPDQOYGAJY-UHFFFAOYSA-N 2-[(2-chloro-4-fluorophenyl)methyl]-2,6-diazaspiro[3.3]heptane Chemical compound ClC1=C(CN2CC3(C2)CNC3)C=CC(=C1)F ZOEWNPDQOYGAJY-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- GOPCBSCOABWUHK-UHFFFAOYSA-N 2-[2-fluoro-4-(trifluoromethyl)phenoxy]-7-azaspiro[3.5]nonane Chemical compound FC1=C(OC2CC3(C2)CCNCC3)C=CC(=C1)C(F)(F)F GOPCBSCOABWUHK-UHFFFAOYSA-N 0.000 description 1
- YDNHMNQYACURIS-UHFFFAOYSA-N 2-[2-fluoro-4-(trifluoromethyl)phenyl]sulfonyl-2,6-diazaspiro[3.3]heptane Chemical compound FC1=C(C=CC(=C1)C(F)(F)F)S(=O)(=O)N1CC2(C1)CNC2 YDNHMNQYACURIS-UHFFFAOYSA-N 0.000 description 1
- GMAZEGHZUXNBAO-UHFFFAOYSA-N 2-[4-(trifluoromethyl)phenyl]sulfonyl-2,7-diazaspiro[3.5]nonane hydrochloride Chemical compound Cl.FC(C1=CC=C(C=C1)S(=O)(=O)N1CC2(C1)CCNCC2)(F)F GMAZEGHZUXNBAO-UHFFFAOYSA-N 0.000 description 1
- UGCMKGZZCQKMPO-UHFFFAOYSA-N 2-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,6-diazaspiro[3.3]heptane Chemical compound FC1=C(CN2CC3(C2)CNC3)C=CC(=C1)C(F)(F)F UGCMKGZZCQKMPO-UHFFFAOYSA-N 0.000 description 1
- PYXVFPFTWWWCTQ-UHFFFAOYSA-N 2-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,7-diazaspiro[3.4]octane Chemical compound FC1=C(CN2CC3(C2)CNCC3)C=CC(=C1)C(F)(F)F PYXVFPFTWWWCTQ-UHFFFAOYSA-N 0.000 description 1
- YHXDLTWRATXSFM-UHFFFAOYSA-N 2-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,7-diazaspiro[3.5]nonane Chemical compound FC1=C(CN2CC3(C2)CCNCC3)C=CC(=C1)C(F)(F)F YHXDLTWRATXSFM-UHFFFAOYSA-N 0.000 description 1
- FSRCEHFVYUWMSF-UHFFFAOYSA-N 2-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,7-diazaspiro[4.4]nonane Chemical compound FC1=C(CN2CC3(CC2)CNCC3)C=CC(=C1)C(F)(F)F FSRCEHFVYUWMSF-UHFFFAOYSA-N 0.000 description 1
- RCRCTBLIHCHWDZ-DOFZRALJSA-N 2-arachidonoylglycerol Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(=O)OC(CO)CO RCRCTBLIHCHWDZ-DOFZRALJSA-N 0.000 description 1
- QPEJAHMNOVMSOZ-UHFFFAOYSA-N 2-azaspiro[3.3]heptane Chemical compound C1CCC21CNC2 QPEJAHMNOVMSOZ-UHFFFAOYSA-N 0.000 description 1
- GSKMWMFOQQBVMI-UHFFFAOYSA-N 2-bromo-5-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=C(Br)N=C1 GSKMWMFOQQBVMI-UHFFFAOYSA-N 0.000 description 1
- ABFPKTQEQNICFT-UHFFFAOYSA-M 2-chloro-1-methylpyridin-1-ium;iodide Chemical compound [I-].C[N+]1=CC=CC=C1Cl ABFPKTQEQNICFT-UHFFFAOYSA-M 0.000 description 1
- GBZFIEBGHKCVEA-UHFFFAOYSA-N 2-chloro-4,5-bis(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CN=C(Cl)C=C1C(F)(F)F GBZFIEBGHKCVEA-UHFFFAOYSA-N 0.000 description 1
- KUAPPJSILOMQPC-UHFFFAOYSA-N 2-chloro-4-fluorobenzenethiol Chemical compound FC1=CC=C(S)C(Cl)=C1 KUAPPJSILOMQPC-UHFFFAOYSA-N 0.000 description 1
- CDSOHIQUELIIIW-UHFFFAOYSA-N 2-hydroxy-7-azaspiro[3.5]nonane-7-carboxylic acid Chemical compound C1C(O)CC21CCN(C(O)=O)CC2 CDSOHIQUELIIIW-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 1
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ONCAZCNPWWQQMW-UHFFFAOYSA-N 3-(trifluoromethyl)benzenesulfonyl chloride Chemical compound FC(F)(F)C1=CC=CC(S(Cl)(=O)=O)=C1 ONCAZCNPWWQQMW-UHFFFAOYSA-N 0.000 description 1
- OIIBRAGQGFLUFI-UHFFFAOYSA-N 3-amino-1h-pyridin-4-one Chemical compound NC1=CNC=CC1=O OIIBRAGQGFLUFI-UHFFFAOYSA-N 0.000 description 1
- PSSWASGEGXCINO-UHFFFAOYSA-N 3-aminopiperidin-4-ol Chemical class NC1CNCCC1O PSSWASGEGXCINO-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- DEGVSFJBDJUTBP-UHFFFAOYSA-N 4-(bromomethyl)-1-methyl-2-(trifluoromethyl)benzene Chemical compound CC1=CC=C(CBr)C=C1C(F)(F)F DEGVSFJBDJUTBP-UHFFFAOYSA-N 0.000 description 1
- XGOCKBMEZPNDPJ-UHFFFAOYSA-N 4-bromo-1-chloro-2-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC(Br)=CC=C1Cl XGOCKBMEZPNDPJ-UHFFFAOYSA-N 0.000 description 1
- UDLRGQOHGYWLCS-UHFFFAOYSA-N 4-bromo-1-methoxy-2-methylbenzene Chemical compound COC1=CC=C(Br)C=C1C UDLRGQOHGYWLCS-UHFFFAOYSA-N 0.000 description 1
- IGMYEVQPXWKFQF-UHFFFAOYSA-N 4-fluoro-2-(trifluoromethyl)benzenesulfonyl chloride Chemical compound FC1=CC=C(S(Cl)(=O)=O)C(C(F)(F)F)=C1 IGMYEVQPXWKFQF-UHFFFAOYSA-N 0.000 description 1
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- LIPILYKZXJUNFI-UHFFFAOYSA-N 6-(2-chloro-4-fluorophenoxy)-2-azaspiro[3.3]heptane Chemical compound ClC1=C(OC2CC3(CNC3)C2)C=CC(=C1)F LIPILYKZXJUNFI-UHFFFAOYSA-N 0.000 description 1
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 1
- PBCADMHKCLFSJK-UHFFFAOYSA-N 6-chloro-2,3-bis(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=C(Cl)N=C1C(F)(F)F PBCADMHKCLFSJK-UHFFFAOYSA-N 0.000 description 1
- AHRJXSOIEPUORI-UHFFFAOYSA-N 6-oxo-2-azaspiro[3.3]heptane-2-carboxylic acid Chemical compound C1N(C(=O)O)CC21CC(=O)C2 AHRJXSOIEPUORI-UHFFFAOYSA-N 0.000 description 1
- BSQKGAVROUDOTE-UHFFFAOYSA-N 7-azaspiro[3.5]nonane Chemical compound C1CCC21CCNCC2 BSQKGAVROUDOTE-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical class [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 102000001381 Arachidonate 5-Lipoxygenase Human genes 0.000 description 1
- 108010093579 Arachidonate 5-lipoxygenase Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- XHAVUEHVXHHNPF-UHFFFAOYSA-N CC1=C(C(F)(F)F)C=C(COC(C2)CC22CNC2)C=C1 Chemical compound CC1=C(C(F)(F)F)C=C(COC(C2)CC22CNC2)C=C1 XHAVUEHVXHHNPF-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical class [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- KSYQOYPOKJWUOB-BTQNPOSSSA-N Cl.CN(S(=O)(=O)C1=CC=CC=C1)[C@H]1COC2(C1)CCNCC2 Chemical compound Cl.CN(S(=O)(=O)C1=CC=CC=C1)[C@H]1COC2(C1)CCNCC2 KSYQOYPOKJWUOB-BTQNPOSSSA-N 0.000 description 1
- QHGYXWXMTGPTDD-UHFFFAOYSA-N Cl.Cl.FC1=NC=CC(=C1)N1CC2(C1)CNCC2 Chemical compound Cl.Cl.FC1=NC=CC(=C1)N1CC2(C1)CNCC2 QHGYXWXMTGPTDD-UHFFFAOYSA-N 0.000 description 1
- ZCWIABUHXBYUND-UHFFFAOYSA-N Cl.ClC(CO)C Chemical compound Cl.ClC(CO)C ZCWIABUHXBYUND-UHFFFAOYSA-N 0.000 description 1
- HYXNAZQWFXYDFT-RFVHGSKJSA-N Cl.ClC1=C(C=CC(=C1)F)S(=O)(=O)N[C@H]1COC2(C1)CCNCC2 Chemical compound Cl.ClC1=C(C=CC(=C1)F)S(=O)(=O)N[C@H]1COC2(C1)CCNCC2 HYXNAZQWFXYDFT-RFVHGSKJSA-N 0.000 description 1
- QYLTULKLPVORBS-UTONKHPSSA-N Cl.O1C[C@@H](CC12CCNCC2)NS(=O)(=O)C2=CC(=CC=C2)C(F)(F)F Chemical compound Cl.O1C[C@@H](CC12CCNCC2)NS(=O)(=O)C2=CC(=CC=C2)C(F)(F)F QYLTULKLPVORBS-UTONKHPSSA-N 0.000 description 1
- VNNBLCRQKUKKOW-UTONKHPSSA-N Cl.O1C[C@@H](CC12CCNCC2)NS(=O)(=O)C2=CC=C(C=C2)C(F)(F)F Chemical compound Cl.O1C[C@@H](CC12CCNCC2)NS(=O)(=O)C2=CC=C(C=C2)C(F)(F)F VNNBLCRQKUKKOW-UTONKHPSSA-N 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 1
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 1
- UUTHPWAAIKJDMU-UHFFFAOYSA-N FC(C(=O)O)(F)F.COC1=C(C=C(C=C1)N1CC2(C1)CNCC2)C Chemical compound FC(C(=O)O)(F)F.COC1=C(C=C(C=C1)N1CC2(C1)CNCC2)C UUTHPWAAIKJDMU-UHFFFAOYSA-N 0.000 description 1
- WDRPNBBEJUGKDV-UHFFFAOYSA-N FC(C(=O)O)(F)F.COC1=C(CC2CC3(CNC3)C2)C=CC(=C1)C(F)(F)F Chemical compound FC(C(=O)O)(F)F.COC1=C(CC2CC3(CNC3)C2)C=CC(=C1)C(F)(F)F WDRPNBBEJUGKDV-UHFFFAOYSA-N 0.000 description 1
- FBVPCXPXTFDXOG-UHFFFAOYSA-N FC(C(=O)O)(F)F.COC1=C(CC2CC3(CNC3)C2)C=CC=C1 Chemical compound FC(C(=O)O)(F)F.COC1=C(CC2CC3(CNC3)C2)C=CC=C1 FBVPCXPXTFDXOG-UHFFFAOYSA-N 0.000 description 1
- AWLVBTBQALCKCW-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(C=C(C=C1)N1CC2(C1)CNCC2)C(F)(F)F Chemical compound FC(C(=O)O)(F)F.ClC1=C(C=C(C=C1)N1CC2(C1)CNCC2)C(F)(F)F AWLVBTBQALCKCW-UHFFFAOYSA-N 0.000 description 1
- XJRRSNUXQCFTHS-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(C=CC(=C1)F)S(=O)(=O)C1CC2(CNC2)C1 Chemical compound FC(C(=O)O)(F)F.ClC1=C(C=CC(=C1)F)S(=O)(=O)C1CC2(CNC2)C1 XJRRSNUXQCFTHS-UHFFFAOYSA-N 0.000 description 1
- PZAWKIVQUNRNFP-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(CC2CC3(CNC3)C2)C=CC(=C1)F Chemical compound FC(C(=O)O)(F)F.ClC1=C(CC2CC3(CNC3)C2)C=CC(=C1)F PZAWKIVQUNRNFP-UHFFFAOYSA-N 0.000 description 1
- XFANGVHYGMSEFT-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(CN(C2COC3(C2)CCNCC3)C)C=CC(=C1)F Chemical compound FC(C(=O)O)(F)F.ClC1=C(CN(C2COC3(C2)CCNCC3)C)C=CC(=C1)F XFANGVHYGMSEFT-UHFFFAOYSA-N 0.000 description 1
- HCAVOKJTQVTZLB-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(CNC2COC3(C2)CCNCC3)C=CC(=C1)F Chemical compound FC(C(=O)O)(F)F.ClC1=C(CNC2COC3(C2)CCNCC3)C=CC(=C1)F HCAVOKJTQVTZLB-UHFFFAOYSA-N 0.000 description 1
- NDCMXSYYDJIHHW-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(OC2CC3(C2)CCNCC3)C=CC(=C1)F Chemical compound FC(C(=O)O)(F)F.ClC1=C(OC2CC3(C2)CCNCC3)C=CC(=C1)F NDCMXSYYDJIHHW-UHFFFAOYSA-N 0.000 description 1
- WEBIEMSGMUSSQM-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(OC2CC3(CNC3)C2)C=CC(=C1)F Chemical compound FC(C(=O)O)(F)F.ClC1=C(OC2CC3(CNC3)C2)C=CC(=C1)F WEBIEMSGMUSSQM-UHFFFAOYSA-N 0.000 description 1
- IQXOVCSHCXYQRH-UHFFFAOYSA-N FC(C(=O)O)(F)F.ClC1=C(OCC2CC3(CNC3)C2)C=CC(=C1)F Chemical compound FC(C(=O)O)(F)F.ClC1=C(OCC2CC3(CNC3)C2)C=CC(=C1)F IQXOVCSHCXYQRH-UHFFFAOYSA-N 0.000 description 1
- WJKBJXPSZGQLIS-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC(C1=C(C=C(C=C1)C(F)(F)F)N1CC2(C1)CNC2)(F)F Chemical compound FC(C(=O)O)(F)F.FC(C1=C(C=C(C=C1)C(F)(F)F)N1CC2(C1)CNC2)(F)F WJKBJXPSZGQLIS-UHFFFAOYSA-N 0.000 description 1
- JNFHYOIIPSJBSU-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC(C1=C(C=CC(=C1)C(F)(F)F)S(=O)(=O)N1CC2(C1)CNC2)(F)F Chemical compound FC(C(=O)O)(F)F.FC(C1=C(C=CC(=C1)C(F)(F)F)S(=O)(=O)N1CC2(C1)CNC2)(F)F JNFHYOIIPSJBSU-UHFFFAOYSA-N 0.000 description 1
- BBMFPLOSXIIXMN-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC(C1=CC(=NC=C1C(F)(F)F)OC1CC2(CNC2)C1)(F)F Chemical compound FC(C(=O)O)(F)F.FC(C1=CC(=NC=C1C(F)(F)F)OC1CC2(CNC2)C1)(F)F BBMFPLOSXIIXMN-UHFFFAOYSA-N 0.000 description 1
- LJSALDDOKOWXDI-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC(C=1C=C(C=CC1)N1CC2(C1)CNC2)(F)F Chemical compound FC(C(=O)O)(F)F.FC(C=1C=C(C=CC1)N1CC2(C1)CNC2)(F)F LJSALDDOKOWXDI-UHFFFAOYSA-N 0.000 description 1
- GWCUABVBGIACEH-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC(C=1C=CC(=NC1C(F)(F)F)OC1CC2(CNC2)C1)(F)F Chemical compound FC(C(=O)O)(F)F.FC(C=1C=CC(=NC1C(F)(F)F)OC1CC2(CNC2)C1)(F)F GWCUABVBGIACEH-UHFFFAOYSA-N 0.000 description 1
- OCGASKDBHJKRGV-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=C(C=CC(=C1)C(F)(F)F)C1CC2(CNC2)C1 Chemical compound FC(C(=O)O)(F)F.FC1=C(C=CC(=C1)C(F)(F)F)C1CC2(CNC2)C1 OCGASKDBHJKRGV-UHFFFAOYSA-N 0.000 description 1
- CWHBTYZULJLVEJ-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=C(C=CC(=C1)F)CC1CC2(CNC2)C1 Chemical compound FC(C(=O)O)(F)F.FC1=C(C=CC(=C1)F)CC1CC2(CNC2)C1 CWHBTYZULJLVEJ-UHFFFAOYSA-N 0.000 description 1
- RPPJNFVGQPBSEO-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C(=CC=C1)C(F)(F)F Chemical compound FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C(=CC=C1)C(F)(F)F RPPJNFVGQPBSEO-UHFFFAOYSA-N 0.000 description 1
- OCKCUPIGQNCGMP-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C(=CC=C1)F Chemical compound FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C(=CC=C1)F OCKCUPIGQNCGMP-UHFFFAOYSA-N 0.000 description 1
- RKLREVBWBKESGM-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C(=CC=C1)OC Chemical compound FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C(=CC=C1)OC RKLREVBWBKESGM-UHFFFAOYSA-N 0.000 description 1
- WDAYPDSBIRSLEA-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C=CC(=C1)C(F)(F)F Chemical compound FC(C(=O)O)(F)F.FC1=C(CC2CC3(CNC3)C2)C=CC(=C1)C(F)(F)F WDAYPDSBIRSLEA-UHFFFAOYSA-N 0.000 description 1
- NKPYSZRHNBMBIX-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=C(OC2CC3(C2)CCNCC3)C=CC(=C1)F Chemical compound FC(C(=O)O)(F)F.FC1=C(OC2CC3(C2)CCNCC3)C=CC(=C1)F NKPYSZRHNBMBIX-UHFFFAOYSA-N 0.000 description 1
- MDCQRLPPBMHPNQ-UHFFFAOYSA-N FC(C(=O)O)(F)F.FC1=CC(=C(C=C1)S(=O)(=O)N1CC2(C1)CNC2)C(F)(F)F Chemical compound FC(C(=O)O)(F)F.FC1=CC(=C(C=C1)S(=O)(=O)N1CC2(C1)CNC2)C(F)(F)F MDCQRLPPBMHPNQ-UHFFFAOYSA-N 0.000 description 1
- XHILFXRSWXEPHM-OAHLLOKOSA-N FC(C1=CC=C(C=C1)S(=O)(=O)N[C@H]1COC2(C1)CCN(CC2)C(=O)OC(C)(C)C)(F)F Chemical compound FC(C1=CC=C(C=C1)S(=O)(=O)N[C@H]1COC2(C1)CCN(CC2)C(=O)OC(C)(C)C)(F)F XHILFXRSWXEPHM-OAHLLOKOSA-N 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 101000929834 Homo sapiens Monoacylglycerol lipase ABHD6 Proteins 0.000 description 1
- 101000650817 Homo sapiens Semaphorin-4D Proteins 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229910010084 LiAlH4 Inorganic materials 0.000 description 1
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical class [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 108020002496 Lysophospholipase Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical class [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 101150114843 Mgll gene Proteins 0.000 description 1
- 238000005654 Michaelis-Arbuzov synthesis reaction Methods 0.000 description 1
- 102100035912 Monoacylglycerol lipase ABHD6 Human genes 0.000 description 1
- 229940122357 Monoacylglycerol lipase inhibitor Drugs 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 102000006386 Myelin Proteins Human genes 0.000 description 1
- 108010083674 Myelin Proteins Proteins 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N N,N′-Dicyclohexylcarbodiimide Substances C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- VHIAUARLGIJNLC-GFCCVEGCSA-N N-[(3R)-1-oxa-8-azaspiro[4.5]decan-3-yl]benzenesulfonamide Chemical compound O1C[C@@H](CC11CCNCC1)NS(=O)(=O)C1=CC=CC=C1 VHIAUARLGIJNLC-GFCCVEGCSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 208000001294 Nociceptive Pain Diseases 0.000 description 1
- URZDDIQGJPWAHY-UHFFFAOYSA-N OC(C(F)(F)F)=O.FC(C(C=C1)=CN=C1OC(C1)CC11CNC1)(F)F Chemical compound OC(C(F)(F)F)=O.FC(C(C=C1)=CN=C1OC(C1)CC11CNC1)(F)F URZDDIQGJPWAHY-UHFFFAOYSA-N 0.000 description 1
- DDPLSYTUKJNESW-UHFFFAOYSA-N OC(C(F)(F)F)=O.FC(C1=CC(F)=C(COC(C2)CC22CNC2)C=C1)(F)F Chemical compound OC(C(F)(F)F)=O.FC(C1=CC(F)=C(COC(C2)CC22CNC2)C=C1)(F)F DDPLSYTUKJNESW-UHFFFAOYSA-N 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 229910020667 PBr3 Inorganic materials 0.000 description 1
- 208000004983 Phantom Limb Diseases 0.000 description 1
- 206010056238 Phantom pain Diseases 0.000 description 1
- 206010053395 Progressive multiple sclerosis Diseases 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 102100027744 Semaphorin-4D Human genes 0.000 description 1
- 241000872198 Serjania polyphylla Species 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 238000006161 Suzuki-Miyaura coupling reaction Methods 0.000 description 1
- 239000012317 TBTU Substances 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 206010054094 Tumour necrosis Diseases 0.000 description 1
- 238000007239 Wittig reaction Methods 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- NPIHYFCXNIDUJZ-UHFFFAOYSA-N [2-chloro-6-[2,6-di(propan-2-yloxy)phenyl]phenyl]-dicyclohexylphosphane Chemical group ClC=1C(=C(C=CC=1)C1=C(C=CC=C1OC(C)C)OC(C)C)P(C1CCCCC1)C1CCCCC1 NPIHYFCXNIDUJZ-UHFFFAOYSA-N 0.000 description 1
- LNXCKLNHUWBTJW-UHFFFAOYSA-N [2-fluoro-4-(trifluoromethyl)phenyl]methyl methanesulfonate Chemical compound CS(=O)(=O)OCC1=C(C=C(C=C1)C(F)(F)F)F LNXCKLNHUWBTJW-UHFFFAOYSA-N 0.000 description 1
- BKWXZDHOVFZQEX-UHFFFAOYSA-M [Br-].FC1=C(C[P+](C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)C=CC(=C1)C(F)(F)F Chemical compound [Br-].FC1=C(C[P+](C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)C=CC(=C1)C(F)(F)F BKWXZDHOVFZQEX-UHFFFAOYSA-M 0.000 description 1
- COERJHDMQUPDCV-UHFFFAOYSA-N [K].FB(F)F Chemical compound [K].FB(F)F COERJHDMQUPDCV-UHFFFAOYSA-N 0.000 description 1
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 208000038016 acute inflammation Diseases 0.000 description 1
- 230000006022 acute inflammation Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 238000002266 amputation Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000002001 anti-metastasis Effects 0.000 description 1
- 230000003443 anti-oncogenic effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- LGEQQWMQCRIYKG-UHFFFAOYSA-N arachidonic acid ethanolamide Natural products CCCCCC=CCC=CCC=CCC=CCCCC(=O)NCCO LGEQQWMQCRIYKG-UHFFFAOYSA-N 0.000 description 1
- 125000002886 arachidonoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 150000007860 aryl ester derivatives Chemical class 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- VKBBZJXAWNVLSE-UHFFFAOYSA-N benzyl 3-amino-4-hydroxypiperidine-1-carboxylate Chemical compound C1CC(O)C(N)CN1C(=O)OCC1=CC=CC=C1 VKBBZJXAWNVLSE-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- YHNUDLCUIKMNSN-UHFFFAOYSA-N bis(1,2,4-triazol-1-yl)methanone Chemical compound C1=NC=NN1C(=O)N1C=NC=N1 YHNUDLCUIKMNSN-UHFFFAOYSA-N 0.000 description 1
- 210000004958 brain cell Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000011575 calcium Chemical class 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 229930003827 cannabinoid Natural products 0.000 description 1
- 239000003557 cannabinoid Substances 0.000 description 1
- 229940065144 cannabinoids Drugs 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 230000006315 carbonylation Effects 0.000 description 1
- 238000005810 carbonylation reaction Methods 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 230000006720 chronic neuroinflammation Effects 0.000 description 1
- 230000037326 chronic stress Effects 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 231100000858 damage to nervous tissue Toxicity 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000017858 demethylation Effects 0.000 description 1
- 238000010520 demethylation reaction Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- OCMNCWNTDDVHFK-UHFFFAOYSA-L dichloronickel;1,2-dimethoxyethane Chemical compound Cl[Ni]Cl.COCCOC OCMNCWNTDDVHFK-UHFFFAOYSA-L 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 229960002986 dinoprostone Drugs 0.000 description 1
- GPAYUJZHTULNBE-UHFFFAOYSA-N diphenylphosphine Chemical compound C=1C=CC=CC=1PC1=CC=CC=C1 GPAYUJZHTULNBE-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000002996 emotional effect Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 230000009483 enzymatic pathway Effects 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 231100000318 excitotoxic Toxicity 0.000 description 1
- 230000003492 excitotoxic effect Effects 0.000 description 1
- 230000004136 fatty acid synthesis Effects 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013373 food additive Nutrition 0.000 description 1
- 239000002778 food additive Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000021588 free fatty acids Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 description 1
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 1
- 125000002140 imidazol-4-yl group Chemical group [H]N1C([H])=NC([*])=C1[H] 0.000 description 1
- 125000000336 imidazol-5-yl group Chemical group [H]N1C([H])=NC([H])=C1[*] 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 125000000593 indol-1-yl group Chemical group [H]C1=C([H])C([H])=C2N([*])C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000002249 indol-2-yl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([*])=C([H])C2=C1[H] 0.000 description 1
- 125000004531 indol-5-yl group Chemical group [H]N1C([H])=C([H])C2=C([H])C(*)=C([H])C([H])=C12 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 229960004903 invert sugar Drugs 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- MILUBEOXRNEUHS-UHFFFAOYSA-N iridium(3+) Chemical compound [Ir+3] MILUBEOXRNEUHS-UHFFFAOYSA-N 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 201000010901 lateral sclerosis Diseases 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000011777 magnesium Chemical class 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 1
- 208000005264 motor neuron disease Diseases 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000005012 myelin Anatomy 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 239000005445 natural material Substances 0.000 description 1
- 239000006225 natural substrate Substances 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 230000004112 neuroprotection Effects 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 239000002581 neurotoxin Substances 0.000 description 1
- 231100000618 neurotoxin Toxicity 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical group 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 238000006772 olefination reaction Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 150000002902 organometallic compounds Chemical class 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000004287 oxazol-2-yl group Chemical group [H]C1=C([H])N=C(*)O1 0.000 description 1
- 125000003145 oxazol-4-yl group Chemical group O1C=NC(=C1)* 0.000 description 1
- 125000004304 oxazol-5-yl group Chemical group O1C=NC=C1* 0.000 description 1
- 125000004274 oxetan-2-yl group Chemical group [H]C1([H])OC([H])(*)C1([H])[H] 0.000 description 1
- 125000006299 oxetan-3-yl group Chemical group [H]C1([H])OC([H])([H])C1([H])* 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 230000004783 oxidative metabolism Effects 0.000 description 1
- 238000006213 oxygenation reaction Methods 0.000 description 1
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 231100000915 pathological change Toxicity 0.000 description 1
- 230000036285 pathological change Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 210000000578 peripheral nerve Anatomy 0.000 description 1
- 239000000575 pesticide Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 238000001050 pharmacotherapy Methods 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 125000006678 phenoxycarbonyl group Chemical group 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 1
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 230000001107 psychogenic effect Effects 0.000 description 1
- 125000004353 pyrazol-1-yl group Chemical group [H]C1=NN(*)C([H])=C1[H] 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000003014 reinforcing effect Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 230000003238 somatosensory effect Effects 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical group COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 238000006277 sulfonation reaction Methods 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- UISVBUXRNGCRPP-OAHLLOKOSA-N tert-butyl (3R)-3-(benzenesulfonamido)-1-oxa-8-azaspiro[4.5]decane-8-carboxylate Chemical compound C1(=CC=CC=C1)S(=O)(=O)N[C@H]1COC2(C1)CCN(CC2)C(=O)OC(C)(C)C UISVBUXRNGCRPP-OAHLLOKOSA-N 0.000 description 1
- XRTONQMRCAHMSG-MRXNPFEDSA-N tert-butyl (3R)-3-[benzenesulfonyl(methyl)amino]-1-oxa-8-azaspiro[4.5]decane-8-carboxylate Chemical compound CN([C@H]1COC2(C1)CCN(CC2)C(=O)OC(C)(C)C)S(=O)(=O)C1=CC=CC=C1 XRTONQMRCAHMSG-MRXNPFEDSA-N 0.000 description 1
- MYPUIPSALVXCSM-UHFFFAOYSA-N tert-butyl 2-(2,4-difluorophenoxy)-7-azaspiro[3.5]nonane-7-carboxylate Chemical compound FC1=C(OC2CC3(C2)CCN(CC3)C(=O)OC(C)(C)C)C=CC(=C1)F MYPUIPSALVXCSM-UHFFFAOYSA-N 0.000 description 1
- URTIXDXQQGKKNT-UHFFFAOYSA-N tert-butyl 2-azaspiro[3.3]heptane-2-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC11CCC1 URTIXDXQQGKKNT-UHFFFAOYSA-N 0.000 description 1
- GBLUNQGZHFQTPW-UHFFFAOYSA-N tert-butyl 3-amino-1-oxa-8-azaspiro[4.5]decane-8-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC11OCC(N)C1 GBLUNQGZHFQTPW-UHFFFAOYSA-N 0.000 description 1
- QNQRLRKESDJZJK-UHFFFAOYSA-N tert-butyl 6-[5,6-bis(trifluoromethyl)pyridin-2-yl]oxy-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound FC(C=1C=CC(=NC=1C(F)(F)F)OC1CC2(CN(C2)C(=O)OC(C)(C)C)C1)(F)F QNQRLRKESDJZJK-UHFFFAOYSA-N 0.000 description 1
- STAKZRAOMUHPCZ-UHFFFAOYSA-N tert-butyl 6-[[2-fluoro-4-(trifluoromethyl)phenyl]methyl]-2,6-diazaspiro[3.3]heptane-2-carboxylate Chemical compound FC1=C(CN2CC3(CN(C3)C(=O)OC(C)(C)C)C2)C=CC(=C1)C(F)(F)F STAKZRAOMUHPCZ-UHFFFAOYSA-N 0.000 description 1
- ZNCOKAWJXJBPDT-UHFFFAOYSA-N tert-butyl 6-[[2-fluoro-4-(trifluoromethyl)phenyl]methylidene]-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound FC1=C(C=C2CC3(CN(C3)C(=O)OC(C)(C)C)C2)C=CC(=C1)C(F)(F)F ZNCOKAWJXJBPDT-UHFFFAOYSA-N 0.000 description 1
- HKDQMAXFXVWMMK-UHFFFAOYSA-N tert-butyl 6-hydroxy-6-(trifluoromethyl)-2-azaspiro[3.3]heptane-2-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC21CC(O)(C(F)(F)F)C2 HKDQMAXFXVWMMK-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000036962 time dependent Effects 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 229940062627 tribasic potassium phosphate Drugs 0.000 description 1
- LAYPMCGIWDGYKX-UHFFFAOYSA-N trichloromethyl hydrogen carbonate Chemical compound OC(=O)OC(Cl)(Cl)Cl LAYPMCGIWDGYKX-UHFFFAOYSA-N 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- RIOQSEWOXXDEQQ-UHFFFAOYSA-O triphenylphosphanium Chemical compound C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-O 0.000 description 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
La invención proporciona nuevos compuestos heterocíclicos que tienen la fórmula general (I) en la que A, B, L, X, R1, R2, R3 y R4 son como se describen en el presente documento, composiciones que incluyen los compuestos, procesos de fabricación de los compuestos y métodos de uso de los compuestos. . (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Nuevos compuestos heterocíclicos
Campo de la invención
La presente invención se refiere a compuestos orgánicos útiles para el tratamiento o la profilaxis en un mamífero, y en particular a inhibidores de monoacilglicerol lipasa (MAGL) para el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer, trastornos mentales, esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña y/o depresión en un mamífero.
Antecedentes de la invención
Los endocanabinoides (EC) son lípidos de señalización que ejercen sus acciones biológicas al interactuar con los receptores de canabinoides (CBR), CB1 y CB2. Modulan múltiples procesos fisiológicos que incluyen neuroinflamación, neurodegeneración y regeneración de tejidos (Iannotti, F.A., et al., Progress in lipid research 2016, 62, 107-28). En el cerebro, el principal endocanabinoide, 2-araquidonoilglicerol (2-AG), se produce por diacilglicerol lipasas (DAGL) y se hidroliza por la monoacilglicerol lipasa, MAGL. MAGL hidroliza un 85 % de 2-AG; hidrolizándose el 15 % restante por ABHD6 y ABDH12 (Nomura, D.K., et al., Science 2011, 334, 809). MAGL se expresa en todo el cerebro y en la mayoría de los tipos de células cerebrales, incluyendo las neuronas, los astrocitos, los oligodendrocitos y los microgliocitos (Chanda, P.K., et al., Molecular pharmacology 2010, 78, 996; Viader, A., et al., Cell reports 2015, 12, 798). La hidrólisis de 2-AG da como resultado la formación de ácido araquidónico (AA), el precursor de las prostaglandinas (PG) y los leucotrienos (LT). El metabolismo oxidativo del AA se incrementa en los tejidos inflamados. Existen dos vías enzimáticas principales de oxigenación del ácido araquidónico implicadas en procesos inflamatorios, la ciclooxigenasa que produce las PG y la 5-lipoxigenasa que produce los LT. De los diversos productos de la ciclooxigenasa formados durante la inflamación, PGE2 es una de las más importantes. Estos productos se han detectado en sitios de inflamación, por ejemplo, en el líquido cefalorraquídeo de pacientes que padecen trastornos neurodegenerativos y se cree que contribuyen a la respuesta inflamatoria y a la progresión de la enfermedad. Los ratones que carecen de MAGL (Mgll-/-) presentan una actividad hidrolasa de 2-AG drásticamente reducida y niveles elevados de 2-AG en el sistema nervioso, mientras que otras especies de lípidos neutros y fosfolípidos que contienen araquidonoilo, incluyendo anandamida (AEA), así como otros ácidos grasos libres, están inalterados. Por el contrario, los niveles de AA y prostaglandinas derivadas de AA y otros icosanoides, incluyendo prostaglandina E2 (PGE2), D2 (PGD2), F2 (PGF2) y tromboxano B2 (TXB2), están fuertemente disminuidos. Las enzimas fosfolipasa A2 (PLA2) se han visto como la fuente principal de AA, pero los ratones carentes de cPLA2 tienen niveles de AA inalterados en su cerebro, lo que refuerza el papel clave de MAGL en el cerebro para la producción de AA y la regulación de los procesos inflamatorios del cerebro.
La neuroinflamación es un cambio patológico común característico de las enfermedades del cerebro, incluyendo, pero no restringido a, enfermedades neurodegenerativas (por ejemplo, esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia y trastornos mentales tales como ansiedad y migraña). En el cerebro, la producción de icosanoides y prostaglandinas controla el proceso de neuroinflamación. El agente proinflamatorio lipopolisacárido (LPS) produce un incremento consistente y dependiente del tiempo en los icosanoides cerebrales que se atenúa notablemente en ratones Mgll-/-. El tratamiento con LPS también induce una elevación generalizada de las citocinas proinflamatorias, incluyendo interleucina-1-a (IL-1-a), IL-1b, IL-6 y factor de necrosis tumoral-a (TNF-a) que se evita en ratones M gll-/-.
La neuroinflamación se caracteriza por la activación de las células inmunitarias inherentes del sistema nervioso central, la microglia y los astrocitos. Se ha informado de que los fármacos antiinflamatorios pueden suprimir en modelos preclínicos la activación de los neurogliocitos y la progresión de la enfermedad, incluyendo la enfermedad de Alzheimer y la esclerosis múltiple (Lleo A., Cell Mol Life Sci. 2007, 64, 1403). De forma importante, la alteración genética y/o farmacológica de la actividad MAGL también bloquea la activación inducida por LPS de microgliocitos en el cerebro (Nomura, D.K., et al., Science 2011, 334, 809).
Además, se demostró que la alteración genética y/o farmacológica de la actividad MAGL es protectora en varios modelos animales de neurodegeneración, incluyendo, pero no restringidos a, la enfermedad de Alzheimer, la enfermedad de Parkinson y la esclerosis múltiple. Por ejemplo, un inhibidor de MAGL irreversible se ha usado ampliamente en modelos preclínicos de neuroinflamación y neurodegeneración (Long, J.Z., et al., Nature chemical biology 2009, 5, 37). La inyección sistémica de dicho inhibidor da lugar al fenotipo de ratones Mgll-/- en el cerebro, incluyendo un incremento en los niveles de 2-AG, una reducción en los niveles de AA y la producción de icosanoides relacionados, así como la prevención de la producción de citocinas y la activación de la microglia después de la neuroinflamación inducida por LPS (Nomura, D.K., et al., Science 2011, 334, 809), lo que confirma en conjunto que MAGL es una diana farmacológica.
Como consecuencia de la alteración genética y/o farmacológica de la actividad MAGL, se incrementan los niveles endógenos del sustrato natural de MAGL en el cerebro, 2-AG. Se ha informado de que 2-AG muestra efectos beneficiosos sobre el dolor con, por ejemplo, efectos antinocisensibles en ratones (Ignatowska-Jankowska B. et al., J. Pharmacol. Exp. Ther. 2015, 353, 424) y en trastornos mentales, tales como la depresión en modelos de estrés crónico (Zhong P. et al., Neuropsychopharmacology 2014, 39, 1763).
Además, los oligodendrocitos (OL), las células mielinizantes del sistema nervioso central y sus precursores (OPC) expresan el receptor canabinoide 2 (CB2) en su membrana. 2-AG es el ligando endógeno de los receptores CB1 y c B2. Se ha informado de que tanto los canabinoides como la inhibición farmacológica de MAGL atenúan la vulnerabilidad de los OL y los OPC a las agresiones excitotóxicas y, por lo tanto, pueden ser neuroprotectores (Bernal-Chico, A., et al., Glia 2015, 63, 163). Adicionalmente, la inhibición farmacológica de MAGL incrementa el número de OL mielinizantes en el cerebro de ratones, lo que sugiere que la inhibición de MAGL puede promover la diferenciación de los OPC en OL mielinizantes in vivo (Alpar, A., et al., Nature comunicaciones 2014, 5, 4421). También se demostró que la inhibición de MAGL promueve la remielinización y la recuperación funcional en un modelo de ratón de esclerosis múltiple progresiva (Feliu A. et al., Journal of Neuroscience 2017, 37 (35), 8385).
Finalmente, en los últimos años se habla de un metabolismo altamente importante en la investigación contra el cáncer, especialmente del metabolismo de los lípidos. Los investigadores creen que la síntesis de ácidos grasos de novo juega un papel importante en la aparición de tumores. Muchos estudios ilustraron que los endocanabinoides tienen acciones antioncógenas, que incluyen efectos antiproliferación, de inducción de apoptosis y antimetastásicos. MAGL como una enzima de descomposición importante tanto para el metabolismo de los lípidos como para el sistema de endocanabinoides, adicionalmente como parte de una firma de expresión génica, contribuye a diferentes aspectos de la oncogénesis (Qin, H., et al., Cell Biochem. Biophys. 2014, 70, 33; Nomura DK et al., Cell 2009, 140(1), 49-61; Nomura DK et al., Chem. Biol. 2011, 18(7), 846-856).
En conclusión, suprimir la acción y/o la activación de MAGL es una nueva estrategia terapéutica prometedora para el tratamiento o prevención de la neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y trastornos mentales. Además, suprimir la acción y/o la activación de MAGL es una nueva estrategia terapéutica prometedora para proporcionar neuroprotección y regeneración de mielina. En consecuencia, existe una gran necesidad médica no cubierta de nuevos inhibidores de MAGL.
Sumario de la invención
En un primer aspecto, la presente invención proporciona un compuesto de fórmula (I)

en la que A, B, L, X, R1, R2, R3 y R4 son como se describe en el presente documento.
En un aspecto, la presente invención proporciona un procedimiento de fabricación de los compuestos de urea de fórmula (I) descritos en el presente documento y sales farmacéuticamente aceptables de los mismos, que comprende:
a hacer reaccionar una primera amina de fórmula 1, en la que R1 R2 son como se describe en el presente


en presencia de una base y un reactivo formador de urea, para formar dicho compuesto de fórmula (I); y opcionalmente
(b) transformar dicho compuesto de fórmula (I) en una sal farmacéuticamente aceptable del mismo.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, cuando se fabrica de acuerdo con los procedimientos descritos en el presente documento.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, para su uso como sustancia terapéuticamente activa.
En otro aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto de fórmula (I) como se describe en el presente documento y un vehículo terapéuticamente inerte.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento o una composición farmacéutica descrita en el presente documento para su uso en un procedimiento de inhibición de monoacilglicerol lipasa en un mamífero.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento o una composición farmacéutica descrita en el presente documento para su uso en el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento o una composición farmacéutica descrita en el presente documento, para su uso en el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
Descripción detallada de la invención
Definiciones
Se ha de entender que los rasgos característicos, números enteros, características, compuestos, restos o grupos químicos descritos junto con un aspecto, modo de realización o ejemplo particular de la invención son aplicables a cualquier otro aspecto, modo de realización o ejemplo descrito en el presente documento, a menos que sea incompatible con el mismo. Todos los rasgos característicos divulgados en esta memoria descriptiva (incluyendo cualquier reivindicación, el resumen y dibujo adjuntos), y/o todas las etapas de cualquier procedimiento o proceso así divulgado, se pueden combinar en cualquier combinación, excepto combinaciones donde al menos algunos de dichos rasgos característicos y/o etapas son mutuamente excluyentes. La invención no se limita a los detalles de ninguno de los modos de realización anteriores. La invención se extiende a cualquier rasgo característico novedoso o a cualquier combinación novedosa de los rasgos característicos divulgados en esta memoria descriptiva (incluyendo cualquier reivindicación, el resumen y dibujo adjuntos), o a cualquier etapa novedosa o a cualquier combinación novedosa de las etapas de cualquier procedimiento o proceso así divulgado.
El término "alquilo" se refiere a un grupo hidrocarburo saturado lineal o ramificado, mono- o multivalente, por ejemplo, mono- o bivalente, de 1 a 6 átomos de carbono ("alquilo C1-C6"), por ejemplo, 1, 2, 3, 4, 5 o 6 átomos de carbono. En algunos modos de realización, el grupo alquilo contiene de 1 a 3 átomos de carbono, por ejemplo, 1,2 o 3 átomos de carbono. Algunos ejemplos no limitantes de alquilo incluyen metilo, etilo, propilo, 2-propilo (isopropilo), n-butilo, /so-butilo, sec-butilo, tere-butilo y 2,2-dimetilpropilo. Un ejemplo en particular preferente, aunque no limitante, de alquilo es metilo.
El término "alcoxi" se refiere a un grupo alquilo, como se define previamente, unido al resto molecular original por medio de un átomo de oxígeno. A menos que se especifique de otro modo, el grupo alcoxi contiene de 1 a 6 átomos de carbono ("alcoxi C1-C6"). En algunos modos de realización preferentes, el grupo alcoxi contiene de 1 a 4 átomos de carbono. Todavía en otros modos de realización, el grupo alcoxi contiene de 1 a 3 átomos de carbono. Algunos
ejemplos no limitantes de grupos alcoxi incluyen metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi y ferc-butoxi. Un ejemplo de alcoxi en particular preferente, aunque no limitante, es metoxi.
El término "halógeno" o "halo" se refiere a flúor (F), cloro (Cl), bromo (Br) o yodo (I). Preferentemente, el término "halógeno" o "halo" se refiere a flúor (F), cloro (Cl) o bromo (Br). Los ejemplos en particular preferentes, aunque no limitantes, de "halógeno" o "halo" son flúor (F) y cloro (Cl).
El término "espirociclo bicíclico" se refiere a una entidad química que consiste en dos restos heterociclilo o dos cicloalquilo como se define en el presente documento, o a una combinación de un resto heterociclilo y uno cicloalquilo, que tienen un átomo de anillo en común, es decir, los dos anillos se conectan por medio de un átomo de anillo común. Algunos ejemplos preferentes, aunque no limitantes, de espirociclos bicíclicos incluyen 2-azaespiro[3.3]heptano, 2,6-diazaespiro[3.3]heptano, 2,7-diazaespiro[3.5]nonano, 7-azaespiro[3.5]nonano, 1-oxa-8-azaespiro[4.5]decano, 2,7-diazaespiro[4.4]nonano y 2,7-diazaespiro[3.4]octano.
El término "heterociclilo" se refiere a un sistema de anillo monocíclico saturado o parcialmente insaturado de 3 a 14 átomos de anillo, preferentemente de 3 a 10 átomos de anillo, más preferentemente de 3 a 8 átomos de anillo, en el que 1,2 o 3 de dichos átomos de anillo son heteroátomos seleccionados de N, O y S, siendo los átomos de anillo restantes, carbono. Preferentemente, de 1 a 2 de dichos átomos de anillo se seleccionan de N y O, siendo los átomos de anillo restantes, carbono. Algunos ejemplos no limitantes de grupos heterociclilo incluyen acetidin-3-ilo, acetidin-2-ilo, oxetan-3-ilo, oxetan-2-ilo, 2-oxopirrolidi n-1-ilo, 2-oxopi rrolidin-3-ilo, 5-oxopirrolidin-2-ilo, 5-oxopirrolidin-3-ilo, 2-oxo-1-piperidilo, 2-oxo-3-piperidilo, 2-oxo-4-piperidilo, 6-oxo-2-piperidilo, 6-oxo-3-piperidilo, 1- piperidinilo, 2-piperidinilo, 3-piperidinilo, 4-piperidinilo, morfolino, morfolin-2-ilo, morfolin-3-ilo, pirrolidin-1-ilo, pirrolidin-2-ilo y pirrolidin-3-ilo.
El término "cicloalquilo", como se usa en el presente documento, se refiere a un grupo hidrocarburo monocíclico saturado o parcialmente insaturado de 3 a 10 átomos de carbono de anillo ("cicloalquilo C3-10"). En algunos modos de realización preferentes, el grupo cicloalquilo es un grupo hidrocarburo monocíclico saturado de 3 a 8 átomos de carbono de anillo. Preferentemente, el grupo cicloalquilo es un grupo hidrocarburo monocíclico saturado de 3 a 6 átomos de carbono de anillo, por ejemplo, de 3, 4, 5 o 6 átomos de carbono. Algunos ejemplos no limitantes de cicloalquilo incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo y cicloheptilo. Un ejemplo preferente, aunque no limitante, de cicloalquilo es ciclopropilo.
El término "arilo" se refiere a un sistema de anillos carbocíclico monocíclico, bicíclico o tricíclico que tiene un total de 6 a 14 miembros de anillo ("arilo C6-14"), preferentemente, de 6 a 12 miembros de anillo y, más preferentemente, de 6 a 10 miembros de anillo, y en el que al menos un anillo en el sistema es aromático. Un ejemplo en particular preferente, aunque no limitante, de arilo es fenilo.
El término "heteroarilo" se refiere a un sistema de anillos monocíclico o bicíclico, preferentemente bicíclico, mono-0 multivalente, que tiene un total de 5 a 14 miembros de anillo, preferentemente, de 5 a 12 miembros de anillo y, más preferentemente, de 5 a 10 miembros de anillo, en el que al menos un anillo en el sistema es aromático y al menos un anillo en el sistema contiene uno o más heteroátomos. Preferentemente, "heteroarilo" se refiere a un heteroarilo de 5-10 miembros que comprende 1,2, 3 o 4 heteroátomos seleccionados independientemente de O, S y N. Lo más preferentemente, "heteroarilo" se refiere a un heteroarilo de 5-10 miembros que comprende de 1 a 2 heteroátomos seleccionados independientemente de O y N. Algunos ejemplos no limitantes de heteroarilo incluyen 2- piridilo, 3-piridilo, 4-piridilo, indol-1-ilo, 1 H-indol-2-ilo, 1 H-indol-3-ilo, 1 H-indol-4-ilo, 1 H-indol-5-ilo, 1 H-indol-6-ilo, 1 H-indol-7-ilo, 1,2-benzoxazol-3-ilo, 1,2-benzoxazol-4-ilo, 1,2-benzoxazol-5-ilo, 1,2-benzoxazol-6-ilo, 1,2-benzoxazol-7-ilo, 1H-indazol-3-ilo, 1H-indazol-4-ilo, 1H-indazol-5-ilo, 1H-indazol-6-ilo, 1H-indazol-7-ilo, pirazol-1-ilo, 1 H-pirazol-3-ilo, 1 H-pirazol-4-ilo, 1 H-pirazol-5-ilo, imidazol-1-ilo, 1H-imidazol-2-ilo, 1H-imidazol-4-ilo, 1 H-imidazol-5-ilo, oxazol-2-ilo, oxazol-4-ilo y oxazol-5-ilo. Un ejemplo en particular preferente, aunque no limitante, de heteroarilo es indolilo, en particular 1 H-indol-3-ilo.
El término "hidroxi" se refiere a un grupo -OH.
El término "ciano" se refiere a un grupo -CN (nitrilo).
El término "carbonilo" se refiere a un grupo C(O).
El término "oxo" se refiere a un átomo de oxígeno unido a la molécula original a través de un doble enlace (=O).
El término "alcoxicarbonilo" se refiere a un grupo -C(O)-O-alquilo (es decir, un éster de alquilo). Un ejemplo en particular preferente, aunque no limitante, de alcoxicarbonilo es ferc-butoxicarbonilo.
El término "ariloxicarbonilo" se refiere a un grupo -C(O)-O-arilo (es decir, un éster de arilo). Un ejemplo en particular preferente, aunque no limitante, de alcoxicarbonilo es fenoxicarbonilo.
El término "heteroariloxicarbonilo" se refiere a un grupo -C(O)-O-heteroarilo (es decir, un éster de heteroarilo). Un
ejemplo en particular preferente, aunque no limitante, de alcoxicarbonilo es piridiloxicarbonilo.
El término "haloalquilo" se refiere a un grupo alquilo, en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un átomo de halógeno, preferentemente flúor. Preferentemente, "haloalquilo" se refiere a un grupo alquilo en el que 1, 2 o 3 átomos de hidrógeno del grupo alquilo se han reemplazado por un átomo de halógeno, lo más preferentemente flúor. Un ejemplo en particular preferente, aunque no limitante, de haloalquilo es trifluorometilo (CF3).
El término "haloalcoxi" se refiere a un grupo alcoxi, en el que al menos uno de los átomos de hidrógeno del grupo alcoxi se ha reemplazado por un átomo de halógeno, preferentemente flúor. Preferentemente, "haloalcoxi" se refiere a un grupo alcoxi en el que 1,2 o 3 átomos de hidrógeno del grupo alcoxi se han reemplazado por un átomo de halógeno, lo más preferentemente flúor. Un ejemplo en particular preferente, aunque no limitante, de haloalcoxi es trifluorometoxi (-OCF3).
El término "sal farmacéuticamente aceptable" se refiere a las sales que conservan la eficacia biológica y las propiedades de las bases libres o ácidos libres, que no son biológicamente o de otro modo indeseables. Las sales se forman con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares, en particular ácido clorhídrico, y ácidos orgánicos tales como ácido acético, ácido propiónico, ácido glicólico, ácido pirúvico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico, N-acetilcisteína y similares. Además, estas sales se pueden preparar por adición de una base inorgánica o una base orgánica al ácido libre. Las sales derivadas de una base inorgánica incluyen, pero no se limitan a, sales de sodio, potasio, litio, amonio, calcio, magnesio y similares. Las sales derivadas de bases orgánicas incluyen, pero no se limitan a, sales de aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas naturales, aminas cíclicas y resinas de intercambio iónico básicas, tales como resinas de isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, etanolamina, lisina, arginina, N-etilpiperidina, piperidina, poliimina y similares. Las sales farmacéuticamente aceptables particulares de los compuestos de fórmula (I) son las sales clorhidrato.
El término "grupo protector" (PG) indica un grupo que bloquea selectivamente un sitio reactivo en un compuesto multifuncional de modo que se pueda llevar a cabo selectivamente una reacción química en otro sitio reactivo desprotegido en el sentido convencionalmente asociado con él en química de síntesis. Los grupos protectores se pueden retirar en el momento apropiado. Los grupos protectores ejemplares son grupos protectores amino, grupos protectores carboxi o grupos protectores hidroxi. Los grupos protectores particulares son ferc-butoxicarbonilo (Boc), benciloxicarbonilo (Cbz), fluorenilmetoxicarbonilo (Fmoc) y bencilo (Bn). Otros grupos protectores particulares son ferc-butoxicarbonilo (Boc) y fluorenilmetoxicarbonilo (Fmoc). Un grupo protector más particular es ferc-butoxicarbonilo (Boc). Los grupos protectores ejemplares y su aplicación en síntesis orgánica se describen, por ejemplo, en "Protective Groups in Organic Chemistry" de T. W. Greene y P.G.M. Wutts, 5.a ed., 2014, John Wiley & Sons, N.Y.
El término "reactivo formador de urea" se refiere a un compuesto químico que puede convertir una primera amina en una especie que reaccionará con una segunda amina, formando de este modo un derivado de urea. Los ejemplos no limitantes de reactivos formadores de urea incluyen carbonato de bis(triclorometilo), fosgeno, cloroformiato de triclorometilo, carbonato de 4-nitrofenilo y 1,1'-carbonildiimidazol. Los reactivos formadores de urea se describen en G. Sartori ef al., Green Chemisfry 20 O0 , 2, 140.
Los compuestos de fórmula (I) pueden contener varios centros asimétricos y pueden estar presentes en forma de enantiómeros ópticamente puros, mezclas de enantiómeros, tales como, por ejemplo, racematos, diastereoisómeros ópticamente puros, mezclas de diastereoisómeros, racematos diastereoisómeros o mezclas de racematos diastereoisómeros. En un modo de realización preferente, el compuesto de fórmula (I) de acuerdo con la invención es un enantiómero cis de fórmula (la) o (Ib), respectivamente, como se describe en el presente documento.
De acuerdo con la convención de Cahn-Ingold-Prelog, el átomo de carbono asimétrico puede tener la configuración "R" o "S".
La abreviatura "MAGL" se refiere a la enzima monoacilglicerol lipasa. Los términos "MAGL" y "monoacilglicerol lipasa" se usan en el presente documento de manera intercambiable.
El término "tratamiento" como se usa en el presente documento incluye: (1) inhibir el estado, trastorno o afección (por ejemplo, detener, reducir o retrasar la aparición de la enfermedad, o una recidiva de la misma en caso de tratamiento de mantenimiento, de al menos un síntoma clínico o subclínico de la misma); y/o (2) aliviar la afección (es decir, provocar la regresión del estado, trastorno o afección o al menos uno de sus síntomas clínicos o subclínicos). El beneficio para un paciente que se va a tratar es estadísticamente significativo o bien al menos perceptible para el paciente o para el médico. Sin embargo, se apreciará que cuando se administra un medicamento a un paciente para tratar una enfermedad, el resultado puede no ser siempre un tratamiento eficaz.
El término "profilaxis", como se usa en el presente documento, incluye: evitar o retrasar la aparición de síntomas clínicos del estado, trastorno o afección que padece un mamífero y especialmente un ser humano que puede estar aquejado de, o predispuesto a, el estado, trastorno o afección pero aún no experimenta o presenta síntomas clínicos o subclínicos del estado, trastorno o afección.
El término "neuroinflamación", como se usa en el presente documento, se refiere a la inflamación aguda y crónica del tejido nervioso, que es el principal componente tisular de las dos partes del sistema nervioso; el cerebro y la médula espinal del sistema nervioso central (SNC), y los nervios periféricos ramificados del sistema nervioso periférico (SNP). La neuroinflamación crónica está asociada con enfermedades neurodegenerativas tales como enfermedad de Alzheimer, enfermedad de Parkinson y esclerosis múltiple. La neuroinflamación aguda normalmente sigue de inmediato a una lesión del sistema nervioso central, por ejemplo, como resultado de un traumatismo craneoencefálico (TCE).
El término "traumatismo craneoencefálico" ("TCE", también conocido como "lesión intracraneal"), se refiere al daño al cerebro que resulta de una fuerza mecánica externa, tal como una aceleración o desaceleración rápida, un impacto, ondas expansivas o penetración de un proyectil.
El término "enfermedades neurodegenerativas" se refiere a enfermedades que están relacionadas con la pérdida progresiva de la estructura o la función de las neuronas, incluyendo la muerte de las neuronas. Los ejemplos de enfermedades neurodegenerativas incluyen, pero no se limitan a, esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson y esclerosis lateral amiotrófica.
El término "trastornos mentales" (también llamados enfermedades mentales o trastornos psiquiátricos) se refiere a patrones conductuales o mentales que pueden provocar sufrimiento o una capacidad deficiente para funcionar en la vida. Dichos rasgos característicos pueden ser constantes, recidivantes y remisivos, o producirse como un episodio único. Los ejemplos de trastornos mentales incluyen, pero no se limitan a, ansiedad y depresión.
El término "dolor" se refiere a una experiencia sensorial y emocional desagradable asociada con un daño tisular real o potencial. Los ejemplos de dolor incluyen, pero no se limitan a, dolor nocisensible, dolor crónico (incluyendo el dolor idiopático), dolor neuropático que incluye neuropatía inducida por quimioterapia, dolor fantasma y dolor psicogénico. Un ejemplo particular de dolor es el dolor neuropático, que se provoca por daño o enfermedad que afecta a cualquier parte del sistema nervioso implicada en las sensaciones corporales (es decir, el sistema somatosensitivo). En un modo de realización, "dolor" es dolor neuropático resultante de amputación o toracotomía. En un modo de realización, "dolor" es neuropatía inducida por quimioterapia.
El término "neurotoxicidad" se refiere a la toxicidad en el sistema nervioso. Se produce cuando la exposición a sustancias tóxicas naturales o artificiales (neurotoxinas) alteran la actividad normal del sistema nervioso de tal manera que provocan daño al tejido nervioso. Los ejemplos de neurotoxicidad incluyen, pero no se limitan a, la neurotoxicidad resultante de la exposición a sustancias usadas en quimioterapia, radioterapia, farmacoterapias, drogadicción y trasplantes de órganos, así como la exposición a metales pesados, determinados alimentos y aditivos alimentarios, plaguicidas, disolventes industriales y/o de limpieza, cosméticos y algunas sustancias naturales.
El término "cáncer" se refiere a una enfermedad caracterizada por la presencia de una neoplasia o tumor resultante del crecimiento anómalo descontrolado de las células (siendo dichas células "células cancerosas"). Como se usa en el presente documento, el término cáncer incluye explícitamente, pero no se limita a, carcinoma hepatocelular, carcinogénesis de colon y cáncer de ovario.
El término "mamífero" como se usa en el presente documento incluye tanto seres humanos como no humanos e incluye pero no se limita a seres humanos, primates no humanos, caninos, felinos, murinos, bovinos, equinos y porcinos. En un modo de realización en particular preferente, el término "mamífero" se refiere a seres humanos.
Compuestos de la invención
En un primer aspecto, la presente invención proporciona un compuesto de fórmula (I)

o una sal farmacéuticamente aceptable del mismo, en la que:
(i) X es C-R5;
L es un enlace covalente, -(CH2)n-N(alquil C1-6)-, -(CH 2)n-NH-, -N(alquil C1-6)-(CH2)p-, -NH-(CH2)p-, -(CH 2)n-O-, -O-(CH2)p-, -SO 2-N(alquil Ci-a)-, -SO 2-NH-, -N(alquil C1-6)-SO2-, -NH-SO2-, carbonilo, -(CH2)n-, -CHR6-, -CF2-(CH2)n-, -(CH2)p-CF2-, -(CH2)n-S-, -S-(CH2)p-, -SO 2-, -C(O)-NH-, -C(O)-N(alquil C1-6)-, -NH-C(O)- o -N(alquil C1-6)-C(O)-; y
A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5 -i4 miembros sustituido con Ri0, R11 y Ri2; o
(ii) X es N;
L es un enlace covalente, -(CH2)n-, -CHRa-, -SO 2-, carbonilo, -N(alquil C1-6)-(CH2)p-, -NH-(CH2)p-, -0-(CH2)p-, -C F 2-CH2-, -N(alquil C1-6)-SO2-, -NH-SO2-, -NH-C(O)- o -N(alquil C1-6)-C(O)-; y
A es:
(i) arilo C6-14 sustituido con R7, R 8 y R9; o
(ii) heteroarilo de 5 -14 miembros sustituido con R10, R11 y R12; o
(iii) X es N;
L es alcoxicarbonilo C1-6, ariloxicarbonilo C6-14 o heteroariloxicarbonilo de 5 -14 miembros; y
A está ausente;
B es un espirociclo bicíclico;
R1 , R2, R3, R4 y R5 son independientemente hidrógeno, halógeno, hidroxi, alquilo C1-6 o halo-alquilo C1-6;
Ra es arilo C6-14 o heteroarilo de 5 -i4 miembros;
R7, R8, R9, R10, R11 y R12 son cada uno en cada aparición independientemente hidrógeno, hidroxi, alquilo Ci-a, halo-alquilo C1-6, halógeno, alcoxi C1-6, halo-alcoxi C1-6, SFs, alquilsulfonilo C1-6, ciano o un grupo

C es heteroarilo de 5 -i4 miembros, heterociclilo de 3 -14 miembros o cicloalquilo C3-10;
RC1, RC2 y RC3 son cada uno independientemente hidrógeno, alquilo Ci-a, halo-alquilo Ci-a, oxo, halógeno, hidroxi, alcoxi C1-6 o halo-alcoxi C1-6;
cada aparición de n es independientemente 0, 1 , 2 o 3;
cada aparición de p es independientemente i, 2 o 3; y
q es 0, 1 o 2.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, en la que:
(i) X es C-R5;
L es -(CH2)n-N(alquil C1-6 )-, -(CH 2)n-NH-, -N(alquil C1-6 )-(CH2)p-, -NH-(CH2)p-, -(CH2)n-O-, -O-(CH2)p-, -SO 2-N(alquil C1-6 )-, -SO 2-NH-, -N (a lq u il C1-6 )-SO2-, -NH-SO2-, carbonilo, -(CH2)n- , -CHRa-, -C F 2-(CH2)n-, -(CH 2)p-CF2-, -(CH2)n-S-, -S-(CH2)p- , -SO 2-, -C(O)-NH-, -C(O)-N(a lq u il C1-6 )-, -NH-C(O)- o -N(alquil Ci-a )-C(O)-; y
A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5-14 miembros sustituido con R10, R11 y R12; o
(ii) X es N;
L es -(CH2)n-, -CHRa-, -SO 2-carbonilo, -N(alquil C1-6)-(CH2)p-, -NH-(CH2)p- -O-(CH2)p-, -CF2-CH2-, -N(alquil C1-6)-SO2-, -NH-SO2-, -NH-C(O)- o -N(alquil C1-6)-C(O)-; y
A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5-14 miembros sustituido con R10, R11 y R12; o
(iii) X es N;
L es alcoxicarbonilo C1-a , ariloxicarbonilo Ca-14 o heteroariloxicarbonilo de 5-14 miembros; y
A está ausente;
B es un espirociclo bicíclico;
R1 es hidrógeno o alquilo C1-6;
R2 es hidrógeno o alquilo C1-6;
R3 es hidrógeno, alquilo C1-6 , halo-alquilo C1-6 , halógeno o hidroxi;
R4 es hidrógeno, alquilo C1-6 , halo-alquilo C1-6 , halógeno o hidroxi;
R5 es hidrógeno, alquilo C1-6 , halo-alquilo C1-6 , halógeno o hidroxi;
Ra es arilo C6-14 o heteroarilo de 5-14 miembros;
R7, R8, R9, R10, R11 y R12 son cada uno en cada aparición independientemente hidrógeno, alquilo C1-a , halo-alquilo C1-6, halógeno, alcoxi C1-6, halo-alcoxi C1-6, SFs, SO2CH3, ciano,

R13 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, hidroxi o alcoxi C1-6; y
R14 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, hidroxi, alcoxi C1-6; o
R13 y R14, tomados conjuntamente con el átomo de carbono al que están unidos, forman un anillo de 4-a miembros que contiene 0, 1 o 2 heteroátomos seleccionados de oxígeno y NR18;
R15 es hidrógeno, alquilo C1-6 o halo-alquilo C1-6;
R1a es hidrógeno, alquilo C1-6, halo-alquilo C1-6, hidroxi o ciano;
R17 es hidrógeno, hidroxi, ciano, halo-alquilo C1-6 o alquilo C1-6;
R18 es hidrógeno o alquilo C1-6;
cada aparición de n es independientemente 0, 1, 2 o 3;
cada aparición de p es independientemente 1,2 o 3; y
q es 0, 1 o 2.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R1 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R2 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R1 y R2 son ambos hidrógeno. En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R3 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R4 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R3 y R4 son ambos hidrógeno. En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R1, R2, R3 y R4 son todos hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R5 es hidrógeno o halo-alquilo C1-6.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R5 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R6 es arilo C1-6.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R7 es hidrógeno, hidroxi, alquilo
C1-6, halo-alquilo C1-6  , halógeno, alcoxi C1-6, halo-alcoxi C1-6, SFs o un grupo
, halógeno, alcoxi C1-6, halo-alcoxi C1-6, SFs o un grupo
en la que
C es heteroarilo de 5-14 miembros o heterociclilo de 3-14 miembros;
RC1 es alquilo C1-6, halo-alquilo C1-6 u oxo; y
RC2 y RC3 son ambos hidrógeno.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R7 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, alcoxi C1-6, halo-alcoxi C1-6 o SF5.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R7 es hidrógeno, flúor, cloro, CF3, metilo, metoxi, trifluorometoxi o SF5.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R7 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, alcoxi C1-6 o halo-alcoxi C1-6.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R7 es hidrógeno, flúor, cloro, CF3, metilo, metoxi o trifluorometoxi.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R8 es hidrógeno, alcoxi C1-6, halo-alquilo C1-6 o halógeno.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R8 es hidrógeno, halo-alquilo C1-6 o halógeno.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R8 es hidrógeno, CF3, cloro o flúor.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R9 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
R7 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, alcoxi C1-6 o halo-alcoxi C1-6;
R8 es hidrógeno, halo-alquilo C1-6 o halógeno; y
R9 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R10 es halógeno o halo-alquilo C1-6.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R10 es halo-alquilo C1-6.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R10 es CF3.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R11 es hidrógeno o halo-alquilo C1-6.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R11 es hidrógeno o CF3.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R12 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
R10 es halo-alquilo C1-6;
R11 es hidrógeno o halo-alquilo C1-6; y
R12 es hidrógeno.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que A es fenilo o
piridilo.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es un enlace covalente, -(CH2)n-N(alquil C1-6)-, -(CH2)n-NH-, -(CH 2)n-O-, -OCH2- , -CH 2-, -SO 2-, -SO 2-N(alquil C1-6) - o -SO 2-NH-;
n es 0 o 1; y
R5 es como se define en el presente documento.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es un enlace covalente, -CH 2O-, -O -, -OCH2-, -CH 2- o -SO 2-N(alquil C1-6)-; y
R5 es como se define en el presente documento.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que: X es C-R5;
L es un enlace covalente, -CH 2O-, -O -, -OCH2-, -CH 2- o -SO 2-N(metil)-; y
R5 es como se define en el presente documento.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es -CH 2-N(alquil C1-6)-, -CH 2-NH-, -(CH2)n-O-, -O-CH2-, -SO 2-N(alquil C1-6) - o -SO 2-NH-;
n es 0 o 1; y
R5 es como se define en el presente documento.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es -CH 2-N(alquil C1-6)-, -CH 2-NH-, -(CH2)n-O-, -O-CH2-, -SO 2-N(alquil C1-6) - o -SO 2-NH-;
n es 0 o 1; y
R5 es hidrógeno.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es -(CH2)n-O-, -O-CH2- o -SO 2-N(alquil C1-6)-;
n es 0 o 1; y
R5 es hidrógeno.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I)
como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que: X es C-R5;
L es -(CH2)n-O-, -O-CH2- o -SO 2-N(metil)-;
n es 0 o 1; y
R5 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es N;
L es un enlace covalente, -CH 2-, -CHR6- o -SO 2-; y
R6 es como se define en el presente documento.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es N; y
L es -CH 2- o -SO 2-.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es N;
L es -(CH2)n-, -CHR6- o -SO 2-; y
n es 1; y
R6 es como se define en el presente documento.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es N;
L es -(CH2)n-, -CHR6- o -SO 2-; y
n es 1; y
R6 es arilo C6-14.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
X es N;
L es -(CH2)n- o -SO 2- ; y
n es 1.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico que tiene la fórmula (II):
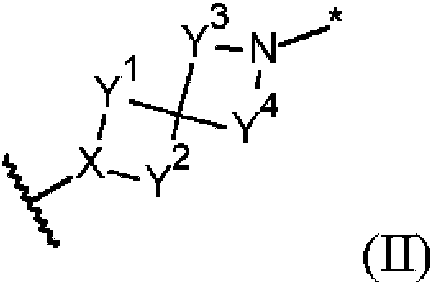
en la que:
X es como se define en el presente documento;
Y1, Y2, Y3 e Y4 son cada uno independientemente -(CH 2)m-, -(CH 2)mO-, -O(CH2)m-, -(CH2)mNH- o -NH(CH2)m-; cada aparición de m es independientemente 1, 2 o 3;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I).
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
X es como se define en el presente documento;
Y1 es -(CH2)m- o -(CH2)mO-, en el que m es 1 o 2;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH2)m- , en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I).
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
X es como se define en el presente documento;
Y1 es -CH 2-;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH 2)m- , en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I).
En otro modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico seleccionado del grupo que consiste en:

(i) una línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
un asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I); o
(ii) una línea ondulada indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I); y
un asterisco indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I).
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico seleccionado del grupo que consiste en:

una línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y un asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I).
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
(i) X es C-R5;
L es un enlace covalente, -CH 2-N(alquil C1 -6 )-, -CH 2-NH-, -O -, -CH 2O-, -OCH2-, -CH 2-, -SO 2-, -SO 2-N(alquil C1-6 ) - o -SO 2-NH-; y
A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5-14 miembros sustituido con R10, R11 y R12; o
(ii) X es N;
L es un enlace covalente, -CH 2-, -CHR6- o -SO 2-; y
A es arilo C6-14 sustituido con R7, R8 y R9; o
(iii) X es N;
L es alcoxicarbonilo C1-6; y
A está ausente;
B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
Y1 es -(CH2)m- o -(CH2)mO-, en el que m es 1 o 2;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH 2)m- , en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I);
cada uno de R1, R2, R3, R4, R9 y R12 es hidrógeno;
R5 es hidrógeno o alquilo C ^ ;
Ra es arilo C6-14;
R7 es hidrógeno, hidroxi, alquilo Ci-e, halo-alquilo Ci-e, halógeno, alcoxi C1-6 o halo-alcoxi C1-6, SFs o un grupo

R8 es hidrógeno, alcoxi C1-6 , halo-alquilo C1-a o halógeno;
R10 es halógeno o halo-alquilo C1-6 ;
R11 es hidrógeno o halo-alquilo C1-6;
RC1 es alquilo C1-6, halo-alquilo C1-6 u oxo;
RC2 y RC3 son ambos hidrógeno; y
C es heteroarilo de 5-14 miembros o heterociclilo de 3-14 miembros.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
(i) X es C-R5;
L es -CH 2O-, -OCH2-, -O -, -CH 2- o -SO 2-N(alquil C1-6)-; y
A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5-14 miembros sustituido con R10, R11 y R12; o
(ii) X es N;
L es -C H 2- o -SO 2- ; y
A es arilo C6-14 sustituido con R7, R8 y R9; o
B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
Y1 es -CH 2-;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH 2)m- , en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I);
cada uno de R1, R2, R3, R4, R5, R9 y R12 es hidrógeno;
R7 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, alcoxi C1-6, halo-alcoxi C1-6 o SF5;
R8 es hidrógeno, halo-alquilo C1-6 o halógeno;
R10 es halo-alquilo C1-6; y
R11 es hidrógeno o halo-alquilo C1-6.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que: (i) X es C-R5;
L es -CH 2O-, -OCH2-, -O -, -CH 2- o -SO 2-N(metil)-; y
A es:
(i) fenilo sustituido con R7, R8 y R9; o
(ii) piridilo sustituido con R10, R11 y R12; o
(ii) X es N;
L es -CH 2- o -SO 2-; y
A es fenilo sustituido con R7, R8 y R9; o
B es un espirociclo bicíclico seleccionado del grupo que consiste en:

la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I);
cada uno de R1, R2, R3, R4, R5, R9 y R12 es hidrógeno;
R7 es hidrógeno, flúor, cloro, CF3, metilo, metoxi, trifluorometoxi o SF5;
R8 es hidrógeno, CF3, cloro o flúor;
R10 es CF3; y
R11 es hidrógeno o CF3.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
(i) X es C-R5;
L es -CH 2-N(alquil C1-6)-, -CH 2-NH-, -(CH2)n-O-, -O-CH2-, -SO 2-N(alquil C1-6) - o -SO 2-NH-; y A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5-14 miembros sustituido con R10, R11 y R12; o
(ii) X es N;
L es -CH 2-, -CHR6- o -SO 2-; y
A es arilo C6-14 sustituido con R7, R8 y R9; o
(iii) X es N;
L es alcoxicarbonilo C1-6; y
A está ausente;
B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
Y1 es -(CH2)m- o -(CH2)mO-, en el que m es 1 o 2;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH 2)m- , en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I);
cada uno de R1, R2, R3, R4, R5, R9 y R12 es hidrógeno;
R6 es arilo C6-14;
R7 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, alcoxi C1-6 o halo-alcoxi C1-6;
R8 es hidrógeno, halo-alquilo C1-6 o halógeno;
R10 es halo-alquilo C1-6;
R11 es hidrógeno o halo-alquilo C1-6; y
n es 0 o 1.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
(i) X es C-R5;
L es -(CH2)n-O-, -O-CH2- o -SO 2-N(alquil C1-6)-; y
A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5-14 miembros sustituido con R10, R11 y R12; o
(ii) X es N;
L es -CH 2- o -SO 2-; y
A es arilo C6-14 sustituido con R7, R8 y R9; o
B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
Y1 es -CH 2-;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH 2)m- , en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I);
cada uno de R1, R2, R3, R4, R5, R9 y R12 es hidrógeno;
R7 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, alcoxi C1-6 o halo-alcoxi C1-6;
R8 es hidrógeno, halo-alquilo C1-6 o halógeno;
R10 es halo-alquilo C1-6;
R11 es hidrógeno o halo-alquilo C1-6; y
n es 0 o 1.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que: (i) X es C-R5;
L es -(CH2)n-O-, -O-CH2- o -SO 2-N(metil)-; y
A es:
(i) fenilo sustituido con R7, R8 y R9; o
(ii) piridilo sustituido con R10, R11 y R12; o
(ii) X es N;
L es -CH 2- o -SO 2-; y
A es fenilo sustituido con R7, R8 y R9; o
B es un espirociclo bicíclico seleccionado del grupo que consiste en:

la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I);
cada uno de R1, R2, R3, R4, R5, R9 y R12 es hidrógeno;
R7 es hidrógeno, flúor, cloro, CF3, metilo, metoxi o trifluorometoxi;
R8 es hidrógeno, CF3, cloro o flúor;
R10 es CF3;
R11 es hidrógeno o CF3; y
n es 0 o 1.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en el que dicho compuesto de fórmula (I)
se selecciona del grupo que consiste en:
(4aR,8aS)-6-(6-(2-doro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-5-(trifluorometil)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][ 1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-cloro-2-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(3-fluoro-5-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b ][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-cloro-4-fluorobencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxa cin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespi ro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b] [1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-cloro-4-fluorofenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(7-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespi ro[4,4]nonano-2-carbonil)hexahidro-2H-pirido[4,3-b] [1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespi ro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][ 1.4] oxacin-3(4H)-ona;
rac-(4aR,8aS)-N-((R)-8-(3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il) bencenosulfonamida;
rac-(4aR,8aS)-N-((S)-8-(3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespi ro[4.5]decan-3-il) bencenosulfonamida;
rac-(4aR,8aS)-6-(2-benzhidrilo-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(4-((4-fluorofenil)sulfonil)-1-oxa-4,9-diazaespiro[5.5]undecano-9-carbonil)hexahidro-2H-pirido[4,3 -b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4,5-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((5,6-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
2-doro-4-fluoro-N-metil-N-((R)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azae spiro[4.5]decan-3-il)bencenosulfonamida;
(4aR,8aS)-6-(6-((5-(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4-metil-3-(trifluorometil)bencil)oxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b] [1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-((2-doro-4-fluorofenil)sulfonil)-2,7-diazaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pi rido[4,3-b][1, 4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il)-4-(t rifluorometil)bencenosulfonamida;
N-metil-N-((R)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan -3-il)bencenosulfonamida;
2-doro-4-fluoro-N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.
5] decan-3-il)bencenosulfonamida;
N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il)-3-(t rifluorometil)bencenosulfonamida;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-((4-(trifluorometil)fenil)sulfonil)-2,7-diazaespi ro[3.5]nonano-7-carbonil)hexahidro-2H-pi rido[4,3-b][1 ,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[ 4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(fenilsulfonil)-2,7-diazaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-o na;
rac-6-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-2,6-diazaespi ro[3.4]octano-2-carboxilato de ferc-butilo;
(4aR,8aS)-6-(6-(4-(1-metil-1H-pirazol-5-il)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-6-hidroxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(6-(2-hidroxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(4-(2-oxopirrolidin-1-il)fenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o
xacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-6-metoxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(pentafluoro-16-sulfanil)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorobencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-doro-4-fluorofenoxi)metil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o xacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)bencil)oxi)-6-(trifluorometil)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidr o-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)fenil)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(2-(trifluorometil)pirrolidin-1-il)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3 -b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-doro-4-fluorobencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-6-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(6-(3-(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(2-(4-(trifluorometil)fenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin -3(4H)-ona;
(4aR,8aS)-6-(2-(3-(trifluorometil)fenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin -3(4H)-ona;
(4aR,8aS)-6-(2-(4-isopropoxifenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(4-isopropoxifenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3 (4H)-ona;
(4aR,8aS)-6-(2-(4-metoxi-3-metilfenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin -3(4H)-ona;
(4aR,8aS)-6-(2-(4-doro-3-(trifluorometil)fenil)-2,6-diazaespi ro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoropiridin-4-il)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3( 4H)-ona;
(4aR,8aS)-6-(6-(2,5-bis(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4-fluoro-2-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirid
o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-doro-4-fluorofenil)sulfonil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o xacin-3(4H)-ona;
(4aR,8aS)-6-(6-((3-doro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido [4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2,4-bis(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4, 3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,6-difluorobencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona; y
(4aR,8aS)-6-(6-(2-metoxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en el que dicho compuesto de fórmula (I) se selecciona del grupo que consiste en:
(4aR,8aS)-6-(6-(2-cloro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-5-(trifluorometil)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometoxi)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][ 1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-cloro-2-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b ][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4,5-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(pentafluoro-16-sulfanil)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorobencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona; y
(4aR,8aS)-6-(6-((2-doro-4-fluorofenoxi)metil)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o xacin-3(4H)-ona.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en el que dicho compuesto de fórmula (I) se selecciona del grupo que consiste en:
(4aR,8aS)-6-(6-(2-cloro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-5-(trifluorometil)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][ 1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-cloro-2-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(3-fluoro-5-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b ][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-cloro-4-fluorobencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxa cin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespi ro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b] [1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-cloro-4-fluorofenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(7-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespi ro[4,4]nonano-2-carbonil)hexahidro-2H-pirido[4,3-b] [1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespi ro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][ 1.4] oxacin-3(4H)-ona;
rac-(4aR,8aS)-N-((R)-8-(3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il) bencenosulfonamida;
rac-(4aR,8aS)-N-((S)-8-(3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespi ro[4.5]decan-3-il) bencenosulfonamida;
rac-(4aR,8aS)-6-(2-benzhidrilo-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(4-((4-fluorofenil)sulfonil)-1-oxa-4,9-diazaespiro[5.5]undecano-9-carbonil)hexahidro-2H-pirido[4,3 -b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4,5-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((5,6-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
2-doro-4-fluoro-N-metil-N-((R)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azae spiro[4.5]decan-3-il)bencenosulfonamida;
(4aR,8aS)-6-(6-((5-(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4-metil-3-(trifluorometil)bencil)oxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b] [1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-((2-doro-4-fluorofenil)sulfonil)-2,7-diazaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pi rido[4,3-b][1, 4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il)-4-(t rifluorometil)bencenosulfonamida;
N-metil-N-((R)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan -3-il)bencenosulfonamida;
2-doro-4-fluoro-N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.
5] decan-3-il)bencenosulfonamida;
N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il)-3-(t rifluorometil)bencenosulfonamida;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-((4-(trifluorometil)fenil)sulfonil)-2,7-diazaespi ro[3.5]nonano-7-carbonil)hexahidro-2H-pi rido[4,3-b][1 ,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[ 4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(fenilsulfonil)-2,7-diazaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-o na; y
rac-6-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-2,6-diazaespi ro[3.4]octano-2-carboxilato de ferc-butilo.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en el que dicho compuesto de fórmula (I) se selecciona del grupo que consiste en:
(4aR,8aS)-6-(6-(2-cloro-4-(trifluorometoxi)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1
,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-5-(trifluorometil)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][ 1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-doro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-doro-2-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b ][1,4]oxacin-3(4H)-ona; y
(4aR,8aS)-6-(6-((4,5-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en el que dicho compuesto de fórmula (I) no es (4aR,8aS)-6-(6-(2-fluoro-6-metoxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona.
En un modo de realización particular, la presente invención proporciona sales farmacéuticamente aceptables de los compuestos de acuerdo con la fórmula (I) como se describe en el presente documento, especialmente sales clorhidrato. En otro modo de realización particular, la presente invención proporciona compuestos de acuerdo con la fórmula (I) como se describe en el presente documento.
En algunos modos de realización, los compuestos de fórmula (I) se marcan isotópicamente reemplazando uno o más átomos por un átomo que tiene una masa atómica o número de masa diferente. Se considera que dichos compuestos de fórmula (I) marcados isotópicamente (es decir, radiomarcados) están dentro del alcance de la presente divulgación. Los ejemplos de isótopos que se pueden incorporar en los compuestos de fórmula (I) incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, azufre, flúor, cloro y yodo, tales como, pero sin limitarse a, 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O,31P, 32P, 35S, 18F, 36Cl,123I y 125I, respectivamente. Determinados compuestos de fórmula (I) marcados isotópicamente, por ejemplo, los que incorporan un isótopo radioactivo, son útiles en estudios de distribución en tejidos de fármacos y/o sustratos. Los isótopos radioactivos tritio, es decir 3H, y carbono 14, es decir, 14C, son en particular útiles para este propósito en vista de su facilidad de incorporación y rápidos medios de detección. Por ejemplo, un compuesto de fórmula (I) se puede enriquecer con un 1,2, 5, 10, 25, 50, 75, 90, 95 o 99 por ciento de un isótopo dado.
La sustitución con isótopos más pesados tales como deuterio, es decir 2H, puede dar determinadas ventajas terapéuticas resultantes de una mayor estabilidad metabólica, por ejemplo, requisitos de semivida en vivo incrementada o de dosificación reducida.
La sustitución con isótopos emisores de positrones, tales como 11C, 18F, 15O y 13N, puede ser útil en los estudios de topografía por emisión de positrones (PET) para examinar la ocupación de receptor de sustrato. Los compuestos de fórmula (I) marcados isotópicamente, en general, se pueden preparar por técnicas convencionales conocidas para los expertos en la técnica o por procedimientos análogos a los descritos en los ejemplos que se exponen a continuación usando un reactivo marcado isotópicamente apropiado en lugar del reactivo no marcado empleado previamente.
Procedimientos de fabricación
La preparación de compuestos de fórmula (I) de la presente invención se puede llevar a cabo en vías sintéticas secuenciales o convergentes. Las síntesis de la invención se muestran en los siguientes esquemas generales. Las habilidades requeridas para llevar a cabo la reacción y purificación de los productos resultantes son conocidas para los expertos en la técnica. Los sustituyentes e índices usados en la siguiente descripción de los procedimientos tienen el significado dado en el presente documento, a menos que se indique lo contrario.
Si uno de los materiales de partida, intermedios o compuestos de fórmula (I) contienen uno o más grupos funcionales que no son estables o son reactivos en las condiciones de reacción de una o más etapas de reacción, se pueden introducir grupos protectores apropiados (como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wutts, 5.a ed., 2014, John Wiley & Sons, N.Y.) antes de la etapa crucial aplicando procedimientos bien conocidos en la técnica. Dichos grupos protectores se pueden retirar en una fase posterior de la síntesis usando procedimientos estándar descritos en la literatura.
Si los materiales de partida o intermedios contienen centros estereogénicos, los compuestos de fórmula (I) se pueden obtener como mezclas de diastereómeros o enantiómeros, que se pueden separar por procedimientos bien conocidos en la técnica, por ejemplo, HPLC quiral, SFC quiral o cristalización quiral. Los compuestos racémicos, por ejemplo, se pueden separar en sus antípodas por medio de sales diastereoméricas por cristalización con ácidos ópticamente puros o por separación de las antípodas por procedimientos cromatográficos específicos usando un adsorbente quiral o bien un eluyente quiral. Es igualmente posible separar materiales de partida e intermedios que contienen centros estereogénicos para dar materiales de partida e intermedios enriquecidos de forma diastereomérica/enantiomérica. El uso de dichos materiales de partida e intermedios enriquecidos de forma diastereomérica/enantiomérica en la síntesis de compuestos de fórmula (I) típicamente dará lugar a los respectivos compuestos de fórmula (I) enriquecidos de forma diastereomérica/enantiomérica.
Un experto en la técnica reconocerá que en la síntesis de compuestos de fórmula (I), en la medida en que no se desee de otro modo, se aplicará una "estrategia de grupo de protección ortogonal", que permite la escisión de varios grupos protectores uno a la vez sin afectar a otros grupos protectores en la molécula. El principio de protección ortogonal es bien conocido en la técnica y también se ha descrito en la literatura (por ejemplo, Barany y R.B. Merrifield, J. Am. Chem. Soc. 1977, 99, 7363; H. Waldmann et al., Angew. Chem. Int. Ed. Engl. 1996, 35, 2056).
Un experto en la técnica reconocerá que la secuencia de reacciones puede variar dependiendo de la reactividad y la naturaleza de los intermedios.
Con más detalle, los compuestos de fórmula (I) se pueden fabricar por los procedimientos dados a continuación, por los procedimientos dados en los ejemplos o por procedimientos análogos. Las condiciones de reacción apropiadas para las etapas de reacción individuales son conocidas para un experto en la técnica. Además, para las condiciones de reacción descritas en la literatura que afectan a las reacciones descritas, véase, por ejemplo: Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2.a edición, Richard C. Larock. John Wiley & Sons, New York, NY. 1999). Se encontró práctico llevar a cabo las reacciones en presencia o ausencia de un disolvente. No existe una restricción particular sobre la naturaleza del disolvente que se va a emplear, siempre que no tenga un efecto adverso sobre la reacción o los reactivos implicados y que pueda disolver los reactivos, al menos hasta cierto punto. Las reacciones descritas pueden tener lugar en un amplio intervalo de temperaturas, y la temperatura de reacción precisa no es crucial para la invención. Es conveniente llevar a cabo las reacciones descritas en un intervalo de temperaturas entre de -78 °C a reflujo. El tiempo requerido para la reacción también puede variar ampliamente, dependiendo de muchos factores, notablemente la temperatura de reacción y la naturaleza de los reactivos. Sin embargo, normalmente será suficiente un período de desde 0,5 horas a varios días para proporcionar los intermedios y compuestos descritos. La secuencia de reacción no se limita a la presentada en los esquemas, sin embargo, dependiendo de los materiales de partida y de su respectiva reactividad, se puede modificar libremente la secuencia de las etapas de reacción. Si
Si los materiales de partida o los intermedios no están disponibles comercialmente o su síntesis no está descrita en la literatura, se pueden preparar de forma análoga a los procedimientos existentes para análogos cercanos o como se explica en la sección experimental.
Las siguientes abreviaturas se usan en el presente texto:
AcOH = ácido acético, ACN = acetonitrilo, Bn = bencilo, Boc = terc-butiloxicarbonilo, CAS RN = número de registro de resúmenes químicos, Cbz = benciloxicarbonilo, Cs2CO3 = carbonato de cesio, CO = monóxido de carbono, CuCl = cloruro de cobre(I), CuCN = cianuro de cobre(I), Cul = yoduro de cobre(I), DAST = trifluoruro de (dietilamino)azufre, DBU = 1,8-diazabiciclo[5,4,0]undec-7-eno, DCM = diclorometano, DEAD = azodicarboxilato de dietilo, DIAD = azodicarboxilato de diisopropilo, DMAP = 4-dimetilaminopiridina, DME = dimetoxietano, DMEDA = N,N'-dimetiletilendiamina, DMF = N,N-dimetilformamida, DIPEA = N,N-diisopropiletilamina, dppf = 1,1-bis(difenilfosfino)ferroceno, EDC.HCl = clorhidrato de N-(3-dimetilaminopropil)-N'-etilcarbodiimida, EI =
impacto de electrones, EM = espectro de masas, ESI = ionización por electropulverización, EtOAc = acetato de etilo, EtOH = etanol, h = hora(s), FA = ácido fórmico, H2O = agua, H2SO4 = ácido sulfúrico, HATU = hexafluorofosfato de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio-3-óxido, HBTU = hexafluoro-fosfato de O-benzotriazol-N,N,N',N'-tetrametil-uronio, HCl = cloruro de hidrógeno, HOBt = 1-hidroxi-1H-benzotriazol; HPLC = cromatografía de líquidos de alto rendimiento, iPrMgCl = cloruro de isopropilmagnesio, I2 = yodo, IPA = 2-propanol, ISP = pulverización iónica positiva (modo), ISN = pulverización iónica negativa (modo), K2CO3 = carbonato de potasio, KHCO3 = bicarbonato de potasio, KI = yoduro de potasio, KOH = hidróxido de potasio, K3PO4 = fosfato de potasio tribásico, LiAlH4 o LAH = hidruro de litio y aluminio, LiHMDS = bis(trimetilsilil)amida de litio, LiOH = hidróxido de litio, MgSO4 = sulfato de magnesio, min = minuto(s), ml = mililitros, MPLC = cromatografía de líquidos de presión media, MTBE = éter metil-terc-butílico, nBuLi = n-butillitio, NaBH3CN = cianoborohidruro de sodio, NaH = hidruro de sodio, NaHCO3 = hidrogenocarbonato de sodio, Na2NO2 = nitrito de sodio, NaBH(OAc)3 = triacetoxiborohidruro de sodio, NaOH = hidróxido de sodio, Na2CO3 = carbonato de sodio, Na2SO4 = sulfato de sodio, Na2S2O3 = tiosulfato de sodio, NBS = N-bromosuccinimida, nBuLi = n-butillitio, NEt3 = trietilamina (TEA), NH4Cl = cloruro de amonio, NMP = N-metil-2-pirrolidona, OAc = acetoxi, T3P = anhídrido propilfosfónico, PE = éter de petróleo, PG = grupo protector, Pd-C = paladio sobre carbón activado, PdCh (dppf)-CH2Ch = complejo 1,1'-bis(difenilfosfino)ferroceno-dicloruro de paladio(II) diclorometano, Pd2(dba)3 = tris(dibencilidenacetona)dipaladio(0), Pd(OAc)2 = acetato de paladio(II), Pd(OH)2 = hidróxido de paladio, Pd(PPh3)4 = tetraquiis(trifenilfosfina)paladio(0), PTSA = ácido p-toluenosulfónico, R = cualquier grupo, SFC = cromatografía de fluidos supercríticos, S-PHOS = 2-diciclohexilfosfino-2',6'-dimetoxibifenilo, TA = temperatura ambiente, TBAI = yoduro de tetrabutilamonio, TEA = trietilamina, TFA = ácido trifluoroacético, THF = tetrahidrofurano, TMEDA = N,N,N',N'-tetrametiletilendiamina, ZnCh = cloruro de cinc, Hal = halógeno.
Los compuestos de fórmula I en la que A, L, X, R1, R2, R3 y R4 son como se describe en el presente documento se pueden sintetizar de forma análoga a los procedimientos de la literatura y/o como se representa, por ejemplo, en el esquema 1a.

Esquema 1a
En consecuencia, las 4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-onas 1 se hacen reaccionar con intermedios 2 en presencia de un reactivo formador de urea tal como carbonato de bis(triclorometilo) usando una base adecuada y un disolvente tal como, por ejemplo, bicarbonato de sodio en DCM, para dar los compuestos de fórmula I (etapa a). Otros reactivos formadores de urea incluyen, pero no se limitan a, fosgeno, cloroformiato de triclorometilo, carbonato de 4-nitrofenilo, 1,1'-carbonildiimidazol o 1, 1 '-carbonil-di-(1,2,4-triazol). Las reacciones de este tipo y el uso de estos reactivos están descritos ampliamente en la literatura (por ejemplo, G. Sartori et al., Green Chemistry 2000, 2, 140). Un experto en la técnica reconocerá que el orden de la adición de los reactivos puede ser importante en este tipo de reacciones debido a la reactividad y estabilidad de los cloruros de carbamαlo intermediarios formados, así como para evitar la formación de subproductos de urea simétricos no deseados.
Los compuestos de fórmula (I) en la que R1, R2, R3 y R4 son como se define en el presente documento y en la que R7 es Cl o Br, se pueden modificar además de acuerdo con el procedimiento general explicado en el esquema 1b.

Esquema 1b
El tratamiento de los compuestos de fórmula I, que contienen un bromo- o cloroarilo como A en condiciones típicas de una reacción de Suzuki-Miyaura, una reacción de Buchwald-Hartwig u otros acoplamientos cruzados C-C o C-N
organometálicos conocidos en la técnica dan lugar a compuestos de fórmula I sustituidos donde el bromo se ha reemplazado por un resto alquilo, heterociclilo, cicloalquilo o heteroarilo. Típicamente, esto requiere un compañero de reacción adecuado, tal como un ácido borónico, un trifluoroborato de potasio, un boronato de pinacol, una amina o un compuesto de organocinc, un catalizador adecuado, por ejemplo, tetraquis(trifenilfosfina)paladio(0), PdCl2(DPPF)-CH2Cl2, Pd2(dba)3 + Xantphos, cataCXium A Pd G2, RuPhos Pd g 2, una base orgánica o inorgánica tal como carbonato de sodio, TEA, TMEDA o carbonato de cesio en un sistema de disolventes tal como dioxano/agua, DMF o tolueno/agua. Las reacciones se llevan a cabo típicamente a temperaturas elevadas entre 100 y 120 °C bajo atmósfera inerte (argón).
Los intermedios 1 se pueden sintetizar como se representa, por ejemplo, en el esquema 2 y/o de forma análoga a los procedimientos descritos en la literatura.

Esquema 2
Por tanto, los derivados de 3-aminopiperidin-4-ol 3 en el que "PG" significa un grupo protector adecuado tal como un grupo protector Cbz o Boc, y R2 es como se define en el presente documento, se pueden acilar, por ejemplo, con cloruros de acilo 4 en el que R1 es como se define en el presente documento y "LG" significa un grupo saliente adecuado (por ejemplo, Cl o Br), usando una base adecuada tal como carbonato de sodio o potasio, hidróxido de sodio o acetato de sodio en un disolvente apropiado tal como THF, agua, acetona o mezclas de los mismos, para proporcionar los intermedios 5 (etapa a). Los intermedios 4 están disponibles comercialmente o bien se pueden preparar de acuerdo con los procedimientos de la literatura en forma aquiral (R1 = H) racémica (R1 distinto de H) o enantioméricamente pura (R1 distinto de H).
Los intermedios 5 se pueden ciclar a los intermedios 6 usando procedimientos bien conocidos en la técnica, por ejemplo, por tratamiento de 5 con hidruro de sodio en THF o terc-butóxido de potasio en IPA y agua (etapa b). Las reacciones de ese tipo se describen en la literatura (por ejemplo, Z. Rafinski et al., J. Org. Chem. 2015, 80, 7468; S. Dugar et al., Synthesis 2015, 47(5), 712; documento WO2005/066187).
La retirada del grupo protector en los intermedios 6 , aplicando procedimientos conocidos en la técnica (por ejemplo, un grupo Boc usando TFA en DCM a temperaturas entre 0 °C y temperatura ambiente, un grupo Cbz usando hidrógeno en presencia de un catalizador adecuado tal como Pd o Pd(OH)2 sobre carbón activado en un disolvente adecuado tal como MeOH, EtOH, EtOAc o mezclas de los mismos y como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wuts, 4.a ed., 2006, Wiley N.Y.), proporciona los intermedios 1 (etapa c).
Los intermedios 1 se pueden obtener como mezclas de diastereómeros y enantiómeros, respectivamente, o como estereoisómeros individuales dependiendo de si se emplean mezclas racémicas o formas enantioméricamente puras de derivados cis- o trans-3-aminopiperidin-4-ol 3 y cloruros de ácido 4 (cuando R1 no es H) en sus síntesis. En caso de que se produzca racemización en un estereocentro que porta R1 durante la conversión de 3 a 5 (etapa a) y/o de 5 a 6 (etapa b), los diastereoisómeros resultantes se pueden separar por cromatografía (por ejemplo, HPLC, HPLC quiral) u otros procedimientos conocidos en la técnica. Los intermedios 3 están disponibles comercialmente y su síntesis también se ha descrito en la literatura (por ejemplo, documento WO2005/066187; documento WO2011/0059118; documento WO2016/185279). Los intermedios configurados en cis ópticamente puros 1B y 1C se pueden obtener, por ejemplo, de acuerdo con el esquema 3 por separación quiral de rac-(4aR,8aS)-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (1A) comercialmente disponible (opcionalmente en forma de una sal tal como, por ejemplo, una sal clorhidrato) usando procedimientos conocidos en la técnica, por ejemplo, por cristalización de sales diastereoméricas o por cromatografía quiral (etapa a).

En algunos modos de realización, los intermedios 2 son intermedios de tipo A . Los intermedios de tipo A en los que A, B, R3 y R4 son como se describe en el presente documento y R5 es hidrógeno, alquilo C1-6 o halo-alquilo C1-6, se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 4.

Esquema 4
Los compuestos espirocíclicos 7 en los que PG significa un grupo protector adecuado tal como un grupo protector Boc, Cbz o Bn (disponible comercialmente o bien preparado como se describe en la literatura, por ejemplo, en Eur. J. Org. Chem. 2oH , 36, 5316; Topics in Het. Chem. 2014, 35, 189; World Journal of Pharmacy and Pharmaceutical Sciences 2014, 3(12), 536; Chem. Rev. 2014, 114(16), 8257-8322) se pueden someter a una reacción de Mitsunobu con derivados de alcohol 8 usando una fosfina apropiada tal como trifenilfosfina y un azodicarboxilato de dialquilo tal como DEAD o DIAD en un disolvente adecuado tal como THF para dar los intermedios 9 (etapa a). Las reacciones de Mitsunobu de ese tipo se describen ampliamente en la literatura (por ejemplo, Org. Chem. Front.
2015, 2, 739; Chem. Rev. 2009, 109 (6), 2551).
La retirada del grupo protector de los intermedios 9 aplicando procedimientos conocidos en la técnica, por ejemplo, un grupo Boc usando TFA en DCM o HCl 4 M en dioxano a temperaturas entre 0 °C y temperatura ambiente, un grupo Bn o Cbz usando hidrógeno en presencia de un catalizador adecuado tal como Pd o Pd(OH)2 sobre carbón activado en un disolvente adecuado tal como MeOH, EtOH, EtOAc o mezclas de los mismos y como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wuts, 4.a ed., 2006, Wiley N.Y.), proporciona los intermedios A (etapa b).
Los intermedios 9 se pueden preparar de forma alternativa por alquilación de los compuestos 8 con derivados espirocíclicos 10 (disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica) en
los que LG significa un grupo saliente adecuado tal como cloro, bromo, yodo, OSO2alquilo (por ejemplo, mesilato (metanosulfonato), OSO2fluoroalquilo (por ejemplo, triflato (trifluorometanosulfonato) u OSO2arilo (por ejemplo, tosilato (p-toluenosulfonato) usando una base adecuada y un disolvente apropiado (por ejemplo, hidruro de sodio en DMF) a temperaturas entre 0 °C y la temperatura de ebullición del disolvente (etapa c).
En algunos modos de realización, los intermedios 2 son intermedios de tipo B. Los intermedios de tipo B en los que A, B, R3 y R4 son como se describe en el presente documento se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 5.

Esquema 5
Los compuestos de tipo 11 disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica y en los que PG significa un grupo protector adecuado tal como, por ejemplo, un grupo protector Boc, Cbz o Bn, se pueden someter a una reacción de aminación reductora con aldehidos de tipo 12 usando un agente reductor adecuado y un disolvente tal como NaBH3CN en MeOH, AcOH o mezclas de los mismos, o NaBH(OAc)3 en DCE, DCM o THF para dar los intermedios 13 (etapa a).
La retirada del grupo protector de los intermedios 13, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios B (etapa b).
Los intermedios 13 se pueden preparar de forma alternativa por alquilación de los compuestos 11 con compuestos 15 (disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica) en los que LG es un grupo saliente adecuado tal como cloro, bromo, yodo, OSO2alquilo (por ejemplo, metanosulfonato), OSO2fluoroalquilo (por ejemplo, trifluorometanosulfonato) u OSO2arilo (por ejemplo, p-toluenosulfonato usando una base adecuada en un disolvente apropiado (por ejemplo, NEt3 o DIPEA en ACN) a temperaturas entre 0 °C y la temperatura de ebullición del disolvente (etapa c). Las reacciones de ese tipo son conocidas en la técnica y se describen ampliamente en la literatura (por ejemplo, ARKIVOC 2005 (vi) 287-292).
En algunos modos de realización, los intermedios 2 son intermedios de tipo C, D o E. Los intermedios de tipo C, D o E en los que A, B, R3 y R4 son como se describe en el presente documento, se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 6.

Los compuestos de tipo 11 disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica y en los que PG significa un grupo protector adecuado tal como, por ejemplo, un grupo protector Boc, Cbz o Bn, se pueden acilar con ácidos carboxílicos 16 para dar los intermedios 17 (etapa a). Los acoplamientos de amida de este tipo están ampliamente descritos en la literatura y se pueden lograr por el uso de reactivos de acoplamiento tales como CDI, DCC, HATU, HBTU, HOBT, TBTU, T3P o reactivo de Mukaiyama (por ejemplo, Angew. Chem., Int. Ed. Engl. 1979, 18, 707) en un disolvente adecuado tal como DMF, DMA, Dc M o dioxano, opcionalmente en presencia de una base, por ejemplo, NEt3, DIPEA (base de Huenig) o DMAP.
De forma alternativa, los ácidos carboxílicos 16 se pueden convertir en sus cloruros de ácido por tratamiento con, por ejemplo, cloruro de tionilo o cloruro de oxalilo, sin diluir u opcionalmente en un disolvente tal como DCM. La reacción del cloruro de ácido con los intermedios 11 en un disolvente apropiado tal como DCM o DMF y una base, por ejemplo, NEt3, base de Huenig, piridina, DMAP o bis(trimetilsilil)amida de litio a temperaturas que varían de 0 °C a la temperatura de reflujo del disolvente o mezcla de disolventes, proporciona los productos intermedios 17 (etapa a).
La retirada del grupo protector de los intermedios 17, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios C (etapa b).
Los compuestos de tipo 11 se pueden sulfonilar, por ejemplo, por tratamiento con cloruros de sulfonilo 18 (disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica o descritos en la literatura) usando una base y un disolvente adecuados tales como NEt3 o piridina en DCM para proporcionar los intermedios 19 (etapa c).
La retirada del grupo protector de los intermedios 19, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios D (etapa d).
Los compuestos de tipo 11 se pueden convertir en los carbamatos 20 correspondientes, por ejemplo, haciéndose reaccionar primero 11 con un reactivo de activación y carbonilación tal como carbonato de bis(triclorometilo) usando una base y un disolvente adecuados tales como, por ejemplo, bicarbonato de sodio en DCM, seguido de la reacción del cloruro de carbamoílo formado de forma intermedia con alcoholes de tipo 8 en presencia de una base adecuada tal como piridina o NEt3, opcionalmente a temperaturas elevadas (etapa e). Otros agentes activadores
incluyen, pero no se limitan a, fosgeno, cloroformiato de triclorometilo, carbonato de 4-nitrofenilo o 1,1'-carbonildiimidazol. La síntesis de carbamatos es bien conocida en la técnica y se describe ampliamente en la literatura (por ejemplo, J. Med. Chem. 2015, 58(7), 2895).
La retirada del grupo protector de los intermedios 20, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios E (etapa f).
En algunos modos de realización, los intermedios 2 son intermedios de tipo F, G y H. Los intermedios de tipo F, G y H en los que A, B, R3 y R4 son como se describe en el presente documento y R5 es hidrógeno; alquilo C1-6, halo-alquilo C1-6, se pueden preparar por procedimientos conocidos en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 7.

Los intermedios 21, disponibles comercialmente o bien preparados por procedimientos de la literatura, se pueden convertir en los intermedios 22, por ejemplo, por aminación reductora usando los aldehídos 12 y aplicando las condiciones descritas en el esquema 5, etapa a (etapa a).
La retirada del grupo protector de los intermedios 22, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios F (etapa b).
Los intermedios 21 se pueden hacer reaccionar con los ácidos carboxílicos 16 usando, por ejemplo, las condiciones descritas en el esquema 6, etapa a, para proporcionar los intermedios 23 (etapa c).
La retirada del grupo protector de los intermedios 23, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios G (etapa d).
Los intermedios 21 se pueden sulfonilar, por ejemplo, por tratamiento con los cloruros de sulfonilo 18 (disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica o descritos en la literatura) usando, por ejemplo, las condiciones de reacción descritas en el esquema 6, etapa c, para proporcionar los productos intermedios 24 (etapa e).
La retirada del grupo protector de los intermedios 24, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios H (etapa f).
En algunos modos de realización, los intermedios 2 son intermedios de tipo J, K y L. Los intermedios de tipo J, K y L en los que A, B, R3 y R4 son como se describe en el presente documento, R5 es hidrógeno, alquilo C1-6 o halo-alquilo C1-6 y R19 es hidrógeno o alquilo C1-6 se pueden preparar por procedimientos conocidos en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 8.

Los intermedios 22 se pueden convertir en los intermedios 27 por ejemplo, por aminación reductora usando los aldehídos 26 y aplicando las condiciones descritas en el esquema 5, etapa a (etapa a).
De forma alternativa, los intermedios 22 se pueden alquilar con compuestos 25 de tipo R20LG en los que LG es un grupo saliente adecuado tal como cloro, bromo, yodo, metanosulfonato, trifluorometanosulfonato o p-toluenosulfonato usando, por ejemplo, las condiciones descritas en el esquema 4, etapa c, para proporcionar los intermedios 27 (etapa a).
La retirada del grupo protector de los intermedios 27, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios J (etapa b).
Los intermedios 22 se pueden alquilar con compuestos 25 de tipo R20LG en los que LG es un grupo saliente adecuado tal como cloro, bromo, yodo, metanosulfonato, trifluorometanosulfonato o p-toluenosulfonato usando, por ejemplo, las condiciones descritas en el esquema 4, etapa c, para proporcionar los intermedios 28 (etapa c). La retirada del grupo protector de los intermedios 28, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios K (etapa d).
Los intermedios 24 se pueden alquilar con compuestos 25 de tipo R20LG en los que LG es un grupo saliente adecuado tal como cloro, bromo, yodo, metanosulfonato, trifluorometanosulfonato o p-toluenosulfonato usando, por ejemplo, las condiciones descritas en el esquema 4, etapa c, para proporcionar los intermedios 29 (etapa e). La retirada del grupo protector de los intermedios 29, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios L (etapa d).
En algunos modos de realización, los intermedios 2 son intermedios de tipo M. Los intermedios de tipo M en los que A, B, R3 y R4 son como se describe en el presente documento y R5 es hidrógeno; alquilo C1-6 o halo-alquilo C1-6 se pueden preparar por procedimientos bien conocidos en la técnica y como se ejemplifica por los procedimientos de síntesis generales explicados en el esquema 9.
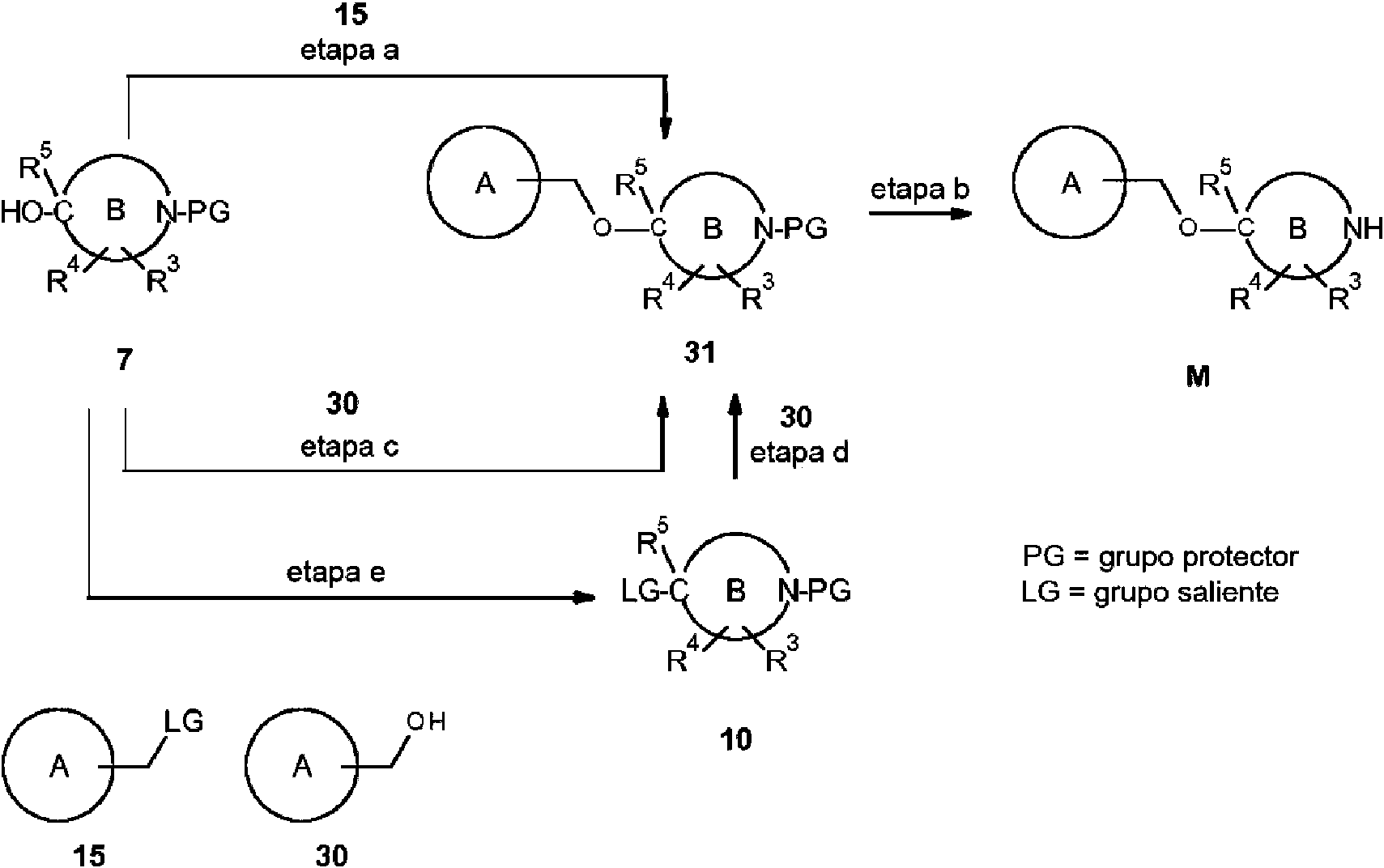
Esquema 9
Los compuestos espirocíclicos 7 en los que PG es un grupo protector adecuado se pueden alquilar con compuestos 15 en los que LG es un grupo saliente adecuado tal como cloro, bromo, yodo, metanosulfonato, trifluorometanosulfonato o p-toluenosulfonato (preparados por procedimientos de la literatura, por ejemplo, a partir de compuestos 30) usando, por ejemplo, las condiciones descritas en el esquema 4, etapa c, para proporcionar los intermedios 31 (etapa a).
La retirada del grupo protector de los intermedios 31, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios M (etapa b).
De forma alternativa, los intermedios 31 se pueden preparar a partir de los intermedios 7 y los compuestos 30 por medio de la reacción de Mitsunobu, aplicando, por ejemplo, las condiciones descritas en el esquema 4, etapa a (etapa c).
Además, los intermedios 31 también se pueden preparar por alquilación de los compuestos 7 con compuestos 10 y usando, por ejemplo, las condiciones descritas en el esquema 4, etapa c (etapa d).
Los intermedios 10, a su vez, se pueden sintetizar a partir de los compuestos 7 convirtiendo la función hidroxi en un grupo saliente adecuado tal como un haluro de alquilo (por ejemplo, bromo usando PBr3, cloro a través del uso de SOCh) o sulfonato de alquilo o arilo tal como metanosulfonato (usando cloruro de mesilo) o p-toluenosulfonato (usando cloruro de tosilo). Las reacciones de ese tipo se describen ampliamente en la literatura y son bien conocidas en la técnica.
En algunos modos de realización, los intermedios 2 son intermedios de tipo N. Los intermedios de tipo N en los que A, B, R3, R4 y R5 son como se describe en el presente documento, se pueden preparar por procedimientos bien conocidos en la técnica y como se ejemplifica por los procedimientos de síntesis generales explicados en el esquema 10.

Los compuestos de tipo 32, disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica, se pueden someter a una reacción de Mitsunobu con compuestos 8 , aplicando, por ejemplo, las condiciones descritas en el esquema 4, etapa a, para proporcionar los intermedios 34 (etapa a).
La retirada del grupo protector de los intermedios 34, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios N (etapa b).
De forma alternativa, los intermedios 34 se pueden preparar por alquilación de los compuestos 32 con compuestos 33 en los que LG es un grupo saliente adecuado aplicando las condiciones explicadas, por ejemplo, en el esquema 4, etapa c (etapa c).
Los intermedios N, de forma alternativa, se pueden preparar como se ejemplifica por los procedimientos de síntesis generales explicados en el esquema 11.

Esquema 11
Los compuestos 8 se pueden someter a una reacción de Mitsunobu con compuestos 32 usando las condiciones descritas en el esquema 4, etapa a, para proporcionar los intermedios 34 (etapa a).
La retirada del grupo protector de los intermedios 34, aplicando procedimientos conocidos en la técnica y, por ejemplo, descritos en el esquema 4, etapa b, proporciona los intermedios N (etapa b).
De forma alternativa, los compuestos 8 se pueden alquilar con compuestos 35 en los que LG significa un grupo saliente adecuado tal como cloro, bromo, yodo, metanosulfonato, trifluorometanosulfonato o p-toluenosulfonato, usando, por ejemplo, las condiciones descritas en el esquema 4, etapa c, para proporcionar los intermedios 34 (etapa c).
En algunos modos de realización, los intermedios 2 son intermedios de tipo P. Los intermedios de tipo P en los que A, B, R3 y R4 son como se describe en el presente documento y R5 es hidrógeno, alquilo C1-6 o halo-alquilo C1-6, se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 12.

Esquema 12
Los compuestos espirocíclicos 38 en los que PG significa un grupo protector adecuado tal como un grupo protector Boc, Cbz o Bn (disponible comercialmente o bien preparado como se describe en la literatura, por ejemplo, en Eur. J. Org. Chem. 2017, 36, 5316; Topics in Het. Chem. 2014, 35, 189; World Journal of Pharmacy and Pharmaceutical Sciences 2014, 3(12), 536; Chem. Rev. 2014, 114(16), 8257-8322) se pueden preparar por alquilación de tiol 37 con derivados espirocíclicos 36 (disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica) en los que LG significa un grupo saliente adecuado tal como cloro, bromo, yodo, OSO2alquilo (por ejemplo, mesilato (metanosulfonato)), OSO2fluoroalquilo (por ejemplo, triflato (trifluorometanosulfonato)) u OSO2arilo (por ejemplo, tosilato (p-toluenosulfonato)) usando una base adecuada y un disolvente apropiado (por ejemplo, K2CO3 en DMF) a temperaturas entre 0 °C y la temperatura de ebullición del disolvente (etapa a).
Los intermedios 38 se pueden oxidar a intermedios 39, usando un reactivo oxidante adecuado, tal como mCPBA, en un disolvente apropiado (por ejemplo, en DCM) a temperaturas entre 0 °C y la temperatura de ebullición del disolvente (etapa b).
La retirada del grupo protector de los intermedios 39, aplicando procedimientos conocidos en la técnica, por ejemplo, un grupo Boc usando TFA en DCM o HCl 4 M en dioxano a temperaturas entre 0 °C y temperatura ambiente, un grupo Bn o Cbz usando hidrógeno en presencia de un catalizador adecuado tal como Pd o Pd(OH)2 sobre carbón activado en un disolvente adecuado tal como MeOH, EtOH, EtOAc o mezclas de los mismos y como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wuts, 4.a ed., 2006, Wiley N.Y.), proporciona los intermedios P (etapa c).
En algunos modos de realización, los intermedios 2 son intermedios de tipo Q. Los intermedios de tipo Q en los que A, B, R3 y R4 son como se describe en el presente documento y R5 es hidrógeno, alquilo C1-6 o halo-alquilo C1-6, se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 13.

Esquema 13
De forma alternativa, los compuestos 40, funcionalizados con un bromuro, se pueden someter a un acoplamiento electrofílico cruzado con bromuros de arilo o heteroarilo 41 bajo irradiación con una lámpara de luz azul de 420 nm usando un fotocatalizador apropiado tal como [Ir{dF(CF3)ppy}2(dtbpy)]PF6 hexafluorofosfato de ([4,4'-bis(1, 1-dimetiletil)-2,2'-bipiridina-N1, N1 ']bis[3,5-difIuoro-2-[5-(trifluorometil)-2-piridinil-N]fenM-C]iridio(III)), un catalizador de níquel como NiCh glyme (dicloro(dimetoxietano)níquel), 4,4'-di-terc-butil-2,2'-dipiridilo y tris(trimetilsilil)silano, en presencia de una base adecuada tal como carbonato de sodio anhidro en un disolvente como DME. Las reacciones de este tipo se describen en la literatura, por ejemplo, J. Am. Chem. Soc. 2016, 138, 8084. (etapa a).
La retirada del grupo protector de los intermedios 42 aplicando procedimientos bien conocidos en la técnica y como se describe, por ejemplo, en el esquema 12, etapa c, proporciona los intermedios Q (etapa b).
En algunos modos de realización, los intermedios 2 son intermedios de tipo R y S . Los intermedios de tipo R y S en los que A, B, R3 y R4 son como se describe en el presente documento y R5 es hidrógeno, se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 14.

Esquema 14
Las cetonas 43, disponibles comercialmente o preparadas por procedimientos conocidos en la técnica, se pueden someter, por ejemplo, a una reacción de Wittig con alquiliden trifenilfosforanos de tipo 44a en un disolvente adecuado tal como, por ejemplo, THF, metil-THF o DMSO para dar los intermedios 45 (etapa a). Los fosforanos 44a se pueden formar tratando las sales de fosfonio correspondientes con una base adecuada tal como BuLi, NaH o KOtBu en un disolvente adecuado tal como THF, dioxano o metil-THF y se pueden aislar o usar in situ. Las sales de fosfonio, a su vez, están fácilmente disponibles a partir de un haluro de alquilo sustituido con arilo/heteroarilo/heterocíclico (siendo el haluro Cl, Br y yodo) y trifenilfosfina en un disolvente adecuado tal como tolueno. Se puede aplicar calentamiento para acelerar la reacción o conducir la reacción hasta su finalización (por
ejemplo, H.J. Cristau, F. Plénat en PATAI'S Chemistry of Functional Groups, Editor(es): Frank R. Hartley, 7 de agosto de 2006, Editor(es) de la serie: Prof. Saul Patai).
De forma alternativa, los intermedios 45 se pueden obtener usando una reacción de Horner-Wadsworth-Emmons (HWE) usando cetonas 43 y fosfonatos 44b, en los que Ra es alquilo, por ejemplo, metilo o etilo. Los fosfonatos 44b se a-metalizan in situ usando una base y un disolvente adecuados tales como NaH, nBuLi o KOtBu en THF (etapa a). Los fosfonatos 44b se preparan fácilmente usando, por ejemplo, la reacción de Arbuzov por alquilación de un haluro de arilo/heteroarilo/heterocíclico (siendo el haluro Cl, Br y yodo) con fosfito de trialquilo disponible comercialmente (por ejemplo, Chem. Rev. 1984, 84, 577).
Las reacciones de olefinación de ambos tipos se describen ampliamente en la literatura (por ejemplo, Current Org. Chem. 2015, 19(9), página 744; Chem. Rev. 1989, 89(4), 863; Org. React. 1977, 25, 73; Liebigs Ann./Recueil 1997, 1283; Acc. Chem. Res. 1983,16, 411).
La reducción del doble enlace en los intermedios 45 usando, por ejemplo, hidrógeno en presencia de un catalizador adecuado tal como paladio sobre carbón activado en un disolvente apropiado o una mezcla de disolventes tal como EtOAc, MeOH o AcOH proporciona los compuestos 46 (etapa b).
La retirada del grupo protector de los intermedios 45 aplicando procedimientos conocidos en la técnica (por ejemplo, un grupo Boc usando TFA en DCM o HCl 4 M en dioxano a temperaturas entre 0 °C y temperatura ambiente, un grupo Cbz usando hidrógeno en presencia de un catalizador adecuado tal como Pd o Pd(OH)2 sobre carbón activado en un disolvente adecuado tal como MeOH, EtOH, EtOAc o mezclas de los mismos y como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wuts, 4.a ed., 2006, Wiley N.Y.), proporciona los intermedios R (etapa c).
Si metoxi está entre los sustituyentes de A (R7, R8 o R9), la desmetilación puede dar lugar a intermedios sustituidos con hidroxi S . Esto requiere un reactivo tal como BBr3 en un disolvente adecuado tal como DCM, a temperaturas entre 0 °C y la temperatura de ebullición del disolvente. Un grupo protector Boc también se escindirá en las condiciones de reacción, dando lugar directamente a los intermedios S (etapa d).
En algunos modos de realización, los intermedios 2 son intermedios de tipo T . Los intermedios de tipo T en los que A, B, R3 y R4 son como se describe en el presente documento y X es nitrógeno, se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por el procedimiento de síntesis general explicado en el esquema 15.

Esquema 15
El tratamiento de las aminas 47 con un bromo-arilo o bromo-heteroarilo 41 en condiciones típicas de una reacción de Buchwald-Hartwig, u otros acoplamientos cruzados C-N organometálicos conocidos en la técnica, da lugar a los intermedios 48. Esto típicamente requiere un sistema de catalizadores adecuado, por ejemplo, PdCl2(DPPF)-CH2Cl2, Pd2(dba)3 + Xantphos, cataCXium A Pd G2, RuPhos Pd G2, una base orgánica o inorgánica tal como carbonato de cesio o terc-butóxido de sodio en un disolvente tal como dioxano o tolueno. Las reacciones se llevan a cabo típicamente a temperaturas elevadas entre 70 y 120 °C bajo atmósfera inerte (etapa a).
La retirada del grupo protector de los intermedios 48 aplicando procedimientos bien conocidos en la técnica y como se describe, por ejemplo, en el esquema 12, etapa c, proporciona los intermedios T (etapa b).
En un aspecto, la presente invención proporciona un procedimiento de fabricación de los compuestos de urea de fórmula (I) descritos en el presente documento y sales farmacéuticamente aceptables de los mismos, que comprende:
(c) hacer reaccionar una primera amina de fórmula 1, en la que R1 y R2 son como se describe en el presente

con una segunda amina 2 , en la que A, B, L, X, R3 y R4 son como se describe en el presente documento

en presencia de una base y un reactivo formador de urea, para formar dicho compuesto de fórmula (I); y opcionalmente
(d) transformar dicho compuesto de fórmula (I) en una sal farmacéuticamente aceptable de los mismos.
En un modo de realización, se proporciona un procedimiento de acuerdo con la invención, en el que dicha base es bicarbonato de sodio.
En un modo de realización, se proporciona un procedimiento de acuerdo con la invención, en el que dicho reactivo formador de urea se selecciona de carbonato de bis(triclorometilo), fosgeno, cloroformiato de triclorometilo, carbonato de 4-nitrofenilo y 1,1-carbonildiimidazol, preferentemente en el que dicho reactivo formador de urea es carbonato de bis(triclorometilo).
En un aspecto, la presente invención proporciona un compuesto de fórmula (I) como se describe en el presente documento, cuando se fabrica de acuerdo con uno cualquiera de los procedimientos descritos en el presente documento.
Actividad inhibidora de MAGL
Los compuestos de la presente invención son inhibidores de MAGL. Por tanto, en un aspecto, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para inhibir MAGL en un mamífero.
En otro aspecto, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en un procedimiento de inhibición de MAGL en un mamífero.
En otro aspecto, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para inhibir MAGL en un mamífero.
En otro aspecto, la presente invención proporciona un procedimiento para inhibir MAGL en un mamífero, comprendiendo dicho procedimiento administrar al mamífero una cantidad eficaz de un compuesto de fórmula (I) como se describe en el presente documento.
Los compuestos se perfilaron para obtener la actividad inhibidora de MAGL midiendo la actividad enzimática de MAGL siguiendo la hidrólisis de acetato de 4-nitrofenilo que da como resultado 4-nitrofenol, que absorbe a 405-412 nm (G.G. Muccioli, G. Labar, D.M. Lambert, Chem. Bio. Chem. 2008, 9, 2704-2710). Este ensayo se abrevia a continuación en el presente documento como "ensayo 4-NPA".
El ensayo 4-NPA se llevó a cabo en placas de ensayo de 384 pocillos (negras con fondo transparente, superficie de
no unión tratada, ref. de Corning 3655) en un volumen total de 40 μl. Las diluciones del compuesto se realizaron en DMSO al 100 % (VWR Chemicals 23500.297) en una placa de polipropileno en etapas de dilución de 3 veces para dar un intervalo de concentración final en el ensayo de 25 μM a 1,7 nM. Se añadieron diluciones del compuesto de 1 μl (DMSO al 100 %) a 19 μl de MAGL (natural recombinante) en tampón de ensayo (TRIS 50 mM (GIBCO, 15567-027), EDTA 1 mM (Fluka, 03690-100 ml)). Se agitó la placa durante 1 min a 2000 rpm (Variomag Teleshake) y a continuación se incubó durante 15 min a TA. Para iniciar la reacción, se añadieron 20 μl de acetato de 4-nitrofenilo (Sigma N-8130) en tampón de ensayo con EtOH al 6 %. Las concentraciones finales en el ensayo fueron 1 nM de MAGL y 300 μM de acetato de 4-nitrofenilo. Después de agitar (1 min, 2000 rpm) y 5 min de incubación a TA, se midió la absorbancia a 405 nm por primera vez (Molecular Devices, SpectraMax Paradigm). A continuación se realizó una segunda medición después de la incubación durante 80 min a TA. A partir de las dos mediciones, se calculó la pendiente restando la primera de la segunda medición.
De forma alternativa, los compuestos se perfilaron para obtener la actividad inhibidora de MAGL determinando la actividad enzimática siguiendo la hidrólisis del sustrato natural 2-araquidonoilglicerol que da como resultado ácido araquidónico, que se puede seguir por espectrometría de masas. Este ensayo se abrevia a continuación en el presente documento como "ensayo 2-AG".
El ensayo 2-AG se llevó a cabo en placas de ensayo de 384 pocillos (PP, cat. de Greiner n.° 784201) en un volumen total de 20 μl. Las diluciones del compuesto se realizaron en DMSO al 100 % (VWR Chemicals 23500.297) en una placa de polipropileno en etapas de dilución de 3 veces para dar un intervalo de concentración final en el ensayo de 12,5 μM a 0,8 μM. Se añadieron diluciones del compuesto de 0,25 μl (DMSO al 100 %) a 9 pl de MAGL en tampón de ensayo (TRIS 50 mM (GIBCO, 15567-027), Ed Ta 1 mM (Fluka, 03690-100 ml), Tween al 0,01 % (v/v). Después de agitar, se incubó la placa durante 15 min a TA. Para iniciar la reacción, se añadieron 10 μl de 2-araquidonoilglicerol en tampón de ensayo. Las concentraciones finales en el ensayo fueron 50 μM de MAGL y 8 μM de 2-araquidonoilglierol. Después de agitar y 30 min de incubación a TA, se detuvo la reacción por la adición de 40 μl de acetonitrilo que contenía 4 μM de ácido d8-araquidónico. Se rastreó la cantidad de ácido araquidónico por un sistema SPE en línea (Agilent Rapidfire) acoplado a un espectrómetro de masas de triple cuadrupolo (Agilent 6460). Se usó un cartucho SPE C18 (G9205A) en una configuración líquida de acetonitrilo/agua. El espectrómetro de masas se hizo funcionar en modo de electronebulización negativa siguiendo las transiciones de masas 303,1 ^ 259,1 para ácido araquidónico y 311,1 ^ 267,0 para ácido d8-araquidónico. Se calculó la actividad de los compuestos en base a la proporción de intensidades [ácido araquidónico/ácido d8-araquidónico].
Tabla 1



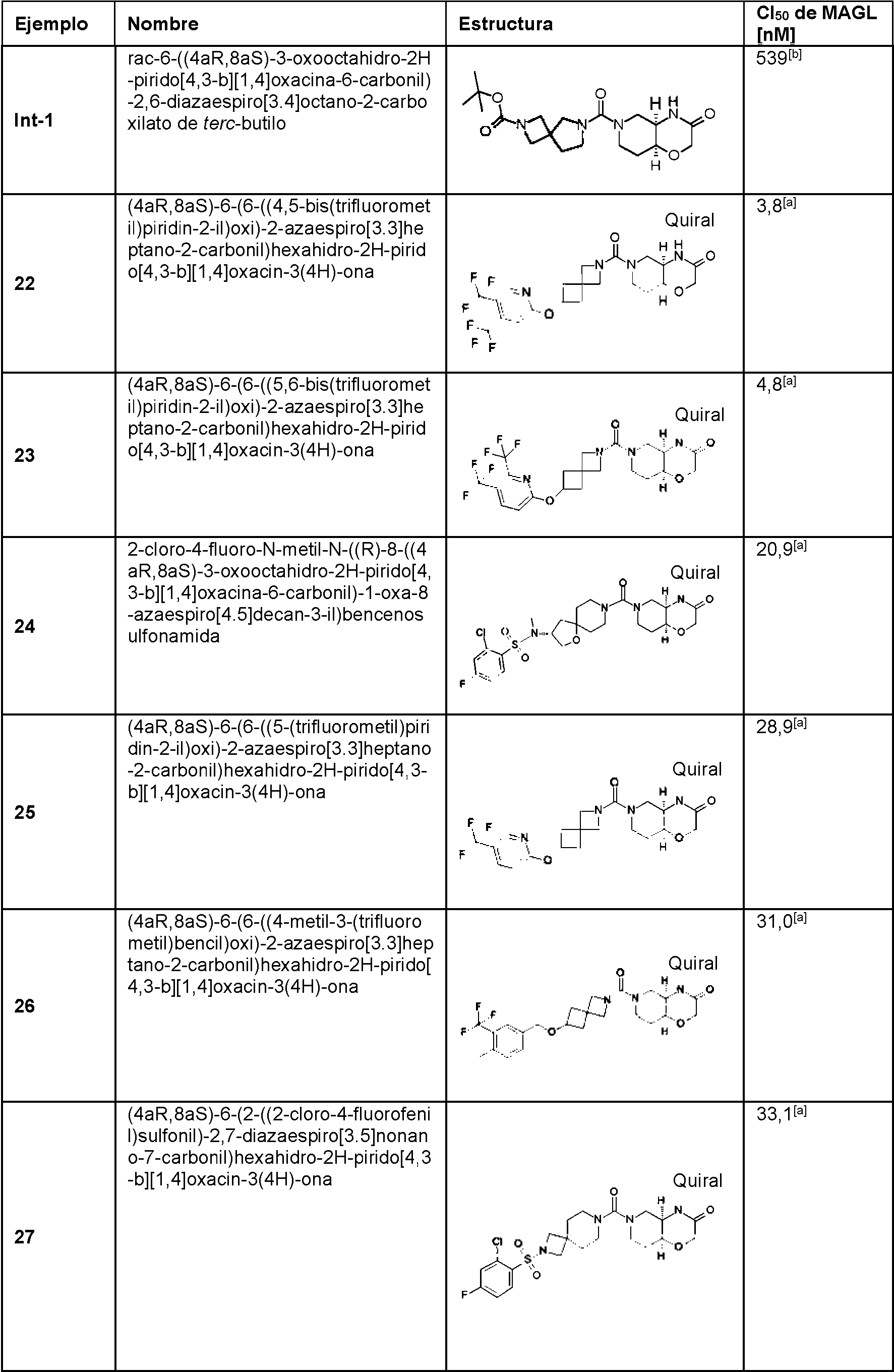









[a]: medida en el ensayo 2-AG; [b]: medida en el ensayo 4-NPA; n/d: no disponible.
En un aspecto, la presente invención proporciona compuestos de fórmula (I) y sus sales farmacéuticamente aceptables como se describe en el presente documento, en los que dichos compuestos de fórmula (I) y sus sales farmacéuticamente aceptables tienen CI50 para la inhibición de MAGL por debajo de 25 |j M, preferentemente por debajo de 10 j M, más preferentemente por debajo de 5 j M como se mide en los ensayos de Ma GL descritos en el presente documento.
En un modo de realización, los compuestos de fórmula (I) y sus sales farmacéuticamente aceptables como se describe en el presente documento tienen valores de CI50 (inhibición de MAGL) entre 0,000001 j M y 25 j M, los compuestos particulares tienen valores de CI50 entre 0,00005 j M y 10 j M, otros compuestos particulares tienen valores de CI50 entre 0,00005 j M y 5 j M, como se mide en los ensayos de MAGL descritos en el presente documento.
En un modo de realización, la presente invención proporciona compuestos de fórmula (I) y sus sales farmacéuticamente aceptables como se describe en el presente documento, en los que dichos compuestos de fórmula (I) y sus sales farmacéuticamente aceptables tienen un CI50 para MAGL por debajo de 25 j M, preferentemente por debajo de 10 j M, más preferentemente por debajo de 5 j M como se mide en un ensayo que comprende las etapas de:
a) proporcionar una solución de un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo, en d Ms O;
b) proporcionar una solución de MAGL (natural recombinante) en tampón de ensayo (tris(hidroximetil)aminometano 50 mM; ácido etilendiaminatetraacético 1 mM);
c) añadir 1 j l de solución del compuesto de la etapa a) a 19 j l de solución de MAGL de la etapa b);
d) agitar la mezcla durante 1 min a 2000 rpm;
e) incubar durante 15 min a TA;
f) añadir 20 j l de una solución de acetato de 4-nitrofenilo en tampón de ensayo (tris(hidroximetil)aminometano 50 mM; ácido etilendiaminatetraacético 1 mM, EtOH al 6 %);
g) agitar la mezcla durante 1 min a 2000 rpm;
h) incubar durante 5 min a TA;
i) medir la absorbancia de la mezcla a 405 nm por primera vez;
j) incubar durante otros 80 min a TA;
k) medir la absorbancia de la mezcla a 405 nm por segunda vez;
l) restar la absorbancia medida en i) de la absorbancia medida en k) y calcular la pendiente de absorbancia; en los que:
i) la concentración del compuesto de fórmula (I), o la sal farmacéuticamente aceptable del mismo en el ensayo después de la etapa f) está en el intervalo de 25 |jM a 1,7 nM;
ii) la concentración de MAGL en el ensayo después de la etapa f) es de 1 nM;
iii) la concentración de acetato de 4-nitrofenilo en el ensayo después de la etapa f) es de 300 j M; y iv) las etapas de a) a 1) se repiten al menos 3 veces, cada vez con una concentración diferente del compuesto de fórmula (I), o la sal farmacéuticamente aceptable del mismo.
Uso de los compuestos de la invención
En un aspecto, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso como sustancia terapéuticamente activa.
En un aspecto, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En un modo de realización, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de neuroinflamación y/o enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de cáncer en un mamífero.
En un modo de realización, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de enfermedades neurodegenerativas en un mamífero.
En un aspecto, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
En un modo de realización preferente, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer y/o enfermedad de Parkinson en un mamífero.
En un modo de realización en particular preferente, la presente invención proporciona compuestos de fórmula (I) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de esclerosis múltiple en un mamífero.
En un aspecto, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero. En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de neuroinflamación y/o enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de cáncer en un mamífero.
En otro aspecto, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
En un modo de realización preferente, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer y/o enfermedad de Parkinson en un mamífero.
En un modo de realización en particular preferente, la presente invención proporciona el uso de compuestos de fórmula (I) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de esclerosis múltiple en un mamífero.
Composiciones farmacéuticas y administración
En un aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto de fórmula (I) como se describe en el presente documento y un vehículo terapéuticamente inerte.
Los compuestos de fórmula (I) y sus sales farmacéuticamente aceptables se pueden usar como medicamentos (por ejemplo en forma de preparaciones farmacéuticas). Las preparaciones farmacéuticas se pueden administrar por vía interna, tal como por vía oral (por ejemplo, en forma de comprimidos, comprimidos recubiertos, grageas, cápsulas de gelatina dura y blanda, soluciones, emulsiones o suspensiones), por vía nasal (por ejemplo, en forma de pulverizadores nasales) o por vía rectal (por ejemplo, en forma de supositorios). Sin embargo, la administración también se puede efectuar por vía parenteral, tal como por vía intramuscular o intravenosa (por ejemplo, en forma de soluciones inyectables).
Los compuestos de fórmula (I) y sus sales farmacéuticamente aceptables se pueden procesar con adyuvantes inorgánicos u orgánicos farmacéuticamente inertes para la producción de comprimidos, comprimidos recubiertos, grageas y cápsulas de gelatina dura. Se pueden usar lactosa, almidón de maíz o derivados del mismo, talco, ácido esteárico o sus sales, etc., por ejemplo, como dichos adyuvantes para comprimidos, grageas y cápsulas de gelatina dura.
Los adyuvantes adecuados para cápsulas de gelatina blanda son, por ejemplo, aceites vegetales, ceras, grasas, sustancias semisólidas y polioles líquidos, etc.
Los adyuvantes adecuados para la producción de soluciones y jarabes son, por ejemplo, agua, polioles, sacarosa, azúcar invertido, glucosa, etc.
Los adyuvantes adecuados para soluciones inyectables son, por ejemplo, agua, alcoholes, polioles, glicerol, aceites vegetales, etc.
Los adyuvantes adecuados para supositorios son, por ejemplo, aceites naturales o hidrogenados, ceras, grasas, polioles semisólidos o líquidos, etc.
Además, las preparaciones farmacéuticas pueden contener conservantes, solubilizantes, sustancias que incrementan la viscosidad, estabilizantes, agentes humectantes, emulsionantes, edulcorantes, colorantes, saborizantes, sales para variar la presión osmótica, tampones, agentes de enmascaramiento o antioxidantes. También pueden contener todavía otras sustancias terapéuticamente valiosas.
La dosificación puede variar en límites amplios y, por supuesto, se ajustará a los requisitos individuales en cada caso particular. En general, en el caso de administración oral, debería ser apropiada una dosificación diaria de aproximadamente 0,1 mg a 20 mg por kg de peso corporal, preferentemente de aproximadamente 0,5 mg a 4 mg por kg de peso corporal (por ejemplo, aproximadamente 300 mg por persona), dividida en preferentemente 1-3 dosis individuales, que pueden consistir, por ejemplo, en las mismas cantidades. Quedará claro, sin embargo, que el límite superior dado en el presente documento se puede exceder cuando se demuestre que está indicado. Ejemplos
La invención se entenderá más completamente por referencia a los siguientes ejemplos. Sin embargo, las
reivindicaciones no se deben interpretar como limitadas al alcance de los ejemplos.
En caso de que los ejemplos preparativos se obtengan como una mezcla de enantiómeros, los enantiómeros puros se pueden separar por procedimientos descritos en el presente documento o por procedimientos conocidos para el experto en la técnica, tales como, por ejemplo, cromatografía quiral (por ejemplo, SFC quiral) o cristalización. Todos los ejemplos de reacción y los intermedios se prepararon bajo una atmósfera de argón si no se especifica de otro modo.
Procedimiento A1
Ejemplo 4
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometoxi)fenoxi)-2-azaespiro[3.3]heptano-2-carbonM)hexahidro-2H-pirido[ 4,3-b][1,4]oxacin-3(4H)-ona
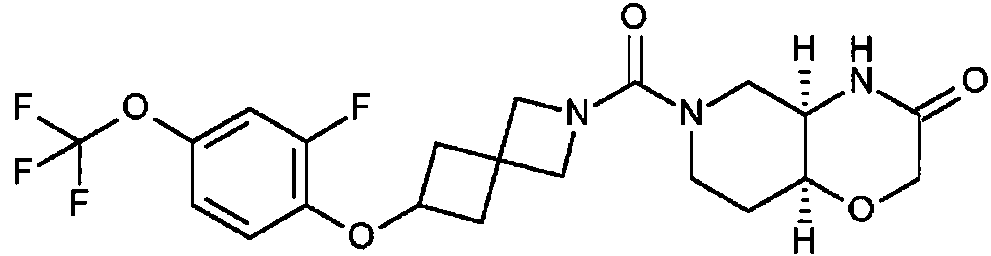
A una solución de BB9 (143,7 mg, 284 |jmol) en ACN (1,42 ml) se le añadió DIPEA (110 mg, 149 jl, 851 |jmol) y (4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (91,1 mg, 284 jmol, BB2a). Se agitó el vial de reacción a 80 °C durante 4 h. Se purificó el material bruto por HPLC de fase inversa para proporcionar el compuesto del título (66,8 mg, 134 jmol, 47,3 %) como un sólido blanco. EM (ESI): m/z = 474,3 [M+H]+.
Procedimiento A2
Ejemplo 1
(4aR,8aS)-6-(6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o xacin-3(4H)-ona

A una solución de trifluoroacetato de 6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano (1026 mg, 2,31 mmol, BB6) en ACN (11,5 ml) se le añadió DIPEA (895 mg, 1,21 ml, 6,92 mmol) y (4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (816 mg, 2,54 mmol, BB2a). Se agitó el vial de reacción a 80 °C durante 2,5 h. Se evaporó la solución. Se disolvió el residuo en solución de NaHCO3 acuosa saturada (30 ml) y EtOAc (30 ml) y se separaron las capas. Se lavó la capa orgánica una vez con solución de NaHCO3 acuosa saturada (30 ml). Se extrajeron las capas acuosas combinadas una vez con EtOAc (100 ml). Se lavaron las capas orgánicas una vez con solución de NaHCO3 acuosa saturada (100 ml) y salmuera, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 40 g usando un sistema de MPLC que se eluyó con un gradiente de n-heptano : EtOAc/EtOH 3/1 (de 70 : 30 a 0 : 100) y se evaporó para dar el compuesto del título como una espuma blanca (841,2 mg, 1,94 mmol, 84,3 %). EM (ESI): m/z = 424,4 [M+H]+.
Procedimiento A3
Ejemplo 18
rac-(4aR,8aS)-N-((R)-8-(3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]deca n-3-il)bencenosulfonamida

A una suspensión helada de carbonato de bis(tridorometilo) (45,3 mg, 153 |jmol) y NaHCO3 (73,3 mg, 873 |jmol) en DCM (1 ml) se le añadió 2,2,2-trifluoroacetato de (R)-N-(1-oxa-8-azaespiro[4.5]decan-3-il)bencenosulfonamida (89,6 mg, 218 jmol, preparado como se describe en el documento US20170029390) en una porción y se agitó la mezcla a TA durante la noche. Se enfrió en un baño de hielo y se añadieron diclorhidrato de rac-(4aR,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (50 mg, 218 jmol, BB1) y DIPEA (113 mg, 152 jl, 873 jmol). Se agitó la suspensión a TA durante 4 h. Se vertió la mezcla de reacción sobre agua y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el producto en una HPLC preparativa (columna Gemini NX) usando un gradiente de ACN : agua (que contenía ácido fórmico al 0,1 %) (de 2o : 80 a 98 : 2) para proporcionar el compuesto deseado como una goma incolora (0,025 g; 23,9 %). EM (ESI): m/z = 479,2 [M+H]+.
Procedimiento A4
Ejemplo 22
Se secó un matraz de fondo redondo con pistola de calor bajo HV, se rellenó con argón y se cargó con carbonato de bis(triclorometilo) (39,9 mg, 134 jmol) y bicarbonato de sodio (64,5 mg, 768 jmol). Se añadió DCM (2 ml) para dar una suspensión. Se añadió (4aR,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (30 mg, 192 jmol) a la suspensión a 0 °C. Se agitó la mezcla a 0 °C durante 5 min y a TA durante 20 horas. Se añadieron BB22 (84,6 mg, 192 jmol) y DIPEA (99,3 mg, 134 jl, 768 jmol). Se agitó la suspensión blanquecina resultante a TA durante 1 hora. Se vertió la mezcla de reacción en 5 ml de H2O y se extrajo con DCM (2x10 ml). Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 10 g, de 0 % a 10 % de MeOH en DCM). Se combinaron las fracciones y se evaporó para proporcionar el producto como espuma blanca, 69 mg, 70 % de rendimiento.
Procedimiento A5
Separación quiral de estereoisómeros con columna NR
Se separaron los dos estereoisómeros de rac-(4aR,8aS)-6-(3-((2-cloro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespi ro[4.5]decano-8-carbonil)hexahidro -2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (ejemplos 33 y 34) por HPLC quiral preparativa (columna NR quiral Reprosil) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAc) : n-heptano (40 : 60). Estereoquímica absoluta de los isómeros no determinada.
Procedimiento A6
Separación quiral de estereoisómeros con columna OD
Se separaron los dos estereoisómeros de rac-(4aR,8aS)-6-(3-((2-cloro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pi rido[ 4,3-b][1,4]oxacin-3(4H)-ona (ejemplo 38) por HPLC quiral preparativa (columna OD quiral Reprosil) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAc) : n-heptano (40 : 60). Estereoquímica absoluta de los isómeros no determinada.
Procedimiento A7
HPLC preparativa: columna Gemini NX, 12 nm, 5 jm , de 100 x 30 mm, tiempo de desarrollo de 15 min, gradiente 25-45-60-100 % de ACN en agua HCOOH al 0,1 %
Procedimiento A8
HPLC preparativa: columna YMC-Triart C18, 12 nm, 5 jm , de 100 x 30 mm, tiempo de desarrollo de 11 min,
gradiente 30-50-60-100 % de ACN en agua HCOOH al 0,1 %
Procedimiento A9
HPLC preparativa: columna Gemini NX, 12 nm, 5 |jm, 100 x 30 mm, gradiente de ACN en agua TEA al 0,1 % Procedimiento A10
HPLC preparativa: columna YMC-Triart C18, 12 nm, 5 |jm, de 100 x 30 mm, tiempo de desarrollo de 11 min, gradiente 20-40-60-100 % de ACN en agua TEA al 0,1 %
Procedimiento A11
HPLC preparativa: columna YMC-Triart C18, 12 nm, 5 jm , de 100 x 30 mm, tiempo de desarrollo de 11 min, gradiente 15-35-50-100 % de ACN en agua HCOOH al 0,1 %
Los siguientes ejemplos se prepararon a partir de los componentes básicos y procedimientos correspondientes, como se explica en la tabla 2.
Tabla 2











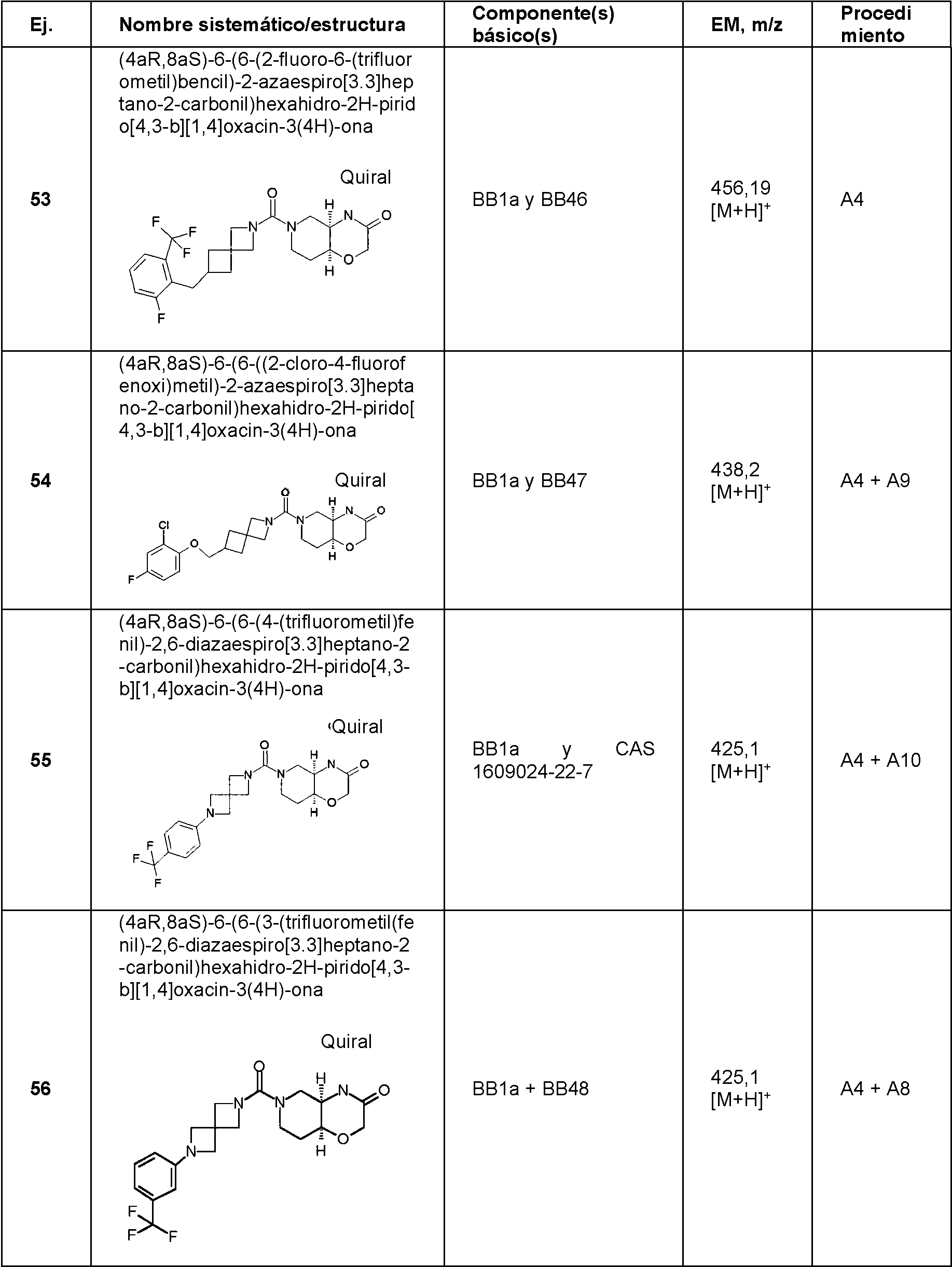





Ejemplo 47
(4aR,8aS)-6-(6-(4-(2-(trifluorometil)pirrolidin-1-il)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pir ido[4,3-b][1,4]oxacin-3(4H)-ona

A una suspensión de (4aR,8aS)-6-(6-(4-bromofenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-on a (0,050 g, 115 |jmol) y 2-(trifluorometil)pirrolidina (16 mg, 115 |jmol) en terc-butanol (1 ml) bajo argón se le añadieron XPhos (4,94 mg, 10,4 jmol, eq: 0,09), Pd2(dba)3 • c Hqb (3,57 mg, 3,45 jmol) y carbonato de cesio (150 mg, 460 jmol) y se calentó la mezcla en un microondas hasta 100 °C durante 30 min. Se filtró la mezcla, se evaporó el filtrado. Se purificó el producto por prep-HPLC (Gemini NX, 12 nm, 5 jm , 100 x 30 mm, gradiente de acetonitrilo/agua TEA al 0,1 %) proporcionando el producto deseado como 2,1 mg de un aceite amarillo. EM (ESI): m/z = 493,4 [M+H]+.
Etapa a) Se obtuvo trifluoroacetato de 6-(4-bromofenil)-2-azaespiro[3.3]heptano de forma análoga a BB41 a partir de 6-bromo-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (1 eq) y 1,4-dibromobenceno (2 eq). EM (ESI): m/z = 298,1 [M-56-H]+.
Etapa b) (4aR,8aS)-6-(6-(4-bromofenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-on a sintetizada de forma análoga al procedimiento general A4 a partir de trifluoroacetato de 6-(4-bromofenil)-2-azaespiro[3.3]heptano y BB1a, purificada por cromatografía ultrarrápida (gel de sílice, de 0 % a
10 % de MeOH en DCM). EM (ESI): m/z = 434,2 [M+H]+.
Ejemplo 48
(4aR,8aS)-6-(6-(4-(1-metiMH-pirazol-5-il)fenM)-2-azaespiro[3.3]heptano-2-carbonM)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
Se disolvieron (4aR,8aS)-6-(6-(4-bromofenil)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3( 4H)-ona (0,050 g, 115 jmol), ácido (1-metil-1H-pirazol-5-il)borónico (14,5 mg, 1 l5 |jmol), carbonato de potasio (79,5 mg, 576 jmol) y tetraquis(trifenilfosfina)paladio(0) (6,65 mg, 5,76 |jmol) en THF (1,5 ml)/agua (0,150 ml) bajo argón y se agitó a 80 °C durante 2 días. Se vertió la mezcla de reacción en 10 ml de H2O y se extrajo con EtOAc (2 x 20 ml). Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el material bruto por HPLC preparativa (YMC-Triart C18, 12 nm, 5 jm , 100 x 30 mm, 15 min de tiempo de desarrollo, gradiente 15-35-50-100 de ACN en agua HCOOH al 0,1 %). Se obtuvo el producto como un polvo blanco liofilizado (15,6 mg, 31 %). EM (ESI): m/z = 436,4 [M+H]+.
Síntesis de componentes básicos
BB1a y BB1b
(+)-(4aR,8aS)-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (BB1a)
y
(-)-(4aS,8aR)-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (BB1b)
Se separaron los enantiómeros de diclorhidrato de rac-(4aR,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (BB1, 500 mg, 2,18 mmol, ChemBridge Corporation) por HPLC quiral preparativa (columna NR quiral Reprosil) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAc) : n-heptano (30 : 70).
Primer enantiómero en eluir: (+)-cis-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (BB1a). Sólido amarillo (0,150 g; 44,0 %). EM (ESI): m/z = 157,1 [M+H]+.
Segundo enantiómero en eluir: (-)-cis-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona. (BB1b). Sólido amarillo (0,152 g; 44,6 %). EM (ESI): m/z = 157,1 [M+H]+.
BB2a y BB2b
(+)-(4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (BB2a)
y
(-)-(4aS,8aR)-3-oxohexahidro-2H-pirido [4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (BB2b)
A una suspensión de rac-(4aR,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; sal diclorhidrato (4,5 g, 19,6 mmol, BB1) en DCM seco (125 ml) a 0 °C se le añadió DiPEA (6,35 g, 8,58 ml, 49,1 mmol) seguido de cloroformiato de 4-nitrofenilo (4,35 g, 21,6 mmol). Se agitó la mezcla de reacción a 0 °C durante 10 min y a TA. durante 2 h. Se diluyó la reacción bruta con DCM y se transfirió a un embudo de separación para su extracción con solución de Na2CO3 ac. sat. Se recogió la fase orgánica y se extrajo de nuevo la fase acuosa con DCM. Se secaron las fases orgánicas combinadas sobre Na2SO4 y se evaporó hasta sequedad para proporcionar 6,62 g de un producto racémico bruto (BB2) como un sólido amarillo. Se sometió el material bruto directamente a separación por SFC quiral para proporcionar el enantiómero BB2b (2,72 g, segundo enantiómero en eluir) como un sólido amarillo
y el enantiómero BB2a (3,25 g, primer enantiómero en eluir) como un sólido beige claro pero contaminado con BB2b. Se llevó a cabo otra separación quiral por SFC para proporcionar 2,71 g de BB2a. EM (ESI): m/z = 322,2 [M+H]+ para ambos enantiómeros.
BB3
(2R,4aR,8aS)-2-metil-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacm-3-ona
A una solución de 6-bencil-2-metil-5,6,7,8-tetrahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (isómero A, 1,10 g, 4.26 mmol) en EtOAc (16 ml) y MeOH (16 ml) bajo argón se le añadió Pd-C (227 mg, 213 jmol) y se agitó la suspensión bajo atmósfera de hidrógeno (globo) a 1 bar durante 24 h. Se filtró la suspensión sobre un filtro de microvidrio y se lavó con 20 ml de EtOAc bajo gas inerte. Se evaporó el filtrado para dar BB4 como un sólido incoloro (715 mg). EM (ESI): m/z = 170,8 [M+H]+. Nota: solo se formó el enantiómero individual durante la reducción. Conformación relativa confirmada por RMN de protón.
Etapa a) 2-metil-4H-pirido[4,3-b][1,4]oxacin-3-ona
A una solución de 3-aminopiridin-4-ol (2,5 g, 22,7 mmol) en DMF (100 ml) se le añadió gota a gota cloruro de 2-cloropropanolo (3,03 g, 2,31 ml, 23,8 mmol) y se agitó la mezcla a TA durante 30 min. Después de la adición de K2CO3 (7,84 g, 56,8 mmol), se calentó la suspensión hasta 100 °C (baño de aceite) durante 20 h. Se retiró la DMF a vacío, a continuación se añadieron 100 ml de EtOAc y se agitó a TA durante 10 min, y se lavó con 50 ml de H2O, se extrajo 3 veces con EtOAc. Se combinaron las fases orgánicas, se secó con MgSO4 y se concentró a vacío para proporcionar 3,72 g de 2-metil-4H-pirido[4,3-b][1,4]oxacin-3-ona que se usó en la siguiente etapa sin purificación adicional.
Etapa b) Bromuro de 6-bencil-2-metil-3-oxo-3,4-dihidro-2H-pirído[4,3-b][1,4]oxadn-6-io
Se trató una suspensión de 2-metil-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (3,72 g, 22,7 mmol) en DCM (32 ml) y MeOH (8 ml) con (bromometil)benceno (4,65 g, 3,23 ml, 27,2 mmol) y se agitó la mezcla a TA durante 60 h. Se formó una suspensión, que se enfrió hasta 0 °C, se añadieron 20 ml de n-hexano y a continuación se filtró el precipitado. Se lavó el residuo con 15 ml de DCM/n-hexano fríos para proporcionar el compuesto como un sólido blanquecino (5,2 g). EM (ESI): m/z = 255 [M+H]+.
Etapa c) (rac)-6-bencil-2-metil-5,6,7,8-tetrahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
A una suspensión de bromuro de 6-bencil-2-metil-3-oxo-3,4-dihidro-2H-pirido[4,3-b][1,4]oxacin-6-io (5,2 g, 15,5 mmol) en EtOH (38 ml) se le añadió en porciones NaBH4 (763 mg, 20,2 mmol) (exotérmica, de 22 °C a 30 °C, suspensión amarilla). Después de que se atenuó la reacción exotérmica, se agitó la mezcla a temperatura ambiente durante 3 h, a continuación a 60 °C durante 1 h y a 22 °C durante 1 h. Se evaporó la mezcla de reacción, se repartió entre H2O y EtOAc y se separaron las capas. Se extrajo una vez la capa acuosa con EtOAc. Se lavaron las capas orgánicas dos veces con H2O, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 120 g usando un sistema de MPLC que se eluyó con un gradiente de n-heptano : EtOAc (de 50 a 100 en 30 min) para proporcionar el compuesto como un sólido amarillo claro (2,48 g) que se pudo usar en la siguiente etapa sin purificación adicional.
Etapa d) 6-bencil-2-metil-5,6,7,8-tetrahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
Se separaron los enantiómeros por HPLC quiral preparativa (columna Chiralcel OD) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAc) : n-heptano (10 : 90). Se evaporaron las fracciones para proporcionar los compuestos deseados como sólidos amarillo claro (1,17 g de isómero A, 1,10 g de isómero B).
BB4
rac-(4aS,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
Se disolvió rac-(4aS,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de bencilo (125 mg, 431 |jmol) en MeOH (5 ml). Se desgasificó la solución de reacción a vacío y se rellenó con argón. Se añadió Pd-C (20 mg, 188 jmol) bajo una atmósfera de argón. Se evacuó el argón de la mezcla de reacción y se rellenó con hidrógeno. Se agitó la mezcla de reacción a TA durante 15 h en atmósfera de hidrógeno. Se filtró la mezcla de reacción a través de un filtro de jeringa y se concentró a vacío para dar el producto deseado como un sólido incoloro (62 mg, 92,2 %). EM (ESI): m/z = 157,098 [M+H]+.
Etapa a) rac-(3S,4S)-3-(2-cloroacetamido)-4-hidroxipiperidina-1-carboxilato de bencilo
A una suspensión agitada de rac-(3S,4S)-3-amino-4-hidroxipiperidina-1-carboxilato de bencilo (317 mg, 1.27 mmol, sintetizado de acuerdo con la patente US 2011/59118 A1) y acetato de sodio (208 mg, 2,53 mmol, CAS
RN 127-09-3) en una mezcla de acetona (4 ml)/H2O (0,5 ml) se le añadió gota a gota una solución de cloruro de cloroacetilo (150 mg, 107 μl, 1,33 mmol, CAS RN 79-04-9) en acetona (3 ml) entre 0-5 °C. Después de la adición, se agitó la mezcla de reacción a TA durante 1 h y posteriormente se evaporó hasta sequedad dando una goma amarilla. Se purificó el producto bruto por cromatografía en gel de sílice para dar el producto deseado como un sólido amarillo (385 mg, 93 %). EM (ESI): m/z = 325,2 [M-H]-.
Etapa b) rac-(4aS,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de bencilo
A una solución agitada de rac-(3S,4S)-3-(2-cloroacetamido)-4-hidroxipiperidina-1-carboxilato de bencilo (385 mg, 1,18 mmol) en THF seco (4 ml) se le añadió NaH (67,9 mg, 1,7 mmol) a 0 °C. Se dejó que la mezcla alcanzara la TA y a continuación se agitó durante 90 min bajo una atmósfera de argón. Se añadió H2O (5 ml) y se continuó la agitación durante 10 min a TA. Se retiró el THF a vacío de la mezcla de reacción. Se trató el residuo con DCM y se lavó la fase orgánica con H2O y salmuera, se secó sobre Na2SO4, se filtró y a continuación se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (columna de fase inversa de 12 g, gradiente 0-100 % de ACN en H2O (que contenía FA al 0,1 %) para dar el producto deseado como un sólido incoloro (133 mg, 38,9 %). EM (ESI): m/z = 291,3 [M+H]+.
BB5
6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano; sal trifluoroacetato
A una solución de 6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (1,5065 g, 4,41 mmol) en DCM (22 ml) se le añadió TFA (4,02 g, 2,72 ml, 35,3 mmol) y se agitó la reacción a TA durante 3,5 h. Se concentró la mezcla de reacción para dar el compuesto del título como un aceite amarillo (2,015 g, 4,42 mmol, 100 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 242,2 [M+H]+.
Etapa a) 6-(2-cloro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo
A una solución de 2-cloro-4-fluorofenol (756 mg, 562 μl, 5,16 mmol, CAS RN 1996-41-4), 6-hidroxi-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (1000 mg, 4,69 mmol, CAS RN 1147557-97-8) y trifenilfosfina (1,48 g, 5,63 mmol, CAS RN 603-35-0) en THF (23,4 ml) se le añadió DIAD (1,14 g, 1,09 ml, 5,63 mmol, c As RN 2446-83-5) gota a gota a 0 °C y se agitó la reacción a Ta durante 18 h. Se añadió trifenilfosfina (738 mg, 2,81 mmol), seguida de DIAD (569 mg, 547 μl, 2,81 mmol) y se agitó la reacción a TA durante 6 h. Se vertió la mezcla de reacción en solución de NaHCO3 ac. sat. (50 ml) y se añadió EtOAc (30 ml). Se separaron las fases y se extrajo la fase ac. con EtOAc. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre sulfato de sodio, se filtró y se concentró para dar un aceite naranja. Se inmovilizó el producto bruto en Isolute y se purificó por cromatografía en columna (40 g, de 0 a 30 % de EtOAc en heptano) para dar el compuesto del título como un sólido amarillo (1,51 g, 4,19 mmol, 89,3 %). EM (ESI): m/z = 286,2 [M-56+H]+.
De forma análoga a la etapa a) de BB5 y BB5, se prepararon los intermedios BB6 - BB13 de la siguiente tabla a partir de los fenoles disponibles comercialmente.


BB14
2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano; sal trifluoroacetato
A una solución de 2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (21,3 mg, 52,8 jmol) en DCM (520 |jl) se le añadió ácido trifluoroacético (48,2 mg, 32,5 jl, 422 jmol) y se agitó la reacción a TA durante 19 h. Se concentró la mezcla de reacción para dar el compuesto del título como un sólido blanquecino (23,2 mg, 52,8 jmol, 100 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 304,2 [M+H]+.
Etapa a) 2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo
A una solución de 2-fluoro-4-(trifluorometil)fenol (41 mg, 28,7 jl, 228 jmol, CAS RN 77227-78-2), 2-hidroxi-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (50 mg, 207 jmol, CAS RN 240401-28-9) y trifenilfosfina (59,8 mg, 228 jmol, CAS RN 603-35-0) en THF (1,04 ml) se le añadió DIAD (46,1 mg, 44,3 jl, 228 jmol, CAS RN 2446-83-5) gota a gota y se agitó la reacción a ta durante 23 h. Se desactivó la mezcla de reacción por la adición de solución de NaHCÜ3 ac. sat. Se separaron las fases y se extrajo la fase ac. con EtOAc. Se secaron las capas orgánicas combinadas sobre sulfato de sodio, se filtró y se concentró para dar un aceite incoloro. Se inmovilizó el producto bruto en Isolute y se purificó por cromatografía en columna (12 g, de 0 a 30 % de EtOAc en n-heptano) para dar el compuesto del título como un aceite incoloro (21,3 mg, 50,2 jmol, 24,2 %). EM (ESI): m/z = 348,2 [M-56+H]+.
BB15
2-(2-cloro-4-fluorobencil)-2,6-diazaespiro[3.3]heptano; sal trifluoroacetato
A una solución de 6-(2-cloro-4-fluorobencil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (65,5 mg, 192 jmol) en DCM (961 jl) se le añadió TFA (175 mg, 118 jl, 1,54 mmol) y se agitó la reacción a TA durante 7 h. Se concentró la mezcla de reacción para dar el compuesto del título como un aceite amarillo que cristalizó tras su reposo (116,8 mg, 191 jmol, 99,4 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 241.1 [M+H]+.
Etapa a) 6-(2-cloro-4-fluorobencil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo
A una suspensión de hemioxalato de 2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (50 mg, 204 jmol, CAS RN 1041026-71-4) y 2-cloro-4-fluorobenzaldehído (32,4 mg, 204 jmol, CAS RN 84194-36-5) en DCE (1,02 ml) se le añadió triacetoxiborohidruro de sodio (64,9 mg, 306 jmol, CAS RN 56553-60-7) y se agitó la mezcla a TA durante 2,5 h. Se diluyó la solución con EtOAc. Se lavaron las capas orgánicas combinadas con solución de NaHCÜ3 sat. ac. Se separaron las fases y se extrajo la capa ac. con EtOAc, se lavó con salmuera, se filtró sobre MgSÜ4 y se evaporó hasta sequedad para dar el compuesto del título como un aceite amarillo claro (65,5 mg, 84,7 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 341,1 [M+H]+.
BB16
2-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.3]heptano; sal trifluoroacetato
A una solución de 6-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (71,0 mg, 190 jmol) en Dc M (948 j l) se le añadió ácido trifluoroacético (173 mg, 117 jl, 1,52 mmol) y se agitó la reacción a TA durante 17 h. Se concentró la mezcla de reacción para dar el compuesto del título como un aceite amarillo (119,5 mg, 188 jmol, 99 %) que se usó sin purificación adicional en la siguiente etapa. EM (ESI): m/z = 275.2 [M+H]+.
Etapa a) 2-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo
A una suspensión de hemioxalato de 2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (50 mg, 204 jmol, CAS RN 1041026-71-4) y 2-fluoro-4-(trifluorometil)benzaldehído (39,2 mg, 27,8 jl, 204 jmol, CAS RN 89763-93-9) en DCE (1,0 ml) se le añadió triacetoxiborohidruro de sodio (64,9 mg, 306 jmol, CAS RN 56553-60-7) y se agitó la mezcla a temperatura ambiente durante 19 h. Se diluyó la solución con EtOAc y se lavó con solución de NaHCO3 sat. ac. Se separaron las fases y se extrajo la capa ac. con EtOAc. Se lavaron las capas orgánicas combinadas con salmuera, se filtró sobre MgSO4 y se evaporó hasta sequedad. Se inmovilizó el residuo en Isolute y se purificó por
cromatografía en columna (4 g, de 0 a 40 % de EtOAc/EtOH 3:1 en n-heptano) para dar el compuesto del título como un aceite amarillo claro (71,0 mg, 180 |jmol, 88,2 %). EM (ESI): m/z = 375,3 [M+H]+.
BB17
2-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespiro[3.5]nonano
A una solución de 2-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo (79 mg, 196 jmol) en DCM (982 j l) se le añadió ácido trifluoroacético (179 mg, 121 jl, 1,57 mmol) y se agitó la reacción a ta durante 7 h. Se concentró la mezcla de reacción y se disolvió el residuo resultante en EtOAc, se lavó con solución de NaHCO3 ac. sat. y se volvió a extraer la fase acuosa con EtOAc tres veces. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar el compuesto del título como un aceite amarillo (57,3 mg, 91,7 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 303,3 [M+H]+.
Etapa a) 2-(2-fluom-4-(trifíuommetil)bendl)-2J-diazaespim[3.5]nonano-7-carboxilato de terc-butilo
A una suspensión de 2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo (50 mg, 221 jmol, CAS RN 896464-16-7) y 2-fluoro-4-(trifluorometil)benzaldehído (42,4 mg, 30,1 jl, 221 jmol, CAS RN 89763-93-9) en DCE (1,1 ml) se le añadió triacetoxiborohidruro de sodio (70,2 mg, 331 jmol, CAS RN 56553-60-7) y se agitó la mezcla a TA durante 4 h . Se diluyó la solución con EtOAc y se lavó con solución de NaHCO3 sat. ac. Se separaron las fases y se extrajo la capa ac. con EtOAc. Se lavaron las capas orgánicas combinadas con salmuera, se filtró sobre MgSO4 y se evaporó hasta sequedad para dar el compuesto del título como un aceite blanquecino (79,0 mg, 80 %) que se usó sin purificación adicional en la siguiente etapa. EM (ESI): m/z = 403,4 [M+H]+.
BB18
2-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespiro[4.4]nonano
A una solución de 7-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (123 mg, 275 jmol) en DCM (1,38 ml) se le añadió ácido trifluoroacético (251 mg, 170 jl, 2,2 mmol) y se agitó la reacción a ta durante 7 h. Se concentró la mezcla de reacción y se disolvió el residuo resultante en EtOAc, se lavó con solución de NaHCO3 ac. sat. y se volvió a extraer la fase acuosa con EtOAc tres veces. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar el compuesto del título como un aceite amarillo (107,7 mg, 99,7%) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 303,3 [M+H]+.
Etapa a) 7-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo
A una suspensión de 2,7-diazaespiro[4.4]nonano-2-carboxilato de terc-butilo (83,1 mg, 367 jmol, CAS RN 236406-49-8) y 2-fluoro-4-(trifluorometil)benzaldehído (70,5 mg, 0,05 ml, 367 jmol, CAS RN 89763-93-9) en DCE (1,1 ml) se le añadió triacetoxiborohidruro de sodio (117 mg, 550 jmol, CAS RN 56553-60-7) y se agitó la mezcla a ta durante 1 h. Se diluyó la solución con EtOAc y se lavó con solución de NaHCO3 sat. ac. Se separaron las fases y se extrajo la capa ac. con EtOAc. Se lavaron las capas orgánicas combinadas con salmuera, se filtró sobre MgSO4 y se evaporó hasta sequedad para dar el compuesto del título como un aceite amarillo claro (123,0 mg, 75 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 403,4 [M+H]+.
BB19
2-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.4]octano
A una solución de 2-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.4]octano-6-carboxilato de terc-butilo (147,6 mg, 361 jmol) en DCM (1,8 ml) se le añadió ácido trifluoroacético (329 mg, 223 jl, 2,89 mmol) y se agitó la reacción a ta durante 7 h. Se concentró la mezcla de reacción y se disolvió el residuo resultante en EtOAc, se lavó con solución de NaHCO3 ac. sat. y se volvió a extraer la fase acuosa con EWtOAc tres veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre sulfato de sodio y se concentró para dar el compuesto del título como un aceite amarillo (84,7 mg, 77,3 %) que se usó sin purificación adicional en la siguiente etapa. EM (ESI): m/z = 289,2 [M+H]+.
Etapa a) 2-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.4]octano-6-carboxilato de terc-butilo
A una suspensión de 2,6-diazaespiro[3.4]octano-6-carboxilato de terc-butilo (85,6 mg, 403 jmol, CAS RN 885270-86-0) y 2-fluoro-4-(trifluorometil)benzaldehído (77,5 mg, 55 jl, 403 jmol, CAS RN 89763-93-9) en DCE (2,0 ml) se le añadió triacetoxiborohidruro de sodio (128 mg, 605 jmol, CAS Rn 56553-60-7) y se agitó la mezcla a ta durante 1 h. Se diluyó la solución con EtOAc y se lavó con solución de NaHCO3 sat. ac. Se separaron las fases y se extrajo la capa ac. con EtOAc. Se lavaron las capas orgánicas combinadas con salmuera, se filtró sobre
MgSO4 y se evaporó hasta sequedad para dar el compuesto del título como un aceite amarillo claro (147,6 mg, 361 |jmol, 89,5 % de rendimiento) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 389,3 [M+H]+.
BB20
2-((2-cloro-4-fluorofeml)sulfoml)-2,6-diazaespiro[3.3]heptano; sal trifluoroacetato
A una solución de 6-((2-cloro-4-fluorofenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (54,7 mg, 140 jmol) en DCM (700 j l) se le añadió ácido trifluoroacético (128 mg, 86,3 jl, 1,12 mmol) y se agitó la reacción a TA durante 22 h. Se concentró la mezcla de reacción para dar el compuesto del título como un aceite blanquecino (75,7 mg, 140 jmol, 100 %) que se usó sin purificación adicional en la siguiente etapa. EM (ESI): m/z = 291,2 [M+H]+.
Etapa a) 6-((2-cloro-4-fluorofenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo
A una suspensión de hemioxalato de 2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (50 mg, 204 jmol, CAS RN 885270-86-0) se le añadió TEA (31 mg, 43 jl, 306 jmol), seguido de cloruro de 2-cloro-4-fluorobencenosulfonilo (58,5 mg, 37 jl, 255 jmol, CAS RN 85958-57-2) y se agitó la solución transparente resultante a TA durante 17,5 h. Se diluyó la mezcla de reacción con DCM y se desactivó con agua. Se separaron las fases y se extrajo la fase ac. con DCM. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre sulfato de sodio, se filtró y se concentró para dar el compuesto del título como un sólido blanco (54,7 mg, 65,1 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 335,1 [M+H]+.
BB21
2-((2-fluoro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano; sal trifluoroacetato
A una solución de 6-((2-fluoro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (67,6 mg, 159 jmol) en Dc M (796 j l) se le añadió TFA (145 mg, 98,2 jl, 1,27 mmol) y se agitó la reacción a TA durante 20 h. Se concentró la mezcla de reacción para dar el compuesto del título como un aceite amarillo (90 mg, 99,3 %) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 325,2 [M+H]+.
Etapa a) 6-((2-fluoro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo
A una suspensión de hemioxalato de 2,6-diazaespiro[3.3]heptano-2-carboxilato de terc-butilo (50 mg, 204 jmol, CAS RN 885270-86-0) en DCM se le añadió TEA (31 mg, 42,7 jl, 306 jmol), seguido de cloruro de 2-fluoro-4-(trifluorometil)bencenosulfonilo (59 mg, 225 jmol, CAS 1177009-38-9) y se agitó la solución transparente resultante a TA durante 18 h. Se diluyó la mezcla de reacción con DCM y se desactivó con agua. Se separaron las fases y se extrajo la fase ac. con DCM. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre sulfato de sodio, se filtró y se concentró para dar el compuesto del título como un sólido blanco (67,6 mg, 74,1%) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 369,2 [M+H]+.
BB22
Sal trifluoroacetato de 6-[[4,5-bis(trifluorometil)-2-piridil]oxi]-2-azaespiro[3.3]heptano
A una solución de 6-((4,5-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,334 g, 783 jmol) en CH2O 2 (5 ml) se le añadió TFA (893 mg, 604 jl, 7,83 mmol). Se agitó la mezcla de reacción resultante a TA durante 1 hora. Se concentró la mezcla de reacción a alto vacío para proporcionar 366 mg del producto deseado como un aceite amarillo claro. EM (ESI): m/z = 327,2 [M+H]+.
Etapa a) 6-((4,5-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo
A una solución de 6-hidroxi-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,2 g, 938 jmol) en THF seco (3 ml) se le añadió una solución de terc-butóxido de potasio 1 M en THF (985 jl, 985 jmol) y se agitó la mezcla de reacción turbia a TA durante 15 min seguido de la adición de 2-cloro-4,5-bis(trifluorometil)piridina (234 mg, 938 jmol). A continuación, se agitó la mezcla de reacción a TA durante 19 horas. Se diluyó la reacción bruta con EtOAc y se extrajo con agua, se recogió la fase orgánica y se volvió a extraer la fase acuosa con EtOAc. Se secaron las fases orgánicas combinadas sobre sulfato de sodio y se evaporó hasta sequedad. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 100 % de EtOAc en heptano). Se obtuvo el producto deseado como un sólido blanco, 334 mg. EM (ESI): m/z = 371,2 [M - 56 H]+
BB23
Sal trifluoroacetato de 6-[[5,6-bis(trifluorometil)-2-piridil]oxi]-2-azaespiro[3.3]heptano
A una solución de 6-((5,6-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,376 g, 882 |jmol) en CH2CI2 (5 ml) se le añadió TFA (1,o1 g, 679 jl, 8,82 mmol). Se agitó la mezcla de reacción resultante a TA durante 1 hora. Se concentró la mezcla de reacción a alto vacío para proporcionar 398 mg del producto deseado como un aceite amarillo claro. EM (ESI): m/z = 327,2 [M+H]+.
Etapa a) 6-((5,6-bis(trifluommetil)pirídin-2-il)oxi)-2-azaespim[3.3]heptano-2-carboxilato de terc-butilo
A una solución de 6-hidroxi-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,2 g, 938 jmol) en THF seco (3 ml) se le añadió una solución 1 M de terc-butóxido de potasio en THF (985 jl, 985 jmol) y se agitó la mezcla de reacción turbia a TA durante 15 min seguido de la adición de 6-cloro-2,3-bis(trifluorometil)piridina (234 mg, 938 jmol). A continuación, se agitó la mezcla de reacción a TA durante 19 horas. Se diluyó la reacción bruta con EtoAc y se extrajo con agua, se recogió la fase orgánica y se volvió a extraer la fase acuosa con EtOAc. Se secaron las fases orgánicas combinadas sobre sulfato de sodio y se evaporó hasta sequedad. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 100 % de EtOAc en heptano). Se obtuvo el producto deseado como un sólido blanco, 376 mg. EM (ESI): m/z = 371,2 [M-56+H]+.
BB24
Sal clorhidrato de 2-cloro-4-fluoro-N-metil-N-[(3R)-1-oxa-8-azaespiro[4.5]decan-3-il]bencenosulfonamida
En un tubo de 10 ml, se disolvió (R)-3-((2-cloro-4-fluoro-N-metilfenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (87 mg, 188 jmol) en DCM (3,61 ml) y se añadió HCl en éter dietílico 2 M (752 jl, 1,5 mmol). Se agitó la reacción a TA durante 6 h. Se retiró el disolvente a vacío, se usó el producto en la siguiente etapa sin purificación. EM (ESI): m/z = 363,1 [M+H]+.
Etapa a) (R)-3-((2-cloro-4-fluorofenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo
En un tubo de 20 ml purgado con argón, se disolvió (R)-3-amino-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (80 mg, 312 jmol) en DCM (2,67 ml). Se añadieron TEA (69,5 mg, 687 jmol) y cloruro de 2-cloro-4-fluorobencenosulfonilo (75,1 mg, 328 jmol), y se agitó la mezcla de reacción durante 2 h a Ta . Se extrajo la mezcla de reacción con DCM/agua, se secó con Na2SO4, se retiró el disolvente a vacío, y se purificó el residuo por HPLC preparativa (columna Gemini NX, gradiente de ACN/agua TEA al 0,1 %). Se obtuvo el producto como un sólido blanco (83 mg). EM (ESI): m/z = 448,9 [M-H]\
Etapa b) (R)-3-((2-cloro-4-fluoro-N-metilfenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo
A una solución de (R)-3-((2-cloro-4-fluorofenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (110 mg, 245 jmol) en DMF (2 ml) a 0 °C se le añadió NaH en aceite de vaselina al 60 % (14,7 mg, 368 jmol.). Se agitó la mezcla de reacción a TA durante 30 minutos, tras lo que se añadió yodometano (104 mg, 46 jl, 735 jmol) y se continuó agitando durante 1 hora.
Se añadió solución de cloruro de amonio acuosa saturada y se extrajo la fase acuosa tres veces con EtOAc. Se secaron las capas orgánicas combinadas sobre sulfato de magnesio, se filtró y se concentró a vacío para proporcionar el producto bruto, que se purificó por HPLC preparativa (columna Gemini NX, gradiente de ACN/agua HCOOH al 0,1 %). Se obtuvieron 87 mg de producto como un sólido blanco. EM (ESI): m/z = 407,2 [M-56+H]+.
BB25
Sal trifluoroacetato de 6-[[5-(trifluorometil)-2-piridil]oxi]-2-azaespiro[3.3]heptano
Se disolvió 6-((5-(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,314 g, 876 jmol) en CH2Cl2 (3 ml) y se añadió TFA (799 mg, 540 jl, 7,01 mmol). Se agitó la mezcla de reacción a TA durante 2 horas. Se retiró el disolvente a vacío, se usó el producto en la siguiente etapa sin purificación. EM (ESI): m/z = 259,2 [M+H]+.
Etapa a) 6-((5-(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo
A una solución de 6-hidroxi-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,200 g, 938 jmol) en THF seco (3 ml) se le añadió una solución 1 M de terc-butóxido de potasio en THF (985 jl, 985 jmol) y se agitó la mezcla de reacción turbia a TA durante 15 min seguido de la adición de 2-bromo-5-(trifluorometil)piridina (212 mg, 938 jmol). A continuación, se agitó la mezcla de reacción a TA durante 19 horas. Se diluyó la reacción bruta con EtOAc y se extrajo con agua, se recogió la fase orgánica y se volvió a extraer la fase acuosa con EtOAc. Se secaron las fases orgánicas combinadas sobre sulfato de sodio y se evaporó hasta sequedad. Purificación: Se purificó el material
bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 100 % de EtOAc en heptano). Se obtuvo el producto como un sólido amarillo claro (314 mg). EM (ESI): m/z = 303,2 [M-56+H]+.
BB26
6-[[4-metil-3-(trifluorometil)fenil]metoxi]-2-azaespiro[3.3]heptano
Se disolvió 6-((4-metil-3-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (170 mg, 441 |jmol) en DCM (2 ml) y TFA (302 mg, 204 jl, 2,65 mmol). Se agitó la mezcla de reacción a TA durante 8 horas. Se retiró el disolvente a vacío, se usó el producto en la siguiente etapa sin purificación. EM (ESI): m/z = 286,3 [M+H]+.
Etapa a) 6-((4-metil-3-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo
En un tubo de 20 ml bajo argón, se disolvió 6-hidroxi-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (142 mg, 664 jmol) en DMF (2,5 ml) y se enfrió a 2-4 °C. Se añadió NaH (31,9 mg, 797 jmol) y se agitó la mezcla durante 20 min. Se retiró el baño de enfriamiento y se añadió 4-(bromometil)-1-metil-2-(trifluorometil)benceno (168 mg, 664 jmol), a continuación se agitó a 22 °C durante 3 h. Se añadieron 3 ml de solución de NH4Cl sat., se extrajo con agua/EtOAc/NaCl sat., se secó sobre MgSO4. Se retiró el disolvente y se purificó el producto bruto por cromatografía ultrarrápida (20 g de sílice con heptano/EtOAc de 0 a 40 % en 30 min a UV 265 nm). EM (ESI): m/z = 330,2 [M-56+H]+.
BB27
Clorhidrato de 2-((2-cloro-4-fluorofenil)sulfonil)-2,7-diazaespiro[3.5]nonano
En un tubo de 10 ml, se disolvió 2-((2-cloro-4-fluorofenil)sulfonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo (140 mg, 334 |jmol) en Dc M (4 ml) y se añadió HCl en éter dietílico 2 M (1 ml, 2 mmol). Se agitó la reacción a TA durante 6 h. Se retiró el disolvente a vacío, se usó el producto en la siguiente etapa sin purificación. EM (ESI): m/z = 319,1 [M+H]+.
Etapa a) 2-((2-cloro-4-fluorofenil)sulfonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo
En un tubo de 20 ml purgado con argón, se disolvió 2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo (80 mg, 353 jmol) en d Cm (3,3 ml). Se añadieron TEA (78,7 mg, 108 jl, 778 jmol) y cloruro de 2-cloro-4-fluorobencenosulfonilo (89,1 mg, 389 jmol), se agitó la mezcla 2 h a TA. Se extrajo la reacción con DCM/agua, se combinó la fracción orgánica y se secó sobre Na2SO4, se retiró el disolvente a vacío, se purificó el residuo por HPLC preparativa (YMC-Triart C18, 12 nm, 5 jm , 100 x 30 mm, 9 min con gradiente de ACN/agua TEA al 0,1 %). Se obtuvo el producto como una espuma blanca. EM (ESI): m/z = 363,1 [M-56+H]+.
BB28
2,2,2-trifluoroacetato de 6-((2-fluoro-4-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano
Se disolvió 6-((2-fluoro-4-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (235 mg, 604 jmol) en Dc M (3 ml) y TFA (344 mg, 232 jl, 3,02 mmol). Se agitó la mezcla de reacción a TA durante 8 horas y se concentró a vacío (azeótropo con toluol, EE+hep). Usado directamente para la siguiente etapa. EM (ESI): m/z = 290,2 [M+H]+.
Etapa a) 6-((2-fluoro-4-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo
A una solución helada de 6-hidroxi-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (250 mg, 1,17 mmol) en DMF (3 ml) se le añadió NaH al 60 % en aceite de vaselina (51,6 mg, 1,29 mmol) en porciones y se agitó la mezcla a la temperatura del baño de hielo durante 5 minutos seguido de agitación a TA durante 40 minutos. Se disolvió una solución de metanosulfonato de 2-fluoro-4-(trifluorometil)bencilo (383 mg, 1,41 mmol) en DMF (1 ml) y se añadió gota a gota a la mezcla a TA. Se continuó agitando la suspensión a TA durante 16 horas. Se vertió la mezcla de reacción sobre solución de NH4CI acuosa saturada (10 ml) y EtOAc (20 ml) y se separaron las capas. Se extrajo la capa acuosa una vez con EtOAC (50 ml). Se lavaron las capas orgánicas dos veces con agua, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 20 g usando un sistema de MPLC que se eluyó con un gradiente de n-heptano : EtOAc (de 100 : 0 a 50 : 40) para obtener el compuesto deseado como un sólido amarillo claro (235 mg). EM (ESI): m/z = 334,2 [M-56+H]+.
BB29
Clorhidrato de (R)-N-(1-oxa-8-azaespiro[4.5]decan-3-il)-4-(trifluorometil)bencenosulfonamida
En un tubo de 10 ml, se disolvió (R)-3-((4-(trifluorometil)fenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (63 mg, 136 |jmol) en DCM (1 ml) y se añadió HCl en éter dietílico 2 M (678 ul, 1,36 mmol). Se agitó la reacción a TA durante 4 h. Se retiró el disolvente a vacío, se usó el producto en la siguiente etapa sin purificación. EM (ESI): m/z = 365,1 [M+H]+.
Etapa a) (R)-3-((4-(trifluorometil)fenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo En un tubo de 20 ml purgado con argón, se disolvió (R)-3-amino-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (60 mg, 234 jmol) en DCM (2 ml). Se añadieron TEA (52,1 mg, 515 jmol) y cloruro de 4-(trifluorometil)bencenosulfonilo (68,7 mg, 281 jmol), se agitó la mezcla durante 2 h a TA. Se extrajo la reacción con DCM/agua, se combinó la fracción orgánica y se secó sobre Na2SO4, se retiró el disolvente a vacío, se purificó el residuo por HPLC preparativa (Gemini NX, 12 nm, 5 |jm, 100 x 30 mm, gradiente de ACN/agua TEA al 0,1 %). Se obtuvo el producto como un sólido blanco (63 mg). EM (ESI): m/z = 463,3 [M-H]-.
BB30
Clorhidrato de (R)-N-metil-N-(1-oxa-8-azaespiro[4.5]decan-3-il)bencenosulfonamida
En un tubo de 10 ml, se disolvió (R)-3-(N-metilfenilsulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (96 mg, 234 jmol) en DCM (1 ml) y se añadió HCl en éter dietílico 2 M (1,75 ml, 3,51 mmol). Se agitó la reacción a TA durante 4 h. Se retiró el disolvente a vacío, se usó el producto en la siguiente etapa sin purificación. EM (ESI): m/z = 311,2 [M+H]+.
Etapa a) (R)-3-(fenilsulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo
En un tubo de 20 ml purgado con argón, se disolvió (R)-3-amino-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (60 mg, 234 jmol) en DCM (2 ml). Se añadieron TEA (52,1 mg, 515 jmol) y cloruro de bencenosulfonilo (49,6 mg, 281 jmol), se agitó la mezcla durante 2 h a TA. Se extrajo la reacción con DCM/agua, se combinó la fracción orgánica y se secó sobre Na2SO4, se retiró el disolvente a vacío, se purificó el residuo por HPLC preparativa (Gemini NX, 12 nm, 5 jm , 100 x 30 mm, gradiente de ACN/agua TEA al 0,1 %). Se obtuvo el producto como un sólido blanco (63 mg). EM (ESI): m/z = 395,3 [M-H]\
Etapa b) (R)-3-(N-metilfenilsulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo
A una solución de (R)-3-(fenilsulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (110 mg, 277 jmol) en DMF (1,2 ml) a 0 °C se le añadió NaH en aceite de vaselina al 60 % (16,6 mg, 416 jmol). Se agitó la mezcla de reacción a TA durante 30 minutos, tras lo que se añadió yodometano (118 mg, 52 jl, 832 jmol) y se continuó agitando durante 1 hora. Se añadió solución de NH4CI acuosa saturada y se extrajo la fase acuosa tres veces con EtOAc. Se secaron las capas orgánicas combinadas sobre sulfato de magnesio, se filtró y se concentró a vacío para proporcionar el producto bruto que se purificó por HPLC preparativa. (Gemini NX, 12 nm, 5 jm , 100 x 30 mm, gradiente de ACN/agua TEA al 0,1 %). Se obtuvo el producto como un aceite incoloro (96 mg). EM (ESI): m/z = 355,1 [M-56+H]+.
BB31
Clorhidrato de (R)-2-cloro-4-fluoro-N-(1-oxa-8-azaespiro[4.5]decan-3-il)bencenosulfonamida
En un tubo de 10 ml, se disolvió (R)-3-((2-cloro-4-fluorofenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (64 mg, 143 jmol) en DCM (2 ml) y se añadió HCl en éter dietílico 2 M (1070 jl, 2,14 mmol). Se agitó la reacción a TA durante 6 h. Se retiró el disolvente a vacío, se usó el producto en la siguiente etapa sin purificación. EM (ESI): m/z = 349,1 [M+H]+.
Etapa a) (R)-3-((2-cloro-4-fluorofenil)sulfonamido)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo Síntesis descrita en BB24.
BB32
Clorhidrato de (R)-N-(1-oxa-8-azaespiro[4.5]decan-3-il)-3-(trifluorometil)bencenosulfonamida Sintetizado como se describe para BB29, comenzando a partir de (R)-3-amino-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (60 mg, 234 umol) y cloruro de 3-(trifluorometil)bencenosulfonilo (68,7 mg, 281 jmol). 53 mg de producto obtenido como un aceite amarillo claro.
EM (ESI): m/z = 365,1 [M+H]+.
BB33
Trifluoroacetato de N-(2-cloro-4-fluorobencil)-N-metil-1-oxa-8-azaespiro[4.5]decan-3-amina
A una solución de 3-((2-cloro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4,5]decano-8-carboxilato de terc-butilo (0,107 g, 259 |jmol) en DCM (2 ml) se le añadió ácido trifluoroacético (236 mg, 160 jl, 2,07 mmol) y se agitó la reacción a TA durante 19 h. Se concentró la mezcla de reacción para dar el compuesto del título como aceite amarillo claro (111 mg) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 313,2 [M+H]+.
Etapa a) 3-((2-cloro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo
A una solución de 3-amino-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (0,5 g, 1,95 mmol) y 2-cloro-4-fluorobenzaldehído (309 mg, 1,95 mmol) en MeOH (12 ml) se le añadió cianoborohidruro de sodio (613 mg, 9,75 mmol), se agitó la mezcla de reacción a TA durante 2 horas. Para el procesamiento, se vertió la mezcla de reacción en NaHCO3 sat. y se extrajo con EtOAc. Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 100 % de EtOAc en heptano) para obtener 231 mg de producto como un aceite incoloro. EM (ESI): m/z = 399,2 [M+H]+.
Etapa b) 3-((2-cloro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo
A una solución de 3-((2-cloro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (0,100 g, 251 jmol) en Dm F (1,5 ml) a 0 °C se le añadió NaH al 60 % en aceite de vaselina (15 mg, 376 jmol). Se agitó la mezcla de reacción a TA durante 30 minutos y, a continuación, se añadió yodometano (107 mg, 47 jl, 752 jmol) y se continuó agitando durante 1 hora. Se añadió solución de cloruro de amonio acuosa saturada y se extrajo la fase acuosa tres veces con EtOAc. Se secaron las capas orgánicas combinadas sobre sulfato de magnesio, se filtró y se concentró a vacío para proporcionar el producto deseado como un aceite amarillo (104 mg), que se usó directamente para la siguiente etapa. EM (ESI): m/z = 413,4 [M+H]+.
BB34
Trifluoroacetato de N-(2-cloro-4-fluorobencil)-1-oxa-8-azaespiro[4.5]decan-3-amina
A una solución de 3-((2-cloro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo (0,088 g, 221 jmol) en DCM (1 ml) se le añadió ácido trifluoroacético (201 mg, 136 jl, 1,76 mmol) y se agitó la reacción a TA durante 2 h. Se concentró la mezcla de reacción para dar el compuesto del título como aceite incoloro (91 mg) que se usó en la siguiente etapa sin purificación adicional.
Etapa a) 3-((2-cloro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo
Se sintetizó 3-((2-cloro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carboxilato de terc-butilo como se describe para BB33.
BB35
Clorhidrato de 2-((4-(trifluorometil)fenil)sulfonil)-2,7-diazaespiro[3.5]nonano
En un tubo de 10 ml, se disolvió 2-((4-(trifluorometil)fenil)sulfonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo (124 mg, 285 jmol) en DCM (4 ml) y se añadió HCl en éter dietílico 2 M (856 jl, 1,71 mmol). Se agitó la reacción a TA durante 3 h. Se retiró el disolvente a vacío, se obtuvo el producto como un sólido blanco (105 mg) y se usó en la siguiente etapa sin purificación. EM (ESI): m/z = 335,1 [M+H]+.
Etapa a) 2-((4-(trifluorometil)fenil)sulfonil)-2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo
En un tubo de 20 ml purgado con argón, se disolvió 2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo (80 mg, 353 jmol, eq: 1) en DCM (3,3 ml). Se añadieron TEA (78,7 mg, 108 jl, 778 jmol) y cloruro de 4-(trifluorometil)bencenosulfonilo (95,1 mg, 389 jmol), se agitó la mezcla durante 2 h a TA. Se extrajo la reacción con DCM/agua, se combinó la fracción orgánica y se secó sobre Na2SO4, se retiró el disolvente a vacío, se purificó el residuo por HPLC preparativa (YMC-Triart C18, 12 nm, 5 jm , 100 x 30 mm, 9 min con gradiente de ACN/agua TEA al 0,1 %). Se obtuvo el producto como una espuma blanca. EM (ESI): m/z = 379,1 [M-56+H]+.
BB36
Clorhidrato de 2-(fenilsulfonil)-2,7-diazaespiro[3.5]nonano
Se obtuvo BB36 a partir de 2,7-diazaespiro[3.5]nonano-7-carboxilato de terc-butilo (80 mg, 353 |jmol) y cloruro de bencenosulfonilo (74,9 mg, 424 jmol), como se describe para BB35. EM (ESI): m/z = 267,2 [M+H]+.
BB37
Trifluoroacetato de 2-(2,4-difluorofenoxi)-7-azaespiro[3.5]nonano
A una solución de 2-(2,4-difluorofenoxi)-7-azaespiro[3.5]nonano-7-carboxilato de ferc-butilo (510 mg, 1,44 mmol) en DCM (3 ml) se le añadió ácido trifluoroacético (823 mg, 556 jl, 7,22 mmol) y se agitó la reacción a TA durante 3 h. Se concentró la mezcla de reacción para dar el compuesto del título como un sólido blanco (510 mg) que se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 254,2 [M+H]+.
Etapa a) 2-(2,4-difíuorofenoxi)-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo
En un matraz de sulfonación de cuatro bocas de 25 ml bajo argón, se disolvió 2-hidroxi-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (401 mg, 1,66 mmol, CAS RN 240401-28-9) en THF (6 ml), se añadieron 2,4-difluorofenol (216 mg, 159 jl, 1,66 mmol) y trifenilfosfina (479 mg, 1,83 mmol). Se agitó la solución transparente a TA durante 5, a continuación se enfrió hasta 0-2 °C y se añadió DEAD (318 mg, 289 jl, 1,83 mmol) lentamente en 10 min, se continuó agitando durante 1 h a 2-4 °C, a continuación se retiró el baño de enfriamiento y se agitó durante la noche a TA. Se añadieron 20 ml de éter dietílico, se lavó la mezcla con agua, NaOH 1 M y salmuera. Se secó la capa orgánica sobre Na2SO4 y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 50 g, de 0 % a 40 % de EtOAc en heptano) para obtener 511 mg de producto como un aceite incoloro. EM (ESI): m/z = 298,3 [M-56+H]+.
BB38
Trifluoroacetato de 2-(2-cloro-4-fluorofenoxi)-7-azaespiro[3.5]nonano
Se obtuvo BB38 a partir de 2-hidroxi-7-azaespiro[3.5]nonano-7-carboxilato de terc-butilo (412 mg, 1,71 mmol) y 2-cloro-4-fluorofenol (250 mg, 1,71 mmol), como se describe para BB37. EM (ESI): m/z = 270,2 [M+H]+.
BB39
Trifluoroacetato de 6-[[2-fluoro-4-(trifluorometil)fenoxi]metil]-2-azaespiro[3.3]heptano
Se obtuvo BB39 de forma análoga a BB37 a partir de 6-[[2-fluoro-4-(trifluorometil)fenoxi]metil]-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo y se usó en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 290,2 [M+H]+.
Etapa a) 6-[[2-fíuoro-4-(trifíuorometil)fenoxi]metil]-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo Se obtuvo el compuesto de forma análoga al ejemplo 37, etapa a, a partir de 6-(hidroximetil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (CAS RN 1363381-93-4) y 2-fluoro-4-(trifluorometil)fenol (Ca S RN 77227-78-2). Después de la extracción, se usó el material en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 334,1 [M-56-H]+.
BB40
Trifluoroacetato de 6-((2-fluoro-4-(trifluorometil)bencil)oxi)-6-(trifluorometil)-2-azaespiro[3.3]heptano Se obtuvo BB40 de forma análoga a BB25 a partir de 6-hidroxi-6-(trifluorometil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (CAS 1251923-04-2) y 1-(bromometil)-2-fluoro-4-(trifluorometil)benceno. EM (ESI): m/z = 358,1 [M-56-H]+.
BB41
Trifluoroacetato de 6-(2-fluoro-4-(trifluorometil)fenil)-2-azaespiro[3.3]heptano
A una solución de 6-(2-fluoro-4-(trifluorometil)fenil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,151 g, 420 jmol) en DCM (4 ml) se le añadió TFA (240 mg, 162 jl, 2,1 mmol).
Se agitó la mezcla de reacción resultante a TA durante 1 hora y a continuación se concentró a vacío (azeótropo con tolueno) proporcionando 143 mg de aceite incoloro, usado en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 260,2 [M+H]+.
Etapa a) 6-(2-fluoro-4-(trifluorometil)fenil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo
A un vial de 20 ml equipado con una barra de agitación se le añadió fotocatalizador (Ir[dF(CF3)ppy]2(dtbpy))PF6 (6,09 mg, 5,43 |jmol), 1-bromo-2-fluoro-4-(trifluorometil)benceno (198 mg, 137 jl, 8 l5 jmol), 6-bromo-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,150 g, 543 jmol), tris(trimetilsilil)silano (135 mg, 168 jl, 543 jmol) y carbonato de sodio anhidro (115 mg, 1,09 mmol). Se selló el vial y se colocó bajo argón antes de añadir DME (3 ml). A un vial separado se le añadió complejo de cloruro de níquel(N)-etilenglicol-éter dimetílico (1,19 mg, 5,43 jm ol) y 4,4'-di-terc-butil-2,2'-bipiridina (1,46 mg, 5,43 jmol). Se selló el vial de precatalizador, se purgó con argón, a continuación, se le añadió DME (2 ml). Se sonicó el vial de precatalizador durante 5 min, después de lo que se inyectó 1 ml (catalizador al 0,5 % mol, 0,005 eq) en el recipiente de reacción. Se desgasificó la solución rociando con argón. Se agitó la reacción y se irradió con una lámpara de 420 nm durante 5 horas. Se desactivó la reacción por exposición al aire y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 50 g, de 0 % a 20 % de EtOAc en heptano), proporcionando 151 mg (77 % de rendimiento, no puro basado en RMN) de un líquido incoloro que se usó para la siguiente etapa. EM (ESI): m/z = 304,2 [M-56-H]+.
BB42
Trifluoroacetato de 6-(2-fluoro-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano
A una solución de 6-(2-fluoro-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,102 g, 273 jmol) en DCM (3 ml) se le añadió TFA (156 mg, 105 jl, 1,37 mmol). Se agitó la mezcla de reacción resultante a TA durante 2 horas y a continuación se concentró a vacío (azeótropo con tolueno) proporcionando 108 mg de aceite incoloro, usado en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 274,2 [M+H]+.
Etapa a) Bromuro de (2-fluoro-4-(trifluorometil)bencil)trifenilfosfonio Bajo argón, se disolvió trifenilfosfina (1,02 g, 3,89 mmol) en acetonitrilo (10 ml) y se añadió 1-(bromometil)-2-fluoro-4-(trifluorometil)benceno (1 g, 3,89 mmol). Se agitó la mezcla a 80 °C durante 3 horas. Se dejó enfriar la suspensión hasta TA. Se añadieron 100 ml de MTBE y se agitó a TA durante 30 min. Se filtró el sólido y se lavó con MTBE. Se secó el sólido bajo HV, se usó el producto directamente para la siguiente etapa. Sólido blanco, 2,02 g (98 %). EM (ESI): m/z = 439,2 [M+H]+.
Etapa b) 6-(2-fíuoro-4-(trifíuorometil)bencilideno)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo Bajo argón a -78 °C, se disolvió bromuro de (2-fluoro-4-(trifluorometil)bencil)trifenilfosfonio (0,5 g, 963 jmol) en THF seco (5 ml) y se añadió LiHMDS (1,93 ml, 1,93 mmol). Se agitó la mezcla de reacción a -78 °C durante 2 horas. A continuación, a TA, se añadió 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (407 mg, 1,93 mmol) y se agitó la mezcla a 85 °C durante la noche. Se añadió MTBE y se separó por filtración el precipitado (óxido de trifenilfosfino). Se concentró el filtrado y se purificó directamente. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 80 % de EtOAc en heptano) proporcionando el producto como un sólido amarillo (119 mg, 33 %). EM (ESI): m/z = 316,2 [M-56+H]+.
Etapa c) 6-(2-fluoro-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo Se disolvió 6-(2-fluoro-4-(trifluorometil)bencilideno)-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo (0,119 g, 320 |jmol) en acetato de etilo (2,5 ml). Se purgó el matraz y se rellenó con argón (3x). Se añadió Pd-C (34,1 mg, 32 jm ol) y se agitó la reacción bajo H2 (globo) durante 2 horas. Se filtró la mezcla de reacción a través de una capa de celite, se lavó con EtOAc y se secó a vacío, proporcionando el producto como un aceite incoloro (108 mg, 90 %). EM (ESI): m/z = 318,2 [M-56+H]+.
BB43
Trifluoroacetato de 6-(2-cloro-4-fluorobencil)-2-azaespiro[3.3]heptano
Se obtuvo BB43 de forma análoga a BB42 comenzando a partir de 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo y 1-(bromometil)-2-cloro-4-fluorobenceno. EM (ESI): m/z = 240,1 [M+H]+.
BB44
Trifluoroacetato de 6-(2,4-difluorobencil)-2-azaespiro[3.3]heptano
Se obtuvo BB44 de forma análoga a BB42 comenzando a partir de 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de terc-butilo y 1-(bromometil)-2,4-difluorobenceno. EM (ESI): m/z = 224,1 [M+H]+.
BB45
Trifluoroacetato de 6-(2-metoxi-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano
Se obtuvo BB45 de forma análoga a BB42 comenzando a partir de 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de
ferc-butilo y 1-(bromometil)-2-metoxi-4-(trifluorometil)benceno. EM (ESI): m/z = 286,2 [M+H]+.
BB46
Trifluoroacetato de 6-(2-fluoro-6-(trifluorometil)bencil)-2-azaespiro[3.3]heptano
Se obtuvo BB46 de forma análoga a BB42 comenzando a partir de 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo y 1-(bromometil)-2-fluoro-6-(trifluorometil)benceno. EM (ESI): m/z = 274,2 [M+H]+.
BB47
Trifluoroacetato de 6-((2-cloro-4-fluorofenoxi)metil)-2-azaespiro[3.3]heptano
Se obtuvo BB47 a partir de 6-(hidroximetil)-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo (300 mg, 1,32 mmol) y 2-cloro-4-fluorofenol (193 mg, 1,32 mmol), como se describe para BB37. EM (ESI): m/z = 256,1 [M+H]+.
BB48
Trifluoroacetato de 2-(3-(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano
A una solución de 6-(3-(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo (230 mg, 672 |jmol) en DCM (2 ml) se le añadió TFA (306 mg, 207 jl, 2,69 mmol). Se agitó la mezcla de reacción resultante a TA durante la noche y a continuación se concentró a vacío (azeótropo con tolueno) proporcionando 245 mg de aceite incoloro, usado en la siguiente etapa sin purificación adicional. Em (ESI): m/z = 243,2 [M+H]+.
Etapa a) 6-(3-(frifluoromefil)fenil)-2,6-diazaespiro[3.3]hepfano-2-carboxilafo de ferc-bufilo A un matraz se le añadió: 1-bromo-3-(trifluorometil)benceno (170 mg, 104 jl, 756 jmol), 2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo (165 mg, 831 jmol), Cs2CO3 (492 mg, 1,51 mmol) y 1,4-dioxano (4 ml), se burbujeó la suspensión con N2 durante 5 min y se añadió cloro(2-diciclohexilfosfino-2',6'-diisopropoxi-1,1'-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio(N) (RuPhos Pd G2, 29.3 mg, 37,8 jmol). Se calentó la mezcla hasta 100 °C durante 2 h. Se diluyó la mezcla con 10 ml de EA y se filtró a través de celite, se concentró el filtrado para dar un aceite amarillo. A continuación, se purificó sobre 20 g de sílice con heptano/EA 0 - 40 %, se concentraron las fracciones de producto para dar el producto deseado como un sólido amarillo (233 mg, 90 %). EM (ESI): m/z = 343,2 [M+H]+.
BB49
2.2.2- trifluoroacetato de 2-(4-isopropoxifenil)-2,6-diazaespiro[3.3]heptano
Se obtuvo BB49 de forma análoga a BB48 comenzando a partir de 2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo y 1-bromo-4-isopropoxibenceno. Se calentó la reacción hasta 120 °C durante 4 h. EM (ESI): m/z = 233,2 [M+H]+.
BB50
2.2.2- trifluoroacetato de 1-(4-(2,6-diazaespiro[3.4]octan-2-il)fenil)pirrolidin-2-ona
Se obtuvo BB50 de forma análoga a BB48 comenzando a partir de 2,6-diazaespiro[3.4]octano-6-carboxilato de ferc-butilo y 1-(4-bromofenil)pirrolidin-2-ona. Se calentó la reacción hasta 125 °C durante 18 h. EM (ESI): m/z = 272.3 [M+H]+.
BB51
Trifluoroacetato de 2-(4-metoxi-3-metilfenil)-2,6-diazaespiro[3.4]octano
Se obtuvo BB51 de forma análoga a BB48 comenzando a partir de 2,6-diazaespiro[3.4]octano-6-carboxilato de ferc-butilo y 4-bromo-1-metoxi-2-metilbenceno. Se calentó la reacción hasta 125 °C durante 18 h. EM (ESI): m/z = 233,2 [M+H]+.
BB52
Trifluoroacetato de 2-(4-cloro-3-(trifluorometil)fenil)-2,6-diazaespiro[3.4]octano
Se obtuvo BB52 de forma análoga a BB48 comenzando a partir de 2,6-diazaespiro[3.4]octano-6-carboxilato de ferc-butilo y 4-bromo-1-cloro-2-(trifluorometil)benceno. Se calentó la reacción hasta 125 °C durante 18 h. EM (ESI):
m/z = 291,1 [M+H]+.
BB53
Diclorhidrato de 2-(2-fluoropiridin-4-il)-2,6-diazaespiro[3.4]octano
Se obtuvo BB53 de forma análoga a BB48 comenzando a partir de 2,6-diazaespiro[3.4]octano-6-carboxilato de ferc-butilo y 4-bromo-2-fluoropiridina. Se calentó la reacción hasta 125 °C durante 18 h. Se logró la desprotección usando HCl 2 M en éter dietílico (16 h, TA). EM (ESI): m/z = 208,2 [M+H]+.
BB54
Trifluoroacetato de 2-(2,5-bis(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano
Se obtuvo BB54 de forma análoga a BB48 comenzando a partir de 2,6-diazaespiro[3.4]octano-6-carboxilato de ferc-butilo y 4-bromo-2-fluoropiridina. Se calentó la reacción hasta 120 °C durante 4 h. EM (ESI): m/z = 311,4 [M+H]+.
BB55
2,2,2-trifluoroacetato de 2-((4-fluoro-2-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano
A una solución de 6-((4-fluoro-2-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo (269 mg, 634 jmol) en DCM (2 ml) se le añadió TFA (434 mg, 293 jl, 3,8 mmol). Se agitó la mezcla de reacción resultante a TA durante 4 h y a continuación se concentró a vacío (azeótropo con tolueno) proporcionando 264 mg de un sólido blanco, usado en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 325,1 [M+H]+. Etapa a) 6-((4-fluoro-2-(frifluoromefil)fenil)sulfonil)-2,6-diazaespiro[3.3]hepfano-2-carboxilafo de ferc-bufilo En un tubo de vidrio de 20 ml bajo argón, se agitó 2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo (190 mg, 958 jmol) en DCM (3 ml) y TEA (145 mg, 200 jl, 1,44 mmol) durante 5 min a TA, a continuación, se añadió cloruro de 4-fluoro-2-(trifluorometil)bencenosulfonilo (315 mg, 1,2 mmol) (ligeramente exotérmica) y se agitó durante la noche a TA. Se añadieron 10 ml de DCM y gel de sílice, se retiró el disolvente a vacío y la cromatografía sobre 20 g de sílice con heptano/EA (de 0 a 50 % en 35 min) proporcionó el producto deseado como un sólido blanco (269 mg, 66 %). EM (ESI): m/z = 369,1 [M-56+H]+.
BB56
Trifluoroacetato de 6-((2-cloro-4-fluorofenil)sulfonil)-2-azaespiro[3.3]heptano
A una solución de 6-((2-cloro-4-fluorofenil)sulfonil)-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo (209 mg, 536 jmol) en DCM (2 ml) se le añadió TFA (306 mg, 207 jl, 2,68 mmol). Se agitó la mezcla de reacción resultante a TA durante 16 h y a continuación se concentró a vacío (azeótropo con tolueno), se añadieron 5 ml de éter dietílico y se puso la suspensión en un baño ultrasónico, la filtración proporcionó el producto deseado como un sólido blanco (195 mg, 100 %), usado en la siguiente etapa sin purificación adicional. EM (ESI): m/z = 290,1 [M+H]+. Etapa a) 6-((2-cloro-4-fluorofenil)sulfonil)-2-azaespiro[3.3]hepfano-2-carboxilafo de ferc-bufilo En un tubo de vidrio de 20 ml, se disolvieron 6-((metilsulfonil)oxi)-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo (538 mg, 1,84 mmol), 2-cloro-4-fluorobencenotiol (250 mg, 1,54 mmol) y K2CO3 (425 mg, 3,07 mmol) en DMF (7 ml) bajo atmósfera de argón. Se calentó la suspensión a 80°C durante 4 h. Se diluyó la mezcla de reacción con acetato de etilo (20 ml), se lavó dos veces con agua (40 ml), a continuación, con salmuera (40 ml). Se secó la capa orgánica sobre MgSO4, se separa por filtración y se retiró el disolvente bajo presión reducida. Se purificó el producto bruto sobre 50 g de sílice con heptano/EA de 0 a 40 % en 40 min, después de retirar el disolvente a vacío se obtuvo el producto como un aceite viscoso incoloro (218 mg, 38 %). EM (ESI): m/z = 302,1 [M-56+H]+.
Etapa b) 6-((2-cloro-4-fluorofenil)sulfonil)-2-azaespiro[3.3]hepfano-2-carboxilafo de ferc-bufilo
En un tubo de vidrio de 25 ml bajo argón, se disolvió 6-((2-cloro-4-fluorofenil)tio)-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo (218 mg, 609 jmol) en DCM (8 ml ), se añadió mCPBA (315 mg, 1,28 mmol) en porciones a 10-12 °C y se agitó a TA durante 3 h. Se añadieron 10 ml de DCM, se lavó la fase orgánica con NaHCO3 al 5 %, agua y salmuera, se secó con MgSO4 y se retiró el disolvente a vacío. La cromatografía sobre 20 g de sílice con heptano/EA (de 0 a 50 %) proporcionó el producto como 209 mg (88 %) de un sólido blanco. EM (ESI): m/z = 334,1 [M-56+H]+.
BB57
Trifluoroacetato de 2-((3-cloro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano
Se obtuvo BB57 de forma análoga a BB55 comenzando a partir de 2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo y cloruro de 3-doro-4-(trifluorometil)bencenosulfonilo. EM (ESI): m/z = 341,0 [M+H]+.
BB58
Trifluoroacetato de 2-((2,4-bis(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano
Se obtuvo BB58 de forma análoga a BB55 comenzando a partir de 2,6-diazaespiro[3.3]heptano-2-carboxilato de ferc-butilo y cloruro de 2,4-bis(trifluorometil)bencenosulfonilo. EM (ESI): m/z = 419,1 [M+H]+.
BB59
Trifluoroacetato de 6-(2,6-difluorobencil)-2-azaespiro[3.3]heptano
Se obtuvo BB59 de forma análoga a BB42 comenzando a partir de 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo y 1-(bromometil)-2,6-difluoro-benceno. EM (ESI): m/z = 224,1 [M+H]+.
BB60
Trifluoroacetato de 6-(2-fluoro-6-metoxibencil)-2-azaespiro [3.3]heptano
Se obtuvo BB60 de forma análoga a BB42 comenzando a partir de 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo y 1-(bromometil)-2-fluoro-6-metoxibenceno. EM (ESI): m/z = 236,2 [M+H]+.
BB61
Trifluoroacetato de 6-(2-metoxibencil)-2-azaespiro[3.3]heptano
Se obtuvo BB61 de forma análoga a BB42 comenzando a partir de 6-oxo-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo y 1-(bromometil)-2-metoxibenceno. EM (ESI): m/z = 218,2 [M+H]+.
BB62
2-((2-azaespiro[3.3]heptan-6-il)metil)-3-fluorofenol
En un matraz de fondo redondo de 10 ml, se combinó 6-(2-fluoro-6-metoxibencil)-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo (0,050 g, 149 |jmol) con DCM (1 ml) para dar una solución incolora. Se añadió BBr3 (37,3 mg, 14,1 jl, 149 jmol) a 0 °C. Se agitó la reacción a TA durante 3 horas. Se añadió BBr3 (37,3 mg, 14,1 jl, 149 jmol) de nuevo y se agitó la reacción a TA durante la noche. Se desactivó la mezcla de reacción por la adición de solución saturada de NaHCO3 y se extrajo con EtOAc/THF. Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. EM (ESI): m/z = 222,2 [M+H]+.
BB63
2-((2-azaespiro[3.3]heptan-6-il)metil)fenol
Se obtuvo BB61 de forma análoga a BB62 comenzando a partir de 6-(2-metoxibencil)-2-azaespiro[3.3]heptano-2-carboxilato de ferc-butilo. EM (ESI): m/z = 204,2 [M+H]+.
Claims (17)
1. Un compuesto de fórmula (I)

o una sal farmacéuticamente aceptable del mismo, en la que:
(i) X es C-R5;
L es un enlace covalente, -(CH 2)n-N(alquil C1-6)-, -(CH 2)n-NH-, -N(alquil C1-6)-(CH2)p-, -NH-(CH2)p-, -(CH 2)n-O-, -O-(CH2)p-, -SO 2-N(alquil C1-6)-, -SO 2-NH-, -N(alquil C1-6)-SO2-, -NH-SO2-, carbonilo, -(CH2)n- -CHR6-, -CF2-(CH2)n-, -(CH2)p-CF2-, -(CH2)n-S-, -S-(CH2)p-, -SO 2-, -C(O)-NH-, -C(O)-N(alquil C1-6)-, -NH-C(O)- o -N(alquil C1-6)-C(O)-; y
A es:
(i) arilo C6-14 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5 -i4 miembros sustituido con R10, R11 y R12; o
(ii) X es N;
L es un enlace covalente, -(CH 2)n-, -CHRa-, -SO 2-, carbonilo, -N(alquil Ci-a)-(CH2)p-, -NH-(CH2)p-, -O-(CH2)p-, -C F 2-CH2-, -N(alquil C1-6)-SO2-, -NH-SO2-, -NH-C(O)- o -N(alquil Ci-a)-C(O)-; y
A es:
(i) arilo Ca-i4 sustituido con R7, R8 y R9; o
(ii) heteroarilo de 5 -14 miembros sustituido con R10, R11 y R12; o
(iii) X es N;
L es alcoxicarbonilo Ci-a, ariloxicarbonilo Ca-i4 o heteroariloxicarbonilo de 5 -14 miembros; y
A está ausente;
B es un espirociclo bicíclico;
Ri , R2, R3, R4 y R5 son independientemente hidrógeno, halógeno, hidroxi, alquilo C1-6 o halo-alquilo C1-6;
Ra es arilo Ca-i4 o heteroarilo de 5 -i4 miembros;
R7, R 8 , R9, R10, R1 1 y R12 son cada uno en cada aparición independientemente hidrógeno, hidroxi, alquilo C1-6,

C es heteroarilo de 5 -14 miembros, heterociclilo de 3 -14 miembros o cicloalquilo C3-10;
RC1, RC2 y RC3 son cada uno independientemente hidrógeno, alquilo C1-6, halo-alquilo C1-6, oxo, halógeno, hidroxi,
alcoxi C1-6 o halo-alcoxi C1-6;
cada aparición de n es independientemente 0, 1, 2 o 3; y
cada aparición de p es independientemente 1, 2 o 3.
2. El compuesto de fórmula (I) de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en la que R1, R2, R3 y R4 son todos hidrógeno.
3. El compuesto de fórmula (I) de acuerdo con la reivindicación 1 o 2, o una sal farmacéuticamente aceptable del mismo, en la que:

C es heteroarilo de 5-14 miembros o heterociclilo de 3-14 miembros;
RC1 es alquilo C1-6, halo-alquilo C1-6 u oxo; y
RC2 y RC3 son ambos hidrógeno;
R8 es hidrógeno, alcoxi C1-6, halo-alquilo C1-6 o halógeno;
R9 y R12 son ambos hidrógeno;
R10 es halógeno o halo-alquilo C1-6; y
R11 es hidrógeno o halo-alquilo C1-6.
4. El compuesto de fórmula (I) de acuerdo con la reivindicación 3, o una sal farmacéuticamente aceptable del mismo, en la que:
R7 es hidrógeno, alquilo C1-6, halo-alquilo C1-6, halógeno, alcoxi C1-6, halo-alcoxi C1-6 o SF5;
R8 es hidrógeno, halo-alquilo C1-6 o halógeno;
R9 y R12 son ambos hidrógeno;
R10 es halo-alquilo C1-6; y
R11 es hidrógeno o halo-alquilo C1-6.
5. El compuesto de fórmula (I) de acuerdo con la reivindicación 4, o una sal farmacéuticamente aceptable del mismo, en la que
R7 es hidrógeno, flúor, cloro, CF3, metilo, metoxi, trifluorometoxi o SF5;
R8 es hidrógeno, CF3, cloro o flúor;
R9 y R12 son ambos hidrógeno;
R10 es CF3; y
R11 es hidrógeno o CF3.
6. El compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo, en la que A es fenilo o piridilo.
7. El compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 6, o una sal
farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es un enlace covalente, -(CH2)n-N(alquil C1-6)-, -(CH2)n-NH-, -(CH 2)n-O-, -OCH2- , -CH 2-, -SO 2-, -SO 2-N(alquil C1-6) - o -SO 2-NH-;
n es 0 o 1; y
R5 es hidrógeno o halo-alquilo C1-6; o
X es N;
L es un enlace covalente, -CH 2-, -CHR6- o -SO 2-; y
R6 es arilo C6-14.
8. El compuesto de fórmula (I) de acuerdo con la reivindicación 7, o una sal farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es un enlace covalente, -CH 2O-, -O -, -OCH2-, -CH 2- o -SO 2-N(alquil C1-6)-; y
R5 es hidrógeno; o
X es N; y
L es -CH 2- o -SO 2-.
9. El compuesto de fórmula (I) de acuerdo con la reivindicación 8, o una sal farmacéuticamente aceptable del mismo, en la que:
X es C-R5;
L es un enlace covalente, -CH 2O-, -O -, -OCH2-, -CH 2- o -SO 2-N(metil)-; y
R5 es hidrógeno; o
X es N; y
L es -CH 2- o -SO 2-.
10. El compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
X es como se define en el presente documento;
Y1, Y2, Y3 e Y4 son cada uno independientemente -(CH2)m-, -(CH 2)mO-, -O(CH2)m-, -(CH2)mNH- o -NH(CH2)m-; cada aparición de m es independientemente 1, 2 o 3;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I).
11. El compuesto de fórmula (I) de acuerdo con la reivindicación 10, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
X es como se define en el presente documento;
Y1 es -(CH2)m- o -(CH2)mO-, en el que m es 1 o 2;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH 2)m-, en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I).
12. El compuesto de fórmula (I) de acuerdo con la reivindicación 11, o una sal farmacéuticamente aceptable del mismo, en la que B es un espirociclo bicíclico que tiene la fórmula (II):

en la que:
X es como se define en el presente documento;
Y1 es -CH 2-;
Y2 es -CH 2- o -CH 2O-;
Y3 e Y4 son cada uno independientemente -(CH 2)m- , en el que m es 1 o 2;
la línea ondulada indica el punto de unión del espirociclo bicíclico B a L en la fórmula (I); y
el asterisco indica el punto de unión del espirociclo bicíclico B al resto de la fórmula (I).
13. El compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 12, o una sal farmacéuticamente aceptable del mismo, en el que dicho compuesto de fórmula (I) se selecciona del grupo que consiste en:
(4aR,8aS)-6-(6-(2-cloro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)fenoxi)-7-azaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-5-(trifluorometil)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometoxi)fenoxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][ 1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-doro-4-fluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-doro-2-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorofenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(3-fluoro-5-(trifluorometil)fenoxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b ][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-doro-4-fluorobencil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxa cin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespi ro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b] [1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-doro-4-fluorofenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1 ,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(7-(2-fluoro-4-(trifluorometil)bencil)-2,7-diazaespi ro[4,4]nonano-2-carbonil)hexahidro-2H-pirido[4,3-b] [1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoro-4-(trifluorometil)bencil)-2,6-diazaespi ro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][ 1.4] oxacin-3(4H)-ona;
rac-(4aR,8aS)-N-((R)-8-(3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il) bencenosulfonamida;
rac-(4aR,8aS)-N-((S)-8-(3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespi ro[4.5]decan-3-il) bencenosulfonamida;
rac-(4aR,8aS)-6-(2-benzhidrilo-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(4-((4-fluorofenil)sulfonil)-1-oxa-4,9-diazaespiro[5.5]undecano-9-carbonil)hexahidro-2H-pirido[4,3 -b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4,5-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((5,6-bis(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
2-cloro-4-fluoro-N-metil-N-((R)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azae spiro[4.5]decan-3-il)bencenosulfonamida;
(4aR,8aS)-6-(6-((5-(trifluorometil)piridin-2-il)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4-metil-3-(trifluorometil)bencil)oxi)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b] [1.4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-((2-doro-4-fluorofenil)sulfonil)-2,7-diazaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1, 4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)bencil)oxi)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il)-4-(t rifluorometil)bencenosulfonamida;
N-metil-N-((R)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan -3-il)bencenosulfonamida;
2-doro-4-fluoro-N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.
5] decan-3-il)bencenosulfonamida;
N-((S)-8-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-1-oxa-8-azaespiro[4.5]decan-3-il)-3-(t rifluorometil)bencenosulfonamida;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)(metil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-((4-(trifluorometil)fenil)sulfonil)-2,7-diazaespi ro[3.5]nonano-7-carbonil)hexahidro-2H-pi rido[4,3-b][1 ,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(3-((2-doro-4-fluorobencil)amino)-1-oxa-8-azaespiro[4.5]decano-8-carbonil)hexahidro-2H-pirido[ 4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(fenilsulfonil)-2,7-diazaespiro[3.5]nonano-7-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-o na;
rac-6-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)-2,6-diazaespi ro[3.4]octano-2-carboxilato de ferc-butilo;
(4aR,8aS)-6-(6-(4-(1-metil-1H-pirazol-5-il)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-6-hidroxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(6-(2-hidroxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(4-(2-oxopirrolidin-1-il)fenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o xacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-6-metoxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(pentafluoro-16-sulfanil)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,4-difluorobencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(2-metoxi-4-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-doro-4-fluorofenoxi)metil)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o xacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-fluoro-4-(trifluorometil)bencil)oxi)-6-(trifluorometil)-2-azaespi ro[3.3]heptano-2-carbonil)hexahidr o-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-4-(trifluorometil)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(2-(trifluorometil)pirrolidin-1-il)fenil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3 -b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-doro-4-fluorobencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2-fluoro-6-(trifluorometil)bencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1, 4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(4-(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(6-(3-(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaci n-3(4H)-ona;
(4aR,8aS)-6-(2-(4-(trifluorometil)fenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin -3(4H)-ona;
(4aR,8aS)-6-(2-(3-(trifluorometil)fenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin -3(4H)-ona;
(4aR,8aS)-6-(2-(4-isopropoxifenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona;
(4aR,8aS)-6-(6-(4-isopropoxifenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3 (4H)-ona;
(4aR,8aS)-6-(2-(4-metoxi-3-metilfenil)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin -3(4H)-ona;
(4aR,8aS)-6-(2-(4-doro-3-(trifluorometil)fenil)-2,6-diazaespi ro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4 ]oxacin-3(4H)-ona;
(4aR,8aS)-6-(2-(2-fluoropiridin-4-il)-2,6-diazaespiro[3.4]octano-6-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3( 4H)-ona;
(4aR,8aS)-6-(6-(2,5-bis(trifluorometil)fenil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((4-fluoro-2-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirid o[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2-doro-4-fluorofenil)sulfonil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]o xacin-3(4H)-ona;
(4aR,8aS)-6-(6-((3-doro-4-(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido [4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-((2,4-bis(trifluorometil)fenil)sulfonil)-2,6-diazaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4, 3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(6-(2,6-difluorobencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4 H)-ona; y
(4aR,8aS)-6-(6-(2-metoxibencil)-2-azaespiro[3.3]heptano-2-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona.
14. Un procedimiento de fabricación de los compuestos de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 13, o sales farmacéuticamente aceptables de los mismos, que comprende:
(a) hacer reaccionar una primera amina de fórmula 1, en la que R1 y R2 son como se describe en una cualquiera de las reivindicaciones 1 a 13,

con una segunda amina 2 , en la que A, B, L, X, R3 y R4 son como se describe en una cualquiera de las reivindicaciones 1 a 13

en presencia de una base y un reactivo formador de urea, para formar dicho compuesto de fórmula (I); y opcionalmente
(b) transformar dicho compuesto de fórmula (I) en una sal farmacéuticamente aceptable del mismo.
15. Un compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 13 para su uso como sustancia terapéuticamente activa.
16. Una composición farmacéutica que comprende un compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 13 y un vehículo terapéuticamente inerte.
17. Un compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 13 o una composición farmacéutica de acuerdo con la reivindicación 16 para su uso en el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero, en particular para su uso en el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18207725 | 2018-11-22 | ||
| PCT/EP2019/081870 WO2020104494A1 (en) | 2018-11-22 | 2019-11-20 | New heterocyclic compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2955037T3 true ES2955037T3 (es) | 2023-11-28 |
Family
ID=64453331
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19809026T Active ES2955037T3 (es) | 2018-11-22 | 2019-11-20 | Nuevos compuestos heterocíclicos |
Country Status (31)
| Country | Link |
|---|---|
| US (1) | US20210387999A1 (es) |
| EP (1) | EP3883936B1 (es) |
| JP (1) | JP7444882B2 (es) |
| KR (1) | KR20210094540A (es) |
| CN (1) | CN113166170A (es) |
| AR (1) | AR117139A1 (es) |
| AU (1) | AU2019383500A1 (es) |
| BR (1) | BR112021009348A2 (es) |
| CA (1) | CA3119506A1 (es) |
| CL (1) | CL2021001330A1 (es) |
| CO (1) | CO2021007056A2 (es) |
| CR (1) | CR20210247A (es) |
| DK (1) | DK3883936T3 (es) |
| ES (1) | ES2955037T3 (es) |
| FI (1) | FI3883936T3 (es) |
| HR (1) | HRP20231083T1 (es) |
| HU (1) | HUE062913T2 (es) |
| IL (1) | IL283333B2 (es) |
| LT (1) | LT3883936T (es) |
| MA (1) | MA55131B1 (es) |
| MX (1) | MX2021005714A (es) |
| PE (1) | PE20211870A1 (es) |
| PH (1) | PH12021551167A1 (es) |
| PL (1) | PL3883936T3 (es) |
| PT (1) | PT3883936T (es) |
| RS (1) | RS64579B1 (es) |
| SG (1) | SG11202104206SA (es) |
| SI (1) | SI3883936T1 (es) |
| TW (1) | TWI825227B (es) |
| UA (1) | UA128505C2 (es) |
| WO (1) | WO2020104494A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR114136A1 (es) | 2017-10-10 | 2020-07-29 | Hoffmann La Roche | Compuestos heterocíclicos |
| CN111386269A (zh) | 2017-11-28 | 2020-07-07 | 豪夫迈·罗氏有限公司 | 新型杂环化合物 |
| CN111566105A (zh) | 2018-01-08 | 2020-08-21 | 豪夫迈·罗氏有限公司 | 作为MAGL抑制剂的八氢吡啶并[1,2-α]吡嗪类化合物 |
| JP2021534139A (ja) | 2018-08-13 | 2021-12-09 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | モノアシルグリセロールリパーゼ阻害剤としての新規の複素環化合物 |
| US20210094971A1 (en) * | 2019-09-09 | 2021-04-01 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
| US11814375B2 (en) * | 2019-09-12 | 2023-11-14 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
| CR20230115A (es) | 2020-09-03 | 2023-04-11 | Hoffmann La Roche | Compuestos heterocíclicos |
| JP2023542908A (ja) * | 2020-09-18 | 2023-10-12 | シャンハイ ファーマシューティカルズ ホールディング カンパニー,リミティド | カルボニル複素環系化合物及びその使用 |
| US12065445B2 (en) | 2021-01-29 | 2024-08-20 | Cedilla Therapeutics, Inc. | CDK2 inhibitors and methods of using the same |
| WO2022272106A1 (en) | 2021-06-26 | 2022-12-29 | Cedilla Therapeutics, Inc. | Cdk2 inhibitors and methods of using the same |
| WO2023130050A1 (en) * | 2021-12-29 | 2023-07-06 | Psy Therapeutics, Inc. | Monoacylglycerol lipase inhibitors and use thereof for the treatment and management of pain |
| WO2023130043A1 (en) * | 2021-12-29 | 2023-07-06 | Psy Therapeutics, Inc. | Monoacylglycerol lipase inhibitors and use thereof for the treatment of anxiety |
| WO2023144160A1 (en) * | 2022-01-25 | 2023-08-03 | F. Hoffmann-La Roche Ag | New heterocyclic compounds |
| WO2023247670A1 (en) * | 2022-06-24 | 2023-12-28 | F. Hoffmann-La Roche Ag | New heterocyclic-carbonyl-cyclic compounds as magl inhibitors |
| WO2024033277A1 (en) | 2022-08-08 | 2024-02-15 | F. Hoffmann-La Roche Ag | New heterocyclic compounds |
| WO2024033479A1 (en) * | 2022-08-11 | 2024-02-15 | Remynd N.V. | (aza)spiroheptane derivatives for the treatment of neurodegenerative disorders |
| WO2024061853A1 (en) * | 2022-09-20 | 2024-03-28 | F. Hoffmann-La Roche Ag | Fluorescent probes for magl |
| WO2024088922A1 (en) * | 2022-10-24 | 2024-05-02 | F. Hoffmann-La Roche Ag | Heterocyclic compounds as inhibitors of monoacylglycerol lipase (magl) |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7579495B2 (en) | 2003-12-19 | 2009-08-25 | Momentive Performance Materials Inc. | Active-releasing cyclic siloxanes |
| BR112012005382A2 (pt) | 2009-09-10 | 2016-03-29 | Hoffmann La Roche | inibidores de jak |
| WO2011059118A1 (ko) | 2009-11-10 | 2011-05-19 | Kim Hyun Jeen | 후각인지능력 검사 시스템 |
| JP5990297B2 (ja) | 2015-03-03 | 2016-09-14 | タイヨーエレック株式会社 | 遊技機 |
| EP3279191B1 (en) * | 2015-03-30 | 2020-09-02 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| BR112017024678A2 (pt) | 2015-05-21 | 2018-07-31 | Glaxosmithkline Intellectual Property Development Limited | derivados de benzoimidazol como inibidores de pad4 |
| WO2017021805A1 (en) | 2015-07-31 | 2017-02-09 | Pfizer Inc. | 1,1,1-trifluoro-3-hydroxypropan-2-yl carbamate derivatives and 1,1,1-trifluoro-4-hydroxybutan-2-yl carbamate derivatives as magl inhibitors |
| US10385057B2 (en) * | 2015-11-20 | 2019-08-20 | Lundbeck La Jolla Research Center, Inc. | Pyrazole compounds and methods of making and using same |
-
2019
- 2019-11-20 AU AU2019383500A patent/AU2019383500A1/en active Pending
- 2019-11-20 UA UAA202103201A patent/UA128505C2/uk unknown
- 2019-11-20 ES ES19809026T patent/ES2955037T3/es active Active
- 2019-11-20 PT PT198090268T patent/PT3883936T/pt unknown
- 2019-11-20 MX MX2021005714A patent/MX2021005714A/es unknown
- 2019-11-20 HU HUE19809026A patent/HUE062913T2/hu unknown
- 2019-11-20 HR HRP20231083TT patent/HRP20231083T1/hr unknown
- 2019-11-20 PL PL19809026.8T patent/PL3883936T3/pl unknown
- 2019-11-20 IL IL283333A patent/IL283333B2/en unknown
- 2019-11-20 BR BR112021009348-6A patent/BR112021009348A2/pt unknown
- 2019-11-20 LT LTEPPCT/EP2019/081870T patent/LT3883936T/lt unknown
- 2019-11-20 CR CR20210247A patent/CR20210247A/es unknown
- 2019-11-20 SG SG11202104206SA patent/SG11202104206SA/en unknown
- 2019-11-20 WO PCT/EP2019/081870 patent/WO2020104494A1/en active Application Filing
- 2019-11-20 FI FIEP19809026.8T patent/FI3883936T3/fi active
- 2019-11-20 RS RS20230788A patent/RS64579B1/sr unknown
- 2019-11-20 DK DK19809026.8T patent/DK3883936T3/da active
- 2019-11-20 MA MA55131A patent/MA55131B1/fr unknown
- 2019-11-20 JP JP2021528972A patent/JP7444882B2/ja active Active
- 2019-11-20 CN CN201980077284.5A patent/CN113166170A/zh active Pending
- 2019-11-20 PE PE2021000714A patent/PE20211870A1/es unknown
- 2019-11-20 KR KR1020217015287A patent/KR20210094540A/ko unknown
- 2019-11-20 EP EP19809026.8A patent/EP3883936B1/en active Active
- 2019-11-20 SI SI201930619T patent/SI3883936T1/sl unknown
- 2019-11-20 CA CA3119506A patent/CA3119506A1/en active Pending
- 2019-11-21 TW TW108142267A patent/TWI825227B/zh active
- 2019-11-21 AR ARP190103417A patent/AR117139A1/es unknown
-
2021
- 2021-05-20 US US17/325,934 patent/US20210387999A1/en active Pending
- 2021-05-20 CL CL2021001330A patent/CL2021001330A1/es unknown
- 2021-05-21 PH PH12021551167A patent/PH12021551167A1/en unknown
- 2021-05-28 CO CONC2021/0007056A patent/CO2021007056A2/es unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2955037T3 (es) | Nuevos compuestos heterocíclicos | |
| CN109715613B (zh) | 杂环化合物 | |
| RU2769507C1 (ru) | Новые гетероциклические соединения как ингибиторы моноацилглицеринлипазы | |
| ES2821821T3 (es) | Derivados de tetrahidroisoquinolina | |
| JP7249950B2 (ja) | ヘテロ環化合物 | |
| BR112020025642A2 (pt) | compostos heterocíclicos como inibidores da monoacilglicerol lipase | |
| EP4028401B1 (en) | 4,4a,5,7,8,8a-hexapyrido[4,3-b][1,4]oxazin-3-one compounds as magl inhibitors | |
| JP2022549446A (ja) | 新規複素環モノアシルグリセロールリパーゼ(magl)阻害剤 | |
| JP2014518853A (ja) | 新規な二環式窒素含有ヘテロアリールtgr5受容体調節因子 | |
| TW202227415A (zh) | 新穎雜環化合物 | |
| RU2809257C2 (ru) | Новые гетероциклические соединения | |
| RU2801190C2 (ru) | Новые гетероциклические соединения в качестве ингибиторов моноацилглицеринлипазы | |
| EP4416154A1 (en) | Heterocyclic compounds |