ES2947748T3 - Radiofármaco dirigido a PSMA para el diagnóstico y tratamiento del cáncer de próstata - Google Patents
Radiofármaco dirigido a PSMA para el diagnóstico y tratamiento del cáncer de próstata Download PDFInfo
- Publication number
- ES2947748T3 ES2947748T3 ES19775196T ES19775196T ES2947748T3 ES 2947748 T3 ES2947748 T3 ES 2947748T3 ES 19775196 T ES19775196 T ES 19775196T ES 19775196 T ES19775196 T ES 19775196T ES 2947748 T3 ES2947748 T3 ES 2947748T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- integer
- mmol
- bond
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010060862 Prostate cancer Diseases 0.000 title claims abstract description 45
- 208000000236 Prostatic Neoplasms Diseases 0.000 title claims abstract description 45
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 title claims abstract description 30
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 title claims abstract description 30
- 238000003745 diagnosis Methods 0.000 title claims abstract description 12
- 230000008685 targeting Effects 0.000 title abstract description 7
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 title abstract 2
- 239000012217 radiopharmaceutical Substances 0.000 title description 6
- 229940121896 radiopharmaceutical Drugs 0.000 title description 6
- 230000002799 radiopharmaceutical effect Effects 0.000 title description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 204
- 239000002738 chelating agent Substances 0.000 claims abstract description 39
- 229910052751 metal Inorganic materials 0.000 claims abstract description 28
- 239000002184 metal Substances 0.000 claims abstract description 28
- 230000002285 radioactive effect Effects 0.000 claims abstract description 27
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 8
- 150000003839 salts Chemical class 0.000 claims description 37
- 229910052739 hydrogen Inorganic materials 0.000 claims description 35
- 239000001257 hydrogen Substances 0.000 claims description 34
- 125000002947 alkylene group Chemical group 0.000 claims description 32
- 239000000203 mixture Substances 0.000 claims description 28
- 150000002431 hydrogen Chemical class 0.000 claims description 22
- 230000027455 binding Effects 0.000 claims description 18
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 16
- 125000004432 carbon atom Chemical group C* 0.000 claims description 16
- 229910052736 halogen Inorganic materials 0.000 claims description 16
- 150000002367 halogens Chemical class 0.000 claims description 16
- 229910052760 oxygen Inorganic materials 0.000 claims description 16
- 239000001301 oxygen Substances 0.000 claims description 16
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 16
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 14
- OHSVLFRHMCKCQY-NJFSPNSNSA-N lutetium-177 Chemical compound [177Lu] OHSVLFRHMCKCQY-NJFSPNSNSA-N 0.000 claims description 13
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 12
- 229910052717 sulfur Inorganic materials 0.000 claims description 12
- 239000011593 sulfur Substances 0.000 claims description 12
- 239000004480 active ingredient Substances 0.000 claims description 11
- 229940125666 actinium-225 Drugs 0.000 claims description 8
- 125000000217 alkyl group Chemical group 0.000 claims description 8
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 4
- 230000002265 prevention Effects 0.000 claims description 4
- 125000004076 pyridyl group Chemical group 0.000 claims description 4
- 239000004472 Lysine Substances 0.000 abstract description 11
- 210000000056 organ Anatomy 0.000 abstract description 7
- 230000000694 effects Effects 0.000 abstract description 6
- 230000008878 coupling Effects 0.000 abstract description 5
- 238000010168 coupling process Methods 0.000 abstract description 5
- 238000005859 coupling reaction Methods 0.000 abstract description 5
- 238000001727 in vivo Methods 0.000 abstract description 5
- 230000001225 therapeutic effect Effects 0.000 abstract description 5
- 125000003118 aryl group Chemical group 0.000 abstract description 2
- 125000006850 spacer group Chemical group 0.000 abstract description 2
- 210000001519 tissue Anatomy 0.000 abstract description 2
- 230000001747 exhibiting effect Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 360
- 238000002360 preparation method Methods 0.000 description 149
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 120
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 78
- 238000003756 stirring Methods 0.000 description 77
- 238000005160 1H NMR spectroscopy Methods 0.000 description 74
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 71
- 239000000243 solution Substances 0.000 description 48
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 45
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 44
- 238000004128 high performance liquid chromatography Methods 0.000 description 43
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 41
- 238000006243 chemical reaction Methods 0.000 description 41
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 41
- 238000004440 column chromatography Methods 0.000 description 39
- 239000007787 solid Substances 0.000 description 34
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 33
- 235000008504 concentrate Nutrition 0.000 description 33
- 239000012141 concentrate Substances 0.000 description 33
- 239000000376 reactant Substances 0.000 description 29
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 27
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 26
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 23
- 239000007788 liquid Substances 0.000 description 23
- 150000002894 organic compounds Chemical class 0.000 description 23
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 22
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 20
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 20
- 239000012317 TBTU Substances 0.000 description 19
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 19
- 239000000706 filtrate Substances 0.000 description 17
- 206010028980 Neoplasm Diseases 0.000 description 16
- UPWPDUACHOATKO-UHFFFAOYSA-K gallium trichloride Chemical compound Cl[Ga](Cl)Cl UPWPDUACHOATKO-UHFFFAOYSA-K 0.000 description 16
- 239000003960 organic solvent Substances 0.000 description 16
- 150000002148 esters Chemical class 0.000 description 14
- 239000007983 Tris buffer Substances 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- 210000004027 cell Anatomy 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000011541 reaction mixture Substances 0.000 description 12
- 239000002904 solvent Substances 0.000 description 12
- -1 aromatic sulfonic acids Chemical class 0.000 description 11
- 239000012153 distilled water Substances 0.000 description 11
- WLWNRAWQDZRXMB-YLFCFFPRSA-N (2r,3r,4r,5s)-n,3,4,5-tetrahydroxy-1-(4-phenoxyphenyl)sulfonylpiperidine-2-carboxamide Chemical compound ONC(=O)[C@H]1[C@@H](O)[C@H](O)[C@@H](O)CN1S(=O)(=O)C(C=C1)=CC=C1OC1=CC=CC=C1 WLWNRAWQDZRXMB-YLFCFFPRSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 10
- 238000000034 method Methods 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 238000002600 positron emission tomography Methods 0.000 description 9
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- ZNSPHKJFQDEABI-NZQKXSOJSA-N Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 Chemical compound Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 ZNSPHKJFQDEABI-NZQKXSOJSA-N 0.000 description 7
- 229940125876 compound 15a Drugs 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 238000002156 mixing Methods 0.000 description 7
- VPMIAOSOTOODMY-KJAPKAAFSA-N (4r)-6-[(e)-2-[6-tert-butyl-4-(4-fluorophenyl)-2-propan-2-ylpyridin-3-yl]ethenyl]-4-hydroxyoxan-2-one Chemical compound C([C@H](O)C1)C(=O)OC1/C=C/C=1C(C(C)C)=NC(C(C)(C)C)=CC=1C1=CC=C(F)C=C1 VPMIAOSOTOODMY-KJAPKAAFSA-N 0.000 description 6
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 5
- VUDZSIYXZUYWSC-DBRKOABJSA-N (4r)-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2-one Chemical compound FC1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N[C@H](O)CC1 VUDZSIYXZUYWSC-DBRKOABJSA-N 0.000 description 5
- QRDAPCMJAOQZSU-KQQUZDAGSA-N (e)-3-[4-[(e)-3-(3-fluorophenyl)-3-oxoprop-1-enyl]-1-methylpyrrol-2-yl]-n-hydroxyprop-2-enamide Chemical compound C1=C(\C=C\C(=O)NO)N(C)C=C1\C=C\C(=O)C1=CC=CC(F)=C1 QRDAPCMJAOQZSU-KQQUZDAGSA-N 0.000 description 5
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 5
- QCMHGCDOZLWPOT-FMNCTDSISA-N COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 Chemical compound COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 QCMHGCDOZLWPOT-FMNCTDSISA-N 0.000 description 5
- CYSWUSAYJNCAKA-FYJFLYSWSA-N ClC1=C(C=CC=2N=C(SC=21)OCC)OC1=CC=C(C=N1)/C=C/[C@H](C)NC(C)=O Chemical compound ClC1=C(C=CC=2N=C(SC=21)OCC)OC1=CC=C(C=N1)/C=C/[C@H](C)NC(C)=O CYSWUSAYJNCAKA-FYJFLYSWSA-N 0.000 description 5
- QBXVXKRWOVBUDB-GRKNLSHJSA-N ClC=1C(=CC(=C(CN2[C@H](C[C@H](C2)O)C(=O)O)C1)OCC1=CC(=CC=C1)C#N)OCC1=C(C(=CC=C1)C1=CC2=C(OCCO2)C=C1)C Chemical compound ClC=1C(=CC(=C(CN2[C@H](C[C@H](C2)O)C(=O)O)C1)OCC1=CC(=CC=C1)C#N)OCC1=C(C(=CC=C1)C1=CC2=C(OCCO2)C=C1)C QBXVXKRWOVBUDB-GRKNLSHJSA-N 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- SRVFFFJZQVENJC-IHRRRGAJSA-N aloxistatin Chemical compound CCOC(=O)[C@H]1O[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)NCCC(C)C SRVFFFJZQVENJC-IHRRRGAJSA-N 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- OSVHLUXLWQLPIY-KBAYOESNSA-N butyl 2-[(6aR,9R,10aR)-1-hydroxy-9-(hydroxymethyl)-6,6-dimethyl-6a,7,8,9,10,10a-hexahydrobenzo[c]chromen-3-yl]-2-methylpropanoate Chemical compound C(CCC)OC(C(C)(C)C1=CC(=C2[C@H]3[C@H](C(OC2=C1)(C)C)CC[C@H](C3)CO)O)=O OSVHLUXLWQLPIY-KBAYOESNSA-N 0.000 description 5
- 238000004445 quantitative analysis Methods 0.000 description 5
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 5
- 235000010378 sodium ascorbate Nutrition 0.000 description 5
- 229960005055 sodium ascorbate Drugs 0.000 description 5
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- DAAXYQZSKBPJOX-FQEVSTJZSA-N (2S)-2-amino-3-[4-[5-[3-(4-hydroxyphenyl)-4-methoxyphenyl]-1,2,4-oxadiazol-3-yl]phenyl]propanoic acid Chemical compound COC1=C(C=C(C=C1)C2=NC(=NO2)C3=CC=C(C=C3)C[C@@H](C(=O)O)N)C4=CC=C(C=C4)O DAAXYQZSKBPJOX-FQEVSTJZSA-N 0.000 description 4
- VGNCBRNRHXEODV-XXVHXNRLSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-6-dodecoxy-4,7-dihydroxy-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@H](O)[C@H](C(O2)(C(O)=O)C(O)(C(O1)C(O)=O)C(O)=O)OCCCCCCCCCCCC)C1=CC=CC=C1 VGNCBRNRHXEODV-XXVHXNRLSA-N 0.000 description 4
- IGVKWAAPMVVTFX-BUHFOSPRSA-N (e)-octadec-5-en-7,9-diynoic acid Chemical compound CCCCCCCCC#CC#C\C=C\CCCC(O)=O IGVKWAAPMVVTFX-BUHFOSPRSA-N 0.000 description 4
- DPRJPRMZJGWLHY-HNGSOEQISA-N (e,3r,5s)-7-[5-(4-fluorophenyl)-3-propan-2-yl-1-pyrazin-2-ylpyrazol-4-yl]-3,5-dihydroxyhept-6-enoic acid Chemical compound OC(=O)C[C@H](O)C[C@H](O)/C=C/C=1C(C(C)C)=NN(C=2N=CC=NC=2)C=1C1=CC=C(F)C=C1 DPRJPRMZJGWLHY-HNGSOEQISA-N 0.000 description 4
- HNFMVVHMKGFCMB-UHFFFAOYSA-N 3-[3-[4-(1-aminocyclobutyl)phenyl]-5-phenylimidazo[4,5-b]pyridin-2-yl]pyridin-2-amine Chemical compound NC1=NC=CC=C1C1=NC2=CC=C(C=3C=CC=CC=3)N=C2N1C1=CC=C(C2(N)CCC2)C=C1 HNFMVVHMKGFCMB-UHFFFAOYSA-N 0.000 description 4
- MWDVCHRYCKXEBY-LBPRGKRZSA-N 3-chloro-n-[2-oxo-2-[[(1s)-1-phenylethyl]amino]ethyl]benzamide Chemical compound N([C@@H](C)C=1C=CC=CC=1)C(=O)CNC(=O)C1=CC=CC(Cl)=C1 MWDVCHRYCKXEBY-LBPRGKRZSA-N 0.000 description 4
- OVDGUTHABMXVMI-UHFFFAOYSA-N 3-nitro-4-(propylamino)benzoic acid Chemical compound CCCNC1=CC=C(C(O)=O)C=C1[N+]([O-])=O OVDGUTHABMXVMI-UHFFFAOYSA-N 0.000 description 4
- TXEBWPPWSVMYOA-UHFFFAOYSA-N 4-[3-[(1-amino-2-chloroethyl)amino]propyl]-1-[[3-(2-chlorophenyl)phenyl]methyl]-5-hydroxyimidazolidin-2-one Chemical compound NC(CCl)NCCCC1NC(=O)N(Cc2cccc(c2)-c2ccccc2Cl)C1O TXEBWPPWSVMYOA-UHFFFAOYSA-N 0.000 description 4
- TZKBVRDEOITLRB-UHFFFAOYSA-N 4-methyl-n-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1h-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2C=C3C=NNC3=NC=2)=C1 TZKBVRDEOITLRB-UHFFFAOYSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- REDUQXCPUSNJOL-UHFFFAOYSA-N C(C1=CC=CC=C1)NC(CN(C(C1=CC=C(C=C1)C(C)C)=O)CC1=CC=C(C=C1)C(NO)=O)=O Chemical compound C(C1=CC=CC=C1)NC(CN(C(C1=CC=C(C=C1)C(C)C)=O)CC1=CC=C(C=C1)C(NO)=O)=O REDUQXCPUSNJOL-UHFFFAOYSA-N 0.000 description 4
- 206010027476 Metastases Diseases 0.000 description 4
- NPUXORBZRBIOMQ-RUZDIDTESA-N [(2R)-1-[[4-[[3-(benzenesulfonylmethyl)-5-methylphenoxy]methyl]phenyl]methyl]-2-pyrrolidinyl]methanol Chemical compound C=1C(OCC=2C=CC(CN3[C@H](CCC3)CO)=CC=2)=CC(C)=CC=1CS(=O)(=O)C1=CC=CC=C1 NPUXORBZRBIOMQ-RUZDIDTESA-N 0.000 description 4
- KSCRVOKQPYZBHZ-IXPOFIJOSA-N benzyl n-[(2s)-1-[[(2s)-1-[[(2s)-1-(1,3-benzothiazol-2-yl)-1-oxo-3-[(3s)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]carbamate Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C[C@H]1C(NCC1)=O)C(=O)C=1SC2=CC=CC=C2N=1)C(C)C)C(=O)OCC1=CC=CC=C1 KSCRVOKQPYZBHZ-IXPOFIJOSA-N 0.000 description 4
- 238000007664 blowing Methods 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 4
- 230000009401 metastasis Effects 0.000 description 4
- MUJNAWXXOJRNGK-UHFFFAOYSA-N n-[3-(6-methyl-1,2,3,4-tetrahydrocarbazol-9-yl)propyl]cyclohexanamine Chemical compound C1=2CCCCC=2C2=CC(C)=CC=C2N1CCCNC1CCCCC1 MUJNAWXXOJRNGK-UHFFFAOYSA-N 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 238000007911 parenteral administration Methods 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000010898 silica gel chromatography Methods 0.000 description 4
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 3
- SHAHPWSYJFYMRX-GDLCADMTSA-N (2S)-2-(4-{[(1R,2S)-2-hydroxycyclopentyl]methyl}phenyl)propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C[C@@H]1[C@@H](O)CCC1 SHAHPWSYJFYMRX-GDLCADMTSA-N 0.000 description 3
- SLTBMTIRYMGWLX-XMMPIXPASA-N (2r)-2-[(4-chloroanilino)carbamoylamino]-3-(1h-indol-3-yl)-n-(2-phenylethyl)propanamide Chemical compound C1=CC(Cl)=CC=C1NNC(=O)N[C@@H](C(=O)NCCC=1C=CC=CC=1)CC1=CNC2=CC=CC=C12 SLTBMTIRYMGWLX-XMMPIXPASA-N 0.000 description 3
- KRULQRVJXQQPQH-SANMLTNESA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-6-(phenylmethoxycarbonylamino)hexanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)CCCNC(=O)OCC1=CC=CC=C1 KRULQRVJXQQPQH-SANMLTNESA-N 0.000 description 3
- KAFZOLYKKCWUBI-HPMAGDRPSA-N (2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-3-amino-2-[[(2s)-2-[[(2s)-2-(3-cyclohexylpropanoylamino)-4-methylpentanoyl]amino]-5-methylhexanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]butanediamide Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](CCC(C)C)C(=O)N[C@@H](CN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC(N)=O)C(N)=O)C(=O)CCC1CCCCC1 KAFZOLYKKCWUBI-HPMAGDRPSA-N 0.000 description 3
- ITOFPJRDSCGOSA-KZLRUDJFSA-N (2s)-2-[[(4r)-4-[(3r,5r,8r,9s,10s,13r,14s,17r)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H](CC[C@]13C)[C@@H]2[C@@H]3CC[C@@H]1[C@H](C)CCC(=O)N[C@H](C(O)=O)CC1=CNC2=CC=CC=C12 ITOFPJRDSCGOSA-KZLRUDJFSA-N 0.000 description 3
- AEVBPXDFDKBGLT-YOUFYPILSA-N (2s,3s,4r,5r)-n-[2-[4-(diethoxyphosphorylmethyl)anilino]-2-oxoethyl]-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolane-2-carboxamide Chemical compound C1=CC(CP(=O)(OCC)OCC)=CC=C1NC(=O)CNC(=O)[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(NC(=O)C=C2)=O)O1 AEVBPXDFDKBGLT-YOUFYPILSA-N 0.000 description 3
- RXNPEQZHMGFNAY-GEALJGNFSA-N (5R)-4-[(1S,6R)-5-[(2S)-2-(4-chlorophenyl)-3-(propan-2-ylamino)propanoyl]-2,5-diazabicyclo[4.1.0]heptan-2-yl]-5-methyl-6,8-dihydro-5H-pyrido[2,3-d]pyrimidin-7-one Chemical compound C[C@@H]1CC(=O)NC2=C1C(=NC=N2)N3CCN([C@H]4[C@@H]3C4)C(=O)[C@H](CNC(C)C)C5=CC=C(C=C5)Cl RXNPEQZHMGFNAY-GEALJGNFSA-N 0.000 description 3
- UXKLQDCALAWFIU-VKNDCNMPSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-4,7-dihydroxy-6-tetradecoxy-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@H](O)[C@H](C(O2)(C(O)=O)C(O)(C(O1)C(O)=O)C(O)=O)OCCCCCCCCCCCCCC)C1=CC=CC=C1 UXKLQDCALAWFIU-VKNDCNMPSA-N 0.000 description 3
- FJZNNKJZHQFMCK-LRDDRELGSA-N 1-[(3S,4R)-4-(2,6-difluoro-4-methoxyphenyl)-2-oxopyrrolidin-3-yl]-3-phenylurea Chemical compound C1(=CC(=CC(=C1[C@H]1[C@@H](C(=O)NC1)NC(=O)NC1=CC=CC=C1)F)OC)F FJZNNKJZHQFMCK-LRDDRELGSA-N 0.000 description 3
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 3
- YKYWUHHZZRBGMG-JWTNVVGKSA-N 1-methyl-2-[[(1r,5s)-6-[[5-(trifluoromethyl)pyridin-2-yl]methoxymethyl]-3-azabicyclo[3.1.0]hexan-3-yl]methyl]benzimidazole Chemical compound C1([C@@H]2CN(C[C@@H]21)CC=1N(C2=CC=CC=C2N=1)C)COCC1=CC=C(C(F)(F)F)C=N1 YKYWUHHZZRBGMG-JWTNVVGKSA-N 0.000 description 3
- RYWCQJDEHXJHRI-XJMXIVSISA-N 2-[3-[5-[6-[3-[3-(carboxymethyl)phenyl]-4-[(2r,3s,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]hexyl]-2-[(2r,3s,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]phenyl]acetic acid Chemical compound O[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC(C(=C1)C=2C=C(CC(O)=O)C=CC=2)=CC=C1CCCCCCC(C=C1C=2C=C(CC(O)=O)C=CC=2)=CC=C1O[C@@H]1[C@@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 RYWCQJDEHXJHRI-XJMXIVSISA-N 0.000 description 3
- CODBZFJPKJDNDT-UHFFFAOYSA-N 2-[[5-[3-(dimethylamino)propyl]-2-methylpyridin-3-yl]amino]-9-(trifluoromethyl)-5,7-dihydropyrimido[5,4-d][1]benzazepine-6-thione Chemical compound CN(C)CCCC1=CN=C(C)C(NC=2N=C3C4=CC=C(C=C4NC(=S)CC3=CN=2)C(F)(F)F)=C1 CODBZFJPKJDNDT-UHFFFAOYSA-N 0.000 description 3
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 3
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 3
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 3
- IXWOVMRDYFFXGI-UHFFFAOYSA-N 4-(4-methylphenyl)butanoic acid Chemical compound CC1=CC=C(CCCC(O)=O)C=C1 IXWOVMRDYFFXGI-UHFFFAOYSA-N 0.000 description 3
- WCDLCPLAAKUJNY-UHFFFAOYSA-N 4-[4-[3-(1h-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidin-6-yl]phenyl]morpholine Chemical compound C1COCCN1C1=CC=C(C2=CN3N=CC(=C3N=C2)C2=CNN=C2)C=C1 WCDLCPLAAKUJNY-UHFFFAOYSA-N 0.000 description 3
- KUZSBKJSGSKPJH-VXGBXAGGSA-N 5-[(9R)-6-[(3R)-3-methylmorpholin-4-yl]-11-oxa-1,3,5-triazatricyclo[7.4.0.02,7]trideca-2,4,6-trien-4-yl]pyrazin-2-amine Chemical compound C[C@@H]1COCCN1c1nc(nc2N3CCOC[C@H]3Cc12)-c1cnc(N)cn1 KUZSBKJSGSKPJH-VXGBXAGGSA-N 0.000 description 3
- MITGKKFYIJJQGL-UHFFFAOYSA-N 9-(4-chlorobenzoyl)-6-methylsulfonyl-2,3-dihydro-1H-carbazol-4-one Chemical compound ClC1=CC=C(C(=O)N2C3=CC=C(C=C3C=3C(CCCC2=3)=O)S(=O)(=O)C)C=C1 MITGKKFYIJJQGL-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 108010088751 Albumins Proteins 0.000 description 3
- 102000009027 Albumins Human genes 0.000 description 3
- QUMCIHKVKQYNPA-RUZDIDTESA-N C1(CCCCC1)CN1[C@@H](C=2N(C=3C=NC(=NC1=3)NC1=C(C=C(C(=O)NC3CCN(CC3)C)C=C1)OC)C(=NN=2)C)CC Chemical compound C1(CCCCC1)CN1[C@@H](C=2N(C=3C=NC(=NC1=3)NC1=C(C=C(C(=O)NC3CCN(CC3)C)C=C1)OC)C(=NN=2)C)CC QUMCIHKVKQYNPA-RUZDIDTESA-N 0.000 description 3
- 229940126657 Compound 17 Drugs 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- HPKJGHVHQWJOOT-ZJOUEHCJSA-N N-[(2S)-3-cyclohexyl-1-oxo-1-({(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino)propan-2-yl]-1H-indole-2-carboxamide Chemical compound C1C(CCCC1)C[C@H](NC(=O)C=1NC2=CC=CC=C2C=1)C(=O)N[C@@H](C[C@H]1C(=O)NCC1)C=O HPKJGHVHQWJOOT-ZJOUEHCJSA-N 0.000 description 3
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 3
- NELWQUQCCZMRPB-UBPLGANQSA-N [(2r,3r,4r,5r)-4-acetyloxy-5-(4-amino-5-ethenyl-2-oxopyrimidin-1-yl)-2-methyloxolan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1[C@H](OC(C)=O)[C@@H](C)O[C@H]1N1C(=O)N=C(N)C(C=C)=C1 NELWQUQCCZMRPB-UBPLGANQSA-N 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 210000000988 bone and bone Anatomy 0.000 description 3
- AEULIVPVIDOLIN-UHFFFAOYSA-N cep-11981 Chemical compound C1=C2C3=C4CNC(=O)C4=C4C5=CN(C)N=C5CCC4=C3N(CC(C)C)C2=CC=C1NC1=NC=CC=N1 AEULIVPVIDOLIN-UHFFFAOYSA-N 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940126142 compound 16 Drugs 0.000 description 3
- 229940125810 compound 20 Drugs 0.000 description 3
- 229940125846 compound 25 Drugs 0.000 description 3
- 229940125851 compound 27 Drugs 0.000 description 3
- 229940127204 compound 29 Drugs 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 229910001873 dinitrogen Inorganic materials 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 239000012669 liquid formulation Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- HBEDNENASUYMPO-LJQANCHMSA-N n-hydroxy-4-[[(2r)-3-oxo-2-(thiophen-2-ylmethyl)-2,4-dihydroquinoxalin-1-yl]methyl]benzamide Chemical compound C1=CC(C(=O)NO)=CC=C1CN1C2=CC=CC=C2NC(=O)[C@H]1CC1=CC=CS1 HBEDNENASUYMPO-LJQANCHMSA-N 0.000 description 3
- 238000011580 nude mouse model Methods 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- TZSZZENYCISATO-WIOPSUGQSA-N rodatristat Chemical compound CCOC(=O)[C@@H]1CC2(CN1)CCN(CC2)c1cc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)nc(N)n1 TZSZZENYCISATO-WIOPSUGQSA-N 0.000 description 3
- DVWOYOSIEJRHKW-UIRZNSHLSA-M sodium (2S)-2-[[(2S)-2-[[(4,4-difluorocyclohexyl)-phenylmethoxy]carbonylamino]-4-methylpentanoyl]amino]-1-hydroxy-3-[(3S)-2-oxopyrrolidin-3-yl]propane-1-sulfonate Chemical compound FC1(CCC(CC1)C(OC(=O)N[C@H](C(=O)N[C@H](C(S(=O)(=O)[O-])O)C[C@H]1C(NCC1)=O)CC(C)C)C1=CC=CC=C1)F.[Na+] DVWOYOSIEJRHKW-UIRZNSHLSA-M 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- FRACPXUHUTXLCX-BELIEFIBSA-N tert-butyl N-{1-[(1S)-1-{[(1R,2S)-1-(benzylcarbamoyl)-1-hydroxy-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]carbamoyl}-2-cyclopropylethyl]-2-oxopyridin-3-yl}carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CN(C1=O)[C@@H](CC2CC2)C(=O)N[C@@H](C[C@@H]3CCNC3=O)[C@H](C(=O)NCC4=CC=CC=C4)O FRACPXUHUTXLCX-BELIEFIBSA-N 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- JQSHBVHOMNKWFT-DTORHVGOSA-N varenicline Chemical compound C12=CC3=NC=CN=C3C=C2[C@H]2C[C@@H]1CNC2 JQSHBVHOMNKWFT-DTORHVGOSA-N 0.000 description 3
- OGOMLUBUDYFIOG-UHFFFAOYSA-N 4-(4-iodophenyl)butanoic acid Chemical compound OC(=O)CCCC1=CC=C(I)C=C1 OGOMLUBUDYFIOG-UHFFFAOYSA-N 0.000 description 2
- JOFDSYLCZIHGGO-UHFFFAOYSA-N 4-[(4-cyclohexylphenyl)methyl-[2-[[5-(dimethylamino)naphthalen-1-yl]sulfonyl-methylamino]acetyl]amino]-2-hydroxybenzoic acid Chemical compound C1=CC=C2C(N(C)C)=CC=CC2=C1S(=O)(=O)N(C)CC(=O)N(C=1C=C(O)C(C(O)=O)=CC=1)CC(C=C1)=CC=C1C1CCCCC1 JOFDSYLCZIHGGO-UHFFFAOYSA-N 0.000 description 2
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- TZCCKCLHNUSAMQ-DUGSHLAESA-N NC(=O)C[C@H](NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](Cc2ccc(F)cc2)NC(=O)[C@H](Cc3c[nH]c4ccccc34)NC(=O)Cc5cccs5)C(=O)N Chemical compound NC(=O)C[C@H](NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](Cc2ccc(F)cc2)NC(=O)[C@H](Cc3c[nH]c4ccccc34)NC(=O)Cc5cccs5)C(=O)N TZCCKCLHNUSAMQ-DUGSHLAESA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 2
- 102000007066 Prostate-Specific Antigen Human genes 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- ZCXUVYAZINUVJD-GLCXRVCCSA-N [18F]fluorodeoxyglucose Chemical compound OC[C@H]1OC(O)[C@H]([18F])[C@@H](O)[C@@H]1O ZCXUVYAZINUVJD-GLCXRVCCSA-N 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N caprylic alcohol Natural products CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 238000013170 computed tomography imaging Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000013399 early diagnosis Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 2
- QAPTWHXHEYAIKG-RCOXNQKVSA-N n-[(1r,2s,5r)-5-(tert-butylamino)-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](NC(C)(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 QAPTWHXHEYAIKG-RCOXNQKVSA-N 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- HVAMZGADVCBITI-UHFFFAOYSA-N pent-4-enoic acid Chemical compound OC(=O)CCC=C HVAMZGADVCBITI-UHFFFAOYSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 239000008213 purified water Substances 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- GGCZERPQGJTIQP-UHFFFAOYSA-N sodium;9,10-dioxoanthracene-2-sulfonic acid Chemical compound [Na+].C1=CC=C2C(=O)C3=CC(S(=O)(=O)O)=CC=C3C(=O)C2=C1 GGCZERPQGJTIQP-UHFFFAOYSA-N 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 230000001954 sterilising effect Effects 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 2
- ISXSCDLOGDJUNJ-UHFFFAOYSA-N tert-butyl prop-2-enoate Chemical compound CC(C)(C)OC(=O)C=C ISXSCDLOGDJUNJ-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- RWRDJVNMSZYMDV-SIUYXFDKSA-L (223)RaCl2 Chemical compound Cl[223Ra]Cl RWRDJVNMSZYMDV-SIUYXFDKSA-L 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- NQEPBRYUGPSVPU-UHFFFAOYSA-N 1-azido-2-(2-azidoethoxy)ethane Chemical compound [N-]=[N+]=NCCOCCN=[N+]=[N-] NQEPBRYUGPSVPU-UHFFFAOYSA-N 0.000 description 1
- 108010066465 177Lu-PSMA-617 Proteins 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- USTXYASKHZDVMV-UHFFFAOYSA-N 2-(2-azidoethoxy)acetic acid Chemical compound OC(=O)COCCN=[N+]=[N-] USTXYASKHZDVMV-UHFFFAOYSA-N 0.000 description 1
- NDKDFTQNXLHCGO-UHFFFAOYSA-N 2-(9h-fluoren-9-ylmethoxycarbonylamino)acetic acid Chemical compound C1=CC=C2C(COC(=O)NCC(=O)O)C3=CC=CC=C3C2=C1 NDKDFTQNXLHCGO-UHFFFAOYSA-N 0.000 description 1
- OCIIYNXOTJRSHW-UHFFFAOYSA-N 2-[2-(2-azidoethoxy)ethoxy]acetic acid Chemical compound OC(=O)COCCOCCN=[N+]=[N-] OCIIYNXOTJRSHW-UHFFFAOYSA-N 0.000 description 1
- RSTDSVVLNYFDHY-IOCOTODDSA-K 2-[4-[2-[[4-[[(2S)-1-[[(5S)-5-carboxy-5-[[(1S)-1,3-dicarboxypropyl]carbamoylamino]pentyl]amino]-3-naphthalen-2-yl-1-oxopropan-2-yl]carbamoyl]cyclohexyl]methylamino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate lutetium-177(3+) Chemical compound [177Lu+3].OC(=O)CC[C@H](NC(=O)N[C@@H](CCCCNC(=O)[C@H](Cc1ccc2ccccc2c1)NC(=O)C1CCC(CNC(=O)CN2CCN(CC([O-])=O)CCN(CC([O-])=O)CCN(CC([O-])=O)CC2)CC1)C(O)=O)C(O)=O RSTDSVVLNYFDHY-IOCOTODDSA-K 0.000 description 1
- OEYIOHPDSNJKLS-SFIIULIVSA-N 2-hydroxy(211C)ethyl(trimethyl)azanium Chemical compound O[11CH2]C[N+](C)(C)C OEYIOHPDSNJKLS-SFIIULIVSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- VLVCDUSVTXIWGW-UHFFFAOYSA-N 4-iodoaniline Chemical compound NC1=CC=C(I)C=C1 VLVCDUSVTXIWGW-UHFFFAOYSA-N 0.000 description 1
- 125000006306 4-iodophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1I 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 235000005979 Citrus limon Nutrition 0.000 description 1
- 244000131522 Citrus pyriformis Species 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 208000007433 Lymphatic Metastasis Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-N Nitrous acid Chemical compound ON=O IOVCWXUNBOPUCH-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZZXDRXVIRVJQBT-UHFFFAOYSA-M Xylenesulfonate Chemical compound CC1=CC=CC(S([O-])(=O)=O)=C1C ZZXDRXVIRVJQBT-UHFFFAOYSA-M 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-L acetylenedicarboxylate(2-) Chemical compound [O-]C(=O)C#CC([O-])=O YTIVTFGABIZHHX-UHFFFAOYSA-L 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- QQINRWTZWGJFDB-YPZZEJLDSA-N actinium-225 Chemical compound [225Ac] QQINRWTZWGJFDB-YPZZEJLDSA-N 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- 159000000032 aromatic acids Chemical class 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- JCXGWMGPZLAOME-RNFDNDRNSA-N bismuth-213 Chemical compound [213Bi] JCXGWMGPZLAOME-RNFDNDRNSA-N 0.000 description 1
- 238000007469 bone scintigraphy Methods 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- JOYKCMAPFCSKNO-UHFFFAOYSA-N chloro benzenesulfonate Chemical compound ClOS(=O)(=O)C1=CC=CC=C1 JOYKCMAPFCSKNO-UHFFFAOYSA-N 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- PBVFROWIWWGIFK-KWCOIAHCSA-N fluoromethylcholine (18F) Chemical compound [18F]C[N+](C)(C)CCO PBVFROWIWWGIFK-KWCOIAHCSA-N 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 210000000548 hind-foot Anatomy 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000008155 medical solution Substances 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N n-Octanol Natural products CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 210000003049 pelvic bone Anatomy 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-M propynoate Chemical compound [O-]C(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-M 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 201000007094 prostatitis Diseases 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- RWRDJVNMSZYMDV-UHFFFAOYSA-L radium chloride Chemical compound [Cl-].[Cl-].[Ra+2] RWRDJVNMSZYMDV-UHFFFAOYSA-L 0.000 description 1
- 229910001630 radium chloride Inorganic materials 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229910001961 silver nitrate Inorganic materials 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- FVAUCKIRQBBSSJ-FXMLPJBTSA-M sodium;iodine-125(1-) Chemical compound [Na+].[125I-] FVAUCKIRQBBSSJ-FXMLPJBTSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- KKEYFWRCBNTPAC-UHFFFAOYSA-L terephthalate(2-) Chemical compound [O-]C(=O)C1=CC=C(C([O-])=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-L 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- VNWKTOKETHGBQD-JQYAHLJZSA-N tetradeuteriomethane Chemical compound [2H]C([2H])([2H])[2H] VNWKTOKETHGBQD-JQYAHLJZSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- CCRMAATUKBYMPA-UHFFFAOYSA-N trimethyltin Chemical compound C[Sn](C)C.C[Sn](C)C CCRMAATUKBYMPA-UHFFFAOYSA-N 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 229940066799 xofigo Drugs 0.000 description 1
- 229940071104 xylenesulfonate Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0474—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group
- A61K51/0487—Metallocenes, i.e. complexes based on a radioactive metal complexed by two cyclopentadienyl anions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0402—Organic compounds carboxylic acid carriers, fatty acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0474—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0497—Organic compounds conjugates with a carrier being an organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/003—Compounds containing elements of Groups 3 or 13 of the Periodic Table without C-Metal linkages
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pyridine Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
La presente invención se refiere a una composición farmacéutica para el diagnóstico y tratamiento del cáncer de próstata, capaz de dirigirse al PSMA, y un compuesto proporcionado por un aspecto de la presente invención tiene un compuesto de glutamina-urea-lisina al que se acopla estructuralmente un quelante acoplado a un metal radiactivo. y al que se acopla un grupo arilo que puede unirse adicionalmente a la proteína PSMA. El acoplamiento entre el compuesto de glutamina-urea-lisina y el quelante incluye un espaciador polar para cumplir la función de reducir el acoplamiento no específico in vivo y exhibir un efecto de ser eliminado rápidamente de los órganos vitales, pero no del cáncer de próstata. Estas características reducen la exposición a la radiación, que es causada por un compuesto acoplado a un radioisótopo terapéutico, en tejidos y órganos normales y, por lo tanto, reducen los efectos secundarios. Además, (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Radiofármaco dirigido a PSMA para el diagnóstico y tratamiento del cáncer de próstata
Antecedentes de la invención
1. Campo de la invención
La presente invención se refiere a un radiofármaco dirigido a PSMA para diagnosticar y tratar el cáncer de próstata.
2. Descripción de la técnica relacionada
El cáncer de próstata es el cáncer masculino más frecuente en el mundo y ocupa el segundo lugar en mortalidad. El cáncer de próstata suele desarrollarse en hombres mayores de 50 años, y el número de pacientes aumenta rápidamente con la edad. Suele progresar lentamente, pero cuando se convierte en una metástasis maligna, es extremadamente difícil de tratar. La metástasis suele comenzar en los ganglios linfáticos, los huesos de la pelvis, las vértebras y la vejiga alrededor del cáncer de próstata y se extiende gradualmente por todo el cuerpo.
En la actualidad, la prueba del antígeno prostático específico ("Prostate-specific antigen", prueba PSA) y el tacto rectal se utilizan principalmente para el diagnóstico del cáncer de próstata, y también se emplean la ecografía transrectal, la TC, la RM y el escáner óseo de cuerpo entero ("Whole body bone scan", WBBS). También se realizan biopsias para el diagnóstico del cáncer de próstata. Sin embargo, en la mayoría de los casos la precisión diagnóstica es baja y el diagnóstico precoz de la enfermedad es difícil. Además, es difícil determinar la metástasis y también es difícil de distinguir de enfermedades benignas, tales como la hiperplasia prostática y la prostatitis.
La PET ("Positron Emission Tomography", tomografía por emisión de positrones) es una técnica médica de diagnóstico por imagen de una enfermedad que utiliza un isótopo radiactivo de semivida corta que emite positrones. Esta técnica puede utilizarse para el diagnóstico precoz de una enfermedad, la evaluación del tratamiento y la confirmación de metástasis/recidiva.
La [18F]FDG es un radiofármaco de PET representativo utilizado para el diagnóstico del cáncer porque puede observar el metabolismo aumentado de la glucosa de las células cancerosas. Sin embargo, el cáncer de próstata tiene la característica de que es difícil detectarlo precozmente o diagnosticar la progresión de la enfermedad porque la captación de [18F]FDG no es elevada. La colina es un material utilizado para la biosíntesis de la fosfatidilcolina, fundamental para la formación de las membranas celulares, y se sabe que la [11C]colina y la [18F]fluorocolina son más adecuadas para diagnosticar el cáncer de próstata que la [18F]FDG. Sin embargo, tienen una baja sensibilidad para diagnosticar el cáncer de próstata precoz, la metástasis en los ganglios linfáticos y la recidiva, y es difícil distinguir el cáncer de próstata de otros tipos de cáncer.
El antígeno de membrana específico de la próstata ("Prostate-Specific Membrane Antigen", PSMA) es una proteína que se sobreexpresa específicamente en el cáncer de próstata y tiene una actividad enzimática que degrada el N-acetil-L-aspartil-glutamato (NAAL). Se sabe que un compuesto que tiene una estructura de ácido glutámico-urea-lisina (GUL) no se descompone en análogos de NAAL y se une a PSMA muy selectivamente. Hasta la fecha, se han desarrollado varios compuestos con GUL como estructura básica y, entre ellos, los compuestos marcados con F-18 (semivida: 110 minutos) se están desarrollando como radiofármacos de PET para el diagnóstico del cáncer de próstata.
Además del F-18, el Ga-68 es un metal radiactivo que emite positrones y tiene la característica de formar complejos con facilidad con un quelante unido a un precursor, y un compuesto GUL marcado con 68Ga también puede utilizarse como radiofármaco de PET para diagnosticar el cáncer de próstata.
En el caso de un compuesto GUL marcado con 68Ga, este puede utilizarse como tratamiento dirigido al cáncer de próstata al sustituir el Ga-68, un isótopo emisor de positrones, por un metal radiactivo terapéutico que emita rayos beta o partículas alfa. En el caso del 68Ga-PSMA-617, un compuesto marcado con 68Ga, está en marcha un estudio clínico en el que se sintetiza 177Lu-PSMA-617 marcado con Lu-177 (lutecio-177), que emite rayos beta, en lugar de Ga-68 y se está utilizando en pacientes con cáncer de próstata. Se ha notificado de que la mayor parte de un cáncer de próstata que se ha extendido por todo el cuerpo se ha eliminado mediante la administración tres veces repetida.
Además, también se está desarrollando un tratamiento dirigido a PSMA marcado con un isótopo emisor de partículas alfa, y dado que emite más energía que los rayos beta, su efecto terapéutico es mejor. Algunos núclidos representativos son Ac-225 (actinio-225), Bi-213 (bismuto-213), At-211 (astatina-211), etc. En la actualidad, se utiliza Xofigo® se utiliza para tratar el cáncer de próstata que ha hecho metástasis en el hueso, pero se trata de una inyección de 223Ra-RaCl2 (dicloruro de radio), que no tiene ningún efecto terapéutico en el cáncer de próstata formado aparte del hueso.
Los agentes dirigidos al antígeno de membrana específico de la próstata se describen, por ejemplo, en el documento WO 2013/022797 A1, M. Eder et al., Bioconjugate Chemistry, 23 (4), 2012, págs. 688-697,o el documento WO 2017/165473 A1.
Los presentes inventores han completado la presente invención después de confirmar que los nuevos compuestos estructurados dirigidos a PSMA marcados con un metal radiactivo tienen una alta fuerza de unión y selectividad para el PSMA, y también excelentes propiedades farmacocinéticas.
Resumen de la invención
Un objetivo de la presente invención consiste en proporcionar un compuesto en el que un quelante radiactivo acoplado a un metal esté acoplado a un compuesto de ácido glutámico-urea-lisina que tiene una excelente fuerza de unión a la proteína PSMA y presenta excelentes propiedades farmacocinéticas in vivo, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo.
Otro objetivo de la presente invención consiste en proporcionar un compuesto en el que un quelante esté acoplado a un compuesto de ácido glutámico-urea-lisina que tiene una excelente fuerza de unión a la proteína PSMA y presenta excelentes propiedades farmacocinéticas in vivo, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo.
Otro objetivo de la presente invención consiste en proporcionar una composición para su uso en el diagnóstico del cáncer de próstata que comprende el compuesto como principio activo.
Otro objetivo de la presente invención consiste en proporcionar una composición farmacéutica para su uso en la prevención o el tratamiento del cáncer de próstata que comprende el compuesto como principio activo.
Para lograr los objetivos anteriores, en un aspecto de la presente invención, la presente invención proporciona un compuesto representado por la siguiente fórmula 1, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo:
[Fórmula 1]
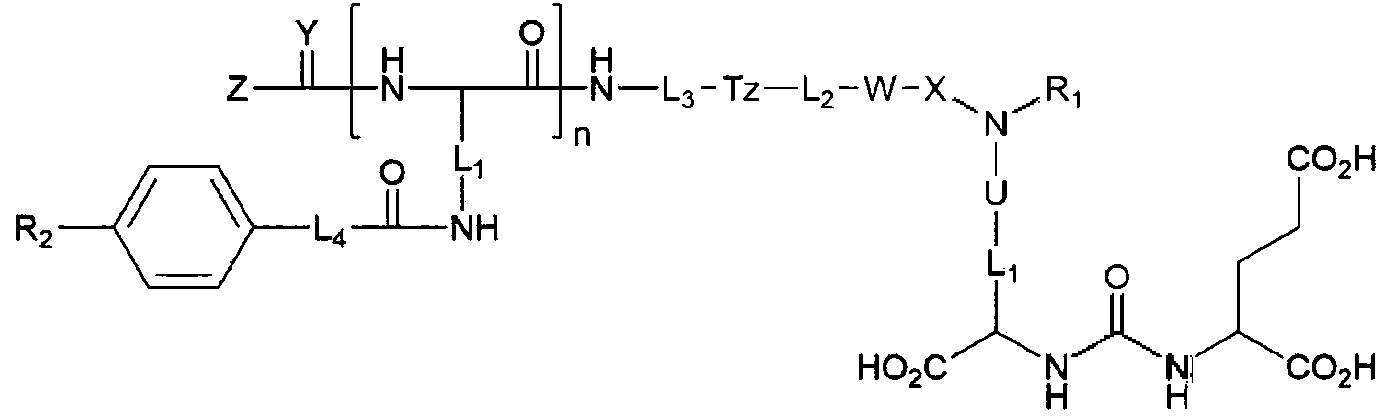
en la que, en la fórmula 1,
L1 es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 6;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, A1 es hidrógeno, o -(CH2)c-piridilo, en el que c es un número entero de 0 a 3;
L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64, Cu-67, Y-90, Sc-47, In-111, Sn-117m, Lu-177, Bi-212, Bi-213, Pb-212, Ra-223, o Ac-225, y el quelante es

En otro aspecto de la presente invención, la presente invención proporciona un compuesto representado por la siguiente fórmula 2, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo:
[Fórmula 2]

en la que, en la fórmula 2,
L1 es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 6;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, A1 es hidrógeno, o -(CH2)c-piridilo, en el que c es un número entero de 0 a 3;
L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6 ;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z' es un quelante, en el que el quelante es

o

En otro aspecto de la presente invención, la presente invención proporciona una composición para su uso en el diagnóstico del cáncer de próstata que comprende el compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo como principio activo.
En otro aspecto de la presente invención, la presente invención proporciona una composición farmacéutica para su uso en la prevención o el tratamiento del cáncer de próstata que comprende el compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo como principio activo.
Efecto ventajoso
Los compuestos proporcionados por un aspecto de la presente invención en los que se introduce el ácido carboxílico unido a la lisina del ácido glutámico-urea-lisina (GUL) forman una fuerte interacción de puente salino con el parche de arginina en el sitio de unión a la proteína PSMA, lo que resulta en un alto poder de unión. Estos compuestos se caracterizan por un rápido efecto de eliminación de la radiación de fondo y una baja unión inespecífica in vivo debido a las características hidrófilas del ácido carboxílico. Además, los compuestos se ingieren en altas concentraciones en tumores o cánceres que expresan la proteína PSMA al mantener un largo tiempo de residencia en sangre.
Breve descripción de los dibujos
La figura 1 es un gráfico que muestra los resultados del análisis cuantitativo de las imágenes MicroPET/CT adquiridas durante 270 minutos tras la administración de [68Ga]1e.
La figura 2 es un gráfico que muestra los resultados del análisis cuantitativo de las imágenes MicroPET/CT adquiridas durante 270 minutos tras la administración de [68Ga]1g.
La figura 3 es un gráfico que muestra los resultados del análisis cuantitativo de las imágenes MicroPET/CT adquiridas durante 390 minutos tras la administración de [68Ga]1h.
La figura 4 es un gráfico que muestra los resultados del análisis cuantitativo de las imágenes MicroPET/CT adquiridas durante 390 minutos tras la administración de [68Ga]1k.
Descripción de las realizaciones preferidas
A continuación se describe en detalle la presente invención.
El alcance de la presente invención está limitado únicamente por las reivindicaciones. Los expertos en la materia entienden que las realizaciones de la presente invención se ofrecen para explicar la presente invención con mayor precisión. Además, la "inclusión" de un elemento a lo largo de la memoria descriptiva no excluye otros elementos, sino que puede incluir otros elementos, a menos que se indique específicamente lo contrario.
En un aspecto de la presente invención, la presente invención proporciona un compuesto representado por la siguiente fórmula 1, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo como principio activo:
[Fórmula 1]

en la que, en la fórmula 1,
L1 es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 6;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, A1 es hidrógeno, o -(CH2)c-piridilo, en el que c es un número entero de 0 a 3; L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64, Cu-67, Y-90, Sc-47, In-111, Sn-117m, Lu-177, Bi-212, Bi-213, Pb-212, Ra-223, o Ac-225, y el quelante es

En otro aspecto de la presente invención,
L1 es -(CH2)a, en el que a es un número entero de 1 a 6;
U es un enlace, o -C(O)-;
R1 es o -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 4;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, A1 es hidrógeno, o -(CH2)c-piridilo, en el que c es un número entero de 0 a 1; L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 6;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 2 a 4;
n es un número entero de 0 a 1;
R2 es hidrógeno, alquilo C1-3 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64, Cu-67, Y-90, Sc-47, In-111, Sn-117m, Lu-177, Bi-212, Bi-213, Pb-212, Ra-223, o Ac-225, y el quelante es

o

En otro aspecto de la presente invención,
L1 es -(CH2)a-, en el que a es un número entero de 2 a 4;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 2;
X es un enlace, o -C(O)-;
W es un enlace, o -NA1-, en el que A1 es hidrógeno o piridilo;
L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 2;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)3-;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, metilo o halógeno;
Y es oxígeno;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64 o Lu-177, y el quelante puede ser

En otro aspecto de la presente invención, el compuesto representado por la fórmula 1 puede ser un compuesto representado por la siguiente fórmula 1-2.
[Fórmula 1-2]

En la fórmula 1-2,
L1 es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
X es un enlace, o -C(O)-;
L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6 ;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64, Cu-67, Y-90, Sc-47, In-111, Sn-117m, Lu-177, Bi-212, Bi-213, Pb-212, Ra-223, o Ac-225, y el quelante es

o
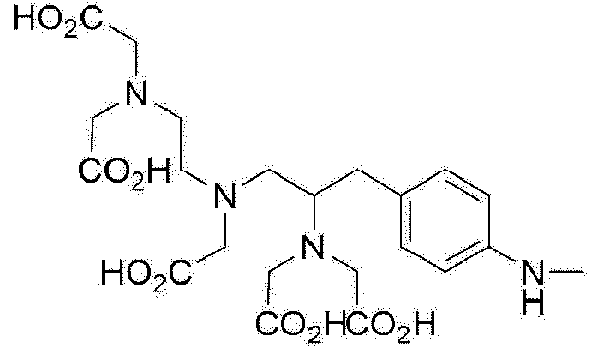
En otro aspecto de la presente invención, el compuesto representado por la fórmula 1 puede ser cualquier compuesto seleccionado del grupo que consiste en los siguientes compuestos.





en las que, en las fórmulas anteriores, M es un metal radiactivo, y el metal radiactivo es como se define en la fórmula 1.
En otro aspecto de la presente invención, la presente invención proporciona un compuesto representado por la siguiente fórmula 2, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo:
[Fórmula 2]

en la que, en la fórmula 2,
Li es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 6;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, y A1 es hidrógeno o -(CH2)c-piridilo, en el que c es un número entero de 0 a 3; L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6 ;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z' es un quelante, y el quelante es

o

En otro aspecto de la presente invención,
L1 es -(CH2)a, en el que a es un número entero de 1 a 6 ;
U es un enlace, o -C(O)-;
R1 es-L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 4;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, y A1 es hidrógeno o -(CH2)c-piridilo, en el que c es un número entero de 0 a 1; L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 6 ;
Tz es un enlace,

L3 es alquileno Cm 2 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 2 a 4;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-3 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z' es un quelante, y el quelante puede ser

o

En otro aspecto de la presente invención,
L1 es -(CH2)a-, en el que a es un número entero de 2 a 4;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 2;
X es un enlace, o -C(O)-;
W es un enlace o-NA1-, y A1 es hidrógeno o piridilo;
L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 2 ;
Tz es

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)3-;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, metilo o halógeno;
Y es oxígeno;
Z' es un quelante, y el quelante puede ser

En otro aspecto de la presente invención, el compuesto representado por la fórmula 2 puede ser un compuesto representado por la siguiente fórmula 2-2:
[Fórmula 2-2]

en la que, en la fórmula 2-2 ,
L1 es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
X es un enlace, o -C(O)-;
L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6 ;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z' es un quelante, y el quelante es

o

En otro aspecto de la presente invención, el compuesto representado por la fórmula 2 puede ser cualquier compuesto seleccionado del grupo que consiste en los siguientes compuestos.

5



El compuesto representado por la fórmula 1 o la fórmula 2 de la presente invención puede utilizarse en forma de una sal farmacéuticamente aceptable, en la que la sal es preferentemente una sal de adición de ácidos formada con ácidos libres farmacéuticamente aceptables. La sal de adición de ácidos del presente documento puede obtenerse a partir de ácidos inorgánicos, tales como ácido clorhídrico, ácido nítrico, ácido fosfórico, ácido sulfúrico, ácido bromhídrico, ácido yodhídrico, ácido nitroso y ácido fosforoso; ácidos orgánicos no tóxicos, tales como mono/dicarboxilato alifático, alcanoato sustituido con fenilo, hidroxialcanoato, alcandioato, ácidos aromáticos y ácidos sulfónicos alifáticos/aromáticos; o ácidos orgánicos, tales como ácido acético, ácido benzoico, ácido cítrico, ácido láctico, ácido maleico, ácido glucónico, ácido metanosulfónico, ácido 4-toluenosulfónico, ácido tartárico y ácido fumárico. Algunos
ejemplos de las sales farmacéuticamente no tóxicas son sulfato, pirosulfato, bisulfato, sulfito, bisulfito, nitrato, fosfato, monohidrógenofosfato, dihidrógenofosfato, metafosfato, pirofosfato, cloruro, bromuro, yoduro, fluoruro, acetato, propionato, decanoato, caprilato, acrilato, formiato, isobutilato, caprato, heptanoato, propiolato, oxalato, malonato, succinato, suberato, cabacato, fumarato, maliato, butino-1,4-dioato, hexano-1,6-dioato, benzoato, clorobenzoato, metilbenzoato, dinitrobenzoato, hidroxibenzoato, metoxibenzoato, ftalato, tereftalato, bencenosulfonato, toluenosulfonato, clorobencenosulfonato, xilenosulfonato, fenilacetato, fenilpropionato, fenilbutilato, citrato, lactato, hidroxibutilato, glicolato, malato, tartrato, metanosulfonato, propanosulfonato, naftaleno-1-sulfonato, naftaleno-2-sulfonato y mandelato.
La sal de adición de ácidos de la presente invención puede prepararse por el procedimiento convencional conocido por los expertos en la materia. Por ejemplo, el derivado representado por la fórmula 1 o la fórmula 2 se disuelve en un disolvente orgánico, tal como metanol, etanol, acetona, cloruro de metilo y acetonitrilo, al que se añade un ácido orgánico o un ácido inorgánico para inducir la precipitación. A continuación, el precipitado se filtra y se seca para obtener la sal, o el disolvente y el exceso de ácido se destilan a presión reducida y se secan para obtener la sal, o el precipitado se cristaliza en un disolvente orgánico para obtener el mismo.
Puede prepararse una sal metálica farmacéuticamente aceptable utilizando una base. La sal de metal alcalino o de metal alcalinotérreo se obtiene mediante los siguientes procesos: disolución del compuesto en una solución en exceso de hidróxido de metal alcalino o de hidróxido de metal alcalinotérreo; filtración de la sal del compuesto insoluble; evaporación de la solución restante y secado de la misma. En este momento, la sal metálica se prepara preferentemente en la forma farmacéuticamente adecuada de sal de sodio, potasio o calcio. La sal de plata correspondiente se prepara mediante la reacción de una sal de metal alcalino o alcalinotérreo con una sal de plata adecuada (por ejemplo, nitrato de plata).
Además, la presente invención incluye no sólo el compuesto representado por la fórmula 1 o la fórmula 2, sino también una sal farmacéuticamente aceptable del mismo, y un solvato, un isómero óptico o un hidrato que puede producirse a partir del mismo.
En otro aspecto de la presente invención, la presente invención proporciona una composición para su uso en el diagnóstico del cáncer de próstata que comprende el compuesto representado por la fórmula 1, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo como principio activo.
La composición para su uso en el diagnóstico del cáncer de próstata puede diagnosticar el cáncer de próstata mediante la unión selectiva del compuesto al PSMA (antígeno de membrana específico de la próstata) sobreexpresado en las células de cáncer de próstata.
En otro aspecto de la presente invención, la presente invención proporciona una composición farmacéutica para su uso en la prevención o el tratamiento del cáncer de próstata que comprende el compuesto representado por la fórmula 1, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo como principio activo.
El compuesto representado por la fórmula 1 y la sal farmacéuticamente aceptable del mismo pueden administrarse por vía oral o parenteral y utilizarse en formas farmacéuticas generales. Es decir, el compuesto representado por la fórmula 1 y la sal farmacéuticamente aceptable del mismo pueden prepararse para la administración oral o parenteral mezclándolo con diluyentes o excipientes de uso general, tales como cargas, extensores, aglutinantes, agentes humectantes, agentes disgregantes y tensioactivos. Las formas farmacéuticas sólidas para la administración oral son comprimidos, píldoras, polvos, gránulos y cápsulas. Estas formas farmacéuticas sólidas se preparan mezclando uno o más compuestos de la presente invención con uno o más excipientes adecuados, tales como almidón, carbonato de calcio, sacarosa o lactosa, gelatina, etc. A excepción de los excipientes sencillos, pueden utilizarse lubricantes, por ejemplo estearato de magnesio, talco, etc. Las formas farmacéuticas líquidas para la administración oral son suspensiones, soluciones, emulsiones y jarabes, y las formas farmacéuticas mencionadas pueden contener diversos excipientes, tales como agentes humectantes, edulcorantes, aromatizantes y conservantes, además de diluyentes sencillos que se emplean habitualmente, tales como agua y parafina líquida. Las formas farmacéuticas para la administración parenteral son soluciones acuosas esterilizadas, excipientes insolubles en agua, suspensiones y emulsiones. Los excipientes y las suspensiones insolubles en agua pueden contener, además del compuesto o compuestos activos, propilenglicol, polietilenglicol, aceite vegetal, tal como aceite de oliva, éster inyectable, tal como etilolato, etc.
La composición farmacéutica que comprende el compuesto representado por la fórmula 1 o la sal farmacéuticamente aceptable del mismo como principio activo de la presente invención puede administrarse por vía parenteral, y la administración parenteral incluye inyección subcutánea, inyección intravenosa, inyección intramuscular o inyección intratorácica.
Para preparar la composición como una forma farmacéutica para la administración parenteral, el compuesto representado por la fórmula 1 o la sal farmacéuticamente aceptable del mismo de la presente invención se mezcla con un estabilizante o un agente tamponante para producir una solución o suspensión, que luego se formula en forma de ampollas o viales. La presente composición puede esterilizarse y contener además conservantes, estabilizantes,
polvos humectables o emulsionantes, sales y/o tampones para la regulación de la presión osmótica y otros materiales terapéuticamente útiles, y la composición puede formularse por el procedimiento convencional de mezclado, granulación o recubrimiento.
Algunos ejemplos de formas farmacéuticas para la administración oral son comprimidos, píldoras, cápsulas duras/blandas, soluciones, suspensiones, emulsiones, jarabes, gránulos, elixires y trociscos, etc. Estas formas farmacéuticas pueden incluir diluyentes (por ejemplo, lactosa, dextrosa, sacarosa, manitol, sorbitol, celulosa y/o glicina) y lubricantes (por ejemplo, sílice, talco, estearato y su sal de magnesio o calcio y/o polietilenglicol), además del principio activo. Los comprimidos pueden incluir agentes aglutinantes, tales como silicato de aluminio y magnesio, pasta de almidón, gelatina, metilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona y, si es necesario, pueden incluirse además agentes disgregantes, tales como almidón, agarosa, ácido algínico o su sal de sodio o mezclas azeotrópicas y/o absorbentes, colorantes, aromatizantes y edulcorantes.
En lo sucesivo, la presente invención se describirá en detalle mediante los siguientes ejemplos.
Ejemplo 1: Preparación de los compuestos 3b y 3c

Preparación del compuesto 3b
El compuesto 3a (5,2 g, 10,66 mmol) se disolvió en diclorometano (100 ml) y se enfrió hasta 0 °C, tras lo cual se añadió lentamente bromoacetato de terc-butilo (1,9 ml, 12,8 mmol). La mezcla se mantuvo a 0 °C, se añadió lentamente trietilamina (2,2 ml, 16 mmol) y la mezcla se agitó mientras se elevaba gradualmente la temperatura hasta la temperatura ambiente. Tras agitar la mezcla durante 3 horas, se añadió agua (50 ml) y el compuesto orgánico se extrajo con diclorometano (50 ml, dos veces). La capa orgánica recogida se trató con sulfato de sodio anhidro, se concentró a presión reducida y se purificó mediante cromatografía en columna (metanol al 5 %metanol/diclorometano) para obtener el compuesto 3b (3,36 g, 52 %).
RMN de 1H (400 MHz, CDCl3) δ 1,39-1,53 (m, 36H), 1,55-1,89 (m, 5H), 2,02-2,10 (m, 1H), 2,22-2,37 (m, 2H), 2,54-2,58 (m, 2H), 3,27 (s, 2H), 4,28-4,36 (m, 2H), 5,07-5,10 (m, 2H);
RMN de 13C (100 MHz, CDCl3) δ 22,6, 27,9, 28,0, 28,1, 28,2, 28,5, 29,6, 31,6, 32,8, 49,0, 51,7, 53,0, 53,5, 80,5, 81,1,81,6, 82,0, 156,8, 171,9, 172,1, 172,4, 172,5;
MS (ESI) m/z 602 [M+H]+
Preparación del compuesto 3c
El compuesto 3a (500 mg, 1,03 mmol) se disolvió en etanol (10 ml), seguido de agitación a 0 °C durante 10 minutos. Se añadió lentamente acrilato de terc-butilo (0,38 ml, 2,58 mmol) y se agitó a 0 °C durante 20 horas. Una vez completada la reacción, se eliminó el disolvente y el concentrado se separó por cromatografía en columna (metanol al 8 %/diclorometano) para obtener el compuesto 3c (0,23 g, 370).
RMN de 1H (400 MHz, CDCl3) δ 1,40 (s, 9H), 1,41 (s, 9H), 1,43 (s, 18H), 1,48-1,65 (m, 3H), 1,70-1,86 (m, 2H), 2,00-2,07 (m, 1H), 2,21-2,36 (m, 2H), 2,48 (t, J = 6,6 Hz, 2H), 2,58- 2,69 (m, 2H), 2,86 (t, J = 6,6 Hz, 2H), 4,26-4,34 (m, 2H), 5,26 (dd, J = 13,0, 8,2 Hz, 2H);
MS (ESI) m/z 616 [M+H]+
Ejemplo 2: Preparación de los compuestos 2a y 2b (no según la invención)

Etapa 1: Preparación del compuesto 5a
Se disolvió trifosgeno (107 mg, 0,36 mmol) en acetonitrilo (5,0 ml) y luego se añadió lentamente a 0 °C el compuesto 3a (500 mg, 1,03 mmol) disuelto en acetonitrilo. A continuación, se añadió trietilamina (0,50 ml, 3,61 mmol) y se agitó durante 30 minutos. Se añadió propagilamina (4a, 0,072 ml, 1,13 mmol) a 0 °C. Después de 15 minutos, la mezcla se agitó a temperatura ambiente durante 1 hora, se concentró a presión reducida y se añadió agua. El compuesto orgánico se extrajo 3 veces con acetato de etilo. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó por cromatografía en columna (metanol al 2 %/diclorometano) para obtener el compuesto 5a (492 mg, 84 %) como un sólido blanco.
RMN de 1H (400 MHz, CDCl3) δ 1,25-1,30 (m, 2H), 1,44 (s, 18H), 1,48 (s, 9H), 1,51-1,60 (m, 3H), 1,67-1,76 (m, 1H), 1,80-1,90 (m, 1H), 2,05-2,13 (m, 1H), 2,18 (t, J = 2,6 Hz, 1H), 2,29-2,40 (m, 2H), 3,06-3,12 (m, 1H), 3,30-3,36 (m, 1H), 3,95-4,06 (m, 2H), 4,08-4,14 (m, 1H), 4,36 (sext., J = 4,4 Hz, 1H), 5,64 (d, J = 7,6 Hz, 1H), 5,69 (t, J = 5,2 Hz, 1H), 5,89 (t, J = 5,4 Hz, 1H), 6,11 (d, J = 8,4 Hz, 1H);
RMN de 13C (100 MHz, CDCla) δ 23,4, 27,7, 27,8, 27,9, 28,0, 29,6, 29,7, 31,7, 32,1, 39,4, 53,3, 54,2, 70,5, 80,7, 81,4, 81,5, 83,1, 158,0, 158,2, 172,0, 172,3, 174,6;
MS (ESI) m/z 569 [M+H]+
Etapa 1: Preparación del compuesto 5b
El compuesto 4b (200 mg, 1,51 mmol) se disolvió en acetonitrilo (5,0 ml) y luego se añadió lentamente cloroformiato de 4-nitrofenilo (305 mg, 1,51 mmol) a 0 °C. Se añadió trietilamina (0,50 ml, 3,61 mmol) y se agitó durante 30 minutos. Se añadió lentamente el compuesto 3a (886 mg, 1,82 mmol) disuelto en acetonitrilo (10 ml) a 0 °C y luego se añadió diisopropiletilamina (0,324 ml, 1,82 mmol). Tras 15 minutos, la mezcla se agitó a 100 °C durante 12 horas. Tras enfriar la mezcla hasta la temperatura ambiente, se le añadió agua. El compuesto orgánico se extrajo 3 veces con acetato de etilo. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 5b (836 mg, 86 %) como un líquido incoloro.
RMN de 1H (400 MHz, CDCl3) δ 1,27-1,37 (m, 2H), 1,43 (s, 9H), 1,45 (s, 18H), 1,50-1,55 (m, 2H), 1,59-1,65 (m, 1H), 1,72-1,88 (m, 2H), 2,01-2,10 (m, 1H), 2,27-2,34 (m, 1H), 2,35 (t, J = 2,4 Hz, 1H), 2,16 (q, J = 6,7 Hz, 2H), 4,25-4,34 (m, 2H), 4,50 (ddd, J = 25,2, 18,0, 2,4 Hz, 2H), 5,21 (t, J = 5,8 Hz, 1H), 5,48 (s, 1H), 5,50 (s, 1H), 7,32 (dd, J = 4,8, 1,6 Hz, 2H), 8,59 (d, J = 6,4 Hz, 2H);
RMN de 13C (100 MHz, CDCl3) δ 22,4, 27,9, 28,0, 28,1, 28,3, 29,4, 31,6, 32,4, 38,2, 40,7, 52,9, 53,3, 72,9, 79,3, 80,5, 81,6, 82,0, 119,5, 149,6, 151,2, 155,3, 157,1, 172,3, 172,4, 172,5;
MS (ESI) m/z 646 [M+H]+
Etapa 2: Preparación del compuesto 6a
El compuesto 5a (0,8 g, 1,4 mmol) y 2-aminoetil 2'-azidoetil éter (0,37 g, 2,81 mmol) se disolvieron en etanol (20 ml) y luego se añadieron CuSO41 M (0,28 ml, 0,28 mmol) y ascorbato de sodio 2 M (0,21 ml, 0,42 mmol), seguido de agitación durante 1 hora. El reactante se filtró y el disolvente se eliminó a presión reducida. El concentrado se separó mediante cromatografía en columna de gel de sílice NH (metanol al 2 %/diclorometano) para obtener el compuesto 6a (0,45 g, 46 %).
MS (ESI) m/z 699 [M+H]+
Etapa 2: Preparación del compuesto 6b
El compuesto 6b (450 mg, 42 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 6a, excepto que se usó el compuesto 5b (880 mg, 1,4 mmol), 2-aminoetil, 2'-azidoetil éter (0,26 g, 2,00 mmol), CuSO41 M (0,27 ml, 0,27 mmol) y ascorbato de sodio 2 M (0,20 ml, 0,41 mmol).
MS (ESI) m/z 776 [M+H]+
Etapa 3: Preparación del compuesto 7a
El éster de DOTA-tris(tBu) (0,44 g, 0,77 mmol) se disolvió en diclorometano (15 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 0,13 g, 0,97 mmol), TBTU (0,31 g, 0,97 mmol) y diisopropiletilamina (0,224 ml, 0,13 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 6a (0,45 g, 0,64 mmol) disuelto en diclorometano (5 ml) y se agitó durante 1 hora. La reacción se terminó añadiendo agua (20 ml) y el compuesto orgánico se extrajo con diclorometano (20 ml x 2). El disolvente orgánico se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 4 %/diclorometano) para obtener el compuesto 7a (0,32 g, 40 %).
RMN de 1H (400 MHz, metanol-d4) δ 1,45-1,50 (m, 69H), 1,52-1,74 (m, 4H), 1,74-1,85 (m, 2H), 1,98-2,09 (m, 4H), 2,25-2,38 (m, 6H), 3,09-3,16 (m, 6H), 3,35-3,43 (m, 4H), 3,47-3,56 (m, 4H), 3,61-3,68 (m, 2H), 3,81-3,86 (m, 4H), 4,11-4,15 (m, 2H), 4,18-4,25 (m, 2H), 4,36-4,38 (m, 4H), 4,55 (t, J = 4,8 Hz, 4H), 7,84 (s, 0,7H), 7,86 (s, 0,3H);
MS (ESI) m/z 1254 [M+H]+
Etapa 3: Preparación del compuesto 7b
El compuesto 7b (0,15 g, 29 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 7a, excepto que se usaron el éster de DOTA-tris(tBu) (270 mg, 0,46 mmol), hidroxibenzotriazol (HOBt, 0,078 g, 0,58 mmol), TBTU (0,19 g, 0,58 mmol), diisopropiletilamina (0,134 ml, 0,77 mmol) y el compuesto 6b (0,30 g, 0,39 mmol).
RMN de 1H (400 MHz, metanol-d4) δ 1,35-1,49 (m, 63H), 1,60-1,68 (m, 1H), 1,73-1,85 (m, 4H), 1,99-2,08 (m, 3H), 2,27-2,34 (m, 4H), 3,20-3,26 (m, 8H), 3,51 (t, J = 5,6 Hz, 4H), 3,69-3,78 (m, 5H), 3,81-3,83 (m, 1H), 4,09 4,23 (m, 1H), 4,46-4,56 (m, 4H), 5,03 (s, 4H), 7,41 (d, J = 6,8 Hz, 2H), 7,94 (s, 1H), 8,43 (d, J = 6,4 Hz, 2H); MS (ESI) m/z 1331 [M+H]+
Etapa 4: Preparación del compuesto 2a
El compuesto 7a (300 mg, 0,24 mmol) se añadió a ácido trifluoroacético al 70 %/diclorometano (6 ml), seguido de agitación durante 5 horas. Se añadió éter dietílico (20 ml) para la precipitación y se separó utilizando una centrifugadora. La mezcla se separó por HPLC y se secó con un liofilizador para obtener el compuesto 2a (115 mg, 52 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,31-1,44 (m, 2H), 1,45-1,55 (m, 2H), 1,66-1,75 (m, 1H), 1,79-1,88 (m, 1H), 1,93-2,02 (m, 1H), 2,14-2,22 (m, 1H), 2,52 (t, J = 7,2 Hz, 2H), 3,11 (t, J = 6,8 Hz, 3H), 3,14-3,55 (m, 26H), 3.58 (t, J = 5,2 Hz, 3H), 3,62-3,93 (m, 6H), 3,96 (t, J = 5,6 Hz, 4H), 4,18 (dd, J = 13,6, 4,8 Hz, 1H), 4,27 (dd, J = 14,4, 5,2 Hz, 1H), 4,39 (s, 2H), 4,61 (t, J = 5,2 Hz, 2H), 7,93 (s, 1H);
MS (ESI) m/z 918 [M+H]+
Etapa 4: Preparación del compuesto 2b
El compuesto 2b (10 mg, 48% ) se obtuvo de la misma manera que la descrita en la preparación del compuesto 2a como un sólido, excepto que se utilizó el compuesto 7a (28 mg, 21 μmol) y ácido trifluoroacético al 70 %/diclorometano (0,4 ml).
RMN de 1H (400 MHz, D2O) δ 1,31-1,44 (m, 4H), 1,51-1,62 (m, 2H), 1,64-1,75 (m, 1H), 1,79-1,87 (m, 1H), 1,90-1,99 (m, 1H), 2,11-2,19 (m, 1H), 2,49 (t, J = 7,6 Hz, 2H), 2,90-3,48 (m, 18H), 5,52 (t, J = 5,2 Hz, 3H), 3,61-3,88 (m, 6H), 3,92 (t, J = 4,8 Hz, 3H), 4,16 (dd, J = 14,0, 5,2 Hz, 1H), 4,25 (dd, J = 14,4, 5,2 Hz, 1H), 4.59 (t, J = 4,4 Hz, 2H), 5,19 (s, 2H), 7,60 (d, J = 7,2 Hz, 2H), 8,03 (s, 1H), 8,42 (d, J = 7,2 Hz, 2H);
MS (ESI) m/z 995 [M+H]+
Ejemplo 3: Preparación de los compuestos 2c y 2d

Etapa 1: Preparación del compuesto 5c
El ácido 4-pentaenoico (82 mg, 0,83 mmol) se disolvió en diclorometano (10 ml) y se enfrió hasta 0 °C, tras lo cual se añadió N,N'-diciclohexilcarbodiimida (190 mg, 0,91 mmol) y el compuesto 3c (0,5 g, 0,83 mmol), seguido de agitación a temperatura ambiente durante 1 hora. La capa orgánica se filtró varias veces y el disolvente se eliminó a presión reducida. El concentrado se separó por cromatografía en columna (acetato de etilo al 30 %/n-hexano) para obtener el compuesto 5c (0,29 g, 52 %).
RMN de 1H (400 MHz, CDCl3) δ 1,34-1,67 (m, 39H), 1,68-2,02 (m, 5H), 2,16-2,32 (m, 2H), 2,37-2,56 (m, 5H), 3,22 (t, J = 7,2 Hz, 1H), 3,29 (t, J = 7,6 Hz, 1H), 3,82-3,90 (m, 2H), 4,17-4,29 (m, 2H), 5,49-5,52 (m, 1,5H), 5,60 (d, J = 8,0 Hz, 0,5 Hz);
MS (ESI) m/z 704 [M+Na]+
Etapa 1: Preparación del compuesto 5d
El compuesto 5d (0,18 g, 79 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 5c, excepto que se utilizaron ácido 4-pentaenoico (32 mg, 0,32 mmol), N,N'-diciclohexilcarbodiimida (74 mg, 0,36 mmol) y el compuesto 3c (0,20 g, 0,32 mmol).
RMN de 1H (400 MHz, CDCla) δ 1,41-1,43(m, 27), 1,44 (s, 9H), 1,51-1,63 (m, 3H), 1,76-1,88 (m, 2H), 1,93 1,96 (m, 1H), 2,01-2,08 (m, 1H), 2,20-2,36 (m, 2H), 2,46-2,53 (m, 5H), 2,57-2,60 (m, 1H), 3,26 (dt, J = 21,2, 7,7 Hz, 2H), 3,52 (q, J = 7,2 Hz, 2H), 4,24-4,35 (m, 2H), 5,05 (dd, J = 16,4, 8,0 Hz, 1H), 5,33 (dd, J = 61,2, 8,0 Hz, 1H);
MS (ESI) m/z 718 [M+Na]+
Etapa 2: Preparación del compuesto 6c
El compuesto 5c (0,26 g, 0,38 mmol) y 2-aminoetil 2'-azidoetil éter (60 mg, 0,46 mmol) se disolvieron en etanol (5 ml), y luego se añadieron CuSO41 M (0,076 ml, 0,076 mmol) y ascorbato de sodio 2 M (0,057 ml, 0,11 mmol), seguido de agitación durante 1 hora. El reactante se filtró y el disolvente se eliminó a presión reducida. El concentrado se separó mediante cromatografía en columna de gel de sílice NH (metanol al 3 %/diclorometano) para obtener el compuesto 6c (0,27 g, 87 %).
RMN de 1H (400 MHz, metanol-d4) δ 1,37-1,55 (m, 36H), 1,56-1,70 (m, 2H), 1,71-1,94 (m, 2H), 1,95-2,14 (m, 2H), 2,24-2,40 (m, 2H), 2,58-2,91 (m, 2H), 2,92-3,12 (m, 2H), 3,33-3,48 (m, 4H), 3,49-3,76 (m, 4H), 3,77-3,92 (m, 2H), 3,96 (s, 1H), 4,45-4,28 (m, 3H), 4,46-4,65 (m, 1H);
MS (ESI) m/z 813 [M+H]+
Etapa 2: Preparación del compuesto 6d
El compuesto 6d (60,0 mg, 50 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 6c, excepto que se usó el compuesto 5d (0,10 g, 0,14 mmol), 2-aminoetil 2'-azidoetil éter (21 mg, 0,16 mmol), CuSO41 M (0,030 ml, 0,030 mmol) y ascorbato de sodio 2 M (0,020 ml, 0,040 mmol).
RMN de 1H (400 MHz, CDCl3) δ 1,23-1,30 (m, 2H), 1,40 (s, 18H), 1,42 (s, 18H), 1,68 (s, 6H), 1,74-1,87 (m, 2H), 1,99-2,09 (m, 1H), 2,24-2,35 (m, 2H), 2,41-2,47 (m, 2H), 2,70-2,75 (m, 1H), 2,96-3,08 (m, 2H), 3,20-3,31 (m, 2H), 3,28-3,54 (m, 3H), 3,81 (t, J = 8,0 Hz, 2H), 4,24-4,42 (m, 2H), 4,47-4,55 (m, 2H), 5,59 (dd, J = 53,4, 7,4 Hz, 1H), 5,77 (dd, J = 37,6, 8,4 Hz, 1H), 7,53 (d, J = 16,4 Hz, 1H);
MS (ESI) m/z 826 (M+H)+
Etapa 3: Preparación del compuesto 7c
El éster de DOTA-tris(tBu) (84 mg, 0,015 mmol) se disolvió en diclorometano (5 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 25 mg, 0,019 mmol), TBTU (59 mg, 0,019 mmol) y diisopropiletilamina (0,042 ml, 0,25 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 6c (100 mg, 0,12 mmol) disuelto en diclorometano (2 ml) y se agitó durante 1 hora. La reacción se terminó añadiendo agua (10 ml) y el compuesto orgánico se extrajo con diclorometano (10 ml x 2). El reactante se secó sobre sulfato de sodio anhidro y se concentró a presión reducida. El concentrado se separó por cromatografía en columna (metanol al 3 %/diclorometano) para obtener el compuesto 7c (95 mg, 56 %).
RMN de 1H (400 MHz, metanol-d4) δ 1,42-1,65 (m, 63H), 1,71-1,85 (m, 2H), 1,95-3,69 (m, 40H), 3,74 (s, 3H), 3,79-3,92 (m, 2H), 3,96 (s, 1H), 4,11-4,20 (m, 3H), 4,50-4,58 (m, 2H);
MS (ESI) m/z 1388 [M+Na]+
Etapa 3: Preparación del compuesto 7d
El compuesto 7d (51 mg, 61 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 7c, excepto que el éster de DOTA-tris(tBu) (29 mg, 0,073 mmol) se disolvió en diclorometano (5 ml) y se usaron hidroxibenzotriazol (HOBt) (12 mg, 0,091 mmol), TBTU (29 mg, 0,091 mmol), diisopropiletilamina (15,86 μl, 91,07 μmol) y el compuesto 6d (50 mg, 60,5 pmol).
RMN de 1H (400 MHz, CDCla) δ 0,75-0,94 (m, 2H), 1,23-1,61 (m, 63H), 1,69 (s, 5H), 1,77-1,87 (m, 2H), 2,00 2,08 (m, 3H), 2,21 (sa, 2H), 2,27-2,37 (m, 3H), 2,41-2,48 (m, 4H), 2,78 (s, 4H), 2,94-3,06 (m, 3H), 3,20-3,38 (m, 5H), 3,43-3,56 (m, 4H), 3,60 (t, J = 5,2 Hz, 2H), 3,66-3,75 (m, 5H), 3,80 (t, J = 4,8 Hz, 2H), 4,19 (d, J = 4,0 Hz, 2H), 4,25-4,34 (m, 2H), 4,50-4,54 (m, 2H), 5,47 (dd, J = 26,8, 8,0 Hz, 1H), 5,66 (dd, J = 12,4, 8,4 Hz, 1H), 7,71 (d, J = 41,2 Hz, 1H)
MS (ESI) m/z 1381 [M+H]+
Etapa 4: Preparación del compuesto 2c
El compuesto 7c (60 mg, 0,044 mmol) se añadió a ácido trifluoroacético al 70 %/diclorometano (2 ml), seguido de agitación durante 4 horas. Se añadió éter dietílico (20 ml) para la precipitación y se separó utilizando una centrifugadora. La mezcla se separó por HPLC y se secó con un liofilizador para obtener el compuesto 2c (25 mg, 58 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,10-1,30 (m, 2H), 1,31-1,50 (m, 2H), 1,52-1,63 (m, 1H), 1,64-1,76 (m, 1H), 1,78-1,89 (m, 1H), 1,99-2,09 (m, 1H), 2,36-2,40 (m, 2H), 2,62-2,65 (m, 1H), 2,77-2,80 (m, 2H), 2,95-2,98 (m, 3H), 3,00-3,19 (m, 7H), 3,21-3,42 (m, 11H), 3,46-3,47 (m, 3H), 3,49-3,72 (m, 4H), 3,82-3,86 (m, 3H), 3,95 (s, 2H), 4,01-4,15 (m, 4H), 4,53-4,56 (m, 2H), 7,84 (s, 1H);
MS (ESI) m/z 974 [M+H]+
Etapa 4: Preparación del compuesto 2d
El compuesto 2d (19 mg, 66% ) se obtuvo de la misma manera que la descrita en la preparación del compuesto 2c como un sólido, excepto que se utilizó el compuesto 7d (40 mg, 0,029 mmol).
RMN de 1H (400 MHz, D2O) δ 1,25-1,42 (m, 2H), 1,44-1,64 (m, 2H), 1,65-1,76 (m, 1H), 1,78-1,91 (m, 1H), 1,92-2,04 (m, 1H), 2,14-2,22 (m, 0,5H), 2,52 (t, J = 7,2 Hz, 2H), 2,59 (t, J = 7,2 Hz, 1,5H), 2,64 (t, J = 7,2 Hz, 1H), 2,81 (t, J = 7,2 Hz, 1H), 2,88 (t, J = 7,2 Hz, 1H), 3,03-3,07 (m, 3H), 3,08-3,54 (m, 19H), 3,55-3,65 (m, 7H), 3,66-3,87 (m, 4H), 3,96 (t, J = 4,8 Hz, 4H), 4,17-4,22 (m, 1H), 4,25-4,28 (m, 1H), 4,62-4,64 (m, 2H), 7,92 (s, 0,6H), 7,93 (s, 0,4H);
MS (ESI) m/z 974 [M+H]+
Ejemplo 4: Preparación del compuesto 2e

Etapa 1: Preparación del compuesto 9a
El compuesto 3b (600 mg, 0,997 mmol) sintetizado en el ejemplo 1 se disolvió en diclorometano (10 ml) y luego se añadió lentamente N,N'-diciclohexilcarbodiimida (DCC, 226 mg, 1,04 mmol) a temperatura ambiente. Se añadió lentamente el ácido 2-(2-(2-azidoetoxi)etoxi)acético 8a (N3-(CH2CH2O)2-CH2COOH, 226 mg, 1,20 mmol) y se agitó durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (acetato de etilo al 60 %/n-hexano) para obtener el compuesto 9a (520 mg, 67 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,44-1,49 (m, 36H), 1,50-1,57 (m, 2H), 1,58-1,71 (m, 2H), 1,73-1,84 (m, 2H), 2,00-2,09 (m, 1H), 2,25-2,38 (m, 2H), 3,33-3,39 (m, 4H), 3,65-3,72 (m, 6H), 3,96 (d, J = 1,2 Hz, 1H), 4,11-4,22 m, 3H), 4,33 (s, 2H), 6,32-6,36 (m, 1H);
MS (ESI) m/z 773 [M+H]+
Etapa 2: Preparación del compuesto 10a
El compuesto 9a (490 mg, 0,634 mmol) sintetizado en la etapa 1 anterior se disolvió en etanol (20 ml) y luego se añadió paladio al 10% sobre carbono (67 mg), seguido de agitación durante 12 horas bajo una atmósfera de hidrógeno. La solución de reacción se filtró, se lavó con etanol y se concentró a presión reducida. El concentrado se separó por cromatografía en columna (metanol al 4 %/diclorometano, gel de sílice NH) para obtener el compuesto 10a (425 mg, 90 %) como un líquido incoloro.
RMN de 1H (400 MHz, metano-d4) δ 1,34-1,39 (m, 2H), 1,44-1,49 (m, 36H), 1,51-1,65 (m, 4H), 1,73-1,84 (m, 2H), 2,00-2,07 (m, 1H), 2,31 (q, J = 6,8 Hz, 2H), 2,80 (t, J = 5,2 Hz, 2H), 3,33-3,40 (m, 1H), 3,52 (q, J = 5,2 Hz, 2H), 3,61-3,66 (m, 3H), 3,69-3,71 (m, 1H), 3,97 (d, J = 1,2 Hz, 1H), 4,11 (s, 1H), 4,13-4,21 (m, 2H), 4,32 (s, 2H);
MS (ESI) m/z 747 [M+H]+
Etapa 3: Preparación del compuesto 11a
El éster de DOTA-tris(tBu) (55 mg, 0,096 mmol) se disolvió en diclorometano (2,0 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 22 mg, 0,160 mmol), TBTU (52 mg, 0,160 mmol) y diisopropiletilamina (0,042 ml, 0,241 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 10a (60 mg, 0,080 mmol) sintetizado en la etapa 2 anterior disuelto en diclorometano (2,0 ml), seguido de agitación a temperatura ambiente durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 3 %/diclorometano) para obtener el compuesto 11a (75 mg, 71 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4 ) δ 1,44-1,50 (m, 63H), 1,54-1,65 (m, 4H), 1,73-1,82 (m, 2H), 2,00-2,09 (m, 3H), 2,15-2,34 (m, 6H), 2,59-3,25 (s a, 16H), 3,38-3,40 (m, 2H), 3,55-3,57 (m, 3H), 3,62 (s, 2H), 3,63-3,69 (m, 3H), 3,97 (s, 2H), 4,08 (s, 2H), 4,09-4,21 (m, 3H), 4,31 (s, 2H);
MS (ESI) m/z 1302 [M+H]+
Etapa 4: Preparación del compuesto 2e
El compuesto 11a (50 mg, 0,038 mmol) sintetizado en la etapa 3 anterior se disolvió en ácido trifluoroacético al 70 %/diclorometano (0,5 ml), seguido de agitación a temperatura ambiente durante 4 horas. El reactante se concentró a presión reducida y se separó por cromatografía de líquidos de alto rendimiento ("high performance liquid chromatography", HPLC) para obtener el compuesto 2e (26 mg, 74 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,16-1,30 (m, 2H), 1,36-1,50 (m, 2H), 1,52-1,62 (m, 1H), 1,64-1,73 (m, 1H), 1,76-1,86 (m, 1H), 1,98-2,06 (m, 1H), 2,35 (td, J = 7,2, 1,6 Hz, 2H), 2,86-3,38 (m, 20H), 3,48-3,60 (m, 10H), 3,70-3,91 (s a, 3H), 3,96 (s, 2H), 4,00-4,12 (m, 3H), 4,25 (s, 2H);
MS (ESI) m/z 910 [M+2H]+
Ejemplo 5: Preparación de los compuestos 2f, 2g y 2h

Etapa 1: Preparación del compuesto 12a
Se disolvió lisina (Fmoc-Lys(Z)-OH, 275 mg, 0,546 mmol) en diclorometano (10 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 123 mg, 0,1910 mmol), TBTU (292 mg, 0,910 mmol) y diisopropiletilamina (0,238 ml, 1,37 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 10a (340 mg, 0,455 mmol) sintetizado en la etapa 2 del ejemplo 4 disuelto en diclorometano (5,0 ml) se añadió, seguido de agitación a temperatura ambiente durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 2 %/diclorometano). Se añadió diclorometano (15 ml) al compuesto obtenido y luego se añadió piperidina (0,043 ml, 0,438 mmol), seguido de agitación a temperatura ambiente durante 24 horas. El reactante se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 3 %/diclorometano, gel de sílice NH) para obtener el compuesto 12a (480 mg, 84 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,34-1,39 (m, 2H), 1,44-1,48 (m, 36H), 1,50-1,70 (m, 10 H), 1,72-1,84 (m, 2H), 2,00-2,08 (m, 1H), 2,24-2,38 (m, 2H), 3,11 (t, J = 6,8 Hz, 2H), 3,33-3,41 (m, 4H), 3,55 (q, J = 5,2 Hz, 2H), 3,61-3,68 (m, 4H), 3,96 (d, J = 1,6 Hz, 1H), 4,08 (d, J = 1,2 Hz, 1H), 4,10-4,21 (m, 3H), 4,31 (s, 1H), 5,06 (s, 2H), 7,27-7,32 (m, 1H), 7,33-7,34 (m, 4H);
MS (ESI) m/z 1010 [M+H]+
Etapa 2: Preparación del compuesto 13a
El éster de DOTA-tris(tBu) (211 mg, 0,369 mmol) se disolvió en diclorometano (10 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 83 mg, 0,614 mmol), Tb Tu (197 mg, 0,614 mmol) y diisopropiletilamina (0,161 ml, 0,921 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 12a (310 mg, 0,307 mmol) sintetizado en la etapa 1 anterior disuelto en diclorometano (5,0 ml), seguido de agitación a temperatura ambiente durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 13a (323 mg, 67 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,36-1,39 (m, 2H), 1,44-1,49 (m, 63H), 1,51-1,73 (m, 8H), 1,75-1,84 (m, 2H), 2,00-2,07 (m, 3H), 2,08-2,26 (m, 4H), 2,28-2,36 (m, 3H), 2,38-3,05 (s a, 12H), 3,11 (t, J = 6,6 Hz, 2H), 3,16-3,28 (m, 4H), 3,36 (t, J = 6,6 Hz, 2H), 3,38-3,52 (s a, 3H), 3,54 (q, J = 4,0 Hz, 2H), 3,58-3,68 (m, 5H), 3,97 (d, J = 4,4 Hz, 1H), 4,07 (s, 1H), 4,09-4,22 (m, 3H), 4,26-4,28 (m, 1H), 4,31 (s, 1H), 5,06 (s, 2H), 7,26 7,32 (m, 1H), 7,33-7,38 (m, 4H);
MS (ESI) m/z 1565 [M+2H]+
Etapa 3: Preparación del compuesto 14a
El compuesto 13a (300 mg, 0,192 mmol) sintetizado en la etapa 2 anterior se disolvió en etanol (20 ml) y luego se añadió paladio al 10 % sobre carbono (20 mg), seguido de agitación durante 2 horas bajo una atmósfera de hidrógeno. La solución de reacción se filtró, se lavó con etanol y se concentró a presión reducida. El concentrado se separó por cromatografía en columna (metanol al 4 %/diclorometano, gel de sílice NH) para obtener el compuesto 14a (260 mg, 95 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,33-1,42 (m, 4H), 1,44-1,49 (m, 63H), 1,51-1,57 (m, 4H), 1,59-1,73 (m, 4H) 1,74-1,85 (m, 3H), 2,00-2,08 (m, 3H), 2,09-2,27 (s a, 4H), 2,29-2,38 (m, 3H), 2,60-2,65 (m, 2H), 2,68 (t, J = 7,2 Hz, 2H), 2,73-2,94 (s a, 7H), 3,05-3,17 (s a, 3H), 3,25-3,28 (m, 2H), 3,34-3,39 (m, 2H), 3,43 (s a, 1H), 3,47-3,39 (m, 1H), 3,53-3,57 (m, 3H), 3,63 (s, 2H), 3,65-3,68 (m, 2H), 3,98 (d, J = 5,6 Hz, 1H), 4,09 (s, 1H), 4,11-4,22 (m, 3H), 4,32 (s, 2H);
MS (ESI) m/z 1430 [M+H]+
Etapa 4: Preparación del compuesto 15a
El ácido 4-fenilbutírico (8,4 mg, 0,050 mmol) se disolvió en diclorometano (1,0 ml) y se añadieron hidroxibenzotriazol (HOBt, 11 mg, 0,084 mmol), TBTU (27 mg, 0,084 mmol) y diisopropiletilamina (0,022 ml, 0,126 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 14a (60 mg, 0,042 mmol) sintetizado en la etapa 3 anterior disuelto en diclorometano (1,0 ml), seguido de agitación a temperatura ambiente durante 2 horas. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 4 %/diclorometano) para obtener el compuesto 15a (17 mg, 26 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,44-1,48 (m, 63H), 1,51-1,71 (m, 6H), 1,74-1,84 (m, 4H), 1,86-1,94 (m, 2H), 1,98-2,15 (m, 6H), 2,19 (t, J = 7,4 Hz, 2H), 2,24-2,34 (m, 3H), 2,36-3,04 (s a, 3H), 2,61 (t, J = 7,8 Hz, 2H), 2,63-3,04 (s a, 10H), 3,12-3,19 (m, 3H), 3,25-3,26 (m, 4H), 3,35-3,37 (m, 4H), 3,47-3,56 (m, 5H), 3,59 3,68 (m, 4H), 3,97 (d, J = 4,0 Hz, 1H), 4,08-4,22 (m, 4H), 4,31 (s, 2H), 7,1-7,18 (m, 3H), 7,24-7,27 (m, 2H);
MS (ESI) m/z 1577 [M+H]+
Etapa 4: Preparación del compuesto 15b
El compuesto 15b (17 mg, 26% ) se obtuvo de la misma manera que la descrita en la preparación del compuesto 15a como un líquido incoloro, excepto que se usaron ácido 4-(p-tolil)butírico (12 mg, 0,063 mmol), hidroxibenzotriazol (HOBt, 14 mg, 0,106 mmol), TBTU (34 mg, 0,106 mmol), diisopropiletilamina (0,028 ml, 0,159 mmol) y el compuesto 14a (76 mg, 0,053 mmol) sintetizado en la etapa 3 anterior.
MS (ESI) m/z 1590 [M+H]+
Etapa 4: Preparación del compuesto 15c
El compuesto 15c (36 mg, 51%) se obtuvo de la misma manera que la descrita en la preparación del compuesto 15a como un líquido incoloro, excepto que se usaron ácido 4-(p-yodofenil)butírico (15 mg, 0,050 mmol), hidroxibenzotriazol (HOBt, 11 mg, 0,084 mmol), t Bt U (27 mg, 0,084 mmol), diisopropiletilamina (0,022 ml, 0,126 mmol) y el compuesto 14a (60 mg, 0,042 mmol) sintetizado en la etapa 3 anterior.
RMN de 1H (400 MHz, metanol-d4) 51,44-1,48 (m, 63H), 1,51-1,57 (m, 4H), 1,58-1,70 (m, 3H), 1,71-1,82 (m, 3H), 1,84-1,92 (m, 3H), 1,93-2,15 (m, 5H), 2,18 (t, J = 7,6 Hz, 2H), 2,20-2,34 (m, 5H), 2,36-2,56 (s a, 3H), 2,58 (t, J = 7,6 Hz, 2H), 2,61-2,76 (s a, 3H), 2,81 (s, 2H), 2,86-3,09 (s a, 5H), 3,11-3,18 (m, 3H), 3,20-3,26 (m, 3H), 3,35-3,39 (m, 2H), 3,42-3,48 (s a, 2H), 3,53 (q, J = 4,0 Hz, 2H), 3,62 (s, 2H), 3,64-3,69 (m, 3H), 3,97 (d, J = 3,6 Hz, 1H), 4,08 (s, 1H), 4,10-4,23 (m, 4H), 4,31 (s, 2H), 6,32-6,36 (m, 1H), 6,99 (d, J = 8,4 Hz, 2H), 7,60 (d, J = 8,0 Hz, 2H);
MS (ESI) m/z 1702 [M+H]+
Etapa 5: Preparación del compuesto 2f
El compuesto 15a (14 mg, 0,0089 mmol) sintetizado en la etapa 4 anterior se disolvió en ácido trifluoroacético al 70 %/diclorometano (0,5 ml), seguido de agitación a temperatura ambiente durante 1 hora. El reactante se concentró a presión reducida y el concentrado se separó por cromatografía de líquidos de alto rendimiento (HPLC) para obtener el compuesto 2f (7 mg, 67 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,22-1,31 (m, 4H), 1,39 (p, J = 7,4 Hz, 2H), 1,42-1,51 (m, 2H), 1,57-1,73 (m, 4H), 1,79 (p, J = 7,6 Hz, 2H), 1,82-1,89 (m, 1H), 2,01-2,09 (m, 1H), 2,13 (t, J = 7,2 Hz, 2H), 2,39 (t, J = 7,2 Hz, 2H), 2,51 (t, J = 7,4 Hz, 2H), 2,53-3,00 (s a, 3H), 3,03 (t, J = 6,8 Hz, 2H), 3,09-3,43 (m, 18H), 3,47 (q, J = 5.2 Hz, 2H), 3,48-3,60 (m, 6H), 3,61-3,91 (s a, 5H), 3,96 (s, 2H), 4,00-4,16 (m, 3H), 4,25 (s, 2H), 7,13-7,17 (m, 3H), 7,22-7,27 (m, 2H);
MS (ESI) m/z 1183 [M+H]+, 1181 [M-H]-
Etapa 5: Preparación del compuesto 2g
El compuesto 2g (10 mg, 36%) se obtuvo de la misma manera que la descrita en la preparación del compuesto 2f como un sólido blanco, excepto que se utilizó el compuesto 15b (37 mg, 0,023 mmol) sintetizado en la etapa 4 anterior y ácido trifluoroacético al 70 %/diclorometano (0,5 ml).
RMN de 1H (400 MHz, D2O) δ 1,14-1,26 (m, 4H), 1,35 (p, J = 6,8 Hz, 2H), 1,39-1,48 (m, 2H), 1,50-1,63 (m, 3H), 1,66-1,69 (m, 1H), 1,72 (p, J = 7,2 Hz, 2H), 1,82 (p, J = 7,2 Hz, 1H), 2,01 (p, J = 7,0 Hz, 1H), 2,08 (t, J = 7.2 Hz, 2H), 2,15 (s, 3H), 2,35 (t, J = 7,2 Hz, 2H), 2,43 (t, J = 7,4 Hz, 2H), 2,66-2,98 (s a, 2H), 2,99 (t, J = 6,6 Hz, 2H), 3,01-3,40 (m, 18H), 3,44 (q, J = 5,0 Hz, 2H), 3,48-3,56 (m, 5H), 3,57-3,88 (s a, 6H), 3,92 (s, 2H), 3,98-4,12 (m, 4H), 4,21 (s, 2H), 7,01 (d, J = 8,0 Hz, 2H), 7,04 (d, J = 8,0 Hz, 2H);
MS (ESI) m/z 1198 [M+H]+, 1196 [M-H]-
Etapa 5: Preparación del compuesto 2h
El compuesto 2h (13 mg, 54%) se obtuvo de la misma manera que la descrita en la preparación del compuesto 15a como un sólido blanco, excepto que se utilizó el compuesto 15c (30 mg, 0,018 mmol) sintetizado en la etapa 4 anterior y ácido trifluoroacético al 70 %/diclorometano (0,5 ml).
RMN de 1H (400 MHz, D2O) δ 1,18-1,30 (m, 4H), 1,37 (p, J = 6,8 Hz, 2H), 1,41-1,52 (m, 2H), 1,54-1,67 (m, 3H), 1,70-1,74 (m, 1H), 1,78 (t, J = 7,4 Hz, 2H), 1,81-1,90 (m, 1H), 1,98-2,07 (m, 1H), 2,11 (t, J = 7,2 Hz, 2H), 2,39 (t, J = 7,2 Hz, 2H), 2,48 (t, J = 7,4 Hz, 2H), 3,00 (t, J = 6,8 Hz, 2H), 3,02-3,26 (m, 17H), 3,29-3,35 (m, 2H), 3,48 (q, J = 4,4 Hz, 2H), 3,52-3,58 (m, 6H), 3,60-3,93 (s a, 7H), 3,97 (s, 2H), 4,03-4,17 (m, 3H), 4,25 (s, 2H), 6,95 (d, J = 8,4 Hz, 2H), 7,59 (d, J = 8,4 Hz, 2H);
MS (ESI) m/z 1309 [M+H]+, 1307 [M-H]-
Ejemplo 6: Preparación del compuesto 2i

Etapa 1: Preparación del compuesto 9b
El compuesto 3b (691 mg, 1,15 mmol) sintetizado en el ejemplo 1 se disolvió en diclorometano (10 ml) y luego se añadió lentamente N,N'-dicidohexilcarbodiimida (DCC, 3710 mg, 1,79 mmol) a temperatura ambiente. Se añadió el ácido 2-(2-azidoetoxi)acético 8b (N3-CH2CH2O-CH2COOH, 200 mg, 1,38 mmol) y se agitó durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (acetato de etilo al 40 %/n-hexano) para obtener el compuesto 9b (670 mg, 80 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,44-1,51 (m, 36H), 1,52-1,67 (m, 4H), 1,71-1,84 (m, 2H), 2,00-2,09 (m, 1H), 2,25-2,38 (m, 2H), 3,36 (q, J = 7,5 Hz, 2H), 3,41-3,45 (m, 2H), 3,66 (t, J = 5,0 Hz, 1H), 3,71 (t, J = 5,0 Hz, 1H), 3,97 (d, J = 0,8 Hz, 1H), 4,11 (d, J = 1,2 Hz, 1H), 4,12-4,23 (m, 3H), 4,32 (s, 2H), 6,34 (p, J = 4,2 Hz, 2H);
MS (ESI) m/z 729 [M+H]+
Etapa 2: Preparación del compuesto 10b
El compuesto 9b (650 mg, 0,892 mmol) sintetizado en la etapa 1 anterior se disolvió en etanol (20 ml) y luego se añadió paladio al 10 % sobre carbono (95 mg), seguido de agitación durante 12 horas bajo hidrógeno. La solución de reacción se filtró, se lavó con etanol y se concentró a presión reducida. El concentrado se separó por cromatografía en columna (metanol al 2 %/diclorometano, gel de sílice NH) para obtener el compuesto 10b (573 mg, 91 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4 ) δ 1,37-1,42 (m, 2H), 1,44-1,49 (m, 36H), 1,51-1,67 (m, 4H), 1,71-1,84 (m, 2H), 1,99-2,09 (m, 1H), 2,29-2,34 (m, 2H), 2,84 (p, J = 5,2 Hz, 2H), 3,35-3,40 (m, 1H), 3,54 (t, J = 5,4 Hz, 1H), 3,59 (t, J = 5,4 Hz, 1H), 3,98 (d, J = 0,8 Hz, 1H), 4,07 (s, 1H), 4,09-4,21 (m, 2H), 4,31 (s, 2H);
MS (ESI) m/z 703 [M+H]+
Etapa 3: Preparación del compuesto 11b
El éster de DOTA-tris(tBu) (82 mg, 0,143 mmol) se disolvió en diclorometano (2,0 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 32 mg, 0,239 mmol), t Bt U (77 mg, 0,239 mmol) y diisopropiletilamina (0,062 ml, 0,358 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 10b (84 mg, 0,120 mmol) sintetizado en la etapa 2 anterior disuelto en diclorometano (2,0 ml), seguido de agitación a temperatura ambiente durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida
y se separó mediante cromatografía en columna (metanol al 2 %/diclorometano) para obtener el compuesto 11b (65 mg, 43 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4 ) δ 1,44-1,49 (m, 63H), 1,51-1,54 (m, 2H), 1,57-1,65 (m, 2H), 1,75-1,84 (m, 2H), 2,04-2,09 (m, 2H), 2,30-2,34 (m, 2H), 2,81-3,25 (s a, 18H), 3,35-3,54 (s a, 7H), 3,55 (t, J = 5,4 Hz, 2H), 3,61 (t, J = 5,2 Hz, 2H), 3,98 (s, 2H), 4,06 (s, 1H), 4,10-4,22 (m, 2H), 4,29 (s, 2H);
MS (ESI) m/z 1258 [M+H]+
Etapa 4: Preparación del compuesto 2i
El compuesto 11b (55 mg, 0,044 mmol) sintetizado en la etapa 3 anterior se disolvió en ácido trifluoroacético al 70 %/diclorometano (0,5 ml), seguido de agitación a temperatura ambiente durante 4 horas. El reactante se concentró a presión reducida y el concentrado se separó por cromatografía de líquidos de alto rendimiento (HPLC) para obtener el compuesto 2i (17 mg, 45 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,20-1,34 (m, 2H), 1,42-1,54 (m, 2H), 1,56-1,65 (m, 1H), 1,70-1,77 (m, 1H), 1,86 (p, J = 7,4 Hz, 1H), 2,05 (p, J = 7,2 Hz, 1H), 2,39 (t, J = 7,2 Hz, 2H), 2,84-3,49 (m, 20H), 3,51 (t, J = 5,0 Hz, 1H), 3,55 (t, J = 4,0 Hz, 1H), 3,58-3,62 (s a, 4H), 3,63-3,95 (s a, 3H), 4,00 (s, 2H), 4,05 (s, 1H), 4,07-4,17 (m, 2H), 4,29 (s, 2H);
MS (ESI) m/z 865 [M+H]+, 863 [M-H]-
Ejemplo 7: Preparación de los compuestos 2j y 2k

Etapa 1: Preparación del compuesto 12b
Se disolvió lisina (Fmoc-Lys(Z)-OH, 386 mg, 0,768 mmol) en diclorometano (10 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 173 mg, 1,28 mmol), TBTU (411 mg, 1,28 mmol) y diisopropiletilamina (0,335 ml, 1,92 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 10b (450 mg, 0,640 mmol) sintetizado en la etapa 2 del ejemplo 6 disuelto en diclorometano (5,0 ml), seguido de agitación a temperatura ambiente durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 2 %/diclorometano). Se añadió diclorometano (15 ml) al compuesto obtenido y luego se añadió piperidina (0,050 ml, 0,505 mmol), seguido de agitación a temperatura ambiente durante 24 horas. El reactante se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 3 %/diclorometano, gel de sílice NH) para obtener el compuesto 12b (380 mg, 61 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,33-1,41 (m, 4H), 1,44-1,48 (m, 36H), 1,51-1,72 (m, 8H), 1,74-1,84 (m, 2H), 1,99-2,08 (m, 1H), 2,24-2,38 (m, 2H), 3,11 (t, J = 6,8 Hz, 2H), 3,24-3,28 (m, 1H), 3,34-3,46 (m, 3H), 3,55
(t, J = 5,4 Hz, 1H), 3,60 (t, J = 5,4 Hz, 1H), 3,96 (s, 1H), 4,05 (d, J = 1,2 Hz, 1H), 4,16-4,22 (m, 3H), 4,29 (s, 1H), 5,05 (s, 2H), 7,26-7,32 (m, 1H), 7,33-7,38 (m, 4H);
MS (ESI) m/z 966 [M+H]+
Etapa 2: Preparación del compuesto 13b
El éster de DOTA-tris(tBu) (271 mg, 0,472 mmol) se disolvió en diclorometano (10 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 106 mg, 0,787 mmol), TBTU (253 mg, 0,787 mmol) y diisopropiletilamina (0,206 ml, 1,18 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 12b (380 mg, 0,394 mmol) sintetizado en la etapa 1 anterior disuelto en diclorometano (5,0 ml), seguido de agitación a temperatura ambiente durante 1 hora. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 3 %/diclorometano) para obtener el compuesto 13b (487 mg, 81 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4 ) δ 1,44-1,49 (m, 63H), 1,51-1,53 (m, 2H), 1,59-1,66 (m, 4H), 1,75-1,84 (m, 4H), 2,01-2,09 (m, 3H), 2,10-2,26 (s a, 4H), 2,27-2,34 (m, 3H), 2,38-2,94 (s a, 12H), 2,95-3,21 (m, 6H), 3,23 3,27 (m, 2H), 3,32-3,64 (m, 8H), 3,99-4,08 (m, 2H), 4,11-4,22 (m, 3H), 4,30 (s, 2H), 5,06 (s, 2H), 7,28-7,31 (m, 1H), 7,33-7,34 (m, 4H);
MS (ESI) m/z 1520 [M+H]+
Etapa 3: Preparación del compuesto 14b
El compuesto 13b (467 mg, 0,307 mmol) sintetizado en la etapa 2 anterior se disolvió en etanol (20 ml) y luego se añadió paladio al 10 % sobre carbono (33 mg), seguido de agitación durante 2 horas bajo hidrógeno. La solución de reacción se filtró, se lavó con etanol y se concentró a presión reducida. El concentrado se separó por cromatografía en columna (metanol al 2 %/diclorometano, gel de sílice NH) para obtener el compuesto 14b (366 mg, 86 %) como un líquido incoloro.
RMN de 1H (400 MHz, metanol-d4) δ 1,44-1,49 (m, 63H), 1,51-1,67 (m, 8H), 1,74-1,84 (m, 3H), 1,89-2,00 (s a, 1H), 2,01-2,08 (m, 2H), 2,09-2,26 (s a, 5H), 2,29-2,34 (m, 2H), 2,36-2,62 (s a, 5H), 2,63-2,68 (m, 2H), 2,70 3,22 (s a, 10H), 3,26 (t, J = 7,6 Hz, 2H), 3,39 (t, J = 7,2 Hz, 2H), 3,42-3,73 (m, 6H), 3,95-4,06 (m, 2H), 4,08 4,21 (m, 4H), 4,32 (s, 2H);
MS (ESI) m/z 1386 [M+H]+
Etapa 4: Preparación del compuesto 15d
Se disolvió ácido 4-(p-tolil)butírico (11 mg, 0,059 mmol) en diclorometano (1,0 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 13 mg, 0,098 mmol), TBTU (32 mg, 0,098 mmol) y diisopropiletilamina (0,026 ml, 0,147 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 14b (68 mg, 0,049 mmol) sintetizado en la etapa 3 anterior disuelto en diclorometano (1,0 ml), seguido de agitación a temperatura ambiente durante 2 horas. Se añadió agua al reactante y, a continuación, el compuesto orgánico se extrajo 3 veces con diclorometano. El disolvente orgánico recogido se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y se separó mediante cromatografía en columna (metanol al 4 %/diclorometano) para obtener el compuesto 15d (60 mg, 42 %) como un líquido incoloro.
Etapa 4: Preparación del compuesto 15e
El compuesto 15e (36 mg, 51%) se obtuvo de la misma manera que la descrita en la preparación del compuesto 15a como un líquido incoloro, excepto que se usaron el ácido 4-(p-yodofenil)butírico (32 mg, 0,104 mmol), hidroxibenzotriazol (HOBt, 23 mg, 0,173 mmol), TBTU (56 mg, 0,173 mmol), diisopropiletilamina (0,045 ml, 0,260 mmol) y el compuesto 14b (120 mg, 0,087 mmol) sintetizado en la etapa 3 anterior.
RMN de 1H (400 MHz, CDCla) δ 0,82-0,96 (m, 2H), 0,97-1,14 (m, 2H), 1,16-1,37 (m, 6H), 1,42-1,50 (m, 63 H), 1,53-1,69 (m, 2H), 1,71-1,96 (m, 5H), 1,99-2,10 (m, 3H), 2,11-2,38 (m, 11H), 2,41-2,68 (m, 6H), 2,81 (s, 3H), 2,82-3,11 (s a, 7H), 3,16-3,33 (m, 5H), 3,35-3,61 (m, 5H), 3,63-3,75 (m, 2H), 3,90 (s, 2H), 4,10 (d, J = 3,2 Hz, 1H), 4,24-4,35 (m, 3H), 5,60 (q, J = 7,6 Hz, 1H), 6,97 (d, J = 8,0 Hz, 2H), 7,55 (d, J = 8,0 Hz, 2H);
MS (ESI) m/z 1658 [M+H]+
Etapa 5: Preparación del compuesto 2j
El compuesto 15d (24 mg, 0,016 mmol) sintetizado en la etapa 4 anterior se disolvió en ácido trifluoroacético al 70 %/diclorometano (0,5 ml), seguido de agitación a temperatura ambiente durante 1 hora. El reactante se concentró a presión reducida y el concentrado se separó por cromatografía de líquidos de alto rendimiento (HPLC) para obtener el compuesto 2j (67 mg 37 %) como un sólido blanco
RMN de 1H (400 MHz, D2O) δ 1,16-1,28 (m, 4H), 1,35-1,48 (m, 4H), 1,52-1,64 (m, 3H), 1,65-1,70 (m, 1H), 1,74 (t, J = 7,6 Hz, 2H), 1,79-1,87 (m, 1H), 2,00-2,05 (m, 1H), 2,10 (t, J = 7,0 Hz, 2H), 2,16 (s, 3H), 2,36 (t, J = 6,6 Hz, 2H), 2,44 (t, J = 7,2 Hz, 2H), 2,82-3,10 (m, 8H), 3,11-3,27 (m, 9H), 3,28-3,38 (m, 4H), 3,39-3,49 (m, 4H), 3,50-3,80 (m, 7H), 3,93 (s, 2H), 3,98-4,12 (m, 3H), 4,19 (s, 2H), 7,02 (d, J = 8,0 Hz, 2H), 7,06 (d, J = 7,2 Hz, 2H) ;
MS (ESI) m/z 1153 [M+H]+, 1151 [M-H]-
Etapa 5: Preparación del compuesto 2k
El compuesto 2k (4,0 mg, 53 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 2j como un sólido blanco, excepto que se utilizó el compuesto 15e (10 mg, 0,0060 mmol) sintetizado en la etapa 4 anterior.
RMN de 1H (400 MHz, D2O) δ 1,15-1,27 (m, 4H), 1,28-1,36 (m, 2H), 1,38-1,48 (m, 2H), 1,54-1,64 (m, 3H), 1,66-1,68 (m, 1H), 1,74 (p, J = 7,4 Hz, 2H), 1,79-1,87 (m, 1H), 1,98-2,03 (m, 1H), 2,08 (t, J = 7,0 Hz, 2H), 2,36 (t, J = 7,4 Hz, 2H), 2,43 (t, J = 7,2 Hz, 2H), 2,71-2,87 (s a, 3H), 2,96 (t, J = 6,6 Hz, 2H), 3,03-3,33 (m, 17H), 3,38-3,49 (m, 3H), 3,52-3,89 (s a, 6H), 3,94 (d, J = 4,4 Hz, 2H), 4,00-4,14 (m, 4H), 4,19 (s, 2H), 6,90 (d, J = 8,4 Hz, 2H), 7,54 (d, J = 8,0 Hz, 2H);
MS (ESI) m/z 1266 [M+H]+, 1264 [M-H]-
Ejemplo 8: Preparación del compuesto 2l

Etapa 1: Preparación del compuesto 16
Se disolvió Fmoc-Gly-OH (0,49 g, 1,66 mmol) en diclorometano (10 ml) y luego se añadió N,N'-diciclohexilcarbodiimida (0,34 g, 1,66 mmol), seguido de agitación a 0 °C durante 10 minutos. Se añadió lentamente el compuesto 3b (0,50 g, 0,83 mmol) disuelto en diclorometano (10 ml) a la mezcla de reacción, seguido de agitación a 0 °C durante 1,5 horas. La mezcla de reacción se filtró, se lavó con diclorometano varias veces y el disolvente se eliminó a presión reducida. El concentrado se separó por cromatografía en columna (acetato de etilo del 35 % al 50 %/n-hexano) para obtener el compuesto 16 (0,45 g, 61 %).
RMN de 1H (400 MHz, CDCl3) δ 1,05-1,21 (m, 2H), 1,38 (s, 9H), 1,40 (s, 9H), 1,43 (s, 9H), 1,45 (s, 9H), 1,56 1,62 (m, 2H), 1,64-1,71 (m, 2H), 1,75-1,86 (m, 2H), 1,88-1,94(m, 2H), 2,19-2,35 (m, 2H), 3,16-3,293,88-4,02 (m, 2H), 4,19-4,28 (m, 2H), 4,29-4,40 (m, 3H), 5,23 (dt, J = 64,4, 12,0 Hz, 2H), 5,96 (dt, J = 105,2, 4,4 Hz, 1H), 7,27-7,30 (m, 2H), 7,37 (t, J = 13,4 Hz, 2H), 7,55 (d, J = 7,2 Hz, 2H), 7,74 (d, J = 7,2 Hz, 2H);
MS (ESI) m/z 881 [M+H]+
Etapa 2: Preparación del compuesto 10c
El compuesto 16 (0,40 g, 0,45 mmol) se disolvió en diclorometano (10 ml) y luego se añadió piperidina (0,1 ml, 1,01 mmol), seguido de agitación a temperatura ambiente durante 1,5 horas. La mezcla de reacción se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 10c (0,19 g, 63 %).
RMN de 1H (400 MHz, CDCla) δ 1,41 (s, 9H), 1,43 (s, 9H), 1,46 (s, 9H), 1,47 (s, 9H), 1,53-1,64 (m, 2H), 1,75 1,89 (m, 2H), 2,02-2,11 (m, 2H), 2,24-2,38 (m, 2H), 3,23 (t, J = 7,4 Hz, 2H), 3,63 (s, 2H), 3,69 (s, 2H), 3,84 3,97 (m, 2H), 4,22-4,37 (m, 2H), 5,46-5,84 (m, 2H);
MS (ESI) m/z 659 [M+H]+
Etapa 3: Preparación del compuesto 11c
El éster de DOTA-tris(tBu) (31 mg, 55 |jmol) se disolvió en diclorometano (0,6 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 9 mg, 68 jmol), TBTU (22 mg, 68 jm ol) y diisopropiletilamina (16 jl, 91 jmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 10c (30 mg, 45,5 umol) disuelto en diclorometano (0,3 ml) y se agitó a temperatura ambiente durante 1 hora. La reacción se terminó añadiendo agua (10 ml), y el compuesto orgánico se extrajo con diclorometano (10 ml x 3). El reactante se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 11c (47,4 mg, 86 %).
RMN de 1H (400 MHz, CDCla) δ 1,46 (s, 9H), 1,47 (s, 27H), 1,48 (s, 27H), 1,68 (s, 6H), 1,73-1,80 (m, 1H), 1,78-1,90 (m, 1H), 2,04-2,15 (m, 3H), 2,20-2,48 (m, 6H), 2,83 (s, 9H), 3,00 (s a, 3H), 3,27 (t, J = 7,4 Hz, 2H), 3,32-3,41 (m, 2H), 3,44-3,54 (m, 2H), 3,91-4,14 (m, 3H), 4,20-4,40 (m, 2H), 5,25-5,34 (m, 1H), 5,46 (dd, J =7,4 Hz, 1H);
MS (ESI) m/z 1213 [M+H]+
Etapa 4: Preparación del compuesto 2l
El compuesto 11c (40 mg, 32,96 μmol) se añadió a ácido trifluoroacético al 70 %/diclorometano (1 ml), seguido de agitación durante 5 horas. Después añadir gota a gota la mezcla de reacción a éter dietílico (40 ml) para formar un precipitado, este se separó utilizando una centrífuga. A continuación, se obtuvo el compuesto 2l (15 mg, 55%) mediante cromatografía de líquidos de alto rendimiento (HPLC).
RMN de 1H (400 MHz, D2O) δ 1,29-1,38 (m, 2H), 1,46 (s a, 1H), 1,54-1,62 (m, 2H), 1,63-1,70 (m, 1H), 1,75 1,82 (m, 1H), 1,83-1,93 (m, 1H), 2,05-2,13 (m, 1H), 2,42 (t, J = 7,2 Hz, 2H), 2,70-3,89 (a, 22H), 3,94 (s, 1H), 3,99 (s, 1H), 4,04 (s, 2H), 4,07-4,12 (1H), 4,12-4,15 (m, 2H), 4,18 (t, J =4,4 Hz, 2H)
MS (ESI) m/z 821 [M+H]+
Ejemplo 9: Preparación del compuesto 2m

Etapa 1: Preparación del compuesto 17
Se disolvió Fmoc-Lys(Z)-OH (183 mg, 0,36 mmol) en diclorometano (1 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 62 mg, 0,46 mmol), TBTU (146 mg, 0,46 mmol) y diisopropiletilamina (106 jl, 0,61 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 10c (0,20 g, 0,30 mmol) disuelto en diclorometano (1 ml) y se agitó durante 4 horas. La reacción se terminó añadiendo agua (10 ml), y el compuesto orgánico se extrajo con diclorometano (10 ml x 3). El reactante se secó sobre sulfato de sodio anhidro, se concentró
a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 17 (268 mg, 77 %) como un sólido blanco.
RMN de 1H (400 MHz, CDCl3) δ 1,19-1,29 (m, 2H), 1,37-1,46 (m, 36H), 1,49-1,55 (m, 2H), 1,57-1,63 (m, 2H), 1,68 (s, 3H), 1,77-1,84 (m, 2H), 2,00-2,10 (m, 2H), 2,22-2,35 (m, 2H), 3,10-3,23 (m, 3H), 3,79-3,99 (m, 3H), 4,13 (s, 1H), 4,18-4,26 (m, 2H), 4,32-4,42 (m, 3H), 5,05 (s, 2H), 5,37-5,87 (m, 3H), 7,02 (d, J = 22 Hz, 1H), 7,23-7,40 (m, 8H), 7,54-7,60 (m, 2H), 7,73 (q, J = 6,1 Hz, 2H)
MS (ESI) m/z 1165 [M+Na]+
Etapa 2: Preparación del compuesto 12c
El compuesto 17 (250 mg, 0,22 mmol) se disolvió en CH2Ch (1 ml) y luego se añadió piperidina (64,80 μl, 0,66 mmol), seguido de agitación a temperatura ambiente durante 5 horas. Tras eliminar el disolvente de la mezcla de reacción a presión reducida, el concentrado se sometió a cromatografía en columna para obtener el compuesto 12c (170 mg, 84 %) como un líquido incoloro.
RMN de 1H (400 MHz, CDCl3) δ 1,24-1,36 (m, 2H), 1,45 (s, 9H), 1,47 (s, 9H), 1,48 (s, 18H), 1,52-1,59 (m, 4H), 1,71 (s, 4H), 1,77-1,89 (m, 3H), 2,04-2,13 (m, 1H), 2,25-2,39 (m, 2H), 3,15-3,25 (m, 2H), 3,26-3,34 (m, 1H), 3,85-4,05 (m, 2H), 4,12-4,20 (m, 2H), 4,26-4,41 (m, 2H), 5,06-5,16 (m, 3H), 5,51 (ddd, J = 49,3, 29,4, 8,1 Hz, 2H), 7,37 (d, J = 4,4 Hz, 4H), 7,92 (s, 1H);
MS (ESI) m/z 922 [M+H]+
Etapa 3: Preparación del compuesto 13c
Se disolvió el éster de DOTA-tris(tBu) (99 mg, 0,119 mmol) en diclorometano (0,5 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 23 mg, 0,261 mmol), TBTU (56 mg, 0,261 mmol) y diisopropiletilamina (30 μl, 347 umol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 12c (160 mg, 0,174 mmol) disuelto en diclorometano (1,6 ml) y se agitó durante 1 hora. La reacción se terminó añadiendo agua (10 ml), y el compuesto orgánico se extrajo con diclorometano (10 ml x 3). El reactante se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 13c (180 mg, 72 %) como un líquido incoloro.
RMN de 1H (400 MHz, CDCla) δ 1,28-1,33 (m, 3H), 1,45 (s, 9H), 1,46 (s, 9H), 1,47 (s, 9H), 1,47 (s, 9H), 1,48 (s, 18H), 1,49 (s, 9H), 1,79 (s, 8H), 2,04-2,15 (m, 5H), 2,25-2,41 (m, 5H), 2,59 (s a, 3H), 2,83 (s a, 4H), 2,95 (s a, 3H), 3,18-3,28 (m, 5H), 3,47 (s a, 4H), 3,92 (dd, J = 44,0, 18,4 Hz, 2H), 4,04-4,24 (m, 2H), 4,33-4,39 (m, 3H), 5,10 (d, J = 3,2 Hz, 2H), 7,15 (d, J = 18,4 Hz, 1H), 7,30-7,37 (m, 4H), 7,46 (s, 1H);
MS (ESI) m/z 1498 [M+Na]+
Etapa 4: Preparación del compuesto 14c
Se introdujo paladio (paladio al 10 % sobre carbono, 6 mg, 5,8 umol) en un matraz de fondo redondo, que luego se cerró con un septo y se rellenó con gas hidrógeno al vacío. El compuesto 13c (170 mg, 115 umol) disuelto en metanol (2 ml) se cargó en un recipiente de reacción, seguido de agitación a temperatura ambiente durante 15 horas. El reactante se filtró con Celite, se concentró a presión reducida y el concentrado se separó mediante cromatografía en columna de gel de sílice NH (metanol al 0-1 %/diclorometano) para obtener el compuesto 14c (117 mg, 76 %) como un líquido incoloro.
RMN de 1H (400 MHz, CDCl3) δ 1,14-1,28 (m, 3H), 1,36 (s, 18H), 1,41 (s, 18H), 1,44 (s, 27H), 1,53-1,62 (m, 4H), 1,68-1,83 (m, 5H), 1,97-2,09 (m, 4H), 2,10-2,37 (m, 7H), 2,40-2,69 (m, 6H), 2,70-3,10 (m, 7H), 3,15-3,26 (m, 2H), 3,31-3,65 (m, 4H), 3,78-3,95 (m, 2H), 4,01-4,13 (m, 2H), 4,21-4,35 (m, 3H), 5,37-5,55 (m, 2H), 7,17 (d, J = 28,8 Hz, 1H), 7,52 (s a, 1H);
MS (ESI) m/z 1365 [M+Na]+
Etapa 5: Preparación del compuesto 15f
Se disolvió ácido 4-(p-tolil)butírico (16 mg, 89 umol) en diclorometano (1 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 15 mg, 112 umol), TBTU (36 mg, 112 umol) y DIEA (26 μl, 149 umol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 14c (100 mg, 75 μmol) disuelto en diclorometano (2 ml) y se agitó durante 1,5 horas. La reacción se terminó añadiendo agua (10 ml), y el compuesto orgánico se extrajo con diclorometano (10 ml x 3). El reactante se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y el concentrado se separó por cromatografía en columna para obtener el compuesto 15f (69 mg, 86 %) como un líquido incoloro.
RMN de 1H (400 MHz, CDCl3) δ 1,26-1,32 (m, 2H), 1,35-1,40 (m, 27H), 1,42-1,47 (m, 36H), 1,52-1,59 (m, 4H), 179-196 (m 6H) 200-208 (m 4H) 210-220 (m 2H) 221-241 (m 14H) 254-262 (m 6H), 2,78 (s,
3H), 2,83-2,95 (m, 2H), 3,20 (s, 3H), 3,28-3,62 (m, 4H), 3,73-3,88 (m, 2H), 3,93-4,03 (m, 2H), 4,13-4,25 (m, 2H), 4,27-4,33 (m, 2H), 5,35-5,61 (m, 2H), 7,05 (d, J = 1,6 Hz, 4H)
MS (ESI) m/z 1524 [M+Na]+
Etapa 6 : Preparación del compuesto 2m
El compuesto 15f (86 mg, 57 umol) obtenido en la etapa 5 anterior se añadió a ácido trifluoroacético al 70 %/diclorometano (1 ml), seguido de agitación durante 6 horas. Después añadir gota a gota la mezcla de reacción a éter dietílico (40 ml) para formar un precipitado, este se separó utilizando una centrífuga. La mezcla se separó por cromatografía de líquidos de alto rendimiento (HPLC) y se secó con un liofilizador para obtener el compuesto 2m (44 mg, 69 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,36-1,41 (m, 4H), 1,46-1,53 (m, 3H), 1,54-1,59 (m, 2H), 1,66-1,75 (m, 2H), 1,77-1,88 (m, 4H), 1,90-1,99 (m, 1H), 2,10-2,16 (m, 1H), 2,21 (t, J = 7,2 Hz, 2H), 2,27 (s, 3H), 2,48 (t, J = 7,6 Hz, 2H), 2,53 (t, J = 7,6 Hz, 2H), 2,93-3,56 (a, 20H), 3,71 (s a, 3H), 3,87 (s, 1H), 3,91 (s, 1H), 3,98 (s, 1H), 4,04 (t, J = 8,6 Hz, 3H), 4,12 (s, 1H), 4,13-4,19 (m, 2H), 4,22-4,26 (m, 1H), 4,30 (s a, 1H), 7,15 (q, J = 7,9 Hz, 4H);
MS (ESI) m/z 1109 [M-H]-Ejemplo 10: Preparación del compuesto 19

Preparación del compuesto 19a
El compuesto 18 (500 mg, 2,87 mmol) se disolvió en diclorometano (5 ml), se enfrió hasta 0 °C y luego se añadió trietilamina (0,6 ml, 4,31 mmol). Se añadió lentamente bromoacetato de terc-butilo (620 mg, 3,16 mmol) disuelto en diclorometano (5 ml) y se agitó a temperatura ambiente durante 18 horas. La reacción se terminó añadiendo agua (10 ml), y el compuesto orgánico se extrajo con diclorometano (10 ml x 2). El reactante se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 2 %/diclorometano) para obtener el compuesto 19a (0,46 g, 55 %).
RMN de 1H (400 MHz, CDCls) δ 1,45 (s, 9H), 2,78 (t, J = 4,8 Hz, 2H), 3,31 (s, 2H), 3,38 (t, J = 5,6 Hz, 2H), 3,68-3,59 (m, 9H);
RMN de 13C (100 MHz, 3) δ 28,1,48,7, 50,6, 51,7, 56,6, 76,7, 77,0, 77,3, 81,0, 171,5;
MS (ESI) m/z 289 [M+H]+
Preparación del compuesto 19b
El compuesto 18 (300 mg, 1,72 mmol) se disolvió en etanol (10 ml), se enfrió hasta 0 °C y luego se añadió lentamente acrilato de terc-butilo (220 mg, 1,72 mmol). Tras elevar gradualmente la temperatura de la mezcla hasta la temperatura ambiente, se agitó durante 18 horas. El disolvente orgánico se eliminó a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 19b (380 mg, 73 %).
RMN de 1H (400 MHz, CDCls) δ 1,43 (s, 9H), 2,42 (t, J = 6,4 Hz, 2H), 2,79 (t, J = 5,6 Hz, 2H), 2,85 (t, J = 6,4 Hz, 2H), 3,38 (t, J = 4,8 Hz, 2H), 3,58 (t, J = 5,2 Hz, 2H), 3,62-3,68 (m, 6H);
RMN de 13C (100 MHz, 3) δ 28,1,35,9, 45,2, 49,1,50,7, 76,7, 77,0, 77,3, 80,4, 172,0;
MS (ESI) m/z 303 [M+H]+
Ejemplo 11: Preparación de los compuestos 2n y 2o

Etapa 1: Preparación del compuesto 21a
El compuesto 20 (180 mg, 0,36 mmol) se disolvió en diclorometano (5 ml), se enfrió hasta 0 °C y luego se añadieron N,N'-dicidohexilcarbodiimida (DCC, 83 mg, 0,40 mmol) y el compuesto 19a (110 mg, 0,36 mmol), seguido de agitación a temperatura ambiente durante 1 hora. La capa orgánica se filtró varias veces y el disolvente se eliminó a presión reducida. El concentrado se separó por cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 21a (250 mg, 91 %).
RMN de 1H (400 MHz, CDCla) δ 1,43 (s, 9H), 1,44 (s, 9H), 1,45 (s, 9H), 1,46 (s, 9H), 1,79-1,86 (m, 1H), 2,01 2,20 (m, 3H), 2,26-2,36 (m, 3H), 2,44-2,55 (m, 1H), 2,77 (a, 1H), 3,40 (t, J = 6,4 Hz, 2H), 3,56-3,74 (m, 9H), 4,03 (dd, J = 44,0, 17,2 Hz, 2H), 4,22-4,35 (m, 2H);
MS (ESI) m/z 759 [M+H]+
Etapa 1: Preparación del compuesto 21b
El compuesto 21b (275 mg, 87% ) se obtuvo de la misma manera que la descrita en la preparación del compuesto 21a, excepto que se utilizaron el compuesto 20 (200 mg, 0,41 mmol), N,N'-diciclohexilcarbodiimida (DCC, 102 mg, 0,49 mmol) y el compuesto 19b (200 mg, 0,41 mmol).
MS (ESI) m/z 773 [M+H]+
Etapa 2: Preparación del compuesto 22a
El compuesto 21a (200 mg, 0,26 mmol) se disolvió en metanol (8 ml) y luego se añadió paladio (paladio al 10 % sobre carbono, 13 mg, 13 umol). Se rellenó el matraz de reacción con gas hidrógeno y se agitó a temperatura ambiente durante 1 hora. Tras hacer pasar el producto de reacción a través de Celite, se eliminó el disolvente a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 8 %/diclorometano) para obtener el compuesto 22a (120 mg, 63 %).
RMN de 1H (400 MHz, metanol-d4) δ 1,45 (s, 9H), 1,47-1,49 (m, 27H), 1,76-1,94 (m, 2H), 2,01-2,13 (m, 2H), 2,27-2,39 (m, 3H), 2,55-2,60 (m, 2H), 2,79-2,81 (m, 2H), 3,35 (s, 1H), 3,52 (t, J = 5,2 Hz, 2H), 3,55-3,65 (m, 8H), 4,04 (dd, J = 17,6, 4,8 Hz, 1H), 4,14-4,22 (m, 3H);
MS (ESI) m/z 733 [M+H]+
Etapa 2: Preparación del compuesto 22b
El compuesto 22b (210 mg, 90 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 22a, excepto que se utilizó el compuesto 21b (240 mg, 0,32 mmol) y paladio (paladio al 10 % sobre carbono, 17 mg, 16 umol).
RMN de 1H (400 MHz, metanol-d4) 51,45 (s, 18H), 1,48 (s, 18H), 1,59-1,63 (m, 1H), 1,68-1,75 (m, 2H), 1,79 1,91 (m, 4H), 2,03-2,14 (m, 2H), 2,30-2,36 (m, 2H), 2,48-2,62 (m, 3H), 2,80-2,82 (m, 2H), 3,43-3,70 (m, 12H), 4,14-4,20 (m, 2H);
MS (ESI) m/z 747 [M+H]+
Etapa 3: Preparación del compuesto 23a
El éster de DOTA-tris(tBu) (38 mg, 65 umol) se disolvió en diclorometano (7 ml) y luego se añadieron hidroxibenzotriazol (HOBt, 110 mg, 82 umol), TBTU (26 mg, 82 umol) y diisopropiletilamina (19 μl, 0,11 mmol), seguido de agitación durante 10 minutos. Se añadió el compuesto 22a (40 mg, 55 μmol) disuelto en diclorometano (3 ml) y se agitó durante 1 hora. La reacción se terminó añadiendo agua (10 ml), y el compuesto orgánico se extrajo con diclorometano (10 ml x 2). El reactante se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 5 %/diclorometano) para obtener el compuesto 23a (60 mg, 70 %) como un sólido blanco.
RMN de 1H (400 MHz, metanol-d4) δ 1,40-1,57 (m, 63H), 1,76-2,87 (m, 26H), 3,36-3,63 (m, 17H), 3,74 (s, 1H), 4,04 (d, J = 8,0 Hz, 2H), 4,14-4,24 (m, 3H), 6,34-6,38 (m, 1H);
MS (ESI) m/z 1288 [M+H]+
Etapa 3: Preparación del compuesto 23b
El compuesto 23b (60 mg, 68 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 23a , excepto que se utilizaron éster de DOTA-tris(tBu) (46 mg, 80 pmol), hidroxibenzotriazol (HOBt, 14 mg, 0,10 mmol), TBTU (32 mg, 0,10 mmol), diisopropiletilamina (24 μl, 0,13 mmol) y el compuesto 22b (50 mg, 67 umol).
RMN de 1H (400 MHz, metanol-d4) δ 1,09-1,24 (m, 12H), 1,29-1,43 (m, 10H), 1,45-1,53 (m, 41H), 1,57-1,63 (m, 4H), 1,66-1,76 (m, 7H), 1,80-1,91 (m, 8H), 2,03-2,12 (m, 2H), 2,25-2,37 (m, 2H), 2,44-2,64 (m, 4H), 2,82 (s, 9H), 3,35-3,49 (m, 5H), 3,52-3,69 (m, 7H), 4,14-4,22 (m, 2H);
MS (ESI) m/z 1302 [M+H]+
Etapa 4: Preparación del compuesto 2n
El compuesto 23a (45 mg, 35 umol) se añadió a ácido trifluoroacético al 70 %/diclorometano (1 ml), seguido de agitación durante 7 horas. Después añadir gota a gota la mezcla de reacción a éter dietílico (20 ml) para formar un precipitado, este se separó utilizando una centrífuga. La mezcla se separó por cromatografía de líquidos de alto rendimiento (HPLC) y se secó con un liofilizador para obtener el compuesto 2n (22 mg, 70 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,93-2,02 (m, 2H), 2,14-2,22 (m, 2H), 2,45-2,54 (m, 3H), 2,63-2,68 (m, 2H), 2,88-3,56 (m, 18H), 3,57-3,72 (m, 11H), 3,73-3,85 (m, 3H), 3,86-4,10 (m, 3H), 4,16 (s, 1H), 4,21-4,31 (m, 3H); MS (ESI) m/z 895 [M+H]+
Etapa 4: Preparación del compuesto 2o
El compuesto 2o (17 mg, 48 %) se obtuvo de la misma manera que la descrita en la preparación del compuesto 2n, excepto que se utilizó el compuesto 23b (50 mg, 38 μmol) y ácido trifluoroacético al 70 %/diclorometano (1 ml).
RMN de 1H (400 MHz, D2O) δ 1,92-2,03 (m, 2H), 2,15-2,25 (m, 2H), 2,48-2,68 (m, 5H), 2,71-2,75 (m, 1H), 2,92-3,53 (m, 18H), 3,55-3,86 (m, 17H), 3,87-4,17 (m, 3H), 4,22-4,31 (m, 2H);
MS (ESI) m/z 909 [M+H]+
Ejemplo 12: Preparación del compuesto 2p

Etapa 1: Preparación del compuesto 25
El compuesto 20 (500 mg, 1,02 mmol) se disolvió en diclorometano (10 ml), se enfrió hasta 0 °C, luego se añadieron N,N'-dicidohexilcarbodiimida (DCC, 230 mg, 1,12 mmol) y el compuesto 24 (170 mg, 1,02 mmol), seguido de agitación a temperatura ambiente durante 1 hora. La capa orgánica se filtró varias veces y el disolvente se eliminó a presión reducida. El concentrado se separó por cromatografía en columna (acetato de etilo al 30 %/n-hexano) para obtener el compuesto 25 (450 mg, 69 %).
RMN de 1H (400 MHz, CDCla) δ 1,37-1,50 (m, 36H), 1,81-1,90 (m, 1H), 1,96-2,25 (m, 4H), 2,28-2,39 (m, 3H), 2,51-2,56 (m, 1H), 4,01-4,10 (m, 2H), 4,14-4,21 (m, 2H), 4,24-4,36 (m, 3H), 5,51 (a, 1H), 5,75 (a, 1H); MS (ESI) m/z 774 [M+H]+
Etapa 2: Preparación del compuesto 26
El compuesto 25 (350 mg, 0,55 mmol) y 2-aminoetil 2'-azidoetil éter (110 mg, 0,82 mmol) se disolvieron en etanol (10 ml) y luego se añadieron CuSO41 M (0,11 ml, 0,11 mmol) y ascorbato de sodio 2 M (0,082 ml, 0,16 mmol), seguido de agitación durante 1 hora. El reactante se filtró y el disolvente se eliminó a presión reducida. El concentrado se separó mediante cromatografía en columna de gel de sílice NH (metanol al 3 %/diclorometano) para obtener el compuesto 26 (250 mg, 59 %).
RMN de 1H (400 MHz, metanol-d4) δ 1,41-1,51 (m, 37H), 1,78-1,90 (m, 2H), 2,03-2,16 (m, 2H), 2,29-2,42 (m, 3H), 2,67-2,71 (m, 1H), 3,12 (q, J = 5,2 Hz, 2H), 3,65-3,70 (m, 2H), 3,88-3,94 (m, 2H), 4,01 (d, J = 6,0 Hz, 1H), 4,16-4,22 (m, 3H), 4,52-4,64 (m, 3H), 4,69-4,75 (m, 1H), 7,95 (s, 0,6H), 8,07 (s, 0,5H);
MS (ESI) m/z 770 [M+H]+
Etapa 3: Preparación del compuesto 27
El éster de DOTA-tris(tBu) (36 mg, 62 umol) se disolvió en diclorometano (5 ml) y luego se añadieron hidroxibenzotriazol (DCC, 11 mg, 78 umol), TBTU (25 mg, 78 umol) y diisopropiletilamina (18 μl, 0,10 mmol), seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió el compuesto 26 (40 mg, 52 umol) disuelto en diclorometano (1 ml) y se agitó durante 1 hora. La reacción se terminó añadiendo agua (10 ml), y el compuesto orgánico se extrajo con diclorometano (10 ml x 2). El reactante se secó sobre sulfato de sodio anhidro, se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 3 %/diclorometano) para obtener el compuesto 27 (50 mg, 72 %).
RMN de 1H (400 MHz, metanol-d4) δ 1,40-1,52 (m, 63H), 1,55-1,66 (m, 1H), 1,76-1,97 (m, 4H), 2,02-2,28 (m, 9H), 2,29-2,48 (m, 8H), 2,68-2,72 (m, 3H), 3,35-3,41 (m, 4H), 3,48-3,71 (m, 4H), 3,81-3,87 (m, 3H), 3,94-4,08 (m, 1H), 4,13-4,23 (m, 4H), 4,52-4,59 (m, 3H), 4,64-4,75 (m, 2H), 7,92 (s, 0,6H), 8,05 (s, 0,4H);
MS (ESI) m/z 1325 [M+H]+
Etapa 4: Preparación del compuesto 2p
El compuesto 27 (43 mg, 32 umol) se añadió a ácido trifluoroacético al 70 %/diclorometano (1 ml), seguido de agitación durante 7 horas. Después añadir gota a gota la mezcla de reacción a éter dietílico (20 ml) para formar un precipitado,
este se separó utilizando una centrífuga. La mezcla se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto 2p (22 mg, 73 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,90-2,03 (m, 2H), 2,12-2,23 (m, 2H), 2,43-2,53 (m, 3H), 2,72-2,78 (m, 1H), 2,99-3,46 (m, 16H), 3,52-3,59 (m, 3H), 3,62-4,10 (m, 9H), 4,14 (s, 1H), 4,19-4,35 (m, 3H), 4,57-4,63 (m, 2H), 4,65-4,74 (m, 2H), 4,77 (s, 2H), 7,95 (s, 0,4H), 8,04 (s, 0,6H);
MS (ESI) m/z 932 [M+H]+
Ejemplo 13: Preparación de Ga-1

Preparación del compuesto Ga-1a (no según la invención)
El compuesto 2a (10 mg, 11 umol) se disolvió en agua (0,6 ml) y luego se añadió tricloruro de galio (19 mg, 0,11 mmol) disuelto en agua (0,6 ml), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1a (6 mg, 56 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,34-1,51 (m, 4H), 1,64-1,73 (m, 1H), 1,77-1,86 (m, 1H), 1,91-2,00 (m, 1H), 2,12-2,20 (m, 1H), 2,50 (t, J = 7,2 Hz, 2H), 3,10 (t, J = 6,8 Hz, 2H), 3,31-3,43 (m, 10H), 3,52-3,57 (m, 6H), 3,65 (s, 2H), 3,78 (s, 2H), 3,84-3,94 (m, 8H), 4,00-4,03 (m, 2H), 4,16 (dd, J = 14,0, 5,2 Hz, 1H), 4,24 (dd, J = 14,0, 5,2 Hz, 1H), 4,38 (s, 2H), 4,57 (t, J = 4,8 Hz, 2H), 7,88 (s, 1H);
MS (ESI) m/z 984 [M+H]+
Preparación del compuesto Ga-1b (no según la invención)
El compuesto 2b (19 mg, 19 umol) se disolvió en agua (0,8 ml) y luego se añadió tricloruro de galio (33 mg, 0,19 mmol) disuelto en agua (0,8 ml), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1b (13 mg, 64 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,28-1,42 (m, 3H), 1,50-1,57 (m, 2H), 1,65-1,73 (m, 1H), 1,77-1,86 (m, 1H), 1,89-1,99 (m, 1H), 2,10-2,20 (m, 1H), 2,48 (t, J = 7,2 Hz, 2H), 3,27-3,42 (m, 11H), 3,49-3,57 (m, 6H), 3,67 (s, 2H), 3,78 (s, 2H), 3,83-3,92 (m, 8H), 4,00-4,03 (m, 2H), 4,15 (dd, J = 14,0, 4,8 Hz, 1H), 4,24 ((dd, J = 14,0, 4,8 Hz, 1H), 4,58 (t, J = 4,8 Hz, 2H), 5,19 (s, 2H), 7,62 (d, J = 7,6 Hz, 2H), 8,01 (s, 1H), 8,42 (d, J = 7,6 Hz, 2H);
MS (ESI) m/z 1061 [M+H]+
Preparación del compuesto Ga-1c
El compuesto 2c (10 mg, 10 umol) se disolvió en agua (1 ml) y luego se añadió tricloruro de galio (18 mg, 100 umol) disuelto en agua (1 ml), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1c (6 mg, 58 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,28-1,42 (m, 2H), 1,44-1,62 (m, 2H), 1,64-1,77 (m, 1H), 1,78-1,90 (m, 1H), 1,93-2,23 (m, 1H), 2,50-2,54 (m, 2H), 2,72-2,75 (m, 0,5H), 2,87-2,90 (m, 1,5H), 3,01-3,07 (m, 2H), 3,36-3,44 (m, 12H), 3,54-3,59 (m, 6H), 3,69 (s, 2H), 3,77 (s, 2H), 3,85-3,94 (m, 8H), 4,00-4,12 (m, 4H), 4,16-4,29 (m, 3H), 4,57-4,60 (m, 2H), 7,81 (s, 1H);
MS (ESI) m/z 1041 [M+H]+
Preparación del compuesto Ga-1d
El compuesto 2d (6 mg, 6 umol) se disolvió en agua (0,8 ml) y luego se añadió tricloruro de galio (14 mg, 80 ^mol) disuelto en agua (0,8 ml), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se
separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1d (4 mg, 62 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,26 -1,43 (m, 2H), 1,44-1,60 (m, 2H), 1,66-1,78 (m, 1H), 1,79-1,91 (m, 1H), 1,94-1,20 (m, 1H), 2,15-2,24 (m, 1H), 2,48-2,67 (m, 4H), 2,79-2,88 (m, 2H), 3,01-3,09 (m, 2H), 3,28-3,49 (m, 11H), 3,52-3,65 (m, 8H), 3,70 (s, 2H), 3,79 (s, 2H), 3,84-3,99 (m, 7H), 4,05 (d, J = 10,8 Hz, 2H), 4,18-4,29 (m, 2H), 4,60 (t, J = 5,2 Hz, 2H), 7,82 (s, 0,6H), 7,84 (s, 0,4H);
MS (ESI) m/z 1055 [M+H]+
Preparación del compuesto Ga-1e
El compuesto 2e (16 mg, 18 umol) se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (16 mg, 91 umol), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1e (15 mg, 86 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,17-1,32 (m, 2H), 1,37-1,51 (m, 2H), 1,53-1,62 (m, 1H), 1,65-1,75 (m, 1H), 1.82 (p, J = 7,4 Hz, 1H), 2,01 (p, J = 7,6 Hz, 1H), 2,35 (t, J = 7,2 Hz, 2H), 3,00-3,30 (m, 12H), 3,36-3,41 (m, 4H), 3,47 (q, J = 4,8 Hz, 2H), 3,51-3,60 (m, 6H), 3,65 (s, 2H), 3,73 (s, 4H), 3,76-3,79 (m, 2H), 3,85-3,88 (m, 2H), 3,97 (s, 2H), 4,01-4,12 (m, 3H), 4,24 (s, 2H);
MS (ESI) m/z 975 [M+H]+
Preparación del compuesto Ga-1f
El compuesto 2f (3,8 mg, 3,2 μmol) sintetizado en la etapa 5 del ejemplo 5 se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (3,0 mg, 17 umol), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1f (2,7 mg, 68 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,20-1,31 (m, 4H), 1,36-1,50 (m, 5H), 1,54-1,74 (m, 5H), 1,76-1,84 (m, 2H), 1,85-1,96 (m, 1H), 1,99-2,04 (m, 2H), 2,13 (t, J = 7,4 Hz, 2H), 2,35 (t, J = 7,2 Hz, 2H), 2,51 (t, J = 7,0 Hz, 2H), 3,03 (t, J = 6,8 Hz, 2H), 3,15-3,28 (m, 9H), 3,33-3,44 (m, 8H), 3,46-3,50 (m, 3H), 3,55-3,58 (m, 3H), 3,65-3,70 (m, 3H), 3,75-3,81 (m, 4H), 3,87 (s, 2H), 3,92-4,12 (m, 5H), 4,26 (s, 2H), 7,14-7,17 (m, 3H), 7,24-7,27 (m, 2H);
MS (ESI) m/z 1252 [M+2H]+, 1248 [M -2H
Preparación del compuesto Ga-1g
El compuesto 2g (4,4 mg, 3,7 umol) sintetizado en la etapa 5 del ejemplo 5 se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (4,0 mg, 23 pmol), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1g (2,0 mg, 43 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,12-1,28 (m, 4H), 1,32-1,37 (m, 2H), 1,40-1,48 (m, 2H), 1,49-1,62 (m, 3H), 1,64-1,70 (m, 1H), 1,73 (p, J = 7,4 Hz, 2H), 1,77-1,86 (m, 1H), 1,99-2,05 (m, 1H), 2,08 (t, J = 7,2 Hz, 2H), 2,15 (s, 3H), 2,36 (t, J = 7,6 Hz, 2H), 2,43 (t, J = 6,8 Hz, 2H), 2,96-3,03 (m, 2H), 3,04-3,28 (m, 1H), 3,30-3,47 (m, 8H), 3,84-3,91 (m, 2H), 3,92-3,40 (m, 2H), 4,03-4,08 (m, 2H), 4,09-4,14 (m, 1H), 4,23 (s, 2H), 7,01 (d, J = 8,0 Hz, 2H), 7,05 (d, J = 8,0 Hz, 2H);
MS (ESI) m/z 1265 [M+H]+, 1263 [M-H]'
Preparación del compuesto Ga-1h
El compuesto 2h (6,0 mg, 4,6 μmol) sintetizado en la etapa 5 del ejemplo 5 se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (6,0 mg, 34,1 umol), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1h (5,4 mg, 86 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,21-1,30 (m, 4H), 1,33-1,38 (m, 2H), 1,40-1,51 (m, 2H), 1,54-1,75 (m, 4H), 1.82 (p, J = 7,4 Hz, 2H), 1,83-1,89 (m, 1H), 2,03-2,08 (m, 1H), 2,12 (t, J = 7,2 Hz, 2H), 2,39 (t, J = 7,2 Hz, 1H), 2,48 (t, J = 7,2 Hz, 1H), 3,00 (t, J = 6,6 Hz, 2H), 3,13-3,33 (m, 11H), 3,39-3,52 (m, 8H), 3,55-3,60 (m, 6H), 3,66-3,74 (m, 3H), 3,97 (s, 2H), 4,00-4,17 (m, 3H), 4,27 (s, 2H), 6,94 (d, J = 8,0 Hz, 2H), 7,59 (d, J = 8,0 Hz, 2H);
MS (ESI) m/z 1375 [M+H]+, 1373 [M-H]
Preparación del compuesto Ga-1i
El compuesto 2i (7,0 mg, 8,1 umol) sintetizado en la etapa 4 del ejemplo 6 se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (7,0 mg, 40 |jmol), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1i (6,0 mg, 79 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,22-1,33 (m, 2H), 1,40-1,52 (m, 2H), 1,56-1,64 (m, 1H), 1,68-1,76 (m, 1H), 1,85 (p, J = 7,2 Hz, 1H), 2,04 (p, J = 7,2 Hz, 1H), 2,38 (t, J = 7,2 Hz, 2H), 3,17-3,34 (m, 11H), 3,41-3,43 (m, 4H), 3,48-3,53 (m, 2H), 3,58 (s, 4H), 3,76 (s, 4H), 3,79 (s, 2H), 3,88-3,91 (m, 2H), 3,97 (s, 2H), 4,06-4,14 (m, 3H), 4,26 (s, 2H);
MS (ESI) m/z 933 [M+2H]+, 929 [M-2H]-Preparación del compuesto Ga-1j
El compuesto 2j (4,4 mg, 3,8 jm ol) sintetizado en la etapa 5 del ejemplo 7 se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (4,0 mg, 23 jmol), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1j (2,3 mg, 49 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,20-1,30 (m, 4H), 1,34-1,50 (m, 4H), 1,56-1,66 (m, 3H), 1,69-1,80 (m, 3H), 1,83-1,87 (m, 1H), 1,97-2,08 (m, 1H), 2,11 (t, J = 6,8 Hz, 2H), 2,18 (s, 3H), 2,37 (t, J = 7,2 Hz, 2H), 2,42-2,48 (m, 2H), 3,02 (t, J = 6,4 Hz, 2H), 3,15-3,30 (m, 9H), 3,34-3,55 (m, 11H), 3,64-3,70 (m, 3H), 3,76-3,79 (m, 4H), 3,88-3,93 (m, 4H), 4,05-4,10 (m, 3H), 4,22 (s, 2H), 7,04 (d, J = 8,0 Hz, 2H), 7,08 (d, J = 8,0 Hz, 2H);
MS (ESI) m/z 1121 [M+H]+, 1119 [M-H]-Preparación del compuesto Ga-1k
El compuesto 2k (2,0 mg, 1,6 jm ol) sintetizado en la etapa 5 del ejemplo 7 se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (2,0 mg, 11 jmol), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1k (0,6 mg, 29 %) como un sólido blanco.
MS (ESI) m/z 1332 [M+H]+, 1330 [M-H]-Preparación del compuesto Ga-1l
El compuesto 2l (7,0 mg, 8,5 jm ol) se disolvió en agua destilada (0,5 ml) y luego se añadió tricloruro de galio (4,5 mg, 25,6 jmol), seguido de agitación a 70 °C durante 2 horas. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1l (5,5 mg, 73 %) como un sólido blanco.
RMN de 1H (400 MHz, CDCla) δ 1,24-1,40 (m, 2H), 1,42-1,51 (m, 1H), 1,54-1,62 (m, 2H), 1,63-1,94 (m, 1H), 2,06-2,14 (m, 1H), 2,44 (t, J = 7,2 Hz, 2H), 3,20-3,40 (m, 9H), 3,49 (d, J = 8,8 Hz, 4H), 3,72-3,86 (m, 6H), 3,87 (d, J = 10,4 Hz, 2H), 3,92-4,00 (m, 3H), 4,04 (s, 2H), 4,10-4,20 (m, 4H);
MS (ESI) m/z 887 [M+H]+
Preparación del compuesto Ga-1m
El compuesto 2m (14 mg, 12,6 jm ol) se disolvió en H2O (0,8 ml) y luego se añadió tricloruro de galio (6,7 mg, 37,9 jmol), seguido de agitación a 70 °C durante 2 horas. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1m (7,1 mg, 48 %) como un sólido blanco.
RMN de 1H (400 MHz, D2O) δ 1,30-1,44 (m, 4H), 1,48-1,54 (m, 2H), 1,58 (sa, 2H), 1,68-1,76 (m, 2H), 1,79 1,89 (m, 4H), 1,93-1,98 (m, 1H), 2,14-2,17 (m, 1H), 2,21 (d, J = 1,2 Hz, 4H), 2,29 (s, 3H), 2,45 (t, J = 7,2 Hz, 2H), 2,54-2,57 (m, 2H), 3,15 (t, J = 6,4 Hz, 2H), 2,78-3,42 (m, 9H), 3,54 (t, J = 10 Hz, 4H), 3,78 (s, 3H), 3,83 3,97 (m, 6H), 4,00-4,11 (m, 4H), 4,16-4,34 (m, 4H), 7,16 (dd, J = 17,6, 7,6 Hz, 4H)
MS (ESI) m/z 1174 [M-H]-Preparación del compuesto Ga-1n
El compuesto 2n (9 mg, 10 jm ol) se disolvió en agua (1 ml) y luego se añadió tricloruro de galio (18 mg, 100 umol) disuelto en agua (1 ml), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se
separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1n (4 mg, 41 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,94-2,03 (m, 2H), 2,15-2,45 (m, 2H), 2,47-2,54 (m, 3H), 2,63-2,71 (m, 2H), 3,32-3,48 (m, 9H), 3,53-3,72 (m, 15H), 3,78 (s, 2H), 3,90 (s, 4H), 3,95 (d, J = 10,4 Hz, 2H), 4,04 (d, J = 11,2 Hz, 2H), 4,22 (s, 2H), 4,23-4,33 (m, 3H);
MS (ESI) m/z 961 [M+H]+
Preparación del compuesto Ga-1o
El compuesto 2o (7 mg, 8 ^mol) se disolvió en agua (0,8 ml) y luego se añadió tricloruro de galio (14 mg, 80 umol) disuelto en agua (0,8 ml), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1o (6 mg, 80 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,95-2,04 (m, 2H), 2,14-2,24 (m, 2H), 2,51-2,68 (m, 5H), 2,73-2,76 (m, 1H), 3,37-3,48 (m, 10H), 3,54-3,79 (m, 20H), 3,91 (s, 4H), 3,96 (d, J = 10,4 Hz, 2H), 4,04 (d, J = 11,2 Hz, 2H), 4,23 4,30 (m, 2H);
MS (ESI) m/z 977 [M+H]+
Preparación del compuesto Ga-1p
El compuesto 2p (9 mg, 10 ^mol) se disolvió en agua (1 ml) y luego se añadió tricloruro de galio (18 mg, 10 umol) disuelto en agua (1 ml), seguido de agitación a 70 °C durante 1 hora. La solución de reacción se filtró. El filtrado se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) y se secó utilizando un liofilizador para obtener el compuesto Ga-1p (5 mg, 51 %) como un sólido.
RMN de 1H (400 MHz, D2O) δ 1,86-2,02 (m, 2H), 2,08-2,21 (m, 2H), 2,44-2,49 (m, 3H), 2,67-2,73 (m, 1H), 3,25-3,42 (m, 9H), 3,45-3,59 (m, 8H), 3,66 (s, 2H), 3,80-3,95 (m, 7H), 4,00-4,10 (m, 4H), 4,15-4,19 (m, 2H), 4,56-4,59 (m, 2H), 4,64-4,73 (m, 2H), 4,77 (s, 2H), 7,93 (s, 0,4H), 8,03 (s, 0,6H);
MS (ESI) m/z 998 [M+H]+
Ejemplo 14: Preparación del compuesto [68Ga]1

Preparación del compuesto [68Ga]1a
Se vertió ácido clorhídrico 0,1 N (5 ml) en un generador de 68Ge/68Ga y se situó en tubos de ensayo (1 ml/tubo). Tras medir la radiactividad de cada tubo de ensayo, se transfirieron al recipiente de reacción dos soluciones de 68Ga (4,6 mCi, 2 ml) de los dos tubos de ensayo que mostraban una radiactividad elevada. El compuesto 2a (200 ^g) se disolvió en acetato de sodio 1,0 M (0,4 ml)-solución acuosa de ácido clorhídrico (pH 4,55), que se cargó en un recipiente de reacción, seguido de la reacción a 80 °C durante 10 minutos. El disolvente de la reacción se filtró y el filtrado se separó mediante cromatografía de líquidos de alto rendimiento. La solución separada se diluyó con agua (10 ml), se hizo pasar a través de C-18 SepPak para la captura y se lavó con agua (5 ml). Tras soplar gas nitrógeno para eliminar la humedad, se eluyó con etanol (1 ml) para obtener el compuesto [68Ga]1a (1,4 mCi) .
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: RESTEK AQ (5 um, 250 mm * 10 mm);
Fase móvil: metanol al 25 %/agua (TFA al 0,1 %);
Caudal: 3 ml/min;
Detector UV: 230 nm;
Tiempo de residencia: 12 min.
Preparación del compuesto [68Ga]1b
El compuesto [68Ga]1b (2,8 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (5,94 mCi, 2 ml) y el compuesto 2b (200 |jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm) ;
Fase móvil: etanol al 5 %/agua (TFA al 0,1 %);
Caudal: 5 ml/min;
Detector UV: 254 nm;
Tiempo de residencia: 32 min.
Preparación del compuesto [68Ga]1c
El compuesto [68Ga]1c (2,5 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (5,7 mCi, 2 ml) y el compuesto 2c (200 jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm) ;
Fase móvil: durante 30 min acetonitrilo al 0-15 %/agua (TFA al 0,1 %);
Caudal: 3 ml/min;
Detector UV: 230 nm;
Tiempo de residencia: 31 min.
Preparación del compuesto [68Ga]1e
El compuesto [68Ga]1e (2,4 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (5,5 mCi, 2 ml) y el compuesto 2e (200 jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm) ;
Fase móvil: etanol al 8 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 14 min.
Preparación del compuesto [68Ga]1f
El compuesto [68Ga]1f (1,3 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (4,6 mCi, 2 ml) y el compuesto 2f (200 jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: Xterra MS C18 (10 um, 250 mm * 10 mm);
Fase móvil: etanol al 20 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 15 min.
Preparación del compuesto [68Ga]1g
El compuesto [68Ga]1g (2,3 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó solución de 68Ga (5,5 mCi, 2 ml) y el compuesto 2g (200 jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm) ;
Fase móvil: etanol al 25 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 17 min.
Preparación del compuesto [68Ga]1h
El compuesto [68Ga]1h (1,3 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (4,6 mCi, 2 ml) y el compuesto 2h (200 |jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm) ;
Fase móvil: etanol al 30 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 14 min.
Preparación del compuesto [68Ga]1k
El compuesto [68Ga]1k(1,1 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (3,9 mCi, 2 ml) y el compuesto 2k (200 jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: Xterra MS C18 (10 um, 250 mm * 10 mm);
Fase móvil: etanol al 25 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 18 min.
Preparación del compuesto [68Ga]1n
El compuesto [68Ga]1n (3,8 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (6,9 mCi, 2 ml) y el compuesto 2n (200 jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm) ;
Fase móvil: acetonitrilo al 7 %/agua (TFA al 0,1 %);
Caudal: 3 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 13 min.
Preparación del compuesto [68Ga]1o
El compuesto [68Ga]1o (2,1 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (5,4 mCi, 2 ml) y el compuesto 2o (200 jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm) ;
Fase móvil: etanol al 7 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 20 min.
Preparación del compuesto [68Ga]1p
El compuesto [68Ga]1p (2,1 mCi) se obtuvo de la misma manera que la descrita en la preparación del compuesto 1a, excepto que se utilizó la solución de 68Ga (5,8 mCi, 2 ml) y el compuesto 2p (200 |jg).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: RESTEK AQ (250 mm * 10 mm);
Fase móvil: metanol al 15 %/agua (TFA al 0,1 %);
Caudal: 3 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 15 min.
Ejemplo 15: Preparación del compuesto [64Cu]1b
La solución acuosa de ácido clorhídrico en la que se disolvió [64Cu]CuCh (7,3 mCi) se calentó a 90 °C y se secó al mismo tiempo que se soplaba gas nitrógeno. Tras el secado, se añadieron 0,1 ml de citrato de sodio 0,1 M (pH 5,5) en el que se disolvió el compuesto 2b (100 jg), seguido de la reacción a 60 °C durante 10 minutos. Una vez completada la reacción, se añadió agua (0,3 ml) a la mezcla de reacción, se filtró y se lavó dos veces con agua (0,3 ml). El filtrado se separó por cromatografía de líquidos de alto rendimiento, se hizo pasar a través de C-18 SepPak para la captura, se lavó con 5 ml de agua y se vertió 1 ml de etanol para obtener el compuesto [64Cu]1b (5,22 mCi).
Condiciones de cromatografía de líquidos de alto rendimiento:
Columna: Xterra MS C18 (10 um, 250 mm * 10 mm);
Fase móvil: acetonitrilo al 50 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 230 nm;
Tiempo de residencia: 17,5 min.
Ejemplo 16: Preparación del compuesto [177Lu]1g
El compuesto 2g (200 jg ) disuelto en acetato de sodio 1,0 M (0,4 ml)-solución acuosa de ácido clorhídrico (pH 4,88) se cargó en un recipiente de reacción que contenía Lu-177 (2,2 mCi), seguido de la reacción a 80°C durante 10 minutos. La reacción se filtró y el filtrado se separó por cromatografía de líquidos de alto rendimiento. La solución separada se diluyó con agua (10 ml), se hizo pasar a través de C-18 SepPak para la captura y se lavó con agua (5 ml). Tras soplar gas nitrógeno para eliminar la humedad, se eluyó con etanol (1 ml) para obtener el compuesto [177Lu]1g (1,36 mCi).
Columna: YMC-Pack ODS-A (S-5 um, 12 nm, 250 mm * 10 mm);
Fase móvil: etanol al 25 %/agua (TFA al 0,1 %);
Caudal: 4 ml/min;
Detector UV: 220 nm;
Tiempo de residencia: 19 min.
Ejemplo comparativo 1: Síntesis del compuesto [125I]30

Etapa 1: Preparación del compuesto 28
Se disolvió trifosgeno (21 mg, 71 vvo j) en diclorometano (5 vM) y luego se añadió lentamente a 0 °C 4-yodoanilina (45 vn, 0,205 vvo j) disuelta en diclorometano (5 vM). Se añadió trietilamina (0,57 vM, 0,410 vvoj), seguido de agitación a 0 °C durante 30 minutos. Se añadió lentamente a 0 °C el compuesto 3a (100 vn, 0,205 vvo j) disuelto en diclorometano (10 vM) y también se añadió trietilamina (0,57 vM, 0,410 vvoj). La mezcla se agitó durante 5 horas mientras se elevaba lentamente la temperatura hasta la temperatura ambiente. La mezcla de reacción se concentró a presión reducida y el concentrado se separó por cromatografía en columna (metanol al 2 %/diclorometano) para obtener el compuesto 28 (66 mg, 44 %) como un líquido blanco.
RMN de 1H (400 MHz, CDCla) 61,20-1,27 (m, 2H), 1,37 (s, 9H), 1,40 (s, 9H), 1,44 (s, 9H), 1,47-1,57 (m, 2H), 1,71-1,81 (m, 2H), 1,83-1,91 (m, 1H), 2,03-2,11 (m, 1H), 2,37 (sext, J = 8,2 Hz, 2H), 3,01-3,07 (m, 1H), 3,51 3,56 (m, 1H), 3,97-4,01 (m, 1H), 4,26-4,32 (m, 1H), 5,75 (d, J = 7,2 Hz, 1H), 6,31 (q, J = 3,4 Hz, 1H), 6,40 (d, J = 8,0 Hz, 1H), 7,27 (d, J = 8,8 Hz, 2H), 7,52 (d, J = 8,8 Hz, 2H), 7,90 (s, 1H);
RMN de 13C (100 MHz, CDCla) 624,5, 27,1, 27,8, 27,9, 28,0, 29,6, 31,7, 32,0, 39,1, 53,8, 54,9, 81,0, 81,8, 83,6, 83,7, 120,2, 137,5, 140,2, 155,6, 158,5, 171,8, 172,0, 175,3;
MS (ESI) m/z 733 [M+H]+
Etapa 2: Preparación del compuesto 29
El compuesto 28 (50 mg, 0,068 mmol) sintetizado en la etapa 1 anterior se disolvió en dioxano (1,0 ml) y luego se añadieron hexametil diestaño ((Me3Sn)2, 043 vM, 0,206 vvo j) y dicloruro de bis(trifenilfosfina)paladio(II) ( ^ 6^ 03^ 2, 4,8 vn, 5 vvo|j) en ese orden, seguido de agitación a 110 °C durante 1,5 horas. La mezcla se enfrió hasta la temperatura ambiente y luego se añadió una solución acuosa de fluoruro de potasio (50 vM), seguida de agitación durante 1 hora. El reactante se filtró y el compuesto orgánico se extrajo con acetato de etilo. La solución orgánica recogida se secó sobre sulfato de sodio anhidro y se concentró a presión reducida. El concentrado se separó por cromatografía en columna (trietilamina:acetato de etilo:n-hexano, 1:40:59) para obtener el compuesto 29 (28 mg, 53 %) como un sólido blanco.
RMN de 1H (400 MHz, CDCls) 60,25 (s, 9H), 1,22-1,29 (m, 2H), 1,38 (s, 9H), 1,41 (s, 9H), 1,43 (s, 9H), 1,48 1,59 (m, 2H), 1,72-1,78 (m, 1H), 1,81-1,91 (m, 1H), 2,05-2,13 (m, 2H), 2,34-2,43 (m, 2H), 3,04-3,09 (m, 1H), 3,51-3,55 (m, 1H), 4,04 (pent, J = 4,9 Hz, 1H), 4,33 (sext, J = 4,5 Hz, 1H), 5,73 (d, J = 6,8 Hz 1H), 6,23 (s a, 1H), 6,32 (d, J = 8,4 Hz, 1H), 7,35 (d, J = 8,0 Hz, 2H), 7,43 (d, J = 8,4 Hz, 2H), 7,73 (s, 1H);
RMN de 13C (100 MHz, CDCl3) δ -9,5, 24,2, 27,4, 27,8, 27,9, 28,0, 29,7, 31,8, 32,1, 39,1, 53,7, 54,7, 80,9, 81,7, 83,5, 118,4, 133,6, 136,2, 140,4, 155,9, 158,3, 171,9, 172,2, 175,1;
MS (ESI) m/z 771 [M+2H]+
Etapa 3: Preparación del compuesto [125I]28
El compuesto 29 (100 vη) obtenido en la etapa 2 anterior se disolvió en etanol (0,250 vM) y luego se añadió una solución de yoduro de sodio [125I] (3,2 mCi, 50 vM), seguido de agitación a temperatura ambiente. Se añadieron una solución acuosa de ácido clorhídrico 1 E (0,10 ml) y H2O2 al 3 %, seguido de agitación a temperatura ambiente durante 10 minutos. Se añadió una solución de tiosulfato de sodio 0,1 M (0,20 ml) a la mezcla de reacción y luego se añadió agua destilada (18 ml). Esta solución se hizo pasar a través de C-18 SepPak y se lavó con agua destilada (20 ml). Tras verter acetonitrilo (2,0 ml) en el C-18 Sep-Pak, se sopló nitrógeno en la solución para eliminar el acetonitrilo. Etapa 4: Preparación del compuesto [125I]30
Se añadieron secuencialmente diclorometano (0,2 ml) y ácido trifluoroacético (0,8 ml) al recipiente de reacción que contenía la mezcla de reacción obtenida en la etapa 3 anterior, seguido de agitación a temperatura ambiente durante 20 minutos. El disolvente de la reacción se eliminó soplando nitrógeno y luego se añadió agua destilada (2,0 ml). Esta solución se separó mediante cromatografía de líquidos de alto rendimiento (HPLC) para obtener el compuesto [125I]20 (1,1 mCi, 24 %).
Condiciones de HPLC:
Columna, XTerra MS C18 (250 mm * 10 mm); Fase móvil, acetonitrilo al 30 %/agua (TFA al 0,1 %); Caudal, 5 ml/min; UV, 254 mm; Tiempo de residencia, 10,4 min.
Ejemplo de referencia 1: Preparación de líneas celulares de cáncer de próstata y ratones atímicos
Una línea celular de cáncer de próstata humano (22RV1) utilizada en el presente documento se adquirió de la American Type Culture Collection (ATCC) . Las líneas celulares de cáncer de próstata humano PC3 PIP (PSMA+) y PC3 flu (PSMA-) fueron proporcionadas por el Dr. Martin G. Pomper (Johns Hopkins Medical School, Baltimore, MD). Las líneas celulares de cáncer de próstata humano se mantuvieron en medio RPMI1640 suplementado con un 10 % de suero bovino fetal ("fetal bovine serum", FBS) y un 1 % de antibiótico/antifúngico. En el cultivo de las líneas celulares PC3 PIP (PSMA+) y PC3 flu (PSMA-), se añadió además puromicina a la concentración de 2 μg/ml.
Como animales de ensayo se utilizaron ratones atímicos macho de 6 semanas de edad (Narabio, Seúl, Corea). Ejemplo experimental 1: Medición de la lipofilia (logP)
Cada uno de los compuestos [68Ga]1 (1a2 mCi) sintetizados en el ejemplo 14 se transfirió a un vial, se retiró el disolvente y luego se añadió 1 ml de n-octanol y 1 ml de PBS, se cerró bien la tapa y, a continuación, se mezcló con una agitadora vorticial durante 1 minuto. Una vez separadas las capas, se tomaron 0,1 ml de cada capa y se midió la dosis de radiación. La dosis de radiación se midió 3 veces y se obtuvo el valor medio.
Tabla 1

Ejemplo experimental 2: Medición de la capacidad de unión
Para confirmar la capacidad de unión de los compuestos de la presente invención a PSMA, se realizó el siguiente experimento.
Como solución tampón se utilizó RPMI1640 suplementado con un 1 % de BSA ("bovine serum albumin", albúmina de suero bovino).
Se añadió [125I]30 (0,1 nM) obtenido en el ejemplo comparativo 1 a un recipiente que contenía células 22RV1 (5 * 104) y luego se cargó el compuesto representado por la fórmula 1 en 9 concentraciones (de 1,00 * 10-4 a 1,00 * 10-12 M), seguido de agitación a 37 °C durante 2 horas. Una vez finalizada la agitación, se lavó el recipiente con solución de PBS (2 ml) tres veces y, a continuación, se midió la radiactividad con un contador gamma (2480 WIZARD2 Gamma Counter PerkinElmer Co., MA). La concentración de inhibición del 50% (CI50) para cada compuesto se calculó utilizando un programa GraphPad Prism (GraphPad Software, Inc., CA)
La siguiente tabla 2 es una tabla que muestra la afinidad de unión (CI50) de cada compuesto y se determinó mediante medición que el valor Kd del compuesto [125I]30 era 0,13 nM (Maresca, K. P et al., 2009, J. Med. Chem., 52, 347-357)
Tabla 2

Como se muestra en la tabla 2, los compuestos Ga-1ay Ga-1bdel ejemplo 13 de la presente invención son compuestos que no tienen un ácido carboxílico en el nitrógeno del residuo de lisina y, por lo tanto, no están de acuerdo con la invención. En concreto, Ga-1a mostró una fuerza de unión relativamente baja a PSMA. Por otra parte, Ga-1c, que tiene una estructura similar a Ga-1a, es un compuesto que tiene un ácido carboxílico en el nitrógeno del residuo de lisina, y puede observarse que la fuerza de unión al PSMA era unas 18,6 veces superior que la de Ga-1a. Esto se debe a que uno (R463) de los tres residuos de arginina, denominado parche de arginina, en la región de unión de
PSMA y el ácido carboxílico unido al nitrógeno del residuo de lisina del compuesto representado por la fórmula 1 de la presente invención forman una fuerte interacción de puente salino.
Además, el ácido carboxílico del residuo de lisina en el compuesto representado por la fórmula 1 de la presente invención no sólo mejoró en gran medida la capacidad de unión a PSMA, sino que también aumentó la hidrofilia del compuesto para disminuir la unión inespecífica in vivo y eliminarla más rápidamente en órganos normales.
Los compuestos Ga-1f, Ga-1g y Ga-1h tienen un grupo fenilo o un grupo fenilo sustituido, y se confirmó que tienen una mayor capacidad de unión que el compuesto Ga-1e, que tiene una estructura similar sin un grupo fenilo. Entre ellos, el compuesto unido a 4-yodofenilo Ga-1h presentaba la mayor capacidad de enlace.
Ejemplo experimental 3: Experimento de obtención de imágenes MicroPET/CT de ratones trasplantados con líneas celulares de cáncer de próstata
Se preparó un modelo tumoral inyectando por vía subcutánea células PSMA+ PC-3 PIP (una línea celular de cáncer de próstata humano) en el lado derecho de la pata trasera de un ratón atímico. Cada uno de los compuestos marcados con 68Ga [68Ga]1e, [68Ga]1g, [68Ga]1h y [68Ga]1kse inyectó por vía intravenosa con 5,5 a 6,5 MBq (l48-175 jCi/200 j l) y se obtuvieron imágenes de PET/CT utilizando INVEON PET/CT para pequeños animales (Siemens Medical Solutions, Knoxville, EE. UU.) durante 60 minutos. Tras 150 minutos, 270 minutos y 390 minutos, también se obtuvieron imágenes de PET/CT durante 30 minutos. Los resultados de las imágenes de PET/CT obtenidos se analizaron cuantitativamente utilizando Inveon™ Research Workplace (IRW).
Las figuras 1, 2, 3 y 4 son gráficos que muestran los resultados del análisis cuantitativo de las imágenes de MicorPET/CT para [68Ga]1e, [68Ga]1g, [68Ga]1h y [68Ga]1ken porcentaje de dosis inyectada (ID)/g y se resumen en las tablas 3, 4, 5 y 6, respectivamente.
Se observó que [68Ga]1e se excretaba rápidamente a través del riñón y la vejiga en la fase inicial tras la inyección y se confirmó que se unía selectivamente a los tumores PSMA+ PC-3 PIP en un nivel de 4,05 ± 0,64 % ID/g a los 270 minutos tras la inyección (figura 1, tabla 3).
En el caso de [68 ]1g, se confirmó que el tiempo de residencia en la sangre aumentaba debido a la capacidad de unión a la albúmina del grupo fenilo, y la captación tumoral aumentaba con el tiempo. En comparación con el compuesto [68Ga]1e, se confirmó la captación tumoral de 13,00 ± 4,95 % ID/g, que aumentó en aproximadamente 3 veces a los 270 minutos (figura 2, tabla 4).
En el caso de [68Ga]1hy [68Ga]1k, debido a la capacidad de unión a la albúmina del grupo fenilo sustituido, se incrementó el tiempo de residencia en la sangre y se confirmó que la captación tumoral aumentó con el tiempo, y la captación tumoral continuó aumentando después de 390 minutos.
El compuesto [68Ga]1h mostró una captación tumoral de 16,75 ± 0,92 % ID/g a los 390 minutos (figura 3, tabla 5) y el compuesto [68Ga]1k mostró una captación tumoral de 18.2514.17 % ID/g a los 390 minutos (figura 4, tabla 6).
Además, se confirmó que todos los compuestos [68Ga]1e, [68Ga]1g, [68Ga]1h y [68Ga]1kse excretaban rápidamente fuera del cuerpo al disminuir la captación en el riñón después de 150 minutos.
Tabla 3


Tabla 4
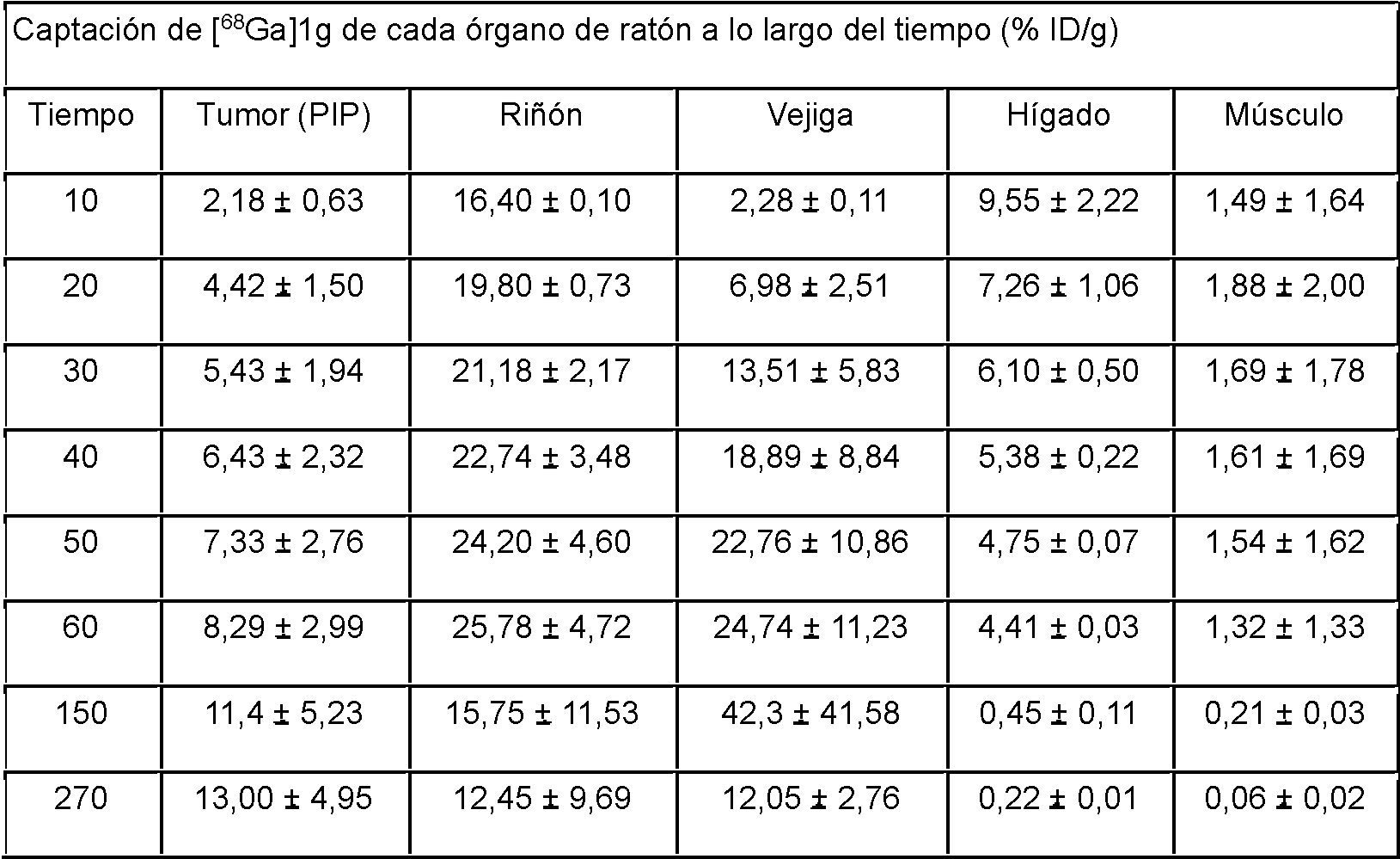
Tabla 5

Tabla 6

Ejemplo experimental 4: Prueba de biodistribución con ratones trasplantados con líneas celulares de cáncer de próstata
Tras 270 minutos de inyección de[68Ga]1e y [68Ga]1g, se obtuvieron imágenes de microPET/CT durante 30 minutos y, a continuación, se extrajo cada órgano (sangre, músculo, grasa, corazón, pulmón, hígado, bazo, estómago, intestino, riñón, hueso y tumor) y se midió su radiactividad con un contador gamma.
La tabla 7 muestra la captación de cada órgano 5 horas después de la inyección de [68Ga]1e o [68Ga]1g.
La biodistribución se confirmó 5 horas después de la inyección del compuesto. Como resultado, [68Ga]1g, que contenía un grupo fenilo, mostró una tasa de captación tumoral (% ID/g) más alta, de más del 10 %, que era aproximadamente 1,4 veces superior a la de [68Ga]1e.
Tabla 7


Por otra parte, el compuesto representado por la fórmula 1 según la presente invención puede formularse en diversas formas dependiendo del propósito de uso. A continuación, se ilustran algunos procedimientos de formulación en los que el compuesto representado por la fórmula 1 según la presente invención está contenido como principio activo, pero la presente invención no se limita a estos.
Ejemplo de fabricación 1: Preparación de formulaciones farmacéuticas
1-1. Preparación de polvos

Se prepararon polvos mezclando todos los componentes anteriores, que se envasaron en paquetes herméticos. 1-2. Preparación de comprimidos

Los comprimidos se prepararon mezclando todos los componentes anteriores por el procedimiento convencional de preparación de comprimidos.
1-3. Preparación de cápsulas

Se prepararon cápsulas mezclando todos los componentes anteriores, que se introdujeron en cápsulas de gelatina según el procedimiento convencional de preparación de cápsulas.
1-4. Preparación de una solución inyectable

Las soluciones inyectables se prepararon mezclando todos los componentes anteriores, introduciendo la mezcla en ampollas de 2 ml y esterilizándolas por el procedimiento convencional de preparación de soluciones inyectables.
1-5. Preparación de formulaciones líquidas

Todos los componentes anteriores se disolvieron en agua purificada. Tras añadir aroma de limón, se ajustó el volumen total a 100 ml añadiendo agua purificada. Las formulaciones líquidas se prepararon introduciendo la mezcla en botellas marrones y esterilizándolas por el procedimiento convencional de preparación de formulaciones líquidas.
Como se mencionó anteriormente, la presente invención se ha descrito en detalle a través de los ejemplos preparativos preferidos, los ejemplos y los ejemplos experimentales, pero el alcance de la presente invención no se limita a los ejemplos específicos y debe interpretarse por medio de las reivindicaciones anexas. Además, los expertos en la materia deben entender que son posibles muchas modificaciones y variaciones sin apartarse del alcance de la presente invención.
Aplicabilidad industrial
La presente invención se refiere a una composición farmacéutica para diagnosticar y tratar el cáncer de próstata, capaz de dirigirse a PSMA, y un compuesto proporcionado por un aspecto de la presente invención contiene un compuesto de glutamina-urea-lisina al que está acoplado estructuralmente un quelante radiactivo acoplado a metal y al que está acoplado un grupo arilo que puede unirse también a la proteína PSMA. El acoplamiento entre el compuesto de glutamina-urea-lisina y el quelante incluye un espaciador polar para cumplir la función de reducir el acoplamiento inespecífico in vivo y presentar un efecto de eliminación rápida de los órganos vitales, pero no del cáncer de próstata. Estas características disminuyen la exposición a la radiación, causada por un compuesto terapéutico acoplado a radioisótopos, de los tejidos y órganos normales y se reducen así los efectos secundarios. Además, un compuesto que contiene un grupo fenilo, que presenta un fuerte acoplamiento con la albúmina, tiene un mayor tiempo de residencia en la sangre, por lo que se acumula más en el cáncer de próstata.
Claims (11)
1. Un compuesto representado por la fórmula 1, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo:
[Fórmula 1]

en la que, en la fórmula 1,
Li es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 6 ;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, A1 es hidrógeno, o -(CH2)c-piridilo, en el que c es un número entero de 0 a 3;
L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6 ;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64, Cu-67, Y-90, Sc-47, In-111, Sn-117m, Lu-177, Bi-212, Bi-213, Pb-212, Ra-223, o Ac-225, y el quelante es


2. El compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo de la reivindicación 1, en el que:
Li es -(CH2)a, en el que a es un número entero de 1 a 6 ;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 4;
X es un enlace, o -C(O)-;
W es un enlace, o -NA1-, A 1 es hidrógeno, o -(CH2)c-piridilo, en el que c es un número entero de 0 a 1; L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 6 ;
Tz es un enlace,

L3 es alquileno C1.10 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 2 a 4;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-3 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64, Cu-67, Y-90, Sc-47, In-111, Sn-117m, Lu-177, Bi-212, Bi-213, Pb-212, Ra-223, o Ac-225, y el quelante es
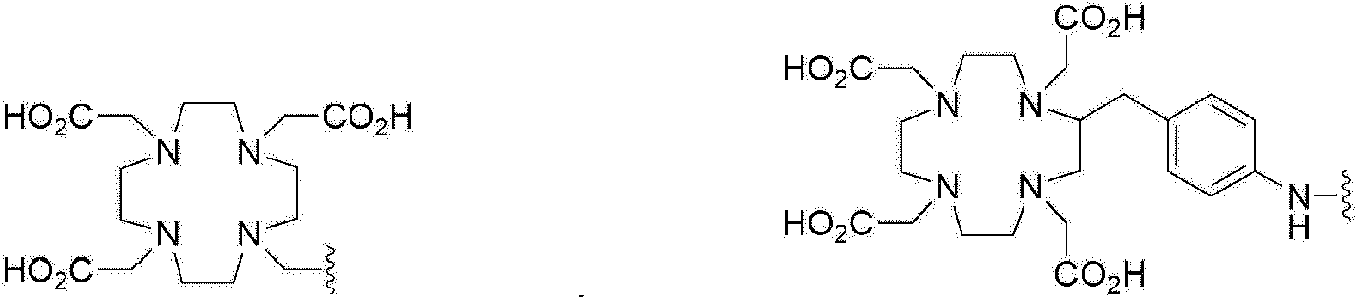

3. El compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo de la reivindicación 1, en el que:
Li es -(CH2)a-, en el que a es un número entero de 2 a 4;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 2;
X es un enlace, o -C(O)-;
W es un enlace, o -NA1-, en el que A1 es hidrógeno o piridilo;
L2 es un enlace, o -(CH2)d-, en el que d es un número entero de 1 a 2;
Tz es un enlace,
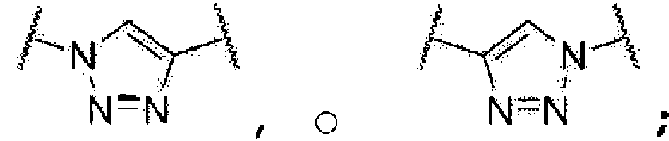
L3 es alquileno lineal C1-8, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)3-;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, metilo o halógeno;
Y es oxígeno;
Z es un quelante que incluye un metal radiactivo, en el que el metal radiactivo es Ga-68, Cu-64 o Lu-177, y el quelante puede ser

4. El compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo de la reivindicación 1, en el que el compuesto representado por la fórmula 1 se selecciona del grupo que consiste en los siguientes compuestos:

5 


en las que, en las fórmulas anteriores, M es un metal radiactivo, y el metal radiactivo es como se define en la reivindicación 1.
5. Un compuesto representado por la fórmula 2, un estereoisómero del mismo, un hidrato del mismo o una sal farmacéuticamente aceptable del mismo:
[Fórmula 2]

en la que, en la fórmula 2 ,
L1 es -(CH2)a, en el que a es un número entero de 1 a 8;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 6 ;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, y A1 es hidrógeno o -(CH2)c-piridilo, en el que c es un número entero de 0 a 3; L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 8;
Tz es un enlace,

L3 es alquileno C1-12 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 1 a 6 ;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-5 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z' es un quelante, y el quelante es
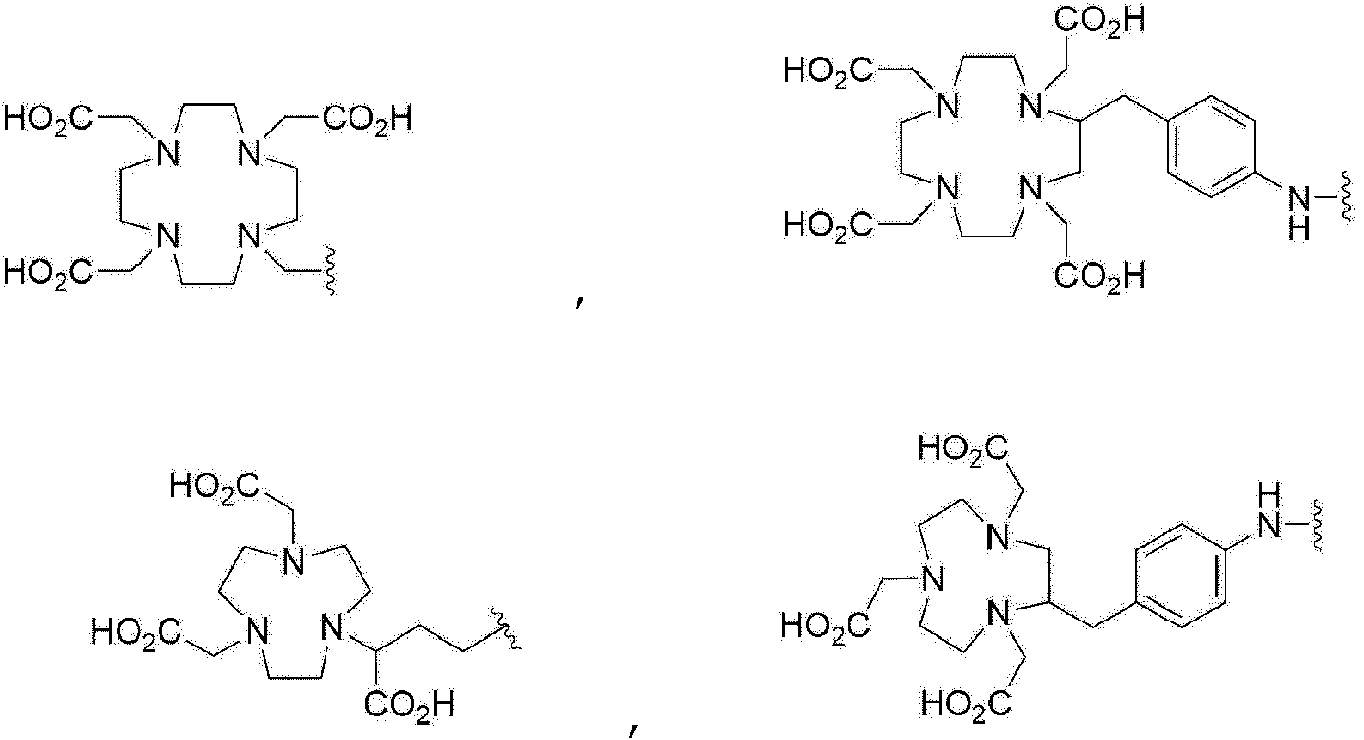
o

6. El compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable de la reivindicación 5, en el que:
L1 es -(CH2)a, en el que a es un número entero de 1 a 6 ;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 4;
X es un enlace, o -C(O)-;
W es un enlace o -NA1-, y A1 es hidrógeno o -(CH2)c-piridilo, en el que c es un número entero de 0 a 1; L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 6 ;
Tz es

L3 es alquileno C1-10 lineal o ramificado, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)e-, en el que e es un número entero de 2 a 4;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, alquilo C1-3 lineal o ramificado, o halógeno;
Y es oxígeno o azufre;
Z' es un quelante, y el quelante es

o

7. El compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo de la reivindicación 5, en el que:
L1 es -(CH2)a-, en el que a es un número entero de 2 a 4;
U es un enlace, o -C(O)-;
R1 es -L5-CO2H, en el que L5 es -(CH2)b-, en el que b es un número entero de 1 a 2;
X es un enlace, o -C(O)-;
W es un enlace o-NA1-, y A1 es hidrógeno o piridilo;
L2 es un enlace o -(CH2)d-, en el que d es un número entero de 1 a 2;
Tz es un enlace,

L3 es alquileno lineal C1-8, en el que uno o más átomos de carbono del alquileno pueden sustituirse por átomos de oxígeno;
L4 es -(CH2)3-;
n es un número entero de 0 a 1 ;
R2 es hidrógeno, metilo o halógeno;
Y es oxígeno;
Z' es un quelante, y el quelante es

8. El compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable de la reivindicación 5, en el que el compuesto representado por la fórmula 2 se selecciona del grupo que consiste en los siguientes compuestos:

5 

5 ( 13 )

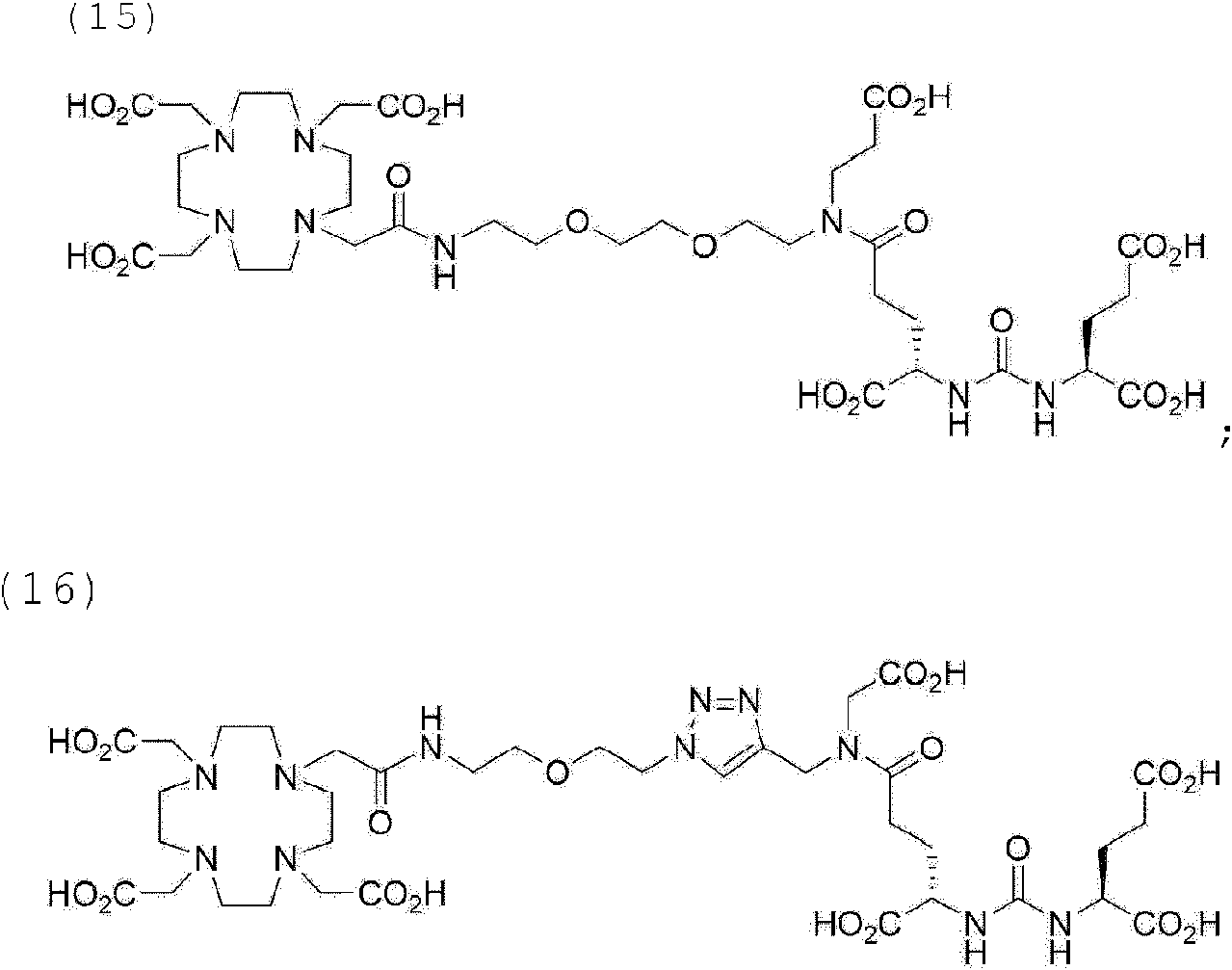
9. Una composición que comprende el compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo de la reivindicación 1 como principio activo para su uso en el diagnóstico del cáncer de próstata.
10. La composición para el uso de la reivindicación 9, en la que la composición diagnostica el cáncer de próstata mediante la unión selectiva del compuesto al PSMA (antígeno de membrana específico de la próstata) sobreexpresado en las células del cáncer de próstata.
11. Una composición farmacéutica que comprende el compuesto, el estereoisómero del mismo, el hidrato del mismo o la sal farmacéuticamente aceptable del mismo de la reivindicación 1 como principio activo para su uso en la prevención o el tratamiento del cáncer de próstata.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20180037226 | 2018-03-30 | ||
| PCT/KR2019/003716 WO2019190266A1 (ko) | 2018-03-30 | 2019-03-29 | 전립선암 진단 및 치료를 위한 psma-표적 방사성의약품 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2947748T3 true ES2947748T3 (es) | 2023-08-17 |
Family
ID=68058285
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19775196T Active ES2947748T3 (es) | 2018-03-30 | 2019-03-29 | Radiofármaco dirigido a PSMA para el diagnóstico y tratamiento del cáncer de próstata |
Country Status (26)
| Country | Link |
|---|---|
| US (1) | US11931431B2 (es) |
| EP (1) | EP3778592B1 (es) |
| JP (1) | JP7094591B2 (es) |
| KR (1) | KR102156385B1 (es) |
| CN (1) | CN112004812B (es) |
| AU (1) | AU2019243408B2 (es) |
| BR (1) | BR112020019566A2 (es) |
| CA (1) | CA3094620C (es) |
| CL (1) | CL2020002510A1 (es) |
| DK (1) | DK3778592T3 (es) |
| EA (1) | EA202092333A1 (es) |
| ES (1) | ES2947748T3 (es) |
| FI (1) | FI3778592T3 (es) |
| HR (1) | HRP20230604T1 (es) |
| HU (1) | HUE062904T2 (es) |
| LT (1) | LT3778592T (es) |
| MX (1) | MX2020010266A (es) |
| MY (1) | MY197419A (es) |
| PH (1) | PH12020551471A1 (es) |
| PL (1) | PL3778592T3 (es) |
| PT (1) | PT3778592T (es) |
| RS (1) | RS64296B1 (es) |
| SG (1) | SG11202009649RA (es) |
| SI (1) | SI3778592T1 (es) |
| WO (1) | WO2019190266A1 (es) |
| ZA (1) | ZA202006008B (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102269315B1 (ko) * | 2019-10-24 | 2021-06-24 | 서울대학교산학협력단 | 전립선 암의 영상 또는 치료를 위한 동위원소 표지 화합물 |
| US20230016265A1 (en) * | 2019-10-29 | 2023-01-19 | The Cleveland Clinic Foundation | Psma-targeting imaging agents |
| KR20220006286A (ko) | 2020-07-08 | 2022-01-17 | 한국원자력의학원 | 전립선암 진단 및 치료를 위한 전립선특이 막 항원 표적 화합물 및 이를 포함하는 전립선암 진단 및 치료용 조성물 |
| KR20230050552A (ko) * | 2021-10-07 | 2023-04-17 | (주)퓨쳐켐 | 가돌리늄 화합물 및 이를 포함하는 전립선암의 진단 및 치료용 약학적 조성물 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013022797A1 (en) * | 2011-08-05 | 2013-02-14 | Molecular Insight Pharmaceuticals | Radiolabeled prostate specific membrane antigen inhibitors |
| US9636413B2 (en) * | 2012-11-15 | 2017-05-02 | Endocyte, Inc. | Conjugates for treating diseases caused by PSMA expressing cells |
| WO2014110372A1 (en) | 2013-01-14 | 2014-07-17 | Molecular Insight Pharmaceuticals | Triazine based radiopharmaceuticals and radioimaging agents |
| MX2016005013A (es) | 2013-10-18 | 2017-02-28 | Deutsches Krebsforsch | Inhibidores marcados de antigeno prostatico especifico de membrana (psma), su uso como agentes formadores de imagenes y agentes farmaceuticos para el tratamiento de cancer de prostata. |
| CA3215134A1 (en) * | 2014-05-06 | 2015-11-12 | The Johns Hopkins University | Metal/radiometal-labeled psma inhibitors for psma-targeted imaging and radiotherapy |
| PT3925952T (pt) * | 2016-03-22 | 2023-12-13 | Univ Johns Hopkins | Agentes alvo de alta afinidade direcionados ao antigénio de membrana específica da próstata para endo radioterapia de cancro da próstata |
| US20210276971A1 (en) * | 2018-06-20 | 2021-09-09 | The Research Foundation For The State University Of New York | Triazamacrocycle-derived chelator compositions for coordination of imaging and therapy metal ions and methods of using same |
-
2019
- 2019-03-29 DK DK19775196.9T patent/DK3778592T3/da active
- 2019-03-29 SG SG11202009649RA patent/SG11202009649RA/en unknown
- 2019-03-29 BR BR112020019566-9A patent/BR112020019566A2/pt active Search and Examination
- 2019-03-29 MY MYPI2020004923A patent/MY197419A/en unknown
- 2019-03-29 PL PL19775196.9T patent/PL3778592T3/pl unknown
- 2019-03-29 RS RS20230503A patent/RS64296B1/sr unknown
- 2019-03-29 EP EP19775196.9A patent/EP3778592B1/en active Active
- 2019-03-29 JP JP2021502679A patent/JP7094591B2/ja active Active
- 2019-03-29 PT PT197751969T patent/PT3778592T/pt unknown
- 2019-03-29 CN CN201980023911.7A patent/CN112004812B/zh active Active
- 2019-03-29 KR KR1020190037293A patent/KR102156385B1/ko active IP Right Grant
- 2019-03-29 CA CA3094620A patent/CA3094620C/en active Active
- 2019-03-29 ES ES19775196T patent/ES2947748T3/es active Active
- 2019-03-29 HR HRP20230604TT patent/HRP20230604T1/hr unknown
- 2019-03-29 WO PCT/KR2019/003716 patent/WO2019190266A1/ko unknown
- 2019-03-29 LT LTEPPCT/KR2019/003716T patent/LT3778592T/lt unknown
- 2019-03-29 US US16/981,432 patent/US11931431B2/en active Active
- 2019-03-29 AU AU2019243408A patent/AU2019243408B2/en active Active
- 2019-03-29 FI FIEP19775196.9T patent/FI3778592T3/fi active
- 2019-03-29 MX MX2020010266A patent/MX2020010266A/es unknown
- 2019-03-29 SI SI201930575T patent/SI3778592T1/sl unknown
- 2019-03-29 EA EA202092333A patent/EA202092333A1/ru unknown
- 2019-03-29 HU HUE19775196A patent/HUE062904T2/hu unknown
-
2020
- 2020-09-15 PH PH12020551471A patent/PH12020551471A1/en unknown
- 2020-09-28 CL CL2020002510A patent/CL2020002510A1/es unknown
- 2020-09-29 ZA ZA2020/06008A patent/ZA202006008B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ZA202006008B (en) | 2021-10-27 |
| KR102156385B1 (ko) | 2020-09-15 |
| BR112020019566A2 (pt) | 2021-01-05 |
| EA202092333A1 (ru) | 2021-01-21 |
| WO2019190266A1 (ko) | 2019-10-03 |
| SG11202009649RA (en) | 2020-10-29 |
| MX2020010266A (es) | 2020-11-06 |
| CA3094620C (en) | 2022-11-22 |
| MY197419A (en) | 2023-06-16 |
| EP3778592A1 (en) | 2021-02-17 |
| EP3778592B1 (en) | 2023-04-26 |
| CN112004812A (zh) | 2020-11-27 |
| SI3778592T1 (sl) | 2023-08-31 |
| LT3778592T (lt) | 2023-06-26 |
| PH12020551471A1 (en) | 2021-09-01 |
| DK3778592T3 (da) | 2023-06-19 |
| AU2019243408A1 (en) | 2020-10-29 |
| US20210106701A1 (en) | 2021-04-15 |
| US11931431B2 (en) | 2024-03-19 |
| CN112004812B (zh) | 2023-05-26 |
| HUE062904T2 (hu) | 2023-12-28 |
| EP3778592A4 (en) | 2021-02-17 |
| FI3778592T3 (fi) | 2023-06-19 |
| JP7094591B2 (ja) | 2022-07-04 |
| CA3094620A1 (en) | 2019-10-03 |
| CL2020002510A1 (es) | 2021-02-19 |
| AU2019243408B2 (en) | 2021-10-14 |
| PL3778592T3 (pl) | 2023-11-27 |
| PT3778592T (pt) | 2023-06-19 |
| KR20190114908A (ko) | 2019-10-10 |
| JP2021519822A (ja) | 2021-08-12 |
| RS64296B1 (sr) | 2023-07-31 |
| HRP20230604T1 (hr) | 2023-09-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2947748T3 (es) | Radiofármaco dirigido a PSMA para el diagnóstico y tratamiento del cáncer de próstata | |
| ES2867814T3 (es) | Compuestos heterocíclicos deuterados y su uso como agentes de formación de imágenes | |
| Chin et al. | First experience with clinical-grade [18 F] FPP (RGD) 2: an automated multi-step radiosynthesis for clinical PET studies | |
| LT4391B (lt) | Trinariai radiofarmaciniai kompleksai | |
| BR112021014933A2 (pt) | Radiomarcador de modo duplo de ligação a psma e terapêutica | |
| JP5690733B2 (ja) | ビスホスホン酸誘導体及びその放射性金属核種標識体 | |
| ES2244608T3 (es) | Complejos de ligandos ternarios utiles como radiofarmacos. | |
| Lee et al. | Synthesis and biological evaluation of RGD peptides with the 99m Tc/188 Re chelated iminodiacetate core: Highly enhanced uptake and excretion kinetics of theranostics against tumor angiogenesis | |
| Kim et al. | Synthesis and characterization of a 68Ga-labeled N-(2-diethylaminoethyl) benzamide derivative as potential PET probe for malignant melanoma | |
| TW202241527A (zh) | 雙模放射性追蹤劑及治療方法 | |
| Gomez et al. | Synthesis and Evaluation of Diastereoisomers of 1, 4, 7-Triazacyclononane-1, 4, 7-tris-(glutaric acid)(NOTGA) for Multimeric Radiopharmaceuticals of Gallium | |
| HUT73665A (en) | Bifunctional-chelating agents braked with calcogene atoms, pharmaceutical compositions containing them , and use of these compositions in radio- diagnosis and radiotherapy | |
| ES2616045T3 (es) | Forma estabilizada de tetrofosmina y su uso | |
| JP5481673B2 (ja) | 放射性標識薬剤 | |
| KR102412174B1 (ko) | 암 또는 염증질환의 진단 및 치료를 위한 사전표적 방사성의약품 | |
| Niu et al. | Synthesis and Preliminary Evaluation of 1‐[18F] Fluoro‐1‐deoxy‐2, 5‐anhydro‐D‐mannitol as a PET Radiotracer for Breast Cancer Imaging | |
| EA041468B1 (ru) | Псма-таргетные радиофармацевтические средства для диагностики и лечения рака простаты | |
| TWI244925B (en) | New imaging agents, precursors thereof and methods of manufacture | |
| CN107847617B (zh) | 含有取代的亚氨基二乙酸配体的金属三羰基复合物以及作为放射性同位素示踪物的用途 | |
| CN104114191A (zh) | 心肌灌注显像造影剂 | |
| JP2019085344A (ja) | 放射性フッ素標識化合物 | |
| KR101427292B1 (ko) | 허혈성 조직 영상을 위한 플루오르―18 표지 트리아자노난 유도체 또는 이의 약학적으로 허용가능한 염 | |
| KR20210048827A (ko) | 전립선 암의 영상 또는 치료를 위한 동위원소 표지 화합물 | |
| WO2022093079A1 (ru) | Комплексное соединение y, содержащее радионуклид 227th и бисфосфонат и его применение | |
| JPWO2020045638A1 (ja) | 放射性イミダゾチアジアゾール誘導体化合物 |