ES2735729T3 - Macrociclos de diarilo como moduladores de proteínas cinasas - Google Patents
Macrociclos de diarilo como moduladores de proteínas cinasas Download PDFInfo
- Publication number
- ES2735729T3 ES2735729T3 ES15740510T ES15740510T ES2735729T3 ES 2735729 T3 ES2735729 T3 ES 2735729T3 ES 15740510 T ES15740510 T ES 15740510T ES 15740510 T ES15740510 T ES 15740510T ES 2735729 T3 ES2735729 T3 ES 2735729T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- fluoro
- ethenopyrazolo
- tetrahydro
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 Diaryl macrocycles Chemical class 0.000 title claims abstract description 102
- 102000001253 Protein Kinase Human genes 0.000 title description 6
- 108060006633 protein kinase Proteins 0.000 title description 6
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 315
- 150000001875 compounds Chemical class 0.000 claims abstract description 184
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims abstract description 99
- 229910052805 deuterium Inorganic materials 0.000 claims abstract description 99
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims abstract description 89
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 66
- 150000003839 salts Chemical class 0.000 claims abstract description 60
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 57
- 150000002367 halogens Chemical group 0.000 claims abstract description 55
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims abstract description 47
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 47
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 45
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 43
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 41
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims abstract description 39
- 125000002950 monocyclic group Chemical group 0.000 claims abstract description 37
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 36
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 33
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 28
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 27
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims abstract description 26
- 125000001153 fluoro group Chemical group F* 0.000 claims abstract description 23
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 23
- 125000006578 monocyclic heterocycloalkyl group Chemical group 0.000 claims abstract description 14
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims abstract description 10
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 44
- 125000003118 aryl group Chemical group 0.000 claims description 43
- 238000011282 treatment Methods 0.000 claims description 43
- 206010028980 Neoplasm Diseases 0.000 claims description 28
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 24
- 201000011510 cancer Diseases 0.000 claims description 21
- 239000008194 pharmaceutical composition Substances 0.000 claims description 20
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 18
- 229910052739 hydrogen Inorganic materials 0.000 claims description 15
- 125000002619 bicyclic group Chemical group 0.000 claims description 14
- 208000002193 Pain Diseases 0.000 claims description 11
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 claims description 10
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 claims description 10
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 9
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 claims description 8
- 206010061218 Inflammation Diseases 0.000 claims description 7
- 230000004054 inflammatory process Effects 0.000 claims description 7
- 208000023275 Autoimmune disease Diseases 0.000 claims description 6
- 208000012902 Nervous system disease Diseases 0.000 claims description 6
- 208000025966 Neurological disease Diseases 0.000 claims description 6
- JEGVYOJIPWPTPT-UHFFFAOYSA-N O1C=CC(N=CC=CN=CN=CC2=C1C=CC=C2)=O Chemical compound O1C=CC(N=CC=CN=CN=CC2=C1C=CC=C2)=O JEGVYOJIPWPTPT-UHFFFAOYSA-N 0.000 claims description 4
- SUEAKNISOMJCOJ-UHFFFAOYSA-N O1ONN=CC=CC=CC=CC2=C1C=CC=C2 Chemical compound O1ONN=CC=CC=CC=CC2=C1C=CC=C2 SUEAKNISOMJCOJ-UHFFFAOYSA-N 0.000 claims description 3
- 125000005843 halogen group Chemical group 0.000 claims description 3
- YMRTYJKGNCAGKP-UHFFFAOYSA-N 5-chloro-6-fluoro-3-methyl-2,10-dioxa-13,17,18,21-tetrazatetracyclo[13.5.2.04,9.018,22]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one Chemical compound CC1OC2=NC3=C(C=NN3C=C2)C(=O)NCCOC2=C1C(Cl)=C(F)C=C2 YMRTYJKGNCAGKP-UHFFFAOYSA-N 0.000 claims description 2
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 abstract 27
- 125000000041 C6-C10 aryl group Chemical group 0.000 abstract 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 abstract 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 abstract 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 223
- 239000000203 mixture Substances 0.000 description 201
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 153
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 147
- 239000000243 solution Substances 0.000 description 141
- 238000006243 chemical reaction Methods 0.000 description 97
- 235000019439 ethyl acetate Nutrition 0.000 description 96
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 83
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 79
- 239000011734 sodium Substances 0.000 description 77
- 238000000034 method Methods 0.000 description 73
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 68
- 239000007787 solid Substances 0.000 description 57
- 239000012074 organic phase Substances 0.000 description 53
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 48
- 210000004027 cell Anatomy 0.000 description 45
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 44
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 43
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 42
- 239000012267 brine Substances 0.000 description 42
- 230000002829 reductive effect Effects 0.000 description 41
- 239000011541 reaction mixture Substances 0.000 description 40
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 35
- 239000000047 product Substances 0.000 description 33
- 239000007858 starting material Substances 0.000 description 30
- 239000000284 extract Substances 0.000 description 29
- 239000012299 nitrogen atmosphere Substances 0.000 description 28
- 239000000741 silica gel Substances 0.000 description 28
- 229910002027 silica gel Inorganic materials 0.000 description 28
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 27
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 26
- 238000004809 thin layer chromatography Methods 0.000 description 26
- 125000001624 naphthyl group Chemical group 0.000 description 24
- 239000000460 chlorine Substances 0.000 description 22
- 238000005481 NMR spectroscopy Methods 0.000 description 21
- 238000003818 flash chromatography Methods 0.000 description 21
- 201000010099 disease Diseases 0.000 description 20
- 239000002904 solvent Substances 0.000 description 19
- 125000001424 substituent group Chemical group 0.000 description 19
- 230000015572 biosynthetic process Effects 0.000 description 18
- 239000000377 silicon dioxide Substances 0.000 description 18
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 description 17
- 102100037236 Tyrosine-protein kinase receptor UFO Human genes 0.000 description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 16
- 101000779641 Homo sapiens ALK tyrosine kinase receptor Proteins 0.000 description 16
- 101000807561 Homo sapiens Tyrosine-protein kinase receptor UFO Proteins 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 239000002253 acid Substances 0.000 description 15
- 239000003814 drug Substances 0.000 description 15
- 238000003786 synthesis reaction Methods 0.000 description 15
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 14
- 150000001299 aldehydes Chemical class 0.000 description 14
- 208000024891 symptom Diseases 0.000 description 14
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 13
- 102100035108 High affinity nerve growth factor receptor Human genes 0.000 description 13
- 101000596894 Homo sapiens High affinity nerve growth factor receptor Proteins 0.000 description 13
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 13
- 108091000080 Phosphotransferase Proteins 0.000 description 13
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 13
- 102000020233 phosphotransferase Human genes 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 230000035772 mutation Effects 0.000 description 12
- 239000003208 petroleum Substances 0.000 description 12
- 229940002612 prodrug Drugs 0.000 description 12
- 239000000651 prodrug Substances 0.000 description 12
- 108090000623 proteins and genes Proteins 0.000 description 12
- 238000010898 silica gel chromatography Methods 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 11
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 11
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 11
- 239000004480 active ingredient Substances 0.000 description 11
- 125000000304 alkynyl group Chemical group 0.000 description 11
- 239000002585 base Substances 0.000 description 11
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 11
- 239000003112 inhibitor Substances 0.000 description 11
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 11
- OOWSDKUFKGVADH-UHFFFAOYSA-N 1-diphenylphosphoryloxy-2,3,4,5,6-pentafluorobenzene Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1OP(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 OOWSDKUFKGVADH-UHFFFAOYSA-N 0.000 description 10
- 238000005160 1H NMR spectroscopy Methods 0.000 description 10
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 10
- 125000002757 morpholinyl group Chemical group 0.000 description 10
- 125000004193 piperazinyl group Chemical group 0.000 description 10
- 125000003386 piperidinyl group Chemical group 0.000 description 10
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- 239000007821 HATU Substances 0.000 description 9
- 101000686031 Homo sapiens Proto-oncogene tyrosine-protein kinase ROS Proteins 0.000 description 9
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- 125000003342 alkenyl group Chemical group 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 239000003446 ligand Substances 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 125000006413 ring segment Chemical group 0.000 description 9
- 229920006395 saturated elastomer Chemical group 0.000 description 9
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 8
- 102100023347 Proto-oncogene tyrosine-protein kinase ROS Human genes 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- UOZKVCQDLXBWBG-UHFFFAOYSA-N ethyl 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate Chemical compound C1=CC(Cl)=NC2=C(C(=O)OCC)C=NN21 UOZKVCQDLXBWBG-UHFFFAOYSA-N 0.000 description 8
- 238000000634 powder X-ray diffraction Methods 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 8
- 101000850794 Homo sapiens Tropomyosin alpha-3 chain Proteins 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 239000007795 chemical reaction product Substances 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 238000010790 dilution Methods 0.000 description 7
- 239000012895 dilution Substances 0.000 description 7
- 238000005516 engineering process Methods 0.000 description 7
- 239000012458 free base Substances 0.000 description 7
- 125000002541 furyl group Chemical group 0.000 description 7
- 238000011534 incubation Methods 0.000 description 7
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 7
- 239000003960 organic solvent Substances 0.000 description 7
- 238000002360 preparation method Methods 0.000 description 7
- 125000005032 thiofuranyl group Chemical group S1C(=CC=C1)* 0.000 description 7
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 230000002159 abnormal effect Effects 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- 230000005754 cellular signaling Effects 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 229960005061 crizotinib Drugs 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 125000001188 haloalkyl group Chemical group 0.000 description 6
- 239000005457 ice water Substances 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 6
- 125000003367 polycyclic group Chemical group 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 238000004007 reversed phase HPLC Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 229920001213 Polysorbate 20 Polymers 0.000 description 5
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 5
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 5
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 125000001309 chloro group Chemical group Cl* 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 235000019441 ethanol Nutrition 0.000 description 5
- 239000012091 fetal bovine serum Substances 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 230000004927 fusion Effects 0.000 description 5
- 208000020816 lung neoplasm Diseases 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 5
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- HOMSZIFULUHWLN-UHFFFAOYSA-N 2-chloroethylcarbamic acid Chemical compound OC(=O)NCCCl HOMSZIFULUHWLN-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 4
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 4
- 239000005909 Kieselgur Substances 0.000 description 4
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 4
- 101150111783 NTRK1 gene Proteins 0.000 description 4
- 229930182555 Penicillin Natural products 0.000 description 4
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical class OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 4
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 229940043355 kinase inhibitor Drugs 0.000 description 4
- 201000005202 lung cancer Diseases 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 4
- 229940049954 penicillin Drugs 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 230000011664 signaling Effects 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 229960005322 streptomycin Drugs 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- NFRQMEOKZASTGF-UHFFFAOYSA-N 4-fluoro-2-(methylaminomethyl)phenol Chemical compound CNCC1=CC(F)=CC=C1O NFRQMEOKZASTGF-UHFFFAOYSA-N 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 101150022345 GAS6 gene Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 3
- 229940122245 Janus kinase inhibitor Drugs 0.000 description 3
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- 239000005089 Luciferase Substances 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- 108010017324 STAT3 Transcription Factor Proteins 0.000 description 3
- 102100024040 Signal transducer and activator of transcription 3 Human genes 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 3
- 208000027418 Wounds and injury Diseases 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 238000009739 binding Methods 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 238000001516 cell proliferation assay Methods 0.000 description 3
- 150000005829 chemical entities Chemical class 0.000 description 3
- 125000003636 chemical group Chemical group 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 238000000113 differential scanning calorimetry Methods 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 229940121647 egfr inhibitor Drugs 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 229960001433 erlotinib Drugs 0.000 description 3
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 3
- AVYSSBABIUPEMY-UHFFFAOYSA-N ethyl 6-chloroimidazo[1,2-b]pyridazine-3-carboxylate Chemical compound C1=CC(Cl)=NN2C(C(=O)OCC)=CN=C21 AVYSSBABIUPEMY-UHFFFAOYSA-N 0.000 description 3
- YCAVTOUTYRMOGQ-UHFFFAOYSA-N ethyl 7-[3-[[3-(trifluoromethyl)benzoyl]amino]phenyl]pyrazolo[1,5-a]pyrimidine-3-carboxylate Chemical compound C=1C=NC2=C(C(=O)OCC)C=NN2C=1C(C=1)=CC=CC=1NC(=O)C1=CC=CC(C(F)(F)F)=C1 YCAVTOUTYRMOGQ-UHFFFAOYSA-N 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000001605 fetal effect Effects 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000007903 gelatin capsule Substances 0.000 description 3
- 230000014509 gene expression Effects 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 208000005017 glioblastoma Diseases 0.000 description 3
- 238000003384 imaging method Methods 0.000 description 3
- 208000014674 injury Diseases 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 125000004043 oxo group Chemical group O=* 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 230000003449 preventive effect Effects 0.000 description 3
- 239000003531 protein hydrolysate Substances 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 3
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical class OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000012265 solid product Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 229940032147 starch Drugs 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- VACLTXTYDFLHJW-UHFFFAOYSA-N tert-butyl n-(2-chloroethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCl VACLTXTYDFLHJW-UHFFFAOYSA-N 0.000 description 3
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 3
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- XEBJNAQDHRTAHP-UHFFFAOYSA-N 2-[C-(cyclopropylmethyl)-N-hydroxycarbonimidoyl]-4-fluorophenol Chemical compound C1(CC1)CC(=NO)C1=C(C=CC(=C1)F)O XEBJNAQDHRTAHP-UHFFFAOYSA-N 0.000 description 2
- BIJQDOONYNJEHP-UHFFFAOYSA-N 2-chloro-3-fluoro-6-hydroxybenzaldehyde Chemical compound OC1=CC=C(F)C(Cl)=C1C=O BIJQDOONYNJEHP-UHFFFAOYSA-N 0.000 description 2
- BLTRISANKPFBEV-UHFFFAOYSA-N 2-cyclopropyl-1-(5-fluoro-2-hydroxyphenyl)ethanone Chemical compound C1(CC1)CC(=O)C1=C(C=CC(=C1)F)O BLTRISANKPFBEV-UHFFFAOYSA-N 0.000 description 2
- JSBQCGLLLJJHCD-UHFFFAOYSA-N 2-cyclopropyl-1-(5-fluoro-2-methoxyphenyl)ethanone Chemical compound COC1=CC=C(F)C=C1C(=O)CC1CC1 JSBQCGLLLJJHCD-UHFFFAOYSA-N 0.000 description 2
- OEHXEZQFNWLCNU-UHFFFAOYSA-N 2-ethenyl-4-fluorophenol Chemical compound OC1=CC=C(F)C=C1C=C OEHXEZQFNWLCNU-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- JTFZLADQJIPIBS-UHFFFAOYSA-N 4-fluoro-2-(N-hydroxy-C-phenylcarbonimidoyl)phenol Chemical compound FC=1C=CC(=C(C1)C(=NO)C1=CC=CC=C1)O JTFZLADQJIPIBS-UHFFFAOYSA-N 0.000 description 2
- ICGLPKIVTVWCFT-UHFFFAOYSA-N 4-methylbenzenesulfonohydrazide Chemical compound CC1=CC=C(S(=O)(=O)NN)C=C1 ICGLPKIVTVWCFT-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- ZHKYCTBLKJTTKA-UHFFFAOYSA-N C(CCC)[Si](CC1=C(C=C(C=C1)F)C=C)(CCCC)CCCC Chemical compound C(CCC)[Si](CC1=C(C=C(C=C1)F)C=C)(CCCC)CCCC ZHKYCTBLKJTTKA-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 102000011727 Caspases Human genes 0.000 description 2
- 108010076667 Caspases Proteins 0.000 description 2
- MHSNEBSYDOXQFX-UHFFFAOYSA-N Cc1c2nc(C)cc[n]2nc1 Chemical compound Cc1c2nc(C)cc[n]2nc1 MHSNEBSYDOXQFX-UHFFFAOYSA-N 0.000 description 2
- KMTNNKBCFIEPSX-UHFFFAOYSA-N Cc1cnc2[n]1nc(C)cc2 Chemical compound Cc1cnc2[n]1nc(C)cc2 KMTNNKBCFIEPSX-UHFFFAOYSA-N 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 230000004544 DNA amplification Effects 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical group FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 201000003803 Inflammatory myofibroblastic tumor Diseases 0.000 description 2
- 206010067917 Inflammatory myofibroblastic tumour Diseases 0.000 description 2
- 239000003810 Jones reagent Substances 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 2
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- 101150117329 NTRK3 gene Proteins 0.000 description 2
- 108010025020 Nerve Growth Factor Proteins 0.000 description 2
- 102000007072 Nerve Growth Factors Human genes 0.000 description 2
- 101150056950 Ntrk2 gene Proteins 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 238000011529 RT qPCR Methods 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical class OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 2
- NVQCRFPCPKQAET-GFCCVEGCSA-N [(2R)-1-[(2-methylpropan-2-yl)oxycarbonylamino]propan-2-yl] 4-methylbenzenesulfonate Chemical compound CC1=CC=C(C=C1)S(=O)(=O)O[C@@H](CNC(=O)OC(C)(C)C)C NVQCRFPCPKQAET-GFCCVEGCSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- JYFJNCCRKBBRKZ-UHFFFAOYSA-N chembl194764 Chemical compound C=1C=CC=C(F)C=1CCN1C(=O)C(CC)=C(C)N=C1C1=CC=CC=C1O JYFJNCCRKBBRKZ-UHFFFAOYSA-N 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 230000001351 cycling effect Effects 0.000 description 2
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 208000037765 diseases and disorders Diseases 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- XHUWDDXMORFULQ-UHFFFAOYSA-N ethyl 2h-pyrimidine-1-carboxylate Chemical compound CCOC(=O)N1CN=CC=C1 XHUWDDXMORFULQ-UHFFFAOYSA-N 0.000 description 2
- LQOKKXPOMXDBOY-UHFFFAOYSA-N ethyl 6-[(5-fluoro-2-hydroxyphenyl)methyl-methylamino]imidazo[1,2-b]pyridazine-3-carboxylate Chemical compound CCOC(=O)C1=CN=C2C=CC(=NN12)N(C)CC1=C(O)C=CC(F)=C1 LQOKKXPOMXDBOY-UHFFFAOYSA-N 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Chemical group 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229940014259 gelatin Drugs 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- 150000002466 imines Chemical class 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229910052740 iodine Chemical group 0.000 description 2
- 239000011630 iodine Chemical group 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical class CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 229960002900 methylcellulose Drugs 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- YCWSUKQGVSGXJO-NTUHNPAUSA-N nifuroxazide Chemical group C1=CC(O)=CC=C1C(=O)N\N=C\C1=CC=C([N+]([O-])=O)O1 YCWSUKQGVSGXJO-NTUHNPAUSA-N 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 230000009038 pharmacological inhibition Effects 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 229960000215 ruxolitinib Drugs 0.000 description 2
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 229960004274 stearic acid Drugs 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- GZEBNTSVSCPKID-UHFFFAOYSA-N tert-butyl N-[2-[4-fluoro-2-(1-hydroxyethyl)phenoxy]ethyl]carbamate Chemical compound FC1=CC(=C(OCCNC(OC(C)(C)C)=O)C=C1)C(C)O GZEBNTSVSCPKID-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- NMQGIZBZUAOIHA-UHFFFAOYSA-N (5-fluoro-2-methoxyphenyl)methanethiol Chemical compound COC1=CC=C(F)C=C1CS NMQGIZBZUAOIHA-UHFFFAOYSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- KOFFXZYMDLWRHX-UHFFFAOYSA-N 1-(5-fluoro-2-hydroxyphenyl)ethanone Chemical compound CC(=O)C1=CC(F)=CC=C1O KOFFXZYMDLWRHX-UHFFFAOYSA-N 0.000 description 1
- YRTFLDFDKPFNCJ-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-2-oxo-4-[3-(3-pyrrolidin-1-ylpropoxy)phenyl]-1,8-naphthyridin-3-yl]urea;dihydrochloride Chemical compound Cl.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C(C=1)=CC=CC=1OCCCN1CCCC1 YRTFLDFDKPFNCJ-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- UNHOPMIDKWXFMF-UHFFFAOYSA-N 1-methylpyrrolidin-3-amine Chemical compound CN1CCC(N)C1 UNHOPMIDKWXFMF-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical class OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- CWLMBDIDXJZTQH-UHFFFAOYSA-N 2-(1-amino-2-cyclopropylethyl)-4-fluorophenol Chemical compound C=1C(F)=CC=C(O)C=1C(N)CC1CC1 CWLMBDIDXJZTQH-UHFFFAOYSA-N 0.000 description 1
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 1
- SSNRPTHWRFURQQ-RXMQYKEDSA-N 2-[(1r)-1-aminoethyl]-4-fluorophenol Chemical compound C[C@@H](N)C1=CC(F)=CC=C1O SSNRPTHWRFURQQ-RXMQYKEDSA-N 0.000 description 1
- FYELSNVLZVIGTI-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-5-ethylpyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1CC)CC(=O)N1CC2=C(CC1)NN=N2 FYELSNVLZVIGTI-UHFFFAOYSA-N 0.000 description 1
- JIQXVIJARQLCOY-UHFFFAOYSA-N 2-bromo-4-fluoro-1-methoxybenzene Chemical compound COC1=CC=C(F)C=C1Br JIQXVIJARQLCOY-UHFFFAOYSA-N 0.000 description 1
- MEYRABVEYCFHHB-UHFFFAOYSA-N 2-bromo-4-fluorophenol Chemical compound OC1=CC=C(F)C=C1Br MEYRABVEYCFHHB-UHFFFAOYSA-N 0.000 description 1
- IKCLCGXPQILATA-UHFFFAOYSA-N 2-chlorobenzoic acid Chemical class OC(=O)C1=CC=CC=C1Cl IKCLCGXPQILATA-UHFFFAOYSA-N 0.000 description 1
- KVVDRQDTODKIJD-UHFFFAOYSA-N 2-cyclopropylacetic acid Chemical compound OC(=O)CC1CC1 KVVDRQDTODKIJD-UHFFFAOYSA-N 0.000 description 1
- AFENDNXGAFYKQO-UHFFFAOYSA-N 2-hydroxybutyric acid Chemical class CCC(O)C(O)=O AFENDNXGAFYKQO-UHFFFAOYSA-N 0.000 description 1
- IXHXXDUTMPLLHR-UHFFFAOYSA-N 2-oxa-5,9,11-triazabicyclo[11.4.0]heptadeca-1(13),3,5,7,9,11,14,16-octaene-15-carboxylic acid Chemical compound C1=CN=CN=CC2=C(C=CC(=C2)C(=O)O)OC=CN=C1 IXHXXDUTMPLLHR-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- HTFZDNFCQIFLAA-UHFFFAOYSA-N 3,5,11-triazabicyclo[11.4.0]heptadeca-1,3,5,7,10,12,14,16-octaen-9-one Chemical compound C=1N=CN=CC=CC(C=NC=C2C=1C=CC=C2)=O HTFZDNFCQIFLAA-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZBQCCTCQUCOXBO-UHFFFAOYSA-N 4-(4-aminophenyl)-2,2,6,6-tetramethylcyclohex-3-en-1-amine Chemical compound CC1(C)C(N)C(C)(C)CC(C=2C=CC(N)=CC=2)=C1 ZBQCCTCQUCOXBO-UHFFFAOYSA-N 0.000 description 1
- RHMPLDJJXGPMEX-UHFFFAOYSA-N 4-fluorophenol Chemical compound OC1=CC=C(F)C=C1 RHMPLDJJXGPMEX-UHFFFAOYSA-N 0.000 description 1
- HSXSASGKNUNRBT-UHFFFAOYSA-N 5-[(5-fluoro-2-hydroxyphenyl)methyl-methylamino]-2-methylpyrazolo[1,5-a]pyrimidine-3-carboxylic acid Chemical compound CN(CC1=C(O)C=CC(F)=C1)C1=NC2=C(C(O)=O)C(C)=NN2C=C1 HSXSASGKNUNRBT-UHFFFAOYSA-N 0.000 description 1
- ODDMWBLZRVLPJZ-UHFFFAOYSA-N 5-[1-(5-fluoro-2-hydroxyphenyl)ethylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid Chemical compound CC(NC1=NC2=C(C=NN2C=C1)C(O)=O)C1=C(O)C=CC(F)=C1 ODDMWBLZRVLPJZ-UHFFFAOYSA-N 0.000 description 1
- VWESHKLNKZLURX-UHFFFAOYSA-N 5-[[2-(2-aminoethoxy)-5-fluorophenyl]methyl-methylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid Chemical compound CN(CC1=C(OCCN)C=CC(F)=C1)C1=NC2=C(C=NN2C=C1)C(O)=O VWESHKLNKZLURX-UHFFFAOYSA-N 0.000 description 1
- HARBXLRPJCOHPA-UHFFFAOYSA-N 5-[[2-chloro-3-fluoro-6-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxy]phenyl]methyl-methylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid Chemical compound CN(CC1=C(Cl)C(F)=CC=C1OCCNC(=O)OC(C)(C)C)C1=NC2=C(C=NN2C=C1)C(O)=O HARBXLRPJCOHPA-UHFFFAOYSA-N 0.000 description 1
- PFYCXOCKGKYXPI-UHFFFAOYSA-N 5-[[5-fluoro-2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxy]phenyl]methyl-methylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid Chemical compound C(C)(C)(C)OC(=O)NCCOC1=C(CN(C2=NC=3N(C=C2)N=CC=3C(=O)O)C)C=C(C=C1)F PFYCXOCKGKYXPI-UHFFFAOYSA-N 0.000 description 1
- ZTFVAKGBLOWFDV-UHFFFAOYSA-N 5-[[6-(2-aminoethoxy)-2-chloro-3-fluorophenyl]methyl-methylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid Chemical compound CN(CC1=C(Cl)C(F)=CC=C1OCCN)C1=NC2=C(C=NN2C=C1)C(O)=O ZTFVAKGBLOWFDV-UHFFFAOYSA-N 0.000 description 1
- FDUBQNUDZOGOFE-UHFFFAOYSA-N 5-fluoro-2-hydroxybenzaldehyde Chemical compound OC1=CC=C(F)C=C1C=O FDUBQNUDZOGOFE-UHFFFAOYSA-N 0.000 description 1
- XMQOCSTWSHBZGY-UHFFFAOYSA-N 6-fluoro-2-methyl-10-oxa-2,13,17,21,22-pentazatetracyclo[13.5.2.04,9.018,22]docosa-1(21),4(9),5,7,15,17,19-heptaen-14-one Chemical compound CN1CC2=C(OCCNC(=O)C3=CN=C4C=CC1=NN34)C=CC(F)=C2 XMQOCSTWSHBZGY-UHFFFAOYSA-N 0.000 description 1
- 239000005725 8-Hydroxyquinoline Substances 0.000 description 1
- BJFRNNPXYZRHMI-UHFFFAOYSA-N 8-oxa-3,5,11-triazabicyclo[11.4.0]heptadeca-1,4,10,12,14,16-hexaen-9-one Chemical compound C=1N=CNCCOC(C=NC=C2C=1C=CC=C2)=O BJFRNNPXYZRHMI-UHFFFAOYSA-N 0.000 description 1
- DYGUMGGMDSQHRH-UHFFFAOYSA-N 8-thia-3,5,11-triazabicyclo[11.4.0]heptadeca-1,4,10,12,14,16-hexaen-9-one Chemical compound C=1N=CNCCSC(C=NC=C2C=1C=CC=C2)=O DYGUMGGMDSQHRH-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 101710168331 ALK tyrosine kinase receptor Proteins 0.000 description 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 102100035080 BDNF/NT-3 growth factors receptor Human genes 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 1
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 1
- LPTVWZSQAIDCEB-UHFFFAOYSA-N Brc1cnc2[nH]ccc2c1 Chemical compound Brc1cnc2[nH]ccc2c1 LPTVWZSQAIDCEB-UHFFFAOYSA-N 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- OSCIWFFJPTYHQH-UHFFFAOYSA-N CC(c(c(OCCN)ccc1F)c1Cl)Oc1cnc2[nH]cc(C)c2c1 Chemical compound CC(c(c(OCCN)ccc1F)c1Cl)Oc1cnc2[nH]cc(C)c2c1 OSCIWFFJPTYHQH-UHFFFAOYSA-N 0.000 description 1
- MAQISEFFLMTIKQ-UHFFFAOYSA-N CC(c(cc(cc1)F)c1OCCN1C)Oc(cc2)n[n]3c2ncc3C1=O Chemical compound CC(c(cc(cc1)F)c1OCCN1C)Oc(cc2)n[n]3c2ncc3C1=O MAQISEFFLMTIKQ-UHFFFAOYSA-N 0.000 description 1
- QWHGFMYWPQWVNK-UHFFFAOYSA-N CC(c(cc(cc1)F)c1OCCN1C)Oc(cc[n]2nc3)nc2c3C1=O Chemical compound CC(c(cc(cc1)F)c1OCCN1C)Oc(cc[n]2nc3)nc2c3C1=O QWHGFMYWPQWVNK-UHFFFAOYSA-N 0.000 description 1
- TUJVMVJWTWPTML-SEYXRHQNSA-N CC(c1c(C)c(F)ccc1OCCN(C(OC(C)(C)C)=O)C(OC(C)(C)C)=O)OC(C=NC1C)=CC1/C=C\N Chemical compound CC(c1c(C)c(F)ccc1OCCN(C(OC(C)(C)C)=O)C(OC(C)(C)C)=O)OC(C=NC1C)=CC1/C=C\N TUJVMVJWTWPTML-SEYXRHQNSA-N 0.000 description 1
- VIYDZECUSYPWCV-UHFFFAOYSA-N CN(Cc(cc(cc1)F)c1OC(CO)CN1)c(cc[n]2nc3)nc2c3C1=O Chemical compound CN(Cc(cc(cc1)F)c1OC(CO)CN1)c(cc[n]2nc3)nc2c3C1=O VIYDZECUSYPWCV-UHFFFAOYSA-N 0.000 description 1
- SEVMDGANWYFSKE-UHFFFAOYSA-N CNCc(cc(cc1)P)c1O Chemical compound CNCc(cc(cc1)P)c1O SEVMDGANWYFSKE-UHFFFAOYSA-N 0.000 description 1
- MAQISEFFLMTIKQ-LLVKDONJSA-N C[C@H](c(cc(cc1)F)c1OCCN1C)Oc(cc2)n[n]3c2ncc3C1=O Chemical compound C[C@H](c(cc(cc1)F)c1OCCN1C)Oc(cc2)n[n]3c2ncc3C1=O MAQISEFFLMTIKQ-LLVKDONJSA-N 0.000 description 1
- QWHGFMYWPQWVNK-LLVKDONJSA-N C[C@H](c(cc(cc1)F)c1OCCN1C)Oc(cc[n]2nc3)nc2c3C1=O Chemical compound C[C@H](c(cc(cc1)F)c1OCCN1C)Oc(cc[n]2nc3)nc2c3C1=O QWHGFMYWPQWVNK-LLVKDONJSA-N 0.000 description 1
- 206010058019 Cancer Pain Diseases 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102000003952 Caspase 3 Human genes 0.000 description 1
- 108090000397 Caspase 3 Proteins 0.000 description 1
- TUUPXVAUUWDJDR-UHFFFAOYSA-N Cc1c[nH]c2c1nc(C)cn2 Chemical compound Cc1c[nH]c2c1nc(C)cn2 TUUPXVAUUWDJDR-UHFFFAOYSA-N 0.000 description 1
- 0 Cc1c[n]c2ncc(*)nc12 Chemical compound Cc1c[n]c2ncc(*)nc12 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 241000701867 Enterobacteria phage T7 Species 0.000 description 1
- 208000007207 Epithelioid hemangioendothelioma Diseases 0.000 description 1
- 208000032027 Essential Thrombocythemia Diseases 0.000 description 1
- VTEYJZZYSCFBFS-UHFFFAOYSA-N FC(=C(F)F)B Chemical compound FC(=C(F)F)B VTEYJZZYSCFBFS-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical class OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 101000596896 Homo sapiens BDNF/NT-3 growth factors receptor Proteins 0.000 description 1
- 101000844245 Homo sapiens Non-receptor tyrosine-protein kinase TYK2 Proteins 0.000 description 1
- 101000582914 Homo sapiens Serine/threonine-protein kinase PLK4 Proteins 0.000 description 1
- 101000617830 Homo sapiens Sterol O-acyltransferase 1 Proteins 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 102100039064 Interleukin-3 Human genes 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- 102000042838 JAK family Human genes 0.000 description 1
- 108091082332 JAK family Proteins 0.000 description 1
- 230000035986 JAK-STAT signaling Effects 0.000 description 1
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 1
- 238000003749 KINOMEscan Methods 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000037196 Medullary thyroid carcinoma Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 102100032028 Non-receptor tyrosine-protein kinase TYK2 Human genes 0.000 description 1
- ILUJQPXNXACGAN-UHFFFAOYSA-N O-methylsalicylic acid Chemical class COC1=CC=CC=C1C(O)=O ILUJQPXNXACGAN-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 206010033701 Papillary thyroid cancer Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- IGVPBCZDHMIOJH-UHFFFAOYSA-N Phenyl butyrate Chemical class CCCC(=O)OC1=CC=CC=C1 IGVPBCZDHMIOJH-UHFFFAOYSA-N 0.000 description 1
- 108010001441 Phosphopeptides Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 239000012083 RIPA buffer Substances 0.000 description 1
- 239000012979 RPMI medium Substances 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 108091005682 Receptor kinases Proteins 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 102100030267 Serine/threonine-protein kinase PLK4 Human genes 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- 206010041660 Splenomegaly Diseases 0.000 description 1
- 102100021993 Sterol O-acyltransferase 1 Human genes 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 101000697584 Streptomyces lavendulae Streptothricin acetyltransferase Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 108091005729 TAM receptors Proteins 0.000 description 1
- 108091077436 Tam family Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 108010030743 Tropomyosin Proteins 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 229940061720 alpha hydroxy acid Drugs 0.000 description 1
- 150000001280 alpha hydroxy acids Chemical class 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000002547 anomalous effect Effects 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 230000035578 autophosphorylation Effects 0.000 description 1
- 108010023337 axl receptor tyrosine kinase Proteins 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000012148 binding buffer Substances 0.000 description 1
- 238000010876 biochemical test Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000007248 cellular mechanism Effects 0.000 description 1
- 210000001638 cerebellum Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 239000012829 chemotherapy agent Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 230000008711 chromosomal rearrangement Effects 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000002435 cytoreductive effect Effects 0.000 description 1
- 125000005534 decanoate group Chemical class 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-M deoxycholate Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-M 0.000 description 1
- 229940009976 deoxycholate Drugs 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000004076 epigenetic alteration Effects 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- NVRLFNAHPUQTHA-UHFFFAOYSA-N ethyl 5-[(5-fluoro-2-hydroxyphenyl)methyl-methylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylate Chemical compound CCOC(=O)C1=C2N=C(C=CN2N=C1)N(C)CC1=C(O)C=CC(F)=C1 NVRLFNAHPUQTHA-UHFFFAOYSA-N 0.000 description 1
- ZHNGVXPXQNOJQX-UHFFFAOYSA-N ethyl 6-[1-[5-fluoro-2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxy]phenyl]ethoxy]imidazo[1,2-b]pyridazine-3-carboxylate Chemical compound C(C)OC(=O)C1=CN=C2N1N=C(C=C2)OC(C)C1=C(C=CC(=C1)F)OCCNC(=O)OC(C)(C)C ZHNGVXPXQNOJQX-UHFFFAOYSA-N 0.000 description 1
- PHZOHCPMSOIQFV-UHFFFAOYSA-N ethyl 6-[[5-fluoro-2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxy]phenyl]methyl-methylamino]imidazo[1,2-b]pyridazine-3-carboxylate Chemical compound CCOC(=O)C1=CN=C2C=CC(=NN12)N(C)CC1=C(OCCNC(=O)OC(C)(C)C)C=CC(F)=C1 PHZOHCPMSOIQFV-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 238000007421 fluorometric assay Methods 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229960002598 fumaric acid Drugs 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 201000006585 gastric adenocarcinoma Diseases 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 229960004275 glycolic acid Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 230000011132 hemopoiesis Effects 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 102000045479 human ROS1 Human genes 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 229940071826 hydroxyethyl cellulose Drugs 0.000 description 1
- 230000037417 hyperactivation Effects 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000000021 kinase assay Methods 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229960000448 lactic acid Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229940033355 lauric acid Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 238000011418 maintenance treatment Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940098895 maleic acid Drugs 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 230000005741 malignant process Effects 0.000 description 1
- 150000002690 malonic acid derivatives Chemical class 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- VHMGYHFYSWICSP-UHFFFAOYSA-N methyl 3-amino-2-hydroxypropanoate;hydrochloride Chemical compound Cl.COC(=O)C(O)CN VHMGYHFYSWICSP-UHFFFAOYSA-N 0.000 description 1
- MGRXCNJFDMCLQH-UHFFFAOYSA-N methyl 3-chlorobenzenecarboperoxoate Chemical compound COOC(=O)C1=CC(Cl)=CC=C1 MGRXCNJFDMCLQH-UHFFFAOYSA-N 0.000 description 1
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical class COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical class C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 208000004296 neuralgia Diseases 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical class CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229960002969 oleic acid Drugs 0.000 description 1
- 238000006384 oligomerization reaction Methods 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000008008 oral excipient Substances 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- 230000008058 pain sensation Effects 0.000 description 1
- 229940098695 palmitic acid Drugs 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 102000013415 peroxidase activity proteins Human genes 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 239000008063 pharmaceutical solvent Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical class CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical class OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 238000003566 phosphorylation assay Methods 0.000 description 1
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 1
- 125000005498 phthalate group Chemical class 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001144 powder X-ray diffraction data Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical class CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- RZWZRACFZGVKFM-UHFFFAOYSA-N propanoyl chloride Chemical compound CCC(Cl)=O RZWZRACFZGVKFM-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229940095574 propionic acid Drugs 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 230000008261 resistance mechanism Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 102200048955 rs121434569 Human genes 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- 235000021391 short chain fatty acids Nutrition 0.000 description 1
- 150000004666 short chain fatty acids Chemical class 0.000 description 1
- 238000002603 single-photon emission computed tomography Methods 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 1
- 229960002218 sodium chlorite Drugs 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 238000011895 specific detection Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical class OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000007755 survival signaling Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- JSGWPDROOLRKIO-UHFFFAOYSA-N tert-butyl N-[2-(2-acetyl-4-fluorophenoxy)ethyl]carbamate Chemical compound C(C)(C)(C)OC(NCCOC1=C(C=C(C=C1)F)C(C)=O)=O JSGWPDROOLRKIO-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- WMXCDAVJEZZYLT-UHFFFAOYSA-N tert-butylthiol Chemical compound CC(C)(C)S WMXCDAVJEZZYLT-UHFFFAOYSA-N 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 238000001757 thermogravimetry curve Methods 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 208000013818 thyroid gland medullary carcinoma Diseases 0.000 description 1
- 208000030045 thyroid gland papillary carcinoma Diseases 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 1
- 238000003325 tomography Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 229940062627 tribasic potassium phosphate Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 229940005605 valeric acid Drugs 0.000 description 1
- 229960003862 vemurafenib Drugs 0.000 description 1
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- GDJZZWYLFXAGFH-UHFFFAOYSA-M xylenesulfonate group Chemical group C1(C(C=CC=C1)C)(C)S(=O)(=O)[O-] GDJZZWYLFXAGFH-UHFFFAOYSA-M 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Abstract
Un compuesto de la siguiente fórmula (IV):**Fórmula** en la que M es CH o N; X1 y X1' son independientemente -C(R1a)(R2a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk')-; cada R1a y R2a es independientemente H, deuterio, alquilo C1-6, cicloalquilo C3-6, arilo C6-10, -C(O)ORa', - C(O)NRa'Rb', -NRa'Rb', -SRa', -S(O)Ra', -S(O)NRa'. -S(O)2Ra', -S(O)2NRa' o -ORa' en el que cada átomo de hidrógeno del alquilo C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1-4), -N(alquilo C1-4)2, NHC(O)alquilo C1-4, -N(alquilo C1-4)C(O)alquilo C1-4, - NHC(O)NHalquilo C1-4, -N(alquilo C1-4)C(O)NHalquilo C1-4, -NHC(O)N(alquilo C1-4)2, -N(alquilo C1- 4)C(O)N(alquilo C1-4)2, -NHC(O)Oalquilo C1-4, -N(alquilo C1-4)C(O)Oalquilo C1-4, -CO2H, -CO2alquilo C1-4, - CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, -Salquilo C1-4, -S(O)alquilo C1-4, -S(O)2alquilo C1-4, - S(O)NH(alquilo C1-4), -S(O)2NH(alquilo C1-4), -S(O)N(alquilo C1-4)2, -S(O)2N(alquilo C1-4)2, cicloalquilo C3-6 o heterocicloalquilo de 3 a 7 miembros; R3a y R3b son cada uno independientemente H, deuterio, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3; R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por deuterio, halógeno, -CN, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1-4), -N(alquilo C1-4)2, -CO2H, -CO2alquilo C1-4, - CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, cicloalquilo o heterocicloalquilo monocíclico; cada Rk' es independientemente H, deuterio, alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk' está independientemente opcionalmente sustituido por deuterio, halógeno, alquilo C1-6, haloalquilo C1-6 u -ORa'; en el que cada Ra' y Rb' es independientemente H, deuterio, alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo; cada Z1, Z2, Z3, Z4, Z5, Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente es independientemente H, deuterio, halógeno, alquilo C1-4, -O-alquilo C1-4, -OH, -NH2, -NH(alquilo C1-4), - NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3, Z4, Z5, Z6 o Z7 sea N o NH; y m' es 2 o 3; o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Macrociclos de diarilo como moduladores de proteínas cinasas
La presente invención se refiere a ciertos derivados macrocíclicos de diarilo, a las composiciones farmacéuticas que los contienen y a su uso en procedimientos para tratar el cáncer, el dolor, las enfermedades neurológicas, las enfermedades autoinmunitarias y la inflamación.
Las proteínas cinasas son reguladores clave para el crecimiento, la proliferación y la supervivencia celulares. Las alteraciones genéticas y epigenéticas se acumulan en las células cancerosas, lo que da lugar a una activación anómala de las vías de transducción de señales que conducen los procesos malignos. Manning, G. et al., Science 2002, 298, 1912-1934. La inhibición farmacológica de estas vías de señalización presenta oportunidades de intervención prometedoras para tratamientos dirigidos contra el cáncer. Sawyers, C., Nature 2004, 432, 294-297.
MET, junto con RON, pertenece a una subfamilia única de tirosina cinasas receptoras, y se produce principalmente en células de origen epitelial o endotelial. Park, M. et al., Cell 1986, 45, 895-904. El factor de crecimiento de hepatocitos (HGF), también conocido como factor de dispersión (SF), es el único ligando natural conocido de alta afinidad de MET, y se expresa principalmente en células de origen mesenquimal. Bottaro, D. P. et al., Science 1991,251, 802-804. La señalización de HGF/MET controla los procesos de proliferación, supervivencia y migración de células dependientes de MET que son críticos para el crecimiento invasivo durante el desarrollo embrionario y la regeneración de órganos postnatal, y están totalmente activos en adultos solo para procesos de cicatrización y regeneración de tejidos. Trusolino, L. et al., Nature Rev. Mol. Cell Biol. 2010, 11, 834-848. El eje HGF/MET está frecuentemente regulado por incremento en muchos cánceres a través de la activación de mutaciones, la amplificación de genes, la señalización paracrina aberrante o la producción de ligandos autocrinos, y está estrechamente vinculado con la oncogénesis, el crecimiento invasivo y la metástasis. Gherardi, E. et al., Nature Rev. Cancer 2012, 12, 89-103. Además, la activación de la señalización de HGF/MET está emergiendo como un mecanismo importante en la resistencia a los tratamientos con inhibidores de EGFR y BRAF a través de la amplificación de MET y/o la regulación por incremento del HGF estromal. Engelman, J. A. et al., Science 2007, 316, 1039-1043; Wilson, T.R. et al., Nature 2012, 487, 505-509. Debido a la función de la señalización aberrante de HGF/MET en la oncogénesis humana, la invasión/metástasis y la resistencia, la inhibición de las vías de señalización de HGF/MET tiene un gran potencial en el tratamiento del cáncer.
ALK, junto con la tirosina cinasa lecucocitaria (LTK), se agrupa dentro de la superfamilia de receptores de insulina (IR) de las tirosina cinasas receptoras. La ALK se expresa principalmente en los sistemas nerviosos central y periférico, lo que sugiere una función potencial en el desarrollo normal y la función del sistema nervioso. Pulford, K. et al., Cell Mol. Life Sci. 2004, 61, 2939. La ALK se descubrió por primera vez como una proteína de fusión, NPM (nucleofosmina)-ALK, codificada por un gen de fusión que surge de la translocación cromosómica t(2;5)(p23;q35) en líneas celulares de linfoma anaplásico de células grandes (LACG). Morris, S.W. et al., Science 1994, 263, 1281. Se han descubierto más de veinte socios distintos de traslocación ALK en muchos cánceres, incluyendo LACG (60-90 % de incidencia), tumores inflamatorios miofibroblásticos (TIM, 50-60 %), carcinomas de pulmón no microcíticos (CPNM, 3-7%), cánceres colorrectales (CCR, 0-2,4%), cánceres de mama (0-2,4%) y otros carcinomas. Grande, E. et al., Mol. Cancer Ther. 2011, 10, 569-579. Las proteínas de fusión ALK están localizadas en el citoplasma, y los socios de fusión con ALK desempeñan una función en la dimerización u oligomerización de las proteínas de fusión a través de una interacción espiral-espiral para generar la activación constitutiva de la función cinasa de ALK. Bischof, D. et al., Mol. Cell Biol., 1997, 17, 2312-2325. EML4-ALK, que comprende porciones del gen similar a la proteína 4 asociada a microtúbulos de equinodermo (EML4) y el gen ALK, se descubrió por primera vez en el CPNM, es altamente oncogénico y se demostró que causa adenocarcinoma pulmonar en ratones transgénicos. Soda, M. et al., Nature 2007, 448, 561-566. Mutaciones oncogénicas puntuales de ALK en casos familiares y esporádicos de neuroblastoma. Mossé, Y. P. et al., Nature 2008, 455, 930-935. La ALK es una diana molecular atractiva para la intervención terapéutica del cáncer debido a las importantes funciones en los tumores hematopoyéticos, sólidos y mesenquimales. Grande, supra.
Las tirosina cinasas receptoras relacionadas con la tropomiosina (Trk) son el receptor de alta afinidad para las neurotrofinas (NT), una familia de proteínas del factor de crecimiento nervioso (FCN). Los miembros de la familia Trk están altamente expresados en células de origen neural. La activación de Trk (TrkA, TrkB y TrkC) por sus neurotrofinas preferentes (FCN para TrkA, factor neurotrófico derivado del cerebro [FCDC] y NT4/5 para TrkB, y NT3 para TrkC) media en la supervivencia y diferenciación de las neuronas durante el desarrollo. La vía de señalización NT/Trk funciona como un sistema endógeno que protege a las neuronas después de agresiones bioquímicas, isquemia transitoria o lesión física. Thiele, C. J. et al., Clin. Cancer Res. 2009, 15, 5962-5967. Sin embargo, Trk se clonó originalmente como un oncogén fusionado con el gen de la tropomiosina en el dominio extracelular. Las mutaciones activadoras causadas por reordenamientos cromosómicos o mutaciones en NTRK1 (TrkA) se han identificado en el carcinoma de tiroides papilar y medular, y recientemente en el carcinoma de pulmón no microcítico. Pierotti, M. A. et al., Cancer Lett. 2006, 232, 90-98; Vaishnavi, A. et al., Nat. Med. 2013, 19, 1469 1472. Debido a la importante función desempeñada por las Trk en la sensación de dolor, así como en el crecimiento de las células tumorales y en la señalización de supervivencia, los inhibidores de las cinasas receptoras Trk pueden proporcionar beneficios como tratamientos para el dolor y el cáncer.
La tirosina cinasa receptora AXL pertenece a la familia de proteínas TAM y se detectó originalmente en pacientes con leucemia mielógena crónica (LMC) como un gen transformador no identificado. Verma, A. et al., Mol. Cancer Ther. 2011, 10, 1763-1773. El ligando principal para los receptores TAM es la proteína 6 específica para la detención del crecimiento (Gas6). El AXL se expresa de forma ubicua y se ha detectado en una amplia variedad de órganos y células, incluyendo el hipocampo y el cerebelo, monocitos, macrófagos, plaquetas, células endoteliales (CE), corazón, músculo esquelético, hígado, riñón y testículos. Se ha informado de la regulación por incremento de Gas6/AXL en muchos cánceres humanos, incluyendo el cáncer de colon, esófago, tiroides, mama, pulmón, hígado y el astrocitoma-glioblastoma. Id. Se ha observado un incremento de la activación de AXL en modelos de cáncer de pulmón con mutación en EGFR in vitro e in vivo con resistencia adquirida a erlotinib en ausencia de alteración de EGFR T790M o activación de MET. Zhang, Z. et al., Nat. Genet. 2012, 44, 852-860. La inhibición genética o farmacológica de AXL restauró la sensibilidad al erlotinib en estos modelos de tumores. Se encontró un incremento de la expresión de AXL y, en algunos casos, de su ligando Gas6 en los cánceres de pulmón con mutación en EGFR obtenidos de individuos con resistencia adquirida a los inhibidores de tirosina cinasa. Id. Por lo tanto, AXL es una diana terapéutica prometedora para los pacientes con cáncer de pulmón con mutación en EGFR que adquirieron resistencia a los inhibidores de EGFR.
Crizotinib (PF-02341066) es un fármaco de tirosina cinasa dirigido a MET/ALK/ROS1/RON con actividad moderada contra Tr K y AXL. Cui, J. J. et al., J. Med. Chem. 2011, 54, 6342-6363. Fue aprobado para tratar a ciertos pacientes con CPNM en fase tardía (localmente avanzado o metastásico) que expresa el gen de fusión ALK anómalo identificado por una prueba diagnóstica complementaria (kit de sonda Vysis ALK Break Apart FISH). De forma similar a imatinib y a otros fármacos inhibidores de cinasa, la resistencia se desarrolla invariablemente después de un cierto tiempo de tratamiento con crizotinib. Los mecanismos de resistencia incluyen la amplificación génica de ALK, mutaciones secundarias de ALK y activación aberrante de otras cinasas, incluyendo KIT y EGFR. Katayama, R. et al., Sci. Transl. Med. 2012, 4, 120ra17. En base al éxito clínico de los inhibidores de ABL de segunda generación para el tratamiento de la resistencia a imatinib en pacientes con LMC, está surgiendo una segunda generación de inhibidores de ALK. Estos fármacos se dirigen al tratamiento del paciente con CPNM resistente o insensible a crizotinib con inhibición más potente contra proteínas ALK tanto naturales como mutantes. Gridelli, C. et al., Cancer Treat Rev. 2014, 40, 300-306.
Mediante la modulación de múltiples dianas del grupo de las tirosina cinasas estructuralmente relacionadas MET, ALK, AXL y TRK, los compuestos descritos en el presente documento abordan la resistencia al crizotinib, la resistencia al fármaco inhibidor de EGFR y otras indicaciones primarias con señalización celular anómala debido a mutaciones en MET, ALK, AXL y/o TRK y a amplificación génica. Los compuestos descritos en el presente documento son inhibidores de MET, ALK naturales y mutantes, AXL y TRK y serán útiles en el tratamiento de pacientes con cáncer con señalización anómala de uno o más de MET, ALK, AXL o TRK.
La familia de cinasas Janus (JAK) incluye JAK1, JAK2, JAK3 y TYK2, y son tirosina cinasas citoplásticas requeridas para la señalización fisiológica de citocinas y factores de crecimiento. Quintas-Cardama, A. et al., Nat. Rev. Drug Discov. 2011, 10(2), 127-40; Pesu, M. et al., Immunol. Rev. 2008, 223, 132-142; Murray, P.J., J. Immunol. 2007, 178(5), 2623-2329. Las JAK se activan mediante la oligomerización inducida por ligando, lo que da como resultado la activación de la vía de señalización transcripcional posterior denominada STAT (transductores de señal y activadores de la transcripción). Las STAT fosforiladas dimerizan y translocan en el núcleo para dirigir la expresión de genes específicos implicados en proliferación, apoptosis, diferenciación, que son esenciales para la hematopoyesis, la inflamación y la respuesta inmunitaria. Murray, supra.
Los estudios en ratones "knockout" han implicado las funciones principales de la señalización JAK-STAT con cierta superposición entre ellos. JAK1 desempeña un papel crítico en la señalización de diversas citocinas proinflamatorias tales como IL-1, IL-4, IL-6 y el factor de necrosis tumoral alfa (TNFa). Muller, M. et al., Nature 1993, 366(6451), 129-135. JAK2 actúa en la señalización de factores de crecimiento hematopoyéticos tales como Epo, IL-3, IL-5, GM-CSF, hormona de crecimiento trombopoyetina y señalización mediada por prolactina. Neubauer, H. et al., Cell 1998 93(3), 397-409. JAK3 desempeña un papel en la mediación de las respuestas inmunitarias, y TYK2 se asocia con JAK2 o JAK3 para transducir la señalización de citocinas, tales como la IL-12. Nosaka, T. et al., Science 1995, 270(5237), 800-802; Vainchenker, W. et al., Semin. Cell. Dev. Biol. 2008, 19(4), 385-393.
La regulación aberrante de las vías JAK/STAT ha estado implicada en múltiples enfermedades patológicas humanas, incluyendo el cáncer (JAK2) y la artritis reumatoide (JAK1, JAK3). Se ha descubierto una mutación de ganancia de función de JAK2 (JAK2V617F) con alta frecuencia en pacientes con MPN. Levine, R.L. et al., Cancer Cell 2005, 7(4), 387-397; Kralovics, R. et al., N. Engl. J. Med. 2005, 253(17), 1779-1790; James, C. et al., Nature 2005, 434(7037), 1144-1148; Baxter, E.J. et al. Lancet 2005, 365(9464), 1054-1061. La mutación en el dominio de pseudocinasa JH2 de JAK2 da lugar a la actividad cinasa constitutiva. Las células que contienen la mutación JAK2V617F adquieren una capacidad de crecimiento independiente de citocinas y, a menudo, se convierten en tumores, lo que proporciona una sólida base racional para el desarrollo de inhibidores de JAK como tratamiento dirigido.
Múltiples inhibidores de JAK en un ensayo clínico mostraron un beneficio significativo en la esplenomegalia y en los síntomas inespecíficos relacionados con la enfermedad en los pacientes con mielofibrosis, incluyendo el primer
inhibidor de JAK2 aprobado por la FDA, ruxolitinib, en 2011. Quintas-Cardama, supra; Sonbol, M.B. et al., Ther. Adv. Hematol. 2013, 4(1), 15-35; LaFave, L.M. et al., Trends Pharmacol. Sci. 2012, 33(11), 574-582. Los datos clínicos recopilados recientemente relacionados con el tratamiento con ruxolitinib indicaron que los inhibidores de JAK funcionan tanto en casos de JAK2 natural como en casos de JAK2 mutados. Verstovsek, S. et al., N. Engl. J. Med. 2012, 366(9), 799-807; Quintas-Cardama, A. et al., Blood 2010, 115(15), 3109-3117. El descubrimiento de inhibidores selectivos de JAK2 vs. JAK1/3 sigue siendo un desafío sin resolver. Además, la hiperactivación de JAK2/transductores de señal y activadores de la transcripción 3 (JAK2/STAT3) es responsable de la diferenciación anómala de células dendríticas que da lugar a la diferenciación anómala de células dendríticas y a la acumulación de células mieloides inmunosupresoras en el cáncer (Nefedova, Y. et al., Cancer Res 2005; 65(20): 9525-35). En los tumores senescentes Pten-null, la activación de la vía JAK2/STAT3 establece un microambiente tumoral inmunosupresor que contribuye al crecimiento del tumor y la quimiorresistencia (Toso, A. et al., Cell Reports 2014, 9, 75-89). Por lo tanto, la inhibición farmacológica de la vía JAK2/STAT3 puede ser una nueva estrategia terapéutica importante para potenciar la actividad antitumoral por medio de la regulación de la inmunidad antitumoral.
La cinasa ROS1 es una tirosina cinasa receptora con un ligando desconocido. Las funciones normales de la cinasa ROS1 humana no se han comprendido completamente. Sin embargo, se ha informado de que la cinasa ROS1 sufre reordenamientos genéticos para crear proteínas de fusión constitutivamente activas en una variedad de cánceres humanos incluyendo glioblastoma, carcinoma de pulmón no microcítico (CPNM), colangiocarcinoma, cáncer de ovario, adenocarcinoma gástrico, cáncer colorrectal, tumor miofibroblástico inflamatorio, angiosarcoma y hemangioendotelioma epitelioide (Davies, K. D. et al., Clin Cancer Res 2013, 19 (15): 4040-4045). Dirigirse a proteínas de fusión ROS1 con crizotinib ha demostrado una eficacia clínica prometedora en pacientes con CPNM cuyos tumores son positivos para anomalías genéticas ROS1 (Shaw, A. T. et al., N Engl J Med. 2014, 371(21): 1963-1971). Se han observado mutaciones resistentes adquiridas en pacientes tratados con crizotinib (Awad, M. M. et al., N Engl J Med. 2013, 368(25): 2396-2401). Es urgente desarrollar la segunda generación de inhibidores de ROS1 para superar la resistencia a ROS1 de crizotinib.
Sigue existiendo la necesidad de inhibidores de moléculas pequeñas de estas dianas de proteínas múltiples o tirosina cinasa con propiedades farmacéuticas deseables. Se han encontrado que ciertos compuestos macrocíclicos de diarilo en el contexto de la presente invención tienen este perfil de actividad ventajoso.
La presente invención proporciona un compuesto de la siguiente Fórmula (IV):

en la que
M es CH o N;
X1 y X 1 ' son independientemente -C(R1 a )(R2a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk )-;
cada R1a y R2a es independientemente H, deuterio, alquilo C1 -6 , cicloalquilo C3-6, arilo C6 -10, -C(O)ORa', -C(O)NRa'Rb', -NRa'Rb', -SRa ’, -S(O)Ra', -S(O)NRa', -S(O)2 Ra', -S(O)2 NRa' o -ORa ’ en el que cada átomo de hidrógeno del alquilo C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo C1.4 J2 , -NHC(O)alquilo C1 -4 , -N(alquilo C1 -4 )C(o)alquilo C1 -4 , -NHC(O)NHalquilo C1 -4 , -N(alquilo C-M )C(O)NHalquilo C1.4 , -NHC(O)N(alquilo ^ .4)2 , -N(alquilo C-M )C(O)N(alquilo ^ .4)2 , -NHC(O)Oalquilo C1 -4 , -N(alquilo C1 -4 )C(O)Oalquilo C1 -4 , -CO2 H, -CO2alquilo C1 -4 , -CONH2 , -CONH(alquilo C1 -4 ), -CON(alquilo ^ -4)2 , -Salquilo C1 -4 , -S(o)alquilo C1 -4 , -S(O)2alquilo C1 -4 , -S(O)NH(alquilo C1 -4 ), -S(O)2 NH(alquilo C1.4 ), -S(O)N(alquilo C1.
4)2 , -S(O)2 N(alquilo C1 -4 )2 , cicloalquilo C3.6 o heterocicloalquilo de 3 a 7 miembros;
R3a y R3b son cada uno independientemente H, deuterio, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3;
R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por deuterio, halógeno, -CN, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo C1 -4)2 , -CO2 H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1.4 ), -CON(alquilo C1 -4 )2 , cicloalquilo o heterocicloalquilo monocíclico;
cada Rk’ es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk’ está independientemente opcionalmente sustituido por deuterio, halógeno, alquilo C1-6 , haloalquilo C1-6 u -ORa ’;
en la que cada Ra' y Rb' es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo;
cada Z1, Z2, Z3 , Z4 , Z5 , Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente es independientemente H, deuterio, halógeno, alquilo C1 -4 , -O-alquilo C1 -4 , -OH, -NH2 , -NH(alquilo C1 -4 ), -NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3 , Z4, Z5 , Z6 o Z7 sea N o NH; y
m' es 2 o 3;
o una sal farmacéuticamente aceptable del mismo.
En otro modo de realización, la presente invención proporciona una composición farmacéutica que comprende (a) al menos un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, y (b) un excipiente farmacéuticamente aceptable.
En otro modo de realización, la presente invención proporciona un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer, el dolor, las enfermedades neurológicas, las enfermedades autoinmunitarias o la inflamación en un sujeto que necesita dicho tratamiento.
La presente memoria descriptiva divulga además un compuesto de la siguiente Fórmula (I-A) (la Fórmula (I-A) forma parte de la invención en la medida en que se superpone con la Fórmula (IV) como se define en la reivindicación 1):

en la que
el Anillo A' y el Anillo B' son cada uno independientemente un arilo o heteroarilo monocíclico o bicíclico; en el que uno del Anillo A' y el Anillo B' es un arilo o heteroarilo monocíclico y el otro es un heteroarilo bicíclico; y al menos uno del Anillo A' y el Anillo B' comprende al menos un elemento de anillo de nitrógeno;
cada L1 y L2 es independientemente -C(R1 ')(R2')-, -O-, -N(Rk')-, -S-, -S(O)- o -S(O)2-;
cada R1 ' y R2' son independientemente H, deuterio, halógeno, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico, -ORa', -OC(O)Ra', -OC(O)NRa'Rb', -OS(O)Ra', -OS(O)2Ra', -SRa', -S(O)Ra', -S(O)2Ra', -S(O)NRa'Rb', -S(O)2NRa'Rb', -OS(O)NRa'Rb', -OS(O)2NRa'Rb', -NRa'Rb', -NRa'C(O)Rb', -NRa'C(O)ORb', -NRa'C(O)NRa'Rb', -NRa'S(O)Rb', -NRa'S(O)2Rb', -NRa'S(O)NRa'Rb', -NRa'S(O)2NRa'Rb', -C(O)Ra', -C(O)ORa', -C(O)NRa'Rb', -PRa'Rb', -P(O)Ra'Rb', -P(O)2Ra'Rb', -P(O)NRa'Rb', -P(O)2NRa'Rb', -P(O)ORa', -P(O)2ORa', -CN o -NO2, o R1' y R2' tomados conjuntamente con el carbono o carbonos a los que están unidos forman un cicloalquilo C3-6 o un heterocicloalquilo de 4 a 6 miembros, en el que cada átomo de hidrógeno del alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico o heterocicloalquilo de 4 a 6 miembros está independientemente y opcionalmente sustituido por deuterio, halógeno, alquilo C1 -6 , cicloalquilo C1-6 , -ORe', -OC(O)Re', -OC(O)NRe'Rf, -OS(O)Re', -OS(O)2Re', -OS(O)NRe'Rf', -OS(O)2NRe'Rf, -SRe', -S(O)Re', -S(O)2Re', -S(O)NRe'Rf', -S(O)2NRe'Rf', -NRe'Rf', -NRe'C(O)Rf', -NRe'C(O)ORf', -NRe'C(O)NRe'Rf', -NRe'S(O)Rf', -NRe'S(O)2Rf', -NRe'S(O)NRe'Rf', -NRe'S(O)2NRe'Rf', -C(O)Re', -C(O)ORe', -C(O)NRe'Rf , -PRe'Rf', -P(O)Re'Rf', -P(O)2Re'Rf', -P(O)NRe'Rf', -P(O)2 NRe'Rf', -P(O)ORe', -P(O)2ORe', -CN o -NO2 ;
cada Rk' es independientemente H, deuterio, alquilo C1-6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3.6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico está independientemente y opcionalmente sustituido por deuterio, halógeno, alquilo C1.6 , haloalquilo C1.6 , -ORe', -OC(O)Re', -OC(O)NRe'Rf, -OS(O)Re', -OS(O)2 Re', -OS(O)NRe'Rf, -OS(O)2 NRe'Rf', -SRe', -S(O)Re', -S(O)2Re', -S(O)NRe'Rf , -S(O)2NRe'Rf', -NRe'Rf', -NRe'C(O)Rf', -NRe'C(O)ORf', -NRe'C(O)NRe'Rf',
NRe'S(O)Rf , -NRe'S(O)2Rf , -NRe'S(O)NRe'Rf , -NRe'S(O)2NRe'Rf , -C(O)Re', -C(O)ORe', -C(O)NRe'Rf', -PRe'Rf', -P(O)Re'Rf , -P(O)2 Re'Rf', -P(O)NRe'Rf', -P(O)2 NRe'Rf', -P(O)ORe', -P(O)2ORe', -CN o -NO2 ;
cada R3' y R4' es independientemente deuterio, halógeno, -ORc', -OC(O)Rc', -OC(O)NRc'Rd', -OC(=N)NRc'Rd', -OS(O)Rc', -OS(O)2Rc', -OS(O)NRc'Rd', -OS(O)2NRc'Rd', -SRc', -S(O)Rc', -S(O)2Rc', -S(O)NRc'Rd', -S(O)2NRc'Rd', -NRc'Rd', -NRc'C(O)Rd', -NRc'C(O)ORd', -NRc'C(O)NRc'Rd", -NRc'C(=N)NRc'Rd', -NRc'S(O)Rd', -NRc'S(O)2Rd', -NRc'S(O)NRc'Rd', -NRc'S(O)2NRc'Rd', -C(O)Rc', -C(O)ORc', -C(O)NRc'Rd', -C(=N)NRc'Rd', -PRc'Rd', -P(O)Rc'Rd', -P(O)2 Rc'Rd', -P(O)NRc'Rd', -P(O)2 NRc'Rd', -P(O)ORc', -P(O)2ORc', -CN, -NO2 , alquilo C1.6 , alquenilo C2-6, alquinilo C2-6 , cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico, o cualquiera de
los dos grupos R3' o cualquiera de los dos grupos R4' tomados conjuntamente con el anillo al que están unidos
forman un cicloalquilo C5-8 o un heterocicloalquilo de 5 a 8 miembros, en el que cada átomo de hidrógeno del
alquilo C1-6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico, cicloalquilo C5-8 o un heterocicloalquilo de 5 a 8 miembros está independientemente
y opcionalmente sustituido por deuterio, halógeno, alquilo C1 -6 , haloalquilo C1 -6 , -ORe', -OC(O)Re', -OC(O)NRe'Rf', -OS(O)Re', -OS(O)2Re', -OS(O)NRe'Rf', -OS(O)2NRe'Rf', -SRe', -S(O)Re', -S(O)2Re', -S(O)NRe'Rf, -S(O)2NRe'Rf', -NRe'Rf', -NRe'C(O)Rf', -NRe'C(O)ORf', -NRe'C(O)NRe'Rf', -NRe'S(O)Rf', -NRe'S(O)2Rf', -NRe'S(O)NRe'Rf, -NRe'S(O)2NRe'Rf, -C(O)Re', -C(O)ORe', -C(O)NRe'Rf', -PRe'Rf', -P(O)Re'Rf', -P(O)2Re'Rf', -P(O)NRe'Rf, -P(O)2NRe'Rf', -P(O)ORe', -P(O)2ORe', -CN o -NO2 ;
R7' es H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros,
arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1-6 , alquenilo C2-6, alquinilo
C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico está independientemente y opcionalmente sustituido por deuterio, halógeno, -OR i', -OC(O)Rf, -OC(O)NRi'Rj ', -OS(O)R i', -OS(O)2Ri', -OS(O)NR i'Rj ', -OS(O)2NRi'Rj ', -SR i', -S(O)R i', -S(O)2Ri', -S(O)NR i'Rj', -S(O)2NRi'Rj ', -NR i'Rj', -NR i'C(O)Rj ',
-NR i'C(O)ORj ', -NR i'C(O)NRi'Rj ', -NR i'S(O)Rj ', -NR i'S(O)2Rj', -NR i'S(O)NRi'Rj', -NR i'S(O)2NRi'Rj ', -C(O)R i', -C(O)ORf, -C(O)NR i'Rj ', -PR i'Rj ', -P(O)R i'Rj', -P(O)2 Ri'Rj ', -P(O)NR i'Rj ', -P(O)2 NRi'Rj ', -P(O)ORf, -P(O)2ORi', -CN o -NO2 ;
cada Ra', Rb', Rc', Rd', Re', Rf', Ri' y Rj ' se selecciona independientemente del grupo que consis alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 y heteroarilo;
m' es 2, 3, 4 o 5;
n' es 2, 3 o 4;
p' es 0, 1, 2, 3 o 4; y
q' es 0, 1, 2, 3 o 4;
o una sal farmacéuticamente aceptable del mismo.
La presente memoria descriptiva divulga además una entidad química de la siguiente Fórmula (I-A) (la Fórmula (I-A) forma parte de la invención en la medida en que se superpone con la Fórmula (IV) como se define en la reivindicación 1):

en la que
el Anillo A' y el Anillo B' son cada uno independientemente un arilo o heteroarilo monocíclico o bicíclico;
en el que uno del Anillo A' y el Anillo B' es un arilo o heteroarilo monocíclico y el otro es un heteroarilo bicíclico; y
al menos uno del Anillo A' y el Anillo B' comprende al menos un elemento de anillo de nitrógeno;
cada R3' y R4' es independientemente deuterio, halógeno, -ORc', -OC(O)Rc', -OC(O)NRc'Rd', -OC(=N)NRc'Rd', -OS(O)0-2Rc', -OS(O)0-2NRc'Rd', -S(O)0-2Rc', -S(O)0-2NRc'Rd', -NRc'Rd', -NRc'C(O)Rd', -NRc'C(O)NRc'Rd', -NRc'C(=N)NRc'Rd', -NRc'S(O)0-2Rd', -NRc'S(O)0-2NRc'Rd', -C(O)Rc', -C(O)ORc', -C(O)NRc'Rd', -C(=N)NRc'Rd', -P(O)0-2 Rc'Rd', -P(O)0 -2NRc'Rd', -P(O)0-2ORc', -c N, -NO2 , alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono- o bicíclico; o cualquiera de los dos R3' o
cualquiera de los dos R4' tomados conjuntamente con el anillo al que están unidos forman un cicloalquilo C5-8 o un heterocicloalquilo de 5 a 8 miembros;
en la que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, fenilo, naftilo y heteroarilo mono- o bicíclico está no sustituido o sustituido con uno o más sustituyentes seleccionados del grupo que consiste de deuterio, halógeno, alquilo C1 -6 , haloalquilo C1-6 , -ORe', -OC(O)Re', -OC(O)NRe'Rf, -OS(O)ü-2Re', -OS(O)ü .2NRe'Rf, -S(O)ü.2Re', -S(O)ü.2NRe'Rf, -NRe'Rf', -NRe'C(O)Rf', -NRe'C(O)NRe'Rf', -NRe'S(O)ü.2Rf , -NRe'S(O)ü-2NRe'Rf', -C(O)Re', -C(O)ORe', -C(O)NRe'Rf', -P(O)o-2Re'Rf , -P(O)ü-2NRe'Rf', -P(O)ü-2ORe', -CN y -NO2 ; y
cada Rc', Rd', Re' y Rf' se selecciona independientemente del grupo que consiste en H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3.6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo y heteroarilo; R7' es H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo y heteroarilo mono- o bicíclico;
en la que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, fenilo, naftilo o heteroarilo está sustituido o no sustituido con uno o más sustituyentes seleccionados del grupo que consiste en deuterio, halógeno, -OR i', -OC(O)R i', -OC(O)NRfRj ', -OS(O)ü-2Ri', -OS(O)ü-2NRi'Rj ', -S(O)ü-2Ri', -S(O)ü-2NRi'Rj ', -NR i'Rj', -NR i'C(O)Rj ', -NRi'C(O)NRi'Rj ', -NR i'S(O)ü-2Rj ', -NR i'S(O)ü-2NRi'Rj ', -C(O)Rf, -C(O)OR i', -C(O)NRfRj ', -P(O)ü-2Ri'Rj ', -P(O)ü-2NRi'Rj ', -P(O)ü-2OR i', -CN y -NO2 ;
en la que cada Ri' y Rj ' es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 , heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo y heteroarilo mono- o bicíclico;
cada L1 y L2 es independientemente -C(Rr )(R2')-, -O-, -N(Rk')- o -S(O)ü-2;
en la que cada R1 ' y R2' son independientemente H, deuterio, halógeno, alquilo C1-6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono- o bicíclico; o R1 ' y R2' tomados conjuntamente con el carbono o los carbonos a los que están unidos forman un cicloalquilo C3-6 o un heterocicloalquilo de 4 a 6 miembros;
cada Rk' es independientemente H, deuterio, alquilo C1-6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono- o bicíclico;
en la que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, fenilo, naftilo o heteroarilo en R1 ', R2' o Rk' está independientemente no sustituido o sustituido con uno o más sustituyentes seleccionados del grupo que consiste de deuterio, halógeno, alquilo C1 -6 , haloalquilo C1-6 , -ORa', -OC(O)Ra', -OC(O)NRa'Rb', -OS(O)ü -2Ra', -OS(O)ü-2NRa'Rb', -S(O)ü-2Ra', -S(O)ü-2NRa'Rb', -NRa'Rb', -NRa'C(O)Rb', -NRa'C(O)NRa'Rb', -NRa'S(O)ü-2Rb', -NRa'S(O)ü-2 NRa'Rb', -C(O)Ra', -C(O)ORa', -C(O)NRa'Rb', -P(O)ü-2Ra'Rb', -P(O)ü-2NRa'Rb', -P(O)ü-2ORa', -CN y -NO2 ;
en la que cada Ra' y Rb' es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo;
m' es 3, 4 o 5;
n' es 2, 3 o 4;
p' es ü, 1, 2, 3 o 4; y
q' es ü, 1, 2, 3 o 4;
o una sal farmacéuticamente aceptable del mismo.
La presente memoria descriptiva divulga además una entidad química de la siguiente Fórmula (I) (la Fórmula (I) forma parte de la invención en la medida en que se superpone con la Fórmula (IV) como se define en la reivindicación 1):

en la que
el Anillo A y el Anillo B son cada uno independientemente un arilo o heteroarilo monocíclico o bicíclico; en donde uno del Anillo A y el Anillo B es monocíclico y el otro es bicíclico; y el Anillo comprende al menos un elemento de anillo de nitrógeno;
R1 y R2 son cada uno independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono- o bicíclico; o R1 y R2 tomados conjuntamente con el carbono al que están unidos forman un cicloalquilo C3-6 o un heterocicloalquilo de 4 a 6 miembros;
en la que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, fenilo, naftilo o heteroarilo está no sustituido o sustituido con uno o más sustituyentes seleccionados del grupo que consiste de deuterio, halógeno, alquilo C1 -6 , haloalquilo C1-6 , -ORa, -OC(O)Ra , -OC(O)NRaRb, -OS(O)ü -2Ra , -OS(O)ü -2NRaRb, -NRaRb, -NRaC(O)Rb, -NRaC(O)NRaRb, -NRaS(O)ü-2Rb, -NRaS(O)ü-2NRaRb, -C(O)Ra , -C(O)ORa, -C(O)NRaRb, -P(O)ü-2RaRb, -P(O)ü-2 NRaRb, -P(O)ü-2ORa, -CN y -NO2 ;
en la que cada Ra y Rb es independientemente H, deuterio, alquilo C1-6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo;
cada R3 y R4 es independientemente deuterio, halógeno, -ORc , -OC(O)Rc , -OC(O)NRcRd, -OC(=N)NRcRd, -OS(O)ü-2Rc , -OS(O)ü-2NRcRd, -NRcRd, -NRcC(O)Rd, -NRcC(O)NRcRd, -NRcC(=N)NRcRd, -NRcS(O)ü-2Rd, -NRcS(O)ü-2NRcRd, -C(O)Rc , -C(O)ORc, -C(O)NRcRd, -C(=N)NRcRd, -P(O)ü-2RcRd, -P(O)ü-2NRcRd, -P(O)ü-2ORc, -CN, -NO2 , alquilo C1-6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono- o bicíclico;
en la que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, fenilo, naftilo y heteroarilo mono- o bicíclico está no sustituido o sustituido con uno o más sustituyentes seleccionados del grupo que consiste de deuterio, halógeno, alquilo C1 -6 , haloalquilo C1-6 , -ORe, -OC(O)Re, -OC(O)NReRf, -OS(O)ü-2Re, -OS(O)ü-2NReRf, -NReRf, -NReC(O)Rf, -NReC(O)NReRf, -NReS(O)ü-2Rf, -NReS(O)ü-2NReRf, -C(O)Re, -C(O)ORe , -C(O)NReRf, -P(O)ü-2 ReRf, -P(O)ü -2NReRf, -P(O)ü-2ORe , -CN y -NO2 ; y
cada Rc , Rd, Re y Rf se selecciona independientemente del grupo que consiste en H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo y heteroarilo;
R5 y R6 son cada uno independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono- o bicíclico; o R5 y R6 tomados conjuntamente con el carbono al que están unidos forman un cicloalquilo C3-6 o un heterocicloalquilo de 4 a 6 miembros;
en la que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, fenilo, naftilo o heteroarilo está sustituido o no sustituido con uno o más sustituyentes seleccionados del grupo que consiste de deuterio, halógeno, alquilo C1.6 , haloalquilo C1.6 , -ORg, -OC(O)Rg, -OC(O)NRgRh, -OS(O)ü-2Rg, -OS(O)ü -2NRgRh, -NRgRh, -NRgC(O)Rh, -NRgC(O)NRgRh, -NRgS(O)ü-2Rh, -NRgS(O)ü-2NRgRh, -C(O)Rg, -C(O)ORg, -C(O)NRgRh, -P(O)ü-2RgRh, -P(O)ü-2NRgRh, -P(O)ü-2ORg, -CN y -NO2 ;
en la que cada Rg y Rh es independientemente H, deuterio, alquilo C1-6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono o bicíclico;
R7 es H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono o bicíclico;
en la que cada alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, fenilo, naftilo o heteroarilo está sustituido o no sustituido con uno o más sustituyentes seleccionados del grupo que consiste en deuterio, halógeno, -OR i, -OC(O)R i, -OC(O)NRiRj , -OS(O)ü-2Ri, -OS(O)ü-2NRiRj , -NR iRj , -NR iC(O)Rj , -NR iC(O)NRiRj , -NR iS(O)ü-2Rj , -NRS(O)ü-2 NRiRj , -C(O)Ri, -C(O)OR i, -C(O)NR iRj , -P(O)ü-2RiRj , -P(O)ü-2NRiRj , -P(O)ü-2OR i, -CN y -NO2 ;
en la que cada Ri y Rj es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 , heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono o bicíclico;
X e Y son cada uno independientemente -C(Rk)(Rk)-, -O- o -N(Rk)-;
en la que cada Rk es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, fenilo, naftilo o heteroarilo mono o bicíclico;
m es 2, 3 o 4;
n es 1, 2 o 3;
p es ü, 1, 2, 3 o 4; y
q es ü, 1, 2, 3 o 4;
o una sal farmacéuticamente aceptable del mismo.
En determinados modos de realización, el compuesto de Fórmula (I) o (I-A) es un compuesto seleccionado de aquellas especies descritas o ejemplificadas en la descripción detallada a continuación. Los compuestos de Fórmula (I) o (I-A) están de acuerdo con la invención en la medida en que se corresponden con la Fórmula (IV) como se define en la reivindicación 1.
En determinados modos de realización, el compuesto de Fórmula (I) o (I-A) es un compuesto que tiene la fórmula

o una sal farmacéuticamente aceptable del mismo.
En determinados modos de realización, el compuesto de Fórmula (I) o (I-A) es un compuesto que tiene la fórmula

o una sal farmacéuticamente aceptable del mismo.
En determinados modos de realización, el compuesto de Fórmula (I) o (I-A) es un compuesto que tiene la fórmula

o una sal farmacéuticamente aceptable del mismo.
En determinados modos de realización, el compuesto de Fórmula (I) o (I-A) es un compuesto que tiene la fórmula

o una sal farmacéuticamente aceptable del mismo.
En determinados modos de realización, el compuesto de Fórmula (I) o (I-A) es un compuesto que tiene la fórmula

o una sal farmacéuticamente aceptable del mismo.
En determinados modos de realización, el compuesto de Fórmula (I) o (I-A) es un compuesto que tiene la fórmula

o una sal farmacéuticamente aceptable del mismo.
En otro aspecto, la invención se refiere a una forma cristalina de la base libre del compuesto de fórmula

que tiene un patrón de difracción de rayos X en polvo sustancialmente igual a la Fig. XX. En algunos modos de realización, la forma polimorfa cristalina 1 de la base libre del compuesto de fórmula

en el que el patrón de difracción de rayos X en polvo tiene un pico a un ángulo de difracción (20) de 21,94. En algunos modos de realización, la forma polimorfa 1 de la base libre del compuesto de fórmula

en el que el patrón de difracción de rayos X en polvo tiene picos a ángulos de difracción (20) de 21,94 y 23,96. En algunos modos de realización, la forma polimorfa 1 de la base libre del compuesto de fórmula

en el que el patrón de difracción de rayos X en polvo tiene picos a ángulos de difracción (20) de 21,94, 23,96 y 19,64.
En otro aspecto, la invención se refiere a una composición farmacéutica que comprende al menos un compuesto de Fórmula (I) o (I-A) o una sal farmacéuticamente aceptable del mismo. Las composiciones farmacéuticas de acuerdo con la invención pueden comprender además un excipiente farmacéuticamente aceptable. La invención también es un compuesto de Fórmula (I) o (I-A) o una sal farmacéuticamente aceptable del mismo para su uso como medicamento.
En otro aspecto, la memoria descriptiva divulga un procedimiento para tratar el cáncer, el dolor, las enfermedades neurológicas, las enfermedades autoinmunitarias o la inflamación, que comprende administrar a un sujeto que necesita dicho tratamiento una cantidad eficaz de al menos un compuesto de Fórmula (I) o (I-A) o una sal farmacéuticamente aceptable del mismo.
En otro aspecto, la memoria descriptiva divulga el uso de un compuesto de Fórmula (I) o (I-A) en la preparación de un medicamento para el tratamiento de dichas enfermedades y afecciones médicas, y el uso de dichos compuestos y sales para el tratamiento de dichas enfermedades. y afecciones médicas.
Aún en otro aspecto, la memoria descriptiva divulga un procedimiento para inhibir proteínas o tirosina cinasas, que incluyen una o más de MET, ALK, ROS1, AXL, TRK y JAK, que comprende poner en contacto una célula que comprende una o más de dichas cinasas con una cantidad eficaz de al menos un compuesto de Fórmula (I) o (I-A) o una sal del mismo, y/o con al menos una composición farmacéutica de la invención, en el que el contacto es in vitro, ex vivo o in vivo.
Modos de realización, características y ventajas adicionales de la invención serán evidentes a partir de la siguiente descripción detallada y a través de la práctica de la invención.
La Fig. 1 muestra un patrón de difracción de rayos X en polvo de la forma polimorfa cristalina 1 de la base libre de 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclo-tridecin-4(5H)-ona (Ejemplo 20).
La Fig. 2 muestra un termograma de calorimetría diferencial de barrido de la forma polimorfa cristalina 1 de la base libre de 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclo-tridecin-4(5H)-ona (Ejemplo 20).
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado como entiende comúnmente uno de los expertos en la técnica a la que pertenece la presente invención.
Como se usa en el presente documento y en las reivindicaciones adjuntas, las formas singulares "un", "una" y "el/la" incluyen referentes plurales a menos que el contexto lo indique claramente de otro modo. Se indica además que las reivindicaciones se pueden redactar para excluir cualquier elemento opcional. Como tal, esta declaración pretende servir como base antecedente para el uso de dicha terminología exclusiva tal como "únicamente", "solamente" y similares en conexión con la indicación de elementos reivindicados, o el uso de una limitación "negativa".
Como se usa en el presente documento, los términos "incluyendo", "que incluye", "que contiene" y "que comprende" se usan en su sentido abierto y no limitante.
Para proporcionar una descripción más concisa, algunas de las expresiones cuantitativas que se ofrecen en el presente documento no están calificadas con el término "aproximadamente". Se entiende que, ya sea que el término "aproximadamente" se use explícitamente o no, cada cantidad dada en el presente documento se refiere al valor real dado, y también se refiere a la aproximación a dicho valor dado que razonablemente se podría inferir basado en la experiencia del experto en la técnica, incluyendo equivalentes y aproximaciones debidas a las condiciones experimentales y/o de medición para dicho valor dado. Cuando un rendimiento se da como un porcentaje, dicho rendimiento se refiere a una masa de la entidad para la cual se proporciona el rendimiento con respecto a la cantidad máxima de la misma entidad que se podría obtener en las condiciones estequiométricas particulares. Las concentraciones que se dan como porcentajes se refieren a relaciones de masa, a menos que se indique lo contrario.
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado como entiende comúnmente uno de los expertos en la técnica a la que pertenece la presente invención. Aunque en la práctica o ensayo de la presente invención se puede usar cualquier procedimiento y material similar o equivalente a los descritos en el presente documento, a continuación se describen los procedimientos y/o materiales preferentes.
Excepto que se indique de otro modo, los procedimientos y técnicas de los presentes modos de realización se realizan en general de acuerdo con procedimientos convencionales bien conocidos en la técnica y como se describe en diversas referencias generales y más específicas que se citan y discuten a lo largo de la presente memoria descriptiva. Ver, por ejemplo, Loudon, Organic Chemistry, 4a edición, Nueva York: Oxford University Press, 2002, pp. 360-361, 1084-1085; Smith y March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001.
La nomenclatura química para los compuestos descritos en el presente documento se ha derivado en general usando el ACD/Name 2014 (ACD/Labs) o ChemBioDraw Ultra 13,0 (Perkin Elmer) disponible comercialmente.
Se aprecia que ciertas características de la invención, que para mayor claridad se describen en el contexto de modos de realización separados, se pueden proporcionar también en combinación en un único modo de realización. Por el contrario, diversas características de la invención, que por motivos de brevedad se describen en el contexto de un único modo de realización, también se pueden proporcionar por separado o en cualquier subcombinación adecuada. Todas las combinaciones de los modos de realización que pertenecen a los grupos químicos representados por las variables están específicamente abarcadas por la presente invención y se divulgan
en el presente documento como si todas y cada una de las combinaciones se hubieran divulgado individual y explícitamente, en la medida en que dichas combinaciones abarcan compuestos que son compuestos estables (es decir, compuestos que se pueden aislar, caracterizar y someter a prueba para la actividad biológica). Además, todas las subcombinaciones de los grupos químicos enumerados en los modos de realización que describen dichas variables también se abarcan específicamente en la presente invención y se divulgan en el presente documento como si todas y cada una de dichas subcombinaciones de grupos químicos se hubieran divulgado individual y explícitamente en el presente documento.
DEFINICIONES QUÍMICAS
El término "alquilo" se refiere a un grupo alquilo de cadena lineal o ramificada que tiene de 1 a 12 átomos de carbono en la cadena. Los ejemplos de grupos alquilo incluyen metilo (Me), etilo (Et), n-propilo, isopropilo, butilo, isobutilo, sec-butilo, terc-butilo (tBu), pentilo, isopentilo, terc-pentilo, hexilo, isohexilo y grupos que en vista del experto en la técnica y las enseñanzas proporcionadas en el presente documento se considerarían equivalentes a cualquiera de los ejemplos anteriores.
El término "alquenilo" se refiere a un grupo hidrocarburo de cadena lineal o ramificada que tiene de 2 a 12 átomos de carbono en la cadena, y que tiene uno o más enlaces dobles. Los ejemplos de grupos alquenilo incluyen etenilo (o vinilo), alilo y but-3-en-1-ilo. Incluidos dentro de este término están los isómeros cis y trans y mezclas de los mismos.
El término "alquinilo" se refiere a un grupo hidrocarburo de cadena lineal o ramificada que tiene de 2 a 12 átomos de carbono en la cadena, y que tiene uno o más enlaces triples. Los ejemplos de grupos alquinilo incluyen acetilenilo (-C=CH) y propargilo (-CH2CECH).
El término "cicloalquilo" se refiere a un carbociclo monocíclico o policíclico, saturado o parcialmente saturado que tiene de 3 a 12 átomos en el anillo. Los carbociclos policíclicos incluyen sistemas policíclicos fusionados, con puentes y espiro. Los ejemplos ilustrativos de grupos cicloalquilo incluyen las siguientes entidades, en forma de restos apropiadamente unidos:

El término "halógeno" representa cloro, flúor, bromo o yodo. El término "halo" representa cloro, fluoro, bromo o yodo.
El término "haloalquilo" se refiere a un grupo alquilo con uno o más sustituyentes halo, o uno, dos o tres sustituyentes halo. Los ejemplos de grupos haloalquilo incluyen -CF3 , -(CH2)F, -CHF2 , -CH2 Br, -CH2CF3 y -CH2CH2 F.
El término "arilo" se refiere a un grupo monocíclico o policíclico de anillos fusionados todo de carbono de 6 a 14 átomos de carbono (C6-C14) que tiene un sistema de electrones pi completamente conjugado. Arilo incluye grupos monocíclicos o policíclicos de anillos fusionados todos de carbono de 6 a 10 átomos de carbono (por ejemplo, "arilo C6-10"). Ejemplos de grupos arilo son fenilo, naftalenilo y antracenilo. El grupo arilo puede estar sustituido como se describe anteriormente para el alquilo o puede estar no sustituido. Los grupos sustituyentes también incluyen aquellos descritos en otra parte en la presente divulgación en relación con arilo.
El término "heterocicloalquilo" se refiere a una estructura de anillo monocíclico o policíclico que está saturada o parcialmente saturada y tiene de 3 a 12 átomos de anillo, con 1 a 5 de los átomos de anillo seleccionados de nitrógeno, oxígeno y azufre. Los sistemas de anillos policíclicos incluyen sistemas fusionados, con puentes y espiro. La estructura de anillo puede contener opcionalmente hasta dos grupos oxo en elementos de anillo de carbono o azufre. Los ejemplos ilustrativos de grupos heterocicloalquilo incluyen las siguientes entidades, en forma de restos apropiadamente unidos:


El término "heteroarilo" se refiere a un heterociclo aromático monocíclico, bicíclico fusionado o policíclico fusionado (estructura de anillo que tiene átomos o elementos de anillo seleccionados de átomos de carbono y hasta cuatro heteroátomos seleccionados de nitrógeno, oxígeno y azufre) que tiene de 3 a 12 átomos de anillo por heterociclo. Los ejemplos ilustrativos de grupos heteroarilo incluyen las siguientes entidades, en forma de restos apropiadamente unidos:

Un heteroarilo "monocíclico" es un heterociclo aromático de cinco o seis miembros. Un heteroarilo de cinco miembros contiene hasta cuatro átomos de anillo de heteroátomo, donde (a) un átomo de anillo es oxígeno y azufre y cero, uno o dos átomos de anillo es nitrógeno, o (b) cero átomos de anillo son oxígeno o azufre y hasta cuatro átomos de anillo son nitrógeno. En algunos modos de realización, un heteroarilo de cinco miembros es furano, tiofeno, pirrol, oxazol, isoxazol, tiazol, isotiazol, pirazol, imidazol, oxadiazol, tiadiazol, triazol o tetrazol. Un heteroarilo de seis miembros contiene uno o dos átomos de anillo de nitrógeno. En algunos modos de realización, un heteroarilo de seis miembros es piridina, pirazina, pirimidina, piridazina o triazina. Un "heteroarilo bicíclico" es un sistema bicíclico fusionado que comprende un anillo de heteroarilo fusionado a un anillo de fenilo u otro heteroarilo.
El término "oxo" representa un oxígeno carbonílico. Por ejemplo, un ciclopentilo sustituido con oxo es ciclopentanona.
El término "sustituido" significa que el grupo o resto especificado tiene uno o más sustituyentes. El término "no sustituido" significa que el grupo especificado no tiene sustituyentes. Cuando el término "sustituido" se usa para describir un sistema estructural, se pretende que la sustitución se produzca en cualquier posición del sistema permitida por la valencia. En algunos modos de realización, "sustituido" significa que el grupo o resto especificado tiene uno, dos o tres sustituyentes. En otros modos de realización, "sustituido" significa que el grupo o resto especificado tiene uno o dos sustituyentes. Todavía en otros modos de realización, "sustituido" significa que el grupo o resto especificado tiene un sustituyente.
Cualquier fórmula descrita en el presente documento pretende representar un compuesto de esa fórmula estructural, así como determinadas variaciones o formas. Por ejemplo, una fórmula dada en el presente documento pretende incluir una forma racémica, o uno o más isómeros enantioméricos, diastereoméricos o geométricos, o una mezcla de los mismos. Adicionalmente, cualquier fórmula dada en el presente documento pretende referirse también a un hidrato, solvato o polimorfo de dicho compuesto, o una mezcla de los mismos.
Cualquier fórmula dada en el presente documento también pretende representar formas no marcadas, así como formas marcadas isotópicamente de los compuestos. Los compuestos marcados isotópicamente tienen estructuras descritas por las fórmulas dadas en el presente documento, excepto que uno o más átomos están reemplazados por un átomo que tiene una masa atómica o número de masa seleccionado. Los ejemplos de isótopos que se pueden incorporar a los compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor, cloro y yodo, tales como 2H, 3H, 11C, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F, 36Cl e 125I, respectivamente. Dichos compuestos marcados isotópicamente son útiles en estudios metabólicos (preferentemente con 14C), estudios cinéticos de reacción (con, por ejemplo, 2H o 3H), técnicas de detección o formación de imágenes [tal como la tomografía por emisión de positrones (PET) o la tomografía de emisión monofotónica (SPECT)], incluyendo los ensayos de distribución de fármacos o sustratos en tejidos, o en el tratamiento radioactivo de pacientes. Además, la sustitución con isótopos más pesados tales como el deuterio (es decir, 2H) puede proporcionar determinadas ventajas terapéuticas que dan como resultado una mayor estabilidad metabólica, por ejemplo, un aumento de la semivida in vivo o requisitos de dosificación reducidos. Los compuestos marcados isotópicamente de la presente invención y los profármacos de los mismos se pueden preparar en general llevando a cabo los procedimientos divulgados en los esquemas o en los ejemplos y preparaciones descritos a continuación sustituyendo un reactivo marcado isotópicamente fácilmente disponible por un reactivo no marcado isotópicamente.
La nomenclatura "(ATOM)i-j" con j > i, cuando se aplica en el presente documento a una clase de sustituyentes, se refiere a modos de realización de la presente invención para los cuales todos y cada uno del número de átomos miembro, de i a j, incluyendo i y j, se realizan de forma independiente. A modo de ejemplo, el término C1-3 se refiere independientemente a modos de realización que tienen un miembro de carbono (C1 ), modos de realización que tienen dos miembros de carbono (C2) y modos de realización que tienen tres miembros de carbono (C3 ).
Cualquier disustituyente al que se hace referencia en el presente documento pretende englobar las diversas posibilidades de unión cuando se permite más de una de dichas posibilidades. Por ejemplo, la referencia a un disustituyente -AB-, donde A t B, se refiere en el presente documento a dicho disubstituyente con A unido a un primer miembro sustituido y B unido a un segundo miembro sustituido, y también se refiere a dicho disustituyente con A unido al segundo miembro sustituido y B unido al primer miembro sustituido.
La invención también incluye sales farmacéuticamente aceptables de los compuestos representados por la Fórmula (I) o (I-A), preferentemente de los compuestos descritos anteriormente y de los compuestos específicos ejemplificados en el presente documento, y composiciones farmacéuticas que comprenden dichas sales, y procedimientos de uso de dichas sales.
Una "sal farmacéuticamente aceptable" pretende significar una sal de un ácido o base libre de un compuesto representado en el presente documento que no es tóxica, es biológicamente tolerable o, de otro modo, es biológicamente adecuada para la administración al sujeto. Ver, en general, S.M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci., 1977, 66, 1-19. Las sales farmacéuticamente aceptables preferentes son aquellas que son farmacológicamente eficaces y adecuadas para el contacto con los tejidos de sujetos sin toxicidad, irritación o respuesta alérgica indebidas. Un compuesto descrito en el presente documento puede poseer un grupo suficientemente ácido, un grupo suficientemente básico, ambos tipos de grupos funcionales, o más de uno de cada tipo y, en consecuencia, reaccionar con un número de bases inorgánicas u orgánicas, y ácidos inorgánicos y orgánicos, para formar una sal farmacéuticamente aceptable.
Los ejemplos de sales farmacéuticamente aceptables incluyen sulfatos, pirosulfatos, bisulfatos, sulfitos, bisulfitos, fosfatos, monohidrogenofosfatos, dihidrogenofosfatos, metafosfatos, pirofosfatos, cloruros, bromuros, yoduros, acetatos, propionatos, decanoatos, caprilatos, acrilatos, formiatos, isobutiratos, caproatos, heptanoatos, propiolatos, oxalatos, malonatos, succinatos, suberatos, sebacatos, fumaratos, maleatos, butino-1,4-dioatos, hexino-1,6-dioatos, benzoatos, clorobenzoatos, metilbenzoatos, dinitrobenzoatos, hidroxibenzoatos, metoxibenzoatos, ftalatos, sulfonatos, metilsulfonatos, propilsulfonatos, besilatos, xilenosulfonatos, naftaleno-1-sulfonatos, naftaleno-2-sulfonatos, fenilacetatos, fenilpropionatos, fenilbutiratos, citratos, lactatos, yhidroxibutiratos, glicolatos, tartrato y mandelatos. Se encuentran listas de otras sales farmacéuticamente aceptables adecuadas en Remington's Pharmaceutical Sciences, 17a edición, Mack Publishing Company, Easton, Pa., 1985.
Para un compuesto de Fórmula (I) o (I-A) que contiene un nitrógeno básico, se puede preparar una sal farmacéuticamente aceptable mediante cualquier procedimiento adecuado disponible en la técnica, por ejemplo, el tratamiento de la base libre con un ácido inorgánico tal como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido sulfámico, ácido nítrico, ácido bórico y ácido fosfórico, o con un ácido orgánico tal como ácido acético, ácido fenilacético, ácido propiónico, ácido esteárico, ácido láctico, ácido ascórbico, ácido maleico, ácido hidroximaleico, ácido isetiónico, ácido succínico, ácido valérico, ácido fumárico, ácido malónico, ácido pirúvico, ácido oxálico, ácido glicólico, ácido salicílico, ácido oleico, ácido palmítico, ácido láurico, un ácido piranosidilo tal como ácido glucurónico o ácido galacturónico, un alfa-hidroxi ácido tal como ácido mandélico, ácido cítrico o ácido tartárico, un aminoácido tal como el ácido aspártico o ácido glutámico, un ácido aromático tal como el ácido benzoico, ácido 2-acetoxibenzoico, ácido naftoico o ácido cinámico, un ácido sulfónico tal como ácido laurilsulfónico, ácido ptoluensulfónico, ácido metanosulfónico o ácido etanosulfónico, o cualquier mezcla compatible de ácidos tales como
los que se dan como ejemplos en el presente documento, y cualquier otro ácido y mezcla de los mismos que se consideren equivalentes o sustitutos aceptables en vista del nivel ordinario de conocimiento en esta tecnología.
La memoria descriptiva también divulga profármacos farmacéuticamente aceptables de los compuestos de Fórmula (I) o (I-A), y procedimientos de tratamiento que emplean dichos profármacos farmacéuticamente aceptables. El término "profármaco" significa un precursor de un compuesto designado que, después de la administración a un sujeto, produce el compuesto in vivo por medio de un proceso químico o fisiológico tal como solvólisis o escisión enzimática, o en condiciones fisiológicas (por ejemplo, un profármaco en el momento en que alcanza el pH fisiológico se convierte en el compuesto de Fórmula (I) o (I-A)). Un "profármaco farmacéuticamente aceptable" es un profármaco que no es tóxico, es biológicamente tolerable y, de otro modo, biológicamente adecuado para la administración al sujeto. Los procedimientos ilustrativos para la selección y preparación de derivados de profármacos adecuados se describen, por ejemplo, en "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
La memoria descriptiva también divulga metabolitos farmacéuticamente activos de compuestos de Fórmula (I) o (I-A), y usos de dichos metabolitos en los procedimientos divulgados en el presente documento. Un "metabolito farmacéuticamente activo" significa un producto farmacológicamente activo del metabolismo en el cuerpo de un compuesto de Fórmula (I) o (I-A) o una sal del mismo. Los profármacos y los metabolitos activos de un compuesto se pueden determinar usando técnicas de rutina conocidas o disponibles en la técnica. Ver, por ejemplo, Bertolini et al., J. Med. Chem. 1997, 40, 2011-2016; Shan et al., J. Pharm. Sci. 1997, 86 (7), 765-767; Bagshawe, Drug Dev. Res. 1995, 34, 220-230; Bodor, Adv. Drug Res. 1984, 13, 255-331; Bundgaard, Design of Prodrugs (Elsevier Press, 1985); y Larsen, Design and Application of Prodrugs, Drug Design and Development (Krogsgaard-Larsen et al., eds., Harwood Academic Publishers, 1991).
MODOS DE REALIZACIÓN REPRESENTATIVOS
En algunos modos de realización de Fórmula (I-A), el Anillo A' es arilo o heteroarilo monocíclico y el Anillo B' es heteroarilo bicíclico. En otros modos de realización, el Anillo A' es heteroarilo bicíclico y el Anillo B' es arilo o heteroarilo monocíclico. En algunos modos de realización, el Anillo A' es fenilo o un heteroarilo de 6 miembros. En otros modos de realización, el Anillo B' es heteroarilo bicíclico que contiene 1,2 o 3 átomos de anillo de nitrógeno. En otros modos de realización, el Anillo A' es fenilo o piridilo.
Todavía en otros modos de realización, el Anillo A' es fenilo. Todavía en otros modos de realización, el Anillo A' sustituido con -(R3')p' es

Todavía en otros modos de realización, el Anillo A' sustituido con -(R3')p' es
En algunos modos de realización, el Anillo B' es:

en el que Z1-Z7 se definen como se describe en el presente documento. Todavía en otros modos de realización, el Anillo B' es:

en los que Z1-7 se definen de otro modo como se describe en el presente documento. Todavía en otros modos de realización, el Anillo B' es:

Todavía en otros modos de realización, el Anillo B' es

Todavía en otros modos de realización, el Anillo B' es

En otros modos de realización de Fórmula (I-A), el Anillo A' es un grupo heteroarilo bicíclico, y es:

en el que Z1-Z7 se definen como se describe en el presente documento. Todavía en otros modos de realización, el Anillo A' es:

en los que Z1-7 se definen de otro modo como se describe en el presente documento. Todavía en otros modos de realización, el Anillo A' es:

Todavía en otros modos de realización, el Anillo A' es

Todavía en otros modos de realización, el Anillo A' es

En algunos modos de realización, el Anillo B' es el arilo o heteroarilo monocíclico. Todavía en otros modos de realización, el Anillo B' es fenilo. Todavía en otros modos de realización, el Anillo B' es piridilo.
En algunos modos de realización, cada R3' es independientemente deuterio, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN, -CF3 , -NH2 , -NH(alquilo C1 -4), -N(alquilo ^ -4)2 , -CO2alquilo C1 -4 , -CO2 H, -NHC(O)alquilo C1.4 , -SO2alquilo C1.4 , -C(O)NH2 , -C(O)NH(alquilo C1.4 ), -C(O)N(alquilo ^ -4)2 , ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, pirrolidinilo, piperidinilo, piperazinilo, morfolinilo o tiomorfolinilo. Todavía en otros modos de realización, cada R3' es independientemente fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3. Todavía en otros modos de realización, cada R3' es fluoro o cloro.
En algunos modos de realización, R7' es H, deuterio, metilo, etilo, propilo, isopropilo, ciclopropilo, ciclobutilo, ciclopentilo, pirrolidinilo, furanilo, tiofuranilo, piperidinilo, piperazinilo, morfolinilo, fenilo o heteroarilo monocíclico, cada uno sustituido o no sustituido como en la Fórmula (I-A). En otros modos de realización, R7' es H, o es metilo, etilo, propilo, isopropilo o ciclopropilo, cada uno no sustituido o sustituido como en la Fórmula (I-A). Todavía en otros modos de realización, R7' es H o es metilo o etilo, cada uno no sustituido o sustituido con halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo ^ -4)2 , -CO2 H, -CO2alquilo C1 -4 , -CONH2 , cicloalquilo o heterocicloalquilo monocíclico. Todavía en otros modos de realización, R7' es H, metilo, hidroxietilo, -CH2CONH2 o 3-pirrolidinilmetilo. Todavía en otros modos de realización, R7' es H o metilo.
En algunos modos de realización, R1 ' y R2' son cada uno independientemente H, deuterio, metilo, etilo, propilo, isopropilo, ciclopropilo, ciclobutilo, ciclopentilo, pirrolidinilo, furanilo, tiofuranilo, piperidinilo, piperazinilo, morfolinilo, fenilo o heteroarilo monocíclico, cada uno sustituido o no sustituido como en la Fórmula (I-A). En otros modos de realización, R1 ' es H. Todavía en otros modos de realización, R2' es deuterio, metilo, etilo, propilo, isopropilo, ciclopropilo, ciclobutilo, ciclopentilo, pirrolidinilo, furanilo, tiofuranilo, piperidinilo, piperazinilo, morfolinilo, fenilo o heteroarilo monocíclico, cada uno sustituido o no sustituido como en la Fórmula (I-A). Todavía en otros modos de realización, R2' es H o es metilo o etilo, cada uno no sustituido o sustituido con halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo ^ -4)2 , -CO2 H, -CO2alquilo C1 -4 , -CONH2 , cicloalquilo o heterocicloalquilo monocíclico. Todavía en otros modos de realización, R2' es H, metilo, fluorometilo, hidroximetilo o ciclopropilo. Todavía en otros modos de realización, R2' es H. Todavía en otros modos de realización, R2' es metilo.
En algunos modos de realización, cada Rk' es independientemente H, metilo, etilo, propilo, isopropilo o ciclopropilo. En otros modos de realización, cada Rk' es independientemente H o metilo.
En algunos modos de realización, cada L1 y L2 es independientemente -CH2- o -CH(metilo)-, -CH(metilo sustituido) , -CH(ciclopropilo C3-6)-, -CH(OH)-, -O-, -n H-, -N(alquilo C1 -4 )-, -N(ciclopropilo C3-6)-, -S-, -S(O)- o -SO2-. En algunos modos de realización, -(L1)n- es -CH2-O-, -CH(alquilo C1 -4 )-O- o -CH(cicloalquilo C3-6)-O-. En otros modos de realización, -(L1)n- es -CH(H o alquilo C1-4 opcionalmente sustituido)-N(H o alquilo C1-4 opcionalmente sustituido)-, -CH(CO2alquilo C1.4 o C(O)N(H o alquilo C1 -4 )2)-N(H o alquilo C1.4 opcionalmente sustituido). Todavía en otros modos de realización, -(L1)n'- es -CH2S(O)ü.2-. En otros modos de realización, -(L1)n'- es -SO2-N(H o alquilo C1 -4 ). En algunos modos de realización, -(L1)n'- es -(CH2 )3-. En algunos modos de realización, -(L1)n'- es -(CH2)2-. En algunos modos de realización, -(L1)n- es -CH(CH3)CH2-.
En algunos modos de realización, -(L2)m' es -O-(C(Rr )(R2'))2-3-. En otros modos de realización, -(L2)m' es -O-(CH2)2-3-. En otros modos de realización, -(L2)m' es -N(Rk')-(C(Rr )(R2'))2-3-. En otros modos de realización, -(L2)m' es -N(H o alquilo C1 -4 )-(CH2)2-3-. En otros modos de realización, -(L2)m' es -S-(C(Rr )(R2'))2-3-. En otros modos de realización, -(L2)m' es -SO2-(C(Rr )(R2'))2-3-. Todavía en otros modos de realización, -(L2)m' es -SO2-N(Rk')-(C(Rr )(R2'))2-. Todavía en otros modos de realización, -(L2)m' es -(C(Rr )(R2'))3-.
En algunos modos de realización, m' es 3. En otros modos de realización, m' es 4. Todavía en otros modos de realización, m' es 5. En algunos modos de realización, n' es 2. En otros modos de realización, n' es 3. Todavía en otros modos de realización, n' es 4. En algunos modos de realización, p' es 0, 1 o 2. En otros modos de realización, p' es 1 o 2. En algunos modos de realización, q' es 0. En otros modos de realización, q' es 1. Todavía en otros modos de realización, q' es 2.
En algunos modos de realización, los compuestos de Fórmula (I-A) son compuestos de Fórmula (I) o sales farmacéuticamente aceptables de los mismos. En otros modos de realización, los compuestos de Fórmula (I-A) son compuestos de Fórmula (I), en los que cada variable se define independientemente como se indica a continuación para la Fórmula (I). En algunos modos de realización, las variables de Fórmula (I-A) se corresponden con la Fórmula (I) de la siguiente manera: A' es A; B' es B; R1 ' es R1; R2' es R2 ; R3' es R3 ; R4' es R4; R7' es R7; Ra -Rf' y Ri'-Rk' se corresponden con Ra-Rf y Ri-Rk, respectivamente; y L1 y L2 son -Y-(C(R5)(R6))m- y -C((R1)(R2))n-X-, respectivamente.
En algunos modos de realización de Fórmula (I), el Anillo A es fenilo o un heteroarilo de 6 miembros. En otros modos de realización, el Anillo A es fenilo o piridilo. Todavía en otros modos de realización, el Anillo A es fenilo. Todavía en otros modos de realización, el Anillo A sustituido con -(R3)p es

Todavía en otros modos de realización, el Anillo A sustituido con -(R3)p es

En algunos modos de realización, cada R3 es independientemente deuterio, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN, -CF3 , -NH2 , -NH(alquilo C1 -4), -N(alquilo ^ -4)2 , -CO2alquilo C1 -4 , -CO2 H, -NHC(O)alquilo C1 -4 , -SO2alquilo C1 -4 , -C(O)NH2 , -C(O)NH(alquilo C1 -4 ), -C(O)N(alquilo ^ -4)2 , ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, pirrolidinilo, piperidinilo, piperazinilo, morfolinilo o tiomorfolinilo. Todavía en otros modos de realización, cada R3 es independientemente fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3. Todavía en otros modos de realización, cada R3 es fluoro o cloro.
Todavía en otros modos de realización, el Anillo A sustituido con -(R3)p es

donde R3a y R3b son cada uno independientemente H, fluoro o cloro y M es CH o N. En algunos modos de realización, R3a es fluoro.
En algunos modos de realización, p es 1 o 2. En otros modos de realización, p es cero. Todavía en otros modos de realización, p es 1. Todavía en otros modos de realización, p es 2.
En algunos modos de realización, el Anillo B es un heteroarilo bicíclico. En otros modos de realización, el Anillo B es un heteroarilo bicíclico de 9 miembros.
En algunos modos de realización, cada R4 es independientemente deuterio, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN, -CF3 , -NH2 , -NH(alquilo C1 -4), -N(alquilo ^ -4)2 , -CO2alquilo C1 -4 , -CO2 H, -NHC(O)alquilo C1 -4 , -SO2alquilo C1 -4 , -C(O)NH2 , -C(O)NH(alquilo C1 -4 ), -C(O)N(alquilo C1 -4 )2 , ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, pirrolidinilo, piperidinilo, piperazinilo, morfolinilo o tiomorfolinilo. Todavía en otros modos de realización, cada R4 es independientemente fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3.
En otros modos de realización, el Anillo B sustituido con -(R4)q es:

en los que Z1, Z2, Z3 y Z6 son cada uno independientemente -C(Rx)- o N;
en los que cada Rx es independientemente H, deuterio, halógeno, alquilo C1 -4 , -O-alquilo C1 -4 , -OH, -NH2 , -NHalquilo C1 -4 , -NH-fenilo, -NH-heteroarilo, CN, o -CF3 ;
Z4 y Z5 son cada uno independientemente -C- o -N-; y
Z7 es -CH-, -N- o -NH-;
En otros modos de realización:
(a) Z1 Z4 y Z7 son cada uno -N-;
(b) Z1 Z5 y Z7 son cada uno -N-;
(c) Z1 y Z3 son cada uno -N- y Z7 es -NH-;
(d) Z3 es -N- y Z7 es -NH-;
(e) Z3 y Z6 son cada uno -N- y Z7 es -NH-;
(f) Z2 Z4 y Z7 son cada uno -N-;
(g) Z1 Z2 , Z4 y Z7 son cada uno -N-;
(h) Z1 Z3 y Z4 son cada uno -N-;
(i) Z3 y Z4 son cada uno -N-;
(j) Z1 Z2 , Z5 y Z7 son cada uno -N-;
(k) Z2 , Z5 y Z7 son cada uno -N-;
(l) Z3 y Z5 son cada uno -N-;
(m) Z3 , Z5 y Z6 son cada uno -N-;
(n) Z1 Z5 , Z6 y Z7 son cada uno -N-;
(o) Z2 , Z5 , Z6 y Z7 son cada uno -N-; o
(p) Z1 Z3 y Z6 son cada uno -N- y Z7 es -NH-Todavía en otros modos de realización de (a)-(p), cada átomo del anillo Z que no está definido expresamente es independientemente -C- o -C(Rx)-(de manera consecuente con la definición de dicho átomo del anillo). Todavía en otros modos de realización, Z3 es -N-. En otro modo de realización, Z7 es -N- o -NH-. Todavía en otros modos de realización, Z3 es -N- y Z7 es -N- o -NH-. Todavía en otros modos de realización, el Anillo B sustituido con -(R4)q es:

en los que Z1-7 se definen de otro modo como anteriormente.
Todavía en otros modos de realización, el Anillo B sustituido con -(R4)q es:

Todavía en otros modos de realización, el Anillo B sustituido con -(R4)q es

Todavía en otros modos de realización, el Anillo B sustituido con -(R4)q es

En algunos modos de realización, q es 0. En otros modos de realización, q es 1.
En algunos modos de realización, R1 y R2 son cada uno independientemente H, deuterio, metilo, etilo, propilo, isopropilo, ciclopropilo, ciclobutilo, ciclopentilo, pirrolidinilo, furanilo, tiofuranilo, piperidinilo, piperazinilo, morfolinilo, fenilo o heteroarilo monocíclico, cada uno sustituido o no sustituido como en la Fórmula (I). En otros modos de realización, R1 es H. Todavía en otros modos de realización, R2 es deuterio, metilo, etilo, propilo, isopropilo, ciclopropilo, ciclobutilo, ciclopentilo, pirrolidinilo, furanilo, tiofuranilo, piperidinilo, piperazinilo, morfolinilo, fenilo o heteroarilo monocíclico, cada uno sustituido o no sustituido como en la Fórmula (I). Todavía en otros modos de realización, R2 es H o es metilo o etilo, cada uno no sustituido o sustituido con halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo ^ -4)2 , -CO2 H, -CO2alquilo C1 -4 , -CONH2 , cicloalquilo o heterocicloalquilo monocíclico. Todavía en otros modos de realización, R2 es H, metilo, fluorometilo, hidroximetilo o ciclopropilo. Todavía en otros modos de realización, R2 es H. Todavía en otros modos de realización, R2 es metilo. Todavía en otros modos de realización, R1 es H y R2 no es H y está en la configuración estereoquímica que se muestra a continuación:

Todavía en otros modos de realización, R1 y R2 se toman conjuntamente para formar un cicloalquilo C3-6. En otros modos de realización, R1 y R2 se toman conjuntamente para formar un heterocicloalquilo de 5 o 6 miembros, opcionalmente sustituido con alquilo C1 -4.
En algunos modos de realización, n es 1 o 2. Todavía en otros modos de realización, n es 1.
En algunos modos de realización, R5 y R6 son cada uno independientemente H, deuterio, metilo, etilo, propilo, isopropilo, ciclopropilo, ciclobutilo, ciclopentilo, pirrolidinilo, furanilo, tiofuranilo, piperidinilo, piperazinilo, morfolinilo, fenilo o heteroarilo monocíclico, cada uno sustituido o no sustituido como en la Fórmula (I). En otros modos de realización, cada R5 es H. Todavía en otros modos de realización, cada R6 es independientemente H, o es metilo, etilo o ciclopropilo, cada uno sustituido o no sustituido como en la Fórmula (I). Todavía en otros modos de realización, cada R6 es independientemente H o metilo, no sustituido o sustituido con -OH. Todavía en otros modos de realización, cada R6 es H o metilo. Todavía en otros modos de realización, R5 y R6 se toman conjuntamente para formar un cicloalquilo C3-6. En otros modos de realización, R5 y R6 se toman conjuntamente para formar un heterocicloalquilo de 5 o 6 miembros, opcionalmente sustituido con alquilo C1 -4.
En algunos modos de realización, m es 2 o 3. En otros modos de realización, m es 2.
En algunos modos de realización, R7 es H, deuterio, metilo, etilo, propilo, isopropilo, ciclopropilo, ciclobutilo, ciclopentilo, pirrolidinilo, furanilo, tiofuranilo, piperidinilo, piperazinilo, morfolinilo, fenilo o heteroarilo monocíclico, cada uno sustituido o no sustituido como en la Fórmula (I). En otros modos de realización, R7 es H, o es metilo, etilo, propilo, isopropilo o ciclopropilo, cada uno no sustituido o sustituido como en la Fórmula (I). Todavía en otros modos de realización, R7 es H o es metilo o etilo, cada uno no sustituido o sustituido con halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo ^ -4)2 , -CO2 H, -CO2alquilo C1 -4 , -CONH2 , cicloalquilo o heterocicloalquilo monocíclico. Todavía en otros modos de realización, R7 es H, metilo, hidroxietilo, -CH2CONH2 o 3-pirrolidinilmetilo. Todavía en otros modos de realización, R7 es H o metilo.
En algunos modos de realización, cada uno de X e Y es independientemente -O- o -N(Rk)-. En algunos modos de realización, X es -O- o -N(Rk)-. En algunos modos de realización, Y es -O-. En algunos modos de realización, cada Rk es independientemente H, metilo, etilo, propilo, isopropilo o ciclopropilo. En otros modos de realización, cada Rk es independientemente H o metilo.
En algunos modos de realización, los compuestos de Fórmula (I) o (I-A) son compuestos de Fórmula (II):

en los que M, R3, q, R2, X, R7 y Z1'7 son cada uno como se definen en cualquiera de las varias formas mencionadas anteriormente;
R5a, R5b, R6a y R6b son cada uno R5 y R6 como se definen en cualquiera de las varias formas mencionadas anteriormente;
o una sal farmacéuticamente aceptable del mismo.
En algunos modos de realización, los compuestos de Fórmula (I) o (I-A) son compuestos de Fórmula (III):

en los que
M es CH o N;
R3a y R3b son cada uno independientemente H, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3 ;
R2a es H o es metilo o etilo, cada uno no sustituido o sustituido con halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo Ci - 4 )2 , -CO2 H, -CO2alquilo C1 -4 , -CONH2 , cicloalquilo o heterocicloalquilo monocíclico;
X1 es O -N(CH3 )-;
R5 a, R6a, R5b y R6b son cada uno independientemente H, o metilo o etilo, cada uno no sustituido o sustituido con halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo Ci - 4 )2 , -CO2 H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1.4), -CON(alquilo Ci -4 ))2 , cicloalquilo, o heterocicloalquilo monocíclico;
R7a es H, o es metilo o etilo, cada uno no sustituido o sustituido con halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1.4 ), -N(alquilo Ci - 4 )2 , -CO2 H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1 -4 ), -CON(alquilo Ci -4 ))2 , cicloalquilo, o heterocicloalquilo monocíclico;
Z1-7 son cada uno como se define en cualquiera de las varias formas mencionadas anteriormente;
o una sal farmacéuticamente aceptable del mismo.
En algunos modos de realización de Fórmula (III), M es CH.
En otros modos de realización, R3a y R3b son cada uno independientemente H, fluoro o cloro. Todavía en otros modos de realización, R3a es H o fluoro. Todavía en otros modos de realización, R3a es fluoro. Todavía en otros modos de realización, R3b es H o cloro.
En algunos modos de realización de Fórmula (III), R2a es H, metilo, fluorometilo o ciclopropilo.
En algunos modos de realización de Fórmula (III), X1 es O. En otros modos de realización, X es -N(CH3)-.
En algunos modos de realización, R7a es H, metilo, hidroxietilo, -CH2CONH2 o 3-pirrolidinilmetilo. En otros modos de realización, R7a es H o metilo.
En algunos modos de realización, los compuestos de Fórmula (I) o (I-A) son compuestos de Fórmula (IV):

en la que
M es CH o N;
X1 y X 1 ' son independientemente -C(R1 a )(R2a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk')-;
cada R1a y R2a es independientemente H, deuterio, alquilo C1 -6 , cicloalquilo C3-6, arilo C6 -10, -C(O)ORa', -C(O)NRa'Rb', -NRa'Rb', -SRa', -S(O)Ra', -S(O)NRa', -S(O)2 Ra', -S(O)2 NRa' u -ORa en los que cada átomo de hidrógeno del alquilo C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo C1.4 J2 , -NHC(O)alquilo C1.4 , -N(alquilo C1 -4 )C(O)alquilo C1.4 , -NHC(O)NHalquilo C1.4 , -N(alquilo C1 -4 )C(O)NHalquilo C1 -4 , -NHC(O)N(alquilo C1 -4 )2 , -N(alquilo C1 -4 )C(O)N(alquilo C1 -4 )2 , -NHC(O)Oalquilo C1 -4 , -N(alquilo C1 -4 )C(O)Oalquilo C1.4 , -CO2 H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1 -4 ), -CON(alquilo C1 -4 )2 , -Salquilo C1.4 , -S(O)alquilo C1.4 , -S(O)2alquilo C1.4 , -S(O)NH(alquilo C1 -4 ), -S(O)2 NH(alquilo C1.4 ), -S(O)N(alquilo C1.
4)2 , -S(O)2 N(alquilo C1 -4 )2 , cicloalquilo C3-6 o heterocicloalquilo de 3 a 7 miembros;
R3a y R3b son cada uno independientemente H, deuterio, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3;
R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por deuterio, halógeno, -CN, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo C1_4)2 , -CO2 H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1 -4 ), -CON(alquilo C1 -4 )2 , cicloalquilo o heterocicloalquilo monocíclico;
cada Rk’ es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk’ está independientemente opcionalmente sustituido por deuterio, halógeno, alquilo C1-6 , haloalquilo C1-6 u -ORa ’;
en el que cada Ra’ y Rb’ es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo;
cada Z1, Z2, Z3 , Z4 , Z5 , Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente es independientemente H, deuterio, halógeno, alquilo C1 -4 , -O-alquilo C1.4 , -OH, -NH2 , -NH(alquilo C1.4 ), -NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3 , Z4, Z5 , Z6 o Z7 sea N o NH; y
m' es 2 o 3;
o una sal farmacéuticamente aceptable del mismo.
En algunos modos de realización, Z1, Z4 y Z7 son N, y Z2 , Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1 y Z3 son N, Z7 es NH y Z2, Z4, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z3 y Z6 son N, Z7 es NH y Z2, Z4 y Z5 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3 es N, Z7 es NH y Z1, Z2, Z4, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3 y Z6 son N, Z7 es NH y Z1, Z2, Z4 y Z5 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z2 , Z4 y Z7 son N y Z1, Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z5 y Z7 son N y Z2 , Z3 , Z4 y Z6 son C(Rx), en los que cada Rx cuando está
presente es H. En algunos modos de realización, Z1, Z2, Z4 y Z7 son N y Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z2, Z5 y Z7 son N y Z3, Z4 y Z6 son C(Rx), en los
que cada Rx cuando está presente es H. En algunos modos de realización, Z3, Z5 y Z6 son N y Z1, Z2, Z4 y Z7 son
C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z5, Z6 y Z7 son N y Z2,
Z3 y Z4 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z2 y Z4
son N y Z3, Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización,
Z1, Z3 y Z4 son N y Z2, Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3 y Z4 son N y Z1, Z2, Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos
modos de realización, Z2, Z5 y Z7 son N y Z1, Z3, Z4 y Z6 son C(Rx), en los que cada Rx cuando está presente es H.
En algunos modos de realización, Z3 y Z5 son N y Z1, Z2, Z4, Z6 y Z7 son C(Rx), en los que cada Rx cuando está
presente es H. En algunos modos de realización, Z2, Z5, Z6 y Z7 son N y Z1, Z3 y Z4 son C(Rx), en los que cada Rx
cuando está presente es H.
En algunos modos de realización, Rk’ se selecciona del grupo que consiste en H, metilo, etilo, propiol, isopropilo, ciclopropilo, 2-hidroxietilo, 2-hidroxi-2-metil-propilo y N-metil-pirrol-3-ilo. En algunos modos de realización, M es
CH. En algunos modos de realización, M es c H, Z1, Z4 y Z7 son N y Z2, Z3, Z5 y Z6 son C(Rx), en los que cada Rx
cuando está presente es H. En algunos modos de realización, M es CH, Z1, Z4 y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx),
en los que cada Rx cuando está presente es H, y X1 es -N(Rk')-. En algunos modos de realización, M es CH, Z1, Z4
y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H, X1 es -N(Rk')- y X1' es -O-. En
algunos modos de realización, M es CH, Z1, Z4 y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando
está presente es H, X1 es -C(R1a)(R2a)- y X1' es -O-.
En algunos modos de realización, los compuestos de Fórmula (I) o (I-A) son compuestos de Fórmula (V):

en la que
M es CH o N;
X1 y X 1 ' son independientemente -C(R1 a )(R2a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk')-;
cada R1a y R2a es independientemente H, deuterio, alquilo C1 -6 , cicloalquilo C3-6, arilo C6 -10, -C(O)ORa', -C(O)NRa'Rb',
-NRa'Rb', -SRa', -S(O)Ra', -S(O)NRa', -S(O)2 Ra', -S(O)2 NRa' u -ORa en los que cada átomo de hidrógeno del alquilo
C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1.4 ), -N(alquilo C1.4 J2 , -NHC(O)alquilo C1.4 , -N(alquilo C1 -4 )c(O)alquilo C1.4 , -NHC(O)NHalquilo C1.4 , -N(alquilo C1 -4 )C(O)NHalquilo C1 -4 , -NHC(O)N(alquilo C1 -4 )2 , -N(alquilo C1 -4 )C(O)N(alquilo C1 -4 )2 , -NHC(O)Oalquilo
C1.4 , -N(alquilo C1 -4 )C(O)Oalquilo C1.4 , -CO2 H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1.4 ), -CON(alquilo ^ -4)2 ,
-Salquilo C1 -4 , -S(O)alquilo C1.4 , -S(O)2alquilo C1.4 , -S(O)NH(alquilo C1.4 ), -S(O)2 NH(alquilo C1.4 ), -S(O)N(alquilo C1.
4)2 , -S(O)2 N(alquilo C1 -4 )2 , cicloalquilo C3-6 o heterocicloalquilo de 3 a 7 miembros;
R3a y R3b son cada uno independientemente H, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3 ;
R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1 -4 ), -N(alquilo C1 -4 )2 , -CO2H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquil CON(alquilo C1 -4 )2 , cicloalquilo o heterocicloalquilo monocíclico;
cada Rk’ es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno
del alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk’ está independientemente opcionalmente sustituido por deuterio, halógeno,
alquilo C1-6 , haloalquilo C1-6 u -ORa ’;
en el que cada Ra’ y Rb’ es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo
C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo;
cada Z1, Z2, Z3 , Z4 , Z5 , Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente es independientemente H, deuterio, halógeno, alquilo C1 -4 , -O-alquilo C1 -4 , -OH, -NH2 , -NH(alquilo C1 -4 ), -NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3 , Z4, Z5 , Z6 o Z7 sea N o NH; y
m' es 2 o 3;
o una sal farmacéuticamente aceptable del mismo.
En algunos modos de realización, Z1, Z4 y Z7 son N, y Z2 , Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1 y Z3 son N, Z7 es NH y Z2, Z4, Z5 y Z6 son C(Rx), en los que
cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z3 y Z6 son N, Z7 es NH y Z2, Z4 y Z5
son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3 es N, Z7 es NH y
Z1, Z2, Z4, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3
y Z6 son N, Z7 es NH y Z1, Z2, Z4 y Z5 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos
de realización, Z2 , Z4 y Z7 son N y Z1, Z3, Z5 y Z6 son C(Rx), en los que cada Rx algunos modos de realización, Z1, Z5 y Z7 son N y Z2 , Z3 , Z4 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z2, Z4 y Z7 son N y Z3, Z5 y Z6 son C(Rx), en los que cada Rx
cuando está presente es H. En algunos modos de realización, Z1, Z2, Z5 y Z7 son N y Z3 , Z4 y Z6 son C(Rx), en los
que cada Rx cuando está presente es H. En algunos modos de realización, Z3 , Z5 y Z6 son N y Z1, Z2 , Z4 y Z7 son
C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z5 , Z6 y Z7 son N y Z2 ,
Z3 y Z4 son c(R x), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z2 y Z4
son N y Z3, Z5 , Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización,
Z1, Z3 y Z4 son N y Z2 , Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3 y Z4 son N y Z1, Z2 , Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos
modos de realización, Z2, Z5 y Z7 son N y Z1, Z3, Z4 y Z6 son C(Rx), en los que cada Rx cuando está presente es H.
En algunos modos de realización, Z3 y Z5 son N y Z1, Z2, Z4 , Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z2 , Z5, Z6 y Z7 son N y Z1, Z3 y Z4 son C(Rx), en los que cada Rx
cuando está presente es H.
En algunos modos de realización, Rk’ se selecciona del grupo que consiste en H, metilo, etilo, propiol, isopropilo, ciclopropilo, 2-hidroxietilo, 2-hidroxi-2-metil-propilo y N-metil-pirrol-3-ilo. En algunos modos de realización, M es
CH. En algunos modos de realización, M es CH, Z1, Z4 y Z7 son N y Z2 , Z3, Z5 y Z6 son C(Rx), en los que cada Rx
cuando está presente es H. En algunos modos de realización, M es CH, Z1, Z4 y Z7 son N, Z2 , Z3, Z5 y Z6 son C(Rx),
en los que cada Rx cuando está presente es H, y X1 es -N(Rk')-. En algunos modos de realización, M es CH, Z1, Z4
y Z7 son N, Z2, Z3 , Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H, X1 es -N(Rk')- y X 1 ' es -O-. En
algunos modos de realización, M es CH, Z1, Z4 y Z7 son N, Z2 , Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando
está presente es H, X1 es -C(R1 a )(R2a)- y X 1 ' es -O-.
En algunos modos de realización, los compuestos de Fórmula (I) o (I-A) son compuestos seleccionado del grupo
que consiste en

en la que
M es CH o N;
X 1 y X 1 ' son independientemente -C(R1 a )(R2 a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk')-; cada R1a y R2a es independientemente H, deuterio, alquilo C1 ’6, cicloalquilo C3-6, arilo C6 -10 , -C(O)ORa’, -C(O)NRa’Rb’, -NRa’Rb’, -SRa ’, -S(O)Ra’, -S(O)NRa’, -S(O)2 Ra’, -S(O)2 NRa’ o -ORa ’ en el que cada átomo de hidrógeno del alquilo C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1.
4), -N(alquilo C1 -4 )2 , -NHC(O)alquilo C1.4 , -N(alquilo C1 -4 )C(O)alquilo C1.4 , -NHC(O)NHalquilo C1.4 , -N(alquilo C1.
4)C(O)NHalquilo C1 -4 , -NHC(O)N(alquilo C1 -4 )2 , -N(alquilo C1 -4 )C(O)N(alquilo C1 -4)2 , -NHC(O)Oalquilo C1 -4 , -N(alquilo C1 -4 )C(O)Oalquilo C1.4 , -CO2 H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1 -4 ), -CON(alquilo C1 -4 )2 , -Salquilo C1.4 , -S(o)alquilo C1.4 , -S(O)2alquilo C1.4 , -S(O)NH(alquilo C1.4 ), -S(O)2 NH(alquilo C1.4 ), -S(O)N(alquilo C1 -4)2 , -S(O)2 N(alquilo C1 -4 )2 , cicloalquilo C3-6 o heterocicloalquilo de 3 a 7 miembros;
R3a y R3b son cada uno independientemente H, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3 ;
R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por halógeno, -OH, -Oalquilo C1 -4 , -NH2 , -NH(alquilo C1.4 ), -N(alquilo C1 -4 )2 , -CO2H, -CO2alquilo C1.4 , -CONH2 , -CONH(alquilo C1.4 ), -CON(alquilo C1 -4 )2 , cicloalquilo o heterocicloalquilo monocíclico;
cada Rk’ es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3.6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk’ está independientemente opcionalmente sustituido por deuterio, halógeno, alquilo C1-6 , haloalquilo C1-6 u -ORa ’;
en el que cada Ra’ y Rb’ es independientemente H, deuterio, alquilo C1 -6 , alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo;
cada Z1, Z2, Z3, Z4, Z5, Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente es independientemente H, deuterio, halógeno, alquilo C1 -4 , -O-alquilo C1.4 , -OH, -NH2 , -NH(alquilo C1.4 ), -NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3, Z4, Z5, Z6 o Z7 sea N o NH; y
m' es 2 o 3;
o una sal farmacéuticamente aceptable del mismo.
En algunos modos de realización, Z1, Z4 y Z7 son N, y Z2, Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1 y Z3 son N, Z7 es NH y Z2, Z4, Z5 y Z6 son C(Rx), en los que
cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z3 y Z6 son N, Z7 es NH y Z2, Z4 y Z5
son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3 es N, Z7 es NH y
Z1, Z2, Z4, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3
y Z6 son N, Z7 es NH y Z1, Z2, Z4 y Z5 son C(Rx), en los que cada Rx cuando está presente es H. de realización, Z2, Z4 y Z7 son N y Z1, Z3, Z5 y Z6 son C(Rx), en los que ca algunos modos de realización, Z1, Z5 y Z7 son N y Z2, Z3, Z4 y Z6 son C(Rx), en los que cada Rx cuando está
presente es H. En algunos modos de realización, Z1, Z2, Z4 y Z7 son N y Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z2, Z5 y Z7 son N y Z3, Z4 y Z6 son C(Rx), en los
que cada Rx cuando está presente es H. En algunos modos de realización, Z3, Z5 y Z6 son N y Z1, Z2, Z4 y Z7 son
C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z5, Z6 y Z7 son N y Z2,
Z3 y Z4 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z1, Z2 y Z4
son N y Z3, Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización,
Z1, Z3 y Z4 son N y Z2, Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos modos de realización, Z3 y Z4 son N y Z1, Z2, Z5, Z6 y Z7 son C(Rx), en los que cada Rx cuando está presente es H. En algunos
modos de realización, Z2, Z5 y Z7 son N y Z1, Z3, Z4 y Z6 son C(Rx), en los que cada Rx cuando está presente es H.
En algunos modos de realización, Z3 y Z5 son N y Z1, Z2, Z4, Z6 y Z7 son C(Rx), en los que cada Rx cuando está
presente es H. En algunos modos de realización, Z2, Z5, Z6 y Z7 son N y Z1, Z3 y Z4 son C(Rx), en los que cada Rx cuando está presente es H.
En algunos modos de realización, Rk’ se selecciona del grupo que consiste en H, metilo, etilo, propiol, isopropilo, ciclopropilo, 2-hidroxietilo, 2-hidroxi-2-metil-propilo y N-metil-pirrol-3-ilo. En algunos modos de realización, M es
CH. En algunos modos de realización, M es c H, Z1, Z4 y Z7 son N y Z2, Z3, Z5 y Z6 son C(Rx), en los que cada Rx
cuando está presente es H. En algunos modos de realización, M es CH, Z1, Z4 y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx),
en los que cada Rx cuando está presente es H, y X1 es -N(Rk')-. En algunos modos de realización, M es CH, Z1, Z4
y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando está presente es H, X1 es -N(Rk')- y X1' es -O-. En
algunos modos de realización, M es CH, Z1, Z4 y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx), en los que cada Rx cuando
está presente es H, X1 es -C(R1a)(R2a)- y X1' es -O-.
En otros modos de realización, el compuesto de Fórmula (I) o (I-A) se selecciona del grupo que consiste en (13R)-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona;
(13R)-12-cloro-11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-5-(2-hidroxietil)-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-5-(2-hidroxietil)-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 2-[(13R)-12-cloro-11-fluoro-13-metil-4-oxo-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-5(4H)-il]acetamida; 2-[12-cloro-11-fluoro-13-metil-4-oxo-6,7-dihidro-13H-1.15- etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-5(4H)-il]acetamida; (13R)-12-cloro-11 -fluoro-13-metil-5-(pirrolidin-2-ilmetil)-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-13-metil-5-(pirrolidin-2-ilmetil)-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-7-(hidroximetil)-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-7-(hidroximetil)-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13S)-11-fluoro-13-(fluorometil)-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 11-fluoro-13-(fluorometil)-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-13-ciclopropil-11-fluoro-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 13-ciclopropil-11 -fluoro-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-11 -fluoro-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 11 -fluoro-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-6-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-7-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (8R)-9-cloro-10-fluoro-8-metil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazaciclotridecin-17(14H)-ona; 9-cloro-10-fluoro-8-metil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazaciclotridecin-17(14H)-ona; (7R)-8-cloro-9-fluoro-7-metil-14.15- dihidro-2H,7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazaciclotridecin-16(13H)-ona; 8-cloro-9-fluoro-7-metil-14,15-dihidro-2H,7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazaciclotridecin-16(13H)-ona; (5R)-3-fluoro-5-metil-14,15-dihidro-5H,10H-9,7-(azenometeno)pirido[2,3-k]pirrolo[3,4-d][1,10,3,7]dioxadiazaciclotridecin-12(13H)-ona; 3-fluoro-5-metil-14,15-dihidro-5H,10H-9,7
(azenometeno)pirido[2,3-k]pirrolo[3,4-d][1,10,3,7]dioxadiazacidotridecin-12(13H)-ona; (5R)-3-fluoro-5,16-dimetil-13.14.15.16- tetrahidro-5H-9,7-(azenometeno)pirido[2,3-k]pin'olo[3,4-d][1,3,7,10]oxatriazaddotndedn-12(10H)-ona; 3-fluoro-5,16-dimetil-13,14,15,16-tetrahidro-5H-9,7-(azenometeno)pirido[2,3-k]pirrolo[3,4-d][1,3,7,10]oxatriazacidotridecin-12(10H)-ona; (13R)-12-doro-11-fluoro-5,13-dimetil-6,7-dihidro-2H,13H-1,15-(azenometeno)pirrolo[3,4-f][1,10,4]benzodioxazacidotridecin-4(5H)-ona; 12-doro-11-fluoro-5,13-dimetil-6,7-dihidro-2H,13H-1,15-(azenometeno)pirrolo[3,4-f][1,10,4]benzodioxazacidotridecin-4(5H)-ona; (7R)-8-cloro-9-fluoro-7,15-dimetil-14,15-dihidro-2H,7H-3,5-(azenometeno)pirazolo[3,4-f][1,10,4]benzodioxazacidotridecin-16(13H)-ona; 8-doro-9-fluoro-7,15-dimetil-14,15-dihidro-2H,7H-3,5-(azenometeno)pirazolo[3,4-f][1,10,4]benzodioxazacidotridecin-16(13H)-ona; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][l,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; (13R)-12-doro-11-fluoro-13,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 12-doro-11-fluoro-13,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 12-doro-11-fluoro-5,14-dimetil-6.7.13.14- tetrahidro-15,1-(azenometeno)pirazolo[4,3-f][1,4,10]benzoxadiazacidotridecin-4(5H)-ona; 12-cloro-11-fluoro-14-metil-6,7,13,14-tetrahidro-15,1-(azenometeno)pirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 12-cloro-11 -fluoro-14-metil-6,7,13,14-tetrahidro-1,15-(azenometeno)pirrolo[3,2-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 12-doro-11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-(azenometeno)pirrolo[3,2-f][1,4,10]benzoxadiazaddotridedn-4(5H)-ona; 9-doro-10-fluoro-7-metil-7,8,15,16-tetrahidro-3,6-etenoimidazo[5,1-f][1,4,7,8,10]benzoxatetraazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metil-7.8.15.16- tetrahidro-6,3-(azenometeno)imidazo[5,1-f][1,4,7,8,10]benzoxatetraazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metil-7,8,15,16-tetrahidro-6,3-(azenometeno)imidazo[5,1-f][1,4,7,10]benzoxatnazaddotndedn-17(14H)-ona; 9-doro-10-fluoro-7-metil-7,8,15,16-tetrahidro-3,6-(azenometeno)pirrolo[2,1-f][1,4,7,10]benzoxatriazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metil-7,8,15,16-tetrahidro-3,6-(azenometeno)imidazo[2,1-f][1,4,7,10]benzoxatriazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metil-7,8,15,16-tetrahidro-3,6-eteno[1,2,4]triazolo[3,4-f][1,4,7,8,10]benzoxatetraazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metil-7,8,15,16-tetrahidro-6,3-(azenometeno)[1,2,4]triazolo[3,4-f][1,4,7,10]benzoxatriazacidotridecin-17(14H)-ona;
8-doro-9-fluoro-6-metil-6,7,14,15-tetrahidro-2H-3,5-(azenometeno)pirrolo[3,4-f][1,4,8,10]benzoxatriazacidotridecin-16(13H)-ona; 8-doro-9-fluoro-6-metil-6,7,14,15-tetrahidro-2H-3,5-(azenometeno)pirazolo[3,4-f][1,4,8,10]benzoxatriazacidotridecin-16(13H)-ona; 8-doro-9-fluoro-6-metil-6,7,14,15-tetrahidro-2H-3,5-(azenometeno)pirazolo[3,4-f][1,4,10]benzoxadiazacidotridecin-16(13H)-ona; 12-cloro-11-fluoro-5.14- dimetil-6,7,l3,14-tetrahidro-2H-1,15-(azenometeno)pirrolo[3,4-f][1,4,10]benzoxadiazacidotridecin-4(5H)-ona; (8R)-10-fluoro-8,16-dimetil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecinl7(14H)-ona; 10-fluoro-8,16-dimetil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecin-17(14H)-ona; (7R)-9-fluoro-7,15-dimetil-14,15-dihidro-2H,7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazacidotridecin-l6(13H)-ona; y 9-fluoro-7,15-dimetil-14,15-dihidro-2H,7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazacidotridecin-16(13H)-ona; o una sal farmacéuticamente aceptable de los mismos.
En otros modos de realización, el compuesto de Fórmula (I) o (I-A) se selecciona del grupo que consiste en 12-cloro-11 -fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-3,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 10-fluoro-8-metil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatnazaddotridedn-17(14H)-ona; 10-fluoro-7-metil-7,8,15,16-tetrahidro-3,6-etenoimidazo[5,1-f][1,4,7,8,10]benzoxatetraazaciclotridecin-17(14H)-ona; 14-etil-11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-14-propil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-14-(propan-2-il)-6,7,13,14-tetrahidro-1.15- etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 14-ciclopropil-11 -fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-14-(2-hidroxietil)-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-6,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 14-metil-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatnazaddotridedn-4(5H)-ona; 11 -fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-13-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; (13R)-11-fluoro-13-metil-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 12-cloro-11-fluoro-13- metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridecin-4(5H)-ona; 11-fluoro-14- metil-4-oxo-4,5,6,7,13,14-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecina-7-carboxamida; 11-fluoro-7-(hidroximetil)-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 11-fluoro-13-metil-4-oxo-4,5,6,7,13,14-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecina-7-carboxamida; 11-fluoro-7-(hidroximetil)-13-metil-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 11-fluoro-4-oxo-4.5.6.7.13.14- hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecina-7-carboxamida; 11-fluoro-7-(hidroximetil)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 11-fluoro-4-oxo-4,5,6,7,13,14-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecina-13-carboxilato de metilo; 11 -fluoro-4-oxo-4,5,6,7,13,14-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecina-13-carboxamida; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f]pirido[3,2-1][1,4,8,10]oxatriazaciclotridecin-4(5H)-ona; 11-fluoro-13-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f]pirido[3,2-1][1,4,8,10]oxatriazaciclotridecin-4(5H)-ona; 11-fluoro-13-(propan-2-il)-6,7,13,14-tetrahidro-1,15etenopirazolo[4,3-f]pirido[3,2-1][1,4,8,10]oxatriazacidotridecin-4(5H)-ona; 13-ciclopropil-11-fluoro-6,7,13,14-tetrahidro-1,l5-etenopirazolo[4,3-f]pirido[3,2-1][1,4,8,10]oxatriazacidotridecin-4(5H)-ona; 13-ciclopropil-11 -fluoro-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 11-fluoro-13-(propan-2- il)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzoxatiadiazacidotridecin-4(5H)-ona; 11-fluoro-6,7-dihidro-13H-1.15- etenopirazolo[4,3-f][1,10,4,8]benzoxatiadiazacidotridecin-4(5H)-ona 14,l4-dióxido; 6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][10,1,4,8]benzoxatiadiazacidotridecin-4(5H)-ona; 14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][l,4,8,10]benzotiatriazacidotridecin-4(5H)-ona; 13-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][l,4,8,10]benzotiatriazacidotridecin-4(5H)-ona; 11-fluoro-6,7-dihidro-5H-1,15-etenopirazolo[3,4-e|11,1,2,4,8]benzoxatiatriazaciclotridecin-4(14H)-ona 13,13-dióxido; 11-fluoro-14-metil-6,7-dihidro-5H-1,15-etenopirazolo[3,4-e][11,1,2,4,8]benzoxatiatriazacidotridecin-4(14H)-ona 13,13-dióxido; 12-fluoro-15-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazacidotetradecin-4-ona; 12-fluoro-14-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazacidotetradecin-4-ona;
(14R)-12-fluoro-14-metil-5,6,7,8,14,15 -hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazaciclotetradecin-4-ona; 11-fluoro-7,14-dimetil-4,5,6,7,13,14-hexahidro-8H-1,15-etenopirazolo[3,4-e][2,4,10]benzotriazaciclotridecin-8-ona; 11-fluoro-7,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[3,4-e][7,2,4,10]benzoxatriazaciclotridecin-8(5H)-ona; 11-fluoro-7,14-dimetil-4,5,6,7,13,14-hexahidro-8H-1,15-etenopirazolo[3,4-e][2,4,7,10]benzotetraazaciclotridecin-8-ona; 11-fluoro-4,7,14-trimetil-4.5.6.7.13.14- hexahidro-8H-1,15-etenopirazolo[3,4-e][2,4,7,10]benzotetraazaciclotridecin-8-ona; 11 -fluoro-7,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[3,4-e]Í7,2,4,10]benzotiatriazaciclotridecin-8(5H)-ona; 11-fluoro-7.14- dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[3,4-e][7,2,4,10]benzotiatriazaciclotridecin-8(5H)-ona 4,4-dióxido; y 12-fluoro-8,15-dimetil-5,6,7,8,14,15-hexahidro-9H-1,16-etenopirazolo[3,4-e] [7,2,4,8,11]benzotiatetraazaciclotetradecin-9-ona 4,4-dióxido; o una sal farmacéuticamente aceptable de los mismos.
En otros modos de realización, el compuesto de Fórmula (I) o (I-A) se selecciona del grupo que consiste en 11-cloro-13-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatnazaciclotridecin-4(5H)-ona; 13-etil-11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-/][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 13-ciclobutil-11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotndecin-4(H)-ona; 11-fluoro-14-metil(6,6,7,7-2 H4)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotndecin-4(5H)-ona; 11-fluoro-13-fenil-6,7,13,14-tetrahidro-1,15-etenopirazoloÍ4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 13-(ciclopropilmetil)-11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f] [1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; (7R,14R)-12-fluoro-7-hidroxi-14-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazaciclotetradecin-4-ona; (7S,14R)-12-fluoro-7-hidroxi-14-metil-5.6.7.8.14.15- hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazaciclotetradecin-4-ona; (7R,13R)-11-fluoro-7,13-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotndecin-4(5H)-ona;
(7S,13R)-11-fluoro-7,13-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; (7R)-11-fluoro-7,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f] [1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; (6R)-11 -fluoro-6,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-fj[1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 12-fluoro-7-hidroxi-15-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazacidotetradecin-4-ona; (7S)-11-fluoro-7,14-dimetil-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 11 -fluoro-13-(hidroximetil)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotndecin-4(5H)-ona; 12-fluoro-14-(hidroximetil)-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g] [1,5,9,11]benzoxatriazaciclotetradecin-4-ona; 11-fluoro-13,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-fj[1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 11-fluoro-14-(2-hidroxi-2-metilpropil)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-fj[1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; 12-fluoro-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9]benzoxadiazacidotetradecin-4-ona; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzotiatriazaddotridedn-4(5H)-ona; 11-fluoro-14-(1-metilpirrolidin-3- il)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazadclotridedn-4(5H)-ona; 11 -fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-/][1,4,8,10]benzotiatriazaciclotridecin-4(5H)-ona 8-óxido; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-/][1,4,8,10]benzotiatriazaddotridedn-4(5H)-ona 8,8-dióxido; (7S)-11-fluoro-7-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8]benzoxadiazaciclotndecin-4(5H)-ona; (6S,13R)-11-fluoro-6,13-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; (6R,13R)-11 -fluoro-6,13-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-fj[1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; (7S,13S)-11-fluoro-13-(hidroximetil)-7-metil-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona; y 11-fluoro-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzoxatiadiazaciclotridecin-4(5H)-ona; o una sal farmacéuticamente aceptable de los mismos.
En otros modos de realización, el compuesto de Fórmula (I) o (I-A) se selecciona del grupo que consiste en (13R)-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-11 -fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona;
(13R)-12-cloro-11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-5-(2-hidroxietil)-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 2-[(13R)-12-cloro-11-fluoro-13metil-4-oxo-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazacidotridecin-5(4H)-il]acetamida; (13R)-12-doro-11-fluoro-13-met¡l-5-(p¡rrol¡d¡n-2-¡lmet¡l)-6,7-d¡h¡dro-13H-1,15-etenop¡razolo[4,3-f][1,10,4,8]benzod¡oxad¡azac¡dotr¡dedn-4(5H)-ona; (13R)-12-doro-11-fluoro-7-(h¡drox¡met¡l)-5,13-d¡met¡l-6,7-d¡h¡dro-13H-1,15-etenop¡razolo[4,3-f][1,10,4,8]benzod¡oxad¡azaddotr¡dedn-4(5H)-ona; (13S)-11 -fluoro-13-(fluoromet¡l)-5-met¡l-6,7-d¡h¡dro-13H-1,15-etenop¡razolo[4,3-f][1,10,4,8]benzod¡oxad¡azaddotr¡dedn-4(5H)-ona; (13R)-13-ddoprop¡l-11-fluoro-5-met¡l-6,7-d¡h¡dro-13H-1,15-etenop¡razolo[4,3-f][1,10,4,8]benzodioxadiazacidotridecin-4(5H)-ona; (13R)-11 -fluoro-13-met¡l-6,7-d¡h¡dro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazacidotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-13-met¡l-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazacidotridecin-4(5H)-ona; (8R)-9-cloro-10-fluoro-8-metil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecin-17(14H)-ona; (7R)-8-cloro-9-fluoro-7-metil-14,15-dihidro-2H,7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazacidotridecin-16(13H)-ona; (5R)-3-fluoro-5-met¡l-14,15-d¡h¡dro-5H,10H-9,7-(azenometeno)p¡ndo[2,3-k]p¡rrolo[3,4-d][1,10,3,7]dioxadiazacidotridecin-12(13H)-ona; (5R)-3-fluoro-5,16-d¡met¡l-13,14,15,16-tetrah¡dro-5H-9,7-(azenometeno)p¡r¡do[2,3-k]p¡rrolo[3,4-d][1,3,7,10]oxatr¡azaddotndedn-12(10H)-ona; (13R)-12-cloro-11-fluoro-5,13-dimetil-6,7-dihidro-2H,13H-1,15-(azenometeno)pirrolo[3,4-f][1,10,4]benzodioxazacidotridecin-4(5H)-ona; (7R)-8-doro-9-fluoro-7,15-d¡met¡l-14,15-d¡h¡dro-2H,7H-3,5-(azenometeno)p¡razolo[3,4-f] [1,10,4]benzod¡oxazac¡dotr¡dedn-16(13H)-ona; (13R)-12-doro-11-fluoro-13,14-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; (8R)-10-fluoro-8,16-d¡met¡l-15,16-d¡h¡dro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecin-17(14H)-ona; (7R)-9-fluoro-7,15-d¡met¡l-14,15-dihidro-2H,7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazacidotridecin-16(13H)-ona; (13R)-11-fluoro-13-met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-t][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona;
(14R)-12-fluoro-14-met¡l-5,6,7,8,14,15-hexah¡dro-4H-1,16-etenop¡razolo[4,3-g] [1,5,9,11]benzoxatr¡azaddotetradec¡n-4-ona; (7R,14R)-12-fluoro-7-h¡drox¡-14-met¡l-5,6,7,8,14,15-hexah¡dro-4H-1,16-etenop¡razolo[4,3-g][1,5,9,11]benzoxatr¡azaddotetradec¡n-4-ona; (7S,14R)-12-fluoro-7-h¡drox¡-14-met¡l-5,6,7,8,14,15-hexah¡dro-4H-1,16-etenop¡razolo[4,3-g][1,5,9,11]benzoxatr¡azaddotetradedn-4-ona; (7R,13R)-11-fluoro-7,13-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-t][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; (7S,13R)-11-fluoro-7,13-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-f][1,4,8,10]benzoxatr¡azaddotndedn-4(5H)-ona; (7R)-11-fluoro-7,14-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-t][1,4,8,10]benzoxatr¡azaddotr¡dedn-4(5H)-ona; (6R)-11-fluoro-6,14-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-fj[1,4,8,10]benzoxatr¡azaddotndedn-4(5H)-ona; (7S)-11-fluoro-7,14-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-t][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; (7S)-11-fluoro-7-met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-t][1,4,8]benzoxad¡azaddotndedn-4(5H)-ona; (6S,13r )-11 -fluoro-6,13-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-t][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; (6R,13R)-11-fluoro-6,13-d¡met¡l-6,7,13,14-tetrah¡dro-1,[4,3-t][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; y (7S,13S)-11-fluoro-13-(h¡drox¡met¡l)-7-met¡l-6,7,13,14-tetrah¡dro-1,15-etenop¡razolo[4,3-t][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; o una sal farmacéut¡camente aceptable de los m¡smos.
Lo s¡gu¡ente representa modos de real¡zac¡ón ¡lustrat¡vos de compuestos de Fórmula (I) o (I-A) (los compuestos son de acuerdo con la ¡nvenc¡ón en la med¡da en que forma parte de la Fórmula (IV) como se def¡ne en la re¡v¡nd¡cac¡ón 1):




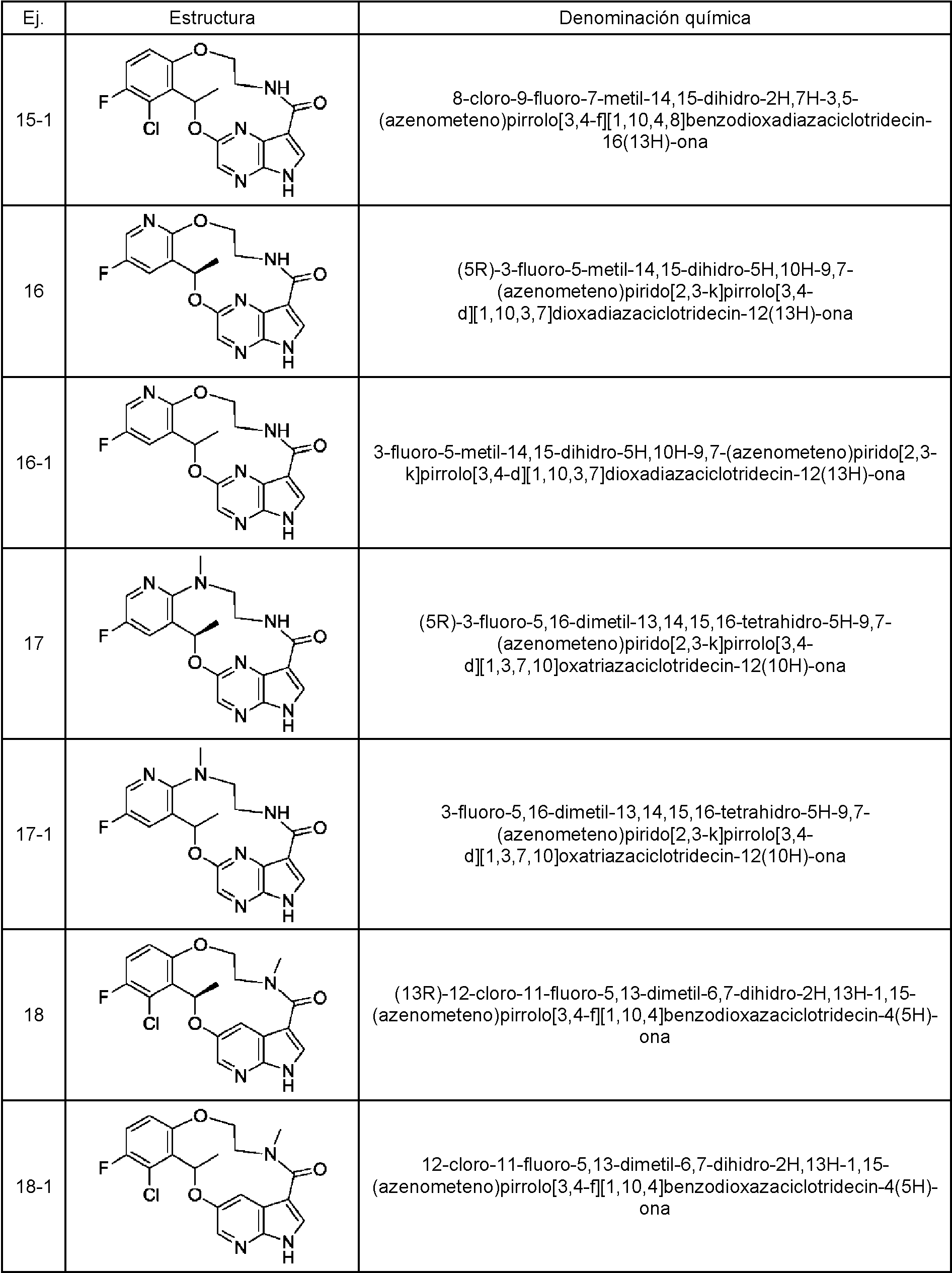



y sales farmacéuticamente aceptables de los mismos.
Lo siguiente representa modos de realización ilustrativos de compuestos de Fórmula (I) o (I-A):


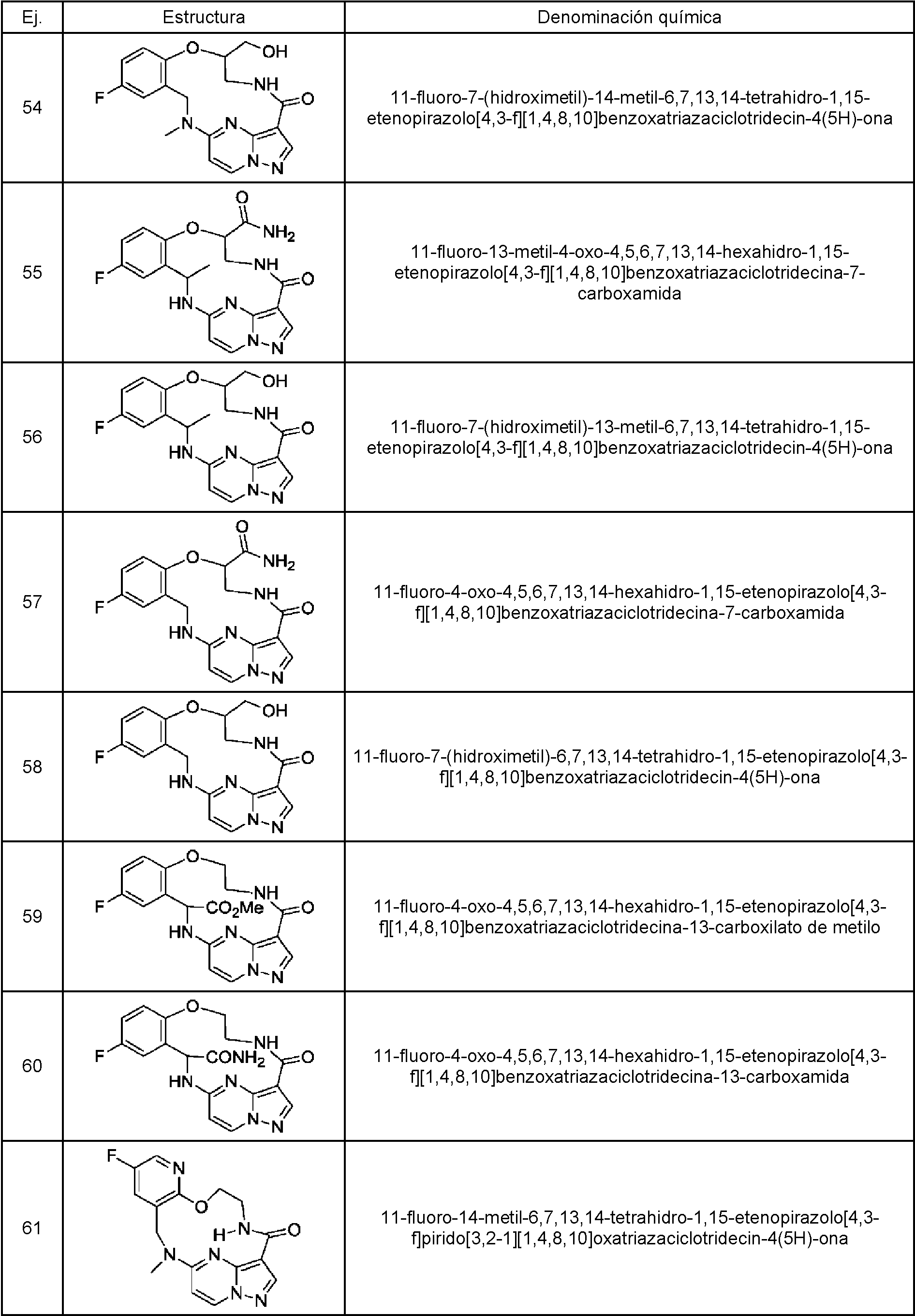




y sales farmacéuticamente aceptables de los mismos.
Lo siguiente representa modos de realización ilustrativos de compuestos de Fórmula (I) o (I-A):




y sales farmacéuticamente aceptables de los mismos.
Los expertos en la técnica reconocerán que las especies enumeradas o ilustradas en el presente documento no son exhaustivas, y que también se pueden seleccionar especies adicionales dentro del alcance de estos términos definidos.
COMPOSICIONES FARMACÉUTICAS
Para propósitos de tratamiento, las composiciones farmacéuticas que comprenden los compuestos descritos en el presente documento pueden comprender además uno o más excipientes farmacéuticamente aceptables. Un excipiente farmacéuticamente aceptable es una sustancia que no es tóxica y es, de otro modo, biológicamente adecuada para la administración a un sujeto. Dichos excipientes facilitan la administración de los compuestos descritos en el presente documento y son compatibles con el ingrediente activo. Los ejemplos de excipientes farmacéuticamente aceptables incluyen estabilizantes, lubricantes, tensioactivos, diluyentes, antioxidantes, aglutinantes, agentes colorantes, agentes voluminizadores, emulsionantes o agentes modificadores del sabor. En modos de realización preferentes, las composiciones farmacéuticas de acuerdo con la invención son composiciones estériles. Las composiciones farmacéuticas se pueden preparar usando técnicas de composición conocidas o que están disponibles para los expertos en la técnica.
La invención también contempla composiciones estériles, que incluyen composiciones que están de acuerdo con las regulaciones nacionales y locales que regulan dichas composiciones.
Los compuestos y composiciones farmacéuticos descritos en el presente documento se pueden formular como soluciones, emulsiones, suspensiones o dispersiones en disolventes o vehículos farmacéuticos adecuados, o como pastillas, comprimidos, pastillas para chupar, supositorios, sobres, grageas, gránulos, polvos, polvos para reconstitución o cápsulas junto con vehículos sólidos de acuerdo con procedimientos convencionales conocidos en la técnica para la preparación de diversas formas farmacéuticas. Las composiciones farmacéuticas de la invención se pueden administrar por una vía de administración adecuada, tal como por vía oral, parenteral, rectal, nasal, tópica u ocular, o por inhalación. Preferentemente, las composiciones se formulan para administración intravenosa u oral.
Para la administración oral, los compuestos de la invención se pueden proporcionar en forma sólida, tal como un comprimido o cápsula, o como una solución, emulsión o suspensión. Para preparar las composiciones orales, los compuestos de la invención se pueden formular para proporcionar una dosis de, por ejemplo, de aproximadamente 0,1 mg a 1 g al día, o de aproximadamente 1 mg a 50 mg al día, o de aproximadamente 50 a 250 mg al día, o de aproximadamente 250 mg a 1 g al día. Los comprimidos orales pueden incluir el ingrediente o los ingredientes activos mezclados con excipientes farmacéuticamente aceptables compatibles, tales como diluyentes, agentes desintegrantes, agentes aglutinantes, agentes lubricantes, agentes edulcorantes, agentes saborizantes, agentes colorantes y agentes conservantes. Los rellenos inertes adecuados incluyen carbonato de sodio y calcio, fosfato de sodio y calcio, lactosa, almidón, azúcar, glucosa, metilcelulosa, estearato de magnesio, manitol y sorbitol. Los excipientes orales líquidos ejemplares incluyen etanol, glicerol y agua. El almidón, la polivinilpirrolidona (PVP), el glicolato sódico de almidón, la celulosa microcristalina y el ácido algínico son agentes disgregantes ejemplares. Los agentes aglutinantes pueden incluir almidón y gelatina. El agente lubricante, si está presente, puede ser estearato de magnesio, ácido esteárico o talco. Si se desea, los comprimidos se pueden recubrir con un material tal como monoestearato de glicerilo o diestearato de glicerilo para retrasar la absorción en el tubo gastrointestinal, o se pueden recubrir con un recubrimiento entérico.
Las cápsulas para administración oral incluyen cápsulas de gelatina dura y blanda. Para preparar cápsulas de gelatina dura, el ingrediente o los ingredientes activos se pueden mezclar con un diluyente sólido, semisólido o líquido. Las cápsulas de gelatina blanda se pueden preparar mezclando el ingrediente activo con agua, un aceite, tal como aceite de cacahuete o aceite de oliva, parafina líquida, una mezcla de mono y diglicéridos de ácidos grasos de cadena corta, polietilenglicol 400 o propilenglicol.
Los líquidos para administración oral pueden estar en forma de suspensiones, soluciones, emulsiones o jarabes, o se pueden liofilizar o presentar como un producto seco para reconstituir con agua u otro vehículo adecuado antes de su uso. Dichas composiciones líquidas pueden contener opcionalmente: excipientes farmacéuticamente aceptables tales como agentes de suspensión (por ejemplo, sorbitol, metilcelulosa, alginato de sodio, gelatina, hidroxietilcelulosa, carboximetilcelulosa y gel de estearato de aluminio); vehículos no acuosos, por ejemplo, aceite (por ejemplo, aceite de almendra o aceite de coco fraccionado), propilenglicol, alcohol etílico o agua; conservantes (por ejemplo, metil o propil p-hidroxibenzoato o ácido sórbico); agentes humectantes tales como lecitina; y, si se desea, agentes aromatizantes o colorantes.
Para uso parenteral, incluyendo las vías intravenosa, intramuscular, intraperitoneal, intranasal o subcutánea, los agentes de la invención se pueden proporcionar en soluciones o suspensiones acuosas estériles, tamponadas a un pH e isotonicidad apropiados o en un aceite parenteralmente aceptable. Los vehículos acuosos adecuados incluyen la solución de Ringer y el cloruro de sodio isotónico. Dichas formas se pueden presentar en forma de dosis unitaria tal como ampollas o dispositivos de inyección desechables, en formas multidosis tales como viales
de los que se puede retirar la dosis apropiada, o en una forma sólida o preconcentrada que se puede usar para preparar una formulación inyectable. Las dosis de infusión ilustrativas varían de aproximadamente 1 a 1000 |jg/kg/minuto de agente mezclado con un vehículo farmacéutico durante un período que varía de varios minutos a varios días.
Para administración nasal, inhalada u oral, las composiciones farmacéuticas según la invención se pueden administrar usando, por ejemplo, una formulación en aerosol que también contiene un vehículo adecuado. Las composiciones según la invención se pueden formular para administración rectal como un supositorio.
Para aplicaciones tópicas, los compuestos de la presente invención se formulan preferentemente como cremas o pomadas o un vehículo similar adecuado para la administración tópica. Para la administración tópica, los compuestos según la invención se pueden mezclar con un vehículo farmacéutico a una concentración de aproximadamente 0,1 % a aproximadamente 10% de fármaco en el vehículo. Otro modo de administrar los agentes de la invención puede utilizar una formulación en parche para efectuar la administración transdérmica.
Como se usa en el presente documento, los términos "tratar" o "tratamiento" engloban tanto el tratamiento "preventivo" como el "curativo". El tratamiento "preventivo" pretende indicar un aplazamiento del desarrollo de una enfermedad, un síntoma de una enfermedad o una afección médica, la supresión de los síntomas que pueden aparecer o la reducción del riesgo de desarrollar o de la recidiva de una enfermedad o síntoma. El tratamiento "curativo" incluye la reducción de la gravedad o la supresión del empeoramiento de una enfermedad, síntoma o afección existente. Por tanto, el tratamiento incluye mejorar o prevenir el empeoramiento de los síntomas de la enfermedad existente, prevenir la aparición de síntomas adicionales, mejorar o prevenir las causas sistémicas subyacentes de los síntomas, inhibir el trastorno o la enfermedad, por ejemplo, detener el desarrollo del trastorno o la enfermedad, aliviar el trastorno o enfermedad, causar la regresión del trastorno o enfermedad, aliviar una afección causada por la enfermedad o trastorno, o detener los síntomas de la enfermedad o trastorno.
El término "sujeto" se refiere a un paciente mamífero que necesita dicho tratamiento, tal como un humano.
Las enfermedades ejemplares incluyen cáncer, dolor, enfermedades neurológicas, enfermedades autoinmunitarias e inflamación. El cáncer incluye, por ejemplo, cáncer de pulmón, cáncer de colon, cáncer de mama, cáncer de próstata, carcinoma hepatocelular, carcinoma de células renales, cánceres gástricos y esófago-gástricos, glioblastoma, cánceres de cabeza y cuello, tumores miofibroblásticos inflamatorios y linfoma anaplásico de células grandes. El dolor incluye, por ejemplo, el dolor de cualquier fuente o etiología, incluyendo el dolor del cáncer, el dolor del tratamiento quimioterápico, el dolor de los nervios, el dolor de una lesión u otras fuentes. Las enfermedades autoinmunitarias incluyen, por ejemplo, artritis reumatoide, síndrome de Sjogren, diabetes tipo I y lupus. Las enfermedades neurológicas ejemplares incluyen la enfermedad de Alzheimer, la enfermedad de Parkinson, la esclerosis lateral amiotrófica y la enfermedad de Huntington. Las enfermedades inflamatorias ejemplares incluyen ateroesclerosis, alergia e inflamación por infección o lesión.
En un aspecto, los compuestos y composiciones farmacéuticas de la invención se dirigen específicamente a las tirosina cinasas receptoras, en particular MET, ALK, AXL, TRK y JAK. Por tanto, estos compuestos y composiciones farmacéuticas se pueden usar para prevenir, revertir, retardar o inhibir la actividad de una o más de estas cinasas. En modos de realización preferentes, los compuestos para su uso en procedimientos de tratamiento se dirigen al cáncer. En otros modos de realización, los compuestos para su uso en procedimientos son para tratar cáncer de pulmón o carcinoma de pulmón no microcítico.
En los procedimientos inhibidores divulgados en el presente documento, una "cantidad eficaz" significa una cantidad suficiente para inhibir la proteína diana. La medición de dicha modulación objetivo se puede realizar mediante procedimientos analíticos de rutina tales como los que se describen a continuación. Dicha modulación es útil en una variedad de entornos, incluyendo los ensayos in vitro. En dichos procedimientos, la célula es preferentemente una célula cancerosa con señalización anómala debido a la regulación por incremento de MET, ALK, AXL, TRK y/o JAK.
En el compuesto para su uso en los procedimientos de tratamiento de acuerdo con la invención, una "cantidad eficaz" significa una cantidad o dosis suficiente para lograr en general aproximadamente el beneficio terapéutico deseado en sujetos que necesitan dicho tratamiento. Las cantidades o dosis eficaces de los compuestos de la invención se pueden determinar mediante procedimientos de rutina, tales como modelado, aumento de la dosis o ensayos clínicos, teniendo en cuenta factores de rutina, por ejemplo, el modo o la vía de administración o la administración del fármaco, la farmacocinética del agente, la gravedad y el curso de la infección, el estado de salud, la afección y el peso del sujeto, y el criterio del médico tratante. Una dosis ejemplar está en el intervalo de aproximadamente 0,1 mg a 1 g al día, o de aproximadamente 1 mg a 50 mg al día, o de aproximadamente 50 a 250 mg al día, o de aproximadamente 250 mg a 1 g al día. La dosificación total se puede administrar en unidades de dosificación únicas o divididas (por ejemplo, BID, TID, QID).
Una vez que se ha producido una mejoría de la enfermedad del paciente, la dosis se puede ajustar para un tratamiento preventivo o de mantenimiento. Por ejemplo, la dosificación o la frecuencia de administración, o ambas, se pueden reducir en función de los síntomas, hasta un nivel en el que se mantiene el efecto terapéutico o
profiláctico deseado. Por supuesto, si los síntomas se han aliviado hasta un nivel adecuado, el tratamiento puede cesar. Sin embargo, los pacientes pueden requerir tratamiento intermitente a largo plazo ante cualquier recidiva de los síntomas. Los pacientes también pueden requerir tratamiento crónico a largo plazo.
COMBINACIONES FARMACOLÓGICAS
Los compuestos según la invención descritos en el presente documento se pueden usar en composiciones farmacéuticas o procedimientos en combinación con uno o más ingredientes activos adicionales en el tratamiento de las enfermedades y trastornos descritos en el presente documento. Otros ingredientes activos adicionales incluyen otras tratamientos o agentes que mitigan los efectos adversos de los tratamiento para los objetivos de la enfermedad previstos. Dichas combinaciones pueden servir para aumentar la eficacia, mejorar otros síntomas de la enfermedad, disminuir uno o más efectos secundarios o disminuir la dosis requerida de un compuesto según la invención. Los ingredientes activos adicionales se pueden administrar en una composición farmacéutica separada de un compuesto de la presente invención o se pueden incluir con un compuesto de la presente invención en una única composición farmacéutica. Los ingredientes activos adicionales se pueden administrar simultáneamente con, antes de o después de la administración de un compuesto de la presente invención.
Los agentes de combinación incluyen ingredientes activos adicionales, son aquellos que se conocen o se descubre que son eficaces en el tratamiento de las enfermedades y trastornos descritos en el presente documento, incluyendo aquellos que son activos contra otra diana asociada con la enfermedad. Por ejemplo, las composiciones y formulaciones de la invención, así como los compuestos para su uso en procedimientos de tratamiento, pueden comprender además otros fármacos o productos farmacéuticos, por ejemplo, otros agentes activos útiles para tratar o paliar las enfermedades diana o síntomas o afecciones relacionados. Para indicaciones de cáncer, dichos agentes adicionales incluyen inhibidores de cinasas, tales como inhibidores de EGFR (por ejemplo, erlotinib, gefitinib), inhibidores de Raf (por ejemplo, vemurafenib), inhibidores de VEGFR (por ejemplo, sunitinib), agentes de quimioterapia estándar tales como agentes alquilantes, antimetabolitos, antibióticos antitumorales, inhibidores de topoisomerasa, fármacos de platino, inhibidores mitóticos, anticuerpos, tratamientos hormonales o corticosteroides. Para indicaciones de dolor, los agentes de combinación adecuados incluyen antiinflamatorios tales como los AINE. Las composiciones farmacéuticas de la invención pueden comprender adicionalmente uno o más de dichos agentes activos, y los compuestos para su uso en procedimientos de tratamiento pueden comprender adicionalmente administrar una cantidad eficaz de uno o más de dichos agentes activos.
SÍNTESIS QUÍMICA
Las entidades químicas ejemplares útiles en compuestos para su uso en procedimientos de la invención se describirán ahora mediante referencia a esquemas sintéticos ilustrativos para su preparación general a continuación y los ejemplos específicos que siguen. Los expertos reconocerán que, para obtener los diversos compuestos en el presente documento, los materiales de partida se pueden seleccionar de manera adecuada de modo que los sustituyentes finalmente deseados se lleven a través del esquema de reacción con o sin protección según sea apropiado para proporcionar el producto deseado. De forma alternativa, puede ser necesario o deseable emplear, en lugar del sustituyente finalmente deseado, un grupo adecuado que se pueda llevar a través del esquema de reacción y reemplazar según sea apropiado con el sustituyente deseado. Además, un experto en la técnica reconocerá que las transformaciones mostradas en los esquemas a continuación se pueden realizar en cualquier orden que sea compatible con la funcionalidad de los grupos salientes particulares. Cada una de las reacciones representadas en los esquemas generales se realiza preferentemente a una temperatura de aproximadamente 0 °C a la temperatura de reflujo del disolvente orgánico usado. A menos que se especifique de otro modo, las variables son como se define anteriormente en referencia a la Fórmula (I). Los compuestos marcados isotópicamente como se describen en el presente documento se preparan de acuerdo con los procedimientos descritos a continuación, usando materiales de partida adecuadamente marcados. Dichos materiales están en general disponibles de proveedores comerciales de reactivos químicos radiomarcados.
Procedimiento general A:

Se apreciará que los compuestos de fórmula A o A-1 se pueden preparar de acuerdo con el Procedimiento general A usando materiales de partida y productos intermedios apropiadamente funcionalizados.
Etapa 1. A una solución de un compuesto apropiadamente funcionalizado A-1 (~1,00 eq.), donde RA y RB son grupos compatibles con las condiciones de reacción descritas en el presente documento y Nu es un grupo nucleofílico tal como un anión o un grupo capaz de formar un nucleófilo, tal como un haluro, en un reactivo capaz de promover la combinación de A-1 y A-2, tal como un ácido (por ejemplo, TfOH (0,6 M)) o un haluro de alquilo (por ejemplo, n-BuLi), se puede añadir A-2, donde RC es un grupo compatible con las condiciones de reacción descritas en el presente documento y X2 es, por ejemplo, un grupo saliente (~1,00eq.), a una temperatura apropiada (por ejemplo, 0 °C). La mezcla se puede agitar a una temperatura apropiada (por ejemplo, 60 °C) hasta que se complete la reacción. A continuación, la reacción se puede devolver a temperatura ambiente, y la mezcla de reacción se puede extinguir, neutralizar, lavar, extraer, secar y/o concentrar a vacío, según sea necesario para obtener A-3.
Etapa 2. Una mezcla de A-3, donde RA, RB y RC son grupos compatibles con las condiciones de reacción descritas en el presente documento (en algunos modos de realización ejemplares descritos en el presente documento, A-3 puede ser un aldehído o cetona disponible comercialmente, o A-3 se puede preparar a partir de la etapa 1 , ~1,00 eq.), y una amina A-4 disponible comercialmente, donde RC es un grupo compatible con las condiciones de reacción descritas en el presente documento, (~1,50 eq.) en un disolvente apropiado (por ejemplo, metanol (0,5 M)) se puede agitar a una temperatura apropiada (por ejemplo, temperatura ambiente) durante un periodo de tiempo apropiado o hasta que la formación de imina se complete por TLC o LC-MS. A la solución de reacción se le puede añadir un agente reductor (por ejemplo, NaBH4 (~2,00 eq.)) en porciones. La mezcla se puede agitar a una temperatura apropiada (por ejemplo, temperatura ambiente) hasta que la TLC o LC-MS muestre que la reducción se ha completado. La reacción se puede extinguir, lavar, extraer, secar y/o concentrar a vacío según sea necesario para proporcionar A-5.
Etapa 3. Una mezcla de un A-5 disponible comercialmente o preparado, donde RA, RB y RC son grupos compatibles con las condiciones de reacción descritas en el presente documento (~1 eq.), 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo disponible comercialmente [(A-6, ~1 eq.) y una base apropiada (por ejemplo, diisopropiletilamina (~5 eq.)) en un disolvente apropiado (por ejemplo, butanol (0,4 M)) se puede agitar a una temperatura adecuada (por ejemplo, 110 °C) durante un periodo de tiempo determinado o hasta que se demuestre que la reacción se ha completado. La reacción se puede devolver a temperatura ambiente y diluir con agua según sea necesario. La mezcla se puede extraer, lavar, secar, concentrar bajo presión reducida y/o purificar por procedimientos cromatográficos según sea necesario para proporcionar A.
En algunos modos de realización ejemplares, el Procedimiento general A se puede llevar a cabo como sigue:

Etapa 1. A una solución de A-1 (1,00 eq.) en TfOH (0,6 M) se puede añadir A-2 (1,00 eq.) a 0 °C. La mezcla se puede agitar a 60 °C durante 4 horas o hasta que se complete la reacción. Después de enfriar a temperatura ambiente, la mezcla de reacción se puede verter en hielo-agua (w/w = 1/1), neutralizar con NaHCO3 a pH ~ 9 y extraer con EtOAc tres veces según sea necesario. Las fases orgánicas combinadas se pueden lavar con salmuera, secar sobre Na2SO4 anhidro según sea necesario y concentrar para dar A-3.
Etapa 2. Una mezcla de A-3 (aldehído o cetona disponible comercialmente o preparado a partir de la etapa 1, 1,00 eq.) y la amina A-4 disponible comercialmente (1,50 eq.) en metanol (0,5 M) se puede agitar a temperatura ambiente durante 2 horas o hasta que se demuestre por TLC o LC-MS que la formación de imina se ha completado. A la solución de reacción se le puede añadir NaBH4 (2,00 eq.) en porciones. La mezcla se puede agitar a temperatura ambiente hasta que la TLC o LC-MS muestre que la reducción se ha completado. La reacción se puede extinguir con agua y extraer tres veces con diclorometano, según sea necesario. La fase orgánica combinada se puede lavar con salmuera, secar con Na2SO4 anhidro, filtrar y concentrar a vacío para dar A-5.
Etapa 3. El A-5 preparado o comercial disponible (1 eq.), 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo (A-6 , 1 eq.) y diisopropiletilamina (5 eq.) en butanol (0,4 M) se puede calentar a 110 °C durante 30 minutos o hasta que se demuestre que la reacción se ha completado. La reacción se puede enfriar y diluir con agua. La mezcla se puede extraer con diclorometano cuatro veces (según sea necesario) y los extractos combinados se pueden secar sobre sulfato de sodio anhidro. Después de la filtración, la mezcla se puede concentrar bajo presión reducida y el residuo se puede purificar mediante cromatografía ultrarrápida para proporcionar A.
Procedimiento general A alternativo:

Etapa 1 de acoplamiento. La mezcla de un AA-1 apropiadamente funcionalizado (~1,00eq.), un reactivo de acoplamiento de vinilo apropiadamente funcionalizado (~1,00-1,50 eq.) y un catalizador de paladio (~0,05 eq.) en condiciones de reacción apropiadas se puede calentar a una temperatura apropiada (por ejemplo, ~90 °C) durante un período de tiempo apropiado bajo atmósfera inerte hasta que la TLC indique que el material de partida se ha consumido completamente. La mezcla de reacción se puede verter en H2O según sea necesario. La mezcla se
puede extraer y la fase orgánica se puede lavar, secar, concentrar y purificar mediante cromatografía en columna en gel de sílice, según sea necesario, para dar AA-2.
Etapa 2 de acoplamiento. La mezcla de un compuesto del tipo AA-2 (~1,00 eq.), 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo (A-6, ~1,00 eq.) y un catalizador de paladio en condiciones de reacción apropiadas se puede calentar a una temperatura apropiada (por ejemplo, 120 °C) durante un período de tiempo apropiado bajo atmósfera inerte hasta que la TLC indique que el material de partida se ha consumido completamente. La mezcla de reacción se puede verter en H2O según sea necesario. La mezcla se puede extraer y la fase orgánica se puede lavar, secar, concentrar y purificar mediante cromatografía en columna en gel de sílice, según sea necesario, para dar AA-3.
Etapa 3. A una mezcla de AA-3 (~1,00 eq.) y 4-metilbencenosulfonohidracida (en exceso molar) en un disolvente adecuado se puede añadir una base apropiada (en exceso molar) a una temperatura apropiada bajo atmósfera inerte. La mezcla se puede calentar a una temperatura apropiada (por ejemplo, 65 °C) y agitar durante un periodo de tiempo apropiado hasta que la TLC indique que la reacción se ha completado. La mezcla se puede enfriar y concentrar bajo presión reducida según sea necesario. La mezcla de reacción concentrada se puede diluir con agua según sea necesario y se puede extraer. La fase orgánica combinada se puede lavar, secar, filtrar, concentrar a vacío y purificar para dar AA-4.
Procedimiento general B:

Etapa 1. Una solución de aldehído B-1 (~1,0 eq.) donde RA y RB son grupos compatibles con las condiciones de reacción descritas en el presente documento, B-2 (~1,0 eq.) donde X1 es un grupo saliente y PG es un grupo protector, una base adecuada (en exceso molar) y un catalizador en un disolvente adecuado se puede calentar y agitar durante un periodo de tiempo apropiado hasta que la reacción se complete. Se puede añadir B-2 adicional y se puede calentar adicionalmente según sea necesario. La mezcla se puede enfriar a temperatura ambiente y diluir con agua según sea necesario. La mezcla se puede extraer y los extractos combinados se pueden lavar, secar y concentrar bajo presión reducida, según sea necesario. El producto de reacción bruto se puede purificar mediante cromatografía ultrarrápida para proporcionar B-3.
Etapa 2. El aldehído B-3 (~1,0 eq.) y una amina apropiadamente funcionalizada (~2,0-4,0 eq.) donde RC es un grupo compatible con las condiciones de reacción descritas en el presente documento en un disolvente apropiado se pueden calentar y agitar durante un periodo de tiempo apropiado. La mezcla se puede enfriar a temperatura ambiente y se puede añadir un agente reductor adecuado (~1,0 eq.). La mezcla se puede agitar durante un periodo de tiempo adecuado y a continuación se puede extinguir mediante la adición de agua según sea necesario. La mezcla se puede extraer con un disolvente orgánico apropiado y los extractos combinados se pueden lavar, secar y concentrar bajo presión reducida, según sea necesario. El producto de reacción bruto se puede purificar mediante cromatografía ultrarrápida según sea necesario para proporcionar B-4.
Etapa 3. El compuesto B-4 (~1,0 eq.), 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo (A-6, ~1,0 eq.) y una base adecuada (en exceso molar) en un disolvente adecuado se pueden calentar durante un periodo de tiempo apropiado. La reacción se puede enfriar y diluir con agua según sea necesario. La mezcla se puede extraer con un disolvente orgánico adecuado y los extractos combinados se pueden secar y concentrar bajo presión reducida según sea necesario. El producto de reacción bruto se puede purificar mediante cromatografía ultrarrápida para proporcionar B1.
En algunos modos de realización ejemplares, el Procedimiento general B se puede llevar a cabo como sigue:
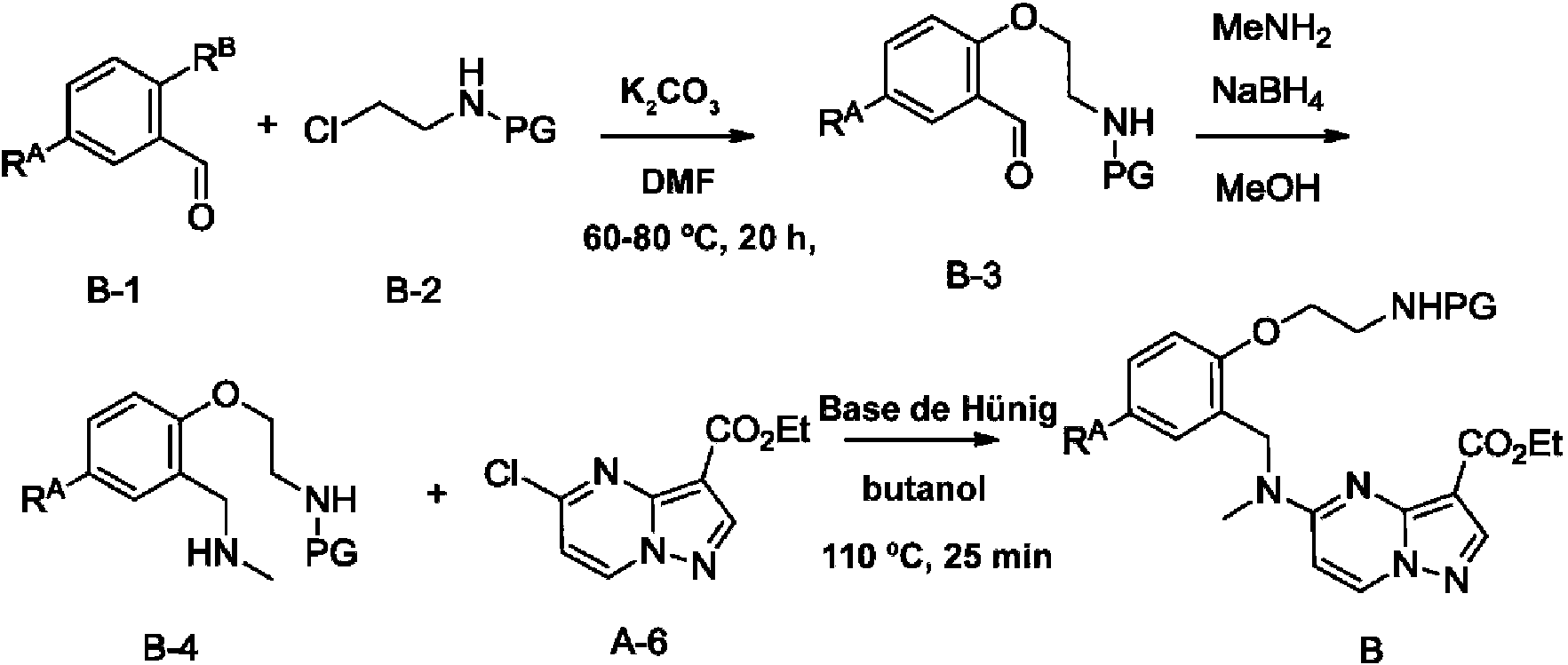
Etapa 1. Una solución de aldehido B-1 (~1,0 eq.) donde RA y RB son grupos compatibles con las condiciones de reacción descritas en el presente documento, B-2 (~1,0 eq.) donde X1 es un grupo saliente y PG es un grupo protector, carbonato de potasio (en exceso molar) y yoduro de potasio (cantidad catalítica) en DMF se puede calentar a 60 °C y agitar durante ~15 horas. Se puede añadir cloruro de B-2 adicional y se puede calentar adicionalmente a 80 °C según sea necesario hasta que se demuestre que la reacción se ha completado. La mezcla se puede enfriar a temperatura ambiente y diluir mediante la adición de agua (250 ml) según sea necesario. La mezcla se puede extraer con acetato de etilo (3 x 300 ml) y los extractos combinados se pueden lavar con agua ( 200 ml) y salmuera ( 100 ml), se pueden secar con sulfato de sodio y se pueden concentrar bajo presión reducida según sea necesario. El producto de reacción bruto se puede purificar mediante cromatografía ultrarrápida para proporcionar B-3.
Etapa 2. El aldehído B-3 (~1,0 eq.) y metilamina (~2,5 eq.) en metanol se pueden calentar a 60 °C y agitar durante ~1 hora. La mezcla se puede enfriar a temperatura ambiente y se puede añadir borohidruro de sodio (~1,0 eq.). La mezcla se puede agitar durante ~30 minutos y a continuación se puede extinguir mediante la adición de agua (200 ml) según sea necesario. La mezcla se puede extraer con diclorometano y los extractos combinados se pueden lavar con salmuera (50 ml), se pueden secar con sulfato de sodio y concentrar bajo presión reducida según sea necesario. El producto de reacción bruto se puede purificar mediante cromatografía ultrarrápida para proporcionar B-4.
Etapa 3. La amina B-4 (~1,0 eq.), 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo (A-6, ~1,0 eq.) y la base de Hünig (en exceso molar) en butanol se pueden calentar a 110 °C durante ~25 minutos. La reacción se puede enfriar y diluir con agua (250 ml). La mezcla se puede extraer con diclorometano y los extractos combinados se pueden secar con sulfato de sodio según sea necesario. La mezcla se puede concentrar bajo presión reducida según sea necesario. El producto de reacción bruto se puede purificar mediante cromatografía ultrarrápida para proporcionar B.
Procedimiento general C:

Etapa 1. A una solución de C-1 (~1,0 eq.) donde RA, RB, RC, RD y RE son grupos compatibles con las condiciones de reacción descritas en el presente documento, X1-AlkNHPG (~1,5-2,0 eq.) donde X1 es un grupo saliente, Alk es un grupo alquilo apropiadamente funcionalizado y PG es un grupo protector en un disolvente adecuado se puede añadir una base adecuada (~3,0 eq.). La mezcla se puede calentar a una temperatura apropiada durante un periodo de tiempo apropiado bajo atmósfera inerte hasta que el LC-MS muestre la conversión completa del material de partida en el producto. La mezcla se puede enfriar a temperatura ambiente, diluir con agua y extraer con un disolvente orgánico adecuado según sea necesario. Los extractos orgánicos combinados se pueden lavar con agua y salmuera, secar sobre Na2SO4 y concentrar según sea necesario. El residuo resultante se puede purificar mediante cromatografía en columna en gel de sílice según sea necesario para dar C-2.
Etapa 2. A una solución de C-2 (1 eq.) donde RA, RC, RD y RE son grupos compatibles con las condiciones de reacción descritas en el presente documento, Alk es un grupo alquilo apropiadamente funcionalizado y PG es un grupo protector en un disolvente adecuado se puede añadir una base adecuada (en exceso molar). La solución se puede calentar a una temperatura adecuada durante un periodo de tiempo apropiado. La reacción se puede neutralizar con un ácido adecuado a pH < 5 y la mezcla de reacción se puede extraer con un disolvente orgánico adecuado. Los extractos orgánicos combinados se pueden lavar y secar según sea necesario. La mezcla bruta del producto de reacción se puede filtrar, concentrar bajo presión reducida y secar a alto vacío según sea necesario para proporcionar C-3.
Etapa 3. A una solución de C-3 (~1,0 eq.) en un disolvente orgánico adecuado se puede añadir un ácido adecuado (~4 eq.) a una temperatura apropiada (por ejemplo, 0 °C). La mezcla de reacción se puede agitar a una temperatura apropiada durante un periodo de tiempo apropiado hasta que se demuestre por LC-MS que la reacción se ha completado. El producto bruto se puede filtrar, lavar y secar a alto vacío para proporcionar un C-4.
Etapa 4a: A una solución de C-4 (~1,0 eq.) en un disolvente adecuado se puede añadir una base adecuada (en exceso molar). La solución se puede enfriar en un baño de agua con hielo y se puede añadir un agente de acoplamiento adecuado (~1,5eq.) para producir un éster activado. La solución se puede llevar a temperatura ambiente lentamente y agitar hasta que se demuestre por LC-MS que el material de partida se ha convertido en el producto deseado. La mezcla se puede diluir con agua y extraer con un disolvente orgánico adecuado según sea necesario. Los extractos orgánicos combinados se pueden lavar, secar y concentrar bajo presión reducida según sea necesario. El residuo resultante se puede purificar mediante una cromatografía en columna en gel de sílice para dar C.
En algunos modos de realización ejemplares, el Procedimiento general C se puede llevar a cabo como sigue:

Etapa 1. A una solución de C-1 (~1,0 eq.) donde RA, RB, RC, RD y RE son grupos compatibles con las condiciones de reacción descritas en el presente documento, X1-AlkNHPG (~1,5-2,0 eq.) donde X1 es un grupo saliente, Alk es un grupo alquilo apropiadamente funcionalizado y PG es un grupo protector en DMF (0,5 M) se puede añadir K2CO3 (~3,0 eq.). La mezcla se puede calentar a ~80 °C durante ~2 horas o hasta que se pueda demostrar por LC-MS la conversión completa del material de partida en el producto. La mezcla se puede enfriar a temperatura ambiente, diluir con agua según sea necesario y extraer tres veces con EtOAc según sea necesario. Las fases orgánicas combinadas se pueden lavar con agua y salmuera, secar sobre Na2SO4 y concentrar según sea necesario. El residuo resultante se puede purificar mediante cromatografía en columna en gel de sílice eluyendo con EtOAc/hexano (5-100 %, 10 V.C.) para dar C-2.
Etapa 2. A una solución de C-2 (~1 eq.) en metanol/THF/H2O (3:1:1, 0,2 M) se puede añadir LOH.H2O (~5,0 eq.). La solución se puede calentar a ~70 °C durante ~2 horas. La reacción se puede neutralizar a ~0 °C con HCl ac. (2 M) a pH < 5 y se puede extraer cuatro veces con CH2Cl2 según sea necesario. Los extractos orgánicos combinados se pueden lavar con salmuera y se pueden secar sobre Na2SO4 según sea necesario. La mezcla bruta del producto se puede filtrar, concentrar bajo presión reducida y secar a alto vacío según sea necesario para proporcionar C-3.
Etapa 3. A una solución de C-3 (~1,0 eq.) en CH2Cl2 (0,25 M) se pueden añadir HCl en dioxano (4 M, ~4 eq.) a ~0 °C. La reacción se puede agitar y dejar que pase de 0 °C a temperatura ambiente durante aproximadamente 27 horas o hasta que se demuestre por LC-MS que la reacción se ha completado. La mezcla de reacción resultante se puede filtrar, lavar con CH2Cl2 y secar a alto vacío según sea necesario para proporcionar C-4.
Etapa 4a: Ciclización con HATU. A una solución de C-4 (~1,0 eq.) en ~10 ml de DMF (~0,005 M) se puede añadir DIPEA (~5,0 eq.). La solución se puede enfriar en un baño de agua con hielo y se puede añadir HATU (~1,5 eq.). Se puede dejar que la solución alcance temperatura ambiente y se puede agitar hasta que se pueda demostrar por LC-MS la conversión completa del material de partida en el producto deseado. La mezcla se puede diluir con agua y extraer tres veces con EtOAc según sea necesario. Los extractos orgánicos combinados se pueden lavar con agua y salmuera, secar sobre Na2SO4 y concentrar bajo presión reducida según sea necesario. El residuo resultante se puede purificar mediante cromatografía en columna en gel de sílice (0-5 % MeOH/DCM) para dar C.
Etapa 4b: Ciclización con FDPP. A una solución de DIPEA (~5 eq.) en DMF/CH2O 2 (3:1, ~0,005 M) se puede añadir C-4 (~1,00eq.). Después de que el C-4 se disuelva completamente, se puede añadir difenilfosfinato de pentafluorofenilo (FDPP, ~1,05 eq.). La reacción de acoplamiento se puede dejar en agitación durante 30 minutos o hasta que la LC-MS demuestre que la reacción se ha completado. La solución de reacción se puede diluir con CH2Cl2, lavar tres veces con agua, solución acuosa de Na2CO3 (2 M) y salmuera, se pueden secar sobre Na2SO4 según sea necesario. Después de la filtración y concentración bajo presión reducida, el residuo se puede purificar por cromatografía en columna en gel de sílice eluyendo con MeOH/CH2Cl2 (0-5 %) para proporcionar C.
EJEMPLOS
Los siguientes ejemplos se ofrecen para ilustrar la invención. Un experto en la técnica reconocerá que las siguientes reacciones y esquemas sintéticos se pueden modificar mediante la elección de materiales de partida y reactivos adecuados para acceder a otros compuestos de Fórmula (I) o (I-A). Estos compuestos se incluyen en la invención en la medida en que están abarcados por la Fórmula (IV) como se define en la reivindicación 1. Los grupos heteroaromáticos bicíclicos con funcionalidad adecuada para su uso en los procedimientos sintéticos están disponibles comercialmente.
Abreviaturas Los ejemplos descritos en el presente documento usan materiales, incluyendo los descritos por las siguientes abreviaturas conocidas por los expertos en la técnica:

Ejemplo A6

Etapa 1. A una solución de 5-fluoro-2-hidroxibenzaldehído (500,00 mg, 3,57 mmol, 1,00 eq.) en MeOH (20,00 ml) se añadió 1-metilpirrolidin-3-amina (357,43 mg, 3,57 mmol, 1,00 eq.) en una porción a 16 °C en atmósfera de N2.
Se agitó la mezcla a 16 °C durante 10 horas en atmósfera de N2. A continuación, se añadió NaBH4 (270,00 mg, 7,14 mmol, 2,00 eq.) y la mezcla se agitó a 16 °C durante 6 horas en atmósfera de N2. La TLC (DCM:MeOH = 15:1) mostró que la reacción se había completado. La mezcla de reacción se concentró bajo presión reducida para eliminar el MeOH. El residuo se diluyó con agua (50 ml) y se extrajo con DCM (20 ml x 3). Las fases orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida para dar A6-5 (350,00 mg, 1,56 mmol, 43,71 % de rendimiento) como un sólido amarillo. 1H RMN (300 MHz, DMSO-cfe) 8 (ppm) 6,94 (dd, J = 2,7, 9,3 Hz, 1H), 6 , 86 (dt, J =3,0, 8 , 6 Hz, 1H), 6,67 (dd, J = 4,7, 8,7 Hz, 1H), 3,71 (s, 2H), 3,24-3,09 (m, 1H), 2,58 (dd, J = 7,1, 8 , 8 Hz, 1H), 2,48-2,32 (m, 2H), 2,30-2,17 (m, 4H), 2,05-1,82 (m, 1H), 1,60-1,43 (m, 1H).
Etapa 2. A una solución de A6-5 (300,00 mg, 1,34 mmol, 1,00 eq.) y 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo (302,34 mg, 1,34 mmol, 1,00 eq.) en n-BuOH (40,00 ml) se añadió DlPEA (1,04 g, 8,04 mmol, 6,00 eq.) a 16 °C en atmósfera de N2. Se agitó la mezcla a 120 °C durante 2 horas. La TLC (éter de petróleo: EtOAc = 1:1) mostró que la reacción se había completado. La mezcla se vertió en agua (50 ml) y se extrajo con DCM (50 ml x 3). La mezcla se purificó mediante PLC preparativa para dar la sal de ácido fórmico de A6 (290,00 mg, 701,43 pmol, 52,35 % de rendimiento) como un sólido blanco.
Ejemplo A8

A una solución de 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo (1,25 g, 5,54 mmol) y la sa1HCl de (R)-2-(1-aminoetil)-4-fluorofenol (adquirida a NetChem, Inc.) en EtOH (15,83 ml) se añadió base de Hunig (3,58 g, 27,70 mmol) y se calentó a 70 °C durante 1,5 horas. La reacción se sometió a evaporación rotatoria hasta sequedad, se suspendió en agua y se extrajo con DCM (5 x 50 ml). Los extractos combinados se secaron con Na2SO4 y se concentraron bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice (40 g), 0-5 % de metanol en diclorometano) proporcionó A8 (1,89 g, 5,49 mmol, 99 % de rendimiento).
Ejemplo A9

Etapa 1. A una solución de 4-fluorofenol (2,00 g, 17,84 mmol, 1,00 eq.) en TfOH (30,00 ml) se añadió cloruro de propanoilo (1,65 g, 17,84 mmol, 1,00 eq.) a 0 °C. Se agitó la mezcla a 60 °C durante 4 horas hasta que la TLC mostró que la reacción se había completado. La mezcla se enfrió a 25 °C, se vertió en hielo-agua (p/p = 1/1) (120 ml), se neutralizó con NaHCO3 para obtener pH ~ 9 y se extrajo con EtOAc (120 ml x 3). Las fases orgánicas combinadas se lavaron con salmuera (50 ml), se secaron con Na2SO4 anhidro y se concentraron para dar A9-3 (1,80 g, 10,70 mmol, 59,98 % de rendimiento) como un aceite incoloro. 1H RMN (400 MHz, CDCh) 8 (ppm) 12,09 (s, 1H), 7,45 (dd, J = 3,0, 9,0 Hz, 1H), 7,26-7,20 (m, 1H), 6,97 (dd, J = 4,5, 9,0 Hz, 1H), 3,02 (q, J = 7,3 Hz, 2H), 1,27 (t, J = 7,2 Hz, 3H).
Etapa 2. Se burbujeó amoniaco gas en MeOH (20 ml) a -78 °C durante 10 minutos. Se añadió A9-3 (1,00 g, 5,95 mmol, 1,00 eq.) a la solución y se agitó a 25 °C durante 1 hora. A la mezcla de reacción se añadió Ti(i-PrO)4 (1,63 g, 7,14 mmol, 1,20 eq.) y la mezcla se agitó durante 1 hora más. A continuación, se añadió NaBH4 (449,93 mg, 11,89 mmol, 2,00 eq.). Se agitó la mezcla a 25 °C durante 12 horas hasta que la TLC mostró que el material de partida se había consumido completamente. El residuo se vertió en agua (50 ml) y se agitó durante 30 minutos. La mezcla se filtró y el filtrado se ajustó con HCl (1 M) a pH ~ 1 y se extrajo con EtOAc (50 ml x 2). Se añadió bicarbonato de sodio a la fase acuosa para ajustar el pH a ~ 9 y se extrajo con DCM (50 ml x 2). Las fases orgánicas combinadas se lavaron con salmuera saturada (50 ml), se secaron con Na2SO4 anhidro, se filtraron y se concentraron a vacío para dar A9-5 (310,00 mg, 1,83 mmol, 30,79 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, CDCla) 8 (ppm) 6 , 86 (dt, J = 3,0, 8,4 Hz, 1H), 6,79-6,74 (m, 1H), 6,67 (dd, J = 2,9, 8,9 Hz, 1H), 3,98 (t, J = 7,0 Hz, 1H), 1,92-1,81 (m, 1H), 1,80-1,68 (m, 1H), 0,95 (t, J = 7,4 Hz, 3H).
Etapa 3. A9-5 se acopló con 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo en presencia de DIPEA en n-BuOH para proporcionar A9 como se describe en el Procedimiento general A.
Ejemplo A13-5: Preparación de 2-(1-am¡no-2-cicloprop¡let¡l)-4-fluorofenol

Etapa 1. A una mezcla de ácido 2-ciclopropilacético (4,47 g, 44,60 mmol, 1,00 eq.) en DCM (150,00 ml) se añadió CDI (7,96 g, 49,10 mmol, 1,10 eq.) en una porción a 25 °C en atmósfera de N2. Se agitó la mezcla a 25 °C durante 1 h. A continuación, se añadió clorhidrato de N-metoximetanamina (4,79 g, 49,06 mmol, 1,10 eq.). Se agitó la mezcla a 25 °C durante otras 12 horas. La reacción se extinguió con ácido clorhídrico acuoso 1 N (50 ml) y se separó en fases. La fase acuosa se extrajo con DCM (30 ml x 2). La fase orgánica combinada se lavó con carbonato de sodio acuoso saturado al 50 % (50 ml) y salmuera saturada (30 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a vacío para dar 2-ciclopropil-Ñ-metoxi-W-metilacetamida (6,00 g, 41,91 mmol, 93,96% de rendimiento) como un aceite. 1H RMN (400 MHz, CDCla) 8 (ppm) 3,65 (s, 1H), 3,18 (s, 1H), 2,33 (d, J = 6 , 8 Hz, 2H), 1,13-1,02 (m, 1H), 0,57-0,49 (m, 2H), 0,19-0,11 (m, 2H).
Etapa 2. A una mezcla de 2-ciclopropil-W-metoxi-W-metilacetamida (6,00 g, 29,27 mmol, 1,00 eq.) en THF (100,00 ml) se añadió n-BuLi (2,5 M, 12,88 ml, 1,10 eq.) gota a gota a -78 °C en atmósfera de N2. Se agitó la mezcla a -78 °C durante 10 min. A continuación, la mezcla se trató con 2-bromo-4-fluoro-1-metoxibenceno (4,19 g, 29,27 mmol, 1,00 eq.) en THF (20 ml) durante un período de 20 min. Después de agitar a -78 °C durante 1 h, se dejó que la mezcla alcanzara 25 °C y se agitó durante una hora más. La TLC mostró que la reacción se había completado. La mezcla se vertió en una solución acuosa de HCl al 10 % (100 ml) y se agitó durante 10 min. La fase acuosa se extrajo con acetato de etilo (300 ml x 3). La fase orgánica combinada se lavó con salmuera (200 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 50/1, 10/1) para dar 2-ciclopropil-1-(5-fluoro-2-metoxifenil)etan-1-ona (2,4 g, 39,38% de rendimiento) como un aceite incoloro. 1H RMN (400 MHz, CDCh) 8 (ppm) 7,42 (dd, J = 3,3, 8 , 8 Hz, 1H), 7,15 (ddd, J = 3,3, 7,5, 9,0 Hz, 1H), 6,91 (dd, J = 4,0, 9,0 Hz, 1H), 3,91-3,85 (m, 3H), 2,89 (d, J = 6 , 8 Hz, 2H), 1,18-1,05 (m, 1H), 0,61-0,50 (m, 2H), 0,20-0,09 (m, 2H).
Etapa 3. A una solución de 2-ciclopropil-1-(5-fluoro-2-metoxifenil)etan-1-ona (500,00 mg, 2,40 mmol, 1,00 eq.) en DCM (10,00 ml) se añadió BCh (1 M, 3,00 ml, 1,25 eq.) gota a gota a -78 °C en atmósfera de N2. Se agitó la mezcla a -78 °C durante 2 h. La TLC mostró que la reacción se había completado. Se dejó que la mezcla alcanzara 25 °C y se vertió en agua con hielo (p/p = 1/1) (10 ml) y se agitó durante 10 min. La fase acuosa se extrajo con acetato de etilo (30 ml x 3). La fase orgánica combinada se lavó con salmuera saturada (30 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para dar 2-ciclopropil-1-(5-fluoro-2-hidroxifenil)etan-1-ona (430,00 mg, 2,21 mmol, 92,3 % de rendimiento) como un aceite. 1H RMN (400 MHz, CDCh) 8 (ppm) 12,12 (s, 1H), 7,40 (dd, J = 3,0, 8 , 8 Hz, 1H), 7,24 (ddd, J = 3,0, 7,8, 9,0 Hz, 1H), 6,98 (dd, J = 4,5, 9,3 Hz, 1H), 2,88 (d, J = 6 , 8 Hz, 2H), 1,23-1,11 (m, 1H), 0,70-0,63 (m, 2H), 0,25 (q, J = 5,0 Hz, 2H).
Etapa 4. A una solución de 2-ciclopropil-1-(5-fluoro-2-hidroxifenil)etan-1-ona (400,00 mg, 1,92 mmol, 1,00 eq.) en MeOH (20,00 ml) se añadió NH2OH.HC (160,18 mg, 2,31 mmol, 1,20 eq.) y AcONa (189,09 mg, 2,31 mmol, 1,20 eq.) a 25 °C en atmósfera de N2 durante 12 horas. La TLC (éter de petróleo/acetato de etilo = 3:1) mostró que el material de partida se había consumido completamente. La reacción se extinguió con agua y a continuación se extrajo con d Cm (30 ml x 3). La fase orgánica combinada se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para dar el producto puro 2-ciclopropil-1-(5-fluoro-2-hidroxifenil)etan-1-ona oxima (400,00 mg, 1,79 mmol, 93,32 % de rendimiento) como un sólido blanco. El sólido se usó en la siguiente etapa sin purificación adicional.
Etapa 5. A una solución de 2-ciclopropil-1-(5-fluoro-2-hidroxifenil)etan-1-ona oxima (260,00 mg, 1,16 mmol, 1,00 eq.) en MeOH/HCl (10,00 ml, 4 N) se añadió Pd-C (10 %, 100 mg) en atmósfera de N2. La suspensión se desgasificó a vacío y se purgó con H2 varias veces. Se agitó la mezcla bajo H2 (50 psi) a 50 °C durante 12 horas. La LC-MS mostró que el material de partida se había consumido completamente. La mezcla de reacción se filtró y el filtrado se concentró para dar 2-(1-amino-2-ciclopropiletil)-4-fluorofenol (200,00 mg, 955,75 |jmol, 82,39 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-cfe) 5 (ppm) 10,44-9,82 (m, 1H), 8,52 (s. a., 2H), 7,36 (dd, J = 2,8, 9,5 Hz, 1H), 7,07-6,93 (m, 2H), 4,49 (d, J = 5,5 Hz, 1H), 1,82-1,72 (m, 2H), 0,67-0,55 (m, 1H), 0,43-0,28 (m, 2H), 0,12-0,06 (m, 1H), (-0,03)-(-0,09) (m, 1H).
Ejemplo A 14-5: Preparación de 2-(amino(fen¡l)met¡l)-4-fluorofenol

Etapa 1. A una solución de A14-3 (2,00 g, 9,25 mmol, 1,00 eq.) y AcOK (1,10 g, 11,20 mmol, 1,20 eq.) en etanol (30,00 ml) se añadió NH2OH.HCl (642,80 mg, 9,25 mmol, 1,00 eq.) en una porción a 25 °C en atmósfera de N2. Se agitó la mezcla a 25 °C durante 30 minutos, a continuación se calentó a 90 °C y se agitó durante 5 horas. La TLC mostró que la reacción se había completado. La mezcla se concentró y se añadió agua (50 ml). La mezcla se extrajo con acetato de etilo (50 ml x 3). La fase orgánica combinada se lavó con salmuera (50 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró para dar (5-fluoro-2-hidroxifenil)(fenil)metanona oxima (1,50 g, 6,49 mmol, 70,13 % rendimiento) como un sólido amarillo. 1H RMN (400 MHz, CDCla) 5 (ppm) 7,50-7,37 (m, 5H), 7,19-7,07 (m, 2H), 6,71 (dd, J = 2,9, 8,9 Hz, 1H).
Etapa 2. A una mezcla de (5-fluoro-2-hidroxifenil)(fenil)metanona oxima (900,00 mg, 4,18 mmol, 1,00 eq.) y Zn en polvo (1,09 g, 16,73 mmol, 4 eq.) en THF (10,00 ml) se añadió NH4O (2,24 g, 41,82 mmol, 10,00 eq.) en una porción a 25 °C en atmósfera de N2. Se agitó la mezcla a 25 °C durante 30 minutos, a continuación se calentó a 60 °C y se agitó durante 15 horas. La mezcla se concentró y se añadió agua (100 ml), seguido de extracción con acetato de etilo (50 ml x 3). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron para dar A14-5 (630,00 mg, 2,90 mmol, 69,38 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, CDCla) 5 (ppm) 7,42 (d, J = 7,5 Hz, 2H), 7,33 (t, J = 7,5 Hz, 2H), 7,27-7,20 (m, 1H), 6,93-6,80 (m, 2H), 6,70 (dd, J = 4,9, 8,7 Hz, 1H), 5,28 (s, 1H).
Ejemplo A17

Etapa 1. A una solución de 5-((2-bromo-5-fluorobencil)(metil)amino)pirazolo[1,5-a]pirimidina-3-carboxilato de etilo (preparada de acuerdo con el Procedimiento general A) (300,00 mg, 0,736 mmol, 1,00 eq.), 2-metilpropano-2-tiol (166,10 mg, 1,84 mmol, 2,50 eq.), Pd2(dba)3 (84,72 mg, 0,147 mmol, 0,20 eq.) en dioxano (8,00 ml) se añadió XantPhos (127,87 mg, 0,221 mmol, 0,30 eq.) y K2CO3 (101,81 mg, 0.736 mmol, 1,00 eq.). La mezcla se desgasificó y se calentó a 120 °C durante 24 horas en atmósfera de N2. La TLC (éter de petróleo/acetato de etilo = 1:1) mostró que el material de partida se había consumido completamente. La mezcla de reacción se vertió
en H2O (20 ml) y se extrajo con acetato de etilo (50 ml x 3). La fase orgánica se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro, se concentró y se purificó por cromatografía en columna en gel de sílice (éter de petróleo/acetato de etilo = 2:1 a 1:1) para dar 5-((2-(terc-butiltio)-5-fluorobencil)(metil)amino)pirazolo[1,5-a]pirimidina-3-carboxilato de etilo (200,00 mg, 0,48 mmol, 65,18% de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, CDCls) 8 (ppm) 8,34 (s, 1H), 8,29 (s. a., 1H), 7,60 (dd, J = 5,9, 8,4 Hz, 1H), 7,00 (t, J = 7,7 Hz, 1H), 6,29 (s. a., 2H), 5,00 (s. a., 2H), 4,37 (d, J = 6 , 8 Hz, 2H), 3,41 (s. a., 3H), 1,36-1,20 (m, 12H).
Etapa 2. A una solución de 5-((2-(terc-butiltio)-5-fluorobencil)(metil)amino)pirazolo[1,5-a]pirimidina-3-carboxilato de etilo (300,00 mg, 0,720 mmol, 1,00 eq.) en DCM (8,00 ml) se añadió BBr3 (902,21 mg, 3,60 mmol, 5,00 eq.) gota a gota a 0 °C en atmósfera de N2. Se agitó la mezcla a 0 °C durante 2,5 horas. La TLC (éter de petróleo: acetato de etilo = 1:1) mostró que la reacción se había completado. La mezcla se vertió en agua (20 ml). La fase acuosa se extrajo con diclorometano (50 ml x 3). La fase orgánica combinada se lavó con salmuera (30 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a vacío. El residuo se purificó por HPLC preparativa (Columna: Phenomenex Synergi C18 150 x 30 mm x 4 pm; condiciones: HCl al 0,05 % en ACN) y se liofilizó para dar la sal de HCl de A17 (38,00 mg, 0,098 mmol, 13,61 % de rendimiento) como un sólido blanco.
Ejemplo A18

Etapa 1. La mezcla de 2-bromo-4-fluorofenol (10,00 g, 52,36 mmol, 1,00 eq.), sal de potasio de trifluoro(vinil)-borano (9,84 g, 66,50 mmol, 1,27 eq.), Cs2CO3 (51,18 g, 157,08 mmol, 3,00 eq.) y Pd(PPh3)2Cl2 (1,84 g, 2,62 mmol, 0,05 eq.) en THF (90,00 ml) y H2O (10,00 ml) se desgasificó y a continuación se calentó a 90 °C durante 12 horas en atmósfera de N2. La TLC (éter de petróleo/acetato de etilo = 10/1) mostró que el material de partida se había consumido completamente. La mezcla de reacción se vertió en H2O (100 ml). La mezcla se extrajo con acetato de etilo (300 ml x 3). La fase orgánica se lavó con salmuera saturada (200 ml), se secó sobre Na2SO4 anhidro, se concentró y se purificó con una cromatografía en columna en gel de sílice (eluyendo con EtOAc/éter de petróleo = 1/30) para dar 4-fluoro-2 -vinilfenol (3,50 g, 25,34 mmol, 48,39 % de rendimiento) como un aceite incoloro. 1H RMN (400 MHz, CDCla) 8 (ppm) 7,12 (dd, J = 3,0, 9,5 Hz, 1H), 6,89-6,81 (m, 1H), 6,79-6,73 (m, 1H), 5,75 (d, J =17,6 Hz, 1H), 5,64 (s, 1H), 5,39 (d, J =11,3 Hz, 1H).
Etapa 2. La mezcla de 4-fluoro-2-vinilfenol (1,95 g, 14,12 mmol, 1,00 eq.), TBSCl (6,38 g, 42,35 mmol, 3,00 eq.) y 1H-imidazol (5,77 g, 84,70 mmol, 6,00 eq.) en DCM (20,00 ml) se agitó a 20 °C durante 5 horas en atmósfera de N2. La TLC (éter de petróleo/acetato de etilo = 10:1) mostró que el material de partida se había consumido completamente. La mezcla de reacción se vertió en H2O (30 ml). La mezcla se extrajo con diclorometano (50 ml x 3). La fase orgánica se lavó con salmuera (50 ml), se secó sobre Na2SO4 anhidro, se concentró y se purificó por cromatografía en columna en gel de sílice eluida con éter de petróleo para dar tri-butil(4-fluoro-2-vinilbencil)silano (2,30 g, 9,11 mmol, 64,54 % de rendimiento) como un aceite incoloro.
Etapa 3. La mezcla de tri-butil(4-fluoro-2-vinilbencil)silano (2,30 g, 9,11 mmol, 1,00 eq.), 5-cloropirazolo[1,5-a]pirimidina-3-carboxilato de etilo (2,06 g, 9,11 mmol, 1,00 eq.), Pd(PhCN)2Cl2 (118,20 mg, 0,455 mmol, 0,05 eq.) y tris-o-tolilfosfano (277,36 mg, 0,911 mmol, 0 , 10 eq.), DIPeA (7,07 g, 54,68 mmol, 6 , 00 eq.) en DMF (25,00 ml) se desgasificó y a continuación se calentó a 120 °C durante 24 horas en atmósfera de N2. La TLC (éter de petróleo/acetato de etilo =1:1) mostró que el material de partida se había consumido completamente. La mezcla de reacción se vertió en H2O (30 ml). La mezcla se extrajo con acetato de etilo (100 ml x 3). La fase orgánica se lavó con salmuera saturada (30 ml), se secó sobre Na2SO4 anhidro, se concentró y se purificó por cromatografía en columna en gel de sílice (EtOAc:éter de petróleo = 1:3) para dar (E)-5-(5-fluoro-2-hidroxiestiril)pirazolo[1,5-a]pirimidina-3-carboxilato de etilo (1,00 g, 2,26 mmol, 24,86 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, CDCl3) 8 (ppm) 9,29 (s. a., 1H), 8,50 (d, J =7,0 Hz, 1H), 8,28 (s. a., 1H), 7,84 (d, J = 16,6 Hz, 1H), 7,20 7,04 (m, 3H), 6,69 (d, J = 5,8 Hz, 2H), 4,20 (q, J = 6,9 Hz, 2H), 1,30-1,19 (m, 3H).
Etapa 4. A una mezcla de ('£)-5-(5-fluoro-2-hidroxiestiril)pirazolo[1,5-a]pirimidina-3-carboxilato de etilo (378,22 mg, 1,04 mmol, 1,00 eq.) y 4-metilbencenosulfonohidracida (3,29 g, 17,68 mmol, 17,00 eq.) en THF (4,00 ml) se añadió
NaOAc (1,71 g, 20,80 mmol, 20,00 eq.) en una porción a 20 °C en atmósfera de N2. La mezcla se calentó a continuación a 65 °C y se agitó durante 12 horas. La TLC mostró que la reacción se había completado. La mezcla se enfrió a 20 °C y se concentró bajo presión reducida a 45 °C. Se añadió agua (100 ml) al residuo. La fase acuosa se extrajo con acetato de etilo (300 ml x 2). La fase orgánica combinada se lavó con salmuera saturada (50 ml), se secó con Na2SO4 anhidro, se filtró, se concentró a vacío y se purificó mediante HPLC preparativa (Columna: Phenomenex Synergi Max-RP 250 x 50 mm x 10 pm, FA al 0,225% en ACN) para dar A18 (120,00 mg, 0,347 mmol, 33,42 % de rendimiento) como un sólido blanco.
Ejemplo A20

A una mezcla de 4-fluoro-2-metilaminometil-fenol (305,2 mg, 1,97 mmol) y éster etílico del ácido 6 -cloroimidazo[1,2-b]piridazina-3-carboxílico (230 mg, 1,02 mmol) en DMSO (5 ml) se añadió KF (180 mg, 3,01 mmol). La mezcla de reacción se agitó a 120 °C durante 18 horas en atmósfera de nitrógeno. A continuación, la solución se enfrió a temperatura ambiente, se diluyó con agua (20 ml) y se extrajo con EtOAc (3 x 50 ml). Las fases orgánicas combinadas se lavaron adicionalmente con agua (3 x 50 ml) y salmuera (50 ml), se secaron sobre Na2SO4 y se concentraron. El residuo se purificó a continuación en una columna de gel de sílice eluyendo con EtOAc/hexano (0-50 %, 10 V.C.) para dar el producto deseado como un sólido blanco (240 mg, 69 %).
Ejemplo A22

Se preparó A21-1 de acuerdo con el Procedimiento general A. A una solución de A22-1 (150 mg, 0,387 mmol) en etanol (2 ml) se añadió HCl 4 M en dioxano (2 ml) y la solución de reacción se calentó a 75 °C durante 2 horas. Se evaporaron los disolventes y el residuo se neutralizó con Et3N y se purificó en un cartucho de gel de sílice eluyendo con metanol/CH2Cl2 (0-12,5 %) para proporcionar A22 (144 mg, 100 %).
Ejemplo A23

Etapa 1. A una mezcla de (5-fluoro-2-metoxifenil)metanotiol (496,1 mg, 2,88 mmol) y éster etílico del ácido 6 -cloroimidazo[1,2-b]piridazina-3-carboxílico (650,0 mg, 2,88 mmol) en etanol (14,4 ml) se añadió DIPEA (1,12 g, 8,64 mmol). La mezcla de reacción se agitó a 80 °C durante 1 hora. La solución se enfrió a temperatura ambiente, se diluyó con agua (50 ml) y se extrajo con DCM (3 x 50 ml). Los extractos combinados se secaron sobre Na2SO4 y se concentraron bajo presión reducida. El residuo se purificó con cromatografía ultrarrápida (sistema ISCO, sílice (120 g) eluyendo con EtOAc/hexano (0-50 %) para dar A23-2 (560 mg, 54 % de rendimiento). A23-2 se perdió en la columna durante la purificación.
Etapa 2. A una solución de A23-2 (498,7 mg, 1,38 mmol) en metanol (100 ml) se añadió HCl 4 M en dioxano (10 ml) y la solución de reacción se calentó a 75 °C durante 2 horas. Los disolventes se evaporaron y el residuo se neutralizó con Et3N y se purificó en un cartucho de gel de sílice eluyendo con metanol/CH2Cl2 (0-12,5%) para proporcionar A23 (470 mg, 98 %).
A1-A24 se prepararon de acuerdo con el Procedimiento general A y los procedimientos descritos en el presente documento.




Ejemplo B7

Etapa 1. A una mezcla de 1-(5-fluoro-2-mdroxi-feml)-etanona (773 mg, 5,0 mmol) y ester terc-butilico del acido (2-cloro-etil)-carbámico (1,80 g, 10,0 mmol) en DMF (20 ml) se añadieron KI (2,0 mg, 0,012 mmol) y Cs2CO3 (3,26 g, 10,0 mmol). Se agitó la mezcla a 80 °C durante la noche. La mezcla se enfrió a continuación a temperatura ambiente, se diluyó con EtOAc y se lavó con NaOH 1 N (5 x 10 ml) hasta que la LC-MS no mostró un pico de 1-(5-fluoro-2-hidroxi-fenil)-etanona. La fase orgánica se secó sobre Na2SO4 y se concentró. El residuo se purificó a continuación en una columna de gel de sílice eluyendo con EtOAc/hexano (0-30 %, 10 V.C.) para dar el producto deseado B7-2 como un sólido amarillento (1,1 g, 73,8 %). LC-MS (ESI) m/z 320,3 (M+Na)+.
Etapa 2. A una solución de B7-2 (1,0 g, 3,36 mmol) en MeOH (10 ml) se añadió NaBH4 (640 mg, 16,8 mmol) en porciones. Se agitó la mezcla a temperatura ambiente durante 2 h hasta que no quedó material de partida por LC-MS. La solución se diluyó a continuación con agua (50 ml) y se extrajo con DCM (3 x 20 ml). Las fases de DCM combinadas se secaron sobre Na2SO4 y se concentraron. El residuo se purificó en una columna de gel de sílice eluyendo con EtOAc/hexano (0-50 %, 10 V.C.) para dar el producto deseado B7-3 como un sólido amarillo pálido (0,75 g, 75 %). LC-MS (ESI) m/z 322,3 (M+Na)+; 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 7,11 (dd, J = 9,2, 3,4 Hz, 1H), 6,89 (ddd, J = 9,0, 7,9, 3,2 Hz, 1H), 6,77 (dd, J = 8,9, 4,4 Hz, 1H), 5,09 (q, J = 6 , 6 Hz, 1H), 4,92 (d, J = 4,4 Hz, 1H), 4,03 (t, J =5,2 Hz, 2H), 3,62-3,50 (m, 2H), 1,49 (d, J = 6,4 Hz, 3H), 1,45 (s, 9H).
Etapa 3: A una solución de B7-3 (600 mg, 2,0 mmol) y ester terc-butílico del ácido {2-[4-fluoro-2-(1 -hidroxietil)-fenoxi]-etil}-carbámico (450 mg, 2,0 mmol) en THF seco (40,0 ml) a -78 °C se añadió NaH (60 %, 80 mg, 2,0 mmol) en porciones. La suspensión se agitó a -78 °C durante 4 h y se dejó que la mezcla alcanzara 0 °C y se agitó durante 4 h adicionales. La mezcla se puso a continuación en el congelador a -20 °C durante la noche. La LC-MS mostró una buena conversión al producto deseado. La mezcla se extinguió a continuación con una mezcla de hielo y HCl 1 N y se extrajo con EtOAc (3 x 20 ml). La fase orgánica se secó sobre Na2SO4, se concentró y purificó dos veces para dar el producto deseado B7 como un sólido amarillo (240 mg, 25 %):
B1-B7 se prepararon de acuerdo con el Procedimiento general B y los procedimientos descritos en el presente documento.


Ejemplos 2 y 2-1

Síntesis A:
El Ejemplo 2 se puede preparar como se muestra en el siguiente esquema, comenzando con materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. A una mezcla de los compuestos 2A (1 equiv.) y 2B (1,2 equiv.) en DMF anhidro (0,2 M) se añade Cs2CO3 (1,5 equiv.) y la reacción se calienta en un baño de aceite a 80 °C en atmósfera de nitrógeno durante la noche. La mezcla se enfría, se vierte en agua y se extrae con EtOAc tres veces. Las fases orgánicas combinadas se lavan con agua cinco veces, se lavan con salmuera y se secan sobre Na2SO4. Después de la condensación, el residuo se purifica en una columna ultrarrápida eluyendo con EtOAc/hexanos para proporcionar el compuesto 2C.
Etapa 2. A una solución del compuesto 2C (1 equiv.) en THF anhidro (0,2 M) se añade NaH (1,2 equiv.). La mezcla de reacción se agita a temperatura ambiente durante 0,5 horas. A la mezcla se añade el compuesto 2D y la reacción se calienta a reflujo en atmósfera de nitrógeno durante la noche. La reacción se enfría a temperatura ambiente y se diluye con una porción de agua (1/3 del volumen de THF) y NaOH (3 equiv.). La mezcla se agita y se calienta a 70 °C durante 2 horas o hasta que el éster se hidroliza completamente al ácido correspondiente. Después del enfriamiento, la fase orgánica se separa y la fase acuosa se neutraliza a pH~5. El precipitado resultante se filtra, se lava con agua tres veces y se seca a vacío para proporcionar el compuesto 2E, que se usa sin purificación adicional.
Etapa 3. A una solución del compuesto 2E (1 equiv.) en CH2Cl2 (0,2 M) se añade HCl 4 M/dioxano (10 equiv.) y la mezcla se agita hasta que el compuesto 2E se convierte completamente en el compuesto 2F. La mezcla se concentra y el residuo se purifica por HPLC preparativa de fase inversa para proporcionar el compuesto 2F.
Etapa 4. Una solución del compuesto 2F (1 equiv.) y DIPEA (10 equiv.) en DMF (0,2 M) se añade gota a gota a una solución de HATU (1,4 equiv.) en DMF (0,1 M) a 0 °C. Una vez completada la adición, la mezcla se agita a 0 °C durante otros 30 minutos. Se agrega agua y la mezcla se extrae con EtOAc tres veces. Las fases orgánicas combinadas se lavan con NaHCO3 saturado dos veces, a continuación con salmuera, se secan sobre Na2SO4 y se concentran. El residuo se purifica en una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar el Ejemplo 2.
Síntesis B:
Los Ejemplos 2 y 2-1 también se pueden preparar de acuerdo con el siguiente esquema usando materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. El compuesto 2C se hace reaccionar con el compuesto 2G en las condiciones descritas en la Síntesis A, Etapa 2, para proporcionar el compuesto 2H.
Etapa 2. El compuesto 2H se convierte en el compuesto 21 en las condiciones descritas en la Síntesis A, Etapa 3.
Etapa 3. A una solución del compuesto 21 (1 equiv.) y DIPEA (2 equiv.) en tolueno (0,01 M) se añade Pd(PtBu3)2 (1 equiv.). La mezcla de reacción se calienta a 100 °C bajo 4 bar de CO durante la noche y, a continuación, se concentra. El residuo se purifica en una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar el Ejemplo 2.
Ejemplos 10 y 10-1.

Los Ejemplos 10 y 10-1 se pueden preparar como se muestra en el siguiente esquema usando materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. El compuesto 10C se prepara a partir de los compuestos 10A y 10B usando el procedimiento descrito en el Ejemplo 2, Síntesis A, Etapa 1.
Etapa 2. El compuesto 10E se prepara a partir de los compuestos 10C y 10D usando el procedimiento descrito en el Ejemplo 2, Síntesis A, Etapa 2.
Etapa 3. Una mezcla de compuesto 10E (1 equiv.) y NH2-NH2 (10 equiv.) en metanol (0,2 M) se calienta a reflujo hasta que el compuesto 10E se convierte completamente en el compuesto 10F. La mezcla se concentra y el residuo se purifica por HPLC preparativa de fase inversa para proporcionar el compuesto 10F.
Etapa 4. El compuesto 10F se convierte en el Ejemplo 10 de acuerdo con el procedimiento descrito para el Ejemplo 2, Síntesis A, Etapa 4.
Ejemplo 11-1

Etapa 1. A una solución de 2-cloro-3-fluoro-6-hidroxi-benzaldehído (175 mg, 1,0 mmol), bis-tos etilenglicol (740 mg, 2,0 mmol) en ACN (5 ml) se añadió K2CO3 (276 mg, 2,0 mmol) y KI (2 mg). La mezcla de reacción se agitó a 120 °C durante 24 horas. El sólido se separó por filtración y el filtrado se concentró y purificó por cromatografía en columna para dar el producto deseado 11-1B como un sólido blanco. El material se usó directamente en la siguiente etapa.
Etapa 2. A una solución de 11-1B (373 mg, 1 mmol) en ACN (5 ml) se añadió NaN3 (650 mg, 10 mmol) y la mezcla se agitó a 120 °C durante 24 horas. El sólido se separó por filtración y el residuo se concentró y purificó por cromatografía en columna para dar 11-1C como un sólido blanco (200 mg, 82 %). 1H RMN (500 MHz, cloroformod) 8 (ppm) 10,49 (d, J = 1,1 Hz, 1H), 7,31 (dd, J = 9,2, 8,2 Hz, 1H), 6 , 88 (dd, J =9,2, 3,7 Hz, 1H), 4,21 (dd, J = 5,4, 4,5 Hz, 2H), 3,67 (dd, J = 5,4, 4,5 Hz, 2H).
Etapa 3. A una solución de 11-1C (100 mg, 0,41 mmol) en THF anhidro (5 ml) a -78 °C, se añadió bromuro de metil magnesio (1 N en Et2O, 0,82 ml, 0,82 mmol). Se dejó que la mezcla alcanzara temperatura ambiente y se agitó durante 2 horas hasta que la TLC no mostró material de partida presente. La solución se enfrió a continuación a 0 °C y se extinguió con una solución acuosa sat. de NH4OAc y se extrajo con EtOAc (20 ml x 3). La fase orgánica combinada se secó sobre Na2SO4 y se concentró. El residuo 11-1D se usó directamente en la siguiente etapa. 1H RMN (500 MHz, cloroformo-d) 8 (ppm) 6,97 (dd, J = 9,2, 8,3 Hz, 1H), 6,77 (dd, J = 9,1, 4,1 Hz, 1H), 5,27 (q, J = 6,7 Hz, 1H), 4,34-4,29 (m, 1H), 4,22-4,16 (m, 1H), 4,04-3,98 (m, 1H), 3,95-3,88 (m, 2H), 1,51 (d, J = 6,7 Hz, 3H).
Etapa 4. A una solución de éster etílico del ácido 5-cloro-pirazolo[1,5-a]pirimidina-3-carboxílico (100 mg, 0,44 mmol) y 11-1D (110 mg, 0,41 mmol) en THF anhidro (5,0 ml) a -78 °C se añadió NaH (60%, 17 mg, 0,44 mmol). Se dejó que la mezcla alcanzara temperatura ambiente y se agitó durante 8 horas hasta que se formó una buena cantidad del producto deseado. La mezcla se diluyó a continuación con agua/hielo y se extrajo con DCM (3 x 20 ml). La fase orgánica se secó sobre Na2SO4, se concentró y purificó por cromatografía en columna en gel de sílice para dar 11-1E como un líquido amarillo (20 mg, 0,045 mmol, 6 %), que se usó directamente en la siguiente etapa.
Etapa 5. A una solución de 11-1E (20 mg, 0,045 mmol) en MeOH (1 ml) se añadió LiOH (16 mg, 0,38 mmol), seguido de 1 ml de H2O. La mezcla se dejó en agitación a 60 °C durante 4 horas hasta que la LC-MS y la TLC mostraron que la reacción se había completado. La solución se enfrió a temperatura ambiente, se concentró parcialmente y se acidificó con HCl 1 N hasta pH 2-3. La mezcla acuosa se extrajo con DCM (3 x10 ml). La fase orgánica se secó sobre Na2SO4 y se concentró. El residuo 11-1F se usó directamente en la siguiente etapa.
Etapa 6. A una solución de 11-1F (20 mg, 0,045 mmol) en DCM (5 ml) se añadió PPh3 (24 mg, 0,09 mmol). La solución se agitó durante 1 hora hasta que la TLC mostró una conversión completa del material de partida en el producto deseado. La mezcla se usó a continuación directamente en la siguiente etapa sin más caracterización.
11-1G MS ESI+ m/z 417,7 (M+Na)+.
Etapa 7. A una solución de 11-1G obtenido en la etapa anterior en DMF (10 ml) se añadió DIPEA (0,20 ml, 1,15 mmol). La solución se enfrió con un baño de hielo seco/acetona y se añadió HATU (40,0 mg, 0,11 mmol). Se dejó que la solución alcanzara temperatura ambiente lentamente y la LC-MS mostró una transformación limpia del material de partida en el producto deseado. La mezcla se diluyó a continuación con agua (50 ml) y se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se lavó con agua (3 x 50 ml) y salmuera (50 ml) y se secó sobre Na2SO4. Se eliminó el disolvente y se purificó el residuo resultante por cromatografía en columna de sílice (0-5 % MeOH/DCM), dando el producto deseado como un sólido blanco (2,6 mg, 20 % de rendimiento).
Ejemplos 14 y 14-1.

Los Ejemplos 14 y 14-1 se pueden preparar de acuerdo con el siguiente esquema usando materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. A una mezcla de los compuestos 14A (1 equiv.) y 14B (1,2 equiv.) en DMF anhidro (0,2 M) se añade Cs2CO3 (1,5 equiv.) y la reacción se calienta en un baño de aceite a 80 °C en atmósfera de nitrógeno durante la noche. La mezcla se enfría, se vierte en agua y se extrae con EtOAc tres veces. Las fases orgánicas combinadas se lavan con agua cinco veces, se lavan con salmuera y se secan sobre Na2SO4. Después de la condensación, el residuo se purifica en una columna ultrarrápida en gel de sílice eluyendo con EtOAc/hexanos para proporcionar 14C.
Etapa 2. A una solución enfriada (-78 °C) de 14C (1 equiv.) en THF anhidro (0,2 M) se añade MeMgBr (3 equiv, 3 M en dietil éter). La reacción se agitó durante 2 h de -78 °C a 0 °C, y se extinguió con solución acuosa saturada de NH4Cl y a continuación se extrajo con EtOAc (2x). Los extractos orgánicos se secan sobre MgSO4, se filtran y se concentran. Este residuo se purifica mediante cromatografía en columna en gel de sílice eluyendo con EtOAc/hexanos para dar 14D.
Etapa 3. A una solución del compuesto 14D (1 equiv.) en THF anhidro (0,2 M) se añade NaH (1,2equiv.). La mezcla de reacción se agita a temperatura ambiente durante 0,5 horas. A la mezcla se añade el compuesto 14E y la reacción se calienta a reflujo en atmósfera de nitrógeno durante la noche. La reacción se enfría a temperatura ambiente y a continuación se vierte en agua. El producto se extrae con EtOAc tres veces. Las fases orgánicas combinadas se lavan con salmuera, se secan sobre Na2SO4 y se concentran. El residuo se purifica con una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar el producto 14F.
Etapa 4. A una solución del compuesto 14F (1 equiv.) en CH2Cl2 (0,2 M) se añade HCl 4 M/dioxano (10 equiv.) y la mezcla se agita hasta que todo el compuesto l4F se convierte en 14G. Después de la concentración, el residuo se purifica en una HPLC preparativa de fase inversa para proporcionar 14G.
Etapa 5. A una solución de 14G (1 equiv.) y DIPEA (2 equiv.) en tolueno (0,01 M) se añade Pd(Pt-Bu3)2 (1 equiv.). La mezcla de reacción se calienta a 100 °C bajo 4 bar de CO durante la noche y, a continuación, se concentra. El residuo se purifica en una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar 14.
Ejemplos 15 y 15-1.

Los Ejemplos 15 y 15-1 se pueden preparar de acuerdo con el siguiente esquema usando materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. A una suspensión de 15A (1,0 equiv.) en THF (0,15 M) se añade una solución acuosa de NaOH 2,0 M (3 equiv.). La mezcla de reacción homogénea se agita durante la noche, y a continuación se eliminan los compuestos orgánicos bajo presión reducida. El residuo acuoso se lleva a pH~4 con HCl acuoso 1,0 M. El precipitado resultante se recoge por filtración y se aclara con H2O para dar un sólido de 15B. El filtrado se extrae
con EtOAc (2x) y los extractos orgánicos se concentran bajo presión reducida para proporcionar una porción adicional de 15B.
Etapa 2. Se prepara una solución madre de reactivo de Jones (2,67 M) añadiendo cuidadosamente H2SO4 concentrado (2,3 ml) a CrO3 (2,67 g) y a continuación diluyendo hasta 10 ml con H2O. A una suspensión de 15B (1,0 equiv.) en acetona (0,067 M) se añade lentamente reactivo de Jones (1,2 equiv.). La mezcla de reacción se agita durante 15 minutos y a continuación se extingue con i-PrOH y se filtra a través de una capa de tierra de diatomeas, enjuagando con acetona. El filtrado se concentra para proporcionar 15C que se usa sin purificación adicional.
Etapa 4. A una solución de 15C (1,0 equiv.) en DMF (0,40 M) a 0 °C se añade NaH (60 % en aceite mineral, 1,5 equiv.). La mezcla de reacción se agita a temperatura ambiente durante 30 minutos y a continuación se enfría nuevamente a 0 °C y se añade lentamente cloruro de 2-(trimetilsilil)etoximetilo (4,3 ml, 1,2 equiv.). Se deja que la mezcla alcance temperatura ambiente, se agita durante 1 h y a continuación se extingue con H2O y se extrae con EtOAc (3x). Los extractos orgánicos combinados se lavan con H2O (3x) y salmuera y, a continuación, se secan sobre MgSO4 y se concentran. El residuo se purifica por cromatografía ultrarrápida en gel de sílice eluyendo con 20-30 % de EtOAc/hexanos para dar 15D.
Etapa 5. A una mezcla de reacción de 14D (1,0 equiv.), yoduro de cobre(I) (0,05 equiv.), 8 -hidroxiquinolina (0,1 equiv.) y fosfato de potasio tribásico (2,0 equiv.) en DMF (0,2 M) en atmósfera de nitrógeno se añade 15D (1,2 equiv.) y la mezcla de reacción se calienta a 120 °C durante 24 h. La mezcla de reacción se enfría a temperatura ambiente y a continuación se diluye con EtOAc. La mezcla se filtra a través de una capa de tierra de diatomeas y el filtrado se evapora a vacío. El residuo bruto se purifica en una columna de gel de sílice eluyendo con EtOAc/heaxanos para dar 15E.
Etapa 6. Una suspensión a 0 °C de 15E (1,0 equiv.) en 1,4-dioxano (0,062 M) y agua (1/3 de THF) se trata con ácido sulfámico (6,0 equiv.). Se añade una solución de clorito de sodio (1,3 equiv.) y dihidrógeno de fosfato de potasio (12 equiv.) en agua (1,2 M) a través de un embudo de decantación durante 20 min. Una vez completada la adición, se retira el baño de hielo y la mezcla de reacción se agita a temperatura ambiente durante 3 h. Se añade THF y la mezcla de reacción se agita a temperatura ambiente durante 3 h adicionales. La mezcla de reacción se diluye con agua y se extrae con EtOAc (2x). Las fases orgánicas combinadas se lavan con agua y salmuera y a continuación se secan sobre Na2SO4, se filtran y se concentran. El residuo se tritura con acetato de etilo/hexanos para dar 15F.
Etapa 7. A una solución del compuesto 15F (1 equiv.) en CH2Cl2 (0,2 M) se añade HCl 4 M/dioxano (10 equiv.) y la mezcla se agita hasta que todo el compuesto 15f se convierte en 15G. Después de la concentración, el residuo se purifica en una HPLC preparativa de fase inversa para proporcionar 15G.
Etapa 8. Una solución del compuesto 15G (1 equiv.) y DIPEA (10 equiv.) en DMF (0,2 M) se añade gota a gota a una solución de HATU (1,4 equiv.) en DMF (0,1 M) a 0 °C. Una vez completada la adición, la mezcla se agita a 0 °C durante otros 30 minutos. Se agrega agua y la mezcla se extrae con EtOAc tres veces. Las fases orgánicas combinadas se lavan con NaHCO3 saturado dos veces, con salmuera, se secan sobre Na2SO4 y se evaporan. El residuo se purifica en una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar 15.
Ejemplos 18 y 18-1.

Los Ejemplos 18 y 18-1 se pueden preparar de acuerdo con el siguiente esquema usando materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. A una mezcla de reacción de 14D (1,0 equiv.), 18A (1,2 equiv.) y yoduro de cobre(I) (0,05 equiv.) en DMF (0,2 M) en atmósfera de nitrógeno se añade NaH (3,0 equiv.). La mezcla de reacción se calienta a 120 °C durante 24 h y, a continuación, se enfría a temperatura ambiente y se diluye con EtOAc. La mezcla se filtra a través de una capa de tierra de diatomeas y el filtrado se evapora a vacío. El residuo bruto se purifica en una columna de gel de sílice eluyendo con EtOAc/heaxanos para dar 18B.
Etapa 2. A una mezcla de reacción de 18B (1,0 equiv.) en DMF (0,2 M) se añaden KOH (2 equiv.) e I2 (1,1 equiv.). La mezcla de reacción se agita a temperatura ambiente durante 1 h y a continuación se extingue con NaHSO3 y se extrae con EtOAc. Las fases orgánicas combinadas se lavan con NaHCO3 saturado dos veces, con salmuera, se secan sobre Na2SO4 y se evaporan. El residuo se purifica en una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar 18C.
Etapa 3. A una solución del compuesto 18C (1 equiv.) en CH2Cl2 (0,2 M) se añade HCl 4 M/dioxano (10 equiv.) y la mezcla se agita hasta que todo el compuesto 18 c se convierte en 18D. Después de la concentración, el residuo se purifica en una HPLC preparativa de fase inversa para proporcionar 18D.
Etapa 4. A una solución de 18D (1 equiv.) y DIPEA (2 equiv.) en tolueno (0,01 M) se añade Pd(Pt-Bu3)2 (1 equiv.). La mezcla de reacción se calienta a 100 °C bajo 4 bar de CO durante la noche y, a continuación, se concentra. El residuo se purifica en una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar 18.
Ejemplo 20.

El Ejemplo 20 se preparó de acuerdo con el siguiente esquema:

Etapa 1. (2-(4-Fluoro-2-formilfenoxi)etil)carbamato de terc-butilo (20C). Una solución de aldehido 20A (1,5 g, 11 mmol), cloruro 20B (2,1 g, 12 mmol), carbonato de potasio (7,4 g, 54 mmol) y yoduro de potasio (36 mg, 0,2 mmol) en DMF (11 ml) se calentó a 60 °C y se agitó durante 15 horas. El cloruro 20b adicional (1,0 g, 6 mmol) y el calentamiento adicional a 80 °C durante 5 horas completaron la reacción. La mezcla se enfrió a temperatura ambiente y se diluyó mediante la adición de agua (250 ml). La mezcla se extrajo con acetato de etilo (3 x 300 ml) y los extractos combinados se lavaron con agua ( 200 ml) y salmuera ( 100 ml), se secaron con sulfato de sodio y se concentraron bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice, 0-20 % de acetato de etilo en hexano) proporcionó 20C (3,0 g, 99 %) como un aceite viscoso. LRESIMS m/z 306,1 [M+Na] , calculado para C^H-ieF-iN-iNa-O 306,1.
Etapa 2. (2-(4-Fluoro-2-((metilamino)metil)fenoxi)etil)carbamato de ferc-butilo (20D). El aldehido 20C (2,5 g, 8 , 8 mmol) y metilamina (0,69 g, 22 mmol) en metanol ( 88 ml) se calentaron a 60 °C y se agitaron durante 1 hora. La mezcla se enfrió a temperatura ambiente y se añadió borohidruro de sodio (0,33 g, 8 , 8 mmol). Se agitó la mezcla durante 30 minutos y a continuación se extinguió mediante la adición de agua (200 ml). La mezcla se extrajo con diclorometano (4x 100 ml) y los extractos combinados se secaron con salmuera (50 ml), sulfato de sodio y se concentraron bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice, 0-100% de (10% de metanol en acetato de etilo) en hexano) proporcionó el compuesto del título (2,1 g, 80 %) como un gel. LRESIMS m/z 299,2 [M+H]+, calculado para C15H24F1N2O3299,2.
Etapa 3. 5-((2-(2-((terc-Butoxicarbonil)amino)etoxi)-5-fluorobencil)(metil)amino)pirazolo[1,5-a]pirimidina-3-carboxilato de etilo (20F). La amina 2 0 D (2 ,1 g, 7,0 mmol), el éster 20 E (1,59 g, 7,0 mmol) y base de Hünig (7,0 ml, 5,2 g, 40 mmol) en butanol (17 ml) se calentaron a 110 °C durante 25 minutos. La reacción se enfrió y se diluyó con agua (250 ml). La mezcla se extrajo con diclorometano (4 x 100 ml) y los extractos combinados se secaron con sulfato de sodio. La mezcla se concentró bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO,
sílice, 20-100 % de acetato de etilo en hexano) proporcionó el compuesto del título (2,1 g, 75 %) como un sólido. LRESIMS m/z 488,3 [M+H]+, calculado para C24H31F1N5O5488,2.
Etapa 4. Ácido 5-((2-(2-((terc-butoxicarbonil)amino)etoxi)-5-fluorobencil)(metil)amino)pirazolo[1,5-a]pirimidina-3-carboxílico (20G). Se añadió hidróxido de sodio (40 ml, 2 M en agua) a una solución agitada de éster 20F (2,1 g, 4,3 mmol) en tetrahidrofurano:metanol (3:2, 100 ml) a temperatura ambiente. La reacción se calentó a 60 °C y se agitó durante 6,5 horas. La mezcla se enfrió a 0 °C y se acidificó con ácido clorhídrico (45 ml, 2 M en agua) y, a continuación, se diluyó con agua (100 ml). La mezcla se extrajo con acetato de etilo (4 x 150 ml) y los extractos combinados se secaron con salmuera (50 ml) y sulfato de sodio. La mezcla se concentró bajo presión reducida para proporcionar el compuesto del título (1,92 g, 97 %) como un sólido. LRESIMS m/z 460,2 [M+H]+, calculado para C22H27F1N5O5460,2.
Etapa 5. Ácido 5-((2-(2-aminoetoxi)-5-fluorobencil)(metil)amino)pirazolo[1,5-a]pirimidina-3-carboxílico (20H). Se añadió ácido clorhídrico (5 ml, 4 M en dioxano) a una solución agitada del ácido carboxílico 20G (1,92 g, 4,2 mmol) en diclorometano (25 ml) a temperatura ambiente. La reacción se agitó durante 2 horas y a continuación se concentró bajo presión reducida para proporcionar el compuesto del título como un sólido. LRESIMS m/z 360,2 [M+H]+, calculado para C17H10F1N5O3360,2.
Etapa 6. Bajo una atmósfera de argón, se añadió HATU (1,67 g, 4,4 mmol) a una solución agitada del ácido carboxílico 20H (1,50 g, 4,2 mmol) y base de Hünig (7,28 ml, 5,40 g, 41,8 mmol) en DMF:diclorometano (5:1, 60 ml) a -78 °C. Se dejó que la reacción alcanzara lentamente temperatura ambiente y se agitó durante 3 horas y, a continuación, se extinguió con agua (300 ml). La mezcla se extrajo con acetato de etilo (3 x 100 ml), a continuación con diclorometano (2 x 100 ml) y los extractos combinados se secaron con salmuera (50 ml) y sulfato de sodio. La mezcla se concentró bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice, 1-4 % de metanol en diclorometano), seguida de recristalización en acetato de etilo/metanol, proporcionó el Ejemplo 20 (0,98 g, 68 %, 2 etapas) como un sólido. LRESIMS m/z 342,2 [M+H]+, calculado para C17H17F1N5O2342,1; 1H RMN (500 MHz, DMSO-cfe) 8 (ppm) 9,43 (dd, J = 6,9, 2,7 Hz, 1H), 8,76 (d, J =7,9 Hz, 1H), 8,10 (s, 1H), 7,19-7,25 (m, 1H), 7,03 7,07 (m, 2H), 6,72 (d, J = 7,9 Hz, 1H), 5,64 (dd, J =14,9, 1,5 Hz, 1H), 4,48 (dt, J =10,2, 4,3 Hz, 1H), 4,04-4,10 (m, 2H), 3,81-3,87 (m, 1H), 3,58 (s, 3H), 3,38-3,46 (m, 1H).
Síntesis alternativa del Ejemplo 20:
El Ejemplo 20 también se preparó por la siguiente ruta alternativa:

Etapa 1. 5-oxo-4H-pirazolo[1,5-a]p¡nm¡d¡na-3-carbox¡lato de etilo (20J). A una mezcla de 20I (150,00 g, 1,08 mmol) y (E)-3-etoxiprop-2-enoato de etilo (292,16 g, 2,03 mol) en DMF (3,2 l) se añadió Cs2CO3 (656,77 g, 2,02 mol) en una porción a 20 °C en atmósfera de N2. Se agitó la mezcla a 110 °C durante 6 horas. La mezcla se enfrió a 20 °C y se filtró a través de una capa de tierra de diatomeas. La torta de filtrado se lavó con acetato de etilo (3 x 30 ml). El filtrado se añadió a H2O (2 l) y se acidificó con HOAc hasta pH = 4. El precipitado resultante se filtró para dar 20J (173,00 g, 834,98 mmol, 86,36% de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSo-cfe) 8 (ppm) 8,54 (d, J = 7,91 Hz, 1H), 8,12 (s, 1H), 6,13 (d, J =7,91 Hz, 1H), 4,27 (q, J = 7,11 Hz, 2H), 1,28 (t, J = 7,09 Hz, 3H).
Etapa 2. 5-clorop¡razolo[1,5-a]p¡r¡m¡d¡na-3-carbox¡lato de etilo (20K). A una mezcla de 20J (158,00 g, 762,59 mmol) en MeCN (1,6 l) se añadió p Oc I3 (584,64 g, 3,81 mol) a 20 °C en atmósfera de N2. Se agitó la mezcla a 100 °C durante 2 horas. La mezcla se enfrió a 20 °C y se vertió en agua con hielo (5000 ml) en porciones a 0 °C y se agitó durante 20 min. El precipitado se filtró y se secó para proporcionar 20K (110,00 g, 487,52 mmol, 63,93 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-cfe) 8 (ppm) 9,33 (d, J = 7,28 Hz, 1H), 8 , 66 (s, 1H), 7,41 (d, J = 7,15 Hz, 1H), 4,31 (q, J = 7,15 Hz, 2H), 1,32 (t, J =7,09 Hz, 3H).
Etapa 3. 4-fluoro-2-metilaminometil-fenol (20M). A una solución de 20L (5,00 g, 35,69 mmol, 1,00 eq.) en MeOH (50,00 ml) se añadió metanamina acuosa (8 , 8 ml, 71,38 mmol, 25%, 2,00 eq.) en una porción a 25 °C en atmósfera de N2. Se agitó la mezcla a 25 °C durante 3 horas, a continuación se añadió NaBH4(2,70 g, 71,38 mmol, 2,00 eq.) en porciones y se agitó la mezcla a 25 °C durante otras 9 horas. La TLC mostró que la reacción se había completado. La mezcla se concentró bajo presión reducida a 45 °C. El residuo se vertió en agua (50 ml). La fase acuosa se extrajo con diclorometano (3 x 200 ml) y la fase orgánica combinada se lavó con salmuera (200 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para dar 20M (5,10 g, 32,87 mmol, 92,09 % de rendimiento) como un sólido incoloro. 1H RMN (400 MHz, CDCI3 ) 8 (ppm) 6 , 86 (dt, J = 3,0, 8,7 Hz, 1H), 6,78-6,69 (m, 2H), 3,93 (s, 2H), 2,48 (s, 3H).
Etapa 4. éster etílico del ácido 5-[(5-fluoro-2-h¡drox¡-benc¡l)met¡l-am¡no]p¡razolo[1,5-a]p¡r¡m¡d¡na-3-carboxíl¡co (A1). A una suspensión de 20 M (33,70 g, 217,17 mmol, 1 , 00 eq.) y 20 K (49,00 g, 217,17 mmol, 1 , 00 eq.) en n-BuoH (740,00 ml) se añadió DIPEA (159,98 g, 1,24 mol, 5,70 eq.). La mezcla de reacción se agitó a 120 °C durante 2 horas en atmósfera de nitrógeno. La TLC mostró la finalización de la reacción. La solución se enfrió a 25 °C y a continuación se eliminó el disolvente. El residuo se diluyó con agua (500 ml) y se extrajo con diclorometano (3 x 500 ml). Los extractos orgánicos combinados se lavaron con salmuera (300 ml), se secaron sobre Na2SO4
anhidro y se concentraron a vacío. El residuo se trituró con EtOAc (100 ml) para dar A1 (60,00 g, 174,25 mmol, 80,24 % de rendimiento) como un sólido blanco. 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 9,71 (s, 1H), 8,32 (d, J = 7,9 Hz, 1H), 8,30 (s, 1H), 6,98-6,87 (m, 3H), 6,37 (d, J = 7,9 Hz, 1H), 4,82 (s, 2H), 4,42 (q, J =7,1 Hz, 2H), 3,21 (s, 3H), 1,39 (t, J = 7,1 Hz, 3H).
Etapa 5. éster etílico del ácido 5-{[2-(2-ferc-butoxicarbonilamino-etoxi)-5-fluoro-bencil]metil-amino}pirazolo[1,5-a]pirimidina-3-carboxílico (B1). A una solución de A1 (102,85 g, 298,6 mmol, 1 eq.) y éster terc-butílico del ácido (2-cloro-etil)-carbámico (56,33 g, 313,5 mmol, 1,05 eq.) en DMF (854 ml) se añadió K2CO3 (206,41 g, 1,493 mmol, 5,0 eq.). La mezcla se calentó a 80 °C durante 20 horas con una conversión del ~85 % del material de partida en el producto por LC-MS. Se añadieron porciones adicionales de éster terc-butílico del ácido (2-cloro-etil)-carbámico (5,633 g, 31,35 mmol, 0,1 eq.) y K2CO3 (41,282 g, 298,6 mmol, 1 eq.) al matraz de reacción. La reacción se mantuvo a 80 °C durante 21 horas adicionales. La mezcla se enfrió a continuación a temperatura ambiente, se extinguió con agua (1000 ml) y se extrajo con EtOAc (3 x 900 ml). Los extractos orgánicos combinados se lavaron a continuación con agua (3 x 700 ml) y salmuera (500 ml), se secaron sobre Na2SO4 y se concentraron. El residuo resultante se purificó en una columna de gel de sílice eluyendo con EtOAc/hexano (0-70 %) para dar B1 como un sólido blanco (128,74 g, 96,7 % de rendimiento). LC-MS (ESI) m/z 510,1 (M+Na)+; 1H RMN (500 MHz, cloroformod) 6 (ppm) 8,30 (s, 1H), 8,26 (s, 1H), 6,92 (td, J = 8 ,6 , 3,3 Hz, 1H), 6,83-6,76 (m, 1H), 6,31 (s, 1H), 4,93 (s, 2H), 4 ,51 -4 , 44 (m, 1H), 4,36 (q, J = 7,2 Hz, 2H), 4,03 (t, J = 4,9 Hz, 2H), 3,69-3,63 (m, 1H), 3,51 (s, 2H), 3,30 (s, 2H), 1,44 (s, 9H), 1,41-1,35 (t, J = 7,2 Hz, 3H).
Etapa 6. 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecin-4(5H)-ona (20). A una solución de B1 (128,74 g, 264,07 mmol, 1 eq.) en metanol (750 ml) y THF (250 ml) se añadió L O H H 2O (55,40 g, 1320 mmol, 5,0 eq.) en H2O (250 ml). La solución transparente se calentó a 70 °C durante 2 horas. La reacción se neutralizó a 0 °C con HCl ac. ( 2 M, 250 ml) hasta pH < 5 y a continuación se extrajo con CH2Cl2 (1 x 1000 ml, 3 x 500 ml). Los extractos orgánicos combinados se lavaron con salmuera (300 ml) y se secaron sobre Na2SO4. Después de la filtración, evaporación y secado a alto vacío, se obtuvo un sólido blanco (126,47 g, 275,25 mmol, 104 % de rendimiento). A una solución del ácido (121,30 g, 264 mmol) en CH2Cl2 (996 ml) se añadió HCl en dioxano (4 M, 204 ml) a 0 °C. Se mantuvo la agitación de 0 °C a temperatura ambiente durante 27 horas hasta que el de-Boc se completó por LC-MS. El sólido blanco se filtró, se lavó con DCM (400 ml) y se secó a alto vacío para proporcionar un sólido blanco de la sal 3HCl de amina (123,55 g) que se usó directamente sin purificación adicional. A una solución de DIPEA (169,4 g, 228 ml, 1310 mmol) en DMF (3,7 l) y CH2O 2 (1,0 l) se añadió la sal de HCl de la amina ácida (22,92 g, 49,0 mmol, 1,00 eq.). Después de que la sal sólida se disolvió completamente, se añadió difenilfosfinato de pentafluorofenilo (FDPP) en CH2Cl2 (1,1 M, 19,76 g, 51,44 mmol, 1.05 eq.). El acoplamiento se completó en 30 minutos según LC-MS y a continuación se añadieron las segundas porciones de la sal y de FDPP siguiendo el mismo procedimiento que con la primera porción. La adición de la sal seguida de FDPP se repitió cada 30 minutos y se monitorizó mediante LC-MS para cada ciclo de adición. Se añadieron al matraz de reacción un total de la sal (123,55 g, 264 mmol, 1,00 eq.) y FDPP (106,44 g, 277 mmol, 1.05 eq.) en porción. La solución de reacción se concentró hasta un volumen de ~500 ml y se formó una gran cantidad de precipitado. El producto sólido 20 se filtró y se lavó con DMF (50 ml x 3). El filtrado se vertió en agua (2 l) y se precipitó producto adicional. El producto sólido se filtró y se lavó con agua (100 ml x 3). El producto sólido combinado se secó y se volvió a disolver en metanol al 10 % en diclorometano (1,5 l) y, a continuación, se añadió acetato de etilo (1 l). La solución se condensó hasta ~500 ml y se formó una gran cantidad de sólido blanco. Después de la filtración y secado a alto vacío, se obtuvo un compuesto 20 como un sólido blanco (74,58 g, 83 % de rendimiento).
Difracción de rayos X en polvo (PXRD) del Ejemplo 20.
Una muestra del Ejemplo 20, la forma cristalina polimorfa 1, se transfirió a una placa de fondo cero para el análisis por PXRD. Los datos de PXRD se obtuvieron usando un difractómetro de rayos X Bruker D8 de acuerdo con los procedimientos recomendados por el fabricante. Parámetros para la exploración: intervalo 2-theta: 4,5 a 39,1 grados; tamaño de paso: 0 , 02 grados; tiempo de paso: 1 segundo; tiempo de análisis: 180 segundos.
Los picos de difracción se miden típicamente con un error de ±0,1 grados (20).
Los resultados se muestran en la Fig. 1. Los datos se resumen en la Tabla 1.
Tabla 1


Calorimetría diferencial de barrido (DSC) del Ejemplo 20
Las mediciones de DSC, que se muestran en la Fig. 2, se llevaron a cabo usando un calorímetro diferencial de barrido Seiko modelo SSC/5200. Una muestra de 7,92 mg del Ejemplo 20, la forma polimorfa cristalina 1, se equilibró a 36 °C y a continuación se elevó la temperatura hasta 380 °C a una velocidad de 10 °C/min. La muestra del Ejemplo 20, la forma polimorfa cristalina 1, mostró un punto de fusión de 298,9 °C.
Ejemplo 26

El Ejemplo 26 se puede preparar de 

Etapa 1. Se añade isopropóxido de titanio(IV) (1,3 equiv.) a una solución de metilamina disponible comercialmente en metanol (2 M, 3 equiv.), seguido de la adición del aldehído de partida 14C (1,0 equiv.). La mezcla de reacción se agita a temperatura ambiente durante 5 h, después de lo cual se añade borohidruro de sodio (1,0 equiv.) y la mezcla resultante se agita adicionalmente durante otro período de 2 h. La reacción se extingue a continuación mediante la adición de agua, el precipitado inorgánico resultante se filtra y se lava con EtOAc. La fase orgánica se separa y la fase acuosa se extrae adicionalmente con EtOAc (x2). Los extractos combinados se secan (K2CO3) y se concentran a vacío para dar 26A.
Etapa 2. Una mezcla del compuesto 26A (1 equiv.) y DIPEA (2 equiv.) en n-BuOH (0,2 M) se calienta a 120 °C durante la noche, se enfría a temperatura ambiente y a continuación se concentra. El residuo se purifica con una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar el producto 26B.
Etapa 3. A una solución del compuesto 26B (1 equiv.) en CH2CI2 (0,2 M) se añade HCl 4 M/dioxano (10 equiv.) y la mezcla se agita hasta que todo el compuesto 26b se convierte en 26C. Después de la concentración, el residuo se purifica en una HPLC preparativa de fase inversa para proporcionar 26C.
Etapa 4. A una solución de 26C (1 equiv.) y DIPEA (2 equiv.) en tolueno (0,01 M) se añade Pd(Pt-Bu3)2 (1 equiv.). La mezcla de reacción se calienta a 100 °C bajo 4 bar de CO durante la noche y, a continuación, se concentra. El residuo se purifica en una columna de gel de sílice eluyendo con EtOAc/hexanos para proporcionar 26.
Ejemplos 37 y 37-1.

Los Ejemplos 37 y 37-1 se pueden preparar de acuerdo con el siguiente esquema usando materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. El compuesto 37B se prepara a partir del compuesto 2C y el compuesto 37A usando el procedimiento descrito para el Ejemplo 2 , Síntesis A, Etapa 2.
Etapa 2. El compuesto 37C se prepara a partir del compuesto 37B usando el procedimiento descrito en el Ejemplo 2, Síntesis A, Etapa 3.
Etapa 3. El Ejemplo 37 se prepara a partir del compuesto 37C usando el procedimiento descrito en el Ejemplo 2, Síntesis A, Etapa 4.
Ejemplos 38 y 38-1.
Los Ejemplos 38 y 38-1 se pueden preparar de acuerdo con el siguiente esquema usando materiales de partida racémicos o enantioméricamente enriquecidos:

Etapa 1. El compuesto 38B se prepara a partir de los compuestos 2C y 38A como se describe en el Ejemplo 2, Síntesis A, Etapa 2.
Etapa 2. El compuesto 38C se prepara a partir del compuesto 38B usando el procedimiento descrito en el Ejemplo 2, Síntesis A, Etapa 3.
Etapa 3. El Ejemplo 38 se prepara a partir del compuesto 38C usando el procedimiento descrito en el Ejemplo 2, Síntesis B, Etapa 4.
Ejemplo 39.

El ejemplo 39 se preparó de acuerdo con los siguientes esquemas:

Etapa 1. éster ferc-butílico del ácido 2-(3-doro-4-fluoro-2-formil-fenoxi)-etil]-carbámico (39B). A una solución de 2-cloro-3-fluoro-6-hidroxi-benzaldehído (39A, 53 mg, 0,3 mmol) y éster ferc-butílico del ácido (2-cloro-etil)-carbámico (135 mg, 0,75 mmol) en DMF (5 ml) se añadieron KI (2,0 mg, 0,012 mmol) y K2CO3 (105 mg, 0,75 mmol). La mezcla se sometió a microondas a 100 °C durante 2 h. La mezcla se diluyó a continuación con agua (20 ml) y se extrajo con EtOAc (3 x 20 ml). Las fases orgánicas combinadas se lavaron con agua (3x20 ml) y salmuera (20 ml), se secaron sobre Na2SO4 y se concentraron para dar 39B. El residuo en bruto se usó directamente en la siguiente etapa. LC-MS (ESI) m/z 340,3 (M+Na)+.
Etapa 2. éster ferc-butílico del ácido {[2-(3-cloro-4-fluoro-2-metilaminometil-fenoxi)-etil]-carbámico (39C). A una solución de 39B (95,4 mg, 0,3 mmol) en MeOH (3 ml) se añadió clorhidrato de metilamina (50,7 mg, 0,75 mmol). Se agitó la mezcla a 60 °C durante 30 min. La solución se enfrió a continuación a temperatura ambiente y se añadió NaBH4 (11,1 mg, 0,3 mmol). Se agitó la mezcla a temperatura ambiente durante 2 h. La solución se diluyó a continuación con agua (50 ml) y se extrajo con DCM (3 x 20 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4 y se concentraron para dar 39C. El residuo en bruto se usó directamente en la siguiente etapa. LC-MS: (ESI) m/z 333,3 (M+H)+.
Etapa 3. éster etílico del ácido 5-{[6-(2-terc-butoxicarbonilamino-etoxi)-2-cloro-3-fluoro-bencil]-metil-amino}-pirazolo[1,5-a]pirimidina-3-carboxílico (39D). A una solución de 20K (67,5 mg, 0,3 mmol) y 39C (99,9 mg, 0,3 mmol) en n-BuOH (2,0 ml) se añadió DIEA (1,0 ml). La mezcla se calentó en microondas a 150 °C durante 2 horas. La mezcla se diluyó con agua y se extrajo con DCM (3 x 20 ml). La fase orgánica se secó sobre Na2SO4, se concentró y purificó por cromatografía en columna en gel de sílice para dar 17 como un líquido amarillo. LC-MS (ESI) m/z 522,5 (M+H)+.
Etapa 4. ácido 5-{[6-(2-terc-butoxicarbonilamino-etoxi)-2-cloro-3-fluoro-bencil]-metil-amino}-pirazolo[1,5-a]pirimidina-3-carboxílico (39E). A una solución de 39D (40 mg, 0,0776 mmol) en MeOH (1 ml) se añadió LiOH (16 mg, 0,38 mmol) y H2O (1 ml). Se agitó la mezcla a 60 °C durante 4 h. La solución se enfrió a temperatura ambiente, se concentró parcialmente y se acidificó con HCl acuoso (1 N) hasta pH 2-3. La mezcla acuosa se extrajo con DCM (3x10 ml). La fase orgánica se secó sobre Na2SO4 y se concentró para dar 39E. El residuo en bruto se usó directamente en la siguiente etapa. LC-MS (ESI) m/z 494,3 (M+H)+.
Etapa 5. ácido 5-{[6-(2-amino-etoxi)-2-cloro-3-fluoro-bencil]-metil-amino}-pirazolo[1,5-a]pirimidina-3-carboxílico (39F). A una solución de 39E (40 mg, 0,0776 mmol) en DCM (2 ml) se añadió TFA (0,4 ml). La solución se agitó durante 1 h. Se eliminó el disolvente en un rotavapor. El residuo se volvió a disolver con DCM y se reconcentró (3x) para dar 39F como un sólido de tipo espuma. LC-MS (ESI) m/z 393,5 (M+H)+.
Etapa 6. A una solución de 39F (36 mg, 0,078 mmol) en 10 ml de DCM se añadió DIEA (0,20 ml, 1,15 mmol). La solución se enfrió con un baño de hielo seco/acetona y se añadió HATU (40,0 mg, 0,11 mmol). Se dejó que la solución alcanzara temperatura ambiente lentamente. La mezcla se diluyó con agua (50 ml) y se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se lavó con agua (3 x 50 ml) y salmuera (50 ml), se secó sobre Na2SO4 y se concentró. El residuo resultante se purificó en una columna de sílice (0-5 % MeOH/DCM), para dar el Ejemplo 39 como un sólido blanco (6,2 mg, 23,4 %). LC-MS (ESI) m/z 376,5 (M+H)+. 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 9,51 (s, 1H), 8,40-8,33 (m, 2H), 7,03 (ddd, J = 8,9, 8,0, 0,7 Hz, 1H), 6,78 (dd, J = 9,3, 4,2 Hz, 1H), 6,40 (d, J = 7,9 Hz, 1H), 5,97 (dd, J =15,0, 2,1 Hz, 1H), 4,49-4,43 (m, 1H), 4,31 (ddd, J =10,9, 6,4, 4,5 Hz, 1H), 4,12-4,03 (m, 1H), 3,91 (d, J = 14,9 Hz, 1H), 3,72-3,63 (m, 1H), 3,56 (s, 3H).
Ejemplo 40.

El Ejemplo 40 se preparó como se muestra en el siguiente esquema:

Etapa 1. ácido 5-[(5-fluoro-2-hidroxi-bencil)-metil-amino]-2-metil-pirazolo[1,5-a]pirimidina-3-carboxílico (40B). A una solución de 19A (75 mg, 0,14 mmol) en MeOH (2 ml) se añadió LiOH (60 mg, 1,4 mmol) y H2O (2 ml). Se agitó la mezcla a 60 °C durante 4 h. La solución se enfrió a temperatura ambiente, se concentró parcialmente y se acidificó con HCl acuoso (1 N) hasta pH 2-3. La suspensión resultante se extrajo con EtOAc (3 x 20 ml). La fase orgánica se secó sobre Na2SO4 y se concentró para dar 40A. LC-MS (ESI) m/z 331,6 (M+H)+.
Etapa 2. (2-hidroxietil)-amida del ácido 5-[(5-fluoro-2-hidroxi-bencil)-metil-amino]-2-metil-pirazolo[1,5-a]pirimidina-3-carboxílico (40B). A una solución de 40A (140 mg, 0,42 mmol) y 2-amino-etanol (244 mg, 4 mmol) en Dc M (5 ml) a 0 °C se añadieron DIEA (0,20 ml, 1,15 mmol) y HATU (380,0 mg, 1,0 mmol). Se dejó que la solución alcanzara
temperatura ambiente lentamente. La mezcla se diluyó a continuación con agua (25 ml) y se extrajo con EtOAc (3 x 25 ml). Las fases orgánicas combinadas se lavaron con HCl (1 N, 3 x 20 ml) y salmuera (50 ml), se secaron sobre Na2SO4 y se concentraron. El residuo resultante se purificó en una columna de gel de sílice eluyendo con 0 5 % de MeOH/DCM (10 V.C.) para dar 40B como un sólido blanco (74 mg, 47 %). LC-MS (ESI) m/z 374,3 (M+H)+.
Etapa 3. A una solución de 40B (74 mg, 0,2 mmol) en THF (3 ml) y DCM (3 ml) a 0 °C se añadieron PPh3 (131 mg, 0,5 mmol) y azodicarboxilato de di-terc-butilo (DTAD) (115 mg, 0,5 mmol). Se dejó que la mezcla alcanzara temperatura ambiente y se agitó durante 4 h adicionales. Se eliminó el disolvente y se purificó el residuo en una columna de gel de sílice eluyendo con 0-10 % de MeOH/DCM (10 V.C.), seguido de TLC preparativa para dar el Ejemplo 40 como un sólido blanco (15 mg). LC-MS (ESI) m/z 356,5 (M+H)+; 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 8,12 (d, J = 7,7 Hz, 1H), 6,93 (ddd, J = 9,0, 3,1, 0,9 Hz, 1H), 6,78 (ddd, J = 9,0, 7,3, 3,0 Hz, 1H), 6,71 (dd, J = 9,1, 4,5 Hz, 1H), 6,28 (d, J = 7,7Hz, 1H), 5,77 (dd, J =15,2, 1,7 Hz, 1H), 4,38-4,33 (m, 1H), 3,98 (s, 1H), 3,91 (d, J = 1,4 Hz, 1H), 3,78 (dd, J =15,1, 0,9 Hz, 1H), 3,45 (s, 3H), 3,43-3,36 (m, 1H), 2,45 (s, 3H).
Ejemplo 41.

El Ejemplo 41 se preparó usando el procedimiento que se muestra en el siguiente esquema:

Etapa 1. Éster terc-butílico del ácido [2-(2-acetil-4-fluoro-fenoxi)-etil]-carbámico (41B). A una mezcla de 1-(5-fluoro-2-hidroxi-fenil)-etanona (41A, 773 mg, 5,0 mmol) y éster terc-butílico del ácido (2-cloroetil)-carbámico (1,80 g, 10,0 mmol) en DMF (20 ml) se añadieron KI (2,0 mg, 0,012 mmol) y Cs2CO3 (3,26 g, 10,0 mmol). Se agitó la mezcla a 80 °C durante la noche. La mezcla se enfrió a continuación a temperatura ambiente, se diluyó con EtOAc y se lavó con NaOH 1 N (5 x 10 ml) hasta que la LC-MS no mostró un pico de 1-(5-fluoro-2-hidroxi-fenil)-etanona. La fase orgánica se secó sobre Na2SO4 y se concentró. El residuo se purificó a continuación en una columna de gel de sílice eluyendo con EtOAc/hexano (0-30 %, 10 V.C.) para dar el producto deseado 41B como un sólido amarillo (1,1 g, 73,8%). LC-MS (ESI) m/z 320,3 (M+Na)+.
Etapa 2. (2-(4-Fluoro-2-(1-hidroxietil)fenoxi)etil)carbamato de terc-butilo (41C). A una solución de 41B (1,0 g, 3,36 mmol) en MeOH (10 ml) se añadió NaBH4 (640 mg, 16,8 mmol) en porciones. Se agitó la mezcla a temperatura ambiente durante 2 h. La solución se diluyó a continuación con agua (50 ml) y se extrajo con DCM (3 x 20 ml). Las fases de DCM combinadas se secaron sobre Na2SO4 y se concentraron. El residuo se purificó en una columna de gel de sílice eluyendo con EtOAc/hexano (0-50 %, 10 V.C.) para dar el producto deseado como un sólido amarillo pálido (0,75 g, 75 %). LC-MS (ESI) m/z 322,3 (M+Na)+; 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 7,11 (dd, J = 9,2, 3,4 Hz, 1H), 6,89 (ddd, J = 9,0, 7,9, 3,2 Hz, 1H), 6,77 (dd, J = 8,9, 4,4 Hz, 1H), 5,09 (q, J = 6 , 6 Hz, 1H), 4,92 (d, J = 4,4 Hz, 1H), 4,03 (t, J = 5,2 Hz, 2H), 3,62-3,50 (m, 2H), 1,49 (d, J = 6,4 Hz, 3H), 1,45 (s, 9H).
Etapa 3. Éster etílico del ácido 6-{1-[2-(2-terc-butoxicarbonilamino-etoxi)-5-fluoro-fenil]-etoxi}-imidazo[1,2-b]piridazina-3-carboxílico (41D). A una solución de 41C (600 mg, 2,0 mmol) y éster terc-butílico del ácido {2-[4-fluoro-2-(1-hidroxietil)-fenoxi]-etil}-carbámico (450 mg, 2,0 mmol) en THF seco (40,0 ml) a -78 °C se añadió NaH
(60 %, 80 mg, 2,0 mmol) en porciones. La suspensión se agitó a -78 °C durante 4 h y se dejó que la mezcla alcanzara 0 °C y se agitó durante 4 h adicionales. La mezcla se puso a continuación en el congelador a -20 °C durante la noche. La mezcla se extinguió a continuación con una mezcla de hielo y HCl 1 N y se extrajo con EtOAc (3 x 20 ml). La fase orgánica se secó sobre Na2SO4, se concentró y purificó dos veces para dar el producto deseado como un sólido amarillo (240 mg, 25 %): LC-MS (ESI) m/z 511,6 (M+Na)+; 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 8,16 (s, 1H), 7,90 (d, J = 9,7 Hz, 1H), 7,16 (dd, J = 9,0, 3,2 Hz, 1H), 0,95 (d, J =9,5 Hz, 1H), 6,90-6,88 (m, 1H), 6,81-6,78 (m, 1H), 6 , 68 (q, J = 6,2 Hz, 1H), 5,84-5,68 (m, 1H), 4,38 (q, J = 7,2 Hz, 2H), 4,15-4,09 (m, 2H), 3.60- 3,52 (m, 2H), 1,65 (d, J = 6,4 Hz, 3H), 1,38 (d, J = 7,2 Hz, 3H), 1,35 (s, 9H).
Etapa 4. El compuesto 41D se convirtió en el Ejemplo 41 usando procedimientos análogos a los descritos en el presente documento. MS: 343,2 (M+H)+; 1H RMN (500 MHz, cloroformo-d; 6 (ppm) 9,82 (d, J = 7,0 Hz, 1H), 8,27 (s, 1H), 8,09 (d, J = 9,5 Hz, 1H), 7,18 (dd, J = 8,9, 3,2 Hz, 1H), 7,01-6,94 (m, 2H), 6,83 (dd, J =9,0, 4,3 Hz, 1H), 6.60- 6,53 (m, 1H), 4,63-4,52 (m, 1H), 4,27-4,16 (m, 1H), 4,16-4,04 (m, 1H), 3,70-3,56 (m, 1H), 1,70 (d, J = 6,4 Hz, 3H)
Ejemplo 42.
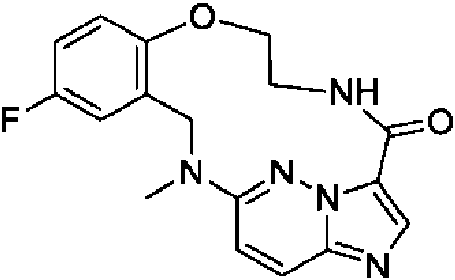
El Ejemplo 42 se preparó usando los procedimientos que se muestran en el siguiente esquema:

Etapa 1. Éster etílico del ácido 6-[(5-fluoro-2-hidroxi-bencil)-metil-amino]-imidazo[1,2-b]piridazina-3-carboxílico (42B). A una mezcla de 4-fluoro-2-metilaminometil-fenol (20L, 305,2 mg, 1,97 mmol) y éster etílico del ácido 6 cloro-imidazo[1,2-b]piridazina-3-carboxílico (42A, 230 mg, 1,02 mmol) en DMSO (5 ml) se añadió KF (180 mg, 3,01 mmol). La mezcla de reacción se agitó a 120 °C durante 18 horas en atmósfera de nitrógeno. A continuación, la solución se enfrió a temperatura ambiente, se diluyó con agua (20 ml) y se extrajo con EtOAc (3 x 50 ml). Las fases orgánicas combinadas se lavaron adicionalmente con agua (3 x 50 ml) y salmuera (50 ml), se secaron sobre Na2SO4 y se concentraron. El residuo se purificó a continuación en una columna de gel de sílice eluyendo con EtOAc/hexano (0-50 %, 10 V.C.) para dar el producto deseado como un sólido blanco (240 mg, 69 %). LC-MS (ESI) m/z 345,2 (M+H)+; 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 8,61 (s, 1H), 8,17 (s, 1H), 7,91 (d, J =10,0 Hz, 1H), 7,00-6,86 (m, 4H), 4,78 (s, 2H), 4,47 (qd, J = 7,2, 0,5 Hz, 2H), 3,17 (s, 3H), 1,41 (td, J = 7,1, 0,5 Hz, 3H).
Etapa 2. Éster etílico del ácido 6-{[2-(2-terc-butoxicarbonilamino-etoxi)-5-fluoro-bencil]-metil-amino}-imidazo[1,2-b]piridazina-3-carboxílico (42C). A una solución de éster etílico del ácido 6-[(5-fluoro-2-hidroxi-bencil)-metil-amino]-imidazo[1,2-b]piridazina-3-carboxílico (2B, 200 mg, 0,58 mmol) y éster terc-butilico del ácido (2-cloro-etil)-carbámico (209 mg, 1,16 mmol) en DMF (5 ml) se añadieron K2CO3 (200 mg, 1,45 mmol) y KI (2,0 mg, 0,012 mmol). La mezcla se calentó a 90 °C durante 4 h en atmósfera de nitrógeno. La mezcla se diluyó a continuación con agua (20 ml) y se extrajo con EtOAc (3x10 ml). Las fases orgánicas combinadas se lavaron a continuación con agua (3 x 5 ml) y salmuera (2 x 5 ml). La fase orgánica se secó sobre Na2SO4 y se concentró. El residuo resultante se purificó en una columna de gel de sílice eluyendo con EtOAc/hexano (0-100 %, 10 V.C.) para dar 42°C como un sólido blanco (203 mg, 76 %). LC-MS (ESI) m/z 510,1 (M+Na)+; 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 8,16 (s, 1H), 7,85 (d, J = 9,9 Hz, 1H), 7,00 (dd, J = 8,9, 3,2 Hz, 1H), 6,95-6,87 (m, 2H), 6,80 (dd, J =8,9,
4,3 Hz, 1H), 4,95 (s, 1H), 4,74 (s, 2H), 4,41 (q, J = 7,2 Hz, 2H), 4,04 (t, J = 5,2 Hz, 2H), 3,56-3,50 (m, 2H), 3,26 (s, 3H), 1,43 (s, 9H), 1,40 (t, J = 7,2 Hz, 3H).
Etapa 3. El compuesto 42C se convirtió en el Ejemplo 42 usando procedimientos análogos a los descritos en el presente documento. MS: 342,5 (M+H)+; 1H RMN (500 MHz, cloroformo-d) 6 (ppm) 10,01 (d, J = 6,9 Hz, 1H), 8,17 (s, 1H), 8,04 (d, J = 10,0 Hz, 1H), 7,07-7,04 (m, 1H), 7,00 (d, J =10,0 Hz, 1H), 6,96-6,92 (m, 1H), 6,84 (dd, J = 9,1, 4,5 Hz, 1H), 5,69 (dd, J = 15,8, 1,6 Hz, 1H), 4,55 (dt, J = 9,9, 3,7 Hz, 1H), 4,20-4,09 (m, 2H), 3,98 (dd, J =15,9, 1,0 Hz, 1H), 3,66-3,62 (m, 1H), 3,61 (s, 3H).
Ejemplo 51-1

Etapa 1. A una solución de A8 (399,4 mg, 1,16 mmol) y (2-cloroetil)carbamato de tere-butilo (260,5 mg, 1,45 mmol) en DMF (5,8 ml) se añadió K2CO3 (801,6 mg, 5,80 mmol) y se calentó a 80 °C con agitación durante 6 horas. La reacción se enfrió a temperatura ambiente y se diluyó con DCM (3 ml), se filtró a través de un filtro de jeringa y se concentró bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice (12 g), 0-70 % de acetato de etilo en hexano) proporcionó 51-1A (407,4 mg, 0,836 mmol, 72 % de rendimiento).
Etapa 2. A una solución de 51-1A (407,4 mg, 0,836 mmol) en MeOH ( 6 ml) y THF (4 ml) se añadió solución acuosa de LiOH (2 M, 4,0 ml) a temperatura ambiente. La solución de reacción se calentó a 70 °C durante 2 horas. El matraz de reacción se enfrió a temperatura ambiente, se diluyó con agua y metanol y, a continuación, se extinguió con una solución acuosa de HCl (2 M, 4 ml) hasta pH < 5. La mezcla se extrajo con DCM (3 x 5 ml), se secó con Na2SO4. se concentró bajo presión reducida y se secó a alto vacío durante la noche. A una solución del producto ácido en DCM ( 6 ml) se añadió HCl 4 M en 1,4-dioxano (2,97 ml). Se agitó la mezcla a temperatura ambiente durante 3 horas y, a continuación, se concentró bajo presión reducida y se secó a alto vacío. A una solución del producto de-Boc y FDPP (352,9 mg, 0,918 mmol) en DMF (21 ml) se añadió base de Hunig (539,5 mg, 0,327 mmol) a temperatura ambiente. Se agitó la mezcla durante 2,5 horas y, a continuación, se extinguió la reacción con solución de Na2CO32 M (21 ml). Se agitó la mezcla durante 15 minutos y a continuación se extrajo con DCM (4 x10 ml). Los extractos combinados se secaron con Na2SO4 y se concentraron bajo presión reducida. El residuo se purificó con cromatografía ultrarrápida (sistema ISCO, sílice (12 g), 0-11,25 % de metanol en diclorometano) para proporcionar 51-1 (164,0 mg, 0,480 mmol, 57,55 % de rendimiento en tres etapas).
Ejemplo 53.

El Ejemplo 53 se preparó usando los procedimientos que se muestran en el siguiente esquema:

Etapa 1. Ácido 5-[1-(5-fluoro-2-hidroxi-fenil)-etilamino]-pirazolo[1,5-a]pirimidina-3-carboxílico (53A). A una solución de éster etílico del ácido 5-[(5-fluoro-2-hidroxi-bencil)-metil-amino]-pirazolo[1,5-a]pirimidina-3-carboxílico (20M, 300 mg, 0,87 mmol) en MeOH (5 ml) se añadió LiOH (420 mg, 10 mmol), seguido por 5 ml de H2O. La mezcla se mantuvo en agitación a 60 °C durante 4 h. La solución se enfrió a temperatura ambiente, se concentró parcialmente y se acidificó con HCl 1 N hasta pH 2-3. La suspensión resultante se extrajo con EtOAc (3 x 20 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4 y se concentraron. El residuo se usó directamente en la siguiente etapa. LC-MS (ESI+) m/z 317,4 (M+H)+.
Etapa 2. Éster metílico del ácido 3-({5-[(5-fluoro-2-hidroxi-bencil)-metil-amino]-pirazolo[1,5-a]pirimidina-3-carbonil}-amino)-2 -hidroxi-propiónico (53B). A una solución de 53A (80 mg, 0,25 mmol) y clorhidrato de éster metílico del ácido 3-amino-2-hidroxi-propiónico (70 mg, 0,5 mmol) en DCM (5 ml) a 0 °C se añadió DIPEA (1,0 ml, 5,7 mmol), seguido de HATU (140,0 mg, 0,5 mmol). Se dejó que la solución alcanzara temperatura ambiente lentamente. La mezcla se diluyó con agua (25 ml) y se extrajo con EtOAc (3 x 25 ml). Las fases orgánicas combinadas se lavaron con HCl 1 N (3 x 20 ml) y salmuera (50 ml) y se secaron sobre Na2SO4. Se eliminó el disolvente y el sólido blanco resultante se usó directamente en la siguiente etapa. LC-MS (ESI+) m/z 418,4 (M+H)+.
Etapa 3. 11-Fluoro-14-metil-4-oxo-4,5,6,7,13,14-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaciclotridecina 7-carboxilato de metilo (53C). A una solución de 53B (83 mg, 0,2 mmol) en DCM (5 ml) se añadió PPh3 (263 mg, 1 ,0 mmol), seguido de CBr4 (332 mg, 1 ,0 mmol). Se agitó la mezcla a temperatura ambiente durante la noche. Se eliminó el disolvente y el residuo se redisolvió en DMF (5 ml), seguido de la adición de K2CO3 (116,8 mg, 0,84 mmol). A continuación, se agitó la mezcla a 80 °C hasta una formación completa del producto deseado. La mezcla se diluyó a continuación con EtOAc y se lavó con agua. La fase orgánica se secó sobre Na2SO4 y se concentró. El residuo se purificó en columna de sílice (0-10 %, MeOH/DCM) para dar 53C como un sólido blanco (40 mg). LC-MS (ESI+) m/z 400,2 (M+H)+.
Etapa 4. Para obtener 53C (20 mg, 0,05 mmol), se añadió NH3 en solución en MeOH (7 N, 2 ml). Se agitó la mezcla a 60 °C durante la noche. Se eliminó el disolvente y el residuo se purificó en columna de sílice (0-10%, MeOH/DCM) para dar el Ejemplo 53 como un sólido blanquecino ( 8 mg). Lc -MS (ESI+) m/z 385,5 (M+H)+; 1H RMN (300 MHz, cloroformo-d) 6 (ppm) 8,41 (s, 1H), 8,34 (d, J =7,9 Hz, 1H), 8,17 (s, 1H), 6,99-6,92 (m, 2H), 6,77 (dd, J = 6,2, 3,5 Hz, 1H), 6,38 (d, J = 7,9 Hz, 1H), 5,63-5,44 (m, 2H), 5,09 (dd, J =11,0, 8,4 Hz, 1H), 4,38 (dd, J = 14,7, 11,0 Hz, 1H), 4,28-4,17 (m, 1H), 4,17-4,07 (m, 2H), 3,22 (s, 3H).
Ejemplo 54.

El Ejemplo 54 se preparó usando el procedimiento que se muestra en el siguiente esquema:

A una solución del compuesto 53C (20 mg, 0,05 mmol) en MeOH (2 ml) se anadio NaBH4 (19 mg, 0,5 mmol) en porciones. Se agitó la mezcla durante 4 h. Se eliminó el disolvente y el residuo se purificó en columna de sílice (0 10 %, MeOH/DCM) para dar el producto deseado como un sólido blanco ( 8 mg). LC-MS (ESI+) m/z 372,5 (M+H)+; 1H RMN (300 MHz, cloroformo-d) 6 (ppm) 8,39 (s, 1H), 8,32 (d, J = 7,9 Hz, 1H), 7,01-6,85 (m, 3H), 6,35 (d, J = 8,0 Hz, 1H), 5,55-5,43 (m, 1H), 4,92-4,82 (m, 1H), 4,09-3,98 (m, 2H), 3,80-3,70 (m, 3H), 3,23 (s, 3H).
Ejemplo 93.

Etapa 1. A una solución de (R)-(2-hidroxipropil)carbamato de ferc-butilo (1,00 g, 5,71 mmol) y cloruro de tosilo (1,14 g, 6,00 mmol) en DCM (29 ml) se anadió trietilamina (1,44 g, 14,28 mmol y se agitó la mezcla a temperatura ambiente durante 48 horas. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía ultrarrápida (sistema ISCO, sílice (40 g), 0-20 % de acetato de etilo en hexano) para proporcionar (R)-1-((terc-butoxicarbonil)amino)propan-2-il-4-metilbencenosulfonato (1,12 g, 3,40 mmol, 59,54% de rendimiento).
Etapa 2. A una solución de A8 (100,00 mg, 0,290 mmol) y (R)-1-((terc-butoxicarbonil)amino)propan-2-il-4-metilbencenosulfonato (143,50 mg, 0,436 mmol) en DMF (1,45 ml) se anadió K2CO3 (200,7 mg, 1,45 mmol) y se calentó a 80 °C con agitación durante 16 horas. La reacción se enfrió a temperatura ambiente y se diluyó con DCM (3 ml), se filtró a través de un filtro de jeringa y se concentró bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice (12 g), 0-60% de acetato de etilo en hexano) proporcionó 93A (32,90 mg, 0,0656 mmol, 22,59% de rendimiento).
Etapa 3. A una solución de 93A (32,90 mg, 0,0656 mmol) en MeOH (3 ml) y THF (2 ml) se anadió solución acuosa de LiOH (2 M, 2 ml) a temperatura ambiente. La solución de reacción se calentó a 70 °C durante 2 horas. El matraz de reacción se enfrió a temperatura ambiente, se diluyó con agua y metanol y, a continuación, se extinguió con una solución acuosa de HCl (2 M, 2 ml) hasta pH < 5. La mezcla se extrajo con DCM (3 x 5 ml), se secó con Na2SO4. se concentró bajo presión reducida y se secó a alto vacío durante la noche. A una solución del producto ácido en DCM (4 ml) se anadió HCl 4 M en 1,4-dioxano (2,0 ml). Se agitó la mezcla a temperatura ambiente durante 3 horas y, a continuación, se concentró bajo presión reducida y se secó a alto vacío. A una solución del producto de-Boc y FDPP (27,62 mg, 0,0719 mmol) en DMF (1,6 ml) se anadió base de Hunig (42,23 mg, 0,327 mmol) a temperatura ambiente. Se agitó la mezcla durante 2,5 horas y, a continuación, se extinguió la reacción con solución de Na2CO32 M (2 ml). Se agitó la mezcla durante 15 minutos y a continuación se extrajo con DCM (4x10 ml). Los
extractos combinados se secaron con Na2SO4 y se concentraron bajo presión reducida. El residuo se purificó con cromatografía ultrarrápida (sistema ISCO, sílice (12 g), 0-10 % de metanol en diclorometano) para proporcionar 93 (10,1 mg, 0,0284 mmol, 43,49 % de rendimiento en tres etapas).
Ejemplos 104, 106 y 107

Etapa 1. A una solución de A17HCl (38 mg, 0,096 mmol) y (2-cloroetil)carbamato de terc-butilo (12,9 mg, 0,072 mmol) en DMF (0,5 ml) se añadió K2CO3 (33,1 mg, 0,24 mmol) y se calientó a 80 °C con agitación durante 1,5 horas. La reacción se enfrió a temperatura ambiente y se diluyó con DCM (3 ml), se filtró a través de un filtro de jeringa y se concentró bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice (12 g), 0-60% de acetato de etilo en hexano) proporcionó 104A (20,8 mg, 0,0413 mmol, 86,3% de rendimiento).
Etapa 2. 104 se preparó de acuerdo con el Procedimiento general C a partir de 104A como un sólido blanco.
Etapa 3. A una solución de 104 (4,6 mg, 0,0129 mmol) en DCM (0,3 ml) se añadió 3-clorobenzoperoxoato de metilo (2,2 mg, 0,0129 mmol) y la reacción se agitó durante 20 minutos, seguido de la adición de una solución saturada de NaHCO3 (3 ml).) y extracción con DCM (4 x 4 ml). Los extractos combinados se secaron con Na2SO4 y se concentraron bajo presión reducida. La cromatografía ultrarrápida (sistema ISCO, sílice (12 g), 0-12,5% de metanol en diclorometano) proporcionó 106 (0,5 mg, 10,4% de rendimiento) y 107 (1,7 mg, 33,9% de rendimiento).
Los siguientes ejemplos se prepararon usando procedimientos análogos a los descritos en el presente documento, especialmente los Procedimientos generales A, B y C, tal como se describe en el presente documento.





Se preparan ejemplos adicionales usando procedimientos análogos a los descritos anteriormente.
Ejemplo biológico 1: Ensayos bioquímicos de cinasas.
La inhibición de enzimas MET/ALK/AXL/TRK se puede medir mediante el ensayo fluorométrico continuo Omnia (Invitrogen Inc.). Las reacciones se llevan a cabo en volúmenes de 50 pl en placas de 96 pocillos a 30 °C. Las mezclas contienen dominio cinasa diana recombinante humano 1 nM, péptido fosfoacceptor 2 pM, compuesto de prueba (11 dosis, diluciones en serie de 3 veces, DMSO final al 2 %) o DMSO solo, Dt T 0,2 mM y MgCh 10 mM en Hepes 20 mM, pH 7,5, y las reacciones se inician mediante la adición de ATP (concentración final de 100 pM) después de una incubación previa de 20 minutos. Las velocidades iniciales de formación de fosfopéptidos se miden durante 20 minutos usando un lector de microplacas Tecan Safire con ajustes de longitud de onda de 360 nm para la excitación y 485 nm para la emisión. Los valores Ki se calculan ajustando los datos a la ecuación para la inhibición competitiva usando un procedimiento de regresión no lineal (GraphPad Prism, GraphPad Software, San Diego, CA).
Ejemplo biológico 2: Ensayos ELISA de fosforilación de cinasa celular
Los experimentos se realizan en base a los procedimientos descritos en la publicación (Christensen, J. et al., "Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma", Mol. Cancer Ther. 2007, 6 (12): 3314-3322.) Todos los
experimentos se realizan en condiciones estándar (37 °C y 5 % de CO2). Los valores de CI50 se calculan mediante el ajuste de la curva de concentración/respuesta usando un procedimiento de cuatro parámetros basado en Microsoft Excel. Las células se siembran en placas de 96 pocillos en medio complementado con un 10 % de suero fetal bovino (FBS) y se transfieren a un medio libre de suero [con 0,04 % de seroalbúmina bovina (BSA)] después de 24 h. En los experimentos que investigan la fosforilación de RTK dependiente de ligando, se añaden los factores de crecimiento correspondientes durante hasta 20 min. Después de la incubación de células con un inhibidor durante 1 hora y/o ligandos apropiados para los tiempos designados, las células se lavan una vez con HBSS complementado con 1 mmol/L de Na3VO4 y se generan lisados de proteínas a partir de las células. Posteriormente, la fosforilación de las proteínas cinasas seleccionadas se evalúa mediante un procedimiento ELISA tipo sándwich usando anticuerpos de captura específicos para cubrir placas de 96 pocillos y un anticuerpo de detección específico para residuos de tirosina fosforilados. Las placas recubiertas con anticuerpo (a) se incuban en presencia de lisados de proteínas a 4 °C durante la noche, (b) se lavan siete veces en Tween 20 al 1 % en PBS, (c) se incuban en un anticuerpo fosfotirosina anti-total conjugada con peroxidasa de rábano picante (PY-20) (1:500) durante 30 min, (d) se lavan siete veces nuevamente, (e) se incuban en sustrato 3,3,5,5-tetrametilbencidina peroxidasa (Bio-Rad) para iniciar una reacción colorimétrica que se detiene añadiendo H2SO40,09 N y (f) se midió la absorbancia a 450 nm usando un espectrofotómetro. Las líneas celulares que se usan para las cinasas individuales incluyen A549 para MET, Karpas 299 para ALK, 293-AXL para AXL, PAET RKA para TRKA y PAE-TRKB para TRKB.
Ejemplo biológico 3: Ensayos de unión de cinasa.
Los ensayos de unión de cinasa se realizaron en DiscoveRx usando el protocolo general KINOMEscan Kd (Fabian, M. A. et al., "A small molecule-kinase interaction map for clinical kinase inhibitors," Nat. Biotechnol. 2005, 23(3):329-36). Para la mayoría de los ensayos, se prepararon cepas de fago T7 marcadas con cinasa en un huésped de E. coli derivado de la cepa BL21. E. coli se cultivaron hasta la fase logarítmica y se infectaron con fago T7 y se incubaron con agitación a 32 °C hasta la lisis. Los lisados se centrifugaron y se filtraron para eliminar los residuos celulares. Las cinasas restantes se produjeron en células HEK-293 y posteriormente se etiquetaron con ADN para la detección de qPCR. Las perlas magnéticas recubiertas con estreptavidina se trataron con ligandos de moléculas pequeñas biotiniladas durante 30 minutos a temperatura ambiente para generar resinas de afinidad para los ensayos de cinasa. Las perlas ligadas se bloquearon con exceso de biotina y se lavaron con tampón de bloqueo (SeaBlock (Pierce), BSA al 1 %, Tween 20 al 0,05 %, DTT 1 mM) para eliminar el ligando no unido y reducir la unión no específica. Las reacciones de unión se ensamblaron combinando cinasas, perlas de afinidad ligadas y compuestos de prueba en tampón de unión 1x (20 % SeaBlock, 0,17x PBS, Tween 20 al 0,05 %, DTT 6 mM). Todas las reacciones se realizaron en placas de poliestireno de 96 pocillos en un volumen final de 0,135 ml. Las placas de ensayo se incubaron a temperatura ambiente con agitación durante 1 hora y las perlas de afinidad se lavaron con tampón de lavado (1x PBS, Tween 20 al 0,05 %). Las perlas se volvieron a suspender en tampón de elución (1x PBS, Tween 20 al 0,05 %, ligando de afinidad no biotinilado 0,5 pM) y se incubaron a temperatura ambiente con agitación durante 30 minutos. La concentración de cinasa en los eluatos se midió mediante qPCR. Los resultados de los compuestos sometidos a prueba en este ensayo se presentan en la Tabla 2. Con este procedimiento, el Ejemplo 20 también tenía una afinidad de unión con PLK4 cinasa (Kd 2,9 nM).
Tabla 2.


Ejemplo biológico 4: Ensayo de proliferación de células Ba/F3.
Los ensayos de proliferación de células TRKA Ba/F3 fueron realizados por ACD (Advanced Cellular Dynamics). Las líneas celulares Ba/F3 se mantuvieron en medios de cultivo RPMI-1640 que contenían un 10 % de suero fetal bovino y antibióticos. Se recogieron células en fase de crecimiento logarítmica y se distribuyeron 5.000 células en cada pocillo de una placa de 384 pocillos en 50 pl de medio de crecimiento. Se añadieron 50 nanolitros de compuesto diluido a los pocillos apropiados, por duplicado, y las células se cultivaron durante 48 horas a 37 °C en una incubadora humidificada con CO2 al 5%. La viabilidad se determinó añadiendo 15 pl de CellTiter-Glo y midiendo la luminiscencia, que se informa como unidades de luz relativas (RLU) medidas en recuentos por segundo. Los datos (RLU) para cada compuesto se normalizaron a la respuesta máxima promedio obtenida en presencia de vehículo (DMSO) solo. Estos datos se usaron para derivar el porcentaje de inhibición (100 - % de respuesta máxima) y el promedio de dos puntos de datos/concentraciones se usó para calcular los valores de CI50 (concentración que causa una inhibición de la supervivencia celular del 50 % del máximo) mediante análisis de regresión no lineal usando el software GraphPad Prism (GraphPad, Inc., San Diego, CA). Con este procedimiento, el Ejemplo 20 inhibió la proliferación celular de células TRKa Ba/F3 con una CI50 de 3,0 nM. Los datos para los compuestos sometidos a prueba en este ensayo se presentan en la Tabla 3.
Ejemplo biológico 5: Ensayo de creación de líneas y proliferación de línea celular estable EML4-ALK Ba/F3
El gen natural EML4-ALK (variante 1) se sintetizó en GenScript y se clonó en el plásmido pCDH-CMV-MCS-EF1-Puro (System Biosciences, Inc). La línea celular natural Ba/F3-EML4-ALK se generó infectando células Ba/F3 con lentivirus que contienen EML4-ALK de tipo natural. Se seleccionaron líneas celulares estables mediante tratamiento con puromicina, seguido de retirada de IL-3. Se sembraron 5000 células en una placa blanca de 384 pocillos durante la noche antes del tratamiento con compuesto. La proliferación celular se midió usando el ensayo de detección de ATP basado en luciferasa CellTiter-Glo (Promega) siguiendo el protocolo del fabricante después de 48 horas de diversas concentraciones de incubación del compuesto. Las determinaciones de CI50 se realizaron usando el software GraphPad Prism (GraphPad, Inc., San Diego, CA.). Los datos para los compuestos sometidos a prueba en este ensayo se presentan en la Tabla 3.
Ejemplo biológico 6 : Ensayos de proliferación celular.
Se cultivaron células de líneas celulares colorrectales KM 12 (que albergan el gen de fusión TPM3-TRKA endógeno) en medio DMEM, complementado con suero fetal bovino al 10% y 100 U/ml de penicilina/estreptomicina. Se sembraron 5000 células en una placa blanca de 384 pocillos durante 24 horas antes del tratamiento con los compuestos. La proliferación celular se midió usando el ensayo de detección de ATP basado en luciferasa CellTiter-Glo (Promega) siguiendo el protocolo del fabricante después de 72 horas de incubación. Las determinaciones de CI50 se realizaron usando el software GraphPad Prism (GraphPad, Inc., San Diego, CA).
Alternativamente: Se cultivaron células de líneas celulares colorrectales KM12 (que albergan el gen de fusión TPM3-TRKA endógeno) en medio DMEM, complementado con suero fetal bovino al 10% y 100 U/ml de penicilina/estreptomicina. Se cultivaron células de la línea celular de trombocitemia esencial SET-2 (que albergan la mutación puntual JAK2 V618F endógena) o la línea celular Karpas-299 del linfoma de linfocitos T (que alberga el gen de fusión ALPM-ALK endógena) en medio RPMI, complementado con 10 % de suero fetal bovino y 100 U/ml de penicilina/estreptomicina. Se sembraron 5000 células en una placa blanca de 384 pocillos durante 24 horas antes del tratamiento con los compuestos. La proliferación celular se midió usando el ensayo de detección de ATP basado en luciferasa CellTiter-Glo (Promega) siguiendo el protocolo del fabricante después de 72 horas de incubación. Las determinaciones de CI50 se realizaron usando el software GraphPad Prism (GraphPad, Inc., San Diego, CA).
Los datos para los compuestos sometidos a prueba en estos ensayos se presentan en la Tabla 3.
Tabla 3



Ejemplo biológico 7: Estudios de mecanismo celular de acción / Ensayos de fosforilación de dianas de señal corriente abaio y TRKA.
Se cultivaron células de líneas celulares colorrectales KM 12 (que albergan el gen de fusión TPM3-TRKA endógeno) en medio DMEM, complementado con suero fetal bovino al 10% y 100 U/ml de penicilina/estreptomicina. Se sembraron un millón de células en una placa de 6 pocillos durante 24 horas antes del tratamiento con los compuestos. Las células se lavaron con IxPBS y se recogieron después de 5 horas de tratamiento y se lisaron en tampón RIPA (Tris 50 mM, pH 7,4, NaCl 150 mM, NP-40 al 1 %, desoxicolato al 0,5 %, SDS al 0,1 %) complementado con EDTA 10 mM, proteasa Halt. e inhibidores de fosfatasa (Thermo Scientific). Los lisados de proteínas (20 |jg) se resolvieron en geles premoldeados Bolt Bis-Tris al 4-12 % con tampón de migración MES (Life Technologies), se transfirieron a membranas de nitrocelulosa usando el sistema de transferencia Trans-Blot Turbo Transfer System (Bio-Rad) y se detectaron con anticuerpos dirigidos a TRKA fosforilados (Cell Signaling Technology, Y496, Y680, Y681, clon C50F3; dilución 1:1000), TRKA total (Santa Cruz Biotechnology, sc-11; clon C-14, dilución 1:2000), AKT fosforilado (Cell Signaling Technology, S473, D9E, #9271; dilución 1:5000), AKT total (Cell Signaling Technology, 40D4; dilución 1:2000), ERK fosforilada (Cell Signaling Technology, Thr 202/204, D13,14.4E, #4370; dilución 1:2000), ERK total (Cell Signaling Technology, dilución 1:1000) y tubulina (Sigma, T4026, dilución 1:5000). Los anticuerpos se incubaron típicamente durante la noche a 4 °C con agitación suave, seguido de lavados e incubación con los anticuerpos secundarios conjugados con HRP apropiados. Las membranas se expusieron a un sustrato quimioluminiscente durante 5 minutos a temperatura ambiente (SuperSignal West Femto, Thermo Scientific). Las imágenes se obtuvieron con un sistema de formación de imágenes C-Digit (LI-COR Biosciences). La densidad relativa de las bandas se obtuvo directamente a través de Image Studio Digits de LICOR. Los valores de concentración inhibitoria del 50 % (CI50) se calcularon usando análisis de regresión no lineal a través de software GraphPad Prism (GraphPad, Inc., San Diego, CA). Con este procedimiento, el Ejemplo 20 inhibió la autofosforilación de TPM3-TRKA con una CI50 de 1,07 nM y la fosforilación de sus dianas de señalización corriente abajo AKT y ERK con una CI50 de 2,80 nM y 2,00 nM, respectivamente, en células KM12.
Ejemplo biológico 8: Ensayos de actividad caspasa.
Las células KM12 se mantuvieron en medio DMEM complementado con un 10% de suero fetal bovino y antibióticos. Se sembraron 500.000 células en una placa de 12 pocillos y se introdujeron diversas concentraciones de compuestos durante 72 horas. Para el tratamiento con estaurosporina, se añadieron 500 nM de STS a las 60 horas y se incubó durante 12 horas como control positivo. Todas las células se recogieron y se lavaron con 1xPBS dos veces y a continuación se lisaron en un tampón de lisis (HEPES 20 mM, NaCl 150 mM, KCl 10 mM, EDTA 5 mM, NP40 al 1 %) complementado con inhibidores de la proteasa Halt y fosfatasa (Thermo Scientific). Para los ensayos de caspasa se incubaron aproximadamente 20 j l (20 jg ) de lisado celular con 20 j l de reactivo caspasa3 glo (Promega), midiendo la actividad enzimática mediante la liberación de luminiscencia después de 20 minutos de incubación a 37 °C. Para la inmunoelectrotransferencia, los lisados celulares se hirvieron y se analizaron por SDS-PAGE/inmunotransferencia usando PARP o anticuerpos de actina. Con este procedimiento, el Ejemplo 20 indujo la apoptosis de células KM 12.
Claims (13)
1. Un compuesto de la siguiente fórmula (IV):

en la que
M es CH o N;
X1 y X1' son independientemente -C(R1a)(R2a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk)-;
cada R1a y R2a es independientemente H, deuterio, alquilo Ci-6, cicloalquilo C3-6, arilo C6-10, -C(O)ORa’, -C(O)NRa’Rb’, -NRa’Rb’, -SRa’, -S(O)Ra’, -S(O)NRa’. -S(O)2Ra’, -S(O)2NRa’ o -ORa’ en el que cada átomo de hidrógeno del alquilo C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1.4), -N(alquilo C i-4)2, NHC(O)alquilo C1.4, -N(alquilo C i-4)C(O)alquilo C1.4, -NHC(O)NHalquilo C1.4, -N(alquilo C i-4)C(O)NHalquilo C1.4, -NHC(O)N(alquilo ^ .4)2, -N(alquilo Ci-4)C(O)N(alquilo C1-4)2, -NHC(O)Oalquilo C1-4, -N(alquilo C1-4)C(O)Oalquilo C1-4, -CO2H, -CO2alquilo C1-4, -CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, -Salquilo C1-4, -S(O)alquilo C1-4, -S(O)2alquilo C1-4, -S(O)NH(alquilo C1-4), -S(O)2NH(alquilo C1-4), -S(O)N(alquilo C1-4)2, -S(O)2N(alquilo C1-4)2, cicloalquilo C3-6 o heterocicloalquilo de 3 a 7 miembros;
R3a y R3b son cada uno independientemente H, deuterio, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3;
R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por deuterio, halógeno, -CN, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1-4), -N(alquilo C i-4)2, -CO2H, -CO2alquilo C1.4, -CONH2, -CONH(alquilo C1-4), -CON(alquilo C i-4)2, cicloalquilo o heterocicloalquilo monocíclico;
cada Rk’ es independientemente H, deuterio, alquilo C1-6, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1-6, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6 , heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk’ está independientemente opcionalmente sustituido por deuterio, halógeno, alquilo C1-6, haloalquilo C1-6 u -ORa’;
en el que cada Ra’ y Rb’ es independientemente H, deuterio, alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo;
cada Z1, Z2, Z3, Z4, Z5, Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente es independientemente H, deuterio, halógeno, alquilo C1-4, -O-alquilo C1-4, -OH, -NH2, -NH(alquilo C1.4), -NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3, Z4, Z5, Z6 o Z7 sea N o NH; y
m' es 2 o 3;
o una sal farmacéuticamente aceptable del mismo.
2. El compuesto de la reivindicación 1 que tiene la Fórmula

en la que
M es CH o N;
X1 y X1' son independientemente -C(R1a)(R2a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk)-;
cada R1a y R2a es independientemente H, deuterio, alquilo C1-6 , cicloalquilo C3-6, arilo C6-10, -C(O)ORa’, -C(O)NRa’Rb’, -NRa’Rb’, -SRa’, -S(O)Ra’, -S(O)NRa’, -S(o)2Ra’, -S(O)2NRa’ o -ORa’ en el que cada átomo de hidrógeno del alquilo C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1-4), -N(alquilo C1-4)2, -NHC(O)alquilo C1-4, -N(alquilo C1-4)C(O)alquilo C1-4 , -NHC(O)NHalquilo C1-4, -N(alquilo C1-4)C(O)NHalquilo C1-4, -NHC(O)N(alquilo C1-4)2, -N(alquilo C1-4)C(O)N(alquilo C1-4)2, -NHC(O)Oalquilo C1-4, -N(alquilo C1-4)C(O)Oalquilo C1-4, -CO2H, -CO2alquilo C1-4, -CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, -Salquilo C1-4, -S(O)alquilo C1-4, -S(O)2alquilo C1-4, -S(O)NH(alquilo C1-4), -S(O)2NH(alquilo C1-4), -S(O)N(alquilo ^ - 4 )2, -S(O)2N(alquilo C1-4)2, cicloalquilo C3-6 o heterocicloalquilo de 3 a 7 miembros;
R3a y R3b son cada uno independientemente H, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3;
R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por halógeno, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1.4), -N(alquilo C1-4)2, -CO2H, -CO2alquilo C1.4, -CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, cicloalquilo o heterocicloalquilo monocíclico;
cada Rk’ es independientemente H, deuterio, alquilo C1-6, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1-6, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6 , heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk’ está independientemente opcionalmente sustituido por deuterio, halógeno, alquilo C1-6, haloalquilo C1-6 u -ORa’;
en el que cada Ra’ y Rb’ es independientemente H, deuterio, alquilo C1-6, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6 , heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo;
cada Z1, Z2, Z3, Z4, Z5, Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente es independientemente H, deuterio, halógeno, alquilo C1-4, -O-alquilo C1-4, -OH, -NH2, -NH(alquilo C1.4), -NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3, Z4, Z5, Z6 o Z7 sea N o NH; y
m' es 2 o 3;
o seleccionado del grupo que consiste en


en la que
M es CH o N;
X1 y X1' son independientemente -C(R1a)(R2a)-, -S-, -S(O)-, -S(O)2-, -O- o -N(Rk)-;
cada R1a y R2a es independientemente H, deuterio, alquilo C1-6 , cicloalquilo C3-6, arilo C6-10, -C(O)ORa’, -C(O)NRa’Rb’, -NRa’Rb’, -SRa’, -S(O)Ra’, -S(O)NRa’, -S(o)2Ra’, -S(O)2NRa’ o -ORa’ en el que cada átomo de hidrógeno del alquilo C1-6 está independientemente y opcionalmente sustituido por deuterio, halógeno, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1-4), -N(alquilo C1-4)2, -NHC(O)alquilo C1-4, -N(alquilo C1-4)C(O)alquilo C1-4 , -NHC(O)NHalquilo C1-4, -N(alquilo C1-4)C(O)NHalquilo C1-4, -NHC(O)N(alquilo C1-4)2, -N(alquilo C1-4)C(O)N(alquilo C1-4)2, -NHC(O)Oalquilo C1-4, -N(alquilo C1-4)C(O)Oalquilo C1-4, -CO2H, -CO2alquilo C1-4, -CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, -Salquilo C1-4, -S(O)alquilo C1-4, -S(O)2alquilo C1-4, -S(O)NH(alquilo C1-4), -S(O)2NH(alquilo C1-4), -S(O)N(alquilo ^ - 4 )2, -S(O)2N(alquilo C1-4)2, cicloalquilo C3-6 o heterocicloalquilo de 3 a 7 miembros;
R3a y R3b son cada uno independientemente H, fluoro, cloro, bromo, metilo, etilo, propilo, isopropilo, metoxi, etoxi, isopropoxi, -CN o -CF3;
R7a es H, alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros, en el que cada átomo de hidrógeno del alquilo C1-6 o heterocicloalquilo de 3 a 7 miembros está independientemente y opcionalmente sustituido por halógeno, -OH, -Oalquilo C1-4, -NH2, -NH(alquilo C1.4), -N(alquilo C1-4)2, -CO2H, -CO2alquilo C1.4, -CONH2, -CONH(alquilo C1-4), -CON(alquilo C1-4)2, cicloalquilo o heterocicloalquilo monocíclico;
cada Rk’ es independientemente H, deuterio, alquilo C1-6, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6, heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo mono- o bicíclico; en el que cada átomo de hidrógeno del alquilo C1-6, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6 , heterocicloalquilo de 3 a 7
miembros, arilo C6-10 o heteroarilo mono- o bicíclico de Rk’ está independientemente opcionalmente sustituido por deuterio, halógeno, alquilo C1-6, haloalquilo C1-6 u -ORa’;
en el que cada Ra’ y Rb’ es independientemente H, deuterio, alquilo Ci-a, alquenilo C2-6 , alquinilo C2-6 , cicloalquilo C3-6 , heterocicloalquilo de 3 a 7 miembros, arilo C6-10 o heteroarilo;
cada Z1, Z2, Z3, Z4, Z5, Z6 o Z7 es independientemente N, NH o C(Rx), en la que cada Rx cuando está presente
es independientemente H, deuterio, halógeno, alquilo C1-4, -O-alquilo C1.4, -OH, -NH2, -NH(alquilo C1.4), -NH(fenilo), -NH(heteroarilo), -CN o -CF3, siempre que al menos uno de Z1, Z2, Z3, Z4, Z5, Z6 o Z7 sea N o NH;
y
m' es 2 o 3;
o una sal farmacéuticamente aceptable del mismo.
3. El compuesto de la reivindicación 1 o 2, o una sal farmacéuticamente aceptable del mismo, en el que Z1,
Z4 y Z7 son N, y Z2, Z3, Z5 y Z6 son C(Rx), en el que cada Rx cuando está presente es H.
4. El compuesto de la reivindicación 1 o 2, o una sal farmacéuticamente aceptable del mismo, en el que M
es CH, Z1, Z4 y Z7 son N, y Z2, Z3, Z5 y Z6 son C(Rx), en el que cada Rx cuando está presente es H.
5. El compuesto de la reivindicación 1 o 2, o una sal farmacéuticamente aceptable del mismo, en el que M
es CH, Z1, Z4 y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx), en el que cada Rx cuando está presente es H y X1 es -N(Rk’)-.
6. El compuesto de la reivindicación 1 o 2, o una sal farmacéuticamente aceptable del mismo, en el que M
es CH, Z1, Z4 y Z7 son N, Z2, Z3, Z5 y Z6 son C(Rx), en el que cada Rx cuando está presente es H, X1' es -O-.
7. El compuesto de la reivindicación 1 o 2, o una sal farmacéuticamente aceptable del mismo, en el que M
es CH, Z1, Z4y Z7 son N, Z2, Z3, Z5y Z6son C(Rx), en el que cada Rx cuando está presente es H, X1 es -C(R1a)(R2a)-y X1' es -O-.
8. El compuesto de una cualquiera de las reivindicaciones 1-7, o una sal farmacéuticamente aceptable del
mismo, en el que Rk’ se selecciona del grupo que consiste en H, metilo, etilo, propiol, isopropilo, ciclopropilo, 2 hidroxietilo, 2-hidroxi-2-metil-propilo y N-metil-pirrol-3-ilo.
9. El compuesto de una cualquiera de las reivindicaciones 1-8, o una sal farmacéuticamente aceptable del
mismo, en el que Rk’ es H o metilo.
10. El compuesto de la reivindicación 1 se selecciona del grupo que consiste en (13R)-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazo!o[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazoío[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona;
(13R)-12-cloro-11-fluoro-5-(2-hidroxietil)-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-5-(2-hidroxietil)-13-metil-6,7-dihidro-13H-1.15- etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 2-[(13R)-12-cloro-11-fluoro-13-metil-4-oxo-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-5(4H)-il]acetamida; 2-[12-cloro-11-fluoro-13-metil-4-oxo-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-5(4H)-il]acetamida; (13R)-12-cloro-11-fluoro-13-metil-5-(pirrolidin-2-ilmetil)-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11 -fluoro-13-metil-5-(pirrolidin-2-ilmetil)-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-7-(hidroximetil)-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-7-(hidroximetil)-5,13-dimetil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13S)-11-fluoro-13-(fluorometil)-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 11-fluoro-13-(fluorometil)-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona;
(13R)-13-ciclopropil-11-fluoro-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 13-ciclopropil-11 -fluoro-5-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-11-fluoro-13-metil-6,7-dihidro-13H-1.15- etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 11-fluoro-13-metil-6,7-dihidro-13H-1.15- etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; (13R)-12-cloro-11-fluoro-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-13-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-6-metil-6,7-dihidro-13H-1, 15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona; 12-cloro-11-fluoro-7-metil-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzodioxadiazaciclotridecin-4(5H)-ona;
(8R)-9-doro-10-fluoro-8-metil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecinl7(14H)-ona; 9-doro-10-fluoro-8-metiM5,16-dihidro-8H-3,6-etenoiiTiidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecin-17(14H)-ona; (7R)-8-doro-9-fluoro-7-metiM4,15-dihidro-2H,7H-3,5-(azenometeno)pimdo[3,4-f][1,10,4,8]benzodioxadiazaddotridedn-16(13H)-ona; 8-doro-9-fluoro-7-metiM4,15-dihidro-2H, 7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazacidotridecin-16(13H)-ona; (5R)-3-fluoro-5-metil-14,15-dihidro-5H,10H-9,7-(azenometeno)pirido[2,3-k]pirrolo[3,4-d][1,10,3,7]dioxadiazacidotridecin-12(13H)-ona; 3-fluoro-5-metiM4,15-dihidro-5H,10H-9,7-(azenoirieteno)pindo[2,3-k]pimdo[3,4-d][1,10,3,7]dioxadiazacidotridecin-12(13H)-ona; (5R)-3-fluoro-5,16-dimetiM3,14,15,16-tetrahidro-5H-9,7-(azenometeno)pirido[2,3-k]pimdo[3,4-d][1,3,7,10]oxatriazaddotndedn-12(10H)-ona; 3-fluoro-5,16-dimetN-13.14.15.16- tetrahidro-5H-9,7-(azenometeno)pirido[2,3-k]pirrolo[3,4-d][1,3,7,l0]oxatriazacidotridecin-12(10H)-ona; (13R)-12-doro-11-fluoro-5,13-dimetN-6,7-dihidro-2H,13H-1,15-(azenoiTieteno)pimdo[3,4-f][1,10,4]benzodioxazaddotridedn-4(5H)-ona; 12-doro-11-fluoro-5,13-dimetil-6,7-dihidro-2H, 13H-1,15-(azenometeno)pirrolo[3,4-f][1,10,4]benzodioxazacidotridecin-4(5H)-ona; (7R)-8-doro-9-fluoro-7,15-dimetiM4,15-dihidro-2H,7H-3,5-(azenoiTieteno)pirazolo[3,4-f][1,10,4]benzodioxazaddotridedn-16(13H)-ona; 8-doro-9-fluoro-7.15- dimetil-14,15-dihidro-2H,7H-3,5-(azenometeno)pirazolo[3,4-f][1,10,4]benzodioxazacidotridecin-16(13H)-ona; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; (13R)-12-cloro-11 -fluoro-13,14-dimetN-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 12-doro-11-fluoro-13,14-dimetN-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 12-doro-11-fluoro-5,14-dimetN-6,7,13,14-tetrahidro-15,1-(azenometeno)pirazolo[4,3-f][1,4,10]benzoxadiazaddotndedn-4(5H)-ona; 12-doro-11-fluoro-14-metil-6,7,13,14-tetrahidro-15,1-(azenometeno)pirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 12-cloro-11 -fluoro-14-metil-6,7,13,l4-tetrahidro-1,15-(azenometeno)pirrolo[3,2-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 12-cloro-11 -fluoro-14-metN-6,7,13,14-tetrahidro-1,15-(azenoiTieteno)pimdo[3,2-f][1,4,10]benzoxadiazacidotridecin-4(5H)-ona; 9-doro-10-fluoro-7-metN-7,8,15,16-tetrahidro-3,6-etenoiiTiidazo[5,1-f][l,4,7,8,10]benzoxatetraazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metN-7,8,15,16-tetrahidro-6,3-(azenometeno)imidazo[5,1-f][1,4,7,8,10]benzoxatetraazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metN-7.8.15.16- tetrahidro-6,3-(azenometeno)imidazo[5,1-f][1,4,7,10]benzoxatriazacidotridecin-17(14H)-ona; 9-cloro-10- fluoro-7-metil-7,8,15,l6-tetrahidro-3,6-(azenometeno)pirrolo[2,1-f][1,4,7,10]benzoxatriazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metN-7,8,15,16-tetrahidro-3,6-(azenoiTieteno)iiTiidazo[2,1-f][1,4,7,10]benzoxatriazaddotridedn-17(14H)-ona; 9-doro-10-fluoro-7-metN-7,8,15,16-tetrahidro-3,6-eteno[1,2,4]triazolo[3,4-f][1,4,7,8,10]benzoxatetraazacidotridecin-17(14H)-ona; 9-doro-10-fluoro-7-metN-7.8.15.16- tetrahidro-6,3-(azenometeno)[1,2,4]triazolo[3,4-f][1,4,7,10]benzoxatriazacidotridecin-17(14H)-ona; 8-doro-9-fluoro-6-metil-6,7,14,15-tetrahidro-2H-3,5-(azenometeno)pirrolo[3,4-f][1,4,8,10]benzoxatriazacidotridecin-16(13H)-ona; 8-doro-9-fluoro-6-metN-6,7,14,15-tetrahidro-2H-3,5-(azenoiTieteno)pirazolo[3,4-f][1,4,8,10]benzoxatriazacidotridecin-16(13H)-ona; 8-doro-9-fluoro-6-metN-6,7,14,15-tetrahidro-2H-3,5-(azenometeno)pirazolo[3,4-f][1,4,10]benzoxadiazacidotridecin-16(13H)-ona; 12-doro-11-fluoro-5,14-dimetN-6.7.13.14- tetrahidro-2H-1,15-(azenometeno)pirrolo[3,4-f][1,4,10]benzoxadiazacidotridecin-4(5H)-ona; (8R)-10-fluoro-8,16-dimetil-15, 16-dihidro-8H-3,6-etenoiiTiidazo[5,1-f][1,10,4,7,8]benzodioxatnazaddotndedn-17(14H)-ona; 10-fluoro-8,16-dimetil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecin-17(14H)-ona; (7R)-9-fluoro-7,15-dimetiM4,15-dihidro-2H,7H-3,5-(azenoiTieteno)pimdo[3,4-f][1,10,4,8]benzodioxadiazacidotridecin-l6(13H)-ona; 9-fluoro-7,15-dimetiM4,15-dihidro-2H,7H-3,5-(azenometeno)pirrolo[3,4-f][1,10,4,8]benzodioxadiazacidotridecin-16(13H)-ona; 12-doro-11-fluoro-14-metN-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 11-fluoro-3,14-dimetil-6,7,13,14-tetrahidro-1,l5-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 10-fluoro-8-metil-15,16-dihidro-8H-3,6-etenoimidazo[5,1-f][1,10,4,7,8]benzodioxatriazacidotridecin-17(l4H)-ona; 10-fluoro-7-metil-7,8,15,16-tetrahidro-3,6-etenoimidazo[5,1-f][1,4,7,8,10]benzoxatetraazacidotridecin-l7(l4H)-ona; 14-etil-11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-14-propN-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 11-fluoro-14-(propan-2-yl)-6,7,13,14-tetrahidro-1,l5-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 14-cidopropil-l1-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11- fluoro-14-(2-hidroxietN)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 11-fluoro-6,14-dimetN-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 14-metN-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][l,4,8,10]benzoxatnazaddotndedn-4(5H)-ona; 11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazob[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 11-fluoro-13-metN-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][l,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; (13R)-11 -fluoro-13-metN-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 12-doro-11-fluoro-13-metN-6,7,13,14-tetrahidro-1,l5-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-14-metil-4-oxo-4.5.6.7.13.14- hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecina-7-carboxamida; 11-fluoro-7-(hidroximetil)-14-metil-6,7,13,14-tetrahidro-1,l5-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-13-metil-4-oxo-4,5,6,7,13,l4-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecina-7-carboxamida; 11-fluoro-7-(hidroximetil)-13-metil-6,7,13,14-tetrahidro-1.15- etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-4-oxo-4,5,6,7,13,14-hexahidro-1.15- etenopirazolo[4,3-f][l,4,8,10]benzoxatnazaddotndedna-7-carboxairiida; 11-fluoro-7-(hidroximetil)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-4-oxo-4,5,6,7,13,14-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedna-13-carboxNato de metilo; 11-fluoro-4-oxo4,5,6,7,13,14-hexahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecina-13-carboxamida; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f]pirido[3,2-1][1,4,8,10]oxatriazacidotridecin-4(5H)-ona; 11-fluoro-13-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f]pirido[3,2-1][1,4,8,10]oxatriazaddotndedn-4(5H)-ona; 11-fluoro-13-(propan-2-yl)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f]pindo[3,2-1][1,4,8,10]oxatriazacidotridecin-4(5H)-ona; 13-ciclopropil-ll -fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f]pirido[3,2-1][1,4,8,10]oxatriazacidotridecin-4(5H)-ona; 13-ddopropil-11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacidotridecin-4(5H)-ona; 11-fluoro-13-(propan-2-yl)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 11-fluoro-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzoxatiadiazacidotridecin-4(5H)-ona; 11-fluoro-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][l,10,4,8]benzoxatiadiazacidotridecin-4(5H)-ona 14,14-dióxido; 6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][l0,1,4,8]benzoxatiadiazacidotridecin-4(5H)-ona; 14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][l,4,8,10]benzotiatriazacidotridecin-4(5H)-ona; 13-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][l,4,8,10]benzotiatriazacidotridecin-4(5H)-ona; 11-fluoro-6,7-dihidro-5H-1,15-etenopirazolo[3,4-e][11,1,2,4,8]benzoxatiatriazacidotndedn-4(14H)-ona 13,13-dióxido; 11-fluoro-14-metil-6,7-dihidro-5H-1,15-etenopirazolo[3,4-e][11,1,2,4,8]benzoxatiatriazacidotridecin-4(14H)-ona 13,13-dióxido; 12-fluoro-15-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazacidotetradecin-4-ona; 12-fluoro-14-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo'[4,3-g][1,5,9,11]benzoxatriazaddotetradedn-4-ona;
(14R)-12-fluoro-14-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazaddotetradedn-4-ona; 11-fluoro-7,14-dimetil-4,5,6,7,13,14-hexahidro-8H-1,15-etenopirazolo[3,4-e][2,4,10]benzotriazaddotndedn-8-ona; 11-fluoro-7,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[3,4-e][7,2,4,10]benzoxatriazacidotridecin-8(5H)-ona; 11-fluoro-7,14-dimetil-4,5,6,7,13,14-hexahidro-8H-1,15-etenopirazolo[3,4-e][2,4,7,10]benzotetraazacidotridecin-8-ona; 11-fluoro-4,7,14-trimetil-4.5.6.7.13.14- hexahidro-8H-1,15-etenopirazolo[3,4-e][2,4,7,10]benzotetraazacidotridecin-8-ona; 11 -fluoro-7,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[3,4-e][7,2,4,10]benzotiatriazaddotndedn-8(5H)-ona; 11-fluoro-7.14- dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[3,4-e][7,2,4,10]benzotiatriazacidotridecin-8(5H)-ona 4,4-dióxido; 12-fluoro-8,15-dimetil-5,6,7,8,14,15-hexahidro-9H-1,16-etenopirazolo[3,4-e] [7,2,4,8,11]benzotiatetraazaddotetradedn-9-ona 4,4-dióxido; 11-doro-13-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 13-etil-11-fluoro-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 13-ddobutil-11-fluoro-6,7,13,14-tetrahidro-1.15- etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 11-fluoro-14-metil(6,6,7,7-2H4)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 11-fluoro-13-phenyl-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 13-(ciclopropilmetil)-11 -fluoro-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; (7R,14R)-12-fluoro-7-hidroxi-14-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazaddotetradedn-4-ona; (7S,14R)-12-fluoro-7-hidroxi-14-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazaciclotetradecin-4-ona; (7R,13R)-11-fluoro-7,13-d¡met¡l-6,7,13,14-tetrah¡dro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazacydotridedn-4(5H)-ona; (7S,13R)-11-fluoro-7,13-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; (7R)-11-fluoro-7,14-dimetil-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; (6R)-11-fluoro-6,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 12-fluoro-7-hidroxi-15-metil-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazaddotetradedn-4-ona; (7S)-11-fluoro-7,14-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; 11-fluoro-13-(hidroximetil)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f] [1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 12-fluoro-14-(hidroximetil)-5,6,7,8,14,15-hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9,11]benzoxatriazacidotetradecin-4-ona; 11-fluoro-13,14-dimetil-6,7,13,14-tetrahidro-1.15- etenopirazolo[4,3-f][1,4,8,10]benzoxatnazaddotridedn-4(5H)-ona; 11-fluoro-14-(2-hidroxi-2-metilpropil)-6.7.13.14- tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotridedn-4(5H)-ona; 12-fluoro-5.6.7.8.14.15- hexahidro-4H-1,16-etenopirazolo[4,3-g][1,5,9]benzoxadiazacidotetradecin-4-ona; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzotiatriazaddotridedn-4(5H)-ona; 11-fluoro-14-(1-metilpirrolidin-3-yl)-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona;
11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzotiatnazaddotndedn-4(5H)-ona 8-óxido; 11-fluoro-14-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzotiatriazaddotridedn-4(5H)-ona 8,8-dióxido; (7S)-11-fluoro-7-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8]benzoxadiazaddotridedn-4(5H)-ona; (6s,13R)-11-fluoro-6,13-dimetil-6,7,13, 14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; (6R,13R)-11-fluoro-6,13-dimetil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxatriazaddotndedn-4(5H)-ona; (7S,13S)-11 -fluoro-13-(hidroximetil)-7-metil-6,7,13,14-tetrahidro-1,15-etenopirazolo[4,3-f][1,4,8,10]benzoxathazaddotndedn-4(5H)-ona;
and 11-fluoro-6,7-dihidro-13H-1,15-etenopirazolo[4,3-f][1,10,4,8]benzoxatiadiazaddotridedn-4(5H)-ona; o una sal farmacéuticamente aceptable de los mismos.
11. El compuesto de la reivindicación 1 que tiene la Fórmula
o 
o
o 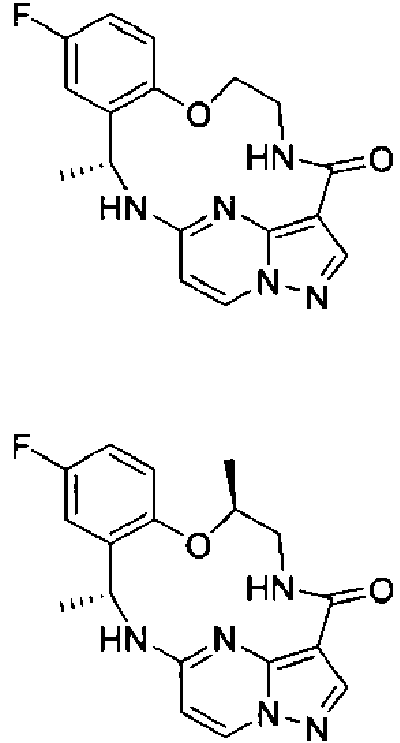
o
o 
o una sal farmacéuticamente aceptable del mismo.
12. Una composición farmacéutica que comprende (a) al menos un compuesto de una cualquiera de las reivindicaciones 1-11, o una sal farmacéuticamente aceptable del mismo, y (b) un excipiente farmacéuticamente aceptable.
13. Un compuesto de una cualquiera de las reivindicaciones 1-11, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer, el dolor, las enfermedades neurológicas, las enfermedades autoinmunitarias o la inflamación en un sujeto que necesita dicho tratamiento.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461931506P | 2014-01-24 | 2014-01-24 | |
| US201462049326P | 2014-09-11 | 2014-09-11 | |
| US201562106301P | 2015-01-22 | 2015-01-22 | |
| PCT/US2015/012597 WO2015112806A2 (en) | 2014-01-24 | 2015-01-23 | Diaryl macrocycles as modulators of protein kinases |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2735729T3 true ES2735729T3 (es) | 2019-12-20 |
Family
ID=53682127
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19169041T Active ES2933350T3 (es) | 2014-01-24 | 2015-01-23 | Macrociclos de diarilo como moduladores de proteína quinasas |
| ES15740510T Active ES2735729T3 (es) | 2014-01-24 | 2015-01-23 | Macrociclos de diarilo como moduladores de proteínas cinasas |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19169041T Active ES2933350T3 (es) | 2014-01-24 | 2015-01-23 | Macrociclos de diarilo como moduladores de proteína quinasas |
Country Status (31)
| Country | Link |
|---|---|
| US (5) | US9714258B2 (es) |
| EP (6) | EP4338739A2 (es) |
| JP (5) | JP6490713B2 (es) |
| KR (1) | KR102402179B1 (es) |
| CN (3) | CN110317214B (es) |
| AP (1) | AP2016009383A0 (es) |
| AU (2) | AU2015209239B2 (es) |
| CA (1) | CA2936079C (es) |
| CL (1) | CL2016001876A1 (es) |
| CY (1) | CY1121850T1 (es) |
| DK (2) | DK3572416T3 (es) |
| EA (2) | EA201892241A1 (es) |
| ES (2) | ES2933350T3 (es) |
| HK (1) | HK1231407A1 (es) |
| HR (2) | HRP20221518T1 (es) |
| HU (2) | HUE060554T2 (es) |
| IL (2) | IL246860B (es) |
| LT (3) | LT3097107T (es) |
| MX (2) | MX2016009588A (es) |
| MY (1) | MY193524A (es) |
| NZ (2) | NZ761094A (es) |
| PE (1) | PE20160931A1 (es) |
| PH (1) | PH12016501463A1 (es) |
| PL (2) | PL3572416T3 (es) |
| PT (2) | PT3097107T (es) |
| RS (2) | RS63829B1 (es) |
| SG (2) | SG11201605929RA (es) |
| SI (2) | SI3097107T1 (es) |
| UA (1) | UA121206C2 (es) |
| WO (1) | WO2015112806A2 (es) |
| ZA (2) | ZA201604654B (es) |
Families Citing this family (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT2350075E (pt) | 2008-09-22 | 2014-06-09 | Array Biopharma Inc | Compostos imidazo[1,2b]piridazina substituídos como inibidores da trk cinase |
| SG10201914059WA (en) | 2008-10-22 | 2020-03-30 | Array Biopharma Inc | Substituted pyrazolo[1,5-a]pyrimidine compounds as trk kinase inhibitors |
| AR077468A1 (es) | 2009-07-09 | 2011-08-31 | Array Biopharma Inc | Compuestos de pirazolo (1,5 -a) pirimidina sustituidos como inhibidores de trk- quinasa |
| MX365251B (es) | 2010-05-20 | 2019-05-28 | Array Biopharma Inc | Compuestos macrocíclicos como inhibidores de trk cinasa. |
| US9714258B2 (en) | 2014-01-24 | 2017-07-25 | Tp Therapeutics, Inc. | Diaryl macrocycles as modulators of protein kinases |
| CN113354649A (zh) | 2014-11-16 | 2021-09-07 | 阵列生物制药公司 | 一种新的晶型 |
| US10758525B2 (en) | 2015-01-22 | 2020-09-01 | MyoKardia, Inc. | 4-methylsulfonyl-substituted piperidine urea compounds |
| MA44936A1 (fr) | 2015-06-01 | 2021-09-30 | Loxo Oncology Inc | Méthodes de diagnostic et de traitement du cancer |
| JP6871903B2 (ja) * | 2015-07-02 | 2021-05-19 | ターニング・ポイント・セラピューティクス・インコーポレイテッドTurning Point Therapeutics,Inc. | プロテインキナーゼのモジュレーターとしてのキラルジアリール大環状分子 |
| EP3319969B1 (en) * | 2015-07-06 | 2024-04-03 | Turning Point Therapeutics, Inc. | Diaryl macrocycle polymorph |
| PL3322706T3 (pl) | 2015-07-16 | 2021-07-19 | Array Biopharma, Inc. | Podstawione związki pirazolo[1,5-a]pirydynowe jako inhibitory kinazy ret |
| EP3325488B1 (en) * | 2015-07-21 | 2020-06-24 | Turning Point Therapeutics, Inc. | Chiral diaryl macrocycle and use thereof in the treatment of cancer |
| CA3003153A1 (en) | 2015-10-26 | 2017-05-04 | Loxo Oncology, Inc. | Point mutations in trk inhibitor-resistant cancer and methods relating to the same |
| US11278521B2 (en) | 2015-12-15 | 2022-03-22 | University Of South Florida | GAS5 binding compounds, formulations, and uses thereof |
| US10045991B2 (en) | 2016-04-04 | 2018-08-14 | Loxo Oncology, Inc. | Methods of treating pediatric cancers |
| KR20210010652A (ko) | 2016-04-04 | 2021-01-27 | 록쏘 온콜로지, 인코포레이티드 | 소아암을 치료하는 방법 |
| PE20181888A1 (es) | 2016-04-04 | 2018-12-11 | Loxo Oncology Inc | Formulaciones liquidas de (s)-n-(5-((r)-2-(2,5-difluorofenil)-pirrolidin-1-il)-pirazolo[1,5-a]pirimidin-3-il)-3-hidroxipirrolidina-1-carboxamida |
| RU2745953C2 (ru) | 2016-05-18 | 2021-04-05 | Локсо Онколоджи, Инк. | Способ получения (s)-n-(5-((r)-2-(2,5-дифторфенил)пирролидин-1-ил)-пиразоло[1,5-a]пиримидин-3-ил)-3-гидроксипирролидин-1-карбоксамида и его солей |
| AU2017302019A1 (en) * | 2016-07-28 | 2019-02-07 | Turning Point Therapeutics, Inc. | Macrocycle kinase inhibitors |
| JOP20190077A1 (ar) | 2016-10-10 | 2019-04-09 | Array Biopharma Inc | مركبات بيرازولو [1، 5-a]بيريدين بها استبدال كمثبطات كيناز ret |
| TWI704148B (zh) | 2016-10-10 | 2020-09-11 | 美商亞雷生物製藥股份有限公司 | 作為ret激酶抑制劑之經取代吡唑并[1,5-a]吡啶化合物 |
| JOP20190092A1 (ar) | 2016-10-26 | 2019-04-25 | Array Biopharma Inc | عملية لتحضير مركبات بيرازولو[1، 5-a]بيريميدين وأملاح منها |
| WO2018136661A1 (en) | 2017-01-18 | 2018-07-26 | Andrews Steven W | SUBSTITUTED PYRAZOLO[1,5-a]PYRAZINE COMPOUNDS AS RET KINASE INHIBITORS |
| WO2018136663A1 (en) | 2017-01-18 | 2018-07-26 | Array Biopharma, Inc. | Ret inhibitors |
| TWI808958B (zh) | 2017-01-25 | 2023-07-21 | 美商特普醫葯公司 | 涉及二芳基巨環化合物之組合療法 |
| JOP20190213A1 (ar) | 2017-03-16 | 2019-09-16 | Array Biopharma Inc | مركبات حلقية ضخمة كمثبطات لكيناز ros1 |
| CN107586796B (zh) * | 2017-07-20 | 2021-08-06 | 暨明医药科技(苏州)有限公司 | (r)-2-(1-氨基乙基)-4-氟苯酚的合成方法 |
| UA126158C2 (uk) | 2017-07-28 | 2022-08-25 | Тьорнінґ Поінт Терапьютикс, Інк. | Макроциклічні сполуки і їх використання |
| TWI791053B (zh) | 2017-10-10 | 2023-02-01 | 美商亞雷生物製藥股份有限公司 | 6-(2-羥基-2-甲基丙氧基)-4-(6-(6-((6-甲氧基吡啶-3-基)甲基)-3,6-二氮雜雙環[3.1.1]庚-3-基)吡啶-3-基)吡唑并[1,5-a]吡啶-3-甲腈之結晶形式及其醫藥組合物 |
| TWI812649B (zh) | 2017-10-10 | 2023-08-21 | 美商絡速藥業公司 | 6-(2-羥基-2-甲基丙氧基)-4-(6-(6-((6-甲氧基吡啶-3-基)甲基)-3,6-二氮雜雙環[3.1.1]庚-3-基)吡啶-3-基)吡唑并[1,5-a]吡啶-3-甲腈之調配物 |
| US20190247398A1 (en) | 2017-10-26 | 2019-08-15 | Array Biopharma Inc. | Formulations of a macrocyclic trk kinase inhibitor |
| CN109516999B (zh) * | 2017-11-01 | 2021-08-17 | 郑州泰基鸿诺医药股份有限公司 | 用作蛋白质激酶调节剂的化合物及其应用 |
| WO2019094143A1 (en) * | 2017-11-10 | 2019-05-16 | Angex Pharmaceutical, Inc. | Macrocyclic compounds as trk kinase inhibitors and uses thereof |
| US20210087206A1 (en) | 2017-12-19 | 2021-03-25 | Turning Point Therapeutics, Inc. | Macrocyclic kinase inhibitors and their use |
| PL3728271T3 (pl) * | 2017-12-19 | 2023-01-23 | Turning Point Therapeutics, Inc. | Związki makrocykliczne do leczenia chorób |
| JP7150356B2 (ja) * | 2017-12-22 | 2022-10-11 | 深▲チェン▼市塔吉瑞生物医薬有限公司 | 置換ピラゾロ[1,5-a]ピリミジン化合物並びにその医薬組成物および使用 |
| US11524963B2 (en) | 2018-01-18 | 2022-12-13 | Array Biopharma Inc. | Substituted pyrazolo[3,4-d]pyrimidines as RET kinase inhibitors |
| TWI802635B (zh) | 2018-01-18 | 2023-05-21 | 美商亞雷生物製藥股份有限公司 | 作為ret激酶抑制劑之經取代吡咯并[2,3-d]嘧啶化合物 |
| TW201932464A (zh) | 2018-01-18 | 2019-08-16 | 美商亞雷生物製藥股份有限公司 | 作為ret激酶抑制劑之經取代吡唑基[4,3-c]吡啶化合物 |
| WO2019144885A1 (zh) * | 2018-01-23 | 2019-08-01 | 深圳市塔吉瑞生物医药有限公司 | 取代的吡唑并[1,5-a]嘧啶类的大环化合物 |
| TW201932472A (zh) * | 2018-01-30 | 2019-08-16 | 大陸商上海吉倍生物技術有限公司 | 具有大環分子結構的化合物及其用途 |
| CN110156813B (zh) * | 2018-02-13 | 2023-07-25 | 北京诺诚健华医药科技有限公司 | 作为trk抑制剂的杂环化合物 |
| CA3093140A1 (en) * | 2018-03-28 | 2019-10-03 | Fochon Pharmaceuticals, Ltd. | Macrocyclic compounds as trk kinases inhibitors |
| US20210023086A1 (en) | 2018-03-29 | 2021-01-28 | Loxo Oncology, Inc. | Treatment of trk-associated cancers |
| JP7092405B2 (ja) * | 2018-04-16 | 2022-06-28 | 深▲チェン▼市塔吉瑞生物医薬有限公司 | キナーゼ活性を阻害するためのジ(ヘテロ)アリール大環状化合物 |
| WO2019206069A1 (zh) * | 2018-04-25 | 2019-10-31 | 北京普祺医药科技有限公司 | 一种二芳基巨环化合物、药物组合物以及其用途 |
| CN111918868B (zh) * | 2018-05-04 | 2022-12-30 | 正大天晴药业集团股份有限公司 | 作为蛋白激酶调节剂的二芳基大环化合物 |
| CN110627812B (zh) | 2018-06-25 | 2022-10-11 | 北京诺诚健华医药科技有限公司 | 作为trk抑制剂的杂环化合物 |
| KR102653681B1 (ko) | 2018-07-31 | 2024-04-03 | 록쏘 온콜로지, 인코포레이티드 | (s)-5-아미노-3-(4-((5-플루오로-2-메톡시벤즈아미도)메틸)페닐)-1-(1,1,1-트리플루오로프로판-2-일)-1h-피라졸-4-카르복스아미드의분무-건조된 분산물 및 제제 |
| ES2922314T3 (es) | 2018-09-10 | 2022-09-13 | Array Biopharma Inc | Compuestos heterocíclicos condensados como inhibidores de cinasa RET |
| CN110950889B (zh) * | 2018-09-27 | 2022-04-05 | 北京赛林泰医药技术有限公司 | 一种多靶点激酶抑制剂及其制备方法和用途 |
| EP3858834A4 (en) * | 2018-09-29 | 2022-06-22 | Shandong Luye Pharmaceutical Co., Ltd. | PYRAZOLOPYRIMIDE DERIVATIVES AS SELECTIVE TRK INHIBITORS |
| EP3870579A4 (en) | 2018-10-22 | 2022-10-19 | Alumis Inc. | TYK2 INHIBITORS AND THEIR USES |
| CN113166155A (zh) * | 2018-11-09 | 2021-07-23 | 山东轩竹医药科技有限公司 | 大环类酪氨酸激酶抑制剂及其用途 |
| CN111171049B (zh) * | 2018-11-09 | 2021-06-04 | 山东轩竹医药科技有限公司 | 酪氨酸激酶抑制剂及其用途 |
| CN113490666A (zh) | 2018-12-19 | 2021-10-08 | 奥瑞生物药品公司 | 作为fgfr酪氨酸激酶的抑制剂的取代的吡唑并[1,5-a]吡啶化合物 |
| EP3898615A1 (en) | 2018-12-19 | 2021-10-27 | Array Biopharma, Inc. | 7-((3,5-dimethoxyphenyl)amino)quinoxaline derivatives as fgfr inhibitors for treating cancer |
| CN111592541B (zh) * | 2019-02-21 | 2021-09-10 | 山东轩竹医药科技有限公司 | 大环类激酶抑制剂及其用途 |
| US20220235070A1 (en) * | 2019-05-21 | 2022-07-28 | Zhejiang Hisun Pharmaceutical Co., Ltd. | Macrolide derivatives, preparation method and application thereof |
| PE20220135A1 (es) * | 2019-06-19 | 2022-01-27 | Turning Point Therapeutics Inc | Polimorfos de un inhibidor de cinasas macrociclicas |
| WO2020257189A1 (en) * | 2019-06-19 | 2020-12-24 | Turning Point Therapeutics, Inc. | Macrocycles for treating disease |
| WO2020257165A1 (en) * | 2019-06-19 | 2020-12-24 | Turning Point Therapeutics, Inc. | Macrocycles for use in treating disease |
| CN112110938B (zh) * | 2019-06-21 | 2021-11-09 | 成都海博为药业有限公司 | 一种作为蛋白质激酶抑制剂的化合物及其制备方法和用途 |
| EP3986902B1 (en) * | 2019-06-21 | 2024-01-24 | Janssen Pharmaceutica NV | Macrocyclic inhibitors of mcl-1 |
| WO2021027503A1 (zh) * | 2019-08-12 | 2021-02-18 | 罗欣药业(上海)有限公司 | 三环类化合物、其制备方法、中间体及应用 |
| WO2021063276A1 (zh) * | 2019-09-30 | 2021-04-08 | 浙江海正药业股份有限公司 | 大环类衍生物及其制备方法和用途 |
| EP4065395A4 (en) | 2019-11-27 | 2024-02-14 | Turning Point Therapeutics Inc | COMBINATION THERAPY WITH DIARYL MACROCYCLIC COMPOUNDS |
| EP4065125A4 (en) * | 2019-11-27 | 2024-01-03 | Turning Point Therapeutics Inc | COMBINATION THERAPY WITH DIARYL MACROCYCLIC COMPOUNDS |
| CN114929710B (zh) | 2019-12-03 | 2024-03-29 | 特普医药公司 | 用于治疗疾病的巨环 |
| WO2021134004A1 (en) | 2019-12-27 | 2021-07-01 | Schrodinger, Inc. | Cyclic compounds and methods of using same |
| CN113045587A (zh) * | 2019-12-27 | 2021-06-29 | 成都倍特药业股份有限公司 | 一种大环结构化合物的晶型及其制备方法 |
| CN113135938B (zh) * | 2020-01-19 | 2022-06-14 | 山东轩竹医药科技有限公司 | 取代的大环类酪氨酸激酶抑制剂及其用途 |
| IL295938A (en) * | 2020-03-02 | 2022-10-01 | Turning Point Therapeutics Inc | Therapeutic uses of macrocyclic compounds |
| EP4148055A4 (en) * | 2020-05-08 | 2023-10-11 | Shandong Xuanzhu Pharma Co., Ltd. | CRYSTAL FORM OF A MACROCYCLIC TYROSINE KINASE INHIBITOR AND PROCESS FOR PRODUCTION THEREOF |
| WO2021244609A1 (zh) * | 2020-06-04 | 2021-12-09 | 成都倍特药业股份有限公司 | 具有大环结构的化合物及其用途 |
| CN116171279A (zh) * | 2020-07-10 | 2023-05-26 | 荣山医药股份有限公司 | 大环化合物及其用途 |
| WO2022055963A1 (en) | 2020-09-10 | 2022-03-17 | Schrödinger, Inc. | Heterocyclic pericondensed cdc7 kinase inhibitors for the treatment of cancer |
| WO2022164789A1 (en) | 2021-01-26 | 2022-08-04 | Schrödinger, Inc. | Tricyclic compounds useful in the treatment of cancer, autoimmune and inflammatory disoders |
| WO2022182845A1 (en) * | 2021-02-25 | 2022-09-01 | Blossomhill Therapeutics, Inc. | Macrocycles and their use |
| TW202300150A (zh) | 2021-03-18 | 2023-01-01 | 美商薛定諤公司 | 環狀化合物及其使用方法 |
| CN116063326A (zh) * | 2021-11-02 | 2023-05-05 | 赛诺哈勃药业(成都)有限公司 | 作为蛋白激酶调节剂的含氨基大环化合物 |
| WO2023133375A1 (en) * | 2022-01-05 | 2023-07-13 | Blossomhill Therapeutics, Inc. | Macrocyclic compounds and use as kinase inhibitors |
| CN115746023A (zh) * | 2022-10-27 | 2023-03-07 | 复旦大学 | 一种作为蛋白激酶抑制剂的含吲唑结构的杂环大环化合物及其制备方法 |
Family Cites Families (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4838925A (en) | 1986-04-25 | 1989-06-13 | E. I. Du Pont De Nemours And Company | Heterocyclic acyl sulfonamides |
| DE3861031D1 (de) | 1987-03-07 | 1990-12-20 | Bayer Ag | Verfahren zur herstellung von 5-amino-4,6-dihalogenpyridinen. |
| DK0690843T3 (da) | 1993-03-25 | 2000-11-13 | Upjohn Co | Fornyl- eller cyanosubstituerede indolderivater med dopaninerg aktivitet |
| FR2746309B1 (fr) | 1996-03-22 | 1998-04-17 | Oreal | Composition de teinture des fibres keratiniques contenant des pyrazolopyrimidineoxo ; leur utilisation pour la teinture comme coupleurs, procedes de teinture |
| GB9800569D0 (en) | 1998-01-12 | 1998-03-11 | Glaxo Group Ltd | Heterocyclic compounds |
| FR2793791B1 (fr) | 1999-05-19 | 2002-01-25 | Univ Paris 7 Denis Diderot | Nouveaux composes inhibiteurs specifiques de phospholipases a2 |
| AU2001283955B2 (en) | 2000-07-31 | 2006-05-18 | F. Hoffmann-La Roche Ag | Piperazine derivatives |
| CA2441080A1 (en) | 2001-03-23 | 2002-10-03 | Eli Lilly And Company | Non-imidazole aryl alkylamines compounds as histamine h3 receptor antagonists, preparation and therapeutic uses |
| WO2004052315A2 (en) | 2002-12-11 | 2004-06-24 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| CN102143750B (zh) | 2008-09-08 | 2014-04-02 | 默克专利有限公司 | 用作蛋白激酶抑制剂的大环嘧啶 |
| PT2350075E (pt) | 2008-09-22 | 2014-06-09 | Array Biopharma Inc | Compostos imidazo[1,2b]piridazina substituídos como inibidores da trk cinase |
| SG10201914059WA (en) | 2008-10-22 | 2020-03-30 | Array Biopharma Inc | Substituted pyrazolo[1,5-a]pyrimidine compounds as trk kinase inhibitors |
| AU2009308675A1 (en) | 2008-10-31 | 2010-05-06 | Genentech, Inc. | Pyrazolopyrimidine JAK inhibitor compounds and methods |
| WO2010063487A1 (en) | 2008-12-05 | 2010-06-10 | Merz Pharma Gmbh & Co. Kgaa | Pyrazolopyrimidines, a process for their preparation and their use as medicine |
| WO2010086040A1 (en) | 2009-01-29 | 2010-08-05 | Biomarin Iga, Ltd. | Pyrazolo-pyrimidines for treatment of duchenne muscular dystrophy |
| AR077468A1 (es) | 2009-07-09 | 2011-08-31 | Array Biopharma Inc | Compuestos de pirazolo (1,5 -a) pirimidina sustituidos como inhibidores de trk- quinasa |
| US20110029480A1 (en) * | 2009-08-03 | 2011-02-03 | IntelliCubes, Inc. | Method of Compiling Multiple Data Sources into One Dataset |
| US9150577B2 (en) | 2009-12-07 | 2015-10-06 | Boehringer Ingelheim International Gmbh | Heterocyclic compounds containing an indole core |
| US9073926B2 (en) | 2009-12-07 | 2015-07-07 | Boehringer Ingelheim International Gmbh | Heterocyclic compounds containing a pyrrolopyridine or benzimidazole core |
| SA111320200B1 (ar) | 2010-02-17 | 2014-02-16 | ديبيوفارم اس ايه | مركبات ثنائية الحلقة واستخداماتها كمثبطات c-src/jak مزدوجة |
| MX365251B (es) * | 2010-05-20 | 2019-05-28 | Array Biopharma Inc | Compuestos macrocíclicos como inhibidores de trk cinasa. |
| WO2012034095A1 (en) | 2010-09-09 | 2012-03-15 | Irm Llc | Compounds and compositions as trk inhibitors |
| US8637516B2 (en) | 2010-09-09 | 2014-01-28 | Irm Llc | Compounds and compositions as TRK inhibitors |
| WO2012075393A2 (en) | 2010-12-02 | 2012-06-07 | President And Fellows Of Harvard College | Activators of proteasomal degradation and uses thereof |
| EP2508607A1 (en) | 2011-04-07 | 2012-10-10 | Helmholtz-Zentrum für Infektionsforschung GmbH | Medicament for liver regeneration and for treatment of liver failure |
| WO2013001310A1 (en) * | 2011-06-30 | 2013-01-03 | Centro Nacional De Investigaciones Oncológicas (Cnio) | Macrocyclic compounds and their use as cdk8 inhibitors |
| WO2013028470A1 (en) | 2011-08-19 | 2013-02-28 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| WO2013045701A2 (en) | 2011-09-29 | 2013-04-04 | L'oreal | Dyeing process using a composition comprising a glycosyl iridoid compound and a nucleophile or an amino or thio polymer, composition and devices therefor |
| WO2013045702A2 (en) | 2011-09-29 | 2013-04-04 | L'oreal | Dye composition comprising a non-glycosyl iridoid compound and a specific nucleophile or an amino or thio polymer, dyeing process, and device therefor |
| CA2849999A1 (en) | 2011-09-30 | 2013-04-04 | Oncodesign S.A. | Macrocyclic flt3 kinase inhibitors |
| KR20140077963A (ko) | 2011-10-20 | 2014-06-24 | 글락소스미스클라인 엘엘씨 | 시르투인 조절제로서의 치환된 비시클릭 아자-헤테로사이클 및 유사체 |
| ES2621220T3 (es) | 2012-03-06 | 2017-07-03 | Pfizer Inc. | Derivados macrocíclicos para el tratamiento de enfermedades proliferativas |
| AU2013230128B2 (en) | 2012-03-09 | 2017-08-17 | Lexicon Pharmaceuticals, Inc. | Pyrazolo[1,5-a]pyrimidine-based compounds, compositions comprising them, and methods of their use |
| WO2013134219A1 (en) * | 2012-03-09 | 2013-09-12 | Lexicon Pharmaceuticals, Inc. | Imidazo [1, 2 - b] pyridazine - based compounds, compositions comprising them, and uses thereof |
| GB201205669D0 (en) | 2012-03-30 | 2012-05-16 | Agency Science Tech & Res | Bicyclic heterocyclic derivatives as mnk2 and mnk2 modulators and uses thereof |
| ES2634014T3 (es) | 2013-01-30 | 2017-09-26 | Bayer Pharma Aktiengesellschaft | Isotiazoles sustituidos con amino |
| WO2014173928A1 (en) | 2013-04-23 | 2014-10-30 | Lek Pharmaceuticals D.D. | Novel synthetic process to 8-chloro-1-methyl-benzo[d]azepine, novel intermediates and the production thereof |
| JPWO2015012328A1 (ja) | 2013-07-24 | 2017-03-02 | 武田薬品工業株式会社 | 複素環化合物 |
| WO2015073267A1 (en) | 2013-11-15 | 2015-05-21 | Calitor Sciences, Llc | Substituted heteroaryl compounds and methods of use |
| US9714258B2 (en) | 2014-01-24 | 2017-07-25 | Tp Therapeutics, Inc. | Diaryl macrocycles as modulators of protein kinases |
| WO2016070241A1 (en) | 2014-11-03 | 2016-05-12 | Ctxt Pty Ltd | Triazine compounds, compositions and synthesis |
| CA2971024C (en) | 2014-12-15 | 2023-09-26 | Handok Inc. | Fused ring heteroaryl compounds and their use as trk inhibitors |
| ES2928084T3 (es) | 2015-02-20 | 2022-11-15 | Rigel Pharmaceuticals Inc | Inhibidores de GDF-8 |
| EP3267996B1 (en) | 2015-03-12 | 2020-11-11 | Merck Sharp & Dohme Corp. | Pyrazolopyrimidine inhibitors of irak4 activity |
| EP3268367B8 (en) | 2015-03-12 | 2022-11-16 | Merck Sharp & Dohme LLC | Carboxamide inhibitors of irak4 activity |
| PE20181024A1 (es) | 2015-05-05 | 2018-06-27 | Bayer Pharma AG | Derivados de ciclohexano sustituido con amido |
| JP6871903B2 (ja) | 2015-07-02 | 2021-05-19 | ターニング・ポイント・セラピューティクス・インコーポレイテッドTurning Point Therapeutics,Inc. | プロテインキナーゼのモジュレーターとしてのキラルジアリール大環状分子 |
| EP3319969B1 (en) | 2015-07-06 | 2024-04-03 | Turning Point Therapeutics, Inc. | Diaryl macrocycle polymorph |
| EP3325488B1 (en) | 2015-07-21 | 2020-06-24 | Turning Point Therapeutics, Inc. | Chiral diaryl macrocycle and use thereof in the treatment of cancer |
| CA3003153A1 (en) | 2015-10-26 | 2017-05-04 | Loxo Oncology, Inc. | Point mutations in trk inhibitor-resistant cancer and methods relating to the same |
| US11596639B2 (en) | 2016-03-04 | 2023-03-07 | Vanderbilt University | Substituted indole Mcl-1 inhibitors |
| AU2017302019A1 (en) | 2016-07-28 | 2019-02-07 | Turning Point Therapeutics, Inc. | Macrocycle kinase inhibitors |
| WO2018112027A1 (en) | 2016-12-14 | 2018-06-21 | Development Center For Biotechnology | Antibody-drug conjugates and uses thereof |
| TWI808958B (zh) | 2017-01-25 | 2023-07-21 | 美商特普醫葯公司 | 涉及二芳基巨環化合物之組合療法 |
| UA126158C2 (uk) | 2017-07-28 | 2022-08-25 | Тьорнінґ Поінт Терапьютикс, Інк. | Макроциклічні сполуки і їх використання |
-
2015
- 2015-01-23 US US15/113,583 patent/US9714258B2/en active Active
- 2015-01-23 RS RS20221161A patent/RS63829B1/sr unknown
- 2015-01-23 PL PL19169041.1T patent/PL3572416T3/pl unknown
- 2015-01-23 MX MX2016009588A patent/MX2016009588A/es active IP Right Grant
- 2015-01-23 EP EP23220719.1A patent/EP4338739A2/en active Pending
- 2015-01-23 DK DK19169041.1T patent/DK3572416T3/da active
- 2015-01-23 SG SG11201605929RA patent/SG11201605929RA/en unknown
- 2015-01-23 HR HRP20221518TT patent/HRP20221518T1/hr unknown
- 2015-01-23 LT LTEP15740510.1T patent/LT3097107T/lt unknown
- 2015-01-23 CN CN201910549748.XA patent/CN110317214B/zh active Active
- 2015-01-23 SG SG10202000191YA patent/SG10202000191YA/en unknown
- 2015-01-23 SI SI201530810T patent/SI3097107T1/sl unknown
- 2015-01-23 EP EP15740510.1A patent/EP3097107B1/en active Active
- 2015-01-23 ES ES19169041T patent/ES2933350T3/es active Active
- 2015-01-23 PT PT15740510T patent/PT3097107T/pt unknown
- 2015-01-23 RS RS20190900A patent/RS59059B1/sr unknown
- 2015-01-23 CN CN201910549014.1A patent/CN110317213B/zh active Active
- 2015-01-23 EA EA201892241A patent/EA201892241A1/ru unknown
- 2015-01-23 CN CN201580005705.5A patent/CN106170289B/zh active Active
- 2015-01-23 SI SI201531908T patent/SI3572416T1/sl unknown
- 2015-01-23 AU AU2015209239A patent/AU2015209239B2/en active Active
- 2015-01-23 PE PE2016000959A patent/PE20160931A1/es unknown
- 2015-01-23 CA CA2936079A patent/CA2936079C/en active Active
- 2015-01-23 NZ NZ761094A patent/NZ761094A/en unknown
- 2015-01-23 HU HUE19169041A patent/HUE060554T2/hu unknown
- 2015-01-23 ES ES15740510T patent/ES2735729T3/es active Active
- 2015-01-23 AP AP2016009383A patent/AP2016009383A0/en unknown
- 2015-01-23 HU HUE15740510A patent/HUE045208T2/hu unknown
- 2015-01-23 DK DK15740510.1T patent/DK3097107T3/da active
- 2015-01-23 EA EA201691492A patent/EA031863B1/ru active IP Right Revival
- 2015-01-23 NZ NZ722375A patent/NZ722375A/en unknown
- 2015-01-23 EP EP19169041.1A patent/EP3572416B1/en active Active
- 2015-01-23 UA UAA201608991A patent/UA121206C2/uk unknown
- 2015-01-23 WO PCT/US2015/012597 patent/WO2015112806A2/en active Application Filing
- 2015-01-23 PT PT191690411T patent/PT3572416T/pt unknown
- 2015-01-23 LT LTEP19169041.1T patent/LT3572416T/lt unknown
- 2015-01-23 MY MYPI2016001385A patent/MY193524A/en unknown
- 2015-01-23 KR KR1020167020188A patent/KR102402179B1/ko active IP Right Grant
- 2015-01-23 JP JP2016566857A patent/JP6490713B2/ja active Active
- 2015-01-23 PL PL15740510T patent/PL3097107T3/pl unknown
- 2015-01-23 EP EP19212110.1A patent/EP3636649B1/en active Active
- 2015-01-23 EP EP19212106.9A patent/EP3636648A1/en not_active Withdrawn
- 2015-01-23 LT LTEP19212110.1T patent/LT3636649T/lt unknown
- 2015-01-23 EP EP19212126.7A patent/EP3636650A1/en not_active Withdrawn
-
2016
- 2016-07-07 ZA ZA2016/04654A patent/ZA201604654B/en unknown
- 2016-07-20 IL IL246860A patent/IL246860B/en active IP Right Grant
- 2016-07-22 MX MX2020001360A patent/MX2020001360A/es unknown
- 2016-07-22 CL CL2016001876A patent/CL2016001876A1/es unknown
- 2016-07-25 PH PH12016501463A patent/PH12016501463A1/en unknown
-
2017
- 2017-05-23 HK HK17105209.7A patent/HK1231407A1/zh unknown
- 2017-05-31 US US15/609,962 patent/US10246466B2/en active Active
-
2018
- 2018-11-29 US US16/204,566 patent/US10618912B2/en active Active
-
2019
- 2019-02-27 JP JP2019034652A patent/JP2019104748A/ja not_active Withdrawn
- 2019-07-16 HR HRP20191283TT patent/HRP20191283T1/hr unknown
- 2019-07-17 CY CY20191100764T patent/CY1121850T1/el unknown
- 2019-08-21 IL IL268820A patent/IL268820B/en unknown
- 2019-12-10 AU AU2019279951A patent/AU2019279951B2/en active Active
-
2020
- 2020-02-21 US US16/797,866 patent/US20200216465A1/en not_active Abandoned
- 2020-07-06 ZA ZA2020/04100A patent/ZA202004100B/en unknown
-
2021
- 2021-01-07 JP JP2021001588A patent/JP2021063121A/ja active Pending
- 2021-09-27 US US17/485,942 patent/US20220112213A1/en not_active Abandoned
-
2022
- 2022-07-08 JP JP2022110613A patent/JP7356546B2/ja active Active
-
2023
- 2023-09-22 JP JP2023158350A patent/JP2023179539A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2735729T3 (es) | Macrociclos de diarilo como moduladores de proteínas cinasas | |
| ES2864839T3 (es) | Macrociclos de diarilo quirales como moduladores de proteína quinasas | |
| TWI736134B (zh) | 作為蛋白質激酶之調節劑的二芳基巨環 | |
| US20240140964A1 (en) | Diaryl macrocycles as modulators of protein kinases | |
| BR112016017101B1 (pt) | Compostos diaril macrociclos como moduladores das proteínas quinases e sua composição farmacêutica | |
| EA041662B1 (ru) | Способ ингибирования протеин- или тирозинкиназы в клетке с применением диарильного макроциклического соединения |