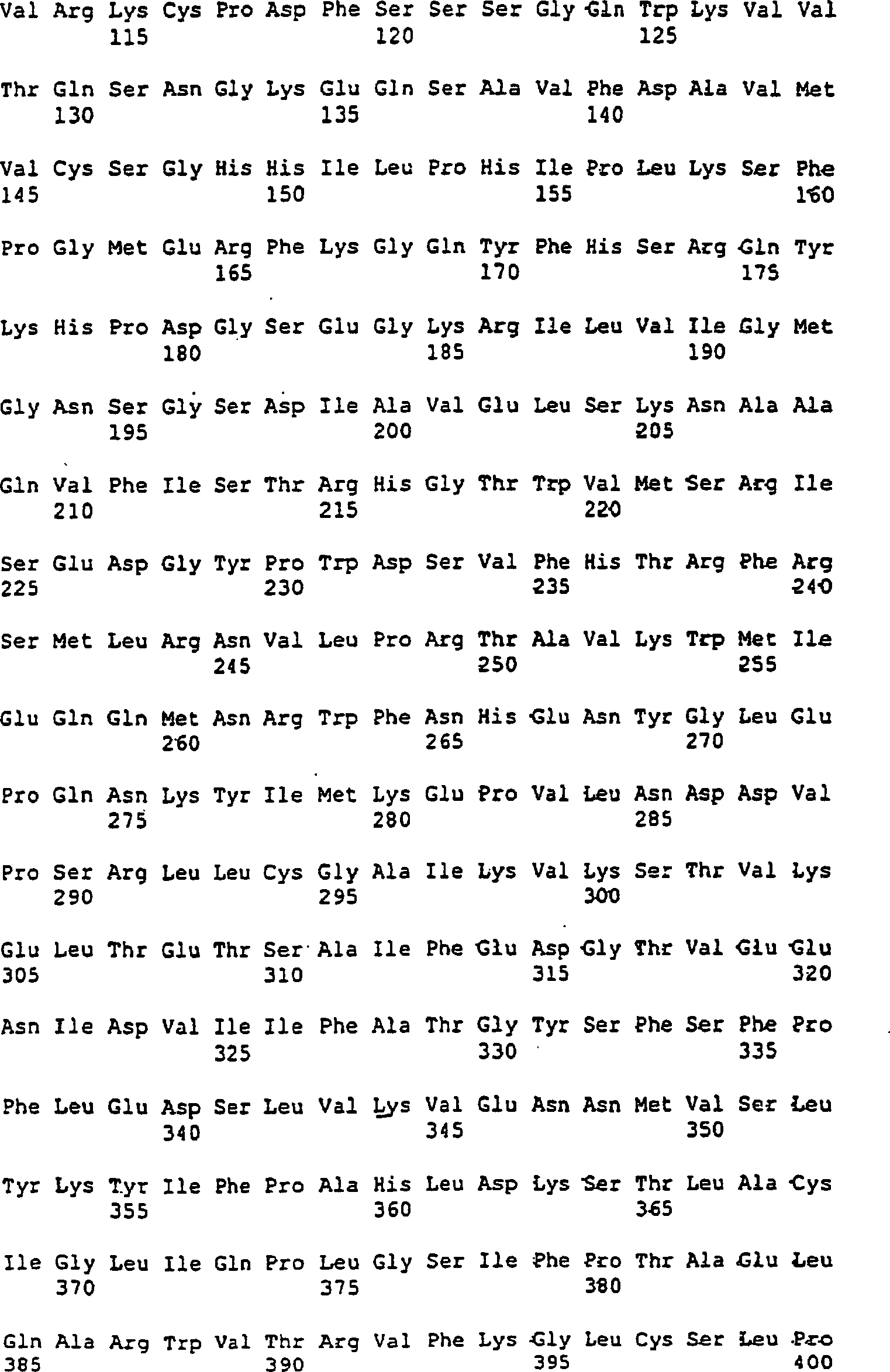-
Die
vorliegende Erfindung betrifft eine mutierte humane Flavin-Monooxygenase
2 (hFMO2), als auch ihre Nukleotid- und Polypeptidsequenzen. Die
vorliegende Erfindung betrifft auch Klonierungs- und/oder Expressionsvektoren,
die die Nukleotidsequenzen enthalten, und durch diese Vektoren transformierte
Zellen als auch Verfahren zur Herstellung der Polypeptide. Die Erfindung
umfasst zudem Verfahren zur Selektion von Zusammensetzungen und
zur Diagnostik der Prädisposition
für Pathologien
und/oder Mängel,
die mit diesem mutierten humanem FMO2 assoziiert sind, als auch
pharmazeutische Zusammensetzungen, die die vorgesehenen Verbindungen
enthalten, zur Behandlung und/oder Verhinderung dieser Pathologien.
-
Die
Flavinmonooxygenasen (FMOs) (Lawton et al., 1994) bilden eine Familie
von mikrosomalen Enzymen, die die NADPH-abhängige Oxidation einer Vielzahl
organischer exogener Verbindungen (Xenobiotika) katalysieren, die
ein nukleophiles Heteroatom, wie im Besonderen das Stickstoff-,
Schwefel-, Phosphor oder Selenatom, enthalten (Ziegler, D.M., 1988;
Ziegler, D.M., 1993), wobei es sich um Medikamente, Pestizide oder
andere potentiell toxische Substanzen handelt. Das Cystamin ist
das einzige endogene Substrat von FMOs, das derzeit bekannt ist.
-
Die
FMOs stellen eine mulitgene Familie dar. Die Expression verschiedener
FMO-Formen ist abhängig von
der Art des Gewebes und der betrachteten Spezies.
-
Die
FMOs sind lokalisiert worden in verschiedenen Gewebetypen, im Besonderen
in der Leber, den Lungen und den Nieren.
-
Bisher
sind fünf
Isoformen von FMOs identifiziert worden in der Referenzspezies,
dem Kaninchen. Ihre Homologie ist 50–60 %. Es ist ebenfalls das
Dokument Yueh al. zu zitieren. (AC W59453, EMBL Datenbank, Heidelberg)
das die Sequenz des FMO2-Proteins von Makak beschreibt.
-
Vier
Isoformen, FMO1, FMO3, FMO4 und FMO5, sind für Menschen identifiziert worden
(GeneBank-Sequenzen M64082, M83772, Z11737 bzw. L37080). Unter den
Säugerspezies
ist die Homologie zwischen orthologen FMO über 80%. Das Vorliegen eines
FMO, neben anderen Isoformen, kann für Menschen vernünftigerweise
postuliert werden.
-
Die
FMOs sind assoziiert mit dem endoplasmatischen Retikulum und sind
impliziert in der Entgiftung von xenobiotischen Verbindungen, wobei
es die Monooxygenierung erlaubt das Xenobiotikum in eine polarere Substanz überzuführen, ein
vorausgehender Schritt vor seiner Ausscheidung. Sie können gleichermaßen impliziert
sein in der metabolischen Aktivierung verschiedener toxischer Verbindungen
und/oder Kanzerogenen, die in der Umwelt vorliegen.
-
Der
Mechanismus der Reaktion von FMO ist detailliert beschrieben worden
(Poulsen, L.L. et al., 1995). Im Gegensatz zu allen anderen bekannten
Oxidasen oder Monooxigenasen haben FMOs die einzigartige Eigenschaft
ein stabiles intermediäres
Enzym zu bilden, 4α-Hydroperoxyflavin,
das NADP(H)- und Sauerstoffabhängig
ist, in der Abwesenheit eines oxidierbaren Substrats. Da die Energie
der Katalyse in dem FMO-Enzym bereits vor dem Kontakt mit seinem
potentiellen Substrat vorliegt, muss die völlige Übereinstimmung nicht so genau
sein wie für
andere Enzymtypen. Diese spezifische Charakteristik von FMO ist
verantwortlich für
die große
Vielzahl von Substraten, die von den FMOs akzeptiert werden (einschließlich zum
Beispiel die tertiären
und sekundären
Alkyl- und Arylamine, zahlreiche Hydrazine, Thiocarbamide, Thioamide,
Sulfide, Disulfide, Thiole).
-
Zahlreiche
Moleküle,
aktive Verbindungen bzw. Wirkstoffe von Medikamenten, sind als Substrate
von FMOs bekannt, entweder für
eine N-Oxidation oder eine S-Oxidation
(Gasser 1996), unter welchen man insbesondere Antidepressiva, Antipsychotika,
Mittel gegen Geschwüre,
Vasodilatatoren und blutdrucksenkende Mittel findet.
-
Obwohl
bestimmte Substrate von FMO zu weniger aktiven Derivaten oxidiert
werden, können
zahlreiche nukleophile Verbindungen metabolisiert werden zu Zwischenprodukten,
die reaktiver und/oder potentiell toxischer sein können; anstatt
ausgeschieden zu werden, könnten
solche Produkte toxische Reaktionen induzieren durch kovalente Fixierung
an zelluläre
Makromoleküle
oder durch andere Mechanismen. Zum Beispiel können die Mercaptopyrimidine
und die Thiocarbamide vorwiegend aktiviert werden durch eine FMO-Aktivität (Hines
et al., 1994). Genauer gesagt, ist gezeigt worden, dass die Nephrotoxizität, die mit
der Konjugation von Glutathion an Acrolein assoziiert ist, auf seinem
Metabolismus beruht, der durch das renale FMO vermittelt wird; das
FMO bildet ein S-Oxid, das danach freigesetzt wird, durch Eliminierungsreaktion,
die im basischen Milieu katalysiert wird, unter Bildung von cytotoxischem
Acrolein (Park, S.B. et al., 1992). So können die FMOs ebenfalls eine
wichtige Rolle spielen in den ersten Schritten der chemischen Toxizität als auch
der Entoxifizierung von xenobiotischen Verbindungen.
-
Wie
hier nachfolgend beschrieben, enthält eine große Anzahl von Medikamenten,
die heute in der klinischen Untersuchungsphase ist oder in großem Ausmaß verschrieben
wird, Funktionen mit nukleophiler Charakteristik des Stickstoff-,
Schwefel-, Phosphor-Typs oder anderer Typen. Die Rolle von FMO in
dem Oxidationsmechanismus von Medikamenten und endogenen chemischen
Verbindungen beim Menschen ist dennoch kaum bekannt.
-
Cashman
et al. (1996) haben kürzlich
die Beiträge
von FMO-Enzymen zum physiologischen Mechanismus des Cimetidins und
des S-Nikotins in vivo untersucht. Der größte Teil ihrer Ergebnisse bestätigt die
Tatsache, dass die FMO3-Aktivität
der Leber eines Erwachsenen verantwortlich ist für die Oxidation des Cimetidins
und des S-Nikotins, wobei diese Oxidation stereospezifisch ist.
Die Autoren zeigten außerdem,
dass die Stereochemie der Hauptmetaboliten des Cimetidins und des
S-Nikotins für
kleine Versuchstiere verschieden von denen sind, die für Menschen
beobachtet werden und legten nahe, dass verschiedene Isoformen von FMOs
entsprechend den Spezies vorherrschen könnten, wobei dies wichtige
Konsequenzen haben könnte
bei der Auswahl der Versuchstiere für die Ausarbeitungsprogramme
und Entwicklungen von Medikamenten für den Menschen.
-
Es
ist bekannt, dass das FMO1 bei Menschen in den Nieren, jedoch nicht
in der Leber exprimiert wird. Das FMO2 wird vorherrschend in den
Lungen aller getesteten Säugerspezies
exprimiert. Das FMO3 ist isoliert worden aus Mensch aus der Leber,
wo es bei Erwachsenen überwiegend
vorliegt. Das FMO3 ist die hauptsächliche Isoform, die bei der
Sulfoxidierung des Methionins und bei der stereospezifischen Oxidation
des Cimetidins und des S-Nikotins beteiligt ist. Das FMO3 zeigt
eine Spezifität
für sein
Substrat, die viel größer ist als
die des FMO1, das in der Leber in den meisten der untersuchten Tierspezies
gefunden wird. Das FMO4 ist eine weniger häufige Isoform, von welcher
die Funktion und die Substratspezifität wenig bekannt sind. Es liegt in
der humanen Leber vor und wird auch exprimiert im Gehirn, wo es
beteiligt sein könnte
bei der Oxidation von Antidepressiva wie Imipramin. Das FMO5 wird
in der humanen Leber in einem geringeren Ausmaß Weise exprimiert als FMO3.
Sein offensichtlicher Effizienzmangel als Enzym, das beteiligt ist
am Metabolismus von Medikamenten, legt nahe, dass es beteiligt sein
könnte
in einer physiologischen Funktion.
-
Die
verschiedenen Expressionsprofile von FMO-Isoformen in den Geweben
und/oder Spezies bilden folglich möglicherweise einen signifikanten
Faktor, der zu Unterschieden von Aktivitäten von FMO beiträgt, die zwischen
den Geweben und/oder zwischen Spezies beobachtet werden. So könnte die
Vielzahl der Formen von FMO seinen signifikanten Einfluss auf die
unterschiedliche Reaktion der Gewebe und/oder Spezies auf die Exposition
an eine xenobiotische Verbindungen haben. Tatsächlich beruhen die beobachteten
Unterschiede zwischen den Geweben und/oder Spezies in der Reaktion
auf xenobiotische Verbindungen und ihrer Toxizität zu einem wichtigen Teil auf
Variationen der Aktivität
und Spezifität,
die impliziert sind im Metabolismus dieser Substrate durch die FMOs.
Genetische Faktoren und Gewebespezifität in der Expression der FMOs
sind wichtige Faktoren dieser Variationen.
-
Bezüglich der
genetischen Faktoren ist zum Beispiel beschrieben worden, dass die
Trimethylaminurie, eine Pathologie, die bei 1 % der weißen Briten
vorliegt, und die sich manifestiert durch einen starken Geruch nach
verdorbenem Fisch in der ausgeatmeten Luft, dem Schweiß oder dem
Urin, auf einer Defizienz genetischen Ursprungs der Funktion eines
hepatischen FMO beruht.
-
Aus
vorstehend genannten Gründen
besteht daher heute ein wichtiger Bedarf zum Identifizieren neuer Isoformen
von FMO als auch der genetischen Polymorphismen, die eventuell damit
verbunden sind, die Spezifitäten
betreffend ihrer Substrate und/oder ihres Gewebeexpressionsprofils
zeigen, die impliziert sein könnten
im Metabolismus von Xenobiotika, als auch dem Metabolismus von Medikamenten
oder exogenen Substanzen, die in der Umwelt vorliegen, wie z.B.
die Pestizide, oder außerdem
die beteiligt sein könnten
an einer physiologischen Funktion. Dies ist genau der Gegenstand
der vorliegenden Erfindung.
-
Mehrere
Gene der Familie der humanen FMOs sind lokalisiert worden in der
Region 1g23-25 des Chromosoms 1 durch in situ-Hybridisierung des
Chromosoms in der Metaphase.
-
Wenn
eine solche Kandidatenregion definiert worden ist, ist es erforderlich
Zugriff auf das Fragment des Genoms zu haben, das das Intervall
abdeckt wo sich das(die) gesuchte(n) Gen(e) befinden. Dieser Schritt erfolgt
durch die Etablierung einer physischen Karte, nämlich die Gewinnung der Region
durch mehrere klonierte und geordnete Fragmente. Heute sind dank
der Daten integrierten CEPH/Généthon-Karte
des humanen Genoms ungefähr
80 % des Genoms durch YAC-Klone abgedeckt, subkloniert in BACS,
wobei die Lokalisierung auf den Chromosomen durch den Zwischenraum
von polymorphen und geordneten genetischen Markern erfolgt (Chumakov
et al., 1995). Diese physiko-genetische Karte erlaubt einen beachtlichen
Zeitgewinn, insbesondere durch die Verwendung von umfangreicher
Sequenzierung der interessierenden Regionen.
-
So
ist gemäß der vorliegenden
Erfindung nach Lokalisierung des BAC 123H04M auf dem genetischen Lokus
1q24-25, der zuvor angegeben wurde, etabliert worden, dass die Insertion,
die es trägt,
die Abschnitte 3' von
hFMO3 und 5' von
hFMO1 sowie die vollständige
Sequenz von hFMO2, und die eines anderen neuen Genmitglieds der
FMO-Familie, dem hFMOx, enthält.
-
Darüber hinaus
kann man dank der Verwendung der Banken mit den 5'-Markierungen die Expression von Kandidatengenen,
wie zuvor identifiziert, verifizieren: Die Identifizierung einer
Markierung, die an eine der Kandidatensequenzen hybridisiert, zeigt,
nachdem diese aus einer cDNA-Bank stammt ist, das Vorliegen von mRNA
und folglich einer Expression der fraglichen Sequenzen in den betrachteten
Geweben.
-
Die
vorliegende Erfindung beschreibt insbesondere ein isoliertes Polynukleotid
dessen Sequenz SEQ ID Nr. 1 teilweise in 2 dargestellt
ist und das codiert für
ein Polypeptid mit der Sequenz SEQ ID Nr. 3, die in 1 dargestellt
ist.
-
Die
vorliegende Erfindung beschreibt auch ein isoliertes Polynukleotid
dessen Sequenz SEQ ID Nr. 4 teilweise in 10 dargestellt
ist und das codiert für
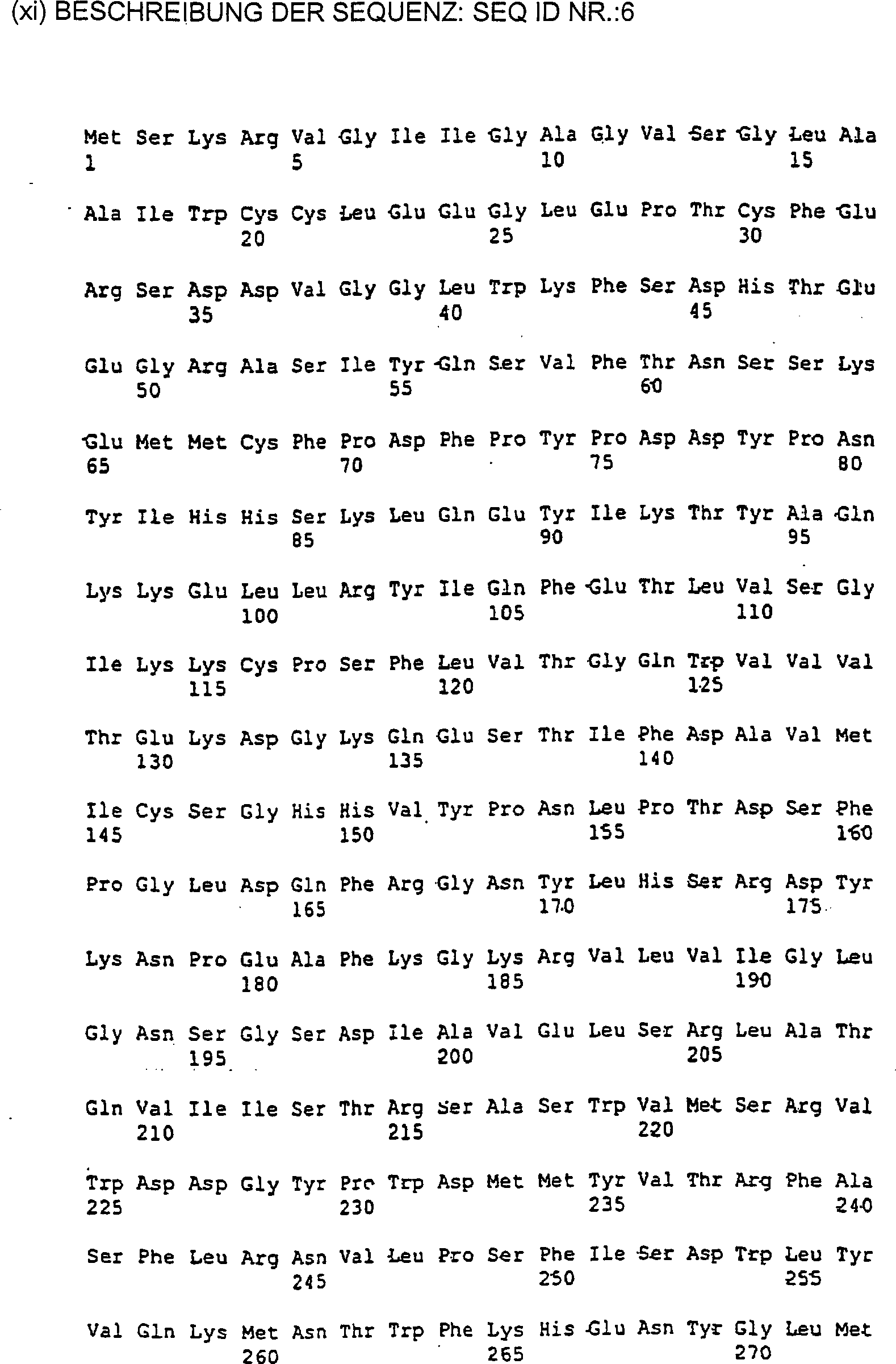
ein Polypeptid mit der Sequenz SEQ ID Nr. 6.
-
Diese
beiden Nukleotidsequenzen sind diejenigen von zwei Genen, die für neue Enzyme
der Familie der humanen Flavin-Monooxygenasen (FMO), hFMO2 bzw.
hFMOx, codieren. Dieses ist festgestellt worden durch Vergleich
der identifizierten Sequenzen mit bereits bekannten Sequenzen von
FMO: sehr starke strukturelle Homologien zwischen den beiden untersuchten
Sequenzen und denen von FMOs, sehr starke Homologien zwischen der
ersten Sequenz und den bekannten von FMO2, insbesondere die vom
Makaken (FMO2 des Makaken: GeneBank Sequenz U59453), gleichwie eine
nicht ausreichende Homologie der zweiten Sequenz mit jeder der bereits
für den
Menschen veröffentlichten
haben eine Schlussfolgerung erlaubt.
-
Die
bereits bekannte Exon-Struktur der Gene der FMO-Familie ist vollständig konserviert
in der hFMO2-Nukleotidsequenz gemäß der Erfindung. Die Sequenzen
von jedem der 9 Exons des Polynukleotids gemäß der Erfindung (3) zeigen Homologiegrade, von 95 % bis
98 % in DNA mit der entsprechenden mRNA-Sequenz des Makaken-FMO2
(4). Die Unterschiede zwischen den beiden Nukleotidsequenzen, gleichwie
ihre Bedeutung gegenüber
der Peptidsequenz, sind in 5 angegeben.
Die Polynukleotidsequenz SEQ ID Nr. 1 gemäß der Erfindung codiert für ein Polypeptid
mit der Sequenz SEQ ID Nr. 3 mit 535 Aminosäuren (1); die
Sequenz SEQ ID Nr. 2 der vorhergesagten mRNA, gleichwie die Polypeptidsequenz
des humanen Proteins, sind zu 97 % homolog mit denen des FMO2 des
Makaken (6 und 7), was
es erlaubte das Polypeptid gemäß der Erfindung
als das humane FMO2 zu identifizieren. Das Polypeptid mit der Sequenz SEQ
ID Nr. 3, das in 3 dargestellt ist,
stellt gleichfalls einen hohen Homologiegrad mit anderen Flavinmonooxygenasen
2 der Säuger
dar; seine Homologiegrade mit anderen Proteinen der Familie der
Flavinmonooxygenasen sind weniger stark.
-
Wie
zuvor angeführt,
hat das Fehlen einer ausreichenden Homologie zwischen den Sequenzen,
die hFMOx entsprechen – genomische
Sensequenz (SEQ ID Nr. 4), mRNA-Sequenz (SEQ ID Nr. 5) und Peptidaequenz
(SEQ ID Nr. 6) – und
den Sequenzen der bekannten FMOs es erlaubt zu schlussfolgern, dass
es sich um eine neue Isoform von FMO handelt.
-
Die
vorliegende Erfindung beschreibt folglich die DNA- oder RNA-Sequenzen,
wobei die DNA genomische, komplementäre oder synthetische DNA sein
kann, von FMOs, insbesondere von hFMO2 und hFMOx, als auch entsprechender
Proteine.
-
Die
vorliegende Erfindung beschreibt außerdem Klonierungsvektoren
und/oder Expressionsvektoren, die die Nukleotidsequenzen enthalten,
durch diese Vektoren transformierte Zellen oder Tiere, die die Zellen enthalten,
als auch Verfahren zur Herstellung der Polypeptide in der Form rekombinanter
Polypeptide.
-
Die
Erfindung beschreibt auch Selektionsverfahren für eine Verbindungen, die in
der Lage zum Modulieren der FMO-Aktivität ist.
-
Die
Erfindung beschreibt auch Diagnoseverfahren einer Prädisposition
für Störungen,
die auf mit FMO assoziiert sind, als auch pharmazeutische Zusammensetzungen,
die zur Behandlung und/oder Verhinderung solcher Störungen vorgesehen
sind.
-
Ein
erstes Beispiel solcher Störungen
könnte
das Weitwinkelprimärglaukom
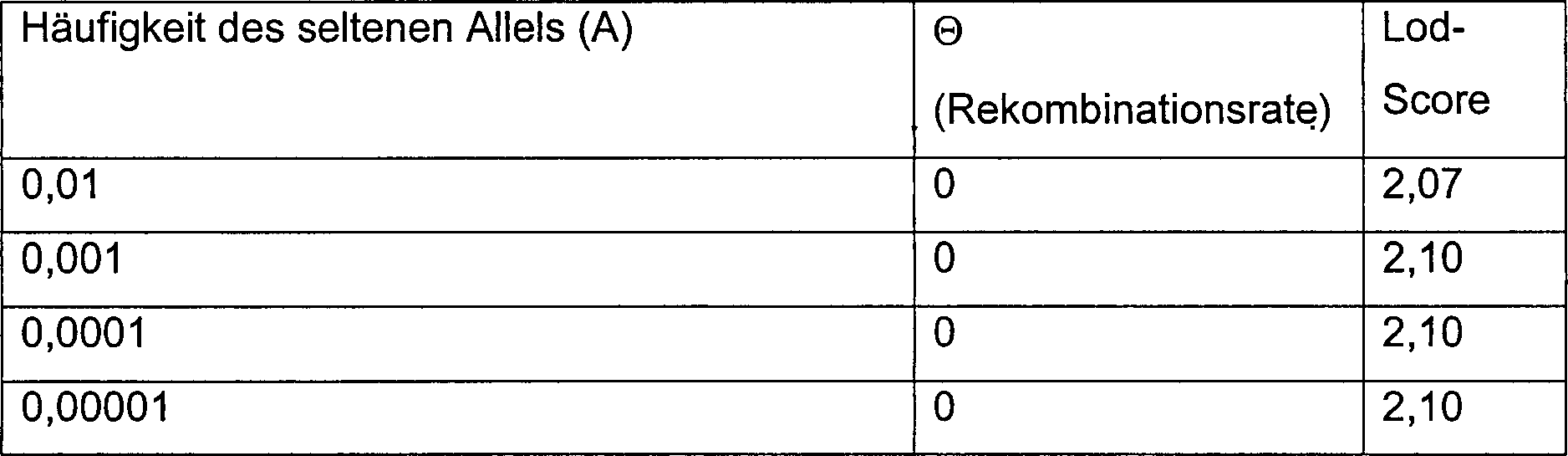
(GPAO). Allerdings haben Sunden et al. (1996), wie auch die Erfinder
(Belmunden et al., 1996), einen Zusammenhang zwischen der Chromosomenregion
GLC1A, die unter anderen Gensequenzen diejenigen trägt, die
von der Familie der FMOs bekannt sind, in 1q23-25, und dem plötzlichen
Auftreten von juvenilem GPAO (GPAO-J) identifiziert. Andererseits
ist eine mögliche
Rolle der Monooxygenasen in der Ethiologie des Glaukoms vorher nahegelegt worden
(Schwartzman et al. 1987). Tatsächlich
trugen die Metaboliten der Oxidationsreaktionen unter Inhibierung
der Aktivität
von Na+-, K+-ATPase in der Hornhaut zur Steuerung der Transparenz
der Hornhaut und der humoralen Augensekretion bei; folglich sind
eine Opazität
der Hornhaut und eine Augenüberdruck
die beiden Hauptkriterien der Glaukomdiagnose.
-
So
ist eine heterozygote Stelle, die eine Segregation in genotypischem
Zusammenhang in einer Familie zeigt, die auf Grund ihrer zahlreichen
von GPAO- J betroffenen
Mitglieder untersucht worden ist, in dem Exon 8 des Polypeptids
hFMO2 gemäß der Erfindung
durch die Erfinder identifiziert worden.
-
Unter
Verfolgung der Untersuchung der Polymorphismen, die in den geeignet
ausgewählten
Populationen vorliegen, und gelegen sind in den Sequenzen, die denjenigen
entsprechen, die von der Insertion von BAC 123H04M getragen werden,
oder allgemeiner durch die FMO-Sequenzen, kann man insbesondere
die Mutationen identifizieren, die assoziiert sind mit Pathologien
oder Störungen,
die auf einer FMO-Veränderung beruhen.
-
Die
verschiedenen Isoformen der FMOs scheinen sich weniger durch die
Gewebespezifität
ihrer Expression als durch ihre Substrate, von welchen sie die Transformation
katalysieren, zu unterscheiden. Wie zuvor angegeben, ist die Expression
der FMOs in der Leber, den Lungen, den Nieren oder dem Gehirn nachgewiesen
worden.
-
Die
pathogene Wirkung eines funktionellen Mangels einer FMO könnte aus
einer verringerten Kapazität
der Gewebe resultieren, wo sie sich ausdrückt oxidativer Belastung zu
widerstehen.
-
Allgemeiner
ausgedrückt,
könnten
die FMOs durch ihre Rolle im oxidativen Metabolismus und ihrer Enttoxifizierungsfunktion
beteiligt sein an der gesamten degenerativen oder toxischen Pathologie,
erwiesenermaßen
oder noch zu beweisen, insbesondere denjenigen, wo ein programmierter
Zelltod nachgewiesen werden kann und den degenerativen Erkrankungen
des zentralen Nervensystems.
-
Allgemein
ausgedrückt,
sind die Pathologien, die mit der Funktion von FMOs assoziiert sind,
zusammengefasst unter dem Ausdruck "mit FMO assoziierte Störungen".
-
Unter
den mit FMO assoziierten Störungen
kann man zum Beispiel, ohne sich darauf zu begrenzen, die Folgenden
nennen:
- – Oxidation
von Medikamenten, Substrate von FMO, zu weniger aktiven Derivaten,
womit eine Verschlechterung der Wirksamkeit des Medikaments in Verbindung
steht;
- – Nicht-Metabolisierung
von Medikamenten, die in der Form von Metaboliten aktiv sind, unter
Verlust der Wirksamkeit des Medikaments;
- – Nicht-Metabolisierung
von toxischen und/oder kanzerogenen Xenobiotika von exogenen Substanzen,
die natürlich
in der Nahrung vorliegen, wie etwa pflanzliche Alkaloide, oder toxischen
Substanzen, die in der Umwelt vorkommen, wie etwa die Pestizide
oder Herbizide;
- – Metabolisierung
von Medikamenten zu Zwischenprodukten, die reaktiver sein könnten, mit
der Folge einer Überdosierung
mit einer möglichen
Nebenwirkung;
- – Metabolisierung
von Xenobiotika, von Medikamenten oder anderen exogenen Substanzen,
zu Zwischenprodukten, die potentiell toxisch sein könnten; und
oder
- – Veränderung
der physiologischen Funktion, an welcher FMO beteiligt ist; im Besonderen
die Veränderung der
Funktion von FMO, die beteiligt sein könnte in der Glaukom-Symptologie.
-
Unter „FMO" wird man die Bezeichnung
eines beliebigen der bekannten humanen FMOs, FMO1, FMO3, FMO4 und
FMO5, oder neu in der Patentanmeldung beschriebenen, nämlich FMO2
oder FMOx, verstehen.
-
Bestimmte
dieser Störungen
können
eine multigenen Ursprung haben, jedoch vor allem tragen die Modifikationen
eines oder mehrerer FMOs zum Fortbestehen einer Störung oder
deren Verschlimmerung bei.
-
Die Nukleotidsequenzen
-
Die
vorliegende Erfindung betrifft vorallem eine isolierte Nukleotidsequenz,
die dadurch gekennzeichnet ist, dass sie für die humane Variante des Proteins
FMO2 der Sequenz SEQ ID Nr. 3 codiert, die einen Lysinrest anstelle
eines Glutaminsäurerests
in Position 402 der Sequenz SEQ ID Nr. 3 aufweist.
-
Die
vorliegende Erfindung betrifft gleichermaßen eine isolierte Nukleotidsequenz
gemäß der Erfindung,
dadurch gekennzeichnet, dass sie für die Variante des humanen
Proteins FMO2 der Sequenz SEQ ID Nr. 3 codiert, die einen Lysinrest
anstelle eines Glutaminsäurerests
in Position 402 der Sequenz SEQ ID Nr. 3 aufweist und dass sie eine
Sequenz umfasst, ausgewählt
aus:
- a) der Nukleotidsequenz von dem Nukleotid
in Position 2001 bis zu dem Nukleotid in Position 2056 der Nukleotidsequenz
SEQ ID Nr. 1,
- b) der Nukleotidsequenz von dem Nukleotid in Position 2405 bis
zu dem Nukleotid in Position 2542 der Nukleotidsequenz SEQ ID Nr.
1,
- c) der Nukleotidsequenz von dem Nukleotid in Position 10026
bis zu dem Nukleotid in Position 10214 der Nukleotidsequenz SEQ
ID Nr. 1,
- d) der Nukleotidsequenz von dem Nukleotid in Position 13341
bis zu dem Nukleotid in Position 13503 der Nukleotidsequenz SEQ
ID Nr. 1,
- e) der Nukleotidsequenz von dem Nukleotid in Position 16036
bis zu dem Nukleotid in Position 16178 der Nukleotidsequenz SEQ
ID Nr. 1,
- f) der Nukleotidsequenz von dem Nukleotid in Position 20558
bis zu dem Nukleotid in Position 20757 der Nukleotidsequenz SEQ
ID Nr. 1,
- g) der Nukleotidsequenz von dem Nukleotid in Position 21972
bis zu dem Nukleotid in Position 22327 der Nukleotidsequenz SEQ
ID Nr. 1,
- h) der Nukleotidsequenz von dem Nukleotid in Position 24411
bis zu dem Nukleotid in Position 24483 der Nukleotidsequenz SEQ
ID Nr. 1, in welcher das Nukleotid in Position 24431 der SEQ ID
Nr. 1 ein Adenosin anstelle eines Guanins ist,
- i) der Nukleotidsequenz von dem Nukleotid in Position 25487
bis zu dem Nukleotid in Position 25899 der Nukleotidsequenz SEQ
ID Nr. 1,
- j) der Nukleotidsequenz SEQ ID Nr. 1, in welcher das Nukleotid
in Position 24431 ein Adenosin anstelle eines Guanins ist, und
- k) der Nukleotidsequenz SEQ ID Nr. 2 in welcher das Nukleotid
in Position 1266 ein Adenosin anstelle eines Guanins ist.
-
Die
Erfindung umfasst darüber
hinaus eine isolierte Nukleotidsequenz, die dadurch gekennzeichnet ist,
dass sie ausgewählt
ist aus den Sequenzen, die für ein
Polypeptid codieren, umfassend das Polypeptid der Sequenz SEQ ID
Nr. 3, das einen Lysinrest anstelle eines Glutaminsäurerests
in Position 402 aufweist, oder komplementär ist zu einer Sequenz gemäß der vorliegenden
Erfindung.
-
Es
ist festzuhalten, dass die vorliegende Erfindung nicht die genomischen
Nukleotidsequenzen in ihrer natürlichen
Chromosomenumgebung betrifft, d.h. im natürlichen Zustand, es handelt
sich um Sequenzen, die isoliert worden sind, d.h., dass sie direkt
oder indirekt entnommen worden sind, zum Beispiel durch Kopieren (cDNA),
wobei ihre Umgebung zumindest teilweise modifiziert worden ist.
-
So
kann es sich ebensogut um cDNA handeln als auch um genomische DNA,
die teilweise modifiziert ist oder von Sequenzen getragen wird,
die mindestens teilweise verschieden sind von den Sequenzen, die
sie natürlich
tragen.
-
Diese
Sequenzen können
ebenfalls als „nicht-natürlich" bezeichnet werden.
-
Unter „Nukleotidsequenz" versteht man ein
Fragment einer isolierten natürlichen
oder synthetischen DNA und/oder RNA, die eine präzise Aneinanderreihung von
Nukleotiden beschreibt, modifiziert oder nicht, womit es möglich ist
ein Fragment, ein Segment oder eine Region einer Nukleinsäure zu definieren.
-
Unter „Allelen" versteht man die
Bezeichnung der natürlichen
mutierten Sequenzen, entsprechend den Polymorphismen, die im Menschen
vorliegen können,
und insbesondere diejenigen, die zur Entwicklung von Störungen,
die mit FMO assoziiert sind, führen
können.
-
Unter „Proteinvariante" versteht man die
Bezeichnung der Gesamtheit der mutierten Proteine, die im Menschen
vorliegen können,
die insbesondere Verkürzungen,
Substitutionen, Deletionen und/oder Additionen von Aminosäureresten
entsprechen, als auch künstliche
Varianten, die gleichermaßen „Proteinvarianten" genannt werden.
Im vorliegenden Fall sind die Varianten zum Teil mit dem Auftreten
von Störungen
assoziiert, die mit FMO assoziiert sind.
-
Gemäß der Erfindung
können
die Fragmente der Nukleotidsequenzen insbesondere für Domänen des Proteins
codieren oder auch als Sonde oder Primer in Verfahren zum Nachweis
oder zur Identifizierung oder Amplifizierung verwendet werden. Diese
Fragmente haben eine Länge
von mindestens 10 Basen und man bevorzugt Fragmente mit 20 Basen,
bevorzugt 30 Basen.
-
Gemäß der Erfindung
ist die Homologie statistisch einzigartig, sie zeigt, dass die Sequenzen
minimal 80 % und vorzugsweise 90 % gemeinsame Nukleotide aufweisen.
-
Die 1 zeigt
die Sequenz SEQ ID Nr. 3, die 2 zeigt
teilweise die Sequenz SEQ ID Nr. 1 von FMO2, die 10 zeigt teilweise die Sequenz SEQ ID Nr. 4 von
FMOx, so wie sie sequenziert worden sind im Genom eines Individuums,
das keine erkennbaren FMO-Störungen
zeigt.
-
Die
Struktur des Gens von hFMO2 ist in der 3 identifiziert.
-
Die
Sequenz SEQ ID Nr. 4 von FMOx ist teilweise in 10 dargestellt.
-
Die
Erfindung beschreibt ebenfalls Fragmente dieser Sequenzen, im Speziellen
Sequenzen, die für Polypeptide
codieren, die das Gesamte oder einen Teil der Aktivität des FMO-Proteins
beibehalten haben.
-
Bestimmte
dieser Sequenzen können
identifiziert werden indem man sich insbesondere 3 auf
bezieht, die die Organisation von hFMO2 schematisch darstellt.
-
Diese
partiellen Sequenzen können
verwendet werden für
zahlreiche Anwendungen, wie nachfolgend hier beschrieben werden
wird, insbesondere um Proteinkonstruktionen des FMO-Typs oder verschiedener FMO-Typen
auszuführen,
jedoch auch zum Realisieren von zum Beispiel FMO-ähnlicher
Proteine.
-
Wenn
die beschriebenen Proteinsequenzen im Allgemeinen die normalen Sequenzen
sind, betrifft die Erfindung somit eine mutierte Sequenz, die eine
Punktmutation aufweist.
-
Die
vorliegende Erfindung betrifft somit eine mutierte Nukleotidsequenz,
in welcher die Punktmutation nicht still ist, d.h., dass sie zu
einer Modifikation der durch die normale Sequenz codierten Aminosäure führt. Die
Erfindung beschreibt bestimmte Mutationen, die vorliegen können hinsichtlich
der Aminosäuren,
die die FMO-Proteine
oder die entsprechenden Fragmente davon bilden, insbesondere in
den Regionen, die katalytischen Stellen entsprechen, an den Regulationsstellen
oder an den Stellen zur Bindung von Cofaktoren; die Mutationen können sich
auch auf den am Transport und an der Adressierung beteiligten Sequenzen
befinden; sie können
auch im Besonderen die Cysteine supprimieren oder, im Gegensatz,
sie sichtbar machen, jedoch ebenfalls den Charakter des Proteins ändern, sei
es bezogen auf die Ladung, sei es bezogen auf die Hydrophobizität.
-
Diese
Mutationen können
ebenfalls auftreten in den Promotor- und/oder Regulationssequenzen
humaner FMO-Gene, welche Wirkungen auf die Expression des Proteins
haben können,
insbesondere auf seinen Expressionsgrad.
-
Unter
den Nukleotidfragmenten, die interessant sein können, insbesondere für die Diagnose,
müssen ebenfalls
genomische Intronsequenzen des FMO-Gens genannt werden, zum Beispiel
die Verbindungssequenzen zwischen Introns und Exons.
-
Die
Erfindung umfasst so die Nukleotidsequenzen gemäß der Erfindung, die dadurch
gekennzeichnet sind, dass sie mindestens die Mutation G.1263mac.A
umfasst, diejenige, die hier nachfolgend in den Beispielen definiert
wird.
-
Die
Erfindung umfasst ebenfalls die Nukleotidsequenzen, wie Nukleotidprimer,
die dadurch gekennzeichnet sind, dass die Sequenzen ausgewählt sind
aus den Sequenzen SEQ ID Nr. 7, SEQ ID Nr. 8, SEQ ID NR. 9 und SEQ
ID Nr. 10.
-
Die
Erfindung betrifft außerdem
die Nukleotidsequenzen als spezifische Sonde, dadurch gekennzeichnet,
das die Sequenzen ausgewählt
sind aus den Sequenzen SEQ ID Nr. 11, SEQ ID Nr. 12, SEQ ID Nr.
13 und SEQ ID Nr. 14.
-
Die
durch die Nukleotidsequenzen gemäß der Erfindung
codierten Polypeptide, insbesondere das Polypeptid mit der Sequenz
SEQ ID Nr. 3, das einen Lysinrest anstelle eines Glutaminsäurerests
in Position 402 aufweist, sind selbstverständlich Teil der Erfindung.
-
In
der vorliegenden Beschreibung sind die Ausdrücke Protein, Polypeptid oder
Peptid untereinander austauschbar.
-
Die
vorliegende Erfindung benennt ebenfalls die Nukleotidsequenzen,
die nicht-natürliche Nukleotide aufweisen
können,
insbesondere Schwefel-Nukleotide oder mit der α- oder β-Struktur.
-
Es
wird in der vorliegenden Erfindung gesagt, dass die Sequenzen selbstverständlich die
DNA- als auch RNA-Sequenzen betreffen, sowie Sequenzen, die mit
ihnen hybridisieren, als auch die entsprechenden doppelsträngen DNAs.
-
Unter
den interessanten Nukleinsäurefragmenten
sind im Speziellen die antisense-Oligonukleotide zu nennen, d.h.
deren Struktur durch Hybridisierung mit der Zielsequenz für eine Inhibierung
der Expression des entsprechenden Produkts sorgt. Ebenfalls müssen die
sense-Oligonukleotide genannt werden, die durch Wechselwirkung mit
Proteinen an der Regulation der Expression des entsprechenden Produkts
beteiligt sind, entweder eine Inhibierung oder eine Aktivierung
dieser Expression induzieren.
-
Wie
es nachfolgend hier beschrieben wird, kann es für bestimmte Anwendungen erforderlich
sein gemischte Konstrukte, Protein/DNA/chemische Verbindung, insbesondere
die Verwendung von z.B. Interkalationsmitteln, vorzusehen.
-
Die
Proteine, Polypeptide oder Peptide, die den zuvor genannten Sequenzen
entsprechen, sind in der nicht-natürlichen Form, d.h., dass sie
nicht in ihrer natürlichen
Umgebung vorliegen, sie jedoch erhalten werden können durch Reinigung, ausgehend
von natürlichen
Quellen, oder erhalten werden können
durch genetische Rekombination, wie diejenige, die hier nachfolgend
beschrieben wird.
-
Die
Erfindung zitiert ebenfalls die gleichen Polypeptide oder Proteine,
die erhalten werden durch chemische Synthese und nicht-natürliche Aminosäuren aufweisen
können.
-
Die
vorliegende Erfindung zitiert auch rekombinante Proteine, die ebenfalls
gut erhalten werden in glykosilierter Form als auch nicht-glykosilierter
Form und die natürliche
Tertiärstruktur
aufweisen können
oder nicht.
-
Die Vektoren und die Zellen
-
Die
vorliegende Erfindung betrifft ebenfalls Klonierungs- und/oder Expressionsvektoren,
die eine Nukleotidsequenz gemäß der Erfindung
aufweisen.
-
Diese
Klonierungs- und/oder Expressionsvektoren könnten Elemente aufweisen, die
die Expression der Sequenz in einer Wirtszelle sicherstellen, insbesondere
Promotorsequenzen und Regulationssequenzen, die in der Zelle wirkungsvoll
sind.
-
Der
fragliche Vektor könnte
autonom replizieren oder auch vorgesehen sein, um die Integration
der Sequenz in Chromosomen der Wirtszelle sicherzustellen.
-
Im
Falle von autonomen Replikationssystemen, in Abhängigkeit von der Wirtszelle,
prokariontisch oder eukariontisch, wird man vorzugsweise Systeme
des Plasmidtyps oder des viralen Typs verwenden, wobei die Virusvektoren
insbesondere Adenoviren (Perricaut et al., 1992), Retroviren, Pockenviren
oder Herpesviren (Epstein et al., 1992) sein können. Der Fachman kennt die
für jeden
dieser Viren verwendbaren Techniken.
-
Ebenso
ist es bekannt, als viralen Vektor defekte Viren zu verwenden, deren
Kultivierung in komplementierenden Zellen realisiert wird, was die
eventuellen Risiken einer Proliferation eines infektiösen viralen Vektors
vermeidet.
-
Wenn
die Integration der Sequenz in die Chromosomen der Wirtszelle gewünscht sein
sollte, wäre
es notwendig auf beiden Seiten der zu integrierenden Nukleotidsequenz
eine oder mehrere Nukleotidsequenzen, die von der Wirtszelle stammen,
vorzusehen, um die Rekombination zu gewährleisten. Es handelt sich
hierbei ebenfalls um Verfahren, die weitestgehend im Stand der Technik
beschrieben sind. Man könnte
zum Beispiel Systeme des Plasmid- oder viralen Typs verwenden; solche
Viren können
zum Beispiel die Retroviren (Temin 1986) oder die AAV, Adenovirus-assoziierte
Viren (Carter 1993), sein.
-
Die
Erfindung betrifft ebenfalls prokariontische oder eukariontische
Zellen, die transformiert sind durch einen Vektor gemäß der Erfindung
und dies, um die Expression eines natürlichen oder varianten FMO-Proteins oder
auch beispielsweise einer seiner Domänen sicherzustellen.
-
Die
Tiere, die dadurch gekennzeichnet sind, dass sie eine transformierte
Zelle gemäß der Erfindung enthalten,
bilden ebenfalls einen Teil der Erfindung.
-
Die
Erfindung umfasst weiterhin ein Verfahren zur Herstellung eines
Polypeptids gemäß der Erfindung, dadurch
gekennzeichnet, dass man eine Zelle gemäß der Erfindung kultiviert
und dass man das erzeugte Protein gewinnt.
-
Wie
es zuvor angegeben wurde, betrifft die vorliegende Erfindung außerdem die
Polypeptide, die erhalten werden durch Kultivieren von derart transformierten
Zellen und die Gewinnung des exprimierten Polypeptids, wobei die
Gewinnung auf intrazelluläre
Art als auch extrazelluläre
Art im Kulturmilieu erfolgen kann, wenn der Vektor so entwickelt
ist, dass er die Ausscheidung des Polypeptids auf einem Umweg, z.B. über eine „Leader-Sequenz,
sicherstellt, wobei das Protein exprimiert wird in der Form eines
Prä-Proteins
oder Prä-Pro-Proteins.
Die Konstrukte, die die Sezernierung von Polypeptiden erlauben,
sind bekannt, sowohl für prokaryontische
als auch eukaryontische Systeme. Bestimmte der FMO-Polypeptide können ihr
eigenes Sezernierungs- oder Membraninsertionssystem aufweisen.
-
Unter
den für
die Herstellung dieser Polypeptide verwendbaren Zellen sind selbstverständlich die
Bakterienzellen (Olins und Lee, 1993), als auch die Hefezellen (Buckholz,
1993), als auch die tierischen Zellen, insbesondere die Säugerzellkulturen
(Edwards und Aruffo, 1993), aber ebenso die Insektenzellen, in welchen man
Verfahren verwenden kann, die zum Beispiel Baculovirus einsetzen
(Luckow, 1993), zu nennen.
-
Die
so erhaltenen Zellen können
die Herstellung natürlicher
oder varianter FMO-Polypeptide
erlauben, jedoch auch von Fragmenten dieser Polypeptide, insbesondere
von Polypeptiden, die verschiedenen fraglichen Domänen entsprechen
können.
-
Die
Erfindung betrifft vorzugsweise ebenfalls die mono- oder polyklonalen
Antikörper,
die gegen die Polypeptide gemäß der Erfindung
gerichtet sind, dadurch gekennzeichnet, dass sie erhalten wurden
durch Immunreaktion eines humanen oder tierischen Organismus mit
einem immunogenen Mittel, bestellend aus einem Polypeptid gemäß der Erfindung,
insbesondere einem rekombinanten oder synthetischen Polypeptid gemäß der Erfindung.
-
Die
Erfindung betrifft ebenfalls die Antikörper gemäß der Erfindung, dadurch gekennzeichnet,
dass es sich um markierte Antikörper
handelt, insbesondere zur Bilddarstellung.
-
Diese
markierten monoklonalen oder polyklonalen Antikörper und welche insbesondere
dem Gesamten oder einem Teil der mutierten Proteine entsprechen,
können
zum Beispiel verwendet werden als Bilddarstellungsmittel, in vivo
oder ex vivo, auf biologischen Proben (Bilddarstellung mit Hilfe
von Antikörpern,
gekoppelt an ein nachweisbares Molekül, z.B. mit einer Bilddarstelllung
des PET-Abtast-Typs).
-
Die zellulären Modelle
-
Gemäß der vorliegenden
Erfindung können
die transformierten oder tierischen, nicht-humanen Zellen gemäß der Erfindung
ebenfalls verwendet werden als Modell für die in vitro-Selektion von
Produkten, die direkt oder indirekt an der Aktivität von mutiertem
FMO2 beteiligt sind, zum Untersuchen der verschiedenen einbezogenen
Wechselwirkungen. Sie können
ebenfalls verwendet werden zur in vitro-Selektion von mit mutiertem FMO2
wechselwirkenden Produkten, als Agonist, insbesondere enzymatisches
Aktivierungsmittel, oder Antagonist, insbesondere enzymatisches
Inhibierungsmittel.
-
Eine
andere potentielle typische Anwendung dder Charakterisierung dieser
Gene ist folglich die Möglichkeit
zum Identifizieren von Verbindungen, insbesondere auf Proteinbasis,
die mit diesen FMOs wechselwirken. Zum Beispiel kann es sich genausogut
um Inhibitoren als auch Aktivatoren, Substrate oder Cofaktoren, handeln.
Ihre Identifizierung wird erlauben sie in Abhängigkeit von ihren Wechselwirkungen
mit dem normalen Protein oder der Proteinvariante zu verwenden.
Im Speziellen wird man versuchen können Mittel zu isolieren, die
verschiedene Wirkungen auf normale und variante FMOs haben.
-
Es
ist ebenso beschrieben, das man diese zellulären Modelle verwenden können wird
zum Untersuchen des Metabolismus von Xenobiotika, Medikamenten oder
anderen, mit einem normalen oder varianten FMO. Dies könnte angewendet
werden bei der Identifizierung des toxischen Verhaltens bestimmter
Verbindungen, bei der Auswahl und Entwicklung von Verbindungen mit
erniedrigter Toxizität
oder mit verstärkter
Aktivität oder
bei den modifizierten FMOs, die ein besseres Vermögen zum
Metabolisieren der interessierenden Verbindungen aufweisen.
-
Dieser
Typ eines zellulären
Modells kann realisiert werden durch Anwendung von Gentechniken.
Es handelt sich unter Beachtung des Zelltyps, den man zu verwenden
beabsichtigt, darum das fragliche Gen in seiner normalen Form oder
in seiner mutierten Form in einen Expressionsvektor zu klonieren,
wobei es sich um einen autonomen Replikationsvektor oder einen Integrationsvektor
handeln kann, wobei der Vektor alle Elemente aufweist, die die Expression
des Gens in der fraglichen Zelle ermöglichen, oder diese alle Elemente aufweist,
die die Expression der fraglichen Sequenz ermöglichen.
-
Man
erhält
so eukaryontische oder prokaryontische Zellen, die das oder die
FMO-Protein(e),
normale oder variant, exprimieren, die dann Modelle bilden können, die
es erlauben auf einmal alle Wechselwirkungen verschiedener Produkte
mit den FMO-Proteinen oder ihren Varianten zu testen, oder Verbindungen
zu testen, insbesondere chemische Synthese-Produkte, die wechselwirken
können
mit dem Produkt des FMO-Gens, normal oder mutiert, und dies wenn
man sie dem Kulturmedium der Zellen zugibt..
-
Es
ist insbesondere anzumerken, dass die in Frage stehenden Produkte
genauso gut Mittel mit Antagonisten- als auch Agonistenaktivität sein können.
-
Die
Verwendung zellulärer
Modelle in Hinblick auf das Testen von pharmazeutischen Verbindungen
ist allgemein bekannt, sodass es wiederum nicht erforderlich ist
diesen Modelltyp detailliert zu beschreiben. Man kann dennoch unter
den verwendeten Techniken die „Phage
Display"-Technik
(Allen et al., 1995), und die Doppelhybrid-Verfahren (Luban und
Goff., 1995) zitieren.
-
Diese
Modelle können
vom in vitro-Typ sein, zum Beispiel Kulturen von humanen Zellen,
entweder in normaler Kultur oder möglicherweise in der Form eines
isolierten Organs.
-
Die
vorliegende Erfindung beschreibt ebenfalls Organismen, wie etwa
die tierischen, im Besonderen Mäuse,
die den Phänotyp
exprimieren, der dem normalen oder varianten FMO humanen Ursprungs
entspricht. Hier können
wiederum diese Tiere verwendet werden als Tiermodelle zum Testen
der Wirksamkeit bestimmter pharmazeutischer Produkte.
-
Diagnoseverfahren
-
Die
vorliegende Erfindung betrifft Diagnoseverfahren einer Prädisposition
für juveniles
Glaukom bei einem Patienten, dadurch gekennzeichnet, dass ausgehend
von einer biologischen Probe Patienten das Vorliegen der Mutation
G24431A in mindestens einer Sequenz, die für FMO2 codiert, bestimmt durch
die Analyse der gesamten oder eines Teils der Nukleinsäuresequenz,
die dem Gen entspricht, wobei das Vorliegen mindestens einer solchen
Mutation eine Prädisposition
des Patienten für
juveniles Glaukom anzeigt, die mit FMO2 assoziiert ist.
-
Es
ist wichtig klarzustellen, dass die vorliegende Erfindung nur hFMO2
und hFMOx im Detail beschreibt, aber es ist hier angegeben, dass
beschriebene Diagnoseverfahren und die für Therapie vorgesehenen Verbindungen,
ebenfalls die früheren
FMOs betreffen, wie etwa FMO1, FMO3, FMO4 und FMO5. Tatsächlich greifen
die FMOs allgemein in den Metabolismus von Xenobiotika und die Störungen,
die damit verbunden sind, ein, wie etwa zum Beispiel die vorstehend
genannten Xenobiotika und die Störungen,
die mit FMO assoziiert sind.
-
Die
Mutation, die für
die Erfindung untersucht wird, ist die Mutation G.1263mac.A (lokalisiert
in 6).
-
Die
analysierten Nukleinsäuresequenzen
könnten
genausogut die genomische DNA, eine cDNA oder eine mRNA sein
-
Wie
zuvor angegeben wurde, versteht man unter den Störungen, die mit FMO assoziiert
sind, welche nachgewiesen werden können, im Spezielleren die Pathologien,
die mit dem xenobiotischen Metabolismus, wie zuvor zitiert, assoziiert
sind, oder assoziiert sind mit der biologischen Funktion von FMO,
wobei es jedoch andere Störungen
geben kann, die mit einer Anomalie der FMOs assoziiert sein können.
-
Die
diagnostischen Werkzeuge, die auf der vorliegenden Erfindung basieren,
werden, wenngleich sie eine positive und differenzierte Diagnose
für einen
isolierten Patienten erlauben können,
vorzugsweise von Interesse sein für eine Präsymtom-Diagnose für ein gefährdetes
Individuum, insbesondere mit familiärer Vorbelastung, und es ist
ebenfalls möglich
eine pränatale
Diagnose vorzusehen.
-
Darüber hinaus
kann der Nachweis einer spezifischen Mutation eine fortschreitende
Diagnose erlauben, insbesondere betreffend die Stärke der
Störung
oder den möglichen
Zeitpunkt ihres Auftretens.
-
Die
Verfahren, die es erlauben die Mutation in einem Gen in Bezug auf
ein natürliches
Gen nachzuweisen, sind selbstverständlich sehr zahlreich. Man
kann sie im Wesentlichen in zwei große Kategorien einteilen, wobei
der erste Verfahrenstyp derjenige ist, worin das Vorliegen einer
Mutation nachgewiesen wird durch Vergleich der mutierten Sequenz
mit der entsprechenden nichtmutierten natürlichen Sequenz und der zweite Typ
derjenige ist, in welchem das Vorliegen der Mutation indirekt nachgewiesen
wird, zum Beispiel durch Nachweis von Fehlpaarungen, die auf das
Vorliegen der Mutation zurückzuführen sind.
-
In
den beiden Fällen
bevorzugt man im Allgemeinen die Verfahren, in welchen die Gesamte
oder ein Teil der FMO entsprechenden Sequenz amplifiziert wird vor
dem Nachweis der Mutation, wobei diese Amplifikationsmethoden durch
Verfahren realisiert werden können,
die als PCR oder PCR-ähnlich
bezeichnet werden. Unter PCR-ähnlich
wird man die Bezeichnung aller Verfahren verstehen, die direkte
oder indirekte Reproduktionen von Nukleinsäuresequenzen verwenden, als
auch in welchen die Marker-Systeme amplifiziert worden sind, wobei
die Techniken allgemein bekannt sind, wobei es sich im Allgemeinen
um die Amplifikation von DNA durch eine Polymerase handelt; wenn
die Ursprungs-Probe eine RNAist, wird geeigneterweise vorab eine reverse
Transkription durchgeführt.
Es gibt derzeit sehr viele Verfahren, die diese Amplifikation erlauben,
zum Beispiel die Verfahren, die als NASBA „Nucleic Acid Sequence Based
Amplification" (Compton
1991), TAS „Transcription
Based Amplification System" (Guatelli
et al., 1990), LCR „Ligase
Chain Reaction" (Landgren
et al., 1988), „Endo
Run Amplification" (ERA), „Cycling
Probe Reaction" (CPR)
und SDA „Strand
Displacement Amplification" (Walker
et al., 1992), bezeichnet werden, die dem Fachmann allgemein bekannt
sind.
-
Tabelle
1 zeigt Primersequenzen, die anwendbar sind zum Amplifizieren der
Sequenzen betreffend die Mutation G.1263mac.A.
-
Das
Reagens, das verwendet wird zum Nachweisen und oder Identifizieren
einer Mutation des FMO-Gens in einer biologischen Probe umfasst
eine Sonde, die Einfangsonde oder Nachweissonde genannt wird, wobei
mindestens eine dieser Sonden eine Sequenz gemäß der vorliegenden Erfindung
umfasst, die zuvor beschrieben wurde.
-
Suche nach Punktmutationen
-
Allgemein
können
mehrere Nachweisverfahren angewendet oder gegebenenfalls angepasst
werden nach der Amplifikation der interessierenden Sequenzen durch
PCR. Als Beispiele kann man nennen:
- 1) Sequenzierung:
Vergleich von Sequenzen mehrerer Individuen und/oder Identifizierung
einer heterozygoten Stelle bei einem einzelnen Individuum.
- 2) „Single
nucleotide primer extension" (Syvanen
et al., 1990). Beispiele von Primern, die verwendbar sind zum Nachweisen
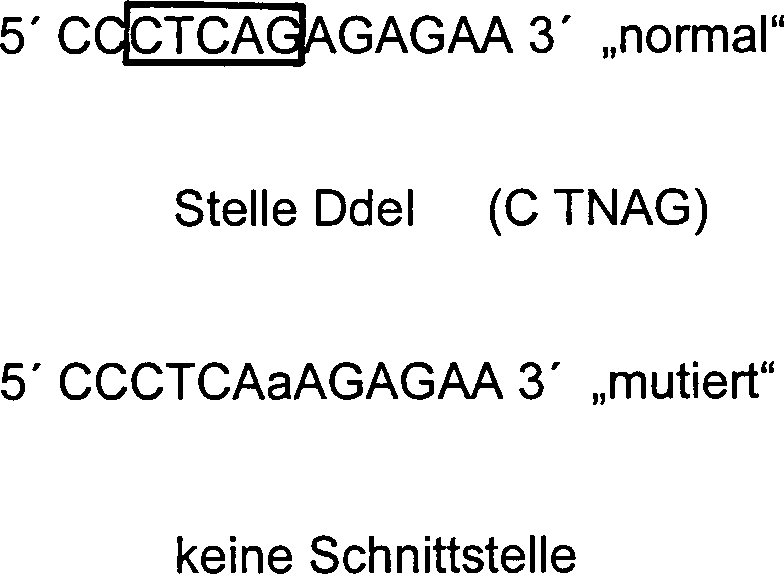
der Mutation G.1263mac.A. Durch das in Tabelle 2 dargestellte Verfahren.
- 3) RFLP „Restriction
Fragment Length Polymorphism".
Ein Beispiel eines Restriktionsenzyms, das verwendbar ist zum Nachweisen
der G.1263mac.A-Mutation durch RFLP wird in Tabelle 3 gezeigt.
- 4) Suche nach „Single
Strand Conformation Polymorphisms" (SSCP).
- 5) Verfahren, die auf einer Abspaltung von falsch gepaarten
Regionen basieren (enzymatische Abspaltung durch die S1-Nuklease,
chemische Abspaltung durch verschiedene Verbindungen, wie etwa Piperidin
oder Osmiumtetroxid, etc.
- 6) Nachweisen eines Heteroduplex durch Elektrophorese.
- 7) Verfahren, basierend auf der Anwendung der Hybridisierung
von spezifischen Oligonukleotidsonden von Allelen: (Allele Specific
Oligonucleotide" (ASO)
(Stoneking et al., 1991). Beispiele für Sonden, die verwendbar sind
für den
Nachweis der Mutation G.1263mac.A durch ASO sind in Tabelle 4 dargestellt.
- 8) OLA-Verfahren (dual color Oligonucleotide Ligation Assay" (Samiotaki et al.,
1994).
- 9) ARMS-Verfahren "Amplification
Refractory Mutation System" oder
ASA-Verfahren "Allele Specific Amplification" oder PASA-Verfahren "PCR Amplification
of Specific Allele" (Wu
et al., 1989).
-
Diese
Liste ist nicht vollständig
und andere gut bekannte Verfahren können verwendet werden.
-
Suche nach Veränderungen,
zum Beispiel vom Deletionstyp
-
Andere
allgemein bekannte Verfahren, die auf Hybridisierungstechniken mit
Hilfe genomischer Sonden, cDNA-Sonden, Oligonukleotid-Sonden oder
Ribosonden basieren, können
verwendet werden zur Untersuchung dieses Typs von Veränderungen.
-
So
bilden ebenfalls einen Gegenstand der Erfindung die Diagnoseverfahren
einer Prädisposition
für juveniles
Glaukom eines Patienten die mit mutiertem FMO2 gemäß der Erfindung
assoziiert sind, dadurch gekennzeichnet, dass die Analyse durch
Hybridisierung realisiert wird, wobei die Hybridisierung vorzugsweise
mit Hilfe mindestens einer Allel-spezifischen Oligonukleotidsonde
realisiert wird oder dadurch, dass das Vorliegen einer Mutation
nachgewiesen wird durch Vergleich mit der entsprechenden nicht-mutierten
natürlichen
Sequenz, oder dadurch, dass die Analyse durch Sequenzieren oder
durch elektrophoretische Migration erfolgt, und im Spezielleren
durch SSCP oder DGGE, oder dadurch, dass die Analyse durch eine
Methodik erfolgt, die darauf ausgerichtet ist eine Verkürzung des
Proteins nachzuweisen.
-
Einen
Teil der Erfindung bilden auch die Diagnoseverfahren einer Prädisposition
für juveniles
Glaukom, das auf mutiertem FMO2 gemäß der Erfindung beruht, bei
einem Patienten dadurch gekennzeichnet, dass die gesamte oder ein
Teil der Nukleotidsequenz des FMO-Gens zuvor unter Nachweis der
mindestens einen Mutation amplifiziert wird, wobei die Amplifikation
vorzugsweise durch PCR oder auf PCR-ähnliche Art realisiert wird,
wobei die zum Durchführen
der Amplifikation ausgewählten
Primer vorzugsweise ausgewählt werden
aus den Primern gemäß der Erfindung.
-
Die
Reagenzien zum Nachweisen und/oder Identifizieren der Gen-Mutation
des humanen FMO2-Gens in einer biologischen Probe, dadurch gekennzeichnet,
dass sie eine Sonde umfassen, die Einfangsonde und/oder Nachweissonde
genannt wird, wobei mindestens eine der Sonden eine Sequenz gemäß der Erfindung
aufweist, oder ein Antikörper
der Erfindung, bilden ebenfalls einen Teil der Erfindung.
-
Verfahren, die auf dem
Nachweis des Genprodukts basieren.
-
Die
Mutation des humanen FMO2-Gens ist verantwortlich für eine Modifikation
des Genprodukts, wobei die Modifikation verwendbar ist für einen
diagnostischen Ansatz. Tatsächlich
können
die antigenen Modifikationen die Entwicklung spezifischer Antikörper erlauben.
Alle diese Modifikationen können
verwendet werden als diagnostischer Ansatz, dank zahlreicher allgemein
bekannter Verfahren, die auf der Verwendung der monoklonalen oder
polyklonalen Antikörper
basieren, die das normale Protein oder mutierte Varianten erkennen,
zum Beispiel das RIA- oder ELISA-Verfahren.
-
Schließlich wird
hier beschrieben, dass es ebenfalls möglich ist eine Prädisposition
für die
mit FMO assoziierten Störungen
eines Patienten zu diagnostizieren, durch Messung der enzymatischen
Aktivität
des oder der FMO(s), ausgehend von biologischen Proben des Patienten.
Die Messung dieser Aktivität(en)
durch Vergleich mit einem internen oder externen Standard wird tatsächlich eine
Prädisposition
für eine
der vorstehend zitierten Störungen
anzeigen.
-
Therapeutische Zusammensetzungen
-
Die
vorliegende Erfindung beschreibt ebenfalls therapeutische, heilende
oder präventive
Behandlungen von Störungen,
die mit FMO assoziiert sind.
-
Man
könnte
die Verbindungen verwenden, die direkt oder indirekt an der FMO-Aktivität beteiligt
sind, und die aus der Verwendung der vorstehend beschriebenen zellulären Modellen
hervorgegangen sind.
-
Man
könnte
im Besonderen die Verbindungen verwenden, die mit den normalen oder
varianten FMOs wechselwirken können,
insbesondere als Agonist oder Antagonist.
-
Die
vorliegende Erfindung beschreibt ebenfalls therapeutische Zusammensetzungen,
die als aktives Mittel eine Verbindungen aufweisen, die in die FMO-Aktivität modulieren
kann, wobei es sich um Zusammensetzungen mit pro-FMO-Aktivität, insbesondere
solche, die zuvor beschrieben sind, oder um Zusammensetzungen mit
anti-FMO-Aktivität
handeln kann.
-
Allgemein
versteht man unter einer Verbindungen mit „pro-FMO-Aktivität" eine Verbindungen,
die die Aktivität
von FMO induzieren wird, im Gegensatz zu einer anti-FMO-Verbindungen,
die die Tendenz zur Verringerung der FMO-Aktivität haben wird. Die tatsächliche
Wirkung dieser Aktivitätstypen
wird vom Typ des exprimierten Enzyms, normal oder pathogen, abhängen.
-
Bevorzugt
könnte
man therapeutische Zusammensetzungen verwenden, deren Aktivität verschieden gegenüber den
normalen und varianten-FMO-Enzymen ist.
-
Es
ist zunächst
möglich
eine Substitutionsbehandlung vorzusehen, d.h. therapeutische Zusammensetzungen,
die dadurch gekennzeichnet sind, dass sie als aktives Mittel eine
Verbindungen mit pro-FMO-Aktivität
aufweisen; es könnte
sich insbesondere vollständig
oder zum Teil um Polypeptide handeln, wie sie vorstehend beschrieben
worden sind, oder auch um einen Expressionsvektor derselben Polypeptide,
oder auch um chemische oder biologische Verbindungen, die eine pro-FMO-Aktivität, eine
FMO-ähnliche
Aktivität
aufweisen oder die Produktion von FMO induzieren.
-
Es
ist ebenfalls möglich
therapeutische Zusammensetzungen zu verwenden, in welchen das aktive Mittel
eine anti-FMO-Wirkung haben wird, im Speziellen eine anti-FMO-Variante.
In diesem Falle handelt es sich um eine supprimierende Behandlung.
Es könnte
sich zum Beispiel um Verbindungen handeln, die wechselwirken mit
den Enzymen, insbesondere Proteinverbindungen, und im Besonderen
anti-FMO-Antikörper,
insbesondere wenn diese Antikörper
die Proteinvarianten erkennen. Es könnte sich ebenfalls um chemische
Produkte handeln, die eine anti-FMO-Aktivität aufweisen, insbesondere Antagonisten
der FMO-Variante.
-
Unter
den zahlreichen verwendbaren pharmazeutischen Zusammensetzungen,
sind ganz besonders die anti-Sense-Sequenzen zu zitieren, die mit
dem normalen oder mutierten FMO-Gen wechselwirken, als auch die
Sense-Sequenzen die auf die Regulation der Expression dieser Gene
wirken, wobei die Produkte ebenfalls wechselwirken können stromabwärts der
durch die FMOs induzierten Expressionsprodukte,.
-
Ebenfalls
zu nennen sind die monoklonalen Antikörper, die die FMOs inhibieren,
inbesondere die mutierten FMOs, und/oder die entsprechenden Liganden
und/oder die Produkte, die durch FMO-Aktivität induziert werden, inhibieren,
die folglich pro- oder anti-Aktivitäten haben können.
-
Es
ist ebenfalls möglich
die Expression von Proteinen oder ihrer Fragmente in vivo vorzusehen,
insbesondere über
den Umweg der Gentherapie und unter Verwendung von den Vektoren,
die vorausgehend beschrieben worden sind.
-
Im
Rahmen der Gentherapie ist es ebenfalls möglich die Verwendung von zuvor
beschriebenen „nackten" Gensequenzen oder
cDNAs vorzusehen, wobei diese Technik insbesondere entwickelt worden
ist durch die Gesellschaft Vical, die gezeigt hat, dass es unter
diesen Bedingungen möglich
ist das Protein in bestimmten Geweben zu exprimieren, insbesondere
ohne auf die Unterstützung
eines viralen Vektors zurückgreifen
zu müssen.
-
Heute
ist es im Rahmen der Gentherapie ebenfalls möglich die Verwendung von ex
vivo transformierten Zellen vorzusehen, welche anschließend wieder
reimplantiert werden könnten,
entweder so wie sie sind oder innerhalb der Systeme des organoiden
Typs, so wie dies auch im Stand der Technik bekannt ist (Danos et
al., 1993). Man kann ebenfalls die Verwendung von Mitteln vorsehen,
die das Abzielen auf einen bestimmten zellulären Typ erleichtern, die Penetration
in die Zellen oder den Transport in Richtung des Kerns.
-
So
beschreibt die Erfindung ebenfalls eine therapeutische Zusammensetzung,
dadurch gekennzeichnet, dass sie als aktives Mittel mindestens eine
Verbindungen enthält,
die in der Lage ist zum Modulieren der FMO-Aktivität, vorzugsweise
der Aktivität
von FMO2 und/oder FMOx.
-
Die
Erfindung beschreibt auch eine therapeutische Zusammensetzung, dadurch
gekennzeichnet, dass sie als aktives Mittel mindestens eine Verbindungen
enthält,
die in der Lage ist zum Wechselwirken mit FMO, vorzugsweise in der
Lage ist zum Wechselwirken mit FMO2 und/oder FMOx, oder eine therapeutische Zusammensetzung,
die dadurch gekennzeichnet ist, dass sie eine verschiedene Aktivität gegenüber normalem
FMO und pathologischem FMO zeigt.
-
Die
vorliegende Erfindung umfasst eine therapeutische Zusammensetzung,
die dadurch gekennzeichnet ist, dass sie als aktives Mittel mindestens
einen Agonisten des mutierten humanen FMO2 aufweist, ausgewählt aus
den folgenden Verbindungen:
- a) einem Protein
oder einem Polypeptid gemäß der Erfindung;
- b) einem Expressionsvektor gemäß der Erfindung;
- c) einer Nukleotidsequenz gemäß der Erfindung, dadurch gekennzeichnet,
dass die Sequenz eine Sense-Sequenz ist, die die Expression des
mutierten FMO2 induziert.
-
Die
Erfindung betrifft weiterhin eine therapeutische Zusammensetzung,
dadurch gekennzeichnet, dass sie als aktives Mittel einen Antagonisten
des mutierten humanen FMO2 umfasst, ausgewählt aus den folgenden Verbindungen:
- a) einem anti-FMO-Antikörper gemäß der Erfindung;
- b) einer Nukleotidsequenz gemäß der Erfindung, dadurch gekennzeichnet,
dass die Sequenz eine Antisense-Sequenz ist, die die Expression
des mutierten FMO2 inhibiert,
- c) einer Nukleotidsequenz gemäß der Erfindung, dadurch gekennzeichnet,
dass die Sequenz eine Sense-Sequenz ist, die die Expression des mutierten
FMO2 inhibiert.
-
Die
Erfindung beschreibt auch eine therapeutische Zusammensetzung gemäß der Erfindung,
dadurch gekennzeichnet, dass das aktive Mittel eine lösliche Sequenz
ist, die mit FMO wechselwirkt.
-
Ein
weiterer Gegenstand der Erfindung ist auch die Verwendung eines
aktiven Mittels, das in der Lage ist zum Modulieren der Aktivität des mutierten
humanen FMO2, zum Realisieren eines Medikaments, das zur Behandlung
und/oder Verhinderung von juvenilem Glaukom vorgesehen ist, dadurch
gekennzeichnet, dass das aktive Mittel ausgewählt ist aus den Agonisten-
oder Antagonisten-Verbindungen
der therapeutischen Zusammensetzungen der Erfindung.
-
Unter
einem anderen Aspekt beschreibt die Erfindung auch einen Bioabbau-
oder Biosyntheseprozess einer organischen oder anorganischen Verbindung,
dadurch gekennzeichnet, dass er ein Polypeptid oder eine Zelle gemäß der Erfindung
verwendet.
-
Die
Polypeptide mit FMO-Aktivität
könnten
tatsächlich
vorteilhafterweise verwendet werden zum Bioabbau, unter Befolgung
von Oxidationsreaktionen, z.B. denjenigen, die von Ziegler (Ziegler
et al., 1993) beschrieben sind, der FMO-Substratverbindungen, im Besonderen
solcher Verbindungen, die in der vorliegenden Beschreibung genannt
sind, oder verwendet werden zur Biosynthese einer interessierenden
Verbindung ausgehend von FMO-Substratverbindungen, insbesondere
zur Biosynthese eines Medikaments, eines Nahrungsergänzungsmittels,
eines Pestizids oder Herbizids.
-
Die
Verarbeitungsverfahren der Verbindung von Interesse, die dadurch
gekennzeichnet sind, dass sie ein Polypeptid oder eine Zelle gemäß der Erfindung
verwenden, sind hier zitiert. Die Polypeptide oder Zellen gemäß der Erfindung
könnten
allerdings vorteilhafterweise in vitro verwendet werden zum Bestimmen
der potentiellen Metabolisierung der Verbindung von Interesse und
zum Analysieren der möglicherweise
erhaltenen Metaboliten, ihrer Toxizität und/oder ihrer Aktivität. Die erhaltenen
Ergebnisse werden es erlauben die Verbindung zu bestätigen oder
sie wieder zu formulieren, so, dass sie Substrat von FMO wird oder
nicht, oder die gebildeten Metaboliten verschieden sind.
-
Schließlich beschreibt
die vorliegende Erfindung die Verwendung des Polypeptids oder der
Zelle gemäß der Erfindung
zur Entgiftung einer xenobiotischen Verbindung, ein Substrat von
FMO. Diese xenobiotischen Verbindungen können in der Umwelt vorliegen,
wie etwa Pestizide oder Herbizide, natürlich vorliegen in den Pflanzen,
wie bestimmte Alkaloide, oder können
pharmazeutischen Verbindungen entsprechen.
-
Andere
Charakteristika und Vorteile der vorliegenden Erfindung werden ersichtlich
aus den nachfolgenden Beispielen, die unter Bezugnahme auf die nachfolgenden
beiliegenden Figuren angegeben werden:
-
1:
Polypeptidsequenz, entsprechend der Sequenz SEQ ID Nr. 3, vorausgesagt
von hFMO2, humanes Homologes des FMO2 des Makaken.
-
2:
Nukleotidsequenz, teilweise entsprechend der Sequenz SEQ ID Nr.
1 des Gens, das für hFMO2
codiert, humanes Homologes des FMO2 des Makaken.
-
In
Hinblick auf die Homologien der mRNA, die bekannt sind von Genen
der Familie der Flavin-Monooxygenasen, teilen diese Gene die gleiche
Exon/intron-Struktur:
Exon
1: nicht übersetzt,
variabel in der Größe und der
Sequenz,
Exon 2: Beginn der codierenden Region, Codierung für die Aminosäuren 1–44,
Exon
3: Aminosäuren
45–107,
Exon
4: Aminosäuren
108–161,
Exon
5: Aminosäuren
162–209,
Exon
6: Aminosäuren
210–275,
Exon
7: Aminosäuren
276–394,
Exon
8: Aminosäuren
395–419,
Exon
9: Aminosäuren
420–535,
Ende der codierenden und nichttranslatierten 3'-Region.
-
Die
Introns sind in der Größe und Komplexität variabel.
Wir haben zunächst
die Sequenz von drei Fragmenten von BAC 123H04M isoliert, die die
gesamten Exons dieses Homologen enthielten.
Fragment 1: enthält die Exons
1 und 2,
Fragment 2: enthält
das Exon 3,
Fragment 3: enthält die Exons 4 bis 9.
-
Die
Sequenzen von zwei Introns sind dann vervollständigt worden.
-
3:
3A:
Beschreibung der Exon/Intron-Struktur des Gens, das für hFMO2
codiert, humanes Homologes von FMO2 des Makaken.
Angegeben
sind die Positionen der Anfänge
und Enden der Exons auf den Nukleotidsequenzen SEQ ID Nr. 1 und
Nr. 2.
3b: Beschreibung der Exon/Intron-Struktur
des Gens, das für
hFMOx codiert. Angegeben sind die Positionen der Anfänge und
Enden der Exons auf den Nukleotidsequenzen SEQ ID Nr. 4 und Nr.
5.
-
4:
Homologie zwischen dem FMO2-Gen des Makaken und seinem humanen Homologen,
wie in 2 angegeben. Die nichttranslatierte
5'-Region divergiert
leicht von der Sequenz von Makak wie in 2 angegeben.
-
5:
Zusammenstellung der varianten Positionen der Sequenz der mRNA von
humanem FMO2, unter Bezugnahme auf die homologe Makaksequenz; Einfluss
von Variationen auf die Proteinsequenz.
-
6:
Homologien zwischen den Sequenzen der mRNA des FMO2 aus Makak und
seinem humanen Homologen.
-
Die
Position der Mutation G.1263mac.A ist durch einen vertikalen Pfeil
gekennzeichnet.
-
7:
Homologien zwischen den Peptidsequenzen des FMO2 aus Makak und seinem
humanen Homologen.
-
8:
Analyse der Segregation des Polymorphismus G.1263 mac.A in der untersuchten
Familie.
-
Die
genomische DNA der Individuen 3, 4 und 7 bis 14 ist amplifiziert
worden durch PCR, und die Sequenz der erhaltenen Fragmente analysiert
worden zum Nachweisen von heterozygoten Stellen, die mit der Krankheit
segregiert werden.
-
Die
ausgefüllten
Symbole zeigen die Individuen, die von juvenilem GPAO befallen sind.
Die durchgestrichenen Symbole zeigen die nichtgenotypischen Individuen.
Die Individuen 11 und 12 sind Zwillinge.
-
G/G
= Homozygote für
die Base in der Position homolog zur Position 1263 der mRNA des
FMO2 aus Makak.
-
G/A
= Heterozygote für
die Base in der Position homolog zur Position 1263 der mRNA des
FMO2 aus Makak.
-
9:
Chromosomenlokalisation von BAC123H04M durch Fluoreszenzhybridisierung
in situ.
-
(A)
Ein spezifisches Signal wird auf den beiden Chromosomen 1 beobachtet.
Auf der Fotografie (B) ist ein einziges der beiden Chromosomen 1
dargestellt. In (C) werden nur die R-Banden dieser Chromosomen beobachtet,
wobei gezeigt wird, dass das Signal der Sonde 123H04M auf der Bande
1q23 lokalisiert ist.
-
10: Nukleotidsequenz, die teilweise der Sequenz
SEQ ID Nr. 4 des Gens entspricht, codierend für die humane hFMOx-Isoform.
-
Dieses
Gen zeigt einen Homologiegrad von 75 % für DNA und von 70 % für Aminosäuren mit
den Messenger-RNAs der Flavon-Monooxygenasen auf BAC 123H04M. Es
ist in dieser vorläufigen
Figur in vier Fragmenten dargestellt:
Fragment 1: codiert für Exon 2
(erstes codierendes Exon),
Fragment 2: Exon 3,
Fragment
3: Exons 4 bis 8,
Fragment 4: Exon 9.
-
Beispiele
-
Isolierung von BAC 123H04M
-
Zum
Identifizieren eines Gens, das für
eine neue FMO codiert, hat man ein BAC („Bacterial Artificial Chromosome") isoliert, das der
Kandidatenregion entspricht, die zuvor auf dem Chromosom 1 lokalisiert
worden ist. Eine Bank von BACs, die das vollständige humane Genom abdeckt,
ist hergestellt worden, ausgehend von DNA einer humanen lymphoblastischen
Linie, die von dem Individuum Nr. 8445 der CEPH-Familien stammt.
Diese Linie ist verwendet worden als Quelle von DNA mit hohem Molekulargewicht.
Die DNA ist partiell durch das Restriktionsenzym BamH1 verdaut worden,
danach in die BamH1-Stelle des Plasmids pBeIoBacII kloniert worden.
Die so erhaltenen Klone sind „gepoolt" worden und gescreent
worden, entsprechend einem dreidimensionalen Analyseverfahren, das
beschrieben ist für
das Screenung von Banken von YACs („Yeast Artificial Chromosome") (Chumakov et al.,
1992). Die erhaltenen dreidimensinalen Pools sind durch PCR gescreent
worden mit Hilfe von Primern, die den Marker D1S3423 (WI-10286)
einrahmen. Diese STS („Sequence
Tagged Site) ist zuvor lokalisiert worden in der Kandidatenregion.
Ein Klon von BAC 123H04M ist so isoliert worden.
-
Nach
Verdau durch das Restriktionsenzym NotI, ist die Länge des
Inserts, das von diesem BAC getragen wird, bestimmt worden auf einem
0,8 % Agarosegel nach Migration durch Elektrophorese im alternierenden
Feld (CHEF) (4 Stunden bei 9 Volt/cm, mit einem Winkel von 100°, bei 11 °C in 0,5 × TAE-Puffer).
Man hat so nachgewiesen, dass das BAC 123H04 ein Insert mit 180
kb trägt.
-
Chromosomenlokalisation
von BAC 123H04M durch in situ-Fluoreszenzhybridisierung
(FISH)
-
Die
Chromosomenlokalisierung des BAC in der Kandidatenregion 1q23-q25
ist bestätigt
worden durch in situ-Fluoreszenzhybridisierung (FISH) auf Metaphase-Chromosomen gemäß dem von
Cherif et al., 1990, beschriebenen Verfahren. Das BAC 123H04M ist
genauer lokalisiert worden in der Bande 1q23 von Chromosom 1 (9).
-
Sequenzierung des Inserts
von BAC 123H04M
-
Zum
Sequenzieren des Inserts von BAC 123H04M hat man drei verschiedene
Banken von Subklonen hergestellt, ausgehend von beschallter DNA
dieses BAC.
-
Nach
Inkubation über
Nacht sind die Zellen, die aus drei Liter Kulturmedium hervorgehen,
durch alkalische Lyse entsprechend klassischer Techniken behandelt
worden. Nach Zentrifugation des erhaltenen Produkts in einem Cäsiumchloridgradienten
sind 52 μg
DNA von BAC 123H04M gereinigt worden. 7 μg DNA sind unter drei verschiedenen
Bedingungen beschallt worden, um Fragmente, von welchen sich die
Größen gleichförmig von
1 bis 9 kb verteilen, zu erhalten. Die erhaltenen Fragmente sind
in einem Volumen von 50 μl
mit 2 Einheiten Vent-Polymerase für 20 Minuten bei 70 °C in Gegenwart
der 4 Deoxytriphosphate (100 μM)behandelt
worden. Die aus diesem Schritt resultierenden Fragmente mit glatten
Enden, sind durch Gelelektrophorese mit 1 % Agarosegel mit niedrigem
Schmelzpunkt getrennt worden (60 Volt während 3 Stunden). Die gemäß ihren
Größen eingeteilten
Gruppen sind ausgeschnitten worden und die erhaltenen Banden durch
Agarase behandelt worden. Nach Extraktion mit Chloroform und Dialyse
auf Microcon 100-Säulen,
ist die DNA in Lösung eingestellt
worden auf eine Konzentration von 100 ng/μl. Eine Ligation ist bewirkt
worden, Inkubation für
eine Nacht, durch Vorlegen von 100 ng DNA-Fragment von BAC 123H04M und 20 ng DNA
des durch enzymatischen Verdau linearisierten und durch alkalische
Phosphatase behandelten Vektors.
-
Diese
Reaktion ist realisiert worden in einem Endvolumen von 10 μl in Gegenwart
von 40 Einheiten/μl T4-DNA-Ligase
(Epizentrum). Die Ligationsprodukte haben dann dazu gedient durch
Elektroporation entweder einen XL-Blue-Stamm (für die Plasmide mit mehreren
Kopien) oder einen D10HB-Stamm (für die Subklone, die von BAC
abstammen) zu transformieren. Die Klone, die lacZ– waren
und gegenüber
dem Antibiotikum resistent waren sind einzeln auf Mikroplatten plattiert
worden zur Aufbewahrung und Sequenzierung.
-
So
erhielt man:
- – 864 Subklone, die aus der
Insertion von Fragmenten mit 2 bis 3 kb in die SmaI-Stelle des Plasmids
puc18 hervorgehen;
- – 1728
Subklone, die der Insertion von Fragmenten mit 1,5 bis 2 kb in die
BamHI-Stelle (glatt gemacht) des Plasmids BluescriptSK entsprechen;
- – 288
Subklone, die Fragmente mit 4 bis 7 kb aufweisen, die insertiert
sind in die PmII-Stelle eines modifizierten BAC-Vektors.
-
Die
Insertionen dieser Subklone sind amplifiziert worden durch PCR von
Bakterienkulturen, die eine Nacht unter Verwendung der Primer der
Vektoren, die die Insertionen flankieren, inkubiert wurden. Die
Sequenzen der Enden dieser Inserts (durchschnittlich 500 Basen an
jeder Seite) sind bestimmt worden durch automatische Fluoreszenzsequenzierung
auf dem Sequenzierer ABI 377, ausgestattet mit der Software ABI Prism
DNA Sequencing Analysis (Version 2.1.2).
-
Die
Fragmente der Sequenz, die von Sub-BACs stammen, sind zusammengesetzt
worden durch die Software Gap4 von R. Staden (Bonfield et al., 1995).
Diese Software erlaubt die Rekonstruktion einer vollständigen Sequenz
ausgehend von Sequenzfragmenten. Die aus der Anordnung verschiedener
Fragmente deduzierte Sequenz ist die Konsenssequenz.
-
Schließlich hat
man gerichtete Sequenzierungstechniken (systematischer Lauf mit
Primern) verwendet, um die Sequenzen zu vervollständigen und
die Contigs zu verbinden.
-
Sequenzanalyse
-
Die
potentiellen Exons von BAC 123H04M sind ausfindig gemacht worden
durch Homologierecherche in allgemein zugänglichen Banken für Proteine,
Nukleinsäuren
und EST (Exprimierte Sequenz-Tags).
-
Datenbanken:
-
Man
verwendete lokale Überarbeitungen
allgemein zugänglicher
Hauptbanken. Die verwendete Proteinbank ist gebildet durch nichtredundante
Fusion der Banken Genpept (Automatische Translation von GenBank,
NCBI; Benson et al., 1996); Swissprot, (George et al., 1996); und
PIR/NBRF (Bairoch et al., 1996). Die Verdopplungen sind eliminiert
worden durch die Software „nrdb" (gemeinfreie Software);
NCBI; Benson et al., 1996). Die internen Wiederholungen sind dann
maskiert worden durch die Software „xnu" (gemeinfreie Software; NCBI; Benson
et al., 1996). Die resultierende Bank, die als NRPU (Non-Redundant-Protein-Unique)
bezeichnet wird, diente als Referenz für Recherchen von Proteinhomologien.
Die mit dieser Bank gefundenen Homologien haben die Lokalisierung
von Regionen erlaubt, die potentiell für ein Proteinfragment codieren,
das zumindest einem bekannten Protein (codierend Exons) ähnlich ist.
Die verwendete EST-Bank besteht aus Unterabschnitten „gbest" (1–9) der
Genbank (NCBI; Benson et al., 1996). Sie enthält alle Fragmente öffentlich zugänglicher
Transkripte.
-
Die
gefundenen Homologien mit dieser Bank haben die Lokalisierung von
potentiell transkribierten Regionen erlaubt (vorhanden auf mRNA).
-
Die
verwendete Bank von Nukleinsäuren
(andere als die EST) enthielt alle anderen Unterabschnitte von Genbank
und EMBL (Rodriguez-Tome et al., 1996) wovon die Verdopplungen wie
zuvor eliminiert wurden.
-
Software:
-
Man
verwendete die komplette Blast-Software (gemeinfreie Software, Altschul
et al., 1990) zur Suche nach Homologien zwischen einer Sequenz und
Protein- oder Nukleinsäure-Datenbanken.
Die Signifikanzschwellen hingen von der Länge und der Komplexizität der getesteten
Region als auch der Größe der Referenzbank
ab. Sie sind eingestellt worden auf und angepasst worden an jede
Analyse.
-
Identifizierung genetischer
Polymorphismen, die assoziiert sind mit FMO in Verbindung mit einem
phänotypischen
Polymorphismus, der assoziiert ist mit dem plötzlichen juvenilen GPAO-J-Glaukom,
eine Krankheit mit autosomaler dominanter Vererbung (Locus GLC1A)
-
Nachweis
von Polymorphismen/Mutationen
-
1) Extraktion der DNA
-
Die
DNA wird extrahiert aus venösem
peripherem Blut nach Zelllyse, Proteinverdau, Abtrennung von organischem
Material und schließlich
alkoholischer Präzipitation.
-
Das
Blut (20 ml) wird durch periphäre
venöse
Punktion entnommen mit einem Röhrchen,
das EDTA enthält.
-
Es
wird verdünnt
mit einem Volumen bidestilliertem Wasser. Nach zehn Minuten werden
die Zellen durch Zentrifugation gesammelt bei 1600 g für 10 Minuten.
Dieses Verfahren wird wiederholt.
-
Die
weißen
Blutkörperchen
werden in Gegenwart von 20 ml CLB-Puffer (Tris 10 mM, pH-Wert 7,6,
5 mM MgCl2, 0,32 M Sucrose, 1 % V/V Trition
X-100) lysiert. Die Zellkerne werden durch Zentrifugation bei 1600 g
für 10
Minuten gesammelt. Dieses Verfahren wird wiederholt.
-
Die
Zellkerne werden einmal in dem RSB-Puffer gewaschen (Tris 10 mM,
pH-Wert 8, NaCl 10 mM, EDTA 10 mM). Der Bodensatz wird in 2 ml RSB-Puffer,
dem Natriumlaurylsulfat (1 %) und die Proteinase K (200 mg/ml) zugegeben
wurden, resuspendiert. Die Mischung wird bei 55 °C mindestens drei Stunden inkubiert
und regelmäßig bewegt.
-
Die
so erhaltene DNA-Lösung
wird dann mit einem Volumen Phenol, äquilibriert mit einem 50 mM Tris-Puffer,
pH-Wert 8, extrahiert. Dieser Vorgang wird wiederholt und abgeschlossen
durch eine Extraktion mit einem Volumen Chloroform/Isoamylalkohol
(24:1 V/V).
-
Die
DNA wird mit einem Volumen Isopropanol präzipitiert, mit Ethanol (70
%) gespült,
getrocknet und schließlich
in einem ml TE-Puffer (Tris 10 mM, pH-Wert 8, EDTA 0,5 mM) resuspendiert.
Die DNA-Konzentration wird beurteilt durch Messen der Absorption
bei 260 nm unter Verwendung der Äquivalenz
von 50 μg/ml DNA
für eine
Absorptionseinheit. Die DNA-Konzentration wird dann auf 200 μg/ml eingestellt.
-
2) Amplifikation der genomischen
DNA
-
Die
zur genomischen Amplifikation der von BAC 123H04M stammenden Exon-Sequenzen
verwendeten Oligonukleotidprimer, wie durch Computeranalyse vorhergesagt,
sind definiert worden mit Hilfe der Software OSP (Hillier et al.,
1991).
-
Alle
diese Primer enthielten stromaufwärts der Basen, die für die Amplifikation
vorgesehen waren, einen gemeinsamen Oligonukleotidstrang, der vorsieht
die Sequenzierung der amplifizierten Fragmente zu erlauben (PU für die stromaufwärtigen Primer
und RP für
die stromabwärtigen
Primer; die Sequenzen sind in Tabelle 5 angegeben).
-
Die
Oligonukleotidprimer sind entsprechend dem Phosphoramiditverfahren
synthetisiert worden auf einem Genset UFPS 24.1-Synthesizer.
-
Die
Amplifikation jeder vorhergesagten Exon-Sequenz wurde durch Polmerasekettenreaktion-Amplifikation
(PCR) durchgeführt
unter den folgenden Bedingungen:
| Endvolumen | 50 μl |
| genomische
DNA | 100
ng |
| MgCl2 | 2
mM |
| dNTP
(jeweils) | 200 μm |
| Primer
(jeweils) | 7,5
pmol |
| AmpliTaq
Gold DNA-Polymerase (Perkin) | 1
Einheit |
| *PCR-Puffer | 1X |
- *: (10X = 0,1 M Tris HCl, pH-Wert 8,3,
0,5 M KCl)
-
Die
Amplifikation wird in einem Perkin Elmer 9600- oder MJ Research
PTC200-Thermocycler mit Heizhaube durchgeführt. Nach Erhitzen auf 94°C für 10 Minuten
wurden 35 Zyclen durchgeführt.
Jeder Zyklus umfasst: 30 Sekunden bei 94 °C, 1 Minute bei 55 °C und 30
Sekunden bei 72 °C.
Ein abschließender
Abschnitt von 7 Minuten bei 72 °C
beendet die Amplfikation.
-
Die
Menge der erhaltenen Amplifikationsprodukte wird auf Mikroplatten
mit 96 Vertiefungen durch Fluorometrie unter Verwendung des Picogreen-Intercalationsmittels
(Molecular Probes) bestimmt.
-
3) Bestimmung der Polymorphismen/Mutationen
-
-
Die
Produkte der genomischen Amplifikation durch PCR wurden sequenziert
auf einem automatischen Sequenzierer ABI 377, unter Verwendung fluoreszierender
Primer, die markiert waren mit den Fluorochromen ABI (Joe, Fam,
Rox und Tamra) und der DNA-Polymerase Thermosequanase (Amersham)
Die Reaktionen sind realisiert worden auf Mikroplatten mit 96 Vertiefungen
auf dem Perkin Elmer Thermocycler 9600, unter den klassischen Temperaturzyklusbedingungen:
- – 8
Zyklen: Denaturierung: 5 sec bei 94 °C; Hybridisierung: 10 sec; Elongation:
30 sec bei 72 °C,
dann
- – 13
Zyklen: Denaturierung: 5 sec bei 94 °C; Elongation: 30 sec bei 72 °C.
-
6
Einheiten Thermosequanase und 5–25
ng Amplifikationsprodukt wurden pro Sequenzierungsreaktion verwendet.
-
Ausgehend
von den Amplifikationszyklen werden die Produkte der Sequenzierungsreaktionen
in Ethanol präzipitiert,
resuspendiert in Aufgabepuffer, der Formamid enthält, denaturiert
und auf 4 % Acrylamidgele gegeben; die Elektrophoresen (2 Stunden
30 bei 3000 Volt) werden auf ABI 377-Sequenzierern, die ausgestattet
sind mit der Aufnahme- und Analyse-Software ABI (ABI Prism DNA Sequencing
Analyses Software, Version 2.1.2) durchgeführt.
-
Da
GPAO-J ist eine autosomal dominante Krankheit ist, wurden die erhaltenen
Sequenzdaten analysiert, um das Vorliegen von heterozygoten Stellen
unter Patienten, die an juvenilem Glaukom erkrankt waren, nachzuweisen.
Die heterozygoten Stellen wurden bestätigt nach Vergleich der Sequenzen
der zwei genomischen DNA-Stränge
von jedem betroffenen Individuum. Eine heterozygote Stelle wird
als Mutationskandidat beibehalten, der verantwortlich ist für das plötzliche
Auftreten von Problemen, die mit FMO verbunden sind, wenn sie in
einer Population von Mitgliedern der gleichen Familie vorliegt,
während
sie im Allgemeinen nicht vorliegt bei Kontrollen, die nicht mit
der Familie verwandt sind.
-
Ergebnisse
-
Unter
allen untersuchten Amplifikationsfragmenten, die von BAC 123H04M
abgeleitet sind, stellt eines unter diesen eine heterozygote Stelle
dar, die mit dem plötzlichen
Auftreten von juvenilem Glaukom in einem Stammbaum, dargestellt
in 8, sezerniert wird.
-
Diese
heterozygote Stelle (G/A) liegt bei 7 Patienten vor, die an GPAO-J
erkrankt sind, während
sie bei 3 gesunden homozygoten Patienten (G/G) nicht vorliegt, wobei
alle von der gleichen Familie stammen. Weiterhin sind 99 nichtverwandte
Kontrollen ebenso homozygot (G/G) für diese Stelle, wodurch angezeigt wird,
dass die Häufigkeit
des Allels A in der allgemeinen Population kleiner als 0,005 ist.
-
Die
Stelle ist enthalten im Exon 8 des Gens, das für das Protein hFMO2 gemäß der Erfindung
codiert; die beschriebene Mutation verändert Glutaminsäure in Position
402 der Sequenz SEQ ID Nr. 1 des hFMO2 zu Lysin (1).
-
Es
ist überraschend
festzustellen, dass die LOD-Score-Berechnung, die die vorhergehenden
Daten für
verschiedene Häufigkeitshypothesen
für jedes
Allel in der allgemeinen Population integriert, eine Wahrscheinlichkeit
von mehr als 100 zu eins angibt, dass die beschriebene Heterozygotie
(G/A) mit GPAO-J verbunden ist (Tabelle 6). Diese Wahrscheinlichkeit
ist signifikant für
die Tatsache, dass die Analyse von einer einzigen Familie getragen
wird.
-
Die
Primer, die die Amplifikation des DNA-Fragments erlaubt haben, das
die heterozygote Stelle enthält,
sind in Tabelle 1 beschrieben.
-
Tabelle 1: Primersequenzen,
die verwendet werden zum Amplifizieren der Exon-Region, die von BAC 123H04M abgeleitet
ist und eine heterozygote Stelle enthält, die mit juvenilem GPAO
verbunden ist.
-
- Locus des Fragments: FMO2/Exon 8
- Größe des amplifizierten
Fragments: 420
- Primer:
PU stromaufwärts
(SEQ ID Nr. 7): 5'TCACATAGAGTGCTATGGGGG
RP
stromabwärts
(SEQ ID Nr. 8): 5'CTTAGGAAGAAGATAAAAATGCAAC
-
Tabelle 2: Beispiele der
Primer zum Nachweisen der Mutation G.1263mac.A durch „Single
Nucleotide Primer Extension"
-
- a) SEQ ID Nr. 9: 5' AATGTCCATCATCATAGTTCTCT 3' (Antisense)
und/oder
- b) SEQ ID Nr. 10: 5' TAGGCTTGTGTAGCCTGCCCTCA
3'(Sense)
-
Tabelle
3: Identifizierung der Mutation G.1263mac.A durch RFLP
-
Tabelle
4: Beispiel von Sonden für
den Nachweis der Mutation G.1263mac.A durch die ASO-Technik
-
Tabelle 5: Sequenz der
Primer, die verwendet wurden zur Sequenzierung der Amplifikationsfragmente,
ausgehend von genomischer DNA
-
- PU 5'TGTAAAACGACGGCCAGT
- RP 5'CAGGAAACAGCTATGACC
-
Tabelle
6: Lod-Score zwischen dem Polymorphismus G.1263mac.A und dem juvenilen
GPAO in der untersuchten Familie als Funktion der Häufigkeit
der beiden Allele in der allgemeinen Population.
-
Sequenzprotokoll
-
- SEQ ID Nr. 1
- SEQ ID Nr. 2
- SEQ ID Nr. 3
- SEQ ID Nr. 4
- SEQ ID Nr. 5
- SEQ ID Nr. 6
- SEQ ID Nr. 7
- SEQ ID Nr. 8
- SEQ ID Nr. 9
- SEQ ID Nr. 10
- SEQ ID Nr. 11
- SEQ ID Nr. 12
- SEQ ID Nr. 13
- SEQ ID Nr. 14
-
LITERATURSTELLEN
-
- Allen J.B., Walberg M.W., Edwards M.C. & Elledge S.J. Finding prospective
partners in the library: the two hybrid system and phage display
find a match.
TIBS 20: 511–516
(1995).
- Altschul, Stephen F., Gish W., Miller W., Myers E. W. & Lipman D.J.
Basic
local alignment search tool.
J. Mol. Biol. 215:403-10 (1990).
- Bairoch A. & Apweiler
R. The SWISS-PROT protein sequence data bank and its new supplement
TREMBL. Nucleic Acids Res. 24: 21–25 (1996).
- Belmouden A., Adam M. F., Dupont de Dinechin S., Brezin A.P.,
Rigault P., Chumakov I., Bach J-F. & Garchon H-J., 1996, Recombinational
and physical mapping of the locus for primary open-angle glaucoma
(GLC1A) on chromosome 1q23-q25. Genomics, im Druck.
- Benson D. A., Boguski M., Lipman D. J. & Ostell J. GenBank. Nucleic Acids
Res. 24: 1–5
(1996).
- Bonfield J. K., Smith K. F. & Staden
R. A new DNA sequence assembly program.
Nucleic Acids Res.
23: 4992-9 (1995).
- Buckholz R.G. Yeast Systems for the Expression of Heterologous
Gene Products. Curr. Op. Biotechnology 4: 538–542 (1993).
- Cashman J. R., Park, B. P., Berkman, C.-E. & Cashman, L.E. Rôle of hepatic flavin-monoxygenase
3 in drug and chemical metabolism in adult humans.
Chemico-Biological
Interactions 96: 33–46
(1995).
- Carter B.J. Adeno-Associated virus vectors. Curr. Op. Biotechnology
3: 533–539
(1993).
- Cherif D., Julier C., Delattre O., Derré J., Lathrop G.M. & Berger R.: Simultaneous
localization of cosmids and chromosome R-banding by fluorescence
microscopy – Applications
to regional mapping of chromosome 11.
Proc.Natl.Acad.Sci. USA.
87: 6639–6643
(1990).
- Chumakov I., Rigault P., Guillou S., Ougen P., Billault A.,
Guasconi G., Gervy P., Le Gall I., Soularue P., Grinas P. et al.
Continuum of overlapping clones spanning the entire human chromosome
21q. Nature 359: 380–386 (1992).
- Chumakov I.M., Rignault P., Le Gall I. et al. A YAC contig map
of the human genome. Nature 377 Suppl.: 175–183 (1995).
- Compton J. Nucleic Acid Sequence-Based Amplification. Nature
350: 91–92
(1991).
- Danos O., Moullier P. & Heard
J.M. Réimplantation
de cellules génetiquément modifiées dans
des néo-organes vascularisés. Médecine/Sciences
9:62–64
(1993).
- Edwards C.P. und Aruffo A. Current applications of COS cell
based transient expression systems. Curr. Op. Biotechnology: 558–563 (1993).
- Epstein A.: Les vecteurs herpétiques pour le transfert
de gènes – Medecine/Sciences
B: 902–911
(1992).
- George D.G., Barker W.C.,. Mewes H.W., Pfeiffer F. &. Tsugita A. The
PIR-International
Protein Sequence Database Nucleic Acids Res. 24: 17–20 (1996).
- Guatelli J.C. et al. Isothermal in vitro amplification of nucleic
acids by a multienzyme reaction modeled after retroviral replication.
Proc. Natl. Acad. Sci.
USA 87: 1874–1878 (1990).
- Hillier L. & Green
P. OSP: a computer program for choosing PCR and DNA sequencing primers.
PCR Methods Appl. 1: 124-8 (1991).
- Hines et al., Toxicol. Appl. pharmacol. 125, 1–6 (1994).
- Landegren U., Kaiser R., Sanders J. & Hood L.A. ligase-mediated gene detection
technique. Science 241: 1077–1080
(1988).
- Lawton M.P., Cashman J.R., Cresteil T., Dolphin C.T., Elfarra
A.A., Hines R.N., Hodgson E., Kimura T., Ozols J., Phillips I.R.,
Philpot R.M., Poulsen L.L., Rettie A.E., Shephard E.A., Williams
D.E & Ziegler
D.M.: A nomenclature for the mammalian flavin-containing monooxygenase
gene family based on amino acid sequence identities. Arch. Biochem.
Biophys. 308:1, 254–257
(1994).
- Luban J. & Goff
S.P. The yeast two-hybride system for studying protein – protein
interactions. Current Op. Biotechnology 1995, 6:59–64.
- Luckow V.A. Baculovirus systems for the expression of human
gene products. Curr. Op. Biotechnology 4: 564–572 (1993).
- Olins P.O. und Lee S.C. Recent advances in heterologous gene
expression in E. coli. Curr. Op. Biotechnology 4:520–525 (1993).
- Park, S.B. et al., Chem. Res. Toxicol. 5, 193–201 (1992).
- Perricaudet M., Stratford-Perricaudet L. & Briand P.: La therapie genique par
adenovirus – La
Recherche 23: 471–473
(1992).
- Poulsen, L.L. et al., Chem. Biol. Interact. 96, 57–73 (1995).
- Rodriguez-Tome P., Stoehr P.J., Cameron G.N., & Flores T.P. The
European Bioinformatics Institute (EBI) databases. Nucleic Acids
Res. 24: 6–12
(1996).
- Samiotaki M., Kwiatkowksi M. Parik J. & Landegren U. Dualcolor detection
of DNA sequence variants through ligase-mediated analysis, Genomics
20: 238–242
(1994).
- Schwartzman M.L., Masferrer J., Dunn M.W., McGiff J.-C., Abracham
N.G., 1987, Curr Eye Res. 6: 623–630.
- Schwartzman M.L., Balazy M., Masferrer J., Abraham, N.G., McGiff
J.C., Murphy R.C., 1987, PNAS USA 84: 8125–8129.
- Stoneking M., Hedgecock D., Higuchi R.G., Vigilant L. & Erlich H.A. Population
variation of human DNA control region sequences by enzymatic amplification
and sequencespecific oligonucleotide probes. Am. J. Hum. Genet..
48: 370–382
(1991).
- Sunden S.L.F., Alward W.L.M., Nichols B.E., Rokhlina T.R., Nystuen
A., Stone E.M. & Sheffield
V.C. Fine mapping of the autosomal dominant juvenile open angle
glaucome (GLC1A) region and evaluation of candidate genes. Genome research
6: 862–869
(1996).
- Syvänen
A.C., Aalto-Setala K., Harju L., Kontula K. & Soderlund H. A primerguided nucleotide
incorporation assay in the genotyping of Apo E. Genomics 8: 684–692 (1990).
- Temin H.M.: Retrovirus vectors for gene transfer. In Kucherlapati
R., Hrsg., Gene Transfer, New York, Plenum Press, 149–167 (1986).
- Walker G.T., Fraiser M.S., Schram J.L., Little M.C., Nadeau
J.G. & Malinowski
D.P. Strand displacement amplification: an isothermal in vitro DNA
amplification technique. Nucleic Acids Res. 20: 1691–1696 (1992).
- Wu D.Y., Ugozzoli L., Pal B.K., Wallace R.B. Allele-specific
amplification of b-globin
genomic DNA for diagnosis of sickle cell anemia. Proc. Natl. Acad.
Sci. USA 86: 2757–2760
(1989).
- Ziegler, D.M., Drug Metab. Rev. 19, 1–32 (1988).
- Ziegler, D.M., Annu. Rev. Pharmacol. Toxicol., 33, 179–199 (1993).
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-