-
TECHNISCHES
GEBIET DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft neue Verbindungsklassen, welche Caspase-Inhibitoren
sind, insbesondere Inhibitoren von Interleukin-1β umwandelndem Enzym („ICE"). Diese Erfindung
betrifft auch Arzneimittel, umfassend diese Verbindungen. Die erfindungsgemäßen Verbindungen
oder Arzneimittel sind besonders gut zur Hemmung der Caspaseaktivität geeignet
und können
infolgedessen vorteilhafterweise als Mittel gegen durch Interleukin-1
(„IL-1"), Apoptose, Interferon-γ-induzierenden
Faktor (IGIF) oder Interferon-γ („INF-γ") vermittelte Erkrankungen,
einschließlich
entzündlicher
Erkrankungen, Autoimmunerkrankungen, destruktiver Knochenerkrankungen,
proliferativer Störungen,
infektiöser
Erkrankungen und degenerativer Erkrankungen verwendet werden. Diese
Erfindung betrifft auch die Verwendung der erfindungsgemäßen Verbindungen
und Zusammensetzungen zur Herstellung eines Arzneimittels zur Hemmung
der Caspaseaktivität
und zur Senkung der IGIF-Produktion und IFN-γ-Produktion und Verfahren zur
Behandlung von durch Interleukin-1, Apoptose und Interferon-γ vermittelten
Erkrankungen. Diese Erfindung betrifft auch Verfahren zum Herstellen
der erfindungsgemäßen Verbindungen.
-
HINTERGRUND
DER ERFINDUNG
-
Interleukin
1 („IL-1") ist ein bedeutendes
proentzündliches
und immunregulatorisches Protein, das Fibroblastendifferenzierung
und -proliferation, die Produktion von Prostaglandinen, Kollagenase
und Phospholipase durch Synovialzellen und Chondrocyten, basophile
und eosinophile Degranulation und neutrophile Aktivierung stimuliert.
Oppenheim, J. H. et al., Immunology Today, 7, Seiten 45 – 56 (1986).
Als derartiges ist es an der Pathogenese chronischer und akuter
entzündlicher
Erkrankungen und Autoimmunerkrankungen beteiligt. Zum Beispiel ist
IL-1 bei rheumatoider Arthritis ein Vermittler sowohl von Entzündungssymptomen
als auch der Zerstörung
des Knorpelproteoglycans in befallenen Gelenken. Wood, D. D. et
al., Arthritis Rheum, 26, 975 (1983); Pettipher, E. J. et al., Proc.
Natl. Acad. Sci. USA, 71, 295 (1986); Arend, W. P. und Dayer, J.
M., Arthritis Rheum, 38, 151 (1995). IL-1 ist auch ein sehr starkes
Knochenresorptionsmittel. Jandiski, J. J., J. Oral Path 17, 145
(1988); Dewhirst, F. E. et al., J. Immunol., 8, 2562 (1985). Bei
destruktiven Knochenerkrankungen wie zum Beispiel Osteoarthritis
und multiplem Myelom wird es alternativ als "Osteoklast aktivierender Faktor" bezeichnet. Bataille,
R. et al., Int. J. Clin. Lab. Res., 21 (4), 283 (1992). Bei bestimmten
proliferativen Störungen wie
zum Beispiel akuter myologener Leukämie und multiplem Myelom kann
IL-1 Tumorzellwachstum und -adhäsion
fördern.
Bani, M. R., J. Natl. Cancer Inst., 83, 123 (1991); Vidal-Vanaclocha,
F., Cancer Res., 54, 2667 (1994). Bei diesen Störungen stimuliert IL-1 auch
die Produktion anderer Cytokine wie zum Beispiel IL-6, das die Tumorentwicklung
modulieren kann (Tartour et al., 25 Cancer Res., 54, S. 6243 (1994).
IL-1 wird vorwiegend durch periphere Blutmonocyten als Teil der
Entzündungsantwort
produziert und kommt in zwei verschiedenen Agonistenformen, IL-1α und IL-1β, vor. Mosely,
B. S. et al., Proc. Nat. Acad. Sci., 84, Seiten 4572 – 4576 (1987);
Lonnemann, G. et al., Eur. J. Immunol., 19, Seiten 1531 – 1536 (1989).
-
IL-1β wird als
biologisch inaktiver Vorläufer,
pIL-1β synthetisiert.
pIL-1β fehlt
eine herkömmliche
Leadersequenz und wird nicht durch eine Signalpeptidase prozeniert.
March, C. J., Nature, 315, Seiten 641 – 647 (1985). Stattdessen wird
pIL-1β durch
das Interleukin-1β umwandelnde
Enzym („ICE") zwischen Asp-116
und Ala-117 gespalten, um das in menschlichem® Serum
und Gelenkschmiere gefundene biologisch aktive C-terminale Fragment
zu produzieren. Sleath, P. R. et al., J. Biol. Chem., 265, Seiten
14526 – 14528
(1992); A. D. Howard et al., J. Immunol., 147, Seiten 2964 – 2969 (1991).
ICE ist eine hauptsächlich
in Monocyten lokalisierte Cysteinprotease. Es wandelt Vorläufer-pIL-1β in die reife
Form um. Black, R. A. et al., FEBS Lett., 247, Seiten 386 – 390 (1989);
Kostura, M. J. et al., Proc. Natl. Acad. Sci USA, 86, Seiten 5227 – 5231 (1989).
Auch ist das Bearbeiten durch ICE für den Transport von reifem
IL-1β durch
die Zellmembran notwendig.
-
ICE
ist ein Mitglied einer Familie von homologen Enzymen, die Caspasen
genannt werden. Diese Homologen weisen Sequenzähnlichkeiten in den Regionen
des aktiven Zentrums der Enzyme auf. Derartige Homologe (Caspasen)
schließen
TX (oder ICErel-II oder ICH-2) (Faucheu
et al., EMBO J., 14, S. 1914 (1995); Kamens J. et al., J. Biol.
Chem., 270, S. 15250 (1995); Nicholson et al., J. Biol. Chem., 270,
15870 (1995)), TY (oder ICErel-III) (Nicholson
et al., J. Biol. Chem., 270, S. 15870 (1995); ICH-1 (oder Nedd-2)
(Wang, L. et al., Cell, 78, S. 739 (1994)), MCH-2 (Fernandes-Alnemri,
T. et al., Cancer Res., 55, S. 2737 (1995), CPP32 (oder YAMA oder
Apopain) (Fernandes-Alnemri, T. et al., J. Biol. Chem., 269, S.
30761 (1994); Nicholson, D. W. et al., Nature, 376, S. 37 (1995))
und CMH-1 (oder MCH-3) (Lippke et al., J. Biol. Chem., (1996); Fernandes-Alnemri,
T. et al., Cancer Res., (1995)) ein.
-
Jedes
dieser ICE-Homologen, ebenso wie ICE selbst, ist, wenn es in transfizierten
Zelllinien überexprimiert
wird, in der Lage, Apoptose hervorzurufen. Hemmung von einem oder
mehreren dieser Homologen mit dem Peptidyl-ICE-Inhibitor Tyr-Val-Ala-Asp-Chlormethylketon
führt zur
Hemmung der Apoptose in primären
Zellen oder Zelllinien. Lazebnik et al., Nature, 371, S. 346 (1994).
-
Caspasen
scheinen auch an der Regulation des programmierten Zelltods oder
Apoptose beteiligt zu sein. Yuan, J. et al., Cell, 75, Seiten 641 – 652 (1993);
Miura, M. et al., Cell 75, Seiten 653 – 660 (1993); Nett-Fiordalisi,
M. A. et al, J. Cell Biochem., 17B, S. 117 (1993). Insbesondere
wird geglaubt, dass ICE oder ICE-Homologe mit der Regulation von
Apoptose bei neurodegenerativen Erkrankungen wie zum Beispiel Alzheimer- und
Parkinson-Krankheit in Zusammenhang stehen. Marx, J. und M. Baringa,
Science, 259, Seiten 760 – 762 (1993);
Gagliardini, V. et al, Science, 263, Seiten 826 – 828 (1994). Therapeutische
Anwendungen zur Hemmung von Apoptose können die Behandlung von Alzheimer-Krankheit,
Parkinson-Krankheit,
Schlaganfall, Myocardinfarkt, spinaler Atrophie und Alterung einschließen.
-
Von
ICE ist gezeigt worden, dass es in bestimmten Gewebetypen Apoptose
(programmierter Zelltod) vermittelt. Steller, H., Science, 267,
5. 1445 (1995); Whyte, M. und Evan, G., Nature, 376, S. 17 (1995);
Martin, S. J. und Green, D. R., Cell, 82, S. 349 (1995); Alnemri,
E. S. et al., J. Biol. Chem., 270, S. 4312 (1995); Yuan, J. Curr.
Opin. Cell Biol., 7, S. 211 (1995). Eine transgene Maus mit einer
Disruption des ICE-Gens hat eine unzureichende Fas-vermittelte Apoptose
(Kuida, K. et al., Science, 267, 2000 (1995)). Diese ICE-Aktivität ist verschieden
von seiner Rolle als Prozenierungsenzym für pro-IL-1β. Es ist denkbar, dass in bestimmten
Gewebetypen die Hemmung von ICE nicht die Sekretion von reifem IL-1β beeinflussen
kann, aber Apoptose hemmen kann.
-
Enzymatisch
aktives ICE ist zuvor als ein aus zwei Untereinheiten, p20 und p10
(Molekulargewicht von 20 kDa bzw. 10 kDa), bestehendes Heterodimer
beschrieben worden. Diese Untereinheiten sind durch einen Aktivierungsmechanismus,
der autokatalytisch ist, aus einem 45 kDa Proenzym (p45) über eine
p30 Form abgeleitet. Thornberry, N. A. et al., Nature, 356, Seiten
768 – 774
(1992). Das ICE Proenzym ist in mehrere funktionelle Domänen eingeteilt
worden: eine Prodomäne
(p14), eine p22/20-Untereinheit, ein Polypeptid-Linker und eine p10-Untereinheit. Thornberry
et al., supra; Casano et al., Genomics, 20, Seiten 474 – 481 (1994).
-
p45
in ganzer Länge
ist durch seine cDNA und Aminosäuresequenzen
gekennzeichnet worden. PCT-Patentanmeldungen WO 91/15577 und WO
94/00154. Die p20- und p10-cDNA und Aminosäuresequenzen sind auch bekannt.
Thornberry et al., supra. ICE aus Maus und Ratte ist auch sequenziert
und cloniert worden. Sie weisen große Aminosäure- und Nucleinsäure-Sequenzhomologie
zu menschlichem ICE auf. Miller, D. K. et al., Ann. N. Y. Acad.
Sci., 696, Seiten 133 – 148
(1993); Molineaux, S. M. et al., Proc. Nat. Acad. Sci., 90, Seiten
1809 – 1813
(1993). Die dreidimensionale Struktur von ICE ist in atomarer Auflösung durch
Röntgenkristallographie
bestimmt worden. Wilson, K. P. et al., Nature, 370, Seiten 270 – 275 (1994).
Das aktive Enzym kommt als ein Tetramer aus zwei p20- und zwei p10-Untereinheiten vor.
-
Kürzlich sind
ICE und andere Mitglieder der ICE/CED-3-Familie mit der Umwandlung
von pro-IGIF in IGIF oder mit der Produktion von IFN-γ in vivo
in Verbindung gebracht worden (PCT-Anmeldung PCT/US96/20843, eingereicht
am 20.12.96, veröffentlicht
am 26.06.97 unter der Veröffentlichungsnr.
WO 97/22619, welche hierin durch Bezugnahme aufgenommen ist). IGIF
wird in vivo als das Vorläuferprotein „pro-IGIF" synthetisiert.
-
Der
Interferon-gamma induzierende Faktor (IGIF) ist ein annähernd 18
kDa-Polypeptid, das die T-Zell-Produktion von Interferon-gamma (IFN-γ) stimuliert.
IGIF wird in vivo durch aktivierte Kupfferzellen und Makrophagen
produziert und wird aus derartigen Zellen nach Stimulierung durch
Endotoxin ausgeführt.
Daher würde
eine Verbindung, welche die IGIF Produktion senkt, als ein Inhibitor
derartiger T-Zell-Stimulierung nützlich
sein, welches wiederum die Mengen der IFN-γ Produktion durch diese Zellen
verringern würde.
-
IFN-γ ist ein
Cytokin mit immunmodulatorischen Wirkungen auf eine Vielfalt von
Immunzellen. Insbesondere ist IFN-γ an der Macrophagenaktivierung
und der Th1-Zellselektion
beteiligt (F. Belardelli, APMIS, 103, S. 161 (1995)). INF-γ übt seine
Wirkungen zum Teil durch das Modulieren der Expression von Genen durch
die STAT- und IRF- Signalwege
aus (C. Schindler und J. E. Darnell, Ann. Rev. Biochem., 64, S.
621 (1995); T. Taniguchi, J. Cancer Res. Clin. Oncol. 121, S. 516
(1995)).
-
Mäuse, denen
INF-γ oder
sein Rezeptor fehlt, haben multiple Defekte in der Immunzellfunktion
und sind resistent gegen endotoxischen Schock (S. Huang et al.,
Science, 259, S. 1742 (1993); D. Dalton et al., Science, 259, S.
1739 (1993); B. D. Car et al., J. Exp. Med., 179, S. 1437 (1994)).
Zusammen mit IL-12 scheint IGIF ein starker Induktor der IFN-γ Produktion
durch T-Zellen zu sein (H. Okamura et al., Infection and Immunity,
63, S. 3966 (1995); H. Okamura et al., Nature, 378, S. 88 (1995);
S. Ushio et al., J. Immunol., 156, S. 4274 (1996)).
US 5,716,926 offenbart neue Verbindungsklassen,
welche ICE-Inhibitoren, Arzneimittel sind, umfassend jene Verbindungen,
und ihre Verwendung zur Behandlung von durch IL-1 vermittelten Erkrankungen.
-
Von
IFN-γ ist
gezeigt worden, dass es zu der Pathologie beiträgt, die mit einer Vielfalt
von entzündlichen,
infektiösen
Autoimmunstörungen
und -erkrankungen in Zusammenhang steht. Daher würden Verbindungen, die zur
Senkung der IFN-γ Produktion
in der Lage sind, nützlich
sein, um die Wirkungen von mit IFN-γ in Beziehung stehenden Pathologien
zu verbessern.
-
Folglich
würden
Zusammensetzungen und Verfahren, die in der Lage sind, die Umwandlung
von pro-IGIF in IGIF zu regulieren, bei der Senkung der IGIF- und
IFN-γ-Produktion
in vivo und daher zur Verbesserung der schädlichen Wirkungen dieser Proteine,
welche zu Störungen
und Erkrankungen beim Menschen beitragen, nützlich sein.
-
Caspase-Inhibitoren
stellen eine Verbindungsklasse dar, die zur Kontrolle von Entzündung oder
Apoptose oder beidem nützlich
ist. Peptid- und Peptidyl-Inhibitoren von ICE sind beschrieben worden.
PCT-Patentanmeldungen WO 91/15577; WO 93/05071; WO 93/09135; WO
93/14777 und WO 93/16710; und Europäische Patentanmeldung 0 547
699. Von derartigen Peptidyl-Inhibitoren von ICE ist beobachtet
worden, dass sie die Produktion von reifem IL-1β in einem Maus-Entzündungmodell
(vide infra blockieren und Wachstum von Leukämiezellen in vitro unterdrücken (Estrov
et al., Blood 84, 380a (1994)). Dennoch sind derartige Inhibitoren auf
Grund ihrer peptischen Natur typischerweise durch unerwünschte pharmakologische
Eigenschaften gekennzeichnet, wie zum Beispiel geringe zelluläre Penetration
und zelluläre
Aktivität,
geringe orale Absorption, geringe Stabilität und schnellen Metabolismus.
Plattner, J. J. und D. W. Norbeck, in Drug Discovery Technologies,
C. R. Clark und W. H. Moos, Hrsg. (Ellis Horwood, Chichester, England,
1990), Seiten 92 – 126.
Dies hat ihre Entwicklung in wirksame Arzneistoffe behindert.
-
Auch
von Nicht-Peptidyl-Verbindungen ist berichtet worden, dass sie ICE
in vitro hemmen. PCT-Patentanmeldung WO 95/26958; US-Patent 5,552,400;
Dolle et al., J. Med. Chem., 39, Seiten 2438 – 2440 (1996). Dennoch ist
nicht klar, ob diese Verbindungen die geeigneten pharmakologischen
Profile, um therapeutisch nützlich
zu sein, aufweisen.
-
Folglich
besteht ein Bedarf für
Verbindungen, die Caspasen wirksam hemmen können, zur Verwendung als Mittel
zur Prävention
und Behandlung chronischer und akuter Formen von durch IL-1 vermittelten
Erkrankungen, Apoptose, durch IGIF oder IFN-γ vermittelten Erkrankungen ebenso
wie von entzündlichen,
Autoimmun-, destruktiven Knochen-, proliferativen, infektiösen oder
degenerativen Erkrankungen.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Die
vorliegende Erfindung stellt neue Verbindungsklassen und pharmazeutisch
verträgliche
Derivate davon bereit, die als Inhibitoren von Caspasen, insbesondere
als Inhibitoren von ICE nützlich
sind. Diese Verbindungen können
allein oder in Kombination mit anderen therapeutischen oder vorbeugenden
Mitteln wie zum Beispiel Antibiotika, Immunmodulatoren oder anderen
entzündungshemmenden
Mitteln zur Behandlung oder zur Vorbeugung von durch IL-1, Apoptose,
IGIF oder IFN-γ vermittelten
Erkrankungen verwendet werden. Gemäß einer bevorzugten Ausführungsform
sind die erfindungsgemäßen Verbindungen
zur Bindung an das aktive Zentrum von ICE und zur Hemmung der Aktivität dieses
Enzyms in der Lage.
-
Es
ist eine Hauptaufgabe dieser Erfindung, neue Verbindungsklassen,
die durch die Formeln
dargestellt sind, bereitzustellen,
wobei
die unterschiedlichen Substituenten hierin beschrieben werden.
-
Diese
Erfindung stellt auch Zusammensetzungen, umfassend die durch die
Formeln (I) und (II) dargestellten Verbindungen, ihre Verwendung
zur Herstellung von Arzneimitteln zur Behandlung oder zur Prävention unterschiedlicher
Störungen
und Verfahren zur Herstellung dieser Verbindungen bereit.
-
DETAILLIERTE
BESCHREIBUNG DER ERFINDUNG
-
Um
die hierin beschriebene Erfindung verständlicher zu machen, wird die
folgende detaillierte Beschreibung dargelegt.
-
Die
folgenden Abkürzungen
und Definitionen werden die Anmeldung hindurch verwendet.
-
Abkürzungen
-
-
- Ac2O
- Essigsäureanhydrid
- n-Bu
- normal-Butyl
- DMF
- Dimethylformamid
- DIEA
- N,N-Diisopropylethylamin
- EDC
- 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
- Et2O
- Diethylether
- EtOAc
- Ethylacetat
- Fmoc
- 9-Fluorenylmethyloxycarbonyl
- HBTU
- O-Benzotriazol-1-yl-N,N,N,N'-tetramethyluroniumhexafluorphosphat
- HOBT
- 1-Hydroxybenzotriazolhydrat
- MeOH
- Methanol
- TFA
- Trifluoressigsäure
-
Der
Begriff „Caspase" bezeichnet ein Enzym,
das ein Mitglied der Enzymfamilie ist, die ICE einschließt (siehe
H. Hara, Natl. Acad. Sci., 94, Seiten 2007 – 2012 (1997)).
-
Die
Begriffe „HBV, „HCV" und „HGV" bezeichnen das Hepatitis-B-Virus,
Hepatitis-C-Virus
bzw. Hepatitis-G-Virus.
-
Der
Begriff „Ki" bezeichnet
ein numerisches Maß der
Wirksamkeit einer Verbindung beim Hemmen der Aktivität eines
Zielenzyms wie zum Beispiel ICE. Niedrige Ki-Werte
spiegeln höhere
Wirksamkeit wider. Der Ki-Wert ist ein durch
das Anpassen experimentell bestimmter Geschwindigkeitsdaten an enzymkinetische Standardgleichungen
abgeleiteter Wert (siehe I. H., Segel, Enzyme Kinetics, Wiley-Interscience,
1975).
-
Der
Begriff „Interferon
gamma induzierender Faktor" oder „IGIF" bezeichnet einen
Faktor, welcher zur Stimulierung der endogenen Produktion von IFN-γ in der Lage
ist.
-
Der
Begriff „Caspase-Inhibitor" bezeichnet eine
Verbindung, die in der Lage ist, eine nachweisbare Hemmung einer
oder mehrerer Caspasen zu zeigen. Der Begriff „ICE Inhibitor" bezeichnet eine
Verbindung, die in der Lage ist, eine nachweisbare Hemmung von ICE
und gegebenenfalls einer oder mehrerer zusätzlicher Caspasen zu zeigen.
Die Hemmung dieser Enzyme kann unter Verwendung der beschriebenen
und hierin durch Bezugnahme aufgenommenen Verfahren bestimmt werden.
Der Fachmann erkennt, dass ein in vivo-Enzyminhibitor nicht notwendigerweise
ein in vitro-Enzyminhibitor ist. Zum Beispiel zeigt eine Prodrugform
einer Verbindung üblicherweise
wenig oder keine Aktivität
bei in vitro-Analysen.
Derartige Prodrugformen können
im Patienten durch metabolische oder andere biochemische Verfahren
verändert
werden, um in vivo einen Enzyminhibitor bereitzustellen. Der Begriff „Cytokin" bezeichnet ein Molekül, welches
Wechselwirkungen zwischen Zellen vermittelt.
-
Der
Begriff „Zustand" bezeichnet jegliche
Erkrankung, Störung
oder Wirkung, die schädliche
biologische Folgen bei einem Untersuchungsobjekt erzeugt.
-
Der
Begriff „Untersuchungsobjekt
bzw. Individuum" bezeichnet
ein Tier oder ein oder mehrere Zellen, die von einem Tier stammen.
Vorzugsweise ist das Tier ein Säuger,
am meisten bevorzugt ein Mensch. Die Zellen können in jeglicher Form sein,
einschließlich
in Geweben zurückgehaltener
Zellen, Zellhaufen, immortalisierter Zellen, transfizierter oder
transformierter Zellen und Zellen, die von einem Tier stammen, das
physisch oder phänotypisch
verändert
worden ist, sind aber nicht darauf beschränkt.
-
Der
Begriff „Patient" bezeichnet, wie
in dieser Anmeldung verwendet, jeglichen Säuger, vorzugsweise Menschen.
-
Der
Begriff „Alkyl" bezeichnet einen
geradkettigen oder verzweigten, gesättigten aliphatischen Kohlenwasserstoff,
der 1 bis 6 Kohlenstoffatome enthält.
-
Der
Begriff „Alkenyl" bezeichnet einen
geradkettigen oder verzweigten, ungesättigten Kohlenwasserstoff,
der 2 bis 6 Kohlenstoffatome und mindestens eine Doppelbindung enthält.
-
Der
Begriff „Alkinyl" bezeichnet einen
geradkettigen oder verzweigten, ungesättigten Kohlenwasserstoff,
der 2 bis 6 Kohlenstoffatome und mindestens eine Dreifachbindung
enthält.
-
Der
Begriff „Cycloalkyl" bezeichnet ein mono-
oder polycyclisches, nicht aromatisches Kohlenwasserstoffringsystem,
das gegebenenfalls ungesättigte
Bindungen in dem Ringsystem enthält.
Beispiele schließen Cyclohexyl,
Adamantyl und Norbornyl ein.
-
Der
Begriff „Aryl" bezeichnet ein mono-
oder polycyclisches Ringsystem, das 6, 10, 12 oder 14 Kohlenstoffe
enthält,
wobei mindestens ein Ring des Ringsystems aromatisch ist. Die Arylreste
dieser Erfindung sind gegebenenfalls einzeln oder mehrfach mit R17 substituiert. Beispiele von Arylringsystemen
schließen
Phenyl, Naphthyl und Tetrahydronaphthyl ein.
-
Der
Begriff „Heteroaryl" bezeichnet ein mono-
oder polycyclisches Ringsystem, das 1 bis 15 Kohlenstoffatome und
1 bis 4 Heteroatome enthält,
und wobei mindestens ein Ring des Ringsystems aromatisch ist. Heteroatome
sind Schwefel, Stickstoff oder Sauerstoff. Die Heteroarylreste dieser
Erfindung sind gegebenenfalls einzeln oder mehrfach mit R17 substituiert.
-
Der
Begriff „heterocyclisch" bezeichnet ein mono-
oder polycyclisches Ringsystem, das 1 bis 15 Kohlenstoffatome und
1 bis 4 Heteroatome enthält,
wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen
enthalten kann, aber nicht aromatisch ist. Heteroatome sind unabhängig voneinander
Schwefel, Stickstoff oder Sauerstoff.
-
Der
Begriff „Alkylaryl" bezeichnet einen
Alkylrest, wobei ein oder mehrere Wasserstoffatome des Alkylrestes
durch ein oder mehrere Arylreste ersetzt sind.
-
Der
Begriff „Alkylheteroaryl" bezeichnet einen
Alkylrest, wobei ein Wasserstoffatom des Alkylrestes durch einen
Heteroarylrest ersetzt ist.
-
Der
Begriff „substituiert" bezeichnet den Ersatz
eines Wasserstoffatoms in einer Verbindung durch einen Substituentenrest.
-
Der
Begriff „gerade
Kette" bezeichnet
eine zusammenhängende
nicht verzweigende Kette kovalent gebundener Atome. Die gerade Kette
kann substituiert sein, diese Substituenten sind aber kein Teil
der geraden Kette.
-
Der
Begriff „Aminosäure-Seitenkette" bezeichnet den an
das α-Kohlenstoffatom
gebundenen Substituenten einer natürlichen oder nicht natürlichen α-Aminosäure.
-
In
chemischen Formeln werden hierin Klammern verwendet, um den Zusammenhang
in Molekülen oder
Gruppen zu kennzeichnen. Insbesondere werden Klammern verwendet,
um anzuzeigen: 1) dass mehr als ein Atom oder Rest an ein bestimmtes
Atom gebunden ist; oder 2) einen Verzweigungspunkt (d. h. das Atom
unmittelbar vor der offenen Klammer ist sowohl an das Atom oder
den Rest in Klammern als auch an das Atom oder den Rest unmittelbar
nach der Klammer gebunden). Ein Beispiel für die erste Verwendungsart
ist „-N(Alkyl)
2",
was anzeigt, dass zwei Alkylreste an ein N-Atom gebunden sind. Ein
Beispiel für
die zweite Verwendungsart ist „-C(O)NH
2",
was anzeigt, dass sowohl eine Carbonylgruppe als auch eine Aminogruppe („NH
2")
an das angezeigte Kohlenstoffatom gebunden sind. Ein Rest „-C(O)NH
2" kann
auf andere Weisen dargestellt werden, einschließlich der folgenden Struktur:
-
Substituenten
können
in unterschiedlichen Formen dargestellt werden. Diese unterschiedlichen
Formen sind Fachleuten bekannt und können untereinander austauschbar
verwendet werden. Zum Beispiel kann ein Methylsubstituent an einem
Phenylring in jeglicher der folgenden Formen dargestellt werden:
-
Unterschiedliche
Formen von Substituenten, zum Beispiel Methyl, werden hierin untereinander
austauschbar verwendet.
-
Wo
es nötig
ist, werden andere Definitionen in der Beschreibung dargelegt.
-
Erfindungsgemäße Verbindungen
-
Die
Verbindungen einer erfindungsgemäßen Ausführungsform
(A) sind jene der Formel (I):
wobei:
Y
mit der Maßgabe, dass
wenn R
5 -OH ist, Y dann auch
m 0 oder 1 ist;
W -CH
2-, -(O)-, S(O)
2 oder
-S(O) – ist;
X
-C(H) -, -C(R
8)- oder
ist;
Z -CH
2-,
-O-, -S- oder -N(R
1)- ist, mit der Maßgabe, dass,
wenn Z -N(R
1)- ist, dann W -C(O)-, -S(O)
2- oder -S(O)- ist;
jeder Rest R
1 unabhängig
-H, -S(O)
2-CH
3,
ist;
R
2 -C(O)R
8, -C(O)C(O)R
8, -S(O)
2R
8, -S(O)R
8, -C(O)OR
8, -C(O)N(H)R
8, -S(O)
2N(H)-R
8, -S(O)N(H)-R
8, -C(O)C(O)N(H)R
8,
-C(O)CH=CHR
8, -C(O)CH
2OR
8, -C(O)CH
2N(H)R
8, -C(O)N(R
8)
2, -S(O)
2N(R
8)
2, -S(O)N(R
8)
2, -(O)C(O)N(R
8)
2, -C(O)CH
2N(R
8)
2,
-CH
2-R
8, -CH
2-Alkenyl-R
8 oder
-CH
2-Alkinyl-R
8 ist;
R
3 -H, eine Aminosäure-Seitenkette,
jeder
Rest R
4 unabhängig -OH, -F, -Cl, -Br, -I,
-NO
2, -CN, -NH
2,
-CO
2H, -C(O)NH
2,
-N(H)C(O)H, -N(H)C(O)NH
2, -Alkyl, -Cycloalkyl,
-Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)
2,
-C(O)N(H)Alkyl, -C(O)N(Alkyl)
2, -N(H)C(O)Alkyl,
-N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)
2,
-S-Alkyl, -S(O)
2Alkyl, -S(O)Alkyl, -C(O)Alkyl,
-CH
2NH
2, -CH
2N(H)Alkyl, -CH
2N(Alkyl)
2 oder -N(H)C(O)O-Alkyl ist;
R
5 -OH, -OR
8 oder
-N(H)OH ist;
R
6 -H, -CH
2OR
9, -CH
2SR
10, -CH
2N(H)R
9, -CH
2N(R
9)R
11, -C(H)N
2, -CH
2F, -CH
2Cl, -C(O)N(R
11)
2, -R
13 oder -R
14 ist;
jeder Rest R
8 unabhängig -Alkyl,
-Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, -Alkylcycloalkyl,
-Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist;
R
9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl,
-C(O)Alkylheteroaryl, -Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl
oder -P(O)(R
15)
2 ist;
R
10-Alkylaryl oder -Alkylheteroaryl ist;
jeder
Rest R
11 unabhängig -H, -Alkyl, -Aryl, -Heteroaryl,
-Cycloalkyl, -Alkylaryl oder -Alkylheteroaryl ist;
R
13 -Alkylaryl oder -Alkylheteroaryl ist;
R
14 wobei
Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls
durch -R
17 ersetzt ist, und jedes Wasserstoffatom
in (ii), (iii) und (iv) gegebenenfalls durch -R
17,
-R
18 oder -Alkyl- R
18 ersetzt
ist;
jeder Rest R
15 unabhängig -H,
-OH, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, Alkylheteroaryl,
-O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroarylist;
jeder
Rest R
17 unabhängig -OH, -F, -Cl, -Br, -I,
-NO
2, -CN, -NH
2,
-CO
2H, -C(O)NH
2,
-N(H)C(O)H, -N(H)C(O)NH
2, -SO
2NH
2, -C(O)H, -Alkyl, -Cycloalkyl, -Perfluoralkyl,
-O-Alkyl, -N(H)Alkyl,
-N(Alkyl)
2, -CO
2Alkyl,
-C(O)N(H)Alkyl, -C(O)N(Alkyl)
2, -N(H)C(O)Alkyl,
-N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)
2,
-S(O)
2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)
2N(Alkyl)
2, -S(O)N(Alkyl)
2, -S-Alkyl, -S(O)
2Alkyl,
-S(O)Alkyl oder -C(O)Alkyl ist; und
jeder Rest R
18 unabhängig -Aryl,
-Heteroaryl, -Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl,
-N(H)Aryl, -N(Aryl)
2, -N(H)Heteroaryl, -N(Heteroaryl)
2, -N(H)Alkylaryl, -N(Alkylaryl)
2, -N(H)Alkylheteroaryl,
-N(Alkylheteroaryl)
2, -S-Aryl, -S-Heteroaryl,
-S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl,
-C(O)Alkylheteroaryl, -CO
2Aryl, -CO
2Heteroaryl, -CO
2Alkylaryl, -CO
2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)
2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)
2, -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)
2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)
2, -S(O)
2-Aryl, -S(O)-Aryl, -S(O)
2-Heteroaryl,
-S(O)-Heteroaryl, -S(O)
2-Alkylaryl, -S(O)-Alkylaryl,
-S(O)
2-Alkylheteroaryl, -S(O)-Alkylheteroaryl,
-S(O)
2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)
2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)
2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)
2N(H)-Alkylheteroaryl,
-S(O)N(H)-Alkylheteroaryl, -S(O)
2N(Aryl)
2, -S(O)N(Aryl)
2, -S(O)
2N(Heteroaryl)
2,
-S(O)N(Heteroaryl)
2, -S(O)
2N(Alkylaryl)
2, -S(O)N(Alkylaryl)
2, -S(O)
2N(Alkylheteroaryl)
2, -S(O)N(Alkylheteroaryl)
2, N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl,
-N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)
2, -N(H)C(O)N(Heteroaryl)
2,
-N(H)C(O)N(Alkylaryl)
2 oder -N(H)C(O)N(Alkylheteroaryl)
2 ist;
jedes Heteroaryl ein mono- oder
polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und
1 bis 4 Heteroatome, ausgewählt
aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein
Ring des Ringsystems aromatisch ist;
jedes Heterocyclyl ein
mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome
und 1 bis 4 Heteroatome, ausgewählt
aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische
Ringsystem gegebenenfalls ungesättigte
Bindungen enthalten kann, aber nicht aromatisch ist; und
jedes
Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das
gegebenenfalls ungesättigte
Bindungen in dem Ringsystem enthalten kann.
-
Die
Verbindungen einer anderen erfindungsgemäßen Ausführungsform (B) sind jene der
Formel (II):
wobei
C ein Arylring
ist, wobei jedes an den C-Ring gebundene Wasserstoffatom gegebenenfalls
mit -R
4 substituiert ist; und
die anderen
Substituenten wie vorstehend in Ausführungsform (A) beschrieben
sind.
-
Die
Verbindungen von zwei anderen erfindungsgemäßen Ausführungsformen, (C) und (D),
sind jene der Formeln (I) bzw. (II), wobei:
Y (c), (d), (e)
oder (f) ist, wenn R
19 ein Wasserstoff ist:
und wenn
R
19 kein Wasserstoff ist, bilden R
19 und Y zusammen mit dem Stickstoffatom,
an welches sie gebunden sind, einen Ring (g):
R
7 -C(O)Alkyl,
-C(O)Cycloalkyl, -C(O)Alkenyl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl,
-C(O)Heterocyclus oder -C(O)Alkylheterocyclus ist;
jeder Rest
R
22 unabhängig -H, -Alkyl, -Aryl, -Heteroaryl,
-Cycloalkyl, -Alkylaryl oder -Alkylheteroaryl ist; und
die
anderen Substituenten wie vorstehend beschrieben sind.
-
Vorzugsweise:
-
- ist m 0;
- ist W -CH2- oder -C(O)-;
- ist X -C(H)- oder
- ist Z -CH2-;
- ist R5 -OH-;
- ist R6 -H- oder -R14;
- ist R7 -C(O)Alkyl;
- ist R8 Methyl, Ethyl, n-Propyl, Isopropyl,
Cyclopentyl, Phenethyl oder Benzyl; oder
- ist R9 -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl,
-C(O)Alkylheteroaryl, -Alkylaryl, -Alkylheteroaryl, -Aryl oder -Heteroaryl;
ist Q O;
- ist R14 (i) substituiert mit -O-Alkyl,
-F oder -Cl oder (ii) substituiert mit Phenyl; oder ist C ein Benzoring,
wobei jede Wasserstoffbindung an den Ring gegebenenfalls durch -R4 ersetzt ist;
Stärker bevorzugt ist R6-H.
-
In
jeder der vorstehenden Ausführungsformen
sind die bevorzugten Formen der Formel (I) jene, wobei:
Z -CH
2- ist, W -C(O)- ist und X -C(H)- ist (Ia);
Z
-CH
2- ist, W -CH
2-
ist und X -C(H)- ist (Ib),
Z -CH
2-
ist, W -C(O)- ist und X
ist (Ic); oder
Z -CH
2- ist, W -CH
2- ist
und X
ist (Id):
und die
anderen Substituenten wie vorstehend beschrieben sind.
-
In
jeder der vorstehenden Ausführungsformen
sind die bevorzugten Formen der Formel (II) jene, wobei:
W
-C(O)- ist und X -C(H)- ist (IIa);
W -CH
2-
ist und X -C(H)- ist (IIb),
W -C(O)- ist und X
ist (IIc); oder
W -CH
2- ist und X
ist (IId); und
C ein
Benzoring ist, wobei jeder an den Ring gebundene Wasserstoff gegebenenfalls
durch – R
4 substituiert ist;
und die
anderen Substituenten wie vorstehend beschrieben sind.
-
Die
Verbindungen einer bevorzugten erfindungsgemäßen Ausführungsform (E) sind jene der
Formel (I):
wobei:
Y
m 0 oder 1 ist;
W -CH
2-, -C(O)-, S(O)
2 oder
-S(O)- ist;
X -C(H) -, -C(R
8)- oder
ist;
Z -CH
2-,
-O-, -S- oder -N(R
1)- ist, mit der Maßgabe, dass,
wenn Z -N(R
1)- ist, darin W -C(O)-, -S(O)
2- oder -S(O)- ist;
jeder Rest R
1 unabhängig
-H, -S(O)
2-CH
3,
R
2 -C(O)R
8, -C(O)C(O)R
8, -S(O)
2R
8, -S(O)R
8, -C(O)OR
8, -C(O)N(H)R
8, -S(O)
2N(H)-R
8, -S(O)N(H)-R
8, -C(O)C(O)N(H)R
8,
-C(O)CH=CHR
8, -C(O)CH
2OR
8, -C(O)CH
2N(H)R
8, -C(O)N(R
8)
2, -S(O)
2N(R
8)
2, -S(O)N(R
8)
2, -C(O)C(O)N(R
8)
2, -C(O)CH
2N(R
8)
2,
-CH
2-R
8, -CH
2-Alkenyl-R
8 oder
-CH
2-Alkinyl-R
8 ist;
R
3-H, eine Aminosäure-Seitenkette,
jeder
Rest R
4 unabhängig -OH, -F, -Cl, -Br, -I,
-NO
2, -ON, -NH
2,
-CO
2H, -C(O)NH
2,
-N(H)C(O)H, -N(H)C(O)NH
2, -Alkyl, -Cycloalkyl,
-Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)
2,
-C(O)N(H)Alkyl, -C(O)N(Alkyl)
2, -N(H)C(O)Alkyl,
-N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)
2,
-S-Alkyl, -S(O)
2Alkyl, -S(O)Alkyl, -C(O)Alkyl,
-CH
2NH
2, -CH
2N(H)Alkyl, -CH
2N(Alkyl)
2 oder -N(H)C(O)O-Alkyl ist;
R
5 -OH, -OR
8, -N(H)OH
oder -N(H)SO
2R
8 ist;
R
6 -H, -CH
2OR
9, -CH
2SR
10, -CH
2N(H)R
9, -OH
2N(R
9)R
11, -C(H)N
2, -CH
2F, -CH
2Cl, -CH
2Br, -CH
2I, -O(O)N(R
11)
2, -R
13 oder -R
14 ist;
jeder Rest R
8 unabhängig -Alkyl,
-Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, -Alkylcycloalkyl,
-Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist;
R
9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl,
-C(O)Alkylheteroaryl, -Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl
oder -P(O)(R
15)
2 ist;
R
10-Alkylaryl oder -Alkylheteroaryl ist;
jeder
Rest R
11 unabhängig -H, -Alkyl, -Aryl, -Heteroaryl,
-Cycloalkyl, -Alkylaryl oder -Alkylheteroaryl ist;
R
13-Alkylaryl oder Alkylheteroaryl ist;
R
14 wobei
Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls
durch -R
17 ersetzt ist, und jedes Wasserstoffatom
in (ii), (iii) und (iv) gegebenenfalls durch -R
17,
-R
18 oder -Alkyl-R
18 ersetzt
ist;
jeder Rest R
15 unabhängig -H,
-OH, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, -Alkylheteroaryl,
-O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl
ist;
jeder Rest R
17 unabhängig -OH,
-F, -Cl, -Br, -I, -NO
2, -CN, -NH
2, -CO
2H, -C(O)NH
2, -N(H)C(O)H, -N(H)C(O)NH
2, -SO
2NH
2, -C(O)H, -Alkyl,
-Cycloalkyl, -Perfluoralkyl, -O-Alkyl,
-N(H)Alkyl, -N(Alkyl)
2, -CO
2Alkyl,
-C(O)N(H)Alkyl, -O(O)N(Alkyl)
2, -N(H)C(O)Alkyl,
-N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)
2,
-S(O)
2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)
2N(Alkyl)
2, -S(O)N(Alkyl)
2, -S-Alkyl, -S(O)
2Alkyl,
-S(O)Alkyl oder -C(O)Alkyl ist; und
jeder Rest R
18 unabhängig -Aryl,
-Heteroaryl, -Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl,
-N(H)Aryl, -N(Aryl)
2, -N(H)Heteroaryl, -N(Heteroaryl)
2, -N(H)Alkylaryl, -N(Alkylaryl)
2, -N(H)Alkylheteroaryl,
-N(Alkylheteroaryl)
2, -S-Aryl, -S-Heteroaryl,
-S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl,
-C(O)Alkylheteroaryl, -CO
2Aryl, -CO
2Heteroaryl, -CO
2Alkylaryl, -CO
2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)
2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)
2, -C(O)N(H)Alkylaryl), -C(O)N(Alkylaryl)
2, -O(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)
2, -S(O)
2Aryl, -S(O)-Aryl, -S(O)
2-Heteroaryl,
-S(O)-Heteroaryl, -S(O)
2-Alkylaryl, -S(O)-Alkylaryl,
-S(O)
2-Alkylheteroaryl, -S(O)-Alkylheteroaryl,
-S(O)
2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)
2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)
2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)
2N(H)- Alkylheteroaryl,
-S(O)N(H)-Alkylheteroaryl, -S(O)
2N(Aryl)
2, -S(O)N(Aryl)
2, -S(O)
2N(Heteroaryl)
2,
-S(O)N(Heteroaryl)
2, -S(O)
2N(Alkylaryl)
2, -S(O)N(Alkylaryl)
2,
-S(O)
2N(Alkylheteroaryl)
2, -S(O)N(Alkylheteroaryl)
2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl,
-N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)
2, -N(H)C(O)N(Heteroaryl)
2,
-N(H)C(O)N(Alkylaryl)
2 oder -N(H)C(O)N(Alkylheteroaryl)
2 ist;
jedes Heteroaryl ein mono- oder
polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und
1 bis 4 Heteroatome, ausgewählt
aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein
Ring des Ringsystems aromatisch ist;
jedes Heterocyclyl ein
mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome
und 1 bis 4 Heteroatome, ausgewählt
aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische
Ringsystem gegebenenfalls ungesättigte
Bindungen enthalten kann, aber nicht aromatisch ist; und
jedes
Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das
gegebenenfalls ungesättigte
Bindungen in dem Ringsystem enthalten kann.
-
Die
Verbindungen einer anderen bevorzugten erfindungsgemäßen Ausführungsform
(F) sind jene der Formel (II):
wobei
C ein Arylring
ist, wobei jedes an den C-Ring gebundene Wasserstoffatom gegebenenfalls
mit -R
4 substituiert ist; und
die anderen
Substituenten wie vorstehend in Ausführungsform (E) beschrieben
sind.
-
Die
Verbindungen von zwei anderen erfindungsgemäßen Ausführungsformen, (G) und (H),
sind jene der Formeln (I) bzw. (II), wobei:
Y (c), (d), (e)
oder (f) ist:
R
7 -C(O)Alkyl, -C(O)Cycloalkyl, -C(O)Alkenyl,
-C(O)Alkylaryl, -C(O)Alkylheteroaryl, -C(O)-Heterocyclus oder -C(O)-Alkylheterocyclus
ist; und die anderen Substituenten sind, wie vorstehend beschrieben.
-
Vorzugsweise:
-
- ist m 0;
- ist W -CH2- oder -C(O)-;
- ist X -C(H) – oder
- ist Z -CH2-;
- ist R5 -OH-;
- ist R6 -H, -R14,
-CH2OR9 oder -CH2F;
- ist R7 -C(O)Alkyl;
- ist R8 Methyl, Ethyl, n-Propyl, Isopropyl,
Cyclopentyl, Phenethyl oder Benzyl;
- ist R9 C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl,
-C(O)Alkylheteroaryl, -Alkylaryl, -Alkylheteroaryl -Aryl oder -Heteroaryl;
- ist Q O;
- ist R14 mit -O-Alkyl, -F oder -Cl substituiert,
oder (ii) mit Phenyl substituiert;
- ist C ein Benzoring, wobei jede an den Ring gebundene Wasserstoffatom
gegebenenfalls durch -R4 ersetzt ist;
- ist R1: ist R3 -H, eine Aminosäureseitenkette,
- Stärker
bevorzugt
ist R2: ist R3: ist R6 -H.
-
In
den Ausführungsformen
ist (C), (D), (G) oder (H) Y:
und V ist vorzugsweise:
-
In
jeder der vorstehenden Ausführungsformen
sind die bevorzugten Formen der Formel (I) jene, wobei:
Z -CH
2- ist, W -C(O)- ist und X -C(H)- ist (Ia);
Z
-CH
2- ist, W -CH
2-
ist und X -C(H)- ist (Ib);
Z -CH
2-
ist, W -C(O)- ist und X
ist (Ic); oder
Z -CH
2- ist, W -CH
2- ist
und X
ist (Id):
und die
anderen Substituenten wie vorstehend beschrieben sind.
-
Stärker bevorzugte
Formen der Formel (I) sind wie folgt:
-
In
jeder der vorstehenden Ausführungsformen
sind die bevorzugten Formen der Formel (II) jene, wobei:
W
-C(O)- ist und X -C(H)- ist (IIa),
W -CH
2-
ist und X -C(H)- ist (IIb),
W -C(O)- ist und X
ist (IIc) oder
W -CH
2- ist und X
ist(IId); und
C ein
Benzoring ist, wobei jedes an den Ring gebundene Wasserstoffatom
gegebenenfalls mit R
4 substituiert ist:
und die
anderen Substituenten wie vorstehend beschrieben sind.
-
Besondere
erfindungsgemäße Verbindungen
schließen
ein, sind
aber nicht darauf beschränkt.
-
Die
erfindungsgemäßen Verbindungen
können
ein oder mehrere „asymmetrische" Kohlenstoffatome enthalten
und können
daher als Racemate und racemische Gemische, einzelne Enantiomere,
diastereomere Gemische und individuelle Diastereomere auftreten.
Jeder stereogene Kohlenstoff kann in der R- oder S-Konfiguration
sein. Obwohl die in dieser Anmeldung beispielhaft erläuterten
spezifischen Verbindungen und Gerüste in einer bestimmten stereochemischen
Konfiguration beschrieben werden können, werden auch Verbindungen
und Gerüste,
die entweder die entgegengesetzte Stereochemie an jedem vorgegebenen
chiralen Zentrum oder Gemische davon aufweisen, vorgesehen.
-
Alle
derartigen isomeren Formen dieser Verbindungen sind, ebenso wie
pharmazeutisch verträgliche Derivate
davon, ausdrücklich
in der vorliegenden Erfindung eingeschlossen.
-
Der
Begriff „pharmazeutisch
verträgliches
Derivat" bedeutet
jedes pharmazeutisch verträgliche
Salz, jedes pharmazeutisch verträglichen
Ester oder das Salz eines derartigen Esters einer erfindungsgemäßen Verbindung
oder jeder anderen Verbindung, welche nach Verabreichung an einen
Empfänger
zur Bereitstellung (direkt oder indirekt) einer erfindungsgemäßen Verbindung
oder eines aktiven anti-ICE-Metaboliten oder -Restes davon in der
Lage ist.
-
Pharmazeutisch
verträgliche
Salze der erfindungsgemäßen Verbindungen
schließen
zum Beispiel jene ein, die aus pharmazeutisch verträglichen
anorganischen und organischen Säuren
und Basen abgeleitet sind. Beispiele geeigneter Säuren schließen Salz-,
Bromwasserstoff-, Schwefel-, Salpeter-, Perchlor-, Fumar-, Malein-,
Phosphor-, Glycol-, Milch-, Salicyl-, Bernstein-, Toluol-p-sulfon-,
Wein-, Essig-, Citronen-, Methansulfon-, Ameisen-, Benzoe-, Malon-,
Naphthalin-2-sulfon- und Benzolsulfonsäuren ein. Andere Säuren wie
zum Beispiel Oxalsäure,
obwohl pharmazeutisch selbst nicht verträglich, können bei der Herstellung von
Salzen als nützliche
Zwischenprodukte für
das Erhalten der erfindungsgemäßen Verbindungen
und ihrer pharmazeutisch verträglichen
Säureadditionssalze
verwendet werden. Von geeigneten Basen abgeleitete Salze schließen Alkalimetall-
(z. B. Natrium), Erdalkalimetall- (z. B. Magnesium), Ammonium- und
N(C1-4-Alkyl)4 +- Salze ein.
-
Diese
Erfindung sieht auch die „Quaternisierung" aller Reste der
hierin offenbarten Verbindungen, welche basischen Stickstoff enthalten,
vor. Der basische Stickstoff kann mit allen dem Fachmann bekannten
Mitteln quaternisiert werden, einschließlich zum Beispiel Niederalkylhalogeniden
wie zum Beispiel Methyl-, Ethyl-, Propyl- und Butylchloriden, -bromiden
und -iodiden; Dialkylsulfaten, einschließlich Dimethyl-, Diethyl-,
Dibutyl- und Diamylsulfaten; langkettigen Halogeniden wie zum Beispiel
Decyl-, Lauryl-, Myristyl- und Stearylchloriden, -bromiden und -iodiden;
und Aralkylhalogeniden, einschließlich Benzyl- und Phenethylbromiden.
Wasser- oder öllösliche oder
dispergierbare Produkte können
durch eine derartige Quaternisierung erhalten werden.
-
Wenn
mehrfach substituiert, kann jeder Substituent unabhängig von
jedem anderen Substituenten genommen werden, solange die Kombination
von Substituenten zur Bildung einer stabilen Verbindung führt.
-
In
dieser Erfindung sind nur jene Kombinationen von Substituenten und
Variablen vorgesehen, die zur Bildung stabiler Verbindungen führen. Wie
hierin verwendet, bezeichnet der Begriff „stabil" Verbindungen, die ausreichende Stabilität besitzen,
um die Herstellung und Verabreichung an einen Säuger mit auf dem Fachgebiet
bekannten Verfahren zu ermöglichen.
Typischerweise sind derartige Verbindungen bei einer Temperatur von
40 °C oder
weniger in Gegenwart oder Abwesenheit von Feuchtigkeit oder anderen
chemisch reaktiven Bedingungen mindestens eine Woche lang stabil.
-
Bevorzugte
erfindungsgemäße Verbindungen
können
nach oraler Verabreichung durch den Blutstrom der Patienten leicht
absorbiert werden. Diese orale Verfügbarkeit macht derartige Verbindungen
zu exzellenten Mitteln für
oral verabreichte Behandlungs- und Präventionsstrategien gegen durch
IL-1, Apoptose, IGIF oder IFN-γ vermittelte
Erkrankungen.
-
Es
sollte selbstverständlich
sein, dass die erfindungsgemäßen Verbindungen,
abhängig
von den Bedingungen, einschließlich
der Wahl des Lösungsmittels,
pH-Werts und anderer, dem Fachmann bekannten Bedingungen, in verschiedenen
Gleichgewichtsformen vorkommen können.
Alle derartigen Formen dieser Verbindungen sind ausdrücklich in
der vorliegenden Erfindung eingeschlossen. Insbesondere können viele
der erfindungsgemäßen Verbindungen,
besonders jene, welche in Y Aldehyd- oder Ketongruppen und Carbonsäuregruppen
enthalten, Hemiacetal- oder Hemiketalformen oder hydratisierte Formen
annehmen. Zum Beispiel sind Verbindungen der Ausführungsform
(A) in einer Hemiacetal- oder Hemiketalform, wenn Y
ist.
-
Abhängig von
der Wahl des Lösungsmittels
und anderen dem Fachmann bekannten Bedingungen, können erfindungsgemäße Verbindungen
auch hydratisierte Formen, Acyloxyketal-, Acyloxyacetal-, Ketal-, Acetal-
oder Enolformen annehmen. Zum Beispiel liegen erfindungsgemäße Verbindungen
in hydratisierten Formen vor, wenn Y
in Acyloxyketal- oder Acylacetalformen,
wenn Y
in Ketal- oder Acetalformen,
wenn Y
und in Enolformen, wenn Y
-
Zusätzlich sollte
es selbstverständlich
sein, dass die Gleichgewichtsformen der erfindungsgemäßen Verbindungen
tautomere Formen enthalten können.
Alle derartigen Formen dieser Verbindungen sind ausdrücklich in
der vorliegenden Erfindung eingeschlossen.
-
Die
Verbindungen der Formeln (I) und (II) können unter Verwendung herkömmlicher
Verfahren synthetisiert werden. Vorteilhafterweise werden diese
Verbindungen zweckmäßigerweise
aus leicht erhältlichen
Ausgangsmaterialien synthetisiert.
-
Die
erfindungsgemäßen Verbindungen
gehören
zu den am leichtesten synthetisierten bekannten Caspase-Inhibitoren.
Viele der vorher beschriebenen Caspase- oder ICE-Inhibitoren enthalten vier oder mehr
chirale Zentren und zahlreiche Peptidbindungen. Die relative Leichtigkeit,
mit der die erfindungsgemäßen Verbindungen
synthetisiert werden können,
stellt einen Vorteil bei der Massenproduktion dieser Verbindungen
dar.
-
Zum
Beispiel können
die erfindungsgemäßen Verbindungen
unter Verwendung der hierin beschriebenen Verfahren hergestellt
werden. Für
den erfahrenen Praktiker ist es ersichtlich, dass diese Verfahren
nicht die einzigen Wege sind, durch welche die beschriebenen und
in dieser Anmeldung beanspruchten Verbindungen synthetisiert werden
können.
Fachleuten werden weitere Verfahren offensichtlich sein. Zusätzlich können die
unterschiedlichen hierin beschriebenen synthetischen Stufen in einer
wechselnden Reihenfolge oder Ordnung durchgeführt werden, um die gewünschten
Verbindungen zu geben.
-
Es
sollte selbstverständlich
sein, dass die erfindungsgemäßen Verbindungen
durch geeignete Funktionalitäten
modifiziert werden können,
um selektive biologische Eigenschaften zu verstärken. Derartige Modifikationen
sind auf dem Fachgebiet bekannt und schließen jene ein, welche die biologische
Penetration in ein vorgegebenes biologisches System (z. B. Blut,
lymphatisches System, Zentralnervensystem) steigern, die orale Verfügbarkeit
steigern, die Löslichkeit,
um die Verabreichung durch Injektion zu ermöglichen, steigern, den Metabolismus ändern und
die Ausscheidungsrate ändern.
Zusätzlich
können
die Verbindungen derartig in eine Prodrugform geändert werden, dass die gewünschte Verbindung
im Körper
des Patienten als das Ergebnis der Wirkung von metabolischen oder
anderen biochemischen Prozessen am Prodrug erzeugt wird. Derartige
Prodrugformen zeigen typischerweise wenig oder keine Aktivität in in
vitro-Analysen. Einige Beispiele für Prodrugformen schließen Ketal-,
Acetal-, Oxim-, Imin- und Hydrazonformen von Verbindungen ein, die
Keton- oder Aldehydreste enthalten, besonders wenn sie im Rest Y
der erfindungsgemäßen Verbindungen
auftreten. Andere Beispiele für
Prodrugformen schließen
die Hemiketal-, Hemiacetal-, Acyloxyketal-, Acyloxyacetal-, Ketal-,
Acetal- und Enolformen, die hierin beschrieben werden, ein.
-
Zusammensetzungen
und Verfahren
-
Die
erfindungsgemäßen Verbindungen
sind Caspase-Inhibitoren, besonders ICE-Inhibitoren. Folglich sind
diese Verbindungen in der Lage, auf Ereignisse in durch IL-1, Apoptose,
IGIF und IFN-γ vermittelten
Erkrankungen und dadurch auf die Endaktivität des Proteins bei entzündlichen
Erkrankungen, Autoimmunerkrankungen, destruktiven Knochenerkrankungen,
proliferativen Störungen,
infektiösen
Erkrankungen und degenerativen Erkrankungen abzuzielen und diese
zu hemmen. Zum Beispiel hemmen die erfindungsgemäßen Verbindungen die Umwandlung
des Vorläufer-IL-1β zu reifem
IL-1β durch
Hemmung von ICE. Weil ICE für
die Produktion von reifem IL-1 essentiell ist, blockiert die Hemmung
dieses Enzyms durch die Hemmung der Erzeugung von reifem IL-1 wirksam
den Beginn von durch IL-1 vermittelten physiologischen Wirkungen
und Symptomen wie zum Beispiel Entzündung. Durch Hemmung der IL-1β-Vorläuferaktivität funktionieren
die erfindungsgemäßen Verbindungen
daher wirksam als IL-1-Inhibitoren.
-
Auch
hemmen erfindungsgemäße Verbindungen
die Umwandlung von pro-IGIF in aktives reifes IGIF durch Hemmung
von ICE. Weil ICE essentiell für
die Erzeugung von reifem IGIF ist, blockiert die Hemmung von ICE
durch Hemmung der Produktion von reifem IGIF wirksam den Beginn
von durch IGIF vermittelten physiologischen Wirkungen und Symptomen.
IGIF wiederum ist essentiell für
die Erzeugung von IFN-γ.
Folglich blockiert ICE wirksam den Beginn von durch IFN-γ vermittelten
physiologischen Wirkungen und Symptomen durch Hemmung der Produktion
von reifem IGIF und daher der Produktion von IFN-γ.
-
Daher
werden die erfindungsgemäßen Arzneimittel
und Verfahren zur Kontrolle der Caspasaktivität in vivo nützlich sein. Die erfindungsgemäßen Zusammensetzungen
und Verfahren werden daher zur Kontrolle der Mengen an IL-1, IGIF
oder IFN-γ in
vivo und zur Behandlung oder Verringerung des Fortschreitens, des Schweregrads
oder der Wirkungen von durch IL-1, Apoptose, IGIF oder IFN-γ vermittelten
Zuständen,
einschließlich
Erkrankungen, Störungen
oder Wirkungen, nützlich
sein.
-
Erfindungsgemäße Arzneimittel
umfassen eine Verbindung der Formel (I) oder (II) oder ein pharmazeutisch
verträgliches
Salz davon und einen pharmazeutisch verträglichen Träger. Derartige Zusammensetzungen
können
gegebenenfalls ein zusätzliches
therapeutisches Mittel umfassen. Derartige Mittel schließen ein
entzündungshemmendes
Mittel, einen Matrix-Metalloprotease-Inhibitor,
einen Lipoxygenase-Inhibitor, einen Cytokin-Antagonisten, einen
Immunsuppressor, ein anti-Krebsmittel, ein anti-Virusmittel, ein
Cytokin, einen Wachstumsfaktor, einen Immunmodulator, ein Prostaglandin
oder eine antivaskuläre
Hyperproliferationsverbindung ein, sind aber nicht darauf beschränkt.
-
Der
Begriff „pharmazeutisch
verträglicher
Träger" bezeichnet einen
nicht toxischen Träger,
der zusammen mit einer erfindungsgemäßen Verbindung an einen Patienten
verabreicht werden kann und welcher die pharmakologische Aktivität davon
nicht zerstört.
-
Pharmazeutisch
verträgliche
Träger,
die in den erfindungsgemäßen Arzneimitteln
verwendet werden können,
schließen
Ionenaustauscher, Aluminiumoxid, Aluminumstearat, Lecithin, Serumproteine
wie zum Beispiel menschliches Serumalbumin, Pufferstoffe wie zum
Beispiel Phosphate, Glycin, Sorbinsäure, Kaliumsorbat, partielle
Glyceridgemische gesättigter
pflanzlicher Fettsäuren,
Wasser, Salze oder Elektrolyte wie zum Beispiel Protaminsulfat,
Dinatriumhydrogenphosphat, Kaliumhydrogenphosphat, Natriumchlorid,
Zinksalze, kolloidale Silicamasse, Magnesiumtrisilicat, Polyvinylpyrrolidon,
auf Cellulose basierende Stoffe, Polyethylenglycol, Natriumcarboxymethylcellulose,
Polyacrylate, Wachse, Polyethylen-Polyoxypropylen-Blockpolymere, Wollfett
und selbst-emulgierende Arzneistoff-Abgabe-Systeme (SEDDS) wie zum Beispiel α-Tocopherol,
Polyethylenglycol-1000-Succinat oder andere ähnliche polymere Abgabematrizen,
ein, sind aber nicht darauf beschränkt.
-
Bei
Arzneimitteln, umfassend nur eine Verbindung der Formel (I) oder
(II) als die aktive Komponente, können Verfahren zur Verabreichung
dieser Zusammensetzungen zusätzlich
den Schritt des Verabreichens eines zusätzlichen Mittels an das Individuum
umfassen. Derartige Mittel schließen entzündungshemmende Mittel, einen
Matrix-Metalloprotease-Inhibitor,
einen Lipoxygenase-Inhibitor, einen Cytokin-Antagonisten, einen Immunsuppressor,
ein anti-Krebsmittel, ein anti-Virusmittel, ein Cytokin, einen Wachstumsfaktor,
einen Immunmodulator, ein Prostaglandin oder eine antivaskuläre Hyperproliferationsverbindung
ein, sind aber nicht darauf beschränkt.
-
Der
Begriff „pharmazeutisch
wirksame Menge" bezeichnet
eine bei der Behandlung oder Verbesserung einer durch IL-1, Apoptose,
IGIF oder IFN-γ vermittelten
Erkrankung in einem Patienten wirksame Menge. Der Begriff „vorbeugend
wirksame Menge" bezeichnet
eine bei der Prävention
oder bei der wesentlichen Verminderung von durch IL-1, Apoptose,
IGIF oder IFN-γ vermittelten
Erkrankungen in einem Patienten wirksame Menge.
-
Die
erfindungsgemäßen Verbindungen
können
auf herkömmliche
Weise zur Kontrolle von IGIF- und IFN-γ-Mengen in vivo und zur Behandlung
von Erkrankungen oder zum Verringern des Fortschreitens oder des Schweregrads
der Wirkungen, die durch IL-1, Apoptose, IGIF oder IFN-γ vermittelt
werden, verwendet werden. Derartige Behandlungsverfahren, ihre Dosierungsmengen
und -anforderungen können
von Fachleuten aus verfügbaren
Verfahren und Techniken ausgewählt
werden.
-
Zum
Beispiel kann eine erfindungsgemäße Verbindung
mit einem pharmazeutisch verträglichen
Adjuvans zur Verabreichung an einen Patienten, der an einer durch
IL-1, Apoptose, IGIF oder IFN-γ vermittelten Erkrankung
leidet, auf eine pharmazeutisch verträgliche Weise und in einer Menge,
die wirksam den Schweregrad der Erkrankung vermindert, kombiniert
werden.
-
In
einer anderen Ausführungsform
können
die erfindungsgemäßen Verbindungen
in Zusammensetzungen und Verfahren zur Behandlung oder zum Schutz
von Individuen gegen durch IL-1, Apoptose, IGIF oder IFN-γ vermittelte
Erkrankungen über
lange Zeiträume
hinweg verwendet werden. Die Verbindungen können in derartigen Zusammensetzungen
entweder allein oder zusammen mit anderen erfindungsgemäßen Verbindungen
auf eine mit der herkömmlichen
Verwendung von Enzyminhibitoren in Arzneimitteln übereinstimmende Weise
verwendet werden. Zum Beispiel kann eine erfindungsgemäße Verbindung
mit pharmazeutisch verträglichen,
in Impfstoffen herkömmlich
angewendeten Adjuvantien kombiniert werden und in vorbeugend wirksamen
Mengen verabreicht werden, um Individuen über lange Zeiträume hinweg
gegen durch IL-1, Apoptose, IGIF oder IFN-γ vermittelte Erkrankungen zu
schützen.
-
Auch
können
die Verbindungen der Formel (I) oder (II) mit anderen Caspase- oder
ICE-Inhibitoren
mitverabreicht werden, um die Wirkung der Therapie oder Vorbeugung
gegen verschiedene durch IL-1, Apoptose, IGIF oder IFN-γ vermittelte
Erkrankungen zu steigern.
-
Zusätzlich können die
erfindungsgemäßen Verbindungen
in Kombination mit entweder herkömmlichen
entzündungshemmenden
Mitteln oder mit Matrix-Metalloprotease-Inhibitoren, Lipoxygenase-Inhibitoren und
Antagonisten von Cytokinen mit Ausnahme von IL-1β verwendet werden.
-
Die
erfindungsgemäßen Verbindungen
können
auch in Kombination mit Immunmodulatoren (z. B. Bropirimin, anti-Mensch-alpha-Interferon-Antikörper, IL-2,
GM-CSF, Methionin-Enkephalin,
Interferon-alpha, Diethyldithiocarbamat, Tumor-Nekrose-Faktor, Naltrexon
und EPO), mit Prostaglandinen oder mit anti-Virusmitteln (z. B.
3TC, polysulfatierte Polysaccharide, Ganciclovir, Ribavirin, Acyclovir,
alpha-Interferon, Trimethotrexat und Fancyclovir) oder Prodrugs
von diesen oder verwandten Verbindungen verabreicht werden, um durch
IL-1 vermittelte Erkrankungssymptome wie zum Beispiel Entzündung zu
vermeiden oder zu bekämpfen.
-
Wenn
die erfindungsgemäßen Verbindungen
in Kombinationstherapien mit anderen Mitteln verabreicht werden,
können
sie aufeinanderfolgend oder gleichzeitig an den Patienten verabreicht
werden. In einer anderen Ausführungsform
umfassen erfindungsgemäße Arzneimittel
oder vorbeugende Zusammensetzungen eine Kombination von einer Verbindung
der Formel (I) oder (II) und einem anderen therapeutischen oder
vorbeugenden Mittel.
-
Die
erfindungsgemäßen Arzneimittel
können
oral, parenteral, durch Sprühinhalation,
topisch, rektal, nasal, buccal; vaginal oder über ein implantiertes Reservoir
verabreicht werden. Wir bevorzugen die orale Verabreichung. Die
erfindungsgemäßen Arzneimittel
können
jegliche herkömmliche
nicht toxische pharmazeutisch verträgliche Träger, Adjuvantien oder Vehikel
enthalten. In einigen Fällen
kann der pH-Wert der Formulierung mit pharmazeutisch verträglichen
Säuren,
Basen oder Puffern eingestellt werden, um die Stabilität der formulierten
Verbindung oder ihrer Abgabeform zu erhöhen. Der Begriff parenteral,
wie hierin verwendet, schließt
subcutane, intracutane, intravenöse,
intramuskuläre,
intraartikuläre,
intrasynoviale, intrasternale, intrathecale, intraläsionale
und intrakranielle Injektions- oder Infusionsverfahren ein.
-
Die
Arzneimittel können
in der Form einer sterilen injizierbaren Herstellung sein, zum Beispiel
als eine sterile injizierbare wässrige
oder ölige
Suspension. Diese Suspension kann gemäß auf dem Fachgebiet bekannten
Verfahren unter Verwendung geeigneter Dispergier- oder Netzmittel (wie zum Beispiel Tween
80) und Suspensionsmittel formuliert werden. Auch kann die sterile
injizierbare Herstellung eine sterile injizierbare Lösung oder
Suspension in einem nicht toxischen, parenteral verträglichen
Verdünnungsmittel
oder Lösungsmittel,
zum Beispiel als Lösung
in 1,3-Butandiol, sein. Unter den verträglichen Vehikeln und Lösungsmitteln,
die angewendet werden können,
sind Mannit, Wasser, Ringersche Lösung und isotonische Natriumchloridlösung. Zusätzlich werden
sterile nicht flüssige Öle herkömmlich als
ein Lösungsmittel
oder Suspensionsmittel angewendet. Zu diesem Zweck kann jedes milde
nicht flüssige Öl angewendet
werden, einschließlich
synthetischer Mono- oder
Diglyceride. Fettsäuren
wie zum Beispiel Ölsäure und
ihre Glyceridderivate sind bei der Herstellung von Injektionsmitteln
nützlich,
wie auch natürliche
pharmazeutisch verträgliche Öle wie zum
Beispiel Olivenöl
oder Rizinusöl,
besonders in ihren polyoxyethylierten Versionen. Diese Öllösungen oder
-suspensionen können
auch ein langkettiges Alkoholverdünnungsmittel oder -dispergiermittel
wie zum Beispiel jene im Arzneibuch Helvetica beschriebenen oder
einen ähnlichen
Alkohol enthalten.
-
Die
erfindungsgemäßen Arzneimittel
können
oral in jeder oral verträglichen
Dosierungsform verabreicht werden, einschließlich Kapseln, Tabletten und
wässriger
Suspensionen und Lösungen,
sind aber nicht darauf beschränkt.
Im Fall von Tabletten zur oralen Verwendung schließen Träger, welche
gewöhnlich
verwendet werden, Lactose und Maisstärke ein. Typischerweise werden
auch Gleitmittel wie zum Beispiel Magnesiumstearat zugegeben. Zur
oralen Verabreichung in einer Kapselform schließen nützliche Verdünnungsmittel Lactose
und getrocknete Maisstärke
ein. Wenn wässrige
Suspensionen oral verabreicht werden, wird der Wirkstoff mit Emulgatoren
und Suspensionsmitteln kombiniert. Falls gewünscht, können bestimmte Süß- und/oder Geschmacks-
und/oder Farbstoffe zugegeben werden.
-
Die
erfindungsgemäßen Arzneimittel
können
auch in Form von Zäpfchen
zur rektalen Verabreichung verabreicht werden. Diese Zusammensetzungen
können
durch Mischen einer erfindungsgemäßen Verbindung mit einem geeigneten
nicht reizenden Exzipienten, welcher bei Raumtemperatur fest, bei
Rektaltemperatur aber flüssig
ist, und daher im Rektum schmelzen wird, um die Wirkstoffe freizusetzen,
hergestellt werden. Derartige Materialien schließen Kakaobutter, Bienenwachs
und Polyethylenglycole ein, sind aber nicht darauf beschränkt.
-
Die
topische Verabreichung der erfindungsgemäßen Arzneimittel ist besonders
nützlich,
wenn die gewünschte
Behandlung mit Arealen und Organen verbunden ist, die der topischen
Verabreichung leicht zugänglich
sind. Zur topischen Anwendung auf der Haut sollte das Arzneimittel
mit einer geeigneten Salbe formuliert werden, welche die Wirkstoffe
in einem Träger
suspendiert oder gelöst
enthält.
Träger
zur topischen Verabreichung der erfindungsgemäßen Verbindungen schließen Mineralöl, flüssiges Petroleum,
weißes
Petroleum, Propylenglycol, Polyoxyethylen-Polyoxypropylen-Verbindung,
emulgierendes Wachs und Wasser ein, sind aber nicht darauf beschränkt. In
einer anderen Ausführungsform
kann das Arzneimittel mit einer geeigneten Lotion oder Creme, welche
den Wirkstoff in einem Träger
suspendiert oder gelöst
enthält,
formuliert werden. Geeignete Träger
schließen
Mineralöl,
Sorbitanmonostearat, Polysorbat 60, Wachse der Cetylester, Cetearylalkohol,
2-Octyldodecanol,
Benzylalkohol und Wasser ein, sind aber nicht darauf beschränkt. Auch
können
die erfindungsgemäßen Arzneimittel
topisch im unteren Intestinaltrakt durch eine Rektalzäpfchenformulierung oder
in einer geeigneten Klistierformulierung angewendet werden. Auch
sind topisch verabreichte Transdermalpflaster in dieser Erfindung
eingeschlossen.
-
Die
erfindungsgemäßen Arzneimittel
können
durch ein Nasenaerosol oder durch Inhalation verabreicht werden.
Derartige Zusammensetzungen werden gemäß von auf dem Fachgebiet der
pharmazeutischen Formulierung wohlbekannter Verfahren hergestellt
und können
als Lösungen
in Salzlösung,
welche Benzylalkohol oder andere geeignete Konservierungsmittel,
Absorptionsförderer,
um die Bioverfügbarkeit
zu erhöhen, Fluorkohlenstoffe
und/oder andere im Fachgebiet bekannte solubilisierende oder dispergierende
Mittel anwenden, hergestellt werden.
-
Dosierungsmengen
der Wirkstoffverbindung zwischen etwa 0,01 und etwa 100 mg/kg Körpergewicht pro
Tag, vorzugsweise zwischen 0,5 und etwa 75 mg/kg Körpergewicht
pro Tag und am meisten bevorzugt zwischen etwa 1 und 50 mg/kg Körpergewicht
pro Tag sind bei einer Monotherapie zur Prävention und Behandlung von
durch IL-1, Apoptose, IGIF und IFN-γ vermittelten Erkrankungen nützlich,
einschließlich
entzündlicher Erkrankungen,
Autoimmunerkrankungen, destruktiver Knochenerkrankungen, proliferativer
Störungen, infektiöser Erkrankungen,
degenerativer Erkrankungen, nekrotischer Erkrankungen, entzündlicher
Peritonitis, Osteoarthritis, akuter Pankreatitis, chronischer Pankreatitis,
Asthma, Schocklunge, Glomerulonephritis, rheumatoider Arthritis,
systemischem Lupus erythematodes, Sclerodermia, chronischer Thyroiditis,
Morbus Basedow, Autoimmungastritis, insulinabhängigem Diabetes mellitus (Typ
I), autoimmunhämolytischer
Anämie,
Autoimmun-Neutropenie, Thrombozytopenie, chronischer aktiver Hepatitis,
Myasthenia gravis, entzündlicher
Darmerkrankung, Morbus Crohn, Psoriasis, Transplantat-Wirt-Erkrankung,
Osteoporose, mit multiplem Myelom verbundener Knochenerkrankungen,
akuter myelogener Leukämie,
chronischer myelogener Leukämie,
metastatischem Melanom, Kaposi-Sarkom, multiplem Myelom, Sepsis,
septischem Schock, Shigellose, Alzheimer-Krankheit, Parkinson-Krankheit, zerebraler
Ischämie,
myokardialer Ischämie,
spinaler Muskelatrophie, multipler Sklerose, mit AIDS verbundener
Enzephalitis, mit HIV verbundener Enzephalitis, Alterung, Alopecia, neurologischer
Schäden
aufgrund eines Schlaganfalls, ulzerativer Kolitis, traumatischer
Gehirnverletzung, Organtransplantatabstoßung, infektiöser Hepatitis,
juvenilem Diabetes, Lichen Planus, akuter Dermatomyositis, Ekzem,
primärer
Zirrhose, Uveitis, Behçet-Erkrankung,
atopischer Hauterkrankung, aregenerativer Anämie, aplastischer Anämie, amyotropher
Lateralsklerose, nephrotischem Syndrom und systemischer Erkrankungen oder
Erkrankungen mit in der Leber oder anderen Organen lokalisierten
Wirkungen, welche eine entzündliche oder
eine apoptotische Komponente aufweisen und durch übermäßige Alkoholaufnahme
mit der Nahrung oder durch Viren wie zum Beispiel HBV, HCV, HGV,
Gelbfieber-Virus, Denguefieber-Virus und japanischen Enzephalitis-Virus
verursacht werden.
-
Typischerweise
werden die erfindungsgemäßen Arzneimittel
von etwa ein- bis fünfmal
pro Tag verabreicht werden, oder in einer anderen Ausführungsform
als kontinuierliche Infusion. Eine derartige Verabreichung kann
zur Dauer- oder Akut-Therapie verwendet werden. Die Wirkstoffmenge,
die mit den Trägermaterialien
kombiniert werden kann, um eine Einzeldosierungsform zu erzeugen,
wird abhängig
vom behandelten Wirt und der besonderen Verabreichungsweise variieren.
Eine typische Herstellung wird von etwa 5 % bis etwa 95 % Wirkstoff
(Gew./Gew.) enthalten. Vorzugsweise enthalten derartige Herstellungen
von etwa 20 % bis etwa 80 % Wirkstoff.
-
Wenn
die erfindungsgemäßen Zusammensetzungen
eine Kombination einer Verbindung der Formel (I) oder (II) und ein
oder mehrere zusätzliche
therapeutische oder vorbeugende Mittel umfassen, sollten sowohl die
Verbindung als auch das zusätzliche
Mittel in Dosierungsmengen von zwischen etwa 10 % bis 80 % der normalerweise
bei einer Monotherapiestrategie verabreichten Dosierung vorliegen.
-
Nach
der Besserung des Zustands eines Patienten kann gegebenenfalls eine
Erhaltungsdosis einer erfindungsgemäßen Verbindung, Zusammensetzung
oder Kombination verabreicht werden. Anschließend kann die Dosierung oder
Frequenz der Verabreichung oder beides als eine Funktion der Symptome
auf eine Menge verringert werden, bei welcher der gebesserte Zustand
aufrecht erhalten wird, wenn die Symptome auf den gewünschten
Wert abgeschwächt
worden sind, sollte die Behandlung enden. Dennoch können Patienten zeitweilige
Behandlung auf einer Langzeitbasis nach jedem Wiederauftreten von
Erkrankungssymptomen benötigen.
-
Für den Fachmann
ist es selbstverständlich,
dass niedrigere oder höhere
Dosen als jene vorstehend vorgetragenen benötigt werden können. Spezielle
Dosierungs- und Behandlungsstrategien für jeden besonderen Patienten
werden von einer Vielfalt von Faktoren abhängen, einschließlich der
Aktivität
der angewendeten speziellen Verbindung, des Alters, des Körpergewichts,
des allgemeinen Gesundheitszustands, des Geschlechts, der Ernährung, der
Zeit der Verabreichung, der Ausscheidungsrate, der Arzneistoffkombination,
des Schweregrads und des Verlaufs der Erkrankung und der Erkrankungsveranlagung
des Patienten und der Beurteilung des behandelnden Arztes.
-
Durch
IL-1 oder Apoptose vermittelte Erkrankungen können durch die erfindungsgemäßen Verbindungen
behandelt oder vermieden werden. Derartige Erkrankungen schließen entzündliche
Erkrankungen, Autoimmunerkrankungen, destruktive Knochenerkrankungen,
proliferative Störungen,
infektiöse
Erkrankungen und degenerative Erkrankungen ein, sind aber nicht
darauf beschränkt.
-
Die
durch IL-1 oder Apoptose vermittelten entzündliche Erkrankungen, welche
behandelt oder vermieden werden können, schließen Osteoarthritis,
akute Pankreatitis, chronische Pankreatitis, Asthma, entzündliche
Peritonitis und Schocklunge ein, sind aber nicht darauf beschränkt. Vorzugsweise
ist die entzündliche
Erkrankung Osteoarthritis oder akute Pankreatitis.
-
Die
durch IL-1 oder Apoptose vermittelten Autoimmunerkrankungen, welche
behandelt oder vermieden werden können, schließen Glomerulonephritis,
rheumatoide Arthritis, systemischen Lupus erythematodes, Sclerodermia,
chronische Thyroiditis, Morbus Basedow, Autoimmungastritis, insulinabhängigen Diabetes mellitus
(Typ I), autoimmunhämolytische
Anämie,
Autoimmun-Neutropenie, Thrombozytopenie, chronisch aktive Hepatitis,
Myasthenia gravis, multiple Sklerose, entzündliche Darmerkrankung, Morbus
Crohn, Psoriasis und Transplantat-Wirt-Erkrankung ein, sind aber
nicht darauf beschränkt.
Vorzugsweise ist die Autoimmunerkrankung rheumatoide Arthritis,
entzündliche
Darmerkrankung, Morbus Crohn oder Psoriasis.
-
Durch
IL-1 oder Apoptose vermittelte destruktive Knochenerkrankungen,
welche behandelt oder vermieden werden können, schließen Osteoporose
und mit multiplem Myelom verbundene Knochenerkrankungen ein, sind
aber nicht darauf beschränkt.
-
Die
durch IL-1 oder Apoptose vermittelten proliferativen Erkrankungen,
welche behandelt oder vermieden werden können, schließen akute
myologene Leukämie,
chronische myologene Leukämie,
metastatisches Melanom, Kaposi-Sarkom und multiples Myelom ein,
sind aber nicht darauf beschränkt.
-
Die
durch IL-1 oder Apoptose vermittelten infektiösen Erkrankungen, welche behandelt
oder vermieden werden können,
schließen
Sepsis, septischen Schock und Shigellose ein, sind aber nicht darauf
beschränkt.
-
Die
durch IL-1 oder Apoptose vermittelten degenerativen oder nekrotischen
Erkrankungen, welche durch die erfindungsgemäßen Verbindungen behandelt
oder vermieden werden können,
schließen
Alzheimer-Krankheit, Parkinson-Krankheit, zerebrale Ischämie und
myokardiale Ischämie
ein, sind aber nicht darauf beschränkt. Vorzugsweise ist die degenerative
Erkrankung die Alzheimer-Krankheit.
-
Die
durch IL-1 oder Apoptose vermittelten degenerativen Erkrankungen,
welche durch die erfindungsgemäßen Verbindungen
behandelt oder vermieden werden können, schließen Alzheimer-Krankheit,
Parkinson-Krankheit, zerebrale Ischämie, myokardiale Ischämie, spinale
Muskelatrophie, multiple Sklerose, mit AIDS verbundene Enzephalitis,
mit HIV verbundene Enzephalitis, Alterung, Alopecia und neurologischen
Schäden aufgrund
eines Schlaganfalls ein, sind aber nicht darauf beschränkt.
-
Andere
Erkrankungen, welche eine entzündliche
oder apoptotische Komponente aufweisen, können durch die erfindungsgemäßen Verbindungen
behandelt oder vermieden werden. Derartige Erkrankungen können systemische
Erkrankungen oder Erkrankungen mit in der Leber oder anderen Organen
lokalisierten Wirkungen sein und können zum Beispiel aufgrund übermäßiger Alkoholaufnahme
mit der Nahrung oder durch Viren wie zum Beispiel HBV, HCV, HGV,
Gelbfieber-Virus, Dengue-Fieber-Virus und japanisches Enzephalitis-Virus
verursacht werden.
-
Durch
IGIF oder IFN-γ vermittelte
Erkrankungen können
auch durch die erfindungsgemäßen Verbindungen
behandelt oder vermieden werden. Derartige Erkrankungen schließen entzündliche,
infektiöse,
Autoimmun-, proliferative, neurodegenerative und nekrotische Zustände ein,
sind aber nicht darauf beschränkt. Durch
IGIF oder IFN-γ vermittelte
entzündliche
Erkrankungen, welche behandelt oder vermieden werden können, schließen Osteoarthritis,
akute Pankreatitis, chronische Pankreatitis, Asthma, rheumatoide
Arthritis, entzündliche
Darmerkrankung, Morbus Crohn, ulzerative Kolitis, zerebrale Ischämie, myokardiale
Ischämie
und Schocklunge ein, sind aber nicht darauf beschränkt. Vorzugsweise
ist die entzündliche
Erkrankung rheumatoide Arthritis, ulzerative Kolitis, Morbus Crohn,
Hepatitis oder Schocklunge.
-
Durch
IGIF oder IFN-γ vermittelte
infektiöse
Erkrankungen, welche behandelt oder vermieden werden können, schließen infektiöse Hepatitis,
Sepsis, septischen Schock und Shigellose ein, sind aber nicht darauf beschränkt.
-
Die
durch IGIF oder IFN-γ vermittelten
Autoimmunerkrankungen, welche behandelt oder vermieden werden können, schließen Glomerulonephritis,
systemischen Lupus erythematodes, Sclerodermia, chronische Thyroiditis,
Morbus Basedow, Autoimmungastritis, insulinabhängigen Diabetes mellitus (Typ
I), juvenilen Diabetes, autoimmunhämolytische Anämie, Autoimmun-Neutropenie,
Thrombozytopenie, Myasthenia gravis, multiple Sklerose, Psoriasis,
Lichen planus, Transplantat-Wirt-Erkrankung, akute Dermatomyositis,
Ekzem, primäre
Zirrhose, Hepatitis, Uveitis, Behçet-Erkrankung, atopische
Hauterkrankung, aregenerative Anämie, aplastische
Anämie,
amyotrophe Lateralsklerose und nephrotisches Syndrom ein, sind aber
nicht darauf beschränkt.
Vorzugsweise ist die Autoimmunerkrankung eine Glomerulonephritis,
insulinabhängiger
Diabetes mellitus (Typ I), juveniler Diabetes, Psoriasis, Transplantat-Wirt-Erkrankung
oder Hepatitis.
-
Stärker bevorzugte
Erkrankungen, welche durch die erfindungsgemäßen Verbindungen behandelt oder
vermieden werden können,
schließen
rheumatoide Arthritis, entzündliche
Darmerkrankung, einschließlich
Morbus Crohn und ulzerativer Kolitis, entzündliche Peritonitis, septischen
Schock, Pankreatitis, traumatische Gehirnverletzung, Organtransplantatabstoßung und
Asthma ein.
-
Folglich
stellt eine erfindungsgemäße Ausführungsform
die Verwendung jeder/jedes hierin beschriebenen Verbindung, Arzneimittels
oder Kombination und eines pharmazeutisch verträglichen Trägers zur Herstellung eines
Arzneimittels zur Behandlung oder Prävention einer durch IL-1 oder
Apoptose vermittelten Erkrankung bei einem Individuums bereit.
-
Eine
andere erfindungsgemäße Ausführungsform
stellt die Verwendung jeder/jedes hierin beschriebenen Verbindung,
Arzneimittels oder Kombination und eines pharmazeutisch verträglichen
Trägers
zur Herstellung eines Arzneimittels zur Senkung der IGIF-Produktion
bei einem Individuum bereit.
-
Eine
andere erfindungsgemäße Ausführungsform
stellt die Verwendung jeder/jedes hierin beschriebenen Verbindung,
Arzneimittels oder Kombination und eines pharmazeutisch verträglichen
Trägers
zur Herstellung eines Arzneimittels zur Senkung der IFN-γ Produktion
bei einem Individuum bereit.
-
Obwohl
diese Erfindung sich auf die Verwendung der hierin offenbarten Verbindungen
zur Prävention und
Behandlung von durch IL-1, Apoptose, IGIF und IFN-γ vermittelten
Erkrankungen konzentriert, können
die erfindungsgemäßen Verbindungen
auch als inhibitorische Mittel für
andere Cysteinproteasen verwendet werden.
-
Die
erfindungsgemäßen Verbindungen
sind auch als handelsübliche
Reagenzien nützlich,
welche wirksam an Caspasen oder andere Cysteinproteasen binden,
einschließlich,
aber nicht beschränkt
auf ICE. Als handelsübliche
Reagenzien können
die erfindungsgemäßen Verbindungen
und ihre Derivate verwendet werden, um die Proteolyse eines Zielpeptids
in biochemischen oder zellulären
Analysen für
ICE und ICE-Homologe zu blockieren, oder können derivatisiert werden,
um an ein stabiles Harz als angebundenes Substrat für Affinitätschromatogaphieanwendungen
zu binden. Diese und andere Verwendungen, welche handelsübliche Cysteinproteaseinhibitoren
kennzeichnen, werden für
Fachleute offensichtlich sein.
-
Damit
diese Erfindung besser verstanden wird, werden die folgenden Beispiele
dargelegt. Diese Beispiele sind nur zum Zweck der Veranschaulichung
und nicht auf irgendeine Weise als Beschränken des Umfangs der Erfindung
aufzufassen.
-
Allgemeine
Verfahren
-
Die
erfindungsgemäßen Verbindungen
können
in unterschiedlichen biologischen Analysen untersucht werden, einschließlich jener
in den Beispielen 2 – 4
beschriebenen.
-
Andere
Analysen, die verwendet werden können,
um die erfindungsgemäßen Verbindungen
zu untersuchen, sind in der PCT-Anmeldung PCT/US96/20843 offenbart,
veröffentlicht
am 26. Juni 1997 unter der Veröffentlichungsnr.
WO 97/22619, welche hierin durch Bezugnahme aufgenommen ist. Derartige
Analysen schließen
Bestimmungen der in vivo-Bioverfügbarkeit,
pharmakokinetische Untersuchungen in Maus, Hemmung von ICE-Homologen,
Hemmung von Apoptose, Akut-Analyse in vivo zur Wirksamkeit der Entzündungshemmung,
Messung von Blutwerten, IGIF-Analysen, peritoneale Carrageen-Entzündungsanalyse
bei der Maus und durch Typ II-Kollagen hervorgerufene Arthritis
ein.
-
Beispiel 1
-
Erfindungsgemäße Verbindungen
können
gemäß veröffentlichter
Verfahren, wie zum Beispiel die in Robl, J. A. et al., Bioorg. Med.
Chem. Lett. 4, Seiten 2055 – 2060
(1994) oder im Patent der Vereinigten Staaten Nr. 4,465,679 beschriebenen
Verfahren, welche hierin durch Bezugnahme aufgenommen sind, hergestellt
werden. Fachleute werden erkennen, dass derartige Verfahren modifiziert
werden können,
um die erfindungsgemäßen Verbindungen
zu erhalten.
-
Zum
Beispiel können
die durch die Formel (Ia) oder (Ib) dargestellten Verbindungen,
wie in Schema 1 beschrieben, hergestellt werden. Schema
1. Synthese von Analoga der Ausführungsformen
Ia und Ib
-
Zum
Beispiel können
die durch die Formel (Ic) oder (Id) dargestellten Verbindungen,
wie in Schema 2 beschrieben, hergestellt werden. Schema
2. Synthese von Analoga der Ausführungsform
Ic und Id
-
Zum
Beispiel können
die durch die Formel (IIa) oder (IIb) dargestellten Verbindungen,
wie in Schema 3 beschrieben, hergestellt werden. Schema
3. Synthese der Ausführungsformen
IIa und IIb
-
Zum
Beispiel können
die durch die Formel (IIc) oder (IId) dargestellten Verbindungen,
wie in Schema 4 beschrieben, hergestellt werden: Schema
4. Synthese der Ausführungsformen
IIc und IId
-
Durch
die Formel (I) oder (II) dargestellte Verbindungen, wobei R
19 und Y zusammen mit dem Stickstoffatom,
an das sie gebunden sind, einen Ring (g) bilden, können, wie
in Schema 5 beschrieben, hergestellt werden. Schema
5. Synthese von Analoga der Ausführungsform
(g)
-
Schema
5 beschreibt die Synthese von Verbindungen, wobei m 0 ist. Verbindungen,
wobei m 1 ist, können
durch ähnliche
Verfahren hergestellt werden. Das N-Alloc geschützte Amin kann mit anderen
Gruppen, die dem Fachmann bekannt sind, geschützt werden. Das Palladiumkupplungsverfahren
wird detaillierter in der PCT-Anmeldung PCT/US96/20843, Veröffentlichungsnr.
WO 97/22619, das hierin unter Bezugnahme aufgenommen ist, beschrieben.
-
Die
Schemata 1 – 5
beschreiben die Synthese bestimmter erfindungsgemäßer Ausführungsformen. Andere
Ausführungsformen
können
durch ähnliche
Verfahren hergestellt werden.
-
Beispiel 2
-
1. Enzym-Analyse mit im
UV sichtbarem Substrat
-
Diese
Analyse wird unter Verwendung eines Succinyl-Tyr-Val-Ala-Asp-p-Nitroanilid-Substrats durchgeführt. Die
Synthese von analogen Substraten wird von L. A. Reiter (Int. J.
Peptide Protein Res., 43, Seiten 87 – 96 (1994)) beschrieben. Das
Analysengemisch enthält:
65 μl Puffer
(10 mM Tris, 1 mM DTT, 0,1 % CHAPS, @pH 8,1)
10 μl ICE (50
nM Endkonzentration, um eine Geschwindigkeit von ~ 1 mOD/min zu
geben)
5 μl
DMSO/Inhibitorgemisch
20 μl
400 μM Substrat
(80 μM Endkonzentration)
100 μl Gesamtumsetzungsvolumen
-
Die
sichtbare ICE-Analyse wird in einer Mikrotiterplatte mit 96 Vertiefungen
durchgeführt.
Puffer, ICE und DMSO (falls Inhibitor vorliegt) werden zu den Vertiefungen
in der aufgelisteten Reihenfolge gegeben. Man lässt die Komponenten stehen,
um sie 15 Minuten lang, ausgehend von dem Zeitpunkt, bei dem alle
Komponenten in allen Vertiefungen vorliegen, bei Raumtemperatur
zu inkubieren. Das Mikrotiterplatten-Lesegerät ist eingestellt, dass es
bei 37 °C
inkubiert. Nach der 15 Minuten langen Inkubation wird Substrat direkt
in die Vertiefungen gegeben, und die Umsetzung wird durch das Folgen
der Freisetzung des Chromophors (pNA) bei 405 – 603 nm 20 Minuten lang bei
37 °C überwacht.
Eine lineare Anpassung der Daten wird durchgeführt, und die Geschwindigkeit
wird in mOD/min berechnet. DMSO liegt nur während der Experimente, die
Inhibitoren einbeziehen, vor, Puffer wird verwendet, um das Volumen
in den anderen Experimenten auf 100 μl zu bringen.
-
2. Enzymanalyse mit fluoreszierendem
Substrat
-
Diese
Analyse wird im Wesentlichen gemäß Thornberry
et al., Nature, 356, Seiten 768 – 774 (1992), unter Verwendung
von Substrat 17, auf das in dem Artikel Bezug genommen wird, durchgeführt. Das
Substrat ist: Acetyl-Tyr-Val-Ala-Asp-amino-4-methylcumarin (AMC). Die folgenden Komponenten
werden gemischt:
65 μl
Puffer (10 mM Tris, 1 mM DTT, 0,1 % CHAPS, pH 8,1)
10 μl ICE (2 – 10 nM
Endkonzentration)
5 μl
DMSO/Inhibitorlösung
20 μl 150 μM Substrat
(30 μM Endk.)
100 μl Gesamtumsetzungsvolumen
-
Die
Analyse wird in einer Mikrotiterplatte mit 96 Vertiefungen durchgeführt. Puffer
und ICE werden zu den Vertiefungen gegeben. Die Komponenten werden
stehen gelassen, um 15 Minuten lang bei 37°C in einer temperaturkontrollierten
Platte mit Vertiefungen zu inkubieren. Nach der 15 Minuten langen
Inkubation wird die Umsetzung durch direkte Zugabe des Substrats
in die Vertiefungen gestartet, und die Umsetzung wird durch Folgen
der Freisetzung des AMC-Fluorophors unter Verwendung einer Anregungswellenlänge von
380 nm und einer Emissionswellenlänge von 460 nm 30 Minuten lang
bei 37 °C überwacht.
Eine lineare Anpassung der Daten wird für jede Vertiefung durchgeführt, und
eine Geschwindigkeit wird in Fluoreszenzeinheiten pro Sekunde bestimmt.
-
Zur
Bestimmung der Enzym-Hemmungs-Konstanten (Ki)
oder der Art der Hemmung (kompetitiv, unkompetitiv oder nicht kompetitiv)
werden die Geschwindigkeitsdaten, die in den Enzymanalysen bei variierenden
Inhibitiorkonzentrationen bestimmt wurden, mit dem Computer an Standard-Enzymkinetikgleichungen
angepasst (siehe I. H. Segel, Enzyme Kinetics, Wiley-Interscience,
1975).
-
Die
Bestimmung der Geschwindigkeitskonstanten zweiter Ordnung für irreversible
Inhibitoren wurde durch das Anpassen der Fluoreszenz- vs Zeitdaten
an die Fortschritts-Gleichungen
von Morrison bestimmt. Morrison, J. F., Mol. Cell. Biophys., 2,
Seiten 347 – 368
(1985). Thornberry et al. veröffentlichten
eine Beschreibung dieser Verfahren zur Messung der Geschwindigkeitskonstanten
irreversibler Inhibitoren von ICE. Thornberry, N. A., et al. Biochemistry,
33, Seiten 3923 – 3940
(1994). Für
Verbindungen, bei denen kinetisch keine vorherige Komplexbildung
beobachtet werden kann, werden die Geschwindigkeitskonstanten zweiter
Ordnung (kinact) direkt aus der Steigung
der linearen Kurven von kbeob. vs Inhibitorkonzentration
[I] abgeleitet. Für
Verbindungen, bei denen eine vorherige Komplexbildung zum Enzym
detektiert werden kann, werden die hyperbolischen Kurven von kbeob. vs [I] an die Gleichung für Sättigungskinetiken
angepasst, um zuerst Ki und k' zu erstellen. Die
Geschwindigkeitskonstante zweiter Ordnung kinact ist
dann durch k'/Ki gegeben.
-
3. PBMC Zellanalyse
-
IL-1β-Analyse
mit einer gemischten Population menschlicher peripherer einkerniger
Blutzellen (PBMC) oder mit angereicherten adhärenten einkernigen Zellen
-
Die
Prozessierung von prä-IL-1β durch ICE
kann in der Zellkultur unter Verwendung einer Vielfalt von Zellquellen
gemessen werden. Von gesunden Spendern erhaltene menschliche PBMC
stellen eine gemischte Population von Lymphocyten-Subtypen und einkernigen
Zellen bereit, die ein Spektrum von Interleukinen und Cytokinen
als Antwort auf viele Klassen physiologischer Stimulatoren produzieren.
Adhärente
einkernige Zellen aus PBMC stellen eine angereicherte Quelle normaler
Monocyten für
selektive Studien der Cytokinproduktion durch aktivierte Zellen
bereit.
-
Experimentelles Verfahren:
-
Eine
Ausgangsverdünnungsreihe
der Testverbindung in DMSO oder Ethanol wird hergestellt, mit einer anschließenden Verdünnung in
RPMI-10 % FBS-Medium (welches 2 mM L-Glutamin, 10 mM HEPES, 50 U und
50 μg/ml
Pen/Strep enthält),
um Arzneistoffe mit jeweils 4x der Test-Endkonzentration zu erbringen,
welche 0,4 % DMSO oder 0,4 % Ethanol enthalten. Die Endkonzentration
an DMSO ist für
alle Arzneistoffverdünnungen
0,1 %. Eine Konzentrationstitration, welche die für eine Testverbindung
in einer ICE-Hemmungsanalyse bestimmte scheinbare Ki umfasst,
wird im Allgemeinen für
die erste Verbindungssichtung verwendet.
-
Im
Allgemeinen werden 5 – 6
Verdünnungen
der Verbindung getestet, und die zelluläre Komponente der Analyse wird
doppelt durchgeführt,
mit doppelten ELISA-Bestimmungen
jedes Zellkulturüberstands.
-
PBMC-Isolation und IL-1-Analyse:
-
Aus
einem Pint menschlichem Blut isolierte Lymphocytenmanschettenzellen
(was ein Endvolumen von 40 – 45
ml Plasma plus Zellen erbringt) werden mit Medium auf 80 ml verdünnt und
LeukoPREP-Trennungsröhrchen
(Becton Dickinson) werden jeweils mit 10 ml Zellsuspension überschichtet.
Nach 15 min langer Zentrifugation bei 1500 – 1800 × g wird die Plasma/Mediumschicht
abgesaugt und dann wird die einkernige Zellschicht mit einer Pasteurpipette
gesammelt und in ein konisches 15 ml-Zentrifugenröhrchen (Corning) überführt. Medium
wird zugegeben, um das Volumen auf 15 ml zu bringen, die Zellen
werden durch Umdrehen sanft gemischt und bei 300 × g 15 min
lang zentrifugiert. Das PBMC-Pellet wird in einem geringen Volumen
an Medium resuspendiert, die Zellen werden gezählt und auf 6 × 106 Zellen/ml eingestellt.
-
Für die zelluläre Analyse
werden 1 ml der Zellsuspension, 0,5 ml der Testverbindungsverdünnung und 0,5
ml LPS Lösung
(Sigma #L-3012; 20 ng/ml Lösung,
hergestellt in RPMI-Komplettmedium; LPS-Endkonzentration 5 ng/ml)
in jede Vertiefung einer Gewebekulturplatte mit 24 Vertiefungen
mit flachem Boden (Corning) gegeben. Die 0,5 ml-Zugaben der Testverbindung
und von LPS sind gewöhnlich
ausreichend, um die Inhalte der Vertiefungen zu mischen. Drei Kontrollgemische
werden pro Experiment durchgeführt,
entweder mit LPS alleine, Lösungsmittelvehikel-Kontrolle und/oder
zusätzlichem
Medium, um das Endvolumen der Kultur auf 2,0 ml einzustellen. Die
Zellkulturen werden 16 – 18
Std. lang bei 37 °C
in Gegenwart von 5 % CO2 inkubiert.
-
Am
Ende des Inkubationszeitraums werden die Zellen geerntet und in
konische 15 ml Zentrifugenröhrchen überführt. Nach
10 min langer Zentrifugation bei 200 × g werden die Überstände geerntet
und in 1,5 ml-Eppendorfröhrchen überführt. Es
sollte beachtet werden, dass das Zellpellet für eine biochemische Bestimmung
des Gehalts an prä-IL-1β und/oder reifem IL-1β in Cytosolextrakten
durch Western-Blot-Verfahren oder ELISA mit prä-IL-β spezifischen Antiseren benutzt
werden kann.
-
Isolierung von adhärenten einkernigen
Zellen:
-
PBMC
werden, wie vorstehend beschrieben, isoliert und hergestellt. Medium
(1,0 ml) wird zuerst zu den Vertiefungen gegeben, gefolgt von 0,5
ml der PBMC-Suspension.
-
Nach
einer Stunde Inkubation werden die Platten sanft geschüttelt und
nicht-adhärente
Zellen aus jeder Vertiefung abgesaugt. Die Vertiefungen werden dann
dreimal sanft mit 1,0 ml Medium gewaschen und letztendlich in 1,0
ml Medium resuspendiert. Die Anreicherung auf adhärente Zellen
ergibt im Allgemeinen 2,5 – 3,0 × 105 Zellen pro Vertiefung. Die Zugabe der Testverbindungen,
von LPS, Inkubationsbedingungen der Zellen und die Bearbeitung des Überstands
läuft,
wie vorstehend beschrieben, ab.
-
ELISA:
-
Quantikine-Kits
(R&D Systems)
können
zur Messung von reifem IL-1β verwendet
werden. Die Analysen werden gemäß den Anweisungen
des Herstellers durchgeführt.
Die Mengen von reifem IL-1β von
etwa 1 – 3
ng/ml werden sowohl in PBMC als auch in adhärenten einkernigen positiven
Zellkontrollen beobachtet. ELISA-Analysen werden an 1:5-, 1:10-
und 1:20-Verdünnungen
der Überstände von
LPS-positiven Kontrollen durchgeführt, um die optimale Verdünnung für Überstände des
Testreihe auszuwählen.
-
Die
inhibitorische Stärke
der Verbindungen kann durch einen IC50-Wert
dargestellt werden, welcher die Konzentration des Inhibitors ist,
bei welcher im Vergleich zu den Positivkontrollen 50 % reifes IL-1β im Überstand
nachgewiesen wird.
-
Der
Fachmann erkennt, dass die in den Zellanalysen erhaltenen Werte
von mehreren Faktoren abhängen
können.
Die Werte mögen
nicht notwendigerweise präzise
quantitative Ergebnisse darstellen.
-
Beispiel 3
-
Gesamtblutanalyse zur
IL-1β-Produktion
-
Gesamtblutanalyse-IC50-Werte für erfindungsgemäße Verbindungen
wurden unter Verwendung des nachstehend beschriebenen Verfahrens
erhalten:
-
Ziel:
-
Die
Gesamtblutanalyse ist ein einfaches Verfahren zur Messung der Produktion
von IL-1β (oder
von anderen Cytokinen) und der Aktivität von möglichen Inhibitoren. Die Komplexität dieses
Analysesystems mit seinem ganzen Komplement an lymphoiden und entzündlichen
Zellarten, dem Spektrum an Plasmaproteinen und roten Blutzellen
ist eine ideale in vitro-Darstellung
menschlicher physiologischer in vivo-Zustände.
-
Materialien:
-
- Pyrogen-freie Spritzen (~ 30 cm3)
- Pyrogen-freie sterile Vakuumröhrchen, welche lyophilisiertes
Na2EDTA (4,5 mg/10 ml Röhrchen) enthalten
- Menschliche Gesamtblutprobe (~ 30 – 50 cm3)
- 1,5 ml Eppendorfröhrchen
- Vorratslösungen
der Testverbindung (~ 25 mM in DMSO oder in einem anderen Lösungsmittel)
- Endotoxin-freie Natriumchloridlösung (0,9 %) und HBSS-Lipopolysaccharide
(Sigma; Kat.# L-3012)-Vorratslösung
mit 1 mg/ml in HBSS
- IL-1β ELISA
Kit (R & D Systems;
Kat # DLB50)
- TNFα ELISA
Kit 8R & D Systems;
Kat # DTA50)
- Wasserbad oder Inkubator
-
Experimentelles Verfahren
zur Gesamtblutanalyse:
-
Man
stellt den Inkubator oder das Wasserbad auf 30 °C ein. Man aliquotiert 0,25
ml Blut in 1,5 ml-Eppendorfröhrchen.
Anmerkung: Man muss sicher sein, dass das Gefäß mit der Gesamtblutprobe nach
jedem zweiten Aliquot umgedreht wird. Unterschiede bei den Wiederholungsversuchen
können
sich ergeben, falls die Zellen sedimentieren und nicht einheitlich
suspendiert sind. Die Verwendung einer positiven Verdrängungspipette
wird ebenfalls Unterschiede zwischen den Aliquoten der Wiederholungsversuche
minimieren.
-
Man
stellt Arzneistoffverdünnungen
in steriler, pyrogen-freier Kochsalzlösung durch serielle Verdünnung her.
Eine Verdünnungsreihe,
welche den scheinbaren Ki-Wert für eine Testverbindung,
die durch eine ICE-Inhibierungsanalyse bestimmt wurde, umfasst,
wird im Allgemeinen für
die primäre
Verbindungsdurchmusterung verwendet. Für extrem hydrophobe Verbindungen
stellt man Verdünnungen
der Verbindung in frischem Plasma, welches vom gleichen Blutspender
erhalten wurde, oder in PBS-enthaltendem 5 % DMSO her, um die Löslichkeit
zu verbessern.
-
Man
gibt 25 μl
Verdünnung
der Testverbindung oder eine Vehikelkontrolle zu und mischt die
Probe sanft. Dann gibt man 5,0 μl
LPS Lösung
(250 ng/ml vorrätig,
frisch hergestellt: 5,0 ng/ml Endkonzentration an LPS) zu und mischt
wieder. Man inkubiert die Röhrchen
unter gelegentlichem Mischen 16 bis 18 Std. lang bei 30 °C in einem
Wasserbad. In einer anderen Ausführungsform
können
die Röhrchen
für den
gleichen Inkubationszeitraum in einen rotierenden Apparator gestellt
werden, der auf 4 U/min eingestellt ist. Diese Analyse sollte in
doppelter oder in dreifacher Ausführung mit den folgenden Kontrollen
aufgestellt werden: Negativkontrolle – kein LPS; Positivkontrolle – kein Testinhibitor;
Vehikelkontrolle – die
höchste
Konzentration an DMSO oder Lösungsmittel
für die
Verbindung, das im Experiment verwendet wird. Zusätzliche
Kochsalzlösung
wird allen Kontrollröhrchen
zugegeben, um das Volumen für
sowohl die Kontrolle als auch die experimentellen Gesamtbluttestproben
zu normalisieren.
-
Nach
dem Inkubationszeitraum werden die Gesamtblutproben 10 Minuten lang
bei ~ 2000 U/min in der Mikrofuge zentrifugiert, das Plasma wird
in ein frisches Mikrofugengefäß überführt und
gegebenenfalls bei 1000 × g
zentrifugiert, um die restlichen Plättchen zu pelletieren. Plasmaproben
können
vor der Analyse der Cytokinmengen durch ELISA bei –70 °C eingefroren
gelagert werden.
-
ELISA:
-
R & D Systems (614
McKinley Place N. E. Minneapolis, MN 55413)-Quantikine-Kits können zur
Messung von IL-1β und
TNF-α verwendet
werden. Die Analysen werden gemäß den Anleitungen
des Herstellers durchgeführt.
IL-1β-Mengen
von ~ 1 – 5
ng/ml in den Positivkontrollen bei einer Auswahl von Individuen
können
beobachtet werden. Eine 1 : 200 Verdünnung des Plasmas aller Proben
reicht gewöhnlicherweise
für die Experimente
aus, damit die Ergebnisse vom ELISA in den linearen Bereich der
ELISA-Standardkurven fallen. Es kann notwendig sein, Standardverdünnungen
zu optimieren, falls man Unterschiede in der Gesamtblutanalyse beobachtet.
Nerad, J. L. et al., J. Leukocyte Biol., 52, Seiten 687 – 692 (1992).
-
Beispiel 4
-
Die
antivirale Wirksamkeit von Verbindungen kann in unterschiedlichen
in vitro- und in vivo-Analysen bestimmt
werden. Zum Beispiel können
Verbindungen in viralen in vitro-Replikationsanalysen
getestet werden. In in vitro-Analysen können ganze Zellen oder isolierte
zelluläre
Komponenten verwendet werden. In vivo-Analysen schließen Tiermodelle
für Viruserkrankungen
ein. Beispiele für
derartige Tiermodelle schließen
Nagetiermodelle für
HBV- oder HCV-Infektion, das Waldmurmeltiermodell für HBV-Infektion
und das Schimpansenmodel für
die HCV-Infektion ein, sind aber nicht darauf beschränkt.
-
Erfindungsgemäße Verbindungen
können
auch in Tiermodellen für
eine durch Alkoholinduzierte-Erkrankung induziert werden, der mit
der Nahrung aufgenommen wird.
-
Beispiel 5
-
Verbindungen
10a – 10d
und 11a – 11d
wurden, wie nachstehend beschrieben, hergestellt:
-
Herstellung
von (N-Benzyloxycarbonyl-hydrazino)-essigsäure-tert-butylester (1). Zu
einem Gemisch aus Benzylcarbamat (25,0 g, 150 mmol), Kaliumcarbonat
(20,78 g, 150 mmol) in 230 ml Dimethylformamid (DMF) wurde tert-Butylbromacetat
(26,4 g, 145 mmol) gegeben. Die Suspension wurde 16 Std. lang bei
Raumtemperatur gerührt.
Das Umsetzungsgemisch wurde mit 1000 ml Ethylacetat verdünnt, mit
Eiswasser, dann dreimal mit Wasser gewaschen. Die organische Schicht
wurde über
wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum eingedampft, um ein klares Öl zu liefern,
welches durch Flash-Chromatographie
unter Verwendung von Hexan/EtOAc (9/1 bis 7/3) gereinigt wurde,
um 23,55 g (62 % Ausbeute) der Titelverbindung zu geben. 1H-NMR (500 MHz, CDCl3) δ 1,45 (s,
9H), 3,55 (s, 2H), 4,20 (br, 1H), 5,15 (s, 2H), 6,70 (br, 1H), 7,40
(s, 5H). Analytische HPLC*: 10,11 min
-
Herstellung
von 2-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)pentandisäure-5-benzylester
(2). Ein Gemisch aus γ-Benzyl-1-glutamat
(11,85 g, 50 mmol) und Phthalsäureanhydrid
(7,40 g, 50 mmol) in Toluol (150 ml) wurde mit einem Dean-Stark
Rohr unter Rückfluss
16 Stunden lang erhitzt. Das Lösungsmittel
wurde unter verringertem Druck entfernt. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von Hexan/Ethylacetat/Essigsäure (90/10/1 bis 50/50/1) gereinigt,
um 14,83 g (80 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR 500 MHz, CDCl3) δ 2,40 – 2,70 (m,
4H), 4,95 – 5,10
(m, 3H), 7,27 – 7,40
(m, 5H), 7,70 – 7,95
(m, 4H). Analytische HPLC: 13,28 min LC-MS (ES+):
m/e = 368 (M+H+).
-
Herstellung
von 5-(N'-Benzyloxycarbonyl-N-tert-butoxycarbonylmethyl-hydrazino)-4-(1,3-diozo-1,3-dihydro-isoindol-2-yl)-5-oxo-pentansäurebenzylester
(3). Zu einer Lösung
aus 2-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-pentandisäure-5-benzylester
(2) (1,84 g, 5 mmol) in 25 ml Dichlormethan mit 0,1 ml Dimethylformamid
(DMF) wurde bei 0 °C
tropfenweise Oxalylchlorid (666 mg, 5,25 mmol) gegeben. Die Lösung wurde
30 min lang bei 0 °C,
dann eine Stunde lang bei Raumtemperatur gerührt. K2CO3 (1,03 g) wurde bei 0 °C zugegeben, gefolgt von einer Lösung aus
(N-Benzyloxycarbonyl-hydrazino)-essigsäure-tert-butylester (1) (1,40
g, 5 mmol) in 5 ml Dichlormethan. Das Gemisch wurde 16 Stunden lang
bei Raumtemperatur gerührt.
Das Lösungsmittel
wurde unter verringertem Druck entfernt, und der Rückstand
wurde in 400 ml Ethylacetat suspendiert, mit Wasser (200 ml × 2), dann
Kochsalzlösung
(200 ml × 2)
gewaschen. Die organische Schicht wurde über wasserfreiem Na2SO4 getrocknet,
filtriert, im Vakuum konzentriert, um 2,8 g eines klaren Öls zu liefern.
Flash-Chromatographie unter Verwendung von Hexan/Ethylacetat (9/1
bis 7/3) gab 1,93 g (61 % Ausbeute) der Titelverbindung. 1H-NMR (500 MHz, CDCl3) δ 1,35 (s,
9H), 2,30 – 2,55
(m, 4H), 4,70 – 5,20
(br, 4H), 5,08 (s, 2H), 5,30 (m, 1H), 7,26 – 7,35 (m, 10H), 7,65 – 7,90 (m,
4H). Analytische HPLC: 14,6 min. LC-MS (ES+)
: m/e = 630 (M+H+).
-
Herstellung
von [6-(1,3-Diozo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]-diazepan-1-yl]-essigsäure-tert-butylester
(4). Eine Suspension aus 5-(N'-Benzyloxycarbonyl-N-tert-butoxycarbonylmethyl-hydrazino)-4-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-5-oxo-pentansäurebenzylester
(3) und 10 % Palladium auf Kohlenstoff (250 mg) in Tetrahydrofuran
(THF) (30 ml) und DMF (3 ml) wurde unter einer Wasserstoffatmosphäre 16 Stunden
lang gerührt.
Das Gemisch wurde durch Celite filtriert, und das Filtrat wurde
im Vakuum eingedampft. Der Rückstand
wurde in 30 ml Dichlormethan gelöst.
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
(EDC) (770 mg, 4 mmol) wurde zugegeben, und die Lösung wurde
3,5 Stunden lang bei Raumtemperatur gerührt. Das Lösungsmittel wurde im Vakuum entfernt,
und der Rückstand
wurde in Ethylacetat (300 ml) gelöst, dann mit Wasser (100 ml × 2) gewaschen. Die
organische Schicht wurde über
wasserfreiem Na2SO4 getrocknet,
filtriert, durch Flash-Chromatographie unter Verwendung von Hexan/Ethylacetat
(85/15 bis 50/50) gereinigt, um 1,06 g (75 % Ausbeute) der Titelverbindung
zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,40
(s, 9H), 2,30 – 2,40
(m, 1H), 2,40 – 2,50
(m, 1H), 3,05 – 3,12
(m, 1H), 3,38 – 3,48
(m, 1H), 3,90 – 4,00
(d, 1H), 4,60 – 4,70
(d, 1H), 5,55 – 5,63
(m, 1H), 7,65 (s, 1H), 7,75 – 7,7,9
(m, 2H), 7,88 – 7,92
(m, 2H). Analytische HPLC: 10,36 min. LC-MS (ES+):
m/e = 388 (M+H+).
-
Herstellung
von [2-Benzyl-6-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(5a). Ein Gemisch aus [6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(4) (200 mg, 0,52 mmol), Benzylbromid (110 mg, 0,64 mmol), K2CO3 (125 mg, 0,9
mmol) und Benzyltriethylammoniumchlorid (15 mg) in THF (5 ml) wurde
40 Stunden lang bei Raumtemperatur gerührt. Das Gemisch wurde mit
Ethylacetat (100 ml) verdünnt
und dreimal mit Wasser gewaschen. Die organische Schicht wurde über wasserfreiem
Na2SO4 getrocknet
und filtriert. Das Filtrat wurde durch Flash-Chromatographie unter
Verwendung von Dichlormethan/Ethylacetat (99,5/0,5 bis 97,5/2,5)
gereinigt, um 166 mg (67 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,40 (s,
9H), 2,30 – 2,40 (m,
1H), 2,50 – 2,57
(m, 1H), 3,30 – 3,40
(m, 1H), 3,60 – 3,68
(m, 1H), 3,72 – 3,80
(d, 1H), 4,48 – 4,52
(d, 1H), 4,80 – 4,92
(q, 2H), 5,15 – 5,20
(m, 1H), 7,35 (s, 5H), 7,70 (d, 2H), 7,82 (d, 2H). Analytische HPLC:
13,43 min. LC-MS (ES+) : m/e = 478 (M+H+).
-
(2-Allyl-6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester (5b)
wurde aus 4 und Allylbromid durch das Verfahren und die Chromatographie,
die verwendet wurden, um 5a herzustellen, synthetisiert, um 388
mg (91 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR
(500 MHz, CDCl3) δ 1,40 (s, 9H), 2,35 – 2,45 (m,
1H), 2,48 – 2,52
(m, 1H), 3,42 – 3,50
(m, 1H), 3,63 – 3,70
(m, 1H), 3,82 – 3,90
(m, 2H), 4,65 – 4,67
(d, 1H), 4,70 – 4,77
(q, 1H), 5,35 – 5,42
(m, 2H), 6,00 – 6,10
(m, 1H), 7,71 – 7,74
(d, 2H), 7,82 – 7,85
(d, 2H). Analytische HPLC: 13,28. LC-MS (ES+)
: m/e = 428 (M+H+).
-
Herstellung
von [6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-2-propyl-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(5e). Ein Gemisch aus [2-Allyl-6-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(5b) (380 mg, 0,89 mmol) und 20 % Palladium(II)hydroxid auf Kohlenstoff
(Pearlman's Katalysator)
(80 mg) in Ethanol (10 ml) wurde unter einer Wasserstoffatmosphäre 3 Stunden lang
gerührt.
Das Umsetzungsgemisch wurde durch Celite filtriert, und das Filtrat
wurde im Vakuum eingedampft, um 380 mg (99,5 % Ausbeute) der Titelverbindung
zu geben. 1H-NMR (500 MHZ, CDCl3) δ 1,05 – 1,09 (t,
3H), 1,47 (s, 9H), 1,71 – 1,82
(m, 2H), 2,33 – 2,46
(m, 1H), 2,46 – 2,53
(m, 1H), 3,00 – 3,09
(m, 1H), 3,43 – 3,60
(m, 2H), 3,95 – 4,00
(d, 1H), 4,10 – 4,18
(m, 1H), 4,54 – 4,58
(d, 1H), 5,40 – 5,45
(m, 1H), 7,70 – 7,76
(m, 2H), 7,80 – 7,85
(m, 2H). Analytische HPLC: 13,53 min. LC-MS (ES+):
m/e = 430 (M+H+)
-
Herstellung
von (6-Amino-2-benzyl-3,7-dioxo-[1,2]diazepan-1-yl)-essigsäure-tert-butylester (6a).
Eine Lösung
aus [2-Benzyl-6-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(5a) (150 mg, 0,31 mmol) und Hydrazinmonohydrat (17,3 mg, 35 mmol)
in Ethanol (1,5 ml) wurde 6 Stunden lang bei Raumtemperatur gerührt. Das
Lösungsmittel
wurde im Vakuum entfernt. Der Rückstand
wurde in Essigsäure
(1,5 ml) aufgenommen und 30 min lang bei Raumtemperatur gerührt. Das
Gemisch wurde im Vakuum eingedampft, und der so erhaltene Rückstand
wurde in Ethylacetat (20 ml) gelöst,
mit 5 % Na2CO3, dann
mit Wasser gewaschen. Die wässrige
Lösung
wurde mit Ethylacetat (20 ml × 2)
extrahiert. Die vereinigten organischen Schichten wurden über Na2SO4 getrocknet und
filtriert. Das Filtrat wurde im Vakuum zur Trockene eingedampft,
um 107 mg (98 % Ausbeute) der Titelverbindung zu liefern. 1H-MMR (500 MHz, CDCl3) δ 1,45 (s,
9H), 1,70 – 1,78
(m, 1H), 2,30 – 2,48
(m, 2H), 3,10 – 3,15
(m 1H), 3,30 – 3,38
(m, 1H), 3,95 – 4,00
(d, 1H), 4,36 – 4,40
(d, 1H), 4,45 – 4,49
(d, 1H), 5,07 – 5,12
(d, 1H), 7,28 – 7,38
(m, 5H). Analytische HPLC: 8,03 min. LC-MS (ES+):
m/e = 348 (M+H+).
-
(6-Amino-3,7-dioxo-2-propyl-(1,2]diazepan-1-yl)-essigsäure-tert-butylester
(6c) wurde aus 5c durch das Verfahren, das verwendet wurde um 6a
herzustellen, hergestellt, um 182 mg (69 % Ausbeute) der Titelverbindung
zu liefern. 1H-NMR (500 MHz, CDCl3) δ 0,94 – 1,01 (t,
3H), 1,50 (s, 9H), 1,55 – 1,67
(m, 2H), 1,70 – 1,80
(m, 1H), 2,30 – 2,37
(m, 1H), 2,50 – 2,60
(m, 1H), 3,10 – 3,18
(m, 1H), 3,21 – 3,28
(m, 1H), 3,81 – 3,87 (m,
1H), 3,91 – 3,98
(m, 1H), 4,10 – 4,15
(d, 1H), 4,39 – 4,43
(d, 1H). Analytische HPLC: 6,10 min. LC-MS (ES+) :
m/e = 300 (M+H+).
-
Herstellung
von {2-Benzyl-6-[(isochinolin-1-carbonyl)-amino]-3,7-diozo-[1,2]diazepan-1-yl}-essigsäure-tert-butylester
(7a). Zu einer Lösung
aus Isochinolin-1-carbonsäure
(173 mg, 1 mmol) in Dichlormethan (5 ml) wurde HOBT (135 mg, 1 mmol),
gefolgt von EDC (192 mg, 1 mmol) bei 0 °C gegeben. Das Gemisch wurde 15
min lang gerührt,
und eine Lösung
aus (6-Amino-2-benzyl-3,7-dioxo-[1,2]diazepan-1-yl)-essigsäure-tert-butylester
(6a) (105 mg, 0,30 mmol) in Dichlormethan (5 ml) wurde zugegeben.
Die Umsetzung wurde 30 min lang bei 0 °C, dann 16 Stunden lang bei
Raumtemperatur gerührt.
Das Gemisch wurde mit Dichlormethan (50 ml) verdünnt und mit Wasser (50 ml × 3) gewaschen.
Die organische Schicht wurde über
wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum eingedampft, um einen hellgelben Feststoff
zu geben, der durch Flash-Chromatographie unter Verwendung von Dichlormethan/Ethylacetat
(95/5 bis 85/15) gereinigt wurde, um 128 mg (84 % Ausbeute) der
Titelverbindung zu liefern. 1H-NMR (500
MHz, CDCl3) δ 1,49 (s, 9H), 2,02 – 2,10 (m,
1H), 2,50 – 2,57
(m, 1H), 2,88 – 2,97
(m, 1H), 3,60 – 3,70
(m, 1H), 3,84 – 3,89
(d, 1H), 4,52 – 4,57
(d, 1H), 4,84 – 4,98
(m, 3H), 7,26 – 7,42
(m, 5H), 7,64 – 7,84
(m, 4H), 8,45 (d, 1H), 8,76 (d, 1H), 9,44 (d, 1H). Analytische HPLC:
14,20 min. LC-MS (ES+) : m/e = 503 (M+H+).
-
[6-(Isochinolin-1-carbonylamino)-3,7-dioxo-2-propyl-[1,2]-diazepan-1-yl]-essigsäure-tert-butylester (7b) wurde
aus (6-Amino-3,7-dioxo-2-propyl-[1,2]-diazepan-1-yl)-essigsäure-tert-butylester (6c) und
Isochinolin-1-carbonsäure
durch das Verfahren und die Chromatographie, die verwendet wurden,
um 7a herzustellen, hergestellt, um 234 mg (86 % Ausbeute) der Titelverbindung
zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,01 – 1,07 (t,
3H), 1,49 (s, 9H), 1,73 – 1,82
(m, 1H), 2,03 – 2,12
(m, 1H), 2,42 – 2,50
(m, 1H), 2,84 – 2,93
(m, 1H), 3,15 – 3,22
(m, 1H), 3,50 – 3,58
(m, 1H), 4,03 – 4,15
(m, 2H), 4,60 – 4,63
(d, 1H), 5,23 – 5,30
(m, 1H), 7,64 – 7,83
(m, 2H), 7,80 – 7,88
(m, 2H), 8,50 (d, 1H), 8,87 (d, 1H), 9,55 (d, 1H). Analytische HPLC:
10,45 nun. LC-MS (ES+) : m/e = 455 (M+H+).
-
[6-(4-Allyloxy-3,5-dichlor-benzoylamino)-3,7-dioxo-2-propyl-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(7c) wurde aus (6-Amino-3,7-dioxo-2-propyl-[1,2]diazepan-1-yl)-essigsäure-tert-butyleseter
(6c) und 3,5-Dichlor-4-allyloxy-benzoylsäure durch das Verfahren und
die Chromatographie, die verwendet wurden, um 7a herzustellen, hergestellt,
um 200 mg (72 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500 MHz, CDCl3) δ 0,96 – 1,00 (t,
3H), 1,50 (s, 9H), 1,64 – 1,73
(m, 2H), 1,90 – 1,97
(m, 1H), 2,40 – 2,45
(m, 1H), 2,83 – 2,92
(m, 1H), 3,10 – 3,17
(m, 1H), 3,40 – 3,49
(m, 1H), 4,02 – 4,10
(m, 2H), 4,48 – 4,51
(d, 1H), 4,60 – 4,63
(m, 2H), 5,11 – 5,18
(m, 1H), 5,30 – 5,34
(d, 1H), 5,40 – 5,44
(d, 1H), 6,10 – 6,18
(m, 1H), 7,86 – 7,89
(d, 1H), 7,73 (s, 2H). Analytische HPLC: 10,47 min. LC-MS (ES+) : m/e = 528, 530 (M+H+).
-
Herstellung
von Isochinolin-1-carbonsäure-{1-benzyl-2-[(2-benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-dioxo-[1,2]-diazepan-4-yl}-amid
(10a). {2-Benzyl-6-[(isochinolin-1-carbonyl)-amino]-3,7-dioxo-[1,2]diazepan-1-yl}-essigsäure-tert-butylester (7a) (115
mg, 0,23 mmol) wurde in 20 % Trifluoressigsäure (TFA) in Dichlormethan
(2 ml) über
Nacht gerührt.
Die Lösung
wurde eingedampft, um {2-Benzyl-6-[(isochinolin-1-carbonyl)-amino]-3,7-dioxo-[1,2]diazepan-1-yl}-essigsäure (8a)
zu liefern. 1H-NMR 500 MHz, CDCl3) δ 2,05 – 2,11 (m,
1H), 2,45 – 2,50
(m, 1H), 2,72 – 2,83
(m, 1H), 3,45 – 3,54
(m, 1H), 4,01 – 4,06
((d, 1H), 4,59 – 4,63
(d, 1H), 4,70 – 4,82
(m, 2H), 4,93 – 4,98
(d, 1H), 7,28 – 7,42
(m, 5H), 7,80 – 8,10
( m, 4H), 8,40 – 8,52
(m, 2H), 8,85 (d, 1H). Die Säure
(8a) wurde in Dichlormethan (2 ml) gelöst, gefolgt von der Zugabe
von HOBT (77 mg, 0,57 mmol) und EDC (110 mg, 0,57 mmol), und 30
min lang gerührt.
Eine Lösung
aus (2-Benzyloxy-5-oxo-tetrahydrofuran-3-yl)-carbaminsäureallylester
(9, anti-Diastereomer) (166 mg, 0,57 mmol) in Dichlormethan/DMF
(3/1 ml), die 30 min lang mit 1,3-Dimethylbarbitursäure (DMBA) (90 mg, 0,57 mmol)
und Pd(PPh3)4 (66
mg, 0,057 mmol) beladen wurde, wurde zugegeben, und das so erhaltene
Gemisch wurde 16 Stunden lang bei Raumtemperatur gerührt. Das
Umsetzungsgemisch wurde mit Ethylacetat (100 ml) verdünnt und
mit Wasser (50 ml × 3)
gewaschen, über
Na2SO4 getrocknet,
filtriert und eingedampft, um einen hellgelben Feststoff zu geben, der
durch Flash-Chromatographie unter Verwendung von Dichlormethan/Methanol
(99,5/0,5 bis 98,5/1,5) gereinigt wurde, um 97,5 mg (67 % Ausbeute)
der Titelverbindung zu liefern. 1H-NMR (500
MHz, CDCl3) δ 2,03 – 2,13 (m, 1H), 2,40 – 2,43 (d,
1H), 2,55 – 2,60
(m, 1H), 2,83 – 2,92
(m, 1H), 3,04 – 3,10
(q, 1H), 3,28 – 3,38 (m,
1H), 4,08 – 4,11
(d, 1H), 4,20 – 4,24
(d, 1H), 4,38 – 4,41
(m, 1H), 4,67 – 4,70
(d, 1H), 4,78 – 4,87
(m, 2H), 4,91 – 5,02
(m, 2H), 5,36 (s, 1H), 6,05 (d, 1H), 7,20 – 7,38 (m, 10H), 7,68 – 7,78 (m,
2H), 7,80 – 7,88
(m, 2H), 8,48 (d, 1H), 8,70 (d, 1H), 9,49 (d, 1H). Analytische HPLC:
12,53 min. LC-MS (ES+) : m/e = 636 (M+H+).
-
Isochinolin-1-carbonsäure-{2-[(2-benzylogy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-diozo-1-propyl-(1,2]diazepan-4-yl}-amid
(10b) wurde aus 7b und den Diastereomeren von 9 durch das Verfahren
und die Chromatographie, die verwendet wurden, um 10a herzustellen,
synthetisiert, um sowohl das syn-Diastereomer (203 mg, 67 % Ausbeute,
höherer
Rf) als auch das anti-Diastereomer (93 mg, 31 % Ausbeute, niedriger
Rf) der Titelverbindung zu liefern. 1H-NMR
(500 MHz, CDCl3) für das anti-Diastereomer: δ 0,96 – 1,05 (t,
3H), 1,72 – 1,82
(m, 2H), 2,10 – 2,20
(m, 1H), 2,48 – 2,56
(m, 2H), 2,78 – 2,86
(m, 1H), 2,92 – 3,18
(m, 3H), 3,25 – 3,39 (m,
1H), 3,95 – 4,02
(m, 1H), 4,06 – 4,17
(m, 1H), 4,41 – 4,50
(m, 1H), 4,62 – 4,66
(d, 1H), 4,82 – 4,89
(d, 1H), 5,10 – 5,20
(m, 1H), 5,5 (s, 1H), 6,68 (d, 1H), 7,28 – 7,40 (m, 5H), 7,70 – 7,77 (m,
2H), 7,82 – 7,89
(m, 2H), 8,51 (d, 1H), 8,77 (d, 1H), 9,52 (d, 1H); für das syn-Diastereomer: δ 0,90 – 0,98 (t,
3H), 1,60 – 1,80
(m, 2H), 2,00 – 2,10
(m, 1H), 2,43 – 2,50
(m, 2H), 2,80 – 2,98
(m, 2H), 3,10 – 3,20
(m, 2H), 4,03 – 4,17
(m, 2H), 4,29 – 4,33
(d, 1H), 4,60 – 4,63
(d, 1H), 4,70 – 4,79
(m, 1H), 4,85 – 4,89
(d, 1H), 5,21 – 5,30
(m, 1H), 5,52 (d, 1H), 6,70 (d, 1H), 7,28 – 7,33 (m, 5H), 7,64 – 7,78 (m,
2H), 7,83 – 7,90
(m, 2H), 8,50 (d, 1H), 8,85 (d, 1H), 9,50 (d, 1H). Analytische HPLC:
10,60 min für
das anti- Diastereomer
und 10,30 min für
das syn-Diastereomer. LC-MS (ES+) für das Produktgemisch:
m/e = 588 (M+H+).
-
4-Allyloxy-N-{2-[(2-benzylogy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-dioxo-1-propyl-[1,2]diazepan-4-yl}-3,5-dichlorbenzamid
(10c) wurde aus 7c und den Diastereomeren von 9 durch das Verfahren
und die Chromatographie, die verwendet wurden, um 10a herzustellen,
synthetisiert, um sowohl das anti-Diastereomer (78 mg, 31 % Ausbeute,
niedriger Rf) als auch das syn-Diastereomer (129 mg, 52 % Ausbeute,
höherer
Rf) der Titelverbindung zu liefern. 1H-NMR
(500 MHz, CDCl3) für das anti-Diastereomer: δ 1,00 – 1,05 (t,
3H), 1,60 – 1,80
(m, 2H), 2,03 – 2,18
(m, 1H), 2,23 – 2,30
(d, 1H), 2,40 – 2,48
(m, 1H), 2,62 – 2,78
(m, 1H), 2,90 – 3,12 (m,
2H), 3,50 – 3,60
(m, 1H), 4,05 – 4,25
(m, 3H), 4,48 – 4,52
(m, 1H), 4,60 – 4,68
(m, 3H), 4,80 – 44,85
(d, 1H), 5,08 – 5,20
(m, 1H), 5,30 – 5,39
(m, 2H), 5,40 – 5,45
(d, 1H), 6,08 – 6,20
(m, 1H), 6,82 – 6,85
(d, 1H), 7,30 – 7,45
(m, 6H), 7,85 (s, 2H); für
das syn-Diastereomer: δ 0,94 – 1,04 (t,
3H), 1,60 – 1,72
(m, 2H), 1,85 – 1,93 (m,
1H), 2,31 – 2,40
(m, 1H), 2,41 – 2,51
(m, 1H), 2,72 – 2,82
(m, 1H), 2,83 – 2,95
(m, 1H), 3,00 – 3,09
(m, 1H), 3,17 – 3,30
(m, 1H), 3,99 – 4,10
(m, 1H), 4,10 – 4,18
(d, 1H), 4,21 – 4,30
(d, 1H), 4,58 – 4,65
(m, 3H), 4,70 – 4,78
(m, 1H), 4,85 – 4,90
(d, 1H), 5,05 – 5,15
(m, 1H), 5,28 – 5,35
(d, 1H), 5,40 – 5,45
(d, 1H), 5,53 (d, 1H), 6,08 – 6,17
(m, 1H), 6,55 – 6,60
(d, 1H), 6,75 – 6,80
(d, 1H), 7,27 – 7,40
(m, 3H), 7,40 – 7,55
(m, 1H), 7,60 – 7,70
(m, 1H), 7,73 (s, 2H). Analytische HPLC für Diastereomere der Titelverbindung:
10,26 min. LC-MS (ES+) für das Produktgemisch: m/e =
661, 663 (M+H+).
-
Herstellung
von N-{2-[(2-Benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-dioxo-1-propyl-[1,2]diazepan-4-yl}-3,5-dichlor-4-hydroxybenzamid
(10d). Zu einer Lösung
aus 4-Allyloxy-N-{2-[(2-benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-dioxo-1-propyl-[1,2]diazepan-4-yl}-3,5-dichlorbenzamid
(10c, Diastereomere) (103 mg, 0,16 mmol) in Dichlormethan (15 ml)
wurde DMBA, gefolgt von Pd(PPh3)4 (30 mg, 0,026 mmol) gegeben, 7 Stunden
lang bei Raumtemperatur gerührt.
Das Umsetzungsgemisch wurde mit Wasser gewaschen, über wasserfreiem
Na2SO4 getrocknet,
filtriert und durch Flash-Chromatographie
unter Verwendung von Dichlormethan/Methanol (99,5/0,5 bis 97/3)
gereinigt, um sowohl das anti-Diastereomer (18 mg, 18 % Ausbeute,
niedriger Rf) als auch das syn-Diastereomer (48 mg, 50 % Ausbeute,
höherer
Rf) der Titelverbindung zu liefern. 1H-NMR (500 MHz, CDCl3) für
das anti-Diastereomer: δ 0,9 – 1,02 (m,
3H), 1,65 – 185
(m, 2H), 2,01 – 2,18
(m, 0,5H), 2,20 – 2,30
(d, 0,5H), 2,37 – 2,47
(m, 1H), 2,58 – 2,70
(m, 1H), 2,90 – 3,00
(m, 1H), 3,00 – 3,10
(m, 1H), 3,25 – 3,50
(m, 1H), 4,0 – 4,15
(m, 2H), 4,15 – 4,25 (d,
1H), 4,40 – 4,55
(m, 1H), 4,55 – 4,67
(m, 1H), 4,72 – 4,82
(d, 1H), 5,05 – 5,17
(m, 1H), 5,27 – 5,38
(m, 2H), 6,90 – 7,00
(d, 1H), 7,25 – 7,40
(m, 6H), 7,60 – 7,70
(m, 1H), 7,80 (s, 2H); für
das syn-Diastereomer: δ 0,9 – 1,00 (m,
3H), 1,50 – 1,70
(m, 2H), 1,8 – 1,92
(m, 1H), 2,38 – 2,42
(m, 1H), 2,48 – 2,58
(m, 1H), 2,74 – 2,87
(m, 1H), 2,88 – 2,98
(m, 1H), 3,00 – 3,13
(m, 1H), 3,17 – 3,30
(m, 1H), 4,00 – 4,12
(m, 1H), 4,14 – 4,30
(d, 1H), 4,22 – 4,32
(m, 1H), 4,62 – 4,46
(d, 1H), 4,75 – 4,80
(m, 1H), 4,90 – 4,95
(d, 1H), 5,10 – 5,20
(m, 1H), 5,53 – 5,57
(d, 1H), 6,24 (s, 1H), 6,49 (d, 1H), 6,70 (d, 1H), 7,25 – 7,40 (m,
5H), 7,71 (s, 2H). Analytische HPLC: 8,55 min für das syn-Diastereomer und
8,57 min für
das anti-Diastereomer. LC-MS (ES+) für das Produktgemisch: m/e
= 621, 623 (M+H+).
-
Herstellung
von 3-(2-{2-Benzyl-6-[(isochinolin-1-carbonyl)-amino]-3,7-dioxo-[1,2]-diazepan-1-yl}-acetylamino)-4-oxo-butansäure (11a). Isochinolin-1-carbonsäure-{1-benzyl-2-[(2-benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-dioxo-[1,2]-diazepan-4-yl}-amid
{10a, Diastereomere) (14 mg, 0,022 mmol) wurde in der Lösung aus
10 %iger HCl (1,5 ml) und Acetonitril (1 ml) 4 Stunden lang gerührt. Das
Umsetzungsgemisch wurde mit Wasser (30 ml) verdünnt, mit Ether (30 ml) zweimal
gewaschen. Die wässrige
Lösung
wurde 30 min lang mit Stickstoff gespült, dann in Trockeneis gekühlt und über Nacht
lyophilisiert, um 10 mg (83 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500 MHz, CD3OD) δ 2,00 – 2,10 (m,
1H), 2,32 – 2,50
(m, 2H), 3,62 – 3,72
(m, 1H), 4,15 – 4,35 (m,
2H), 4,45 – 4,80
(m, 4H), 5,25 – 5,32
(m, 1H), 7,30 – 7,68
(m, 5H), 7,95 – 8,05
(m, 1H), 8,12 – 8,17
(m, 1H), 8,22 – 8,27
(m, 1H), 8,35 – 8,41
(d, 1H), 8,56 (s, 1H), 8,65 – 8,74
(d, 1H). Analytische HPLC: 9,40 min. LC-MS (ES+)
: m/e = 546 (M+H+).
-
3-(2-{6-[(Isochinolin-1-carbonyl)-amino]-3,7-dioxo-2-propyl-[1,2]diazepan-1-yl}-acetylamino)-4-oxo-butansäure (11b)
wurde aus Isochinolin-1-carbonsäure-{2-[(2-benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-dioxo-1-propyl-[1,2]diazepan-4-yl}-amid (10b,
Diastereomere) (95 mg, 0,16 mmol) durch das Verfahren, das verwendet
wurde, um 11a herzustellen, hergestellt, um 28 mg (35 % Ausbeute)
der Titelverbindung zu liefern. 1H-NMR (500 MHz,
CDCl3/CD3OD = 0,5
ml/3 Tropfen) δ 0,90 – 1,00 (t,
3H), 1,65 – 1,80
(m, 2H), 2,10 – 2,28
(m, 1H), 2,32 – 2,40
(m, 1H), 2,47 – 2,80
(m, 3H), 3,05 – 3,18
(m, 1H), 3,30 – 3,51
(m, 2H), 4,00 – 4,55
(m, 3H), 5,10 – 5,20 (m,
1H), 7,75 – 8,05
(m, 4H), 8,49 (s, 1H), 8,90 – 9,00
(m, 1H). Analytische HPLC: 6,26 min. LC-MS (ES+):
m/e = 498 (M+H+).
-
3-{2-[6-(3,5-Dichlor-4-hydroxy-benzoylamino)-3,7-diozo-2-propyl-[1,2]diazepan-1-yl]-acetylamino}-4-oxo-butansäure (11e)
wurde aus N-{2-[(2-Benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-3,7-dioxo-1-propyl-[1,2]diazepan-4-yl}-3,5-dichlor-4-hydroxybenzamid
(10d, Diastereomere) (30 mg, 0,05 mmol) durch das Verfahren, das
verwendet wurde, um 11a herzustellen, hergestellt, um 19 mg (74
% Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500
MHz, CDCl3/CD3OD
= 0,5 ml/3 Tropfen) δ 0,90 – 0,95 (t,
3H), 1,60 – 1,70
(m, 2H), 2,00 – 2,10
(m, 1H), 2,25 – 2,36
(m, 1H), 2,48 – 2,82
(m, 3H), 2,98 – 3,10
(m, 1H), 3,20 – 3,35
(m, 1H), 3,90 – 4,50
(m, 4H), 4,95 – 5,08
(m, 1H), 7,62 – 7,72
(m, 2H). Analytische HPLC: 5,40 min. LC-MS (ES+):
m/e = 531, 533 (M+H+).
-
3-{2-[6-(3,5-Dichlor-4-hydrogybenzoylamino)-3,7-dioxo-2-methyl-[1,2]diazepan-1-yl]-acetylamino}-4-oxo-butansäure (11d)
wurde gemäß des Verfahrens,
dass verwendet wurde, um (11e) herzustellen, nur durch Substituieren
von Iodmethan für
Allylbromid hergestellt.
-
Beispiel 6
-
Verbindungen
19 und 20 wurden, wie nachstehend beschrieben, hergestellt:
-
Herstellung
von 2-(N'-Benzyloxycarbonyl-hydrazino)-propionsäureethylester
(12). Zu einer Lösung aus
Benzylcarbamat (665 mg, 4 mmol), Triethylamin (1,11 ml) in Dichlormethan
(4 ml) wurde tropfenweise Ethyl-O-trifluormethansulfonyl-D-lactat
bei 0 °C
gegeben. Die Lösung
wurde 15 min lang bei 0 °C,
dann 16 Stunden lang bei Raumtemperatur gerührt. Das Gemisch wurde mit
Dichlormethan (100 ml) verdünnt,
mit Wasser (50 ml × 2),
1 %iger HCl (50 ml × 2)
gewaschen. Die organische Schicht wurde über wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum zur Trockene eingedampft, um 570 mg (54
% Ausbeute) der Titelverbindung zu geben. 1H-NMR
(500 MHz, CDCl3) δ 1,25 – 1,40 (m, 6H), 3,68 – 3,78 (m,
1H), 4,12 – 4,20
(m, 3H), 5,07 – 5,15
(m, 1H), 6,45 – 6,57
(m, 1H), 7,30 – 7,45
(m, 5H). Analytische HPLC: 5,56 min. LC-MS (ES+) :
m/e = 267 (M+H+).
-
5-[N'-Benzyloxycarbonyl-N-(1-ethoxycarbonyl-ethyl)-hydrazino]-4-(1,3-diozo-1,3-dihydro-isoindol-2-yl)-5-oxo-pentansäurebenrylester
(13) wurde aus 2-(N'-Benzyloxycarbonyl-hydrazino)-propionsäureethylester
(12) und 2 durch das Verfahren und die Chromatographie, die verwendet
wurden, um 3 herzustellen, hergestellt, um 850 mg (64 % Ausbeute)
der Titelverbindung zu liefern. 1H-NMR (500
MHz, CDCl3) δ 1,10 – 1,50 (m, 6H), 2,40 – 2,70 (m,
5H), 4,05 – 4,30
(m, 2H), 4,65 – 4,70
(d, 0,5H), 4,80 – 4,86
(d, 0,5H), 5,00 – 5,40
(m, 5H), 7,15 – 7,50
(m, 10H), 7,65 – 7,90
(m, 4H). Analytische HPLC: 9,00 min. LC-MS (ES+): m/e
= 616 (M+H+).
-
2-[6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-propionsäure-tert-butylester (14)
wurde aus 5-[N'-Benzyloxycarbonyl-N-(1-ethoxycarbonyl-ethyl)-hydrazino]-4-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-5-oxo-pentansäurebenzylester
(13) durch das Verfahren und die Chromatographie, die verwendet
wurden, um 4 herzustellen, hergestellt, um 318 mg (64 % Ausbeute)
der Titelverbindung zu liefern. 1H-NMR (500
MHz, CDCl3) δ 1,25 – 1,35 (m, 3H), 1,50 – 1,60 (m,
3H), 2,37 – 2,55
(m, 2H), 2,75 – 2,95
(m, 1H), 3,35 – 3,65
(m, 2H), 4,15 – 4,30
(m, 2H), 5,15 – 5,40
(m, 1H), 5,47 – 5,60
(m, 1H), 7,65 – 7,90
(m, 4H). Analytische HPLC: 5,60 min. LC-MS (ES+)
: m/e = (M+H+).
-
Herstellung
von 2-[6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-2-methyl-3,7-dioxo-[1,2]diazepan-1-yl]-propionsäureethylester
(15). Ein Gemisch aus 2-[6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-propionsäure-tert-butylester
(14) (314 mg, 0,84 mmol), Benzyltriethylammoniumchlorid (30 mg,
0,13 mmol), K2CO3 (406
mg, 2,94 mmol) und Iodmethan (360 mg, 2,53 mmol) in THF (8 ml) wurde
5 Tage lang bei Raumtemperatur gerührt. Das Gemisch wurde mit
Dichlormethan (70 ml) verdünnt,
dreimal mit Wasser gewaschen, über
wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum zur Trockene eingedampft, um 296 mg (91
% Ausbeute) der Titelverbindung als ein Gemisch aus Diastereomeren
zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,25 – 1,35 (m,
2,5H), 1,47 – 1,52
(m, 0,5H), 1,60 – 1,75
(m, 3H), 2,37 – 2,52
(m, 2H), 3,05 – 3,14
(m, 0,5H), 3,30 – 3,45
(m, 3,5H), 3,45 – 3,58
(m, 1H), 4,14 – 4,28
(m, 2H), 4,52 – 4,58
(m, 0,5H), 4,80 – 4,87
(m, 0,5H), 5,13 – 5,28
(m, 1H), 7,70 – 7,90
(m, 4H). Analytische HPLC: 6,00 und 6,11 min. LC-MS(ES+): m/e
= 388 (M+H+).
-
2-(6-Amino-2-methyl-3,7-dioxo-[1,2]-diazepan-1-yl)-propionsäureethylester
(16) wurde aus 2-[6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-2-methyl-3,7-dioxo-[1,2]diazepan-1-yl)-propionsäureethylester
(15) durch das Verfahren, das verwendet wurde, um 6a herzustellen,
hergestellt, um 143 mg (73 % Ausbeute) der Titelverbindung als ein
Gemisch aus Diastereomeren zu liefern. 1H-NMR
(500 MHz, CDCl3) δ 1,22 – 1,30 (m, 2,5H), 1,47 – 1,50 (d,
0,5H), 1,52 – 1,70
(m, 3H), 1,70 – 1,82
(m, 1H), 2,30 – 2,40
(m, 1H), 2,48 – 2,69
(m, 1H), 2,75 – 2,82
(m, 0,5H), 3,03 – 3,11
(m, 0,5H), 3,22 (d, 3H), 3,61 – 3,75
(m, 1H), 4,18 – 4,30
(m, 2H), 4,49 – 4,54 (m,
0,5H), 4,85 – 4,90
(m, 0,5H). Analytische HPLC: 3,90 und 4,06 min. LC-MS (ES+)
: m/e = 258 (M+H+).
-
2-[6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methyl-3,7-dioxo-[1,2]diazepan-1-yl]-propionsäureethylester
(17) wurde aus 2-(6-Amino-2-methyl-3,7-dioxo-[1,2]-diazepan-1-yl)-propionsäureethylester
(16) und 3,5-Dichlor-4-allyloxy-benzoylsäure durch das Verfahren und
die Chromatographie, die verwendet wurden, um 7a herzustellen, hergestellt,
um 216 mg (80 % Ausbeute) der Titelverbindung als ein Gemisch aus
Diastereomeren zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,22 – 1,40 (m,
3H), 1,50 – 1,80
(m, 3H), 1,80 – 1,95
(m, 1H), 2,40 – 2,50
(m, 1H), 2,82 – 3,02
(m, 2H), 3,10 – 3,40
(m, 4H), 4,20 – 4,35
(m, 2H), 4,50 – 5,01
(m, 3H), 5,25 – 5,45
[m, 2H), 6,05 – 6,20
(m, 1H), 6,90 – 7,00
(m, 2H), 7,70 – 7,80
(d, 2H). Analytische HPLC: 6,97 und 7,06 min. LC-MS (ES+)
: m/e = 486, 488 (M+H+).
-
Herstellung
von 2-[6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methyl-3,7-dioxo-[1,2]diazepan-1-yl]-propionsäure (18).
2-[6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methyl-3,7-dioxo-[1,2]diazepan-1-yl]-propionsäureethylester
(17) (216 mg, 0,44 mmol) wurde in 1 N NaOH (2 ml) und MeOH (2 ml)
45 min lang bei Raumtemperatur gerührt. Das Gemisch wurde mit
Wasser (30 ml) verdünnt,
dreimal mit Dichlormethan extrahiert. Die vereinigten organischen
Schichten wurden über
wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum zur Trockene eingedampft, um 201 mg (99
% Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,58 – 1,68 (m,
3H), 1,88 – 2,00
(m, 1H), 2,40 – 2,48
(m, 1H), 2,80 – 3,18
(m, 2H), 3,25 – 3,48
(d, 3H), 4,47 – 4,65
(m, 2,5H), 4,74 – 4,81
(m, 0,5H), 4,90 – 5,05
(m, 1H), 5,27 – 5,31
(d, 1H), 5,38 – 5,43
(d, 1H), 6,08 – 61,8
(m, 1H), 7,07 – 7,22
(m, 2H), 7,71 (d, 2H). Analytische HPLC: 5,85 min. LC-MS (ES+) : m/e = 458, 460 (M+H+).
-
Herstellung
von N-{2-[1-(2-Benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-ethyl]-1-methyl-3,7-dioxo-[1,2]diazepan-4-yl}-3,5-dichlor-4-hydroxybenzamid
(19). Zu einer Lösung
aus 2-[6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methyl-3,7-dioxo-[1,2]diazepan-1-yl]-propionsäure (18)
(183 mg, 0,40 mmol) in Dichlormethan wurde HOBT (65 mg, 0,48 mmol),
gefolgt von EDC (123 mg, 0,64 mmol) gegeben. Das Gemisch wurde 30
min lang bei 0 °C gerührt, dann
wurde eine Lösung
aus (2-Benzyloxy-5-oxo-tetrahydrofuran-3-yl)-carbaminsäureallylester (9, anti-Diastereomer)
(140 mg, 0,48 mmol) in Dichlormethan, die 30 min lang mit 1,3-Dimethylbarbitursäure (DMBA)
(75 mg, 0,48 mmol) und Pd(PPh3)4 (60
mg, 0,05 mmol) beladen wurde, zugegeben, und das so erhaltene Gemisch
wurde 5 Stunden lang bei Raumtemperatur gerührt. Die zweite Portion DMBA
(63 mg, 0,40 mmol) wurde zugegeben, und das Umsetzungsgemisch wurde
kontinuierlich 16 Stunden lang bei Raumtemperatur gerührt. Die
Umsetzung wurde mit Wasser gequencht und mit Dichlormethan extrahiert.
Die organische Schicht wurde über
wasserfreiem Na2SO4 getrocknet,
filtriert und eingedampft, um einen hellgelben Feststoff zu geben,
der durch Flash-Chromatographie unter Verwendung von Dichlormethan/Methanol
(99/1 bis 95/5) gereinigt wurde, um 70 mg (29 % Ausbeute) der Titelverbindung
als ein Gemisch aus Diastereomeren zu liefern. 1H-NMR
(500 MHz, CDCl3) δ 1,39 – 1,45 (m, 3H), 2,60 – 2,80 (m,
2H), 2,80 – 3,10
(m, 1H), 3,12 – 3,32 (m,
4H), 4,20 – 4,62
(m, 2H), 4,70 – 4,88
(m, 2H), 5,25 – 5,45
(m, 1H), 6,65 – 6,95
(m, 3H), 7,20 – 7,50
(m, 5H), 7,61 (s, 1H), 7,72 (d, 1H). Analytische HPLC: 10,86 und
10,98 min. LC-MS (ES+) : m/e = 607, 609
(M+H+).
-
3-{2-[6-(3,5-Dichlor-4-hydroxy-benzoylamino)-2-methyl-3,7-dioxo-[1,2]diazepan-1-yl]-propionylamino}-4-oxo-butansäure (20)
wurde aus N-{2-[1-(2-Benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-ethyl]-1-methyl-3,7-dioxo-[1,2]diazepan-4-yl}-3,5-dichlor-4-hydroxy-benzamid
(19) (28 mg, 0,046 mmol) durch das Verfahren, das verwendet wurde,
um 11a herzustellen, hergestellt, um 17 mg (71 % Ausbeute) der Titelverbindung
zu liefern. 1H-NMR (500 MHz, CDCl3/CD3OD = 0,5 ml/3 Tropfen) δ 1,20 – 1,50 (m, 3H), 1,90 – 2,10 (m,
1H), 2,25 – 2,85
(m, 4H), 3,10 – 3,40 (m,
3H), 4,05 – 4,50
(m, 1H), 4,55 – 4,65
(m, 1H), 4,75 – 5,05
(m, 2H), 7,72 – 7,76
(m, 2H). Analytische HPLC: 7,51 min. LC-MS (ES+)
: m/e = 517, 519 (M+H+).
-
Beispiel 7
-
Verbindung
27 wurde, wie nachstehend beschrieben, hergestellt;
-
Herstellung
von [6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-7-oxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(21). Zu einer Lösung
aus [6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-3,7-dioxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(4) (775 mg, 2,0 mmol) in THF (4 ml) wurde Boran-THF-Komplex in
THF (1 M, 4 ml) gegeben. Die Umsetzung wurde 30 min lang bei 0 °C, dann 3
Stunden lang bei Raumtemperatur gerührt. Das Gemisch wurde mit
Eiswasser (60 ml) verdünnt,
mit Ethylacetat (60 ml × 3)
extrahiert. Die vereinigten organischen Schichten wurden über wasserfreiem
Na2SO4 getrocknet,
filtriert und im Vakuum eingedampft, um einen Feststoff zu geben,
der durch Flash-Chromatographie unter Verwendung von Hexan/Ethylacetat
(95/5 bis 70/30) gereinigt wurde, um 585 mg (78 % Ausbeute) der
Titelverbindung zu liefern. 1H-NMR (500
MHz, CDCl3) δ 1,43 (s, 9H), 1,78 – 1,90 (m, 1H), 2,00 – 2,15 (m,
2H), 2,65 – 2,78
(m, 1H), 2,95 – 3,07
(m, 1H), 3,22 – 3,30 (m,
1H), 3,85 – 3,92
(d, 1H), 4,30 – 4,42
(m, 2H), 5,39 – 5,42
(m, 1H), 7,62 – 7,71
(m, 2H), 7,79 – 7,86
(m, 2H). Analytische HPLC: 7,86 min. LC-MS (ES+):
m/e = 374 (M+H+).
-
Herstellung
von [6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-2-methansulfonyl-7-oxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(22). Zu einer Lösung
aus [6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-7-oxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(21) (155 mg, 0,42 mmol), Triethylamin (0,5 ml) und 4-N,N-Dimethylaminopyridin
(DMAP) (101 mg, 0,50 mmol) in Dichlormethan wurde tropfenweise Methansulfonylchlorid
(95 mg, 0,83 mmol) gegeben. Das Gemisch wurde 5 min lang bei 0 °C, dann 16
Stunden lang bei Raumtemperatur gerührt. Das Gemisch wurde mit
Wasser gequencht, mit Dichlormethan extrahiert. Die organische Lösung wurde
mit Wasser, dann Kochsalzlösung
gewaschen, über
wasserfreiem Na2SO4 getrocknet,
filtriert und durch Flash-Chromatographie unter Verwendung von Dichlormethan/Ethylacetat
(99/1 bis 95/5) gereinigt, um 154 mg (82 % Ausbeute) der Titelverbindung
zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,47 (s,
9H), 2,05 – 2,21
(m, 2H), 2,83 – 2,87
(m, 1H), 3,27 (s, 3H), 3,53 – 3,58
(m, 1H), 4,07 – 4,11
(d, 1H), 4,29 – 4,33
(m, 1H), 4,50 – 4,53
(d, 1H), 5,42 – 5,44
(m, 1H), 7,70 – 7,85
(m, 4H). Analytische HPLC: 12,27 min. LC-MS (ES+):
m/e = 452 (M+H+).
-
(6-Amino-2-methansulfonyl-7-oxo-[1,2]-diazepan-1-yl)-essigsäure-tert-butylester
(23) wurde aus [6-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-2-methansulfonyl-7-oxo-[1,2]-diazepan-1-yl]-essigsäure-tert-butylester
(22) durch das Verfahren, das verwendet wurde, um 6a herzustellen,
hergestellt, um 95 mg (89 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500 MHz; CDCl3) δ 1,48 (s,
9H), 1,66 – 1,71
(m, 1H), 1,83 – 1,87 (m,
1H), 1,94 – 198
(m, 1H), 2,09 – 2,13
(m, 1H), 3,07 (s, 3H), 3,45 – 3,51
(m, 1H), 3,87 – 3,91
(d, 1H), 4,00 – 4,11
(m, 2H), 4,59 – 4,63
(d, 1H). Analytische HPLC: 5,87 min. LC-MS (ES+)
: m/e = 322 (M+H+).
-
[6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methansulfonyl-7-oxo-[1,2]diazepan-1-yl]-essigsäure-tert-butylester
(24) wurde aus (6-Amino-2-methansulfonyl-7-oxo-[1,2]diazepan-1-yl)-essigsäure-tert-butylester
(23) und 3,5-Dichlor-4-allyloxybenzoylsäure durch das Verfahren und
die Chromatographie, die verwendet wurden, um 7c herzustellen, hergestellt,
um 193 mg (87 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,50 (s,
9H), 1,78 – 1,93
(m, 2H), 2,22 – 2,25
(m, 2H), 3,16 (s, 3H), 3,52 (m, 1H), 4,03 – 4,06 (d, 1H), 4,22 – 4,25 (d,
1H), 4,60 – 4,64
(m, 3H), 5,11 – 5,13
(m, 1H), 5,28 – 5,29
(d, 1H), 5,40 – 5,44
(m, 1H), 6,10 – 6,16
(m, 1H), 7,21 – 7,30
(m, 1H), 7,75 (s, 2H). Analytische HPLC: 7,39 min. LC-MS (ES+) : m/e = 550, 552 (M+H+).
-
[6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methansulfonyl-7-oxo-[1,2]diazepan-1-yl]-essigsäure (25) wurde
aus [6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methansulfonyl-7-oxo-[1,2]-diazepan-1-yl]-essigsäure-tert-butylester
(24) durch das Verfahren, das verwendet wurde, um 8a herzustellen,
hergestellt, um 173 mg (100 % Ausbeute) der Titelverbindung zu liefern. 1H-NMR (500 MHz, CDCl3) δ 1,74 – 1,77 (m,
1H), 1,91 – 1,93 (m,
1H), 2,22 – 2,25
(m, 2H), 3,19 (s, 3H), 3,39 – 3,44
(m, 1H), 4,24 – 4,28
(m, 2H), 4,61 – 4,63
(m, 2H), 4,71 – 4,75
(d, 1H), 5,12 – 5,15
(m, 1H), 5,28 – 5,31
(m, 1H), 5,40 – 5,44
(m, 1H), 6,10 – 6,16
(m, 1H), 7,31 – 7,33 (d,
1H), 7,79 (s, 2H). Analytische HPLC: 5,70 min. LC-MS (ES+) : m/e = 494, 496 (M+H+).
-
4-Allyloxy-N-{2-[(2-benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-1-methansulfonyl-3-oxo-[1,2]-diazepan-4-yl}-3,5-dichlorbenzamid
(26a) wurde aus [6-(4-Allyloxy-3,5-dichlor-benzoylamino)-2-methansulfonyl-7-oxo-[1,2]diazepan-1-yl]-essigsäure (25)
und (2-Benzyloxy-5-oxo-tetrahydrofuran-3-yl)-carbaminsäureallylester
(9, ein Gemisch aus Diastereomeren) durch das Verfahren und die
Chromatographie, die verwendet wurden, um 10a herzustellen, hergestellt,
um 178 mg (74 % Ausbeute) der Titelverbindung als ein Gemisch aus
Diastereomeren zu liefern. 1H-NMR (500 MHz,
CDCl3) δ 1,58 – 1,70 (m,
2H), 1,89 – 1,97
(m, 1H), 2,20 – 2,32
(m, 2H), 2,40 – 2,60
(m, 1H), 2,86 – 3,05
(m, 1H), 3,28 – 3,40
(m, 4H), 4,05 – 4,15
(m, 2H), 4,30 – 4,37 (m,
1H), 4,42 – 4,48
(m, 0,5H), 4,60 – 4,66
(m, 3H), 4,66 – 4,71
(m, 0,5H), 4,78 – 4,88
(m, 1H), 5,05 – 5,15
(m, 1H), 5,28 – 5,31
(d, 1H), 5,40 – 5,44
(m, 1H), 6,07 – 6,18
(m, 1H), 6,75 – 7,15
(m, 1H), 7,25 – 7,3
8 (m, 3H), 7,43 – 7,49
(m, 1H), 7,52 – 7,58
(m, 0,5H), 7,65 – 7,70
(m, 2H), 7,78 (s, 0,5H). Analytische HPLC (Cyansäule): 7,12 min. LC-MS (ES+): m/e = 683, 685 (M+H+).
-
Herstellung
von N-{2-[(2-Benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-1-methansulfonyl-3-oxo-[1,2]-diazepan-4-yl}-3,5-dichlor-4-hydroxybenzamid
(26b). 4-Allyloxy-N-{2-[(2-benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-1-methansulfonyl-3-oxo-[1,2]diazepan-4-yl}-3,5-dichlorbenzamid
(26a) (178 mg, 0,26 mmol) in Dichlormethan (6 ml) wurde 16 Stunden
lang mit DMBA (45 mg, 0,29 mmol) und Pd(PPh3)4 (20 mg, 0,017 mmol) bei Raumtemperatur
behandelt. Das Gemisch wurde mit Dichlormethan (40 ml) verdünnt, dreimal
mit Wasser gewaschen. Die organische Schicht wurde über wasserfreiem
Na2SO4 getrocknet,
filtriert und eingedampft, um einen hellgelben Feststoff zu geben,
der durch Flash-Chromatographie unter Verwendung von Dichlormethan/Methanol
(99,2/0,8 bis 97,5/2,5) gereinigt wurde, um 78 mg (47 % Ausbeute)
des syn-Diastereomers (höherer
Rf) der Titelverbindung und 59 mg (35 % Ausbeute) des anti-Diastereomers (niedriger
Rf) zu liefern. 1H-NMR (500 MHz, CDCl3) für
das syn-Diastereomer: δ 1,60 – 1,70 (m,
1H), 1,88 – 1,91
(m, 1H), 2,22 – 2,32
(m, 2H), 2,48 – 2,58
(m, 1H), 2,84 – 2,90
(m, 1H), 3,00 (s, 3H), 3,27 – 3,40
(m, 1H), 4,05 – 4,18
(m, 2H), 4,22 – 4,35
(m, 1H), 4,55 – 4,80
(m, 2H), 4,86 – 4,89
(d, 1H), 5,10 – 5,15
(m, 1H), 5,59 – 5,61
(d, 1H), 6,75 – 6,77
(d, 1H), 7,05 – 7,07
(d, 1H), 7,25 – 7,37
(m, 5H), 7,70 (s, 2H); und für
das anti-Diastereomer: δ 1,60 – 1,80 (m,
1H), 1,85 – 1,95
(m, 1H), 2,20 – 2,39
(m, 2H), 2,39 – 2,50 (m,
1H), 3,00 – 3,10
(m, 1H), 3,12 (s, 3H), 3,28 – 3,42
(m, 1H), 4,05 – 4,15
(m, 2H), 4,28 – 4,47
(m, 2H), 4,56 – 4,62
(m, 1H), 4,77 – 4,83
(d, 1H), 5,10 – 5,20
(m, 1H), 5,43 (s, 1H), 6,93 – 6,94
(d, 1H), 7,09 – 7,11
(d, 1H), 7,25 – 7,39
(m, 5H), 7,80 (s, 2H). Analytische HPLC: 11,13 und 11,36 min. LC-MS
(ES+) für
das Gemisch der Diastereomeren: m/e = 643, 645 (M+H+).
-
Herstellung
von 3-{2-[6-(3,5-Dichlor-4-hydroxy-benzoylamino)-2-methansulfonyl-7-oxo-[1,2]diazepan-1-yl]-acetylamino}-4-oxo-butansäure (27). N-{2-[(2-Benzyloxy-5-oxo-tetrahydrofuran-3-ylcarbamoyl)-methyl]-1-methansulfonyl-3-oxo-[1,2]diazepan-4-yl}-3,5-dichlor-4-hydroxybenzamid
(26b) wurde in CH3CN (0,5 ml) und 2 N HCl
(1 ml) 7 Stunden lang gerührt.
Die Lösung
wurde im Vakuum auf die Hälfte
des Volumens konzentriert und mit Ether (2 ml × 4) extrahiert. Die vereinigten
Etherschichten wurden mit Ethylacetat/Hexan (1/9, 3 ml) verdünnt, mit
1 M Na2CO3 (2 ml)
gewaschen. Die wässrige
Lösung
wurde mit Ethylacetat/Hexan (1/9, 2 ml × 2) gewaschen, mit 6 N HCl
auf pH ~3 angesäuert,
mit Ethylacetat (1,5 ml × 3)
extrahiert. Die vereinigten Extrakte wurden über wasserfreiem Na2SO4 getrocknet,
filtriert und im Vakuum zur Trockene konzentriert, um 20 mg (47
% Ausbeute) der Titelverbindung zu liefern. 1H-NMR
(500 MHz, CDCl3 + CD3OD) δ 0,75 – 0,90 (m,
1H), 1,18 – 1,30
(m, 3H), 1,67 – 1,88
(m, 1H), 1,93 – 2,30
(m, 2H), 2,40 – 2,61 (m,
1H), 3,10 – 3,16
(m, 3H), 3,95 – 4,30
(m, 2H), 4,30 – 4,50
(m, 1H), 5,00 – 5,08
(d, 1H), 7,81 (s, 2H). Analytische HPLC: 8,10 min. LC-MS (ES+): m/e = 553, 555 (M+H+).
-
*Analytische HPLC Bedingungen:
-
- Säule:
MicrosorbTM C-18, 5μ, 4,6 × 150 mm (außer es ist
anders angegeben).
- Lösungsmittel
A: 0,1 % TFA/1 % MeCN/98,9 % Wasser
- Lösungsmittel
B: 0,1 % TFA/99,9 % MeCN
- Gradient: A zu B über
20 min hinweg bei einer Fließgeschwindigkeit
von 1 ml/min
-
Beispiel 8
-
Tabelle
1. In vitro Daten
-
Insofern
die erfindungsgemäßen Verbindungen
in der Lage sind Caspasen, insbesondere ICE, in vitro zu inhibieren,
und außerdem
an Säuger
oral abgegeben werden können,
sind sie von offensichtlichem klinischem Nutzen für die Behandlung
von durch IL-1, Apoptose, IGIF und IFN-γ vermittelten Erkrankungen.
 dargestellt sind, bereitzustellen,
dargestellt sind, bereitzustellen,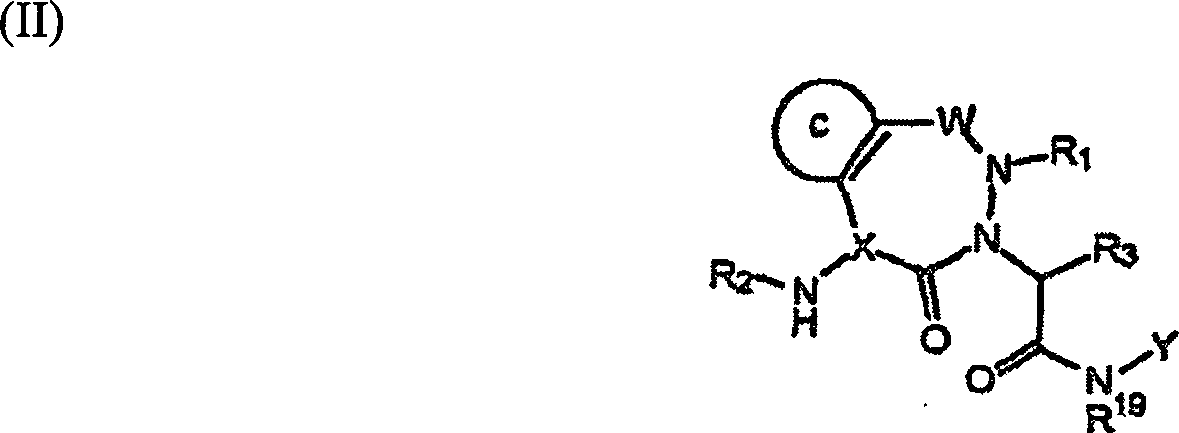
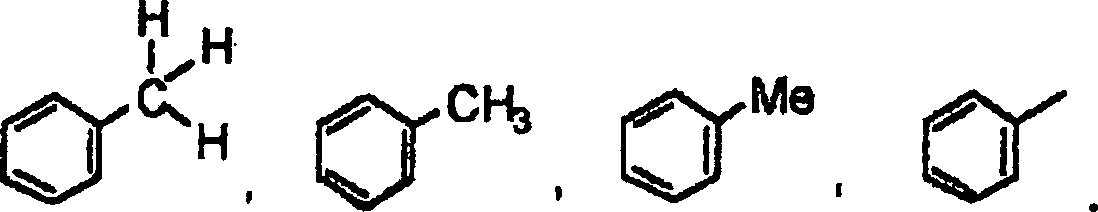
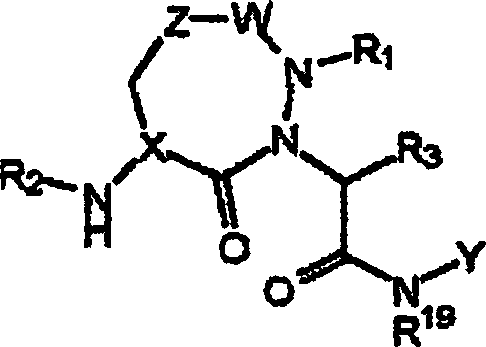
 m 0 oder 1 ist;
m 0 oder 1 ist;




 R7 -C(O)Alkyl, -C(O)Cycloalkyl, -C(O)Alkenyl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -C(O)Heterocyclus oder -C(O)Alkylheterocyclus ist;
R7 -C(O)Alkyl, -C(O)Cycloalkyl, -C(O)Alkenyl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -C(O)Heterocyclus oder -C(O)Alkylheterocyclus ist;


 und die anderen Substituenten wie vorstehend beschrieben sind.
und die anderen Substituenten wie vorstehend beschrieben sind.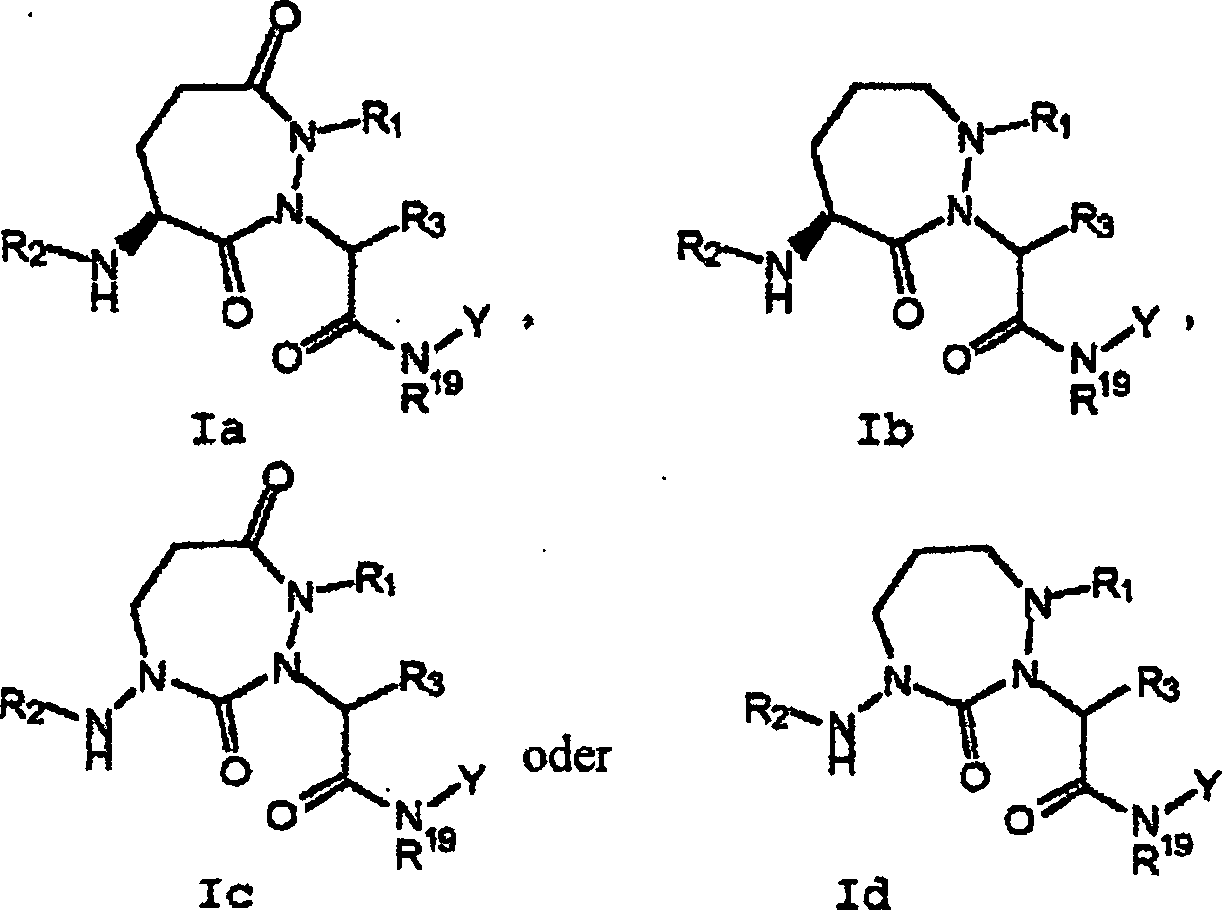

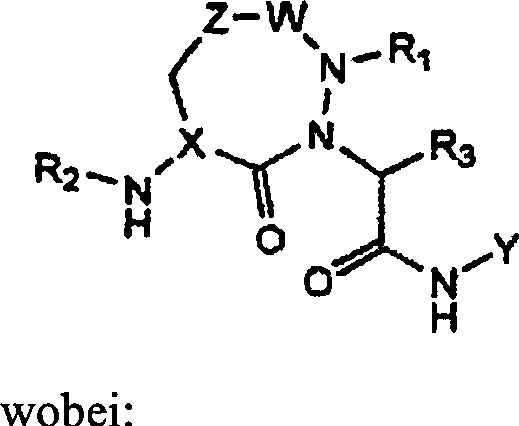
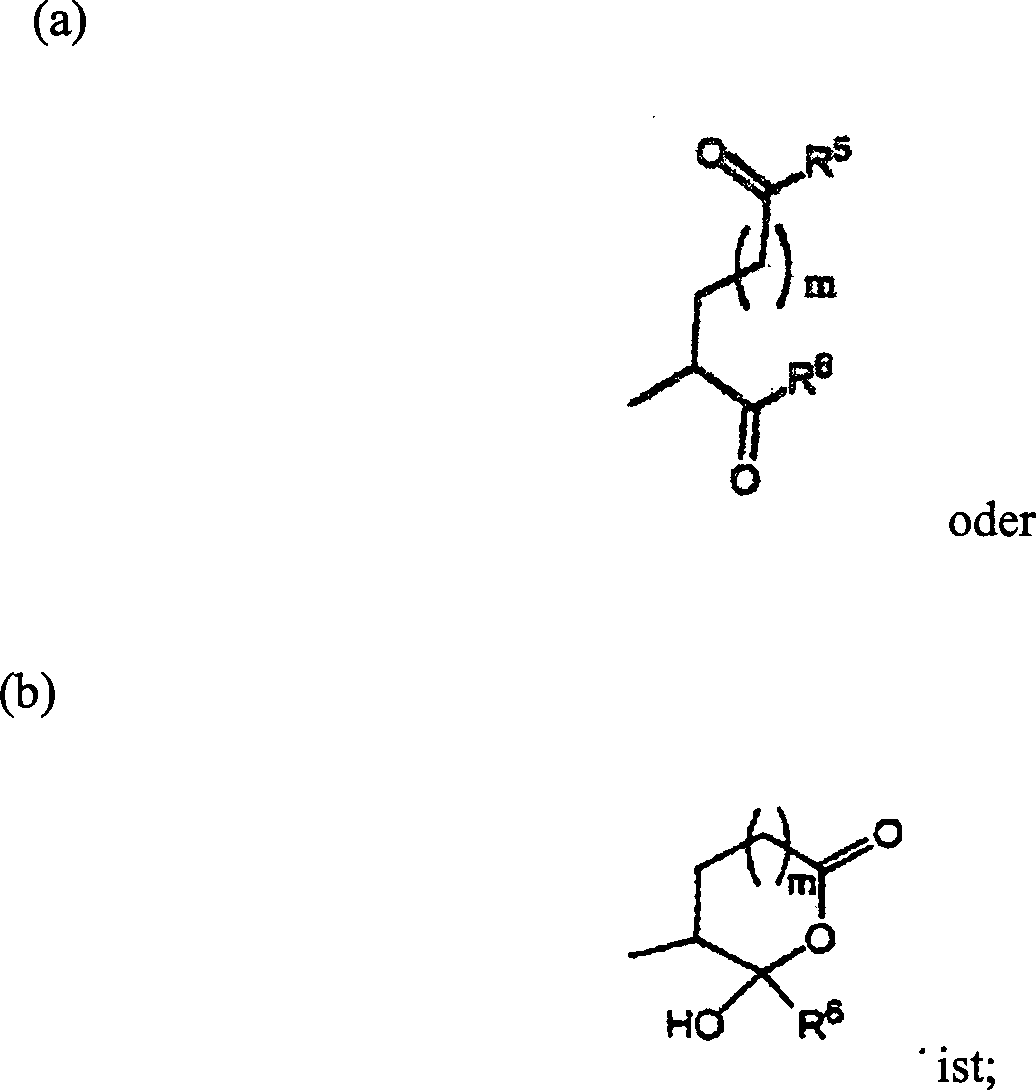

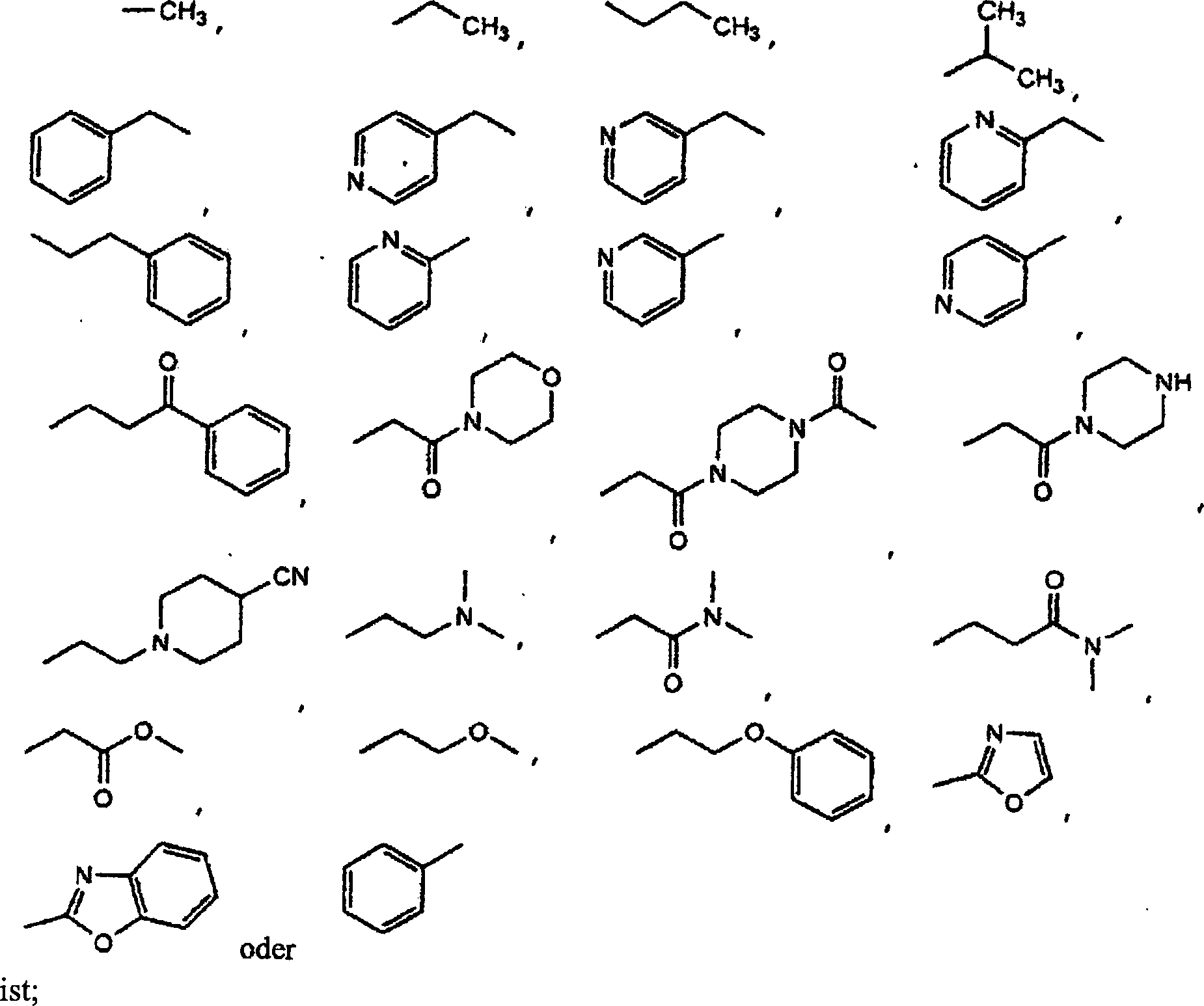






 ist R3:
ist R3: ist R6 -H.
ist R6 -H.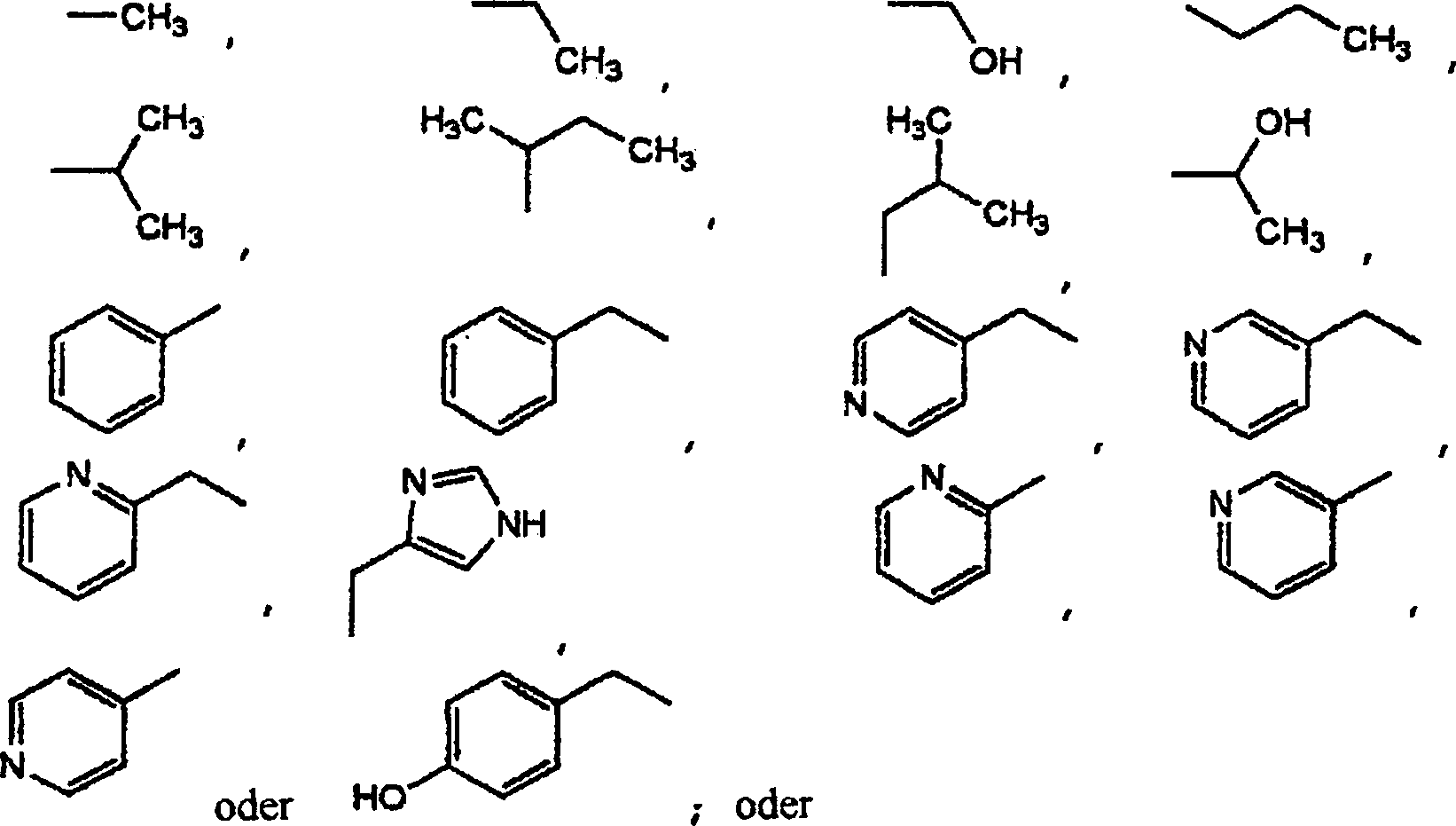
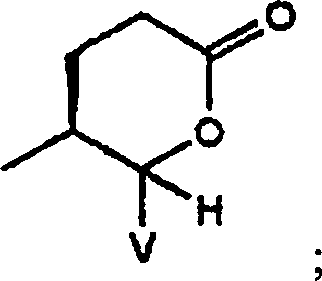

 und die anderen Substituenten wie vorstehend beschrieben sind.
und die anderen Substituenten wie vorstehend beschrieben sind.

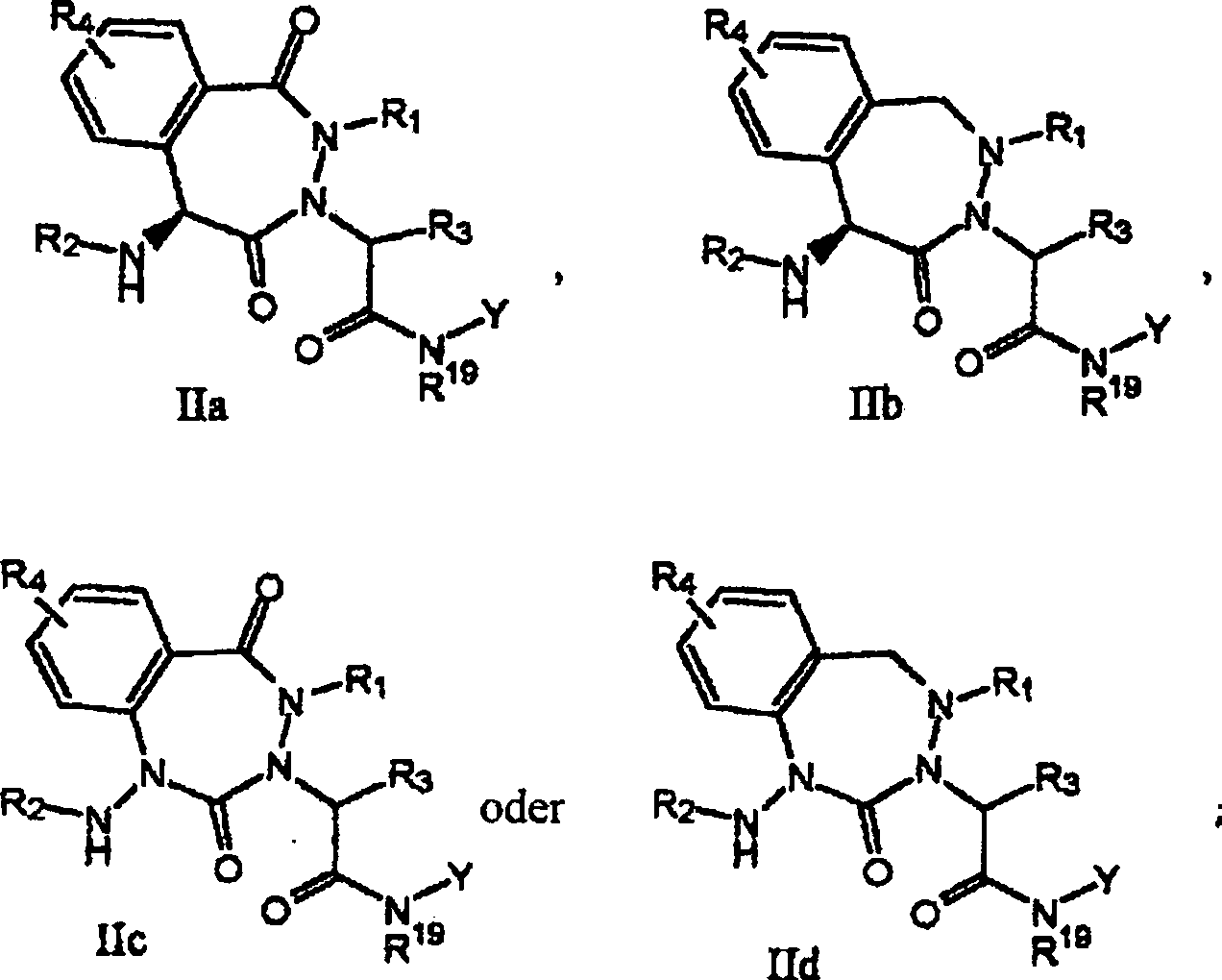
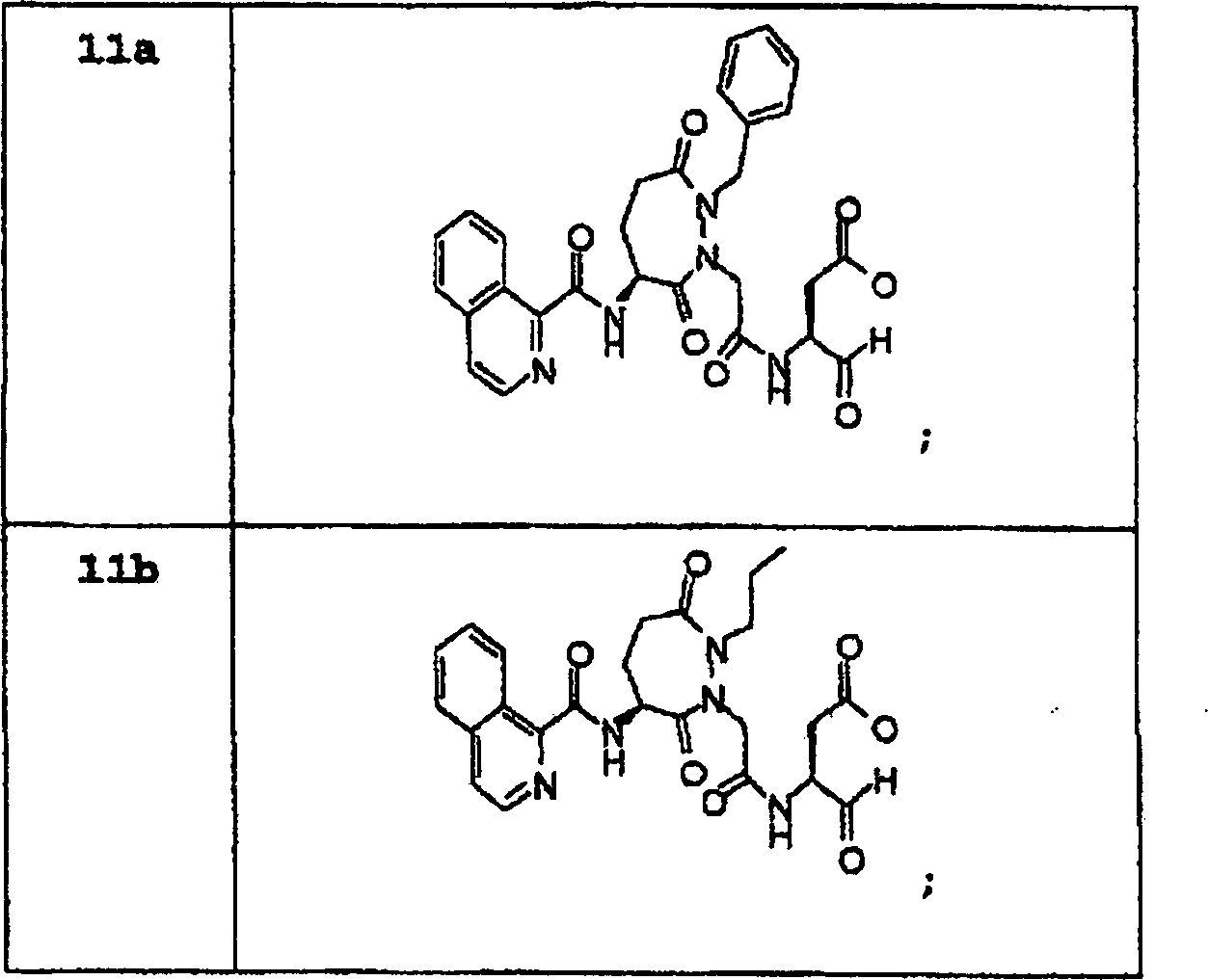 ein, sind aber nicht darauf beschränkt.
ein, sind aber nicht darauf beschränkt.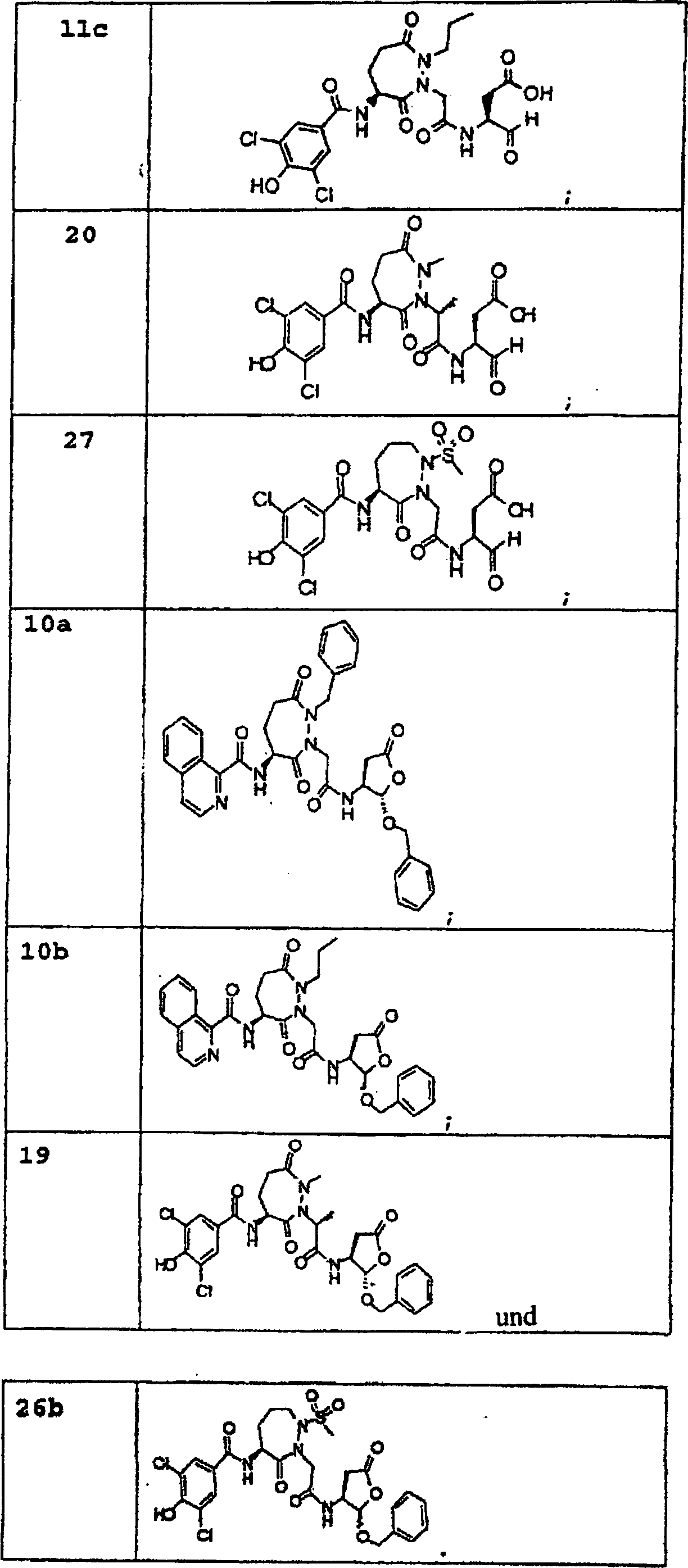

 in Ketal- oder Acetalformen, wenn Y
in Ketal- oder Acetalformen, wenn Y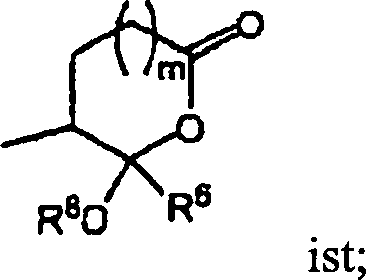 und in Enolformen, wenn Y
und in Enolformen, wenn Y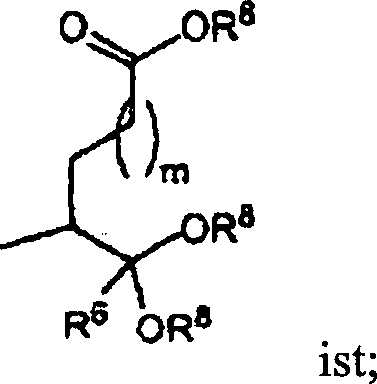
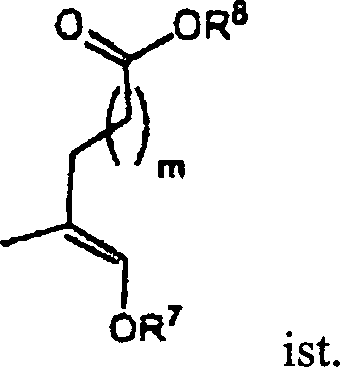

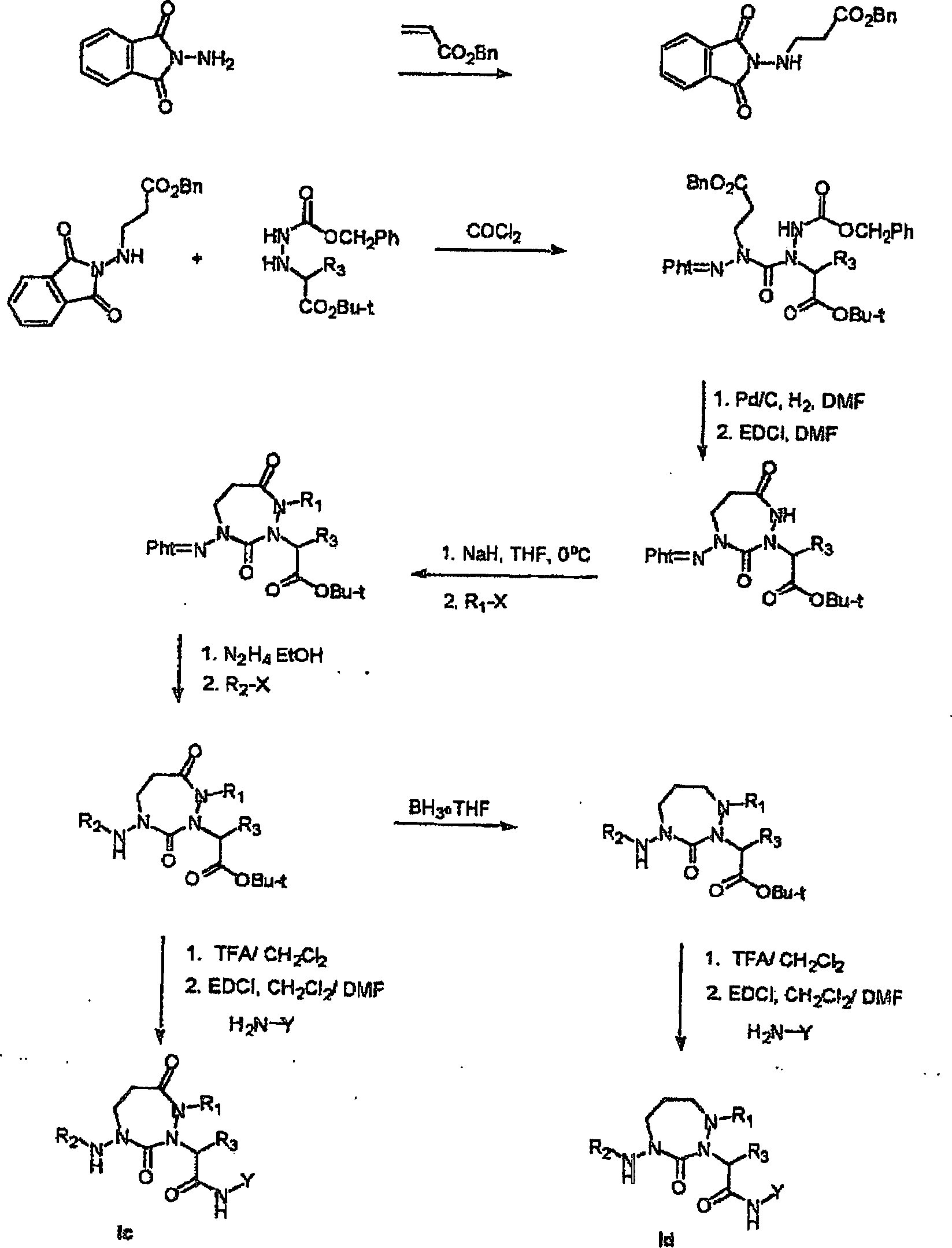
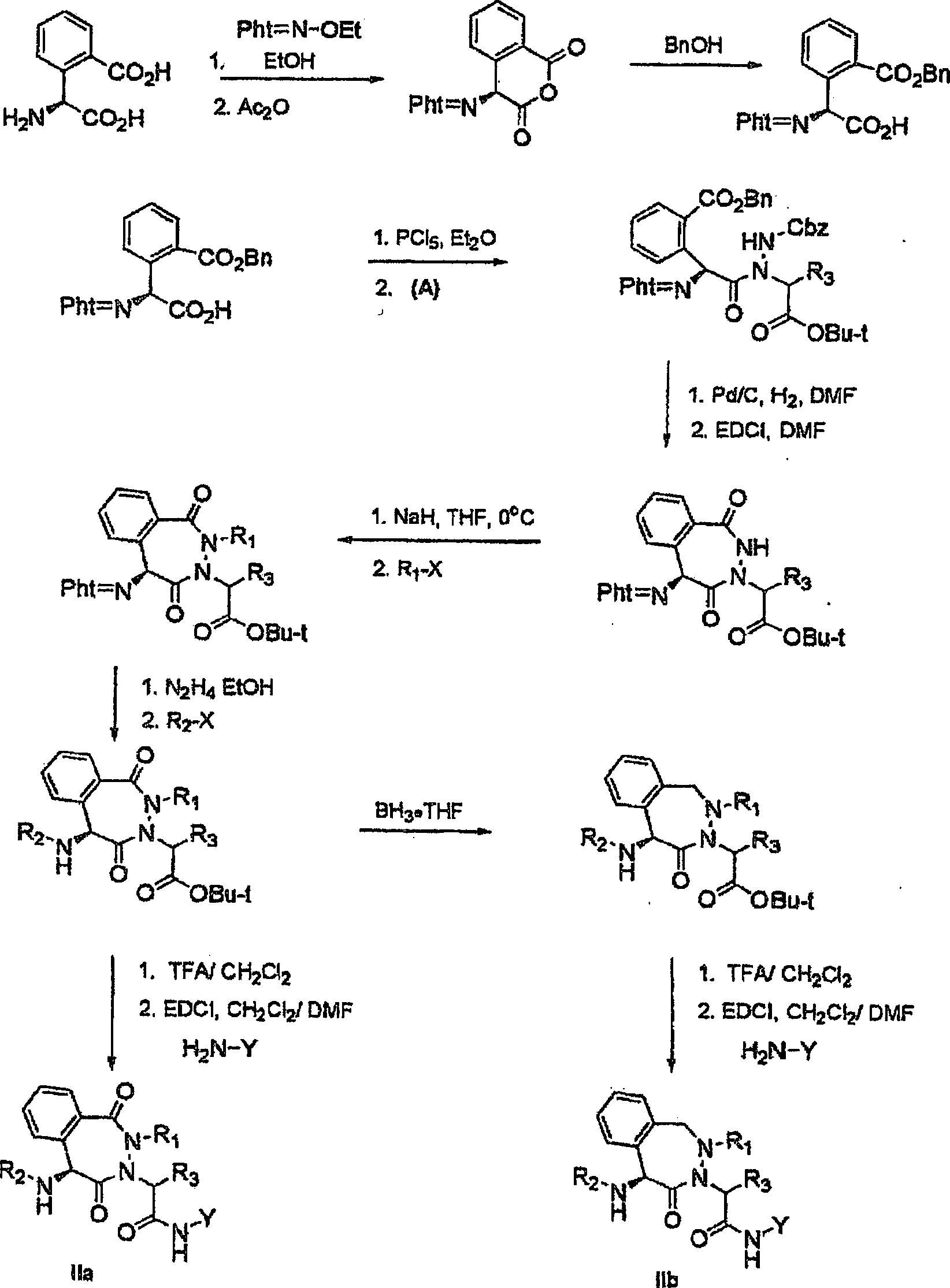


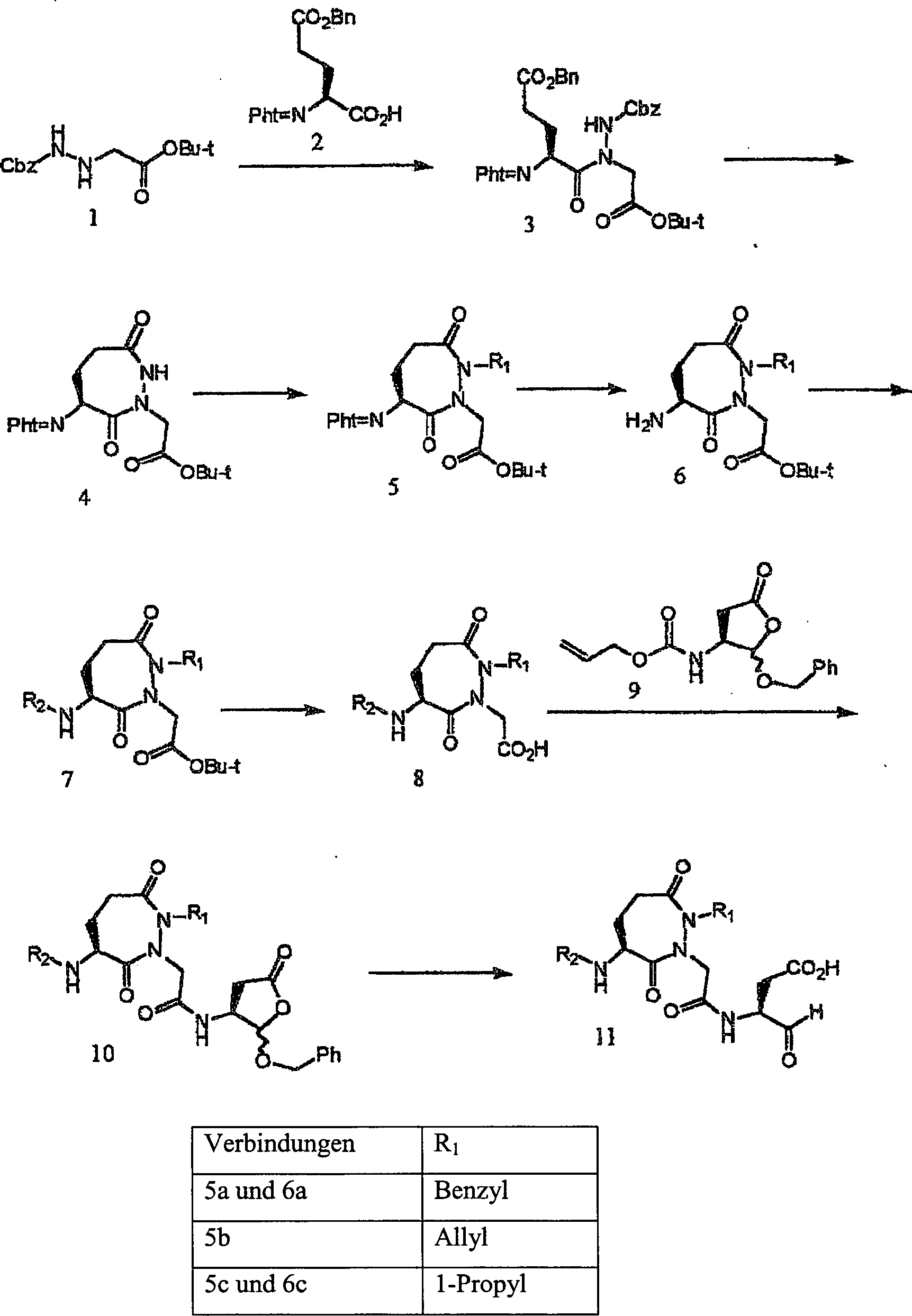

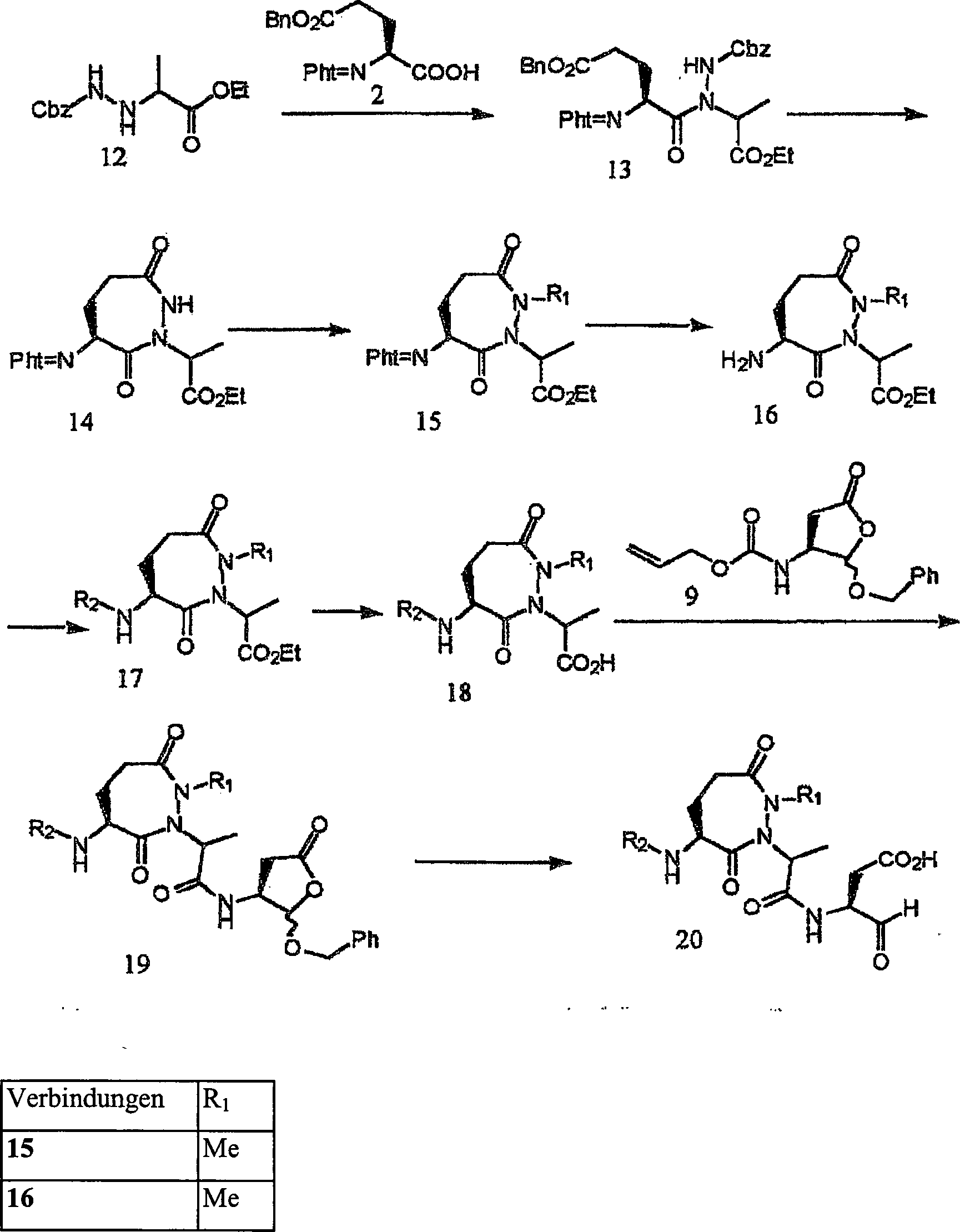
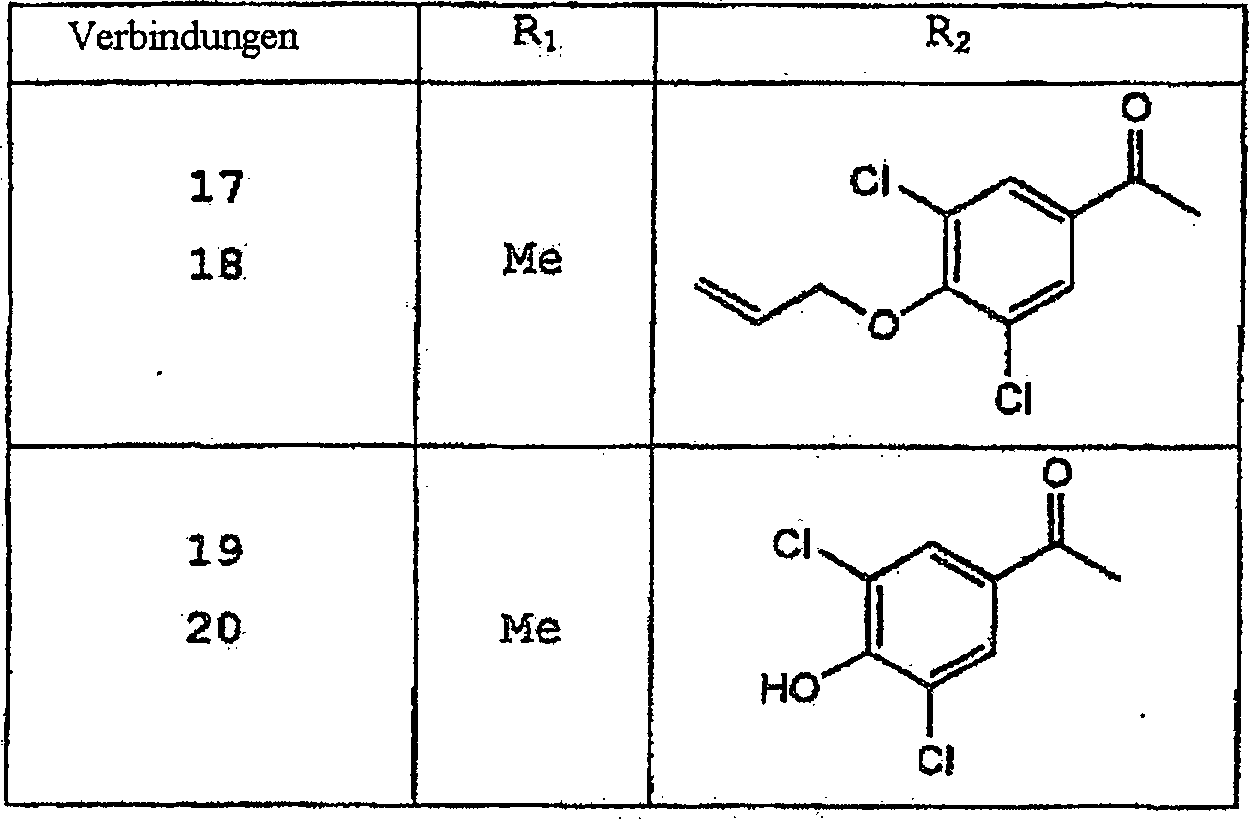


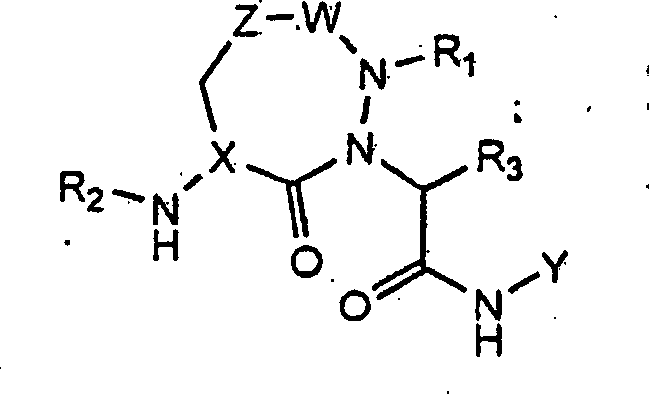 ist; m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder
ist; m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder ist; Z -CH2-, -O-, -S- oder -N(R1)- ist, mit der Maßgabe, daß wenn Z -N(R1)- ist; dann W -C(O)-, -S(O)2- oder -S(O)- ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3,
ist; Z -CH2-, -O-, -S- oder -N(R1)- ist, mit der Maßgabe, daß wenn Z -N(R1)- ist; dann W -C(O)-, -S(O)2- oder -S(O)- ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3, R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -CH2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette,
R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -CH2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette,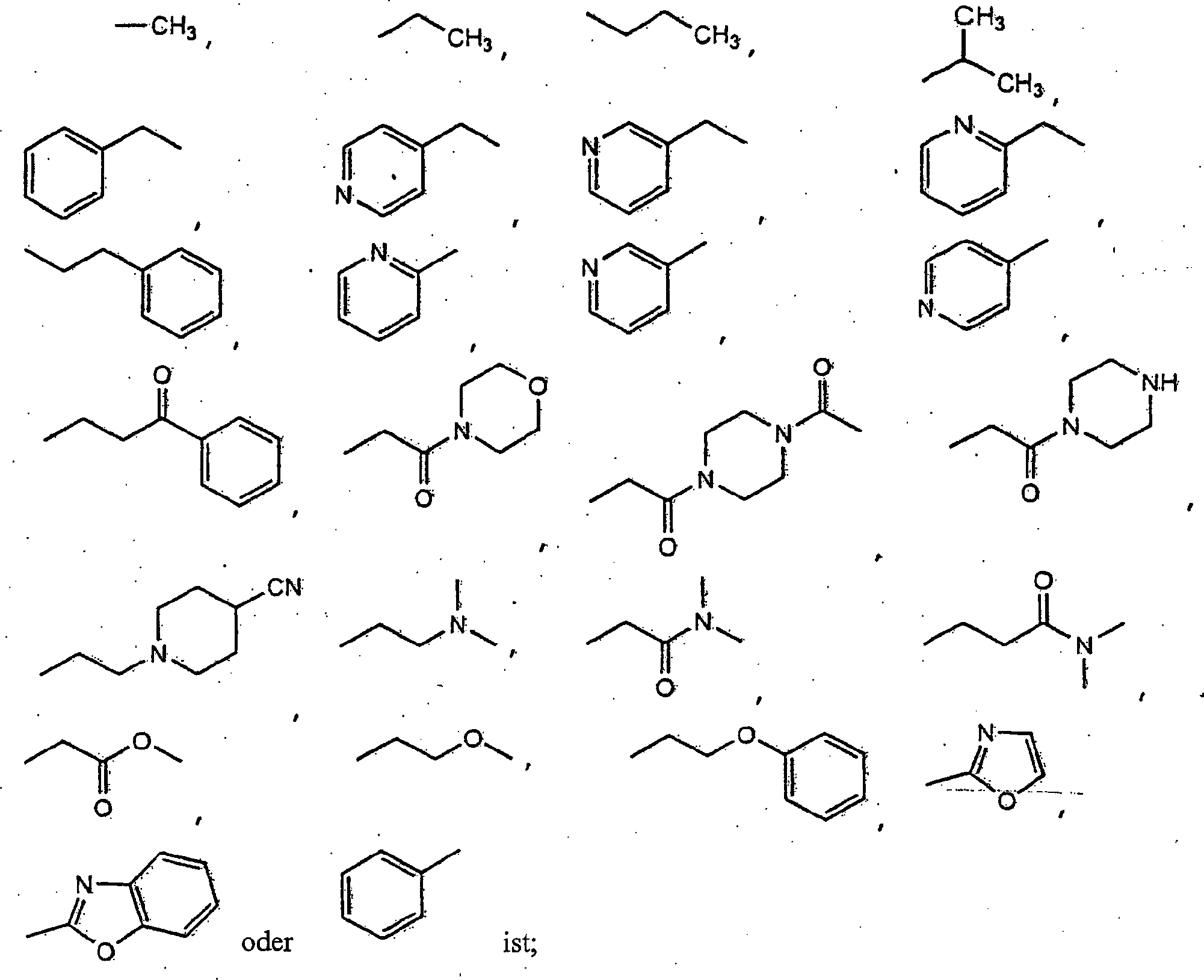 jeder Rest R4 unabhängig -OH, -F; -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2; -N(H)C(O)H, -N(H)C(O)NH2, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R5 -OH, -OR8, -N(H)OH oder -N(H)SO2R8 ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R13 oder -R14 ist; jeder Rest R8 unabhängig Alkyl, -Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, -Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder -P(O)(R15)2 ist; R10-Alkylaryl oder -Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, Alkyl, Aryl, -Heteroaryl, -Cycloalkyl, Alkylaryl oder -Alkylheteroaryl ist; R13 Alkylaryl oder Alkylheteroaryl ist; R14
jeder Rest R4 unabhängig -OH, -F; -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2; -N(H)C(O)H, -N(H)C(O)NH2, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R5 -OH, -OR8, -N(H)OH oder -N(H)SO2R8 ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R13 oder -R14 ist; jeder Rest R8 unabhängig Alkyl, -Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, -Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder -P(O)(R15)2 ist; R10-Alkylaryl oder -Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, Alkyl, Aryl, -Heteroaryl, -Cycloalkyl, Alkylaryl oder -Alkylheteroaryl ist; R13 Alkylaryl oder Alkylheteroaryl ist; R14 wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist, und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, -R18 oder Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig -H, -OH, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, -Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder -C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, Heteroaryl, -Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -O(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2, -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2-Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.
wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist, und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, -R18 oder Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig -H, -OH, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, -Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder -C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, Heteroaryl, -Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -O(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2, -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2-Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.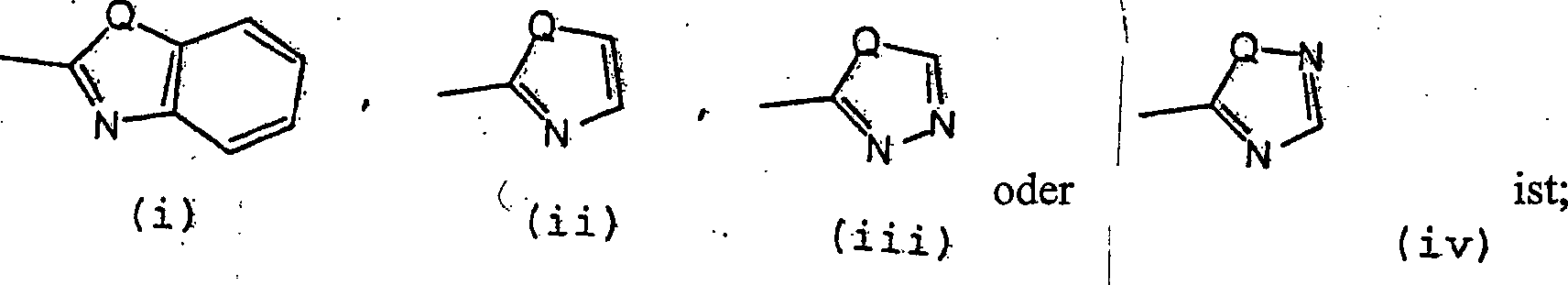
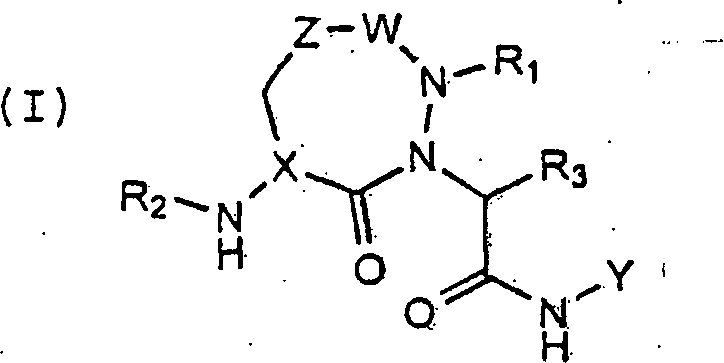 m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder
m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder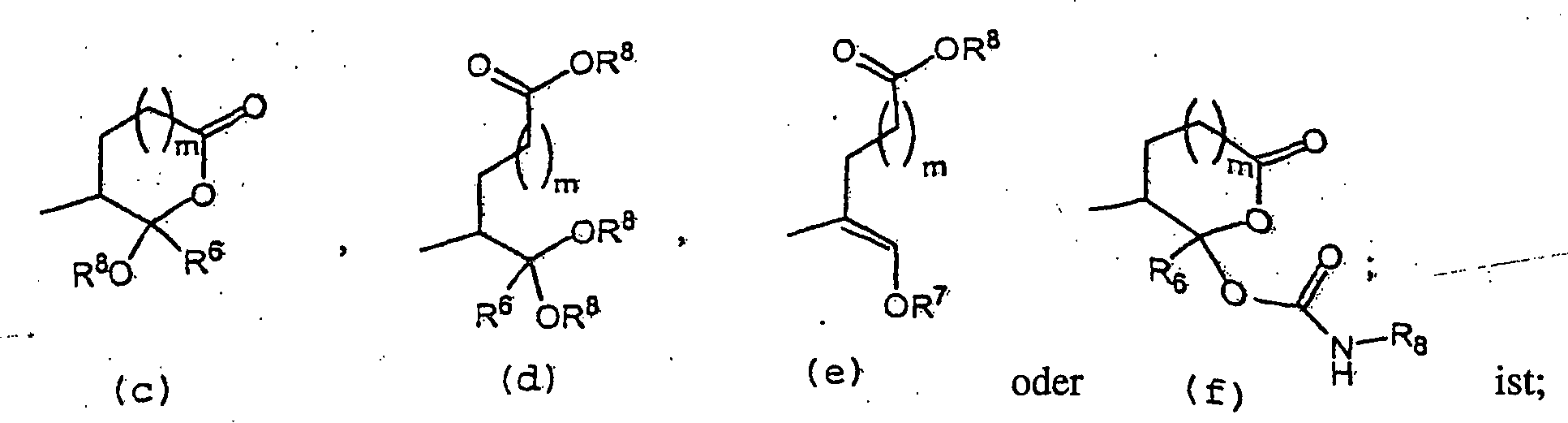 ist; Z -CH2-, -O-, -S- oder -N(R1)- ist, mit der Maßgabe, daß wenn Z -N(R1)- ist, dann W -C(O)-, -S(O)2- oder -S(O)- ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3,
ist; Z -CH2-, -O-, -S- oder -N(R1)- ist, mit der Maßgabe, daß wenn Z -N(R1)- ist, dann W -C(O)-, -S(O)2- oder -S(O)- ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3, R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -CH2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette,
R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -CH2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette, jeder Rest R4 unabhängig -OH, -F, -Cl, Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H; -N(H)C(O)NH2, Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R17 oder -R14 ist; R7 -C(O)Alkyl, -C(O)Cycloalkyl, -C(O)Alkenyl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -C(O)Heterocyclus oder -C(O)Alkylheterocyclus ist; jeder Rest R8 unabhhängig -Alkyl, -Cycloalkyl, -Aryl, Heteroaryl, -Heterocyclyl, Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder P(O)(R15)2 ist; R10 Alkylaryl oder Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, Alkylaryl oder Alkylheteroaryl ist; R13 Alkylaryl oder Alkylheteroaryl ist; R14
jeder Rest R4 unabhängig -OH, -F, -Cl, Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H; -N(H)C(O)NH2, Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R17 oder -R14 ist; R7 -C(O)Alkyl, -C(O)Cycloalkyl, -C(O)Alkenyl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -C(O)Heterocyclus oder -C(O)Alkylheterocyclus ist; jeder Rest R8 unabhhängig -Alkyl, -Cycloalkyl, -Aryl, Heteroaryl, -Heterocyclyl, Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder P(O)(R15)2 ist; R10 Alkylaryl oder Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, Alkylaryl oder Alkylheteroaryl ist; R13 Alkylaryl oder Alkylheteroaryl ist; R14 wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist; und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, R18 oder Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig H, -OH, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder -C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, Heteroaryl, Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, -N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2, -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2-Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist; das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.
wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist; und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, R18 oder Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig H, -OH, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder -C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, Heteroaryl, Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, -N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2, -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2-Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist; das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.
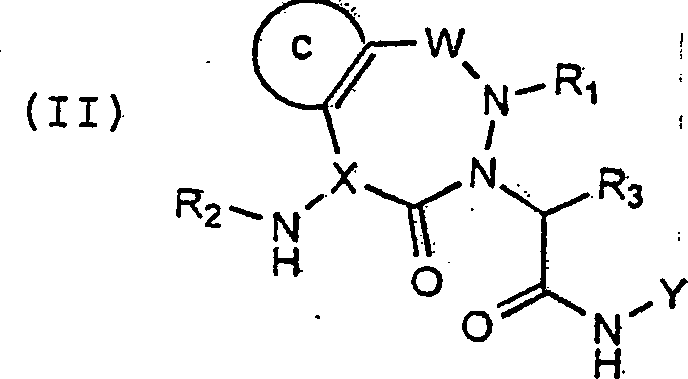 C ein Arylring ist, wobei jedes an den C-Ring gebundene Wasserstoffatom gegebenenfalls mit -R4 substituiert ist; m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder
C ein Arylring ist, wobei jedes an den C-Ring gebundene Wasserstoffatom gegebenenfalls mit -R4 substituiert ist; m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder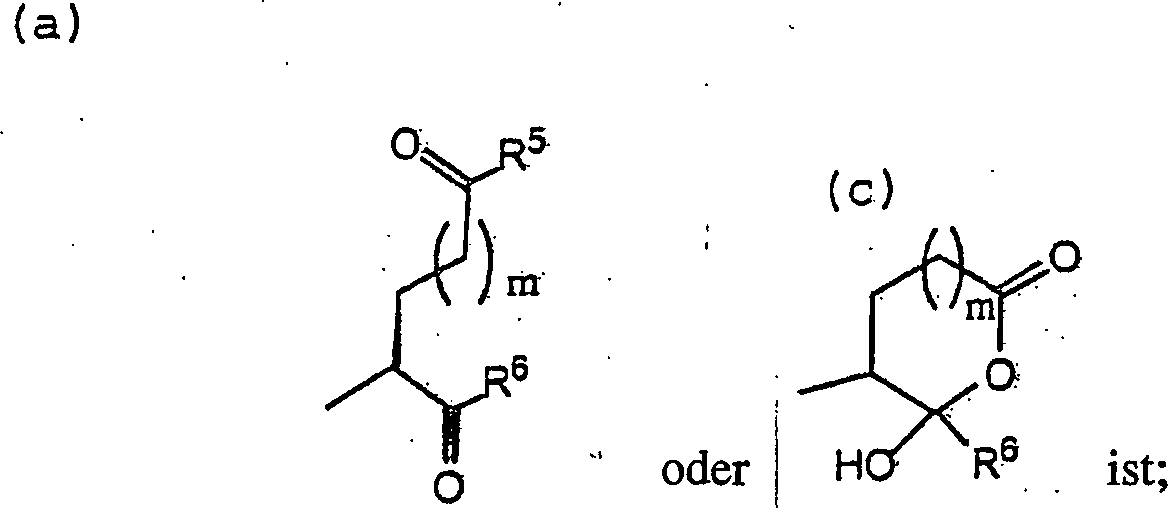 ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3,
ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3, R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -CH2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette,
R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -CH2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette, jeder Rest R4 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O- Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R5 -OH, -OR8, -N(H)OH oder -N(H)SO2R8 ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R13 oder -R14 ist; jeder Rest R8 unabhhängig -Alkyl, -Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, -Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder -P(O)(R15)2 ist; R10 Alkylaryl oder -Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl oder -Alkylheteroaryl ist; R13 -Alkylaryl oder -Alkylheteroaryl ist; R14
jeder Rest R4 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O- Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R5 -OH, -OR8, -N(H)OH oder -N(H)SO2R8 ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R13 oder -R14 ist; jeder Rest R8 unabhhängig -Alkyl, -Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, -Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder -P(O)(R15)2 ist; R10 Alkylaryl oder -Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, -Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl oder -Alkylheteroaryl ist; R13 -Alkylaryl oder -Alkylheteroaryl ist; R14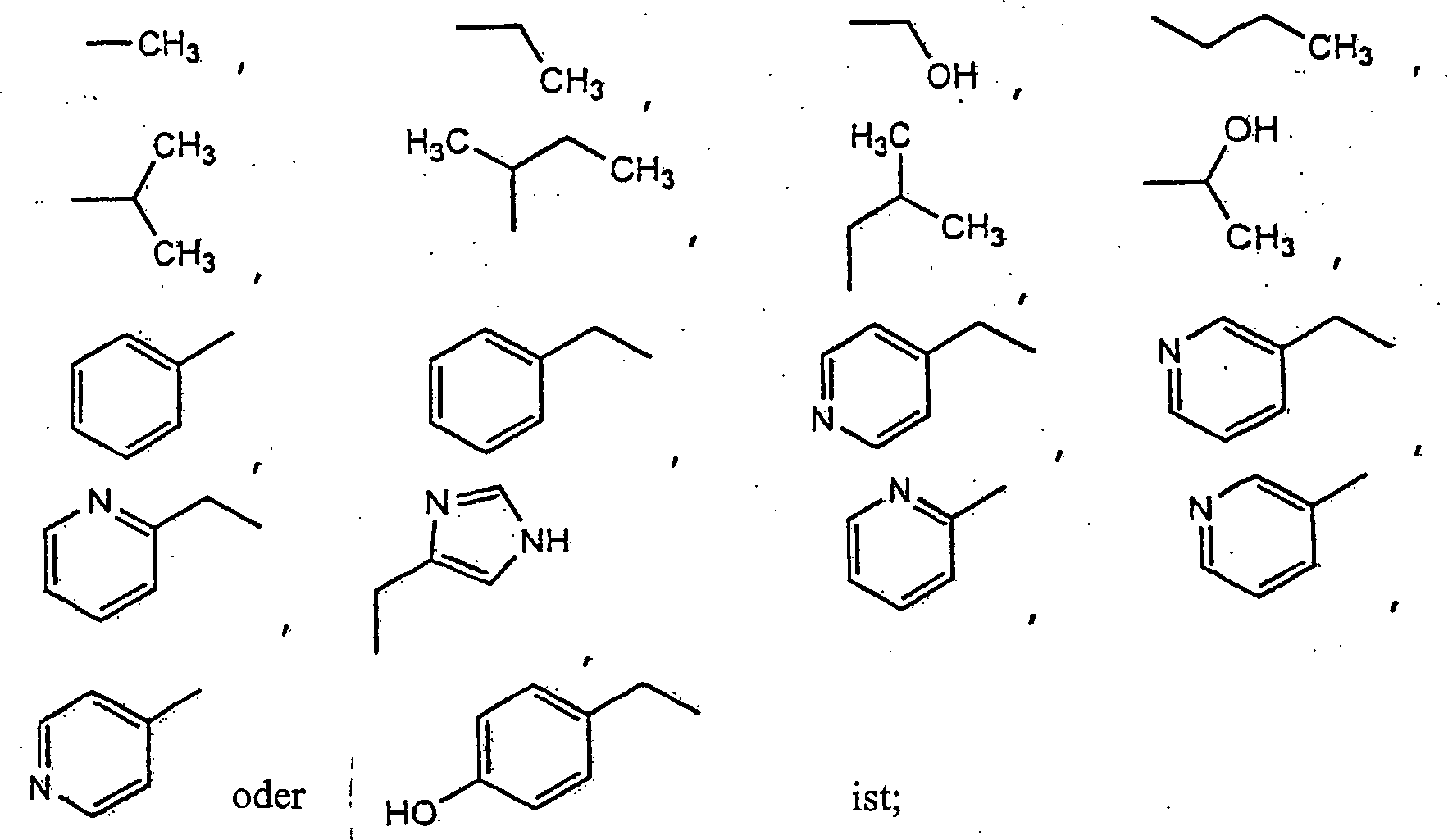 wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist, und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, -R18 oder -Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig -H, -OH, Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, -Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder, Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder -C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, -Heteroaryl, Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, -N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2; -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2-Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.
wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist, und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, -R18 oder -Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig -H, -OH, Alkyl, -Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, -Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder, Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)Alkyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder -C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, -Heteroaryl, Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, -N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2; -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2-Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.
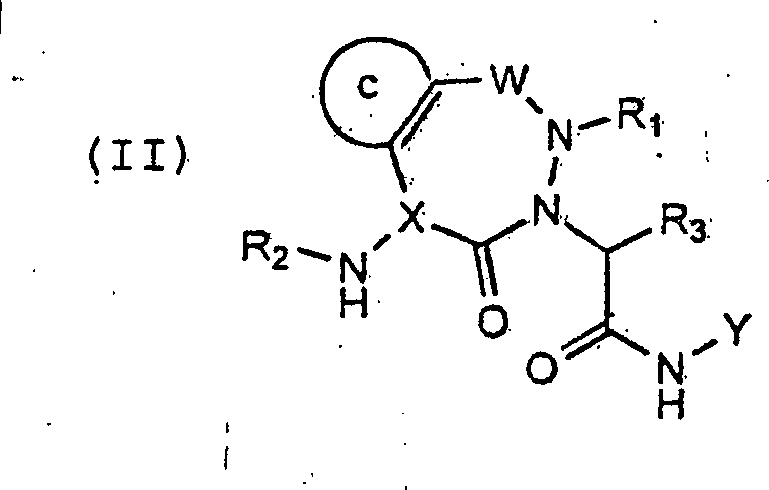 C ein Arylring ist, wobei jedes an den C-Ring gebundene Wasserstoffatom gegebenenfalls mit -R4 substituiert ist; m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder
C ein Arylring ist, wobei jedes an den C-Ring gebundene Wasserstoffatom gegebenenfalls mit -R4 substituiert ist; m 0 oder 1 ist; W -CH2-, -C(O)-, S(O)2 oder -S(O)- ist; X -C(H)-, -C(R8)- oder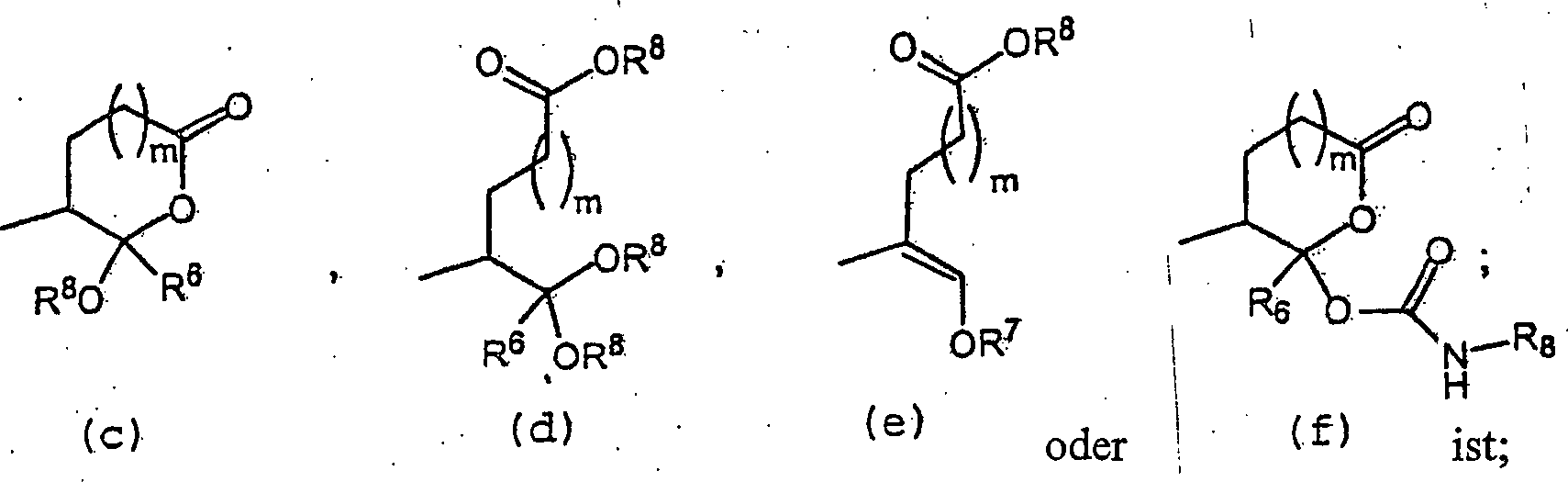 ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3,
ist; jeder Rest R1 unabhängig -H, -S(O)2-CH3,
 R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -H2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette,
R2 -C(O)R8, -C(O)C(O)R8, -S(O)2R8, -S(O)R8, -C(O)OR8, -C(O)N(H)R8, -S(O)2N(H)-R8, -S(O)N(H)-R8, -C(O)C(O)N(H)R8, -C(O)CH=CHR8, -C(O)CH2OR8, -C(O)CH2N(H)R8, -C(O)N(R8)2, -S(O)2N(R8)2, -S(O)N(R8)2, -C(O)C(O)N(R8)2, -C(O)CH2N(R8)2, -H2-R8, -CH2-Alkenyl-R8 oder -CH2-Alkinyl-R8 ist; R3 -H, eine Aminosäure-Seitenkette, jeder Rest R4 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2; -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R13 oder R14 ist; R7 -C(O)Alkyl, -C(O)Cycloalkyl, -C(O)Alkenyl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -C(O)Heterocyclus oder -C(O)Alkylheterocyclus ist; jeder Rest R8 unabhhängig -Alkyl, -Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder -P(O)(R15)2 ist; R10-Alkylaryl oder -Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, -Alkyl, Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl oder -Alkylheteroaryl ist; R13 -Alkylaryl oder -Alkylheteroaryl ist; R14
jeder Rest R4 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2; -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl, -C(O)Alkyl, -CH2NH2, -CH2N(H)Alkyl, -CH2N(Alkyl)2 oder -N(H)C(O)O-Alkyl ist; R6 -H, -CH2OR9, -CH2SR10, -CH2N(H)R9, -CH2N(R9)R11, -C(H)N2, -CH2F, -CH2Cl, -CH2Br, -CH2I, -C(O)N(R11)2, -R13 oder R14 ist; R7 -C(O)Alkyl, -C(O)Cycloalkyl, -C(O)Alkenyl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -C(O)Heterocyclus oder -C(O)Alkylheterocyclus ist; jeder Rest R8 unabhhängig -Alkyl, -Cycloalkyl, -Aryl, -Heteroaryl, -Heterocyclyl, Alkylcycloalkyl, -Alkylaryl, -Alkylheteroaryl oder -Alkylheterocyclyl ist; R9 -H, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, Alkylaryl, -Alkylheteroaryl, -Aryl, -Heteroaryl oder -P(O)(R15)2 ist; R10-Alkylaryl oder -Alkylheteroaryl ist; jeder Rest R11 unabhängig -H, -Alkyl, Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl oder -Alkylheteroaryl ist; R13 -Alkylaryl oder -Alkylheteroaryl ist; R14 wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist, und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, -R18 oder -Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig -H, -OH, A1kyl, Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, -Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)A1kyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder-C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, -Heteroaryl, -Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, -N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2, -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.
wobei Q -O- oder -S- ist, jedes Wasserstoffatom in (i) gegebenenfalls durch -R17 ersetzt ist, und jedes Wasserstoffatom in (ii), (iii) und (iv) gegebenenfalls durch -R17, -R18 oder -Alkyl-R18 ersetzt ist; jeder Rest R15 unabhängig -H, -OH, A1kyl, Aryl, -Heteroaryl, -Cycloalkyl, -Alkylaryl, -Alkylheteroaryl, -O-Alkyl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl oder -O-Alkylheteroaryl ist; jeder Rest R17 unabhängig -OH, -F, -Cl, -Br, -I, -NO2, -CN, -NH2, -CO2H, -C(O)NH2, -N(H)C(O)H, -N(H)C(O)NH2, -SO2NH2, -C(O)H, -Alkyl, -Cycloalkyl, -Perfluoralkyl, -O-Alkyl, -N(H)Alkyl, -N(Alkyl)2, -CO2Alkyl, -C(O)N(H)Alkyl, -C(O)N(Alkyl)2, -N(H)C(O)Alkyl, -N(H)C(O)N(H)Alkyl, -N(H)C(O)N(Alkyl)2, -S(O)2N(H)Alkyl, -S(O)N(H)A1kyl, -S(O)2N(Alkyl)2, -S(O)N(Alkyl)2, -S-Alkyl, -S(O)2Alkyl, -S(O)Alkyl oder-C(O)Alkyl ist; und jeder Rest R18 unabhängig -Aryl, -Heteroaryl, -Alkylaryl, -Alkylheteroaryl, -O-Aryl, -O-Heteroaryl, -O-Alkylaryl, -O-Alkylheteroaryl, -N(H)Aryl, -N(Aryl)2, -N(H)Heteroaryl, -N(Heteroaryl)2, -N(H)Alkylaryl, -N(Alkylaryl)2, -N(H)Alkylheteroaryl, -N(Alkylheteroaryl)2, -S-Aryl, -S-Heteroaryl, -S-Alkylaryl, -S-Alkylheteroaryl, -C(O)Aryl, -C(O)Heteroaryl, -C(O)Alkylaryl, -C(O)Alkylheteroaryl, -CO2Aryl, -CO2Heteroaryl, -CO2Alkylaryl, -CO2Alkylheteroaryl, -C(O)N(H)Aryl, -C(O)N(Aryl)2, -C(O)N(H)Heteroaryl, -C(O)N(Heteroaryl)2, -C(O)N(H)Alkylaryl, -C(O)N(Alkylaryl)2, -C(O)N(H)Alkylheteroaryl, -C(O)N(Alkylheteroaryl)2, -S(O)2Aryl, -S(O)-Aryl, -S(O)2-Heteroaryl, -S(O)-Heteroaryl, -S(O)2Alkylaryl, -S(O)-Alkylaryl, -S(O)2-Alkylheteroaryl, -S(O)-Alkylheteroaryl, -S(O)2N(H)-Aryl, -S(O)N(H)-Aryl, -S(O)2NH-Heteroaryl, -S(O)NH-Heteroaryl, -S(O)2N(H)-Alkylaryl, -S(O)N(H)-Alkylaryl, -S(O)2N(H)-Alkylheteroaryl, -S(O)N(H)-Alkylheteroaryl, -S(O)2N(Aryl)2, -S(O)N(Aryl)2, -S(O)2N(Heteroaryl)2, -S(O)N(Heteroaryl)2, -S(O)2N(Alkylaryl)2, -S(O)N(Alkylaryl)2, -S(O)2N(Alkylheteroaryl)2, -S(O)N(Alkylheteroaryl)2, -N(H)C(O)N(H)Aryl, -N(H)C(O)N(H)Heteroaryl, -N(H)C(O)N(H)Alkylaryl, -N(H)C(O)N(H)Alkylheteroaryl, -N(H)C(O)N(Aryl)2, -N(H)C(O)N(Heteroaryl)2, -N(H)C(O)N(Alkylaryl)2 oder -N(H)C(O)N(Alkylheteroaryl)2 ist; jedes Heteroaryl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält und bei dem mindestens ein Ring des Ringsystems aromatisch ist; jedes Heterocyclyl ein mono- oder polycyclisches Ringsystem ist, das 1 bis 15 Kohlenstoffatome und 1 bis 4 Heteroatome, ausgewählt aus Schwefel, Stickstoff und Sauerstoff, enthält, wobei das mono- oder polycyclische Ringsystem gegebenenfalls ungesättigte Bindungen enthalten kann, aber nicht aromatisch ist; und jedes Cycloalkyl ein mono- oder polycyclisches, nicht aromatisches Kohlenwasserstoff-Ringsystem ist, das gegebenenfalls ungesättigte Bindungen in dem Ringsystem enthalten kann.