-
Bereich der
Erfindung
-
Die
vorliegende Erfindung betrifft allgemein neuartige Formulierungen,
welche Faktor IX umfassen, einschließlich sowohl hochgradig konzentrierte,
als auch lyophilisierte und flüssige
Faktor IX umfassende Formulierungen, welche zur Verabreichung auf
verschiedenen Wegen, einschließlich
zum Beispiel Wegen wie intravenös,
subkutan, intramuskulär
und intradermal geeignet sind.
-
Hintergrund
der Erfindung
-
Eine
Vielfalt an Faktoren, welche im Blutgerinnungsprozess einbezogen
sind, sind identifiziert worden, einschließlich Faktor IX, einem Plasma-Glycoprotein.
Ein Mangel an Faktor IX kennzeichnet einen Hämophilie-Typ (Typ B). Behandlung
dieser Krankheit hat traditionell intravenöse Infusion von Proteinkonzentraten
von Faktor IX einbezogen, welche von menschlichem Plasma abgeleitet
wurden. Infusion von Blutkonzentraten bezieht das Risiko der Übertragung
verschiedener infektiöser
Wirkstoffe ein, wie virale Hepatitis und HIV, oder thromboembolische
Faktoren. Ein alternatives Verfahren zum Herstellen von Faktor IX
durch rekombinante DNA-Techniken ist in USPN 4770999, Kaufman et
al., 13. September 1988 beschrieben worden. Die cDNA, welche für den humanen
Faktor IX codiert, ist isoliert, gekennzeichnet und in Expressionsvektoren
kloniert worden. Siehe zum Beispiel Choo et al., Nature 299: 178–180 (1982);
Fair et al., Blood 64: 194–204
(1984) und Kurachi et al., Proc. Nat. Acad. Sci., USA 79: 6461–6464 (1982).
So ist es durch Fortschritte in der rekombinanten DNA-Technologie
möglich
geworden, Faktor IX Protein herzustellen.
-
Es
ist wünschenswert,
sowohl Masse- als auch Fertigformen von Faktor IX zu haben, welche
sowohl zur Lagerung, als auch zur Abgabe geeignet sind. Typischerweise
ergibt ein Reinigungsverfahren für
ein Protein Konzentrieren des Proteins. Dieses konzentrierte Protein,
auch als Masseprotein bekannt, kann sich in einem Formulierungspuffer
befinden. Masseprotein kann dann typischerweise bei einer Konzentration
von etwa 2 bis mindestens 20 mg/ml gefroren zu einer Füll/Fertig-Anlage
transportiert werden, wo es an eine passende Dosierungskonzentration
angepasst und in Dosierungsvialen oder eine Vorrichtung platziert
wird, welche zur Verabreichung geeignet ist, z. B. eine vorgefüllte Spritze.
Idealerweise wird das Arzneiprodukt in flüssigem Zustand belassen und
als Flüssigkeit
gelagert und verabreicht. Alternativ wird das Arzneiprodukt lyophilisiert,
also gefriergetrocknet. Idealerweise hat das lyophilisierte Arzneiprodukt
eine ausreichende Stabilität,
um in Langzeitlagerung, also mehr als sechs Monate, gehalten zu
werden: lyophilisiertes Arzneiprodukt wird zu einem späteren Zeitpunkt
durch Zugeben eines geeigneten Verabreichungsverdünnungsmittels
gerade kurz vor der Patientenverwendung rekonstituiert.
-
Die
Entscheidung, das fertige Arzneiprodukt entweder als Flüssigkeit
zu halten oder zu gefriertrocknen, basiert üblicherweise auf der Stabilität der Proteinarznei
in jenen Formen. Die Proteinstabilität kann unter anderem durch
solche Faktoren wie Ionenstärke,
pH, Temperatur, wiederholte Frost/Tau-Wechsel und Aussetzung unter
Scherkräfte
beeinflusst werden. Wirksames Protein kann als Folge physikalischer
Instabilitäten
einschließlich
Denaturierung und Aggregation (sowohl lösliche, als auch nicht lösliche Aggregatbildung),
als auch chemischer Instabilitäten,
einschließlich
zum Beispiel Hydrolyse, Entamidierung und Oxidation, um nur ein paar
zu nennen, verloren werden. Für
eine allgemeine Übersicht über Stabilität von Protein-Pharmazeutika siehe
zum Beispiel Manning et al., Pharmaceutical Research 6: 903–918 (1989).
-
Während das
mögliche
Auftreten von Protein-Instabilitäten
weit anerkannt ist, ist es unmöglich,
bestimmte Instabilitätsprobleme
eines bestimmten Proteins vorherzusehen. Jede dieser Instabilitäten kann
die Bildung eines Proteins, Protein-Nebenprodukts oder Derivats
mit verringerter Wirksamkeit, erhöhter Toxizität und/oder
erhöhter
Immunogenizität
zur Folge haben. Tatsächlich
kann Proteinausfällung
zu Thrombose, Nicht-Homogenität
von Dosierungsform und Menge, als auch verklumpten Spritzen führen. Auch
gibt es Faktor IX spezifisch mehrere post-translationale Modifikationen
(zum Beispiel die Gamma-Carboxylierung von bestimmten Glutaminsäureresten
im N-Terminus und die Addition von Kohlenhydrat) welche alle potentielle
Stellen vorsehen, die anfällig
für Modifikation
bei Lagerung sein können.
Somit steht die Sicherheit und Wirksamkeit jeder pharmazeutischen
Formulierung aus einem Protein in direkter Beziehung zu ihrer Stabilität. Beibehalten
dieser Stabilität
in einer flüssigen
Do sierungsform unterscheidet sich im Allgemeinen von einer lyophilisierten
Dosierungsform aufgrund stark erhöhtem Potential für molekulare
Bewegung und daher erhöhter Wahrscheinlichkeit
molekularer Interaktionen. Beibehalten der Stabilität in einer
hochgradig konzentrierten Form ist aufgrund der Neigung zur Aggragatbildung
bei hohen Proteinkonzentrationen ebenfalls unterschiedlich.
-
Bei
der Entwicklung einer flüssigen
Formulierung werden viele Faktoren in Betracht gezogen. Kurzfristige,
also weniger als sechs Monate, Flüssigkeitstabilität hängt im Allgemeinen
vom Vermeiden von groben strukturellen Veränderungen ab wie Denaturierung
und Aggregation. Diese Verfahren werden in der Literatur für eine Anzahl
an Proteinen beschrieben und es gibt viele Beispiele für Stabilisatoren
(„Strategies
to Suppress Aggregation of Recombinant Keratinocyte Growth Factor
during Liquid Formulation Development", B. L. Chen et al., J. Pharm. Sci.
83(12): 1657–1661,
(1994); „Formulation
Design of Acidic Fibroblast Growth Factor", P. K. Tsai et al., Pharm. Res. 10(5):
649–659
(1993); „The
Stabilization of Beta-Lactoglobulin by Glycine and NaCl", Tsutomu Arakawa,
Biopolymers 28: 1397–1401
(1989); „Structural
stability of lipase from wheat germ", A. N. Rajeshwara und V. Prakash, Internat.
J. of Peptide & Prot.
Res. 44: 435–440
(1994); „Thermal
Stability of Human Immunoglobulins with Sorbitol", M. Gonzalez et al., Vox Sang 68: 1–4 (1995)).
Es ist gut bekannt, dass ein Wirkstoff, welcher beim Stabilisieren
eines Proteins wirksam ist, tatsächlich
so wirkt, dass er ein anderes destabilisiert. Sobald das Protein
vor groben Strukturveränderungen
stabilisiert worden ist, hängt
Entwickeln einer flüssigen
Formulierung für
Langzeitstabilität
(zum Beispiel mehr als sechs Monate) vom weiteren Stabilisieren
des Proteins vor Abbauarten ab, die für das Protein spezifisch sind.
Spezifischere Abbauarten können
zum Beispiel Disulfid-Bindungsverwürfelung, Oxidation von Oligosacchariden
und/oder bestimmten Resten, Entamidierung, Cyclisierung und Ähnliches
einschließen.
Obwohl es nicht immer möglich
ist, die einzelnen Abbauarten genau aufzuzeigen, werden Tests entwickelt,
um feine Veränderungen
zu überwachen,
um so die Fähigkeit
spezifischer Exzipienten zu überwachen,
das Protein von Interesse einzigartig zu stabilisieren.
-
Zusätzlich zu
Stabilitätsüberlegungen
wählt man
im Allgemeinen Exzipienten aus, welche der Billigung verschiedener,
weltweiter, medizinischer Aufsichtsbehörden entsprechen werden. Es
ist hochgradig wünschenswert,
dass die Formulierung annähernd
isotonisch ist, und dass sich der pH der Formulierung in einer physiologisch
geeigneten Bandbreite bei Injektion/Infusion befindet, weil ansonsten
Schmerz und Unbehagen für
den Patienten die Folge sein kann. Die Wahl und Menge des verwendeten
Puffers ist wichtig, um die gewünschte
pH-Bandbreite zu erreichen. Die Wahl und Menge von Wirkstoffen,
welche verwendet werden, um die Tonizität zu modifizieren, ist wichtig,
um Leichtigkeit der Verabreichung sicherzustellen.
-
Traditionell
werden große,
labile Proteine wie Faktor IX intravenös, entweder prophylaktisch
oder als Antwort auf Blutungsepisoden verabreicht. Wenn es intravenös gegeben
wird, ist das Protein direkt im Blutstrom verfügbar. Unglücklicherweise kann es Nebenwirkungen
in Verbindung mit wiederholten Injektionen geben, einschließlich Okklusion
und/oder Fibrinbildung, insbesondere bei Älteren. Darüber hinaus kann es, wo die
Venen des Patienten besonders klein sind, z. B. bei kleinen Kindern,
schwierig sein, die erforderliche therapeutische Dosis zu erreichen.
-
EP-0317376
offenbart ein chromatographisches Verfahren zum Trennen einer Fraktion
aus humanem Plasma, welche Faktor IX enthält, bei einer Konzentration
von 25 U/ml. Die Zugabe von Arginin zum Elutionspuffer, um den Faktor
IX zu stabilisieren, wird beschrieben.
-
Gegenwärtig gibt
es keine hochgradig konzentrierten Faktor IX Formulierungen, welche
im Handel erhältlich
sind. Die einzigen beiden im Handel erhältlichen (in den US), Träger-Protein-freien, Plasma-abgeleiteten
Faktor IX Formulierungen sind gefriergetrocknete Produkte, welche
zur Verwendung rekonstituiert werden und auf niedrige Faktor IX
Konzentrationen beschränkt
sind, z. B. etwa 100 U/ml oder weniger als 1 mg/ml. Solche niedrigen
Konzentrationen sind primär
für intravenöse Verabreichung
angezeigt und nicht für
subkutane, intramuskuläre
oder intradermale Verwendung beabsichtigt. Alpha Therapeutic Corporation
sieht lyophilisiertes AlphaNine® SD
vor, welches Heparin, Dextrose, Polysorbat 80 und Tri(n-butyl)phosphat
umfasst.
-
Von
diesem Präparat
ist beabsichtigt, dass es bei Temperaturen zwischen 2° und 8°C gelagert
wird. Heparin ist zu vermeiden, da es ein Anti-Koagulans ist, und
Tri(n-butyl)phosphat reizt Schleimhäute; somit ist diese Formulierung
weniger als ideal. Armour Pharmaceutical Company sieht lyophilisiertes
Mononine® vor, welches
Histidin, Natriumchlorid und Mannitol umfasst, welches ähnlicherweise
bei 2° bis
8°C gelagert
werden muss. Die Packungsbeilage empfiehlt kein Lagern dieser Formulierung
für mehr
als einen Monat bei Raumtemperatur. Es gibt weder flüssige, noch
hochgradig konzentrierte Faktor IX Produkte, welche gegenwärtig im Handel
erhältlich
sind. Schwinn, PCT/EP90/02238 offenbart Faktor IX, 0,9 M Saccharose,
0,5 M Lysin und 0,003 M Calciumchlorid, gelagert bei 4–8°C, stabil
nur für
einen Zeitraum von Wochen und daher zur kommerziellen Herstellung
ungeeignet; diese Formulierung ist unglücklicherweise hypertonisch
und der pH ist außerhalb
der Bandbreite für
angenehme Verabreichung und daher zur Injektion ungeeignet.
-
Für den Patienten
leichter zu handhaben sind Verabreichungsformen wie subkutan, intramuskulär oder intradermal.
Subkutane Verabreichung von Faktor IX wird in Berrettini, Am. J.
Hematol. 47: 61 (1994) und in WO-93/07890, BTG, Brownlee (veröffentlicht
am 29. April 1993) beschrieben. In Berrettini wurde ein Immuno-Produkt verwendet:
ImmunineTM, Faktor IX, Heparin, Natriumcitrat,
Natriumchlorid und Antithrombin III bei einer Konzentration von
118 U/mg. Von dem Produkt wurde berichtet, dass es schlecht und
langsam in die Zirkulation transportiert wurde und die Autoren schlossen,
dass subkutane Verabreichung nicht verlässlich zum Behandeln oder Verhindern
von Blutung bei Hämophilie
B Patienten war und dass noch konzentriertere Formen bezüglich klinischer
Wirksamkeit unannehmbar sein würden.
Brownlee, supra, offenbart eine MononineTM Faktor
IX Formulierung bei einer Konzentration von 10–500 U/ml. Es wurden nur niedrige
zirkulierende Gehalte erhalten und auf Seite 9 wird vermerkt, dass
sich nach vier Stunden große
Gerinsel unter der Haut an jeder Stelle gebildet hatten, wo der
Faktor IX injiziert worden war, was schwere Prellungen ergab. So
etwas kann beobachtet werden, wenn ein Produkt verwendet wird, das
unrein ist.
-
Bei
Hämophilie
B Hunden (Brinkhous et al., FASEB 7. 117 (1993)) und bei einem Hämophilie
B Patienten (Liles et al., Thromb. Haemost. 73: 1986a (1995)) wurde
Plasma-abgeleiteter Faktor IX (pFIX) subkutan (bei Dosen von 15–47 U/kg
bei Hunden und bei einer Dosis von 30 U/kg beim Patienten) verabreicht.
Dies ergab Plasma Faktor IX bei Hunden, welches Dosis abhängig war
und von 0,8 bis 7,6% reicht, wobei intramuskulär höhere Gehalte ergab. Beim Hämophilie-Patienten
erreicht Plasma Faktor IX Wirksamkeit nur 1% innerhalb von sechs
Stunden; dieser Wirksamkeitsgrad hielt 36 Stunden an; die niedrige
Konzentration an Plasma-abgeleitetem Faktor IX erforderte Injektionen
mit hohem Volumen an multiplen (10) Stellen.
-
Eines
der Hauptprobleme in Verbindung mit Formulieren einer geeigneten
subkutanen Formulierung ist das Erreichen einer ausreichend hohen
Konzentration des Proteins, ohne Aggragatbildung des Proteins zu verursachen
und ohne gleichzeitigem Konzentrieren von Verunreinigungen in dem
Präparat.
Sowohl die Aggregatbildung, als auch Verunreinigungen führen zu
erhöhter
Immunogenizität.
Bei gegenwärtig
erhältlichen Produkten
erfordert das Geben einer angemessenen Dosis an Faktor IX subkutan
die Verwendung von multiplen Injektionsstellen. Dieses verursacht
großes
Unbehagen und Unbequemlichkeit beim Patienten. Um Faktor IX subkutan
praktisch abzugeben ist es notwendig, Faktor IX auf mindestens 1000
U/ml oder höher
zu konzentrieren und es in einer stabilen, nicht-aggregierenden
Dosierungsform vorzusehen. Solch eine konzentrierte Form ist gegenwärtig nicht
erhältlich.
-
Idealerweise
sollten Formulierungen eine Faktor IX Stabilität für mehr als ein Jahr und eine
Kompatibilität über eine
große
Bandbreite an Proteinkonzentrationen (0,1 mg/ml bis mehr als 160
mg/ml, also 20 U/ml bis mehr als 56000 U/ml beispielsweise) vorsehen.
Dies ermöglicht
Flexibilität
bei Verabreichungsverfahren, welche höhere Proteinkonzentrationen
erfordern können,
z. B. subkutane, intradermale oder intramuskuläre Verabreichung, oder jene,
welche niedrige Proteinkonzentration nutzen, z. B. intravenöse Verabreichung.
Im Allgemeinen ermöglichen
höher konzentrierte
Formen die Verabreichung von kleineren Volumina, was aus Sicht des
Patienten hochgradig wünschenswert
ist. Flüssige
Formulierungen können
gegenüber
gefriergetrockneten Produkten bezüglich Einfachheit der Verabreichung
und Verwendung viele Vorteile haben. Entsprechend bleibt weiterhin ein
Bedarf nach Stand der Technik für
Verfahren zum Verbessern der Faktor IX Proteinstabilität, Erhöhen der
Konzentration, Beibehalten von wirksamkeitsgraden und Vorsehen stabiler
flüssiger Formulierungen,
welche zur verlängerten
Lagerung für
mehr als ein Jahr bei 2 bis 8°C
geeignet sind.
-
Kurze Zusammenfassung
der Erfindung
-
Die
vorliegende Erfindung betrifft Zusammensetzungen und hochgradig
konzentrierte, lyophilisierte und flüssige Präparate von Faktor IX, welche
als Masseprotein brauchbar sind oder zur Verabreichung brauchbar
sind. Die Erfindung sieht eine Zusammensetzung vor, welche hochgradig
konzentrierten Faktor IX und Arginin, einen Puffer und Sucrose oder
Mannitol umfasst, wobei die Faktor IX Konzentration etwa 2 bis etwa
160 mg/ml beträgt.
Diese Zusammensetzungen sind entweder gefroren oder flüssig für mindestens
sechs Monate und vorzugsweise bis zu 36 und 60 Monate stabil und
können
bei Temperaturen im Bereich von –100°C bis 40°C von –80°C bis 0°C und von –20°C bis 10°C gelagert werden. Die Zusammensetzungen
umfassen Faktor IX, Tonizitätsmodifikatoren,
Kryoschutzmittel und Puffer und/oder weitere Exzipienten, welche
Faktor IX weiter stabilisieren. Die Faktor IX Konzentration reicht
von etwa 2 bis etwa 160 mg/ml (wobei 160 mg/ml mit mindestens 56000
U/ml äquivalent
sind), wobei 2 bis 10 mg/ml (2500 U/ml) bevorzugt werden, wobei
die am meisten bevorzugte Bandbreite vom Verabreichungsweg abhängt. Tonizitätmodifikatoren
schließen
ein, aber sind nicht beschränkt
auf Salze, Zucker, Polyole und Aminosäuren. Geeignete Aminosäuren schließen Arginin,
Glycin und Histidin bei einer Konzentration von etwa 10 bis 500
mM ein, wobei etwa 10 bis 300 mM und etwa 10 bis 200 mM bevorzugt
werden. Die Argininkonzentration beträgt vorzugsweise etwa 130 bis
160 mM, etwa 130 bis 235 mM oder etwa 60 bis 70 mM. Insbesondere
bevorzugt beträgt
die Argininkonzentration etwa 160 mM. Kroyschutzmittel Polyole,
Mannitol und Sucrose haben eine Konzentrationsbandbreite von etwa
1 bis 400 mM, wobei etwa 5 bis 200 mM und 20 bis 100 mM bevorzugt
werden. Sucrosekonzentration beträgt vorzugsweise etwa 3 bis
60 mM. Gegebenenfalls können
die Zusammensetzungen auch einen Surfactanten oder Detergens enthalten,
wie Polysorbat (z. B. Tween) oder Polyethylenglycol (PEG), welcher
auch als Kryoschutzmittel während
des Gefrierens dienen kann. Der Surfactant reicht von etwa 0,005
bis 1%, wobei etwa 0,005 bis 0,1% und etwa 0,005 bis 0,02% bevorzugt
werden. Die Zusammensetzungen enthalten einen passenden Puffer,
um einen physiologisch geeigneten pH zu halten, z. B. im Bereich
von etwa 5,8 bis 8,0, wobei etwa 6,2 bis 7,2 und etwa 6,5 bis 7,0
bevorzugt werden. Puffer schließen
vorzugsweise Histidin, Natriumcitrat, Kaliumcitrat, Maleinsäure, Ammoniumacetat,
Tris, Natriumphosphat, Kaliumphosphat und Diethanolamin ein, wobei
Natrium-/Kaliumcitrat bevorzugt werden, mit einem bevorzugten pH
von etwa 6,5 bis 7,5 und einer Konzentrationsbandbreite von etwa
1–100
mM, wobei 5 bis 50 mM und 10 bis 25 mM bevorzugt werden. Die bevorzugte
Citratkonzentration beträgt
etwa 1 bis 40 mM, insbesondere bevorzugt etwa 15 mM. Gegebenenfalls
werden kleine Mengen eines Chelatbildners wie EDTA bei Konzentrationen
von 0,05 bis 50 mM oder 0,05 bis 10 mM oder 0,1 bis 5 mM eingeschlossen,
wobei etwa 1 bis 5 mM bevorzugt werden.
-
Ein
weiterer Aspekt der vorliegenden Erfindung sieht Formulierungen
von Faktor IX vor, welche zur Verabreichung in einer Enddosierungsform
geeignet sind, zum Beispiel durch intravenöse, subkutane, intradermale
oder intramuskuläre
Verabreichungswege. Typischerweise werden große Mengen an Massearznei gefroren
und können,
falls notwendig, zu einem Herstellungsort transportiert werden,
wo die Massearznei in kleine Vialen gefüllt wird; falls gewünscht, ist
die Enddosierungsform eine verdünnte,
pH-angepasste Form; die Massearznei umfasst typischerweise eine
höhere
Proteinkonzentration als Fertigarznei und muss nicht isotonisch
sein. Die Fertigarzneizusammensetzungen umfassen Faktor IX, Tonizitätsmodifikatoren,
Kryoschutzmittel und einen Puffer und/oder weitere Exzipienten,
welche Faktor IX weiter stabilisieren, wie supra beschrieben. Die
Fertigarzneiformulierungen sind für mindestens sechs Monate und
vorzugsweise bis zu 36 und 60 Monate stabil und können bei
Temperaturen im Bereich von –100°C bis 40°C, von –20°C und 37°C und von
2°C bis 8°C gelagert
werden. Die Konzentrationen der Exzipienten sehen eine kombinierte
Osmolalität
von etwa 250 bis 420 milliosmolal vor. Bevorzugte Formulierungen
schließen
Faktor IX Konzentrationen im Bereich von etwa 2 bis größer als
160 mg/ml (bis größer als
56000 U/ml) ein, mit Natriumcitrat als Puffer; einer Kombination
aus Mannitol, Sucrose, Arginin und Glycin als Kryoschutzmittel und
Tonizitätsmodifikatoren
und gegebenenfalls kleinen Mengen eines Chelatbildners wie EDTA
(ca. 1 bis 5 mM) und/oder kleinen Mengen Polysorbat (0,005% bis
0,02%.
-
Die
Zusammensetzungen der vorliegenden Erfindung können eine Argininkonzentration
von etwa 130 bis 160 mM, eine Sucrosekonzentration von etwa 0 bis
60 mM und eine Surfactantkonzentration von etwa 0 bis 0,005% umfassen;
alternativ beträgt
besagte Argininkonzentration etwa 60 bis 70 mM, besagter Puffer
ist etwa 0 bis 20 mM Sucrose und 165 mM Mannitol und besagte Surfactantkonzentration
beträgt
0 bis 0,005%. In einer weiteren Zusammensetzung beträgt die Argininkonzentration
etwa 160 mM, besagte Sucrosekonzentration beträgt etwa 0 mM, besagte Citratkonzentration
beträgt
etwa 15 mM und besagte Surfactantkonzentration beträgt etwa
0%; ober besagte Argininkonzentration beträgt etwa 70 mM, besagte Mannitolkonzentration beträgt etwa
165 mM, besagte Citratkonzentration beträgt etwa 15 mM und besagter
Surfactant oder Chelatbildner beträgt etwa 0%.
-
Weitere
Zusammensetzungen umfassen hochgradig konzentrierten Faktor IX und
Arginin, einen Puffer und Sucrose oder Mannitol in Formulierungen,
welche umfassen:
etwa: 2 bis 160 mg/ml Faktor IX,
130
bis 235 mM Arginin und
7,5 bis 40 mM Citrat.
etwa: 2 bis
160 mg/ml Faktor IX,
66 bis 90 mM Arginin,
110 bis 165
mM Mannitol und
7,5 bis 40 mM Citrat.
etwa: 2 bis 160
mg/ml Faktor IX,
70 mM Arginin,
165 mM Mannitol und
15
mM Citrat.
etwa: 2 bis 160 mg/ml Faktor IX,
160 mM Arginin
und
15 mM Citrat.
-
Wo
der Surfactant Polysorbat ist, umfasst eine alternative Zusammensetzung
etwa:
2 bis 160 mg/ml Faktor IX,
130 bis 235 mM Arginin,
0
bis 60 mM Sucrose
0 bis 0,005% Polysorbat und
7,5 bis
40 mM Citrat.
-
Die
Erfindung sieht ebenfalls die Verwendung einer Zusammensetzung wie
oben beschrieben für
die Herstellung einer pharmazeutischen Zusammensetzung zum Erhöhen der
Faktor IX Konzentration vor, wobei vorzugsweise die besagte pharmazeutische
Zusammensetzung so entworfen ist, dass sie sowohl intravenös, als auch
subkutan verabreicht werden kann.
-
Auch
werden Verfahren zur Verabreichung von hochgradig konzentriertem
Faktor IX unter Verwendung von sowohl intravenösen, als auch subkutanen Wegen
vorgesehen, z. B. eine intravenöse
Dosis, gefolgt von einer subkutanen Dosis.
-
Detaillierte
Beschreibung der Erfindung
-
Wie
hierin verwendet, schließt
Faktor IX sowohl von Plasma hergeleitetes, als auch rekombinant
oder synthetisch hergestelltes ein. Die Faktor IX Konzentration
wird bequem als mg/ml oder als U/ml ausgedrückt, wobei 1 mg üblicherweise > 150 U ± 100 U
oder mehr repräsentiert.
Eine Wirksamkeitseinheit wird definiert als Menge von Faktor IX
Gerinnungswirksamkeit in einem Milliliter von normalem menschlichen
Plasma. Die spezifische Wirksamkeit ist das Verhältnis von Gerinnungswirksamkeitskonzentration
zu Proteinkonzentration, ausgedrückt
als U/mg Protein. Patienten mit Hämophilie haben im Allgemeinen
von < 1 bis 25%
des Faktor IX Gerinnungsfaktors, wie er in normalem menschlichen
Plasma gefunden wird.
-
Wie
hierin verwendet verstehen sich die spezifizierten Mengen als ± etwa
10%, z. B. etwa 50 mM schließt
50 mM ± 5
mM ein; z. B. 4% schließt
4% ± 0,4%
ein usw.
-
Wie
hierin verwendet schließt
der Begriff „Tonizitätsmodifikator" Wirkstoffe ein,
welche zur Osmolalität der
Lösung
beitragen. Beispiele für
Tonizitätsmodifikatoren
schließen
ein, aber sind nicht beschränkt
auf Aminosäuren
wie Arginin, Histidin und Glycin, Salze wie Natriumchlorid, Kaliumchlorid
und Natriumcitrat und Saccharide wie Sucrose, Glucose und Mannitol
und Ähnliches.
-
Der
Begriff „Kryoschutzmittel" schließt allgemein
Wirkstoffe ein, welche dem Protein Stabilität vor durch Gefrieren herbeigeführte Spannung
verleiht; jedoch können
Kroyschutzmittel auch allgemeine Stabilität vorsehen, zum Beispiel für Massearznei-Formulierungen während der
Lagerung vor nicht durch Gefrieren herbeigeführte Spannungen. Bespielhafte
Kryoschutzmittel schließen
Polyole und Saccharide wie Mannitol und Sucrose ein, als auch Surfactanten
wie Polysorbat oder Polyethylenglycol und Ähnliches. Während bevorzugte Konzentrationen
an Kroyschutzmitteln von etwa 0,2 bis 4% (Gewicht/Volumen) reichen,
sind relativ hohe Konzentrationen, zum Beispiel größer als
5% ebenfalls geeignet; die verwendeten Grade werden nur durch jene begrenzt,
welche üblicherweise
in der klinischen Praxis verwendet werden. Die oberen Konzentrationsgrenzen für Massearznei
können
höher sein,
als für
Fertigdosierungen, z. B. größer als
5%. „Surfactanten" schließt allgemein
jene Wirkstoffe ein, welche das Protein vor durch Luft/Lösung-Grenzfläche herbeigeführte Spannungen
und durch Lösung/Oberfläche herbeigeführte Spannungen
schützt
(was z. B. Proteinaggregation ergibt) und kann Detergenzien wie
Polysorbat-80 (Tween), zum Beispiel etwa 0,005 bis 1% (Volumen/Volumen)
oder Polyethylenglycol (PEG) wie zum Beispiel PEG8000 einschließen. Gegebenenfalls
sind relativ hohe Konzentrationen, z. B. bis zu 0,5% zum Beibehalten
von Proteinstabilität
geeignet, jedoch werden die in der tatsächlichen Praxis verwendeten
Grade üblicherweise
durch die klinische Praxis eingeschränkt.
-
Der
Begriff „Puffer" umfasst jene Wirkstoffe,
welche den pH der Lösung
in einer annehmbaren Bandbreite halten, und kann Histidin, Phosphat
(Natrium oder Kalium), Citrat (Natrium oder Kalium), Maleinsäure, Ammoniumacetat,
Tris (Tris(hydroxymethyl)aminomethan), Diethanolamin und Ähnliches
einschließen.
Die oberen Konzentrationsgrenzen können für Massenprotein höher sein,
als für
Fertigdosierung-Proteinformen, wie leicht durch einen Fachmann erkannt
wird. Zum Beispiel während
Pufferkonzentrationen von mehreren millimolar bis zur oberen Grenze
ihrer Löslichkeit
reichen können,
z. B. Citrat könnte
so hoch wie 200 mM sein, würde
es der Fachmann ebenfalls bedenken, eine physiologisch angemessene
Konzentration sowohl zu erreichen, als auch zu halten. Die Prozentsätze sind
Gewicht/Volumen, wenn sie sich auf Feststoffe beziehen, die in Lösung gelöst sind,
und Volumen/Volumen, wenn sie sich auf Flüssigkeiten beziehen, die in
Lösungen gemischt
sind. Zum Beispiel ist es für
Sucrose das Trockengewicht Sucrose/Lösungsvolumen und für Tween ist
es das Volumen von 100 Ausgangsmatrial/Lösungsvolumen. Der Begriff „isotonisch
mit Serum" 300 ± 50 milliosmolal
bedeutet ein Maß der
Osmolalität
der Lösung
vor Verabreichung. Beibehalten der physiologischen Osmolalität ist wichtig,
damit die Dosierungsformulierungen ohne vorherige Verdünnung injizierbar
sind. Jedoch können
für Masseformulierungen
viel höhere
Osmolaliltäten
wirksam genutzt werden, solange die Lösung vor der Verwendung isotonisch
gemacht wird. Der Begriff „Exzipienten" schließt pharmazeutisch
annehmbare Reagenzien ein, um angemessene Tonizität, Kryoschutz
des Proteins, Beibehaltung des pH und richtige Konformation des
Proteins während
der Lagerung vorzusehen, so dass wesentliche Erhaltung von biologischer
Wirksamkeit und Proteinstabilität
beibehalten bleibt.
-
Wirkung von
Calciumionen
-
Die
Herstellung von rekombinantem Faktor IX ist in USPN 4770999, Kaufman
et al., beschrieben worden. Ein geeignetes Reinigungsverfahren ist
das, welches in Hrinda et al., Preclinical Studies of a Monoclonal Antibody-Purified
factor IX, MononineTM Seminars in Hematology,
28(3): 6 (Juli 1991) beschrieben wurde. Weiter Herstellungsverfahren
schließen
die Verwendung von Konformations-spezifischen monoklonalen Antikörper ein,
wie durch Tharakan et al., „Physical
and biochemical properties of five commercial resins for immunoaffinity
purification of factor IX",
Journal of Chromatograpy 595. 103–111 (1992); und durch Liebman
et al., „Immunoaffinity
purification of factor IX (Christmas factor) by using conformation-specific
antibodies directed against the factor IX-metal complex" Proc. Nat. Acad.
Sci., USA 82: 3879–3883
(1985) beschrieben; als auch durch herkömmliche chromatographische
Verfahren wie zum Beispiel durch Hashimoto et al., „A Method
for Systematic Purification from Bovine Plasma of Six Vitamin K-Dependent
Coagulation Factors: Prothrombin, Factor X, Factor IX, Protein C
and Protein Z",
J. Biochem. 97: 1347–1355
(1985) und Bajaj, P. et al., Prep. Biochem. 11: 397 (1981), „Large-scale
preparation and biochemical characterization of a new high purity
factor IX concentrate prepared by metal chelate affinity chromatography", P. A. Feldman et
al., Blood Coagulation and Fibrinolysis 5: 939–948 (1994) beschrieben. Noch
ein weiteres Reinigungsverfahren wird in USSN 08/472823, eingereicht
7. Juni 1995 beschrieben und hierin durch Verweis eingeschlossen.
-
Eine
gut gekennzeichnete Eigenschaft von Faktor IX ist seine Fähigkeit,
Ca2+ Ionen zu binden. Strukturstudien zeigen
an, dass Ca2+-Bindung eine stabilere Struktur
verleihen kann, wobei die Möglichkeit
molekularer Bewegung verringert wird („Structure of the Metal-free γ-Carboxyglutamic
Acid-rich Membrane Binding Region of Factor IX by Two-dimensional
NMR Spectroscopy",
S. J. Freedman, B. C. Furie, B. Furie und J. D. Baleja, J. Biol.
Chem. 270(14): 7980–7989
(1995); „Structure
of the Calcium Ion-Bound γ-Carboxyglutamic Acid-Rich
Domain of Factor IX.",
S. J. Freedman, B. C. Furie, B. Furie, und J. D. Baleja, Biochemistry
34: 12126–12137
(1994): „The
Structure of a Ca2+-Binding Epidermal Growth
Factor-like Domain: Its Role in Protein-Protein Interactions", S. Rao., P. Handford,
M. Mayhew, V. Knott, G. Brownlee und D. Stuart, Cell 82: 131–141 (1995); „Structure
of Ca2+ Prothrombin Fragment 1 Including
the Conformation of the Gla Domain", M. Soriano-Garcia, C. H. Park, A.
Tulinsky, K. G. Ravichandran und E. Skrzypczak-Jankun, Biochem.
28: 6805–6810
(1989)). Vermutlich entspricht weniger Mobilität einer niedrigeren Wahrscheinlichkeit
von molekularer Interaktion, wodurch sich die Wahrscheinlichkeit
von Abbauprozessen verringert. Überraschend
stellt sich heraus, dass dies nicht der Fall ist.
-
Proben
werden in den in Tabelle 1 unten dargestellten Formulierungen bei
einer Konzentration von rekombinantem Faktor IX Protein von –0,5 mg/ml
(100 U/ml) und eine Osmolalität
von 300 ± 50
milliosmolal hergestellt. Alle Proben enthalten eine rekombinante
Form von Faktor IX. Um den potentiellen Nutzen von Ca2+ als
Stabilisator zu untersuchen, wurde ein Probensatz in den in Tabelle
1 aufgelisteten Formulierungen hergestellt. Die Formulierung von
Probe A ist die Formulierung, welche für kommerziell erhältlichen,
von Plasma abgeleiteten, lyophilisierten Faktor IX (MononineTM) verwendet wird. Alle Proben enthalten
eine rekombinante Form von Faktor IX.
-
Tabelle
1
Probenformulierungen
-
Proben
von Faktor IX in jeder Formulierung wurden bei 4°C für 2,5 Monate gelagert. Die
Proben wurden bezüglich
Proteinkonzentration und Gerinnungswirksamkeit getestet. Faktor
IX Wirksamkeit wird gemäß dem Verfahren
von Pittman, D. et al., Blood 79: 389–397 (1992) unter Nutzung von
Faktor IX armem Blut bestimmt. Das Verhältnis von Gerinnungswirksamkeit
zu Proteinkonzentration, die spezifische Wirksamkeit, welche in
Einheiten/mg Protein ausgedrückt
wird, wird in Tabelle 2 angegeben. Eine annehmbare spezifische Wirksamkeit
ist eine, die im Allgemeinen nicht mehr als 20% höher ist,
als die anfängliche
spezifische Wirksamkeit, weil eine ungewöhnlich hohe spezifische Wirksamkeit
ein Aktivierungs-ähnliches
Ereignis anzeigen kann, was thrombotische Implikationen haben kann.
-
Tabelle
2
Faktor IX spezifische Wirksamkeit
-
Die
Proben, welche Calcium enthalten, also Proben C, E und G, haben
höhere
spezifische Wirksamkeit nach 2,5 Monaten Lagerung. Dies ist Folge
der Inklusion von Ca2+ und zeigt an, dass
sich der Faktor IX einer Umwandlung zu einem aktivierten-ähnlichen
Molekül
unterzogen hat. Aktivierter Faktor IX ist Faktor IX, der bei Resten
R145-A146 und R180-V181 gespalten
wurde und dann in der Lage ist, Gerinnung zu katalysieren. Normalerweise
zirkuliert Faktor IX als intaktes Protein und wird nicht in seine
aktivierte Form umgewandelt, sofern es keine Initiation der Gerinnungskaskade
gibt. Wenn man jemandem aktiviertes rhFIX injiziert, könnte dies
thrombotische Implikationen haben. Daher ist Inklusion von Ca2+ bei einer Konzentration von 5 mM destabilisierend
und muss vermieden werden.
-
Wirkungen der Pufferwahl
auf BMW-Bildung
-
Die
durchschnittliche spezifische Wirksamkeit nach acht Monaten Lagerung
bei 40°C
von Proben, formuliert in Puffer/Exzipient Kombinationen ähnlich und
einschließlich
jenen in Tabelle 1, aber ohne Calcium, bei pH 7,0 bis 112,5 ± 10,5
U/mg aber bei pH 7,5 ist nur 84,0 ± 22,1 U/mg, was anzeigt,
dass feine Verschiebungen beim pH für das Beibehalten der Langzeit
Faktor IX Stabilität
signifikant sind.
-
Faktor
IX wird in einem Satz isotonischer experimenteller Formulierungen
wie in Tabelle 3 zusammengefasst hergestellt, einschließlich mehrerer
unterschiedlicher Exzipient-Kombina tionen für jeden Puffer und einigen,
welche weniger als 5 mM EDTA einschließen. Die Faktor IX Konzentrationen
betragen etwa 1 mg/ml (Durchschnitt 161 U/ml). Die Proben werden
bezüglich
der Menge an vorhandenem Material mit hohem Molekulargewicht (HMW)
und bezüglich
Gerinnungswirksamkeit getestet. Die Bildung von signifikanten (> 3%) Mengen an HMW
ist unerwünscht
und weist auf physikalischen Abbau von Faktor IX mit möglichem
Einfluss auf Produktsicherheit und Wirksamkeit hin. HMW ist insbesondere
für subkutane
Verabreichung unerwünscht, da
aggregierte Proteine immunogener sind, wenn sie subkutan gegeben
werden. Morein, B. und K. Simons, Vaccine 3: 83. Subunit vaccines
against enveloped viruses: virosomes, micelles, and other protein
complexes (1985); und Antibodies: A laboratory manual, (Seite 100),
E. Harlow und D. Lane, Cold Spring Harbor Laboratory, 1988.
-
Tabelle
3
Probenformulierung
-
Tabelle
4 zeigt die Wirkungen der unterschiedlichen Puffer auf HMW Erzeugung,
wie durch Größen-Exklusionschromatographie
(SEC-HPLC) gemessen. Die Proben wurden bei 3 sechs Wochen gelagert. Tabelle
4 gibt den durchschnittlichen Anstieg an, ausgedrückt als
(HMW/Gesamtprotein × 100%)
bei sechs Wochen minus dem bei Zeit Null.
-
Tabelle
4
Prozentualer Anstieg der HMW-Erzeugung
-
Die
mit Citrat gepufferten Proben hatten im Durchschnitt die kleinste
Menge an erzeugtem HMW, ungeachtet der weiteren eingeschlossenen
Exzipienten. Ein angemessener Puffer erlaubt keinen Anstieg größer als
2%.
-
Aggregate
sind gewöhnlich
als immunogener, als monomere Proteine bekannt und sind allgemein
für eine
intravenöse
Formulierung nicht annehmbar. Jedoch sind Aggregate für subkutane,
intradermale oder intramuskuläre
Formulierung noch unerwünschter,
weil diese Verabreichungswege noch wahrscheinlicher eine Immunresponse
erzeugen.
-
Alle
Proben werden für
sechs Monate bei 4°C
weiter gelagert und bezüglich
Gerinnungswirksamkeit getestet. Die durchschnittliche Menge an Wirksamkeit,
welche für
Proben verblieb, welche die verschiedenen Saccharide enthielten,
variierte stark; Sucrose-enthaltende Proben behielten durchschnittlich
71% der Anfangswirksamkeit, Mannitol 53%, Glucose 52% und Mannose
nur 27%. Überraschend
sind nicht alle Saccharide gleich wirksam beim Erhalten der Faktor
IX Wirksamkeit, trotz Zugabe weiterer Exzipienten.
-
Die
folgenden Beispiele veranschaulichen die Praxis der Erfindung. Diese
Beispiele dienen nur veranschaulichenden Zwecken und sind nicht
beabsichtigt, den Umfang der beanspruchten Erfindung in irgend einer Weise
einzuschränken.
Beispiel 1 veranschaulicht die Verwendung der Erfindung für höhere Konzentrationen von
Faktor IX. Beispiel 2 veranschaulicht die Komplexität von Exzipient-Interaktionen
beim Stabilisieren von Faktor IX. Beispiel 3 beschreibt Faktor IX
in verschiedenen Formulierungen bezogen auf Frost/Tau-Stabilität. Beispiel
4 beschreibt die Wirkungen von Langzeitlagerung und Beispiele 5 und
6 veranschaulichen hochgradig konzentrierte Formen und ihre Verwendung.
-
Beispiel 1: Stabilität bei hoher
Konzentration
-
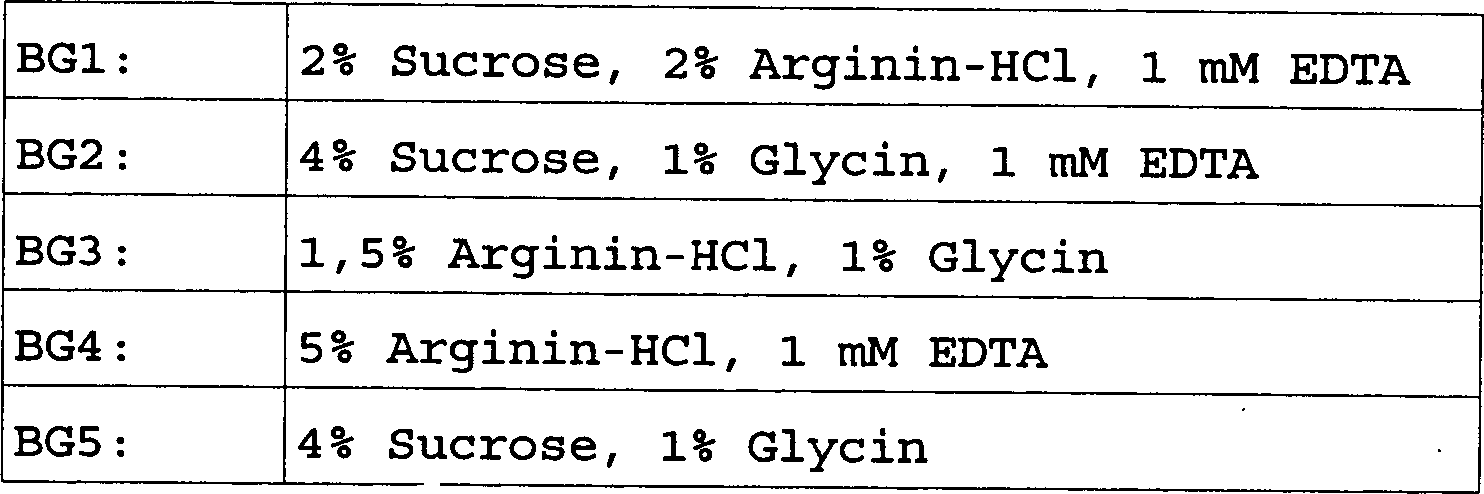
Ein
weiterer Satz Formulierungen wird hergestellt, welcher hohe Konzentrationen
an Faktor IX umfasst; die Proben werden in den in Tabelle 5 aufgelisteten
Formulierungen bei einer Konzentration von 8 mg/ml (2000 U/ml) hergestellt.
Alle enthalten 15 mM Natriumcitrat und sind bei pH 6,8 gepuffert,
ohne Surfactant. BG4 ist leicht hypertonisch, der Rest ist isotonisch.
-
Tabelle
5
Probenformulierungen
-
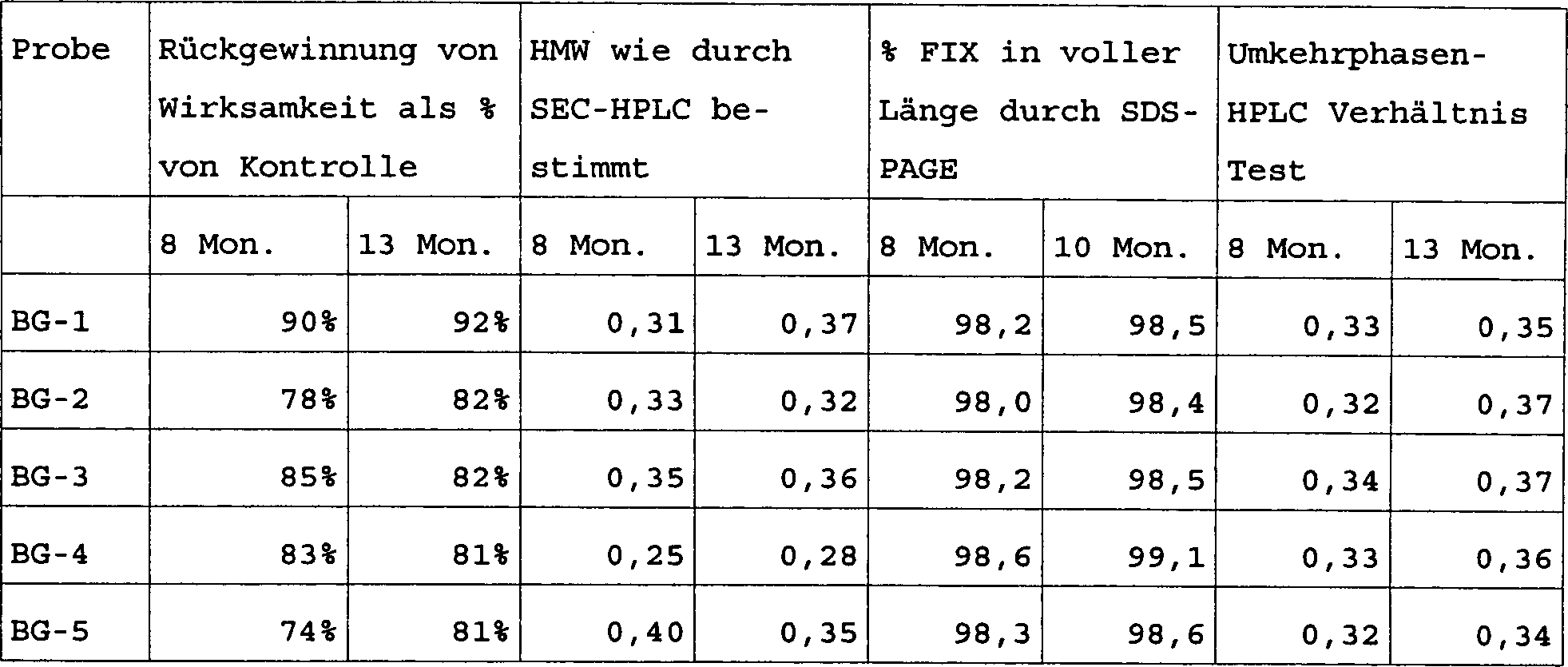
Die
Proben werden in sowohl Glasvialen, als auch vorgefüllten Glasspritzen
für acht,
zehn und dreizehn Monate bei 4°C
gelagert, um zu bestimmen, ob die Menge an Luft/Lösung Grenzfläche oder
siliconisierte Stopper/Lösung
Grenzfläche
die Stabilität
des Produkts beeinträchtigen
würde.
Zu allen drei Zeitpunkten werden keine signifikanten Unterschiede
durch irgend einen Stabilitätstest
zwischen den Vialen und Spritzen gesehen. Die Ergebnisse von mehreren
analytischen Verfahren werden in Tabelle 6 gezeigt. „Rückgewinn
an Wirksamkeit" betrifft
die Menge an Gerinnungswirksamkeit, welche in der Probe verbleibt,
ausgedrückt
als Prozentsatz der Menge an Gerinnungswirksamkeit zur „Zeit Null", dem Beginn der
Studie. „HMW" wird supra beschrieben. „SDS-PAGE" ist Polyacrylamid-Gel-Elektrophorese;
Gels werden gescannt und Bänder
quantifiziert. Umkehrphasen-HPLC wird verwendet, um Produktheterogenität zu bewerten
und Veränderungen
in Peak-Verhältnissen
können
Veränderungen
im Produkt anzeigen, zum Beispiel Oxidation von Oligosacchariden.
-
-
Selbst
bei dieser hohen Konzentration von Faktor IX (8 mg/ml; 2000 U/ml)
sind diese Formulierungen wirksam. Der Anstieg in beobachteter Wirksamkeit
für bestimmte
Proben bei 13 Monaten bezogen auf die Kontrolle ist innerhalb der
Variabilität
des Tests.
-
Beispiel 2: Exzipienten-Interaktionen
-
Ein
weiterer Satz an Faktor IX Formulierungen, welche alle Citrat enthalten,
wird wie in Tabelle 7 zusammengefasst hergestellt. Alle Formulierungen
sind isotonisch, enthalten Faktor IX bei Konzentrationen von 1 bis
2 mg/ml (durchschnittlich 208 bis 481 U/ml), verwenden 10 mM bis
15 mM Natriumcitrat als pH Puffer und sind auf pH 6,8 eingestellt.
-
Tabelle
7
Probenformulierung
-
Die
Proben werden bei 4°C
gelagert und zu mehreren Zeitpunkten getestet. Nach acht Monaten
bei 4°C
Lagerung behielten neun Proben 100% der Gerinnungswirksamkeit des
Ausgangsmaterials. Die Formulierungen dieser Neun werden in Tabelle
8 gezeigt (alle schließen
15 mM Natriumcitrat ein, haben einen pH von 6,8 und sind isotonisch).
-
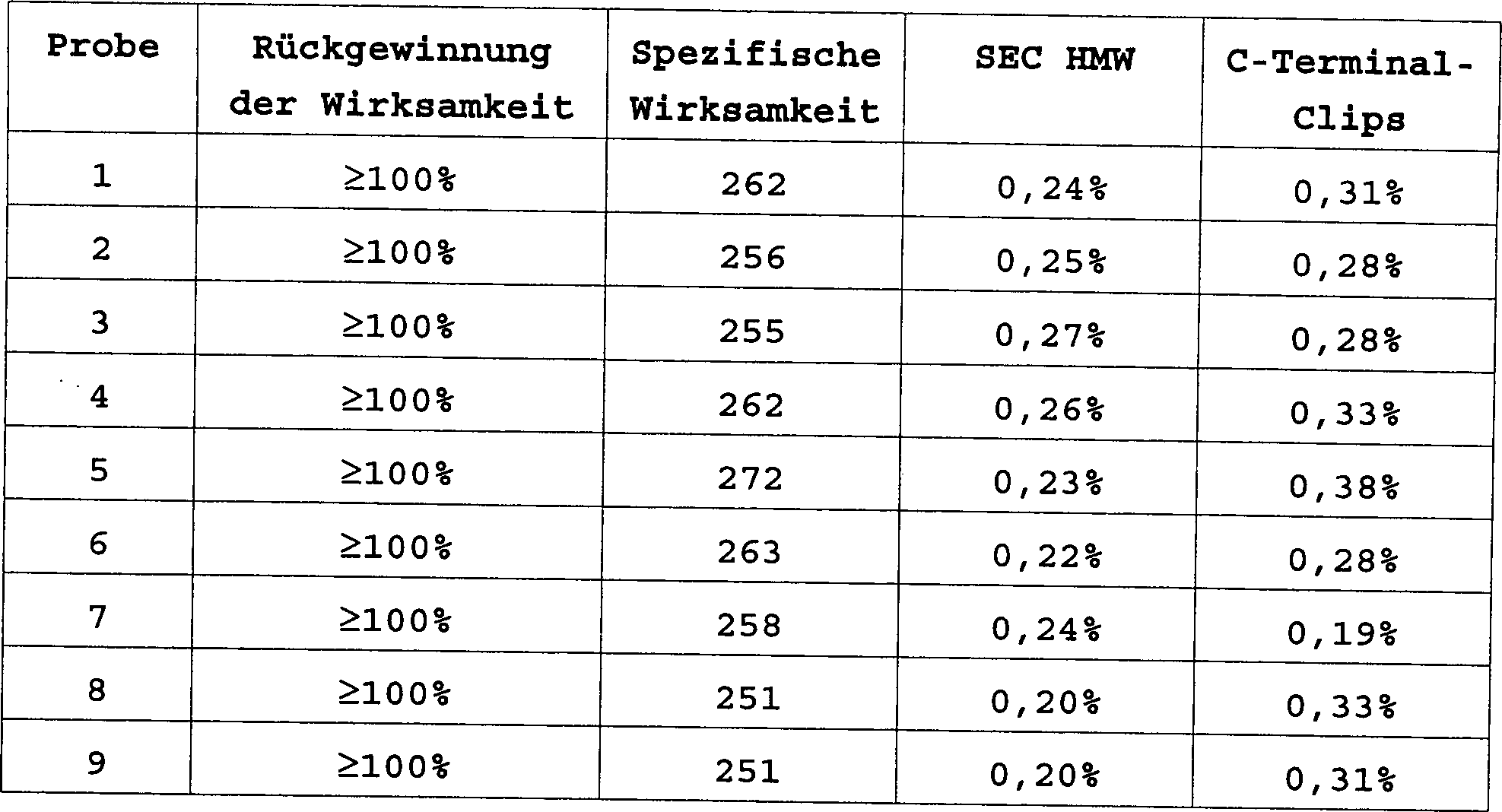
-
Mehrere
Formulierungen, welche ähnliche
Exzipienten in ähnlichen
Verhältnissen
enthalten, behalten dennoch überraschenderweise
ihr Gerinnungswirksamkeit nicht annähernd so gut. Zum Beispiel
ergab 2,3% Glycin allein nur 86%; und 4% Sucrose, 2% Arginin, beide
mit und ohne Tween und mit und ohne EDTA ergaben 87–89% Gerinnungswirksamkeit.
-
Für die neun
Formulierungen von Tabelle 8 werden die Ergebnisse von weiteren
Stabilität-anzeigenden
Tests gezeigt. Spezifische Wirksamkeit wird als U/mg ausgedrückt und
ein annehmbarer Bereich ist 200 bis 350 U/mg. SEC-HMW ist ein Maß für Aggregate
mit hohem Molekulargewicht, wie durch Größen-Exklusionschromatographie
bestimmt; weniger als 1% wird für
eine subkutane, intradermale oder intramuskuläre Formulierung bevorzugt. „C-Terminal-Clips" ist ein Maß für Abbauarten
wie durch Umkehrphasenchromatographie bestimmt; weniger als 1% wird
bevorzugt.
-
-
Basierend
auf den in Tabellen 8 und 9 dargestellte bevorzugten Formulierungen
schließen
zwei bevorzugtere Formulierungen Folgendes ein: (beide sind bei
pH 6,8 mit 15 mM Citrat gepuffert und sind isotonisch), 3% Mannitol,
1,5% Arginin-HCl und 3,3% Arginin-HCl.
-
Beispiel 3: Wirkungen
von Frost/Tau-Wechsel
-
Idealerweise
wird eine ähnliche
Formulierung für
Masseprotein genutzt, wie sie für
die Fertigdosierungsform verwendet wird. Dies erfordert, dass die
gleiche Formulierung, die Faktor IX vor Langzeitlagerungsspannungen
stabilisiert, auch zum Stabilisieren von Faktor IX gegenüber den
Spannungen angemessen ist, welche normalerweise durch Masseprotein
angetroffen werden, wie Gefrieren und Tauen.
-
Die
Proben werden in den Formulierungen hergestellt, welche in Tabelle
10 unten dargestellt werden, bei einer Proteinkonzentration von
~2 mg/ml (500 U/ml) und Osmolalität von 300 ± 50 milliosmolal. Alle schließen 10 mM
Natriumcitrat, pH 6,8 ein und alle werden sowohl mit, als auch ohne
0,005% Tween-80 (Polysorbat) hergestellt.
-
Tabelle
10
Probenformulierungen
-
Proben
von Faktor IX in jeder Formulierung wurden fünf Frost/Tau-Wechseln unterworfen,
um Anfälligkeit
gegenüber
durch Forst herbeigeführte
Denaturierung zu bestimmen, welche Bildung von Proteinaggregaten
ergeben kann. Eine Folge von Frost/Tau-Wechseln ist ein brauchbarer Indikator
für die
Anfälligkeit
eines Proteins, Aggragatbildung zu erhöhen, wie es während Gefrieren
und Langzeitlagerung beobachtet werden kann. Die Proben werden hinsichtlich
der Menge an vorhandenem HMW getestet. Proben mit und ohne Tween-80
(0,005%) haben eine minimale Aggregation (weniger als 0,15% HMW
Anstieg).
-
Basierend
auf den Daten hierin werden die folgenden beiden Formulierungen
(ausgedrückt
als Bandbreiten an Bestandteilen) ebenfalls bevorzugt.
-
-
Beispiel 4: Wirkung von
Langzeitlagerung bei 4°C
-
Faktor
IX wird bei 2 mg/ml (500 U/ml) in 15 mM Natriumcitrat (0,38%), 0,16
M Arginin (3,3%), pH 6,8 formuliert und für ein Jahr bei 4°C gelagert.
Die Rückgewinnung
der Wirksamkeit beträgt
95% und das %-HMW ist 0,32%. Faktor IX wird bei 2 mg/ml in 15 ml
Natriumcitrat, 3% Mannitol, 1,5% Arginin, pH 6,8 formuliert und für ein Jahr
bei 4°C
gelagert. Die Rückgewinnung
der Wirksamkeit beträgt
76% und das %-HMW war 0,36%. Der Wirksamkeitsverlust wird Entamidierung
zugeschrieben.
-
Faktor
IX wird bei 2 mg/ml in 15 mM Natriumcitrat, 1% = 29 mM Sucrose,
3% = 0,14 M Arginin HCl formuliert und für ein Jahr bei 4°C gelagert.
Die Rückgewinnung
der Wirksamkeit beträgt
86% und das %-HMW ist 0,27.
-
Beispiel 5: Wirkungen
von hoher Proteinkonzentration und von Frost-Tau
-
Faktor
IX wird bei 4000 U/ml, 8000 U/ml, 16000 U/ml und mehr als 30000
U/ml (also 16 bis mehr als 120 mg/ml) in 10 mM Histidin, 260 mM
Glycin, 1% Sucrose, 0,005% Tween-80, pH 7,0 formuliert. Faktor IX wird
durch Zentrifugalkonzentration in einem Centricon-10 und durch Rühr-Zellkonzentration
in einer Amicon Rührzelle
unter Verwendung einer YM-10 Membran konzentriert. Weitere Verfahren,
welche zum Konzentrieren von Proteinen verwendet werden, insbesondere
jene, welche Membranen verwenden, welche Arten zurückhalten
und ausschließen,
basierend auf Molekulargewicht, wie Filtration mit Tangentialströmung, können ebenfalls
verwendet werden. Zusätzlich
kann Sprühtrocknen
ohne ungünstige
Wirkungen verwendet werden.
-
Überraschenderweise
wird kein nachweisbares aggregiertes Protein (HMW wie durch SEC-HPLC
bestimmt) erzeugt, selbst bei diesen außergewöhnlich hohen Proteinkonzentrationen.
-
Die
Proben werden nachfolgend wiederholt gefroren und getaut und behalten überraschenderweise noch
annehmbare Grade von HMW (≤ 1%).
Dies ist in Anbetracht der kommerziell erhältlichen, von Plasma abgeleiteten
Faktor IX Produkte wie MononineTM und AlphanineTM (supra auf Seite 3, Zeilen 20–29) überraschend,
welche häufig
10% oder mehr HMW enthalten, obwohl die Faktor IX Konzentration
ziemlich niedrig ist. Solche ein hoher % HMW ist für subkutane,
intradermale oder intramuskuläre
Verabreichung aufgrund des Potentials für Immunogenizität unannehmbar.
-
Ferner
werden, wenn Faktor IX in der gleichen Formulierung wie MononineTM formuliert wird und wiederholten Frost/Tau-Wechseln unterworfen
wird, signifikante Mengen (~15%) HMW erzeugt. Diese Daten zeigen
zusammen mit den in Beispiel 3 gezeigten Daten die überraschenden
und unvorhergesehenen Wirkungen von Formulierung auf die Stabilität von Faktor
IX.
-
Beispiel 6: Verwendung
von hochgradig konzentriertem Faktor IX
-
Hochgradig
konzentrierter Faktor IX ist wirksam, wenn er subkutan, intradermal
oder intramuskulär verabreicht
wird. Unter Nutzung einer hochgradig konzentrierten Formulierung
von Faktor IX, also 4000 U/ml bis mehr als 56000 U/ml ist eine subkutane
Injektion an einer einzelnen Stelle mit niedrigem Volumen möglich, wie
unten beschrieben.
-
Drei
experimentelle Gruppen wurden unter Verwendung von Faktor IX bei
einer Konzentration von 4000 IU/ml in 260 mM Glycin, 10 mM Histidin,
29 mM (1%) Sucrose und 0,005% Polysorbat bewertet. In Gruppe I wurde
Hunden 200 U/kg (0,05 ml/kg) Faktor IX intravenös gegeben. In Gruppe II wurde
Hunden 200 U/kg (0,05 ml/kg) Faktor IX subkutan gegeben. In Gruppe
III wurde Hunden eine Faktor IX intravenöse Initialdosis von 50 U/kg
(0,0125 ml/kg) gegeben, gefolgt von einer 200 U/kg (0,05 ml/kg)
subkutanen Dosis 24 Stunden später.
Intravenöser
Faktor IX erzeugte eine 240 Faktor IX Wirksamkeit (wo 100% = Standard
von gepooltem menschlichen Plasma) innerhalb von fünf Minuten
nach der Injektion, welche auf 6,4% bis Tag 5 sank. Subkutane Faktor
IX Wirksamkeit betrug 0,9% bei 5 Minuten, 10% bei drei Stunden und
5,8% an Tag 5. Die Kombination einer intravenösen Ladungsdosis, gefolgt von
einer subkutanen Dosis 24 Stunden später, ergab eine Plasma Faktor
IX Wirksamkeit von 25% drei Stunden nach der subkutanen Dosis und
eine Faktor IX Wirksamkeit von 9,1% an Tag 5 nach der subkutanen
Injektion. Die biologische Verfügbarkeit
der subkutanen Dosis wurde als 43% errechnet. Subkutanes Faktor
IX erzeugt therapeutische Grade von Faktor IX Wirksamkeit in weniger
als drei Stunden nach Verabreichung. Die Kombinationsdosis aus einer
intravenösen
mit einer subkutanen Dosis sieht sofortigen Koagulanzschutz vor
und verbessert die Wirksamkeit der subkutanen Dosis. Auch können hochgradig
konzentrierte Formen von Faktor IX in den supra beschriebenen Formulierungen
in Beispielen 1–3
formuliert und wirksam für
Verabreichung verwendet werden.
-
Während die
vorliegende Erfindung bezüglich
spezifischer Verfahren, Formulierungen und Zusammensetzungen beschrieben
worden ist, versteht es sich, dass den Fachleuten bei Betrachtung
der vorliegenden Erfindung Variationen und Modifikationen einfallen
werden.
-
Es
wird erwartet, dass den Fachleuten zahlreiche Modifikationen und
Variationen der Erfindung wie in den obigen veranschaulichenden
Beispielen beschrieben, einfallen werden, und demzufolge sollten
darauf nur solche Einschränkungen
auferlegt werden, wie sie in den angehängten Ansprüchen erscheinen. Entsprechend ist
es in den angehängten
Ansprüchen
beabsichtigt, alle solchen äquivalenten
Variationen abzudecken, welche innerhalb des Umfangs der Erfindung
wie beansprucht fallen.