ES2719093T3 - Composiciones de fármacos poco solubles en agua con mayor estabilidad y métodos para su preparación - Google Patents
Composiciones de fármacos poco solubles en agua con mayor estabilidad y métodos para su preparación Download PDFInfo
- Publication number
- ES2719093T3 ES2719093T3 ES06802739T ES06802739T ES2719093T3 ES 2719093 T3 ES2719093 T3 ES 2719093T3 ES 06802739 T ES06802739 T ES 06802739T ES 06802739 T ES06802739 T ES 06802739T ES 2719093 T3 ES2719093 T3 ES 2719093T3
- Authority
- ES
- Spain
- Prior art keywords
- composition
- docetaxel
- citrate
- cancer
- albumin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5169—Proteins, e.g. albumin, gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
- A61K38/13—Cyclosporins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/42—Proteins; Polypeptides; Degradation products thereof; Derivatives thereof, e.g. albumin, gelatin or zein
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y5/00—Nanobiotechnology or nanomedicine, e.g. protein engineering or drug delivery
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/902—Specified use of nanostructure
- Y10S977/904—Specified use of nanostructure for medical, immunological, body treatment, or diagnosis
- Y10S977/906—Drug delivery
Abstract
Una composición que comprende a) nanopartículas que comprenden docetaxel recubierto con albúmina, y b) citrato, en la que la estabilidad de la composición se aumenta comparada con la de la composición sin citrato.
Description
DESCRIPCIÓN
Composiciones de fármacos poco solubles en agua con mayor estabilidad y métodos para su preparación Antecedentes de la invención
Existe un número cada vez mayor de fármacos farmacéuticos formulados que son poco solubles o insolubles en disoluciones acuosas. Dichos fármacos presentan retos para su administración en una forma inyectable tal como mediante administración parenteral. Una formulación bien diseñada debe, como mínimo, poder presentar una cantidad terapéuticamente efectiva del fármaco poco soluble en el sitio de absorción deseado, en una forma absorbible. Además, estas composiciones tienden a ser inestables, produciéndose sedimentación y/o precipitación en 24 horas después de la rehidratación o reconstitución.
Los taxanos, en particular los dos fármacos taxanos disponibles actualmente, paclitaxel y docetaxel, son agentes antitumorales potentes. El paclitaxel es muy poco soluble en agua (menos de 10 |jg/ml) y, como resultado, no puede formularse de forma práctica con un medio acuoso para la administración IV. Actualmente, el paclitaxel se formula para la administración IV a pacientes con cáncer en una disolución con aceite de ricino polioxietilado (Polioxil 35 o Cremophor®) como el disolvente/tensioactivo principal, empleando altas concentraciones de etanol como codisolvente. Una de las mayores dificultades en la administración de paclitaxel es la aparición de reacciones de hipersensibilidad. Estas reacciones, que incluyen erupciones cutáneas graves, urticarias, eritema, disnea, taquicardia y otros, pueden atribuirse, al menos en parte, a las altas concentraciones de etanol y Cremophor usadas como disolventes en la formulación. El docetaxel, un análogo del paclitaxel, se produce de forma semisintética a partir de 10-deacetil bacatina III, un precursor no citotóxico extraído de las agujas de Taxus baccata y esterificado con una cadena lateral sintetizada químicamente (Cortes y Pazdur, 1995, J. Clin. Oncol. 13(10):2643-55). Como el paclitaxel, el docetaxel es muy poco soluble en agua. Actualmente, el disolvente/tensioactivo más preferido usado para disolver el docetaxel es polisorbato 80 (Tween 80) (Bissery et al. 1991 Cáncer Res. 51(18):4845-52; Tomiak et al. 1992). Como el Cremophor, el Tween causa frecuentemente reacciones de hipersensibilidad en los pacientes. Además, el Tween 80 no puede usarse con aparatos de administración de PVC debido a su tendencia a lixiviar ftalato de dietilhexilo, que es altamente tóxico.
La purificación de paclitaxel y docetaxel semisintéticos es un problema desafiante debido a la formación de varios productos de degradación a lo largo de la ruta sintética. Además, se encuentra que los taxanos purificados experimentan degradación, incluso en condiciones de almacenamiento controladas. Por lo tanto, es deseable desarrollar formas estables de estas moléculas que retengan las propiedades anticancerosas deseables. Los esfuerzos previos para obtener docetaxel adecuado se han centrado en procesos para preparar formar trihidratadas de docetaxel, que se creía que tenían una estabilidad sustancialmente mayor que la del producto anhidro. Véanse, p. ej., las Pat. de EEUU Nos. 6.022.985; 6.838.569.
Con el fin de conseguir los efectos terapéuticos esperados de agentes poco solubles en agua tales como paclitaxel y docetaxel, habitualmente se requiere que se administre a un paciente una forma solubilizada o forma nanodispersada del agente.
Así, se han desarrollado varios métodos que están basados en el uso de: disolventes auxiliares; tensioactivos; formas solubles del fármaco, p. ej., sales y solvatos; formas químicamente modificadas del fármaco, p. ej., profármacos; complejos solubles polímero-fármaco; vehículos especiales de fármacos tales como liposomas; y otros. De hecho, el uso de micelas de copolímeros en bloque anfifílicos ha atraído gran parte del interés como un vehículo de fármacos potencialmente efectivo que es capaz de solubilizar un fármaco hidrofóbico en un entorno acuoso.
Cada uno de los métodos anteriores está obstaculizado por uno o más problemas particulares. Por ejemplo, el método basado en el uso de micelas de tensioactivo para solubilizar fármacos hidrofóbicos tiene problemas ya que algunos de los tensioactivos son relativamente tóxicos y se produce la precipitación de los fármacos hidrofóbicos cuando se someten a dilución.
Previamente, se han desarrollado formulaciones de liposomas basadas en fosfolípidos para paclitaxel, Taxotere, y otros taxanos activos (Straubinger et al. 1993, J. Natl. Cáncer Inst. Monogr. (15):69-78; Straubinger et al. 1994; Sharma et al. 1993, Cancer Res. 53(24):557-81; Sharma y Straubinger 1994, Pharm. Res. 11(6):889-96; A. Sharma et al. 1995, J. Pharm. Sci. 84(12):1400-4) y se han estudiado las propiedades físicas de estas y otras formulaciones de taxanos (Sharma y Straubinger 1994, Pharm. Res. 11(6):889-96; de EEUU Sharma et al. 1995, J. Pharm. Sci.
84(10):1223-30; Balasubramanian y Straubinger 1994, Biochemistry 33(30):8941-7; Balasubramanian et al. 1994, J. Pharm. Sci. 83(10):1470-6). La utilidad principal de estas formulaciones es la eliminación de la toxicidad relacionada con el excipiente Cremophor EL y una reducción en la toxicidad del taxano en sí mismo, como se demuestra en varios modelos tumorales en animales (Sharma et al. 1993, Cancer Res. 53(24):557-81; A. Sharma et al. 1995, J. Pharm. Sci. 84(12):1400-4; Sharma et al. 1996, Cancer Lett. 107(2):265-272). Esta observación se aplica a varios taxanos además de paclitaxel (A. Sharma et al. 1995, J. Pharm. Sci. 84(12):1400-4). En algunos casos, la potencia antitumoral del fármaco parece ser ligeramente mayor para las formulaciones basadas en liposomas (Sharma et al.
1993, Cancer Res. 53(24):557-81).
Estas formulaciones liposomales comprenden fosfolípidos y otros aditivos, además del taxano, y pueden almacenarse en un estado seco. Después de la adición de una fase acuosa a la mezcla, se forman partículas espontáneamente y pueden adoptar la forma de liposomas (Straubinger et al. 1993). Los liposomas son estructuras vesiculares cerradas que consisten en una membrana en bicapa limitante que rodea un núcleo acuoso. Una composición de formulación preferida (Sharma y Straubinger 1994) contiene un fosfolípido neutro (zwitteriónico) tal como lecitina (fosfatidilcolina, 80-90 % en relación molar), junto con un fosfolípido cargado negativamente tal como fosfatidilglicerol (10-20 %). El último previene la agregación de las partículas mediante repulsión electrostática. El contenido en taxano más estable está en el intervalo de 3-4 % en moles (respecto al contenido total de fosfolípidos); dichos liposomas pueden ser físicamente y/o químicamente estables durante 2 meses después de la hidratación. En la mayor parte de las condiciones, las formulaciones de paclitaxel que contienen concentraciones mayores de fármaco (p. ej., 8 % en moles) son muy inestables y pueden precipitar en minutos después de la preparación (Sharma y Straubinger 1994).
La mayor preocupación respecto a estas formulaciones ha sido el contenido relativamente bajo en taxano de formulaciones aceptablemente estables (3-5 % en moles), lo que necesita la administración de una gran cantidad de fosfolípido (5-10 g) a pacientes con el fin de proporcionar la dosis anticipada del fármaco. Aunque a los seres humanos se les proporcionan frecuentemente grandes cantidades de lípidos por vía intravenosa para Nutrición Parenteral Total (TPN), un objetivo de desarrollo importante ha sido producir liposomas de taxanos que tengan un mayor contenido en taxanos.
Otras estrategias para formular fármacos poco solubles para administración oral o parenteral incluyen, por ejemplo, formulaciones en las que el fármaco poco soluble es una emulsión de aceite en agua, una microemulsión, o una disolución de micelas u otras partículas vehiculares multilamelares. Aunque dichas estrategias pueden ser apropiadas para algunos agentes terapéuticos hidrofóbicos ionizables así como no ionizables, no se aprovechan de las propiedades químicas de ácido-base únicas, y propiedades de solubilidad asociadas, de los compuestos ionizables.
Los fármacos que son insolubles en agua pueden tener unos beneficios significativos cuando se formulan como una suspensión estable de partículas submicrométricas. El control exacto del tamaño de las partículas es esencial para el uso seguro y eficaz de estas formulaciones. Las partículas deben ser menores de siete micrómetros de diámetro para pasar de forma segura a través de los capilares sin causar embolia (Allen et al., 1987; Davis y Taube, 1978; Schroeder et al., 1978;Yokel et al., 1981, Toxicol. Lett. 9(2):165-70).
Otra estrategia se describe en la Patente de EEUU No. 5.118.528 que describe un proceso para preparar nanopartículas. El proceso incluye las etapas de: (1) preparar una fase líquida de una sustancia en un disolvente o una mezcla de disolventes a la que puede añadirse uno o más tensioactivos, (2) preparar una segunda fase líquida de un no disolvente o una mezcla de no disolventes, siendo el no disolvente miscible con el disolvente o mezcla de disolventes para la sustancia, (3) añadir conjuntamente las disoluciones de (1) y (2) con agitación y (4) retirar los disolventes no deseados para producir una suspensión coloidal de nanopartículas. La patente '528 describe que produce partículas de la sustancia menores de 500 nm sin el suministro de energía. En particular, la patente '528 afirma que es indeseable el uso de equipamiento de alta energía tal como sonicadores y homogeneizadores.
La Patente de EEUU No. 4.826.689 describe un método para preparar partículas con tamaño uniforme de fármacos u otros compuestos orgánicos insolubles en agua. En primer lugar, se disuelve un compuesto orgánico sólido adecuado en un disolvente orgánico y la disolución puede diluirse con un no disolvente. Después, se infunde un líquido que precipita lo acuoso, precipitando partículas no agregadas con un diámetro medio sustancialmente uniforme. Las partículas se separan entonces del disolvente orgánico. Dependiendo del compuesto orgánico y del tamaño deseado de las partículas, los parámetros de temperatura, relación de no disolvente a disolvente orgánico, velocidad de infusión, velocidad de agitación y volumen pueden variarse según la patente. La patente '689 describe que este proceso forma un fármaco en un estado metaestable que es termodinámicamente inestable y que se convierte eventualmente en un estado cristalino más estable. La patente '689 describe el atrapamiento del fármaco en un estado metaestable en el que la energía libre se encuentra entre la de la disolución del fármaco de partida y de la forma cristalina estable. La patente '689 describe la utilización de inhibidores de la cristalización (p. ej., polivinilpirrolidinona) y agentes tensioactivos (p. ej., poli(oxietileno-co-oxipropileno)) para hacer que el precipitado sea lo suficientemente estable como para aislarse por centrifugación, filtración en membrana u ósmosis inversa.
Otra estrategia para proporcionar fármacos insolubles para administración parenteral se describe en la Patente de EEUU No. 5.145.684. La patente '684 describe la molienda en húmedo de un fármaco insoluble en presencia de un modificador de la superficie para proporcionar una partícula de fármaco que tiene un tamaño de partícula promedio efectivo de menos de 400 nm. La patente '684 describe que el modificador de la superficie se adsorbe en la superficie de la partícula de fármaco en una cantidad suficiente como para prevenir la aglomeración en partículas mayores. Las nanopartículas de fármacos insolubles preparadas en condiciones de altas fuerzas de cizalladura (p. ej., sonicación, homogeneización con alta presión, o semejantes) con polímeros biocompatibles (p. ej., albúmina) se describen, por ejemplo, en las Patentes de EEUU Nos. 5.916.596, 6.506.405 y 6.537.579 y también en la Publicación de Patente de EEUU 2005/0004002 A1.
El documento US 2005/004002 se refiere a una composición farmacéutica que comprende un agente farmacéutico y
un vehículo farmacéuticamente aceptable, cuyo vehículo comprende una proteína, por ejemplo, albúmina de suero humano y/o deferoxamina. La albúmina de suero humano está presente en una cantidad eficaz para reducir uno o más efectos secundarios asociados con la administración de la composición farmacéutica. También se proporcionan métodos para reducir uno o más efectos secundarios de la administración de la composición farmacéutica, métodos para inhibir el crecimiento microbiano y la oxidación en la composición farmacéutica y métodos para potenciar el transporte y la unión de un agente farmacéutico a una célula.
A la vista de lo anterior, existe una necesidad de composiciones farmacéuticas que comprendan fármacos poco solubles en agua con estabilidad física y química incrementada, que eliminen el uso de disolventes y excipientes fisiológicamente dañinos y métodos de producción de estas. Es deseable que dichas composiciones farmacéuticas no se degraden, permanezcan estables en condiciones de almacenamiento y permanezcan físicamente y/o químicamente estables después de la rehidratación. También sería deseable tener una composición farmacéutica que comprenda una forma anhidra del fármaco poco soluble en agua que tenga una mayor solubilidad en disolventes y excipientes usados tradicionalmente, así como en disolventes y excipientes que no sean fisiológicamente dañinos. La presente invención proporciona dichas composiciones farmacéuticas y métodos.
Resumen breve de la invención
La presente invención proporciona una composición que comprende a) nanopartículas que comprenden docetaxel recubierto con albúmina, y b) citrato, y en la que se aumenta la estabilidad de la composición comparada con la de la composición sin citrato.
En una realización, las nanopartículas en la composición tienen un tamaño promedio o medio de las partículas no mayor de 200 nm.
En una realización, la albúmina es albúmina de suero humano.
En una realización, la relación en peso de albúmina a docetaxel es 18:1 o menos.
En una realización, la composición es una suspensión líquida con al menos 1 mg/ml de docetaxel.
En una realización, la composición es una suspensión líquida con al menos 15 mg/ml de docetaxel.
En una realización, la composición es una composición seca que se puede reconstituir a una suspensión líquida con al menos 1 mg/ml de docetaxel.
En una realización, la composición seca es una composición liofilizada.
En una realización, el citrato es citrato de sodio.
En una realización, la composición comprende además cloruro de sodio.
En una realización, la composición comprende citrato de sodio 200 mM y cloruro de sodio 300 mM.
En una realización, el docetaxel usado para preparar la composición está en una forma anhidra.
En una realización, la composición seca es una composición farmacéutica.
En una realización, la composición comprende además un azúcar.
En una realización, el azúcar es sacarosa, manitol, fructosa, lactosa, maltosa o trehalosa.
En una realización, la composición es una composición farmacéutica estéril para administración parenteral.
En una realización, la composición es para inyección y tiene un pH no inferior a 6,5, 7 u 8.
La presente invención también proporciona una composición según la presente invención para usar en un método de tratamiento del cáncer en un individuo.
En una realización, el cáncer es cualquiera de cáncer de próstata, cáncer de colon, cáncer de mama, cáncer de cabeza y cuello, cáncer de páncreas, cáncer de pulmón, y cáncer de ovario.
La presente invención también proporciona un vial sellado que comprende una composición de la presente invención.
La presente invención también proporciona un método de estabilización de una composición que comprende nanopartículas de docetaxel recubiertas con albúmina, en el que el método comprende combinar la composición con un citrato, en el que la composición resultante es estable en las mismas condiciones en las que la composición es inestable antes de la adición del citrato.
En una realización, el citrato es citrato de sodio.
En una realización, la composición comprende además cloruro de sodio.
En una realización, la invención proporciona formulaciones farmacéuticas de docetaxel como se reivindica, que comprenden citrato o derivados de este. En un ejemplo, se describen formulaciones farmacéuticas de docetaxel que comprenden pirofosfato de sodio. En un ejemplo, se describen formulaciones farmacéuticas de docetaxel que comprenden EDTA o derivados de este. En un ejemplo, se describen formulaciones farmacéuticas de docetaxel que comprenden gluconato de sodio. En una realización, la invención proporciona formulaciones farmacéuticas de docetaxel como se reivindican, que comprenden citrato y cloruro de sodio. En una realización, la invención proporciona una formulación de docetaxel como se reivindica que comprende un tensioactivo, en la que el docetaxel usado para preparar la formulación está en una forma anhidra antes de ser incorporado en la formulación.
Por consiguiente, en un aspecto, la invención proporciona composiciones (tales como composiciones farmacéuticas) que comprenden un agente farmacéutico poco soluble en agua (docetaxel) y un agente estabilizante como se reivindica en las que se aumenta la estabilidad de la composición comparada con la de una composición sin el agente estabilizante. El agente estabilizante también descrito en la presente memoria agentes quelantes (tales como ácido málico, edetato, y pentetato), pirofosfato de sodio, y gluconato de sodio.
En otro aspecto, se proporcionan varias composiciones (tales como composiciones farmacéuticas), que comprenden docetaxel como se reivindica, en las que el docetaxel usado para la preparación de la composición está en forma anhidra (por ejemplo, el docetaxel puede ser anhidro antes de ser incorporado en la composición). En algunas realizaciones, la invención proporciona composiciones (tales como composiciones farmacéuticas), que comprenden docetaxel como se reivindica y un tensioactivo, en las que el docetaxel usado para la preparación de la composición está en forma anhidra (por ejemplo, el docetaxel puede ser anhidro antes de ser incorporado en la composición). También se proporcionan formas de dosificación unitarias de composiciones proporcionadas en la presente memoria, artículos de fabricación que comprenden las composiciones o formas de dosificación unitarias inventivas en un envasado adecuado y kits que comprenden las composiciones. La invención también proporciona métodos de preparación y usos de estas composiciones como se proporcionan en la presente memoria.
Debe entenderse que una, alguna, o todas las propiedades de las varias realizaciones descritas en la presente memoria pueden combinarse para formar otras realizaciones de la presente invención.
Descripción breve de las figuras
La Figura 1 muestra la pérdida de peso corporal de ratas a una dosis de 5 mg/kg de docetaxel para una formulación de nanopartículas de albúmina de docetaxel (Nab-docetaxel) y Tween 80-docetaxel (Taxotere®). La dosificación se produjo en los días 0, 4 y 8.
La Figura 2 muestra la comparación de neutropenia en ratas a una dosis de 5 mg/kg para Nab-docetaxel y Tween 80-docetaxel (Taxotere®). La dosificación se produjo en los días 0, 4 y 8.
Las Figuras 3A-3D muestran la comparación farmacocinética de Nab-docetaxel y Taxotere. Las Figuras 3A-3C muestran la concentración plasmática de Nab-docetaxel y Taxotere® a dosis de 10 mg/kg, 20 mg/kg y 30 mg/kg, respectivamente. La Figura 3D muestra la relación lineal entre la AUC (Área Bajo la Curva) y la dosis para Nab-docetaxel y la relación no lineal entre la AUC y la dosis para Taxotere. El Nab-docetaxel presentó una relación lineal ajustada por la ecuación AUC=218*Dosis; el Taxotere presentó una curva exponencial ajustada por la ecuación AUC=722*exp(0,10*Dosis).
La Figura de referencia 4 muestra la inhibición de la unión del fármaco a albúmina en presencia de tensioactivo Tween 80 y Cremophor EL®/EtOH.
Las Figuras 5A y 5B muestran la actividad antitumoral (5A) y pérdida de peso corporal (5B) con Nab-docetaxel en ratones con xenoinjerto de tumor de colon H29. Los ratones se dosificaron con Nab-docetaxel a 15 mg/kg, q4dx3. Las Figuras 6A y 6B muestran la actividad antitumoral (6A) y pérdida de peso corporal (6B) en un ratón con xenoinjerto de tumor de colon HCT116 dosificado con disolución salina, Nab-docetaxel (22 mg/kg), y Taxotere (15 mg/kg).
Las Figuras 7A y 7B muestran la pérdida de peso corporal (7A) y actividad antitumoral (7B) en un ratón con xenoinjerto de tumor de próstata PC3 dosificado con disolución salina, Nab-docetaxel (10, 15, 20, 30 mg/kg) y Tween 80-docetaxel (10 mg/kg).
Descripción detallada de la invención
Se describen composiciones y métodos de preparación de docetaxel y otros fármacos o agentes farmacéuticos poco solubles en agua que retienen los efectos terapéuticos deseables y permanecen física y/o químicamente estables tras la exposición a determinadas condiciones tales como el almacenamiento prolongado, temperatura elevada o
dilución para administración parenteral.
Una composición estable, es, por ejemplo, una que permanece físicamente y/o químicamente estable y, por lo tanto, que no muestra evidencia de precipitación o sedimentación durante al menos aproximadamente 8 horas, incluyendo por ejemplo al menos aproximadamente cualquiera de 24 horas, 48 horas, o hasta aproximadamente 96 horas después de la reconstitución o rehidratación. Por ejemplo, las composiciones pueden permanecer estables durante al menos 24 horas después de la reconstitución o rehidratación.
La estabilidad de una suspensión se evalúa generalmente (pero no necesariamente) en las condiciones habituales de transporte y almacenamiento esperadas durante la distribución del producto (tal como temperatura ambiente (tal como 20-25 °C) o condiciones refrigeradas (tal como 4 °C)). Por ejemplo, una suspensión es estable a una temperatura de almacenamiento si no presenta floculación o aglomeración de partículas visible a simple vista o cuando se observa en el microscopio óptico con un aumento de 1.000 veces (u otras técnicas adecuadas para la caracterización de partículas), aproximadamente a los quince minutos después de la preparación de la suspensión. La estabilidad también puede evaluarse en condiciones exageradas de temperatura, humedad, luz, y/u otras, para ensayar la estabilidad de las composiciones en un ensayo acelerado. Por ejemplo, la estabilidad puede evaluarse a una temperatura que es mayor de aproximadamente 40 °C. La estabilidad de la composición también puede evaluarse, por ejemplo, por la capacidad de la composición de permanecer suspendida sin mostrar evidencia de sedimentación o cremosidad, o por la capacidad de la composición de permanecer inalterada (es decir, sin diferencia visible) en términos de color o consistencia.
La estabilidad de una composición seca (tal como una liofilizada) puede evaluarse sobre la base del comportamiento de la suspensión líquida resultante de la reconstitución o rehidratación de la composición seca.
Es un objeto de la invención proporcionar composiciones farmacéuticas capaces de mantener físicamente y/o químicamente estabilizadas, cantidades terapéuticamente efectivas de agentes farmacéuticos poco solubles en agua. Es otro objeto de la invención proporcionar composiciones farmacéuticas capaces de mantener físicamente y/o químicamente estabilizados agentes farmacéuticos poco solubles en agua después de la dilución para administración a un paciente. Es un objeto adicional de la invención proporcionar composiciones farmacéuticas capaces de mantener físicamente y/o químicamente estabilizadas, cantidades terapéuticamente efectivas de agentes farmacéuticos poco solubles en agua con toxicidades reducidas. Es un objeto adicional de la invención proporcionar formulaciones farmacéuticas estables usando docetaxel anhidro, así como composiciones que son el resultado del uso de docetaxel anhidro.
Es un objeto adicional de la invención proporcionar métodos mejorados de preparación de composiciones farmacéuticas capaces de mantener físicamente y/o químicamente estabilizadas, cantidades terapéuticamente efectivas de agentes farmacéuticos poco solubles en agua. Un objeto adicional de la invención es proporcionar métodos mejorados de preparación de composiciones farmacéuticas capaces de mantener un agente farmacéutico poco soluble en agua físicamente y/o químicamente estabilizado tras la dilución para la administración a un paciente. Es un objeto adicional de la invención proporcionar métodos mejorados de preparación de composiciones farmacéuticas capaces de mantener físicamente y/o químicamente estabilizadas, cantidades terapéuticamente efectivas de agentes farmacéuticos poco solubles en agua con toxicidades reducidas.
En una realización la invención proporciona una composición farmacéutica estéril para la administración parenteral compuesta de un agente farmacéutico poco soluble en agua, que es estabilizado físicamente y/o químicamente por la adición de excipientes a la composición. Antes de la presente invención, la estabilidad relativa de determinados agentes farmacéuticos poco solubles en agua ha limitado su uso en composiciones farmacéuticas parenterales debido a la degradación en las condiciones de almacenamiento y/o precipitación después de la dilución. Muchos agentes farmacéuticos diferentes no se podían preparar satisfactoriamente como parenterales debido a la ausencia de una composición estable.
La presente invención implica el descubrimiento sorprendente de que el excipiente común citrato es capaz de estabilizar el agente farmacéutico poco soluble en agua docetaxel. Por lo tanto un objeto principal de la invención es proporcionar composiciones que comprenden docetaxel y excipientes para obtener composiciones farmacéuticas parenterales, estables. Por lo tanto, en una realización, la invención proporciona una composición farmacéutica que comprende docetaxel y citrato como se reivindica. En otra realización, la invención proporciona una composición farmacéutica que comprende docetaxel, citrato y cloruro de sodio como se reivindica.
Varias realizaciones de la invención
La invención proporciona composiciones (tales como composiciones farmacéuticas) que comprenden un agente farmacéutico poco soluble en agua y un agente estabilizante como se reivindica, en las que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. Por ejemplo, la composición puede comprender docetaxel y un agente estabilizante, en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En la presente invención, el polímero biocompatible es una albúmina, por ejemplo, albúmina de suero humano (HSA). En algunas realizaciones, la estabilidad de la composición es al menos 1,5 veces (incluyendo, por ejemplo, al menos aproximadamente
cualquiera de 2x, 3x, 4x, 5x, 6x, 7x, 8x, 9x, 10x, 15x, 20x, 25x, 30x, o más) mayor comparada con la de la composición sin el agente estabilizante. En algunas realizaciones, el agente farmacéutico poco soluble en agua es inestable en una composición que no comprende el agente estabilizante.
Se describe una composición que comprende un agente farmacéutico poco soluble en agua y un agente estabilizante, en la que el agente estabilizante es un agente quelante, y en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. Se proporciona una composición que comprende docetaxel y un agente estabilizante como se reivindica, en la que el agente estabilizante es un agente quelante, y en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En la presente invención, el polímero biocompatible es albúmina, por ejemplo, HSA. En algunos ejemplos, el agente estabilizante es un agente quelante polidentado. En algunos ejemplos, el agente estabilizante comprende uno o más grupos ácido carboxílico. En algunos ejemplos, el agente quelante no es deferoxamina (es decir, es otro distinto de deferoxamina). En algunos ejemplos, el agente quelante es cualquiera de (y en algunos ejemplos seleccionado del grupo que consiste en) edetato, ácido málico, pentetato, trometamina, derivados de estos y mezclas de estos. En la presente invención, el agente estabilizante es un citrato o uno de sus derivados (tal como citrato de sodio y en algunas realizaciones ácido cítrico). En algunas realizaciones, la composición comprende citrato de sodio y cloruro de sodio. En algunas realizaciones, la composición comprende citrato aproximadamente 200 mM y cloruro de sodio aproximadamente 300 mM. En algunos ejemplos, el agente estabilizante es un edetato o uno de sus derivados (tal como EDTA).
En algunos ejemplos, hay una composición que comprende un agente farmacéutico poco soluble en agua y un agente estabilizante, en la que el agente estabilizante es gluconato de sodio, y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunos ejemplos, hay una composición que comprende docetaxel y un agente estabilizante, en la que el agente estabilizante es gluconato de sodio, y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunos ejemplos, la composición comprende además un polímero biocompatible. En algunos ejemplos, el polímero biocompatible es una proteína vehicular (tal como albúmina, por ejemplo, hSa ).
En algunos ejemplos, hay una composición que comprende un agente farmacéutico poco soluble en agua y un agente estabilizante, en la que el agente estabilizante es pirofosfato de sodio, y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunos ejemplos, hay una composición que comprende docetaxel y un agente estabilizante, en la que el agente estabilizante es pirofosfato de sodio, y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunos ejemplos, la composición comprende además un polímero biocompatible. En algunos ejemplos, el polímero biocompatible es una proteína vehicular (tal como albúmina, por ejemplo, hSa ).
En algunas realizaciones, la composición comprende un agente farmacéutico poco soluble en agua, una albúmina, y un agente estabilizante como se reivindica, en la que la relación en peso de la albúmina al agente farmacéutico poco soluble en agua en la composición es de aproximadamente 0,01:1 a aproximadamente 100:1, y en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunas realizaciones, la composición comprende un agente farmacéutico poco soluble en agua, una albúmina, y un agente estabilizante como se reivindica, en la que la relación en peso de la albúmina al agente farmacéutico poco soluble en agua en la composición es de aproximadamente 18:1 o menos (incluyendo por ejemplo cualquiera de aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, y aproximadamente 9:1), y en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunas realizaciones, la composición comprende docetaxel, una albúmina, y un agente estabilizante como se reivindica, en la que la relación en peso de la albúmina al docetaxel en la composición es de aproximadamente 18:1 o menos (incluyendo por ejemplo cualquiera de aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, y aproximadamente 9:1), y en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. Como se describe en la presente memoria, el agente estabilizante es un agente quelante, tal como cualquiera de (y en algunos ejemplos seleccionado del grupo que consiste en) edetato, ácido málico, pentetato, trometamina, derivados de estos, y mezclas de estos. En la presente invención, el agente estabilizante es un citrato o uno de sus derivados (tal como citrato de sodio). En algunas realizaciones, la composición comprende citrato de sodio y cloruro de sodio. En algunos ejemplos, el agente estabilizante es un edetato o uno de sus derivados (tal como EDTA). En algunos ejemplos, el agente estabilizante es gluconato de sodio. En algunos ejemplos, el agente estabilizante es pirofosfato de sodio.
En algunos ejemplos, la proteína/agente farmacéutico está en forma(s) de partículas, que en varias realizaciones puede ser de los diámetros medios como se describen en la presente memoria.
En algunos ejemplos, la composición comprende un agente farmacéutico poco soluble en agua asociado a proteína y un agente estabilizante, en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunas realizaciones, la composición comprende docetaxel asociado a una proteína y un agente estabilizante como se reivindica, en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunos ejemplos, el agente estabilizante es un
agente quelante, tal como cualquiera de (y en algunos ejemplos seleccionado del grupo que consiste en) edetato, ácido mélico, pentetato, trometamina, derivados de estos, y mezclas de estos. En la presente invención, el agente estabilizante es un citrato o uno de sus derivados (tal como citrato de sodio). En algunas realizaciones, la composición comprende citrato de sodio y cloruro de sodio. En algunos ejemplos, el agente estabilizante es un edetato o uno de sus derivados (tal como EDTA). En algunos ejemplos, el agente estabilizante es gluconato de sodio. En algunos ejemplos, el agente estabilizante es pirofosfato de sodio.
En algunos ejemplos, la composición comprende (1) partículas (tales como nanopartículas) que comprenden (en varias realizaciones que consisten en o que consisten esencialmente en) un agente farmacéutico poco soluble en agua y polímero biocompatible (tal como proteína vehicular, que puede ser albúmina tal como HSA); y (2) un agente estabilizante, en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunas realizaciones, la composición comprende nanopartículas que comprenden (en varias realizaciones que consisten en o que consisten esencialmente en) (1) docetaxel y albúmina tal como HSA; y (2) un agente estabilizante como se reivindica, en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En la presente invención, el docetaxel está recubierto con el polímero biocompatible. En algunos ejemplos, el agente estabilizante es un agente quelante, tal como cualquiera de (y en algunos ejemplos seleccionado del grupo que consiste en) edetato, ácido mélico, pentetato, trometamina, derivados de estos, y mezclas de estos. En la presente invención, el agente estabilizante es un citrato o uno de sus derivados (tal como citrato de sodio). En algunas realizaciones, la composición comprende citrato de sodio y cloruro de sodio. En algunos ejemplos, el agente estabilizante es un edetato o uno de sus derivados (tal como EDTA). En algunos ejemplos, el agente estabilizante es un ácido mélico. En algunos ejemplos, el agente estabilizante es gluconato de sodio. En algunos ejemplos, el agente estabilizante es pirofosfato de sodio.
En algunos ejemplos, la composición comprende (1) partículas (tales como nanopartículas) que comprenden (en varios ejemplos que consisten en o que consisten esencialmente en) un agente farmacéutico poco soluble en agua y albúmina; y (2) un agente estabilizante, en la que la relación en peso de la albúmina al agente farmacéutico poco soluble en agua en la composición es de aproximadamente 0,01:1 a aproximadamente 100:1, y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunos ejemplos, el agente poco soluble en agua está recubierto con albúmina. En algunos ejemplos, la composición comprende (1) partículas (tales como nanopartículas) que comprenden (en varios ejemplos que consisten en o que consisten esencialmente en) un agente farmacéutico poco soluble en agua y albúmina; y (2) un agente estabilizante, en la que la relación en peso de la albúmina al agente farmacéutico poco soluble en agua en la composición es de aproximadamente 18:1 o menos (incluyendo por ejemplo cualquiera de aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, y aproximadamente 9:1), y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunos ejemplos, el agente poco soluble en agua está recubierto con albúmina.
En algunas realizaciones, la composición comprende (1) nanopartículas que comprenden (en varias realizaciones que consisten en o que consisten esencialmente en) docetaxel y albúmina; y (2) un agente estabilizante como se reivindica, en la que la relación en peso de la albúmina al docetaxel en la composición es de aproximadamente 0,01:1 a aproximadamente 100:1, y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En algunas realizaciones, la composición comprende (1) nanopartículas que comprenden (en varias realizaciones que consisten en o que consisten esencialmente en) docetaxel y albúmina; y (2) un agente estabilizante como se reivindica, en la que la relación en peso de la albúmina y el docetaxel en la composición es de aproximadamente 18:1 o menos (incluyendo por ejemplo cualquiera de aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, y aproximadamente 9:1), y en la que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. En la presente invención, el docetaxel se recubre con albúmina. En algunas realizaciones, la composición carece sustancialmente (tal como carece) de tensioactivo. En algunas realizaciones, la composición comprende una suspensión acuosa estable de nanopartículas que comprenden docetaxel recubierto con albúmina, en la que la composición comprende además un agente estabilizante como se reivindica, en la que la relación en peso de la albúmina y el docetaxel en la composición es de aproximadamente 18:1 o menos (incluyendo por ejemplo de aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, y aproximadamente 9:1), y en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunas realizaciones, la composición comprende una composición seca (tal como liofilizada) que se puede reconstituir, resuspender, o rehidratar para formar en general una suspensión acuosa estable de nanopartículas que comprende docetaxel recubierto con albúmina, en la que la composición comprende además un agente estabilizante como se reivindica, en la que la relación en peso de la albúmina y el docetaxel en la composición es de aproximadamente 18:1 o menos (incluyendo por ejemplo cualquiera de aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, y aproximadamente 9:1); y en la que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunos ejemplos, el agente estabilizante es un agente quelante, tal como cualquiera de (y en algunos ejemplos seleccionado del grupo que consiste en) edetato, ácido málico,
pentetato, trometamina, derivados de estos, y mezclas de estos. En la presente invención, el agente estabilizante es un citrato o uno de sus derivados (tal como citrato de sodio). En algunas realizaciones, la composición comprende citrato de sodio y cloruro de sodio. En algunos ejemplos, el agente estabilizante es un edetato o uno de sus derivados (tal como EDTA). En algunos ejemplos, el agente estabilizante es gluconato de sodio. En algunos ejemplos, el agente estabilizante es pirofosfato de sodio.
En algunas realizaciones, las partículas (nanopartículas) en la composición tienen un diámetro promedio o medio no mayor de aproximadamente cualquiera de 1.000, 900, 800, 700, 600, 500, 400, 300, 200 y 100 nm. En algunas realizaciones, el diámetro promedio o medio de las partículas es entre aproximadamente 20 a aproximadamente 400 nm. En algunas realizaciones, el diámetro promedio o medio de las partículas es entre aproximadamente 40 a aproximadamente 200 nm. En algunas realizaciones, las partículas o gotitas se pueden esterilizar por filtración. Las composiciones descritas en la presente memoria pueden ser una suspensión acuosa estable del agente farmacéutico poco soluble en agua, tal como una suspensión acuosa estable del agente farmacéutico poco soluble en agua a una concentración de cualquiera de aproximadamente 0,1 a aproximadamente 100 mg/ml, incluyendo, por ejemplo, de aproximadamente 0,1 a aproximadamente 50 mg/ml, aproximadamente 0,1 a aproximadamente 20 mg/ml, aproximadamente 1 a aproximadamente 15 mg/ml, aproximadamente 1 a aproximadamente 10 mg/ml, aproximadamente 2 a aproximadamente 8 mg/ml, aproximadamente 4 a aproximadamente 6 mg/ml y aproximadamente 5 mg/ml. En algunas realizaciones, la concentración del agente farmacéutico poco soluble en agua es al menos aproximadamente cualquiera de 1 mg/ml, 1,3 mg/ml, 1,5 mg/ml, 2 mg/ml, 3 mg/ml, 4 mg/ml, 5 mg/ml, 6 mg/ml, 7 mg/ml, 8 mg/ml, 9 mg/ml, 10 mg/ml, 15 mg/ml, 20 mg/ml, 25 mg/ml, 30 mg/ml, 40 mg/ml y 50 mg/ml.
En algunas realizaciones, la composición es una composición seca (tal como liofilizada) que puede reconstituirse, resuspenderse, o rehidratarse generalmente para formar una suspensión acuosa estable del agente farmacéutico poco soluble en agua. En algunas realizaciones, la composición es una composición líquida (tal como acuosa) obtenida mediante la reconstitución o resuspensión de una composición seca. En algunas realizaciones, la composición es una composición líquida intermedia (tal como acuosa) que puede secarse (tal como liofilizarse). En algunas realizaciones, la composición es adecuada para administración parenteral (tal como intravenosa). En algunas realizaciones, la composición es adecuada para administración en múltiples dosis. En algunas realizaciones, la composición se puede esterilizar por filtración. En algunas realizaciones, la composición no causa efectos secundarios significativos en un individuo (tal como un ser humano) cuando se administra al individuo. En algunas realizaciones, las composiciones descritas en la presente memoria carecen sustancialmente (tal como carecen) de tensioactivos. Las composiciones que contienen un agente estabilizante descritas en la presente memoria pueden comprender además un azúcar (incluyendo, por ejemplo, sacarosa, manitol, fructosa, lactosa, maltosa y trehalosa) u otros auxiliares de liofilización o reconstitución.
En algunas realizaciones, la cantidad del agente estabilizante en la composición está por debajo del nivel que induce un efecto toxicológico (es decir, por encima de un nivel de toxicidad clínicamente aceptable) o está a un nivel en el que un efecto secundario potencial puede controlarse o tolerarse cuando la composición se administra a un individuo.
En otro aspecto, se proporcionan composiciones (tales como composición farmacéutica), que comprenden docetaxel como se reivindica, en las que el docetaxel usado para la preparación de la composición está en forma anhidra (por ejemplo el docetaxel puede ser anhidro antes de ser incorporado en la composición). Las composiciones que incluyen el uso de docetaxel anhidro se describen adicionalmente en una sección más adelante.
En algunos ejemplos, la composición comprende docetaxel y un polímero biocompatible (tal como una proteína vehicular, por ejemplo, albúmina), en la que el docetaxel usado para la preparación de la composición está en forma anhidra. En algunos ejemplos, la composición comprende partículas (tales como nanopartículas) que comprenden docetaxel y un polímero biocompatible (tal como una proteína vehicular, por ejemplo albúmina), en la que el docetaxel usado para la preparación de la composición está en forma anhidra.
En algunas realizaciones, la composición comprende nanopartículas que comprenden docetaxel y albúmina como se reivindica, en las que el docetaxel usado para la preparación de la composición está en forma anhidra. En algunas realizaciones, la relación en peso de albúmina y docetaxel en la composición es menor de aproximadamente 18:1, incluyendo, por ejemplo, cualquiera de aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, aproximadamente 9:1. En algunas realizaciones, el docetaxel se recubre con albúmina. En algunas realizaciones, las nanopartículas en la composición tienen un tamaño de partícula promedio o medio que no es mayor de aproximadamente 200 nm. En algunas realizaciones, las partículas en la composición se pueden esterilizar por filtración. En algunas realizaciones, las nanopartículas en las composiciones tienen dos o más de estas propiedades.
En algunas realizaciones, la composición como se reivindica comprende docetaxel y un tensioactivo, en la que el docetaxel usado para la preparación de la composición está en forma anhidra. En algunas realizaciones, el
tensioactivo usado en la preparación de la composición es anhidro. En algunas realizaciones, el tensioactivo es un polisorbato (tal como Tween 80). En algunas realizaciones, el tensioactivo es Cremophor.
Las composiciones preparadas con docetaxel anhidro pueden ser composiciones secas (tal como liofilizadas). En algunas realizaciones, la composición es una composición líquida (tal como acuosa) obtenida mediante la reconstitución o resuspensión de una composición seca. En algunas realizaciones, la composición es una composición líquida intermedia (tal como acuosa) que puede secarse (tal como liofilizarse).
También se proporcionan formas de dosificación unitarias de composiciones descritas en la presente memoria, artículos de fabricación que comprenden las composiciones o formas de dosificación unitarias inventivas en un envase adecuado (tal como viales o recipientes (incluyendo viales o recipientes sellados y viales o recipientes estériles)), y kits que comprenden las composiciones. La invención también proporciona métodos para hacer las composiciones como se describen en la presente memoria.
También se describen métodos para estabilizar un agente farmacéutico poco soluble en agua en una composición. En algunos ejemplos, hay un método para estabilizar un agente farmacéutico poco soluble en agua en una composición (tal como una composición de nanopartículas), que comprende combinar la composición (tal como una composición de nanopartículas) que comprende un agente farmacéutico poco soluble en agua con un agente estabilizante, en el que la composición resultante es estable en la misma condición en la que la composición es inestable antes de la adición del agente estabilizante. En algunos ejemplos, el método comprende además identificar y seleccionar una composición que es inestable en una o más condiciones. En algunos ejemplos, la composición para la selección comprende un agente farmacéutico poco soluble en agua y una proteína vehicular (tal como albúmina).
También se proporcionan lusos de las composiciones descritas en la presente memoria. Por ejemplo, en algunas realizaciones, se proporciona una composición de la presente invención para usar en un método para tratar el cáncer en un individuo (tal como un ser humano), que comprende administrar al individuo una cantidad efectiva de una composición que comprende un agente antineoplásico poco soluble en agua, albúmina, y un agente estabilizante como se reivindica, en el que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunas realizaciones, se proporciona una composición de la presente invención para usar en un método para tratar el cáncer en un individuo (tal como un ser humano), que comprende administrar al individuo una cantidad efectiva de una composición que comprende docetaxel, albúmina, y un agente estabilizante como se reivindica, en el que la estabilidad de la composición se aumenta comparada con la de la composición sin el agente estabilizante. En algunas realizaciones, la composición comprende nanopartículas que comprenden docetaxel y albúmina (tal como formulaciones de nanopartículas que comprenden albúmina de docetaxel o Nab-docetaxel). En algunas realizaciones, la composición comprende Nab-docetaxel y citrato. En algunas realizaciones, la composición comprende Nab-docetaxel, citrato y cloruro de sodio (tal como cloruro de sodio aproximadamente 200 mM y citrato de sodio aproximadamente 300 mM). En algunas realizaciones, el cáncer es cualquiera de: cáncer de próstata, cáncer de colon, cáncer de cabeza y cuello, cáncer de mama, cáncer de páncreas, cáncer de pulmón y cáncer de ovario. En algunas realizaciones, el cáncer es tumor sólido. En algunas realizaciones, la composición se administra al menos aproximadamente cualquiera de una vez cada tres semanas, una vez cada dos semanas, una vez a la semana, dos veces a la semana, tres veces a la semana, cuatro veces a la semana, cinco veces a la semana, seis veces a la semana, o diariamente. En algunas realizaciones, la composición se administra (con o sin descansos) durante al menos aproximadamente cualquiera de 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, o más meses. En algunas realizaciones, la composición se administra a través de cualquiera de ruta intravenosas, intraarterial, oral, tópica, o inhalatoria.
La referencia general a "las composiciones" o "composiciones" incluye y es aplicable a composiciones de la invención. La invención también proporciona composiciones farmacéuticas que comprenden los componentes proporcionados en la presente memoria.
La referencia al docetaxel en la presente memoria se aplica al docetaxel o sus derivados (o análogos) y por consiguiente la invención contempla e incluye ambas realizaciones. La referencia a "docetaxel" es para simplificar la descripción y es ilustrativa. Los derivados o análogos del docetaxel incluyen, pero no se limitan a compuestos que son estructuralmente similares al docetaxel o están en la misma clase química general que el docetaxel, p. ej., taxanos. En algunas realizaciones, el derivado o análogo de docetaxel retiene similares propiedades biológica, farmacológica, química y/o física (incluyendo, por ejemplo, funcionalidad) del docetaxel. Los ejemplos de derivados o análogos del docetaxel incluyen paclitaxel y ortataxel. Este principio de descripción se aplica a otros agentes descritos en la presente memoria tales como incluyendo, por ejemplo, agentes estabilizantes y agentes farmacéuticos poco solubles en agua (tales como taxano (que incluye paclitaxel, ortataxel, u otros taxanos), geldanamicina, 17-alil amino geldanamicina, tiocolchicina y sus dímeros, rapamicina, ciclosporina, epotilona, radicicol, y combretastatina).
Se entiende que los aspectos y realizaciones de la invención descritos en la presente memoria incluyen "que consiste" y/o "que consiste esencialmente en" aspectos y realizaciones.
Agentes estabilizantes
Varias composiciones descritas en la presente memoria comprenden un agente estabilizante. "Agente estabilizante" tal y como se usa en la presente memoria se refiere a un agente que aumenta la estabilidad de la composición comparada con una composición sin la adición del agente estabilizante. En algunas realizaciones, la estabilidad de la composición que contiene agente estabilizante es al menos aproximadamente 1,5x (incluyendo por ejemplo al menos aproximadamente cualquiera de 2x, 3x, 4x, 5x, 6, 7x, 8x, 9x, 10x, 15x, 20x, 25x, 30x, o más) mayor comparada con la de la composición sin el agente estabilizante.
Como se ha descrito anteriormente, la estabilidad de una composición puede evaluarse por la capacidad del agente farmacéutico poco soluble en agua de permanecer en forma no precipitada o no sedimentada (por ejemplo, sobre la base de una observación visual y/u observación microscópica) en una suspensión líquida durante un determinado periodo de tiempo. La estabilidad de una composición seca (tal como una liofilizada) puede evaluarse sobre la base del comportamiento de la suspensión líquida resultante de la reconstitución o rehidratación de la composición seca.
En algunas realizaciones, el agente estabilizante retrasa o previene la precipitación o sedimentación del agente farmacéutico poco soluble en agua en una suspensión líquida. En algunas realizaciones, el agente estabilizante retrasa o previene la cristalización del agente farmacéutico poco soluble en agua en la composición. En algunas realizaciones, cuando la composición comprende partículas de agentes poco solubles en agua, el agente estabilizante puede prevenir o retrasar los cambios en los tamaños de las partículas en la composición.
Los agentes estabilizantes son particularmente útiles para composiciones que de otra manera presentarían una inestabilidad significativa. Por ejemplo, en algunas realizaciones, la composición antes de la adición del agente estabilizante es estable durante menos de aproximadamente 24 horas (incluyendo, por ejemplo, menos de aproximadamente cualquiera de 12, 10, 8, 6, 4, o 2 horas). En algunas realizaciones, el agente farmacéutico poco soluble en agua en una suspensión líquida antes de la adición del estabilizante precipita o sedimenta en menos de aproximadamente 24 horas (incluyendo, por ejemplo, menos de aproximadamente cualquiera de 12, 10, 8, 6, 4, o 2 horas). En algunas realizaciones, la composición antes de la adición del agente estabilizante precipita o sedimenta en menos de aproximadamente 24 horas cuando la concentración del agente farmacéutico poco soluble en agua es mayor de aproximadamente 0,1 mg/ml (incluyendo, por ejemplo, mayor de aproximadamente cualquiera de 0,5 mg/ml, 1 mg/ml, 1,3 mg/ml, 1,5 mg/ml, 2 mg/ml, 3 mg/ml, 4 mg/ml, 5 mg/ml, o 10 mg/ml). En algunas realizaciones, la composición antes de la adición del agente estabilizante precipita o sedimenta después de la dilución de la composición para administración parental. La adición de agente estabilizante a estas composiciones permite que las composiciones permanezcan estables (por ejemplo, que no precipiten o sedimenten) en condiciones similares. De acuerdo con esto, en algunas realizaciones, se proporciona una composición que comprende un agente farmacéutico poco soluble en agua y un agente estabilizante, en el que la composición (tal como una composición de nanopartículas) es estable en la misma condición en la que la composición sin el agente estabilizante es inestable. En algunas realizaciones, el agente estabilizante retrasa o previene la precipitación o sedimentación del agente farmacéutico poco soluble en agua en una suspensión líquida de una composición en una condición en la que de otra manera el agente farmacéutico poco soluble en agua precipitaría o sedimentaría.
Los agentes estabilizantes adecuados incluyen, pero no están limitados a, citrato de sodio (todas las formas, 0,01-20 % p/v), pirofosfato de sodio (0,1-10 % p/v), EDtA (todas las formas, 0,01-20 %), pentetato (todas las formas, 0,01-20 %), gluconato de sodio (0,1-10 % p/v) y combinaciones adecuadas de estos. El porcentaje en peso (p/v) se refiere al porcentaje del agente estabilizante en una composición líquida, o, en el caso de una composición sólida, el porcentaje en peso (p/v) del agente estabilizante después de la reconstitución o rehidratación. El agente estabilizante debería usarse en una cantidad suficiente como para incrementar la estabilidad de la formulación. Preferiblemente, la cantidad usada del agente estabilizante proporcionará una composición estable que no muestra evidencia de precipitación o sedimentación durante al menos aproximadamente 8 horas, más preferiblemente al menos aproximadamente 24 horas después de la reconstitución o rehidratación, más preferiblemente durante al menos aproximadamente 48 horas, lo más preferiblemente durante al menos aproximadamente 72 horas.
En algunos ejemplos, el agente estabilizante es un agente quelante. Estos agentes quelantes son bien específicos para un ion metálico particular (tal como calcio, cinc, magnesio, etc.), o muestran un amplio espectro de especificidad para iones metálicos. En algunos ejemplos, el agente quelante es un polidentato. En algunos ejemplos, el agente quelante comprende uno o más grupos ácido carboxílico. En algunos ejemplos, el agente quelante no es deferoxamina. Los agentes quelantes adecuados incluyen, pero no están limitados a, edetato, citrato, ácido málico, pentetato, trometamina y derivados de estos.
Un agente estabilizante descrito en la presente memoria es un edetato, es decir, ácido etilendiaminotetraacético (EDTA) y derivados de este. Los edetatos adecuados incluyen edetato de disodio, edetato de trisodio, edetato de tetrasodio y edetato de calcio disodio. En algunos ejemplos, el edetato está presente en las composiciones en una concentración de aproximadamente 0,01 mg/ml a aproximadamente 200 mg/ml, incluyendo, por ejemplo, aproximadamente 0,05 mg/ml a aproximadamente 150 mg/ml, aproximadamente 0,1 mg/ml a aproximadamente 100 mg/ml, aproximadamente 0,2 a aproximadamente 50 mg/ml, aproximadamente 0,5 mg/ml a aproximadamente 20 mg/ml, aproximadamente 1 mg/ml a aproximadamente 10 mg/ml y aproximadamente 1 mg/ml a aproximadamente 5 mg/ml. En algunos ejemplos, la relación en peso del edetato y el agente farmacéutico poco soluble en agua (tal como docetaxel) en la composición es de aproximadamente 0,002:1 a aproximadamente 40:1, incluyendo por ejemplo de aproximadamente 0,01:1 a aproximadamente 30:1, aproximadamente 0,02:1 a aproximadamente 20:1,
aproximadamente 0,04:1 a aproximadamente 10:1, aproximadamente 0,1:1 a aproximadamente 4:1, aproximadamente 0,2:1 a aproximadamente 2:1, aproximadamente 0,2:1 a aproximadamente 1:1.
El agente estabilizante proporcionado en la presente memoria es citrato o un derivado de este (es decir, ácido cítrico o derivados de este), tal como citrato de sodio. Las concentraciones adecuadas de citrato incluyen, por ejemplo, de aproximadamente 0,1 mg/ml a aproximadamente 200 mg/ml, incluyendo por ejemplo cualquiera de aproximadamente 0,2 mg/ml a aproximadamente 100 mg/ml, aproximadamente 0,3 mg/ml a aproximadamente 50 mg/ml, aproximadamente 0,5 mg/ml a aproximadamente 10 mg/ml y aproximadamente 1 mg/ml a aproximadamente 5 mg/ml. En algunas realizaciones, la concentración de citrato es menor de aproximadamente 200 mg/ml, tal como menor de aproximadamente cualquiera de 100, 50, 30, 20, 10, 9, 8, 7, 6, 5, 4, 3, 2, 1, 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3, o 0,2 mg/ml. En algunas realizaciones, la relación en peso del citrato y el agente farmacéutico poco soluble en agua en la composición es de aproximadamente 0,02:1 a aproximadamente 40:1, incluyendo, por ejemplo, cualquiera de aproximadamente 0,04:1 a aproximadamente 20:1, aproximadamente 0,06:1 a aproximadamente 10:1, aproximadamente 0,1:1 a aproximadamente 2:1 y aproximadamente 0,2:1 a aproximadamente 1:1. En algunas realizaciones, la relación en peso del citrato y el agente farmacéutico poco soluble en agua en la composición es menor de aproximadamente cualquiera de 20:1, 10:1, 8:1, 5:1, 2:1, 1:1, 0,8:1, 0,5:1, 0,2:1 y 0,1:1.
Cualquier forma de citrato es aceptable para uso en la presente invención, e incluyen, por ejemplo, ácido cítrico y citrato de sodio. El citrato de sodio se prefiere particularmente. Cuando se utiliza el citrato de sodio, las concentraciones adecuadas incluyen de aproximadamente 1 a 600 mM. Cuando se utilizan citrato y cloruro de sodio, las concentraciones adecuadas incluyen de aproximadamente 1 a 600 mM y 1 a 1.000 mM, respectivamente. En algunas realizaciones, las concentraciones de citrato y cloruro de sodio son aproximadamente 50 a aproximadamente 200 mM y aproximadamente 300 a aproximadamente 500 mM, respectivamente. En algunas realizaciones, la composición comprende citrato aproximadamente 50 mM (tal como citrato de sodio) y cloruro de sodio aproximadamente 500 mM. En algunas realizaciones, la composición comprende citrato aproximadamente 200 mM (tal como citrato de sodio) y cloruro de sodio aproximadamente 300 mM. En algunas realizaciones, la composición es una composición seca (tal como liofilizada), en la que la relación en peso del citrato a docetaxel en la composición es aproximadamente 17:1 y, cuando está presente cloruro de sodio, la relación en peso del cloruro de sodio a docetaxel es aproximadamente 3,5:1. En otras realizaciones, el agente estabilizante no es un citrato (es decir, distinto de citrato).
El agente estabilizante también puede ser un pentetato (incluyendo pentetato de calcio y trisodio). En algunos ejemplos, la cantidad de pentetato está en una concentración de aproximadamente 0,01 mg/ml a aproximadamente 200 mg/ml, incluyendo, por ejemplo, cualquiera de aproximadamente 0,05 mg/ml a aproximadamente 150 mg/ml, aproximadamente 0,1 mg/ml a aproximadamente 100 mg/ml, aproximadamente 0,2 a aproximadamente 50 mg/ml, aproximadamente 0,5 mg/ml a aproximadamente 20 mg/ml, aproximadamente 1 mg/ml a aproximadamente 10 mg/ml y aproximadamente 1 mg/ml a aproximadamente 5 mg/ml. En algunos ejemplos, la relación en peso del pentetato y el agente farmacéutico poco soluble en agua (tal como docetaxel) en la composición es de aproximadamente 0,002:1 a aproximadamente 40:1, incluyendo, por ejemplo, cualquiera de aproximadamente 0,01:1 a aproximadamente 30:1, aproximadamente 0,02:1 a aproximadamente 20:1, aproximadamente 0,04:1 a aproximadamente 10:1, aproximadamente 0,1:1 a aproximadamente 4:1, aproximadamente 0,2:1 a aproximadamente 2:1, aproximadamente 0,2:1 a aproximadamente 1:1.
Otro agente estabilizante descrito en la presente memoria es trometamina. La trometamina tal y como se usa en la presente memoria, se refiere a 2-amino-2-hidroximetil-1,3-propanodiol, también conocido como TRIS. En algunos ejemplos, la trometamina está en una concentración de aproximadamente 0,1 mg/ml a aproximadamente 100 mg/ml, incluyendo, por ejemplo, de aproximadamente 0,5 mg/ml a aproximadamente 50 mg/ml, aproximadamente 1 mg/ml a aproximadamente 10 mg/ml y aproximadamente 2 mg/ml a aproximadamente 5 mg/ml. En algunos ejemplos, la relación en peso de la trometamina y el agente farmacéutico poco soluble en agua en la composición es de aproximadamente 0,02:1 a aproximadamente 20:1, incluyendo por ejemplo de 0,1:1 a aproximadamente 10:1, aproximadamente 0,2:1 a aproximadamente 2:1, y aproximadamente 0,4:1 a aproximadamente 1:1.
Otros agentes estabilizantes quelantes de metales adecuados y su cantidad ejemplar incluyen, pero no están limitados a, sorbato de potasio (0,5 mg/ml), ascorbato de sodio (1mg/ml), sulfoxilato de formaldehído de sodio (0,1 mg/ml) y monotiolglicerol (5 mg/ml).
En algunos ejemplos, el agente estabilizante es pirofosfato de sodio. La concentración adecuada de pirofosfato de sodio incluye cualquiera de aproximadamente 0,1 a aproximadamente 10 % (p/v), aproximadamente 0,5 a aproximadamente 5 % y aproximadamente 1 a aproximadamente 2 %. En algunos ejemplos, la relación en peso del pirofosfato de sodio y el agente farmacéutico poco soluble en agua en la composición es cualquiera de aproximadamente 0,2:1 a aproximadamente 20:1, aproximadamente 1:1 a aproximadamente 10:1, aproximadamente 2:1 a aproximadamente 4:1.
En algunos ejemplos, el agente estabilizante es gluconato de sodio. La concentración adecuada de gluconato de sodio incluye cualquiera de aproximadamente 0,1 a aproximadamente 10 % (p/v), aproximadamente 0,5 a aproximadamente 5 % y aproximadamente 1 a aproximadamente 2 %. En algunos ejemplos, la relación en peso del gluconato de sodio y el agente farmacéutico poco soluble en agua en la composición es cualquiera de
aproximadamente 0,2:1 a aproximadamente 20:1, aproximadamente 1:1 a aproximadamente 10:1, aproximadamente 2:1 a aproximadamente 4:1.
En algunos ejemplos, las composiciones descritas en la presente memoria comprenden al menos dos (incluyendo, por ejemplo, al menos cualquiera de 2, 3, 4, 5, 6, 7, 8, 9, o 10) agentes estabilizantes diferentes (tales como los agentes estabilizantes descritos en la presente memoria).
Agentes farmacéuticos poco solubles en agua
Las composiciones descritas en la presente memoria comprenden agentes farmacéuticos poco solubles en agua. Por ejemplo, la solubilidad en agua del agente poco soluble en agua a 20-25°C puede ser menor de aproximadamente 10 mg/ml, incluyendo por ejemplo menor de aproximadamente cualquiera de 5, 2, 1, 0,5, 0,2, 0,1, 0,05, 0,02, y 0,01 mg/ml.
Los agentes farmacéuticos poco solubles en agua descritos en la presente memoria incluyen agentes farmacéuticamente activos poco solubles en agua, agentes de diagnóstico, agentes de valor nutricional y semejantes. Los agentes farmacéuticos poco solubles en agua pueden ser, por ejemplo, analgésicos/antipiréticos, anestésicos, antiasmáticos, antibióticos, antidepresivos, antidiabéticos, agentes antifúngicos, agentes antihipertensivos, antiinflamatorios, antineoplásicos, agentes antiansiedad, agentes inmunosupresores, agentes antimigraña, sedantes, agentes antianginas, agentes antipsicóticos, agente antimaníacos, antiarrítmicos, agentes antiartríticos, agentes antigota, anticoagulantes, agentes trombolíticos, agentes antifibrinolíticos, agentes hemorreológicos, agentes antiplaquetarios, anticonvulsivos, agentes antiparkinsonianos, antihistamínicos antipruríticos, agentes útiles para la regulación del calcio, agentes antibacterianos, agentes antivíricos,,antimicrobianos, antiinfecciosos, broncodilatadores, hormonas, agentes hipoglucémicos, agentes hipolipidémicos, agentes antiúlcera/antireflujo, antináuseas/antiheméticos, vitaminas solubles en aceite (p. ej., vitaminas A, D, E, K, y semejantes).
En algunos ejemplos, el agente farmacéutico poco soluble en agua es un agente antineoplásico. En algunos ejemplos, el agente farmacéutico poco soluble en agua es un agente quimioterapéutico.
Los agentes farmacéuticos poco solubles en agua adecuados incluyen, pero no se limitan a, taxanos (tales como paclitaxel, docetaxel, ortataxel y otros taxanos), epotilonas, camptotecinas, colchicinas, geladanamicinas, amiodarones, hormonas tiroideas, amfotericina, corticosteroides, propofol, melatonina, ciclosporina, rapamicina (sirolimus) y derivados, tacrolimus, ácidos micofenólicos, ifosfamida, vinorelbina, vancomicina, gemcitabina, SU5416, tiotepá, bleomicina, agentes de radiocontraste para diagnóstico, y derivados de estos. Otros agentes farmacéuticos poco solubles en agua que son útiles se describen, por ejemplo, en las patentes de EEUU N° 5.916.596, 6.096.331, 6.749.868, y 6.537.539. Ejemplos adicionales de agentes farmacéuticos poco solubles en agua incluyen los compuestos que son poco solubles en agua y que se citan en el "Therapeutic Category and Biological Activity Index" de The Merck Index (12a Edición, 1996).
En algunos ejemplos, el agente farmacéutico poco soluble en agua es cualquiera de (y en algunas realizaciones se selecciona del grupo que consiste en) paclitaxel, ortataxel u otro taxano o análogo de taxano, 17-alil amino geldanamicina (17-AAG), geldanamicina 18-derivatizada, camptotecina, propofol, amiodarona, ciclosporina, epotilona, radicicol, combretastatina, rapamicina, anfotericina, liotironina, epotilona, colchicina, tiocolchicina y sus dímeros, hormona tiroidea, péptido intestinal vasoactiva, corticosteroides, melatonina, tacrolimus, ácidos micofenólicos, epotilonas, radicicoles, combretastatinas, y análogos o derivados de estos. En algunos ejemplos, el agente farmacéutico poco soluble en agua es cualquiera de (y en algunas realizaciones se selecciona del grupo que consiste en) paclitaxel, ortataxel u otros taxanos, geldanamicina, 17-alil amino geldanamicina, tiocolquicina y sus dímeros, rapamicina, ciclosporina, epotilona, radicicol, y combretastatina. En algunos ejemplos, el agente farmacéutico poco soluble en agua es rapamicina. En algunos ejemplos, el agente farmacéutico poco soluble en agua es 17-AAG.En algunos ejemplos, el agente farmacéutico poco soluble en agua es un dímero de tiocolchicina (tal como IDN5404).
En algunos ejemplos, el agente farmacéutico poco soluble en agua es un taxano o derivado de este, que incluye, pero no se limita a, paclitaxel, y IDN5109 (ortataxel), o uno de sus derivados. En algunos ejemplos, la composición comprende un taxano no cristalino y/o amorfo (tal como paclitaxel o uno de sus derivados). En algunas realizaciones, la composición se prepara usando un docetaxel anhidro o uno de sus derivados.
En la presente invención, el agente farmacéutico poco soluble en agua es docetaxel o uno de sus derivados. En algunas realizaciones, el docetaxel en la composición no es cristalino o es amorfo. En algunas realizaciones, el docetaxel está en una cualquiera o más de las siguientes formas: formas de anhidrato, hemihidrato, dihidrato, y trihidrato. Se ha mostrado que el docetaxel anhidro produce una formulación más estable que las preparadas con un docetaxel hidratado tal como trihidrato o hemihidrato de docetaxel, y es particularmente útil para la preparación de las composiciones de docetaxel descritas en la presente memoria.
Polímeros biocompatibles y proteínas vehiculares
Las composiciones descritas en la presente memoria también pueden comprender polímeros biocompatibles, tales
como proteínas vehiculares descritas adicionalmente en la presente memoria.
Tal y como se usa en la presente memoria, el término "biocompatible" describe una sustancia que no altera o afecta apreciablemente de ninguna manera adversa, el sistema biológico en el que se introduce. Un polímero biocompatible incluye materiales biocompatibles naturales o sintéticos tales como proteínas, polinucleótidos, polisacáridos (p. ej., almidón, celulosa, dextranos, alginatos, quitosán, pectina, ácido hialurónico y semejantes) y lípidos. Los polímeros biocompatibles adecuados incluyen, por ejemplo, proteínas naturales o sintéticas tales como albúmina, insulina, hemoglobina, lisozima, inmunoglobulinas, a-2-macroglobulina, fibronectina, vitronectina, fibrinógeno, caseína y semejantes, así como combinaciones de cualesquiera dos o más de estas. Los polímeros sintéticos incluyen, por ejemplo, polialquilenglicoles (p. ej., cadena lineal o ramificada), poli(alcohol vinílico), poliacrilatos, poli(metacrilato de hidroxietilo), poli(ácido acrílico), polietiloxazolina, poliacrilamidas; poliisopropilacrilamidas, polivinilpirrolidona, polilactida/glicólido y semejantes, y combinaciones de estos.
El término "proteínas" se refiere a polipéptidos o polímeros de aminoácidos de cualquier longitud (incluyendo de longitud completa o fragmentos), que pueden ser lineales o ramificados, que comprenden aminoácidos modificados y/o que pueden estar interrumpidos por no aminoácidos. El término también engloba un polímero de aminoácidos que se ha modificado de forma natural o por intervención; por ejemplo, formación de puente disulfuro, glicosilación, lipidación, acetilación, fosforilación, o cualquier otra manipulación o modificación. También se incluyen en este término, por ejemplo, polipéptidos que contienen uno o más análogos de un aminoácido (incluyendo, por ejemplo, aminoácidos no naturales, etc.), así como otras modificaciones conocidas en la técnica. Las proteínas descritas en la presente memoria pueden ser naturales, es decir, se obtienen o derivan de una fuente natural (tal como la sangre), o pueden sintetizarse (tal como sintetizarse químicamente o sintetizarse por técnicas de ADN recombinante).
Los ejemplos de proteínas adecuadas incluyen proteínas que se encuentran de forma natural en la sangre o plasma, que incluyen, pero no están limitadas a, albúmina, inmunoglobulina incluyendo IgA, lipoproteínas, apolipoproteína B, a-ácido glicoproteína, p-2-macroglobulina, tiroglobulina, transferrina, fibronectina, factor VII, factor VIII, factor IX, factor X y semejantes. En algunos ejemplos, la proteína vehicular es una proteína que no se encuentra en la sangre, tal como caseína, a-lactalbúmina, p-lactoglobulina. Las proteínas pueden tener un origen natural o pueden prepararse sintéticamente. En la presente invención, la proteína es albúmina, tal como HSA. La HSA es una proteína globular altamente soluble con Mr 65K y consiste en 585 aminoácidos. La HSA es la proteína más abundante en el plasma y es responsable del 70-80 % de la presión osmótica coloidal del plasma humano. La secuencia de aminoácidos de la HSA contiene un total de 17 puentes disulfuro, un tiol libre (Cys 34) y un único triptófano (Trp 214). El uso intravenoso de disolución de HSA se ha indicado para la prevención y tratamiento de choque hipovolémico (véase, p. ej., Tullis, JAMA, 237, 355-360, 460-463, (1977)) y Houser et al., Surgery, Gynecology and Obstetrics, 150, 811-816 (1980)) y conjuntamente con transfusión de intercambio en el tratamiento de hiperbilirrubinemia neonatal (véase, p. ej., Finlayson, Seminars in Thrombosis and Hemostasis, 6, 85-120, (1980)). Se contemplan otras albúminas, tal como albúmina de suero bovino. El uso de dichas albúminas no humanas podría ser apropiado, por ejemplo, en el contexto del uso de estas composiciones en un mamífero no humano, tal como los animales veterinarios (incluyendo mascotas domésticas y animales agrícolas).
La albúmina de suero humano (HSA) tiene múltiples sitios de unión hidrofóbicos (un total de ocho para ácidos grasos, un ligando endógeno de la HSA) y se une a un conjunto diverso de agentes farmacéuticos, especialmente compuestos hidrofóbicos neutros y cargados negativamente (Goodman et al., The Pharmacological Basis of Therapeutics, 9a ed, McGraw-Hill Nueva York (1996)). Se han propuesto dos sitios de unión de alta afinidad en los subdominios IIA y IIIA de HSA, que son bolsillos hidrofóbicos altamente elongados con residuos cargados de lisina y arginina cerca de la superficie que funcionan como puntos de unión para características de ligandos polares (véase, p. ej., Fehske et al., Biochem. Pharmcol., 30, 687-92 (1981), Vorum, Dan. Med. Bull., 46, 379-99 (1999), Kragh-Hansen, Dan. Med. Bull., 1441, 131-40 (1990), Curry et al., Nat. Struct. Biol, 5, 827-35 (1998), Sugio et al., Protein. Eng, 12, 439-46 (1999), He et al., Nature, 358, 209-15 (1992) y Carter et al., Adv. Protein. Chem, 45, 153-203 (1994)). Se ha mostrado que el paclitaxel y propofol se unen a HSA (véase, p. ej., Paal et al., Eur. J. Biochem., 268(7), 2187-91 (2001), Purcell et al., Biochim. Biophys. Acta, 1478(1), 61-8 (2000), Altmayer et al., Arzneimittelforschung, 45, 1053-6 (1995) y Garrido et al., Rev. Esp. Anestestiol. Reanim., 41, 308-12 (1994)). Además, se ha mostrado que el docetaxel se une a proteínas de plasma humano (véase, p. ej., Urien et al., Invest. New Drugs, 14(2), 147-51 (1996)).
Para proporcionar un ejemplo, las proteínas vehiculares se describen adicionalmente más adelante. Se entiende que esta descripción se aplica generalmente a polímeros biocompatibles.
La proteína vehicular (tal como albúmina) en la composición sirve generalmente como un vehículo para el agente farmacéutico poco soluble en agua, es decir, la proteína vehicular en la composición hace que el agente farmacéutico poco soluble en agua sea más fácil de suspender en un medio acuoso o ayuda a mantener la suspensión comparado con composiciones que no comprenden una proteína vehicular. Esto puede evitar el uso de disolventes tóxicos para solubilizar el agente farmacéutico poco soluble en agua y, de esta manera, puede reducir uno o más efectos secundarios de la administración del agente farmacéutico poco soluble en agua en un individuo (tal como un ser humano). Así, en algunas realizaciones, la composición proporcionada en la presente memoria carece sustancialmente (tal como carece) de tensioactivos (tal como Tween 20). Una composición "carece sustancialmente de tensioactivo" si la cantidad de tensioactivo en la composición no es suficiente para causar uno o
más efectos secundarios en un individuo cuando la composición se administra al individuo.
En algunos ejemplos, la proteína vehicular está asociada con el agente farmacéutico poco soluble en agua, es decir, la composición comprende el agente farmacéutico poco soluble en agua asociado con la proteína vehicular. "Asociación" o "asociado" se usa en la presente memoria en un sentido general y se refiere a que la proteína vehicular afecta el comportamiento y/o propiedad del agente farmacéutico poco soluble en agua en una composición acuosa. Por ejemplo, se considera que la proteína vehicular y el agente farmacéutico poco soluble en agua están "asociados" si la proteína vehicular hace que el agente farmacéutico poco soluble en agua se pueda suspender más fácilmente en un medio acuoso comparado con una composición sin la proteína vehicular. Como otro ejemplo, la proteína vehicular y el agente farmacéutico poco soluble en agua están asociados si la proteína vehicular estabiliza al agente farmacéutico poco soluble en agua en una suspensión acuosa. Por ejemplo, la proteína vehicular y el agente farmacéutico poco soluble en agua pueden estar presentes en una partícula o una nanopartícula, que se describen adicionalmente en la presente memoria.
Un agente farmacéutico poco soluble en agua se "estabiliza" por una proteína vehicular en una suspensión acuosa si permanece suspendido en un medio acuoso (tal como sin precipitación o sedimentación visible) durante un periodo de tiempo prolongado, tal como durante al menos aproximadamente cualquiera de 0,1, 0,2, 0,25, 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 24, 36, 48, 60, o 72 horas. La suspensión es generalmente, pero no necesariamente, adecuada para administración a un individuo (tal como un ser humano). Como se ha descrito anteriormente, la estabilidad de la suspensión se evalúa, en algunas realizaciones, a temperatura ambiente (tal como 20-25 °C) o condiciones refrigeradas (tal como 4 °C). La estabilidad también puede evaluarse en condiciones de ensayo aceleradas, tal como a una temperatura que es mayor de aproximadamente 40 °C. Como se ha descrito anteriormente, la estabilidad de la suspensión puede aumentarse adicionalmente por la adición de los agentes estabilizantes descritos en la presente memoria.
La proteína vehicular y el agente farmacéutico poco soluble en agua en la composición pueden estar asociados de varias maneras. Por ejemplo, en algunos ejemplos, la proteína vehicular está mezclada con el agente farmacéutico poco soluble en agua. En la presente invención, la proteína vehicular encapsula o atrapa al agente farmacéutico poco soluble en agua. En algunas realizaciones, la proteína vehicular está unida (tal como unida no covalentemente) al agente farmacéutico poco soluble en agua. En algunas realizaciones, la composición puede presentar uno o más de los aspectos anteriores.
En la presente invención, la composición comprende nanopartículas que comprenden (en varias realizaciones que consisten esencialmente en) un agente farmacéutico poco soluble en agua y una proteína vehicular como se reivindica. Cuando el agente farmacéutico poco soluble en agua está en forma líquida, las partículas o nanopartículas también se refieren como gotitas o nanogotitas. En la presente invención, el agente poco soluble en agua se recubre con la proteína vehicular. Las partículas (tales como nanopartículas) de agentes farmacéuticos poco solubles en agua se han descrito, por ejemplo, en las Pat. de EEUU Nos. 5.916.596; 6.506.405; y 6.537.579 y también en la Pub. de Solic. de Pat. de EEUU No. 2005/0004002A1.
En algunas realizaciones, la composición comprende partículas (nanopartículas) con un diámetro promedio o medio que no es mayor de aproximadamente 1.000 nanómetros (nm), tal como menor de aproximadamente cualquiera de 900, 800, 700, 600, 500, 400, 300, 200 y 100 nm. En algunas realizaciones, el diámetro promedio o medio de las partículas no es mayor de aproximadamente 200 nm. En algunas realizaciones, el diámetro promedio o medio de las partículas es entre aproximadamente 20 a aproximadamente 400 nm. En algunas realizaciones, el diámetro promedio o medio de las partículas es entre aproximadamente 40 a aproximadamente 200 nm. En algunas realizaciones, las nanopartículas en la composición tienen un tamaño de partícula promedio o medio que no es mayor de aproximadamente 200 nm. En algunas realizaciones, las partículas se pueden esterilizar por filtración.
Las partículas (nanopartículas) proporcionadas en la presente memoria pueden estar presentes en una formulación seca (tal como composición liofilizada) o suspendidas en un medio biocompatible. Los medios biocompatibles adecuados incluyen, pero no están limitados a, agua, medio acuoso tamponado, disolución salina, disolución salina tamponada, opcionalmente disoluciones tamponadas de aminoácidos, opcionalmente disoluciones tamponadas de proteínas, opcionalmente disoluciones tamponadas de azúcares, opcionalmente disoluciones tamponadas de vitaminas, opcionalmente disoluciones tamponadas de polímeros sintéticos, emulsiones que contienen lípidos y semejantes.
La cantidad de proteína vehicular en la composición descrita en la presente memoria variará dependiendo del agente farmacéutico poco soluble en agua y otros componentes en la composición. En algunas realizaciones, la composición comprende una proteína vehicular en una cantidad que es suficiente para estabilizar el agente farmacéutico poco soluble en agua en una suspensión acuosa, por ejemplo, en la forma de una suspensión coloidal estable (tal como una suspensión estable de nanopartículas). En algunas realizaciones, la proteína vehicular está en una cantidad que reduce la tasa de sedimentación del agente farmacéutico poco soluble en agua en un medio acuoso. Para composiciones que contienen partículas, la cantidad de la proteína vehicular también depende del tamaño y densidad de las partículas del agente farmacéutico poco soluble en agua.
En algunas realizaciones, la proteína vehicular está presente en una cantidad que es suficiente para estabilizar el
agente farmacéutico poco soluble en agua en una suspensión acuosa a una determinada concentración. Por ejemplo, la concentración del agente farmacéutico poco soluble en agua en la composición es aproximadamente 0,1 a aproximadamente 100 mg/ml, incluyendo, por ejemplo, cualquiera de aproximadamente 0,1 a aproximadamente 50 mg/ml, aproximadamente 0,1 a aproximadamente 20 mg/ml, aproximadamente 1 a aproximadamente 10 mg/ml, aproximadamente 2 a aproximadamente 8 mg/ml y aproximadamente 4 a aproximadamente 6 mg/ml. En algunas realizaciones, la concentración del agente farmacéutico poco soluble en agua es al menos aproximadamente cualquiera de 1,3 mg/ml, 1,5 mg/ml, 2 mg/ml, 3 mg/ml, 4 mg/ml, 5 mg/ml, 6 mg/ml, 7 mg/ml, 8 mg/ml, 9 mg/ml, 10 mg/ml, 15 mg/ml, 20 mg/ml, 25 mg/ml, 30 mg/ml, 40 mg/ml y 50 mg/ml. En algunas realizaciones, la proteína vehicular está presente en una cantidad que evita el uso de tensioactivos (tales como Tween 80 o Cremophor), de manera que la composición carece o carece sustancialmente de tensioactivo (tal como Tween 80 o Cremophor).
En algunas realizaciones, la composición, en forma líquida, comprende de aproximadamente 0,1 % a aproximadamente 50 % (p/v) (p. ej., aproximadamente 0,5 % (p/v), aproximadamente 5 % (p/v), aproximadamente 10 % (p/v), aproximadamente 15 % (p/v), aproximadamente 20 % (p/v), aproximadamente 30 % (p/v), aproximadamente 40 % (p/v), aproximadamente 50 % (p/v)) de la proteína vehicular. En algunas realizaciones, la composición, en forma líquida, comprende aproximadamente 0,5 % a aproximadamente 5 % (p/v) de la proteína vehicular.
En algunas realizaciones, la relación en peso de albúmina al agente farmacéutico poco soluble en agua es tal que una cantidad suficiente del agente farmacéutico poco soluble en agua se une a, o es transportada por, la célula. Aunque la relación en peso de la proteína vehicular al agente farmacéutico tendrá que optimizarse para diferentes combinaciones de proteína vehicular y fármaco, generalmente la relación en peso de albúmina a agente farmacéutico (p/p) es de aproximadamente 0,01:1 a aproximadamente 100:1, incluyendo, por ejemplo, cualquiera de aproximadamente 0,02:1 a aproximadamente 50:1, aproximadamente 0,05:1 a aproximadamente 20:1, aproximadamente 0,1:1 a aproximadamente 20:1, aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, aproximadamente 5:1 a aproximadamente 9:1 y aproximadamente 9:1. En algunas realizaciones, la relación en peso de proteína vehicular a agente farmacéutico es aproximadamente cualquiera de 18:1 o menos, tal como aproximadamente cualquiera de 15:1 o menos, 14:1 o menos, 13:1 o menos, 12:1 o menos, 11:1 o menos, 10:1 o menos, 9:1 o menos, 8:1 o menos, 7:1 o menos, 6:1 o menos, 5:1 o menos, 4:1 o menos y 3:1 o menos.
En algunas realizaciones, la proteína vehicular permite que la composición se administre a un individuo (tal como un ser humano) sin efectos secundarios significativos. En algunas realizaciones, la proteína vehicular (albúmina) está en una cantidad que es efectiva para reducir uno o más efectos secundarios de la administración del agente farmacéutico poco soluble en agua a un ser humano. El término "reducir uno o más efectos secundarios de la administración del agente farmacéutico poco soluble en agua" se refiere a una reducción, alivio, eliminación, o evitación de uno o más efectos indeseables causados por el agente farmacéutico poco soluble en agua, así como los efectos secundarios causados por los vehículos de la administración (tales como disolventes que hacen que los agentes farmacéuticos poco solubles en agua sean adecuados para inyección) usados para administrar el agente farmacéutico poco soluble en agua. Dichos efectos secundarios incluyen, por ejemplo, mielosupresión, neurotoxicidad, hipersensibilidad, inflamación, irritación venosa, flebitis, dolor, irritación cutánea, neuropatía periférica, fiebre neutropénica, reacción anafiláctica, trombosis venosa, extravasación y combinaciones de estos. Estos efectos secundarios, sin embargo, son meramente ejemplares y pueden reducirse otros efectos secundarios, o combinación de efectos secundarios, asociados con varios agentes farmacéuticos.
En algunos ejemplos, la composición comprende partículas (tales como nanopartículas) que comprenden (en varios ejemplos que consisten en o que consisten esencialmente en) un agente farmacéutico poco soluble en agua y una albúmina, en el que la relación en peso de la albúmina al agente farmacéutico poco soluble en agua en la composición (p/p) es de aproximadamente 0,01:1 a aproximadamente 100:1, incluyendo, por ejemplo, cualquiera de aproximadamente 0,02:1 a aproximadamente 50:1, aproximadamente 0,05:1 a aproximadamente 20:1, aproximadamente 0,1:1 a aproximadamente 20:1, aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1, aproximadamente 4:1 a aproximadamente 10:1, aproximadamente 5:1 a aproximadamente 9:1 y aproximadamente 9:1. En algunos ejemplos, la relación en peso de proteína vehicular al agente farmacéutico es aproximadamente cualquiera de 18:1 o menos, 15:1 o menos, 14:1 o menos, 13:1 o menos. 12:1 o menos, 11:1 o menos, 10:1 o menos, 9:1 o menos, 8:1 o menos, 7:1 o menos, 6:1 o menos, 5:1 o menos, 4:1 o menos, y 3:1 o menos. En algunos ejemplos, el agente farmacéutico poco soluble en agua está recubierto con la albúmina. En algunos ejemplos, las partículas (tales como nanopartículas) que comprenden un agente farmacéutico poco soluble en agua y albúmina están suspendidas en un medio acuoso (tal como un medio acuoso que contiene albúmina). Por ejemplo, la composición puede ser una suspensión coloidal de partículas (tales como nanopartículas) del agente farmacéutico poco soluble en agua. En algunos ejemplos, la composición es una composición seca (tal como liofilizada) que puede reconstituirse o resuspenderse para formar una suspensión estable de partículas o descrita en la presente memoria. La concentración del agente farmacéutico poco soluble en agua en la composición líquida o composición reconstituida puede ser diluida (0,1 mg/ml) o concentrada (100 mg/ml), incluyendo por ejemplo cualquiera de aproximadamente 0,1 a aproximadamente 50 mg/ml, aproximadamente 0,1 a aproximadamente 20 mg/ml, aproximadamente 1 a aproximadamente 10 mg/ml, aproximadamente 2 mg/ml a aproximadamente 8 mg/ml, aproximadamente 4 a
aproximadamente 6 mg/ml y 5 mg/ml. En algunos ejemplos, la concentración del agente farmacéutico poco soluble en agua (tal como docetaxel) es mayor de aproximadamente 0,1 mg/ml. En algunos ejemplos, la concentración del agente farmacéutico poco soluble en agua es mayor de aproximadamente cualquiera de 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, o 50 mg/ml. En algunos ejemplos, el agente farmacéutico poco soluble en agua es un taxano o un derivado de este (tal como docetaxel o un derivado de este).
En algunas realizaciones, la composición comprende nanopartículas que comprenden docetaxel como se reivindica, tales como nanopartículas con un diámetro promedio o medio de entre aproximadamente 20 a aproximadamente 400 nm. En algunas realizaciones, las partículas tienen un diámetro promedio o medio de entre aproximadamente 40 a aproximadamente 200 nm. En la presente invención, la composición comprende nanopartículas que comprenden (en varias realizaciones que consisten esencialmente en) docetaxel recubierto con albúmina. En algunas realizaciones, la relación en peso de la albúmina al docetaxel (p/p) en la composición es de aproximadamente 0,01:1 a aproximadamente 100:1, incluyendo por ejemplo cualquiera de aproximadamente 0,02:1 a aproximadamente 50:1, aproximadamente 0,05:1 a aproximadamente 20:1, aproximadamente 1:1 a aproximadamente 18:1, aproximadamente 2:1 a aproximadamente 15:1, aproximadamente 3:1 a aproximadamente 12:1. En algunas realizaciones, la relación de la albúmina al docetaxel (p/p) es aproximadamente cualquiera de 18:1 o menos, 15:1 o menos, 14:1 o menos, 13:1 o menos, 12:1 o menos, 10:1 o menos, 9:1 o menos, 8:1 o menos, 7:1 o menos, 6:1 o menos, 5:1 o menos, 4:1 o menos, y 3:1 o menos.
En algunas realizaciones, las partículas (nanopartículas) que comprenden docetaxel y albúmina se suspenden en un medio acuoso (tal como un medio acuoso que contiene la albúmina). Por ejemplo, la composición puede ser una suspensión coloidal de las partículas (nanopartículas) que contienen docetaxel. En algunas realizaciones, la composición es una composición seca (tal como composición liofilizada) que puede reconstituirse para formar una suspensión acuosa de partículas que contienen docetaxel. En algunas realizaciones, la concentración del docetaxel en la composición es entre aproximadamente 0,1 mg/ml y aproximadamente 100 mg/ml, incluyendo, por ejemplo, cualquiera de aproximadamente 0,1 a aproximadamente 50 mg/ml, aproximadamente 0,1 a aproximadamente 20 mg/ml, aproximadamente 1 a aproximadamente 10 mg/ml, aproximadamente 2 a aproximadamente 8 mg/ml, aproximadamente 4 a aproximadamente 6 mg/ml y aproximadamente 5 mg/ml. En algunas realizaciones, la concentración de docetaxel es al menos aproximadamente cualquiera de 1,3 mg/ml, 1,5 mg/ml, 2 mg/ml, 3 mg/ml, 4 mg/ml, 5 mg/ml, 6 mg/ml, 7 mg/ml, 8 mg/ml, 9 mg/ml, 10 mg/ml, 15 mg/ml, 20 mg/ml, 25 mg/ml, 30 mg/ml, 40 mg/ml, y 50 mg/ml.
Docetaxel anhidro
Además del uso de agentes estabilizantes descritos en la presente memoria (tales como citrato de sodio y citrato de sodio/cloruro de sodio), se ha encontrado sorprendentemente que el uso de docetaxel anhidro da como resultado una formulación más estable que la que puede prepararse con un docetaxel hidratado tal como trihidrato o hemihidrato de docetaxel. Las formulaciones de docetaxel anhidro de la presente invención mejoran adicionalmente la estabilidad de las suspensiones de nanopartículas acuosas de manera que la estabilidad de las suspensiones, bien antes o después de la liofilización, supera 1 día. Además, los beneficios de la estabilidad añadida del docetaxel anhidro también se extienden a formulaciones convencionales tales como una formulación en Tween 80, Cremophor u otros tensioactivos conocidos.
Así, según la presente invención, el docetaxel puede disolverse en un disolvente o disolventes farmacéuticamente aceptables a una concentración final en el intervalo de aproximadamente 1-99 % v/v, más preferiblemente en el intervalo de aproximadamente 5-25 % v/v. Los disolventes incluyen, por ejemplo, disolventes clorados, acetato de etilo, etanol, tetrahidrofurano, dioxano, acetonitrilo, acetona, dimetilsulfóxido, dimetilformamida, metilpirrolidinona, aceites tales como aceite de soja, aceite de alazor y otros aceites inyectables y semejantes.
En algunos ejemplos, hay una composición que comprende docetaxel, en la que el docetaxel usado para preparar la composición está en una forma anhidra. En algunos ejemplos, hay una composición que comprende docetaxel, en la que al menos algo del docetaxel en la composición está en una forma anhidra. Por ejemplo, en algunos ejemplos, al menos aproximadamente 10 % (tal como al menos aproximadamente cualquiera de 20 %, 30 %, 40 %, y 50 %) del docetaxel en la composición está en una forma anhidra. En algunos ejemplos, la composición comprende además un agente estabilizante (tal como el agente estabilizante descrito en la presente memoria).
En algunos ejemplos, la composición comprende docetaxel y un polímero biocompatible (tal como una proteína vehicular descrita en la presente memoria, por ejemplo albúmina), en la que el docetaxel usado para la preparación de la composición está en una forma anhidra. En algunas realizaciones, la composición comprende docetaxel, albúmina y un agente estabilizante como se reivindica, en la que el docetaxel usado para la preparación de la composición está en forma anhidra. En algunas realizaciones, la composición carece sustancialmente (tal como carece) de tensioactivos. En algunas realizaciones, la composición comprende tensioactivo.
En algunas realizaciones, la invención proporciona una composición que comprende docetaxel y albúmina como se reivindica, en la que al menos algo del docetaxel en la composición está en una forma anhidra. Por ejemplo, en algunas realizaciones, al menos aproximadamente 10 % (tal como al menos aproximadamente cualquiera de 20 %, 30 %, 40 %, y 50 %) del docetaxel en la composición está en una forma anhidra.
En algunas realizaciones, la invención proporciona una composición que comprende docetaxel como se reivindica y un tensioactivo (tal como tensioactivo anhidro), en la que el docetaxel usado para la preparación de la composición está en una forma anhidra. En algunas realizaciones, el tensioactivo usado para preparar la composición está en una forma anhidra. Los tensioactivos adecuados incluyen, por ejemplo, polisorbato (tal como Tweens) y Cremophor. En algunas realizaciones, la invención proporciona una composición que comprende docetaxel como se reivindica y un tensioactivo, en la que al menos algo del docetaxel en la composición está en una forma anhidra. Por ejemplo, en algunas realizaciones, al menos aproximadamente 10% (tal como al menos aproximadamente cualquiera de 20 %, 30 %, 40 %, y 50 %) del docetaxel en la composición está en una forma anhidra.
En algunas realizaciones, la composición descrita en la presente memoria es una composición seca (tal como liofilizada) que puede reconstituirse, resuspenderse, o rehidratarse generalmente para formar una suspensión acuosa estable del docetaxel. En algunas realizaciones, la composición es una composición líquida (tal como acuosa) obtenida mediante la reconstitución o resuspensión de una composición seca. En algunas realizaciones, la composición es una composición líquida intermedia (tal como acuosa) que puede secarse (tal como liofilizarse). En algunas realizaciones, se proporciona un método de preparación de composiciones que comprenden docetaxel como se reivindica y un tensioactivo, en las que el método comprende combinar un docetaxel anhidro con el tensioactivo. En algunas realizaciones, el tensioactivo usado para la preparación de la composición es anhidro. En alguna realización, se proporciona un método de preparación de una composición que comprende docetaxel y albúmina, en la que el método comprende combinar un docetaxel anhidro con albúmina como se reivindica. También se proporcionan composiciones producidas por métodos proporcionados en la presente memoria.
Otros componentes en las composiciones
Las composiciones descritas en la presente memoria pueden incluir otros agentes, excipientes, o estabilizantes para mejorar las propiedades de la composición. Los ejemplos de excipientes y diluyentes adecuados incluyen, pero no están limitados a, lactosa, dextrosa, sacarosa, sorbitol, manitol, almidones, goma arábiga, fosfato de calcio, alginatos, tragacanto, gelatina, silicato de calcio, celulosa microcristalina, polivinilpirrolidona, celulosa, agua, disolución salina, jarabe, metilcelulosa, metil- y propilhidroxibenzoatos, talco, estearato de magnesio y aceite mineral. Las formulaciones pueden incluir adicionalmente agentes lubricantes, agentes humectantes, agentes emulsionantes y de suspensión, agentes conservantes, agentes edulcorantes o agentes saporíferos. Los ejemplos de agentes emulsionantes incluyen ésteres de tocoferol tales como succinato de tocoferil polietilenglicol y semejantes, pluronic®, emulsionantes basados en compuestos de polioxi etileno, Span 80 y compuestos relacionados y otros emulsionantes conocidos en la técnica y aprobados para uso en animales o formas de dosificación para seres humanos. Las composiciones pueden formularse de manera que proporcionen una liberación rápida, sostenida o retardada del ingrediente activo después de la administración al paciente mediante el empleo de procedimientos muy conocidos en la técnica.
Las composiciones preferidas para administración por inyección incluyen las que comprenden un agente farmacéutico poco soluble en agua como el ingrediente activo en asociación con un agente tensioactivo (o agente humectante o surfactante), o en forma de una emulsión (p. ej., como una emulsión de agua en aceite o aceite en agua). Se pueden añadir otros ingredientes, por ejemplo, manitol u otros vehículos farmacéuticamente aceptables. En algunas realizaciones, la composición es adecuada para administración a un ser humano. En algunas realizaciones, la composición es adecuada para administración a un mamífero tal como, en el contexto veterinario, incluyendo mascotas domésticas y animales agrícolas. Existe una amplia variedad de formulaciones adecuadas de la composición inventiva (véase, p. ej., las Pat. de EEUU Nos. 5.916.596 y 6.096.331). Las siguientes formulaciones y métodos son meramente ejemplares y no son de ninguna manera limitantes. Las formulaciones adecuadas para administración oral pueden consistir en (a) disoluciones líquidas, tal como una cantidad efectiva del compuesto disuelta en diluyentes, tales como agua, disolución salina, o zumo de naranja, (b) cápsulas, sobres o comprimidos, conteniendo cada uno una cantidad predeterminada del ingrediente activo, como sólidos o gránulos, (c) suspensiones en un líquido apropiado, (d) emulsiones adecuadas, y (e) polvos. Las formas de comprimidos pueden incluir uno o más de lactosa, manitol, almidón de maíz, almidón de patata, celulosa microcristalina, goma arábiga, gelatina, dióxido de silicio coloidal, croscarmelosa de sodio, talco, estearato de magnesio, ácido esteárico y otros excipientes, colorantes, diluyentes, agentes tamponadores, agentes humectantes, conservantes, agentes saporíferos, y excipientes farmacológicamente compatibles. Las formas de comprimidos para chupar pueden comprender el ingrediente activo en un sabor, habitualmente sacarosa y goma arábiga o de tragacanto, así como pastillas que comprenden el ingrediente activo en una base inerte, tal como gelatina y glicerina, o sacarosa y goma arábiga, emulsiones, geles, y semejantes que contienen, además del ingrediente activo, dichos excipientes como se conoce en la técnica.
Las formulaciones adecuadas para administración parenteral incluyen disoluciones para inyección estériles isotónicas, acuosas y no acuosas, que pueden contener antioxidantes, tampones, bacteriostáticos, y solutos que hacen que la formulación sea compatible con la sangre del receptor pretendido, y suspensiones estériles, acuosas y no acuosas, que pueden incluir agentes de suspensión, solubilizantes, agentes espesantes, estabilizantes, y conservantes. Las formulaciones pueden presentarse en contenedores sellados de dosis unitarias o múltiples dosis, tales como ampollas y viales, y pueden almacenarse en una condición secada por congelación (liofilizada)
requiriendo sólo la adición del excipiente líquido estéril, por ejemplo, agua, para inyecciones, inmediatamente antes de su uso. Las disoluciones y suspensiones para inyección extemporáneas pueden prepararse a partir de polvos estériles, gránulos, y comprimidos de la clase descrita previamente. Se prefieren las formulaciones inyectables. Las formulaciones adecuadas para administración en aerosol que comprenden la composición inventiva incluyen disoluciones estériles isotónicas, acuosas y no acuosas, que pueden contener antioxidantes, tampones, bacteriostáticos y solutos, así como suspensiones estériles, acuosas y no acuosas, que pueden incluir agentes de suspensión, solubilizantes, agentes espesantes, estabilizantes y conservantes, solos o en combinación con otros componentes adecuados, que pueden prepararse en formulaciones de aerosol para ser administradas mediante inhalación. Estas formulaciones en aerosol pueden ponerse en propelentes aceptables presurizados, tales como diclorodifluorometano, propano, nitrógeno, y semejantes. También pueden formularse como farmacéuticos para preparaciones no presurizadas, tales como en un nebulizador o un atomizador.
En algunas realizaciones, la composición se formula para tener un pH en el intervalo de aproximadamente 4,5 a aproximadamente 9,0, incluyendo por ejemplo un pH en los intervalos de cualquiera de aproximadamente 5,0 a aproximadamente 8,0, aproximadamente 6,5 a aproximadamente 7,5, y aproximadamente 6,5 a aproximadamente 7,0. En algunas realizaciones, el pH de la composición se formula para que no sea menor de aproximadamente 6, incluyendo, por ejemplo, no menor de aproximadamente cualquiera de 6,5, 7, u 8 (tal como aproximadamente 7,5 o aproximadamente 8). La composición también puede prepararse para ser isotónica con la sangre por la adición de un modificador de la tonicidad adecuado, tal como glicerol.
También se proporcionan artículos de fabricación que comprenden las composiciones proporcionadas en la presente memoria en un envase adecuado. Los envases adecuados para las composiciones proporcionadas en la presente memoria son conocidos en la técnica e incluyen, por ejemplo, viales (tales como viales sellados), recipientes (tales como recipientes sellados), ampollas, botellas, frascos, envases flexibles (p. ej., Mylar sellado o bolsas de plástico), y semejantes. Estos artículos de fabricación además pueden esterilizarse y/o sellarse. También se proporcionan formas de dosificación unitarias que comprenden las composiciones proporcionadas en la presente memoria. Estas formas de dosificación unitarias pueden almacenarse en un envase adecuado en dosificación de una única o múltiples unidades y además también pueden esterilizarse y sellarse.
La presente invención también proporciona kits que comprenden composiciones (o formas de dosificación unitarias y/o artículos de fabricación) proporcionadas en la presente memoria y además pueden comprender instrucciones sobre los métodos para usar la composición, tal como los usos descritos adicionalmente en la presente memoria. En algunas realizaciones, el kit de la invención comprende el envase descrito anteriormente. En otras realizaciones, el kit de la invención comprende el envase descrito anteriormente y un segundo envase que comprende un tampón. Puede incluir además otros materiales deseables desde un punto de vista comercial y de usuario, incluyendo otros tampones, diluyentes, filtros, agujas, jeringas, y prospectos con instrucciones para llevar a cabo cualquiera de los métodos descritos en la presente memoria.
También pueden proporcionarse kits que contienen dosificaciones suficientes del agente farmacéutico poco soluble en agua (docetaxel) como se proporciona en la presente memoria para proporcionar un tratamiento efectivo para un individuo durante un periodo prolongado, tal como cualquiera de una semana, 2 semanas, 3 semanas, 4 semanas, 6 semanas, 8 semanas, 3 meses, 4 meses, 5 meses, 6 meses, 7 meses, 8 meses, 9 meses o más. Los kits también pueden incluir múltiples dosis unitarias del agente farmacéutico poco soluble en agua y composiciones farmacéuticas e instrucciones para el uso y envasados en cantidades suficientes para el almacenamiento y uso en farmacias, por ejemplo, farmacias hospitalarias y farmacias formuladoras.
Método para preparar y usar las composiciones
También se proporcionan métodos para hacer y usar composiciones proporcionadas en la presente memoria. Por ejemplo, se describe un método para preparar una composición que comprende un agente farmacéutico poco soluble en agua (tal como un taxano, por ejemplo, paclitaxel, docetaxel, u ortataxel), opcionalmente un polímero biocompatible (tal como una proteína vehicular, por ejemplo, albúmina), y un agente estabilizante, en el que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante, que comprende combinar (tal como mezclar) una composición que comprende un agente farmacéutico poco soluble en agua y opcionalmente un polímero biocompatible (tal como una proteína vehicular) con un agente estabilizante. También se describen métodos para la formación de nanopartículas de docetaxel preparadas en condiciones de altas fuerzas de cizalladura (p. ej., sonicación, homogeneización a alta presión o semejantes). La preparación de nanopartículas de polímeros biocompatibles (p. ej., albúmina) se describe, por ejemplo, en las Patentes de EEUU Nos. 5.916.596; 6.506.405 y 6.537.579 y también en la Publicación de Patente de EEUU 2005/0004002 A1.
Brevemente, el agente farmacéutico poco soluble en agua (tal como docetaxel) se disuelve en un disolvente orgánico y la disolución puede añadirse a una disolución acuosa de albúmina. La mezcla se somete a homogeneización a alta presión. El disolvente orgánico puede eliminarse entonces por evaporación. La dispersión obtenida además puede liofilizarse. Los disolventes orgánicos adecuados incluyen, por ejemplo, cetonas, ésteres, éteres, disolventes clorados y otros disolventes conocidos en la técnica. Por ejemplo, el disolvente orgánico puede
ser cloruro de metileno o cloroformo/etanol (por ejemplo, con una relación de 1:9, 1:8, 1:7, 1:6, 1:5, 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, o 9:1).
Se encontró, sorprendentemente, que las composiciones de docetaxel, tales como las preparadas en las referencias citadas anteriormente, tienen una estabilidad que dura menos de 1 día. De hecho, cuando se ensayaron, muchas de las composiciones fueron estables sólo durante 4 a 8 horas. La presente invención permite incrementar la estabilidad en líquido y la estabilidad después de la reconstitución por la adición de determinados estabilizantes antes de la formación de las nanopartículas o después de que se hayan formado las nanopartículas.
Por lo tanto, se proporcionan métodos como se reivindican para estabilizar una composición que comprende un agente farmacéutico poco soluble en agua, que comprenden combinar la composición con un agente estabilizante, en el que la composición resultante es estable en la misma condición en la que la composición es inestable antes de la adición del agente estabilizante. En algunas realizaciones, el método comprende además identificar y seleccionar una composición que es inestable en determinadas condiciones. En la presente invención, la composición para seleccionar comprende nanopartículas que comprenden el agente farmacéutico poco soluble en agua y albúmina como se reivindica.
También pueden añadirse a la composición excipientes farmacéuticamente aceptables. Los excipientes farmacéuticamente aceptables pueden ser una disolución, emulsión o suspensión. Por ejemplo, una emulsión de propofol en aceite y estabilizado por lecitina, es muy conocido en la técnica. También pueden prepararse otras formulaciones en emulsión o de nanopartículas de la invención. Una emulsión se forma por la homogeneización a alta presión y altas fuerzas de cizalladura. Dicha homogeneización se lleva a cabo convenientemente en un homogeneizador a alta presión, operado típicamente a presiones en el intervalo de aproximadamente 210,9 hasta 2109,2 kg/cm2 (3.000 hasta 30.000 psi). Preferiblemente, dichos procesos se llevan a cabo a presiones en el intervalo de aproximadamente 421,8 hasta 1757,7 kg/cm2 (6.000 hasta 25.000 psi). La emulsión resultante comprende nanogotitas muy pequeñas del disolvente no acuoso que contienen el agente farmacológicamente activo disuelto y nanogotitas muy pequeñas del agente estabilizante proteico. Los métodos aceptables de homogeneización incluyen procesos que confieren una alta cizalladura y cavitación tales como, por ejemplo, homogeneización a alta presión, mezcladores de alta cizalladura, sonicación, propulsores de alta cizalladura y semejantes.
Los sistemas coloidales preparados según la presente invención pueden convertirse además en forma de polvo por la eliminación del agua, p. ej., por liofilización a un perfil de temperatura-tiempo apropiado. La proteína (p. ej., hSa ) en sí misma actúa como un crioprotector y el polvo se reconstituye fácilmente por la adición de agua, disolución salina o tampón, sin la necesidad de usar crioprotectores convencionales tales como manitol, sacarosa, glicina y semejantes. Aunque no se requiere, se entiende por supuesto que pueden añadirse crioprotectores convencionales a las composiciones farmacéuticas si así se desea.
El agente estabilizante puede bien mezclarse con el agente farmacéutico poco soluble en agua y/o la proteína vehicular durante la preparación de la composición de agente farmacéutico poco soluble en agua/proteína vehicular, o añadirse después de que se prepare la composición de agente farmacéutico poco soluble en agua/proteína vehicular. Por ejemplo, el agente estabilizante puede estar presente en una disolución de proteína antes de la formación de la composición de agente farmacéutico poco soluble en agua /proteína vehicular. El agente estabilizante también puede añadirse junto con un medio acuoso usado para reconstituir/suspender la composición de agente farmacéutico poco soluble en agua/proteína vehicular o añadirse a una suspensión acuosa del agente farmacéutico poco soluble en agua asociado con la proteína vehicular. En algunas realizaciones, el agente estabilizante se mezcla con la composición de agente farmacéutico poco soluble en agua/proteína vehicular antes de la liofilización. En algunas realizaciones, el agente estabilizante se añade como un componente seco a la composición liofilizada de agente farmacéutico/proteína vehicular. En algunas realizaciones el agente estabilizante se puede añadir antes o después de formar las nanopartículas.
En algunas realizaciones cuando la adición del agente estabilizante cambia el pH de la composición, el pH de la composición se ajusta generalmente (pero no necesariamente) a un pH deseado. Los valores de pH ejemplares de las composiciones incluyen, por ejemplo, aproximadamente 5 a aproximadamente 8,5. En algunas realizaciones, el pH de la composición se ajusta para no ser menor de aproximadamente 6, incluyendo, por ejemplo, no menor de aproximadamente cualquiera de 6,5, 7, u 8 (tal como aproximadamente 7,5 u 8).
También se proporcionan métodos para preparar composiciones farmacéuticas que comprenden combinar cualquiera de las composiciones descritas en la presente memoria (incluyendo las anteriores) con un excipiente farmacéuticamente aceptable.
También se proporcionan en la presente memoria usos de las composiciones de la presente invención. En algunas realizaciones, se proporciona una composición de la presente invención para usar en un método para tratar una enfermedad o afección. Por ejemplo, en algunas realizaciones, se proporciona una composición de la presente invención para usar en un método de tratamiento del cáncer en un individuo (tal como ser humano). En algunas realizaciones, la cantidad del agente estabilizante en la composición no causa ningún efecto toxicológico cuando la composición se administra a un individuo (tal como un ser humano). En algunas realizaciones, la invención
proporciona una composición de la presente invención para usar en un método para tratar el cáncer en un individuo (tal como ser humano), en la que el docetaxel usado para preparar la composición está en forma anhidra. Por ejemplo, el docetaxel puede ser anhidro antes de ser incorporado en la composición.
El término "cantidad efectiva" usado en la presente memoria se refiere a una cantidad de un compuesto o composición suficiente para tratar un trastorno, afección o enfermedad específico tal como mejorar, paliar, disminuir y/o retrasar uno o más de sus síntomas. En referencia a los cánceres u otra proliferación celular no deseada, una cantidad efectiva comprende una cantidad suficiente para causar que un tumor se encoja y/o disminuya la velocidad de crecimiento del tumor (tal como suprimir el crecimiento tumoral). En algunas realizaciones, una cantidad efectiva es una cantidad suficiente para retrasar el desarrollo. En algunas realizaciones, una cantidad efectiva es una cantidad suficiente para prevenir la ocurrencia y/o recurrencia. Una cantidad efectiva puede administrarse en una o más administraciones.
Los cánceres que se van a tratar mediante las composiciones proporcionadas en la presente memoria incluyen, pero no se limitan a, carcinoma, linfoma, blastoma, sarcoma, y leucemia. Los ejemplos de cánceres que pueden tratarse con composiciones proporcionadas en la presente memoria incluyen, pero no están limitados a, cáncer de células escamosas, cáncer de pulmón (incluyendo cáncer de pulmón de células pequeñas, cáncer de pulmón de células no pequeñas, adenocarcinoma del pulmón y carcinoma escamoso del pulmón), cáncer del peritoneo, cáncer hepatocelular, cáncer gástrico o de estómago (incluyendo cáncer gastrointestinal), cáncer de páncreas, glioblastoma, cáncer de cuello uterino, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, cáncer de mama, cáncer de colon, melanoma, carcinoma endometrial o uterino, carcinoma de la glándula salivar, cáncer de riñón o renal, cáncer de hígado, cáncer de próstata, cáncer vulvar, cáncer de tiroides, carcinoma hepático, cáncer de cabeza y cuello, cáncer colorrectal, cáncer rectal, sarcoma de tejido blando, sarcoma de Kaposi, linfoma de células B (incluyendo linfoma de bajo grado/folicular no de Hodgkin (NHL), NHL linfocítico pequeño (SL), NHL de grado intermedio/folicular, NHL de grado intermedio difuso, NHL de grado alto inmunoblástico, NHL de grado alto linfoblástico, NHL de grado alto de células no escindidas pequeñas, enfermedad voluminosa de NHL, linfoma de células del manto, linfoma relacionado son SIDA y macroglobulinemia de Waldenstrom), leucemia linfocítica crónica (CLL), leucemia linfoblástica aguda (ALL), mieloma, leucemia de células pilosas, leucemia mieloblástica crónica y trastorno linfoproliferativo posterior a trasplante (PTLD), así como proliferación vascular anormal asociada con facomatosis, edema (tal como el asociado con tumores cerebrales) y síndrome de Meigs. En algunas realizaciones, se proporciona una composición de la presente invención para usar en un método de tratamiento de cáncer metastásico (es decir, cáncer que ha metastatizado desde el tumor primario). En algunas realizaciones, se proporciona una composición de la presente invención para usar en un método de reducción de la proliferación celular y/o migración celular. En algunas realizaciones, se proporciona una composición de la presente invención para usar en un método de tratamiento de la hiperplasia.
En algunas realizaciones, se proporciona una composición de la presente invención para usar en métodos de tratamiento del cáncer en fase(s) avanzada(s). En algunas realizaciones, se proporciona una composición de la presente invención para usar en métodos de tratamiento del cáncer de mama (que puede ser HER2 positivo o HER2 negativo), incluyendo, por ejemplo, cáncer de mama avanzado, cáncer de mama en fase IV, cáncer de mama localmente avanzado, y cáncer de mama metastásico. En algunas realizaciones, el cáncer es cáncer de pulmón, incluyendo, por ejemplo, cáncer de pulmón de células no pequeñas (NSCLC, tal como NSCLC avanzado), cáncer de pulmón de células pequeñas (SCLC, tal como SCLC avanzado) y tumor maligno sólido avanzado en el pulmón. En algunas realizaciones, el cáncer es cáncer de ovario, cáncer de cabeza y cuello, neoplasias malignas gástricas, melanoma (incluyendo melanoma metastásico), cáncer colorrectal, cáncer de páncreas y tumores sólidos (tales como tumores sólidos avanzados). En algunas realizaciones, el cáncer es cualquiera de (y, en algunas realizaciones, se selecciona del grupo que consiste en) cáncer de mama, cáncer colorrectal, cáncer rectal, cáncer de pulmón de células no pequeñas, linfoma no de Hodgkins (NHL), cáncer de células renales, cáncer de próstata, cáncer de hígado, cáncer de páncreas, sarcoma de tejido blando, sarcoma de Kaposi, carcinoma carcinoide, cáncer de cabeza y cuello, melanoma, cáncer de ovario, mesotelioma, gliomas, glioblastomas, neuroblastomas y mieloma múltiple. En algunas realizaciones, el cáncer es un tumor sólido. En algunas realizaciones, el cáncer es cualquiera de (en algunas realizaciones, se selecciona del grupo que consiste en) cáncer de próstata, cáncer de colon, cáncer de mama, cáncer de cabeza y cuello, cáncer de páncreas, cáncer de pulmón y cáncer de ovario.
Los individuos adecuados para recibir estas composiciones dependen de la naturaleza del agente farmacéutico poco soluble en agua, así como de la enfermedad/afección/trastorno que se va a tratar y/o prevenir. De acuerdo con esto, el término individuo incluye cualquiera de vertebrados, mamíferos y seres humanos dependiendo del uso adecuado pretendido. En algunas realizaciones, el individuo es un mamífero. En algunas realizaciones, el individuo es uno cualquiera o más de ser humano, bovino, equino, felino, canino, roedor, o primate. En algunas realizaciones, el individuo es un ser humano.
En otro aspecto, se proporciona una composición de la presente invención para usar en un método de tratamiento de carcinoma (tal como carcinoma de colon) en un individuo. En algunas realizaciones, el docetaxel usado para la preparación de la composición que se administra al individuo está en una forma anhidra.
Las composiciones proporcionadas en la presente memoria pueden administrarse solas o en combinación con otros agentes farmacéuticos, incluyendo agentes farmacéuticos poco solubles en agua. Por ejemplo, puede
coadministrarse con uno o más de otros agentes quimioterapéuticos incluyendo, pero no limitados a, carboplatino, navelbine® (vinorelbina), antraciclina (Doxil), lapatinib (GW57016), Herceptina, gemcitabina (Gemzar®), capecitabina (Xeloda®), alimta, cisplatino, 5-fluorouracilo, epirrubicina, ciclofosfamida, avastina, velcade®, etc. En algunas realizaciones, la composición de taxano se coadministra con un agente quimioterapéutico seleccionado del grupo que consiste en antimetabolitos (incluyendo análogos de nucleósidos), agentes basados en platino, agentes alquilantes, inhibidores de tirosina quinasa, antibióticos de antraciclina, alcaloides de vinca, inhibidores del proteasoma, macrólidos, e inhibidores de topoisomerasa. Estos otros agentes farmacéuticos pueden estar presentes en la misma composición que el fármaco, o en una composición separada que se administra simultáneamente o secuencialmente con la composición que contiene el fármaco. Los métodos de terapia de combinación que usan formulaciones de nanopartículas de taxano con otros agentes (o métodos terapéuticos) se han descrito en la Solicitud de Patente Internacional No. PCT/US2006/006167.
La dosis de la composición inventiva administrada a un individuo (tal como un ser humano) variará con la composición particular, el método de administración y la enfermedad particular que se está tratando. La dosis debería ser suficiente para incitar una respuesta deseable, tal como una respuesta terapéutica o profiláctica frente a una enfermedad o afección particular. Por ejemplo, la dosificación de docetaxel administrada puede ser de aproximadamente 1 a aproximadamente 300 mg/m2, incluyendo, por ejemplo, aproximadamente 10 a aproximadamente 300 mg/m2, aproximadamente 30 a aproximadamente 200 mg/m2 y aproximadamente 70 a aproximadamente 150 mg/m2. Típicamente, la dosificación de docetaxel en la composición puede estar en el intervalo de aproximadamente 50 a aproximadamente 200 mg/m2 cuando se proporciona en un esquema de 3 semanas, o aproximadamente 10 a aproximadamente 100 mg/m2 cuando se proporciona en un esquema semanal. Además, si se proporciona en un régimen metronómico (p. ej., diariamente o unas pocas veces a la semana), la dosificación puede estar en el intervalo de aproximadamente 1-50 mg/m2.
La frecuencia de la dosificación para la composición incluye, pero no está limitada a, al menos aproximadamente cualquiera de una vez cada tres semanas, una vez cada dos semanas, una vez a la semana, dos veces a la semana, tres veces a la semana, cuatro veces a la semana, cinco veces a la semana, seis veces a la semana, o diariamente. En algunas realizaciones, el intervalo entre cada administración es menor de aproximadamente una semana, tal como menor de aproximadamente cualquiera de 6, 5, 4, 3, 2, o 1 día. En algunas realizaciones, el intervalo entre cada administración es constante. Por ejemplo, la administración puede llevarse a cabo diariamente, cada dos días, cada tres días, cada cuatro días, cada cinco días, o semanalmente. En algunas realizaciones, la administración puede llevarse a cabo dos veces al día, tres veces al día, o de manera más frecuente.
La administración de la composición puede prolongarse durante un periodo de tiempo prolongado, tal como de aproximadamente un mes hasta aproximadamente tres años. Por ejemplo, el régimen de la dosificación puede prolongarse durante un periodo de tiempo de cualquiera de aproximadamente 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 18, 24, 30 y 36 meses. En algunas realizaciones, no hay descanso en el esquema de dosificación. En algunas realizaciones, el intervalo entre cada administración no es mayor de aproximadamente una semana.
Las composiciones proporcionadas en la presente memoria pueden administrarse a un individuo (tal como un ser humano) a través de varias rutas, incluyendo, por ejemplo, intravenosa, intraarterial, intraperitoneal, intrapulmonar, oral, inhalación, intravesicular, intramuscular, intratraqueal, subcutánea, intraocular, intratecal, transmucosal y transdérmica. Por ejemplo, la composición inventiva puede administrarse por inhalación para tratar afecciones del tracto respiratorio. La composición puede usarse para tratar afecciones respiratorias tales como fibrosis pulmonar, bronqueolitis obliterante, cáncer de pulmón, carcinoma broncoalveolar, y semejantes. En una realización de la invención, las nanopartículas se pueden administrar por cualquier vía aceptable que incluye, pero no se limita a, oral, intramuscular, transdérmica, intravenosa, mediante un inhalador u otros sistemas de suministro por aire y semejantes.
Cuando se prepara la composición para inyección, particularmente para administración intravenosa, la fase continua comprende preferiblemente una disolución acuosa de modificadores de la tonicidad, tamponada a un intervalo de pH de aproximadamente 5 a aproximadamente 8,5. El pH también puede ser menor de 7 o menor de 6. En algunas realizaciones, el pH de la composición no es menor de aproximadamente 6, incluyendo, por ejemplo, no menor de aproximadamente cualquiera de 6,5, 7, u 8 (tal como aproximadamente 7,5 u 8).
Las nanopartículas de esta invención pueden incluirse en una cápsula dura o blanda, pueden comprimirse en comprimidos, o pueden incorporarse con bebidas o alimento o incorporarse de otra forma en la dieta. Las cápsulas pueden formularse mezclando las nanopartículas con un diluyente farmacéutico inerte e insertando la mezcla en una cápsula de gelatina dura del tamaño apropiado. Si se desean cápsulas blandas, puede encapsularse una suspensión de sólidos de las nanopartículas con un aceite vegetal aceptable, petróleo ligero u otro aceite inerte mediante una máquina en una cápsula de gelatina.
También se describen en la presente memoria métodos para reducir los efectos secundarios asociados con la administración de un agente farmacéutico poco soluble en agua a un ser humano, que comprenden administrar a un ser humano una composición farmacéutica que comprende el agente farmacéutico poco soluble en agua, un polímero biocompatible (tal como una proteína vehicular) y un agente estabilizante, en el que la estabilidad de la composición se aumenta comparada con la de una composición sin el agente estabilizante. Por ejemplo, se
describen métodos para reducir varios efectos secundarios asociados con la administración del agente farmacéutico poco soluble en agua, incluyendo, pero no limitado a, mielosupresión, neurotoxicidad, hipersensibilidad, inflamación, irritación venosa, flebitis, dolor, irritación cutánea, neuropatía periférica, fiebre neutropénica, reacción anafiláctica, toxicidad hematológica y toxicidad cerebral o neurológica y combinaciones de estas. En algunos ejemplos, hay un método para reducir las reacciones de hipersensibilidad asociadas con la administración del agente farmacéutico poco soluble en agua, incluyendo, por ejemplo, erupciones cutáneas graves, urticarias, eritema, disnea, taquicardia y otros.
Además, se proporciona un método para aumentar la estabilidad de una composición que comprende un agente farmacéutico poco soluble en agua y un polímero biocompatible como se reivindica, que comprende añadir a la composición un agente estabilizante en una cantidad que es efectiva para aumentar la estabilidad de la composición. La presente invención proporciona un método de preparación de una composición que comprende un agente poco soluble en agua (docetaxel), un polímero biocompatible (albúmina), y un agente estabilizante como se reivindica, que comprende combinar (tal como mezclar) el agente poco soluble en agua y el polímero biocompatible con el agente estabilizante. En algunas realizaciones, la composición es una composición líquida. En algunas realizaciones, la composición es una composición obtenida después de la reconstitución.
El agente estabilizante puede bien mezclarse con el agente farmacéutico poco soluble en agua y/o la proteína vehicular durante la preparación de la composición de agente farmacéutico poco soluble en agua/proteína vehicular, o añadirse junto con un medio acuoso usado para reconstituir la composición de agente farmacéutico/proteína vehicular.
En un aspecto adicional de la invención, se proporciona el uso de las composiciones proporcionadas en la presente memoria en la fabricación de un medicamento. Particularmente, la fabricación de un medicamento para uso en el tratamiento de las afecciones descritas en la presente memoria. Además, la composición farmacéutica de este, descritas diversamente en la presente memoria, también se pretenden para uso en la fabricación de un medicamento para uso en el tratamiento de las afecciones y, según los métodos, descritos en la presente memoria, a no ser que se indique otra cosa.
La invención se describirá ahora con mayor detalle con referencia a los siguientes ejemplos no limitantes. A no ser que se indique otra cosa, las estabilidades de las composiciones en los ejemplos siguientes se evalúan bien a 25°C o 4°C.
Ejemplo 1 (como referencia)
Este ejemplo demuestra la inestabilidad de una preparación de composiciones farmacéuticas que comprende docetaxel y albúmina preparada como se describe en la Publicación de Patente de EEUU 2005/0004002 A1.
Se disolvieron 30 mg de docetaxel en 2 ml de cloroformo/etanol. La disolución se añadió entonces en 27,0 ml de disolución de HSA (3 % p/v). La mezcla se homogeneizó durante 5 minutos a baja RPM (homogeneizador Vitris modelo Tempest I.Q.) con el fin de formar una emulsión bruta y después se transfirió a un homogeneizador de alta presión (Avestin). La emulsificación se realizó a 632,8-2812,3 kg/cm2 (9.000-40.000 psi). El sistema resultante se transfirió a un rotavapor y el disolvente se eliminó rápidamente a presión reducida. La dispersión resultante fue translúcida y el diámetro promedio típico de las partículas resultantes estuvo en el intervalo 50-220 nm (promedio Z, Malvern Zetasizer). La dispersión se liofilizó además durante 48 horas. La torta resultante se reconstituyó fácilmente a la dispersión original por la adición de agua o disolución salina estéril. El tamaño de las partículas después de la reconstitución fue el mismo que antes de la liofilización. Cuando la suspensión líquida antes de la liofilización se almacenó, se encontró sorprendentemente que aunque la suspensión fue estable a aproximadamente 4-8 horas después de la preparación, a las 24 horas, hubo alguna sedimentación indicando inestabilidad. De forma similar, para la suspensión liofilizada reconstituida, se encontró sorprendentemente que aunque la suspensión fue estable a aproximadamente 4-8 horas después de la preparación, a las 24 horas, hubo alguna sedimentación indicando inestabilidad. Esta inestabilidad a las 24 horas no se había observado previamente por los inventores ya que el periodo de monitorización después de la preparación y reconstitución de la formulación inicial fue típicamente de sólo 4-8 horas.
Ejemplo 2 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por sonicación.
Se añadieron 25,9 mg de docetaxel a un vial de centelleo de 20-ml y se disolvieron en 0,3 ml de cloroformo. Se añadieron 4,7 ml de HSA (3,0 %, p/v) a la mezcla disuelta de docetaxel. La composición se sonicó (Sonic Dismembrator, modelo 550, Fisher Scientific Company, Pittsburgh, PA 155275) al 50 % de potencia durante 1 min. La mezcla se transfirió a un evaporador giratorio y se eliminó rápidamente el cloroformo-etanol a 45 °C, a presión reducida. El diámetro de las partículas de docetaxel resultantes fue 250-300 nm (promedio Z, Malvern Zetasizer). La suspensión precipitó en menos de 1 día.
Ejemplo 3 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por sonicación, ensayando aceite de soja como un estabilizante.
Se añadieron 18,0 mg de docetaxel a un vial de centelleo de 20-ml y se disolvieron en 0,1 ml de mezcla de cloroformo-etanol. Se añadieron 0,05 ml de aceite de soja y 2,35 ml de HSA (5,0 %, p/v) al disolvente orgánico anterior. La muestra se sonicó (Sonic Dismembrator, modelo 550, Fisher Scientific Company, Pittsburgh, PA 155275) durante 2 min. La mezcla se transfiere a un evaporador giratorio y se elimina rápidamente el cloroformoetanol a 45 °C, a presión reducida. El diámetro de las partículas de docetaxel resultantes fue 270 nm (promedio Z, Malvern Zetasizer). La suspensión precipitó en menos de 1 día.
Ejemplo 4 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por sonicación usando una mezcla de acetato de etilo-acetato de n-butilo.
Se añadieron 22,7 mg de docetaxel a un vial de centelleo de 20-ml y se disolvieron en 1,0 ml de mezcla de acetato de etilo-acetato de n-butilo. Se añadieron 2,4 ml de HSA (5,0 %, p/v) al docetaxel disuelto en el disolvente orgánico. La muestra se sonicó (Sonic Dismembrator, modelo 550, Fisher Scientific Company, Pittsburgh, PA 155275) al 50 % de potencia durante 1 min. La mezcla se transfirió a un evaporador giratorio y se eliminan el acetato de etilo-acetato de n-butilo a presión reducida. La composición precipitó en una hora.
Ejemplo 5 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por homogeneización a alta presión.
Se disolvieron 49,0 mg de docetaxel en 0,56 ml de cloroformo. La disolución se añadió a 9,6 ml de HSA (5 % p/v). La mezcla se pre-homogeneizó para formar una emulsión bruta y entonces se transfirió a un homogeneizador de alta presión (Avestin). La emulsificación se realizó a 1265,5-1406,1 kg/cm2 (18.000-20.000 psi). El sistema resultante se transfirió a un evaporador giratorio y se eliminaron el cloroformo y el alcohol t-butílico a presión reducida. El diámetro de las partículas de docetaxel resultantes fue 160-175 nm (promedio Z, Malvern Zetasizer). Se observó precipitación en < 1 día. En el examen microscópico se observaron los precipitados cristalinos.
Ejemplo 6 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por homogeneización a alta presión usando lecitina.
Se disolvieron 55,3 mg de docetaxel y 48,8 mg de lecitina de huevo en 0,56 ml de una mezcla de cloroformo y alcohol t-butílico. La disolución se añadió a 9,6 ml de HSA (5 % p/v). La mezcla se pre-homogeneizó para formar una emulsión bruta y se transfirió a un homogeneizador de alta presión (Avestin). La emulsificación se realizó a 1265,5-1406,1 kg/cm2 (18.000-20.000 psi). El sistema resultante se transfirió a un evaporador giratorio y se eliminaron el cloroformo y el alcohol t-butílico a presión reducida. El diámetro de las partículas de docetaxel resultantes fue 190-220 nm (promedio Z, Malvern Zetasizer). Se observó precipitación en < 24 horas.
Ejemplo 7 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por homogeneización a alta presión ensayando ácido poliláctico-glicólico (PLGA).
Se disolvieron 56,3 mg de docetaxel y 40,8 mg de PLGA (50:50) en 0,56 ml de cloroformo. La disolución se añadió a 9,6 ml de HSA (5 % p/v). La mezcla se pre-homogeneizó para formar una emulsión bruta y se transfirió a un homogeneizador de alta presión (Avestin). La emulsificación se realizó a 1265,5-1406,1 kg/cm2 (18.000-20.000 psi). El sistema resultante se transfirió a un evaporador giratorio y se eliminaron el cloroformo y el alcohol t-butílico a presión reducida. El diámetro de las partículas de docetaxel resultantes fue 575 nm (promedio Z, Malvern Zetasizer). Se observó precipitación en < 24 horas.
Ejemplo 8 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por homogeneización a alta presión ensayando ácido benzoico.
Se disolvieron 50,3 mg de docetaxel y 3,0 mg de ácido benzoico en 0,56 ml de una mezcla de cloroformo y alcohol tbutílico. La disolución se añadió a 10,0 ml de HSA (5 % p/v). La mezcla se pre-homogeneizó para formar una emulsión bruta y se transfirió a un homogeneizador de alta presión (Avestin). La emulsificación se realizó a 1265,5 1406,1 kg/cm2 (18.000-20.000 psi). La emulsión resultante se transfirió a un evaporador giratorio y se eliminaron el cloroformo y el alcohol t-butílico a presión reducida. El diámetro de las partículas de docetaxel resultantes fue 160 nm (promedio Z, Malvern Zetasizer). La composición precipitó en < 24 horas.
Ejemplo 9 (como referencia)
Este ejemplo demuestra la inestabilidad de nanopartículas de docetaxel preparadas por homogeneización a alta presión ensayando colesterol.
Se disolvieron 51,0 mg de docetaxel y 16,5 mg de colesterol en 0,56 ml de una mezcla de cloroformo y alcohol tbutílico. La disolución se añadió a 10,0 ml de HSA (5 % p/v). La mezcla se pre-homogeneizó para formar una emulsión bruta y se transfirió a un homogeneizador de alta presión (Avestin). La emulsificación se realizó a 1265,5 1406,1 kg/cm2 (18.000-20.000 psi). La emulsión resultante se transfirió a un evaporador giratorio y se eliminaron el cloroformo y el alcohol t-butílico a presión reducida. La precipitación ocurrió en < 24 horas.
Ejemplo 10
Este ejemplo demuestra la estabilidad de nanopartículas de docetaxel preparadas por homogeneización a alta presión ensayando citrato de sodio.
Se disolvieron 50,0 mg de docetaxel en 0,56 ml de una mezcla de cloroformo y alcohol t-butílico (10,2:1 (v/v)). La disolución se añadió a 9,6 ml de HSA (5 %, p/v) que contenía citrato de trisodio 100 mM (2,94 % p/v). La mezcla se pre-homogeneizó para formar una emulsión bruta y se transfirió a un homogeneizador de alta presión (Avestin). La emulsificación se realizó a 1265,5-1406,1 kg/cm2 (18.000-20.000 psi). La emulsión resultante se transfirió a un evaporador giratorio y se eliminaron el cloroformo y el alcohol t-butílico a presión reducida. El diámetro de las partículas de docetaxel resultantes fue 150-225 nm (promedio Z, Malvern Zetasizer). La formulación fue sorprendentemente estable >24 horas sin precipitado observable.
Ejemplo 11
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato (3,9 %, 133 mM) y cloruro de sodio (1,75 %, 300 mM).
La fase acuosa se preparó añadiendo HSA (5 % en peso), citrato de sodio (3,9 % en peso) y cloruro de sodio (1,75 % en peso) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (7 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló con un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). El cloroformo y el etanol en la emulsión se eliminaron entonces usando un evaporador giratorio a una presión reducida. La suspensión se filtró por filtración seriada (1,2 pm, 0,8 pm y 0,45 pm) y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida es homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de 165,6 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5pm. La suspensión se almacenó tanto a 4° C como a 25° C. Sorprendentemente, la suspensión fue estable hasta 3 días a 4° C y > 1 día a 25° C. La suspensión no presentó ninguna sedimentación o precipitación y no cambió de color o consistencia. Además, el producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5min. Después de la reconstitución, las partículas tenían un tamaño de partículas promedio de 164,6nm. La suspensión reconstituida se almacenó a 4 °C y, sorprendentemente, permaneció estable > 1 día.
Ejemplo 12
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato (2,9 %, 100 mM) y cloruro de sodio (1,75 %, 300 mM).
La fase acuosa se preparó añadiendo HSA (5 % en peso), citrato de sodio (2,9 % en peso) y cloruro de sodio (1,75 % en peso) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (7 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. La fase orgánica se añadió a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). El cloroformo y el etanol en la emulsión se eliminaron entonces usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada (1,2 pm, 0,8 pm, y 0,45 pm) y se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de 157,1 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 pm. La suspensión se almacenó tanto a 4° C como a 25° C. Sorprendentemente, la suspensión fue estable hasta 3 días a 4° C y > 1 día a 25° C. La suspensión no presentó ninguna sedimentación o cremosidad y no cambió de color o consistencia.
El producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5 min. Después de la reconstitución, las partículas tenían un tamaño de partículas promedio de 150,9 nm. La suspensión
reconstituida se almacenó a 4°C y permaneció estable > 1 día.
Ejemplo 13
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato (3,9 %, 133 mM).
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (3,9 % en peso) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). El cloroformo y el etanol en la emulsión se eliminaron entonces usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada (1,2 pm, 0,8 pm, 0,45 pm y 0,22 pm) y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de 131 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 pm. Sorprendentemente, la suspensión de nanopartículas fue estable durante > 1 día.
Ejemplo 14
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato (11,7 %, 400 mM).
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (11,7 % en peso) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). El cloroformo y el etanol en la emulsión se eliminaron entonces usando un evaporador giratorio a una presión reducida. La suspensión se filtró por filtración seriada (1,2 pm, 0,8 pm, 0,45 pm y 0,22 pm) y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de 143,5 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5pm. La suspensión se almacenó tanto a 4° C como a 25° C. Sorprendentemente, la suspensión fue estable hasta 3 días a 4° C y > 1 día a 25° C. La suspensión no presentó ninguna sedimentación o cremosidad y no cambió de color o consistencia.
El producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5 min. Después de la reconstitución, las partículas tenían un tamaño de partículas promedio de 151,8 nm. Sorprendentemente, la suspensión reconstituida se almacenó a 4°C y permaneció estable > 1 día.
Ejemplo 15
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato (7,7 %, 200 mM).
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (7,7 % en peso) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). El cloroformo y el etanol en la emulsión se eliminaron entonces usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada (1,2 pm, 0,8 pm, 0,45 pm y 0,22 pm) y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida es homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tienen un tamaño promedio de 226,4 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 pm. La suspensión se almacenó tanto a 4° C como a 25° C. Sorprendentemente, la suspensión fue estable hasta 3 días a 4° C y > 1 día a 25° C. La suspensión no presentó ninguna sedimentación o cremosidad y no cambió de color o consistencia.
El producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5 min. Después de la reconstitución, las partículas tenían un tamaño de partículas promedio de 211,4 nm. La suspensión reconstituida se almacenó a 4°C y permaneció estable > 1 día.
Ejemplo 16
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato/NaCl.
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (5,88 %, 200 mM) y NaCI (1,75 %, 300 mM) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de <200 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 pm. La suspensión se almacenó y, sorprendentemente, la suspensión fue estable sin precipitados o sedimento durante > 1 día.
El producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5 min. La suspensión reconstituida se almacenó y, sorprendentemente, permaneció estable > 1 día.
Ejemplo 17
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato/NaCl.
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (2,94 %, 100 mM) y NaCl (2,9 %, 500 mM) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de <200 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 pm. La suspensión se almacenó y, sorprendentemente, la suspensión fue estable sin precipitados o sedimento durante > 1 día.
El producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5 min. La suspensión reconstituida se almacenó y, sorprendentemente, permaneció estable > 1 día.
Ejemplo 18
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato/NaCl.
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (2,94 %, 100 mM) y NaCl (3,5 %, 600 mM) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de <200 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 pm. La suspensión se almacenó y, sorprendentemente, la suspensión fue estable sin precipitados o sedimento durante > 1 día.
El producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5 min. La suspensión reconstituida se almacenó y, sorprendentemente, permaneció estable > 1 día.
Ejemplo 19
Este ejemplo demuestra la estabilidad de una preparación de nanopartículas de docetaxel con citrato/NaCl.
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (1,47 %, 50 mM) y NaCl (2,9 %, 500 mM) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 20 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un
Malvern Zetasizer. Las partículas tenían un tamaño promedio de <200 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 |jm. La suspensión se almacenó y, sorprendentemente, la suspensión fue estable sin precipitados o sedimento durante > 1 día.
El producto liofilizado tenía la apariencia de una torta sólida. La reconstitución de la torta liofilizada duró < 5 min. La suspensión reconstituida se almacenó y, sorprendentemente, permaneció estable > 1 día.
Ejemplo 20
Este ejemplo demuestra el efecto de una preparación de nanopartículas de docetaxel anhidro frente a hidratado con citrato/NaCl.
La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (200 mM) y NaCl (300 mM) a agua para inyección y se agitó hasta que se disolvió. Se preparó la fase orgánica para tres formulaciones diferentes disolviendo bien docetaxel anhidro, trihidrato de docetaxel o hemihidrato de docetaxel (hidrato parcial) (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tienen un tamaño promedio de <200 nm. Las tres suspensiones se almacenaron y, sorprendentemente, la suspensión que contenía el docetaxel anhidro era la más estable sin precipitados o sedimento durante > 1 día. Ambas preparaciones de docetaxel hidratado mostraron precipitado o sedimento en < 1 día. La misma observación se observó para la suspensión liofilizada después de la reconstitución. Así, se determinó que la forma anhidra de docetaxel era la más adecuada para una preparación de nanopartículas de docetaxel. Ejemplo 20A
Este ejemplo proporciona una comparación de docetaxel anhidro, trihidrato de docetaxel y hemihidrato de docetaxel por Calorimetría Diferencial de Barrido (DSC).
Los tres tipos diferentes de docetaxel se sometieron a DSC usando técnicas estándar. Todos mostraron una endoterma de fusión a aproximadamente 162-166 °C. Sin embargo, sólo los 2 materiales hidratados mostraron una endoterma de deshidratación de agua entre aproximadamente 74-80 °C.
Ejemplo 20B
Este ejemplo proporciona una comparación de docetaxel anhidro, trihidrato de docetaxel y hemihidrato de docetaxel por Difracción de Rayos X en Polvo (XRD).
Los tres tipos diferentes de docetaxel se sometieron a XRD usando técnicas estándar. Los tres materiales mostraron todos una variedad de picos agudos indicando cristalinidad. Sin embargo, el material anhidro mostró un espectro diferente comparado con los dos materiales hidratados. En particular, hubo un pico que apareció a 2-theta de 7-8 para la muestra anhidra que estaba ausente en el material hidratado. Esto indicó una estructura de cristal diferente para el docetaxel anhidro frente a los docetaxeles hidratados.
Ejemplo 21 (como referencia)
Este ejemplo demuestra que el grado de hidratación afecta la solubilidad de docetaxel y proporciona una comparación de la solubilidad de docetaxel anhidro, trihidrato de docetaxel y hemihidrato de docetaxel.
Para comparar si los diferentes tipos de material de docetaxel tenían diferentes perfiles de solubilidad como resultado de sus diferentes estructuras, sus velocidades de solubilidad se compararon en el disolvente acetonitrilo. Se añadió acetonitrilo a una cantidad fija de docetaxel de diferentes proveedores para obtener una concentración de 5 mg/ml (base anhidra). Se observó la velocidad a la que se producía la disolución de los diferentes docetaxeles. Se observó que los docetaxeles anhidros (de 2 proveedores diferentes) se disolvieron completamente en menos de 1 minuto. Por el contrario, los materiales hidratados (trihidrato e hidrato parcial de 2 proveedores diferentes) no se disolvieron fácilmente y tuvo que añadirse disolvente adicional hasta una concentración final de 2,5 mg/ml. En estas condiciones más diluidas, el tiempo para la disolución fue entre 5 y 10 minutos para los materiales hidratados. Se hizo una observación similar cuando se usó el disolvente cloroformo. Así, se encontró sorprendentemente que el grado de hidratación o la naturaleza anhidra pueden afectar sustancialmente la solubilidad de docetaxel.
Ejemplo 22 (como referencia)
Este ejemplo demuestra que el grado de hidratación de docetaxel afecta la estabilidad y proporciona una comparación de las formulaciones de trihidrato, hemihidrato de docetaxel y docetaxel anhidro en Tween 80.
Es muy conocido que el docetaxel se formula con Tween 80 como un solubilizante o emulsionante para el producto comercial Taxotere. Los diferentes docetaxeles se disolvieron en Tween 80 a una concentración de 40 mg/ml (en base anhidra). Se observaron 2 ml de estas disoluciones para determinar la estabilidad con el tiempo. Se encontró, sorprendentemente, que después de unos pocos días, se observó un precipitado o sedimento para el docetaxel hidratado pero no se observó precipitado con el docetaxel anhidro. Así, el docetaxel anhidro se prefiere en la formulación de Tween. Además, puede ser útil usar un tensioactivo Tween 80 o equivalente que es anhidro o con muy bajo contenido en agua ya que la forma anhidra de docetaxel puede absorber agua para formar la forma hidratada que podría dar como resultado la precipitación.
Ejemplo 23 (como referencia)
Este ejemplo de muestra la estabilidad de una preparación de nanopartículas de docetaxel con docetaxel anhidro y sin estabilizantes añadidos.
La fase acuosa se preparó añadiendo HSA (5 % en peso) a agua para inyección. La fase orgánica se preparó disolviendo docetaxel anhidro (5 % en peso) en una mezcla de disolventes (6 % en volumen) que contenía cloroformo y etanol y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de <200 nm. La muestra también se examinó por microscopía y la mayor parte de las partículas eran < 0,5 pm. La suspensión se almacenó y, sorprendentemente, la suspensión fue estable sin precipitados o sedimento durante aproximadamente 1 día. Así, en ausencia de estabilizantes, el Nab-docetaxel preparado con docetaxel anhidro parece ser más estable que cuando se prepara con una forma hidratada de docetaxel para la que la estabilidad es mucho menor de 1 día, típicamente sólo unas pocas horas.
Ejemplo 24
Este ejemplo demuestra la preparación de una preparación de nanopartículas de docetaxel anhidro con citrato/NaCl. La fase acuosa se preparó añadiendo HSA (8,5 % en peso) y citrato de sodio (200 mM) y NaCl (300 mM) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel anhidro (133 mg/ml) en una mezcla de disolventes que contenía cloroformo y etanol (1:1) y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica (6 % en volumen) a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 200 ml. La emulsión bruta se homogeneizó a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de <200 nm. La suspensión se almacenó y, sorprendentemente, no mostró precipitados o sedimento durante > 1 día. La misma observación se observó para la suspensión liofilizada después de la reconstitución.
Ejemplo 25
Este ejemplo demuestra la preparación de una preparación de nanopartículas de docetaxel anhidro con citrato/NaCl. La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (200 mM) y NaCl (300 mM) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel anhidro (160 mg/ml) en una mezcla de disolventes que contenía cloroformo y etanol (1:1) y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica (8 % en volumen) a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 200 ml. La emulsión bruta se sometió a homogeneización a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de menos de 200 nm. La suspensión se almacenó y, sorprendentemente, no mostró precipitación o sedimentación durante más de un día. La misma observación se observó para la suspensión liofilizada después de la reconstitución.
Ejemplo 26
Este ejemplo demuestra la preparación de una preparación de nanopartículas de docetaxel anhidro con citrato/NaCl. La fase acuosa se preparó añadiendo HSA (5 % en peso) y citrato de sodio (200 mM) y NaCl (300 mM) a agua para inyección y se agitó hasta que se disolvió. La fase orgánica se preparó disolviendo docetaxel anhidro (160 mg/ml) en
una mezcla de disolventes que contenía cloroformo y etanol (1:1) y se agitó hasta que se disolvió. Lentamente, se añadió la fase orgánica (8 % en volumen) a la fase acuosa y se mezcló usando un mezclador de rotor y estátor. El tamaño del lote fue 200 ml. La emulsión bruta se sometió a homogeneización a alta presión a 1406,1 kg/cm2 (20.000 psi). Los disolventes en la emulsión se eliminaron usando un evaporador giratorio a presión reducida. Se añadió albúmina adicional a la suspensión evaporada para incrementar la relación albúmina: fármaco hasta 8:1 en peso. La suspensión se filtró por filtración seriada y después se liofilizó (Liofilizador de Bandeja FTS).
La suspensión líquida fue homogénea y blanquecina. El análisis del tamaño de las partículas se realizó usando un Malvern Zetasizer. Las partículas tenían un tamaño promedio de menos de 200 nm. La suspensión se almacenó y, sorprendentemente, no mostró precipitados o sedimento durante más de un día. La misma observación se observó para la suspensión liofilizada después de la reconstitución.
Ejemplo 27 (como referencia)
Este ejemplo demuestra el efecto del pH en la estabilidad de la suspensión de nanopartículas, así como en la degradación química del docetaxel.
Se prepararon formulaciones de nanopartículas de docetaxel como se describe en los ejemplos anteriores. El efecto del pH en estas formulaciones se ensayó entre pH 4 y pH 9. Se encontró que el incremento del pH por encima de pH 6 incrementaba la estabilidad física medida en términos de tamaño de las nanopartículas y sedimentación de la formulación mientras, al mismo tiempo, se incrementaba la cantidad de degradación de docetaxel a 7-epi docetaxel a temperatura ambiente. Se encontró así que un intervalo de pH óptimo en el que tanto la estabilidad física como la estabilidad química eran aceptables estaba entre 6-8,5. Un intervalo de pH más preferible fue 6,5-8 y se encontró que un intervalo lo más preferible era pH 7,25 a 7,75.
Ejemplo 28
Este ejemplo compara la estabilidad de Nab-docetaxel preparado bien con formas hidratadas de docetaxel o docetaxel anhidro en presencia o ausencia de estabilizantes adecuados.
La estabilidad de estas preparaciones se examinó visualmente antes de la liofilización y también después de la reconstitución de las preparaciones liofilizadas. Además, la estabilidad de las preparaciones liofilizadas (que contienen estabilizantes) después de la reconstitución se evaluó a diferentes concentraciones de docetaxel en la suspensión reconstituida. Los resultados se muestran en las Tablas 1-3 siguientes.
Tabla 1. Evaluación de la estabilidad de suspensiones de nanopartículas de Nab-docetaxel antes de la liofilización

Como se muestra en la Tabla 1, la estabilidad de Nab-docetaxel preparado usando docetaxel anhidro fue significativamente mejor que Nab-docetaxel preparado usando formas hidratadas de docetaxel estuvieran o no presentes estabilizantes en la formulación.
La adición de estabilizantes (citrato 200mM/NaCl 300mM) mejoró significativamente la estabilidad de las preparaciones de Nab-docetaxel que no contenían estabilizante.
La adición de estabilizantes mejoró la estabilidad de las preparaciones de Nab-docetaxel hechas a partir de trihidrato de docetaxel.
Tabla 2. Estabilidad en la reconstitución de polvo liofilizado de Nab-docetaxel que contenía estabilizante (citrato 200mM/NaCl 300mM) reconstituido a 5 mg/ml de docetaxel en agua para inyección

Código: 1 - Sin sedimentación
2 - Sedimentación ligera
3 - Más sedimentación
4 - Sedimentación espesa
5 - Sedimentación completa
Tabla 3. Estabilidad en la reconstitución de polvo liofilizado de Nab-docetaxel que contenía estabilizante (citrato 200mM/NaCl 300mM) reconstituido a 1 mg/ml de docetaxel en agua para inyección

Código: 1 - Sin sedimentación
2 - Sedimentación ligera
3 - Más sedimentación
4 - Sedimentación espesa
5 - Sedimentación completa
Como se muestra en las Tablas 2 y 3, la estabilidad de Nab-docetaxel reconstituido que contenía estabilizantes se mejora significativamente a una concentración mayor de 5 mg/ml de docetaxel frente a una concentración menor de aproximadamente 1 mg/ml de docetaxel. La estabilidad de la formulación de Nab-docetaxel reconstituido que contenía estabilizantes preparado con docetaxel anhidro es significativamente mejor que Nab-docetaxel preparado con trihidrato de docetaxel.
Ejemplo 29 (como referencia)
Este ejemplo demuestra los perfiles de toxicidad de una formulación de nanopartículas de albúmina de docetaxel (Nab-docetaxel) frente a Taxotere.
Se determinó la dosis máxima tolerada (MTD) para Nab-docetaxel y Taxotere® (Tween 80- docetaxel) durante un estudio de escalada de la dosis en ratones desnudos. Se trataron ratones desnudos, tamaño del grupo de 10 por grupo, con dosis crecientes de Taxotere (0 mg/kg, 7 mg/kg, 15 mg/kg, 22 mg/kg, 33 mg/kg y 50 mg/kg) usando un
esquema q4dx3. Se trataron ratones desnudos, tamaño del grupo de 6 por grupo, con dosis crecientes de Nabdocetaxel (0 mg/kg, 15 mg/kg, 22 mg/kg, 33 mg/kg, 50 mg/kg y 75 mg/kg) usando un esquema q4dx3. Los animales se pesaron en días alternos. La pérdida máxima de peso corporal se representó frente a la dosis y se ajustó usando una ecuación de Hill. Se calculó MTD, definida como la pérdida de peso igual al 20 %, usando los datos ajustados. La MTD fue 2,3 veces mayor para Nab-docetaxel frente a Taxotere (Tween 80 docetaxel). Las MTD fueron 47,2 mg/kg y 20,6 mg/kg para Nab-docetaxel y Taxotere, respectivamente.
Ejemplo 30
Este ejemplo demuestra la eficacia antitumoral de las nanopartículas de docetaxel de la invención (Nab-docetaxel) con estabilizante frente a Taxotere.
La eficacia de Nab-docetaxel (preparado con citrato 200mM y NaCl 300mM) se comparó frente a Taxotere en un modelo de xenoinjerto tumoral en ratones desnudos que portaban tumor de colon humano HCT-116. Se usaron 10 ratones por grupo para el estudio. El Taxotere se dosificó a 15mg/kg y el Nab-docetaxel se dosificó a 22 mg/kg, ambos en un esquema q4dx3. El Nab-docetaxel (22 mg/kg) fue más efectivo en la supresión tumoral que el Taxotere (15 mg/kg, MTD) con p < 0,0001. Además, el Nab-Docetaxel presentó un índice terapéutico mayor que el Taxotere ya que la pérdida máxima de peso en el grupo de Taxotere fue 20 %, mientras que para el Nab-docetaxel fue aproximadamente 17 %, a pesar de una dosis un 50 % más alta.
Ejemplo 31
Este ejemplo demuestra un estudio de infusión de Nab-docetaxel (citrato 200 mM/NaCl 300 mM).
Se llevó a cabo un estudio en ratas con una infusión de 5 min de Nab-docetaxel, con velocidades de infusión crecientes de una formulación de Nab-docetaxel que contenía aproximadamente 200 mM de citrato/300 mM de NaCl. Una infusión de 5 min en ratas puede considerarse equivalente a una infusión de 30 min en seres humanos. La velocidad máxima de la infusión segura fue ~0,5 ml/min. Esta es equivalente a 0,23 mmol/kg/min o 68 mg/kg/min de citrato para una infusión de 5 min en ratas. Trasladado a la dosis humana, esta era equivalente a aproximadamente 170 mg de docetaxel/m2 en una infusión de 30 min.
Ejemplo 32
Este ejemplo demuestra la biocompatibilidad con la sangre de 5 mg/ml de Nab-docetaxel (citrato 200 mM/NaCl 300 mM).
Se llevó a cabo un estudio de hemolisis in vitro en sangre de rata usando una formulación placebo (todos los componentes excepto el docetaxel) y la formulación de Nab-docetaxel. El placebo no causó hemolisis incluso a la relación mása alta de sangre de rata:placebo de 1:1. La formulación de Nab-docetaxel interfirió con la lectura de absorción debido a la dispersión de la luz característica de las nanopartículas, pero cuando se realizaron controles de línea base apropiados, no se detectó hemolisis a la relación más alta de sangre de rata: Nab-docetaxel de 1:1. Esto demuestra que el Nab-docetaxel con estabilizante como se indica es compatible con la sangre de rata.
Ejemplo 33
Este ejemplo proporciona un estudio piloto de escalada de dosis múltiple en ratas. Todas las formulaciones de Nabdocetaxel descritas en la presente memoria contienen citrato 200 mM y NaCl 300 mM.
Para comparar la seguridad de la formulación de Nab-docetaxel con la formulación de Taxotere, las ratas se dosificaron bien con Tween 80 docetaxel (misma formulación que Taxotere) o Nab-docetaxel a 5,0, 10,0, 15,0, 30,0 y 50 mg/kg usando una infusión de 10 minutos a través de catéteres permanentes en la yugular en los días 0, 4 y 8 para un total de tres tratamientos. Se usó como un control disolución salina (1ml/kg/min).
Cada animal se observó y se pesó diariamente durante los días 0-25. El peso corporal se registró diariamente para cada animal tratado. Los signos de distrés clínico se registraron diariamente. La sangre se recogió en los días 13, 16 y en el día 25 en tubos tratados con EDTA y se sometió a análisis diferencial. La necropsia se llevó a cabo en el día 25.
El resultado del estudio se muestra en la Tabla 4. Como se muestra en la tabla, los animales en todos los grupos de dosis toleraron el primer tratamiento sin toxicidades agudas o relacionadas con la infusión incluso a la dosis más alta de 50 mg/kg. Sin embargo, sólo los animales en el nivel de dosis más baja de 5 mg/kg y el control de disolución salina sobrevivieron hasta el final del experimento (los tres tratamientos). Todos los animales que recibieron dosis mayores, murieron bien entre la segunda y tercera dosis o después de la tercera dosis.
Tabla 4. Pérdida de peso y Mortalidad en el estudio de escalada de dosis

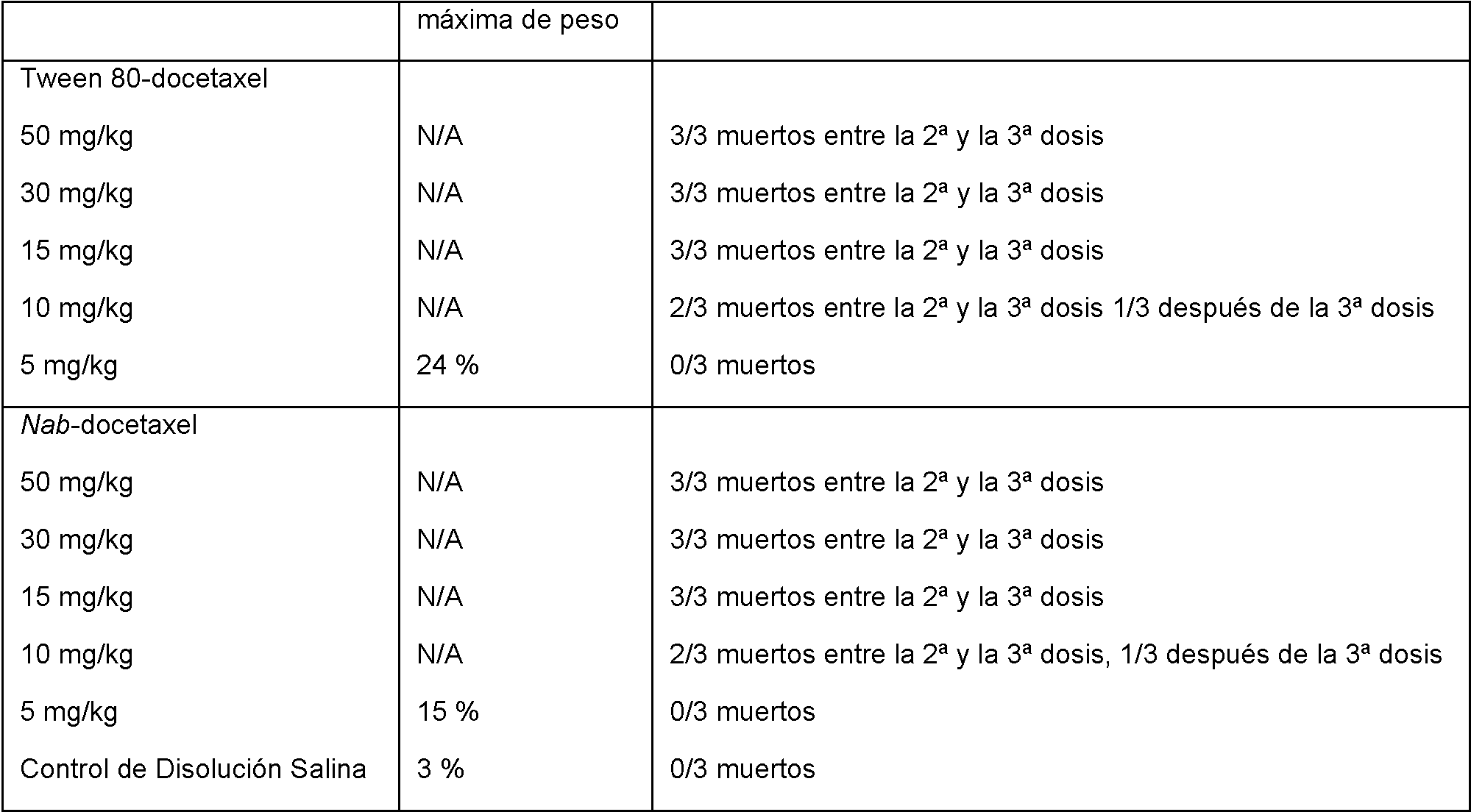
La pérdida de peso de los animales tratados se muestra en la Figura 1. La comparación de neutropenia a una dosis de 5 mg/kg de docetaxel para Nab-docetaxel y Tween 80-docetaxel se muestra en la Figura 2. La pérdida de peso para el grupo superviviente de 5 mg/kg de Nab-docetaxel fue significativamente menor que la del grupo de 5 mg/kg de Tween-docetaxel (p=0,02+, ANOVA). Esto fue paralelo a una neutropenia grave significativamente mayor para Tween-docetaxel (5 mg/kg) frente a Nab-docetaxel (5 mg/kg), p<0,0001, ANOVA, Figura 2) en el día 13. La necropsia al final del experimento (día 25) en los grupos supervivientes de 5 mg/kg reveló anormalidades en 2/3 animales en el grupo de Tween-docetaxel (un caso de acumulación de fluido lechoso en la cavidad torácica y un caso de adherencia anormal del bazo a la pared abdominal, estómago y páncreas). Los animales en Nab-docetaxel (5 mg/kg) y disolución salina eran normales.
Este estudio piloto mostró una mejoría significativa en la seguridad para Nab-docetaxel en términos de pérdida de peso corporal global. La neutropenia fue significativamente mayor para Tween-docetaxel.
Ejemplo 34
Este ejemplo demuestra las cinéticas en sangre de Nab-docetaxel. Todas las formulaciones de Nab-docetaxel descritas en la presente memoria contienen citrato 200 mM y NaCl 300 mM.
Las ratas se dividieron en seis grupos (3 por grupo). El día 1, cada animal se pesó y se le administró una única dosis intravenosa del artículo apropiado mostrado a continuación:
Grupo A: Taxotere, 10 mg/kg
Grupo B: Taxotere, 20 mg/kg
Grupo C: Taxotere, 30 mg/kg
Grupo D: Nab-docetaxel, 10 mg/kg
Grupo E: Nab-docetaxel, 20 mg/kg
Grupo F: Nab-docetaxel, 30 mg/kg
Los artículos de ensayo se administraron durante un periodo de infusión de 10 /-1 minutos. Se recogieron muestras de sangre (200 jl) de la vena de la cola de cada rata en los siguientes intervalos: Antes de la infusión (línea base); durante la infusión (5 minutos en la infusión, t = -5 minutos); y a la compleción de la infusión (t = 0). También se recogió sangre a los siguientes puntos de tiempo después de la compleción de la infusión: 5, 10 y 20 minutos; 40 ± 3 minutos; 2 horas ± 5 minutos; 3 horas ± 10 minutos; 4 horas ± 10 minutos; 8 horas ± 10 minutos; 24 ± 1 hora; 48 ± 1 hora; y 120 ± 2 horas. Se recogieron muestras de sangre en tubos con el tapón verde (heparina de sodio) y se procesaron para recogida de plasma mediante centrifugación a aproximadamente 2.000 rpm durante
aproximadamente 10 minutos. Las muestras de plasma se almacenaron congeladas hasta su transporte en nieve carbónica a ALTA Analytical (El Dorado Hills, CA) para análisis por LC/MS de los niveles de docetaxel.
Los resultados de los experimentos se muestran en la Figura 3 y la Tabla 5. No hubo diferencias significativas entre los perfiles de PK de Nab-docetaxel frente a Taxotere a 10 mg/kg. Sin embargo, las diferencias entre Nab-docetaxel y Taxotere fueron significativas a 20 mg/kg con Cmáx y AUC, 53 % y 70 % de Taxotere respectivamente y Vz y Vss fueron 177 % y 243 % de Taxotere respectivamente. A 30 mg/kg, de nuevo, las diferencias fueron significativas con Cmáx y AUC para Nab-docetaxel de 46 % y 47 % de Taxotere respectivamente y Vz y Vss fueron 225 % y 375 % de Taxotere respectivamente.
Tabla 5: Parámetros de PK para Nab-docetaxel y Taxotere

Cuando la AUC se representó frente a la dosis, la ausencia de linealidad para Taxotere fue claramente evidente, con la AUC de Nab-docetaxel siendo lineal respecto a la dosis (Figura 3D). Esto puede explicarse por la propiedad de formación de micelas de Tween 80, la alta solubilidad de docetaxel en el núcleo hidrofóbico de las micelas y el secuestro correspondiente de docetaxel en el plasma (6). Además, la rápida distribución tisular para Nab-docetaxel también puede explicarse por la utilización de la transcitosis mediada por albúmina/caveolas a través de las células endoteliales, un proceso descrito previamente para el Abraxano (Nab-paclitaxel).
Los datos de PK sugieren que el Tween 80 en Taxotere presentaba secuestro de docetaxel en plasma similar al observado con Cremophor EL en el caso de Taxol. Esto resultó en mayores Cmáx y AUC y volúmenes menores de
distribución para Taxotere que para Nab-docetaxel. La PK de Nab-docetaxel es lineal mientras que la de Tween 80-docetaxel (Taxotere) no es lineal respecto a la dosis. Las dosificaciones descritas en la presente memoria, es decir, 10 mg/kg, 20 mg/kg y 30 mg/kg, son equivalentes a una dosificación humana de aproximadamente 60 mg/m2, aproximadamente 120 mg/m2, y aproximadamente 180 mg/m2. Típicamente, el intervalo lineal de PK del Nabdocetaxel es aproximadamente 10-180 mg/m2.
Ejemplo 35 (como referencia)
Este ejemplo demuestra la inhibición de la interacción fármaco-albúmina por tensioactivos tales como Tween 80. El experimento se hizo usando un paclitaxel marcado con fluorescencia (Flutax) como un suplente para paclitaxel/docetaxel. Se mostró que Flutax tiene una unión similar a albúmina que paclitaxel.
Se inmovilizó HSA en una microplaca de plástico de 96 pocillos. La albúmina inmovilizada se hizo reaccionar durante 1 h con concentración constante de Flutax y concentración creciente de disolventes (Cremophor EL/EtOH, Tween 80 y TPGS). Los ligandos no unidos se eliminaron por lavado con tampón. Los ligandos unidos se cuantificaron usando un fluorímetro. La CI50 se determinó usando una ecuación de desintegración exponencial. Los resultados del experimento se mostraron en la Figura 4. Como se muestra en la Figura 4, la interacción albúmina-paclitaxel se inhibió por el disolvente usado comúnmente en la formulación de fármaco insoluble en agua tal como Cremophor EL/EtOH, Tween 80 y TPGS (CI50 de 0,009 %, 0,003 % y 0,008 %, respectivamente). La inhibición completa ocurrió a 0,02 % o 0,2 pl de Tween 80/ml. Esto es clínicamente relevante ya que los pacientes tratados con Taxotere presentaron 0,07-0,41 pl de Tween 80/ml de sangre al final de la infusión del fármaco.
Este experimento demuestra que el Tween 80 en la formulación de Taxotere puede inhibir la unión de docetaxel a albúmina y prevenir su transcitosis endotelial a través del mecanismo gp60/caveolar. Los datos de PK en los estudios anteriores también apoyan esta observación.
Ejemplo 36
Este ejemplo proporciona una evaluación de la actividad antitumoral de Nab-docetaxel frente a xenoinjerto de carcinoma de colon H29 en ratones desnudos atímicos. Los ratones se dividieron en el grupo control y el grupo de Nab-docetaxel (N = 4 ratones por grupo, cada uno con tumores bilaterales). Todas las formulaciones de Nabdocetaxel descritas en la presente memoria contienen citrato 200 mM y NaCl 300 mM.
Brevemente, se implantaron tumores H29 subcutáneamente en ratones desnudos atímicos, se dejó que crecieran hasta 100 mm3 y entonces se trataron bien con el control (sin fármaco) o Nab-docetaxel (15 mg/kg, q4dx3, bolo iv). Las mediciones del tamaño tumoral y peso corporal se obtuvieron tres veces a la semana y se representan en la Figura 5.
Como se muestra en la Figura 5, hubo una inhibición significativa del tumor HT29 in vivo, p<0,0001 frente al control, ANOVA. A la dosis de 15 mg/kg de Nab-docetaxel, la pérdida media de peso fue entre 10-20 % lo que sugiere que esta dosis puede estar cerca de la MTD para Nab-docetaxel. Se ha reportado que la MTD para Taxotere es 15 mg/kg en este esquema.
Ejemplo 37
Este ejemplo compara la actividad antitumoral de Nab-docetaxel y Taxotere usando el xenoinjerto de carcinoma de colon HCT116 en ratones desnudos atímicos con una dosis un 50 % mayor de Nab-docetaxel comparado con Taxotere. Los ratones se dividieron en el grupo control, el grupo de Nab-docetaxel y el grupo de Taxotere (N = 10 ratones por grupo). Todas las formulaciones de Nab-docetaxel descritas en la presente memoria contienen citrato 200 mM y NaCl 300 mM.
Brevemente, la actividad antitumoral de Nab-docetaxel y Taxotere se compararon a dosis de 22 mg/kg q4x3 y 15 mg/kg q4x3, respectivamente en el xenoinjerto de carcinoma de colon HCT116. Los resultados de los experimentos se muestran en la Figura 6.
Como se muestra en la Figura 6, tanto Nab-docetaxel como Taxotere mostraron inhibición tumoral respecto al control. Como se muestra más adelante, la inhibición tumoral se mejoró con Nab-docetaxel frente a Taxotere (p=0,03, ANOVA) y la pérdida de peso fue en cierto modo menor pero no estadísticamente significativa (p=ns, ANOVA) entre los dos grupos.
En este estudio piloto, la actividad antitumoral de Nab-docetaxel fue superior a la de Taxotere. Los ratones toleraron una dosis de docetaxel un 50 % mayor para Nab-docetaxel con una pérdida de peso corporal global en cierto modo menor comparada con Taxotere.
Ejemplo 38
Este ejemplo compara la toxicidad de una preparación de Nab-docetaxel con estabilizantes (citrato/NaCl) frente a Taxotere (Tween-docetaxel) en ratas a las que se proporcionó una única dosis de cada preparación.
Se infundió a ratas Sprague-Dawley macho (160-180 g, n=3/grupo) Taxotere, o Nab-docetaxel (citrato/NaCI). El tiempo de la infusión fue 10 minutos y se usaron los siguientes niveles de dosis de docetaxel: 25, 50, 75, 100 y 125 mg/kg. Los animales se pesaron y monitorizaron diariamente para signos de toxicidad/mortalidad. El porcentaje de mortalidad (%) a los 7 días después del tratamiento se muestra en la Tabla 6.
Tabla 6. Porcentaje de mortalidad en ratas tratadas con Taxotere y Nab-docetaxel.

Como se muestra en la Tabla 6, la formulación de Nab-docetaxel fue significativamente menos tóxica que Taxotere (Tween-docetaxel). Este efecto fue particularmente pronunciado a dosis de 25 y 50 mg/kg. Se calculó que la LD50 era 63 mg/kg para Nab-docetaxel frente a aproximadamente 12,5 mg/kg para Tween-docetaxel.
Ejemplo 39
Este ejemplo muestra la eficacia de Nab-docetaxel en el tratamiento de cáncer de próstata en un modelo de tumor de xenoinjerto de próstata PC3.
Se implantaron tumores PC3 subcutáneamente en ratones desnudos atímicos, se dejó que crecieran hasta 100 mm3 y entonces se trataron q4 x 3, i.v. bien con disolución salina o Nab-docetaxel (10, 15, 20, o 30 mg/kg) o Tweendocetaxel (10 mg/kg). Se evaluaron seis ratones en cada grupo.
Los resultados del estudio se muestran en la Figura 7. Los seis ratones tratados con Tween-docetaxel murieron durante el curso del estudio. Por el contrario, el Nab-docetaxel se toleró bien a todos los niveles de dosis. Sólo hubo una muerte a 15 mg/kg y no se observó ninguna a los niveles de dosis más altos de 20 mg/kg y 30 mg/kg. Se observó supresión tumoral a todos los niveles de dosis de Nab-docetaxel. En particular, a la dosis de 30 mg/kg, hubo seis de seis regresiones completas.
Aunque la invención anterior se ha descrito con algún detalle a modo de ilustración y ejemplo con fines de claridad de la comprensión, está claro para los expertos en la técnica que se practicarán algunos cambios y modificaciones poco importantes. Por lo tanto, la descripción y ejemplos no deben considerarse como limitantes del alcance de la invención.
Claims (23)
1. Una composición que comprende a) nanopartículas que comprenden docetaxel recubierto con albúmina, y b) citrato, en la que la estabilidad de la composición se aumenta comparada con la de la composición sin citrato.
2. La composición según la reivindicación 1, en la que las nanopartículas en la composición tienen un tamaño de partículas promedio o medio no mayor que 200 nm.
3. La composición según la reivindicación 1 o 2, en la que la albúmina es albúmina de suero humano.
4. La composición según una cualquiera de las reivindicaciones 1 a 3, en la que la relación en peso de albúmina a docetaxel es 18:1 o menos.
5. La composición según una cualquiera de las reivindicaciones 1 a 4, en la que la composición es una suspensión líquida con al menos 1 mg/ml de docetaxel.
6. La composición según la reivindicación 5, en la que la composición es una suspensión líquida con al menos 15 mg/ml de docetaxel.
7. La composición según una cualquiera de las reivindicaciones 1 a 6, en la que la composición es una composición seca que se puede reconstituir a una suspensión líquida con al menos 1 mg/ml de docetaxel.
8. La composición según la reivindicación 7, en la que la composición seca es una composición liofilizada.
9. La composición según una cualquiera de las reivindicaciones 1 a 8, en la que el citrato es citrato de sodio.
10. La composición según una cualquiera de las reivindicaciones 1 a 9, en la que la composición comprende además cloruro de sodio.
11. La composición según la reivindicación 10, en la que la composición comprende citrato de sodio 200 mM y cloruro de sodio 300 mM.
12. La composición según una cualquiera de las reivindicaciones 1 a 11, en la que el docetaxel usado para la preparación de la composición está en una forma anhidra.
13. La composición según una cualquiera de las reivindicaciones 1 a 12, en la que la composición es una composición farmacéutica.
14. La composición según una cualquiera de las reivindicaciones 1 a 13, en la que la composición comprende además un azúcar.
15. La composición según la reivindicación 14, en la que el azúcar es sacarosa, manitol, fructosa, lactosa, maltosa, o trehalosa.
16. La composición según una cualquiera de las reivindicaciones 1 a 15, en la que la composición es una composición farmacéutica estéril para administración parenteral.
17. La composición según una cualquiera de las reivindicaciones 1 a 16, en la que la composición es para inyección y tiene un pH no menor de 6,5, 7 u 8.
18. La composición según una cualquiera de las reivindicaciones 1 a 17 para usar en un método de tratamiento de cáncer en un individuo.
19. La composición para usar según la reivindicación 18, en la que el cáncer es cualquiera de cáncer de próstata, cáncer de colon, cáncer de mama, cáncer de cabeza y cuello, cáncer de páncreas, cáncer de pulmón, y cáncer de ovario.
20. Un vial sellado que comprende la composición de una cualquiera de las reivindicaciones 1 a 17.
21. Un método de estabilización de una composición que comprende nanopartículas de docetaxel recubiertas con albúmina, en la que el método comprende combinar la composición con un citrato, en la que la composición resultante es estable en las mismas condiciones en las que la composición es inestable antes de la adición del citrato.
22. El método de la reivindicación 21, en el que el citrato es citrato de sodio.
23. El método de la reivindicación 21 o 22, en el que la composición comprende además cloruro de sodio.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71286505P | 2005-08-31 | 2005-08-31 | |
| US73693105P | 2005-11-14 | 2005-11-14 | |
| US73696205P | 2005-11-14 | 2005-11-14 | |
| PCT/US2006/034103 WO2007027941A2 (en) | 2005-08-31 | 2006-08-30 | Compositions and methods for preparation of poorly water soluble drugs with increased stability |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2719093T3 true ES2719093T3 (es) | 2019-07-08 |
Family
ID=41404560
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES06802739T Active ES2719093T3 (es) | 2005-08-31 | 2006-08-30 | Composiciones de fármacos poco solubles en agua con mayor estabilidad y métodos para su preparación |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US8034765B2 (es) |
| EP (2) | EP3527202A1 (es) |
| JP (1) | JP5368093B2 (es) |
| KR (2) | KR101457834B1 (es) |
| CN (1) | CN101291658B (es) |
| AU (1) | AU2006284657B2 (es) |
| BR (1) | BRPI0615292A8 (es) |
| CA (1) | CA2620389C (es) |
| ES (1) | ES2719093T3 (es) |
| IL (3) | IL189600A (es) |
| NO (3) | NO343793B1 (es) |
| NZ (2) | NZ592132A (es) |
| WO (1) | WO2007027941A2 (es) |
Families Citing this family (158)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5439686A (en) * | 1993-02-22 | 1995-08-08 | Vivorx Pharmaceuticals, Inc. | Methods for in vivo delivery of substantially water insoluble pharmacologically active agents and compositions useful therefor |
| US20030133955A1 (en) * | 1993-02-22 | 2003-07-17 | American Bioscience, Inc. | Methods and compositions useful for administration of chemotherapeutic agents |
| US20070117863A1 (en) * | 1993-02-22 | 2007-05-24 | Desai Neil P | Novel formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| WO1999000113A1 (en) * | 1997-06-27 | 1999-01-07 | Vivorx Pharmaceuticals, Inc. | Novel formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| US8137684B2 (en) | 1996-10-01 | 2012-03-20 | Abraxis Bioscience, Llc | Formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| US8853260B2 (en) | 1997-06-27 | 2014-10-07 | Abraxis Bioscience, Llc | Formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| US20030199425A1 (en) * | 1997-06-27 | 2003-10-23 | Desai Neil P. | Compositions and methods for treatment of hyperplasia |
| GB0011903D0 (en) * | 2000-05-18 | 2000-07-05 | Astrazeneca Ab | Combination chemotherapy |
| TR200502189T1 (tr) * | 2002-12-09 | 2007-01-22 | American Bioscience, Inc. | Kompozisyonlar ve farmakolojik etken maddelerin aktarımı için yöntemler. |
| US8735394B2 (en) | 2005-02-18 | 2014-05-27 | Abraxis Bioscience, Llc | Combinations and modes of administration of therapeutic agents and combination therapy |
| MX2007009960A (es) * | 2005-02-18 | 2008-03-11 | Abraxis Bioscience Inc | Combinaciones y metodos de administracion de agentes terapeuticos y terapia de combinacion. |
| US20070166388A1 (en) * | 2005-02-18 | 2007-07-19 | Desai Neil P | Combinations and modes of administration of therapeutic agents and combination therapy |
| NZ592132A (en) | 2005-08-31 | 2012-12-21 | Abraxis Bioscience Llc | Composition comprising nanoparticles of docitaxel and a citrate |
| EP1931321B1 (en) * | 2005-08-31 | 2018-12-26 | Abraxis BioScience, LLC | Compositions comprising poorly water soluble pharmaceutical agents and antimicrobial agents |
| BRPI0600194A (pt) | 2006-01-30 | 2007-10-23 | Quiral Quimica Do Brasil S A | composições farmacêuticas contendo docetaxel e um inibidor de degradação e processo de obtenção das mesmas |
| US20080280987A1 (en) * | 2006-08-31 | 2008-11-13 | Desai Neil P | Methods of inhibiting angiogenesis and treating angiogenesis-associated diseases |
| US8168661B2 (en) * | 2006-11-06 | 2012-05-01 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| US8178564B2 (en) * | 2006-11-06 | 2012-05-15 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| US8173686B2 (en) | 2006-11-06 | 2012-05-08 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| US8168662B1 (en) | 2006-11-06 | 2012-05-01 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| WO2008076373A1 (en) | 2006-12-14 | 2008-06-26 | Abraxis Bioscience, Llc | Breast cancer therapy based on hormone receptor status with nanoparticles comprising taxane |
| US20110033528A1 (en) * | 2009-08-05 | 2011-02-10 | Poniard Pharmaceuticals, Inc. | Stabilized picoplatin oral dosage form |
| KR101943230B1 (ko) * | 2007-03-07 | 2019-01-28 | 아브락시스 바이오사이언스, 엘엘씨 | 항암제로서 라파마이신 및 알부민을 포함하는 나노입자 |
| US20100290982A1 (en) * | 2007-04-13 | 2010-11-18 | University Of North Texas Health Science Center At Fort Worth | Solid in oil/water emulsion-diffusion-evaporation formulation for preparing curcumin-loaded plga nanoparticles |
| CA2683777C (en) | 2007-04-13 | 2016-08-23 | University Of North Texas Health Science Center At Fort Worth | Formulation of active agent loaded activated plga nanoparticles for targeted cancer nano-therapeutics |
| EP2146695A4 (en) * | 2007-04-23 | 2010-05-19 | Sun Pharmaceuticals Ind Ltd | PHARMACEUTICAL COMPOSITIONS |
| EP3326630A3 (en) | 2007-05-03 | 2018-08-29 | Abraxis BioScience, LLC | Methods and compositions for treating pulmonary hypertension |
| CA2689914C (en) * | 2007-06-01 | 2016-08-16 | Abraxis Bioscience, Llc | Use of protein-bound taxane nanoparticles in the treatment of recurrent gynecological cancers |
| AU2008269179A1 (en) * | 2007-06-22 | 2008-12-31 | Scidose Llc | Solubilized formulation of docetaxel without Tween 80 |
| TW200916094A (en) * | 2007-06-27 | 2009-04-16 | Poniard Pharmaceuticals Inc | Stabilized picoplatin dosage form |
| US20100260832A1 (en) * | 2007-06-27 | 2010-10-14 | Poniard Pharmaceuticals, Inc. | Combination therapy for ovarian cancer |
| WO2009011861A1 (en) * | 2007-07-16 | 2009-01-22 | Poniard Pharmaceuticals, Inc. | Oral formulations for picoplatin |
| WO2009078755A1 (en) * | 2007-12-19 | 2009-06-25 | Ardenia Investments, Ltd. | Drug delivery system for administration of a water soluble, cationic and amphiphilic pharmaceutically active substance |
| WO2009078754A1 (en) * | 2007-12-19 | 2009-06-25 | Ardenia Investments, Ltd. | Drug delivery system for administration of poorly water soluble pharmaceutically active substances |
| WO2009078756A1 (en) | 2007-12-19 | 2009-06-25 | Ardenia Investments, Ltd. | Drug delivery system for administration of a water soluble, cationic and amphiphilic pharmaceutically active substance |
| US9801818B2 (en) | 2007-12-31 | 2017-10-31 | Samyang Biopharmaceuticals Corporation | Method for stabilizing amphiphilic block copolymer micelle composition containing poorly water-soluble drug |
| CN101485629B (zh) | 2008-01-16 | 2013-01-23 | 沈阳药科大学 | 一种给药系统及其制备方法 |
| WO2009099649A1 (en) * | 2008-02-08 | 2009-08-13 | Poniard Pharmaceuticals, Inc. | Use of picoplatin and bevacizumab to treat colorectal cancer |
| WO2009125858A1 (ja) * | 2008-04-07 | 2009-10-15 | 大鵬薬品工業株式会社 | 2-インドリノン誘導体を含有する脂質分散体製剤 |
| MX2010011165A (es) * | 2008-04-10 | 2011-02-22 | Abraxis Bioscience Llc | Composiciones de derivados de taxano hidrofobos y sus usos. |
| KR101100867B1 (ko) * | 2008-05-14 | 2012-01-02 | 주식회사 운화 | 주목의 형성층 또는 전형성층 유래 식물줄기세포주를 유효성분으로 함유하는 항산화, 항염증 또는 항노화용 조성물 |
| JP2012501966A (ja) | 2008-06-16 | 2012-01-26 | バインド バイオサイエンシズ インコーポレイテッド | ビンカアルカロイド含有治療用ポリマーナノ粒子並びにその製造方法及び使用方法 |
| US8420123B2 (en) | 2008-06-16 | 2013-04-16 | Bind Biosciences, Inc. | Drug loaded polymeric nanoparticles and methods of making and using same |
| US8613951B2 (en) * | 2008-06-16 | 2013-12-24 | Bind Therapeutics, Inc. | Therapeutic polymeric nanoparticles with mTor inhibitors and methods of making and using same |
| US8324426B2 (en) * | 2008-08-28 | 2012-12-04 | Telik, Inc. | Formulations of canfosfamide and their preparation |
| US20100056825A1 (en) * | 2008-08-28 | 2010-03-04 | Telik, Inc. | Formulations of canfosfamide and their preparation |
| US8541360B2 (en) * | 2008-11-19 | 2013-09-24 | Ben Venue Laboratories, Inc. | Parenteral formulations comprising sugar-based esters and ethers |
| MX2011006167A (es) | 2008-12-11 | 2011-07-28 | Abraxis Bioscience Llc | Combinaciones y modos de administracion de agentes terapeuticos y terapia de combinacion. |
| US8563041B2 (en) * | 2008-12-12 | 2013-10-22 | Bind Therapeutics, Inc. | Therapeutic particles suitable for parenteral administration and methods of making and using same |
| WO2010075072A2 (en) * | 2008-12-15 | 2010-07-01 | Bind Biosciences | Long circulating nanoparticles for sustained release of therapeutic agents |
| CA2755121A1 (en) | 2009-03-13 | 2010-09-16 | Neil P. Desai | Combination therapy with thiocolchicine derivatives |
| BRPI1014160A2 (pt) * | 2009-04-10 | 2015-08-25 | Abraxis Bioscience Llc | Formulações de nanopartículas e aplicações das mesmas |
| BRPI1012525A2 (pt) * | 2009-04-15 | 2018-07-10 | Abraxis Bioscience, Llc. | composições de nanopatículas isentas de príons e métodos para sua fabricação |
| CN105214143A (zh) | 2009-04-28 | 2016-01-06 | 苏尔莫迪克斯公司 | 用于递送生物活性剂的装置和方法 |
| CN102497858B (zh) * | 2009-06-19 | 2015-03-04 | 纳米模型匈牙利有限公司 | 纳米颗粒替米沙坦组合物及其制备方法 |
| WO2011017835A1 (en) | 2009-08-11 | 2011-02-17 | Nanjing University | Preparation method of protein or peptide nanoparticles for in vivo drug delivery by unfolding and refolding |
| KR20120053052A (ko) | 2009-08-25 | 2012-05-24 | 아브락시스 바이오사이언스, 엘엘씨 | 헤지호그 억제제 및 탁산의 나노입자 조성물로의 조합 요법 |
| US8476310B2 (en) | 2009-10-19 | 2013-07-02 | Scidose Llc | Docetaxel formulations with lipoic acid |
| US7772274B1 (en) | 2009-10-19 | 2010-08-10 | Scidose, Llc | Docetaxel formulations with lipoic acid |
| US8912228B2 (en) | 2009-10-19 | 2014-12-16 | Scidose Llc | Docetaxel formulations with lipoic acid |
| US8541465B2 (en) * | 2009-10-19 | 2013-09-24 | Scidose, Llc | Docetaxel formulations with lipoic acid and/or dihydrolipoic acid |
| US20110092579A1 (en) * | 2009-10-19 | 2011-04-21 | Scidose Llc | Solubilized formulation of docetaxel |
| CA2783535C (en) | 2009-12-11 | 2017-11-28 | Greg Troiano | Stable formulations for lyophilizing therapeutic particles |
| ES2780156T3 (es) | 2009-12-15 | 2020-08-24 | Pfizer | Composiciones terapéuticas de nanopartículas poliméricas con alta temperatura de transición vítrea o copolímeros de alto peso molecular |
| EP2528601A1 (en) | 2010-01-26 | 2012-12-05 | Yissum Research Development Company of the Hebrew University of Jerusalem Ltd. | Compositions and methods for prevention and treatment of pulmonary hypertension |
| US9333189B2 (en) * | 2010-02-03 | 2016-05-10 | Oncbiomune, Inc. | Taxane- and taxoid-protein compositions |
| MX359413B (es) | 2010-03-26 | 2018-09-27 | Abraxis Bioscience Llc | Metodos de tratamiento de carcinoma hepatocelular. |
| CA2794147A1 (en) | 2010-03-29 | 2011-10-06 | Abraxis Bioscience, Llc | Use of a composition comprising nanoparticles comprising a taxane and an albumin to improve uptake of chemotherapeutics by tumors and for treating a cancer that is highly fibrotic and/or has a dense stroma |
| NZ602385A (en) | 2010-03-29 | 2014-08-29 | Abraxis Bioscience Llc | Methods of treating cancer |
| EP3279319B1 (en) | 2010-04-26 | 2020-06-10 | Novozymes A/S | Enzyme granules |
| KR101752944B1 (ko) | 2010-05-03 | 2017-07-03 | 테이코쿠 팔마 유에스에이, 인코포레이티드 | 비수성의 탁산 프로에멀젼 제제, 및 그의 제조 및 사용 방법 |
| NZ708506A (en) | 2010-06-02 | 2016-08-26 | Abraxis Bioscience Llc | Methods of treating bladder cancer |
| CN103140225A (zh) | 2010-06-04 | 2013-06-05 | 阿布拉科斯生物科学有限公司 | 治疗胰腺癌的方法 |
| MY166014A (en) | 2010-06-07 | 2018-05-21 | Abraxis Bioscience Llc | Combination therapy methods for treating proliferative diseases |
| TWI511725B (zh) * | 2010-11-10 | 2015-12-11 | Tasly Holding Group Co Ltd | A taxane-containing drug solution containing chelating agent and a preparation method thereof |
| WO2012101242A1 (en) * | 2011-01-27 | 2012-08-02 | Capsulution Pharma Ag | Novel pharmaceutical suspension for parenteral application |
| CA2831848C (en) | 2011-04-01 | 2020-12-22 | Astrazeneca Ab | Combination comprising (s)-4-amino-n-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide and a taxane |
| JP6038123B2 (ja) | 2011-04-28 | 2016-12-07 | アブラクシス バイオサイエンス, エルエルシー | ナノ粒子組成物の脈管内送達およびそれらの使用 |
| ES2792062T3 (es) | 2011-05-09 | 2020-11-06 | Mayo Found Medical Education & Res | Tratamientos contra el cáncer |
| US10213529B2 (en) | 2011-05-20 | 2019-02-26 | Surmodics, Inc. | Delivery of coated hydrophobic active agent particles |
| US9861727B2 (en) | 2011-05-20 | 2018-01-09 | Surmodics, Inc. | Delivery of hydrophobic active agent particles |
| US8883177B2 (en) * | 2011-06-28 | 2014-11-11 | Nian Wu | Pharmaceutical compositions for parenteral administration |
| JP6162709B2 (ja) | 2011-11-01 | 2017-07-12 | セルジーン コーポレイション | シチジンアナログの経口製剤を使用して癌を治療する方法 |
| FI2785349T4 (fi) | 2011-11-30 | 2023-02-09 | Syövän yhdistelmähoito | |
| WO2013085902A1 (en) | 2011-12-05 | 2013-06-13 | The University Of Texas M.D. | Combination therapy methods for treating an inflammatory breast cancer |
| PT2790675T (pt) | 2011-12-14 | 2019-09-23 | Abraxis Bioscience Llc | Utilização de excipientes poliméricos para a liofilização ou a congelação de partículas |
| WO2013115559A1 (ko) * | 2012-01-30 | 2013-08-08 | 성균관대학교 산학협력단 | 수난용성 약물을 내부에 포함하는 알부민 나노입자 제조방법 |
| WO2014002108A1 (en) | 2012-06-27 | 2014-01-03 | Amrita Vishwa Vidyapeetham University | A core-shell nanostructure on the basis of proteins with corresponding therapeutic agents |
| ES2673093T3 (es) | 2012-04-04 | 2018-06-19 | Halozyme, Inc. | Terapia combinada con hialuronidasa y un taxano dirigido a un tumor |
| AU2013204533B2 (en) | 2012-04-17 | 2017-02-02 | Astrazeneca Ab | Crystalline forms |
| US20150140109A1 (en) * | 2012-05-18 | 2015-05-21 | Unichempharm Ltd | Docetaxel-based prolonged-release cancer treatment drug |
| JP6039256B2 (ja) * | 2012-06-15 | 2016-12-07 | 高砂香料工業株式会社 | 可溶化促進剤 |
| SI2895156T1 (sl) | 2012-09-17 | 2019-08-30 | Pfizer Inc. | Postopek za pripravo terapevtskih nanodelcev |
| EP2903610B1 (en) | 2012-10-01 | 2021-11-03 | Mayo Foundation For Medical Education And Research | Cancer treatments |
| JO3685B1 (ar) * | 2012-10-01 | 2020-08-27 | Teikoku Pharma Usa Inc | صيغ التشتيت الجسيمي للتاكسين غير المائي وطرق استخدامها |
| US11246963B2 (en) | 2012-11-05 | 2022-02-15 | Surmodics, Inc. | Compositions and methods for delivery of hydrophobic active agents |
| US9149455B2 (en) | 2012-11-09 | 2015-10-06 | Abraxis Bioscience, Llc | Methods of treating melanoma |
| EA022182B1 (ru) * | 2012-12-24 | 2015-11-30 | Общество С Ограниченной Ответственностью "Технология Лекарств" | Способ получения липосомальной формы доцетаксела |
| CN103908430A (zh) * | 2013-01-07 | 2014-07-09 | 苏州纳晶医药技术有限公司 | 一种稳定的包裹紫杉醇固体蛋白纳米粒及其质量控制方法 |
| US9511046B2 (en) | 2013-01-11 | 2016-12-06 | Abraxis Bioscience, Llc | Methods of treating pancreatic cancer |
| EP2950797B1 (en) | 2013-02-01 | 2019-09-11 | Glialogix, Inc. | Compositions and methods for the treatment of neurodegenerative and other diseases |
| KR102148551B1 (ko) | 2013-02-11 | 2020-08-26 | 아브락시스 바이오사이언스, 엘엘씨 | 흑색종을 치료하는 방법 |
| MX362905B (es) | 2013-03-04 | 2019-02-25 | Astrazeneca Ab | Tratamiento de combinacion. |
| EP2968254B1 (en) | 2013-03-12 | 2020-04-22 | Abraxis BioScience, LLC | Methods of treating lung cancer |
| EP2968253A4 (en) | 2013-03-13 | 2016-11-02 | Abraxis Bioscience Llc | METHODS OF TREATING PEDIATRIC SOLID TUMOR |
| ES2881851T3 (es) | 2013-03-14 | 2021-11-30 | Abraxis Bioscience Llc | Métodos para tratar el cáncer de vejiga |
| KR20160024985A (ko) * | 2013-06-28 | 2016-03-07 | 바인드 쎄라퓨틱스, 인크. | 암 치료를 위한 도세탁셀 중합체성 나노입자 |
| SI3116547T1 (sl) | 2014-03-14 | 2019-08-30 | Pfizer, Inc. | Terapevtski nanodelci, ki obsegajo terapevtsko stredstvo, in postopek izdelave ter uporabe le-teh |
| EP3154595A4 (en) | 2014-06-16 | 2018-01-24 | Mayo Foundation for Medical Education and Research | Treating myelomas |
| US20150359810A1 (en) | 2014-06-17 | 2015-12-17 | Celgene Corporation | Methods for treating epstein-barr virus (ebv) associated cancers using oral formulations of 5-azacytidine |
| US9446148B2 (en) | 2014-10-06 | 2016-09-20 | Mayo Foundation For Medical Education And Research | Carrier-antibody compositions and methods of making and using the same |
| SI3215133T1 (sl) * | 2014-11-05 | 2021-07-30 | Selecta Biosciences, Inc. | Postopki in sestavki v zvezi z uporabo površinsko aktivnih snovi z nizkim HLB v proizvodnji sintetičnih nanonosilcev, ki vsebujejo rapalog |
| US10705070B1 (en) | 2015-03-05 | 2020-07-07 | Abraxis Bioscience, Llc | Methods of assessing suitability of use of pharmaceutical compositions of albumin and poorly water soluble drug |
| US10527604B1 (en) | 2015-03-05 | 2020-01-07 | Abraxis Bioscience, Llc | Methods of assessing suitability of use of pharmaceutical compositions of albumin and paclitaxel |
| CN106137969B (zh) * | 2015-04-03 | 2020-05-15 | 四川科伦药物研究院有限公司 | 多西他赛白蛋白纳米粒药物组合物及其制备方法及应用 |
| US20160346219A1 (en) | 2015-06-01 | 2016-12-01 | Autotelic Llc | Phospholipid-coated therapeutic agent nanoparticles and related methods |
| EP3302431B1 (en) | 2015-06-04 | 2020-11-11 | Crititech, Inc. | Taxane particles and their use |
| ES2897991T3 (es) | 2015-06-29 | 2022-03-03 | Abraxis Bioscience Llc | Nanopartículas que comprenden sirolimus y una albúmina para uso en tratamiento de tumores de células epitelioides |
| TW201707725A (zh) | 2015-08-18 | 2017-03-01 | 美國馬友醫藥教育研究基金會 | 載體-抗體組合物及其製造及使用方法 |
| CN105012251A (zh) * | 2015-08-24 | 2015-11-04 | 吉林大学 | 注射用紫杉烷类药物白蛋白纳米粒冻干制剂及制备方法 |
| CA2998483C (en) | 2015-09-16 | 2022-09-06 | Dfb Soria, Llc | Delivery of drug nanoparticles and methods of use thereof |
| TW201713360A (en) | 2015-10-06 | 2017-04-16 | Mayo Foundation | Methods of treating cancer using compositions of antibodies and carrier proteins |
| EP3399861A4 (en) | 2016-01-07 | 2019-08-07 | Mayo Foundation for Medical Education and Research | INTERFERON CANCER TREATMENT METHODS |
| AU2017217881B2 (en) | 2016-02-12 | 2022-11-17 | Mayo Foundation For Medical Education And Research | Hematologic cancer treatments |
| CN108700566A (zh) | 2016-02-19 | 2018-10-23 | 河谷控股Ip有限责任公司 | 免疫原性调节的方法 |
| AU2017238119A1 (en) | 2016-03-21 | 2018-10-11 | Mayo Foundation For Medical Education And Research | Methods for reducing toxicity of a chemotherapeutic drug |
| WO2017165439A1 (en) | 2016-03-21 | 2017-09-28 | Mayo Foundation For Medical Education And Research | Methods for improving the therapeutic index for a chemotherapeutic drug |
| US11154597B2 (en) | 2016-03-24 | 2021-10-26 | Nantcell, Inc. | Sequence arrangements and sequences for neoepitope presentation |
| CN109475492B (zh) | 2016-04-04 | 2022-04-29 | 克里蒂泰克公司 | 实体肿瘤治疗方法 |
| US10618969B2 (en) | 2016-04-06 | 2020-04-14 | Mayo Foundation For Medical Education And Research | Carrier-binding agent compositions and methods of making and using the same |
| CA3027911A1 (en) | 2016-06-30 | 2018-01-04 | Nant Holdings Ip, Llc | Coordinated treatment regimen to treat a tumor |
| KR20190053203A (ko) | 2016-09-01 | 2019-05-17 | 메이오 파운데이션 포 메디칼 에쥬케이션 앤드 리써치 | T-세포 암을 표적화하는 방법 및 조성물 |
| EP3506942B1 (en) | 2016-09-01 | 2022-11-16 | Mayo Foundation for Medical Education and Research | Carrier-pd-l1 binding agent compositions for treating cancers |
| US11311631B2 (en) * | 2016-09-06 | 2022-04-26 | Mayo Foundation For Medical Education And Research | Paclitaxel-albumin-binding agent compositions and methods for using and making the same |
| CA3035655A1 (en) | 2016-09-06 | 2018-03-15 | Mayo Foundation For Medical Education And Research | Methods of treating pd-l1 expressing cancer |
| WO2018048815A1 (en) | 2016-09-06 | 2018-03-15 | Nantibodyfc, Llc | Methods of treating triple-negative breast cancer using compositions of antibodies and carrier proteins |
| TW201818933A (zh) | 2016-10-21 | 2018-06-01 | 美商吉利亞洛吉克斯公司 | 用於神經退化性及其他疾病的治療之組成物和方法 |
| US11564944B2 (en) | 2016-11-21 | 2023-01-31 | Nant Holdings Ip, Llc | Fractal combination therapy |
| US10898446B2 (en) | 2016-12-20 | 2021-01-26 | Surmodics, Inc. | Delivery of hydrophobic active agents from hydrophilic polyether block amide copolymer surfaces |
| AU2018219862B2 (en) | 2017-02-07 | 2020-09-17 | Nant Holding IP, LLC | Maximizing T-cell memory and compositions and methods therefor |
| ES2955884T3 (es) | 2017-03-15 | 2023-12-07 | Dfb Soria Llc | Terapia tópica para el tratamiento de malignidades de la piel con nanoparticulas de taxanos |
| JP7051898B2 (ja) | 2017-04-24 | 2022-04-11 | ナントセル,インコーポレイテッド | 標的型ネオエピトープベクター及びそのための方法 |
| JP2020523285A (ja) | 2017-06-09 | 2020-08-06 | クリチテック,インコーポレイテッド | 抗腫瘍粒子の嚢胞内注入による上皮嚢胞の治療 |
| SG11201909837YA (en) | 2017-06-14 | 2019-11-28 | Crititech Inc | Methods for treating lung disorders |
| CN111278436A (zh) | 2017-10-03 | 2020-06-12 | 克里蒂泰克公司 | 局部递送抗肿瘤颗粒与全身递送免疫治疗剂相结合用于治疗癌症 |
| BR112020009055A2 (pt) | 2017-11-06 | 2020-11-03 | Rapt Therapeutics, Inc. | moduladores de receptor de quimiocina para tratamento de câncer positivo para vírus epstein barr |
| EP3740497A4 (en) | 2018-01-17 | 2021-10-20 | NantBio, Inc. | INCREASED IMMUNOGENICITY FOR GPI ANCHORED ANTIGENS |
| US11497726B2 (en) | 2018-03-16 | 2022-11-15 | Dfb Soria, Ll. | Topical therapy for the treatment of cervical intraepithelial neoplasia (CIN) and cervical cancer using nanoparticles of taxanes |
| CN112188892A (zh) | 2018-03-20 | 2021-01-05 | 阿布拉科斯生物科学有限公司 | 通过mTOR抑制剂和白蛋白的纳米颗粒的施用治疗中枢神经系统障碍的方法 |
| US11823773B2 (en) | 2018-04-13 | 2023-11-21 | Nant Holdings Ip, Llc | Nant cancer vaccine strategies |
| WO2019222435A1 (en) | 2018-05-16 | 2019-11-21 | Halozyme, Inc. | Methods of selecting subjects for combination cancer therapy with a polymer-conjugated soluble ph20 |
| CN110496103B (zh) * | 2018-05-18 | 2022-05-10 | 上海维洱生物医药科技有限公司 | 一种多西他赛棕榈酸酯脂质体及其制备方法 |
| WO2019231499A1 (en) * | 2018-05-31 | 2019-12-05 | Crititech, Inc. | Use of antineoplastic agents to stimulate the immune system for treatment of cancer |
| KR20220106758A (ko) | 2019-10-28 | 2022-07-29 | 아브락시스 바이오사이언스, 엘엘씨 | 알부민 및 라파마이신의 제약 조성물 |
| EP4054550A4 (en) * | 2019-11-05 | 2024-01-17 | Luminus Biosciences Inc | NANOPARTICLES WITH PRODRUGS STABILIZED BY ALBUMIN FOR THE TREATMENT OF CANCER AND OTHER DISEASES |
| KR102576559B1 (ko) * | 2021-06-14 | 2023-09-08 | 충북대학교 산학협력단 | 도세탁셀 및 아파티닙이 봉입된 알부민 나노입자 및 이의 용도 |
| WO2023194441A1 (en) | 2022-04-05 | 2023-10-12 | Istituto Nazionale Tumori Irccs - Fondazione G. Pascale | Combination of hdac inhibitors and statins for use in the treatment of pancreatic cancer |
| WO2024081674A1 (en) | 2022-10-11 | 2024-04-18 | Aadi Bioscience, Inc. | Combination therapies for the treatment of cancer |
Family Cites Families (117)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4826689A (en) | 1984-05-21 | 1989-05-02 | University Of Rochester | Method for making uniformly sized particles from water-insoluble organic compounds |
| FR2601675B1 (fr) * | 1986-07-17 | 1988-09-23 | Rhone Poulenc Sante | Derives du taxol, leur preparation et les compositions pharmaceutiques qui les contiennent |
| FR2608988B1 (fr) * | 1986-12-31 | 1991-01-11 | Centre Nat Rech Scient | Procede de preparation de systemes colloidaux dispersibles d'une substance, sous forme de nanoparticules |
| US5334582A (en) * | 1988-06-22 | 1994-08-02 | Applied Microbiology, Inc. | Pharmaceutical bacteriocin compositions and methods for using the same |
| US4960799A (en) * | 1988-09-13 | 1990-10-02 | Ciba-Geigy Corporation | Stabilized aqueous solutions of pharmaceutically acceptable salts of ortho-(2,6-dichlorophenyl)-aminophenylacetic acid for opthalmic use |
| US5580575A (en) * | 1989-12-22 | 1996-12-03 | Imarx Pharmaceutical Corp. | Therapeutic drug delivery systems |
| US6120805A (en) * | 1990-04-06 | 2000-09-19 | Rhone-Poulenc Rorer Sa | Microspheres, process for their preparation and their use |
| US5725804A (en) * | 1991-01-15 | 1998-03-10 | Hemosphere, Inc. | Non-crosslinked protein particles for therapeutic and diagnostic use |
| US5145684A (en) | 1991-01-25 | 1992-09-08 | Sterling Drug Inc. | Surface modified drug nanoparticles |
| PT835657E (pt) | 1992-11-27 | 2004-11-30 | Mayne Pharma Usa Inc | Composicao injectavel estavel de paclitaxel |
| US6096331A (en) * | 1993-02-22 | 2000-08-01 | Vivorx Pharmaceuticals, Inc. | Methods and compositions useful for administration of chemotherapeutic agents |
| US5916596A (en) * | 1993-02-22 | 1999-06-29 | Vivorx Pharmaceuticals, Inc. | Protein stabilized pharmacologically active agents, methods for the preparation thereof and methods for the use thereof |
| US6528067B1 (en) * | 1993-02-22 | 2003-03-04 | American Bioscience, Inc. | Total nutrient admixtures as stable multicomponent liquids or dry powders and methods for the preparation thereof |
| US6749868B1 (en) * | 1993-02-22 | 2004-06-15 | American Bioscience, Inc. | Protein stabilized pharmacologically active agents, methods for the preparation thereof and methods for the use thereof |
| US5665383A (en) * | 1993-02-22 | 1997-09-09 | Vivorx Pharmaceuticals, Inc. | Methods for the preparation of immunostimulating agents for in vivo delivery |
| US5665382A (en) * | 1993-02-22 | 1997-09-09 | Vivorx Pharmaceuticals, Inc. | Methods for the preparation of pharmaceutically active agents for in vivo delivery |
| PT693924E (pt) * | 1993-02-22 | 2004-09-30 | American Biosciences | Processos para administracao (in vivo) de substancias biologicas e composicoes utilizadas nestes processos |
| US5439686A (en) * | 1993-02-22 | 1995-08-08 | Vivorx Pharmaceuticals, Inc. | Methods for in vivo delivery of substantially water insoluble pharmacologically active agents and compositions useful therefor |
| US20070117863A1 (en) * | 1993-02-22 | 2007-05-24 | Desai Neil P | Novel formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| US5362478A (en) | 1993-03-26 | 1994-11-08 | Vivorx Pharmaceuticals, Inc. | Magnetic resonance imaging with fluorocarbons encapsulated in a cross-linked polymeric shell |
| US5997904A (en) | 1993-02-22 | 1999-12-07 | American Bioscience, Inc. | Total nutrient admixtures as stable multicomponent liquids or dry powders and methods for the preparation thereof |
| US6537579B1 (en) * | 1993-02-22 | 2003-03-25 | American Bioscience, Inc. | Compositions and methods for administration of pharmacologically active compounds |
| US5650156A (en) * | 1993-02-22 | 1997-07-22 | Vivorx Pharmaceuticals, Inc. | Methods for in vivo delivery of nutriceuticals and compositions useful therefor |
| US6753006B1 (en) * | 1993-02-22 | 2004-06-22 | American Bioscience, Inc. | Paclitaxel-containing formulations |
| WO1999000113A1 (en) * | 1997-06-27 | 1999-01-07 | Vivorx Pharmaceuticals, Inc. | Novel formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| DE69435002T2 (de) * | 1993-07-19 | 2008-03-20 | Angiotech Pharmaceuticals, Inc., Vancouver | Anti-angiogene Mittel enthaltend Taxol und einen nicht biodegradierbaren Träger und deren Verwendung |
| GB9405593D0 (en) * | 1994-03-22 | 1994-05-11 | Zeneca Ltd | Pharmaceutical compositions |
| US5731356A (en) * | 1994-03-22 | 1998-03-24 | Zeneca Limited | Pharmaceutical compositions of propofol and edetate |
| FR2722191B1 (fr) | 1994-07-08 | 1996-08-23 | Rhone Poulenc Rorer Sa | Procede de preparation du trihydrate du (2r,3s)-3-tertbutoxycarbonylamino-2-hydroxy-3-phenylpropionate de 4-acetoxy2alpha-benzoyloxy-5beta,20epoxy-1,7beta,10beta trihydroxy-9-oxo-tax-11-en-13alpha-yle |
| DE4438577A1 (de) * | 1994-10-28 | 1996-05-02 | Basf Ag | Selbsttragende Dübelmasse für die chemische Befestigungstechnik |
| US5681846A (en) * | 1995-03-17 | 1997-10-28 | Board Of Regents, The University Of Texas System | Extended stability formulations for paclitaxel |
| US6964947B1 (en) * | 1995-11-07 | 2005-11-15 | Genentech, Inc. | Stabilizing formulation for NGF |
| US6537539B2 (en) * | 1996-01-11 | 2003-03-25 | Human Genome Sciences, Inc. | Immune cell cytokine |
| US6441025B2 (en) * | 1996-03-12 | 2002-08-27 | Pg-Txl Company, L.P. | Water soluble paclitaxel derivatives |
| EP0932399B1 (en) * | 1996-03-12 | 2006-01-04 | PG-TXL Company, L.P. | Water soluble paclitaxel prodrugs |
| US5637625A (en) * | 1996-03-19 | 1997-06-10 | Research Triangle Pharmaceuticals Ltd. | Propofol microdroplet formulations |
| KR100330373B1 (ko) * | 1996-05-28 | 2002-11-07 | 주식회사한국신약 | 탁솔을 함유한 주사용 약제 조성물 |
| US5731556A (en) * | 1996-09-30 | 1998-03-24 | Ingersoll-Rand Company | Muffler for pneumatic device |
| US8137684B2 (en) * | 1996-10-01 | 2012-03-20 | Abraxis Bioscience, Llc | Formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| US20050019266A1 (en) * | 1997-05-06 | 2005-01-27 | Unger Evan C. | Novel targeted compositions for diagnostic and therapeutic use |
| US20020039594A1 (en) * | 1997-05-13 | 2002-04-04 | Evan C. Unger | Solid porous matrices and methods of making and using the same |
| US6416740B1 (en) | 1997-05-13 | 2002-07-09 | Bristol-Myers Squibb Medical Imaging, Inc. | Acoustically active drug delivery systems |
| CH692322A5 (it) | 1997-05-26 | 2002-05-15 | Westy Ag | Formulazione iniettabile limpida di Propofol. |
| US20030199425A1 (en) | 1997-06-27 | 2003-10-23 | Desai Neil P. | Compositions and methods for treatment of hyperplasia |
| US8853260B2 (en) * | 1997-06-27 | 2014-10-07 | Abraxis Bioscience, Llc | Formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof |
| US6217886B1 (en) * | 1997-07-14 | 2001-04-17 | The Board Of Trustees Of The University Of Illinois | Materials and methods for making improved micelle compositions |
| DK1052975T3 (da) | 1998-02-10 | 2007-12-27 | Sicor Inc | Propofolsammensætning indeholdende sulfit |
| US6914130B2 (en) * | 1998-06-17 | 2005-07-05 | Genentech, Inc. | Compositions and methods for the diagnosis and treatment of tumor |
| AU5241199A (en) * | 1998-07-30 | 2000-02-21 | Novopharm Biotech Inc. | Pharmaceutically acceptable composition comprising an aqueous solution of paclitaxel and albumin |
| DE69912441T2 (de) * | 1998-08-19 | 2004-08-19 | Skyepharma Canada Inc., Verdun | Injizierbare wässerige propofoldispersionen |
| US6150423A (en) | 1998-10-15 | 2000-11-21 | Phoenix Scientific, Inc. | Propofol-based anesthetic and method of making same |
| US6028108A (en) * | 1998-10-22 | 2000-02-22 | America Home Products Corporation | Propofol composition comprising pentetate |
| US6140374A (en) | 1998-10-23 | 2000-10-31 | Abbott Laboratories | Propofol composition |
| US6140373A (en) * | 1998-10-23 | 2000-10-31 | Abbott Laboratories | Propofol composition |
| KR100638150B1 (ko) * | 1998-12-23 | 2006-10-26 | 이데아 악티엔게젤샤프트 | 생체내에서 국부적인 비침습성 용도로 사용되는 개선된 제제 |
| PT1143962E (pt) * | 1999-01-28 | 2005-03-31 | Kurani Shashikant Prabhudas | Solucao parenterica de propofol (2,6-di-isopropilfenol) e 2,5-di-o-metil-1,4;3,6-di-anidro-d-glucitol, como dissolvente |
| US6177477B1 (en) * | 1999-03-24 | 2001-01-23 | American Home Products Corporation | Propofol formulation containing TRIS |
| US6100302A (en) * | 1999-04-05 | 2000-08-08 | Baxter International Inc. | Propofol formulation with enhanced microbial characteristics |
| WO2000064437A1 (en) * | 1999-04-22 | 2000-11-02 | American Biosciences, Inc. | Long term administration of pharmacologically active agents |
| US6610317B2 (en) * | 1999-05-27 | 2003-08-26 | Acusphere, Inc. | Porous paclitaxel matrices and methods of manufacture thereof |
| EP1202719B1 (en) * | 1999-06-21 | 2006-05-31 | Kuhnil Pharmaceutical Co., Ltd. | Anesthetic composition for intravenous injection comprising propofol |
| GB9920548D0 (en) * | 1999-08-31 | 1999-11-03 | Rhone Poulenc Rorer Sa | Treatment of hepatocellular carcinoma |
| ITMI20001107A1 (it) | 2000-05-18 | 2001-11-18 | Acs Dobfar Spa | Metodo per il trattamento di tumori solici mediante microparticelle di albumina incorporanti paclitaxel |
| IL153457A0 (en) * | 2000-06-16 | 2003-07-06 | Skyepharma Canada Inc | Improved injectable dispersions of propofol |
| AU2001270310A1 (en) * | 2000-07-07 | 2002-01-21 | Guilford Pharmaceuticals Inc. | Compositions for sustained release of antineoplastic taxanes, and methods of making and using the same |
| DE10036871A1 (de) * | 2000-07-28 | 2002-02-14 | Pharmasol Gmbh | Dispersionen zur Formulierung wenig oder schwer löslicher Wirkstoffe |
| US6623765B1 (en) | 2000-08-01 | 2003-09-23 | University Of Florida, Research Foundation, Incorporated | Microemulsion and micelle systems for solubilizing drugs |
| JP2004536026A (ja) * | 2000-11-28 | 2004-12-02 | トランスフォーム ファーマシューティカルズ,インコーポレーティッド. | パクリタキセル、その誘導体、および薬剤として許容される塩を含む医薬品製剤 |
| US6399087B1 (en) * | 2000-12-20 | 2002-06-04 | Amphastar Pharmaceuticals, Inc. | Propofol formulation with enhanced microbial inhibition |
| US6972448B2 (en) * | 2000-12-31 | 2005-12-06 | Texas Instruments Incorporated | Sub-lithographics opening for back contact or back gate |
| US20030157161A1 (en) * | 2001-05-01 | 2003-08-21 | Angiotech Pharmaceuticals, Inc. | Compositions and methods for treating inflammatory conditions utilizing protein or polysaccharide containing anti-microtubule agents |
| US20030099674A1 (en) * | 2001-08-11 | 2003-05-29 | Chen Andrew X. | Lyophilized injectable formulations containing paclitaxel or other taxoid drugs |
| KR100774366B1 (ko) * | 2001-09-10 | 2007-11-08 | 주식회사 중외제약 | 파클리탁셀 주사제 조성물 |
| US20040210289A1 (en) | 2002-03-04 | 2004-10-21 | Xingwu Wang | Novel nanomagnetic particles |
| US20040143004A1 (en) * | 2002-02-26 | 2004-07-22 | Joseph Fargnoli | Metronomic dosing of taxanes |
| ITMI20020680A1 (it) | 2002-03-29 | 2003-09-29 | Acs Dobfar Spa | Composizione antitumorale migliorata a base di paclitaxel e metodo per il suo ottenimento |
| ITMI20020681A1 (it) * | 2002-03-29 | 2003-09-29 | Acs Dobfar Spa | Procedimento per la produzione di nanoparticelle di paclitaxel ed albumina |
| US20040009168A1 (en) * | 2002-04-05 | 2004-01-15 | Elizabet Kaisheva | Multidose antibody formulation |
| EP2305710A3 (en) | 2002-06-03 | 2013-05-29 | Genentech, Inc. | Synthetic antibody phage libraries |
| AU2003279070A1 (en) * | 2002-09-30 | 2004-04-23 | Acusphere Inc | Sustained release porous microparticles for inhalation |
| US20040126360A1 (en) * | 2002-10-09 | 2004-07-01 | Manning Mark C. | Oral formulations for proteins and polypeptides |
| TR200502189T1 (tr) * | 2002-12-09 | 2007-01-22 | American Bioscience, Inc. | Kompozisyonlar ve farmakolojik etken maddelerin aktarımı için yöntemler. |
| KR20050095826A (ko) * | 2002-12-09 | 2005-10-04 | 아메리칸 바이오사이언스, 인크. | 약리학적 물질의 조성물 및 그 전달방법 |
| US6838569B2 (en) | 2002-12-16 | 2005-01-04 | Dabur India Limited | Process for preparation of paclitaxel trihydrate and docetaxel trihydrate |
| US20040171560A1 (en) * | 2002-12-23 | 2004-09-02 | Dabur Research Foundation | Stabilized pharmaceutical composition |
| UY28213A1 (es) | 2003-02-28 | 2004-09-30 | Bayer Pharmaceuticals Corp | Nuevos derivados de cianopiridina útiles en el tratamiento de cáncer y otros trastornos. |
| US6900184B2 (en) | 2003-04-14 | 2005-05-31 | Wyeth Holdings Corporation | Compositions containing pipercillin and tazobactam useful for injection |
| CN1268619C (zh) * | 2003-05-08 | 2006-08-09 | 上海迪赛诺化学制药有限公司 | 多烯紫杉醇三水化合物的制备方法 |
| KR20160114727A (ko) | 2003-05-30 | 2016-10-05 | 제넨테크, 인크. | 항-vegf 항체를 사용한 치료 |
| CN1838942A (zh) * | 2003-07-11 | 2006-09-27 | 普罗医药公司 | 递送疏水性药物的组合物和方法 |
| US20050152979A1 (en) * | 2003-09-05 | 2005-07-14 | Cell Therapeutics, Inc. | Hydrophobic drug compositions containing reconstitution enhancer |
| SG195524A1 (en) | 2003-11-06 | 2013-12-30 | Seattle Genetics Inc | Monomethylvaline compounds capable of conjugation to ligands |
| EP1692279A4 (en) | 2003-11-19 | 2006-12-27 | Merck & Co Inc | VIRUS PREPARATIONS CONTAINING A CONSERVATIVE |
| WO2006055008A2 (en) | 2003-11-20 | 2006-05-26 | Angiotech International Ag | Implantable sensors and implantable pumps and anti-scarring agents |
| ES2325506T3 (es) * | 2003-12-12 | 2009-09-07 | Quiral Quimica Do Brasil | Procedimiento para la preparacion de principios activos farmaceuticos (api) anhidros e hidratados; composiciones farmaceuticas estables preparadas a partir de los mismos y usos de dichas composiciones. |
| KR20050099311A (ko) * | 2004-04-09 | 2005-10-13 | 에이엔에이치 케어연구소(주) | 주사제용 항암제 조성물 |
| US8420603B2 (en) * | 2004-05-14 | 2013-04-16 | Abraxis Bioscience, Llc | SPARC and methods of use thereof |
| US20060079672A1 (en) * | 2004-10-07 | 2006-04-13 | Paul Glidden | Kits for modulating angiogenesis |
| KR101320936B1 (ko) | 2004-12-21 | 2013-10-23 | 넥타르 테라퓨틱스 | 안정화된 중합체성 티올 시약 |
| MX2007009960A (es) * | 2005-02-18 | 2008-03-11 | Abraxis Bioscience Inc | Combinaciones y metodos de administracion de agentes terapeuticos y terapia de combinacion. |
| US8735394B2 (en) | 2005-02-18 | 2014-05-27 | Abraxis Bioscience, Llc | Combinations and modes of administration of therapeutic agents and combination therapy |
| JP2008535475A (ja) * | 2005-02-18 | 2008-09-04 | アブラクシス バイオサイエンス、インコーポレイテッド | Q3sparc欠失変異体及びその使用 |
| US20070166388A1 (en) * | 2005-02-18 | 2007-07-19 | Desai Neil P | Combinations and modes of administration of therapeutic agents and combination therapy |
| BRPI0608173A2 (pt) * | 2005-02-24 | 2010-11-09 | Elan Pharma Int Ltd | composição, uso da mesma, e, método de produzir uma composição de docetaxel nanoparticulada ou análogo do mesmo |
| EP1862183B1 (en) | 2005-03-14 | 2010-11-03 | Otsuka Pharmaceutical Factory, Inc. | Kit comprising drug and fat emulsion |
| CA2611592A1 (en) | 2005-06-17 | 2006-12-21 | Hospira Australia Pty Ltd | Liquid pharmaceutical formulations of docetaxel |
| AU2006257718A1 (en) * | 2005-06-17 | 2006-12-21 | Hospira Australia Pty Ltd | Liquid pharmaceutical formulations of docetaxel |
| US20070025910A1 (en) * | 2005-07-29 | 2007-02-01 | Norenberg Jeffrey P | Anticancer therapy |
| NZ592132A (en) | 2005-08-31 | 2012-12-21 | Abraxis Bioscience Llc | Composition comprising nanoparticles of docitaxel and a citrate |
| EP1931321B1 (en) | 2005-08-31 | 2018-12-26 | Abraxis BioScience, LLC | Compositions comprising poorly water soluble pharmaceutical agents and antimicrobial agents |
| EP1819688A2 (en) | 2005-10-12 | 2007-08-22 | Sicor, Inc. | Crystalline forms of docetaxel and processes for their preparation |
| WO2007096900A1 (en) | 2006-02-21 | 2007-08-30 | Dabur Pharma Limited | Stable pharmaceutical composition of taxanes |
| AU2007226937A1 (en) | 2006-03-21 | 2007-09-27 | Dr. Reddy's Laboratories Ltd. | Docetaxel polymorphs and processes |
| US20080280987A1 (en) | 2006-08-31 | 2008-11-13 | Desai Neil P | Methods of inhibiting angiogenesis and treating angiogenesis-associated diseases |
| US8909947B2 (en) * | 2006-09-28 | 2014-12-09 | Dell Products L.P. | System and method for remote use of information handling system audio components |
| WO2008076373A1 (en) * | 2006-12-14 | 2008-06-26 | Abraxis Bioscience, Llc | Breast cancer therapy based on hormone receptor status with nanoparticles comprising taxane |
| MX2010011165A (es) | 2008-04-10 | 2011-02-22 | Abraxis Bioscience Llc | Composiciones de derivados de taxano hidrofobos y sus usos. |
-
2006
- 2006-08-30 NZ NZ592132A patent/NZ592132A/xx unknown
- 2006-08-30 ES ES06802739T patent/ES2719093T3/es active Active
- 2006-08-30 NZ NZ566705A patent/NZ566705A/en unknown
- 2006-08-30 AU AU2006284657A patent/AU2006284657B2/en active Active
- 2006-08-30 EP EP19150684.9A patent/EP3527202A1/en not_active Withdrawn
- 2006-08-30 WO PCT/US2006/034103 patent/WO2007027941A2/en active Application Filing
- 2006-08-30 KR KR1020137019918A patent/KR101457834B1/ko active IP Right Grant
- 2006-08-30 US US11/513,756 patent/US8034765B2/en active Active
- 2006-08-30 CN CN200680038640.5A patent/CN101291658B/zh active Active
- 2006-08-30 KR KR1020087007603A patent/KR101643416B1/ko active IP Right Grant
- 2006-08-30 EP EP06802739.0A patent/EP1928435B8/en active Active
- 2006-08-30 CA CA2620389A patent/CA2620389C/en active Active
- 2006-08-30 JP JP2008529288A patent/JP5368093B2/ja active Active
- 2006-08-30 BR BRPI0615292A patent/BRPI0615292A8/pt not_active Application Discontinuation
-
2008
- 2008-02-19 IL IL189600A patent/IL189600A/en active IP Right Grant
- 2008-03-28 NO NO20081576A patent/NO343793B1/no unknown
-
2009
- 2009-03-11 US US12/402,358 patent/US7981445B2/en active Active
-
2016
- 2016-08-25 IL IL247486A patent/IL247486B/en active IP Right Grant
-
2019
- 2019-04-24 NO NO20190526A patent/NO344945B1/no unknown
-
2020
- 2020-01-26 IL IL272236A patent/IL272236A/en unknown
- 2020-03-17 NO NO20200320A patent/NO20200320A1/no not_active Application Discontinuation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2719093T3 (es) | Composiciones de fármacos poco solubles en agua con mayor estabilidad y métodos para su preparación | |
| ES2663324T3 (es) | Composiciones que comprenden docetaxel con estabilidad incrementada | |
| ES2746057T3 (es) | Uso de excipientes poliméricos para liofilización o congelación de partículas | |
| AU2013370955B2 (en) | Nanoparticle compositions of albumin and paclitaxel | |
| MX2008002742A (es) | Composiciones y metodos para la preparacion de farmacos escasamente solubles en agua con estabilidad incrementada |