-
Die
vorliegende Erfindung betrifft ein Verfahren zur Herstellung von
leichten Olefinen, die reich an Ethylen sind, aus Methanol und Dimethylether.
-
In
den letzten Jahrzehnten hat bei der Herstellung von synthetischen
Fasern, Kunststoffen und Kautschuk ein beachtliches Wachstum stattgefunden.
Dieses Wachstum beruht zu einem sehr großen Maße und wird gefördert durch
das sich vergrößernde Angebot
an billigen petrolchemischen Rohmaterialien wie Ethylen, Propylen
und anderen Olefinen mit 4 und 5 Kohlenstoffatomen. Mit diesem Wachstum
geht ein steigender Bedarf an Alkylat einher, das durch Umsetzung
von Olefinen mit Isobutan hergestellt wird, zur Verwendung als Komponente
von Benzin mit hoher Oktanzahl.
-
Der
aufkeimende Bedarf an Olefinen, insbesondere Ethylen, Propylen und
Butenen, hat natürlich
zu Zeiträumen
der Verknappung geführt,
was zu wesentlichen Preiserhöhungen
bei den Einsatzmaterialien für
die kommerzialisierten Technologien geführt hat. Diese Einsatzmaterialien
sind vornehmlich C2- bis C4-Paraffine, die
zusammen mit Erdgas und/oder paraffinischem Straight Run-Naphtha
hergestellt werden. Diese Einsatzmaterialien können wesentlich teurer als
Methan sein, was es wünschenswert
macht, wirksame Mittel zur Umsetzung von Methan in Olefine bereitzustellen.
-
Die
Umsetzung von Methan zu Methanol gefolgt von der Umsetzung von Methanol
zu leichten Olefinen gehört
zu den ökonomischsten
Wegen zur Herstellung von leichten Olefinen aus Methan. Dabei ist
bekannt, dass Methanol oder Methylether durch Kontakt bei bestimmten
Bedingungen mit bestimmten Typen von kristallinen Zeolithmaterialien
katalytisch zu olefinhaltigen Kohlenwasserstoffmischungen umgesetzt
werden können.
Die US-A-4 025 575
und US-A-4 038 889 offenbaren z.B. Verfahren, durch die Methanol
und/oder Methylether über
einem Zeolithkatalysator mit einem Einschränkungsindex (Constraint Index)
1-12, insbesondere ZSM-5, zu einem olefinhaltigen Produkt umgesetzt
werden können.
Tatsächlich
setzt ZSM-5 mit einer verlängerten
Katalysatorlebensdauer, bevor eine Katalysatorregenerierung notwendig
wird, Methanol und/oder Methylether zu Kohlenwasserstoffen um, die
eine relativ hohe Konzentration an leichten Olefinen enthalten.
-
Es
ist auch berichtet worden, dass andere Typen von Zeolithkatalysatoren
verwendet werden können, um
Methanol und/oder Methylether zu olefinhaltigen Kohlenwasserstoffprodukten
umzusetzen, die noch höhere
Anteile an leichten Olefinen enthalten, als sie bereits mit ZSM-5
erhalten wurden. Zum Beispiel offenbart die US-A-4 079 095, dass
Zeolithe des Erionit-Offretit-Chabazit-Typs
und insbesondere ZSM-34 brauchbar eingesetzt werden können, um
die Umsetzung von Methanol und/oder Methylether zu Produkten zu
fördern,
die eine größere Menge
von Ethylen und Propylen umfassen. Obwohl Katalysatoren vom Typ
Erionit-Offretit-Chabazit für
die Herstellung von leichten Olefinen hoch selektiv sind, neigen
Zeolithe mit kleineren Poren dazu, im Vergleich mit ZSM-5 schnell
zu altern, wenn sie für
die Methanol/Methylether-Umsetzung verwendet werden.
-
Die
US-A-4 677 242 und die US-A-4 752 651 offenbaren die Umsetzung von
Methanol zu C2- bis C4-Olefinen über verschiedenen
Silicoaluminophosphaten und "nicht-zeolithischen
Molekularsieben" (wie
Metallaluminophosphaten) und lehren, dass die Zugabe von Verdünnungsmitteln,
wie aromatischen Materialien, die eine kinetischen Durchmesser größer die
Porengröße des Molekularsiebs
haben, das Ethylen-zu-Propylen-Verhältnis in dem Produkt erhöht.
-
T.
Mole, G. Bett und D.J. Seddon, Journal of Catalysis 84, 435 (1983)
offenbaren, dass die Gegenwart von aromatischen Verbindungen die
Zeolith-katalysierte Umsetzung von Methanol zu Kohlenwasserstoffen
beschleunigen kann. Der Artikel berichtet von Ethylenausbeuten von
5 bis 22 %, wenn Methanol katalytisch in der Gegenwart von Benzol
oder Toluol über
ZSM-5 bei Subatmos phärendruck,
279 bis 350 °C
und 100%igem Methanolumsatz umgesetzt wird.
-
Die
US-A-4 499 314 offenbart, dass die Zugabe von verschiedenen Promotoren,
einschließlich
aromatischen Verbindungen wie Toluol, die Umsetzung von Methanol
zu Kohlenwasserstoffen über
Zeolithen wie ZSM-5, die eine ausreichende Porengröße haben,
um die Sorption und Diffusion des Promotors zu ermöglichen,
beschleunigt. Insbesondere lehrt die US-A-4 499 314, dass der erhöhte Umsatz,
der sich aus der Zugabe des Promotors ergibt, die Verwendung von
weniger strengen Bedingungen ermöglicht,
insbesondere niedrigeren Temperaturen, die die Ausbeute an niederen
Olefinen erhöhen
(Spalte 4, Zeilen 17 bis 22). Somit vermindert in Beispiel 1 der
Patentschrift die Zugabe von Toluol als Promotor die Temperatur,
die notwendig ist, um einen vollständigen Umsatz von Methanol
zu erreichen, von 295 auf 288 °C,
während
die Ethylenausbeute von 11 auf 18 Gew.-% erhöht wird. In den Beispielen
der US-A-4 499 314 wird das Methanoleinsatzmaterial mit Wasser und
Stickstoff so verdünnt,
dass der Methanolpartialdruck kleiner als 2 psia (13,8 kPa) ist.
-
Trotz
der Kenntnis von Methanolumsetzungsverfahren, die eine Vielzahl
von Zeolithkatalysatoren und Verfahrensbedingungen einsetzen, besteht
ein anhaltender Bedarf an der Entwicklung neuer Verfahren, die zur
selektiven Umsetzung einer organischen Charge, die Methanol und/oder
Dimethylether umfasst, zu Leichtolefinprodukten und insbesondere
Ethylen geeignet sind. Eine Aufgabe der vorliegenden Erfindung besteht
darin, diesen Bedarf zu befriedigen.
-
Die
vorliegende Erfindung besteht in einem Verfahren zur Umsetzung von
Methanol oder Dimethylether im Gemisch mit Methanol zu einem Produkt,
das C2- bis C4-Olefine
enthält,
das den Schritt umfasst, bei dem Einsatzmaterial, das Methanol oder
Dimethylether im Gemisch mit Methanol umfasst, mit Katalysator in Kontakt
gebracht wird, der poröses
kristallines Material umfasst, wobei der Kontaktierschritt in Anwesenheit von
co-zuge führter
aromatischer Verbindung bei Umsetzungsbedingungen durchgeführt wird,
die eine Temperatur von 350 bis 480 °C und einen Methanolpartialdruck über 10 psia
(70 kPa) einschließen,
das poröse
kristalline Material eine Porengröße größer als der kritische Durchmesser
der aromatischen Verbindung hat und die aromatische Verbindung der
Alkylierung durch Methanol oder Dimethylether im Gemisch mit Methanol
bei den Umsetzungsbedingungen zugänglich ist.
-
Vorzugsweise
ist das molare Verhältnis
von Methanol oder Dimethylether im Gemisch mit Methanol zu aromatischer
Verbindung größer als
5 : 1 und kleiner als 300 : 1. Bevorzugter ist das molare Verhältnis von Methanol
oder Dimethylether im Gemisch mit Methanol zu aromatischer Verbindung
von 5 : 1 bis 250 : 1. Insbesondere ist das molare Verhältnis von
Methanol oder Dimethylether im Gemisch mit Methanol zu aromatischer
Verbindung von 10 : 1 bis 150 : 1.
-
Vorzugsweise
schließen
die Umsetzungsbedingungen eine Temperatur von 400 bis 480 °C ein.
-
Vorzugsweise
sind die Umsetzungsbedingungen so, dass die Methanolumsatzrate kleiner
als 90 % und bevorzugter kleiner als 80 % ist.
-
Vorzugsweise
weist das poröse
kristalline Material eine Porengröße von 5 bis 7 Å auf.
-
Vorzugsweise
ist das poröse
kristalline Material ein Alumosilikatzeolith und insbesondere ZSM-5.
Vorzugsweise hat der Katalysator einen α-Wert von weniger als 10 und
bevorzugter weniger als 2.
-
Vorzugsweise
hat das poröse
kristalline Material einen Diffusionsparameter für 2,2-Dimethylbutan von 0,1
bis 20 s–1,
bestimmt bei einer Temperatur von 120 °C und einem 2,2-Dimethylbutandruck
von 60 Torr (8 kPa).
-
Vorzugsweise
hat das poröse
kristalline Material einen Diffusionsparameter von 0,2 bis 5 s–1.
-
Vorzugsweise
enthält
der Katalysator Koks oder Oxidmodifizierungsmittel ausgewählt aus
Oxiden von Bor, Magnesium, Silicium und insbesondere Phosphor.
-
Vorzugsweise
enthält
der Katalysator 0,05 bis 20 Gew.-% und bevorzugter 1 bis 10 Gew.-%
des Koks oder des Oxidmodifizierungsmittels auf elementarer Basis.
-
Die
vorliegende Erfindung stellt ein Verfahren zur selektiven Umsetzung
von Methanol oder Dimethylether im Gemisch mit Methanol zu C2- bis C4-Olefinen,
insbesondere Ethylen, über
porösem
kristallinem Katalysator und in Anwesenheit von co-zugeführter aromatischer
Verbindung, die einen kritischen Durchmesser kleiner als die Porengröße des Katalysators
hat und die der Alkylierung durch das Methanol oder den Dimethylether
im Gemisch mit Methanol bei den Umsetzungsbedingungen zugänglich ist,
als Co-Einsatzmaterial. Das erfindungsgemäße Verfahren unterscheidet
sich von dem in der US-A-4 499 314 oben diskutierten dadurch, dass
ein im Wesentlichen wasserfreies Methanoleinsatzmaterial mit einem
Zeolithkatalysator wie ZSM-5 in Anwesenheit von reaktiver aromatischer
Verbindung bei einer relativ hohen Temperatur von 350 bis 480 °C und einem
relativ hohen Methanolpartialdruck über 10 psia (70 kPa) in Kontakt
gebracht wird. Das Methanol kann verschiedene Mengen Wasser enthalten,
im Gegensatz zu anderen Verfahren ist aber Dampf als Co-Einsatzmaterial
nicht notwendig und hat keine schädliche Wirkung. Außerdem werden
die Verfahrensbedingungen vorzugsweise so eingestellt, dass der
Methanolumsatz kleiner als 90 % und bevorzugter kleiner als 80 ist.
Auf diese Weise ist gefunden worden, dass Ethylenselektivitäten über 30 Gew.-%
erreicht werden können,
im Vergleich mit den in der US-A-4 499 314 beschriebenen Ethylenselektivitäten von
18 bis 25 Gew.-%.
-
Ohne
an eine beliebige Theorie der Betriebsweise gebunden sein zu wollen,
scheint sich die Ethylenselektivität des erfindungsgemäßen Verfahrens
gemäß unserer
Beobachtung dadurch zu ergeben, dass praktisch das gesamte Ethylen,
das über
den katalytischen partiellen Umsatz von Methanol zu leichten Olefinen unter
Verwendung von Zeolithkatalysatoren hergestellt wird, durch Backcracking
von ethylaromatischen Intermediaten abgeleitet ist. Es wird angenommen,
dass die Bildung von solchen ethylaromatischen Intermediaten bei
dem vorliegenden Verfahren durch einen Mechanismus gefördert wird,
bei dem die aromatische Verbindung quasi als Katalysator bei der
Umsetzung von zwei Molekülen
Methanol in ein Molekül
Ethylen wirkt. Somit ist die Methylierung von Aromaten mit Methanol
in Zeolithen wie ZSM-5 eine gut bekannte, schnelle Reaktion. Die
Produktpolymethylbenzole sind stabil, jedoch zu groß, um die
Poren des Katalysators leicht zu verlassen. Obwohl sie relativ langsam
ist, ist die nächste
erwartete Reaktion eines Polymethylaromaten die Skelettisomerisierung
zu einem gemischten Methyl-Ethyl-Aromaten. Sobald sie gebildet sind,
unterliegen Ethylaromaten einer schnellen Crackreaktion unter Bildung
von Ethylen und dem co-katalytischen aromatischen Ring.
-
In
dem in der US-A-4 499 314 beschriebenen Verfahren ist das Toluol-Co-Einsatzmaterial
lediglich ein Promotor, um die Reaktion zu starten. Wenn Methanol über dem
gleichen Katalysator bei 288 °C
in Anwesenheit oder in Abwesenheit von Toluol umgesetzt wird, gibt
es wenig Unterschied in der erhaltenen Ethylenselektivität. Außerdem ergibt
ein Verfahren, das den gleichen Katalysator, wie er in der US-A-4
499 314 verwendet wird, bei der gleichen Temperatur (288 bis 295 °C), bei hohem
Druck (90 psia, 620 kPa) und größer 50 %
Methanolumsatz verwendet, wenig oder keine Olefine. Im Gegensatz
dazu hält
das erfindungsgemäße Verfahren infolge
Reaktordrücken
nahe 100 psia (690 kPa) unerwarteterweise eine fast konstante Ethylenselektivität bei 80
% Methanolumsatz aufrecht.
-
Zusätzlich zeigt
die hier beschriebene Erfindung unerwarteterweise, dass Co-Zufuhrmaterialaromaten als
neuartige Kontrollmechanismen für
die Ethylenausbeute verwendet werden können. Eine Erhöhung der Menge
von Co-Zufuhrmaterialaromaten von 0 auf 5 Gew.-% erhöht die Selektivität zu Ethylen
bei konstanten Bedingungen und konstantem Umsatz. Weil nur eine
geringe Menge notwendig ist, wird mit geringen Kosten für den Betreiber
die Verwendung eines aromatischen Co-Einsatzmaterials als Kontrollmechanismus
einfach erreicht.
-
Jedes
beliebige Methanoleinsatzmaterial, das mindestens 60 Gew.-% Methanol
enthält,
kann verwendet werden, um Methanol zur erfindungsgemäßen Verwendung
bereitzustellen. Im Wesentlichen reines Methanol, wie wasserfreies
Methanol mit Industriereinheit, ist in hohem Grade geeignet. Rohmethanol,
das üblicherweise
12 bis 20 Gew.-% Wasser enthält,
oder eine noch verdünntere
Lösung,
könnte
ebenfalls verwendet werden. Die Anwesenheit von Wasser als Verdünnungsmittel,
um den Methanolpartialdruck zu vermindern, ist jedoch nicht notwendig.
Spurenmengen (< 1
Gew.-%) von nicht-aromatischen organischen Verunreinigungen, wie
höheren
Alkoholen, Aldehyden oder anderen sauerstoffhaltigen Verbindungen
haben eine geringe Wirkung auf die erfindungsgemäße Umsetzungsreaktion und können in
dem Methanoleinsatzmaterial enthalten sein.
-
Neben
Methanol kann das nicht-aromatische Reaktanteinsatzmaterial Dimethylether
umfassen. Wenn diese Komponente anwesend ist, kann sie bis 100%
des nicht-aromatischen organischen Reaktanteinsatzmaterials ausmachen,
oder Dimethylether kann mit Methanol gemischt werden, um das nicht-aromatische
Reaktanteinsatzmaterial zu bilden. Für die Zwecke dieser Erfindung
ist vorgesehen, Methanol oder Dimethylether im Gemisch mit Methanol
in dem Einsatzmaterial direkt zu einer Kohlenwasserstoffmischung
umzusetzen, die durch einen hohen Gehalt an leichten Olefinen, insbesondere
Ethylen, gekennzeichnet ist. Mengen von Dimethylether, die bei der
Umsetzungsreaktion gebil det werden, können jedoch auch gewonnen und
mit frischem organischen Reaktanteinsatzmaterial zurückgeführt werden.
-
Das
aromatische Co-Einsatzmaterial kann aus einer Vielzahl von Quellen
stammen, und selbst wesentliche Mengen von nicht-aromatischen organischen Komponenten
haben einen geringen Einfluss auf die katalytische Rolle des aromatischen
Co-Einsatzmaterials. Aus diesem Grunde ist jeder beliebige organische Einsatzmaterialstrom,
der > 10 Gew.-% Aromaten
enthält,
die einen kritischen Durchmesser kleiner als die Porengröße des Katalysators
haben, um so in der Lage zu sein, leicht in die Katalysatorporen
zu diffundieren, zur Verwendung als das aromatische Co-Einsatzmaterial bei
dem erfindungsgemäßen Verfahren
geeignet. Diese schließen
ein, sind jedoch nicht beschränkt
auf, Benzol, Toluol, Xylol, C9 +-Reformatströme, leichte
Reformate, Vollbereichsreformate oder jede beliebige destillierte
Fraktion derselben, Coker-Naphtha oder jede beliebige destillierte
Fraktion desselben, FCC-Naphtha oder jede beliebige destillierte
Fraktion desselben, und von Kohle abgeleitete Aromaten. Ein Teil
der notwendigen aromatischen Verbindung kann auch in-situ durch
Aromatisierung des Methanol-Einsatzmaterials hergestellt werden.
Die Gegenwart von Verunreinigungen, wie Stickstoff- und Schwefelverbindungen,
Dienen und Styrolen in der aromatischen Komponente kann mit geringem Einfluss
toleriert werden, wenn Wirbel- oder Bewegtbettausführungen
der Erfindung eingesetzt werden.
-
In
einer bevorzugten Ausführungsform
macht Toluol etwas von dem oder den gesamten aromatischen Anteil
des Einsatzmaterials aus.
-
Das
molare Verhältnis
von Methanol oder Dimethylether im Gemisch mit Methanol zu aromatischer Verbindung
ist normalerweise größer als
5 : 1, weil höhere
Konzentrationen an aromatischer Verbindung zu übermäßigem Verkoken, erhöhten Volumina
der Abtrenn- und Rückführströme und minimalem
Gewinn der chemischen Gesamtselektivitäten führen. Darüber hinaus wird das molare
Verhältnis
von Methanol oder Dimethylether im Gemisch mit Methanol zu aromatischer
Verbindung normalerweise unter 300 : 1 gehalten, weil geringere
Konzentrationen von aromatischer Verbindung zu einer sehr geringen
oder keiner spürbaren
Verbesserung der Ethylenselektivität des Verfahrens führen. Die
notwendige Menge von aromatischer Verbindung verringert sich jedoch
mit höherem
Druck, sodass es bei hohem Druck möglich ist, eine signifikante
Verbesserung der Ethylenselektivität mit molaren Verhältnissen
von Methanol oder Dimethylether im Gemisch mit Methanol zu aromatischer
Verbindung oberhalb von 300 : 1 zu erreichen. Vorzugsweise ist das
molare Verhältnis von
Methanol oder Dimethylether im Gemisch mit Methanol zu aromatischer
Verbindung von 5 : 1 bis 250 : 1, bevorzugter von 10 : 1 bis 150
: 1.
-
Der
bei dem erfindungsgemäßen Verfahren
eingesetzte Katalysator ist ein porösen kristallines Material,
das eine Porengröße größer als
der der kritische Durchmesser der aromatischen Verbindung als Co-Einsatzmaterial
hat. Bevorzugte Katalysatoren sind poröse kristalline Materialien
mit einer Porengröße von 5
bis 7 Å,
und insbesondere Aluminosilikatzeolithe mit mittlerer Porengröße. Eine übliche Definition
für Zeolithe
mit mittlerer Porengröße ist der
Constraint Index-Test, der in der US-A-4 016 218 beschrieben ist. Dabei haben Zeolithe
mit mittleren Poren eine Constraint Index von 1 bis 12, bestimmt
bei dem Zeolithen allein ohne die Einführung von Modifizierungsmitteln
und vor jedweder Behandlung, um die Diffusionsfähigkeit des Katalysators einzustellen.
Neben Aluminosilikaten mit mittlerer Porengröße können andere saure Metallosilikate
mit mittlerer Porengröße, wie
Silicoaluminophosphate (SAPOs) bei dem erfindungsgemäßen Verfahren
verwendet werden.
-
Besondere
Beispiele für
geeignete Zeolithe mit mittlerer Porengröße schließen ZSM-5, ZSM-11, ZSM-12,
ZSM-22, ZSM-23, ZSM-35, ZSM-48 und MCM-22 ein, wobei ZSM-5 und ZSM-11
besonders bevorzugt sind.
-
Der
Zeolith ZSM-5 und dessen konventionelle Herstellung sind in der
US-A-3 702 886 beschrieben. Zeolith ZSM-11 und dessen konventionelle
Herstellung sind in der US-A-3 709 979 beschrieben. Zeolith ZSM-12
und dessen konventionelle Herstellung sind in der US-A-3 832 449
beschrieben. Zeolith ZSM-23 und dessen konventionelle Herstellung
sind in der US-A-4 076 842 beschrieben. Zeolith ZSM-35 und dessen
konventionelle Herstellung sind in der US-A-4 016 245 beschrieben.
ZSM-48 und dessen konventionelle Herstellung werden in der US-A-4
375 573 gelehrt. MCM-22 ist in den US-A-5 304 698, US-A-5 250 277,
US-A-5 095 167 und US-A-5 043 503 offenbart.
-
Um
die Konzentration von Aromaten in den Katalysatorporen ohne eine
Erhöhung
des molaren Verhältnisses
von Aromat zu Methanol zu erhöhen,
kann es gewünscht
sein, einen Katalysator mit erhöhtem
Diffusionswiderstand zu verwenden. Insbesondere kann es gewünscht sein,
einen Katalysator einzusetzen, der ein poröses kristallines Material mit
einem Diffusionsparameter für
2,2-Dimethylbutan von 0,1 bis 20 s–1 hat, vorzugsweise
0,1 bis 15 s–1 und
insbesondere 0, 2 bis 5 s–1, bestimmt bei einer
Temperatur von 120 °C
und einem 2,2-Dimethylbutan-Druck von 60 Torr (8 kPa).
-
Der
Diffusionsparameter des bestimmten porösen kristallinen Materials
ist hier definiert als D/r2 × 106, wobei D der Diffusionskoeffizient (cm2/sec) ist und r der Kristallradius (cm)
ist. Die notwendigen Diffusionsparameter können durch Sorptionsmessungen
abgeleitet werden, wenn man von der Annahme ausgeht, dass das Glattlagenmodell
das Diffusionsverfahren beschreibt. Das heißt für eine angegebene Sorbatbeladung
Q der Wert Q/Q∞, wobei Q∞ die
Gleichgewichtssorbatbeladung ist und mathematisch mit (Dt/r2)½) in Beziehung steht,
wobei t die Zeit (sec) ist, die notwendig ist, um die Sorbatbeladung
Q zu erreichen. Graphische Lösungen für das Glattlagenmodell
werden von J. Crank in "Mathematics
of Diffusion", Oxford
University Press, Ely House, London, 1967 angegeben.
-
Die
oben als für
das erfindungsgemäße Verfahren
bevorzugt beschriebenen Zeolithe mit mittlerer Porengröße können Diffusionsparameterwerte über dem
notwendigen Bereich von 0,1 bis 20 sec–1 haben.
Der Diffusionsparameter kann jedoch durch eine Vielzahl von Methoden
auf den notwendigen Wert eingestellt oder modifiziert werden. Zum
Beispiel kann der notwendige Diffusionskoeffizient durch Verwendung
von großen Kristallformen
(größer als
1 μm) des
porösen
kristallinen Materials erreicht werden, indem vor der Verwendung in
dem Verfahren Koks auf dem Material abgelagert wird (wie in der
US-A-4 097 543 beschrieben) und/oder indem das Material mit mindestens
einem Oxidmodifizierungsmittel kombiniert wird, vorzugsweise ausgewählt aus
der Gruppe bestehend aus Oxiden von Bor, Magnesium, Calcium, Silicium,
Lanthan und insbesondere Phosphor. Die Gesamtmenge an Koks oder
Oxidmodifizierungsmittel, bestimmt auf elementarer Basis, kann bei
0,5 bis 20 Gew.-% liegen, vorzugsweise 1 bis 10 Gew.-%, bezogen
auf das Gewicht des Endkatalysators.
-
Alternativ
kann die notwendige Diffusionsgrenze erreicht werden, indem der
Katalysator so stark dampfbehandelt wird, dass eine kontrollierte
Verminderung des Mikroporenvolumens des Katalysators auf nicht weniger
als 50 % und vorzugsweise 50 bis 90% desjenigen des nicht dampfbehandelten
Katalysators bewirkt wird. Die Verminderung des Mikroporenvolumens
wird bestimmt, indem die n-Hexan-Adsorptionskapazität des Katalysators
vor und nach Dampfbehandlung bei 90 °C und 75 Torr (10 kPa) n-Hexan-Druck
gemessen wird. Die Dampfbehandlung des porösen kristallinen Materials
wird bei einer Temperatur von mindestens 850 °C, vorzugsweise 950 °C bis 1075 °C und insbesondere
1000 bis 1050 °C
für 10
Minuten bis 10 Stunden, vorzugsweise 30 Minuten bis 5 Stunden bewirkt.
Um die gewünschte
kontrollierte Verminderung des Diffusionskoeffizienten und des Mikroporenvolumens
zu bewirken, kann es gewünscht
sein, das poröse
kristalline Material vor der Dampfbehandlung mit Phosphormodifizierungsmittel
zu kombinieren. Die Gesamtmenge von Phosphormodifizierungsmittel,
die normalerweise in dem Katalysator in Oxidform vorhanden ist,
bestimmt auf elementarer Basis, kann von 0,05 bis 20 Gew.-% betragen,
vorzugsweise 1 bis 10 Gew.-%, bezogen auf das Gewicht des Endkatalysators.
-
Wenn
das Modifizierungsmittel Phosphor ist, wird ein Einschluss des Modifizierungsmittels
in den erfindungsgemäßen Katalysator
einfach durch die in den US-A-4 356 338, US-A-5 110 776 und US-A-5
231 064 beschriebenen Methoden erreicht. Ähnliche in der Technik bekannte
Verfahren können
verwendet werden, um andere modifizierende Oxide in den erfindungsgemäßen Katalysator
einzuschließen.
-
Das
bei dem erfindungsgemäßen Verfahren
eingesetzte poröse
kristalline Material kann mit einer Vielzahl von Bindemittel- oder Matrixmaterialien
kombiniert werden, die gegenüber
den bei dem Verfahren eingesetzten Temperaturen und anderen Bedingungen
beständig
sind. Solche Materialien schließen
aktive und inaktive Materialien ein, wie Tone, Siliciumdioxid und/oder
Metalloxide wie Aluminiumoxid. Die Letztgenannten können entweder
natürlich
vorkommend oder in Form von gallertartigen Niederschlägen oder
Gelen sein, einschließlich
Mischungen von Siliciumdioxid und Metalloxiden. Die Verwendung eines
Materials, das aktiv ist, neigt dazu, den Umsatz und/oder die Selektivität des Katalysators
zu verändern
und ist demzufolge im Allgemeinen nicht bevorzugt. Inaktive Materialien
dienen geeigneterweise als Verdünnungsmittel,
um das Ausmaß der
Umsetzung bei einem bestimmten Verfahren so zu steuern, dass Produkte ökonomisch
und gleichmäßig erhalten
werden können,
ohne andere Mittel zur Steuerung der Geschwindigkeit der Reaktion
einzusetzen. Diese Materialien können
in natürlich
vorkommende Tone, z.B. Bentonit und Kaolin, eingeschlossen werden, um
die Druckfestigkeit des Katalysators bei kommerziellen Betriebsbedingungen
zu verbessern. Diese Materialien, d.h. Tone, Oxide etc., wirken
als Bindemittel für
den Katalysator. Es ist bevorzugt, einen Katalysator mit guter Druckfestigkeit
bereitzustellen, weil es bei der kommerziellen Verwendung verhindert
werden soll, dass der Katalysator zu pulverartigen Materialien zerbricht.
Diese Ton- und/oder Oxidbin demittel sind normalerweise nur zum Zwecke
der Verbesserung der Druckfestigkeit des Katalysators eingesetzt
worden.
-
Natürlich vorkommende
Tone, die mit dem porösen
kristallinen Material in Verbund gebracht werden können, schließen die
Familie Montmorillonit und Kaolin ein, wobei diese Familien die
Subbentonite einschließen,
und die Kaoline, die üblicherweise
als Dixie-, McNamee-, Georgia- und Florida-Tone bekannt sind, oder andere,
bei denen der Hauptmineralbestandteil Halloysit, Kaolinit, Dickit,
Nakrit oder Anauxit ist. Solche Tone können im Rohzustand wie ursprünglich bergmännisch gewonnen
verwendet werden oder eingangs Calcinierung, Säurebehandlung oder chemischer
Modifizierung unterworfen werden.
-
Neben
den genannten Materialien kann das poröse kristalline Material in
Verbund mit porösem
Matrixmaterial wie Siliciumdioxid-Aluminiumoxid, Siliciumdioxid-Magnesiumoxid,
Siliciumdioxid-Zirkondioxid, Siliciumdioxid-Thoriumoxid, Siliciumdioxid-Berylloxid, Siliciumdioxid-Titandioxid
als auch ternären
Zusammensetzungen wie Siliciumdioxid-Aluminiumoxid-Thoriumoxid,
Siliciumdioxid-Aluminiumoxid-Zirkondioxid, Siliciumdioxid-Aluminiumoxid-Magnesiumoxid
und Siliciumdioxid-Magnesiumoxid-Zirkondioxid gebracht werden.
-
Die
relativen Anteile von porösem
kristallinen Material und anorganischer Oxidmatrix variieren breit, wobei
der Gehalt an dem Erstgenannten im Bereich von 1 bis 90 Gew.-% und
mehr liegt, und normalerweise, insbesondere wenn der Verbund in
Form von Perlen hergestellt wird, im Bereich von 2 bis 80 Gew.-%
des Verbunds.
-
Vorzugsweise
umfasst das Bindemittelmaterial Siliciumdioxid oder Kaolinton.
-
Verfahren
zur Herstellung von Siliciumdioxid-gebundenen Zeolithen wie ZSM-5
sind in den US-A-4 582 815, US-A-5 053 374 und US-A-5 182 242 beschrieben.
Ein besonderes Verfahren zur Verbindung von ZSM-5 mit Siliciumdioxidbindemittel
schließt
ein Extrudierverfahren ein.
-
Das
porösen
kristalline Material kann mit Bindemittel in Form eines Wirbelbettkatalysators
kombiniert werden. Dieser Wirbelbettkatalysator kann Ton in dessen
Bindemittel umfassen und kann durch ein Sprühtrocknungsverfahren gebildet
werden, um Katalysatorteilchen mit einer Teilchengröße von 20
bis 200 μm zu
bilden.
-
Der
erfindungsgemäß eingesetzte
Katalysator hat vorzugsweise eine sehr niedrige Säureaktivität. Unter
Verwendung des in Journal of Catalysis, Band 61, Seite 395 (1980)
offenbarten α-Tests
für die
Säureaktivität hat der
erfindungsgemäße Katalysator
einen α-Wert
von weniger als 10, vorzugsweise weniger als 2.
-
Das
erfindungsgemäße Verfahren
wird vorzugsweise in einem Bewegt- oder Wirbelkatalysatorbett mit kontinuierlicher
oxidativer Regenerierung durchgeführt. Das Ausmaß der Koksbeladung
kann dann kontinuierlich gesteuert werden, indem die Schärfe und/oder
die Häufigkeit
der Regenerierung variiert werden.
-
Das
erfindungsgemäße Verfahren
wird bei eine relativ hohen Temperatur von 350 bis 500 °C durchgeführt, vorzugsweise
400 bis 480 °C,
weil, wie in den nachfolgenden Beispielen entgegen der Lehre der US-A-4
499 314 gezeigt ist, gefunden wurde, dass ein solcher Temperaturbereich
für die
selektive Herstellung von niederen Olefinen entscheidend ist. Ohne
an eine beliebige Theorie der Wirkweise gebunden sein zu wollen,
glauben wir, dass eine relativ hohe Temperatur für die Skelettisomerisierung
und das Cracken des hergestellten Polymethylbenzolintermediats wichtig
ist, während
höhere
Temperaturen zu übermäßigem Verkoken führen.
-
Das
erfindungsgemäße Verfahren
ist dadurch vorteilhaft, dass gefunden wurde, dass die Selektivität zu niederem
Olefin des Produkts im Allgemeinen vom Methanolpartialdruck unabhängig ist,
sodass die Notwendigkeit von Verfahren des Standes der Technik,
den Methanoldruck durch die Zugabe von Verdünnungsmittel oder durch einen
Betrieb bei vermindertem Druck zu vermindern, vermieden werden kann.
Die Möglichkeit,
bei höherem
Methanolpartialdruck vorzugehen, ermöglicht auch, dass die absolute
Ausbeute pro Durchgang des Olefinprodukts erhöht werden kann. Ein geeigneter
Methanolpartialdruck zum Einsetzen bei dem erfindungsgemäßen Verfahren
liegt über
10 psia (70 kPa), vorzugsweise 15 bis 150 psia (103 kPa bis 1030
kPa).
-
Außerdem ist
gewünscht,
dass die Umsetzungsbedingungen so gesteuert werden, dass das Methanolumsatzniveau
niedriger als 90 % und vorzugsweise niedriger als 80 % ist, weil
bei höheren
Umsatzniveaus Konkurrenzreaktionen zur Aromatenmethylierung, wie
Olefinalkylierung und/oder Oligomerisierung unter Herstellung von
C5+-Isoolefinen und/oder Olefinumsatz zu
Aromaten und Paraffinen die Selektivität zu Ethylen und Propylen vermindert.
Eine geeignete Steuerung des Methanolumsatzes kann natürlich durch
Variation des stündlichen
Massendurchsatzes erreicht werden, der typischerweise von 0,1 bis
100, vorzugsweise 0,1 bis 10 variiert werden kann.
-
Das
erfindungsgemäße Verfahren
setzt Methanol oder Dimethylether im Gemisch mit Methanol zu einem
Leichtolefinstrom um, in dem Ethylen über 40 Gew.-% und typischerweise über 50 Gew.-%
der C2- bis C4-Olefine
ausmacht und in dem Ethylen mehr als 90 Gew.-%, vorzugsweise mehr
als 95 Gew.-% der C2-Komponente ausmacht.
Neben den Leichtolefinen ist ein Produkt des Verfahrens Xylole,
die einen hohen Anteil des p-Isomers umfassen, insbesondere wenn
ein diffusionsbeschränkter
Katalysator wie oben beschrieben eingesetzt wird.
-
Die
Erfindung wird nun genauer in den folgenden Beispielen und den angefügten Zeichnungen
beschrieben, wobei:
-
1 eine
graphische Darstellung ist, in der Ethylenselektivität gegen
Methanolumsatz für
den Katalysator von Beispiel 1 bei der Verwendung zur Umsetzung
von Methanol in Anwesenheit von variierenden Mengen Wasser und Toluol
aufgetragen ist,
-
2 eine
graphische Darstellung ist, die die Ethylenselektivität mit der
Temperatur für
den Katalysator von Beispiel 1 bei der Verwendung zur Umsetzung
von Methanol in Anwesenheit von Toluol bei molaren Verhältnissen
von Methanol zu Toluol von 10 : 1 und 155 : 1 vergleicht,
-
3 und 4 graphische
Darstellungen ähnlich 2 sind,
die jedoch den Toluolumsatz bzw. Gew.-% Ethylen/p-Xylol mit der
Temperatur unter Verwendung des gleichen Katalysators und der gleichen
molaren Verhältnisse
von Methanol zu Toluol vergleichen und
-
5 eine
graphische Darstellung ist, die Ethylenselektivität mit Methanolumsatz
für den
Katalysator von Beispiel 1 bei einem molaren Verhältnis von
Methanol zu Toluol von 55 : 1 mit der des Katalysators von Beispiel
4 bei einem molaren Verhältnis
von Methanol zu Toluol von 26 : 1 vergleicht.
-
6 ist
eine graphische Darstellung, die Olefinselektivität mit der
Temperatur bei Methanolpartialdrücken
von 15 und 90 psia (103 kPa und 620 kPa) für den Katalysator von Beispiel
5 vergleicht.
-
7 ist
eine graphische Darstellung, die die Wirkungen von Methanolpartialdruck
auf Olefinselektivität
bei verschiedenen Temperaturen unter Verwendung des Katalysators
von Beispiel 5 vergleicht.
-
In
den Beispielen wurden Mikroporenvolumen (n-Hexan)-Messungen auf
einem Computer-gesteuerten (Vista/Fortran) thermogravimetrischen
Analysegerät
duPont 951 durchgeführt.
Isothermen wurden bei 90 °C
gemessen und Adsorptionswerte bei 75 Torr (10 kPa) n-Hexan genommen.
Die Diffusionsmessungen wurden auf einem thermogravimetrischen Analysegerät TA Instruments
2950 durchgeführt,
das mit einer Thermal Analysis 2000-Steuerung, einer Gasumschaltvorrichtung
und einem automatischen Probenwechsler ausgerüstet war. Diffusionsmessungen
wurden bei 120 °C
und 60 Torr (8 kPa) 2,2-Dimethylbutan durchgeführt. Die Daten wurden als die
Aufnahme gegen die Quadratwurzel der Zeit aufgetragen. Das Testen
mit Festbettkatalyse wurde unter Verwendung eines Reaktors mit Strömung nach
unten und 3/8" (0,95
cm) Außendurchmesser durchgeführt, der
mit einem Thermoelement ausgerüstet
war. Methanol, Wasser und Aromaten wurden mittels eines Verdampfers,
der mit einem statischen Mischer ausgerüstet war, um die Einsatzmaterialien
stromaufwärts
von dem Reaktor sorgfältig
zu vergasen und zu mischen, zu dem Reaktor gepumpt. Der Reaktor
war mit einem Gegendruckregler ausgerüstet, um eine Untersuchung
der Produkte bei einer breiten Vielzahl von Temperaturen, Drücken und
WHSVs zu ermöglichen.
Der Gesamtreaktorausfluss wurde online durch Gaschromatographie
analysiert. Der Methanolumsatz wurde nur auf der Basis von Kohlenwasserstoffbildung
berechnet. Selektivitäten
zu Kohlenwasserstoffprodukt wurden auf "wasserfreier" Basis berechnet.
-
Beispiel 1
-
Phosphorsäure, Kaolinton
und 450 : 1 SiO2/Al2O3-ZSM-5 wurden in Wasser aufgeschlämmt und
sprühgetrocknet,
um einen typischen wirbelbettkatalysator herzustellen. Der Katalysator
wurde in Luft bei 510 °C calciniert.
Der fertige Katalysator enthielt 40 Gew.-% ZSM-5 und 4,5 Gew.-%
Phosphor. Dieses Material hatte eine n-Hexan-Sorption von 33,5,
eine Diffusionsparameter von 27 und einen α-Wert von 7. Der Katalysator wurde
dann bei 1050 °C
0,75 Stunden lang in 1 Atmosphäre
(101,3 kPa) Dampf mit Dampf behandelt, um einen Endkatalysator mit
einem Diffusionsparameter von 0,46 s–1 und
einer n-Hexan-Sorption von 30,6 mg/g herzustellen.
-
Beispiel 2
-
Eine
erste Probe (0,5 g) des mit Dampf behandelten Katalysators von Beispiel
1 wurde verwendet, um ein reines Methanoleinsatzmaterial bei 0,5
bis 10 WHSV, 430 °C
und 1 atm (101,3 kPa) Druck umzusetzen. Ein breiter Bereich von
Methanolumsätzen
wurde erhalten. Kohlenwasserstoffproduktethylenselektivität ist in 1 gegen
Methanolumsatz aufgetragen.
-
Eine
zweite Probe (0,5 g) des mit Dampf behandelten Katalysators von
Beispiel 1 wurde zur Umsetzung eines Einsatzmaterials mit 3 : 1
mol Methanol zu Wasser bei 0,5 bis 10 WHSV, 430 °C und 1 atm (101,3 kPa) Druck
verwendet. Ein breiter Bereich von Methanolumsätzen wurde erhalten. Kohlenwasserstoffproduktethylenselektivität ist in 1 gegen
Methanolumsatz aufgetragen.
-
Eine
dritte Probe (0,5 g) des mit Dampf behandelten Katalysators von
Beispiel 1 wurde verwendet, um ein Einsatzmaterial mit 26 : 1 mol
Methanol zu Toluol bei 0,5 bis 5 WHSV, 430 °C und 1 atm (101,3 kPa) Druck umzusetzen.
Ein breiter Bereich von Methanolumsätzen wurde erhalten. Kohlenwasserstoffproduktethylenselektivität ist in 1 gegen
Methanolumsatz aufgetragen.
-
Eine
vierte Probe (0,5 g) des mit Dampf behandelten Katalysators von
Beispiel 1 wurde verwendet, um ein Einsatzmaterial mit 3 : 1 mol
Methanol zu Toluol bei 0,5 bis 5 WHSV, 430 °C und 1 atm (101,3 kPa) Druck
umzusetzen. Ein breiter Bereich von Methanolumsätzen wurde erhalten. Kohlenwasserstoffproduktethylenselektivität ist in 1 gegen
Methanolumsatz aufgetragen.
-
1 zeigt
deutlich, dass die Zugabe von 1 mol Dampf je 3 mol Methanol zu dem
Einsatzmaterial zu einer vernachlässigbaren Änderung der Ethylenselektivität führt. Demgegenüber führt die
Zugabe von Toluol in einer Menge von nur 1 mol Toluol je 26 mol
Methanol zu einer starken Verbesserung der Ethylenselektivität, insbesondere
wenn der Methanolumsatz unter 90% gehalten wird.
-
Beispiel 3
-
Eine
weitere Probe (0,5 g) des mit Dampf behandelten Katalysators von
Beispiel 1 wurde verwendet, um ein Einsatzmaterial mit 12 : 1 mol
Methanol zu Toluol bei 1 atm (101,3 kPa) Druck, 0,5 bis 5 WHSV und unterschiedlichen
Temperaturen von 380 bis 480 °C
umzusetzen. Ein breiter Bereich von Methanolumsätzen wurde erhalten. Aus diesen
Daten wurden Ethylenselektivität,
Toluolumsatz und Gew.-% Ethylen/p-Xylol bei 70 % Methanolumsatz
bei jeder Temperatur berechnet, und die Ergebnisse sind in den 2 bis 4 aufgetragen.
Der Test wurde dann mit einem Einsatzmaterial mit einem molaren
Verhältnis
Methanol:Toluol 155 : 1 wiederholt und die Daten wiederum in 2 bis 4 auf
getragen.
-
Die 2 bis 4 belegen,
dass das Einsatzmaterial mit Methanol:Toluol 155 : 1 nicht genug
aromatisches Co-Einsatzmaterial bei jeder beliebigen Temperatur
hat, um so viel Ethylen wie das Einsatzmaterial mit Methanol zu
Toluol 12 : 1 herzustellen. Ferner fällt mit steigender Temperatur
die Ethylenselektivität
mit dem Einsatzmaterial mit Methanol:Toluol 155 : 1 rasch ab. Die 2 bis 4 zeigen
auch, dass für
das Einsatzmaterial mit Methanol:Toluol 12 : 1 die Ethylenselektivität mit der
Temperatur konstant ist, das Verhältnis Ethylen:p-Xylol in dem
Produkt aber mit steigender Temperatur rasch abfällt. Deshalb ermöglicht es
eine Steuerung von Katalysator, Einsatzmaterial und Temperaturen
einem Hersteller, aus einem breiten Bereich von Ethylen:p-Xylol-Verhältnissen
im Produkt auszuwählen.
-
Beispiel 4
-
Eine
Probe des Phosphor-behandelten, nicht mit Dampf behandelten Katalysators
von Beispiel 1 wurde bei 870 °C
6 Stunden lang in 1 atm (101,3 kPa) Dampf mit Dampf behandelt, um
einen Katalysator mit einem Diffusionsparameter von 31 s–1 und
einer n-Hexan-Sorption von 34,9 mg/g herzustellen. 0,5 g dieses
Katalysators wurden zur Umsetzung eines Einsatzmaterials mit 26
: 1 mol Methanol zu Toluol bei 0,5 bis 10 WHSV, 430 °C und 1 atm
(101,3 kPa) Druck verwendet. Ein breiter Bereich von Methanolumsätzen wurde
erhalten, und Ethylenselektivität
des Kohlenwasserstoffprodukts ist in 5 gegen
Methanolumsatz aufgetragen.
-
Zum
Vergleich wurde eine weitere Probe des mit Dampf behandelten Katalysators
von Beispiel 1 verwendet, um ein Einsatzmaterial mit 55 : 1 mol
Methanol zu Toluol bei den gleichen Bedingungen umzusetzen, und
wiederum ist die in 5 die Ethylenselektivität gegen
den Methanolumsatz aufgetragen.
-
5 belegt
die Beziehung zwischen der Menge von aromatischem Co-Einsatzmaterial
und dem Diffusionskoeffizienten des Katalysators. Die Daten mit
55 : 1-Einsatzmaterial mit dem diffusionsbegrenzten Katalysator
von Beispiel 1 sind im Wesentlichen die Gleichen wie die Daten mit
dem Einsatzmaterial mit 26 : 1 molar mit dem weniger diffusionsbeschränkten Katalysator
von Beispiel 4. Dies zeigt, dass weniger aromatisches Co-Einsatzmaterial
notwendig ist, um eine bestimmte Produktverteilung mit einem stärker diffusionsbeschränkten Katalysator
zu erreichen, als mit einem weniger diffusionsbeschränkten Katalysator
notwendig ist.
-
Beispiel 5
-
Ein
kommerziell erhältlicher
FCC-Additivkatalysator, der 25 Gew.-% ZSM-5 mit einem Silicium-zu-Aluminium-Molverhältnis von
26 : 1 und 3 Gew.-% Phosphor enthielt, wurde bei diesem Beispiel
verwendet. Der Katalysator war bei 1450 °F (790 °C) 4 Stunden lang mit Dampf
vorbehandelt worden und besaß einen α-Wert von
3, einen Diffusionsparameter von 25 und eine n-Hexan-Sorption von 25 mg/g.
Der Katalysator wurde verwendet, um eine Mischung von 90 Gew.-%
Methanol und 10 Gew.-% Toluol (Methanol zu Toluol-Molverhältnis von
26 : 1) bei Methanolpartialdrücken
von 15 psia (103 kPa) und 90 psia (620 kPa) und bei einem Methanolumsatzniveau
von 80 % umzusetzen. Verschiedene Temperaturen von 210 bis 490 °C wurden
verwendet, und die Ergebnisse sind in 6 aufgetragen.
Aus 6 ist ersichtlich, dass die Umsetzung von Methanol
in Anwesenheit von Toluol bei Temperaturen von 350 bis 480 °C hochselektiv
zu Olefinen ist, und dass die Olefinselektivität im Allgemeinen vom Methanolpartialdruck
unabhängig
ist, insbesondere bei Temperaturen von 400 und 450 °C.
-
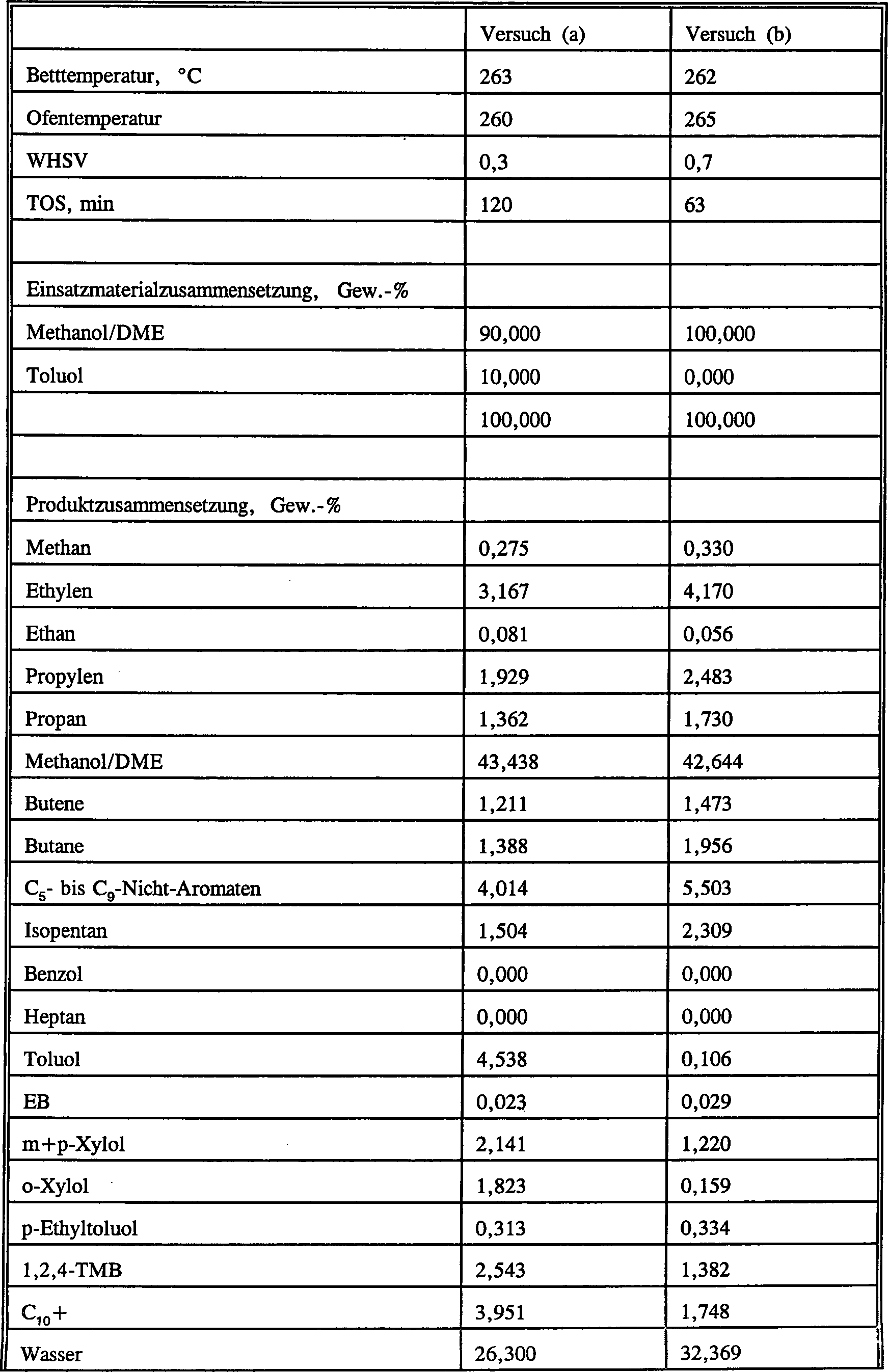
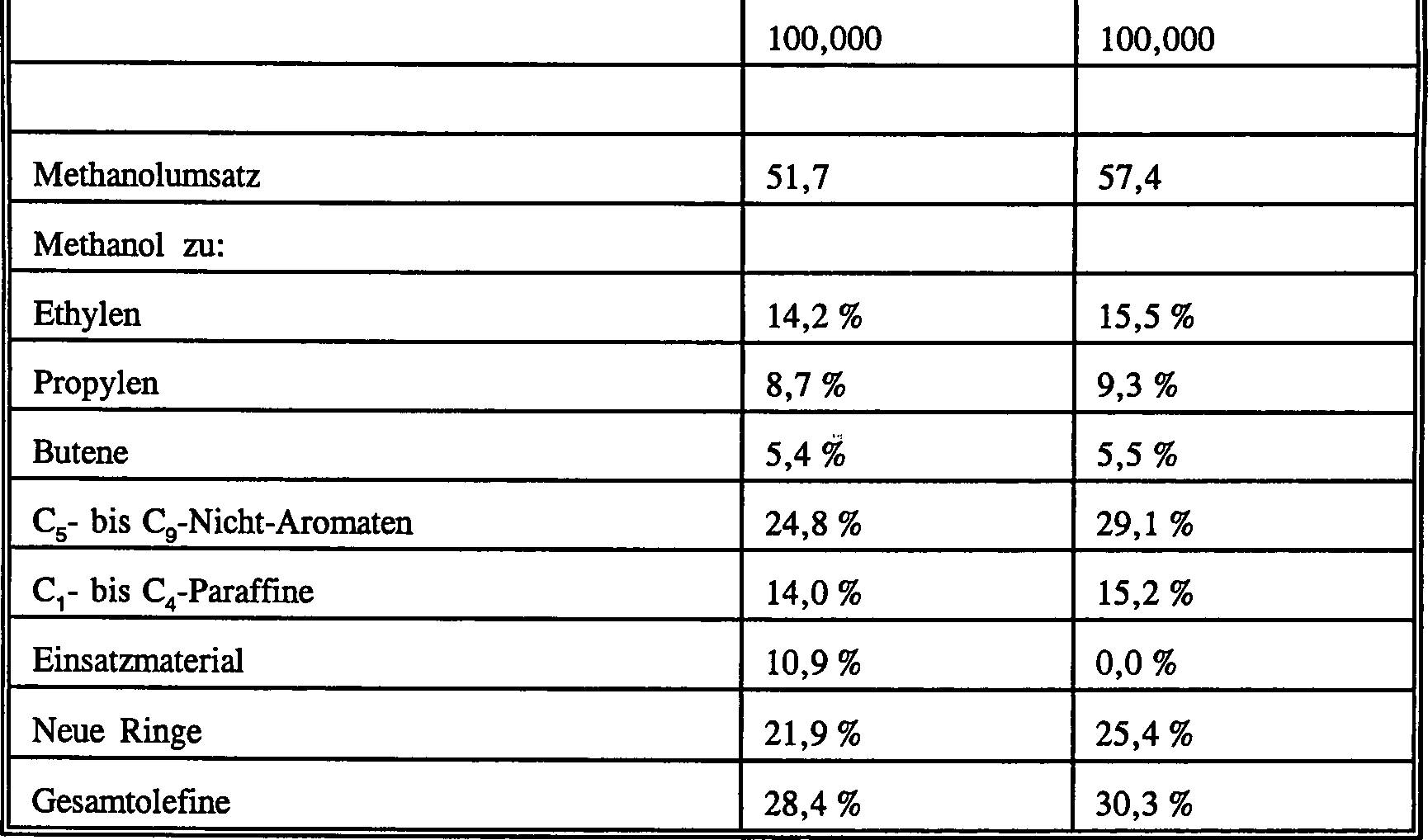
Beispiel 6 (Vergleich)
-
Bei
diesem Beispiel wurde ein Aluminiumoxid-gebundener ZSM-5-Katalysator
verwendet, der 65 Gew.-% ZSM-5 mit einem Siliciumdioxid-zu-Aluminiumoxid-Molverhältnis von
26 : 1 enthielt, der bei 515 °C
1 Stunde lang dampfbehandelt worden war. Der Katalysator besaß einen α-Wert von
100, einen Diffusionsparameter von 1000 und eine n-Hexan-Sorption
von 77 mg/g. Der Katalysator wurde verwendet, um (a) eine Mischung
von 90 Gew.-% Methanol und 10 Gew.-% Toluol (Methanol:Toluol-Molverhältnis von
26 : 1) und (b) reines Methanol bei einer Temperatur von 270 °C und einem
Methanolumsatzniveau von 50% umzusetzen, und die Ergebnisse sind
in Tabelle 1 aufgelistet. Aus Tabelle 1 ist ersichtlich, dass die
Leichtolefinausbeute bei dieser niedrigen Temperatur mit dem Toluol-Co-Einsatzmaterial
nur wenig geringer ist als mit dem reinen Methanoleinsatzmaterial.
-
-
-
Beispiel 7
-
Der
Katalysator von Beispiel 5 wurde verwendet, um eine Mischung von
90 Gew.-% Methanol und 10 Gew.-% Toluol (Methanol:Toluol-Molverhältnis von
26 : 1) bei verschiedenen Temperaturen und Drücken umzusetzen. Die Gesamtolefinselektivität bei jeder
Bedingung bei einem Methanolumsatzniveau von 80 % ist in 7 gezeigt.
Die Figur zeigt klar, dass bei niedrigeren Temperaturen die Wirkung
des Drucks auf die Gesamtolefinselektivität mehr und mehr ausgeprägt ist.
Zusätzlich
heben diese Ergebnisse wiederum hervor, dass die Gesamtolefinselektivität eine deutliche
Funktion der Temperatur ist.