DE60031154T2 - Verfahren zur herstellung von clarithromycin - Google Patents
Verfahren zur herstellung von clarithromycin Download PDFInfo
- Publication number
- DE60031154T2 DE60031154T2 DE60031154T DE60031154T DE60031154T2 DE 60031154 T2 DE60031154 T2 DE 60031154T2 DE 60031154 T DE60031154 T DE 60031154T DE 60031154 T DE60031154 T DE 60031154T DE 60031154 T2 DE60031154 T2 DE 60031154T2
- Authority
- DE
- Germany
- Prior art keywords
- oxide
- clarithromycin
- methylerythromycin
- mixture
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 229960002626 clarithromycin Drugs 0.000 title claims abstract description 42
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 title claims abstract description 39
- 238000000034 method Methods 0.000 title claims description 31
- 238000002360 preparation method Methods 0.000 title claims description 20
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 18
- LUIPOVCSQJTWHA-RWJQBGPGSA-N (2s,3r,4s,6r)-2-[[(3r,4s,5s,6r,7r,9r,11r,12r,13s,14r)-14-ethyl-7,12,13-trihydroxy-4-[(2r,4r,5s,6s)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy-3,5,7,9,11,13-hexamethyl-2,10-dioxo-oxacyclotetradec-6-yl]oxy]-3-hydroxy-n,n,6-trimethyloxan-4-amine oxide Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)[N+](C)(C)[O-])O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 LUIPOVCSQJTWHA-RWJQBGPGSA-N 0.000 claims abstract description 11
- 239000012022 methylating agents Substances 0.000 claims abstract description 7
- 239000000203 mixture Substances 0.000 claims description 44
- MWBJRTBANFUBOX-SQYJNGITSA-N (3r,4s,5s,6r,7r,9r,10e,11s,12r,13s,14r)-6-[(2s,3r,4s,6r)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-14-ethyl-12,13-dihydroxy-10-hydroxyimino-4-[(2r,4r,5s,6s)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy-7-methoxy-3,5,7,9,11,13-hexamethyl-oxacyclot Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=N/O)/[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MWBJRTBANFUBOX-SQYJNGITSA-N 0.000 claims description 36
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 30
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 24
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 claims description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 14
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 12
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 claims description 12
- 229960003276 erythromycin Drugs 0.000 claims description 12
- 239000011734 sodium Substances 0.000 claims description 11
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 10
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 claims description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 8
- 239000002585 base Substances 0.000 claims description 8
- 239000003054 catalyst Substances 0.000 claims description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 7
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 claims description 6
- 229910000838 Al alloy Inorganic materials 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 6
- 229910000564 Raney nickel Inorganic materials 0.000 claims description 6
- 229910021626 Tin(II) chloride Inorganic materials 0.000 claims description 6
- 239000001257 hydrogen Substances 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 239000003960 organic solvent Substances 0.000 claims description 6
- 239000001119 stannous chloride Substances 0.000 claims description 6
- 235000011150 stannous chloride Nutrition 0.000 claims description 6
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 5
- 239000007868 Raney catalyst Substances 0.000 claims description 5
- 235000019253 formic acid Nutrition 0.000 claims description 5
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 4
- 229910003310 Ni-Al Inorganic materials 0.000 claims description 4
- 229910045601 alloy Inorganic materials 0.000 claims description 4
- 239000000956 alloy Substances 0.000 claims description 4
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 claims description 4
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 claims description 4
- 238000005984 hydrogenation reaction Methods 0.000 claims description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 claims description 4
- ZUHZGEOKBKGPSW-UHFFFAOYSA-N tetraglyme Chemical compound COCCOCCOCCOCCOC ZUHZGEOKBKGPSW-UHFFFAOYSA-N 0.000 claims description 4
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 claims description 3
- REDSKZBUUUQMSK-UHFFFAOYSA-N tributyltin Chemical compound CCCC[Sn](CCCC)CCCC.CCCC[Sn](CCCC)CCCC REDSKZBUUUQMSK-UHFFFAOYSA-N 0.000 claims description 3
- LZDKZFUFMNSQCJ-UHFFFAOYSA-N 1,2-diethoxyethane Chemical compound CCOCCOCC LZDKZFUFMNSQCJ-UHFFFAOYSA-N 0.000 claims description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 2
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 claims description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 2
- LLJZKKVYXXDWTB-UHFFFAOYSA-N acetic acid;sodium Chemical compound [Na].[Na].CC(O)=O LLJZKKVYXXDWTB-UHFFFAOYSA-N 0.000 claims description 2
- 229910000102 alkali metal hydride Inorganic materials 0.000 claims description 2
- 150000008046 alkali metal hydrides Chemical class 0.000 claims description 2
- 150000004703 alkoxides Chemical class 0.000 claims description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 claims description 2
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 claims description 2
- 239000011790 ferrous sulphate Substances 0.000 claims description 2
- 235000003891 ferrous sulphate Nutrition 0.000 claims description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 claims description 2
- 150000004679 hydroxides Chemical class 0.000 claims description 2
- BAUYGSIQEAFULO-UHFFFAOYSA-L iron(2+) sulfate (anhydrous) Chemical compound [Fe+2].[O-]S([O-])(=O)=O BAUYGSIQEAFULO-UHFFFAOYSA-L 0.000 claims description 2
- 229910000359 iron(II) sulfate Inorganic materials 0.000 claims description 2
- VUQUOGPMUUJORT-UHFFFAOYSA-N methyl 4-methylbenzenesulfonate Chemical compound COS(=O)(=O)C1=CC=C(C)C=C1 VUQUOGPMUUJORT-UHFFFAOYSA-N 0.000 claims description 2
- 229940102396 methyl bromide Drugs 0.000 claims description 2
- MBABOKRGFJTBAE-UHFFFAOYSA-N methyl methanesulfonate Chemical compound COS(C)(=O)=O MBABOKRGFJTBAE-UHFFFAOYSA-N 0.000 claims description 2
- 239000012285 osmium tetroxide Substances 0.000 claims description 2
- 229910000489 osmium tetroxide Inorganic materials 0.000 claims description 2
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 claims description 2
- 229910000059 tellane Inorganic materials 0.000 claims description 2
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 claims description 2
- XQKBFQXWZCFNFF-UHFFFAOYSA-K triiodosamarium Chemical compound I[Sm](I)I XQKBFQXWZCFNFF-UHFFFAOYSA-K 0.000 claims description 2
- 229910052725 zinc Inorganic materials 0.000 claims description 2
- 239000011701 zinc Substances 0.000 claims description 2
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 claims 1
- -1 6-O-methylerythromycin A N-oxide Chemical class 0.000 abstract 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 34
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 23
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 14
- 239000002904 solvent Substances 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 11
- 239000000843 powder Substances 0.000 description 11
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 9
- 235000019439 ethyl acetate Nutrition 0.000 description 9
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 8
- 238000007069 methylation reaction Methods 0.000 description 8
- 239000006227 byproduct Substances 0.000 description 7
- 239000013078 crystal Substances 0.000 description 7
- 229930006677 Erythromycin A Natural products 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Chemical compound O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 6
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 5
- 238000009835 boiling Methods 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 229910000029 sodium carbonate Inorganic materials 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 4
- 230000011987 methylation Effects 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 230000003472 neutralizing effect Effects 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 150000002923 oximes Chemical group 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- KYTWXIARANQMCA-RWJQBGPGSA-N (3r,4s,5s,6r,7r,9r,11s,12r,13s,14r)-6-[(2s,3r,4s,6r)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-14-ethyl-7,12,13-trihydroxy-10-hydroxyimino-4-[(2r,4r,5s,6s)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy-3,5,7,9,11,13-hexamethyl-oxacyclotetradecan-2 Chemical class O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=NO)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 KYTWXIARANQMCA-RWJQBGPGSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- 241000590002 Helicobacter pylori Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 241000204031 Mycoplasma Species 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 238000006972 Polonovski rearrangement reaction Methods 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000002026 chloroform extract Substances 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 230000010485 coping Effects 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 238000003386 deoximation reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- FGRVOLIFQGXPCT-UHFFFAOYSA-L dipotassium;dioxido-oxo-sulfanylidene-$l^{6}-sulfane Chemical compound [K+].[K+].[O-]S([O-])(=O)=S FGRVOLIFQGXPCT-UHFFFAOYSA-L 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- RDYMFSUJUZBWLH-UHFFFAOYSA-N endosulfan Chemical compound C12COS(=O)OCC2C2(Cl)C(Cl)=C(Cl)C1(Cl)C2(Cl)Cl RDYMFSUJUZBWLH-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229940037467 helicobacter pylori Drugs 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000003120 macrolide antibiotic agent Substances 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- 230000001035 methylating effect Effects 0.000 description 1
- 238000006146 oximation reaction Methods 0.000 description 1
- 125000003544 oxime group Chemical group 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- DJEHXEMURTVAOE-UHFFFAOYSA-M potassium bisulfite Chemical compound [K+].OS([O-])=O DJEHXEMURTVAOE-UHFFFAOYSA-M 0.000 description 1
- 229940099427 potassium bisulfite Drugs 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- 235000010259 potassium hydrogen sulphite Nutrition 0.000 description 1
- KAQHZJVQFBJKCK-UHFFFAOYSA-L potassium pyrosulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OS([O-])(=O)=O KAQHZJVQFBJKCK-UHFFFAOYSA-L 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000005956 quaternization reaction Methods 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 238000011935 selective methylation Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- JXAZAUKOWVKTLO-UHFFFAOYSA-L sodium pyrosulfate Chemical compound [Na+].[Na+].[O-]S(=O)(=O)OS([O-])(=O)=O JXAZAUKOWVKTLO-UHFFFAOYSA-L 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000006104 solid solution Substances 0.000 description 1
- XTQHKBHJIVJGKJ-UHFFFAOYSA-N sulfur monoxide Chemical class S=O XTQHKBHJIVJGKJ-UHFFFAOYSA-N 0.000 description 1
- 229910052815 sulfur oxide Inorganic materials 0.000 description 1
- VTLHPSMQDDEFRU-UHFFFAOYSA-N tellane Chemical compound [TeH2] VTLHPSMQDDEFRU-UHFFFAOYSA-N 0.000 description 1
- 241001148471 unidentified anaerobic bacterium Species 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Saccharide Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Description
- Gebiet der Erfindung
- Die vorliegende Erfindung betrifft ein Verfahren zur Herstellung von Clarithromycin unter Verwendung von Erythromycin A-N-oxid; und die Verwendung von 6-O-Methylerythromycin A-N-oxid in dem Verfahren.
- Hintergrund der Erfindung
- Clarithromycin, 6-O-Methylerythromycin A, ist ein semisynthetisches Makrolidantibiotikum, welches einen großen Bereich an antibakterieller Aktivität gegen grampositive Bakterien, einige gramnegative Bakterien, anaerobe Bakterien, Mycoplasma, Chlamidia und Helicobacter pylori zeigt.
- Clarithromycin kann hergestellt werden durch die Methylierung der 6-Hydroxygruppe von Erythromycin A. Gleichwohl ist eine selektive Methylierung der 6-Hydroxygruppe schwierig zu erreichen, da Erythromycin A vier reaktive Hydroxygruppen neben der 6-Hydroxygruppe sowie eine Dimethylaminogruppe, welche eine Quaternisierung während der Methylierungsreaktion erfahren kann, besitzt.
- Um mit einem solchen Problem fertig zu werden, sind verschiedene Verfahren zur Herstellung von Clarithromycin entwickelt worden.
- Zum Beispiel wurde ein allgemeines Verfahren zur Herstellung von Clarithromycin unter Verwendung eines Erythromycin A-9-oxim-Derivats als ein Intermediat beschrieben (siehe EP-Patent Nr. 0 158 467, 0 195 960, 0 260 938 und 0 272 110 und internationale Veröffentlichung Nr. WO 97/36912 und WO 97/36913). Obgleich dieses Verfahren zu einer relativ hohen Ausbeute führt, leidet es unter einer niedrigen Produktivität aufgrund der Tatsache, dass das Verfahren eine Vielzahl von Reaktionsschritten erfordert, einschließlich der Schritte einer Oximierung, dem Schutz der Oximgruppe, der Entfernung der Oximschutzgruppe und der Desoximierung.
- Ein weiteres allgemeines Verfahren zur Herstellung von Clarithromycin wurde in dem EP-Patent Nr. 0 147 062 und 0 177 696 beschrieben. Das Verfahren, welches in dem EP-Patent Nr. 0 147 062 beschrieben ist, umfasst folgende Schritte: Schützen der 2-Hydroxy- und Aminogruppen von Erythromycin A mit Benzyloxycarbonylgruppen; Methylierung der 6-Hydroxygruppe; Entfernen der zwei Benzyloxycarbonylschutzgruppen und Methylieren der Aminogruppe zum Erhalt von Clarithromycin. Gleichwohl leidet dieses Verfahren unter dem Problem einer niedrigen Ausbeute von 13,1 bis 18,5% und erfordert die Verwendung von überschüssigem Benzyloxycarbonylchlorid, neben der Schwierigkeit der Anwendung der säulenchromatographischen Abtrennung des Produktes bei der Massenherstellung.
- Die WO 98/35976 beschreibt ein Verfahren zur Herstellung von Clarithromycin ausgehend von Erythromycin A, Schützen der 9-Oxogruppe durch Umwandlung zu einem Oxim, Oxidieren des 3'-N zum Erhalt eines 3'-N-Oxid-9-O-oxim-Derivats, Methylieren der 6-Hydroxygruppe, gefolgt von einer Eliminierung der 3'-N-Oxid- und 9-O-Oxim-Gruppen.
- Y.-Y. Ku. et al. beschreiben eine Behandlung von 6-O-Methylerythromycin A-N-oxid mit Essigsäureanhydrid in EtOAc zur Herstellung von Des-N-Methyl-N-acetylclarithromycin, welche die Polonovsky-Reaktion involviert [Y.-Y. Ku et al., Bioorg. Med. Chem. Lett., 7(9), 1 203 (1997)]. Dabei wird die Oxidation von Clarithromycin zum Erhalt von 6-O-Methylerythromycin A-N-oxid erwähnt, wie bereits früher von D. R. Hill et al. [D. R. Hill et al., Tetrahedron Lett., 37(6), 787 (1996)].
- Demzufolge bestand der Bedarf nach der Entwicklung eines verbesserten Verfahrens zur Herstellung von Clarithromycin.
- Zusammenfassung der Erfindung
- Es ist mithin ein Ziel der vorliegenden Erfindung, ein Verfahren zur Herstellung von Clarithromycin bereitzustellen.
- Es ist ein weiteres Ziel der vorliegenden Erfindung, die Verwendung von 6-O-Methylerythromycin A-N-oxid in dem Verfahren bereitzustellen.
- In Übereinstimmung mit einem Aspekt der vorliegenden Erfindung wird ein Verfahren zur Herstellung von Clarithromycin der Formel (I) bereitgestellt, umfassend die Schritte:
- (a) Umsetzen von Erythromycin A-N-oxid der Formel (II) mit einem Methylierungsmittel unter Erhalt von 6-O-Methylerythromycin A-N-oxid der Formel (III); und
- (b) Behandeln des in Schritt (a) erhaltenen 6-O-Methylerythromycin
A-N-oxids mit einem Reduktionsmittel unter Erhalt von Clarithromycin:


- In Übereinstimmung mit einem anderen Aspekt der vorliegenden Erfindung wird die Verwendung von 6-O-Methylerythromycin A-N-oxid der Formel (III) für ein Verfahren zur Herstellung von Clarithromycin der Formel (I) bereitgestellt.
- Detaillierte Beschreibung der Erfindung
- Die Verbindung der Formel (I) kann wie folgt ausgehend von Erythromycin A-N-oxid hergestellt werden.
- Schritt (a)
- 6-O-Methylerythromycin A-N-oxid der Formel (III) wird durch die Umsetzung von Erythromycin A-N-oxid der Formel (II) hergestellt, erzeugt in einer Ausbeute von mehr als 98% aus Erythromycin A gemäß einem allgemein bekannten Verfahren [E. H. Flynn et al., J. Am. Chem. Soc., 76, 3 121 (1954) und P. H. Jones und E. K. Rowley, J. Org. Chem., 33, 665 (1968)], und zwar mit einem Methylierungsmittel in einem organischen Lösungsmittel in Gegenwart einer Base.
- Beispielhafte Methylierungsmittel, welche in geeigneter Weise in der vorliegenden Erfindung verwendet werden können, sind Methylbromid, Methyliodid, Dimethylsulfat, Methyl-p-toluolsulfonat, Methylmethansulfonat und eine Mischung davon. Das Methylierungsmittel kann in einer Menge im Bereich von 1 bis 3 Äquivalenten, bezogen auf die Menge an Erythromycin A-N-oxid der Formel (II), verwendet werden.
- Beispielhafte Lösungsmittel, welche in der obigen Methylierungsreaktion verwendet werden können, schließen N,N-Dimethylformamid, N,N-Dimethylacetamid, N-Methyl-2-pyrrolidon, Dimethylsulfoxid, Sulfolan, N,N,N',N',N'',N''-Hexamethylphosphoramid, Tetrahydrofuran, 1,4-Dioxan, 1,2-Dimethoxyethan, 1,2-Diethoxyethan, 2-Methoxyethylether, 2-Ethoxyethylether, 1,2-Bis(2-methoxyethoxy)ethan, Tetraethylenglykoldimethylether, Aceton, Acetonitril oder eine Mischung davon ein.
- Ferner kann eine Base, die aus der Gruppe gewählt wird, welche aus Alkalimetallhydriden, -hydroxiden und -alkoxiden, zum Beispiel Natriumhydrid, Kaliumhydrid, Natriumhydroxid, Kaliumhydroxid und Kalium-t-butoxid, besteht, in geeigneter Weise in diesem Verfahren verwendet werden. Die Base kann in einer Menge verwendet werden, die im Bereich von 0,9 bis 2 Äquivalenten, basierend auf der Menge an Erythromycin A-N-oxid der Formel (II), liegt. Die Methylierungsreaktion kann bei einer Temperatur im Bereich von –15 bis 40°C, vorzugsweise von 0°C bis Raumtemperatur, durchgeführt werden.
- Nach der Beendigung der Methylierungsreaktion wird Wasser zu der resultierenden Mischung hinzugesetzt, und die Mischung wird mit Chloroform extrahiert. Der Extrakt wird konzentriert, und Aceton wird dem Rückstand hinzugesetzt, und dann wird er gerührt, um Nebenprodukte zu präzipitieren. Die resultierende Mischung wird filtriert, um Nebenprodukte zu entfernen, und das Filtrat wird konzentriert, wodurch man rohes 6-O-Methylerythromycin A-N-oxid erhielt (Ausbeute: 67 bis 73%, und Reinheit: 50 bis 57%).
- Das oben erhaltene rohe 6-O-Methylerythromycin A-N-oxid kann wie in Schritt (b) verwendet werden, oder durch Umkristallisation aus Ethylacetat gereinigt werden, wodurch man verbessertes 6-O-Methylerythromycin A-N-oxid erhielt (Reinheit von 83% bis 88% und Ausbeute von 40 bis 44%), welches durch Umkristallisation aus Chloroform weiter gereinigt werden kann, um 6-O-Methylerythromycin A-N-oxid-Kristalle mit einer Reinheit von über 95% zu erhalten.
- Schritt (b)
- Clarithromycin der Formel (I) wird hergestellt, indem das in Schritt (a) erhaltene 6-O-Methylerythromycin A-N-oxid mit einem Reduktionsmittel in einem organischen Lösungsmittel zur Entfernung eines N-Oxids umgesetzt wird.
- Beispielhafte Reduktionsmittel, welche in geeigneter Weise in der vorliegenden Erfindung verwendet werden können, sind Wasserstoff in Gegenwart eines Hydrierungskatalysators, wie Palladium, Raney-Nickel oder Platinoxid (PtO2); eine Nickel-Aluminium-Legierung (Ni-Al-Legierung) kombiniert mit Kaliumhydroxid; metallisches Zink in Gegenwart von Ameisensäure oder Essigsäure; Natriumhydrogentellurid (NaTeH); Samariumiodid (SmI2); Zinn(II)-chlorid (SnCl2); Hexabutyldizinn (Bu3SnSnBu3); Cyclohexen und Osmiumtetroxid (OsO4); und Eisen(II)-sulfat, und bevorzugte Reduktionsmittel bei der Ausführung der vorliegenden Erfindung sind Zinn(II)-chlorid (SnCl2), Hexabutyldizinn (Bu3SnSnBu3), eine Nickel-Aluminium-Legierung kombiniert mit Kaliumhydroxid und Wasserstoff in Gegenwart eines Raney-Nickel- oder Platinoxid (PtO2)-Katalysators.
- Im Fall der Verwendung von Zinn(II)-chlorid als ein Reduktionsmittel können Lösungsmittel wie Methanol, Ethanol, Isopropanol, Ethylacetat, Acetonitril, Aceton, Tetrahydrofuran, 1,2-Dimethoxyethan, Dichlormethan, Chloroform und eine Mischung davon verwendet werden. Die Menge an verwendetem Zinn(II)-chlorid liegt im Bereich von 1 bis 3 Moläquivalenten, bezogen auf die Menge an 6-O-Methylerythromycin A-N-oxid der Formel (III), und die Reaktion kann bei einer Temperatur im Bereich von 0°C bis zum Siedepunkt des verwendeten Lösungsmittels, vorzugsweise von 10 bis 45°C, durchgeführt werden. Nach Beendigung der Reaktion kann Clarithromycin durch (1) Neutralisieren der Reaktionsmischung mit einer Base wie Triethylamin; Hinzusetzen von Wasser; und Extrahieren der Mischung mit einem organischen Lösungsmittel; oder (2) durch Hinzusetzen von Wasser zur Reaktionsmischung; Neutralisieren der resultierenden Mischung mit einer Base wie Natriumbicarbonat, Natriumcarbonat, Natriumhydroxidlösung und wässrigem Ammoniak; und Extrahieren der Mischung mit einem organischen Lösungsmittel isoliert werden.
- In dem Fall, dass Hexabutyldizinn als ein Reduktionsmittel verwendet wird, können Lösungsmittel wie Ethylacetat, Acetonitril, Aceton, Tetrahydrofuran, 1,2-Dimethoxyethan und eine Mischung verwendet werden. Hexabutyldizinn wird in einer Menge verwendet, die im Bereich von 1 bis 3 Moläquivalenten, bezogen auf die Menge an 6-O-Methylerythromycin A-N-oxid der Formel (III), liegt, und die Reaktion kann bei einer Temperatur durchgeführt werden, die zwischen Raumtemperatur und dem Siedepunkt des verwendeten Lösungsmittels liegt.
- Wenn eine Ni-Al-Legierung zusammen mit Kaliumhydroxid als ein Reduktionsmittel verwendet wird, wird eine Mischung aus Wasser und einem niederen Alkohol, z. B. Methanol und Ethanol, als ein Lösungsmittel angewandt. Die Mengen der verwendeten Ni-Al-Legierung und des verwendeten Kaliumhydroxids liegen im Bereich von 0,5 bis 3 g bzw. 2 bis 20 Mol, bezogen auf ein Mol an 6-O-Methylerythromycin A-N-oxid der Formel (III). Die Reaktion kann bei einer Temperatur im Bereich von Raumtemperatur bis zum Siedepunkt des verwendeten Lösungsmittels durchgeführt werden.
- Wenn eine katalytische Hydrierung als ein Mittel der Durchführung der Reaktion des Schrittes (b) durchgeführt wird, kann ein niederer Alkohol oder eine Mischung aus Wasser und einem niederen Alkohol als ein Lösungsmittel verwendet werden, und ein Hydrierungskatalysator wie Raney-Nickel als Katalysator bei einer Temperatur im Bereich von Raumtemperatur bis zum Siedepunkt des Lösungsmittels unter einer Wasserstoffatmosphäre.
- Die Reaktion vom Schritt (b) kann ebenfalls unter Verwendung eines anorganischen Reduktionsmittels wie Natriumbisulfit (NaHSO3), Natriumsulfit (Na2SO3), Natriumthiosulfat (Na2S2O3), Natriumhydrogensulfit (Na2S2O4), Natriumpyrosulfat (Na2S2O5), Natriumthionat (Na2S2O6), Kaliumbisulfit (KHSO3), Kaliumthiosulfat (K2S2O3) und Kaliumpyrosulfat (K2S2O5) durchgeführt werden. Die Reaktion kann in einer Mischung von Wasser und einem niederen Alkohol wie Methanol, Ethanol und Isopropanol bei einer Temperatur im Bereich von 0°C bis zum Siedepunkt des verwendeten Lösungsmittels durchgeführt werden. Das Reduktionsmittel kann in einer Menge verwendet werden, die im Bereich von 1 bis 20 Äquivalenten liegt, vorzugsweise von 1 bis 4 Äquivalenten, bezogen auf die Menge an 6-O-Methylerythromycin A-N-oxid der Formel (III).
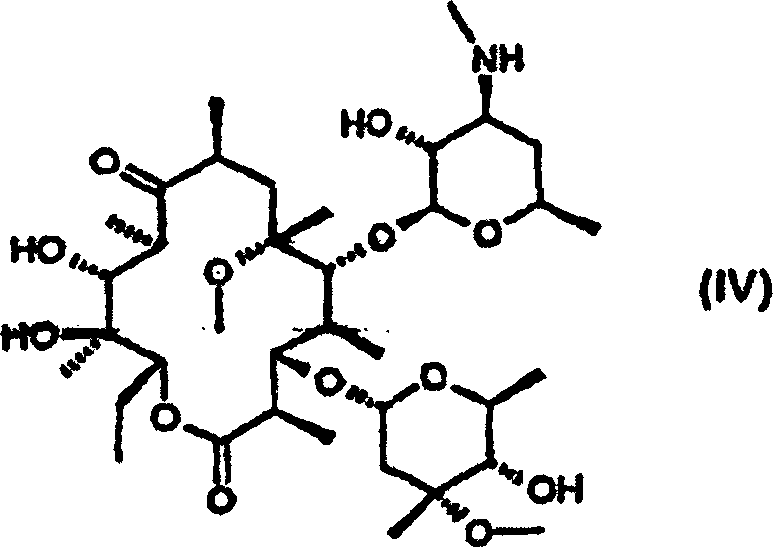
- Bei der Durchführung der Reaktion des Schrittes (b) unter Verwendung von Schwefeloxiden als ein Reduktionsmittel können 3'-N-Desmethyl-6-O-methylerythromycin A der Formel (IV) als ein Nebenprodukt neben Clarithromycin der Formel (I) gebildet werden. In einem solchen Fall können die Nebenprodukte zu Clarithromycin durch Methylierung der sekundären Amingruppe des Nebenproduktes unter Verwendung von Ameisensäu re und Formaldehyd in Übereinstimmung mit einem bekannten Verfahren umgewandelt werden [siehe Eschweiler & Clarke, Org. React., 5, 290 (1945)].
-
- Das folgende Referenzbeispiel und die Beispiele sollen die vorliegende Erfindung weiter veranschaulichen, ohne ihren Umfang zu beschränken; und die experimentellen Verfahren, die in der vorliegenden Erfindung verwendet werden können, können gemäß dem Referenzbeispiel und den Beispielen, die hierin unten angeführt werden, ausgeführt werden, wenn nichts anderes angegeben ist.
- Ferner basieren die unten angeführten Prozentangaben für Feststoff in fester Mischung, Flüssigkeit in Flüssigkeit und Feststoff in Flüssigkeit auf w/w, vol/vol bzw. w/vol, wenn nicht spezifisch in anderer Weise angegeben.
- Referenzbeispiel: Herstellung von Erythromycin A-N-oxid
- 220,2 g (0,3 Mol) Erythromycin A wurden in einer Mischung aus 1 500 ml Methanol und 1 000 ml Wasser gelöst. 79 ml (0,9 Mol) 35%iges Wasserstoffperoxid wurden tropfenweise der Lösung bei einer Temperatur im Bereich von 15 bis 20°C hinzugesetzt und bei Raumtemperatur 20 Stunden lang in einem ähnlichen Verfahren, das in E. H. Flynn et al., J. Am. Chem. Soc., 76, 3 121 (1954) und P. H. Jones et al., J. Org. Chem., 33, 665 (1968) beschrieben ist, gerührt. Die resultierende Mischung wurde auf die Hälfte des Volumens unter einem reduzierten Druck konzentriert und der Reihe nach mit 1 000 ml und 500 ml großen Portionen an Chloroform extrahiert. Der vereinigte Chloroformextrakt wurde mit Salzlösung gewaschen, über wasserfreiem Magnesiumsulfat getrocknet und dann unter einem reduzierten Druck konzentriert, um einen schaumförmigen Rückstand zu erhalten. 1 000 ml Aceton wurden dem Rückstand hinzugesetzt, und es wurde bei Raumtemperatur 3 Stunden lang gerührt. Das gebildete kristalline Material wurde filtriert und über Nacht bei 45°C getrocknet, um 220 g Erythromycin A-N-oxid in einer Ausbeute von 98% zu erhalten.
Schmp.: 218~221°C (Schmp. in der Literatur: 222~224°C)
1H-NMR (CDCl3, ppm): δ 5,00 (dd, 1H, 13-H), 4,86 (d, 1H, 1''-H), 4,50 (d, 1H, 1'-H), 3,34 (s, 3H, Cladinose-OCH3), 3,17 (d, 6H, Desoxamin-N(CH3)2), 1,43 (s, 3H, 18-H), 0,82 (t, 3H, 15-H).
MS (m/z): ESI 750 [M + 1]+ - Beispiel 1: Herstellung von 6-O-Methylerythromycin A-N-oxid
- 75,0 g (0,1 Mol) des im Referenzbeispiel erhaltenen Erythromycin A-N-oxids wurden in einer Mischung aus 450 ml Dimethylsulfoxid und 450 ml Tetrahydrofuran suspendiert und auf 5°C gekühlt. 8,1 ml Iodmethan und 7,26 g 85%iges Kaliumhydroxidpulver wurden hinzugesetzt, und die Mischung wurde 1 Stunde lang gerührt. 1 000 ml kaltes Wasser wurden der resultierenden Mischung hinzugesetzt und der Reihe nach mit 1 000 ml und 500 ml großen Portionen an Chloroform extrahiert. Der vereinigte Extrakt wurde zweimal mit 500 ml Wasser gewaschen, über wasserfreiem Magnesiumsulfat getrocknet und dann unter einem reduzierten Druck konzentriert. 500 ml Aceton wurden dem schaumartigen Rückstand hinzugesetzt, und es wurde bei Raumtemperatur 5 Stunden lang gerührt. Die resultierende Mischung wurde filtriert, um präzipitierte Nebenprodukte zu entfernen. Das Filtrat wurde unter einem reduzierten Druck konzentriert, wodurch man 52,7 g rohes 6-O-Methylerythromycin A-N-oxid mit einer Reinheit von 57% erhielt.
- 300 ml Ethylacetat wurden dem rohen Produkt hinzugesetzt, und es wurde zum Erhalt von Kristallen gerührt. Die Kristalle wurden filtriert und über Nacht bei 45°C getrocknet, wodurch man 30,3 g 6-O-Methylerythromycin A-N-oxid als ein kristallines Pulver mit einer Reinheit von 88% erhielt.
- Das kristalline Pulver wurde aus Chloroform umkristallisiert, wodurch man 20,1 g der Titelverbindung mit einer Reinheit von mehr als 95% in einer Ausbeute von 27% erhielt.
Schmp.: 204~207°C
IR (KBr, cm–1): 3 446, 2 972, 2 938, 1 733, 1 693, 1 462, 1 379, 1 169, 1 111, 1 079.
1H-NMR (CDCl3, ppm): δ 5,06 (dd, 1H, 13-H), 4,93 (d, 1H, 1''-H), 4,55 (d, 1H, 1'-H), 4,02 (dq, 1H, 5''-H), 3,76 (d, 1H, 11-H), 3,73 (dd, 1H, 3-H), 3,70 (d, 1H, 5-H), 3,65 (ddq, 1H, 5'-H), 3,38 (s, 3H, 3''-OCH3), 3,19 (d, 6H, 3'-N(CH3)2), 3,05 (s, 3H, 6-OCH3), 2,91 (dq, 1H, 2-H); 2,59 (ddq, 1H, 8-H), 1,41 (s, 3H, 18-H), 1,12 (s, 3H, 6-CH3), 0,83 (t, 3H, 14-CH3).
13C-NMR (CDCl3, ppm): δ 221,4 (C1=O), 176,2 (C9=O), 102,9 (C1'), 96,5 (C1''), 81,6 (C5), 79,0 (C6), 78,7 (C3), 78,2 (C4''), 77,6 (C3'), 77,0 (C13), 76,7 (C2'), 74,7 (C12), 73,1 (C3''), 69,5 (C11), 67,4 (C5'), 66,3 (C5''), 59,1 (C8'), 52,8 (C7'), 51,0 (C22), 50,1 (C8''), 45,7 (C8), 45,4 (C2), 39,7 (C7), 39,6 (C4), 37,7 (C10), 35,3 (C2''), 32,0 (C4'), 22,0 (C6'), 21,7 (C7''), 21,4 (C14), 20,2 (C18), 19,1 (C6''), 18,4 (C19), 16,4 (C21), 16,3 (C16), 12,7 (C20), 11,0 (C15), 9,4 (C17).
MS (m/z): ESI 764 [M + 1]+. - Beispiel 2: Herstellung von 6-O-Methylerythromycin A-N-oxid
- 75,0 g (0,1 Mol) des im Referenzbeispiel erhaltenen Erythromycin A-N-oxids wurden in einer Mischung 450 ml Dimethylsulfoxid und 450 ml Tetrahydrofuran suspendiert und auf 5°C gekühlt. 8,1 ml Iodmethan und 7,26 g 85%iges Kaliumhydroxidpulver wurden hinzugesetzt, und die Mischung wurde 1,5 Stunden lang gerührt. 1 000 ml kaltes Wasser wurden der resultierenden Mischung hinzugesetzt, und es wurde der Reihe mit 1 000 ml und 500 ml großen Portionen an Chloroform extrahiert. Der vereinigte Extrakt wurde zweimal mit 500 ml Wasser gewaschen, über wasserfreiem Magnesiumsulfat getrocknet und dann unter einem reduzierten Druck konzentriert, um einen schaumförmigen Rückstand zu erhalten. 500 ml Aceton wurden dem Rückstand hinzugesetzt, und es wurde bei Raumtemperatur 5 Stunden lang gerührt. Die resultierende Mischung wurde filtriert, um präzipitierte Nebenprodukte zu entfernen. Das Filtrat wurde unter reduziertem Druck konzentriert, wodurch man 50,4 g rohes 6-O-Methylerythromycin A-N-oxid mit einer Reinheit von 55% erhielt.
- Beispiel 3: Herstellung von Clarithromycin unter Verwendung von Zinn(II)-chlorid-dihydrat als ein Reduktionsmittel
- 3,82 g (5 mMol) des in Beispiel 1 erhaltenen 6-O-Methylerythromycin A-N-oxids wurden in 30 ml Isopropanol suspendiert. 2,26 g (2,0 mMol) Zinn(II)-chlorid-dihydrat wurden hinzugesetzt, und die Mischung wurde bei einer Temperatur im Bereich von 30 bis 40°C 2 Stunden lang gerührt. Eine gesättigte Natriumbicarbonatlösung wurde zu der resultierenden Mischung hinzugesetzt, und es wurde zweimal mit Ethylacetat extrahiert. Die vereinigte organische Schicht wurde mit Wasser gewaschen, über wasserfreiem Magnesiumsulfat getrocknet und dann unter einem reduzierten Druck konzentriert, wodurch man 3,60 g Clarithromycin als ein weißes Pulver in einer Ausbeute von 96% erhielt.
Schmp.: 220~223°C (Schmp. in der Literatur: 222~225°C)
1H-NMR (CDCl3, ppm): δ 5,06 (dd, 1H, 13-H), 4,92 (d, 1H, 1''-H), 4,44 (d, 1H, 1'-H), 4,02 (dq, 1H, 5''-H), 3,78 (dd, 1H, 3-H), 3,77 (d, 1H, 11-H), 3,67 (d, 1H, 5-H), 3,57 (ddq, 1H, 5'-H), 3,33 (s, 3H, 3''-OCH3), 3,20 (dd, 1H, 2'-H), 3,07~2,95 (m, 2H, 10-H und 4''-H), 3,03 (s, 3H, 6-OCH3), 2,87 (dq, 1H, 2-H), 2,58 (ddq, 1H, 8-H), 2,40 (ddd, 1H, 3'-H), 2,37 (d, 1H, 2''-Heq), 2,28 (s, 6H, 3'-N(CH3)2), 2,00~1,80 (m, 3H, 4-H und 7-H2), 1,41 (s, 3H, 18-H), 1,13 (s, 3H, 6-CH3), 0,85 (t, 3H, 14-CH3). - Beispiel 4: Herstellung von Clarithromycin unter Verwendung von Hexabutyldizinn als ein Reduktionsmittel
- 3,82 g (5 mMol) an im Beispiel 1 erhaltenem 6-O-Methylerythromycin A-N-oxid wurden in 50 ml Tetrahydrofuran suspendiert. 5,6 ml (2,2 mMol) Hexabutyldizinn wurden hinzugesetzt, und die Mischung wurde 24 Stunden am Rückfluss gehalten. Das Lösungsmittel wurde unter einem reduzierten Druck konzentriert, und Isopropylether und Hexan wurden dem Rückstand hinzugesetzt. Die gebildeten Kristalle wurden filtriert und mit Hexan gewaschen, wodurch man 3,55 g Clarithromycin als ein weißes Pulver in einer Ausbeute von 95% erhielt.
- Beispiel 5: Herstellung von Clarithromycin unter Verwendung einer Nickel-Aluminium-Legierung als ein Reduktionsmittel
- 3,82 g (5 mMol) des in Beispiel 1 erhaltenen 6-O-Methylerythromycin A-N-oxid wurden in einer Mischung aus 100 ml Methanol und 50 ml 1 N Kaliumhydroxid suspendiert. 5 g einer Nickel-Aluminium-Legierung wurden während eines Zeitraums von 30 Minuten hinzugesetzt, während die Temperatur bei 35 bis 40°C gehalten wurde und 3 Stunden lang gerührt wurde. Die resultierende Mischung wurde mit Methanol verdünnt und durch ein Celite-Kissen filtriert, um Feststoffe zu entfernen. Das Filtrat wurde unter einem reduzierten Druck konzentriert, um Methanol zu entfernen, und der Rückstand wurde mit Wasser verdünnt. Gebildete Kristalle wurden filtriert, mit Wasser gewaschen und dann getrocknet, wodurch man 3,44 g Clarithromycin als ein weißes Pulver in einer Ausbeute von 92% erhielt.
- Beispiel 6: Herstellung von Clarithromycin durch Hydrierung unter Verwendung eines Raney-Nickel-Katalysators
- 3,82 g (5 mMol) des in Beispiel 1 erhaltenen 6-O-Methylerythromycin A-N-oxid wurden in 100 ml Ethanol suspendiert. 0,25 g W4 Raney-Nickel wurden hinzugesetzt, und es wurde bei einer Temperatur im Bereich von 40 bis 50°C unter einer Wasserstoffatmosphäre 3 Stunden lang gerührt. Die resultierende Mischung wurde durch ein Celite-Kissen bei 50°C oder mehr filtriert, um den verbrauchten Katalysator zu entfernen. Das Filtrat wurde unter einem reduzierten Druck auf 30 ml konzentriert und auf Raumtemperatur gekühlt. Gebildete Kristalle wurden filtriert und getrocknet, wodurch man 3,44 g Clarithromycin als ein weißes Pulver in einer Ausbeute von 92% erhielt.
- Beispiel 7: Herstellung von Clarithromycin unter Verwendung von Natriumbisulfit als ein Reduktionsmittel
- 3,82 g (5 mMol) des in Beispiel 1 erhaltenen 6-O-Methylerythromycin A-N-oxid wurden in einer Mischung aus 15 ml Ethanol und 15 ml Wasser suspendiert. 1,05 g (10 mMol) Natriumbisulfit wurden hinzugesetzt, und es wurde bei Raumtemperatur 1 Stunde lang gerührt. Die resultierende Mischung wurde konzentriert, Wasser wurde hinzugesetzt, und dann wurde mit 10%igem Natriumcarbonat auf einen pH-Wert von 10 eingestellt. Die resultierende Mischung wurde dreimal mit Ethylacetat extrahiert, die organischen Schichten wurden vereinigt, mit Wasser und Salzlösung gewaschen und dann über wasserfreiem Magnesiumsulfat getrocknet. Das Lösungsmittel wurde unter einem reduzierten Druck entfernt, und der erhaltene Rückstand wurde in 20 ml Ethanol gelöst. Nach dem Hinzusetzen von 0,55 ml Ameisensäure und 0,8 ml 35%igem Formaldehyd wurde die Mischung 2 Stunden am Rückfluss gehalten. Die Mischung wurde auf die Hälfte des Volumens konzentriert, und 20 ml Wasser wurden hinzugesetzt, und dann wurde die resultierende Mischung mit 10%igem Natriumcarbonat auf einen pH-Wert von 11 eingestellt, wodurch man 3,44 g Clarithromycin als ein weißes Pulver in einer Ausbeute von 92% erhielt.
- Beispiel 8: Herstellung von Clarithromycin
- Die Prozedur vom Beispiel 1 wurde wiederholt unter Verwendung von 75,0 g (0,1 Mol) des im Referenzbeispiel erhaltenen Erythromycin A-N-oxid, wodurch man 54,0 g an rohem 6-O-Methylerythromycin A-N-oxid mit einer Reinheit von etwa 54% erhielt.
- 54,0 g des so erhaltenen rohen 6-O-Methylerythromycin A-N-oxid wurden in 400 ml Dichlormethan gelöst, und 32,6 g (144 mMol) Zinn(II)-chlorid-dihydrat wurden hinzugesetzt, und dann wurde bei Raumtemperatur 1,5 Stunden lang gerührt. 300 ml 10%iges Natriumcarbonat wurden der resultierenden Mischung hinzugesetzt, und Dichlormethan wurde unter einem reduzierten Druck entfernt. Der Rückstand wurde der Reihe nach mit 400 ml und 200 ml großen Portionen an Ethylacetat extrahiert, die organischen Schichten wurden vereinigt, zweimal mit 300 ml Wasser gewaschen, und dann wurden sie über wasserfreiem Magnesiumsulfat getrocknet. Das Lösungsmittel wurde unter einem reduzierten Druck entfernt, 100 ml Ethanol wurden dem Rückstand hinzugesetzt, es wurde 30 Minuten lang refluxiert und 3 Stunden lang bei Raumtemperatur gerührt. Die gebildeten Feststoffe wurden gesammelt und getrocknet, wodurch man 24,0 g Clarithromycin als einen weißen Kristall in einer Ausbeute von 32% erhielt.
- Beispiel 9: Herstellung von Clarithromycin
- Die Prozedur von Beispiel 1 wurde unter Verwendung von 75,0 g (0,1 Mol) des im Referenzbeispiel erhaltenen Erythromycin A-N-oxid wiederholt, wodurch man 33,2 g an 6-O-Methylerythromycin A-N-oxid als ein kristallines Pulver mit einer Reinheit von etwa 85% erhielt.
- 33,2 g des zu 85% reinen 6-O-Methylerythromycin A-N-oxid wurden in einer Mischung aus 70 ml Ethanol und 70 ml Wasser gelöst, 6,52 g (62,6 mMol) Natriumbisulfit wurden hinzugesetzt, und dann wurde bei Raumtemperatur 1 Stunde lang gerührt. Die resultierende Mischung wurde auf ein kleines Volumen konzentriert, Wasser wurde dem Konzentrat hinzugesetzt, und dann wurde mit 10%igem Natriumcarbonat auf einen pH-Wert von 10 eingestellt. Die resultierende Mischung wurde dreimal mit Ethylacetat extrahiert, die organischen Schichten wurden vereinigt, mit Wasser und Salzlösung gewaschen und dann über wasserfreiem Magnesiumsulfat getrocknet. Das Lösungsmittel wurde unter einem reduzierten Druck entfernt, und der Rückstand wurde in 80 ml Ethanol gelöst. 5,0 ml Ameisensäure und 7,1 ml 35%iger Formaldehyd wurden der Ethanollösung hinzugesetzt, und dann wurde 2 Stunden lang refluxiert. 200 ml Wasser wurden der resultierenden Mischung hinzugesetzt. Es wurde mit konzentriertem wässrigem Ammoniak auf einem pH-Wert von 11 eingestellt, und dann wurde auf Raumtemperatur gekühlt. Die gebildeten Feststoffe wurden filtriert und getrocknet, wodurch man 22,5 g Clarithromycin als ein weißes Pulver in einer Ausbeute von 30% erhielt.
Claims (10)
- Verfahren zur Herstellung von Clarithromycin der Formel (I), umfassend die Schritte: (a) Umsetzen von Erythromycin A-N-oxid der Formel (II) mit einem Methylierungsmittel unter Erhalt von 6-O-Methylerythromycin A-N-oxid der Formel (III); und (b) Behandeln des in Schritt (a) erhaltenen 6-O-Methylerythromycin A-N-oxids mit einem Reduktionsmittel unter Erhalt von Clarithromycin:


- Verfahren nach Anspruch 1, wobei der Schritt (a) in einem organischen Lösungsmittel bei einer Temperatur im Bereich von –15 bis 40°C in Gegenwart einer Base durchgeführt wird.
- Verfahren nach Anspruch 1, wobei das Methylierungsmittel gewählt ist aus der Gruppe bestehend aus Methylbromid, Methyliodid, Dimethylsulfat, Methyl-p-toluolsulfonat, Methylmethansulfonat und einer Mischung davon.
- Verfahren nach Anspruch 1, wobei die Menge an Methylierungsmittel im Bereich von 1 bis 3 Äquivalenten auf Basis der Menge von Erythromycin A-N-oxid liegt.
- Verfahren nach Anspruch 2, wobei das organische Lösungsmittel N,N-Dimethylformamid, N,N-Dimethylacetamid, N-Methyl-2-pyrrolidon, Dimethylsulfoxid, Sulfolan, N,N,N',N',N'',N''-Hexamethylphosphoramid, Tetrahydrofuran, 1,4-Dioxan, 1,2-Dimethoxyethan, 1,2-Diethoxyethan, 2-Methoxyethylether, 2-Ethoxyethylether, 1,2-Bis(2-methoxyethoxy)ethan, Tetraethylenglykoldimethylether, Aceton, Acetonitril oder eine Mischung davon ist.
- Verfahren nach Anspruch 2, wobei die Base gewählt ist aus der Gruppe bestehend aus Alkalimetallhydriden, -hydroxiden, -alkoxiden und einer Mischung davon.
- Verfahren nach Anspruch 2, wobei die Menge der Base im Bereich von 0,9 bis 2 Äquivalenten auf Basis der Menge von Erythromycin A-N-oxid liegt.
- Verfahren nach Anspruch 1, wobei das Reduktionsmittel gewählt ist aus der Gruppe bestehend aus Wasserstoff in Gegenwart eines Hydrierungskatalysators; einer Nickel-Aluminium-Legierung (Ni-Al-Legierung), kombiniert mit Kaliumhydroxid; metallischem Zink in Gegenwart von Ameisensäure oder Essigsäure; Natriumhydrogentellurid (NaTeH); Samariumiodid (SmI2); Zinn(II)-chlorid (SnCl2); Hexabutyldizinn (Bu3SnSnBu3); Cyclohexen und Osmiumtetroxid (OsO4); und Eisen(II)-sulfat; NaHSO3; Na2SO3; Na2S2O3; Na2S2O4; Na2S2O5; Na2S2O6; KHSO3; K2S2O3; K2S2O5; und einer Mischung davon.
- Verfahren nach Anspruch 1, wobei das Reduktionsmittel Zinn(II)-chlorid (SnCl2); Hexabutyldizinn (Bu3SnSnBu3), eine Nickel-Aliuminium-Legierung, kombiniert mit Kaliumhydroxid oder Wasserstoff in Gegenwart eines Raney-Nickel- oder Platinoxid-(PtO2)-Katalysators ist.
- Verwendung von 6-O-Methylerythromycin A-N-oxid der Formel (III):in einem Verfahren zur Herstellung von Clarithromycin.

Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1019990052371A KR100336447B1 (ko) | 1999-11-24 | 1999-11-24 | 클라리스로마이신의 개선된 제조방법 |
| KR9952371 | 1999-11-24 | ||
| PCT/KR2000/001349 WO2001038340A1 (en) | 1999-11-24 | 2000-11-23 | Method of preparing clarithromycin |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60031154D1 DE60031154D1 (de) | 2006-11-16 |
| DE60031154T2 true DE60031154T2 (de) | 2007-01-11 |
Family
ID=19621549
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60031154T Expired - Fee Related DE60031154T2 (de) | 1999-11-24 | 2000-11-23 | Verfahren zur herstellung von clarithromycin |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US6809188B1 (de) |
| EP (1) | EP1232171B1 (de) |
| JP (1) | JP4040877B2 (de) |
| KR (1) | KR100336447B1 (de) |
| CN (1) | CN1399642A (de) |
| AT (1) | ATE341559T1 (de) |
| AU (1) | AU755838C (de) |
| BR (1) | BR0015780A (de) |
| CA (1) | CA2391145A1 (de) |
| DE (1) | DE60031154T2 (de) |
| ES (1) | ES2272342T3 (de) |
| RU (1) | RU2225413C1 (de) |
| TR (1) | TR200201382T2 (de) |
| WO (1) | WO2001038340A1 (de) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100336447B1 (ko) * | 1999-11-24 | 2002-05-15 | 민경윤 | 클라리스로마이신의 개선된 제조방법 |
| EP1313486A1 (de) * | 2000-02-29 | 2003-05-28 | Teva Pharmaceutical Industries Ltd. | Verfahren zur herstellung des clarithromycins und eines clarithromycin-zwischenprodukt; clarithromycin wesentlich frei von oxime; und pharmazeutische zusammensetzung die sie enthalten |
| CA2529817C (en) | 2003-03-10 | 2013-02-12 | Optimer Pharmaceuticals, Inc. | Novel antibacterial agents |
| AU2004251712A1 (en) * | 2003-06-23 | 2005-01-06 | Auspex Pharmaceuticals, Inc. | Novel therapeautic agents for the treatment of cancer, metabolic diseases and skin disorders |
| WO2007143507A2 (en) * | 2006-06-05 | 2007-12-13 | Auspex Pharmaceuticals, Inc. | Preparation and utility of substituted erythromycin analogs |
| JP5698979B2 (ja) * | 2007-10-25 | 2015-04-08 | センプラ ファーマシューティカルズ,インコーポレイテッド | マクロライド系抗菌剤の調製プロセス |
| JP5602748B2 (ja) | 2008-10-24 | 2014-10-08 | センプラ ファーマシューティカルズ,インコーポレイテッド | トリアゾール含有マクロライドを用いた生体防御 |
| US9937194B1 (en) | 2009-06-12 | 2018-04-10 | Cempra Pharmaceuticals, Inc. | Compounds and methods for treating inflammatory diseases |
| ES2608285T3 (es) | 2009-09-10 | 2017-04-07 | Cempra Pharmaceuticals, Inc. | Procedimientos para el tratamiento de paludismo, tuberculosis y enfermedades por MAC |
| DK2550286T3 (en) | 2010-03-22 | 2016-02-29 | Cempra Pharmaceuticals Inc | CRYSTALLINE FORMS OF A MACROLID AND APPLICATIONS THEREOF |
| BR112012029586A2 (pt) | 2010-05-20 | 2016-08-02 | Cempra Pharmaceuticals Inc | processos para preparar macrolídeos e cetolídeos e intermediários dos mesmos |
| JP6042334B2 (ja) | 2010-09-10 | 2016-12-14 | センプラ ファーマシューティカルズ,インコーポレイテッド | 疾患治療のための水素結合形成フルオロケトライド |
| US10188674B2 (en) | 2012-03-27 | 2019-01-29 | Cempra Pharmaceuticals, Inc. | Parenteral formulations for administering macrolide antibiotics |
| JP6426696B2 (ja) | 2013-03-14 | 2018-11-21 | センプラ ファーマシューティカルズ,インコーポレイテッド | 呼吸器疾患の治療のための方法および製剤 |
| US9751908B2 (en) | 2013-03-15 | 2017-09-05 | Cempra Pharmaceuticals, Inc. | Convergent processes for preparing macrolide antibacterial agents |
| CN103172686B (zh) * | 2013-04-08 | 2015-06-24 | 山东大学 | 克拉霉素-n-氧化物的制备方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5872229A (en) * | 1995-11-21 | 1999-02-16 | Abbott Laboratories | Process for 6-O-alkylation of erythromycin derivatives |
| US5858986A (en) * | 1996-07-29 | 1999-01-12 | Abbott Laboratories | Crystal form I of clarithromycin |
| US5864023A (en) * | 1997-02-13 | 1999-01-26 | Abbott Laboratories | 3'-N'oxide, 3'-n-dimethylamine, 9-oxime erythromycin a derivatives |
| IL135792A0 (en) * | 1997-12-01 | 2001-05-20 | Abbott Lab | 6-o-alkyl derivatives of erythronolide b |
| US5892008A (en) * | 1997-12-16 | 1999-04-06 | Abbott Laboratories | Process for the preparation of 6-O-methyl erythromycin a using 9-hydroxy erythromycin derivatives |
| KR100336447B1 (ko) * | 1999-11-24 | 2002-05-15 | 민경윤 | 클라리스로마이신의 개선된 제조방법 |
| KR20000037126A (ko) * | 2000-04-08 | 2000-07-05 | 김용규 | 6-메틸 에리스로마이신 a의 제조방법 |
-
1999
- 1999-11-24 KR KR1019990052371A patent/KR100336447B1/ko not_active IP Right Cessation
-
2000
- 2000-11-23 ES ES00981874T patent/ES2272342T3/es not_active Expired - Lifetime
- 2000-11-23 CA CA002391145A patent/CA2391145A1/en not_active Abandoned
- 2000-11-23 EP EP00981874A patent/EP1232171B1/de not_active Expired - Lifetime
- 2000-11-23 RU RU2002116698/04A patent/RU2225413C1/ru not_active IP Right Cessation
- 2000-11-23 WO PCT/KR2000/001349 patent/WO2001038340A1/en active IP Right Grant
- 2000-11-23 US US10/129,419 patent/US6809188B1/en not_active Expired - Fee Related
- 2000-11-23 TR TR2002/01382T patent/TR200201382T2/xx unknown
- 2000-11-23 DE DE60031154T patent/DE60031154T2/de not_active Expired - Fee Related
- 2000-11-23 AT AT00981874T patent/ATE341559T1/de not_active IP Right Cessation
- 2000-11-23 JP JP2001540103A patent/JP4040877B2/ja not_active Expired - Fee Related
- 2000-11-23 AU AU18983/01A patent/AU755838C/en not_active Ceased
- 2000-11-23 BR BR0015780-5A patent/BR0015780A/pt not_active IP Right Cessation
- 2000-11-23 CN CN00816126A patent/CN1399642A/zh active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| BR0015780A (pt) | 2002-07-23 |
| US6809188B1 (en) | 2004-10-26 |
| KR20010047939A (ko) | 2001-06-15 |
| RU2225413C1 (ru) | 2004-03-10 |
| DE60031154D1 (de) | 2006-11-16 |
| AU755838C (en) | 2004-07-08 |
| CA2391145A1 (en) | 2001-05-31 |
| JP2003514915A (ja) | 2003-04-22 |
| ATE341559T1 (de) | 2006-10-15 |
| EP1232171A4 (de) | 2002-10-31 |
| KR100336447B1 (ko) | 2002-05-15 |
| TR200201382T2 (tr) | 2002-10-21 |
| EP1232171B1 (de) | 2006-10-04 |
| CN1399642A (zh) | 2003-02-26 |
| AU755838B2 (en) | 2002-12-19 |
| AU1898301A (en) | 2001-06-04 |
| ES2272342T3 (es) | 2007-05-01 |
| EP1232171A1 (de) | 2002-08-21 |
| WO2001038340A1 (en) | 2001-05-31 |
| JP4040877B2 (ja) | 2008-01-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60031154T2 (de) | Verfahren zur herstellung von clarithromycin | |
| DE60122791T2 (de) | Verfahren zur Herstellung von Clarythromycin mit nicht pharmazeutischer Qualität | |
| DE69818977T2 (de) | 3'-n-oxid,3'-n-dimethylamin,9-oxime erythromycin a derivate | |
| DE69627381T2 (de) | Vefahren zur herstellung von erythrmycinderivate | |
| DE3012533C2 (de) | ||
| DE69913413T2 (de) | 2-Fluor 3-de (2,6-dideoxy 3-C-methyl-3-0-methyl-alpha-L-ribohexopyranosyl oxy) 6-1-methyl 3-oxo Erythromycin, ihrer Verfahren zur Herstellung und ihrer Verwendung zur Synthese von Arzneimitteln | |
| DE69814422T2 (de) | 9-hydrazon- und 9-azynerythromycinderivate und verfahren zu deren herstellung | |
| DD216017A5 (de) | Verfahren zur herstellung von 4"-epi-9-desoxo-9a-methyl-9a-aza-9a-homoerythromycin a | |
| US5869629A (en) | Synthesis of 9-deoxo-9a-aza-11,12-deoxy-9a-methyl-9a-homoerythromycin A 11,12 Hydrogenorthoborate dihydrate and a process for the preparation of azitromicin dihydrate | |
| DE69618607T2 (de) | Derivate von 5-0-desosaminyl-6-0-methyl erythronolid a, ihre verfahren zur herstellung und verwendung zur herstellung von arzneimitteln | |
| JPS6360032B2 (de) | ||
| DE2740950A1 (de) | Verfahren zur herstellung von flavonen | |
| DE2366288C3 (de) | Verfahren zur Herstellung von 3' -Desoxykanamycin B und 3' -Desoxyneamin | |
| DE60205377T2 (de) | Verfahren zur arylierung von funktionalisierten o-allyl erythromycin derivaten | |
| DE2756057C2 (de) | ||
| WO1990012796A1 (de) | 8β-SUBSTITUIERTE ERGOLINE, VERFAHREN ZU IHRER HERSTELLUNG UND IHRE VERWENDUNG | |
| EP1444244B1 (de) | Verfahren zur herstellung von desclarithromycin, sowie zwischenprodukte | |
| DE2535755C2 (de) | Verfahren zur Herstellung von Streptamin | |
| DE1804331A1 (de) | Derivate von (25R)-3ss-Amino-5alpha-22alpha-spirostan und Verfahren zu deren Herstellung | |
| KR20010100197A (ko) | 에리스로마이신 에이 9-오-트로필옥심 유도체를 이용한클라리스로마이신의 제조방법 | |
| DE19627067A1 (de) | Verfahren zur Herstellung von N-(2-Aminoacylamino-2-desoxy-hexosyl)-amiden | |
| EP0548590A2 (de) | N4-substituierte 1-Alkoxy-2-acylamino-4-aminobenzole, Verfahren zu ihrer Herstellung und ihre Verwendung | |
| DE2742955A1 (de) | 1-subst.-glycopyranoside mit insulinaehnlicher aktivitaet und verfahren zu ihrer herstellung | |
| DE2535754A1 (de) | 1,3-didesoxy-1,3- eckige klammer auf n,n'-(1',2',3',4'-tetrahydro-1',4'-dioxo)- phthalazino eckige klammer zu -inosit-verbindungen | |
| CS241099B2 (cs) | Způsob přípravy 4“-epi-9-deoxo-9a-methyl-9a-aza- -9a-bomoerythromycinu A |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |