-
Diese
Patentanmeldung ist eine teilweise Fortsetzung der ebenfalls anhängigen US-Patentanmeldung 09/871,772,
eingereicht am 01.06.2001 von Nikolay E. Nifantiev als Erfinder,
mit dem Titel: "Wasserlösliche Porphyrinderivate
und Methoden zu ihrer Herstellung". Diese Patentanmeldung ist als Referenz
in diesen Patentantrag eingeschlossen.
-
Die
Erfindung bezieht sich auf die Chemie biologisch aktiver Verbindungen,
insbesondere auf eine neue Methode, um wasserlösliche Porphyrinderivate, speziell
Chlorin-, Bakteriochlorin-, Phäophorbid-
und Bakteriophäophorbid-Derivate
der Typen 1 und 2 herzustellen. Die Substanzen, die Gegenstand der
vorliegenden Erfindung sind, können
als Photosensibilisatoren für
die photodynamische Therapie von Tumorerkrankungen, von Infektionen
und anderen Erkrankungen wie auch für eine Behandlung unter Lichtanregung
in anderen Fällen
genutzt werden.
-
-
Worin
B ein Ring mit folgender Struktur ist:
-
Wobei:
R1 = -CH=CH2, -CH(OAlk)CH3, -CHO, -C(O)CH3,
-CH2CH3, -CH(Alk)CH(COAlk)2, -CH2CH(COAlk)2, -CH(Alk)CH2COAlk,
-CH(Alk)CH2CH(OH)CH3,
oder -CH2CH2CH(OH)CH3
R2 = -CH3, -CHO, -CH(OH)Alk, -CH=CHAlk, CH2OH, oder CH2OAlk;
R3 = -OH, -OAlk, -NH-Alk, -NH-X-COO–(HG)+, -NH-Y-NR8R9, oder -NH-Y-OH;
R4 =
-O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+;
R5 = -O–(HG)+, -OAlk, -NH-Alk, oder NH-X-COO–(HG)+;
R6 = -H oder
-COOAlk;
R7 = -O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+;
R8 = -H oder
-Alk;
R9 = -H oder -Alk
-
Wobei:
-NH-X-COO– =
ein organischer Aminosäurerest;
X
= Alkyliden, Peptide, Oligopeptide oder -(CH2CH2O)nCH2CH2-, wobei n = 1 – 30;
Y = Alkyliden oder
-(CH2CH2O)nCH2CH2-, wobei n =
1 – 30;
G
= ein hydrophiles organisches Amin (z. B. N-Methyl-D-glucamin oder
andere Aminogruppen-enthaltende Kohlenhydrat-Derivate, TRIS, Aminosäuren, Oligopeptide);
und
Alk = ein Alkyl-Substituent.
-
Die
Photodynamische Therapie (PDT) ist eine der vielversprechendsten
neuen Techniken, die auf ihre Eignung für eine Vielzahl von medizinischen
Anwendungen hin überprüft wird
(Photodynamic therapy, basic principles and clinical applications.
Eds. B.W. Henderson, Th.J. Dougherty, Marcel Dekker, 1992, New York). Sie
ist insbesondere anerkannt für
die Zerstörung
von Tumorgewebe (Photodynamic tumor therapy. 2nd and
3rd generation photosensitizers. Ed. J.G.
Moser, Harwood Academic Publishers, 1998, Amsterdam). Porphyrine sind
Substanzen, die häufig
für die
PDT genutzt werden. Ein zentrales Problem für die pharmazeutische Anwendung
von Porphyrinen ist ihre geringe Löslichkeit in physiologischen
Lösungen.
Dies macht es beinahe unmöglich,
effektive injizierbare Lösungen
in pharmazeutischer Qualität
für PDT
und andere Anwendungen herzustellen.
-
Methoden
zur Herstellung wasserlöslicher
Porphyrinderivate für
die PDT sind bereits Stand der Technik bekannt. Das U.S.-Patent
Nr. 5,330,741 von Smith et al. beschreibt eine Methode, um Trinatrium-lysyl-chlorin
p6 darzustellen, indem man Purpurin-18-methlyester,
hergestellt durch Modifikation von Methylphäophorbid a, mit wässerigem
Lysin in Dichlormethan in Gegenwart von Pyridin umsetzt. Das Gemisch
wird für
12 Stunden bei Raumtemperatur gerührt, anschließend werden
die Lösungsmittel
im Hochvakuum entfernt. Das auf diese Weise hergestellte Rohprodukt
wird mittels Reversed-Phase-HPLC gereinigt und anschließend lyophylisiert. Um
hieraus eine injizierbare Lösung
für die
PDT-Tumorbehandlung herzustellen, wird das Produkt zuerst in Phosphatpufferlösung gelöst und anschließend wird
0,1 N Natriumhydroxid hinzugefügt.
Der pH-Wert der Lösung
wird durch Zusatz von 0,1 N HCl auf pH 7,35 eingestellt, dann folgt
eine Sterilfiltration durch einen mikroporösen Filter.
-
Die
Nachteile der oben erwähnten
Methode umfassen u. a. eine mangelnde Reproduzierbarkeit, Schwierigkeiten
bei der Aufarbeitung und die Verwendung toxischer Reagenzien. Daher
ist diese Methode nicht geeignet für die pharmazeutische Produktion.
Darüber
hinaus ist das auf diese Weise hergestellte wasserlösliche Produkt
in wässeriger
Lösung
lediglich für
24 h bei 4°C
im Dunkeln stabil, in fester Form beträgt die Stabilität nur bis
zu 4 Monaten bei 4°C
im Dunkeln [M.W. Leach, R.J. Higgins, J.E. Boggan, S.-J. Lee, S.
Autry, K.M. Smith, Effectiveness of a Lysylchlorin p6/Chlorin
p6 mixture in Photodynamic Therapy of the
Subcutaneous 9L Glioma in the Rat. Cancer Res., 1992, 52, 1235 – 1239;
U.S. Patent Nr. 5,330,741].
-
Im
U.S. Patent Nr. 5,378,835 von Nakazato et al. wird eine Methode
zur Darstellung eines wasserlöslichen
Natriumsalzes von Phäophorbid
a (3) beschrieben. Nach der Beschreibung dieser Erfindung wird Phäophorbid
a (4) in Diethylether gelöst.
Hierzu wird eine sehr verdünnte
Lösung
von Alkali in n-Propanol, iso-Propanol oder einer Mischung dieser
beiden Lösungsmittel
sehr langsam hinzugetropft. Die Reaktion wird so lange fortgeführt, bis
das Phäophorbid-a-Salz
vollständig
ausgefallen ist, dieses wird dann durch Zentrifugieren abgetrennt
und in vacuo getrocknet. Das Produkt wird in Wasser gelöst, wobei
eine Lösung
mit einer Konzentration von 0,5% und einem pH-Wert von 9,2 – 9,5 entsteht,
diese wird dann mit Phosphatpuffer verdünnt auf einen pH-Wert von 7,4 – 7,8.
-
Der
Nachteil der von Nakazato beschriebenen Methode liegt in der Tatsache,
dass es mit dieser Methode nicht möglich ist, eine konzentrierte
(> 1%) injizierbare
Lösung
von Phäophorbid
a in Wasser herzustellen. Darüber
hinaus haben die Autoren der vorliegenden Erfindung gezeigt, dass
diese Salze bei trockener Lagerung chemisch instabil sind, und dass
sie sich nur unvollständig
in Wasser auflösen,
nachdem sie in trockenem Zustand gelagert worden sind.
-
-
Die
dem vorliegenden Patent ähnlichste
Methode ist in dem russischen Patent Nr
RU 2144538 von G.V. Ponomarev et al.
offen gelegt. Darin wird die Darstellung wasserlöslicher Komplexe von Chlorin
e
6 beschrieben (7) und zwar mit Hilfe raumerfüllender
organischer Amine unter Einschluss von N-Methyl-D-glucosamin in
einer Multischritt-Reaktionssequenz: Gewinnung von Chlorophyll a
aus der Biomasse von Cyanobakterien der Art Spirulina Platensis,
Umwandlung in Chlorin e
6 mit Hilfe von Standardmethoden
[S. Lötjönen, P.H. Hynninen,
An improved method for the preparation of (10R)- and (10S)- pheophytins a and
b. Synthesis, 1983, 705 – 708;
P.H. Hynninen, S. Lötjönen, Preparation
of phorbin derivatives from chlorophyll mixture utilizing the principle
of selective hydrolysis. Synthesis, 1980, 539 – 541; S. Lötjönen, P.H. Hynninen, A convenient
method for the preparation of wet chlorin e
6 and
rhodin g
7 trimethyl esters. Synthesis, 1980,
541 – 543]
mit einer Gesamtausbeute von über
50% nach Ausfällung
des Chlorins e
6 durch schrittweise Zugabe
von Wasser zu dessen Lösung
in Aceton, anschließende
Abtrennung durch Zentrifugieren, dreimaliges Waschen mit Wasser,
gefolgt von der Umsetzung des feuchten Chlorins e
6 mit
einer wässrigen
Lösung
von 2 g-eq. des raumerfüllenden organischen
Amins.
-
Die
entscheidenden Nachteile dieser Methode, die zu deutlichen Schwierigkeiten
bei der präparativen Synthese
wasserlöslicher
Chlorine, insbesondere für
industrielle Synthesen und für
die Arzneimittelherstellung führen,
sind die folgenden:
- 1. Das als Zwischenprodukt
auftretende Chlorin e6 fällt als feuchte Masse mit einem
nicht genau bestimmbaren Gehalt an Chlorin e6 an.
Diese Schwankung im Chlorin-e6-Gehalt führt zu Unsicherheiten, die
es sehr schwierig machen, die daraus gewonnenen Lösungen zu
standardisieren.
- 2. Das wichtigste Zwischenprodukt in der Synthesesequenz ist
Phäophorbid
a (4), welches wegen seiner sauren Eigenschaften schwer für Reinigung
und Standardisierung zu handhaben ist. Die Abtrennung von Phäophorbid
a (4) durch wiederholtes Ausfällen
(wie bei Ponomarev) gelingt nicht quantitativ und ist daher für die Darstellung
großer
Mengen ungeeignet.
- 3. Phäophorbid
a (4), das auf diese Weise gewonnen wird, enthält Verunreinigungen, die schwierig
abzutrennen sind. Dieser Nachteil führt zu Unsicherheiten bei der
Quantifizierung von Phäophorbid
a (4) und stört
bei der Öffnung
des Cyclopentanon-Ringes im Rahmen der Umwandlung von Phäophorbid
a (4) zu Chlorinen.
- 4. Es muss darauf hingewiesen werden, dass die Proben wasserlöslicher
Chlorin-e6-Salze, die entsprechend der Vorschrift
von Ponomarev hergestellt wurden, eine Vielzahl porphyrinartiger
und nicht-porphyrinartiger Verunreinigungen enthalten, die sich
mit den im Patent beschriebenen Methoden nicht von der Zielverbindung
Chlorin e6 abtrennen ließen. Unter den auftretenden
Verunreinigungen konnten mittels Dünnschichtchromatographie und
HPLC unter anderem Phäophorbid
a (4), Purpurin 18 (8), Chlorin p6 (9) und andere
begleitende Verunreinigungen nachgewiesen werden.
- Es wurde festgestellt, dass Verbindungen der Typen (4), (8)
und (9) der oben erwähnten
Erfindung als Salze mit hydrophilen Aminen durch eine, verglichen
mit ihren entsprechenden Chlorin-e6-Salzen,
deutlich geringere Wasserlöslichkeit
charakterisiert sind. Dennoch sind die Verbindungen des Typs (4),
(8) und (9) als Salze mit hydrophilen Aminen, wie in der oben erwähnten Erfindung,
in Gegenwart von Chlorin-e6-Salzen deutlich
besser wasserlöslich,
als man dies mit einer möglichen
Komplexbildung mit den Chlorin-e6-Salzen erklären könnte. Dieses
Phänomen
macht eine Abtrennung der Chlorin-e6-Produkte
von Verunreinigungen wie den Verbindungen der Typen (4), (8) und
(9) etwa unter Ausnutzung ihrer unterschiedlichen Wasserlöslichkeit
unmöglich.
- 5. Die organischen Amine, die von Ponomarev für die Darstellung
wasserlöslicher
Chlorine verwendet wurden, sind für eine praktische Anwendung
nicht optimal. Insbesondere D-Glucosamin, welches Komplexe mit Chlorinen
bildet, die durch eine höhere
Löslichkeit
gekennzeichnet sind, ist aufgrund der möglichen Oxidation seiner Aldehydgruppe
nicht stabil genug. Gleichzeitig kann D-Glucosamin in der Lösung in
mehreren isomeren Formen vorliegen, was Unsicherheiten in Bezug
auf die Struktur mit sich bringt und dementsprechend auch Schwierigkeiten
bei der genauen Charakterisierung der Struktur, was dazu führt, dass
die Qualitätskontrollanforderungen
für eine
pharmazeutische Herstellung nicht eingehalten werden können. Ein weiteres
raumerfüllendes
Amin, das von Ponomarev benutzt wird, N-Methyl-D-glucosamin, besitzt dieselben
Nachteile wie das oben erwähnte
D-Glucosamin und
ist darüber
hinaus wegen seiner aufwendigen Herstellung nur schlecht verfügbar.
- 6. Ponomarev behauptet die Bildung wasserlöslicher Salze von Chlorin e6 mit raumerfüllenden organischen Aminen,
was als sehr fragwürdig
angesehen werden muss, weil übliche
raumerfüllende
organische Amine, z. B. solche, die tert-Butyl-, Neopentyl-, Adamantyl-
oder Cyclohexyl-Gruppen enthalten, wegen der hohen Hydrophobie der
raumerfüllenden
organischen Reste nicht für
die Darstellung wasserlöslicher
Chlorin-e6-Salze verwendet werden können.
- Dieser Aspekt zusammen mit den anderen oben erwähnten Gründen macht
die von Ponomarev beanspruchte Methode ungeeignet für die Herstellung
von Zubereitungen in pharmazeutischer Qualität nach den GMP-Anforderungen.
-
Geeignete
Porphyrinderivate, die als Edukte für die im Rahmen der hier vorliegenden
Erfindung interessierenden Synthesen dienen können, werden herkömmlicherweise
aus den gereinigten bzw. Standard-Rohmaterialien Methylphäophorbid
a (5) oder Ethylphäophorbid
a (6) gewonnen. Die heute allgemein bekannten Methoden zur Abtrennung
von Porphyrinen von biologischem Rohmaterial bestehen aus einer
langen Abfolge von mühsamen
Waschvorgängen
mit organischen Lösungsmitteln
und/oder dem Einfrieren der Zubereitung, um die Zellwände des
Biomaterials zu zerstören,
und wiederholten Extraktionen in Verbindung mit der Behandlung der
Biomasse mit verschiedenen Chemikalien, um zuerst Chlorophyll zu
erhalten, welches dann in Phäophytin überführt wird,
das dann zu Phäophorbid
a hydrolysiert wird (K.M. Smith, D.A. Goff and D.J. Simpson, J.
Amer. Chem. Soc., 1985, 107, 4946 – 4954; R.K. Pandey, D.A. Bellnier,
K.M. Smith and T.J. Dougherty, Photochem. Photobiol., 1991, 53,
65 – 72).
-
Somit
gibt es Bedarf, eine einfache und effiziente Methode zur Herstellung
reiner und chemisch stabiler wasserlöslicher Porphyrinderivate mit
einem standardisierten Anteil der gewünschten Substanz zur Verfügung zu
stellen, die sich für
medizinische Anwendungen, insbesondere die photodynamische Therapie,
eignet. Die vorliegende Erfindung löst dieses Problem und besitzt
darüber
hinaus noch weitere Vorteile.
-
Gegenstand
der vorliegenden Erfindung ist es, chemisch stabile wasserlösliche Porphyrinderivate
mit einen standardisierten Anteil der gewünschten Substanz zur Verfügung zu
stellen, die für
verschiedenste medizinische Anwendungen, insbesondere die PDT, geeignet
sind.
-
Ein
weiterer Gegenstand der vorliegenden Erfindung ist es, hochreine,
wasserlösliche
Porphyrinderivate in pharmazeutischer Qualität zur Verfügung zu stellen, die in pharmazeutischen
Zubereitungen wirksam sind.
-
Ein
weiterer Gegenstand der vorliegenden Erfindung ist es, eine Methode
zur Darstellung chemisch-stabiler wasserlöslicher Porphyrinderivate zur
Verfügung
zu stellen.
-
Noch
ein weiterer Gegenstand der vorliegenden Erfindung ist es, eine
einfache und zeitsparende Methode zur Darstellung chemisch stabiler,
wasserlöslicher
Porphyrinderivate ausgehend von biologischen Rohmaterialien zur
Verfügung
zu stellen, die gleichzeitig die Nachteile des bisherigen Standes
der Technik vermeidet.
-
Ein
weiterer Gegenstand der vorliegenden Erfindung ist es, chemisch-stabile,
wasserlösliche
Porphyrinderivate in einer pharmazeutisch akzeptablen Zubereitung
zur Verfügung
zu stellen, die für
medizinische Anwendungen, wie z. B. die Behandlung von Tumorerkrankungen
und anderen hyperproliferativen Erkrankungen, von Infektionen und
anderen Erkrankungen, benutzt werden kann.
-
Eine
Ausführungsform
der vorliegenden Erfindung besteht aus einer Methode zur Darstellung
wasserlöslicher
Porphyrinderivate, die folgende Schritte umfasst: ein- oder zweischrittige
direkte saure Alkoholyse von biologischem Rohmaterial, die zu kristallinem
Alkylphäophorbid
führt,
Umwandlung des erhaltenen Alkylphäophorbids in ein saures Porphyrin
und die Reaktion des sauren Porphyrins mit einem hydrophilen organischen
Amin in Wasser oder einer wässerigen
organischen Lösung.
-
Eine
weitere Ausführungsform
der vorliegenden Erfindung besteht aus einer Methode zur Darstellung wasserlöslicher
Porphyrinderivate, die die Reaktion eines sauren Porphyrins in Wasser
oder einer wässerigen organischen
Lösung
mit einem hydrophilen organischen Amin umfasst.
-
Noch
eine weitere Ausführungsform
der vorliegenden Erfindung besteht aus einer Methode zur Darstellung
wasserlöslicher
Porphyrinderivate, die folgende Schritte umfasst: ein- oder zweischrittige
direkte saure Alkoholyse von biologischem Rohmaterial, die zu kristallinem
Alkylphäophorbid
führt,
Umwandlung des erhaltenen Alkylphäophorbids in ein saures Porphyrin,
Reaktion des sauren Porphyrins mit einem hydrophilen organischen
Amin in Wasser oder einer wässerigen
organischen Lösung
und Reinigung des entstandenen wasserlöslichen Porphyrinderivates
durch Reversed-Phase-Chromatographie mit Hilfe flüchtiger
organischer Lösungsmittel.
-
In
noch einer weiteren Ausführungsform
stellt die vorliegende Erfindung eine Methode zur Darstellung wasserlöslicher
Porphyrinderivate zur Verfügung,
die folgende Schritte umfasst: Reaktion des sauren Porphyrins mit
einem hydrophilen organischen Amin in Wasser oder einer wässerigen
organischen Lösung
und Reinigung eines wasserlöslichen
Porphyrinderivates durch Reversed-Phase-Chromatographie mit Hilfe
flüchtiger organischer
Lösungsmittel.
Darüber
hinaus stellt die vorliegende Erfindung wasserlösliche Porphyrinderivate der
Formeln (1) und (2) zur Verfügung,
die für
pharmazeutische Zubereitungen nützlich sind,
die durch die in der vorliegenden Erfindung beschriebenen Methoden
erhalten werden können
und die in der photodynamischen Therapie und anderen medizinischen
Anwendungen benutzt werden können.
-
1: Bestimmung der Dunkeltoxizität (Cytotoxizität, Beispiel
14) des wasserlöslichen
Salzes von Chlorin e
6 (7) mit N-Methyl-D-glucamin
(10) hergestellt (A) entsprechend der vorliegenden Erfindung (Beispiel 9)
und (B) nach Ponomarev (
RU 2144538 );
der Test wurde mit OV2774-Zellen unter Zusatz der jeweils angegebenen
Photosensibilisator-Konzentrationen durchgeführt.
-
2: Bestimmung der Phototoxizität (Beispiel
15) des wasserlöslichen
Salzes von Chlorin e
6 (7) mit N-Methyl-D-glucamin
(10) hergestellt (A) entsprechend der vorliegenden Erfindung (Beispiel
9) und (B) nach Ponomarev (
RU
2144538 ); der Test wurde mit OV2774-Zellen unter Zusatz
der jeweils angegebenen Photosensibilisator-Konzentrationen und
einer Bestrahlung bei 670 nm durchgeführt.
-
Vor
einer Beschreibung der Erfindung mag es hilfreich sein, hier einige
Definitionen bestimmter Begriffe zugeben, die in der folgenden Patentschrift
benutzt werden.
-
Porphyrine
sind makrozyklische Verbindungen mit Brücken von einem Kohlenstoffatom
bzw. einem Stickstoffatom, die Pyrrole so verbinden, dass eine charakteristische
Tetrapyrrol-Ringstruktur entsteht. Es gibt viele verschiedene Klassen
von Porphyrinderivaten, eingeschlossen Derivate mit Dihydropyrrol-Einheiten.
Der Ausdruck Porphyrine wird hier so verwendet, dass er Porphyrine,
Phthalocyanine, Chlorine, Phäophorbide
und die jeweiligen Metallkomplexe sowie andere porphyrinartige Verbindungen
einschließt,
die für
die PDT und pharmazeutische Anwendungen geeignet sind.
-
In
der hier verwendeten Bedeutung sind biologische Rohmaterialien Materialien
zur Darstellung von Verbindungen aus der vorliegenden Erfindung.
Diese umfassen z. B. Pflanzen, Algen, Blutkomponenten und Insektenabsonderungen.
-
Das
Ziel der vorliegenden Erfindung wird durch die beschriebene Methode
erreicht, welche die Reaktion eines sauren Porphyrins in Wasser
oder wässeriger
organischer Lösung
mit einem hydrophilen organischen Amin umfasst, vorzugsweise N-Methyl-D-glucamin
(10), welches eine polyhydroxylierte stabile und nicht-toxische
Verbindung für
die Arzneimittelherstellung ist, oder mit Aminoalkyl- und Aminoarylglykosiden
wie den Maltosederivaten (11) und (12) oder anderen Aminogruppen-enthaltenden
Kohlenhydratderivaten. Weitere mögliche
Reagenzien schließen
folgende Verbindungen ein, sind aber nicht auf diese be schränkt: Tris(hydroxymethyl)aminomethan
(auch bekannt als TRIST) (13), ebenfalls
eine stabile und nicht-toxische Verbindung, die für die Arzneimittelherstellung
geeignet ist, oder TRIS-Derivate, wie die Verbindungen (14) und
(15), wie auch andere Arten hydrophiler Amine, wie z. B. Bis(2-hydroxyethyl)amin
(16). Aminosäuren
oder Oligopeptide wie z. B. Oligolysine, vorzugsweise Penta- und
Hexalysine, können
ebenfalls als hydrophile organische Amine genutzt werden, die für die Darstellung
wasserlöslicher
Porphyrinderivate gemäß der vorliegenden Erfindung
geeignet sind.
-
-
Entsprechend
einer bevorzugten Ausführungsform
der vorliegenden Erfindung, wie sie in Beispiel 9 beschrieben ist,
wird die quantitative stöchiometrische
Reaktion eines chemisch reinen Porphyrins (als freie Säure) und
eines geeigneten hydrophilen aromatischen Amins bei Raumtemperatur
unter Inertgas und im Dunkeln durchgeführt. Das benutzte Lösungsmittel
ist entweder chemisch reines Wasser, das unter Inertgas (z. B. Argon,
Helium oder ähnliche
Gase) entgast worden ist, oder aber, falls dies notwendig ist, ein
Gemisch aus Wasser mit einem geeigneten chemisch reinen und entgasten
organischen Lösungsmittel.
Das organische Lösungsmittel
wird anschließend
ohne Erhitzen im Vakuum entfernt (um eine mögliche Zerstörung des
Ausgangsporphyrins zu verhindern) und das Produkt wird dann gefriergetrocknet.
In manchen Fällen
ist es notwendig, ein organisches Lösungsmittel hinzuzufügen, um
die Reaktion durch Lösung
des Ausgangsporphyrins zu unterstützen, so dass die Reaktion
des Porphyrins (als freie Säure)
mit dem hydrophilen organischen Amin stattfindet. Beispiele für mögliche organische
Lösungsmittel
sind Aceton oder eine Mischung aus Dichlormethan und Methanol. Das
erhaltene gefriergetrocknete, wasserlösliche Porphyrin ist chemisch
rein und bedarf für
medizinische und biologische Anwendungen keiner weiteren Reinigung
außer
der Sterilisierung.
-
Entsprechend
einer weiteren bevorzugten Ausführungsform
der vorliegenden Erfindung, können nicht-reine
Ausgangsstoffe einschließlich
feuchter Rohporphyrin-Pasten benutzt werden. Die Reaktion wird genauso
wie oben beschrieben durchgeführt,
aber es wird ein Überschuss
des hydrophilen organischen Amins benutzt, um alle Porphyrinkomponenten
zur Reaktion zu bringen. Nachdem das Reaktionsgemisch im Vakuum aufkonzentriert
worden ist, wird das erhaltene wasserlösliche Porphyrin dann chromatographisch
gereinigt und zwar auf einer Säule
mit einem geeigneten Reversed-Phase-Adsorbens, vorzugsweise vom
RP C-8- oder RP C-18-Typ.
Die Fraktionen mit den Zielprodukten werden gesammelt, im Vakuum
ohne Erhitzen eingeengt, um das organische Lösungsmittel zu entfernen, und
dann gefriergetrocknet, um das gewünschte wasserlösliche Porphyrinderivat
zu erhalten. Hierfür
wurden Protokolle entwickelt, die es erlauben, wasserlösliche Porphyrinderivate
mit Reversed-Phase-Chromatographie
unter Zuhilfenahme flüchtiger
organischer Lösungsmittel
zu reinigen und so Produkte mit einem hohen qualitativen Standard
zu erhalten, wie er für
die Herstellung medizinischer Zubereitungen erforderlich ist.
-
Eine
weitere bevorzugte Ausführungsform
der vorliegenden Erfindung ist eine einfache und effiziente Methode,
um Porphyrinverbindungen aus biologischen Rohmaterialien zu erhalten.
Die Methode umfasst folgende Schritte: ein- oder zweischrittige
direkte saure Alkoholyse von biologischem Rohmaterial, vorzugsweise Methanolyse
oder Ethanolyse, die zu kristallinen Alkylphäophorbiden führt (vorzugsweise
Methyl- oder Ethylphäophorbid).
Diese sind als entscheidende Zwischenprodukte für eine Vielzahl von chemischen
Transformationen geeignet, um so die Ziel-Porphyrinderivate zu erhalten.
Diese einfache und vergleichsweise schnelle Methode erlaubt die
Darstellung von Porphyrinderivaten aus biologischen Rohmaterialien,
ohne dass auf mühsame
Waschvorgänge
mit organischen Lösungs mitteln,
das Einfrieren (um die Zellwände
zu zerstören)
und wiederholte Extraktionen zurückgegriffen
werden muss, wie sie bei den bisher bekannten Prozeduren erforderlich
sind.
-
Der
Einsatz kristalliner Alkylphäophorbide
(vorzugsweise Methyl- oder Ethylphäophorbid) als synthetische
Zwischenprodukte ermöglicht
eine einfache Reinigung und Standardisierung, wie sie für die Herstellung pharmazeutischer
Zubereitungen zum Einsatz in der Medizin wie etwa in der PDT notwendig
ist.
-
Die
Durchführung
der Alkoholyse hängt
von der Qualität
des biologischen Rohmaterials ab, von dem ausgegangen wird, insbesondere
von seiner Trockenheit, die wichtig ist, um bei der Alkoholyse die
notwendige Säurekonzentration
aufrecht zu erhalten. Im Falle ausreichend trockenen Materials,
z. B. von getrockneten Spirulina- oder Chlorella-Biomassen oder
getrockneten, pulverisierten Brennnesselblättern (vgl. Beispiele 1 – 5) ist
es möglich,
eine direkte einschrittige Herstellung von Alkylphäophorbiden
durchzuführen.
-
Falls
nur ungenügend
getrocknetes Rohmaterial zur Verfügung steht, wird die Herstellung
der Alkylphäophorbide
mit einer zweischrittigen Alkoholyse durchgeführt, hier beispielhaft gezeigt
durch die Herstellung der Methylphäophorbide a (5) und b (17)
aus Spinat (siehe Beispiel 6). In diesen Fällen verhindert das Vorhandensein
einer zu großen
Menge Wasser die Einhaltung einer Säurekonzentration, wie sie für die Spaltung
des Phytolesters notwendig ist. Dennoch sind die nach dem ersten
Alkoholyseschritt erhaltenen Phäophytine
trocken genug, um in einem zweiten Alkoholyseschritt zur Herstellung
kristalliner Alkylphäophorbide
benutzt zu werden.
-
Die
Herstellung saurer Porphyrine, die für eine weitere Umwandlung in
wasserlösliche
Formen geeignet sind, erfolgt durch chemische Umwandlung der Porphyrin-Rohmaterialien,
wie z. B. kristalline Alkylphäophorbide,
die wiederum aus biologischen Rohmaterialien mit Hilfe der in dieser
Erfindung beschriebenen Vorgehensweise erhalten werden.
-
-
Speziell
2-Devinyl-2-(1-alkoxyethyl)-chlorine e6 können auf
folgende Weise erhalten werden: Durch Hydrobromierung von Methyl-
oder Ethylphäophorbiden
a mit einer gesättigten
Lösung
von HBr in Essigsäure erhält man die
entsprechenden 2-Devinyl-2-(1-bromethyl)-phäophorbide a, aus denen man
durch weitere Alkoholyse die 2-Devinyl-2-(1-alkoxyethyl)-phäophorbide
a erhält
(z. B. Verbindung 18) (C. Rimington, A. Roennestad, A. Western,
and J. Moan, Int. J. Biochem., 1988, 20, 1139 – 1149; K.R. Adams, C.R. Berembaum,
R. Bonnett, A.N. Nizhnik, A. Salgado, and M.A. Valles, J. Chem.
Soc. Perkin Trans. 1, 1992, 1465 – 1470), anschließende Verseifung
führt zu
den entsprechenden 2-Devinyl-2-(1-alkoxyethyl)-chlorinen e6 in Form der Tricarbonsäuren (19) oder ihren wasserlöslichen
Salzen, z. B. mit N-Methyl-D-glucamin (10) (Beispiel 10).
-
In
gleicher Weise führt
die Reaktion der 2-Devinyl-2-(1-bromethyl)-phäophorbide mit anderen Nukleophilen
statt mit Alkoholen zu einer Vielzahl von möglichen Porphyrinderivaten,
die sich mit den in dieser Erfindung beschriebenen Methoden in ihre
wasserlöslichen
Formen umwandeln lassen.
-
Die
Methode aus der vorliegenden Erfindung zur Herstellung wasserlöslicher
Formen verschiedener Chlorin- und Phäophorbidderivate wird auch
durch die Beispiele 15 – 30
(unten aufgeführt)
für die
Herstellung der wasserlöslichen
Salze der Säuren
(4) und (20) – (33)
mit dem Amin (10) illustriert.
-
-
-
Die
Reaktion saurer Porphyrinderivate mit hydrophilen Aminen gemäß der vorliegenden
Erfindung führt
zur Bildung der entsprechenden wasserlöslichen Salze. Speziell im
Fall der Phäophorbide
sind dies Mono-Salze, während
Bis-Salze von Tricarbonsäure-Chlorinen
(34) (über
die Bildung eines der möglichen
Mono-Salze als Zwischenprodukt, wie das Zwischenprodukt (35), vgl.
Schema 1) gebildet werden, da die Carboxylgruppe an Position 6 (Die
Atomnummerierung ist für
die Verbindungen 4 – 6
gezeigt.) nicht genügend
Azididät
für eine
Salzbildung besitzt.
-
-
Da
die wasserlöslichen
Porphyrine der vorliegenden Erfindung in wässeriger Lösung der Hydrolyse unterliegen
können
(wie für
die Chlorine in Schema 2 beispielhaft gezeigt), ist es wünschenswert,
wasserlösliche
Salze gemäß der vorliegenden
Erfindung herzustellen und sie in gelöstem Zustand in Gegenwart eines Überschusses
von hydrophilem Amin zu lagern.
-
Die
wasserlösliche
Chlorin-Bis-Salze aus der vorliegenden Erfindung können in
gereinigter Form durch Reinigung mittels Säulenchromatographie erhalten
werden, wie dies in Beispiel 9(B) gezeigt ist. Das Auflösen der
gereinigten Bis-Salze in Wasser ist von einer reversiblen Hydrolyse
begleitet, die zu Mono-Salzen führt
(z. B. vom Typ 35, vgl. Schema 2), die eine verringerte Wasserlöslichkeit
verglichen mit Bis-Salzen besitzen, oder sogar dem Ausgangschlorin
(34), welches nur eine sehr geringe Wasserlöslichkeit aufweist. Diese Prozesse
können
zur Bildung geringer Mengen von Präzipitat während der Lagerung der Lösungen führen.
-
-
Um
diese unerwünschten
Prozesse zu vermeiden und um klare und homogene Lösungen aufrecht
zu erhalten, die unbedingt notwendig sind, wenn diese Lösungen für medizinische
Anwendungen wie die PDT benutzt werden sollen, ist es vorteilhaft,
die Bis-Salze in Wasser in Gegenwart kleiner, bekannter Mengen des entsprechenden
hydrophilen Amins (z. B. kleiner als 2 Moläquivalente, besser zwischen
0,05 und 0,5 Äquivalenten),
so dass die gewünschten
Porphyrinderivate weiter in Form ihrer Bis-Salze vorliegen und auf
diese Weise die Bildung der Mono-Salze und der Ausgangschlorine
durch Hydrolyse der Bis-Salze verhindert wird.
-
Die
Salze von Chlorin- und Phäophorbidderivaten,
die in dieser Erfindung beschrieben sind, besitzen eine Wasserlöslichkeit
in einer Größenordnung
von einigen 10 mg/l, wodurch sie für eine Vielzahl von Anwendungen
geeignet sind. Ein weiterer Gegenstand der vorliegenden Erfindung
ist es, Phäophorbidderivate
herzustellen, die eine durch eine Peptidbindung verknüpfte Aminosäureneinheit
enthalten, und die Salze mit hydrophilen Aminen bilden. Diese Salze
sind im Vergleich mit den Ausgangsphäophorbidderivaten ohne Aminosäureeinheiten
durch eine mehr als hundertfach höhere Wasserlöslichkeit
gekennzeichnet. Speziell das Salz des Phäophorbidderivates (36) mit
N-Methyl-D-glucamin (10) besitzt eine Löslichkeit von 40 mg/l, wohingegen die
Derivate, die über
eine Peptidbindung mit Aminosäureresten
verknüpft
sind, eine Löslichkeit
größer als
5 g/l besitzen.
-
-
Es
ist ein weiterer Gegenstand der vorliegenden Erfindung, chemisch
stabile und wasserlösliche
Porphyrinderivate der Formeln (1) und (2) für verschiedenste medizinische
Anwendungen zu nutzen. Solche Verbindungen sind besonders geeignet
für eine
Anwendung in der PDT, um damit Tumorerkrankungen und andere hyperproliferative
Krankheiten, Infektionen, Psoriasis, Arteriosklerose, AMD und andere
Krankheiten und Infektionen, die mit der photodynamischen Therapie
behandelt werden können,
zu behandeln. Wegen der Wasserlöslichkeit
können
besagte Verbindungen in verschiedensten pharmazeutisch akzeptablen
und aktiven Zubereitungen für
verschiedene Verabreichungsformen, wie z. B. Injektionen, hergestellt
werden.
-
Die
vorliegende Erfindung erlaubt außerdem den Einsatz hochreiner
wasserlöslicher
Porphyrinderivate, die nach den Ausführungen in der vorliegenden
Erfindung hergestellt wurden, für
die photodynamische Therapie von Tumorerkrankungen und anderen hyperproliferativen
Erkrankungen und von Infektionen. Die PDT wird durchgeführt, indem
die Derivate zuerst in ein pharmazeutisch akzeptables Vehikel für eine Abgabe am
Ort der Behandlung überführt werden.
In einer Ausführungsform,
für Erkrankungen
wie z. B. Hautkrebs oder anderen dermatologischen Erkrankungen,
ist dieses Vehikel zur Verabreichung im allgemeinen eine dermatologische
Creme, ein Gel oder manchmal ein Aerosol-Flüssigkeitsdispergiermittel.
Nachdem die Derivate mit Hilfe dieses Vehikels am Behandlungsareal
verabreicht worden sind, wird lange genug gewartet, so dass sich
die Porphyrinderivate bevorzugt im erkrankten Gewebe anreichern.
Zum Schluss wird das Behandlungsareal mit Licht geeigneter Wellenlänge und
ausreichender Leistung bestrahlt, so dass die Porphyrinderivate aktiviert
werden, um die Zellen des besagten Gewebes zu nekrotisieren.
-
Die
Bestimmung der Dunkeltoxizität
(Beispiel 31,
1) und der Phototoxizität (Beispiel
32,
2) eines der Porphyrinderivate
der vorliegenden Erfindung, speziell des wasserlöslichen Salzes von Chlorin
e
6 (7) mit N-Methyl-D-glucamin (10), hergestellt
ent sprechend dem Beispiel 9(B), zeigte in Zellkulturexperimenten beispielhaft
die hervorragenden Eigenschaften der Verbindungen für einen
Einsatz in der PDT. Die Experimente wurden auch deshalb durchgeführt, um
die deutlich schlechteren Eigenschaften (höhere Dunkeltoxizität, geringere
Phototoxizität)
derselben Verbindung zu zeigen, wenn sie nach der von Ponomarev
beschriebenen Technologie hergestellt wird (
RU 2144538 ).
-
In
besonderen Fällen
jedoch kann die die Verabreichung von definierten Mischungen der
Salze hydrophiler Amine mit verschiedenen Porphyrinderivaten vorteilhaft
sein, wenn diese Mischungen gegenüber erkranktem Gewebe eine
höhere
Phototoxizität
aufweisen. Diese erhöhte
Phototoxizität
könnte
durch das hier beschriebene Phänomen
der erhöhten
Wasserlöslichkeit
des Gemisches der Verbindungen (4), (7) und (8) (als Salze mit hydrophilen
Aminen) in Gegenwart derselben Chlorin-e6-Salze
verursacht sein.
-
Weitere
Beispiele der guten phototoxischen Eigenschaften der wasserlöslichen
Formen entsprechend der hier vorliegenden Erfindung sind in den
Tabellen 1 und 2 zusammengestellt, die die Ergebnisse von Dunkel-
und Phototoxizitätsexperimenten
mit HeLa-Zellen
mit den wasserlöslichen
Salzen der Säuren
(4), (7), (22) und (25) mit N-Methyl-D-glucamin zusammenfassen (10).
-
Ein
weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung
der beschriebenen wasserlöslichen
Formen von Chlorin- und Phäophorbidderivaten
in der antimikrobiellen photodynamischen Therapie. Dies wird durch
die Daten in Tabelle 3 veranschaulicht, die die Ergebnisse der photodynamischen
Behandlung einer Reihe von Mikroorganismen mit den wasserlöslichen
Salzen der Säuren
(4), (7), (22) und (25) mit N-Methyl-D-glucamin (10) zusammenfasst.
-
Die
im folgenden gezeigten Beispiele sollen dem Fachmann eine umfassende
und anschauliche Beschreibung davon geben, wie wasserlösliche Porphyrinderivate
gemäß der vorliegenden
Erfindung hergestellt werden können,
um für
die Herstellung pharmazeutischer Zubereitungen genutzt zu werden;
die Beispiele sind nicht dazu bestimmt, den Umfang dessen, was der
Erfinder als Gegenstand der Erfindung erachtet, zu beschränken. Es
wurde nach Möglichkeit
dafür Sorge
getragen, dass die angegebenen Zahlenwerte (z. B. Mengenangaben,
Temperatur etc.) korrekt sind, aber dennoch können einige experimentelle
Abweichungen auftreten.
-
Beispiel 1: Gewinnung
von Methylphäophorbid
(5) aus Spirulina platensis
-
- (A) Ein Gemisch von 20 g Spirulina platensis,
60 ml Methanol und 10 ml konzentrierter Schwefelsäure wird 3
h bei Raumtemperatur gerührt,
mit 30 ml Methanol verdünnt und
dann durch eine Schicht Celite filtriert. Der Inhalt des Filtertrichters
wird mit Methanol gewaschen (70 ml). Die erhaltene Lösung wird
mit Hexan extrahiert (2 × 30
ml), mit Chloroform verdünnt
(100 ml) und dann zu einer gesättigten
Natriumchloridlösung hinzugegeben
(300 ml). Das entstandene Gemisch wird durch eine Schicht Celite
filtriert und die wässerige Phase
mit Chloroform extrahiert (2 × 50
ml). Die vereinigten Extrakte werden mit Wasser gewaschen, durch Baumwolle
filtriert und aufkonzentriert. Der Rückstand wird in einem Gemisch
aus Chloroform-Hexan (1:1, 30 ml) gelöst und dann durch eine Schicht
Aluminiumoxid filtriert, wobei zuerst mit Hexan gewaschen wird (um
unpolare Nicht-Chlorin-Komponenten zu entfernen) und dann mit Dichlormethan
(um Methylphäophorbid
zu extrahieren). Die Methylenchloridlösung wird aufkonzentriert und
der Rückstand
wird zuerst aus Dichlormethan-Methanol (3 ml + 7 ml), dann noch
einmal aus Dichlormethan-Methanol (1 ml + 10 ml) umkristallisiert
und anschließend
mit Methanol (10 ml) gewaschen, um 113 mg reines Methylphäophorbid
a (5) zu erhalten. 1H-NMR-Spektrum: 9,41,
9,23, 8,56 (3H, alle s, meso-H); 7,88 (1H,
q, -CH=CH2),
6,25 – 6,10
(2H, -CH=CH 2),
6,28 (1H, s, Cyclopentanon-H), 4,50, 4,25 (2H, m, 7-H, 8-H); 3,55
(2H, q, 4-CH 2CH3); 3,93, 3,70, 3,63, 3,39, 3,15 (15H, alle
s, 5 × -CH3); 2,75 – 2,20 (4H, m, -CH 2CH 2COOCH3); 1,85 (3H, d, 8-CH 3); 1,71 (3H,
m, 4-CH2CH 3); 0,55 und –1,68 ppm (2H, 2 breite s,
2 × -NH-).
Das Produkt ist identisch mit derselben Verbindung, die von Porphyrin
Products Inc., USA bezogen wurde.
- (B) Ein Gemisch von 10 g Spirulina platensis, 30 ml Methanol
und 5 ml konzentrierter Schwefelsäure wird 3 h bei Raumtemperatur
gerührt,
mit kaltem Wasser (70 ml) verdünnt
und dann durch eine Schicht Celite filtriert. Der Inhalt des Filtertrichters
wird mit Wasser bis pH 7 gewaschen, dann mit Ethanol (50 ml), Hexan (4 × 30 ml)
und das gewünschte
Methylphäophorbid
wird aus dem Inhalt des Filtertrichters mit Aceton extrahiert (120
ml). Die erhaltene Lösung
wird aufkonzentriert, in Chloroform gelöst, durch eine Schicht Natriumsulfat
filtriert und erneut aufkonzentriert. Der Rückstand wird zuerst aus Dichlormethan-Methanol
(1.5 ml + 5 ml), dann noch einmal aus Dichlormethan-Methanol (1.5
ml + 10 ml) umkristallisiert und anschließend mit Methanol (15 ml) gewaschen,
um 60 mg reines Methylphäophorbid
a (5) zu erhalten.
- (C) Schwefelsäure
(konz., 250 ml) wurde zu einer Suspension von 500 g Spirulina platensis
in Methanol (1500 ml) bei Raumtemperatur unter Rühren hinzugegeben. Das gebildete
Gemisch wurde 3 h bei Raumtemperatur gehalten, dann in Wasser (6
l) eingegossen und durch eine Schicht Celite filtriert (Durchmesser 12
cm, Höhe
2 cm, Filter Schott N3). Der Rückstand
auf dem Filter wurde mit Wasser gewaschen (3 × 800 ml, bis pH 6), dann mit Ethanol
(3 × 300
ml) und mit Petrolether (Sdp. 40 – 60°C, 3 × 250 ml). Als nächstes wurde
das Zielprodukt vom Filter mit Aceton (insgesamt 1,21) herunter
gewaschen, aufkonzentriert, in Chloroform gelöst (200 ml), durch Baumwolle
filtriert und wieder im Vakuum aufkonzentriert. Der Rückstand wurde
aus einem Gemisch aus Dichlormethan (40 ml) und Methanol (200 ml)
umkristallisiert, um 2,45 g Methylphäophorbid a in 90%iger Reinheit
(TLC in CHCl3/Aceton, 95:5) zu erhalten.
Dieses wurde in Chloroform gelöst
(20 ml) und durch eine Schicht Aluminiumoxid (neutral, Grad II,
Durchmesser 8 cm, Höhe
5 cm) filtriert (Nachwaschen mit Chloroform), aufkonzentriert und
dann aus einem Gemisch aus Dichlormethan (50 ml) und Methanol (250
ml) umkristallisiert. Es wurden 2,38 g reines (TLC Kontrolle) Methylphäophorbid a
erhalten. Zusätzliche
Mengen an Methylphäophorbid
a können
durch Standard-Aufarbeitung (Chromatographie und Umkristallisation)
der Mutterlaugen der beiden Umkristallisationen erhalten werden.
-
Beispiel 2: Gewinnung
von Ethylphäophorbid
(6) aus Spirulina platensis
-
Die
Ethanolyse von 20 g Spirulina platensis in 60 ml 96%igem Ethanol
und 10 ml konzentrierter Schwefelsäure und anschließender Aufarbeitung
wie in Beispiel 1(A) für
die Darstellung von Methylphäophorbid
a (5) beschrieben, nur dass in allen Schritten Ethanol statt Methanol
verwendet wurde, ergab 110 mg kristallines Ethylphäophorbid
a (6). 1H-NMR-Spektrum: 9,57, 9,42, 8,61 (3H, alle
s, meso-H); 7,99 (1H, q, -CH=CH2), 6,32, 6,26 (2H, dd, -CH=CH 2), 6,27 (1H,
s, Cyclopentanon-H), 4,51, 4,28 (2H, m, 7-H, 8-H); 4,07 (2H, q,
-COOCH 2CH3); 3,71 (2H, q, 4-CH 2CH3);
3,89, 3,72, 3,41, 3,27 (12H, alle s, 4 × -CH3);
2,69, 2,47, 2,37, 2,22 (4H, m, -CH 2CH 2COOCH2CH3); 1,83 (3H, d, 8-CH 3); 1,73 (3H,
t, 4-CH2CH 3); 1,12 (3H, t, -COOCH2CH 3); 0,57, –1,46 ppm
(2H, 2 breite s, 2 × -NH-).
-
Beispiel 3: Gewinnung
von Methylphäophorbid
a (5) aus Spirulina maxima
-
Die
Behandlung von 10 g Spirulina maxima mit 30 ml Methanol und 5 ml
konzentrierter Schwefelsäure und
anschließender
Aufarbeitung wie für
die Darstellung von Methylphäophorbid
a (5) in Beispiel 1B beschrieben ergab 64 mg reines Methylphäophorbid
a (5).
-
Beispiel 4: Gewinnung
von Methylphäophorbid
a (5) and b (17) aus Chlorella
-
10
g trockene Chlorella-Biomasse wurden einer Methanolyse und anschließenden Aufarbeitung
gemäß dem Beispiel
3 unterworfen, um ein Gemisch (140 mg) der Methylphäophorbide
a und b in einem Verhältnis
von 20:7 zu erhalten (Bestimmung des Verhält nisses mit 1H-NMR-Spektroskopie).
Eine Säulenchromatographie
des erhaltenen Gemisches mit Silicagel 60 (Fluka, 70 – 230 mesh),
Eluierung mit Chloroform-Toluen-Aceton 15:30:1.5 ergab die beiden
einzelnen Methylphäophorbide
a (5) und b (17). Das Methylphäophorbid a
(5) war mit dem in den vorhergehenden Beispielen beschriebenen Produkt
identisch. 1H-NMR-spektroskopische Daten von Methylphäophorbid
b (17): 11,0 (1H, s, CHO), 10,22, 9,50, 8,55 (3H, alle s, meso-H);
7,98 (1H, q, -CH=CH2), 6,40 – 6,15 (2H, -CH=CH 2), 6,25 (1H,
s, Cyclopentanon-H), 4,48, 4,20 (2H, m, 7-H, 8-H); 3,60 (2H, q,
4-CH 2CH3); 3,93, 3,78, 3,75, 3,40 (12H, alle s,
4 × -CH3); 2,75 – 2,20 (4H, m, -CH 2CH 2COOCH3); 1,85 (3H, d, 8-CH 3); 1,71 (3H,
m, 4-CH2CH 3); 0,48 und –1,60 ppm (2H, 2 breite s,
2 × -NH-).
-
Beispiel 5: Gewinnung
von Methylphäophorbid
a (5) und b (17) aus pulverisierten trockenen Brennnesselblättern
-
Die
Methanolyse von 500 g trockenen und pulverisierten Brennnesselblättern und
anschließende
Aufarbeitung wie in Beispiel 1C beschrieben ergab ein Gemisch (1,74
g) der Methylphäophorbide
a (5) und b (17) in einem Verhältnis
von 6.5:1 (Bestimmung des Verhältnisses
mit 1H-NMR-Spektroskopie).
-
Beispiel 6: Gewinnung
von Methylphäophorbid
a (5) und b (17) aus gefrorenen Spinatblättern
-
Schwefelsäure (konz.,
5 ml) wurde bei Raumtemperatur zu einer Mischung von 100 g gefrorenen
Spinatblättern
und Methanol (100 ml) unter Rühren
hinzugegeben. Das erhaltene Gemisch wurde 16 h bei Raumtemperatur
gehalten, dann mit Wasser verdünnt
(100 ml) und durch Celite filtriert. Der Rückstand wurde mit Aceton gewaschen
(3 × 50
ml), die Aceton-Extrakte
wurden mit Dichlormethan-Wasser (1:1, 100 ml) verdünnt, die
organische Phase wurde abgetrennt und aufkonzentriert. Der Rückstand
wurde einer Methanolyse in 100 ml 5%iger Schwefelsäure in Methanol
unterworfen, und die anschließende
Aufarbeitung gemäß dem Beispiel 1(C)
ergab 40 mg eines Gemisches der Methylphäophorbide a (5) und b (17)
in einem Verhältnis
von 2:1 (Bestimmung des Verhältnisses
mit 1H-NMR-Spektroskopie).
-
Beispiel 7: Darstellung
von Methyl 2-devinyl-2-(1-ethoxyethyl)-phäophorbid a (18)
-
3,5
g (5,8 mmol) Methylphäophorbid
a (5) wurden in einem Gemisch von Bromwasserstoff und Essigsäure (d 1,44,
50 ml) gelöst
und für
18 h stehen gelassen. Das Gemisch wurde dann im Vakuum bei 50°C bis zur
Trockne eingeengt, dann wurden 100 ml absolutes Ethanol hinzugegeben.
Nach 18 h wurde das Reaktionsgemisch unter Rühren auf zerstoßenes Eis
gegossen und mit Dichlormethan extrahiert (3 × 40 ml). Die vereinigten Extrakte
wurden mit Wasser gewaschen (4 × 70
ml) und dann im Vakuum bis zur Trockne eingeengt. Säulenchromatographie
auf Kieselgel (40 – 63 μm, Merck)
mit Dichlormethan als Elutionsmittel ergab 2,95 g des Produktes
(18), Ausbeute 77%. 1H-NMR-Spektrum: 9,82,
9,58, 8,55 (3H, alle s, meso-H); 6,31 (1H, s, 10-H), 5,96 (1H, q,
2-CHCH3);
4,53, 4,25 (2H, m, 7-H, 8-H);
3,71 (4H, dq, 4-CH 2CH3, -OCH 2CH3); 3,93, 3,87, 3,63,
3,41, 3,28 (15H, alle s, 5 × -CH3); 2,67, 2,51, 2,37, 2,23 (4H, m, -CH 2CH 2COOCH3); 2,11 (3H, d, 2-CHCH 3); 1,80 (3H,
d, 8-CH 3);
1,75 (3H, t, 4-CH2CH 3); 1,36 (3H,
t, -OCH2CH3); 0,55, –1,41 (2H,
2 breite s, 2 × -NH-).
-
Beispiel 8: Darstellung
von Chlorin e6 (7)
-
Eine
wässerige
KOH-Lösung
(entgast, 10%ige Lösung,
10 ml) wurde unter Argon zu einer gerührten Lösung von 140 mg (231 μmol) Methylphäophorbid
a (5) in mit Helium entgastem Aceton (12 ml) hinzugegeben. Das Gemisch
wurde 40 min bei 40°C
gerührt,
auf 65°C
erwärmt,
und dann wurde eine 3%ige Lösung
von Kaliumhydroxid (hergestellt aus 5 ml entgaster 10%iger wässeriger
Kaliumhydroxidlösung
und 11 ml entgastem Wasser) hinzugegeben. Die entstandene Lösung wurde
für 2,5
h unter Argon gerührt
und auf 65°C
erwärmt,
dann auf Raumtemperatur abgekühlt,
mit 100 ml Wasser verdünnt
und mit 2 N Salzsäure
(12 ml) angesäuert.
Der Niederschlag wurde durch Zentrifugieren abgetrennt (3 min bei
5000 U/min), bei erneutem Zentrifugieren mit Wasser gewaschen (3 × 30 ml),
in 10 ml Wasser resuspendiert und gefriergetrocknet, um Rohchlorin
e6 als freie Säure zu erhalten (120 mg, 87%,
~ 90% Reinheit, überprüft durch
TLC). TLC: RP-18-TLC-Platten (Merck), MeOH-CH2Cl2 (3:1), Rf 0,6; Begleiter: polarere Verunreinigungen
mit Rf < 0,3. Endreinigung:
20 mg Rohchlorin e6 wurden in MeOH-CH2Cl2-Wasser (3:1:1,
4 ml) gelöst
und per MPLC auf einer RP-18-Säule
chromatographiert (Merck, #11447, 240 × 10, 40 – 63 μm), Eluierung mit MeOH-CH2Cl2-Wasser (4:3:1,
2 ml/min), um reines Chlorin e6 (7) (15
mg, 75%) zu erhalten. Eine begleitende Verunreinigung wurde mit
MeOH-CH2Cl2 (3:1)
eluiert. 1H-NMR-Spektrum (DMSO-d6):
9,88, 9,78, 9,18 (3H, alle s, meso-H); 8,33 (1H, dd, -CH=CH2); 6,47 (1H,
d, cis-CH=CH 2);
6,22 (1H, d, trans-CH=CH 2); 5,38 (2H, m, -CH 2COOH); 4,62 (1H,
m, -CHCH3);
4,48 (1H, m, -CHCH2);
3,83 (2H, m, -CH 2CH3); 3,59, 3,53, 3,33 (9H, all s, -CH 3);
2,62, 2,27, 2,14, (4H, m, -CH 2CH2COOH); 1,70,
1,66 (6H, m, -CHCH 3 +
-CH2CH 3); 1,64, –1,90 (2H, 2 breite s mit unterschiedlicher
Intensität,
2 × -NH-). 13C-NMR-Spektrum (DMSO-d6)
nur charakteristische Signale: 174,15, 173,46, 172,34 (COOH); 129,21
(-CH=CH2);
122,25 (-CH=CH2);
103,74 (γ);
101,12 (β);
98,09 (α); 94,67
(δ); 52,66 (CHCH2);
48,19 (CHCH3);
37,80 (-CH2COOH);
30,82, 29,50 (-CHCH2 CH2COOH); 22,92 (CHCH3);
18,91 (CH2 CH3); 17,58 (CH2CH3); 11,00, 10,98 (ArMe).
-
Beispiel 9: Herstellung
des wasserlöslichen
Salzes von Chlorin e6 (7) mit N-Methyl-D-glucamin (10)
-
- (A) Eine Lösung
von N-Methyl-D-glucamin (10) 4 mg, 21 μmol) in Wasser (4 ml) wurde
zu einer Lösung
von 5 mg (8.4 μmol)
Chlorin e6 (7) in MeOH-CH2Cl2 (3:1, 20 ml) (oder in Aceton) hinzugegeben,
und die organischen Lösungsmittel
wurden im Vakuum entfernt. Die erhaltene wässerige Lösung wurde durch eine Membran
filtriert (20 μm)
und gefriergetrocknet, um quantitativ das wasserlösliche Salz
zu erhalten (9 mg, dies schließt
einen Überschuss
von N-Methyl-D-glucamin ein).
- (B) Eine 10%ige wässerige
entgaste Lösung
von Kaliumhydroxid (10 ml) wurde zu einer Lösung von 50 mg Methylphäophorbid
a (5) in entgastem Aceton (12 ml) hinzugegeben. Das Gemisch wurde
30 min bei 40°C unter
Inertgasatmosphäre
(Argon) gerührt,
anschließend
wurden 15 ml einer 3%igen wässerigen
entgasten Lösung
von Kaliumhydroxid hinzugegeben. Das erhaltene Gemisch wurde 2 h
bei 65°C
unter Inertgasatmosphäre
(Argon) gerührt,
mit Wasser verdünnt
(100 ml), und das Chlorin e6 (7) wurde durch
Zugabe von 2 N wässeriger
HCl-Lösung
(bis zu pH 6) ausgefällt.
Der Niederschlag wurde durch Zentrifugieren (5 min bei 3000 U/min)
abgetrennt, mit Wasser gewaschen (3 × 10 ml), wobei eine feuchte
Masse von Chlorin e6 erhalten wurde, die
für die
nächste
Stufe ohne weitere Reinigung eingesetzt wurde. Die gesamte erhaltene Menge
an Chlorin e6 (7) wurde unter Argon mit
N-Methyl-D-glucamin (10) (30 mg, 2 Äquiv.) und Wasser (10 ml) gemischt,
um eine etwa 5%ige Lösung
des wasserlöslichen
Salzes zu erhalten. Das erhaltene Gemisch wurde gerührt, bis
es sich vollständig
auflöste,
bis zur Trockne eingeengt und dann per HPLC auf einer RP C-8-Säule mit
einem Wasser-Methanol-Gradienten (von 40% bis 80%) chromatographiert.
Die Fraktionen mit dem Zielprodukt wurden gesammelt und lyophilisiert,
um das wasserlösliche
Bis-Salz mit etwa 75 – 80% Ausbeute
zu erhalten. Die Struktur wurde durch 1H-NMR-Spektroskopie
bestätigt.
Speziell das 1:2 Verhältnis
von Chlorin e6 (7) zu den N-Methyl-D-glucamin-(10)-Resten
wurde durch die Integration der Signale der N-Me-Gruppe der Glucaminkomponente
(bei 2,70 ppm) und der 8-Me-Gruppe der Chlorineinheit (bei 1,75 ppm)
bestätigt.
- (C) Das Gemisch aus 50 mg (84 μmol) gepulvertem Chlorin e6 (7), 40 mg (0,21 mmol), N-Methyl-D-glucamin (10)
und Wasser (50 ml, vorher mit Inertgas entgast) wurde unter Argon
im Dunkeln gerührt,
bis es sich vollständig
auflöste.
Die entstandene Lösung
wurde durch eine Membran (20 μm)
filtriert und gefriergetrocknet, um quantitativ das wasserlösliche Salz
zu erhalten (90 mg, dies schließt
einen Überschuss
von N-Methyl-D-glucamin
ein).
-
Beispiel 10: Herstellung
von 2-Devinyl-2-(1-ethoxyethyl)-chlorin e6 (19).
-
Methyl-2-devinyl-2-(1-ethoxyethyl)-phäophorbid
a (18) (2,8 g, 4,1 mmol) wurde wie für die Herstellung von Chlorin
e6 beschrieben verseift (Beispiel 8), um
nach Säulenchromatographie
2,1 g des Produktes (19) zu erhalten, Ausbeute 79%. 1H-NMR-Spektrum
(DMSO-d6):
9,88, 9,78, 9,18 (3H, alle s, meso-H); 6,47 (1H, d, cis-CH=CH 2);
6,22 (1H, d, trans-CH=CH 2); 5.5.1 (2H, m, -OCH 2CH3);
5,38 (2H, m, -CH 2COOH);
4,62 (1H, m, -CHCH3);
4,48 (1H, m, -CHCH2);
4,29 (1H, q, -CHCH3);
3,83 (2H, m, -CH 2CH3); 3,59, 3,53, 3,33 (9H, alle s, -CH 3);
2,62, 2,27, 2,14, (4H, m, -CH 2CH 2COOH); 1,83 (1H, d, -CH(O-)CH 3); 1,70, 1,66
(6H, m, -CHCH 3 + -CH2CH 3); 1,54 (3H, t, -OCH2CH 3); –1,90 (2H,
2 breite s mit unterschiedlicher Intensität, 2 × -NH-). 13C-NMR-Spektrum
(DMSO-d6) nur charakteristische Signale:
174,15, 173,46, 172,34 (COOH); 129,21 (-CH=CH2); 103,74 (γ); 101,12 (β); 98,09 (α); 94,67 (δ); 67,97 (-CH(O-)CH3); 63,49
(-OCH2CH3); 52,66 (CHCH2); 48,19 (CHCH3); 37,80 (-CH2COOH); 30,82, 29,50 (-CHCH2 CH2COOH);
22,92 (CHCH3);
20,20 (-CH(O-)CH3);
18,91 (CH2CH3); 17,58, 15,23 (CH2CH3 + -OCH2 CH3); 11,00, 10,98
(ArMe).
-
Beispiel 11: Herstellung
des wasserlöslichen
Salzes des Chlorin-e6-Derivates (19) mit
N-Methyl-D-glucamin (10)
-
Die
Reaktion von 30 mg 2-Devinyl-2-(1-etoxyethyl)-chlorin e6 (19)
mit 20 mg N-Methyl-D-glucamin
(10) wie in Beispiel 9(C) beschrieben ergab quantitativ 50 mg des
gefriergetrockneten Salzes.
-
Beispiel 12: Herstellung
von Bis[2-(β-maltosyloxy)ethyl]amin
(11)
-
Eine
Lösung
von Per-O-acetyl-maltosylbromid (500 mg, 0,7 mmol), N-Benzyloxycarbonyl-N,N-bis(2-hydroxyethyl)amin
(56 mg, 0,233 mmol) und 2,4,6-Trimethylpyridin (80 μl, 0,605
mmol) wurde tropfenweise zu einem gerührten Gemisch von Silbertrifluormethanesulfonat
(208 mg, 0,805 mmol) und 1,5 ml absolutem CH2Cl2 bei –20°C hinzugegeben.
Man ließ das
Gemisch sich auf Raumtemperatur erwärmen, dann wurde es mit 0,5
ml Triethylamin behandelt, mit 200 ml Dichlormethan verdünnt, mit
einer gesättigten
wässerigen
Lösung
von Na2S2O3 (50 ml) und Wasser (50 ml) gewaschen, aufkonzentriert
und in Ethyl acetat-Petrolether (1:1) Flash-chromatogaphiert, um
das rohe N-Benzyloxycarbonyl-N,N-bis[2-(hepta-O-acetyl-β-maltosyloxy)ethyl]amin
(57 mg) zu erhalten. Ausgewählte
strukturspezifische 13C-NMR-Daten (500 MHz,
CDCl3): 20,41 (CH3CO); 46,92, 47,24, 48,00 und 48,02 (OCH2CH2N und OCH2 CH2N);
61,30, 61,50, 61,98, und 62,52 (C-6 der Glucoseeinheiten); 67,18
(NCOOCH2C6H5); 67,81, 68,33,
69,14, 69,84, 72,02, 72,55, 75,10 (C-2 – C-5 der Glucoseeinheiten);
95,38 (C-1 der α-Glucoseeinheiten);
100,18 und 101,10 (C-1 der β-Glucoseeinheiten); 127,73,
128,01 und 128,40 (OCH2 C 6H5);
164,00 (NCOO); 169,24, 169,42,
169,76, 169,98, 170,35 (CH3 CO). [α]D 38,9° (c1,
Ethylacetat). Das erhaltene Produkt wurde in 0,1 M Natriummethylat
in absolutem Methanol gelöst
und 2 Stunden gerührt,
dann wurde es mit einem Ionenaustauscher-Harz KU-2(H+)
neutralisiert und filtriert. Das Filtrat wurde über Nacht einer Hydrierung
mit Pd/C unterworfen, filtriert und gefriergetrocknet, um 23 mg von
Verbindung (11) zu erhalten, [α]D 71° (c1,
Wasser). Ausgewählte
strukturspezifische 13C-NMR-Daten (500 MHz,
D O): 48,74 (OCH2 CH2N), 50,85 (OCH2CH2N); 61,72 (C-6 der α-Glucoseeinheiten), 61,92 (C-6
der β-Glucoseeinheiten),
77,28 (C-4 der β-Glucoseeinheiten);
100,83 (C-1 der α-Glucoseeinheiten);
103,40 (C-1 der β-Glucoseeinheiten).
-
Beispiel 13: Herstellung
des wasserlöslichen
Salzes von Chlorin e6 (7) mit Bis[2-(β-maltosyloxy)ethyl]amin (11).
-
Die
Reaktion von 5 mg Chlorin e6 (7) mit 18,6
mg des Amins (11), wie in Beispiel 9(C) beschrieben, ergab quantitativ
nahezu 23 mg des gefriergetrockneten wasserlöslichen Salzes.
-
Beispiel 14: Gewinnung
von Methylmesophäophorbid
a (37)
-
Methylphäophorbid
a (5) (130 mg, 0,2 mmol) wurde in 5 ml Tetrahydrofuran gelöst und bei
~ 1 atm H2 über 30 mg 10% Pd(OH)2 auf Kohlenstoff für 1 Stunde bei Raumtemperatur
hydriert. Der Katalysator wurde abflitriert und die Lösung wurde
aufkonzentriert, um reines Methylmesophäophorbid a (37) zu erhalten
(125 mg, quantitativ).
-
Beispiel 15: Gewinnung
von Mesochlorin e6 (20)
-
Alkalische
Hydrolyse von Methylmesophäophorbid
a (37) (20 mg, 0,033 mmol), wie in Beispiel 9(B) beschrieben, ergab
Mesochlorin e6 (17 mg, 86%). MS(–)(Elektrospray):
597,3 (M-H).
-
Beispiel 16: Herstellung
des wasserlöslichen
Salzes von Mesochlorin e6 (20) mit N-Methyl-D-glucamin (10)
-
Die
Reaktion von 15 mg Mesochlorin e6 (20) mit
12 mg N-Methyl-D-glucamin (10), wie in Beispiel 9(C) beschrieben,
ergab quantitativ 27 mg des gefriergetrockneten wasserlöslichen
Salzes.
-
-
Beispiel 17: Herstellung
des wasserlöslichen
Salzes von Phäophorbid
a (4) mit N-Methyl-D-glucamin
(10)
-
Methylphäophorbid
a (5) (30 mg, 0,049 mmol) wurde in 2 ml 50%iger Schwefelsäure gelöst, und
das Gemisch wurde für
2 Stunden bei Raumtemperatur gerührt.
Es wurde mit eiskaltem Wasser verdünnt und mit Chloroform extrahiert.
Die gesammelten Extrakte wurden mit Wasser gewaschen, über wasserfreiem
Na2SO4 getrocknet
und aufkonzentriert, um rohes Phäophorbid
a (4) zu erhalten. Letzteres wurde in 3 ml einer wässerigen
N-Methyl-D-glucamin
(10) (11,6 mg, 1,2 Äquiv.)
Lösung
gelöst.
Die erhaltene Lösung
wurde durch einen 45 μm
Filter filtriert und gefriergetrocknet, um 29 mg des gefriergetrockneten
wasserlöslichen
Salzes zu erhalten. MS(–)(Elektrospray)
(Säurekomponente
4): 591,2 (M-H).
-
Beispiel 18: Herstellung
des wasserlöslichen
Salzes von Dimethylchlorin-e6-dimethylester
(21) mit N-Methyl-D-glucamin (10)
-
Saure
Hydrolyse der drei Methylestergruppen von Chlorin e6 Trimethylester
(38) [hergestellt nach S. Lötjönen, P.H.
Hynninen, Synthesis, 1980, 541 – 543]
(25 mg, 0,039 mmol) mit 50%iger Schwefelsäure und Reaktion mit 9 mg N-Methyl-D-glucamin
(10) wie in Beispiel 17 beschrieben, ergab 32 mg des gefriergetrockneten
wasserlöslichen
Salzes. MS(–)(Elektrospray)
(Säurekomponente
21): 623,5 (M-H).
-
Beispiel 19: Herstellung
des wasserlöslichen
Salzes des Mesopyrophäophorbid-a-Derivates
(25) mit N-Methyl-D-glucamin (10)
-
Methylphäophorbid
a (5) (1,3 g, 2,09 mmol) wurde in 100 ml Pyridin gelöst und 8
Stunden unter Rückfluss
im Dunkeln unter Stickstoff gerührt.
Das Pyridin wurde im Vakuum abdestilliert, und der verbleibende Rückstand
wurde aus Dichlormethan/Methanol umkristallisiert, um 1,08 g (92%)
Methylpyrophäophorbid
a (39) zu erhalten. UV/vis λmax (ε)
(CH2Cl2): 410 (112500),
509 (11400), 537 (9800), 611 (8500), 669 (47000) nm.
-
Methylpyrophäophorbid
a (39) (1,0 g, 1,78 mmol) wurde 100 ml Aceton gelöst und für 2 Stunden
bei Raumtemperatur bei ~ 8 –10
atm H2 über
300 mg 10% Pd auf Kohlenstoff hydriert. Der Katalysator wurde abfiltriert,
und die Lösung
wurde aufkonzentriert, um reines Methylmesopyrophäophorbid
a (40) zu erhalten (1,0 g, quant.). UV/vis λmax (ε) (CH2Cl2): 408 (136000),
501 (12400), 534 (11800), 603 (10500), 656 (54600) nm; 1H-NMR
(200 MHz, CDCl3): 9,40 (s, 1H, β-meso H),
9,16 (s, 1H, α-meso
H), 8,46 (s, 1H, δ-meso
H), 5,26 und 5,08 (AB, J = 19,7 Hz, 10-CH2),
4,46 (dq, 1H, J7,8 = 2,0 Hz, 8-H), 4,27
(dt, 1H, 7-H), 3,80 und 3,64 (jeweils q, 4H, J = 7,5 Hz, 2a- und
4a-CH2), 3,63, 3,52, 3,28 und 3,21 (jeweils
s, 12H, 1-, 3-, 5-Me und COOMe), 2,80 – 2,20 (m, 4H, 7a,b-CH2CH2), 1,80 (d, 3H,
J = 7,3 Hz, 8-Me), 1,71 und 1,63 (jeweils t, 6H, J = 7,5 Hz, 2b-
und 4b-Me), 0,60 und –1,60
(jeweils breites s, 2H, NH).
-
Methylmesopyrophäophorbid
a (40) (250 mg, 0,443 mmol) wurde in 5 ml 50%iger Schwefelsäure gelöst, und
das Gemisch wurde 2 Stunden bei Raumtemperatur gerührt. Es wurde
mit eiskaltem Wasser verdünnt und
mit Chloroform extrahiert. Diese Extrakte wurden mit Wasser gewaschen, über wasserfreiem
Na2SO4 getrocknet
und aufkonzentriert, um rohes Mesopyrophäophorbid a (36) zu erhalten.
Letzteres wurde in 20 ml Dichlormethan gelöst, dann wurden 0,3 ml (2,22
mmol, ~ 5 eq.) Triethylamin hinzugegeben gefolgt von 0,1 ml (0,576
mmol, 1,3 eq.) Pentafluorphenyltrifluoracetat. Das Gemisch wurde
20 min bei Raumtemperatur gerührt (um
Verbindung 41 zu erhalten), 0,5 ml Wasser wurden hinzugegeben gefolgt
von der Zugabe der Verbindung 42 (200 mg, Produkt der G1ycoSense
AG, Jena, Germany) in 5 ml Dichlormethan. Das Gemisch wurde 30 min bei
Raumtemperatur gerührt,
mit Dichlormethan verdünnt,
dann mit Wasser und 5%iger Schwefelsäure gewaschen, getrocknet und
aufkonzentriert. Der Rückstand
wurde durch Flash-Chromatographie auf Kieselgel gereinigt. Eluierung
mit Aceton-Dichlormethan (20:80) ergab 290 mg (88%) des Amids 42.
UV/vis λmax (ε) (CH2Cl2): 408 (136500),
501 (12300), 534 (11800), 603 (10000), 656 (54500) nm; 1H-NMR
(300 MHz, CDCl3): 9,41 (s, 1H, β-meso H),
9,20 (s, 1H, α-meso
H), 8,47 (s, 1H, δ-meso
H), 6,08 (br s, 1H, CONH), 5,26 und 5,08 (AB, J = 19,7 Hz, 10-CH2), 4,51 (br q, 1H, 8-H), 4,32 (br d, 1H,
7-H), 3,82 und 3,66 (jeweils q, 4H, J = 7,5 Hz, 2a- und 4a-CH2), 3,78 (s, 2H, OCH 2COOMe), 3,63,
3,32 und 3,21 (jeweils s, 9H, 1-, 3- und 5-Me), 3,52 (s, 3H, COOMe), 3,28 (br s, 12H, NHCH 2CH 2OCH2CH 2OCH 2CH 2O), 2,80 – 1,90 (m, 4H, 7a,b-CH2CH2), 1,80 (d, 3H,
J = 7.3 Hz, 8-Me), 1,72 und 1,68 (jeweils t, 6H, J = 7.5 Hz, 2b-
und 4b-Me), 0,70 und –1,60
(jeweils br s, 2H, NH). 13C-NMR (75 MHz,
CDCl3): 196,3, 172,4, 172,3, 160,2, 155,2,
150,3, 149,0, 145,0, 142,3, 141,6, 137,3, 136,9, 135,6, 131,3, 130,1,
127,8, 105,9, 104,1, 95,8, 92,5, 70,5, 70,2, 69,9, 69,6, 68,2, 51,6,
50,1, 48,0, 39,1, 39,0, 32,6, 30,2, 23,1, 19,5, 19,4, 17,5, 17,0,
12,0, 11,3, 11,0.
-
Das
Amid 42 (195 mg, 0.263 mmol) wurde in 4 ml 50%iger Schwefelsäure gelöst und das
Gemisch wurde 2 Stunden bei Raumtemperatur gelöst, anschließend wurde
mit eiskaltem Wasser verdünnt.
Der entstandene Niederschlag wurde durch Zentrifugieren abgetrennt
(3 min bei 5000 U/min), mit Wasser bis pH 7gewaschen (2 × 50 ml)
(Bildung von 25) und in einem Gemisch aus Aceton (50 ml), Methanol
(15 ml) und Wasser gelöst
(6 ml). N-Methyl-D-glucamin
(10) (56 mg, 0,29 mmol, 1.1 Äquiv.)
wurde zu dieser Lösung
hinzugegeben. Die entstandene Lösung
wurde bei 40°C
im Vakuum aufkonzentriert (~ 20 mmHg), um die organischen Lösungsmittel
zu entfernen. Der Rückstand
des Aufkonzentrierens wurde in 40 ml Wasser gelöst, durch einen 45 μm-Filter
filtriert und gefriergetrocknet, um 239 mg des wasserlöslichen
Salzes zu erhalten. UV/vis λmax (ε)
(H2O): 375 (46550), 514 (5750), 548 (5750),
608 (6320), 660 (21260) nm. MS(–)(Elektrospray) (Säurekomponente
25): 724,7 (M-H).
-
-
Beispiel 20: Herstellung
des wasserlöslichen
Salzes des Bakteriophäophorbidderivates
(29) mit N-Methyl-D-glucamin (10)
-
0,5
ml Pyridin wurden zu einer gerührten
Lösung
von Methylmesopyrophäophorbid
a (40) (650 mg, 1,152 mmol) in 130 ml Dichlormethan hinzugegeben,
gefolgt von der Zugabe von 600 mg (2,361 mmol, 2,05 Äquiv.) Osmiumtetroxid.
Das Reaktionsgemisch wurde 48 Stunden bei Raumtemperatur gerührt, dann
wurde für
5 min H2S durch die Lösung geleitet, um das Osmat
in das freie Diol zu überführen (44).
Das Gemisch wurde auf Kieselgel getrennt. Eluierung mit Aceton-Chloroform
(5:95) ergab das Startmaterial 40 (190 mg, 29%). Eluierung mit Aceton-Chloroform
(15:85) ergab das Diol (44) (350 mg, 51%). Letzteres wurde bei Raumtemperatur
für 30
min mit 20 ml konzentrierter H2SO4 behandelt. Das Reaktionsgemisch wurde mit
eiskaltem Wasser verdünnt
und mit Chloroform extrahiert. Die Extrakte wurden mit wässerigem
Natriumbicarbonat und Wasser gewaschen, über wasserfreiem Na2SO4 getrocknet und
aufkonzentriert. Der Rückstand
wurde durch Säulenchromatographie
auf Kieselgel gereinigt. Eluierung mit Aceton-Chloroform (1:99)
ergab das Keton (45) (180 mg, 27% bezogen auf (40)) [vgl. R.K. Pandey
et al., J. Org. Chem., 1997, 62, 1463 – 1472]. Das letztere wurde in
5 ml 50%iger Schwefelsäure
gelöst,
das Gemisch wurde 2 Stunden bei Raumtemperatur gerührt, mit
eiskaltem Wasser verdünnt
und mit Chloroform extrahiert. Die Extrakte wurden mit Wasser gewaschen, über wasserfreiem
Na2SO4 getrocknet
und aufkonzentriert, um das Rohprodukt 46 zu erhalten. Dieses wurde
in 20 ml Dichlormethan gelöst,
0,2 ml (1,58 mmol, ~ 5 Äquiv.)
Triethylamin wurden hinzugegeben, gefolgt von der Zugabe von 70 μL (0,411
mmol, 1,3 Äquiv.)
Pentafluorphenyltrifluoracetat. Das Gemisch wurde 20 min bei Raumtemperatur
gerührt
(um 47 zu erhalten), und dann wurden 0,5 ml Wasser hinzugegeben,
gefolgt von der Zugabe einer Lösung
der Verbindung 42 (150 mg, Produkt der G1ycoSense AG, Jena, Germany)
in 5 ml Dichlormethan. Das Gemisch wurde 30 min bei Raumtemperatur
gerührt,
mit Dichlormethan verdünnt,
mit Wasser und 5%iger Schwefelsäure
gewaschen, getrocknet und aufkonzentriert. Der Rückstand wurde durch Flash-Chromatographie
auf Kieselgel gereinigt. Eluierung mit Aceton-Dichlormethan (20:80)
ergab 205 mg (85%) des Amids (48). UV/vis λmax (ε) (CH2Cl2): 394 (42300),
412 (44100), 506 (7650), 539 (9100), 597 (4500), 652 (15750), 712
(30600) nm; 1H-NMR (250 MHz, CDCl3): 9,18, 8,72, 8,50 (jeweils s, 3H, meso
H), 6,10 (br s, 1H, CONH), 5,20 (m, 2H, 10-CH2),
4,50 und 4,30 (beide m, 2H, 8-H und 7-H), 3,88 (s, 2H, OCH 2COOMe), 3,58, 3,41
und 3,21 (jeweils s, 9H, 1-, 3- und 5-Me), 3,51 (s, 3H, COOMe), 3,30 (br s, 12H, NHCH 2CH 2OCH 2CH 2OCH 2CH 2O),
2,80 – 1,90
(m, 4H, 7a,b-CH2CH2),
1,90 – 1,65
(m, 6H, 2 × CH2CH 3), 0,5 (m, 3H, CH2CH 3), –0,10 und –1,70 (jeweils
br s, 2H, NH). 13C-NMR (75 MHz, CDCl3): 201,4, 199,2, 172,3, 171,7, 153,3, 141,0,
137/2, 98,2, 95,0, 91,3, 89,7, 87,1, 79,4, 70,7, 70,4, 70,1, 69,8,
68,4, 53,4, 51,8, 49,9, 48,0, 39,3, 36,2, 32,9, 32,1, 30,5, 28,5,
25,9, 23,2, 19,3, 16,9, 11,9, 11,0, 9,0, 8,1.
-
Das
Amidderivat (47) (205 mg, 0,270 mmol) wurde in 4 ml 50%iger Schwefelsäure gelöst und das
Gemisch wurde 2 Stunden bei Raumtemperatur gerührt und dann mit eiskaltem
Wasser verdünnt.
Der entstandene Niederschlag wurde durch Zentrifugieren abgetrennt
(3 min bei 5000 U/min), mit Wasser bis pH 7 gewaschen (2 × 50 ml)
(um 29 zu erhalten) und in einem Gemisch aus Aceton (50 ml), Methanol
(15 ml) und Wasser (6 ml) gelöst.
N-Methyl-D-glucamin
(10) (56 mg, 0,29 mmol, 1,1 Äquiv.)
wurde zu dieser Lösung
hinzugegeben. Die entstandene Lösung
wurde bei 40°C
im Vakuum (~ 20 mmHg) aufkonzentriert, um die organischen Lösungsmittel
zu entfernen. Der Rückstand
wurde in 40 ml Wasser gelöst,
durch einen 45 μm-Filter
filtriert und die Lösung
wurde bei 40°C
im Vakuum (~ 20 mmHg) aufkonzentriert (~ 1 mmHg), um 240 mg des
wasserlöslichen
Salzes zu erhalten. UV/vis λmax (ε)
(H2O): 420 (40400), 515 (5380), 561 (6150),
668 (9610), 776 (51200) nm. MS(–)(Elektrospray)
(Säurekomponente
29): 740,7 (M-H).
-
Beispiel 21: Herstellung
des wasserlöslichen
Salzes des Phäophorbid-a-derivates
(22) mit N-Methyl-D-glucamin
(10)
-
Phäophorbid
a (4) (17 mg, 0,028 mmol) wurde mit Pentafluorphenyltrifluoracetat
(15 μl)
aktiviert, gefolgt von der Reaktion mit Verbindung 42 (25 mg, Produkt
der GlycoSense AG, Jena, Germany) in Gegenwart eines Überschusses
von Triethylamin. Anschließende
saure Hydrolyse mit 50%iger Schwefelsäure und Reaktion mit 6,5 mg
N-Methyl-D-glucamin
(10), wie in Beispiel 19 beschrieben, ergab 23 mg des gefriergetrockneten wasserlöslichen
Salzes. MS(–)(Elektrospray)
(Säurekomponente
22): 780,1 (M-H).
-
Beispiel 22: Herstellung
des wasserlöslichen
Salzes des Pyrophäophorbid-a-Derivates
(23) mit N-Methyl-D-glucamin (10)
-
Saure
Hydrolyse von Methylpyrophäophorbid
a (39) (15 mg) mit 50%iger Schwefelsäure, Aktivierung des Produktes
mit Pentafluorphenyltrifluoracetat (15 μl) und Reaktion mit Verbindung
42 (25 mg, Produkt der GlycoSense AG, Jena, Germany) in Gegenwart
eines Überschusses
von Triethylamin, gefolgt von saurer Hydrolyse mit 50%iger Schwefelsäure und
Reaktion mit 6 mg N-Methyl-D-glucamin (10), wie in Beispiel 19 beschrieben,
ergab 19 mg des gefriergetrockneten wasserlöslichen Salzes. MS(–)(Elektrospray)
(Säurekomponente
23): 722,1 (M-H).
-
Beispiel 23: Herstellung
des wasserlöslichen
Salzes des Mesophäophorbid-a-Derivates
(24) mit N-Methyl-D-glucamin (10)
-
Saure
Hydrolyse von Methylmesophäophorbid
a (37) (16 mg) mit 50%iger Schwefelsäure, Aktivierung des Produktes
mit Pentafluorphenyltrifluoracetat (15 μl) und Reaktion mit Verbindung
42 (25 mg, Produkt der GlycoSense AG, Jena, Germany) in Gegenwart
eines Überschusses
von Triethylamin, gefolgt von saurer Hydrolyse mit 50%iger Schwefelsäure und
Reaktion mit 6 mg N-Methyl-D-glucamin (10), wie in Beispiel 19 beschrieben,
ergab 18 mg des gefriergetrockneten wasserlöslichen Salzes. MS(–)(Elektrospray)
(Säurekomponente
24): 782,2 (M-H).
-
Beispiel 24: Herstellung
des wasserlöslichen
Salzes des Mesopyrophäophorbid-a-Derivates
(26) mit N-Methyl-D-glucamin (10)
-
Aktivierung
von Mesopyrophäophorbid
a (36) (20 mg, 0,034 mmol) mit Pentafluorphenyltrifluoracetat (20 μl), gefolgt
von der Reaktion mit Verbindung 49 (40 mg, Produkt der G1ycoSense
AG, Jena, Germany) in Gegenwart eines Überschusses von Triethylamin
und Reaktion mit 7 mg N-Methyl-D-glucamin (10), wie in Beispiel
19 beschrieben, ergab 32 mg gefriergetrockneten wasserlöslichen
Salzes. MS(–)(Elektrospray)
(Säurekomponente
26): 856,7 (M-H).
-
Beispiel 25: Herstellung
des wasserlöslichen
Salzes des Mesopyrophäophorbid-a-Derivates
(27) mit N-Methyl-D-glucamin (10)
-
Aktivierung
von Mesopyrophäophorbid
a (36) (15 mg, 0,026 mmol) mit Pentafluorphenyltrifluoracetat (15 μl) gefolgt
von der Reaktion mit 20 mg Glycinmethylester in Gegenwart eines Überschusses
von Triethylamin, gefolgt von Hydrolyse mit 50%iger Schwefelsäure und
Reaktion mit 5 mg N-Methyl-D-glucamin (10), wie in Beispiel 19 beschrieben,
ergab 19 mg des gefriergetrockneten wasserlöslichen Salzes. MS(–)(Elektrospray) (Säurekomponente
27): 592,6 (M-H).
-
Beispiel 26: Herstellung
des wasserlöslichen
Salzes des Mesopyrophäophorbid-a-Derivates
(28) mit N-Methyl-D-glucamin (10)
-
Aktivierung
von Mesopyrophäophorbid
a (36) (14 mg, 0,024 mmol) mit Pentafluorphenyltrifluoracetat (15 μl), anschließende Reaktion
mit 25 mg ε-Aminocapronsäuremethylester
in Gegenwart eines Überschusses von
Trietylamin, gefolgt von saurer Hydrolyse mit 50%iger Schwefelsäure und
Reaktion mit 5 mg N-Methyl-D-glucamin (10), wie in Beispiel 19 beschrieben,
ergab 20 mg des gefriergetrockneten wasserlöslichen Salzes. MS(–)(Elektrospray)
(Säurekomponente
28): 648,5 (M-H).
-
Beispiel 27: Herstellung
des wasserlöslichen
Salzes des Chlorin-e6-Derivates (30) mit
N-Methyl-D-glucamin (10)
-
Ein
Gemisch von Methylphäophorbid
a (5) (38 mg, 0,063 mmol) und N,N-Dimethyl-1,3-diaminopropan (100 μl) in 1,4-Dioxan
(3 ml) wurde 10 Stunden bei Raumtemperatur gerührt (um Verbindung 50 zu erhalten). Zu
dieser Lösung
wurde eine 6M wässerige
NaOH-Lösung (0,1
ml) gegeben, und das entstandene Gemisch wurde 10 min am Rückfluss
erhitzt. Es wurden 5 ml Wasser hinzugegeben, und das Gemisch wurde
weitere 10 min am Rückfluss
erhitzt. Das Gemisch wurde abgekühlt,
mit 30 ml Wasser verdünnt
und mit Essigsäure auf
pH 4,5 angesäuert.
Der entstandene Niederschlag wurde durch Zentrifugieren abgetrennt
(3 min bei 5000 U/min) und mit Wasser gewaschen, um das Disäurederivat
(30) zu erhalten. Letzteres wurde in 7 ml einer wässerigen
N-Methyl-D-glucamin-Lösung
(10) (30 mg, 1,2 Äquiv.)
gelöst.
Die erhaltene Lösung
wurde durch einen 45 μm-Filter
filtriert und gefriergetrocknet, um 62 mg des wasserlöslichen
Salzes zu erhalten. MS(+)(Elektrospray) (Säurekomponente 30): 681,3 (M+H).
-
Beispiel 28: Herstellung
des wasserlöslichen
Salzes des Chlorin-e6-derivates (32) mit
N-Methyl-D-glucamin (10)
-
Eine
Lösung
von Phäophorbid
a (4) (20 mg, 0,033 mmol) und ε-Aminocapronsäuremethylester
(50 mg, ~ 10 Äquiv.)
in 1,4-Dioxan (3 ml) wurde 120 Stunden bei Raumtemperatur gerührt. Das
Gemisch wurde mit Dichlormethan verdünnt, mit 0,5 N wässeriger
HCl-Lösung und
Wasser gewaschen, getrocknet, aufkonzentriert und durch Flash-Chromatographie
auf Kieselgel gereinigt. Eluierung mit 3% MeOH-Dichlormethan ergab 22
mg (88%) des Amidderivates (51). Saure Hydrolyse des Letzteren mit
50%iger Schwefelsäure
und anschließende
Umsetzung mit 13,5 mg N-Methyl-D-glucamin (10), wie in Beispiel
17 beschrieben, ergab 33 mg des gefriergetrockneten wasserlöslichen
Salzes. MS(–)(Elektrospray)
(Säurekomponente
32): 722,3 (M-H).
-
Beispiel 29: Herstellung
des wasserlöslichen
Salzes des Chlorin-e6-Derivates (33) mit
N-Methyl-D-glucamin (10)
-
Eine
Lösung
von Phäophorbid
a (4) (20 mg, 0,034 mmol) und 2-Aminoethanol (50 μl) in 1,4-Dioxan
(3 ml) wurde 1 Stunde bei Raumtemperatur gerührt. Das Gemisch wurde mit
Dichlormethan verdünnt,
mit 0,5 N wässeriger
HCl-Lösung
und Wasser gewaschen, getrocknet, aufkonzentriert, und durch Flash-Chromatographie
auf Kieselgel gereinigt. Eluierung mit 3% MeOH-Dichlormethan ergab
20 mg (91%) des Amidderivates 33. Umsetzung mit 8 mg N-Methyl-D-glucamin
(10), wie in Beispiel 17 beschrieben, ergab 28 mg des gefriergetrockneten
wasserlöslichen
Salzes. MS(–)(Elektrospray)
(Säurekomponente
33): 652,2 (M-H).
-
Beispiel 30: Bestimmung
der Dunkeltoxizität
(Cytotoxizität)
des wasserlöslichen
Chlorin-e
6-Salzes (7) mit N-Methyl-D-glucamin (10)
(in OV2774-Zellen), hergestellt gemäß der vorliegenden Erfindung
im Vergleich mit dem nach Ponomarev et al. (
RU 2144538 ) hergestellten Salz
-
Um
die Cytotoxizität
(Dunkeltoxizität)
des wasserlöslichen
Chlorin-e6-Salzes (7) mit N-Methyl-D-glucamin
(10), hergestellt gemäß Beispiel
9(B) der vorliegenden Erfindung (Substanz A) und gemäß dem russischen Patent
Nr. 2144538 von Ponomarev (Substanz B), zu bestimmen, wurden OV2774-Zellen
(Besiedlungsdichte: 50 – 75
Zellen/mm2 in RPMI-1640 w. P. (mit Phenolrot), 5% fötalem Kälberserum,
2 mM Glutamax I, 100 μg/ml Penicillin/Streptomycin)
mit zunehmenden Konzentrationen von bis zu 50 μM für 24 Stunden inkubiert. Der Zell-Survival
wurde nach weiteren 24 Stunden in Sensitizer-freiem Medium unter
Verwendung des Neutralrot-Assays bestimmt. Die Werte sind als Prozentsatz
bezogen auf die nicht-inkubierten Kontrollproben angegeben. Für jede inkubierte
Konzentration wurden drei voneinander unabhängige Experimente mit vier
Proben durchgeführt.
Die Daten der Experimente sind in 1 wiedergegeben.
Substanz A zeigte im Vergleich zu Substanz B eine geringere Cytotoxizität gegenüber OV2774-Zellen.
Der Zell-Survival wurde durch Konzentrationen von bis zu 25 μM (Substanz
B: 10 μM)
nicht wesentlich verringert. Auf der Basis dieser Daten konnte der
IC50-Wert (Die Inkubationskonzentration,
die das Zellwachstum, verglichen mit den Kontrollen, auf 50% herabsetzt)
noch nicht bestimmt werden. Bei der höchsten getesteten Inkubationskonzentration
ergab sich ein Zell-Survival größer 80%.
-
Beispiel 31: Bestimmung
der Phototoxizität
des wasserlöslichen
Chlorin-e
6-Salzes (7) mit N-Methyl-D-glucamin
(10) (in OV2774-Zellen, unter Bestrahlung bei 670 nm), hergestellt
gemäß der vorliegenden
Erfindung im Vergleich mit dem nach Ponomarev et al. (
RU 2144538 ) hergestellten Salz
-
Um
die Phototoxizität
des wasserlöslichen
Chlorin-e6-Salzes (7) mit N-Methyl-D-glucamin (10), hergestellt
gemäß Beispiel
9 der vorliegenden Erfindung (Substanz A) und gemäß dem russischen
Patent Nr. 2144538 von Ponomarev (Substanz B), zu bestimmen, wurden
OV2774-Zellen (Besiedlungsdichte: 50 – 75 Zellen/mm2 in
RPMI-1640 w/o P. (mit Phenolrot), 5% fötalem Kälberserum, 2 mM Glutamax I,
100 μg/ml
Penicillin/Streptomycin) mit derselben Konzentration wie in den
vorhergehenden Experimenten für
Substanz B inku biert (10 μM,
24 h). Die Bestrahlung erfolgte bei 670 nm (10 – 25 mW/cm2,
0,1 – 2,5
J/cm2) nach einer zweiten Inkubationsperiode
im Medium ohne Photosensibilisator und ohne Phenolrot. Der Zell-Survival
wurde unter Verwendung des Neutralrot-Assays bestimmt. Die Werte
sind als Prozentsätze
bezogen auf die inkubierten, aber nicht bestrahlten Kontrollproben
angegeben. Es wurden fünf
voneinander unabhängige
Experimente mit vier Proben durchgeführt. Die ID50-Werte
[Der Energiefluss (die Energiedichte), die das Zellwachstum, verglichen
mit den Kontrollen, auf 50% reduziert] der Proben dienten als quantitatives
Maß für die Phototoxizität. Die Daten
der Experimente sind in 2 wiedergegeben.
Die Ergebnisse zeigten eine etwas höhere Phototoxizität von Substanz
A verglichen mit Substanz B. Der ID50-Wert
war etwa halb so groß wie
der von Substanz B (0,1 J/cm2 im Vergleich
zu 0,2 J/cm2).
-
Beispiel 32: Bestimmung
der Dunkeltoxizität
(Cytotoxizität)
der wasserlöslichen
Salze von Chlorin- (7) und Phäophorbidderivaten
[(4), (22) und (25)] mit N-Methyl-D-glucamin (10) in HeLa-Zellen.
-
Um
die Dunkeltoxizität
der wasserlöslichen
Salze von Chlorin e6 (7) und den Phäophorbid-a-Derivaten (4,
22, 25) mit N-Methyl-D-glucamin (10) (hergestellt gemäß den Beispielen
9, 17, 19 und 21) zu bestimmen, wurden HeLa- (Humane-Zervixkarzinom-)
Zell-Monoschichtkulturen
in 96-well-Platten (Besiedelungsdichte: 7,000 Zellen pro well in
Dulbeco-modified essentional medium (DMEM) mit 10% fötalem Kälberserum)
mit zunehmenden Konzentrationen der Photosensibilisatoren (von 2
bis 500 μg/ml)
für 48
Stunden inkubiert.
-
Die
Zellen wurden mit reinem DMEM gewaschen und 15 Minuten bei Raumtemperatur
mit 10% Formalin behandelt. Die Zellen wurden zweimal mit Wasser
gewaschen, 15 Minuten mit einer 0,1%igen Kristallviolettlösung (50 μl/well) inkubiert,
dann mit Wasser gewaschen und mit Ethanol behandelt (100 μl/well).
Die optischen Dichten der erhaltenen ethanolischen Lösungen wurden
mit einem Specord 100 (Analytik Jena AG, Germany) Spektrophotometer
bei 594 nm gemessen, um den Zell-Survival zu verfolgen.
-
Die
Werte sind als Prozentsätze
bezogen auf die nicht-inkubierten Kontrollproben angegeben. Für jede Inkubationskonzentration
wurden acht Experimente durchgeführt.
Die Daten der Experimente sind in Tabelle 1 angegeben, welche die
IC50- und IC80-Werte
angibt (Die Inkubationskonzentrationen, die das Zellwachstum, verglichen
mit den Kontrollen, auf 50% bzw. 20% herabsetzen).
-
Tabelle
1: Daten zur Dunkeltoxizität
(Cytotoxizität)
der wasserlöslichen
Salze von Chlorin-(7)
und Phäophorbidderivaten
[(4), (22) und (25)] mit N-Methyl-D-glucamin (10) in HeLa-Zellen Beisiel 33)
-
Beispiel 33: Bestimmung
der Phototoxizität
der wasserlöslichen
Salze von Chlorin- (7) und Phäophorbidderivaten
[(4), (22) und (25)] mit N-Methyl-D-glucamin (10) in HeLa-Zellen
unter Bestrahlung bei 662 nm.
-
Um
die Phototoxizität
der wasserlöslichen
Salze von Chlorin e6 (7) und den Phäophorbid-a-Derivaten (4,
22, 25) mit N-Methyl-D-glucamin (10) (hergestellt gemäß den Beispielen
9, 17, 19 und 21) zu bestimmen, wurden HeLa-(Humane-Zervixkarzinom-)Zell-Monoschichtkulturen
in 96-well-Platten (Besiedelungsdichte: 30,000 Zellen pro well in
DMEM mit 10% fötalem
Rinderserum) mit zunehmenden Photosensibilisator-Konzentrationen
von 0,01 bis 40 μg/ml
inkubiert. Die Bestrahlung wurde bei 662 nm [Ceralas PDT Laser,
biolitec AG, Deutschland; 150 mW/cm2, 5 – 20 J/cm2) nach einer 30-minütigen Inkubationsperiode durchgeführt. Der Zell-Survival
wurde unter Verwendung des MTT-Assays gemessen. Die Werte sind als
Prozentsätze
bezogen auf die bestrahlten, aber nicht-inkubierten Kontrollproben
angegeben. Die Experimente wurden jeweils mit acht Proben durchgeführt. Die
Daten der Experimente sind in Tabelle 2 angegeben; diese zeigt die
beobachteten IC50- und IC90-Werte
nach Bestrahlung mit 10 J/cm2.
-
Tabelle
2: Daten zur Phototoxizität
der wasserlöslichen
Salze von Chlorin- (7) und Phäophorbidderivaten
[(4), (22) und (25)] mit N-Methyl-D-glucamin (10) in HeLa-Zellen
(Beispiel 34)
-
Beispiel 34: Bestimmung
der Phototoxizität
der wasserlöslichen
Salze von Chlorin- (7) und Phäophorbidderivaten
[(4), (22) und (25)] mit N-Methyl-D-glucamin (10) in Experimenten
zur antimikrobiellen photodynamischen Therapie.
-
Die
verwendeten Mikroorganismen waren historische und klinische Bakterienzelllinien
von Staphylococcus aureus (ATCC25923), Enterococcus faecalis (ATCC29212)
und Candida spp (klinisch).
-
Die
Bakterienkolonien wurden auf folgenden festen Medien kultiviert:
Staphylococcus aureus auf Staphylococcus Agar 110, Enterococcus
faecalis auf der Basis von Columbia Agar mit 5% Pferdeblut, Candida spp
auf Saburo Dextrose Agar.
-
Die
Ausgangsbakteriensuspension (IBS) wurde in isotonischer NaCl-Lösung gemäß dem McFarland-Standard
mit einem Wert von 0,5 – 1,0
(1,5 – 5 × 108 Bakterienzellen in 1 ml) hergestellt, gefolgt
von einer Verdünnung
1:100.
-
Zur
Kontrolle des Bakterienzell-Counts wurde ein Teil der IBS 1:1000
mit isotonischer NaCl-Lösung verdünnt (auf
10–3 der
Ausgangskonzentration), dann wurden drei Teilproben (jeweils 0,1
ml) entnommen und auf separate Petrischalen mit dem für die Mikroorganismen
geeigneten Medium aufgebracht. Die Bakterienkolonien wurden 24 Stunden
(48 Stunden für
Staphylococcus aureus) bei 37°C
inkubiert, dann wurden die Kolonien gezählt und ein Mittelwert von
jeder der drei Petrischalen wurde als Kontrolle verwendet.
-
A) Kontrolle von Temperatur
und Laser-Bestrahlung.
-
2,0
ml der Ausgangsbakteriensuspension (IBS) wurden 30 min bei 37°C gelagert
und dann 1:1000 mit isotonischer NaCl-Lösung verdünnt (auf 10–3 der
Ausgangskonzentration), dann wurden drei Teilproben (jeweils 0,1
ml) entnommen und auf separate Petrischalen mit dem für die Mikroorganismen
geeigneten Medium aufge bracht. Die Bakterienkolonien wurden 24 Stunden
bei 37°C
gelagert, dann gezählt
und ein Mittelwert von jeder der drei Petrischalen wurde bestimmt
und als Kontrolle verwendet.
-
Eine
weitere Probe der IBS (2,0 ml) wurde 30 min bei 37°C gelagert
und dann mit Laserlicht in 40 mm-Petrischalen (Dicke der Suspensionsschicht:
2 mm) mit einem „Ceralas
PDT"-Laser für 210 Sekunden
mit einer Ausgangsleistung von 3 W bestrahlt, so dass die Dosis
der abgegebenen Lichtenergie etwa 50 J/cm2 betrug.
Das Gemisch wurde 1:1000 mit isotonischer NaCl-Lösung verdünnt (auf 10–3 der
Ausgangskonzentration), dann wurden drei Teilproben (jeweils 0.1
ml) entnommen und auf separate Petrischalen aufgebracht, kultiviert
und dann wie oben beschrieben gezählt.
-
Die
Daten in Tabelle 3 [Behandlungsregime B und C gezeigt für (7)] zeigen,
dass die Lagerung der IBS für
30 min bei 37°C
ohne Photosensibilisator und unter Laserbestrahlung mit einer Dosis
von 50 J/cm2 die Bakterienviabilität praktisch
nicht beeinflusst.
-
B) Bestimmung der Dunkeltoxizität.
-
0,5
ml einer Vorratslösung
des Photosensibilisators mit einer Konzentration von 500 μg/ml wurden
zu 2,0 ml IBS hinzugegeben, um eine Konzentration des Photosensibilisators
von 100 μg/ml
zu erreichen. Die gebildete Suspension wurde 30 min bei 37°C gelagert,
und dann 1:1000 mit isotonischer NaCl-Lösung verdünnt (auf 103 der
Ausgangskonzentration). Drei Anteile von jeweils 0,1 ml wurden entnommen,
auf drei Petrischalen aufgebracht und dann wie oben beschrieben
gezählt.
-
Die
Daten in Tabelle 3 zeigen, dass die Photosensibilisatoren der vorliegenden
Erfindung unterhalb einer Konzentration von 100 μg/ml bei 30 Minuten Inkubation
nahezu keinen dunkeltoxischen Effekt besitzen.
-
C) Bestimmung der Phototoxizität.
-
0,125
oder 0.5 ml einer Vorratslösung
des Photosensibilisators mit einer Konzentration von 500 μg/ml wurden
zu 2,0 ml IBS hinzugegeben, um eine Konzentration des Photosensibilisators
von 25 bzw. 100 μg/ml zu
erreichen. Die gebildeten Suspensionen wurden 30 min bei 37°C gelagert
und dann mit einer Dosis von 50 J/cm2 bestrahlt.
Drei Teilproben von jeweils 0,1 ml wurden entnommen und auf drei
Petrischalen aufgebracht. Anteile der verbliebenen Suspensionen
wurden 1:100 oder 1:1000 mit isotonischer NaCl-Lösung verdünnt (auf 10–2 oder
10–3 der
Ausgangskonzentration), dann wurden drei Anteile von jeweils 0,1
ml von jeder Verdünnung auf
drei Petrischalen aufgebracht und wie oben beschrieben gezählt.
-
Die
Daten in Tabelle 3 zeigen die mögliche
Anwendbarkeit der wasserlöslichen
Salze aus der vorliegenden Erfindung als Photosensibilisatoren für die antimikrobielle
photodynamische Therapie. Tabelle
3: Anwendung der wasserlöslichen
Salze von Chlorin- (7) und Phäophorbidderivaten
[(4), (22) und (25)] mit N-Methyl-D-glucamin (10) in Experimenten
zur antimikrobiellen photodynamischen Therapie (Beispiel 34)
- * Behandlungsregime:
- A Kontrollbakterien-Count in der verwendeten Ausgangsbakteriensuspension
(IBS);
- B Bakterien-Count nach der Lagerung der IBS für 30 Minuten
bei 37°C;
- C Bakterien-Count nach Bestrahlung mit Laser mit einer Dosis
von 50 J/cm2 („Ceralas PDT", 3 W, 210 Sekunden);
- D Bakterien-Count nach 30-minütiger Inkubation mit Photosensibilisator
(Konzentration of 100 μg/ml);
- E Bakterien-Count nach photodynamischer Behandlung mit einer
Konzentration des Photosensibilisators von 25 μg/ml;
- F Bakterien-Count nach photodynamischer Behandlung mit einer
Konzentration des Photosensibilisators von 100 μg/ml.
-
Nachdem
bevorzugte Ausführungsformen
der vorliegenden Erfindung mit Bezug auf die Beispiele beschrieben
worden sind, versteht es sich von selbst, dass die Erfindung nicht
auf diese speziellen Ausführungsformen
beschränkt
ist, und dass verschiedene Änderungen
und Modifikationen von Fachleuten daran vorgenommen werden können, ohne
vom Geltungsbereich der Erfindung, so wie er in den beigefügten Patentansprüchen definiert
ist, abzuweichen.
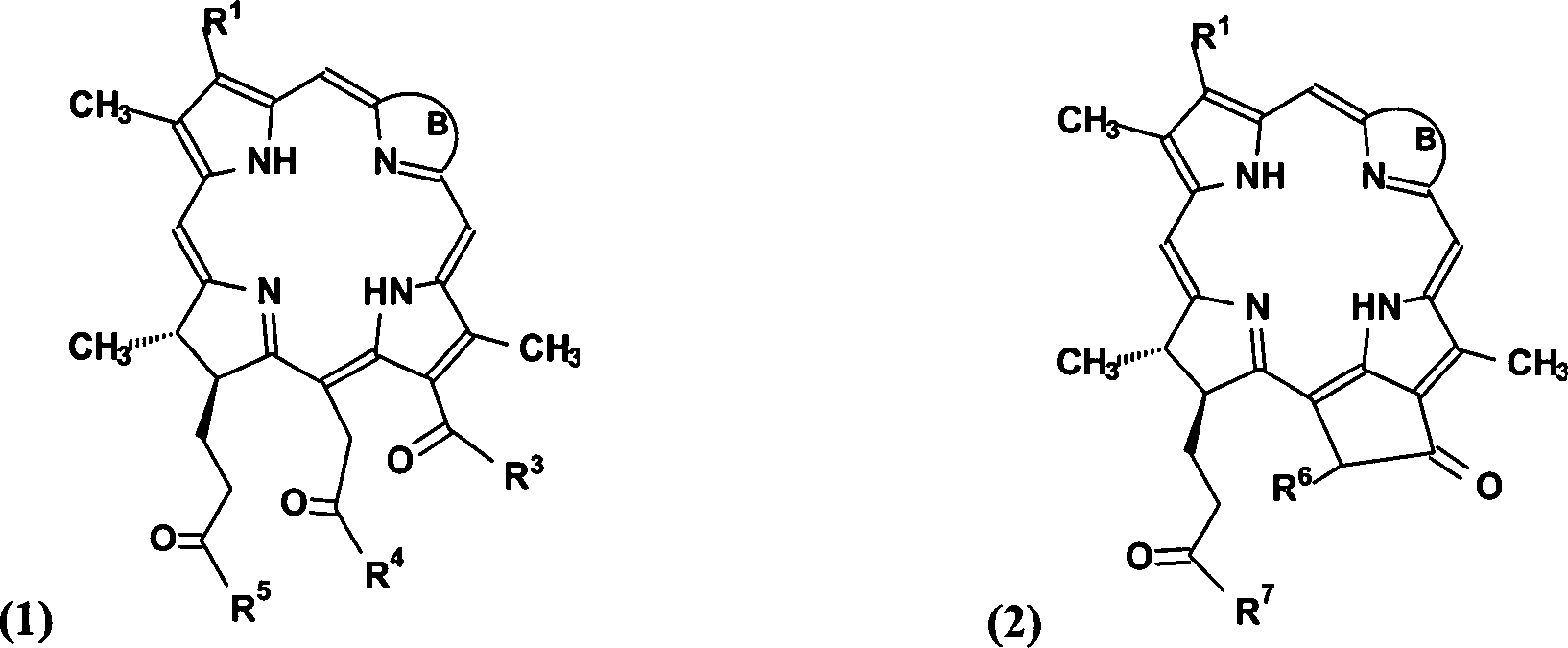















 wobei: R1 = -CH=CH2, -CH(OAlk)CH3, -CHO, -C(O)CH3, -CH2CH3, -CH(Alk)CH(COAlk)2, -CH2CH(COAlk)2, -CH(Alk)CH2COAlk, -CH(Alk)CH2CH(OH)CH3, oder -CH2CH2CH(OH)CH3; R2 = -CH3, -CHO, -CH(OH)Alk, -CH=CHAlk, -CH2OH, oder -CH2OAlk; R3 = -OH, -OAlk, -NH-Alk, NH-X-COO–(HG)+, -NH-Y-NR8R9, oder -NH-Y-OH; R4 = -O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+; R5 = -O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+; R6 = -H oder -COOAlk; R7 = -O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+; R8 = -H oder -Alk; R9 = -H oder -Alk mit: -NH-X-COO– = Reste organischer Aminosäuren; X = Alkylidene, Peptide, Oligopeptide oder -(CH2CH2O)nCH2CH2-, mit n = 1 – 30; Y = Alkylidene oder -(CH2CH2O)nCH2CH2-, mit n = 1 – 30; G = ein hydrophiles organisches Amin (z. B. N-Methyl-D-glucamin oder andere Aminogruppen-enthaltende Kohlenhydrat-Derivate, TRIS, Aminosäuren, Oligopeptide); und Alk = einem Alkyl-Substituenten; beinhaltend die Schritte: a. ein- oder zweistufige direkte saure Alkoholyse von biologischem Ausgangsmaterial, die kristalline Alkyl-Pheophorbide ergibt; b. Umwandlung der erhaltenen Alkyl-Pheophorbide in ein acidisches Porphyrin und c. Reaktion des acidischen Porphyrins mit einem hydrophilen organischen Amin in einem Medium, gewählt aus der Gruppe Wasser und wässrige organische Lösungen.
wobei: R1 = -CH=CH2, -CH(OAlk)CH3, -CHO, -C(O)CH3, -CH2CH3, -CH(Alk)CH(COAlk)2, -CH2CH(COAlk)2, -CH(Alk)CH2COAlk, -CH(Alk)CH2CH(OH)CH3, oder -CH2CH2CH(OH)CH3; R2 = -CH3, -CHO, -CH(OH)Alk, -CH=CHAlk, -CH2OH, oder -CH2OAlk; R3 = -OH, -OAlk, -NH-Alk, NH-X-COO–(HG)+, -NH-Y-NR8R9, oder -NH-Y-OH; R4 = -O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+; R5 = -O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+; R6 = -H oder -COOAlk; R7 = -O–(HG)+, -OAlk, -NH-Alk, oder -NH-X-COO–(HG)+; R8 = -H oder -Alk; R9 = -H oder -Alk mit: -NH-X-COO– = Reste organischer Aminosäuren; X = Alkylidene, Peptide, Oligopeptide oder -(CH2CH2O)nCH2CH2-, mit n = 1 – 30; Y = Alkylidene oder -(CH2CH2O)nCH2CH2-, mit n = 1 – 30; G = ein hydrophiles organisches Amin (z. B. N-Methyl-D-glucamin oder andere Aminogruppen-enthaltende Kohlenhydrat-Derivate, TRIS, Aminosäuren, Oligopeptide); und Alk = einem Alkyl-Substituenten; beinhaltend die Schritte: a. ein- oder zweistufige direkte saure Alkoholyse von biologischem Ausgangsmaterial, die kristalline Alkyl-Pheophorbide ergibt; b. Umwandlung der erhaltenen Alkyl-Pheophorbide in ein acidisches Porphyrin und c. Reaktion des acidischen Porphyrins mit einem hydrophilen organischen Amin in einem Medium, gewählt aus der Gruppe Wasser und wässrige organische Lösungen.