ES2697902T3 - Derivados de feniltriazol sustituido con hidroxialquilo y sus usos - Google Patents
Derivados de feniltriazol sustituido con hidroxialquilo y sus usos Download PDFInfo
- Publication number
- ES2697902T3 ES2697902T3 ES15788389T ES15788389T ES2697902T3 ES 2697902 T3 ES2697902 T3 ES 2697902T3 ES 15788389 T ES15788389 T ES 15788389T ES 15788389 T ES15788389 T ES 15788389T ES 2697902 T3 ES2697902 T3 ES 2697902T3
- Authority
- ES
- Spain
- Prior art keywords
- triazol
- chlorophenyl
- methyl
- triazole
- hydroxypropyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C*1C=CC([n]2nc(CN3N=C(c(cc4)ccc4Cl)N(CCC(F)(F)F)C3=O)nc2C(*)O)=C*(*)C=C1 Chemical compound C*1C=CC([n]2nc(CN3N=C(c(cc4)ccc4Cl)N(CCC(F)(F)F)C3=O)nc2C(*)O)=C*(*)C=C1 0.000 description 3
- KVWMXBUHFJPNKO-PKDNWHCCSA-N CC(c1nc(CN2N=C(c(cc3)ccc3Cl)N(C[C@@H](C(C=C)(F)F)O)C2=O)n[n]1-c1cccc(Cl)c1)O Chemical compound CC(c1nc(CN2N=C(c(cc3)ccc3Cl)N(C[C@@H](C(C=C)(F)F)O)C2=O)n[n]1-c1cccc(Cl)c1)O KVWMXBUHFJPNKO-PKDNWHCCSA-N 0.000 description 1
- KACFYNFZUWEXMY-KUHUBIRLSA-N C[C@H](c1nc(CN2N=C(c(cc3)ccc3Cl)N(C[C@@H](C(C=C)(F)F)O)C2=O)n[n]1-c1cc(F)ccc1)O Chemical compound C[C@H](c1nc(CN2N=C(c(cc3)ccc3Cl)N(C[C@@H](C(C=C)(F)F)O)C2=O)n[n]1-c1cc(F)ccc1)O KACFYNFZUWEXMY-KUHUBIRLSA-N 0.000 description 1
- QIFYQCSEJVGOAB-JTQLQIEISA-N OCc1nc(CN2N=C(c(cc3)ccc3Cl)N(C[C@@H](C(F)(F)F)O)C2=O)n[nH]1 Chemical compound OCc1nc(CN2N=C(c(cc3)ccc3Cl)N(C[C@@H](C(F)(F)F)O)C2=O)n[nH]1 QIFYQCSEJVGOAB-JTQLQIEISA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
- C07D249/10—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D249/12—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
Abstract
Un compuesto de fórmula (I) **Fórmula** en la que R1 es hidrógeno o metilo, y R2A y R2B se seleccionan independientemente del grupo que consiste en hidrógeno, fluoro, cloro, ciano, metilo, fluorometilo, difluorometilo, trifluorometilo, etilo, metoxi, difluorometoxi y trifluorometoxi, o una sal, hidrato y/o solvato farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Derivados de feniltriazol sustituido con hidroxialquilo y sus usos
La presente invención se refiere a derivados novedosos de 5-(hiroxialquilo)-1-fenil-1,2,4-triazol, a procedimientos para la preparación de dichos compuestos, a composiciones farmacéuticas que contienen dichos compuestos, y al uso de dichos compuestos o composiciones para el tratamiento y/o prevención de enfermedades, en particular para el tratamiento y/o prevención de enfermedades cardiovasculares y renales.
El contenido líquido del cuerpo humano está sujeto a diversos mecanismos de control fisiológico, cuyo fin es mantenerlo constante (homeostasis de volumen). En el procedimiento, tanto el relleno de volumen del sistema vascular y también la osmolaridad del plasma se registran constantemente por sensores apropiados (barorreceptores y osmorreceptores). La información que estos sensores suministran a los centros relevantes en el cerebro regula el comportamiento de beber y controla la excreción de fluidos por los riñones mediante señales humorales y neurales. La hormona peptídica vasopresina tiene gran importancia en esto [Schrier RW, Abraham WT, New Engl. J. Med. 341, 577-585 (1999)].
La vasopresina se produce en neuronas endócrinas especializadas en el Nucleus supraopticus y N. paraventricularis en la pared del tercer ventrículo (hipotalamo) y es transportada desde allí a través de los procesdimientos neurales hacia los lóbulos posteriores de la hipófisis (neurohipófisis). Allí la hormona es liberada en el torrente sanguíneo en respuesta a estímulos. Una pérdida de volumen, por ejemplo, como resultado de sangrado agudo, mucha sudoración, sed o diarrea prolongada, es un estímulo para la liberación intensificada de la hormona. Por el contrario, la secreción de vasopresina es inhibida por un aumento del volumen intravascular, por ejemplo, como resultado de un mayor consumo de fluidos.
La vasopresina ejerce su acción principalmente uniéndose a tres receptores, que se clasifican como receptores V1a, V1b y V2, y que pertenecen a la familia de los receptores acoplados a la proteína G. Los receptores V1a se ubican principalmente en las células de la musculatura lisa vascular. Su activación da lugar a la vasoconstricción, como resultado de la cual aumenta la resistencia periférica y la presión sanguínea. Aparte de esto, los receptores V1a también se pueden detectar en el hígado. Los receptores V1b (también denominaos receptores V3) son detectables en el sistema nervioso central. Junto con la hormona liberadora de corticotropina (CRH), la vasopresina regula la secreción basal e inducida por estrés de la hormona adrenocorticotropa (ACTH) mediante el receptor V1b. Los receptores V2 se ubican en el epitelio tubular distal y el epitelio de los túbulos recolectores del riñón. Su activación hace que estos epitelios sean permeables al agua. Este fenómeno se debe a la incorporación de acuaporinas (canales especiales de agua) en la membrana luminal de las células epiteliales.
La importancia de la vasopresina para la reabsorción de agua de la orina en el riñón se vuelve evidente a partir del panorama clínico de la diabetes insipidus, que es provocada por una deficiencia de la hormona, por ejemplo, debido a daño a la hipófisis. Los pacientes que padecen esta enfermedad excretan hasta 20 litros de orina cada 24 horas si no se les proporciona hormona de reemplazo. Este volumen corresponde a alrededor del 10 % de la orina primaria. Debido a su gran importancia para la reabsorción del agua de la orina, la vasopresina también se denomina hormona antidiurética (ADH). Por consiguiente, la inhibición farmacológica de la acción de la vasopresina/ADH sobre el receptor V2 resulta en una mayor excreción de orina. Sin embargo, en contraste a la acción de otros diuréticos (tiazidas y diuréticos de bucle), los antagonistas del receptor V2 provocan una mayor excreción de agua, sin aumentar sustancialmente la excreción de electrolitos. Esto significa que con fármacos antagonistas de V2, la homeostasis de volumen se puede restaurar sin afectar la homeostasis de electrolitos. Por lo tanto, los fármacos con actividad antagonista de V2 parecen ser particularmente adecuados para el tratamiento de todas las patologías asociadas con una sobrecarga del cuerpo con agua, sin que los electrolitos aumenten adecuadamente en paralelo.
Una anormalidad considerable de electrolitos se puede medir en química clínica como hiponatremia (concentración de sodio <135 mmol/l); es la anormalidad de electrolitos más importante en pacientes internados, con una incidencia de alrededor del 5 % o 250 000 casos por año en Estados Unidos solamente. Si la concentración de sodio en plasma es menor que 115 mmol/l, los estados comatosos y la muerte son inminentes. Dependiendo de la causa subyacente, se hace una distinción entre hiponatremia hipovolémica, euvolémica e hipervolémica. Las formas de hipervolemia con formación de edema son clínicamente significativas. Ejemplos típicos de estas son el síndrome de secreción inapropiada de ADH/vasopresina (SIADH) (por ejemplo, después del traumatismo cráneocerebral o como una paraneoplasia en carcinomas) y la hiponatremia hipervolémica en cirrosis hepática, diversas enfermedades renales e insuficiencia cardíaca [De Luca L. y col., Am. J. Cardiol. 96 (suppl.), 19L-23L (2005)]. En particular, los pacientes con insuficiencia cardíaca, pese a su hiponatremia o hipervolemia relativa, con frecuencia muestran niveles elevados de vasopresina, que se ven como consecuencia de una regulación neurohumoral generalmente alterada en la insuficiencia cardíaca [Francis G.S. y col., Circulation 82, 1724-1729 (1990)].
La regulación neurohormonal alterada se manifiesta esencialmente por sí misma en una elevación del tono simpático y una activación inapropiada del sistema renina-angiotensina-aldosterona. Mientras la inhibición de
estos componentes por bloqueantes del receptor beta por un lado y por inhibidores ACE o bloqueantes del receptor de angiotensina por el otro es ahora una parte inherente del tratamiento farmacológico de la insuficiencia cardíaca, la elevación inapropiada de la secreción de vasopresina en la insuficiencia cardíaca avanzada actualmente no se puede tratar adecuadamente. Aparte de la retención de agua mediada por receptores V2 y las consecuencias hemodinámicas desfavorables asociadas con esto en términos de mayor retraso, el vaciado del ventrículo izquierdo, la presión en los vasos sanguíneos pulmonares y el rendimiento cardíaco también se ven afectados negativamente por una vasoconstricción mediada por V1a. Además, en función de los datos experimentales en animales, una acción promotora de hipertrofia directa sobre el músculo cardíaco se atribuye también a la vasopresina. En contraste al efecto renal de la expansión de volumen, que es mediada por la activación de los receptores V2, la acción directa sobre el músculo cardíaco es desencadenada por la activación de los receptores V1a.
Por estas razones, los agentes que inhiben la acción de la vasopresina sobre el receptor V2 y/o V1a parecen ser adecuados para el tratamiento de la insuficiencia cardíaca. En particular, los compuestos con actividad combinada en ambos receptores de vasopresina (V1a y V2) deberían tener tanto efectos renales como hemodinámicos deseables y por lo tanto ofrecer un perfil especialmente ideal para el tratamiento de pacientes con insuficiencia cardíaca. El suministro de tales antagonistas de vasopresina combinados también parece tener sentido en la medida en que una disminución de volumen mediada solamente por el bloqueo del receptor V2 puede provocar la estimulación de los osmorreceptores y, como resultado, puede llevar a un aumento compensatorio adicional en la liberación de vasopresina. A través de esto, en ausencia de un componente que simultáneamente bloquea el receptor V1a, los efectos nocivos de la vasopresina, tales como por ejemplo vasoconstricción e hipertrofia del músculo cardíaco, se podrían intensificar aún más [Saghi P. y col., Europ. Heart J. 26, 538-543 (2005)].
Determinados derivados de 4-fenil-1,2,4-triazol-3-ilo se han descrito en el documento WO 2005/063754-A1 y el documento WO 2005/105779-A1 por actuar como antagonistas del receptor V1a de vasopresina que son útiles para el tratamiento de trastornos ginecológicos, particularmente trastornos menstruales tales como dismenorrea. En el documento WO 2011/104322-A1, un grupo particular de 1,2,4-triazol-3-onas unidas por bis-arilo, incluyendo 5fenil-1,2,4-triazol-3-ilo y derivados de 1-fenil-1,2,3-triazol-4-ilo de este, se han desvelado como antagonistas de los receptores V1a y/o V2 de vasopresina útiles para el tratamiento y/o prevención de enfermedades cardiovasculares. Durante una investigación adicional de esta clase estructural surgió, sin embargo, que los compuestos candidatos se veían comprometidos frecuentemente por una potencia acuarética insatisfactoria cuando se evaluaban in vivo después de la administración peroral a ratas conscientes. Sin embargo, como se destacó anteriormente, una eficacia acuarética sólida es un requisito previo deseable para el tratamiento de patologías que se asocian con una sobrecarga del cuerpo con agua, tal como, por ejemplo, en la insuficiencia cardíaca congestiva.
Un aumento considerable en la potencia acuarética también ayudaría a reducir la cantidad de sustancia que será necesaria para lograr y mantener el efecto terapéutico deseado, limitando así la posibilidad de efectos secundarios no aceptables y/o interacciones fármaco-fármaco no deseadas durante el tratamiento de pacientes que ya podrían estar en alto riesgo, tales como, por ejemplo, en la insuficiencia cardíaca aguda o crónica o insuficiencia renal. Por lo tanto, el problema técnico a resolver de acuerdo con la presente invención puede ser la identificación y suministro de nuevos compuestos que actúen como potentes antagonistas tanto de los receptores V1A como v 2 de vasopresina y, además, presenten, un aumento considerable en la potencia acuarética in vivo.Sorprendentemente, se ha descubierto que determinados derivados de 5-(hidroxialquil)-1-fenil-1,2,4-triazolo representan antagonistas duales altamente potentes de los receptores V1a y V2 de vasopresina que presentan una potencia acuarética in vivo significativamente mejorada después de la aplicación oral. Esta mejora en el perfil de actividad hace que los compuestos de la presente invención sean particularmente útiles para el tratamiento y/o prevención de enfermedades cardiovasculares y renales.
En un aspecto, la presente invención se refiere a derivados de 5-(hidroxialquil)-1-fenil-1,2,4-triazolo de fórmula general (I)

en la que
R1 es hidrógeno o metilo,
y
R2A y R2B se seleccionan independientemente del grupo que consiste en hidrógeno, fluoro, cloro, ciano, metilo, fluorometilo, difluorometilo, trifluorometilo, etilo, metoxi, difluorometoxi y trifluorometoxi.
Los compuestos de acuerdo con la presente invención también pueden estar presentes en la forma de sus sales, solvatos y/o solvatos de las sales.
Los compuestos de acuerdo con la invención son los compuestos de fórmula (I) y sus sales, solvatos y solvatos de las sales, los compuestos incluidos en la fórmula (I) de las fórmulas mencionadas a continuación y sus sales, solvatos y solvatos de las sales y los compuestos incluidos en la fórmula (I) y mencionados a continuación como productos de procedimiento y/o ejemplos de realizaciones y sus sales, solvatos y solvatos de las sales, en los que los compuestos incluidos en la fórmula (I) y mencionados a continuación ya no son sales, solvatos y solvatos de las sales.
Sales a efectos de la presente invención son preferentemente sales farmacéuticamente aceptables de los compuestos de acuerdo con la invención (por ejemplo, véase S. M. Berge y col., "Pharmaceutical Salts", J. Pharm. Sci. 1977, 66, 1 19). También se incluyen las sales que en sí mismas no son adecuadas para usos farmacéuticos, pero se pueden usar por ejemplo para aislar, purificar o almacenar los compuestos de acuerdo con la invención.
Las sales farmacéuticamente aceptables incluyen sales de adición de ácidos de ácidos minerales, ácidos carboxílicos y ácidos sulfónicos, por ejemplo sales de ácido clorhídrico, ácido bromhírico, ácido sulfúrico, ácido fosfórico, ácido metanosulfónico, ácido etanosulfónico, ácido bencenosulfónico, ácido toluenosulfónico, ácido naftalenosulfónico, , ácido fórmico, ácido acético, ácido trifluoroacético, ácido propiónico, ácido láctico, ácido tartárico, ácido málico, ácido cítrico, ácido fumárico, ácido maleico y ácido benzoico.
Las sales farmacéuticamente aceptables también incluyen sales de bases habituales, tales como por ejemplo sales de metal alcalino (por ejemplo, sales de sodio y potasio), sales de metal alcalinotérreo (por ejemplo, sales de calcio y magnesio), sales de amonio derivadas de amoníaco o aminas orgánicas, tales como a modo de ejemplo y preferentemente, etilamina, dietilamina, trietilamina, N,N-diisopropiletilamina, monoetanolamina, dietanolamina, trietanolamina, dimetilaminoetanol, dietilaminoetanol, procaina, diciclohexilamina, dibencilamina, N-metilmorfolina, N-metilpiperidina, arginina, lisina y 1,2-etilendiamina.
Los solvatos en el contexto de la invención se designan como las formas de los compuestos de acuerdo con la invención que forman un complejo en el estado sólido o líquido por coordinación estoiquiométrica con moléculas de disolvente. Los hidratos son una forma específica de solvatos, en los que la coordinación tiene lugar con agua. Los hidratos son solvatos preferentes en el contexto de la presente invención.
Los compuestos de la presente invención pueden, ya sea por naturaleza de centros asimétricos o por rotación limitada, estar presentes en forma de isómeros (enantiómeros, diastereómeros). Cualquier isómero puede estar presente en el que el centro asimétrico está en la configuración (R)-, (S)-, o (R,S).
También se apreciará que cuando dos o más centros asimétricos están presentes en los compuestos de la invención, varios diastereómeros y enantiómeros de los ejemplos de estructuras con frecuencia son posibles, y que diastereómeros puros y enantiómeros puros representan realizaciones preferentes. Se pretende que los estereoisómeros puros, diaestereómeros puros, enantiómeros puros y mezclas de estos, estén dentro del ámbito de la invención.
Todos los isómeros, ya sea separados, puros, parcialmente puros o en mezcla racémica de los compuestos de la presente invención se abarquen dentro del ámbito de la presente invención. La purificación de dichos isómeros y la separación de dichas mezclas isoméricas se puede lograr por técnicas convencionales conocidas en la técnica. Por ejemplo, las mezclas diastereómericas se pueden separar en isómeros individuales por procedimientos cromatográficos o cristalización, y los racematos se pueden separar en los enantiómeros correspondientes por procedimientos cromatográficos en fases quirales o por resolución.
Además, todas las formas tautoméricas posibles de los compuestos descritos anteriormente se incluyen de acuerdo con la presente invención.
La presente invención también abarca todas las variantes isotópicas adecuadas de los compuestos de acuerdo con la invención. Se entiende que una variante isotópica de un compuesto de acuerdo con la invención significa un compuesto en el que al menos un átomo dentro del compuesto de acuerdo con la invención se ha intercambiado por otro átomo del mismo número atómico, pero con una masa atómica diferente que la masa atómica que general o predominantemente se produce en la naturaleza. Ejemplos de isótopos que se pueden incorporar en un compuesto de acuerdo con la invención son los de hidrógeno, carbono, nitrógeno, oxígeno, flúor, cloro, bromo y yodo, tales como 2H (deuterio), 3H (tritio), 3C, 14C, 15N, l7O, 18O, 18F, 36Cl, 82Br, 123I, 12\ 129I y 131I. Las variantes isotópicas particulares de un compuesto de acuerdo con la invención, especialmente aquellas en las que se incorporó uno o más isótopos radioactivos, pueden ser beneficiosas, por ejemplo, para el examen del mecanismo de acción o de la distribución del compuesto activo en el cuerpo. Debido a una capacidad de preparación y detectabilidad comparativamente fácil, especialmente los compuestos etiquetados con isótopos 3H, 14C y/o 18F son adecuados a estos efectos. Además, la incorporación de isótopos, por ejemplo de deuterio, puede llevar a beneficios terapéuticos particulares como consecuencia de una mayor estabilidad metabólica del compuesto, por ejemplo una extensión de la semivida en el cuerpo o una reducción de la dosis activa requerida. Tales modificaciones de los compuestos de acuerdo con la invención pueden, por lo tanto, en algunos casos constituir una realización preferente de la presente invención. Las variantes isotópicas de los compuestos de acuerdo con la invención se pueden preparar por procedimientos conocidos por los expertos en la técnica, por ejemplo, por los procedimientos descritos a continuación y los procedimientos descritos en los ejemplos de trabajo, mediante el uso de modificaciones isotópicas correspondientes de los reactivos particularesy/o compuestos de partida en la misma..
En una realización diferente, la presente invención se refiere a compuestos de fórmula (I), en la que R1 es metilo. En una realización diferente adicional, la presente invención se refiere a compuestos de fórmula (I), en la que al menos uno de R2A y R2B es diferente de hidrógeno.
En otra realización diferente, la presente invención se refiere a compuestos de fórmula (I), en la que
R1 es hidrógeno o metilo,
R2A y R2B se seleccionan independientemente del grupo que consiste en hidrógeno, fluoro, cloro, metilo y metoxi, en la que al menos uno de R2A y R2B es diferente de hidrógeno.
En una realización preferente, la presente invención se refiere a compuestos de acuerdo con la fórmula (I) que se seleccionan del grupo que consiste en los siguientes compuestos
5-(4-clorofenil)-2-{[1-(3-clorofenil)-5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-{[1-(3-fluorofenil)-5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-{[5-(hidroximetil)-1-(2-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
2-({1-(2-cloro-4-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
2-{[1-(2-cloro-4-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona
(diastereómero 1);
2-{[1-(2-cloro-4-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona
(diastereómero 2);
2-({1-(2-doro-5-fluorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l}met¡l)-5-(4-dorofen¡l)-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
2-{[1-(2-doro-5-fluorofen¡l)-5-(1-h¡drox¡et¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-5-(4-dorofen¡l)-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona
(diastereómero 1);
2-{[1-(2-doro-5-fluorofen¡l)-5-(1-h¡drox¡et¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-5-(4-dorofen¡l)-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona
(diastereómero 2);
5-(4-dorofen¡l)-2-({1-(3-fluorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(3-fluorofenM)-5-[(1R)-1-hídrox¡et¡l]-1 W-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡ l ]-2,4-d¡h¡dro-3H-1,2,4-tr¡azo l -3-ona;
5-(4-dorofen¡l)-2-({1-(3-fluorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(3-dorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(3-dorofen¡l)-5-[(1R)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(3-dorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(2-dorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(2-dorofen¡l)-5-[(1R)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
y
5-(4-dorofen¡l)-2-({1-(2-dorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona.
En una real¡zac¡ón part¡cularmente preferente, la presente ¡nvenc¡ón se ref¡ere a compuestos de acuerdo con la fórmula (I) que se selecc¡onan del grupo que cons¡ste en los s¡gu¡entes compuestos
5-(4-dorofen¡l)-2-({1-(3-fluorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(3-fluorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(3-dorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(3-dorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-dorofen¡l)-2-({1-(2-dorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
y
5-(4-dorofen¡l)-2-({1-(2-dorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona.
En una real¡zac¡ón ad¡c¡onal, la presente ¡nvenc¡ón se ref¡ere a un proced¡m¡ento para preparar los compuestos de fórmula general (I), caracter¡zado porque un compuesto de fórmula (II)

se hace reaccionar primero con hidrazina para proporcionar la hidrazida de fórmula (III)

luego se condensa con una amidina de fórmula (IV)

o una sal de la misma,
en la que R1 tiene el significado descrito anteriormente,
en presencia de una base para proporcionar un derivado de 1,2,4-triazol de fórmula (V)

y/o un tautómero del mismo,
en la que R1 tiene el significado descrito anteriormente,
y posteriormente se acopla con un ácido fenilborónico de fórmula (VI)

en la que R2A y R2B tienen los significados descritos anteriormente,
en presencia de un catalizador de cobre y una base amina para proporcionar el compuesto diana de fórmula (I)

en la que R1, R2A y R2B tienen los significados descritos anteriormente,
seguido de, opcionalmente, cuando fuera adecuado, (i) separar los compuestos de fórmula (I) obtenidos así en sus respectivos diastereómeros, preferentemente usando procedimientos cromatográficos, y /o (ii) convertir los compuestos de fórmula (I) en sus respectivos hidratos, solvatos, sales y/o hidrato o solvatos de las sales por tratamiento con los correspondientes disolventes y/o ácidos o bases.
Los compuestos de fórmula (I), en la que R1 representa metilo, también se pueden obtener en forma diastereoméricamente pura empleando el enantiómero apropiado de amidina (IV) [R1 = metilo], es decir (IV-A) o (IV-B)

o una sal del mismo, en la reacción de condensación descrita anteriormente.
La transformación (II) ^ (III) se realiza en la forma habitual tratando éster metílico (II) con hidrazina o hidrato de hidrazina en un disolvente alcohólico, tal como metanol, etanol, n-propanol, isopropanol o n-butanol, a una temperatura en el intervalo de 20 °C a 100 °C.
La reacción de condensación (III) (IV) ^ (V) se realiza habitualmente en un disolvente dipolar-aprótico inerte, tal como W,W-dimetilformamida (DMF), W,W-dimetilacetamida (DMA), dimetilsulfóxido (DMSO), /V-metilpirrolidinona (NMP) o WV-dimetilpropileno urea (DMPU), en presencia de una base suficientemente fuerte, tal como hidruro de sodio o un alcóxido de sodio o potasio, por ejemplo metóxido de sodio o potasio, etóxido de sodio o potasio, o ferc-butóxido de sodio o potasio. La amidina (IV) se puede emplear como tal en esta reacción o en forma de sal, por ejemplo como la sal clorhidrato. En el último caso, se usa un exceso proporcional de base. La reacción se realiza generalmente a una temperatura entre 80 °C y 150 °C. El calentamiento por medio de un dispositivo de reactor de microondas puede tener un efecto beneficioso para esta reacción de condensación.
El derivado de 1,2,4-triazol de fórmula (V) producido por esta reacción también puede estar presente en otras formas tautoméricas, tales como (V-A) o (V-B)
o como una mezcla de tautómeros.
La reacción de acoplamiento (V) (VI) ^ (I) se realiza típicamente con la ayuda de un catalizador de cobre y una base amina [condiciones de "acoplamiento Chan-Lam"; véase, por ejemplo, DMT Chan y col., Tetrahedron Lett.
44 (19), 3863-3865 (2003); J. X. Qiao y PYS Lam, Synthesis, 829-856 (2011); K.S. Rao y T-S. Wu, Tetrahedron 68, 7735-7754 (2012)]. Los catalizadores de cobre adecuados para este procedimiento son en particular sales de cobre (II), tales como acetato de cobre (II), trifluorometanosulfonato de cobre (II) o bromuro de cobre (II). Las bases amina prácticas incluyen, por ejemplo, trietilamina, N,N-diisopropiletilamina, piridina y 4-(N,N-dimetilamino)piridina. La reacción se realiza en un disolvente orgánico inerte, tal como diclorometano, 1,2-dicloroetano, éter terc-butílico de metilo, tetrahidrofurano, 1,4-dioxano, 1,2-dimetoxietano, tolueno, piridina, acetato de etilo, acetonitrilo o N,N-dimetilformamida, o en una mezcla de estos disolventes. Preferentemente, se usa piridina como disolvente y base. El acoplamiento se realiza generalmente a una temperatura en el intervalo de 20° C a 120 °C, preferentemente a 20 °C a 70 °C. La irradiación de microondas concomitante puede tener un efecto beneficioso en esta reacción también.
Los derivados de feniltriazol regioisoméricos que pueden surgir de una reacción de acoplamiento que se produce en otros átomos de triazol nitrógeno [cf. tautómeros (V-A), (V-B)] pueden, en el caso, separarse fácilmente del producto diana (I) por cromatografía HPLC convencional.
El compuesto de fórmula (II) se puede sintetizar por los procedimientos descritos en la solicitud de patente internacional WO 2011/104322-A1 (véase también los esquemas de síntesis 1a y 1b a continuación).
Los compuestos de fórmulas (IV), (IV-A), (IV-B) y (VI) están disponibles en el mercado, conocidos de la literatura, o se pueden preparar con materiales de partida fácilmente disponibles mediante la adaptación de procedimientoss convencionales descritos en la literatura. Los procedimientos detallados y las referencias de literatura para preparar los materiales de partida también se pueden encontrar en la parte experimental en la sección de la preparación de los materiales de partida e intermedios.
La preparación de los compuestos de la invención se puede ilustrar mediante los siguientes esquemas de síntesis:
Esquema 1a

[cf. solicitud de patente internacional WO 2011/104322-A1].
Esquema 1b
[cf. solicitud de patente internacional WO 2011/104322-A1].
Esquema 2

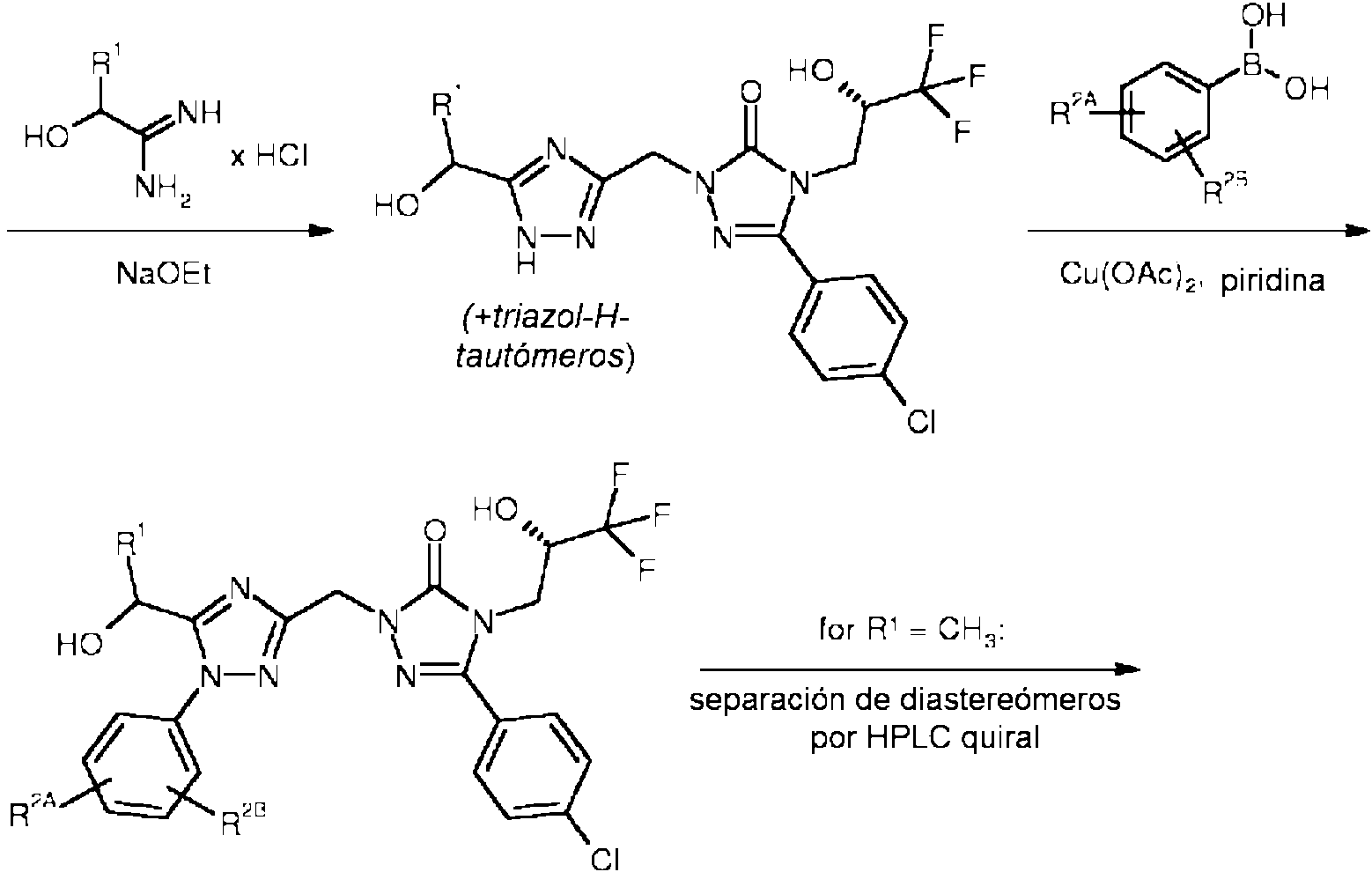

Los compuestos de la presente invención tienen propiedades farmacológicas valiosas y se pueden usar para evitar y/o tratar diversas enfermedades y estados inducidos por enfermedad en seres humanos y otros mamíferos. En el contexto de la presente invención, el término "tratamiento" o "tratar" incluye inhibir, retrasar, aliviar, mitigar, detener, reducir, o provocar la regresión de una enfermedad, trastorno, afección, o estado, el desarrollo y/o evolución del mismo y/o los síntomas del mismo. El término "prevención" o "prevenir" incluye reducir el riesgo de tener, contraer o experimentar una enfermedad, trastorno, afección o estado, el desarrollo y/o evolución del mismo y/o los síntomas del mismo. El término prevención incluye profilaxis. El tratamiento o prevención de un trastorno, enfermedad, afección o estado puede ser parcial o completo.
En todo este documento con el fin de simplicidad, se prefiere el uso de lenguaje singular en vez de lenguaje plural, pero generalmente se pretende incluir el lenguaje plural si no se indica lo contrario. Por ejemplo, la expresión "Un procedimiento para tratar una enfermedad en un paciente, que comprende administrar a un paciente una cantidad eficaz de un compuesto de fórmula (I)" pretende incluir el tratamiento simultáneo de más de una enfermedad así
como la administración de más de un compuesto de fórmula (I).
Los compuestos de la presente invención son antagonistas duales altamente potentes de receptores de vasopresina V1a y V2. Además, los compuestos de la invención muestran un efecto acuerético pronunciado in vivo después de la aplicación oral. Por lo tanto, los compuestos de la invención se espera que sean altamente valiosos como agentes terapéuticos para el tratamiento y/o prevención de enfermedades, en particular para el tratamiento y/o prevención de enfermedades cardiovasculares y renales.
Las enfermedades cardiovasculares en este contexto que se pueden tratar y/o evitar con los compuestos de la invención incluyen, pero sin limitación, las siguientes: insuficiencia cardíaca aguda y crónica que incluye empeoramiento de la insuficiencia cardíaca crónica (u hospitalización para insuficiencia cardíaca) e insuficiencia cardíaca congestiva, hipertensión arterial, hipertensión resistente, hipertensión pulmonar arterial, enfermedad cardíaca coronaria, angina de pecho estable e inestable, arritmias auriculares y ventriculares, trastornos del ritmo auricular y ventricular y trastornos de conducción, por ejemplo bloques aurioventriculares de grado I-III (AVB I-III), taquiarritmia supraventricular, fibrilación auricular, aleteo auricular, fibrilación ventricular, aleteo ventricular, taquiarritmia ventricular, taquicardia torsade-de-pointes, extrasístoles auriculares y ventriculares, extrasístoles de unión AV, síndrome del seno enfermo, síncopes, taquicardia de reingreso de nódulo AV y síndrome Wolff-Parkinson-White, síndrome coronario agudo (ACS), enfermedades cardíacas autoinmunitarias (pericarditis, endocarditis, valvulitis, aortitis, cardiomiopatías), choque tal como choque cardiogénico, choque séptico y choque anafiláctico, aneurismas, cardiomiopatía de Boxer (contracción ventricular prematura), otras enfermedades tromboembólicas e isquemias tales como trastornos de perfusión periférica, lesión por reperfusión, trombosis arteriales y venosas, insuficiencia del miocardio, disfunción endotelial, daño micro- y macrovascular (vasculitis) y para evitar restenosis tales como terapias de trombólisis, angioplastia transluminal percutánea (PTA), angioplastia coronaria transluminal percutánea (PTCA), operaciones de bypass y transplante de corazón, arteriesclerosis, trastornos del metabolismo lipídico, hipolipoproteinemias, dislipidemias, hipertrigliceridemias, hiperlipidemias e hiperlipidemias combinadas, hipercolesterolemias, abetalipoproteinemia, sitosterolemia, xantomatosis, enfermedad de Tangier, adipositas, obesidad, síndrome metabólico, ataques transitorios e isquémicos, apoplejía, enfermedades cardiovasculares inflamatorias, enfermedades vasculares periféricas y cardíacas, trastornos de circulación periférica, espasmos de las arterias coronarias y arterias periféricas, y edema tale como, por ejemplo, edema pulmonar, edema cerebral, edema renal y edema relacionado con insuficiencia cardíaca.
En el sentido de la presente invención, la expresión insuficiencia cardíaca también incluye formas de enfermedades más específicas o relacionadas tales como insuficiencia cardíaca derecha, insuficiencia cardíaca izquierda, insuficiencia global, cardiomiopatía isquémica, cardiomiopatía dilatativa, defectos cardíacos congénitos, defectos de válvula cardíaca, insuficiencia cardíaca con defectos de válvula cardíaca, estenosis de válvula mitral, insuficiencia de válvula mitral, estenosis de válvula aórtica, insuficiencia de válvula aórtica, estenosis tricúspide, insuficiencia tricúspide, estenosis de válvula pulmonar, insuficiencia de válvula pulmonar, defectos de válvula cardíaca combinada, inflamación del músculo cardíaco (miocarditis), miocarditis crónica, miocarditis aguda, miocarditis viral, insuficiencia cardíaca diabética, cardiomiopatía tóxica alcohólica, enfermedades cardíacas por almacenamiento , insuficiencia cardíaca con fracción de eyección conservada (HFpEF o insuficiencia cardíaca diastólica), e insuficiencia cardíaca con fracción de eyección reducida (HFrEF o insuficiencia cardíaca sistólica). Los compuestos de acuerdo con la invención también son adecuados para el tratamiento y/o prevención de enfermedades renales, en particular de insuficiencia renal aguda y crónica y de fallo renal agudo y crónico. En el sentido de la presente invención, la expresión insuficiencia renal comprende manifestaciones agudas y crónicas de insuficiencia renal, así como enfermedades renales subyacentes o relacionadas tales como hipoperfusión renal, hipotensión intradialítica, uropatía obstructiva, glomerulopatías, glomerulonefritis, glomerulonefritis aguda, glomeruloesclerosis, enfermedades tubulointersticiales, enfermedades nefropáticas tales como enfermedad renal primaria y congénita, nefritis, enfermedades renales inmunológicas tales como rechazo de transplante de riñón, enfermedades renales inducidas por complejo inmune, nefropatía inducida por sustancias tóxicas, nefropatía inducida por medio de contraste, nefropatía diabética y no diabética, pielonefritis, quistes renales, nefroesclerosis, nefroesclerosis hipertensa y síndrome nefrótico, que se pueden caracterizar por diagnóstico, por ejemplo, por excreción anormalmente reducida de creatinina y/o agua, concentraciones anormalmente aumentadas en sangre de urea, nitrógeno, potasio y/o creatinina, actividad alterada de enzimas renales tales como, por ejemplo, glutamil sintetasa, osmolaridad de orina o volumen de orina alterado, microalbuminuria aumentada, macroalbuminuria, lesiones de glomérulos y arteriolas, dilatación tubular, hiperfosfatemia y/o la necesidad de diálisis. La presente invención también comprende el uso de los compuestos de acuerdo con la invención para el tratamiento y/o prevención de secuelas de insuficiencia renal, por ejemplo edema pulmonar, insuficiencia cardíaca, uremia, anemia, trastornos de electrolitos (por ejemplo, hiperpotasemia, hiponatremia) y trastornos en los huesos y metabolismo de carbohidratos.
Los compuestos de la presente invención pueden ser particularmente útiles para el tratamiento y/o prevención del síndrome cardiorenal (CRS) y sus diversos subtipos. Esta expresión adopta determinados trastornos del corazón y los riñones por lo cual la disfunción aguda o crónica en un órgano puede inducir la disfunción aguda o crónica del otro. CRS se ha subclasificado en cinco tipos basados en el órgano que inició el insulto así como la agudeza y
cronicidad de la enfermedad (tipo 1: desarrollo de insuficiencia renal que resulta de la insuficiencia cardíaca descompensada aguda; tipo 2: insuficiencia cardíaca congestiva crónica que resulta en disfunción renal progresiva; tipo 3: disfunción cardíaca aguda que resulta de una caída brusca de la función renal; tipo 4: enfermedad de riñón crónica que lleva a remodelación cardíaca; tipo 5: enfermedad sistémica que implica el corazón y los riñones) [véase, por ejemplo, MR Kahn y col., Nature Rev. Cardiol. 10, 261-273 (2013)].
Los compuestos de acuerdo con la invención también son adecuados para el tratamiento y/o prevención de enfermedad de riñón poliquístico (PCKD) y del síndrome de secreción de ADH inadecuada (SIADH). Además, los compuestos de la invención son adecuados para su uso como un diurético para el tratamiento de edemas y en trastornos de electrolitos, en particular en hiponatremia hipervolémica y euvolémica.
Además, los compuestos de acuerdo con la invención se pueden usar para el tratamiento y/o prevención del fenómeno de Raynaud primario o secundario, trastornos de microcirculación, claudicación, neuropatías periféricas y autonómicas, microangiopatías diabéticas, retinopatía diabética, úlceras de miembro diabético, gangrena, síndrome CREST, trastornos eritematosos, onicomicosis, enfermedades reumáticas y para promover la cicatrización.
Además, los compuestos de la invención son adecuados para tratar enfermedades urológicas y enfermedades del sistema urogenital masculino y femenino tales como, por ejemplo, síndrome prostático benigno (BPS), hiperplasia prostática benigna (BPH), agrandamiento de próstata benigno (BPE), obstrucción de salida de vejiga (BOO), síndromes del tracto urinario inferior (LUTS), vejiga hiperactiva neurogénica (OAB), cistitis intersticial (IC), incontinencia urinaria (UI), por ejemplo, incontinencia mixta, de urgencia, de esfuerzo y por sobrecarga (MUI, UUI, SUI, OUI), dolores pélvicos, disfunción eréctil y disfunción sexual femenina.
Los compuestos de acuerdo con la invención también se pueden usar para el tratamiento y/o prevención de enfermedades inflamatorias, enfermedades asmáticas, enfermedad pulmonar obstructiva crónica (COPD), síndrome de alteración respiratoria aguda (ARDS), lesión pulmonar aguda (ALI), deficiencia de alfa-1-antitripsina (AATD), fibrosis pulmonar, enfisema pulmonar (por ejemplo, enfisema pulmonar inducido por tabaco) y fibrosis quística (CF). Además, los compuestos de la invención se pueden usar para el tratamiento y/o prevención de la hipertensión arterial pulmonar (PAH) y otras formas de hipertensión pulmonar (PH), que incluyen hipertensión pulmonar asociada con enfermedad del ventrículo izquierdo, infección por VIH, anemia de células falciformes, tromboembolismo (CTEPH), sarcoidosis, enfermedad pulmonar obstructiva crónica (COPD) o fibrosis pulmonar. Además, los compuestos de acuerdo con la invención se pueden usar para el tratamiento y/o prevención de cirrosis hepática, ascitis, diabetes mellitus y complicaciones diabéticas tales como, por ejemplo, neuropatía y nefropatía. Además, los compuestos de la invención son adecuados para el tratamiento y/o prevención de trastornos del sistema nervioso tales como estados de ansiedad y depresión, de glaucoma y de cáncer, en particular de tumores pulmonares, y para el tratamiento del desajuste del ritmo circadiano tal como jet lag y trabajos en turnos.
Además, los compuestos de acuerdo con la invención pueden ser útiles para el tratamiento y/o prevención de condiciones de dolor, enfermedades de las glándulas adrenales tales como, por ejemplo, feocromocitoma y apoplexia adrenal, enfermedades del intestino tales como, por ejemplo, enfermedad de Crohn y diarrea, trastornos menstruales tales como, por ejemplo, dismenorrea, o de endometriosis, parto prematuro y para tocolisis.
Debido a su actividad y perfil de selectividad, los compuestos de la presente invención se cree que son particularmente adecuados para el tratamiento y/o prevención de insuficiencia cardíaca aguda y crónica, síndrome cardiorrenal (tipo 1-5), hiponatremia hipervolémica y euvolémica, cirrosis hepática, ascitis, edema y el síndrome de secreción inadecuada de ADH (SIADH).
Las enfermedades mencionadas anteriormente han sido bien caracterizadas en los seres humanos, pero también existen con una etiología comparable en otros mamíferos y se pueden tratar en aquellos con los compuestos y procedimientos de la presente invención.
Por lo tanto, la presente invención se refiere además al uso de los compuestos de acuerdo con la invención para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades anteriormente mencionadas. La presente invención se refiere además al uso de los compuestos de acuerdo con la invención para preparar una composición farmacéutica para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades anteriormente mencionadas.
La presente invención se refiere además al uso de los compuestos de acuerdo con la invención en un procedimiento para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades anteriormente mencionadas.
La presente invención se refiere además a un procedimiento para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades anteriormente mencionadas, usando una cantidad eficaz de al menos uno de
los compuestos de acuerdo con la invención.
Los compuestos de la presente invención se pueden administrar como el agente farmacéutico solo o en combinación con uno o más agentes terapéuticos adicionales mientras esta combinación no lleve a efectos secundarios no deseados y/o inaceptables. Dicha terapia de combinación incluye la administración de una formulación de dosificación farmacéutica individual que contiene un compuesto de fórmula (I), como se define anteriormente, y uno o más agentes terapéuticos adicionales, así como la administración de un compuesto de fórmula (I) y cada agente terapéutico adicional en su propia formulación de dosificación farmacéutica separada. Por ejemplo, un compuesto de fórmula (I) y un agente terapéutico se pueden administrar al paciente juntos en una composición de dosificación oral individual (fija) tal como un comprimido o cápsula, o cada agente se puede administrar en formulaciones de dosificación separada.
Cuando se usan formulaciones de dosificación separada, el compuesto de fórmula (I) y uno o más agentes terapéuticos adicionales se pueden administrar esencialmente al mismo tiempo (es decir, simultáneamente) o en momentos escalonados por separado (es decir, secuencialmente).
En particular, los compuestos de la presente invención se pueden usar en combinación fija o separada con
• nitratos orgánicos y donantes de NO, por ejemplo nitroprusiato de sodio, nitroglicerina, mononitrato de isosorbida, dinitrato de isosorbida, molsidomina o SIN-1, NO por inhalación;
• compuestos que inhiben la degradación de guanosina monofosfato cíclica (cGMP), por ejemplo inhibidores de fosfodiesterasas (PDE) 1,2 y/o 5, en particular inhibidores de PDE-5 tales como sildenafil, vardenafil, tadalafil, udenafil, dasantafil, avanafil, mirodenafil o lodenafil;
• agentes inotrópicos positivos, tales como por ejemplo glucósidos cardíacos (digoxina) y agonistas betaadrenérgicos y dopaminérgicos tales como isoproterenol, adrenalina, noradrenalina, dopamina o dobutamina;
• péptidos natriuréticos, tales como por ejemplo péptido natriurético atrial (ANP, anaritida), péptido natriurético tipo B o péptido natriurético de cerebro (BNP, nesiritida), péptido natriurético tipo C (CNP) o urodilatina;
• sensibilizantes de calcio, tales como por ejemplo y preferentemente levosimendán;
• activadores independientes de NO y heme de guanilato ciclasa soluble (sGC), tales como en particular cinaciguat y también los compuestos descritos en los documentos WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 y WO 02/070510;
• estimuladores independientes de NO pero dependientes de heme de guanilato ciclasa (sGC), tales como en particular riociguat y vericiguat y también los compuestos descritos en los documentos WO 00/06568, WO 00/06569, WO 02/42301, WO 03/095451, WO 2011/147809, WO 2012/004258, WO 2012/028647 y WO 2012/059549;
• inhibidores de elastasa de neutrófilo humana (HNE), tales como por ejemplo sivelestat o DX-890 (reltrán); • compuestos que inhiben la cascada de transducción de señal, en particular inhibidores de tirosina y/o serina/treonina cinasa, tales como por ejemplo, nintedanib, dasatinib, nilotinib, bosutinib, regorafenib, sorafenib, sunitinib, cediranib, axitinib, telatinib, imatinib, brivanib, pazopanib, vatalanib, gefitinib, erlotinib, lapatinib, canertinib, lestaurtinib, pelitinib, semaxanib o tandutinib;
• compuestos que influyen en el metabolismo de energía del corazón, tales como por ejemplo y preferentemente etomoxir, dicloroacetato, ranolazina o trimetazidina, o agonistas del receptor de adenosina parciales o completos A1;
• compuestos que influyen en el ritmo cardíaco, tales como por ejemplo y preferentemente ivabradina;
• activadores de miosina cardíaca, tales como por ejemplo y preferentemente omecamtiv mecarbil (CK-1827452);
• agentes antitrombóticos, por ejemplo y preferentemente del grupo de inhibidores de agregación plaquetaria, anticoagulantes y sustancias profibrinolíticas;
• agentes de reducción de presión sanguínea, por ejemplo y preferentemente del grupo de antagonistas de calcio, antagonistas de angiotensina AII, inhibidores de ACE, inhibidores de vasopeptidasa, antagonistas de endotelina, inhibidores de renina, bloqueantes alfa, bloqueantes beta, antagonistas del receptor mineralocorticoide y diuréticos; y/o
• agentes de alteración del metabolismo de grasa, por ejemplo y preferentemente del grupo de agonistas del
receptor de la tiroides, inhibidores de la síntesis de colesterol, tales como por ejemplo y preferentemente inhibidores de síntesis de escualeno o HMG-CoA-reductasa, inhibidores de ACAt , inhibidores de CETP, inhibidores de MTP, agonnistas PPAR-alfa, PPAR-gamma y/o PPAR-delta, inhibidores de la absorción de colesterol, inhibidores de lipasa, absorbentes de ácido biliar polimérico, inhibidores de reabsorsción de ácido biliar y antagonistas de lipoproteína(a).
Agentes antitrombóticos se entenderán preferentemente como compuestos del grupo de inhibidores de agregación plaquetaria, anticoagulantes y sustancias profibrinolíticas.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de agregación plaquetaria, por ejemplo y preferentemente aspirina, clopidogrel, ticlopidina o dipiridamol.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de trombina, por ejemplo y preferentemente ximelagatrán, dabigatrán, melagatrán, bivalirudina o enoxaparina.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de GPIIb/IIIa, por ejemplo y preferentemente tirofiban o abciximab.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor del factor Xa, por ejemplo y preferentemente rivaroxaban, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, DU-176b, Pm D-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 o SSR-128428.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con heparina o un derivado de heparina de bajo peso molecular (LMW).
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de vitamina K, por ejemplo y preferentemente coumarina.
Agentes de reducción de presión sanguínea se entenderán preferentemente como compuestos del grupo de antagonistas de calcio, antagonistas de angiotensina AII, inhibidores de ACE, inhibidores de vasopeptidasa, antagonistas de endotelina, inhibidores de renina, bloqueantes alfa, bloqueantes beta, antagonistas del receptor mineralocorticoide y diuréticos.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de calcio, por ejemplo y preferentemente nifedipina, amlodipina, verapamil o diltiazem.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un bloqueante del receptor alfa-1, por ejemplo y preferentemente prazosina o tamsulosina. En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un bloqueante beta, por ejemplo y preferentemente propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipranolol, nadolol, mepindolol, carazolol, sotalol, metoprolol, betaxolol, celiprolol, bisoprolol, carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol o bucindolol.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista del receptor de angiotensina AII, por ejemplo y preferentemente losartán, candesartán, valsartán, telmisartán, irbesartán, olmesartán, eprosartán o azilsartán.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de vasopeptidasa o inhibidor de endopeptidasa neutra (NEP), tal como por ejemplo y preferentemente sacubitril, omapatrilat o AVE-7688.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista del receptor de angiotensina doble AII/inhibidor de NEP (ARNI), por ejemplo y preferentemente LCZ696.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de ACE, por ejemplo y preferentemente enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril o trandopril.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de endotelina, por ejemplo y preferentemente bosentán, darusentán,
ambrisentán, tezosentán o sitaxsentán.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de renina, por ejemplo y preferentemente aliskiren, SPP-600 o SPP-800.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista del receptor mineralocorticoide, por ejemplo y preferentemente finerenona, espironolactona, canrenona, canrenoato de potasio o eplerenona.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un diurético, tal como por ejemplo y preferentemente furosemida, bumetanida, piretanida, torsemida, bendroflumetiazida, clorotiazida hidroclorotiazida, xipamida, indapamida, hidroflumetiazida, meticlotiazida, politiazida, triclorometiazida, clorotalidona, metolazona, quinetazona, acetazolamida, diclorofenamida, metazolamida, glicerina, isosorbida, manitol, amilorida o triamtereno.
Agentes para la alteración del metabolismo de grasa se entienden preferentemente como compuestos del grupo de inhibidores de CETP, agonistas del receptor de tiroides, inhibidores de la síntesis del colesterol tales como inhibidores de síntesis de escualeno o HMG-CoA-reductasa, inhibidores de ACAT, inhibidores de MTP, agonistas PPAR-alfa, PPAR-gamma y/o PPAR-delta, inhibidores de la absorción de colesterol, absorbentes de ácido biliar polimérico, inhibidores de la reabsorción de ácido biliar, inhibidores de lipasa y antagonistas de lipoproteína(a). En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de CETP, por ejemplo y preferentemente dalcetrapib, anacetrapib, BAY 60-5521 o CETP-vacuna (Avant).
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agonista del receptor de tiroides, por ejemplo y preferentemente D-tiroxina, 3,5,3'-triiodotironina (T3), CGS 23425 o axitirome (CGS 26214).
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de HMG-CoA-reductasa de la clase estatinas, por ejemplo y preferentemente lovastatina, simvastatina, pravastatina, fluvastatina, atorvastatina, rosuvastatina o pitavastatina.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de síntesis de escualeno, por ejemplo y preferentemente BMS-188494 o TAK-475. En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de ACAT, por ejemplo y preferentemente avasimiba, melinamida, pactimiba, eflucimiba o SMP-797.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de MTP, por ejemplo y preferentemente implitapida, R-103757, BMS-201038 o JTT-130.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agonista de PPAR-gamma, por ejemplo y preferentemente pioglitazona o rosiglitazona. En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agonista de PPAR-delta, por ejemplo y preferentemente GW 501516 o BAY 68-5042.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de la absorción del colesterol por ejemplo y preferentemente ezetimiba, tiquesida o pamaquesida.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de lipasa, por ejemplo y preferentemente orlistat.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un adsorbente de ácido biliar polimérico, por ejemplo y preferentemente colestiramina, colestipol, colesolvam, CholestaGel o colestimida.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de la reabsorción de ácido biliar, por ejemplo y preferentemente inhibidores de ASBT (= IBAT) tales como AZD-7806, S-8921, AK-105, BARI-1741, SC-435 o SC-635.
En una realización preferente de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de lipoproteína(a), por ejemplo y preferentemente gemcabeno calcio (CI-1027) o ácido nicotínico.
En una realización particularmente preferente , los compuestos de la presente invención se administran en combinación con uno o más agentes terapéuticos adicionales que se seleccionan del grupo que consiste en diuréticos, antagonistas de angiotensina AII, inhibidores de ACE, bloqueantes del receptor beta, antagonistas del receptor mineralocorticoide, nitratos orgánicos, donantes de NO, activadores de la guanilato ciclasa soluble (sGC), estimuladores de la guanilato ciclasa soluble y agentes inotrópicos positivos.
Por lo tanto, en una realización adicional, la presente invención se refiere a composiciones farmacéuticas que comprenden al menos uno de los compuestos de acuerdo con la invención y uno o más agentes terapéuticos adicionales para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades anteriormente mencionadas.
Además, los compuestos de la presente invención se pueden utilizar, como tales o en composiciones, en investigaciones o diagnósticos, o como patrones de referencia analítica y similares, bien conocidos en la técnica. Cuando los compuestos de la presente invención se administran como productos farmacéuticos a seres humanos u otros mamíferos, se pueden administrar en sí o como una composición farmacéutica que contiene, por ejemplo, 0,1 % a 99,5 % (más preferentemente, 0,5 % a 90 %) de principio activo con uno o más excipientes farmacéuticamente aceptables.
Por lo tanto, en otro aspecto, la presente invención se refiere a composiciones farmacéuticas que comprenden al menos uno de los compuestos de acuerdo con la invención, de forma convencional junto con uno o más excipientes farmacéuticamente adecuados, inertes y no tóxicos, y al uso de los mismos para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades anteriormente mencionadas.
Los compuestos de acuerdo con la invención pueden actuar de forma sistémica y/o local. Con este fin, se pueden administrar de una forma adecuada tal como, por ejemplo, por una vía oral, parenteral, pulmonar, nasal, lingual, sublingual, bucal, rectal, dérmica, transdérmica, conjuntival, ótica o tópica, o como un implante o stent.
Para estas vías de administración, los compuestos de la invención se pueden administrar en formas de aplicación adecuadas.
Son adecuadas para la administración oral las formas de aplicación que funcionan de acuerdo con el estado de la técnica y entregan los compuestos de acuerdo con la invención rápidamente y/o de forma modificada, y que contienen los compuestos de acuerdo con la invención en forma cristalina, amorfa y/o disuelta, tal como, por ejemplo comprimidos (comprimidos sin revestimiento o revestidos, por ejemplo con revestimientos entéricos o revestimientos que son insolubles o se disuelven con una retraso y controlan la liberación del compuesto de acuerdo con la invención), comprimidos que se desintegran rápidamente en la boca, o películas/obleas, películas/liofilizados, cápsulas (por ejemplo cápsulas de gelatina dura o blanda), comprimidos recubiertos con azúcar, gránulos, sedimentos, polvos, emulsiones, suspensiones, aerosoles o soluciones.
La aplicación parenteral se puede realizar evitando una etapa de absorción (de forma intravenosa, intraarterial, intracardíaca, intraespinal o intralumbar) o con inclusión de una absorción (de forma intramuscular, subcutánea, intracutánea, percutánea o intraperitoneal). Las formas de aplicación parenteral adecuadas incluyen preparaciones de inyección e infusión en la forma de soluciones, suspensiones, emulsiones, liofilizados y polvos estériles.
Las formas adecuadas para otras vías de aplicación incluyen, por ejemplo, formas farmacéuticas por inhalación (por ejemplo, inhaladores de polvo, nebulizadores), gotas nasales, soluciones o pulverizadores, comprimidos o cápsulas a ser administrados de forma lingual, sublingual o bucal (por ejemplo, e.g. trociscos, pastillas para chupar), supositorios, preparaciones para oídos y ojos (por ejemplo, gotas, ungüentos), cápsulas vaginales, suspensiones acuosas (lociones, mezclas de agitación), suspensiones lipofílicas, ungüentos, cremas, leches, pastas, espumas, polvos de uso externo, sistemas terapéuticos transdérmicos (por ejemplo, parches), implantes y stents.
En una realización preferente, la composición farmacéutica que comprende un compuesto de fórmula (I) como se define anteriormente se proporciona en una forma adecuada para la administración oral. En otra realización preferente, la composición farmacéutica que comprende un compuesto de fórmula (I) como se define anteriormente se proporciona en una forma adecuada para la administración intravenosa.
Los compuestos de acuerdo con la invención se pueden convertir en las formas de aplicación mencionadas de forma conocida en sí mezclando con excipientes farmacéuticamente aceptables inertes y no tóxicos. Estos excipientes incluyen, entre otros, portadores (por ejemplo, celulosa microcristalina, lactosa, manitol), disolventes (por ejemplo, polietilenglicoles líquidos), emulsionantes (por ejemplo, dodecil sulfato de sodio), tensioactivos (por ejemplo, polioxisorbitán oleato), dispersantes (por ejemplo, polivinilpirrolidona), polímeros sintéticos y naturales (por ejemplo, albúmina), estabilizantes (por ejemplo, antioxidantes tales como, por ejemplo, ácido ascórbico), colorantes (por ejemplo, pigmentos inorgánicos tales como, por ejemplo, óxidos de hierros), y agentes enmascarantes de sabor y/o olor.
Una dosis preferente del compuesto de la presente invención es el máximo que un paciente pueda tolerar y no desarrollar efectos secundarios graves. De forma ilustrativa, el compuesto de la presente invención se puede administrar de forma parenteral a una dosis de alrededor de 0,001 mg/kg a alrededor de 10 mg/kg, preferentemente de alrededor de 0,01 mg/kg a alrededor de 1 mg/kg de peso corporal. En la administración oral, un ejemplo de intervalo de dosis es alrededor de 0,01 a 100 mg/kg, preferentemente alrededor de 0,01 a 20 mg/kg, y más preferentemente alrededor de 0,1 a 10 mg/kg de peso corporal. Los intervalos intermedios de los valores anteriormente mencionados también se pretenden como parte de la invención.
Sin embargo, los niveles de dosificación reales y el transcurso de tiempo de la administración de los principios activos en las composiciones farmacéuticas de la invención pueden variar para obtener una cantidad del principio activo que sea eficaz para alcanzar la respuesta terapéutica deseada para un paciente, composición y modo de administración particulares, sin ser tóxica para el paciente. Por lo tanto puede ser necesario cuando fuera apropiado desviarse de las cantidades mencionadas, en particular en función de la edad, sexo, peso corporal, dieta y estado general de salud del paciente, las características de biodisponibilidad y farmacodinámica del compuesto particular y su modo y vía de administración, el tiempo o intervalo en el cual ocurre la administración, el régimen de dosificación seleccionado, la respuesta del paciente individual al principio activo, la enfermedad específica implicada, el grado o la implicación o gravedad de la enfermedad, el tipo de tratamiento concurrente (es decir, la interacción del compuesto de la invención con otros productos terapéuticos coadministrados) y otras circunstancias relevantes.
Por lo tanto, puede ser satisfactorio en algunos casos arreglarse con menos de la cantidad mínima mencionada anteriormente, mientras en otros casos se debe exceder el límite superior establecido. El tratamiento se puede iniciar con dosificaciones más pequeñass que son menores que la dosis óptima del compuesto. Posteriormente, la dosificación se puede aumentar en incrementos pequeños hasta alcanzar el efecto óptimo según las circunstancias. Por conveniencia, la dosificación diaria total se puede dividir y administrar en partes individuales repartidas en el día.
Las siguientes realizaciones ejemplares ilustran la invención. La invención no se limita a los ejemplos.
Los porcentajes en las pruebas y ejemplos a continuación, salvo a menos que se indique lo contrario, son en peso; las partes son en peso. Las relaciones de disolvente, relaciones de dilución y concentraciones indicadas para líquidos/soluciones líquidas se basan cada una en volumen.
A. Ejemplos
Abreviaturas v acrónimos:
Ac acetilo
ac. acuoso (solución)
a. amplio (señal 1H NMR)
cat. catalítico
conc. concentrado
d doblete (señal 1H NMR)
DCI ionización química directa (MS)
d.e. exceso diastereomérico
DMF W,W-dimetilformamida
DMSO dimetilsulfóxido
EI ionización por impacto de electrones (MS)
eq. equivalente(s)
ESI ionización por electro-pulverización (MS)
Et etilo
h hora(s)
1H NMR espectroscopía de resonancia magnética nuclear de protón
HPLC cromatografía líquida de alto rendimiento
LC/MS espectroscopía de masa acoplada con cromatografía líquida
m multiplete (señal 1H NMR)
Me metilo
min minuto(s)
MS espectroscopía de masas
MTBE metil ferc-butil éter
m/z relación masa a carga (MS)
de teoría de teoría (rendimiento químico)
c cuarteto (señal 1H NMR)
cuant. cuantitativo (rendimiento)
rac racémico
Rf factor de retención TLC
RP fase inversa (HPLC)
ta temperatura ambiente
Rt tiempo de retención (HPLC)
s singlete (señal 1H NMR)
sat. saturado (solución)
SFC cromatografía de fluidos supercríticos
t triplete (señal 1H NMR)
tBu tere-butilo
tere terciario
TFA ácido trifluoroacético
THF tetrahidrofurano
TLC cromatografía de capa fina
UV ultravioleta
Procedimientos LC/MS y HPLC
Procedimiento 1 (LC/MS):
Instrumento: Waters Acquity SQD UPLC System; columna: Waters Acquity UPLC HSS T31,8 ^, 50 mm x 1 mm; eluyente A: 1 l agua+ 0,25 ml 99 % ácido fórmico, eluyente B: 1 l acetonitrilo 0,25 ml 99 % ácido fórmico; gradiente: 0,0 min 90 % A ^ 1,2 min 5 % A ^ 2,0 min 5 % A; horno: 50 °C; velocidad de flujo: 0,40 ml/min; detección UV: 208-400 nm.
Procedimiento 2 (LC/MS):
Instrumento: Waters Acquity SQD UPLC System; columna: Waters Acquity UPLC HSS T31,8 ^, 50 mm x 1 mm; eluyente A: 1 l agua+ 0,25 ml 99 % ácido fórmico, eluyente B: 1 l acetonitrilo 0,25 ml 99 % ácido fórmico; gradiente: 0,0 min 95 % A ^ 6,0 min 5 % A ^ 7,5 min 5 % A; horno: 50 °C; velocidad de flujo: 0,35 ml/min; detección UV: 210-400 nm.
Procedimiento 3 (LC/MS):
Instrumento MS: Agilent MS Quad 6150; Instrumento HPLC: Agilent 1290; columna: Waters Acquity UPLC HSS T3 1,8 ^, 50 mm x 2,1 mm; eluyente A: 1 l agua+ 0,25 ml 99 % ácido fórmico, eluyente B: 1 l acetonitrilo 0,25 ml 99 % ácido fórmico; gradiente: 0,0 min 90 % A ^ 0,3 min 90 % A ^ 1,7 min 5 % A ^ 3,0 min 5 % A; horno: 50 °C; velocidad de flujo: 1,20 ml/min; detección UV: 205-305 nm.
Procedimiento 4 (HPLC preparativa):
Columna: Chromatorex C18 10 ^m, 125 mm x 30 mm; eluyente A: agua+ 0,05 % TFA, eluyente B: acetonitrilo 0,05 % TFA; gradiente: 20 % B ^ 45 % B, 45 % B isocrático, 45 % B ^ 80 % B; temperatura de columna: temperatura ambiente; velocidad de flujo: 50 ml/min; detección UV: 210 nm.
Materiales de partida e intermedios:
Ejemplo 1A
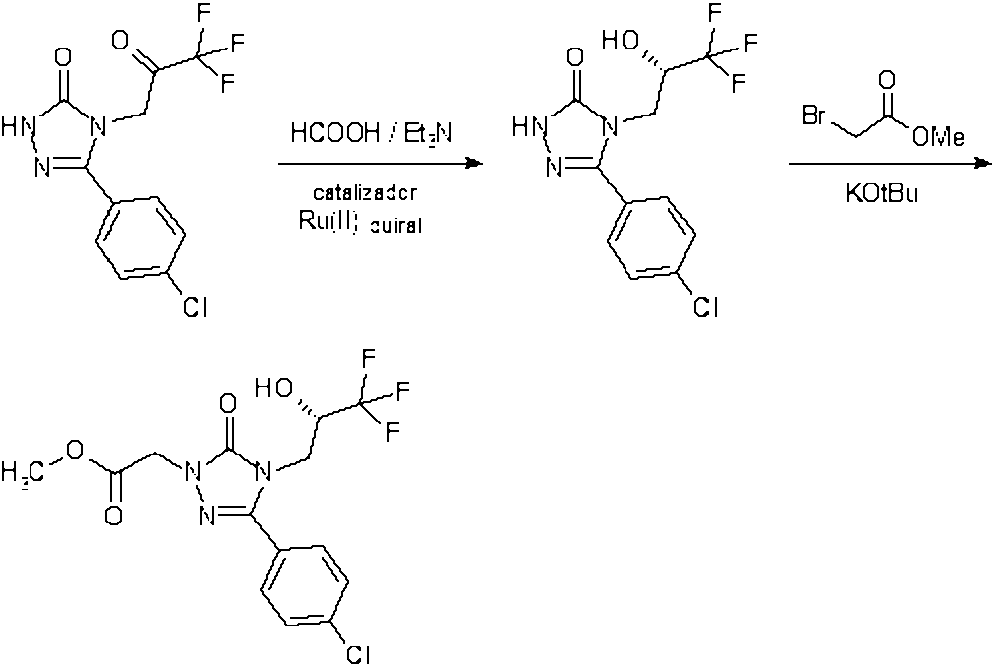
{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acetato de metilo

Bajo argón, se agregó en porciones terc-butóxido de potasio (9,118 g, 81,26 mmol) a temperatura ambiente a una solución de 5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (Ejemplo 5A en el documento WO 2011/104322-A1; 20 g, 65,01 mmol) en THF (40 ml). A esta solución se agregó bromoacetato de metilo (10,939 g, 71,51 mmol), y la mezcla se agitó a temperatura ambiente durante una noche. La mezcla de reacción luego se diluyó con agua y se extrajo con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. Se obtuvo 16,4 g (30,23 mmol) del compuesto deseado (46,5 % de rendimiento, 70 % de pureza).
LC/MS [procedimiento 1]: Rt = 0,90 min; MS [ESlpos]: m/z = 380 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 3,70 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,19-4,33 (m, 1H), 4,72 (s, 2H), 6,92 (d, 1H), 7,60-7,69 (m, 2H), 7,73-7,81 (m, 2H).
El compuesto del título también se puede sintetizar mediante el procedimiento descrito en el documento WO 2011/104322-A1 (Ejemplo 7A).
Ejemplo 2A
2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acetohidrazida

Se disolvió 7,2 g (18,96 mmol) de {3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acetato de metilo en 60 ml de etanol absoluto. A esta solución se agregó 2,088 g (41,71 mmol) de hidrato de hidrazina, y la mezcla se agitó bajo reflujo durante 5 h y luego a temperatura ambiente durante una noche. La mezcla resultante se concentró parcialmente al vacío y luego se diluyó con agua y se extrajo con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El residuo se disolvió en diclorometano y después de la cristalización el sólido blanco se retiró por filtración y se secó a alto vacío. Se obtuvo 7,02 g (18,49 mmol) del compuesto deseado (97,5 % de rendimiento).
LC/MS [procedimiento1]: Rt = 0,73 min; MS [ESIpos]: m/z = 380 (M+H)+
1H NMR (400 MHz, DMSO-cfe): L [ppm] 3,82 (dd, 1H), 3,96 (dd, 1H), 4,24-4,34 (m, 3H), 4,38 (d, 2H), 6,90 (d, 1H), 7,61-7,66 (m, 2H), 7,73-7,78 (m, 2H), 9,23 (t, 1H).
Ejemplo 3A
-(4-clorofenil)-2-{[5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

Bajo argón, se agregó en porciones etóxido de sodio (2,987 g, 42,14 mmol, 96 % de pureza) a temperatura ambiente a una solución de 2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acetohidrazida (8,0 g, 21,07 mmol) y clorhidrato de 2-hidroxiacetamidina (2,329 g, 21,07 mmol) en DMF (200 ml). La mezcla de reacción se agitó a 100 °C durante una noche. Después de enfriarse, la mezcla de reacción se concentró parcialmente al vacío y luego se diluyó con acetato de etilo. La mezcla resultante se lavó con agua, y después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron bajo presión reducida. El sólido resultante se secó bajo alto vacío para proporcionar 8,69 g (89 % de pureza, 18,47 mmol) del compuesto deseado que se usó sin purificación adicional (~88 % de rendimiento).
LC/MS [procedimiento 1]: Rt = 0,74 min; MS [ESIpos]: m/z = 419 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 3,83 (dd, 1H), 3,98 (dd, 1H), 4,24-4,36 (m, 1H), 4,53 (a, s, 2H), 4,96 (br, s, 2H), 5,64 (a, s, 1H), 6,91 (d, 1H), 7,58-7,67 (m, 2H), 7,72-7,78 (m, 2H), 13,75 (a, s, 1H).
Ejemplo 4A
5-(4-clorofenil)-2-({5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

Bajo argón, se agregó en porciones etóxido de sodio (1,531 g, 21,59 mmol, 96 % de pureza) a temperatura ambiente a una solución de 2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acetohidrazida (4,1 g, 10,80 mmol) y clorhidrato de 2-hidroxipropanimidamida (1,480 g, 11,88 mmol) en DMF (110 ml). La mezcla de reacción se agitó a 120 °C durante 4,5 h. Después de enfriarse, la mezcla de reacción se concentró parcialmente al vacío y luego se diluyó con acetato de etilo. La mezcla resultante se lavó con agua, y después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron bajo presión reducida. El sólido resultante se secó bajo alto vacío para proporcionar 4,90 g (92 % de pureza, 10,42 mmol) del compuesto deseado como una mezcla de diastereómeros que se usó sin purificación adicional.
LC/MS [procedimiento 1]: Rt = 0,82 min; MS [ESIpos]: m/z = 433 (M+H)+
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,39 (d, 3H), 3,79-3,88 (m, 1H), 3,93-4,02 (m, 1H), 4,24-4,36 (m, 1H), 4,80 (quin, 1H), 4,89-5,00 (m, 2H), 5,73 (d, 1H), 6,93 (d, 1H), 7,58-7,65 (m, 2H), 7,70-7,77 (d, 2H), 13,68 (s, 1H).
Ejemplo 5A
5-(4-clorofenil)-2-({5-[(1 S)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

El compuesto del título se sintetizó de forma análoga al Ejemplo 4A comenzando con clorhidrato de (2S)-2-hidroxipropanimidamida (1,1 eq.), usando solo 1,1 eq. de etóxido de sodio como base (temperatura de reacción: 100°C).
LC/MS [procedimiento 1]: Rt = 0,81 min; MS [ESIpos]: m/z = 433 (M+H)+.
Clorhidrato de (2S)-2-hidroxipropanimidamida, que está disponible en el mercado, también se sintetizó a partir de (2S)-2-hidroxipropanamida [L-(-)-lactamida] mediante el procedimiento descrito en el documento WO 00/ 59510-A1 (Preparación 11, etapa A) y el documento WO 2013/138860-A1 (intermedio del precursor 82, p. 101 -102).
Ejemplo 6A
5-(4-clorofenil)-2-({5-[(1 R)-1 -hidroxietil]-1 H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

El compuesto del título se sintetizó de forma análoga al ejemplo 4A comenzando con clorhidrato de (2R)-2-hidroxipropanimidamida (1,1 eq.), usando solo 1,1 eq. de etóxido de sodio como base (temperatura de reacción: 100 °C).
LC/MS [procedimiento 3]: Rt = 1,00 min; MS [ESIpos]: m/z = 433 (M+H)+.
Clorhidrato de (2R)-2-hidroxipropanimidamida, que está disponible en el mercado, también se sintetizó a partir de (2R)-2-hidroxipropanamida [R-(+)-lactamida] mediante el procedimiento descrito en el documento WO 00/ 59510-A1 (Preparación 11, etapa A) y el documento WO 2013/138860-A1 (intermedio del precursor 82, p. 101-102).
Ejemplos de preparaciones:
Los ejemplos de compuestos que presentan un sustituyente 1-hidroxietilo [R1 en la fórmula (I) = CH3] que se denominan a continuación "diastereómero 1" y "diastereómero 2", respectivamente, representan pares de diastereómeros separados cuya configuración absoluta con respecto al resto 1-hidroxietilo (1R o 1S) no se ha determinado.
Los valores de exceso diastereomérico (d.e.) se determinaron de la forma habitual por análisis de áreas de pico de HPLC de acuerdo con la fórmula siguiente:
d.e. - diastereómero 1 ( % área pico ) — diastereómero 2 (% área pico )
diastereómero 1 (% área pico ) diastereómero 2 (% área pico ) x 100%.
Ejemplo 1
5-(4-clorofenil)-2-{[1-(3-clorofenil)-5-(hidroximetil)-1 W-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

A una solución de 5-(4-clorofenil)-2-{[5-(mdroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,96 mmol) en piridina (12 ml) se agregó ácido (3-clorofenil)borónico (298,74 mg, 1,91 mmol) y acetato de cobre (II) (347 mg, 1,91 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (74,7 mg, 0,48 mmol, 0,5 eq.) debido a conversión incompleta. Después de agitar durante dos días adicionales, la mezcla de reacción se diluyó con MTBE y luego se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (113 mg, 0,21 mmol) (rendimiento 22,4 %).
LC/MS [procedimiento 2]: Rt = 3,07 min; MS [ESIpos]: m/z = 529 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,58 (s, 2H), 5,08 (s, 2H), 6,90 (a, d, 1H), 7,55-7,64 (m, 4H), 7,65-7,69 (m, 1H), 7,73-7,78 (m, 2H), 7,78-7,81 (m, 1H).
Ejemplo 2
5-(4-clorofen¡l)-2-{[1-(3-fluorofen¡l)-5-(h¡drox¡met¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona

A una soluc¡ón de 5-(4-clorofen¡l)-2-{[5-(h¡drox¡met¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (100 mg, 0,24 mmol) en p¡r¡d¡na (3 ml) se agregó ác¡do (3-fluorofen¡l)borón¡co (66,83 mg, 0,48 mmol) y acetato de cobre (II) (86,75 mg, 0,48 mmol). La mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante 5 días, después de lo cual se agregó ác¡do borón¡co ad¡c¡onal (16,72 mg, 0,12 mmol, 0,5 eq.) deb¡do a convers¡ón ¡ncompleta. Después de ag¡tar durante dos días ad¡c¡onales, la mezcla de reacc¡ón se d¡luyó con MTBE y luego se ¡nact¡vó con ác¡do clorhídr¡co acuoso (0,5 M). Después de la separac¡ón de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgán¡cas comb¡nadas se secaron en sulfato de sod¡o, se f¡ltraron y concentraron al vacío. El producto bruto se pur¡f¡có por HPLC preparat¡va [proced¡m¡ento 4], y se obtuvo el compuesto deseado (25 mg, 0,05 mmol) (rend¡m¡ento 20,2 %).
LC/MS [proced¡m¡ento 2]: Rt = 2,87 m¡n; MS [ESlpos]: m/z = 513 (M+H)+
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,59 (s, 2H), 5,08 (s, 2H), 6,90 (a, d, 1H), 7,37 (td, 1H), 7,51-7,66 (m, 5H), 7,72-7,79 (m, 2H).
Ejemplo 3
5-(4-clorofen¡l)-2-{[5-(h¡drox¡met¡l)-1-(2-metox¡fen¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona

A una soluc¡ón de 5-(4-clorofen¡l)-2-{[5-(h¡drox¡met¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (300 mg, 0,72 mmol) en p¡r¡d¡na (9 ml) se agregó ác¡do (2-metox¡fen¡l)borón¡co (217,72 mg, 1,43 mmol) y acetato de cobre (II) (260,25 mg, 1,43 mmol). La mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante 5 días, después de lo cual se agregó ác¡do borón¡co ad¡c¡onal (54,4 mg, 0,36 mmol, 0,5 eq.) deb¡do a convers¡ón ¡ncompleta. Después de ag¡tar durante dos días ad¡c¡onales, la mezcla de reacc¡ón se d¡luyó con MTBE y luego se ¡nact¡vó con ác¡do clorhídr¡co acuoso (0,5 M). Después de la separac¡ón de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgán¡cas comb¡nadas se secaron en sulfato de sod¡o, se f¡ltraron y concentraron al vacío. El producto bruto se pur¡f¡có por HPLC preparat¡va [proced¡m¡ento 4], y se obtuvo el compuesto deseado (73 mg, 0,14 mmol) (rend¡m¡ento 22,4 %, 98 % de pureza). Lc /MS [proced¡m¡ento 3]: Rt = 1.18 m¡n; MS [ESIpos]: m/z = 525 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 3,77 (s, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,31 (a, s, 1H), 4,35 (s, 2H), 5,04 (s, 2H), 6,91 (a, s, 1H), 7,09 (t, 1H), 7,25 (d, 1H), 7,37 (dd, 1H), 7,52 (td, 1H), 7,63 (d, 2H), 7,76 (d, 2H).
Ejemplo 4
5-(4-clorofen¡l)-2-{[1-(2-clorofen¡l)-5-(h¡drox¡met¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]
2,4-dihidro-3H-1,2,4-tr¡azol-3-ona

A una solución de 5-(4-clorofen¡l)-2-{[5-(h¡drox¡met¡l)-1H-1,2,4-tr¡azol-3-¡l]metil}-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (3 g, 5,946 mmol, 83 % de pureza) en piridina (75 ml) se agregó ácido (2-clorofen¡l)borónico (930 mg, 5,946 mmol) y acetato de cobre (II) (2,16 g, 11,89 mmol). La mezcla de reacción se agitó a temperatura ambiente durante una noche, después de lo cual se agregó ácido borónico adicional (500 mg, 3,20 mmol) debido a conversión incompleta. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 9 días. En este tiempo, se agregaron tres porciones adicionales de ácido borónico (1,5 g en total, 9,6 mmol). Después de esto, la mezcla de reacción se diluyó con MTBE y luego se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (1,44 g, 2,72 mmol) (rendimiento 45,7 %).
LC/MS [procedimiento 2]: Rt = 2,79 min; MS [ESlpos]: m/z = 529 (M+H)+
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 3,85 (dd, 1H), 4,01 (dd, 1H), 4,31 (a, s, 1H), 4,41 (s, 2H), 5,07 (s, 2H), 6,91 (d, 1H), 7,49-7,67 (m, 5H), 7,68-7,80 (m, 3H).
Ejemplo 5
5-(4-clorofen¡l)-2-{[5-(h¡drox¡met¡l)-1-(2-met¡lfen¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-trifluoro-2-h¡drox¡prop¡l]-2,4-dihidro-3H-1,2,4-tr¡azol-3-ona

A una solución de 5-(4-clorofen¡l)-2-{[5-(h¡drox¡met¡l)-1H-1,2,4-tr¡azol-3-¡l]metil}-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (400 mg, 0,96 mmol) en piridina (12 ml) se agregó ácido (2-metilfenil)borón¡co (259,7 mg, 1,91 mmol) y acetato de cobre (II) (347 mg, 1,91 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (64,9 mg, 0,48 mmol, 0,5 eq.) debido a conversión incompleta. Después de agitar durante dos días adicionales, la mezcla de reacción se diluyó con MTBE y luego se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (58 mg, 0,11 mmol) (rendimiento 11,9 %).
LC/MS [procedimiento 2]: Rt = 2,85 min; MS [ESIpos]: m/z = 509 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 2,01 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,34 (s, 2H), 5,07 (s, 2H), 6,90 (a, s, 1H), 7,32-7,50 (m, 4H), 7,62 (a, d, 2H), 7,75 (a. d, 2H).
Ejemplo 6
2-{[1-(3-cloro-5-fluorofen¡l)-5-(h¡drox¡met¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-5-(4-clorofenil)-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-dih¡dro-3H-1,2,4-tr¡azol-3-ona

A una solución de 5-(4-clorofenil)-2-{[5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,96 mmol) en piridina (12 ml) se agregó ácido (3-cloro-5-fluorofenil)borónico (333,1 mg, 1,91 mmol) y acetato de cobre (II) (347 mg, 1,91 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (83,2 mg, 0,48 mmol, 0,5 eq.) debido a conversión incompleta. Después de agitar durante dos días adicionales, la mezcla de reacción se diluyó con MTBE y luego se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (91 mg, 0,17 mmol) (rendimiento 17,4 %).
LC/MS [procedimiento 3]: Rt = 1.31 min; MS [ESIpos]: m/z = 547 (M+H)+
1H NMR (400 MHz, DMSO-cfe): S [ppm] 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,63 (s, 2H), 5,08 (s, 2H), 6,90 (a, s, 1H), 7,59-7,66 (m, 4H), 7,67-7,71 (m, 1H), 7,72-7,78 (m, 2H).
Ejemplo 7
5-(4-clorofenil)-2-{[1-(3,5-difluorofenil)-5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

El compuesto del título se preparó de forma análoga al ejemplo 1 comenzado con 360 mg (0,86 mmol) de 5-(4-clorofenil)-2-{[5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona Se obtuvo 61 mg (0,11 mmol) del compuesto deseado (13,4 % de rendimiento).
LC/MS [procedimiento 3]: Rt = 1,25 min; MS [ESIpos]: m/z = 531 (M+H)+
1H NMR (500 MHz, DMSO-cfe): S [ppm] 3,86 (dd, 1H), 4,01 (dd, 1H), 4,25-4,38 (m, 1H), 4,64 (d, 2H), 5,08 (s, 2H), 5,94 (t, 1H), 6,92 (d, 1H), 7,46 (tt, 1H), 7,49-7,55 (m, 2H), 7,62 (a, d, 2H), 7,76 (a, d, 2H).
Ejemplo 8
5-(4-clorofenil)-2-{[5-(hidroximetil)-1-(2-(trifluorometil)fenil)-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

El compuesto del título se preparó de forma análoga al ejemplo 1 comenzado con 500 mg (1,19 mmol) de 5-(4-dorofeml)-2-{[5-(h¡drox¡met¡l)-1H-1,2,4-triazol-3-¡l]met¡l}-4-[(2S)-3,3,3-tnfluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-triazol-3-ona Se obtuvo 104 mg (0,18 mmol) del compuesto deseado (15,5 % de rend¡m¡ento).
LC/MS [proced¡m¡ento 3]: Rt = 1,24 m¡n; MS [ESlpos]: m/z = 563 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,39 (s, 2H), 5,01-5,12 (m, 2H), 5,53 (a, s, 1H), 6,92 (d, 1H), 7,63 (d, 2H), 7,70 (d, 1H), 7,75 (d, 2H), 7,78-7,91 (m, 2H), 7,97 (d, 1H).
Ejemplo 9
2-{[1-(2-doro-5-fluorofeml)-5-(h¡drox¡met¡l)-1H-1,2,4-triazol-3-¡l]met¡l}-5-(4-dorofeml)-4-[(2S)-3,3,3-tnfluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona

El compuesto del título se preparó de forma análoga al ejemplo 1 comenzado con 500 mg (1,19 mmol) de 5-(4-dorofeml)-2-{[5-(h¡drox¡met¡l)-1H-1,2,4-triazol-3-¡l]met¡l}-4-[(2S)-3,3,3-tnfluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona Se obtuvo 8,7 mg (0,02 mmol) del compuesto deseado (1,3 % de rend¡m¡ento, 95 % de pureza). LC/MS [proced¡m¡ento 3]: Rt = 1,23 m¡n; MS [ESlpos]: m/z = 547 (M+H)+
1H NMR (500 MHz, DMSO-cfe): 8 [ppm] 3,86 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,46 (s, 2H), 5,03-5,11 (m, 2H), 6,92 (a, d, 1H), 7,53 (td, 1H), 7,61-7,65 (m, 2H), 7,68 (dd, 1H), 7,73-7,80 (m, 3H).
Ejemplo 10
5-(4-clorofen¡l)-2-({5-[(1RS)-1-h¡drox¡et¡l]-1-(2-metox¡fen¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (mezcla diastereomérica )
A una solución de 5-(4-clorofen¡l)-2-({5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l}metil)-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (500 mg, 1,16 mmol) en piridina (15 ml) se agregó ácido (2-metoxifen¡l)borónico (351,1 mg, 2,31 mmol) y acetato de cobre (II) (419,7 mg, 2,31 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (87 mg, 0,58 mmol, 0,5 eq.) debido a conversión incompleta. Después de agitar durante dos días adicionales, la mezcla de reacción se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (132 mg, 0,22 mmol) como una mezcla de diastereómeros (rendimiento 19,1 %, 90 % de pureza).
LC/MS [procedimiento 2]: Rt = 2,87 min; MS [ESlpos]: m/z = 539 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,36 (d, 3H), 3,76 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,47-4,61 (m, 1H), 4,98-5,10 (m, 2H), 6,90 (a, s, 1H), 7,09 (td, 1H), 7,24 (dd, 1H), 7,35 (dd, 1H), 7,49-7,55 (m, 1H), 7,60-7,65 (m, 2H), 7,73-7,79 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 128 mg disuelto en 10 ml metanol; volumen de inyección: 0,3 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/metanol 70:30; velocidad de flujo: 80 ml/min; temperatura: 40 °C; detección UV: 210 nm]. Después de la separación, se aislaron 43,6 mg de diastereómero 1 (Ejemplo 11), que eluyó primero, y 45,1 mg de diastereómero 2 (Ejemplo 12), que eluyó después.
Ejemplo 11
5-(4-clorofen¡l)-2-{[5-(1-hidrox¡et¡l)-1-(2-metox¡fen¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica (SFC): Rt = 3,08 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-3250 x 4 mm; eluyente: dióxido de carbono/metanol (5 % ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,36 (d, 3H), 3,76 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,52 (quin, 1H), 4,96-5,11 (m, 2H), 5,37 (d, 1H), 6,90 (d, 1H), 7,09 (td, 1H), 7,24 (d, 1H), 7,35 (dd, 1H), 7,52 (td, 1H), 7,60-7,65 (m, 2H), 7,73-7,79 (m, 2H).
Ejemplo 12
5-(4-clorofen¡l)-2-{[5-(1-hidrox¡et¡l)-1-(2-metox¡fen¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡propil]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (diastereómero 2)
HPLC quiral analítica (SFC): Rt = 3,38 min, d.e. = 91,1 % [columna: Daicel Chiralcel® OX-3 250 x 4 mm; eluyente: dióxido de carbono/metanol (5 % ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,36 (d, 3H), 3,76 (s, 3H), 3,84 (dd, 1H), 4,00 (dd, 1H), 4,31 (a, s, 1H), 4,52 (quin, 1H), 4,99-5,09 (m, 2H), 5,37 (d, 1H), 6,91 (d, 1H), 7,09 (td, 1H), 7,24 (d, 1H), 7,35 (dd, 1H), 7,52 (td, 1H), 7,60-7,65 (m, 2H), 7,74-7,79 (m, 2H).
Ejemplo 13
2-({1-(3-cloro-5-fluorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l}met¡l)-5-(4-clorofenil)-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica)

A una solución de 5-(4-clorofen¡l)-2-({5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l}metil)-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-triazol-3-ona (430 mg, 0,99 mmol) en piridina (12,5 ml) se agregó ácido (3-cloro-5-fluorofen¡l)borónico (346,49 mg, 1,99 mmol) y acetato de cobre (II) (360,9 mg, 1,99 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (86,7 mg, 0,497 mmol, 0,5 eq.) debido a conversión incompleta. Después de agitar durante dos días adicionales, la mezcla de reacción se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (148 mg, 0,26 mmol) como una mezcla de diastereómeros (rendimiento 26,5 %).
LC/MS [procedimiento 2]: Rt = 3,28 min; MS [ESlpos]: m/z = 561 (M+H)+
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,29 (a, s, 1H), 4,83-4,91 (m, 1H), 5,01-5,13 (m, 2H), 6,89 (a, s, 1H), 7,55-7,70 (m, 5H), 7,72-7,78 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa (SFC) [preparación de muestras: 143 mg disuelto en 15 ml metanol; volumen de inyección: 0,5 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: dióxido de carbono/metanol 80:20; velocidad de flujo: 80 ml/min; temperatura: 40 °C; detección UV: 210 nm]. Después de la separación, se aislaron 70 mg de diastereómero 1 (Ejemplo 14), que eluyó primero, y 60 mg de diastereómero 2 (Ejemplo 15), que eluyó después.
Ejemplo 14
2-{[1-(3-cloro-5-fluorofen¡l)-5-(1-h¡drox¡et¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-5-(4--clorofen¡l)-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-triazol-3-ona
(diastereómero 1)
HPLC quiral analítica (SFC): Rt = 4,45 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-3 250 x 4 mm; eluyente: dióxido de carbono/metanol (5 % ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,36 (m, 1H), 4,87 (quin, 1H), 5,07 (s, 2H), 5,83 (d, 1H), 6,89 (d, 1H), 7,58-7,69 (m, 5H), 7,71-7,79 (m, 2H).
Ejemplo 15
2-{[1-(3-cloro-5-fluorofen¡l)-5-(1-h¡drox¡et¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-5-(4--clorofen¡l)-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica (SFC): Rt = 4,80 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-3 250 x 4 mm; eluyente: dióxido de carbono/metanol ( 5% ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,23-4,36 (m, 1H), 4,87 (quin, 1H), 5,02-5,12 (m, 2H), 5,83 (d, 1H), 6,89 (d, 1H), 7,57-7,70 (m, 5H), 7,71-7,79 (m, 2H).
Ejemplo 16
5-(4-clorofen¡l)-2-({5-[(1RS)-1-h¡drox¡et¡l]-1-fen¡l-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-trifluoro-2-h¡drox¡prop¡l]-2,4-dihidro-3H-1,2,4-tr¡azol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofen¡l)-2-({5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l}metil)-4-[(2S)-3,3,3-tr¡fluoro-2-hidrox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-triazol-3-ona (250 mg, 0,462 mmol, 80 % de pureza) en piridina (6 ml) se agregó ácido fenilborónico (112,69 mg, 0,92 mmol) y acetato de cobre (II) (167,9 mg, 0,92 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 4 días, después de
lo cual se agregó ácido borónico adicional (28,2 mg, 0,23 mmol, 0,5 eq.) debido a conversión incompleta. Después de 4 h a 60 °C seguro de agitación a temperatura ambiente durante dos días adicionales, la mezcla de reacción se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (42,5 mg, 0,08 mmol) como una mezcla de diastereómeros (rendimiento 18,1 %).
LC/MS [procedimiento 3]: Rt = 1.21 min; MS [ESIpos]: m/z = 509 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 6 [ppm] 1,45 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,77 (q, 1H), 4,99-5,13 (m, 2H), 6,90 (a, s, 1H), 7,47-7,66 (m, 7H), 7,72-7,79 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 38,9 mg disuelto en 1 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 75:25; velocidad de flujo: 15 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 13 mg de diastereómero 1 (Ejemplo 17), que eluyó primero, y 14 mg de diastereómero 2 (Ejemplo 18), que eluyó después.
Ejemplo 17
5-(4-clorofenil)-2-{[5-(1 -hidroxietil)-1 -fenil-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 8,18 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 6 [ppm] 1,45 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (br, s, 1H), 4,77 (quin, 1H), 4,98-5,14 (m, 2H), 5,68 (d, 1H), 6,89 (d, 1H), 7,48-7,65 (m, 7H), 7,72-7,80 (m, 2H).
Ejemplo 18
5-(4-clorofenil)-2-{[5-(1 -hidroxietil)-1 -fenil-1 H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 11,40 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 6 [ppm] 1.45 (d, 3H), 3.85 (dd, 1H), 4.00 (dd, 1H), 4.30 (a. s, 1H), 4.77 (quin, 1H), 5.07 (s, 2H), 5.68 (d, 1H), 6.90 (d, 1H), 7.47-7.65 (m, 7H), 7.72-7.79 (m, 2H).
Ejemplo 19
5-(4-clorofenil)-2-({1 -[3-difluorometil)fenil]-5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,74 mmol, 80 % de pureza) en piridina (9,6 ml) se agregó ácido [3-(difluorometil)fenil]borónico (254,26 mg, 1,48 mmol) y acetato de cobre (Il) (268,6 mg, 1,48 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 5 días. La mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con
acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (47 mg, 0,08 mmol) como una mezcla de diastereómeros (rendimiento 11,3 %).
Lc /MS [procedimiento 1]: Rt = 1,04 min; MS [ESIpos]: m/z = 559 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,36 (m, 1H), 4,81 (q, 1H), 5,02-5,13 (m, 2H), 5,74 (a, s, 1H), 6,89 (a, s, 1H), 7,14 (t, 1H), 7,59-7,65 (m, 2H), 7,69-7,78 (m, 4H), 7,81-7,87 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 45 mg disuelto en 1 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 75:25; velocidad de flujo: 15 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 20 mg de diastereómero 1 (Ejemplo 20), que eluyó primero, y 20 mg de diastereómero 2 (Ejemplo 21), que eluyó después.
Ejemplo 20
5-(4-clorofenil)-2-({1 -[3-(difluorometil)fenil]-5-(1 -hidroxietil)-1 H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 6,72 min, d.e. = 99 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSOdO: 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,23-4,36 (m, 1H), 4,81 (quin, 1H), 5,02-5,14 (m, 2H), 5,74 (d, 1H), 6,88 (d, 1H), 7,14 (t, 1H), 7,59-7,64 (m, 2H), 7,69-7,79 (m, 4H), 7,80-7,87 (m, 2H).
Ejemplo 21
5-(4-clorofenil)-2-({1 -[3-(difluorometil)fenil]-5-(1 -hidroxietil)-1 H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 9,36 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,37 (m, 1H), 4,81 (quin, 1H), 5,08 (s, 2H), 5,74 (d, 1H), 6,89 (d, 1H), 7,13 (t, 1H), 7,58-7,66 (m, 2H), 7,69-7,79 (m, 4H), 7,81 7,87 (m, 2H).
Ejemplo 22
5-(4-clorofenil)-2-({5-[(1 RS)-1 -hidroxietil]-1 -[3-trifluorometil)fenil]-1 H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,74 mmol, 80 % de pureza) en piridina (9,6 ml) se agregó ácido [3-(trifluorometil)fenil]borónico (280,86 mg, 1,48 mmol) y acetato de cobre (II) (268,6 mg, 1,48 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (70,2 mg, 0,37 mmol, 0,5 eq.) debido a conversión incompleta. Después de 4 h a 60 °C seguido de agitación a temperatura ambiente durante una noche, la mezcla de reacción se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M).
Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (58,2 mg, 0,10 mmol) como una mezcla de diastereómeros (rendimiento 13,6 %).
LC/MS [procedimiento 1]: Rt = 1,06 min; MS [ESIpos]: m/z = 577 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,83 (q, 1H), 5,02-5,15 (m, 2H), 5,79 (a, s, 1H), 6,85-6,94 (m, 1H), 7,58-7,66 (m, 2H), 7,71-7,85 (m, 3H), 7,86-7,92 (m, 1H), 7,94-8,01 (m, 1H), 8,04 (s, 1H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 58 mg disuelto en 2 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 75:25; velocidad de flujo: 15 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 19,5 mg de diastereómero 1 (Ejemplo 23), que eluyó primero, y 19,2 mg de diastereómero 2 (Ejemplo 24), que eluyó después.
Ejemplo 23
5-(4-clorofenil)-2-({5-(1-hidroxietil)-1 -[3-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 4,94 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,83 (q, 1H), 5,03-5,13 (m, 2H), 5,79 (a, s, 1H), 6,88 (d, 1H), 7,59-7,65 (m, 2H), 7,72-7,85 (m, 3H), 7,86-7,92 (m, 1H), 7,95 8,01 (m, 1H), 8,04 (a, s, 1H).
Ejemplo 24
5-(4-clorofenil)-2-({5-(1-hidroxietil)-1 -[3-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 6,13 min, d.e. = 98,6 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,36 (m, 1H), 4,83 (quin, 1H), 5,09 (s, 2H), 5,78 (d, 1H), 6,89 (d, 1H), 7,62 (d, 2H), 7,71-7,77 (m, 2H), 7,78-7,85 (m, 1H), 7,86 7,92 (m, 1H), 7,94-8,01 (m, 1H), 8,04 (a, s, 1H).
Ejemplo 25
5-(4-clorofenil)-2-({1 -(3,5-difluorofenil)-5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (3,5-difluorofenil)borónico (291,9 mg, 1,85 mmol) y acetato de cobre (II) (335,7 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 4 días, después de lo cual se agregó ácido borónico adicional (72,98 mg, 0,46 mmol, 0,5 eq.) debido a conversión incompleta. Después de 2 h a 60 °C seguido de agitación a temperatura ambiente durante tres días adicionales, la mezcla de reacción se
concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (114,3 mg, 0,21 mmol) como una mezcla de diastereómeros (rendimiento 22,7 %).
LC/MS [procedimiento 3]: Rt = 1.28 min; MS [ESIpos]: m/z = 545 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,49 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,88 (c, 1H), 5,02-5,13 (m, 2H), 6,89 (a, s, 1H), 7,41-7,54 (m, 3H), 7,62 (d, 2H), 7,75 (d, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 110 mg disuelto en 7 ml etanol/isohexano (1:1); volumen de inyección: 0,6 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 20 ml/min; temperatura: 40 °C; detección UV: 220 nm]. Después de la separación, se aislaron 42 mg de diastereómero 1 (Ejemplo 26), que eluyó primero, y 44 mg de diastereómero 2 (Ejemplo 27), que eluyó después.
Ejemplo 26
5-(4-clorofenil)-2-{[1-(3,5-difluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimento 2]: Rt = 3,10 min; MS [ESIpos]: m/z = 545 (M+H)+
HPLC quiral analítica: Rt = 1,09 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,49 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,23-4,36 (m, 1H), 4,88 (quin, 1H), 5,02-5,13 (m, 2H), 5,84 (d, 1H), 6,89 (d, 1H), 7,41-7,54 (m, 3H), 7,59-7,65 (m, 2H), 7,73-7,76 (m, 2H).
Ejemplo 27
5-(4-clorofenil)-2-{[1-(3,5-difluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 3,09 min; MS [ESIpos]: m/z = 545 (M+H)+
HPLC quiral analítica: Rt = 1,28 min, d.e. = 99 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,36 (m, 1H), 4,88 (quin, 1H), 5,07 (s, 2H), 5,83 (d, 1H), 6,90 (d, 1H), 7,42-7,54 (m, 3H), 7,59-7,65 (m, 2H), 7,72-7,79 (m, 2H). Ejemplo 28
5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1-(3-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (3-metilfenil)borónico (251,32 mg, 1,85 mmol) y acetato de cobre (II) (335,7 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante tres días. La mezcla de
reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (59,6 mg, 0,11 mmol) como una mezcla de diastereómeros (rendimiento 12,3 %).
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,44 (d, 3H), 2,38 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,29 (a, s, 1H), 4,76 (c, 1H), 5,01-5,11 (m, 2H), 5,66 (a, s, 1H), 6,90 (t, 1H), 7,29-7,35 (m, 1H), 7,38-7,47 (m, 3H), 7,59-7,65 (m, 2H), 7,72-7,78 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 56 mg disuelto en 2 ml etanol/isohexano (1:1); volumen de inyección: 0,5 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 15 ml/min; temperatura: 25 °C; detección UV: 220 nm]. Después de la separación, se aislaron 22 mg de diastereómero 1 (Ejemplo 29), que eluyó primero, y 24 mg de diastereómero 2 (Ejemplo 30), que eluyó después.
Ejemplo 29
5-(4-clorofenil)-2-{[5-(1-hidroxietil)-1-(3-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 7,97 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,44 (d, 3H), 2,38 (s, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,36 (m, 1H), 4,76 (quin, 1H), 5,00-5,11 (m, 2H), 5,67 (d, 1H), 6,89 (d, 1H), 7,30-7,35 (m, 1H), 7,38-7,47 (m, 3H), 7,59 7,65 (m, 2H), 7,72-7,78 (m, 2H).
Ejemplo 30
5-(4-clorofenil)-2-{[5-(1-hidroxietil)-1-(3-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 11,44 min, d.e. = 99,1 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,44 (d, 3H), 2,38 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,77 (quin, 1H), 5,01-5,10 (m, 2H), 5,67 (d, 1H), 6,90 (d, 1H), 7,29-7,35 (m, 1H), 7,38-7,47 (m, 3H), 7,59 7,65 (m, 2H), 7,73-7,78 (m, 2H).
Ejemplo 31
5-(4-clorofenil)-2-({1-(2-etilfenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (600 mg, 1,39 mmol) en piridina (18 ml) se agregó ácido (2-etilfenil)borónico (415,87 mg, 2,77 mmol) y acetato de cobre (II) (503,6 mg, 2,77 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante tres días. La mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se
purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (69,4 mg, 0,13 mmol) como una mezcla de diastereómeros (rendimiento 9,1 %).
LC/MS [procedimiento 2]: Rt = 3,16 min; MS [ESIpos]: m/z = 537 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 0,98 (t, 3H), 1,37 (d, 3H), 2,27 (cd, 2H), 3,84 (dd, 1H), 4,00 (dd, 1H), 4,21
4,37 (m, 1H), 4,52 (c, 1H), 5,00-5,13 (m, 2H), 5,48 (a, s, 1H), 6,90 (dd, 1H), 7,32-7,40 (m, 2H), 7,41-7,54 (m, 2H),
7,58-7,65 (m, 2H), 7,70-7,77 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 65 mg disuelto en 4 ml etanol/isohexano (1:1); volumen de inyección: 0,5 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 50:50; velocidad de flujo: 20 ml/min; temperatura: 40 °C; detección UV: 220 nm].
Después de la separación, se aislaron 25 mg de diastereómero 1 (Ejemplo 32), que eluyó primero, y 25 mg de diastereómero 2 (Ejemplo 33), que eluyó después.
Ejemplo 32
5-(4-clorofenil)-2-{[1-(2-etilfenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2.4- dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 2]: Rt = 3,15 min; MS [ESIpos]: m/z = 537 (M+H)+
HPLC quiral analítica: Rt = 0,96 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 50:50; velocidad de flujo: 1 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 0,98 (t, 3H), 1,37 (d, 3H), 2,27 (cd, 2H), 3,84 (dd, 1H), 4,00 (dd, 1H), 4.23- 4,35 (m, 1H), 4,52 (quin, 1H), 5,00-5,12 (m, 2H), 5,48 (d, 1H), 6,89 (d, 1H), 7,32-7,40 (m, 2H), 7,42-7,53 (m, 2H), 7,59-7,66 (m, 2H), 7,71-7,78 (m, 2H).
Ejemplo 33
5-(4-clorofenil)-2-{[1-(2-etilfenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2.4- dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 3,15 min; MS [ESIpos]: m/z = 537 (M+H)+
HPLC quiral analítica: Rt = 1,09 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 50:50; velocidad de flujo: 1 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 0,98 (t, 3H), 1,37 (d, 3H), 2,27 (cd, 2H), 3,84 (dd, 1H), 4,00 (dd, 1H), 4.23- 4,36 (m, 1H), 4,52 (quin, 1H), 5,07 (s, 2H), 5,48 (d, 1H), 6,90 (d, 1H), 7,32-7,40 (m, 2H), 7,42-7,54 (m, 2H), 7,59-7,66 (m, 2H), 7,71-7,78 (m, 2H).
Ejemplo 34
2-({1-(2-cloro-4-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica)

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (600 mg, 1,39 mmol) en piridina (18 ml) se agregó ácido (2-cloro-4-fluorofenil)borónico (483 mg, 2,77 mmol) y acetato de cobre (II) (503,6 mg, 2,77 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (242 mg, 1,39 mmol) debido a conversión incompleta. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 4 días. En este tiempo, se agregaron dos porciones
adicionales de ácido borónico (483 mg en total, 2,77 mmol). Después de esto, la mezcla de reacción se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (107 mg, 0,19 mmol) como una mezcla de diastereómeros (rendimiento 13,7 %).
LC/MS [procedimiento 2]: Rt = 3,02 min; MS [ESIpos]: m/z = 561 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,38 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,36 (m, 1H), 4,57-4,67 (m, 1H), 5,00-5,12 (m, 2H), 6,90 (a, s, 1H), 7,42 (td, 1H), 7,57-7,79 (m, 6H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 104 mg disuelto en 5 ml etanol/isohexano (1:1); volumen de inyección: 0,5 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 20 ml/min; temperatura: 40 °C; detección UV: 220 nm]. Después de la separación, se aislaron 56 mg de diastereómero 1 (Ejemplo 35), que eluyó primero, y 29 mg de diastereómero 2 (Ejemplo 36), que eluyó después.
Ejemplo 35
2-{[1-(2-cloro-4-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 1,36 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1.38 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,36 (m, 1H), 4,61 (quin, 1H), 5,01-5,11 (m, 2H), 5,51 (d, 1H), 6,89 (d, 1H), 7,42 (td, 1H), 7,60-7,65 (m, 2H), 7,66-7,72 (m, 1H), 7,72-7,78 (m, 3H).
Ejemplo 36
2-{[1-(2-cloro-4-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 1,72 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,38 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,37 (m, 1H), 4,61 (quin, 1H), 5,06 (s, 2H), 5,51 (d, 1H), 6,90 (d, 1H), 7,42 (td, 1H), 7,60-7,65 (m, 2H), 7,66-7,72 (m, 1H), 7,72 7,78 (m, 3H).
Ejemplo 37
5-(4-clorofenil)-2-({1-(5-fluoro-2-metoxifenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (600 mg, 1,39 mmol) en piridina (18 ml) se agregó ácido (5-fluoro-2-metoxifenil)borónico (471,22 mg, 2,77 mmol) y acetato de cobre (II) (503,6 mg, 2,77 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante tres días. La mezcla
de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (62,2 mg, 0,11 mmol) como una mezcla de diastereómeros (rendimiento 8,1 %).
LC/MS [procedimiento 2]: Rt = 2,93 min; MS [ESIpos]: m/z = 557 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,37 (d, 3H), 3,75 (s, 3H), 3,80-3,89 (m, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,52-4,62 (m, 1H), 4,99-5,04 (m, 2H), 6,90 (t, 1H), 7,26 (dd, 1H), 7,33 (dd, 1H), 7,37-7,44 (m, 1H), 7,60-7,65 (m, 2H), 7,73-7,78 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 55,4 mg disuelto en 6 ml etanol/isohexano (1:1); volumen de inyección: 2 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 20 ml/min; temperatura: 40 °C; detección UV: 220 nm]. Después de la separación, se aislaron 23 mg de diastereómero 1 (Ejemplo 38), que eluyó primero, y 21 mg de diastereómero 2 (Ejemplo 39), que eluyó después.
Ejemplo 38
5-(4-clorofenil)-2-{[1-(5-fluoro-2-metoxifenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 2]: Rt = 2,91 min; MS [ESIpos]: m/z = 557 (M+H)+
HPLC quiral analítica: Rt = 2,13 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 30 °C; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,37 (d, 3H), 3,75 (s, 3H), 3,81-3,89 (m, 1H), 4,00 (dd, 1H), 4,25-4,35 (m, 1H), 4,53-4,62 (m, 1H), 4,99-5,10 (m, 2H), 5,40 (d, 1H), 6,89 (d, 1H), 7,26 (dd, 1H), 7,33 (dd, 1H), 7,41 (td, 1H), 7,60-7,66 (m, 2H), 7,73-7,79 (m, 2H).
Ejemplo 39
5-(4-clorofenil)-2-{[1-(5-fluoro-2-metoxifenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 2,90 min; MS [ESIpos]: m/z = 557 (M+H)+
HPLC quiral analítica: Rt = 2,75 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 30 °C; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1.37 (d, 3H), 3,75 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,57 (quin, 1H), 4,99-5,10 (m, 2H), 5,40 (d, 1H), 6,91 (d, 1H), 7,26 (dd, 1H), 7,33 (dd, 1H), 7,41 (td, 1H), 7,60-7,65 (m, 2H), 7,73-7,78 (m, 2H).
Ejemplo 40
5-(4-clorofenil)-2-({1-(2-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (600 mg, 1,39 mmol) en piridina (18 ml) se agregó ácido (2-fluorofenil)borónico (387,96 mg, 2,77 mmol) y acetato de cobre (II) (503,6 mg, 2,77 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 3 días. En este tiempo, se agregaron dos porciones adicionales de ácido borónico (387,96 mg en total, 2,77 mmol). Después de esto, la
mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (30,1 mg, 0,06 mmol) como una mezcla de diastereómeros (rendimiento 4,1 %).
LC/MS [procedimiento 2]: Rt = 2,84 min; MS [ESIpos]: m/z = 527 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,29 (a, s, 1H), 4,69 (c, 1H), 5,01-5,12 (m, 2H), 6,89 (a, s, 1H), 7,38 (t, 1H), 7,48 (t, 1H), 7,57-7,66 (m, 4H), 7,71-7,80 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 26 mg disuelto en 4 ml etanol/isohexano (1:1); volumen de inyección: 2 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 25 ml/min; temperatura: 40 °C; detección UV: 220 nm]. Después de la separación, se aislaron 11 mg de diastereómero 1 (Ejemplo 41), que eluyó primero, y 9 mg de diastereómero 2 (Ejemplo 42), que eluyó después.
Ejemplo 41
5-(4-clorofenil)-2-{[1-(2-fluorofenil)-5-(1-hidroxietil)-1 W-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 2]: Rt = 2,83 min; MS [ESIpos]: m/z = 527 (M+H)+
HPLC quiral analítica: Rt = 2,32 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 30 °C; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,69 (quin, 1H), 5,01-5,12 (m, 2H), 5,53 (d, 1H), 6,89 (d, 1H), 7,38 (t, 1H), 7,48 (t, 1H), 7,57-7,66 (m, 4H), 7,72-7,78 (m, 2H).
Ejemplo 42
5-(4-clorofenil)-2-{[1-(2-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 2,82 min; MS [ESIpos]: m/z = 527 (M+H)+
HPLC quiral analítica: Rt = 3,23 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 pm, 50 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 30 °C; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,40 (d, 3H), 3,84 (dd, 1H), 4,00 (dd, 1H), 4,24-4,35 (m, 1H), 4,69 (quin, 1H), 5,07 (s, 2H), 5,53 (d, 1H), 6,90 (d, 1H), 7,38 (t, 1H), 7,48 (t, 1H), 7,57-7,67 (m, 4H), 7,72-7,79 (m, 2H).
Ejemplo 43
2-({1-(3-cloro-4-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica)

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (3-cloro4-fluorofenil)borónico (322,6 mg, 1,85 mmol) y acetato de cobre (II) (335,7 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 6 días. La mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (99,1 mg, 0,18 mmol) como una mezcla de diastereómeros (rendimiento 19,1 %).
LC/MS [procedimiento 3]: Rt = 1.31 min; MS [ESIpos]: m/z = 561 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,46 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,80 (c, 1H), 5,01-5,13 (m, 2H), 6,90 (a, s, 1H), 7,59-7,70 (m, 4H), 7,72-7,78 (m, 2H), 7,93 (dd, 1H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 97,1 mg disuelto en 3 ml etanol; volumen de inyección: 0,3 ml; columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 25 °C; detección UV: 220 nm]. Después de la separación, se aislaron 40 mg de diastereómero 1 (Ejemplo 44), que eluyó primero, y 42 mg de diastereómero 2 (Ejemplo 45), que eluyó después.
Ejemplo 44
2-{[1-(3-cloro-4-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 2]: Rt = 3,22 min; MS [ESIpos]: m/z = 561 (M+H)+
HPLC quiral preparativa: Rt = 9,97 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 25 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,46 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,22-4,36 (m, 1H), 4,80 (quin, 1H), 5,01-5,12 (m, 2H), 5,75 (d, 1H), 6,89 (d, 1H), 7,58-7,70 (m, 4H), 7,71-7,78 (m, 2H), 7,93 (dd, 1H).
Ejemplo 45
2-{[1-(3-cloro-4-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 3,22 min; MS [ESIpos]: m/z = 561 (M+H)+
HPLC quiral preparativa: Rt = 11,35 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 25 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,46 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,25-4,35 (m, 1H), 4,81 (quin, 1H), 5,06 (s, 2H), 5,74 (d, 1H), 6,90 (d, 1H), 7,59-7,70 (m, 4H), 7,73-7,78 (m, 2H), 7,93 (dd, 1H).
Ejemplo 46
5-(4-clorofenil)-2-({1-(3,5-diclorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica)

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (3,5-diclorofenil)borónico (352,73 mg, 1,85 mmol) y acetato de cobre (II) (335,7 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 6 días. La mezcla de
reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (105,5 mg, 0,18 mmol) como una mezcla de diastereómeros (rendimiento 19,8 %).
LC/MS [procedimiento 3]: Rt = 1,39 min; MS [ESIpos]: m/z = 577 (M+H)+
1H NMR (400 MHz, DMSO-da): S [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,23-4,36 (m, 1H), 4,82-4,90 (m, 1H), 5,02-5,12 (m, 2H), 6,84-6,94 (m, 1H), 7,59-7,65 (m, 2H), 7,72-7,82 (m, 5H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 103,5 mg disuelto en 14 ml etanol/isohexano (1:1); volumen de inyección: 2 ml; columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 20 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 29,2 mg de diastereómero 1 (Ejemplo 47), que eluyó primero, y 28,9 mg de diastereómero 2 (Ejemplo 48), que eluyó después.
Ejemplo 47
5-(4-clorofenil)-2-{[1-(3,5-diclorofenil)-5-(1-hidroxietil)-1R-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3R-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 1,49 min, d.e. = 100 % [columna: Daicel Chiralpack OX-3 3 ^m, 50 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 30°C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): S [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,35 (m, 1H), 4,86 (quin, 1H), 5,01-5,12 (m, 2H), 5,82 (d, 1H), 6,88 (d, 1H), 7,58-7,66 (m, 2H), 7,72-7,82 (m, 5H).
Ejemplo 48
5-(4-clorofenil)-2-{[1-(3,5-diclorofenil)-5-(1-hidroxietil)-1R-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3R-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 2,02 min, d.e. = 99,8 % [columna: Daicel Chiralpack OX-3 3 ^m, 50 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 30 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-da): S [ppm] 1,48 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,23-4,36 (m, 1H), 4,86 (quin, 1H), 5,07 (s, 2H), 5,82 (d, 1H), 6,89 (d, 1H), 7,59-7,65 (m, 2H), 7,72-7,81 (m, 5H).
Ejemplo 49
5-(4-clorofenil)-2-({1-(2,5-diclorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3R-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3R-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (2,5-diclorofenil)borónico (352,73 mg, 1,85 mmol) y acetato de cobre( II) (335,75 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante tres días, después de lo cual se agregó ácido borónico adicional (100 mg, 0,52 mmol) debido a conversión incompleta. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 6 días. En este tiempo, se agregó otra porción de ácido borónico (100 mg, 0,52 mmol). Después de esto, la mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación
de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (52,8 mg, 0,09 mmol, 97 % de pureza) como una mezcla de diastereómeros (rendimiento 9,6 %).
LC/MS [procedimiento 3]: Rt = 1,33 min; MS [ESIpos]: m/z = 577 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,35 (m, 1H), 4,63-4,72 (m, 1H), 5,01-5,12 (m, 2H), 6,89 (a, s, 1H), 7,60-7,65 (m, 2H), 7,67-7,78 (m, 4H), 7,81 (a, d, 1H).
Los dos diastereómeros se separaron por HPLC quiral preparativa (SFC) [preparación de muestras: 50 mg disuelto en 10 ml metanol; volumen de inyección: 0,5 ml; columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: dióxido de carbono/metanol 82:18; velocidad de flujo: 80 ml/min; temperatura: 40 °C; detección UV: 210 nm]. Después de la separación, se aislaron 20,3 mg de diastereómero 1 (Ejemplo 50), que eluyó primero, y 24,1 mg de diastereómero 2 (Ejemplo 51), que eluyó después.
Ejemplo 50
5-(4-clorofenil)-2-{[1-(2,5-diclorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 3]: Rt = 1,30 min; MS [ESIpos]: m/z = 577 (M+H)+
HPLC quiral analítica (SFC): Rt = 2,87 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-3 250 x 4 mm; eluyente: dióxido de carbono/metanol (5 % ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-cC6): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,36 (m, 1H), 4,67 (quin, 1H), 5,01-5,12 (m, 2H), 5,53 (d, 1H), 6,89 (d, 1H), 7,60-7,65 (m, 2H), 7,67-7,78 (m, 4H), 7,81 (a, d, 1H).
Ejemplo 51
5-(4-clorofenil)-2-{[1-(2,5-diclorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 3]: Rt = 1,30 min; MS [ESIpos]: m/z = 577 (M+H)+
HPLC quiral analítica (SFC): Rt = 3,11 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-3 250 x 4 mm; eluyente: dióxido de carbono/metanol (5 % ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm]. 1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,35 (m, 1H), 4,67 (quin, 1H), 5,01-5,12 (m, 2H), 5,53 (d, 1H), 6,89 (d, 1H), 7,59-7,65 (m, 2H), 7,66-7,78 (m, 4H), 7,81 (a, d, 1H).
Ejemplo 52
2-({1 -(3-cloro-2-fluorofenil)-5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica)

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (3-cloro-2-fluorofenil)borónico (322,31 mg, 1,85 mmol) y acetato de cobre (II) (335,75 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 12 días. En este tiempo, se agregó ácido borónico adicional (322,31 mg en total, 1,85 mmol) en porciones diariamente. Después de esto, la mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con
acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (15,9 mg, 0,03 mmol, 97 % de pureza) como una mezcla de diastereómeros (rendimiento 3 %).
LC/MS [procedimiento 2]: Rt = 3,12 min; MS [ESIpos]: m/z = 561 (M+H)+
1H NMR (500 MHz, DMSO-de): 8 [ppm] 1,42 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,35 (m, 1H), 4,75 (c, 1H), 5,02-5,12 (m, 2H), 5,57 (a, s, 1H), 6,89 (a, d, 1H), 7,40 (td, 1H), 7,60-7,65 (m, 3H), 7,72-7,77 (m, 2H), 7,81 (ddd, 1H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 14 mg disuelto en 1 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 25 °C; detección UV: 220 nm]. Después de la separación, se aislaron 6 mg de diastereómero 1 (Ejemplo 53), que eluyó primero, y 6 mg de diastereómero 2 (Ejemplo 54), que eluyó después.
Ejemplo 53
2-{[1-(3-cloro-2-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 2]: Rt = 3,14 min; MS [ESIpos]: m/z = 561 (M+H)+
HPLC quiral analítica: Rt = 5,49 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2% TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (500 MHz, DMSO-d6): 8 [ppm] 1,42 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,34 (m, 1H), 4,75 (quin, 1H), 5,03-5,11 (m, 2H), 5,57 (d, 1H), 6,89 (d, 1H), 7,40 (td, 1H), 7,60-7,65 (m, 3H), 7,73-7,77 (m, 2H), 7,81 (ddd, 1H).
Ejemplo 54
2-{[1-(3-cloro-2-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 3,13 min; MS [ESIpos]: m/z = 561 (M+H)+
HPLC quiral analítica: Rt = 6,16 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (500 MHz, DMSO-d6): 8 [ppm] 1,42 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,25-4,34 (m, 1H), 4,75 (quin, 1H), 5,07 (s, 2H), 5,57 (d, 1H), 6,89 (d, 1H), 7,40 (td, 1H), 7,60-7,65 (m, 3H), 7,73-7,77 (m, 2H), 7,81 (ddd, 1H).
Ejemplo 55
5-(4-clorofenil)-2-({1-[3-difluorometoxi)fenil]-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido [3(difluorometoxi)fenil]borónico (347,40 mg, 1,85 mmol) y acetato de cobre (II) (335,75 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 6 días, después de lo cual se agregó ácido borónico adicional (100 mg, 0,53 mmol) debido a conversión incompleta. La mezcla de reacción se agitó a temperatura ambiente durante dos días adicionales. La mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (60,3 mg, 0,10 mmol) como una mezcla de diastereómeros (rendimiento 11,4 %).
LC/MS [procedimiento 3]: Rt = 1,28 min; MS [ESIpos]: m/z = 575 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,25-4,35 (m, 1H), 4,78-4,85 (m, 1H), 5,03-5,12 (m, 2H), 6,89 (a, s, 1H), 7,33 (t, 1H), 7,31-7,35 (m, 1H), 7,48-7,56 (m, 2H), 7,59-7,65 (m, 3H), 7,72-7,78 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 58 mg disuelto en 2 ml etanol; volumen de inyección: 0,7 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 35 °C; detección UV: 220 nm]. Después de la separación, se aislaron 20,7 mg de diastereómero 1 (Ejemplo 56), que eluyó primero, y 17,7 mg de diastereómero 2 (Ejemplo 57), que eluyó después.
Ejemplo 56
5-(4-clorofenil)-2-({1-[3-(difluorometoxi)fenil]-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 5,57 min, d.e. = 98,7 % [columna: Daicel Chiralcel® OX-H 5, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,81 (c, 1H), 5,02-5,13 (m, 2H), 6,88 (a, s, 1H), 7,33 (t, 1H), 7,30-7,35 (m, 1H), 7,48-7,56 (m, 2H), 7,59-7,65 (m, 3H), 7,72 7,78 (m, 2H).
Ejemplo 57
5-(4-clorofenil)-2-({1-[3-(difluorometoxi)fenil]-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 6,70 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,23-4,35 (m, 1H), 4,82 (quin, 1H), 5,07 (s, 2H), 5,75 (d, 1H), 6,90 (d, 1H), 7,33 (t, 1H), 7,30-7,35 (m, 1H), 7,48-7,56 (m, 2H), 7,59 7,65 (m, 3H), 7,72-7,78 (m, 2H).
Ejemplo 58
2-({1-(2-cloro-5-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica)
A una solución de 5-(4-clorofenil)-2-({5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-M}metM)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (2-cloro-5-fluorofenil)borónico (322,31 mg, 1,85 mmol) y acetato de cobre (II) (335,75 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 10 días. En este tiempo, se agregó ácido borónico adicional (322,31 mg en total, 1,85 mmol) en porciones diariamente. La mezcla de reacción resultante se concentró al vacío, luego se diluyó con acetato de etilo y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con acetato de etilo. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento4], y se obtuvo el compuesto deseado (61 mg, 0,11 mmol, 98 % de pureza) como una mezcla de diastereómeros (rendimiento 11,5 %).
LC/MS [procedimiento 2]: Rt = 3,02 min; MS [ESlpos]: m/z = 561 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,63-4,72 (m, 1H), 5,01-5,13 (m, 2H), 5,52 (a, s, 1H), 6,90 (dd, 1H), 7,52 (td, 1H), 7,60-7,69 (m, 3H), 7,73-7,79 (m, 3H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 58 mg disuelto en 3 ml etanol/isohexano (2:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 25 °C; detección UV: 220 nm]. Después de la separación, se aislaron 25 mg de diastereómero 1 (Ejemplo 59), que eluyó primero, y 25 mg de diastereómero 2 (Ejemplo 60), que eluyó después.
Ejemplo 59
2-{[1-(2-cloro-5-fluorofeml)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-N]metN}-5-(4--clorofenil)-4-[(2S)-3,3,3-tnfluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 2]: Rt = 3,02 min; MS [ESlpos]: m/z = 561 (M+H)+
HPLC quiral analítica: Rt = 5,43 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,35 (m, 1H), 4,67 (quin, 1H), 5,01-5,12 (m, 2H), 5,53 (d, 1H), 6,89 (d, 1H), 7,52 (td, 1H), 7,60-7,68 (m, 3H), 7,72-7,79 (m, 3H).
Ejemplo 60
2-{[1-(2-cloro-5-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 3,02 min; MS [ESlpos]: m/z = 561 (M+H)+
HPLC quiral analítica: Rt = 6,11 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,67 (quin, 1H), 5,07 (s, 2H), 5,53 (d, 1H), 6,90 (d, 1H), 7,52 (td, 1H), 7,60-7,68 (m, 3H), 7,73-7,79 (m, 3H).
Ejemplo 61
5-(4-clorofenil)-2-({1-(2,3-diclorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2
hidrox¡prop¡l]-2,4-d¡hidro-3H-1,2,4-triazol-3-ona (500 mg, 0,92 mmol, 80 % de pureza) en piridina (12 ml) se
agregó ácido (2,3-diclorofeml)borómco (176,36 mg, 0,92 mmol) y acetato de cobre (II) (335,75 mg, 1,85 mmol).
La mezcla de reacción se calentó hasta 60 °C durante 1 h y luego se agitó a temperatura ambiente durante 24 h,
después de lo cual se agregó ácido borónicoadicional (80 mg, 0,42 mmol) debido a conversión incompleta. La
mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 5 días. En este tiempo, se agregaron
dos porciones adicionales de ácido borónico (160 mg en total, 0,84 mmol). Después de esto, la mezcla de
reacción resultante se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso
(0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas
combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por
HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (148 mg, 0,25 mmol, 97,3 % de pureza)
como una mezcla de diastereómeros (rendimiento 27 %).
LC/MS [procedimiento 2]: Rt = 3,19 min; MS [ESIpos]: m/z = 577 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,39 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,35 (m, 1H), 4,60-4,71 (m,
1H), 5,02-5,13 (m, 2H), 5,52 (br, s, 1H), 6,89 (dd, 1H), 7,55 (t, 1H), 7,59-7,66 (m, 3H), 7,73-7,78 (m, 2H), 7,87
(dd, 1H).
Los dos diastereómeros se separaron por HPLC quiral preparativa (SFC) [preparación de muestras: 141 mg
disuelto en 18 ml metanol; volumen de inyección: 0,3 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm;
eluyente: dióxido de carbono/metanol 70:30; velocidad de flujo: 80 ml/min; temperatura: 40 °C; detección UV: 210
nm]. Después de la separación, se aislaron 58,5 mg de diastereómero 1 (Ejemplo 62), que eluyó primero, y 53 mg
de diastereómero 2 (Ejemplo 63), que eluyó después.
Ejemplo 62
5-(4-clorofen¡l)-2-{[1-(2,3-d¡clorofen¡l)-5-(1-h¡drox¡et¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-t^fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (diastereómero 1)
LC/MS [procedimiento 2]: Rt = 3,21 min; MS [ESIpos]: m/z = 577 (M+H)+; 95 % de pureza
HPLC quiral analítica (SFC): Rt = 3,09 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-3250 x 4 mm;
eluyente: dióxido de carbono/metanol (5 % ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,39 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,65 (a, s, 1H), 5,01-5,13 (m, 2H), 5,52 (d, 1H), 6,89 (d, 1H), 7,55 (t, 1H), 7,59-7,66 (m, 3H), 7,72-7,78 (m, 2H), 7,87 (dd,
1H).
Ejemplo 63
5-(4-clorofen¡l)-2-{[1-(2,3-d¡clorofen¡l)-5-(1-h¡drox¡et¡l)-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-t^fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 3,20 min; MS [ESIpos]: m/z = 577 (M+H)+; 95 % de pureza
HPLC quiral analítica (SFC): Rt = 3,38 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-3250 x 4 mm;
eluyente: dióxido de carbono/metanol (5 % ^ 60 %); velocidad de flujo: 3 ml/min; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,39 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,35 (m, 1H), 4,65 (a, s, 1H), 5,07 (s, 2H), 5,52 (d, 1H), 6,90 (d, 1H),  7,55 (t, 1H), 7,59-7,66 (m, 3H), 7,72-7,79 (m, 2H), 7,87 (dd, 1H).
7,55 (t, 1H), 7,59-7,66 (m, 3H), 7,72-7,79 (m, 2H), 7,87 (dd, 1H).
Ejemplo 64
5-(4-clorofen¡l)-2-({1-(2,3-d¡fluorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona (mezcla diastereomérica )
A una solución de 5-(4-clorofenil)-2-({5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-M}metM)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (430 mg, 0,99 mmol) en piridina (12,5 ml) se agregó ácido (2,3-difluorofenil)borónico (156,89 mg, 0,99 mmol) y acetato de cobre (II) (360,94 mg, 1,99 mmol). La mezcla de reacción se calentó hasta 60 °C durante 1 h y luego se agitó a temperatura ambiente durante 24 h, después de lo cual se agregó ácido borónico adicional (80 mg, 0,51 mmol) debido a conversión incompleta. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante cinco días. En este tiempo, se agregaron cinco porciones adicionales de ácido borónico (400 mg en total, 2,54 mmol). Después de esto, la mezcla de reacción resultante se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (44 mg, 0,08 mmol) como una mezcla de diastereómeros (rendimiento 8,1 %).
LC/MS [procedimiento 2]: Rt = 2,97 min; MS [ESlpos]: m/z = 545 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,42 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,76 (c, 1H), 5,02-5,13 (m, 2H), 6,89 (a, s, 1H), 7,35-7,43 (m, 1H), 7,45-7,51 (m, 1H), 7,59-7,71 (m, 3H), 7,72-7,79 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 40 mg disuelto en 1 ml etanol; volumen de inyección: 0,5 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 35 °C; detección UV: 220 nm]. Después de la separación, se aislaron 18 mg de diastereómero 1 (Ejemplo 65), que eluyó primero, y 16 mg de diastereómero 2 (Ejemplo 66), que eluyó después.
Ejemplo 65
5-(4-clorofeml)-2-{[1-(2,3-difluorofeml)-5-(1-hidroxietN)-1H-1,2,4-triazol-3-N]metN}-4-[(2S)-3,3,3-tnfluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
HPLC quiral analítica: Rt = 5,74 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,42 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,35 (m, 1H), 4,76 (quin, 1H), 5,02-5,13 (m, 2H), 5,58 (d, 1H), 6,89 (d, 1H), 7,35-7,44 (m, 1H), 7,45-7,52 (m, 1H), 7,59-7,72 (m, 3H), 7,72-7,78 (m, 2H).
Ejemplo 66
5-(4-clorofenil)-2-{[1-(2,3-difluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 6,59 min, d.e. = 99,2 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,42 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,36 (m, 1H), 4,76 (quin, 1H), 5,07 (s, 2H), 5,58 (d, 1H), 6,90 (d, 1H), 7,35-7,43 (m, 1H), 7,44-7,51 (m, 1H), 7,59-7,72 (m, 3H), 7,72-7,78 (m, 2H).
Ejemplo 67
2-{[1-(2-cloro-3-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4--clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
A una solución de 5-(4-clorofenil)-2-({5-[(1 RS)-1 -hidroxietil]-1 H-1,2,4-triazol-3-M}metM)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (500 mg, 0,92 mmol, 80 % de pureza) en piridina (12 ml) se agregó ácido (2-cloro-3-fluorofenil)borónico (161,15 mg, 0,92 mmol) y acetato de cobre (Il) (335,75 mg, 1,85 mmol). La mezcla de reacción se calentó hasta 60 °C durante 1 h y luego se agitó a temperatura ambiente durante 24 h, después de lo cual se agregó ácido borónico adicional (75 mg, 0,43 mmol) debido a conversión incompleta. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante seis días. En este tiempo, se agregaron cinco porciones adicionales de ácido borónico (375 mg en total, 2,15 mmol). Después de esto, la mezcla de reacción resultante se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se aisló 91 mg del compuesto deseado como una mezcla diastereomérica que todavía contiene algunas impurezas.
Una purificación adicional por HPLC quiral preparativa proporcionó los dos diastereómeros puros y separados [preparación de muestras: 90 mg disuelto en 3 ml etanol; volumen de inyección: 0,3 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 35 °C; detección UV: 220 nm]. Después de la separación, se aislaron 20 mg de diastereómero 1 (Ejemplo 67), que eluyó primero, y 21 mg de diastereómero 2 (Ejemplo 68), que eluyó después.
LC/m S [procedimiento 2]: Rt = 3,01 min; MS [Eslpos]: m/z = 561 (M+H)+
HPLC quiral analítica: Rt = 6,22 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1.39 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,36 (m, 1H), 4,66 (quin, 1H), 5,01-5,14 (m, 2H), 5,53 (d, 1H), 6,89 (d, 1H), 7,48-7,70 (m, 5H), 7,72-7,78 (m, 2H).
Ejemplo 68
2-{[1-(2-cloro-3-fluorofeml)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-N]metN}-5-(4--clorofenil)-4-[(2S)-3,3,3-tnfluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
LC/MS [procedimiento 2]: Rt = 3,01 min; MS [ESlpos]: m/z = 561 (M+H)+
HPLC quiral analítica: Rt = 7,94 min, d.e. = 100 % [columna: Daicel Chiralcel® OX-H 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30 0,2 % TFA y 1 % agua; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,40 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,66 (quin, 1H), 5,07 (s, 2H), 5,53 (d, 1H), 6,90 (d, 1H), 7,49-7,70 (m, 5H), 7,72-7,78 (m, 2H).
Ejemplo 69
5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1-(2-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (400 mg, 0,92 mmol) en piridina (12 ml) se agregó ácido (2-metilfenil)borónico (251,32 mg, 1,85 mmol) y acetato de cobre (II) (335,75 mg, 1,85 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (62,8 mg, 0,46 mmol, 0,5 eq.) debido a conversión incompleta. Después de agitar durante dos días adicionales, la mezcla de reacción se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por
HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (100 mg, 0,17 mmol) como una mezcla de diastereómeros (rendimiento 17,2 %, 90 % de pureza).
LC/MS [procedimiento 3]: Rt = 1,24 min; MS [ESIpos]: m/z = 523 (M+H)+
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 98 mg disuelto en 2 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 pm, 250 x 20 mm; eluyente: isohexano/etanol 75:25; velocidad de flujo: 15 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 37 mg de diastereómero 1 (Ejemplo 70), que eluyó primero, y 39 mg de diastereómero 2 (Ejemplo 71), que eluyó después.
Ejemplo 70
5-(4-clorofenil)-2-{[5-(1-hidroxietil)-1-(2-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1)
LC/MS [procedimiento 3]: Rt = 1,24 min; MS [ESIpos]: m/z = 523 (M+H)+
HPLC quiral analítica: Rt = 7,65 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,36 (d, 3H), 1,98 (s, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,35 (m, 1H), 4,54 (quin, 1H), 5,00-5,12 (m, 2H), 5,48 (d, 1H), 6,89 (d, 1H), 7,31-7,49 (m, 4H), 7,59-7,65 (m, 2H), 7,71 7,77 (m, 2H).
Ejemplo 71
5-(4-clorofenil)-2-{[5-(1-hidroxietil)-1-(2-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 10,27 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,37 (d, 3H), 1,98 (s, 3H), 3,84 (dd, 1H), 4,00 (dd, 1H), 4,24-4,35 (m, 1H), 4,54 (quin, 1H), 5,06 (s, 2H), 5,48 (d, 1H), 6,90 (d, 1H), 7,31-7,49 (m, 4H), 7,58-7,66 (m, 2H), 7,71-7,78 (m, 2H).
Ejemplo 72
5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1-[2-trifluorometil)fenil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (600 mg, 1,11 mmol, 80 % de pureza) en piridina (14,5 ml) se agregó ácido [2-(trifluorometil)fenil]borónico (421,30 mg, 2,22 mmol) y acetato de cobre (II) (402,9 mg, 2,22 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (105 mg, 0,55 mmol, 0,5 eq.) debido a conversión incompleta. Después de agitar adicionalmente a temperatura ambiente durante una noche, la mezcla de reacción resultante se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (80 mg) como una mezcla de diastereómeros (rendimiento 12,4 %).
LC/MS [procedimiento 2]: Rt = 3,19 min; MS [ESlpos]: m/z = 577 (M+H)+
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 78 mg disuelto en 2 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 |jm, 250 x 20 mm; eluyente: isohexano/etanol 75:25; velocidad de flujo: 15 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 34 mg de diastereómero 1 (Ejemplo 73), que eluyó primero, y 30 mg de diastereómero 2 (Ejemplo 74), que eluyó después.
Ejemplo 73
5-(4-clorofenil)-2-({5-(1-hidroxietil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona
(diastereómero 1)
HPLC quiral analítica: Rt = 6,16 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 jm , 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,36 (d, 3H), 3,84 (dd, 1H), 4,00 (dd, 1H), 4,24-4,35 (m, 1H), 4,57 (quin, 1H), 4,99-5,12 (m, 2H), 5,50 (d, 1H), 6,89 (d, 1H), 7,59-7,65 (m, 2H), 7,66-7,71 (m, 1H), 7,72-7,76 (m, 2H), 7,77-7,90 (m, 2H), 7,93-7,99 (m, 1H).
Ejemplo 74
5-(4-clorofenil)-2-({5-(1-hidroxietil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2)
HPLC quiral analítica: Rt = 8,67 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 jm , 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,36 (d, 3H), 3,84 (dd, 1H), 3,99 (dd, 1H), 4,24-4,35 (m, 1H), 4,54-4,62 (m, 1H), 5,05 (s, 2H), 5,50 (d, 1H), 6,90 (d, 1H), 7,60-7,65 (m, 2H), 7,67-7,71 (m, 1H), 7,72-7,90 (m, 4H), 7,93 7,98 (m, 1H).
Ejemplo 75
5-(4-clorofenil)-2-({1-(3-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (430 mg, 0,795 mmol, 80 % de pureza) en piridina (10 ml) se agregó ácido (3-fluorofenil)borónico (222,432 mg, 1,59 mmol) y acetato de cobre (II) (288,75 mg, 1,59 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (55,6 mg, 0,40 mmol) debido a conversión incompleta. La mezcla de reacción se calentó nuevamente hasta 60 °C durante 2 h seguido de agitación a temperatura ambiente durante una noche. La mezcla de reacción resultante se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (100 mg, 0,19 mmol) como una mezcla de diastereómeros (rendimiento 23,9 %).
LC/MS [procedimiento 2]: Rt = 2,99 min; MS [ESlpos]: m/z = 527 (M+H)+
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,83 (c, 1H),
5,02-5,13 (m, 2H), 6,89 (a, s, 1H), 7,38 (td, 1H), 7,48-7,66 (m, 5H), 7,72-7,78 (m, 2H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 97 mg disuelto en 4 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 36 mg de (1 S)-diastereómero (Ejemplo 76), que eluyó primero, y 40 mg de (1R)-diastereómero (Ejemplo 77), que eluyó después.
Ejemplo 76
5-(4-clorofenil)-2-({1-(3-fluorofenil)-5-[(1 S)-1 -hidroxietil]-1 R-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifiuoro-2-hidroxipropil]-2,4-dihidro-3R-1,2,4-triazol-3-ona;

LC/MS [procedimiento 3]: Rt = 1,24 min; MS [ESlpos]: m/z = 527 (M+H)+
HPLC quiral analítica: Rt = 9,71 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 ^m, 250 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-da): S [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,23-4,37 (m, 1H), 4,82 (quin, 1H), 5,01-5,13 (m, 2H), 5,76 (d, 1H), 6,89 (d, 1H), 7,38 (td, 1H), 7,48-7,66 (m, 5H), 7,72-7,79 (m, 2H). La estereoquímica absoluta del compuesto se determinó realizando adicionalmente la misma reacción con diastereómero enantiopuro 5-(4-clorofenil)-2-({5-[(1 S)-1-hidroxietil]-1R-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3R-1,2,4-triazol-3-ona (Ejemplo 5A) como el material de partida y la comparación de los dos productos correspondientes por HPLC quiral analítica.
Ejemplo 77
5-(4-clorofenil)-2-({1-(3-fluorofenil)-5-[(1R)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3R-1,2,4-triazol-3-ona

LC/MS [procedimiento 2]: Rt = 2,93 min; MS [ESlpos]: m/z = 527 (M+H)+
HPLC quiral analítica: Rt = 13,60 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 ^m, 250 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-da): S [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,36 (m, 1H), 4,83 (quin, 1H), 5,07 (s, 2H), 5,76 (d, 1H), 6,90 (d, 1H), 7,38 (td, 1H), 7,48-7,65 (m, 5H), 7,72-7,78 (m, 2H).
La estereoquímica absoluta del compuesto se determinó realizando adicionalmente la misma reacción con
diastereómero enantiopuro 5-(4-clorofenil)-2-({5-[(1R)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (Ejemplo 6A) como el material de partida y la comparación de los dos productos correspondientes por HPLC quiral analítica.
Ejemplo 78
5-(4-clorofenil)-2-({1-(3-diclorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifiuoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (430 mg, 0,795 mmol, 80 % de pureza) en piridina (10 ml) se agregó ácido (3-clorofenil)borónico (248,59 mg, 1,59 mmol) y acetato de cobre (II) (288,75 mg, 1,59 mmol). La mezcla de reacción se calentó hasta 60 °C durante 2 h y luego se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (62,1 mg, 0,40 mmol) debido a conversión incompleta. La mezcla de reacción se calentó nuevamente hasta 60 °C durante 2 h seguido de agitación a temperatura ambiente durante una noche. La mezcla de reacción resultante se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (130 mg, 0,24 mmol) como una mezcla de diastereómeros (rendimiento 30,1 %).
Lc /MS [procedimiento 2]: Rt = 3,19 min; MS [ESIpos]: m/z = 543 (M+H)+
1H NMR (400 MHz, DMSO-cfe): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,30 (a, s, 1H), 4,81 (c, 1H), 5,02-5,13 (m, 2H), 6,89 (a, s, 1H), 7,56-7,67 (m, 5H), 7,72-7,79 (m, 3H).
Los dos diastereómeros se separaron por HPLC quiral preparativa [preparación de muestras: 128 mg disuelto en 4 ml etanol/isohexano (1:1); volumen de inyección: 1 ml; columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 15 ml/min; temperatura: 30 °C; detección UV: 220 nm]. Después de la separación, se aislaron 52 mg de (1 S)-diastereómero (Ejemplo 79), que eluyó primero, y 49 mg de (1R)-diastereómero (Ejemplo 80), que eluyó después.
Ejemplo 79
5-(4-clorofenil)-2-({1-(3-clorofenil)-5-[(1S)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

LC/MS [procedimiento 2]: Rt = 3,14 min; MS [ESIpos]: m/z = 543 (M+H)
HPLC quiral analítica: Rt = 9,96 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 ^m, 250 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,23-4,36 (m, 1H), 4,81 (quin, 1H), 5,01-5,13 (m, 2H), 5,76 (d, 1H), 6,89 (d, 1H), 7,56-7,66 (m, 5H), 7,71-7,79 (m, 3H).
13C NMR (125 MHz, DMSO-d6): 6 [ppm] 21,3, 42,1, 42,2, 59,6, 65,5, 123,0, 124,5, 124,6, 125,3, 128,5, 128,9 (2x), 130,0 (2x), 130,7, 133,0, 135,2, 138,2, 144,8, 153,1, 157,8, 158,6.
La estereoquímica absoluta del compuesto se determinó realizando adicionalmente la misma reacción con diastereómero enantiopuro 5-(4-clorofenil)-2-({5-[(1S)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (Ejemplo 5A) como el material de partida y la comparación de los dos productos correspondientes por HPLC quiral analítica.
Ejemplo 80
5-(4-clorofenil)-2-({1-(3-clorofenil)-5-[(1R)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

LC/MS [procedimiento 2]: Rt = 3,15 min; MS [ESlpos]: m/z = 543 (M+H)+
HPLC quiral analítica: Rt = 14,41 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 pm, 250 x 4,6 mm; eluyente: isohexano/etanol 80:20; velocidad de flujo: 1 ml/min; temperatura: 35 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-d6): 8 [ppm] 1,47 (d, 3H), 3,85 (dd, 1H), 4,01 (dd, 1H), 4,24-4,37 (m, 1H), 4,81 (quin, 1H), 5,07 (s, 2H), 5,76 (d, 1H), 6,90 (d, 1H), 7,56-7,66 (m, 5H), 7,71-7,79 (m, 3H).
La estereoquímica absoluta del compuesto se determinó realizando adicionalmente la misma reacción con diastereómero enantiopuro 5-(4-clorofenil)-2-({5-[(1R)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (Ejemplo 6A) como el material de partida y la comparación de los dos productos correspondientes por HPLC quiral analítica.
Ejemplo 81
5-(4-clorofenil)-2-({1-(2-diclorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (mezcla diastereomérica )

A una solución de 5-(4-clorofenil)-2-({5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (2,10 g, 3,88 mmol, 80 % de pureza) en piridina (50 ml) se agregó ácido (2-clorofenil)borónico (1,214 g, 7,76 mmol) y acetato de cobre (II) (1,410 g, 7,76 mmol). La mezcla de reacción se calentó hasta 60 °C durante 1 h y luego se agitó a temperatura ambiente durante 5 días, después de lo cual se agregó ácido borónico adicional (303 mg, 1,94 mmol) debido a conversión incompleta. Después de
agitar a temperatura ambiente durante dos días adicionales, la mezcla de reacción resultante se concentró al vacío, luego se diluyó con MTBE y se inactivó con ácido clorhídrico acuoso (0,5 M). Después de la separación de fases, la fase acuosa se extrajo dos veces con MTBE. Las fases orgánicas combinadas se secaron en sulfato de sodio, se filtraron y concentraron al vacío. El producto bruto se purificó por HPLC preparativa [procedimiento 4], y se obtuvo el compuesto deseado (580 mg, 1,01 mmol, 95 % de pureza) como una mezcla de diastereómeros (rendimiento 26,1 %).
LC/MS [método 3]: Rt = 1.24 min; MS [ESIpos]: m/z = 543 (M+H)+
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,38 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,55-4,64 (m, 1H), 5,01-5,13 (m, 2H), 6,85-6,94 (m, 1H), 7,50-7,65 (m, 5H), 7,67-7,78 (m, 3H).
Los dos diastereómeros se separaron por HPLC quiral preparativa (SFC) [preparación de muestras: 575 mg disuelto en 35 ml metanol; volumen de inyección: 0,4 ml; columna: Daicel Chiralcel® OX-H 5 ^m, 250 x 20 mm; eluyente: dióxido de carbono/metanol 70:30; velocidad de flujo: 80 ml/min; temperatura: 40 °C; detección UV: 210 nm]. Después de la separación, se aislaron 206 mg de (1S)-diastereómero (Ejemplo 82), que eluyó primero, y 189 mg de (1R)-diastereómero (Ejemplo 83), que eluyó después.
Ejemplo 82
5-(4-clorofenil)-2-({1-(2-clorofenil)-5-[(1S)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona

LC/MS [procedimiento 3]: Rt = 1,24 min; MS [ESIpos]: m/z = 543 (M+H)+
HPLC quiral analítica: Rt = 8,34 min, d.e. = 100 % [columna: LUX Cellulose-4, 5 ^m, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-^): 8 [ppm] 1,38 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,30 (a, s, 1H), 4,59 (c, 1H), 5,01-5,13 (m, 2H), 5,50 (a, s, 1H), 6,90 (d, 1H), 7,50-7,65 (m, 5H), 7,67-7,78 (m, 3H).
La estereoquímica absoluta del compuesto se determinó realizando adicionalmente la misma reacción con diastereómero enantiopuro 5-(4-clorofenil)-2-({5-[(1S)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (Ejemplo 5A) como el material de partida y la comparación de los dos productos correspondientes por HPLC quiral analítica.
Ejemplo 83
5-(4-clorofenil)-2-({1-(2-clorofenil)-5-[(1ft)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona
HPLC quiral analítica: Rt = 11,88 min, d.e. = 98,1 % [columna: LUX Cellulose-4, 5 |jm, 250 x 4,6 mm; eluyente: isohexano/etanol 70:30; velocidad de flujo: 1 ml/min; temperatura: 40 °C; detección UV: 220 nm].
1H NMR (400 MHz, DMSO-da): 8 [ppm] 1,38 (d, 3H), 3,85 (dd, 1H), 4,00 (dd, 1H), 4,24-4,36 (m, 1H), 4,54-4,65 (m, 1H), 5,07 (s, 2H), 5,51 (a, s, 1H), 6,90 (d, 1H), 7,50-7,65 (m, 5H), 7,68-7,79 (m, 3H).
La estereoquímica absoluta del compuesto se determinó realizando adicionalmente la misma reacción con diastereómero enantiopuro 5-(4-clorofenil)-2-({5-[(1R)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (Ejemplo 6A) como el material de partida y la comparación de los dos productos correspondientes por HPLC quiral analítica.
B. Evaluación de actividad biológica
Abreviaturas y acrónimos:
Acc. n.° número de referencia
AVP arginina vasopresina
Bmáx máxima capacidad de unión al ligando
ASB albúmina en suero bovino
AMPc adenosina monofosfato cíclica
Cat. n.° número de catálogo
ADNc ácido desoxirribonucleico complementario
OHC ovario de hámster chino
CRE elemento de respuesta a AMPc
Ct umbral de ciclo
DMEM/F12 medio de Eagle modificado por Dulbecco/medio Ham's F12 (1:1)
ADN ácido desoxirribonucleico
DTT ditiotreitol
CE50 concentración eficaz media máxima
EDTA ácido etilendiamina tetraacético
FAM carboxifluoresceína succinimidil éster
f.c. concentración final
STF suero de ternero fetal
HEPES ácido 4-(2-hidroxietil)piperazina-1-etanosulfónico
CI50 concentración inhibidora media máxima
Kd constante de disociación
Ki constante de disociación de un inhibidor
ARNm ácido ribonucleico mensajero
PBS solución salina tamponada con fosfato
p.o. per os, peroral
ARN ácido ribonucleico
RTPCR reacción en cadena de la polimerasa en tiempo real
SPA ensayo de proximidad de centelleo
TAMRA carboxitetrametilrodamina
TRIS 2-amino-2-hidroximetilpropano-1,3-diol
La demostración de la actividad de los compuestos de la presente invención se puede lograr a través de ensayos in vitro, ex vivo, e in vivo que son conocidos en la técnica. Por ejemplo, para demostrar la actividad de los compuestos de la presente invención, se pueden usar los siguientes ensayos.
B-1. Ensayo celular in vitro para determinar actividad del receptor de vasopresina
La identificación de agonistas y antagonistas de los receptores de vasopresina V1a y V2 de seres humanos, ratas y perros así como la cuantificación de la actividad de los compuestos de la invención se realizan usando líneas celulares recombinantes. Estas líneas celulares derivan originalmente de una célula epitelial de ovario de hámster (ovario de hámster chino, OHC K1, ATCC: American Type Culture Collection, Manassas, VA 20108, EE.UU.). Estas líneas celulares de prueba expresan constitutivamente los receptores V1a y V2 de seres humanos, ratas o perros. En el caso de de receptores V1a acoplados a Gaq, las células también se transfectan de forma estable con una forma modificada de fotoproteínas sensibles a calcio aequorina (V1a de ser humano y rata) u obelina (V1a de perro), que, después de la reconstitución con el cofactor coelenterazina, emiten luz cuando hay aumentos en las concentraciones de calcio libre [Rizzuto R, Simpson AW, Brini M, Pozzan T, Nature 358, 325-327 (1992); Illarionov BA, Bondar VS, Illarionova VA, Vysotski ES, Gene 153 (2), 273-274 (1995)]. Las células del receptor de vasopresina resultantes reaccionan con la estimulación de receptores V1a expresados recombinantemente mediante la liberación intracelular de iones de calcio, que se pueden cuantificar por la luminiscencia de fotoproteína resultante. Los receptores V2 acoplados a Gs se transfectan de forma estable en líneas celulares que expresan el gen para luciferasa de luciérnaga bajo el control de un promotor responsable de CRE. La activación
de receptores V2 induce la activación del promotor de respuesta a CRE mediante el aumento de AMPc, induciendo así la expresión de luciferasa de luciérnaga. La luz emitida por fotoproteínas de líneas celulares V1a así como la luz emitida por luciferasa de luciérnaga de líneas celulares V2 corresponden a la activación o inhibición del receptor de la vasopresina respectivo. La bioluminiscencia de las líneas celulares se detecta usando un luminómetro adecuado [Milligan G, Marshall F, Rees S, Trends in Pharmacological Sciences 17, 235-237 (1996)].
Procedimiento de prueba:
Líneas celulares del receptor de vasopresina V1a:
El día previo al ensayo, las células se colocan en medio de cultivo (DMEM/F12, 2 % STF, glutamina 2mM, HEPES 10 mM, 5 |jg/ml coelenterazina) en placas de mimcrotitulación den 384 pocillos y se mantienen en una incubadora celular (96 % de humedad, 5 % v/v CO2, 37 °C). El día del ensayo, los compuestos de prueba en diversas concentraciones se colocan durante 10 minutos en los pocillos de la placa de microtitulación antes de agregar el agonista [Arg8]-vasopresina en una concentración de CE50. La señal de luz resultante se mide inmediatamente en el luminómetro.
Líneas celulares del receptor de vasopresina V2:
El día previo al ensayo, las células se colocan en medio de cultivo (DMEM/F12, 2 % STF, glutamina 2mM, HEPES 10 mM) en placas de mimcrotitulación de 384 pocillos y se mantienen en una incubadora celular (96 % de humedad, 5 % v/v CO2, 37 °C). El día del ensayo, los compuestos de prueba en diversas concentraciones y el agonista [Arg8]-vasopresina en una concentración de CE50 se agregan juntos a los pocillos, y las placas se incuban durante 3 horas en una incubadora celular. Tras la adición del reactivo de lisis celular Triton™ y la luciferina sustrato, se mide la luminiscencia de luciferasa de luciérnaga en un luminómetro.
La Tabla 1A a continuación enumera los valores individuales de CI50 para los compuestos de la invención (que incluyen mezclas diastereoméricas así como diastereómeros separados enantiopuros) que se obtuvieron de líneas celulares transfectadas con el receptor V1a o V2 humano:
Tabla 1A




Los datos de CI50 enumerados en la Tabla 1A demuestran que los compuestos de la presente invención actúan como antagonistas duales altamente potentes de los receptores de vasopresina V1a y V2.
Con fines comparativos, los derivados seleccionados de fenil-triazol e imidazol que se suponía representaban de la forma más cercana la técnica anterior (cf. patente internacional publicada WO 2011/104322-A1 y ejemplos de compuestos descritos en la misma) también se analizaron en los ensayos celulares de V1a y v 2 descritos anteriormente. Los valores de CI50 para estos compuestos obtenidos de líneas celulares transfectadas con el receptor V1a o V2 humano se enumeran en la Tabla 1B a continuación:
Tabla 1B

B-2. Ensayo de unión radioactivo
Los valores de IC50 y K se determinaron en un ensayo SPA de competencia de unión radioactiva usando fracciones de membrana de líneas celulars OHC recombinantes que expresan los receptores V1a y V2 de vasopresina respectivos de ser humano, rata o perro. Estas células derivan de una célula epitelial de ovario de hámster (ovario de hámster chino, OHC K1, ATCC: American Type Culture Collection, Manassas, VA 20108, EE.UU.). Además, las células se transfectan de forma estable con el receptor V1a o V2 de ser humano, rata o perro. Las preparaciones de membrana se someten al ensayo de competencia de unión del receptor radioactivo descrito a continuación.
Las células OHC transfectadas con receptor de vasopresina se cultivaron en una cantidad adecuada en matraces T-175 con DMEM/F12, 10 % STF, HEPES 15 mM, 1 mg/ml G418 y se mantuvieron en una incubadora celular (96 % de humedad, 5 % v/v CO2, 37 °C). Después de alcanzar la confluencia adecuada, las células se cosecharon para preparar la membrana. Las células se rasparon en PBS y se sedimentaron por centrifugación suave a 200 x g durante 5 min a temperatura ambiente. Los sedimentos se resuspendieron en PBS y se centrifugaron nuevamente. Después de repetir esta etapa una vez más, los sedimentos resultantes se congelaron a- 80 °C durante 30 min. Los sedimentos congelados se resuspendieron en tampón de preparación helada (TRIS 50 mM,
EDTA 2 mM, DTT 2 mM, cocktail de inhibidor de proteasa completo) y se homogeneizaron a 2000 rpm durante 35 segundos (Polytron PT3000, Kinematica). El homogenato se enfrió durante 2 min en hielo, y la homogeneización se repitió dos veces. El homogenato resultante se centrifugó a 500 x g durante 10 min a 4 °C. Las membranas se sedimentaron a 4500 x g durante 20 min a 4 °C, se resuspendieron en tampón de almacenamiento (TRIS 7,5 mM, MgCh 12,5 mM, EDTA 0,3 mM, sucrosa 250 mM, cocktail de inhibidor de proteasa completo) y se homogeneizaron a 2000 rpm durante 2 segundos (Polytron PT3000, Kinematica). La concentración de proteína se determinó usando el ensayo de proteína BCA (Thermo Scientific Pierce), y las preparaciones de membrana se almacenaron a -80 °C. El día de uso, se descongelaron partes alícuotas y se sometieron a agitación con formación de vórtice brevemente.
Para la determinación de la afinidad de unión al receptor de compuestos de prueba, se configuró un ensayo SPA de la siguiente manera. Para cada preparación de membrana, se determinaron valores Kd yBmax. A partir de estos datos, el número de perlas SPA (perlasWGA PVT, PerkinElmer, 200 pg/pocillo), la concentración de ligando radioactivo (3H-AVP, PerkinElmer, 2,431 TBq/mmol, f.c. 1-2 x Kd) y la cantidad de la preparación de membrana respectiva (10 pg proteína/pocillo) se correspondieron con el volumen de ensayo (100 pl) en tampón de unión (TRIS 50 mM, 0,2 % ASB) en una placa de 96 pocillos. Los compuestos de prueba se diluyeron en tampón de unión (f.c. 10-4 M a 10-12 M) y se sometieron al ensayo. Las placas se agitaron suavemente durante 1-3 horas a temperatura ambiente y se incubaron adicionalmente durante 1-2 horas. Las señales generadas por 3H-AVP unido se midieron usando un contador p (1450 Microbeta Trilux). A partir de estos resultados, los valores CI50 y Ki para los compuestos probados se calcularon usando GraphPad Prism.
B-3. Ensayo celular in vitro para detectar la acción de antagonistas del receptor de vasopresina V1a en la regulación de genes profibróticas
La línea celular H9C2 (American Type Culture Collection n.°. CRL-1446), descrita como un tipo de cardiomiocito aislado de tejido cardíaco de rata, expresa de forma endógena el receptor de vasopresina V1A AVPR1A en gran cantidad de copias, mientras la expresión de AVPR2 no se puede detectar. Para los ensayos celulares para la inhibición de la regulación dependiente del receptor AVPR1A de expresión génica por antagonistas del receptor, el procedimiento es el siguiente:
Las células H9C2 se siembran en placas de microtitulación de 6 pocillos para cultivo celular a una densidad celular de 50000 células/pocillo en 2,0 ml de medio Opti-MEM (Invitrogen Corp., Carlsbad, CA, EE.UU., n.° de catálogo 11058-021) y se mantienen en una incubadora celular (96 % de humedad, 8 % v/v CO2, 37 °C). Después de 24 horas, se cargan conjuntos de tres pocillos (triplicado) con solución vehicular (control negativo) y solución de vasopresina ([Arg8]-vasopresina acetato, Sigma, n.° de catálogo V9879), o compuesto de prueba (disuelto en vehículo: agua con 20 % v/v etanol) y solución de vasopresina. En el cultivo celular, la concentración de vasopresina final es 1 nM. La solución del compuesto de prueba se agrega al cultivo celular en pequeños volúmenes, de modo que no se exceda una concentración final de 0,03 % de etanol en el ensayo celular. Después de un tiempo de incubación de 5 horas, el sobrenadante de cultivo se quita bajo succión, las células adherentes se lisan en 350 pl de tampón RLT (Qiagen, n.° de catálogo 79216), y el ARN se aísla del lisado usando el kit RNeasy (Qiagen, n.° de catálogo 74104). A esto le sigue la digestión de ADNasa (Invitrogen, n.° de catálogo 18068-015), síntesis de ADNc (Promaga, ImProm-II Reverse Transcription System, n.° de catálogo A3800) y TIRCP (RCPp MasterMix TI-QP2X-03-075, Eurogentec, Seraing, Bélgica). Todos los procedimientos se realizan de acuerdo con los protocolos de trabajo de los fabricantes de losreactivos de prueba. Los conjuntos de cebadores para laTIRCP se seleccionan según las secuencias génicas de ARNm (NCBI GenBank Entrez Nucleotide Data Base) usando el programa Primer3Plus con sondas etiquetadas con 6-FAM TAMRA. El TIRCP para determinar la expresión de ARNm relativa de células de diversos lotes de ensayo se realiza usando el detector de secuencia Applied Biosystems ABI Prism 7700 en formato de placa de microtitulación de 384 pocillos de acuerdo con las instrucciones operativas del instrumento. La expresión génica relativa se representa por el valor delta-delta Ct [Applied Biosystems, User Bulletin n.°. 2 ABI Prism 7700 SDS, 11 de diciembre de 1997 (actualizado 10/2001)] con referencia al nivel de expresión del gen L-32 de proteína ribosómica (n.° de rerferencia GenBank NM_013226) y el valor Ct umbral de Ct = 35.
B-4. Ensayo in vivo para detectar efectos cardiovasculares: medición de presión sanguínea en ratas anestesiadas (modelo de 'desafío' de vasopresina)
En ratas macho Sprague-Dawley (250-350 g de peso corporal) bajo anestesia de inyección de ketamina/xilazina/pentobarbital, tubos de polietileno (PE-50, Intramedic®), que se precargan con solución de cloruro de sodio isótónica que contiene heparina (500 UI/ml), se introducen en la vena yugular y la vena femoral y luego se relacionan. Mediante un acceso venoso, con la ayuda de una jeringa, se inyecta Arg-vasopresina; la sustancia de prueba se administra mediante el segundo acceso venoso. Para la determinación de la presión sanguínea sistólica, se relaciona un catéter de presión (Millar SPR-3202F) en la arteria carótida. El catéter arterial está conectado a un transductor de presión que alimenta sus señales a una computadora de registro equipada con software de registro adecuado. En un experimento típico, al animal experimental se le administran 3-4 inyecciones de bolo sucesivas en intervalos de de 10-15 min con una cantidad definida de Arg-vasopresina (30 ng/kg) en
solución de cloruro de sodio isotónica. Cuando la presión sanguínea alcanzó nuevamente niveles iniciales, la sustancia de prueba se administra como un bolo con infusión continua posterior en un disolvente adecuado. Después de esto, en intervalos definidos (1015 min) se administra nuevamente la misma cantidad de Argvasopresina que al comienzo. En función de los valores de presión sanguínea, se hace una determinación de la medida con la que la sustancia de prueba contrarresta el efecto hipertenso de Arg-vasopresina. Los animales de control solo reciben disolvente en vez de la sustancia de prueba.
Después de la administración intravenosa, los compuestos de la invención en comparación con los controles del solvente ocasionan una inhibición del aumento de presión sanguínea por Arg-vasopresina.
B-5. Ensayo in vivo para detectar efectos cardiovasculares: investigaciones de diuresis en ratas conscientes mantenidas en jaulas de metabolismo
Se mantienen ratas Wistar (220-450 g de peso corporal) con acceso libre a alimento (Altromin) y agua potable. Durante el experimento, los animales se mantienen con acceso libre a agua potable durante 4 a 8 o hasta 24 horas individualmente en jaulas de metabolismo adecuadas para ratas de esta clase de peso (Tecniplast Deutschland GmbH, D-82383 HohenpeilJenberg). Al comienzo del experimento, se les administra a los animales la sustancia de prueba en un volumen de 1 a 3 ml/kg de peso corporal de un disolvente adecuado mediante sonda en el estómago. Los animales de control solo recibieron disolvente. Las pruebas de control y sustancia se realizan en paralelo el mismo día. Los grupos de control y los grupos de dosis de sustancia consisten en 4 a 8 animales. Durante el experimento, la orina excretada por los animales se recoge continuamente en un receptor en la base de la jaula. El volumen de orina por unidad de tiempo se determina de forma separada para cada animal, y la concentración de electrolitos urinarios se mide por procedimientos convencionales de fotometría de llama. Antes de comenzar el experimento, se determina el peso corporal de los animales individuales.
Después de la administración oral, en comparación con las aplicaciones de control de disolvente, los compuestos de la invención ocasionan una excreción aumentada de orina que se basa esencialmente en una excreción aumentada de agua (acuaresis).
La Tabla 2A a continuación muestra los cambios observados en la excreción urinaria con relación al control de disolvente (= 100 %) para ejemplos de compuestos de la invención en dosificaciones diferentes:
Tabla 2A

Con fines comparativos, los derivados seleccionados de fenil-triazol e imidazol que se suponía representaban de la forma más cercana la técnica anterior (cf. patente internacional publicada WO 2011/104322-A1 y ejemplos de compuestos descritos en la misma) también se analizaron para determinar el efecto diurético en este ensayo. Los cambios observados en la excreción urinaria con relación al control de disolvente (= 100 %) con dos dosificaciones diferentes se muestran en la Tabla 2B a continuación:
Tabla 2B

Los resultados que se muestran en la Tabla 2A y 2B demuestran que los compuestos de la presente invención son significativamente más potentes in vivo: Los ejemplos analizados de la presente invención provocan más de un aumento triple, en algunos casos más de un aumento de diez veces de volumen urinario versus el grupo de control de vehículo a una dosis p.o. de 3 mg/kg y la mayoría de los ejemplos mostró actividad acuarética sustancial ya en dosis p.o. de 0,3 mg/kg o 1 mg/kg. Esto es opuesto a los derivados de feniltriazol e imidazol que se supone representan de la forma más cercana la técnica anterior que no son activos en dosis p.o. inferiores a 3 mg/kg y algo activos a 3 mg/kg.
B-6. Ensayo in vivo para detectar efectos cardiovasculares: investigaciones hemodinámicas en perros anestesiados
Se anestesian perros beagle machos (Beagle, Marshall BioResources) con un peso de entre 10 y 15 kg con pentobarbital (30 mg/kg i.v., Narcoren®, Merial, Alemania) para las intervenciones quirúrgicas y exámenes hemodinámicos y funcionales. EL bromuro de pancuronio (2 mg/animal i.v., Ratiopharm, Alemania) sirve además como relajante muscular. Los perros se intuban y ventilan con una mezcla de aire ambiente/oxígeno (40/60 %, alrededor de 3-4 l/min). La ventilación se realiza usando un ventilador de GE Healthcare (Avance) y se monitorea usando un analizador (Datex-Ohmeda, GE). Se mantiene la anestesia por infusión continua de pentobarbital (50 pg/kg/min); se usa fentanilo como un analgésico (10-40 pg/kg/h). Una alternativa al pentobarbital es usar isoflurano (1-2 % por volumen).
En intervenciones preparatorias, los perros se equipan con un marcapasos cardíaco. A los 21 días antes de la primera prueba de fármaco (es decir, al comienzo del experimento), se implanta un marcapasos cardíaco de Biotronik (Logos®) en un bolsillo de piel subcutáneo y se pone en contacto con el corazón mediante un electrodo de marcapasos que se hace avanzar a través de la vena yugular externa con transiluminación en el ventrículo derecho. Posteriormente todos los accesos se eliminan y los perros se despiertan espontáneamente de la anestesia. Después de 7 días adicionales (es decir, 14 días antes de la primera prueba de fármaco), se activa el marcapasos descrito anteriormente y el corazón se estimula a una frecuencia de 220 latidos por minuto.
Los experimentos de prueba de sustancia real se realizan 14 y 28 días después del comienzo de la estimulación del marcapasos usando la siguiente instrumentación:
• introducción de un catéter en la vejiga para liberación de la vejiga y para medir el flujo de orina;
• la unión de ECG lleva a las extremidades para la medición de ECG.
• introducción de un tubo Fluidmedic® PE 300 relleno con solución de cloruro de sodio en la arteria femoral; este tubo se conecta con un sensor de presión (Braun Melsungen, Alemania) para medir la presión sanguínea sistémica;
• introducción de un catéter Millar Tip (tipo 350 PC, Millar Instruments, Houston, EE.UU.) a través del atrio izquierdo o a través de un puerto asegurado en la arteria carótida, para medir la hemodinámica cardíaca; • introducción de un catéter Swan-Ganz (CCOmbo 7.5F, Edwards, Irvine, EE.UU.) mediante la vena yugular en la arteria pulmonar, para medir la salida cardíaca, saturación de oxígeno, presiones arteriales pulmonares y presión venosa central;
• colocación de un catéter venoso en la vena cefálica para infundir pentobarbital para el remplazo líquido y para muestreo de sangre (determinación de niveles de plasma de la sustancia de prueba o de otros valores sanguíneos clínicos);
• colocación de un catéter venoso en la vena safena para infundir fentanilo y para la administración de la sustancia de prueba;
• infusión continua de vasopresina (Sigma, 4 mU/kg/min); los compuestos de prueba luego se administran y evalúan a diferentes dosificaciones bajo esta infusión de vasopresina.
Las señales primarias se amplían si es necesario (amplificador ACQ 7700, DataSciences Inc., Minneapolis, EE.UU., o Edwards-Vigilance-Monitor, Edwards, Irvine, EE.UU.) y se introducen posteriormente en el sistema Ponemah (DataSciences Inc., Minneapolis, EE.UU) para evaluación. Las señales se registran continuamente a lo largo del período experimental y se procesan además de forma digital mediante el software y se promedian durante 30 segundos.
Aunque la invención se ha desvelado con referencia a realizaciones específicas, es evidente que otras realizaciones y variaciones de la invención se pueden encontrar por parte de otros expertos en la técnica sin apartarse del espíritu y alcance verdaderos de la invención. Las reivindicaciones pretenden entenderse como que incluyen todas estas variaciones derealizaciones y equivalentes.
C. Ejemplos relacionados con composiciones farmacéuticas
Las composiciones farmacéuticas de acuerdo con la presente invención se pueden ilustrar de la siguiente forma: Solución i.v. estéril:
Se puede fabricar una solución de 5 mg/ml del compuesto deseado de la invención usando agua inyectable estéril, y el pH se ajusta si es necesario. La solución se diluye para administración a 1-2 mg/ml con 5 % de dextrosa estéril y se administra como una infusión i.v. durante alrededor de 60 minutos.
Polvo liofilizado para administración i.v.:
Se puede preparar una preparación estéril con (i) 100-1000 mg del compuesto deseado de la invención como un polvo liofilizado, (ii) 32-327 mg/ml de citrato de sodio, y (iii) 300-3000 mg de Dextrano 40. La formulación se reconstituye con solución salina inyectable estéril o 5 % de dextrosa hasta una concentración de 10 a 20 mg/ml, que se diluye adicionalmente con solución salina o 5 % de dextrosa hasta 0,2 a 0.4 mg/ml, y se administra como bolo i.v. o por infusión i.v. durante 15-60 minutos.
Suspensión intramuscular:
La siguiente solución o suspensión se puede preparar para inyección intramuscular:
50 mg/ml del compuesto insoluble en agua deseado de la invención; 5 mg/ml de carboximetilcelulosa de sodio; 4 mg/ml de Tween 80; 9 mg/ml de cloruro de sodio; 9 mg/ml de alcohol bencílico.
Cápsulas de corteza dura:
Se prepara un gran número de cápsulas unitarias llenando cápsulas de gelatina dura en dos partes convencionales cada una con 100 mg del compuesto en polvo deseado de la invención, 150 mg de lactosa, 50 mg de celulosa y 6 mg de estearato de magnesio.
Cápsulas de gelatina blanda:
Se prepara e inyecta una mezcla del compuesto deseado de la invención en un aceite digerible, tal como aceite de soja, aceite de semilla de algodón o aceite de oliva, mediante una bomba de desplazamiento positivo en gelatina derretida para formar cápsulas de gelatina blanda que contienen 100 mg del principio activo. Las cápsulas se lavan y secan. El compuesto deseado de la invención se puede disolver en una mezcla de polietilenglicol, glicerina y sorbitol para preparar una mezcla de medicinas miscible en agua.
Comprimidos:
Se prepara un gran número de comprimidos mediante procedimientos convencionales de modo que la unidad de dosificación sea 100 mg del compuesto deseado de la invención, 0,2 mg de dióxido de silicio coloidal, 5 mg de estearato de magnesio, 275 mg de celulosa microcrostalina, 11 mg de almidón, y 98,8 mg de lactosa. Se pueden aplicar revestimientos acuosos y no acuosos apropiados para aumentar la palatabilidad, mejorar la elegancia y estabilidad o retrasar la absorción.
Claims (13)
1. Un compuesto de fórmula (I)

en la que
R1 es hidrógeno o metilo,
R2A y R2B se seleccionan independientemente del grupo que consiste en hidrógeno, fluoro, cloro, ciano, metilo, fluorometilo, difluorometilo, trifluorometilo, etilo, metoxi, difluorometoxi y trifluorometoxi,
o una sal, hidrato y/o solvato farmacéuticamente aceptable del mismo.
2. El compuesto de fórmula (I) de acuerdo con la reivindicación 1, en el que
R1 es hidrógeno o metilo,
R2A y R2B se seleccionan independientemente del grupo que consiste en hidrógeno, fluoro, cloro, metilo y metoxi, en la que al menos uno de R2A y R2B es diferente de hidrógeno,
o una sal, hidrato y/o solvato farmacéuticamente aceptable del mismo.
3. El compuesto de fórmula (I) de acuerdo con la reivindicación 1 o 2, en el que el compuesto se selecciona del grupo que consiste en
5-(4-clorofenil)-2-{[1-(3-clorofenil)-5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-{[1-(3-fluorofenil)-5-(hidroximetil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-{[5-(hidroximetil)-1-(2-metilfenil)-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
2-({1-(2-cloro-4-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
2-{[1-(2-cloro-4-fluorofenil)-5-(1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1);
2-{[1-(2-cloro-4-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2);
2-({1-(2-cloro-5-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il}metil)-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
2-{[1-(2-cloro-5-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 1);
2-{[1-(2-cloro-5-fluorofenil)-5-(1-hidroxietil)-1H-1,2,4-triazol-3-il]metil}-5-(4-clorofenil)-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona (diastereómero 2);
5-(4-clorofenil)-2-({1-(3-fluorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-({1-(3-fluorofenil)-5-[(1R)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-({1-(3-fluorofenil)-5-[(1S)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-({1-(3-clorofenil)-5-[(1RS)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-ona;
5-(4-clorofenil)-2-({1-(3-clorofenil)-5-[(1R)-1-hidroxietil]-1H-1,2,4-triazol-3-il]metil}-4-[(2S)-3,3,3-trifluoro-2
h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(3-clorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(2-clorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(2-clorofen¡l)-5-[(1R)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
y
5-(4-clorofen¡l)-2-({1-(2-clorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
o una sal, h¡drato y/o solvato farmacéut¡camente aceptable de los m¡smos.
4. El compuesto de fórmula (I) de acuerdo con la re¡v¡nd¡cac¡ón 1, 2 o 3, en el que el compuesto se selecc¡ona del grupo que cons¡ste en
5-(4-clorofen¡l)-2-({1-(3-fluorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(3-fluorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(3-clorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(3-clorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
5-(4-clorofen¡l)-2-({1-(2-clorofen¡l)-5-[(1RS)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
y
5-(4-clorofen¡l)-2-({1-(2-clorofen¡l)-5-[(1S)-1-h¡drox¡et¡l]-1H-1,2,4-tr¡azol-3-¡l]met¡l}-4-[(2S)-3,3,3-tr¡fluoro-2-h¡drox¡prop¡l]-2,4-d¡h¡dro-3H-1,2,4-tr¡azol-3-ona;
o una sal, h¡drato y/o solvato farmacéut¡camente aceptable de los m¡smos.
5. El proced¡m¡ento para preparar un compuesto de fórmula (I) como se def¡ne en las re¡v¡nd¡cac¡ones 1 a 4, caracterizado porque un compuesto de fórmula (II)

se hace reacc¡onar pr¡mero con h¡draz¡na para proporc¡onar la h¡draz¡da de fórmula (III)

luego se condensa con una am¡d¡na de fórmula (IV)

o una sal de la misma,
en la que R1 tiene el significado que se indica en las reivindicaciones 1 a 4,
en presencia de una base para proporcionar un derivado de 1,2,4-triazol de fórmula (V)

en la que R1 tiene el significado que se indica en las reivindicaciones 1 a 4,
y posteriormente se acopla con un ácido fenilborónico de fórmula (VI)

en la que R2A y R2B tienen los significados que se indican en las reivindicaciones 1 a 4,
en presencia de un catalizador de cobre y una base amina para proporcionar el compuesto diana de fórmula (I)

en la que R1, R2A y R2B tienen los significados que se indican en las reivindicaciones 1 a 4,
seguido de, opcionalmente, cuando fuera adecuado, (i) separar los compuestos de fórmula (I) obtenidos así en sus correspondientes diastereómeros, y /o (ii) convertir los compuestos de fórmula (I) en sus correspondientes hidratos, solvatos, sales y/o hidrato o solvatos de las sales por tratamiento con los correspondientes disolventes y/o ácidos o bases.
6. Un compuesto como se define en cualquiera de las reivindicaciones 1 a 4 para su uso en el tratamiento y/o prevención de enfermedades.
7. Un compuesto como se define en cualquiera de las reivindicaciones 1 a 4 para su uso en un procedimiento para el tratamiento y/o prevención de insuficiencia cardíaca aguda y crónica, síndrome cardiorrenal, hiponatremia hipervolémica y euvolémica, cirrosis hepática, ascitis, edema y el síndrome de secreción inadecuada de ADH
(SIADH).
8. Uso de un compuesto como se define en cualquiera de las reivindicaciones 1 a 4 para la fabricación de una composición farmacéutica para el tratamiento y/o prevención de insuficiencia cardíaca aguda y crónica, síndrome cardiorrenal, hiponatremia hipervolémica y euvolémica, cirrosis hepática, ascitis, edema y el síndrome de secreción inadecuada de ADH (SIADH).
9. Una composición farmacéutica que comprende un compuesto como se define en cualquiera de las reivindicaciones 1 a 4 y uno o más excipientes farmacéuticamente aceptables.
10. La composición farmacéutica de la reivindicación 9 que comprende además uno o más agentes terapéuticos adicionales que se seleccionan del grupo que consiste en diuréticos, antagonistas de angiotensina AII, inhibidores de ACE, bloqueadores del receptor beta, antagonistas del receptor mineralocorticoide, nitratos orgánicos, donantes de NO, activadores de la guanilato ciclasa soluble, estimuladores de la guanilato ciclasa soluble y agentes inotrópicos positivos.
11. La composición farmacéutica como se define en la reivindicación 9 o 10 para el tratamiento y/o prevención de insuficiencia cardíaca aguda y crónica, síndrome cardiorrenal, hiponatremia hipervolémica y euvolémica, cirrosis hepática, ascitis, edema y el síndrome de secreción inadecuada de ADH (SIADH).
12. 5-(clorofenil)-2({1-(3-clorofenil)-5-[(1S)-1-hidroxietil]-1H-1,2,4-triazol-3il}metil)-4[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H,2,4-triazol-3-ona de acuerdo con la reivindicación 1 de la siguiente fórmula

o una sal, hidrato y/o solvato farmacéuticamente aceptable del mismo.
13. El compuesto de la reivindicación 12, en el que el compuesto es 5-(clorofenil)-2({1-(3-clorofenil)-5-[(1S)-1-hidroxietil]-1H-1,2,4-triazol-3il}metil)-4[(2S)-3,3,3-trifluoro-2-hidroxipropil]-2,4-dihidro-3H-1,2,4-triazol-3-onade la siguiente fórmula
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14191491 | 2014-11-03 | ||
| PCT/EP2015/075200 WO2016071212A1 (en) | 2014-11-03 | 2015-10-30 | Hydroxyalkyl-substituted phenyltriazole derivatives and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2697902T3 true ES2697902T3 (es) | 2019-01-29 |
Family
ID=51897095
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15788389T Active ES2697902T3 (es) | 2014-11-03 | 2015-10-30 | Derivados de feniltriazol sustituido con hidroxialquilo y sus usos |
Country Status (40)
Families Citing this family (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PE20130683A1 (es) | 2010-02-27 | 2013-06-20 | Bayer Ip Gmbh | Ariltriazolonas ligadas a bisarilo y su uso |
| US10138236B2 (en) | 2014-09-24 | 2018-11-27 | Bayer Pharma Aktiengesellschaft | Factor xia-inhibiting pyridobenzazepine and pyridobenzazocine derivatives |
| CN107949562B (zh) | 2015-06-09 | 2021-07-23 | 拜耳制药股份公司 | 毒蕈碱性m2受体的正性变构调节剂 |
| EP3452472A1 (en) | 2016-05-03 | 2019-03-13 | Bayer Aktiengesellschaft | Hydroxyalkyl-substituted heteroaryltriazole derivatives and uses thereof |
| UY37222A (es) * | 2016-05-03 | 2017-11-30 | Bayer Pharma AG | PROCEDIMIENTO PARA LA PREPARACIÓN DE DERIVADOS DE 1-FENILO-1,2,4-TRIAZOL SUSTITUIDOS POR 5-HIdroxIalQUILO |
| WO2017191105A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted aryltriazole derivatives and uses thereof |
| US9988367B2 (en) * | 2016-05-03 | 2018-06-05 | Bayer Pharma Aktiengesellschaft | Amide-substituted pyridinyltriazole derivatives and uses thereof |
| JP6911052B2 (ja) * | 2016-05-03 | 2021-07-28 | バイエル ファーマ アクチエンゲゼルシャフト | オキソアルキル置換フェニルトリアゾール誘導体およびその使用 |
| EP3452470B1 (en) | 2016-05-03 | 2020-03-18 | Bayer Pharma Aktiengesellschaft | Amide-substituted phenyltriazole derivatives and uses thereof |
| WO2017191117A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | V1a receptor antagonists for use in the treatment of renal diseases |
| US10525041B2 (en) | 2016-05-03 | 2020-01-07 | Bayer Pharma Aktiengesellschaft | Fluoroalkyl-substituted aryltriazole derivatives and uses thereof |
| CA3030204A1 (en) | 2016-07-11 | 2018-01-18 | Bayer Pharma Aktiengesellschaft | 7-substituted 1-pyridyl-naphthyridine-3-carboxylic acid amides and use thereof |
| EP3296298A1 (de) | 2016-09-14 | 2018-03-21 | Bayer Pharma Aktiengesellschaft | 7-substituierte 1-aryl-naphthyridin-3-carbonsäureamide und ihre verwendung |
| JOP20190045A1 (ar) | 2016-09-14 | 2019-03-14 | Bayer Ag | مركبات أميد حمض 1- أريل-نفثيريدين-3-كربوكسيليك مستبدلة في الموضع 7 واستخدامها. |
| JOP20190080A1 (ar) | 2016-10-14 | 2019-04-11 | Bayer Pharma AG | مركبات مشتقة من 6-(1h-بيرازول-1-يل) بيريميدين-4- أمين مستبدل واستخداماتها |
| EP3529244A1 (en) | 2016-10-20 | 2019-08-28 | Bayer Pharma Aktiengesellschaft | Hydroxyalkyl-substituted triazole derivatives and uses thereof |
| WO2018149929A1 (en) * | 2017-02-16 | 2018-08-23 | Bayer Pharma Aktiengesellschaft | Combination of reporter gene assays and transcriptional analysis |
| EP3700899A1 (en) | 2017-10-24 | 2020-09-02 | Bayer Pharma Aktiengesellschaft | Substituted triazole derivatives and uses thereof |
| WO2019081291A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | PRODRUGS OF SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| CA3084411A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | Substituted triazole derivatives and uses thereof |
| US11331314B2 (en) | 2017-10-24 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Amine substituted triazole derivatives and uses thereof |
| US11298367B2 (en) | 2017-10-24 | 2022-04-12 | Bayer Aktiengesellschaft | Prodrugs of substituted triazole derivatives and uses thereof |
| US11149023B2 (en) | 2017-10-24 | 2021-10-19 | Bayer Pharma Aktiengesellschaft | Substituted triazole derivatives and uses thereof |
| US11173151B2 (en) | 2017-10-24 | 2021-11-16 | Bayer Aktiengesellschaft | Substituted triazole derivatives and uses thereof |
| AU2018354785A1 (en) | 2017-10-24 | 2020-04-23 | Bayer Aktiengesellschaft | Substituted imidazopyridine amides and use thereof |
| EP3740236A1 (de) | 2018-01-16 | 2020-11-25 | Bayer Aktiengesellschaft | Unterstützung bei der behandlung von herzinsuffizienz |
| EP3553079A1 (en) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | C-type natriuretic peptide engrafted antibodies |
| EP3553082A1 (en) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Brain natriuretic peptide engrafted antibodies |
| EP3553081A1 (en) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Atrial natriuretic peptide engrafted antibodies |
| EA202092680A1 (ru) | 2018-05-15 | 2021-04-13 | Байер Акциенгезельшафт | 1,3-тиазол-2-ил-замещенные бензамиды для лечения заболеваний, ассоциированных с сенситизацией нервных волокон |
| EA202092779A1 (ru) | 2018-05-17 | 2021-02-02 | Байер Акциенгезельшафт | Замещенные дигидропиразолопиразинкарбоксамидные производные |
| WO2020216669A1 (de) | 2019-04-23 | 2020-10-29 | Bayer Aktiengesellschaft | Phenylsubstituierte imidazopyridinamide und ihre verwendung |
| CN110082451B (zh) * | 2019-05-23 | 2021-12-24 | 南京趣酶生物科技有限公司 | 一种2-氯-1-(6-氟-色满-2-)乙醇手性醇中间体制备过程的sfc检测方法 |
| WO2021094209A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted pyrrolo triazine carboxamide derivatives as prostaglandin ep3 receptor antagonists |
| WO2021094210A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted pyrazine carboxamide derivatives as prostaglandin ep3 receptor antagonists |
| WO2021094208A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted imidazo pyrimidine ep3 antagonists |
| EP3822265A1 (en) | 2019-11-15 | 2021-05-19 | Bayer AG | Substituted hydantoinamides as adamts7 antagonists |
| EP3822268A1 (en) | 2019-11-15 | 2021-05-19 | Bayer Aktiengesellschaft | Substituted hydantoinamides as adamts7 antagonists |
| EP3900722A1 (en) | 2020-04-22 | 2021-10-27 | Bayer AG | Combination of finerenone and pecavaptan for the treatment and/or prevention of cardiovascular and/or renal diseases |
| EP4138826A1 (en) | 2020-04-22 | 2023-03-01 | Bayer Aktiengesellschaft | Combination of finerenone and a sglt2 inhibitor for the treatment and/or prevention of cardiovascular and/or renal diseases |
| WO2021259852A1 (en) | 2020-06-25 | 2021-12-30 | Bayer Aktiengesellschaft | Process for preparing 5-(alkoxycarbonyl)-and 5-(carboxamide)-1-aryl-1,2,4-triazole derivatives |
| WO2022112213A1 (en) | 2020-11-30 | 2022-06-02 | Bayer Aktiengesellschaft | Crystalline forms of 3-[[3-(4-chlorophenyl)-5-oxo-4-((2s)-3,3,3-trifluoro- 2-hydroxypropyl)-4,5-dihydro-1h-1,2,4-triazol-1-yl]methyl]-1-[3- (trifluoromethyl)pyridin-2-yl]-1h-1,2,4-triazole-5-carboxamide |
| CN115175681A (zh) | 2020-12-10 | 2022-10-11 | 拜耳公司 | sGC活化剂用于治疗眼科疾病的用途 |
| WO2022122916A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolyl piperidine carboxylic acids |
| CA3204495A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| EP4011873A1 (en) | 2020-12-10 | 2022-06-15 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| EP4011874A1 (en) | 2020-12-10 | 2022-06-15 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| WO2022214206A1 (en) | 2021-04-08 | 2022-10-13 | Bayer Aktiengesellschaft | Combination of finerenone and pecavaptan for the treatment and/or prevention of cardiovascular and/or renal diseases |
| WO2023237577A1 (en) | 2022-06-09 | 2023-12-14 | Bayer Aktiengesellschaft | Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in women |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3042466A1 (de) | 1980-11-11 | 1982-06-16 | A. Nattermann & Cie GmbH, 5000 Köln | N-substituierte (omega) -(2-oxo-4-imidazolin-l-yl)-alkansaeure-derivate, verfahren zu ihrer herstellung und diese enthaltende pharmazeutische praeparate |
| US5468448A (en) | 1989-12-28 | 1995-11-21 | Ciba-Geigy Corporation | Peroxide disinfection method and devices therefor |
| DE4107857A1 (de) | 1991-03-12 | 1992-09-17 | Thomae Gmbh Dr K | Cyclische harnstoffderivate, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| WO1992020662A1 (en) | 1991-05-10 | 1992-11-26 | Merck & Co., Inc. | Acidic aralkyl triazole derivatives active as angiotensin ii antagonists |
| WO1993017681A1 (en) | 1992-03-02 | 1993-09-16 | Abbott Laboratories | Angiotensin ii receptor antagonists |
| SK94393A3 (en) | 1992-09-11 | 1994-08-10 | Thomae Gmbh Dr K | Cyclic derivatives of urea, process for their production and their pharmaceutical agents with the content of those |
| GB9301192D0 (en) | 1993-06-09 | 1993-06-09 | Trott Francis W | Flower shaped mechanised table |
| US6844005B2 (en) | 2002-02-25 | 2005-01-18 | Trutek Corp | Electrostatically charged nasal application product with increased strength |
| FR2708608B1 (fr) | 1993-07-30 | 1995-10-27 | Sanofi Sa | Dérivés de N-sulfonylbenzimidazolone, leur préparation, les compositions pharmaceutiques en contenant. |
| US5897858A (en) | 1994-02-03 | 1999-04-27 | Schering-Plough Healthcare Products, Inc. | Nasal spray compositions exhibiting increased retention in the nasal cavity |
| US20010020100A1 (en) | 1994-06-14 | 2001-09-06 | G.D. Searle & Co. | N-substituted-1, 2, 4-triazolone compounds for treatment of cardiovascular disorders |
| AU741769B2 (en) | 1997-12-17 | 2001-12-06 | Merck Sharp & Dohme Corp. | Integrin receptor antagonists |
| DE19816882A1 (de) | 1998-04-17 | 1999-10-21 | Boehringer Ingelheim Pharma | Triazolone mit neuroprotektiver Wirkung |
| JP2002521373A (ja) | 1998-07-24 | 2002-07-16 | バイエル アクチェンゲゼルシャフト | 置換されたベンゾイルシクロヘキサンジオン類 |
| DE19834047A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Substituierte Pyrazolderivate |
| DE19834044A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Neue substituierte Pyrazolderivate |
| DE19914140A1 (de) | 1999-03-27 | 2000-09-28 | Bayer Ag | Substituierte Benzoylpyrazole |
| SK13532001A3 (sk) | 1999-04-01 | 2003-02-04 | Pfizer Products Inc. | Aminopiridíny ako inhibítory sorbitoldehydrogenázy |
| DE19920791A1 (de) | 1999-05-06 | 2000-11-09 | Bayer Ag | Substituierte Benzoylisoxazole |
| DE19921424A1 (de) | 1999-05-08 | 2000-11-09 | Bayer Ag | Substituierte Benzoylketone |
| DE19929348A1 (de) | 1999-06-26 | 2000-12-28 | Bayer Ag | Verfahren zur Herstellung von 2-Heterocyclylmethyl-benzoesäurederivaten |
| US6531142B1 (en) | 1999-08-18 | 2003-03-11 | The Procter & Gamble Company | Stable, electrostatically sprayable topical compositions |
| DE19943635A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943634A1 (de) | 1999-09-13 | 2001-04-12 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943636A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943639A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Dicarbonsäurederivate mit neuartigen pharmazeutischen Eigenschaften |
| AR027575A1 (es) | 2000-03-06 | 2003-04-02 | Bayer Ag | Benzoilciclohexenonas substituidas |
| US7080644B2 (en) | 2000-06-28 | 2006-07-25 | Microdose Technologies, Inc. | Packaging and delivery of pharmaceuticals and drugs |
| AR031176A1 (es) | 2000-11-22 | 2003-09-10 | Bayer Ag | Nuevos derivados de pirazolpiridina sustituidos con piridina |
| WO2002066447A1 (en) | 2001-02-21 | 2002-08-29 | Ono Pharmaceutical Co., Ltd. | 4h-1,2,4-triazole-3(2h)-thione deratives as sphingomyelinase inhibitors |
| DE10110749A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Substituierte Aminodicarbonsäurederivate |
| DE10110750A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| US20040071757A1 (en) | 2001-11-20 | 2004-04-15 | David Rolf | Inhalation antiviral patch |
| DE10220570A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Ag | Carbamat-substituierte Pyrazolopyridine |
| JP4673295B2 (ja) | 2003-03-14 | 2011-04-20 | メルク・シャープ・エンド・ドーム・コーポレイション | 単環式アニリドスピロヒダントインcgrp受容体拮抗物質 |
| EP1689252A2 (en) | 2003-06-27 | 2006-08-16 | Berglin Corporation of Washington | Machine for precision low stress coring and slicing of apples and other soft-cored or pitted fruits |
| AU2004309164B2 (en) * | 2003-12-22 | 2007-11-15 | Pfizer Inc. | Triazole derivatives as vasopressin antagonists |
| WO2005086836A2 (en) | 2004-03-08 | 2005-09-22 | Wyeth | Ion channel modulators |
| CN1933832A (zh) | 2004-03-08 | 2007-03-21 | 惠氏公司 | 离子通道调节剂 |
| WO2005105779A1 (en) | 2004-04-28 | 2005-11-10 | Pfizer Limited | 3-heterocyclyl-4-phenyl-triazole derivatives as inhibitors of the vasopressin v1a receptor |
| US7642275B2 (en) | 2004-12-16 | 2010-01-05 | Takeda San Diego, Inc. | Histone deacetylase inhibitors |
| WO2006117657A1 (en) | 2005-05-03 | 2006-11-09 | Ranbaxy Laboratories Limited | Triazolone derivatives as anti-inflammatory agents |
| DE102006024024A1 (de) | 2006-05-23 | 2007-11-29 | Bayer Healthcare Aktiengesellschaft | Substituierte Arylimidazolone und -triazolone sowie ihre Verwendung |
| EP2150534B1 (en) * | 2007-04-26 | 2012-01-11 | H. Lundbeck A/S | Isoquinolinone derivatives as nk3 antagonists |
| DE102008060967A1 (de) | 2008-12-06 | 2010-06-10 | Bayer Schering Pharma Aktiengesellschaft | Substituierte Phenylsulfonyltriazolone und ihre Verwendung |
| DE102010001064A1 (de) * | 2009-03-18 | 2010-09-23 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 2-Acetamido-5-Aryl-1,2,4-triazolone und deren Verwendung |
| DE102009013642A1 (de) | 2009-03-18 | 2010-09-23 | Bayer Schering Pharma Aktiengesellschaft | Substituierte Phenylalaninderivate und deren Verwendung |
| DE102009028929A1 (de) | 2009-08-27 | 2011-07-07 | Bayer Schering Pharma Aktiengesellschaft, 13353 | Heterocyclisch-substituierte 2-Acetamido-5-Aryl-1,2,4-triazolone und deren Verwendung |
| PE20130683A1 (es) * | 2010-02-27 | 2013-06-20 | Bayer Ip Gmbh | Ariltriazolonas ligadas a bisarilo y su uso |
| DE102010021637A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung |
| MX2013000198A (es) | 2010-07-09 | 2013-01-28 | Bayer Ip Gmbh | Pirimidinadas y triazinas condensadas y su uso. |
| DE102010040187A1 (de) | 2010-09-02 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Substituierte N-Phenethyl-triazolonacetamide und ihre Verwendung |
| DE102010040233A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclische Aza-Heterocyclen und ihre Verwendung |
| DE102010043379A1 (de) | 2010-11-04 | 2012-05-10 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 6-Fluor-1H-Pyrazolo[4,3-b]pyridine und ihre Verwendung |
| US9604976B2 (en) | 2012-03-22 | 2017-03-28 | Spero Gyrase, Inc. | Antibacterial compounds |
| AU2014334040A1 (en) * | 2013-10-07 | 2016-04-28 | Bayer Pharma Aktiengesellschaft | Cyclic thienouracil-carboxamides and use thereof |
-
2015
- 2015-10-30 SG SG11201703199UA patent/SG11201703199UA/en unknown
- 2015-10-30 ES ES15788389T patent/ES2697902T3/es active Active
- 2015-10-30 CN CN201580059512.8A patent/CN107074783B/zh not_active Expired - Fee Related
- 2015-10-30 BR BR112017009204-2A patent/BR112017009204A2/pt not_active Application Discontinuation
- 2015-10-30 HU HUE15788389A patent/HUE040696T2/hu unknown
- 2015-10-30 AR ARP150103529A patent/AR102487A1/es unknown
- 2015-10-30 PT PT15788389T patent/PT3215498T/pt unknown
- 2015-10-30 MA MA40893A patent/MA40893B1/fr unknown
- 2015-10-30 UY UY0001036383A patent/UY36383A/es not_active Application Discontinuation
- 2015-10-30 SI SI201530445T patent/SI3215498T1/sl unknown
- 2015-10-30 AU AU2015342017A patent/AU2015342017B2/en not_active Ceased
- 2015-10-30 KR KR1020177011653A patent/KR20170081178A/ko not_active Application Discontinuation
- 2015-10-30 MX MX2017005479A patent/MX2017005479A/es active IP Right Grant
- 2015-10-30 RS RS20181378A patent/RS58049B1/sr unknown
- 2015-10-30 CU CU2017000057A patent/CU24462B1/es unknown
- 2015-10-30 PE PE2017000788A patent/PE20170945A1/es unknown
- 2015-10-30 TW TW104135707A patent/TWI694072B/zh not_active IP Right Cessation
- 2015-10-30 US US15/523,061 patent/US20170313665A1/en not_active Abandoned
- 2015-10-30 LT LTEP15788389.3T patent/LT3215498T/lt unknown
- 2015-10-30 MY MYPI2017701506A patent/MY186908A/en unknown
- 2015-10-30 PL PL15788389T patent/PL3215498T3/pl unknown
- 2015-10-30 EA EA201790951A patent/EA032137B1/ru not_active IP Right Cessation
- 2015-10-30 CA CA2966256A patent/CA2966256A1/en not_active Abandoned
- 2015-10-30 JP JP2017523872A patent/JP6623220B2/ja not_active Expired - Fee Related
- 2015-10-30 EP EP15788389.3A patent/EP3215498B1/en active Active
- 2015-10-30 DK DK15788389.3T patent/DK3215498T3/en active
- 2015-10-30 WO PCT/EP2015/075200 patent/WO2016071212A1/en active Application Filing
- 2015-11-02 US US14/930,157 patent/US9771352B2/en active Active
- 2015-11-02 JO JOP/2015/0271A patent/JO3503B1/ar active
-
2017
- 2017-03-23 IL IL251354A patent/IL251354B/en active IP Right Grant
- 2017-04-26 SV SV2017005426A patent/SV2017005426A/es unknown
- 2017-04-27 SA SA517381429A patent/SA517381429B1/ar unknown
- 2017-04-27 PH PH12017500802A patent/PH12017500802A1/en unknown
- 2017-04-28 DO DO2017000109A patent/DOP2017000109A/es unknown
- 2017-04-28 EC ECIEPI201726733A patent/ECSP17026733A/es unknown
- 2017-05-02 CL CL2017001087A patent/CL2017001087A1/es unknown
- 2017-05-03 CO CONC2017/0004471A patent/CO2017004471A2/es unknown
- 2017-05-03 GT GT201700094A patent/GT201700094A/es unknown
- 2017-06-02 ZA ZA2017/03802A patent/ZA201703802B/en unknown
-
2018
- 2018-10-23 HR HRP20181742TT patent/HRP20181742T1/hr unknown
- 2018-11-19 CY CY181101217T patent/CY1120941T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2697902T3 (es) | Derivados de feniltriazol sustituido con hidroxialquilo y sus usos | |
| US10800746B2 (en) | Oxoalkyl-substituted phenyltriazole derivatives and uses thereof | |
| TWI772295B (zh) | 經醯胺-取代的吡啶基三唑衍生物及其用途 | |
| US10526314B2 (en) | Hydroxyalkyl-substituted heteroaryltriazole derivatives and uses thereof | |
| US10525041B2 (en) | Fluoroalkyl-substituted aryltriazole derivatives and uses thereof | |
| US10927098B2 (en) | Hydroxyalkyl-substituted triazole derivatives and uses thereof | |
| WO2017191105A1 (en) | Amide-substituted aryltriazole derivatives and uses thereof | |
| US11230540B2 (en) | Substituted triazole derivatives and uses thereof | |
| EP3700913A1 (en) | Prodrugs of substituted triazole derivatives and uses thereof | |
| EP3452468A1 (en) | Amide-substituted aryltriazole derivatives and uses thereof |