EP0503748A2 - Verfahren zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen, nichtflüchtigen grossen Molekülen - Google Patents
Verfahren zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen, nichtflüchtigen grossen Molekülen Download PDFInfo
- Publication number
- EP0503748A2 EP0503748A2 EP92250055A EP92250055A EP0503748A2 EP 0503748 A2 EP0503748 A2 EP 0503748A2 EP 92250055 A EP92250055 A EP 92250055A EP 92250055 A EP92250055 A EP 92250055A EP 0503748 A2 EP0503748 A2 EP 0503748A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- molecules
- ionization
- electron
- carrier gas
- photon
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images





Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01J—ELECTRIC DISCHARGE TUBES OR DISCHARGE LAMPS
- H01J49/00—Particle spectrometers or separator tubes
- H01J49/02—Details
- H01J49/04—Arrangements for introducing or extracting samples to be analysed, e.g. vacuum locks; Arrangements for external adjustment of electron- or ion-optical components
- H01J49/0459—Arrangements for introducing or extracting samples to be analysed, e.g. vacuum locks; Arrangements for external adjustment of electron- or ion-optical components for solid samples
- H01J49/0463—Desorption by laser or particle beam, followed by ionisation as a separate step
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01J—ELECTRIC DISCHARGE TUBES OR DISCHARGE LAMPS
- H01J49/00—Particle spectrometers or separator tubes
- H01J49/02—Details
- H01J49/10—Ion sources; Ion guns
- H01J49/14—Ion sources; Ion guns using particle bombardment, e.g. ionisation chambers
- H01J49/147—Ion sources; Ion guns using particle bombardment, e.g. ionisation chambers with electrons, e.g. electron impact ionisation, electron attachment
Definitions
- the invention relates to a method for generating ions, in particular for a mass spectrometer, such as time-of-flight mass spectrometer, from thermally unstable, non-volatile large molecules, in which a sample substance containing the molecules is exposed to energy pulses, by means of which molecules are released from the sample substance, and in which the released substances Molecules entrained by a jet of a carrier gas and cooled during its expansion and subsequently ionized in an ionization space, and a device for generating ions, in particular for a mass spectrometer, such as time-of-flight mass spectrometer, made of thermally unstable, non-volatile large molecules, with a device for generating a carrier gas jet, an energy source for desorbing molecules from the sample material and a device for introducing sample material into the carrier gas jet, in particular for carrying out the method specified above.
- a mass spectrometer such as time-of-flight mass spectrometer
- a method of the generic type is known in which the molecules are desorbed by a laser beam. It is used to convert, in particular, large molecules into the gas phase before the molecules are brought into a chemical state by means of a subsequent ionization process, in which they are only accessible for mass spectrometric analysis. This takes advantage of the fact that the internal energy absorbed by the molecules through the desorption in the carrier gas jet is greatly reduced, so that the molecules are strongly cooled and their thermal decomposition is largely prevented.
- This desorption method is suitable for liquid and solid sample substances, and it has proven to be advantageous to accommodate the molecules of the sample substance in a matrix that decomposes easily thermolytically.
- Single-photon or multiphoton ionization has proven itself for the mass spectrometric analysis of the large molecules under consideration. Since the wavelength of the irradiated photons can be matched to the energy difference between the ground state and an excited state of the neutral molecule, it is possible to selectively ionize only the molecules to be examined; the carrier gas particles remain in their neutral state and do not influence the subsequent test results.
- Ionization processes which act non-selectively. This includes electron impact ionization.
- electron impact ionization cannot be used for large molecules, as in the present case, since they lead to severe fragmentation of the molecule.
- the carrier gas particles would also be ionized, leading to saturation effects, electrostatic repulsion and thus to poor resolution and insufficient Sensitivity of the analysis led. Such influences cannot be neglected, because the carrier gas particles are at least a thousand times higher than the molecules to be examined.
- the object of the invention is to further develop the generic method in such a way that ions can be provided from thermally unstable, non-volatile large molecules, a non-selective ionization method being able to be used.
- this object is achieved in a further development of the generic method in that the molecules are ionized by electron impact; that the radiation density of the electrons used for the ionization is selected so that a potential well is generated in the focus of the electron beam, the depth of which is greater than the translation energy of the molecular ions in the carrier gas stream; that the molecular ions generated by the electron impact ionization are each collected in the potential well for a certain period of time; and that the molecular ions collected in each case in the potential well are pulsed out of the ionization chamber.
- the device according to the invention is characterized in that an ionization chamber is provided which has an inlet opening and an outlet opening for a particle beam, an electron source being arranged such that the electron beam generated by the latter is focused within the ionization chamber on the path of the particle beam; that the device for generating the carrier gas jet has an outlet opening for the carrier gas jet; and that a sample carrier is arranged in the vicinity of the outlet opening, on which the sample material is attached.
- the invention is based on the surprising finding that, contrary to the widespread prejudice of the professional world, it is also possible to ionize unstable molecules by means of electron impact, this possibility being created by the fact that the "jet" beam, which is generated by the generic method, a such cooling of the heavy molecules, which only carry out very small relative movements within the beam and which are later to be ionized, brings about that they do not disintegrate during electron impact ionization.
- the additional measure of pulsed attraction of the ionized molecules which is made possible by the generation of the potential well of adjustable depth, promotes the detection sensitivity in the mass spectrometer and thus the resolution in a manner essential to the invention.
- molecules are thus ionized by electron impact, helium and / or neon preferably being used as the carrier gas; the energy of the electrons in the electron beam is of course preferably below the ionization energy of the carrier gas.
- the entire advantages of the known technique of laser evaporation with subsequent cooling can be used to investigate thermally unstable, non-volatile molecules that would otherwise not be accessible to such analysis methods. Since the internal energies of the molecules are significantly reduced by cooling, the subsequent electron impact ionization also leads to fewer fragments than is usually expected.
- the advantages of the spatial separation of evaporation and ionization can be maintained - flexibility in the design of the desorption and ionization system without the need for structural compromises; no staining of the ion source by desorbed sample material; Ions formed during desorption (in contrast to neutral sample molecules) do not reach the ion source - whereby good yields can be achieved.
- noble gases helium or neon as carrier gas
- Both noble gases have a very high ionization potential (24.6 eV or 21.6 eV), so that they are practically not ionized at electron energies below these ionization potentials, as are preferably used.
- a mass spectrometric detection method that uses the pulse structure of ion generation is time-of-flight (TOF) mass spectrometry.
- TOF time-of-flight
- a time-of-flight mass spectrometer has the advantage that a complete mass spectrum is registered with each pulse.
- time-of-flight mass spectrometry has a physical property that makes it particularly suitable for the study of large molecules. The resolution increases with increasing mass.
- the sensitivity of the arrangement can be increased if the radiation density of the electrons used for the ionization is chosen such that a potential well is generated in the focus of the beam.
- the neutral molecules to be examined fly into the focus of the electron beam, are ionized there, but can then - in the ionized state - no longer leave the focus. They are collected in a spatially limited volume over a relatively long period of up to 100 ⁇ s. The ionized molecules collected in this way can each be subtracted as a "package" with a precisely defined start time.
- Light pulses generated by laser can also be used as energy pulses for desorption; continuously operating lasers are also suitable.
- the wavelengths used range from micrometers down to a few tens of nanometers.
- the energy pulses can also be applied by bombardment with ions or neutral particles.
- the sample is optionally ionized by electron impact or by photon excitation in the same ionization space.
- the sample to be examined therefore only has to be prepared once and let into the ionization space and can then be examined using the advantages of both ionization methods. This ensures that one and the same sample is examined using both measurement methods. It is advantageous if both the photon beam is pulsed and the gaseous sample is supplied in a pulsed manner, the ionization processes having to be switched in a correspondingly synchronized manner.
- the switching frequency is only limited by the time required to record the spectrum and evaluate it.
- an electron source and a photon source are provided for one and the same ionization chamber and can be operated optionally for ionizing the gaseous sample within the ionization chamber, the ionization preferably taking place in a pulsed manner.
- the photon ionization was carried out in a constant electric field, the necessary, precisely defined starting time of the ions being determined by means of laser pulses which were measured over time. Such a procedure is impractical for electron impact ionization. If one would like to switch easily between electron impact ionization and photon ionization, this can only be done by carrying out the photon ionization not in a standard manner but in a manner adapted to the apparatus for electron impact ionization.
- the adjustment and thus also the mass calibration of the downstream mass spectrometer do not have to be changed. Only like that Repetition rates of the order of 20 Hz can be achieved, so that it is possible to switch from molecules to be examined for each pulse packet.
- the ions must be ionized at exactly the same time, in the same volume and at exactly the same potential.
- the electron beam emitted by the electron source and the photon beam emitted by the photon source are focused essentially on the same area of the ionization chamber. If the electron beam and the laser beam are set up to be adjustable, the necessary settings can be carried out relatively easily during the operation of the device.
- the photon ionization is also carried out under the same conditions as the electron impact ionization.
- the ionization chamber is advantageously an ionization chamber set to a positive potential, the end plate of which, ie the area in which the ionized molecules leave the ionization chamber, can be acted upon separately.
- the starting time for the ions released from the ionization chamber is then determined by the fact that this end plate is at ground potential, i.e. is switched to 0 V.
- the end plate is switched to the acceleration potential, the ions start their flight in the acceleration field thus created in the mass spectrometer connected downstream of the ionization chamber.
- Electron source and / or photon source operated in a pulsed manner so that a time-of-flight mass spectrometer can be used as the mass spectrometer, with which a complete mass spectrum can be recorded for each ion packet.
- the first exemplary embodiment of the device for generating ions from thermally unstable, non-volatile large molecules by the method according to the invention comprises a device 1 for generating a carrier gas jet, from which the carrier gas jet is controlled by a pulsed valve with a nozzle 10 , exits in a vacuum.
- a gas pulse with a length of 1 ⁇ s to 10 ms is generated, a pulse length of 500 ⁇ s or less being optimal for most purposes.
- a helium inlet pressure of about 2 bar is set on the high pressure side of the valve; in principle, it can be advisable to keep the pressure between 0.2 bar and 200 bar depending on the requirements.
- the nozzle 10 has an opening with a diameter of 0.2 mm, which can also be varied in the size range from 0.01 to 1 mm.
- the valve or nozzle 10 is opened electromagnetically.
- a gas pulse generated in this way is also called a supersonic jet.
- the carrier gas atoms move at approximately the same speed, the relative thermal movement of the atoms being relatively low.
- the beam has a low temperature in the order of 1 K.
- a sample carrier 3 In the immediate vicinity of the opening of the nozzle 10 there is a sample carrier 3 with a sample attached to it, which sample can either be solid or liquid, it also being possible to incorporate this sample into a matrix.
- pulsed infrared light is emitted from a suitable light source, for example from a CO2 laser, as a photon beam 2 onto the sample carrier 3 or the sample located thereon.
- a lens 20 is provided for focusing the photon beam 2.
- the pulse of this photon beam 2 is synchronized in time with the pulse of the carrier gas emerging from the nozzle 10.
- a suitable pulse length for a CO2 laser with a wavelength of 10.6 ⁇ m is 10 ⁇ s.
- the material to be examined is preferably desorbed into the space in front of the nozzle 10 by the incident photon beam 2. First of all, all degrees of freedom of the molecules, namely degrees of freedom of rotation, vibration and translation, are excited; the energy contained therein will then cool down sharply in the particle beam, the supersonic jet. This largely prevents the thermally unstable molecules from decaying.
- the molecules desorbed from the sample carrier 3 are now in the gaseous state and are largely in the carrier gas jet emerging from the nozzle 10. Together with the carrier gas, the molecules are transported as a particle beam 4 to a wiper or skimmer 5, which only allows the central area of the particle beam 4 to pass.
- the "peeled" portion of the particle beam 4 must be pumped out for reasons of vacuum technology and is therefore no longer available for analysis.
- the skimmer 5 consists essentially of a hollow cone placed on a flat wall 50, the tip of which is formed into an opening 51, the diameter of which is selected in accordance with the cross section of the particle beam 4 to be masked out. It is thus achieved that a particle beam aligned almost exactly in a preselected direction finally enters the ionization region.
- the ionization takes place within an ionization chamber 7.
- the front wall 70 of the ionization chamber 7 is aligned with the nozzle 10 and the opening 51 of the skimmer 5, an inlet opening 71 through which the particle beam 4 enters.
- Impinging the particle beam 4 perpendicularly, a pulsed electron beam 6 is introduced into the ionization chamber 7, the focus 61 of which is set so that it lies on the path of the particle beam 4.
- the electron beam is pulsed with a length of 10 ns to 100 microseconds, the pulse being synchronized with the time span in which the particle "packet" is flying by.
- the ionization chamber 7 is at a positive potential of approximately 1000 V.
- the energy of the electrons introduced in the electron beam 6 can be regulated from a few eV to 100 eV. These electrons now ionize the molecules to be examined by electron impact. If the energy of the electrons is selected in the order of magnitude of 25 eV, the particles of the carrier gas are not ionized, so that there is no later falsification of the result in the mass spectrometric analysis.
- the radiance of the electrons is so high that in the focus 61 of the electron beam 6 a potential well or potential can be built up that is deep enough for the ionized molecules, i.e. molecular cations, which initially move at the speed of the particle beam 4 for a short time catch. In this way, the molecular ions to be examined are collected in a spatially limited volume.
- the pulse duration of the electron beam 6 is adjusted so that the pulse is ended when the collection is also finished. A few 10 ns later, the end plate 73 closing the ionization chamber 7 is switched to 0 V in less than 5 ns. At this point in time, the molecular ions start their flight in the resulting acceleration field from the collection point in focus 61 to the outlet opening 72 in the end plate 73 Time of flight mass spectrometer.
- Figure 2 shows the space in front of the nozzle 10 of the device for generating a carrier gas jet.
- the jet 4 emerges as a pulse packet from the nozzle 10 and passes the sample substance 30 on the sample carrier 3 from the molecules to be examined.
- a photon pulse 2 is irradiated, which causes the molecules to be desorbed from the sample substance or from the sample carrier 3.
- the molecules diffuse into the particle beam and are carried by it in the direction of the skimmer or the ionization chamber.
- FIG. 3 shows the intensity-time diagram of the electron beam which effects the ionization of the molecules in the ionization chamber.
- the pulse has steep edges and is kept constant over the period of time required for ionization.
- the potential of the end plate of the Faraday cage is switched off within a very short time, so that there is also a steep-edged pulse, which over a period of z. B. 20 ⁇ s is kept at 0 V, which is sufficient to generate the field required for the acceleration of the molecular ions; of course, instead of 0 V, the end plate can also be switched to another potential suitable for accelerating the molecular ions.
- Figure 6 shows a raw data spectrum of the thermally unstable peptide Trp-Met-Asp-Phe-NH2.
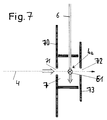
- the further exemplary embodiment of the device according to the invention shown in FIG. 7 comprises an ionization chamber 7, the front wall or plate 70 of which is provided with an inlet opening 71 through which the molecules 4 to be examined can enter in the form of a continuous beam or as a particle packet.
- An end plate 73 is provided opposite the front plate 70 and has an outlet opening 72 aligned with the inlet opening 71.
- the molecules to be examined do not flow in on the axis defined by the inlet opening 71 and outlet opening 72, but instead reach the ionization chamber 7 from all sides by diffusion.
- ionization by electron impact can initially respectively.
- an electron beam 6 is spatially focused on the center of the ionization chamber, the energy of the electrons being adjustable from a few eV up to 100 eV.
- the electron beam 6 is also switched on in pulsed operation, the pulse duration being from 10 ns to approximately 100 ⁇ s.
- the molecular ions In order to achieve a good resolution when examining the molecular ions with a time-of-flight mass spectrometer, the molecular ions must start at a point (t ⁇ 5 ns) that is as precisely defined as possible in a space that is as small as possible ( ⁇ 1 mm). In general, it is not possible to meet these conditions and to use the entire sample contained in a gas jet. However, it has been shown that the sensitivity of the arrangement can be increased if the radiation density of the electrons used for the ionization is selected such that a potential well is generated in the focus 61 of the beam.
- the end plate 73 of the ionization chamber 7 is switched to 0 V approximately 10 ns later, this switching taking place in less than 5 ns. This provides the start pulse for the ions for their flight in the time-of-flight mass spectrometer.
- the ionization chamber 7 is again placed at positive potential overall, for example at 600 V.
- the photon ionization can then be carried out.
- a pulsed laser beam 4a is irradiated into the ionization chamber 1.
- the laser pulses used have a typical duration of 5 ns.
- the short duration of the laser pulses alone would ensure a precisely defined starting time for the ions, so that the ionization chamber 7 with the closing plate 73, which can be acted upon separately, would not be required.
- the focus of the electron beam 6 and the focus of the photon beam 4a coincide in a region 61 which lies on the path of the molecules to be examined.
- FIG. 9 shows the raw data spectra of Pro-Phe-Gly-Lys acetate, the spectra again being recorded on the one hand by multi-photon ionization (MPI) and on the other hand by electron impact ionization (EI).
- MPI multi-photon ionization
- EI electron impact ionization
Landscapes
- Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Analytical Chemistry (AREA)
- Optics & Photonics (AREA)
- Engineering & Computer Science (AREA)
- Plasma & Fusion (AREA)
- Other Investigation Or Analysis Of Materials By Electrical Means (AREA)
- Electron Tubes For Measurement (AREA)
Abstract
Description
- Die Erfindung betrifft ein Verfahren zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen nichtflüchtigen großen Molekülen, bei dem eine die Moleküle aufweisende Probensubstanz Energiepulsen ausgesetzt wird, durch welche Moleküle aus der Probensubstanz freigesetzt werden, und bei dem die freigesetzten Moleküle von einem Strahl eines Trägergases mitgenommen und bei dessen Expansion gekühlt sowie anschließend in einem Ionisationsraum ionisiert werden, sowie eine Vorrichtung zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen nichtflüchtigen großen Molekülen, mit einer Einrichtung zum Erzeugen eines Trägergasstrahles, einer Energieguelle zum Desorbieren von Molekülen aus dem Probenmaterial und einer Einrichtung zum Einbringen von Probenmaterial in den Trägergasstrahl, insbesondere zur Durchführung des vorstehend angegebenen Verfahrens.
- Aus der DE-PS 38 00 504 ist ein Verfahren der gattungsgemäßen Art bekannt, bei dem die Desorption der Moleküle durch einen Laserstrahl erfolgt. Es dient zum Überführen insbesondere großer Moleküle in die Gasphase, bevor die Moleküle mittels eines anschließend durchgeführten Ionisationsvorganges in einen chemischen Zustand gebracht werden, in dem sie erst einer massenspektrometrischen Analyse zugänglich werden. Dabei wird ausgenutzt, daß die von den Molekülen durch die Desorption aufgenommene innere Energie im Trägergasstrahl stark reduziert wird, so daß die Moleküle stark abgekühlt werden und deren thermische Zersetzung weitgehend verhindert wird. Dieses Desorptionsverfahren ist für flüssige und feste Probensubstanzen geeignet, wobei es sich als günstig herausgestellt hat, die Moleküle der Probensubstanz in einer Matrix unterzubringen, die sich thermolytisch leicht zersetzt.
- Eine Vorrichtung, mit der dieses Desorptionsverfahren durchgeführt werden kann, ist in der Zeitschrift "ANGEWANDTE CHEMIE" 1988, Seiten 461 ff., in dem Übersichtsartikel "Die Multiphotonen-Ionisation (MUPI)-Massenspektrometrie", beschrieben. Die Probensubstanz wird dabei vor die Öffnung einer Düse gebracht, aus der das Trägergas austritt. Durch Verwendung von Infrarot-Laserlicht werden die Moleküle der Probensubstanz in den expandierenden Strahl des Trägergases desorbiert. Dadurch werden die inneren Freiheitsgrade der Moleküle abgekühlt, und die Moleküle werden von dem Trägergasstrahl weitertransportiert. Üblicherweise wird diese Vorrichtung als gepulstes System betrieben, das aus einem gepulsten Ventil zum Erzeugen des Trägergasstrahles und einem Laser zur Desorption der neutralen Moleküle besteht. Da die Moleküle als Strahl, bzw. im Pulsbetrieb als Teilchenpaket, weitertransportiert werden, ist es möglich, diesen Desorptionsprozeß räumlich getrennt von dem nunmehr nachfolgenden Ionisationsprozeß zu halten.
- Für die massenspektrometrische Untersuchung der betrachteten großen Moleküle hat sich die Einphotonen- oder Multiphotonen-Ionisation bewährt. Da die Wellenlänge der eingestrahlten Photonen auf die Energiedifferenz zwischen dem Grundzustand und einem angeregten Zustand des neutralen Moleküls abgestimmt werden kann, ist es möglich, die Ionisation selektiv nur der zu untersuchenden Moleküle vorzunehmen; die Trägergasteilchen bleiben dabei in ihrem neutralen Zustand und beeinflussen die nachfolgenden Untersuchungsergebnisse nicht.
- Obwohl die Multiphotonen-Ionisations-Massenspektrometrie erfolgreich betrieben wird, ist sie doch für einige Problemstellungen nicht anwendbar, da oftmals eine lediglich selektive Anregung der neutralen Moleküle nicht genügend Information zur gewünschten Strukturaufklärung des Moleküls liefern kann, weil bei unbekannten Molekülen die zu wählende Anregungswellenlänge nicht hinreichend vorherbestimmbar ist. Auch hat sich herausgestellt, daß einige Substanzen auf die dargestellte Weise nur schwer ionisiert werden können.
- Es sind Ionisationsverfahren bekannt, die nicht-selektiv wirken. Dazu zählt die Elektronenstoß-Ionisation. Für große Moleküle, wie im vorliegenden Fall, sind derartige Verfahren jedoch nicht anwendbar, da sie zu einer starken Fragmentierung des Moleküls führen. Zudem würden auch die Trägergasteilchen ionisiert, was zu Sättigungseffekten, elektrostatischer Abstoßung und damit zu schlechter Auflösung und ungenügender Empfindlichkeit der Analyse führte. Solche Einflüsse sind schon deswegen nicht vernachlässigbar, da die Trägergasteilchen gegenüber den zu untersuchenden Molekülen in mindestens tausendfach höherer Konzentration vorliegen.
- Aus der DE-PS 873 765 ist die Verwendung der Elektronenstoßionisation bekannt, jedoch führt die Kombination dieser Vorgehensweise mit einem Verfahren, wie es aus der DE-OS 36 19 886 vorbekannt ist, lediglich zu stark fragmentierten Ionen im niedrigen Massenbereich, so daß sich hiermit also keine großen, thermisch labile, nicht-flüchtige Moleküle, wie z. B. Peptide, untersuchen lassen.
- Der Erfindung liegt die Aufgabe zugrunde, das gattungsgemäße Verfahren dahingehend weiterzubilden, daß Ionen aus thermisch instabilen, nicht-flüchtigen großen Molekülen bereitgestellt werden können, wobei eine nicht-selektive Ionisationsmethode eingesetzt werden kann.
- Erfindungsgemäß wird diese Aufgabe in Weiterbildung des gattungsgemäßen Verfahrens dadurch gelöst, daß die Moleküle durch Elektronenstoß ionisiert werden; daß die Strahlungsdichte der für die Ionisation verwendeten Elektronen so gewählt wird, daß im Fokus des Elektronenstrahles eine Potentialmulde erzeugt wird, deren Tiefe größer als die Translationsenergie der Molekülionen im Trägergasstrom ist; daß die durch die Elektronenstoßionisation erzeugten Molekülionen in der Potentialmulde jeweils für einen bestimmten Zeitraum gesammelt werden; und daß die jeweils in der Potentialmulde gesammelten Molekülionen gepulst aus der Ionisationskammer herausbeschleunigt werden.
- Besonders bevorzugte Ausführungsformen des Verfahrens nach der Erfindung sind Gegenstand der entsprechenden Unteransprüche.
- Die Vorrichtung nach der Erfindung ist dadurch gekennzeichnet, daß eine Ionisationskammer vorgesehen ist, die eine Eintrittsöffnung und eine Austrittsöffnung für einen Teilchenstrahl aufweist, wobei eine Elektronenquelle so angeordnet ist, daß der von dieser erzeugte Elektronenstrahl innerhalb der Ionisationskammer auf die Bahn des Teilchenstrahles fokussiert ist; daß die Einrichtung zum Erzeugen des Trägergasstrahles eine Austrittsöffnung für den Trägergasstrahl aufweist; und daß in der Nähe der Austrittsöffnung ein Probenträger angeordnet ist, auf dem das Probenmaterial angebracht ist.
- Bevorzugte Ausführungsformen der Vorrichtung nach der Erfindung sind Gegenstand der Unteransprüche.
- Der Erfindung liegt die überraschende Erkenntnis zugrunde, daß es gelingt, entgegen dem verbreiteten Vorurteil der Fachwelt auch instabile Moleküle mittels Elektronenstoß zu ionisieren, wobei diese Möglichkeit dadurch geschaffen wird, daß der "Jet"-Strahl, der mittels des gattungsgemäßen Verfahrens erzeugt wird, eine derartige Abkühlung der innerhalb des Strahles nur sehr geringe Relativbewegungen ausführenden schweren Moleküle, die später ionisiert werden sollen, herbeiführt, daß sie bei der Elektronenstoßionisation nicht zerfallen. Die zusätzliche Maßnahme des gepulsten Anziehens der ionisierten Moleküle, welche durch die Erzeugung der Potentialmulde einstellbarer Tiefe ermöglicht wird, fördert in erfindungswesentlicher Weise die Nachweisempfindlichkeit im Massenspektrometer und damit die Auflösung.
- Erfindungsgemäß werden Moleküle also durch Elektronenstoß ionisiert, wobei als Trägergas vorzugsweise Helium und/oder Neon verwendet wird/werden; die Energie der Elektronen im Elektronenstrahl liegt dabei natürlich vorzugsweise unterhalb der Ionisationsenergie des Trägergases.
- Mit dem erfindungsgemäßen Verfahren können die gesamten Vorteile der bekannten Technik der Laserverdampfung mit anschließender Kühlung genutzt werden, um thermisch instabile, nicht flüchtige Moleküle zu untersuchen, die derartigen Analyseverfahren sonst nicht zugänglich wären. Da die inneren Energien der Moleküle durch die Kühlung erheblich verringert sind, führt auch die anschließende Elektronenstoß-Ionisation zu weniger Fragmenten, als üblicherweise erwartet werden. Die Vorteile der räumlichen Trennung von Verdampfung und Ionisation können beibehalten werden - Flexibilität in der Konstruktion von Desorptions- und Ionisationssystem ohne Notwendigkeit struktureller Kompromisse; kein Verdrecken der Ionenquelle durch desorbiertes Probenmaterial; bei der Desorption gebildete Ionen (im Gegensatz zu neutralen Probenmolekülen) erreichen nicht die Ionenquelle -, wobei gute Ausbeuten zu erzielen sind. Durch Verwendung der Edelgase Helium oder Neon als Trägergas wird die Entstehung von Trägergasionen verhindert oder zumindest sehr stark unterdrückt. Beide Edelgase haben ein sehr hohes Ionisierungspotential (24.6 eV bzw. 21.6 eV), so daß sie bei Elektronenenergien unterhalb dieser Ionisierungspotentiale, wie sie vorzugsweise zur Anwendung kommen, praktisch nicht ionisiert werden. Die zu untersuchenden Moleküle, mit einem Ionisierungspotential in der Größenordnung von etwa 10 eV, werden bei den genannten Elektronenenergien hingegen bereits sehr gut ionisiert.
- Ein massenspektrometrisches Nachweisverfahren, das die Pulsstruktur der Ionenerzeugung nutzt, ist die Flugzeit- oder Time-Of-Flight (TOF)-Massenspektrometrie. Grundsätzlich hat ein Flugzeit-Massenspektrometer den Vorteil, daß mit jedem Puls ein vollständiges Massenspektrum registriert wird. Darüber hinaus verfügt die Flugzeitmassenspektrometrie über eine physikalische Eigenschaft, die sie zur Untersuchung von großen Molekülen besonders geeignet macht. Die Auflösung nimmt nämlich mit wachsender Masse zu.
- Allerdings kann eine gute Auflösung bei der Untersuchung der Molekülionen mit einem Flugzeitmassenspektrometer auch nur dann erreicht werden, wenn die Molekülionen zu einem möglichst genau definierten Zeitpunkt ( t < 5ns) auf einem möglichst kleinen Raum ( < 1mm) starten. Im allgemeinen ist es deshalb nicht möglich, die gesamte in einem Gasstrahl enthaltene Probe auszunutzen.
- Es hat sich aber gezeigt, daß die Empfindlichkeit der Anordnung gesteigert werden kann, wenn die Strahlungsdichte der für die Ionisation verwendeten Elektronen so gewählt wird, daß im Fokus des Strahles eine Potentialmulde erzeugt wird. Die zu untersuchenden neutralen Moleküle fliegen in den Fokus des Elektronenstrahles, werden dort ionisiert, können jedoch dann - im ionisierten Zustand - den Fokus nicht mehr verlassen. Sie werden also in einem räumlich begrenzten Volumen über einen relativ langen Zeitraum von bis zu 100 µs gesammelt. Die so gesammelten ionisierten Moleküle lassen sich jeweils als "Paket" mit exakt definierter Startzeit abziehen, wobei z. B. 50 ns nach dem Ende des Sammelns (Abschalten des Elektronenstrahles) die Abschlußplatte auf 0 V geschaltet wird und hierdurch die ionisierten Moleküle in das Massenspektrometer beschleunigt werden. Durch diese gepulste "Paketübertragung" läßt sich eine gute Ausbeute mit hoher Auflösung (wie für Laser-Ionisation üblich) bei gleichzeitig hoher Empfindlichkeit (wie für Elektronen-Stoßionisation charakteristisch) erzielen.
- Als Energiepulse für die Desorption können im übrigen mit Laser erzeugte Lichtpulse verwendet werden; dabei sind auch kontinuierlich arbeitende Laser geeignet. Die eingesetzten Wellenlängen liegen dabei im Bereich von Mikrometern bis hinab zu einigen zehn Nanometern. Die Energiepulse können auch durch Beschuß mit Ionen oder Neutralteilchen aufgebracht werden.
- Bei einer besonderen Ausführungsform der Erfindung wird wahlweise eine Ionisation der Probe durch Elektronenstoß oder durch Photonenanregung in demselben Ionisationsraum durchgeführt. Die zu untersuchende Probe muß daher nur einmal präpariert und in den Ionisationsraum eingelassen werden und kann anschließend unter Ausnutzen der Vorteile beider Ionisationsverfahren untersucht werden. Dabei ist also sichergestellt, daß ein- und dieselbe Probe mit beiden Meßmethoden untersucht wird. Vorteilhaft ist es dabei, wenn sowohl der Photonenstrahl gepulst ist als auch die gasförmige Probe gepulst zugeführt wird, wobei die Ionisationsverfahren entsprechend synchronisiert geschaltet werden müssen. Die Umschaltfrequenz ist dabei lediglich durch die für die Aufnahme des Spektrums und dessen Auswertung benötigte Zeit begrenzt.
- Im Gegensatz zum Stand der Technik sind, wie ausgeführt, für ein und dieselbe Ionisationskammer eine Elektronenquelle und eine Photonenquelle vorgesehen, die zur Ionisierung der gasförmige Probe innerhalb der Ionisationskammer wahlweise betreibbar sind, wobei die Ionisierung vorzugsweise pulsförmig erfolgt. In den bisher bekannten Apparaturen wurde die Photonen-Ionisation in einem konstanten elektrischen Feld durchgeführt, wobei die notwendige genau definierte Startzeit der Ionen durch zeitlich entsprechend bemessene Laserpulse festgelegt wurde. Für die Elektronenstoß-Ionisation ist eine solche Vorgehensweise unzweckmäßig. Wenn man zwischen Elektronenstoß-Ionisation und Photonen-Ionisation problemlos umschalten möchte, kann dies nur dadurch geschehen, daß die Photonen-Ionisation nicht standardmäßig, sondern an die Apparatur für die Elektronenstoß-Ionisation angepaßt durchgeführt wird.
- Für die Umschaltung ist es weiterhin wichtig, daß die Justierung und damit auch die Masseneichung des nachgeschalteten Massenspektrometers nicht verändert werden müssen. Nur so können Wiederholraten in der Größenordnung von 20 Hz erreicht werden, so daß bei jedem Pulspaket aus zu untersuchenden Molekülen umgeschaltet werden kann. Um dies zu erreichen, müssen die Ionen exakt zur vergleichbaren Zeit, exakt in demselben Volumen und exakt auf demselben Potential ionisiert werden. Neben dem Durchführen der Ionisation innerhalb einer Ionisationskammer für die unterschiedlichen Ionisationsverfahren ist dazu vorteilhaft, daß der von der Elektronenquelle ausgesandte Elektronenstrahl und der von der Photonenquelle ausgesandte Photonenstrahl im wesentlichen auf denselben Bereich der Ionisationskammer fokussiert sind. Sind der Elektronenstrahl und der Laserstrahl justierbar eingerichtet, so kann man die erforderlichen Einstellungen während des Betriebes der Vorrichtung relativ einfach durchführen.
- Es wurde weiter oben schon ausgeführt, daß auch die Photonen-Ionisation unter denselben Bedingungen wie die Elektronenstoß-Ionisation durchgeführt wird. Vorteilhaft ist dazu die Ionisationskammer eine auf positives Potential gelegte Ionisationskammer, dessen Abschlußplatte, also der Bereich, in dem die ionisierten Moleküle die Ionisationskammer verlassen, separat beaufschlagbar ist. Die Startzeit für die aus der Ionisationskammer entlassenen Ionen wird dann dadurch festgelegt, daß diese Abschlußplatte innerhalb sehr kurzer Zeit auf Erdpotential, d.h. auf 0 V geschaltet wird. Natürlich ist es innerhalb des Erfindungsgedankens auch möglich, die Abschlußplatte auf ein von 0 V verschiedenes Potential zu schalten, sofern dieses nur so gewählt wird, daß es zur Beschleunigung der Ionen in das Massenspektrometer geeignet ist. Zum Zeitpunkt des Schaltens der Abschlußplatte auf das Beschleunigungspotential starten die Ionen in dem somit entstandenen Beschleunigungsfeld ihren Flug im der Ionisationskammer nachgeschalteten Massenspektrometer.
- Nach einer bevorzugten Ausführungsform der Erfindung werden Elektronenquelle und/oder Photonenquelle, wie bereits dargelegt, gepulst betrieben, so daß als Massenspektrometer ein Flugzeitmassenspektrometer eingesetzt werden kann, mit dem zu jedem Ionenpaket ein komplettes Massenspektrum aufgezeichnet werden kann.
- Weitere Merkmale und Ausführungsformen der Erfindung ergeben sich aus den Ansprüchen und aus der nachfolgenden Beschreibung, in der Ausführungsbeispiele anhand der schematischen Zeichnung im einzelnen erläutert sind. Dabei zeigt:
- Fig. 1
- den schematischen Aufbau eines ersten Ausführungsbeispieles einer Vorrichtung zur Durchführung des erfindungsgemäßen Verfahrens;
- Fig. 2
- eine Detailansicht der Vorrichtung im Bereich der zu verdampfenden Probensubstanz;
- Fig. 3
- ein Intensitäts-Zeit-Diagramm für den verwendeten Elektronenstrahl;
- Fig. 4
- ein Spannungs-Zeit-Diagramm für die Beschleunigungsplatte aus der Vorrichtung nach Figur 1;
- Fig. 5
- ein Rohdatenspektrum der nichtflüchtigen Substanz Mesoporphyrin, wobei dem Trägergasstrahl (Helium) zu Testzwecken Luft und Benzol beigemischt wurde;
- Fig. 6
- das Rohdatenspektrum des nicht-flüchtigen, thermisch instabilen Peptids Trp-Met-Asp-Phe-NH₂;
- Fig. 7
- den schematischen Aufbau eines zweiten Ausführungsbeispieles einer Vorrichtung zur Durchführung einer weiteren Ausführungsform des erfindungsgemäßen Verfahrens;
- Fig. 8
- mit der Vorrichtung von Fig. 7 erhaltene Rohdatenspektren für das thermisch instabile Peptid Trp-Pro-Leu-Gly-Amid, wobei sowohl das Multiphotonen-Ionisationsspektrum (MPI) als auch das Elektronenstoß-Ionisations-Spektrum (EI) dargestellt sind; und
- Fig. 9
- ein Rohdatenspektrum, erhalten mit der Vorrichtung von Fig. 7, des thermisch instabilen Peptides Pro-Phe-Gly-Lys-Acetat, wobei wiederum sowohl das Multiphotonen-Ionisations-Spektrum (MPI) als auch das Elektronenstoß-Ionisations-Spektrum (EI) dargestellt sind.
- Das erste Ausführungsbeispiel der Vorrichtung zum Erzeugen von Ionen aus thermisch instabilen, nichtflüchtigen großen Molekülen nach dem erfindungsgemäßen Verfahren, die in Figur 1 dargestellt ist, umfaßt eine Einrichtung 1 zur Erzeugung eines Trägergasstrahles, aus der der Trägergasstrahl, gesteuert von einem gepulsten Ventil mit Düse 10, in ein Vakuum austritt. Bei Verwendung von Helium als Trägergas wird dabei ein Gaspuls der Länge 1 µs bis 10 ms erzeugt, wobei eine Pulslänge von 500 µs oder weniger für die meisten Zwecke optimal ist. Auf der Hochdruckseite des Ventils wird ein Helium-Vordruck von etwa 2 bar eingestellt; grundsätzlich kann es zweckmäßig sein, den Druck je nach Anforderung zwischen 0,2 bar bis 200 bar zu halten. Die Düse 10 hat eine Öffnung mit einem Durchmesser von 0,2 mm, der aber auch im Größenbereich von 0,01 bis 1 mm variiert werden kann. Das Öffnen des Ventils beziehungsweise der Düse 10 geschieht elektromagnetisch.
- Ein auf diese Weise erzeugter Gaspuls wird auch Überschallstrahl genannt. Dabei bewegen sich die Trägergasatome mit etwa gleicher Geschwindigkeit, wobei die relative thermische Bewegung der Atome verhältnismäßig gering ist. Demzufolge hat der Strahl eine niedrige Temperatur in der Größenordnung von 1 K.
- In unmittelbarer Nähe der Öffnung der Düse 10 befindet sich ein Probenträger 3 mit darauf angebrachter Probe, die entweder fest oder flüssig sein kann, wobei es auch möglich ist, diese Probe in eine Matrix einzubauen. Etwa senkrecht zum aus der Düse 10 austretenden Strahl wird aus einer geeigneten Lichtquelle, beispielsweise aus einem CO₂-Laser, gepulstes Infrarotlicht als Photonenstrahl 2 auf den Probenträger 3 bzw. die darauf befindliche Probe gestrahlt.
- Zum Fokussieren des Photonenstrahls 2 ist eine Linse 20 vorgesehen. Der Puls dieses Photonenstrahls 2 ist zeitlich mit dem aus der Düse 10 austretenden Puls des Trägergases synchronisiert. Eine geeignete Pulslänge für einen CO₂-Laser mit der Wellenlänge des Lichtes von 10,6 µm ist 10 µs. Durch den auftreffenden Photonenstrahl 2 wird das zu untersuchende Material bevorzugt in den Raum vor der Düse 10 desorbiert. Zunächst sind sämtliche Freiheitsgrade der Moleküle, nämlich Rotations-, Vibrations- und Translationsfreiheitsgrade, angeregt; die darin enthaltene Energie wird anschließend im Teilchenstrahl, dem Überschallstrahl, stark abkühlen. Dadurch wird ein Zerfall der thermisch instabilen Moleküle weitgehend verhindert.
- Die vom Probenträger 3 desorbierten Moleküle liegen nun im gasförmigen Zustand vor und befinden sich zum größten Teil im aus der Düse 10 austretenden Trägergasstrahl. Zusammen mit dem Trägergas werden die Moleküle als Teilchenstrahl 4 auf einen Abstreifer oder Skimmer 5 transportiert, der lediglich den zentralen Bereich des Teilchenstrahles 4 durchläßt. Der "abgeschälte" Anteil des Teilchenstrahles 4 muß aus vakuumtechnischen Gründen abgepumpt werden und steht somit für die Analyse nicht mehr zur Verfügung.
- Der Skimmer 5 besteht im wesentlichen aus einem auf eine ebene Wand 50 aufgesetzten Hohlkegel, dessen Spitze zu einer Öffnung 51 ausgebildet ist, deren Durchmesser entsprechend dem Querschnitt des auszublendenden Teilchenstrahles 4 gewählt ist. Somit wird erreicht, daß ein nahezu genau in einer vorgewählten Richtung ausgerichteter Teilchenstrahl schließlich in den Ionisationsbereich eintritt.
- Die Ionisation findet innerhalb einer Ionisationskammer 7 statt. Die Frontwand 70 der Ionisationskammer 7 weist, ausgerichtet mit der Düse 10 und der Öffnung 51 des Skimmers 5, eine Eintrittsöffnung 71 auf, durch die der Teilchenstrahl 4 eintritt. Senkrecht auf den Teilchenstrahl 4 auftreffend wird ein gepulster Elektronenstrahl 6 in die Ionisationskammer 7 eingeführt, deren Fokus 61 so eingestellt ist, daß er auf der Bahn des Teilchenstrahles 4 liegt. Der Elektronenstrahl ist zeitlich mit einer Länge von 10 ns bis 100 µs gepulst, wobei der Puls auf die Zeitspanne, in der das Teilchen-"Paket" vorbeifliegt, synchronisiert ist. Die Ionisationskammer 7 befindet sich auf einem positiven Potential von etwa 1000 V.
- Die Energie der im Elektronenstrahl 6 eingebrachten Elektronen ist von einigen eV bis 100 eV regelbar. Diese Elektronen ionisieren nun die zu untersuchenden Moleküle durch Elektronenstoß. Wenn die Energie der Elektronen in der Größenordnung von 25 eV gewählt wird, werden die Teilchen des Trägergases nicht ionisiert, so daß sich später keine Verfälschungen des Ergebnisses bei der massenspektrometrischen Analyse ergeben.
- Die Strahldichte der Elektronen ist so hoch, daß sich im Fokus 61 des Elektronenstrahles 6 eine Potentialmulde oder Potentialkuhle aufbauen kann, die tief genug ist, um die ionisierten Moleküle, also Molekülkationen, die sich zunächst mit der Geschwindigkeit des Teilchenstrahles 4 bewegen, für kurze Zeit zu fangen. So werden die zu untersuchenden Molekülionen in einem räumlich begrenzten Volumen gesammelt.
- Die Pulsdauer des Elektronenstrahls 6 ist so angepaßt, daß der Puls beendet ist, wenn auch das Sammeln beendet ist. Einige 10 ns später wird die die Ionisationskammer 7 verschließende Abschlußplatte 73 in weniger als 5 ns auf 0 V geschaltet. Zu diesem Zeitpunkt starten die Molekülionen vom Sammelpunkt im Fokus 61 zur Austrittsöffnung 72 in der Abschlußplatte 73 hin in dem entstandenen Beschleunigungsfeld ihren Flug im Flugzeitmassenspektrometer.
- Figur 2 zeigt den Raum vor der Düse 10 der Einrichtung zur Erzeugung eines Trägergasstrahles. Der Strahl 4 tritt als Pulspaket aus der Düse 10 aus und passiert die auf dem Probenträger 3 befindliche Probensubstanz 30 aus den zu untersuchenden Molekülen. Synchronisiert mit dem Trägergas-Pulspaket wird ein Photonenpuls 2 eingestrahlt, der die Desorption der Moleküle aus der Probensubstanz bzw. vom Probenträger 3 bewirkt. Die Moleküle diffundieren in den Teilchenstrahl hinein und werden von diesem in Richtung auf den Skimmer bzw. auf die Ionisationskammer getragen.
- Figur 3 zeigt das Intensitäts-Zeitdiagramm des Elektronenstrahls, der die Ionisierung der Moleküle in der Ionisationskammer bewirkt. Der Puls ist steilflankig und wird über den für die Ionisation benötigten Zeitraum konstant gehalten.
- Nach Abschalten des Elektronenstrahl-Pulses (Figur 3) wird das Potential der Abschlußplatte des Faraday-Käfigs, wie in Figur 4 gezeigt, innerhalb sehr kurzer Zeit abgeschaltet, so daß sich hier ebenfalls ein steilflankiger Puls ergibt, der über einen Zeitraum von z. B. 20 µs auf 0 V gehalten wird, welcher ausreicht, das für die Beschleunigung der Molekülionen erforderliche Feld zu erzeugen; natürlich kann die Abschlußplatte statt auf 0 V auch auf ein anderes zur Beschleunigung der Molekülionen geeignetes Potential geschaltet werden.
- Figur 5 zeigt ein Rohdatenspektrum der nichtflüchtigen Substanz Mesoporphyrin. Dabei sind dem Trägergas Helium (mit dem Masse-Ladung-Verhältnis m/z = 4) Luft und Benzol beigemischt. Diese Beimischungen ergeben Peaks im Bereich von m/z = 28 sowie einen relativ genau definierten Peak bei m/z = 78.
- Obwohl bei der Elektronenstoß-Ionisation üblicherweise ein hoher Fragmentanteil auftritt, während das ionisierte Molekül in der Regel nicht beobachtet werden kann, erhält man hier einen gut ausgebildeten Peak bei m/z = 566,3, dessen Existenz durch die zuvor durchgeführte Kühlung der zu untersuchenden Moleküle hervorgerufen wird. Das Massespektrum (Isotopenverteilung) in der Umgebung des Molekülion-Peaks ist in höherer Auflösung derselben Figur im Detail dargestellt.
- Figur 6 zeigt ein Rohdatenspektrum des thermisch instabilen Peptides Trp-Met-Asp-Phe-NH₂. Auch hier ist ein Peak beim entsprechenden Molekülion (m/z = 596,4) festzustellen, der zuvor ohne die Kombination der Elektronenstoß-Ionisation mit vorangehender Kühlung niemals aufgetreten ist.
- Das in Figur 7 dargestellte weitere Ausführungsbeispiel der Vorrichtung nach der Erfindung umfaßt eine Ionisationskammer 7, deren Frontwand oder -platte 70 mit einer Eintrittsöffnung 71 versehen ist, durch den die zu untersuchenden Moleküle 4 in Form eines kontinuierlichen Strahles oder als Teilchenpaket eintreten können. Der Frontplatte 70 gegenüberliegend ist eine Abschlußplatte 73 vorgesehen, die eine mit der Eintrittsöffnung 71 ausgerichtete Austrittsöffnung 72 aufweist.
- Für manche Anwendungen kann es vorteilhaft sein, wenn die zu untersuchenden Moleküle nicht auf der von Eintrittsöffnung 71 und Austrittsöffnung 72 definierten Achse einströmen, sondern durch Diffusion von allen Seiten in die Ionisationskammer 7 gelangen.
- Sobald sich die zu untersuchenden Moleküle in der Ionisationskammer 7 befinden, wird der Ionisationsvorgang eingeleitet.
- Beispielsweise kann zunächst die Ionisation durch Elektronenstoß erfolgen. Dazu wird ein Elektronenstrahl 6 auf das Zentrum der Ionisationskamner räumlich fokussiert, wobei die Energie der Elektronen von einigen eV bis hinauf zu 100 eV regelbar ist.
- Wenn der Einlaß der zu untersuchenden Moleküle gepulst erfolgt, wird auch der Elektronenstrahl 6 im Pulsbetrieb zugeschaltet, wobei die Pulsdauer von 10 ns bis etwa 100 µs betragen kann.
- Um eine gute Auflösung bei der Untersuchung der Molekülionen mit einem Flugzeitmassenspektrometer zu erreichen, müssen die Molekülionen zu einem möglichst genau definierten Zeitpunkt ( t < 5 ns) auf einem möglichst kleinen Raum (< 1 mm) starten. Im allgemeinen ist es nicht möglich, diese Bedingungen einzuhalten und die gesamte, in einem Gasstrahl enthaltene Probe auszunutzen. Es hat sich aber gezeigt, daß die Empfindlichkeit der Anordnung gesteigert werden kann, wenn die Strahlungsdichte der für die Ionisation verwendeten Elektronen so gewählt wird, daß im Fokus 61 des Strahles eine Potentialmulde erzeugt wird. Die zu untersuchenden neutralen Moleküle fliegen in den Fokus des Elektronenstrahles 6, werden dort ionisiert, können jedoch dann - im ionisierten Zustand - den Fokus 61 nicht mehr verlassen. Sie werden also in einem räumlich begrenzten Volumen über einen relativ langen Zeitraum von bis zu 100 µs gesammelt.
- Nachdem die Elektronenstoß-Ionisation beendet ist, wird die Abschlußplatte 73 der Ionisationskammer 7 etwa 10 ns später auf 0 V geschaltet, wobei dieses Umschalten in weniger als 5 ns stattfindet. Damit wird der Startimpuls für die Ionen für ihren Flug im Flugzeitmassenspektrometer geliefert.
- Einige µs später wird die Ionisationskammer 7 wieder insgesamt auf positives Potential gelegt, beispielsweise auf 600 V. Anschließend kann die Photonen-Ionisation vorgenommen werden. Dazu wird ein gepulster Laserstrahl 4a in die Ionisationskammer 1 eingestrahlt. Die verwendeten Laserpulse haben eine typische Dauer von 5 ns.
- Bei der Photonen-Ionisation würde allein die kurze Dauer der Laserpulse für eine genau definierte Startzeit der Ionen sorgen, so daß es der Ionisationskammer 7 mit der separat beaufschlagbaren Abschlußplatte 73 nicht bedürfte. Allerdings wäre eine problemlose Umschaltung zwischen Elektronenstoß-Ionisation und Photonen-Ionisation nicht möglich, wenn man für beide Ionisationsverfahren unterschiedliche räumliche Anordnungen verwenden müßte. Auch wäre es an sich möglich, bei Photonen-Ionisation die Abschlußplatte auf konstantem Potential zu halten, wobei sich aber in der Praxis veränderliche Potentialverteilungen ergeben, die eine Umjustierung des Massenspektrometers notwendig machen. Daher wird auch für die Photonen-Ionisation der Startimpuls für die ionisierten Moleküle durch das Schalten der Abschlußplatte 73 auf 0 V gegeben, was unter denselben Bedingungen wie zuvor im Zusammenhang mit der Elektronenstoß-Ionisation beschrieben geschieht.
- Bei der Vorrichtung nach Fig. 1 decken sich der Fokus des Elektronenstrahles 6 und der Fokus des Photonenstrahles 4a in einem Bereich 61, der auf der Bahn der zu untersuchenden Moleküle liegt.
- Fig. 8 zeigt Rohdatenspektren für das thermisch instabile Peptid Trp-Pro-Leu-Gly-Amid. Das Multiphotonen-Ionisations-Spektrum (MPI) zeigt einen gut ausgebildeten Peak beim entsprechenden Molekülion (m/z = 447,4), der beim Elektronenstoß-Ionisations-Spektrum (EI) weniger stark ausgebildet ist. Die beiden sich gegenüberstehenden Spektren zeigen deutlich, daß jeweils unterschiedliche Fragmente in unterschiedlichen Anteilen erhalten werden. Beide Spektren wurden unter exakt denselben experimentellen Bedingungen mit derselben Probe aufgenommen, wobei das erfindungsgemäße schnelle Umschalten zwischen der Photonen-Ionisation und der Elektronenstoß-Ionisation vorgenommen wurde. Als Einlaßsystem wurde die für thermisch instabile Moleküle geeignete Laser-Desorption in einem Überschallstrahl benutzt.
- Fig. 9 zeigt die Rohdatenspektren von Pro-Phe-Gly-Lys-Acetat, wobei die Spektren wiederum einerseits durch Multi-Photonen-Ionisation (MPI) und andererseits durch Elektronenstoß-Ionisation (EI) aufgenommen worden sind. Dabei wurden dieselben experimentellen Bedingungen unter Verwendung derselben Probe eingehalten. Wieder wurde zwischen Photonen-Ionisation und Elektronenstoß-Ionisation schnell umgeschaltet. Man erkennt, daß mit der Elektronenstoß-Ionisation kleinere Fragmente erhalten werden, so daß gerade hier deutlich wird, daß sich die beiden mit unterschiedlichen Ionisationsverfahren erhaltenen Spektren vorteilhaft ergänzen.
- Die in der vorstehenden Beschreibung, in den Zeichnungen sowie in den Ansprüchen offenbarten Merkmale der Erfindung können sowohl einzeln als auch in beliebiger Kombination für die Verwirklichung der Erfindung in ihren verschiedenen Ausführungsformen wesentlich sein.
- 1
- Einreichung zur Erzeugung eines Trägergasstrahles
- 2
- Photonenstrahl
- 3
- Probenträger
- 4
- Teilchenstrahl
- 4a
- Photonenstrahl
- 5
- Abstreifer (Skimmer)
- 6
- gepulster Elektronenstrahl
- 7
- Ionisationskammer
- 10
- gepulstes Ventil mit Düse
- 20
- Linse
- 30
- Probe
- 50
- Wand
- 51
- Öffnung
- 61
- Fokus
- 70
- Frontwand
- 71
- Eintrittsöffnung
- 72
- Austrittsöffnung
- 73
- Abschlußplatte
Claims (17)
- Verfahren zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen nichtflüchtigen großen Molekülen, bei dem eine die Moleküle aufweisende Probensubstanz Energiepulsen ausgesetzt wird, durch welche Moleküle aus der Probensubstanz freigesetzt werden, und bei dem die freigesetzten Moleküle von einem Strahl eines Trägergases mitgenommen und bei dessen Expansion gekühlt sowie anschließend in einem Ionisationsraum ionisiert werden, dadurch gekennzeichnet, daß die Moleküle durch Elektronenstoß ionisiert werden; daß die Strahlungsdichte der für die Ionisation verwendeten Elektronen so gewählt wird, daß im Fokus des Elektronenstrahles eine Potentialmulde erzeugt wird, deren Tiefe größer als die Translationsenergie der Molekülionen im Trägergasstrom ist; daß die durch die Elektronenstoßionisation erzeugten Molekülionen in der Potentialmulde jeweils für einen bestimmten Zeitraum gesammelt werden; und daß die jeweils in der Potentialmulde gesammelten Molekülionen gepulst aus der Ionisationskammer herausbeschleunigt werden.
- Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß die Energie der ionisierenden Elektronen niedriger gewählt wird, als dies für die Ionisierung des Trägergases notwendig wäre.
- Verfahren nach Anspruch 1 oder 2, dadurch gekennzeichnet, daß als Trägergas Helium und/oder Neon verwendet wird/werden.
- Verfahren nach einem der vorangehenden Ansprüche, dadurch gekennzeichnet, daß die zum Freisetzen der Moleküle aus der Probensubstanz verwendeten Energiepulse mittels Laser erzeugte Lichtpulse sind.
- Verfahren nach einem der Ansprüche 1 bis 3, dadurch gekennzeichnet, daß die zum Freisetzen der Moleküle aus der Probensubstanz verwendeten Energiepulse durch Beschuß mit Ionen oder Neutralteilchen aufgebracht werden.
- Verfahren nach einem der vorangehenden Ansprüche, dadurch gekennzeichnet, daß die Moleküle wahlweise durch Elektronenstoß oder durch Photonenanregung in demselben Ionisationsraum ionisiert werden, wobei die Elektronen und/oder die Photonen gepulst aufgegeben werden.
- Verfahren nach Anspruch 6, dadurch gekennzeichnet, daß die Probenmoleküle gepulst zugeführt werden.
- Verfahren nach Anspruch 6 oder 7, dadurch gekennzeichnet, daß das Herausbeschleunigen der in der Potentialmulde gesammelten Molekülionen bei Elektronenstoß- und Photonenionisation im selben Rhythmus erfolgt.
- Verfahren nach einem der Ansprüche 6 bis 8, dadurch gekennzeichnet, daß eine Multiphotonenanregung verwendet wird.
- Vorrichtung zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen nichtflüchtigen großen Molekülen, mit einer Einrichtung zum Erzeugen eines Trägergasstrahles, einer Energiequelle zum Desorbieren von Molekülen aus dem Probenmaterial und einer Einrichtung zum Einbringen von Probenmaterial in den Trägergasstrahl, insbesondere zur Durchführung des Verfahrens nach einem der vorangehenden Ansprüche, dadurch gekennzeichnet, daß eine Ionisationskammer (7) vorgesehen ist, die eine Eintrittsöffnung (71) und eine Austrittsöffnung (72) für einen Teilchenstrahl aufweist, wobei eine Elektronenquelle so angeordnet ist, daß der von dieser erzeugte Elektronenstrahl (6) innerhalb der Ionisationskammer (7) auf die Bahn des Teilchenstrahles fokussiert ist; daß die Einrichtung zum Erzeugen des Trägergasstrahles (1) eine Austrittsöffnung für den Trägergasstrahl aufweist; und daß in der Nähe der Austrittsöffnung ein Probenträger (3) angeordnet ist, auf dem das Probenmaterial angebracht ist.
- Vorrichtung nach Anspruch 10 zur Durchführung des Verfahrens nach einem der Ansprüche 6 bis 9, dadurch gekennzeichnet, daß eine Elektronenquelle und eine Photonenquelle vorgesehen sind, die zur Ionisierung der gasförmigen Probe (4) innerhalb der Ionisationskammer (7) wahlweise betreibbar sind.
- Vorrichtung nach Anspruch 11, dadurch gekennzeichnet, daß die Elektronenquelle und die Photonenquelle zur Ionisierung der gasförmigen Probe (4) innerhalb der Ionisationskammer (7) wahlweise alternierend betreibbar sind.
- Vorrichtung nach Anspruch 11 oder 12, dadurch gekennzeichnet, daß der von der Elektronenquelle ausgesandte Elektronenstrahl (6) und der von der Photonenquelle ausgesandte Photonenstrahl (4a) im wesentlichen auf denselben Bereich (61) der Ionisationskammer (7) fokussiert sind.
- Vorrichtung nach Anspruch 11 oder 12, dadurch gekennzeichnet, daß der von der Elektronenquelle ausgesandte Elektronenstrahl (6) und der von der Photonenquelle ausgesandte Photonenstrahl (4a) auf einander benachbarte Bereiche (61) der Ionisationskammer (7) fokussiert sind.
- Vorrichtung nach einem der Ansprüche 11 bis 14, dadurch gekennzeichnet, daß die Ionisationskammer (7) auf positives Potential gelegt ist und eine separat beaufschlagbare Abschlußplatte (73) aufweist.
- Vorrichtung nach einem der Ansprüche 11 bis 15, dadurch gekennzeichnet, daß die Elektronenquelle und/oder die Photonenquelle gepulst betreibbar sind.
- Vorrichtung nach Anspruch 15 und 16, dadurch gekennzeichnet, daß die Abschlußplatte (73) bei Elektronenstoß- und Photonenionisation im selben Rhythmus schaltbar ist.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP95104288A EP0669638A1 (de) | 1991-03-13 | 1992-03-07 | Verfahren und Vorrichtung zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeugmassenspektrometer, aus thermisch instabilien, nichtflüchtigen Molekülen |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE4108462A DE4108462C2 (de) | 1991-03-13 | 1991-03-13 | Verfahren und Vorrichtung zum Erzeugen von Ionen aus thermisch instabilen, nichtflüchtigen großen Molekülen |
| DE4108463 | 1991-03-13 | ||
| DE4108463 | 1991-03-13 | ||
| DE4108462 | 1991-03-13 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP95104288.6 Division-Into | 1992-03-07 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0503748A2 true EP0503748A2 (de) | 1992-09-16 |
| EP0503748A3 EP0503748A3 (en) | 1993-04-28 |
| EP0503748B1 EP0503748B1 (de) | 1996-12-11 |
Family
ID=25901910
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP92250055A Expired - Lifetime EP0503748B1 (de) | 1991-03-13 | 1992-03-07 | Verfahren zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen, nichtflüchtigen grossen Molekülen |
| EP95104288A Withdrawn EP0669638A1 (de) | 1991-03-13 | 1992-03-07 | Verfahren und Vorrichtung zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeugmassenspektrometer, aus thermisch instabilien, nichtflüchtigen Molekülen |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP95104288A Withdrawn EP0669638A1 (de) | 1991-03-13 | 1992-03-07 | Verfahren und Vorrichtung zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeugmassenspektrometer, aus thermisch instabilien, nichtflüchtigen Molekülen |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US5294797A (de) |
| EP (2) | EP0503748B1 (de) |
| DE (2) | DE4108462C2 (de) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0633602A3 (de) * | 1993-07-02 | 1995-11-22 | Bergmann Eva Martina | Flugzeit-Massenspektrometer mit Gasphasen-Ionenquelle, mit hoher Empfindlichkeit und grossem dynamischem Bereich. |
| DE19822672A1 (de) * | 1998-05-20 | 1999-12-09 | Gsf Forschungszentrum Umwelt | Verfahren und Vorrichtung zur Erzeugung eines gerichteten Gasstrahls |
| DE19822674A1 (de) * | 1998-05-20 | 1999-12-09 | Gsf Forschungszentrum Umwelt | Gaseinlaß für eine Ionenquelle |
| DE102005005333A1 (de) * | 2005-01-28 | 2006-08-17 | Deutsches Zentrum für Luft- und Raumfahrt e.V. | Verfahren zur Aerosol-Analyse, Sondenvorrichtung und Analysevorrichtung |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19608963C2 (de) * | 1995-03-28 | 2001-03-22 | Bruker Daltonik Gmbh | Verfahren zur Ionisierung schwerer Moleküle bei Atmosphärendruck |
| GB2310950A (en) * | 1996-03-08 | 1997-09-10 | Bruker Franzen Analytik Gmbh | Method for the ionization of heavy molecules at atmospheric pressure |
| DE19630547C2 (de) * | 1996-07-17 | 1998-05-28 | Forschungsverbund Berlin Ev | Verfahren zur Matrix-unterstützten Laserdesorption und Ionisierung von Analytmolekülen, insbesondere von fragilen bzw. labilen Molekülen |
| US5742050A (en) * | 1996-09-30 | 1998-04-21 | Aviv Amirav | Method and apparatus for sample introduction into a mass spectrometer for improving a sample analysis |
| DE19649201A1 (de) * | 1996-11-27 | 1998-05-28 | Max Planck Gesellschaft | Elektronenstrahlanalyse |
| DE19705762C2 (de) * | 1997-02-14 | 2001-06-07 | Rainer Edmund Weinkauf | Verfahren und Vorrichtung zum Erzeugen von Gasstrahlen |
| DE19734460A1 (de) * | 1997-08-11 | 1999-02-18 | Gsf Forschungszentrum Umwelt | Verfahren und Vorrichtung zum analytischen Nachweis von Spuren |
| US7109476B2 (en) | 1999-02-09 | 2006-09-19 | Syagen Technology | Multiple ion sources involving atmospheric pressure photoionization |
| US7119342B2 (en) * | 1999-02-09 | 2006-10-10 | Syagen Technology | Interfaces for a photoionization mass spectrometer |
| US6211516B1 (en) * | 1999-02-09 | 2001-04-03 | Syagen Technology | Photoionization mass spectrometer |
| US6630664B1 (en) | 1999-02-09 | 2003-10-07 | Syagen Technology | Atmospheric pressure photoionizer for mass spectrometry |
| DE19911801C1 (de) | 1999-03-17 | 2001-01-11 | Bruker Daltonik Gmbh | Verfahren und Vorrichtung zur matrixunterstützten Laserdesorptions-Ionisierung von Substanzen |
| DE10014847A1 (de) * | 2000-03-24 | 2001-10-04 | Gsf Forschungszentrum Umwelt | Verfahren und Vorrichtung zum Nachweis von Verbindungen in einem Gasstrom |
| DE10042394B4 (de) * | 2000-08-29 | 2006-06-29 | Dräger Safety AG & Co. KGaA | Ionenquelle für Ionenmobilitätsspektrometer mit optischer und Teilchenstrahlun gsionisation |
| US6737642B2 (en) | 2002-03-18 | 2004-05-18 | Syagen Technology | High dynamic range analog-to-digital converter |
| US20080223409A1 (en) * | 2003-12-12 | 2008-09-18 | Horsky Thomas N | Method and apparatus for extending equipment uptime in ion implantation |
| WO2005059942A2 (en) * | 2003-12-12 | 2005-06-30 | Semequip, Inc. | Method and apparatus for extending equipment uptime in ion implantation |
| DE102004002729B4 (de) * | 2004-01-20 | 2008-11-27 | Bruker Daltonik Gmbh | Ionisierung desorbierter Analytmoleküle bei Atmosphärendruck |
| US7291845B2 (en) * | 2005-04-26 | 2007-11-06 | Varian, Inc. | Method for controlling space charge-driven ion instabilities in electron impact ion sources |
| JP4958258B2 (ja) * | 2006-03-17 | 2012-06-20 | 株式会社リガク | ガス分析装置 |
| JP4825028B2 (ja) * | 2006-03-17 | 2011-11-30 | 浜松ホトニクス株式会社 | イオン化装置 |
| EP2017875A1 (de) * | 2007-07-16 | 2009-01-21 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Verfahren und Vorrichtung zur Bereitstellung einer Probe für eine nachfolgende Analyse |
| US7875125B2 (en) * | 2007-09-21 | 2011-01-25 | Semequip, Inc. | Method for extending equipment uptime in ion implantation |
| US7750291B2 (en) * | 2008-02-25 | 2010-07-06 | National Sun Yat-Sen University | Mass spectrometric method and mass spectrometer for analyzing a vaporized sample |
| US8742335B2 (en) * | 2009-12-18 | 2014-06-03 | Sri International | Rotator sample introduction interface |
| CN102903597A (zh) * | 2012-10-19 | 2013-01-30 | 山东省科学院海洋仪器仪表研究所 | 一种热解吸与电子轰击相结合的离子源 |
| GB2518122B (en) * | 2013-02-19 | 2018-08-08 | Markes International Ltd | An electron ionisation apparatus |
| CN105190829B (zh) | 2013-04-17 | 2018-04-03 | 富鲁达加拿大公司 | 用于质谱流式细胞术的样品分析 |
| GB201721700D0 (en) | 2017-12-22 | 2018-02-07 | Micromass Ltd | Ion source |
| JP7039439B2 (ja) * | 2018-10-26 | 2022-03-22 | 株式会社堀場製作所 | ガス分析装置及びガス分析方法 |
| CN116031138B (zh) * | 2023-01-10 | 2025-10-10 | 中国科学院大学 | 一种气相污染物全物种高灵敏在线质谱仪及检测方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1024654A (fr) * | 1950-09-16 | 1953-04-03 | Csf | Spectromètre de masse à sélection par temps de transit utilisant la modulation de fréquence |
| US3182190A (en) * | 1962-07-31 | 1965-05-04 | Gulf Research Development Co | Magnetic field free ion source with adjustable electron gun |
| US3296434A (en) * | 1964-05-26 | 1967-01-03 | Martin H Studier | Method of operating an ion source for a time of flight mass spectrometer |
| DE3224801C2 (de) * | 1982-07-02 | 1986-04-30 | Edward William Prof. Dr. 8000 München Schlag | Verfahren und Einrichtung zum Erzeugen von gepulsten Molekularstrahlen, die große, thermisch instabile Moleküle enthalten |
| GB2177507B (en) * | 1985-06-13 | 1989-02-15 | Mitsubishi Electric Corp | Laser mass spectroscopic analyzer |
| DE3809504C1 (de) * | 1988-03-22 | 1989-09-21 | Bruker - Franzen Analytik Gmbh, 2800 Bremen, De | |
| DE3920566A1 (de) * | 1989-06-23 | 1991-01-10 | Bruker Franzen Analytik Gmbh | Ms-ms-flugzeit-massenspektrometer |
| GB8928917D0 (en) * | 1989-12-21 | 1990-02-28 | Vg Instr Group | Method and apparatus for surface analysis |
-
1991
- 1991-03-13 DE DE4108462A patent/DE4108462C2/de not_active Expired - Fee Related
-
1992
- 1992-03-07 EP EP92250055A patent/EP0503748B1/de not_active Expired - Lifetime
- 1992-03-07 EP EP95104288A patent/EP0669638A1/de not_active Withdrawn
- 1992-03-07 DE DE59207642T patent/DE59207642D1/de not_active Expired - Fee Related
- 1992-03-12 US US07/849,886 patent/US5294797A/en not_active Expired - Fee Related
Non-Patent Citations (6)
| Title |
|---|
| ANALYTICAL INSTRUMENTATION, Band 16, Nr. 1, 1987, Seiten 93-115, New York, US; J.K. OLTHOFF et al.: "Modification of Wiley-McLaren TOF analysers for laser desorption" * |
| APPLIED PHYSICS B. PHOTOPHYSICS AND CHEMISTRY, Band 51, Nr. 6, Dezember 1990, Seiten 395-403, Heidelberg, DE; G. MEIJER et al.: "Laser desorption jet-cooling of organic molecules" * |
| APPLIED PHYSICS B. PHOTOPHYSICS AND CHEMISTRY, Band B41, Nr. 6, Dezember 1990, Seiten 395-403, Heidelberg, DE; G. MEIJER et al.: "Laser desorption jet-cooling of organic molecules" * |
| PHYSICAL REVIEW, B. CONDENSED MATTER, Band 38, Nr. 5, 15. August 1988, Seiten 3517-3520, New York, US; C.W.S. CONOVER et al.: "Laser vaporisation of solids into an inert gas: a measure of high-temperature cluster stability" * |
| REVIEW OF SCIENTIFIC INSTRUMENTS, Band 58, Nr. 1, Januar 1987, Seiten 32-37, New York, US; J.E. POLLARD et al.: "Electron-impact ionization time-of-flight mass spectrometer for molecular beams" * |
| REVIEW OF SCIENTIFIC INSTRUMENTS, Band 59, Nr. 4, April 1988, Seiten 557-561, New York, US; L. LI et al.: "Pulsed laser desorption method for volatilizing thermally labile molecules for supersonic jet spectroscopy" * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0633602A3 (de) * | 1993-07-02 | 1995-11-22 | Bergmann Eva Martina | Flugzeit-Massenspektrometer mit Gasphasen-Ionenquelle, mit hoher Empfindlichkeit und grossem dynamischem Bereich. |
| DE19822672A1 (de) * | 1998-05-20 | 1999-12-09 | Gsf Forschungszentrum Umwelt | Verfahren und Vorrichtung zur Erzeugung eines gerichteten Gasstrahls |
| DE19822674A1 (de) * | 1998-05-20 | 1999-12-09 | Gsf Forschungszentrum Umwelt | Gaseinlaß für eine Ionenquelle |
| DE19822672B4 (de) * | 1998-05-20 | 2005-11-10 | GSF - Forschungszentrum für Umwelt und Gesundheit GmbH | Verfahren und Vorrichtung zur Erzeugung eines gerichteten Gasstrahls |
| DE102005005333A1 (de) * | 2005-01-28 | 2006-08-17 | Deutsches Zentrum für Luft- und Raumfahrt e.V. | Verfahren zur Aerosol-Analyse, Sondenvorrichtung und Analysevorrichtung |
| DE102005005333B4 (de) * | 2005-01-28 | 2008-07-31 | Deutsches Zentrum für Luft- und Raumfahrt e.V. | Verfahren zur Probennahme und Aerosol-Analyse |
Also Published As
| Publication number | Publication date |
|---|---|
| DE59207642D1 (de) | 1997-01-23 |
| US5294797A (en) | 1994-03-15 |
| EP0503748A3 (en) | 1993-04-28 |
| DE4108462C2 (de) | 1994-10-13 |
| DE4108462A1 (de) | 1992-09-17 |
| EP0503748B1 (de) | 1996-12-11 |
| EP0669638A1 (de) | 1995-08-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0503748B1 (de) | Verfahren zum Erzeugen von Ionen, insbesondere für ein Massenspektrometer, wie Flugzeitmassenspektrometer, aus thermisch instabilen, nichtflüchtigen grossen Molekülen | |
| DE69132461T2 (de) | Verfahren und vorrichtung zur spurenanalyse | |
| DE69631556T2 (de) | Biomolekülenanalyse mittels Flugzeitmassenspektrometrie | |
| DE19511333C1 (de) | Verfahren und Vorrichtung für orthogonalen Einschuß von Ionen in ein Flugzeit-Massenspektrometer | |
| EP1200984B1 (de) | Verfahren und vorrichtung zur clusterfragmentation | |
| DE19822672B4 (de) | Verfahren und Vorrichtung zur Erzeugung eines gerichteten Gasstrahls | |
| DE4441972C2 (de) | Verfahren und Vorrichtung zum Nachweis von Probenmolekülen in einem Trägergas | |
| DE4202123A1 (de) | Verfahren und vorrichtung fuer die massenspektrometrische untersuchung schneller organischer ionen | |
| DE102021105327B3 (de) | Desorptions-Ionenquelle mit Postdesorptions-Ionisierung in Transmissionsgeometrie | |
| DE69031062T2 (de) | Verfahren und Vorrichtung zur massenspektrometrischen Analyse | |
| DE3733853C2 (de) | ||
| EP0112858B1 (de) | Verfahren und einrichtung zum erzeugen von molekularstrahlen und verwendung dieses verfahrens | |
| DE1922871B2 (de) | Ionenquelle | |
| WO2005117062A2 (de) | Verfahren und vorrichtung zur massenspektroskopischen untersuchung von analyten | |
| DE19637480C2 (de) | Vorrichtung zur massenspektrometrischen Analyse von Oberflächen | |
| DE2048862A1 (de) | Vorrichtung zur spektralphotometri sehen Analyse | |
| DE4317749A1 (de) | Massenspektrometer mit Einrichtungen zum Überwachen der Strahlung, die ausgesendet wird, wenn Ionen mit einem Zielgas kollidieren | |
| DE19547949C2 (de) | Flugzeitmassenspektrometer | |
| DE102017218456B3 (de) | Vorrichtung und Verfahren zum Erzeugen von Ionenpulsen sowie deren Verwendung | |
| DE102018106412B4 (de) | Teilchenspektrometer und Teilchenspektrometrieverfahren | |
| DE102016112328B3 (de) | Ionenmikroskopievorrichtung | |
| DE19756444C1 (de) | Verfahren und Vorrichtung zum Nachweis von Probenmolekülen in einem Trägergas | |
| DE19705762C2 (de) | Verfahren und Vorrichtung zum Erzeugen von Gasstrahlen | |
| DE19630547C2 (de) | Verfahren zur Matrix-unterstützten Laserdesorption und Ionisierung von Analytmolekülen, insbesondere von fragilen bzw. labilen Molekülen | |
| DE10015415C2 (de) | Verfahren und Vorrichtung zur Erzeugung hochenergetischer Ionenstrahlen und/oder kurzwelliger elektromagnetischer Strahlung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): DE FR GB |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): DE FR GB |
|
| 17P | Request for examination filed |
Effective date: 19930524 |
|
| 17Q | First examination report despatched |
Effective date: 19941123 |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| RTI1 | Title (correction) | ||
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): DE FR GB |
|
| DX | Miscellaneous (deleted) | ||
| ET | Fr: translation filed | ||
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 19961211 |
|
| REF | Corresponds to: |
Ref document number: 59207642 Country of ref document: DE Date of ref document: 19970123 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: IF02 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20020306 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20020312 Year of fee payment: 11 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20020318 Year of fee payment: 11 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20030307 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031001 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20030307 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20031127 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |