ES2717181T3 - Amino triazoles sustituidos útiles como inhibidores de la quitinasa humana - Google Patents
Amino triazoles sustituidos útiles como inhibidores de la quitinasa humana Download PDFInfo
- Publication number
- ES2717181T3 ES2717181T3 ES16767368T ES16767368T ES2717181T3 ES 2717181 T3 ES2717181 T3 ES 2717181T3 ES 16767368 T ES16767368 T ES 16767368T ES 16767368 T ES16767368 T ES 16767368T ES 2717181 T3 ES2717181 T3 ES 2717181T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- aryl
- arylalkyl
- compound
- nhc
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 Amino substituted triazoles Chemical class 0.000 title claims description 169
- 102000012286 Chitinases Human genes 0.000 title claims description 68
- 108010022172 Chitinases Proteins 0.000 title claims description 68
- 239000003112 inhibitor Substances 0.000 title claims description 26
- 241000282414 Homo sapiens Species 0.000 title description 13
- 150000001875 compounds Chemical class 0.000 claims description 325
- 125000000217 alkyl group Chemical group 0.000 claims description 218
- 125000003118 aryl group Chemical group 0.000 claims description 167
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 161
- 230000015572 biosynthetic process Effects 0.000 claims description 131
- 238000003786 synthesis reaction Methods 0.000 claims description 120
- 125000001072 heteroaryl group Chemical group 0.000 claims description 113
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 105
- 125000001188 haloalkyl group Chemical group 0.000 claims description 103
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims description 102
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 95
- 201000010099 disease Diseases 0.000 claims description 61
- 125000000623 heterocyclic group Chemical group 0.000 claims description 53
- 125000005843 halogen group Chemical group 0.000 claims description 52
- 125000003545 alkoxy group Chemical group 0.000 claims description 48
- 239000003814 drug Substances 0.000 claims description 47
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 44
- 102100037328 Chitotriosidase-1 Human genes 0.000 claims description 39
- 239000002253 acid Substances 0.000 claims description 37
- 208000006673 asthma Diseases 0.000 claims description 37
- 150000003839 salts Chemical class 0.000 claims description 37
- 239000008194 pharmaceutical composition Substances 0.000 claims description 35
- 238000011282 treatment Methods 0.000 claims description 35
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 34
- 125000004414 alkyl thio group Chemical group 0.000 claims description 31
- 208000035475 disorder Diseases 0.000 claims description 31
- 125000001424 substituent group Chemical group 0.000 claims description 28
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 27
- 230000014509 gene expression Effects 0.000 claims description 27
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 25
- 206010028980 Neoplasm Diseases 0.000 claims description 24
- 210000001519 tissue Anatomy 0.000 claims description 24
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 22
- 125000002947 alkylene group Chemical group 0.000 claims description 22
- 201000011510 cancer Diseases 0.000 claims description 22
- 239000003795 chemical substances by application Substances 0.000 claims description 21
- 210000004027 cell Anatomy 0.000 claims description 20
- 229940124597 therapeutic agent Drugs 0.000 claims description 20
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 claims description 19
- 208000026935 allergic disease Diseases 0.000 claims description 19
- 208000036971 interstitial lung disease 2 Diseases 0.000 claims description 19
- 229940079593 drug Drugs 0.000 claims description 17
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 16
- 229910052760 oxygen Inorganic materials 0.000 claims description 16
- 241000124008 Mammalia Species 0.000 claims description 15
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 15
- 125000004432 carbon atom Chemical group C* 0.000 claims description 14
- 206010016654 Fibrosis Diseases 0.000 claims description 12
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 12
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 12
- 208000023275 Autoimmune disease Diseases 0.000 claims description 11
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 11
- 201000010105 allergic rhinitis Diseases 0.000 claims description 11
- 229910052799 carbon Inorganic materials 0.000 claims description 11
- 239000012453 solvate Substances 0.000 claims description 11
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 10
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 10
- 208000029523 Interstitial Lung disease Diseases 0.000 claims description 10
- 201000008937 atopic dermatitis Diseases 0.000 claims description 10
- 230000004761 fibrosis Effects 0.000 claims description 10
- 230000003176 fibrotic effect Effects 0.000 claims description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- 125000004429 atom Chemical group 0.000 claims description 9
- 239000000460 chlorine Chemical group 0.000 claims description 9
- 208000037976 chronic inflammation Diseases 0.000 claims description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 9
- 201000010065 polycystic ovary syndrome Diseases 0.000 claims description 9
- 230000002265 prevention Effects 0.000 claims description 9
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 claims description 8
- 208000024827 Alzheimer disease Diseases 0.000 claims description 8
- 208000011231 Crohn disease Diseases 0.000 claims description 8
- 206010036049 Polycystic ovaries Diseases 0.000 claims description 8
- 206010039085 Rhinitis allergic Diseases 0.000 claims description 8
- 230000001594 aberrant effect Effects 0.000 claims description 8
- 208000019423 liver disease Diseases 0.000 claims description 8
- 230000001404 mediated effect Effects 0.000 claims description 8
- 201000006417 multiple sclerosis Diseases 0.000 claims description 8
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 8
- 239000003381 stabilizer Substances 0.000 claims description 8
- 125000004455 (C1-C3) alkylthio group Chemical group 0.000 claims description 7
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 claims description 7
- 208000015943 Coeliac disease Diseases 0.000 claims description 7
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 7
- 201000009273 Endometriosis Diseases 0.000 claims description 7
- 229910052801 chlorine Chemical group 0.000 claims description 7
- 230000002327 eosinophilic effect Effects 0.000 claims description 7
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 7
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 7
- 208000017169 kidney disease Diseases 0.000 claims description 7
- 208000030159 metabolic disease Diseases 0.000 claims description 7
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 7
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 7
- 201000008482 osteoarthritis Diseases 0.000 claims description 7
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 7
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 6
- 230000001154 acute effect Effects 0.000 claims description 6
- 108010057052 chitotriosidase Proteins 0.000 claims description 6
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 6
- 208000037893 chronic inflammatory disorder Diseases 0.000 claims description 6
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 6
- 125000005358 mercaptoalkyl group Chemical group 0.000 claims description 6
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 claims description 6
- 150000003431 steroids Chemical class 0.000 claims description 6
- 208000030090 Acute Disease Diseases 0.000 claims description 5
- 201000001320 Atherosclerosis Diseases 0.000 claims description 5
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 5
- 208000012902 Nervous system disease Diseases 0.000 claims description 5
- 208000025966 Neurological disease Diseases 0.000 claims description 5
- 208000014151 Stomatognathic disease Diseases 0.000 claims description 5
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 5
- 230000001476 alcoholic effect Effects 0.000 claims description 5
- 208000010668 atopic eczema Diseases 0.000 claims description 5
- 208000027157 chronic rhinosinusitis Diseases 0.000 claims description 5
- 239000012528 membrane Substances 0.000 claims description 5
- 201000000306 sarcoidosis Diseases 0.000 claims description 5
- 238000003860 storage Methods 0.000 claims description 5
- 208000035408 type 1 diabetes mellitus 1 Diseases 0.000 claims description 5
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 4
- 206010006187 Breast cancer Diseases 0.000 claims description 4
- 208000026310 Breast neoplasm Diseases 0.000 claims description 4
- 229940124638 COX inhibitor Drugs 0.000 claims description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 4
- 206010010744 Conjunctivitis allergic Diseases 0.000 claims description 4
- 208000003556 Dry Eye Syndromes Diseases 0.000 claims description 4
- 206010013774 Dry eye Diseases 0.000 claims description 4
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 claims description 4
- 208000031888 Mycoses Diseases 0.000 claims description 4
- 208000000592 Nasal Polyps Diseases 0.000 claims description 4
- 239000012826 P38 inhibitor Substances 0.000 claims description 4
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 claims description 4
- 206010039710 Scleroderma Diseases 0.000 claims description 4
- 230000002378 acidificating effect Effects 0.000 claims description 4
- 239000000556 agonist Substances 0.000 claims description 4
- 208000002205 allergic conjunctivitis Diseases 0.000 claims description 4
- 229940125715 antihistaminic agent Drugs 0.000 claims description 4
- 239000000739 antihistaminic agent Substances 0.000 claims description 4
- 208000024998 atopic conjunctivitis Diseases 0.000 claims description 4
- 229910052794 bromium Inorganic materials 0.000 claims description 4
- 125000002837 carbocyclic group Chemical group 0.000 claims description 4
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 claims description 4
- 125000001153 fluoro group Chemical group F* 0.000 claims description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 4
- 206010023332 keratitis Diseases 0.000 claims description 4
- 201000010666 keratoconjunctivitis Diseases 0.000 claims description 4
- 230000001394 metastastic effect Effects 0.000 claims description 4
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 4
- 229960000485 methotrexate Drugs 0.000 claims description 4
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 claims description 4
- 230000001932 seasonal effect Effects 0.000 claims description 4
- 239000002750 tryptase inhibitor Substances 0.000 claims description 4
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 3
- BSYNRYMUTXBXSQ-FOQJRBATSA-N 59096-14-9 Chemical compound CC(=O)OC1=CC=CC=C1[14C](O)=O BSYNRYMUTXBXSQ-FOQJRBATSA-N 0.000 claims description 3
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 3
- 206010009944 Colon cancer Diseases 0.000 claims description 3
- 206010010741 Conjunctivitis Diseases 0.000 claims description 3
- 206010012442 Dermatitis contact Diseases 0.000 claims description 3
- 208000004262 Food Hypersensitivity Diseases 0.000 claims description 3
- 208000015872 Gaucher disease Diseases 0.000 claims description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 3
- 206010050017 Lung cancer metastatic Diseases 0.000 claims description 3
- 206010025323 Lymphomas Diseases 0.000 claims description 3
- 206010027406 Mesothelioma Diseases 0.000 claims description 3
- 206010033128 Ovarian cancer Diseases 0.000 claims description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 3
- 208000030852 Parasitic disease Diseases 0.000 claims description 3
- 206010060862 Prostate cancer Diseases 0.000 claims description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 3
- 206010038389 Renal cancer Diseases 0.000 claims description 3
- 206010038979 Retroperitoneal fibrosis Diseases 0.000 claims description 3
- 208000036284 Rhinitis seasonal Diseases 0.000 claims description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 3
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 3
- 208000002029 allergic contact dermatitis Diseases 0.000 claims description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 3
- 125000001246 bromo group Chemical group Br* 0.000 claims description 3
- 201000010881 cervical cancer Diseases 0.000 claims description 3
- 230000007882 cirrhosis Effects 0.000 claims description 3
- 208000029742 colonic neoplasm Diseases 0.000 claims description 3
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 claims description 3
- 235000020932 food allergy Nutrition 0.000 claims description 3
- 206010017758 gastric cancer Diseases 0.000 claims description 3
- 208000005017 glioblastoma Diseases 0.000 claims description 3
- 201000010982 kidney cancer Diseases 0.000 claims description 3
- 208000032839 leukemia Diseases 0.000 claims description 3
- 201000007270 liver cancer Diseases 0.000 claims description 3
- 208000014018 liver neoplasm Diseases 0.000 claims description 3
- 201000001441 melanoma Diseases 0.000 claims description 3
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 claims description 3
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 3
- 208000008275 microscopic colitis Diseases 0.000 claims description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 3
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 claims description 3
- 201000008968 osteosarcoma Diseases 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 208000017022 seasonal allergic rhinitis Diseases 0.000 claims description 3
- 208000017520 skin disease Diseases 0.000 claims description 3
- 201000011549 stomach cancer Diseases 0.000 claims description 3
- 125000003107 substituted aryl group Chemical group 0.000 claims description 3
- 125000001140 1,4-phenylene group Chemical group [H]C1=C([H])C([*:2])=C([H])C([H])=C1[*:1] 0.000 claims description 2
- 208000017667 Chronic Disease Diseases 0.000 claims description 2
- 206010009208 Cirrhosis alcoholic Diseases 0.000 claims description 2
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 2
- 208000024720 Fabry Disease Diseases 0.000 claims description 2
- 208000006933 Hermanski-Pudlak Syndrome Diseases 0.000 claims description 2
- 206010071775 Hermansky-Pudlak syndrome Diseases 0.000 claims description 2
- 208000014919 IgG4-related retroperitoneal fibrosis Diseases 0.000 claims description 2
- 208000015439 Lysosomal storage disease Diseases 0.000 claims description 2
- 208000014060 Niemann-Pick disease Diseases 0.000 claims description 2
- 208000005141 Otitis Diseases 0.000 claims description 2
- 201000004681 Psoriasis Diseases 0.000 claims description 2
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 2
- 208000010002 alcoholic liver cirrhosis Diseases 0.000 claims description 2
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 2
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 2
- 208000016097 disease of metabolism Diseases 0.000 claims description 2
- 208000019258 ear infection Diseases 0.000 claims description 2
- 201000009580 eosinophilic pneumonia Diseases 0.000 claims description 2
- 201000005206 focal segmental glomerulosclerosis Diseases 0.000 claims description 2
- 150000002617 leukotrienes Chemical class 0.000 claims description 2
- 230000002580 nephropathic effect Effects 0.000 claims description 2
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 claims description 2
- 201000001245 periodontitis Diseases 0.000 claims description 2
- 201000009732 pulmonary eosinophilia Diseases 0.000 claims description 2
- 108091006082 receptor inhibitors Proteins 0.000 claims description 2
- 208000037999 tubulointerstitial fibrosis Diseases 0.000 claims description 2
- 208000027004 Eosinophilic disease Diseases 0.000 claims 1
- 206010064212 Eosinophilic oesophagitis Diseases 0.000 claims 1
- 208000005176 Hepatitis C Diseases 0.000 claims 1
- 206010019728 Hepatitis alcoholic Diseases 0.000 claims 1
- 201000000708 eosinophilic esophagitis Diseases 0.000 claims 1
- 238000000034 method Methods 0.000 description 152
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 141
- 239000000203 mixture Substances 0.000 description 101
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 100
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 98
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical group CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 89
- 239000000243 solution Substances 0.000 description 78
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 68
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 58
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 57
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 57
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 56
- 239000011541 reaction mixture Substances 0.000 description 50
- 238000006243 chemical reaction Methods 0.000 description 42
- 239000002904 solvent Substances 0.000 description 42
- 239000000047 product Substances 0.000 description 40
- 235000002639 sodium chloride Nutrition 0.000 description 39
- 238000009472 formulation Methods 0.000 description 38
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 35
- 235000019439 ethyl acetate Nutrition 0.000 description 35
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 34
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 33
- 239000011575 calcium Substances 0.000 description 32
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 32
- 239000012267 brine Substances 0.000 description 31
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 31
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 30
- 230000000694 effects Effects 0.000 description 30
- 101710132290 Chitotriosidase-1 Proteins 0.000 description 28
- 239000011734 sodium Substances 0.000 description 26
- 230000001225 therapeutic effect Effects 0.000 description 26
- 239000012043 crude product Substances 0.000 description 24
- 238000003818 flash chromatography Methods 0.000 description 20
- 239000007858 starting material Substances 0.000 description 20
- 210000004072 lung Anatomy 0.000 description 19
- 239000012044 organic layer Substances 0.000 description 19
- 239000007787 solid Substances 0.000 description 19
- 239000000725 suspension Substances 0.000 description 19
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 18
- 150000001412 amines Chemical class 0.000 description 18
- 239000010410 layer Substances 0.000 description 18
- 239000000463 material Substances 0.000 description 18
- 239000002245 particle Substances 0.000 description 18
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 17
- 206010061218 Inflammation Diseases 0.000 description 17
- 108090000176 Interleukin-13 Proteins 0.000 description 17
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 17
- 230000004054 inflammatory process Effects 0.000 description 17
- 238000000746 purification Methods 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 16
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 15
- 238000004440 column chromatography Methods 0.000 description 15
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 15
- 235000019341 magnesium sulphate Nutrition 0.000 description 15
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- 230000005764 inhibitory process Effects 0.000 description 14
- 108090000623 proteins and genes Proteins 0.000 description 14
- 238000004809 thin layer chromatography Methods 0.000 description 14
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 13
- 239000000284 extract Substances 0.000 description 13
- 125000006239 protecting group Chemical group 0.000 description 13
- 102000004169 proteins and genes Human genes 0.000 description 13
- 102000004190 Enzymes Human genes 0.000 description 12
- 108090000790 Enzymes Proteins 0.000 description 12
- 150000001299 aldehydes Chemical group 0.000 description 12
- 125000003342 alkenyl group Chemical group 0.000 description 12
- 239000003153 chemical reaction reagent Substances 0.000 description 12
- 239000000843 powder Substances 0.000 description 12
- 238000002360 preparation method Methods 0.000 description 12
- 235000018102 proteins Nutrition 0.000 description 12
- 238000006268 reductive amination reaction Methods 0.000 description 12
- 239000000758 substrate Substances 0.000 description 12
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 11
- 206010020751 Hypersensitivity Diseases 0.000 description 11
- 235000019441 ethanol Nutrition 0.000 description 11
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 11
- 108010010803 Gelatin Proteins 0.000 description 10
- 229920002472 Starch Polymers 0.000 description 10
- 238000010828 elution Methods 0.000 description 10
- 239000008273 gelatin Substances 0.000 description 10
- 229920000159 gelatin Polymers 0.000 description 10
- 229940014259 gelatin Drugs 0.000 description 10
- 235000019322 gelatine Nutrition 0.000 description 10
- 235000011852 gelatine desserts Nutrition 0.000 description 10
- 125000005842 heteroatom Chemical group 0.000 description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 10
- 230000002401 inhibitory effect Effects 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- 125000002950 monocyclic group Chemical group 0.000 description 10
- 238000010898 silica gel chromatography Methods 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 10
- 229920002101 Chitin Polymers 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 9
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 9
- 238000002425 crystallisation Methods 0.000 description 9
- 230000008025 crystallization Effects 0.000 description 9
- 229910052739 hydrogen Inorganic materials 0.000 description 9
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- 235000019698 starch Nutrition 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- 229910052717 sulfur Inorganic materials 0.000 description 9
- 208000024891 symptom Diseases 0.000 description 9
- 238000012384 transportation and delivery Methods 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 239000007884 disintegrant Substances 0.000 description 8
- 150000002148 esters Chemical group 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 230000009467 reduction Effects 0.000 description 8
- 238000006722 reduction reaction Methods 0.000 description 8
- 229910000104 sodium hydride Inorganic materials 0.000 description 8
- 239000004094 surface-active agent Substances 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- 239000000443 aerosol Substances 0.000 description 7
- 230000007815 allergy Effects 0.000 description 7
- 150000001408 amides Chemical class 0.000 description 7
- 229940024606 amino acid Drugs 0.000 description 7
- 235000019270 ammonium chloride Nutrition 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 239000000969 carrier Substances 0.000 description 7
- 238000004587 chromatography analysis Methods 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 230000000875 corresponding effect Effects 0.000 description 7
- 125000004122 cyclic group Chemical group 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- 210000003979 eosinophil Anatomy 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 230000002757 inflammatory effect Effects 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 7
- 230000002685 pulmonary effect Effects 0.000 description 7
- 230000004044 response Effects 0.000 description 7
- 235000017557 sodium bicarbonate Nutrition 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 239000008107 starch Substances 0.000 description 7
- 229940032147 starch Drugs 0.000 description 7
- 210000002784 stomach Anatomy 0.000 description 7
- PSGQCCSGKGJLRL-UHFFFAOYSA-N 4-methyl-2h-chromen-2-one Chemical group C1=CC=CC2=C1OC(=O)C=C2C PSGQCCSGKGJLRL-UHFFFAOYSA-N 0.000 description 6
- WDYVUKGVKRZQNM-UHFFFAOYSA-N 6-phosphonohexylphosphonic acid Chemical compound OP(O)(=O)CCCCCCP(O)(O)=O WDYVUKGVKRZQNM-UHFFFAOYSA-N 0.000 description 6
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 6
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 239000004215 Carbon black (E152) Substances 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 125000000304 alkynyl group Chemical group 0.000 description 6
- 238000010171 animal model Methods 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- 125000002619 bicyclic group Chemical group 0.000 description 6
- 230000004071 biological effect Effects 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 150000001721 carbon Chemical group 0.000 description 6
- 239000001768 carboxy methyl cellulose Substances 0.000 description 6
- 238000000576 coating method Methods 0.000 description 6
- 239000012230 colorless oil Substances 0.000 description 6
- 239000003085 diluting agent Substances 0.000 description 6
- 230000002255 enzymatic effect Effects 0.000 description 6
- 210000002919 epithelial cell Anatomy 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 229930195733 hydrocarbon Natural products 0.000 description 6
- 150000002894 organic compounds Chemical class 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- 230000008506 pathogenesis Effects 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 6
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 6
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 6
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 6
- 210000002966 serum Anatomy 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 238000013268 sustained release Methods 0.000 description 6
- 239000012730 sustained-release form Substances 0.000 description 6
- 230000009885 systemic effect Effects 0.000 description 6
- 239000003039 volatile agent Substances 0.000 description 6
- YZNCNMQLYFVHNO-ZYMOGRSISA-N (2R)-4-[1-(5-amino-1H-1,2,4-triazol-3-yl)piperidin-4-yl]-5-[(4-chlorophenyl)methyl]-N-methylmorpholine-2-carboxamide Chemical compound CNC(=O)[C@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 YZNCNMQLYFVHNO-ZYMOGRSISA-N 0.000 description 5
- RPHQMRWKOPBZQY-VIFPVBQESA-N (2s)-2-amino-3-(4-chlorophenyl)propan-1-ol Chemical compound OC[C@@H](N)CC1=CC=C(Cl)C=C1 RPHQMRWKOPBZQY-VIFPVBQESA-N 0.000 description 5
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 5
- KUQCBVPEFGKXTQ-MUMRKEEXSA-N 3-[4-[(2R)-5-[(4-chlorophenyl)methyl]-2-(ethoxymethyl)morpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound CCOC[C@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 KUQCBVPEFGKXTQ-MUMRKEEXSA-N 0.000 description 5
- ITISUZUNOJIUPF-RBUKOAKNSA-N 3-[4-[(2R,5S)-5-[(4-chlorophenyl)methyl]-2-(2-methoxypropan-2-yl)morpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound ClC1=CC=C(C[C@@H]2N(C[C@@H](OC2)C(C)(C)OC)C2CCN(CC2)C=2NC(=NN=2)N)C=C1 ITISUZUNOJIUPF-RBUKOAKNSA-N 0.000 description 5
- WQWHBQYUWSARDT-UHFFFAOYSA-N 3-[4-[3-[(4-chlorophenyl)methyl]morpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound ClC1=CC=C(CC2COCCN2C2CCN(CC2)C=2NC(=NN=2)N)C=C1 WQWHBQYUWSARDT-UHFFFAOYSA-N 0.000 description 5
- NBTTYMBZXDHFCP-UHFFFAOYSA-N 4-(4-methylphenyl)sulfonylmorpholine Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1CCOCC1 NBTTYMBZXDHFCP-UHFFFAOYSA-N 0.000 description 5
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 5
- YDEVMFFTVHMZIL-UHFFFAOYSA-N [1-[1-(5-amino-1H-1,2,4-triazol-3-yl)piperidin-4-yl]piperidin-2-yl]-(4-chlorophenyl)methanol Chemical compound NC=1NC(=NN=1)N1CCC(CC1)N1C(CCCC1)C(O)C1=CC=C(C=C1)Cl YDEVMFFTVHMZIL-UHFFFAOYSA-N 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 239000000427 antigen Substances 0.000 description 5
- 102000036639 antigens Human genes 0.000 description 5
- 108091007433 antigens Proteins 0.000 description 5
- 150000001540 azides Chemical group 0.000 description 5
- 125000005605 benzo group Chemical group 0.000 description 5
- 239000011230 binding agent Substances 0.000 description 5
- 239000000090 biomarker Substances 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Natural products C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 5
- 239000000945 filler Substances 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 150000002367 halogens Chemical group 0.000 description 5
- 239000000314 lubricant Substances 0.000 description 5
- 238000012544 monitoring process Methods 0.000 description 5
- 239000006199 nebulizer Substances 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 239000003586 protic polar solvent Substances 0.000 description 5
- 230000002441 reversible effect Effects 0.000 description 5
- 238000007363 ring formation reaction Methods 0.000 description 5
- 239000012047 saturated solution Substances 0.000 description 5
- 150000003335 secondary amines Chemical class 0.000 description 5
- 235000010356 sorbitol Nutrition 0.000 description 5
- 239000000600 sorbitol Substances 0.000 description 5
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 5
- 239000011593 sulfur Substances 0.000 description 5
- 125000001425 triazolyl group Chemical group 0.000 description 5
- 229910052721 tungsten Inorganic materials 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- KFEHVUCMKWANPT-ZWKOTPCHSA-N (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(methoxymethyl)-4-piperidin-4-ylmorpholine Chemical compound ClC1=CC=C(C[C@H]2CO[C@H](CN2C2CCNCC2)COC)C=C1 KFEHVUCMKWANPT-ZWKOTPCHSA-N 0.000 description 4
- QPNRFXNVCIRUAH-ZVWHLABXSA-N (2R,5S)-5-[(4-chlorophenyl)methyl]-N-methylmorpholine-2-carboxamide hydrochloride Chemical compound Cl.CNC(=O)[C@H]1CN[C@@H](Cc2ccc(Cl)cc2)CO1 QPNRFXNVCIRUAH-ZVWHLABXSA-N 0.000 description 4
- QDEYVNGMQPTFLE-JMFXEUCVSA-N (2S)-2-[(4-chlorophenyl)methyl]-3-methoxyazetidine hydrochloride Chemical compound Cl.COC1CN[C@H]1Cc1ccc(Cl)cc1 QDEYVNGMQPTFLE-JMFXEUCVSA-N 0.000 description 4
- BZGVVLPYBNZSPN-UXQCFNEQSA-N (2S,3R)-2-[(4-chlorophenyl)methyl]-3-fluoroazetidine hydrochloride Chemical compound Cl.F[C@@H]1CN[C@H]1Cc1ccc(Cl)cc1 BZGVVLPYBNZSPN-UXQCFNEQSA-N 0.000 description 4
- AOONNEHLWNWOIX-CJRXIRLBSA-N (2S,4R)-2-[(4-chlorophenyl)methyl]-4-methoxy-1-piperidin-4-ylpiperidine hydrochloride Chemical compound Cl.CO[C@@H]1CCN([C@@H](Cc2ccc(Cl)cc2)C1)C1CCNCC1 AOONNEHLWNWOIX-CJRXIRLBSA-N 0.000 description 4
- QUPLJPIJRLSDFC-CABZTGNLSA-N (2S,5S)-5-[(4-chlorophenyl)methyl]-2-methylmorpholine Chemical compound ClC1=CC=C(C[C@H]2CO[C@H](CN2)C)C=C1 QUPLJPIJRLSDFC-CABZTGNLSA-N 0.000 description 4
- MONMFXREYOKQTI-UWTATZPHSA-N (2r)-2-bromopropanoic acid Chemical compound C[C@@H](Br)C(O)=O MONMFXREYOKQTI-UWTATZPHSA-N 0.000 description 4
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 4
- SDWHBDLCXPZZKY-UHFFFAOYSA-N (4-chlorophenyl)-piperidin-2-ylmethanol Chemical compound C=1C=C(Cl)C=CC=1C(O)C1CCCCN1 SDWHBDLCXPZZKY-UHFFFAOYSA-N 0.000 description 4
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 4
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 4
- CKGIHYVZWVUSHR-ZWKOTPCHSA-N 3-[4-[(2S,4R)-2-[(4-chlorophenyl)methyl]-4-methoxypiperidin-1-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound ClC1=CC=C(C[C@@H]2N(CC[C@H](C2)OC)C2CCN(CC2)C=2NC(=NN=2)N)C=C1 CKGIHYVZWVUSHR-ZWKOTPCHSA-N 0.000 description 4
- UQQKVCFHEQIIGA-UHFFFAOYSA-N 3-[4-[2-[(4-chlorophenyl)methyl]piperidin-1-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound NC1=NN=C(N1)N1CCC(CC1)N1CCCCC1CC1=CC=C(Cl)C=C1 UQQKVCFHEQIIGA-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 4
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 4
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 4
- 229920002307 Dextran Polymers 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- 208000019693 Lung disease Diseases 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- KBHCPIJKJQNHPN-UHFFFAOYSA-N N=NP(O)=O Chemical group N=NP(O)=O KBHCPIJKJQNHPN-UHFFFAOYSA-N 0.000 description 4
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- WBTCZXYOKNRFQX-UHFFFAOYSA-N S1(=O)(=O)NC1=O Chemical group S1(=O)(=O)NC1=O WBTCZXYOKNRFQX-UHFFFAOYSA-N 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- 239000012317 TBTU Substances 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 4
- 230000009285 allergic inflammation Effects 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 125000004103 aminoalkyl group Chemical group 0.000 description 4
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 235000010980 cellulose Nutrition 0.000 description 4
- 239000007810 chemical reaction solvent Substances 0.000 description 4
- 239000011248 coating agent Substances 0.000 description 4
- 125000000392 cycloalkenyl group Chemical group 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 230000003111 delayed effect Effects 0.000 description 4
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 4
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical group C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 4
- 125000003709 fluoroalkyl group Chemical group 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 4
- 239000007943 implant Substances 0.000 description 4
- 210000004969 inflammatory cell Anatomy 0.000 description 4
- 208000027866 inflammatory disease Diseases 0.000 description 4
- 210000000936 intestine Anatomy 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 229960001375 lactose Drugs 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 210000004379 membrane Anatomy 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000004005 microsphere Substances 0.000 description 4
- VSEAAEQOQBMPQF-UHFFFAOYSA-N morpholin-3-one Chemical compound O=C1COCCN1 VSEAAEQOQBMPQF-UHFFFAOYSA-N 0.000 description 4
- 238000010172 mouse model Methods 0.000 description 4
- 244000045947 parasite Species 0.000 description 4
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical group [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 description 4
- IWELDVXSEVIIGI-UHFFFAOYSA-N piperazin-2-one Chemical compound O=C1CNCCN1 IWELDVXSEVIIGI-UHFFFAOYSA-N 0.000 description 4
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 description 4
- 239000003880 polar aprotic solvent Substances 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 230000002035 prolonged effect Effects 0.000 description 4
- 239000003380 propellant Substances 0.000 description 4
- 238000007634 remodeling Methods 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 150000003456 sulfonamides Chemical class 0.000 description 4
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 238000001665 trituration Methods 0.000 description 4
- BXSGINJUMJWYKC-UNTBIKODSA-N (2R)-2-[(4-chlorophenyl)methyl]-1-piperidin-4-ylpiperidine hydrochloride Chemical compound Cl.Clc1ccc(C[C@H]2CCCCN2C2CCNCC2)cc1 BXSGINJUMJWYKC-UNTBIKODSA-N 0.000 description 3
- HGLIHLLTNYZFMP-GRTNUQQKSA-N (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(ethoxymethyl)-4-piperidin-4-ylmorpholine hydrochloride Chemical compound Cl.CCOC[C@H]1CN([C@@H](Cc2ccc(Cl)cc2)CO1)C1CCNCC1 HGLIHLLTNYZFMP-GRTNUQQKSA-N 0.000 description 3
- FCXDDCKBHIQKMQ-RBUKOAKNSA-N (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(methoxymethyl)-4-(4-methylphenyl)sulfonylmorpholine Chemical compound ClC1=CC=C(C[C@H]2CO[C@H](CN2S(=O)(=O)C2=CC=C(C)C=C2)COC)C=C1 FCXDDCKBHIQKMQ-RBUKOAKNSA-N 0.000 description 3
- VCOALGIHIMEPHE-JHEYCYPBSA-N (2S,4R)-2-[(4-chlorophenyl)methyl]-4-methoxypiperidine hydrochloride Chemical compound Cl.CO[C@@H]1CCN[C@@H](Cc2ccc(Cl)cc2)C1 VCOALGIHIMEPHE-JHEYCYPBSA-N 0.000 description 3
- VCOALGIHIMEPHE-QNTKWALQSA-N (2S,4S)-2-[(4-chlorophenyl)methyl]-4-methoxypiperidine hydrochloride Chemical compound Cl.CO[C@H]1CCN[C@@H](Cc2ccc(Cl)cc2)C1 VCOALGIHIMEPHE-QNTKWALQSA-N 0.000 description 3
- NQQWHCGVPQELTR-KYLFUZKPSA-N (2S,5S)-5-[(4-chlorophenyl)methyl]-2-methyl-4-piperidin-4-ylmorpholine hydrochloride Chemical compound Cl.C[C@H]1CN([C@@H](Cc2ccc(Cl)cc2)CO1)C1CCNCC1 NQQWHCGVPQELTR-KYLFUZKPSA-N 0.000 description 3
- HQVUQRZWYHAOGK-UHFFFAOYSA-N (4-chlorophenyl)-(1-piperidin-4-ylpiperidin-2-yl)methanol Chemical compound N1(C(CCCC1)C(O)C1=CC=C(C=C1)Cl)C1CCNCC1 HQVUQRZWYHAOGK-UHFFFAOYSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- PMEIRBOZYDXMLT-JHEYCYPBSA-N 2-[(2R,5S)-5-[(4-chlorophenyl)methyl]morpholin-2-yl]propan-2-ol 2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CC(C)(O)[C@H]1CN[C@@H](Cc2ccc(Cl)cc2)CO1 PMEIRBOZYDXMLT-JHEYCYPBSA-N 0.000 description 3
- JTOFILOVFYOIGD-HOTGVXAUSA-N 3-[4-[(2S,3S)-2-[(4-chlorophenyl)methyl]-3-methoxyazetidin-1-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound ClC1=CC=C(C[C@@H]2N(C[C@@H]2OC)C2CCN(CC2)C=2NC(=NN=2)N)C=C1 JTOFILOVFYOIGD-HOTGVXAUSA-N 0.000 description 3
- CKGIHYVZWVUSHR-ROUUACIJSA-N 3-[4-[(2S,4S)-2-[(4-chlorophenyl)methyl]-4-methoxypiperidin-1-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound ClC1=CC=C(C[C@@H]2N(CC[C@@H](C2)OC)C2CCN(CC2)C=2NC(=NN=2)N)C=C1 CKGIHYVZWVUSHR-ROUUACIJSA-N 0.000 description 3
- 125000006283 4-chlorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Cl)C([H])([H])* 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 101710178876 Acidic mammalian chitinase Proteins 0.000 description 3
- 102100037839 Acidic mammalian chitinase Human genes 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- 241000271566 Aves Species 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 description 3
- 101710203678 Chitinase-like protein Proteins 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 208000000059 Dyspnea Diseases 0.000 description 3
- 206010013975 Dyspnoeas Diseases 0.000 description 3
- 102100023688 Eotaxin Human genes 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 241000233866 Fungi Species 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000711549 Hepacivirus C Species 0.000 description 3
- 241000238631 Hexapoda Species 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 241000287531 Psittacidae Species 0.000 description 3
- 201000009594 Systemic Scleroderma Diseases 0.000 description 3
- 206010042953 Systemic sclerosis Diseases 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 239000013566 allergen Substances 0.000 description 3
- 125000003368 amide group Chemical group 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 150000008064 anhydrides Chemical class 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 125000005129 aryl carbonyl group Chemical group 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 3
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 3
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 3
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 3
- 208000013677 cerebrovascular dementia Diseases 0.000 description 3
- 238000007385 chemical modification Methods 0.000 description 3
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 3
- 230000006020 chronic inflammation Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 230000008021 deposition Effects 0.000 description 3
- 230000002074 deregulated effect Effects 0.000 description 3
- 239000003599 detergent Substances 0.000 description 3
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000008298 dragée Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 3
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 3
- UQEAIHBTYFGYIE-UHFFFAOYSA-N hexamethyldisiloxane Chemical compound C[Si](C)(C)O[Si](C)(C)C UQEAIHBTYFGYIE-UHFFFAOYSA-N 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 3
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 3
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- 239000012678 infectious agent Substances 0.000 description 3
- 230000000968 intestinal effect Effects 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 3
- 230000003902 lesion Effects 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000007170 pathology Effects 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 3
- 208000005069 pulmonary fibrosis Diseases 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 229910052705 radium Inorganic materials 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 229910052701 rubidium Inorganic materials 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 3
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 208000011580 syndromic disease Diseases 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 3
- 238000000844 transformation Methods 0.000 description 3
- 150000003852 triazoles Chemical group 0.000 description 3
- HBENZIXOGRCSQN-VQWWACLZSA-N (1S,2S,6R,14R,15R,16R)-5-(cyclopropylmethyl)-16-[(2S)-2-hydroxy-3,3-dimethylpentan-2-yl]-15-methoxy-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11-trien-11-ol Chemical compound N1([C@@H]2CC=3C4=C(C(=CC=3)O)O[C@H]3[C@@]5(OC)CC[C@@]2([C@@]43CC1)C[C@@H]5[C@](C)(O)C(C)(C)CC)CC1CC1 HBENZIXOGRCSQN-VQWWACLZSA-N 0.000 description 2
- GVVGVIWQBGVCLW-GRTNUQQKSA-N (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(2-methoxypropan-2-yl)-4-piperidin-4-ylmorpholine 2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.COC(C)(C)[C@H]1CN([C@@H](Cc2ccc(Cl)cc2)CO1)C1CCNCC1 GVVGVIWQBGVCLW-GRTNUQQKSA-N 0.000 description 2
- KSQQLBZTZAAXRQ-LYKKTTPLSA-N (2S)-4-[1-(5-amino-1H-1,2,4-triazol-3-yl)piperidin-4-yl]-5-[(4-chlorophenyl)methyl]morpholine-2-carboxamide Chemical compound NC(=O)[C@@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 KSQQLBZTZAAXRQ-LYKKTTPLSA-N 0.000 description 2
- NUJWKQSEJDYCDB-GNRVTEMESA-N (3s)-1-[(1s,2r,4r)-4-[methyl(propan-2-yl)amino]-2-propylcyclohexyl]-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-2-one Chemical compound CCC[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 NUJWKQSEJDYCDB-GNRVTEMESA-N 0.000 description 2
- XHNLLTGZBXJRGH-LBPRGKRZSA-N (3s)-4-(4-chlorophenyl)-3-[(2-methylpropan-2-yl)oxycarbonylamino]butanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](CC(O)=O)CC1=CC=C(Cl)C=C1 XHNLLTGZBXJRGH-LBPRGKRZSA-N 0.000 description 2
- VIMMECPCYZXUCI-MIMFYIINSA-N (4s,6r)-6-[(1e)-4,4-bis(4-fluorophenyl)-3-(1-methyltetrazol-5-yl)buta-1,3-dienyl]-4-hydroxyoxan-2-one Chemical compound CN1N=NN=C1C(\C=C\[C@@H]1OC(=O)C[C@@H](O)C1)=C(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 VIMMECPCYZXUCI-MIMFYIINSA-N 0.000 description 2
- RXNPEQZHMGFNAY-GEALJGNFSA-N (5R)-4-[(1S,6R)-5-[(2S)-2-(4-chlorophenyl)-3-(propan-2-ylamino)propanoyl]-2,5-diazabicyclo[4.1.0]heptan-2-yl]-5-methyl-6,8-dihydro-5H-pyrido[2,3-d]pyrimidin-7-one Chemical compound C[C@@H]1CC(=O)NC2=C1C(=NC=N2)N3CCN([C@H]4[C@@H]3C4)C(=O)[C@H](CNC(C)C)C5=CC=C(C=C5)Cl RXNPEQZHMGFNAY-GEALJGNFSA-N 0.000 description 2
- ZYBYHZYNINTXOC-MBSJSRAVSA-N (NZ,S)-N-[2-(4-chlorophenyl)ethylidene]-2-methylpropane-2-sulfinamide Chemical compound ClC1=CC=C(C=C1)C\C=N/[S@@](=O)C(C)(C)C ZYBYHZYNINTXOC-MBSJSRAVSA-N 0.000 description 2
- HHJVFVAOLMGAKZ-KUHUBIRLSA-N (S)-N-[(2R)-1-(4-chlorophenyl)pent-4-en-2-yl]-2-methylpropane-2-sulfinamide Chemical compound ClC1=CC=C(C=C1)C[C@@H](CC=C)N[S@@](=O)C(C)(C)C HHJVFVAOLMGAKZ-KUHUBIRLSA-N 0.000 description 2
- DPRJPRMZJGWLHY-HNGSOEQISA-N (e,3r,5s)-7-[5-(4-fluorophenyl)-3-propan-2-yl-1-pyrazin-2-ylpyrazol-4-yl]-3,5-dihydroxyhept-6-enoic acid Chemical compound OC(=O)C[C@H](O)C[C@H](O)/C=C/C=1C(C(C)C)=NN(C=2N=CC=NC=2)C=1C1=CC=C(F)C=C1 DPRJPRMZJGWLHY-HNGSOEQISA-N 0.000 description 2
- KKRLKLQTDMUQTR-UHFFFAOYSA-N 1-(4-chlorophenyl)-1,5,6,7,8,8a-hexahydro-[1,3]oxazolo[3,4-a]pyridin-3-one Chemical compound ClC1=CC=C(C=C1)C1OC(N2C1CCCC2)=O KKRLKLQTDMUQTR-UHFFFAOYSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- SZCBDIVMCGFVPW-UHFFFAOYSA-N 1-[4-(aminomethyl)-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-(3-methoxyphenyl)-2-oxo-1,8-naphthyridin-3-yl]urea;hydrochloride Chemical compound Cl.CC(C)C=1C=C(CN)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OC)=C1 SZCBDIVMCGFVPW-UHFFFAOYSA-N 0.000 description 2
- OXTVBHDILDPYAS-UHFFFAOYSA-N 1-[4-(aminomethyl)-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(3-hydroxypropoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;hydrochloride Chemical compound Cl.CC(C)C=1C=C(CN)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OCCCO)=C1 OXTVBHDILDPYAS-UHFFFAOYSA-N 0.000 description 2
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical compound C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 2
- WNUUCOYSNFCNTL-CJRXIRLBSA-N 2-[(2R,5S)-5-[(4-chlorophenyl)methyl]-4-piperidin-4-ylmorpholin-2-yl]propan-2-ol 2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CC(C)(O)[C@H]1CN([C@@H](Cc2ccc(Cl)cc2)CO1)C1CCNCC1 WNUUCOYSNFCNTL-CJRXIRLBSA-N 0.000 description 2
- JSIAIROWMJGMQZ-UHFFFAOYSA-N 2h-triazol-4-amine Chemical class NC1=CNN=N1 JSIAIROWMJGMQZ-UHFFFAOYSA-N 0.000 description 2
- CASGKNKGRHYUAE-QRWMCTBCSA-N 3-[4-[(2R)-5-[(4-chlorophenyl)methyl]-2-(methoxymethyl)morpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound COC[C@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 CASGKNKGRHYUAE-QRWMCTBCSA-N 0.000 description 2
- STWVLEKJQQRGMO-CWQZNGJJSA-N 3-[4-[(2S)-5-[(4-chlorophenyl)methyl]-2-methylmorpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound C[C@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 STWVLEKJQQRGMO-CWQZNGJJSA-N 0.000 description 2
- TXEBWPPWSVMYOA-UHFFFAOYSA-N 4-[3-[(1-amino-2-chloroethyl)amino]propyl]-1-[[3-(2-chlorophenyl)phenyl]methyl]-5-hydroxyimidazolidin-2-one Chemical compound NC(CCl)NCCCC1NC(=O)N(Cc2cccc(c2)-c2ccccc2Cl)C1O TXEBWPPWSVMYOA-UHFFFAOYSA-N 0.000 description 2
- KUZSBKJSGSKPJH-VXGBXAGGSA-N 5-[(9R)-6-[(3R)-3-methylmorpholin-4-yl]-11-oxa-1,3,5-triazatricyclo[7.4.0.02,7]trideca-2,4,6-trien-4-yl]pyrazin-2-amine Chemical compound C[C@@H]1COCCN1c1nc(nc2N3CCOC[C@H]3Cc12)-c1cnc(N)cn1 KUZSBKJSGSKPJH-VXGBXAGGSA-N 0.000 description 2
- FCBOUJYKAGWYQM-DEOSSOPVSA-N 6-[[(2s)-1-hydroxy-3-phenylpropan-2-yl]amino]-n-(2-phenoxyethyl)-2-(3,4,5-trimethoxyphenyl)pyridine-3-carboxamide Chemical compound COC1=C(OC)C(OC)=CC(C=2C(=CC=C(N[C@H](CO)CC=3C=CC=CC=3)N=2)C(=O)NCCOC=2C=CC=CC=2)=C1 FCBOUJYKAGWYQM-DEOSSOPVSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 241000272517 Anseriformes Species 0.000 description 2
- 241000726096 Aratinga Species 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- NZSQBRZWARZNQH-ZWOACCQCSA-N C1(CC1)NC(=O)O[C@H]1C(C2CC[C@]3([C@@]4(CC[C@@]5(C(C4CCC3[C@]2(CC1)C)[C@@H](CC5)[C@H](C)O)C(=O)O)C)C)(C)C Chemical compound C1(CC1)NC(=O)O[C@H]1C(C2CC[C@]3([C@@]4(CC[C@@]5(C(C4CCC3[C@]2(CC1)C)[C@@H](CC5)[C@H](C)O)C(=O)O)C)C)(C)C NZSQBRZWARZNQH-ZWOACCQCSA-N 0.000 description 2
- QCMHGCDOZLWPOT-FMNCTDSISA-N COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 Chemical compound COC1=C(CC[C@@H]2CCC3=C(C2)C=CC(=C3)[C@H]2CC[C@](N)(CO)C2)C=CC=C1 QCMHGCDOZLWPOT-FMNCTDSISA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 241000282836 Camelus dromedarius Species 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 206010011224 Cough Diseases 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 2
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 101710139422 Eotaxin Proteins 0.000 description 2
- 241000283073 Equus caballus Species 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- 241000699694 Gerbillinae Species 0.000 description 2
- XLYOFNOQVPJJNP-ZSJDYOACSA-N Heavy water Chemical compound [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 2
- 206010019668 Hepatic fibrosis Diseases 0.000 description 2
- 108090000978 Interleukin-4 Proteins 0.000 description 2
- 239000003810 Jones reagent Substances 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- HPKJGHVHQWJOOT-ZJOUEHCJSA-N N-[(2S)-3-cyclohexyl-1-oxo-1-({(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino)propan-2-yl]-1H-indole-2-carboxamide Chemical compound C1C(CCCC1)C[C@H](NC(=O)C=1NC2=CC=CC=C2C=1)C(=O)N[C@@H](C[C@H]1C(=O)NCC1)C=O HPKJGHVHQWJOOT-ZJOUEHCJSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- 206010036790 Productive cough Diseases 0.000 description 2
- 208000037656 Respiratory Sounds Diseases 0.000 description 2
- 241000219061 Rheum Species 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 206010047924 Wheezing Diseases 0.000 description 2
- NPUXORBZRBIOMQ-RUZDIDTESA-N [(2R)-1-[[4-[[3-(benzenesulfonylmethyl)-5-methylphenoxy]methyl]phenyl]methyl]-2-pyrrolidinyl]methanol Chemical compound C=1C(OCC=2C=CC(CN3[C@H](CCC3)CO)=CC=2)=CC(C)=CC=1CS(=O)(=O)C1=CC=CC=C1 NPUXORBZRBIOMQ-RUZDIDTESA-N 0.000 description 2
- NELWQUQCCZMRPB-UBPLGANQSA-N [(2r,3r,4r,5r)-4-acetyloxy-5-(4-amino-5-ethenyl-2-oxopyrimidin-1-yl)-2-methyloxolan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1[C@H](OC(C)=O)[C@@H](C)O[C@H]1N1C(=O)N=C(N)C(C=C)=C1 NELWQUQCCZMRPB-UBPLGANQSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 230000036428 airway hyperreactivity Effects 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 150000001335 aliphatic alkanes Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 2
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- 201000009961 allergic asthma Diseases 0.000 description 2
- 125000000266 alpha-aminoacyl group Chemical group 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 125000005199 aryl carbonyloxy group Chemical group 0.000 description 2
- 125000000732 arylene group Chemical group 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 229960000686 benzalkonium chloride Drugs 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- XTEOJPUYZWEXFI-UHFFFAOYSA-N butyl n-[3-[4-(imidazol-1-ylmethyl)phenyl]-5-(2-methylpropyl)thiophen-2-yl]sulfonylcarbamate Chemical compound S1C(CC(C)C)=CC(C=2C=CC(CN3C=NC=C3)=CC=2)=C1S(=O)(=O)NC(=O)OCCCC XTEOJPUYZWEXFI-UHFFFAOYSA-N 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- AEULIVPVIDOLIN-UHFFFAOYSA-N cep-11981 Chemical compound C1=C2C3=C4CNC(=O)C4=C4C5=CN(C)N=C5CCC4=C3N(CC(C)C)C2=CC=C1NC1=NC=CC=N1 AEULIVPVIDOLIN-UHFFFAOYSA-N 0.000 description 2
- 229910052729 chemical element Inorganic materials 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 238000011097 chromatography purification Methods 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 239000007979 citrate buffer Substances 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 238000004040 coloring Methods 0.000 description 2
- 238000004891 communication Methods 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 150000004985 diamines Chemical class 0.000 description 2
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 2
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 2
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 208000037765 diseases and disorders Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 239000013583 drug formulation Substances 0.000 description 2
- 210000001198 duodenum Anatomy 0.000 description 2
- 239000003974 emollient agent Substances 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 238000007824 enzymatic assay Methods 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- 230000005713 exacerbation Effects 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 230000000893 fibroproliferative effect Effects 0.000 description 2
- 238000005187 foaming Methods 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 239000007902 hard capsule Substances 0.000 description 2
- 125000004475 heteroaralkyl group Chemical group 0.000 description 2
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 2
- 210000003630 histaminocyte Anatomy 0.000 description 2
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 description 2
- 206010020718 hyperplasia Diseases 0.000 description 2
- 230000004047 hyperresponsiveness Effects 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000015788 innate immune response Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007913 intrathecal administration Methods 0.000 description 2
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 102000003835 leukotriene receptors Human genes 0.000 description 2
- 108090000146 leukotriene receptors Proteins 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 229940057995 liquid paraffin Drugs 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 230000004199 lung function Effects 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 201000004792 malaria Diseases 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 229940071648 metered dose inhaler Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 239000003094 microcapsule Substances 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000003595 mist Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 210000000440 neutrophil Anatomy 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 125000001715 oxadiazolyl group Chemical group 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 125000003367 polycyclic group Polymers 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 description 2
- 238000010837 poor prognosis Methods 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 2
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 230000008521 reorganization Effects 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 208000013220 shortness of breath Diseases 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 235000010413 sodium alginate Nutrition 0.000 description 2
- 239000000661 sodium alginate Substances 0.000 description 2
- 229940005550 sodium alginate Drugs 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 210000003802 sputum Anatomy 0.000 description 2
- 208000024794 sputum Diseases 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- RPRMPBGHQUNDOV-JKSUJKDBSA-N tert-butyl (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(2-hydroxypropan-2-yl)morpholine-4-carboxylate Chemical compound ClC1=CC=C(C[C@H]2CO[C@H](CN2C(=O)OC(C)(C)C)C(C)(C)O)C=C1 RPRMPBGHQUNDOV-JKSUJKDBSA-N 0.000 description 2
- VSZMMSCSLKSGIH-LSDHHAIUSA-N tert-butyl (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(hydroxymethyl)morpholine-4-carboxylate Chemical compound ClC1=CC=C(C[C@H]2CO[C@H](CN2C(=O)OC(C)(C)C)CO)C=C1 VSZMMSCSLKSGIH-LSDHHAIUSA-N 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 125000003396 thiol group Chemical class [H]S* 0.000 description 2
- 230000007838 tissue remodeling Effects 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- QQJLHRRUATVHED-UHFFFAOYSA-N tramazoline Chemical compound N1CCN=C1NC1=CC=CC2=C1CCCC2 QQJLHRRUATVHED-UHFFFAOYSA-N 0.000 description 2
- 229960001262 tramazoline Drugs 0.000 description 2
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 2
- 229940029284 trichlorofluoromethane Drugs 0.000 description 2
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- BFDBKMOZYNOTPK-UHFFFAOYSA-N vonoprazan Chemical compound C=1C=CN=CC=1S(=O)(=O)N1C=C(CNC)C=C1C1=CC=CC=C1F BFDBKMOZYNOTPK-UHFFFAOYSA-N 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- IZUAHLHTQJCCLJ-UHFFFAOYSA-N (2-chloro-1,1,2,2-tetrafluoroethyl) hypochlorite Chemical compound FC(F)(Cl)C(F)(F)OCl IZUAHLHTQJCCLJ-UHFFFAOYSA-N 0.000 description 1
- KSQQLBZTZAAXRQ-OEMAIJDKSA-N (2R)-4-[1-(5-amino-1H-1,2,4-triazol-3-yl)piperidin-4-yl]-5-[(4-chlorophenyl)methyl]morpholine-2-carboxamide Chemical compound NC(=O)[C@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 KSQQLBZTZAAXRQ-OEMAIJDKSA-N 0.000 description 1
- SIHMHZAQHLFZKY-QWHCGFSZSA-N (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(methoxymethyl)morpholine Chemical compound ClC1=CC=C(C[C@H]2CO[C@H](CN2)COC)C=C1 SIHMHZAQHLFZKY-QWHCGFSZSA-N 0.000 description 1
- XOJUVYFPNNCKBJ-NWDGAFQWSA-N (2R,5S)-5-[(4-chlorophenyl)methyl]-2-(methylcarbamoyl)morpholine-4-carboxylic acid Chemical compound CNC(=O)[C@H]1CN([C@@H](Cc2ccc(Cl)cc2)CO1)C(O)=O XOJUVYFPNNCKBJ-NWDGAFQWSA-N 0.000 description 1
- IONQHATYKFGSFN-RGURZIINSA-N (2S)-2-[(4-chlorophenyl)methyl]-3-hydroxyazetidine-1-carboxylic acid Chemical compound OC1CN([C@H]1Cc1ccc(Cl)cc1)C(O)=O IONQHATYKFGSFN-RGURZIINSA-N 0.000 description 1
- DAAXYQZSKBPJOX-FQEVSTJZSA-N (2S)-2-amino-3-[4-[5-[3-(4-hydroxyphenyl)-4-methoxyphenyl]-1,2,4-oxadiazol-3-yl]phenyl]propanoic acid Chemical compound COC1=C(C=C(C=C1)C2=NC(=NO2)C3=CC=C(C=C3)C[C@@H](C(=O)O)N)C4=CC=C(C=C4)O DAAXYQZSKBPJOX-FQEVSTJZSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- VRKNREMRDSDPTQ-NWDGAFQWSA-N (2S,4R)-2-[(4-chlorophenyl)methyl]-4-hydroxypiperidine-1-carboxylic acid Chemical compound O[C@@H]1CCN([C@@H](Cc2ccc(Cl)cc2)C1)C(O)=O VRKNREMRDSDPTQ-NWDGAFQWSA-N 0.000 description 1
- FLRZFNOWIPFTKS-QWHCGFSZSA-N (2S,4R)-2-[(4-chlorophenyl)methyl]-4-methoxypiperidine-1-carboxylic acid Chemical compound CO[C@@H]1CCN([C@@H](Cc2ccc(Cl)cc2)C1)C(O)=O FLRZFNOWIPFTKS-QWHCGFSZSA-N 0.000 description 1
- WLWOLADTSSQZOA-WMZOPIPTSA-N (2S,5S)-5-[(4-chlorophenyl)methyl]-4-(4-methylphenyl)sulfonylmorpholine-2-carboxamide Chemical compound ClC1=CC=C(C[C@H]2CO[C@@H](CN2S(=O)(=O)C2=CC=C(C)C=C2)C(=O)N)C=C1 WLWOLADTSSQZOA-WMZOPIPTSA-N 0.000 description 1
- BAMZFZCCPHNFRF-WMZOPIPTSA-N (2S,5S)-5-[(4-chlorophenyl)methyl]-4-(4-methylphenyl)sulfonylmorpholine-2-carboxylic acid Chemical compound ClC1=CC=C(C[C@H]2CO[C@@H](CN2S(=O)(=O)C2=CC=C(C)C=C2)C(=O)O)C=C1 BAMZFZCCPHNFRF-WMZOPIPTSA-N 0.000 description 1
- UEBARDWJXBGYEJ-SCSAIBSYSA-N (2r)-2-bromo-3-methylbutanoic acid Chemical compound CC(C)[C@@H](Br)C(O)=O UEBARDWJXBGYEJ-SCSAIBSYSA-N 0.000 description 1
- NNFDHJQLIFECSR-RXMQYKEDSA-N (2r)-2-bromo-4-methylpentanoic acid Chemical compound CC(C)C[C@@H](Br)C(O)=O NNFDHJQLIFECSR-RXMQYKEDSA-N 0.000 description 1
- YAQLSKVCTLCIIE-GSVOUGTGSA-N (2r)-2-bromobutanoic acid Chemical compound CC[C@@H](Br)C(O)=O YAQLSKVCTLCIIE-GSVOUGTGSA-N 0.000 description 1
- PTLWNCBCBZZBJI-CRCLSJGQSA-N (2r,3s)-piperidine-2,3-dicarboxylic acid Chemical group OC(=O)[C@H]1CCCN[C@H]1C(O)=O PTLWNCBCBZZBJI-CRCLSJGQSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- PEMUHKUIQHFMTH-QMMMGPOBSA-N (2s)-2-amino-3-(4-bromophenyl)propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(Br)C=C1 PEMUHKUIQHFMTH-QMMMGPOBSA-N 0.000 description 1
- YEYDDJFAYMBQTE-GRHHLOCNSA-N (2s)-2-amino-3-(4-chlorophenyl)propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(Cl)C=C1.OC(=O)[C@@H](N)CC1=CC=C(Cl)C=C1 YEYDDJFAYMBQTE-GRHHLOCNSA-N 0.000 description 1
- LJIOTBMDLVHTBO-CUYJMHBOSA-N (2s)-2-amino-n-[(1r,2r)-1-cyano-2-[4-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]phenyl]cyclopropyl]butanamide Chemical compound CC[C@H](N)C(=O)N[C@]1(C#N)C[C@@H]1C1=CC=C(C=2C=CC(=CC=2)S(=O)(=O)N2CCN(C)CC2)C=C1 LJIOTBMDLVHTBO-CUYJMHBOSA-N 0.000 description 1
- ZWVMLYRJXORSEP-LURJTMIESA-N (2s)-hexane-1,2,6-triol Chemical compound OCCCC[C@H](O)CO ZWVMLYRJXORSEP-LURJTMIESA-N 0.000 description 1
- TWYYFYNJOJGNFP-CUXYNZQBSA-N (2s,4r,5s,6s)-2-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-2-carbamoyl-4-[[(e,4s,6s)-4,6-dimethyloct-2-enoyl]oxymethyl]-5-hydroxy-1,3-dioxane-4,5,6-tricarboxylic acid Chemical compound O1[C@H](C(O)=O)[C@](C(O)=O)(O)[C@](COC(=O)/C=C/[C@@H](C)C[C@@H](C)CC)(C(O)=O)O[C@]1(C(N)=O)CCC(=C)[C@@H](OC(C)=O)[C@H](C)CC1=CC=CC=C1 TWYYFYNJOJGNFP-CUXYNZQBSA-N 0.000 description 1
- PHDIJLFSKNMCMI-ITGJKDDRSA-N (3R,4S,5R,6R)-6-(hydroxymethyl)-4-(8-quinolin-6-yloxyoctoxy)oxane-2,3,5-triol Chemical compound OC[C@@H]1[C@H]([C@@H]([C@H](C(O1)O)O)OCCCCCCCCOC=1C=C2C=CC=NC2=CC=1)O PHDIJLFSKNMCMI-ITGJKDDRSA-N 0.000 description 1
- YXSZAZSRXHKOBE-INIZCTEOSA-N (3S)-3-[(4-chlorophenyl)methyl]-4-piperidin-4-ylmorpholine Chemical compound ClC1=CC=C(C[C@@H]2N(CCOC2)C2CCNCC2)C=C1 YXSZAZSRXHKOBE-INIZCTEOSA-N 0.000 description 1
- OYBVODNMGYFPHN-NSHDSACASA-N (3S)-3-[(4-chlorophenyl)methyl]morpholine Chemical compound ClC1=CC=C(C[C@H]2COCCN2)C=C1 OYBVODNMGYFPHN-NSHDSACASA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- DMQYDVBIPXAAJA-VHXPQNKSSA-N (3z)-5-[(1-ethylpiperidin-4-yl)amino]-3-[(3-fluorophenyl)-(5-methyl-1h-imidazol-2-yl)methylidene]-1h-indol-2-one Chemical compound C1CN(CC)CCC1NC1=CC=C(NC(=O)\C2=C(/C=3NC=C(C)N=3)C=3C=C(F)C=CC=3)C2=C1 DMQYDVBIPXAAJA-VHXPQNKSSA-N 0.000 description 1
- FRJJJAKBRKABFA-TYFAACHXSA-N (4r,6s)-6-[(e)-2-[6-chloro-4-(4-fluorophenyl)-2-propan-2-ylquinolin-3-yl]ethenyl]-4-hydroxyoxan-2-one Chemical compound C(\[C@H]1OC(=O)C[C@H](O)C1)=C/C=1C(C(C)C)=NC2=CC=C(Cl)C=C2C=1C1=CC=C(F)C=C1 FRJJJAKBRKABFA-TYFAACHXSA-N 0.000 description 1
- LXEJHPLLFBTVKJ-JTQLQIEISA-N (5S)-5-[(4-chlorophenyl)methyl]morpholin-3-one Chemical compound ClC1=CC=C(C[C@H]2COCC(N2)=O)C=C1 LXEJHPLLFBTVKJ-JTQLQIEISA-N 0.000 description 1
- KGYKHCSCMZQEMA-OQHSHRKDSA-N (6R)-1-[1-(5-amino-1H-1,2,4-triazol-3-yl)piperidin-4-yl]-6-[(4-chlorophenyl)methyl]-3-methylpiperidine-3-carboxylic acid Chemical compound NC=1NC(=NN=1)N1CCC(CC1)N1CC(CC[C@@H]1CC1=CC=C(C=C1)Cl)(C(=O)O)C KGYKHCSCMZQEMA-OQHSHRKDSA-N 0.000 description 1
- QOLHWXNSCZGWHK-BWBORTOCSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-4,7-dihydroxy-6-(11-phenoxyundecylcarbamoyloxy)-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@@H]([C@@H](OC(=O)NCCCCCCCCCCCOC=3C=CC=CC=3)C(O1)(C(O)=O)C(O)(C(O2)C(O)=O)C(O)=O)O)C1=CC=CC=C1 QOLHWXNSCZGWHK-BWBORTOCSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- LVHOHZHTZXRVRJ-CMDGGOBGSA-N (e)-3-(3-methoxyphenyl)-n-(3,4,5-trimethoxyphenyl)prop-2-enamide Chemical compound COC1=CC=CC(\C=C\C(=O)NC=2C=C(OC)C(OC)=C(OC)C=2)=C1 LVHOHZHTZXRVRJ-CMDGGOBGSA-N 0.000 description 1
- JNPGUXGVLNJQSQ-BGGMYYEUSA-M (e,3r,5s)-7-[4-(4-fluorophenyl)-1,2-di(propan-2-yl)pyrrol-3-yl]-3,5-dihydroxyhept-6-enoate Chemical compound CC(C)N1C(C(C)C)=C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)C(C=2C=CC(F)=CC=2)=C1 JNPGUXGVLNJQSQ-BGGMYYEUSA-M 0.000 description 1
- VAVHMEQFYYBAPR-ITWZMISCSA-N (e,3r,5s)-7-[4-(4-fluorophenyl)-1-phenyl-2-propan-2-ylpyrrol-3-yl]-3,5-dihydroxyhept-6-enoic acid Chemical compound CC(C)C1=C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)C(C=2C=CC(F)=CC=2)=CN1C1=CC=CC=C1 VAVHMEQFYYBAPR-ITWZMISCSA-N 0.000 description 1
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 1
- ZCXLTWVZYXBHJS-UHFFFAOYSA-N 1,2-benzoxazepine Chemical compound O1N=CC=CC2=CC=CC=C12 ZCXLTWVZYXBHJS-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- IGERFAHWSHDDHX-UHFFFAOYSA-N 1,3-dioxanyl Chemical group [CH]1OCCCO1 IGERFAHWSHDDHX-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- ILWJAOPQHOZXAN-UHFFFAOYSA-N 1,3-dithianyl Chemical group [CH]1SCCCS1 ILWJAOPQHOZXAN-UHFFFAOYSA-N 0.000 description 1
- FLOJNXXFMHCMMR-UHFFFAOYSA-N 1,3-dithiolanyl Chemical group [CH]1SCCS1 FLOJNXXFMHCMMR-UHFFFAOYSA-N 0.000 description 1
- GUJAGMICFDYKNR-UHFFFAOYSA-N 1,4-benzodiazepine Chemical class N1C=CN=CC2=CC=CC=C12 GUJAGMICFDYKNR-UHFFFAOYSA-N 0.000 description 1
- UWXIKAZQXAYFPT-UHFFFAOYSA-N 1,4-benzoxazepine Chemical compound O1C=CN=CC2=CC=CC=C12 UWXIKAZQXAYFPT-UHFFFAOYSA-N 0.000 description 1
- WXLCDTBTIVJDCE-UHFFFAOYSA-N 1,4-oxazepine Chemical compound O1C=CC=NC=C1 WXLCDTBTIVJDCE-UHFFFAOYSA-N 0.000 description 1
- JQAOHGMPAAWWQO-UHFFFAOYSA-N 1-[(2-methylpropan-2-yl)oxycarbonyl]piperidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCCCC1C(O)=O JQAOHGMPAAWWQO-UHFFFAOYSA-N 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- GWQSENYKCGJTRI-UHFFFAOYSA-N 1-chloro-4-iodobenzene Chemical compound ClC1=CC=C(I)C=C1 GWQSENYKCGJTRI-UHFFFAOYSA-N 0.000 description 1
- PZNPLUBHRSSFHT-RRHRGVEJSA-N 1-hexadecanoyl-2-octadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[C@@H](COP([O-])(=O)OCC[N+](C)(C)C)COC(=O)CCCCCCCCCCCCCCC PZNPLUBHRSSFHT-RRHRGVEJSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- OHMHBGPWCHTMQE-UHFFFAOYSA-N 2,2-dichloro-1,1,1-trifluoroethane Chemical compound FC(F)(F)C(Cl)Cl OHMHBGPWCHTMQE-UHFFFAOYSA-N 0.000 description 1
- 125000004565 2,3-dihydrobenzofuran-4-yl group Chemical group O1CCC2=C1C=CC=C2* 0.000 description 1
- 125000004563 2,3-dihydroindol-5-yl group Chemical group N1CCC2=CC(=CC=C12)* 0.000 description 1
- FJMQJSUOOGOWBD-UHFFFAOYSA-N 2-(2-chlorophenyl)-3-(4-chlorophenyl)-7-(2,2-difluoropropyl)-5,6-dihydropyrazolo[3,4-f][1,4]oxazepin-8-one Chemical compound O=C1N(CC(F)(F)C)CCOC=2C1=NN(C=1C(=CC=CC=1)Cl)C=2C1=CC=C(Cl)C=C1 FJMQJSUOOGOWBD-UHFFFAOYSA-N 0.000 description 1
- MWSNYBOKRSGWAN-UHFFFAOYSA-N 2-(4-chlorophenyl)acetaldehyde Chemical compound ClC1=CC=C(CC=O)C=C1 MWSNYBOKRSGWAN-UHFFFAOYSA-N 0.000 description 1
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 description 1
- RYWCQJDEHXJHRI-XJMXIVSISA-N 2-[3-[5-[6-[3-[3-(carboxymethyl)phenyl]-4-[(2r,3s,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]hexyl]-2-[(2r,3s,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]phenyl]acetic acid Chemical compound O[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC(C(=C1)C=2C=C(CC(O)=O)C=CC=2)=CC=C1CCCCCCC(C=C1C=2C=C(CC(O)=O)C=CC=2)=CC=C1O[C@@H]1[C@@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 RYWCQJDEHXJHRI-XJMXIVSISA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- 125000006056 2-methyl-4-pentenyl group Chemical group 0.000 description 1
- CESUXLKAADQNTB-ZETCQYMHSA-N 2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](N)=O CESUXLKAADQNTB-ZETCQYMHSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- HNFMVVHMKGFCMB-UHFFFAOYSA-N 3-[3-[4-(1-aminocyclobutyl)phenyl]-5-phenylimidazo[4,5-b]pyridin-2-yl]pyridin-2-amine Chemical compound NC1=NC=CC=C1C1=NC2=CC=C(C=3C=CC=CC=3)N=C2N1C1=CC=C(C2(N)CCC2)C=C1 HNFMVVHMKGFCMB-UHFFFAOYSA-N 0.000 description 1
- JUBVZHROMXPRJA-ANYOKISRSA-N 3-[4-[(2S)-5-[(4-chlorophenyl)methyl]-2-(2-methylpropyl)morpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound CC(C)C[C@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 JUBVZHROMXPRJA-ANYOKISRSA-N 0.000 description 1
- GTWHZKOJJZOTCX-ZVAWYAOSSA-N 3-[4-[(2S)-5-[(4-chlorophenyl)methyl]-2-ethylmorpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound CC[C@H]1CN(C(Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)c1n[nH]c(N)n1 GTWHZKOJJZOTCX-ZVAWYAOSSA-N 0.000 description 1
- TVNHFKAPQIMECM-CABCVRRESA-N 3-[4-[(2S,3R)-2-[(4-chlorophenyl)methyl]-3-fluoroazetidin-1-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound ClC1=CC=C(C[C@@H]2N(C[C@H]2F)C2CCN(CC2)C=2NC(=NN=2)N)C=C1 TVNHFKAPQIMECM-CABCVRRESA-N 0.000 description 1
- CKGIHYVZWVUSHR-ZVAWYAOSSA-N 3-[4-[(4S)-2-[(4-chlorophenyl)methyl]-4-methoxypiperidin-1-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound CO[C@H]1CCN(C(Cc2ccc(Cl)cc2)C1)C1CCN(CC1)c1n[nH]c(N)n1 CKGIHYVZWVUSHR-ZVAWYAOSSA-N 0.000 description 1
- CDYQBZJWGZGZKD-UHFFFAOYSA-N 3-[4-[3-[(4-bromophenyl)methyl]morpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound Nc1nc(n[nH]1)N1CCC(CC1)N1CCOCC1Cc1ccc(Br)cc1 CDYQBZJWGZGZKD-UHFFFAOYSA-N 0.000 description 1
- GAIOPWBQKZMUNO-UHFFFAOYSA-N 3-[[5-fluoro-4-[4-methyl-2-(methylamino)-1,3-thiazol-5-yl]pyrimidin-2-yl]amino]benzenesulfonamide Chemical compound S1C(NC)=NC(C)=C1C1=NC(NC=2C=C(C=CC=2)S(N)(=O)=O)=NC=C1F GAIOPWBQKZMUNO-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- QOXOZONBQWIKDA-UHFFFAOYSA-N 3-hydroxypropyl Chemical group [CH2]CCO QOXOZONBQWIKDA-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- JWWKSAIWNUHSAO-RBUKOAKNSA-N 4-[(2R,5S)-5-[(4-chlorophenyl)methyl]-2-(2-methoxypropan-2-yl)morpholin-4-yl]piperidine-1-carboxylic acid Chemical compound COC(C)(C)[C@H]1CN([C@@H](Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)C(O)=O JWWKSAIWNUHSAO-RBUKOAKNSA-N 0.000 description 1
- KXRDITPVTSUWEL-RBUKOAKNSA-N 4-[(2R,5S)-5-[(4-chlorophenyl)methyl]-2-(ethoxymethyl)morpholin-4-yl]piperidine-1-carboxylic acid Chemical compound CCOC[C@H]1CN([C@@H](Cc2ccc(Cl)cc2)CO1)C1CCN(CC1)C(O)=O KXRDITPVTSUWEL-RBUKOAKNSA-N 0.000 description 1
- WCDLCPLAAKUJNY-UHFFFAOYSA-N 4-[4-[3-(1h-pyrazol-4-yl)pyrazolo[1,5-a]pyrimidin-6-yl]phenyl]morpholine Chemical compound C1COCCN1C1=CC=C(C2=CN3N=CC(=C3N=C2)C2=CNN=C2)C=C1 WCDLCPLAAKUJNY-UHFFFAOYSA-N 0.000 description 1
- NIGWMJHCCYYCSF-QMMMGPOBSA-N 4-chloro-L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(Cl)C=C1 NIGWMJHCCYYCSF-QMMMGPOBSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- SRSGVKWWVXWSJT-ATVHPVEESA-N 5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-n-(2-pyrrolidin-1-ylethyl)-1h-pyrrole-3-carboxamide Chemical compound CC=1NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C(C)C=1C(=O)NCCN1CCCC1 SRSGVKWWVXWSJT-ATVHPVEESA-N 0.000 description 1
- HIHOEGPXVVKJPP-JTQLQIEISA-N 5-fluoro-2-[[(1s)-1-(5-fluoropyridin-2-yl)ethyl]amino]-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyridine-3-carbonitrile Chemical compound N([C@@H](C)C=1N=CC(F)=CC=1)C(C(=CC=1F)C#N)=NC=1NC=1C=C(C)NN=1 HIHOEGPXVVKJPP-JTQLQIEISA-N 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- XKFBUSDGBIEDDM-UHFFFAOYSA-N 6,7-dihydro-2,1,3-benzoxadiazole Chemical compound C1=CCCC2=NON=C21 XKFBUSDGBIEDDM-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- JYSLFQTWNRYWJT-UHFFFAOYSA-N 8-(3,5-dichlorophenyl)sulfanyl-9-[3-(propan-2-ylamino)propyl]purin-6-amine Chemical compound N=1C2=C(N)N=CN=C2N(CCCNC(C)C)C=1SC1=CC(Cl)=CC(Cl)=C1 JYSLFQTWNRYWJT-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 241000238876 Acari Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000036065 Airway Remodeling Diseases 0.000 description 1
- 208000007082 Alcoholic Fatty Liver Diseases 0.000 description 1
- 208000022309 Alcoholic Liver disease Diseases 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 241000218157 Aquilegia vulgaris Species 0.000 description 1
- 201000002909 Aspergillosis Diseases 0.000 description 1
- 208000036641 Aspergillus infections Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 229910015900 BF3 Inorganic materials 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- 241001674044 Blattodea Species 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 208000009079 Bronchial Spasm Diseases 0.000 description 1
- 208000014181 Bronchial disease Diseases 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- 102100021935 C-C motif chemokine 26 Human genes 0.000 description 1
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 1
- LERNTVKEWCAPOY-VOGVJGKGSA-N C[N+]1(C)[C@H]2C[C@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)C(O)(c1cccs1)c1cccs1 Chemical compound C[N+]1(C)[C@H]2C[C@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)C(O)(c1cccs1)c1cccs1 LERNTVKEWCAPOY-VOGVJGKGSA-N 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- 241001494315 Cacatuidae Species 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241000700190 Caviidae Species 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108010082548 Chemokine CCL11 Proteins 0.000 description 1
- 108010083698 Chemokine CCL26 Proteins 0.000 description 1
- 206010008469 Chest discomfort Diseases 0.000 description 1
- 241000700112 Chinchilla Species 0.000 description 1
- 101710151413 Chitinase 1 Proteins 0.000 description 1
- 229940125842 Chitinase inhibitor Drugs 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 229940126559 Compound 4e Drugs 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241000938605 Crocodylia Species 0.000 description 1
- 201000007336 Cryptococcosis Diseases 0.000 description 1
- 241000221204 Cryptococcus neoformans Species 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 101710107327 Endochitinase 1 Proteins 0.000 description 1
- 102400000686 Endothelin-1 Human genes 0.000 description 1
- 101800004490 Endothelin-1 Proteins 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 241000283074 Equus asinus Species 0.000 description 1
- 241001331845 Equus asinus x caballus Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 229920003136 Eudragit® L polymer Polymers 0.000 description 1
- 229920003137 Eudragit® S polymer Polymers 0.000 description 1
- 208000004930 Fatty Liver Diseases 0.000 description 1
- RRSNDVCODIMOFX-MPKOGUQCSA-N Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O Chemical compound Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O RRSNDVCODIMOFX-MPKOGUQCSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- NIGWMJHCCYYCSF-UHFFFAOYSA-N Fenclonine Chemical compound OC(=O)C(N)CC1=CC=C(Cl)C=C1 NIGWMJHCCYYCSF-UHFFFAOYSA-N 0.000 description 1
- 206010016946 Food allergy Diseases 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 239000007818 Grignard reagent Substances 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 241000264877 Hippospongia communis Species 0.000 description 1
- 101000738085 Homo sapiens Acidic mammalian chitinase Proteins 0.000 description 1
- 101100059969 Homo sapiens CHIT1 gene Proteins 0.000 description 1
- 102000002265 Human Growth Hormone Human genes 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- 239000000854 Human Growth Hormone Substances 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 108010002616 Interleukin-5 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 206010023203 Joint destruction Diseases 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 206010054949 Metaplasia Diseases 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical class COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 241001504654 Mustela nivalis Species 0.000 description 1
- 241000282341 Mustela putorius furo Species 0.000 description 1
- 241000282331 Mustelidae Species 0.000 description 1
- 238000007126 N-alkylation reaction Methods 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- MPKOJZAEXLLVEI-XPKAQORNSA-N NC=1NC(=NN1)N1CCC(CC1)N1CC(CC[C@@H]1CC1=CC=C(C=C1)Cl)C Chemical compound NC=1NC(=NN1)N1CCC(CC1)N1CC(CC[C@@H]1CC1=CC=C(C=C1)Cl)C MPKOJZAEXLLVEI-XPKAQORNSA-N 0.000 description 1
- ZNSPHKJFQDEABI-NZQKXSOJSA-N Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 Chemical compound Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 ZNSPHKJFQDEABI-NZQKXSOJSA-N 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- 208000022873 Ocular disease Diseases 0.000 description 1
- 206010030216 Oesophagitis Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 206010033078 Otitis media Diseases 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 241000286209 Phasianidae Species 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920001363 Polidocanol Polymers 0.000 description 1
- 229920001305 Poly(isodecyl(meth)acrylate) Polymers 0.000 description 1
- 229920002319 Poly(methyl acrylate) Polymers 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920002701 Polyoxyl 40 Stearate Polymers 0.000 description 1
- 229920000037 Polyproline Polymers 0.000 description 1
- 229920001219 Polysorbate 40 Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 229920002642 Polysorbate 65 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 241000243142 Porifera Species 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000001280 Prediabetic State Diseases 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 108010026552 Proteome Proteins 0.000 description 1
- 208000003251 Pruritus Diseases 0.000 description 1
- 208000007400 Relapsing-Remitting Multiple Sclerosis Diseases 0.000 description 1
- 208000002200 Respiratory Hypersensitivity Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 208000036071 Rhinorrhea Diseases 0.000 description 1
- 206010039101 Rhinorrhoea Diseases 0.000 description 1
- 241000282849 Ruminantia Species 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- 241000287219 Serinus canaria Species 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 235000015125 Sterculia urens Nutrition 0.000 description 1
- 240000001058 Sterculia urens Species 0.000 description 1
- 241001504624 Streptopelia Species 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 230000029662 T-helper 1 type immune response Effects 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 208000024780 Urticaria Diseases 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 208000006903 Wheat Hypersensitivity Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- 206010000269 abscess Diseases 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000012387 aerosolization Methods 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 208000037883 airway inflammation Diseases 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 208000026594 alcoholic fatty liver disease Diseases 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000005079 alkoxycarbonylmethyl group Chemical group 0.000 description 1
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 229960004977 anhydrous lactose Drugs 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 239000003831 antifriction material Substances 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 238000000889 atomisation Methods 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 150000001538 azepines Chemical class 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 125000004045 azirinyl group Chemical group 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 238000007630 basic procedure Methods 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- PNPBGYBHLCEVMK-UHFFFAOYSA-N benzylidene(dichloro)ruthenium;tricyclohexylphosphanium Chemical compound Cl[Ru](Cl)=CC1=CC=CC=C1.C1CCCCC1[PH+](C1CCCCC1)C1CCCCC1.C1CCCCC1[PH+](C1CCCCC1)C1CCCCC1 PNPBGYBHLCEVMK-UHFFFAOYSA-N 0.000 description 1
- FCDPQMAOJARMTG-UHFFFAOYSA-M benzylidene-[1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]-dichlororuthenium;tricyclohexylphosphanium Chemical compound C1CCCCC1[PH+](C1CCCCC1)C1CCCCC1.CC1=CC(C)=CC(C)=C1N(CCN1C=2C(=CC(C)=CC=2C)C)C1=[Ru](Cl)(Cl)=CC1=CC=CC=C1 FCDPQMAOJARMTG-UHFFFAOYSA-M 0.000 description 1
- 235000013361 beverage Nutrition 0.000 description 1
- 125000002527 bicyclic carbocyclic group Chemical group 0.000 description 1
- GPRLTFBKWDERLU-UHFFFAOYSA-N bicyclo[2.2.2]octane Chemical group C1CC2CCC1CC2 GPRLTFBKWDERLU-UHFFFAOYSA-N 0.000 description 1
- SHOMMGQAMRXRRK-UHFFFAOYSA-N bicyclo[3.1.1]heptane Chemical group C1C2CC1CCC2 SHOMMGQAMRXRRK-UHFFFAOYSA-N 0.000 description 1
- GNTFBMAGLFYMMZ-UHFFFAOYSA-N bicyclo[3.2.2]nonane Chemical group C1CC2CCC1CCC2 GNTFBMAGLFYMMZ-UHFFFAOYSA-N 0.000 description 1
- WNTGVOIBBXFMLR-UHFFFAOYSA-N bicyclo[3.3.1]nonane Chemical group C1CCC2CCCC1C2 WNTGVOIBBXFMLR-UHFFFAOYSA-N 0.000 description 1
- KVLCIHRZDOKRLK-UHFFFAOYSA-N bicyclo[4.2.1]nonane Chemical group C1C2CCC1CCCC2 KVLCIHRZDOKRLK-UHFFFAOYSA-N 0.000 description 1
- 239000000227 bioadhesive Substances 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- IULFXBLVJIPESI-UHFFFAOYSA-N bis(methylsulfanyl)methylidenecyanamide Chemical compound CSC(SC)=NC#N IULFXBLVJIPESI-UHFFFAOYSA-N 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 238000009395 breeding Methods 0.000 description 1
- 230000001488 breeding effect Effects 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 229910000394 calcium triphosphate Inorganic materials 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 125000001721 carboxyacetyl group Chemical group 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000003729 cation exchange resin Substances 0.000 description 1
- 229940023913 cation exchange resins Drugs 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 102000021178 chitin binding proteins Human genes 0.000 description 1
- 108091011157 chitin binding proteins Proteins 0.000 description 1
- 230000001794 chitinolytic effect Effects 0.000 description 1
- KYKAJFCTULSVSH-UHFFFAOYSA-N chloro(fluoro)methane Chemical compound F[C]Cl KYKAJFCTULSVSH-UHFFFAOYSA-N 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 229940117975 chromium trioxide Drugs 0.000 description 1
- WGLPBDUCMAPZCE-UHFFFAOYSA-N chromium trioxide Inorganic materials O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 description 1
- GAMDZJFZMJECOS-UHFFFAOYSA-N chromium(6+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Cr+6] GAMDZJFZMJECOS-UHFFFAOYSA-N 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000007881 chronic fibrosis Effects 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125876 compound 15a Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000003271 compound fluorescence assay Methods 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 150000003997 cyclic ketones Chemical class 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 125000005959 diazepanyl group Chemical group 0.000 description 1
- ZFTFAPZRGNKQPU-UHFFFAOYSA-N dicarbonic acid Chemical compound OC(=O)OC(O)=O ZFTFAPZRGNKQPU-UHFFFAOYSA-N 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 235000019329 dioctyl sodium sulphosuccinate Nutrition 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 125000004276 dioxalanyl group Chemical group 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- YHAIUSTWZPMYGG-UHFFFAOYSA-L disodium;2,2-dioctyl-3-sulfobutanedioate Chemical compound [Na+].[Na+].CCCCCCCCC(C([O-])=O)(C(C([O-])=O)S(O)(=O)=O)CCCCCCCC YHAIUSTWZPMYGG-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000009513 drug distribution Methods 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 238000000119 electrospray ionisation mass spectrum Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 230000000470 endochitinolytic effect Effects 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 208000017338 epidermoid cysts Diseases 0.000 description 1
- 208000006881 esophagitis Diseases 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- JEBURGBMPSDSFT-LRTDYKAYSA-N ethyl (6R)-1-[1-(5-amino-1H-1,2,4-triazol-3-yl)piperidin-4-yl]-6-[(4-chlorophenyl)methyl]piperidine-3-carboxylate Chemical compound NC=1NC(=NN=1)N1CCC(CC1)N1CC(CC[C@@H]1CC1=CC=C(C=C1)Cl)C(=O)OCC JEBURGBMPSDSFT-LRTDYKAYSA-N 0.000 description 1
- SUPCQIBBMFXVTL-UHFFFAOYSA-N ethyl 2-methylprop-2-enoate Chemical class CCOC(=O)C(C)=C SUPCQIBBMFXVTL-UHFFFAOYSA-N 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- GDCRSXZBSIRSFR-UHFFFAOYSA-N ethyl prop-2-enoate;2-methylprop-2-enoic acid Chemical compound CC(=C)C(O)=O.CCOC(=O)C=C GDCRSXZBSIRSFR-UHFFFAOYSA-N 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- 238000013265 extended release Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 208000030533 eye disease Diseases 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 230000003328 fibroblastic effect Effects 0.000 description 1
- 230000001497 fibrovascular Effects 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229910021485 fumed silica Inorganic materials 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 125000004612 furopyridinyl group Chemical group O1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- SYUXAJSOZXEFPP-UHFFFAOYSA-N glutin Natural products COc1c(O)cc2OC(=CC(=O)c2c1O)c3ccccc3OC4OC(CO)C(O)C(O)C4O SYUXAJSOZXEFPP-UHFFFAOYSA-N 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 125000003147 glycosyl group Chemical group 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 150000004795 grignard reagents Chemical class 0.000 description 1
- 239000011984 grubbs catalyst Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 208000016354 hearing loss disease Diseases 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 210000004024 hepatic stellate cell Anatomy 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 102000044398 human CHIA Human genes 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- BTJRKNUKPQBLAL-UHFFFAOYSA-N hydron;4-methylmorpholine;chloride Chemical compound Cl.CN1CCOCC1 BTJRKNUKPQBLAL-UHFFFAOYSA-N 0.000 description 1
- JXYZHMPRERWTPM-UHFFFAOYSA-N hydron;morpholine;chloride Chemical compound Cl.C1COCCN1 JXYZHMPRERWTPM-UHFFFAOYSA-N 0.000 description 1
- VEIWYFRREFUNRC-UHFFFAOYSA-N hydron;piperidine;chloride Chemical compound [Cl-].C1CC[NH2+]CC1 VEIWYFRREFUNRC-UHFFFAOYSA-N 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 210000003405 ileum Anatomy 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 1
- 125000003114 inden-1-yl group Chemical group [H]C1=C([H])C([H])(*)C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000004125 inden-2-yl group Chemical group [H]C1=C(*)C([H])([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000004126 inden-3-yl group Chemical group [H]C1=C(*)C2=C([H])C([H])=C([H])C([H])=C2C1([H])[H] 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 238000009434 installation Methods 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 208000028774 intestinal disease Diseases 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000005969 isothiazolinyl group Chemical group 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000003971 isoxazolinyl group Chemical group 0.000 description 1
- 230000007803 itching Effects 0.000 description 1
- 210000001630 jejunum Anatomy 0.000 description 1
- 208000018937 joint inflammation Diseases 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 229950006462 lauromacrogol 400 Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 208000002780 macular degeneration Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000014380 magnesium carbonate Nutrition 0.000 description 1
- 229940037627 magnesium lauryl sulfate Drugs 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- HBNDBUATLJAUQM-UHFFFAOYSA-L magnesium;dodecyl sulfate Chemical compound [Mg+2].CCCCCCCCCCCCOS([O-])(=O)=O.CCCCCCCCCCCCOS([O-])(=O)=O HBNDBUATLJAUQM-UHFFFAOYSA-L 0.000 description 1
- IJMWREDHKRHWQI-UHFFFAOYSA-M magnesium;ethene;chloride Chemical compound [Mg+2].[Cl-].[CH-]=C IJMWREDHKRHWQI-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229940127554 medical product Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 230000015689 metaplastic ossification Effects 0.000 description 1
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 230000002025 microglial effect Effects 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000002062 molecular scaffold Substances 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- LSNOAQQZECGQNS-UHFFFAOYSA-N morpholine-2-carboxamide Chemical compound NC(=O)C1CNCCO1 LSNOAQQZECGQNS-UHFFFAOYSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- 210000000651 myofibroblast Anatomy 0.000 description 1
- NYHHTEVXEUTBMH-AJPXZRRQSA-N n-[(2s,3r,4s,5s,6r)-2-[(2r,3s,4s,5r,6r)-6-[[(3ar,4r,5r,6s,6as)-4-hydroxy-6-(hydroxymethyl)-2-(methylamino)-4,5,6,6a-tetrahydro-3ah-cyclopenta[d][1,3]oxazol-5-yl]oxy]-5-acetamido-4-hydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-4,5-dihydroxy-6-(hydroxymethyl)oxan- Chemical compound O([C@@H]1[C@@H](CO)O[C@H]([C@@H]([C@@H]1O)NC(C)=O)O[C@H]1[C@H](O)[C@H]2N=C(O[C@H]2[C@@H]1CO)NC)[C@@H]1O[C@H](CO)[C@@H](O)[C@@H](O)[C@H]1NC(C)=O NYHHTEVXEUTBMH-AJPXZRRQSA-N 0.000 description 1
- MUJNAWXXOJRNGK-UHFFFAOYSA-N n-[3-(6-methyl-1,2,3,4-tetrahydrocarbazol-9-yl)propyl]cyclohexanamine Chemical compound C1=2CCCCC=2C2=CC(C)=CC=C2N1CCCNC1CCCCC1 MUJNAWXXOJRNGK-UHFFFAOYSA-N 0.000 description 1
- YRCHYHRCBXNYNU-UHFFFAOYSA-N n-[[3-fluoro-4-[2-[5-[(2-methoxyethylamino)methyl]pyridin-2-yl]thieno[3,2-b]pyridin-7-yl]oxyphenyl]carbamothioyl]-2-(4-fluorophenyl)acetamide Chemical compound N1=CC(CNCCOC)=CC=C1C1=CC2=NC=CC(OC=3C(=CC(NC(=S)NC(=O)CC=4C=CC(F)=CC=4)=CC=3)F)=C2S1 YRCHYHRCBXNYNU-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- DOWVMJFBDGWVML-UHFFFAOYSA-N n-cyclohexyl-n-methyl-4-(1-oxidopyridin-1-ium-3-yl)imidazole-1-carboxamide Chemical compound C1=NC(C=2C=[N+]([O-])C=CC=2)=CN1C(=O)N(C)C1CCCCC1 DOWVMJFBDGWVML-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004706 n-propylthio group Chemical group C(CC)S* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical group C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000005963 oxadiazolidinyl group Chemical group 0.000 description 1
- 125000005882 oxadiazolinyl group Chemical group 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- RFWLACFDYFIVMC-UHFFFAOYSA-D pentacalcium;[oxido(phosphonatooxy)phosphoryl] phosphate Chemical compound [Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O.[O-]P([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O RFWLACFDYFIVMC-UHFFFAOYSA-D 0.000 description 1
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 239000012466 permeate Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000011422 pharmacological therapy Methods 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- ZLHGCTVLGRGYER-UHFFFAOYSA-N piperidin-4-amine 2H-triazol-4-amine Chemical compound Nc1cn[nH]n1.NC1CCNCC1 ZLHGCTVLGRGYER-UHFFFAOYSA-N 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001490 poly(butyl methacrylate) polymer Polymers 0.000 description 1
- 229920000212 poly(isobutyl acrylate) Polymers 0.000 description 1
- 229920000205 poly(isobutyl methacrylate) Polymers 0.000 description 1
- 229920000196 poly(lauryl methacrylate) Polymers 0.000 description 1
- 229920000184 poly(octadecyl acrylate) Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 description 1
- 229920000129 polyhexylmethacrylate Polymers 0.000 description 1
- 229920000197 polyisopropyl acrylate Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010483 polyoxyethylene sorbitan monopalmitate Nutrition 0.000 description 1
- 239000000249 polyoxyethylene sorbitan monopalmitate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010988 polyoxyethylene sorbitan tristearate Nutrition 0.000 description 1
- 239000001816 polyoxyethylene sorbitan tristearate Substances 0.000 description 1
- 229940099429 polyoxyl 40 stearate Drugs 0.000 description 1
- 229920000182 polyphenyl methacrylate Polymers 0.000 description 1
- 108010026466 polyproline Proteins 0.000 description 1
- 229940101027 polysorbate 40 Drugs 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 229940099511 polysorbate 65 Drugs 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920002744 polyvinyl acetate phthalate Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- 201000009104 prediabetes syndrome Diseases 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002206 pro-fibrotic effect Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 201000007914 proliferative diabetic retinopathy Diseases 0.000 description 1
- UFUASNAHBMBJIX-UHFFFAOYSA-N propan-1-one Chemical group CC[C]=O UFUASNAHBMBJIX-UHFFFAOYSA-N 0.000 description 1
- OVARTBFNCCXQKS-UHFFFAOYSA-N propan-2-one;hydrate Chemical compound O.CC(C)=O OVARTBFNCCXQKS-UHFFFAOYSA-N 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical class CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- SVOOVMQUISJERI-UHFFFAOYSA-K rhodium(3+);triacetate Chemical compound [Rh+3].CC([O-])=O.CC([O-])=O.CC([O-])=O SVOOVMQUISJERI-UHFFFAOYSA-K 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- TZSZZENYCISATO-WIOPSUGQSA-N rodatristat Chemical compound CCOC(=O)[C@@H]1CC2(CN1)CCN(CC2)c1cc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)nc(N)n1 TZSZZENYCISATO-WIOPSUGQSA-N 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000013049 sediment Substances 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 description 1
- KZJPVUDYAMEDRM-UHFFFAOYSA-M silver;2,2,2-trifluoroacetate Chemical compound [Ag+].[O-]C(=O)C(F)(F)F KZJPVUDYAMEDRM-UHFFFAOYSA-M 0.000 description 1
- 235000021309 simple sugar Nutrition 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- DVWOYOSIEJRHKW-UIRZNSHLSA-M sodium (2S)-2-[[(2S)-2-[[(4,4-difluorocyclohexyl)-phenylmethoxy]carbonylamino]-4-methylpentanoyl]amino]-1-hydroxy-3-[(3S)-2-oxopyrrolidin-3-yl]propane-1-sulfonate Chemical compound FC1(CCC(CC1)C(OC(=O)N[C@H](C(=O)N[C@H](C(S(=O)(=O)[O-])O)C[C@H]1C(NCC1)=O)CC(C)C)C1=CC=CC=C1)F.[Na+] DVWOYOSIEJRHKW-UIRZNSHLSA-M 0.000 description 1
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical group [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000012089 stop solution Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 125000001010 sulfinic acid amide group Chemical group 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 238000005694 sulfonylation reaction Methods 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 210000002437 synoviocyte Anatomy 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 238000012385 systemic delivery Methods 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- WGRQANOPCQRCME-PMACEKPBSA-N teneligliptin Chemical compound O=C([C@H]1NC[C@H](C1)N1CCN(CC1)C1=CC(=NN1C=1C=CC=CC=1)C)N1CCSC1 WGRQANOPCQRCME-PMACEKPBSA-N 0.000 description 1
- FRACPXUHUTXLCX-BELIEFIBSA-N tert-butyl N-{1-[(1S)-1-{[(1R,2S)-1-(benzylcarbamoyl)-1-hydroxy-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]carbamoyl}-2-cyclopropylethyl]-2-oxopyridin-3-yl}carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CN(C1=O)[C@@H](CC2CC2)C(=O)N[C@@H](C[C@@H]3CCNC3=O)[C@H](C(=O)NCC4=CC=CC=C4)O FRACPXUHUTXLCX-BELIEFIBSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000005888 tetrahydroindolyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 125000005304 thiadiazolidinyl group Chemical group 0.000 description 1
- 125000005305 thiadiazolinyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000002769 thiazolinyl group Chemical group 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- 125000001395 thiirenyl group Chemical group 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- 229960000257 tiotropium bromide Drugs 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 230000002094 transglycosylational effect Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 208000037972 tropical disease Diseases 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 229940070384 ventolin Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 201000006520 wheat allergy Diseases 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/14—Antitussive agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/02—Antidotes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Biomedical Technology (AREA)
- Oncology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Obesity (AREA)
- Communicable Diseases (AREA)
- Otolaryngology (AREA)
- Hospice & Palliative Care (AREA)
- Emergency Medicine (AREA)
- Transplantation (AREA)
- Psychiatry (AREA)
- Toxicology (AREA)
- Vascular Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Heart & Thoracic Surgery (AREA)
Description
DESCRIPCIÓN
Amino triazoles sustituidos útiles como inhibidores de la quitinasa humana
Solicitudes relacionadas
Esta solicitud reivindica el beneficio de prioridad de la Solicitud Provisional de Patente Estadounidense N.° de serie 62/214.299, presentada el 4 de septiembre de 2015.
Antecedentes de la invención
La quitinasa de mamífero ácida (AMCasa; Mr = ~52,2 kD) es una enzima segregada, que se encuentra típicamente en el estómago, glándulas salivales y pulmones. Única entre las enzimas de mamífero porque tiene un pH ácido óptimo, la enzima cataliza la hidrólisis de sustratos tipo quitina artificiales. Se induce durante la inflamación de Th2 a través de un mecanismo dependiente de IL-13. Se cree que las quitinasas cumplen una función clave en la inmunidad innata con parásitos y otros agentes infecciosos. Cuando se producen de forma desregulada, las enzimas también pueden cumplir una función importante en la patogénesis de alergia y/o asma.
El asma es una enfermedad inflamatoria crónica de las vías respiratorias caracterizado por episodios recurrentes de obstrucción reversible de las vías respiratorias e hiperreactividad de las vías respiratorias (AHR). Las manifestaciones clínicas típicas incluyen falta de aliento, sibilancia, tos y opresión torácica que se tornan letales o graves. Mientras existen terapias enfocadas en la reducción de broncoespasmos sintomáticos e inflamación pulmonar, existe una preocupación creciente acerca de la función del remodelado de vías respiratorias a largo plazo en el deterioro acelerado de pulmones en asmáticos. La remodelación de las vías respiratorias se refiere a una cantidad de rasgos patológicos que incluyen músculo liso epitelial e hiperplasia de miofibroblastos y/o metaplasia, fibrosis subepitelial y deposición de matriz.
Generalmente se acepta que el asma alérgica se inicia por una reacción inflamatoria inapropiada a alérgenos en el aire. Los pulmones se asmáticos demuestran una infiltración intensa de linfocitos, mastocitos y especialmente eosinófilos. AMCasa se expresa prominentemente en pulmones de ratones transgénicos IL-13 y expuestos y sensibles al antígeno. El ARNm de AMCasa no se detecta fácilmente en tejido de pulmones de pacientes sin enfermedad pulmonar conocida, pero se detectó, histológica y morfométricamente, en las células epiteliales y células subepiteliales en tejidos de pacientes con asma.
Los estudios publicados de forma preliminar (Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, y Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science 304: 1678-1682, 2004; Peterson et al. Demethylallosamidin, a chitinase inhibitor, suppresses airway inflammation and hyperresponsiveness. Biochem Biophys Res Commun 390: 103-108, 2009) sugieren que AMCasa cumple una función en la respuesta inflamatoria provocada por Th-2 en un modelo de murino de asma alérgico. Las respuestas de Th-1 no parecen estar implicadas. No se observaron efectos terapéuticos en un modelo de ratón que expresa Th-1, pero no Th-2 (Fitz LJ, et al. Acidic mammalian chitinase is not a critical target for allergic airway disease. Am J Respir Cell Mol Biol 46: 71-9, 2011). Este resultado sería el esperado dado que las células Th-1 están implicadas principalmente en la defensa del hospedador contra patógenos.
La quitotriosidasa 1 (CHIT1, Mr = ~52 kD o ~39kDa) es una quitinasa expresada predominantemente en las células mieloides y células epiteliales de los pulmones como un mediador inmunitario innato que digiere las paredes celulares de patógenos eucariotas que contienen quitina. CHIT1 es una enzima en circulación con actividad endoquitinolítica y transglicosilante. Además de su función en el reconocimiento de quitina y respuesta inmunitaria innata, la CHIT1 está implicada en la patogénesis de enfermedades pulmonares fibróticas. La fibrosis pulmonar se redujo considerablemente en ratones con inactivación de CHIT1 en modelo de animal con fibrosis pulmonar inducida por bleomicina y se sugiere que esta quitinasa cumpla una función en la remodelación de tejido y fibrinogénesis en el pulmón.
La fibrosis pulmonar idiopática (IPF) es un trastorno fibroproliferativo progresivo refractario a las terapias farmacológicas actuales con una supervivencia media de solo 3-5 años después del diagnóstico. La IPF es una enfermedad devastadora caracterizada por el depósito excesivo de matriz que altera la arquitectura normal del parénquima del pulmón. Los rasgos patológicos clave del IPF incluyen focos fibroblásticos, áreas de quistes epiteliales asociados con la apariencia de panales de los pulmones, e inflamación intersticial linfoplasmacítica que está asociada con áreas de hiperplasia celular de tipo II. La patogénesis de cada forma de fibrosis pulmonar todavía se entiende muy poco. Cada una resulta en una pérdida progresiva de función pulmonar con disnea en aumento y la mayoría de las formas eventualmente resultan en mortalidad.
Un mal pronóstico de pacientes con IPF genera una gran necesidad de nuevas dianas que puedan usarse como estrategia terapéutica para mejorar los resultados clínicos en IPF con CHIT 1 entre estos. La sobreexpresión de CHIT 1 se mostró en fibrotic Interstitial Lung Disease (ILD), incluyendo IPF (Bargagli W et al. Chitotriosidase activity in patients with interstitial lung diseases. Respir Med. 101(10):2176-81, 2007) y enfermedad pulmonar obstructiva crónica (EPOC) (Letuve S et al. Lung chitinolytic activity and chitotriosidase are elevated in chronic obstructive pulmonary disease and
contribute to lung inflammation. Am J Pathol. 176(2):638-49, 2010) caracterizado por inflamación y remodelación de tejidos y demostró de forma interesante ser amplificador potente de la señalización de TGFp (Lee CG et al. Chitinase 1 is a biomarker for and therapeutic target in scleroderma-associated interstitial lung disease that augments TGF-P1 signaling. J Immunol. 189(5):2635-44, 2012). Un estudio mostró que la actividad de CHIT1 estaba elevada en BAL de pacientes con IPF en comparación con controles que sugieren que puede ser la responsable de la remodelación y daño del tejido observado en el pulmón de pacientes con IPF. Como tal, se puede imaginar que CHIT1 podría estar implicada en la fibrogénesis de otros ILD, tales como esclerosis sistémica (SSc), donde el grupo de pacientes con implicancia pulmonar muestra altos niveles de actividad de CHIT1 en circulación que se correlaciona con la gravedad de la enfermedad.
Las enfermedades, trastornos y afecciones mediados por AMCasa y CHIT1 se describen en más detalle a continuación.
Los amino triazoles sustituidos que inhiben AMCasa y CHIT1 se describieron (véase publicación de solicitud de patente internacional n.° WO 2015/095701, y solicitud de patente provisional estadounidense n.° 62/094.446).
Existe una necesidad continua de investigar la inhibición de AMCasa y CHIT 1, y descubrir tratamientos de afecciones asociadas con la expresión elevada de AMCasa o CHIT 1, tales como asma y respuestas alérgicas o EPOC y trastornos fibroproliferativos. En particular, existe una necesidad de nuevos andamiajes moleculares que inhiban de forma eficaz AMCasa y CHIT1, y por lo tanto, puedan actuar como agentes terapéuticos para el tratamiento de estas afecciones.
Sumario de la invención
En un aspecto, la invención proporciona un compuesto representado por la fórmula (I),

donde:
W es halo, alquilo(C1-C3), alcoxi(C1-C3), o alquiltio(C1-C3);
X es un enlace sencillo, -CH2-, -CH2CH2-, -c H=CH-, -C(alquilo(C1-C3))2-, o -C(O)-;
Y es un enlace sencillo, -CH-, -CHCH2-, -CH2CH-, -C=CH-, -CH=C-, -N-, -O-, -OCH2-, -S(O)-, o -S(O)2-;
si Y es un enlace sencillo, -O-, -OCH2-, -S(O)-, o -S(O)2-, entonces R1 está ausente;
R1 es H, OH, alquilo(C1-C6), haloalquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), arilalquilo(C1-C6), heteroarilalquilo(C1-C6) -C(O)alquilo(C1-C6), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1-C6), -C(O)heteroarilalquilo(C1-C6), -S(O)2 alquilo(C1-C6), -S(O)2arilo, -S(O)2heteroarilo, -S(O)2arilalquilo(C1-C6), -S(O)2heteroarilalquilo(C1-C6), -CO2H, -C(O)O alquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroaril), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NH2, -C(O)NHOH, -C(O)NHCN, -C(O)NH(alquilo(C1-C6)), -C(O)N(alquilo(C1-C6))2, -C(O)NH(arilalquilo(C1-C6)), -C(O)N(arilalquilo(C1-C6))2, -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6)), -C(O)N(arilo)2, -C(O)N alquil(C1-C6))(arilalquilo(C1-C6)), -C(O)N(aril)(arilalquilo(C1-C6)), -C(O)NH(haloalquilo(C1-C6)), -C(O)N(haloalquilo(C1-C6))2, -S(O)2NH2, -S(O)2NH(alquilo(C1-C6)), -S(O)2NH(haloalquilo(C1-C6)), -S(O)2NH(aril), -S(O)2NH(heteroarilalquilo(C1-C6)), -S(O)2NH(heteroarilo), -S(O)2N(alquilo(C1-C6))2, -S(O)2NHC(O)alquilo(C1-C6), -S(O)2NHC(O)haloalquilo(C1-C6), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(C1-C6), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(C1-C6), -NHS(O)2 alquilo(C1-C6), -NHS(O)2aryl, -NHS(O)2 haloalquilo(C1-C6), -NHS(O)2arilalquilo(C1-C6), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(C1-C6), -NHC(O)(alquilo(C1-C6)), -NHC(O)(haloalquilo(C1-C6)), -NHC(O)(arilo), -NHC(O)(arilalquilo(C1-C6)), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(C1-C6)), -NHC(O)NH alquilo(C1-C6), -NHC(O)NH(arilo), -NHC(O)NH(arilalquilo(C1-C6)), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(C1-C6)), -C(O)NHS(O)2 alquilo(C1-C6), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(C1-C6)), -C(O)NHS(O)2(arilalquilo(C1-C6)), -C(O)NHS(O)2heteroarilo, -C(O)NHS(O)2(heteroarilalquilo(C1-C6)), -P(O)(OH)2, -(alquileno(C1-C6))C(O)OH, -(alquileno(C1-C6))C(O)O alquilo(C1-C6), -NH2, -NH alquilo(C1-C6), -N(alquilo(C1-C6))2, -NC, -CN, -C(S)NH2, -NHC(O)NH2, -CeCH, alquiltio(C1-C6)-, mercaptoalquilo(C1-C6)-, o -C(O)heterociclilo;
Z es -CH-, -C(O)-, -C(alquilo(C1-C3))- o -C(=CH2)-;
si Z es -C(O)-, entonces R2 está ausente,
R2 es H, Oh , halo, alquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), -CO2H, -C(O)O alquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NHOH, -C(O)NHCN, -C(O)NH2, -C(O)NH(alquilo(C1-C6)), -C(O)N(alquilo(C1-C6))2, -C(O)NH(arilalquilo(C1-C6)), -C(O)N(arilalquilo(C1-C6))2, -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6)), -C(O)N(arilo)2, -C(O)N alquil(C1C6))(arilalquilo(Ci-C6)), -C(O)N(aril)(arilalquilo(Ci-C6)), -C(O)NH(haloalquilo(Ci-C6)), -C(O)N(haloalquilo(Ci-C6))2, -S(O)2NH2, -S(O)2NH(alquilo(Ci-C6)), -S(O)2NH(haloalquilo(Ci-C6)), -S(O)2NH(arilo), -S(O)2NH(heteroarilalquilo(Ci-C6)), -S(O)2NH(heteroarilo), -S(O)2NH(heteroarilalquilo(Ci-C6)), -S(O)2N(alquilo(Ci-C6))2, -S(O)2NHC(O)alquilo(Ci-C6), -S(O)2NHC(O)haloalquilo(Ci-C6), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(Ci-C6), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(Ci-C6), -NHS(O)2 alquilo(Ci-C6), -NHS(O)2arilo, NHS(O)2 haloalquilo(Ci-C6), -NHS(O)2arilalquilo(Ci-C6), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(Ci-C6), -NHC(O)(alquilo(Ci-C6)), -NHC(O)(haloalquilo(Ci-C6)), -NHC(O)(arilo), -NHC(O)(arilalquilo(Ci-C6)), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(Ci-C6)), -NHC(O)NH alquilo(Ci-C6), -NHC(O)NHarilo, -NHC(O)NH(arilalquilo(Ci-C6)), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(Ci-C6)), -C(O)NHS(O)2 alquilo(Ci-C6), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(Ci-C6)), -C(O)NHS(O)2(arilalquilo(Ci-C6)), -C(O)NHS(O)2heteroarilo, -C(O)NHS(O)2(heteroarilalquilo(Ci-C6)), -P(O)(OH)2, arilalquilo(Ci-C6), cicloalquil(C3-C7)alquilo(Ci-C6)-, -NC, -CN, -C(S)NH2, -NHC(O)NH2, -CeCH, alquiltio(Ci-C6)-, mercaptoalquilo(Ci-C6)-, o -C(O)heterociclilo;
o Ri y R2, tomados juntos con los átomos que son parte forman un anillo carbocíclico o heterocíclico;
R3 es H, alquilo(Ci-C3), alcoxi(Ci-C3)alquilo(Ci-C3), alquiltio(Ci-C3)alquilo(Ci-C3), haloalquilo(Ci-C6), -NC, -CN, -C(S)NH2, -NHC(O)NH2, o -CeCH;
R4 es H, alquilo(Ci-C3), alquiltio(Ci-C3)alquilo(Ci-C3), haloalquilo(Ci-C6), alcoxi(Ci-C3)alquilo(Ci-C3), o hidroxialquilo(Ci-C4);
R5 es H, halo, -NO2, -CN, alquilo(Ci-C6), haloalquilo(Ci-C6), -NH2, -NH(alquilo(Ci-C6)), -N(alquilo(Ci-C6))2, -OH, alcoxi(Ci-C6), hidroxialquilo(Ci-C6), alcoxi(Ci-C6)alquilo(Ci-C6), haloalcoxi(Ci-C6), -SH, alquiltio(Ci-C3)alquilo(Ci-C3), -NC, -C(S)NH2, -NHC(O)NH2, o -CeCH; y
R6 es H, halo, -OH, -NH2, o -SH; o
R6, tomado junto con el átomos de carbono que lo contiene, representa C(O)-;
donde:
cualquier aparición de alquilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo, o hidroxialquilo está sustituida opcionalmente con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2, -NH(alquilo(Ci-C6)), -N(alquilo(Ci-C6))2, -CN, -NO2, alquilo(Ci-C6), haloalquilo(Ci-C6), alcoxi(Ci-C6), arilo, heteroarilo, arilalquilo(Ci-C6), heteroarilalquilo(Ci-C6), cicloalquilo(C3-C7), heterociclilo, -C(O)OH, -c (o )0 alquilo(Ci-C6), -C(O)NH2, -C(O)NH alquilo(Ci-C6), y -C(O)N(alquilo(Ci-C6))2;
o una sal, solvato o tautómero, estereoisómero o polimorfo farmacéuticamente aceptable de estos.
En determinadas realizaciones, la invención proporciona un compuesto representado por la fórmula (II),

donde:
W es halo, alquilo(Ci-C3), alcoxi(Ci-C3), o alquiltio(Ci-C3);
X es un enlace sencillo, -CH2-, -CH2CH2-, -CH=CH-, -C(alquilo(Ci-C3))2-, -C(O)-, -CH2O-, -CH2NH-, o -CH2N(alquilo(Ci-C3))-, donde cuando X es -CH2O-, -CH2NH-, o -CH2N(alquilo(Ci-C3))-, entonces el átomo de O o N está unido covalentemente con el Anillo A ;
El Anillo A representa un anillo arilo o heteroarilo opcionalmente sustituido;
R3 es H, alquilo(Ci-C3), alcoxi(Ci-C3)alquilo(Ci-C3), alquiltio(Ci-C3)alquilo(Ci-C3), haloalquilo(Ci-C6), -NC, -CN, -C(S)NH2, -NHC(O)NH2, o -CeCH;
R4 es H, alquilo(Ci-C3), alquiltio(Ci-C3)alquilo(Ci-C3), haloalquilo(Ci-C6), alcoxi(Ci-C3)alquilo(Ci-C3), o hidroxialquilo(Ci-C4);
R5 es H, halo, -NO2, -CN, alquilo(Ci-C6), haloalquilo(Ci-C6), -NH2, -NH(alquilo(Ci-C6)), -N(alquilo(Ci-C6))2, -OH, alcoxi(Ci-C6), hidroxialquilo(Ci-C6), alcoxi(Ci-C6)alquilo(Ci-C6), haloalcoxi(Ci-C6), -SH, alquiltio(Ci-C3)alquilo(Ci-C3), -NC, -C(S)NH2, -NHC(O)NH2, o -CeCH; y
R6 es H, halo, -OH, -NH2, o -SH; o
R6, tomado junto con el átomos de carbono que lo contiene, representa C(O)-;
donde:
cualquier aparición de alquilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo, o hidroxialquilo está sustituida opcionalmente con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2, -NH(alquilo(Ci-C6)), -N(alquilo(CiC6))2, -CN, -NO2, alquilo(Ci-C6), haloalquilo(C1-C6), alcoxi(Ci-C6), arilo, heteroarilo, arilalquilo(C1-C6), heteroarilalquilo(C1-C6), cicloalquilo(C3-C7), heterociclilo, -C(O)0h , -C(O)O alquilo(Ci-C6), -C(O)NH2, -C(0)n H alquilo(Ci-C6), y -C(O)N(alquilo(Ci-C6))2;
o una sal, solvato o tautómero, estereoisómero o polimorfo farmacéuticamente aceptable de estos.
También se describen en el presente documento composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de un compuesto de la invención y un portador farmacéuticamente aceptable.
En determinados aspectos, la composición farmacéutica también incluye uno o más segundos agentes terapéuticos seleccionados del grupo que consiste en esteroides, estabilizadores de membrana, inhibidores de 5LO, inhibidores del receptor y la síntesis de leucotrieno, inhibidores de la conmutación del isotipo IgE o la síntesis de IgE, conmutación del isotipo IgG o síntesis de IgG, p-agonistas, inhibidores de triptasa, ácido acetilsalicílico, inhibidores de la COX, metotrexato, fármacos anti-TNF, rituxina, inhibidores de PD4, inhibidores de p38, inhibidores de PDE4 y antihistaminas.
En otro aspecto, la invención proporciona métodos para inhibir la quitinasa de mamífero ácida en una célula o un tejido, que comprende poner en contacto la célula o el tejido con al menos un compuesto de acuerdo con la invención o con una composición farmacéutica de la invención.
También se desvelan métodos para inhibir la quitotriosidasa 1 (CHIT1) en una célula o un tejido, que comprende poner en contacto la célula o el tejido con al menos un compuesto de acuerdo con la invención o con una composición farmacéutica de la invención.
También se desvelan métodos para el tratamiento o la prevención de una enfermedad, trastorno o afección asociada con la actividad o expresión aberrante de la quitinasa de mamífero ácida, que comprenden administrarle a un sujeto que lo necesita una cantidad terapéuticamente eficaz de al menos un compuesto de la invención o con una composición farmacéutica de la invención.
También se desvelan métodos para el tratamiento o la prevención de una enfermedad, trastorno o afección asociada con la actividad o expresión aberrante de la CHIT1, que comprenden administrarle a un sujeto que lo necesita una cantidad terapéuticamente eficaz de al menos un compuesto de la invención o con una composición farmacéutica de la invención.
También se desvelan métodos para el tratamiento o la prevención de una enfermedad, trastorno o afección seleccionado del grupo que consiste en enfermedades alérgicas, enfermedades inflamatorias agudas y crónicas, enfermedades autoinmunes, enfermedades dentales, enfermedades neurológicas, enfermedades metabólicas, enfermedades hepáticas, enfermedades renales, enfermedades de la piel, síndrome de ovario poliquístico, endometriosis, enfermedades fibróticas, enfermedades de almacenamiento, y cáncer, que comprenden administrarle a un sujeto que lo necesita una cantidad terapéuticamente eficaz de al menos un compuesto de la invención, o una composición farmacéutica de la invención.
T ambién se desvelan métodos para inhibir quitotriosidasa y quitinasa de mamífero ácida en una célula o un tejido, que comprende poner en contacto una célula o un tejido con al menos un compuesto de la invención o con una composición farmacéutica de la invención.
Descripción detallada
La presente invención se basa en el descubrimiento inesperado de que la modificación química de los compuestos de molécula pequeña de amino triazol 4-amino piperidina con un anillo heterocíclico (por ejemplo una morfolina o piperazina sustituida) aumenta la rigidez de la molécula, fijando así su geometría molecular. Esta rigidez geométrica cambia de forma beneficiosa varias propiedades moleculares y proporciona una efectividad inhibidora inesperada hacia la quitinasa de mamífero ácida.
Los compuestos de amino triazol de acuerdo con la invención entonces son útiles en el tratamiento de trastornos asociados con actividad de AMCasa regulada positivamente y desregulada, tales como asma y reacciones alérgicas, así como los trastornos asociados con la actividad de CHIT1 regulada positivamente y desregulada.
Definiciones
En el presente documento, los artículos “uno” y “una” se utilizan para hacer referencia a uno o más (es decir, al menos uno) del objeto gramatical del artículo. A modo de ejemplo, “un elemento” significa un elemento o más de un elemento.
Los términos “grupo” y “radical” se usan de forma intercambiable en el presente documento y denotan una porción de una molécula en cuestión que está unida al resto de la molécula por un enlace covalente (o enlaces, como resulta del párrafo anterior).
Los términos usados en el presente documento pueden estar precedidos y/o seguidos por un único guion, “-”, o un guion doble, “=“, para indicar el orden de unión del enlace entre el sustituyente nombrado y su resto principal; un único guion indica un enlace sencillo y un guion doble indica un enlace doble. En caso de no haber un guion único o doble se comprende que se forma un enlace sencillo entre el sustituyente y su resto principal; además, se pretende que los sustituyentes se lean de “izquierda a derecha” salvo que haya un guion que indique lo contrario. Por ejemplo, alcoxicarboniloxi (C1-C6) y -OC(O)alquil(C1-C6) indican la misma funcionalidad; de manera similar, arilalquilo y alquilarilo indican la misma funcionalidad.
La frase “enlace sencillo” como se usa en el presente documento denota un enlace covalente similar entre dos átomos, tales como C-C, C-H, o C-O.
El término “alquilo” como se usa en el presente documento es un término de la técnica y se refiere a grupos alifáticos saturados, que incluyen grupos alquilo de cadena recta y grupos alquilo de cadena ramificada. Los subíndices que le siguen a C indican el número (o intervalo de números) de átomos de carbono en el alquilo de cadena recta o cadena ramificada. En determinadas realizaciones, un alquilo de cadena lineal o de cadena ramificada tiene aproximadamente 30 o menos átomos de carbono en su estructura (por ejemplo, C1-C30 para cadena lineal, C3-C30 para cadena ramificada), y de manera alternativa aproximadamente 20 o menos, 10 o menos o preferentemente 1-6 carbonos. Los ejemplos representativos de (alquilo C1-C6) incluyen, pero no se limita a, metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, isobutilo, terc-butilo, n-pentilo, isopentilo, neopentilo y n-hexilo. Los ejemplos de alquilo C1-C3 incluyen metilo, etilo, n-propilo e isopropilo. Alquilo puede representar un grupo, como se define anteriormente, o una porción de un resto más grande, tales como alcoxi(C1-C3)alquilo (C1-C3). Un alcoxi (C1-C3)alquilo(C1-C3) está unido al resto de la molécula a través del resto alquilo(C1-C3).
El término “cicloalquilo” se refiere a anillos carbocíclicos saturados o parcialmente saturados ramificados o mono o bicíclicos, cada uno con 3 a 12 átomos de carbono. Determinados cicloalquilos tienen de 3-8, o de 3-6 átomos de carbono en su estructura de anillo. Determinados cicloalquilos tienen de 5-12 átomos de carbono en la estructura del anillo y pueden tener 6-10 carbonos en la estructura del anillo. Preferentemente, cicloalquilo es cicloalquilo (C3-C7), que representa un anillo carbocíclico saturado monocíclico, con de 3 a 7 átomos de carbono. Los ejemplos de cicloalquilos monocíclicos incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclopentenilo, ciclohexilo, ciclohexenilo, cicloheptilo y ciclooctilo. Los sistemas anulares cicloalquilo bicíclicos incluyen anillos monocíclicos puenteados y anillos bicíclicos fusionados. Los anillos monocíclicos puenteados contienen un anillo cicloalquilo monocíclico donde dos átomos de carbono no adyacentes del anillo monocíclico están unidos mediante un puente alquileno de entre uno y tres átomos de carbono adicionales (es decir, un grupo puente de la forma -(CH2)w-, donde w es 1,2 o 3). Los ejemplos representativos de sistemas de anillos bicíclicos incluyen, pero no se limita a, biciclo[3.1.1]heptano, biciclo[2.2.1]heptano, biciclo[2.2.2]octano, biciclo[3.2.2]nonano, biciclo[3.3.1]nonano y biciclo[4.2.1]nonano. Los sistemas anulares cicloalquilo bicíclicos fusionados contienen un anillo cicloalquilo monocíclico fusionado con un fenilo, un cicloalquilo monocíclico, un cicloalquenilo monocíclico, un heterociclilo monocíclico o un heteroarilo monocíclico. El cicloalquilo bicíclico puenteado o fusionado se une al resto molecular principal a través de cualquier átomo de carbono dentro del anillo cicloalquilo monocíclico. Los grupos cicloalquilo están opcionalmente sustituidos. En determinadas realizaciones, el cicloalquilo bicíclico fusionado es un anillo cicloalquilo monocíclico de 5 o 6 miembros fusionado a un anillo fenilo, un cicloalquilo monocíclico de 5 o 6 miembros, un cicloalquenilo monocíclico de 5 o 6 miembros, un heterociclilo monocíclico de 5 o 6 miembros o un heteroarilo monocíclico de 5 o 6 miembros, donde el cicloalquilo bicíclico fusionado está opcionalmente sustituido.
El término “cicloalquilalquilo”, tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con uno o más grupos cicloalquilo. Un ejemplo de cicloalquilalquilo es grupo ciclohexilmetilo.
El término “heterociclilo” como se usa en el presente documento se refiere a un radical de un sistema de anillo no aromático, que incluye pero no se limita a, anillos monocíclicos, bicíclicos, y tricíclicos, que pueden estar completamente saturados o que pueden contener una o más unidades de insaturación, para evitar dudas, el grado de insaturación no resulta en un sistema de anillo aromático, y con 3 a 14, o 3 a 12 átomos que incluyen al menos un heteroátomo tal como nitrógeno, oxígeno o azufre. Los grupos heterocicloalquilo más preferidos tienen de 5-10 miembros de anillo donde de 1-4 de los miembros del anillo son heteroátomos seleccionados del grupo que consiste en O, N, y S, el resto de los átomos del anillo son C. Para ejemplificar, lo que se no debe entenderse como limitativo del alcance de esta invención, estos son ejemplos de anillos heterocíclicos: aziridinilo, azirinilo, oxiranilo, tiiranilo, tiirenilo, dioxiranilo, diazirinilo, diazepanilo, 1,3-dioxanilo, 1,3-dioxolanilo, 1,3-ditiolanilo, 1,3-ditianilo, imidazolidinilo, isotiazolinilo, isotiazolidinilo, isoxazolinilo, isoxazolidinilo, azetilo, oxetanilo, oxetilo, tietanilo, tietilo, diazetidinilo, dioxetanilo, dioxetenilo, ditietanilo, ditieilo, dioxalanilo, oxazolilo, tiazolilo, triazinilo, isotiazolilo, isoxazolilo, azepinas, azetidinilo, morfolinilo, oxadiazolinilo, oxadiazolidinilo, oxazolinilo, oxazolidinilo, oxopiperidinilo, oxopirrolidinilo, piperazinilo, piperidinilo, piranilo, pirazolinilo, pirazolidiil, pirrolinilo, pirrolidinilo, quinuclidinilo, tiomorfolinilo, tetrahidropiranilo, tetrahidrofuranilo, tetrahidrotienilo, tiadiazolinilo, tiadiazolidinilo, tiazolinilo, tiazolidinil, tiomorfolinilo, 1,1-dioxidotiomorfolinilo (tiomorfolino sulfona), tiopiranilo, y tritianilo. Un grupo heterociclilo está opcionalmente sustituido por uno o más sustituyentes como se describe a continuación.
Tal como se usa en el presente documento, el término “heterociclileno” se refiere a un grupo (heterocicloalquilo) heterociclilo bivalente, es decir, un grupo alquileno cíclico, con de 3-10 miembros y de 1-4 heteroátomos seleccionados
de S, O, y N. Un ejemplo es ácido piperidina-2,3-dicarboxílico, es decir, ene se compuesto, el anillo de piperidina es un grupo heterociclileno.
El término “heteroátomo” está reconocido en la técnica e incluye un átomo de cualquier elemento diferente a carbono o hidrógeno. Heteroátomos ilustrativos incluyen boro, nitrógeno, oxígeno, fósforo, azufre y selenio, y de manera alternativa oxígeno, nitrógeno y azufre.
El término “heterocicloalquilalquilo”, tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con uno o más grupos heterocicloalquilo (es decir heterociclilo).
El término “alquenilo”, como se usa en el presente documento, se refiere a un radical hidrocarburo de cadena lineal o ramificada que contiene entre 2 y 10 carbonos, y que contiene al menos un enlace doble carbono-carbono formado por la retirada de dos hidrógenos. Preferentemente, alquenilo contiene de 2 a 6 carbonos. Los ejemplos representativos de alquenilo incluyen, pero no se limita a, etenilo, 2-propenilo, 2-metil-2-propenilo, 3-butenilo, 4-pentenilo, y 5-hexenilo. Los uno o más enlaces insaturados del grupo alquenilo pueden estar ubicados en cualquier parte del resto y pueden tener la configuración (Z) o (E) aproximadamente los uno o más enlaces dobles. Las moléculas que difieren solo en su configuración alrededor del enlace doble se denominan isómeros geométricos.
El término “alquinilo”, como se usa en el presente documento, se refiere a un radical hidrocarburo de cadena lineal o ramificada que contiene entre 2 y 10 átomos de carbono y que contiene al menos un enlace triple carbono-carbono. Los ejemplos representativos de alquinilo incluyen, pero no se limita a, acetilenilo, 1-propinilo, 2-propinilo, 3-butinilo, 2-pentinilo y 1-butinilo.
El término “alquileno” está reconocido en la técnica y como se usa en el presente documento se refiere a un diradical obtenido retirando dos átomos de hidrógeno de un grupo alquilo como se define anteriormente. En una realización un alquileno se refiere a un alcano disustituido es decir un alcano sustituido en dos posiciones con sustituyentes tales como halógeno, azida, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, hidroxilo, alcoxilo, amino, nitro, sulfhidrilo, imino, amido, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, sulfonamido, cetona, aldehído, éster, heterocicliclo, restos aromáticos o heteroaromáticos, fluoroalquilo (tales como triflurometilo), ciano, o similares. Es decir, en una realización un “alquilo sustituido” es un “alquileno”.
El término “amino” es un término de la técnica y como se usa en el presente documento se refiere tanto a aminas no sustituidas como sustituidas, por ejemplo, un resto que puede representarse mediante las fórmulas generales:

donde cada uno de Ra, Rb, y Rc representa de forma independiente, un hidrógeno, un alquilo, un alquenilo, (CH2)x-Rd, o Ra y Rb tomados junto con el átomo N al cual se unen completan un heterociclo que tiene de 4 a 8 átomos en la estructura de anillo; Rd representa un arilo, un cicloalquilo, un cicloalquenilo, un heterociclilo o un policiclilo; y x es cero o un entero en el intervalo de 1 a 8. En determinadas realizaciones, solo uno de Ra o Rb puede ser un carbonilo, por ejemplo, Ra, Rb y el nitrógeno juntos no forman una imida. En algunas realizaciones, Ra y Rb (y opcionalmente Rc) cada uno representa independientemente un hidrógeno, un alquilo, un alquenilo o -(CH2)x-Rd. En determinadas realizaciones, el término “amino” se refiere a -NH2.
El término “amido”, tal como se usa en el presente documento, se refiere a -NHC(=O), donde el grupo amido está unido con el resto molecular principal a través del nitrógeno. Los ejemplos de amido incluyen alquilamido tal como CHaC(=O)N(H) y CH3CH2C(=O)N(H).
El término “acilo” es un término de la técnica y como se usa en el presente documento se refiere a cualquier grupo o radical de la forma RCO- donde R es cualquier grupo orgánico, por ejemplo, alquilo, arilo, heteroarilo, aralquilo, y heteroaralquilo. Los grupos acilo representativos incluyen acetilo, benzoílo y malonilo.
El término “aminoalquilo”, tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con uno o más grupos amino. En una realización, el término “aminoalquilo” se refiere a un grupo aminometilo.
El término “aminoacilo” es un término de la técnica y tal como se usa en el presente documento, se refiere a un grupo acilo sustituido con uno o más grupos amino.
El término “aminotionilo” tal como se usa en el presente documento se refiere a un análogo de un aminoacilo donde el O de RC(O)- fue remplazado por azufre, por tanto es de la forma RC(S)-.
El término “azida” o “azido”, como se usa en el presente documento, se refiere a un grupo -N3.
El término “carbonilo”, tal como se utiliza en el presente documento, se refiere a -C(=O)-.
El término “tiocarbonilo”, tal como se utiliza en el presente documento, se refiere a -C(=S)-.
El término “alquiltio”, tal como se utiliza en el presente documento, se refiere a alquilo-S-. Ejemplos representativos de alquiltio (C1-C6) incluyen metiltio, etiltio, n-propiltio, y ferc-butiltio. Ejemplos representativos de alquiltio (C1-C3) incluyen metiltio, etiltio, y n-propiltio.
El término “mercaptoalquilo”, tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido por un resto -SH. Ejemplos representativos de mercaptoalquilo (C1-C6) incluyen mercaptometilo, mercaptoetilo, y mercapto-n-propilo.
El término “carboxi”, como se usa en el presente documento, se refiere a un grupo CO2H. Este grupo puede formar una porción de otro sustituyente tal como carboximetilo, es decir, HO2C-CH2-.
El término “arilo” es un término de la técnica y como se usa en el presente documento se refiere a incluir grupos hidrocarburo aromáticos monocíclicos, bicíclicos y policíclicos, por ejemplo, benceno, naftaleno, 1,2,3,4-tetrahidronaftaleno, indeno, 2,3-dihidroindeno, y pireno. El anillo aromático se puede sustituir en una o más posiciones del anillo con uno o más sustituyentes, tales como halógeno, azida, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, cicloalquilalquilo, (cicloalquil)alcoxilo, hidroxilo, alcoxilo, amino, nitro, sulfhidrilo, imino, amido, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, aminosulfonilo, sulfonamido, cetona, aldehído, éster, heterociclilo, heterociclilalquilo, restos aromáticos o heteroaromáticos, aminoalquilo, haloalquilo, fluoroalquilo (tal como trifluorometilo), haloalcoxilo, ciano o similares. El término “arilo” también incluye sistemas de anillo policíclicos con dos o más anillos cíclicos donde dos o más carbonos son comunes a dos anillos contiguos (los anillos son “anillos fusionados”) donde al menos uno de los anillos es un hidrocarburo aromático, por ejemplo, los demás anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos, heteroarilos y/o heterociclilos. Los ejemplos representativos de los sistemas de anillo arilo policíclicos incluyen, pero no se limita a, azulenilo, naftilo, dihidroinden-1-ilo, dihidroinden-2-ilo, dihidroinden-3-ilo, dihidroinden-4-ilo, 2,3-dihidroindol-4-ilo, 2,3-dihidroindol-5-ilo, 2,3-dihidroindol-6-ilo, 2,3-dihidroindol-7-ilo, inden-1-ilo, inden-2-ilo, inden-3-ilo, inden-4-ilo, dihidronaftalen-2-ilo, dihidronaftalen-3-ilo, dihidronaftalen-4-ilo, dihidronaftalen-1-ilo, 5,6,7,8-tetrahidronaftalen-1-ilo, 5,6,7,8-tetrahidronaftalen-2-ilo, 2,3-dihidrobenzofuran-4-ilo, 2,3-dihidrobenzofuran-5-ilo, 2,3-dihidrobenzofuran-6-ilo, 2,3-dihidrobenzofuran-7-ilo, benzo[d][1,3]dioxol-4-ilo, benzo[d][1,3]dioxol-5-ilo, 2H-cromen-2-on-5-ilo, 2H-cromen-2-on-6-ilo, 2H-cromen-2-on-7-ilo, 2H-cromen-2-on-8-ilo, isoindolina-1,3-dion-4-ilo, isoindolina-1,3-dion-5-ilo, inden-1-on-4-ilo, inden-1-on-5-ilo, inden-1-on-6-ilo, inden-1-on-6-ilo, 2,3-dihidrobenzo[b][1,4]dioxan-5-ilo, 2,3-dihidrobenzo[b][1,4]dioxan-6-ilo,2H-benzo[b][1,4]oxazin3(4H)-on-5-ilo,2H-benzo[b][1,4]oxazin3(4H)-on-6-ilo,2H-benzo[b][1,4]oxazin3(4H)-on-7-ilo,2H-benzo[b][1,4]oxazin3(4H)-on-8-ilo, benzo[d]oxazin-2(3H)-on-5-ilo, benzo[d]oxazin-2(3H)-on-6-ilo, benzo[d]oxazin-2(3H)-on-7-ilo, benzo[d]oxazin-2(3H)-on-8-ilo, quinazolin-4(3H)-on-5-ilo, quinazolin-4(3H)-on-6-ilo, quinazolin-4(3H)-on-7-ilo, quinazolin-4(3H)-on-8-ilo,quinoxalin-2(1H)-on-5-ilo, quinoxalin-2(1H)-on-6-ilo,quinoxalin-2(1H)-on-7-ilo, quinoxalin-2(1H)-on-8-ilo,benzo[d]tiazol-2(3H)-on-4-ilo, benzo[d]tiazol-2(3H)-on-5-ilo, benzo[d]tiazol-2(3H)-on-6-ilo, y, benzo[d]tiazol-2(3H)-on-7-ilo. En determinadas realizaciones, el arilo bicíclico es (i) naftilo o (ii) un anillo fenilo fusionado a un cicloalquilo monocíclico de 5 o 6 miembros, un cicloalquenilo monocíclico de 5 o 6 miembros o un heterociclilo monocíclico de 5 o 6 miembros, donde los grupos heterociclilo, cicloalquenilo y cicloalquilo fusionados se sustituyen opcionalmente. En determinadas realizaciones, el término “arilo” se refiere a un grupo fenilo.
El término “heteroarilo” es un término de la técnica y como se usa en el presente documento se refiere a un grupo aromático monocíclico o bicíclico con 3 a 14, 5 a 14, 3 a 12, o 3 a 10 átomos totales que incluyen uno o más heteroátomos tales como nitrógeno, oxígeno o azufre en la estructura de anillo. Los grupos heteroarilo más preferidos tienen 5-10 miembros de anillo donde de 1-4 de los miembros de anillo son heteroátomos seleccionados del grupo que consiste en O, N, y S. Ejemplos de grupos heteroarilo incluyen, por ejemplo, azaindolilo, benzo(b)tienilo, bencimidazolilo, benzofuranilo, benzoxazolilo, benzotiazolilo, benzotiadiazolilo, benzotriazolilo, benzoxadiazolilo, furanilo, imidazolilo, imidazopiridinilo, indolilo, indolinilo, indazolilo, isoindolinilo, isoxazolilo, isotiazolilo, isoquinolinilo, oxadiazolilo, oxazolilo, purinilo, piranilo, pirazinilo, pirazolilo, piridinilo, pirimidinilo, pirrolilo, pirrolo[2,3-d]pirimidinilo, pirazolo[3,4-d]pirimidinilo, quinolinilo, quinazolinilo, triazolilo, tiazolilo, tiofenilo, tetrahidroindolilo, tetrazolilo, tiadiazolilo, tienilo, tiomorfolinilo, triazolilo o tropanilo, y similares. Cualquier heteroarilo puede estar opcionalmente sustituido en una o más posiciones del anillo con uno o más sustituyentes tales como halógeno, azida, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, hidroxilo, alcoxilo, amino, nitro, sulfhidrilo, imino, amido, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, sulfonamido, cetona, aldehído, éster, heterociclilo, restos aromáticos o heteroaromáticos, fluoroalquilo (tal como trifluorometilo), ciano o similares. El término “heteroarilo” también incluye sistemas de anillo policíclicos con dos o más anillos cíclicos donde dos o más carbonos son comunes a dos anillos contiguos (los anillos son “anillos fusionados”) donde al menos uno de los anillos es un grupo aromático con uno o más heteroátomos en la estructura de anillo, por ejemplo, los demás anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos, heteroarilos y/o heterociclilos. Los ejemplos representativos de heteroarilo bicíclico incluyen, pero no se limita a, bencimidazolilo, benzofuranilo, benzotienilo, benzoxadiazolilo, benzoxatiadiazolilo, benzotiazolilo, cinnolinilo, 5,6
dihidroquinolin -2 ilo, 5,6 dihidroisoquinolin -1 ilo, furopiridinilo, indazolilo, indolilo, isoquinolinilo, naftiridinilo, quinolinilo, purinilo, 5,6,7,8 tetrahidroquinolin -2 ilo, 5,6,7,8 tetrahidroquinolin -3 ilo, 5,6,7,8 tetrahidroquinolin -4 ilo, 5,6,7,8 tetrahidroisoquinolin -1 ilo, tienopiridinilo, 4,5,6,7 tetrahidrobenzo[c][1,2,5]oxadiazolilo y 6,7 dihidrobenzo[c][1,2,5]oxadiazol -4(5H) onilo. Cualquier heteroarilo o heteroarilo bicíclico puede estar opcionalmente sustituido como se detalla a continuación.
El término “aralquilo”, “arilalquilo”, o “arilalquilo(C1-C6)” es un término de la técnica y como se usa en el presente documento se refiere a un grupo alquilo, por ejemplo un grupo alquilo C1-C6, sustituido con un grupo arilo, donde el resto está unido con la molécula principal a través del grupo alquilo. Un ejemplo de aralquilo es el grupo bencilo, es decir, el grupo fenilo-metilo.
El término “arileno” está reconocido en la técnica y como se usa en el presente documento se refiere a un diradical obtenido retirando dos átomos de hidrógeno de un grupo arilo como se define anteriormente. Un ejemplo de grupo arileno es 1,4-fenileno.
El término “heteroaralquilo”, “heteroarilalquilo”, o “heteroarilalquilo(C1-C6” es un término de la técnica y como se usa en el presente documento se refiere a un grupo alquilo, por ejemplo un grupo alquilo C1-C6, sustituido con un grupo heteroarilo unido con el resto molecular principal a través del grupo alquilo.
El término “alcoxi” o “alcoxilo”, como se usa en el presente documento, se refiere a un grupo alquilo, como se define en el presente documento, unido al resto molecular principal a través de un átomo de oxígeno. Preferentemente, el grupo alcoxi es alcoxi (C1-C6). Los ejemplos representativos incluyen, pero no se limita a, metoxi, etoxi, propoxi, 2-propoxi, butoxi, terc-butoxi, pentiloxi y hexiloxi. Ejemplos representativos de alcoxi (C1-C3) incluyen metoxi, etoxi, y propoxi.
El término “alcoxicarbonilo” se refiere a un grupo alcoxi, como se define en el presente documento, unido al resto molecular principal a través de un grupo carbonilo, representado por C(=O), como se define en el presente documento. Ejemplos representativos de alcoxicarbonilo incluyen pero no se limita a, metoxicarbonilo, etoxicarbonilo y terc-butoxicarbonilo. Alcoxicarbonilo puede formar una porción de otro resto, por ejemplo metoxicarbonilmetilo.
El término “alquilcarbonilo”, como se usa en el presente documento, se refiere a un grupo alquilo, como se define en el presente documento, unido al resto molecular principal a través de un grupo carbonilo, como se define en el presente documento. Los ejemplos representativos de alquenilo incluyen, pero no se limita a, acetilo, 1-oxopropilo, 2,2-dimetil-1-oxopropilo, 1-oxobutilo, y 1-oxopentilo.
El término “arilcarbonilo”, como se usa en el presente documento, se refiere a un grupo arilo, como se define en el presente documento, unido al resto molecular principal a través de un grupo carbonilo, como se define en el presente documento. Ejemplos representativos de arilcarbonilo incluyen pero no se limita a benzoílo y (2-piridinil)carbonilo.
El término “alquilcarboniloxi” y “arilcarboniloxi”, como se usa en el presente documento, se refiere a un grupo alquilcarbonilo o arilcarbonilo, como se define en el presente documento, unido al resto molecular principal a través de un átomo de oxígeno. Ejemplos representativos de alquilcarboniloxi incluyen pero no se limita a, acetiloxi, etilcarboniloxi y terc-butilcarboniloxi. Los ejemplos representativos de arilcarboniloxi incluyen pero no se limita a, fenilcarboniloxi.
El término “alquenoxi” o “alquenoxilo”, como se usa en el presente documento, se refiere a un grupo alquenilo, como se define en el presente documento, unido al resto molecular principal a través de un átomo de oxígeno. Ejemplos representativos de alquenoxilo incluyen pero no se limita a 2-propen-1-oxilo (es decir, CH2=CH-CH2-O-) y viniloxi (es decir, CH2=CH-O-).
El término “ariloxi”, como se usa en el presente documento, se refiere a un grupo arilo, como se define en el presente documento, unido al resto molecular principal a través de un átomo de oxígeno.
El término “heteroariloxi”, como se usa en el presente documento, se refiere a un grupo heteroarilo, como se define en el presente documento, unido al resto molecular principal a través de un átomo de oxígeno.
Los términos “ciano” y “nitrilo” son términos de la técnica y como se usan en el presente documento se refieren a -CN.
El término “nitro” como se usa en el presente documento se refiere a -NO2.
Los términos “halo” o “halógeno”, son términos de la técnica y tal como se usan en el presente documento, se refieren a F, Cl, Br o I.
El término “haloalquilo”, como se utiliza en el presente documento, se refiere a un grupo alquilo tal como se define en el presente documento, donde algunos o toros los hidrógenos están remplazados por átomos de halógeno. El término “haloalcoxi” se refiere a un grupo alcoxi tal como se define en el presente documento, donde algunos o toros los
hidrógenos están remplazados por átomos de halógeno. Un ejemplo de grupo haloalquilo (C1-C6) es trifluorometilo.
El término “hidroxi” es un término de la técnica y como se usa en el presente documento se refiere a -OH.
El término “hidroxialquilo”, como se usa en el presente documento, se refiere al menos a un grupo hidroxi, como se define en el presente documento, unido al resto molecular principal a través de un grupo alquilo, como se define en el presente documento. Ejemplos representativos de hidroxialquilo(C1-C6) incluyen pero no se limita a, hidroximetilo, 2-hidroxietilo, 3-hidroxipropilo, y 2,3-dihidroxipentilo.
El término “sulfonilo”, como se usa en el presente documento se refiere al grupo -S(O)2- que puede formar una porción de restos más grandes, tales como metanosulfonilo o p-toluenosulfonilo.
El término “sililo”, como se usa en el presente documento incluye derivados de hidrocarbilo del grupo sililo (H3Si) (es decir, (hidrocarbil)3Si-), donde un radical hidrocarburo son grupos univalentes formados retirando un átomo de hidrógeno de un hidrocarburo, por ejemplo, etilo, fenilo. Los radicales hidrocarburos pueden ser combinaciones de diferentes grupos que pueden variar para proporcionar una cantidad de grupos sililo, tales como trimetilsililo (TMS), ferc-butildifenilsililo (TBDPS), ferc-butildimetilsililo (TBS/TBDMS), triisopropilsililo (TIPS), y [2-(trimetilsilil)etoxi]metilo (SEM).
El término “sililoxi”, como se usa en el presente documento, se refiere a un grupo sililo, como se define en el presente documento, unido a la molécula principal a través de un átomo de oxígeno.
Determinados compuestos contenidos en las composiciones de la presente invención pueden existir en formas geométricas o estereoisoméricas particulares. Además, los compuestos de la presente invención también pueden estar ópticamente activos. La presente invención contempla todos dichos compuestos, incluyendo isómeros cis y trans, enantiómeros (R) y (S), diastereómeros, isómeros (d), isómeros (l), las mezclas racémicas de los mismos y otras mezclas de los mismos, que se encuentran comprendidos en el alcance de la invención. Puede haber átomos de carbono asimétricos adicionales en un sustituyente, tal como un grupo alquilo. Se pretende que todos dichos isómeros, así como mezclas de estos, estén incluidos en la presente invención.
El término “mezcla racémica” se refiere a una mezcla que contienen proporciones iguales del primer enantiómero de la molécula y del segundo enantiómero de esta molécula, donde el segundo enantiómero es la imagen idéntica del primero.
El término “mezcla escalémica” se refiere a cualquier mezcla no racémica de estereoisómeros de la molécula.
Si, por ejemplo, se desea un enantiómero particular de un compuesto de la presente invención, puede prepararse mediante síntesis asimétrica, o mediante derivación con un auxiliar quiral, donde la mezcla diastereomérica resultante se separa y el grupo auxiliar se escinde para proporcionar los enantiómeros deseados puros. De manera alternativa, cuando la molécula contiene un grupo funcional básico, tal como amino, o un grupo funcional ácido, tal como carboxilo, se forman sales diastereoméricas con un ácido o base ópticamente activos adecuados, seguido de resolución de los diastereómeros formados de esta manera mediante cristalización fraccionaria o medios cromatográficos conocidos en la técnica, y la posterior recuperación de los enantiómeros puros.
Los compuestos orgánicos frecuentemente ocurren en más de una forma cristalina que puede diferir en sus propiedades físicas y biológicas, tales como punto de fusión, estabilidad, solubilidad, biodisponibilidad. Dichas formas cristalinas se denominan polimorfos. Todos los polimorfos de los compuestos inventivos de fórmula (I) y de sus sales pretenden estar comprendidos en el alcance de esta invención.
Dado que muchos elementos químicos pueden ocurrir como isótopos, su abundancia en la molécula del compuesto inventivo de fórmula (I) puede ser idéntica a la naturaleza o estar alterada. Algunos isótopos muestran diferentes propiedades espectrales o biológicas y este fenómeno se puede usar para análisis de distribución y metabolismo de fármacos en el cuerpo del receptor. Todas las formas de los compuestos de fórmula (I) con una abundancia natural o no natural de isótopos de cualquiera de sus elementos integrantes pretenden estar dentro del alcance de esta invención.
Se entenderá que “sustitución” o “sustituido con” incluye la condición implícita de que dicha sustitución concuerda con la valencia permitida del átomo sustituido y el sustituyente, y que la sustitución da como resultado un compuesto estable, por ejemplo, que no se somete espontáneamente a una transformación tal como por reorganización, fragmentación, descomposición, ciclación, eliminación, u otra reacción.
El término “sustituido” también se contempla que incluye todos los sustituyentes permitidos de compuestos orgánicos. En un aspecto amplio, los sustituyentes admisibles incluyen sustituyentes acíclicos y cíclicos, ramificados y no ramificados, carbocíclicos y heterocíclicos, aromáticos y no aromáticos de compuestos orgánicos. Sustituyentes ilustrativos incluyen por ejemplo halógeno, azida, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, cicloalquilalquilo, (cicloalquil)alcoxilo, hidroxilo, alcoxilo, amino, nitro, sulfhidrilo, imino, amido, fosfonato, fosfinato, carbonilo, carboxilo,
sililo, éter, alquiltio, sulfonilo, aminosulfonilo, sulfonamido, cetona, aldehido, éster, heterociclilo, heterociclilalquilo, restos aromáticos o heteroaromáticos, aminoalquilo, haloalquilo, fluoroalquilo (tal como trifluorometilo), haloalcoxilo, ciano u otros sustituyentes descritos anteriormente. Los sustituyentes permitidos pueden ser uno o más e iguales o diferentes para los compuestos orgánicos apropiados. Para los fines de esta invención, los heteroátomos tales como nitrógeno pueden tener sustituyentes de hidrógeno y/o cualquier sustituyente permitido de compuestos orgánicos descritos en el presente documento que satisfacen las valencias de los heteroátomos. Esta invención no pretende estar limitada de ninguna manera por los sustituyentes permitidos de los compuestos orgánicos.
La frase "grupo protector” se refiere a sustituyentes temporales que protegen un grupo funcional potencialmente reactivo de transformaciones químicas no deseados. Ejemplos de dichos grupos protectores incluyen ésteres de ácidos carboxílicos, silil éteres de alcoholes y acetales y cetales de aldehidos y cetonas respectivamente. El campo de química de grupo protector fue revisado (Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2a ed.; Wiley: New York, 1991). Las formas protegidas formas de los compuestos inventivos están incluidas dentro del alcance de la presente invención.
Un compuesto "saturado” o "completamente saturado” se refiere a que la estructura química mencionada no contiene ningún enlace carbono-carbono múltiple. Por ejemplo, un grupo cicloalquilo saturado como se define en el presente documento incluye ciclohexilo, ciclopropilo y similares.
Un compuesto "insaturado” o "parcialmente insaturado” se refiere a que la estructura química mencionada puede contener uno o más enlaces carbono-carbono múltiples, pero no es aromática. Por ejemplo, un grupo cicloalquilo no saturado como se define en el presente documento incluye ciclohexenilo, ciclopentenilo, ciclohexadienilo y similares.
Para los fines de esta invención, los elementos químicos se identifican de acuerdo con la Tabla Periódica de los Elementos, versión IUPAC, The Merck Index, 12a edición, 1996, interior de portada.
Otros términos químicos en el presente documento se utilizan de acuerdo con el uso convencional en la técnica, como se ejemplifica en The McGraw-Hill Dictionary of Chemical Terms (ed. Parker, S., 1985), McGraw-Hill, San Francisco). A menos que se defina de otro modo, todos los términos técnicos y científicos utilizados en el presente documento tienen el mismo significado entendido normalmente por un experto en la materia a la que pertenece esta invención.
Los expertos en la técnica sabrán que los compuestos de esta descripción pueden existir en formas tautoméricas. Por ejemplo, las siguientes estructuras ilustran algunas formas tautoméricas del grupo triazol.

En esta memoria descriptiva solo se describe una forma tautomérica para cada compuesto, pero todas dichas formas tautoméricas de los compuestos se encuentran dentro del alcance de la descripción.
A menos que se indique lo contrario, también se pretende que las estructuras representadas en el presente documento incluyan todas las formas estereoquímicas de la estructura, es decir, las configuraciones R y S para cada centro asimétrico. Por lo tanto, los isómeros estereoquímicos individuales, así como las mezclas enantioméricas y diastereoméricas de los compuestos del presente documento, se encuentran dentro del alcance de la descripción. Los isómeros estereoquímicos R y S, así como todas las mezclas de estos, se incluyen dentro del alcance de la descripción.
La frase "farmacéuticamente aceptable” se usa en el presente documento para hacer referencia a los compuestos, materiales, composiciones y/o formas de dosificación que, según un criterio médico sensato, son adecuados para su uso en contacto con los tejidos de seres humanos y animales sin provocar una toxicidad excesiva, irritación, respuesta
alérgica u otros problemas o complicaciones, con una relación beneficio/riesgo correspondiente razonable.
El término “sal farmacéuticamente aceptable” como se usa en el presente documento incluye sales derivadas de ácidos inorgánicos u orgánicos que incluyen por ejemplo, clorhídrico, bromhídrico, sulfúrico, nítrico, perclórico, fosfórico, fórmico, acético, láctico, maleico, fumárico, succínico, tartárico, glicólico, salicílico, cítrico, metansulfónico, bencensulfónico, benzoico, malónico, trifluoroacético, tricloroacético, naftaleno-2-sulfónico, y otros ácidos. Las formas de sale farmacéuticamente aceptables pueden incluir formas donde la relación de moléculas que comprenden la sal no es 1:1. Por ejemplo, la sal puede comprender más de una molécula de ácido inorgánico u orgánico por molécula de base, tal como dos moléculas de ácido clorhídrico por molécula de compuesto de Fórmula I. Como otro ejemplo, la sal puede comprender menos de una molécula de ácido inorgánico u orgánico por molécula de base, tal como dos moléculas de compuesto de Fórmula I por molécula de ácido tartárico.
Como se usa en el presente documento, un disolvente prótico es un disolvente que tiene un átomo de hidrógeno unido a un oxígeno (como en un grupo hidroxilo) o un nitrógeno (como en un grupo amina). En términos generales, cualquier disolvente que contiene H+ lábil se denomina disolvente prótico. Las moléculas de dichos disolventes donan fácilmente protones (H+) a los reactivos. Por el contrario, un disolvente aprótico es un disolvente que no tiene un átomo de hidrógeno unido a un oxígeno (como en un grupo hidroxilo) o un nitrógeno (como en un grupo amina) y no puede donar hidrógeno.
Como se usa en el presente documento, un disolvente prótico polar es un disolvente prótico que disolverá muchas sales. En general, estos disolventes tienen constantes dieléctricas altas y polaridad alta. Ejemplos no taxativos de disolventes próticos polares incluyen ácido acético, amoníaco, etanol, ácido fórmico, isopropanol, metanol, n-butanol, nitrometano, n-propanol, t-butanol, y agua.
Como se usa en el presente documento, un disolvente aprótico polar es un disolvente que disolverá muchas sales, pero carece un hidrógeno ácido; estos disolventes generalmente tienen polaridad y constantes dieléctricas intermedias a altas. Ejemplos no taxativos de disolventes apróticos polares incluyen acetona, acetonitrilo, diclorometano (DCM), dimetil sulfóxido (DMSO), acetato de etilo, triamida hexametilfosfórico (HMPT), W,W-dimetilformamida (DMF), y tetrahidrofurano (THF).
Como se usa en el presente documento, un disolvente aprótico no polar es un disolvente que disolverá muchas sales, pero carece un hidrógeno ácido; estos disolventes generalmente tienen polaridad y constantes dieléctricas bajas. Ejemplos no taxativos de disolventes apróticos no polares incluyen benceno, cloroformo, ciclohexano, dietil éter, hexano, pentano, y tolueno.
Un médico o veterinario con experiencia en la técnica puede determinar e indicar fácilmente la cantidad terapéuticamente eficaz de la composición farmacéutica necesaria. Por ejemplo, el médico o veterinario puede comenzar las dosis de la composición farmacéutica o compuesto en niveles inferiores a los requeridos para alcanzar el efecto terapéutico deseado y aumentar gradualmente la dosificación hasta alcanzar el efecto deseado. “Cantidad terapéuticamente eficaz” se refiere a la concentración de un compuesto que es suficiente para provocar el efecto terapéutico deseado. Generalmente se entiende que la cantidad eficaz del compuesto variará de acuerdo con el peso, sexo, edad e historia clínica del sujeto. Otros factores que influyen la cantidad eficaz pueden incluir pero no se limita a la gravedad de la afección del paciente, el trastorno a ser tratado, la estabilidad del compuesto, el modo de administración, la biodisponibilidad del compuesto particular y si se desea, otro tipo de agente terapéutico que se administra con el compuesto de la invención. Se puede administrar una dosis total mayor mediante múltiples administraciones del agente. Métodos para determinar la eficacia y dosificación son conocidos por los expertos en la técnica (Isselbacher et al. (1996) Harrison's Principies of Infernal Medicine 13a ed., 1814-1882).
“Modular” o “que modula” se refiere al tratamiento, prevención, supresión, mejora o inducción de una función, afección o trastorno.
El término “tratar” incluye tratamientos profilácticos y/o terapéuticos. La expresión tratamiento “profiláctico” o “terapéutico”, se reconoce en la técnica e incluye administrarle al hospedador una o más de las composiciones del presente documento. Si se administra antes de la manifestación clínica de la afección no deseada (por ejemplo, enfermedad u otro estado no deseado del animal hospedador), el tratamiento es profiláctico (es decir, protege al hospedador contra el desarrollo de la afección no deseada), mientras que, si se administra después de la manifestación de la afección no deseada, el tratamiento es terapéutico (es decir, pretende disminuir, paliar o estabilizar la afección no deseada existente, o los efectos secundarios de la misma).
Como se usa en el presente documento “sujeto” se refiere a un animal de sangre caliente tal como un mamífero, preferentemente un humano, o un niño humano, que padece o tiene el potencial de padecer una o más enfermedades y trastornos descritos en el presente documento.
“EC50” se refiere a una dosificación, concentración o cantidad de un compuesto de prueba particular que provoca una respuesta dependiente de la dosis al 50 % de la expresión máxima de una respuesta particular que es inducida, provocada o potenciada por el compuesto de prueba particular.
“ IC50” se refiere a una cantidad, concentración o dosificación de un compuesto de prueba particular que alcanza un 50 % de inhibición de una respuesta máxima en un ensayo que mide dicha respuesta.
Compuestos de la invención
En determinadas realizaciones, la invención se refiere a un compuesto representado por la fórmula (I),

donde:
W es halo, alquilo(C1-C3), alcoxi(C1-C3), o alquiltio(C1-C3);
X es un enlace sencillo, -CH2-, -CH2CH2-, -CH=CH-, -C(alquilo(C1-C3))2-, o -C(O)-;
Y es un enlace sencillo, -CH-, -CHCH2-, -CH2CH-, -C=CH-, -CH=C-, -N-, -O-, -OCH2-, -S(O)-, o -S(O)2-;
si Y es un enlace sencillo, -O-, -OCH2-, -S(O)-, o -S(O)2-, entonces R1 está ausente;
R1 es H, OH, alquilo(C1-C6), haloalquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), arilalquilo(C1-C6), heteroarilalquilo(C1-C6), -C(O)alquilo(C1-C6), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1 -C6), -C(O)heteroarilalquilo(C1-C6), -S(O)2 alquilo(C1-C6), -S(O)2arilo, -S(O)2heteroarilo, -S(O)2arilalquilo(C1-C6), -S(O)2heteroarilalquilo(C1-C6), -CO2H, -C(O)O alquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NH2, -C(O)NHOH, -C(O)NHCN, -C(O)NH(alquilo(C1-C6)), -C(O)N(alquilo(C1-C6))2, -C(O)NH(arilalquilo(C1-C6)), -C(O)N(arilalquilo(C1-C6))2, -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6)), -C(O)N(arilo)2, -C(O)N alquil(C1 -C6))(arilalquilo(C1 -Ce)), -C(O)N(aril)(arilalquilo(C1-C6)), -C(O)NH(haloalquilo(C1-Ce)), -C(O)N(haloalquilo(C1-Ce))2, -S(O)2NH2, -S(O)2NH(alquilo(C1-Ce)), -S(O)2NH(haloalquilo(C1-Ce)), -S(O)2NH(arilo), -S(O)2NH(heteroarilalquilo(C1-Ce)), -S(O)2NH(heteroarilo), -S(O)2N(alquilo(C1-Ce))2, -S(O)2NHC(O)alquilo(C1-Ce), -S(O)2NHC(O)haloalquilo(C1-Ce), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(C1-Ce), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(C1-Ce), -NHS(O)2 alquilo(C1-Ce), -NHS(O)2arilo, -NHS(O)2 haloalquilo(C1-Ce), -NHS(O)2arilalquilo(C1-Ce), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(C1-C6), -NHC(O)(alquilo(C1-Ce)), -NHC(O)(haloalquilo(C1-Ce)), -NHC(O)(arilo), -NHC(O)(arilalquilo(C1-Ce)), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(C1-Ce)), -NHC(O)NH alquilo(C1-Ce), -NHC(O)NH(arilo), -NHC(O)NH(arilalquilo(C1-Ce)), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(C1-Ce)), -C(O)NHS(O)2 alquilo(C1-Ce), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(C1-Ce)), -C(O)NHS(O)2(arilalquilo(C1-Ce)), -C(O)NHS(O)2heteroarilo, -C(O)NHS(O)2(heteroarilalquilo(C1-Ce)), -P(O)(OH)2, -(alquileno(C1-Ce))C(O)OH, -(alquileno(C1-Ce))C(O)O alquilo(C1-Ce), -NH2, -NH alquilo(C1-Ce), -N(alquilo(C1-Ce))2, -NC, -CN, -C(S)NH2, -NHC(O)NH2, -C=CH, alquiltio(C1-C6)-, mercaptoalquilo(C1-C6)-, o -C(O)heterociclilo;
Z es -CH-, -C(O)-, -C(alquilo(C1-C3))-, o -C(=CH2)-;
si Z es -C(O)-, entonces R2 está ausente,
R2 es H, Oh , halo, alquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), -CO2H, -C(O)O alquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NHOH, -C(O)NHCN, -C(O)NH2, -C(O)NH(alquilo(C1-Ce)), -C(O)N(alquilo(C1-Ce))2, -C(O)NH(arilalquilo(C1-Ce)), -C(O)N(arilalquilo(C1-Ce))2, -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-Ce)), -C(O)N(arilo)2, -C(O)N alquil(C1-Ce))(arilalquilo(C1-Ce)), -C(O)N(aril)(arilalquilo(C1-Ce)), -C(O)NH haloalquilo((C1-Ce)), -C(O)N(haloalquilo(C1-Ce))2, -S(O)2NH2, -S(O)2NH(alquilo(C1-Ce)), -S(O)2NH(haloalquilo(C1-Ce)), -S(O)2NH(arilo), -S(O)2NH(heteroarilalquilo(C1-C6)), -S(O)2NH(heteroarilo), -S(O)2NH(heteroarilalquilo(C1-C6)), -S(O)2N(alquilo(C1-Ce))2, -S(O)2NHC(O)alquilo(C1-Ce), -S(O)2NHC(O)haloalquilo(C1-Ce), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(C1-Ce), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(C1-Ce), -NHS(O)2 alquilo(C1-Ce), -NHS(O)2aryl, NHS(O)2 haloalquilo(C1-Ce), -NHS(O)2arilalquilo(C1-C6), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(C1-Ce), -NHC(O)(alquilo(C1-Ce)), -NHC(O)(haloalquilo(C1-Ce)), -NHC(O)(arilo), -NHC(O)(arilalquilo(C1-Ce)), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(C1-Ce)), -NHC(O)NH alquilo(C1-Ce), -NHC(O)NHarilo, -NHC(O)NH(arilalquilo(C1-Ce)), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(C1-Ce)), -C(O)NHS(O)2 alquilo(C1-Ce), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(C1-Ce)), -C(O)NHS(O)2(arilalquilo(C1-Ce)), -C(O)NHS(O)2heteroarilo, -C(O)NHS(O)2(heteroarilalquilo(C1-Ce)), -P(O)(OH)2, arilalquilo(C1-Ce), cicloalquil(C3-C7)alquilo(C1-C6)-, -NC, -CN, -C(s )n H2, -NHC(O)NH2, -C=CH, alquiltio(C1 -Ce)-, mercaptoalquilo(C1-C6)-, o -C(O)heterociclilo;
o R1 y R2, tomados juntos con los átomos que son parte forman un anillo carbocíclico o heterocíclico;
R3 es H, alquilo(C1-C3), alcoxi(C1-C3)alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-Ce), -NC, -CN, -C(S)NH2, -NHC(O)NH2, o -C=CH;
R4 es H, alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-Ce), alcoxi(C1-C3)alquilo(C1-C3), o hidroxialquilo(C1-C4);
R5 es H, halo, -NO2, -CN, alquilo(Ci-C6), haloalquilo(0i-06), -NH2, -NH(alquilo(Ci-C6)), -N(alquilo(Ci-C6))2, -OH, alcoxi(Ci-C6), hidroxialquilo(0i-06), a lcoxi(0i-06)alquilo(0i-06), haloalcoxi(Ci-C6), -SH, alquiltio(Ci-03)alquilo(Ci-C3), -NC, -C(S)NH2, -NHC(O)NH2, o -0E0H; y
R6 es H, halo, -OH, -NH2, o -SH; o
R6, tomado junto con el átomos de carbono que lo contiene, representa C(O)-;
donde:
cualquier aparición de alquilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo, o hidroxialquilo está sustituida opcionalmente con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2, -NH(alquilo(Ci-C6)), -N(alquilo(0i-0 6))2, -0N, -NO2, alquilo(0i-06), haloalquilo(0i-06), alcoxi(0i-06), arilo, heteroarilo, arilalquilo(0i-06), heteroarilalquilo(0i-06), cicloalquilo(03-07), heterociclilo, -C(O)OH, -C(O)O alquilo(Ci-C6), -C(O)NH2, -0 (O)n H alquilo(0i-06), y -C(O)N(alquilo(Ci-C6))2;
o una sal, solvato o tautómero, estereoisómero o polimorfo farmacéuticamente aceptable de estos.
En realizaciones adicionales, la invención se dirige a un compuesto de fórmula (I) donde:
W es halo, alquilo(Ci-C3), alcoxi(Ci-C3), o alquiltio(Ci-C3);
X es un enlace sencillo, -OH2-, -OH2OH2-, -c H=0 H-, o -C(O)-;
Y es un enlace sencillo, -0H-, -OHOH2-, -OH2OH-, -0=0H-, -0H=0-, -N-, -O-, -S(O)-, o -S(O)2-;
si Y es un enlace sencillo, -O-, -S(O)-, o -S(O)2-, entonces R1 está ausente;
R1 es H, alquilo(Ci-C6), haloalquilo(0i-06), alcoxi(0i-06), hidroxialquilo(0i-06), alcoxi(0i-06)alquilo(0i-06), arilalquilo(0i-06), heteroarilalquilo(0i-06), -0(O)alquilo(Ci-C6), -C(O)arilo, -0(O)heteroarilo, -0(O)arilalquilo(Ci-C6), -C(O)heteroarilalquilo(Ci-C6), -S(O)2 alquilo(0i-06), -S(O)2arilo, -S(O)2heteroarilo, -S(O)2arilalquilo(Ci-C6), -S(O)2heteroarilalquilo(Ci-C6), -OO2H, -C(O)O alquilo(0i-06), -C(O)O(arilo), -C(O)O(heteroarilo), -0(O)O(arilalquilo(0i-06)), -C(O)O(heteroarilalquilo(Ci-C6)), -0(O)NH2, -C(O)NHOH, -0(O)NH0N, -0(O)NH(alquilo(0i-06)), -C(O)N(alquilo(Ci-C6))2, -0(O)NH(arilalquilo(0i-06)), -0(O)N(arilalquilo(0i-06))2, -0(O)NH(arilo), -0(O)N(aril)(alquilo(0i-06)), -0(O)N(arilo)2, -0(O)N alquil(0i-06))(arilalquilo(0i-06)), -0(O)N(aril)(arilalquilo(0i-06)), -C(O)NH(haloalquilo(Ci-C6)), -0(O)N(haloalquilo(0i-06))2, -S(O)2NH2, -S(O)2NH(alquilo(0i-06)), -S(O)2NH(haloalquilo(Ci-C6)), -S(O)2NH(arilo), -S(O)2NH(heteroarilalquilo(0i-06)), -S(O)2NH(heteroarilo), -S(O)2N(alquilo(Ci-C6))2, -S(O)2NHC(O)alquilo(Ci-C6), -S(O)2NHC(O)haloalquilo(Ci-C6), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(Ci-C6), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(Ci-C6), -NHS(O)2 alquilo(Ci-06), -NHS(O)2arilo, -NHS(O)2 haloalquilo(0i-06), -NHS(O)2arilalquilo(0i-06), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(0i-06), -NH0(O)(alquilo(0i-06)), -NHC(O)(haloalquilo(Ci-C6)), -NH0(O)(arilo), -NH0(O)(arilalquilo(0i-06)), -NH0(O)(heteroarilo), -NH0(O)(heteroarilalquilo(0i-06)), -NH0(O)NH alquilo(0i-06), -NH0(O)NH(arilo), -NH0(O)NH(arilalquilo(0i-06)), -NH0(O)NH(heteroarilo), -NH0(O)NH(heteroarilalquilo(0i-06)), -C(O)NHS(O)2 alquilo(Ci-C6), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(Ci-C6)), -C(O)NHS(O)2(arilalquilo(Ci-C6)), -C(O)NHS(O)2heteroarilo, -C(O)NHS(O)2(heteroarilalquilo(Ci-C6)), -P(O)(OH)2, -(alquileno(Ci-C6))C(O)OH, -(alquileno(Ci-C6))C(O)O alquilo(Ci-C6), -NH2, -NH alquilo(0i-06), -N(alquilo(0i-06))2, -N0, -0N, -0(S)NH2, -NH0 (O)NH2, -Ce c H, alquiltio(0i-06)-, mercaptoalquilo(0i-06)-, o -0(O)heterociclilo;
Z es -0H-, -0(O)-, o -C(alquilo(Ci-C3))-;
si Z es -0(O)-, entonces R2 está ausente,
R2 es H, halo, alquilo(Ci-C6), a lcoxi(0i-06), hidroxialquilo(0i-06), alcoxi(0i-06)alquilo(0i-06), -OO2H, -C(O)O alquilo(0i-06), -C(O)O(arilo), -C(O)O(heteroarilo), -0(O)O(arilalquilo(Ci-C6)), -0(O)O(heteroarilalquilo(Ci-C6)), -C(O)NHOH, -0(O)NH0N, -0(O)NH2, -0(O)NH(alquilo(0i-06)), -0(O)N(alquilo(0i-06))2, -C(O)NH(arilalquilo(Ci-C6)), -0(O)N(arilalquilo(0i-06))2, -0(O)NH(arilo), -0(O)N(aril)(alquilo(0i-06)), -0(O)N(arilo)2, -0(O)N alquil(0i-06))(arilalquilo(0i-06)), -0(O)N(aril)(arilalquilo(0i-06)), -C(O)NH(haloalquilo(Ci-C6)), -0(O)N(haloalquilo(0i-06))2, -S(O)2NH2, -S(O)2NH(alquilo(0i-06)), -S(O)2NH(haloalquilo(0i-06)), -S(O)2NH(arilo), -S(O)2NH(heteroarilalquilo(0i-06)), -S(O)2NH(heteroarilo), -S(O)2NH(heteroarilalquilo(0i-06)), -S(O)2N(alquilo(0i-06))2, -S(O)2NHC(O)alquilo(Ci-C6), -S(O)2NHC(O)haloalquilo(Ci-C6), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(Ci-C6), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(Ci-C6), -NHS(O)2 alquilo(Ci-C6), -NHS(O)2arilo, NHS(O)2 haloalquilo(0i-06), -NHS(O)2arilalquilo(0i-06), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(0i-06), -NH0(O)(alquilo(0i-06)), -NHC(O)(haloalquilo(Ci-C6)), -NH0(O)(arilo), -NH0(O)(arilalquilo(0i-06)), -NH0(O)(heteroarilo), -NH0(O)(heteroarilalquilo(0i-06)), -NH0(O)NH alquilo(0i-06), -NH0(O)NHarilo, -NH0(O)NH(arilalquilo(0i-06)), -NHC(O)NH(heteroarilo), -NH0(O)NH(heteroarilalquilo(0i-06)), -C(O)NHS(O)2alquilo(Ci-C6), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(Ci-C6)), -C(O)NHS(O)2(arilalquilo(Ci-C6)), -C(O)NHS(O)2heteroarilo, -c(O)NHs(O)2(heteroarilalquilo(Ci-C6)), -P(O)(OH)2, arilalquilo(0i-06), cicloalquil(03-07)alquilo(0i-06)-, -N0, -0N, -c (s )NH2, -NH0(O)NH2, -0 e0 H, alquiltio(0i-06)-, mercaptoalquilo(Ci-C6)-, o -0(O)heterociclilo;
R3 es H, alquilo(Ci-C3), a lcoxi(0i-03)alquilo(0i-03), alquiltio(0i-03)alquilo(0i-03), haloalquilo(0i-06), -N0, -0N, -0(S)NH2, -NH0(O)NH2, o -0E0H;
R4 es H, alquilo(Ci-C3), alquiltio(0i-03)alquilo(0i-C3), haloalquilo(0i-06), alcoxi(0i-03)alquilo(0i-03), o hidroxialquilo(0i-04);
R5 es H, halo, -NO2, -0N, alquilo(0i-06), haloalquilo(0i-06), -NH2, -NH(alquilo(0i-06)), -N(alquilo(0i-06))2, -OH, alcoxi(0i-06), hidroxialquilo(0i-06), alcoxi(Ci-C6)alquilo(Ci-C6), haloalcoxi(0i-06), -SH, alquiltio(0i-03)alquilo(0i-C3), -N0, -C(S)NH2, -NH0(O)NH2, o -0E0H; y
R6 es H, halo, -OH, -NH2, o -SH; o
R6, tomado junto con el átomo de carbono que lo contiene, representa C(O)-;
donde:
i4
cualquier aparición de alquilo, arilo, arilalquMo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo, o hidroxialquilo está sustituida opcionalmente con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2, -NH(alquilo(C1-C6)), -N(alquilo(C1-C6))2, -CN, -NO2, alquilo(C1-C6), haloalquilo(C1-C6), alcoxi(C1-C6), arilo, heteroarilo, arilalquilo(C1-C6), heteroarilalquilo(C1-C6), cicloalquilo(C3-C7), heterociclilo, -C(O)o H, -C(O)O alquilo(C1-C6), -C(O)Nh 2, -C(O)NH alquilo(C1-C6), y -C(O)N(alquilo(C1-C6))2. En determinadas realizaciones, W es fluoro, cloro, bromo, metilo o metoxi.
En determinadas realizaciones, W es cloro o bromo.
En determinadas realizaciones, W es cloro.
En determinadas realizaciones, R6 es H o -OH.
En determinadas realizaciones, R6 es H.
En determinadas realizaciones, X es un enlace sencillo, -CH2-, o -C(O)-.
En determinadas realizaciones, X es -CH2-.
En determinadas realizaciones, Y es un enlace sencillo, -CH-, -N-, -O-, o -S(O)2-.
En determinadas realizaciones, Y es un enlace sencillo, -O-, o -S(O)2-.
En determinadas realizaciones, Y es un enlace sencillo.
En determinadas realizaciones, Y es -O-.
En determinadas realizaciones, Y es -CH- o -N-.
En determinadas realizaciones, Y es -CH-.
En determinadas realizaciones, Y es -N-.
En determinadas realizaciones, R1 es H, alquilo(C1-C6), haloalquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), arilalquilo(C1-C6), heteroarilalquilo(C1-C6), -C(O)alquilo(C1-C6), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1-C6), -C(O)heteroarilalquilo(C1-C6), -S(o )2 alquilo(C1-C6), -S(O)2arilo, -S(O)2heteroarilo, -S(O)2arilalquilo(C1-C6), -S(O)2heteroarilalquilo(C1-C6), -CO2H, -C(O)O alquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NH2, -C(O)NHOH, -C(O)NHCN, -C(O)NH(alquilo(C1-C6)), -C(O)N(alquilo(C1-C6))2, -C(O)NH(arilalquilo(C1-C6)), -C(O)N(arilalquilo(C1-C6))2, -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6)), -C(O)N(arilo)2, -C(O)N alquil(C1-C6))(arilalquilo(C1-C6)), -C(O)N(aril)(arilalquilo(C1-C6)), -(alquileno(C1-C6))C(O)OH, -(alquileno(C1-C6)C(O)O alquilo(C1-C6), -NH2, -NH alquilo(C1-C6), -N(alquilo(C1-C6))2, o -C(O)heterociclilo.
En determinadas realizaciones, R1 es H, alquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), -C(O)alquilo(C1-C6), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1-C6), -C(O)heteroarilalquilo(C1-C6), -S(O)2alquilo(C1-C6), -S(O)2arilo, -C(O)O alquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -(alquileno(C1-C6))C(O)OH, -(alquileno(C1-C6))C(O)O alquilo(C1-C6), o -C(O)heterociclilo.
En determinadas realizaciones, R1 es H, alquilo(C1-C6), alcoxi(C1-C6), -C(O)alquilo(C1-C6), -S(O)2 alquilo(C1-C6), -S(O)2arilo, -C(O)O alquilo(C1-C6), -(alquileno(C1-C6))C(O)OH, o -(alquileno(C1-C6))C(O)Oalquilo(C1-C6).
En determinadas realizaciones, R1 es H o alquilo(C1-C6).
En determinadas realizaciones, R1 es alquilo (C1-C4).
En determinadas realizaciones, R1 es -C(O)heterociclilo que se selecciona del grupo que consiste en


En determinadas realizaciones, R1 es -CH2CO2H o -CH2C(O)O alquilo(Ci-C6).
En determinadas realizaciones, Z es -C(O)-.
En determinadas realizaciones, Z es -C(alquilo(Ci-C3))-.
En determinadas realizaciones, Z es -CH-.
En determinadas realizaciones, R2 es H, halo, alquilo(Ci-C6), alcoxi(Ci-C6), hidroxialquilo(Ci-C6), alcoxi(Ci-Ca)alquilo(Ci-Ca), -CO2H, -C(O)O alquilo(Ci-Ce), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(Ci-C6)), -C(O)O(heteroarilalquilo(Ci-Ca)), -C(O)NH2, -C(O)NH(alquilo(Ci-Ca)), -C(O)N(alquilo(Ci-Ca))2, -C(O)NH(arilalquilo(Ci-Ca)), -C(O)N(arilalquilo(Ci-Ca))2, -C(O)NH(aril), -C(O)N(aril)(alquilo(Ci-Ca)), -C(O)N(aril)2, -C(O)N alquil(Ci-Ca))(arilalquilo(Ci-Ca)), -C(O)N(aril)(arilalquilo(Ci-Ca)), -C(O)NH(haloalquilo(Ci-Ca)), -C(O)N(haloalquilo(Ci-Ca))2, arilalquilo(Ci-Ca), cicloalquil(C3-C7)alquilo(Ci-Ca)-, o -C(O)heterociclilo.
En determinadas realizaciones, R2 es H, halo, alquilo(Ci-Ca), alcoxi(Ci-Ca), hidroxialquilo(Ci-Ca), alcoxi(Ci-Ca)alquilo(Ci-Ca), -CO2H, -C(O)O alquilo(Ci-Ca), -C(O)NH2, -C(O)NH(alquilo(Ci-Ca)), arilalquilo(Ci-Ca), o cicloalquil(C3-C7)alquilo(Ci-Ca)-.
En determinadas realizaciones, R2 es arilalquilo(Ci-Ca) o cicloalquil(C3-C7)alquilo(Ci-Ca)-, opcionalmente sustituido en cualquier posición por -OH, halo, -NH2, alquilo(Ci-Ca), haloalquilo(Ci-Ca), o alcoxi(Ci-Ca).
En determinadas realizaciones, R2 es H, alquilo(Ci-Ca), o alcoxi(Ci-Ca)alquilo(Ci-Ca).
En determinadas realizaciones, R2 es H, alquilo(Ci-C4), alcoxi(Ci-C3)alquilo(Ci-C3), carboxi, alcoxicarbonilo(Ci-C3), carboximetilo, o alcoxicarbonilmetilo(Ci-C3).
En determinadas realizaciones, R2 es -C(O)heterociclilo que se selecciona del grupo que consiste en

En determinadas realizaciones, R2 es halo(i,4 -fenilen)metilo.
En determinadas realizaciones, R3 es H o alquilo(Ci-C3).
En determinadas realizaciones, R3 es H.
En determinadas realizaciones, R4 es H o alquilo(Ci-C3).
En determinadas realizaciones, R4 es H.
En determinadas realizaciones, R5 es H o alquilo(Ci-C3).
En determinadas realizaciones, R5 es H.
En algunas realizaciones, el compuesto está representado por la Fórmula (I'):
ia

En determinadas realizaciones del compuesto de fórmula (I):
W es bromo o cloro;
X es un enlace sencillo, -CH2-, o -C(O)-;
Y es un enlace sencillo, -CH-, -N-, -O-, o -S(O)2-;
si Y es un enlace sencillo, -O- o -S(O)2-, entonces R1 está ausente,
R1 es H, metilo, isobutilo, metoxi, acetilo, metoxicarbonilo, metanosulfonilo, p-toluenosulfonilo, metoxicarbonilmetilo, o carboximetilo;
Z es -CH-, -C(O)-, o -C(CH)-;
si Z es -C(O)-, entonces R2 está ausente,
R2 es H, metilo, etilo, isopropilo, isobutilo, -C(O)NH2, -C(O)NHMe, -CH2OH, -CH2OCH3, -CH2OCH2CH3, -C(CH3)2OH, -C(CH3)2OCH3, -CO2H, -CO2CH2CH3, -OCH3, -F, -CH2-(p-clorofenilo), o -CH2-ciclohexilo;
Cada uno de R3, R4 , y R5 es H; y
R6 es H o OH.
En determinadas realizaciones, la invención proporciona un compuesto representado por la fórmula (II),

donde:
W es halo, alquilo(C1-C3), alcoxi(C1-C3), o alquiltio(C1-C3);
X es un enlace sencillo, -CH2-, -CH2CH2-, -CH=CH-, -C(alquilo(C1-C3))2-, -C(O)-, -CH2O-, -CH2NH-, o -CH2N(alquilo(C1-C3))-, donde cuando X es -CH2O-, -CH2NH-, o -CH2N(alquilo(C1-C3))-, entonces el átomo de O o N está unido covalentemente con el Anillo A ;
El Anillo A representa un anillo arilo o heteroarilo opcionalmente sustituido;
R3 es H, alquilo(C1-C3), alcoxi(C1-C3)alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-C6), -NC, -CN, -C(S)NH2, -NHC(O)NH2, o -CeCH;
R4 es H, alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-C6), alcoxi(C1-C3)alquilo(C1-C3), o hidroxialquilo(C1-C4);
R5 es H, halo, -NO2, -CN, alquilo(C1-C6), haloalquilo(C1-C6), -NH2, -NH(alquilo(C1-C6)), -N(alquilo(C1-C6))2, -OH, alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), haloalcoxi(C1-C6), -SH, alquiltio(C1-C3)alquilo(C1-C3), -NC, -C(S)NH2, -NHC(O)NH2, o -CeCH; y
R6 es H, halo, -OH, -NH2, o -SH; o
R6 , tomado junto con el átomos de carbono que lo contiene, representa C(O)-;
donde:
cualquier aparición de alquilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo, o hidroxialquilo está sustituida opcionalmente con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2, -NH(alquilo(C1-C6)), -N(alquilo(C1-C6))2, -CN, -NO2, alquilo(C1-C6), haloalquilo(C1-C6), alcoxi(C1-C6), arilo, heteroarilo, arilalquilo(C1-C6), heteroarilalquilo(C1-C6), cicloalquilo(C3-C7), heterociclilo, -C(O)OH, -C(O)O alquilo(C1-C6), -C(O)NH2, -C(O)NH alquilo(C1-C6), y -C(O)N(alquilo(C1-C6))2;
o una sal, solvato o tautómero, estereoisómero o polimorfo farmacéuticamente aceptable de estos.
En determinadas realizaciones del compuesto de fórmula (II), W es fluoro, cloro, bromo, metilo o metoxi, preferentemente cloro.
En determinadas realizaciones del compuesto de fórmula(II), R6 es H.
En determinadas realizaciones, X es -CH2O-, -CH2NH-, o -CH2N(alquilo(Ci-C3))-, preferentemente -CH2O- o -CH2N(CH3)-.
En determinadas realizaciones, R3 es H.
En determinadas realizaciones, R4 es H.
En determinadas realizaciones, R5 es H.
En determinadas realizaciones, el Anillo A representa un anillo heteroarilo, tal como un anillo piridilo.
En determinadas realizaciones, el Anillo A representa un anillo fenilo opcionalmente sustituido. Por ejemplo, el Anillo A puede representar un anillo fenilo sustituido por una o más apariciones de halo, tales como cloro.
En determinadas realizaciones, la invención se refiere a un compuesto de cualquiera de las siguientes fórmulas estructurales:






Las sales, hidratos y solvatos de los compuestos de la invención son preferentemente sales, hidratos y solvatos farmacéuticamente aceptables. Los solvatos pueden contener una cantidad estequiométrica o no estequiométrica de uno o más disolventes, tales como agua, etanol o acetato de etilo, además de la molécula del compuesto de la invención. Los solvatos formados con agua se llaman hidratos.
Los compuestos descritos en el presente documento son útiles para tratar enfermedades inflamatorias, tales como inflamación eosinofílica esofágica, queratoconjuntivitis, conjuntivitis alérgica estacional, síndrome del ojo seco, o rinosinusitis crónica con o sin pólipos nasales. Los compuestos se pueden usar en el tratamiento de enfermedades causadas por agentes infecciosos, tales como hongos, gusanos o parásitos. Los compuestos se pueden usar en el tratamiento de enfermedad pulmonar obstructiva crónica (EPOC) o enfermedades autoinmunes pero no se limitan a enfermedad intestinal inflamatoria o artritis reumatoide.
Composiciones farmacéuticas de la invención
Otro aspecto de la invención proporciona una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de la invención (por ejemplo un compuesto de fórmula (I)) y un portador farmacéuticamente aceptable.
La naturaleza exacta del portador, o por ejemplo del excipiente o diluyente, dependerá del uso deseado para la composición y puede ser adecuada o aceptable para uso veterinario y/o adecuada o aceptable para uso humano. La composición puede incluir opcionalmente uno o más compuestos adicionales, que incluyen uno o más agentes terapéuticos adicionales.
Los compuestos de la invención se pueden combinar con otros agentes terapéuticos. El compuesto de la invención y otro agente terapéutico se pueden administrar simultánea o secuencialmente. Cuando los otros agentes terapéuticos se administrar simultáneamente, se pueden administrar en las mismas formulaciones o separadas pero se administrar sustancialmente al mismo tiempo. Los otros agentes terapéuticos se administran secuencialmente entre sí y con un compuesto de la invención, cuando la administración de los demás agentes terapéuticos y el compuesto de la invención está separada temporalmente. La separación en el tiempo entre la administración de estos compuestos puede ser cuestión de minutos o puede ser de más tiempo.
Algunos ejemplos de otros agentes terapéuticos que se pueden administrar con los compuestos de la invención incluyen esteroides, estabilizadores de membrana, inhibidores de 5LO, inhibidores del receptor y la síntesis de leucotrieno, inhibidores de la conmutación del isotipo IgE o la síntesis de IgE, conmutación del isotipo IgG o síntesis de IgG, p-agonistas, inhibidores de triptasa, aspirina, inhibidores de la COX, metotrexato, fármacos anti-TNF, rituxina, inhibidores de PD4, inhibidores de p38, inhibidores de PDE4 y antihistaminas.
De esta manera, otro aspecto de la invención proporciona una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de la invención y un segundo agente terapéutico seleccionado del grupo que consiste en esteroides, estabilizadores de membrana, inhibidores de 5LO, inhibidores del receptor y la síntesis de leucotrieno, inhibidores de la conmutación del isotipo IgE o la síntesis de IgE, conmutación del isotipo IgG o síntesis de IgG, p-agonistas, inhibidores de triptasa, ácido acetilsalicílico, inhibidores de la COX, metotrexato, fármacos anti-TNF, rituxina, y otros agentes dirigidos a las células B, agentes dirigidos a TNF, inhibidores de PD4, inhibidores de p38, inhibidores de PDE4 y antihistaminas.
Tal como se menciona anteriormente una “cantidad eficaz” se refiere a cualquier cantidad suficiente como para alcanzar un efecto biológico deseado. Combinado con las enseñanzas proporcionadas en el presente documento, eligiendo entre los diversos compuestos activos y factores de ponderación tales como potencia, biodisponibilidad relativa, peso corporal del paciente, gravedad de efectos adversos y modo de administración preferido, se puede planificar un régimen de tratamiento profiláctico o terapéutico que no provoque toxicidad no deseada sustancial y aun sea eficaz para tratar al sujeto particular. La cantidad eficaz para cualquier aplicación puede variar según dichos factores como la enfermedad o condición tratada, el compuesto de la invención particular administrado, el tamaño del sujeto o la gravedad de la enfermedad o afección. Un experto en la materia puede determinar empíricamente la cantidad eficaz de un compuesto particular de la invención y/u otro agente terapéutico sin la necesidad de experimentar de forma indebida. Generalmente, se prefiere que se utilice una dosis máxima, es decir, la mayor dosis segura de acuerdo con la opinión médica bien fundada. Múltiples dosis por día pueden contemplarse para alcanzar niveles sistémicos apropiados de compuestos. Niveles sistémicos apropiados se pueden determinar por ejemplo, por la medición del nivel en plasma del fármaco pico o sostenido del paciente. “Dosis” y “dosificación” se usan de manera intercambiable en el presente documento.
Generalmente, las dosis orales diarias de compuestos activos serán para sujeto humanos, de aproximadamente 0.0001 miligramos/kg por día, 0,001 miligramos/kg por día, o 0,01 miligramos/kg por día a aproximadamente 100 miligramos/kg por día o 1000 miligramos/kg por día. Se espera que las dosis orales en el intervalo de 0,5 a 50 miligramos/kg, en una o más administraciones por día, proporcione los resultados deseados. La dosificación se puede ajustar de forma adecuada para alcanzar los niveles de fármaco deseados suficientes para alcanzar o mantener un efecto terapéutico deseado, local o sistémico, dependiendo del modo de administración. Por ejemplo, se espera que la administración intravenosa sea de un orden a varios órdenes de dosis menor de magnitud por día. En el caso de que la respuesta en un sujeto sea insuficiente con dichas dosis, se pueden usar dosis incluso más altas (o dosis mayores eficaces por una vía de administración más localizada diferente) en la medida permitida por la tolerancia del paciente. Múltiples dosis por día pueden contemplarse para alcanzar niveles sistémicos apropiados de compuestos. Los compuestos se pueden administrar una vez por semana, varias veces por semana (por ejemplo día por medio), una vez al día o múltiples veces al día, dependiendo de entre otras cosas el modo de administración, siendo la indicación específica que se trata y la opinión del médico tratante.
En una realización, la administración intravenosa de un compuesto de la invención puede ser típicamente de 0,1 mg/kg/día a 20 mg/kg/día.
La determinación de una dosificación eficaz de un compuesto para un uso y modo de administración particulares se
encuentra dentro de las capacidades de los expertos en la técnica. Dosificaciones eficaces se pueden estimar inicialmente a partir de ensayos de metabolismo y actividad in vitro. Por ejemplo una dosificación inicial del compuesto para su uso en animales se puede formular para alcanzar una concentración en suero o sangre en circulación del compuesto activo metabolito que es de una IC50 del compuesto particular o mayor según se mide por ensayo in vitro. Calcular las dosificaciones para alcanzar dichas concentraciones en suero o sangre en circulación tomando en cuenta la biodisponibilidad del compuesto particular mediante la vía de administración deseada se encuentra dentro de las capacidades de los expertos en la técnica. Las dosificaciones iniciales del compuesto también se pueden estimar a partir de datos in vivo tales como modelos animales. Para cualquier compuesto que se describió en el presente documento, la cantidad terapéuticamente eficaz se puede determinar inicialmente a partir de modelos animales. Una dosis terapéuticamente eficaz también se puede determinar a partir de datos humanos para compuestos de la invención que se analizaron en humanos y para compuestos que se conocen por exhibir actividades farmacológicas similares, tales como otros agentes activos relacionados. Pueden ser necesarias dosis mayores para administración parenteral. La dosis aplicada se puede ajustar en función de la potencia y la biodisponibilidad relativa del compuesto administrado. El ajuste de la dosis para lograr la eficacia máxima en función de los métodos que se describieron anteriormente y otros métodos que se conocen en la técnica se encuentra dentro de las capacidades del experto en la materia.
Las formulaciones de la invención se pueden administrar en soluciones farmacéuticamente aceptables que pueden contener de forma rutinaria concentraciones farmacéuticamente aceptables de sal, agentes amortiguadores, conservantes, portadores compatibles, adyuvantes y opcionalmente otros ingredientes terapéuticos.
Las composiciones farmacéuticas comprenden el compuesto de la invención que puede ser elaborado mediante procedimientos de mezcla convencional, disolución, granulación, fabricación de grageas, levigado, emulsión, encapsulación, atrapamiento o liofilización. Las composiciones se pueden formular de manera convencional utilizando uno o más portadores, diluyentes, excipientes o auxiliares fisiológicamente aceptables que facilitan el procesamiento de los compuestos en preparaciones que se pueden utilizar farmacéuticamente.
Para su uso en terapia, una cantidad eficaz del compuesto de la invención se puede administrar a un sujeto por cualquier modo que administra el compuesto de la invención a la superficie deseada. Administrar la composición farmacéutica de la presente invención puede lograrse mediante cualquier medio conocido para el experto. Las vías de administración incluyen pero no se limita a oral, bucal, nasal, rectal, vaginal, ocular, tópica, intravenosa, intramuscular, intraperitoneal, subcutánea, transdérmica, intratecal, inyección directa (por ejemplo, en un absceso), mucosa, inhalación e insuflación.
Para administración oral, los compuestos (es decir compuestos dela invención y otros agentes terapéuticos) se pueden formular fácilmente al combinar el o los compuestos activos con portadores farmacéuticamente aceptables conocidos en la técnica. Tales vehículos permiten que los compuestos de la invención se formulen como comprimidos, pastillas, grageas, píldoras, cápsulas, líquidos, geles, jarabes, lechadas, suspensiones y similares, para ingestión oral por un sujeto a ser tratado. Las preparaciones farmacéuticas para uso oral se pueden obtener mediante un excipiente sólido, opcionalmente la mezcla resultante es molida y se procesa la mezcla de gránulos, después de la adición de auxiliares adecuados, si se desea, para obtener comprimidos o centros de grageas. Excipientes adecuados son en particular agentes aglutinantes, rellenos, lubricantes, disgregantes y agentes humectantes. Los rellenos adecuados incluyen azúcares que incluyen lactosa, sacarosa, manitol o sorbitol; preparaciones de celulosa tales como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de papa, gelatina, goma tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona (PVP). Si se desea, se pueden añadir agentes disgregantes, tales como polivinilpirrolidona reticulada, agar o ácido algínico o una sal de estos, tal como alginato de sodio. Opcionalmente las formulaciones orales también se pueden formular en solución salina o amortiguadores, por ejemplo, EDTA para neutralizar condiciones de ácido interno o se pueden administrar sin portadores.
También se contemplan específicamente formas de dosificación oral del componente o los componentes que anteceden. El componente o los componentes pueden modificarse químicamente de manera que el suministro oral del derivado sea eficaz. Generalmente, la modificación química contemplada es la unión de al menos un resto a la molécula del componente en sí, donde dicho resto permite (a) la inhibición de la hidrólisis ácida y (b) la absorción en el torrente sanguíneo del estómago o el intestino. También es deseable el aumento en la estabilidad general del componente o los componentes y el aumento en el tiempo de circulación en el cuerpo. Ejemplos de estos restos incluyen: polietilenglicol, copolímeros de etilenglicol y propilenglicol, carboximetilcelulosa, dextrano, alcohol polivinílico, polivinil pirrolidona y poliprolina. Abuchowski y Davis, “Soluble Polymer-Enzyme Adducts”, en: Enzymes as Drugs, Hocenberg and Roberts, eds., Wiley-Interscience, Nueva York, N.Y., pp. 367-383 (1981); Newmark et al., J Appl Biochem 4:185-9 (1982). Otros polímeros que se podrían usar son poli-1,3-dioxolano y poli-1,3,6-tioxocano. Como se indicó anteriormente, los restos de polietilenglicol se prefieren para el uso farmacéutico.
Para el componente (o derivado), la ubicación de la liberación puede ser el estómago, el intestino delgado (el duodeno, el yeyuno o el íleo) o el intestino grueso. Un experto en la materia tiene formulaciones disponibles que no se disolverán en el estómago pero igual liberará el material en el duodeno o en otra parte del intestino. Preferentemente, la liberación evitará los efectos perjudiciales del ambiente estomacal, ya sea por protección del compuesto de la invención (o
derivado) o por la liberación del material biológicamente activo más allá del ambiente del estómago, como en el intestino.
Para asegurar una resistencia gástrica completa, un recubrimiento impermeable al menos a pH 5,0 es esencial. Ejemplos de los ingredientes inertes más comunes que se usan como recubrimientos entéricos son acetato trimelitato (Ca T), hidroxipropilmetilcelulosa ftalato (HPMCP), Hp MCP 50, HPMCP 55, polivinil acetato ftalato (PVAP), Eudragit L30D, Aquateric, celulosa acetato ftalato (CAP), Eudragit L, Eudragit S y laca. Estos recubrimientos pueden usarse como películas mezcladas.
Un recubrimiento o mezcla de recubrimientos también puede usarse sobre comprimidos, que no se desean para la protección contra el estómago. Esto puede incluir recubrimientos de azúcar o recubrimientos que hacen que sea más fácil tragar el comprimido. Las cápsulas pueden consistir en una cubierta dura (como gelatina) para el suministro de un compuesto terapéutico seco (por ejemplo, polvo); para las formas líquidas se puede usar una cubierta de gelatina blanda. El material de cubierta de las obleas podría ser almidón grueso u otro papel comestible. Para las píldoras, grageas, comprimidos moldeados o triturados, se pueden usar técnicas de concentración en húmedo.
El compuesto terapéutico puede incluirse en la formulación como múltiples partículas finas en la forma de gránulos o sedimentos de un tamaño de partículas de aproximadamente 1 mm. La formulación del material para la administración de cápsulas también puede ser como un polvo, tapones ligeramente comprimidos o incluso como comprimidos. El compuesto terapéutico se podría preparar mediante compresión.
Se pueden incluir agentes colorantes y saborizantes. Por ejemplo, el compuesto de la invención (o el derivado) se puede formular (como mediante encapsulación en liposomas o microesferas) y después puede estar contenido adicionalmente en un producto comestible, como una bebida refrigerada que contenga agentes colorantes y saborizantes.
Se puede diluir o aumentar el volumen del compuesto terapéutico con un material inerte. Tales diluyentes podrían incluir carbohidratos, especialmente, manitol, a-lactosa, lactosa anhidra, celulosa, sacarosa, dextranos modificados y almidón. Algunas sales inorgánicas también pueden ser útiles como rellenos, incluyendo trifosfato de calcio, carbonato de magnesio y cloruro de sodio. Algunos diluyentes comercialmente disponibles son Fast-Flo, Emdex, STA-Rx 1500, Emcompress y Avicell.
Se pueden incluir disgregantes en la formulación del producto terapéutico en una forma de dosificación sólida. Los materiales utilizados como disgregantes incluyen, pero no se limita a, almidón, incluyendo el disgregante comercial con base en almidón, Explotab. Se pueden usar glicolato de almidón sódico, Amberlite, carboximetilcelulosa de sodio, ultramilopectina, alginato de sodio, gelatina, cáscara de naranja, carboximetilcelulosa ácida, esponja natural y bentonita. Otra forma de los disgregantes son las resinas de intercambio catiónico insolubles. Las gomas en polvo se pueden utilizar como disgregantes y como aglutinantes y pueden incluir gomas en polvo como agar, Karaya o tragacanto. El ácido algínico y su sal de sodio también son útiles como disgregantes.
Los aglutinantes se pueden usar para mantener el agente terapéutico junto para formar un comprimido duro e incluyen materiales de productos naturales como acacia, tragacanto, almidón y gelatina. Otros incluyen metilcelulosa (MC), etilcelulosa (EC) y carboximetilcelulosa (CMC). La polivinilpirrolidona (PVP) y la hidroxipropilmetilcelulosa (HPMC) se podrían usar en soluciones alcohólicas para granular el compuesto terapéutico.
Se puede incluir un agente antifricción en la formulación del compuesto terapéutico para evitar que se pegue durante el proceso de formulación. Los lubricantes se pueden usar como una capa entre el compuesto terapéutico y la pared de tinte y estos pueden incluir, pero no se limita a, ácido esteárico, incluyendo sus sales de magnesio y calcio, politetrafluoroetileno (PTFE), parafina líquida, ceras y aceites vegetales. También se pueden usar lubricantes solubles como laurilsulfato de sodio, laurilsulfato de magnesio, polietilenglicol de diversos pesos moleculares, Carbowax 4000 y 6000.
Se pueden añadir emolientes que puedan mejorar las propiedades de flujo del fármaco durante la formulación y asistir la reorganización durante la compresión. Los emolientes pueden incluir almidón, talco, sílice pirogénica y silicoaluminato hidratado.
Para ayudar a la disolución del compuesto terapéutico en el ambiente acuoso, se puede añadir un tensioactivo como un agente humectante. Los tensioactivos pueden incluir detergentes aniónicos como laurilsulfato de sodio, dioctilsulfosuccinato de sodio y dioctilsulfonato de sodio. Se pueden usar detergentes catiónicos y pueden incluir cloruro de benzalconio y cloruro de bencetonio. Los posibles detergentes no iónicos que se podrían incluir en la formulación como tensioactivos son lauromacrogol 400, estearato de polioxil 40, aceite de ricino hidrogenado polioxietilenado 10, 50 y 60, monoestearato de glicerol, polisorbato 40, 60, 65 y 80, éster del ácido graso de sacarosa, metilcelulosa y carboximetilcelulosa. Estos tensioactivos podrían estar presentes en la formulación del compuesto de la invención o el derivado solos o como una mezcla en diferentes proporciones.
Las preparaciones farmacéuticas que se pueden usar oralmente incluyen cápsulas duras hechas de gelatina, así como
cápsulas suaves, selladas hechas de gelatina y un plastificante tal como glicerol o sorbitol. Las cápsulas duras pueden contener los ingredientes activos mezclados con un relleno como lactosa, aglutinantes como almidones y/o lubricantes como talco o estearato de magnesio y, opcionalmente, estabilizadores. En las cápsulas blandas, los compuestos activos se pueden disolver o suspender en líquidos adecuados como aceites grasos, parafina líquida o polietilenglicoles líquidos. Además, se pueden añadir estabilizadores. También se pueden usar microesferas formuladas para la administración oral. Estas microesferas se han definido bien en la técnica. Todas las formulaciones destinadas a la administración oral se deben encontrar en dosificaciones adecuadas para tal administración.
Las preparaciones líquidas para administración oral pueden adoptar la forma de, por ejemplo, elíxires, soluciones, jarabes o suspensiones o pueden presentarse como un producto seco para su constitución con agua u otro vehículo adecuado antes de su uso. Tales preparaciones líquidas se pueden preparar mediante medios convencionales con aditivos farmacéuticamente aceptables tales como agentes de suspensión (por ejemplo, jarabe de sorbitol, derivados de celulosa o grasas comestibles hidrogenadas); agentes emulsionantes (por ejemplo, lecitina o acacia); vehículos no acuosos (por ejemplo, aceite de almendras, ésteres aceitosos, alcohol etílico o aceites vegetales fraccionados); y conservantes (por ejemplo, metil o propil-p-hidroxibenzoatos o ácido sórbico). Las preparaciones también pueden contener sales amortiguadoras, conservantes, agentes saborizantes, colorantes y endulzantes según corresponda.
Las composiciones farmacéuticas, si se desea, pueden presentarse en un envase o un dispositivo dispensador que puede contener una o más formas de dosificación unitaria que contienen el o los compuestos. Por ejemplo, el envase puede comprender una lámina de plástico o metálica, tal como un blíster. El envase o dispositivo dispensador puede estar acompañado por instrucciones para su administración.
Para la administración bucal, las composiciones pueden adoptar la forma de comprimidos o pastillas para chupar formulados de manera convencional.
Para la administración tópica, el compuesto puede formularse como soluciones, geles, ungüentos, cremas, suspensiones, etc. tal como se conoce en la técnica. Las formulaciones sistémicas incluyen las diseñadas para su administración mediante inyección, por ejemplo, inyección subcutánea, intravenosa, intramuscular, intratecal o intraperitoneal, así como también aquellas diseñadas para su administración transdérmica, transmucosa, oral o pulmonar.
Para la administración por inhalación, los compuestos que serán utilizados conforme a la presente invención se pueden administrar convenientemente en forma de una presentación en aerosol pulverizador a partir de paquetes presurizados o un nebulizador, con el uso de un propulsor adecuado, por ejemplo, diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas adecuado. En el caso de un aerosol presurizado, la unidad de dosificación puede determinarse mediante la provisión de una válvula para administrar una cantidad medida. Las cápsulas y los cartuchos de por ejemplo, gelatina para usarse en un inhalador o insuflador pueden formularse de forma que contengan una mezcla del compuesto y una base de polvo adecuada tal como lactosa o almidón.
En el presente documento también se contempla la administración pulmonar de los compuestos de la invención (o los derivados de los mismos). El compuesto de la invención (o el derivado) se suministra a los pulmones de un mamífero mientras que inhala y atraviesa el recubrimiento epitelial pulmonar hasta el torrente sanguíneo. Otros informes de moléculas inhaladas incluyen Adjei et al., Pharm Res 7:565-569 (1990); Adjei et al., Int J Pharmaceutics 63:135-144 (1990) (leuprolida acetato); Braquet et al., J Cardiovasc Pharmacol 13(supl. 5):143-146 (1989) (endotelina-1); Hubbard et al., Annal Int Med 3:206-212 (1989) (a1-antitripsina); Smith et al., 1989, J Clin Invest 84:1145-1146 (a-1-proteinasa); Oswein et al., 1990, "Aerosolization of Proteins", Proceedings of Symposium on Respiratory Drug Delivery II, Keystone, Colorado, marzo, (hormona de crecimiento humana recombinante); Debs et al., 1988, J Immunol 140:3482-3488 (interferon-gamma y factor de necrosis tumoral alfa) y Platz et al., patente estadounidense n.° 5.284.656 (factor de estimulación de colonias de granulocitos). Un método y una composición para el suministro pulmonar de fármacos para lograr un efecto sistémico se describe en la patente estadounidense n.° 5.451.569, emitida el 19 de setiembre de 1995 a Wong et al.
Se contempla para el uso en la práctica de la presente invención una amplia variedad de dispositivos mecánicos diseñados para el suministro pulmonar de productos terapéuticos, incluyendo, pero no se limita a, nebulizadores, inhaladores con dosis medida e inhaladores de polvo, los cuales son conocidos por los expertos en la materia.
Algunos ejemplos específicos de dispositivos comercialmente disponibles adecuados para la práctica de la presente invención son el nebulizador Ultravent, fabricado por Mallinckrodt, Inc., St. Louis, Mo.; el nebulizador Acorn II, fabricado por Marquest Medical Products, Englewood, Colo.; el inhalador de dosis medida Ventolin, fabricado por Glaxo Inc., Research Triangle Park, Carolina del Norte o el inhalador de polvo Spinhaler, fabricado por Fisons Corp., Bedford, Mass. y el inhalador de niebla fina Respimat, fabricado por Boehringer Ingelheim, Alemania. Otros dispositivos inhaladores manuales o accionados por un ser humano también se pueden aplicar.
Todos esos dispositivos necesitan el uso de formulaciones adecuadas para la dispersión del compuesto de la invención (o el derivado). Típicamente, cada formulación es específica para el tipo de dispositivo utilizado y puede implicar el uso de un material propulsor apropiado, además de diluyentes, adyuvantes y/o portadores habituales útiles
en el tratamiento. Asimismo, también está contemplado el uso de liposomas, microcápsulas o microesferas, complejos de inclusión u otros tipos de portadores. El compuesto químicamente modificado de la invención también se puede preparar en diferentes formulaciones dependiendo del tipo de modificación química o el tipo de dispositivo empleado.
Las formulaciones adecuadas para uso con un nebulizador, ya sea de tipo a chorro, ultrasónico o de niebla suave, comprenderán típicamente el compuesto de la invención (o el derivado) disuelto en agua a una concentración de aproximadamente 0.1 a 25 mg de compuesto biológicamente activo de la invención por ml de solución. La formulación también puede incluir un amortiguador y un azúcar simple (por ejemplo, para la estabilización del compuesto de la invención y regulación de la presión osmótica). La formulación de nebulizador también puede contener un tensioactivo para reducir o prevenir la agregación inducida por la superficie del compuesto de la invención ocasionada por atomización de la solución en la formación del aerosol.
Las formulaciones para uso con el dispositivo inhalador de dosis medida generalmente comprenden un polvo finamente dividido que contiene el compuesto de la invención (o el derivado) suspendido en un propulsor con la ayuda de un tensioactivo. El propulsor puede ser cualquier material convencional utilizado a tales efectos, como clorofluorocarbono, un hidroclorofluorocarbono, un hidrofluorocarbono o un hidrocarbono, incluyendo triclorofluorometano, diclorodifluorometano, diclorotetrafluoroetanol y 1,1,1,2-tetrafluoroetano o combinaciones de estos. Los tensioactivos adecuados incluyen trioleato de sorbitán y lecitina de soja. También puede ser útil el ácido oleico como tensioactivo.
Las formulaciones para dispensar desde un dispositivo inhalador de polvo comprenderán un polvo seco finamente dividido que contiene el compuesto de la invención (o el derivado) y también pueden incluir un agente de volumen, como lactosa, sorbitol, sacarosa o manitol, en cantidades que facilitan la dispersión del polvo desde el dispositivo, por ejemplo, de 50 a 90 % en peso de la formulación. El compuesto de la invención (o el derivad) debería preparase, para lograr una mayor ventaja, en forma de partículas con un tamaño de partícula promedio menor a los 10 micrómetros (|im), más preferiblemente, de 0.5 a 5 |im, para un suministro más eficaz al pulmón distal.
También se contempla el suministro nasal de una composición farmacéutica de la presente invención. El suministro nasal permite el pasaje de una composición farmacéutica de la presente invención al torrente sanguíneo directamente después de administrar el producto terapéutico en la nariz, sin la necesidad de depositar el producto en el pulmón. Las formulaciones para el suministro nasal incluyen las que tienen dextrano o ciclodextrano.
Para la administración nasal, un dispositivo útil es una botella rígida y pequeña unida a un pulverizador de dosis medida. En una realización, la dosis medida se suministra mediante extracción de la composición farmacéutica de la solución de la presente invención e introducción en una cámara de volumen definido, que tiene una abertura dimensionada para suministrar una formulación en aerosol mediante la formación de un rocío cuando se comprime un líquido en la cámara. La cámara se comprime para administrar la composición farmacéutica de la presente invención. En una realización específica, la cámara es una disposición de pistones. Estos dispositivos se encuentran comercialmente disponibles.
De manera alternativa, se usa una botella comprimible de plástico con una abertura o brecha dimensionada para suministrar una formulación en aerosol mediante la formación de un rocío al apretarse. La abertura se encuentra habitualmente en la parte superior de la botella y la parte superior está generalmente ahusada para ajustarse parcialmente a los pasajes nasales para una administración eficaz de la formulación en aerosol. Preferentemente, el inhalador nasal proporcionará una cantidad medida de la formulación en aerosol para la administración de una dosis medida del fármaco.
Los compuestos, cuando se desea su suministro sistémico, pueden formularse para administración parenteral por inyección, por ejemplo, mediante inyección de bolo o infusión continua. Las formulaciones para inyección se pueden presentar en formas de dosificación unitaria, por ejemplo, en ampollas o en recipientes de múltiples dosis, con un conservante agregado. Las composiciones pueden tomar formas tales como suspensiones, soluciones o emulsiones en vehículos oleaginosos o acuosos y pueden contener agentes de formulación tales como agentes de suspensión, estabilización y/o dispersión.
Las formulaciones farmacéuticas para administración parenteral incluyen soluciones acuosas de los compuestos activos en forma soluble en agua. De manera adicional, las suspensiones de los compuestos activos se pueden preparar como suspensiones de inyección oleosas apropiadas. Los disolventes o vehículos lipofílicos adecuados incluyen aceites grasos, tales como aceite de sésamo o ésteres de ácido graso sintético, tales como oleato de etilo o triglicéridos o liposomas. Las suspensiones de inyección acuosas pueden contener sustancias que aumentan la viscosidad de la suspensión, tal como carboximetilcelulosa sódica, sorbitol o dextrano. Opcionalmente, la suspensión también puede contener estabilizadores o agentes adecuados que aumenten la solubilidad de los compuestos para permitir la preparación de soluciones altamente concentradas.
De manera alternativa, el ingrediente activo se puede encontrar en forma de polvo para su constitución con un vehículo adecuado, por ejemplo, agua estéril libre de pirógenos, amortiguador, solución de dextrosa, antes de su uso. Con este fin, el compuesto activo puede secarse por cualquier técnica conocida tal como liofilización y reconstituirse antes de
su uso.
Los compuestos también se pueden formular en composiciones rectales o vaginales tales como supositorios o enemas de retención, por ejemplo, que contienen bases de supositorios convencionales tales como manteca de cacao u otros glicéridos.
En el caso de la administración transmucosal, se utilizan en la formulación penetrantes adecuados para permear la barrera. Estos penetrantes se conocen en la técnica.
Para la administración ocular, el o los compuestos pueden formularse como una solución, suspensión, etc. adecuada para la administración en el ojo. En la técnica se conoce una variedad de vehículos adecuados para administrar compuestos en el ojo.
Además de las formulaciones descritas anteriormente, para el suministro prolongado, los compuestos pueden formularse también como una preparación de depósito para la administración, por ejemplo, por implante o inyección intramuscular. T ales formulaciones de acción prolongada se pueden formular con materiales poliméricos o hidrofóbicos adecuados (por ejemplo, como una emulsión en un aceite aceptable) o resinas de intercambio iónico o como derivados de solubilidad limitada, por ejemplo, como una sal de solubilidad limitada. De manera alternativa, se pueden usar sistemas de suministro transdérmico fabricados como un parche o disco adhesivo que liberan lentamente el compuesto para absorción percutánea. Con este fin, los potenciadores de la permeación pueden usarse para facilitar la penetración transdérmica del compuesto.
Las composiciones farmacéuticas también pueden comprender portadores o excipientes de fase sólida o gel adecuados. Ejemplos de dichos portadores o excipientes incluyen pero no se limita a, carbonato de calcio, fosfato de calcio, varios azúcares, almidones, derivados de celulosa, gelatina y polímeros tales como polietilenglicoles.
Las formas de preparación farmacéutica líquidas o sólidas adecuadas son, por ejemplo, soluciones acuosas o salinas para inhalación, microencapsuladas, formando cocleatos, recubiertas sobre partículas microscópicas de oro, contenidas en liposomas, nebulizadas, aerosoles, sedimentos para implante en la piel o secadas sobre un objeto afilado que se debe raspar sobre la piel. Las composiciones farmacéuticas también incluyen gránulos, polvos, comprimidos, comprimidos recubiertos, (micro)cápsulas, supositorios, jarabes, emulsiones, suspensiones, cremas, gotitas o preparaciones con liberación prolongada de compuestos activos, en cuya preparación se usan habitualmente excipientes y aditivos y/o auxiliares como disgregantes, aglutinantes, agentes de recubrimiento, agentes de volumen, lubricantes, saborizantes, endulzantes o solubilizantes como se describe anteriormente. Las composiciones farmacéuticas son adecuadas para su uso en diversos sistemas de administración de fármacos. Se puede encontrar una breve reseña de los métodos para administración de fármacos en Langer R, Science 249:1527-33 (1990).
Los compuestos de la invención y opcionalmente otros compuestos terapéuticos pueden administrarse en sí mismos (puros) o en la forma de una sal farmacéuticamente aceptable. Cuando se usan en medicina, las sales deberían ser farmacéuticamente aceptables, pero las sales farmacéuticamente no aceptables pueden usarse para preparar sales farmacéuticamente aceptables de estas. Estas sales incluyen, pero no se limita a, las preparadas a partir de los siguientes ácidos: clorhídrico, bromhídrico, sulfúrico, nítrico, fosfórico, maleico, acético, salicílico, p-toluenosulfónico, tartárico, cítrico, metanosulfónico, fórmico, malónico, succínico, naftaleno-2-sulfónico y bencenosulfónico. Asimismo, estas sales pueden prepararse como sales de metales alcalinos o alcalinotérreas, tales como sales de sodio, potasio o calcio del grupo de ácido carboxílico. Típicamente, estas sales son más solubles en soluciones acuosas que las bases y ácidos libres correspondientes pero también se pueden formar sales con menor solubilidad que las bases y ácidos libes correspondientes.
Los compuestos pueden formularse alternativamente en la composición farmacéutica en sí misma o en la forma de un hidrato, solvato o N-óxido.
Los agentes amortiguadores adecuados incluyen: ácido acético y una sal (1-2 % p/v); ácido cítrico y una sal (1-3 % p/v); ácido bórico y una sal (0,5-2,5 % p/v) y ácido fosfórico y una sal (0,8-2 % p/v). Los conservantes adecuados incluyen cloruro de benzalconio (0,003-0,03 % p/v); clorobutanol (0,3-0,9 % p/v); parabenos (0,01-0,25 % p/v) y timerosal (0,004-0,02 % p/v).
Las composiciones farmacéuticas de la invención contienen una cantidad eficaz de un compuesto de la invención y opcionalmente agentes terapéuticos incluidos en un portador farmacéuticamente aceptable. El término “portador farmacéuticamente aceptable” se refiere a uno o más rellenos sólidos o líquidos compatibles, diluyentes o sustancias encapsulantes que son adecuadas para la administración a un ser humano u otro animal vertebrado. El término “portador” denota un ingrediente orgánico o inorgánico, natural o sintético, con el que se combina el ingrediente activo para facilitar la aplicación. Los componentes de las composiciones farmacéuticas también son capaces de mezclarse con los compuestos de la presente invención, y unos con otros, en una forma tal que no haya interacción que perjudicaría sustancialmente la eficacia farmacéutica deseada.
El o los agentes terapéuticos, incluyendo de manera específica pero sin limitarse a, el compuesto de la invención,
pueden proporcionarse en partículas. Las partículas como se usan en el presente documento se refieren a nanopartículas o micropartículas (o en algunos casos partículas más grandes) que pueden consistir en su totalidad o en parte en el compuesto de la invención o el o los otros agentes terapéuticos como se describen en el presente documento. Las partículas pueden contener el o los agentes terapéuticos en un núcleo rodeado de un recubrimiento que incluye, pero no se limita a, un recubrimiento entérico. El o los agentes terapéuticos también pueden estar dispersados a través de las partículas. El o los agentes terapéuticos también pueden ser absorbidos por las partículas. Las partículas pueden tener una cinética de liberación de cualquier orden, incluyendo liberación de orden cero, liberación de primer orden, liberación de segundo orden, liberación retrasada, liberación sostenida, liberación inmediata y cualquier combinación de estas, etc. La partícula puede incluir, además del o los agentes terapéuticos, cualquiera de los materiales usados rutinariamente en la técnica de farmacia y medicina incluyendo, pero no se limita a, material erosionable, no erosionable, biodegradable o no biodegradable o combinaciones de estos. Las partículas pueden ser microcápsulas que contienen el compuesto de la invención en una solución o en un estado semisólido. Las partículas pueden tener casi cualquier forma.
Se pueden usar tanto materiales poliméricos no biodegradables como biodegradables en la fabricación de partículas para suministrar el o los agentes terapéuticos. Estos polímeros pueden ser polímeros naturales o sintéticos. El polímero se selecciona en función del período de tiempo en el que se desea la liberación. Los polímeros bioadhesivos de interés particular incluyen hidrogeles bioerosionables descritos en Sawhney H S et al. (1993) Macromolecules 26:581-7, cuyas instrucciones se incorporan en el presente documento. Estos incluyen ácidos polihialurónicos, caseína, gelatina, glutina, polianhídridos, ácido poliacrílico, alginato, chitosano, poli(metil metacrilatos), poli(etil metacrilatos), poli(butilmetacrilato), poli(isobutil metacrilato), poli(hexilmetacrilato), poli(isodecil metacrilato), poli(lauril metacrilato), poli(fenil metacrilato), poli(metil acrilato), poli(isopropil acrilato), poli(isobutil acrilato) y poli(octadecil acrilato).
El o los agentes terapéuticos pueden estar contenidos en sistemas de liberación controlada. La expresión “liberación controlada” se refiere a cualquier formulación que contenga fármacos en la cual la forma y el perfil de liberación del fármaco de la formulación están controlados. Esto se refiere a formulaciones de liberación inmediata, así como no inmediata, donde las formulaciones de liberación no inmediata incluyen, pero no se limita a, formulaciones de liberación sostenida y liberación retardada. La expresión “liberación sostenida” (también denominada “ liberación extendida”) se utiliza en su sentido convencional para referirse a una formulación de fármaco que proporciona la liberación gradual de un fármaco en un período de tiempo prolongado, y que preferentemente, aunque no necesariamente, tiene como resultado niveles de sangre sustancialmente constantes de un fármaco durante un período de tiempo prolongado. La expresión “ liberación retardada” se utiliza en su sentido convencional para referirse a una formulación de fármaco en la que hay un retraso en el tiempo entre la administración de la formulación y la liberación del fármaco de esta. La “ liberación retardada” puede implicar o no la liberación gradual del fármaco durante un período de tiempo extendido y, por lo tanto, puede ser o no “liberación sostenida”.
El uso de un implante de liberación sostenida a largo plazo puede ser particularmente adecuado para el tratamiento de afecciones crónicas. Liberación “de largo plazo", como se usa en el presente documento, significa que el implante se construye y dispone para suministrar niveles terapéuticos del ingrediente activo por al menos 7 días y preferentemente 30-60 días. Los implantes de liberación sostenida a largo plazo son conocidos por los expertos en la técnica e incluyen algunos de los sistemas de liberación descritos anteriormente.
Un experto en la materia relevante comprenderá que otras modificaciones y adaptaciones adecuadas a las composiciones y métodos descritos en el presente documento son fácilmente aparentes a partir de la descripción de la invención contenida en el presente documento en vista de la información conocida por el experto y que estos pueden llevarse a cabo sin alejarse del alcance de la invención o cualquier realización de esta.
Métodos y usos
Como se usa en el presente documento, los compuestos de la invención son útiles para inhibir la actividad enzimática y biológica de la quitinasa de mamífero ácida (“AMCasa”) y quitotriosidasa 1 (“CHIT1”).
Por consiguiente, se desvelan métodos para inhibir la quitinasa de mamífero ácida en una célula o un tejido, que comprende poner en contacto una célula o un tejido con al menos un compuesto de acuerdo con la invención o con una composición farmacéutica de acuerdo con la invención.
De manera similar, se desvelan métodos para inhibir la quitotriosidasa 1 en una célula o un tejido, que comprende poner en contacto una célula o un tejido con al menos un compuesto de acuerdo con la invención o con una composición farmacéutica de acuerdo con la invención.
También se desvelan métodos para el tratamiento o la prevención de una enfermedad, trastorno o afección asociada con la actividad o expresión aberrante de la quitinasa de mamífero ácida, que comprenden administrarle a un sujeto que lo necesita una cantidad terapéuticamente eficaz de al menos un compuesto de acuerdo con la invención o con una composición farmacéutica de acuerdo con la invención.
De manera similar, también se desvelan métodos para el tratamiento o la prevención de una enfermedad, trastorno o afección asociada con la actividad o expresión aberrante de la quitotriosidasa 1, que comprenden administrarle a un sujeto que lo necesita una cantidad terapéuticamente eficaz de al menos un compuesto de acuerdo con la invención o con una composición farmacéutica de acuerdo con la invención.
En determinadas realizaciones, las enfermedades, trastornos o afecciones asociadas con la actividad o expresión aberrante de la quitinasa de mamífero ácida incluyen enfermedades alérgicas, enfermedades inflamatorias agudas y crónicas, enfermedades autoinmunes, enfermedades dentales, enfermedades neurológicas, enfermedades metabólicas, enfermedades hepáticas, síndrome de ovario poliquístico, endometriosis y cáncer.
En realizaciones adicionales, las enfermedades, trastornos o afecciones asociadas con la actividad o expresión aberrante de quitotriosidasa 1 incluyen asma o trastornos fibróticos como fibrosis pulmonar idiopática (IPF). En otras realizaciones, estas enfermedades y trastornos incluyen enfermedades pulmonares intersticiales fibróticas como IPF o enfermedad pulmonar obstructiva crónica (EPOC).
También se desvelan métodos para tratar enfermedades provocadas por agentes infecciosos como hongos, gusanos y parásitos; el método comprende administrarle a un sujeto que necesita tal tratamiento una cantidad eficaz de uno o más compuestos de la invención.
También se desvelan métodos para tratar alergias, que comprende administrarle a un sujeto que necesita tal tratamiento una cantidad eficaz de uno o más compuestos de la invención. En determinadas realizaciones, estas alergias son provocadas por cualquiera de una variedad de antígenos incluyendo fuentes biológicas como ácaros de polvo, moho, cucarachas y otros insectos, caspa de mascotas u otros mamíferos, polen, esporas, moho, otras fuentes fúngicas y otros antígenos vegetales o fuentes no biológicas como productos químicos (por ejemplo, isocianatos).
También se desvela un método para tamizar agentes terapéuticos útiles para tratar asma en un mamífero, que comprende: (a) poner en contacto una proteína quitinasa de mamífero ácida con un compuesto (por ejemplo, un compuesto de la invención) y un sustrato de dicha quitinasa y (b) determinar si el compuesto inhibe la actividad de la quitinasa; si el compuesto inhibe la actividad de la quitinasa entonces el compuesto es un agente terapéutico útil para tratar asma.
También se desvelan métodos para monitorizar la efectividad de un tratamiento para el asma, que comprende (a) administrar un compuesto de la invención a un mamífero y (b) monitorizar la expresión de la quitinasa de mamífero ácida en el mamífero después de la administración del compuesto, donde una reducción de la expresión de la quitinasa de mamífero ácida indica que el compuesto es útil para tratar asma, enfermedades alérgicas como fiebre del heno, rinitis alérgica, dermatitis atópica u otras enfermedades asociadas con o mediadas por Th-2.
También se desvela un método para detectar agentes terapéuticos útiles para tratar asma en un mamífero, que comprende: (a) poner en contacto una proteína quitotriosidasa 1 con un compuesto (por ejemplo, un compuesto de la invención) y un sustrato de dicha proteína y (b) determinar si el compuesto inhibe la actividad de la quitotriosidasa 1; si el compuesto inhibe la actividad de la quitotriosidasa 1 entonces el compuesto es un agente terapéutico útil para tratar asma.
También se desvelan métodos para monitorizar la eficacia de un tratamiento para el asma y otras enfermedades alérgicas, que comprenden (a) administrar un compuesto de la invención a un mamífero y (b) monitorizar la expresión de mediadores inflamatorios como IL-13, IL-5, IL-4, eotaxina o IgE o células inflamatorias tales como eosinófilos, neutrófilos o linfocitos en lavados bronqueoalveolares, esputo o tejidos obtenidos del mamífero después de la administración del compuesto, donde una reducción de la expresión indica que el compuesto es útil para tratar asma o enfermedades alérgicas como fiebre del heno, rinitis alérgica, dermatitis atópica u otras enfermedades asociadas con o mediadas por Th-2.
También se desvelan métodos para evaluar la eficacia de un agente para tratar asma en un sujeto, que comprende las etapas de:
a) detectar en una muestra de sujeto recogida en un primer punto de tiempo el nivel de expresión de la proteína quitinasa de mamífero ácida;
b) repetir la etapa a) en uno o más puntos posteriores de tiempo después de la administración del agente y c) comparar el nivel de expresión de la proteína quitinasa de mamífero ácida detectada en la etapa a) con uno o más niveles de expresión detectados en la etapa b),
donde un mayor nivel de expresión de proteína quitinasa de mamífero ácida en el primer punto de tiempo respecto de al menos un punto posterior en el tiempo indica que el agente es eficaz para tratar asma.
Un agente identificado por tal método es eficaz para tratar asma, fiebre del heno, rinitis alérgica, dermatitis atópica, reacciones alérgicas o un trastorno asociado con Th-2.
De manera alternativa, la eficacia de un agente para tratar asma o una reacción alérgica puede evaluarse mediante la medición del nivel de expresión de un mediador inflamatorio tal como IL-13, IL-5, IL-4, eotaxina, IgE o medir la cantidad de células inflamatorias como eosinófilos, neutrófilos o linfocitos en lavados bronqueoalveolares, esputo o tejidos obtenidos de un mamífero. En algunas de estas realizaciones, el nivel de expresión puede medirse antes y después de la administración de un agente. Cuando el nivel de expresión del mediador inflamatorio o el nivel de células inflamatorias se reduce después de la administración de un agente, tal agente es eficaz para tratar asma, fiebre del heno, rinitis alérgica, dermatitis atópica, reacciones alérgicas o un trastorno asociado con Th-2.
También se desvelan métodos para identificar un agente para tratar asma, que comprenden:
a) poner en contacto una muestra que comprende una proteína quitinasa de mamífero ácida con el agente y b) determinar la capacidad del agente de inhibir la actividad de la proteína quitinasa de mamífero ácida, donde la menor actividad de la proteína quitinasa de mamífero ácida identifica un agente para tratar el asma.
En determinadas realizaciones, la actividad de la proteína quitinasa de mamífero ácida se evalúa con un ensayo de fluorescencia usando un reactivo que es hidrolizado por la proteína quitinasa de mamífero ácida. En determinadas realizaciones, el reactivo es 4-metilumbeliferil B-D-N,N'-diacetilquitobiosida hidrato.
Aplicaciones terapéuticas
Los compuestos de la invención son útiles para inhibir la actividad enzimática y biológica de la quitinasa de mamífero ácida (AMCasa) y quitotriosidasa 1 (CHIT1). Se ha demostrado que AMCasa es inducida en modelos animales de asma y en seres humanos que han muerto de asma, mientras que la inhibición de AMCasa con antisuero de AMCasa o por alosamidina (Zhu et al. Science 304:1678-1682, 2004) o desmetilalosamidina (Matsumoto et al., Biochemical and Biophysical Research Communications 390:103-108, 2009) reduce la inflamación en ratones. Además, estos estudios claramente establecieron un vínculo entre IL-13 y la inducción de AMCasa y que la inflamación alérgica dependía de la actividad enzimática de la AMCasa. La sobreexpresión de CHIT1 se ha vinculado con la enfermedad pulmonar intersticial fibrótica, incluyendo fibrosis pulmonar idiopática y enfermedad pulmonar obstructiva crónica (EPOC).
Más específicamente, la invención proporciona métodos para inhibir AMCasa en una célula, que comprende poner en contacto una célula con al menos un compuesto de acuerdo con la presente invención o una composición de este como se describe en el presente documento.
En algunas realizaciones, la invención proporciona métodos para el tratamiento o la prevención de una enfermedad o afección asociada con la expresión o actividad de AMCasa en un sujeto. Por ejemplo, la enfermedad, trastorno o afección se selecciona del grupo que consiste en enfermedades alérgicas, enfermedades inflamatorias agudas y crónicas, enfermedades autoinmunes, enfermedades dentales, enfermedades neurológicas, enfermedades metabólicas, enfermedades hepáticas, síndrome de ovario poliquístico, endometriosis y cáncer.
De acuerdo con determinadas realizaciones, los compuestos de la invención son útiles para tratar enfermedades alérgicas, como asma, rinitis alérgica, rinitis alérgica estacional, rinosinusitis crónica con o sin pólipos nasales, conjuntivitis, queratoconjuntivitis, conjuntivitis alérgica estacional, síndrome del ojo seco, esofagitis eosinofílica, enfermedad celíaca, alergias a los alimentos, síndrome del intestino irritable, enfermedad del intestino irritable, eczema atópico, dermatitis atópica, dermatitis de contacto alérgica, otitis eosinofílica media, neumonía eosinofílica y enfermedad mediada por IgG4.
En determinadas realizaciones, la reacción provocada por un alérgeno es rinitis alérgica o dermatitis atópica.
En determinadas realizaciones, la reacción provocada por un alérgeno se caracteriza por la aparición de uno o más síntomas, que puede incluir ojos rojos, picazón, rinorrea, eczema, deterioro de la audición, urticaria, un ataque de asma, aumento de la producción de mocos en los pulmones, tos, sibilancia y falta de aliento.
Ejemplos de trastornos inflamatorios agudos y crónicos que pueden tratarse usando los compuestos de la invención incluyen enfermedades fúngicas, infección parasítica, enfermedad celíaca, colitis microscópica, enfermedad pulmonar obstructiva crónica (EPOC), fibrosis pulmonar idiopática, enfermedad pulmonar intersticial, fibrosis quística (CF), enfermedad de Hermansky-Pudlak y Alzheimer (AD).
En determinadas realizaciones, la enfermedad o afección tratada por los métodos de la invención es un trastorno autoinmune que se selecciona del grupo que consiste en enfermedad inflamatoria intestinal, colitis ulcerativa (UC), enfermedad de Crohn (CD), artritis reumatoide (RA), osteoartritis, psoriasis, escleroderma, esclerosis múltiple (MS), síndrome de Sjogren, aterosclerosis y sarcoidosis.
Los compuestos de acuerdo con la presente invención también son útiles para tratar enfermedades dentales como periodontitis y enfermedades metabólicas como diabetes mellitus insulinodependiente (IDDM) y diabetes mellitus no insulinodependiente (NIDDM).
En determinadas realizaciones, la invención proporciona métodos para tratar una enfermedad hepática que se selecciona del grupo que consiste en enfermedad del hígado graso no alcohólico, esteatohepatitis no alcohólica, cirrosis y fibrosis inducida por el virus de la hepatitis C y fibrosis alcohólica.
En algunas realizaciones, los métodos de la invención se usan en el tratamiento de cáncer, donde el cáncer se selecciona del grupo que consiste en glioblastoma, cáncer de mama, cáncer de colon, cáncer pulmonar primario y metastásico, mesotelioma, osteosarcoma, melanoma maligno, cáncer de ovario, cáncer de cuello de útero, cáncer de próstata, cáncer de hígado, cáncer gástrico, cáncer renal metastásico, leucemia y linfoma.
En algunas realizaciones, el sujeto que recibe tratamiento es un mamífero. Por ejemplo, los métodos y usos descritos en el presente documento son adecuados para su uso médico en seres humanos. De manera alternativa, los métodos y usos también son adecuados en un contexto veterinario, donde la invención puede administrarse a animales de sangre caliente, aves y reptiles. Los animales de sangre caliente incluyen, por ejemplo, todos los primates no humanos (por ejemplo, chimpancé y mono), rumiantes (por ejemplo, vaca, oveja y cabra), porcinos (por ejemplo, cerdos), equinos (por ejemplo, caballo, mula y burro), camélidos (por ejemplo, camello y dromedario), caninos (por ejemplo, perro), felinos (por ejemplo, gato), leporinos (por ejemplo, conejo), murinos (por ejemplo, ratón y rata), cávidos (por ejemplo, cobayo), gerbilinos (por ejemplo, jerbo), cricetinos (por ejemplo, hámster), mustélidos (por ejemplo, hurón y comadreja) y chinchillos (por ejemplo, chinchilla). Las aves incluyen animales de la clase aviar, por ejemplo, todos los fasianinos (por ejemplo, gallina y codorniz), anserinos (por ejemplo, gansos), anatinos (por ejemplo, patos), meleágridos (por ejemplo, pavo), daruduelines (por ejemplo, canario), psitaciformes (por ejemplo, loro, guacamayo, perico y periquito), cacatuidae (por ejemplo, cacatúa) y colombinas (por ejemplo, paloma y tórtola).
En determinadas realizaciones, la invención se administra preferentemente a animales de compañía domesticados y a animales productivos y de cría.
Asma
Como se describe anteriormente, se ha demostrado que la proteína AMCasa se induce en los pulmones de modelos animales de asma y en seres humanos que han muerto de asma (Zhu et al. Science 304:1678-1682, 2004, Matsumoto et al., Biochemical and Biophysical Research Communications 390:103-108, 2009, Sutherland et al. Chemistry and Biology 18:569-579, 2011). Además, estos investigadores también demostraron que la inhibición de AMCasa con antisuero de AMCasa, o por inhibidores de la actividad enzimática de AMCasa, reduce la inflamación e hiperrespuesta de las vías respiratorias en ratones. Estos estudios también establecieron claramente un vínculo entre la interleucina de Th-2 IL-13, que se ha descrito en detalle, y la inducción de la expresión de proteína AMCasa. Estos estudios también han demostrado que la inflamación alérgica mediada por IL-13 puede reducirse o eliminarse mediante la inhibición de la actividad enzimática de AMCasa estableciendo evidencias de que la inflamación alérgica depende total o parcialmente de la actividad enzimática de AMCasa. En una realización de la invención, los compuestos que inhiben la actividad enzimática de AMCasa o la actividad biológica de AMCasa pueden administrarse a sujetos con asma o síntomas asmáticos para inhibir la inflamación relacionada y aliviar los síntomas de la enfermedad.
Rinitis
En otra realización, los compuestos de la invención pueden administrarse a sujetos con rinitis alérgica, rinitis alérgica estacional o rinosinusitis crónica para tratar la enfermedad debido a que estos síndromes están vinculados con IL-13 (Akdis et al., J. Allergy and Clinical Immunology 131:1479-1490, 2013) y se sabe que la AMCasa se produce por células epiteliales de rinosinusitis crónica con pólipos nasales (Ramanathan et al., Am. J. Rhinol. 20: 330-335, 2006., Lalaker et al., Am. J. Rhinol. Allergy 23(1):8-14, 2009., Gu et al. J. Otolaryngol. Head Neck Surg. 40(1):64-69, 2011).
Enfermedades oculares
La AMCasa se ha demostrado claramente como un mediador inflamatorio en la conjuntivitis, queratoconjuntivitis, conjuntivitis alérgica estacional y síndrome del ojo seco (Bucolo et al., Frontiers in Pharmacology 2(43):1-4, 2011; Musumeci et al., Cornea 28(6):667-672, 2009.) Además, se ha demostrado que la inhibición de AMCasa en un modelo animal de enfermedades inflamatorias del ojo alivia la inflamación (Bucolo et al. Pharmacol Res. 3:247-252, 2008). T ambién se ha demostrado que las proteínas de quitinasa aumentan en el tejido ocular de pacientes con degeneración macular (Rakic et al., Invest. Opthalmol. Vis. Sci. 44(4)1740-1746, 2003). Por lo tanto, un aspecto de la presente invención es tratar enfermedades inflamatorias y otras enfermedades oculares usando una preparación de uno o más de los compuestos descritos en el presente documento.
Otras enfermedades alérgicas
La otitis media eosinofílica es conocida por implicar la citocina IL-13 que induce la AMCasa (Ohta et al., Allergology International 63:171-180, 2014) y que AMCasa media aspectos de la inflamación y patología de IL-13. Eczema atópica es otra enfermedad alérgica en la que la gravedad clínica se ha correlacionado con niveles de IL-13 en la piel de la lesión (Szeqedi et al., J. Eur. Acad. Dermatol. Venereol., epub 2015).
La dermatitis de contacto alérgica también implica IL-13 que también se propone como una prueba complementaria para la enfermedad (Martins and Reis, J. Eur. Acad. Dermatol. Venereol., 27(3):390-393, 20l3).
Debido a que todas estas afecciones están asociadas con IL-13, un estimulante potente de la regulación por aumento de AMCasa y que se ha demostrado que la inhibición de AMCasa reduce la inflamación mediada por IL-13, se espera que el tratamiento de sujetos con uno o más compuestos descritos en el presente documento lleve a la mejora de estas enfermedades.
Inflamación eosinofílica esofágica (EoE)
EoE es una afección mediada por la inflamación de Th-2 incluyendo la presencia de eosinófilos, linfocitos CD8+, FcepsilonRI, mastocitos, depósito de colágeno y ARNm de eotaxina-3 (CCL26). EoE está asociada con otra enfermedad alérgica en la que los antígenos son frecuentemente alimentos pero también polen. Los tratamientos existentes tienen baja eficacia (intentar eliminar la exposición al antígeno) y efectos laterales (esteroides tópicos seguido de esteroides orales después dilatación mecánica). En un modelo de ratón de EoE, los niveles de AMCasa aumentan en el tejido esofágico. Cabe destacar que el inhibidor de AMCasa alosamidina inhibe la inflamación eosinofílica incluyendo la cantidad de eosinófilos, remodelación esofágica y proteína eotaxina-1 (Cho et al., Int. Immunopharmacol. 18(1):35-42, 2014). Se espera que el tratamiento de sujetos con EoE por uno o más compuestos descritos en el presente documento reduzca o cure los síntomas de la enfermedad.
Enfermedad celíaca, alergia a los alimentos, síndrome del intestino irritable, enfermedad inflamatoria intestinal
Una forma de la enfermedad celíaca se asocia frecuentemente con EoE y también tiene marcas distintivas de la enfermedad alérgica incluyendo eosinófilos tisulares aumentados (Mehta and Furuta, Immunol. Allergy Clin. North Am.
35(3):413-437, 2015). La enfermedad celíaca también se ha definido como un trastorno autoinmune diferente a la alergia al trigo y tiene características similares al síndrome del intestino irritable (Elli et al., World J. Gastroenterology 21(27):8221-8226, 2015). Mehta y Furuta también incluyen a las enfermedades intestinales inflamatorias dentro de las que implican eosinófilos en la patogénesis de la enfermedad. Por lo tanto, debido a que la AMCasa se expresa constitutivamente en el tracto gastrointestinal (Boot et al., J. Histochem. Cytochem. 53:1283-1292, 2005) y que hay al menos una implicancia parcial de la inflamación alérgica en estas enfermedades, la inhibición de la AMCasa en sujetos con estas enfermedades o alguna similar puede tratarse con los inhibidores de AMCasa descritos en el presente documento.
Enfermedades autoinmunes
Para las enfermedades autoinmunes que incluyen enfermedad inflamatoria intestinal (IBD), artritis reumatoide (RA), esclerosis múltiple (MS) y diabetes mellitus insulinodependiente (IDDM) o diabetes tipo I hay pruebas la regulación por aumento de la AMCasa y otras proteínas en la familia de la 18 glicosil hidrolasa. También hay pruebas de que estas proteínas pueden activar la autoinmunidad (Sekine et al., Ann. Rheum. Dis., 60(1)49-54, 2001, Tsuruha et al., J. Rheumatol., 29(7):1459-1466, 2002, Du et al., Rheumatol. Int., 26(1):35-41, 2005)
Enfermedad inflamatoria intestinal (colitis ulcerativa y enfermedad de Crohn)
Se ha demostrado que las quitinasas tienen una función en la patogénesis de la enfermedad inflamatoria intestinal (IBD) y modelos de IBD. Hay cada vez más pruebas de que los síntomas de IBD pueden modificarse alterando el bioma intestinal (Strober et al., J. Clin. Invest.117:514-521,2007., Zatorski and Fichna, 1:15-16, 2014). También se ha establecido que las cepas patogénicas de bacterias se unen a células epiteliales del colon mediante la proteína de unión a la quitina bacteriana y moléculas tipo quitinasa (Kawanda et al., Lab. Invest. 88:883-895, 2008) particularmente en la enfermedad de Crohn (Chassing et al., Gastroenterology 140:1720-1728, 2011). Las cepas patogénicas de bacterias en CD invaden la mucosa intestinal mediante la unión a células epiteliales y los estudios en un modelo de ratón de IBD mostraron que la colitis mejoraba cuando se infectaba a los ratones con bacterias con mejor capacidad de unirse a las células epiteliales del colon mediante las proteínas quitinasa (Low et al. Gastroenterology 145(3):602-612, 2013). El tratamiento de mamíferos con inflamación intestinal (UC, CD, enfermedad intestinal irritable, colitis microscópica y otras enfermedades intestinales) con una preparación de uno o más compuestos de esta invención pueden usarse para tratar enfermedad y síntomas de enfermedad.
Artritis reumatoide (RA) y osteoartritis (OA)
Las proteínas tipo quitinasa se sobreexpresan en condrocitos articulares y fibroblastos sinoviales y el suero de pacientes con Ra y OA (Hakala et al., J. Biol. Chem., 268(34):25803-25810, 1993, Hu et al., J. Biol. Chem., 271(32):19415-19420, 1996, Volck et al., Scand. J. Rheumatol. 28(3):171-179, 1999, Connor et al., Osteoarthritis Cartilage 8(2):87-95, 2000). Las concentraciones en suero de proteínas tipo quitinasa se correlacionan con la inflamación y destrucción de articulaciones en RA y OA (Kzyshkowska et al., Biomarker Insights 2:128-146, 2007). IL-6, una citocina prominente y objetivo del tratamiento de RA se conoce por regular por aumento la expresión de al menos una de las proteínas tipo quitinasa (Johansen et al. Can. Epidemiol. Biomarkers Prev. 15(2)194-202, 2006). Además, se ha demostrado que las proteínas quitinasa inducen la respuesta inmunitaria de Th1 en leucocitos de RA
y estimulan el crecimiento de células sinoviales (Kzyshkowska et al., Biomarker Insights 2:128-146, 2007) y por lo tanto pueden aumentar y perpetuar la inflamación crónica en RA.
Esclerosis múltiple (MS)
Las proteínas quitinasa están elevadas en el líquido espinal central en pacientes con MS remitente recidivante y en la nueromielitis óptica. Estas quitinasas aumentaron la liberación del mediador inflamatorio y estimularon la migración de células inflamatorias en una barrera hematoencefálica in vitro (Correale and Fiol, Mult. Scler. 17(5):521-31,2011). Los compuestos descritos en el presente documento pueden usarse para tratar la esclerosis múltiple y enfermedades neurológicas relacionadas.
Diabetes mellitus
En pacientes con retinopatía diabética proliferativa, existe un aumento de IL-13 en el humor vítreo en comparación con los individuos saludables y niveles especialmente elevados en áreas que desarrollaron membranas fibrovasculares y contribuye a la retinopatía (Yoshida et al., Br. J. Opthalmol., 99(5):629-634, 2014). La aplicación de compuestos de esta invención inhibe la AMCasa, un mediador corriente abajo de los efectos de IL-13 y que se puede esperar que interfiera con la retinopatía diabética. En la diabetes tipo II, también conocida como diabetes mellitus no insulinodependiente (NIDDM), la concentración en plasma de moléculas tipo quitinasa está asociada con la resistencia a la insulina (Rathcke et al., Inflamm. Res., 55(2):53-59, 2006) y niños con riesgo de padecer diabetes presentan niveles aumentados de proteínas quitinasa en contraste con los niños normales (Kyrgios et al., Metabolism 61(4)562-568, 2012). Puede esperarse de manera razonable que el tratamiento de sujetos con diabetes o prediabetes con uno o más compuestos de la presente invención pueda tratar o prevenir la diabetes.
Síndrome de Siogren (SS)
La expresión de quitinasa aumenta en pacientes con SS y los niveles se correlacionan con la gravedad de la enfermedad (Greenwell-Wild et al., Arthritis Rheum. 63(10):3103-3115, 2011), lo que indica que los sujetos con este síndrome pueden tratarse con los compuestos del presente documento.
Ateroesclerosis
Las quitinasas y proteínas de familias asociadas son indicadores de los macrófagos activados en la placa ateroesclerótica y el nivel enzimático aumenta hasta 55 veces en la placa ateroesclerótica (Boot et al., J. Biol. Chem., 273(40):25680-25685, 1998., Artieda et al., Arterioscler. Thromb. Vasc. Biol., 23(9):1645-1652, 2003) y provoca la migración de células de músculo liso vascular. Debido a que las quitinasas están implicadas en la patogénesis de la ateroesclerosis, puede predecirse de manera razonable que los inhibidores de las quitinasas descritos en la presente invención tratan, previenen o resuelven la ateroesclerosis en sujetos afectados.
Sarcoidosis
Las quitinasas también están elevadas en el suero de pacientes con sarcoidosis (Grosso et al., Scand. J. Clin. Lab. Invest., 64(1):57-62, 2004) y se producen en el granuloma sarcoidal en el pulmón (Johansen et al., Resp. Med.
99(4):396-402, 2005). Los inhibidores de quitinasa descritos en el presente documento se pueden usar para tratar sujetos con sarcoidosis.
Enfermedades hepáticas
El aumento de quitinasa se sintetiza en células Kupffer de pacientes con esteatohepatitis de hígado graso no alcohólico (NASH) y estimula la activación de las células estrelladas hepáticas, lo que sugiere una función de las proteínas quitinasa en la evolución de la fibrosis hepática (Malaguarnera et al., Gut 55(9):1313-1320, 2006, Malaguarnera et al., Am. J. Gastroenterol., 10(9):2060-2069, 2006). Las proteínas de familia quitinasa también están asociadas con la fibrosis y cirrosis inducida por el virus de la hepatitis C (HCV) y con la fibrosis de hígado alcohólico y no alcohólico (Shakel et al., Hepatology 38(3):577-588, 2003, Tran et al., Eur. J. Gastroenterol. Hepatol. 12(9):989-993, 2000). Además, la esteatohepatitis alcohólica; esteatohepatitis no alcohólica y enfermedad hepática de ácido graso no alcohólico, que ocurren en un entorno de enfermedad metabólica y cardiovascular; fibrosis hepática inducida viralmente y cirrosis biliar primaria, que tiene una base autoinmune, se asocian con la inflamación crónica y fibrosis y, como resultado, los compuestos descritos en esta invención se pueden usar para tratar varias enfermedades hepáticas.
Enfermedades renales
La inflamación y fibrosis acompañan muchas enfermedades renales como nefropatía, incluyendo nefropatía diabética, glomeruloesclerosis segmentaria y focal, fibrosis tubulointersticial, fibrosis postrasplante, así como fibrosis retroperitoneal/enfermedad de Ormond. Los compuestos descritos en el presente documento pueden usarse para tratar sujetos con estos trastornos para aliviar síntomas y reducir exacerbaciones y la evolución de la enfermedad.
Enfermedades cutáneas
El aumento de fibrosis está asociado con la lesión dérmica e inflamación crónica que acompaña la formación de lesiones excesivas o hipertróficas, queloides, enfermedades autoinmunes como escleroderma, lupus eritematoso sistémico (SLE). Se espera de manera razonable que los compuestos descritos en la presente invención sean eficaces para prevenir o tratar estas enfermedades.
Enfermedad pulmonar obstructiva crónica (EPOC)
Las proteínas de la familia quitinasa aumentan por la exposición al humo del cigarrillo y están presentes en niveles muy altos en pacientes con EPOC (Nikota et al., Resp. Res., 12:39-50, 2011; Letuve et al., Am. J. Pathol., 176:638-649, 2010) y las quitinasas estimulan la liberación de otros mediadores proinflamatorios que median la destrucción del tejido pulmonar. También se ha hecho una asociación genética entre la función pulmonar y la expresión de quitinasa (Aminuddin et al., Hum. Genet. 131(7):1105-1114, 2012). Los compuestos descritos en el presente documento pueden usarse para tratar sujetos con EPOC para aliviar síntomas y reducir las exacerbaciones y la evolución de la enfermedad.
Enfermedades pulmonares intersticiales, escleroderma y síndrome de Hermansky-Pudlak
La fibrosis pulmonar idiopática (IPF) y otras enfermedades pulmonares intersticiales están asociadas con el aumento de quitinasas en el tejido pulmonar y el plasma y aumentan la actividad profibrótica de TGFbeta (Cho et al., Allergy Asthma Immunol. Res. 7(1):14-21, 2015; Lee et al. J Immunol. 189(5):2635-44, 2012) y las proteínas quitinasa contribuyen a la lesión (Zhou et al. J Clin Invest. 125(8):3178-3192, 2015; Zhou et al., Sci. Transl. Med., 6(240): 240ra76, 2014) de manera que pueda esperarse de manera razonable que los inhibidores de quitinasas traten sujetos con cambios fibróticos pulmonares.
Fibrosis quística (CF)
Las proteínas tipo quitinasa tienen están elevadas en la CF y se correlacionan con la gravedad de la enfermedad (Hector et al., Plos One, 6(9):e24399-24405, 2011). Por lo tanto, puede esperarse que el tratamiento de pacientes con CF con uno o más compuestos de la presente invención mejores lo síntomas y la gravedad o evolución de la enfermedad.
Enfermedad de Alzheimer (AD)
En la enfermedad de Alzheimer, las proteínas y el ARNm de la familia quitinasa están altamente elevados en el cerebro de pacientes y estas proteínas también están asociadas con células microgliales alternativamente activadas patogénicas en modelos AD de ratón (Colton et al. J. Neuroimflamm. 3:27-382006). La expresión de quitinasa también estaba elevada en la demencia cerebrovascular isquémica (CvD) (DiRosa et al., Eur. J. Neurosci., 23(10)2648-2656, 2006). Se espera que el tratamiento de sujetos con AD o CvD por uno o más compuestos descritos en el presente documento reduzca la patología y evolución de la enfermedad.
Síndrome de ovario poliquístico (POCS) y endometriosis
El síndrome de ovario poliquístico (PCOS) es un estado inflamatorio crónico de bajo grado con un aumento significativo de la actividad de quitinasa en suero (Alanbay et al. Arch Gynecol Obstet. 2012;286:1065; Aydogdu et al. Exp Clin Endocrinol Diabetes. 2012;120:261). La actividad de quitinasa en el plasma de pacientes con endometriosis también aumenta significativamente (Alanbay et al. Gynecol Endocrinol. 2012;28:220). Se espera que el tratamiento de sujetos con POCS o endometriosis por uno o más compuestos descritos en el presente documento reduzca la patología y evolución de la enfermedad.
Otras enfermedades y aplicaciones
Debido a que la quitina es necesaria para el crecimiento de la mayoría de los hongos e insectos y la remodelación de la quitina es necesaria durante el crecimiento de estos organismos que incluyen la actividad de quitinas para degradar la quitina así como la síntesis de quitina, los inhibidores de la actividad de la quitinasa y, por lo tanto, la capacidad de estos organismos de remodelar, suprimir el ectoesqueleto, etc., los compuestos descritos en la presente invención también pueden ser útiles en el procesamiento y producción médica, agrícola y alimenticia u otras aplicaciones donde la inhibición de la quitinasa resultaría en una menor supervivencia de los organismos que contienen quitina. Estos incluyen, pero no se limita a, enfermedades fúngicas de mamíferos como aspergilosis, criptococosis y enfermedades vegetales provocadas por infección fúngica o daño de insectos, enfermedades trópicas incluyendo, pero no se limita a, malaria y otras enfermedades parasíticas. De hecho, la actividad de la quitinasa aumenta en la malaria (Barone et al., Clin. Chim. Acta. 331(1-2):79-85, 2003) y por lo tanto la inhibición de la quitinasa puede ser útil para inactivar o de otro modo volver al parásito inútil.
Enfermedades de almacenamiento
Las quitinasas están fuertemente reguladas por aumento en la enfermedad de Gaucher (Bussink et al. Int Rev Cytol.
2006;252:71-128). Por lo tanto, se espera que la inhibición de las quitinasas con los compuestos descritos en el presente documento reduzca la evolución de enfermedades de almacenamiento como enfermedad de Gaucher, enfermedad de Fabry, trastornos de almacenamiento lisosomal, enfermedad de Niemann-Pick, cisteinosis nefropática, globotiaosilceramidosis ligada a X.
Cáncer
La quitinasa y las proteínas tipo quitinasa se sobreexpresan en muchos cánceres incluyendo tumores cerebrales como glioblastoma (Francescone et al. J Biol Chem 2011;286:15332-43; Ku et al. Int J Cancer 2011;128:1316-26) o astrocitoma (Zhang et al. Cancer. 2010;116:2688), cáncer de mama (Johansen et al. Breast Cancer Res Treat 2003;80:15-21), cáncer de colon (Nutt et al., 2005, Pelloski et al., 2005; Fijneman et al. Clin Cancer Res. 2012;18:2613; Chen et al. Am J Pathol. 2011;179:1494), cáncer pulmonar primario y metastásico (Wang et al. Tumour Biol 2015;36:901-7; Johansen et al. Lung Cancer 2004;46:333-40), mesotelioma (Corradi et al. Anticancer Res. 2013 Dec;33(12):5517), osteosarcoma, melanoma maligno (Ma et al. Cancer Res 2015;75:487-96), cáncer de ovarios (Hogdall et al. BMC Cancer 2009;9:8; Dupont et al. J Clin Oncol. 2004;22:3330), cáncer de cuello de útero (Ngernyuang et al. Int J Biochem Cell Biol 2014;51:45-52.), cáncer de próstata (Jeet et al. Endocr Relat Cancer. 2014;21:723), cáncer de hígado (Pan et al. J Cancer Res Clin Oncol 2013;139:1043-54), cáncer gástrico (Li et al. Chin Med J 2012;125:1777), cáncer renal metastásico (Zhangg et al. Tumour Biol 2014;35:12131-7), neoplasias malignas hematológicas seleccionadas de leucemia o linfoma (Mactier et al. J Proteome Res. 2011;10:1030; Marchesi et al. Vet Pathol. 2006;43:773-6; Marchesi et al. J Vet Med A Physiol Pathol Clin Med. 2003;50:103) y otros tipos de cánceres con antecedentes inflamatorios (Quershi et al. Genes Cancer. 2011;2:74; Eurich et al. World J Gastroenterol.
2009;15:5249; Roslind and Johansen, Methods of Mol Biol. 2009;511:159). De hecho, los mayores niveles de plasma indican un mal pronóstico y un mayor potencial metastásico para varios cánceres (Johansen et al., Cancer Epidemiol. Biomarkers Prev., 15(2):194-202, 2006). Se prevé que la inhibición de la quitinasa y la función biológica de la proteína tipo quitinasa con uno o más compuestos descritos en la presente invención tengan utilidad terapéutica en sujetos con cáncer.
Ejemplos
La presente invención se ilustra de manera adicional por los siguientes ejemplos que de ningún modo deberán considerarse como que limitan el alcance de la invención reivindicada.
Materiales y métodos de preparación y caracterización
Los compuestos del presente documento descripción pueden prepararse usando reacciones y procedimientos químicos conocidos. A continuación se presentan los métodos representativos para sintetizar compuestos de la descripción. Por lo tanto, se entiende que la naturaleza de los sustituyentes necesarios para el compuesto diana deseado frecuentemente determina el método preferido de síntesis. Todos los grupos variables de estos métodos son como se describe en la descripción genérica si no se definen específicamente a continuación.
Los expertos en la técnica reconocerán que los materiales de partida y las condiciones de reacción pueden variar, se puede alterar la secuencia de las reacciones y se pueden emplear etapas adicionales para producir los compuestos comprendidos por el presente documento descripción, como los demuestran los siguientes ejemplos. Muchas referencias generales que proporcionan esquemas sintéticos químicos comúnmente conocidos y condiciones útiles para la síntesis de los compuestos descritos están disponibles (véase, por ejemplo, Smith and March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, quinta edición, Wiley-lnterscience, 2001; o Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, cuarta edición, Nueva York: Longman, 1978).
Las reacciones se realizan en un disolvente apropiado para los reactivos y materiales empleados y adecuado para las transformaciones que se están efectuando. Los expertos en la técnica de la síntesis orgánica comprenderán que la funcionalidad presente en la molécula debe ser compatible con las transformaciones propuestas. Esto a veces requerirá una decisión de modificar el orden de las etapas sintéticas o de seleccionar un esquema de proceso particular en vez de otro para obtener un compuesto deseado de la descripción.
En algunos casos, la protección de determinadas funcionalidades de reacción puede ser necesaria para lograr algunas de las transformaciones que anteceden. En general, la necesidad de estos grupos de protección así como las condiciones necesarias para unir y retirar tales grupos será evidente para los expertos en la técnica de la síntesis orgánica. Un informe acreditado que describe las diversas alternativas para el practicante cualificado son J. F. W. McOmie, "Protective Groups in Organic Chemistry," Plenum Press, Londres y Nueva York 1973, en T. W. Greene y P. G. M. Wuts, "Protective Groups in Organic Synthesis," tercera edición, Wiley, Nueva York 1999, en "The Peptides;" tomo 3 (editores: E. Gross y J. Meienhofer), Academic Press, Londres y Nueva York 1981, en "Methoden der organischen Chemie," Houben-Weyl, 4a edición, Vol. 15/1, Georg Thieme Verlag, Stuttgart 1974, en H.-D. Jakubke
and H. Jescheit, "Aminosauren, Peptide, Proteine," Verlag Chemie, Weinheim, Deerfield Beach, y Basilea 1982, y/o en Jochen Lehmann, "Chemie der Kohlenhydrate: Monosaccharide und Derivate," Georg Thieme Verlag, Stuttgart 1974. Los grupos protectores se pueden eliminar en una etapa posterior conveniente usando métodos conocidos en la técnica.
A continuación se describen los procedimientos sintéticos representativos para la preparación de los compuestos de la presente invención. Los sustituyentes tienen el mismo significado que se definió anteriormente, a menos que se indique otra cosa.
Los materiales de partida pueden obtenerse de fuentes comerciales o prepararse mediante métodos establecidos de la bibliografía conocidos por los expertos en la técnica.
Todos los disolventes, sustratos y reactivos que estaban comercialmente disponibles se usaron sin purificación adicional. Se llevó a cabo un análisis TLC usando placas de vidrio recubiertas previamente (0,2 ±0,03 mm de espesor, GF-254, tamaño de partículas de 0,01-0,04 mm) de Fluorochem Ltd, Reino Unido. La cromatografía en columna se llevó a cabo usando gel de sílice de grado de alta pureza (tamaño de poro de 60 A, 220-440 mesh de tamaño de partícula, 35-75 pm de tamaño de partícula) de Fluka.
Los espectros 1H RMN se registraron en un espectrómetro Agilent Mercury 400 MHz y espectrómetros Bruker Avance 500, 600 y 700 MHz (DRX400, DXR500, DRX600 y DXR700, respectivamente).
Todos los espectros se registraron en disolventes deuterados apropiados (CDCb, DMSO-d6, D2O, CD3OD, etc.) que estaban comercialmente disponibles.
Las resonancias se dan en partes por millón respecto de tetrametilsilano. Los datos de observan tal como sigue: cambio químico (8), multiplicidad (s = singlete, d = doblete, t = triplete, m = multiplete, bs = singlete amplio), constantes de acoplamiento (J en Hz) e integración.
Los espectros ESI-MS se obtuvieron en un módulo de separación de Waters Alliance 2695 con un detector de UV PDA 1996 y un detector de masa Waters Micromass ZQ 2000 equipado con una columna Kinetex 2,1/50 mm, 2,6 pm C18 eluida con 0,3 ml/min de flujo de gradiente al 3-100 % (durante 6 min) de acetonitrilo en agua y un módulo de separación aShimadzu Prominence LC-20AD con un detector SPD-M20A PDA y detector de masa Shimadzu LCMS-2020 equipado con una columna Luna, C18, 2um, 100A, 150x3 mm eluida con 0,5 ml/min de flujo de gradiente al 15 90 % (durante 13 min) de acetonitrilo en agua.
Ensayo de actividad de AMCasa humana
Un ensayo enzimático con AMCasa humana recombinante se usó para establecer la actividad de inhibición de los compuestos (Boot et al., 2001, JBC: 276). El ensayo se ejecutó en un formato de placa de 96 pocillos, cada reacción en un volumen total de 100 pl. Se usó 4-metilumbeliferil B-D-N,N'-diacetilquitobiosida hidrato como sustrato de la enzima. Tras la hidrólisis por AMCasa, el sustrato libera 4-metilumbeliferil (4MU) que, al ionizarse en pH básico, emite fluorescencia a 460 nm.
Brevemente, se añadieron 40 pl de un sustrato a cada pocillo, seguido de 10 pl de dilución del compuesto y 50 pl de una solución enzimática recombinante de hAMCasa. La reacción se llevó a cabo en amortiguador de citrato, pH 5,2, al oscuro, a 37 °C durante 60 minutos con agitación. Después de ese tiempo la reacción se detuvo agregando 195 pl de amortiguador de parada (pH 10,5) a cada pocillo. La fluorescencia del producto de reacción se midió en un lector de placa fluorescente Perkin Elmer Envision a una longitud de onda de excitación de 355 nm. Los valores de IC50 se calcularon usando GraphPad Prism.
Ensayo de actividad de CHIT1 humana
Un ensayo enzimático con CHIT1 humana recombinante se usó para establecer la actividad de inhibición de los compuestos (Boot et al., 2001, JBC: 276). El ensayo se ejecutó en un formato de placa de 96 pocillos, cada reacción en un volumen total de 100 pl. Se usó 4-metilumbeliferil p-D-W,W,W"-triacetilquitotriosa como sustrato para la enzima. Tras la hidrólisis por CHlT 1, el sustrato libera 4-metilumbeliferil (4MU) que, al ionizarse en pH básico, emite fluorescencia a 460 nm.
Brevemente, se añadieron 40 pl de un sustrato a cada pocillo, seguido de 10 pl de dilución del compuesto y 50 pl de una solución enzimática recombinante de CHIT1. La reacción se llevó a cabo en amortiguador de citrato, pH 5,2, al oscuro, a 37 °C durante 60 minutos con agitación. Después de ese tiempo la reacción se detuvo agregando 195 pl de solución de parada (pH 10,5) a cada pocillo. La fluorescencia del producto de reacción se midió en un lector de placa fluorescente Perkin Elmer Envision a una longitud de onda de excitación de 355 nm. Los valores de IC50 se calcularon usando GraphPad Prism.
Los compuestos descritos en la T abla 1 tienen los valores de IC50 que generalmente varían de aproximadamente 0,01
|jM a aproximadamente 100 jM . Sus intervalos de actividad se asignaron de la siguiente manera: A: <0,1 jM ;
B: 0,1-1 jM ;
C: 1-10 jM ; y
D: 10-100 jM .





Procedimientos sintéticos generales
Procedimiento general I
Reducción de a-aminoácido al aminoalcohol correspondiente.

A una suspensión de aminoácidos en tetrahidrofurano anhidro (THF) (3 ml/mmol) se añadió un complejo de boranodimetilsulfuro (BH3.DMS; 3 equivalentes) gota a gota a 0 °C (Advertencia: ¡formación de espuma!) Se retira el baño de enfriamiento y la mezcla de reacción se somete a reflujo durante la noche, después de lo cual la cromatografía no revela presencia de material de partida. La mezcla se enfría hasta temperatura ambiente y 6 M HCl (8 equivalentes respecto del material de partida) se añade cuidadosamente (Advertencia: ¡formación de espuma!) y la mezcla se vuelve a someter a reflujo durante 1,5 horas. La mezcla se enfría a temperatura ambiente y el pH se lleva a 10 con NaOH 4 M. El producto se extrae varias veces con acetato de etilo (AcOEt), los extractos se combinan, se secan en MgSO4, se filtran y concentran al vacío. El producto bruto se tritura con etil éter (Et2O) y se filtra.
Procedimiento general II
Acilación aminoselectiva de aminoalcohol o N-Boc diamina con cloruro de cloroacetilo.

A la solución de aminoalcohol o N-Boc diamina en THF (6 ml/mmol) se añade trietilamina (1,2 equivalentes respecto del aminoalcohol) y la solución se enfría hasta 0 °C. Se añade cloruro de cloroacetilo (1 equivalente respecto del aminoalcohol) lentamente de manera de que la temperatura interna de la reacción no exceda los 5 °C. Después se retira el baño de enfriamiento y la mezcla se agita durante 20 minutos más. La TLC muestra el consumo completo del material de partida a esta altura. Después se añade éter dietílico (doble del volumen del THF usado para la reacción) y toda la mezcla de reacción se lava secuencialmente con 1 M HCl, 1 M NaOH, salmuera, se seca en MgSO4, se filtra y evapora a sequedad para dar el producto bruto. La cristalización a partir de éter dietílico caliente habitualmente proporciona el amido alcohol o amido amina con una pureza de 98 %+.
Procedimiento general III
Acilación aminoselectiva de aminoalcohol con a-bromoácido con el uso de un reactivo formador de amida.

A la solución de a-bromoácido en diclorometano (7 ml/mmol) se añade secuencialmente diisopropiletilamina (DIPEA, 1 equivalente respecto del a-bromoácido de partida), reactivo de acoplamiento (1 equivalente; típicamente TBTU o HATU, pero también pueden usarse otros reactivos de acoplamiento comúnmente usados) y aminoalcohol (1 equivalente) y la mezcla de reacción se agita durante 1,5 horas a temperatura ambiente. Después de este tiempo, el control de TLC muestra el consumo completo de los materiales de partida por lo que la mezcla de reacción se transfiere al embudo de separación y se lava secuencialmente con 1 M HCl, 1 M NaOH y salmuera. La fase orgánica se seca sobre MgSO4, se filtra y evapora a sequedad para dar el producto bruto que se purifica adicionalmente por cristalización o cromatografía de gel de sílice.
Procedimiento general IV
Ciclación de a-haloamida a morfolin-3-ona

A la solución de a-haloamida (es decir, a-cloro- o a-bromoamida) en THF (10 ml/mmol) se añaden 3 equivalentes de hidruro de sodio (NaH) de una vez (puede ser aconsejable enfriar la solución antes de la adición de NaH cuando se trabaja a mayor escala) y la mezcla de reacción se deja agitar a temperatura ambiente durante 2 horas. Después se inactiva cuidadosamente el exceso de NaH mediante la adición gota a gota de salmuera y después se añade un volumen adicional de salmuera (igual al volumen inicial de THF) provocando la separación de fases. La capa orgánica se separa y la capa acuosa se extrae adicionalmente con éter dietílico. Los extractos orgánicos combinados se secan después sobre MgSO4, se filtran y los disolventes se evaporan. En la mayoría de los casos el producto bruto es lo suficientemente puro como para usarse en la siguiente etapa sin purificación adicional.
Procedimiento general V
Reducción de morfolin-3-ona a morfolina o 2-piperazinona a piperazina o amida en amina.

A la solución de morfolin-3-ona o 2-piperazinona o amida en THF (3 ml/mmol) se añaden 3 equivalentes de complejo
de BH3.DMS y la mezcla de reacción se somete a reflujo durante 3 horas, después de lo cual el control de TLC muestra el consumo completo del material de partida. La mezcla de reacción se enfría a temperatura ambiente y se añade cuidadosamente 2 M HCl (6 equivalentes respecto del material de partida). La mezcla de reacción resultante se somete a reflujo durante 2 horas y se enfría hasta temperatura ambiente. Después el pH de la mezcla se ajusta hasta fuertemente alcalino (~10) mediante una adición gota a gota de 6 M NaOH. La capa orgánica se separa y la capa acuosa se extrae adicionalmente con éter dietílico. Los extractos orgánicos combinados se secan después sobre MgSO4, se filtran y los disolventes se evaporan. En la mayoría de los casos, el producto bruto obtenido es lo suficientemente puro como para usarse en la siguiente etapa sin purificación adicional.
Procedimiento general VI
Aminación reductiva de la cetona cíclica (es decir, piperid-4-ona N-protegida) con amina cíclica secundaria (es decir, morfolina, piperazina, piperazin-3-ona, etc.).

La amina secundaria cíclica se disuelve en 1,2-dicloroetano (DCE, 0,65 ml/mmol) y se añade N-Boc-piperid-4-ona (1,5 equivalentes respecto de la amina cíclica) y ácido acético glacial (AcOH) (2 equivalentes respecto de la amina cíclica) y la mezcla se agita durante 4 horas. Después se añade triacetoxiborohidruro de sodio [NaBH(OAc)3, 2 equivalentes] en una porción y la mezcla espesa se agita durante la noche a temperatura ambiente. Después de este tiempo se añade una solución acuosa al 5 % de bicarbonato de sodio (NaHCO3) (dos veces el volumen del DCE usado) y la mezcla bifásica se agita durante 30 minutos. Las capas se separan y la capa acuosa se extrae adicionalmente con diclorometano. Después los extractos orgánicos combinados se secan sobre MgSO4, se filtran y los disolventes se evaporan proporcionando un producto bruto que típicamente necesita purificación adicional por cromatografía de gel de sílice.
Procedimiento general VII
Retirada de grupo terc-butoxicarbonil (Boc-) a partir de amina
La amina N-Boc protegida se trata con una solución 4 M de HCl (5 ml/mmol de material de partida) en un disolvente orgánico apropiado (por ejemplo, AcOEt, 1,4-dioxano, MeOH, DCM) por el tiempo necesario para el consumo completo del material de partida (típicamente 30 minutos - 2 horas). Los volátiles se retiran después al vacío proporcionando el compuesto deseado en la forma de su sal de clorhidrato. El producto bruto es habitualmente lo suficientemente puro para usarse en la siguiente etapa pero la trituración adicional con éter dietílico puede ayudar a retirar cualquier impureza coloreada.
Procedimiento general VIII
Instalación del anillo de 2,5-d¡am¡no-1.2.4-tr¡azol en la sal de clorhidrato de la amina secundaria

Se añaden la sal de clorhidrato de la amina secundaria, carbonato de potasio anhidro (K2CO3) (2 equivalentes) y S,S'-dimetil-N-ciano-ditioiminocarbonato (1,2 equivalente molar) a acetonitrilo (2 ml/mmol del material de partida) y la solución resultante se somete a reflujo durante 1 - 7 horas (monitoreando por TLC). Después se añade monohidrato de hidrazina (3-5 equivalentes) y la reacción se somete a reflujo adicionalmente durante otras 2-5 horas, tiempo después del cual se enfría hasta temperatura ambiente y los sólidos se filtran. El filtrado se concentra al vacío y el producto bruto se purifica por cristalización a partir del disolvente apropiado o cromatografía en gel de sílice regular o gel de sílice C-18 de fase inversa.
Procedimiento general IX
Activación del grupo carboxílico por anhídrido mezclado seguido de formación de una amida.

El ácido carboxílico se disuelve en diclorometano (DCM) (3-8 ml/mmol dependiendo de la solubilidad) y se añade N-metilmorfolina (1,2 equivalentes). La solución se enfría hasta - 15 °C y se añade cloroformiato de alquilo (típicamente metilo, etilo o isobutilo) y la mezcla se agita durante 10 minutos más, tiempo en el cual se añade la amina apropiada (pura, 1,2 equivalentes) o amoníaco acuoso. La mezcla de reacción se deja calentar hasta temperatura ambiente y se agita típicamente durante la noche, pese a que en los casos de amina reactiva el acoplamiento se completa habitualmente dentro de minutos. El producto bruto se aísla lavando la fase orgánica (DCM) posteriormente con 1 M HCl, 1 M NaOH y salmuera. La fase orgánica se seca sobre MgSO4, se filtra y evapora a sequedad para dar el producto bruto que se purifica adicionalmente por cristalización o cromatografía de gel de sílice.
Procedimiento general X
Retirada del terc-butoxicarbonil (Boc-) de amina seguido de ciclación al anillo de seis miembros (es decir, morfolin-3-ona, piperazin-2-ona, etc.) y anillo de siete miembros (1,4-oxazepina, 1,4-benzoxazepina, 1,4-benzodiazepinas, etc.).

X, Y, W pueden ser CH2, CH(R), C=O, O, NH, NH(alquilo), S, SO, SO2, etc.
La sal de clorhidrato de amina bruta del procedimiento de retirada de grupo Boc típico (véase Procedimiento General VII) se suspende en metanol (3 ml/mmol), se añade trietilamina (5 equivalentes) y la mezcla se somete a reflujo por el tiempo apropiado (control de TLC). Se retira el metanol y el exceso de trietilamina al vacío, el residuo se absorbe en acetato de etilo y un lavado de ácido/base acuoso, se seca en MgSO4, se filtra y concentra al vacío. El producto bruto se purifica mediante cristalización o cromatografía de gel de sílice.
Procedimiento general XI
Reducción del aminoácido protegido con Boc al aminoalcohol protegido con Boc correspondiente mediante anhídrido mezclado

El aminoácido protegido con Boc de partida se disuelve en THF (4 ml/mmol) y el grupo carboxílico se activa por la formación del anhídrido mezclado como se describe en el Procedimiento General IX. El clorhidrato de N-metilmorfolina precipitada se filtra rápidamente y el filtrado se transfiere a un matraz de fondo redondo más grande. La suspensión de NaBH4 (2 equivalentes) en agua (1 ml/mmol) se añade cuidadosamente (Advertencia: ¡formación de espuma!) y la mezcla de reacción se deja agitar a temperatura ambiente durante la noche. Se añade 1 M NaOH en un volumen igual al del THF usado y la mezcla se agita por otros 30 minutos, tiempo después del cual se extrae 3 veces con acetato de etilo. Se seca la solución y se retira el disolvente para proporcionar el producto que en general es suficientemente puro como para usarse directamente en la siguiente etapa, pese a que la cristalización o trituración con el disolvente apropiado puede mejorar adicionalmente su pureza.
Procedimiento general XII
Oxidación del aminoalcohol protegido con Boc al aminoaldehído protegido con N correspondiente con peryodinano de Dess-Martin

A la solución de aminoalcohol protegido con Boc en diclorometano (1,5 ml/mmol), se añade peryodinano de Dess-Martin (1,1 equivalente) y la evolución de la reacción se sigue por TLC. Típicamente después de 2 horas se logra la conversión completa y la reacción se inactiva con una solución acuosa de tiosulfato de sodio al 10 % para retirar cualquier especie oxidativa. La mezcla de reacción se diluye 5 veces con diclorometano y el producto se aísla por un procedimiento básico con 1 M NaOH. La fase orgánica se seca en MgSO4, se filtra y evapora hasta sequedad para dar el producto bruto que en general es suficientemente puro como para usarse directamente en la siguiente etapa, pese a que la cristalización o trituración con el disolvente apropiado puede mejorar adicionalmente su pureza.
Procedimiento general XIII
Aminación reductiva del aminoaldehído protegido con Boc con metil éster de aminoácido

A la suspensión de sal de clorhidrato de metil éster de aminoácido en DCE (3 ml/mmol) se añaden aminoaldehído protegido con Boc (1 equivalente) y AcOH glacial (1-2 equivalentes) y la mezcla se agita durante 2 horas a temperatura ambiente antes de añadir NaBH(OAc)3 (3 equivalentes) en una porción. La mezcla resultante se agita durante la noche a temperatura ambiente y el producto se aísla de la misma forma que la descrita en el Procedimiento General VI. Con la condición de que los materiales de partida se usen razonablemente puros, el producto bruto en la mayoría de los casos no requiere purificación adicional.
Procedimiento general XIV
N-metilación de amina secundaria (lineal o cíclica) con formaldehído.

a c
La amina secundaria (o su sal) se disuelve en diclorometano (3 ml/mmol) y se añaden 3 equivalentes de formalina acuosa seguido de NaBH(OAc)3 (4 equivalentes) a esta solución. La mezcla de reacción se agita vigorosamente durante 2-3 horas a temperatura ambiente (control de TLC), tiempo después del cual el producto bruto se aísla de la misma forma que la descrita en el Procedimiento General VI.
Procedimiento general XV
N-sulfonilación de amina secundaria

Se disuelven amina de partida, trietilamina (6 equivalentes), cloruro de sulfonilo (3 equivalentes) en piridina anhidra (5 ml/mmol de material de partida) y la solución resultante de color naranja rojizo se agita a temperatura ambiente durante 2 horas. Después se añade etilendiamina (3 equivalentes respecto del material de partida) y la mezcla de reacción se agita otras 12 horas a temperatura ambiente, tiempo después del cual se diluye con acetato de etilo (4 veces la cantidad de piridina usada) y se lava con bicarbonato de sodio saturado y salmuera. Se seca adicionalmente con MgSO4 anhidro, se filtra y el disolvente se retira al vacío. El producto bruto habitualmente necesita purificación adicional mediante cristalización o cromatografía de gel de sílice.
Procedimiento general XVI
Desprotección del grupo aliloxicarbonilo (Alloc-)

A una solución de amina protegida con Alloc en diclorometano anhidro y desgasificado (5 ml/mmol), se añaden 5 %
en moles de Pd(PPh3)4 y fenilsMano (10 equivalentes). La mezcla de reacción se agita vigorosamente durante 1-3 horas a temperatura ambiente (control de TLC). Después de esto se añaden 2 M HCl a la reacción y las fases se separan. La fase acuosa se alcaliniza a pH 12 con 6 M NaOH y se extrae 3 veces con diclorometano. Los extractos orgánicos combinados se secan en MgSO4 anhidro, se filtran y el disolvente se retira al vacío proporcionando la amina bruta que en general es lo suficientemente pura como para usarse en la siguiente etapa.
Procedimiento general XVII
Formación de la sulfinimina a partir de aldehido y sulfonamina quiral

Una solución de aldehído (S o R)-2-metil-2-propanosulfinamida (1 equivalente), Ti(OEt)4 (2 equivalentes) en diclorometano anhidro (2 ml/mmol) se somete a reflujo durante 2 horas, después se calienta un poco durante la noche con agitación. Se añade MgSO4 anhidro (200 mg/1 ml de disolvente usado) y después de 15 minutos la reacción se filtra a través de un lecho de Celite. El filtrado se concentra y el producto bruto se purifica por cromatografía en columna en un sistema de disolvente de AcOEt/hexanos.
Procedimiento general XVIII
Adición de reactivo de un Grignard-alilo a la sulfinimina quiral

A una solución enfriada (-20 °C) de (S)-terc-butilsulfimina ópticamente pura en diclorometano (2 ml/mmol), se añade una solución de un reactivo de Grignard apropiado (1,5 equivalentes, 1 M en éter dietílico o THF) gota a gota. Tras completar la adición, el baño de enfriamiento se retira y la reacción se permite calentar hasta temperatura ambiente. Después de que esta mezcla de reacción se vierte en una solución saturada de cloruro de amonio y se extrae 3 veces con éter dietílico. Los extractos orgánicos combinados se lavan con salmuera, se secaron con MgSO4 anhidro, se filtraron y concentraron. El producto bruto se purifica por cromatografía en columna en un sistema de disolvente de AcOEt/hexanos.
Procedimiento general XIX
N-alquilación de homoalilsulfinamida quiral con bromuro alil(sustituido)

A una solución de sulfinamida en DMF (2 ml/mmol), se añade hidruro de sodio (2 equivalentes). Después de 20 minutos se añade bromuro alil(sustituido) (1,5 equivalentes) gota a gota y la mezcla de reacción se deja agitar durante 1 hora. Después de este tiempo, la mezcla de reacción se vierte en una solución saturada de cloruro de amonio y se extrae varias veces con éter dietílico. Los extractos orgánicos combinados se lavan con salmuera, se secaron con MgSO4 anhidro, se filtraron y concentraron. El producto bruto se purifica por cromatografía en columna en un sistema de disolvente de AcOEt/hexanos.
Procedimiento general XX
Metátesis de cierre de anillo de homoalilsulfinamida quiral N-alilada

A una solución del material de partida en diclorometano (20 ml/mmol) se añade una 1a y 2a generación de catalizador de Grubbs (5 % en moles) y la mezcla de reacción se somete a reflujo durante 1,5 horas. Después de este tiempo, la mezcla de reacción se enfría y concentra. El producto deseado se aísla por cromatografía en columna de gel de sílice en un sistema de disolvente de AcOEt/hexanos.
Procedimiento general XXI
Alquilación del grupo hidroxilo con alquil haluro

A una solución de alcohol en THF seco (10 ml/mmol), se añade hidruro de sodio (3 equivalentes). Después de 5 minutos, se añade un alquil haluro apropiado (1,5 equivalentes) y la mezcla de reacción se agita a temperatura ambiente hasta el consumo total del material de partida (control de TLC). Después se vierte en una solución saturada de cloruro de amonio y se extrae 3 veces con éter dietílico. Los extractos orgánicos combinados se lavan con salmuera, se secaron con MgSO4 anhidro, se filtraron y concentraron. El producto bruto se purifica por cromatografía en columna en un sistema de disolvente de AcOEt/hexanos.
Procedimiento general XXII
Formación de amida de Weinreb a partir de ácido carboxílico y clorhidrato de N.O-dimetilhidroxilamina con el uso de reactivo formador de amida

A una solución de ácido carboxílico en diclorometano (2 ml/mmol), se añade DIPEA (2,1 equivalentes), seguido de clorhidrato de W,O-dimetilhidroxilamina (1,1 equivalentes). Se añade TBTU (1,1 equivalentes) a la mezcla de reacción y la reacción se agita a temperatura ambiente por el tiempo necesario para el consumo completo del material de partida (habitualmente 16-24 horas) según lo indique la t Lc . La mezcla de reacción se diluye con diclorometano, después se lava con 2 M HCl y salmuera. La capa orgánica se seca en MgSO4 anhidro, se filtra y concentra. El producto se purifica por cromatografía en columna de gel de sílice en un sistema de AcOEt/hexanos.
Procedimiento general XXIII
Acoplamiento de aminoalcohol protegido con Boc con fenol mediante reacción de Mitsunobu

A una solución de fenol (1 mmol) y aminoalcohol protegido con Boc (1,25 equivalentes) en THF seco (3 ml/mmol) en una atmósfera de gas inerte (habitualmente argón), se añade trifenilfosfina (1,5 equivalentes) y la mezcla se enfría hasta -15 °C. Se añade DIAD (1,5 equivalentes) gota a gota a esta temperatura, después se retira el baño de enfriamiento y la reacción se agita a temperatura ambiente durante 24 horas. La mezcla de reacción se concentra y el residuo oleaginoso se purifica por cromatografía en columna de gel de sílice en un sistema de AcOEt/hexanos.
Procedimiento general XXIV
Reducción de amida de Weinreb al aldehído correspondiente

A una solución de amida de Weinreb (1 mmol) en THF seco (5 ml/mmol) en una atmósfera de gas inerte, se añade hidruro de aluminio y litio a 0 °C y la mezcla se agita a esta temperatura hasta que el material de partida se consume
(habitualmente 30 minutos - 2 horas) según lo indique la TLC. La reacción se inactiva con una solución acuosa saturada de sulfato de hidrógeno de potasio y el producto se extrae con éter dietílico (4 x 10 ml). Las capas orgánicas combinadas se lavan con salmuera, se secaron con MgSO4 anhidro, se filtraron y concentraron. El aldehído bruto es suficientemente puro como para usarse directamente en la siguiente etapa.
Procedimiento general XXV
Retirada de terc-butoxicarbonilo (Boc-) a partir de amina seguido de ciclación de un anillo de siete miembros (es decir, benzoxazepina) y la reducción posterior de ¡mina a amina

La sal de clorhidrato de amina bruta del procedimiento de retirada de grupo Boc típico (véase Procedimiento General VII) se suspende 1,2-diclorometano (2 ml/mmol), se añade trietilamina (1,1 equivalentes) y la mezcla se calienta hasta 70 °C durante 1-2 horas. Después de enfriar hasta temperatura ambiente, se añade triacetoxiborohidruro de sodio (2,5 equivalentes) y la mezcla se agita durante la noche. El exceso del agente reductor se descompone con una solución acuosa al 5 % de carbonato de hidrógeno y sodio (dos veces el volumen de DCE usado) y el producto se extrae con diclorometano. Las capas se separan y la capa acuosa se extrae adicionalmente con diclorometano. Los extractos orgánicos combinados se secaron con MgSO4 anhidro, se filtraron y concentraron. La amina bruta es suficientemente pura como para usarse en la siguiente etapa sin purificación adicional.

Síntesis del Ejemplo 1
Ejemplo 1
(S)-3-(4-(3-(4-dorobencil)morfolino)piperidin-1-il)-1 H-1,2,4-triazol-5-amina (1)

Etapa 1
Síntesis de (2S)-2-amino-3-(4-clorofenil)propan-1-ol (1b)

El compuesto del título (1b) se obtuvo como se describe en el Procedimiento General I comenzando con L-pclorofenalanina ópticamente pura (ácido (2S)-2-amino-3-(4-clorofenil)propanoico) - 1a. Se sintetizaron 8,6 g del compuesto 1b (87 % de rendimiento, 46,5 mmol).
ESI-MS m/z para C9H12ClNO encontrado 185,7/187,7 (M+1)+
1H RMN (CDCla, 500 MHz) 57,28 (d, J = 8,3 Hz, 2H), 7,11 (d, J = 8,3 Hz, 2H), 3,62 (dd, J = 10,6, 3,8 Hz, 1H), 3,37 (dd, J = 10,5, 6,9 Hz, 1H), 3,11-3,07 (s a, 1H), 2,76 (dd, J = 13,6, 5,4Hz, 1H), 2,50 (dd, J = 13,6, 8,6 Hz, 1H).
Etapa 2
Síntesis de 2-cloro-N-[(1S)-1-(4-clorobencil)-2-hidroxietil]acetamida (1c)

El compuesto del título se obtuvo a partir del compuesto 1b como se describe en el Procedimiento General II. Se obtuvieron 85 % de rendimiento.
ESI-MS m/z para CnH 1sCl2NO2, encontrado 262,2/264,2 (M+1)+
1H RMN (CDCls, 500 MHz) 58,05 (d, J = 8,5 Hz, 1H), 7,27 (d, J = 8,3 Hz, 2H), 7,18 (d, J = 8,3 Hz, 2H), 4,85 (t, J = 5,5 Hz, 1H), 3,95 (s, 2H), 3,88-3,81 (m, 1H), 3,36-3,27 (m, 2H), 2,8 (dd, J = 5,5,13,7 Hz, 1H), 2,6 (dd, J = 8,6,13,7 Hz, 1H) Etapa 3
Síntesis de (5S)-5-(4-clorobencil)morfolin-3-ona (1d)

El compuesto del título (1d) se obtuvo a partir del compuesto 1c de acuerdo con el Procedimiento General IV en 59 % de rendimiento.
ESI-MS m/z para C11H12CINO2; encontrado 226,2 / 228,2 (M+1)+
1H RMN (CDCla, 500 MHz) 57,30 (d, J = 8,3 Hz, 2H), 7,11 (d, J = 8,3 Hz, 2H), 4,15 (d, J = 2,8 Hz, 2H), 3,87 (dd, J = 3,5,11,8 Hz, 1H), 3,73-3,67 (m, 1H), 3,57-3,52 (m, 1H), 2,85 (dd, J = 6,0, 13,7 Hz, 1H), 2,71 (dd, J = 8,6, 13,7 Hz, 1H) Etapa 4
Síntesis de (5S)-5-(4-clorobencil)morfolina (1e)

El compuesto del título (1e) se obtuvo a partir del compuesto 1d de acuerdo con el Procedimiento General V en 64 % de rendimiento.
ESI-MS m/z para C11H14CINO; encontrado 212,2 / 214,2 (M+1)+
1H RMN (CDCla, 500 MHz) 57,28 (d, J = 8,3 Hz, 2H), 7,11 (d, J = 8,3 Hz, 2H), 3,81-3,75 (m, 2H), 3,55-3,49 (m, 1H), 3,24 (t, J = 10 Hz, 1H), 3,0-2,98 (m, 1H), 2,89-2,8 (m, 2H), 2,62 (dd,J = 4,9, 13,5 Hz, 1H), 2,44 (dd, J = 9,2, 13,5 Hz, 1H)
Etapa 5
Síntesis de (S)-3-(4-clorobencil)-4-(piperidin-4-il)morfolina (1f)

El producto del Etapa 4 (compuesto 1e) se sometió a la reacción de aminación reductiva con N-Boc-piperid-4-ona como se describe en el Procedimiento General VI. El producto bruto de aminación reductiva se sometió directamente a la retirada del grupo Boc de acuerdo con el Procedimiento General VII. La sal de clorhidrato bruta del compuesto del título obtenida de este modo se tomó entre acetato de etilo y 2 M NaOH y la capa orgánica se separó y lavó con 2 M NaOH, agua y salmuera. Se secó sobre MgSO4 anhidro y se filtró y el disolvente se retiró al vacío proporcionando el producto (1f) suficientemente puro para usarse en la siguiente etapa. Se obtuvo 78 % de rendimiento en dos etapas. ESI-MS m/z para C16H23CIN2O; encontrado 295,1 / 297,1 (M+1)+
1H RMN (CDCI3, 500 MHz) 57,26 (d, J = 8,1 Hz, 2H), 7,11 (d, J = 8,1 Hz, 2H), 3,75-3,71 (m, 2H), 3,49 (dd, J = 2,6, 11,2 Hz, 1H), 3,41 (dd, J = 5,5, 11,2 Hz, 1H), 3,2-3,14 (m, 2H), 2,97-2,93 (m, 1H), 2,86 (dd, J = 4,0, 13,5 Hz, 1H), 2,84 2,8 (m, 1H), 2,76-2,7 (m, 2H), 2,68-2,6 (m, 2H), 2,58-2,53 (m, 1H), 1,88-1,84 (m, 1H), 1,82-1,77 (m, 1H), 1,51 (dq, J = 3,8, 11,8 Hz, 1H), 1,41 (dq, J = 3,8, 11,8 Hz, 1H)
Etapa 6
Síntesis de (S)-5-(4-(3-(4-clorobencil)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (1).
El compuesto del título se sintetizó de acuerdo con el Procedimiento General VIII comenzando por el compuesto 1f. Se obtuvieron 56 % de rendimiento.
ESI-MS m/z para C18H25CIN6O; encontrado 377,1 / 379,1 (M+1)+
1H RMN (DMSO-d6, 700 MHz) 57,31 (d, J = 8,3 Hz, 2H), 7,20 (d, J = 8,3 Hz, 2H), 5,70 (s a, 2H), 3,80 (s a, 2H), 3,59 3,53 (m, 2H), 3,36-3,33 (m, 1H), 3,24-3,21 (m, 1H), 2,89-2,83 (m, 2H), 2,80 (s a, 1H), 2,69-2,62 (m, 3H), 2,53-2,51 (m, 2H), 1,77-1,73 (m, 1H), 1,69-1,65 (m, 1H), 1,47 (dq, J = 4,0, 12,0 Hz, 1H), 1,34 (dq, J = 4,2, 12,0Hz, 1H).
Ejemplo 2
Síntesis de 5-(4-(3-(4-clorobencil)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (2)

El compuesto del título 2 (51 % de rendimiento global) se obtuvo de manera similar al Ejemplo 1 comenzando con pclorofenilalanina racémica (ácido 2-amino-3-(4-clorofenil)propanoico).
ESI-MS m/z para C18H25ON6O; 376,9 encontrado 377,1 / 379,1 (M+1)+
1H RMN (DMSO-d6, 700 MHz) 57,31 (d, J = 8,3 Hz, 2H), 7,20 (d, J = 8,3 Hz, 2H), 5,70 (s a, 2H), 3,80 (s a, 2H), 3,59 3,53 (m, 2H), 3,36-3,33 (m, 1H), 3,24-3,21 (m, 1H), 2,89-2,83 (m, 2H), 2,80 (s a, 1H), 2,69-2,62 (m, 3H), 2,53-2,51 (m, 2H), 1,77-1,73 (m, 1H), 1,69-1,65 (m, 1H), 1,47 (dq, J = 4,0, 12,0 Hz, 1H), 1,34 (dq, J = 4,2, 12,0 Hz, 1H).
Ejemplo 3
Síntesis de (S)-5-(4-(3-(4-bromobencil)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (3)
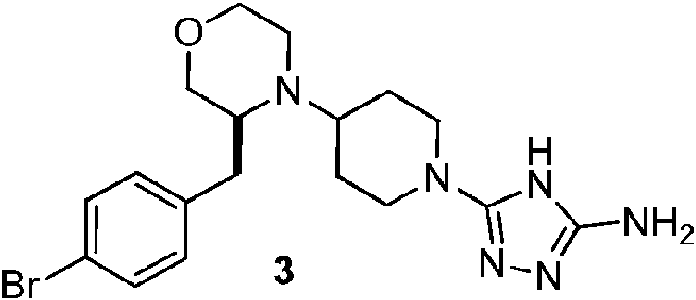
El compuesto del título 3 (62 % de rendimiento global) se obtuvo de acuerdo con un procedimiento similar al ejemplo 1, comenzando con L-p-bromofenilalanina (ácido (2S)-2-amino-3-(4-bromofenil)propanoico).
ESI-LCMS m/z para C18H25BrN6O encontrado 421,5/423,5 (M+1)
1H RMN (DMSO-d6, 500 MHz) 57,50 (d, J = 8,3 Hz, 2H), 7,21 (d, J = 8,3Hz, 2H), 3,84 (s a, 2H), 3,73-3,70 (m, 1H), 3,69 (s a, 1H), 3,67 (s a, 1H), 3,58-3,53 (m, 1H), 3,36-3,31 (m, 1H), 3,26 (s a, 1H), 3,17-3,13 (m, 1H), 2,96-2,88 (m, 2H), 2,81-2,76 (m, 1H), 2,02 (s a, 2H), 1,75-1,62 (m, 2H).
Ejemplo 4
Síntesis de 5-(4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (4)
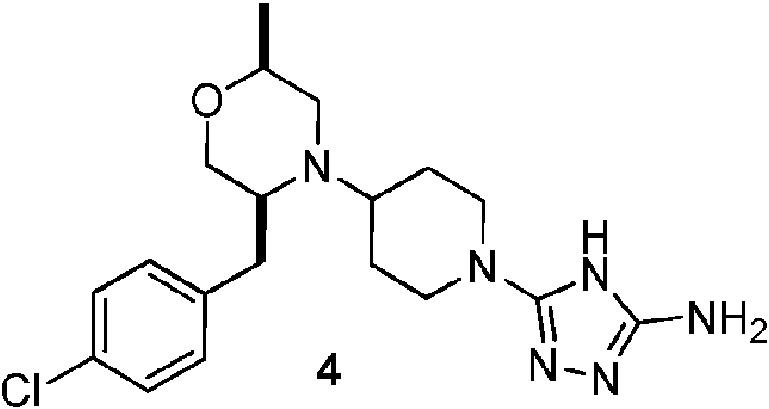
Etapa 1
Síntesis de (R)-2-bromo-N-((S)-1-(4-clorofenil)-3-hidroxipropan-2-il)propanamida (4a)

(2S)-2-amino-3-(4-clorofenil)propan-1-ol (1b) se acopló con ácido (R)-2-bromopropionico en una forma descrita en el Procedimiento General III usando TBTU como un reactivo formador de enlace amida. 1,1 g del compuesto del título 4a se obtuvo de 1,0 g del material de partida (64 % de rendimiento, sólido blanco después de cromatografía de gel de
sílice usando DCM/MeOH 100:1 de sistema disolvente.
ESI-MS m/z para Ci2Hi5BrClNO2 encontrado 320,7/322,7(M+1)+
1H RMN (CDCb, 500 MHz) 57,25 (d, J = 8,2 Hz, 2H), 7,14 (d, J = 8,2 Hz, 2H), 6,58 (d, J = 6,7Hz, 1H), 4,33 (dd, J = 14,1, 7,1 Hz, 1H), 4,13-4,06 (m, 1H), 3,69-3,64 (m, 1H), 3,61-3,57 (m, 1H), 2,90-2,81 (m, 2H), 2,17 (s a, 1H), 1,82 (d, J = 6,9 Hz, 3H).
Etapa 2
Síntesis de (2S,5S)-5-(4-dorobencil)-2-metilmorfolin-3-ona (4b)

El compuesto del título (4b) se obtuvo a partir del 1,1 g del compuesto 4a de acuerdo con el Procedimiento General IV en 79 % de rendimiento (2,71 mmol, 650 mg). El producto bruto de 92 % de pureza por HPLC se usó directamente en una etapa posterior.
ESI-LCMS m/z para C12HmCIN02 encontrado 240,1/242,1(M+1)+
1H RMN (CDCI3, 500 MHz) 57,27 (d, J = 8,2 Hz, 2H), 7,10 (d, J = 8,2 Hz, 2H), 6,13 (s a, 1H), 4,18 (m, 1H), 3,76 (d, J = 3,0 Hz, 2H), 3,55-3,50 (m, 1H), 2,91-2,83 (m, 2H), 1,46 (d, J = 6,9 Hz, 3H)
Etapa 3
Síntesis de (2S,5S)-5-(4-clorobencil)-2-metilmorfolina (4c)

El compuesto del título (4c) se obtuvo a partir de 1,1 g de morfolina 4b de acuerdo con el Procedimiento General V en 90 % de rendimiento (>95 % de pureza).
ESI-MS m/z para C12H16CINO encontrado 226,4/ 228,4 (M+1)+
1H RMN (DMSO-d6+D2O; 500 MHz) 57,33 (d, J = 8,3 Hz, 2H), 7,23 (d, J = 8,3 Hz, 2H), 3,50-3,48 (m, 1H), 3,49-3,47 (m, 2H), 2,87-2,81 (m, 1H), 2,80-2,76 (m, 2H), 2,65 (dd, J = 12,4 Hz, 8,3 Hz, 1H), 2,58 (dd, J = 12,4 Hz, 3,0 Hz, 1H), 1,09 (d, J=6,2Hz, 3H)
Etapa 4
Síntesis de 4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)piperidina-1-carboxilato de tere-butilo (4d)

El compuesto del título (4d) se obtuvo de 4c (6,02 mmol, 1,36 g) de acuerdo con el Procedimiento General VI. Después de la cromatografía en DCM/MeOH 200:1 de sistema de disolvente 4d se obtuvo en 45 % de rendimiento (2,71 mmol, 1,1g).
ESI-LCMS m/z para C22H33ClN2O3 encontrado 409,2/ 411,2 (M+1)+
1H RMN (DMSO-d6+D2O; 500 MHz) 57,33 (d, J = 8,3 Hz, 2H), 7,21 (d, J = 8,3 Hz, 2H), 3,84 (s a, 2H), 3,51-3,46 (m, 1H), 3,44-3,40 (m, 1H), 3,35-3,33 (m, 1H), 2,90-2,85 (m, 3H), 2,82 (s a, 1H), 2,73-2,69 (m, 2H), 2,64-2,60 (m, 1H), 2,28-2,25 (m, 1H), 1,90-1,85 (m, 2H), 1,40 (s, 9H), 1,26-1,21 (m, 1H), 1,20-1,14 (m, 1H), 1,1 (d, J = 6,2 Hz, 3H)
Etapa 5
Síntesis de clorhidrato de (2S,5S)-5-(4-clorobencil)-2-metil-4-(piperidin-4-il)morfolina (4e)

El compuesto del título (4e) se obtuvo a partir del compuesto 4b de acuerdo con el Procedimiento General VII en 79 % de rendimiento, después de la trituración del producto bruto con éter dietílico.
ESI-MS m/z para C17H25ClN2O encontrado 309,9/311,9 (M+1)+
1H RMN (DMSO-d6, 500 MHz): 57,35 (d, J = 8,4 Hz, 2H), 7,33 (d, J = 8,4 Hz, 2H), 4,04 (s a, 2H), 3,59 (s a, 2H), 3,45 (s a, 1H), 3,40 (m, 1H), 3,39-3,36 (m, 2H), 3,10 (s a, 2H), 2,98 (s a, 1H), 2,87 (s a, 2H), 2,31 (s a, 2H), 2,17 (s a, 2H), 1,21 (d, J = 6,2Hz, 3H)
Etapa 6
Síntesis de 5-(4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (4)

El compuesto del título (4) se obtuvo a partir del compuesto 4e (1,86 mmol, 640 mg) de acuerdo con el Procedimiento General VIII en 72 % de rendimiento (1,34 mmol, 455 mg), después de la cromatografía de gel de sílice en AcOEt/MeOH (100:1) de sistema de disolvente seguido de cristalización de MeOH/Et2 O.
ESI-MS m/z para C19H27ClN6O encontrado 341,8/343,8 (M+1)+
1H RMN (DMSO-d6, 500 MHz): 57,39 (d, J = 8,3 Hz, 2H), 7,30 (d, J = 8,3 Hz, 2H), 3,87-3,79 (m, 3H), 3,66 (s a, 2H), 3,58 (s a, 1H), 3,52-3,45 (m, 2H), 3,09 (s a, 2H), 3,00-2,93 (m, 2H), 2,92-2,88 (m, 1H), 2,20 (s a, 2H), 1,67 (s a, 2H), 1,20 (d, J = 6,2 Hz, 3H)
Ejemplo 5
Síntesis de 5-(4-((2S,5S)-5-(4-clorobencil)-2-etilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (5)

El compuesto del título se preparó de la misma forma que el Ejemplo 4 con la excepción de que se usó ácido (2R)-2-bromobutanoico en vez de ácido (2R)-2-bromopropanoico en la primera etapa sintética.
ESI-MS m/z para C20H29ClN6O encontrado 405,1/407,0 (M+1)+, 403,1/405,1 (M-1)
1H RMN (DMSO-d6 +D2 O, 500 MHz): 57,38 (d, Jaabb' = 8,5 Hz, 2H), 7,27 (d, Jaabb' = 8,3Hz, 2H), 3,81 (d a, J = 12,8 Hz, 2H), 3,67-3,69 (m, 1H), 3,52-3,56 (m, 4H), 3,38-3,41 (m, 1H), 2,99-3,06 (m, 3H), 2,85-2,93 (m, 2H), 2,15-2,21 (m, 2H), 1,48-1,60 (m, 4H), 0,89 (t, J = 7,5 Hz, 3H).
Ejemplo 6
Síntesis de 5-(4-((2S,5S)-5-(4-dorobencil)-2-isopropilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (6)

El compuesto del título 6 se preparó de la misma forma que el Ejemplo 4 con la excepción de que se usó ácido (2R)-2-bromo-3-metilbutanoico en vez de ácido (2R)-2-bromopropanoico en la primera etapa sintética.
ESI-MS m/z para C21H31ClNsO encontrado 419,0/ 421,0 (M+1)+
1H RMN (DMSO-da, 700 MHz): 510,15 (s, 1H), 7,45-7,41 (m, 2H), 7,36-7,31 (m, 2H), 7,05 (s a, 2H), 3,88 (t, J = 16,8 Hz, 2H), 3,83-3,75 (m, 1H), 3,63-3,53 (m, 3H), 3,49-3,37 (m, 2H), 3,17-3,07 (m, 1H), 3,03 (t, J = 12,5 Hz, 1H), 2,94 (t, J = 12,8 Hz, 1H), 2,84 (t, J = 12,5 Hz, 1H), 2,28 (d, J = 12,0 Hz, 1H), 2,22 (d, J = 12,2 Hz, 1H), 1,82 (ddd, J = 13,7, 9,4, 5,9 Hz, 1H), 1,69-1,59 (m, 2H), 0,97 (d, J = 6,9 Hz, 3H), 0,96 (d, J= 6,9Hz, 3H).
Ejemplo 7
Síntesis de 5-(4-((2S,5S)-5-(4-clorobencil)-2-isobutilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (7)

El compuesto del título 7 se preparó de la misma forma que el Ejemplo 4 con la excepción de que se usó ácido (2R)-2-bromo-4-metilpentanoico en vez de ácido (2R)-2-bromopropanoico en la primera etapa sintética.
ESI-MS m/z para C22H33ClNsO encontrado 433,1 /434,9 (M+1)+
1H RMN (DMSO-da, 700 MHz): 57,43 (d, J = 8,4 Hz, 2H), 7,34 (d, J = 8,4 Hz, 2H), 6,99 (s a, 1H), 3,89-3,85 (m, 3H), 3,79-3,47 (m, 7H), 3,12 (d, J = 7 Hz, 2H), 3,00 (s, 1H), 2,95-2,91 (m, 1H), 2,86-2,83 (m, 1H), 2,23-2,18 (m, 2H), 1,82 1,76 (m, 1H), 1,64-1,59 (m, 2H), 1,53-1,49 (m, 1H), 1,31-1,27 (m, 1H), 0,91-0,87 (m, 6H).
Ejemplo 8
Síntesis de (2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)morfolina-2-carboxamida (8)

Etapa 1
Síntesis de (S)-2-(((S)-3-((ferc-butildimetilsilil)oxi)-2-hidroxipropil)amino)-3-(4-dorofenil)propan-1-ol (8a)

Una solución e (S)-ferc-butildimetil(oxiran-2-ilmetoxi)silano (4,5 ml, 26,54 mmol) y (2S)-2-amino-3-(4-clorofenil)propan-1-ol (1b) (4,9 g, 26,39 mmol) en 1-propanol se calentó a reflujo durante 15 horas. La solución amarilla resultante se concentró al vacío y el compuesto del título se aisló por cromatografía ultrarrápida en gel de sílice (gradiente de 0 a 10 % de MeOH en AcOEt). Se obtuvieron 6,06 g (15,6 mmol) del compuesto 8a (75 % de rendimiento).
ESI-MS m/z para C18Ha2ClNOaSi encontrado 374,0 /376,0 (M+1)+
1H RMN (CDCla, 500 MHz) 87,25 (AA'BB', J = 8,3 Hz, 2H), 7,12 (AA'BB', J = 8,1Hz, 2H), 3,72-3,65 (m, 1H), 3,65-3,52 (m, 3H), 3,39-3,31 (m, 1H), 2,9-2,83 (m, 1H), 2,83-2,65 (m, 3H), 2,64-2,58 (m, 2H), 0,88 (s, 9H), 0,05 (s, 6H).
Etapa 2
Síntesis de (S)-3-((ferc-butildimetilsilil)oxi)-N-((S)-1-(4-clorofenil)-3-((trimetilsilil)oxi)propan-2-il)-2-((trimetilsilil)oxi)propan-1-amina (8b)

Se añadieron hexametildisilazano (HMDS, 6,9 ml, 33,21 mmol) y cloruro de trimetilsililo (TMSCl, 0,4 ml, 3,24 mmol) secuencialmente a una solución de amino diol 8a (6,06 g, 16,2 mmol) en THF (160 ml) a 0 °C. Después de 2 minutos, se retiró el baño de enfriamiento y la suspensión blanca resultante se agitó a TA durante 70 minutos, después se añadió la porción adicional de TMSCl (0,8 ml, 6,44 mmol) y la suspensión se agitó durante 30 minutos más. La mezcla de reacción se dividió entre éter y una mezcla 1/1 de una solución de amortiguador de fosfato acuosa (0,05 M) y salmuera (200 ml). Se separó la capa orgánica y se extrajo la capa acuosa con éter. Los extractos orgánicos combinados se secaron sobre MgSO4 y se concentraron, proporcionando bis-trimetilsilil éter 8b como un líquido amarillo claro (8,3 g, 99 % de rendimiento).
1H RMN (CDCb, 500 MHz) 87,24 (AA'BB', J = 8,1 Hz, 2H), 7,13 (AA'BB', J = 7,7Hz, 2H), 3,78-3,72 (m, 1H), 3,65-3,40 (m, 4H), 2,92-2,51 (m, 4H), 1,87-1,82 (m, 1H), 1,70-1,68 (m, 1H), 0,88 (s, 9H), 0,11 (s, 9H), 0,07 (s, 9H), 0,05 (s, 6H)
Etapa 3
Síntesis de N-((S)-3-((ferc-butildimetilsilil)oxi)-2-((trimetilsilil)oxi)propil)-N-((S)-1-(4-clorofenil)-3-((trimetilsilil)oxi)propan-2-il)-4-metilbencenosulfonamida (8c)

El compuesto del título (8c) se obtuvo a partir del compuesto 8b (8,3 g, 16,01 mmol) de acuerdo con el Procedimiento General XV en 71 % de rendimiento (7,6 g, aceite incoloro), después de la cromatografía de gel de sílice en AcOEt/hexano 1:10 de sistema de disolvente. (8,3 g, 16,01 mmol)
1H RMN (CDCb, 500 MHz) 87,65 (d, 2H, J = 8,2 Hz), 7,19 (m, 4H), 7,08 (d, J = 8,2 Hz, 2H), 4,14-4,05 (m, 1H), 4,00 3,90 (m, 1H), 3,62-3,52 (m, 2H), 3,51-3,45 (m, 1H), 3,41 (dd, J = 5,2, 15,2 Hz, 1H), 3,36 (dd, J = 5,1, 11,0 Hz, 1H), 3,14 (dd, J = 6,9, 15,4 Hz, 1H), 2,95-2,83 (m, 2H), 2,41 (s, 3H), 0,91 (s, 9H), 0,16 (s, 9H), 0,06 (s, 9H), -0,11 (s, 6H).
Etapa 4
Síntesis de N-((S)-3-((ferc-butildimetilsilil)oxi)-2-hidroxipropil)-N-((S)-1-(4-clorofenil)-3-hidroxipropan-2-il)-4-metilbencenosulfonamida (8d)

Se añadió metóxido de sodio (122 mg, 2,26 mmol) en una porción a una solución de sulfonamida 8c (7,6 g, 11,3 mmol) en metanol (112 ml) a TA. La solución resultante se agitó durante 40 minutos y después se concentró al vacío. El concentrado se dividió entre acetato de etilo y una mezcla 1/1 de una solución acuosa saturada de cloruro de amonio y salmuera. La capa orgánica se separó y la capa acuosa se extrajo con una porción adicional de acetato de etilo. Los orgánicos combinados se secaron sobre MgSO4 y se filtraron en seco y se concentraron para proporcionar diol 8d como un sólido blanco (5,47 g, 92 % de rendimiento).
ESI-MS m/z para C25H38ClNO5SSi encontrado 528.3 /530.3 (M+1)+
1H RMN (CDCl3, 500 MHz) 87.60 (d, J = 8,3 Hz, 2H), 7,23 (d, J = 8,1 Hz, 2H), 7,14 (d, J = 8,3 Hz, 2H), 6,93 (d, J = 8,3 Hz, 2H), 4,09-4,00 (m, 1H), 3,91-3,83 (m, 1H), 3,72-3,57 (m, 4H), 3,51-3,27 (m, 4H), 3,04-2,96 (m, 1H), 2,74 (dd, J = 8,2, 13,7 Hz, 1H), 2,58 (dd, J= 6,5, 13,7 Hz, 1H), 2,42 (s, 3H), 0,91 (s, 9H), 0,09 (s, 6H).
Etapa 5
Síntesis de (S)-2-(N-((S)-3-((ferc-butildimetilsilil)oxi)-2-hidroxipropil)-4-metilfenilsulfonamido)-3-(4-clorofenil)propil 4-metilbencenosulfonato (8e)
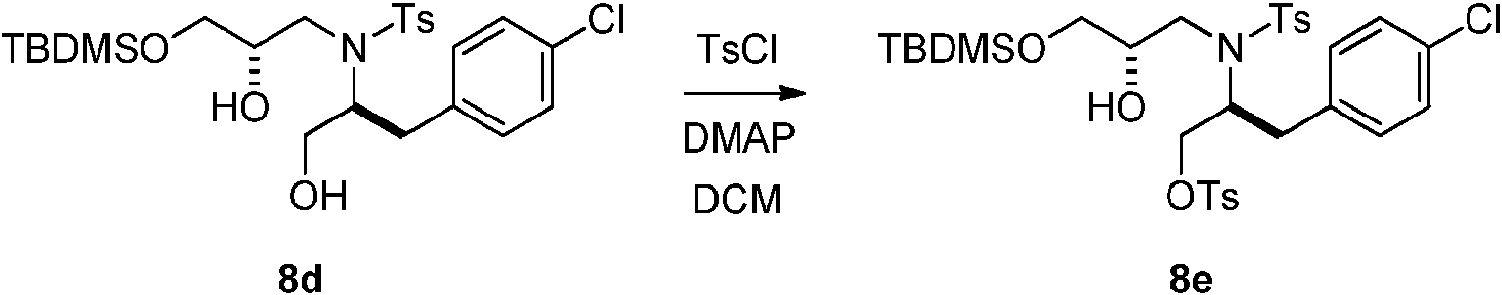
A una solución de 8d (5,47 g, 10,35 mmol), se añadieron secuencialmente trietilamina (Et3N, 5,9 ml, 41,4 mmol) en DCM, 4-dimetilaminopiridina (DMAP, 0,5 g, 4,14 mmol) y cloruro de tosilo (TsCl, 2,0 g, 10,86 mmol) y la solución resultante se agitó a TA durante 1 hora. La reacción se lavó con solución saturada de cloruro de amonio y salmuera. Los orgánicos se secaron sobre MgSO4 y se concentraron. El residuo oleaginoso se purificó mediante cromatografía ultrarrápida (acetato de etilo / hexanos), para proporcionar 8e como un aceite transparente (5,6 g, 80 %).
ESI-MS m/z para C32H44ClNO7S2Si encontrado 705,4 /707,4 (M+Na)+
1H RMN (CDCl3, 500 MHz) 87,63 (d, J = 8,3 Hz, 2H), 7,53 (d, J = 8,3 Hz, 2H), 7,3 (d, J = 7,9 Hz, 2H), 7,19 (d, J = 8,1 Hz, 2H), 7,09 (d, J = 8,3Hz, 2H), 6,91 (d, J = 8,3 Hz, 2H), 4,22 (dd, J = 6,7, 10,3 Hz, 1H), 4,13-4,04 (m, 1H), 3,96 (dd, J = 5,4, 10,3 Hz, 1H), 3,80-3,72 (m, 1H), 3,58-3,47 (m, 2H), 3,38-3,33 (m, 1H), 3,13 (dd, J = 8,1, 15,8 Hz, 1H), 2,95 2,83 (m, 3H), 2,45 (s, 3H), 2,42 (s, 3H), 0,9 (s, 9H), 0,08 (s, 6H)
Etapa 6
Síntesis de (2S,5S)-2-(((ferc-butildimetilsilil)oxi)metil)-5-(4-clorobencil)-4-tosilmorfolina (8f)

Se añadió carbonato de potasio (2,3 g, 16,41 mmol) a una solución de tosilato 8e (5,6 g, 8,2 mmol) en ferc-butil alcohol (300 ml). La mezcla resultante se calentó a reflujo durante 2 horas y después se dividió entre acetato de etilo y una mezcla 1/1 de solución acuosa saturada de cloruro de amonio y salmuera. La capa orgánica se separó y la capa acuosa se extrajo con una porción adicional de acetato de etilo. Los extractos orgánicos combinados se secaron en MgSO4 y los extractos secos se concentraron. El residuo se purificó por cromatografía en columna ultrarrápida (AcOEt/hexanos 1/20) para proporcionar N-tosil morfolina 8f como un sólido blanco (2,7 g, 66 % de rendimiento).
ESI-MS m/z para C25H36CINO4SSÍ encontrado 533,4 /535,4 (M+Na)+
1H RMN (CDCI3, 500 MHz,) 57,65 (d, J = 8,3 Hz, 2H), 7,29 (d, J = 8,1 Hz, 2H), 7,22 (d, J = 8,3 Hz, 2H), 7,08 (d, J = 8,3 Hz, 2H), 3,84-3,77 (m, 1H), 3,70-3,61 (m, 3H), 3,54 (dd, J = 2,6, 12,0 Hz, 1H), 3,49 (dd, J = 3,9, 12,8 Hz, 1H), 3,40 (dd, J = 3,7, 12,7 Hz, 1H), 3,35 (dd, J = 2,4, 12,0 Hz, 1H), 3,03 (dd, J = 11,0, 13,3 Hz, 1H), 2,89 (dd, J = 4,1, 13,3 Hz, 1H), 2,42 (s, 3H), 0,85 (s, 9H), 0,02 (s, 3H), 0,01 (s, 3H).
Etapa 7
Síntesis de ((2S,5S)-5-(4-clorobencil)-4-tosilmorfolin-2-il)metanol (8g)

Una solución de N-tosil morfolina 8f (0,63 g, 1,23 mmol) en THF (2 ml) se trató con fluoruro de tetrabutilamonio (TBAF) (2,5 ml, 2,46 mmol, 1 M en THF) a temperatura ambiente durante 2 horas. La mezcla de reacción se absorbió en gel de sílice y se purificó por cromatografía en columna (AcOEt/hexanos, después AcOEt puro) para dar alcohol 8g (0,56 g, 99 % de rendimiento).
ESI-MS m/z para C19H22ONO4S encontrado 396,0 /398,0 (M+1)+
1H RMN (CDCla, 500 MHz) 57,58 (d, J = 8,1 Hz, 2H), 7,27 (d, 8,4 Hz, 2H), 7,18 (d, 8,3 Hz, 2H), 7,03 (d, J = 8,3 Hz, 2H), 3,93-3,86 (m, 1H), 3,81-3,66 (m, 4H), 3,53 (dd, J = 3,9, 13,1 Hz, 1H), 3,45-3,37 (m, 2H), 3,00-2,87 (m, 2H), 2,43 (s, 3H).
Etapa 8
Síntesis de ácido (2S,5S)-5-(4-clorobencil)-4-tosilmorfolina-2-carboxílico (8h)
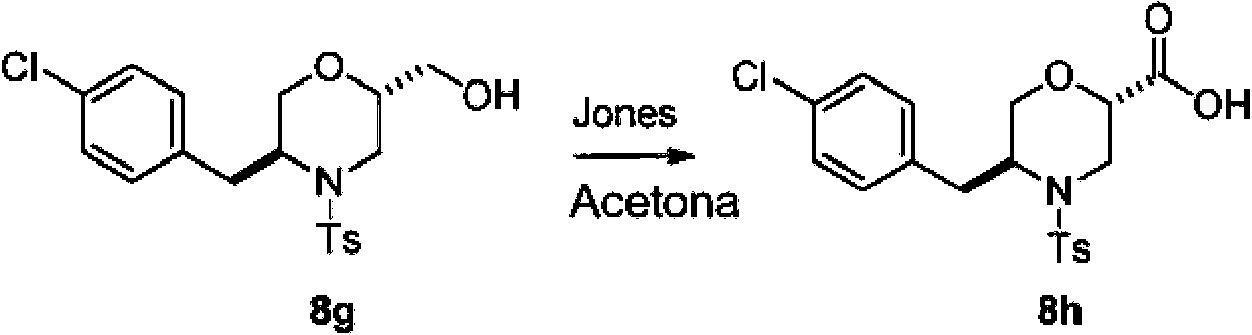
A una solución enfriada a 0 °C de alcohol 8g (0,97 g, 2,45 mmol) en acetona, se añadió reactivo de Jones (solución de trióxido de cromo en ácido sulfúrico diluido y acetona) gota a gota hasta que se consumió todo el sustrato (4,3 ml de 2,6 M de reactivo de Jones). Después la reacción se diluyó con AcOEt, se lavó posteriormente con agua y una mezcla de salmuera/0,5 M EDTA, se secó y concentró para proporcionar ácido 8h como una espuma blanca (0,92 g, 92 % de rendimiento).
ESI-MS m/z para C19H20ONO5S encontrado 432,0 /434,0 (M+Na)+
1H RMN (CDCla, 500 MHz) 57,65 (d, J = 7,9 Hz, 2H), 7,30-7,22 (m, 4H), 7,11 (d, J = 8,3 Hz, 2H), 4,55-4,49 (m, 1H), 4,08-4,00 (m, 1H), 3,90-3,83 (m, 2H), 3,63-3,50 (m, 2H), 3,14-3,04 (m, 1H), 2,83-2,76 (m, 1H), 2,41 (s, 3H).
Etapa 9
Síntesis de (2S,5S)-5-(4-clorobencil)-4-tosilmorfolina-2-carboxamida (8i)

El compuesto del título (8i) se obtuvo a partir del compuesto 8h (0,9 g, 2,19 mmol) de acuerdo con el Procedimiento General IX en 92 % de rendimiento (2,01 mmol, 0,82 g) como una espuma blanca.
ESI-MS m/z para C19H21CIN2O4S encontrado 409,0/ 411,0 (M+1)+, 431,0 /433,0 (M+Na)+
1H RMN (CDCI3, 500 MHz) 57,72 (d, J = 8,1 Hz, 2H), 7,30 (d, J = 7,9 Hz, 2H), 7,26 (d, J = 7,3 Hz, 2H), 7,13 (d, J = 8,3 Hz, 2H), 6,35 (s a, 1H), 5,63 (s a, 1H), 4,30-4,25 (m, 1H), 3,95-3,87 (m, 1H), 3,69-3,58 (m, 2H), 3,57-3,45 (m, 2H), 3,08-3,01 (m, 2H), 2,42 (s, 3H).
Etapa 10
Síntesis de (2S,5S)-5-(4-dorobencil)morfolina-2-carboxamida (8j)

Se añadió naftaleno (1,4 g, 10,95 mmol) en una porción a una suspensión vigorosamente agitada de sodio (0,31 g, 13,68 mmol) en THF seco (6,8 ml, 0,5 ml / 1 mmol de Na). La suspensión verde resultante se agitó durante 2 horas a temperatura ambiente. Después la solución verde se añadió gota a gota en una solución de amida 8i (0,82 g, 2,00 mmol) en THF (30 ml) a -70 °C hasta que la solución de reacción cambió a verde oscura (se añadieron 5 ml de NaC10H8). La reacción se inactivó después de 10 minutos a -70 °C con solución saturada de cloruro de amonio y se dejó calentar hasta TA. Después la mezcla se dividió entre éter y una mezcla de NaHCO3 y salmuera. La fase orgánica se separó, se secó sobre MgSO4 y se concentró. El residuo se purificó por cromatografía en columna de AcOEt/hexanos 1/2 después AcOEt puro después AcOEt/ MeOH 2/1 de sistema de disolvente. La morfolina 8j se obtuvo como un sólido blanco (0,29 g, 56 % de rendimiento).
ESI-MS m/z para C12H15ClN2O2 encontrado 255,6/ 257,6 (M+1)+
1H RMN (CDCl3, 500 MHz) 57,28 (d, J = 8,3 Hz, 2H), 7,12 (d, 8,1 Hz, 2H), 6,46 (s a, 1H), 5,65 (s a, 1H), 3,98-3,93 (m, 1H), 3,91-3,86 (m, 1H), 3,39-3,29 (m, 2H), 2,99-2,90 (m, 1H), 2,70- 2,59 (m, 2H), 2,54-2,45 (m, 1H).
Etapa 11
Síntesis de 4-((2S,5S)-2-carbamoil-5-(4-clorobencil)morfolino)piperidina-1-carboxilato de ferc-butilo (8k)

El compuesto del título (8k) se obtuvo a partir del compuesto 8j (0,29 g, 1,13 mmol) de acuerdo con el Procedimiento General VI en 32 % de rendimiento (3,65 mmol, 0,16 g), después de la cromatografía de gel de sílice (elución de gradiente AcOEt/hexano 1/1, después AcOEt puro, después AcOEt/ MeOH 10/1).
ESI-MS m/z para C22H32ClN3O4 encontrado 438,2/ 440,2 (M+1)+
Etapa 12
Síntesis de clorhidrato de (2S,5S)-5-(4-clorobencil)-4-(piperidin-4-il)morfolina-2-carboxamida (8l)

El grupo protector de Boc se retiró del compuesto 8k (0,37 mmol, 160 mg) de acuerdo con el Procedimiento General VII proporcionando el compuesto del título bruto 8l en 87 % de rendimiento (120 mg) que se usó directamente en la siguiente etapa.
Etapa 13
Síntesis de (2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)morfolina-2-carboxamida (8)

El anillo de triazol se instaló en la piperidina 8l de acuerdo con el Procedimiento General VIII y el producto deseado (8) se aisló por cromatografía HPLC preparativa. Las fracciones que contienen el producto se combinaron y liofilizaron proporcionando el compuesto del título 8 como un polvo blanco (28 % de rendimiento).
ESI-MS m/z para C-igH26ClN7O2 encontrado 420,1/ 422,1 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 57,36 (AA'BB', J = 8,3 Hz, 2H), 7,30 (AA'BB', J = 8,3Hz, 2H), 7,21 (s a, 1H), 7,08 (s a, 1H), 3,89-3,84 (m, 1H), 3,81-3,74 (m, 2H), 3,57-3,53 (m, 1H), 3,21-3,18 (m, 1H), 3,11-3,04 (m, 1H), 3,02-2,92 (m, 2H), 2,87-2,80 (m, 1H), 2,73-2,64 (m, 1H), 2,50-2,44 (m, 2H), 2,27-2,19 (m, 1H), 1,67-1,57 (m, 2H), 1,59-1,49 (m, 1H), 1,35 1,27 (m, 1H).
Ejemplo 9
(2R,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)morfolina-2-carboxamida (9)

Con la excepción de que TBDMS-(R)-glicidil éter (en vez de su (S)-enantiómero) se hizo reaccionar con (2S)-2-amino-3-(4-clorofenil)propan-1-ol (2b) en la primera etapa sintética, el compuesto del título 9 se preparó de la misma forma que el Ejemplo 8.
ESI-MS m/z para C19H26GN7O2 encontrado 420,0/ 422,0 (M+1)+
1H RMN (DMSO-d6+D2O, 700 MHz) 57,38 (d, J = 8,5 Hz, 2H), 7,34 (d, J = 8,4 Hz, 2H), 4,20 (s a, 2H), 3,83 (d, J = 13,8 Hz, 2H), 3,60-3,47 (m,5H), 3,65 (s, 2H), 3,30-3,20 (m, 1H), 3,09-3,02 (m, 2H), 2,91-2,81 (m, 2H), 2,16-2,08(m, 2H), 1,64-1,54(m, 2H).
Ejemplo 10
5-(4-((2R,5S)-5-(4-clorobencil)-2-(metoximetil)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (10)

Etapa 1
Síntesis de (2R,5S)-5-(4-clorobencil)-2-(metoximetil)-4-tosilmorfolina (10a)

El compuesto del título (10a) se obtuvo a partir del compuesto 8g (0,8 g, 2,02 mmol) de acuerdo con el Procedimiento General XXI en 95 % de rendimiento (1,9 mmol, 0,78 g, sólido blanco), después de la cromatografía de gel de sílice (elución de gradiente AcOEt/hexanos 1:10 a 1:1).
ESI-MS m/z para C2üH24CINO4S encontrado 410,0/ 412,0 (M+1)+
1H RMN (CDCla, 500 MHz,) 57,57 (d, J = 8,3 Hz, 2H,), 7,24 (d, J = 8,1 Hz, 2H), 7,20 (d, J = 8,4 Hz, 2H), 7,08 (d, J = 8,4 Hz, 2H), 3,97-3,91 (m, 1H), 3,70-3,66 (m, 1H), 3,62 (dd, J = 2,8, 13,3 Hz, 1H), 3,59-3,54 (m, 1H), 3,48-3,42 (m, 2H), 3,47-3,41 (m, 1H), 3,37 (s, 3H), 3,06 (dd, J = 11,1, 13,1 Hz, 1H), 2,97 (dd, J = 9,6, 13,3 Hz, 1H), 2,74 (dd, 5,6, 13,3 Hz, 1H), 2,40 (s, 3H).
Etapa 2
Síntesis de (2R,5S)-5-(4-clorobencil)-2-(metoximetil)morfolina (10b)

El grupo protector de tosilo se retiró del compuesto 10a (780 mg, 1,9 mmol) en una manera descrita anteriormente para el compuesto 8i. Se obtuvieron 340 mg del compuesto del título 10b en 70 % de rendimiento.
ESI-MS m/z para C13H18CINO2 encontrado 255,8/ 257,8 (M+1)+
1H RMN (CDCI3, 500 MHz,) 57,27 (AA'BB', J = 8,3 Hz, 2H), 7,14 (AA'BB', J = 8,3Hz, 2H), 3,83-3,70 (m, 3H), 3,56 (dd, J = 6,4, 10,1 Hz, 1H), 3,45 (dd, J = 3,9, 10,1 Hz, 1H), 3,39 (s, 3H), 3,09-3,03 (m, 1H), 3,02-2,95 (m, 2H), 2,90 (dd, J =7,1, 13,3 Hz, 1H), 2,81 (dd, J = 3,0, 12,2 Hz, 1H).
Etapa 3
Síntesis de 4-((2R,5S)-5-(4-clorobencil)-2-(metoximetil)morfolino)piperidina-1-carboxilato de tere-butilo (10c)

El compuesto del título (10c) se obtuvo a partir del compuesto 10b (1,51 mmol, 340 mg) de acuerdo con el Procedimiento General VI en 87 % de rendimiento.
ESI-MS m/z para C23H35CIN2O4 encontrado 439,2/ 441,2 (M+1)+
Etapa 4
Síntesis de (2R,5S)-5-(4-clorobencil)-2-(metoximetil)-4-(piperidin-4-il)morfolina (10d)

El grupo protector de Boc se retiró del compuesto 10c (1,03 mmol, 450 mg) de acuerdo con el Procedimiento General VII proporcionando el compuesto del título 10d (99 % de rendimiento).
ESI-m S m/z para C18H27ClN2O2 encontrado 339,0/ 341,0 (M+1)+
Etapa 5
Síntesis de 5-(4-((2R,5S)-5-(4-clorobencil)-2-(metoximetil)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (10)
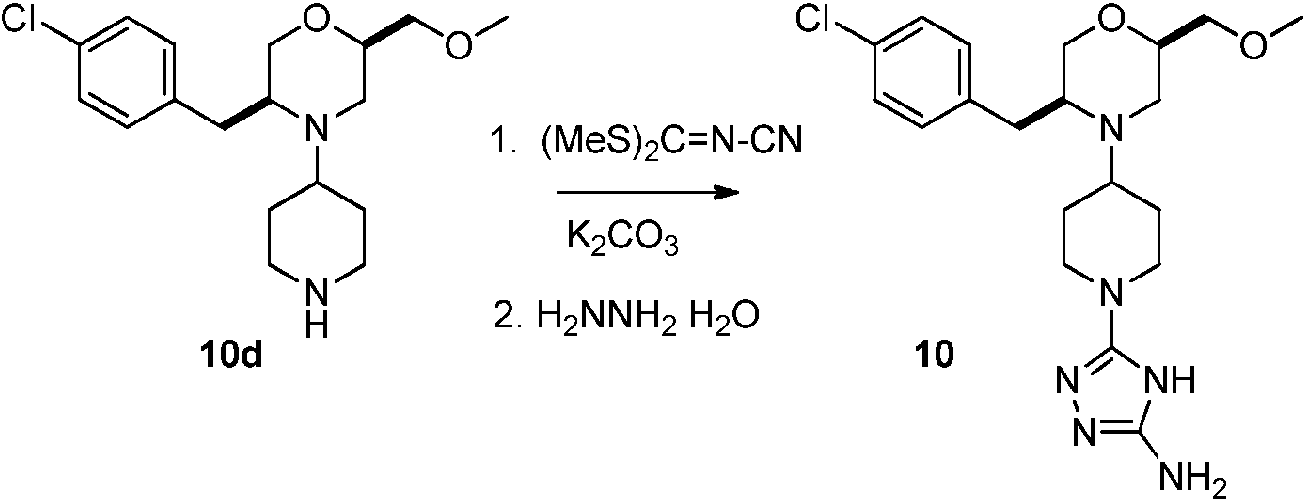
El compuesto del título (10) se sintetizó a partir del compuesto 10d (1,03 mmol, 410 mg) usando el Procedimiento General VIII en 55 % de rendimiento (0,56 mmol, 237 mg).
ESI-MS m/z para C2üH2gClN6O2 encontrado 421,1/ 423,1 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 57,42 (AA'BB', J = 8,3Hz, 2H), 7,33 (AA'BB', J = 8,2 Hz, 2H),3,91-3,80 (m, 4H), 3,64 3,53 (m, 3H), 3,53-3,44 (m, 3H), 3,30 (s, 3H), 3,15-3,02 (m, 3H), 2,94-2,86 (m, 1H), 2,85-2,79 (m, 1H), 2,21-2,12 (m, 2H), 1,66-1,55 (m, 2H).
Ejemplo 11
5-(4-((2R,5S)-5-(4-clorobencil)-2-(ethoximetil)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (11)

Etapa 1
Síntesis de (R)-2-bromo-3-(ferc-butoxi)-N-((S)-1-(4-clorofenil)-3-hidroxipropan-2-il)propanamida (11a)

(2S)-2-amino-3-(4-clorofenil)propan-1-ol (2b) (10,2 mmol, 1,89 g) se acopló con ácido (2R)-2-bromo-3-fercbutoxipropanoico de acuerdo con el Procedimiento General III usando TBTU como un reactivo formador de enlace amida. El compuesto del título 11a se obtuvo en 70 % de rendimiento, 2.96 g).
ESI-MS m/z para C16H23BrClNO3 encontrado 415,3/ 417,3 (M+Na)+
Etapa 2
Síntesis de (2S,5S)-2-(ferc-butoximetil)-5-(4-clorobencil)morfolin-3-ona (11b)

A una solución del compuesto 11a (1,2 g, 3,05 mmol) en THF seco (30 ml), se añadió hidruro de sodio (NaH) (0,44 g, 9,16 mmol) en una porción y después la mezcla de reacción se agitó a temperatura ambiente durante 1 hora. La reacción se inactivó cuidadosamente con 2 M HCl y se extrajo con éter etílico. Los orgánicos se lavaron con agua, salmuera, se secaron sobre MgSO4 y se concentraron para dar el compuesto del título 11b como un aceite amarillo (0.85 g, 89 % de rendimiento), que era lo suficientemente puro como para llevarse a la siguiente etapa.
ESI-MS m/z para C16H22CIn O3 encontrado 334,1/336,1 (M+Na)+
Etapa 3
Síntesis de ((2R,5S)-5-(4-clorobencil)morfolin-2-il)metanol (11c)

El compuesto 11b (0,85 g, 2,72 mmol) se disolvió en 27 ml de THF seco y se añadió cuidadosamente un complejo de borano-dimetilsulfuro (0,8 ml, 8,17 mmol) y la mezcla de reacción se calentó con agitación por aprox. 24 horas. Después de este tiempo, la TLC reveló el consumo completo del material de partida. La mezcla de reacción se inactivó cuidadosamente con 2 M HCl y se siguió calentando durante 1 hora. La mezcla de reacción se enfrió y se lavó con éter. La fase de agua se alcalizó hasta pH 12 y se extrajo con éter. La fase orgánica se secó sobre MgSO4 y se concentró. Se obtuvieron 0,48 g (74 % de rendimiento) del compuesto del título 11c.
ESI-MS m/z para C12H16CINO2 encontrado 242,2/ 244,2 (M+1)+
Etapa 4
Síntesis de 4-((2R,5S)-5-(4-dorobencil)-2-(hidroximetil)morfolino)piperidina-1-carboxilato de ferc-butilo (11d)

La aminación reductiva del compuesto 11c (0,45 g, 1,86 mmol) y Boc-piperid-4-ona se logró de acuerdo con el Procedimiento General VI. El compuesto del título 11d se obtuvo en 56 % de rendimiento (0,44 g, 1,04 mmol).
ESI-MS m/z para C22H33ClN2O4 encontrado 425,1/ 427,1 (M+H)+
1H RMN (CDCl3, 700 MHz) 57,28 (AA'BB', J = 8,4Hz, 2H), 7,21 (AA'BB', J = 8,3 Hz, 2H), 4,14-3,98 (m, 2H), 3,72-3,61 (m, 4H), 3,53-3,48 (m, 1H), 3,00-2,91 (m, 2H), 2,91-2,80 (m, 2H), 2,72-2,62 (m, 3H), 2,55-2,49 (m, 1H), 1,97-1,85 (m, 2H), 1,47 (s, 9H).
Etapa 5
Síntesis de 4-((2R,5S)-5-(4-clorobencil)-2-(etoximetil)morfolino)piperidina-1-carboxilato de ferc-butilo (11e)

El compuesto 11d se O-alquiló de acuerdo con el Procedimiento General XXI. El compuesto del título 11e se obtuvo en 74 % de rendimiento (0,34 g, 0,75 mmol).
ESI-MS m/z para C24H37ClN2O4 encontrado 453,1/ 455,1 (M+1)+
Etapa 6
Clorhidrato de (2R,5S)-5-(4-clorobencil)-2-(ethoximetil)-4-(piperidin-4-il)morfolina (11f)

El grupo protector de Boc se retiró del compuesto 11e de acuerdo con el Procedimiento General VII proporcionando el compuesto del título (10f), suficientemente puro para llevarse a la siguiente etapa, en 95 % de rendimiento.
ESI-m S m/z para C19H29ClN2O2 encontrado 353,2/ 355,2 (M+1)+
Etapa 7
Síntesis de 5-(4-((2R,5S)-5-(4-clorobencil)-2-(etoximetil)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (11)

El compuesto del título (11) se obtuvo a partir del compuesto 11f usando el Procedimiento General VIII en 45 % de rendimiento (185 mg, 0,43 mmol).
ESI-MS m/z para C21H31GN6O2 encontrado 435,1/ 437,1 (M+1)+
1H RMN (DMSO-d6+D2O, 700 MHz) 57,35 (AA'BB', J = 8,3 Hz, 2H), 7,25 (AA'BB', J = 8,5 Hz, 2H), 3,86-3,74 (m, 3H), 3,71-3,66 (m, 1H), 3,64-3,38 (m, 8H), 3,16-3,00 (m, 3H), 2,98-2,88 (m, 2H), 2,22-2,14 (m, 2H), 2,67-2,49 (m, 2H), 1,07 (t, J = 7,1 Hz, 3H).
Ejemplo 12
(2R,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-N-metilmorfolina-2-carboxamida (12)

Etapa 1
Síntesis de (2R,5S)-5-(4-clorobencil)-2-(hidroximetil)morfolina-4-carboxilato de tere-butilo (12a)

A una solución de aminoalcohol 11c (2,87 g, 11,9 mmol) en diclorometano (110 ml), se añadió di-fere-butil dicarbonato (Boc2O) (2,46 g, 11,3 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 2 horas, tiempo después del cual la TLC mostró un consumo casi completo del material de partida. Los volátiles se retiraron al vacío y el residuo se purificó por cromatografía en columna en AcOEt/hexanos 1:1 proporcionando aminoalcohol protegido con N-Boc 12a (3,14 g, 77 % de rendimiento) como un aceite incoloro.
ESI-Ms m/z para C17H24ClNO4 encontrado 242,1/ 246,1 (M+1-Boc)+
Etapa 2
Síntesis de ácido (2R,5S)-4-(ferc-butoxicarbonil)-5-(4-clorobencil)morfolina-2-carboxílico (12b)

A una solución enfriada a 0 °C de alcohol 12a (1,8 g, 5,26 mmol) en acetona (40 ml), se añadió reactivo de Jones (12 ml, 2,6 M) gota a gota. La mezcla de reacción se agitó a 0 °C durante 1 hora, y después se añadió isopropanol (iPrOH) (5 ml). Después de 10 minutos, se añadió acetato de etilo (150 ml) y la mezcla se filtró a través de una almohadilla de Celite. El filtrado se lavó con salmuera, se secó sobre MgSO4 y se evaporó proporcionando el compuesto del título 12b como una espuma blanca (1,7 g, 91 % de rendimiento).
ESI-MS m/z para C17H22ClNO5 encontrado 378,3/ 380,3 (M+Na)+, 256,1/258,1 (M+1-Boc)+
Etapa 3
Síntesis de (2R,5S)-5-(4-clorobencil)-2-(metilcarbamoil)morfolina-4-carboxilato de ferc-butilo (12c)

El compuesto del título (12c) se obtuvo a partir del compuesto 12b (0,24 g, 0,8 mmol) de acuerdo con el Procedimiento General IX en 79 % de rendimiento (0,23 g, 0,59 mmol).
ESI-MS m/z para C18H25ClN2O4 encontrado 391,7/ 393,7 (M+Na)+
Etapa 4
Síntesis de clorhidrato de (2R,5S)-5-(4-clorobencil)-N-metilmorfolina-2-carboxamida (12d)

Morfolina protegida con Boc 12c (0,33 g, 0,89 mmol) se trató con 4 M HCl (gas) en acetato de etilo como se describe en el Procedimiento General VII. Después de 1 hora la reacción se concentró al vacío y la sal de clorhidrato bruta de morfolina 12d se usó en la siguiente etapa.
ESI-MS m/z para C13H17ClN2O2 encontrado 269,2/ 271,2 (M+Na)+
Etapa 5
Síntesis de 4-((2R,5S)-5-(4-clorobencil)-2-(metilcarbamoil)morfolino)piperidina-1-carboxilato de ferc-butilo (12e)

La aminación reductiva se logró de acuerdo con el Procedimiento General VI, comenzando por la amina 12d (0,89 mmol, 239 mg) y N-Boc-piperid-4-ona. Después de la purificación cromatográfica, se obtuvo el compuesto del título 12e en 62 % de rendimiento (0,55 mmol, 249 mg).
ESI-MS m/z para C23H34ClN3O4 encontrado 452,2/ 454,2 (M+1)+
Etapa 6
Síntesis de clorhidrato de (2R,5S)-5-(4-clorobencil)-N-metil-4-(piperidin-4-il)morfolina-2-carboxamida (12f)

La piperidina protegida con Boc 12e (0,25 g, 0,55 mmol) se trató con 4 N HCl (gas) en acetato de etilo como se describe en el Procedimiento General VII. Después de 1 hora, la mezcla de reacción se concentró al vacío y la sal de clorhidrato bruta de piperidina 12f se usó directamente en la siguiente etapa.
ESI-MS m/z para C18H26ClN3O2 encontrado 352,4/ 354,4 (M+1)+
Etapa 7
Síntesis de (2R,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-N-metilmorfolina-2-carboxamida (12)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 12f. El compuesto final 12 se obtuvo por purificación mediante cromatografía de fase inversa en 67 % de rendimiento (0,16 g, 0,37 mmol).
ESI-MS m/z para C20H28ClN7O2 encontrado 434,0/ 436,0 (M+1)+
1H RMN (DMSO-d6, 75°C, 700 MHz) 57,62 (s a, 1H), 7,35 (AA'BB', J = 8,5 Hz, 2H), 7,30 (AA'BB', J = 8,5 Hz, 2H), 4,05-3,97 (m, 1H), 3,82-3,74 (m, 2H), 3,63-3,54 (m, 2H), 3,30-3,11 (m, 2H), 3,02-2,82 (m, 6H), 2,66 (d, J = 4,7 Hz, 3H), 1,98-1,92 (m, 2H), 1,57-1,47 (m, 2H).
Ejemplo 13
2-((2R,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-N)piperidin-4-N)-5-(4-dorobencil)morfoNn-2-N)propan-2-ol (13)

Etapa 1
Síntesis de (2R,5S)-2-metil 5-(4-clorobencil)morfolina-2,4-dicarboxilato de 4-terc-butilo (13a)

A una solución de aminoácido protegido con Boc 12b (1 g, 2,81 mmol) en acetonitrilo, se añadió carbonato de potasio (0,77 g, 5,62 mmol) seguido de yoduro de metilo (MeI) (0,26 ml, 4,21 mmol) a temperatura ambiente. Después de que la reacción se consideraba completa por TLC, la mezcla de reacción se filtró y el disolvente se evaporó. El residuo se disolvió en acetato de etilo, se lavó con salmuera y se secó sobre MgSO4. El disolvente se evaporó al vacío para dar 0,4 g (40 % de rendimiento) del compuesto del título 13a como un aceite amarillo lo suficientemente puro como para usarse en la siguiente etapa.
ESI-MS m/z para C1sH24ClNO5 encontrado 393,1/ 395,1 (M+Na)+
Etapa 2
Síntesis de (2R,5S)-5-(4-clorobencil)-2-(2-hidroxipropan-2-il)morfolina-4-carboxilato de terc-butilo (13b)

A una solución de éster 13a (0,4 g, 1,08 mmol) en THF seco, se añadió una solución de bromuro de metilmagnesio (1,1 ml, 3,24 mmol, 3 M en Et2O) gota a gota a TA. Después de 10 minutos la mezcla de reacción se inactivó con una solución saturada de cloruro de amonio y se extrajo con éter. La fase orgánica se lavó con salmuera, se secó sobre MgSO4 y se concentró para proporcionar 0,4 g (1,08 mmol, 100 % de rendimiento) de alcohol bruto 13b que se usó en la siguiente etapa sin purificación adicional.
ESI-MS C19H28ClNO4 encontrado 393,2/ 395,2 (M+Na)+, 270,0/ 272,0 (M+1-Boc)+
Etapa 3
Síntesis de 2-((2R,5S)-5-(4-clorobencil)morfolin-2-il)propan-2-ol 2,2,2-trifluoroacetato (13c)

Se trataron 0,4 g (1,08 mmol) del compuesto 13b con 3 ml de 50 % de ácido trifluoroacético (TFA) en diclorometano durante 30 minutos a temperatura ambiente, tiempo después del cual los volátiles se retiraron al vacío y el compuesto
del título bruto 13c (0,37 g; 90 % de rendimiento) se usó en la siguiente etapa sin purificación adicional.
ESI-MS m/z para C14H20CWO2 encontrado 270,1/ 272,1 (M+1)+
1H RMN (DMSO-d6+D2O, 500 MHz) S 7,34 (AA'BB', J = 6,2 Hz, 2H), 7,25 (AA'BB', J = 6,4 Hz, 2H), 3,65-3,55 (m, 2H), 3,52-3,44 (m, 1H), 2,42-3,36 (m, 1H), 3,15-3,04 (m, 2H), 2,94-2,87 (m, 1H), 2,48 (m, 3H), 1,10 (s, 3H), 1,08 (s, 3H).
Etapa 4
Síntesis de 4-((2R,5S)-5-(4-clorobencil)-2-(2-hidroxipropan-2-il)morfolino)piperidina-1-carboxilato de ferc-butilo (13d)

La aminación reductiva se logró de acuerdo con el Procedimiento General VI, comenzando por la amina 13c (0,97 mmol) y N-Boc-piperid-4-ona. El compuesto del título 13d se obtuvo después de la purificación cromatográfica en 79 % de rendimiento.
ESI-MS m/z para C24H37ClN2O4 encontrado 454,1/ 456,1 (M+1)+
1H RMN (DMSO D2O, 500 MHz) S 7,29 (AA'BB', J = 8,3 Hz, 2H), 7,19 (AA'BB', J = 8,1Hz, 2H), 3,92-3,80 (m, 2H), 3,66-3,55 (m, 3H), 3,45-3,39 (m, 1H), 3,35-3,29 (m, 1H), 3,20-3,13 (m, 1H), 2,84-2,75 (m, 3H), 2,72-2,65 (m, 2H), 1,97 1,84 (m, 2H), 1,69-1,61 (m, 2H), 1,33 (s, 9H), 1,09 (s, 3H), 1,07 (s, 3H).
Etapa 5
Síntesis de 2-((2R,5S)-5-(4-clorobencil)-4-(piperidin-4-il)morfolin-2-il)propan-2-ol 2,2,2-trifluoroacetato (13e)

Se trataron 0,38 g (0,77 mmol) del compuesto 13d con 3 ml de 50 % de ácido trifluoroacético en diclorometano durante 30 minutos a temperatura ambiente, tiempo después del cual los volátiles se retiraron al vacío y el compuesto del título bruto 13e (125 mg; 58 % de rendimiento) se usó en la siguiente etapa sin purificación adicional.
ESI-MS m/z para C19H29ClN2O2 encontrado 353,4/ 355,1 (M+1)+
1H RMN (DMSO-d6+D2O, 600 MHz) S 7,39 (AA'BB', J = 8,3 Hz, 2H), 7,30 (AA'BB', J = 8,1 Hz, 2H), 3,75-3,52 (m, 2H), 3,49-3,33 (m, 4H), 3,20-2,82 (m, 6H), 2,41-2,32 (m, 1H), 1,87-1,68 (m, 2H), 1,57-1,48 (m, 2H), 1,15 (s, 3H), 1,14 (s, 3H).
Etapa 6
Síntesis de 2-((2R,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)morfolin-2-il)propan-2-ol (13)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 13e. El compuesto final 13 se obtuvo después de la purificación mediante cromatografía de fase inversa en rendimiento (120 mg, 0,22 mmol).
ESI-MS m/z para C21H31ClN6O2 encontrado 435,1/ 437,1 (M+1)+
1H RMN (DMSO-d6+D2O, 600 MHz) 57,39 (AA'BB', J = 8,5 Hz, 2H), 7,31 (AA'BB', J = 8,5 Hz, 2H), 3,89-3,77 (m, 2H), 3,75-3,69 (m, 1H), 3,67-3,53 (m, 3H), 3,46-3,37 (m, 2H), 3,21-2,97 (m, 3H), 2,97-2,86 (m, 2H), 2,25-2,16 (m, 2H), 1,64 1,55 (m, 2H), 1,15 (s, 3H), 1,14 (s, 3H).
Ejemplo 14
5-(4-((2R,5S)-5-(4-clorobencil)-2-(2-metoxipropan-2-il)morfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (14)
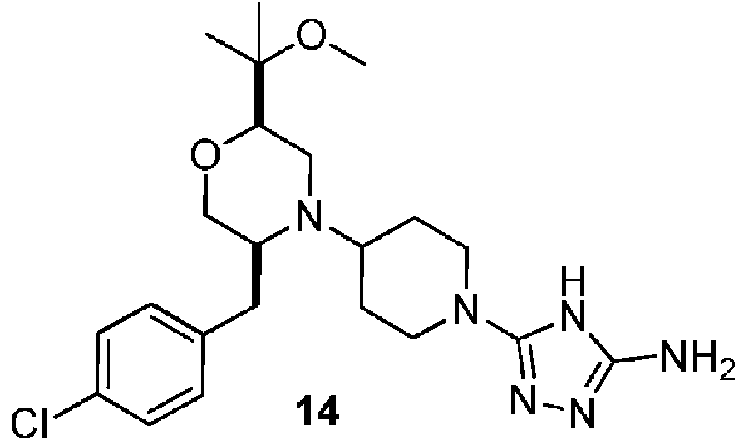
Etapa 1
Síntesis de 4-((2R,5S)-5-(4-clorobencil)-2-(2-metoxipropan-2-il)morfolino)piperidina-1-carboxilato de tere-butilo (14a)

El compuesto 14a (0,18 g, 0,4 mmol) se obtuvo de acuerdo con el Procedimiento General XXI en 86 % de rendimiento (0,16 g, 0,34 mmol).
ESI-MS m/z para C25H3gClN2O4 encontrado 467,2/ 469,2 (M+1)+
Etapa 2
Síntesis de 2,2,2-trifluoroacetato de (2R,5S)-5-(4-clorobencil)-2-(2-metoxipropan-2-il)-4-(piperidin-4-il)morfolina (14b)

Se trataron 0,16 g (0,34 mmol) del compuesto 14a con 1,5 ml de 50 % de ácido trifluoroacético a temperatura ambiente, tiempo después del cual los volátiles se retiraron al vacío y el compuesto del título bruto 14b se usó directamente en la siguiente etapa.
ESI-MS m/z para C20H31CW2O2 encontrado 367,2/ 369,2 (M+1)+
1H RMN (DMSO-d6+D2O, 600 MHz) 57,37 (AA'BB', J = 8,3 Hz, 2H), 7,25 (AA'BB', J = 8,3Hz, 2H), 3,76-3,51 (m, 4H), 3,48-1,39 (m, 2H), 3,35-3,28 (m, 1H), 3,24-3,16 (m, 1H), 3,12 (s, 3H), 3,09-2,81 (m, 5H), 2,42-2,30 (m, 2H), 1,81-1,68 (m, 2H), 1,14 (s, 6H).
Etapa 3
Síntesis de 5-(4-((2R,5S)-5-(4-clorobencil)-2-(2-metoxipropan-2-il)morfolino)piperidin-1 -il)-4H-1,2,4-triazol-3-amina (14)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 14b. El compuesto final 14 se obtuvo después de la purificación mediante cromatografía de fase inversa en 29 % de rendimiento (45 mg, 0,1 mmol).
ESI-MS m/z para C22H33QN6O2 encontrado 449,1/ 451,1 (M+1)+
1H RMN (DMSO-d6+D2O, 600 MHz) 57,40 (AA'BB', J = 8,4 Hz, 2H), 7,29 (AA'BB', J = 8,4 Hz, 2H), 3,88-3,81 (m, 2H), 3,66-3,51 (m, 5H), 3,36-3,30 (m, 1H), 3,23-3,16 (m, 1H), 3,15 (s, 3H), 3,13-3,08 (m, 1H), 3,03-2,96 (m, 1H), 2,94-2,83 (m, 2H), 2,24-2,16 (m, 2H), 1,63-1,54 (m, 2H), 1,17 (s, 6H).
Ejemplo 15
(R)-5-(2-(4-clorobencil)-[1,4'-bipiperidin]-1'-il)-4H-1,2,4-triazol-3-amina (15)

Etapa 1
Síntesis de (S,Z)-N-(2-(4-clorofenil)etiliden)-2-metilpropano-2-sulfinamida (15a)

El compuesto 15a se obtuvo a partir de p-clorofenilacetaldehído (6,5 g, 42,04 mmol), (S)-2-metil-2-propanosulfinamida (5,09 g, 42,04 mmol), de acuerdo con el Procedimiento General XVII en 67 % de rendimiento (7,35 g, 28,5 mmol). ESI-MS m/z para C12H16GNOS encontrado 258,1/ 260,1 (M+1)+
Etapa 2
Síntesis de (S)-N-((R)-1-(4-clorofenil)pent-4-en-2-il)-2-metilpropan-2-sulfinamida (15b)

El compuesto 15b se obtuvo a partir de 15a (7,35 g, 28,5 mmol) de acuerdo con el Procedimiento General XVIII en 58 % de rendimiento (5,0 g, 16,7 mmol), después de la cromatografía en columna con AcOEt/hexano 1:4. ESI-MS m/z
para C15H22CINOS encontrado 300,1/ 302,1 (M+1)+
1H RMN (CDCI3, 500 MHz) 67,21 (AA'BB1, J = 8,3 Hz, 2H), 7,08 (AA'BB1, J = 8,5 Hz, 2H), 5,81-5,71 (m, 1H), 5,18-5,10 (m, 2H), 3,54-3,46 (m, 1H), 3,33-3,28 (m, 1H), 2,78 (dd, J = 7,1, 13,7 Hz, 1H), 2,69 (dd, J = 6,4, 13,7 Hz, 1H), 2,40 2,32 (m, 1H), 2,31-2,23 (m, 1H), 1,08 (s, 9H).
Etapa 3
Síntesis de (S)-N-alil-N-((R)-1-(4-dorofenil)pent-4-en-2-il)-2-metilpropan-2-sulfinamida (15c)

El compuesto 15c se obtuvo a partir de 15b (1 g, 3,33 mmol) de acuerdo con el Procedimiento General XIX en 71 % de rendimiento (0,78 g, 2,29 mmol), después de la cromatografía en columna con AcOEt/hexano 1:3.
ESI MS m/z para C18H26ClNOS encontrado 340,2/ 342,2 (M+1)+
Etapa 4
Síntesis de (R)-1-((S)-terc-butilsulfinil)-2-(4-clorobencil)-1,2,3,6-tetrahidropiridina (15d)

El compuesto 15d se obtuvo a partir de 15c (0,78 g, 2,29 mmol) de acuerdo con el Procedimiento General XX en 94 % de rendimiento (0,67 g, 2,15 mmol), después de la cromatografía en columna con AcOEt/hexano 1:10.
ESI-MS m/z para C16H22ClNOS encontrado 312,2/ 314,2 (M+1)+
Etapa 5
Síntesis de (R)-1-((S)-terc-butilsulfinil)-2-(4-clorobencil)piperidina (15e)

A una solución del sustrato 15d (0,62 g, 1,98 mmol) en metanol (5 ml), se añadió una cantidad catalítica de paladio sobre carbono. La mezcla de reacción se agitó bajo hidrógeno durante la noche. Después de este tiempo, el catalizador se filtró a través de una almohadilla de Celite y el filtrado se concentró hasta sequedad y se purificó por cromatografía en columna usando AcOEt/hexano (1:4), proporcionando 0,48 g (1,53 mmol, 77 % de rendimiento) del producto 15e. ESI-MS m/z para C16H24ClNOS encontrado 314,2/ 316,2 (M+1)+
Etapa 6
Síntesis de (R)-2-(4-dorobencil)piperidina (15f)

El compuesto 15e (0,48 g, 1,5 mmol) se trató con HCl/ MeOH a TA durante 1 hora y después la reacción se concentró hasta sequedad. El residuo se absorbió entre DCM/1 N NaOH y las fases se separaron. La fase acuosa se extrajo con DCM, después los orgánicos se combinaron, se secaron y concentraron para dar 0,23 g del compuesto 15f (1,1 mmol; 72 % de rendimiento) como una amina libre.
ESI-MS m/z para C-i2H-i6ClN encontrado 210,2/ 212,2 (M+1)+
1H RMN (CDCh, 500 MHz) S 7,27 - 7,21 (m, 2H), 7,14 - 7,08 (m, 2H), 3,00 (dp, J = 11,8, 2,0 Hz, 1H), 2,71 - 2,60 (m, 2H), 2,54 (m, 2H), 1,80 - 1,73 (m, 1H), 1,66 (s, 1H), 1,61 - 1,53 (m, 1H), 1,43 (m, 1H), 1,34 - 1,23 (m, 1H), 1,22 - 1,13 (m, 1H).
Etapa 7
Síntesis de (R)-2-(4-clorobencil)-[1,4'-bipiperidina]-1'-carboxilato de ferc-butilo (15g)

La aminación reductiva del compuesto 15f y N-Boc-piperid-4-ona se logró de acuerdo con el Procedimiento General VI. El compuesto del título se obtuvo en 40 % de rendimiento (0,17 g, 0,43 mmol).
ESI-MS m/z para C22H33ClN2O2 encontrado 393,1/ 395,1 (M+1)+
Etapa 8
Síntesis de clorhidrato de (R)-2-(4-clorobencil)-1,4'-bipiperidina (15h)

La retirada del grupo protector de Boc del compuesto 15g se logró de acuerdo con el Procedimiento General VII. El compuesto del título (15h) se obtuvo en 91 % de rendimiento como una sal HCl (0,13 g, 0,39 mmol).
ESI-MS m/z para C17H25GN2 encontrado 293,1/ 295,1 (M+1)+
Etapa 9
Síntesis de (R)-5-(2-(4-dorobencil)-[1,4'-bipiperidin]-1'-il)-4H-1,2,4-triazol-3-amina (15)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 15h. El compuesto del título (15) se obtuvo en 28 % de rendimiento (0,10 mmol; 40 mg).
ESI-MS m/z para C1gH27ClN6 encontrado 391,0/ 393,0 (M+1)+
1H RMN (DMSO-d6,500 MHz) 5: 7,47-7,37 (m, 2H), 7,34-7,29 (m, 2H), 3,91-3,72 (m, 3H), 3,65 -2,34 (m, 5H), 3,02 -2,81 (m, 3H), 2,71-2,60 (m, 1H), 2,10-2,00 (m, 1H), 1,92-1,22 (m, 2H), 1,71-1,52 (m, 3H), 1,55-1,32 (m, 2H).
Ejemplo 16
(6R)-1'-(5-amino-4H-1,2,4-triazol-3-il)-6-(4-clorobencil)-[1,4'-bipiperidina]-3-carboxilato de etilo (16)

Etapa 1
Síntesis de 2-(((S)-N-((R)-1-(4-clorofenil)pent-4-en-2-il)-2-metilpropan-2-ilsulfinamido)metil)acrilato de etilo (16a)

El compuesto 16a se obtuvo a partir de 15b (2,0 g, 6,9 mmol) de acuerdo con el Procedimiento General XIX en 84 % de rendimiento (2,4 g, 5,82 mmol), después de la cromatografía en columna con un gradiente de AcOEt/hexano de 1:20 a 1:8.
ESI-MS m/z para C21H30ClNOaS encontrado 434,1/ 436,1 (M+Na)+
1H RMN (CDCla, 500 MHz) 5: 7,21 (AA'BB', J = 8,1 Hz, 2H), 7,12 (AA'BB', J = 7,9 Hz, 2H), 6,32 (s a, 1H), 5,81 (s a, 1H), 5,70-5,58 (m, 1H), 5,01-4,92 (m, 2H), 4,24-4,12 (m, 3H), 3,38-3,30 (m, 1H), 3,22-3,09 (m, 2H), 2,85-2,77 (m, 1H), 2,46-2,36 (m, 1H), 2,30-2,21 (m, 1H), 1,26 (t, J = 7,1 Hz, 3H), 1,18 (s, 9H).
Etapa 2
Síntesis de (R)-1-((S)-terc-butilsulfinil)-6-(4-clorobencil)-1,2,5,6-tetrahidropiridina-3-carboxilato de etilo (16b)

El compuesto 16b se obtuvo a partir de 16a (2,4 g, 5,82 mmol) y catalizador de Grubbs de segunda generación de acuerdo con el Procedimiento General XX en 81 % de rendimiento (1,8 g, 4,69 mmol), después de la cromatografía en columna con AcOEt/hexano 1:10.
ESI-MS m/z para C19H26ClNO3S encontrado 384,1/ 386,1 (M+1)+
1H RMN (CDCl3, 500 MHz) 6 : 7,24 (AA'BB1, J = 8,3 Hz, 2H), 7,10 (AA'BB1, J = 8,3 Hz, 2H), 7,03-6,98 (m, 1H), 4,23 4,11 (m, 3H), 3,70-3,58 (m, 2H), 2,88 (dd, J = 7,9, 13,5 Hz, 1H), 2,65 (dd, J = 7,7, 13,7 Hz, 1H), 2,61-2,53 (m, 1H), 2,15-2,07 (m, 1H), 1,29 (t, J = 7,1 Hz, 3H), 0,96 (s, 9H).
Etapa 3
Síntesis de (6R)-1-((S)-terc-butilsulfinil)-6-(4-clorobencil)piperidina-3-carboxilato de etilo (16c)

A una solución del compuesto 16b (1,0 g, 2,6 mmol) y NiCl2x6H2O (61 mg, 0,26 mmol) en 100 ml de etanol absoluto, se añadió borohidruro de sodio (NaBH4) (100 mg, 2,6 mmol) en porciones. La reacción se agitó durante 1 hora y después se concentró hasta 1/3 de su volumen inicial, se añadió DCM (40 ml) y la suspensión resultante se filtró a través de una almohadilla de Celite. El filtrado se lavó con 1 M HCl, agua, salmuera, se secó sobre MgSO4 y se concentró para proporcionar 1,0 g (2,57 mmol; 99 % de rendimiento) del compuesto del título 16c.
ESI-MS m/z para C19H2sClNO3S encontrado 408,9/ 410,9 (M+Na)+
Etapa 4
Síntesis de clorhidrato de (6R)-6-(4-clorobencil)piperidina-3-carboxilato de etilo (16d)

El compuesto 16c (1,0 g, 2,6 mmol) se trató con una solución de HCl (gas)/1,4-dioxano 1 N a TA durante 1 hora. Después la mezcla de reacción se concentró hasta sequedad, proporcionando 0,82 g (2,58 mmol; 99 % de rendimiento) de piperidina 16d en una forma de sal de clorhidrato.
ESI-MS m/z para C15H2üClNO2 encontrado 282,2/ 284,2 (M+1)+
Etapas 5-7
Síntesis de (6R)-1l-(5-amino-4H-1,2,4-triazol-3-il)-6-(4-clorobencil)-[1,4'-bipiperidina]-3-carboxilato de etilo (16)
La piperidina 16d se llevó a los etapas sintéticos restantes como se describe en el Procedimiento General VI (aminación reductora con N-Boc-piperid-4-ona), el Procedimiento General VII (desprotección de Boc) y el Procedimiento General VIII (formación de anillo de triazol).
Se sintetizaron 180 mg (0,40 mmol; 16 % de rendimiento) del compuesto del título 16.
ESI-MS C22H31ClN6O2 encontrado 447,1/ 449,1 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 57,36 (AA'BB', J = 8,3 Hz, 2H), 7,31 (AA'BB', J = 8,3 Hz, 2H), 4,17-4,07 (m, 2H), 3,92 3,82 (m, 2H), 3,82-3,12 (m, 4H), 3,20-3,06 (m, 1H), 3,06-2,96 (m, 1H), 2,91-2,79 (m, 3H), 2,16-1,99 (m, 2H), 1,93-1,50 (m, 5H), 1,55-1,43 (m, 1H), 1,21 (t, J = 7,1 Hz, 3H).
Ejemplo 17
Ácido (6R)-1'-(5-amino-4H-1,2,4-triazol-3-il)-6-(4-dorobencil)-[1,4'-bipiperidina]-3-carboxílico (17)

El compuesto 16 (50 mg, 0,11 mmol) se disolvió en 6 M HCl (2 ml) y se sometió a reflujo durante 1 hora, tiempo después del cual los volátiles se retiraron al vacío y el residuo se purificó por cromatografía en fase inversa. Se obtuvieron 25 mg (0,059 mmol; 54 % de rendimiento) del compuesto diana 17.
ESI-MS C20H27ClN6O2 encontrado 419,1/ 421,1 (M+1)+
1H RMN (DMSO-d6, 75 °C, 500 MHz) 57,36 (AA'BB', J = 8,3Hz, 2H), 7,31 (AA'BB', J = 8,3Hz, 2H), 3,94-3,84 (m, 2H), 3,82-3,71 (m, 1H), 3,66-3,56 (m, 1H), 3,52-3,19 (m, 3H), 3,17-3,10 (m, 1H), 3,06-2,97 (m, 1H), 2,87-2,76 (m, 3H), 2,13 1,97 (m, 2H), 1,92-1,60 (m, 5H), 1,56-1,46 (m, 1H).
Ejemplo 18
Ácido (6R)-1'-(5-amino-4H-1,2,4-triazol-3-il)-6-(4-clorobencil)-3-metil-[1,4'-bipiperidina]-3-carboxílico (18)

Etapa 1
Síntesis de (6R)-1-((S)-terc-butilsulfinil)-6-(4-clorobencil)-3-metilpiperidina-3-carboxilato de etilo (18a)

A una solución recién preparada de LDA (2.32 mmol en 5 ml de THF) se añadió una solución del compuesto 16c (0,62 g, 1,6 mmol) en THF (5 ml) gota a gota a -78 °C. Después de 1 hora a -78 °C, se añadió yoduro de metilo (0,34 g, 2,4 mmol) gota a gota y la reacción se dejó calentar hasta TA y se agitó durante la noche. La mezcla de reacción se vertió en una solución saturada de cloruro de amonio y se extrajo con éter dietílico. La fase orgánica se lavó con HCl 1 N, salmuera, se secó sobre MgSO4 y se concentró. La cromatografía en columna en EtOAc/ hexanos 1:6 proporcionó 0,14 g (0,35 mmol; 22 % de rendimiento) del compuesto del título 18a.
ESI-MS m/z para C20H30ClNO3S encontrado 422,1/ 424,1 (M+Na)+
1H RMN (CDCl3, 500 MHz) 57,24 (AA'BB', J = 8,3 Hz, 2H), 7,16 (AA'BB', J = 8,3 Hz, 2H), 4,23-4,12 (m, 1H), 4,04-3,95
(m, 1H), 3,65-3,58 (m, 1H), 3,56-3,37 (m, 1H), 3,32-3,23 (m, 2H), 2,88-2,80 (m, 1H), 2,10-2,03 (m, 1H), 1,77-1,55 (m, 1H), 1,55 (s, 3H), 1,24 (t, J = 7,1 Hz, 2H), 1,21 (m, 2H), 1,18 (s, 9H).
Etapa 2
Síntesis de (6R)-6-(4-dorobencil)-3-metilpiperidina-3-carboxilato de etilo (18b)

El compuesto 18a (0,14 g, 0,35 mmol) se trató con una solución de HCl (gas)/1,4-dioxano 1 N a TA durante 1 hora. Después la mezcla de reacción se concentró hasta sequedad, proporcionando 0,11 g (0,34 mmol; 97 % de rendimiento) de piperidina 18b en una forma de sal de clorhidrato.
ESI-MS m/z para C i6H22ClNO2 encontrado 296,1/ 298,1 (M+1)+
Etapas 3-5
Síntesis de (6R)-1l-(5-amino-4H-1,2,4-triazol-3-il)-6-(4-clorobencil)-3-metil-[1,4'-bipiperidina]-3-carboxilato de etilo (18c)

La piperidina 18b se llevó a los siguientes tres etapas sintéticas como se describe en el Procedimiento General VI (aminación reductora con N-Boc-piperid-4-ona), el Procedimiento General VII (desprotección de Boc) y el Procedimiento General VIII (formación de anillo de triazol). Se sintetizaron 30 mg (0,065 mmol; 19 % de rendimiento en 3 etapas) del compuesto 18c.
ESI-MS m/z para C23H33ClN6O2 encontrado 461,0/ 463,0 (M+1)+
Etapa 6
Síntesis de ácido (6R)-1'-(5-amino-4H-1,2,4-triazol-3-il)-6-(4-clorobencil)-3-metil-[1,4'-bipiperidina]-3-carboxílico (18).

Una solución del compuesto 18c (30 mg, 0,065 mmol) en MeOH (1 ml) se trató con NaOH 1 M (2 ml) y se calentó a 50 °C durante 2 horas. Después la reacción se enfrió hasta temperatura ambiente, se acidificó con HCl 2 M a pH neutro y los disolventes se retiraron al vacío. El residuo se purificó por cromatografía de fase inversa. Se obtuvieron 2 mg (0,005 mmol; 7 % de rendimiento) del compuesto del título 18.
ESI-MS m/z para C21H29ClN6O2 encontrado 433,1/ 435,1 (M+1)+
1H RMN (CDCla, 500 MHz), 57,40 (AA'BB', J = 8,3 Hz, 2H), 7,34 (AA'BB', J = 8,3 Hz, 2H), 3,99-3,89 (m, 2H), 3,71 2,90 (m, 5H), 2,90-2,81 (m, 2H), 2,26-2,13 (m, 1H), 2,04-1,97 (m, 1H), 1,94-1,58 (m, 6H), 1,49-1,38 (m, 1H), 1,26 (s,
3H).
Ejemplo 19
5-((2S,4R)-2-(4-clorobencil)-4-metoxi-[1,4-bipiperidin]-1'-il)-4H-1,2,4-triazol-3-amina (19)

Etapa 1
Síntesis de ácido (S)-2-((ferc-butoxicarbonil)amino)-3-(4-clorofenil)propanoico (19a)

A una solución de p-cloro-L-fenilalanina (18,0 g, 75 mmol) en acetona-agua (150 ml : 150 ml) se añadió hidróxido de sodio (6 g, 150 mmol) a 0 °C seguido de dicarbonato de di-ferc-butilo (16,4 g, 75 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. Se evaporó la acetona. La capa acuosa se acidificó hasta pH 2 con HCl 2 M y se extrajo con acetato de etilo. La capa orgánica se secó en sulfato de magnesio, se filtró y se concentró a presión reducida. El producto bruto se cristalizó a partir de hexanos para obtener 18,0 g de producto 19a como un sólido blanco (80 % de rendimiento).
ESI-MS m/z para C14H1sClNO4 encontrado 299,8 / 301,8 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 8: 7,29(d, J = 8,3 Hz, 2H), 7,21 (d, J = 8,3 Hz, 2H), 7,02 (d, J = 7,3 Hz, 1H), 4,08-3,99 (m, 1H), 2,96 (dd, J = 4,3, 13,7Hz, 1H), 2,76 (dd, J = 10,5, 13,6 Hz, 1H).
Etapa 2
Síntesis de (S)-(1-(4-clorofenil)-4-diazo-3-oxobutan-2-il)carbamato de ferc-butilo (19b)

A una solución de ácido 19a (17,2 g, 57 mmol) en tetrahidrofurano (200 ml) se añadió trietilamina (17 ml, 120 mmol) y cloroformiato de metilo (4,87 ml, 63 mmol) a -10 °C. Después de 15 min, se añadió una solución de diazometano (342 mmol) en éter dietílico (400 ml) a - 30°C. La mezcla de reacción se agitó durante la noche a temperatura ambiente. El exceso de diazometano se destruyó con ácido acético (15 ml). La mezcla se diluyó con éter dietílico y se lavó con 5 % de NaHCO3, NH4Cl saturado y salmuera. La capa orgánica se secó sobre sulfato de magnesio, se filtró y concentró a presión reducida para dar el producto 19b como un sólido naranja (18,0 g, 96 % de rendimiento).
ESI-MS m/z para C^H^ClNaOs encontrado 324,1/ 326,1 (M+1)+
1H RMN (CDCla, 700 MHz) 87,28 (d, J = 8,3 Hz, 2H), 7,13 (d, J = 8,2 Hz, 2H), 5,26 (s a, 1H), 5,07 (s a, 1H), 4,40 (s a, 1H), 3,03 (dd, J = 7,0, 14,0 Hz, 1H), 2,97 (dd, J = 6,1, 13,5 Hz, 1H), 1,42 (s, 9H).
Etapa 3
Síntesis de ácido (S)-3-((terc-butoxicarbonil)amino)-4-(4-clorofenil)butanoico (19c)

A una solución del compuesto 19b (18 g, 65 mmol) en tetrahidrofurano : agua (135 : 15 ml) se añadió una solución de trifluoroacetato de plata (1,57 g, 7,1 mmol) en trietilamina (25 ml, 182 mmol) a -5 °C. La mezcla de reacción se agitó durante 1 hora. Después de este tiempo, el disolvente se retiró a presión reducida. El residuo se diluyó con una solución acuosa de NaHCO3 y la mezcla se extrajo con éter dietílico. Se añadió 1 M HCl a la capa acuosa a 0 °C hasta pH 2-3 y la mezcla se extrajo tres veces con acetato de etilo. Las capas orgánicas se recogieron, se secaron sobre sulfato de magnesio y se evaporaron a presión reducida. El producto bruto se cristalizó a partir de éter dietílico para obtener 7 g de 19c como un sólido blanco en 40 % de rendimiento.
ESI-MS m/z para C15H20ClNO4 encontrado 312,3/ 314,3 (M-1)-1H RMN (DMSO-d6, 500 MHz) 812,14 (s, 1H), 7,28 (d, J = 8,3Hz, 2H), 7,14 (d, J = 8,3 Hz, 2H), 6,76 (d, J = 8,7 Hz, 1H), 3,90-3,86 (m, 1H), 2,68 (dd, J = 5,27, 13,4 Hz, 1H), 2,60 (dd, J = 8,5, 13,4 Hz, 1H), 2,30 (t, J = 7,0Hz, 2H), 1,25 (s, 9H).
Etapa 4
Síntesis de (S)-(1-(4-clorofenil)-4-(metoxi(metil)amino)-4-oxobutan-2-il)carbamato de terc-butilo (19d)

El compuesto 19d se obtuvo a partir de 19c (7 g, 22.3 mmol) de acuerdo con el Procedimiento General XXII (para mejorar la solubilidad de 19c, se usó una mezcla de DCM/DMF 10:1 como un disolvente de reacción) en 93 % de rendimiento (7.4 g), después de cromatografía ultrarrápida usando hexano-acetato de etilo (elución de gradiente de 20:1 a 1:1).
ESI-MS m/z para C17H25ClN2O4 encontrado 380,1/ 382,1 (M+Na)+
1H RMN (CDCla, 500 MHz) 87,25 (d, J = 8,5 Hz, 2H), 7,13 (d, J = 8,3 Hz, 2H), 5,48 (s a, 1H), 4,14-4,10 (m, 1H), 3,57 (s, 3H), 3,17 (s, 3H), 3,00-2,94 (m, 1H), 2,84 (dd, J = 7,9, 13,6 Hz, 1H), 2,58 (qd, J=3,8, 16,4 Hz, 2H), 1,39 (s, 9H).
Etapa 5
Síntesis de (S)-(1-(4-clorofenil)-4-oxohex-5-en-2-il)carbamato de terc-butilo (19e)

A una solución de 19d (6,9 g, 19,3 mmol) en tetrahidrofurano seco (50 ml) se añadió cloruro de vinilmagnesio en THF (48 ml, 77,3 mmol) a 0 °C. La mezcla se llevó a tetrahidrofurano y se agitó durante 3 horas. La mezcla de reacción se vertió en NH4Cl acuoso saturado y se extrajo con éter dietílico. La capa orgánica se lavó con 1 M HCl, salmuera, se secó sobre sulfato de magnesio y se concentró a presión reducida. El producto 19e se purificó por cromatografía ultrarrápida usando hexanos-acetato de etilo (elución de gradiente de 10:1 a 5:1). Se obtuvieron 2,0 g como un sólido blanco (32 % de rendimiento).
ESI-MS m/z para C1yH22ClNOa encontrado 323,8 / 325,8 (M+1)+
Etapa 6
Síntesis de (S)-2-(4-dorobencil)-4-oxopiperidina-1-carboxilato de tere-butilo (19f)

A una solución de 19e (1,1 g, 3,4 mmol) en tetrahidrofurano (10 ml) se añadió un complejo de trifluoruro de boro y éter dietílico (4,27 ml, 34 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. Después la mezcla se diluyó con acetato de etilo y se lavó con 4 M NaOH. La capa orgánica se secó en sulfato de magnesio, se filtró y se concentró a presión reducida. El producto 19f se purificó por cromatografía ultrarrápida usando hexanosacetato de etilo (elución de gradiente de 6:1 a 2:1) para proporcionar 480 mg como un aceite incoloro (43 % de rendimiento).
ESI-MS m/z para C17H22GNO3 encontrado 346,1 / 348,1 (M+Na)+
1H RMN (CDCl3, 500 MHz) S 7,26 (d, J = 8,3 Hz, 2H), 7,09 (d, J = 6,6 Hz, 2H), 4,74 (s a, 1H), 4,37 (s a, 1H), 3,30 (qd, J = 3,7, 11,5 Hz, 1H), 2,81 (dd, J = 7,3, 13,6 Hz, 1H), 2,68 (dd, J = 7,9, 13,7Hz, 1H), 2,60 (dd, J = 6,8, 14,5 Hz, 1H), 2,54-2,49 (m, 1H), 2,39-2,34 (m, 2H), 1,40 (s, 9H).
Etapa 7
Síntesis de (2S,4R)-2-(4-clorobencil)-4-hidroxipiperidina-1-carboxilato de tere-butilo (19g) y (2S,4S)-2-(4-clorobencil)-4-hidroxipiperidina-1-carboxilato de tere-butilo (19h).

A una solución de 19f (470 mg, 1,45 mmol) en metanol (5 ml) se añadió borohidruro de sodio (66 mg, 1,75 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas, después se añadió 1 N NaOH. La capa acuosa se extrajo con acetato de etilo. Se secó la capa orgánica combinada en sulfato de magnesio, se filtró y se concentró a presión reducida. Los productos se purificaron por cromatografía ultrarrápida usando hexanos-acetato de etilo (de 6:1 a 1:1). Se obtuvieron 187 mg (19g diastereómero simple con configuración desconocida en el grupo hidroxi) y 200 mg (19h diastereómero simple con configuración desconocida en el grupo hidroxilo) de productos 19g y 19h como aceite incoloro en 40 % y 42 % de rendimiento respectivamente.
19g
ESI-MS m/z para C17H24GNO3 encontrado 349,0/ 351,0 (M+Na)+
1H RMN (CDCl3, 500 MHz) S 7,23 (d, J = 8,5 Hz, 2H), 7,15 (d, J = 8,3Hz, 2H), 4,34-4,31 (m, 1H), 4,20 (t, J = 3,0 Hz, 1H), 3,96 (d, J = 12,4 Hz, 1H), 3,31 (td, J = 3,8, 13,4 Hz, 1H), 3,10 (dd, J = 7,1, 13,1 Hz, 1H), 3,01 (dd, J = 8,1, 13,2 Hz, 1H), 1,71-1,68 (m, 4H), 1,34 (s, 9H).
19h
ESI-MS m/z para C17H24GNO3 encontrado 349.0/ 351.0 (M+Na)+
1H RMN (CDCl3, 500 MHz) S 7,25 (d, J = 8,5 Hz, 2H), 7,09 (d, J = 6,2 Hz, 2H), 4,50 (s a, 1H), 4,24-4,17 (m, 1H), 4,05 4,00 (m, 1H), 2,94 (td, J = 2,6, 13,6Hz, 1H), 2,82 (dd, J = 8,3, 13,4 Hz, 1H), 2,68 (dd, J = 7,5, 13,0 Hz, 1H), 2,00 (d, 11,9 Hz, 1H), 1,87 (d, 12,4 Hz, 1H), 1,45-1,40 (m, 2H), 1,31 (s, 9H).
Etapa 8
Síntesis de (2S,4R)-2-(4-clorobencil)-4-metoxipiperidina-1-carboxilato de tere-butilo (19i)

El compuesto 19i se obtuvo a partir de 19g (190 mg, 0,58 mmol) de acuerdo con el Procedimiento General XXI (para mejorar la solubilidad, se usó DMF como un disolvente de reacción) en 97 % de rendimiento (190 mg, 0,56 mmol, sólido blanco), después de cromatografía ultrarrápida usando hexano/EtOAc (elución de gradiente de 10:1 a 5:1). ESI-MS m/z para C1sH26ClNO3 encontrado 362,1/364,1 (M+Na)+
1H RMN (CDCl3, 700 MHz) 57,25 (d, J = 8,5 Hz, 2H), 7,16 (d, J = 8,1 Hz, 2H), 4,33-4,31 (m, 1H), 3,96-3,94 (m, 1H), 3,59-3,57 (m, 1H), 3,38 (s, 3H), 3,20 (td, J = 2,6, 13,3 Hz, 1H), 3,06 (dd, J = 8,0, 13,3 Hz, 1H), 2,94 (dd, 7,6, 13,2 Hz, 1H), 1,93-1,91 (m, 1H), 1,86-1,83 (m, 1H), 1,64-1,60 (m, 2H), 1,35 (s, 9H).
Etapa 9
Síntesis de clorhidrato de (2S,4R)-2-(4-clorobencil)-4-metoxipiperidina (19j)

El compuesto 19j se obtuvo a partir de 19i (190 mg, 0,56 mmol) de acuerdo con el Procedimiento General VII en 93 % de rendimiento (150 mg, 0,54 mmol) como un sólido blanco.
ESI-MS m/z para C13H1sClNO encontrado 240,1/ 242,2 (M+1)+
Etapa 10
Síntesis de clorhidrato de (2S,4R)-2-(4-clorobencil)-4-metoxi-1,4'-bipiperidina (19k)

El compuesto 19k se obtuvo a partir de 19j (150 mg, 0,54 mmol) de acuerdo con el Procedimiento General VI seguido de cromatografía ultrarrápida usando hexanos/AcOEt (elución de gradiente de 10:1 a 1:4). En la siguiente etapa, el grupo protector de Boc se retiró disolviendo el material obtenido después de la cromatografía ultrarrápida en acetato de etilo y transfiriéndolo a 6 M de HCl acuoso. La capa acuosa fuertemente ácida se basificó con 4 M NaOH para llevar el pH hasta 10 y después se extrajo tres veces con acetato de etilo. Los orgánicos combinados se secaron sobre sulfato de magnesio, se filtraron y concentraron a presión reducida para proporcionar 90 mg del producto 19k como un aceite incoloro (0,28 mmol; 51 % de rendimiento).
ESI-MS m/z para C18H27ClN2O encontrado 323,3/325,2 (M+1)+
Etapa 11
Síntesis de 5-((2S,4R)-2-(4-clorobencil)-4-metoxi-[1,4'-bipiperidin]-1'-il)-4H-1,2,4-triazol-3-amina (19)

El compuesto 19 se obtuvo a partir de 19k (90 mg, 0,28 mmol) de acuerdo con el Procedimiento General VIII en 35 % de rendimiento (50 mg, 0,1 mmol, sólido blanco), después de la purificación por cromatografía de fase inversa preparativa.
Diastereómero simple con configuración desconocida en el grupo metoxi.
1H RMN (DMSO-da+D2O, 700 MHz) 57,40 (d, J = 8,4 Hz, 2H), 7,31 (d, J = 8,2 Hz, 2H), 3,84-3,78(m, 3H), 3,70-3,67 (m, 1H), 3,50-3,48 (m, 1H), 3,42 (dd, J = 4,6, 13,6 Hz, 1H), 3,32-3,30 (m, 1H), 3,15 (s, 3H), 3,02-2,99 (m, 1H), 2,93 (td, J = 1,8, 13,4 Hz, 1H), 2,87 (td, J = 1,3, 13,0 Hz, 1H), 2,71 (dd, J = 9,7, 13,1 Hz, 1H), 2,19 (d, 12,1 Hz, 1H), 1,93-1,85 (m, 4H), 1,65 (qd, J = 4,6, 12,7Hz, 1H), 1,48 (qd, J = 3,8, 13,5Hz, 1H), 1,43-1,38 (m, 1H).
Ejemplo 20
5-((2S,4S)-2-(4-clorobencil)-4-metoxi-[1,4'-bipiperidin]-1'-il)-4H-1,2,4-triazol-3-amina (20)

Etapa 1
Síntesis de (2S,4S)-2-(4-clorobencil)-4-metoxipiperidina-1-carboxilato de tere-butilo (20a)

El compuesto 20a se obtuvo a partir de 19h (190 mg, 0,58 mmol) de acuerdo con el Procedimiento General XXI (para mejorar la solubilidad, se usó DMF como un disolvente de reacción) en 78 % de rendimiento (0,46 mmol; 150 mg, sólido blanco), después de cromatografía ultrarrápida usando hexanos/EtOAc (elución de gradiente de 10:1 a 5.1). ESI-MS m/z para C18H26ClNO3 encontrado 362,1/364,1 (M+Na)+
1H RMN (CDCla, 500 MHz) 57,25 (d, J = 8,5 Hz, 2H), 7,10 (d, J = 6,2 Hz, 2H), 4,51 (s a, 1H), 4,21 (s a, 1H), 3,57-3,52 (m, 1H), 3,48 (d, J = 5,5 Hz, 2H), 3,35 (s, 3H), 2,92 (td, J = 2,6, 13,7 Hz, 1H), 2,83 (dd, J = 8,1, 13,4 Hz, 1H), 2,68 (dd, J = 7,5, 13,2 Hz, 1H), 2,06 (d, J = 10,4 Hz, 1H), 1,94-1,90 (m, 1H), 1,32 (s, 9H).
Etapa 2
Síntesis de clorhidrato de (2S,4S)-2-(4-clorobencil)-4-metoxipiperidina (20b)

El compuesto 20b se obtuvo a partir de 20a (165 mg, 0,48 mmol) de acuerdo con el Procedimiento General VII en 90 % de rendimiento (0,43 mmol; 165 mg).
ESI-MS m/z para C13H1aClNO encontrado 240,2/ 242,2 (M+1)+.
Etapa 3
Síntesis de clorhidrato de (2S,4S)-2-(4-dorobencil)-4-metoxi-1,4'-bipiperidina (20c)

El compuesto 20c se obtuvo a partir de 20b (120 mg, 0,43 mmol) de acuerdo con el Procedimiento General VI seguido de cromatografía ultrarrápida usando hexanos/AcOEt (elución de gradiente de 10:1 a 1:4). En la siguiente etapa, el grupo protector de Boc se retiró disolviendo el material obtenido después de la cromatografía ultrarrápida en acetato de etilo y transfiriéndolo a 6 M de HCl acuoso. La capa acuosa fuertemente ácida se basificó con 4 M NaOH para llevar el pH hasta 10 y después se extrajo tres veces con acetato de etilo. Los orgánicos combinados se secaron sobre sulfato de magnesio, se filtraron y concentraron a presión reducida para proporcionar 65 mg del producto 20c como un aceite incoloro (0,20 mmol; 46 % de rendimiento).
ESI-MS m/z para C18H27ClN2O encontrado 323,2/ 325,2 (M+1)+
Etapa 4
Síntesis de 5-((2S,4S)-2-(4-clorobencil)-4-metoxi-[1,4'-bipiperidin]-1'-il)-4H-1,2,4-triazol-3-amina (20)

El compuesto 20 se obtuvo a partir de 20c (65 mg, 0,20 mmol) de acuerdo con el Procedimiento General VIII en 56 % de rendimiento (58 mg, 0,14 mmol, sólido blanco), después de la purificación por cromatografía de fase inversa preparativa.
Diastereómero simple con configuración desconocida del grupo metoxi.
ESI-MS m/z para C20H29ClNsO encontrado 405,1/ 407,1 (M+1)+
1H RMN (DMSO-d6+D2O, 500 MHz) 57.36 (d, J = 7.9 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 3.62-3.57(m, 1H), 3.49-3.45 (m, 1H), 3.39 (d, J = 13.9 Hz, 1H), 3.26 (d, J = 12.2 Hz, 1H), 3.11-3.07 (m, 1H), 3.04 (s, 3H), 2.99-2.89 (m, 4H), 2.60 (t, J = 13.0 Hz, 1H), 2.23-2.13 (m, 1H), 2.03-1.93 (m, 2H), 1.90-1.93 (m, 2H), 1.77-1.74 (m, 1H), 1.71-1.56 (m, 3H).
Ejemplo 21
(1'-(5-amino-4H-1,2,4-triazol-3-il)-[1,4'-bipiperidin]-2-il)(4-clorofenil)metanol (21)

Etapa 1
Síntesis de 2-(hidroximetil)piperidina-1-carboxilato de tere-butilo (21a)

El compuesto 21a se obtuvo a partir de ácido 1-Boc-pipecolínico (25 g, 109 mmol) de acuerdo con el Procedimiento General XI en 73 % de rendimiento (17 g, 79,1 mmol, sólido blanco).
ESI-MS m/z para C-nH21NO3encontrado 216,1 (M+1)+
Etapa 2
Síntesis de 2-formilpiperidina-1-carboxilato de tere-butilo (21b)

Se disolvió alcohol 21a (2,3 g, 10,68 mmol) en DMSO (10 ml), se añadió trietilamina (4,44 ml, 32,05 mmol) seguido de complejo de trióxido de azufre y piridina (3,4 g, 21,37 mmol). La mezcla de reacción se agitó a TA durante 4 horas y después se separó entre acetato de etilo y 2 M HCl. La capa orgánica se lavó con 2 M HCl, salmuera, se secó en MgSO4, se filtró y se evaporó hasta sequedad. El producto se purificó por cromatografía ultrarrápida (en gel de sílice) para dar 1,17 g (5,49 mmol; 51 % de rendimiento) de aldehído 21b que se llevó inmediatamente a la siguiente reacción. Etapa 3
Síntesis de 1-(4-clorofenil)tetrahidro-1H-oxazolo[3,4-a]piridin-3(5H)-ona (21c)

A una solución de 1-cloro-4-yodobenceno (1,57 g, 6,58 mmol) en THF seco (10 ml) bajo argón a -78 °C, se añadió una solución de complejo de cloruro de isopropilmagnesio y cloruro de litio (iPrMgCl.LiCl) (1,3M en THF) (5,1 ml, 6,58 mmol) gota a gota y después se agitó durante 1 hora a -78 °C. Se añadió aldehído 21b (1.17 g, 5.49 mmol) en 5 ml de THF seco gota a gota a -78 °C. Después de 20 minutos, la mezcla de reacción se inactivó con NH4Cl sat. y se dejó durante la noche. El producto se extrajo con acetato de etilo, se secó en MgSO4, se filtró y evaporó hasta sequedad para dar 1,55 g (6,18 mmol; 94 % de rendimiento) de producto bicíclico 21c.
ESI-MS m/z para C ^H m C ^ encontrado 252,2 / 254,2 (M+1)+
Etapa 4
Síntesis de (4-clorofenil)(piperidin-2-il)metanol (21d)

Se suspendieron 1,55 g (6,18 mmol) de compuesto 21c en 6 M HCl, se sometió a reflujo durante la noche y después se evaporó hasta sequedad. El residuo se absorbió entre 1 M K2CO3 y AcOEt. La capa orgánica se lavó con salmuera,
se secó en MgSO4, se filtró y evaporó hasta sequedad para dar 0,80 g de producto de aminoalcohol 21d (3,56 mmol; 58 % de rendimiento).
ESI-MS m/z para C12H16ClNO; 225,1 encontrado 226,2 (M+1)+
Etapa 5
Síntesis de 2-((4-clorofenil)(hidroxi)metil)-[1,4'-bipiperidina]-1'-carboxilato de tere-butilo (21e)

Una mezcla de aminoalcohol 21d (0,4 g, 1,77 mmol), tere-butil 4-[(metilsulfonil)oxi]piperidina-1-carboxilato (2,47 g, 8,86 mmol) y K2CO3 (1,22 g, 8,86 mmol) en MeCN se sometió a reflujo durante 3 días. La mezcla de reacción se enfrió hasta TA y se separó entre acetato de etilo y agua. La capa orgánica se lavó con agua, 1 M HCl, salmuera, se secó en MgSO4, se filtró y se evaporó hasta sequedad. El producto bruto se purificó en cromatografía ultrarrápida de gel de sílice para dar 0,21 g (0,51 mmol; 29 % de rendimiento) del compuesto 21e.
ESI-MS m/z para C22H33ON2O3 encontrado 409,6/ 411,6 (M+1)+
Etapa 6
Síntesis de [1,4'-bipiperidin]-2-il(4-clorofenil)metanol (21f)

El compuesto 21e (0,21 g, 0,51 mmol) se disolvió en 3 N HCl/AcOEt (3 ml), la mezcla de reacción se agitó durante 30 min y después se evaporó hasta sequedad. El residuo se absorbió entre 1 N NaOH y acetato de etilo. La capa acuosa se lavó con AcOEt tres veces. Las capas orgánicas se combinaron, se secaron en MgSO4, se filtraron y evaporaron hasta sequedad para dar 0,13 g (0,42 mmol; 83 % de rendimiento) de amina libre 21f.
ESI-MS m/z para C17H25ON2O encontrado 309,4 / 310,4 (M+1)+
Etapa 7
Síntesis de (1'-(5-amino-4H-1,2,4-triazol-3-il)-[1,4'-bipiperidin]-2-il)(4-clorofenil)metanol (21)

El compuesto 21 se obtuvo a partir de 21f (0,13 g, 0,42 mmol) de acuerdo con el Procedimiento General VIII. El producto se purificó por cromatografía preparativa de fase inversa. Las fracciones que contenían el producto puro se combinaron y se evaporaron hasta sequedad. Se añadió 2 M HCl (3 ml) y se evaporó hasta sequedad para proporcionar el producto 21 en 6 % de rendimiento (10 mg, 0,023 mmol) como sal de clorhidrato.
ESI-MS m/z para C19H27ON6O encontrado 391,5 / 393,5 (M+1)+
1H RMN (CD3OD, 500 MHz) 57,44 (d, J = 8,3 Hz, 2H), 7,39 (d, J = 8,3 Hz, 2H), 5,45 (s, 1H), 4,17-4,11 (m, 1H), 4,073,98 (m, 2H), 3,67 (d, J = 11,3 Hz, 1H), 3,59-3,53 (m, 1H), 3,3-3,25 (m, 1H), 3,24-3,19 (m, 1H), 3,1-3,02 (m, 1H), 2,34 2,27 (m, 1H), 2,18-2,08 (m, 2H), 2,03-1,97 (m, 1H), 1,92-1,86 (m, 1H), 1,84-1,77 (m, 1H), 1,77-1,71 (m, 2H), 1,42-1,33 (m,2H)
Ejemplo 22
5-(4-((2S,3S)-2-(4-dorobencil)-3-metoxiazetidin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (22)

Etapa 1
Síntesis de (S)-2-(4-dorobencil)-3-oxoazetidina-1-carboxilato de ferc-butilo (22a)

A una solución del compuesto 19b (3,6 g, 11 mmol) en diclorometano (55 ml) se añadió trietilamina (16 |l, 0,11 mmol). La mezcla de reacción se enfrió hasta 0 °C y se añadió acetato de rodio (25 mg, 0,056 mmol). El baño de enfriamiento se retiró y la reacción se agitó a temperatura ambiente durante la noche. La mezcla se diluyó con agua (50 ml), las fases se separaron y la capa acuosa se extrajo con diclorometano (3 x 25 ml). Los orgánicos combinados se lavaron con salmuera, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. El producto del título (22a) se purificó por cromatografía ultrarrápida usando un sistema de disolvente 1:15 de AcOEt / hexanos. Se obtuvieron 1,5 g (5,1 mmol; 46 % de rendimiento) del compuesto 22a.
ESI-MS m/z para C15H18ClNO3 encontrado 296,1/ 298,1 (M+1)+
1H RMN (CDCl3, 500 MHz) 57,26 (d, J = 8,5 Hz, 2H), 7,12 (d, J = 8,5 Hz, 2H), 5,11-5,08 (m, 1H), 4,54 (d, J = 16,8 Hz, 1H), 4,06 (dd, J = 4,3, 16,8 Hz, 1H), 3,16 (dd, J = 6,2, 14,3 Hz, 1H), 3,07 (dd, J = 4,1, 14,3 Hz, 1H), 1,47 (s, 9H).
Etapa 2
Síntesis de (2S)-2-(4-clorobencil)-3-hidroxiazetidina-1-carboxilato de ferc-butilo (22b)
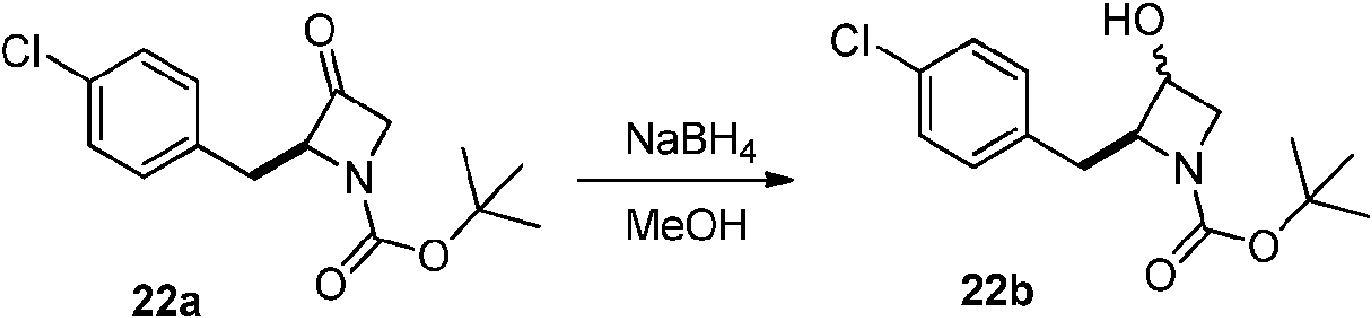
El compuesto 22a (1,5 g, 5,07 mmol) se disolvió en metanol (20 ml) y se añadió borohidruro de sodio (0,23 g, 6,1 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y después se añadieron 15 ml de 1 M NaOH y el metanol se evaporó a presión reducida. La capa acuosa se extrajo con acetato de etilo (3x 25 ml). Las capas orgánicas combinadas se secaron en sulfato de magnesio, se filtraron y concentraron a presión reducida. El producto del título se purificó por cromatografía ultrarrápida usando un sistema de disolvente 1:5 de AcOEt / hexanos.
Se obtuvieron 1,3 g (4,36 mmol; 86 % de rendimiento) del compuesto 22b (mezcla de diastereómeros en una relación 10:1)
ESI-MS m/z para C15H20ClNO3 encontrado 320,2/ 322,2 (M+Na)+
Diastereoisómero A:
1H RMN (DMSO-da, 75 °C, 700 MHz) 57,29 (d, J = 8,5 Hz, 2H), 7,24 (d, J = 8,6 Hz, 2H), 5,42 (d, J = 6,3 Hz, 1H), 4,49 4,45 (m, 1H), 4,00 (dd, J = 7,0, 9,0 Hz, 1H), 3,53 (ddd, J = 0,9, 4,3, 9,0 Hz, 1H), 3,15 (dd, J = 8,6, 14,1 Hz, 1H), 2,92 (dd, J = 5,0, 14,0 Hz, 1H), 1,32 (s, 9H).
Diastereoisómero B:
1H RMN (DMSO-da, 75 °C, 700 MHz) 67,33 (d, J = 8,5 Hz, 2H), 7,24 (d, 2H), 5,33 (d, J = 6,7 Hz, 1H), 4,30-4,27 (m, 1H), 4,04-4,02 (m, 1H), 3,76 (ddd, J = 0,9, 6,6, 8,8 Hz, 1H), 3,44 (dd, J = 4,6, 8,8 Hz, 1H), 2,89 (dd, J=8,3, 14,1 Hz, 1H), 1,39 (s, 9H).
Etapa 3
Síntesis de (2S)-2-(4-dorobencil)-3-metoxiazetidina-1-carboxilato de tere-butilo (22c)

El compuesto 22c se obtuvo a partir de 22b (0,40 g, 1,34 mmol) de acuerdo con el Procedimiento General XXI (para mejorar la solubilidad, se usó DMF como un disolvente de reacción) en >99 % de rendimiento (1,34 mmol; 418 mg, mezcla de diastereómeros en una relación 9:1), después de cromatografía ultrarrápida usando hexanos/EtOAc (elución de gradiente de 10:1 a 5:1).
ESI-MS m/z para C16H22ClNO3 encontrado 334,1/ 336,1 (M+Na)+
Diastereoisómero A:
1H RMN (DMSO-d6, 75 °C, 700 MHz) 67,29 (d, J = 8,4 Hz, 2H), 7,23 (d, J = 8,5 Hz, 2H), 4,45-4,42 (m, 1 H), 4,18 (td, J = 4,3, 6,7 Hz, 1H), 3,96 (dd, J = 6,7, 9,2 Hz, 1H), 3,61 (ddd, J = 0,9, 4,3, 9,2 Hz, 1H), 3,20 (s, 3H), 3,11-3,06 (m, 1H), 2,97-2,95 (m, 1H), 1,31 (s, 9H).
Diastereoisómero B:
1H RMN (DMSO-de, 75 °C, 700 MHz) 67,34 (d, J = 8,4 Hz, 2H), 7,26 (d, J = 8,3 Hz, 2H), 4,08-4,06 (m, 1H), 3,81-3,77 (m, 2H), 3,50-3,49 (m, 1H), 3,12-3,10 (m, 1H), 2,95 (s, 3H), 2,90 (dd, J = 8,7, 13,9 Hz, 1H), 1,40 (s, 9H).
Etapa 4
Síntesis de clorhidrato de (2S)-2-(4-clorobencil)-3-metoxiazetidina (22d)

De acuerdo con el Procedimiento General VII el compuesto 22c (418 mg, 1,34 mmol) se trató con 4 M HCl (gas) en 1,4-dioxano (2,2 ml, 9,04 mmol). El producto bruto se trituró con éter dietílico proporcionando 256 mg (1,03 mmol, 77 % de rendimiento) de azetidina 22d como su sal de clorhidrato (una mezcla de diastereómeros en una relación 9:1). ESI-MS m/z para C11H14CINO encontrado 212,1/ 214,1 (M+1)+
Diastereoisómero A:
1H RMN (DMSO-de, 75 °C, 600 MHz) 67,35-7,33 (m, 4H), 4,73 (dd, J = 7,3, 14,1 Hz, 1H), 4,25 (td, J =4,0, 6,2 Hz, 1H), 4,08 (ddd, J = 0,8, 6,2, 11,3 Hz, 1H), 3,72 (ddd, J = 0,8, 3,9, 11,3 Hz, 1H), 3,18 (d, J =7,7 Hz, 2H), 3,06 (s, 3H). Diastereoisómero B:
1H RMN (DMSO-da, 75 °C, 600 MHz) 67,39-7,37 (m, 4H), 4,33- 4,29 (m, 1H), 4,14 (dd, J = 6,6, 13,2 Hz, 1H), 4,02 (ddd, J = 0,6, 7,2, 10,5 Hz, 1H), 3,96 (dd, J = 5,1, 12,2 Hz, 1H), 3,80 (dd, J = 4,7, 12,1 Hz, 1H), 3,64 (dd, J = 6,4, 10,7 Hz, 1H), 3,45 (s, 3H).
Etapas 5-7
Síntesis de 5-(4-((2S,3S)-2-(4-clorobencil)-3-metoxiazetidin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (22)
La azetidina 22d se llevó a las etapas sintéticas restantes como se describe en el Procedimiento General VI (aminación reductora con N-Boc-piperid-4-ona), el Procedimiento General VII (desprotección de Boc) y el Procedimiento General VIII (formación de anillo de triazol). Se sintetizaron 142 mg (0.37 mmol, 37 % de rendimiento en 3 etapas) del compuesto del título 22 en una forma de diastereómero simple (configuración desconocida; supuestamente eis) (el diastereómero secundario se retiró en una purificación final por cromatografía de fase inversa).
ESI-MS m/z para C18H25CIN6O encontrado 377,0/ 378,9 (M+1)+, 375,1/ 377,0 (M-1)
1H RMN (DMSO-da+D2 O, 500 MHz) 57,39 (d, J = 8,6 Hz, 2H), 7,33 (d, J = 8,5, 2H), 4,88 (s a, 1H), 4,11 (d, J = 5,2, 2H), 4,02-4,00 (m, 1H), 3,81 (d, J = 13,2 Hz, 1H), 3,77 (d, J = 13,2 Hz, 1H), 3,38-3,35 (m, 2H), 3,27 (s, 3H), 2,96 (dd, J = 4,8, 13,9 Hz, 1H), 2,78 (q, J = 10,5 Hz, 2H), 1,96 (d, J = 12,2 Hz, 1H), 1,88 (d, J = 11,8 Hz, 1H), 1,43-1,35 (m, 2H). Ejemplo 23
5-(4-((2S,3R)-2-(4-dorobencil)-3-fluoroazetidin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (23)
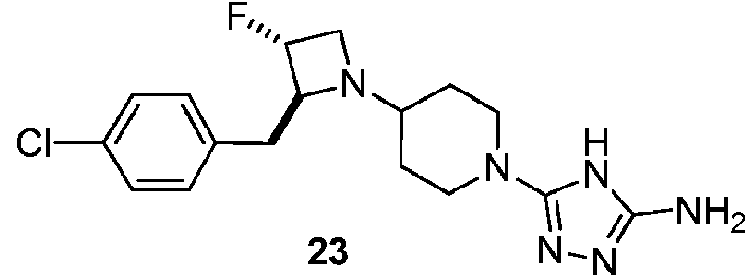
Etapa 1
Síntesis de (2S,3R)-2-(4-dorobencil)-3-fluoroazetidina-1-carboxilato de tere-butilo (23a)

A una solución enfriada (-78°C) del compuesto 22b (400 mg, 1,34 mmol) en diclorometano (10 ml) se añadieron 350 ^l de (2,68 mmol) de trifluoruro de (dietilamino)azufre (DAST) en argón. La mezcla de reacción se dejó calentar hasta temperatura ambiente y se agitó a temperatura ambiente durante la noche. Se inactivó con 5 % de NaHCO3 ac. (20 ml) y se extrajo con acetato de etilo (3 x 15 ml). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. La purificación por cromatografía ultrarrápida en AcOEt / hexanos 1:15 proporcionó 200 mg (0,67 mmol; 50 % de rendimiento) el compuesto del título (23a) como un diastereómero simple (la configuración no está establecida; supuestamente trans).
ESI-MS m/z para C15H19CFNO 2 encontrado 300,2/ 302,2 (M+1)+
1H RMN (DMSO-d6, 75 °C, 600 MHz) 57,31 (d, J = 8,5 Hz, 2H), 7,22 (d, J = 8,3 Hz, 2H), 5,31(dtd, J = 3,0, 6,2 Hz, Jhf = 60 Hz, 1H), 4,52-4,47 (m, 1H), 4,13-4,07 (m, 1H), 3,82-3,76 (m, 1H), 3,10-3,04 (m, 2H), 1,34 (s, 9H).
Etapa 2
Síntesis de clorhidrato de (2S,3R)-2-(4-clorobencil)-3-fluoroazetidina (23b)

De acuerdo con el Procedimiento General VII el compuesto 23a (190 mg, 0.63 mmol) se trató con 4 N HCl (gas) en 1,4-dioxano (2.2 ml, 9.04 mmol). El producto bruto se trituró con éter dietílico proporcionando 150 mg (0.63 mmol, 100 % de rendimiento) de azetidina 23b como una sal de clorhidrato.
ESI-MS m/z para C10H11CFN encontrado 200,1/ 202,1 (M+1)+
Etapas 3-5
Síntesis de 5-(4-((2S,3R)-2-(4-clorobencil)-3-fluoroazetidin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (23)
La azetidina 23b (0,63 mmol, 150 mg) se llevó a las etapas sintéticas restantes como se describe en el Procedimiento General VI (aminación reductora con N-Boc-piperid-4-ona), el Procedimiento General VII (desprotección de Boc) y el Procedimiento General VIII (formación de anillo de triazol). Se sintetizaron 134 mg (0,37 mmol) del compuesto del título 23 en 59 % de rendimiento en 3 etapas.
ESI-MS m/z para C17H22 CFN 6 encontrado 365,0/ 367,0 (M+1)+, 363,0/ 365,0 (M-1)
1H RMN (DMSO-d6, 700 MHz) 57,41 (d, J = 8,5 Hz, 2H), 7,35 (d, J = 8,5 Hz, 2H), 5,34 (dt, Jhf = 56 Hz, J = 4,95 Hz, 1H), 5,0 (s a, 1H), 4,40-4,36 (m, 1H), 4,26 (dd, J = 13,1 Hz, Jhf = 23,3 Hz, 1H), 3,80 (dd, J = 12,7 Hz, Jhf = 22,0 Hz,
2H), 3,39-3,37 (m, 1H), 3,33 (dd, J = 11,2, 13,9 Hz, 1H), 3,11 (dd, J = 3,3, 13,5 Hz, 1H), 2,84-2,80 (m, 2H), 2,02 (d, J = 12,7 Hz, 1H), 1,92 (d, J = 12,1 Hz, 1H), 1,42 (qd, J = 4,4, 12,2 Hz, 2H).
Ejemplo 24
(R)-5-(4-(2-(4-clorobencM)pirroMdin-1-M)piperidin-1-M)-4H-1,2,4-triazol-3-amina (24)

Etapa 1
Síntesis de ácido (R)-1-(fe/-c-butoxicarboml)pirrolidma-2-carboxflico (24a)

A una solución de D-prolina 20 g (173,7 mmol) en acetona/agua 1:1, se añadió hidróxido de sodio 13,9 g (347,44 mmol), seguido de Boc2O 39,8 g (182,41 mmol). La reacción se agitó durante 2 días a pH ~9. La LCMS indicó que la reacción se había completado. La acetona se retiró por evaporación. La fase acuosa restante se lavó con éter dietílico, se acidificó hasta pH~3 y se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de magnesio anhidro y se evaporaron hasta sequedad. El residuo sólido se lavó con hexano y se secó. Se obtuvieron 31,1 g (144 mmol; 83 % de rendimiento) del producto 24a como un sólido blanco.
ESI-MS m/z para C10H17NO4 encontrado 216,1 (M+1)+
Etapa 2
Síntesis de (R)-2-(metoxi(metil)carbamoil)pirrolidina-1-carboxilato de fe/c-butilo (24b)

A una solución de ácido 24a se añadieron 10 g (46,5 mmol) en diclorometano carbonildiimidazol 11,3 g (69,7 mmol) Después de 30 minutos se añadió W,0-dimetilhidroxilamina 6,8 g (69,7 mmol) y la reacción se agitó durante la noche. La LC/MS indicó que la reacción se había completado. La mezcla de reacción se lavó con ácido clorhídrico, una solución acuosa al 5 % de bicarbonato de sodio y salmuera. La capa orgánica se secó en sulfato de magnesio anhidro y se concentró. El producto se purificó mediante cromatografía en columna con hexano/EtOAc 4/1. Se obtuvieron 6,19 g (24,2 mmol; 52 % de rendimiento) del compuesto 24b como un aceite incoloro.
ESI-MS m/z para C12H22NO4 encontrado 259,1 (M+1)+, 281,0 (M+Na)+
1H RMN (CDCla, 600 MHz) S 4,70, 4,60 (m, m, 1H, 2 confórmeros), 3,79, 3,72 (s, s, 3H, 2 confórmeros), 3,58 (m, 1H), 3,48, 3,40 (m, m, 1H, 2 confórmeros), 3,19 (s, 3H), 2,18 (m, 1H), 1,91 (m, 3H), 1,46, 1,41 (s,s, 9H, 2 confórmeros). Etapa 3
Síntesis de (R)-2-(4-clorobenzoil)pirrolidina-1-carboxilato de fe/c-butilo (24c)

La amida 24b 3,0 g (11,61 mmol) se disolvió en éter dietílico seco en una atmósfera de argón y se enfrió hasta -70 °C. Se añadió bromuro de p-clorofenilmagnesio, previamente generado a partir de magnesio 875 mg (36,0 mmol) y pbromoclorobenceno 6,67 g (34,8 mmol) gota a gota a una solución de amida 24b a -70 °C. Después de la adición de reactivo de Grignard, la reacción se agitó a temperatura ambiente durante 2 horas. La LCMS indicó que la reacción se había completado. La reacción se inactivó con solución de cloruro de amonio saturada durante 15 minutos y se extrajo con éter dietílico. La capa orgánica se lavó con 1 M de ácido clorhídrico y salmuera. El disolvente se evaporó y el residuo aceitoso se purificó por cromatografía en columna con hexano/EtOAc 10/1. Se obtuvieron 2,47 g (8,0 mmol; 69 % de rendimiento) del producto 26c como un sólido blanco cristalino.
ESI-MS m/z para C16H20ClNO3 encontrado 310,0/ 312,0 (M+1)+
1H RMN (CDCl3, 600 MHz) 57,94, 7,90 (AA'BB', J= 8,4Hz, AA'BB', J= 8,4Hz, 2H, 2 confórmeros), 7,47, 7,43 (AA'BB', J= 8,4Hz, AA'BB', J= 8,4Hz, 2H, 2 confórmeros), 5,28, 5,27 (dd, J1= 9,0Hz, J2= 3,6Hz, dd, J1= 9,6Hz, J2= 4,8Hz, 1H, 2 confórmeros), 3,68, 3,62 (m, m, 1H, 2 confórmeros), 3,56, 3,48 (m, m, 1H, 2 confórmeros), 2,30 (m, 1H), 1,92 (m, 3H), 1,46, 1,26 (s, s, 9H, 2 confórmeros).
Etapa 4
Síntesis de (R)-2-(4-clorobencil)pirrolidina (24d)

A una solución de cetona 24c 1 g (3,23 mmol) en diclorometano se añadieron cloruro de aluminio anhidro 1,29 g (9,68 mmol) y trietilsilano 1,55 ml (9,68 mmol). Después de 1 hora de agitación, la LCMS indicó que la reacción se había completado. La reacción se inactivó con una mezcla de 4 M NaOH y salmuera. La fase acuosa se extrajo con diclorometano, se secó en sulfato de magnesio anhidro y se concentró. Se obtuvieron 632 mg (3,22 mmol; 99 % de rendimiento) del producto 24d como un aceite amarillo y se llevaron directamente a la siguiente etapa.
ESI-MS m/z para CnH^ClN encontrado 195,9/ 197,9 (M+1)+
Etapa 5
Síntesis de (R)-4-(2-(4-clorobencil)pirrolidin-1-il)piperidina-1-carboxilato de ferc-butilo (24e)

El compuesto 24e se obtuvo a partir de 24d (632 mg, 3,23 mmol) de acuerdo con el Procedimiento General VI seguido de cromatografía ultrarrápida usando metanol/diclorometano 1:50, en 44 % de rendimiento (540 mg, 1,42 mmol, aceite amarillo).
ESI-MS m/z para C21H31ClN2O2 encontrado 379,1/ 381,1 (M+1)+
1H RMN (CDCl3, 600 MHz) 57,24 (AA'BB', J = 8,4 Hz, 2H), 7,11 (AA'BB', J = 8,4 Hz, 2H), 3,03 (s a, 1H), 2,94 (s a, 1H), 2,86 (d, J = 12,6 Hz, 1H), 2,70 (s a, 4H), 2,58 (q, J = 7,2 Hz, 1H), 2,42 (m, 1H), 1,84 (d, J = 7,8 Hz, 1H), 1,78 (d, J = 12,6 Hz, 1H), 1,72 (d, J = 7,2 Hz, 1H), 1,65 (m, 3H), 1,55 (m, 3H), 1,46 (s, 9H).
Etapa 6
Síntesis de clorhidrato de (R)-4-(2-(4-dorobencil)pirrolidin-1-il)piperidina (24f)

De acuerdo con el Procedimiento General VII el compuesto 24e (540 mg, 1,42 mmol) se trató con 2N HCl (gas) en acetato de etilo (6,5 ml, 12,81 mmol). El producto bruto se trituró con éter dietílico proporcionando 447 mg (1,42 mmol, >99 % de rendimiento, cristales amarillos) de 24f como una sal de clorhidrato.
ESI-MS m/z para C1sH23ClN2 encontrado 279,2/ 281,2 (M+1)+
Etapa 7
Síntesis de (R)-5-(4-(2-(4-clorobencil)pirrolidin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (24)

El compuesto 24 se obtuvo a partir de 24f (447 mg, 1,42 mmol) de acuerdo con el Procedimiento General VIII. El producto se purificó por cromatografía preparativa de fase inversa para dar 201 mg (0,56 mmol; 39 % de rendimiento) del compuesto del título 24.
ESI-MS m/z para C1sH25ClN6 encontrado 361,0/ 363,0 (M+1)+
1H RMN (DMSO-d6+D2O, 600 MHz): 57,36 (AA'BB', J = 8,4 Hz, 2H), 7,30 (AA'BB', J = 8,4 Hz, 2H), 3,83 (m, 1H), 3,78 (d, J = 12,6 Hz, 2H), 3,48 (t, J = 12,0 Hz, 1H), 3,40 (q, J = 5,4 Hz, 1H), 3,16 (m, 2H), 2,90 (q, J = 12,0 Hz, 2H), 2,72 (t, J = 12,0 Hz, 1H), 2,10 (d, J = 11,4 Hz, 1H), 2,02 (d, J = 11,4 Hz, 1H), 1,85 (m, 3H), 1,66 (m, 3H).
Ejemplo 25
(S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)piperazin-2-ona (25)

Etapa 1
Síntesis de (S)-(1-amino-3-(4-clorofenil)-1-oxopropan-2-il)carbamato de tere-butilo (25a)

El compuesto del título (25) se obtuvo a partir de Boc-L-p-dorofenilalanina (1,4 g, 4,67 mmol) de acuerdo con el Procedimiento General IX en 97 % de rendimiento (1,36 g, 4,54 mmol) como un polvo blanco.
ESI-MS m/z para C14H19GN2O3 encontrado 299,0 (M+1)+, 321,0 (M+Na)+
Etapa 2
Síntesis de (S)-(1-amino-3-(4-clorofenil)propan-2-il)carbamato de tere-butilo (25b)

La reducción de la amida 25a al compuesto del título se logró de acuerdo con el Procedimiento General V. La purificación en columna de gel de sílice ultrarrápida en un sistema de disolvente de CH2Cl2/MeOH (elución de gradiente de 70:1 a 20:1) proporcionó 360 mg (1,26 mmol, 28 % de rendimiento) del producto puro 25b.
ESI-MS m/z para C14H21CW2O2 encontrado 285,3/287,3 (M+1)+
Etapa 3
Síntesis de (S)-(1-(2-cloroacetamido)-3-(4-clorofenil)propan-2-il)carbamato de tere-butilo (25c)

Se hicieron reaccionar 360 mg (1,26 mmol) de amina 25b con cloruro de cloroacetilo como se describe en el Procedimiento General II. Después del procesamiento acuoso se descubrió que el producto bruto 25c era lo suficientemente puro como para usarse en la siguiente etapa sin purificación adicional.
ESI-MS m/z para C16H22Cl2N2Os encontrado 361,3/363,3 (M+1)+, 383,3/385,3 (M+Na)+
Etapa 4
Síntesis de clorhidrato de (S)-N-(2-amino-3-(4-clorofenil)propil)-2-cloroacetamida (25d)

La retirada del grupo protector de Boc se logró de acuerdo con el Procedimiento General VII. Se obtuvieron 355 mg (1,2 mmol) del compuesto del título 25d (90 % de rendimiento en dos etapas). El producto bruto 25d en una forma de sal de clorhidrato se llevó a la siguiente etapa.
ESI-MS m/z para C nH u C h ^O encontrado 261,2/ 263,2 (M+1)+
Etapa 5
Síntesis de (S)-5-(4-dorobencil)piperazin-2-ona (25e)

Se disolvieron 355 mg (1.2 mmol) del compuesto 25d en 50 ml de acetonitrilo y se añadieron carbonato de potasio anhidro (437 mg; 3,16 mmol) y yoduro de sodio (20 mg) secuencialmente. La reacción se agitó a 50 °C durante 5 horas y después a temperatura ambiente durante la noche. Los sólidos se filtraron, el filtrado se concentró y el producto se purificó por cromatografía ultrarrápida en columna de gel de sílice usando un eluyente de gradiente de sistema de disolvente de CH2Ch/MeOH (elución de gradiente de 40:1 a 5:1). Se obtuvieron 160 mg (0,71 mmol; 60 % de rendimiento) del compuesto del título (25e).
ESI-MS m/z para C11H13CW2O encontrado 225,2/ 227,2 (M+1)+
Etapa 6
Síntesis de (S)-4-(2-(4-clorobencil)-5-oxopiperazin-1-il)piperidina-1-carboxilato de tere-butilo (25f)

La aminación reductiva del compuesto 25e y N-Boc-piperid-4-ona se logró de acuerdo con el Procedimiento General VI. Se obtuvieron 175 mg (0,43 mmol; 60 % de rendimiento) del compuesto del título 25f después de la purificación por cromatografía ultrarrápida en columna de gel de sílice usando un sistema de disolvente de CH2Ch/MeOH 20:1. ESI-MS m/z para C21H30ClN3O3 encontrado 408,4/410,4 (M+1)+
Etapa 7
Síntesis de clorhidrato de (S)-5-(4-clorobencil)-4-(piperidin-4-il)piperazin-2-ona (25g)
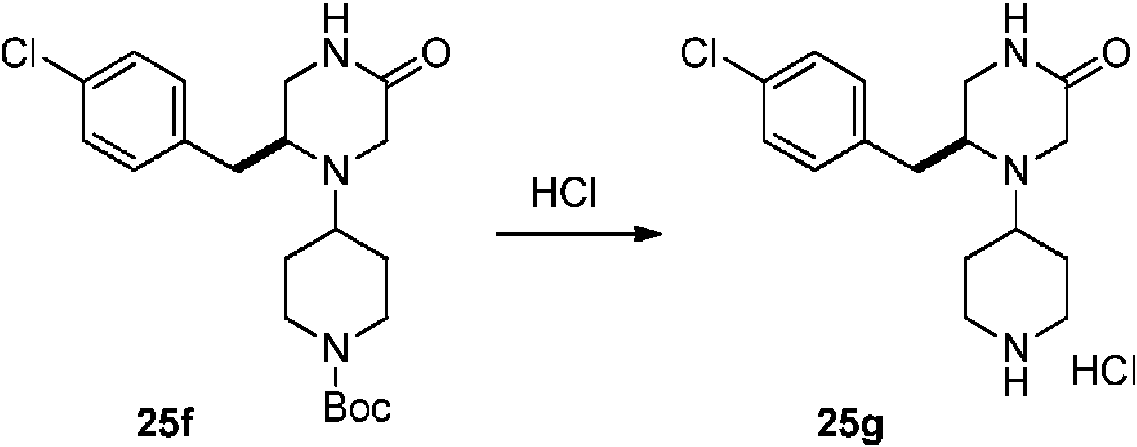
La desprotección de Boc del compuesto 25f se logró de acuerdo con el Procedimiento General VII proporcionando 96 mg (0,31 mmol, 72 % de rendimiento) del compuesto del título 25g.
ESI-MS m/z para C16H22ClN3O encontrado 308,4/310,3 (M+1)+
1H RMN (CDCl3, 500 MHz) 57,27 (AA'BB', J ■= 8,5 Hz, 2H), 7,08 (AA'BB', J = 8,5Hz, 2H), 6,01 (s a, 1H), 3,19-3,30 (m, 4H), 3,00-3,06 (m, 1H), 2,82-2,88 (m, 1H), 2,64-2,71 (m, 3H), 2,01-2,13(m, 4H), 1,88-1,95 (m, 2H), 1,47-1,57 (m, 2H).
Etapa 8
Síntesis de (S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)piperazin-2-ona (25)

La síntesis de aminotriazol se logró de acuerdo con el Procedimiento General VII. A partir de 90 mg (0,292 mmol) del material de partida 25g, se obtuvieron 43 mg del compuesto diana 25 después de la purificación por la cromatografía de fase inversa.
ESI-MS m/z para C16H22CW3O encontrado 390,0/392,0 (M+1)+, 388,0/390,1 (M-1)
1H RMN (D2O, 500 MHz) 57,28 (AA'BB', J = 7,3 Hz, 2H), 7,14 (AA'BB', J = 7,7 Hz, 2H), 4,07-4,12 (m, 1H), 4,01 (d, J = 16,7 Hz, 1H), 3,86 (d, J = 16,9 Hz, 1H), 3,73-3,77 (m, 2H), 3,32-3,36 (m, 1H), 3,17-3,22 (m, 2H), 2,81-3,04 (m, 4H), 2,09-2,15 (m, 2H), 1,68-1,75 (m, 2H).
Ejemplo 26
(S)-5-(4-(2-(4-clorobencil)piperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (26)

La reducción de 2-piperazinona 25 al compuesto del título 26 se logró de acuerdo con el Procedimiento General V. El producto bruto se purificó por la HPLC preparativa de fase inversa en columna. Las fracciones que contenían el producto se liofilizaron para proporcionar 13 mg (34 % de rendimiento) del compuesto del título 26.
ESI-MS m/z para C18H26ClN7 encontrado 376,0/ 378,0 (M+1)+
1H RMN (DMSO-d6+D2O, 700 MHz) 57,40 (AA'BB', J = 8,5 Hz, 2H), 7,32 (AA'BB', J = 8,5 Hz, 2H), 3,74 (t, J = 15,1 Hz, 2H), 3,46 (m, 2H), 3,24 (m, 3H), 3,03 (m, 3H), 2,88 (m, 3H), 2,74 (dd, J= 9,3, 14,1 Hz, 1H), 1,82 (d, J = 12,2 Hz, 1H), 1,78 (d, J = 11,9 Hz, 1H), 1,70 (dq, J = 4,1, 11,9Hz, 1H), 1,51 (dq, J = 4,2, 12,2 Hz, 1H).
Ejemplo 27
5-(4-((2S,5S)-2-(4-clorobencil)-4,5-dimetilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (27)

Etapa 1
Síntesis de (S)-2-((S)-2-((ferc-butoxicarbonil)amino)-3-(4-clorofenil)-N-metilpropanamido)propanoato de metilo (27a)

La Boc-L-p-clorofenilalanina se acopló con clorhidrato de metil éster de N-Me-L-alanina como se describe en el Procedimiento General IX. Se obtuvieron 1,3 g del compuesto del título 27a (99 % de rendimiento) después de la cristalización a partir de acetato de etilo.
ESI-MS m/z para C1gH27ClN2O5 encontrado 399,1/ 401,1 (M+1)+
1H RMN (DMSO-da, 250 MHz) 7,20 (AA'BB', 2H, J=8,4Hz), 7,01 (AA'BB', 2H, J=8,4Hz), 4,87- 4,64 (m, 1H), 4,54-4,32 (m, 1H), 3,48 (s, 3H), 2,80 (s, 3H), 2,75-2,54 (m, 2H), 1,19 (s, 9H), 1,14 (d, 3H, J= 6Hz).
Etapa 2
Síntesis de (3S,6S)-3-(4-clorobencil)-1,6-dimetilpiperazina-2,5-diona (27b)

Comenzando por éster metílico 27a, el compuesto del título se preparó por desprotección ácida del grupo amino seguido del cierre inmediato del anillo de acuerdo con el Procedimiento General X. Se obtuvieron 0,64 g del compuesto del título 27b (73 % de rendimiento) después de la cristalización a partir de acetato de etilo.
ESI-MS m/z para C13H15ClN2O2 encontrado 266,9/ 268,9 (M+1)+
1H RMN (DMSO-d6, 250 MHz) 58,17 (bs, 1H), 7,25 (AA'BB', J = 8,4 Hz, 2H), 6,99 (AA'BB', J = 8,4 Hz, 2H), 4,14-4,05 (m, 1H), 3,57 (q, J = 7,0 Hz, 1H), 2,99 (dd, J = 4,1, 13,2 Hz, 1H), 2,72 (dd, J = 4,8, 13,2 Hz, 1H), 2,59 (s, 3H), 1,04 (d, J=7Hz, 3H).
Etapa 3
Síntesis de (2S,5S)-5-(4-clorobencil)-1,2-dimetilpiperazina (27c)

A la solución de 0,62 g (2,32 mmol) de 2,5-dicetopiperazina 27b en THF (7 ml/mmol) se añaden 2 ml de complejo de BH3.DMS y la mezcla de reacción se sometió a reflujo durante 5 horas, después de lo cual el control de TLC mostró el consumo completo del material de partida. Se enfrió a temperatura ambiente y se añadieron cuidadosamente 8 ml de 2 M HCl (Advertencia: ¡formación de espuma!) y la mezcla de reacción se volvió a someter a reflujo durante 2 horas y se volvió a enfriar hasta temperatura ambiente. Después el pH de la solución se volvió llevó a 13 mediante la adición gota a gota de 6 M NaOH. La capa orgánica se separó y la capa acuosa se extrajo adicionalmente con acetato de etilo. Después los extractos orgánicos combinados se secaron en MgSO4, se filtraron y los disolventes se evaporaron para proporcionar 0,53 g de producto 27c (2,26 mmol, 94 % de rendimiento) que se usó en la siguiente etapa sin purificación.
ESI-MS m/z para C13H19GN2 encontrado 238,6/ 240,6 (M+1)+
Etapa 4
Síntesis de 4-((2S,5S)-2-(4-dorobencil)-4,5-dimetilpiperazin-1-il)piperidina-1-carboxilato de tere-butilo (27d)

La aminación reductiva del compuesto 27c y N-Boc-piperid-4-ona se logró de acuerdo con el Procedimiento General VI. Se obtuvieron 0,36 g (0,85 mmol; 38 % de rendimiento) del compuesto del título 27d después de la purificación por cromatografía ultrarrápida en columna de gel de sílice usando un sistema de disolvente de CH2Cl2/MeOH 15:1. ESI-MS m/z para C23H36ClN3O2 encontrado 422,1/ 424,1 (M+1)+
Etapa 5
Síntesis de (2S,5S)-2-(4-clorobencil)-4,5-dimetil-1-(piperidin-4-il)piperazina (27e)

La desprotección con Boc del compuesto 27d se logró de acuerdo con el Procedimiento General VII. La sal de clorhidrato bruta se absorbió entre acetato de etilo y una solución de 2 M K2CO3. La capa acuosa se extrajo varias veces con acetato de etilo, los productos orgánicos combinados se secaron en MgSO4, se filtraron y el disolvente se evaporó al vacío. Se obtuvieron 0,18 g (0,56 mmol; 66 % de rendimiento) del compuesto del título 27e.
ESI-MS m/z para C18H28ClN3 encontrado 322,1/ 324,1 (M+1)+
Etapa 6
Síntesis de 5-(4-((2S,5S)-2-(4-clorobencil)-4,5-dimetilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (27)

La síntesis de aminotriazol se logró de acuerdo con el Procedimiento General VII.
A partir de 180 mg (0,56 mmol) de material de partida 27e, se obtuvieron 80 mg (0,20 mmol; 35 % de rendimiento) del compuesto diana 27 después de la purificación por cromatografía en gel de sílice en un sistema de disolvente de CHCb/MeOH 20:1.
ESI-MS m/z para C20H30GN7 encontrado 404,2/ 406,2 (M+1)+
1H RMN (DMSO-d6, 250 MHz) S 7,38 (AA'BB', J = 8,4 Hz, 2H), 7,24 (AA'BB', J = 8,4 Hz, 2H), 5,58 (s a, 1H), 3,84-3,71
(m, 2H), 3,11-2,99 (m, 2H), 2,86-2,59 (m, 5H), 2,48-2,25 (m, 2H), 2,13 (s, 3H), 2,06-1,88 (m, 4H), 1,49-1,26 (m, 2H), 1,04 (d, J = 6,1 Hz, 3H).
Ejemplo 28
(S)-5-(4-(2-(4-dorobencil)-4-metilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (28)

Con la excepción de que Boc-L-p-clorofenilalanina se acopló con clorhidrato de metil (metilamino)acetato (clorhidrato de sarcosina metil éster) en vez de N-metil-L-alanina metil éster en la primera etapa sintética, el compuesto del título se preparó de la misma forma que en el Ejemplo 27 y sus intermediarios.
Se sintetizaron 270 mg del compuesto del título 28.
ESI-MS m/z para C19H28ClN7 encontrado 390,2/392,3 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 510,86 (s a, 1H), 7,32-7,26 (m, 2H), 7,17 (d, J = 8,0 Hz, 2H), 5,59 (s, 2H), 3,77-3,67 (m, 2H) 2,91-2,71 (m, 3H), 2,70-2,51 (m, 7H), 2,00 (s, 3H), 1,98-1,90 (m, 2H),1,75-1,55 (m, 2H), 1,49-1,22 (m,2H). Ejemplo 29
(S)-5-(4-(2-(4-clorobencil)-4-isobutilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (29)

Con la excepción de que Boc-L-p-clorofenilalanina se acopló con clorhidrato de metil (isobutilamino)acetato (clorhidrato de N-isobutilglicina metil éster) en vez de N-metil-L-alanina metil éster en la primera etapa sintética, el compuesto del título se preparó de la misma forma que en el Ejemplo 27 y sus intermediarios.
Se sintetizaron 50 mg del compuesto del título 29.
ESI MS m/z para C22H34ClN7 encontrado 432,2/434,2 (M+1)+
1H RMN (DMSO-d6, 250 MHz) 57,45-7,33 (m, 2H), 7,34-7,21 (m, 2H), 5,57 (s a, 1H), 3,83 (d, 2H, J = 12,5 Hz), 3,10 2,63 (m, 7H), 2,45-2,03 (m, 4H), 2,01-1,24 (m, 7H), 0,89 (d, J = 6,4 Hz, 6H).
Ejemplo 30
((2R,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-1-metilpiperazin-2-il)metanol (30)

Etapa 1
Síntesis de (S)-(1-(4-dorofenil)-3-hidroxipropan-2-il)carbamato de tere-butilo (30)
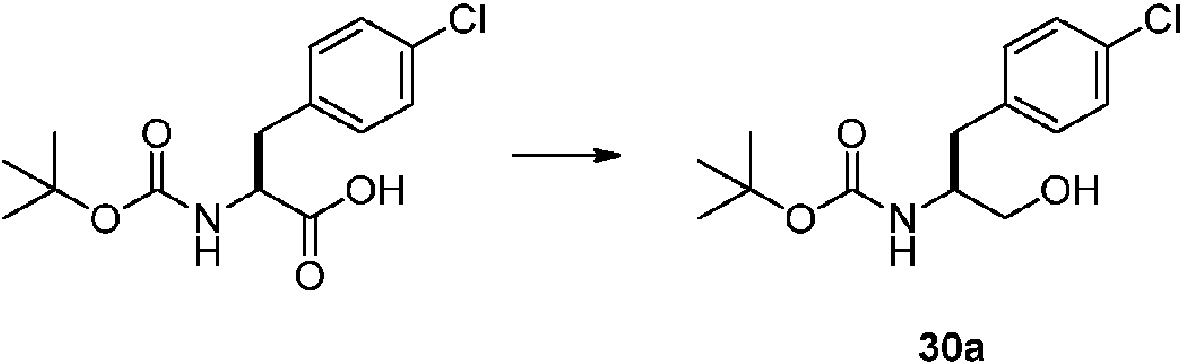
Boc-L-p-clorofenilalanina se redujo al compuesto del título como se describe en el Procedimiento General XI. A partir de 15 g (50 mmol) del material de partida, se obtuvieron 11,5 g (40,2 mmol, 81 % de rendimiento) del compuesto 30a. ESI-MS m/z para C14H2üCINO3 encontrado 285,9/ 287,9 (M+1)+
Etapa 2
Síntesis de (S)-(1-(4-clorofenil)-3-oxopropan-2-il)carbamato de tere-butilo (30b)

El compuesto 30a se oxidó al compuesto del título de acuerdo con el Procedimiento General XII. A partir de 10 g (35 mmol) del material de partida, se obtuvieron 8,5 g (29,9 mmol, 85 % de rendimiento) del compuesto 30b.
ESI-MS m/z para C14 H18CINO3 encontrado 283,9/ 285,9 (M+1)+
Etapa 3
Síntesis de (S)-2-(((S)-2-((tere-butoxicarbonil)amino)-3-(4-clorofenil)propil)amino)-3-hidroxipropanoato de metilo (30c)

La aminación reductiva del compuesto 30b (2,8 g, 9,87 mmol) con clorhidrato de L-serina metil éster (2,3 g, 14,8 mmol) se logró de acuerdo con el Procedimiento General XIII. El producto bruto 30c (3,0 g, 7,8 mmol; 79 % de rendimiento) era lo suficientemente puro para usarse en la siguiente etapa sin purificación adicional.
ESI-MS m/z para C18 H27 CIN2 O5 encontrado 387,1/ 389,1 (M+1)+
Etapa 4
Síntesis de (S)-2-(((S)-2-((tere-butoxicarbonil)amino)-3-(4-clorofenil)propil)(metil)amino)-3-hidroxipropanoato de metilo (30d)

La N-metilación de amina 30c (3,0 g, 7,75 mmol) con HCHO acuoso (0,72 ml, 9,3 mmol) se logró de acuerdo con el Procedimiento General XIV. El producto bruto se purificó por cromatografía ultrarrápida de gel de sílice (sistema de disolvente de cloroformo / MeOH 20:1) para dar 1,2 g (2,99 mmol; 39 % de rendimiento) del compuesto del título 30d. ESI-MS m/z para C19H29ON2O5 encontrado 401,1/403,1 (M+1)+
Etapa 5
Síntesis de (3S,6S)-6-(4-clorobencil)-3-(hidroximetil)-4-metilpiperazin-2-ona (30e)

La ciclación del compuesto 30d (1,2 g, 3 mmol) se logró de acuerdo con el Procedimiento General X. Se obtuvieron 0,61 g (2,27 mmol; 76 % de rendimiento) del producto 30e, lo suficientemente puro como para llevarse a la siguiente etapa.
ESI-MS m/z para C13H17ClN2O2 encontrado 269,1/ 271,1 (M+1)+
Etapa 6
Síntesis de ((2R,5S)-5-(4-clorobencil)-1-metilpiperazin-2-il)metanol (30f)

La reducción de 30e (0,61 g, 2,23 mmol) se logró de acuerdo con el Procedimiento General V. Se obtuvieron 0,45 g del producto 30f (1,76 mmol; 79 % de rendimiento).
ESI-MS m/z para C-i3H-i9ClN2O encontrado 255,3 / 257,3 (M+1)+
Etapa 7
Síntesis de 4-((2S,5R)-2-(4-clorobencil)-5-(hidroximetil)-4-metilpiperazin-1-il)piperidina-1-carboxilato de ferc-butilo (30g)
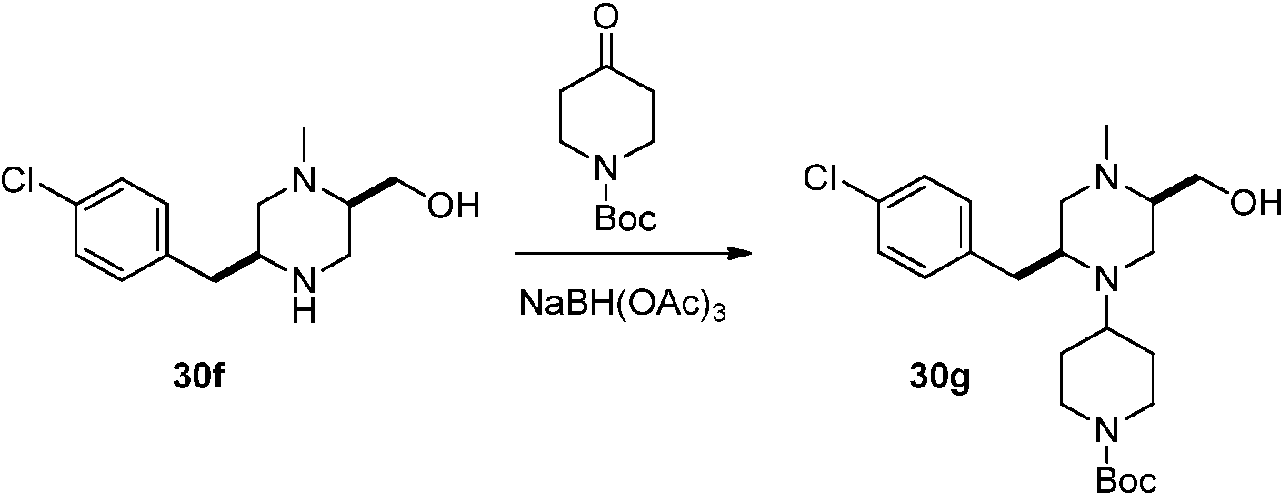
La aminación reductiva del compuesto 30f (0,15 g, 0,59 mmol) y N-Boc-piperid-4-ona se logró de acuerdo con el Procedimiento General VI. Se obtuvieron 71 mg (0,16 mmol; 27 % de rendimiento) del compuesto del título 30g después de la purificación por cromatografía ultrarrápida en columna de gel de sílice usando un sistema de disolvente de CH2Cl2/MeOH 20:1.
ESI-MS m/z para C23H36ON3O3 encontrado 438,0/ 440,0 (M+1)+
Etapa 8
Síntesis de clorhidrato de ((2R,5S)-5-(4-dorobencil)-1-metil-4-(piperidin-4-il)piperazin-2-il)metanol (30h)

De acuerdo con el Procedimiento General VII, se agitaron 70 mg (0,16 mmol) del compuesto 30g durante 1 h en 5 ml de una solución 4 M de HCl en 1,4-dioxano. Los volátiles se retiraron al vacío para dar 71 mg (100 %) del compuesto del título 30h en una forma de sal de clorhidrato.
ESI-MS m/z para C18H28ClN3O encontrado 338,2/ 340,2 (M+1)+
Etapa 9
Síntesis de 5-(4-((2S,5S)-2-(4-clorobencil)-4,5-dimetilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (30)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 30h. Se obtuvieron 30 mg (0,07 mmol) del compuesto final 30 después de la purificación mediante cromatografía de fase inversa (44 % de rendimiento).
ESI-MS m/z para C20H30CW7O encontrado 420,1/ 422,1 (M+1)+
1H RMN (D2O, 700 MHz) S 7,35-7,33 (m, 2H), 7,27-7,25 (m, 2H), 4,03-3,99 (m, 1H), 3,81 (d, J = 13 Hz, 2H), 3,73 (s a, 1H), 3,6-3,56 (m, 1H), 3,45-3,4 (m, 1H), 3,37-3,33 (m, 1H), 3,3 (s a, 3H), 3,28-3,25 (m, 1H), 3,21-3,16 (m, 2H), 2,99 2,94 (m, 1H), 2,92-2,88 (m, 2H), 2,84 (s, 3H), 2,01-1,97 (m, 1H), 1,93-1,89 (m, 1H), 1,62-1,55 (m, 2H).
Ejemplo 31
5-(4-((2S,5S)-2,5-bis(4-clorobencil)-4-metilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (31)

Con la excepción de que el compuesto 30b se aminaba reductivamente con L-p-clorofenilalanina metil éster en vez de L-serina metil éster en la primera etapa sintética, el compuesto del título 31 se preparó de la misma forma que en el Ejemplo 30 y sus intermediarios. Se prepararon 83 mg del compuesto del título 31.
ESI-MS m/z para C26H33G 2N7 encontrado 514,2/ 516,1 (M+1)+
1H RMN (DMSO-d6, 700 MHz) S 7,34-7,26 (m, 4H), 7,22 (d, J = 8,3 Hz, 2H) 7,17 (d, J = 8,3 Hz, 2H),5,44 (s a, 2H),3,69
3,60 (m, 2H), 2,95-2,84 (m, 2H), 2,83-2,75 (m, 1H), 2,71-2,52 (m, 5H), 2,38-2,31 (m, 1H), 2,27-2,21 (m, 2H), 2,19 (s, 3H), 2,01-1,98 (m, 1H), 1,75-1,68 (m, 1H), 1,60-1,52 (m, 1H), 1,35-1,07 (m, 2H).
Ejemplo 32
5-(4-((2S,5S)-2-(4-dorobencil)-5-(cidohexilmetil)-4-metilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (32)

Con la excepción de que el compuesto 30b se aminaba reductivamente con L-cicIohexilalanina metil éster en vez de L-serina metil éster en la primera etapa sintética, el compuesto del título se preparó de la misma forma que en el Ejemplo 30 y sus intermediarios. Se prepararon 59 mg del compuesto del título 32.
ESI-MS m/z para C26H40ON7 encontrado 486,3/ 488,3 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 57,34 (d, J = 7,2 Hz, 2H), 7,28 (d, J = 8,1 Hz, 2H), 5,55 (s a, 1H), 3,92-3,59 (m,2H), 3,17-2,49 (m, 12H), 1,79 (m, 7H), 1,45-1,00 (m, 7H), 0,99-0,71 m, 2H).
Ejemplo 33
5-(4-((2S,5S)-2-(4-clorobencil)-5-isobutil-4-metilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (33)

Con la excepción de que el compuesto 30b se aminaba reductivamente con L-leucina metil éster en vez de L-serina metil éster en la primera etapa sintética, el compuesto del título se preparó de la misma forma que en el Ejemplo 30 y sus intermediarios. Se prepararon 37 mg del compuesto del título 33.
ESI-MS m/z para C2aH36ClN7 encontrado 446,2/ 448,2 (M+1)+
1H RMN (DMSO-d6, 250 MHz) 57,38 (d, J = 8,4 Hz, 2H), 7,25 (d, J = 8,4 Hz, 2H), 5,58 (s, 1H), 3,79 (dd, J = 9,7, 6,6 Hz, 2H), 3,13-2,60 (m, 6H), 2,54-2,30 (m, 2H), 2,18 (s, 3H), 2,17-1,2 (m, 9H), 0,93 (dd, J = 11,6, 6,4 Hz, 6H).
Ejemplo 34
(S)-5-(4-(2-(4-clorobencil)-4-(metilsulfonil)piperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (34)

Etapa 1
Síntesis de (S)-2-((2-((ferc-butoxicarbonil)amino)-3-(4-clorofenil)propil)amino)acetato de metilo (34a)

El compuesto del título se sintetizó a partir de aldehido 30b y clorhidrato de glicina metil éster de acuerdo con el Procedimiento General XIII. Se obtuvieron 2,49 g (7,0 mmol) del compuesto 34a (99 % de rendimiento).
ESI-MS m/z para C17H25ClN2O4 encontrado 357,2/ 359,2 (M+1)+
Etapa 2
Síntesis de (S)-6-(4-clorobencil)piperazin-2-ona (34b)

El compuesto del título se sintetizó a partir del compuesto 34a de acuerdo con el Procedimiento General X. Se obtuvieron 0,97 g (4,3 mmol) del compuesto 34b (64 % de rendimiento).
ESI-MS m/z para CnH 1aClN2O encontrado 225,2/ 227,2 (M+1)+
Etapa 3
Síntesis de (S)-6-(4-clorobencil)-4-(metilsulfonil)piperazin-2-ona (34c)

El compuesto del título se sintetizó a partir del compuesto 34b de acuerdo con el Procedimiento General XV. Se obtuvieron 0,45 g (1,5 mmol) del compuesto 34c (37 % de rendimiento).
ESI-MS m/z para C12H15ClN2O3S encontrado 303,1/ 305,1 (M+1)+
Etapa 4
Síntesis de (S)-3-(4-clorobencil)-1-(metilsulfonil)piperazina (34d)

El compuesto del título se sintetizó a partir del compuesto 34c de acuerdo con el Procedimiento General VI. Se obtuvieron 0,29 g (1,0 mmol) del compuesto 34d (87 % de rendimiento).
ESI-MS m/z para C-i2H-i7ClN2O2S encontrado 289,2/ 291,2 (M+1)+
Etapa 5
Síntesis de (S)-4-(2-(4-clorobencil)-4-(metilsulfonil)piperazin-1-il)piperidina-1-carboxilato de tere-butilo (34e)

La síntesis del compuesto del título se logró por aminación reductiva de N-Bocpiperid-4-ona con el compuesto 34d de acuerdo con el Procedimiento General VII. Se obtuvieron 0,36 g (0,76 mmol) del compuesto 34e (87 % de rendimiento). ESI-MS m/z para C22H34CW3O4S encontrado 472,2/474,2 (M+1)+
Etapa 6
Síntesis de clorhidrato de (S)-2-(4-clorobencil)-4-(metilsulfonil)-1-(piperidin-4-il)piperazina (34f)

El compuesto del título se sintetizó a partir del compuesto 34e de acuerdo con el Procedimiento General VII. Se obtuvieron 0,26 g (0,7 mmol) del compuesto 34f (92 % de rendimiento).
ESI-MS m/z para C17H26ClN3O2S encontrado 372,2/374,2 (M+1)+
Etapa 7
Síntesis de (S)-5-(4-(2-(4-clorobencil)-4-(metilsulfonil)piperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (34)
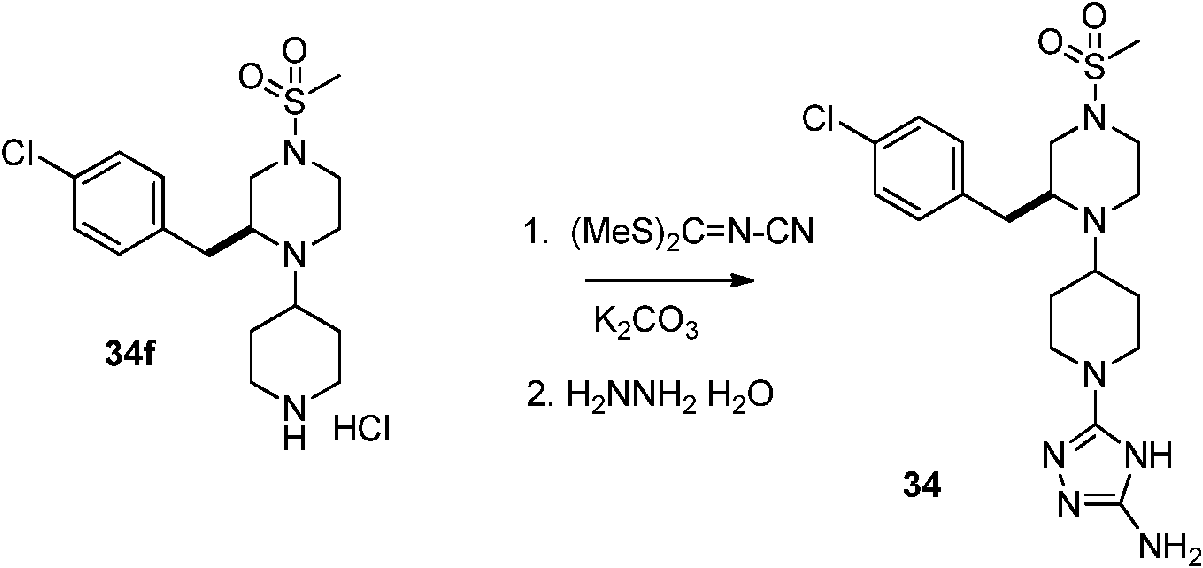
La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 34f. Se sintetizaron 170 mg (0,37 mmol) del compuesto final (54 % de rendimiento).
ESI-MS m/z para C19H2sClN7O2S encontrado 454.2/ 456.2 (M+1)+
1H RMN (DMSO-d6, 250 MHz) S 10,86 (s, 1H), 7,33-7,06 (m, 4H), 5,42 (s, 2H), 3,67 (dd, J= 13,1, 3,7 Hz, 2H), 3,13
2,71 (m,6H), 2,70 (s, 3H), 2,682,48 (m, 6H), 1,79-1,56 (m, 2H), 1,46-1,14 (m, 2H).
Ejemplo 35
(S)-5-(4-(2-(4-dorobencil)-4-tosilpiperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (35)

Con la excepción de que el compuesto 34b se sulfonilaba con cloruro de tosilo en vez de cloruro de mesilo en la tercera etapa sintética; el compuesto del título se preparó de la misma forma que en el Ejemplo 34 y sus intermediarios. Se prepararon 37 mg del compuesto del título 35.
ESI-MS m/z para C25H32CIN7O2S encontrado 530,1/ 532,1 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 510,91 (s, 1H), 7,52-7,47 (m, 2H), 7,41-7,30 (m, 4H), 7,25-7,19 (m, 2H), 5,48 (s a, 1H), 3,68 (d, J= 12,5 Hz, 2H), 3,06-2,93 (m, 2H), 2,83-2,47 (m, 9H), 2,54-2,47 (m, 1H), 2,36 (s, 3H), 1,74-1,57 (m, 2H), 1,33 1,22 (m, 2H).
Ejemplo 36
5-(4-((2S,5S)-2,5-bis(4-clorobencil)-4-(metilsulfonil)piperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (36)

Etapa 1
Síntesis de (3S,6S)-3,6-bis(4-clorobencil)piperazin-2-ona (36a)

El compuesto 31c (intermedio en la síntesis del Ejemplo 31) que se obtuvo por aminación reductiva de L-pclorofenalanina metil éster y aminoaldehído N-protegido 30b de acuerdo con el Procedimiento General XIII, se desprotegió y cicló de acuerdo con el Procedimiento General X proporcionando piperazinona 36a. Se sintetizaron 1,0 g (2,86 mmol) del compuesto 36a (83 % de rendimiento).
ESI-MS m/z para C18H18CI2N2O encontrado 349,3/ 351,3 (M+1)+
Etapa 2
Síntesis de (3S,6S)-3,6-bis(4-dorobencil)-4-(metilsulfonil)piperazin-2-ona (36b)

El compuesto del título se sintetizó a partir del compuesto 36a de acuerdo con el Procedimiento General XV. Se obtuvieron 0,21 g (0,49 mmol) del compuesto 36b (50 % de rendimiento).
ESI-MS m/z para C19H2üCl2N2O3S encontrado 427,5/ 429,5 (M+1)+
Etapa 3
Síntesis de (2S,5S)-2,5-bis(4-clorobencil)-1-(metilsulfonil)piperazina (36c)

El compuesto del título se sintetizó a partir del compuesto 36b de acuerdo con el Procedimiento General VI. Se obtuvieron 0,20 g (0,48 mmol) del compuesto 36c (99 % de rendimiento).
ESI-MS m/z para C ^H 22ChN2O2S encontrado 413,5/ 415,5 (M+1)+
1H RMN (CDCl3, 500 MHz) 57,33-7,22 (m, 4H), 7,23-7,05 (m, 4H), 4,08-4,00 (m, 1H), 3,71-3,53 (m, 1H), 3,15-2,98 (m, 2H), 2,95-2,70 (m, 4H), 2,56-2,44 (m, 1H), 2,47 (s, 3H), 1,58-1,49 (m, 1H), 1,46-1,32 (m, 1H).
Etapa 4
Síntesis de 4-((2S,5S)-2,5-bis(4-clorobencil)-4-(metilsulfonil)piperazin-1-il)piperidina-1-carboxilato de ferc-butilo (36d)

El compuesto del título se sintetizó a partir del compuesto 36c de acuerdo con el Procedimiento General VII. Se obtuvieron 0,30 g (0,50 mmol) del compuesto 36d (83 % de rendimiento).
ESI-MS m/z para C29H39ChN3O4S encontrado 597,1/ 599,1 (M+1)+
Etapa 5
Síntesis de clorhidrato de (2S,5S)-2,5-bis(4-dorobencil)-1-(metilsulfonil)-4-(piperidin-4-il)piperazina (36e)

El compuesto del título se sintetizó a partir del compuesto 36d de acuerdo con el Procedimiento General VII. Se obtuvieron 0,16 g (0,32 mmol) del compuesto 36e (66 % de rendimiento).
ESI-MS m/z para C24H31ChN3O2S encontrado 497,1/ 499,1 (M+1)+
Etapa 6
Síntesis de 5-(4-((2S,5S)-2,5-bis(4-clorobencil)-4-(metilsulfonil)piperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (36)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 36e. Se sintetizaron 25 mg (0,043 mmol) del compuesto final 36 (20 % de rendimiento).
ESI-MS m/z para C26H33Cl2N7O2S encontrado 578,0/ 580,0 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 57,34 (m, 4H), 7,19 (m, 4H), 3,97-3,82 (m, 1H), 3,78 (d, J = 12,4 Hz, 1H), 3,68 (d, J = 12,4 Hz, 1H), 3,24-2,94 (m, 6H), 2,93-2,72 (m, 3H), 2,65 (s, 3H), 2,61-2,51 (m, 2H), 2,36-2,19 (m, 1H), 1,70-1,39 (m, 3H), 1,31-1,15 (m, 1H).
Ejemplo 37
5-(4-((2S,5S)-2-(4-clorobencil)-5-metil-4-(metilsulfonil)piperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (37)

Etapa 1
Síntesis de (S)-2-(((S)-2-((fere-butoxicarbonil)amino)-3-(4-clorofenil)propil)amino)propanoato de metilo (37)

El compuesto del título se sintetizó a partir del intermedio 30b y clorhidrato de L-alanina metil éster de acuerdo con el Procedimiento General XIII. Se obtuvieron 10,2 g (27,5 mmol) del compuesto 37a después de la purificación por cromatografía en columna en un sistema de disolvente de AcOEt/hexanos 1:6 (72 % de rendimiento).
ESI-MS m/z para C18H27ON2O4 encontrado 371,2/ 373,2 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 57,26 (AA'BB', J = 8,3 Hz, 2H), 7,15 (AA'BB', J = 8,5 Hz, 2H), 3,57 (s, 3H), 3,55-3,18 (m, 2H), 2,76 (dd, J = 5,4, 13,3 Hz, 1H), 2,55-2,46 (m, 2H), 2,38-2,30 (m, 1H), 1,26 (s, 9H), 1,13 (d, J= 7Hz, 3H).
Etapa 2
Síntesis de (3S,6S)-6-(4-clorobencil)-3-metilpiperazin-2-ona (37b)

El compuesto del título se sintetizó a partir del compuesto 37a de acuerdo con el Procedimiento General X. Se obtuvieron 5,8 g (24,3 mmol) del producto 37b (89 % de rendimiento).
ESI-MS m/z para Ci2H15ClN2O encontrado 239,0/ 241,0 (M+1)+
Etapa 3
Síntesis de (2S,5S)-5-(4-clorobencil)-2-metil-3-oxopiperazina-1-carboxilato de tere-butilo (37c)

A una solución del compuesto 37b (3,74 g, 15,6 mmol) en DCM (60 ml) se anadió di-rere-butil dicarbonato (Boc2O) (5.1 g, 23.5 mmol) Después de 1 hora, la mezcla de reacción se concentró y se cargó directamente en gel de sílice y se purificó por cromatografía en columna en un sistema de disolvente de AcOEt/hexanos (elución de gradiente de 1:8 a 1:1) proporcionando 3,70 g (10,9 mmol; 70 % de rendimiento) del compuesto del título 37c.
ESI-Ms m/z para C17H23ON2O3 encontrado 339,1/ 341,1 (M+1)+
Etapa 4
Síntesis de (2S,5S)-5-(4-dorobencil)-2-metilpiperazina-1-carboxilato de tere-butilo (37d)

El compuesto del título se sintetizó de acuerdo con el Procedimiento General V, con la excepción de que no se añadió 2 M HCl, sino que la mezcla de reacción se inactivó cuidadosamente con agua (30 ml), después se añadieron 150 ml de 1 M NaOH y la reacción se extrajo varias veces con éter dietílico. Los orgánicos se secaron en MgSO4, se filtraron y concentraron para proporcionar 5,5 g (16,9 mmol; 99 % de rendimiento) del compuesto 37d. Se encontró que tenía una pureza suficiente para usarse en la siguiente etapa.
ESI-MS m/z para Ci7H25ClN2O2 encontrado 269,2/ 271,2 (M+1-tBu)+, 225,1/ 227,1 (M+1- Boc)+
Etapa 5
Síntesis de (2S,5S)-4-(1-((aliloxi)carbonil)piperidin-4-il)-5-(4-clorobencil)-2-metilpiperazina-1-carboxilato de tere-butilo (37e)

La síntesis del compuesto del título se logró mediante la reacción del compuesto 37d con N-Alloc-piperid-4-ona de acuerdo con el Procedimiento General VI. A partir de 5,5 g (16,9 mmol) de material de partida 37d, se obtuvieron 2,64 g (5,36 mmol; 31 % de rendimiento) del compuesto 37e después de la purificación por cromatografía en un sistema de disolvente de AcOEt/hexanos 1:2.
ESI-MS m/z para C26H38ClN3O4 encontrado 492,2/ 494,2 (M+1)+
Etapa 6
Síntesis de clorhidrato de 4-((2S,5S)-2-(4-clorobencil)-5-metilpiperazin-1-il)piperidina-1-carboxilato de alilo (37f)

La retirada del grupo protector de Boc del compuesto 37e se logró de acuerdo con el Procedimiento General VII. Se obtuvieron 2,1 g (4,91 mmol) del compuesto del título 37f (92 % de rendimiento).
ESI-MS m/z para C21H30CIN3O2 encontrado 392,0/ 394,0 (M+1)+
1H RMN (DMSO-da, 500 MHz) 57,32 (AA'BB', J = 8,5 Hz, 2H), 7,28 (AA'BB', J = 8,5Hz, 2H), 5,92-5,83 (m, 1H), 5,25 5,19 (m, 1H), 5,16-5,12 (m, 1H), 4,48-4,44 (m, 2H), 4,05-3,90 (m, 2H), 3,77-3,70 (m, 1H), 3,12-2,98 (m, 4H), 2,80-2,71 (m, 4H), 2,66-2,60 (m, 1H), 2,53-2,47 (m, 1H), 2,43-2,37 (m, 1H), 1,65-1,57 (m, 1H), 1,49-1,42 (m, 2H), 1,14 (d, J = 6,4 Hz, 3H).
Etapa 7
Síntesis de 4-((2S,5S)-2-(4-dorobencil)-5-metil-4-(metilsulfonil)piperazin-1-il)piperidina-1-carboxilato de alilo (37g)

El compuesto del título se sintetizó a partir del intermedio 37f de acuerdo con el Procedimiento General XV. Se obtuvieron 0,34 g (0,72 mmol) del compuesto 37g (85 % de rendimiento).
ESI-MS m/z para C22H32ClN3O4S encontrado 470,1/ 472,1 (M+1)+
Etapa 8
Síntesis de (2S,5S)-2-(4-clorobencil)-5-metil-4-(metilsulfonil)-1-(piperidin-4-il)piperazina (37h)

El compuesto del título se sintetizó a partir del intermedio 37g de acuerdo con el Procedimiento General XVI.
Se obtuvieron 260 mg del compuesto del título 37h (0,67 mmol; 93 % de rendimiento), que eran lo suficientemente puros para usarse en la siguiente reacción.
ESI-MS m/z para C18H28ClN3O2S encontrado 386,2/ 388,2 (M+1)+
1H RMN (DMSO-da, 500 MHz), 57,35-7,22 (m, 4H), 3,81-3,69 (m, 1H), 3,07-2,89 (m, 3H), 2,86-2,72 (m, 5H), 2,78 (s, 3H), 2,68-2,61 (m, 1H), 2,60-2,47 (m, 1H), 1,90-1,81 (m, 1H), 1,72-1,64 (m, 1H), 1,62-1,49 (m, 2H), 1,46-1,37 (m, 1H), 1,30-1,16 (m, 1H), 1,14 (d, J = 6,6Hz, 3H).
Etapa 9
Síntesis de 5-(4-((2S,5S)-2-(4-clorobencil)-5-metil-4-(metilsulfonil)piperazin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (37)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 37h. Se sintetizaron 32 mg (0,07 mmol) del compuesto final 37.
ESI-MS m/z para C2üH3üCIN7O2S encontrado 468,3/ 470,3 (M+1)+
1H RMN (DMSO-da, 500 MHz) 57,38 (AA'BB', J = 8,3 Hz, 2H), 7,33 (AA'BB', J=8,3 Hz, 2H), 3,85-3,72 (m, 6H), 3,37 3,00 (m, 5H), 2,89 (s, 3H), 2,80-2,64 (m, 2H), 2,03-1,65 (m, 3H), 1,60-1,41 (m, 1H), 1,26 (d, J = 4,7 Hz, 3H).
Ejemplo 38
1-((2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-2-metilpiperazin-1-il)etanona (38)

Etapa 1
Síntesis de 4-((2S,5S)-4-acetil-2-(4-clorobencil)-5-metilpiperazin-1-il)piperidina-1-carboxilato de alilo (38a)

A una solución del intermedio 37f (0,35 g, 0,75 mmol) y trietilamina (Et3N) (0,2 ml, 1,5 mmol) en 5 ml de diclorometano, se añadió anhídrido acético (0,11 g, 1,12 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente y después se lavó con 0.1 M HCl, NaHCO3 ac. al 5 %, salmuera, se secó y concentró para proporcionar 0,23 g (0,53 mmol; 71 % de rendimiento) del compuesto del título 38a como un aceite incoloro.
ESI-MS m/z para C23H32ClN3O3 encontrado 434,1/ 436,1 (M+1)+
Etapa 2 y 3
Síntesis de 1-((2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-dorobencil)-2-metilpiperazin-1-il)etanona (38)
La retirada del grupo Alloc y la formación del anillo 1,2,4-triazol se lograron de acuerdo con el Procedimiento General XVI y el Procedimiento General VIII respectivamente comenzando por el compuesto 38a. Se sintetizaron 28 mg (0,06 mmol) del compuesto final 38.
ESI-MS m/z para C21H30ClN7O encontrado 432,1/ 434,1 (M+1)+
1H RMN (DMSO-da, 75 °C, 500 MHz) 57,34 (AA'BB', J = 8,3 Hz, 2H), 7,31 (AA'BB', J = 8,5Hz, 2H), 3,88-3,71 (m, 4H), 3,48-3,28 (m, 2H), 3,19-3,08 (m, 2H), 3,00-2,64 (m, 5H), 1,86 (s, 3H), 1,82-1,62 (m, 3H), 1,55-1,38 (m, 1H), 1,15 (d, J = 5,6Hz, 3H).
Ejemplo 39
(2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-2-metilpiperazina-1-carboxilato de metilo (39)

Etapa 1
Síntesis de (2S,5S)-4-(1-((aliloxi)carbonil)piperidin-4-il)-5-(4-clorobencil)-2-metilpiperazina-1-carboxilato de etilo (39a)

A una solución del intermedio 37f (0.35 g, 0,75 mmol) y trietilamina (Et3N) (0,2 ml, 1,5 mmol) en 5 ml de diclorometano, se añadió cloroformiato de metilo (0,11 g, 1,2 mmol). La reacción se agitó durante 2 horas a temperatura ambiente y después se lavó con 0,1 M HCl, 5 % de NaHCO3, salmuera, se secó y concentró para proporcionar 0,26 g (0,58 mmol, 77 % de rendimiento) del compuesto del título 39a como un aceite incoloro.
ESI-MS m/z para C23H32ClN3O4 encontrado 449,1/ 451,1 (M+1)+
1H RMN (CDCl3, 500 MHz), 57,23 (AA'BB', J = 8,3 Hz, 2H), 7,11 (AA'BB', J = 8,3 Hz, 2H), 5,96-5,84 (m, 1H), 5,30 5,13 (m, 3H), 4,55-4,51 (m, 2H), 4,30-4,04 (m, 4H), 3,80-3,73 (m, 1H), 3,60 (s, 3H), 3,02-2,86 (m, 3H), 2,85-2,65 (m, 3H), 2,62-2,39 (m, 5H), 1,12 (d, J = 6,6 Hz, 3H)
Etapas 2 y 3
Síntesis de (2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-2-metilpiperazina-1-carboxilato de metilo (39)
La retirada del grupo Alloc y la formación del anillo 1,2,4-triazol se lograron de acuerdo con el Procedimiento General XVI y el Procedimiento General VIII respectivamente comenzando por el compuesto 39a. Se sintetizaron 57 mg (0,06 mmol) del compuesto final 39.
ESI-MS m/z para C21H30ClN7O2 encontrado 448,0/ 450,0 (M+1)+
1H RMN (DMSO-da, 500 MHz) 57,44 (AA'BB', J = 7,9 Hz, 2H), 7,33 (AA'BB', J = 8,1 Hz, 2H), 4,30-4,19 (m, 1H), 3,87 3,82 (m, 4H), 3,68-3,54 (m, 3H), 3,50 (s, 3H), 3,40-3,28 (m, 2H), 3,03-2,87 (m, 3H), 2,82-2,70 (m, 1H), 2,02-188 (m,
1H), 1,83-1,72 (m, 1H), 1,67-1,49 (m, 1H), 1,18 (d, J = 6,2 Hz, 3H).
Ejemplo 40
2-((2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-dorobencil)-2-metilpiperazin-1-il)acetato de metilo (40)

Etapa 1
Síntesis de 4-((2S,5S)-2-(4-clorobencil)-4-(2-metoxi-2-oxoetil)-5-metilpiperazin-1-il)piperidina-1-carboxilato de alilo (40a)

La mezcla del compuesto 37f (0,8 g, 1,72 mmol), se calentaron bromoacetato de metilo (0,47 ml, 5,16 mmol) y carbonato de potasio (K2CO3) (0,72 g, 5,16 mmol) en acetonitrilo (10 ml) a 60 °C durante 2 h. El sólido se retiró por filtración y el filtrado se absorbió en gel de sílice y el producto se purificó por cromatografía en columna en AcOEt/hexanos 1:2 para proporcionar 0,79 g (1,70 mmol) del compuesto del título 40a (99 % de rendimiento).
ESI-MS m/z para C24H34ClN3O4 encontrado 464,1/ 466,1 (M+1)+
Etapa 2 y 3.
Síntesis de 2-((2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-2-metilpiperazin-1-il)acetato de metilo (40)
La retirada del grupo Alloc y la formación del anillo 1,2,4-triazol se lograron de acuerdo con el Procedimiento General XVI y el Procedimiento General VIII respectivamente comenzando por el compuesto 40a. Se sintetizaron 125 mg (0,27 mmol) del compuesto final 40.
ESI-MS m/z para C22H32ClN7O2 encontrado 462,2/ 464,2 (M+1)+
1H RMN (DMSO-da, 500 MHz) 59,65 (s a, 1H), 7,40 (AA'BB', J = 8,5 Hz, 2H), 7,33 (AA'BB', J = 8,3 Hz, 2H), 3,92-3,76 (m, 2H), 3,58 (s, 3H), 3,56-3,16 (m, 6H), 3,07-2,96 (m, 2H), 2,95-2,77 (m, 4H), 2,64-2,50 (m, 1H), 2,26-2,10 (m, 2H), 1,69-1,51 (m, 2H), 1,08 (d, J = 5,6 Hz, 3H).
Ejemplo 41
Ácido 2-((2S,5S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-2-metilpiperazin-1-il)acético (41)

El compuesto 40 (110 mg, 0,24 mmol) se sometió a reflujo en 2 ml de 3 N HCl durante 3 horas, tiempo después del cual los volátiles se retiraron al vacío y el residuo se purificó por cromatografía en fase inversa. Se obtuvieron 65 mg (0,14 mmol; 60 % de rendimiento) del compuesto del título 41.
ESI-MS m/z para C21H30ClNyO2 encontrado 448,1/ 450,1 (M+1)+
1H RMN (DMSO-da, 500 MHz) 59,60 (s a, 1H), 7,39 (AA'BB', J = 8,3 Hz, 2H), 7,34 (AA'BB', J = 8,5 Hz, 2H), 3,94-3,34 (m, 6H), 3,31-3,18 (m, 2H), 3,12-2,96 (m, 2H), 2,96-2,76 (m, 4H), 2,65-2,54 (m, 1H), 2,27-2,08 (m, 2H), 1,70-1,51 (m, 2H), 1,09 (d, J = 5 Hz, 3H).
Ejemplo 42
(S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-3-(4-clorobencil)piperazin-2-ona (42)

Etapa 1
Síntesis de (S)-(3-(4-clorofenil)-1-((2,2-dietoxietil)amino)-1-oxopropan-2-il)carbamato de tere-butilo (42a)

El compuesto 19a (1,00 g, 3,34 mmol) se disolvió en DCM (13,4 ml) y se añadió diisopropiletilamina (0,87 ml, 5,0 mmol) a temperatura ambiente seguido de la adición de aminoacetaldehído dietilacetal (0,54 ml, 3,67 mmol) y O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio tetrafluoroborato (TBTU) (1,13 g, 3,50 mmol). La mezcla de reacción se agitó durante 3 horas a TA, se diluyó con cloruro de metileno y se lavó con 1 M K2CO3ac y 1 M HClac, salmuera, se secó en MgSO4 anhidro, se filtró y concentró al vacío. El residuo se purificó por cromatografía ultrarrápida usando diclorometano (DCM), después un sistema de disolvente de DCM/MeOH 100:1. Se obtuvieron 1,27 g (3,07 mmol; 92 % de rendimiento) del compuesto del título 42a.
ESI-MS m/z para C2üH31ClN2O5 encontrado 415,4/ 417,4(M+1)+
1H RMN (CDCl3, 500 MHz) 57,23 (m, 2H), 7,11 (m, 2H), 5,92 (s a, 1H), 5,02 (s a, 1H), 4,28 (s a, 2H), 3,64-3,56 (m, 2H), 3,47-3,43 (m, 1H), 3,40-3,30 (m, 2H), 3,25 (s a, 1H), 2,99 (s a, 2H), 1,38 (s, 9H), 1,14 (q, J = 6,9Hz, 6H)
Etapa 2
Síntesis de (S)-2-(4-clorobencil)-3-oxo-3,4-dihidropirazina-1(2H)-carboxilato de ferc-butilo (42b)

El compuesto 42a (1,38 g, 3,33 mmol) se disolvió en acetona (33 ml, 10 ml/mmol) después se añadió I2 (85 mg, 0,33 mmol) y la mezcla se agitó durante la noche a TA. El disolvente se retiró al vacío y un residuo oleaginoso se disolvió en Et2O y después se lavó dos veces con 10 % de Na2S2Ü4. La capa orgánica se secó en MgSCM, se filtró y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida usando DCM puro, después un sistema de disolvente de DCM/MeOH 100:1. Se obtuvieron 0,88 g (2,72 mmol; 82 % de rendimiento) del compuesto del título. ESI-MS m/z para Ci6H19ClN2O3 encontrado 322,7/324,7 (M+1)+
1H RMN (CDCl3, 500 MHz) - dos confórmeros presentes debido a la rotación perjudicada 8
{[7,62 (1er isómero, s a), 7,58 (2° isómero, s a)], 1H}, {[7,25 (2° isómero, d, J = 8,0Hz), 7,20 (1er isómero, d, J = 8,0Hz)], 2H}, {[7,08 (1er isómero, d, J = 8,0Hz), 7,06 (2° isómero, d, J = 8Hz)], 2H}, {[6,36 (2° isómero, d, J = 5,8 Hz), 6,10 (1er isómero, d, J = 5,8 Hz)], 1H}, {[5,67 (2° isómero, t, J = 5,1 Hz), 5,42 (1er isómero, t, J = 5,1 Hz)], 1H}, {[4,99-4,95 (1er isómero, m), 4,80-4,76 (2° isómero, m)], 1H}, {[3,02-2,96 (1er isómero, m), 2,90-2,86 (2° isómero, m)], 2H}, {[1,35(1er isómero, s), 1,17 (2° isómero, s)], 9H}
Etapa 3
Síntesis de (S)-3-(4-clorobencil)piperazin-2-ona (42c)
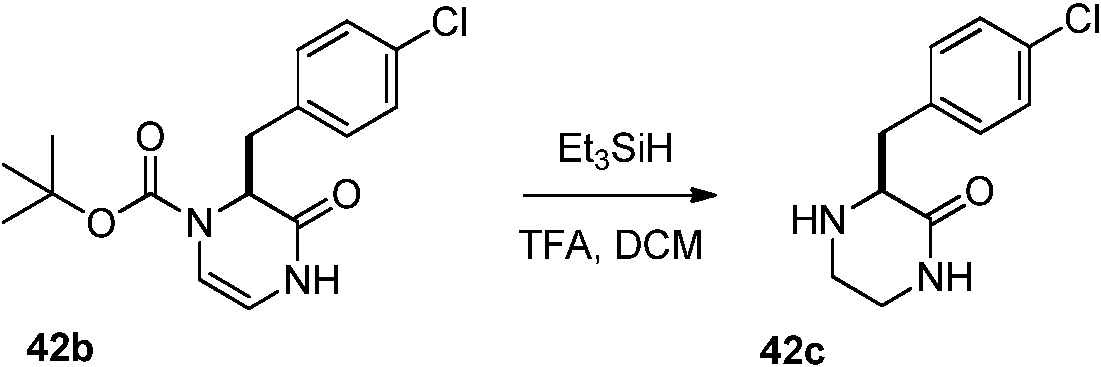
A una solución del compuesto 42b (0,58 g, 1,80 mmol) en diclorometano (DCM) (5 ml), se añadió trietilsilana Et3SiH (1,4 ml, 8,9 mmol) seguido de la adición lenta de ácido trifluoroacético (TFA) (1,3 ml, 17,8 mmol) y la mezcla de reacción se agitó durante la noche a TA. Después los volátiles se retiraron al vacío y el residuo se absorbió entre 1 M NaOH/ y DCM. La fase orgánica se lavó con salmuera, se secó y concentró para proporcionar 0,32 g (1,42 mmol; 79 % de rendimiento) del compuesto del título 42c.
ESI-MS m/z para CnH13ClN2O encontrado 225,2/ 227,2 (M+1)+
1H RMN (CDCl3, 500 MHz) 87,29-7,23 (m, 2H), 7,18 (d, J = 8,3 Hz, 2H), 6,11 (s, 1H), 3,59 (dd, J = 9,6, 3,6 Hz, 1H), 3,41-3,30 (m, 2H), 3,24 (dq, J = 11,2, 3,6 Hz, 1H), 3,06 (dt, J = 12,6, 3,9 Hz, 1H), 2,93-2,82 (m, 2H).
Etapas 4-6
Síntesis de (S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-3-(4-clorobencil)piperazin-2-ona (42)
La piperazinona 42c se llevó a los etapas sintéticos restantes como se describe en el Procedimiento General VI (aminación reductora con N-Boc-piperid-4-ona), el Procedimiento General VII (desprotección de Boc) y el Procedimiento General VIII (formación de anillo de triazol). Se obtuvieron 150 mg (0,38 mmol) del compuesto del título 42.
ESI-MS m/z para C1sH24ClN7O encontrado 390,1/ 392,1 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 87,88 (s, 1H), 7,32-7,22 (m, 4H), 3,85-3,59 (m, 4H), 3,15-2,75 (m, 8H), 1,73 (d, J= 12,2 Hz, 1H), 1,66-1,53 (m, 1H), 1,51-1,28(m, 2H).
Ejemplo 43
1,1-dióxido de (S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-3-(4-dorobencil)tiomorfolina (43)

Etapa 1
Síntesis de metansulfonato de (S)-2-((ferc-butox¡carbon¡l)am¡no)-3-(4-clorofen¡l)prop¡l (45a)

A una soluc¡ón del sustrato 30a (1,8 g, 6,29 mmol) y tr¡et¡lam¡na (1,4 ml, 9,44 mmol), en d¡clorometano, se añad¡ó cloruro de mes¡lo (0,73 ml, 9,44 mmol) gota a gota. Después de 1 hora de ag¡tac¡ón, la mezcla de reacc¡ón se d¡luyó con d¡clorometano, se lavó con 2 M HCl, 5 % de NaHCÜ3 ac., salmuera, se secó y concentró. El res¡duo se lavó con éter para proporc¡onar 2,09 g del producto 43a como un sól¡do blanco (5,76 mmol; 92 % de rend¡m¡ento).
ESI-MS m/z para C19H28ClN3Ü encontrado 386,1/ 388,1 (M+Na)+
Etapa 2
Síntes¡s de (S)-2-((2-((ferc-butox¡carbon¡l)am¡no)-3-(4-clorofen¡l)prop¡l)t¡o)acetato de met¡lo (43b)

Mes¡lato 43a (2,09 g, 5,74 mmol), K2CO3 (1,58 g, 11,48 mmol) y t¡ogl¡colato de met¡lo (0,65 ml, 11,48 mmol) en aceton¡tr¡lo se calentaron a reflujo durante 30 m¡nutos, después la mezcla de reacc¡ón se d¡luyó con agua y el producto se extrajo con éter d¡etíl¡co. Los orgán¡cos se lavaron con 2 M HCl, 5 % de NaHCÜ3 ac., salmuera, se secaron y concentraron para proporc¡onar 2,1 g de producto 43b como un ace¡te amar¡llo (5.61 mmol; 98 % de rend¡m¡ento). ESI-MS m/z para C17H24ClNÜ4S encontrado 374,1/ 376,1 (M+1)+
Etapa 3
Síntes¡s de (S)-2-((2-((ferc-butox¡carbon¡l)am¡no)-3-(4-clorofen¡l)prop¡l)sulfon¡l)acetato de met¡lo (43c)

A una soluc¡ón de 43b (2,1 g, 5,74 mmol) en acetato de et¡lo, se añad¡ó ác¡do peracét¡co (2,1 ml, 12,83 mmol, 39 % en AcÜH) gota a gota, después la reacc¡ón se ag¡tó durante la noche a TA y se concentró hasta sequedad. El res¡duo
se trituró con éter dietílico y se usó 43c bruto en la siguiente etapa.
ESI-MS m/z para C17H24 CINO6 S encontrado 406,1/ 408,1 (M+1)+
Etapa 4
Síntesis de 1,1 -dióxido de (S)-5-(4-clorobencil)tiomorfolin-3-ona (43d)
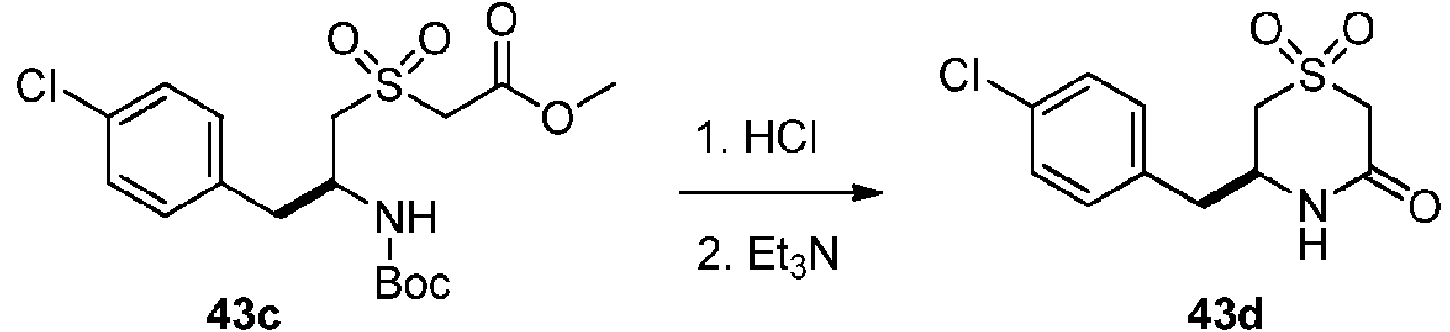
El compuesto 43c (5,74 mmol) se trató con HCl/ dioxano y se agitó durante 1 hora, después se concentró hasta sequedad. El residuo se disolvió en MeOH y se trató con Et3N (1,8 ml, 11,48 mmol). Después de 30 minutos, la reacción se concentró, el residuo se absorbió en diclorometano, se lavó con 2 M HCl, 5 % de NaHCO3 ac., salmuera, se secó con MgSO4 anhidro y se concentró para proporcionar un sólido naranja. Las impurezas coloreadas se retiraron por trituración con éter dietílico proporcionando 0,52 g (1,90 mmol; 33 % de rendimiento en dos etapas) del compuesto 43d como un sólido blanco.
ESI-MS m/z para CnH^ClNOaS encontrado 274,1/ 276,1 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 58,43 (s a, 1H), 7,35 (AA'BB', J = 8,3 Hz, 2H), 7,26 (AA'BB', J = 8,3 Hz, 2H), 4,17-4,11 (m, 1H), 4,00 (dd, J = 2,6, 16,1 Hz, 1H), 3,97-3,90 (m, 1H), 3,23-3,17 (m, 1H), 3,09 (dd, J = 11,1, 13,7 Hz, 1H), 2,97 (dd, J = 4,7, 13,5 Hz, 1H), 2,82 (dd, J = 7,5, 13,5 Hz, 1H).
Etapa 5
Síntesis de 1,1 -dióxido de (S)-3-(4-clorobencil)tiomorfolin (43e)

El compuesto 43d (0,42 g, 1,53 mmol) se disolvió en 15 ml de THF seco y se añadió cuidadosamente un complejo de borano-tetrahidrofurano (4,6 ml, 4,6 mmol) y la reacción se calentó con agitación por 1 hora. Después de este tiempo, la TLC reveló el consumo completo del material de partida. La mezcla de reacción se inactivó cuidadosamente con agua. Se añadió 1 N NaOH y la mezcla de reacción se extrajo con éter dietílico. Los orgánicos se secaron en MgSO4 y se concentraron para proporcionar 0,4 g de producto 43e (1,53 mmol; 99 % de rendimiento).
ESI-MS m/z para Ch HmCIN02S encontrado 260,1/ 262,1 (M+1)+
1H RMN (DMSO-d6, 500 MHz) 57,31 (AA'BB', J = 8,3 Hz, 2H), 7,21 (AA'BB', J = 8,3 Hz, 2H), 3,37-3,17 (m, 2H), 3,09 3,00 (m, 1H), 2,97-2,74 (m, 4H), 2,72-2,63 (m, 2H).
Etapa 6
Síntesis de (S)-4-(3-(4-clorobencil)-1,1-dioxidotiomorfolino)piperidina-1-carboxilato de tere-butilo (43f)

La aminación reductiva se logró de acuerdo con el Procedimiento General VI, comenzando por la amina 43e (0,4 g, 1,53 mmol) y N-Boc-piperid-4-ona para dar 0,61 g del producto 43f (1,38 mmol; 90 % de rendimiento).
ESI-MS m/z para C21H31CIN2O4S encontrado 443,1/ 445,1 (M+1)+
1H RMN (CDCI3, 500 MHz) 57,24 (AA'BB', J = 8,3 Hz, 2H), 7,10 (AA'BB', J = 8,3 Hz, 2H), 3,85-3,72 (m, 2H), 3,52-3,42 (m, 1H), 3,41-3,32 (m, 1H), 3,14-3,01 (m, 2H), 3,02-2,57 (m, 6H), 2,26-1,98 (m, 2H), 1,84-1,73 (m, 2H), 1,73-1,63 (m, 2H), 1,42 (s, 9H).
Etapa 7
Síntesis de 1,1 -dióxido de (S)-3-(4-clorobencil)-4-(piperidin-4-il)tiomorfolina (43g)
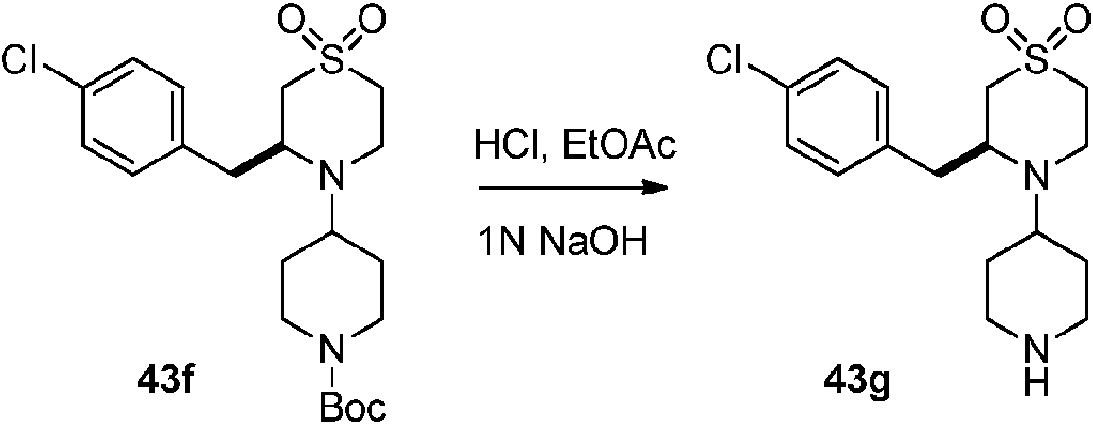
La desprotección de Boc se logró como se describe en el Procedimiento General VII. La sal de clorhidrato bruta del compuesto 43g se transfirió a una base libre absorbiéndolo entre 2 M NaOH y acetato de etilo. Las fases se separaron y la fase orgánica se secó de la manera habitual. La evaporación del disolvente proporcionó 0,42 g (1,22 mmol; 89 % de rendimiento) del compuesto del título 43g.
ESI-MS m/z para C16H23ClN2O2S encontrado 343,0/ 345,0 (M+1)+
Etapa 8
Síntesis de 1,1 -dióxido de (S)-4-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-3-(4-clorobencil)tiomorfolina (43)

La formación del anillo de 1,2,4-triazol se logró de acuerdo con el Procedimiento General VIII comenzando por el compuesto 43g. Se obtuvieron 75 mg (0,18 mmol; 15 % de rendimiento) del compuesto del título.
ESI-MS m/z para C18H25ClN6O2S encontrado 425,0/ 427,0 (M+1)+
1H RMN (DMSO-d6, 500 MHz,) 57,31 (AA'BB', J = 8,5 Hz, 2H), 7,24 (AA'BB', J = 8,5 Hz, 2H), 5,48 (s a, 2H), 3,71-3,64 (m, 2H), 3,52-3,43 (m, 1H), 3,11-2,96 (m, 3H), 2,94-2,79 (m, 4H), 2,78-2,70 (m, 1H), 2,64-2,52 (m, 2H), 1,65-1,55 (m, 2H), 1,37-1,18 (m, 3H).
Ejemplo 44
5-(4-((2R,5S)-5-(4-clorobencil)-2-metilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (44)

El compuesto del título se preparó de la misma forma que el Ejemplo 4 con la excepción de que se usó ácido (2S)-2-bromopropiónico en vez de ácido (2R)-2-bromopropiónico en la primera etapa sintética.
ESI-lCm S m/z para C19H27ClN6Oencontrado 391,1/393,1 (M 1 )+
1H RMN (700 MHz, DMSO-d6): 57,41 (d, J = 8,4 Hz, 2H), 7,35 (d, J = 8,4 Hz, 2H), 4,08 (s a, 1H), 3,91 - 3,84 (m, 2H), 3,83 - 3,80 (m, 2H), 3,70 - 3,67 (m, 2H), 3,48 (d, J = 12,7 Hz, 2H), 2,87 (t, J = 11,6 Hz, 1H), 2,75 (t, J = 11,5 Hz, 1H),
2,70 (dd, J = 14,5, 7,7 Hz, 1H), 2,65 (t, J = 12,0 Hz, 1H), 1,80-1,90 (m, 2H), 1,63 (qd, J = 12,3, 4,2 Hz, 1H), 1,11 (d, J = 6,2 Hz, 3H).
Ejemplo 45
5-(4-((2R,5R)-5-(4-dorobencil)-2-metilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (45)

El compuesto del título se preparó de la misma forma que el Ejemplo 4 con la excepción de que se usó (2R)-2-amino-3-(4-clorofenil)propan-1-ol y ácido (2S)-2-bromopropiónico en vez de (2S)-2-amino-3-(4-clorofenil)propan-1-ol y ácido (2R)-2-bromopropiónico en la primera etapa sintética.
ESI-LCMS: m/z para C19H27ClN6O encontrado: 391,2/ 392,9 (M+1)+.
1H RMN (DMSO-d6, 700 MHz): 87,34 (d, J = 8,4 Hz, 2H,), 7,21 (d, J = 8,4 Hz, 2H), 5,46 (s a, 2H), 4,12 (s a, 1H), 3,74 (br d, J = 12,6 Hz, 2H), 3,51-3,47 (m, 1H), 3,43-3,32 (m, 2H), 2,91-2,87 (m, 2H), 2,75-2,68 (m, 4H), 2,60-2,56 (m, 1H), 2,29-2,26 (m, 1H), 1,92-1,88 (m, 2H), 1,38-1,27 (m, 2H), 1,11 (d, J = 6,3 Hz, 3H).
Ejemplo 46
5-(4-((2S,4S)-2-(4-clorobencil)-4-metoxipirrolidin-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (46)

Etapa 1
Síntesis de (2R,4S)-1-terc-butil 2-metil 4-hidroxipirrolidina-1,2-dicarboxilato (46a)

A una solución de W-Boc-frans-4-hidroxi-D-prolina (10,00 g; 43,25 mmol) en MeCN (100 ml), se añadió carbonato de potasio (11,95 g; 86,50 mmol) seguido de yoduro de metilo (5,40 ml, 86,50 mmol) y la mezcla resultante se agitó durante la noche. La LCMS indicó la presencia de sustrato. Se añadió otra porción de carbonato de potasio (5,98 g; 43,25 mmol) y yoduro de metilo (2,7 ml; 43,25 mmol) y la reacción se agitó durante 2 días, tiempo después del cual la LCMS indicó que la reacción se había completado. La mezcla de reacción se filtró y el residuo sólido se lavó con EtOAc. Después de la evaporación de la fase orgánica, se obtuvieron 9,37 g (38,22 mmol; 88 % de rendimiento) del producto como un aceite amarillento.
ESI-MS m/z para C11H19NO5 encontrado 145,9 (M-Boc+1)+, 268,0 (M+Na)+.
Etapa 2
Síntesis de (2R,4S)-1-terc-butil 2-metil 4-((terc-butildimetilsilil)oxi)pirrolidina-1,2-dicarboxilato (46b)

A una solución de 46b (5 g; 20,38 mmol) en DMF (60 ml), se añadió imidazol (6,94 g; 101,90 mmol) seguido de TBDMSCI (4,61 g; 30,57 mmol) y la reacción se agitó durante la noche. Después de que la LCMS indicó que la reacción se había completado, la mezcla de reacción se extrajo entre agua y EtOAc. La capa orgánica se lavó con agua y salmuera, se secó en MgSO4 anhidro y se concentró. El producto bruto se purificó por cromatografía en columna (hexano/EtOAc 8:1) para dar 6,70 g de 46b como un aceite incoloro (18,63 mmol; 92 % de rendimiento).
ESI-MS m/z para C17H33NO5Si encontrado 260,2 (M-Boc+1)+, 382,1 (M+Na)+.
1H RMN (CDCl3, 700MHz) 5 [4,43-4,40 (m); 4,33 (d, J=7,7Hz); 2H], [3,74 (s); 3,72 (s); 3H], [3,61 (dd, J=11,2, 4,6Hz); 3,57 (dd, J=11,0, 4,8Hz); 1H], [3,40 (dd, J=11,4, 1,3Hz); 3,37 (dd, J=11,2, 2,4Hz); 1H], 2,20-2,14 (m, 1H), 2,04-1,98 (m, 1H), [1,46(s); 1,41(s); 9H], 087 (s, 9H), 0,06 (s, 6H).
Etapa 3
Síntesis de ácido (2R,4S)-1-(terc-butoxicarbonil)-4-((terc-butildimetilsilil)oxi)pirrolidina-2-carboxílico (46c)

El compuesto 46b (6,70 g; 18,63 mmol) se disolvió en una mezcla de 200 ml de THF y 100 ml de MeOH. Una solución de hidrato de hidróxido de litio en 100 ml de agua se añadió a la mezcla de reacción y la mezcla resultante se agitó durante la noche. Después de que el control por LCMS indicara que la reacción se había completado, la mezcla de reacción se concentró. El residuo acuoso se acidificó hasta pH 4 con 2 N HCl a 0 °C y el producto se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó en MgSO4 y se evaporó para dar 5,13 g del producto 46c como un aceite amarillento (14,85 mmol; 80 % de rendimiento).
ESI-MS m/z para C16H31NO5Si encontrado 246,1 (M-Boc+1)+, 368,1 (M+Na)+, 344,1 (M-1)-.
Etapa 4
Síntesis de 4-((terc-butildimetilsilil)oxi)-2-(metoxi(metil)carbamoil)pirrolidina-1-carboxilato de (2R,4S)-terc-butilo (46d)

A una solución del compuesto 46c (10 g; 14.85 mmol) en DCM (40 ml), se añadió trietilamina (5,2 ml; 37,12 mmol), seguido de carbonildiimidazol (CDI; 3,61 g; 22,28 mmol) y la reacción se agitó durante 1 hora. Se añadió clorhidrato de W,0-dimetilhidroxiloamina (2,17 g; 22,28 mmol) y la reacción se agitó durante la noche, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La mezcla de reacción se lavó con agua y salmuera. La capa orgánica se secó en MgSO4 anhidro y se concentró. El producto bruto se purificó por cromatografía en columna (hexano/EtOAc 5:1) para dar 4,15 g de 46d como un aceite incoloro (10,68 mmol; 72 % de rendimiento).
ESI-MS m/z para C18H36N2O5Si encontrado 289.7/290.3 (M-Boc+1)+, 411,1/412,3 (M+Na)+.
1H RMN (CDCl3, 700MHz) 5 [4,82 (s a); 4,73 (t, J = 7,0 Hz); 1H], 4,45 (dq, 1H, J = 18,0, 4,8 Hz), [3,79 (s); 3,72 (s); 3H], [3,68 (dd, J = 11,0, 5,3 Hz); 3,64 (dd, J = 11,0, 5,3 Hz); 1H], [3,40 (dd, J=11,0, 3,1 Hz); 3,32 (dd, J = 10,8, 3,7 Hz); 1H], 3,20 (s, 3H), 2,18-2,12 (m, 1H), 2,00-1,95 (m, 1H), [1,45 (s); 1,41 (s); 9H], 0,88 (s, 9H), 0,06 (s, 6H).
Etapa 5
Síntesis de 4-((terc-butildimetilsilil)oxi)-2-(4-clorobenzoil)pirrolidina-1-carboxilato de (2R,4S)-terc-butilo (46e)

A una solución del compuesto 46d (1.00 g; 2.57 mmol) en Et2O seco (8 ml), se añadió bromuro de pclorofenilmagnesio, previamente generado a partir de p-bromoclorobenceno (6,67 g; 34,84 mmol) y magnesio (875 mg; 36,00 mmol) en Et2O (4 ml), a -70 °C bajo argón. La reacción se agitó a -70 °C durante 15 minutos y 2 horas a temperatura ambiente, tiempo después del cual la LCMS mostró que la reacción se había completado. La reacción se inactivó con NH4Cl saturada y el producto se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua, salmuera y se secaron en MgSO4 anhidro y se concentraron. El producto bruto se purificó por cromatografía en columna (hexano/EtOAc 10:1) para obtener 970 mg de 46e como un aceite incoloro (2,20 mmol; 86 % de rendimiento). ESI-MS m/z para C22H34ClNO4Si encontrado 340,0/341,9 (M-Boc+1)+, 462,1 (M+Na)+.
1H RMN (DMSO-da D2O, 700MHz) 57,97-7,95 (m, 2H), 7,61-7,59 (m, 2H), 5,27-5,22 (m, 1H), 4,44-4,39 (m, 1H), [3,52 (dd, J = 11,4, 4,4 Hz); 3,48 (dd, J = 11,4, 4,4 Hz); 1H], [3,34 (m); 3,30 (m); 1H], 2,24-2,19 (m, 1H), [1,92 (ddd, J = 12,9, 7,8, 4,8 Hz); 1,85 (ddd, J = 13,0, 7,7, 4,8 Hz); 1H], [1,34 (s); 1,11 (s); 9H], 0,83 (s, 9H), [0,04 (s); 0,03 (s); 6H]. Etapa 6
Síntesis de (3S,5S)-5-(4-clorobencil)pirrolidin-3-ol (46f)

A una solución del compuesto 46e (970 mg; 2,20 mmol) en DCM (11 ml), se añadió AlCh (880 mg; 6,60 mmol) bajo argón, seguido de trietilsilano (1,05 ml; 6,60 mmol). La reacción se agitó durante 45 minutos, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La reacción se inactivó con 4 M NaOH, se saturó con cloruro de sodio y se filtró a través de Celite. El producto se extrajo de la fase acuosa con DCM 3 veces 20 ml. Las capas orgánicas combinadas se secaron sobre MgSO4 anhidro y se evaporaron para dar 1,04 g de producto 46f (>99 % de rendimiento) como un aceite amarillento.
ESI-MS m/z para CnHuClNO encontrado 211,9 (M+1)+.
Etapa 7
Síntesis de 2-(4-clorobencil)-4-hidroxipirrolidina-1-carboxilato de (2S,4S)-terc-butilo (46g)

A una solución del compuesto 46f (840 mg; 3,97 mmol) en acetona (8 ml), se añadió agua (8 ml) y el pH se ajustó hasta 12 con K2CO3 (1,1 g; 7,94 mmol). Se añadió Boc2O (954 mg; 4,37 mmol) en una porción y la reacción se agitó durante la noche, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La mezcla de reacción se concentró para retirar la acetona. El residuo acuoso se saturó con NaCl y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó en MgSO4 anhidro y se concentró. El producto bruto se purificó por cromatografía en columna (hexano/EtOAc 1:1) para dar 71 mg de 46g como un aceite incoloro (0,23 mmol; 6 % de rendimiento).
ESI-MS m/z para C16H22ClNO3 encontrado 211,0/213,9 (M-Boc+1)+, 256,0/257,8 (M-fBu+1)+, 333,9 (M+Na)+.
Etapa 8
Síntesis de 2-(4-dorobencN)-4-metoxipirroNdina-1-carboxNato de (2S,4S)-terc-butilo (46h)

El compuesto del título (46h) se obtuvo a partir del compuesto 46g (71 mg, 0,23 mmol) de acuerdo con el Procedimiento General XXI en 99 % de rendimiento (87mg, 0,27 mmol, aceite transparente).
ESI-MS m/z para C17H24ClNOs encontrado 226,0 (M-Boc+1)+, 270,0 (M-fBu+1)+, 349,9 (M+Na)+.
Etapa 9
Síntesis de clorhidrato de (2S,4S)-2-(4-clorobencil)-4-metoxipirrolidina (46i)

El compuesto del título se preparó a partir del compuesto 46h (87 mg; 0,27 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un sólido blanco (70 mg; 99 % de rendimiento).
ESI-MS m/z para C12H16ClNO encontrado 226,0/227,9 (M+1)+.
Etapa 10
Síntesis de 4-((2S,4S)-2-(4-clorobencil)-4-metoxipirrolidin-1-il)piperidina-1-carboxilato de terc-butilo (46j)

El compuesto del título se preparó a partir del compuesto 46i (70 mg; 0,27 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un sólido amarillento (129 mg; 99 % de rendimiento).
ESI-MS m/z para C22H33GN2O3 encontrado 409,1 (M+1)+.
Etapa 11
Síntesis de diclorhidrato de 4-((2S,4S)-2-(4-clorobencil)-4-metoxipirrolidin-1-il)piperidina (46k)
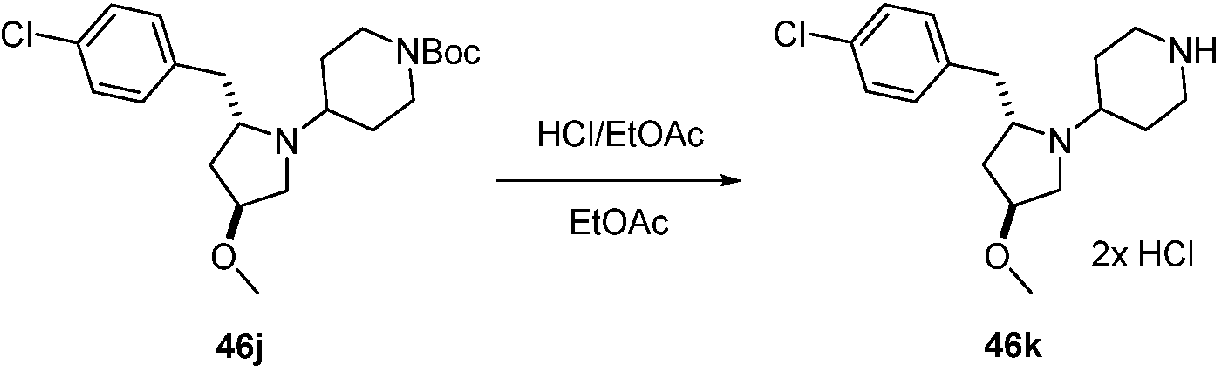
El compuesto del título se preparó a partir del compuesto 46j (110 mg; 0,27 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un sólido blanco (103 mg; 99 % de rendimiento).
ESI-MS m/z para C17H25ClN2O encontrado 309,2 (M+1)+.
Etapa 12
Síntesis de 3-(4-((2S,4S)-2-(4-clorobencil)-4-metoxipirrolidin-1-il)piperidin-1 -il)-1H-1,2,4-triazol-5-amina (46)

El compuesto del título 46 se preparó a partir del compuesto 46k (103 mg; 0,27 mmol) de acuerdo con el Procedimiento General VIII. El producto bruto se purificó por cromatografía de fase inversa para dar 46 como una sal de TFA (11 mg; 8 % de rendimiento).
ESI-MS m/z para C1gH27ClNaO encontrado 391,2 (M+1)+.
1H RMN (DMSO-da+D2O, 700MHz) 57,40 (d, J = 8,5 Hz, 2H), 7,35 (d, J = 8,5 Hz, 2H), 4,01 (s, 1H), 3,94 - 3,80 (m, 3H), 3,56 - 3,41 (m, 3H), 3,31 (d, J = 10,9 Hz, 1H), 3,17 (s, 3H), 2,89 - 2,77 (m, 3H), 2,02 (s a, 2H), 1,92 (s a, 1H), 1,83 - 1,76 (m, 1H), 1,65 (dd, J = 5,1,9,7 Hz, 2H).
Ejemplo 47
(3S,5S)-1-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)pirrolidin-3-ol (47)

Etapa 1
Síntesis de clorhidrato de (3S,5S)-5-(4-clorobencil)pirrolidin-3-ol (47a)
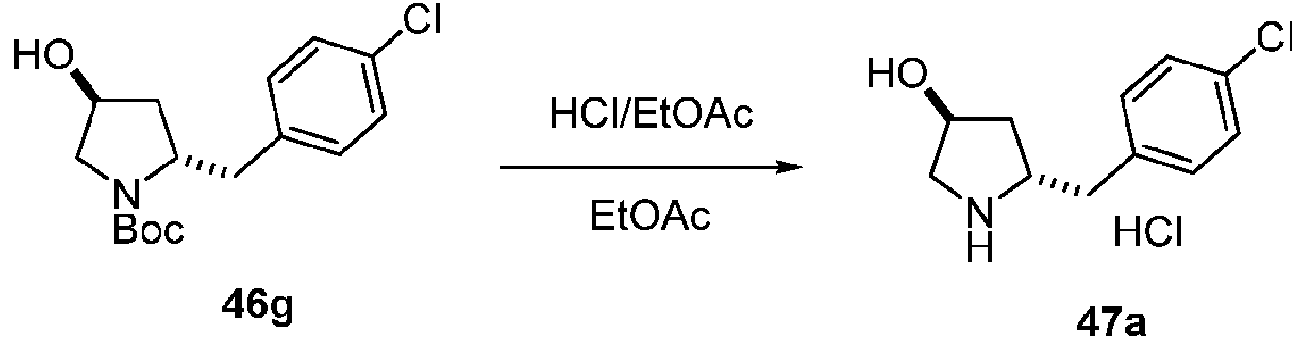
El compuesto del título se sintetizó a partir del compuesto 46g (276 mg; 0,88 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un polvo blanco (204 mg; 0,82 mmol, 93 % de rendimiento).
ESI-MS m/z para C16H23ClN encontrado 212,0/213,9 (M+1)+.
Etapa 2
Síntesis de 4-((2S,4S)-2-(4-clorobencil)-4-hidroxipirrolidin-1-il)piperidina-1-carboxilato de terc-butilo (47b)

El compuesto del título se preparó a partir del compuesto 47a (204 mg; 0,82 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un aceite transparente (147 mg; 0,37 mmol; 45 % de rendimiento).
Etapa 3
Síntesis de diclorhidrato de (3S,5S)-5-(4-clorobencil)-1-(piperidin-4-il)pirrolidin-3-ol (47c)

El compuesto del título se preparó a partir del compuesto 47b (147 mg; 0,37 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un polvo blanco (135 mg; 99 % de rendimiento).
ESI-MS m/z para C16H23ClN2O encontrado 295,1 (M+1)+.
Etapa 4
Síntesis de (3S,5S)-1-(1-(5-amino-1H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)pirrolidin-3-ol (47)

El compuesto del título 47 se sintetizó a partir del compuesto 47c (135 mg; 0,37 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de fase inversa y se obtuvo como una sal de TFA (80 mg, 44 % de rendimiento).
ESI-MS m/z para C18H25GN6O encontrado 377,2 (M+1)+.
Ejemplo 48
(5S)-1-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-3-metilpirrolidin-3-ol (48)

Etapa 1
Síntesis de 2-(4-dorobendl)-4-oxopirrolidina-1-carboxNato de (S)-terc-butilo (48a)

El compuesto del título se preparó a partir del compuesto 46g (1,00 g; 3,21 mmol) de acuerdo con el Procedimiento General XII y se obtuvo como un sólido blanco (839 mg; 2,71 mmol, 84 % de rendimiento).
ESI-MS m/z para C16H2üCINO3 encontrado 210,1 (M-Boc+1)+, 254,2 (MJBu+1)+.
Etapa 2
Síntesis de 2-(4-clorobencil)-4-hidroxi-4-metilpirrolidina-1-carboxilato de (2S)-terc-butilo (48b)

A una solución del compuesto 48a (400 mg; 1,29 mmol) en Et2O seco (20 ml), se añadió bromuro de metilmagnesio (solución 3,0 M en Et2O; 860 pl; 2,58 mmol) gota a gota a -78 °C en argón. La reacción se agitó durante 30 minutos a -78 °C y se dejó calentar a temperatura ambiente y se agitó durante 2 días. La reacción se inactivó con una solución acuosa saturada de NH4Cl y el producto se extrajo con EtOAc. La capa orgánica se secó en MgSO4 anhidro y se concentró. El producto se purificó por cromatografía en columna (hexano/EtOAc 4:1) y se obtuvo como un aceite transparente (152 mg; 0,47 mmol; 36 % de rendimiento).
ESI-Ms m/z para C17H24ClNO3 encontrado 270,2 (MJBu+1)+.
1H RMN (DMSO-d6 D2O, 700 MHz) 67,30 (AA’BB’, J = 8,4 Hz, 2H), 7,18 (AA’BB’, J = 8,4 Hz, 2H), 3,87-3,83 (m, 1H), 3,22 (d, J = 11,4 Hz, 1H), 3,15 (d, J = 11,0 Hz, 1H), 3,07 (dd, J = 13,2, 4,0 Hz, 1H), 2,86 (dd, J = 12,8, 9,2 Hz, 1H), 1,72 (dd, J = 13,2, 8,8 Hz, 1H), 1,67 (dd, J = 12,8, 4,0 Hz, 1H), 1,39 (s, 9H), 1,19 (s, 3H).
Etapa 3
Síntesis de trifluoroacetato de (5S)-5-(4-clorobencil)-3-metilpirrolidin-3-ol (48c)

A una solución del compuesto 48b (150 mg; 0,46 mmol) en DCM (15ml), se añadió TFA (5 ml; 67,09 mmol). La reacción se completó después de 1 hora como se indicó por LCMS. La mezcla se diluyó con DCM y se evaporó hasta sequedad. El producto se obtuvo como un aceite amarillo oscuro (155 mg, 0,46 mmol, 99 % rendimiento).
ESI-MS m/z para C12 H16ClNO encontrado 226,2 (M+1)+.
Etapa 4
Síntesis de 4-((2S)-2-(4-dorobencil)-4-hidroxi-4-metilpirrolidin-1-il)piperidina-1-carboxilato de terc-butilo (48d)
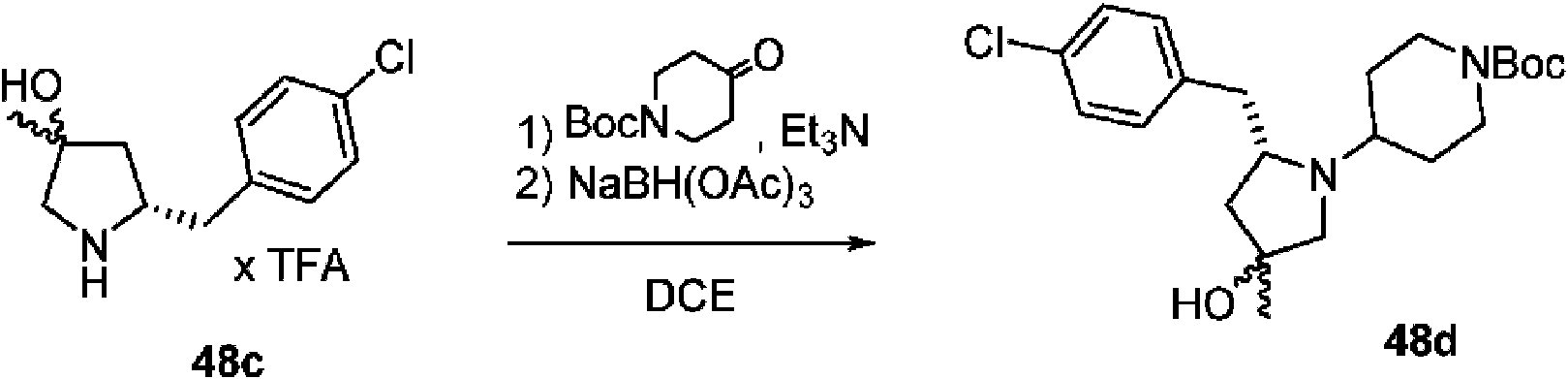
A una solución del compuesto 48c (156 mg; 0,46 mmol) en DCE (1 ml), se añadió Et3N (64 pl; 0,46 mmol) seguido de 1-Boc-piperid-4-ona (l02 mg; 0.51 mmol). La mezcla de reacción se calentó hasta 70 °C y se agitó a temperatura ambiente durante 1 hora. Se añadió triacetoxiborohidruro de sodio (195 mg; 0,92 mmol) y la reacción se agitó durante la noche, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La reacción se inactivó con una solución de NaHCO3 al 5 % durante 30 minutos y las capas se separaron. La capa orgánica se lavó con una solución de NaHCO3 al 5 %, salmuera, se secó en MgSO4 anhidro y se concentró. El producto bruto se purificó por cromatografía en columna (EtOAc/MeOH 20:1) para obtener 48d como un aceite transparente (126 mg; 0,31 mmol; 67 % de rendimiento).
ESI-MS m/z para C22H33ClN2O3 encontrado 409,3 (M+1)+.
1H RMN (700 MHz, DMSO-d6 D2O) 87,25 (d, J = 8,4 Hz, 2H), 7,18 (d, J = 8,5 Hz, 2H), 3,93 - 3,87 (m, 2H), 3,41 (s, 1H), 3,04 (tt, J = 9,6, 4,9 Hz, 1H), 2,85 (dd, J = 13,2, 4,1 Hz, 1H), 2,74 (d, J = 9,1 Hz, 1H), 2,72 - 2,63 (m, 3H), 2,62 -2,57 (m, 1H), 1,72 - 1,65 (m, 2H), 1,58 - 1,48 (m, 2H), 1,36 (s, 9H), 1,28 (qd, J = 112,3, 4,2 Hz, 1H), 1,20 (qd, J = 12,3, 4,3 Hz, 1H), 1,13 (s, 3H).
Etapa 5
Síntesis de difluoroacetato de (5S)-5-(4-clorobencil)-3-metil-1-(piperidin-4-il)pirrolidin-3-ol (48e)

A una solución del compuesto 48d (150 mg; 0,46 mmol) en DCM (1,2 ml), se añadió TFA (231 pl; 3,10 mmol) y la mezcla se agitó a temperatura ambiente. Después de 1 hora, la LCMS indicó que la reacción se había completado. La mezcla se diluyó con DCM y se evaporó hasta sequedad para proporcionar el producto del título como un aceite amarillo (166 mg; 99 % de rendimiento).
ESI-MS m/z para C17H25ClN2O encontrado 309,3 (M+1)+.
Etapa 6
Síntesis de (5S)-1-(1-(5-amino-4H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)-3-metilpirrolidin-3-ol (48)

El compuesto del título 48 se sintetizó a partir del compuesto 48e (166 mg; 0,31 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de fase inversa y se obtuvo como una sal de TFA (69 mg, 0,14 mmol, 44 % de rendimiento).
ESI-MS m/z para C19H27CIN6O encontrado 391,3 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 57,39 (AA'BB', J = 8,4 Hz, 2H), 7,31 (AA'BB', J = 8,4 Hz, 2H), 4,05-4,01 (m, 1H), 3,85-3,80 (m, 2H), 3,48-3,43 (m, 1H), 3,36 (dd,J = 11,4, 1,8 Hz, 2H), 3,16 (dd, J = 12,8, 3,5 Hz, 1H), 3,11-3,05 (m, 2H), 2,85 (dq, J = 11,4, 1,8 Hz, 2H), 2,26-2,24 (m, 1H), 2,02-2,00 (m, 1H), 1,94 (dd, J = 13,2, 10,1 Hz, 1H), 1,82-1,66 (m, 3H), 1,29 (s, 3H).
Ejemplo 49
5-(4-((3S,8aS)-3-(4-clorobencil)hexahidropirrolo[1,2-a]pirazin-2(1H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (49)

Etapa 1
Síntesis de 2-(((S)-3-(4-dorofenil)-1-metoxi-1-oxopropan-2-il)carbamoil)pirrolidina-1-carboxilato de (S)-terc-butilo (49a)

El compuesto del título 49a se sintetizó a partir de W-Boc-L-prolina (1,5 g; 6,968 mmol) y clorhidrato de 4-cloro-L-fenilalanina metil éster (1,92 g; 7,665 mmol) de acuerdo con el Procedimiento General IX con la excepción de que se usó Et3N (2,5 equiv.) en vez de N-metilmorfolina. El producto 49a se obtuvo como un aceite incoloro (1,42 g; 3,46 mmol; 49 % de rendimiento) y se usó en la siguiente etapa sin purificación adicional.
ESI-LCMS m/z para C20H27ClN2O5 encontrado 311,0 (M-Boc+1)+, 433,0 (M+Na)+.
Etapas 2-4
Síntesis de 4-((3S,8aS)-3-(4-clorobencil)hexahidropirrolo[1,2-a]pirazin-2(1H)-il)piperidina-1-carboxilato de terc-butilo (49b)

El compuesto 49b se preparó a partir del compuesto 49a (1,42 g; 3,46 mmol) en la secuencia de las reacciones de acuerdo con el Procedimiento General X (desprotección de Boc seguido de ciclación a dicetopiperazina), el Procedimiento General V (reducción de grupos amida) y el Procedimiento General VI (aminación reductiva con Bocpiperid-4-ona) y se obtuvo como un aceite incoloro (720 mg; 1,66 mmol; 48 % de rendimiento).
ESl-LCMS m/z para C24H36ClN3O2 encontrado 434,3/436,3 (m 1)+.
1H RMN (CDCl3, 400 MHz) 5: 7,24 (AA'BB', J = 8,4 Hz, 2H), 7,11 (AA'BB', J = 8,4 Hz, 2H), 4,07 (s a, 2H), 3,17-3,09 (m, 1H), 3,04 (dd, J = 12,5, 10,1 Hz, 1H), 2,99 (dd, J = 11,6, 3,0 Hz, 1H), 2,93 (td, J = 8,5, 1,7 Hz, 1H), 2,84-2,65 (m, 5H), 2,45 (dd, J = 11,5, 9,7 Hz, 1H), 2,22-2,09 (m, 3H), 1,91-1,65 (m, 5H), 1,56-1,33 (m, 3H), 1,45 (s, 9H).
Etapa 5
Síntesis de (3S,8aS)-3-(4-clorobencil)-2-(piperidin-4-il)octahidropirrolo[1,2-a]pirazina (49c)

A la solución del compuesto 49b (698 mg; 1,61 mmol) en AcOEt (6 ml) HCl en una solución de AcOEt 2,67 M (6 ml; 16,08 mmol) se añadió y la mezcla se agitó a temperatura ambiente durante 2 horas. Debido a la mala conversión del sustrato como se indica por TLC (DCM/MeOH 10:1) se añadió una solución 6 N acuosa de HCl (5 ml) y la mezcla se agitó durante 10 minutos. Las fases se separaron y la fase orgánica se lavó con 6 M HCl (5 ml). Las fases acuosas combinadas se alcalinizaron con 4 M NaOH hasta alcanzar pH ~14 y el producto se extrajo con AcOEt (4 x 20 ml). Las soluciones orgánicas combinadas se secaron en MgSO4 anhidro y se concentraron. El producto bruto (654 mg, 1,607 mmol) se utilizó en la siguiente etapa sin purificación adicional.
Etapa 6
Síntesis de 5-(4-((3S,8aS)-3-(4-clorobencil)hexahidropirrolo[1,2-a]pirazin-2(1 H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (49)

El compuesto del título se preparó a partir del compuesto 49c (654 mg; 1,607 mmol) de acuerdo con el Procedimiento General VIII y se obtuvo como un sólido blanco (259 mg; 0,622 mmol, 38 % de rendimiento).
ESI-LCMS m/z para C21H30ON7 encontrado 208,7 (M+2)2+, 416,2 (M+1)+.
1H RMN (MeOH-d4, 400 MHz) 5: 7,27 (AA'BB', J = 8,5 Hz, 2H), 7,21 (AA'BB', J = 8,4 Hz, 2H), 3,83 (d, J = 13,1 Hz, 2H), 3,23-3,14 (m, 1H), 3,08 (dd, J = 11,5, 3,0 Hz, 1H), 3,04 (dd, J = 12,9, 10,1 Hz, 1H), 2,94 (td, J = 8,6, 2,0 Hz, 1H), 2,89-2,73 (m, 3H), 2,82 (dd, J = 12,8, 4,0 Hz, 1H), 2,77 (dd, J = 11,1,2,3 Hz, 1H), 2,45 (dd, J = 11,4, 10,2 Hz, 1H), 2,10-1,93 (m, 5H), 1,90-1,66 (m, 3H), 1,58-1,40 (m, 3H).
Ejemplo 50
(3R,5S)-1-(1-(5-amino-1H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-clorobencil)pirrolidin-3-ol (50)

Etapa 1
Síntesis de (2R,4R)-1-terc-butil 2-metil 4-hidroxipirrolidina-1,2-dicarboxilato (50a)

A una solución de W-Boc-c/s-4-hidroxi-D-prolina (15,00g, 64,88 mmol) en 325 ml de MeCN, se añadió K2CO3 (17,93 g, 129,76 mmol), seguido de MeI (8,1 ml, 129,76 mmol) y la reacción se agitó durante la noche, tiempo después del cual el control de LCMS indicó la presencia del sustrato. Se añadió otra porción de MeI (4,0 ml, 64,88 mmol) y la reacción se sometió a reflujo durante la noche. Cuando otro control de LCMS indicó que la reacción se había completado, la mezcla se filtró y el residuo sólido se lavó con EtOAc. Después de la evaporación del disolvente, se obtuvieron 16,06 g (99 % de rendimiento) del producto 50a como un aceite amarillento.
ESI-MS m/z para C11H19NO5 encontrado 146,2 (M-Boc+1)+, 268,2 (M+Na)+.
Etapa 2
Síntesis de (2R,4R)-1-terc-butil 2-metil 4-(terc-butoxi)pirrolidina-1,2-dicarboxilato (50b)

A una solución del compuesto 50a (16,06 g; 65,48 mmol) en DMF (100 ml), se añadió Mg(ClO4)2 (1,46 g; 6,55 mmol) seguido de Boc2O (32,87 g; 150,60 mmol) y la reacción se agitó durante la noche. Al día siguiente, se añadió otra porción de Mg(ClO4)2 y Boc2O (44,30 g; 203,03 mmol) y la reacción se agitó durante la noche, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La mezcla de reacción se filtró y evaporó hasta sequedad. El producto se purificó por cromatografía en columna (hexano/EtOAc 9/1) y se obtuvo como un sólido cristalino blanco (13,51 g; 68 % de rendimiento).
ESI-MS m/z para C15H27NO5 encontrado 302,4,2 (M+1)+, 324,3 (M+Na)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 8: 4,24-4,19 (m, 1H), 4,17-4,13 (m, 1H), [3,61 (s); 3,58 (s); 3H], 3,54-3,51 (m, 1H), 2,99-2,94 (m, 1H), 2,38-2,30 (m, 1H), 1,75-1,71 (m, 1H), [1,36 (s); 1,29 (s); 9H], [1,08 (s); 1,07 (s); 9H].
Etapa 3
Síntesis de ácido (2R,4R)-4-(terc-butoxi)-1-(terc-butoxicarbonil)pirrolidina-2-carboxílico (50c)

El compuesto 50b (13,51 g; 44,83 mmol) se disolvió en una mezcla de THF/MeOH (450 ml/220 ml). Después se añadió la solución de hidrato de hidróxido de litio (9,97 g; 237,60 mmol) en agua (220 ml) y la mezcla se agitó durante la noche, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La mezcla de reacción se concentró para retirar el THF y MeOH y el residuo acuoso se acidificó hasta pH 4 con 2 M HCl a 0 °C. El producto se extrajo con EtOAc y la capa orgánica se lavó con salmuera, se secó en MgSO4 anhidro. Después de la evaporación del disolvente, se obtuvieron 12,13 g (94 % de rendimiento) del producto 50c como un aceite sólido blanco.
ESI-MS m/z para C14H25NO5 encontrado 310,3 (M+Na)+, 286,0 (M-1)-.
Etapa 4
Síntesis de 4-(terc-butoxi)-2-(metoxi(metil)carbamoil)pirrolidina-1-carboxilato de (2R,4R)-terc-butilo (50d)

A una solución del compuesto 50c (12,13 g; 42,21 mmol) en DCM (100 ml), se añadió Et3N (14,7 ml; 105,52 mmol), seguido de CDI (10,27 g; 63,32 mmol) y la reacción se agitó durante 30 minutos. Se añadió clorhidrato de N,O-dimetilhidroxiloamina (6,18 g; 63,32 mmol) y la reacción se agitó durante la noche, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La mezcla de reacción se lavó con agua y salmuera, la capa orgánica se secó en MgSO4 anhidro y se concentró. El producto bruto se purificó por cromatografía en columna (hexano/EtOAc 5/1) para obtener 50d como un sólido cristalino blanco (9,34 g; 67 % de rendimiento).
ESI-MS m/z para C16H30N2O5 encontrado 331.4 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 8: 4,49 (t, J = 8,4 Hz, 1H), 4,20 (q, J = 7,7 Hz, 1H), 3,66-3,63 (m, 4H), 3,09 (s, 3H), 2,91 (s a, 1H), 2,47 (dt, J = 2,3, 7,9 Hz, 1H), 1,54-1,50 (m, 1H), 1,31 (s a, 9H), 1,11 (s, 9H).
Etapa 5
Síntesis de 4-(terc-butoxi)-2-(4-clorobenzoil)pirrolidina-1-carboxilato de (2R,4R)-terc-butilo (50e)

A una solución de 50d (4,50 g; 13,62 mmol) en Et2O seco (30 ml), se añadió bromuro de p-clorofenilmagnesio, previamente generado a partir de p-bromoclorobenceno (5,22 g; 27,24 mmol) y magnesio (695 mg; 28,60 mmol) en Et2O (25 ml), a -70 °C bajo argón. La reacción se agitó a -70 °C durante 30 minutos y 2 horas a temperatura ambiente, tiempo después del cual la LCMS mostró que la reacción se había completado. La reacción se inactivó con NH4Cl saturada y el producto se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 anhidro y se concentraron. El producto bruto se purificó por cromatografía en columna (hexano/EtOAc 9/1) para obtener 50e como cristales blancos (2,11 g; 40 % de rendimiento).
ESI-MS m/z para C20H2sClNO4 encontrado 326,3 (M-(Bu+1)+,
1H RMN (DMSO-d6+D2O, 700 MHz) 8: 7,94 (AA'BB', J = 8,8 Hz, 2H), 7,58 (AA'BB', J = 8,4 Hz, 2H), 5,14-5,11 (m, 1H), 4,30-4,26 (m, 1H), 3,65 (dt, J = 7,0, 10,6 Hz, 1H), 3,03-2,99 (m, 1H), 2,60-2,54 (m, 1H), 1,58-1,51 (m, 1H), [1,35 (s); 1,12 (s); 9H], [1,05 (s); 1,02 (s); 9H].
Etapa 6
Síntesis de 2-(4-clorobencil)-4-hidroxipirrolidina-1-carboxilato de (2S,4R)-terc-butilo (50f)

A una solución del compuesto 50e (2,11 g; 5,52 mmol) en DCM (30 ml), se añadió AlCb (2,95 g; 22,08 mmol) bajo argón, seguido de trietilsilano (3,5 ml; 22,08 mmol). La reacción se agitó durante 30 minutos. La LCMS indicó que la reacción se había completado. La reacción se inactivó con hielo y las capas se separaron. La capa orgánica se extrajo con 2 M HCl. Las capas acuosas combinadas se lavaron con hexano. El pH del residuo acuoso se ajustó a 8 con KHCO3 y se añadió Boc2O (1,44 g; 6,62 mmol) en acetona (100 ml). La reacción se agitó durante la noche, tiempo después del cual la LCMS indicó que la reacción se había completado. La mezcla de reacción se concentró y se saturó con NaCl. El producto se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre MgSO4 anhidro. El producto se purificó por cromatografía en columna (hexano/EtOAc 4:1) y se obtuvo como un
sólido cristalino blanco (928 mg; 54 % de rendimiento).
ESI-MS m/z para C16H22ClNO3 encontrado 256,2 (M-fBu+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 5: 7,30 (AA'BB', J = 8,6 Hz, 2H), 7,18 (AA'BB', J = 8,2 Hz, 2H), 4,21-4,18 (m, 1H), 3,87-3,83 (m, 1H), 3,50 (dd, J = 5,6, 11,6 Hz, 1H), 3,10-3,05 (m, 2H), 2,81 (dd, J = 9,5, 12,9 Hz, 1H), 1,88 (ddd, J = 5,6, 8,1, 13,4 Hz, 1H), 1,59 (d, J = 12,9 Hz, 1H), 1,39 (s, 9H).
Etapa 7
Síntesis de clorhidrato de (3R,5S)-5-(4-dorobencil)pirrolidin-3-ol (50g)

El compuesto del título 50g se sintetizó a partir del compuesto 50f (250 mg; 0,80 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un sólido blanco (195 mg; 98 % de rendimiento).
ESI-MS m/z para CnHuClNo encontrado 212,2 (M+1)+.
Etapa 8
Síntesis de 4-((2S,4R)-2-(4-clorobencil)-4-hidroxipirrolidin-1-il)piperidina-1-carboxilato de terc-butilo (50h)

El compuesto del título 50h se preparó a partir del compuesto 47a (195 mg; 0,78 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un sólido transparente (212 mg; 68 % de rendimiento).
ESI-MS m/z para C21H31ClN2O3 encontrado 295,4 (M-Boc+1)+, 395,4 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 5: 7,27 (AA'BB', J = 2,6, 8,6 Hz, 2H), 7,19 (AA'BB', J=2,6, 8,2 Hz, 2H), 4,08-4,05 (m, 1H), 3,92-3,90 (m, 2H), 3,03-2,99 (m, 1H), 2,85 (dd, J = 4,7, 13,3 Hz, 1H), 2,76 (dd, J = 6,2, 10,1 Hz, 1H), 2,71 2,62 (m, 4H), 2,57 (dd, J = 9,0, 12,9 Hz, 1H), 1,81 (dt, J = 7,1, 12,9 Hz, 1H), 1,70-1,65 (m, 2H), 1,37 (s, 9H), 1,36-1,34 (m, 1H), 1,29-1,18 (m, 3H).
Etapa 9
Síntesis de diclorhidrato de (3R,5S)-5-(4-clorobencil)-1-(piperidin-4-il)pirrolidin-3-ol (50i)

El compuesto del título 50i se preparó a partir del compuesto 50h (212 mg; 0,54 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un sólido blanco (193 mg; 98 % de rendimiento).
ESI-MS m/z para C16H23ClN2O encontrado 295,4 (M+1)+.
Etapa 10
Síntesis de (3R,5S)-1-(1-(5-amino-1H-1,2,4-triazol-3-il)piperidin-4-il)-5-(4-dorobencil)pirrolidin-3-ol (50)

El compuesto del título 50 se sintetizó a partir del compuesto 50i (193 mg; 0,52 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de fase inversa y se obtuvo como una sal de TFA (105 mg, 33 % de rendimiento).
ESI-MS m/z para C18H25ClN6O encontrado 377,4 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 5: 7,38 (AA’BB’, J = 2,6, 8,2 Hz, 2H), 7,31 (AA’BB’, J = 8,6 Hz, 2H), 4,37 (s a, 1H), 3,99-3,96 (m, 1H), 3,84-3,80 (m, 3H), 3,47 (t, J = 11,6 Hz, 2H), 3,38 (d, J = 11,6 Hz, 1H), 3,21 (m, 1H), 2,99 (t, J = 12,5Hz, 1H), 2,92-2,85 (m, 2H), 2,22 (d, J = 11,6 Hz, 1H), 2,07-2,01 (m, 2H), 1,78 (dq, J = 4,3, 12,0 Hz, 1H), 1,72-1,66 (m, 2H).
Ejemplo 51
3-(4-((2S,4R)-2-(4-clorobencil)-4-metoxipirrolidin-1-il)piperidin-1-il)-1 H-1,2,4-triazol-5-amina (51)

El compuesto del título 51 se sintetizó de la misma forma que el Ejemplo 46 con la excepción de que se usó el compuesto 50f en vez del 46g en la primera etapa sintética.
ESI-MS m/z para C19H27ClN6O encontrado 391,4 (M+1)+
1H RMN (DMSO-de+D2O, 700 MHz) 5: 7,37 (AA’BB’, J = 2,6, 8,2 Hz, 2H), 7,29 (AA’BB’, J = 2,6, 8,6 Hz, 2H), 4,07-4,06 (m, 1H), 4,03-3,98 (m, 1H), 3,89-3,85 (m, 2H), 3,57 (d, J = 12,5 Hz, 1H), 3,50 (tt, J = 3,0, 12,0 Hz, 2H), 3,28 (s, 3H), 3,22 (dd, J = 4,5, 13,6 Hz, 1H), 2,92-2,82 (m, 3H), 2,17-2,11 (m, 2H), 2,30-2,00 (m, 1H), 1,83-1,67 (m, 3H).
Ejemplo 52
(S)-5-(4-(3-(4-clorobencil)-2,2-dimetilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (52)

Etapa 1
Síntesis de (S)-3-amino-4-(4-dorofenil)-2-metilbutan-2-ol (52a)

La reacción se llevó a cabo en una atmósfera de argón. A la solución de clorhidrato de 4-cloro-L-fenilalanina metil éster (1 g; 4,69 mmol) en THF seco (13 ml), se añadió una solución de bromuro de metilmagnesio 3,0 M en Et2O (11,6 ml; 32,9 mmol) gota a gota a temperatura ambiente y se agitó durante la noche. La reacción se inactivó con una solución saturada de NH4Cl y el producto se extrajo con AcOEt. La capa orgánica se secó en Na2SO4 anhidro y se concentró para obtener el producto 52a (0,82 g; 82 % de rendimiento) que se usó en la siguiente etapa sin purificación adicional.
ESI-MS m/z para CnH^ClNO encontrado 214,2 [M+H]+; 196,2 [M+m-hO]+.
Etapa 2-5
Síntesis de 4-(3-(4-clorobencil)-2,2-dimetilmorfolino)piperidina-1-carboxilato de (S)-terc-butilo (52b)

El compuesto 52b se preparó a partir del compuesto 52a (0,82 g; 3,85 mmol) en la secuencia de las reacciones de acuerdo con el Procedimiento General II (acilación selectiva de aminoalcohol con cloruro de cloroacetilo), el Procedimiento General IV (ciclación de a-haloamida a morfolin-3-ona), el Procedimiento General V (reducción de morfolin-3-ona a morfolina) y el Procedimiento General VI (aminación reductiva con 1-Boc-piperid-4-ona) y se obtuvo como un sólido ceroso (0,471 g; 29 % de rendimiento en cuatro etapas).
ESI-MS m/z para C23H35ON2O3 encontrado 423,3 [M+1]+.
Etapa 6-7
Síntesis de (S)-5-(4-(3-(4-clorobencil)-2,2-dimetilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (52)

El compuesto 52 se preparó a partir del compuesto 52b (0,471 g; 1,115 mmol) en la secuencia de reacciones de acuerdo con el Procedimiento General VII y el Procedimiento General VIII y se obtuvo como un sólido blanco (0,181 g; 40 % de rendimiento en dos etapas).
ESI-MS m/z para C20H29ON6O encontrado 405,3 [M+1]+.
1H RMN (400 MHz, CD3OD) 5: 7,35 - 7,27 (m, 4H), 3,82 - 3,70 (m, 2H), 3,70 - 3,59 (m, 1H), 3,54 - 3,44 (m, 1H), 2,96 - 2,90 (m, 2H), 2,87 (d, J = 3,0 Hz, 1H), 2,85 - 2,78 (m, 1H), 2,74 - 2,65 (m, 3H), 2,52 - 2,44 (m, 2H), 1,71 - 1,59 (m, 2H), 1,37 (s, 3H), 1,29 (dd, J = 11,6, 4,2 Hz, 1H), 1,21 (s, 3H).
Ejemplo 53
5-(4-((3S,6S)-3-(4-dorobencil)-2,2,6-trimetilmorfolino)piperidin-1-il)-4H-1,2,4-triazol-3-amina (53)

El compuesto del título se preparó de la misma forma que el Ejemplo 52 con la excepción de que se usó cloruro de cloropropionilo racémico en vez de cloruro de cloroacetilo en la primera etapa sintética.
ESI-Ms m/z para C21H31ClN6O encontrado 419,4 [M+1]+.
1H RMN (400 MHz, CD3OD) 6: 7,28 - 7,21 (m, 4H), 3,99 - 3,89 (m, 1H), 3,73 - 3,62 (m, 2H), 3,03 - 2,89 (m, 3H), 2,81 - 2,76 (m, 1H), 2,72 (dd, J = 12,0, 3,3 Hz, 1H), 2,56 - 2,49 (m, 1H), 2,40 - 2,31 (m, 1H), 1,89 - 1,72 (m, 2H), 1,52 -1,44 (m, 3H), 1,43 (s, 3H), 1,10 (d, J = 6,2 Hz, 3H), 0,99 (s, 3H).
Ejemplo 54
3-((3S,4S)-4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)-3-metoxipiperidin-1-il)-1 H-1,2,4-triazol-5-amina (54)

Etapa 1
Síntesis de mezcla diastereomérica de (3R,4R)-1-bencil-4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)piperidin-3-ol (54a-I) y (3S,4S)-1-bencil-4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)piperidin-3-ol (54a-N)

A la solución de rac-1-bencil-3,4-epoxipiperidina (1,5 g, 8,0 mmol) (preparada de acuerdo con el procedimiento descrito en la bibliografía: lan S. Young et al. Org. Process. Res. Dev. 2012, 16, 1558-1565.) y bromuro de litio (1,044 g, 5,0 mmol) en acetonitrilo seco (30 ml), (2S,5S)-5-(4-clorobencil)-2-metilmorfolina (4c) (1,8g, 8,0 mmol, 1,0 equiv.) se añadió y la mezcla de reacción se agitó durante 24 horas a temperatura ambiente, después se calentó a 50 °C durante 4 horas. La evolución de la reacción se monitoreó por análisis de LCMS de pequeñas muestras de la mezcla de reacción. Después de 28 horas, la mezcla de reacción se enfrió a temperatura ambiente y se añadieron 20 ml de agua. Se evaporó acetonitrilo a presión reducida y el residuo se extrajo con acetato de etilo (3 * 100 ml). Las capas orgánicas se combinaron, se evaporaron a presión reducida y el producto bruto se purificó por cromatografía ultrarrápida usando hexano-acetato de etilo (elución de gradiente hasta 10:1 a 1:10) para proporcionar 2,3 g de (54a-I) y (54a-II) como un sólido blanco (75 % de rendimiento, mezcla 1:1 de diastereómeros).
Se obtuvieron pequeñas fracciones de diastereómeros puros por separación del producto en TLC preparativa usando diclorometano-metanol 25:1 como fase móvil.
ESI-MS m/z para C24Hs1ClN2O2 encontrado 415,2/ 417,2 (M+1)+
Diastereómero menos polar: 1H RMN (400 MHz, CD3OD) 67,34- 7,29 (m, 5H), 7,27 - 7,22 (m, 2H), 7,21 - 7,15 (m, J
= 8,4 Hz, 2H), 3,64 - 3,42 (m, 6H), 3,14 - 3,05 (m, 1H), 2,95 - 2,80 (m, 3H), 2,80 - 2,74 (m, 1H), 2,67 (dd, J = 11,9, 10,2 Hz, 1H), 2,55 (dd, J = 12,0, 2,8 Hz, 1H), 2,40 (ddd, J = 13,4, 9,4, 4,0 Hz, 1H), 2,09 (td, J = 11,7, 2,4 Hz, 1H), 1,92 (t, J = 10,4 Hz, 1H), 1,87 - 1,80 (m, 1H), 1,70 (ddd, J = 25,1, 12,2, 4,0 Hz, 1H), 1,15 (d, J = 6,0 Hz, 3H).
Diastereómero más polar: 1H RMN (400 MHz, CD3OD) S 7,32 - 7,29 (m, 5H), 7,27 - 7,23 (m, 2H), 7,21 - 7,13 (m, J = 7,3 Hz, 2H), 3,66 -3,44 (m, 6H), 2,99 (ddd, J = 10,7, 4,2, 1,9 Hz, 1H), 2,95 - 2,73 (m, 5H), 2,69 (dd, J = 13,0, 3,1 Hz, 1H), 2,46 - 2,36 (m, 1H), 1,99 (td, J = 11,4, 2,3 Hz, 1H), 1,92 - 1,79 (m, 2H), 1,59 (ddd, J = 24,6, 11,6, 3,9 Hz, 1H), 1,14 (d, J = 6,1 Hz, 3H).
Etapa 2
Síntesis de mezcla diastereomérica de (2S,5S)-4-((3R,4R)-1-bencil-3-metoxipiperidin-4-il)-5-(4-clorobencil)-2-metilmorfolina (54b-I) y (2S,5S)-4-((3S,4S)-1-bencil-3-metoxipiperidin-4-il)-5-(4-clorobencil)-2-metilmorfolina (54b-II).

El compuesto del título se sintetizó a partir de una mezcla diastereomérica de compuestos (54a-I) y (54a-II) en una escala de 3,0 mmol de acuerdo con el Procedimiento General XXI, usando hidruro de sodio (NaH) y yoduro de metilo en THF. El producto bruto se purificó por cromatografía ultrarrápida usando diclorometano-metanol (elución de gradiente de 50:1 a 10:1) para proporcionar 600 mg de (54b-I) y (54b-II) como un aceite amarillento (47 % de rendimiento, mezcla 1:1 de diastereómeros).
ESI-MS m/z para C25H33ClN2O2 encontrado 429,2/ 431,2 (M+1)+.
Etapa 3
Síntesis de mezcla diastereomérica de 4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)-3-metoxipiperidina-1-carboxilato de (3R,4R)-alilo (54c-I) y 4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)-3-metoxipiperidina-1-carboxilato de (3S,4S)-alilo (54c-II).

A una solución de una mezcla diastereomérica de (54c-I) y (54c-II) (600 mg, 1,4 mmol, 1,0 equiv.) en diclorometano (10 ml) a 0 °C, se añadió alil cloroformiato de alilo (224 ^l, 2,1 mmol). Después de que la adición se había terminado,
la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 16 horas. Después de esto, el análisis de LCMS de la mezcla de reacción bruta mostró la conversión total y la reacción se inactivó con una solución acuosa al 10 % de NaHCO3 (5 ml). La fase orgánica se separó y la fase acuosa se extrajo con diclorometano (2 x 20 ml). Las fases orgánicas combinadas se evaporaron y el producto bruto se purificó por cromatografía de fase inversa usando agua-acetonitrilo-0,1 % de TFA (elución de gradiente de 10:1 a 1:5) para proporcionar 400 mg de (54c-I-TFA) y (54c-II-TFA) como un aceite amarillento (53 % de rendimiento, mezcla 1:1 de diastereómeros, sal de TFA).
ESI-MS m/z para C22H31ClN2O4 encontrado 423,2/ 425,2 (M+1)+
Etapa 4-5
Síntesis de 3-((3R,4R)-4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)-3-metoxipiperidin-1 -il)-1H-1,2,4-triazol-5-amina (54-I) y 3-((3S,4S)-4-((2S,5S)-5-(4-clorobencil)-2-metilmorfolino)-3-metoxipiperidin-1-il)-1 H-1,2,4-triazol-5-amina (54)

La retirada del grupo Alloc y la formación del anillo 1,2,4-triazol se lograron de acuerdo con el Procedimiento General XVI y el Procedimiento General VIII respectivamente comenzando por 400 mg, 0,75 mmol del compuesto (54c-I-TFA) y (54c-IITFA). Los diastereómeros se separaron por cromatografía en fase inversa usando agua-acetonitrilo-0,1 % de TFA (elución de gradiente de 5:1 a 2:1) 30 mg de 54 (dr >95:5, diastereómero más polar). La purificación del diastereómero 54-I menos polar no fue exitosa.
Diastereómero más polar 54:
ESI-MS m/z para C2üH29ClN6O2 encontrado 423,2/ 425,2 (M+1)+
1H RMN (400 MHz, CD3OD) 67,41 - 7,34 (m, 2H), 7,34 - 7,26 (m, 2H), 4,29 (dd, J = 12,9, 2,6 Hz, 1H), 3,98 (ddd, J = 10,5, 6,3, 2,4 Hz, 1H), 3,95 - 3,88 (m, 1H), 3,83 (td, J = 9,5, 4,7 Hz, 2H), 3,77 - 3,62 (m, 2H), 3,61 - 3,55 (m, 2H), 3,52 (d, J = 10,6 Hz, 3H), 3,33 (dd, J = 11,9, 3,7 Hz, 1H), 3,29 - 3,26 (m, 1H), 3,24 - 3,15 (m, 1H), 3,11 (ddd, J = 14,8, 9,0, 2,6 Hz, 1H), 2,96 - 2,82 (m, 1H), 2,32 (t, J = 19,5 Hz, 1H), 2,07 - 1,86 (m, 1H), 1,32 (t, J = 6,5 Hz, 3H).
Ejemplo 55
(S)-5-(4-(3-(4-clorobencil)-6-metilen-1,4-oxazepan-4-il)piperidin-1 -il)-4H-1,2,4-triazol-3-amina (55)

Etapa 1
Síntesis de 3-(4-dorobencil)-6-metilen-1,4-oxazepan-4-carboxilato de (S)-terc-butilo (55a)

El compuesto del título 55a se sintetizó a partir de los compuestos 30a y 3-cloro-2-clorometil-1-propeno (0,20 ml; 1,910 mmol) (520 mg; 1,819 mmol) de acuerdo con el Procedimiento General XXI y se obtuvo después como un sólido transparente (317 mg; 0,938 mmol; 51 % de rendimiento).
ESI-Lc MS m/z para C18H24CWO3 encontrado (M-Boc+1)+ 238,1.
Etapa 2-3
Síntesis de 4-(3-(4-clorobencil)-6-metilen-1,4-oxazepan-4-il)piperidina-1-carboxilato de (S)-terc-butilo (55b)

El compuesto del título 55b se preparó a partir del compuesto 55a (210 mg; 0,621 mmol) en un procedimiento de dos etapas que implicaba la retirada del grupo Boc (el Procedimiento General VII) y la aminación reductiva con 1-Bocpipierid-4-ona (el Procedimiento General VI) y se obtuvo como un aceite amarillo (236 mg; 0,560 mmol; 90 % de rendimiento).
ESI-LCMS m/z para C23H33ON2O3 encontrado (M-Boc+1)+ 421,3.
Etapa 4-5
Síntesis de (S)-5-(4-(3-(4-clorobencil)-6-metilen-1,4-oxazepan-4-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (55)

El compuesto del título 55 se sintetizó a partir del compuesto 55b (236 mg; 0,560 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía en columna (AcOEt/MeOH 100:1 ^ 100:5) y se obtuvo una espuma blanca (106 mg; 0,263 mmol; 47 % de rendimiento),
Ejemplo 56
(S)-5-(4-(2-(4-clorobencil)-4-metil-1,4-diazepan-1-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (56)

Etapa 1-2
Síntesis de 3-((2-((terc-butoxicarbonil)amino)-3-(4-dorofenil)propil)(metil)amino)propanoato de (S)-metilo (56a)

El compuesto del título se sintetizó a partir del compuesto 30b (960 mg; 3,38 mmol) de acuerdo con los Procedimientos Generales XIII y XIV y se obtuvo como un aceite transparente (1,12 g; 2,91 mmol; 86 % de rendimiento).
ESI-MS m/z para C-i9H29ClN2O4 encontrado 285,1 (M-Boc+1)+, 385,2/386,9 (M+1)+.
Etapa 3-4
Síntesis de (S)-3-(4-clorobencil)-1-metil-1,4-diazepan-5-ona (56b)

El compuesto del título 56b se preparó a partir del compuesto 56a (1,04 g; 2,70 mmol) en un procedimiento de dos etapas que incluía la retirada del grupo Boc (el Procedimiento General VII) y la ciclación del anillo de 7 miembros (el Procedimiento General X) con la excepción de que se usó W,W-diisopropiletilamina en vez de Et3N. El producto 56b se obtuvo como un sólido cristalino marrón (698 mg, 99 % de rendimiento).
ESI-MS m/z para C13H17CW2O encontrado 253,1/254,9 (M+1)+.
Etapa 5
Síntesis de (S)-3-(4-clorobencil)-1-metil-1,4-diazepan (56c)

El compuesto del título 56c se preparó a partir del compuesto 56b (695 mg; 2,75 mmol) de acuerdo con el Procedimiento General V y se obtuvo como un aceite amarillo (562 mg; 2,36 mmol, 86 % de rendimiento).
ESI-MS m/z para C-iaH^ClN encontrado 239,1/241,0 (M+1)+.
Etapa 6
Síntesis de 4-(2-(4-clorobencil)-4-metil-1,4-diazepan-1-il)piperidina-1-carboxilato de (S)-terc-butilo (56d)

El compuesto del título 56d se preparó a partir del compuesto 56c (558 mg; 2,34 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un aceite amarillo (885 mg; 2,10 mmol, 90 % de rendimiento).
ESI-MS m/z para C23 H36 CIN3 O2 encontrado 322,2 (M-Boc+1)+, 422,2/423,6 (M+1)+.
Etapa 7
Síntesis de diclorhidrato de (S)-2-(4-dorobencil)-4-metil-1-(piperidin-4-il)-1,4-diazepan (50e)

El compuesto del título 56e se preparó a partir del compuesto 56d (880 mg; 2,08 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un polvo blanco (788 mg; 2,00 mmol, 88 % de rendimiento).
ESI-MS m/z para C18H28ClN3 encontrado 322,2 (M+1)+.
Etapa 8
Síntesis de trifluoroacetato de (S)-3-(4-(2-(4-clorobencil)-4-metil-1,4-diazepan-1-il)piperidin-1-il)-1H-1,2,4-triazol-5-amina (56)

El compuesto del título 56 se sintetizó a partir del compuesto 56e (555 mg; 1,29 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de fase inversa y se obtuvo como una sal de TFA (26 mg, 0,05 mmol, 5 % de rendimiento).
ESI-MS m/z para C2üH3üCIN7 encontrado 404,2 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) S 7,24 (AA'BB', J = 8,4 Hz, 2H), 7,19 (AA'BB', J = 8,4 Hz, 2H), 3,64 (m, 2H), 3,47 (ddd, J = 13,8, 8,7, 5,4 Hz, 1H), 3,16 (s a, 1H), 3,08 (dd, J = 14,5, 8,2 Hz, 1H), 3,01 (dd, J = 14,2, 5,4 Hz, 2H), 2,91 2,87 (m, 1H), 2,85-2,77 (m, 6H), 2,65 (s, 3H), 1,75-1,73 (m, 3H), 1,70-1,66 (m, 1H), 1,44 (dq, J=12,3, 4,4Hz, 1H), 1,36 (dq, J = 12,1,4,2 Hz, 1H).
Ejemplo 57
(S)-5-(4-(3-(4-clorobencil)-1-metil-2,3-dihidro-1H-benzo[e][1,4]diazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (57)

Etapa 1
Síntesis de 2-((2-((terc-butoxicarbonil)amino)-3-(4-clorofenil)propil)amino)benzoato de (S)-metilo (57 a)

A una solución de 30b 1,50 g (5,29 mmol) en 5 ml de 1,2-dicloroetano, se añadió 2-aminobenzoato de metilo 800 mg (5,29 mmol). La mezcla de reacción se calentó hasta 70 °C y se enfrió a temperatura ambiente durante 1 hora. Se añadió triacetoxiborohidruro de sodio 2,80 g (13,22 mmol) y la reacción se agitó durante 2 días, tiempo después del cual el control de LCMS indicó que la reacción se había completado. La reacción se inactivó con una solución de NaHCO3 al 5 % durante 30 minutos y las capas se separaron. La capa orgánica se lavó con una solución de NaHCO3 al 5 %, salmuera, se secó en MgSO4 anhidro y se concentró. El producto bruto se purificó por cromatografía en columna hexano/EtOAc 8/1 para obtener 755 mg de 57a como un sólido blanco (1,80 mmol; 34 % de rendimiento).
ESI-MS m/z para C22H27ClN2O4 encontrado 319,1 (M-Boc+1)+, 363,1/364,9 (M -B j+1)+.
1H RMN (CDCla, 700 MHz) 57,93 (m, 1H), 7,37 (m, 1H), 7,30, (s, 1H), 7,18 (d, J = 8,0 Hz, 2H), 6,75 (d, J = 7,7 Hz, 2H), 6,65 (t, J = 7,5 Hz, 1H), 4,59 (s a, 1H), 4,13 (s a, 1H), 3,89 (s, 3H), 3,33 (dd, J = 13,5, 5,1 Hz, 1H), 3,26 (dd, J = 13,0, 5,4 Hz, 1H), 2,92 (d, J = 6,3 Hz, 1H), 1,44 (s, 9H).
Etapa 2
Síntesis de 2-((2-((terc-butoxicarbonil)amino)-3-(4-clorofenil)propil)(metil)amino)benzoato de (S)-metilo (57b)

El compuesto del título se sintetizó a partir del compuesto 57a (500 mg; 1,19 mmol de acuerdo con el Procedimiento General XIV y se obtuvo como un aceite transparente (461 mg; 1,07 mmol; 89 % de rendimiento). ESI-MS m/z para C23H29ClN2O4 encontrado 333,2 (M-Boc+1)+, 433,2/435,0 (M+1)+.
Etapas 3-4
Síntesis de (S)-3-(4-clorobencil)-1-metil-3,4-dihidro-1H-benzo[e][1,4]diazepin-5(2H)-ona (57c)

El compuesto del título 57c se preparó a partir del compuesto 57b (461 mg; 1,06 mmol) en un procedimiento de dos etapas que incluía la retirada del grupo Boc (el Procedimiento General VII) y la ciclación del anillo de 7 miembros (el Procedimiento General X) con la excepción de que se usó W,W-diisopropiletilamina en vez de Et3N. El producto bruto se purificó por cromatografía en columna (hexano/EtOAc 1:2) para obtener 57c como un aceite transparente (137 mg; 0,46 mmol; 44 % de rendimiento).
ESI-MS m/z para C17H17ClN2O encontrado 301,2/303,0 (M+1)+.
1H RMN (DMSO-d6, 700 MHz) 58,15 (d, J = 5,7 Hz, 1H), 7,38 (dd, J = 7,6, 1,7 Hz, 1H), 7,36 (m, 1H,), 7,32 (AA'BB', J = 8,7 Hz, 2H), 7,30 (AA'BB', J = 8,7 Hz, 2H), 6,93-6,90 (m, 2H), 3,49 (m, 1H), 3,25 (t, J = 11,0 Hz, 1H), 2,90 (dd, J = 10,8, 3,6 Hz, 1H), 2,80 (dd, J = 13,8, 8,3 Hz, 1H), 2,75 (s, 3H).
Etapa 5
Síntesis de (S)-3-(4-dorobencil)-1-metil-2,3,4,5-tetrahidro-1H-benzo[e][1,4]diazepina (57d)

El compuesto del título 57d se preparó a partir del compuesto 57c (130 mg; 0,43 mmol) de acuerdo con el Procedimiento General V y se obtuvo como un aceite amarillento (122 mg; 0,42 mmol, 98 % de rendimiento).
ESI-MS m/z para C17H19GN2 encontrado 287,1 (M+1)+.
1H RMN (DMSO-da+D2O, 700 MHz) 57,33 (AA'BB', J = 8,4Hz, 2H), 7,26 (AA'BB', J = 8,4Hz, 2H), 7,13 (td, J = 7,7, 1,5 Hz, 1H), 7,06 (dd, J = 7,3, 1,2 Hz, 1H), 6,86 (d, J = 7,7 Hz, 1H), 6,80 (m, 1H), 3,83 (d, J = 14,2 Hz, 1H), 3,62 (d, J = 14,2 Hz, 1H), 3,07 (dd, J = 13,5, 2,3 Hz, 2H), 2,78 (s, 3H), 2,63-2,57 (m, 3H).
Etapa 6
Síntesis de 4-(3-(4-clorobencil)-1-metil-2,3-dihidro-1H-benzo[e][1,4]diazepin-4(5H)-il)piperidina-1-carboxilato de (S)-terc-butilo (57e)

El compuesto del título 57e se preparó a partir del compuesto 57d (122 mg; 0,42 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un aceite transparente (78 mg; 0,16 mmol, 38 % de rendimiento).
ESI-MS m/z para C27H36ClN3O2 encontrado 470,6/471,6/472,4 (M+1)+.
Etapa 7
Síntesis de diclorhidrato de (S)-3-(4-clorobencil)-1-metil-4-(piperidin-4-il)-2,3,4,5-tetrahidro-1H-benzo[e][1,4]diazepina (57f)

El compuesto del título 57f se preparó a partir del compuesto 57e (78 mg; 0,16 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un polvo blanco (76 mg; 0,16 mmol, 99 % de rendimiento).
ESI-MS m/z para C22H28ClN3 encontrado 370,1 (M+1)+.
Etapa 8
Síntesis de trifluoroacetato de (S)-3-(4-(3-(4-clorobencil)-1-metil-2,3-dihidro-1H-benzo[e][1,4]diazepin-4(5H)-il)piperidin-1-il)-1H-1,2,4-triazol-5-amina (57)

El compuesto del título 57 se sintetizó a partir del compuesto 57f (76 mg; 0,19 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de fase inversa y se obtuvo como una sal de TFA (22 mg, 0,04 mmol, 23 % de rendimiento).
ESI-MS m/z para C24H30ClN7 encontrado 452,1 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 57,35 (d, J = 8,3 Hz, 2H), 7,28-7,24 (m, 4H), 6,84-6,81 (m, 2H), 4,47-4,37 (m, 2H), 3,86 (s a, 1H), 3,74 (d, J = 12,6 Hz, 2H), 3,40 (s a, 2H), 3,08-3,06 (m, 2H), 2,82-2,76 (m, 2H), 2,70 (s, 3H), 2,65 2,63 (m, 1H), 2,12 (s a, 1H), 2,02 (s a, 1H), 1,68 (m, 2H).
Ejemplo 58
(S)-5-(4-(7-cloro-3-(4-clorobencil)-1-metil-2,3-dihidro-1H-benzo[e][1,4]diazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (58)

Etapa 1
Síntesis de 6-cloro-1 H-benzo[d][1,3]oxazina-2,4-diona (58a)

A una suspensión de ácido 5-cloroantranílico (1000 mg; 5,828 mmol) en 1,2-dicloroetano (5 ml) se añadió trifosgeno (692 mg; 2,331 mmol) en una solución de 1,2-dicloroetano (10 ml) y la mezcla se calentó hasta reflujo durante 4 horas. Después de enfriar hasta temperatura ambiente, el precipitado se filtró, se lavó con DCM frío y se secó para proporcionar un sólido blanco (1,128 g; 5,709 mmol; 98 % de rendimiento).
Etapa 2
Síntesis de 6-cloro-1-metil-1H-benzo[d][1,3]oxazina-2,4-diona (58b)
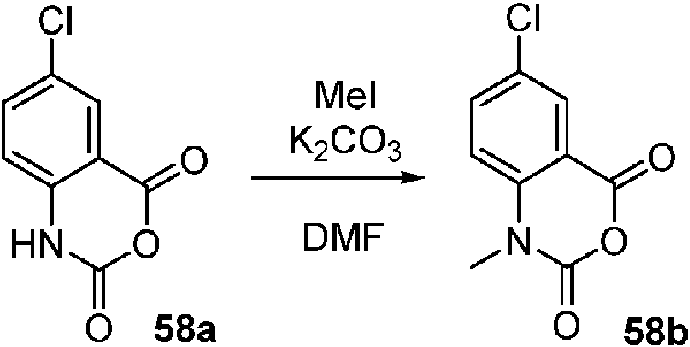
A una solución de 58a (1125 mg; 5,694 mmol) en DMF (6 ml) se añadió carbonato de sodio en polvo (724 mg; 6,833 mmol) seguido de yoduro de metilo (0,53 ml; 8,541 mmol) y la mezcla se agitó a temperatura ambiente durante 18 horas. Después de este tiempo la mezcla se enfrió hasta 0 °C y se añadió cuidadosamente hielo molido (5 g) y 1 M HCl (5 ml). El sólido precipitado se filtró, se lavó con agua fría (2x 10 ml) y se secó al vacío. El producto se obtuvo como un sólido amarillo (960 mg; 4,536 mmol; 80 % de rendimiento) y se usó en la siguiente etapa sin purificación adicional.
1H RMN (DMSO-d6, 400 MHz): 57,95 (d, J = 2,3 Hz, 1H), 7,89 (dd, J = 9,0, 2,6 Hz, 1H), 7,47 (d, J = 8,9 Hz, 1H), 3,44 (s, 3H).
Etapa 3
Síntesis de (S)-7-cloro-3-(4-clorobencil)-1-metil-3,4-dihidro-1 H-benzo[e][1,4]diazepina-2,5-diona (58c)

Una mezcla de 58b (1140 mg; 5,387 mmol) y L-4-clorofenilalanina (1129 mg; 5,656 mmol) en ácido acético glacial (11 ml) se calentó hasta reflujo durante 18 horas. Después de este tiempo, TLC (Hexanos/AcOEt 1:1) mostró que la reacción se había completado. La mezcla se concentró y el residuo se agitó vigorosamente con Na2CO3 saturado (10 ml). Después el producto se extrajo con AcOEt (3x 50 ml). Las soluciones orgánicas combinadas se lavaron con Na2CO3 saturado (10 ml), salmuera (10 ml) y se concentraron. El producto se purificó por cromatografía en columna (éter de petróleo/AcOEt 10:1 ^ 1:1) y se obtuvo como un sólido blanco (1262 mg; 3,613 mmol; 67 % de rendimiento). Es I-LCMS: m/z para C17H14Cl2N2O2 encontrado 349,1(M+1)+.
1H RMN (CDCl3, 400 MHz): 57,81 (d, J = 2,5 Hz), 7,52 (dd, J = 8,7, 2,6 Hz, 1H), 7,26 (AA’BB’, J = 8,5 Hz, 2H), 7,21 (AA’BB’, J = 8,6 Hz, 2H), 7,17 (d, J = 8,9 Hz, 1H), 6,93 (d, J = 5,3 Hz, 1H), 3,95 (dd, J = 13,4, 6,6 Hz, 1H), 3,39 (s, 3 H), 3,39 (dd, J = 14,4, 6,7 Hz, 1H), 3,02 (dd, J = 14,5, 7,7 Hz, 1H).
Etapa 4
Síntesis de (S)-7-cloro-3-(4-clorobencil)-1-metil-2,3,4,5-tetrahidro-1 H-benzo[e][1,4]diazepina (58d)

El compuesto del título 58d se preparó a partir del compuesto 58c (1245 mg; 3,565 mmol) de acuerdo con el Procedimiento General V y se obtuvo como un aceite amarillo (1,140 g; 3,548 mmol, 99 % de rendimiento).
ESI-LCMS: m/z para C17H18CI2N2 encontrado 321,2 (M+1)+.
Etapa 5
Síntesis de 4-(7-cloro-3-(4-clorobencil)-1-metil-2,3-dihidro-1H-benzo[en1,4]diazepin-4(5H)-il)piperidina-1-carboxilato de (S)-terc-butilo (58e)

El compuesto del título 58e se preparó a partir del compuesto 58d (1140 mg; 3,548 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un aceite amarillo (1237 mg; 2,451 mmol, 63 % de rendimiento después de dos etapas).
ESI-LCMS: m/z para C27H35Cl2N3O2 encontrado 504,3 (M+1)+.
1H RMN (MeOH-d4, 400 MHz): 57,27 (AA'BB', J = 8,5 Hz, 2H), 7,21 (AA'BB', J = 8,5 Hz, 2H), 7,06 (dd, J = 8,5, 2,5 Hz, 1H), 7,03 (d, J = 2,5 Hz, 1H), 6,69 (d, J = 8,6 Hz, 1H), 4,41 (d, J = 16,3 Hz, 1H), 3,84-3,67 (m, 2H), 3,74 (d, J = 16,5 Hz, 1H), 3,39-3,32 (m, 1 H), 3,25 (dd, J = 14,3, 7,3 Hz, 1H), 3,06 (dd, J = 14,3, 3,7, 1H), 2,85 (s, 3H), 2,81 (d, J = 6,9 Hz, 2H), 2,73-2,56 (m, 3H), 1,73-1,64 (m, 1H), 1,62-1,52 (m, 1H), 1,41 (s, 9H), 1,31-1,01 (m, 2H).
Etapa 6
Síntesis de clorhidrato de (S)-7-cloro-3-(4-clorobencil)-1-metil-4-(piperidin-4-il)-2,3,4,5-tetrahidro-1H-benzo[e][1,4]diazepina (58f)

El compuesto del título 58f se preparó a partir del compuesto 58e (1,22 g; 2,418 mmol) de acuerdo con el Procedimiento General VII y se usó en la siguiente etapa sin purificación adicional.
Etapa 7
Síntesis de (S)-5-(4-(7-cloro-3-(4-clorobencil)-1-metil-2,3-dihidro-1H-benzo[e][1,4]diazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (58)

El compuesto del título 58 se sintetizó a partir del compuesto 58f (1,066 g; 2,418 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de gel de sílice (sistema de AcOEt/MeOH) y se obtuvo como un sólido amarillo claro (930 mg, 1,916 mmol, 79 % de rendimiento).
ESI-LCMS: m/z para C24H29CI2N7 encontrado 243,8 (M+2)2+, 486,3 (M+1)+.
1H RMN (MeOH-d4, 400 MHz): 57,26 (AA'BB', J = 8,5 Hz, 2H), 7,21 (AA'BB', J = 8,5 Hz, 2H), 7,06 (dd, J = 8,5, 2,5 Hz, 1H), 7,04 (d, J = 2,4 Hz, 1H), 6,70 (d, J = 8,5 Hz, 1H), 4,39 (d, J = 16,2 Hz, 1H), 3,76 (d, J = 16,3 Hz, 1H), 3,69 3,46 (m, 2H), 3,39-3,33 (m, 1H), 3,22 (dd, J = 14,4, 6,9 Hz, 1H), 3,06 (dd, J = 14,4, 3,6 Hz, 1H), 2,84 (s, 3H), 2,82 (d, J = 7,0 Hz, 2H), 2,74-2,50 (m, 3H), 1,78-1,68 (m, 1H), 1,68-1,58 (m, 1H), 1,44-1,20 (m, 2H).
Ejemplo 59
(S)-5-(4-(3-(4-clorobencil)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (59)

Etapa 1
Síntesis de 2-hidroxi-N-metoxi-N-metilbenzamida (59a)

El compuesto del título se preparó a partir de ácido salicílico (10,00 g; 72,35 mmol) de acuerdo con el Procedimiento General XXII y se obtuvo como un aceite incoloro (6,98 g; 38,54 mmol; 53 % de rendimiento).
ESI-MS m/z para C9H11NO3 encontrado 182,0 (M+1)+.
Etapa 2
Síntesis de (1-(4-clorofenil)-3-(2-(metoxi(metil)carbamoil)fenoxi)propan-2-il)carbamato de (S)-terc-butilo (59b)

El compuesto del título se sintetizó a partir de los compuestos 59a (1,27 g; 7,00 mmol) y 30a (2,50 g; 8,75 mmol) de acuerdo con el Procedimiento General XXIII y se obtuvo como un sólido blanco (260 mg; 0,58 mmol, 8 % de rendimiento).
ESI-MS m/z para C23H29ClN2O5 encontrado 471,1 (M+Na)+-Etapa 3
Síntesis de (1-(4-clorofenil)-3-(2-formilfenoxi)propan-2-il)carbamato de (S)-terc-butilo (59c)

El compuesto del título se preparó a partir de 59b (260 mg; 0,58 mmol) de acuerdo con el Procedimiento General XXIV y se obtuvo como un sólido transparente (217 mg; 0,56 mmol; 96 % de rendimiento).
ESI-MS m/z para C21H24GNO4 encontrado 290,0/292,0 (M+1)+-1H RMN (DMSO-da, 700 MHz) 510,38 (s, 1H), 7,66 (dd, , 1H), 7,62-7,60 (m, 1H), 7,29 (AA'BB', J = 8,3Hz, 2H), 7,24 (AA'BB', J = 8,4 Hz, 2H), 7,14 (d, J = 7,1 Hz, 1H), 7,06 (t, J = 7,6 Hz, 1H), 4,10 (dd, J = 9,5, 4,0 Hz, 1H), 4,07-4,03 (m, 1H), 3,99-3,97 (m, 1H), 3,57 (ddd, J = 6,2, 4,2, 2,5 Hz, 1H), 2,89 (dd, J = 13,9, 5,3 Hz, 1H), 1,74-1,72 (m, 1H), 1,26 (s, 9H).
Etapa 4
Síntesis de (S)-3-(4-clorobencil)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepina (59d)

El compuesto del título se sintetizó a partir de 59c (217 mg; 0,56 mmol) de acuerdo con el Procedimiento General XXV y se obtuvo como un sólido amarillento (150 mg; 0,55 mmol; 98 % de rendimiento).
ESI-MS m/z para C16H16ClNO encontrado 274,1/276,0 (M+1)+.
1H RMN (DMSO-d6, 700 MHz) 57,35 (AA'BB', J = 8,3Hz, 2H), 7,28 (AA'BB', J = 8,3Hz, 2H), 7,16-7,14 (m, 2H), 6,97 (t, J = 7,4 Hz, 1H), 6,94 (d, J = 7,9 Hz, 1H), 4,20 (dd, J = 12,3, 2,5 Hz, 1H), 3,89-3,81 (m, 2H), 3,49 (s a, 1H), 3,07 (s a, 3H), 2,68-2,63 (m, 1H).
Etapa 5
Síntesis de 4-(3-(4-clorobencil)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)piperidina-1-carboxilato de (S)-terc-butilo (59e)

El compuesto del título se preparó a partir del compuesto 59d (150 mg; 0,55 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un sólido transparente (46 mg; 0,10 mmol; 18 % de rendimiento).
ESI-MS m/z para C26H33GN2O3 encontrado 457,1 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 57,30 (AA'BB', J = 8,4 Hz, 2H), 7,25 (AA'BB', J = 7,2 Hz, 2H), 7,13-7,10 (m, 2H), 6,92 (td, J = 7,4, 1,1 Hz, 1H), 6,82 (d, J = 7,9 Hz, 1H), 4,40 (d, J = 16,8 Hz, 1H), 4,13-4,08 (m, 2H), 3,78 (s, 1H), 3,72 (d, J = 17,0 Hz, 1H), 3,62-3,59 (m, 1H), 3,47 (s a, 1H), 3,39-3,35 (m, 1H), 2,71 (d, J = 6,4 Hz, 2H), 2,58-2,54 (m, 2H), 1,54 (d, J = 12,9 Hz, 1H), 1,47 (d, J = 11,8 Hz, 1H), 1,32 (s, 9H), 1,03 (m, 1H), 0,88 (s a, 1H).
Etapa 6
Síntesis de diclorhidrato de (S)-3-(4-clorobencil)-4-(piperidin-4-il)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepina (59f)

El compuesto del título 59f se preparó a partir del compuesto 59e (46 mg; 0,10 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un polvo blanco (43 mg; 0,10 mmol, 99 % de rendimiento).
ESI-MS m/z para C21H25GN2O encontrado 357,1 (M+1)+.
Etapa 7
Síntesis de (S)-3-(4-(3-(4-clorobencil)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)piperidin-1-il)-1 H-1,2,4-triazol-5-amina (59)

El compuesto del título 59 se sintetizó a partir del compuesto 59f (43 mg; 0,10 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de fase inversa y se obtuvo como una sal de TFA (28 mg, 0,06 mmol, 63 % de rendimiento).
ESI-MS m/z para C23H27GN6O encontrado 439,0 (M+1)+.
1H RMN (DMSO-d6+D2O, 700 MHz) 57,34 (m, 3H), 7,29-7,27 (m, 3H), 7,02 (t, J = 7,2 Hz, 1H), 6,97 (d, J = 8,0 Hz, 1H), 4,57-4,51 (m, 2H), 4,28 (s a, 1H), 3,91-3,87 (m, 2H), 3,74-3,72 (m, 2H), 3,46 (s a, 1H), 3,21 (s a, 1H), 3,10-3,06 (m, 1H), 2,81 (t, J = 12,2 Hz, 1H), 2,72 (s, 1H), 2,08-1,94 (m, 2H), 1,70-1,62 (m, 2H).
Ejemplo 60
(S)-5-(4-(3-(4-clorobencil)-7-fluoro-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (60)

El compuesto del título 60 se preparó de la misma forma que el Ejemplo 59 con la excepción de que se usó ácido 5-fluorosalicílico en vez de ácido salicílico en la primera etapa sintética.
ESI-MS m/z para C23H26CFN6O encontrado 457,1/459,1 (m 1)+.
1H RMN (MeOH-d4, 400 MHz) 5: 7,27 (AA'BB', J = 8,7 Hz, 2H), 7,24 (AA'BB', J = 8,7 Hz, 2H), 6,90 (dd, J = 8,5, 2,2 Hz, 1H), 6,87-6,80 (m, 2H), 4,49 (d, J = 16,6 Hz, 1H), 4,16-4,07 (m, 2H), 3,81 (d, J = 16,7, 1H), 3,67-3,59 (m, 1H), 3,58-3,50 (m, 1H), 3,48-3,40 (m, 1H), 2,86 (dd, J = 13,4, 6,5 Hz, 1H), 2,81 (dd, J = 13,3, 7,7 Hz, 1H), 2,73-2,55 (m, 3H), 1,78-1,70 (m, 1H), 1,68-1,60 (m, 1H), 1,44-1,21 (m, 2H).
Ejemplo 61
(S)-5-(4-(7-cloro-3-(4-clorobencil)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (61)

El compuesto del título 59 se preparó de la misma forma que el Ejemplo 59 con la excepción de que se usó ácido 5-dorosalicílico en vez de ácido salicílico en la primera etapa sintética.
ESI-MS m/z para C23H26O 2N6O encontrado 473,1/475,1 (M+1)+.
1H RMN (MeOH-d4, 400 MHz) 5: 7,26 (AA'BB', J = 8,7 Hz, 2H), 7,22 (AA'BB', J = 8,7 Hz, 2H), 7,13 (d, J = 2,6 Hz, 1H), 7,08 (dd, J = 8,5, 2,6 Hz, 1H), 6,82 (d, J = 8,5 Hz, 1H), 4,47 (d, J = 16,8 Hz, 1H), 4,20-4,12 (m, 2H), 3,81 (d, J = 16,8 Hz, 1H), 3,66-3,58 (m, 1H), 3,58-3,51 (m, 1H), 3,50-3,42 (m, 1H), 2,80 (dd, J = 13,4, 6,4 Hz, 1H), 2,76 (dd, J = 13,4, 7,7 Hz, 1H), 2,71-2,53 (m, 3H), 1,74-1,66 (m, 1H), 1,66-1,58 (m, 1H), 1,44-1,19 (m, 2H).
Ejemplo 62
(S)-5-(4-(7,9-dicloro-3-(4-clorobencil)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (62)

El compuesto del título se preparó de la misma forma que el Ejemplo 59 con la excepción de que se usó ácido 3,5-diclorosalicílico en vez de ácido salicílico en la primera etapa sintética.
ESI-LCMS: m/z para C23H25CbN6O encontrado 255,2 (M+2)2+, 509,2 (M+1)+.
1H RMN (MeOH-d4, 400 MHz) 5: 7,30-7,20 (m, 5H), 7,12 (d, J = 2,5 Hz, 1H), 4,52 (d, J = 16,7 Hz, 1H), 4,26 (dd, J = 13,1,7,2 Hz, 1H), 4,21 (dd, J = 13,2, 4,5 Hz, 1H), 3,85 (d, J = 16,8 Hz, 1H), 3,67-3,44 (m, 3H), 2,81 (d, J = 7,0 Hz, 2H), 2,75-2,48 (m, 3H), 1,70-1,63 (m, 1H), 1,63-1,55 (m, 1H), 1,38-1,22 (m, 2H).
Ejemplo 63
(S)-5-(4-(3-(4-clorobencil)-2,3-dihidropirido[4,3-f][1,4]oxazepin-4(5H)-il)piperidin-1 -il)-4H-1,2,4-triazol-3-amina (63)

Etapa 1
Síntesis de 3-(2-((terc-butoxicarbonil)amino)-3-(4-clorofenil)propoxi)isonicotinato de (S)-metilo (63a)

El compuesto del título se preparó a partir de 3-hidroxi-4-piridinacarboxilato de metilo (495 mg; 3,232 mmol) y el compuesto 30a (967 mg; 3,394 mmol) de acuerdo con el Procedimiento General XXII. Para aumentar el rendimiento, se usaron 2 equivalentes de trifenilfosfina (1,98 g; 6,788 mmol) y DIAD (1,34 ml; 6,788 mmol). La purificación en cromatografía en columna de gel de sílice (sistema de éter de petróleo/AcOEt) proporcionó el producto del título como un sólido blanco (1,0 g; 2,375 mmol; 73 % de rendimiento).
1H RMN (CDCla, 400 MHz) 8: 8,37 (d, J = 4,8 Hz, 1H), 8,34 (s, 1H), 7,66 (d, J = 4,6 Hz, 1H), 7,25 (AA'BB', J = 6,7 Hz, 2H), 7,17 (AA'BB', J = 6,8 Hz, 2H), 5,36 (d, J = 7,8 Hz, 1H), 4,21-4,01 (m, 3H), 3,98 (s, 3H), 3,07-2,93 (m, 2H), 1,42 (s, 9H).
Etapa 2
Síntesis de 3-(2-amino-3-(4-clorofenil)propoxi)isonicotinato de (S)-metilo (63b)

El compuesto del título se sintetizó a partir del compuesto 63a (990 mg; 2,352 mmol) de acuerdo con el Procedimiento General VII y se obtuvo como un sólido (840 mg; 2,351 mmol; 99 % de rendimiento) que se usó en la siguiente etapa sin purificación adicional.
ESI-LCMS: m/z para C16H17ClN2O3 encontrado 321,1 (M+1)+.
Etapa 3
Síntesis de (S)-3-(4-clorobencil)-3,4-dihidropirido[4,3-f][1,4]oxazepin-5(2H)-ona (63c)

A la suspensión del 63b bruto (840 mg; 2,351 mmol) en xileno (23 ml), se añadió trietilamina (8 ml; 58,78 mmol) y la mezcla se calentó hasta reflujo durante 4 horas. Después de este tiempo, la reacción se completó como se indica por TLC (DCM/MeOH 10:1). La mezcla se concentró y el producto, después de la cristalización a partir de MeOH, se obtuvo como un sólido blanco (354 mg; 1,226 mmol). El filtrado se concentró y se sometió a ciclación y cristalización de la misma forma que la descrita anteriormente para proporcionar un segundo cultivo del producto (96 mg; 0,332 mmol) con un rendimiento total de 66 % (440 mg; 1,558 mmol).
ESI-LCMS: m/z para C15H13ClN2O2 encontrado 289,1 (M+1)+.
1H RMN (CDCla, 400 MHz) 8: 8,49 (s, 1H), 8,35 (d, J = 5,2 Hz, 1H), 8,00 (d, J = 5,2 Hz, 1H), 7,32 (d, J = 8,3 Hz, 2H), 7,16 (d, J = 8,2 Hz, 2H), 6,77 (s a, 1 H), 4,40-4,29 (m, 2H), 3,89-3,78(m, 1H), 2,94 (dd, J = 13,8, 7,0 Hz, 1H), 2,84 (dd, J = 13,9, 8,3 Hz, 1H).
Etapa 4
Síntesis de (S)-3-(4-dorobencil)-2,3,4,5-tetrahidropirido[4,3-f][1,4]oxazepina (63d)

El compuesto del título se sintetizó a partir del compuesto 63c (441 mg; 1,527 mmol) de acuerdo con el Procedimiento General V y se obtuvo como un aceite (415 mg; 1,510 mmol, 99 % de rendimiento).
ESI-LCMS: m/z para C15H15ClN2O encontrado 275,1 (M+1)+.
Etapa 5
Síntesis de 4-(3-(4-clorobencil)-2,3-dihidropirido[4,3-f][1,4]oxazepin-4(5H)-il)piperidina-1-carboxilato de (S)-terc-butilo (63e)

El compuesto del título se preparó a partir del compuesto 63e (410 mg; 1,492 mmol) de acuerdo con el Procedimiento General VI y se obtuvo como un vidrio (207 mg; 0,451 mmol; 29 % de rendimiento).
ESI-LCMS: m/z para C25H32CW3O3 encontrado 402,2 (M+1-fBu)+, 458,3 (M+1)+.
1H RMN (CDCl3, 400 MHz) 5: 8,21 (s, 1H), 8,16 (d, J = 4,8 Hz, 1H), 7,27 (AA'BB', J = 8,8 Hz, 2H), 7,14 (AA'BB', J = 8,4 Hz, 2H), 6,95 (d, J = 4,8 Hz, 1H), 4,34 (d, J = 17,1 Hz, 1H), 4,20-4,10 (m, 2H), 3,78 (d, J = 17,2 Hz, 1H), 3,51-3,41 (m, 1H), 2,79 (dd, J = 13,5, 6,8 Hz, 1H), 2,72 (dd, J = 13,5, 7,0 Hz, 1H), 2,66-2,47 (m, 3H), 1,65-1,50 (m, 3H), 1,42 (s, 9H), 1,29-1,11 (m, 3H).
Etapa 6
Síntesis de (S)-3-(4-clorobencil)-4-(piperidin-4-il)-2,3,4,5-tetrahidropirido[4,3-f][1,4]oxazepina (63f)

A la solución del compuesto 63e (201 mg; 0,438 mmol) en MeOH (1 ml), se añadió una solución acuosa de 6 M HCl (2 ml) se añadió y la mezcla se agitó durante 16 horas. Después de este tiempo, TLC (DCM/MeOH 10:1) mostró que la reacción se había completado. La mezcla se alcalinizó con 4 N NaOH para ajustar el pH hasta ~14 y el producto se extrajo con DCM (4x 10 ml). Las soluciones orgánicas combinadas se secaron en MgSO4 anhidro y se concentraron. El producto bruto se obtuvo como un aceite (154 mg; 0,430 mmol; 98 % de rendimiento) y se usó en la siguiente etapa sin purificación adicional.
Etapa 7
Síntesis de (S)-5-(4-(3-(4-dorobencil)-2,3-dihidropirido[4,3-f][1,4]oxazepin-4(5H)-il)piperidin-1-il)-4H-1,2,4-triazol-3-amina (63)

El compuesto del título 63 se sintetizó a partir del compuesto 63f (154 mg; 0,430 mmol) de acuerdo con el Procedimiento General VIII. Se purificó por cromatografía de gel de sílice en un sistema de AcOEt/MeOH) y se obtuvo como un sólido blanco (140 mg, 0,318 mmol, 72 % de rendimiento).
ESI-LCMS: m/z para C22H26ClN7O encontrado 220,7 (M+2)2+, 440,2 (M+1)+.
1H RMN (MeOH-d4, 400 MHz) 5: 8,07 (s, 1H), 8,06 (d, J = 5,0 Hz, 1H), 7,27 (AA’BB’, J = 8,7 Hz, 2H), 7,24 (AA’BB’, J = 8,7 Hz, 2H), 7,19 (d, J = 5,0 Hz, 1H), 4,52 (d, J = 17,3 Hz, 1H), 4,29 (dd, J = 13,1, 5,4 Hz, 1H), 4,25 (dd, J = 13,1, 7,4 Hz, 1H), 3,92 (d, J = 17,2 Hz, 1H), 3,67-3,49 (m, 3H), 2,78 (dd, J = 13,4, 6,3 Hz, 1H), 2,74 (dd, J = 13,4, 7,5 Hz, 1H), 2,70-2,44 (m, 3H), 1,69-1,55 (m, 2H), 1,35-1,19 (m, 2H).
EQUIVALENTES
Se considera que la memoria descriptiva anterior es suficiente para hacer posible que un experto en la materia ponga en práctica la invención. Los ejemplos provistos no deben limitar el alcance de la presente invención, dado que se pretende que los ejemplos representen una ilustración de un aspecto de la invención y el alcance de la invención abarca otras realizaciones funcionalmente equivalentes. Las ventajas y los objetivos de la invención no están necesariamente comprendidos en cada realización de la invención.
Claims (73)
1. Un compuesto representado por la fórmula (I),

donde:
W es halo, alquilo(C1-C3), alcoxi(C1-C3) o alquiltio(C1 -C3);
X es un enlace sencillo, -CH2-, -CH2CH2-, -CH=c H-, -C(alquilo(C1-C3))2-, o -C(O)-;
Y es un enlace sencillo, -CH-, -CHCH2-, -CH2CH-, -C=CH-, -CH=C-, -N-, -O-, -OCH2-, -S(O)-, o -S(O)2-;
si Y es un enlace sencillo, -O-, -OCH2-, -S(O)-, o -S(O)2-, entonces R1 está ausente;
R1 es H, OH, alquilo(C1-C6), haloalquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), ariloalquilo(C1-C6), heteroarilalquilo(C1-C6), -C(O)alquilo(C1-C6), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1-C6), -C(O)heteroarilalquilo(C1-C6), -S(O)2alquilo(C1-C6), -S(O)2arilo, -S(O)2heteroarilo, -S(O)2arilalquilo(C1-C6), -S(O)2heteroarilalquilo(C1-C6), -CO2H, -C(O)Oalquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NH2, -C(O)NHOH, -C(O)NHCN, -C(O)NH(alquilo(C1-C6)), -C(O)N(alquilo(C1-C6))2, -C(O)NH(arilalquilo(C1-C6)), -C(O)N(arilalquilo(C1-C6))2, -C(O)NH(arilo), -C(O)N(arilo)(alquilo(C1-C6)), -C(O)N(arilo)2, -C(O)Nalquilo(C1-C6))(arilalquilo(C1-C6)), -C(O)N(arilo)(arilalquilo(C1-C6)), -C(O)NH(haloalquilo(C1-C6)), -C(O)N(haloalquilo(C1-C6))2, -S(O)2NH2, -S(O)2NH(alquilo(C1-C6)), -S(O)2NH(haloalquilo(C1-C6)), -S(O)2NH(arilo), -S(O)2NH(heteroarilalquilo(C1-C6)), -S(O)2NH(heteroarilo), -S(O)2N(alquilo(C1-C6))2, -S(O)2NHC(O)alquilo(C1-C6), -S(O)2NHC(O)haloalquilo(C1-C6), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(C1-C6), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(C1-C6), -NHS(O)2alquilo(C1-C6), -NHS(O)2arilo, -NHS(O)2haloalquilo(C1-C6), -NHS(O)2arilalquilo(C1-C6), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(C1-C6), -NHC(O)(alquilo(C1-C6)), -NHC(O)(haloalquilo(C1-C6)), -NHC(O)(arilo), -NHC(O)(arilalquilo(C1-C6)), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(C1-C6)), -NHC(O)NHalquilo(C1-C6), -NHC(O)NH(arilo), -NHC(O)NH(arilalquilo(C1-C6)), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(C1-C6)), -C(O)NHS(O)2alquilo(C1-C6), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(C1-C6)), -C(O)NHS(O)2(arilalquilo(C1-C6)), -C(O)NHS(O)2heteroarilo, -C(O)NHS(O)2(heteroarilalquilo(C1-C6)), -P(O)(OH)2, -(alquileno(C1-C6))C(O)OH, -(alquileno(C1-C6))C(O)Oalquilo(C1-C6), -NH2, -NHalquilo(C1-C6), -N(alquilo(C1-C6))2, -NC, -CN, -C(S)NH2, -NHC(O)NH2, -C=CH, alquiltio(C1-C6)-, mercaptoalquilo(C1-C6)- o -C(O)heterociclilo;
Z es -CH-, -C(O)-, -C(alquilo(C1-C3))- o -C(=CH2)-;
si Z es -C(O)-, entonces R2 está ausente,
R2 es H, OH, halo, alquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), -CO2H, -C(O)Oalquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NHOH, -C(O)NHCN, -C(O)NH2, -C(O)NH(alquilo(C1-C6)), -C(O)N(alquilo(C1-C6))2, -C(O)NH(arilalquilo(C1-C6)), -C(O)N(arilalquilo(C1-C6))2, -C(O)NH(arilo), -C(O)N(arilo)(alquilo(C1-C6)), -C(O)N(arlo)2, -C(O)Nalquil(C1-C6))(arialquilol(C1-C6)), -C(O)N(aril)(arilalquilo(C1-C6)), -C(O)NH(haloalquilo(C1-C6)), -C(O)N(haloalquilo(C1-C6))2, -S(O)2NH2, -S(O)2NH(alquilo(C1-C6)), -S(O)2NH(haloalquilo(C1-C6)), -S(O)2NH(arilo), -S(O)2NH(heteroarilalquilo(C1-C6)), -S(O)2NH(heteroarilo), -S(O)2NH(heteroarilalquilo(C1-C6)), -S(O)2N(alquilo(C1-C6))2, -S(O)2NHC(O)alquilo(C1-C6), -S(O)2NHC(O)haloalquilo(C1-C6), -S(O)2NHC(O)arilo, -S(O)2NHC(O)arilalquilo(C1-C6), -S(O)2NHC(O)heteroarilo, -S(O)2NHC(O)heteroarilalquilo(C1-C6), -NHS(O)2alquilo(C1-C6), -NHS(O)2arilo, NHS(O)2haloalquilo(C1-C6), -NHS(O)2arilalquilo(C1-C6), -NHS(O)2heteroarilo, -NHS(O)2heteroarilalquilo(C1-C6), -NHC(O)(alquilo(C1-C6)), -NHC(O)(haloalquilo(C1-C6)), -NHC(O)(arilo), -NHC(O)(arilalquilo(C1-C6)), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(C1-C6)), -NHC(O)NHalquilo(C1-C6), -NHC(O)NHarilo, -NHC(O)NH(arilalquilo(C1-C6)), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(C1-C6)), -C(O)NHS(O)2alquilo(C1-C6), -C(O)NHS(O)2arilo, C(O)NHS(O)2(haloalquilo(C1-C6)), -C(O)NHS(O)2(arilalquilo(C1-C6)), -C(O)NHS(O)2heteroarilo, -C(O)NHS(O)2(heteroarilalquilo(C1-C6)), -P(O)(OH)2, arilalquilo(C1-C6), (C3-C7)cicloalquilalquilo(C1-C6)-, -NC, -CN, -C(S)NH2, -NHC(O)NH2, -C=CH, (C1 -C6)alquiltio-, mercaptoalquilo(C1-C6)- o -C(O)heterociclilo;
o R1 y R2, tomados juntos con los átomos que son parte forman un anillo carbocíclico o heterocíclico;
R3 es H, alquilo(C1-C3), alcoxi(C1-C3)alquilo(C1-C3), alquiltio(C1 -C3)alquilo(C1 -C3), haloalquilo(C1-C6), -NC, -CN, -C(S)NH2, -NHC(O)NH2 o -C=CH;
R4 es H, alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-C6), alcoxi(C1-C3)alquilo(C1-C3)- o hidroxialquilo(C1-C4);
R5 es H, halo, -NO2, -CN, alquilo(C1-C6), haloalquilo(C1-C6), -NH2, -NH(alquilo(C1-C6)), -N(alquilo(C1-C6))2, -OH, alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), haloalcoxi(C1-C6), -SH, alquiltio(C1-C3)alquilo(C1-C3), -NC, -C(S)NH2, -NHC(O)NH2 o -C=CH; y
R6 es H, halo, -OH, -NH2 , o -SH; o
R6, tomado junto con el átomo de carbono que lo contiene, representa -C(O)-;
donde:
cualquier aparición de alquilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo o hidroxialquilo está opcionalmente sustituida con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2 , -NH(alquilo(C1-C6 )), -N(alquilo(C1-C6 ))2 , -CN, -NO2 , alquilo(C1-C6 ), haloalquilo(C1-C6 ), alcoxi(C1-C6 ), arilo, heteroarilo, arilalquilo(C1-C6 ), heteroarilalquilo(C1-C6 ), cicloalquilo(C3-C7), heterociclilo, -C(O)OH, -C(O)Oalquilo(C1-C6 ), -C(O)Nh 2 , -C(O)NHalquilo(C1-C6 ) y -C(O)N(alquilo(C1-C6 ))2 ;
o una sal, solvato o tautómero, estereoisómero o polimorfo farmacéuticamente aceptable de estos.
2. El compuesto de la reivindicación 1, donde:
W es halo, alquilo(C1-C3), alcoxi(C1-C3) o alquiltio(C1-C3);
X es un enlace sencillo, -CH2-, -CH2 CH2 -, -CH=c H-, o -C(O)-;
Y es un enlace sencillo, -CH-, -CHCH2-, -CH2 CH-, -C=CH-, -CH=C-, -N-, -O-, -S(O)-, o -S(O)2-;
si Y es un enlace sencillo, -O-, -S(O)-, o -S(O)2-, entonces R1 está ausente;
R1 es H, alquilo(C1-C6 ), haloalquilo(C1-C6 ), alcoxi(C1-C6 ), hidroxialquilo(C1-C6 ), alcoxi(C1-C6 )alquilo(C1-C6 ), arilalquilo(C1-C6 ), heteroarilalquilo(C1-C6 ), -C(O)alquilo(C1-C6 ), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1-C6), -C(O)heteroarilalquilo(C1-C6 ), -S(O)2 alquilo(C1-C6 ), -S(O)2 arilo, -S(O)2 heteroarilo, -S(O)2 arilalquilo(C1-C6 ), -S(O)2 heteroarilalquilo(C1-C6 ), -CO2 H, -C(O)Oalquilo(C1-C6 ), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6 )), -C(O)O(heteroarilalquilo(C1-C6 )), -C(O)NH2 , -C(O)NHOH, -C(O)NHCN, -C(O)NH(alquilo(C1-C6 )), -C(O)N(alquilo(C1-C6 ))2 , -C(O)NH(arilalquilo(C1-C6 )), -C(O)N(arilalquilo(C1-C6 ))2 , -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6 )), -C(O)N(arilo)2 , -C(O)Nalquil(C1-C6 ))(arilalquilo(C1-C6 )), -C(O)N(aril)(arilalquilo(C1-C6 )), -C(O)NH(haloalquilo(C1-C6 )), -C(O)N(haloalquilo(C1-C6 ))2 , -S(O)2 NH2 , -S(O)2 NH(alquilo(C1-C6 )), -S(O)2 NH(haloalquilo(C1-C6 )), -S(O)2 NH(arilo), -S(O)2 NH(heteroarilalquilo(C1-C6 )), -S(O)2 NH(heteroarilo), -S(O)2 N(alquilo(C1-C6 ))2 , -S(O)2 NHC(O)alquilo(C1-C6 ), -S(O)2 NHC(O)haloalquilo(C1-C6 ), -S(O)2 NHC(O)arilo, -S(O)2 NHC(O)arilalquilo(C1-C6 ), -S(O)2 NHC(O)heteroarilo, -S(O)2 NHC(O)heteroarilalquilo(C1-C6), -NHS(O)2 alquilo(C1-C6 ), -NHS(O)2 arilo, -NHS(O)2 haloalquilo(C1-C6 ), -NHS(O)2 arilalquilo(C1-C6 ), -NHS(O)2 heteroarilo, -NHS(O)2 heteroarilalquilo(C1-C6 L -NHC(O)(alquilo(C1-C6 )), -NHC(O)(haloalquilo(C1-C6 )), -NHC(O)(arilo), -NHC(O)(arilalquilo(C1-C6 )), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(C1-C6 )), -NHC(O)NHalquilo(C1-C6 ), -NHC(O)NH(arilo), -NHC(O)NH(arilalquilo(C1-C6 )), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(C1-C6 )), -C(O)NHS(O)2 alquilo(C1-C6 ), -C(O)NHS(O)2 arilo, C(O)NHS(O)2 (haloalquilo(C1-C6 )), -C(O)NHS(O)2 (arilalquilo(C1-C6 )), -C(O)NHS(O)2 heteroarilo, -C(O)NHS(O)2 (heteroarilalquilo(C1-C6 )), -P(O)(OH)2 , -(alquileno(C1-C6 ))C(O)OH, -(alquileno(C1-C6 ))C(O)Oalquilo(C1-C6 ), -NH2 , -NHalquilo(C1 -C6 ), -N(alquilo(C1-C6 ))2 , -NC, -CN, -C(S)NH2 , -NHC(O)NH2 , -CECH, alquiltio(C1-C6 )-, mercaptoalquilo(C1-C6 )-, o -C(O)heterociclilo;
Z es -CH-, -C(O)-, o -C(alquilo(C1-C3))-;
si Z es -C(O)-, entonces R2 está ausente,
R2 is H, halo, alquilo(C1-C6 ), alcoxi(C1-C6 ), hidroxialquilo(C1-C6 ), alquil(C1-C6 )alquilo(C1-C6 ), -CO2 H, -C(O)Oalquilo(C1-C6 ), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6 )), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NHOH, -C(O)NHCN, -C(O)NH2 , -C(O)NH(alquilo(C1-C6 )), -C(O)N(alquilo(C1-C6 ))2 , -C(O)NH(arilalquilo(C1-C6 )), -C(O)N(arilalquilo(C1-C6 ))2 , -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6 )), -C(O)N(arilo)2 , -C(O)Nalquil(C1-C6 ))(arilalquilo(C1-C6 )), -C(O)N(aril)(arilalquilo(C1-C6 )), -C(O)NH(haloalquilo(C1-C6 )), -C(O)N(haloalquilo(C1-C6 ))2 , -S(O)2 NH2 , -S(O)2 NH(alquilo(C1-C6 )), -S(O)2 NH(haloalquilo(C1-C6 )), -S(O)2 NH(arilo), -S(O)2 NH(heteroarilalquilo(C1-C6 )), -S(O)2 NH(heteroarilo), -S(O)2 NH(heteroarilalquilo(C1-C6 ) ), -S(O)2 N(alquilo(C1-C6 ))2 , -S(O)2 NHC(O)alquilo(C1-C6 ), -S(O)2 NHC(O)haloalquilo(C1-C6 ), -S(O)2 NHC(O)arilo, -S(O)2 NHC(O)arilalquilo(C1-C6 ), -S(O)2 NHC(O)heteroarilo, -S(O)2 NHC(O)heteroarilalquilo(C1-C6 ), -NHS(O)2 alquilo(C1-C6 ), -NHS(O)2 arilo, NHS(O)2 haloalquilo(C1-C6 ), -NHS(O)2 arilalquilo(C1-C6 ), -NHS(O)2 heteroarilo, -NHS(O)2 heteroarilalquilo(C1-C6 ), -NHC(O)(alquilo(C1-C6 )), -NHC(O)(haloalquilo(C1-C6 )), -NHC(O)(arilo), -NHC(O)(arilalquilo(C1-C6 )), -NHC(O)(heteroarilo), -NHC(O)(heteroarilalquilo(C1-C6 )), -NHC(O)NHalquilo(C1-C6 ), -NHC(O)NHarilo, -NHC(O)NH(arilalquilo(C1-C6 )), -NHC(O)NH(heteroarilo), -NHC(O)NH(heteroarilalquilo(C1-C6 )), -C(O)NHS(O)2 alquilo(C1-C6 ), -C(O)NHS(O)2 arilo, C(O)NHS(O)2 (haloalquilo(C1-C6 )), -C(O)NHS(O)2 (arilalquilo(C1-C6 )), -C(O)NHS(O)2 heteroarilo, -C(O)NHs(O)2 (heteroarilalquilo(C1-C6 )), -P(O)(OH)2 , arilalquilo(C1-C6 ), (C3-C7)cicloalquilalquilo(C1-C6)-, -NC, -CN, -c (s )NH2 , -NHC(O)NH2 , -CeCH, aiquiltio(C1-C6 )-, mercaptoalquilo(C1-C6 )-, o -C(O)heterociclilo;
R3 es H, alquilo(C1-C3), alcoxi(C1-C3)alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-C6 ), -NC, -CN, -C(S)NH2 , -NHC(O)NH2 o -CECH;
R4 es H, alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-C6 ), alcoxi(C1-C3)alquilo(C1-C3)- o hidroxialquilo(C1-C4);
R5 es H, halo, -NO2 , -CN, alquilo(C1-C6 ), haloalquilo(C1-C6 ), -NH2 , -NH(alquilo(C1-C6 )), -N(alquilo(C1-C6 ))2 , -OH, alcoxi(C1-C6 ), hidroxialquilo(C1-C6 ), alcoxi(C1-C6 )alquilo(C1-C6 ), haloalcoxi(C1-C6 ), -SH, alquiltio(C1-C3)alquilo(C1-C3 ), -NC, -C(S)NH2 , -NHC(O)NH2 o -CECH; y
R6 es H, halo, -OH, -NH2 , o -SH; o
R6, tomado junto con el átomo de carbono que lo contiene, representa -C(O)-;
donde:
cualquier aparición de alquilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo o hidroxialquilo está opcionalmente sustituida con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2 , -NH(alquilo(C1-C6 )), -N(alquilo(C1-C6 ))2 , -CN, -NO2 , alquilo(C1-C6 ), haloalquilo(C1-C6 ), alcoxi(C1-C6 ), arilo, heteroarilo, arilalquilo(C1-C6 ), heteroarilalquilo(C1-C6 ), cicloalquilo(C3-C7), heterociclilo, -C(O)OH, -C(O)Oalquilo(C1-C6 ), -C(O)Nh 2 , -C(O)NHalquilo(C1-C6 ) y -C(O)N(alquilo(C1-C6 ))2 ;
3. El compuesto de la reivindicación 1 o 2, donde W es fluoro, cloro, bromo, metilo o metoxi.
4. El compuesto de la reivindicación 3, donde W es cloro o bromo.
5. El compuesto de la reivindicación 4, donde W es cloro.
6. El compuesto de una cualquiera de las reivindicaciones 1-5, donde R6 es H o -OH.
7. El compuesto de una cualquiera de las reivindicaciones 1-5, donde R6 es H.
8. El compuesto de una cualquiera de las reivindicaciones 1-7, donde X es un enlace sencillo, -CH2-, o -C(O)-.
9. El compuesto de una cualquiera de las reivindicaciones 1-7, donde X es -CH2-.
10. El compuesto de una cualquiera de las reivindicaciones 1-9, donde Y es un enlace sencillo, -CH-, -N-, -O-, o -S(O)2-.
11. El compuesto de una cualquiera de las reivindicaciones 1-9, donde Y es un enlace sencillo, -O-, o -S(O)2-.
12. El compuesto de una cualquiera de las reivindicaciones 1-9, donde Y es un enlace sencillo.
13. El compuesto de una cualquiera de las reivindicaciones 1-9, donde Y es -O-.
14. El compuesto de una cualquiera de las reivindicaciones 1-9, donde Y es -CH- o -N-.
15. El compuesto de una cualquiera de las reivindicaciones 1-9, donde Y es -CH-.
16. El compuesto de una cualquiera de las reivindicaciones 1-9, donde Y es N.
17. El compuesto de una cualquiera de las reivindicaciones 14-16, donde R1 es H, alquilo(C1-C6 ), haloalquilo(C1-C6 ), alcoxi(C1-C6 ), hidroxialquilo(C1-C6 ), alcoxi(C1-C6 )alquilo(C1-C6 ), arilalquilo(C1-C6 ), heteroarilalquilo(C1-C6 ), -C(O)alquilo(C1-C6 ), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1-C6 ), -C(O)heteroarilalquilo(C1-C6 ), -S(O)2 alquilo(C1-C6 ), -S(O)2 arilo, -S(O)2 heteroarilo, -S(O)2 arilalquilo(C1-C6 ), -S(O)2 heteroarilalquilo(C1-C6 ), -CO2 H, -C(O)Oalquilo(C1-C6 ), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6 )), -C(O)O(heteroarilalquilo(C1-C6 )), -C(O)NH2 , -C(O)NHOH, -C(O)NHCN, -C(O)NH(alquilo(C1-C6 )), -C(O)N(alquilo(C1-C6 ))2 , -C(O)NH(arilalquilo(C1-C6 )), -C(O)N(arilalquilo(C1-C6 ))2 , -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6 )), -C(O)N(arilo)2 , -C(O)Nalquil(C1-C6 ))(arilalquilo(C1-C6 )), -C(O)N(aril)(arilalquilo(C1-C6 )), -(alquileno(C1-C6 ))C(O)OH, -(alquilen(C1-C6 ))C(O)Oalquilo(C1-C6), -NH2 , -NHalquilo(C1-C6 ), -N(alquilo(C1-C6 ))2 , o -C(O)heterociclilo.
18. El compuesto de cualquiera de las reivindicaciones 14-16, donde R1 es H, alquilo(C1-C6 ), alcoxi(C1-C6 ), hidroxialquilo(C1-C6 ), alcoxi(C1-C6 )alquilo(C1-C6 ), -C(O)alquilo(C1-C6 ), -C(O)arilo, -C(O)heteroarilo, -C(O)arilalquilo(C1 -C6), -C(O)heteroarilalquilo(C1-C6 ), -S(O)2 alquilo(C1-C6 ), -S(O)2 arilo, -C(O)Oalquilo(C1-C6 ), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6 )), -C(O)O(heteroarilalquilo(C1-C6 )), -(alquileno(C1-C6 ))C(O)OH, -(alquileno(C1-C6 ))C(O)Oalquilo(C1-C6 ) o -C(O)heterociclilo.
19. El compuesto de cualquiera de las reivindicaciones 14-16, donde R1 es H, alquilo(C1-C6 ), alcoxi(C1-C6 ), -C(O)alquilo(C1-C6 ), -S(O)2 alquilo(C1-C6 ), -S(O)2 arilo, -C(O)Oalquilo(C1-C6 ), -(alquileno(C1-C6 ))C(O)OH, o -(alquilen(C1-C6 ))C(O)Oalquilo(C1-C6 ).
20. El compuesto de una cualquiera de las reivindicaciones 14-16, donde R1 es H o alquilo(C1-C6 ).
21. El compuesto de una cualquiera de las reivindicaciones 14-16, donde R1 es -C(O)heterociclilo seleccionado del grupo que consiste en

154

22. El compuesto de una cualquiera de las reivindicaciones 14-16, donde R1 es -CH2CO2H o -CH2C(O)Oalquilo(C1-C6).
23. El compuesto de una cualquiera de las reivindicaciones 1-22, donde Z es -C(O)-.
24. El compuesto de una cualquiera de las reivindicaciones 1-22, donde Z es -C(alquilo(C1-C3))-.
25. El compuesto de una cualquiera de las reivindicaciones 1-22, donde Z es -CH-.
26. El compuesto de la reivindicación 24 o 25, donde R2 es H, halo, alquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), -CO2H, -C(O)Oalquilo(C1-C6), -C(O)O(arilo), -C(O)O(heteroarilo), -C(O)O(arilalquilo(C1-C6)), -C(O)O(heteroarilalquilo(C1-C6)), -C(O)NH2, -C(O)NH(alquilo(C1-C6)), -C(O)N(alquilo(C1-C6))2, -C(O)NH(arilalquilo(C1-C6)), -C(O)N(arilalquilo(C1-C6))2, -C(O)NH(arilo), -C(O)N(aril)(alquilo(C1-C6)), -C(O)N(arilo)2, -C(O)Nalquil(C1-C6))(arilalquilo(C1-C6)), -C(O)N(aril)(arilalquilo(C1-C6)), -C(O)NH(haloalquilo(C1-C6)), -C(O)N(haloalquilo(C1-C6))2, arilalquilo(C1-C6), cicloalquil(C3-C7)alquilo(C1-C6)-, o -C(O)heterociclilo.
27. El compuesto de la reivindicación 24 o 25, donde R2 es H, halo, alquilo(C1-C6), alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), -CO2H, -C(O)Oalquilo(C1-C6), -C(O)NH2, -C(O)NH(alquilo(C1-C6)), arilalquilo(C1-C6), o cicloalquil(C3-C7)alquilo(C1-C6)-.
28. El compuesto de la reivindicación 24 o 25, donde R2 es arilalquilo(C1-C6) o cicloalquil(C3-C7)alquilo(C1-C6)-, opcionalmente sustituido en cualquier posición por -OH, halo, -NH2, alquilo(C1-C6), haloalquilo(C1-C6) o alcoxi(C1-C6).
29. El compuesto de la reivindicación 24 o 25, donde R2 es H, alquilo(C1-C6) o alcoxi(C1-C6)alquilo(C1-C6).
30. El compuesto de la reivindicación 24 o 25, donde R2 es -C(O)heterociclilo seleccionado del grupo que consiste en

31. El compuesto de la reivindicación 24 o 25, donde R2 es halo(1,4-fenilen)metilo.
32. El compuesto de una cualquiera de las reivindicaciones 1-31, donde R3 es H o alquilo(C1-C3).
33. El compuesto de una cualquiera de las reivindicaciones 1-31, donde R3 es H.
34. El compuesto de una cualquiera de las reivindicaciones 1-33, donde R4 es H o alquilo(C1-C3).
35. El compuesto de una cualquiera de las reivindicaciones 1-33, donde R4 es H.
36. El compuesto de una cualquiera de las reivindicaciones 1-35, donde R5 es H o alquilo(C1-C3).
37. El compuesto de una cualquiera de las reivindicaciones 1-35, donde R5 es H.
38. El compuesto de una cualquiera de las reivindicaciones 1-37, representado por la fórmula (I'):

39. El compuesto de la reivindicación 1, donde:
W es bromo o cloro;
X es un enlace sencillo, -CH2-, o -C(O)-;
Y es un enlace sencillo, -CH-, -N-, -O-, o -S(O)2-;
si Y es un enlace sencillo, -O-, o -S(O)2-, entonces R1 está ausente;
R1 es H, metilo, isobutilo, metoxi, acetilo, metoxicarbonilo, metanosulfonilo, p-toluenosulfonilo, metoxicarbonilmetilo o carboximetilo;
Z es -CH-, -C(O)-, o -C(CHa)-;
si Z es -C(O)-, entonces R2 está ausente,
R2 es H, metilo, etilo, isopropilo, isobutilo, -C(O)NH2, -C(O)NHMe, -CH2OH, -CH2OCH3, -CH2OCH2CH3, -C(CH3)2OH, -C(CH3)2OCH3, -CO2H, -CO2CH2CH3, -OCH3, -F, -CH2-(p-clorofenilo), o -CH2-ciclohexilo;
R3, R4 y R5 son cada uno H; y
R6 es H u OH.
40. Un compuesto representado por la fórmula (II),

donde:
W es halo, alquilo(C1-C3), alcoxi(C1-C3) o alquiltio(C1 -C3);
X es un enlace sencillo, -CH2-, -CH2CH2-, -CH=CH-, -C(alquilo(C1-C3))2-, -C(O)-, -CH2O-, -CH2NH-, o -CH2N(alquilo(C1-C3))-, donde cuando X es -CH2O-, -CH2NH-, o -CH2N(alquilo(C1-C3))-, entonces el átomo de O o N está unido covalentemente con el Anillo A;
el Anillo A representa un anillo arilo o heteroarilo opcionalmente sustituido;
R3 es H, alquilo(C1-C3), alcoxi(C1-C3)alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-C6), -NC, -CN, -C(S)NH2, -NHC(O)NH2 o -C=CH;
R4 es H, alquilo(C1-C3), alquiltio(C1-C3)alquilo(C1-C3), haloalquilo(C1-C6), alcoxi(C1-C3)alquilo(C1-C3)- o hidroxialquilo(C1-C4);
R5 es H, halo, -NO2, -CN, alquilo(C1-C6), haloalquilo(C1-C6), -NH2, -NH(alquilo(C1-C6)), -N(alquilo(C1-C6))2, -OH, alcoxi(C1-C6), hidroxialquilo(C1-C6), alcoxi(C1-C6)alquilo(C1-C6), haloalcoxi(C1-C6), -SH, alquiltio(C1-C3)alquilo(C1-C3), -NC, -C(S)NH2, -NHC(O)NH2 o -C=CH; y
R6 es H, halo, -OH, -NH2, o -SH; o
R6, tomado junto con el átomo de carbono que lo contiene, representa -C(O)-;
donde:
cualquier aparición de alquilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alquileno, heterociclilo, cicloalquilo, alcoxi, alquiltio, haloalquilo o hidroxialquilo está opcionalmente sustituida con uno o más sustituyentes que se seleccionan independientemente del grupo que consiste en -OH, halo, -NH2, -NH(alquilo(C1-C6)), -N(alquilo(C1-C6))2, -CN, -NO2, alquilo(C1-C6), haloalquilo(C1-C6), alcoxi(C1-C6), arilo, heteroarilo, arilalquilo(C1-C6), heteroarilalquilo(C1-C6), cicloalquilo(C3-C7), heterociclilo, -C(O)OH, -C(O)Oalquilo(C1-C6), -C(O)NH2, -C(O)NHalquilo(C1-C6) y -C(O)N(alquilo(C1-C6))2;
o una sal, solvato, tautómero, estereoisómero o polimorfo farmacéuticamente aceptable de estos.
41. El compuesto de la reivindicación 40, donde W es fluoro, cloro, bromo, metilo o metoxi.
42. El compuesto de la reivindicación 41, donde W es cloro.
43. El compuesto de una cualquiera de las reivindicaciones 40-42, donde R6 es H.
44. El compuesto de una cualquiera de las reivindicaciones 40-43, donde X es -CH2O-, -CH2NH-, o -CH2N(alquilo(Ci-C3))-.
45. El compuesto de la reivindicación 44, donde X es -CH2O- o -CH2N(CH3)-.
46. El compuesto de una cualquiera de las reivindicaciones 40-45, donde R3 es H.
47. El compuesto de una cualquiera de las reivindicaciones 40-46, donde R4 es H.
48. El compuesto de una cualquiera de las reivindicaciones 40-47, donde R5 es H.
49. El compuesto de una cualquiera de las reivindicaciones 40-48, donde el Anillo A representa un anillo heteroaril
50. El compuesto de la reivindicación 49, donde el Anillo A representa un anillo de piridilo.
51. El compuesto de una cualquiera de las reivindicaciones 40-48, donde el Anillo A representa un anillo de fenilo opcionalmente sustituido.
52. El compuesto de la reivindicación 51, donde el Anillo A representa un anillo de fenilo sustituido con uno o más apariciones de halo.
53. El compuesto de la reivindicación 52, donde el Anillo A representa un anillo de fenilo sustituido con uno o más apariciones de cloro.
54. El compuesto de la reivindicación 1 o reivindicación 40 representado por cualquiera de las siguientes fórmulas estructurales:




Ċ

55. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de una cualquiera de las reivindicaciones 1-54; y un portador farmacéuticamente aceptable.
56. La composición farmacéutica de la reivindicación 55, que comprende además una cantidad terapéuticamente eficaz de un agente terapéutico que se selecciona de un grupo que consiste en esteroides, estabilizadores de membrana, inhibidores de 5LO, inhibidores del receptor y la síntesis de leucotrieno, inhibidores de la conmutación del isotipo IgE o la síntesis de IgE, conmutación del isotipo IgG o síntesis de IgG, p-agonistas, inhibidores de triptasa, ácido acetilsalicílico, inhibidores de la COX, metotrexato, fármacos anti-TNF, rituxina, y otros agentes dirigidos a las células B, agentes dirigidos a TNF, inhibidores de PD4, inhibidores de p38, inhibidores de PDE4 y antihistaminas.
57. Compuesto de acuerdo con una cualquiera de las reivindicaciones 1-54 para su uso como un inhibidor de la quitinasa de mamífero ácida en una célula o un tejido.
58. Compuesto de acuerdo con una cualquiera de las reivindicaciones 1-54 para su uso como inhibidor de la quitotriosidasa en una célula o un tejido.
59. Compuesto de acuerdo con una cualquiera de las reivindicaciones 1-54, o una composición farmacéutica de acuerdo con la reivindicación 55 para su uso en el tratamiento o la prevención de una enfermedad, trastorno o afección asociada con la actividad o expresión aberrante de la quitinasa de mamífero ácida.
60. Compuesto de acuerdo con una cualquiera de las reivindicaciones 1-54, o una composición farmacéutica de acuerdo con la reivindicación 55 para su uso en el tratamiento o la prevención de una enfermedad, trastorno o afección asociada con la actividad o expresión aberrante de la quitotriosidasa.
61. Compuestos para su uso de acuerdo con la reivindicación 59 o 60, donde la enfermedad, trastorno o afección se selecciona del grupo que consiste en enfermedades alérgicas, enfermedades inflamatorias agudas y crónicas, enfermedades autoinmunes, enfermedades dentales, enfermedades neurológicas, enfermedades metabólicas, enfermedades hepáticas, enfermedades renales, enfermedades cutáneas, síndrome de ovario poliquístico, endometriosis, trastornos fibróticos, enfermedades de almacenamiento y cáncer.
62. Compuestos para su uso de la reivindicación 61, donde la enfermedad, trastorno o afección es una enfermedad alérgica que se selecciona del grupo que consiste en asma, rinitis alérgica, rinitis alérgica estacional, rinosinusitis crónica con o sin pólipos nasales, conjuntivitis, keratoconjuntivitis, conjuntivitis alérgica estacional, síndrome del ojo seco, esofagitis eosinofílica, enfermedad celíaca, alergias a los alimentos, síndrome del intestino irritable, enfermedad del intestino irritable, eczema atópico, dermatitis atópica, dermatitis de contacto alérgica, otitis eosinofílica media, neumonía eosinofílica y enfermedad mediada por IgG4.
63. Compuestos para su uso de la reivindicación 61, donde la enfermedad, trastorno o afección es una enfermedad aguda o crónica que se selecciona del grupo que consiste en enfermedades fúngicas, infecciones parasíticas, enfermedad celíaca, colitis microscópica, enfermedad pulmonar obstructiva crónica (EPOC), fibrosis pulmonar idiopática, enfermedades pulmonares intersticiales, fibrosis quística, síndrome de Hermansky-Pudlak y enfermedad de Alzheimer.
64. Compuestos para su uso de la reivindicación 61, donde la enfermedad, trastorno o afección es un trastorno autoinmune que se selecciona del grupo que consiste en enfermedad inflamatoria intestinal, colitis ulcerativa, enfermedad de Crohn, artritis reumatoide, osteoartritis, psoriasis, escleroderma, esclerosis múltiple, síndrome de Sjogren, aterosclerosis y sarcoidosis.
65. Compuestos para su uso de la reivindicación 61, donde la enfermedad, trastorno o afección es periodontitis.
66. Compuestos para su uso de la reivindicación 61, donde la enfermedad, trastorno o afección es una enfermedad metabólica que se selecciona del grupo que consiste en diabetes mellitus insulinodependiente y diabetes mellitus no insulinodependiente.
67. Compuestos para su uso de la reivindicación 61, donde la enfermedad, trastorno o afección es una enfermedad hepática que se selecciona del grupo que consiste en enfermedad del hígado graso no alcohólico, esteatohepatitis no alcohólica, cirrosis y fibrosis inducida por el virus de la hepatitis C y fibrosis alcohólica.
68. Compuestos para su uso de la reivindicación 61, donde la enfermedad es un cáncer que se selecciona del grupo que consiste en glioblastoma, cáncer de mama, cáncer de colon, cáncer pulmonar primario y metastásico, mesotelioma, osteosarcoma, melanoma maligno, cáncer de ovario, cáncer de cuello de útero, cáncer de próstata, cáncer de hígado, cáncer gástrico, cáncer renal metastásico, leucemia y linfoma.
69. Compuestos para su uso de la reivindicación 61, donde la enfermedad es una enfermedad renal que se selecciona del grupo que consiste en nefropatía (por ejemplo, nefropatía diabética), glomerulosclerosis segmentaria y focal, fibrosis tubulointersticial, fibrosis postrasplante y fibrosis retroperitoneal (enfermedad de Ormond).
70. Compuestos para su uso de la reivindicación 61, donde la enfermedad es un trastorno fibrótico.
71. Compuestos para su uso de la reivindicación 70, donde el trastorno fibrótico es fibrosis pulmonar idiopática (IPF).
72. Compuestos para su uso de la reivindicación 61, donde la enfermedad es una enfermedad de almacenamiento que se selecciona del grupo que consiste en enfermedad de Gaucher, enfermedad de Fabry, trastornos de almacenamiento lisosomal, enfermedad de Niemann-Pick, cisteinosis nefropática y globotiaosilceramidosis ligada a X.
73. Compuesto de acuerdo con una cualquiera de las reivindicaciones 1-54 para su uso como un inhibidor de la quitotriosidasa y quitinasa de mamífero ácida en una célula o un tejido.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562214299P | 2015-09-04 | 2015-09-04 | |
| PL415078A PL415078A1 (pl) | 2015-09-04 | 2015-12-04 | Podstawione aminotriazole przydatne jako inhibitory kwaśnej chitynazy ssaków |
| PCT/IB2016/055269 WO2017037670A1 (en) | 2015-09-04 | 2016-09-02 | Substituted amino triazoles useful as human chitinase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2717181T3 true ES2717181T3 (es) | 2019-06-19 |
Family
ID=58190067
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16767368T Active ES2717181T3 (es) | 2015-09-04 | 2016-09-02 | Amino triazoles sustituidos útiles como inhibidores de la quitinasa humana |
Country Status (28)
| Country | Link |
|---|---|
| US (3) | US9944624B2 (es) |
| EP (1) | EP3344616B1 (es) |
| JP (2) | JP6859325B2 (es) |
| KR (1) | KR20180080190A (es) |
| CN (1) | CN108290863B (es) |
| AU (1) | AU2016314345B2 (es) |
| CA (1) | CA2997382C (es) |
| CL (1) | CL2018000564A1 (es) |
| CO (1) | CO2018003484A2 (es) |
| CY (1) | CY1121432T1 (es) |
| DK (1) | DK3344616T3 (es) |
| ES (1) | ES2717181T3 (es) |
| HK (1) | HK1249510B (es) |
| HR (1) | HRP20190223T1 (es) |
| HU (1) | HUE042726T2 (es) |
| IL (1) | IL257545B (es) |
| LT (1) | LT3344616T (es) |
| ME (1) | ME03308B (es) |
| MX (1) | MX368674B (es) |
| PE (1) | PE20181048A1 (es) |
| PH (1) | PH12018500286A1 (es) |
| PL (2) | PL415078A1 (es) |
| PT (1) | PT3344616T (es) |
| SA (1) | SA518391033B1 (es) |
| SI (1) | SI3344616T1 (es) |
| TR (1) | TR201903826T4 (es) |
| WO (1) | WO2017037670A1 (es) |
| ZA (1) | ZA201801978B (es) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ702334A (en) | 2012-06-13 | 2016-11-25 | Hoffmann La Roche | New diazaspirocycloalkane and azaspirocycloalkane |
| DK2900669T3 (da) | 2012-09-25 | 2019-11-04 | Hoffmann La Roche | Hexahydropyrrolo[3,4-C]pyrrolderivater og relaterede forbindelser som autotaxin (ATX)-inhibitorer og som inhibitorer af lysophosphatidsyre (LPA)-produktion til behandling af f.eks. nyresygdomme |
| AR095079A1 (es) | 2013-03-12 | 2015-09-16 | Hoffmann La Roche | Derivados de octahidro-pirrolo[3,4-c]-pirrol y piridina-fenilo |
| WO2015078803A1 (en) | 2013-11-26 | 2015-06-04 | F. Hoffmann-La Roche Ag | NEW OCTAHYDRO-CYCLOBUTA [1,2-c;3,4-c']DIPYRROL-2-YL |
| JP6554481B2 (ja) | 2014-03-26 | 2019-07-31 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | オートタキシン(atx)及びリゾホスファチジン酸(lpa)産生の阻害剤としての縮合[1,4]ジアゼピン化合物 |
| AU2015238537B2 (en) | 2014-03-26 | 2019-08-01 | F. Hoffmann-La Roche Ag | Bicyclic compounds as autotaxin (ATX) and lysophosphatidic acid (LPA) production inhibitors |
| MA41898A (fr) | 2015-04-10 | 2018-02-13 | Hoffmann La Roche | Dérivés de quinazolinone bicyclique |
| PL415078A1 (pl) | 2015-09-04 | 2017-03-13 | Oncoarendi Therapeutics Spółka Z Ograniczoną Odpowiedzialnością | Podstawione aminotriazole przydatne jako inhibitory kwaśnej chitynazy ssaków |
| RU2746481C1 (ru) | 2015-09-04 | 2021-04-14 | Ф. Хоффманн-Ля Рош Аг | Феноксиметильные производные |
| RU2018112230A (ru) | 2015-09-24 | 2019-10-30 | Ф. Хоффманн-Ля Рош Аг | Бициклические соединения в качестве ингибиторов atx |
| KR20180054634A (ko) | 2015-09-24 | 2018-05-24 | 에프. 호프만-라 로슈 아게 | 이중 오토탁신(atx)/탄산 무수화효소(ca) 억제제로서 신규한 이환형 화합물 |
| MX2018002217A (es) | 2015-09-24 | 2018-03-23 | Hoffmann La Roche | Nuevos compuestos biciclicos como inhibidores de autotaxina (atx). |
| RU2725138C2 (ru) | 2015-09-24 | 2020-06-30 | Ф. Хоффманн-Ля Рош Аг | Новые бициклические соединения в качестве двойных ингибиторов аутотаксина (atx)/карбоангидразы (ca) |
| JP7090099B2 (ja) | 2017-03-16 | 2022-06-23 | エフ.ホフマン-ラ ロシュ アーゲー | Atxインヒビターとしての新規二環式化合物 |
| WO2018167001A1 (en) | 2017-03-16 | 2018-09-20 | F. Hoffmann-La Roche Ag | Heterocyclic compounds useful as dual atx/ca inhibitors |
| CN109503449A (zh) * | 2018-12-17 | 2019-03-22 | 天津药明康德新药开发有限公司 | 一种3-硝基吖丁啶-1-甲酸叔丁酯的合成方法 |
| KR20220042379A (ko) | 2019-07-15 | 2022-04-05 | 온코아렌디 테라퓨틱스 에스.에이. | 키티나제 억제제로서 유용한 치환된 아미노 트리아졸 |
| TW202237594A (zh) | 2019-09-25 | 2022-10-01 | 波蘭商昂科艾倫迪治療法股份公司 | 製造5-(4-((2s,5s)-5-(4-氯芐基)-2-甲基嗎啉基)哌啶-1-基)-1h-1,2,4-三唑-3-胺的方法 |
| EP3854789A1 (en) | 2020-01-22 | 2021-07-28 | Cygnet Biosciences B.V. | Macrocyclic compounds useful as chitinase inhibitors |
| CN114057714B (zh) * | 2021-11-22 | 2023-07-14 | 苏州健雄职业技术学院 | 杂环基取代的氨基三唑的衍生物及其应用 |
| EP4269400A1 (en) * | 2022-04-29 | 2023-11-01 | Molecure SA | Ykl-40 inhibitors and their therapeutic applications |
| CN114957098B (zh) * | 2022-06-02 | 2024-03-29 | 和鼎(南京)医药技术有限公司 | 一种制备喷他佐辛中间体的方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2082951C (en) | 1991-03-15 | 1999-12-21 | Robert M. Platz | Pulmonary administration of granulocyte colony stimulating factor |
| US5451569A (en) | 1994-04-19 | 1995-09-19 | Hong Kong University Of Science And Technology R & D Corporation Limited | Pulmonary drug delivery system |
| SG11201605757SA (en) * | 2013-12-20 | 2016-08-30 | Inst Drug Discovery | Substituted amino triazoles, and methods using same |
| PL415078A1 (pl) | 2015-09-04 | 2017-03-13 | Oncoarendi Therapeutics Spółka Z Ograniczoną Odpowiedzialnością | Podstawione aminotriazole przydatne jako inhibitory kwaśnej chitynazy ssaków |
-
2015
- 2015-12-04 PL PL415078A patent/PL415078A1/pl unknown
-
2016
- 2016-09-02 PT PT16767368T patent/PT3344616T/pt unknown
- 2016-09-02 EP EP16767368.0A patent/EP3344616B1/en active Active
- 2016-09-02 US US15/255,226 patent/US9944624B2/en active Active
- 2016-09-02 KR KR1020187009490A patent/KR20180080190A/ko not_active Application Discontinuation
- 2016-09-02 WO PCT/IB2016/055269 patent/WO2017037670A1/en active Application Filing
- 2016-09-02 CA CA2997382A patent/CA2997382C/en active Active
- 2016-09-02 LT LTEP16767368.0T patent/LT3344616T/lt unknown
- 2016-09-02 TR TR2019/03826T patent/TR201903826T4/tr unknown
- 2016-09-02 HU HUE16767368A patent/HUE042726T2/hu unknown
- 2016-09-02 SI SI201630168T patent/SI3344616T1/sl unknown
- 2016-09-02 AU AU2016314345A patent/AU2016314345B2/en active Active
- 2016-09-02 ME MEP-2019-33A patent/ME03308B/me unknown
- 2016-09-02 JP JP2018512339A patent/JP6859325B2/ja active Active
- 2016-09-02 MX MX2018002659A patent/MX368674B/es active IP Right Grant
- 2016-09-02 DK DK16767368.0T patent/DK3344616T3/en active
- 2016-09-02 PL PL16767368T patent/PL3344616T3/pl unknown
- 2016-09-02 ES ES16767368T patent/ES2717181T3/es active Active
- 2016-09-02 PE PE2018000314A patent/PE20181048A1/es unknown
- 2016-09-02 CN CN201680062288.2A patent/CN108290863B/zh active Active
-
2018
- 2018-02-08 PH PH12018500286A patent/PH12018500286A1/en unknown
- 2018-02-15 IL IL257545A patent/IL257545B/en active IP Right Grant
- 2018-02-28 US US15/908,249 patent/US10208020B2/en active Active
- 2018-02-28 SA SA518391033A patent/SA518391033B1/ar unknown
- 2018-03-01 CL CL2018000564A patent/CL2018000564A1/es unknown
- 2018-03-26 ZA ZA2018/01978A patent/ZA201801978B/en unknown
- 2018-03-29 CO CONC2018/0003484A patent/CO2018003484A2/es unknown
- 2018-07-12 HK HK18109046.5A patent/HK1249510B/zh unknown
-
2019
- 2019-02-04 HR HRP20190223TT patent/HRP20190223T1/hr unknown
- 2019-02-15 US US16/277,321 patent/US10538508B2/en active Active
- 2019-03-15 CY CY20191100310T patent/CY1121432T1/el unknown
-
2021
- 2021-03-25 JP JP2021050972A patent/JP2021100966A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2717181T3 (es) | Amino triazoles sustituidos útiles como inhibidores de la quitinasa humana | |
| ES2718552T3 (es) | Compuestos de heteroarilo o arilo bicíclicos fusionados y su uso como inhibidores de IRAK4 | |
| JP2020164542A (ja) | Jak阻害剤である5−クロロ−2−ジフルオロメトキシフェニルピラゾロピリミジン化合物 | |
| ES2834462T3 (es) | Compuestos heterocíclicos bicíclicos y sus usos en terapia | |
| KR20070007867A (ko) | 선택된 cgrp 길항제, 이의 제조방법 및 약제로서의이의 용도 | |
| JP2020515575A (ja) | ヘテロ環化合物 | |
| JPH11508553A (ja) | ピペリジン及びモルホリン誘導体並びに治療剤としてのそれらの使用 | |
| MX2014011373A (es) | Inhibidores de diacilglicerol aciltransferasa 2. | |
| KR20210040085A (ko) | Tlr7/8 안타고니스트 및 이의 용도 | |
| EA015529B1 (ru) | 3-(имидазолил)пиразоло[3,4-b]пиридины | |
| ES2247408T3 (es) | 1-arilpiperazinas 2-sustituidas como antangonistas de taquiquinina y/o inhibidores de la recaptacion de serotonina. | |
| US11638707B2 (en) | Substituted amino triazoles useful as chitinase inhibitors | |
| ES2310295T3 (es) | Beta-lactamas para el tratamiento de trastornos del snc. | |
| US20230348516A1 (en) | Ykl-40 inhibitors and their therapeutic applications | |
| JP2003313173A (ja) | ピペラジン誘導体 | |
| DE102004028751A1 (de) | Ausgewählte CGRP-Antagonisten, Verfahren zu deren Herstellung sowie deren Verwendung als Arzneimittel |