-
Die
vorliegende Erfindung betrifft lumineszente, insbesondere fotolumineszente
Nanopartikel mit einem Kern aus einem Metallsalz oder -oxid, umgeben
von einer lumineszenten Schale, die Synthese dieser Partikel sowie
deren Verwendung in (F)RET-Assayverfahren, insbesondere in Bioassayverfahren.
-
Hintergrund
der vorliegenden Anmeldung
-
Im
Laufe der letzten Dekade haben Nanopartikel, d.h. Partikel mit einer
Größe unterhalb
1 μm, großes Interesse
in Forschung und Industrie wegen ihrer einzigartigen Eigenschaften
auf sich gezogen. Forschung und Entwicklung auf optoelektronischem
Gebiet haben sich auf lumineszente Partikel im Hinblick auf deren mögliche Anwendung
Licht emittierenden Dioden (LED), Displays, optoelektronischen Vorrichtungen
in Nanometer-Abmessungen
oder als Lichtquelle in niederschwelligen Lasern konzentriert.
-
Unter
den Lumineszenzmaterialien wird oft zwischen Halbleiter- und Nicht-Halbleiter-Materialien
unterschieden.
-
Halbleiter-Nanopartikel
(oft bezeichnet als "Quanten-Punkte"), wie II-VI- oder
III-V-Halbleiter, die dotiert sein können oder nicht, sind durch
eine Quantenbegrenzung von sowohl dem Elektron als auch dem Loch in
allen drei Raumdimensionen gekennzeichnet, welche zu einer Steigerung
bei der effektiven Bandenlücke des
Materials bei sinkender Kristallgröße führt. Infolgedessen ist es möglich, sowohl
die optische Absorption als auch die Emission der Halbleiter-Nanopartikel
nach blau (zu höheren
Energien) mit zunehmender Verkleinerung der Nanopartikel zu verschieben.
-
Wasserlösliche Kern/Schale-Halbleiter-Nanopartikel
sind z.B. in WO 00/17 655 beschrieben.
-
Im
Vergleich mit Quanten-Punkten beruht die besondere Attraktivität nanokristalliner
Nicht-Halbleiter-basierter Lumineszenzmaterialien, insbesondere
Lanthanid-dotierter Metalloxide oder -salze, darauf, dass deren
Fluoreszenzemission relativ eng ist und nicht im größeren Ausmaß vom Wirtsmaterial
und der Größe der Nanopartikel
abhängt.
Es ist vielmehr lediglich der Typ des Lanthanid-Metalls, welcher
die Emissionsfarbe bestimmt. In PCT/DE 01/03 433 von denselben Anmelderinnen
ist ein allgemein anwendbares Syntheseverfahren für Lanthanid-dotierte
Nanopartikel dieses Typs offenbart. Diese Nanopartikel lassen sich
in Größen (unterhalb
30 nm) herstellen, ohne noch länger
mit der Wellenlänge
des sichtbaren Lichts wechselzuwirken, um dadurch transparente Dispersionen,
z.B. in organischen oder wässrigen
Lösungsmitteln,
zu ergeben.
-
Weitere
Publikationen, betreffend Lanthanid-dotierte Nicht-Halbleiter-basierte
Lumineszenz-Nanopartikel, sind z.B. die folgenden:
- K. Riwotzki
et al., Angewandte Chemie, Int. Ed. 40, 2001, Seiten 573–576, bezüglich LaPO4:Ce,Tb;
- K. Riwotzki, M. Haase, J. Phys. Chem. B; Band 102, 1998, Seiten
10129–10135,
bezüglich
YVO4:Eu, YVO4:Sm und
YVO4:Dy;
- H. Meyssamy et al., Advanced Materials, Band 11, Ausgabe 10,
1999, Seiten 840–844,
bezüglich
LaPO4:Eu, LaPO4:Ce
und LaPO4:Ce,Tb;
- K. Riwotzki et al., J. Phys. Chem. B 2000, Band 104, Seiten
2824–2828, "Liquid phase synthesis
doped nanoparticles: colloids of luminescent LaPO4:Eu
and CePO4:Tb particles with a narrow particle
size distribution";
- M. Haase et al., Journal of Alloys and Compounds, 303–304 (2000)
191–197, "Synthesis and properties
of colloidal lanthanide-doped nanocrystals";
- Jan W. Stouwdam und Frank C. J. M. van Veggel, Nano Letters,
ASAP-Artikel, web-Freigabe: 15. Mai 2002, "Near-infrared emission of redispersible
Er3+, Nd3+ and Ho3+ doped LaF3 nanoparticles"; und
- G. A. Hebbink et al., Advanced Materials 2002, 14, Nr. 16, Seiten
1147–1150, "Lanthanide(III)-doped
nanoparticles that emit in the nearinfrared".
-
Halbleiter-basierte
Nanopartikel ("Quanten-Punkte") sind zur Verwendung
in Bioassayverfahren bereits in Betracht gezogen worden. Bawendi
et al., Physical Review Letters, 76, 1996, Seiten 1517–1520, berichten
z.B. über
FRET-Effekte in spezifisch markierten biologischen Systemen. Ferner
ist in WO 00/29 617 offenbart, dass Proteine oder Nucleinsäuren mittels "Quanten-Punkten" als Markierung in
(F)RET-Assayverfahren detektiert werden können. In
US 6,468,808 B1 und
US 6,326,144 B1 sind
biomolekulare Konjugate von Quanten-Punkten und deren Verwendung
in der Fluoreszenzspektroskopie ebenfalls beschrieben.
-
(F)RET
((Fluoreszenz)-Resonanzenergietransfer) und der verwandte Resonanzenergietransfer
(RET) beruhen auf dem Transfer von Anregungsenergie aus einem Donor
mit der Befähigung
zum Ausstrahlen von Fluoreszenz auf einen Akzeptor in enger Nachbarschaft.
Mit dieser Technik ist es z.B. mit geeigneten fluoreszenten Markierungen
in biologischen Systemen möglich, Abstände auf
molekularer Ebene im Bereich von ca. 1 bis 8 nm zu bestimmen. Die
auf den Akzeptor übertragene
Energie kann ohne Emission durch innere Umwandlung (RET) wieder
nachlassen und führt
dann nur zur Streichung (Löschung)
der Donorfluoreszenz. Alternativ dazu, emittiert der Akzeptor die
aufgenommene Energie auch in der Form von Fluoreszenz (FRET). Diese
Phänomene
sind gut verstanden und lassen sich im Fall einer Dipol-Dipol-Wechselwirkung zwischen Donor
und Akzeptor mit der Theorie von Förster erklären (siehe z.B. J. R. Lakowicz,
Principles of Fluorescence Spectroscopy, Kluwer Academic Press,
New York, 1990, Seiten 368–445).
Der Energietransfer verringert die Intensität der Donorfluoreszenz sowie
deren Lebensdauer und initiiert, sensibilisiert oder steigert gleichzeitig die
Akzeptorfluoreszenz. Die Effizienz bzw. der Wirkungsgrad des Energietransfer
hängen
von der inversen 6. Potenz der intermolekularen Separation ab und
sinken proportional zu R0 6/(R0 6 + R6)
ab. R0, der sogenannte Förster-Radius, kennzeichnet
diesen Abstand zwischen Donor und Akzeptor, für welchen die Effizienz des
Energietransfer 50% beträgt.
-
Die
F(RET)-Effizienz kann entweder über
die Fluoreszenzintensität
des Donor mit Akzeptor (QDA) bzw. ohne Akzeptor
(QD) mittels der Gleichung: 1 – (QDA/QD) oder durch
Vergleich der Lebensdauern des Donor in der Gegenwart (TDA) und der Abwesenheit (TD)
der Akzeptor-Sonde auf Basis der Gleichung: 1 – (TDA/TD) bestimmt werden.
-
Die
Verwendung von "Quanten-Punkten" in Bioassayverfahren
leidet allerdings an verschiedenen Nachteilen. Da die Emissionswellenlängen der
fluoreszenten "Quanten-Punkte" von der Größe der Partikel
abhängen,
kann eine nur sehr enge Größenverteilung
angewandt werden. Dies stellt eine Herausforderung für Synthese-
und/oder Größenauswahlverfahren
dar. Außerdem
ergeben "Quanten-Punkte" im Normalfall relativ niedrige
Quanten-Wirkungsgrade,
was durch emissionsfreie Elektron-Loch-Paarrekombinationen verursacht ist.
Zur Überwindung
dieses Nachteils sind CdSe/CdS-Kern/Schale-Strukturen vorgeschlagen worden, worin der
CdS-Überzug
die Fotostabilität
des lumineszenten CdSe-Kerns schützt
und steigert (X. Peng et al., J. Am. Chem. Soc. 119, 1997, Seiten
7019–7029).
-
In
typischer Weise werden (F)RET-basierte Assayverfahren mit organischen
Farbstoff-Molekülen,
wie mit Fluorescein oder Rhodamin, durchgeführt. Für viele Anwendungen beruht
ein genereller Nachteil, der mit diesen organischen Fluoreszenzfarbstoffen
zusammenhängt,
auf deren unzureichender Stabilität gegenüber einfallendem Licht. Deren
Fototoxizität
kann ferner biologisches Material in der nahen Umgebung beschädigen. Weitere unerwünschte Eigenschaften
beruhen auf deren breiten Emissionsbanden und den kleinen Stokes-Verschiebungen,
d.h. auf der Differenz zwischen Anregungs- und Emissionsmaximum
sowie auf den relativ engen spektralen Anregungsbanden, die oft
den Einsatz mehrerer Lichtquellen und/oder komplizierter Foto-Systeme
erforderlich machen.
-
Demnach
ist es eine Aufgabe der vorliegenden Erfindung, fluoreszente anorganische
Materialien bereitzustellen, die sich besonders für (F)RET-Assayverfahren,
wie insbesondere für
Bioassayverfahren, eignen und mit denen die obigen Nachteile überwunden
werden.
-
Eine
weitere Aufgabe der vorliegenden Erfindung ist es, die (F)RET-Effizenz bzw. den
Wirkungsgrad davon zu steigern. Eine höhere (F)RET-Effizienz erhöht die Empfindlichkeit
des Verfahrens und verbessert z.B. das Signal/Rausch-Verhältnis.
-
Außerdem benötigen (F)RET-basierte
Assayverfahren Donor-Moleküle
mit hohen Quantenausbeuten (dem Verhältnis von emittierten zu absorbierten
Fotonen) zur Steigerung der Gesamtempfindlichkeit des Assay. Deshalb
ist es eine weitere Aufgabe der vorliegenden Erfindung, anorganische
fluoreszente Partikel mit hohen Quantenausbeuten bereitzustellen,
welche diese dann auch besonders attraktiv für andere Anwendungen als in
Bioassayverfahren machen.
-
Gemäß einer
weiteren Aufgabe der vorliegenden Erfindung wird ein spezifisches
Verfahren zur Herstellung dieser fluoreszenten Materialien angegeben
und zur Verfügung
gestellt.
-
Schließlich ist
es noch eine Aufgabe, einen Bioassay auf Basis anorganischer nanoteilchenförmiger Materialien
anzugeben und zur Verfügung
zu stellen.
-
DE 10 131 173 A betrifft
ein Verfahren zur Herstellung nanoteilchenförmiger Kern/Schale-Partikel,
worin der Kern aus einem anorganischen Oxid-Nanopartikel mit einer Größe unterhalb
100 nm besteht und die Schalenmaterialien aus anorganischen Oxid/Hydroxiden,
einem Metall, einem Polymer oder aus einem Glasmaterial ausgewählt sein
können.
Dieses Dokument enthält
keinen Beschreibungshinweis dahingehend, dass dieses Kernmaterial
mit oxidischen Schalen zu überziehen
wäre. In
den Beispielen wird von Polyvinylalkohol als Überzug Gebrauch gemacht.
-
US
2003/0 032 192 A1 offenbart ein Verfahren zur Herstellung homogener
Nicht-Halbleiter-Nanopartikel wie aus Phosphaten.
-
US
2003/0 017 264 A1 offenbart ein Verfahren zur Synthese lumineszenter
Nanopartikel vom Kern/Schale-Typ aus mindestens einem Halbleiter-Kern. Ein bevorzugtes
Kernmaterial ist CdSe.
-
Zusammenfassung
der vorliegenden Erfindung
-
Die
obigen technischen Aufgaben sind mit lumineszenten anorganischen
Nanopartikeln, umfassend
- (a) einen Kern aus
einem ersten Metallsalz oder -oxid, welcher umgeben ist von
- (b) einer Schale aus einem zweiten Metallsalz, das lumineszent
ist und Nicht-Halbleiter-Eigenschaften aufweist,
sowie
mit dem Verfahren zur im Folgenden dargelegten Herstellung gelöst worden.
-
Kurze Beschreibung
der Figuren
-
1 zeigt
die Fluoreszenzspektren homogener CePO4-Kernpartikel
und von CePO4/TbPO4-Kern/Schale-Partikeln
gemäß der vorliegenden
Erfindung.
-
2 zeigt
verschiedene Bilder, erhalten mit Energiefilter-Transmissionselektronenmikroskopie von
1 CePO4:Tb/LaPO4-Kern/Schale-Partikel.
-
3 zeigt
2 Fluoreszenzabfallkurven von CePO4/TbPO4-Kern/Schale-Partikeln gemäß der Erfindung, die modifiziert
bzw. chemisch an Fluorescein gekuppelt wurden. Als Bezugsvergleich
ist die Fluoreszenz-Abfallkurve von CePO4/TbPO4-Kern/Schale-Partikeln, die nicht an Fluorescein
gekuppelt wurden, ebenfalls dargestellt.
-
4 zeigt
die Fluoreszenzabfallkurven Fluorescein-gekuppelter, homogener LaPO4:Ce,Tb-Partikel (Vergleichsbeispiel 1).
-
5a zeigt
2 Fluoreszenzspektren, gemessen im Zeit-geschleusten (time gated
= TGF)-Modus, von CePO4/TbPO4-Kern/Schale-Partikeln
gemäß der Erfindung,
die nicht weiter modifiziert bzw. an Fluorescein gekuppelt worden
waren.
-
5b zeigt
1 Fluoreszenzspektrum, gemessen im Zeit-geschleusten Modus, homogener LaPO4:Ce,Tb-Nanopartikel (Vergleichsbeispiel
1) bei 520 bzw. 542 nm.
-
6a:
Homogener Kinase-Assay mit (F)RET-Partnern, gekuppelt an 1 Molekül
-
6b:
Homogener Immunoassay mit (F)RET-Partnern, gekuppelt an 1 Molekül
-
7:
Kompetitiver Immunassay mit (F)RET-Partnern, gekuppelt an 1 Molekül (Epitop)
-
8:
Homogener Sättigungsimmunoassay
mit (F)RET-Partnern, gekuppelt an getrennte Moleküle
-
9:
Homogener kompetitiver Immunoassay mit (F)RET-Partnern, gekuppelt
an getrennte Moleküle
-
10:
Homogener Assay mit (F)RET-Partnern, gekuppelt an 1 Molekül
-
11:
Assay gemäß einem
Verfahren molekularer Leuchtsignale
-
Detaillierte Beschreibung
der vorliegenden Erfindung
-
I. Lumineszente
Nanopartikel
-
Die
lumineszenten, insbesondere fotolumineszenten Partikel der vorliegenden
Erfindung umfassen (a) einen Kern aus einem ersten Metallsalz oder
-oxid, welcher von (b) einer lumineszenten Nicht-Halbleiter-Schale
aus einem zweiten Metallsalz umgeben ist.
-
"Lumineszenz" kennzeichnet die
Eigenschaft der beanspruchten Nanopartikel, Energie (z.B. in der Form
von Fotonen (IR, sichtbar, UV), Elektronenstrahlen, Röntgenstrahlen
usw.) zu absorbieren, welche dann als Licht niedrigerer Energie
wieder ausgestrahlt wird. Es sollte klar sein, dass der Begriff "lumineszent" über die Beschreibung und die
Ansprüche
hinweg auch die spezifischere und bevorzugtere Bedeutung "fotolumineszent" einschließt.
-
Als "Fotolumineszenz" wird die Befähigung der
anorganischen Metallsalze verstanden, Fotonen einer spezifischen
Energie (z.B. UV, sichtbares Licht) zu absorbieren und Licht niedrigerer
Energie (längerer
Wellenlänge,
z.B. UV, sichtbares Licht, IR) über
eine bestimmte Zeitdauer wieder auszustrahlen. Die Dauer der Lichtemission
kann der Lebensdauer des angeregten Zustands bis zu 10–7 oder
10–8 s,
was in typischer Weise als Fluoreszenz bezeichnet wird, aber auch
einer viel längeren
Zeit entsprechen. Für
Lanthanid-dotierte Salze, z.B. für
Sulfate, Phosphate oder Fluoride, werden in typischer Weise Zeiten
für die
Lebensdauer des angeregten Zustands in der Größenordnung von ms (z.B. von
1 bis 20 ms) beobachtet.
-
Gemäß der vorliegenden
Erfindung ist es bevorzugt, dass weder das Schalen- noch das Kernmaterial Halbleitereigenschaften
zeigen und ergeben.
-
Auch
sind sowohl Schale als auch Kern bevorzugt aus kristallinen Materialien
dargestellt. Dies lässt sich
durch Röntgen-Pulverbeugungsmuster
belegen.
-
Die
Formgestalt der beanspruchten Kern/Schale-Partikel kann z.B. nadelförmig, ellipsoid
oder kugelförmig
sein, wobei die letzteren beiden Optionen bevorzugt sind.
-
Die
beanspruchten Kern/Schale-Nanopartikel weisen vorzugsweise eine
entlang ihrer längsten
Achse gemessene Durchschnittsgröße von 1
bis 100 und bevorzugter von 1 bis 50 nm auf. Durchschnittsgrößen von maximal
30 nm, maximal 20 nm, maximal 10 nm, z.B. von 2 bis 8 oder 4 bis
6 nm sind sogar noch erwünschter. In
jedem Fall beträgt
die Standardabweichung bevorzugt weniger als 30% und insbesondere
weniger als 10%.
-
Die
Partikelgröße und -verteilung
können
gemäß Techniken,
die noch weiter in den bereits zitierten Artikeln von K. Riwotzki
et al. und M. Haase et al. beschrieben sind, z.B. mit Transmissionselektromikrografien (TEM)
gemessen werden. Gelpermeationschromatografie und Ultra-Zentrifugation ermöglichen
ebenfalls die Bestimmung der Größe.
-
Die
Dicke der Schale besteht bevorzugt aus mindestens 2 Monoschichten.
Die bevorzugte Obergrenze für
die Schalendicke beträgt
2 Durchmesser des Kerns (für
nicht-kugelige Partikel, gemessen entlang der längsten Achse) und bevorzugter
1 Kerndurchmesser, z.B. ca. 2/3 davon).
-
Gemäß einer
ersten Ausführungsform
der vorliegenden Erfindung sind der Kern (a) aus einem Metallsalz
oder -oxid, welcher keine Energie aus der Schale nach ihrer elektronischen
Anregung aufnimmt, insbesondere aus einem nicht-lumineszenten Metallsalz
oder -oxid, und (b) die Schale aus einem lumineszenten, insbesondere
dotierten Metallsalz hergestellt.
-
Über die
vorliegende Erfindung hinweg soll "Dotierung" in einem breiten Sinn verstanden sein.
Die Obergrenze des zu verwendenden Dotiermittels sollte niedrig
genug liegen, um die erzeugte Lumineszenz nicht durch Konzentrations-Löschphänomene zu
verringern. Dementsprechend hängt
die Obergrenze von Faktoren wie vom Typ des Dotierions und dem Abstand
zwischen den Dotier-Metallionen im Gitter ab, welche spezifisch
für jedes
Kernmaterial sind. Vorzugsweise ist das Wirtsmaterial durch das
Dotiermittel in einer Menge bis zu 50 und bevorzugt von 0,1 bis
45 mol-%, z.B. von 0,5 bis 40 oder von 1 bis 20 mol-% ersetzt.
-
Es
bestehen auch keine besonderen Einschränkungen bezüglich des Typs des Dotiermetalls,
das eingebaut wird, solange dieses befähigt ist, absorbierte Fotonen
in lumineszente Strahlung umzuwandeln. Somit sind beispielsweise
Metalle wie Ag, Cu, Co oder Mn (z.B. in Kombination mit Zink als
Wirtsmetall) verwendbar. Eine Dotierung mit Lanthanidmetallen ist
allerdings bevorzugt, da die Lumineszenz von Lanthanidmetallen insbesondere
von deren Gitterumgebung unabhängig
ist. Im Allgemeinen ist die Verwendung zwei- oder dreiwertiger Dotiermittel,
insbesondere solcher Lanthanid-Dotiermittel,
bevorzugt. Zweiwertige Lanthaniden (+II-Oxidationszustand) sind
durch eine relativ starke Absorption, aber relativ breite Emissionsbanden
gekennzeichnet. Aus diesem Grund können sie in geeigneter Weise
als Sensibilisierer verwendet werden, welcher die Energie auf andere
lumineszierende Metalle (z.B. auf Eu2+ bis
Mn2+) überträgt. Das
Vermögen
dreiwertiger Lanthaniden (Oxidationszustand: +III) zur Lichtemission
in der Form relativ scharfer Banden macht sie zu besonders attraktiven
Dotiermitteln zur Einzelverwendung, obwohl es, wie später noch
genauer erläutert
wird, geeignete Kombinationen dreiwertiger Lanthaniden als Dotiersysteme
ebenfalls gibt.
-
Geeignete
Dotiermaterialien für
die Schale schließen
Al, Cr, Tl, Mn, Ag, Cu, As, Nb, Ni, Ti, In, Sb, Ga, Si, Pb, Bi,
Zn und Co ein, die, in Abhängigkeit
vom eingesetzten Wirtsmaterial, Lumineszenzeigenschaften aufweisen,
was insbesondere für
Mn, Ag, Cu, Bi, Cr, Sn, Sb und vorzugsweise für die Lanthaniden und insbesondere
für Ce(58),
Pr(59), Nd(60), Sm(62), Eu(63), Gd(64), Tb(65), Dy(66), Ho(67),
Er(68), Tm(69) oder Yb(70) oder für Kombinationen davon zutrifft.
-
Eine
Dotierung mit Lanthanidmetall ist bevorzugt, da die Lumineszenz
der Lanthanidmetalle ganz besonders unabhängig von deren Gitterumgebung
ist.
-
Im
Hinblick auf die Praxis (den Typ der Fluoreszenz, die Intensität usw.)
zeigen und ergeben Ce, Tb, Eu, Nd, Dy, Tm, Sm, Gd, Ho, Er und Yb
die interessantesten Lumineszenzeigenschaften.
-
Er3+, Nd3+ und Ho3+ sind von besonderem Interesse für den Telekommunikationsbereich,
da sie von 1300 bis 1600 nm ausstrahlen. Ce wird bevorzugt in Kombination
mit einem weiteren Dotiermaterial, wie mit Nd, Dy oder Tb, verwendet.
Ce ist dafür
bekannt, UV-Strahlung mit einer Wellenlänge von 250 bis 300 nm stark zu
absorbieren, zeigt aber eine ziemlich breite Lumineszenzbande bei
ca. 330 nm, abhängig
vom Wirtsgitter (z.B. von Phosphat). Bei Verwendung in Kombination
mit weiteren Dotiermitteln, auf die die absorbierte Energie übertragen
werden kann, können
sehr wirkungsvolle Lumineszenzsysteme erzeugt werden. Eine weitere attraktive
Kombination von Dotiermetallen ist Yb und Er, welche von großer Bedeutung
in Er3+-dotierten optischen Verstärkern ist,
in denen Er3+ indirekt über Yb3+ gepumpt
wird, das einen um das 10-Fache höheren Absorptionsquerschnitt
und einen viel breiteren Peak bei 980 nm als Er3+ aufweist.
Nd3+ und Gd3+ sind
ebenfalls kombinierbar.
-
Wie
vorher bereits angegeben, ist es nicht nur möglich, diese Lanthanidmetallkombinationen
als Dotiermittel für
die Schale zu verwenden, sondern es ist gleichermaßen wirkungsvoll,
als Wirtsmetall diese Lanthanidmetallionen (z.B. Ce3+,
Yb3, Nd3+) mit dem
höheren
Absorptions querschnitt einzusetzen und einen Teil davon durch geringere
Mengen weiterer Metalle (z.B. von Tb3+,
Er3+, Gd3+) zu ersetzen.
Aus diesem Grund sind Lanthanidsalze (z.B. Salze von Ce3+,
Yb3+, Nd3+) ebenfalls
als das Wirtsmaterial der Schale verwendbar.
-
Für Anwendungen
in wässrigen
Medien wie für
biologische Assayverfahren sind die am meisten bevorzugten Dotiermittel
diejenigen (z.B. Tb, Dy, Tm, Sm), die eine Lumineszenz im sichtbaren
Bereich zeigen und ergeben, um Wechselwirkungen mit Wasser zu minimieren,
wodurch andernfalls das emittierte Licht absorbiert werden könnte.
-
Das
Wirtsmaterial für
die Schale ist nicht spezifisch eingeschränkt und kann aus bekannten
nicht-lumineszenten Metallsalzen, wie aus Sulfiden, Seleniden, Sulfoseleniden,
Oxisulfiden, Phosphaten, Halophosphaten, Arsenaten, Sulfaten, Boraten,
Aluminaten, Gallaten, Silikaten, Germanaten, Oxiden, Vanadaten,
Niobaten, Tantalaten, Woframaten, Molybdaten, Alkalihalogenaten,
weiteren Halogeniden, insbesondere aus Fluoriden, Phosphiden oder
aus Nitriden, ausgewählt
sein. Die Verwendung von Sulfaten, Phosphaten oder Fluoriden ist
besonders bevorzugt.
-
Die
Metalle dieser Salze gehören
bevorzugt zu den Hauptgruppen 1, 2, 13 oder 14, den Untergruppen 3,
4, 5, 6, 7 oder zu den Lanthaniden. Da die meisten lumineszenten
Dotiermittel zwei- oder dreiwertige Metallionen sind, ist es bevorzugt,
als Gegenion für
die Schale nicht-lumineszente zwei- oder dreiwertige Metallatome
wie die Metalle der Gruppe 2 (Erdalkalimetalle, wie Mg, Ca, Sr oder
Ba) oder der Gruppe 3 (Sc, V oder La) oder der Gruppe 13 (z.B. Al,
Ga oder In) oder Zn zu verwenden.
-
Bevorzugte
Ausgestaltungen der Wirtsmetallsalze umfassen:
Phosphate der
entsprechenden Zahl von Metallatomen (zur Gewährleistung der Ladungsneutralität), ausgewählt aus
der Hauptgruppe 2 (z.B. aus Mg, Ca, Sr, Ba), der Gruppe 3 (z.B.
aus Sc, Y, La) oder aus den Lanthaniden (aus den Elementen 58 bis
71, d.h. aus Ce, Pr, Nd, Pm, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb
und aus Lu);
Sulfate der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben);
Borate der entsprechenden
Zahl von Metallatomen, ausgewählt
aus Hauptgruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus
Sc, Y, La) oder der Gruppe 13 (Al, Ga, In, Tl) oder aus Lanthaniden
(wie oben);
Fluoride der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), der Untergruppe 3 (z.B.
aus Sc, Y, La) oder aus Lanthaniden (wie oben);
Aluminate (z.B.
Al5O12 oder AlO4) der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben);
Gallate (z.B. Ga5O12) der entsprechenden
Zahl von Metallatomen, ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben);
Silikate (z.B. SiO3 oder SiO4) der
entsprechenden Zahl von Metallatomen, ausgewählt aus Gruppe 2 (z.B. aus Mg,
Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y, La), Gruppe 12 (z.B. aus
Zn, Cd) oder aus Lanthaniden (wie oben);
Vanadate (z.B. VO4) der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben);
Wolfamate (z.B. WO4) der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben);
Molybdate (z.B. MoO4) der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben);
Tantalate (z.B. TaO4) der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben); oder
Arsenate (z.B. AsO4) der entsprechenden Zahl von Metallatomen,
ausgewählt
aus Gruppe 2 (z.B. aus Mg, Ca, Sr, Ba), Gruppe 3 (z.B. aus Sc, Y,
La) oder aus Lanthaniden (wie oben).
-
Bei
Auswahl eines geeigneten Wirtsmaterials für ein spezifisches Dotiermittel
ist ferner, wie dies im Stand der Technik bekannt ist, zu bedenken,
dass Wirts- und Dotiermetall vorzugsweise die gleiche Wertigkeit und ähnliche
(Toleranz: z.B. ±20%)
oder identische Ionendurchmesser aufweisen sollten. Gleichzeitig
wird in typischer Weise die Kompatibilität des Dotiermittels mit dem
Wirtsmetall erhöht,
falls diese dazu befähigt
sind, mit einem spezifischen Anion Kristalle des gleichen oder ähnlichen
Gittertyps mit der oder den gleichen oder ähnlichen Gitterkonstante(n)
(Toleranz: z.B. ±20%)
zu bilden.
-
Die
obigen Kriterien können
oft mit Ba und La als Wirtsmaterialmetall für den Kern erfüllt werden,
da diese Metalle Ionendurchmesser ergeben, die denen der zweiwertigen
(+II)-Lanthaniden sehr ähneln.
Aus dem gleichen Grund stellen La- und Y-Salze geeignete Wirtsmaterialien
für dreiwertige
(+III)-Lanthanid-Dotiermittel dar.
-
Spezifische
Beispiele lumineszenter Schalenmaterialien sind z.B.: LiJ:Eu; NaJ:Tl;
CsJ:Tl; CsJ:Na; LiF:Mg; LiF:Mg,Ti; LiF:Mg,Na; KMgF3:Mn;
BaFCl:Eu; BaFCl:Sm; BaFBr:Eu; BaFCl0,5Br0,5:Sm; BaY2F8:A (A = Pr, Tm, Er, Ce); BaSi2O5:Pb; BaMg2Al16O27:Eu; BaMgAl14O23:Eu; BaMgAl10O17:Eu; BaMgAl2O3:Eu; Ba2P2O7:Ti; (Ba,Zn,Mg)3Si2O7:Pb;
Ce(Mg,Ba)Al11O19;
Ce0,65Tb0,35MgAl11O19:Ce,Tb; MgAl11O19:Ce,Tb; MgF2:Mn; MgS:Eu; MgS:Ce; MgS:Sm; MgS:(Sm,Ce);
(Mg,Ca)S:Eu; MgSiO3:Mn; 3,5MgO × 0,5MgF2 × GeO2:Mn; MgWO4:Sm; MgWO4:Pb; 6MgO × As2O5:Mn; (Zn,Mg)F2:Mn;
(Zn4Be)SO4:Mn; Zn2SiO4:Mn; Zn2SiO4:Mn,As; Zn3(PO4)2:Mn; CdBO4:Mn; CaF2:Mn; CaF2:Dy; CaS:A (A = Lanthanid, Bi); (Ca,Sr)S:Bi;
CaWO4:Pb; CaWO4:Sm;
CaSO4:A (A = Mn, Lanthanid); 3Ca3(PO4)2 × Ca(F,Cl)2:Sb,Mn; CaSiO3:Mn,Pb; Ca2Al2Si2O7:Ce;
(Ca,Mg)SiO3:Ce; (Ca,Mg)SiO3:Ti;
2SrO × 6(B2O3) × SrF2:Eu; 3Sr3(PO4)2 × CaCl2:Eu; A3(PO4)2 × ACl2:Eu (A = Sr, Ca, Ba); (Sr,Mg)2P2O7:Eu; (Sr,Mg)3(PO4)2Sn;
SrS:Ce; SrS:Sm,Ce; SrS:Sm; SrS:Eu; SrS:Eu,Sm; SrS:Cu,Ag; Sr2P2O7:Sn;
Sr2P2O7:Eu;
Sr4Al14O25:Eu; SrGa2S4:A (A = Lanthanid, Pb); SrGa2S4:Pb; Sr3Gd2Si6O18:Pb,Mn; YF3:Yb,Er; YF3:Ln (Ln
= Lanthanid); YLiF4:Ln (Ln = Lanthanid);
Y3Al5O12:Ln
(Ln = Lanthanid); YAl3(BO4)3Nd,Yb; (Y,Ga)BO3:Eu;
(Y,Gd)BO3:Eu; Y2Al3Ga2O12:Tb;
Y2SiO5:Ln (Ln =
Lanthanid); Y2O2S:Ln
(Ln = Lanthanid); YVO4A (A = Lanthanid,
In); Y(P,V)O4:Eu; YTaO4:Nb;
YAlO3:A (A = Pr, Tm, Er, Ce); YOCl:Yb,Er;
LnPO4:Ce,Tb (Ln = Lanthanid oder Mischung
von Lanthaniden); LuVO4:Eu; GdVO4:Eu; Gd2O2S:Tb; GdMgB5O10:Ce,Tb; LaOBr:Tb; La2O2S:Tb; LaF3:Nd,Ce;
BaYb2F8:Eu; NaYF4:Yb,Er; NaGdF4:Yb,Er;
NaLaF4:Yb,Er; LaF3:Yb,Er,Tm; BaYF5:Yb,Er; GaN:A (A = Pr, Eu, Er, Tm); Bi4Ge3O12;
LiNbO3:Nd,Yb; LiNbO3:Er;
LiCaAlF6:Ce; LiSrAlF6:Ce; LiLuF4:A (A = Pr, Tm, Er, Ce); Li2B4O7:Mn; Y2O2Eu; Y2SiO5:Eu; CaSiO3:Ln,
worin Ln = 1, 2 oder mehr Lanthaniden.
-
Bei
Klassifizierung gemäß dem Wirts-Gittertyp
können
die folgenden bevorzugten Ausgestaltungen ebenfalls aufgezählt werden:
- 1. Halogenide: z.B. XY2 (X
= Mg, Ca, Sr, Ba; Y = F, Cl, J); CaF2:Eu(II);
BaF2:Eu; BaMgF4:Eu;
LiBaF3Eu; SrF2Eu;
SrBaF2Eu; CaBr2Eu-SiO2; CaCJ2:Eu; CaCJ2:Eu-SiO2; CaCJ2:Eu,Mn-SiO2; CaJ2:Eu; CaJ2:Eu,Mn; KMgF3:Eu; SrF2:Eu(II);
BaF2:Eu(II); YF3;
NaYF4; MgF2:Mn;
MgF2:Ln (Ln = Lanthanid(en)).
- 2. Erdalkalisulfate: z.B. XSO4 (X =
Mg, Ca, Sr, Ba); SrSO4:Eu; SrSO4:Eu,Mn;
BaSO4:Eu; BaSO4:Eu,Mn; CaSO4; CaSO4:Eu; CaSO4:Eu,Mn; sowie gemischte Erdalkalisulfate,
auch in Kombination mit Magnesium, z.B. Ca,MgSO4:Eu,Mn.
- 3. Phosphate und Halophosphate: z.B. CaPO4:Ce,Mn;
Ca5(PO4)3Cl:Ce,Mn; Ca5(PO4)3F:Ce,Mn; SrPO4:Ce,Mn; Sr5(PO4)3Cl:Ce,Mn; Sr5(PO4)3F:Ce,Mn;
die letzteren auch codotiert mit Eu(II) oder codotiert mit Eu,Mn; α-Ca3(PO4)2:Eu; β-Ca3(PO4)2:Eu,Mn;
Ca5(PO4)3Cl:Eu; Sr5(PO4)3Cl:Eu; Ba10(PO4)6Cl:Eu; Ba10(PO4)6Cl:Eu,Mn;
Ca2Ba3(PO4)3Cl:Eu; Ca5(PO4)3F:Eu2+X3+; Sr5(PO4)3F:Eu2+X3+ (X = Nd, Er,
Ho, Tb); Ba5(PO4)3Cl:Eu; β-Ca3(PO4)2:Eu;
CaB2P2O9:Eu;
CaB2P2O9:Eu;
Ca2P2O7:Eu;
Ca2P2O7:Eu,Mn; Sr10(PO4)6Cl2:Eu; (Sr,Ca,Ba,Mg)10(PO4)6Cl2:Eu;
LaPO4:Ce; CePO4;
LaPO4:Eu; LaPO4:Ce;
LaPO4:Ce,Tb; CePO4:Tb.
- 4. Borate: z.B. LaBO3; LaBO3:Ce;
ScBO3:Ce; YAlBO3:Ce;
YBO3:Ce; Ca2B5O9Cl:Eu; xEuO × yNa2O × zB2O3.
- 5. Vanadate: z.B. YVO4; YVO4:Eu;
YVO4:Dy; YVO4:Sm;
YVO4:Bi; YVO4:Bi,Eu;
YVO4:Bi,Dy; YVO4:Bi,Sm; YVO4:Tm; YVO4Bi,Tm;
GdVO4; GdVO4:Eu;
GdVO4:Dy; GdVO4:Sm;
GdVO4:Bi; GdVO4:Bi,Eu;
GdVO4:Bi,Dy; GdVO4:Bi,Sm;
YVO4:Eu; YVO4:Sm;
YVO4:Dy.
- 6. Aluminate: z.B. MgAl2O4:Eu;
CaAl2O4:Eu; SrAl2O4Eu; BaAl2O4:Eu; LaMgAl11O19:Eu; BaMgAl10O17:Eu; BaMgAl10O17:Eu,Mn; CaAl12O19:Eu; SrAl12O19:Eu; SrMgAl10O17:Eu; Ba(Al2O3)6:Eu;
(Ba,Sr)MgAl10O17:Eu,Mn; CaAl2O4:Eu,Nd; SrAl2O4Eu,Dy; Sr4Al14O25:Eu,Dy.
- 7. Silikate: z.B. BaSrMgSi2O7:Eu; Ba2MgSiO7:Eu; BaMg2Si2O7:Eu; CaMgSi2O6:Eu; SrBaSiO4:Eu; Sr2Si3O8 × SrCl2:Eu; Ba5SiO4Br6:Eu; Ba5SiO4Cl6:Eu;
Ca2MgSi2O7:Eu; CaAl2Si2O8:Eu; Ca1,5Sr0,5MgSi2O7:Eu; (Ca,Sr)2MgSi2O7:Eu;
Sr2LiSiO4F:Eu.
- 8. Wolframate und Molybdate: z.B. X3WO6 (X = Mg, Ca, Sr, Ba); X2WO4 (X = Li, Na, K, Rb, Cs); XMoO4 (X =
Mg, Ca, Sr, Ba); sowie Polymolybdate oder Polywolframate oder die
Salze der entsprechenden Hetero- oder Isopolysäuren.
- 9. Germanate: z.B. Zn2GeO4.
- 10. Außerdem
die folgenden Klassen: ALnO2:Yb,Er (A =
Li, Na; Ln = Gd, Y, Lu); LnAO4:Yb,Er (Ln
= La, Y; A = P, V, As, Nb); Ca3Al2Ge3O12:Er;
Gd2O2S:Yb,Er; La2S:Yb,Er.
-
Gemäß der ersten
Ausgestaltung der vorliegenden Erfindung nehmen das Kernmaterial,
d.h. ein Metallsalz oder -oxid, keinen Energietransfer aus der lumineszenten
Schale in ihrem elektronischen Zustand auf.
-
Diese
Bedingung lässt
sich immer mit Kern-Metallsalzen oder -oxiden erfüllen, die
nur elektronische Zustände
aufweisen, worin der energetische Abstand zwischen dem elektronischen
Grundzustand und dem ersten elektronisch angeregten Zustand größer als
der Abstand zwischen dem ersten elektronisch angeregten Zustand
der ausgewählten
lumineszenten Schale und deren Grundzustand ist. Unter diesen Umständen kann die
Energie (z.B. UV, sichtbares Licht, IR), die von der Schale absorbiert
wird, nicht auf die Kern-Metallatome oder -anionen übertragen
werden. Die Lokalisierung der in der Schale erstellten Energie steigert
dadurch Oberflächenlöschphänomene und
soll die Gesamt-(F)RET-Effizienz der Partikel erhöhen. Gemäß einer
bevorzugten Ausgestaltung sind das Kernsalz oder -oxid nicht-lumineszent,
weshalb ihnen Absorptionsbanden (UV-vis oder IR) fehlen, auf die
die Energie aus der angeregten Schale übertragen werden könnte. Da
nicht-lumineszente
Materialien billiger als lumineszente Materialien sind, ist dies
auch wirtschaftlich von Vorteil.
-
Vorzugsweise
entspricht das Kernmaterial dem Wirtsmaterial der dotierten Schale.
-
Geeignete
den Kern bildende Anionen sind somit die gleichen wie die oben angegebenen
und beinhalten, ohne darauf eingeschränkt zu werden, Phosphate, Halophosphate,
Arsenate, Sulfate, Borate, Aluminate, Gallate, Silikate, Germanate,
Oxide, Vanadate, Niobate, Tantalate, Wolframate, Molybdate, Alkalihalogenate,
weitere Halogenide oder Nitride. Nanoteilchenförmige Metallsalze dieses Typs
sind in PCT/DE 01/03 433 offenbart.
-
Die
einzigen Kriterien, die die Auswahl der Kern-Metallatome bestimmen,
ist deren fehlende Befähigung,
Lumineszenz aus der Schale nach Bestrahlung mit Fotonen aufzunehmen.
Bevorzugte Metallionen, die zu diesem Zweck verwendet werden können, sind
die gleichen wie die oben für
das Wirtsmaterial der Schale genannten. Sie schließen, ohne
darauf eingeschränkt
zu sein, Metalle der Gruppe 2 (Erdalkalimetalle wie Mg, Ca, Sr oder
Ba), Metalle der Gruppe 3 (wie Sc, Y oder La), Zink oder Metalle
der Gruppe 13 (wie Al, Ga oder In) ein. Zur Steigerung der Eignung
des Schalenmaterials zum Wachstum auf der Oberfläche des Kernmaterials ist es
ferner bevorzugt, aber nicht absolut notwendig, als Kernmaterial
die gleichen Salze auszuwählen, die
den Wirt der dotierten Schale darstellen. Wird diese Bedingung nicht
erfüllt,
ist es bevorzugt, dass das Wirtsmaterial des Kerns und das Schalenmaterial
zum gleichen Gittertyp gehören
und sehr ähnliche
(Toleranz: z.B. ±20%)
oder identische Gitterkonstanten aufweisen.
-
Gemäß einer
zweiten Ausgestaltung umfasst (a) der Kern ein erstes Metallsalz
oder -oxid ("Donor"), die nach Anregung
befähigt
sind, die Anregungsenergie auf (b) ein zweites Schalen-bildendes
lumineszentes Metallsalz ("Akzeptor") zu übertragen,
das dann diese Energie als Lumineszenz ausstrahlt.
-
Geeignete
Donor-Akzeptor-Metallkombinationen können z.B. unter den oben identifizierten
Dotiermitteln und insbesondere aus Lanthaniden ausgewählt sein
und im Allgemeinen einen Abstand zwischen dem elektronischen Grundzustand
und dem ersten angeregten Zustand des Donormetalls benötigen, welcher
eine höhere
Energie als den entsprechenden Abstand des Akzeptormetalls beinhaltet.
-
Beispiele
für geeignete
Fotonenenergieabsorber (Donoren), die als Kernmaterialien der zweiten
Ausgestaltung der Erfindung verwendet werden können, sind Lanthanidionen mit
relativ hohen Absorptionsquerschnitten wie Ce3+,
Yb3+, Nd3+ oder
Eu2+. Ce3+ wird
bevorzugt in Kombination mit Tb3+ Dy3+ oder mit Nd3+ als Schalenmaterialmetall
und Akzeptor, z.B. in der Form der entsprechenden Sulfate, Phosphate
oder Fluoride, verwendet.
-
Yb3+-Salze, wie die Phosphate, Sulfate oder
Fluoride, werden bevorzugt als Kernmaterial mit Er3+-Salzen,
wie den Sulfaten, Phosphaten bzw. Fluoriden, als Schalenmaterial
kombiniert. Dadurch wird es ermöglicht,
Er3+ indirekt über Yb3+ zu
pumpen.
-
Bezüglich des
Schalenaufbaus können
die Akzeptoratome als hochkonzentrierte Dotiermaterialien der im
Zusammenhang der ersten Ausgestaltung der vorliegenden Erfindung
beschriebenen Wirtsmaterialien verwendet werden. Allerdings ist
es ebenfalls möglich,
dass die gesamte Schale aus dem entsprechenden Akzeptorsalz, z.B.
dem Metallsulfat, -phosphat oder -fluorid, besteht, um den Wirkungsgrad
des Energietransfers aus dem Kern zur Schale zu steigern.
-
Das
Kernmaterial der zweiten Ausgestaltung kann das Donormetall als
hochkonzentriertes Dotiermittel eines oben beschriebenen Wirtsmaterials
umfassen. Alternativ dazu und bevorzugt, besteht der Kern aus dem
entsprechenden Donormetallsalz.
-
Das
Anion des Kernsalzes kann frei unter kompatiblen Anionen ausgewählt werden,
die das Wachstum des ausgewählten
Schalenmaterials ermöglichen.
Beispiele geeigneter Anionen sind die für die erste Ausgestaltung angegeben,
wobei Sulfat, Phosphat oder Fluorid bevorzugt sind.
-
Ein
besonders bevorzugtes Beispiel für
die sogenannte zweite Ausgestaltung betrifft CePO4/TbPO4-Kern/Schale-Partikel.
-
Gemäß der zweiten
Ausgestaltung ist es ebenfalls möglich,
Vanadate, Molybdate, Wolframate oder Germanate als Kernmaterialien
(den Donor) zu verwenden, da die entsprechenden Anionen ebenfalls
befähigt sind,
Energie zu absorbieren und diese auf ein geeignetes Schalenmaterial
(den Akzeptor) zu übertragen,
welche dann die Energie als Lumineszenz wieder ausstrahlen. Diese
können
auch mit Dotiermetallen kombiniert sein, die ihrerseits als Lumineszenzzentren
wirken und somit die Lumineszenz wie Bi3+ und/oder
Eu3+ für
Vanadate verstärken.
Der Kern kann z.B. Vanadate, Molybdate, Wolframate oder Germanate
von Metallen der Gruppe 3 (wie von Sc, Y oder La) oder Metalle der
Gruppe 13 (wie von Al, Ga oder In) umfassen oder daraus bestehen.
Er wird bevorzugt mit Lanthanidsalzen, vorzugsweise mit Phosphaten,
Vanadaten, Molybdaten, Wolframaten oder mit Germanaten, als Schalenmaterial
kombiniert, worin das Lanthanid als Energieakzeptor wirkt. Spezifische
Beispiele schließen
Kern/Schale-Kombinationen des Typs LaVO4/EuPO4, LaVO4/NdPO4 oder YVO4/DyPO4 ein.
-
II. Synthese von Kern/Schale-Nanopartikeln
-
Die
oben beschriebenen Kern/Schale-Nanopartikel der vorliegenden Erfindung
werden in einem unten und in den Ansprüchen dargelegten Verfahren
synthetisiert, welches mindestens die folgenden zwei Stufen umfasst,
in denen man:
- 1. eine sogenannte "erste Mischung" aus Nanopartikeln
eines ersten Metallsalzes oder -oxids, z.B. aus Metallsulfat-, -phosphat-
oder -fluorid-Nanopartikeln
(Kernen), in einem organischen Medium zubereitet und man
- 2. die genannte erste Mischung, eine Anionquelle für die zu
bildende Schale, insbesondere eine Phosphat-, Sulfat- oder Fluoridquelle,
mit einer "zweiten
Mischung" aus Schale-bildenden
Metallionen und einem organischen Komplexiermittel für die genannten
Metallionen bei einer Temperatur von 50 bis 350°C umsetzt, bis die Schale um
die genannten Nanopartikelkerne herum gebildet worden ist.
-
II.1 Erste Verfahrensstufe
und Synthese der Nanopartikel
-
Die
Nanopartikel, die als Kernmaterial vorgesehen und in der sogenannten "ersten Mischung" vorhanden sind,
können
gemäß Verfahren
synthetisiert werden, die im Stand der Technik bekannt sind.
-
Im
Allgemeinen sind Nass-Syntheseverfahren gegenüber trockenen Bildungsverfahren
bevorzugt, da die ersteren eine bessere Steuerung der Partikelgrößen ermöglichen.
Ferner kann die Aggregation der gebildeten Nanopartikel leichter
in Nass-Syntheseverfahren unterdrückt werden.
-
Unter
den bekannten Nass-Syntheseverfahren sind z.B. Sol-Gel-Verfahren, hydrothermische
Synthesen oder organische Synthesen mit Komplexiermitteln, die das
Kristallwachstum regulieren, anwendbar. Ferner ist es möglich, ganz
spezifisch die Fluoride in einem Syntheseverfahren zu erzeugen,
beschrieben im bereits erwähnten
Artikel von J. W. Stouwdam und F. C. J. M. Van Veggel. Demgemäß sind LaF3-Nanopartikel und weitere Fluoride durch
Erhitzen einer Lösung
von Ammoniumdi-n-octadecyldithiophosphat und NaF in Ethanol/Wasser
herstellbar. Anschließend
werden Lösungen
der entsprechenden Metallnitrate in Wasser zugetropft, worauf die
Lösung
2 h lang bei 75°C
gerührt
und dann auf Raumtemperatur abgekühlt wird. Der Nachteil dieses
Verfahrens besteht allerdings darin, dass die erzeugten Partikel
noch eine relativ breite Partikelgrößenverteilung ergeben, was
weitere Reinigungsstufen durch Zentrifugation erforderlich macht.
-
Die "hydrothermische Synthese" Lanthanid-dotierter
Phosphate ist z.B. beschrieben in "Wet-chemical synthesis of doped colloidal
nanomaterials: particles and fibres of LaPO4:Eu,
LaPO4:Ce and LaPO4:Ce,Tb" von H. Meyssamy
et al., Advanced Materials (1999), Band 11, Nr. 10, Seiten 840 ff.
Als Ausgangsmaterialien für Sulfat-,
Phosphat- oder Fluorid-Nanopartikel werden vorzugsweise Metallchloride,
-nitrate oder -acetate verwendet. Die Reaktion wird in Wasser als
Reaktionsmedium in einem Autoklav durchgeführt, um hohe Drücke, vorzugsweise
Drücke
von 10 bis 20 bar, während
der Reaktion einzuhalten.
-
Die
hydrothermische Synthese führt
zu relativ großen
Partikeln, die nadelförmig
sind. Ferner kennzeichnet eine relativ breite Verteilung der Partikelgröße in typischer
Weise das Produkt. Im oben genannten Verfahren von H. Meyssamy et
al. beträgt
der Prozentsatz von Nanopartikeln mit Durchmessern von weniger als
25 nm z.B. lediglich ca. 20%. Diese können durch anschließende Zentrifugationsstufen
isoliert werden.
-
Weitere
Beispiele für
die hydrothermische Synthese sind in PCT/DE 01/03 433 zu finden.
Dieses Dokument offenbart, auf allgemeinerer Ebene und anhand konkreter
Beispiele, die Synthese nanoteilchenförmiger Silikate, Vanadate,
Wolframate, Molybdate, Tantalate usw. in Wasser unter hohen Drücken (im
Autoklav). Ferner bezieht sich dieses Dokument auf eine verwandte
Technik für
die Synthese von Aluminaten oder Gallaten in 1,6-Hexandiol (darin
auch bezeichnet als "glycothermische
Snythese").
-
Ferner
ist es möglich,
gegebenenfalls dotierte Sulfate unter Umgebungsdruck in organischen
Medien, ausgewählt
aus Polyolen und Sulfoxiden, zu erzeugen, welche das Kristallwachstum
durch Metall-Komplexieraktivität regulieren
sollen. Dieses Verfahren wird im Folgenden als "Polyol- oder Sulfoxid-Synthese" bezeichnet.
-
Die
einzusetzenden Polyole weisen vorzugsweise 2 oder 3 Hydroxygruppen
auf, und es können
als Beispiele Glycerin, Ethylenglykol oder Polyethylenglykol genannt
werden, wobei Polyethylenglykol mit niedrigem Molekulargewicht (bevorzugte
Durchschnittszahl der Ethylenglykol-Einheiten bis zu 4) vorzugsweise verwendet
wird. Als Sulfoxid kann Dimethylsulfoxid (DMSO) verwendet werden.
Dieses Syntheseverfahren wird bevorzugt zur Herstellung von Erdalkalimetallsulfaten,
wie von Magnesium-, Calcium-, Strontrium- oder Bariumsulfat, als
dotiertes Wirtsmaterial angewandt.
-
Bevorzugte
Metallatomquellen sind die entsprechenden Chloride und deren Hydrate.
Als Ausgangsmaterial für
das Sulfat werden bevorzugt Alkalimetallsulfate, Ammoniumsulfate
oder Sulfate mit einem organischen Kation herangezogen. Die entsprechenden
Hydrogensulfate sind gleichermaßen
geeignet.
-
Das
organische Kation ist bevorzugt aus basischen N-haltigen aliphatischen,
aromatischen und aliphatisch/aromatischen Substanzen ausgewählt, welche
vorzugsweise 4 bis 30 und bevorzugter 4 bis 20 Kohlenstoffatome
aufweisen. Geeignete Kationen sind z.B.:
- – quartäres Ammonium
oder Phosphonium, worin die 4 Substituenten unabhängig ausgewählt sein
können aus
Alkyl mit vorzugsweise 1 bis 10 Kohlenstoffatomen (bevorzugter mit
1 bis 5) oder aus Benzyl, oder
- – protonierte
aromatische Basen, wie von Hydrazin, Amantadin, Pyridin oder von
Collidin.
-
Ganz
entsprechend können
Sulfat-Nanopartikel aus Ausgangsmaterialien wie aus Tetrabutylammoniumhydrogensulfat,
Tetramethylammoniumsulfat, Bistetrabutylammoniumsulfat oder aus
Triethylammoniumhydrogensulfat hergestellt werden. Weitere geeignete
Ausgangsmaterialien sind Ammoniumhydrogensulfat, Ammoniumsulfat,
Alkalimetallhydrogensulfate, Amantadinsulfate, Ethylendiammoniumsulfat
und Hydraziniumsulfat.
-
Zur
Dotierung des Sulfat-Wirtsmaterials sind Nitrate oder Halogenide
des entsprechenden Dotiermittels und insbesondere des entsprechenden
Metallchlorids verwendbar.
-
Sind
Hydrogensulfate im Ausgangsmaterial enthalten, werden organische
Basen wie Imidazol bevorzugt als Säure-Fänger zum Reaktionsmedium gegeben.
Die Reaktion wird vorzugsweise bei einer Temperatur von 50 bis 240°C durchgeführt, wobei
der niedrigere Temperaturbereich von 50 bis 100°C für Glycerin bevorzugt ist und
höhere
Temperaturen im Bereich von 160 bis 240 und insbesondere von 160
bis 180°C
für die anderen
Polyol- oder Sulfoxid-Lösungsmittel
am meisten geeignet sind. Die erhaltenen Partikel weisen Durchschnittsdurchmesser
in der Größenordnung
von 0,2 bis 50 nm auf und sind gut in wässrigen Medien dispergierbar.
-
Nanopartikelkerne,
erhalten mit Sol-Gel-Verfahren, hydrothermischen Synthesen, glycothermischen Synthesen
oder mit der sogenannten "Polyol- oder Sulfoxid-Synthese", sind manchmal nicht
im organischen Medium, das in der ersten Stufe des beanspruchten
Verfahrens verwendet wird, dispergierbar, besonders wenn sich das
Reaktionsmedium für
den Kern bzw. das Verfahren der Erfindung (die Schale-Synthese)
deutlich bezüglich
der Polarität
unterscheiden. Aus diesem Grund kann es notwendig werden, die Nanopartikel
einer Nachbehandlung mit einer geeigneten polaren organischen Verbindung
zur Steigerung ihrer Dispergierbarkeit zu unterziehen. Vorzugsweise
wird diese Nachbehandlung mit dem und im gleichen organischen Medium
(Komplexiermittel), welche in der Schale-Synthese zur Anwendung
gelangen, oder mit und in organischen Medien ähnlicher Polarität durchgeführt.
-
Wird
z.B. die Schale-Synthese in N- oder P-haltigen Medien durchgeführt, kann
die Nachbehandlung in geeigneter Weise beinhalten, dass man die
Partikel, erhalten in Sol-Gel-Verfahren, glyco- oder hydrothermischen
Synthesen oder in der sogenannten "Polyol- oder Sulfoxid-Synthese" einer Nachbehandlung
mit den N- oder P-haltigen Medien unterzieht.
-
In
dieser Nachbehandlung werden die Nanopartikel in der entsprechenden
organischen Verbindung erhitzt. Dies hat den Effekt, dass Wasser
oder weitere hydrophile Reste, die an der Oberfläche der Nanopartikel gebunden
werden, durch die polare organische Verbindung ersetzt werden. Aus
den oben genannten Gründen
wird die polare organische Verbindung bevorzugt aus N- oder P-haltigen
Komplexiermitteln für
Metallionen ausgewählt,
wie dies weiter unten im Zusammenhang der "organischen Synthese" und der zweiten Verfahrensstufe beschrieben
wird. Allerdings können
auch weitere funktionalisierte polare organische Verbindungen verwendet
werden.
-
Diese
Nachbehandlung ist für
in der "Polyol-
oder Sulfoxid"-Synthese
erzeugte Sulfate nicht erforderlich, falls die anschließenden Herstellstufen
in Polyolen und/oder Sulfoxiden durchgeführt werden.
-
Gemäß einem
weiteren und bevorzugten Verfahren, das nachfolgend als "organische Synthese" bezeichnet ist,
umfasst das Verfahren zur Herstellung der Nanopartikelkerne Stufen,
in denen man:
- a) in einem organischen Reaktionsmedium
aus mindestens einem Metall-Komplexiermittel und gegebenenfalls
mindestens einem weiteren Lösungsmittel
eine im Reaktionsmedium lösliche
oder dispergierbare Metallquelle mit einer im Reaktionsmedium löslichen
oder dispergierbaren Anionquelle, insbesondere mit einer Phosphat-,
Sulfat- oder Fluoridquelle, umsetzt,
- b) gegebenenfalls das Reaktionsmedium aus dem dabei gebildeten
nanoteilchenförmigen
Metallsalz (z.B. aus dem Phosphat, Sulfat oder Fluorid) entfernt
und man
- c) gegebenenfalls das nanoteilchenförmige Salz gewinnt.
-
Als "organisches Medium" werden organische
Lösungsmittel
verstanden, die, neben unvermeidbaren Spuren, kein Wasser enthalten.
Der Siedepunkt des organischen Medium liegt vorzugsweise höher als
die unten angegebenen Reaktionstemperaturen. Er beträgt z.B.
150 bis 400, vorzugsweise mehr als 180 und insbesondere mehr als
210°C (bei
Umgebungsdruck).
-
Abhängig von
der Empfindlichkeit der Metallquelle gegenüber einer Oxidation ist es
bevorzugt, die Reaktion unter Inertgas wie unter Stickstoff oder
Argon durchzuführen.
-
Bezüglich des
Reinheitsgrades der Ausgangsmaterialien ist es empfehlenswert, Metallsalze
mit einer Reinheit von mindestens 99,9% zu verwenden. Alle Reaktionsteilnehmer
und die Lösungsmittel
werden vorzugsweise wasserfrei verwendet und/oder vor Gebrauch getrocknet.
Allerdings sollten Metallchloride, die häufig als Hydrate eingesetzt
werden, bevorzugt keinen längeren
Trocknungsprozeduren unterzogen werden, da dies die Bildung von
im Reaktionsmedium unlöslichen
Oxichloriden begünstigen
kann.
-
Die
Reaktion wird vorzugsweise bei Temperaturen von 50 bis 350°C, z.B. von
120 bis 320 und insbesondere von 180 bis 190°C, durchgeführt. Die geeignete Temperatur
kann der Durchschnittsfachmann ganz leicht und einfach durch Verfolgung
der Reaktion der Reaktionsteilnehmer bei graduell steigenden Temperaturen
ermitteln, um dadurch die Syntheseminimaltemperatur zu bestimmen,
bei der die Reaktion mit hinreichender Geschwindigkeit fortschreitet.
Für diesen
Zweck können
die Nanopartikel z.B. aus Proben des Reaktionsmedium ausgefällt werden,
wodurch es ermöglicht
wird, das Partikelwachstum bei steigenden Reaktionszeiten zu untersuchen.
-
Geeignete
Reaktionszeiten sind in gleicher Weise bestimmbar und liegen vorzugsweise
im Bereich von 10 min bis 48 h und insbesondere von 30 min bis 20
h.
-
Nach
Beendigung der Reaktion kann die Reaktionsmischung auf Raumtemperatur
abgekühlt
werden. Sind die Nanopartikel während
der Reaktion oder nach der Abkühlung
noch nicht vollständig
ausgefällt
worden, ist es möglich,
Methanol zum Reaktionsmedium oder umgekehrt zum Erhalt maximaler
Ausbeuten zu geben.
-
Ohne
an eine Theorie gebunden zu sein, wird davon ausgegangen, dass das
in der "organischen
Synthese" verwendete
Metall-Komplexiermittel an Oberfläche-Metallatome der gebildeten
Nanopartikel koordinert und dadurch deren Wachstum nach der Reaktion
der Ausgangsmaterialien beendet werden. Es wird davon ausgegangen,
dass das Metall-Komplexiermittel an der Partikeloberfläche gebunden
bleibt und auf diese Weise Agglomerations- und Austauschprozesse
zwischen den Partikeln wie eine Oswald-Reifung verhindert oder verringert
werden. Das Verfahren gemäß der "organischen Synthese" führt somit
zu ziemlich kleinen Partikeln, in denen der an der längsten Achse
gemessene Durchschnittsdurchmesser vorzugsweise 1 bis 10 und insbesondere
2 bis 8 nm, z.B. 4 bis 6 nm, mit engen Größenverteilungen (Standardabweichung < 30%, insbesondere < 10%) beträgt. Das
Metall-Komplexiermittel
ist durch das Vorliegen einer polaren Gruppe mit der Befähigung zur
Koordination des Metallions und mindestens eines zweiten Molekül-Teils
(eines weniger polaren, vorzugsweise hydrophoben Teils), z.B. eines
aliphatischen, aromatisch/aliphatischen oder eines rein aromatischen
Molekül-Teils
mit bevorzugt 4 bis 20 und insbesondere 6 bis 14 Kohlenstoffatomen,
gekennzeichnet.
-
Das
Metall-Komplexiermittel ist vorzugsweise eine phosphororganische
Verbindung oder ein mono- oder disubstituiertes Amin.
-
Unter
den letzteren sind die am meisten bevorzugten Ausgestaltungen Mono-
oder Dialkylamine, in denen der Alkylrest vorzugsweise 4 bis 20
und insbesondere 6 bis 14 Kohlenstoffatome, wie Dodecylamin oder Bis(ethylhexyl)amin,
aufweist.
-
Bezüglich der
phosphororganischen Verbindungen, ist es bevorzugt, mindestens eine
der folgenden Substanzen zu verwenden:
- a) Ester
der Phosphinsäure:
- b) Diester der Phosphonsäure:
- c) Triester der Phosphorsäure,
am meisten bevorzugt Trialkylphosphate wie Tributylphosphat oder Tris(ethylhexyl)phosphat:
- d) Trialkylphosphine, wie Trioctylphosphin (TOP): oder
- e) Trialkylphosphinoxide, wie Trioctylphosphinoxid (TOPO):
worin R1, R2 und R3 unabhängig
aus verzweigten oder linearen aliphatischen (bevorzugt Alkyl), aliphatisch/aromatischen
oder aromatischen Resten mit 4 bis 20, bevorzugter 4 bis 14 und
insbesondere mit 4 bis 10 Kohlenstoffatomen, ausgewählt sind.
Die aromatischen Reste können
z.B. Phenyl und die aliphatisch/aromatischen Reste z.B. Tolyl, Xylyl
oder Benzyl sein.
-
Die
Verwendung der phosphororganischen Verbindungen (a) bis (c) und
(e), insbesondere (a) bis (c), ist besonders bevorzugt.
-
Das
Metall-Komplexiermittel kann das einzige Lösungsmittel im organischen
Reaktionsmedium sein. Es wird bevorzugt in einer Menge von mindestens
10 mol, bezogen auf die molare Menge des oder der als Metallquelle
verwendeten Metallatom(e), eingesetzt, wenn es das einzige Lösungsmittel
darstellt. Die bevorzugte Obergrenze beträgt annähernd 1000 mol.
-
Abhängig von
der Wahl des Metall-Komplexiermittels und insbesondere von der Länge des
hydrophoben Molekül-Teils,
kann die Verwendung größerer Mengen
nachteilig sein, da dies eine vollständige Ausfällung der gebildeten Nanopartikel
beeinträchtigen
kann.
-
Daher
ist es bevorzugt, zusätzlich "mindestens ein weiteres
Lösungsmittel" einzusetzen. In
dieser Ausgestaltung wird das Metall-Komplexiermittel (das "erste Lösungsmittel") bevorzugt in einer
molaren Menge von weniger als 10 mol und bevorzugter von 0,9 bis
6 mol eingesetzt, bezogen auf 1 mol Metallionen (verwendet als Metallquelle).
Die Menge des bzw. der "weiteren
Lösungsmittel(s)" beträgt vorzugsweise
5 bis 100 mol, bezogen auf 1 mol Metallatome (verwendet als Metallquelle).
-
Das
oder die "weitere(n)
Lösungsmittel" sollten mit dem
Metall-Komplexiermittel
mischbar sein und einen Siedepunkt oberhalb der Syntheseminimaltemperatur,
vorzugsweise oberhalb 150, bevorzugter oberhalb 180 und am meisten
bevorzugt oberhalb 210°C
aufweisen. Siedepunkte oberhalb 400°C können unerwünscht sein.
-
Das
oder die "weitere(n)
Lösungsmittel" kann/können eines
auf Basis von Kohlenwasserstoffen sein oder mindestens eine polare
Gruppe aufweisen. Die Verwendung der letzteren ist bevorzugt, falls
Kristallisationswasser in den Metallsalz-Ausgangsmaterialien vorhanden
ist und das genannte Wasser durch ein Lösungsmittel ersetzt werden
soll, das zur Koordination an das Metall befähigt ist. Das oder die "weitere(n) Lösungsmittel" ist (sind) vorzugsweise
ausgewählt
aus:
- – Lösungsmitteln
mit mindestens einer Ether-Funktionalität, insbesondere aus Dialkylether
mit 5 bis 10 Kohlenstoffatomen pro Alkylgruppe, wie aus Dipentyl-,
Dihexyl-, Diheptyl-, Dioctyl- oder aus Diisoamylether, Diaryl- oder
Diaralkylethern mit insbesondere 12 bis 18 Kohlenstoffatomen, wie
aus Diphenyl- oder Dibenzylether, aus Mono- oder Polyethylenglykol(PEG)-dialkylethern
(worin jedes Alkyl vorzugsweise 1 bis 4 Kohlenstoffatome aufweist
und die Durchschnittszahl der PEG-Einheiten vorzugsweise 4 bis 10
beträgt),
wie aus Diethylenglykoldibutylether, Triethylenglykoldibutylether
und/oder aus Tetraethylenglykoldimethylether;
- – verzweigten
oder unverzweigten Alkanen, die vorzugsweise 10 bis 18 und insbesondere
12 bis 16 Kohlenstoffatome aufweisen, wie aus Dodecan oder Hexadecan;
und/oder aus
- – einer
organischen hochsiedenden Base, vorzugsweise einer N-haltigen aliphatischen
Base, am meisten bevorzugt aus einem dreifach substituierten Amin,
insbesondere aus Trialkylaminverbindungen mit 5 bis 10 Kohlenstoffatomen
pro Alkylgruppe, wie aus Trioctylamin oder Tris(2-ethylhexyl)amin,
oder aus einer N-haltigen aromatischen Base mit vorzugsweise 3 bis
20 Kohlenstoffatomen, wie aus Imidazol.
-
Diese
Lösungsmittel
können
auch in Kombination eingesetzt werden. Die organische hochsiedende Base
kann nicht nur als Lösungsmittel
dienen, sondern auch als Säure-Fänger fungieren.
Bei Verwendung von z.B. einer Säure,
wie von Phosphorsäure
oder HF, als Anionquelle ist es dann bevorzugt, die Base in annähernd äquimolarer
Menge (z.B. von ca. 0,6 bis 1,4 mol) zu verwenden, bezogen auf den/das
oder die Wasserstoff(e)/atom(e) der Säure.
-
Die "Kationquelle" kann aus jedem geeigneten
(hinreichend reaktiven) Metallsalz ausgewählt sein und ist bevorzugt
ein Metallchlorid, ein Metallalkoxid (worin das Alkoxid vorzugsweise
1 bis 6 und insbesondere 1 bis 4 Kohlenstoffatome aufweist), ein
Metallnitrat oder Metallacetat. Die Verwendung von Metallchloriden
ist besonders bevorzugt. Hydratisierte Metallsalze können ebenfalls
verwendet werden. Allerdings ist es dann bevorzugt, das Kristallisationswasser
vor der Reaktion zu entfernen.
-
Die "Anionquelle" wird bevorzugt aus
in PCT/DE 01/03 433 offenbarten Ausgangsmaterialien ausgewählt. Zur
Synthese der nanoteilchenförmigen
Sulfate, Phosphate, Borate, Fluoride, Sulfide, Arsenate oder Silikate
eigenen sich die folgenden Verbindungen:
- a.
Schwefelsäure,
Phosphorsäure,
Borsäure
oder HF,
- b. Sulfid-, Arsenat-, Phosphat-, Borat-, Sulfat-, Silikat- oder
Fluoridsalze, die in der Synthesemischung löslich oder zumindest dispergierbar
sind, insbesondere Salze mit einem organischen Kation oder Alkalimetallsalze,
oder
- c. Ester, die bei höheren
Temperaturen zersetzt werden, wie Borsäurealkylester, Schwefelsäurealkylester, Arsensäurealkylester
oder Kieselsäurealkylester
(z.B. Tetraethylorthosilikate).
-
Bezüglich Option
b, ist das Kation vorzugsweise aus basischen N-haltigen aliphatischen,
aromatischen und aliphatisch/aromatischen Substanzen ausgewählt, die
bevorzugt 4 bis 30 und bevorzugter 4 bis 20 Kohlenstoffatome aufweisen.
Geeignete Kationen sind z.B. quartäres Ammonium oder Phosphonium,
wie oben beschrieben für
die protonierten aromatischen Basen, wie Pyridin oder Collidin.
Zur Herstellung von Phosphat-Nanopartikeln können Tetrabutylammoniumdihydrogenphosphat,
Tetramethylammoniumdihydrogenphosphat oder Triethylammoniumdihydrogenphosphat
als Anionquelle verwendet werden. Entsprechend können Sulfat-Nanopartikel aus Ausgangsmaterialien
wie aus Tetrabutylammoniumhydrogensulfat, Tetramethylammoniumhydrogensulfat,
Bistetrabutylammoniumsulfat oder aus Triethylammoniumhydrogensulfat
hergestellt werden. Zur Herstellung von Nanopartikeln mit fluorhaltigen
Anionen können
Triethylamin-Trishydrofluorid, Tetrabutylammoniumfluorid, Tetrabutylammoniumhydrogendifluorid,
Dodecylaminhydrofluorid oder die weniger löslichen Pyridin- oder Collidinhydrofluoride
verwendet werden.
-
Werden
das Metallion (die Kationquelle) zu langsam im organischen Medium
aufgelöst,
ist es bevorzugt, diese in einem niedrigen Alkohol, vorzugsweise
in Methanol, vor der Zugabe des Metall-Komplexiermittels und des
Reaktionslösungsmittels
aufzulösen.
Methanol und Kristallisationswasser werden dann abdestilliert, worauf
getrocknet wird, bevor die weiteren Reaktionsteilnehmer zugegeben
werden.
-
Gemäß dem beanspruchten
Verfahren werden die gemäß einem
der obigen Syntheseverfahren erhältlichen
Nanopartikel als Dispersion in einem organischen Medium (als sogenannte "erste Mischung") bereitgestellt.
-
Das
organische Medium beruht vorzugsweise auf einem oder mehreren polaren
Lösungsmitteln
mit einem Siedepunkt von mehr als 120°C und insbesondere von mehr
als 180, aber weniger als 400°C.
Es wird bevorzugt, aus "Metall-Komplexiermitteln", insbesondere aus
den genannten Mono- oder Dialkylaminen, worin die Alkylreste 4 bis
20 C-Atome aufweisen, aus phosphororganischen Verbindungen, Polyolen
und aus Sulfoxiden ausgewählt.
Vorzugsweise enthält
das organische Medium das Metall-Komplexiermittel und gegebenenfalls "mindestens ein weiteres
Lösungsmittel", die im Zusammenhang
des Verfahrens gemäß der "organischen Synthese" beschrieben sind.
-
Entsprechend
ist es möglich
und bevorzugt, die in der "organischen" Synthese oder in "Polyol oder Sulfoxid" in der ersten Stufe
des beanspruchten Verfahrens erzeugten Nanopartikel ohne deren Isolierung
zu verwenden.
-
Es
sei angemerkt, dass das organische Medium als Dispersionsmedium
für die
Nanopartikelkerne dient. Somit werden wegen der Fähigkeit
des organischen Medium, an das Metallatom koordiniert zu werden, die
Nanopartikel in ihrem kolloidalen (nicht-gelösten) Zustand gehalten, bevor
eine Schale darauf wachsen kann.
-
II.2. Zweite Verfahrensstufe
-
In
der zweiten Stufe werden
- – die oben beschriebene erste
Mischung,
- – eine
Anionquelle für
die zu bildende Schale, insbesondere eine Phosphat-, Sulfat- oder
Fluoridquelle, und
- – eine
sogenannte "zweite
Mischung" aus die
Schale bildenden Metallionen (und deren Gegenion) und einem organischen
Komplexiermittel für
die genannten Metallionen
bei einer Temperatur von 50
bis 350°C
umgesetzt, bis eine lumineszente Schale um die genannten Nanopartikel
herum gebildet worden ist.
-
Generell
ist es bevorzugt, die Anionquelle und die erste Mischung getrennt
zu halten, um eine vorzeitige Reaktion zu vermeiden.
-
Die
zweite Verfahrensstufe kann gemäß den folgenden
drei Ausgestaltungen (A), (B) und (C) durchgeführt werden:
Verfahren
(A) umfasst Stufen, in denen man
ein erste Mischung aus Metallsalz-
oder -oxid-Nanopartikel, z.B. aus Metallsulfat-, -phosphat- oder
aus -fluorid-Nanopartikeln, in einem organischen Medium zubereitet,
die
genannte erste Mischung auf eine Temperatur von 50 bis 350°C erhitzt,
zu
dieser ersten Mischung bei dieser Temperatur getrennt eine Anionquelle
für die
zu bildende Schale und eine zweite Mischung aus die Schale bildenden
Metallionen und einem organischen Komplexiermittel für die genannten
Metallionen tropft und man
die entstandene Mischung bei dieser
Temperatur umsetzt, bis eine lumineszente Schale um die genannten
Nanopartikel herum gebildet worden ist.
Durch die getrennte,
aber gleichzeitige Zugabe der Anionquelle und der zweiten Mischung,
z.B. mittels zwei Tropftrichtern, werden die Konzentration der Wirk-Ausgangsmaterialien
für die
Schale und somit die Selektivität
der Reaktion durch Absenkung eines unabhängigen Partikelwachstums aus
den Ausgangsmaterialien für die
Schale gesteigert.
Verfahren (B) umfasst Stufen, in denen man:
eine
erste Mischung aus Nanopartikeln eines ersten Metallsalzes oder
-oxids, z.B. aus Metallsulfat-, -phosphat- oder -fluorid-Nanopartikeln,
in einem organischen Medium zubereitet,
eine die Schale bildende
Anionquelle zur genannten ersten Mischung gibt,
die entstandene
Mischung auf eine Temperatur von 50 bis 350°C erhitzt,
dazu eine zweite
Mischung aus die Schale bildenden Metallionen und einem organischen
Komplexiermittel für die
genannten Metallionen tropft und man
die entstandene Mischung
bei dieser Temperatur umsetzt, bis eine lumineszente Schale um die
genannten Nanopartikel herum gebildet worden ist.
Die Verfahren
(A) und (B) neigen dazu, einheitlichere Partikel zu bilden, die
außerdem
einen geringeren Prozentsatz unabhängig gewachsener Partikel des
Schale-bildenden Materials enthalten.
Verfahren (C) umfasst
Stufen, in denen man
eine erste Mischung aus Nanopartikeln
eines ersten Metallsalzes oder -oxids, z.B. aus Metallsulfat-, -phosphat-
oder -fluorid-Nanopartikeln, in einem organischen Medium zubereitet,
eine
erste Mischung, eine Anionquelle für die zu bildende Schale und
eine zweite Mischung aus die Schale bildenden Metallionen und einem
organischen Komplexiermittel für
die genannten Metallionen vorzugsweise durch Zugabe der genannten
ersten Mischung und der genannten Anionenquelle zur genannten zweiten
Mischung zusammenbringt und
die entstandene Mischung auf eine
Temperatur von 50 bis 350°C
erhitzt, bis eine lumineszente Schale um die genannten Nanopartikel
gebildet worden ist.
-
In überraschender
Weise wurde herausgefunden, dass eine graduelle Zugabe, z.B. durch
Zutropfen, der Ausgangsmaterialien nicht absolut erforderlich ist.
Obwohl, gemäß Verfahren
(C), die Ausgangsmaterialien durch Vermischen der kompletten Anteile
zusammengebracht werden können,
wird das gewünschte Kern/Schale-Material
mit hoher Selektivität
und geringem unabhängigen
Partikelwachstum gebildet. Das Verfahren (C) ist somit leichter
zu handhaben als die Verfahren (A) und (B).
-
Falls
nichts Anderes ausgesagt wird, gelten die folgenden bevorzugten
Ausgestaltungen für
alle drei Verfahren (A), (B) und (C).
-
Als
Metallionquelle kann jedes hinreichend reaktive Metallsalz, vorzugsweise
Chloride oder Alkoxide des Schalen-Metallions, verwendet werden.
Die Alkoxidgruppe weist vorzugsweise 1 bis 4 Kohlenstoffatome auf.
-
Jede
geeignete Anionquelle kann verwendet werden, solange sie befähigt ist,
eine Schale um die in der ersten Stufe erhaltenen Kernpartikel herum
zu bilden.
-
Geeignete
Anionen für
die Schalenbildung schließen,
ohne darauf eingeschränkt
zu sein, Phosphate, Halophosphate, Arsenate, Sulfate, Borate, Aluminate,
Gallate, Silikate, Germanate, Oxide, Vanadate, Niobate, Tantalate,
Wolframate, Molybdate, Alkalihalogenate, weitere Halogenide, Nitride,
Sulfide, Selenide, Sulfoselenide oder Oxisulfide ein.
-
Es
ist bevorzugt, für
die Schalenbildung Anionen zu verwenden, die in organischen Medien
unter ähnlichen
oder identischen Bedingungen wie den in PCT/DE 01/03 433 beschriebenen
in geeigneter Weise reagieren. Beispiele schließen Silikate, Borate, Arsenate,
Sulfide, Sulfate, Phosphate und Fluoride und insbesondere Sulfate,
Phosphate und Fluoride ein. Dieses Dokument enthält auch die technische Lehre,
welche Anionquellen zur Erzeugung des entsprechenden nanoteilchenförmigen Materials
verwendet werden können.
-
Bezüglich einer
geeigneten Quelle für
die Silikate, Borate, Arsenate, Sulfide, Sulfate, Phosphate und die
Fluoride, sind auch Anionquellen zu nennen, die oben für die erste
Stufe des beanspruchten Verfahrens beschrieben sind, insbesondere
diejenigen, die in der "Polyol-
oder Sulfoxid"-
und/oder der "organischen" Synthese verwendet
wurden.
-
Die
Anionquelle, insbesondere die Quelle für die Phosphate, Fluoride oder
Sulfate, wird bevorzugt in Mengen von 0,75 bis 3 und insbesondere
von 0,75 bis 2 mol verwendet, bezogen auf die stöchiometrisch zur Reaktion mit
allen Schale-bildenden zugegebenen Metallatomen benötigte molare
Menge. Bei binären
Salzen (AB) liegt das Verhältnis
B (Anion) zu A (Metall) somit im Bereich von 0,75:1 bis 2:1.
-
Phosphat-
und Fluoridquellen, wie Phosphorsäure oder HF, werden bevorzugt
in überschüssigen Mengen
in der "organischen" Synthese der aus
Phosphat oder Fluorid hergestellten Kern- oder Kern/Schale-Partikel
eingesetzt. Die überschüssige molare
Menge beträgt
vorzugsweise mindestens 1,05 mol, bevorzugter 1,1 bis 2 und insbesondere
1,2 bis 1,6 mol, bezogen auf die stöchiometrisch erforderliche
molare Menge. Ähnlich bevorzugt
ist es, Sulfatquellen, wie quartäre
Ammonium(hydrogen)sulfatsalze, in überschüssigen Mengen in der "Polyol- oder Sulfoxid"-Synthese von Sulfat-Kern- oder Kern/Schale-Partikeln
zu verwenden. Die überschüssige molare
Menge beträgt
vorzugsweise 1,05 mol, bevorzugter 1,1 bis 3 und insbesondere 1,2
bis 2 mol, bezogen auf die stöchiometrisch
erforderliche molare Menge.
-
Das
in der zweiten Mischung enthaltene organische Komplexiermittel kann
ebenfalls aus den organischen Komplexiermitteln ausgewählt sein,
wie bereits oben im Zusammenhang des Verfahrens gemäß der "organischen" Synthese der Nanopartikel
oder der für
die "Polyol- oder
Sulfoxid"-Synthese
beschriebenen Lösungsmittel
erläutert
worden sind.
-
Generell
ist es erwünscht,
die effektive Konzentration der Schale-bildenden Ionen so niedrig wie möglich zu
halten. Gemäß der vorliegenden
Erfindung wird dies durch die Verwendung des vorliegenden Metall-Komplexiermittels
bewerkstelligt. Ohne an eine Theorie gebunden zu sein, wird angenommen,
dass nur eine kleine Konzentration reaktiver (unkomplexierter) Metallionen
das Schalenwachstum gegenüber
der unabhängigen
Bildung neuer Partikel begünstigt.
-
Gemäß einer
bevorzugten Ausgestaltung sind das für die erste Mischung verwendete
organische Medium und das in der zweiten Mischung vorliegende Komplexiermittel
durch eine bzw. eines der vorgenannten phosphororganischen Verbindungen,
mono/disubstituierten Amine, Polyole oder Sulfoxide dargestellt.
Es ist ferner bevorzugt, dieselbe polare organische Verbindung als
organisches Medium und Komplexiermittel zu verwenden.
-
Außerdem ist
es bevorzugt, das vorgenannte "mindestens
eine weitere Lösungsmittel" im gleichen Verhältnis zum
organischen Komplexiermittel einzusetzen. Dadurch wird es ermöglicht,
niedrigere Mengen Metall-Komplexiermittel
als im Fall der Verwendung von nur einem einzigen Lösungsmittel
zu verwenden. Dann beträgt
das Molverhältnis
des Metall-Komplexiermittels
und der die Schale bildenden Metallionen vorzugsweise wiederum 0,9:1
bis 6:1.
-
Besitzt
die Anionquelle für
das Schalenmaterial saure Wasserstoffatome, ist es bevorzugt, die
oben beschriebenen Basen zu verwenden. Die oben beschriebene organische
hochsiedende Base (z.B. Trialkylamin) wird z.B. bevorzugt als Säure-Fänger für Anionquellen
wie Phosphorsäure
oder HF unter den oben beschriebenen Bedingungen verwendet. Diese
organische hochsiedende Base kann auch in der Synthese der Silikate,
Borate, Arsenate oder Sulfate in typischer Weise bei Anwendung von
Anionquellen mit sauren Wasserstoffatomen zugegeben werden. Gemäß Verfahren
(A) oder (B) wird die Base bevorzugt als Bestandteil der "zweiten Mischung" aus der Metallquelle
und dem Komplexiermittel zugegeben.
-
Die
Gesamtmenge an Lösungsmittel(n),
einschließlich
des Metall-Komplexiermittels,
ist vom Durchschnittsfachmann leicht bestimmbar, da es generell
bevorzugt ist, alle Ausgangsmaterialien homogen aufzulösen oder
zu dispergieren. In Verfahren (A) und (B) ist es bevorzugt, ungefähr die gleichen
Mengen an Lösungsmitteln
zur Auflösung
der Anionquelle und der Metallquelle (der zweiten Mischung) zu verwenden.
-
Allgemein
gesprochen, schreitet die Reaktion bevorzugt unter den gleichen
oder ähnlichen
Bedingungen voran, wie dies vorher unter Punkt II.1 für die "Polyol- oder Sulfoxid-" oder die "organische" Synthese diskutiert
wurde, falls nichts Anderes ausgesagt ist. Dies gilt ebenso für die Verwendung
eines inerten Schutzgases und die Trocknung der Reaktionsteilnehmer.
-
Die
Menge an Nanopartikelkernen, die mit den verbleibenden Ausgangsmaterialien
kombiniert wird, ist nicht besonders eingeschränkt und hängt in erster Linie von der
beabsichtigten Schalendicke ab.
-
Gemäß dem Verfahren
der vorliegenden Erfindung wird das Reaktionsmedium auf eine Temperatur von
50 bis 350 und insbesondere von 120 bis 320°C erhitzt, bis eine lumineszente
Schale um die in der ersten Verfahrensstufe hergestellten Nanopartikelkerne
herum gebildet worden ist.
-
Die
Reaktion wird vorzugsweise bei einer Temperatur von 160 bis 240
und insbesondere von 180 bis 220°C
für die
Fluoride und Phosphate und von 160 bis 180°C für die Sulfate durchgeführt. Die
Bildung von Sulfat-Schalen in Glycerin kann auch viel niedrigere
Temperaturen (z.B. von 50 bis 100°C)
zulassen. Die geeigneten Temperaturen können vom Durchschnittsfachmann
ganz leicht durch Verfolgen des Schalenwachstums bei graduell steigenden
Temperaturen ermittelt werden, um dadurch die Syntheseminimaltemperatur
zu bestimmen, bei der die Reaktion mit hinreichender Geschwindigkeit,
aber ohne unerwünschte
Nebenreaktionen, wie die Bildung neuer Partikel aus den für die Schale
vorgesehenen Ausgangsmaterialien, abläuft.
-
In
diesen Verfahren (A) und (B), in denen die Ausgangsmaterialien zugetropft
werden, liegt die Zugabezeit vorzugsweise im Bereich von 0,5 bis
10 und insbesondere von 1 bis 5 h.
-
Bevorzugte
Reaktionszeiten liegen im Bereich von 30 min bis 48 h, insbesondere
von 1 bis 20 h und ganz spezifisch von 1,5 bis 16 h. Wiederum wird
durch Verfolgen der Reaktion, z.B. durch Ausfällen der Nanopartikel aus dem
Reaktionsmedium entnommenen Proben und durch Untersuchen der Partikelgrößenverteilung
in TEM-Mikrografien die Bestimmung der geeigneten Reaktionszeiten
ermöglicht.
Die Reaktion muss, z.B. durch Abkühlen, beendet werden, sobald
eine Oswald-Reifung beobachtet wird, d.h., wenn die größeren Partikel
auf Kosten der kleineren Partikel zu wachsen beginnen.
-
Nach
Beendigung der Reaktion wird das Reaktionsmedium auf Raumtemperatur
abgekühlt.
Dies verstärkt
bereits die Ausfällung
der gebildeten Kern/Schale-Nanopartikel. Bei unvollständiger Ausfällung ermöglicht die
Zugabe von Fällungslösungsmitteln
(z.B. von Methanol) zum Reaktionsmedium oder umgekehrt die vollständige Gewinnung
des Reaktionsprodukts. Alternativ dazu, ist es möglich, die überschüssigen organischen Lösungsmittel,
einschließlich
des organischen Komplexiermittels, abzudestillieren oder eine Ultra-Filtration
durch Membranen mit einer bevorzugten Porengröße durchzuführen, die Dalton-Werten in
der Größenordnung
von 5.000 bis 10.000 entspricht. Diese Werte entsprechen einem Schnitt
bei ca. 3 nm, welcher in vielen Fällen groß genug ist, um das Lösungsmittel
durchlaufen zu lassen, und klein genug ist, um das Durchdringen
und einen Verlust von Nanopartikeln zu verhindern. In typischer
Weise ist ein Druck von 2 bis 5 bar zum Austausch der Lösungsmittel
in den entsprechenden Ultra-Filtrationszellen notwendig.
-
Ferner
ist es bevorzugt, die erhaltenen Nanopartikel z.B. mit Methanol,
Ethanol oder mit Isopropanol zu waschen.
-
Auf
die unten und in den Beispielen angegebenen Weisen kann bestätigt werden,
dass das Schalenwachstum tatsächlich
stattgefunden hat.
-
Eine
Möglichkeit
betrifft das kontinuierliche Verfolgen der Reaktion durch Ausfällen kleiner
Proben und Analysieren von deren Partikelgrößenverteilung, z.B. in TEM-Mikrografien.
Die auf diese Weise gezogenen Proben zeigen, ob ein Schalenwachstum über die
gesamte Reaktionszeit hinweg erfolgt ist oder ob die unabhängige Bildung
kleinerer Partikel ebenfalls beobachtet werden kann. EDX-Analyse
(Energie-dispersive Röntgenanalyse)
kann die Gesamtzusammensetzung der Nanopartikel belegen. XPS-Spektroskopie
vermag weitere Informationen bezüglich
der Verteilung der Zusammensetzung von den äußeren bis zu den inneren Anteilen
der Partikel zu liefern, wenn die XPS bei unterschiedlichen Anregungsenergien
durchgeführt
wird. Außerdem
können
die Lumineszenzspektren der Kern/Schale-Partikel oft ganz leicht
von denen der in der Reaktion eingesetzten Kern-Nanopartikel unterschieden
werden, wie sich dies auch in den Beispielen zeigen lässt.
-
III. Verwendung der Kern/Schale-Partikel
-
III.1 Anwendung in Bioassayverfahren
-
Die
Kern/Schale-Partikel der vorliegenden Erfindung können in
vorteilhafter Weise in Bioassayverfahren zur Anwendung gelangen,
in denen von deren Lumineszenzeigenschaften Gebrauch gemacht wird.
Eine besonders interessante Anwendung für die vorliegenden Kern/Schale-Partikel
sind (F)RET-basierte Assayverfahren ("(Fluoreszenz)Resonanzenergietransfer", wie oben bereits
angedeutet).
-
In
biologischen Systemen wird der (F)RET oft zur Bestimmung der räumlichen
Nachbarschaft entsprechend markierter Biomoleküle oder -molekülgruppen
angewandt. Das Verfahren kann als Beleg für verschiedene biologische
Reaktionen oder Wechselwirkungen von Interesse, z.B. von Protein-Protein-Wechselwirkungen,
Antigen-Antikörper-Reaktionen
während
Immunreaktionen, Rezeptor-Ligand-Wechselwirkungen, eines Hybridismus
von Nucleinsäure
oder der Bindung von Proteinen an Nucleinsäuren, dienen.
-
Die
Bestimmung, dass ein (F)RET aufgetreten ist, erfolgt über die
Messung einer Intensitätsänderung oder
einer spektralen Änderung
der Donor- oder
Akzeptorlumineszenz oder über
die Messung von Änderungen
bei der Abbauzeit der Donorlumineszenz.
-
Viele
Anwendungen dieser Techniken und Verfahrensweisen sind in der Literatur
beschrieben und auch auf die vorliegende Erfindung anwendbar, wie
z.B., ohne darauf eingeschränkt
zu sein, auf: die Bestimmung spezifischer Antigene in Immunofluoreszenz-Assayverfahren
(
US 3,996,345 ;
US 4,160,016 ;
US 4,174,384 ;
US 4,199,559 ), die Bestimmung elektrostatischer
Potenziale in spezifischen lokalisierten Flächen auf der Oberfläche von
Proteinen (Yamamoto et al., J. Mol. Biol. 241, 1994, Seiten 714–731) oder
auf Hochdurchsatz-Siebungsverfahren (Boisclair et al., J. of Biomolecular
Screening 5, 2000, Seiten 319–328).
-
Außerdem kann
mit (F)RET-Systemen auch der absolute Abstand zwischen zwei Biomolekülen bzw. innerhalb
von Teilen von 1 Biomolekül
bestimmt werden. Diese Technik ist bereits erfolgreich auf die Protein- oder
DNA-Strukturanalyse (Heyduk et al., SPIE, Band 3256, 1998, Seiten
218–222),
auf die Messung von Abständen
innerhalb Polypeptiden (Lakowicz et al., Biophys. Chem. 36, 1990,
Seiten 99–115),
auf Proteine (K. Cai et al., J. Biol. Chem. 271, 1996, Seiten 27311–27320),
auf Polynucleotide (Hochstrasser et al., Biophys. Chem. 45, 1992,
Seiten 133–141,
und Ozaki et al., Nucl. Acids Res. 20, 1992, Seiten 5205–5214) oder
auf weitere Makromoleküle,
auf die Analyse von Membranen und Membran-Proteinen und deren Konstruktion
(S. Wang et al., Biochemistry 27, 1988, Seiten 2033–2039),
auf die Detektion (
US 4,996,143 ;
US 5,532,129 ;
US 5,565,332 ) und die Quantifizierung
amplifizierter Nucleinsäuren
durch PCR (Polymerase Chain Reaction) (
US 5,538,848 ;
US 5,723,591 ), z.B. für in vitro-Diagnostika,
genetische Analysen, forensische Analysen, Nahrungsmittel- und agrochemische
Tests oder für
Abstammungstests, angewandt worden. Die DNA oder RNA werden direkt,
d.h. ohne zusätzliche
Trennstufen, detektiert oder quantifiziert.
-
Eine
quantitative Nucleinsäure-Bestimmung
durch Realzeit-PCR mit (F)RET-Systemen liefert der als TaqMan
®-Assay
(Applied Biosystems Division of Perkin-Elmer Corp., Foster City,
USA) bekannte 5'-Nuclease-Assay
(
US 5,538,848 ;
US 5,210,015 ; Holland et
al., Proc. Natl. Acad. Sci. USA 88, 1991, Seiten 7276–7280; Lee
et al., Nucleic Acids Res. 21, 1993, Seiten 3761–3766). Ein Verfahren molekularer
Leuchttürme
(Signale) (Tyagi und Krammer, Nature Biotechnology 14, 1996, Seiten
303–306;
US 5,312,728 ) beruht auf
einem ähnlichen
Mechanismus.
-
Kürzlich wurde
eine Übersicht über "FRET in biochemistry" von S. Brakmann
und N. Nöbel
in Nachrichten aus der Chemie, 51, März 2003, Seiten 319–322, veröffentlicht,
worin weitere Alternativen für
FRET-basierte Bioassayverfahren
beschrieben sind, in denen die Kern/Schale-Partikel der vorliegenden Erfindung ebenfalls
zur Anwendung gelangen können.
-
Demzufolge
können
die Kern/Schale-Partikel der vorliegenden Erfindung in (F)RET-basierten
Bioassayverfahren verwendet werden, welche eine erste Molekülgruppe
A, die mit mindestens einem Energiedonor (Donor) markiert ist, und
mindestens eine zweite Molekülgruppe
B umfassen, die mit mindestens einem Energieakzeptor (Akzeptor)
markiert ist, worin
der Donor ein Molekül oder Partikel umfasst, die
durch Energietransfer aus dem Donor unter teilweiser oder vollständiger Löschung der
Donorlumineszenz angeregt werden können, und worin
Donor
und/oder Akzeptor die Kern/Schale-Partikel der vorliegenden Erfindung,
vorzugsweise diejenigen mit einem entlang ihrer längsten Achse
gemessenen Durchschnittsdurchmesser von nicht mehr als 50 und insbesondere
von nicht mehr als 30 nm usw., wie hierin vorher bereits beschrieben,
umfassen.
-
Dieser
Assay kann auf 2 Weisen durchgeführt
werden. (F)RET-basierte Assayverfahren machen es erforderlich, dass
der Akzeptor ebenfalls zur Lumineszenzemission befähigt ist.
RET-Systeme funktionieren dagegen auch, wenn der Akzeptor ohne Strahlungsemission
relaxiert.
-
Bevorzugt
werden die Kern/Schale-Partikel der vorliegenden Erfindung als Donor
verwendet. Da diese elektromagnetische Strahlung mit Stokes- oder
anti-Stokes-Verschiebung nach einer energetischen Anregung ausstrahlen,
ist eine spektroskopische Unterscheidung zwischen Anregungsquelle
und emittierter Strahlung leicht möglich.
-
Die
Kern/Schale-Partikel der vorliegenden Erfindung zeigen und ergeben
ein überlegenes
Verhalten in Bioassayverfahren des obigen Typs, da ihre Lumineszenz
wirkungsvoller gelöscht
werden kann. Ohne an eine Theorie gebunden zu sein, wird davon ausgegangen,
dass der höhere
Prozentsatz an Lumineszenzzentren an oder in enger Nachbarschaft
zur Oberfläche
im Vergleich mit homogenen Partikeln dieser Beobachtung Rechnung
trägt.
Die höhere
Eignung zur Löschung
bringt verschiedene wichtige Vorteile in (F)RET-basierten Bioassayverfahren,
wie eine höhere
Empfindlichkeit, mit sich.
-
Die
höhere
Eignung zur Löschung
schlägt
sich in den Abbau/Abfallkurven (Intensität der Lumineszenz gegen die
Zeit) als kürzere
Halbwerte (Halbwertszeit) des Donor nieder. In Zeit-geschleuster
Fluoreszenzspektroskopie (TGF-Modus) wird ein nahezu vollständiges Verschwinden
der Donorlumineszenz (aus den Kern/Schale-Partikeln) wegen eines
sehr wirkungsvollen Energietransfer zum Akzeptorsystem erhalten,
wie dies noch detaillierter in den Beispielen erläutert wird.
-
Bezüglich bevorzugter
Ausgestaltungen der Kern/Schale-Partikel, die als Donor und/oder
Akzeptor, vorzugsweise nur als Donor, verwendet werden, wird auf
Punkt I der vorliegenden Beschreibung Bezug genommen.
-
Bei
Auswahl eines geeigneten Donor/Akzeptor-Paars ist es im Allgemeinen
empfehlenswert, Donor-Sonden mit hohen Quantenausbeuten einzusetzen.
Ferner ist es erforderlich, dass das Emissionsspektrum der Donor-Sonde
deutlich mit dem Absorptionsspektrum der Akzeptor-Sonde überlappt.
Eine weitere Bedingung, die angepasste Anordnung (annähernd parallel)
der Donor- und Akzeptor-Übergangsdipol-Orientationen,
stellt im Allgemeinen kein Problem in biologischen Systemen dar,
was eine uneingeschränkte
isotrope Bewegung von Donor und Akzeptor ermöglicht. Ferner ist, wie bereits
erwähnt,
der Förster-Abstand
in Rechnung zu stellen, insofern Donor und Akzeptor vorzugsweise
innerhalb 1 ± 0,5R0 (Förster-Abstand)
voneinander vorliegen. Der Förster-Abstand
ist derjenige Abstand, bei welchem der Energietransfer zu 50% wirkungsvoll ist.
Er lässt
sich, wie im Stand der Technik bekannt, aus den spektralen Eigenschaften
von Donor und Akzeptor berechnen.
-
Typische
Donor- und Akzeptor-Systeme, die sich von Kern/Schale-Partikeln der vorliegenden
Erfindung unterscheiden, sind organische Farbstoffe wie Fluorescein,
Tetramethylrhodamin, IAEDANS, EDANS, Dabcyl, BODIPY FL, QSY 7 und
QSY 9. Weitere im Handel verfügbare
lumineszente organische Farbstoffe, die sich für den Spektralbereich von ca.
350 bis 750 nm und darüber
eignen, sind die Alexa Fluor-Farbstoffe (hergestellt von Molecular
Probes) oder CyDyes (Amersham Pharmacia). Unter diesen Farbstoffen
sind diejenigen, die bei höheren
Wellenlängen
(sichtbar bis Nah-IR) absorbieren und ausstrahlen, besonders attraktiv, da
sie biologische Systeme nicht schädigen.
-
Gemäß der vorliegenden
Erfindung ist es bevorzugt, die Kern/Schale-Partikel der vorliegenden Erfindung
als Donor in Kombination mit einem geeigneten Akzeptor, ausgewählt aus
den obigen organischen Fluoreszenzfarbstoffen, zu verwenden.
-
Partikel
mit einer Eu3+-dotierten Schale können z.B.
als Donor mit Alexa Fluor 680 als Akzeptor oder Tb3+-haltige
Partikel können
mit Dabcyl oder Fluorescein kombiniert werden. Beispiele für diese
Tb-haltigen Kern/Schale-Partikel sind z.B. Kern/Schale-Systeme mit
einem inerten (nicht-lumineszenten) Kern, umgeben von einer Tb3+- oder Ce3+-, Tb3b-dotierten
Metallsalz- oder -oxid-Schale, sowie Kern/Schale-Systeme auf Basis eines
Cer(Cer3+)-Salz- oder -Oxid-Kerns, umgeben
von einer Schale aus Terbium(Tb3+)-Salz
oder -Oxid.
-
Kern/Schale-Partikel
mit einem Durchschnittsdurchmesser unterhalb 50 nm zeigen und ergeben
ein kleineres Potenzial für
unerwünschte
sterische Wechselwirkungen oder Sedimentation in Bioassayverfahren als
größere Partikel.
Außerdem
ist zu erwarten, dass die Kinetik der Bindungsreaktion (z.B. einer
Immunreaktion oder DNA-Hybridisierung) des biochemischen Verfahrens,
das untersucht wird, weniger beeinflusst wird.
-
2
unterschiedliche spektroskopische Modi werden in typischer Weise
zur Messung des Energietransfer in (F)RET-basierten Systemen (WO
87/07 955;
EP 0 242 527 ;
EP 0 439 036 ; WO 92/01 225;
US 4,822,733 ;
US 5,279,943 ;
US 5,622,821 ;
US 5,656,433 ;
US 5,998,146 ;
US 6,239,271 ), d.h. Zeit-geschleuste Fluorimetrie (TGF)
und/oder Zeit-aufgelöste
Fluorimetrie (time resolved fluoremetry = TRF), angewandt. Gemäß TGF-Modus
wird der fluoreszente Donor mit einer gepulsten Lichtquelle (z.B.
mit Laser, Blitzlicht) angeregt, worauf die Lichtemission nach einem
vorbestimmten Verzug innerhalb eines spezifischen Zeitfensters gemessen
wird. Der relativ kurze Verzug ermöglicht immer noch die Messung
mit hinreichend hoher Intensität
der langzeitigen Lumineszenz von Lanthanidionen. Die relativ kurzzeitige
Hintergrundfluoreszenz (in typischer Weise kürzer als 1 μs), wie verursacht durch innewohnende
Autofluoreszenz des biologischen Materials, Verunreinigungen von Lösungsmitteln
oder durch umgebendes biologisches Material, wird durch den Verzug
fast völlig
unterschieden.
-
Im
Gegensatz zum TGF-Modus wird im TRF-Modus die Lumineszenz als Funktion
der Zeit bei konstanter Wellenlänge
gemessen. Der Donor wird ebenfalls durch eine gepulste Lichtquelle
oder in unterschiedlicher Weise modulierte Lichtquellen angeregt.
-
Kern/Schale-Partikel
mit einem Durchmesser von nicht mehr als 50 nm können in geeigneter Weise im
TRF-Modus verwendet werden, wobei für größere Partikel ein Hauptteil
des Partikelvolumen nicht nah genug am Akzeptor vorliegt, um am
Energietransfer teilzunehmen, um dadurch die Intensität des Effekts
zu erniedrigen.
-
Gemäß der vorliegenden
Erfindung zeigt und ergibt mindestens einer der (F)RET-Partner,
d.h. der Donor oder Akzeptor, eine relativ lange Lumineszenzabfallzeit,
wogegen der andere (F)RET-Partner durch kurze Abfallzeiten gekennzeichnet
ist.
-
Bevorzugt
werden Kern/Schale-Partikel mit Lumineszenz-Halbwertszeiten von
1 μs bis
50 ms und bevorzugter von 100 μs
bis 10 ms als Donor verwendet.
-
Werden
diese Donoren mit herkömmlichen
organischen Fluoreszenzfarbstoffen, die in typischer Weise kürzere Abfallzeiten
aufweisen, kombiniert, sensibilisiert und verlängert der Donor die Lumineszenz
des Akzeptor über
seine innewohnende Lumineszenz hinaus. Die Messung solcher Systeme
im TGF-Modus ermöglicht
den Ausschluss der kurzzeitigen innewohnenden Akzeptor-Lumineszenz
und die Bestimmung der sensibilisierten Akzeptor-Lumineszenz mit
hoher Empfindlichkeit.
-
Weitere
geeignete Akzeptoren können
aus elektrisch leitenden Materialien, wie aus Gold, Silber, Platin oder
aus leitfähigen
Metalloxiden, wie aus In-Sn-Oxid (ITO), oder aus leitfähigen Polymeren
ausgewählt
sein.
-
Zur
Bindung der Kern/Schale-Partikel der vorliegenden Erfindung an das
oder die biologische(n) Molekül(e),
auf welcher der Assay beruht, sind die folgenden Techniken und Verfahrensweisen
anwendbar.
-
Die
Bindung kann erzeugt werden durch:
- – chemische
Modifizierung der Kern/Schale-Partikel, wobei in typischer Weise "funktionelle Gruppen" auf der Oberfläche erzeugt
werden, die zur Bindung an ein biologisches Molekül befähigt sind,
und/oder durch
- – Verbinden
der gegebenenfalls chemisch modifizierten Oberfläche des Kern/Schale-Partikels
mit kovalent oder nicht-kovalent gebundenen sogenannten "verbindenden Molekülen",
worauf
das oder die Biomolekül(e)
mit den dadurch erhaltenen Partikeln umgesetzt werden.
-
Der
Begriff "verbindendes
Molekül" bedeutet eine Substanz
mit der Befähigung
zur Verbindung mit den Kern/Schale-Partikeln der Erfindung und ebenso
zur Verbindung an ein Affinitätsmolekül oder an
ein Molekül oder
einen Molekülteil,
welche im Wettstreit um die gleichen Bindungsstellen des Affinitätsmoleküls als Ziel-Molekül, z.B.
um ein Epitop, liegen.
-
Der
Begriff "Ziel-Molekül" bedeutet eine Ganzheit
oder Gruppe, deren An- oder Abwesenheit in einem Material wie einer
biologischen Probe durch Verwendung der Kern/Schale-Partikel der
Erfindung gesichert festgestellt werden soll.
-
Der
Begriff "Affinitätsmolekül" bedeutet ein Biomolekül, das selektiv
an das Ziel-Molekül
(falls vorhanden) in dem Material (z.B. einem biologischen Material),
das analysiert wird, gebunden wird.
-
Der
hierin vorab verwendete Begriff "funktionelle
Gruppen" ist nicht
auf reaktive Gruppen, die kovalente Bindungen bilden, eingeschränkt, sondern
schließt
auch chemische Gruppen ein, die zu einer ionischen Wechselwirkung
oder zu Wasserstoffbindungen mit dem oder den Biomolekül(en) führen. Außerdem sei
angemerkt, dass eine strikte Unterscheidung zwischen an der Oberfläche erzeugten "funktionellen Gruppen" und verbindenden Molekülen, die "funktionelle Gruppen" tragen, nicht möglich ist,
da manchmal die Modifikation der Oberfläche eine Reaktion kleinerer
verbindender Moleküle
wie von Ethylenglykol mit der Nanopartikeloberfläche erforderlich macht.
-
Die
funktionellen Gruppen oder die verbindenden Moleküle, die
diese tragen, können
aus Aminogruppen, Carbonsäuregruppen,
Thiolen, Thioethern, Disulfiden, Guanidinogruppen, Hydroxylgruppen,
Amingruppen, vicinalen Diolen, Aldehyden, α-Haloacetylgruppen, Quecksilberorganylen,
Estergruppen, Säurehalogeniden,
Säurethioestern,
Säureanhydriden,
Isocyanaten, Isothiocyanaten, Sulfonsäurehalogeniden, Imidoestern, Diazoacetaten,
Diazoniumsalzen, 1,2-Diketonen, Phosphonsäuren, Phosphorsäureestern,
Sulfonsäuren, Azoliden,
Imidazolen, Indolen, N-Maleimiden, α-β-ungesättigten Carbonylverbindungen,
Arylhalogeniden oder aus deren Derivaten ausgewählt sein.
-
Nicht-einschränkende Beispiele
für weitere
verbindende Moleküle
mit höheren
Molekulargewichten sind Nucleinsäure-Moleküle, Polymere,
Copolymere, polymerisierbare Kupplungsmittel, Silika, Proteine und kettenartige
Moleküle
mit einer Oberfläche
mit der entgegengesetzten Polarität zu den Kern/Schale-Partikeln. Nucleinsäuren können eine
Verbindung zu Affinitätsmolekülen ergeben,
die selber Nucleinsäure-Moleküle, obwohl
mit einer komplementären
Sequenz bezüglich
des verbindenden Moleküls
enthalten. Als Beispiele für
polymerisierbare Kupplungsmittel können Diacetylen, Styrolbutadien,
Vinylacetat, Acrylat, Acrylamid, Vinylverbindungen, Styrol, Siliziumoxid,
Boroxid, Phosphoroxid, Borate, Pyrrol, Polypyrrol und Phosphate
genannt werden.
-
Verbindungsverfahren
werden nun detaillierter beschrieben:
- 1. Die
Oberfläche
der Kern/Schale-Nanopartikel kann z.B. durch die Bindung von Phosphorsäurederivaten mit
funktionellen reaktiven Gruppen chemisch modifiziert werden. Ein
Beispiel dieser Phosphor- oder Phosphonsäureetherderivate ist Iminobis(methylenphosphono)carbonsäure, die
gemäß einer "Mannich-Moedritzer"-Reaktion synthetisiert
werden kann (Moedritzer und Irani, J. Org. Chem. 1966, 31, 1603).
Diese Bindungsreaktion kann mit Kern/Schale-Partikeln durchgeführt werden,
wie sie direkt aus dem Herstellverfahren der vorliegenden Erfindung
oder nach einer Vorbehandlung (z.B. mit Trimethylsilylbromid) erhalten
werden. Im ersten Fall kann das Phosphonsäure(ester)derivat z.B. Komponenten
des Reaktionsmediums verdrängen,
die noch an der Oberfläche
gebunden sind. Diese Verdrängung
kann bei höheren
Temperaturen verstärkt
werden. Trimethylsilylbromid soll andererseits Alkylgruppen-haltige
Phosphor-basierte
Komplexiermittel, wie sie im Verfahren der Erfindung verwendet werden,
entalkylieren, um dadurch neue Bindungsstellen für das Phosphonsäure(ester)derivat
zu erzeugen. Das Phosphonsäure(ester)derivat
oder daran gebundene verbindende Moleküle können die gleichen funktionellen
Gruppen wie oben ergeben.
- 2. Ein weiteres Beispiel einer Oberflächenbehandlung von Kern/Schale-Nanopartikeln
beinhaltet, dass die Partikel in einem Diol, wie in Ethylenglykol,
erhitzt werden. Es sei angemerkt, dass diese Behandlung überflüssig sein
kann, wenn die Synthese der Kern/Schale-Partikel bereits in einem
Diol erfolgte. Unter diesen Bedingungen weist das direkt erhaltene
Syntheseprodukt wahrscheinlich die notwendigen funktionellen Gruppen
bereits auf. Diese Behandlung ist allerdings auf Kern/Schale-Partikel
anwendbar, die in den oben beschriebenen N- oder P-haltigen Komplexiermitteln
erzeugt wurden. Werden solche Kern/Schale-Partikel einer Nachbehandlung
mit Ethylenglykol unterzogen, können
Bestandteile des Reaktionsmedium (z.B. Komplexiermittel), die noch
an der Oberfläche
gebunden sind, durch das Diol ersetzt und/oder entalkyliert werden.
Die Behandlung mit Diolen führt
zu wasserlöslichen
Partikeln. In analoger Weise können
primäre Alkohole
mit einer zweiten funktionellen reaktiven Gruppe, wie oben angegeben,
für die
Nachbehandlung verwendet werden. Es ist auch möglich, N-haltige Komplexiermittel,
die noch an die Partikeloberfläche
gebunden sind, durch primäre
Aminderivate mit einer zweiten funktionellen Gruppe, ausgewählt aus
den obigen Beispielen, zu ersetzen.
- 3. Die Oberfläche
der Kern/Schale-Partikel der vorliegenden Erfindung kann auch mit
Silika überzogen
werden. Silika ermöglicht
dabei eine relativ einfache chemische Konjugation organischer Moleküle, da Silika leicht
mit organischen Verbindungsmitteln, wie mit Triethoxysilan oder
Chlorsilan, reagiert. Die Partikeloberfläche kann auch mit Homo- oder
Copolymeren überzogen
werden. Beispiele für
polymerisierbare Kupplungsmittel sind N-(3-Aminopropyl)-3-mercaptobenzamidin,
3-(Trimethoxysilyl)propylhydrazid und 3-(Trimethoxysilyl)propylmaleimid.
Weitere Beispiele polymerisierbarer Kupplungsmittel wurden bereits
oben genannt. Diese Kupplungsmittel können einzeln oder in Kombination
in Abhängigkeit
vom Typ des Copolymer verwendet werden, das als Nanopartikelüberzug erzeugt
wird.
- 4. Gemäß einem
weiteren Oberflächenmodifikationsverfahren
können
Kern/Schale-Partikel, die oxidische Übergangsmetallverbindungen
enthalten, durch Chlorglas oder organisches Chlorierungsmittel in
die entsprechenden Oxichloride überführt werden.
Diese Oxichloride sind befähigt,
mit Nukleophilen, wie mit Hydroxy- oder Aminogruppen, die oft in
Biomolekülen
vorgefunden werden, zu reagieren. Dieses Verfahren ermöglicht die
Erzeugung einer direkten Konjugation mit Proteinen, z.B. über die
Aminogruppe von Lysin-Seitenketten. Die Konjugation mit Proteinen
nach Oberflächenmodifikation
mit Oxichloriden kann auch durch Verwendung eines bifunktionellen
Verbindungsmittels, wie von Maleimidopropionsäurehydrazid, bewerkstelligt
werden.
- 5. Für
nicht-kovalente Verbindungsverfahren sind Kette-Typ-Moleküle mit einer
Polarität
oder Ladung besonders geeignet, die zu denen der Kern/Schale-Partikeloberfläche entgegengesetzt
sind. Beispiele für verbindende
Moleküle,
die mit Kern/Schale-Nanopartikeln nicht-kovalent verbunden werden
können, schließen anionische,
kationische oder zwitterionische oberflächenaktive Mittel, saure oder
basische Proteine, Polyamine, Polyamide, Polysulfone oder Polycarbonsäuren ein.
Die hydrophobe Wechselwirkung zwischen Nanopartikel und amphiphilem
Reagenz mit einer funktionellen reaktiven Gruppe kann die notwendige
Verbindung erzeugen. Insbesondere eignen sich Kette-Typ-Moleküle mit amphiphilem
Charakter, wie Phospholipe oder derivatisierte Polysaccharide, die
miteinander vernetzt werden können.
Die Absorption dieser Moleküle
auf der Oberfläche
des Kern/Schale-Partikels kann durch Coinkubation bewerkstelligt werden.
Die Bindung zwischen Affinitätsmolekül und Kern/Schale-Partikel
kann auch auf nicht-kovalenten, selbst-organisierenden Bindungen
beruhen. Ein Beispiel betrifft einfache Detektionssonden mit Biotin
als verbindendes Molekül
und Avidin- oder Streptavidin-gekuppelte Affinitätsmoleküle.
-
Protokolle
für Kupplungsreaktionen
funktioneller Gruppen an biologische Moleküle sind in der Literatur, z.B.
in "Bioconjugate
Techniques" (Greg
T. Hermanson, Academic Press 1996) zu finden. Das biologische Molekül, insbesondere
ein Affinitätsmolekül, kann
an das verbindende Molekül
kovalent oder nicht-kovalent in Linie mit Standardverfahren der
organischen Chemie wie einer Oxidation, Halogenierung, Alkylierung,
Acylierung, Addition, Substitution oder einer Amidierung gekuppelt
werden. Diese Verfahren zur Kupplung eines biologischen Moleküls an das
kovalent- oder nicht-kovalent-gebundene verbindende Molekül kann vor
der Kupplung des verbindenden Moleküls an die Kern/Schale-Nanopartikel
oder danach durchgeführt
werden. Ferner ist es mittels Inkubation möglich, eine direkte Bindung
von Affinitätsmolekülen an entsprechend
vorbehandelte Kern/Schale-Nanopartikel (z.B. mit Trimethylsilylbromid)
zu bewerkstelligen, welche dann eine modifizierte Oberfläche aufgrund
dieser Vorbehandlung (z.B. eine Oberfläche mit höherer Ladung oder Polarität) ergeben. Die
Molekülgruppen
A und B, die mit einem Donor bzw. Akzeptor markiert wurden, können einen
Teil des gleichen Moleküls
darstellen und z.B. an das gleiche Affinitätsmolekül gekuppelt sein. Eine Änderung
beim räumlichen
Abstand dieser Molekülgruppen
kann z.B. durch eine Konformationsänderung oder eine Molekülspaltung
verursacht sein. Diese Konfirmationsänderung oder Molekülspaltung
können
das Ergebnis einer Wechselwirkung zwischen dem Affinitätsmolekül und einem
Ziel-Molekül
sein.
-
Alternativ
dazu, können
die Molekülgruppen
A und B auf unterschiedlichen Molekülen angeordnet vorliegen, wobei
die genannten Molekülgruppen
A und B jeweils an ihre eigenen Affinitätsmoleküle gekuppelt sind. Eine Änderung
beim räumlichen
Abstand kann durch eine Wechselwirkung der Affinitätsmoleküle, die
an Molekülgruppen
A und B angeordnet sind, mit einem verbundenen Ziel-Molekül oder miteinander
herbeigeführt sein.
Diese Wechselwirkung kann z.B. eine Wechselwirkung zwischen Proteinen,
wie eine Immunreaktion von Antigen mit Antikörper, ein Nucleinsäure-Hybridismus
oder die Wechselwirkung zwischen Nucleinsäuren und Proteinen sein.
-
Der
Bioassay kann z.B. ein homogener Immunoassay zum Detektieren eines
Analyts in einer Körperprobe
(z.B. Abstrich, Speichel, Organpunktat, Biopsie, Sekretion, Flüssigkeit,
Galle, Blut, Lymphe, Urin, Faeces) sein. Homogene Assayverfahren
machen keine Wasch- oder Trennstufen erforderlich.
-
Der
Bioassay mit den Kern/Schale-Partikeln der vorliegenden Erfindung
kann auch ein heterogener Assay sein.
-
Der
Analyt (in der Regel das Ziel-Molekül), welche im Assay detektiert
werden, können
z.B. ein mono- oder polyklonaler Antikörper, ein Protein, ein Peptid,
ein Oligonucleotid, eine Nucleinsäure, ein Oligo- oder Polysaccharid,
ein Hapten oder ein niedermolekulares synthetisches oder natürliches
Antigen sein.
-
Ebenso
sind nicht-einschränkende
Beispiele für
Affinitätsmoleküle Proteine,
Peptide, Oligonucleotide oder weitere Nucleinsäure-Moleküle oder verwandte Spezies,
wie PNAs oder Morpholinos sowie Oligo- oder Polysaccharide, Haptene
wie Biotin oder Digoxin oder niedermolekulare synthetische oder
natürliche
Antigene oder Epitope.
-
Der
Assay kann in Lösung
sowie in Festphase- oder Array-basierten Systemen zur Anwendung
gelangen, worin Oligo- oder Polynucleotidketten oder Antikörper oder
Antigene jeweils auf der Oberfläche
immobilisiert werden.
-
Assayverfahren
mit den Kern/Schale-Partikeln der vorliegenden Erfindung können auf
verschiedene Weisen angewandt und genutzt werden.
-
Gemäß einem
Anwendungstyp sind die (F)RET-Partner auf demselben Molekül angeordnet,
d.h., beide (F)RET-Partner werden über entsprechende verbindende
Moleküle
(teilweise nicht dargestellt) mit demselben Affinitätsmolekül gebunden
(6a, 6b, 7, 10 und 11).
Die Bindung eines Ziel-Moleküls an
das Affinitätsmolekül induziert
eine Konfirmationsänderung
des Affinitätsmoleküls, um dadurch
eine Änderung
der räumlichen
Position der Markierungen zueinander und somit eine messbare Differenz
im (F)RET zu ergeben.
-
Für die weiteren
Anwendungen werden die (F)RET-Partner auf verschiedenen Molekülen angeordnet und
jeweils mit ihrem eigenen Affinitätsmolekül (8) oder
mit dem Analyt und dem Affinitätsmolekül (9) gekuppelt.
Die jeweiligen Affinitätsmoleküle können in
einer Weise ausgewählt
werden, um eine Wechselwirkung zwischen Donor und Akzeptor zu ergeben,
die durch die Reaktion mit dem Ziel-Molekül entweder erzeugt oder gelöscht wird,
um dadurch eine Änderung
des Energietransfers zu induzieren.
-
Die
Verwendungen der Kern/Schale-Nanopartikel gemäß der vorliegenden Erfindung
in (F)RET-basierten Bioassayverfahren werden nun noch weiter an
Hand der 6 bis 11 erläutert.
-
6a zeigt
schematisch die Wechselwirkung der (F)RET-Partner auf demselben
Molekül
in einem homogenen Kinase-Assay. Der lad-Nanopartikel 1 (lad
= luminescent anorganic doped/lumineszent anorganisch dotiert) und
Chromophor 2 werden mittels einer Peptidsequenz 3 verbunden.
Die Peptidsequenz enthält eine
Kinase-spezifische Identifikationssequenz 4. Wird die Peptidsequenz 3 an
dieser Position durch eine Kinase 5 phosphoryliert, verändert das
Vorliegen von Phosphat 6 die Konformation der Peptidsequenz 3.
Somit wird die Wechselwirkung zwischen den (F)RET-Partnern, und zwar
dem Nanopartikel 1 und dem Chromophor 2, messbar.
-
6b zeigt
schematisch einen homogenen Immunoassay mit (F)RET-Partnern auf 1 Molekül, wofür Protein-Protein-Wechselwirkungen,
z.B. Antigen-Antikörper-Reaktionen,
zu bestimmen sind. Nanopartikel 1 und Chromophor 2 werden
mittels der Peptidsequenz 3 verbunden. Die Peptidsequenz
enthält
ein Epitop 14. Wird ein Antikörper 15, der in spezifischer
Weise das Epitop 14 erkennt, an dieses gebunden, ändert sich
die Konformation der Peptidsequenz 3. Dadurch wird die Wechselwirkung
zwischen den (F)RET-Partnern, dem Nanopartikel 1 und dem
Chromophor 2, messbar.
-
Das
zu detektierende Molekül
kann direkt an das Affinitätsmolekül gebunden
werden, wie gemäß 6a und 6b beschrieben.
Allerdings kann das erstere auch indirekt für die Bindung eines Moleküls an das
Affinitätsmolekül verantwortlich
sein. Ein entsprechendes Beispiel dafür stellt die Messung von Ca2+-Konzentrationen in lebenden Zellen dar.
Zu diesem Zweck wird die Calcium-abhängige Bindung von Calmodulin an
Myosin-Leichtkette-Kinase
(myosin-light-chain kinase = MLCK) in ungestreiften Muskeln genutzt.
Die Calmodulin-Bindungsdomäne
der MLCK wirkt als Affinitätsmolekül und wird
an (F)RET-Partner gekuppelt. Abhängig
von der Ca2+-Konzentration, wird Calmodulin
an die Bindungsdomäne
gebunden und bewirkt eine Konformationsänderung der Detektiersonde.
Dies ergibt eine Änderung
des messbaren (F)RET.
-
7 zeigt
schematisch einen kompetitiven Immunoassay mit (F)RET-Partnern auf
1 Molekül,
welcher zur Bestimmung der Konzentration eines Analyt 26 in
einer Körperprobe
angewandt wird. Nanopartikel 1 und Chromophor 2 werden über ein
verbindendes Molekül 29 verbunden,
welches an ein Epitop 27 gebunden wird. Das Epitop 27 wird
gemäß einem
Epitop des zu detektierenden Analyt 26 erstellt. Das Affinitätsmolekül 28 wird
spezifisch an das Epitop 27 gebunden. Durch Zugabe einer
Probe (z.B. einer Körperprobe),
die den zu detektierenden Analyt 26 enthält, wird
das noch an das Epitop 27 gebundene Affinitätsmolekül 28 aus
diesem Epitop 27 verdrängt.
Dies führt
zu einer Konformationsänderung
eines Affinitätsmoleküls 29 und
somit zu einer messbaren Änderung
der Wechselwirkung zwischen den (F)RET-Partnern, nämlich dem
Nanopartikel 1 und dem Chromophor 2. Diese (F)RET-Änderung
wird zur Bestimmung der Konzentration des Analyt 26 herangezogen.
-
8 zeigt
schematisch einen homogenen Sättigungsimmunoassay
mit (F)RET-Partnern auf unterschiedlichen Molekülen. Die Affinitätsmoleküle von lad-Nanopartikel 110 bzw.
Chromophor 120 vermögen
unterschiedliche Epitope desselben Ziel-Moleküls 130 zu erkennen,
was zu einem messbaren Energietransfer in der Gegenwart des Ziel-Moleküls 130 führt. Ein
Beispiel eines homogenen Immunassay, bei welchem Donor und Akzeptor
auf unterschiedlichen Molekülen
angeordnet werden, stellt die Detektion von hCG (human chorional
Gonadotropin) in Serum dar. Dabei werden Donor und Akzeptor an Antikörper gekuppelt,
die unterschiedliche Epitope des hCG erkennen. Bei Vorliegen von
hCG in einer Körperprobe
werden Donor- und Akzeptor-Sonden an den Analyt gebunden. Der messbare
FRET kann zur Bestimmung der Konzentration des Analyt in der Körperprobe
mittels einer Eichkurve herangezogen werden.
-
9 zeigt
schematisch einen homogenen, kompetitiven Immunoassay mit (F)RET-Partnern 210 und 220 auf
unterschiedlichen Molekülen.
Ein oder mehr Chromophore 220 werden mit Molekül 222 verbunden, welches
teilweise oder vollständig
einem zu detektierenden Molekül 224 entspricht.
Der lad-Nanopartikel 210 wird
an ein Affinitätsmolekül 212 gekuppelt,
welches spezifisch mit dem Molekül 222 und
dem zu detektierenden Molekül 224 wechselwirkt.
Eine Bindung entsteht zwischen dem Affinitätsmolekül 212 und dem Molekül 222,
um dadurch einen (F)RET zu ermöglichen.
Wird nun eine Probe (z.B. eine Körperprobe),
die das zu detektierende Molekül 224 enthält, zugegeben,
erfolgt eine Verdrängungsreaktion
in Abhängigkeit
von der Konzentration des in der genannten Probe zu detektierenden
Moleküls 224.
Dies erzeugt eine messbare Änderung,
in diesem Fall eine Verringerung des (F)RET, wodurch die Bestimmung
der Konzentration des zu detektierenden Moleküls mittels einer Eichkurve
ermöglicht
wird.
-
10 zeigt
schematisch einen homogenen Assay mit (F)RET-Partnern auf 1 Molekül. Ein lad-Nanopartikel 310 und
ein Chromophor 320 werden über ein Peptid 330 als
Affinitätsmolekül verbunden.
Dieses Peptid kann durch ein zu detektierendes Enzym 340 gespalten
werden. Nach dieser Spaltung kann der (F)RET nicht länger beobachtet
werden.
-
Der
Assay von 10 kann zur Bestimmung einer
spezifischen Enzymaktivität,
z.B. einer für
einen HI-Virus spezifischen Protease, herangezogen werden. Beide
(F)RET-Partner werden durch die kurze Identifikationssequenz dieser
Protease verbunden und räumlich
voneinander durch die Aktivität
dieser Protease getrennt, was zur Peptidspaltung führt. Die
zu detektierende Enzymaktivität
kann auch aus einer Restriktionsendonuclease stammen. Dann werden
beide (F)RET-Partner durch Nucleinsäure verbunden.
-
11 zeigt
schematisch einen Assay gemäß einem
Verfahren molekularer Leuchttürme
(Signale). Molekulare Leuchttürme
sind DNA-Moleküle, die
die Befähigung
zur Selbstfaltung durch intermolekulare komplementäre Sequenzen
zu einer sogenannten Stamm-Schleife- oder Haar-Pin-Struktur aufweisen.
1 lad-Nanopartikel 410 wird an 1 Terminus einer DNA-Sequenz 430 gekuppelt.
Der andere Terminus wird an ein Chromophor 420 als Fluoreszenz-Löschungsmittel
oder -Löscher
gebunden. In der Haar-Pin-Struktur
sind beide (F)RET-Partner 410 und 420 in enger
Nachbarschaft angeordnet. Die Fluoreszenz des Donors 410 wird
deshalb vollständig gelöscht. Ein
Ziel-Molekül 440 zeigt
Sequenzen, die zur Schleifenregion der DNA-Sequenz 430 komplementär sind.
Da die Bindung des Ziel-Moleküls 440 energetisch
günstiger
ist, werden die Haar-Pin-Konformation aufgelöst, der Chromophor 420 und
der lad-Nanopartikel 410 voneinander getrennt und eine
messbare Fluoreszenz ausgestrahlt, da der (F)RET nicht länger die
Fluoreszenzlöschung
verursacht. Die Hybridismuseigenschaften können in einer solchen Weise
angepasst werden, dass eine einzelne Basenpaar-Fehlordnung zwischen dem molekularen
Leuchtturm 430 und der Ziel-DNA 440 nicht zu einer Öffnung der
Haar-Pin-Struktur führt.
Somit wird es ermöglicht,
sogar einzelne Basenunterschiede (z.B. SNPs, einzelne Nucleotid-Polymorphismen)
zu detektieren.
-
Die
in 11 dargestellte Verfahrenstechnik kann auch zum
Markenschutz und/oder zur Sicherheitsmarkierungen von Produkten
angewandt werden. Wird ein Produkt mit DNA (Fragmenten) markiert,
die an beiden Enden kurze komplementäre Strukturen aufweisen, von
denen die eine mit einem Kern/Schale-Partikel der vorliegenden Erfindung
und die andere mit oben erläuterten
Akzeptoren verbunden werden, ist ein (F)RET im entstandenen molekularen
Leuchtturm (der Haar-Pin-Struktur) feststellbar. Sobald diese DNA
(das Fragment) mit der komplementären Struktur in Kontakt gebracht
werden, löst
der Hybridismus die Haar-Pin-Struktur auf, um dadurch den (F)RET
zu verhindern. Dies ermöglicht
eine spezifische Identifikation sowie den Schutz von Handelsprodukten.
Markenschutz auf Basis einer Identifikation mit synthetischer DNA
wurde bereits, z.B. von November AG, Deutschland, in den Handel
gebracht.
-
III.2 Weitere Anwendungen
-
Unabhängig von
ihrer Anwendung in Bioassayverfahren ermöglichen die beanspruchten Kern/Schale-Partikel
ganz allgemein eine (F)RET-basierte Messung von Nanometer-Abständen in
biologischen oder weiteren Systemen, wenn jene mit einem geeigneten
Lumineszenz-Akzeptor kombiniert werden. Derartige Messungen können z.B.
von Interesse für
spektroskopische Zwecke in den Nanomaterial-Wissenschaften sein.
-
Außerdem können die
beanspruchten Kern/Schale-Partikel für verschiedene industrielle
Vorrichtungen und Produkte verwendet werden, in denen ausgezeichnete
(Foto-)Lumineszenzeigenschaften gefordert werden.
-
Zu
diesem Zweck werden sie in typischer Weise als Dispersion in fluiden
oder festen Medien zubereitet.
-
Geeignete
fluide Medien umfassen z.B. ein organisches oder wässriges
Dispersionsmedium, eine Überzugszusammensetzung,
eine Tinte oder einen Farbstoff, eine Polymerzusammensetzung oder
ein Aerosol. Geeignete organische Dispersionsmedien schließen, ohne
darauf eingeschränkt
zu sein, Toluol, CHCl3 oder CH2Cl2 ein.
-
Die
Synthese mit N- oder P-haltigen Medien/Komplexiermitteln, wie oben
beschrieben, gewährleistet die
rasche Dispergierbarkeit der Kern/Schale-Partikel gemäß der vorliegenden
Erfindung in organischen Medien.
-
Die
Zubereitung einer wässrigen
Dispersion kann eine Nachbehandlung erforderlich machen, wobei die
Rückstände von
in der Synthese verwendeten organischen Materialien durch Lösungsmittel
mit einer Funktionalitätsbindung
an der Oberfläche
der Partikel und an einem Molekül-Teil ersetzt werden,
wodurch die notwendige Kompatibilität in Wasser, gegebenenfalls
in Kombination mit Wasser-mischbaren Lösungsmitteln, gewährleistet
wird.
-
Das
feste Dispersionsmedium kann aus einem Überzug, einer Tinte oder einem
Farbstoff, einer Polymerzusammensetzung und insbesondere aus einem
Polymerfilm ausgewählt
sein.
-
Die
Nanopartikel als solche oder in typischer Weise ein fluides oder
festes Medium, die diese enthalten, können z.B. zur Lichterzeugung,
für Druck-
oder Markierungserzeugnisse und -materialien verwendet werden.
-
Derartige
Anwendungen sind z.B. Licht-emittierende Dioden, Displays, optoelektronische
Vorrichtungen, z.B. Verstärker
in nm-Abmessungen und Lichtquellen in Zero-Schwellen-Lasern. Sie
können
auch eine Tinte in Druckvorrichtungen betreffen, was von großer Bedeutung
in der Sicherheitsmarkierung von Dokumenten oder Banknoten ist.
-
IV. Beispiele
-
Beispiel 1: CePO4-Nanopartikelkerne mit einer TbPO4-Schale
-
In
einem 100 mL-Rundkoben mit Hochleistungs-Rückflusskühler, Temperatursonde und Heizmantel werden
3,72 g (10 mmol) CeCl3 × 7H2O
in ca. 4 mL Methanol gelöst,
worauf 40 mL Tris-2-ethylhexylphosphat (TEHP) zur entstandenen Lösung gegeben
werden. Ein Vakuum wird an den Rundkolben angelegt, um Methanol
und Kristallisationswasser zuerst bei Raumtemperatur (1 bis 2 h
lang) und dann bei 50°C
(ca. 1,5 h lang) zu entfernen.
-
In
einem zweiten Kolben wird trockene Orthophosphorsäure (20
mmol) in 5 mL Tetraethylenglykoldimethylether gelöst.
-
Unter
einer Stickstoff-Atmosphäre
werden bei 50°C
13,1 mL (30,0 mmol) Trioctylamin und 2,5 mL Orthophosphorsäure/Tetraethylenglykoldimethylether-Mischung zur CeCl3-Lösung
in TEHP gegeben. Danach wird die Mischung 15 h lang bei 200°C erhitzt.
Nach dieser Zeitspanne wird eine klare Dispersion ("erste Mischung") aus CePO4-Partikeln (Durchschnittsdurchmesser: 5
mm) erhalten.
-
In
einem zweiten 100 mL-Rundkolben mit Hochleistungs-Rückflusskühler, Temperatursonde
und Heizmantel werden 3,72 g (10 mmol) TbCl3 × 6H2O in ca. 4 mL Methanol aufgelöst, worauf
40 mL Tris-2-ethylhexylphosphat (TEHP) zur Lösung gegeben werden. Nach Anlegen
eines Vakuum an diesen Rundkolben werden Methanol und Kristallisationswasser
zuerst bei Raumtemperatur (1 bis 2 h lang) und dann bei 50°C (1,5 h
lang) entfernt. Unter einer Stickstoff-Atmosphäre weden bei 50°C 13,1 mL
(30 mmol) Trioctylamin und 2,5 mL (10 mmol Phosphorsäure) Orthophosphorsäure/Tetraethylenglykoldimethylether-Lösung sowie 14 mL der CePO4-Dispersion (abgekühlt auf ca. 20 bis 30°C) zur TbCl3-Lösung
(zur "zweiten Mischung") gegeben, worauf
bei 200°C
12 h lang erhitzt wird. Nach Abkühlen
auf Raumtemperatur wird die Reaktionsmischung zur Ausfällung der
Kern/Schale-Nanopartikel in Methanol gegossen. Der Niederschlag
wird zentrifugiert (bei 5500 U/min), worauf die entstandenen Partikel
mit Methanol gewaschen und getrocknet werden.
-
Das
Fotolumineszenzspektrum dieser Partikel, dargestellt in 2,
belegte deren Kern/Schale-Struktur.
-
TEM-Messungen
zeigten ferner an, dass die Partikel einen Durchschnittsdurchmesser
(entlang ihrer längsten
Achse) von ca. 6 nm aufwiesen.
-
1 zeigt
die Fluoreszenzspektren homogener CePO4-Partikel
(Linie 1), sowie Kern/Schale-Partikel gemäß der vorliegenden
Erfindung (Linien 2 und 3), worin eine TbPO4-Schale um die CePO4-Partikel
herum gewachsen ist. Das durch die Linie 2 reflektierte
Spektrum wurde nach einer Reaktionszeit von 0,5 h aufgenommen, wogegen
die Linie 3 die Fluoreszenz von CePO4-Kernen
mit einer vollständig
entwickelten TbPO4-Schale zeigt (Reaktionszeit
von 18 h). Die Spektren wurden bei derselben optischen Dichte (10–3 Gew.-%)
in i-Propanol (λexc = 274 nm) aufgenommen.
-
Wie
aus 1 ersichtlich, sind die homogenen CePO4-Partikel durch eine starke Fluoreszenzemission
bei ca. 330 nm gekennzeichnet, zeigen aber keine Emission im sichtbaren
Bereich. Diese Situation wird dramatisch durch den TbPO4-Überzug verändert. Ce3+ absorbiert ganz stark die Anregungsstrahlung, überträgt die absorbierte
Energie auf Tb3+, das diese dann in der
Form von 4 starken charakteristischen Banden bei 488, 545, 586 und
617 nm wieder ausstrahlt. Dieser Energietransfer führt zu einer
erniedrigten Ce-Emission und lässt
die Tb-Emission stark ansteigen.
-
Beispiel 2: LaPO4-Nanopartikelkerne mit einer TbPO4-Schale
-
In
einem 100 mL-Rundkolben mit Hochleistungs-Rückflusskühler, Temperatursonde und Heizmantel werden
3,2 g (8,6 mmol) LaCl3 × 7H2O
in ca. 10 mL Methanol gelöst,
worauf 39 mL Tris-2-ethylhexylphosphat (TEHP) zur entstandenen Lösung gegeben
werden. Ein Vakuum wird an den Kolben zur Entfernung des Methanols
und des Kristallwassers zuerst bei Raumtemperatur (1 bis 2 h lang)
und dann bei 50°C
(einige h lang) angelegt.
-
Unter
einer Stickstoff-Atmosphäre
werden bei 50°C
11,5 mL (26,3 mmol) Trioctylamin und 2,3 mL Orthophosphorsäure/Tetraethylenglykoldimethylether-Mischung zur LaCl3-Lösung
in TEHP gegeben. Danach wird die Mischung 16 h lang bei 200°C erhitzt.
Nach dieser Zeitspanne wird eine klare Dispersion (eine "erste Mischung") aus LaPO4-Partikeln erhalten.
-
In
einem zweiten 100 mL-Rundkolben mit Hochleistungs-Rückflusskühler, Temperatursonde
und Heizmantel werden 2,97 g (8 mmol) TbCl3 × 6H2O in ca. 10 mL Methanol aufgelöst, worauf
35,2 mL Tris-2-ethylhexylphosphat (TEHP) zur Lösung gegeben werden. Nach Anlegen
eines Vakuum am Rundkolben werden das Methanol und das Kristallwasser
zuerst bei Raumtemperatur (1 bis 2 h lang) und dann bei 50°C (einige
h lang) entfernt. Unter einer Stickstoff-Atmosphäre werden bei 50°C 10,5 mL
(24 mmol) Trioctylamin und 2,0 mL (8 mmol Phosphorsäure) Orthophosphorsäure/Tetraethylenglykoldimethylether-Lösung sowie
die gesamte Menge der LaPO4-Dispersion (abgekühlt auf
ca. 20 bis 30°C)
zur TbCl3-Lösung (zur "zweiten Mischung") gegeben, worauf bei 200°C 16 h lang
erhitzt wird. Nach Abkühlen
auf Raumtemperatur wird die Reaktionsmischung in Methanol (300 mL)
zur Ausfällung
der Kern/Schale-Nanopartikel gegossen. Der Niederschlag wird zentrifugiert (bei
5500 U/min), worauf die entstandenen Partikel 2 Mal mit Methanol
gewaschen und dann getrocknet werden.
-
Referenzbeispiel 1: Analyse
der Kern/Schale-Partikel
-
Dieses
Referenzbeispiel beschreibt die Messung der chemischen Zusammensetzung,
des Kerndurchmessers sowie der Schalendicke von CePO4:Tb-Nanopartikelkernen
mit einer LaPO4-Schale. Obwohl diese Partikel
nicht durch die Ansprüche
gedeckt sind, ist das vorliegende Analyseverfahren auf die vorliegende
Erfindung voll anwendbar.
-
Zu
diesem Zweck wurden die Partikel auf einen mit Löchern versehenen Kohlefilm
montiert und unter einem Philipps CM300UT-Mikroskop untersucht.
-
EELS
(Elektronenenergieverlustspektroskopie) zeigte, dass die chemische
Durchschnittszusammensetzung der Kationen war:
Ce/La = 0,34 ± 0,05,
Tb/La = 0,12 ± 0,03,
was bedeutet, dass
La/Ce = 3,0 ± 0,4 und Ce/Tb = 2,8 ± 0,8,
wobei der letztere Wert in etwa dem angewandten Molverhältnis Ce/Tb (3,14/1)
entspricht.
-
HREM
(Hochauflösungselektronenmikroskopie)
bestätigte
die Kristallinität
der erhaltenen Kern/Schale-Partikel.
-
Außerdem wurde
ein Hellfeldbild mit einem leichten Unterbrennpunkt bei einer Rasterrate
von 0,48 nm/Bildpunkt aufgenommen, um auch kleinere Partikel abzudecken.
Die Analyse dieses Bildes zeigte, dass die Hauptpartikelklasse bezüglich des
Volumens Durchmesser von 5 bis 9 nm ergab. 6 dieser Partikel wurden mit
EFTEM (Energie-Filtertransmissionselektronenmikroskopie), in spezifischer
Weise mit dem sogenannten "Spektrum-Bildverfahren", untersucht, das
vom "Landeszentrum
für Hochleistungsspektroskopie,
Institut für Anorganische
Chemie" in Bonn,
Deutschland, zur quantitativen Analyse entwickelt wurde. Zu diesem
Zweck wurden 6 kristalline Partikel bei sehr hoher Vergrößerung auf
einer CCD-Kamera
hinter einem Abbildungsenergiefilter zentriert. Dann wurden der
kleinste Objektivschirm (4,6 mrad) und der größte Eingangsschirm (3 mm) eingesetzt
und der Energiefilter in seinem Spektroskopiemodus angewandt. Dadurch
wird die komplette Intensität,
die durch den Eingangsschirm geht, Linie für Linie auf den Detektor abgebildet.
Wegen der chromatischen Aberration der Linse bildet dieses Verfahren
nur einen Ausschnitt von ca. ±40
eV mit hoher Schärfe (unterhalb
nm) so ab, dass dieser auf den LaM5- und
CeM5,4-Kanten bei 832, 849, 884 und 902
eV fokussiert wird. Bei der ausgewählten Primärvergrößerung von 99 K betrug der
Durchmesser des Eingangsschirms immer 11,2 nm.
-
2 zeigt
(D) das Hellfeldbild (mit Eingangsschirm) von 1 CePO4:Tb-Nanopartikel,
umgeben von LaPO4-Schale (Durchmesser: ca.
7 nm), (E) das Spektrumbild bei 860 eV Energieverlust sowie Profile
durch die Partikeloberfläche
(A, C) und das Zentrum (B). Die Profile (A, B und C) zeigen die
LaM5,4- und CeM5,4-Peaks, deren
relative Intensität
in etwa der lokalen Zusammensetzung entspricht. Die unterschiedlichen
Profile bestätigen
das Vorliegen der Kern/Schale-Struktur eines an Ce reichen Kerns
und einer an La reichen Schale. Die 6 ausgewählten Partikel wiesen einen
Durchschnittsdurchmesser von 7,5 ± 1,9 nm aus einem Ce-reichen
Kern mit einem Durchmesser von 4,0 ± 1,1 nm und einer La-reichen
Schale mit einer Dicke von 1,9 ± 0,7 nm auf (Tb wurde in
dieser Analyse nicht bestimmt).
-
Beispiel 3: Kupplung von
Fluorescein an die Kern/Schale-Partikel aus Beispiel 1 (CePO4/TbPO4)
-
3-1: Amino-Funktionalisierung
der Kern(CePO4)-Schale(TbPO4)-Nanopartikel
mit Iminobis(methylenphosphono)undecansäure und 1,4-Bis(3-aminopropoxy)butan
-
0,388
g (1 mmol) Iminobis(methylenphosphono)undecansäure werden mit 1,5 mL Ethylenglykol
und 0,51 g (2,5 mmol) 1,4-Bis(3-aminopropoxy)butan durch Erwärmen auf
50°C über 30 min
gelöst.
Die entstandene Lösung
ist leicht gelblich. 25 mg (= 71,5 nmol) in Beispiel erhaltene Kern(CePO4)-Schale(TbPO4)-Nanopartikel werden zur Lösung gegeben,
gerührt
und bei 120°C
4 h lang erhitzt. Die Dispersion ist trüb und leicht gelb. Nach Dialyse
(Dialyseleitung: Spectra/Por, 5–6.000
MWCO, Spektrum, Niederlande) über
Nacht gegen 2 × 2
L 10 mM Na-Carbonat-Puffer, pH = 8,5, fallen die Partikel aus.
-
3-2: Kupplung von Fluorescein
an die Amino-funktionalisierten Kern(CePO4)-Schale(TbPO4)-Nanopartikel
-
5
mg (25 nmol) der Amino-funktionalisierten Nanopartikel (oben beschrieben)
werden 10 min bei 5000 U/min zentrifugiert, worauf das Pellet in
500 μL 0,2
M Na-Carbonat-Puffer, pH = 8,5, resuspendiert wird, um eine Konzentration
von 10 mg/mL zu ergeben. FITC (Fluoresceinisothiocyanat) wird in
einer 1:1-Lösung
von DMF und 0,2 m Na-Carbonat-Puffer, pH = 8,5, auf eine Konzentration
von 5 mmol/mL gelöst.
Ein 17-facher Überschuss
(87 μL =
429 nmol) wird zu den Partikeln gegeben, worauf die Mischung unter
Rotieren bei Raumtemperatur 4,5 h lang inkubiert wird. Das ungebundene
FITC wird an einer Sephadex G-25 M PD 10-Säule (Amersham BioScience) mit
10 mM Na-Carbonat-Puffer, pH = 8,5, als Elutionspuffer abgetrennt.
Die eluierte Fraktion von 3,9 mL enthält Fluorescein-gekuppelte Nanopartikel.
-
Vergleichsbeispiel (Comparative
Example = CE) 1: Kupplung von Fluorescein an homogene LaPO4:Ce,Tb-Partikel
-
CE1-1: Herstellung von
LaPO4:Ce,Tb-Nanopartikeln
-
300
mL TEHP (Tris-(2-ethylhexyl)phosphat) wurden in einem trockenen
Stickstoff-Strom entgast. Danach werden 7,43 g LaCl3 × 7H2O (20 mmol), 8,38 g CeCl3 × 7H2O (22,5 mmol) und 2,8 g TbCl3 × 6H2O (7,5 mmol) in 100 mL Methanol gelöst und zum
TEHP gegeben. Danach werden das Wasser und das Methanol unter Vakuum
bei einer Temperatur von 30 bis 40°C entfernt. Dann wird eine frisch
zubereitete Lösung
von 4,9 g trockener Orthophosphorsäure (50 mmol), gelöst in einer
Mischung aus 66,5 mL Trioctylamin und 150 mL TEHP, zugegeben. An
die erhaltene klare Lösung
wird sofort ein Vakuum angelegt, und es wird mit Stickstoff gespült, um die
Oxidation von Ce3+ bei Temperaturerhöhung zu
minimieren. Danach wird die Lösung
auf 200°C erhitzt.
Die Erhitzungsphase wird beendet, wenn die Siedetemperatur auf 175°C abgesunken
ist (nach ca. 30 bis 40 h). Nach Abkühlen auf Raumtemperatur wird
ein 4-facher Überschuss
von Methanol zugegeben, um die Partikel auszufällen. Die Partikel werden abgetrennt,
mit Methanol gewaschen und getrocknet.
-
CE1-2: Entalkylierung
mit Bromtrimethylsilan
-
300
mg der LaPO4:Ce,Tb-Nanopartikel (ca. 850
nmol), erhalten in CE1-1, werden 4 h lang mit 2,3 g Bromtrimethylsilan
(15 mmol) in 100 mL Chloroform am Rückfluss gehalten. Die Hauptmenge
des Bromtrimethylsilan-Überschusses
sowie flüchtige
Zwischenprodukte werden unter Vakuum entfernt und abgetrennt. Der Nanopartikel-haltige
Rückstand
wird über
Nacht unter Rühren
in einer Mischung aus 6 mL Wasser und 100 μL Ammoniak (25%ig) hydrolysiert.
Die entstandenen Partikel bilden eine milchige Suspension und sedimentieren teilweise
nach einigen h. Sie können
durch Zentrifugation abgetrennt werden.
-
CE1-3: Amino-Funktionalisierung
der entalkylierten LaPO4:Ce,Tb-Nanopartikeln mit
Iminobis(methylenphosphono)capronsäure
-
0,5
g (1,75 mmol) Iminobis(methylenphosphono)capronsäure (KBD 9267) werden mit 0,894
g (4,375 mmol) 1,4-Bis(3-aminopropoxy)butan (Fluka) in 2 mL Ethylenglykol
unter Erwärmen
bei 50°C über 30 min
gelöst.
Die entstandene Lösung
ist leicht gelblich. 35 mg (= 175 nmol) in CE1-2 erhaltene LaPO4:Ce,Tb-Nanopartikel werden zur Lösung gegeben,
gerührt
und bei ca. 120°C
4 h lang erhitzt. Die Dispersion ist auch nach Abkühlung auf
Raumtemperatur klar und bräunlich.
Nach Dialyse gegen 2 × 2
L 10 mM Na-Carbonat-Puffer, pH
= 8,5, ist die Lösung
leicht gelb und klar.
-
CE1-4: Kupplung von Fluorescein
an die Amino-funktionalisierten LaPO4:Ce,Tb-Nanopartikel
-
Die
Lösung
der Amino-funktionalisierten LaPO4:Ce,Tb-Nanopartikel
aus CE1-3 wird im Vakuum auf ca. 4,8 mg/mL eingeengt. FITC (Fluoresceinisothiocyanat)
wird in einer 1:1-Lösung
aus DMF und 0,2 m Na-Carbonat-Puffer, pH = 8,5, auf eine Konzentration
von 5 mmol/mL gelöst.
Ein 17-facher Überschuss
(87 μL =
429 nmol) wird zu 5 mg (ca. 1 mL = 25 nmol) der Nanopartikel gegeben,
worauf die Mischung unter Rotieren bei Raumtemperatur 4,5 h lang
inkubiert wird. Ungebundenes FITC wird an einer Sephadex G-25 M
PD 10-Säule
(Amersham Bioscience) mit 10 mM Na-Carbonat-Puffer, pH = 8,5, als
Elutionspuffer abgetrennt. Die eluierte Fraktion von 3,5 mL enthält Fluorescein-gekuppelte
Nanopartikel.
-
Beispiel 4: Messung des
FRET mit den Fluorescein-gekuppelten Nanopartikeln
-
Die
Fluorescein-gekuppelten Kern(CePO4)-Schale(TbPO4)-Partikel des Beispiels 3 sowie die homogenen
LaPO4:Ce,Tb-Partikel des Vergleichsbeispiels
1 wurden mit verschiedenen spektroskopischen Analysen zur Bestimmung
des FRET-Wirkungsgrads untersucht.
-
Die
Messungen wurden mit einem FL3-22-Spektrometer von Jobin Yvon mit
wässrigen
Dispersionen der Probe in optischen Zellen mit einer Breite und
Tiefe von jeweils 1 cm durchgeführt.
Die Konzentration wurde so ausgewählt, dass die optische Dichte
0,3 nicht überstieg.
-
3 zeigt
die Abfallkurven von 2 Partikel-Typen, die im TRF-Modus nach gepulster
Anregung bei 280 nm gemessen wurden:
- – Die gestrichelte
Linie stellt die Abfallkurve bei 542 nm (Tb-Emission) der in Beispiel
1 erhaltenen unmodifizierten Kern(CePO4)-Schale(TbPO4)-Partikel, Halbwertszeit: 1,4 ms, dar;
- – die
fett gedruckte Linie stellt die Abfallkurve bei 542 nm der in Beispiel
3 erhaltenen Fluorescein-gekuppelten Kern(CePO4)-Schale(TbPO4)-Partikel,
Halbwertszeit: 0,02 bis 0,1 ms, dar;
- – die
dünne Linie
stellt die Abfallkurve bei 520 nm (Fluoresceinemission) der in Beispiel
3 erhaltenen Fluorescein-gekuppelten Kern(CePO4)-Schale(TbPO4)-Partikel, Halbwertszeit von 0,02 bis 0,06
ms, dar.
-
4 zeigt
die Abfallkurven der Fluorescein-gekuppelten, homogenen LaPO4:Ce,Tb-Partikel des Vergleichsbeispiels
1, die im TRF-Modus nach gepulster Anregung bei 280 nm gemessen
wurden:
- – Die
fett gedruckte Linie stellt die Abfallkurve bei 542 nm (Halbwertszeit
ca. 1,7 ms) dar;
- – die
dünne Linie
stellt die Abfallkurve der gleichen Partikel bei 520 nm (Halbwertszeit:
ca. 1,0 bis 1,5 ms) dar.
-
Die
Fluorescein-gekuppelten Kern(CePO4)-Schale(TbPO4)-Partikel des Beispiels 3 zeigen viel kürzere Fluoreszenz-Halbwertszeiten
(bei 542 nm: 0,02 bis 0,1 ms) als die Partikel des Vergleichsbeispiels
1 (ca. 1,7 ms). Dies zeigt an, dass die Kern/Schale-Struktur der
Partikel gemäß der Erfindung
einen viel effizienteren Energietransfer zum Akzeptormolekül (zum Fluorescein)
ermöglichen,
um dadurch den FRET-Wirkungsgrad zu steigern. Die Fluoreszenz-Halbwertszeit
(ca. 1,4 ms) der unmodifizierten Kern(CePO4)- Schale(TbPO4)-Partikel ist somit um das ca. 14-Fache
höher als
die für
die Fluorescein-gekuppelten Kern/Schale-Partikel des Beispiels 3
beobachtete.
-
Die
Fluoreszenzhalbwertszeit der unmodifizierten homogenen LaPO4:Ce,Tb-Nanopartikel (nicht gekuppelt an
Fluorescein) beträgt
ca. 2,4 ms, d.h., sie ist um das ca. 1,5-Fache höher als die für die entsprechenden
Fluorescein-gekuppelten Partikel des Vergleichsbeispiels 1 (bei
542 nm: ca. 1,7 ms) beobachtete. Bei Vergleich dieses Verhältnisses
(von ca. 1,5/1) mit dem in 3 dargestellten
Verhältnis
(ca. 14/1) zeigt sich die Verbesserung des mit der vorliegenden
Erfindung erzielten FRET-Wirkungsgrads ganz klar.
-
5a zeigt
Fluoreszenzspektren, gemessen im TGF-Modus nach gepulster Anregung
bei 280 nm und einem Messverzug von 40 μs nach dem letzten Anregungspuls:
- – die
fett gedruckte Linie stellt das Spektrum der in Beispiel 1 erhaltenen
unmodifizierten Kern(CePO4)-Schale(TbPO4)-Partikel dar; und
- – die
dünne Linie
stellt das Spektrum der in Beispiel 3 erhaltenen Fluorescein-gekuppelten Kern(CePO4)-Schale(TbPO4)-Partikel
dar.
-
Der
linke Intensitätsmaßstab entspricht
der fett gedruckten Linie und der rechte entspricht der dünnen Linie.
Das Emissionsspektrum der modifizierten Kern/Schale-Partikel (aus
Beispiel 3) ist durch eine sehr niedrige Intensität der charakteristischen
Tb3+-Bande bei 545 nm (erniedrigt auf ca.
1/40) und durch das Auftreten einer neuen breiten Bande bei 520
nm gekennzeichnet, die aus der Fluoresceinemission stammt. Nach
Energietransfer der Anregungsenergie (bei 280 nm) von den Ce3+(Kern)- zu den Tb3+(Schale)-Lumineszenzzentren lösen die
letzteren eine Fluoresceinlumineszenz durch FRET aus.
-
5b zeigt
1 Fluoreszenzspektrum, gemessen in TGF-Modus nach gepulster Anregung
bei 280 nm und einem Messverzug von 40 μs nach dem letzten Anregungspuls,
von
- – den
Fluorescein-gekuppelten, homogenen LaPO4:Ce,Tb-Partikeln
des Vergleichsbeispiels 1.
-
Dieses
Spektrum bestätigt
ebenfalls das Auftreten des FRET, da eine relativ breite Emissionsbande bei
ca. 520 nm, die aus Fluorescein stammt, beobachtbar ist. Allerdings
ist die durch die charakteristische Tb3+-Bande
bei 545 nm reflektierte Donoremission noch viel stärker als
in 4a (dünne Linie), was den niedrigeren
FRET-Wirkungsgrad belegt.
-
Referenzbeispiel 2 (Reference
Example 2 = RE2): Bioassayverfahren
-
In
den folgenden Referenzbeispielen sind spezifische Verfahrenstechniken
zur Bindung homogener Nanopartikel an Biomoleküle mit den entsprechend markierten
Biomolekülen
in biologischen Assayverfahren dargestellt. Obwohl die eingesetzten
Nanopartikel keine erfindungsgemäßen sind,
sind diese Referenzbeispiele auf die beanspruchten Kern/Schale-Partikel vollkommen übertragbar.
In diesem Zusammenhang ist sich der Fachmann bewusst, dass Oberflächenmodifikationsverfahren,
wie sie im Folgenden für
homogene Nanopartikel dargestellt sind, bevorzugt auch auf Kern/Schale-Partikel,
die in ähnlicher
Weise synthetisiert wurden, insbesondere bezüglich der Wahl des Lösungsmittels
angewandt werden, das in typischer Weise an der Oberfläche der
erhaltenen Nanopartikel gebunden wird.
-
RE2-1: Carboxy-Funktionalisierung
der LaPO4:Ce,Tb-Nanopartikel
-
LaPO4:Ce,Tb-Partikel wurden wie in Vergleichsbeispiel
CE1-1 hergestellt und zubereitet.
-
50
mg dieser Nanopartikel (ca. 140 nmol) werden 3 h lang bei 110°C zusammen
mit 5 mL Ethylenglykol (ca. 180 mmol) und 5 μL Schwefelsäure (96 bis 98%ig) unter Rühren und
Inertgas erhitzt. Alternativ dazu, ist es möglich, weitere Diole, vorzugsweise
Polyethylenglykole verschiedener Kettenlängen, am meisten bevorzugt
HO-(CH2-CH2O)n-OH, worin n = 2 bis 9, zu verwenden. Die
Partikel beginnen sich in Ethylenglykol bei ca. 135°C aufzulösen. Nach
Beendigung der Behandlung werden ein Vakuum von ca. 1,5 mbar angelegt
und ca. die Hälfte
der Ethylenglykolmenge entfernt. Dies ergibt einen klaren Rückstand.
Danach wird der Rückstand gegen
Wasser über
Nacht dialysiert (Dialyseleitung: Spectra/Por, 5–6.000 MWCO, Spectrum, Niederlande).
-
0,5
mL Schwefelsäure
(96 bis 98%ig) werden dann zu einer Lösung von 100 mg (ca. 300 nmol)
der erhaltenen Nanopartikel in 20 mL Wasser gegeben. 1 mM KMnO4-Lösung
wird zur entstandenen Mischung getropft, bis die Entfärbung der
violetten Farbe nicht länger
beobachtbar ist. Danach wird die gleiche Menge KMnO4-Lösung erneut
zugegeben und dann über
Nacht gerührt
(> 12 h lang). Überschüssiges Permanganat wird
durch Zutropfen einer frisch zubereiteten 1 mM Natriumsulfit-Lösung reduziert.
Die entstandene Mischung wird über
Nacht gegen 0,1 M MES, 0,5 M NaCl, pH = 6,0, dialysiert (Dialyseleitung:
Spectra/Por, 5–6.000
MWCO, Spectrum, Niederlande).
-
RE2-2: Entalkylierung
mit Bromtrimethylsilan
-
300
mg (ca. 850 nmol) in Vergleichsbeispiel CE1-1 hergestellte LaPO4:Ce,Tb-Nanopartikel wurden genauso wie in
Vergleichsbeispiel CE1-2 behandelt.
-
RE2-3: Kupplung der LaPO4:Ce,Tb-Nanopartikel mit 11-Bis(phosphorylmethyl)aminoundecansäure und 1,4-Bis(3-aminopropoxy)butan
-
11-Bis(phosphorylmethyl)aminoundecansäure wird
durch Erhitzen einer Mischung aus 201 g 11-Aminoundecansäure, 170
g Phosphorsäure,
200 mL konzentrierter Salzsäure
und aus 200 mL Wasser bei 100°C und
anschließendes
Zutropfen von 324 g (Formalin) (37%ig) über 1 h und durch Rühren 1 h
lang bei 100°C hergestellt.
Nach Abkühlen
auf Raumtemperatur wird das ausgefallene Produkt durch Vakuum-gestützte Filtration
isoliert und unter Vakuum getrocknet. Dadurch werden 334 g 11-Bis(phosphorylmethyl)aminoundecansäure erhalten. Ähnlich geeignet
sind die entsprechenden Säuren
mit 2 bis 18 Kohlenstoffatomen.
-
0,5
g (1,85 mol) 11-Bis(Phosphorylmethyl)aminoundecansäure werden
in 2 mL Ethylenglykol gelöst, worauf
0,894 g (4,375 mmol) 1,4-Bis(3-aminopropoxy)butan zugegeben werden.
Nach Bildung einer klaren Lösung
(exotherme Reaktion) werden 35 mg (100 nmol) in RE2-2 erhaltene
LaPO4:Ce,Tb-Nanopartikel bei 50°C zugegeben und dann bei 125°C erhitzt.
Bei ca. 120°C
lösen sich
die Partikel vollständig
auf. Nach 4 h wird eine klare, leicht bräunliche Lösung erhalten, die sogar nach
Abkühlen
auf Raumtemperatur klar bleibt. Die Reaktionsmischung wird gegen
2 × 2
L 10 mM Natriumcarbonat-Puffer,
pH = 8,5, dialysiert (Dialyseleitung: Spectra/Por, 5–6000 MWCO,
Spectrum, Niederlande). Das erhaltene Dialysat enthält die ausgefällten Nanopartikel.
-
RE2-4: Biotinylierung
der LaPO4:Ce,Tb-Nanopartikel aus RE2-3
-
6,2
mL (= 5 mg oder ca. 15 nmol) der in RE2-3 erhaltenen Nanopartikelmischung
werden in einem Rotationsverdampfer auf 4,81 mg/mL eingeengt. Unter
weiterem Rotieren wird die erhaltene Dispersion 4 h lang mit einem
20-fach molaren Überschuss
von Biotin-X-NHS (Sulfobiotinaminocapronsäure-N-hydroxysuccinimidester,
Calbiochem, Schwalbach, Deutschland) inkubiert und dann gegen PBS-Puffer
(8 mM K2HPO4; 150
mM NaCl; 2 mM Na2HPO4;
pH = 7,4) dialysiert (Dialyseleitung: Spectra/Por, 5–6000 MWCO,
Spectrum, Niederlande). Das erhaltene Dialysat ist leicht neblig.
-
RE2-5: Kupplung von DNA-Oligonucleotid
an die LaPO4:Ce,Tb-Nanopartikel von RE2-3
-
Die
in RE2-3 erhaltenen Nanopartikel werden mit einem 40-fachen Überschuss
von Sulfo-SIAB (Sulfosuccinimidyl(4-jodacetyl)aminobenzoat, Perbio
Science Deutschland GmbH, Bonn, Deutschland) aktiviert: 7,5 mg (ca.
25 nmol) Amino-funktionalisierte Nanopartikel werden erneut in einer
Centricon-Filtriereinheit (MW-Ausschluss bei 50.000, Millipore,
Eschborn, Deutschland) in TSMZ-Puffer, pH = 7,3 (0,1 M NaCl; 0,1
M Triethanolamin-HCl;
0,02 M NaOH; 0,27 mM ZnCl2; 0,1% Tween 20;
1 mM MgCl2) gepuffert und auf eine Konzentration
von ca. 7 mg/mL eingestellt. 50 μL
20 mM Sulfo-SIAB-Lösung in
Wasser werden zur Partikeldispersion gegeben und dann 15 min lang
bei 25°C
inkubiert. Die Reaktion wird durch die Zugabe von 12 μL 1 M Glycin
(12-facher Überschuss)
beendet, worauf das freie Sulfo-SIAB an einer Sephadex G25 PD 10-Säule (Amersham
Pharmacia Biotech, Freiburg, Deutschland) abgetrennt wird. Ein DNA-Oligonucleotid
mit der Sequenz 5'-CCACGCTTGTGGGTCAACCCCCGTGG-3' und einer Thiol-Modifikation
am 5'-Terminus und
einer Dabcyl-Modifikation (4-(4-Dimethylaminophenylazo)benzoyl)
am 3'-Terminus sowie als
Vergleich ein DNA-Oligonucleotid, das sich nur beim fehlenden Dabcyl-Molekül am 3'-Terminus von der
Sonde unterschied, wurden von Interactiva (Ulm, Deutschland) bestellt. Äquimolare
Mengen des DNA-Oligonucleotids
und der SIAB-aktivierten Nanopartikel wurden vermischt und bei 25°C 3 h lang
und über
Nacht bei 4°C
inkubiert. Die an das DNA-Oligonucleotid
gekuppelten Nanopartikel wurden von nicht-gekuppelten Partikeln
und vom freien DNA-Oligonucleotid mittels FPLC (Fast Performance
Liquid Chromatography) abgetrennt. Die gekuppelten Partikel wurden
in 50 mM Tris-HCl, pH = 7,4, und in 0,1% BSA bei 4°C aufbewahrt.
Solange keine Ziel-DNA vorhanden ist, faltet sich das erhaltene
Molekül
in einer Haar-Pin-Struktur,
worin beide Termini des Moleküls
in enger Nachbarschaft zueinander vorliegen und FRET auftreten kann.
Unter diesen Bedingungen wird die Nanopartikel-Fluoreszenz durch
Dabcyl gelöscht.
-
RE2-6: Kupplung eines
anti-β-hCG-Monoklonalantikörpers an
die LaPO4:Ce,Tb-Nanopartikel aus RE2-3
-
Zuerst
werden in RE2-3 erhaltenen LaPO4:Ce,Tb-Nanopartikel
mit einem 30-fach molaren Überschuss
von 2-Iminothiolan (2-IT, Traut's
Reagens, Perbio Science Deutschland GmbH, Bonn) aktiviert: 2 mL (ca.
25 nmol, 4 mg/mL) dieser Partikel wurden in TSE-Puffer, pH = 8,5
(0,04 M NaCl; 0,05 M Triethanolamin-HCl; 0,04 M NaOH; 0,5 mM EDTA;
0,1% Tween 20; pH = 8,5) überführt. Zu
diesem Zweck werden sie 3 Mal 15 min lang bei 3000 g zentrifugiert,
der Überstand
wird abgegossen und der jeweils verbleibende Rückstand wird in 700 μL TSE-Puffer,
pH = 8,5, aufgenommen. Diese Partikel werden mit 75 μl 10 mM 2-IT
(in TSE-Puffer, pH = 8,5) bei 25°C
1 h lang inkubiert, worauf die Reaktion mit 9 μL (12-fachem Überschuss)
1 M Glycin beendet wird. Zur Abtrennung des 2-IT-Überschusses
wird die entstandene Mischung erneut 3 Mal 15 min lang bei 3000
g zentrifugiert, worauf der Überstand
abgegossen und der Niederschlag 2 Mal in 1 mL TSE-Puffer, pH = 7,3
(0,1 M NaCl; 0,1 M Triethanolamin-HCl; 0,02 M NaOH; 1 mM EDTA; 0,1%
Tween 20; pH = 7,3) und nach der 3. Zentrifugation in 250 μL TSE-Puffer,
pH = 7,3, resuspendiert werden. Gleichzeitig wird eine äquimolare
Menge von monoklonalem Maus-Antikörper, der für β-hCG spezifisch ist (Klon F199C1,
Perkin-Elmer Life Sciences-Wallac Oy, Finnland), mit einem 40-fachen Überschuss
von SMCC (N-Succinimidyl-4-(N-maleimidomethyl)cyclohexan-1-carboxylat,
Perbio Science Deutschland GmbH, Bonn) aktiviert. 750 μL anti-β-hCG-Antikörper (=
25 nmol bei einer Konzentration von 5 mg/mL) werden in einer Centricon-Filtriereinheit
(MG-Ausschluss bei 50.000) in TSMZ-Puffer, pH = 7,3 (0,1 M NaCl;
0,1 M Triethanolamin-HCl; 0,02 M NaOH; 0,27 mM ZnCl2;
0,1% Tween 20; 1 mM MgCl2) erneut gepuffert
und auf eine Konzentration von 7 mg/mL eingestellt. 50 μL 20 mM SMCC-Lösung in
DMF (= 1 mmol) werden zu dieser Antikörper-Lösung gegeben und dann bei 25°C 30 min
lang inkubiert. Die Reaktion wird durch Zugabe von 12 μL 1 M Glycin
(12-facher Überschuss)
beendet und das freie SMCC wird an einer gebrauchsfertigen Sephadex
G25 PD 10-Säule (Amersham
Pharmacia Biotech, Freiburg, Deutschland) abgetrennt. Schließlich werden äquimolare
Mengen der 2-IT-aktivierten
Nanopartikel-Dispersion und der SMCC-aktivierten Antikörper-Lösung vermischt und 3 h lang
bei 25°C
und dann über
Nacht bei 4°C
inkubiert. Die Antikörper-gekuppelten
Nanopartikel werden von nicht-gekuppelten
Partikeln und freien Antikörpern
mit Gelpermeationschromatografie an Superdex 200 (Amersham Pharmacia
Biotech, Freiburg, Deutschland) gereinigt. 0,1 M MES, 0,5 M NaCl,
pH = 6,0, werden als Puffer-Eluierungsmittel
verwendet. Die Retentionszeit für
die gekuppelten Nanopartikel beträgt ca. 2 h.
-
RE2-7: Kupplung der LaPO4:Eu3+-Nanopartikel
mit hIL-2
-
LaPO4:Eu3+-Nanopartikel
wurden in TEHP wie in der Literatur (J. Phys. Chem. B 2000, 104, 2824–2828) mit
dem einzigen Unterschied erzeugt, dass 1,76 g LaCl3 × 7H2O anstatt des diesbezüglich genannten Nitrats verwendet
wurden. 300 mg (ca. 1 μmol)
dieser Nanopartikel wurden unter Rückfluss zusammen mit 2,23 g
(15 mmol) Bromtrimethylsilan in 125 mL Chloroform 4 h lang erwärmt. Der
Hauptteil des Bromtrimethylsilan-Überschusses und der gebildeten
Zwischenprodukte wird abdestilliert, worauf der Rückstand
unter leichten alkalischen Bedingungen hydrolysiert wird. Zu diesem
Zweck wird der Rückstand
mit 6 mL Wasser, wozu 100 μL
Ammoniak (25%ig) gegeben wurden, behandelt und über Nacht gerührt. Die
entstandenen Partikel bilden eine milchige Dispersion und sedimentieren
teilweise nach einigen h. 5 mg (= 25 mmol, 106 μL) dieser Bromtrimethylsilan-behandelten
Nanopartikel wurden 1 h lang bei 37°C unter Schütteln mit rekombinantem menschlichen
IL-2-Protein (R&D
Systems, Mineapolis, MN, USA) in 10 mM Natriumcarbonat-Puffer, pH = 8,5,
in einem Molverhältnis
von 2:1 inkubiert. Anschließend
wird überschüssiges Protein
durch Zentrifugieren der entstandenen Mischung 6 Mal 10 min lang
bei 3.000 g abgetrennt und dann jeweils in 1 mL Natriumcarbonat-Puffer,
pH = 8,5, resuspendiert. Das LaPO4:Eu3+/IL-2-Konjugat
wird bei 4°C
aufbewahrt.
-
RE2-8: Homogener Energietransfer-Assay
zum Detektieren von β-hCG
mit den Antikörper-gekuppelten
Nanopartikeln aus RE2-6 als Donor und mit Fluoreszenz-gekuppelten
Antikörpern
als Akzeptor
-
Kupplung von anti-β-hCG-Antikörpern an
Fluorescein:
-
Fluororeporter®-FITC-Protein-Markierungskit,
hergestellt von Molecular Probes, wurde zur Kupplung von Fluorescein
an anti-β-hCG-Antikörper (M15294,
Perkin-Elmer Life Sciences, Wallac Oy, Finnland) gemäß den Instruktionen
des Herstellers angewandt. 0,5 mg Antikörper wurden in einer Centricon-Filtriereinheit (MG-Ausschluss
bei 50.000) in 0,2 M Hydrogencarbonat-Puffer, pH = 9,0, erneut gepuffert.
Die Antikörperlösung wird
dann mit einem 25-fachen Überschuss
von 5 mM Fluoresceinisothiocyanat (FITC)-Lösung (gelöst in einer Mischung des gleichen
Volumens aus DMF und 0,2 M Hydrogencarbonat-Puffer, pH = 9,0) 3
h lang bei Raumtemperatur inkubiert. Der FITG-Überschuss wird an einer gebrauchsfertigen
Sephadex G25 PD 10-Säule
(Amersham Pharmacia Biotech, Freiburg, Deutschland) abgetrennt,
und die Antikörper-Konzentration
und das Verhältnis
Fluorescein/Antikörper
werden spektroskopisch bestimmt. 0,01% Natriumazid und 0,1% BSA werden
zum Konjugat gegeben, das bei 4°C
aufbewahrt wird.
-
Durchführung des Assay:
-
50 μL β-hCG-Standards
aus einem im Handel erhältlichen
Kit zur Messung von freiem β-hCG
in Serum (A007-101, Perkin-Elmer Life Sciences, Wallac Oy, Finnland)
werden 60 min lang bei 25°C
zusammen mit 100 nmol in RE2-6 erhaltenen Nanopartikel-Antikörper-Konjugaten
und 100 nmol Fluorescein-gekuppelten anti-β-hCG-Antikörpern in 200 μL Tris-HCl-Puffer,
pH = 7,4, in einer UV-durchlässigen
96-Lochvertiefungen-Mikrotiterplatte (UVStar, Greiner) inkubiert.
Die 2 anti-β-hCG-Antikörper sind
gegen unterschiedliche Epitope der β-hCG-Untereinheit gerichtet.
Danach werden die Proben in einem Fluoreszenz-Spektrometer (von
Jobin Yvon, Fluorolog 3) unter den folgenden Bedingungen gemessen:
gepulste Anregung bei einer Wellenlänge von 280 nm, Emission: 542
nm, Spaltbreite: 5 nm, Integrationszeit: 0,1 ms. Die für die individuellen β-hCG-Konzentrationen
erhaltenen Ergebnisse werden in eine Eichkurve eingegeben. Der β-hCG-Gehalt
von Körperproben
kann in analoger Weise wie in Serumproben durch Bestimmung der Konzentration
auf Basis dieser Eichkurve gemessen werden.
-
RE2-9: Homogener kompetitiver
Energietransferassay zur Bestimmung von hIL-2 mit den hIL-2-gekuppelten Nanopartikeln
(LaPO4:Eu3+) aus
RE2-7 und mit Alexa Fluor 680-gekuppelten anti-hIL-2Rα-Kette-Antikörpern
-
Kupplung monoklonaler
anti-hIL-2Rα-Kette-Antikörpern mit
Alexa Fluor 680:
-
1
mg monoklonaler Antikörper
7G7B6, der spezifisch die α-Kette
von menschlichem Interleukin-2-Rezeptor (hIL-2α-Kette) (ATCC, Rockville, USA)
erkennt, wurde gegen PBS dialysiert, auf eine Konzentration von 2
mg/mL eingestellt und mit Alexa Fluor 680-Protein-Markierungskit
(Molecular Probes Europe BV, Niederlande) gemäß den Instruktionen des Herstellers
markiert. Mit 0,1 M Natriumbicarbonat-Puffer (pH = 8,3) als Reaktionspuffer
wurde die Inkubation bei Raumtemperatur 1 h lang durchgeführt. Der
gekuppelte Antikörper
wird an einer Säule,
die im Kit enthalten ist, mit PBS-Puffer und 0,2 mM Na-Azid als
Eluierungspuffer gereinigt. In einer 1 cm optischen Zelle wird die
Absorption (A) bei 280 und 679 nm zur Bestimmung der Protein-Konzentration
des gekuppelten Antikörpers
gemessen, wobei diese mit der folgenden Gleichung berechnet wird:
worin 203000 cm
–1M
–1 den
molaren Extinktionskoeffizient von IgG darstellt und 0,05 der Korrekturfaktor
für die Absorption
des Farbstoffs bei 280 nm ist. Die Konzentration des gekuppelten
Antikörpers
beträgt
1,27 und wird auf 1 mg/mL (ca. 6,5 μM) mit PBS und 0,2 mM Na-Azid
eingestellt. Der gekuppelte Antikörper wird bei 4°C aufbewahrt.
Der Markierungswirkungsgrad wird wie folgt berechnet:
worin 184000 cm
–1m
–1 den
molaren Extinktionskoeffizient des Alexa Fluor 680-Farbstoffs bei
679 nm darstellt. Das Verhältnis
von Antikörper/Farbstoff-Konjugat
beträgt
3,2.
-
Durchführen des Assay:
-
Die
notwendigen Verdünnungen
der verschiedenen Komponenten werden mit 50 mM TSA-Puffer (50 mM
Tris-HCl, pH = 7,75; 0,9% NaCl; 0,05% NaN3)
erhalten. 40 Lochvertiefungen von UV-durchlässigen Mikrotiterplatten (UVStar,
Greiner) werden zuerst mit MSA-Lösung
(0,5%) 1 h lang bei Raumtemperatur inkubiert, um unspezifische Bindung
abzusättigen,
worauf eine Mischung des LaPO4:Eu3+/IL-2-Konjugats aus Beispiel RE2-7, Alexa
Fluor 680-markiertem anti-hIL-2α-Kette-Antikörper und
aus rekombinantem hIL-2sRα-Protein (aus
menschlichem IL-2-löslichen-Rezeptor-α, R&D Systems, Mineapolis,
MN, USA) jeweils mit einer Endkonzentration von 40 nM zugegeben
wurde. 20 dieser Lochvertiefungen werden mit nicht-markiertem hIL-2-Protein
in unterschiedlichen Konzentrationen beladen, wobei 20 mit einem
Protein mit keiner Relevanz für
diesen Assay zurückbleiben.
Jede Konzentration wird mit 50 nM zum serienmäßigen Test einer Konzentration
von 0 bis 950 nM gesteigert. Das Endvolumen der Reaktion beträgt in jedem
Fall 200 μL.
Die Inkubation wird im Dunkeln 45 min lang bei Raumtemperatur auf
einem Schüttler
durchgeführt.
Die Signale werden mit einem Wallac 1420 VictorTM Multilabel-Zähler (Perkin-Elmer
Life Sciences, Wallac Oy, Finnland) unter den folgenden Bedingungen
gemessen: Anregung: 340 nm, Emission: 665 nm, Zeitverzug: 50 μs, Zeitfenster:
200 μs und
Kreislaufzeit: 1000 μs.
Jeder Wert wird 2 Mal bestimmt und auf Basis der Ergebnisse für unspezifische
Bindung, erhalten mit dem irrelevanten Protein, korrigiert. Die
gemessenen Werte werden gegen die Proteinkonzentrationen in einem
Diagramm aufgetragen, um eine Eichkurve zu ergeben, mittels derer
die Konzentrationen des menschlichen Interleukin-2 bestimmbar sind.
Dies ist in analoger Weise für
menschliche Körperproben
genauso möglich.
-
RE2-10 Quantitative PCR-Bestimmung
bakterieller DNA durch intramolekularen Energietransfer mit den DNA-Oligonucleotid-gekuppelten
LaPO4:Ce,Tb-Nanopartikeln aus RE2-5
-
Der
Primer und die Sonde zur quantitativen DNA-Bestimmung wurden spezifisch
für das
RNA-Polymerase-Gen von Mycobacterium tuberculosis selektiert und
mit Interactiva (Ulm, Deutschland) erzeugt. Der Primer wies die
folgende Sequenz auf: Vorwärts:
5'-GGCCGGTGGTCGCCGCG-3', Rückwärts: 5'-ACGTGACAGACCGCCGGC-3'.
-
Assay zur
quantitativen Bestimmung bakterieller DNA
-
Für 50 μL-PCR-Reaktionen
wurden 50 nM der Nanopartikel (Dabcyl-Oligonucleotid-gekuppelt) aus RE2-5
als Sonde, 500 nM von jeweils beiden Primern, 2 E Amplitaq Gold-DNA-Polymerase
(Perkin-Elmer), 250 μM
dATP, 250 μM
dCTP, 250 μM
dGTP, 500 μM
dUTP, 4 mM MgCl2, 50 mM KCl und 10 mM Tris-HCl
(pH = 8,0) vermischt. Genomische M. tuberculosis-DNA wird als DNA-Templat
mit den gleichen Primern amplifiziert und in ein Plasmid mit dem
Invitrogen Zero Blunt TOPO PCR-Klonier-Kit (Invitrogen BV/NOVEX,
Niederlande) kloniert. Zum Erhalt einer Standardkurve werden 5 unterschiedliche
Konzentrationen des DNA-Plasmids von 1 pg bis 100 ng sowie eine
Reaktion ohne DNA-Templat verwendet. 30 Reaktionen wurden für jede Konzentration
so zubereitet, dass, beginnend nach dem 15. Zyklus, eine Probe nach
jedem zusätzlichen
Zyklus zur spektrometrischen Messung derselben gezogen werden konnte.
Das Reaktionsvolumen betrug 50 μL,
und die Amplifikation wurde mit einem Thermocycler (PCR-System 2400,
Perkin-Elmer) unter den folgenden Reaktionsbedingungen durchgeführt: 10
min bei 95°C;
15 bis 45 Zyklen von 30 s bei 95°C,
45 s bei 56°C
und 30 s bei 72°C.
Die Proben wurden in einem Fluoreszenz-Spektrometer (von Jobin Yvon,
Fluorolog 3) unter den folgenden Bedingungen gemessen: gepulste
Anregung bei einer Wellenlänge
von 280 nm, Emission: 542 nm, Spaltbreite: 4 nm, Zeitverzug: 50 μs, Wiederholungsrate
von ca. 25 Hz. In gleicher Weise ist es möglich, die Halbwertszeit der
Terbium-Emissionslinie zu bestimmen. Für diesen Zweck wurden die folgenden
Bedingungen angewandt: Anregung: 280 nm, Emission: 542 nm, Spaltbreite:
5 nm, Integrationszeit: 0,1 ms. Während des Hybridismus von Sonde
und Ziel-DNA tritt kein intramolekularer FRET zwischen dem gekoppelten
Nanopartikel und Dabcyl auf. Mit steigender Ziel-DNA-Konzentration steigt die Tb-Fluoreszenz
des Nanopartikels deshalb gegenüber
der Vergleichsprobe ohne Templat an. Gleichzeitig verlängert sich
die Halbwertszeit der Nanopartikel-Fluoreszenz-Lebensdauer gegenüber der
Vergleichsprobe ohne Templat-DNA. Diese Differenzen beider Parameter
können
gegen die Zyklenzahl aufgetragen werden, um eine Eichkurve für jede DNA-Templat-Konzentration
zu erstellen.





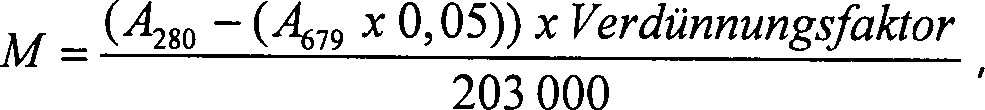 worin 184000 cm–1m–1 den molaren Extinktionskoeffizient des Alexa Fluor 680-Farbstoffs bei 679 nm darstellt. Das Verhältnis von Antikörper/Farbstoff-Konjugat beträgt 3,2.
worin 184000 cm–1m–1 den molaren Extinktionskoeffizient des Alexa Fluor 680-Farbstoffs bei 679 nm darstellt. Das Verhältnis von Antikörper/Farbstoff-Konjugat beträgt 3,2.