ES2799582T3 - Proceso para la síntesis de un inhibidor de indoleamina 2,3-dioxigenasa - Google Patents
Proceso para la síntesis de un inhibidor de indoleamina 2,3-dioxigenasa Download PDFInfo
- Publication number
- ES2799582T3 ES2799582T3 ES14812015T ES14812015T ES2799582T3 ES 2799582 T3 ES2799582 T3 ES 2799582T3 ES 14812015 T ES14812015 T ES 14812015T ES 14812015 T ES14812015 T ES 14812015T ES 2799582 T3 ES2799582 T3 ES 2799582T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- amino
- tert
- reducing agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *C(N(*)S(N(*)CCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)(=O)=O)=O Chemical compound *C(N(*)S(N(*)CCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)(=O)=O)=O 0.000 description 3
- HCNUSGQZCXEZIN-UHFFFAOYSA-N NCCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O Chemical compound NCCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O HCNUSGQZCXEZIN-UHFFFAOYSA-N 0.000 description 2
- QFKFEXBPEHSIEV-UHFFFAOYSA-N CC(C)(C)OC(N(CC=C)S(N(CCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)CC=C)(=O)=O)=O Chemical compound CC(C)(C)OC(N(CC=C)S(N(CCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)CC=C)(=O)=O)=O QFKFEXBPEHSIEV-UHFFFAOYSA-N 0.000 description 1
- PNHGBESJPKQIJY-UHFFFAOYSA-N CC(C)(C)OC(N(Cc(cc1)ccc1OC)S(N(CCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)Cc(cc1)ccc1OC)(=O)=O)=O Chemical compound CC(C)(C)OC(N(Cc(cc1)ccc1OC)S(N(CCNc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)Cc(cc1)ccc1OC)(=O)=O)=O PNHGBESJPKQIJY-UHFFFAOYSA-N 0.000 description 1
- ZKVKUHMVSLYWKV-UHFFFAOYSA-N Cc1cc(N(C(c2n[o]nc2N)=NO2)C2=O)ccc1F Chemical compound Cc1cc(N(C(c2n[o]nc2N)=NO2)C2=O)ccc1F ZKVKUHMVSLYWKV-UHFFFAOYSA-N 0.000 description 1
- KOWPUNQBGWIERF-UHFFFAOYSA-N Nc(cc1Br)ccc1F Chemical compound Nc(cc1Br)ccc1F KOWPUNQBGWIERF-UHFFFAOYSA-N 0.000 description 1
- LVNWHKAEEDWAGF-KXFIGUGUSA-N Nc1n[o]nc1/C(/Cl)=N/O Chemical compound Nc1n[o]nc1/C(/Cl)=N/O LVNWHKAEEDWAGF-KXFIGUGUSA-N 0.000 description 1
- ORHZJUSHZUCMKR-UHFFFAOYSA-N Nc1n[o]nc1/C(/Nc(cc1Br)ccc1F)=N/O Chemical compound Nc1n[o]nc1/C(/Nc(cc1Br)ccc1F)=N/O ORHZJUSHZUCMKR-UHFFFAOYSA-N 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N O=C([n]1cncc1)[n]1cncc1 Chemical compound O=C([n]1cncc1)[n]1cncc1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C307/00—Amides of sulfuric acids, i.e. compounds having singly-bound oxygen atoms of sulfate groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C307/04—Diamides of sulfuric acids
- C07C307/06—Diamides of sulfuric acids having nitrogen atoms of the sulfamide groups bound to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/04—1,2,3-Oxadiazoles; Hydrogenated 1,2,3-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/08—1,2,5-Oxadiazoles; Hydrogenated 1,2,5-oxadiazoles
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Indole Compounds (AREA)
Abstract
Un proceso que comprende hacer reaccionar un compuesto de Fórmula F5: **(Ver fórmula)** con un aldehído de Fórmula F6: **(Ver fórmula)** en donde Pg1 es un grupo protector de amino, para proporcionar un compuesto de Fórmula F7: **(Ver fórmula)**
Description
DESCRIPCIÓN
Proceso para la síntesis de un inhibidor de indoleamina 2,3-dioxigenasa
Campo de la invención
La presente solicitud se refiere a procesos e intermedios para preparar 4-({2-[(aminosulfonilo)amino]etilo}amino)-N-(3-bromo-4-fluorofenilo)-N'-hidroxi-1,2,5-oxadiazol-3-carboximidamida, que es un inhibidor de la 2,3-dioxigenasa de indoleamina útil en el tratamiento del cáncer y otros trastornos.
Antecedentes de la invención
El triptófano (Trp) es un aminoácido esencial requerido para la biosíntesis de proteínas, niacina y el neurotransmisor 5-hidroxitriptamina (serotonina). La enzima indoleamina 2,3-dioxigenasa (también conocida como INDO o IDO) cataliza el primer paso y limita la velocidad en la degradación de L-triptófano a N-formilo-kinurenina. En las células humanas, un agotamiento de Trp resultante de la actividad IDO es un prominente mecanismo efector antimicrobiano inducible por interferón gamma (IFN-y). La estimulación de IFN-y induce la activación de IDO, lo que conduce a un agotamiento de Trp, deteniendo así el crecimiento de patógenos intracelulares dependientes de Trp como Toxoplasma gondii y Chlamydia trachomatis. La actividad de IDO también tiene un efecto antiproliferativo en muchas células tumorales, y se ha observado inducción de IDO in vivo durante el rechazo de tumores alogénicos, lo que indica un posible papel para esta enzima en el proceso de rechazo de tumores (Daubener, et al., 1999, Adv. Exp. Exp. Med. Biol., 467: 517-24; Taylor, et al., 1991, FASEB J , 5: 2516-22).
Se ha observado que las células HeLa cocultivadas con linfocitos de sangre periférica (PBL) adquieren un fenotipo inmunoinhibidor a través de la regulación positiva de la actividad IDO. Se creía que una reducción en la proliferación de PBL tras el tratamiento con interleucina-2 (IL2) era el resultado de IDO liberado por las células tumorales en respuesta a la secreción de IFNG por los PBL. Este efecto fue revertido por el tratamiento con 1-metilo-triptófano (1MT), un inhibidor de IDO específico. Se propuso que la actividad IDO en las células tumorales puede servir para perjudicar las respuestas antitumorales (Logan, et al., 2002, Immunology, 105: 478-87).
Recientemente, un papel inmunorregulador del agotamiento de Trp ha recibido mucha atención. Varias líneas de evidencia sugieren que IDO está involucrado en la inducción de la tolerancia inmune. Los estudios de embarazo en mamíferos, resistencia tumoral, infecciones crónicas y enfermedades autoinmunes han demostrado que las células que expresan IDO pueden suprimir las respuestas de las células T y promover la tolerancia. Se ha observado un catabolismo acelerado de Trp en enfermedades y trastornos asociados con la activación inmune celular, como infección, malignidad, enfermedades autoinmunes y SIDA, así como durante el embarazo. Por ejemplo, se han observado niveles elevados de IFN y niveles elevados de metabolitos Trp urinarios en enfermedades autoinmunes; Se ha postulado que el agotamiento sistémico o local de Trp que ocurre en las enfermedades autoinmunes puede estar relacionado con la degeneración y los síntomas de desgaste de estas enfermedades. En apoyo de esta hipótesis, se observaron altos niveles de IDO en células aisladas de la sinovia de las articulaciones artríticas. Los IFN también están elevados en pacientes con virus de inmunodeficiencia humana (VIH) y el aumento de los niveles de IFN se asocia con un peor pronóstico. Por lo tanto, se propuso que IDO es inducida crónicamente por la infección por VIH, y aumenta aún más por infecciones oportunistas, y que la pérdida crónica de Trp inicia mecanismos responsables de caquexia, demencia y diarrea y posiblemente inmunosupresión de pacientes con SIDA (Brown, et al., 1991, Adv. Exp. Med. Biol., 294: 425-35). Con este fin, se ha demostrado recientemente que la inhibición de IDO puede mejorar los niveles de células T específicas de virus y, concomitantemente, reducir el número de macrófagos infectados por virus en un modelo de VIH de ratón (Portula et al., 2005, Blood, 106: 2382-90).
Se cree que IDO desempeña un papel en los procesos inmunosupresores que evitan el rechazo fetal en el útero. Hace más de 40 años, se observó que, durante el embarazo, el conceptus mamífero genéticamente dispar sobrevive a pesar de lo que predeciría la inmunología del trasplante de tejidos (Medawar, 1953, Symp. Soc. Exp. Biol. 7: 320-38). La separación anatómica de la madre y el feto y la inmadurez antigénica del feto no pueden explicar completamente la supervivencia del aloinjerto fetal. La atención reciente se ha centrado en la tolerancia inmunológica de la madre. Debido a que la IDO se expresa por las células sincitiotrofoblasto humanas y la concentración sistémica de triptófano disminuye durante el embarazo normal, se planteó la hipótesis de que la expresión de IDO en la interfaz materno-fetal es necesaria para prevenir el rechazo inmunológico de los aloinjertos fetales. Para probar esta hipótesis, los ratones gestantes (portadores de fetos singénicos o alogénicos) fueron expuestos a 1MT, y se observó un rápido rechazo inducido por las células T de todos los conceptos alogénicos. Por lo tanto, al catabolizar el triptófano, el concepto de mamífero parece suprimir la actividad de las células T y se defiende contra el rechazo, y el bloqueo del catabolismo del triptófano durante el embarazo murino permite que las células T maternas provoquen el rechazo del aloinjerto fetal (Munn, et al., 1998, Science, 281:1191-3).
La evidencia adicional de un mecanismo de resistencia inmune tumoral basado en la degradación de triptófano por IDO proviene de la observación de que la mayoría de los tumores humanos expresan IDO constitutivamente, y que la expresión de IDO por células tumorales de ratón inmunogénicas evita su rechazo por ratones preinmunizados. Este efecto se acompaña de una falta de acumulación de células T específicas en el sitio del tumor y puede revertirse en
parte mediante el tratamiento sistémico de ratones con un inhibidor de IDO, en ausencia de una toxicidad notable. Por lo tanto, se sugirió que la eficacia de la vacunación terapéutica de pacientes con cáncer podría mejorarse mediante la administración concomitante de un inhibidor de IDO (Uyttenhove et al., 2003, Nature Med., 9:1269-74). También se ha demostrado que el inhibidor de IDO, 1-MT, puede sinergizar con agentes quimioterapéuticas para reducir el crecimiento tumoral en ratones, lo que sugiere que la inhibición de IDO también puede mejorar la actividad antitumoral de las terapias citotóxicas convencionales (Muller et al., 2005, Nature Med., 11: 312-9).
Un mecanismo que contribuye a la falta de respuesta inmunológica hacia los tumores puede ser la presentación de antígenos tumorales por las APC tolerogénicas del huésped. También se ha descrito un subconjunto de células presentadoras de antígeno (APC) que expresan IDO humano que coexpresaron CD123 (IL3RA) y c CR6 e inhibieron la proliferación de células T. Tanto las células dendríticas positivas para CD123 maduras como las inmaduras suprimieron la actividad de las células T, y esta actividad supresora de IDO fue bloqueada por 1MT (Munn, et al., 2002, Science, 297:1867-70). También se ha demostrado que los ganglios linfáticos de drenaje tumoral de ratón (TDLN) contienen un subconjunto de células dendríticas plasmacitoides (pDC) que expresan constitutivamente niveles inmunosupresores de IDO. A pesar de comprender solo el 0,5% de las células de los ganglios linfáticos, in vitro, estos pDC suprimieron potentemente las respuestas de las células T a los antígenos presentados por los propios pDC y también, de manera dominante, suprimieron las respuestas de las células T a los antígenos de terceros presentados por las APC no supresoras. Dentro de la población de pDC, la mayoría de la actividad supresora mediada por IDO funcional se segregó con un nuevo subconjunto de pDC que coexpresan el marcador de linaje B CD19. Por lo tanto, se planteó la hipótesis de que la supresión mediada por IDO por pDC en TDLN crea un microambiente local que es poderosamente supresor de las respuestas de las células T antitumorales del huésped (Munn, et al., 2004, J. Clin. Invest., 114(2):280-90).
IDO degrada el resto indol del triptófano, la serotonina y la melatonina e inicia la producción de metabolitos neuroactivos e inmunorreguladores, conocidos colectivamente como kinureninas. Al agotar localmente el triptófano y aumentar las kinureninas proapoptóticas, la IDO expresada por las células dendríticas (CD) puede afectar en gran medida la proliferación y supervivencia de las células T. La inducción de IDO en CD podría ser un mecanismo común de tolerancia delecional impulsado por las células T reguladoras. Debido a que se puede esperar que tales respuestas tolerogénicas operen en una variedad de condiciones fisiopatológicas, el metabolismo del triptófano y la producción de kinurenina podrían representar una interfaz crucial entre el sistema inmune y nervioso (Grohmann, et al., 2003, Trends Immunol., 24: 242-8) En estados de activación inmune persistente, la disponibilidad de Trp sérico libre disminuye y, como consecuencia de la producción reducida de serotonina, las funciones serotoninérgicas también pueden verse afectadas (Wirleitner, et al., 2003, Curr. Med. Chem., 10:1581-91).
Curiosamente, se ha observado que la administración de interferón-alfa induce efectos secundarios neuropsiquiátricos, como síntomas depresivos y cambios en la función cognitiva. La influencia directa sobre la neurotransmisión serotoninérgica puede contribuir a estos efectos secundarios. Además, debido a que la activación de IDO conduce a niveles reducidos de triptófano, el precursor de la serotonina (5-HT), IDO puede desempeñar un papel en estos efectos secundarios neuropsiquiátricos al reducir la síntesis central de 5-HT. Además, los metabolitos de kinurenina, como la 3-hidroxi-kinurenina (3-OH-KYN) y el ácido quinolínico (QUIN) tienen efectos tóxicos sobre la función cerebral. 3-OH-KYN puede producir estrés oxidativo al aumentar la producción de especies reactivas de oxígeno (ROS), y QUIN puede producir una sobreestimulación de los receptores de N-metilo-D-aspartato (NMDA) del hipocampo, lo que conduce a la apoptosis y la atrofia del hipocampo. Tanto la sobreproducción de ROS como la atrofia del hipocampo causada por la sobreestimulación de NMDA se han asociado con la depresión (Wichers y Maes, 2004, J. Psychiatry Neurosci., 29:11-17). Por lo tanto, la actividad IDO puede desempeñar un papel en la depresión.
Se están desarrollando inhibidores de molécula pequeña de IDO para tratar o prevenir enfermedades relacionadas con IDO como las descritas anteriormente. Por ejemplo, se informan oxadiazol y otros inhibidores heterocíclicos de IDO en los documentos US 2006/0258719 y US 2007/0185165. La publicación Pc T WO 99/29310 informa métodos para alterar la inmunidad mediada por células T que comprende alterar las concentraciones extracelulares locales de triptófano y metabolitos de triptófano, usando un inhibidor de IDO como 1-metilo-DL-triptófano, p-(3-benzofuranilo)-DL-alanina, p-[3-benzo(b)tienilo]-DL-alanina y 6-nitro-L-triptófano) (Munn, 1999). En el documento WO 03/087347, también publicado como Patente Europea 1501918, se describen métodos para fabricar células presentadoras de antígeno para mejorar o reducir la tolerancia de las células T (Munn, 2003). Los compuestos que tienen actividad inhibidora de indoleamina-2,3-dioxigenasa (IDO) se describen adicionalmente en el documento W o 2004/094409; y la Publicación de Solicitud de Patente de los Estados Unidos N° 2004/0234623 está dirigida a métodos para tratar a un sujeto con un cáncer o una infección mediante la administración de un inhibidor de indoleamina-2,3-dioxigenasa en combinación con otras modalidades terapéuticas.
A la luz de los datos experimentales que indican un papel para IDO en la inmunosupresión, resistencia y/o rechazo tumoral, infecciones crónicas, infección por VIH, SIDA (incluidas sus manifestaciones como caquexia, demencia y diarrea), enfermedades o trastornos autoinmunes (como artritis reumatoide), y la tolerancia inmunológica y la prevención del rechazo fetal en el útero, son deseables los agentes terapéuticos destinados a la supresión de la degradación del triptófano mediante la inhibición de la actividad IDO. Los inhibidores de IDO pueden usarse para activar las células T y, por lo tanto, mejorar la activación de las células T cuando las células T son suprimidas por el embarazo, malignidad o un virus como el VIH. La inhibición de IDO también puede ser una estrategia de tratamiento
importante para pacientes con enfermedades o trastornos neurológicos o neuropsiquiátricos como la depresión. WO 2010/005958 describe derivados de 1,2,5-oxadiazol que son inhibidores de IDO y procesos e intermedios para fabricar tales derivados de 1,2,5-oxadiazol.
Debido a la utilidad de los inhibidores de IDO, existe la necesidad de desarrollar nuevos procesos para fabricar inhibidores de IDO. Esta aplicación está dirigida a esta necesidad y a otras.
Resumen de la invención
El compuesto 4-({2-[(aminosulfonilo)amino]etilo}amino)-N-(3-bromo-4-fluorofenilo)-N-hidroxM,2,5-oxadiazol-3-carboximidamida que tiene la Fórmula I:

es un inhibidor de la enzima indoleamina 2,3-dioxigenasa (también conocida como IDO). El compuesto de Fórmula I, así como su preparación y uso, se ha descrito en la patente de EE.UU. N° 8,088,803, que se incorpora aquí como referencia en su totalidad. Los intermedios y procesos proporcionados en este documento ayudan a satisfacer la necesidad continua de desarrollar inhibidores de IDO para el tratamiento de enfermedades graves.
La presente solicitud proporciona, entre otros, productos intermedios y procesos para preparar un compuesto de Fórmula I:

Por consiguiente, la presente solicitud proporciona un proceso que comprende hacer reaccionar un compuesto de Fórmula F5:

con un aldehído de Fórmula F6:
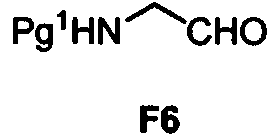
para proporcionar un compuesto de Fórmula F7:

en donde Pg1 se define más adelante.
La presente solicitud proporciona además un proceso que comprende hacer reaccionar un compuesto de Fórmula F15:

con un compuesto de Fórmula F5 para proporcionar un compuesto de Fórmula F16:

en donde R1 y R3 se definen abajo.
La presente solicitud proporciona además un proceso que comprende hacer reaccionar un compuesto de Fórmula F17:

con un compuesto de Fórmula F5 para proporcionar un compuesto de Fórmula F18:

en donde R4 se define más adelante.
Descripción detallada
Si bien algunos de los pasos del proceso se ilustran en los Esquemas que se muestran a continuación, se pretende que los pasos individuales del proceso puedan reclamarse individualmente o en cualquier combinación (por ejemplo, en el Esquema I, pasos E, F, G, H e I puede ser reclamado individualmente o en combinación). No se pretende que los procesos se limiten a un proceso general que tenga todos y cada uno de los pasos en los Esquemas a continuación. Por consiguiente, el esquema general para la preparación del compuesto de Fórmula I se describe en el Esquema 1.

Por consiguiente, la presente solicitud proporciona un proceso que comprende hacer reaccionar un compuesto de Fórmula F5:

con un aldehído de Fórmula F6:

F6
en donde Pg1 es un grupo protector de amino, para proporcionar un compuesto de Fórmula F7:

Los grupos protectores de amino Pg1 pueden usarse para prevenir reacciones no deseadas de un grupo amino mientras realiza una transformación deseada. Los grupos protectores de amino permiten una fácil unión covalente a un átomo de nitrógeno, así como una escisión selectiva del átomo de nitrógeno. "Grupos protectores de amino" adecuados, tales como alcoxicarbonilo (tal como etoxicarbonilo, terc-butoxicarbonilo (Boc), benciloxicarbonilo (Cbz),
9-fluorenilmetiloxicarbonilo (Fmoc) y similares), acilo (tal como acetilo (Ac), benzoilo (Bz), y similares), sulfonilo (como metanosulfonilo, trifluorometanosulfonilo y similares), arilalquilo (como bencilo, 4-metoxibencilo, difenilmetilo, trifenilmetilo (tritilo) y similares), alquenilalquilo (como alilo, prenilo y similares), diarilmetilenilo (como (C6Hs)2C=N, y similares) y sililo (como terc-butildimetilsililo, triisopropilsililo y similares), son conocidos por un experto en la materia. La química de los grupos protectores de amino se puede encontrar en Wuts and Greene, Greene's Protective Groups in Organic Synthesis, 4a Ed., pp 696-926, John Wiley & Sons: Nueva York, 2006.
En algunas realizaciones, Pg1 es etoxicarbonilo, terc-butoxicarbonilo, benciloxicarbonilo o 9-fluorenilmetiloxicarbonilo. En algunas realizaciones, Pg1 es Ci-6 alcoxicarbonilo.
En algunas realizaciones, Pg1 es terc-butoxicarbonilo.
Los solventes apropiados para el Paso E incluyen, pero no se limitan a, metanol o tetrahidrofurano (THF), acetonitrilo y similares. También se pueden usar disolventes de hidrocarburos halogenados (es decir, alcanos halogenados, tales como diclorometano, cloroformo, dicloroetano o tetracloroetano).
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende tetrahidrofurano. Como se usa en el presente documento, un componente disolvente puede referirse a un disolvente o una mezcla de disolventes. En algunas realizaciones, el componente disolvente es un disolvente orgánico. En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende un disolvente hidrocarbonado halogenado. En algunas realizaciones, dicho disolvente de hidrocarburo halogenado es diclorometano.
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende acetonitrilo.
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende diclorometano y acetonitrilo.
En algunas realizaciones, dicha reacción se realiza en presencia de un agente reductor.
El agente reductor puede ser cualquier compuesto capaz de reducir un compuesto orgánico a un estado de oxidación más bajo. La reducción generalmente implica la adición de átomos de hidrógeno o la eliminación de átomos de oxígeno de un grupo. Por ejemplo, los aldehidos como F6 pueden reducirse en presencia de una amina de Fórmula F5 (Paso E, Esquema 1) mediante la adición de hidrógeno, ya sea en forma de hidrógeno gaseoso (H2) o usando un hidruro reactivo (tal como NaB(OAc)3H, NaBH4, LiAlH4 y similares); usando trifenilfosfina; o usando una combinación de yoduro de sodio, clorotrimetilsilano y metanol. En algunas realizaciones, este paso puede realizarse bajo condiciones ácidas en presencia de un ácido (tal como ácido trifluoroacético). En algunas realizaciones, este paso se puede realizar a una temperatura de aproximadamente -15°C a aproximadamente 30°C, p. ej., de aproximadamente -15°C a aproximadamente 0°C, desde aproximadamente -5°C a aproximadamente 5°C, desde aproximadamente -5°C a aproximadamente 0°C, o desde aproximadamente 0°C a aproximadamente 45°C.
En algunas realizaciones, dicho agente reductor es un agente reductor de borohidruro (por ejemplo, NaB(OAc)3H, NaBH4 u otro agente reductor de hidruro que contiene boro).
En algunas realizaciones, dicho agente reductor de borohidruro es triacetoxiborohidruro de sodio.
En algunas realizaciones, dicha reacción se realiza en presencia de ácido trifluoroacético.
En algunas realizaciones, el procedimiento comprende además desproteger dicho compuesto de Fórmula F7 para proporcionar un compuesto de Fórmula F8:

Los agentes de desprotección de amino útiles para este Paso F son conocidos por los expertos en la técnica, tales como los de Wuts y Greene (supra). En particular, los grupos protectores de amino descritos anteriormente se pueden eliminar convenientemente usando muchos agentes desprotectores de amino disponibles que son específicos para los diversos grupos mencionados anteriormente sin afectar otras porciones deseadas del compuesto. El grupo tercbutoxicarbonilo se puede eliminar (por ejemplo, hidrolizar) del átomo de nitrógeno, p. ej., mediante tratamiento con un ácido (tal como ácido clorhídrico, ácido trifluoroacético, ácido toluenosulfónico y similares); una combinación de reactivos (por ejemplo, mezcla de cloruro de acetilo y metanol) que se sabe que generan un ácido; o un ácido de Lewis
(por ejemplo, BF3Et2O). El grupo benciloxicarbonilo se puede eliminar (por ejemplo, hidrogenolizar) del átomo de nitrógeno, p. ej., mediante tratamiento con hidrógeno y un catalizador (como el paladio sobre carbono).
En algunas realizaciones, el agente desprotector de amino es ácido trifluoroacético. En algunas realizaciones, el agente desprotector de amino contiene ácido trifluoroacético y >0,5% en volumen de agua, por ejemplo, >1,0% en volumen de agua, >1,5% en volumen de agua, >2,0% en volumen de agua, de aproximadamente 2% a aproximadamente 10% en volumen de agua, de aproximadamente 10% a aproximadamente 20% en volumen de agua, o de aproximadamente 20% a aproximadamente 50% en volumen de agua. En algunas realizaciones, el agente desprotector de amino puede ser una mezcla de ácido trifluoroacético y agua en una relación volumétrica de aproximadamente 98:2. En algunas realizaciones, el agente desprotector de amino puede ser ácido clorhídrico, opcionalmente en un disolvente (por ejemplo, agua, THF, dioxano, acetato de etilo, etc.). En algunas realizaciones, el componente disolvente es acetato de etilo. En algunas realizaciones, el agente desprotector de amino puede ser ácido clorhídrico opcionalmente en un disolvente tal como un alcohol (tal como isopropanol, metanol o etanol). También se pueden usar disolventes de hidrocarburos halogenados (p. ej., diclorometano, cloroformo, dicloroetano o tetracloroetano). En algunas realizaciones, la relación molar de ácido clorhídrico y el compuesto de Fórmula F7 es aproximadamente 6,0, aproximadamente 5,0, aproximadamente 4,0, aproximadamente 3,0 aproximadamente 2,0, aproximadamente 1,0 o aproximadamente 1,1. En algunas realizaciones, el Paso F se puede realizar a una temperatura de aproximadamente -10°C a aproximadamente 60°C, p. ej., de aproximadamente -10°C a aproximadamente 0°C, de aproximadamente 0°C a aproximadamente 25°C, de aproximadamente 25°C a aproximadamente 45°C, o de aproximadamente 45°C a aproximadamente 60°C.
En algunas realizaciones, dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F7 con ácido clorhídrico.
En algunas realizaciones, dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F7 con ácido clorhídrico en un componente disolvente que comprende isopropanol.
En algunas realizaciones, dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F7 con ácido clorhídrico en un componente disolvente que comprende un disolvente hidrocarbonado halogenado.
En algunas realizaciones, dicho disolvente de hidrocarburo halogenado es diclorometano.
En algunas realizaciones, la invención comprende además hacer reaccionar dicho compuesto de Fórmula F8, con Pg2-NH-SO2-X, en presencia de una base orgánica para proporcionar un compuesto de Fórmula F9:

en donde:
Pg2 es un grupo protector de amino; y
X es halo.
En algunas realizaciones, se puede preparar Pg2-NH-SO2Cl y usarlo inmediatamente en la reacción con el compuesto de Fórmula F8. El grupo protector Pg2 podría seleccionarse de cualquiera de los grupos protectores conocidos en la técnica para proteger aminas o sulfonamidas (tales como las descritas anteriormente para Pg1). En algunas realizaciones, Pg2 puede ser un grupo alcoxicarbonilo (tal como terc-butoxicarbonilo).
Los disolventes apropiados incluyen, pero no se limitan a, disolventes de hidrocarburos halogenados tales como diclorometano y similares. La base orgánica puede ser cualquier base que sirva para neutralizar e1HCl generado durante la reacción del compuesto de Fórmula F8 y el cloruro de amino-sulfonilo protegido. La base orgánica puede incluir aminas terciarias acíclicas tales como tri(C1-6)alquilamina (por ejemplo, trietilamina, diisopropiletilamina (DIPEA) y similares), aminas terciarias cíclicas (por ejemplo, n-metilo piperidina, 1,4-diazabiciclo[2,2,2]octano (DABCO) y similares). En algunas realizaciones, la base orgánica puede ser trietilamina. En algunas realizaciones, este paso se puede realizar a una temperatura de aproximadamente -15°C a aproximadamente 60°C, p. ej., de aproximadamente -15°C a aproximadamente 0°C, de aproximadamente 0°C a aproximadamente 25°C, de aproximadamente 25°C a aproximadamente 45°C, o de aproximadamente 45°C a aproximadamente 60°C.
En tales realizaciones, el Pg2-NH-SO2Cl puede obtenerse por reacción de un alcohol (tal como etanol, alcohol terc-butílico y similares) con isocianato de clorosulfonilo (ClS(O)2NCO).
En algunas realizaciones, Pg2 es etoxicarbonilo, terc-butoxicarbonilo, benciloxicarbonilo o 9-fluorenilmetiloxicarbonilo. En algunas realizaciones, Pg2 es C1-6 alcoxicarbonilo.
En algunas realizaciones, Pg2 es terc-butoxicarbonilo.
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende un disolvente hidrocarbonado halogenado.
En algunas realizaciones, dicho disolvente de hidrocarburo halogenado es diclorometano.
En algunas realizaciones, dicha base orgánica comprende una tri(C1-6)alquilamina.
En algunas realizaciones, dicha base orgánica es trietilamina.
En algunas realizaciones, X es cloro.
En algunas realizaciones, la invención comprende además desproteger dicho compuesto de Fórmula F9 para proporcionar un compuesto de Fórmula F10:

En algunas realizaciones, los agentes desprotectores adecuados pueden incluir los descritos anteriormente para desproteger el compuesto de Fórmula F7.
En algunas realizaciones, dicha desprotección comprende hacer reaccionar un compuesto de Fórmula F9 con ácido clorhídrico. En algunas realizaciones, dicha desprotección comprende hacer reaccionar un compuesto de Fórmula F9 con ácido clorhídrico en un componente disolvente que comprende un alcohol. En algunas realizaciones, dicho alcohol es etanol. En algunas realizaciones, dicha desprotección comprende hacer reaccionar un compuesto de Fórmula F9 con ácido clorhídrico en un componente disolvente que comprende acetato de etilo.
En algunas realizaciones, la invención comprende además hacer reaccionar dicho compuesto de Fórmula F10 con una base para proporcionar un compuesto de Fórmula I:

Se puede usar una base para la conversión (por ejemplo, hidrólisis) del anillo de oxadiazolona en F10 para revelar la amidoxima en el compuesto de Fórmula I, opcionalmente en un disolvente (Paso I, Esquema 1). La protección de la amidoxima como la oxadiazolona puede ser útil para prevenir reacciones adversas del grupo hidroxilo o de la amidoxima en su conjunto. La base puede ser una base inorgánica tal como hidróxido de metal alcalino (por ejemplo, NaOH, LiOH, KOH, Mg(OH)2, etc.); o una base orgánica tal como una amina acíclica (por ejemplo, trietilamina, diisopropiletilamina (DIPEA), etc.) o una amina cíclica (por ejemplo, pirrolidina, piperidina, etc.). La base puede estar disponible en forma de resina (como Amberlite® y similares). En algunas realizaciones adicionales, la base puede proporcionarse en forma de una solución en agua (por ejemplo, aproximadamente 0,5 N de solución, aproximadamente 1 N de solución, aproximadamente 1,5 N de solución, aproximadamente 2,5 N de solución, de aproximadamente 3 N a aproximadamente 5 N de solución, de aproximadamente 5 N a aproximadamente 10 N de solución). En algunas realizaciones, la base es un hidróxido de metal alcalino (tal como hidróxido de sodio). En algunas realizaciones, la base puede ser una solución de NaOH2N en agua. En algunas realizaciones, el disolvente puede ser etanol o tetrahidrofurano (THF). En algunas realizaciones, el disolvente puede ser una mezcla de etanol y agua. En algunas realizaciones, la reacción del compuesto de Fórmula F10 con una base para proporcionar el compuesto de
Fórmula I se puede realizar a una temperatura de aproximadamente -10°C a aproximadamente 60°C, p. ej., de aproximadamente - 10°C a aproximadamente 20°C, de aproximadamente 0°C a aproximadamente 30°C, de aproximadamente 0°C a aproximadamente 10 °C, o de aproximadamente 0°C a aproximadamente 5 °C.
En algunas realizaciones, dicha base comprende un hidróxido de metal alcalino.
En algunas realizaciones, dicho hidróxido de metal alcalino es hidróxido de sodio.
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende tetrahidrofurano, agua y etanol.
En algunas realizaciones, el compuesto de Fórmula F5, se puede obtener en una secuencia de pasos mostrados en el Esquema 2. La preparación del intermedio, 4-amino-N-hidroxi-1,2,5-oxadiazol-3-carboximidamida F2, ha sido descrito en J. Heterocycl. Chem (1965), 2, 253, que se incorpora aquí como referencia en su totalidad, y su conversión a la cloroxima F3 se ha descrito en Synth. Commun. (1988), 18, 1427, que se incorpora aquí como referencia en su totalidad. En algunas realizaciones, la cloroxima de Fórmula F3 se puede acoplar a 3-bromo-4-fluoroanilina, opcionalmente en un disolvente (tal como agua), seguido de la adición de bicarbonato de sodio, para proporcionar una amidoxima de Fórmula F4. La funcionalidad amidoxima del compuesto de F4 se puede convertir en una oxadiazolona o Fórmula F5 usando N,N-carbonildiimidazol (DCI) en un solvente (como acetato de etilo, dioxano, THF y similares), a temperaturas elevadas, como aproximadamente 50°C, aproximadamente 60°C, aproximadamente 70°C, aproximadamente 80°C, aproximadamente 90°C, o aproximadamente 100°C.
Esquema 2

Alternativamente, el compuesto de Fórmula F10 se puede obtener a través de una secuencia de pasos representados en el Esquema 3.
Esquema 3
En algunas realizaciones, la presente solicitud proporciona un proceso, que comprende hacer reaccionar un compuesto de Fórmula F15:

con un compuesto de Fórmula F5:

para proporcionar un compuesto de Fórmula F16:

en donde:
cada R1 es independientemente un grupo protector de amino; y
R3 es C1-6 alquilo o bencilo.
En algunas realizaciones, R1 es C2-4 alquenilo-C1-3 alquenilo o fenilo-C1-3 alquilo, en donde dicho fenilo-C1-3 alquilo está opcionalmente sustituido con 1,2 o 3 grupos C1-4 alcoxi seleccionados independientemente.
En algunas realizaciones, R1 es C2-4 alquenilo-C1-3 alquenilo o fenilo-C1-3 alquilo, en donde dicho fenilo-C1-3 alquilo está opcionalmente sustituido con 1, 2 o 3 grupos metoxi.
En algunas realizaciones, R1 es alilo.
En algunas realizaciones, R1 es 4-metoxibencilo.
En algunas realizaciones, R3 es C1-6 alquilo.
En algunas realizaciones, R3 es ferc-butilo.
En algunas realizaciones, R3 es C1-4 alquilo.
En algunas realizaciones, R3 es butilo.
Preferiblemente, la reacción se realiza en presencia de un agente reductor. El agente reductor puede ser cualquier compuesto capaz de reducir un compuesto orgánico a un estado de oxidación más bajo. En algunas realizaciones, el agente reductor puede ser hidrógeno gaseoso en presencia de un catalizador o un reactivo de hidruro (tal como NaB(OAc)3H, NaBH4, LiAlH4 y similares); usando trifenilfosfina; o usando una combinación de yoduro de sodio, clorotrimetilsilano y metanol. En algunas realizaciones, este paso puede realizarse en presencia de un ácido tal como ácido trifluoroacético. Los disolventes adecuados para este paso incluyen alcohol isopropílico, THF, dioxano o similares. En algunas realizaciones, este paso se puede realizar a una temperatura de aproximadamente -15°C a aproximadamente 30°C, p. ej., de aproximadamente -15°C a aproximadamente 0°C, desde aproximadamente -5°C a aproximadamente 5 °C, desde aproximadamente -5°C a aproximadamente 0°C, de aproximadamente 0 a 5 °C, o desde aproximadamente 0°C a aproximadamente 45°C.
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende tetrahidrofurano. En algunas realizaciones, dicha reacción se realiza en presencia de un agente reductor.
En algunas realizaciones, dicho agente reductor es un agente reductor de borohidruro.
En algunas realizaciones, dicho agente reductor de borohidruro es triacetoxiborohidruro de sodio.
En algunas realizaciones, dicha reacción se realiza en presencia de ácido trifluoroacético.
En algunas realizaciones, la invención comprende además desproteger dicho compuesto de Fórmula F16 para proporcionar un compuesto de Fórmula F10:

El tratamiento de un compuesto F16 para reemplazar RN con NH2 se puede lograr por métodos para la desprotección de grupos protectores de aminas particulares conocidos por un experto en la técnica, tales como los de Wuts y Greene, Greene’s Protective Groups in Organic Synthesis, 4a edición, pp 696-926, John Wiley & Sons: Nueva York, 2006. En algunas realizaciones, cuando R1 es alilo, el agente desprotector puede ser un catalizador de paladio (por ejemplo, Pd(Ph3P)4, Pd/C o Pd(dba)DPPB). En algunas realizaciones, cuando R1 es 4-metoxibencilo, el agente desprotector puede incluir un ácido orgánico (tal como ácido trifluoroacético o ácido metanosulfónico, y similares); un ácido inorgánico (tal como ácido clorhídrico); hidrógeno y paladio; o sodio en amoníaco líquido. La desprotección se puede realizar a una temperatura de aproximadamente 30°C a aproximadamente 90°C, p. ej., de aproximadamente 50°C a aproximadamente 100°C, o de aproximadamente 60°C a aproximadamente 80°C.
En algunas realizaciones, dicha desprotección comprende hacer reaccionar un compuesto de Fórmula F16 con ácido trifluoroacético.
En algunas realizaciones, dicha desprotección comprende hacer reaccionar un compuesto de Fórmula F16 con ácido clorhídrico.
El compuesto F15 puede prepararse mediante un proceso de tres pasos (Pasos J, K y L) a partir de isocianato de clorosulfonilo, como se muestra en el Esquema 4.
Esquema 4

Por consiguiente, la presente solicitud proporciona además un proceso en donde dicho compuesto de Fórmula F15 se obtiene mediante un proceso que comprende tratar un compuesto de Fórmula F14:

con un agente reductor para proporcionar dicho compuesto de Fórmula F15; en donde R2 es C1-4 alquilo; y R3 se define supra.
En algunas realizaciones, R2 es metilo.
En algunas realizaciones, R2 es etilo.
En algunas realizaciones, la reducción se puede llevar a cabo con hidruro de diisobutilaluminio (DIBAL-H). Los disolventes adecuados incluyen disolventes de hidrocarburos halogenados tales como diclorometano, cloroformo, dicloroetano, tetracloroetano y similares. En algunas realizaciones, la reducción puede realizarse a aproximadamente temperatura ambiente, p. ej., desde aproximadamente -80°C a aproximadamente 30°C, desde aproximadamente -78°C a aproximadamente 0°C, de aproximadamente 0°C a aproximadamente 30°C, o de aproximadamente 25°C a aproximadamente 30°C.
En algunas realizaciones, dicho tratamiento se realiza en un disolvente hidrocarbonado halogenado.
En algunas realizaciones, dicho disolvente de hidrocarburo halogenado es diclorometano.
En algunas realizaciones, dicho agente reductor es hidruro de diisobutilaluminio.
En algunas realizaciones, dicho compuesto de Fórmula F14 se obtiene mediante un proceso que comprende proteger un compuesto de Fórmula F13:

con uno o más agentes protectores de amino seleccionados independientemente para proporcionar un compuesto de Fórmula F14.
El grupo protector R1 en F14 se puede seleccionar de los diversos grupos protectores de amino conocidos en la técnica (supra). En algunas realizaciones, el agente protector de amino es bromuro de alilo o cloruro de 4-metoxibencilo. En algunas realizaciones, dicho uno o más agentes protectores de amino se seleccionan de bromuro de alilo y cloruro de 4-metoxibencilo.
En algunas realizaciones, dicha protección se realiza en presencia de una base.
En algunas realizaciones, dicha base es carbonato de potasio.
En algunas realizaciones, dicha protección se realiza en un componente disolvente que comprende acetonitrilo. En algunas realizaciones, la preparación del compuesto de F13 puede obtenerse tratando el isocianato de clorosulfonilo con un alcohol R3OH (donde R3 se define anteriormente) y un éster de glicina H2NCH2CO2R2, en donde R2 es C1-4 alquilo. En algunas realizaciones, este Paso J se lleva a cabo en presencia de un ácido orgánico (tal como ácido acético, ácido benzoico, ácido trifluoroacético). Los disolventes adecuados para este paso incluyen diclorometano, cloroformo, dicloroetano, tetracloroetano y similares.
En algunas realizaciones, la presente solicitud proporciona un compuesto de Fórmula F13:

en donde:
R2 es C1-4 alquilo; y
R3 es Ci-6 alquilo o bencilo.
En algunas realizaciones, R2 es metilo.
En algunas realizaciones, R2 es etilo.
En algunas realizaciones, R3 es C1-6 alquilo.
En algunas realizaciones, R3 es ferc-butilo.
En algunas realizaciones, el compuesto de Fórmula F13 es acetato de etilo-2-((N-(fercbutoxicarbonilo)sulfamoilo)amino):

En algunas realizaciones, la invención proporciona además un compuesto de Fórmula F14:

F14
en donde:
cada R1 es independientemente un grupo protector de amino;
R2 es C1-4 alquilo; y
R3 es C1-6 alquilo o bencilo.
En algunas realizaciones, R1 es C2-4 alquenilo-C1-3 alquenilo o fenilo-C1-3 alquilo, en donde dicho fenilo-C1-3 alquilo está opcionalmente sustituido con 1,2 o 3 grupos C1-4 alcoxi seleccionados independientemente.
En algunas realizaciones, R1 es alilo.
En algunas realizaciones, R1 es 4-metoxibencilo.
En algunas realizaciones, R2 es metilo.
En algunas realizaciones, R2 es etilo.
En algunas realizaciones, R3 es C1-6 alquilo.
En algunas realizaciones, R3 es ferc-butilo.
En algunas realizaciones, R3 es C1-4 alquilo.
En algunas realizaciones, R3 es butilo.
En algunas realizaciones, R3 es C1-4 alquilo.
En algunas realizaciones, R3 es butilo.
En algunas realizaciones, el compuesto de Fórmula F14 es etilo-2-(alilo(N-alilo-N-(fercbutoxicarbonilo)sulfamoilo)amino)acetato:

En algunas realizaciones, el compuesto de Fórmula F14 es etilo-2-(4-metoxibencilo(N-4-metoxibencilo-N-(tercbutoxicarbonilo)sulfamoilo)amino)acetato:

En algunas realizaciones, la presente solicitud proporciona un compuesto de Fórmula F15:

en donde:
R3 es C1-6 alquilo o bencilo; y
cada R1 es independientemente un grupo protector de amino.
En algunas realizaciones, R1 es C2-4 alquenilo-C1-3 alquenilo o fenilo-C1-3 alquilo, en donde dicho fenilo-C1-3 alquilo está opcionalmente sustituido con 1,2 o 3 grupos C1-4 alcoxi seleccionados independientemente.
En algunas realizaciones, R1 es alilo.
En algunas realizaciones, R1 es 4-metoxibencilo.
En algunas realizaciones, R3 es C1-6 alquilo.
En algunas realizaciones, R3 es terc-butilo.
En algunas realizaciones, el compuesto de Fórmula F15 es terc-butilo alilo {[alilo(2-oxoetilo)amino]sulfonilo}carbamato:

En algunas realizaciones, el compuesto de Fórmula F15 es ferc-butilo(4-metoxibencilo){[(4-metoxibencilo)(2-oxoetilo)amino]sulfonilo}carbamato:

En algunas realizaciones, la invención proporciona un compuesto de Formula F16:

en donde R3 es C1-6 alquilo o bencilo y cada R1 es independientemente un grupo protector de amino.
En algunas realizaciones, R1 es C2-4 alquenilo-C1-3 alquenilo o fenilo-C1-3 alquilo, en donde dicho fenilo-C1-3 alquilo está opcionalmente sustituido con 1,2 o 3 grupos C1-4 alcoxi seleccionados independientemente.
En algunas realizaciones, R1 es alilo.
En algunas realizaciones, R1 es 4-metoxibencilo.
En algunas realizaciones, R3 es C1-6 alquilo.
En algunas realizaciones, R3 es ferc-butilo.
En algunas realizaciones, R3 es C1-4 alquilo.
En algunas realizaciones, R3 es butilo.
En algunas realizaciones, el compuesto de Fórmula F16 es ferc-butilo alilo(W-alilo-W-(2-(4-(4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4--oxadiazol-3-ilo)-1,2,5-oxadiazol-3-ilamino)etilo)sulfamoilo)carbamato:

En algunas realizaciones, el compuesto de Formula F16 es ferc-butilo (4-metoxibencilo)-(A/-(4-metoxibencilo)-W-(2-(4-(4-(3-bromo-4-fluorofenilo-)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo)-1,2,5-oxadiazol-3-ilamino)etilo)-sulfamoilo)carbamato:

El esquema 5 delinea una ruta alternativa para la preparación del compuesto de Formula F10.
Esquema 5

La presente solicitud también proporciona un proceso que comprende hacer reaccionar un compuesto de Fórmula F17:

en donde R4 es C-ue alquilo, C-ue haloalquilo, bencilo, o 9/-/-fluoren-9-ilmetilo con un compuesto de Fórmula F5:

para proporcionar un compuesto de Fórmula F18:

En algunas realizaciones, R4 es ferc-butilo.
En algunas realizaciones, R4 es bencilo.
En algunas realizaciones, R4 es etilo.
En algunas realizaciones, R4 es C1-3 haloalquilo.
En algunas realizaciones, R4 es 2,2,2-tricloroetilo.
En algunas realizaciones, R4 es 9H-fluoren-9-ilmetilo.
En este Paso Q, los compuestos F18 pueden prepararse, en algunas realizaciones, haciendo reaccionar F17 con el compuesto de amina de Fórmula F5 en presencia de un agente reductor.
En algunas realizaciones, dicha reacción se lleva a cabo en presencia de un agente reductor.
El agente reductor puede ser cualquier compuesto capaz de reducir un compuesto orgánico a un estado de oxidación inferior, por ejemplo mediante el uso de un organosilano tal como tri(C-i-3 alquilo)silano (por ejemplo, trietilsilano); hidrógeno elemental o usando un reactivo de hidruro (tal como NaB(OAc)3H, NaBH4, LiAlH4 y similares); usando trifenilfosfina; o usando una combinación de yoduro de sodio, clorotrimetilsilano y metanol. En algunas realizaciones, este paso puede realizarse en presencia de un ácido tal como ácido trifluoroacético. Los disolventes adecuados incluyen, pero sin limitación, disolventes de hidrocarburos halogenados (por ejemplo, diclorometano, cloroformo, dicloroetano o tetracloroetano). En algunas realizaciones, el disolvente de hidrocarburo halogenado es 1,2-dicloroetano.
En algunas realizaciones, dicho agente reductor es un organosilano.
En algunas realizaciones, dicho agente reductor es tri(C1-3 alquilo)silano.
En algunas realizaciones, dicho agente reductor es trietilsilano.
En algunas realizaciones, dicha reacción se lleva a cabo en presencia de un ácido orgánico.
En algunas realizaciones, dicho ácido orgánico es ácido trifluoroacético.
En algunas realizaciones, dicho ácido orgánico es ácido metanosulfónico.
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende un disolvente hidrocarbonado halogenado.
En algunas realizaciones, dicho disolvente de hidrocarburo halogenado es diclorometano.
En algunas realizaciones, dicho disolvente de hidrocarburo halogenado es 1,2-dicloroetano.
En algunas realizaciones, el proceso comprende además desproteger dicho compuesto de Fórmula F18 para proporcionar un compuesto de Fórmula F10:

En algunas realizaciones, un experto en la materia conoce métodos para la desprotección de grupos protectores de amina particulares (tales como carbamatos) en la técnica, como los de Wuts y Greene, Greene’s Protective Groups in Organix Synthesis, 4a edición, pp 696-926, John Wiley & Sons: Nueva York, 2006. Por ejemplo, el grupo terc-butoxicarbonilo (por ejemplo, cuando R4 es terc-butilo) puede eliminarse (por ejemplo, hidrolizarse) del átomo de nitrógeno, p. ej., mediante tratamiento con un ácido (tal como ácido clorhídrico, ácido trifluoroacético, ácido toluenosulfónico y similares); una combinación de reactivos (por ejemplo, mezcla de cloruro de acetilo y metanol) que se sabe que generan un ácido; o un ácido de Lewis (por ejemplo, BF3'Et2O). El grupo benciloxicarbonilo (por ejemplo, cuando R4 es bencilo) puede eliminarse (por ejemplo, hidrogenolizarse) del átomo de nitrógeno, p. ej., mediante tratamiento con hidrógeno y un catalizador (tal como paladio sobre carbono). Los grupos metoxicarbonilo y etoxicarbonilo (es decir, cuando R4 es metilo o etilo) pueden eliminarse mediante tratamiento con una base inorgánica (como KOH o K2CO3); una combinación de reactivos (p. ej., mezcla de cloruro de acetilo, yoduro de sodio y acetonitrilo); o por tratamiento con un ácido (p. ej., HBr, AcOH). El grupo 2,2,2-tricloroetoxicarbonilo se puede eliminar, p. ej., mediante tratamiento con un catalizador (por ejemplo, Zn/AcOH o Cd/AcOH). Los disolventes adecuados para este paso incluyen, pero no se limitan a, metanol o tetrahidrofurano (THF), acetonitrilo y similares. En algunas realizaciones, el tratamiento se realiza a una temperatura de aproximadamente 30°C a aproximadamente 90°C, p. ej., de aproximadamente 50°C a aproximadamente 100°C, o de aproximadamente 60°C a aproximadamente 80°C.
En algunas realizaciones, dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F18 con zinc en presencia de ácido acético.
En algunas realizaciones, dicha desprotección se realiza en un componente disolvente que comprende tetrahidrofurano.
En algunas realizaciones, el proceso comprende además hacer reaccionar dicho compuesto de Fórmula F10 con una base para proporcionar un compuesto de Fórmula I:

En algunas realizaciones, dicha base comprende un hidróxido de metal alcalino.
En algunas realizaciones, dicho hidróxido de metal alcalino es hidróxido de sodio.
En algunas realizaciones, dicha reacción se realiza en un componente disolvente que comprende tetrahidrofurano, agua y etanol.
En algunas realizaciones, el proceso comprende además convertir dicho compuesto de Fórmula F18 en un compuesto de Fórmula I:

en donde dicha conversión comprende combinar el compuesto de Fórmula F18 con una base para formar una primera mezcla. En algunas realizaciones, la base es N,N-bis(2-aminoetilo)etano-1,2-diamina.
En algunas realizaciones, la conversión comprende además añadir un ácido a la primera mezcla. En algunas realizaciones, dicho ácido es un ácido fuerte acuoso. En algunas realizaciones, dicho ácido acuoso fuerte es ácido clorhídrico acuoso.
En algunas realizaciones, dicha conversión se realiza en un componente disolvente que comprende tetrahidrofurano y acetato de etilo.
La presente solicitud también proporciona un proceso que comprende:
i) hacer reaccionar un compuesto de Fórmula F19:

con un compuesto de Fórmula F5:

en presencia de trietilsilano y ácido metanosulfónico para proporcionar un compuesto de Fórmula F20:
y
ii) convertir dicho compuesto de Fórmula F20 en un compuesto de Fórmula I:

en donde dicha conversión comprende combinar dicho compuesto de Fórmula F20 con W,A/-bis(2-aminoetilo)etano-1,2-diamina. En algunas realizaciones, dicha conversión comprende además añadir ácido clorhídrico acuoso después de dicha combinación.
El compuesto F17 se puede preparar mediante un proceso de un paso (Paso P) a partir de isocianato de clorosulfonilo, como se muestra en el Esquema 6.
Esquema 6

En algunas realizaciones, la preparación del compuesto de Fórmula F17 se puede obtener tratando el isocianato de cloro sulfonilo con 2,2-dimetoxietanamina y alcohol R4OH (R4 se define como anteriormente), opcionalmente en un solvente (por ejemplo, un solvente de hidrocarburo halogenado tal como diclorometano, cloroformo, dicloroetano, tetracloroetano). En algunas realizaciones, este paso se lleva a cabo en presencia de una base. La base puede ser una base orgánica tal como una amina acíclica (por ejemplo, trietilamina, diisopropiletilamina (DIPEA), etc.) o una amina cíclica (por ejemplo, pirrolidina, piperidina, etc.); o una base inorgánica tal como álcali (por ejemplo, NaOH, LiOH, KOH, Mg(OH)2, etc.). En algunas realizaciones, la reacción se lleva a cabo en un disolvente, p. ej., un disolvente hidrocarbonado halogenado tal como diclorometano, cloroformo, dicloroetano o tetracloroetano.
En algunas realizaciones, la presente solicitud proporciona además un compuesto de Fórmula F17:

en donde R4 es C1-6 haloalquilo o bencilo.
En algunas realizaciones, R4 es tere-butilo.
En algunas realizaciones, R4 es bencilo.
En algunas realizaciones, R4 es etilo.
En algunas realizaciones, R4 es C1-3 haloalquilo.
En algunas realizaciones, R4 es 2,2,2-tricloroetilo.
En algunas realizaciones, R4 es 9H-fluoren-9-ilmetilo.
En algunas realizaciones, el compuesto de Fórmula F17 es N-(2,2-dimetoxietilo)sulfamoilcarbamato de tere-butilo. En algunas realizaciones, el compuesto de Fórmula F17 es bencilo N-(2,2-dimetoxietilo)sulfamoilcarbamato.
En algunas realizaciones, el compuesto de Fórmula F17 es N-(2,2-dimetoxietilo)sulfamoilcarbamato de etilo.
En algunas realizaciones, el compuesto de Fórmula F17 es 2,2,2-tricloroetilo N-(2,2-dimetoxietilo)sulfamoilcarbamato.
En algunas realizaciones, el compuesto de Fórmula F17 es (9H-fluoren-9-ilo)metilo N-(2,2-dimetoxietilo)sulfamoilcarbamato.
En algunas realizaciones, la presente solicitud proporciona además un compuesto de Fórmula F18:

en donde R4 es C1-6 alquilo, C1-6 haloalquilo o bencilo.
En algunas realizaciones, R4 es ferc-butilo.
En algunas realizaciones, R4 es bencilo.
En algunas realizaciones, R4 es etilo.
En algunas realizaciones, R4 es C1-3 haloalquilo.
En algunas realizaciones, R4 es 2,2,2-tricloroetilo.
En algunas realizaciones, R4 es 9H-fluoren-9-ilmetilo.
En algunas realizaciones, el compuesto de Fórmula F18 es bencilo ({[2-({[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo-]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato:

En algunas realizaciones, el compuesto de Fórmula F18 es etilo ({[2-({[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo-]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato:

En algunas realizaciones, el compuesto de Fórmula F18 es 2,2,2-tricloroetilo ({[2-({[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo-]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato:

En algunas realizaciones, el compuesto de Fórmula F18 es (9H-fluoren-9-ilo)metilo N-(2-((4-(4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo)-1,2,5-oxadiazol-3-ilo)amino)etilo)sulfamoilcarbamato:

Como se usa en el presente documento, el término "alquilo," cuando se usa solo o junto con términos de resto adicionales, se refiere a un grupo hidrocarbonado saturado de cadena lineal o ramificada que tiene de 1 a 6 átomos de carbono, 1 a 4 átomos de carbono o 1 a 3 átomos de carbono. Los ejemplos de grupos alquilo incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, terc-butilo y similares.
Como se usa en el presente documento, "alquenilo" se refiere a un grupo alquilo que tiene uno o más enlaces dobles carbono-carbono. En algunas realizaciones, dicho grupo alquilo tiene de 2 a 6 átomos de carbono, de 2 a 4 átomos de carbono o de 2 a 3 átomos de carbono. Los grupos alquenilo de ejemplo incluyen etenilo (vinilo), propenilo y similares.
Como se usa en el presente documento, "alquenilalquilo" se refiere a un grupo de fórmula -alquilo-alquenilo. En algunas realizaciones, el grupo alquenilalquilo es alilo.
Como se usa en el presente documento, el término "haloalquilo", cuando se usa solo o junto con un resto adicional, se refiere a un grupo alquilo sustituido por uno o más átomos de halógeno seleccionados independientemente de F, Cl, Br e I. Los grupos haloalquilo de ejemplo incluyen CF3, CHF2, CH2CF3, y similares.
Como se usa en el presente documento, el término "alcoxi" se refiere a un grupo -O-alquilo. En algunas realizaciones, el grupo alquilo tiene 1 a 6 átomos de carbono, 1 a 4 átomos de carbono o 1 a 3 átomos de carbono. Los grupos alcoxi de ejemplo incluyen metoxi, etoxi, propoxi (por ejemplo, n-propoxi e isopropoxi), t-butoxi y similares.
Como se usa en el presente documento, "trialquilamina" se refiere a un átomo de nitrógeno sustituido por tres grupos alquilo seleccionados independientemente. En algunas realizaciones, cada grupo alquilo tiene de 2 a 6 átomos de carbono, de 2 a 4 átomos de carbono o de 2 a 3 átomos de carbono. Los ejemplos de grupos de trialquilamina incluyen trimetilamina, trietilamina y similares.
Como se usa en el presente documento, el término "alcoxicarbonilo" se refiere a un grupo de fórmula -C(O)-O-alquilo. En algunas realizaciones, el grupo alquilo tiene de 2 a 6 átomos de carbono, de 2 a 4 átomos de carbono o de 2 a 3 átomos de carbono. Los ejemplos de grupos alcoxicarbonilo incluyen etoxicarbonilo, ferc-butoxicarbonilo (Boc) y similares.
Los disolventes de hidrocarburos halogenados se refieren a alcanos halogenados, tales como diclorometano, cloroformo, dicloroetano o tetracloroetano, en los que el alcano puede ser de cadena ramificada o lineal con 1 a 12 átomos de carbono, 1 a 6 átomos de carbono, o 1 a 4 átomos de carbono con uno o más átomos de halo. En algunas realizaciones, el disolvente de hidrocarburo halogenado es un alcano clorado de 1 a 12 átomos de carbono, 1 a 6 átomos de carbono o 1 a 4 átomos de carbono.
En diversos lugares de la presente memoria descriptiva, los sustituyentes de los compuestos de la invención pueden describirse en grupos o en intervalos. Se pretende específicamente que la invención incluya todas y cada una de las subcombinaciones individuales de los miembros de tales grupos y rangos.
Se pretende que los compuestos de la invención sean estables. Como se usa en el presente documento, "estable" se refiere a un compuesto que es suficientemente robusto para sobrevivir al aislamiento hasta un grado útil de pureza de una mezcla de reacción, y preferiblemente capaz de formularse en un agente terapéutico eficaz.
Se aprecia además que ciertas características de la invención, que se describen, por claridad, en el contexto de realizaciones separadas, también se pueden proporcionar en combinación en una sola realización. A la inversa, varias características de la invención que, por brevedad, se describen en el contexto de una sola realización, también se pueden proporcionar por separado o en cualquier subcombinación adecuada.
Los compuestos de la invención pretenden además incluir todos los isómeros geométricos posibles. Los isómeros geométricos cis y trans de los compuestos se describen y pueden aislarse como una mezcla de isómeros o como formas isoméricas separadas.
Los compuestos de la invención también incluyen formas tautoméricas. Las formas tautoméricas resultan del intercambio de un enlace simple con un enlace doble adyacente junto con la migración concomitante de un protón.
Los compuestos de la invención también pueden incluir todos los isótopos de átomos que se encuentran en los compuestos intermedios o finales. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico pero diferentes números de masa. Por ejemplo, los isótopos de hidrógeno incluyen tritio y deuterio.
En algunas realizaciones, los compuestos de la invención, y sus sales, están sustancialmente aislados. Por "sustancialmente aislado" se entiende que el compuesto está al menos parcial o sustancialmente separado del entorno en donde se formó o detectó. La separación parcial puede incluir, p. ej., una composición enriquecida en el compuesto de la invención. La separación sustancial puede incluir composiciones que contienen al menos aproximadamente 50%, al menos aproximadamente 60%, al menos aproximadamente 70%, al menos aproximadamente 80%, al menos aproximadamente 90%, al menos aproximadamente 95%, al menos aproximadamente 97% o al al menos aproximadamente 99% en peso del compuesto de la invención, o sal del mismo. Los métodos para aislar compuestos y sus sales son rutinarios en la técnica.
La presente solicitud también incluye sales de los compuestos descritos aquí. Como se usa en el presente documento, "sales" se refiere a derivados de los compuestos descritos en los que el compuesto original se modifica convirtiendo un resto ácido o base existente en su forma de sal. Los ejemplos de sales incluyen, entre otros, sales minerales de ácido (como HCl, HBr, H2SO4) o ácido orgánico (como ácido acético, ácido benzoico, ácido trifluoroacético) de residuos básicos como aminas; sales alcalinas (como Li, Na, K, Mg, Ca) u orgánicas (como trialquilamonio) de residuos ácidos como los ácidos carboxílicos; y similares. Las sales de la presente solicitud se pueden sintetizar a partir del compuesto original que contiene un resto básico o ácido por métodos químicos convencionales. Generalmente, tales sales pueden prepararse haciendo reaccionar las formas de ácido o base libres de estos compuestos con una cantidad estequiométrica de la base o ácido apropiado en agua o en un disolvente orgánico, o en una mezcla de los dos; generalmente, se prefieren medios no acuosos como éter, acetato de etilo, etanol, isopropanol o acetonitrilo (ACN).
La presente solicitud también incluye sales farmacéuticamente aceptables de los compuestos descritos en este documento. Las "sales farmacéuticamente aceptables" incluyen un subconjunto de las "sales" descritas anteriormente que son sales no tóxicas convencionales del compuesto original formado, p. ej., a partir de ácidos inorgánicos u orgánicos no tóxicos. Las listas de sales adecuadas se encuentran en Remington's Pharmaceutical Sciences, 17a edición, Mack Publishing Company, Easton, Pa., 1985, p. ej. 1418 y Journal of Pharmaceutical Science, 66 , 2 (1977). La frase "farmacéuticamente aceptable" se emplea en el presente documento para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que, dentro del alcance del buen juicio médico, son adecuados para su uso en contacto con los tejidos de seres humanos y animales sin excesivo toxicidad, irritación, respuesta alérgica u otro problema o complicación, proporcional a una relación beneficio/riesgo razonable.
Los procesos descritos en el presente documento pueden monitorizarse de acuerdo con cualquier método adecuado conocido en la técnica. Por ejemplo, la formación del producto se puede controlar por medios espectroscópicos, como la espectroscopia de resonancia magnética nuclear (p. ej., 1H o 13C), espectroscopia infrarroja, espectrofotometría (p. ej., UV-visible) o espectrometría de masas; o por cromatografía tal como cromatografía líquida de alto rendimiento (HPLC) o cromatografía en capa fina. Los compuestos obtenidos por las reacciones se pueden purificar por cualquier método adecuado conocido en la técnica. Por ejemplo, cromatografía (presión media) sobre un adsorbente adecuado (p. ej., gel de sílice, alúmina y similares), HPLC o cromatografía preparativa de capa fina; destilación; sublimación, trituración o recristalización. La pureza de los compuestos, en general, se determina mediante métodos físicos como medir el punto de fusión (en el caso de un sólido), obtener un espectro de RMN o realizar una separación por HPLC. Si el punto de fusión disminuye, si se reducen las señales no deseadas en el espectro de RMN, o si se eliminan los picos extraños en una traza de HPLC, se puede decir que el compuesto se ha purificado. En algunas realizaciones, los compuestos están sustancialmente purificados.
La preparación de compuestos puede implicar la protección y desprotección de varios grupos químicos. Un experto en la materia puede determinar fácilmente la necesidad de protección y desprotección, y la selección de grupos protectores apropiados. La química de los grupos protectores se puede encontrar, p. ej., en Wuts y Greene, Greene's Protective Groups in Organic Synthesis, 4.th Ed., John Wiley & Sons: Nueva York, 2006.
Las reacciones de los procesos descritos en la presente memoria pueden llevarse a cabo en disolventes adecuados que pueden ser seleccionados fácilmente por un experto en la técnica de la síntesis orgánica. Los disolventes adecuados pueden ser sustancialmente no reactivos con los materiales de partida (reactivos), los intermedios o los productos a las temperaturas a las que se llevan a cabo las reacciones, es decir, temperaturas que pueden variar desde la temperatura de congelación del disolvente hasta la temperatura de ebullición del disolvente. Una reacción dada puede llevarse a cabo en un disolvente o una mezcla de más de un disolvente. Dependiendo del paso de reacción, se pueden seleccionar los solventes adecuados para ese paso de reacción particular. Los solventes apropiados incluyen agua, alcanos (como pentanos, hexanos, heptanos, ciclohexano, etc., o una mezcla de los mismos), solventes aromáticos (como benceno, tolueno, xileno, etc.), alcoholes (como metanol, etanol, isopropanol, etc.), éteres (como dialquiléteres, metilo terc-butilo éter (MTBE), tetrahidrofurano (THF), dioxano, etc.), ésteres (como acetato de etilo, acetato de butilo, etc.), solventes de hidrocarburos halogenados (como como diclorometano (DCM), cloroformo, dicloroetano, tetracloroetano), dimetilformamida (DMF), dimetilsulfóxido (DMSO), acetona, acetonitrilo (ACN), hexametilfosforamida (HMPA) y N-metilpirrolidona (NMP). Dichos disolventes pueden usarse en sus formas húmedas o anhidras.
La resolución de mezclas racémicas de compuestos puede llevarse a cabo mediante cualquiera de los numerosos métodos conocidos en la técnica. Un método de ejemplo incluye la recristalización fraccionada usando un "ácido de resolución quiral" que es un ácido orgánico formador de sales ópticamente activo. Los agentes de resolución adecuados para los métodos de recristalización fraccionada son, p. ej., ácidos ópticamente activos, tales como las formas D y L de ácido tartárico, ácido diacetiltartárico, ácido dibenzoiltartárico, ácido mandélico, ácido málico, ácido láctico o los diversos ácidos canforsulfónicos ópticamente activos. La resolución de mezclas racémicas también se puede llevar a cabo mediante elución en una columna empaquetada con un agente de resolución ópticamente activo (por ejemplo, dinitrobenzoilfenilglicina). La composición solvente de elución adecuada puede ser determinada por un experto en la materia.
Métodos de uso
El compuesto de Fórmula I puede inhibir la actividad de la enzima indoleamina-2,3-dioxigenasa (IDO). Por ejemplo, el compuesto de Fórmula I puede usarse para inhibir la actividad de IDO en la célula o en un individuo que necesita modulación de la enzima administrando una cantidad inhibidora del compuesto de Fórmula I.
Los compuestos de Fórmula I pueden usarse en métodos para inhibir la degradación del triptófano en un sistema que contiene células que expresan IDO, como un tejido, un organismo vivo o un cultivo celular. En algunas realizaciones, la presente solicitud proporciona métodos para alterar (por ejemplo, aumentar) los niveles de triptófano extracelular en un mamífero mediante la administración de una cantidad eficaz del compuesto de Fórmula I. Los métodos para medir los niveles de triptófano y la degradación de triptófano son rutinarios en la técnica.
Los compuestos de Fórmula I pueden usarse en métodos para inhibir la inmunosupresión, tales como la inmunosupresión mediada por IDO en un paciente mediante la administración al paciente de una cantidad efectiva del compuesto de Fórmula I. La inmunosupresión mediada por IDO se ha asociado con, p. ej., cánceres, crecimiento tumoral, metástasis, infección viral, replicación viral, etc.
Los compuestos de Fórmula I también se pueden usar en métodos para tratar enfermedades asociadas con la actividad o expresión, incluyendo actividad anormal y/o sobreexpresión, de IDO en un individuo (por ejemplo, paciente) administrando al individuo que necesita dicho tratamiento una cantidad o dosis terapéuticamente efectiva del compuesto de Fórmula I o una composición farmacéutica del mismo. Los ejemplos de enfermedades pueden incluir cualquier enfermedad, trastorno o afección que esté directa o indirectamente vinculada a la expresión o actividad de la enzima IDO, como la sobreexpresión o la actividad anormal. Una enfermedad asociada a IDO también puede incluir cualquier enfermedad, trastorno o afección que pueda prevenirse, mejorarse o curarse modulando la actividad enzimática. Los ejemplos de enfermedades asociadas con IDO incluyen cáncer, infección viral como infección por VIH, infección por VHC, depresión, trastornos neurodegenerativos como la enfermedad de Alzheimer y la enfermedad de Huntington, traumatismos, cataratas relacionadas con la edad, trasplante de órganos (por ejemplo, rechazo de trasplante de órganos) y enfermedades autoinmunes como asma, artritis reumatoide, esclerosis múltiple, inflamación alérgica, enfermedad inflamatoria intestinal, psoriasis y lupus eritematoso sistémico. Los cánceres de ejemplo tratables por los métodos de la presente invención incluyen cáncer de colon, páncreas, mama, próstata, pulmón, cerebro, ovario, cuello uterino, testículos, renal, cabeza y cuello, linfoma, leucemia, melanoma y similares. El compuesto de Fórmula I también puede ser útil en el tratamiento de la obesidad y la isquemia.
Como se usa en el presente documento, el término "célula" se refiere a una célula que es in vitro, ex vivo o in vivo. En algunas realizaciones, una célula ex vivo puede ser parte de una muestra de tejido extirpada de un organismo tal como un mamífero. En algunas realizaciones, una célula in vitro puede ser una célula en un cultivo celular. En algunas realizaciones, una célula in vivo es una célula que vive en un organismo tal como un mamífero.
Como se usa en el presente documento, el término "contacto" se refiere a la unión de restos indicados en un sistema in vitro o un sistema in vivo. Por ejemplo, "poner en contacto" la enzima IDO con el compuesto de Fórmula I incluye la administración del compuesto de Fórmula I a un individuo o paciente, como un ser humano, que tiene IDO, así como, p. ej., introducir el compuesto de Fórmula I en una muestra que contiene una preparación celular o purificada que contiene la enzima IDO.
Como se usa en el presente documento, el término "individuo" o "paciente", usado indistintamente, se refiere a cualquier animal, incluidos mamíferos, preferiblemente ratones, ratas, otros roedores, conejos, perros, gatos, cerdos, vacas, ovejas, caballos o primates, y lo más preferiblemente humanos.
Como se usa en este documento, la frase "cantidad terapéuticamente efectiva" se refiere a la cantidad de compuesto activo o agente farmacéutico que provoca la respuesta biológica o medicinal en un tejido, sistema, animal, individuo o humano que está buscando un investigador, veterinario, médico u otro clínico.
Como se usa en el presente documento, el término "tratar" o "tratamiento" se refiere a 1) prevenir la enfermedad; por ejemplo, prevenir una enfermedad, afección o trastorno en un individuo que puede estar predispuesto a la enfermedad, afección o trastorno pero que aún no experimenta o muestra la patología o sintomatología de la enfermedad; 2) inhibir
la enfermedad; por ejemplo, inhibir una enfermedad, afección o trastorno en un individuo que está experimentando o mostrando la patología o sintomatología de la enfermedad, afección o trastorno (es decir, detener el desarrollo adicional de la patología y/o sintomatología), o 3) mejorar la enfermedad; por ejemplo, mejorar una enfermedad, afección o trastorno en un individuo que está experimentando o mostrando la patología o sintomatología de la enfermedad, afección o trastorno (es decir, revertir la patología y/o sintomatología).
Terapia combinada
Uno o más agentes farmacéuticos adicionales o métodos de tratamiento como, p. ej., agentes antivirales, quimioterapéuticos u otros agentes anticancerígenos, potenciadores inmunes, inmunosupresores, radiación, vacunas antitumorales y antivirales, terapia con citocinas (p. ej., IL2, GM-CSF, etc.), y/o inhibidores de tirosina quinasa pueden usarse en combinación con el compuesto de Fórmula I para el tratamiento de enfermedades, trastornos o afecciones asociadas con IDO. Los agentes se pueden combinar con el compuesto de Fórmula I en una sola forma de dosificación, o los agentes se pueden administrar simultáneamente o secuencialmente como formas de dosificación separadas.
Los agentes antivirales adecuados contemplados para su uso en combinación con el compuesto de Fórmula I pueden comprender inhibidores de la transcriptasa inversa nucleótidos y nucleótidos (INTI), inhibidores de la transcriptasa inversa no nucleósidos (INNTI), inhibidores de la proteasa y otros fármacos antivirales.
Los NRTI adecuados de ejemplo incluyen zidovudina (AZT); didanosina (ddl); zalcitabina (ddC); estavudina (d4T); lamivudina (3TC); abacavir (1592U89); adefovir dipivoxil[bis(POM)-PMEA]; lobucavir (BMS-180194); BCH-10652; emitricitabina[(-)-FTC]; beta-L-FD4 (también llamado beta-L-D4C y denominado beta-L-2',3'-dicleoxi-5-fluoro-citideno); DAPD, ((-)-beta-D-2,6,-diamino-purinadioxolano); y lodenosina (FddA). Los NNRTI adecuados típicos incluyen nevirapina (BI-RG-587); delaviradina (BHAP, U-90152); efavirenz (DMP-266); PNU-142721; AG-1549; MKC-442 (1-(etoxi-metilo)-5-(1-metiíetilo)-6-(fenilmetilo)-(2,4(1H,3H)-pirimidinadiona); y (+)-calanolida A (NSC-675451) y B. Los inhibidores de proteasa adecuados típicos incluyen saquinavir (Ro 31-8959); ritonavir (ABT-538); indinavir (MK-639); nelfnavir (AG-1343); amprenavir (141 W94); lasinavir (BMS-234475); DMP-450; BMS-2322623; ABT-378; y AG-1 549. Otros agentes antivirales incluyen hidroxiurea, ribavirina, IL-2, IL-12, pentafusida y Yissum Project N° 11607.
Quimioterapia adecuada u otros agentes anti-cancerígenos incluyen, p. ej., agentes alquilantes (que incluyen, sin limitación, mostazas nitrogenadas, derivados de etilenimina, sulfonatos de alquilo, nitrosoureas y triazenos) tales como mostaza de uracilo, clormetina, ciclofosfamida (Cytoxan™), ifosfamida, melfalan, clorambucilo, pipobromano, trietileno-melamina, trietilentiofosforamina, busulfano, carmustina, lomustina, estreptozocina, dacarbazina y temozolomida.
En el tratamiento del melanoma, agentes adecuados para su uso en combinación con el compuesto de Fórmula I incluyen: dacarbazina (DTIC), opcionalmente, junto con otros medicamentos de quimioterapia como carmustina (BCNU) y cisplatino; el "régimen de Dartmouth", que consiste en DTIC, BCNU, cisplatino y tamoxifeno; una combinación de cisplatino, vinblastina y DTIC; o temozolomida. Los compuestos de acuerdo con la invención también se pueden combinar con fármacos de inmunoterapia, que incluyen citocinas tales como interferón alfa, interleucina 2 y factor de necrosis tumoral (TNF) en el tratamiento del melanoma.
El compuesto de Fórmula I también se puede usar en combinación con la terapia de vacuna en el tratamiento del melanoma. Las vacunas antimelanoma son, de alguna manera, similares a las vacunas antivirus que se usan para prevenir enfermedades causadas por virus como la poliomielitis, el sarampión y las paperas. Se pueden inyectar células de melanoma debilitadas o partes de células de melanoma llamadas antígenos en un paciente para estimular el sistema inmunitario del cuerpo para destruir las células de melanoma.
Los melanomas que están confinados a los brazos o las piernas también se pueden tratar con una combinación de agentes que incluyen el compuesto de Fórmula I, usando una técnica de perfusión de extremidad aislada hipertérmica. Este protocolo de tratamiento separa temporalmente la circulación de la extremidad involucrada del resto del cuerpo e inyecta altas dosis de quimioterapia en la arteria que alimenta la extremidad, proporcionando así altas dosis al área del tumor sin exponer los órganos internos a estas dosis que de lo contrario podrían causar efectos secundarios graves. Por lo general, el fluido se calienta a 102°C. a 104°F. Melfalán es el medicamento más utilizado en este procedimiento de quimioterapia. Esto se puede administrar con otro agente llamado factor de necrosis tumoral (TNF) (ver sección sobre citocinas).
Los agentes quimioterapéuticos u otros agentes anticancerígenos adecuados incluyen, p. ej., antimetabolitos (que incluyen, sin limitación, antagonistas de ácido fólico, análogos de pirimidina, análogos de purina e inhibidores de adenosina desaminasa) tales como metotrexato, 5-fluorouracilo, floxuridina, citarabina, 6-mercaptopurina, 6-tioguanina, fosfato de fludarabina, pentostatina y gemcitabina.
Los agentes quimioterapéuticos u otros agentes anticancerígenos adecuados incluyen además, p. ej., ciertos productos naturales y sus derivados (por ejemplo, alcaloides de la vinca, antibióticos antitumorales, enzimas, linfocinas y epipodofilotoxinas) tales como vinblastina, vincristina, vindesina, bleomicina, dactinomicina, daunorrubicina, doxorrubicina, epirrubicina, idarrubicina, ara-C, paclitaxel (TAXOL™), mitramicina, desoxicoformicina, mitomicina-C,
L-asparaginasa, interferones (especialmente IFN-a), etopósido y tenipósido.
Otros agentes citotóxicos incluyen navelbeno, CPT-11, anastrazol, letrazol, capecitabina, reloxafina, ciclofosfamida, ifosamida y droloxafina.
También son adecuados los agentes citotóxicos como la epidofilotoxina; una enzima antineoplásica; un inhibidor de topoisomerasa; procarbazina; mitoxantrona; complejos de coordinación de platino tales como cisplatino y carboplatino; modificadores de respuesta biológica; inhibidores del crecimiento; agentes terapéuticos antihormonales; leucovorina; tegafur; y factores de crecimiento hematopoyético.
Otros agentes anticancerígenos incluyen terapias de anticuerpos como trastuzumab (Herceptin), anticuerpos contra moléculas coestimuladoras como CTLA-4, 4-1BB y PD-1, o anticuerpos contra citocinas (IL-10, TGF-p)., etc.).
Otros agentes anticancerígenos también incluyen aquellos que bloquean la migración de las células inmunes, como los antagonistas de los receptores de quimiocinas, incluidos CCR2 y CCR4.
Otros agentes anticancerígenos también incluyen aquellos que aumentan el sistema inmunitario, como los adyuvantes o la transferencia de células T adoptivas.
Las vacunas contra el cáncer incluyen células dendríticas, péptidos sintéticos, vacunas de ADN y virus recombinantes.
Los expertos en la técnica conocen métodos para la administración segura y efectiva de la mayoría de estos agentes quimioterapéuticos. Además, su administración se describe en la literatura estándar. Por ejemplo, la administración de muchos de los agentes quimioterapéuticos se describe en la "Physicians' Desk Reference" (PDR, p. ej., edición de 1996, Medical Economics Company, Montvale, NJ).
Formulaciones farmacéuticas y formas de dosificación
Cuando se emplean como productos farmacéuticos, el compuesto de fórmula I se puede administrar en forma de composiciones farmacéuticas que es una combinación del compuesto de fórmula I y un vehículo farmacéuticamente aceptable. Estas composiciones pueden prepararse de una manera bien conocida en la técnica farmacéutica, y pueden administrarse por una variedad de rutas, dependiendo de si se desea un tratamiento local o sistémico y del área a tratar. La administración puede ser tópica (incluidas las membranas oftálmicas y mucosas, incluidas la administración intranasal, vaginal y rectal), pulmonar (por ejemplo, por inhalación o insuflación de polvos o aerosoles, incluso por nebulizador; intratraqueal, intranasal, epidérmico y transdérmico), ocular, oral o transdérmica parenteral. Los métodos para el suministro ocular pueden incluir administración tópica (gotas para los ojos), inyección subconjuntival, periocular o intravítrea o introducción por catéter con balón o insertos oftálmicos colocados quirúrgicamente en el saco conjuntival. La administración parenteral incluye inyección o infusión intravenosa, intraarterial, subcutánea, intraperitoneal o intramuscular; o administración intracraneal, p. ej., intratecal o intraventricular. La administración parenteral puede ser en forma de una sola dosis en bolo, o puede ser, p. ej., mediante una bomba de perfusión continua. Las composiciones y formulaciones farmacéuticas para administración tópica pueden incluir parches transdérmicos, pomadas, lociones, cremas, geles, gotas, supositorios, aerosoles, líquidos y polvos. Los portadores farmacéuticos convencionales, bases acuosas, en polvo o aceitosas, espesantes y similares pueden ser necesarios o deseables.
Las composiciones farmacéuticas que contienen el compuesto de Fórmula I se pueden preparar en combinación con uno o más vehículos farmacéuticamente aceptables. Al hacer las composiciones de la invención, el ingrediente activo se mezcla típicamente con un excipiente, se diluye con un excipiente o se encerra dentro de dicho vehículo en forma de, p. ej., una cápsula, bolsita, papel u otro recipiente. Cuando el excipiente sirve como diluyente, puede ser un material sólido, semisólido o líquido, que actúa como vehículo, portador o medio para el ingrediente activo. Por lo tanto, las composiciones pueden estar en forma de tabletas, píldoras, polvos, pastillas, sobres, sellos, elixires, suspensiones, emulsiones, soluciones, jarabes, aerosoles (como un medio sólido o líquido), ungüentos que contienen, p. ej., hasta 10% en peso del compuesto activo, cápsulas de gelatina blandas y duras, supositorios, soluciones inyectables estériles y polvos envasados estériles.
Al preparar una formulación, el compuesto activo se puede moler para proporcionar el tamaño de partícula apropiado antes de combinarlo con los otros ingredientes. Si el compuesto activo es sustancialmente insoluble, se puede moler a un tamaño de partícula de menos de 200 mesh. Si el compuesto activo es sustancialmente soluble en agua, el tamaño de partícula se puede ajustar mediante molienda para proporcionar una distribución sustancialmente uniforme en la formulación, p. ej., aproximadamente 40 mesh.
Algunos ejemplos de excipientes adecuados incluyen lactosa, dextrosa, sacarosa, sorbitol, manitol, almidones, goma de acacia, fosfato de calcio, alginatos, tragacanto, gelatina, silicato de calcio, celulosa microcristalina, polivinilpirrolidona, celulosa, agua, jarabe y metilcelulosa. Las formulaciones pueden incluir adicionalmente: agentes lubricantes tales como talco, estearato de magnesio y aceite mineral; agentes humectantes; agentes emulsionantes y de suspensión; agentes conservantes tales como metilo y propilhidroxibenzoatos; agentes edulcorantes; y agentes
aromatizantes. Las composiciones de la invención pueden formularse para proporcionar una liberación rápida, sostenida o retardada del ingrediente activo después de la administración al paciente empleando procedimientos conocidos en la técnica.
Las composiciones pueden formularse en una forma de dosificación unitaria, conteniendo cada dosificación de aproximadamente 5 a aproximadamente 100 mg, más habitualmente de aproximadamente 10 a aproximadamente 30 mg, del ingrediente activo. El término "formas de dosificación unitarias" se refiere a unidades físicamente discretas adecuadas como dosificaciones unitarias para sujetos humanos y otros mamíferos, conteniendo cada unidad una cantidad predeterminada de material activo calculada para producir el efecto terapéutico deseado, en asociación con un excipiente farmacéutico adecuado.
El compuesto activo puede ser efectivo en un amplio intervalo de dosificación y generalmente se administra en una cantidad farmacéuticamente efectiva. Sin embargo, se entenderá que la cantidad del compuesto administrado realmente será determinada generalmente por un médico, de acuerdo con las circunstancias relevantes, incluida la afección a tratar, la ruta de administración elegida, el compuesto real administrado, la edad, peso y respuesta del paciente individual, la gravedad de los síntomas del paciente y similares.
Para preparar composiciones sólidas tales como tabletas, el ingrediente activo principal se mezcla con un excipiente farmacéutico para formar una composición sólida de preformulación que contiene una mezcla homogénea del compuesto de Fórmula I. Cuando se hace referencia a estas composiciones de preformulación como homogéneas, el ingrediente activo se dispersa típicamente de manera uniforme en toda la composición, de modo que la composición se puede subdividir fácilmente en formas de dosificación unitarias igualmente eficaces, tales como tabletas, píldoras y cápsulas. Esta formulación previa sólida se subdivide luego en formas de dosificación unitarias del tipo descrito anteriormente que contienen, p. ej., de 0,1 a aproximadamente 500 mg del ingrediente activo de la presente solicitud.
Las tabletas o píldoras que contienen el compuesto de Fórmula I pueden recubrirse o combinarse para proporcionar una forma de dosificación que proporcione la ventaja de una acción prolongada. Por ejemplo, la tableta o píldora puede comprender una dosificación interna y un componente de dosificación externa, esta última en forma de una envoltura sobre la primera. Los dos componentes pueden estar separados por una capa entérica que sirve para resistir la desintegración en el estómago y permite que el componente interno pase intacto al duodeno o se retrase su liberación. Se puede usar una variedad de materiales para tales capas o recubrimientos entéricos, tales materiales que incluyen varios ácidos poliméricos y mezclas de ácidos poliméricos con materiales tales como goma laca, alcohol cetílico y acetato de celulosa.
Las formas líquidas en las que los compuestos y composiciones de la presente solicitud pueden incorporarse para administración oral o por inyección incluyen soluciones acuosas, jarabes con sabor adecuado, suspensiones acuosas o oleosas y emulsiones aromatizadas con aceites comestibles tales como aceite de semilla de algodón, aceite de sésamo, coco. aceite o aceite de maní, así como elixires y vehículos farmacéuticos similares.
Las composiciones para inhalación o insuflación incluyen soluciones y suspensiones en disolventes acuosos u orgánicos farmacéuticamente aceptables, o mezclas de los mismos, y polvos. Las composiciones líquidas o sólidas pueden contener excipientes farmacéuticamente aceptables adecuados como se describe anteriormente. En algunas realizaciones, las composiciones se administran por vía respiratoria oral o nasal para un efecto local o sistémico. Las composiciones se pueden nebulizar mediante el uso de gases inertes. Las soluciones nebulizadas se pueden respirar directamente desde el dispositivo de nebulización o el dispositivo de nebulización se puede conectar a una tienda de máscaras faciales o a una máquina de respiración de presión positiva intermitente. Las composiciones de solución, suspensión o polvo pueden administrarse por vía oral o nasal desde dispositivos que administran la formulación de manera apropiada.
La cantidad de compuesto o composición administrada a un paciente variará dependiendo de lo que se esté administrando, el propósito de la administración, tal como profilaxis o terapia, el estado del paciente, la forma de administración y similares. En aplicaciones terapéuticas, las composiciones se pueden administrar a un paciente que ya padece una enfermedad en una cantidad suficiente para curar o al menos detener parcialmente los síntomas de la enfermedad y sus complicaciones. Las dosis efectivas dependerán de la condición de la enfermedad que se está tratando, así como del juicio del médico tratante, dependiendo de factores tales como la gravedad de la enfermedad, la edad, el peso y el estado general del paciente, y similares.
Las composiciones administradas a un paciente pueden estar en forma de composiciones farmacéuticas descritas anteriormente. Estas composiciones pueden esterilizarse mediante técnicas de esterilización convencionales, o pueden filtrarse estérilmente. Las soluciones acuosas pueden envasarse para su uso tal cual, o liofilizarse, la preparación liofilizada se combina con un vehículo acuoso estéril antes de la administración. El pH de las preparaciones de compuestos típicamente estará entre 3 y 11, más preferiblemente de 5 a 9 y lo más preferiblemente de 7 a 8. Se entenderá que el uso de ciertos excipientes, vehículos o estabilizadores anteriores dará como resultado la formación de sales farmacéuticas.
La dosificación terapéutica del compuesto de Fórmula I puede variar según, p. ej., el uso particular para el que se
realiza el tratamiento, la forma de administración del compuesto, la salud y el estado del paciente, y el juicio del médico que prescribe. La proporción o concentración del compuesto de Fórmula I en una composición farmacéutica puede variar dependiendo de una serie de factores que incluyen dosis, características químicas (por ejemplo, hidrofobicidad) y la vía de administración. Por ejemplo, el compuesto de Fórmula I puede proporcionarse en una solución de tampón fisiológica acuosa que contiene aproximadamente 0,1 a aproximadamente 10% p/v del compuesto para administración parenteral. Algunos rangos de dosis típicos son de aproximadamente 1 pg/kg a aproximadamente 1 g/kg de peso corporal por día. En algunas realizaciones, el intervalo de dosis es de aproximadamente 0,01 mg/kg a aproximadamente 100 mg/kg de peso corporal por día. Es probable que la dosis dependa de variables tales como el tipo y el grado de progresión de la enfermedad o trastorno, el estado general de salud del paciente en particular, la eficacia biológica relativa del compuesto seleccionado, la formulación del excipiente y su vía de administración. Las dosis efectivas se pueden extrapolar a partir de curvas de dosis-respuesta derivadas de sistemas de prueba in vitro o de modelos animales.
El compuesto de Fórmula I también puede formularse en combinación con uno o más ingredientes activos adicionales que pueden incluir cualquier agente farmacéutico tal como agentes antivirales, vacunas, anticuerpos, potenciadores inmunes, supresores inmunes, agentes antiinflamatorios y similares.
La presente solicitud también incluye kits farmacéuticos útiles, p. ej., en el tratamiento o prevención de enfermedades o trastornos asociados a IDO, obesidad, diabetes y otras enfermedades a las que se hace referencia en el presente documento que incluyen uno o más recipientes que contienen una composición farmacéutica que comprende una cantidad terapéuticamente efectiva del compuesto de Fórmula I. Tales kits pueden incluir además, si se desea, uno o más de varios componentes de kit farmacéutico convencionales, tales como, p. ej., recipientes con uno o más vehículos farmacéuticamente aceptables, recipientes adicionales, etc., como será fácilmente evidente para los expertos en la materia. Las instrucciones, ya sea como insertos o como etiquetas, que indican las cantidades de los componentes a administrar, las pautas para la administración y/o las pautas para mezclar los componentes, también se pueden incluir en el kit.
Ejemplos
La invención se describirá con mayor detalle por medio de ejemplos específicos. Los siguientes ejemplos se ofrecen con fines ilustrativos, y no pretenden limitar la invención de ninguna manera. Los expertos en la materia reconocerán fácilmente una variedad de parámetros no críticos que se pueden cambiar o modificar para obtener esencialmente los mismos resultados.
Ejemplo 1. Síntesis de 4-({2-[(Ammosulfomlo)ammo]etMo}ammo)-W-(3-bromo-4-fluorofemlo)-W-hidroxM,2,5-oxadiazol-3-carboximidamida
Paso A: 4-Amino-N'-hidroxi-1,2,5-oxadiazol-3-carboximidamida (2)

Malononitrilo [Aldrich, producto n° M1407] (320,5 g, 5 mol) se añadió a agua (7 L), precalentado a 45°C y se agitó a 45°C por 5 min. La solución resultante se enfrió en un baño de hielo y se añadió nitrito de sodio (380 g, 5,5 mol, 1,1 equiv.). Cuando la temperatura alcanzó los 10 °C, se añadió ácido clorhídrico 6 N (55 ml). Se produjo una reacción exotérmica leve con una temperatura que alcanzó 16°C después de 15 minutos, se retiró el baño frío y la mezcla de reacción se agitó durante 1,5 h a 16-18°C La mezcla de reacción se enfrió a 13°C y 50% de hidrocloruro de hidroxilamina acuoso (990 g, 15 mol, 3,0 equiv.) se añadieron de una vez. La temperatura subió a 26°C Cuando la reacción exotérmica disminuyó, se retiró el baño frío y se continuó la agitación durante 1 hora a 26-27°C, luego se llevó lentamente a reflujo. El reflujo se mantuvo durante 2 h y luego la mezcla de reacción se dejó enfriar gradualmente durante la noche. La mezcla de reacción se agitó en un baño de hielo y se añadió ácido clorhídrico 6 N (800 ml) en porciones durante 40 minutos para ajustar a pH 7,0. La agitación continuó en el baño de hielo a 5°C. El precipitado se recogió por filtración, se lavó bien con agua y se secó en un horno de vacío (50°C) para dar el producto deseado (644 g, 90%) como un sólido blanquecino. 13C RMN (75 MHz, CD3OD): 8156,0, 145,9, 141,3; C3H5N5N5O2 (MW 143,10), LCMS (EI) m/e 144,0 (M++ H).
Paso B: Cloruro de 4-amino-N-hidroxi-1,2,5-oxadiazol-3-carboximidoilo (3)

Se añadió 4-amino-N'-hidroxi-1,2,5-oxadiazol-3-carboximidamida (422 g, 2,95 mol) a una mezcla de agua (5,9 L), ácido acético (3 L) y ácido clorhídrico 6 N (1,475 L, 3,0 equiv.) y la suspensión se agitó a 42-45°C hasta que se logró una solución clara. Se añadió cloruro de sodio (518 g, 3,0 equiv.) y esta solución se agitó en un baño de hielo/agua/metanol. Se añadió una solución de nitrito de sodio (199,5 g, 0,98 equiv.) en agua (700 ml) durante 3,5 h mientras se mantenía la temperatura por debajo de 0°C. Después de completar la adición, se continuó la agitación en el baño de hielo durante 1,5 h y luego la mezcla de reacción se dejó calentar a 15°C. El precipitado se recogió por filtración, se lavó bien con agua, se tomó en acetato de etilo (3,4 L), se trató con sulfato de sodio anhidro (500 g) y se agitó a ta durante 1 h. Esta suspensión se filtró a través de sulfato de sodio (200 g) y el filtrado se concentró en un evaporador rotativo. El residuo se disolvió en metilo ferc-butilo éter (5,5 l), se trató con carbón activado (40 g), se agitó a ta durante 40 min y se filtró a través de Celite. El disolvente se eliminó en un evaporador rotatorio y el producto resultante se secó en un horno de vacío (45°C) para dar el producto deseado (256 g, 53,4%) como un sólido blanquecino. 13C RMN (100 MHz, CD3OD) 8155,8, 143,4, 129,7; C3^ C lN 4O2 (MW 162,53), LCMS (EI) m/e 163/165 (M+ H).
Paso C: 4-Amino-N-(3-bromo-4-fluorofenilo)-N'-hidroxi-1,2,5-oxadiazol-3-c-arboximidamida (4)

Se mezcló cloruro de 4-amino-N-hidroxi-1,2,5-oxadiazol-3-carboximidoilo (33,8 g, 208 mmol) con agua (300 ml). A 60 °C, se añadió 3-bromo-4-fluoroanilina (Sigma-Aldrich) (43,6 g, 229 mmol, 1,1 equiv.) a la suspensión con agitación durante 10 minutos. Se añadió una solución de bicarbonato de sodio (26,3 g, 313 mmol, 1,5 equiv.) en agua (300 ml) durante 15 minutos con agitación a 60°C. Después de agitar durante 20 minutos, LCMS indicó la finalización de la reacción. La mezcla de reacción se enfrió luego a temperatura ambiente y se extrajo con acetato de etilo (2 x 300 ml). La solución combinada de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró para dar el producto deseado (65 g, 99%) como un sólido blanquecino, que se usó en la reacción posterior sin purificación adicional. CgHyBrFN5O2 (MW 316,09), LCMS (EI) m/e 316/318 (M++ H).
Paso D: 3-(4-Amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (5)

Una mezcla de 4-am¡no-A/-(3-bromo-4-fluorofen¡lo)-W-h¡drox¡-1,2,5-oxad¡azol-3-carbox¡m¡da¡dam¡da (65,7 g, 208 mmol), W,W-carbon¡ld¡¡m¡dazol (S¡gma-Aldr¡ch)(50,6 g, 312 mmol, 1,5 equiv.) y acetato de etilo (750 ml) se calentó a 60°C y se ag¡tó durante 20 m¡n. LCMS ¡ndícó reacc¡ón completada. La reacc¡ón se enfr¡ó a temperatura amb¡ente, se lavó con ác¡do clorhídr¡co 1 N (2 x 750 ml), se secó sobre sulfato de sod¡o y se concentró. El producto bruto se trituró con una mezcla de d¡clorometano, acetato de etilo y éter d¡etíl¡co para dar el producto deseado (60,2 g, 85%) como un sólido blanquec¡no. 1H RMN (300 MHz, DMSO-da) 88,05 (m, 1H), 7,69 (m, 1H), 7,57 (t, 1H, J = 8,7 Hz), 6,58 (s, 2H); CiüHsBrFNsOa (MW 342,08), LCMS (EI) m/e 342/344 (M+ H).
Paso E: [2-({4-[2-(3-bromo-4-fluorofenilo)-5-oxo-2,5-dihidro-1,2,4-oxadiazol-3-ilo-]-1,2,5-oxadiazol-3-ilo}amino)etilo]carbamato de terc-butilo (7)
A una soluc¡ón de ác¡do tr¡fluoroacét¡co (20,0 ml) y tetrah¡drofurano (10,0 ml) se le añad¡ó tr¡acetox¡boroh¡druro de sod¡o (10,59 g, 49,97 mmol, 10,0 equ¡v.) en porc¡ones con ag¡tac¡ón bajo n¡trógeno. Esta mezcla se ag¡tó durante 10 m¡nutos a temperatura amb¡ente y luego se enfr¡ó a -5°C. Una soluc¡ón de 3-(4-am¡no-1,2,5-oxad¡azol-3-¡lo)-4-(3-bromo-4-fluorofen¡lo)-1,2,4-oxad¡azol-ol-5(4H)-ona (1,71 g, 5,0 mmol) y (2-oxoet¡lo)carbamato de terc-but¡lo (S¡gma-Aldrích) (1,99 g, 12,5 mmol, 2,5 equ¡v.) en THF (15,0 mL) se añad¡ó gota a gota durante 30 m¡nutos con ag¡tac¡ón m¡entras se mant¡ene la temperatura por debajo de 0°C. La reacc¡ón se agitó a -5 a 0°C y porc¡ones ad¡c¡onales de (2-oxoet¡lo)carbamato de terc-but¡lo (0,20 g, 1,2 mmol, 0,24 equ¡v.) en THF (1,0 ml) se agregaron gota a gota a los 20 m¡nutos, 40 m¡nutos a ¡ntervalos de 4 horas. La HPLC ¡ndícó que la reacc¡ón se completó después de 5,25 h. La mezcla de reacc¡ón se vert¡ó en el b¡carbonato de sod¡o helado (500 ml) y esta soluc¡ón se ag¡tó a ta durante la noche. El prec¡p¡tado se recog¡ó por f¡ltrac¡ón y se lavó con salmuera. El res¡duo resultante se mezcló con heptano (40 ml) y éter d¡etíl¡co (40 ml) y se ag¡tó a ta durante 5 h. El prec¡p¡tado se recog¡ó por f¡ltrac¡ón, se lavó con éter d¡etíl¡co y se secó en un horno de vacío para dar el producto deseado (1953 mg, 80,5%) como un sól¡do blanquec¡no. 1H RMN (300 MHz, d m so d ) 88,08 (m, 1H), 7,71 (m, 1H), 7,59 (t, 1H, J = 8,7 Hz), 6,94 (m, 1H), 6,52 (m, 1H), 3,32 (m, 2H), 3,15 (m, 2H), 1,36 (s, 9H); C1yH1aBrFN6O5 (MW 485,26); LCMS (EI) m/e 507/509 (M+ Na).
Paso F: 3-{4-[(2-aminoetilo)amino]-1,2,5-oxadiazol-3-ilo}-4-(3-bromo-4-fluoro-fenilo)-1,2,4-oxadiazol-5(4H)-ona hidrocloruro (8)

Método A (preparado a partir de [2-({4-[2-(3-bromo-4-fluorofenilo)-5-oxo-2,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]carbamato de terc-butilo; Paso E)
En un matraz de 500 ml se cargó [2-({4-[2-(3-bromo-4-fluorofen¡lo)-5-oxo-2,5-d¡h¡dro-1,2,4-oxad-¡azol-3-¡lo]-1,2,5-oxad¡azol-3-¡lo}am¡no)et¡lo]carbamato de terc-but¡lo (20 g, 41,2 mmol) e ¡sopropanol (255 ml). La suspens¡ón se ag¡tó a ta. Se añad¡ó cloruro de h¡drógeno gaseoso (7,55 g, 207 mmol, 5,0 equ¡v.) a la suspens¡ón con un tubo de v¡dr¡o subsuperf¡c¡al durante 16 m¡nutos. Luego se añad¡ó acetato de et¡lo (111 ml) al lote y la reacc¡ón se calentó a 43°C y se ag¡tó durante 7,5 h. El lote se enfr¡ó a 19°C y se añad¡ó acetato de et¡lo (44 ml). La suspens¡ón se f¡ltró y el res¡duo resultante se lavó con acetato de et¡lo (2 x 55 ml). El sól¡do a¡slado se secó a pres¡ón reduc¡da a 45°C durante 15 h para proporc¡onar el producto deseado (16,61 g, 95,5% de rend¡m¡ento) como un sól¡do blanquec¡no a blanco. 1H RMN (300 MHz, DMSO-d6) 88,11 (bs, 3H), 7,78 (m, 1H), 7,73 (m, 1H), 7,59 (t, 1H, J = 8,7 Hz), 6,74 (t, 1H, J = 6,1 Hz), 3,50
(m, 2H), 3,02 (m, 2H); C^H-nBrClF^Os (MW 421,61; C^H-ioBrFNaOa para base libre, MW 385,15), LCMS (EI) m/e 385/387 (M+ H).
Método B (preparado directamente a partir de 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiaz-ol-5(4H)-ona; Paso D):
Se mezcló triacetoxiborohidruro de sodio (2,33 g, 11,0 mmol, 11,0 equiv.) con ácido trifluoroacético (12,0 mL, 155,8 mmol, 155,8 equiv.). La solución resultante se mezcló a temperatura ambiente durante 30 minutos. Una solución de 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (5, 0,342 g, 1,0 mmol) y carbamato de terc-butilo (2-oxoetilo) (Sigma-Aldrich) (1,04 g, 6,51 mmol, 6,5 equiv.) en diclorometano (10,0 mL) y acetonitrilo (6,0 mL) bajo N2. Esta solución se enfrió a -5°C y la solución de triacetoxiborohidruro de sodio y ácido trifluoroacético se añadió gota a gota durante 5 min. La reacción se agitó a ta durante 4 h. HPLC y LC-MS (M+ - Boc H: 385/387, patrón de bromuro) indicaron una proporción del producto deseado y el material de partida fue de 4 a 1. La mezcla se concentró y se diluyó con diclorometano (10 ml). La solución se enfrió a 0°C y se añadió lentamente hidróxido de sodio 4 N mientras se mantenía la temperatura a 0 - 5°C para ajustar el pH a 8-9. La capa acuosa se extrajo con diclorometano (3 x 10 ml). La solución de diclorometano combinada se lavó con bicarbonato de sodio y salmuera, se secó sobre sulfato de sodio y se concentró. El residuo bruto se disolvió luego en diclorometano (6,0 ml) y la solución resultante se enfrió a 0°C. Se añadió gota a gota ácido clorhídrico 4 N en dioxano (3,0 ml) a 0 - 5°C. La mezcla se agitó a ta durante 20 min. El precipitado se recogió por filtración, se lavó con éter dietílico y se secó al vacío para proporcionar el producto deseado (289 mg, 54%) como un sólido blanquecino. 1H RMN (300 MHz, DMSO-d6) 8 8,11 (bs, 3H), 7,78 (m, 1H), 7,73 (m, 1H), 7,59 (t, 1H, J = 8,7 Hz), 6,74 (t, 1H, J = 6,1 Hz), 3,50 (m, 2H), 3,02 (m, 2H); C12HnBrClFN6O3 (MW 421,61; C12H10BrFN6O3 para base libre, MW 385,15), LCMS (EI) m/e 385/387 (M+ H).
Paso G: ({[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato de terc-butilo (9)

Se cargó un reactor de vidrio de 20 L con hidrocloruro de 3-{4-[(2-aminoetilo)amino]-1,2,5-oxadiazol-3-ilo}-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (1200 g, 2,846 mol) y diclorometano (6,5 L) a temperatura ambiente. Se añadió trietilamina (950 g, 9,39 mol, 3,3 equiv.) al lote durante 7 minutos. El lote se enfrió luego a -14,6°C.
Un matraz de fondo redondo de 5 L se cargó con terc-butanol (253 g, 3,41 mol, 1,2 equiv.) y diclorometano (2,6 L). La solución se enfrió a 0,9°C. A esta solución se le añadió isocianato de clorosulfonilo (463 g, 3,27 mol, 1,15 equiv.) durante 43 minutos mientras se mantenía la temperatura del lote por debajo de 10°C. La solución resultante de (clorosulfonilo)carbamato de terc-butilo se mantuvo a 3-5°C durante 1 h.
La solución de (clorosulfonilo)carbamato de terc-butilo se añadió al reactor durante 73 minutos mientras se mantenía la temperatura del lote por debajo de 0°C. El lote se calentó a 10°C durante 1 h y luego se agitó a 10 - 14°C durante 1 h. Se añadió agua (4,8 L) al lote y la mezcla de reacción enfriada se agitó a ta durante 14,5 h. El lote se dejó sedimentar y las fases se separaron. La solución de diclorometano se aisló, se mantuvo en el reactor y se cargó con ácido acético (513 g) durante 25 minutos para precipitar el producto. La suspensión resultante se agitó a 20°C por 2,5 h. El producto se aisló por filtración y se lavó con diclorometano (1,8 L). El producto se secó a presión reducida (-30 inHg) a 45°C durante 16 h para proporcionar el producto deseado (1342 g, 83,5% de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-d6): 810,90 (s, 1H), 8,08 (dd, J = 6,2, 2,5 Hz, 1 H), 7,72 (m, 1 H), 7,59 (t, J = 8,6 Hz, 1 H), 6,58 (t, J = 5,7 Hz, 1 H), 3,38 (dd, J = 12,7, 6,2 Hz, 2 H), 3,10 (dd, J = 12,1, 5,9 Hz, 2 H), 1,41 (s, 9 H). C-i/H-iaB^N/O/S (MW 564,34), LCMS (EI) m/e 585,9/587,9 (M+ Na).
Paso H: N-[2-({4-[4-(3-Bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]sulfamida (10)

A un matraz de 20 L que contiene ({[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato de tere-butilo (1200 g, 2,126 mol) se añadió etanol (12 L) a 20°C La mezcla resultante se agitó a ta y se cargó con cloruro de hidrógeno gaseoso (472 g, 12,9 mol, 6,07 equiv.). El lote se calentó a 50°C y la temperatura se mantuvo durante 3 h hasta que se completó la reacción. El disolvente se eliminó por destilación al vacío a 33-39°C y se recogieron 6 kg de destilado. Se añadió acetato de etilo (6,8 L, 6,1 kg) al lote y se destiló para recoger 5,1 kg de destilado. Se añadió acetato de etilo (7,2 L, 6,48 kg) al lote y se destiló para recoger 5,1 kg de destilado. Se añadió acetato de etilo (2,4 L, 2,14 kg) al lote para ajustar la relación de disolventes. 1H RMN indicó que la relación molar de acetato de etilo a etanol era 1,0: 0,1. La solución se calentó a 65°C. Se añadió nheptano (4,1 kg) a la solución a 60-65°C durante 43 min. La suspensión resultante se agitó a 65°C durante 1 h. La suspensión se enfrió a 20°C durante 2,5 h y se agitó a esa temperatura durante 15 h. El producto se recogió por filtración y se lavó con n-heptano (2,42 L). El producto se secó a presión reducida a 45°C durante 65 h para proporcionar el producto deseado (906 g, 91,8% de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, dmso-d6) 88,08 (dd, J = 6,2) 7,72 (m, 1 H), 7,59 (t, J = 8,7 Hz, 1 H), 6,67 (t, J = 5,9 Hz, 1H), 6,55 (s, 2H) 6,52 (t, J = 6,0 Hz, 1 H), 3,38 (dd, J = 12,7, 6,3 Hz, 2 H), 3,11 (dd, J = 12,3, 6,3 Hz, 2H); C-^H-nBrFNyOsS (MW 464,23), LCMS (EI) m/e 485,8/487,8 (M+ - C5H8O2 Na).
Paso 1: 4-({2-[(Aminosulfonilo)amino]etilo}amino)-N-(3-bmmo-4-fluorofenilo-)-N'-hidmxi-1,2,5-oxadiazol-3-carboximidamida (11)

A un reactor de vidrio de 20 L se le añadió N-[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]sulfamida (799,4 g, 1,72 mol) y THF (3,2 L). La solución resultante se agitó a 20°C durante 7 min. y luego se carga con agua (1,6 L). El lote se enfrió a 2°C y se cargó con solución de hidróxido de sodio al 30% en peso (475 ml, 666,4 g, 4,99 mol, 2,9 equiv.) durante 8 minutos. El lote se calentó a 20°C y la temperatura se mantuvo durante 16 h. Luego se cargó el lote con metilo tere-butilo éter (8,0 L) durante 23 minutos. Se añadió agua (2,7 L) y el lote se enfrió a aproximadamente 0°C. Luego se cargó el lote con 85% en peso de ácido fosfórico (370,7 g, 3,22 mol, 1,9 equiv.) durante 9 minutos. El lote se calentó a 20°C y se agitó durante 1 h. El lote se dejó sedimentar y las fases se separaron. La capa orgánica se retuvo en el reactor y se cargó con agua (2,9 L) y 85% en peso de ácido fosfórico (370,7 g, 3,22 mol) y se agitó a 20°C durante 1 h. El lote se dejó sedimentar y las fases se separaron. La capa orgánica se retuvo en el reactor y se cargó con agua (3,2 L) y se agitó a 20°C durante 1 h. El lote se dejó sedimentar y las fases se separaron. La solución orgánica se retuvo en el reactor y se destiló a presión reducida a 20°C para eliminar 3,4 Kg de destilado. Se cargó etanol (4,8 L) en el lote y el lote se destiló a un volumen de 3,2 l. Este proceso de destilación se repitió una vez más. Se añadió etanol (0,6 L) al lote para ajustar el volumen del lote a 4 L. El lote se agitó a 20°C durante 16 h y luego se cargó con agua (6,39 L). La suspensión resultante se agitó a 20°C por 5 h. El producto se recogió por filtración y se lavó dos veces con una mezcla de etanol (529 ml) y agua (1059 ml). El producto se secó a presión reducida a 45°C durante 65 h para proporcionar el producto deseado (719,6 g, 95,4%) como un sólido blanco. 1H RMN (400 MHz, DMSO-d6): 8 11,51 (s, 1H), 8,90 (s, 1H), 7,17 (t, J = 8,8 Hz, 1 H), 7,11 (dd, J = 6,1, 2,7 Hz, 1 H), 6,76 (m, 1 H), 6,71 (t, J = 6,0 Hz, 1 H), 6,59 (s, 2 H), 6,23 (t, J = 6,1 Hz, 1 H), 3,35 (dd, J = 10,9, 7,0 Hz, 2 H), 3,10 (dd, J = 12,1, 6,2 Hz, 2 H); C-i1H13BrFN7O4S (MW 438,23), LCMS (EI) m/e 437,9/439,9 (M+ H).
Ejemplo 2. Preparación alternativa de N-[2-({4-[4-(3-Bromo-4-fluorofenMo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]sulfamida

Una solución de clorosulfonilisocianato (Sigma-Aldrich) (5,0 ml, 57,4 mmol) en diclorometano (100 ml) se enfrió a 0°C. Se añadió diclorometano de alcohol terc-butílico (4,26 g, 57,4 mmol, 1,0 equiv.) a través de un embudo de adición. Esta solución se agitó a 0°C durante 30 min. Se añadió clorhidrato de éster etílico de glicina (8,82 g, 63,2 mmol, 1,1 equiv.) seguido de la adición gota a gota de trietilamina (20,0 ml, 144 mmol, 2,5 equiv.) a 0°C. Esta mezcla de reacción se agitó a ta durante 4 h. La reacción se diluyó con diclorometano (100 ml) y se lavó con ácido clorhídrico 0,1 N y salmuera. La capa orgánica se secó sobre sulfato de sodio y se concentró para dar el producto deseado (13,2 g, 81,4%) como un sólido crudo blanco, que se usó en la reacción posterior sin purificación adicional. 1H RMN (300 MHz, DMSO-da) 610,88 (s, 1H), 8,07 (t, 1H, J = 6,1 Hz), 4,08 (q, 2H, J = 7,1 Hz), 3,78 (d, 2H, J = 6,1 Hz), 1,40 (s, 9H), 1,18 (t, 3H J = 7,1 Hz).
Paso 2a (alilo{[alilo(terc-butoxicarbonilo)amino]sulfonilo}amino)acetato de etilo (14a)

({[(terc-butoxicarbonilo)amino]sulfonilo}aminoacetato de etilo (1,0 g, 3,54 mmol) se mezcló con carbonato de potasio (2,45 g, 17,7 mmol, 5,0 equiv.) y acetonitrilo (23,0 mL) bajo N2 a temperatura ambiente. Se añadió gota a gota bromuro de alilo (1,84 mL, 21,2 mmol, 6,0 equiv.). Esta mezcla de reacción se calentó a 70°C y se agitó a esa temperatura durante 14 h. La HPLC y la LCMS indicaron la finalización de la reacción. La reacción se filtró y el filtrado se concentró. El residuo se disolvió en diclorometano y se lavó con bicarbonato de sodio y salmuera. La capa orgánica se secó sobre sulfato de sodio y se concentró para dar el producto deseado (1,11 g, 87%) como un sólido crudo blanco, que se usó en la reacción posterior sin purificación adicional. 1H RMN (300 MHz, DMSo)-d6) 65,75 (m, 2H), 5,20 (m, 4H), 4,12 (m, 6H), 3,89 (m, 2H), 1,43 (s, 9H), 1,18 (t, 3H, J = 8,7 Hz).
Paso 2b. [{[(terc-butoxicarbonilo)(4-metoxibencilo)amino]sulfonilo}(4-metoxibencilo)-amino]acetato de etilo (14b)

({[(terc-butoxicarbonilo)amino]sulfonilo}amino)acetato de etilo (1,00 g, 4,0 mmol) se mezclo con N,N-dimetilformamida (DMF, 6,0 ml) y se agitó a ta. Se añadieron a la mezcla yoduro de sodio (0,01 g, 0,1 mmol, 0,025 equiv.), carbonato de potasio (2,40 g, 20 mmol, 5,0 equiv.) y cloruro de para-metoxibencilo (2,64 mL, 19,5 mmol, 4,875 equiv.). Esta reacción se calentó a 80°C y se agitó a 80°C durante 2 h. LCMS indicó la finalización de la reacción. La reacción se enfrió a temperatura ambiente y se filtró a través de Celite. El lecho de Celite se lavó con diclorometano y los filtrados orgánicos combinados se concentraron. El residuo concentrado se disolvió en diclorometano (20 ml) y se lavó con bicarbonato de sodio (5 x 12 ml) y salmuera. La capa orgánica se secó sobre sulfato de sodio y se concentró. El residuo se purificó en gel de sílice (0-40% de acetato de etilo/elución en gradiente de hexano) para dar el producto deseado (1,39 g, 80%) como un sólido blanquecino. 1H RMN (300 MHz, DMSO-da) 87,22 (m, 2H), 7,14 (m, 2H), 6,88 (m, 4H), 4,64 (s, 2H), 4,33 (s, 2H), 4,03 (q, 2H, J = 7,1 Hz), 3,92 (s, 2H), 3,72 (s, 3H), 3,71 (s, 3H), 1,39 (s, 9H), 1,14 (t, 3H J = 7,1 Hz).
Paso 3a. Alilo{[alilo(2-oxoetilo)amino]sulfonilo}carbamato de terc-butilo (15a)

Una solución de (alilo{[alilo(terc-butoxicarbonilo)amino]sulfonilo}amino)acetato de etilo (1,11 g, 3,05 mmol) en diclorometano (15 ml) a -78°C bajo N2 se trató con hidruro de diisobutilaluminio 1,0 M en diclorometano (3,66 ml, 3,66 mmol, 1,2 equiv.). La mezcla de reacción se agitó a -78°C durante 1 h y luego se enfrió con metanol (1,5 ml) y se trató con una solución saturada de tartrato de sodio y potasio (65 ml). Esta solución se agitó a ta durante la noche. La capa acuosa se extrajo luego con diclorometano (3x 20 ml). La solución de diclorometano combinada se lavó con salmuera, se secó sobre sulfato de sodio, se filtró y se concentró para dar el producto deseado (0,62 g, 64%) como un aceite incoloro grueso y crudo, que se usó en una reacción posterior sin purificación adicional. 1H RMN (300 MHz, DMSO-d6) 89,45 (s, 1H), 5,76 (m, 2H), 5,18 (m, 4H), 4,15 (m, 4H), 3,72 (m, 2H), 1,43 (s, 9H).

Una solución de [{[(terc-butoxicarbonilo)(4-metoxibencilo)amino]sulfonilo}(4-metoxibencilo)-amino]acetato de etilo (5,30 g, 10 mmol) en diclorometano (20,0 ml) a -78°C bajo N2 se trató con hidruro de diisobutilaluminio 1,0 M en diclorometano (12,2 ml, 12,2 mmol, 1,22 equiv.). La mezcla de reacción se agitó a -78°C por 3 h. La reacción se enfrió luego con metanol (3 ml) y se trató con diclorometano (100 ml) y una solución saturada de tartrato de sodio y potasio (150 ml). Esta solución se agitó a ta durante la noche. La capa acuosa se extrajo luego con diclorometano (3 x 20 ml). La solución de diclorometano combinada se lavó con salmuera, se secó sobre sulfato de sodio y se concentró. El residuo se purificó luego sobre gel de sílice (elución con gradiente de acetato de etilo/hexano al 0-30%) para dar el producto deseado (3,45 g, 71%) como un sólido blanquecino. 1H RMN (300 MHz, DMSO-d6) 89,24 (s, 1H), 7,23 (m, 4H), 6,88 (m, 4H), 4,68 (s, 2H), 4,31 (s, 2H), 4,07 (s, 2H), 3,72 (s, 3H), 3,71 (s, 3H), 1,40 (s, 9H).
Paso 4a alilo(n-alilo-N-(2-(4-(4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4--oxadiazol-3-ilo)-1,2,5-oxadiazol-3-ilamino)etilo)sulfamoilo)carbamato de terc-butilo (16a)

A un matraz de 50 ml se le añadió triacetoxiborohidruro de sodio (1,06 g, 5,0 mmol, 1,0 equiv.), ácido trifluoroacético (TFA, 2,0 mL, 26 mmol) y tetrahidrofurano (THF, 1,0 mL) a temperatura ambiente. Esta mezcla se enfrió a -5°C bajo N2 y se agitó a 0 - 5°C durante 10 min. A esta solución se le añadió 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (0,171 g, 5,0 mmol; Paso D) y alilo{[alilo(2-oxoeti)amino]sulfonilo}carbamato de ferc-butilo (0,398 g, 2,5 mmol, 0,5 equiv.) en THF (1,5 mL) gota a gota a 0-5 °C durante 5 min. La mezcla de reacción resultante se agitó bajo N2 a 0 - 5°C. A 20 min, 40 min y 2,5 h, una solución de alilo{[alilo(2-oxoeti)amino]sulfonilo}carbamato de ferc-butilo (0,040 g, 0,125 mmol, 0,25 equiv.) en THF (0,20 ml) se añadió gota a gota a 0 - 5°C. A las 2,5 h, se añadió una solución de triacetoxiborohidruro de sodio (0,211 g, 1,0 mmol, 0,2 equiv.) en ácido trifluoroacético (TFA, 1,5 mL, 9,5 mmol) a 0 - 5°C. La reacción se calentó a temperatura ambiente y se agitó durante la noche. La mezcla de reacción se vertió luego en una solución saturada de carbonato de sodio enfriada con hielo (50 ml) y se extrajo con diclorometano (3 x 20 ml). Los extractos de diclorometano combinados se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron. El residuo se purificó luego sobre gel de sílice (0-75% de acetato de etilo/elución en gradiente de hexano) para dar el producto deseado (0,239 g, 74,2%) como un sólido blanquecino. 1H RMN (300 MHz, DMSO-da) 88,07 (m, 1H), 7,71 (m, 1H), 7,58 (t, 1H, J = 8,7 Hz), 6,62 (m, 1H), 5,77 (m, 2H), 5,19 (m, 4H), 4,17 (m, 2H), 3,89 (m, 2H), 3,44 (m, 2H), 3,38 (m, 2H), 1,42 (s, 9H); C23H27BrFN7OyS (MW 644,47), LCMS (EI) m/e 544/546 (M+ - Boc H).
Paso 4b. N-(2-(4-(4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-y-1)-1, 2,5-oxadiazol-3-ilamino)etilo)-N-(4-metoxibencilo)sulfamoílo(4-metoxibencilo)carbamato de ferc-butilo (16b)

A un matraz de 50 ml se le añadió triacetoxiborohidruro de sodio (0,50 g, 2,37 mmol, 4,74 equiv.), ácido trifluoroacético (TFA, 1,0 ml, 13 mmol) y tetrahidrofurano a temperatura ambiente. Esta mezcla se enfrió a 0 - 5°C bajo N2 y se agitó a 0 - 5°C durante 10 min. A esta solución se le añadió (4-metoxibencilo){[(4-metoxibencilo)(2-oxoetilo)amino]sulfonilo}-carbamato de ferc-butilo (0,40 g, 0,84 mmol, 1,68 equiv) y 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiaz-ol-5(4H)-ona (0,17 g, 0,50 mmol; Paso D) en tetrahidrofurano (THF, 1,50 mL) a 0 - 5°C La reacción se agitó a 0 - 5°C durante 45 minutos y una solución de (4-metoxibencilo){[(4-metoxibencilo)(2-oxoetilo)amino]sulfonilo}-carbamato de ferc-butilo (0,12 g, 0,20 mmol, 0,4 equiv.) en THF (0,50 ml) se añadió luego a 0 - 5°C. Después de agitar a 0 - 5°C durante 1 h, la reacción se calentó gradualmente a temperatura ambiente con agitación. A los puntos de tiempo de 2,5 h y de 4,5 h, se añadió ácido trifluoroacético (0,25 ml). A las 5 h, una solución de (4-metoxibencilo){[(4-metoxibencilo)(2-oxoetilo)amino]sulfonilo}carbamato de ferc-butilo (0,060 g, 0,1 mmol, 0,2 equiv.) en THF (0,20 mL) adicional. A las 6,5 h, se añadió una solución de triacetoxiborohidruro de sodio (0,060 g, 0,24 mmol, 0,48 equiv.) en ácido trifluoroacético (0,25 ml). La HPLC indicó aproximadamente el 4% del material de partida 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (del Paso D) aún restante. La mezcla de reacción se agitó a ta durante la noche. La HPLC indicó la finalización de la reacción. La mezcla de reacción se vertió en una solución saturada de carbonato de sodio enfriada con hielo (50 ml) y la mezcla se extrajo con diclorometano (3 x 20 ml). Los extractos de diclorometano combinados se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron. El residuo se purificó luego sobre gel de sílice (elución con gradiente de acetato de etilo/hexano al 0-30%) para dar el producto deseado (0,33 g, 82,5%) como un sólido blanquecino. 1H RMN (300 MHz, DMSO-cfe) 8 8,06 (m, 1H), 7,69 (m, 1H), 7,57 (t, 1H, J = 8,7 Hz), 7,22 (m, 4H), 6,87 (m, 4H), 6,48 (m, 1H), 4,72 (s, 2H), 4,36 (s, 2H), 3,70 (S, 6H), 3,39 (m, 2H), 3,31 (m, 2H), 1,37 (s, 9H); CaHaHasBrFNyOgS (MW 804,64), LCMS (EI) m/e 826/828 (M+ -Boc Na).
Paso 5: N-[2-({4-[4-(3-Bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)efilo]sulfamida (10)

A un matraz de 25 ml se le añadió {[[2-({4-[4-(3--bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo](4-metoxibencilo)amino]sulfonilo}(4-metoxibencilo)carbamato de terc-butilo (40,2 mg, 0,050 mmol) en ácido trifluoroacético (TFA, 0,50 ml, 6,5 mmol) a temperatura ambiente. Esta mezcla de reacción se calentó a 70°C bajo N2 y se agitó durante 1 h. HPLC indicó reacción completada. La mezcla de reacción se enfrió a temperatura ambiente y el TFA se evaporó. El TFA residual se eliminó por tratamiento con diclorometano (3 x 10 ml) seguido de evaporación al vacío. El residuo se trituró luego con diclorometano y metanol para dar el producto deseado (20 mg, 87%) como un sólido crudo de color blanquecino. 1H RMN (400 MHz, d m so d ) 58,08 (dd, J = 6,2, 2,5 Hz, 1 H), 7,72 (m, 1 H), 7,59 (t, J = 8,7 Hz, 1 H), 6,67 (t, J = 5,9 Hz, 1H), 6,55 (s, 2H) 6,52 (t, J = 6,0 Hz, 1 H), 3,38 (dd, J = 12,7, 6,3 Hz, 2 H), 3,11 (dd, J = 12,3, 6,3 Hz, 2H); C^H-nBrFNyOsS (MW 464,23), LCMS (EI) m/e 487,8/489,8 (M+ Na).
Ejemplo 3. Preparación alternativa de N-[2-({4-[4-(3-Bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo)amino)etilo]sulfamida

Se enfrió una solución de clorosulfonilisocianato (11,32 g, 80 mmol) en diclorometano (120 ml) a 0°C. Se añadió alcohol terc-butílico (7,65 ml, 80,0 mmol, 1,0 equiv.) mediante un embudo de adición. La mezcla se agitó a 0°C durante 1,5 h. A esta mezcla, se añadió gota a gota una solución de aminoacetaldehído dimetilo acetal (8,76 ml, 80,0 mmol, 1,0 equiv.) y trietilamina (TEA, 33,4 ml, 240 mmol, 3,0 equiv.) en cloruro de metileno (DCM 120,0 ml) embudo adicional. La reacción se calentó a temperatura ambiente y se agitó durante la noche. La reacción se trató con ácido clorhídrico 0,1 N y la capa orgánica se lavó con salmuera, se secó sobre sulfato de sodio y se concentró para dar el producto deseado (15,6 g, 68,5%) como un sólido crudo blanco, que se usó para la subsiguiente reacción sin purificación adicional: 1H RMN (300 MHz, DMSO-d6) 510,84 (s, 1H), 7,62 (t, 1H, J = 6,0 Hz), 4,38 (t, 1H, J = 5,5 Hz), 3,24 (s, 6H), 2,96 (dd, 2H, J = 5,8 Hz), 1,41 (s, 9 H).
Paso 1b N-(2,2-dimetaxietilo)sulfamoilcarbamato de bencilo (17b)

Una solución de clorosulfonilisocianato (16,26 g, 114,9 mmol) en diclorometano (100 ml) se enfrió a 0°C. Se añadió alcohol bencílico (12,44 g, 115,0 mmol, 1,0 equiv.) mediante un embudo de adición. La mezcla se agitó a 0°C por 0,5 h. A esta mezcla se le añadió una mezcla de aminoacetaldehído dimetilo acetal (13,25 g, 126,0 mmol, 1,1 equiv.) y trietilamina (TEA, 17,4 g, 172 mmol, 1,5 equiv.) gota a gota a través de un embudo de adición a menos de 15°C. La reacción se calentó a temperatura ambiente y se agitó durante la noche. La mezcla de reacción se trató con ácido clorhídrico 0,5 N (100 ml) y la fase orgánica recogida se lavó con salmuera, se secó sobre sulfato de sodio y se concentró al vacío para dar el producto deseado (23,5 g, 64,3%) como un crudo crudo. sólido. 1H RMN (300 MHz, DMSO-da) 8 11,29 (s, 1H), 7,90 (t, 1H, J = 6,0 Hz), 7,37 (m, 5H), 5,12 (s, 2H), 4,35 (t, 1H, J = 5,5 Hz), 3,21 (s, 6H), 2,97 (dd, 2H, J = 5,8 Hz).
Paso 1c. N-(2,2-dimetoxietilo)sulfamoilcarbamato de etilo (17c)

Una solución de clorosulfonilisocianato (11,32 g, 80 mmol) en diclorometano (120 ml) se enfrió a 0°C. Se añadió etanol (4,67 ml, 80,0 mmol, 1,0 equiv.) mediante un embudo de adición. La mezcla se agitó a 0°C durante 1,5 h. A esta mezcla se le añadió una solución de aminoacetaldehído dimetilo acetal (8,76 ml, 80,0 mmol, 1,0 equiv.), trietilamina (TEA, 33,4 ml, 240 mmol, 3,0 equiv.) en diclorometano (DCM, 120,0 ml) gota a gota mediante un embudo de adición 0°C. La reacción se calentó a temperatura ambiente y se agitó durante la noche. La reacción se trató con ácido clorhídrico 0,1 N y la fase orgánica recogida se lavó con salmuera, se secó sobre sulfato de sodio y se concentró al vacío para dar el producto deseado (11,2 g, 55%) como un sólido crudo de color blanquecino. 1 H RMN (300 MHz, DMSO-d6 ) 811,13 (s, 1H), 7,81 (t, 1H, J = 6,0 Hz), 4,37 (t, 1H, J = 5,5 Hz), 4,09 (q, 2H, J = 7,1 Hz), 3,23 (s, 6H), 2,97 (dd, 2H, J = 5,8 Hz), 1,19 (t, 3H, J = 7,1 Hz).
Paso 1d. 2,2,2-Tricloroetilo N-(2,2-dimetoxietilo)sulfamoilcarbamato (17d)

Una solución de clorosulfonilisocianato (6,96 ml, 80 mmol) en diclorometano (120 ml) se enfrió a 0°C. Se añadió 2,2,2-tricloroetanol (7,67 ml, 80,0 mmol, 1,0 equiv.) mediante un embudo de adición a 0°C. Esta mezcla se agitó a 0°C durante 1,5 h. Luego, a esta mezcla se le añadió una solución de aminoacetaldehído dimetilo acetal (8,76 ml, 80,0 mmol, 1,0 equiv.) y trietilamina (TEA, 33,4 ml, 240 mmol, 3,0 equiv.) en diclorometano (DCM, 120,0 ml) añadido gota a gota mediante embudo de adición a 0°C. La reacción se calentó a temperatura ambiente y se agitó a ta durante la noche. La reacción se trató con ácido clorhídrico 0,1 N y la fase orgánica recogida se lavó con salmuera, se secó sobre Na2SO4 y se concentró para dar el producto deseado (28,01 g, 97%) como un sólido blanquecino bruto, que se usó en la reacción posterior sin purificación adicional. 1H Rm N (300 MHz, d m so d ) 8 11,79 (s, 1H), 8,08 (t, 1H, J = 5,9 Hz), 4,90 (s, 2H), 4,37 (t, 1H, J = 5,5 Hz), 3,23 (s, 6H), 3,00 (dd, 2H, J = 5,7 Hz).
Paso 2a ({[2-({[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo-]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato de terc-butilo (18a)

Una mezcla de 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (103 mg, 0,302 mmol, 1,5 equiv.; Paso D) y N-(2,2-dimetoxietilo)sulfamoilcarbamato de terc-butilo (57,2 mg, 0,201 mmol) en diclorometano (1,0 ml) se agitó bajo N2 a temperatura ambiente. A esta mezcla se le añadió ácido trifluoroacético (0,50 ml, 6,5 mmol) y trietilsilano (80,2 pL, 0,502 mmol, 2,5 equiv.) gota a gota. Esta mezcla de reacción se agitó a ta durante 2 h. HPLC indicó aproximadamente 30% de conversión. La mezcla de reacción se enfrió a 0°C y se apagó con
bicarbonato de sodio saturado a pH. La mezcla se extrajo en acetato de etilo (3 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron. El residuo se purificó por TLC preparativa (50% acetato de etilo/hexano) para dar el producto deseado (27,5 mg, 29,5%) como un sólido blanquecino. 1H RMN (D M S O d 400 MHz): 6 10,90 (s, 1H), 8,08 (dd, J = 6,2, 2,5 Hz, 1H), 7,72 (m, 1H), 7,59 (t, J = 8,6 Hz, 1H), 6,58 (t, J = 5,7 Hz, 1H), 3,38 (dd, J = 12,7, 6,2 Hz, 2H), 3,10 (dd, J = 12,1, 5,9 Hz, 2H), 1,41 (s, 9H). C-iyH-igBrFNyOyS (MW 564,34), LCMS (EI) m/e 485,8/487,8 (M+ - C5H8O2 Na).
Paso 2b ({[2-({[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo-]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato de bencilo (18b)

Una mezcla de 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (68 mg, 0,20 mmol; del Paso D) y {[(2,2-dimetoxietilo)amino]sulfonilo}carbamato de bencilo (191 mg, 0,60 mmol, 3,0 equiv.) en 1,2-dicloroetano (3,0 ml) se enfrió a 0°C A esta mezcla se le añadió ácido trifluoroacético (1,0 ml, 13,0 mmol) y trietilsilano (105 |jL, 0,66 mmol, 3,3 equiv.) gota a gota. Esta mezcla de reacción se agitó a 0°C durante 2 h. La HPLC indicó la finalización de la reacción. La mezcla de reacción se enfrió a 0°C y se apagó con bicarbonato de sodio saturado a pH y la mezcla de reacción apagada se extrajo con EtOAc (3 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron. El residuo se agitó luego en una mezcla de heptano y éter dietílico durante la noche. Los sólidos se recogieron por filtración, se lavaron con heptano y se secaron al vacío para dar el producto deseado (125 mg, 99%) como un sólido crudo de color blanquecino. 1H RMN (300 MHz, DMSO-cfe) 6 11,31 (s, 1H), 8,05 (m, 1H), 7,87 (m, 1H), 7,68 (m, 1H), 7,56 (m, 1H), 7,32 (m, 5H), 6,54 (m, 1H), 5,07 (s, 2H), 3,29 (m, 2H), 3,09 (m, 2H); C20H1yBrFNyOyS (MW 598,36), LCMS m/e 598/600 (M+ H).
Paso 2c. ({[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilamino}sulfonilo)carbamato de etilo (18c)

Una mezcla de 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (68 mg, 0,20 mmol; del Paso D) y {[(2,2-dimetoxietilo)amino]sulfonilo}carbamato de etilo (154 mg, 0,600 mmol, 3,0 equiv.) en 1,2-dicloroetano (2,50 mL, 31,7 mmol) se agitó a 0°C. A esta mezcla se añadió ácido trifluoroacético (1,00 ml, 13,0 mmol) y trietilsilano (105 j L, 0,66 mmol, 3,3 equiv.) = gota a gota. La mezcla de reacción se agitó a 0°C por 3 h. La HPLC indicó una conversión del 97,5% en el producto deseado. La mezcla de reacción se enfrió a 0°C y se apagó con bicarbonato de sodio saturado a pH. La mezcla se extrajo en acetato de etilo (3 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron. El residuo se agitó en una mezcla de heptano y éter dietílico durante la noche. Los sólidos se recogieron por filtración, se lavaron con heptano para dar el producto deseado (95 mg, 88%) como un sólido crudo de color blanquecino. 1H RMN (300 MHz, DMSO-d6) 6 11,18 (s, 1H), 8,08 (m, 1H), 7,70 (m, 2H), 7,59 (t, 1H, J = 8,7 Hz), 6,56 (s, 1H), 4,04 (d, 2H, J = 7,2 Hz), 3,35 (m, 2H), 3,11 (m, 2H), 1,15 (t, 3H, J = 7,2 Hz); C^H^BrFNyOyS (MW 536,29), LCMS (EI) m/e 536/538 (M+ H).

Una suspensión de 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5(4H)-ona (5 , 0,680 g, 1,99 mmol) y 2,2,2-tricloroetilo {[(2,2-dimetoxietilo)amino]sulfonilo}carbamato (17d, 2,22 g, 6,17 mmol, 3,1 equiv.) en diclorometano (DCM, 6,0 ml) se agitó a ta. A esta mezcla se le añadió trietilsilano (1,27 ml, 7,95 mmol, 4,0 equiv.) y
una solución de ácido trifluoroacético (TFA, 3,0 ml, 39,0 mmol) en diclorometano (DCM, 2,0 ml) mientras se mantenía la temperatura de reacción por debajo de 30°C La mezcla de reacción se volvió homogénea después de 5 min con agitación a temperatura ambiente y se agitó a ta durante 1 h. La HPLC indicó la finalización de la reacción. La reacción se filtró y el precipitado se suspendió en una mezcla de diclorometano y heptano (la relación de diclorometano a heptano fue de 1 a 9 en volumen). La suspensión se agitó a ta durante la noche. El precipitado se recogió por filtración y se lavó con diclorometano al 10% en heptano y se secó al vacío para dar el producto deseado (1,15 g, 90,4%) como un sólido blanquecino, que se usó en la reacción posterior sin purificación adicional. 1H RMN (300 MHz, DMSO-cfe) 6 11,85 (s, 1H), 8,07 (m, 2H), 7,70 (m, 1H), 7,57 (t, 1H, J = 8,7 Hz), 6,56 (m, 1H), 4,88 (m, 2H), 3,37 (m, 2H), 3,16 (m, 2H); C-isH^BrClaFNyOyS (MW 639,62), LCMS (EI) m/e 638/640/642 (M+ H).
Paso 3. N-[2-({4-[4-(3-Bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]sulfamida (10)

Una solución de 2,2,2-tricloroetilo ({[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato (320 mg, 0,50 mmol; del Paso Q, Método D) en tetrahidrofurano (THF, 4,0 ml) se agitó a ta. Se añadieron secuencialmente ácido acético (0,30 ml, 5,3 mmol) y escamas de zinc (160 mg, 2,5 mmol, 5,0 equiv.). Esta mezcla de reacción se agitó a ta durante 3 h. La HPLC indicó la finalización de la reacción. La mezcla de reacción se filtró a través de Celite y la Celite se lavó con THF. El filtrado combinado se concentró al vacío y el residuo resultante se disolvió en acetato de etilo (20 ml). La solución de acetato de etilo se lavó con carbonato de sodio saturado y salmuera, se secó sobre sulfato de sodio y se concentró. El material bruto se cristalizó en acetato de etilo y éter dietílico para dar el producto deseado (147 mg, 63%) como un sólido blanquecino.
Ejemplo 4. Preparación alternativa de 4-({2-[(Aminosulfonilo)amino]etilo}amino)-N-(3-bromo-4-fluorofenilo)-N'-hidroxi-1,2,5-oxadiazol-3-carboximidamida

Paso 1. (9H- fluoren-9-ilo)metilo N-(2,2-dimetoxietilo)sulfamoilcarbamato

En un matraz de fondo redondo de 4 bocas y 2 L, secado al horno, se cargó 9-fluorenilmetanol (50,0 g, 255 mmol) y DCM anhidro (382 ml) a temperatura ambiente. La suspensión resultante se enfrió en un baño de hielo a aproximadamente 0 - 5°C. Se añadió gota a gota una solución de isocianato de clorosulfonilo (CSI, 23,0 ml, 264 mmol) en DCM anhidro (127 ml) a la suspensión a través de un embudo de adición durante 22 minutos, manteniendo la temperatura de la mezcla de reacción a <5°C La mezcla resultante se agitó a 0 - 5°C durante 1,75 h, produciendo una suspensión espesa y blanca. Se añadió a la mezcla una solución de aminoacetaldehído dimetilo acetal (27,9 ml, 255
mmol) en DCM anhidro (382 ml) y 4-metilmorfolina (84,0 ml, 764 mmol) a aproximadamente 0 - 5°C durante 71 minutos. La mezcla de reacción resultante se agitó luego en el baño de hielo durante 1,5 horas. Cuando la HPLC mostró que la reacción se había completado, la mezcla de reacción se acidificó mediante la adición gota a gota de un ácido fosfórico 1,0 M (H3PO4, acuoso, 640 ml) durante 22 minutos a pH 1-2. Luego se añadieron agua (300 ml), EtOAc (2150 ml) y heptano (250 ml) y la mezcla resultante se agitó durante 10 minutos. Las dos fases se separaron y la fase orgánica se lavó secuencialmente con agua (500 ml), heptano (300 ml) y agua (2 x 500 ml) y se secó sobre MgSO4. El filtrado se concentró al vacío hasta sequedad. Los sólidos resultantes se redisolvieron en EtOAc (600 ml) a 65°C y la solución caliente se filtró en un matraz limpio de fondo redondo de 3 L. El filtrado se enfrió a temperatura ambiente y se agitó durante 2,5 h antes de añadir heptano (1200 ml) a través de un embudo de adición durante 80 min. Después de agitar durante la noche a temperatura ambiente, la mezcla se enfrió en un baño de hielo durante 1 h. Los sólidos resultantes se recogieron por filtración, se lavaron con EtOAc al 25%/heptano (250 ml) y se secaron durante la noche a aproximadamente 40-45°C a vacío para proporcionar 9H-fluoren-9-ilmetilo {[(2,2-dimetoxietilo)amino]sulfonilo}carbamato (91,3 g, 88% de rendimiento) como un polvo blanco. 1H RMN (300 MHz, DMSO-da) 8 11,43 (s, 1H), 7,98-7,85 (m, 3H), 7,76 (d, J = 7,5 Hz, 2H), 7,43 (t, J = 7,2 Hz, 2H), 7,33 (td, J = 7,4, 1,1 Hz, 2H), 4,44-4,33 (m, 3H), 4,33-4,22 (m, 1H), 3,23 (s, 6H), 2,99 (t, J = 5,8 Hz, 2H) ppm.
Paso 2. 9H-fluoren-9-ilmetilo ({[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato

A una suspensión agitada de 3-(4-amino-1,2,5-oxadiazol-3-ilo)-4-(3-bromo-4-fluorofenilo)-1,2,4-oxadiazol-5-4-(4H)-ona (10,00 g, 29,23 mmol) en d Cm (160 ml) se añadió ácido metanosulfónico (MeSOsH, 8,46 g, 88,04 mmol) y trietilsilano (EtsSiH, 8,37 g, 71,96 mmol) a temperatura ambiente durante 10 minutos para dar una suspensión. Se añadió en porciones 9H-fluoren-9-ilmetilo {[(2,2-dimetoxietilo)amino]sulfonilo}carbamato sólido (12,25 g, 30,14 mmol) en porciones (1 g/3-4 min; durante 1 h) mientras se mantenía la temperatura interna a menos de 20°C utilizando un baño de agua. Después de la adición, la mezcla resultante se agitó a aproximadamente 13 a 22°C por 3 días. Se añadieron trietilsilano adicional (EtsSiH, 0,1755 g, 1,51 mmol) y 9H-fluoren-9-ilmetilo {[(2,2-dimetoxietilo)amino]sulfonilo}carbamato (0,3082 g, 0,76 mmol) y la mezcla resultante se agitó a ta durante 23 h adicionales. Se añadió alcohol isopropílico (IPA, 15 ml) y la mezcla resultante se agitó a ta durante 1 h. Se añadió heptano (100 ml) y la mezcla se agitó a ta durante 2 h adicionales. Los sólidos se recogieron por filtración, se lavaron con IPA/heptano (1/5; 2 x 30 ml) y heptano (2 x 30 ml), y se secaron al vacío para proporcionar 9H-fluoren-9-ilmetilo ({[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato como un sólido blanco (18,30 g, 91,1% de rendimiento). 1H RMN (500 MHz, DMSO-d6) 811,44 (s, 1H), 8,07 (dd, J = 6,2, 2,5 Hz, 1H), 7,90 (t, J = 5,6 Hz, 1H), 7,88 (d, J = 7,6 Hz, 2H), 7,72 (d, J = 7,0 Hz, 2H), 7,71 (ddd, J = 8,9, 4,3, 2,6 Hz, 1H), 7,57 (dd, J = 8,7, 8,7 Hz, 1H), 7,40 (t, J = 7,4 Hz, 2H), 7,31 (td, J = 7,4, 1,0 Hz, 2H), 6,55 (t, J = 6,0 Hz, 1H), 4,35 (d, J = 7,3 Hz, 2H), 4,25 (t, J = 7,2 Hz, 1H), 3,39 (q, J = 6,4 Hz, 2H), 3,15 (q, J = 6,3 Hz, 2H); 13C RMN (126 MHz, DMSO-d6) 8 159,03 (d, J = 248,7 Hz), 156,61 (s), 155,22 (s), 151,55 (s), 148,67 (s), 143,29 (s), 140,68 (s), 133,82 (s), 133,39 (s), 130,05 (d, J = 8,5 Hz), 128,54 (d, J = 3,2 Hz), 127,73 (s), 127,07 (s), 125,24 (s), 120,11 (s), 117,42 (d, J = 24,0 Hz), 108,19 (d, J = 22,5 Hz), 66,70 (s), 46,17 (s), 43,34 (s), 40,79(s) ppm; 19F RMN (376 MHz, d m so d ) 8 -103,99 - -107,39 (m) ppm.
Paso 3. 4-({2-[(Aminosulfonilo)amino]etilo}amino)-N-(3-bromo-4-fluorofenilo-)-N'-hidroxi-1,2,5-oxadiazol-3-carboximidamida

En un matraz de fondo redondo de 4 bocas de 1 L se cargó 9H-fluoren-9-ilmetilo ({[2-({4-[4-(3-bromo-4-fluorofenilo)-5-oxo-4,5-dihidro-1,2,4-oxadiazol-3-ilo]-1,2,5-oxadiazol-3-ilo}amino)etilo]amino}sulfonilo)carbamato (25,0 g, 36,4 mmol) y THF anhidro (250 ml) a temperatura ambiente para producir una solución homogénea. La solución se enfrió luego a 0 - 5°C en un baño de hielo antes de añadir N,N-bis(2-aminoetilo)etano-1,2-diamina (114 ml, 728 mmol) gota a gota durante 35 minutos a través de un embudo de adición. El embudo de adición se enjuagó con THF anhidro (50 ml) y el
enjuague se añadió a la mezcla de reacción. El baño frío se retiró y la reacción se calentó gradualmente a temperatura ambiente y se agitó a ta durante 2,5 h. Se añadió EtOAc (400 ml) y la mezcla resultante se transfirió a un matraz de fondo redondo de 4 bocas y 2 litros y se enfrió a aproximadamente 0 - 5°C en un baño de hielo. Se añadió gota a gota una solución de HCl acuoso 2,0 M (400 ml, 800,0 mmol) a través de un embudo de adición, mientras se mantenía la temperatura interna por debajo de 10°C. Las dos fases se separaron y la fase acuosa se extrajo con EtOAc (200 ml). Las fracciones orgánicas se combinaron y se enfriaron a aproximadamente 6-7°C. Se añadió gota a gota una solución de HCl acuoso 2,0 M (200,0 ml, 400,0 mmol) a la fracción orgánica fría, manteniendo la temperatura interna por debajo de 10°C Las dos fases se separaron y la fase orgánica se lavó con agua (2 x 400 ml), se secó sobre MgSO4 y se concentró a presión reducida hasta un jarabe amarillo claro. El jarabe se disolvió en EtOAc (60,0 ml) para producir una solución homogénea. A la solución se le añadió una solución de DCM (250,0 ml) y tere-butilo metilo éter (TBME, 100,0 ml) gota a gota. La suspensión resultante se agitó durante la noche a temperatura ambiente, luego se enfrió en un baño de hielo durante 1 h. Los sólidos se recogieron por filtración, se lavaron con una solución helada de 250 ml de DCM (150 ml) y TBME (100 ml), y se secaron al vacío para dar 14,4 g del producto bruto deseado como sólidos blancos.
El producto bruto se disolvió en EtOAc (140,0 ml) a 60°C y la solución tibia se filtró. El filtrado se enfrió a temperatura ambiente antes de agregar heptano (100,0 ml) gota a gota durante 55 minutos. La mezcla resultante se agitó luego durante la noche a temperatura ambiente. Los sólidos se recogieron por filtración, se lavaron con una mezcla 2:1 de heptano y EtOAc (75 ml) y se secaron al vacío a 40-50°C a peso constante para proporcionar 4-({2-[(aminosulfonilo)amino]etilo}amino)-N-(3-bromo-4-fluorofenilo)-N'-hidroxi-1,2,5-oxadiazol-3-carboximidamida (12,9 g, 81% de rendimiento) como un sólido blanco.
Ejemplo A: Ensayo enzimático de 2,3-dioxigenasa humana (IDO)
Indoleamina 2,3-dioxigenasa humana de indoleamina humana (IDO) con una etiqueta His N-terminal se expresó en E. coli y se purificó hasta homogeneidad. IDO cataliza la escisión oxidativa del anillo de pirrol del núcleo de indol de triptófano para producir N'-formilquinurenina. Los ensayos se realizaron a temperatura ambiente como se describe en la literatura utilizando IDO 95 nM y D-Trp 2 mM en presencia de ascorbato 20 mM, azul de metileno 5 pM y catalasa de 0,2 mg/ml en tampón de fosfato de potasio 50 mM (pH 6,5). Las velocidades de reacción iniciales se registraron siguiendo continuamente el aumento de la absorbancia a 321 nm debido a la formación de W-formilquilquinurenina (Ver: Sono, M., et al., 1980, J. Biol. Chem. 255, 1339-1345). El compuesto de Fórmula I se probó en el ensayo del Ejemplo A y se encontró que tenía un CI50 de <200 nM.
Ejemplo B: Determinación de la actividad inhibidora en ensayo de indoleamina 2,3-dioxigenasa (IDO)/kynurenina basado en células HeLa
Las células HeLa (n° CCL-2) se obtuvieron de la American Type Tissue Culture Collection (ATCC, Manassas, Va.) y se mantuvieron de forma rutinaria en medio esencial mínimo (eagle) con L-glutamina 2 mM y el BSS de Earle ajustado para contener 1,5 g/L de bicarbonato de sodio, aminoácidos no esenciales 0,1 mM, piruvato de sodio 1 mM y suero bovino fetal al 10% (todo de Invitrogen). Las células se mantuvieron a 37°C en una incubadora humidificada con 5% de CO2. El ensayo se realizó como sigue: las células HeLa se sembraron en una placa de cultivo de 96 pocillos a una densidad de 5 x 103 por pocillo y se cultivaron durante la noche. Al día siguiente, IFN-y (concentración final de 50 ng/ml) y diluciones en serie de compuestos (en un volumen total de 200 pL de medio de cultivo) se añadieron a las células. Después de 48 horas de incubación, se transfirieron 140 pl del sobrenadante por pocillo a una nueva placa de 96 pocillos. Se mezclaron 10/l de ácido tricloroacético 6,1 N (n° T0699, Sigma) en cada pocillo y se incubaron a 50°C durante 30 minutos para hidrolizar la N-formilquinnurenina producida por la 2,3-dioxigenasa de indoleamina a kinurenina. La mezcla de reacción se centrifugó luego durante 10 minutos a 2500 rpm para eliminar los sedimentos. Se transfirieron 100/l del sobrenadante por pocillo a otra placa de 96 pocillos y se mezcló con 100/l de pdimetilaminobenzaldehído al 2% (p/v) (n° 15647-7, Sigma-Aldrich) en ácido acético. El color amarillo derivado de kinurenina se midió a 480 nm usando un lector de microplacas SPECTRAmax 250 (Molecular Devices). Se usó L-kinurenina (n° K8625, Sigma) como estándar. Los estándares (240, 120, 60, 30, 15, 7,5, 3,75, 1,87 pM) se prepararon en medios de cultivo de 100 pL y se mezclaron con un volumen igual de p-dimetilaminobenzaldehído al 2% (p/v). Se determinó el porcentaje de inhibición a concentraciones individuales y se obtuvieron los valores promedio de duplicados. Los datos se analizaron mediante el uso de regresión no lineal para generar valores CI50 (Prism Graphpad). Ver: Takikawa 0, et al., 1988, J. Biol. Chem., 263 (4): 2041-8.
Ejemplo C: Determinación del efecto de los inhibidores de IDO sobre la proliferación de células T que se suprime mediante células dendríticas que expresan IDO
Se recogieron monocitos de células mononucleares periféricas humanas mediante leucoforesis. A continuación, los monocitos se sembraron a una densidad de 1 x 106 células/pocillo en una placa de 96 pocillos, usando medio RPMI 1640 suplementado con suero bovino fetal al 10% y L-glutamina 2 mM (todo de Invitrogen). Las células adherentes fueron retenidas en la placa después del cultivo nocturno a 37°C. Los monocitos adherentes fueron estimulados durante 5-7 días con 100 ng/ml de GM-CSF (n° 300-03, PeproTech) y 250 ng/ml de IL-4 (n° 200-04, PeproTech), seguido de activación con 5 pg/mL LPS de Salmonella typhimurium (n° 437650, Sigma) y 50 ng/mL de IFN-y. (n° 285-IF, R&D Systems) durante 2 días adicionales para inducir la maduración de las células dendríticas.
Después de la activación de las células dendríticas, el medio se reemplazó con RPMI 1640 completo suplementado con 100-200 U/ml de ILO-2 (n° CYT-209, ProSpec-Tany TechnoGene) y 100 ng/ml de anticuerpo anti-CD3 (n° 555336, PharMingen), Células T (2-3 x 105 células/pocillo) y diluciones en serie de compuestos IDO. Después de la incubación durante 2 días más, se midió la proliferación de células T mediante el ensayo de incorporación de BrdU, usando un kit ELISA de proliferación celular colorimétrico según las instrucciones del fabricante (n° 1647229, Roche Molecular Biochemicals). Las células se cultivaron continuamente durante 16-18 horas en presencia de solución de marcaje BrdU 10 |jM. A continuación, se retiró el medio de marcado y se añadieron a las células 200 j l de FixDenat por pocillo y se incubaron durante 30 minutos a temperatura ambiente. Se eliminó la solución FixDenat y se añadió una solución de trabajo conjugada de anticuerpo anti-BrdU-POD de 100 jL/pocillo. La reacción se llevó a cabo durante 90 minutos a temperatura ambiente. Luego se eliminó el conjugado de anticuerpo, y las células se enjuagaron tres veces con una solución de lavado de 200 jL/pocillo. finalmente, se añadieron 100 jL/pocillo de solución de sustrato y los resultados se obtuvieron usando un lector de microplacas (Spectra Max PLUS, Molecular Devices) durante el desarrollo del color. Se obtuvieron múltiples lecturas en varios puntos de tiempo para garantizar que los datos estuvieran dentro del rango lineal. Los datos se obtuvieron rutinariamente de experimentos replicados, y se incluyeron controles apropiados. Ver: Terness P, et al. 2002, J. Exp. Med., 196 (4): 447-57; y Hwu, P y col. 2000, J. Immunol., 164 (7): 3596-9.
Ejemplo D: Prueba in vivo de inhibidores de IDO para actividad antitumoral
La eficacia antitumoral in vivo se puede probar usando protocolos modificados de aloinjerto/xenoinjerto tumoral modificado. Por ejemplo, se ha descrito en la literatura que la inhibición de IDO puede sincronizarse con quimioterapia citotóxica en ratones inmunocompetentes (Muller, AJ, et al. 2005, Nat. Med. 11: 312-319). Se demostró que esta sinergia dependía de las células T en comparación con los efectos sinérgicos de un inhibidor de IDO en investigación en modelos de xenoinjerto de tumor murino (por ejemplo, B16 y variantes relacionadas, CT-26, LLC) cultivados en ratones singénicos inmunocompetentes a los observados en ratones singénicos tratados con anticuerpos neutralizantes anti-CD4, o los mismos tumores cultivados en ratones inmunocomprometidos (por ejemplo, nu/nu).
El concepto de efectos antitumorales diferenciales en ratones inmunocompetentes frente a inmunocomprometidos también puede permitir la prueba de inhibidores de IDO en investigación como agentes únicos. Por ejemplo, los tumores LLC crecen bien en su cepa hospedadora singénica, C57B1/6. Sin embargo, si estos ratones se tratan con el inhibidor IDO 1-MT (versus placebo), la formación de tumores se retrasa notablemente, lo que implica que la inhibición IDO era inhibidora del crecimiento (Friberg, M., et al. 2002, Int. J. Cancer 101:151-155). Siguiendo esta lógica, se puede examinar la eficacia de la inhibición de IDO en el modelo de tumor de xenoinjerto LLC cultivado en ratones inmunes inmunes C57B1/6 y compararlo con los efectos de los inhibidores de IDO sobre el crecimiento tumoral LLC en ratones desnudos o SCID (o ratones C57B1/6 tratados con anticuerpos que neutralizan la actividad de las células T). Como los efectos de aliviar la actividad inmunosupresora de IDO mediada por el tumor probablemente diferirán dependiendo del potencial inmunogénico de diferentes modelos tumorales, se pueden realizar modificaciones genéticas en las células tumorales para aumentar su potencial inmunogénico. Por ejemplo, la expresión de GM-CSF en células B16.F10 aumenta su potencial inmunogénico (Dranoff, G., et al. 1993, Proc. Natl. Acad. Sci., EE.UU., 90: 3539-3543). Como tal, en algunos modelos tumorales (p. ej., B16.F10) se pueden generar [policlones] que expresan proteínas inmunoestimulantes como GM-CSF y probar los efectos inhibidores del crecimiento de los inhibidores de IDO contra tumores establecidos a partir de estas células tumorales en ratones tanto competentes como comprometidos.
Una tercera vía para evaluar la eficacia de los inhibidores de IDO in vivo emplea modelos de aloinjerto/xenoinjerto de tumor murino de preinmunización. En estos modelos, los ratones inmunocompetentes se sensibilizan a un antígeno o antígenos tumorales específicos para imitar una vacunación antitumoral terapéutica. Esto prepara a los ratones para una respuesta antitumoral mediada por el sistema inmune cuando los ratones son desafiados posteriormente con líneas celulares tumorales murinas (que poseen antígenos tumorales similares a los utilizados para la inmunización) en experimentos de xenoinjerto. Se ha demostrado que la expresión de IDO reduce la respuesta antitumoral y permite que los xenoinjertos crezcan más rápidamente. Es importante destacar que el crecimiento de tumores en este modelo es inhibido por el inhibidor IDO 1-MT (Uyttenhove, C., et al. 2003, Nat. Med. 9:1269-1274). Este modelo es particularmente atractivo ya que la actividad IDO es permisiva para el crecimiento tumoral de P815 y, por lo tanto, la inhibición específica de IDO debería inhibir el crecimiento.
Por último, la inmunización terapéutica puede usarse para evaluar el impacto de los inhibidores de IDO in vivo. Por ejemplo, se ha demostrado usando células B16-BL6 que se puede desafiar a los ratones Blk/6 con una inyección intravenosa de células tumorales seguido de tratamiento con un péptido inmunogénico bien caracterizado (por ejemplo, TRP-2) expresado por las células tumorales (Ji, et al., 2005, J. Immunol, 175:1456-63). Es importante destacar que los modificadores del sistema inmunitario, como el anticuerpo anti-CTL-4, pueden mejorar las respuestas a tales inmunizaciones terapéuticas. El impacto de los inhibidores de IDO puede evaluarse de manera similar: inmunización con péptido tumoral con o sin inhibidor de IDO. La eficacia se evalúa mediante la supervivencia de los animales (tiempo hasta la morbilidad) o mediante la medición de metástasis tumorales en los pulmones y/u otros órganos en puntos de tiempo definidos.
En cualquiera/todos los modelos mencionados anteriormente, también puede ser posible medir directa y/o
indirectamente el número y/o actividad de las células inmunes reactivas tumorales. Los métodos para medir el número y/o la actividad de las células inmunes reactivas tumorales están bien establecidos y se pueden realizar utilizando técnicas familiares para los expertos en la materia (Current Protocols in Immunology, Vol. 4, Coligan, JE, et al.; Immunotherapy of Cancer, Human Press, 2006, Disis, ML y referencias allí). Conceptualmente, una reducción en los efectos inmunosupresores de IDO puede dar como resultado un aumento en el número o la reactividad de las células inmunes específicas del tumor. Además, la inhibición de IDO puede aumentar aún más el número o la reactividad de las células inmunes reactivas al tumor cuando se combina con otras terapias, por ejemplo quimioterapéuticas y/o moduladores inmunes (por ejemplo, anticuerpo anti-CTLA4).
Todos los experimentos de aloinjerto/xenoinjerto se pueden realizar utilizando técnicas tumorales estándar (revisadas por Corbett, et al., In Cancer Drug Discovery and Development: Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval, 2a Ed. Teicher, BA y Andrews, PA, Gumana Press Inc.: Totowa, NJ, 2004). La clonación e introducción de genes (por ejemplo, IDO, g M-CSF) en líneas celulares tumorales, se puede realizar utilizando técnicas familiares para los expertos en la materia (revisado en Sambrook, J. y Russel, D., Molecular Cloning: A Laboratory Manual). (3a edición), Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2001).
Ejemplo E: Prueba in vivo de inhibidores de IDO en el modelo de encefalitis de virus de la inmunodeficiencia humana-1 (HW-1)
1. Aislamiento celular e infección viral
Los monocitos y PBL se pueden obtener mediante elutriación centrífuga a contracorriente de paquetes de leucoféresis de donantes de VIH-1, 2 y hepatitis B seronegativos. Los monocitos se cultivan en cultivo en suspensión usando matraces de teflón en medio Eagle modificado de Dulbecco (DMEM, Sigma-Aldrich) suplementado con suero humano agrupado inactivado por calor al 10%, glutamina al 1%, 50 pg/ml de gentamicina, 10 pg/ml de ciprofloxacina (Sigma) y factor estimulante de colonias de macrófagos humanos recombinantes altamente purificados de 1000 U/ml. Después de siete días en cultivo, los MDM se infectan con VIH-1ada con una multiplicidad de infección de 0,01.
2. Ratones Hu-PBL-NOD/SCID HIVE
Se pueden comprar ratones macho NOD/CB-17 SCID de cuatro semanas de edad (Jackson Laboratory). Los animales se mantienen en jaulas de microisoladores estériles en condiciones libres de patógenos. Todos los animales se inyectan intraperitonealmente con anti-CD122 de rata (0,25 mg/ratón) tres días antes del trasplante de PBL y dos veces con anticuerpos asialo-GM1 de conejo (0,2 mg/ratón)(Wako) un día antes y tres días después de la inyección de PBL (20 x 106 células/ratón). Se inyectan MDM infectados con VIH-1a d a (3 x 105 células en 10 pl) intracranealmente (i.c.) ocho días después de la reconstitución de PBL generando ratones hu-PBL-NOD/SCID HIVE. Inmediatamente después de la inyección i.c. de MDM infectado con VIH-1, los ratones hu-PBL-NOD/SCID HIVE se implantan subcutáneamente (s.c.) con gránulos de control (vehículo) o compuestos (liberación lenta de 14 o 28 días, Innovative Research). Los experimentos iniciales están diseñados para confirmar la inducción de CTL específicos de virus en los animales hu PBL-NOD/SCID HIVE tratados con compuestos IDO. Esto se confirma mediante tinción con tetrámero y análisis neuropatológicos de la eliminación de MDM del tejido cerebral. A continuación, el experimento está diseñado para analizar la reconstitución de linfocitos humanos, las respuestas inmunes humorales y las alteraciones neuropatológicas. En estos experimentos, los animales se sangran el día 7 y se sacrifican a los 14 y 21 días después de la inyección i.c. de MDM humano. La sangre recolectada en tubos que contienen EDTA se usa para citometría de flujo y el plasma se usa para la detección de VIH-1 p24 usando ELISA (Beckman Coulter™). Los anticuerpos específicos para VIH-1 se detectan mediante pruebas de transferencia Western de acuerdo con las instrucciones del fabricante (kit de transferencia Western Cambridge Biotech VIH-1, Calypte Biomedical). Se detecta una cantidad similar de anticuerpos específicos de virus en animales de control y tratados con compuestos. Se pueden realizar un total de tres experimentos independientes utilizando tres donantes de leucocitos humanos diferentes.
3. FACScan de sangre periférica y bazo en ratones hu PBL-NOD/SCID HIVE
Se pueden realizar análisis FACS de dos colores en sangre periférica en las semanas 1-3 y en los esplenocitos en las semanas 2 y 3 después de la inyección i.c. de MDM humano. Las células se incuban con Abs monoclonales conjugados con fluorocromo (mAbs) a CD4, CD8, CD56, CD3, IFN-y humanos. (eBioscience) durante 30 minutos a 4°C. Para evaluar la respuesta inmune celular, la tinción intracelular IFN-y se realiza en combinación con CD8 antihumano y CD45 anti-ratón conjugado con FITC para excluir células murinas. Para determinar la CTL específica de Ag, tinción de tetrámero conjugado con aloficocianina para VIH-1gag (p17 (aa77-85) SLYNTVATL, SL-9) y VIH-1pDl [(aa476-485) ILKEPVHGV, IL-9] se realiza en esplenocitos estimulados con fitohemaglutinina/interleucina-2 (PHA/IL-2). Las células se tiñen siguiendo la recomendación del NIH/National Institute of Allergy and Infections Disease, National Tetramer Core Facilities. Los datos se analizaron con un FACS Calibur™ utilizando el software CellQuest (Becton Dickinson Immunocytometry System).
4. Histopatología y análisis de imágenes
Se recolecta tejido cerebral en los días 14 y 21 después de la inyección i.c. de MDM, se fija en paraformaldehído
tamponado con fosfato al 4% y se incrusta en parafina o se congela a -80°C para uso posterior. Se cortan secciones coronales de los bloques incrustados para identificar el sitio de inyección. Para cada ratón, se cortan 30-100 (5 |jm de espesor) secciones en serie del sitio de inyección de MDM humano y se analizan 3-7 portaobjetos (con 10 secciones separadas). Las secciones del cerebro se desparafinan con xileno y se hidratan en alcoholes de gradiente. La tinción inmunohistoquímica sigue un protocolo indirecto básico, usando recuperación de antígeno calentando a 95°C en 0,01 mol/L de tampón de citrato durante 30 minutos para la recuperación de antígeno. Para identificar células humanas en cerebros de ratones, se usa mAb a vimentina (1:50, clon 3B4, Dako Corporation), que identifica todos los leucocitos humanos. Los linfocitos MDM y CD8+ humanos se detectan con los anticuerpos CD68 (dilución 1:50, clon KP 1) y CD8 (dilución 1:50, clon 144B), respectivamente. Las células infectadas por virus se marcan con mAb a VIH-1 p24 (1:10, clon Kal-1, todas de Dako). Las células microgliales murinas reactivas se detectan con el anticuerpo Iba-1 (1:500, Wako). La expresión de IDO humana (hulDO) se visualiza con Abs obtenido del Departamento de Farmacología Celular, Instituto de Investigación Central, Escuela de Graduados de Medicina, Universidad de Hokkaido, Sapporo, Japón. Los anticuerpos primarios se detectan con los anticuerpos secundarios biotinilados apropiados y se visualizan con complejos de avidina-biotina (kit Vectastain Elite ABC, Vector Laboratories) y polímero de dextrano acoplado a peroxidasa de rábano picante (HRP) (EnVision, Dako Corporation) Las secciones inmunotinuladas se contratiñen con hematoxilina de Mayer. Las secciones de las que se elimina el anticuerpo primario o se incorpora el isotipo IgG irrelevante sirvieron como controles. Dos observadores independientes en forma ciega cuentan el número de linfocitos CD8+, células CD68+ MDM y VIH-1 p24+ en cada sección de cada ratón. El examen microscópico ligero se realiza con un microscopio Nikon Eclipse 800 (Nikon Instruments Inc). El análisis semicuantitativo para Ibal (porcentaje del área ocupada por inmunotinción) se lleva a cabo mediante análisis de imagen asistido por computadora (Image-Pro®Plus, Media Cybernetics) como se describió anteriormente.
5. Análisis estadístico
Los datos pueden analizarse utilizando Prism (Graph EAP) con la prueba t de Student para comparaciones y ANOVA. Los valores de p < 0,05 se consideraron significativos.
6. Referencia
Poluektova LY, Munn DH, Persidsky Y y Gendelman HE (2002). Generation of cytotoxic T cells against virus-infected human brain macrophages in a murine model of HIV-1 encephalitis. J. Immunol. 168 (8): 3941-9.
Claims (1)
- REIVINDICACIONES1. Un proceso que comprende hacer reaccionar un compuesto de Fórmula F5:
 con un aldehido de Fórmula F6:
con un aldehido de Fórmula F6: F6 9en donde Pg1 es un grupo protector de amino, para proporcionar un compuesto de Fórmula F7:
F6 9en donde Pg1 es un grupo protector de amino, para proporcionar un compuesto de Fórmula F7: 2. El proceso de la reivindicación 1, en donde Pg1 es:(a) etoxicarbonilo, ferc-butoxicarbonilo, benciloxicarbonilo o 9-fluorenilmetiloxicarbonilo; o(b) ferc-butoxicarbonilo.3. El proceso de una cualquiera de las reivindicaciones 1-2, en el que dicha reacción se realiza en presencia de un agente reductor, en el que dicho agente reductor es:(a) un agente reductor de borohidruro; o(b) un agente reductor de borohidruro que es triacetoxiborohidruro de sodio.4. El proceso de una cualquiera de las reivindicaciones 1-3, que comprende además desproteger dicho compuesto de Fórmula F7 para proporcionar un compuesto de Fórmula F8:
2. El proceso de la reivindicación 1, en donde Pg1 es:(a) etoxicarbonilo, ferc-butoxicarbonilo, benciloxicarbonilo o 9-fluorenilmetiloxicarbonilo; o(b) ferc-butoxicarbonilo.3. El proceso de una cualquiera de las reivindicaciones 1-2, en el que dicha reacción se realiza en presencia de un agente reductor, en el que dicho agente reductor es:(a) un agente reductor de borohidruro; o(b) un agente reductor de borohidruro que es triacetoxiborohidruro de sodio.4. El proceso de una cualquiera de las reivindicaciones 1-3, que comprende además desproteger dicho compuesto de Fórmula F7 para proporcionar un compuesto de Fórmula F8: opcionalmente en donde dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F7 con ácido clorhídrico.5. El proceso de la reivindicación 4, que comprende además hacer reaccionar dicho compuesto de Fórmula F8, con Pg2-NH-SO2-X, en presencia de una base orgánica para proporcionar un compuesto de Fórmula F9:
opcionalmente en donde dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F7 con ácido clorhídrico.5. El proceso de la reivindicación 4, que comprende además hacer reaccionar dicho compuesto de Fórmula F8, con Pg2-NH-SO2-X, en presencia de una base orgánica para proporcionar un compuesto de Fórmula F9: en donde:Pg2 es un grupo protector de amino; yX es halo.6. El proceso de la reivindicación 5, en donde Pg2 es:(a) etoxicarbonilo, ferc-butoxicarbonilo, benciloxicarbonilo o 9- fluorenilmetiloxicarbonilo; o(b) ferc-butoxicarbonilo; y en donde X es cloro.7. El proceso de una cualquiera de las reivindicaciones 5-6, que comprende además desproteger dicho compuesto de Fórmula F9 para proporcionar un compuesto de Fórmula F10:
en donde:Pg2 es un grupo protector de amino; yX es halo.6. El proceso de la reivindicación 5, en donde Pg2 es:(a) etoxicarbonilo, ferc-butoxicarbonilo, benciloxicarbonilo o 9- fluorenilmetiloxicarbonilo; o(b) ferc-butoxicarbonilo; y en donde X es cloro.7. El proceso de una cualquiera de las reivindicaciones 5-6, que comprende además desproteger dicho compuesto de Fórmula F9 para proporcionar un compuesto de Fórmula F10: opcionalmente en donde dicha desprotección comprende hacer reaccionar un compuesto de Fórmula F9 con ácido clorhídrico.8. El proceso de la reivindicación 7, que comprende además hacer reaccionar dicho compuesto de Fórmula F10 con una base para proporcionar un compuesto de Fórmula I:
opcionalmente en donde dicha desprotección comprende hacer reaccionar un compuesto de Fórmula F9 con ácido clorhídrico.8. El proceso de la reivindicación 7, que comprende además hacer reaccionar dicho compuesto de Fórmula F10 con una base para proporcionar un compuesto de Fórmula I: opcionalmente en el que dicha base es hidróxido de sodio.9. Un proceso que comprende hacer reaccionar un compuesto de Fórmula F15:
opcionalmente en el que dicha base es hidróxido de sodio.9. Un proceso que comprende hacer reaccionar un compuesto de Fórmula F15: con un compuesto de Fórmula F5:
con un compuesto de Fórmula F5: para proporcionar un compuesto de Fórmula F16:
para proporcionar un compuesto de Fórmula F16: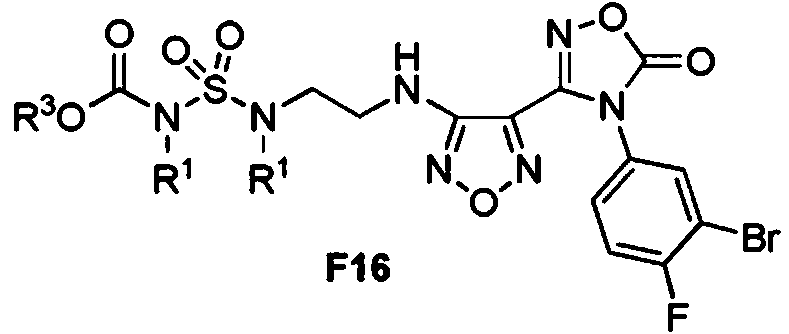 en donde:cada R1 es independientemente un grupo protector de amino seleccionado entre C2-4 alquenilo-C1-3 alquilo o fenilo-C1-3 alquilo, en donde dicho fenilo-C1-3 alquilo está opcionalmente sustituido con 1, 2 o 3 grupos C1-4 alcoxi seleccionados independientemente; yR3 es C1-6 alquilo o bencilo.10. El proceso de la reivindicación 9, en donde:(a) cada R1 es alilo;(b) cada R1 es 4-metoxibencilo;(c) R3 es C1-6 alquilo; o(d) R3 es ferc-butilo.11. El proceso de una cualquiera de las reivindicaciones 9-10, en el que dicha reacción se realiza en presencia de un agente reductor, en el que dicho agente reductor es:(a) un agente reductor de borohidruro; o(b) un agente reductor de borohidruro que es triacetoxiborohidruro de sodio; y en el que dicha reacción se realiza en presencia de ácido trifluoroacético.12. El proceso de una cualquiera de las reivindicaciones 9-11, que comprende además desproteger dicho compuesto de Fórmula F16 para proporcionar un compuesto de Fórmula F10:
en donde:cada R1 es independientemente un grupo protector de amino seleccionado entre C2-4 alquenilo-C1-3 alquilo o fenilo-C1-3 alquilo, en donde dicho fenilo-C1-3 alquilo está opcionalmente sustituido con 1, 2 o 3 grupos C1-4 alcoxi seleccionados independientemente; yR3 es C1-6 alquilo o bencilo.10. El proceso de la reivindicación 9, en donde:(a) cada R1 es alilo;(b) cada R1 es 4-metoxibencilo;(c) R3 es C1-6 alquilo; o(d) R3 es ferc-butilo.11. El proceso de una cualquiera de las reivindicaciones 9-10, en el que dicha reacción se realiza en presencia de un agente reductor, en el que dicho agente reductor es:(a) un agente reductor de borohidruro; o(b) un agente reductor de borohidruro que es triacetoxiborohidruro de sodio; y en el que dicha reacción se realiza en presencia de ácido trifluoroacético.12. El proceso de una cualquiera de las reivindicaciones 9-11, que comprende además desproteger dicho compuesto de Fórmula F16 para proporcionar un compuesto de Fórmula F10: en donde dicha desprotección comprende:(a) hacer reaccionar un compuesto de Fórmula F16 con ácido trifluoroacético; o(b) hacer reaccionar un compuesto de Fórmula F16 con ácido clorhídrico.13. El proceso de una cualquiera de las reivindicaciones 9-12, en el que dicho compuesto de Fórmula F15 se obtiene mediante un proceso que comprende tratar un compuesto de Fórmula F14:
en donde dicha desprotección comprende:(a) hacer reaccionar un compuesto de Fórmula F16 con ácido trifluoroacético; o(b) hacer reaccionar un compuesto de Fórmula F16 con ácido clorhídrico.13. El proceso de una cualquiera de las reivindicaciones 9-12, en el que dicho compuesto de Fórmula F15 se obtiene mediante un proceso que comprende tratar un compuesto de Fórmula F14: con un agente reductor para proporcionar dicho compuesto de Fórmula F15; en donde R2 es C1-4 alquilo; opcionalmente en el que dicho agente reductor es hidruro de diisobutilaluminio.14. El proceso de la reivindicación 13, en el que dicho compuesto de Fórmula F14 se obtiene mediante un proceso que comprende proteger un compuesto de Fórmula F13:
con un agente reductor para proporcionar dicho compuesto de Fórmula F15; en donde R2 es C1-4 alquilo; opcionalmente en el que dicho agente reductor es hidruro de diisobutilaluminio.14. El proceso de la reivindicación 13, en el que dicho compuesto de Fórmula F14 se obtiene mediante un proceso que comprende proteger un compuesto de Fórmula F13: con uno o más agentes protectores de amino seleccionados independientemente para proporcionar un compuesto de Fórmula F14; opcionalmente en donde dicho uno o más agentes protectores de amino se seleccionan de bromuro de alilo y cloruro de 4-metoxibencilo, y en donde dicha protección se realiza en presencia de una base.15. Un compuesto de Fórmula F15:
con uno o más agentes protectores de amino seleccionados independientemente para proporcionar un compuesto de Fórmula F14; opcionalmente en donde dicho uno o más agentes protectores de amino se seleccionan de bromuro de alilo y cloruro de 4-metoxibencilo, y en donde dicha protección se realiza en presencia de una base.15. Un compuesto de Fórmula F15: en donde:R3 es C1-6 alquilo o bencilo; ycada R1 es independientemente un grupo protector de amino.16. El compuesto de la reivindicación 15, que es:(a) alilo{[alilo(2-oxoetilo)amino]sulfonilo}carbamato de ferc-butilo; o(b) (4-metoxibencilo){[(4-metoxibencilo)(2-oxoetilo)amino]sulfonilo}carbamato de ferc-butilo.17. Un proceso que comprende hacer reaccionar un compuesto de Fórmula F17:
en donde:R3 es C1-6 alquilo o bencilo; ycada R1 es independientemente un grupo protector de amino.16. El compuesto de la reivindicación 15, que es:(a) alilo{[alilo(2-oxoetilo)amino]sulfonilo}carbamato de ferc-butilo; o(b) (4-metoxibencilo){[(4-metoxibencilo)(2-oxoetilo)amino]sulfonilo}carbamato de ferc-butilo.17. Un proceso que comprende hacer reaccionar un compuesto de Fórmula F17: en donde R4 es C1-6 alquilo, C1-6 haloalquilo, bencilo o 9H-fluoren-9-ilmetilo con un compuesto de Fórmula F5:
en donde R4 es C1-6 alquilo, C1-6 haloalquilo, bencilo o 9H-fluoren-9-ilmetilo con un compuesto de Fórmula F5: para proporcionar un compuesto de Fórmula F18:
para proporcionar un compuesto de Fórmula F18: (a) ferc-butilo;(b) bencilo;(c) etilo; o(d) 2,2,2-tricloroetilo.19. El proceso de la reivindicación 18, en el que dicha reacción se lleva a cabo en presencia de un agente reductor; opcionalmente en donde dicho agente reductor es trietilsilano, y en donde dicha reacción se lleva a cabo en presencia de un ácido orgánico; opcionalmente en donde dicho ácido orgánico es ácido trifluoroacético.20. El proceso de una cualquiera de las reivindicaciones 18-19, que comprende además desproteger dicho compuesto de Fórmula F18 para proporcionar un compuesto de Fórmula F10:
(a) ferc-butilo;(b) bencilo;(c) etilo; o(d) 2,2,2-tricloroetilo.19. El proceso de la reivindicación 18, en el que dicha reacción se lleva a cabo en presencia de un agente reductor; opcionalmente en donde dicho agente reductor es trietilsilano, y en donde dicha reacción se lleva a cabo en presencia de un ácido orgánico; opcionalmente en donde dicho ácido orgánico es ácido trifluoroacético.20. El proceso de una cualquiera de las reivindicaciones 18-19, que comprende además desproteger dicho compuesto de Fórmula F18 para proporcionar un compuesto de Fórmula F10: opcionalmente en el que dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F18 con zinc en presencia de ácido acético.21. El proceso de la reivindicación 20, que comprende además hacer reaccionar dicho compuesto de Fórmula F10 con una base para proporcionar un compuesto de Fórmula I:
opcionalmente en el que dicha desprotección comprende hacer reaccionar el compuesto de Fórmula F18 con zinc en presencia de ácido acético.21. El proceso de la reivindicación 20, que comprende además hacer reaccionar dicho compuesto de Fórmula F10 con una base para proporcionar un compuesto de Fórmula I: opcionalmente en el que dicha base es hidróxido de sodio.22. El proceso de la reivindicación 17, en el que R4 es 9H-fluoren-9-ilmetilo.23. El proceso de la reivindicación 22, en el que dicha reacción se lleva a cabo en presencia de un agente reductor; opcionalmente en donde dicho agente reductor es trietilsilano; y en el que dicha reacción se lleva a cabo en presencia de un ácido orgánico; opcionalmente en el que dicho ácido orgánico es ácido metanosulfónico.24. El proceso de una cualquiera de las reivindicaciones 22-23, que comprende además convertir dicho compuesto de Fórmula F18 en un compuesto de Fórmula I:
opcionalmente en el que dicha base es hidróxido de sodio.22. El proceso de la reivindicación 17, en el que R4 es 9H-fluoren-9-ilmetilo.23. El proceso de la reivindicación 22, en el que dicha reacción se lleva a cabo en presencia de un agente reductor; opcionalmente en donde dicho agente reductor es trietilsilano; y en el que dicha reacción se lleva a cabo en presencia de un ácido orgánico; opcionalmente en el que dicho ácido orgánico es ácido metanosulfónico.24. El proceso de una cualquiera de las reivindicaciones 22-23, que comprende además convertir dicho compuesto de Fórmula F18 en un compuesto de Fórmula I: en donde dicha conversión comprende combinar el compuesto de Fórmula F18 con una base para formar una primera mezcla; opcionalmente en el que dicha base es W,A/-bis(2-aminoetilo)etano-1,2-diamina.25. El proceso de la reivindicación 24, en el que dicha conversión comprende además añadir ácido clorhídrico acuoso a dicha primera mezcla.26. Un compuesto de Fórmula F17:
en donde dicha conversión comprende combinar el compuesto de Fórmula F18 con una base para formar una primera mezcla; opcionalmente en el que dicha base es W,A/-bis(2-aminoetilo)etano-1,2-diamina.25. El proceso de la reivindicación 24, en el que dicha conversión comprende además añadir ácido clorhídrico acuoso a dicha primera mezcla.26. Un compuesto de Fórmula F17: en donde R4 es C1-6 haloalquilo, bencilo, o 9H-fluoren-9-ilmetilo.27. El compuesto de la reivindicación 26, que es:(a) N-(2,2-dimetoxietilo)sulfamoilcarbamato de bencilo;(b) 2,2,2-tricloroetilo N-(2,2-dimetoxietilo)sulfamoilcarbamato; o(c) (9H-fluoren-9-ilo)metilo N-(2,2-dimetoxietilo)sulfamoilcarbamato.28. Un compuesto que es:(a) N-(2,2-dimetoxietilo)sulfamoilcarbamato de terc-butilo; o(b) N-(2,2-dimetoxietilo)sulfamoilcarbamato de etilo.29. Un proceso que comprende:i) hacer reaccionar un compuesto de Fórmula F19:
en donde R4 es C1-6 haloalquilo, bencilo, o 9H-fluoren-9-ilmetilo.27. El compuesto de la reivindicación 26, que es:(a) N-(2,2-dimetoxietilo)sulfamoilcarbamato de bencilo;(b) 2,2,2-tricloroetilo N-(2,2-dimetoxietilo)sulfamoilcarbamato; o(c) (9H-fluoren-9-ilo)metilo N-(2,2-dimetoxietilo)sulfamoilcarbamato.28. Un compuesto que es:(a) N-(2,2-dimetoxietilo)sulfamoilcarbamato de terc-butilo; o(b) N-(2,2-dimetoxietilo)sulfamoilcarbamato de etilo.29. Un proceso que comprende:i) hacer reaccionar un compuesto de Fórmula F19: con un compuesto de Fórmula F5:
con un compuesto de Fórmula F5: en presencia de trietilsilano y ácido metanosulfónico para proporcionar un compuesto de Fórmula F20:
en presencia de trietilsilano y ácido metanosulfónico para proporcionar un compuesto de Fórmula F20:
 en donde dicha conversión comprende combinar dicho compuesto de Fórmula F20 con N, N-bis(2-aminoetNo)etano-1,2-diamina.
en donde dicha conversión comprende combinar dicho compuesto de Fórmula F20 con N, N-bis(2-aminoetNo)etano-1,2-diamina.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361901689P | 2013-11-08 | 2013-11-08 | |
| PCT/US2014/064531 WO2015070007A1 (en) | 2013-11-08 | 2014-11-07 | Process for the synthesis of an indoleamine 2,3-dioxygenase inhibitor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2799582T3 true ES2799582T3 (es) | 2020-12-18 |
Family
ID=52023608
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14812015T Active ES2799582T3 (es) | 2013-11-08 | 2014-11-07 | Proceso para la síntesis de un inhibidor de indoleamina 2,3-dioxigenasa |
Country Status (31)
| Country | Link |
|---|---|
| US (3) | US9321755B2 (es) |
| EP (2) | EP3066085B1 (es) |
| JP (4) | JP6461953B2 (es) |
| KR (2) | KR102617531B1 (es) |
| CN (2) | CN105899498B (es) |
| AR (1) | AR098343A1 (es) |
| AU (2) | AU2014346647B2 (es) |
| BR (2) | BR122020009912B1 (es) |
| CA (1) | CA2929552C (es) |
| CL (1) | CL2016001082A1 (es) |
| CR (2) | CR20160252A (es) |
| CY (1) | CY1123164T1 (es) |
| DK (1) | DK3066085T3 (es) |
| EA (2) | EA201991770A1 (es) |
| ES (1) | ES2799582T3 (es) |
| HR (1) | HRP20201089T1 (es) |
| HU (1) | HUE049337T2 (es) |
| IL (2) | IL245314B (es) |
| LT (1) | LT3066085T (es) |
| ME (1) | ME03792B (es) |
| MX (2) | MX366874B (es) |
| MY (1) | MY174254A (es) |
| PE (2) | PE20220430A1 (es) |
| PH (2) | PH12016500818B1 (es) |
| PL (1) | PL3066085T3 (es) |
| PT (1) | PT3066085T (es) |
| RS (1) | RS60598B1 (es) |
| SG (2) | SG11201603433UA (es) |
| SI (1) | SI3066085T1 (es) |
| TW (3) | TWI696618B (es) |
| WO (1) | WO2015070007A1 (es) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006122150A1 (en) | 2005-05-10 | 2006-11-16 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| LT2824100T (lt) | 2008-07-08 | 2018-05-10 | Incyte Holdings Corporation | 1,2,5-oksadiazolai, kaip indolamino 2,3-dioksigenazės inhibitoriai |
| GB201311891D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compound |
| GB201311888D0 (en) | 2013-07-03 | 2013-08-14 | Glaxosmithkline Ip Dev Ltd | Novel compounds |
| AR098343A1 (es) | 2013-11-08 | 2016-05-26 | Incyte Holdings Corp | Proceso para la síntesis de un inhibidor de indolamina 2,3-dioxigenasa |
| MX2016010080A (es) * | 2014-02-04 | 2016-10-07 | Incyte Corp | Combinacion de un antagonista de muerte programada-1 y un inhibidor de indolamina 2,3-dioxigenasa 1 para el tratamiento del cancer. |
| KR20180025897A (ko) * | 2015-07-14 | 2018-03-09 | 교와 핫꼬 기린 가부시키가이샤 | 항체와 조합 투여되는 ido 억제제를 포함하는 종양 치료제 |
| MX2018005600A (es) | 2015-11-04 | 2018-11-09 | Incyte Corp | Composiciones farmaceuticas y metodos para la inhibicion de indolamina 2,3-dioxigenasa e indicaciones para ello. |
| WO2017106062A1 (en) * | 2015-12-15 | 2017-06-22 | Merck Sharp & Dohme Corp. | Novel compounds as indoleamine 2,3-dioxygenase inhibitors |
| US9624185B1 (en) * | 2016-01-20 | 2017-04-18 | Yong Xu | Method for preparing IDO inhibitor epacadostat |
| CN105646389B (zh) * | 2016-01-28 | 2019-06-28 | 中国科学院上海有机化学研究所 | 一种作为吲哚胺-2,3-双加氧酶抑制剂的氨基磺酸脂及其制备方法和用途 |
| CN107304191B (zh) * | 2016-04-20 | 2023-09-29 | 上海翰森生物医药科技有限公司 | 吲哚胺2,3-双加氧酶抑制剂及其制备方法与应用 |
| WO2017181849A1 (zh) * | 2016-04-20 | 2017-10-26 | 江苏豪森药业集团有限公司 | 吲哚胺2,3-双加氧酶抑制剂及其制备方法与应用 |
| WO2018036414A1 (zh) | 2016-08-23 | 2018-03-01 | 北京诺诚健华医药科技有限公司 | 稠杂环类衍生物、其制备方法及其在医学上的应用 |
| EP3515914A4 (en) | 2016-09-24 | 2020-04-15 | BeiGene, Ltd. | NEW IMIDAZO [1,5-A] PYRIDINES SUBSTITUTED IN POSITION 5 OR 8 AS INDOLEAMINE AND / OR TRYPTOPHANE 2,3-DIOXYGENASES |
| WO2018095432A1 (en) | 2016-11-28 | 2018-05-31 | Shanghai Fochon Pharmaceutical Co., Ltd. | Sulfoximine, sulfonimidamide, sulfondiimine and diimidosulfonamide compounds as inhibitors of indoleamine 2, 3-dioxygenase |
| PT3559009T (pt) | 2016-12-22 | 2021-05-04 | Calithera Biosciences Inc | Composições e métodos para inibir a atividade da arginase |
| CN109206380A (zh) * | 2017-07-03 | 2019-01-15 | 上海时莱生物技术有限公司 | 吲哚胺2,3-双加氧酶抑制剂化合物及其制备方法和用途 |
| CN110066253B (zh) * | 2018-01-24 | 2023-06-23 | 江苏柯菲平医药股份有限公司 | 1,2,5-噁二唑类衍生物,其制备方法及其在医药中的应用 |
| CN108101899B (zh) * | 2018-02-11 | 2021-01-26 | 中国药科大学 | IDO1抑制剂Epacadostat中间体的制备方法 |
| CN109180603A (zh) * | 2018-10-10 | 2019-01-11 | 中国药科大学 | Epacadostat关键中间体的制备方法 |
| JP2022543062A (ja) | 2019-08-01 | 2022-10-07 | インサイト・コーポレイション | Ido阻害剤の投与レジメン |
| AU2020383618A1 (en) * | 2019-11-14 | 2022-06-16 | Nova Southeastern University | Methods and compositions for treatment of solid tumors using F16 isoindole small molecules |
| CN112341403B (zh) * | 2020-11-10 | 2022-06-28 | 南京工业大学 | 一种利用微流场反应技术制备3-氨基-4-偕胺肟基呋咱的方法 |
| WO2022184930A2 (en) | 2021-03-05 | 2022-09-09 | Universität Basel | Compositions for the treatment of ebv associated diseases or conditions |
| EP4052705A1 (en) | 2021-03-05 | 2022-09-07 | Universität Basel Vizerektorat Forschung | Compositions for the treatment of ebv associated diseases or conditions |
Family Cites Families (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3236855A (en) | 1964-01-17 | 1966-02-22 | Merck & Co Inc | Certain n-phenyl(thiazole-hydroxamidine) compounds and their preparation |
| US3354174A (en) | 1964-02-19 | 1967-11-21 | Sterling Drug Inc | 1-and 2-benzimidazolyl-lower-alkylamidoximes, amidines-, and guanidines |
| US3553228A (en) | 1967-09-26 | 1971-01-05 | Colgate Palmolive Co | 3-substituted-4h(1)benzopyrano(3,4-d) isoxazoles |
| DE2040628A1 (de) | 1970-08-17 | 1972-02-24 | Fahlberg List Veb | Neue herbizide Mittel |
| US3948928A (en) | 1972-03-17 | 1976-04-06 | Dainippon Pharmaceutical Co., Ltd. | 3-Substituted-1,2-benzisoxazoles and pharmaceutically acceptable acid addition salts thereof |
| JPS5621033B2 (es) | 1973-09-05 | 1981-05-16 | ||
| FR2323683A1 (fr) | 1975-09-11 | 1977-04-08 | Philagro Sa | Nouveaux derives d'amidoximes, leur preparation et les compositions qui les contiennent |
| DD128918A5 (de) | 1975-09-11 | 1977-12-21 | Philagro Sa | Phythohormonale und herbizide zusammensetzungen |
| US4323681A (en) | 1980-09-29 | 1982-04-06 | American Home Products Corporation | 4-Amino-2-substituted-5-pyrimidinecarboxamidoximes and carbothioamides |
| JPS58208275A (ja) | 1982-05-20 | 1983-12-03 | Lion Corp | 5−アミノ−ピラゾ−ル誘導体及び該化合物を含有する抗腫瘍剤 |
| DE3462259D1 (de) | 1983-07-22 | 1987-03-05 | Bayer Ag | Substituted furazans |
| US4507485A (en) | 1984-01-23 | 1985-03-26 | Bristol-Myers Company | 3,4-Disubstituted-1,2,5-oxadiazoles having histamine H2 -receptor antagonist activity |
| JPS60193968A (ja) | 1984-03-13 | 1985-10-02 | Toyo Jozo Co Ltd | シクロペンテン環を有するイミダゾ−ルアミドオキシムおよびその製造法 |
| JPS6259283A (ja) | 1985-09-10 | 1987-03-14 | Kaken Pharmaceut Co Ltd | セフアロスポリン化合物 |
| JP2696342B2 (ja) | 1988-06-27 | 1998-01-14 | 日本曹達株式会社 | アミジン誘導体、その製造方法及び殺ダニ剤・農園芸用殺菌剤 |
| DE68901424D1 (de) | 1988-07-05 | 1992-06-11 | Akzo Nv | Verbindungen mit bronchodilatatorischer aktivitaet. |
| JPH04297449A (ja) | 1991-03-27 | 1992-10-21 | Hokko Chem Ind Co Ltd | N−ヒドロキシベンジルグアニジン誘導体および農園芸用殺菌剤 |
| JPH05186458A (ja) | 1991-04-26 | 1993-07-27 | Japan Tobacco Inc | 新規なベンゾピラン誘導体 |
| FR2677019B1 (fr) | 1991-05-27 | 1994-11-25 | Pf Medicament | Nouvelles piperidines disubstituees-1,4, leur preparation et leur application en therapeutique. |
| JP2709677B2 (ja) | 1992-06-19 | 1998-02-04 | 株式会社大塚製薬工場 | ホスホン酸ジエステル誘導体 |
| FR2720396B1 (fr) | 1994-05-27 | 1996-06-28 | Adir | Nouveaux N-pyridyl carboxamides et dérivés leur procédé de préparation et les compositions pharmaceutiques qui les contiennent. |
| US5731315A (en) | 1995-06-07 | 1998-03-24 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Substituted sulfonic acid n- (aminoiminomethyl)phenylalkyl!-azaheterocyclamide compounds |
| US5883102A (en) | 1995-10-17 | 1999-03-16 | Astra Pharmaceuticals Limited | Pharmaceutically active compounds |
| AU1730497A (en) | 1996-02-17 | 1997-09-02 | Agrevo Uk Limited | Fungicidal 1,2,4-oxadiazoles and analogues |
| US5955495A (en) | 1996-05-03 | 1999-09-21 | Hoffmann-La Roche Inc. | Method of treating diseases of the CNS |
| JPH11171702A (ja) | 1997-09-24 | 1999-06-29 | Takeda Chem Ind Ltd | 害虫防除方法 |
| WO1999029310A2 (en) | 1997-12-05 | 1999-06-17 | Medical College Of Georgia Research Institute, Inc. | Regulation of t cell-mediated immunity by tryptophan and its analogs |
| WO1999029689A1 (fr) | 1997-12-10 | 1999-06-17 | Dainippon Ink And Chemicals, Inc. | Derives d'oxime et produits chimiques agricoles les contenant |
| DE69902387T2 (de) | 1998-06-02 | 2003-02-13 | Takeda Chemical Industries, Ltd. | Oxadiazolinderivate und ihre verwendung als insektizide |
| FR2784678B1 (fr) | 1998-09-23 | 2002-11-29 | Sod Conseils Rech Applic | Nouveaux derives de n-(iminomethyl)amines, leur preparation, leur application a titre de medicaments et les compositions pharmaceutiques les contenant |
| KR100396738B1 (ko) | 1999-03-03 | 2003-09-03 | 삼진제약주식회사 | 피페라진 유도체 및 그 제조방법 |
| WO2000061609A2 (de) | 1999-04-09 | 2000-10-19 | Basf Aktiengesellschaft | Prodrugs von thrombininhibitoren |
| CA2716369C (en) | 1999-05-24 | 2013-05-14 | Mitsubishi Tanabe Pharma Corporation | Phenoxypropylamine compounds |
| JP2001158786A (ja) | 1999-11-30 | 2001-06-12 | Takeda Chem Ind Ltd | 哺乳動物の外部寄生虫防除剤 |
| JP2001158785A (ja) | 1999-11-30 | 2001-06-12 | Takeda Chem Ind Ltd | 農薬組成物 |
| ES2293980T3 (es) | 2000-01-13 | 2008-04-01 | Amgen Inc. | Agentes antibacterianos. |
| JP2001233861A (ja) | 2000-02-22 | 2001-08-28 | Ube Ind Ltd | ピラゾールオキシム化合物、その製法及び用途 |
| HU230396B1 (hu) | 2000-06-28 | 2016-04-28 | Smithkline Beecham Plc | Nedves őrlési eljárás |
| GB0108102D0 (en) | 2001-03-30 | 2001-05-23 | Pfizer Ltd | Compounds |
| SK15552003A3 (sk) | 2001-06-18 | 2004-05-04 | Applied Research Systems Ars Holding N. V. | Oxadiazolové a tiadiazolové deriváty pyrolidínu, spôsob ich výroby, farmaceutický prostriedok s ich obsahom a ich použitie |
| AU2003218989A1 (en) | 2002-02-19 | 2003-09-09 | Pharmacia Italia S.P.A. | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents |
| GB0208224D0 (en) | 2002-04-10 | 2002-05-22 | Celltech R&D Ltd | Chemical compounds |
| AU2002307243B2 (en) | 2002-04-12 | 2008-01-03 | Medical College Of Georgia Research Institute, Inc. | Antigen-presenting cell populations and their use as reagents for enhancing or reducing immune tolerance |
| MY137509A (en) | 2002-05-28 | 2009-02-27 | Dimensional Pharm Inc | Thiophene amidines, compositions thereof, and methods of treating complement-mediated diseases and conditions |
| US7157462B2 (en) | 2002-09-24 | 2007-01-02 | Euro-Celtique S.A. | Therapeutic agents useful for treating pain |
| US7015321B2 (en) * | 2002-10-12 | 2006-03-21 | The Scripps Research Institute | Synthesis of non-symmetrical sulfamides using burgess-type reagents |
| EP2260846B1 (en) | 2003-03-27 | 2018-11-28 | Lankenau Institute for Medical Research | Novel methods for the treatment of cancer |
| US7598287B2 (en) | 2003-04-01 | 2009-10-06 | Medical College Of Georgia Research Institute, Inc. | Use of inhibitors of indoleamine-2,3-dioxygenase in combination with other therapeutic modalities |
| JP2007512230A (ja) | 2003-08-20 | 2007-05-17 | バーテックス ファーマシューティカルズ インコーポレイテッド | プロテインキナーゼ阻害剤として有用な(4−アミノ−1,2,5−オキサジアゾール−4−イル)−ヘテロ芳香族化合物 |
| DE10348023A1 (de) | 2003-10-15 | 2005-05-19 | Imtm Gmbh | Neue Alanyl-Aminopeptidasen-Inhibitoren zur funktionellen Beeinflussung unterschiedlicher Zellen und zur Behandlung immunologischer, entzündlicher, neuronaler und anderer Erkrankungen |
| DE10348022A1 (de) | 2003-10-15 | 2005-05-25 | Imtm Gmbh | Neue Dipeptidylpeptidase IV-Inhibitoren zur funktionellen Beeinflussung unterschiedlicher Zellen und zur Behandlung immunologischer, entzündlicher, neuronaler und anderer Erkrankungen |
| US7151097B2 (en) * | 2003-11-07 | 2006-12-19 | Pfizer Inc. | Bicyclic pyrazolyl and imidazolyl compounds and uses thereof |
| PT1801108E (pt) | 2004-09-08 | 2012-12-03 | Mitsubishi Tanabe Pharma Corp | Composto de morfolina para o tratamento de inflamações |
| JP5065908B2 (ja) | 2004-12-24 | 2012-11-07 | プロシディオン・リミテッド | Gタンパク質結合受容体作動薬 |
| US7429667B2 (en) | 2005-01-20 | 2008-09-30 | Ardea Biosciences, Inc. | Phenylamino isothiazole carboxamidines as MEK inhibitors |
| WO2006122150A1 (en) | 2005-05-10 | 2006-11-16 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| WO2006133417A1 (en) | 2005-06-07 | 2006-12-14 | Valeant Pharmaceuticals International | Phenylamino isothiazole carboxamidines as mek inhibitors |
| JP2008544818A (ja) | 2005-06-30 | 2008-12-11 | アントフロゲネシス コーポレーション | 胎盤由来コラーゲンバイオ線維を用いた鼓膜の修復 |
| DE102005060466A1 (de) | 2005-12-17 | 2007-06-28 | Bayer Cropscience Ag | Carboxamide |
| EP1971583B1 (en) | 2005-12-20 | 2015-03-25 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| US20070203140A1 (en) | 2006-02-09 | 2007-08-30 | Combs Andrew P | N-hydroxyguanidines as modulators of indoleamine 2,3-dioxygenase |
| JP4297449B2 (ja) | 2006-05-12 | 2009-07-15 | 株式会社サクラクレパス | 墨汁 |
| CL2007002650A1 (es) | 2006-09-19 | 2008-02-08 | Incyte Corp | Compuestos derivados de heterociclo n-hidroxiamino; composicion farmaceutica, util para tratar cancer, infecciones virales y desordenes neurodegenerativos entre otras. |
| US20080119491A1 (en) | 2006-09-19 | 2008-05-22 | Incyte Corporation | Amidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| JP5319532B2 (ja) | 2006-09-19 | 2013-10-16 | インサイト・コーポレイション | インドールアミン2,3−ジオキシゲナーゼのモジュレーターとしてのn−ヒドロキシアミジノヘテロサイクル |
| US20080146624A1 (en) | 2006-09-19 | 2008-06-19 | Incyte Corporation | Amidines as modulators of indoleamine 2,3-dioxygenase |
| WO2008058178A1 (en) * | 2006-11-08 | 2008-05-15 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| CN101679297B (zh) | 2006-12-08 | 2012-01-11 | 埃克塞利希斯股份有限公司 | Lxr和fxr调节剂 |
| CL2008000066A1 (es) | 2007-01-12 | 2008-08-01 | Smithkline Beecham Corp | Compuestos derivados de (5-hidroxi-3-oxo-2,3-dihidropiridazina-4-carbonil)glicina, inhibidores de hif prolil hidroxilasas; procedimiento de preparacion; composicion farmaceutica que comprende a dichos compuestos; y su uso en el tratamiento de la anem |
| JP2011507910A (ja) | 2007-12-21 | 2011-03-10 | ユニバーシティー オブ ロチェスター | 真核生物の寿命を変更するための方法 |
| WO2009128521A1 (ja) * | 2008-04-15 | 2009-10-22 | 帝人ファーマ株式会社 | システインプロテアーゼ阻害剤 |
| LT2824100T (lt) | 2008-07-08 | 2018-05-10 | Incyte Holdings Corporation | 1,2,5-oksadiazolai, kaip indolamino 2,3-dioksigenazės inhibitoriai |
| AR098343A1 (es) | 2013-11-08 | 2016-05-26 | Incyte Holdings Corp | Proceso para la síntesis de un inhibidor de indolamina 2,3-dioxigenasa |
-
2014
- 2014-11-07 AR ARP140104196A patent/AR098343A1/es unknown
- 2014-11-07 TW TW108102217A patent/TWI696618B/zh active
- 2014-11-07 TW TW103138838A patent/TWI651311B/zh active
- 2014-11-07 EA EA201991770A patent/EA201991770A1/ru unknown
- 2014-11-07 ES ES14812015T patent/ES2799582T3/es active Active
- 2014-11-07 RS RS20200798A patent/RS60598B1/sr unknown
- 2014-11-07 HU HUE14812015A patent/HUE049337T2/hu unknown
- 2014-11-07 CN CN201480071825.0A patent/CN105899498B/zh active Active
- 2014-11-07 PE PE2021001747A patent/PE20220430A1/es unknown
- 2014-11-07 MY MYPI2016000827A patent/MY174254A/en unknown
- 2014-11-07 US US14/535,781 patent/US9321755B2/en active Active
- 2014-11-07 BR BR122020009912-7A patent/BR122020009912B1/pt active IP Right Grant
- 2014-11-07 KR KR1020217032616A patent/KR102617531B1/ko active IP Right Grant
- 2014-11-07 CR CR20160252A patent/CR20160252A/es unknown
- 2014-11-07 MX MX2016005954A patent/MX366874B/es active IP Right Grant
- 2014-11-07 SG SG11201603433UA patent/SG11201603433UA/en unknown
- 2014-11-07 PT PT148120157T patent/PT3066085T/pt unknown
- 2014-11-07 BR BR112016009786-6A patent/BR112016009786B1/pt active IP Right Grant
- 2014-11-07 EP EP14812015.7A patent/EP3066085B1/en active Active
- 2014-11-07 EA EA201690959A patent/EA033667B1/ru unknown
- 2014-11-07 ME MEP-2020-146A patent/ME03792B/me unknown
- 2014-11-07 JP JP2016528055A patent/JP6461953B2/ja active Active
- 2014-11-07 LT LTEP14812015.7T patent/LT3066085T/lt unknown
- 2014-11-07 CN CN201910026055.2A patent/CN109810104B/zh active Active
- 2014-11-07 SG SG10201803874PA patent/SG10201803874PA/en unknown
- 2014-11-07 CA CA2929552A patent/CA2929552C/en active Active
- 2014-11-07 CR CR20190351A patent/CR20190351A/es unknown
- 2014-11-07 PE PE2016000604A patent/PE20160863A1/es unknown
- 2014-11-07 KR KR1020167015077A patent/KR102370067B1/ko active IP Right Grant
- 2014-11-07 DK DK14812015.7T patent/DK3066085T3/da active
- 2014-11-07 EP EP20169098.9A patent/EP3744715A1/en active Pending
- 2014-11-07 WO PCT/US2014/064531 patent/WO2015070007A1/en active Application Filing
- 2014-11-07 AU AU2014346647A patent/AU2014346647B2/en active Active
- 2014-11-07 SI SI201431604T patent/SI3066085T1/sl unknown
- 2014-11-07 PL PL14812015T patent/PL3066085T3/pl unknown
- 2014-11-07 TW TW109116698A patent/TWI775079B/zh active
-
2016
- 2016-04-07 US US15/093,486 patent/US9873688B2/en active Active
- 2016-04-27 IL IL245314A patent/IL245314B/en active IP Right Grant
- 2016-05-02 PH PH12016500818A patent/PH12016500818B1/en unknown
- 2016-05-05 CL CL2016001082A patent/CL2016001082A1/es unknown
- 2016-05-06 MX MX2019008378A patent/MX2019008378A/es unknown
-
2017
- 2017-12-22 US US15/852,995 patent/US10280157B2/en active Active
-
2018
- 2018-10-02 JP JP2018187218A patent/JP6633163B2/ja active Active
-
2019
- 2019-01-21 AU AU2019200404A patent/AU2019200404B2/en active Active
- 2019-04-10 PH PH12019500770A patent/PH12019500770A1/en unknown
- 2019-07-01 IL IL267773A patent/IL267773B/en active IP Right Grant
- 2019-10-07 JP JP2019184589A patent/JP6883079B2/ja active Active
- 2019-10-07 JP JP2019184577A patent/JP6913725B2/ja active Active
-
2020
- 2020-07-13 HR HRP20201089TT patent/HRP20201089T1/hr unknown
- 2020-08-05 CY CY20201100726T patent/CY1123164T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2799582T3 (es) | Proceso para la síntesis de un inhibidor de indoleamina 2,3-dioxigenasa | |
| ES2524266T3 (es) | 1,2,5-Oxadiazoles como inhibidores de la indoleamina 2,3-dioxigenasa | |
| ES2444574T3 (es) | N-hidroxiamidinoheterociclos como moduladores de la indolamina 2,3-dioxigenasa | |
| ES2540561T3 (es) | N-hidroxiamidinoheterociclos como moduladores de indolamina 2,3-dioxigenasa | |
| ES2704641T3 (es) | Compuestos N-hidroxiamidinoheterocíclicos como moduladores de la indoleamina 2,3-dioxigenasa |