CN1255419C - 乙烯低聚生产直链α烯烃用配位体及其催化剂体系 - Google Patents
乙烯低聚生产直链α烯烃用配位体及其催化剂体系 Download PDFInfo
- Publication number
- CN1255419C CN1255419C CNB028162595A CN02816259A CN1255419C CN 1255419 C CN1255419 C CN 1255419C CN B028162595 A CNB028162595 A CN B028162595A CN 02816259 A CN02816259 A CN 02816259A CN 1255419 C CN1255419 C CN 1255419C
- Authority
- CN
- China
- Prior art keywords
- hydrogen
- optional
- replaces
- independent separately
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/02—Iron compounds
- C07F15/025—Iron compounds without a metal-carbon linkage
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/06—Cobalt compounds
- C07F15/065—Cobalt compounds without a metal-carbon linkage
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F17/00—Metallocenes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Abstract
分子式(I)的混合双亚胺吡啶配位体,其中Z1与Z2不同,为任选取代的芳基;以及Z2为任选取代的杂烃基部分,或与金属组合的任选取代的π健共配位到金属上的芳基;分子式(I);含有分子式(I)的混合双亚胺吡啶MXn配合物,其中M为选自Fe或Co的金属,n为2或3,以及X为卤化物、任选取代的烃基、烷氧化物、酰胺或氢化物;含有分子式(I)配位体的混合[双亚胺吡啶MYp·Ln+][NC-]q配合物,其中Y为一种可插入烯烃的配位体;M为选自Fe或Co的金属,NC-为非配位的阴离子,以及p+q为2或3,以便与所述金属原子的形式氧化相适合;L为中性路易斯施主分子和n为0、1或2;以及用所述的配合物由乙烯生产α烯烃的方法。
Description
本发明涉及配位体、各种催化剂前体以及由这些配位体制得的用于乙烯低聚高产率和高选择性生产直链α烯烃的催化剂体系,以及涉及一种生产所述直链α烯烃的方法。
生产高级直链α烯烃的各种方法是已知的(例如D.Vogt,乙烯在有机金属化合物的应用均相催化作用中低聚生产高级直链α烯烃,B.Cornils,W.A.Herrmann编辑,第1卷,第2.3.1.3章,第245页,VCH 1996)。这些商业方法得到泊松或舒茨-弗洛里低聚产物分布。
为了得到泊松分布,在低聚过程中不必发生链中止。
但是相反,在舒茨-弗洛里方法中,链中止的确发生,并与链长度无关。壳牌高级烯烃法(SHOP)的镍催化的乙烯低聚步骤是舒茨-弗洛里方法的一个典型实例。
在舒茨-弗洛里方法中,通常制得各种低聚物,其中每种烯烃的分数可通过基于K-因子的计算来决定。K-因子为烯烃产物相对比例的标志,它为log[Cnmol%]对n作图的斜率计算的[Cn+2]/[Cn]摩尔比,其中n为特定烯烃产物中的碳原子数。对于每一n来说,K-因子定义是相同的。通过改变配位体和调节反应参数,可将K-因子调节到或高或低的数值。用这一方法,可这样操作所述的方法,以便以最佳的经济性生产各种产物。
因为C6-C18馏分的需要大大高于C>20馏分,所以希望使这些方法适合于生产较低碳原子数的烯烃。但是,较高碳原子数烯烃的生成是不可避免的,如果不经进一步加工,这些产物的生成对所述方法的效益是不利的。为了降低较高碳原子数烯烃以及低价值C4馏分的负面影响,已开发了另外的技术来再加工这些物流并将它们转化成更有价值的化学品,例如C6-C18内烯烃,正如在壳牌高级烯烃法中实施的。
但是,从投资和操作观点看,这一技术是昂贵的,因此增加了额外的费用。所以,为了使较高碳原子数烯烃的产量减少,即不大于固有与舒茨-弗洛里K-因子有关的数值,人们已作了相当大的努力。
WO-A-99/12981公开了用于1-烯烃特别是乙烯聚合的催化剂体系,所述的催化剂体系含有分子式(B)骨架单元的含氮过渡金属化合物,
式中M为Fe[II]、Fe[III]、Ru[II]、Ru[III]或Ru[IV];x为与过渡金属M共价键联或离子键联的原子或基团;T为过渡金属M的氧化态,而b为原子或基团X的化合价;R1、R2、R3、R4和R6各自独立选自氢、卤素、烃基、取代的烃基、杂原子烃基或取代的杂原子烃基,R5和R7各自独立选自氢、卤素、烃基、取代的烃基、杂原子烃基或取代的杂原子烃基。
WO-A-00/50470公开了这样的催化剂组合物,据说它们可用于烯烃的聚合或低聚。
所述的催化剂组合物为被单配位基、二配位基、三配位基或四配位基配位体配位的金属配合物,其中至少一种配位体的施主原子为被1-吡咯基或取代的1-吡咯基取代的氮原子;其中配位体的其余施主原子选自C、N、P、As、O、S和Se;以及其中所述金属配合物中的所述金属选自Se、Ta、Ti、Zr、Hf、V、Nb、Cr、Mo、W、Mn、Re、Fe、Ru、Os、Co、Rn、Ir、Ni、Cu、Pd、Pt、Al和Ga。
据说配位体h1为可用于WO-A-00/50470优选催化剂组合物的许多中性配位基配位体中的一个:
特别是,具体公开了许多对称双吡咯基亚胺配位体,即h16-h17、h19-h25、h28、h30和h32。此外,h29、h31和h33为混合双吡咯基亚胺配位体。
WO-A-00/50470的实施例59、60和62说明在含有对称双吡咯基亚胺配位体h16的含铁催化剂组合物存在下乙烯的聚合。
同样,实施例74涉及在含有对称双吡咯基亚胺配位体h17的含铁催化剂组合物存在下乙烯的聚合。
在WO-A-00/50470中,在含铁催化剂中使用某些上述配位体的另一些乙烯聚合的实施例包括其中的实施例99-125、128、131、133和135。
正如在实施例4中举例说明的,WO-A-00/50470的配位体h16的较低同系物即配位体(
11)得到这样一种含铁的双吡咯基亚胺催化剂组合物,它有很低的乙烯转化率,即使有的话。
现令人吃惊地发现了一类新型的催化剂,它们在烯烃特别是乙烯低聚中有高活性,并生产C6-C30范围的直链α烯烃,所述的直链α烯烃有高的纯度。
此外,本发明的一些催化剂还得到舒茨-弗洛里产物分布。
本发明提供一种分子式(I)的混合双亚胺吡啶配位体,其中R1-R5各自独立为氢、任选取代的烃基、惰性官能基,或任何两个彼此相邻的R1-R3可形成一个环;与Z2不同的Z1为任选取代的芳基;以及Z2为任选取代的杂原子烃基部分,或任选取代的与金属组合的芳基,所述的任选取代的芳基为与金属π键共价配位的。

本发明还提供一种含有分子式(I)的混合双亚胺吡啶配位体的混合双亚胺吡啶MXn配合物,其中M为选自Fe或Co的金属原子,n为2或3,以及X为卤化物、任选取代的烃基、烷氧化物、酰胺或氢化物。
在另一方面,本发明提供这样一种生产α烯烃的方法,所述的方法包括本发明的一种或多种MXn配合物在与乙烯和第二种化合物-100至+300℃下接触,后者能将任选取代的烃基或氢化物基团转移到选自Fe或Co的金属原子M上,以及也能从所述的金属原子上取出X-基。
在另一方面,本发明提供这样一种生产α烯烃的方法,所述的方法包括本发明的一种或多种MXn配合物与乙烯和第二种化合物和第三种化合物在-100至+300℃下接触,所述的第一种化合物能将任选取代的烃基或氢化物基团转移到选自Fe或Co的金属原子M上,而第三种化合物能从所述的金属原子上取出X-基。
本发明还提供一种含有分子式(I)配位体的混合[双亚胺吡啶MYp·Ln +][NC-]q配合物,其中Y为可插入烯烃的配位体;M为选自Fe或Co的金属原子;NC-为未配位的阴离子和p+q为2或3,以便与所述金属原子的形式氧化相适合;L为中性路易斯施主分子和n为0、1或2。
本发明还提供一种生产α烯烃的方法,所述的方法包括本发明的一种或多种混合[双亚胺吡啶MYp·Ln +][NC-]q配合物与乙烯在-100至+300℃下接触。
在本发明中,某些术语使用如下:
术语“混合的”表示亚胺部分Z1和Z2彼此不同。
术语“芳基”指芳族环状烃类单基。其实例包括苯基、萘基、蒽基、苯并蒽基等及其取代的衍生物。
术语“与金属的组合的任选取代的芳基,所述的任选取代的芳基为与金属π共配位的任选取代的芳基”包括金属茂部分和夹层和金属芳烃配合物。因此,熟悉本专业的技术人员应当理解,任选地,金属可另外π键共配位到另外的任选取代的芳基上,后者可与Z2中的任选取代的芳基不同,Z2直接键联到亚胺氮原子上和/或共配位到本专业中共知的其他配位体上。还应当理解,在Z2中任选取代的芳基可含有环中的一个或多个杂原子,即所述的任选取代的芳基为任选取代的杂原子芳基,Z2直接键联到亚胺氮原子上和也π键共配位到金属上。同样,金属还π键共配位的另外的任选取代的芳基可在环中含有一个或多个杂原子。所述的金属原子可按常规为铁、钴、镍、铬、钛和钒。这样部分的实例包括铁茂、钴茂、镍茂、铬茂、钛茂、钒茂、双π键芳烃钒配合物、单π键芳烃铬三羰基配合物和类似的杂原子芳烃金属配合物,即双或单π键噻吩或吡咯铁或铬配合物。
术语“杂原子烃基”指另外还含有一个或多个杂原子的烃基。所述的杂原子优选键联到杂原子烃基中的至少两个碳上。
所述的杂原子烃基可为任选取代的芳族杂环部分;任选取代的多芳族杂环部分;任选取代的脂族杂环部分;或任选取代的脂族杂烃基部分。
杂原子烃基的实例包括1-吡咯基、2-吡咯基、3-吡咯基、呋喃基、噻吩基、茚基、嘧唑基、三唑基、噁唑基、异噁唑基、咔唑基、噻唑基、苯并噻唑基、噻二唑基、嘧啶基、吡啶基、哒嗪基等及其取代的衍生物。
烃基:一种只含碳和氢的基团。除非另加说明,碳原子的数目优选为1-30。
在本发明中,短语“任选取代的烃基”用来描述任选含有一个或多个“惰性的”的含杂原子官能基的烃基。所谓的“惰性的”指不以任何实际程度干扰低聚过程的官能基。这些惰性基的非限制性实例为氟化物、氯化物、硅烷、锡烷、有足够空间屏蔽的醚类和胺类;对于熟悉本专业的技术人员来说,这些都是大家熟悉的。
惰性官能基:除了任选取代的烃基外在工艺条件下为惰性的基团。所谓“惰性”指不以任何实际程度干扰低聚过程的官能基。惰性官能基的实例包括卤化物、醚类和胺类,特别是叔胺。
伯碳原子基团:-CH2-R,其中R可为氢、任选取代的烃基、惰性官能基。伯碳原子基团的实例包括-CH3、-C2H5、-CH2Cl、-CH2OCH3、-CH2N(C2H5)2、-CH2Ph。
仲碳原子基团:-CH-R2,其中R可为任选取代的烃基、惰性官能基。仲碳原子基团的实例包括-CH(CH3)2、-CHCl2、CHPh2、-CH=CH2、环己基。
叔碳原子基团:-C-R3,其中R可为任选取代的烃基、惰性官能基。叔碳原子基团的实例包括-C(CH3)3、-CCl3、C≡CPh、l-金刚烷基、-C(CH3)2(OCH3)
所谓“可插入烯烃的配位体”指配位到乙烯分子可插入的金属离子的配位体,以便引发或增长低聚反应。在本发明的混合的[芳基-,(杂)芳基-亚胺吡啶MYp·Ln +][NC-]q配合物中,Y可为氢化物、烷基或任何可插入烯烃的其他阴离子配位体。
所谓“未配位的阴离子”指基本上未配位到金属原子M上的阴离子。
可适合应用的未配位阴离子(NC-)包括大型阴离子,例如四[3,5-双(三氟甲基)苯基]硼酸根(BAF-)、(C6F5)4B-和包括R3AlX-、R2AlClX-、RAlCl2X-和“RAlOX-”在内的烷基铝化合物的阴离子,其中R为氢、任选取代的烃基或惰性官能基,而X为卤化物、烷氧化合物或氧。
熟悉本专业的技术人员应当理解,在上述边界条件内,可很容易选择取代基R1-R15,以便优化催化剂体系的性能及其经济应用。
(II)
本发明提供分子式(II)的混合双亚胺吡啶配位体,其中A1-A6各自独立为碳、氮、氧或硫;以及可任选不含下式原子基团

,以致A1直接键联到A5上;以及R1-R12、R14-R15以及R13(如果存在)各自独立为氢、任选取代的烃基和惰性的官能基,或R1-R15中任何两个彼此相邻的基团可形成环;条件是当A1-A5以及A6(如果存在)都为碳,所述的原子构成环戊二烯基或π键共配位金属的芳基部分。
在本发明一优选的实施方案中,在分子式(II)中,R1-R3、R7-R9、R12R14以及R13(如果存在)各自独立为氢、任选取代的烃基、惰性官能基,或R1-R3、R7-R9、R12-R14中任何两个彼此相邻的基团可形成环;以及
a)R6为惰性官能基或任选取代的烃基,以及R10、R11和R15各自独立为氢或卤化物;或
b)R11为惰性官能基或任选取代的烃基,以及R6、R10和R15各自独立为氢或卤化物;或
c)R6和R10各自独立为惰性官能基或伯或仲碳原子基团,条件是R6和R10不都为仲碳原子基团以及R11和R15各自独立为氢或卤化物;或
d)R11和R15各自独立为惰性官能基或伯或仲碳原子基团,条件是R11和R15不都为仲碳原子基团以及R6和R10各自独立为氢或卤化物;或
e)R6与R7形成环,R10为伯碳原子基团、惰性官能基或氢,R11和R15各自独立为氢或卤化物;或
f)R11与R12形成环,R15为伯碳原子基团、惰性官能基或氢,R6和R10各自独立为氢或卤化物;或
g)R6和R10与R7和R9各自形成环,R11和R15各自独立为氢或卤化物;或
h)R11和R15与R12和R14各自形成环,R6和R10各自独立为氢或卤化物。
取代基R1-R15(如果存在)可各自独立相连并形成环状结构物。这样的结构物的实例例如包括R6与R7相连形成基本的萘基骨架或四氢萘基单元。
此外,任何熟悉均相催化作用基本原理的技术人员都很容易理解,可选择各种不同的取代基R1-R5、R7-R9以及R12-R14(如果存在),以便提高催化剂前体和催化剂体系的其他所需性能,例如在非极性溶剂中的溶解度或扩大在合成中适合原料的范围。
本发明优选的实施方案为(I)的配位体及其衍生物,其中以下的R和Z基团为:
R1-R3为氢;和/或
Z1为任选取代的苯基和Z2为任选取代的二茂铁基或任选取代的1-吡咯基。
本发明优选的实施方案为(II)的配位体及其衍生物,其中以下的R基团为:
R1-R3为氢;和/或
R4和R5为甲基、氢或苯基、优选甲基;和/或
不存在以及A1-A5为碳原子,从而构成二茂铁部分的环戊二烯基部分;或
A3为氮原子,

不存在和A1、A2、A4、A5为碳原子,从而构成吡咯基环;和/或
邻位取代基组合,其中R6为甲基、乙基、异丙基、苯基、叔丁基,或与R7相连形成萘基骨架;R10为氢、氟化物或氯化物;R11和R15各自独立为氢、氟化物或氯化物和/或
邻位取代基组合,其中R6和R10各自独立甲基、乙基或与R7和R9分别相连形成蒽骨架,优选甲基;R11和R15各自独立为氢、氟化物或氯化物。
特别优选的是,R11和R15各自独立为氢或氟化物。
在一优选的实施方案中,提供这样一种分子式(II)的配位体,其中R1-R3为氢;A6-R13不存在和A1-A5为碳原子,从而构成二茂铁部分的亚环戊二烯基部分;R4、R5、R6、R8和R10为甲基;R7、R9、R11、R12、R14和R15为氢。
在另一优选的实施方案中,提供这样一种分子式(II)的配位体,其中R1-R3为氢;A6-R13不存在和A1-A5为碳原子,从而构成二茂铁部分的亚环戊二烯基部分;R4和R5为甲基;R6和R10为乙基;R7、R8、R9、R11、R12、R14和R15为氢。
在另一优选的实施方案中,提供这样一种分子式(II)的配位体,其中R1-R3为氢;A3为氮原子,A6-R13不存在和A1、A2、A4、A5为碳原子,从而构成1-吡咯基环;R4和R5为甲基;R6和R10为乙基;R7、R8、R9、R11、R12、R14和R15为氢。
在一特别优选的实施方案中,提供这样一种分子式(II)的配位体,其中R1-R3为氢;A3为氮原子,A6-R13不存在和A1、A2、A4和A5为碳原子,从而构成1-吡咯基环;R4、R5、R6、R8和R10为甲基;R7、R9、R11、R12、R14和R15为氢。
在得到的混合双亚胺吡啶MXn配合物中,X可方便为卤化物,特别是氯化物。
在混合双亚胺吡啶MXn配合物的优选的实施方案中,金属原子M为Fe和n为2。在另一优选的实施方案中,金属原子M为Fe和n为3。
能将任选取代的烃基或氢化物基团转移到金属原子M上,还能从金属原子M上取出X-基团的化合物包括烷基铝化合物,例如烷基铝噁烷和烷基铝卤化物。优选的化合物为甲基铝噁烷。
能将任选取代的烃基或氢化物基团转移到金属原子M上的化合物包括烷基铝噁烷在内的烷基铝化合物、烷基锂化合物、格利雅试剂、烷基锡和烷基锌化合物。
能从金属原子M上取出X-基团的化合物包括强中性路易斯酸,例如SbF5、BF3和Ar3B,其中Ar为强吸电子芳基,例如C6F5或3,5-(CF3)2C6H3。
中性路易斯酸施主分子为一种可适合起路易斯碱作用的化合物,例如醚类、胺类、硫化物和有机腈类。
施主分子(路易斯碱)例如三乙胺或2,6-二叔丁基吡啶和/或受主分子(路易斯酸)例如二乙基锌的使用可能对乙烯低聚生产1-烯烃方法的选择性有正影响。
此外,与MAO活化的和增溶的催化剂溶液相比,路易斯酸例如三异丁基铝(TIBA)可通过制备稳定且透明的催化剂前体溶液来提高Fe或Co催化的乙烯低聚的连续操作性,前者放置时可能变得混浊。
在本发明的混合[双亚胺吡啶MYp·Ln +][NC-]q配合物中,L可为能被乙烯取代的中性路易斯施主分子,或者空配位中心。
在本发明的混合[双亚胺吡啶MYp·Ln +][NC-]q配合物中,金属原子M优选为Fe,而所述金属原子的氧化态可为2或3。
催化剂体系可通过配合物和任选另外的化合物优选在溶剂例如甲苯或异辛烷中混合来制成。
在低聚反应混合物中通常使用这样的催化剂体系数量,以致每摩尔要反应的乙烯含有10-4-10-9克原子、更优选10-6-10-7克原子金属原子M,特别是Fe[II]或[III]金属。
低聚反应最宜在-100至+300℃、优选0-200℃、更优选50-150℃下进行。
低聚反应宜在0.01-15兆帕(0.1-150巴(绝对))、更优选1-10兆帕(10-100巴(绝对))、最优选1.5-5兆帕(15-50巴(绝对))下进行。
熟悉本专业的技术人员很容易确定用于特定催化剂体系的最佳温度和压力条件,以便使低聚物的产率最大和使竞争反应例如二聚和聚合反应最少。
优选这样选择温度和压力条件,以便使产物的K-因子为0.50-0.90、最优选0.60-0.80。在本发明中,当产物的K-因子大于0.9时,聚合反应出现。
低聚反应可在气相或液相或气-液混合相中进行,这取决于进料和烯烃产物的挥发性。
低聚反应在惰性溶剂存在下进行,所述的溶剂也可为催化剂和/或烯烃进料的载体。适合的溶剂包括烷烃、链烯烃、环烷烃和芳烃。例如,可适合用于本发明的溶剂包括己烷、异辛烷、苯、甲苯和二甲苯。
已发现0.1-10小时的反应时间是适合的,视催化剂的活性而定。反应优选在没有空气或水汽的条件下进行。
低聚反应可按传统的方式进行。可在搅拌釜反应器中进行,其中将烯烃和催化剂或催化剂前体连续加到搅拌釜反应器和反应物中,并将产物、催化剂和未利用的反应物从搅拌釜反应器中除去,分离产物并将催化剂和未利用的反应物循环回搅拌釜反应器。
另一方面,反应可在间歇反应器中进行,其中将催化剂前体和烯烃反应物送入高压釜,反应适当时间以后,用传统的方法例如蒸馏,将产物从反应混合物中分离。
在适宜的反应时间以后,可通过迅速排出乙烯以便使催化剂体系失活的方法来中止低聚反应。
生成α烯烃的链长为4-100个碳原子、优选4-30个碳原子、最优选4-20个碳原子。
宜通过蒸馏来回收烯烃产物,如果需要还可用蒸馏进一步分离,视烯烃的最终用途而定。
现在参考附图,用以下实施例说明本发明,不应以任何方式将这些实施例作为对本发明范围的限制,其中:
图1为实施例2的回归分析;
图2为实施例5的回归分析;
图3为实施例8的回归分析;以及
图4为实施例10的回归分析。
实施例
一般步骤和表征
催化剂体系的所有操作都在氮气气氛下进行。
将无水甲苯(99.8%纯度)(ex.Aldrich)在4分子筛上干燥(最终水含量为约3ppm)。
将乙烯(纯度99.5%)在装有4分子筛和BTS催化剂(ex.BASF)的柱上纯化,以便使水和氧含量降到<1ppm。
2,6-二乙酰基吡啶、2,4,6-三甲基苯胺、4-叔丁基苯胺、2,6-二乙基苯胺和无水氯化铁(II)由ex.Aldrich提供。1-氨基吡咯由日本TCI购买。
二茂铁基胺按文献所述的方法制备(D.Van Leusen和B.Hessen,有机金属化合物,2001,20,224-226)。
制得的低聚物用气相色谱(GC)表征,以便用HP 5890系列II设备和以下的色谱条件评价低聚物分布:
色谱柱:HP-1(交联甲基硅氧烷),薄膜厚度=0.25微米,内径=0.25毫米,长度60米(Hewlett Packard);注射器温度:325℃;检定器温度:325℃;初始温度:40℃,10分钟;程序升温速率:10.0℃/分;最终温度:325℃,41.5分钟;内标:正己基苯。用标准校正混合物,相对于正己基苯(内标),对于偶数直链α烯烃、内己烯:顺-和反-2-己烯和顺-和反-3-己烯,以及对于支链己烯:3-甲基-1-戊烯和2-乙基-1-丁烯测定了响应因子。假设支链十二内烯的响应因子与对应的直链α烯烃相等。
从GC分析得到C4-C30烯烃的产率,由此通过回归分析,用C6-C28的数据得到C4-C100烯烃即全部低聚产物(全部产物)的K-因子和理论产率。
由GC分析发现在所有己烯异构体中直链1-己烯的相对数量以及在所有十二烯异构体中直链1-十二烯的相对数量可用作催化剂生成直链α烯烃选择性的量度。
在室温下,用Varian300MHz或400MHz仪得到NMR数据。
催化剂的组分
1.2-[1-(2,4,6-三甲基苯基亚胺基)乙基1-6-乙酰基吡啶(1)的制备

将2,6-二乙酰基吡啶(7.3克,44.8毫摩尔)和2,4,6-三甲基苯胺(5.74克,42.55毫摩尔)溶于450毫升甲苯中。将4分子筛和少量对甲苯磺酸(0.22毫摩尔)加到这一溶液中。将混合物回流16小时。过滤以后,在真空下除去溶剂。从乙醇中几次结晶得到3.42克(28.7%)单亚胺(
1)。
1H-NMR(CDCl3)δ8.55(d,1H,Py-Hm),8.11(d,1H,Py-Hm),7.92(t,1H,Py-Hp),6.89(s,2H,ArH),2.77(s,3H,Me),2.27(s,3H,Me),2.22(s,3H,Me),1.99(s,6H,Me).
2.2-[1-(2,4,6-三甲基苯基亚胺基)乙基1-6-(1-(4-叔丁基苯基亚胺基)乙基)吡啶(
2)的制备

将单胺(
1,2.8克,10毫摩尔)和4-叔丁基苯胺(1.49克,10毫摩尔)溶解于100毫升甲苯中。将4分子筛和少量对甲苯磺酸(0.1毫摩尔)加到这一溶液中。放置5天并加入更多4分子筛以后,将混合物回流2小时。过滤以后,在真空下除去溶剂。用甲醇洗涤残留物并从乙醇中再结晶。得到2.4克(58%)混合二亚胺(
2)。1H-NMR(CDCl3)δ8.42(d,1H,Py-Hm),8.34(d,1H,Py-Hm),7.86(t ,1H ,Py-Hp),7.38(d,2H,ArH),6.89(s ,2H,ArH),6.78(d,2H,ArH),2.42(s,3H,Me),2.29(s,3H,Me),2.22(s,3H,Me),2.00(s,6H,Me),1.34(s,9H,But).
3.2-[1-(2,4,6-三甲基苯基亚胺基)乙基1-6-[1-(4-叔丁基苯基亚胺基)乙基]吡啶氯化铁[II]配合物(
3)的制备
在惰性气氛中,将1.5克二亚胺(2,3.6毫摩尔)在100毫升二氯甲烷中的溶液加到420毫克FeCl2(3.3亳摩尔)在150毫升二氯甲烷中的溶液中。将混合物搅拌1周。用过滤法分离生成的兰色沉淀,然后在真空下干噪。生成1.5克(84%)铁配合物(
3)。1H-NMR(Cl2CDCDCl2,宽信号)d79.3(1H,Py-Hm),77.7(1H,Py-Hm),27.0(1H,Py-Hp),20.7(3H,Me), 17.3(6H,Me),15.0(2H,ArH),14.3(2H,ArH),1.2(9H,But),-2.6(3H,MeC=N),-17.9(2H,o-ArH),-32.1(3H,MeC=N).
4.2,6-双[1-(二茂铁基亚胺基)乙基]吡啶(
4)的制备

在惰性气氛中,将2,5二乙酰基吡啶(50毫克,0.30毫摩尔)和二茂铁基胺(123.4毫克,0.61毫摩尔)溶解于50毫升甲苯中。将4分子筛加到这一溶液中。在室温下放置65小时后,将混合物过滤。在真空下除去溶剂。使残留物从乙醇中结晶。生成75毫克(46%)二亚胺
4红色晶体。
1H-NMR(CDCl3)δ8.26(d,2H,Py-Hm),7.78(t,1H,Py-Hp),4.43(t,4H,CpH),4.22(t,4H,CpH),4.21(s,10H,CpH),2.54(s,6H,Me).
5.2,6-双[1-(二茂铁基亚胺基)乙基]吡啶氯化铁[II]配合物(
5)的制备
在惰性气氛中,将390毫克二亚胺(
4,0.737毫摩尔)在10毫升二氯甲烷中的红色溶液加到89毫克FeCl2(0.702毫摩尔)在10毫升二氯甲烷中的溶液中。将混合物搅拌16小时。加入6毫升己烷以后,用离心分离法分离生成的兰色沉淀,用己烷洗涤然后在真空下干噪。生成200毫克(44%)铁配合物
5。1H-NMR(CD2Cl2,宽信号)δ83.0(2H,Py-Hm),9.3(4H,CpH),3.3(10H,CpH),2.7(6H,MeC=N),-1.5(4H,CpH),-5.2(1H,Py-Hp).
6.2-[1-(2,4,6-三甲基苯基亚胺基)乙基]-6-[1-(二茂铁基亚胺基)乙基]吡啶(
6)的制备
将单胺2-[1-(2,4,6-三甲基苯基亚胺基)乙基1-6-[1-乙酰基吡啶(1,263毫克,0.94毫摩尔)和二茂铁基胺(280毫克,1.03毫摩尔)溶解于40毫升甲苯中。将4分子筛加到这一溶液中。放置16小时后,将混合物过滤。在真空下除去溶剂。使残留物从乙醇中结晶。生成180毫克(41%)混合二亚胺
6。
1H-NMR(CD2Cl2)δ8.36(dd,2H,Py-Hm),7.85(t,1H,Py-Hp),6.88(s,2H,ArH),4.46(t,2H,CpH),4.25(t,2H,CpH),4.20(s,5H,CpH),2.55(s,3H,Me),2.27(s,3H,Me),2.20(s,3H,Me),1.98(s,6H,Me).
7.2-[1-(2,4,6-三甲基苯基亚胺基)乙基1-6-[1-(二茂铁基亚胺基)乙基]吡啶氯化铁[II]配合物(
7)的制备

在惰性气氛中,将153毫克二亚胺(6,0.33毫摩尔)在5毫升二氯甲烷中的溶液加到41毫克FeCl2(0.32毫摩尔)在5毫升二氯甲烷中的溶液中。将混合物搅拌16小时。用离心分离法分离生成的兰灰色沉淀,用己烷洗涤,然后在真空下干噪。生成170毫克(89%)铁配合物
7。
1H-NMR(CD2Cl2,宽信号,选择的数据)δ88.6(1H,Py-Hm),76.7(1H,Py-Hm),21.3(3H,Me),16.3(6H,Me),2.8(5H,CpH),-11.5(3H,MeC=N)。
8.2-[1-(2,6-二乙基苯基亚胺基)乙基1-6-乙酰基吡啶(
8)的制备
将2,6-二乙酰基吡啶(8.15克,50毫摩尔)和2,6-二乙基苯胺(7.46克,50毫摩尔)溶解于甲苯(150毫升)中。将4分子筛加到这一溶液中。加入两滴浓硫酸,接着将混合物回流16小时,得到73%的转化率。在真空下除去溶剂。
将生成的2,6-二乙酰基吡啶、2,6-双-(1-(2,6-二乙基苯基亚胺基)乙基)吡啶和2-[1-(2,6-二乙基苯基亚胺基)乙基1-6-乙酰基吡啶的混合物从乙醇中结晶,得到2-[1-(2,6-二乙基苯基亚胺基)乙基1-6-乙酰基吡啶和2,6-双-[1-(2,6-二乙基苯基亚胺基)乙基]吡啶为3∶1的混合物。
将这一混合物溶解于THF(75毫升)中,通过与金属卤化物选择性配位来除去二亚胺副产物。结束后在惰性气氛中加入等摩尔量的FeCl2(0.75克,5.93毫摩尔)。在室温下搅拌16小时后,在真空下除去溶剂。
将甲苯(75毫升)加到生成的混合物中。在薄层氧化硅上过滤出沉淀的配合物,得到黄色溶液。在真空下除去溶剂。
从乙醇中结晶得到3.05克2-[1-(2,6-二乙基苯基亚胺基)乙基1-6-乙酰基吡啶(21%)。
1H-NMR(CDCl3)δ8.55(dd,1H,Py-Hm),8.12(dd,1H,Pym),7.93(t,1H,Pyp),7.11(d,2H,ArHm),7.03(dd,1H,ArHp),2.78(s,3H,Me),2.36(m,4H,CH2),2.24(s,3H,Me),1.13(t,6H,Me).
9.2-[1-(2,6-二乙基苯基亚胺基)乙基]-6-[1-(二茂铁基亚胺基)乙基]吡啶(
9)的制备
在惰性气氛中,将单亚胺2-[1-(2,6-二乙基苯基亚胺基)乙基1-6-乙酰基吡啶(
8,368毫克,1.25毫摩尔)和二茂铁基胺(268毫克,1.33毫摩尔)溶解于50毫升甲苯中。将4分子筛加到这一溶液中。放置40小时后,将混合物过滤。在真空下除去溶剂。使残留物从乙醇中结晶。生成160毫克(27%)混合二亚胺
9的红色晶体。
1H-NMR(CD2C12)δ8.38(d,1H,Py-Hm),8.35(d,1H,Py-Hm),7.87(t,1H,Py-Hp),7.10(d,2H,ArH),7.01(t,1H,ArH),4.46(t,2H,CpH),4.26(t,2H,CpH),4.21(s ,5H,CpH),2.56(s,3H,Me),2.36(m,4H,CH2),2.22(s,3H,Me),1.11(t,6H,Me).
10.2-[1-(2,6-二乙基苯基亚胺基)乙基1-6-[1-(二茂铁基亚胺基)乙基]吡啶氯化铁[II]配合物(
10)的制备
在惰性气氛中,将100毫克二亚胺(9,0.21毫摩尔)在5毫升二氯甲烷中的溶液加到25.7毫克FeCl2(0.20毫摩尔)在5毫升二氯甲烷中的溶液中。将混合物搅拌65小时。在加入5毫升己烷后,用离心分离法分离生成的兰灰色沉淀,用己烷洗涤,然后在真空下干噪。生成100毫克(82%)铁配合物
10。
1H-NMR(CD2Cl2,宽信号,选择的数据)δ88.5(1H,Py-Hm),75.3(1H,Py-Hm),16.3(2H,CHaHb),13.2(2H,CHaHb),2.5(5H,CpH),0.8(6H,Me),-4.6(1H,ArH),-12.5(3H,MeC=N),-14.5(2H,CpH)。
11.2,6-双[1-(1-吡咯基亚胺基)乙基]吡啶(
11)的制备

将2,6-二乙酰基吡啶(345毫克,2.11毫摩尔)和1-氨基吡咯(400毫克,4.87毫摩尔)溶解于50毫升甲苯中。将4分子筛加到这一溶液中。在室温下放置2天后,将混合物过滤。在真空下除去溶剂。使残留物从乙醇中结晶。生成350毫克(57%)二亚胺
11。
1H-NMR(CDCl3)δ8.26(d,2H,Py-Hm),7.82(t,1H,Py-Hp),6.93(m,2H,PyrH),6.25(m,2H,PyrH),2.66(s,6H,Me).
12.2,6-双[1-(1-吡咯基亚胺基)乙基]吡啶氯化铁[II]配合物(
12)的制备
在惰性气氛中,将26毫克FeCl2(0.27毫摩尔)在0.4毫升乙醇中的溶液缓慢加到80毫克二亚胺(
11,0.27毫摩尔)在3毫升二氯甲烷中的溶液中。用离心分离法分离生成的兰色沉淀,用甲苯洗涤3次,然后在真空下干噪。生成75毫克铁配合物
12。NMR未显示这一配合物的任何信号。
13.2-[1-(2,6-二乙基苯基亚胺基)乙基]-6-[1-(1-吡咯基亚胺基)乙基]吡啶(
13)的制备

将单亚胺2-[1-(2,6-二乙基苯基亚胺基)乙基]-6-乙酰基吡啶(8,1.5克,5.1毫摩尔)和1-氨基吡咯(460毫克,5.6毫摩尔)溶解于25毫升甲苯中。将4分子筛加到这一溶液中。在放置16小时后,将混合物过滤。在真空下除去溶剂.使残留物从乙醇中结晶。生成845毫克(46%)二亚胺
13。
1H-NMR(CDCl3)δ8.41(d,1H,Py-Hm),8.29(d,1H,Py-Hm),7.86(t,1H,Py-Hp),6.98-7.14(m,3H,ArH),6.93(m,2H,PyrH),6.25(m,2H,PyrH),2.67(s,3H,Me),2.36(m,4H,CH2),2.21(s,3H,Me),1.12(t,6H,Me).
14.2-[1-(2,6-二乙基苯基亚胺基)乙基1-6-[1-(1-吡咯基亚胺基)乙基]吡啶氯化铁[II]配合物
14)的制备
在惰性气氛中,将211毫克二亚胺(
13,0.59毫摩尔)在5毫升二氯甲烷中的溶液加到70毫克FeCl2(0.55毫摩尔)在10毫升二氯甲烷的溶液中。将混合物搅拌60小时。在加入15毫升己烷后,用离心分离法分离生成的兰色沉淀,用己烷洗涤,然后在真空下干噪。生成250毫克克(93%)铁配合物
14。
1H-NMR(CD2Cl2,宽信号)δ87.3(1H,Py-Hm),72.2(1H,Py-Hm),27.9(3H,Me),18.3(2H,CHaHb),14.8(2H,CHaHb),14.4(2H,ArH),8.5(2H,PyrH),4.6(2H,PyrH),1.2(1H,Py-Hp),0.2(6H,Me),-10.8(1H,ArH),-43.4(1H,MeC=N).
15.2-[1-(2,4,6-三甲基苯基亚胺基)乙基1-6-[1-(1-吡咯基亚胺基)乙基]吡啶(
15)的制备
将单亚胺(
1,3.0克,10.7毫摩尔)和1-氨基吡咯(1.0克,12.18毫摩尔)溶解于50毫升甲苯中。将4分子筛加到这一溶液中。在放置40小时后,将混合物过滤。在真空下除去溶剂。使残留物从乙醇中结晶。生成1.85克(50%)混合二亚胺
15。
1H-NMR(CDCl3)δ8.42(d,1H,Py-Hm),8.29(d,1H,Py-Hm),7.86(t,1H,Py-Hp),6.93(m,2H,Pyrrole-H),6.88(s,2H,ArH),6.26(m,2H,Pyrrole-H),2.67(s,3H,Me),2.28(s,3H,Me),2.20(s,3H,Me),2.00(s,6H,Me).
16.2-[1-(2,4,6-三甲基苯基亚胺基)乙基]-6-[1-(1-吡咯基亚胺基)乙基]吡啶氯化铁[II]配合物(
16)的制备
在惰性气氛中,将103毫克FeCl2(0.81毫摩尔)在0.7毫升乙醇中的溶液缓慢加到400毫克二亚胺(
15,1.16毫摩尔)在10毫升甲苯和6毫升戊烷的混合溶剂中。用离心分离法分离生成的绿褐色沉淀,用甲苯洗涤3次,然后在真空下干噪。生成375毫克(98%)铁配合物
16。
1H-NMR(CD2Cl2,宽信号,未确定的)δ88.1(1H),72.4(1H),29.9(3H),19.5(3H),16.9(6H),13.5(2H),8.8(2H),5.8(2H),2.9(1H),-45.1(3H)。
17.2-[1-(2,4,6-三甲基苯基亚胺基)乙基]-6-[1-(1-吡咯基亚胺基)乙基]吡啶氯化铁[II]配合物(
16′)的另一制备

在惰性气氛中,将204毫克混合二亚胺(
15,0.59毫摩尔)在5毫升二氯甲烷中的溶液加到70毫克FeCl2(0.55毫摩尔)在10毫升二氯甲烷中的溶液中。将混合物搅拌65小时。用离心分离法分离生成的绿褐色沉淀,用戊烷洗涤,然后在真空下干噪。生成200毫克(77%)铁配合物
16′。
1H-NMR(CD2Cl2,宽信号,未确定的)δ88.1(1H),72.5(1H),29.8(3H),19.6(3H),16.9(6H),13.6(2H),8.8(2H),5.7(2H),3.6(1H),-45.2(3H)。
18.2,6-[1-(2-甲基苯基亚胺基)乙基]吡啶氯化铁[II]配合物(
X)的制备
按WO-A-99/02472公开的方法制备配合物
x。
19.甲基铝噁烷(MAO)
用于这一实验的MAO甲苯溶液(Eurecen AL 5100/10T,批号:B7683;[Al]=4.88%(重量),TMA=35.7%(重量)(计算的),分子量=900克/摩尔和[Al]=4.97%(重量))为ex.Witco GmbH,Bergkamen,Germany。
20.三乙基铝甲苯溶液(25%(重量)TEA和三异丁基铝甲苯溶液(25%(重量)TIBA)从Aldrich得到。
催化剂体系的制备
在氮气下在Braun MB 200-G干噪箱中进行催化剂的制备。
将铁配合物(通常约10毫克)装在用隔膜密封的玻璃瓶中;然后将上述等级的MAO溶液(4.0克)或表1和2所列数量的替代共催化剂(参见表2中“预混合共催化剂/Al∶Fe摩尔比”)加入,并搅拌2分钟。这样通常生成深色溶液,它有时含有一些沉淀。此后,将甲苯(9.0克)加入,然后将溶液再搅拌10分钟。此后立即将部分混合物即催化剂预混合物通过注射体系加到1升或0.5升钢制高压釜中,并用于低聚反应(用量参见表1和2)。
低聚实验
低聚实验在1升或0.5升钢制高压釜中进行,所述的高压釜装有套管冷却,有加热/冷却浴(ex.Julabo,型号ATS-2)和透平/气体搅拌器和折板。
为了从反应器中除去微量水,在<10Pa和70℃下将反应器抽空过夜。用以下步骤清洗反应器:将250毫升甲苯和MAO(0.3-1.2克溶液)送入,然后在0.4-0.5兆帕氮压和70℃下搅拌30分钟。通过反应器底部的旋塞排放反应器的物料。将反应器抽空到0.4千帕,然后装入250毫升甲苯,加热到40℃并用乙烯加压到表1和2中或实验的说明中所列的压力。
然后借助甲苯(注入的总体积为30毫升,使用类似催化剂预混合物注入的步骤;参见下文)将MAO溶液(对于1升高通常为0.5克,而对于0.5升高压釜通常为0.25克)加到反应器中,并在800转/分下继续搅拌30分钟。
借助甲苯将上述制备的催化剂体系送入搅拌反应器,其数量如表1和2中所列(甲苯的注入总体积为30毫升:将甲苯稀释到10毫升的催化剂预混合物注入,再用甲苯清洗注射体系两次,每次10毫升)。
在活性催化剂体系的情况下,催化剂预混合物的加入引起放热(通常5-20℃),通常在1分钟内达到最高值,随后迅速达到表1和2中所列的温度和压力。
在整个反应中监测温度和压力以及乙烯耗量,同时使乙烯的压力保持不变。
乙烯耗量达到某一体积后,通过以下步骤来中止低聚:迅速排放乙烯,用高压釜底的旋塞将产物混合物卸入收集器。将混合物暴露到空气中,使催化剂迅速失活。
将正己基苯(0.5-3.5克)作为内标加到粗产物中以后,用气相色谱测定C4-C30烯烃的数量,由此用回归分析法测定(表观)舒茨-弗洛里K-因子,通常使用C6-C28直链α烯烃数据。
所谓“表观 K-因子”指在舒茨-弗洛里分布有一些变化的情况中的K-因子。从这一回归分析,计算了C30-C100组分即固体蜡的理论含量。数据列入表1。
在产物中固体的数量测定如下。将粗反应产物在4000转/分下离心分离30分钟,此后将透明上层液倾析。用高剪切混合器(Ultra-Turrax,type TP 18-10)将固体烯烃、甲苯和少量液体烯烃组成的下层液与500毫升丙酮混合。将所述的混合物在上述条件下离心分离。将下层液与200毫升丙酮混合,然后在玻璃过滤器(孔隙率P3)上过滤出。在70℃和<1千帕下将固体产物干噪24小时,称重,用气相色谱测定固体的1,2-二氯苯或1,2,4-三氯苯溶液的<C30含量。表1所列的固体数量为分离的>C28固体。
在所有己烯异构体中直链1-己烯的相对数量以及在所有十二烯异构体中直链1-十二烯的相对数量用GC分析法评估,并列入表1和2。
实施例1(对比例)
实施例1a在乙烯平均压力为1.7兆帕即1.6兆帕(表压)下用铁配合物
5进行,后者不属本发明。实验详情在表1中给出。活化的铁配合物按5份(270、550、1510、4570和9100纳摩尔)在时间为0、3、11、13、23分钟下加入,并加入另一份MAO溶液(在时间为25分钟时,1.0克MAO溶液)。14分钟后,温度从50℃升到70℃,并在实验的其余时间内保持在这一温度下。得到很小(如果有的话)的乙烯转化率,甚至在加入相当大数量对称铁双亚胺吡啶催化剂和MAO以后以及升温以后。
为了检验高压釜体系不受空气或水汽进入的影响,通过在1.5兆帕乙烯压力和40℃下加入待审专利申请书PCT/EP01/01506的活化非对称铁催化剂
3(参见表1的实施例1b)接着迅速升温到70℃来扩展实施例1a。催化剂
3的活性、产物分布和产物纯度与上述待审专利申请书中
3观测到的一致,尽管相对大量的催化剂
5和MAO存在。
实施例2
实施例2在乙烯平均压力为1.7兆帕下进行,使用本发明的混合芳基-,二茂铁基亚胺铁配合物
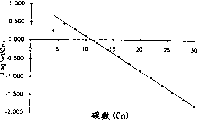
7。实验详情在表1中给出。催化剂的转换频率(TOF)为2.45E+06摩尔乙烯/摩尔Fe*小时,得到高l-己烯和1-十二烯纯度的产物。注意,产物分布与舒茨-弗洛里分布有明显差别,特别是在低碳数下,正如图1所示(回归统计:R2=0.98;对于12个数据点,标准误差=0.08)。
实施例3
实施例3在乙烯平均压力为1.6兆帕下进行,使用本发明的混合芳基-,二茂铁基亚胺铁配合物
10。实验详情在表1中给出。催化剂的转换频率(TOF)为1.97E+06摩尔乙烯/摩尔Fe*小时,得到比实施例2低的l-己烯和1-十二烯纯度的产物。注意,产物分布与舒茨-弗洛里分布比实施例2有更明显差别(回归统计:R2=0.81;对于12个数据点,标准误差=0.25)。这一点被实施例3分离出的占总产物8.4%(重量)的>C28固体蜡证实,而K-因子得到占总低聚产物6.5%(重量)的C30-C100馏分。
实施例4
实施例4a在乙烯平均压力为1.7兆帕下用双-[1-吡咯基亚胺]铁配合物
12进行,后者不属本发明。实验详情在表1中给出。活化的铁配合物按4份(3100、3100、6200和13600纳摩尔)在时间为0、3、4、11分钟下加入。10分钟时,温度从50℃升到70℃,并在这一温度下保持10分钟。在20分钟时温度降到40℃,并在实验的其余时间内保持这一温度。得到很小(如果有的话)的乙烯转化率,甚至在加入相当大数量这种铁双亚胺吡啶催化剂以后以及升温到70℃以后。
为了检验高压釜体系不受空气或水汽进入的影响,通过在1.5兆帕乙烯压力和40℃下加入待审专利申请书PCT/EP01/01506的活化非对称铁催化剂
3(参见表1的实施例4b)接着升温到70℃来扩展实施例4a。催化剂
3的活性、产物分布和产物纯度与上述待审专利申请书中3观测到的一致,尽管相对大量的催化剂
12和MAO存在。
注意,由2,5-二甲基氨基吡咯得到的铁双亚胺吡啶催化剂也不属于本发明,正如颁发给Eastman Chemical Company的专利申请书WO 00/50470实施例58中公开的,据认为它是催化剂
12的更高同系物,有高的乙烯转化活性,T.O.F=4.14E+06摩尔/摩尔*小时,但生成聚乙烯,Mn(NMR)=1154(分别为WO 00/50470的实施例60和59),而不是生成C4-C30的α烯烃。
实施例5
实施例5在乙烯平均压力为1.6兆帕即1.5兆帕(表压)下用非对称铁配合物
14进行,后者属于本发明。实验详情在表1中给出。正如图2中所示,用C6-C28含量的回归分析得到与舒茨-弗洛里分布有明显差别。K-因子为0.678(12个观测结果的回归统计:R2=0.98和标准误差=0.08)。这一点被实施例5分离出的占总产物13.8%(重量)的>C28固体蜡证实,而K-因子得到占总低聚产物2.6%(重量)的C30-C100馏分。催化剂的T.O.F=1.45E+07摩尔乙烯/摩尔Fe*小时,1-己烯和1-十二烯的纯度分别为99.5%和98.4%。
实施例6
实施例6在更高的[Al]/[Fe]比下重复实施例5。列入表1的结果类似实施例5的结果。也观测到与舒茨-弗洛里分布有明显差别(R2=0.98和12个数据点的标准误差=0.09)。这一点被分离出的占总产物12.2%(重量)的>C28固体蜡证实,而K-因子得到占总低聚产物1.9%(重量)的C30-C100馏分。
实施例7
实施例7在较低的乙烯进料量下重复实施例6。列入表1的结术表明在这些条件下有较低表观K-因子趋势。也观测到与舒茨-弗洛里分布有明显差别(R2=0.99和12个数据点的标准误差=0.09)。这一点被实施例7分离出的占总产物5.1%(重量)的>C28固体蜡证实,而K-因子得到占总低聚产物1.0%(重量)的C30-C100馏分。
实施例8
实施例8在乙烯平均压力为1.5兆帕即1.4兆帕(表压)下用混合芳基-,1-吡咯基亚胺铁配合物
16进行,后者属于本发明。实验详情在表1中给出。正如图3中所示,用C6-C28含量的回归分析令人吃惊地得到在整个低聚物范围内有几乎理想的舒茨-弗洛里分布。K-因子为0.649(12个观测结果的回归统计:R2=1.00和标准误差=0.01)。这一点被实施例8分离出的占总产物1.0%(重量)的>C28固体蜡证实,而K-因子得到占总低聚产物1.6%(重量)的C30-C100馏分(分离出比理论预期少的>C28固体是由于它们在<C28低聚物的甲苯溶液中的溶解性)。催化剂的T.O.F=2.95E+07摩尔乙烯/摩尔Fe*小时。己烯馏分有以下组成:1-己烯=99.0%(重量),顺-2-己烯=0.0%(重量),反-2-己烯=0.2%(重量),3-己烯=0.2%(重量),支链己烯=0.5%(重量)。
总之,可以说用这一混合双亚胺铁催化剂体系令人吃惊地观测到与舒茨-弗洛里分布没有差别,与实施例3、5、6和7相比,它说明生成较少高分子量产物。其优点是可更直接的加工(固体在装置中及其处理管线中堵塞少)以及不大需要再加工高分子量烯烃(使工艺在经济上更可行)。
实施例9
实施例9重复实施例8,但在较低的乙烯平均压力和乙烯进料量下进行。实验详情在表1中给出。用C6-C28含量的回归分析再次得到几乎理想的舒茨-弗洛里分布,K-因子为0.671,12个观测结果的回归统计:R2=1.00和标准误差=0.01。舒茨-弗洛里分布再次被分离出的>C28固体蜡证实,它低于由K-因子计算的数量。
实施例10
实施例10重复实施例8,但使用催化剂前体
16′代替
16以及较低的乙烯进料量,即进料量类似实施例9的进料量。实验详情列入表1。用C6-C28含量的回归分析再次得到几乎理想的舒茨-弗洛里分布,K-因子为0.645,12个观测结果有以下回归统计:R2=1.00和标准误差=0.02(参见图4)。舒茨-弗洛里分布再次被分离出的>C28固体蜡数量证实,它低于由K-因子计算的数量。己烯馏分有以下组成:1-己烯=99.5%(重量),顺-2-己烯=0.0%(重量),反-2-己烯=0.1%(重量),3-己烯=0.1%(重量),支链己烯=0.3%(重量)。
实施例10中催化剂
16′的活性高于实施例8中
16的活性,而K-因子在误差范围内仍然相同,1-己烯和1-十二烯的纯度甚至更高。
总之,可以说在使用这一混合双亚胺铁配合物的情况下令人吃惊地观测到与舒茨-弗洛里分布没有差别,这对整个方法的经济性是有利的,因为在这一情况下不生成更多的需要通过异构化和歧化加工固体即重质蜡(由于装置和/或处理管线的堵塞等原因,所述的固体处理本身是麻烦的),例如2-丁烯得到在经济上有吸引力范围(C8-C20)的内烯。此外,这些新型非对称催化剂体系的催化剂活性与待审专利申请书PCT/EP01/01506催化剂不相上下,且1-己烯和1-十二烯的纯度类似。
这些实施例证明本发明催化剂体系可达到的各种有利的效果。正如上面举例说明的,这些改进对于所述方法的经济吸引力是十分重要的。
表1
| 实施例No. | 实施例1a | 实施例1b1,3 | 实施例2 | 实施例3 | 实施例4a | 实施例4b1,3 |
| 铁配合物/(进料量,纳摩尔) | 5(16000) | 5+ 3(539) | 7(1320) | 10(1860) | 12(26000) | 12+ 3(220) |
| [Al]/[Fe](摩尔/摩尔) | 700 | 22000(700)4 | 1500 | 900 | 400 | 48000(400)4 |
| 反应时间(分) | 28 | 44 | 47 | 41 | 34 | 8 |
| 乙烯压力,兆帕(巴(绝对)) | 1.7(17) | 1.5(15) | 1.7(17) | 1.6(16) | 1.7(17) | 1.5(15) |
| 乙烯耗量(总产物)(克) | <2.0 | 115.1 | 70.4 | 70.4 | <3.9 | 47.32 |
| 分离的<C30产物(克) | 0.0 | 82.4 | 35.6 | 23.9 | 0.0 | 41.4 |
| 分离的>C28固体(克) | 0.0 | n.d. | 2.3 | 6.0 | 0.0 | 0.9 |
| 按乙烯计>C28固体(%(重量)) | n.d. | n.d. | 3.3 | 8.4 | n.d. | 1.8 |
| 按总产物计C30-100(计算的)(%重量) | n.d. | 4.3 | 5.3 | 6.5 | n.d. | 3.1 |
| T.O.F(摩尔C2=/摩尔Fe*小时) | <1E+04 | 1.04E+07 | 2.45E+06 | 1.97E+06 | <1E+04 | 5.61E+07 |
| K-因子 | n.d. | 0.706 | 0.720 | 0.732 | n.d. | 0.687 |
| 1-C6=纯度(%(重量)) | n.d. | 98.5 | 96.9 | 92.1 | n.d. | 97.7 |
| 1-C12=纯度(%(重量)) | n.d. | 96.8 | 97.7 | 92.7 | n.d. | 97.5 |
表1(续)
| 实施例No. | 实施例5 | 实施例6 | 实施例7 | 实施例8 | 实施例9 | 实施例10 |
| 铁配合物/(进料量,纳摩尔) | 14294) | 14(332) | 14(153) | 16(180) | 16(330) | 16′(111) |
| [Al]/[Fe](摩尔/摩尔) | 4500 | 11300 | 11400 | 7000 | 4100 | 11500 |
| 反应时间(分) | 38 | 38 | 12 | 114 | 21 | 36 |
| 乙烯压力,兆帕(巴(绝对)) | 1.6(16) | 1.6(16) | 1.6(16) | 1.5(15) | 1.0(10) | 1.5(15) |
| 乙烯耗量(总产物)(克) | 76.3 | 69.3 | 29.4 | 281.8 | 117.5 | 117.4 |
| 分离的<C30产物(克) | 58.2 | 47.4 | 25.8 | 222.6 | 98.8 | 88.3 |
| 分离的>C28固体(克) | 10.5 | 8.4 | 1.5 | 2.7 | 2.1 | 0.4 |
| 按乙烯计>C28固体(%(重量)) | 13.8 | 12.2 | 5.1 | 1.0 | 1.8 | 0.5 |
| 按总产物计C30-100(计算的)(%(重量)) | 2.6 | 1.9 | 1.0 | 1.6 | 2.4 | 1.5 |
| T.O.F摩尔C2=/摩尔Fe*小时) | 1.45E+07 | 1.19E+07 | 3.52E+07 | 2.95E+07 | 3.63E+07 | 6.34E+07 |
| K-因子 | 0.678 | 0.659 | 0.623 | 0.649 | 0.671 | 0.645 |
| 1-C6=纯度(%(重量)) | 99.5 | 99.3 | 99.6 | 99.0 | 98.7 | 99.5 |
| 1-C12=纯度(%(重量)) | 98.4 | 97.6 | 98.2 | 95.6 | 95.6 | 97.4 |
除非另加说明,实验都在50℃下在甲苯中使用1升钢制高压釜进行。
n.d.=未测定。
1在70℃下进行。
2由全部产物(C4-C100烯烃回归分析)得到乙烯耗量。
3按铁催化剂3计算的。
4按铁的总含量计算的。
实施例11-19(表2)
实施例11-19通常在1.6兆帕乙烯压力下在钢制高压釜中进行,使用表2所列的铁催化剂前体、共催化剂和替代共催化剂数量。
从实施例11-13可清楚看出,当MAO或三异丁基铝(TIBA)用于制备铁催化剂预混合物,而三乙基铝(TEA)不用于制备所述预混合物时,发生乙烯低聚。
路易斯酸例如TIBA的使用对Fe催化剂体系在甲苯中的溶解性有利,同时基本上保持在乙烯低聚中的催化活性和选择性。
值得注意的是,在加入相当少量(Al∶Fe=0.5)的三乙基铝(TEA)的情况(实施例12)下,催化活性完全丧失,甚至在逐步增加MAO的数量使Al/Fe摩尔比高达250000以后。
但是,相当少量TIBA(实施例13;Al∶Fe=5)的使用得到非均匀的催化剂预混合物,它有与实施例11可比的活性和选择性。
从实施例14-19可以看出,如果用催化剂前体
3、
7和
16′得到的Fe催化剂体系代替由
X得到的双-(邻甲苯基亚胺)吡啶Fe催化剂体系,TIBA也可有利地应用。
少量TIBA(Al∶Fe=1)的使用是有利的,特别是在连续操作中,因为在甲苯中形成浓缩且仍透明的催化剂预混合物溶液,特别是在催化剂前体
3、
7和
16′的情况下。这就使相应Fe催化剂预混合物易于用泵送计量,而不会出现堵塞问题。
从实施例16可得出,用TIBA制备的铁催化剂前体
3的预混合物在惰性气氛中在室温下至少5天仍然是稳定的,而用MAO制备的相应铁催化剂预混合物或者一开始就出现麻烦和/或在相同条件下在贮存过程中有生成沉淀的倾向。
因此,TIBA的使用在连续操作中是特别有利的,此时优选必需用泵计量将浓缩且透明的溶液送入反应器。
而且,正如表2所示,在乙烯低聚中,这些相当少量的TIBA(Al∶Fe=1-5)的使用不会损害铁催化剂的活性、K-因子和生成α烯烃的选择性。
表2
| 实施例No. | 实施例12 | 实施例13 | 实施例14 | 实施例15 | 实施例16 | 实施例17 | 实施例18 | 实施例19 | |
| 反应器中的铁配合物/数量(纳摩尔) | X 1)/309 | X 1)/73 | X 1)/396 | 3 2)/119 | 3 2)/138 | 3 2,3)/152 | 16 4)/56 | 16 4)/71 | 7 4)/599 |
| 预混合共催化剂/Al/Fe摩尔比 | MAO/236 | TEA/0.5 | TIBA/5 | MAO/359 | TIBA/1 | TIBA/1 | MAO/418 | TIBA/1 | TIBA/1 |
| 反应器共催化剂 | MAO | MAO | MAO | MAO | MAO | MAO | MAO | MAO | MAO |
| Al/Fe总摩尔比 | 2000 | >26005) | 2400 | 4200 | 3400 | 3200 | 8900 | 6700 | 800 |
| 反应时间(分) | 34 | 60 | 35 | 53 | 58 | 60 | 31 | 39 | 37 |
| 乙烯压力兆帕(巴(表压)) | 1.6(16) | 1.6(16) | 1.6(16) | 3.1(31) | 3.1(31) | 3.1(31) | 1.6(16) | 1.6(16) | 1.6(16) |
| 乙烯耗量(克) | 124.4 | 0.0 | 117.4 | 85.36) | 78.76) | 64.76) | 52.36) | 51.36) | 58.7 |
| 分离的<C30产物(克) | 101.7 | 0.0 | 102.8 | 74.4 | 70.5 | 59.5 | 47.8 | 47.9 | 33.9 |
| T.O.F(摩尔C2=/摩尔Fe*小时 | 2.54E+07 | 0.0 | 1.83E+07 | 225E+07 | 1.82E+07 | 1.24E+07 | 3.61E+07 | 5.71E+07 | 5.63E+06 |
| K-因子 | 0.72 | n.a. | 0.72 | 0.67 | 0.70 | 0.69 | 0.63 | 0.64 | 0.72 |
| 1-C6=纯度(%(重量)) | 99.4 | n.a. | 99.1 | 99.3 | 99.1 | 99.2 | 99.4 | 99.4 | 95.0 |
| 1-C12=纯度(%(重量)) | 96.7 | n.a. | 94.0 | 97.2 | 96.2 | 96.4 | 97.4 | 97.3 | 96.8 |
除非另加说明,实验都在50℃下在甲苯中使用1升钢制高压釜进行。
n.a.=没有可利用的;n.d.=未测定;TEA=三乙基铝和TIBA=三异丁基铝
1)按WO-A-99/02472制备的催化剂
2)在90℃下在0.5升钢制高压釜中进行。
3)催化剂预混合物在20℃下在氮气下贮存5天
4)在70℃下在0.5升钢制高压釜中进行。
5)逐步增加MAO,使Al/Fe比为250000
6)由全部产物(C4-C100)得到,假设等于乙烯进料量。
Claims (9)
1.一种分子式(I)的混合双亚胺吡啶配位体,其中R1-R5各自独立为氢、任选取代的烃基、惰性官能基,或R1-R3中彼此相邻的任何两个可形成环;Z1与Z2不同,为任选取代的芳基;以及Z2包括任选取代的杂烃基部分,或与金属组合的任选取代的芳基,所述的任选取代的芳基π健配位到金属上。

2.根据权利要求1的配位体,其中Z2为任选取代的芳族杂环部分、任选取代的多芳族杂环部分、任选取代的脂族杂环部分或任选取代的脂族杂烃基部分。
3.根据权利要求1或2的分子式(II)的配位体,其中A1-A6各自独立为碳、氮、氧或硫;

上式的原子基团可任选不存在,以致A1直接键联到A5上;以及R1-R12、R14-R15和R13(如果存在)各自独立为氢、任选取代的烃基和惰性官能基,或R1-R15中彼此相邻的任何两个基团可形成环;前提条件是当A1-A5以及A6(如果存在)都为碳,所述的原子构成环戊二烯基或π键配位金属的芳基部分。

4.根据权利要求3的配位体,其中R1-R3、R7-R9、R12、R14和R13(如果存在)各自独立为氢、任选取代的烃基、惰性官能基,或R1-R3、R7-R9、R12-R14中彼此相邻的任何两个基团可形成环;以及
a)R6为惰性官能基或任选取代的烃基,以及R10、R11和R15各自独立为氢或卤化物;或
b)R11为惰性官能基或任选取代的烃基,以及R6、R10和R15各自独立为氢或卤化物;或
c)R6和R10各自独立为惰性官能基或伯或仲碳原子基团,条件是R6和R10不都为仲碳原子基团以及R11和R15各自独立为氢或卤化物;或
d)R11和R15各自独立为惰性官能基或伯或仲碳原子基团,条件是R11和R15不都为仲碳原子基团以及R6和R10各自独立为氢或卤化物;或
e)R6与R7形成环,R10为伯碳原子基团、惰性官能基或氢,R11和R15各自独立为氢或卤化物;或
f)R11与R12形成环,R15为伯碳原子基团、惰性官能基或氢,R6和R10各自独立为氢或卤化物;或
g)R6和R10分别与R7和R9形成环,R11和R15各自独立为氢或卤化物;或
h)R11和R15分别与R12和R14形成环,R6和R10各自独立为氢或卤化物。
5.一种含有权利要求1或2的配位体的混合双亚胺吡啶MXn配合物,其中M为选自Fe或Co的金属原子,n为2或3,以及X为卤化物、任选取代的烃基、烷氧化物、酰胺或氢化物。
6.一种含有权利要求1或2的配位体的混合[双亚胺吡啶MYp·Ln+][NC-]q配合物,其中Y为一种可插入烯烃的配位体;M为选自Fe或Co的金属原子,NC-为非配位的阴离子,以及p+q为2或3,以便与所述金属原子的氧化态相适合;L为中性路易斯施主分子和n为0、1或2。
7.一种生产α烯烃的方法,所述的方法包括使权利要求5的一种或多种配合物与乙烯和第二种化合物在-100至+300℃的温度下接触,所述的第二种化合物能将任选取代的烃基或氢化物基团转移到选自Fe或Co的金属原子M上,以及它还能从所述金属原子上吸取X-基团。
8.一种生产α烯烃的方法,所述的方法包括权利要求5的一种或多种配合物与乙烯和第二种化合物和第三种化合物在-100至+300℃的温度下接触,所述的第二种化合物能将任选取代的烃基或氢化物基团转移到选自Fe或Co的金属原子M上,以及所述的第三种化合物能从所述金属原子上吸取X-基团。
9.一种生产α烯烃的方法,所述的方法包括使权利要求6的一种或多种配合物与乙烯在-100至+300℃的温度下接触。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01306607 | 2001-08-01 | ||
| EP01306607.1 | 2001-08-01 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1545520A CN1545520A (zh) | 2004-11-10 |
| CN1255419C true CN1255419C (zh) | 2006-05-10 |
Family
ID=8182162
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB028162595A Expired - Fee Related CN1255419C (zh) | 2001-08-01 | 2002-07-29 | 乙烯低聚生产直链α烯烃用配位体及其催化剂体系 |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US7049442B2 (zh) |
| EP (1) | EP1427740B1 (zh) |
| JP (1) | JP2004536154A (zh) |
| CN (1) | CN1255419C (zh) |
| AT (1) | ATE344268T1 (zh) |
| CA (1) | CA2455853A1 (zh) |
| CZ (1) | CZ2004163A3 (zh) |
| DE (1) | DE60215830T2 (zh) |
| ES (1) | ES2272770T3 (zh) |
| RU (1) | RU2292343C2 (zh) |
| WO (1) | WO2003011876A1 (zh) |
| ZA (1) | ZA200400405B (zh) |
Families Citing this family (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6710006B2 (en) | 2000-02-09 | 2004-03-23 | Shell Oil Company | Non-symmetrical ligands and catalyst systems thereof for ethylene oligomerization to linear alpha olefins |
| US7037988B2 (en) * | 2000-10-03 | 2006-05-02 | Shell Oil Company | Process for the co-oligomerisation of ethylene and alpha olefins |
| CZ2004163A3 (cs) * | 2001-08-01 | 2004-09-15 | Shell Internationale Research Maatschappij B.V. | Ligandy a katalytické systémy s jejich obsahem pro oligomeraci ethylenu na lineární alfa-olefiny |
| US8426535B2 (en) * | 2001-08-06 | 2013-04-23 | Ineos Europe Limited | Chain growth reaction process |
| US7087686B2 (en) * | 2001-08-06 | 2006-08-08 | Bp Chemicals Limited | Chain growth reaction process |
| US6911506B2 (en) | 2001-12-10 | 2005-06-28 | Chevron Phillips Chemical Company Lp | Catalyst composition and olefin polymerization using same |
| EP1542797B1 (en) * | 2002-09-25 | 2006-05-31 | Shell Internationale Researchmaatschappij B.V. | Catalyst systems for ethylene oligomerisation to linear alpha olefins |
| KR20050085108A (ko) * | 2002-11-21 | 2005-08-29 | 피나 테크놀러지, 인코포레이티드 | 올레핀 중합을 위한 새로운 촉매 구조 |
| US7045632B2 (en) | 2003-03-04 | 2006-05-16 | Chevron Phillips Chemical Company, Lp | Composition and method for a hexadentate ligand and bimetallic complex for polymerization of olefins |
| US20050014983A1 (en) * | 2003-07-07 | 2005-01-20 | De Boer Eric Johannes Maria | Process for producing linear alpha olefins |
| US7034157B2 (en) * | 2003-10-23 | 2006-04-25 | Fina Technology, Inc. | Catalyst components, process for their preparation and their use as catalyst components in polymerization of olefins |
| US20050187418A1 (en) | 2004-02-19 | 2005-08-25 | Small Brooke L. | Olefin oligomerization |
| US9550841B2 (en) | 2004-02-20 | 2017-01-24 | Chevron Phillips Chemical Company Lp | Methods of preparation of an olefin oligomerization catalyst |
| US7384886B2 (en) | 2004-02-20 | 2008-06-10 | Chevron Phillips Chemical Company Lp | Methods of preparation of an olefin oligomerization catalyst |
| US20070043181A1 (en) | 2005-08-19 | 2007-02-22 | Knudsen Ronald D | Methods of preparation of an olefin oligomerization catalyst |
| US20050187098A1 (en) | 2004-02-20 | 2005-08-25 | Knudsen Ronald D. | Methods of preparation of an olefin oligomerization catalyst |
| ATE388158T1 (de) | 2004-03-24 | 2008-03-15 | Shell Int Research | Übergangsmetallkomplexe |
| AR049714A1 (es) * | 2004-07-13 | 2006-08-30 | Shell Int Research | Proceso de preparacion de alfa olefinas lineales |
| US7456284B2 (en) | 2004-12-10 | 2008-11-25 | Chevron Phillips Chemical Company Lp | Methods for producing a hexadentate bimetallic complex |
| US7129304B1 (en) | 2005-07-21 | 2006-10-31 | Chevron Phillips Chemical Company Lp | Dimine metal complexes, methods of synthesis, and methods of using in oligomerization and polymerization |
| US7727926B2 (en) | 2005-07-21 | 2010-06-01 | Chevron Phillips Chemical Company Lp | Diimine metal complexes, methods of synthesis, and method of using in oligomerization and polymerization |
| US7268096B2 (en) | 2005-07-21 | 2007-09-11 | Chevron Phillips Chemical Company Lp | Diimine metal complexes, methods of synthesis, and methods of using in oligomerization and polymerization |
| US7271121B2 (en) | 2005-07-21 | 2007-09-18 | Chevron Phillips Chemical Company Lp | Diimine metal complexes, methods of synthesis, and methods of using in oligomerization and polymerization |
| SG163603A1 (en) * | 2005-07-21 | 2010-08-30 | Chevron Phillips Chemical Co | Diimine metal complexes, methods of synthesis, and methods of using in oligomerization and polymerization |
| US7378537B2 (en) | 2006-07-25 | 2008-05-27 | Chevron Phillips Chemical Company Lp | Olefin oligomerization catalysts and methods of using same |
| US7902415B2 (en) | 2007-12-21 | 2011-03-08 | Chevron Phillips Chemical Company Lp | Processes for dimerizing or isomerizing olefins |
| FR2926078B1 (fr) | 2008-01-04 | 2009-12-18 | Inst Francais Du Petrole | Nouveaux composes organiques azotes utilisables comme precurseurs de composition catalytique. |
| US7858817B2 (en) * | 2008-03-10 | 2010-12-28 | Exxonmobil Chemical Patents Inc. | Metallocene-substituted pyridyl amines, their metal complexes, and processes for production and use thereof |
| EP2552863A1 (en) | 2010-03-29 | 2013-02-06 | E.I. Du Pont De Nemours And Company | Lubricant component |
| WO2012106694A2 (en) * | 2011-02-05 | 2012-08-09 | Bridgestone Corporation | Lanthanide complex catalyst and polymerization method employing same |
| US9586872B2 (en) | 2011-12-30 | 2017-03-07 | Chevron Phillips Chemical Company Lp | Olefin oligomerization methods |
| US10513473B2 (en) | 2015-09-18 | 2019-12-24 | Chevron Phillips Chemical Company Lp | Ethylene oligomerization/trimerization/tetramerization reactor |
| US10519077B2 (en) | 2015-09-18 | 2019-12-31 | Chevron Phillips Chemical Company Lp | Ethylene oligomerization/trimerization/tetramerization reactor |
| US9944661B2 (en) | 2016-08-09 | 2018-04-17 | Chevron Phillips Chemical Company Lp | Olefin hydroboration |
| US10604457B2 (en) | 2016-12-29 | 2020-03-31 | Chevron Phillips Chemical Company Lp | Ethylene oligomerization processes |
| US10407360B2 (en) | 2017-12-22 | 2019-09-10 | Chevron Phillips Chemical Company Lp | Ethylene oligomerization processes |
| US20180186708A1 (en) | 2016-12-29 | 2018-07-05 | Chevron Phillips Chemical Company Lp | Ethylene Oligomerization Processes |
| RU2672868C1 (ru) * | 2017-12-07 | 2018-11-20 | Федеральное государственное бюджетное учреждение науки Институт катализа им. Г.К. Борескова Сибирского отделения Российской академии наук (ИК СО РАН) | Способ получения производных 2,6-бис[1-(фенилимино)этил]пиридина с электроноакцепторными заместителями |
| RU2729622C1 (ru) * | 2019-12-20 | 2020-08-11 | Федеральное государственное бюджетное учреждение науки Новосибирский институт органической химии им. Н.Н. Ворожцова Сибирского отделения Российской академии наук (НИОХ СО РАН) | Компонент катализатора для полимеризации этилена в высоколинейный полиэтилен, термостабильный катализатор и способ его приготовления |
| AR124325A1 (es) * | 2020-12-15 | 2023-03-15 | Shell Int Research | UN PROCESO PARA PRODUCIR a-OLEFINAS |
| AR124332A1 (es) | 2020-12-15 | 2023-03-15 | Shell Int Research | UN PROCESO PARA PRODUCIR a-OLEFINAS |
Family Cites Families (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU418462A1 (ru) | 1971-08-16 | 1974-03-05 | СПОСОБ ПОЛУЧЕНИЯ СМЕСИ ОЛЕФИНОВ С^—С^1Изобретение относитс к способу получени смеси олефинов С4—Се с преобладанием альфа-изомеров, которые наход т широкое применение в качестве мономеров и промежуточных продуктов при синтезе пластмасс и кау- чуков.Известен способ получени смеси олефииов С4—Сб содимеризацией этилена с пропиленом при температуре 100—160°С и давлении 40 атм в присутствии триалкилалюмини , например триэтилалюмини или диэтилалюми- нийхлорида, и соединени переходного металла, например хлорида никел или его окиси.К недостаткам известного способа относ тс низкий выход альфа-олефинов и недостаточно высока степень конверсии пропилена.С целью повышени выхода альфа-олефинов предлагаетс в качестве соединени переходного .металла использовать пальмитат никел .Пример. 250 мл (180 г) катализатора, состо щего из 3%-ного раствора триэтилалю-5 мини в н-декапе и пальмитата никел (А1 : N1 = 500 : 1), помещают в предварительно освобожденный от влаги и кислорода автоклав емкостью 2 л, снабженный магнитной мешалкой (1400 об/мин). Затем подают 235 г10 пропилена (чистота 99,5%) и 157 г этилена (чистота 99,8%). Мольное соотношение 1:1. Нагревают до нужной температуры и перемешивают 20 час. Вместе с альфа-олефинами С4—Сб образуетс небольшое количество аль-15 фа-олефинов Cs—Сэ | |
| US4728583A (en) * | 1984-08-31 | 1988-03-01 | Fuji Photo Film Co., Ltd. | Radiation image storage panel and process for the preparation of the same |
| DE3762689D1 (de) * | 1986-01-21 | 1990-06-13 | Fuji Photo Film Co Ltd | Strahlungsbildspeicherplatte. |
| EP0308728B1 (de) | 1987-09-21 | 1991-06-05 | Siemens Aktiengesellschaft | Verfahren zum Betreiben eines Durchlaufdampferzeugers |
| US4822911A (en) * | 1987-09-25 | 1989-04-18 | Shell Oil Company | Process for the selective separation of vinylidene olefins from mixtures with other olefins |
| US4912333A (en) * | 1988-09-12 | 1990-03-27 | Eastman Kodak Company | X-ray intensifying screen permitting an improved relationship of imaging speed to sharpness |
| JPH042999A (ja) * | 1990-04-20 | 1992-01-07 | Fuji Photo Film Co Ltd | 放射線像変換パネル |
| ES2085000T3 (es) * | 1990-12-27 | 1996-05-16 | Exxon Chemical Patents Inc | Un compuesto de amido-metal de transicion y un sistema catalitico para la produccion de polipropileno isotactico. |
| JP3115345B2 (ja) | 1991-04-26 | 2000-12-04 | 三菱化学株式会社 | オレフィン重合体の製造法 |
| EP0813447B1 (en) * | 1995-03-08 | 1998-10-21 | Shell Internationale Researchmaatschappij B.V. | Bridged bis-amido group 4 metal compounds in a catalyst composition |
| JP3479574B2 (ja) * | 1995-07-04 | 2003-12-15 | 富士写真フイルム株式会社 | フロント側用放射線増感スクリーン及び放射線増感スクリーン組体 |
| US5830629A (en) * | 1995-11-01 | 1998-11-03 | Eastman Kodak Company | Autoradiography assemblage using transparent screen |
| DE19547210A1 (de) * | 1995-12-18 | 1997-06-19 | Basf Ag | 1,7-Disubstituierte Perylen-3,4,9-10-tetracarbonsäuren, deren Dianhydride und Diimide |
| KR100469185B1 (ko) * | 1996-07-23 | 2005-01-31 | 이.아이,듀우판드네모아앤드캄파니 | 올레핀의 중합 방법 |
| DE69728677T2 (de) | 1996-09-06 | 2005-03-31 | Hyundai Pertrochemical Co., Ltd. | Katalysatorsystem für die (co)polymerisierung von olefinen und verfahren zur herstellung von olefin-(co)polymeren unter verwendung des katalysatorsystems |
| US6432862B1 (en) * | 1996-12-17 | 2002-08-13 | E. I. Du Pont De Nemours And Company | Cobalt catalysts for the polymerization of olefins |
| IL129929A0 (en) * | 1996-12-17 | 2000-02-29 | Du Pont | Polymerization of ethylene with specific iron or cobalt complexes novel pyridinebis (imines) and novel complexes of pyridinebis(imines) with iron and cobalt |
| US6417305B2 (en) * | 1996-12-17 | 2002-07-09 | E. I. Du Pont De Nemours And Company | Oligomerization of ethylene |
| US6214761B1 (en) * | 1996-12-17 | 2001-04-10 | E. I. Du Pont De Nemours And Company | Iron catalyst for the polymerization of olefins |
| US6423848B2 (en) * | 1996-12-17 | 2002-07-23 | E. I. Du Pont De Nemours And Company | Tridentate ligand |
| AU6018198A (en) * | 1997-01-13 | 1998-08-03 | E.I. Du Pont De Nemours And Company | Polymerization of propylene |
| US5905014A (en) * | 1997-03-19 | 1999-05-18 | Agfa-Gevaert, N.V. | Radiation image storage panel comprising a colorant |
| US6103946A (en) * | 1997-07-15 | 2000-08-15 | E. I. Du Pont De Nemours And Company | Manufacture of α-olefins |
| US6740715B2 (en) * | 1997-07-15 | 2004-05-25 | E. I. Du Pont De Nemours And Company | Manufacture of alpha-olefins |
| SK2712000A3 (en) * | 1997-09-05 | 2001-07-10 | Bp Chem Int Ltd | Polymerisation catalysts |
| US6559252B1 (en) * | 1997-10-29 | 2003-05-06 | Basell Technology Company Bv | Catalysts and processes for the polymerization of olefins |
| JP4467181B2 (ja) * | 1998-03-12 | 2010-05-26 | イネオス ユーロープ リミテッド | 重合触媒 |
| ID27209A (id) * | 1998-03-12 | 2001-03-08 | Bp Chem Int Ltd | Katalis-katalis polimerisasi |
| ATE244268T1 (de) * | 1998-03-12 | 2003-07-15 | Bp Chem Int Ltd | Äthylen-homopolymer |
| US6232259B1 (en) * | 1998-03-31 | 2001-05-15 | E. I. Du Pont De Nemours And Company | Preparation of transition metal imine complexes |
| US6063881A (en) | 1998-04-02 | 2000-05-16 | E. I. Du Pont De Nemours And Company | Oligomerization of propylene |
| EP0952165A1 (en) * | 1998-04-24 | 1999-10-27 | Fina Research S.A. | Production of polyethylene having improved mechanical properties |
| CN1203098C (zh) | 1998-05-29 | 2005-05-25 | 纳幕尔杜邦公司 | 烯烃的共聚合 |
| GB9817004D0 (en) * | 1998-08-06 | 1998-09-30 | Bp Chem Int Ltd | Preparation of polymerisation catalysts |
| GB9819847D0 (en) * | 1998-09-12 | 1998-11-04 | Bp Chem Int Ltd | Novel compounds |
| ATE230410T1 (de) * | 1998-10-02 | 2003-01-15 | Bp Chem Int Ltd | Polymerisationskatalysatoren |
| AU6354399A (en) * | 1998-10-26 | 2000-05-15 | Bp Chemicals Limited | Polymerisation catalysts |
| AU1169200A (en) * | 1998-11-30 | 2000-06-19 | Bp Chemicals Limited | Polymerisation process |
| TWI226337B (en) * | 1998-12-02 | 2005-01-11 | Idemitsu Petrochemical Co | Catalyst for alkene polymerization and the alkene polymerization method by using this catalyst |
| WO2000035974A1 (en) * | 1998-12-15 | 2000-06-22 | Basell Technology Company B.V. | Catalyst system for olefin polymerization |
| DE60018870T2 (de) * | 1999-01-22 | 2006-04-13 | Mitsui Chemicals, Inc. | Verfahren zur herstellung eines olefinpolymers und olefinpolymer |
| US6545108B1 (en) * | 1999-02-22 | 2003-04-08 | Eastman Chemical Company | Catalysts containing N-pyrrolyl substituted nitrogen donors |
| US6559091B1 (en) * | 1999-02-22 | 2003-05-06 | Eastman Chemical Company | Catalysts containing N-pyrrolyl substituted nitrogen donors |
| GB9906296D0 (en) * | 1999-03-18 | 1999-05-12 | Bp Chem Int Ltd | Polymerisation catalysts |
| DE19931873A1 (de) * | 1999-04-14 | 2000-10-19 | Bayer Ag | Katalysatorsystem zur Olefinpolymerisation |
| US6365690B1 (en) * | 1999-05-06 | 2002-04-02 | E. I. Du Pont De Nemours And Company | Polymerization of ethylene |
| US6825297B1 (en) * | 1999-05-14 | 2004-11-30 | The Dow Chemical Company | Transition metal complexes and olefin polymerization process |
| US6479601B1 (en) * | 1999-08-06 | 2002-11-12 | The Goodyear Tire & Rubber Company | Transition metal catalysts for diene polymerizations |
| US6458905B1 (en) * | 1999-08-16 | 2002-10-01 | Phillips Petroleum Company | Complexes of pyridldiimines with vanadium and other transition metals, and their use as olefin oligomerization and polymerization catalysts |
| US6441117B1 (en) * | 1999-09-01 | 2002-08-27 | E. I. Du Pont De Nemours And Company | High density polyethylene packaging |
| JP4674023B2 (ja) | 1999-09-02 | 2011-04-20 | 出光興産株式会社 | 遷移金属化合物、α−オレフィン製造用触媒及びα−オレフィンの製造方法 |
| US6407188B1 (en) * | 1999-09-29 | 2002-06-18 | E. I. Du Pont De Nemours And Company | Polymerization of olefins |
| JP4659322B2 (ja) | 1999-11-11 | 2011-03-30 | 出光興産株式会社 | 遷移金属化合物、オレフィン重合触媒及びオレフィンの重合方法 |
| EP2267044B1 (en) * | 2000-01-26 | 2015-07-08 | Mitsui Chemicals, Inc. | Olefin polymer of very low polydispersity and process for preparing the same |
| US6710006B2 (en) * | 2000-02-09 | 2004-03-23 | Shell Oil Company | Non-symmetrical ligands and catalyst systems thereof for ethylene oligomerization to linear alpha olefins |
| TW572969B (en) | 2000-02-10 | 2004-01-21 | Hayashibara Biochem Lab | Trimethine cyanine dye, light absorbent, light-resistant improver and optical recording medium containing same, and process for producing same |
| US6605677B2 (en) * | 2000-02-18 | 2003-08-12 | Eastman Chemical Company | Olefin polymerization processes using supported catalysts |
| GC0000291A (en) * | 2000-06-30 | 2006-11-01 | Shell Int Research | Ligands and catalyst systems thereof for ethylene oligomerisation to linear alpha olefins |
| US6534691B2 (en) * | 2000-07-18 | 2003-03-18 | E. I. Du Pont De Nemours And Company | Manufacturing process for α-olefins |
| US6555723B2 (en) * | 2000-07-18 | 2003-04-29 | E. I. Du Pont De Nemours And Company | Continuous manufacturing process for alpha-olefins |
| US7037988B2 (en) * | 2000-10-03 | 2006-05-02 | Shell Oil Company | Process for the co-oligomerisation of ethylene and alpha olefins |
| US6706891B2 (en) * | 2000-11-06 | 2004-03-16 | Eastman Chemical Company | Process for the preparation of ligands for olefin polymerization catalysts |
| US6521329B2 (en) * | 2001-06-18 | 2003-02-18 | Eastman Kodak Company | Radiographic phosphor panel having reflective polymeric supports |
| CA2451121A1 (en) * | 2001-06-20 | 2003-01-03 | Shell Internationale Research Maatschappij B.V. | Process for the preparation of oligomers |
| CZ2004163A3 (cs) * | 2001-08-01 | 2004-09-15 | Shell Internationale Research Maatschappij B.V. | Ligandy a katalytické systémy s jejich obsahem pro oligomeraci ethylenu na lineární alfa-olefiny |
| EP1542797B1 (en) * | 2002-09-25 | 2006-05-31 | Shell Internationale Researchmaatschappij B.V. | Catalyst systems for ethylene oligomerisation to linear alpha olefins |
| US20050014983A1 (en) * | 2003-07-07 | 2005-01-20 | De Boer Eric Johannes Maria | Process for producing linear alpha olefins |
-
2002
- 2002-07-29 CZ CZ2004163A patent/CZ2004163A3/cs unknown
- 2002-07-29 ES ES02767309T patent/ES2272770T3/es not_active Expired - Lifetime
- 2002-07-29 JP JP2003517067A patent/JP2004536154A/ja active Pending
- 2002-07-29 DE DE60215830T patent/DE60215830T2/de not_active Expired - Fee Related
- 2002-07-29 CN CNB028162595A patent/CN1255419C/zh not_active Expired - Fee Related
- 2002-07-29 AT AT02767309T patent/ATE344268T1/de not_active IP Right Cessation
- 2002-07-29 RU RU2004105958/04A patent/RU2292343C2/ru not_active IP Right Cessation
- 2002-07-29 CA CA002455853A patent/CA2455853A1/en not_active Abandoned
- 2002-07-29 EP EP02767309A patent/EP1427740B1/en not_active Expired - Lifetime
- 2002-07-29 WO PCT/EP2002/008636 patent/WO2003011876A1/en active IP Right Grant
- 2002-07-30 US US10/208,535 patent/US7049442B2/en not_active Expired - Fee Related
-
2004
- 2004-01-20 ZA ZA200400405A patent/ZA200400405B/en unknown
-
2005
- 2005-03-15 US US11/080,170 patent/US7304159B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| US20030119921A1 (en) | 2003-06-26 |
| CZ2004163A3 (cs) | 2004-09-15 |
| ZA200400405B (en) | 2004-10-19 |
| US7049442B2 (en) | 2006-05-23 |
| DE60215830T2 (de) | 2007-06-06 |
| CA2455853A1 (en) | 2003-02-13 |
| DE60215830D1 (de) | 2006-12-14 |
| ES2272770T3 (es) | 2007-05-01 |
| US20050159601A1 (en) | 2005-07-21 |
| RU2292343C2 (ru) | 2007-01-27 |
| US7304159B2 (en) | 2007-12-04 |
| CN1545520A (zh) | 2004-11-10 |
| ATE344268T1 (de) | 2006-11-15 |
| WO2003011876A1 (en) | 2003-02-13 |
| EP1427740B1 (en) | 2006-11-02 |
| RU2004105958A (ru) | 2005-03-27 |
| JP2004536154A (ja) | 2004-12-02 |
| EP1427740A1 (en) | 2004-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1255419C (zh) | 乙烯低聚生产直链α烯烃用配位体及其催化剂体系 | |
| CN1231487C (zh) | 金属茂和烯烃聚合催化剂 | |
| CN100340568C (zh) | 茂金属一卤化物 | |
| CN1178916C (zh) | 乙烯低聚生成线型α烯烃的配体及其催化剂体系 | |
| CN1208357C (zh) | 烯烃聚合催化剂组分和催化剂体系和使用这类催化剂体系的聚合方法 | |
| CN1050135C (zh) | 烯烃聚合催化剂和烯烃聚合方法 | |
| CN1116314C (zh) | 烯烃聚合的催化剂及方法 | |
| CN1110467C (zh) | α-烯烃的制造 | |
| CN1240455A (zh) | 用铁或钴配合物进行的乙烯聚合反应、新的吡啶双(亚胺)、以及吡啶双(亚胺)与铁钴的新配合物 | |
| CN1098110A (zh) | 烯烃聚合催化体的固体组分及其制备和该催化剂及其应用 | |
| CN1432027A (zh) | 烯烃的聚合 | |
| CN1784431A (zh) | 高活性烯烃聚合催化剂和方法 | |
| CN1270595A (zh) | 改性铝氧烷催化剂活化剂 | |
| CN1492847A (zh) | 用于烯烃三聚反应的催化剂体系 | |
| CN1832970A (zh) | 用于烯烃聚合的催化剂体系 | |
| CN1468205A (zh) | 乙烯与α-烯烃的共低聚方法 | |
| CN1202909A (zh) | 含有束缚阳离子生成活化剂的负载型催化剂 | |
| CN1127524C (zh) | 载体上的催化剂组分、载体上的催化剂、它们的制备方法和加聚方法 | |
| CN1934121A (zh) | 过渡金属配合物 | |
| CN1589284A (zh) | 用于烯烃聚合的亚氨基-氨基催化剂组合物 | |
| CN1168745C (zh) | 烯烃聚合方法 | |
| CN1144808A (zh) | 过渡金属化合物 | |
| CN1268951A (zh) | 茂金属,配位体和烯烃聚合方法 | |
| CN1261221C (zh) | 催化剂负载方法及用负载催化剂的聚合 | |
| CN1518556A (zh) | 非金属茂,其制备方法及其在烯烃聚合中的应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20060510 Termination date: 20100729 |