CN103642882A - 二肽的制造方法 - Google Patents
二肽的制造方法 Download PDFInfo
- Publication number
- CN103642882A CN103642882A CN201310593697.3A CN201310593697A CN103642882A CN 103642882 A CN103642882 A CN 103642882A CN 201310593697 A CN201310593697 A CN 201310593697A CN 103642882 A CN103642882 A CN 103642882A
- Authority
- CN
- China
- Prior art keywords
- gene
- dna
- strain
- amino acid
- microorganism
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/93—Ligases (6)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06026—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atom, i.e. Gly or Ala
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/0606—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing heteroatoms not provided for by C07K5/06086 - C07K5/06139, e.g. Ser, Met, Cys, Thr
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/0606—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing heteroatoms not provided for by C07K5/06086 - C07K5/06139, e.g. Ser, Met, Cys, Thr
- C07K5/06069—Ser-amino acid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
根据本发明,可以提供二肽的制造方法,其特征是将具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力、而且具有生产该1种以上的氨基酸中的至少1种氨基酸能力的微生物在培养基中进行培养,使二肽该培养基中生成、蓄积,从该培养基回收二肽。
Description
本申请是2005年6月24日提交的200580020690.6号发明专利申请“二肽的制造方法”的分案申请。
【技术领域】
本发明涉及二肽的制造方法,其特征是将具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸能力的微生物在培养基中进行培养,使二肽在培养基中生成、蓄积,从该培养基回收二肽。
【背景技术】
目前,很多氨基酸都是通过所谓的发酵法制造的(参照非专利文献1和2)。这里所谓的发酵法指的是在由葡萄糖、醋酸、甲醇、氨、硫酸铵、玉米浸渍液等便宜的物质构成的培养基中培养微生物,利用微生物的代谢活性获得目的氨基酸的方法。作为由便宜的原料通过环境负荷小的方法制造氨基酸的方法,发酵法是很好的制造方法。
有关肽的大量合成法已知有化学合成法(液相法、固相法)、酶促合成法和使用DNA重组法的生物合成法。现在,对于50残基以上的长链肽使用酶促合成法或生物合成法,对于二肽主要使用化学合成法和酶促合成法。
使用化学合成法合成二肽需要对官能团进行保护和去保护等操作,由于也合成消旋体,化学合成法不能说是经济、有效的方法。另外由于化学合成法使用大量的有机溶剂等,从环境卫生方面看也不是一个好的方法。
通过酶法合成二肽已知有利用蛋白分解酶(蛋白水解酶)的逆反应的方法(参照非专利文献3)、利用耐热性氨酰t-RNA合成酶的方法(参照专利文献1~4)、利用脯氨酸亚氨基肽酶的逆反应的方法(参照专利文献5)、利用非核糖体肽合成酶(以下称为NRPS)的方法(参照非专利文献4、5和专利文献6、7)。
然而,就利用蛋白水解酶的逆反应的方法来说,需要对底物氨基酸的官能团进行保护和去保护,存在着提高肽形成反应效率和阻止肽分解反应困难的问题。至于利用耐热性氨酰t-RNA合成酶的方法,存在着酶的表达、阻止目的产物以外的副生物反应困难的问题。而利用脯氨酸亚氨基肽酶的方法存在着需要作为底物一方的氨基酸酰胺化的缺点。关于利用NRPS的方法需要供给作为辅酶的4’-磷酸泛酰巯基乙胺(4'-phosphopantetheine),不能说是有效的制造方法。
除了这些缺点外,这些方法无论那一种由于作为底物都使用氨基酸或它的衍生物,具有制造成本增加的缺点。
另外,已知酶分子量比NRPS小、不需要作为辅酶的4’-磷酸泛酰巯基乙胺(4'-phosphopantetheine)的γ-谷氨酰半胱氨酸合成酶(γ-glutamylcysteine synthetase)、谷胱甘肽合成酶(glutathionesynthetase)、D-丙氨酰-D-丙氨酸(D-Ala-D-Ala)连接酶(D-Ala-D-Alaligase)、聚-γ-谷氨酸合成酶(poly-γ-glutamate synthetase)等一组肽合成酶。几乎所有这些酶由于都具有使用D-氨基酸作为底物催化在γ位羧基形成肽键等特征,所以不能用于在L-氨基酸的α位羧基形成肽键的二肽的合成。
另外,据报道在已知作为抗生物质アルボノルシン(albonoursin)的生产株的诺尔斯氏链霉菌(Streptomyces noursei)ATCC11455株中,与NRPS酶完全不相似的蛋白质(albC基因产物)负责环(L-苯丙氨酰-L-亮氨酸)[环(L-phenylalanyl-L-leucine)]结构的合成,如果使环二肽氧化酶作用于导入了albC基因的大肠杆菌(Escherichia coli)和浅青紫链霉菌(Streptomyces lividans)的培养液,可以检测到albonoursin(参照非专利文献6),但还没有albC基因产物生成直链状二肽的报告。
已知通过在L-氨基酸的α位羧基形成肽键活性生成二肽只有来自属于芽孢杆菌属的微生物的作为二肽抗生物质的杆菌溶素合成酶。已知杆菌溶素合成酶具有合成杆菌溶素(L-丙氨酰-L-anticapsin、L-Ala-L-anticapsin)和L-丙氨酰-L-丙氨酸(L-Ala-L-Ala)的活性,至于其它的二肽的合成活性还不知道(参照非专利文献7和8)。
另外,有关全基因组情报已阐明的枯草芽孢杆菌(Bacillus subtilis)168株(参照非专利文献9)中的杆菌溶素生物合成酶基因组,已知如果对含有ywfA~F的ORF的杆菌溶素操纵子进行扩增,可增加杆菌溶素的产率(参照专利文献8)。然而,在这些ORF中还不知道是否含有编码具有通过肽键连接2种以上的氨基酸的活性的蛋白质的ORF,如果含有,哪一个ORF编码该蛋白质也不清楚。
即,到目前为止也不了解通过发酵生产制造由1种以上的氨基酸构成的二肽的方法。
【非专利文献1】氨基酸发酵、学会出版中心、相田浩等(1986)
【非专利文献2】Biotechnology2nd ed.,Vol.6,Products ofPrimary Metabolism,VCH Verlagsgesellschaft mbH,Weinheim(1996)
【非专利文献3】J.Biol.Chem.,119,707-720(1937)
【非专利文献4】Chem.Biol.,7,373-384(2000)
【非专利文献5】FEBS Lett.,498,42-45(2001)
【非专利文献6】Chemistry&Biol.,9,1355-1364(2002)
【非专利文献7】J.Ind.Microbiol.,2,201-208(1987)
【非专利文献8】Enzyme.Microbial.Technol.,29,400-406(2001)
【非专利文献9】Nature,390,249-256(1997)
【专利文献1】特开昭58-146539号公报
【专利文献2】特开昭58-209991号公报
【专利文献3】特开昭58-209992号公报
【专利文献4】特开昭59-106298号公报
【专利文献5】国际公开专利第03-010307号小册子
【专利文献6】美国专利第5795738号
【专利文献7】美国专利第5652116号
【专利文献8】国际公开专利第00-03009号小册子
【发明内容】
【发明要解决的课题】
本发明的目的在于提供供二肽的制造方法,其特征是将具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力,而且具有生产、蓄积该1种以上的氨基酸中的至少1种氨基酸的能力的微生物在培养基中进行培养,使二肽该培养基中生成、蓄积,从该培养基回收二肽。
【用于解决课题的手段】
本发明涉及到以下(1)~(15)内容。
(1)二肽的制造方法,其特征是将具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸的能力的微生物在培养基中进行培养,使二肽在该培养基中生成、蓄积,从该培养基回收二肽
(2)上述(1)所述的制造方法,其中具有由1种以上的氨基酸生成二肽活性的蛋白质是选自以下[1]~[11]中的蛋白质:
[1]具有序列号1~8中的任一个表示的氨基酸序列的蛋白质
[2]由在序列号1~8中的任一个表示的氨基酸序列缺失、置换或附加1个以上的氨基酸的氨基酸序列构成、而且具有由1种以上的氨基酸生成二肽活性的蛋白质
[3]由与序列号1~8中的任一个表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽活性的蛋白质
[4]具有与序列号17表示的氨基酸序列有80%以上同源性的氨基酸序列,而且具有由1种以上的氨基酸生成二肽活性的蛋白质
[5]具有由序列号37或38表示的氨基酸序列的蛋白质
[6]由在序列号37或38表示的氨基酸序列中缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽活性的蛋白质
[7]由与序列号37或38表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽活性的蛋白质
[8]具有非核糖体肽合成酶(以下称为NRPS)活性的蛋白质
[9]具有序列号43表示的氨基酸序列的蛋白质
[10]由在序列号43表示的氨基酸序列中缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽活性的蛋白质
[11]由与序列号43表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽活性的蛋白质。
(3)上述(1)或(2)所述的制造方法,其中具有由1种以上的氨基酸生成二肽活性的蛋白质是由选自以下[1]~[8]中的DNA编码的蛋白质:
[1]具有序列号9~16和36中的任一个表示的碱基序列的DNA
[2]和具有与序列号9~16和36中的任一个表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA
[3]和具有与序列号18表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA
[4]具有序列号39或40表示的碱基序列的DNA
[5]和具有与序列号39或40表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA
[6]编码具有NRPS活性的蛋白质的DNA
[7]具有序列号44表示的碱基序列的DNA
[8]和具有与序列号44表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA。
(4)上述(1)所述的制造方法,其中具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力的微生物是带有重组DNA的微生物,该重组DNA含有上述(3)的[1]~[8]中任一个DNA。
(5)上述(1)~(4)中的任一个制造方法,其中生产氨基酸的能力是通过选自以下[1]~[5]中的方法获得的:
[1]缓和或解除至少一个控制该氨基酸生物合成的结构的方法
[2]对至少一个参与该氨基酸生物合成的酶表达增强的方法
[3]使至少一个参与该氨基酸生物合成的酶基因的拷贝数增加的方法
[4]对至少一个由该氨基酸生物合成途径到该氨基酸以外的代谢产物旁路的代谢途径进行弱化或阻断的方法
[5]与野生型株相比,选择对该氨基酸的类似物抗性高的细胞株的方法。
(6)上述(1)~(5)中的任一个制造方法,其中微生物是属于埃希氏菌属、棒杆菌属、芽孢杆菌属、沙雷氏菌属、假单孢菌属或链霉菌属的微生物。
(7)上述(6)所述的制造方法,其中属于埃希氏菌属、棒杆菌属、芽孢杆菌属、沙雷氏菌属、假单孢菌属或链霉菌属的微生物是大肠杆菌、谷氨酸棒杆菌、产氨棒杆菌、乳酸发酵棒杆菌、黄色棒杆菌、Corynebacterium efficiens、枯草芽孢杆菌、巨大芽孢杆菌、粘质沙雷氏菌、恶臭假单胞菌、铜绿假单胞菌、天蓝色链霉菌或浅青紫链霉菌。
(8)上述(1)~(5)中的任一个制造方法,其中微生物是1种以上的肽酶和具有1种以上肽转运活性的蛋白质(以下也称为肽转运蛋白质)的活性低下或丧失的微生物。
(9)上述(1)~(5)中的任一个制造方法,其中微生物是3种以上肽酶的活性低下或丧失的微生物。
(10)上述(8)或(9)所述的制造方法,其中肽酶是具有序列号45~48中的任一个表示的氨基酸序列的蛋白质、或具有与序列号45~48中的任一个表示的氨基酸序列有80%以上同源性的氨基酸序列,而且具有肽酶活性的蛋白质。
(11)上述(8)或(10)所述的制造方法,其中肽转运蛋白质是具有序列号49~53中的任一个表示的氨基酸序列的蛋白质、或由与序列号49~53中的任一个表示的氨基酸序列有80%以上同源性的氨基酸序列构成的蛋白质,而且具有肽转运活性的蛋白质。
(12)上述(8)~(11)中的任一个制造方法,其中微生物是属于埃希氏菌属、芽孢杆菌属或棒杆菌属的微生物。
(13)上述(12)所述的制造方法,其中属于埃希氏菌属、芽孢杆菌属或棒杆菌属的微生物是大肠杆菌、谷氨酸棒杆菌、产氨棒杆菌、乳酸发酵棒杆菌、黄色棒杆菌、Corynebacterium efficiens、枯草芽孢杆菌或巨大芽孢杆菌。
(14)上述(1)~(13)中的任一个制造方法,其中氨基酸是选自L-丙氨酸、L-谷氨酰胺、L-谷氨酸、甘氨酸、L-缬氨酸、L-亮氨酸、L-异亮氨酸、L-脯氨酸、L-苯丙氨酸、L-色氨酸、L-蛋氨酸、L-丝氨酸、L-苏氨酸、L-半胱氨酸、L-天冬酰胺、L-酪氨酸、L-赖氨酸、L-精氨酸、L-组氨酸、L-天冬氨酸、L-α-氨基丁酸、L-4-羟基脯氨酸、L-3-羟基脯氨酸、L-鸟氨酸和L-瓜氨酸的氨基酸。
(15)上述(1)~(14)中的任一个制造方法,其中二肽是式(I)表示的二肽;
R1-R2(I)
(式中,R1和R2可以相同或不同,表示选自L-丙氨酸、L-谷氨酰胺、L-谷氨酸、甘氨酸、L-缬氨酸、L-亮氨酸、L-异亮氨酸、L-脯氨酸、L-苯丙氨酸、L-色氨酸、L-蛋氨酸、L-丝氨酸、L-苏氨酸、L-半胱氨酸、L-天冬酰胺、L-酪氨酸、L-赖氨酸、L-精氨酸、L-组氨酸、L-天冬氨酸、L-α-氨基丁酸、L-4-羟基脯氨酸、L-3-羟基脯氨酸、L-鸟氨酸和L-瓜氨酸的氨基酸)。
【发明的效果】
根据本发明可以提供二肽的制造方法,其特征是将具有生产具有由1种以上的氨基酸生成二肽的活性的蛋白质的能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸能力的微生物在培养基中进行培养,使二肽在培养基中生成、蓄积,从该培养基回收二肽。
【附图说明】
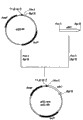
【图1】图1是表示质粒pPE43的构建过程的图。
【图2】图2是表示质粒pQE60ywfE的构建过程的图。
【图3】图3是表示作为具有直链二肽的合成活性的蛋白质的表达质粒载体pAL-nou和pAL-alb的构建过程的图。
【图4】图4是表示作为ywfE基因表达强化型载体的pPE56的构建过程的图。
【图5】图5是表示作为ywfE基因和ald基因表达载体的pPE86的构建过程的图。
【图6】图6是表示作为抗反馈型pheA基因表达载体的pPHEA2、以及抗反馈型pheA基因和抗反馈型aroF基因表达质粒载体的pPHEAF2的构建过程的图。
【符号说明】
ywfE:来自枯草芽孢杆菌168株的ywfE基因
Ptrp:色氨酸启动子基因
PT5:T5启动子
Ampr:氨苄青霉素抗性基因
lacI q:乳糖阻遏蛋白基因
albC:albC基因或albC类似基因
ald:ald基因
pheA fbr :抗反馈型pheA基因
aroF fbr :抗反馈型aroF基因
【具体实施方式】
本发明制造方法中使用的具有由1种以上的氨基酸生成二肽的活性的蛋白质只要是具有由1种以上的氨基酸生成同一或不同氨基酸通过肽键生成二肽的活性的蛋白质,可以是任一种蛋白质,作为该蛋白质,例如、
[1]具有序列号1~8中任一个序列表示的氨基酸序列的蛋白质
[2]由在序列号1~8中的任一个表示的氨基酸序列缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[3]由与序列号1~8中的任一个表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[4]具有与序列号17表示的氨基酸序列有80%以上同源性的氨基酸序列,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[5]具有由序列号37或38表示的氨基酸序列的蛋白质
[6]由在序列号37或38表示的氨基酸序列中缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[7]由与序列号37或38表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[8]具有非核糖体肽合成酶(以下称为NRPS)活性的蛋白质
[9]具有序列号43表示的氨基酸序列的蛋白质
[10]由在序列号43表示的氨基酸序列中缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[11]由与序列号43表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质。
本发明中所说的氨基酸,是下面本发明制造方法中使用的微生物生产的氨基酸,优选L-氨基酸和甘氨酸,更优选L-丙氨酸、L-谷氨酰胺、L-谷氨酸、L-缬氨酸、L-亮氨酸、L-异亮氨酸、L-脯氨酸、L-苯丙氨酸、L-色氨酸、L-蛋氨酸、L-丝氨酸、L-苏氨酸、L-半胱氨酸、L-天冬酰胺、L-酪氨酸、L-赖氨酸、L-精氨酸、L-组氨酸、L-天冬氨酸、L-α-氨基丁酸、L-4-羟基脯氨酸、L-3-羟基脯氨酸、L-鸟氨酸、L-瓜氨酸和甘氨酸,再优选L-丙氨酸、L-谷氨酰胺、L-谷氨酸、L-缬氨酸、L-亮氨酸、L-异亮氨酸、L-脯氨酸、L-苯丙氨酸、L-色氨酸、L-蛋氨酸、L-丝氨酸、L-苏氨酸、L-半胱氨酸、L-天冬酰胺、L-酪氨酸、L-赖氨酸、L-精氨酸、L-组氨酸、L-天冬氨酸、L-α-氨基丁酸和甘氨酸。
在上述中,由缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质可以通过使用Molecular Cloning,A Laboratory Manual,Third Edition,Cold Spring Harbor Laboratory Press(2001)(以下、略为MolecularCloning第3版)、Current Protocols in Molecular Biology,John Wiley&Sons(1987-1997)(以下、略为Current Protocols in MolecularBiology)、Nucleic Acids Research,10,6487(1982)、Proc.Natl.Acad.Sci.USA,79,6409(1982)、Gene,34,315(1985)、Nucleic AcidsResearch,13,4431(1985)、Proc.Natl.Acad.Sci.USA,82,488(1985)等所述的定点突变导入法,例如通过在编码序列号1~8、37、38或43中的任一个表示的氨基酸序列构成的蛋白质的DNA中导入定点突变,可以取得。
缺失、置换或附加的氨基酸的数目没有特别限定,是通过上述的定点突变法等众所周知的方法能够缺失、置换或附加程度的数目,从1个至数十个、优选1~20个、更优选1~10个、再优选1~5个。
所谓在序列号1~8、37、38或43中的任一个表示的氨基酸序列缺失、置换或附加1个以上的氨基酸可以在同一序列中的任意位置缺失、置换或附加1个或多个氨基酸。
作为可置换的氨基酸,例如对序列号1~8、序列号37和38、或众所周知的NRPS与序列号43表示的氨基酸序列分别使用众所周知的序列比对软件进行比较时,在比较的所有的氨基酸序列中不保守的氨基酸。作为众所周知的序列比对软件,例如基因解析软件Genetyx(软件开发株式会社)中含有的序列比对解析软件。作为该解析软件的解析参数,可以使用default值。
另外作为可进行氨基酸缺失或附加的氨基酸位置,例如序列号1~8、37、38和43中的任一个表示的氨基酸序列的N末端侧和C末端侧。
缺失、置换或附加可以同时出现,被置换或附加的氨基酸无论天然型,还是非天然型都可以。作为天然型氨基酸,如L-丙氨酸、L-天冬酰胺、L-天冬氨酸、L-谷氨酰胺、L-谷氨酸、甘氨酸、L-精氨酸、L-组氨酸、L-异亮氨酸、L-亮氨酸、L-赖氨酸、L-蛋氨酸、L-苯丙氨酸、L-脯氨酸、L-丝氨酸、L-苏氨酸、L-色氨酸、L-酪氨酸、L-缬氨酸、L-半胱氨酸等。
以下给出了可相互置换的氨基酸的例子。同一组中含有的氨基酸可相互置换。
A群:亮氨酸、异亮氨酸、正亮氨酸、缬氨酸、正缬氨酸、丙氨酸、2-氨基丁酸、蛋氨酸、O-甲基丝氨酸、t-丁基甘氨酸、t-丁基丙氨酸、环己基丙氨酸
B群:天冬氨酸、谷氨酸、异天冬氨酸、异谷氨酸、2-氨基己二酸、2-氨基辛二酸
C群:天冬酰胺、谷氨酰胺
D群:赖氨酸、精氨酸、鸟氨酸、2,4-二氨基丁酸、2,3-二氨基丙酸
E群:脯氨酸、3-羟基脯氨酸、4-羟基脯氨酸
F群:丝氨酸、苏氨酸、同型丝氨酸
G群:苯丙氨酸、酪氨酸
另外,为了使本发明的蛋白质具有由1种以上的氨基酸生成二肽的活性,希望是序列号1~8、37、38和43中的任一个表示的氨基酸序列,优选与序列号1表示的氨基酸序列的同源性具有65%以上、优选75%以上、更优选85%以上、再优选90%以上、特别优选95%以上、最好优选98%以上的同源性。
氨基酸序列或碱基序列的同源性可以使用Karlin and Altschul的算法BLAST[Pro.Natl.Acad.Sci.USA,90,5873(1993)]或FASTA[Methods Enzymol.,183,63(1990)]决定。通过根据这个算法BLAST开发了称为BLASTN和BLASTX的程序[J.Mol.Biol.,215,403(1990)]。当根据BLAST通过BLASTN对碱基序列进行解析时,参数定为例如Score=100、wordlength=12。另外当根据BLAST通过BLASTX对氨基酸序列进行解析时,参数定为例如score=50、wordlength=3。当使用BLAST和Gapped BLAST程序时,使用各程序的default参数。这些解析方法的具体手法众所周知(http://www.ncbi.nlm.nih.gov.)。
序列号17表示的氨基酸序列是被保存在具有序列号1~7表示的氨基酸序列的蛋白质间的区域,而且是对应于各种微生物的具有Ala-Ala连接酶活性的蛋白质公有序列的区域。
作为具有与序列号17表示的氨基酸序列有80%以上、优选90%以上、再优选95%以上同源性的氨基酸序列的蛋白质,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质也是通过本发明的制造方法中使用的微生物生产的蛋白质。
具有与序列号17表示的氨基酸序列有80%以上、优选90%以上、再优选95%以上同源性的氨基酸序列的蛋白质为了作为具有由1种以上的氨基酸生成二肽的活性的蛋白质,希望该蛋白质的氨基酸序列与序列号1~8中的任一个表示的氨基酸序列的同源性至少具有80%以上、通常为90%以上、最好是具有95%以上的同源性。
氨基酸序列的同源性可以象上述那样使用BLAST或FASTA决定。
作为确认上述[1]~[11]的蛋白质是具有由1种以上的氨基酸生成二肽活性的蛋白质的手段,例如使用DNA重组法制作表达该蛋白质的转化体,使用该转化体制造本发明的蛋白质后,使本发明的蛋白质、1种以上的氨基酸、和ATP存在于水性介质中,通过HPLC等分析二肽是否在该水性介质中生成、蓄积的方法。
本发明的制造方法使用的DNA只要是编码具有由1种以上的氨基酸生成同一或不同氨基酸通过肽键结合形成二肽的活性的蛋白质的DNA,任一种都可以,例如
[12]具有序列号9~16和36中任一个表示的碱基序列的DNA、
[13]和具有与序列号9~16和36任一个表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA、
[14]和具有与序列号18表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA、
[15]具有序列号39或40表示的碱基序列的DNA、
[16]和具有与序列号39或40表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA、
[17]编码具有NRPS活性的蛋白质的DNA、
[18]具有序列号44表示的碱基序列的DNA、和
[19]和具有与序列号44表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA。
所谓在上述严格的条件下可杂交的DNA指的是以具有与序列号9~16、18、36、39、40或44中的任一个表示的碱基序列互补的碱基序列的DNA的一部分、或全部作为探针,通过使用菌落杂交法、噬斑杂交法或Southern blot杂交法等得到的DNA,具体来说,使用固定了来自菌落或噬斑的DNA的滤器,在0.7~1.0mol/L、优选0.9mol/L氯化钠存在下、于65℃下进行杂交后、使用0.1~2倍、优选0.1倍浓度的SSC溶液(1倍浓度的SSC溶液组成由150mmol/l氯化钠、15mmol/l柠檬酸钠构成),于65℃条件下,通过清洗滤器能够鉴定的DNA。杂交可以根据Molecular Cloning第3版、Current Protocols inMolecular Biology、Current Protocols in Molecular Biology、DNACloning1:Core Techniques,A Practical Approach,Second Edition,Oxford University(1995)等记载的方法进行。作为可杂交的DNA,例如使用上述的BLAST和FASTA等,在根据上述参数计算时,与序列号9~16、18、36、39、40或44中的任一个表示的碱基序列至少具有75%以上、优选85%以上、再优选90%以上、特别优选95%以上同源性的DNA。
另外,作为供杂交的DNA样品,例如与在其染色体DNA上具有序列号9~16、18、36、39、40或44中的任一个表示的碱基序列的微生物同属、优选属于同种的微生物的染色体DNA。
在严格的条件下与具有序列号9~16、18、36、39、40或44中的任一个表示的碱基序列的DNA杂交的DNA是编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA,例如象上述那样可以通过使用重组DNA法制造该DNA编码的蛋白质,测定该蛋白质的活性来确认。
(i)本发明制造方法中使用的DNA的制备
本发明的制造方法中使用的DNA可以通过以下(a)~(c)取得:
(a)使用根据序列号9~16和36表示的碱基序列设计的探针,可对微生物、优选属于芽孢杆菌属的微生物的染色体DNA文库进行Southern杂交、或使用根据序列号9~16和36表示的碱基序列设计的引物DNA,以微生物、优选属于芽孢杆菌属的微生物的染色体DNA作为模板的PCR[PCR Protocols,Academic Press(1990)]、
(b)使用根据序列号39或40表示的碱基序列设计的探针可对微生物、优选属于链霉菌属的微生物的染色体DNA文库进行Southern杂交、或使用可根据序列号3或4表示的碱基序列设计的引物DNA,以微生物、优选属于链霉菌属的微生物的染色体DNA作为模板的PCR、和
(c)使用众所周知的编码NRPS的DNA、例如Eur.J.Biochem.,270,4555(2003)、特表2003-512835、美国专利5795738或美国专利5652116所述的编码NRPS的DNA、或可根据序列号44表示的碱基序列设计的探针对微生物、优选属于芽孢杆菌属、链霉菌属、假单孢菌属或黄单孢菌属等的微生物的染色体DNA文库进行Southern杂交、或使用根据上述的编码NPRS的DNA的碱基序列设计的引物DNA,以微生物、优选属于芽孢杆菌属、链霉菌属、假单孢菌属或黄单孢菌属等的微生物的染色体DNA作为模板的PCR取得。
另外,对于各种基因序列数据库,与编码序列号1~8、17、37、38和43中的任一个表示的氨基酸序列的DNA的碱基序列具有75%以上、优选85%以上、更优选90%以上、再优选95%以上、特别优选98%以上同源性的序列进行检索,根据通过该检索得到的碱基序列,由具有该碱基序列的生物的染色体DNA、cDNA文库等通过上述方法也可以取得本发明制造方法中使用的DNA。
取得的DNA可以直接、或用适当的限制酶等进行酶切,通过常法转运到载体,然后将得到的的重组DNA导入宿主细胞后,通过使用通常用的碱基序列解析方法、例如双脱氧法[Proc.Natl.Acad.Sci.,USA,74,5463(1977)]或373A·DNA序列仪(Perkin-Elmer公司生产)等碱基序列分析装置进行分析,可以决定该DNA的碱基序列。
决定碱基序列的结果表明当取得的DNA为部分长时,可以通过使用该部分长DNA作为探针,对染色体DNA文库的Southern杂交法等取得全长DNA。
然后根据决定的DNA的碱基序列,通过使用PerSeptiveBiosystems公司制造的8905型DNA合成装置等进行化学合成,也可以制备目的DNA。
作为象上述那样取得的DNA,例如、具有序列号9~16、36、39、40和44表示的碱基序列的DNA。
作为转运上述的DNA的载体,如pBluescriptII KS(+)(Stratagene公司生产)、pDIRECT[Nucleic Acids Res.,18,6069(1990)]、pCR-Script Amp SK(+)(Stratagene公司生产)、pT7Blue(Novagen公司生产)、pCR II(Invitrogen公司生产)和pCR-TRAP(Genhunter公司生产)等。
作为上述宿主细胞,如属于埃希氏菌属的微生物等。作为属于埃希氏菌属的微生物,例如大肠杆菌XL1-Blue、大肠杆菌XL2-Blue、大肠杆菌DH1、大肠杆菌MC1000、大肠杆菌ATCC12435、大肠杆菌W1485、大肠杆菌JM109、大肠杆菌HB101、大肠杆菌No.49、大肠杆菌W3110、大肠杆菌NY49、大肠杆菌MP347、大肠杆菌NM522、大肠杆菌ME8415等。
作为重组DNA的导入方法,只要是将DNA导入到上述宿主细胞的方法,可以使用任一种方法,例如使用钙离子的方法[Proc.Natl.Acad.Sci.,USA,69,2110(1972)]、原生质体法(特开昭63-248394)、电穿孔法[Nucleic Acids Res.,16,6127(1988)]等。
作为通过上述方法得到的用于本发明制造方法的带有DNA的微生物,例如带有重组DNA的微生物的大肠杆菌NM522/pPE43,该重组DNA含有序列号1表示的序列的DNA。
(ii)具有生产氨基酸能力的微生物的制备
本发明的二肽的制造方法中使用的具有生产氨基酸能力的微生物只要是具有生产1种以上的氨基酸的能力的微生物,无论那一种都可以,为该微生物如从自然界分离的菌株自身具有该能力的该菌株本身、通过众所周知方法人为赋予生产构成目的二肽的氨基酸中的至少1种氨基酸能力的微生物等。
作为众所周知的方法,如
(a)缓和或解除至少一个控制氨基酸生物合成的结构的方法、
(b)对至少一个参与氨基酸生物合成的酶表达增强的方法、
(c)使至少一个参与氨基酸生物合成的酶基因的拷贝数增加的方法、
(d)对至少一个由氨基酸生物合成途径向该氨基酸以外的代谢产物的旁路代谢途径进行弱化或阻断的方法,和
(e)野生型株相比,选择对氨基酸的类似物抗性高的细胞株的方法等,上述众所周知的方法可以单独或组合使用。
关于上述(a),例如Agric.Biol.Chem.,43,105-111(1979)、J.Bacteriol.,110,761-763(1972)和Appl.Microbiol.Biotechnol.,39,318-323(1993)等所述,关于上述(b),例如Agric.Biol.Chem.,43,105-111(1979)和J.Bacteriol.,110,761-763(1972)等所述、关于上述(c),例如Appl.Microbiol.Biotechnol.,39,318-323(1993)和Agric.Biol.Chem.,39,371-377(1987)等所述,关于上述(d),例如Appl.Environ.Micribiol.,38,181-190(1979)和Agric.Biol.Chem.,42,1773-1778(1978)等所述,关于上述(e),例如Agric.Biol.Chem.,36,1675-1684(1972)、Agric.Biol.Chem.,41,109-116(1977)、Agric.Biol.Chem.,37,2013-2023(1973)和Agric.Biol.Chem.,51,2089-2094(1987)等所述。参考上述文献等可以制备具有生产各种氨基酸能力的微生物。
另外,有关通过上述(a)~(e)中的任一个、或组合的方法制备具有生产氨基酸能力的微生物的制备方法,在Biotechnology2nd ed.,Vol.6,Products of Primary Metabolism(VCH VerlagsgesellschaftmbH,Weinheim,1996)section14a,14b和Advances in BiochemicalEngineering/Biotechnology79,1-35(2003)、氨基酸发酵、学会出版中心、相田浩等(1986)中记载了很多例子,另外除了上述以外,其它的具体的具有生产氨基酸能力的微生物的制备方法有特开2003-164297、Agric.Biol.Chem.,39,153-160(1975)、Agric.Biol.Chem.,39,1149-1153(1975)、特开昭58-13599、J.Gen.Appl.Microbiol.,4,272-283(1958)、特开昭63-94985、Agric.Biol.Chem.,37,2013-2023(1973)、WO97/15673、特开昭56-18596、特开昭56-144092和特表2003-511086等很多报告,通过参照上述文献等可以制备具有生产1种以上的氨基酸能力的微生物。
作为通过上述方法可以制备的具有生产氨基酸能力的微生物,例如作为L-谷氨酰胺生产菌,可例举glnE基因和/或glnB基因缺失的微生物,作为L-丙氨酸生产菌,可例举丙氨酸脱氢酶基因(ald基因)的表达被增强的微生物,作为L-脯氨酸生产微生物,可例举表达苯丙氨酸的抗反馈型pheA基因和/或酪氨酸的抗反馈型aroF基因的微生物等。
作为上述的生成、蓄积氨基酸的微生物,只要是可以适用上述(a)~(e)方法的微生物或具有上述遗传性状的微生物,可以是任一种微生物,优选原核生物、更优选细菌。
作为原核生物,如属于埃希氏菌(Escherichia)属、沙雷氏菌(Serratia)属、芽孢杆菌属、短杆菌(Brevibacterium)属、棒杆菌(Corynebacterium)属、微芽孢杆菌属(Microbacterium)、假单胞菌(Pseudomonas)属、土壤杆菌(Agrobacterium)属、脂环酸芽孢杆菌属(Alicyclobacillus)、鱼腥蓝细菌(Anabaena)属、组囊蓝细菌(Anacystis)属、节杆菌(Arthrobacter)属、固氮菌(Azotobacter)属、着色菌(Chromatium)属、欧文氏菌(Erwinia)属、甲基杆菌(Methylobacterium)属、席蓝细菌(Phormidium)属、红细菌(Rhodobacter)属、红假单胞菌(Rhodopseudomonas)属、红螺菌(Rhodospirillum)属、セネデスムス(Scenedesmus)属、链霉菌(Streptomyces)属、聚球蓝细菌(Synechoccus)属、发酵单胞菌(Zymomonas)属等的微生物、例如、大肠杆菌、枯草芽孢杆菌(Bacillus subtilis)、巨大芽孢杆菌(Bacillus megaterium)、解淀粉芽孢杆菌(Bacillus amyloliquefaciens)、凝结芽孢杆菌(Bacilluscoagulans)、地衣芽孢杆菌(Bacillus licheniformis)、短小芽孢杆菌(Bacillus pumilus)、产氨短杆菌(Brevibacterium ammoniagenes)、ブレビバクテリウム·イマリオフィルム(Brevibacterium immariophilum)、解糖短杆菌(Brevibacterium saccharolyticum)、黄色短杆菌(Brevibacterium flavum)、乳酸发酵短杆菌(Brevibacterium lactofermentum)、谷氨酸棒杆菌(Corynebacterium glutamicum)、嗜乙酰乙酸棒杆菌(Corynebacterium acetoacidophilum)、嗜氨微杆菌(Microbacterium ammoniaphilum)、无花果沙雷氏菌(Serratia ficaria)、居泉沙雷氏菌(Serratiafonticola)、液化沙雷氏菌(Serratia liquefaciens)、粘质沙雷氏菌(Serratia marcescens)、铜绿假单胞菌(Pseudomonas aeruginosa)、恶臭假单胞菌(Pseudomonas putida)、放射形土壤杆菌(Agrobacterium radiobacter)、发根土壤杆菌(Agrobacterium rhizogenes)、悬钩子土壤杆菌(Agrobacterium rubi)、柱孢鱼腥蓝细菌(Anabaena cylindrica)、桶形鱼腥蓝细菌(Anabaena doliolum)、水华鱼腥蓝细菌(Anabaenaflos-aquae)、金黄节杆菌(Arthrobacter aurescens)、柠檬节杆菌(Arthrobacter citreus)、球形节杆菌(Arthrobacter globformis)、アースロバクター·ヒドロカーボグルタミカス(Arthrobacter hydrocarboglutamicus)、迈索尔节杆菌(Arthrobacter mysorens)、烟草节杆菌(Arthrobacter nicotianae)、石蜡节杆菌(Arthrobacter paraffineus)、原玻璃蝇节杆菌(Arthrobacter protophormiae)、玫瑰色石蜡节杆菌(Arthrobacter roseoparaffinus)、硫磺节杆菌(Arthrobacter sulfureus)、产尿节杆菌(Arthrobacter ureafaciens)、巴氏着色菌(Chromatium buderi)、微温着色菌(Chromatium tepidum)、酒色着色菌(Chromatium vinosum)、沃氏着色菌(Chromatium warmingii)、クロマチウム·フルビアタティレ(Chromatium fluviatile)、噬夏孢欧文氏菌(Erwinia uredovora)、胡萝卜软腐欧文氏菌(Erwinia carotovora)、波萝欧文氏菌(Erwinia ananas)、草生欧文氏菌(Erwinia herbicola)、エルビニア·パンクタタ(Erwinia punctata)、エルビニア·テレウス(Erwinia terreus)、罗得西亚甲基杆菌(Methylobacterium rhodesianum)、扭脱甲基杆菌(Methylobacterium extorquens)、フォルミディウム·エスピー(Phormidium sp.)ATCC29409、夹膜红细菌(Rhodobacter capsulatus)、类球红细菌(Rhodobacter sphaeroides)、生芽红细菌(Rhodopseudomonas blastica)、海红菌(Rhodopseudomonas marina)、血色红假单胞菌(Rhodopseudomonas palustris)、深红红螺菌(Rhodospirillum rubrum)、需盐红螺菌(Rhodospirillum salexigens)、盐场红螺菌(Rhodospirillum salinarum)、产二素链霉菌(Streptomyces ambofaciens)、金霉素链霉菌(Streptomyces aureofaciens)、金色链霉菌(Streptomyces aureus)、杀真菌素链霉菌(Streptomyces fungicidicus)、灰产色链霉菌(Streptomyces griseochromogenes)、灰色链霉菌(Streptomyces griseus)、浅青紫链霉菌(Streptomyces lividans)、橄榄灰链霉菌(Streptomyces olivogriseus)、枝链霉菌(Streptomyces rameus)、田无链霉菌(Streptomyces tanashiensis)、酒红链霉菌(Streptomyces vinaceus)、运动发酵单胞菌(Zymomonas mobilis)等;作为理想的原核生物,如属于埃希氏菌属、沙雷氏菌属、芽孢杆菌属、短芽孢杆菌属、棒杆菌属、假单孢菌属或链霉菌属等的细菌,例如上述的属于埃希氏菌属、沙雷氏菌属、芽孢杆菌属、短芽孢杆菌属、棒杆菌属、假单孢菌属或链霉菌属等细菌;作为更理想的细菌,如大肠杆菌、谷氨酸棒杆菌、产氨棒杆菌、乳酸发酵棒杆菌、黄色棒杆菌、Corynebacterium efficiens、枯草芽孢杆菌、巨大芽孢杆菌、粘质沙雷氏菌、恶臭假单胞菌、铜绿假单胞菌、天蓝色链霉菌或浅青紫链霉菌,特别优选大肠杆菌。
作为生产氨基酸的微生物的具体例子,作为L-谷氨酰胺生产株,如大肠杆菌JGLE1和大肠杆菌JGLBE1等、作为L-丙氨酸生产株,如保持ald基因表达质粒的大肠杆菌JM101株等、作为L-苯丙氨酸生产株,如带有pPHEA2和/或aroF基因表达质粒的大肠杆菌JM101株等、作为L-谷氨酰胺和L-丙氨酸生产株,如保持ald基因表达质粒的大肠杆菌JGLE1和大肠杆菌JGLBE1等、作为L-丙氨酸和L-苯丙氨酸生产株,如保持ald基因表达质粒和抗反馈型pheA基因和/或抗反馈型aroF基因表达质粒的大肠杆菌JM101等、作为L-苏氨酸和L-苯丙氨酸生产株,如保持抗反馈型pheA基因和/或抗反馈型aroF基因表达质粒的ATCC21277株等。
另外,作为具有生产氨基酸能力的微生物的具体例子,作为L-谷氨酸生产株,如FERM BP-5807和ATCC13032等、作为L-谷氨酰胺生产株,如FERM P-4806和ATCC14751等、作为L-苏氨酸生产株,如ATCC21148、ATCC21277和ATCC21650等、作为L-赖氨酸生产株,如FERM P-5084和ATCC13286等、作为L-蛋氨酸生产株,如FERM P-5479、VKPM B-2175和ATCC21608等、作为L-异亮氨酸生产株,如FERM BP-3757和ATCC14310等、作为L-缬氨酸生产株,如ATCC13005和ATCC19561等、作为L-亮氨酸生产株,如FERMBP-4704和ATCC21302等、作为L-丙氨酸生产株,如FERM BP-4121和ATCC15108等、作为L-丝氨酸生产株,如ATCC21523和FERMBP-6576等、作为L-脯氨酸生产株,如FERM BP-2807和ATCC19224等、作为L-精氨酸生产株,如FERM P-5616和ATCC21831等、作为L-鸟氨酸生产株,如ATCC13232等、作为L-组氨酸生产株,如FERM BP-6674和ATCC21607等、作为L-色氨酸生产株,如DSM10118、DSM10121、DSM10123和FERM BP-1777等、作为L-苯丙氨酸生产株,如ATCC13281和ATCC21669等、作为L-酪氨酸生产株,如ATCC21652等、作为L-半胱氨酸生产株,如W3110/pHC34(特表2003-511086记载)等、作为L-4-羟基脯氨酸生产株,如WO96/27669记载的大肠杆菌SOLR/pRH71等、作为L-3-羟基脯氨酸生产株,如FERM BP-5026和FERM BP-5409等、作为L-瓜氨酸生产株,如FERM P-5643和FERM P-1645等。
而上述的FERM编号表示的菌株可以从独立行政法人产业技术综合研究所专利生物寄托中心(日本)、ATCC编号表示的菌株可以从American Type Culture Collection(美国)、VKPM编号表示的菌株可以从Russian National Collection of Industrial Microorganisms(俄罗斯)、DSM编号表示的菌株可以从Deutsche Sammlung vonMikroorganismen und Zellkulturen(德国)分别获得。
(iii)具有生产具有由1种以上的氨基酸生成二肽的活性的蛋白质能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸的能力的微生物的制备
具有生产具有由1种以上的氨基酸生成二肽的活性的蛋白质能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸的能力的微生物可以通过以下(a)~(d)的方法等可以制备。
(a)在可用上述(ii)方法制备的具有生产1种以上的氨基酸能力的微生物导入可以通过上述(i)方法制备的编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA的方法、
(b)通过上述(ii)的方法赋予导入了通过上述(i)方法制备的编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA微生物生产1种以上的氨基酸的能力的方法、
(c)通过上述(i)的方法向原来具有生产1种以上的氨基酸能力的微生物导入编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA的方法、或
(d)通过上述(ii)方法赋予原来具有生产具有由1种以上的氨基酸生成二肽的活性的蛋白质能力的微生物生产1种以上的氨基酸能力的方法。
通过将能够用上述(i)的方法制备的编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA导入微生物,可以赋予该微生物生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力。作为赋予微生物生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力的方法,可以使用Molecular Cloning第3版、Current Protocols inMolecular Biology等记载的方法等,例如通过以下方法使在用上述(i)的方法制备的DNA在宿主细胞中表达的方法。
以用上述(i)的方法制备的DNA作为基础,根据需要,制备含有编码蛋白质的部分的适当长度的DNA片段。另外,通过将编码该蛋白质部分的碱基序列置换成最适于宿主表达的密码,可以使该蛋白质的产率提高。
通过将该DNA片段插入到适当的表达载体的启动子的下游,可以制作重组DNA。
通过将该重组DNA导入适合该表达载体的宿主细胞可以获得生产该蛋白质的转化体。
作为宿主细胞,只要是能够表达目的基因的微生物,可以使用任一种,优选原核生物、更优选细菌。作为理想的原核生物如上述(ii)的原核生物。
该微生物可以具有生产1种以上的氨基酸的能力,也可以不具有该能力。当使用不具有该能力的微生物作为宿主细胞时,通过下面的方法将用上述方法得到的重组DNA导入该微生物制备转化体后,通过用上述(ii)的方法赋予该转化体生产1以上的氨基酸的能力,可以获得本发明制造方法中使用的微生物。
作为表达载体,使用可在微生物细胞中自我复制或转运到染色体中,在能够转录本发明制造方法中使用的DNA的位置含有启动子的载体。
使用原核生物作为宿主细胞时,具有本发明制造方法中使用的DNA的重组DNA最好是在原核生物中可自我复制,同时由启动子、核糖体结合序列、本发明的制造方法中使用的DNA、转录终止序列构成的重组DNA。也可以含有调控启动子的基因。
作为表达载体,例如pBTrp2、pBTac1、pBTac2(都是BoehringerMannheim公司生产)、pHelix1(Roche Diagnostics公司生产)、pKK233-2(Amersham Pharmacia Biotech公司生产)、pSE280(Invitrogen公司生产)、pGEMEX-1(Promega公司生产)、pQE-8(Qiagen公司生产)、pET-3(Novagen公司生产)、pKYP10(特开昭58-110600)、pKYP200[Agric.Biol.Chem.,48,669(1984)]、pLSA1[Agric.Biol.Chem.,53,277(1989)]、pGEL1[Proc.Natl.Acad.Sci.,USA,82,4306(1985)]、pBluescriptII SK(+)、pBluescript II KS(-)(Stratagene公司生产)、pTrS30[由大肠杆菌JM109/pTrS30(FERMBP-5407)制备]、pTrS32[由大肠杆菌JM109/pTrS32(FERM BP-5408)制备]、pPAC31(WO98/12343)、pUC19[Gene,33,103(1985)]、pSTV28(Takara Bio公司生产)、pUC118(Takara Bio公司生产)、pPA1(特开昭63-233798)、pWH1520(MoBiTec社制)、pCS299P(WO00/63388)、pVLT31[Gene,123,17(1993)]和pIJ702(GeneticManipulation of Streptomyces:a Laboratory Manual:John InnesFoundation)等。
使用属于埃希氏菌属的微生物作为宿主细胞时,作为启动子只要是可在大肠杆菌内发挥功能的,无论那一种都可以,例如来自大肠菌的trp启动子(P trp )、lac启动子(P lac )、PL启动子、PR启动子、
PSE启动子等,或来自噬菌体等的启动子,例如SPO1启动子、SPO2启动子、penP启动子等。也可以使用人为设计使Ptrp改变为两个串联的启动子、tac启动子、lacT7启动子、let I启动子那样的启动子等。
另外也可以使用用于使基因在属于芽孢杆菌属的微生物中表达的xylA启动子[Appl.Microbiol.Biotechnol.,35,594-599(1991)]、用于使基因在属于棒杆菌属的微生物中表达的P54-6启动子[Appl.Microbiol.Biotechnol.,53,674-679(2000)]、用于使基因在属于假单孢菌属的微生物中表达的tac启动子[Gene,123,17-24(1993)]、用于使基因在属于链霉菌属的微生物中表达的xylA启动子(Genetic Manipulation ofStreptomyces:a Laboratory Manual:John Innes Foundation)等。
优选使用将作为核糖体结合序列的Shine-Dalgarno序列与起始密码子之间距离调节到适当距离(例如6~18碱基)的质粒。
在使本发明制造方法中使用的DNA与表达载体结合的重组DNA中,不一定需要转录终止序列,但最好是在紧靠结构基因的下面配置转录终止序列。
作为这样的重组DNA,例如pPE43。
作为将重组DNA导入微生物细胞的导入方法,只要是将DNA导入该细胞的方法,可以使用任一种方法,例如、使用钙离子的方法[Proc.Natl.Acad.Sci.,USA,69,2110(1972)]、原生质体法(特开昭63-248394)、电穿孔法[Nucleic Acids Res.,16,6127(1988)]等。
另外,作为在上述(c)方法中使用的原来具有生产1种以上的氨基酸能力的微生物,可例举具有生产上述(ii)的氨基酸能力的众所周知的菌株等。
作为在上述(d)方法中使用的原来具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质能力的微生物,如(A)属于芽孢杆菌属的微生物、更优选具有杆菌溶素合成活性的属于芽孢杆菌属的微生物、再优选枯草芽孢杆菌、解淀粉芽孢杆菌、凝结芽孢杆菌、地衣芽孢杆菌、巨大芽孢杆菌和短小芽孢杆菌等属于各种属的微生物、最好是枯草芽孢杆菌ATCC15245、枯草芽孢杆菌ATCC6633、枯草芽孢杆菌IAM1213、枯草芽孢杆菌IAM1107、枯草芽孢杆菌IAM1214、枯草芽孢杆菌ATCC9466、枯草芽孢杆菌IAM1033、枯草芽孢杆菌ATCC21555、解淀粉芽孢杆菌IFO3022和短小芽孢杆菌NRRLB-12025中的任一菌株的微生物、和(B)属于链霉菌属的微生物、优选具有生产albonoursin能力的属于链霉菌属的微生物、更优选属于小白链霉菌(Streptomyces albulus)或诺尔斯氏链霉菌(Streptomyces noursei)的微生物。
(iv)肽酶和具有肽转运活性的蛋白质的活性低下或丧失的微生物
作为本发明的制造方法使用的微生物,如在通过上述(iii)的方法制备的微生物中,1种以上肽酶和1种以上肽转运蛋白质的活性低下或丧失的微生物、或3种以上肽酶的活性低下或丧失的微生物。
该微生物可以通过例如如下的方法获得,(a)通过上述(iii)的方法赋予1种以上肽酶和1种以上肽转运蛋白质的功能低下或丧失的微生物、或3种以上肽酶的功能低下或丧失的微生物具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质能力、和生产该1种以上的氨基酸中的至少1种氨基酸的能力的方法,(b)使通过上述(iii)的方法能够制备的微生物的a)1种以上肽酶和1种以上肽转运蛋白质、或b)3种以上肽酶的功能低下或丧失的方法,该微生物具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力、和具有生产该1种以上的氨基酸中的至少1种氨基酸的能力。
作为1种以上肽酶和1种以上肽转运蛋白质的活性低下或丧失的微生物,只要是该微生物能正常生长繁殖,如任意1种以上肽酶和任意1种以上肽转运蛋白质的活性低下或丧失的微生物,优选1种以上9种以下、更优选1种以上7种以下、再优选1种以上4种以下的肽酶的活性低下或丧失,而且优选1种以上5种以下、更优选1种以上3种以下、再优选1种以上2种以下、特别优选1种的肽转运蛋白质活性低下或丧失的微生物。
作为该微生物,更具体讲,如在该微生物的基因组DNA上存在的编码肽酶的基因(以下、略为肽酶基因)和编码肽转运蛋白质的基因(以下、肽转运蛋白质基因)中,由于在1种以上肽酶基因的碱基序列和1种以上肽转运蛋白质基因的碱基序列中缺失该碱基序列的全部或一部分,或由于在该碱基序列中存在碱基的置换或附加,该肽酶和该肽转运蛋白质的活性低下或丧失的微生物。
所谓肽酶的活性低下指的是与上述的碱基没有缺失、置换或附加的基因编码的肽酶相比,肽分解活性低下,通常低至80%以下、优选50%以下、更优选30%以下、再优选20%以下、特别优选10%以下、最好优选5%以下。
微生物的肽分解活性可以使作为底物的肽和微生物菌体在水性介质中共存,进行肽分解反应后,通过众所周知的方法、例如使用HPLC等的分析法对残存的肽量进行定量来测定。
作为上述肽酶,只要是具有肽分解活性的蛋白质就可以,优选二肽分解活性高的蛋白质、更优选二肽酶。
作为更具体的肽酶,例如存在于大肠杆菌的具有序列号45表示的氨基酸序列的PepA、具有序列号46表示的氨基酸序列的PepB、具有序列号47表示的氨基酸序列的PepD、具有序列号48表示的氨基酸序列的PepN、PepP[GenBank accession no.(以下略为GenBank)AAC75946]、PepQ(GenBank AAC76850)、PepE(GenBankAAC76991)、PepT(GenBank AAC74211)、Dcp(GenBank AAC74611)和IadA(GenBank AAC77284)等;存在于枯草芽孢杆菌的AmpS(GenBank AF012285)、PepT(GenBank X99339)、YbaC(GenBankZ99104)、YcdD(GenBank Z99105)、YjbG(GenBank Z99110)、YkvY(GenBank Z99111)、YqjE(GenBank Z99116)、YwaD(GenBankZ99123)等;存在于谷氨酸棒杆菌的具有BAB97732、BAB97858、BAB98080、BAB98880、BAB98892、BAB99013、BAB99598和BAB99819(都是日本DNA数据库的登录号)表示的氨基酸序列的蛋白质等;作为二肽酶,如具有序列号45~48表示的氨基酸序列的PepA、PepB、PepD和PepN、以及PepQ、PepE、IadA。另外与序列号45~48中的任一个氨基酸序列具有80%以上、优选90%以上、更优选95%以上同源性,而且具有肽酶活性的蛋白质也可以作为二肽分解活性高的蛋白质。氨基酸序列或碱基序列的同源性可以使用上述的BLAST和FASTA等来决定。
而所谓肽转运蛋白质的活性低下指的是与编码上述的没有进行碱基缺失、置换或附加的基因的肽转运蛋白质相比,肽转运活性变低通常指的是降低到80%以下、优选50%以下、更优选30%以下、再优选20%以下、特别优选10%以下、最好优选5%以下。
微生物的肽转运活性可以通过使作为底物的肽和微生物菌体共存于水性介质中进行肽转运反应后,利用众所周知的方法、例如使用HPLC等分析法对残存于水性介质中的肽量进行定量来测定。
作为上述肽转运蛋白质,是由染色体DNA上形成操纵子的基因编码的蛋白质,在细胞膜上形成复合体后表达肽转运活性的蛋白质、和作为单独的蛋白质具有肽转运活性的蛋白质等只要是参与微生物的肽转运的蛋白质,任一种都可以,优选二肽转运活性高的蛋白质。
作为更具体的肽转运蛋白质,例如存在于大肠杆菌的具有序列号49表示的氨基酸序列的DppA、具有序列号50表示的氨基酸序列的DppB、具有序列号51表示的氨基酸序列的DppC、具有序列号52表示的氨基酸序列的DppD、具有序列号53表示的氨基酸序列的DppF、OppA(GenBank AAC76569)、OppB(GenBank AAC76568)、OppC(GenBank AAC76567)、OppD(GenBank AAC76566)、OppF(GenBank AAC76565)、YddO(GenBank AAC74556)、YddP(GenBank AAC74557)、YddQ(GenBank AAC74558)、YddR(GenBank AAC74559)、YddS(GenBank AAC74560)、YbiK(GenBank AAC73915)、MppA(GenBank AAC74411)、SapA(GenBank AAC74376)、SapB(GenBank AAC74375)、SapC(GenBank AAC74374)、SapD(GenBank AAC74373)、和SapF(GenBank AAC74372)等;存在于枯草芽孢杆菌的DppA(GenBankCAA40002)、DppB(GenBank CAA40003)、DppC(GenBankCAA40004)、DppD(GenBank CAA40005)、DppE(GenBankCAA40006)、OppA(GenBank CAA39787)、OppB(GenBankCAA39788)、OppC(GenBank CAA39789)、OppD(GenBankCAA39790)、OppF(GenBank CAA39791)、AppA(GenBankCAA62358)、AppB(GenBank CAA62359)、AppC(GenBankCAA62360)、AppD(GenBank CAA62356)、AppF(GenBankCAA62357)、YclF(GenBank CAB12175)和YkfD(GenBankCAB13157)等;存在于谷氨酸棒杆菌的具有BAB99048、BAB99383、BAB99384、BAB99385、BAB99713、BAB99714、BAB99715、BAB99830、BAB99831、BAB99832(都是日本DNA数据库的登录号)表示的氨基酸序列的蛋白质等;另外作为二肽转运活性高的蛋白质,如具有序列号49~53表示的氨基酸序列的DppA、DppB、DppC、DppD、DppF和与它们中任一个的氨基酸序列具有80%以上、优选90%以上、更优选95%以上同源性的蛋白质也可作为肽转运活性高的肽转运蛋白质。
上述氨基酸序列的同源性可以使用上述的BLAST或FASTA等程序决定。
作为3种以上肽酶的活性低下或丧失的微生物,以该微生物能正常生长繁殖为限,如任意3种以上肽酶的活性低下或丧失的微生物,优选3种以上9种以下、更优选3种以上6种以下、再优选3种或4种肽酶的活性低下或丧失的微生物。
作为具体的肽酶,如存在于上述的大肠杆菌、枯草芽孢杆菌和谷氨酸棒杆菌的肽酶和二肽酶。而由与序列号45~48中的任一个氨基酸序列有80%以上、优选90%以上、更优选95%以上同源性的氨基酸序列构成、而且具有肽酶活性的蛋白质也可以作为二肽分解活性高的蛋白质。
上述氨基酸序列的同源性可以使用上述的BLAST或FASTA等程序决定。
(v)肽酶和肽转运蛋白质的活性低下或丧失的微生物的制备
肽酶和肽转运蛋白质的活性低下或丧失的微生物只要是能够取得该微生物的方法,其取得方法没有限制,例如通过在以下所示的微生物的染色体DNA的肽酶基因或肽转运蛋白质基因导入碱基的缺失、置换或附加的方法可以取得。
作为在微生物的染色体DNA的基因导入碱基缺失、置换或附加的方法,如利用同源重组的方法。作为利用一般的同源重组的方法,如使用将导入了碱基缺失、置换或附加的突变基因与在希望导入碱基缺失等的宿主细胞内含有不能自我复制的抗药性基因的质粒DNA连接制作的同源重组用质粒的方法。
通过常法将该同源重组用质粒导入宿主细胞后,以抗药性作为指标,选择通过同源重组在染色体DNA上转运了该同源重组用质粒的转化株。通过将得到的转化株于不含有该药剂的培养基中培养数小时~1天后,涂布于含有该药剂琼脂培养基和不含有该药剂的琼脂培养基,选择在前者培养基中不生长繁殖,而在后者培养基能够生长繁殖的株,可以取得在染色体DNA上发生第2次同源重组的株。通过决定染色体DNA上导入缺失等的基因存在区域的碱基序列,可以确认染色体DNA上的目的基因中导入碱基的缺失、置换或附加。
作为通过上述方法,能够在染色体DNA上的目的基因中导入碱基的缺失、置换或附加的微生物,例如属于埃希氏菌属、芽孢杆菌属或棒杆菌属的微生物。
而作为利用向多个基因有效导入碱基的缺失、置换或附加的同源重组的方法,如使用直链DNA的方法。
具体来说,是使含有希望导入碱基缺失、置换或附加的基因的直链DNA转运到细胞内,在染色体DNA与导入的直链DNA之间发生同源重组的方法。本方法只要是可有效地转运直链DNA的微生物,也可以适用于任一种微生物,作为理想的微生物,如属于埃希氏菌属或芽孢杆菌属的微生物、更优选大肠杆菌、再优选表达来自λ噬菌体的重组蛋白质组(Red重组系)的大肠杆菌。
作为表达λRed重组系的大肠杆菌,如带有含λRed重组系基因的质粒DNA的pKD46[可从大肠杆菌基因储备中心(美国耶鲁大学)获得]的大肠杆菌JM101株等。
作为可用于同源重组的DNA,如可例举:
(a)在抗药性基因的两端含有与位于存在于作为碱基的缺失、置换或附加的导入对象的染色体DNA上区域的两外侧的DNA具有同源性的DNA的直链DNA、
(b)直接连接了与存在于作为碱基的缺失、置换或附加的导入对象的染色体DNA上区域的两外侧的DNA具有同源性的DNA的直链DNA、
(c)在含有抗药性基因和能够用于负选择的基因的DNA的两端含有与存在于作为碱基的缺失、置换或附加的导入对象的染色体DNA上的区域的两外侧的DNA具有同源性的DNA的直链DNA、
(d)在上述(a)的直链DNA中,在抗药性基因和与存在于染色体DNA上区域的两外侧的DNA具有同源性的DNA之间,还含有来自酵母的Flp重组酶〔Proc.Natl.Acad.Sci.USA.,82,5875(1985)〕识别碱基序列的DNA。
作为抗药性基因,只要是对于宿主微生物表现出敏感性的药剂,赋予抗药性的抗药性基因,也可以使用任一种抗药性基因。作为宿主微生物使用大肠杆菌时,作为抗药性基因,例如、卡那霉素抗性基因、氯霉素抗性基因、庆大霉素抗性基因、大观霉素抗性基因、四环素抗性基因和氨苄青霉素抗性基因等。
所谓能够用于负选择的基因指的是当在宿主微生物中使该基因表达时,在一定培养条件下,使该微生物致死的的基因,作为该基因,例如来自属于芽孢杆菌属的微生物的sacB基因〔Appl.Environ.Microbiol.,59,1361-1366(1993)〕、和来自属于埃希氏菌属的微生物的rpsL基因〔Genomics,72,99-104(2001)〕等。
存在于上述的直链DNA的两末端的、与存在于成为染色体DNA上的置换或缺失的导入对象的区域两端外侧的DNA具有同源性的DNA在直链DNA中被配置在与染色体DNA上的方向相同方向,其长度优选10bp~100bp左右,更优选bp~50bp左右、进一步优选30~40bp左右。
所谓来自酵母的Flp重组酶识别的碱基序列只要是该蛋白质识别、催化同源重组的碱基序列,没有特别限定,优选具有序列号54表示的碱基序列的DNA、和具有在该DNA中缺失、置换或附加了1个~数个碱基的碱基序列、而且来自酵母的Flp重组酶识别的、催化同源重组的碱基序列的DNA。
所谓具有同源性是上述直链DNA在染色体DNA上的目的区域,具有发生同源重组的程度的同源性,作为具体的同源性,如80%以上、优选90%以上、更优选95%以上、最优选100%的同源性。
上述碱基序列的同源性可以使用上述的BLAST或FASTA等程序决定。
上述直链DNA可以通过PCR制作。另外将含有上述直链DNA的DNA构建在质粒上后,经限制酶处理也可以得到目的直链DNA。
作为在微生物的染色体DNA中导入碱基的缺失、置换或附加的方法,如以下的方法1~4。
方法1:将上述(a)或(d)的直链DNA导入宿主微生物,以抗药性为指标选择通过同源重组该直链DNA被插入到染色体DNA上的转化株的方法。
方法2:向通过上述方法1取得的转化株导入上述(b)的直链DNA,通过消除经该方法插入到染色体DNA上的药剂基因,使微生物的染色体DNA上的区域置换或缺失的方法。
方法3:
[1]将上述(c)的直链DNA导入宿主微生物,以抗药性为指标选择通过同源重组该直链DNA被插入到染色体DNA上的转化株、
[2]合成在与染色体DNA上方向同一方向连接了与位于染色体DNA上的置换或缺失的对象区域两端外侧的DNA具有同源性的DNA的DNA,导入到上述[1]中得到的转化株,
[3]在可用于负选择的基因表达的条件下,对进行了上述[2]操作的转化株进行培养,选择在该培养条件下可生长繁殖的株作为从染色体DNA上削除了抗药性基因和可用于负选择的基因的菌株的方法。
方法4:
[1]将上述(d)的直链DNA导入宿主微生物,以抗药性为指标选择通过同源重组该直链DNA被插入到染色体DNA上的转化株、
[2]向上述[1]中得到的转化株导入Flp重组酶基因表达质粒,使该基因表达后,取得对上述[1]中使用的药剂有敏感性的菌株的方法。
作为将在上述方法中使用的直链DNA导入宿主微生物的方法,只要是将DNA导入该微生物的方法,可以使用任一种方法,例如、使用钙离子的方法〔Proc.Natl.Acad.Sci.,USA,69,2110(1972)〕、原生质体法(特开昭63-248394)、电穿孔法〔Nucleic Acids Res.,16,6127(1988)〕等。
在方法2或方法3[2]中使用的直链DNA中,通过使用在该DNA的中央部附近转运希望插入到染色体DNA上的任意基因的直链DNA,在削除抗药性基因等的同时,可以将任意基因插入到染色体DNA上。
上述方法2~4由于是对于最终得到的转化株的染色体DNA上不保留抗药性基因和可用于负选择的基因等外源基因的方法,所以通过使用同一抗药性基因和可用于负选择的基因,反复进行该方法的操作,容易制造在染色体DNA上位置不同的2个以上区域带有碱基的缺失、置换或附加的微生物。
(vi)本发明的二肽的制造方法
通过对用上述(iii)和(v)的方法得到的微生物在培养基中培养,使二肽在培养物中生成、蓄积,从该培养物中回收二肽,可以制造二肽。
将该微生物在培养基中进行培养的方法可以根据微生物培养中使用的通常方法来进行。
即,只要是含有该微生物可利用的碳源、氮源、无机盐类等的可有效进行该微生物培养的培养基,可以使用天然培养基、合成培养基中的任一种。
在该培养基中虽然不需要含有构成目的二肽的氨基酸,但在天然培养基、或用于培养氨基酸需求性菌株的培养基中有时也含有该氨基酸。在本发明制造方法中使用的培养基中也可以含有本发明使用的微生物生长繁殖所必需的量的氨基酸。即,通常培养基中含有的氨基酸量由于与通过本发明制造方法中使用的微生物生产的该氨基酸量相比非常少,所以通常培养基中含有的该氨基酸量并不是由于该氨基酸的有无导致本申请发明制造的二肽产量变化那样程度的量,因此在本发明的制造方法中使用的培养基中也可以含有如此程度的该氨基酸。
作为本发明中使用的培养基中含有的氨基酸量,例如如果是天然培养基,通常含不足2.5g/L、优选0.5g/L以下、更优选0.1g/L以下、再优选20mg/L以下的该氨基酸;如果是合成培养基,通常含1g/L以下、优选50mg/L以下、更优选1mg/L以下、再优选0.5mg/L以下的该氨基酸。但是,当通过本申请发明的制造方法制造2种不同氨基酸构成的二肽时,当使用的微生物只有生产构成该二肽的氨基酸中的1种氨基酸的能力时,也可以向本发明中使用的培养基中添加该微生物不能生产的另外1种氨基酸。此时添加的氨基酸量通常为0.5g/L~100g/L、优选2g/L~50g/L。
作为碳源,只要是该微生物可利用的就可以,可以使用葡萄糖、乳糖、蔗糖、含有这些糖的糖蜜、淀粉或淀粉水解物等碳水化合物,醋酸、丙酸等有机酸,乙醇、丙醇等醇类等
作为氮源可以使用氨、氯化铵、硫酸铵、醋酸铵、磷酸铵等无机酸或有机酸的铵盐、其它的含氮化合物、以及胨、肉提取物、酵母提取物、玉米浸渍液、酪蛋白水解物、大豆粕和大豆粕加水分解物、各种发酵菌体、及其消化物等。
作为无机盐,可以使用磷酸二氢钾、磷酸氢二钾、磷酸镁、硫酸镁、氯化钠、硫酸亚铁、硫酸锰、硫酸铜、碳酸钙等。
培养在通常振荡培养或深部通气搅拌培养等好氧条件下进行。培养温度为15~40℃比较好、培养时间通常为5小时~7日间。培养中pH保持3.0~9.0。pH的调整使用无机或有机的酸、碱性溶液、尿素、碳酸钙、氨等进行。
另外根据培养中需要,也可以向培养基中添加氨苄青霉素或四环素等抗生素。
当对用作为启动子使用诱导性的启动子的表达载体转化的微生物进行培养时,根据需要也可以向培养基中添加诱导物。例如、对用使用了lac启动子的表达载体转化的微生物进行培养时,可以添加异丙基-β-D-硫代半乳糖苷等,当用使用了trp启动子的表达载体转化的微生物进行培养时,也可以向培养基添加吲哚丙烯酸等。
作为用上述方法制造的二肽,如1种或2种的氨基酸经α键形成的二肽,优选该氨基酸是L-氨基酸或甘氨酸的二肽、更优选在式(I)
R1-R2 (I)
表示的二肽中,R1和R2为相同或不同、从L-丙氨酸(L-Ala)、L-谷氨酰胺(L-Gln)、L-谷氨酸(L-Glu)、甘氨酸(Gly)、L-缬氨酸(L-Val)、L-亮氨酸(L-Leu)、L-异亮氨酸(L-Ile)、L-脯氨酸(L-Pro)、L-苯丙氨酸(L-Phe)、L-色氨酸(L-Trp)、L-蛋氨酸(L-Met)、L-丝氨酸(L-Ser)、L-苏氨酸(L-Thr)、L-半胱氨酸(L-Cys)、L-天冬酰胺(L-Asn)、L-酪氨酸(L-Tyr)、L-赖氨酸(L-Lys)、L-精氨酸(L-Arg)、L-组氨酸(L-His)、L-天冬氨酸(L-Asp)、L-α-氨基丁酸(L-α-AB)、L-4-羟基脯氨酸(L-4-HYP)、L-3-羟基脯氨酸(L-3-HYP)、L-鸟氨酸(L-Orn)和L-瓜氨酸(L-Cit)选出来的氨基酸的二肽、再优选R1为L-Ala、Gly、L-Met、L-Ser或L-Thr时、R2为L-Gln、L-Glu、Gly、L-Val、L-Leu、L-Ile、L-Pro、L-Phe、L-Trp、L-Met、L-Ser、L-Thr、L-Cys、L-Asn、L-Tyr、L-Lys、L-Arg、L-His、L-Asp、L-α-AB、L-4-HYP、L-3-HYP、L-Orn或L-Cit的二肽、特别优选R1为L-Ala时,R2为L-Gln、Gly、L-Val、L-Leu、L-Ile、L-Phe、L-Trp、L-Met、L-Ser、L-Thr、L-Cys、L-Asn、L-Tyr、L-Lys、L-Arg、L-His、L-α-AB或L-Cit的二肽、R1为Gly时,R2为L-Gln、Gly、L-Phe、L-Trp、L-Met、L-Ser、L-Thr、L-Cys、L-Tyr、L-Lys、L-Arg、L-α-AB或L-Cit的二肽、R1为L-Met时,R2是L-Phe、L-Met、L-Cys、L-Tyr、L-Lys或L-His的二肽、R1为L-Ser时,R2是L-Gln、Gly、L-Phe、L-Met、L-Ser、L-Thr、L-Tyr、L-His或L-α-AB的二肽、R1为L-Thr时,R2是L-Gln、L-Leu、L-Phe、L-Met、L-Ser、L-Thr或L-α-AB的二肽、R1为L-Gln时,R2是L-Phe或L-α-AB的二肽、R1为L-Phe时,R2是L-Gln的二肽、R1为L-Trp时,R2是Gly的二肽、R1是L-Cys时,R2是L-Ala、L-Gln、Gly、或L-Met的二肽、R1为L-Lys时,R2是L-Ala、Gly或L-Met的二肽、R1为L-Arg时,R2是L-α-AB的二肽、R1为L-His时,R2是L-Met的二肽、和R1为L-α-AB时,R2是L-Ala、L-Gln、Gly、L-Ser、L-Thr、L-Arg或L-α-AB的二肽,最好优选L-丙氨酰-L-丙氨酸(L-Ala-L-Ala)、L-丙氨酰-L-谷氨酰胺(L-Ala-L-Gln)、L-丙氨酰-L-苯丙氨酸(L-Ala-L-Phe)、L-苏氨酰-L-苯丙氨酸(L-Thr-L-Phe)、L-丙氨酰-L-酪氨酸(L-Ala-L-Tyr)、L-丙氨酰-L-蛋氨酸(L-Ala-L-Met)、L-丙氨酰-L-缬氨酸(L-Ala-L-Val)、L-丙氨酰-异亮氨酸(L-Ala-L-Ile)、L-丙氨酰-L-亮氨酸(L-Ala-L-Leu)和L-丝氨酰-L-苯丙氨酸(L-Ser-L-Phe)。
培养物中生成、蓄积的二肽的回收可以通过使用活性炭或离子交换树脂等通常的方法、或通过利用有机溶剂萃取、结晶、薄层层析、高效液相层析等进行。
以下给出了编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA取得方法等实验例,该DNA的取得方法等并不限定于该实验例。
【实验例1利用数据库检索具有二肽合成活性的蛋白质】
以来自枯草芽孢杆菌168株的D-Ala-D-Ala连接酶基因的氨基酸序列[Nature,390,249-256(1997)]作为质询,使用作为枯草芽孢杆菌168株的基因组DNA的数据库的Subtilist(http://genolist.pasteur.fr/SubtiList/)的同源性检索功能,对存在于枯草芽孢杆菌168株的基因组DNA序列中的编码具有同源性的蛋白质的基因进行检索。
结果在提取的序列中,将编码作为D-Ala-D-Ala连接酶基序[Biochemistry,30,1673(1991)]的序列号33、34或35表示的氨基酸序列,同时编码其功能已经被鉴定的蛋白质的基因排除后,选择表现出与D-Ala-D-Ala连接酶基序最高同源性(29.1%)的序列作为功能未知基因ywfE。
ywfE的碱基序列如序列号9所示,由该碱基序列编码的蛋白质的氨基酸序列如序列号1所示。
【实验例2ywfE基因表达株的构建】
根据实验例1得到的碱基序列信息,象以下那样操作可以取得枯草芽孢杆菌的ywfE基因片段。
首先将枯草芽孢杆菌168株(ATCC23857)接种于LB培养基[10g/l细菌培养用胰化蛋白胨(Difco公司生产)、5g/l酵母提取物(Difco公司生产)、5g/l氯化钠],于30℃下静置培养过夜。培养后通过使用Current Protocols in Molecular Biology所述的饱和酚方法,对该微生物的染色体DNA进行分离纯化。
使用Perseptive Biosystems公司制造8905型DNA合成仪,合成具有序列号19~22表示的碱基序列的DNA(以下分别称之为引物A、引物B、引物C和引物D)。引物A是附加在枯草芽孢杆菌染色体DNA的含有ywfE起始密码子的区域5'端的含有XhoI识别序列的碱基序列。引物B是附加在与含有ywfE的终止密码子的序列互补的碱基序列号5'端的含有BamHI识别序列的碱基序列。引物C是附加在含有trp启动子的表达载体pTrS30[从大肠杆菌JM109/pTrS30(FERM BP-5407)制备]的trp启动子区域的碱基序列号5'端的含有EcoRI识别序列的碱基序列。引物D是附加在与含有trp启动子的表达载体pTrS30的trp启动子区域的序列互补的序列号5'端的含有XhoI识别序列的碱基序列。
在ywfE基因片段的扩增中使用上述引物A和引物B、枯草芽孢杆菌的染色体DNA作为模板,在trp启动子区域的片段扩增中使用引物C和引物D、pTrS30作为模板进行PCR。PCR通过制备含有作为模板的0.1μg的染色体DNA或10ng的pTrS30、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶(Stratagene公司生产)、4μL的Pfu DNA聚合酶用×10缓冲液(Stratagene公司生产)、各200μmol/L的dNTP(dATP、dGTP、dCTP和dTTP)的反应液40μL,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认在使用引物A和引物B的PCR中扩增了相当于ywfE基因片段的约1.4kb、在使用引物C和引物D的反应中扩增了相当于trp启动子区域的DNA片段的约0.3kb的DNA片段后,添加与剩下反应液等量的TE[10mmol/LTris-HCl(pH8.0)、1mmol/L EDTA]饱和酚/氯仿(1vol/1vol)溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离使DNA沉淀后,将该DNA溶解于20μL的TE。
使用各个该溶液各5μL,将用引物A和引物B扩增的DNA用限制酶XhoI和BamHI进行酶切,而使用引物C和引物D扩增的DNA用限制酶EcoRI和XhoI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,使用geneclean II试剂盒(GENECLEAN II kit、BIO101社制),分别回收含有ywfE的1.4kb和含有trp启动子区域的0.3kb的DNA片段。
将含有trp启动子的表达载体pTrS30[由大肠杆菌JM109/pTrS30(FERM BP-5407)制备]0.2μg用限制酶EcoRI和BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段,通过与上述同样的方法,回收4.5kb的DNA片段。
使用连接试剂盒(Takara Bio公司生产),使上述得到的含有ywfE的1.4kb片段、含有trp启动子区域的0.3kb片段和4.5kb的片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法[Proc.Natl.Acad.Sci.,USA,69,2110(1972)]用该反应液转化大肠杆菌NM522株(Stratagene公司生产)后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖下来的转化体菌落提取质粒,通过使用限制酶解析其构造,确认取得了在trp启动子下游连接了ywfE的表达载体pPE43(图1)。
【实验例3二肽的生产】
将实验例2得到的带有pPE43的大肠杆菌NM522(大肠杆菌NM522/pPE43株)接种在加入了含有50μg/ml氨苄青霉素的8ml LB培养基的大型试管中,于28℃下培养17小时。对该培养液进行离心分离、取得湿菌体。
制备由终浓度60mg/ml的该湿菌体、120mmol/L的磷酸钾缓冲液(pH7.4)、60mmol/L的氯化镁、60mmol/L的ATP、30mmol/L的L-Ala、30mmol/L的L-Gln、0.4%的NymeenS-215构成的0.1ml的反应液,于37℃下反应3分钟。
反应结束后,将反应生成物用二硝基酚化法衍生后,通过HPLC法进行分析。在通过HPLC法进行的分析中,分离柱使用关东化学社制的Lichrosorb-RP-18柱,洗脱液使用1%(v/v)磷酸、25%(v/v)乙腈,以0.7ml/分钟的流速进行。其结果确认反应液中生成蓄积120mg/L的L-丙氨酰-L-谷氨酰胺(L-Ala-L-Gln)。
在作为对照菌株只含有载体的大肠杆菌NM522/pTrS30株的菌体中没有确认L-Ala-L-Gln的生成。
【实验例4C末端附加His-tag重组型二肽合成酶的纯化】
使用上述DNA合成仪,合成具有序列号23和24表示的碱基序列的DNA(以下、分别称为引物E、引物F)。引物E是含有将ywfE的起始密码子(atg)置换为NcoI识别序列(ccatgg)的区域的碱基序列。引物F是含有将ywfE的终止密码子置换为BamHI识别序列(ggatcc)的区域的碱基序列。
以枯草芽孢杆菌168株(ATCC23857)的染色体DNA作为模板,使用上述引物E和引物F作为引物组进行PCR。PCR通过制备含有0.1μg的染色体DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP的反应液40μL,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认扩增了相当于ywfE片段的约1.4kb后,添加与剩下反应液等量的TE饱和酚/氯仿溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA沉淀溶解于20μL的TE。
使用各个该溶液各5μL,将扩增的DNA用限制酶NcoI和BamHI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,使用GENECLEAN II试剂盒回收含有ywfE的1.4kb的DNA片段。
将C末端附加His-tag重组体表达载体pQE60(Qiagen公司生产)0.2g用限制酶NcoI和BamHI酶切后,通过琼脂糖凝胶电泳将DNA片段分离后,使用与上述同样的方法回收3.4kb的DNA片段。
使用连接试剂盒,使上述得到的含有ywfE的1.4kb DNA片段和3.4kb DNA片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该连接反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
通过众所周知的方法从生长繁殖的转化体菌落提取质粒,使用限制酶对其构造进行解析,确认取得了C末端附加His-tag型ywfE表达载体的pQE60ywfE(图2)。
将带有pQE60ywfE的大肠杆菌NM522(大肠杆菌NM522/pQE60ywfE株)接种在加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的大型试管,于28℃下培养17小时。将该培养液接种在加入了含有50μg/ml的氨苄青霉素的50ml的LB培养基的250ml容量三角烧瓶,于30℃下培养3小时后,按照终浓度达到1mmol/L那样添加异丙醇-β-D-硫代半乳糖苷(IPTG),再于30℃下培养4小时。对该培养液进行离心分离取得湿菌体,使用HisTrap(附加His-tag蛋白质纯化试剂盒、Amersham Pharmasia Biotech社制),根据说明书从该湿菌体纯化附加His-tag重组型酶。
【实验例5使用附加His-tag重组型酶生产二肽(1)】
(i)制备由实验例4中取得的纯化了的附加His-tag重组型酶0.04mg、100mmol/L的Tris-HCl(pH8.0)、60mmol/L的氯化镁、60mmol/L的ATP、30mmol/L的L-Ala、30mmol/L的L-Gln构成的0.1ml反应液,于37℃进行16小时反应。
反应结束后,通过与上述实验例3同样的方法分析反应生成物,确认反应液中生成蓄积3.7g/L的L-Ala-L-Gln和0.3g/L的L-丙氨酰-L-丙氨酸(L-Ala-L-Ala)。
(ii)除了酶为0.01mg、取代L-Gln含有L-Phe、L-Met、L-Leu或L-Val以外,制备与上述(i)的反应液组成相同的反应液,于上述(i)的反应条件下进行反应。
反应结束后,通过与上述实验例3同样的方法对反应生成物进行分析,确认反应液中分别生成蓄积7.0g/L的L-丙氨酰-L-苯丙氨酸(L-Ala-L-Phe)、7.0g/L的L-丙氨酰-L-蛋氨酸(L-Ala-L-Met)和0.03g/L的L-Ala-L-Ala、5.0g/L的L-丙氨酰-L-亮氨酸(L-Ala-L-Leu)和0.2g/L的L-Ala-L-Ala、或1.6g/L的L-丙氨酰-L-缬氨酸(L-Ala-L-Val)和0.3g/L的L-Ala-L-Ala。
(iii)除了酶为0.01mg、取代L-Ala含有Gly、取代L-Gln含有L-Phe或L-Met以外,制备与上述(i)的反应液的组成相同的反应液,在上述(i)的反应条件使其反应。
反应结束后,通过与上述实验例3同样的方法对反应生成物进行分析,确认反应液中分别生成蓄积5.2g/L的甘氨酰-L-苯丙氨酸(Gly-L-Phe)或1.1g/L的甘氨酰-L-蛋氨酸(Gly-L-Met)。
如果从上述反应液组成中去除ATP,完全不能生成二肽。
以上结果表明ywfE基因产物在ATP存在下具有生成L-Ala和L-Gln、由L-Phe、L-Met、L-Leu或L-Val生成L-Ala-L-Gln和L-Ala-L-Ala、L-Ala-L-Phe、L-Ala-L-Met和L-Ala-L-Ala、L-Ala-L-Leu和L-Ala-L-Ala、或L-Ala-L-Val和L-Ala-L-Ala的活性、由Gly和L-Phe或L-Met生成Gly-L-Phe或Gly-L-Met的活性。
【实验例6使用附加His-tag重组型酶生产二肽(2)】
制备由实验例4中得到的纯化了的附加His-tag重组型酶0.04mg、100mmol/L的Tris-HCl(pH8.0)、60mmol/L的氯化镁、60mmol/L的ATP构成的0.1ml反应液,按照分别达到30mmol/L那样向反应液中添加由表1的第1行和最左列的氨基酸的组合构成的各种L-氨基酸、Gly或β-Ala,于37℃下进行16小时反应。反应结束后,对反应生成物进行HPLC分析,确认生成表1所示的二肽。
【表1-1】

【表1-2】

【表1-3】
以表1的第1行和最左列所述的2种(或1种)L-氨基酸、Gly或β-Ala作为底物反应时生成的二肽分别表示在表格内。○表示生成了二肽,但序列未确定,×表示不能确认二肽的生成,而空栏表示未实施。
【实验例7使用附加His-tag重组型酶表达株生产二肽】
将实验例4得到的大肠杆菌NM522/pQE60ywfE株接种在加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的大型试管,于28℃下培养17小时。将该培养液接种在加入了含有50μg/ml的氨苄青霉素的50ml的LB培养基的250ml容量三角烧瓶,于30℃下培养3小时后,按照终浓度达到1mmol/L那样添加IPTG,再于30℃下培养4小时。对该培养液进行离心分离取得湿菌体。
将由200g/L的湿菌体、50g/L的葡萄糖、5g/L的肌醇六磷酸(使用33%的浓氢氧化钠溶液稀释成中性)、15g/L的磷酸二氢钾、5g/L的硫酸镁·7水合物、4g/L的NymeenS-215、10ml/L的二甲苯、200mmol/L的L-Ala、200mmol/L的L-Gln构成的20ml反应液(pH7.2)加入到50ml容量的烧杯、于32℃、900rpm的条件下进行2小时反应。反应中使用2mol/L的氢氧化钾将反应液的pH保持在7.2。
将反应生成物用与实验例3所述方法同样的方法进行分析,确认25mg/L的L-Ala-L-Gln的蓄积。
【实验例8从属于芽孢杆菌属的各种微生物中克隆相当于ywfE基因的基因和该基因的解析】
根据序列号9表示的碱基序列,象以下那样操作取得相当于存在于枯草芽孢杆菌ATCC15245、ATCC6633、IAM1213、IAM1107、IAM1214、ATCC9466、IAM1033、ATCC21555、解淀粉芽孢杆菌IFO3022、和短小芽孢杆菌NRRL B-12025的ywfE基因的基因。
首先,将枯草芽孢杆菌ATCC15245、ATCC6633、IAM1213、IAM1107、IAM1214、ATCC9466、IAM1033、ATCC21555、解淀粉芽孢杆菌IFO3022、和短小芽孢杆菌NRRL B-12025分别接种于LB培养基,于30℃下静置培养过夜。培养后,通过使用Current Protocolsin Molecular Biology所述的饱和酚的方法,对于各个微生物的染色体DNA分别进行分离纯化。
使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成具有序列号25和26表示的碱基序列的DNA(以下、分别称为引物G、引物H)。引物G是含有存在于枯草芽孢杆菌168株的染色体DNA上的ywfE的起始密码子的上游区域的序列。引物H是与含有ywfE的终止密码子的下游序列互补的序列。
以枯草芽孢杆菌ATCC15245、ATCC6633、IAM1213、IAM1107、IAM1214、ATCC9466、IAM1033、ATCC21555、或解淀粉芽孢杆菌IFO3022的染色体DNA作为模板,使用上述引物G和引物H作为引物组进行PCR。PCR通过制备含有0.1μg的染色体DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP的反应液40μL,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认扩增了相当于ywfE基因片段的约1.4kb后,添加与剩下反应液等量的TE饱和酚/氯仿溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA沉淀溶解于20μL的TE。
使用连接试剂盒,将上述得到的来自各菌株染色体DNA的1.4kb片段和pCR-blunt(Invitrogen公司生产)于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
通过根据众所周知的方法从生长繁殖的转化体的菌落提取质粒,用限制酶对各个构造进行解析,确认取得含有相当于ywfE基因的基因的质粒pYWFE1(来自ATCC15245株,具有序列号36表示的碱基序列的DNA)、pYWFE2(来自ATCC6633株,具有序列号10表示的碱基序列的DNA)、pYWFE3(来自IAM1213株,具有序列号11表示的碱基序列的DNA)、pYWFE4(来自IAM1107株,具有序列号12表示的碱基序列的DNA)、pYWFE5(来自IAM1214株,具有序列号13表示的碱基序列的DNA)、pYWFE6(来自ATCC9466株,具有序列号9表示的碱基序列的DNA)、pYWFE7(来自IAM1033株,具有序列号36表示的碱基序列的DNA)、pYWFE8(来自ATCC21555株,具有序列号14表示的碱基序列的DNA)、pYWFE9(来自IFO3022株,具有序列号15表示的碱基序列的DNA)。
而相当于来自短小芽孢杆菌NRRL B-12025的ywfE的基因(具有序列号16表示的碱基序列的DNA)象以下那样取得。
以上述制备的NRRL B-12025株的染色体DNA作为模板,使用序列号27和28表示的碱基序列构成的DNA作为引物组,进行PCR。PCR通过制备含有0.1μg的染色体DNA、0.5μmol/L的各引物、2.5units的Z-taq聚合酶(Takara Bio公司生产)、5μL的Z-taq聚合酶用×10缓冲液(Takara Bio公司生产)、200μmol/L的各dNTP的反应液50μL,按照98℃下5秒钟、55℃下30秒钟、72℃下1分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认约0.8kb的片段扩增后,添加与剩下反应液等量的TE饱和酚/氯仿溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA沉淀溶解于20μL的TE。
使用连接试剂盒将上述得到的0.8kb片段和pGEM T-easy(Promega公司生产)于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌DH5α株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
从上述得到的转化体提取质粒,决定约0.8kb的插入DNA片段的碱基序列,确认是由序列号16表示的碱基序列中的第358~1160位碱基构成的碱基序列。
然后将该质粒用EcoRI进行酶切后,通过琼脂糖凝胶电泳将DNA片段分离。使用geneclean II试剂盒纯化该DNA片段。使用DIG-HighPrime DNA Labeling&Detection Starter Kit I(Roche Diagnostics公司生产)对约0.5μg该纯化DNA片段进行DIG标记。DIG标记根据该试剂盒附带的说明书进行。
使用上述得到的DIG标记的DNA,进行NRRL B-12025株的染色体DNA的Southern解析。
将NRRL B-12025株的染色体DNA分别使用BamHI、EcoRI、HindIII、KpnI、PstI、SacI、SalI和SphI完全消化,通过琼脂糖电泳将DNA片段分离后,根据常法转移到nylon membrane plus charge(Roche Diagnostics公司生产)。
通过照射UV,将DNA片段固定在该尼龙膜后,使用上述探针DNA和该尼龙膜进行Southern杂交。
杂交通过使该探针DNA和该尼龙膜在65℃下接触16小时,然后使用0.1%SDS和2×SSC构成的溶液将该尼龙膜于室温下清洗5分钟,清洗2次,再用0.1%SDS和0.5×SSC构成的溶液,于65℃下15分钟、清洗2次进行,其它操作、条件和杂交的DNA的检测根据上述的DIG-High Prime DNA Labeling&Detection Starter Kit I附带的说明书进行。
该结果表明在HindIII和PstI完全消化片段3.5kbp附近看到发色。
然后将NRRL B-12025株的染色体DNA使用HindIII和PstI分别完全消化,通过琼脂糖凝胶电泳分离DNA片段。使用geneclean II试剂盒从各个限制酶消化的DNA中纯化3-4kbp片段,使用连接试剂盒使其自环化。
根据上述决定的0.8kb的DNA片段的碱基序列,设计、合成序列号29和30表示的碱基序列,以上述取得的环化DNA作为模板进行PCR。PCR通过制备含有10ng的环化DNA、0.5μmol/L的各引物、2.5units的pyrobest聚合酶(Takara Bio公司生产)、5μL的pyrobest聚合酶用×10缓冲液(Takara Bio公司生产)、200μmol/L的各dNTP的反应液50μL,按照98℃下5秒钟、55℃下30秒钟、72℃下3分30秒钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认约3.0kb片段扩增后,添加与剩下反应液等量的TE饱和酚/氯仿溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA沉淀溶解于20μL的TE。
使用连接试剂盒对上述得到的DNA片段和Zero Blunt PCRCloning Kit(Invitrogen公司生产)进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过使用限制酶对其构造进行解析,确认得到了含有相当于ywfE基因的基因的质粒pYWFE10(来自NRRL B-12025株、具有序列号16表示的碱基序列的DNA)。
使用碱基序列分析装置373A·DNA序列仪决定上述得到的相当于含有pYWFE1~pYWFE10的ywfE基因的各基因的碱基序列。
虽然由pYWFE1、pYWFE6和pYWFE7中含有的基因编码的蛋白质的氨基酸序列与ywfE基因编码的蛋白质的氨基酸序列相同,但由pYWFE2、pYWFE3、pYWFE4、pYWFE5、pYWFE8、pYWFE9和pYWFE10中含有的基因编码的蛋白质的氨基酸序列与ywfE基因编码的蛋白质的氨基酸序列却不同。
由相当于pYWFE2、pYWFE3、pYWFE4、pYWFE5、pYWFE8、pYWFE9、pYWFE10以及pYWFE1和pYWFE7中含有的ywfE基因的基因编码的蛋白质的氨基酸序列如序列号2~8以及1所示,而该基因的碱基序列分别如序列号10~16以及36所示。
【实验例9C末端附加His-tag型重组型二肽合成酶的纯化】
以枯草芽孢杆菌ATCC15245、ATCC6633、IAM1213、IAM1107、IAM1214、ATCC9466、IAM1033、ATCC21555、或解淀粉芽孢杆菌IFO3022的染色体DNA为模板,使用实验例2所述的引物A和引物B作为引物组进行PCR。PCR通过制备含有0.1μg的染色体DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP的反应液40μL,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
当以短小芽孢杆菌NRRL B-12025的染色体DNA作为模板时,使用具有序列号31和32表示的碱基序列的DNA作为引物组,在与上述同样的条件下进行PCR。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认分别扩增了相当于ywfE片段的约1.4kb的DNA片段后,添加与剩下反应液等量的TE饱和酚/氯仿溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA沉淀溶解于20μL的TE。
使用各个该溶液5μL,将扩增的DNA用限制酶NcoI和BamHI酶切,通过琼脂糖凝胶电泳分离DNA片段后,使用geneclean II试剂盒,回收含有相当于ywfE基因的基因的1.4kb的DNA片段。
然后将C末端附加His-tag型重组体表达载体pQE60 0.2μg用限制酶NcoI和BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段,通过与上述同样的方法回收3.4kb的DNA片段。
使用连接试剂盒使上述得到的含有相当于枯草芽孢杆菌168株的ywfE基因的基因的1.4kb的DNA片段和3.4kb的DNA片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过使用限制酶对它们的构造进行解析,确认取得了作为C末端附加His-tag型基因表达载体的pQE60ywfE1(含有来自ATCC15245的基因的载体)、pQE60ywfE2(含有来自ATCC6633的基因的载体)、pQE60ywfE3(含有来自IAM1213的基因的载体)、pQE60ywfE4(含有来自IAM1107的基因的载体)、pQE60ywfE5(含有来自IAM1214的基因的载体)、pQE60ywfE6(含有来自ATCC9466的基因的载体)、pQE60ywfE7(含有来自IAM1033的基因的载体)、pQE60ywfE8(含有来自ATCC21555的基因的载体)、pQE60ywfE9(含有来自IFO3022的基因的载体)、和pQE60ywfE10(含有来自NRRL B-12025的基因的载体)。
将上述得到的大肠杆菌NM522/pQE60ywfE1~NM522/pQE60ywfE10株分别接种在加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的大型试管,于28℃下培养17小时。将该培养液接种于加入了含有50μg/ml的氨苄青霉素的50ml的LB培养基的250ml容量的三角烧瓶,于30℃下培养3小时后,按照终浓度达到1mmol/L那样添加IPTG,再于30℃下培养4小时。使用HisTrap,根据其使用说明书,从对该培养液进行离心分离得到的湿菌体纯化附加His-tag重组型酶。
【实验例10使用纯化酶生产二肽】
制备由实验例9得到的重组型酶0.04mg、100mmol/L的Tris-HCl(pH8.0)、60mmol/L的氯化镁、60mmol/L的ATP、30mmol/L的L-Ala和30mmol/L的L-Gln构成的0.1ml反应液,于37℃下反应16小时。
反应结束后,通过实验例3所述的方法对反应液进行分析,结果确认3.0~3.5g/L的L-Ala-L-Gln和0.25~0.3g/L的L-Ala-L-Ala生成蓄积。
而如果从上述反应液组成除去ATP,完全不能生成L-Ala-L-Gln和L-Ala-L-Ala。
以上结果表明实验例8得到的基因产物无论那一个都具有在ATP存在下由L-Ala和L-Gln生成L-Ala-L-Gln和L-Ala-L-Ala的活性。
【实验例11albC基因和其类似基因的取得】
根据诺尔斯氏链霉菌的albC基因的碱基序列[Chemistry&Biol.,9,1355(2002)],通过以下方法从诺尔斯氏链霉菌和小白链霉菌取得albC基因和其类似基因。
首先将诺尔斯氏链霉菌IFO15452株和小白链霉菌IFO14147株分别接种于添加了1%甘氨酸的KM73培养基[2g/l酵母提取液(Difco公司生产)、10g/l可溶性淀粉(和光纯药工业社制)]、KP培养基[15g/l葡萄糖、10g/l甘油、10g/l多胨(日本制药株式会社制)、10g/l肉提取物(极东制药工业株式会社生产)、4g/l碳酸钙],于28℃下振荡培养过夜。而诺尔斯氏链霉菌IFO15452株和小白链霉菌IFO14147株购自独立行政法人制品评价技术基盘机构生物资源部门[National Instituteof Technology and Evaluation(NITE)Biological Resource Center(BRC)](〒292-0818千葉県木更津市かずさ鎌足2-5-8)。
培养后根据Genetic Manipulation of Streptomyces:a LaboratoryManual:John Innes Foundation所述的方法对该微生物的染色体DNA进行分离纯化。
根据albC基因的碱基序列,使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成具有序列号41和42表示的碱基序列的DNA(以下、分别称为引物J、引物K)。引物J是附加在诺尔斯氏链霉菌染色体DNA的含有albC基因起始密码子的区域5'端的含有NcoI识别序列的碱基序列。引物K是附加在与含有albC基因的终止密码子的序列互补的碱基序列的5'端的含有BglII识别序列的碱基序列。
使用上述的引物J和引物K作为引物组,使用诺尔斯氏链霉菌或小白链霉菌的染色体DNA作为模板进行PCR。PCR通过制备含有作为模板的0.1μg的染色体DNA、0.5μmol/L的各引物、2.5units的Ex Taq DNA聚合酶(Takara Bio公司生产)、5μL的ExTaq DNA聚合酶用×10缓冲液(Takara Bio公司生产)、各200μmol/L的dNTP、5μL的二甲基亚砜的反应液50μL,按照94℃下1分钟、55℃下30秒钟、72℃下1分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认约0.7kb的DNA片段扩增后,添加与剩下反应液等量的TE饱和酚/氯仿溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA沉淀分别溶解于20μL的TE。
使用各个该溶液各5μL,将扩增了的DNA用限制酶NcoI和BglII进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,使用geneclean II试剂盒分别回收700bp的DNA片段。
将0.2μg含有噬菌体T5启动子的表达载体pQE60用限制酶NcoI和BglII酶切后,通过琼脂糖凝胶电泳将DNA片段分离后,使用与上述同样的方法回收3.4kb的DNA片段。
使用连接试剂盒,使含有上述得到的来自各放线菌的0.7kb片段和来自pQE60的3.4kb片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该连接反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
通过众所周知的方法从生长繁殖的转化体菌落提取质粒,使用限制酶对其构造进行解析,确认取得了作为在噬菌体T5启动子下游连接了来自诺尔斯氏链霉菌的DNA的表达载体pAL-nou、和连接了来自小白链霉菌的DNA的表达载体pAL-alb(图3)。
当使用碱基序列决定装置373A·DNA序列仪决定插入到各个质粒的来自放线菌的DNA部分的碱基序列时,确认pAL-alb中含有编码具有序列号37表示的氨基酸序列的蛋白质的DNA、即含有序列号39表示的碱基序列的DNA,在pAL-nou含有编码具有序列号38表示的氨基酸序列的蛋白质的DNA、即具有序列号40表示的碱基序列的DNA。
【实验例12使用菌体作为酶源制造二肽】
将实验例11得到的带有pAL-nou或pAL-alb的大肠杆菌NM522(大肠杆菌NM522/pAL-nou株或NM522/pAL-alb株)和不带有质粒的NM522株接种于加入了含有50μg/ml的氨苄青霉素的10ml的LB培养基的试管(对于不带有质粒的株的情况,不添加氨苄青霉素,以下相同),于30℃下培养17小时。将该培养液0.5ml分别接种于加入了50ml的LB培养基的250ml容量的三角烧瓶,于30℃下振荡培养1小时后,按照终浓度达到1mmol/L那样添加IPTG,再继续培养4小时。对该培养液进行离心分离,取得湿菌体。
制备由终浓度100mg/ml的该湿菌体、60mmol/L的磷酸钾缓冲液(pH7.2)、10mmol/L的氯化镁、10mmol/L的ATP、1g/L的L-Leu、1g/L的L-Phe构成的3.0ml反应液,于30℃下进行反应。反应1小时后取样,按照终浓度达到20%(v/v)那样加乙腈后,使用HPLC分析反应生成物。通过HPLC进行的分析,分离柱使用ODS-HA柱(YMC社制)、作为洗脱液使用30%(v/v)乙腈,以流速0.6ml/min、测定215nm的紫外吸收的条件进行。
分析结果确认在大肠杆菌NM522/pAL-nou株的反应液中有36.7mg/l的环(L-亮氨酰-L-苯丙氨酸)[环(L-Leu-L-Phe)]的蓄积,但在大肠杆菌NM522株的反应液中完全没有检测到环(L-Leu-L-Phe)。将同样反应液在以下的条件下用HPLC进行分析,测定到作为直链二肽(以下、“直链二肽”简单称为“二肽”)的L-亮氨酰-L-苯丙氨酸(L-Leu-L-Phe)和L-苯丙氨酰-L-亮氨酸(L-Phe-L-Leu)。
两二肽用F-moc化法进行衍生后,使用HPLC进行分析。通过HPLC进行的分析,分离柱使用ODS-HG5(野村化学社制)、作为洗脱液使用A液(醋酸6ml/l、20%(v/v)乙腈、用三乙胺调整至pH4.8)和B液(醋酸6ml/l、70%(v/v)乙腈、用三乙胺调整到pH4.8),分析开始后5分钟之前A液:B液=8:2、5分钟~20分钟之前加线性梯度,在达到20分钟时使A液:B液=1:1,在流速0.6ml/分钟、激发波长254nm、荧光波长630nm检测二肽的条件下进行。
结果确认在大肠杆菌NM522/pAL-nou株的反应液中有21.9mg/L的L-Leu-L-Phe和12.0mg/L的L-Phe-L-Leu蓄积。而在作为对照株使用的大肠杆菌NM522株的反应液中没有检测到任何二肽。
这表明实验例11中取得的环二肽合成酶具有合成二肽的能力。
【实验例13使用纯化酶制造二肽(1)】
与实验例12同样培养大肠杆菌NM522/pAL-nou株。培养结束后通过离心分离取得湿菌体,用60mmol/L的磷酸钾缓冲液(pH7.2)清洗后,悬浮于含有10mmol/L咪唑的20mmol/L磷酸钾缓冲液。将该悬浮液于4℃下进行超声处理,取得菌体裂解液。将该菌体裂解液(10ml、含蛋白质0.863mg)加到Amersham公司生产的His-tag纯化柱,通过加含有10mmol/L的咪唑的20mmol/L的磷酸钾缓冲液15ml进行清洗,于柱内对附带His-tag的albC蛋白质进行纯化。然后向保持这个附带His-tag的albC蛋白质的柱加与实验例12同组成的反应液(反应液组成:60mmol/L的磷酸钾缓冲液(pH7.2)、10mmol/L的氯化镁、10mmol/L的ATP、1g/L的L-Leu、1g/L的L-Phe构成的反应液)2ml,在柱内保持底物的状态下,于30℃进行温育。24小时后用同组成的反应液3ml对柱内的反应液进行洗脱,通过与实验例12同样的方法对反应液中的环二肽和二肽进行定量。
由结果可知生成了环(L-Leu-L-Phe)6.8mg/L、L-Leu-L-Phe28.7mg/L和L-Phe-L-Leu18.5mg/L。当用不含有ATP的反应液进行同样温育时,环二肽、二肽都没有检测到。
【实验例14使用纯化酶制造二肽(2)】
除了将底物氨基酸换成其它的氨基酸以外,用与实验例13同样的方法进行酶反应,对生成物进行分析。反应液除了将底物氨基酸换成为1g/L的L-Ala、L-Leu或L-Phe以外,使用与实验例13同样组成的溶液。
由该结果可知在反应开始后24小时内,分别生成了9.41mg/L的L-Ala-L-Ala、7.85mg/L的L-Leu-L-Leu、或5.20mg/L的L-Phe-L-Phe。
【实验例15强化ywfE基因表达的大肠菌的制备】
使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成分别具有序列号84~87所述的序列的DNA(以下、分别称为引物L、引物M、引物N、引物O)。序列号84的序列是附加在质粒pQE60ywfE的含有作为ywfE基因的核糖体结合序列的Shine-Dalgarno序列的区域的5'端的含有XhoI识别序列的序列。序列号85的序列是附加在与含有ywfE基因的终止密码子的序列互补的序列的5'端的含有BamHI识别序列的序列。而序列号86的序列是附加在含有trp启动子的表达载体pTrS30的trp启动子区域序列的5'端的含有EcoRI识别序列的序列。序列号87的序列是附加在与含有trp启动子的表达载体pTrS30的trp启动子区域的序列互补的序列的5'端的含有XhoI识别序列的序列。
以质粒pQE60ywfE作为模板,在ywfE基因片段的扩增中,使用上述的引物L和引物M,在trp启动子区域的片段的扩增中使用引物N和引物O作为引物组进行PCR。PCR通过制备含有10ng的pQE60ywfE、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP的40μL的反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认在使用引物L和引物M的PCR中扩增了相当于ywfE基因片段的约1.4kb、在使用引物N和引物O的反应中扩增了相当于trp启动子区域的片段的约0.3kb的片段后,添加与剩下反应液等量的TE饱和酚/氯仿溶液,并混合。向该溶液经离心分离后得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA溶解于20μL的TE。
使用上述得到的各个DNA溶液5μL,将用引物L和引物M扩增了的DNA用限制酶XhoI和BamHI进行酶切,而使用引物N和引物O扩增了的DNA用限制酶EcoRI和XhoI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,使用geneclean II试剂盒,分别回收含有ywfE的1.4kb和含有trp启动子区域的0.3kb的DNA片段。
将含有0.2μg的trp启动子的表达载体pTrS30用限制酶EcoRI和BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段,同样回收4.5kb的DNA片段。
使用连接试剂盒,使上述得到的含有ywfE的1.4kb片段、含有trp启动子区域的0.3kb片段和4.5kb的片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖下来的转化体菌落提取质粒,得到在trp启动子下游含有ywfE基因的表达载体pPE56。通过限制酶消化确认了该载体的结构(图4)。
【实验例16pepD基因、pepN基因、pepB基因、pepA基因和dpp操纵子缺失株的制作】
大肠杆菌染色体DNA上的特定基因缺失的菌株根据利用λ噬菌体的同源重组系的方法[Proc.Natl.Acad.Sci.USA,97,6641-6645(2000)]制作。
以下所述的质粒pKD46、pKD3和pCP20是从大肠杆菌基因贮存中心(美国耶鲁大学)获得保持该质粒的大肠杆菌株,通过众所周知的方法从该株提取使用。
(1)基因缺失用DNA片段的克隆
以使存在于大肠杆菌K12株的染色体DNA上的具有序列号55表示的碱基序列的pepD基因、具有序列号56表示的碱基序列的pepN基因、具有序列号57表示的碱基序列的pepB基因、具有序列号58表示的碱基序列的pepA基因和具有序列号59表示的碱基序列的dppA基因、具有序列号60表示的碱基序列的dppB、具有序列号61表示的碱基序列的dppC基因、具有序列号62表示的碱基序列的dppD基因和具有序列号63表示的碱基序列的dppF基因缺失为目的,使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成与位于大肠杆菌K12株的染色体DNA上的各个缺失靶基因的上游和下游的由36bp构成的碱基序列同源的碱基序列、和具有序列号54表示的来自酵母的Flp重组酶识别碱基序列的DNA。但是dppA基因、dppB基因、dppC基因、dppD基因和dppF基因因为形成操纵子,合成与位于该操纵子的上游和下游的碱基序列同源的碱基序列的DNA。
即,分别合成由作为pepD基因缺失用DNA片段扩增用引物组的序列号64和65、作为pepN基因缺失用DNA片段扩增用引物组的序列号66和67、作为pepA基因缺失用DNA片段扩增用引物组的序列号68和69、作为pepB基因缺失用DNA片段扩增用引物组的序列号70和71、作为dpp操纵子缺失用DNA片段扩增用引物组的序列号72和73表示的碱基序列构成的DNA。
然后使用上述合成DNA作为引物组,以pKD3DNA作为模板进行PCR。PCR通过使用含有10ng的质粒DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将各个反应液的1/10量进行琼脂糖凝胶电泳,确认目的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并进行混合。
将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,取得pepD基因、pepN基因、pepB基因、pepA基因和dpp操纵子缺失用的含有氯霉素抗性基因的DNA片段。
(2)pepD基因缺失大肠杆菌JM101的制作
用pKD46转化大肠杆菌JM101株后,涂布于含有100mg/L的氨苄青霉素的LB琼脂培养基,通过于30℃下进行培养,选择保持pKD46的大肠杆菌JM101株(以下、称为大肠杆菌JM101/pKD46)。
质粒pKD46含有λRed重组酶基因,该基因的表达可以通过L-阿拉伯糖诱导。因此如果将在L-阿拉伯糖存在下能够生长繁殖的带有pKD46的大肠菌用直链状DNA进行转化,会发生高频率同源重组。另外pKD46由于具有温度敏感性的复制起点,通过在42℃下使其生长繁殖,可以很容易地使质粒脱落。
通过电脉冲法将上述取得的pepD基因缺失用含有氯霉素抗性基因的DNA片段导入在10mmol/L的L-阿拉伯糖和50μg/ml的氨苄青霉素存在下培养得到的大肠杆菌JM101/pKD46,将pepD基因缺失用含有氯霉素抗性基因的DNA片段通过同源重组转运在大肠菌JM101的染色体DNA上的转化株涂布于含有25mg/L的氯霉素的LB琼脂培养基(细菌培养用胰化蛋白胨10g/L、细菌培养用酵母提取物5g/L、氯化钠5g/L、琼脂15g/L),通过于30℃下培养进行选择。
将选择的氯霉素抗性株接种于含有25mg/L的氯霉素的LB琼脂培养基,于42℃下培养14小时后,进行单菌落分离。将得到的各菌落影印在含有25mg/L的氯霉素的LB琼脂培养基、以及含有100mg/l的氨苄青霉素的LB琼脂培养基,于37℃培养,通过选择表现出氯霉素抗性,而且表现出氨苄青霉素敏感性的菌落,取得pKD46脱落株。
然后用pCP20转化上述得到的pKD46脱落株,通过在含有100mg/l的氨苄青霉素的LB琼脂培养基上进行选择,取得保持pCP20的pKD46脱落株。
质粒pCP20具有来自酵母的Flp重组酶基因,该基因的表达可以在42℃下进行诱导。
另外,由于在上述制作的pepD基因、pepN基因、pepB基因、pepA基因以及dpp操纵子缺失用的含有氯霉素抗性基因的DNA片段的氯霉素抗性基因的两端存在Flp重组酶识别的碱基序列,通过Flp重组酶催化的同源重组可以很容易地使该抗性基因脱落。
另外,由于pCP20含有温度敏感性的复制起点,通过使pCP20保持株在42℃下生长繁殖,可以同时诱导Flp重组酶的表达和pCP20的脱落。
将上述取得的保有pCP20的pKD46脱落株接种于没有添加药剂的LB琼脂培养基,于42℃下培养14小时后,进行单菌落分离。将得到的各菌落影印在没有添加药剂的LB琼脂培养基、含有25mg/L氯霉素的LB琼脂培养基和含有100mg/L的氨苄青霉素的LB琼脂培养基,于30℃下培养,选择表现出氯霉素敏感性,而且表现出氨苄青霉素敏感性的菌落。
根据常法(生物工学实验书、日本生物工学会编97~98页、培风馆、1992年)从上述选择的各株制备各个染色体DNA。使用根据缺失靶基因的pepD基因的内部碱基序列设计的具有序列号74和75表示的碱基序列的DNA作为引物组,以染色体DNA作为模板进行PCR。PCR通过使用含有0.1g的染色体DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
在上述PCR中,将没有检测到扩增DNA片段的菌株作为pepD基因缺失株,命名为大肠杆菌JPD1株。
(3)大肠杆菌JM101的染色体DNA上的pepD基因和pepN基因缺失株的制作
将上述(2)得到的大肠杆菌JPD1株用pKD46转化后,涂布于含有100mg/l的氨苄青霉素的LB琼脂培养基,通过于30℃下进行培养,选择保持pKD46的大肠杆菌JPD1(以下、称为大肠杆菌JPD1/pKD46)。通过电脉冲法将pepN基因缺失用含有氯霉素抗性基因的DNA片段导入大肠杆菌JPD1/pKD46,通过同源重组取得在大肠杆菌JPD1/pKD46的染色体DNA上转运了pepN基因缺失用含有氯霉素抗性基因的DNA片段的转化株。
然后通过进行与上述(2)同样的操作,取得氯霉素抗性基因从染色体DNA上脱落的菌株,将该株命名为大肠杆菌JPDN2。
(4)大肠杆菌JM101的染色体DNA上的pepN基因、pepA基因、pepB基因或dpp操纵子缺失株、和多重基因缺失株的制作pepN基因、pepA基因、pepB基因或dpp操纵子缺失株使用上述(1)制作的各基因或操纵子缺失用的含有氯霉素抗性基因的DNA片段,通过与上述(2)同样的方法进行制作。
通过上述方法取得的各个基因缺失株可以通过使用根据各个缺失基因的内部碱基序列设计、合成的具有序列号76~83表示的碱基序列的DNA作为引物组,通过与上述(2)同样的PCR进行确认。其中由序列号76和77表示的碱基序列构成的DNA是确认pepN缺失用的引物组、由序列号78和79表示的碱基序列构成的DNA是确认pepA缺失用的引物组、由序列号80和81表示的碱基序列构成的DNA是确认pepB缺失用的引物组、由序列号82和83表示的碱基序列构成的DNA是确认dpp操纵子缺失用的引物组。
将上述方法取得的dpp操纵子缺失株取名为大肠杆菌JDPP1株、pepN基因缺失株取名为大肠杆菌JPN1株、pepA基因缺失株取名为大肠杆菌JPA1株、pepB基因缺失株取名为大肠杆菌JPB7株。
另外根据上述(3)的方法,制作由pepD基因、pepN基因、pepA基因、pepB基因和dpp操纵子选出的2个以上基因或操纵子的多重缺失株。能够取得的多重缺失株的确认通过与上述(2)同样的PCR进行确认。将用上述方法取得的pepD基因和dpp操纵子缺失的二重基因缺失株命名为大肠杆菌JPDP49株,将pepB基因、pepD基因和pepN基因缺失的三重基因缺失株命名为大肠杆菌JPDNB43株,将pepD基因、pepN基因和dpp操纵子缺失的三重基因缺失株命名为大肠杆菌JPNDDP36株,将pepA基因、pepD基因、pepN基因和dpp操纵子缺失的四重基因缺失株命名为大肠杆菌JPNDAP5株,将pepB基因、pepD基因、pepN基因和dpp操纵子缺失的四重基因缺失株命名为大肠杆菌JPNDBP7株。表2给出了各基因缺失株中缺失基因名。
【表2】
| 菌株名称 | 缺失基因 |
| JM101 | 无 |
| JDPP1 | dpp操纵子 |
| JPN1 | pepN |
| JPA1 | pepA |
| JPB7 | pepB |
| JPD1 | pepD |
| JPDN2 | pepD,pepN |
| JPNDB43 | pepB,pepD,pepN |
| JPDP49 | pepD,dpp操纵子 |
| JPNDDP36 | pepD,pepN,dpp操纵子 |
| JPNDAP5 | pepA,pepD,pepN,dpp操纵子 |
| JPNDBP7 | pepB,pepD,pepN,dpp操纵子 |
【实验例17使用肽酶和二肽转运活性丧失的大肠杆菌生产的L-Ala-L-Gln和L-Ala-L-Ala产率的评价】
用实验例15制作的质粒pPE56转化实验例16得到的编码各种肽酶和二肽转运蛋白质的基因缺失株,取得表现出氨苄青霉素抗性的转化株。
将得到的转化株接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。该培养液1%添加到加人了含有100μg/ml的氨苄青霉素和氨基酸的8ml的水性介质(磷酸氢二钾16g/L、磷酸二氢钾14g/L、硫酸铵5g/L、柠檬酸(无水)1g/L、Casamino acid(Difco社制)0.5g/L、L-Pro1g/L、L-Ala2.5g/L、L-Gln2.5g/L、葡萄糖10g/l、维生素B110mg/L、硫酸镁·7水合物25mg/l、硫酸铁7水合物50mg/l、使用10mol/l的氢氧化钠溶液调整到pH7.2。L-Gln在将10倍浓缩液经滤膜过滤灭菌后,葡萄糖、维生素B1、硫酸镁·7水合物、硫酸铁·7水合物分别蒸气灭菌后添加)的试管中,于30℃下反应24小时。将该水性介质进行离心分离,取得上清。
该上清中的产物用F-moc化法进行衍生后,使用HPLC法进行分析。通过HPLC法进行的分析,分离柱使用ODS-HG5(野村化学社制)、作为洗脱液使用A液(醋酸6ml/l、20%(v/v)乙腈、用三乙胺调整到pH4.8)和B液(醋酸6ml/l、70%(v/v)乙腈、用三乙胺调整到pH4.8),直至分析开始后5分钟之前A液:B液=8:2、5分钟~20分钟之前进行线性梯度洗脱,到达20分钟时使A液:B液=1:1的条件下进行。分析结果如表3所示。
【表3】
| 菌株名称 | 缺失基因 | L-Ala-L-Gln(g/l) | L-Ala-L-Aln(g/l) |
| JM101 | 无 | 0 | 0 |
| JDPP1 | dpp操纵子 | 0.02 | 0.01 |
| JPN1 | pepN | 0.01 | 0.01 |
| JPA1 | pepA | 0.01 | 0.01 |
| JPB7 | pepB | 0.01 | 0.01 |
| JPD1 | pepD | 0.01 | 0.01 |
| JPDN2 | pepD,pepN | 0.02 | 0.03 |
| JPNDB43 | pepB,pepD,pepN | 0.05 | 0.12 |
| JPDP49 | pepD,dpp操纵子 | 0.11 | 0.08 |
| JPNDDP36 | pepD,pepN,dpp操纵子 | 0.16 | 0.21 |
| JPNDAP5 | pepA,pepD,pepN,dpp操纵子 | 0.28 | 0.26 |
| JPNDBP7 | pepB,pepD,pepN,dpp操纵子 | 0.43 | 0.22 |
由表3可知在2种以下编码肽酶的基因缺失的微生物、仅1种编码肽转运蛋白质的操纵子缺失的微生物中,二肽的生成、蓄积量低,而在1种以上编码肽酶的基因和1种编码肽转运蛋白质的操纵子缺失的微生物、或3种以上编码肽酶的基因的活性丧失的微生物中,二肽的生成、蓄积量大幅度增加。
【实验例18使用肽酶和肽转运蛋白质活性丧失的大肠菌株生产的L-丙氨酰-L-缬氨酸(以下称为L-Ala-L-Val)产率的评价】
与实验例17同样,用pPE56转化编码各种肽酶的基因和编码肽转运蛋白质的操纵子缺失的大肠菌株。将得到的转化株接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,与28℃下培养17小时。将该培养液1%添加到加入了含100μg/ml氨苄青霉素和氨基酸的8ml的水性介质(磷酸氢二钾16g/l、磷酸二氢钾14g/l、硫酸铵5g/l、柠檬酸(无水)1g/l、Casamino acid(Difco社制)0.5g/l、L-Pro1g/l、L-Ala2.5g/l、L-Val2.5g/l、葡萄糖10g/l、维生素B110mg/l、硫酸镁·7水合物25mg/l、硫酸铁·7水合物50mg/l、用10mol/l的氢氧化钠溶液调整到pH7.2。葡萄糖、维生素B1、硫酸镁·7水合物、硫酸铁·7水合物分别蒸煮后添加)的试管,于30℃下反应24小时。对该水性介质进行离心分离,取得上清。
通过实验例17所述的方法对该上清中的生成物进行分析。结果如表4所示。
【表4】
| 菌株名称 | 缺失基因 | L-Ala-L-Val(g/l) |
| JM101 | 无 | 0 |
| JDPP1 | dpp操纵子 | 0 |
| JPN1 | pepN | 0 |
| JPA1 | pepA | 0 |
| JPB7 | pepB | 0 |
| JPD1 | pepD | 0 |
| JPDN2 | pepD,pepN | 0 |
| JPNDB43 | pepB,pepD,pepN | 0.04 |
| JPDP49 | pepD,dpp操纵子 | 0.11 |
| JPNDDP36 | pepD,pepN,dpp操纵子 | 0.22 |
| JPNDBP7 | pepB,pepD,pepN,dpp操纵子 | 0.20 |
由表4可知2种以下编码肽酶的基因缺失的微生物、和只1种编码肽转运蛋白质的操纵子缺失的微生物不生产二肽,但3种以上的肽酶基因缺失的微生物、或1种以上的编码肽酶的基因和1种编码肽转运蛋白质的操纵子缺失的微生物生产二肽。
【实验例19使用肽酶和二肽转运系蛋白质活性丧失的大肠菌株生产的甘氨酰-L-谷氨酰胺(以下、称为Gly-L-Gln)的产率的评价】
与实验例17同样,用pPE56转化编码各种肽酶的基因和编码二肽转运蛋白质的操纵子的缺失株。将得到的转化株接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。
将该培养液1%添加到加入了含100μg/ml氨苄青霉素和氨基酸的8ml的水性介质(磷酸氢二钾16g/l、磷酸二氢钾14g/l、硫酸铵5g/l、柠檬酸(无水)1g/l、Casamino acid(Difco社制)0.5g/L、L-Pro1g/L、Gly2.5g/L、L-Gln2.5g/L、葡萄糖10g/l、维生素B110mg/l、硫酸镁·7水合物25mg/l、硫酸铁·7水合物50mg/l、用10mol/l的氢氧化钠溶液调整到pH7.2。L-Gln在将10倍浓缩液经滤膜过滤灭菌后添加,葡萄糖、维生素B1、硫酸镁·7水合物、硫酸铁·7水合物分别蒸煮后添加)的试管,于30℃下反应24小时。对该水性介质进行离心分离,取得上清。
通过实验例17所述的方法对该该上清中的生成物进行分析。结果如表5所示。
【表5】
| 菌株名称 | 缺失基因 | Gly-L-Gln(g/l) |
| JM101 | 无 | 0 |
| JDPP1 | dpp操纵子 | 0 |
| JPDN2 | pepD,pepN | 0 |
| JPNDB43 | pepB,pepD,pepN | 0.01 |
| JPNDDP36 | pepD,pepN,dpp操纵子 | 0.02 |
| JPNDBP7 | pepB,pepD,pepN,dpp操纵子 | 0.03 |
由表5可知2种以下的编码肽酶的基因缺失的微生物、和只1种编码肽转运蛋白质的操纵子缺失的微生物不生产二肽,但3种以上的编码肽酶的基因缺失的微生物、以及2种以上的编码肽酶的基因和1种编码肽转运蛋白质的操纵子缺失的微生物生产二肽。
以下给出了实施例,但本发明并不受下面实施例限定。
【实施例1】
参与L-谷氨酰胺的生物合成调节的glnE基因、glnB基因缺失的微生物的制作
大肠杆菌的染色体DNA上的特定基因的缺失根据利用λ噬菌体的同源重组系的方法[Proc.Natl.Acad.Sci.USA.,97,6641-6645(2000)]进行。
(1)基因缺失用抗药性基因片段的克隆
大肠杆菌K12株的glnE、glnB的各基因的碱基序列已阐明[Science,5331,1453-1474(1997)]。根据报告的碱基序列,使用PerSeptive Biosystems公司制造8905型DNA合成仪合成作为glnE基因缺失用的引物DNA,具有由序列号88和89表示的碱基序列构成的DNA、合成作为glnB基因缺失用的引物DNA,由序列号90和91表示的碱基序列构成的DNA。合成的引物DNA是根据由位于各个缺失的靶基因的上游和下游的36bp构成的碱基序列设计的。
分别使用上述合成DNA作为引物组,以pKD3DNA作为模板进行PCR。PCR通过使用含有质粒DNA10ng、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认目的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。
将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,使DNA沉淀后,将该DNA溶解于20μL的TE。通过上述操作,取得glnE基因、glnB基因缺失用氯霉素抗性基因片段。
(2)染色体DNA上的glnE基因缺失的大肠杆菌JM101株的制作
用pKD46转化大肠杆菌JM101株后,于含有100mg/l的氨苄青霉素的LB琼脂培养基上选择保持pKD46的大肠杆菌JM101(以下、称为大肠杆菌JM101/pKD46)。通过电脉冲法用glnE基因缺失用氯霉素抗性基因片段转化在10mmol/L的L-阿拉伯糖和50μg/ml的氨苄青霉素存在下培养的大肠杆菌JM101/pKD46,在含有25mg/L的氯霉素的LB琼脂培养基上选择已经在JM101株的染色体DNA上的glnE基因插入了氯霉素抗性基因,glnE结构基因缺失的重组的株。
将取得的氯霉素抗性株影印在含有25mg/L的氯霉素的LB琼脂培养基,于42℃保温的状态下实施单菌落分离。将得到的各菌落影印于含有25mg/L的氯霉素的LB琼脂培养基和含有100mg/L的氨苄青霉素的LB琼脂培养基,选择既表现出氯霉素抗性,而且又表现出氨苄青霉素敏感性的菌落。然后用pCP20转化该pKD46脱落株,涂布于含有100mg/L的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
将生长繁殖的氨苄青霉素抗性株影印到没有添加药剂的LB琼脂培养基,在42℃保温的状态下实施单菌落分离。将得到的各菌落分别影印到没有添加药剂、含有25mg/L的氯霉素的LB琼脂培养基和含有100mg/L的氨苄青霉素的LB琼脂培养基,选择既表现出氯霉素敏感性,又表现出氨苄青霉素敏感性的菌落。通过常法(生物工学实验书、日本生物工学会编97~98页、培风馆、1992年)从得到的各株制备各个染色体DNA。使用以缺失的靶glnE基因的内部序列为基础设计的由序列号92和93表示的碱基序列构成的引物DNA,进行菌落PCR。菌落PCR是通过使用含有用200μl的移液管头接触菌落取得的菌体量、引物各0.5μmol/L、PfuDNA聚合酶2.5units、Pfu DNA聚合酶用×10缓冲液4μL、deoxyNTP各200μmol/L的反应液40μl,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。在进行PCR的菌株中,没有发现基因扩增的菌株确认是glnE基因缺失株,命名为大肠杆菌JGLE1。
(3)染色体DNA上的glnE基因和glnB基因缺失的大肠杆菌JM101株的制作
用pKD46转化上述(2)得到的大肠杆菌JGLE1株后,涂布于含有100mg/L的氨苄青霉素的LB琼脂培养基,通过于30℃下培养过夜,取得保持pKD46的大肠杆菌JGLE1株(以下、称为大肠杆菌JGLE1/pKD46)。通过电脉冲法使用glnB基因缺失用氯霉素抗性基因片段转化大肠杆菌JGLE1/pKD46,取得在染色体DNA上的glnB基因中已经插入了氯霉素抗性基因,glnB结构基因缺失的重组株。使用以glnB基因的内部序列为基础设计的由序列号94和95表示的碱基序列构成的引物DNA,在上述(2)的条件下进行菌落PCR。通过该PCR确认没有看到基因扩增的菌株是glnB基因缺失株,命名为大肠杆菌JGLBE1。
【实施例2】
ywfE基因和来自枯草芽孢杆菌的丙氨酸脱氢酶基因(ald基因)表达质粒的构建
以实验例15制作的ywfE基因表达质粒pPE56为基础,通过图5所示方法构建同时组成型表达来自枯草芽孢杆菌的丙氨酸脱氢酶基因(ald基因)表达质粒。
使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成具有序列号96和97表示的碱基序列的DNA(以下、分别称为引物P、引物Q)。序列号96表示的碱基序列是附加在含有作为ald基因的核糖体结合序列的Shine-Dalgarno序列的区域5'端的含有BamHI识别序列的序列。序列号97表示的碱基序列是附加在与含有ald基因的终止密码子的序列互补的碱基序列的5'端的含有BamHI识别序列的序列。
使用实验例2取得的枯草芽孢杆菌的染色体DNA作为模板,用上述引物P和引物Q作为引物组进行PCR。PCR通过制备含有0.1μg的染色体DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认相当于ald基因片段的约1.2kb的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,取得DNA沉淀,溶解于20μL的TE。
使用该溶液5μL,将扩增了的DNA用限制酶BamHI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,通过geneclean II试剂盒回收含有ald基因的1.2kb的DNA片段。
将pPE560.2μg用限制酶BamHI酶切后,通过琼脂糖凝胶电泳将DNA片段分离后,使用与上述同样的方法回收6.3kb的DNA片段。于60℃通过碱性磷酸酶(E.coli C75、Takara Bio公司生产)处理30分钟将该6.3kb的DNA片段的末端去磷酸化。添加与反应液等量的TE饱和酚/氯仿,混合,离心分离后,向得到的上层加2倍容量的冷乙醇并混合,于-80℃下放置30分钟。将该溶液离心分离,将得到的DNA沉淀溶解于20μL的TE。
使用连接试剂盒使上述得到的含有ald基因1.2kb的DNA片段和碱性磷酸酶处理的6.3kb的DNA片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过限制酶消化确认取得了ald基因与ywfE基因顺向插入的质粒,将该质粒命名为pPE86(图5)。
【实施例3】
来自大肠杆菌的抗反馈型pheA基因和抗反馈型aroF基因表达质粒的构建
(1)抗反馈型pheA基因表达质粒的造成
从通过苯丙氨酸类似物抗性突变导入得到的表达苯丙氨酸的抗反馈型pheA基因的质粒pE pheA22(特开昭61-260892)取得抗反馈型pheA基因,从通过酪氨酸抗性突变导入得到的表达酪氨酸的抗反馈型aroF基因的质粒pE aroF18(特开昭62-65691)取得抗反馈型aroF基因,通过以下的方法构建表达质粒。
使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成具有序列号98和99表示的碱基序列的DNA(以下、分别称为引物R、引物S)。序列号98表示的碱基序列是附加在含有pheA基因的核糖体结合序列的Shine-Dalgarno序列的区域5'末端的含有ClaI识别序列的序列。序列号99表示的碱基序列是附加在与含有pheA基因的终止密码子的序列互补的序列的5'末端的含有BamHI识别序列的序列。使用质粒pE pheA22作为模板,用上述引物R和引物S作为引物组进行PCR。PCR通过制备含有10ng的质粒DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认相当于pheA基因片段的约1.1kb的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,取得DNA沉淀,溶解于20μL的TE。
使用该溶液5μL,将扩增了的DNA用限制酶ClaI和BamHI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,通过geneclean II试剂盒回收含有pheA基因的1.1kb的DNA片段。
将含有trp启动子的表达载体pTrS30〔可从大肠菌JM109/pTrS30(FERM BP-5407)制备〕0.2μg用限制酶ClaI和BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段,通过与上述同样的方法,回收4.6kb的DNA片段。
使用连接试剂盒,使上述得到的含有pheA基因的1.1kb的DNA片段和4.6kb的DNA片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氯霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过限制酶消化确认取得了抗反馈型pheA基因表达质粒,将该质粒命名为pPHEA1。
将上述得到的pPHEA1 0.2μg用限制酶EcoRI和BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段后,通过geneclean II试剂盒合成含有trp启动子和抗反馈型pheA基因的1.5kb的DNA片段。
然后将带pACYC184的复制起点,含有氯霉素抗性基因的质粒载体pSTV28(Takara Bio公司生产)0.2μg用限制酶EcoRI和BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段,用与上述同样的方法回收3.0kb的DNA片段。
使用连接试剂盒,使上述得到的含有trp启动子和抗反馈型pheA基因的1.5kb的DNA片段与3.0kb的DNA片段在16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有30μg/ml的氯霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过限制酶消化确认取得了抗反馈型pheA基因表达质粒,将该质粒命名为pPHEA2(图6)。
(2)抗反馈型pheA基因和抗反馈型aroF基因表达质粒的构建
使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成具有序列号100和101表示的序列的DNA(以下、分别称为引物T、引物U)。序列号100表示的碱基序列是附加在含有作为aroF基因的核糖体结合序列的Shine-Dalgarno序列的区域5'端的含有BglII识别序列的序列。序列号101表示的碱基序列是附加在与含有aroF基因的终止密码子的序列互补的序列的5'端的含有BamHI识别序列的序列。以质粒pE aroF18为模板,使用上述引物T和引物U作为引物组进行PCR。PCR通过制备含有10ng的质粒pE aroF18、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP反应液40μL,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认相当于aroF基因片段的约1.1kb的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,将得到的DNA沉淀溶解于20μL的TE。
使用该溶液5μL,将扩增了的DNA用限制酶BglII和BamHI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,通过geneclean II试剂盒回收含有抗反馈型aroF基因的1.1kb的DNA片段。
然后将上述(1)取得的抗反馈型pheA基因表达质粒pPHEA20.2μg用限制酶BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段,用与上述同样的方法回收4.5kb的DNA片段。通过于60℃下碱性磷酸酶处理30分钟,进行该4.5kb的DNA片段的末端去磷酸化。添加与反应液等量的TE饱和酚/氯仿并混合,离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。将该溶液离心分离得到的DNA的沉淀溶解于20μL的TE。
使用连接试剂盒,使上述得到的含有抗反馈型aroF基因的1.1kb的DNA片段与碱性磷酸酶处理的4.5kb的DNA片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有30μg/ml的氯霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过限制酶消化确认取得了抗反馈型aroF基因与抗反馈型pheA基因顺向插入的抗反馈型aroF基因和抗反馈型pheA基因表达质粒,将该质粒命名为pPHEAF2(图6)。
【实施例4】
来自大肠杆菌的表现出酪氨酸抗性的aroF-tyrA操纵子表达质粒的构建
(1)表现出酪氨酸抗性的aroF-tyrA操纵子表达质粒的制作
由通过酪氨酸抗性突变导入得到的表达aroF-tyrA操纵子的质粒pKm1aroFm-18(特开昭60-034197)取得表现出酪氨酸抗性的aroF-tyrA操纵子,用以下的方法制作表达质粒。
使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成具有序列号102和103表示的碱基序列的DNA。序列号102表示的碱基序列是附加在含有作为aroF基因的核糖体结合序列的Shine-Dalgarno序列的区域5'端的含有ClaI识别序列的序列。序列号103表示的序列是附加在与含有tyrA基因的终止密码子的序列互补的序列的5'端的含有SphI识别序列的序列。
以质粒pKm1aroFm-18作为模板,使用由序列号102和103表示的碱基序列构成的DNA作为引物组进行PCR。
PCR通过制备含有10ng的质粒DNA、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各dNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认相当于aroF-tyrA基因片段的约2.2kb的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,将得到的DNA的沉淀溶解于20μL的TE。
使用该溶液5μL,将扩增了的DNA用限制酶ClaI和SphI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,通过geneclean II试剂盒回收含有aroF-tyrA操纵子的2.2kb的DNA片段。
将含有0.2μg的trp启动子的表达载体pTrS30用限制酶ClaI和SphI酶切后,通过琼脂糖凝胶电泳分离DNA片段,用与上述同样的方法回收4.6kb的DNA片段。
使用连接试剂盒,使上述得到的含有aroF-tyrA操纵子的2.2kbDNA片段和4.6kb的DNA片段于16℃下进行16小时反应,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过限制酶消化确认取得了表现出酪氨酸抗性的aroF-tyrA操纵子表达质粒,将该质粒命名为pTY1。
将上述得到的pTY1 0.2μg用限制酶EcoRI和BamHI酶切后,通过琼脂糖凝胶电泳分离DNA片段后,使用geneclean II试剂盒回收含有trp启动子和含有表现出酪氨酸抗性的aroF-tyrA操纵子的2.6kb的DNA片段。
然后将带有pACYC184的复制起点,含有氯霉素抗性基因的质粒载体pSTV28(Takara Bio公司生产)0.2μg用限制酶EcoRI和SphI酶切后,通过琼脂糖凝胶电泳分离DNA片段,用与上述同样的方法回收3.0kb的DNA片段。
使用连接试剂盒,使上述得到的trp启动子和表现出酪氨酸抗性的aroF-tyrA操纵子的2.6kb的DNA片段和3.0kb的DNA片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有30μg/ml的氯霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过限制酶消化确认取得了表现出酪氨酸抗性的aroF-tyrA操纵子表达载体,将该质粒命名为pTY2。
【实施例5】
metJ基因缺失株的制作
(1)metJ基因缺失用抗药性基因片段的克隆
大肠杆菌K12株的metJ基因的碱基序列已经阐明[Science,5331,1453-1474(1997)]。
metJ基因编码大肠杆菌的L-蛋氨酸生物合成系的阻遏物,已知通过导入不能产生该阻遏物的突变,可以提高L-蛋氨酸生产能能力(特开2000-139471)。
根据报告的碱基序列使用PerSeptive Biosystems公司制造8905型DNA合成仪,作为metJ基因缺失株制作用的引物组,合成由序列号104和105表示的碱基序列构成的DNA。
该DNA根据由位于各个缺失的靶基因的上游和下游的36bp构成的碱基序列设计。
使用该DNA作为引物组,以pKD3DNA为模板,通过PCR扩增metJ基因缺失株制作用氯霉素抗性基因片段。
PCR通过使用含有质粒DNA10ng、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认目的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,将得到的DNA的沉淀溶解于20μL的TE。
(2)在染色体DNA上的metJ基因插入了抗药性基因的大肠杆菌JM101株的制作
使用大肠杆菌JM101和上述(1)得到的metJ基因缺失株制作用氯霉素抗性基因片段,通过与实施例1(2)同样的操作,制作在大肠杆菌JM101的染色体DNA上的metJ基因插入了氯霉素抗性基因的重组体。
氯霉素抗性基因插入到染色体上的确认通过使用由具有位于氯霉素抗性基因插入部位的上游和下游的大约400bp的碱基序列的序列号106和107表示的碱基序列构成的DNA作为引物组的菌落PCR进行。菌落PCR在与实施例1(2)相同的条件下进行。
确认在进行菌落PCR的菌株中得到的含有氯霉素抗性基因的大约2kb大小的扩增片段的菌株是metJ基因缺失株后,通过与实施例7(3)同样的操作,使用表达Flp重组酶的pCP20,制作从染色体DNA脱落了氯霉素抗性基因的菌株,命名为大肠杆菌JMJ1。
【实施例6】
ywfE基因和来自大肠杆菌的抗反馈型3-磷酸甘油酸脱氢酶基因(serA基因)表达质粒的构建
在来自大肠杆菌的3-磷酸甘油酸脱氢酶基因(serA基因)中,已知将结构基因的第1096~1098位的密码子置换为终止密码子(TAA)的突变可给出编码3-磷酸甘油酸脱氢酶的C末端的45个氨基酸残基缺失的由丝氨酸导致的实质性抑制被解除的突变型3-磷酸甘油酸脱氢酶的基因(以下、称为抗反馈型serA基因)(日本专利第2584409号)。
对于抗反馈型serA基因扩增用引物使用由序列号108表示的碱基序列构成的DNA和由含有密码子置换突变序列的序列号109表示的碱基序列构成的DNA。
序列号108表示的碱基序列是附加在含有作为serA基因的核糖体结合序列的Shine-Dalgarno序列区域5'端的含有ClaI识别序列的序列,序列号109表示的碱基序列是附加在与含有使serA基因的C末端45氨基酸残基缺失的终止密码子的序列互补的序列的5'端的含有SphI识别序列的序列。
分别使用上述合成DNA作为引物组,以大肠杆菌W3110株的染色体DNA为模板进行扩增抗反馈型serA基因的PCR。PCR通过制备含有0.1μg的染色体DNA、0.5μmol/L的各引物、2.5units的PfuDNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将该反应液的1/10量进行琼脂糖凝胶电泳,确认相当于抗反馈型serA基因片段的约1.1kb的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,将得到的DNA的沉淀溶解于20μL的TE。
使用该溶液5μL,将扩增了的DNA用限制酶ClaI和SphI进行酶切,通过琼脂糖凝胶电泳将DNA片段分离后,通过geneclean II试剂盒回收含有serA的1.1kb的DNA片段。
然后含有0.2μg的trp启动子的表达载体pTrS30〔可从大肠菌JM109/pTrS30(FERM BP-5407)制备〕限制酶ClaI和SphI酶切后,通过琼脂糖凝胶电泳分离DNA片段,同样回收4.3kb的DNA片段。
使用连接试剂盒,使上述得到的含有serA基因的1.1kb的DNA片段和4.3kb的DNA片段于16℃下反应16小时,使它们连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,将该质粒命名为pSE15。通过限制酶消化确认该载体的构造。
以上述得到的大肠杆菌由来抗反馈型serA基因表达质粒pSE15为模板,使用由序列号110和109表示的碱基序列构成的DNA作为引物组,进行抗反馈型serA基因片段的扩增。
以实验例15制作的ywfE基因表达质粒pPE56为模板,使用由序列号111和112表示的碱基序列构成的DNA引物组进行含有trp启动子的ywfE基因片段的扩增。PCR通过制备含有任一种质粒DNA10ng、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的PfuDNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将各反应液的1/10量进行琼脂糖凝胶电泳,确认目的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。
将该混合液离心分离后,向得到的上层中加2倍容量的冷乙醇,并混合,于-80℃下放置30分钟。对该溶液进行离心分离,使DNA沉淀后,溶解于20μL的TE中。
通过上述操作取得了抗反馈型serA基因片段和含有trp启动子的ywfE基因片段。抗反馈型serA基因片段用限制酶BglII和SphI进行酶切,含有trp启动子的ywfE基因片段用限制酶EcoRI和BamHI进行酶切,分别通过琼脂糖凝胶电泳分离DNA片段后,使用genecleanII试剂盒回收含有serA基因的1.1kb和含有trp启动子和ywfE基因的1.8kb的DNA片段。
将0.2μg含有trp启动子的表达载体pTrS30用限制酶EcoRI和SphI酶切后,通过琼脂糖凝胶电泳分离DNA片段,用与上述同样的方法回收3.9kb的DNA片段。
使用连接试剂盒,使上述得到的含有serA基因的1.1kb的DNA片段、含有ywfE基因的1.8kb的DNA片段和3.9kb的DNA片段于16℃下反应16小时,进行连接。
通过使用钙离子的方法用该反应液转化大肠杆菌NM522株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。
根据众所周知的方法从生长繁殖的转化体菌落提取质粒,通过限制酶消化确认取得了抗反馈型serA基因与ywfE基因顺向插入的质粒,将该质粒命名为pPE212。
【实施例7】
ilvL基因缺失株和回复型ilvG基因置换株的制作
(1)ilvL基因缺失株制作用抗药性基因片段和回复型ilvG基因置换株制作用DNA片段的克隆
大肠杆菌K12株的ilvL、ilvG的各基因的碱基序列已经阐明[Science,5331,1453-1474(1997)]。
调节大肠杆菌K12株的ilvGMEDA操纵子表达的衰减区域位于该操纵子的5’端上游区,该碱基序列记载于Nucleic.Acids Res.15,2137(1987)。而由于已知通过除去这个衰减区域,衰减功能丧失,ilvGMEDA操纵子进行组成型表达(特开平8-473979),所以通过以下方法制作ilvGMEDA操纵子组成型表达型的大肠杆菌K12株。
而大肠杆菌K12株的野生株由于ilvG基因带有移码突变,具有活性的乙酰羟酸合酶的同功酶II(AHASII)不能表达[Proc.Natl.Acad.Sci.USA,78,922,(1981)]。因此,参考存在于AHASII正常发挥功能的大肠杆菌O157:H7株的染色体DNA(http://www.genome.wisc.edu/sequencing/o157.htm)上的ilvG基因序列,用以下的方法通过在大肠杆菌K12株的ilvG基因的第981位和982位之间插入称为AA的2个碱基,导入读框恢复的突变,做成乙酰羟酸合酶的活性恢复的大肠杆菌K12株。
根据报告的碱基序列,使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成由序列号113和114表示的碱基序列构成的DNA作为用于扩增ilvL基因缺失株制作用抗药性基因的引物组。
该DNA含有位于各个缺失靶基因的上游和下游的36bp的同源序列。
另外作为回复型ilvG基因上游区域扩增用引物组,合成由序列号115表示的碱基序列构成的DNA、和含有二碱基插入突变序列的由序列号116表示的碱基序列构成的DNA、作为回复型ilvG基因下游区域扩增用引物组合成含有二碱基突变序列的由序列号117表示的碱基序列构成的DNA、和由序列号118表示的碱基序列构成的DNA。
分别使用上述DNA作为引物组,以pKD3DNA作为模板通过PCR对ilvL基因缺失用氯霉素抗性基因片段进行扩增,以大肠杆菌W3110株的染色体DNA为模板通过PCR对回复型ilvG上游区域、和回复型ilvG下游区域进行扩增。PCR通过使用含有染色体DNA0.1μg、或质粒DNA10ng、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将各反应液的1/10量进行琼脂糖凝胶电泳,确认目的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。
向该混合液经离心分离后得到的上层加2倍容量的冷乙醇并混合,于-80℃下放置30分钟。对该溶液进行离心分离,使DNA沉淀后,将该DNA溶解于20μL的TE。通过上述操作取得ilvL基因缺失株制作用氯霉素抗性基因片段、和回复型ilvG基因上游区、回复型ilvG基因下游区。
然后以回复型ilvG基因上游区和回复型ilvG基因下游区作为模板,使用具有序列号115和118表示的碱基序列的DNA作为引物组进行交叉PCR[A.J.Link,D.Phillips,G.M.Church,J.Bacteriol.,179,6228-6237(1997)]。PCR在与上述同样的条件下进行。
通过上述PCR取得了连接了回复型ilvG基因上游区和回复型ilvG基因下游区的回复型ilvG置换株制作用DNA片段。
(2)染色体DNA上的ilvG基因置换为回复型ilvG基因的大肠杆菌JM101株的制作
通过常法用pKD46转化大肠杆菌JM101株后,涂布于含有100mg/l的氨苄青霉素的LB琼脂培养基,通过于30℃下培养过夜,选择保持pKD46的大肠杆菌JM101株(以下、称为大肠杆菌JM101/pKD46)。
通过电脉冲法用上述(1)中取得的回复型ilvG基因置换株制作用DNA片段转化在10mmol/L L-阿拉伯糖和50μg/ml的氨苄青霉素存在下培养的大肠杆菌JM101/pKD46,在含有200mg/L的L-缬氨酸的M9培养基添加了葡萄糖的琼脂培养基上选择染色体DNA上的ilvG基因置换为回复型ilvG的菌株。
将取得的L-缬氨酸抗性株再度影印在含有200mg/L的L-缬氨酸的M9培养基中添加了葡萄糖的琼脂培养基,于42℃保温的状态下实施单菌落分离。将得到的各菌落影印在含有200mg/L的L-缬氨酸的M9培养基中添加了葡萄糖的琼脂培养基和含有100mg/L的氨苄青霉素的LB琼脂培养基,选择表现出L-缬氨酸抗性,而且氨苄青霉素敏感性的菌落,将取得的回复型ilvG基因置换株命名为大肠杆菌JM101G+1。
(3)染色体DNA上的ilvG基因置换为回复型ilvG基因、ilvL基因缺失的大肠杆菌JM101株的制作
用pKD46转化上述(2)得到的大肠杆菌JM101G+1株后,涂布于含有100mg/L的氨苄青霉素的LB琼脂培养基,通过于30℃下培养过夜,取得保持pKD46的大肠杆菌JM101G+1株(以下、称为大肠杆菌JM101G+1/pKD46)。
然后,通过电脉冲法用上述(1)中取得的ilvL基因缺失株制作用氯霉素抗性基因片段转化大肠杆菌JM101G+1/pKD46,在含有25mg/L的氯霉素的LB琼脂培养基上在选择JM101株的染色体DNA上的ilvL基因插入了氯霉素抗性基因的重组株。
将取得的氯霉素抗性株落影印在含有25mg/L的L-氯霉素的LB琼脂培养基,于42℃保温的状态下实施单菌落分离。将得到的各菌落影印在含有25mg/L的氯霉素的LB琼脂培养基和含有100mg/L的氨苄青霉素的LB琼脂培养基,选择表现出氯霉素抗性和氨苄青霉素敏感性的pKD46脱落株。
合成具有位于大肠杆菌的染色体DNA上的氯霉素抗性基因插入部位的上游和下游的大约400bp碱基序列的序列号119和120表示的碱基序列,通过使用该合成DNA作为引物组进行的菌落PCR,确认上述取得的转化株的染色体DNA的结构。菌落PCR通过使用含有用200μl的移液管吸头接触菌落取得的菌体量、引物各0.5μmol/L、PfuDNA聚合酶2.5units、Pfu DNA聚合酶用×10缓冲液4μL、deoxyNTP各200μmol/L的反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
在进行菌落PCR的菌株中得到的含有氯霉素抗性基因大约2kb大小的扩增片段的菌株确认是ilvL基因缺失株,命名为大肠杆菌JILG+Cm1。
然后用pCP20转化上述得到的大肠杆菌JILG+Cm1,通过在含有100mg/l的氨苄青霉素的LB琼脂培养基上进行选择,取得了保持pCP20的大肠杆菌JILG+Cm1。
质粒pCP20含有来自酵母的Flp重组酶基因,该基因的表达可以在42℃进行诱导。
另外在上述(1)中制作的ilvL基因缺失株制作用氯霉素抗性基因片段中的氯霉素抗性基因的两端,由于存在Flp重组酶识别的碱基序列,所以通过Flp重组酶催化的同源重组容易使该抗性基因脱落。
另外由于pCP20含有温度敏感性的复制起点,所以通过使pCP20保持株在42℃下生长繁殖,可以同时诱导Flp重组酶的表达和pCP20的脱落。
将上述取得的大肠杆菌JILG+Cm1接种在没有添加药剂的LB琼脂培养基,于42℃下培养14小时后,进行单菌落分离。将得到的各菌落影印在没有添加药剂的LB琼脂培养基、含有25mg/L的氯霉素的LB琼脂培养基和含有100mg/L的氨苄青霉素的LB琼脂培养基,于30℃下培养,选择表现出氯霉素敏感性和氨苄青霉素敏感性的菌落。
对于上述选择的各菌株,使用由序列号119和120表示的碱基序列构成的DNA作为引物组,进行菌落PCR。菌落PCR通过使用通过200μl的移液管吸头接触菌落取得的菌体量、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
在进行PCR的株中得到的氯霉素抗性基因脱落的大约0.7kb大小扩增片段的菌株确认是ilvL基因缺失株,命名为大肠杆菌JILG+1。
【实施例8】
抗反馈型ilvA置换株的制作
(1)ilvA基因缺失株制作用抗药性基因片段和抗反馈型ilvA基因置换株制作用DNA片段的克隆
大肠杆菌K12株的ilvA基因的碱基序列已经阐明[Science,5331,1453-1474(1997)]。
已知编码由L-异亮氨酸导致的抑制实质上解除的苏氨酸脱氨酶的ilvA219基因(以下、称为抗反馈型ilvA基因)具有第447位的亮氨酸置换为苯丙氨酸的突变[Biochemistry,34,9403(1995)]。
根据报告的碱基序列,使用PerSeptive Biosystems公司制造8905型DNA合成仪,合成由序列号121和122表示的碱基序列构成的DNA作为用于扩增ilvL基因缺失株制作用抗药性基因的引物组。
该DNA含有与位于各个缺失靶基因的上游和下游的36bp同一的碱基序列。
另外作为抗反馈型ilvA基因上游区扩增用的引物组合成由序列号123表示的碱基序列构成的DNA、含有密码子置换突变序列的具有序列号124表示的碱基序列的DNA,作为抗反馈型ilvA基因下游区扩增用引物组,合成含有密码子置换突变序列的由序列号125表示的碱基序列构成的DNA、由序列号126表示的碱基序列构成的DNA。
分别使用上述合成DNA作为引物组,以pKD3DNA作为模板通过PCR扩增ilvA基因缺失株制作用氯霉素抗性基因片段,以大肠杆菌W3110株的染色体DNA为模板通过PCR扩增抗反馈型ilvA基因上游区、和抗反馈型ilvA基因下游区。PCR通过使用含有染色体DNA0.1μg、或质粒DNA10ng、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
将各反应液的1/10量进行琼脂糖凝胶电泳,确认目的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。
将该混合液离心分离后,向得到的上层加2倍容量的冷乙醇并混合,于-80℃下放置30分钟。对该溶液进行离心分离,使DNA沉淀后,将该DNA溶解于20μL的TE中。通过上述操作取得了ilvA基因缺失用氯霉素抗性基因片段、抗反馈型ilvA基因上游区、和抗反馈型ilvA基因下游区。
然后在上述PCR扩增片段中,以抗反馈型ilvA基因上游区、和抗反馈型ilvA基因下游区为模板,使用由序列号123和126表示的碱基序列构成的DNA作为引物组进行交叉PCR。PCR在与上述同样的条件下进行。
通过上述PCR,取得了连接了抗反馈型ilvA基因上游区和抗反馈型ilvA基因下游区的抗反馈型ilvA基因置换株制作用DNA片段。
(2)在大肠杆菌的染色体DNA上的ilvA基因插入抗药性基因的大肠杆菌JM101株的制作
通过电脉冲法用上述(1)中取得的ilvA基因缺失用氯霉素抗性基因片段转化在10mmol/L的L-阿拉伯糖和50μg/ml的氨苄青霉素存在下培养的大肠杆菌JM101/pKD46,在含有25mg/L的氯霉素的LB琼脂培养基上选择在JM101株的染色体DNA上的ilvA基因插入了氯霉素抗性基因、ilvA结构基因缺失的重组株。
将取得的氯霉素抗性株影印在含有25mg/L的氯霉素的LB琼脂培养基,于30℃保温的状态下实施单菌落分离。将得到的各菌落影印在含有25mg/L的氯霉素和100mg/L的氨苄青霉素的LB琼脂培养基,选择表现出氯霉素抗性且氨苄青霉素抗性的菌落。
对于得到的各株,以具有位于染色体DNA上的氯霉素抗性基因插入部位的上游和下游的大约400bp的碱基序列的序列号123和具有126表示的碱基序列的DNA作为引物组,进行菌落PCR。
菌落PCR通过使用含有通过200μl的移液管吸头接触菌落取得的菌体量、0.5μmol/L的各引物、2.5units的Pfu DNA聚合酶、4μL的Pfu DNA聚合酶用×10缓冲液、200μmol/L的各deoxyNTP的40μL反应液,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。
在进行PCR的菌株中得到的含有氯霉素抗性基因的大约2kb大小的扩增片段的菌株确认是ilvA基因缺失株,命名为大肠杆菌JIACm1/pKD46。
(3)染色体DNA上的ilvA基因置换为抗反馈型ilvA基因的大肠杆菌JM101株的制作
将上述(2)中制作的大肠杆菌JIACm1/pKD46株在10mmol/L的L-阿拉伯糖和50μg/ml的氨苄青霉素存在下培养后,通过电脉冲法用上述(1)中取得的抗反馈型ilvA基因置换株制作用DNA片段进行转化,以异亮氨酸需求性作为回复指標,在M9培养基中添加了葡萄糖的琼脂培养基上选择JIACm1株的染色体DNA上的ilvA基因置换为抗反馈型ilvA基因的菌株。
将生长繁殖的氨苄青霉素抗性株影印在没有添加药剂的M9培养基中添加了葡萄糖的琼脂培养基,于42℃保温的状态下实施单菌落分离。将得到的各菌落分别影印在没有添加药剂、含有25mg/L的氯霉素的LB琼脂培养基、和含有100mg/L的氨苄青霉素的LB琼脂培养基,选择表现出氯霉素敏感性且氨苄青霉素敏感性的菌落。确认得到的株是抗反馈型ilvA基因置换株,命名为大肠杆菌JIA1。
(4)染色体DNA上的ilvA基因置换为抗反馈型ilvA基因的大肠杆菌JILG+1的制作
取代大肠杆菌JM101作为亲本株使用实施例7中制作的大肠杆菌JILG+1,通过进行上述(1)~(3)的操作,取得ilvL基因缺失、ilvG基因置换为回复型ilvG基因,而且ilvA基因置换为抗反馈型ilvA基因的株,命名为大肠杆菌JILG+IA1。
【实施例9】
突变型leuA基因置换株的制作
(1)leuA基因缺失株制作用抗药性基因片段和突变型leuA基因置换株制作用DNA片段的克隆
大肠杆菌K12株的leuA基因碱基序列已经阐明[Science,5331,1453-1474(1997)]。
大肠杆菌FERM BP-4704是通过亮氨酸类似物(4-氮亮氨酸)抗性选拔的亮氨酸生产菌(特开平8-70879),认为具有编码实质上可解除L-亮氨酸导致的抑制的异丙基苹果酸合酶的突变型leuA基因。
使用PerSeptive Biosystems公司制造8905型DNA合成仪,根据报告的碱基序列,作为用于扩增leuA基因缺失株制作用抗药性基因片段的引物组,合成序列号127和128表示的碱基序列构成的DNA。
该DNA具有与位于各个缺失的靶基因的上游和下游的36bp同一的碱基序列。
另外作为用于扩增突变型leuA基因置换株制作用DNA片段的引物组,合成具有位于从leuA基因的起始密码子开始上游大约200bp的碱基序列的序列号129表示的DNA、和与位于从终止密码子开始的下游大约200bp碱基序列反方向的序列的序列号130表示的DNA。
分别将上述的DNA作为引物组使用,以pKD3DNA作为模板通过PCR扩增leuA基因缺失株制作用氯霉素抗性基因片段,另外以通过常法制备的FERM BP-4704染色体DNA作为模板通过PCR扩增突变型leuA基因置换株制作用DNA片段。
PCR通过使用含有染色体DNA0.1μg、或质粒DNA10ng、引物各0.5μmol/L、Pfu DNA聚合酶2.5units、Pfu DNA聚合酶用×10缓冲液4μL、deoxyNTP各200μmol/L的反应液40μL,按照94℃下1分钟、55℃下2分钟、72℃下3分钟为一个循环,反复进行30次来实施。。
将各反应液的1/10量进行琼脂糖凝胶电泳,确认目的片段扩增后,添加与剩下的反应液等量的TE饱和酚/氯仿,并混合。
将该混合液离心分离后,向得到的上层中加2倍容量的冷乙醇并混合,于-80℃放置30分钟。将该溶液离心分离,使DNA沉淀后,将该DNA溶解于20μL的TE。通过上述操作可取得leuA基因缺失株制作用氯霉素抗性基因片段、和突变型leuA基因置换株制作用DNA片段。
(2)在染色体DNA上的leuA基因插入了抗药性基因的大肠杆菌JM101株的制作
通过与实施例8(2)同样的操作,制作在大肠杆菌JM101株的染色体DNA上的leuA基因插入了氯霉素抗性基因的大肠杆菌突变株。
染色体DNA上插入了氯霉素抗性基因的确认通过使用具有位于氯霉素抗性基因插入部位的上游和下游的大约200bp的碱基序列的序列号131和132表示的碱基序列构成的DNA作为引物组的菌落PCR进行。
PCR在与实施例8(2)同样的条件下进行。在进行菌落PCR的菌株中得到含有氯霉素抗性基因的大约2kb大小扩增片段的菌株确认是氯霉素抗性基因插入到leuA基因中的leuA基因缺失株,命名为JLACm1/pKD46。
(3)染色体DNA上的leuA基因置换为来自大肠杆菌H-9070株的突变型基因的大肠杆菌JM101株的制作
使用上述(1)制作的突变型leuA基因置换株制作用DNA片段和上述(2)制作的大肠杆菌JLACm1/pKD46株,通过与实施例8(3)同样的操作制作大肠杆菌JLACm1/pKD46的染色体DNA上插入了氯霉素抗性基因的leuA基因置换为突变型leuA基因的重组株,命名为大肠杆菌JLA1。
(4)染色体DNA上的leuA基因置换为突变型leuA基因的大肠杆菌JILG+1的制作
取代大肠杆菌JM101使用实施例7制作的大肠杆菌JILG+1作为亲本株,通过上述(1)~(3)的操作,取得ilvL基因缺失、ilvG基因置换为回复型ilvG基因,而且leuA基因置换为突变型leuA基因的菌株,命名为大肠杆菌JILG+LA1。
【实施例10】
使用具有生产L-丙氨酸能力的微生物发酵生产L-Ala-L-Ala
用实施例2得到的来自枯草芽孢杆菌的ywfE基因和来自枯草芽孢杆菌的ald基因表达质粒pPE86转化大肠杆菌JM101株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,对该质粒进行限制酶处理,确认取得了保持质粒pPE86的大肠杆菌JM101株(以下、称为大肠杆菌JM101/pPE86)。用同样的方法也取得了保持质粒pTrS30的大肠杆菌JM101株(以下、称为大肠杆菌JM101/pTrS30)、保持质粒pPE56的大肠杆菌JM101株(以下、称为大肠杆菌JM101/pPE56)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素的生产培养基[16g/L磷酸氢二钾、14g/L磷酸二氢钾、5g/L硫酸铵、1g/L柠檬酸(无水)、5g/L Casamino acid(Difco公司生产)、10g/L葡萄糖、10mg/L维生素B1、25mg/L硫酸镁·7水合物、50mg/L硫酸铁·7水合物、用10mol/L的氢氧化钠调整到pH7.2、葡萄糖、维生素B1、硫酸镁·7水合物、硫酸铁·7水合物分别蒸气灭菌后添加]的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC法分析用与实验例17同样的方法进行。结果表示在表6中。
【表6】

【实施例11】
利用具有生产L-Ala和L-Gln能力的微生物发酵生产L-Ala-L-Gln
用实施例2得到的pPE86转化实施例1得到的glnE基因和glnB基因双重缺失株大肠杆菌JGLBE1后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,对该质粒进行限制酶处理,确认取得了保持质粒pPE86的大肠杆菌JGLBE1株,将该株命名为大肠杆菌JGLBE1/pPE86。用同样的方法也取得了保持质粒pTrS30的大肠杆菌JGLBE1株(以下、称为大肠杆菌JGLBE1/pTrS30)、保持质粒pPE56的大肠杆菌JGLBE1株(称为大肠杆菌JGLBE1/pPE56)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素的实施例10所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC法分析用与实验例17同样的方法进行。结果表示在表7中。
【表7】

【实施例12】
使用具有生产L-Ala和L-Phe的能力的微生物发酵生产L-Ala-L-Phe
用实施例3中制作的来自大肠杆菌的抗反馈型pheA基因表达质粒pPHEA2、或来自大肠杆菌的抗反馈型pheA基因和抗反馈型aroF基因表达质粒pPHEAF2转化实施例10取得的大肠杆菌JM101/pPE86后,涂布于含有50μg/ml的氨苄青霉素和30μg/ml的氯霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,对该质粒进行限制酶处理,确认取得了保持pPHEA2或pPHEAF2的大肠杆菌JM101/pPE86(分别称为大肠杆菌JM101/pPE86/pPHEA2、大肠杆菌JM101/pPE86/pPHEAF2)。用同样的方法通过用pPHEA2或pPHEAF2转化实施例10中取得的大肠杆菌JM101/pTrS30和大肠杆菌JM101/pPE56,取得了保持pPHEA2的大肠杆菌JM101/pTrS30(以下称为大肠杆菌JM101/pTrS30/pPHEA2)、保持pPHEAF2的大肠杆菌JM101/pTrS30(以下称为大肠杆菌JM101/pTrS30/pPHEAF2)、保持pPHEA2的大肠杆菌JM101/pPE56(以下称为大肠杆菌JM101/pPE56/pPHEA2)、保持pPHEAF2的大肠杆菌JM101/pPE56(以下称为大肠杆菌JM101/pPE56/pPHEAF2)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素和30μg/mL的氯霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素和50μg/ml的氯霉素的上述实施例10所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC分析用与实验例17同样的方法进行。结果表示在表8中。
【表8】

【实施例13】
使用具有生产L-Thr和L-Phe能力的微生物发酵生产L-苏氨酰-L-苯丙氨酸(L-Thr-L-Phe)
用实验例15得到的强化来自枯草芽孢杆菌的ywfE基因表达的质粒pPE56转化表现出脯氨酸、蛋氨酸、异亮氨酸和硫胺素需求性、赋予α-氨基-β-羟基吉草酸抗性的具有生产L-Thr能力的大肠杆菌βIM-4株(ATCC21277)后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过限制酶处理确认取得了保持pPE56的大肠杆菌ATCC21277(以下称为大肠杆菌ATCC21277/pPE56)。
然后用pSTV28(Takara Bio公司生产)、实施例3得到的pPHEA2或pPHEAF2转化大肠杆菌ATCC21277/pPE56后,涂布于含有50μg/ml的氨苄青霉素和30μg/ml的氯霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,对该质粒进行限制酶处理,确认取得了保持pSTV28、pPHEA2或pPHEAF2的大肠杆菌ATCC21277/pPE56(以下分别称为大肠杆菌ATCC21277/pPE56/pSTV28、大肠杆菌ATCC21277/pPE56/pPHEA2和大肠杆菌ATCC21277/pPE56/pPHEAF2)。通过同样方法,取得了保持pTrS30和pSTV28的大肠杆菌ATCC21277(以下称为大肠杆菌ATCC21277/pTrS30/pSTV28)、保持pTrS30和pPHEA2的大肠杆菌ATCC21277(以下称为大肠杆菌ATCC21277/pTrS30/pPHEA2)、保持pTrS30和pPHEAF2的大肠杆菌ATCC21277(以下称为大肠杆菌ATCC21277/pTrS30/pPHEAF2)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素和30μg/mL的氯霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素和50μg/ml的氯霉素的上述实施例4所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC分析用与实验例17同样的方法进行。结果表示在表9中。
【表9】

【实施例14】
利用具有生产L-Ala和L-Tyr能力的微生物发酵生产L-Ala-L-Tyr
用实施例4中制作的来自大肠杆菌的酪氨酸抗性突变型aroF-tyrA操纵子表达质粒pTY2转化实施例10中取得的大肠杆菌JM101/pPE86后,涂布于含有50μg/ml的氨苄青霉素和30μg/ml的氯霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,确认取得了保持pTY2的大肠杆菌JM101/pPE86(以下称为大肠杆菌JM101/pPE86/pTY2)。用同样的方法通过用pTY2转化实施例10中取得的大肠杆菌JM101/pTrS30和大肠杆菌JM101/pPE56,取得了保持pTY2的大肠杆菌JM101/pTrS30(以下称为大肠杆菌JM101/pTrS30/pTY2)、保持pTY2的大肠杆菌JM101/pPE56(以下称为大肠杆菌JM101/pPE56/pTY2)。
将上述的各转化株分别接种于加入了含有50μg/ml的氨苄青霉素和30μg/ml的氯霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素和50μg/ml的氯霉素的实施例10所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC分析用与实验例17同样的方法进行。结果表示在表10中。
【表10】

【实施例15】
利用具有生产L-Ala和L-Met能力的微生物发酵生产L-丙氨酰-L-蛋氨酸(L-Ala-L-Met)
用实施例2得到的pPE86基因转化实施例5中取得的大肠杆菌JMJ1后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过限制酶处理确认取得了保持pPE86的大肠杆菌JMJ1株,将该株命名为大肠杆菌JMJ1/pPE86。用同样的方法也取得了保持质粒pTrS30的大肠杆菌JMJ1株(以下称为大肠杆菌JMJ1/pTrS30)、保持质粒pPE56的大肠杆菌JMJ1株(以下称为大肠杆菌JMJ1/pPE56)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素的实施例10所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC法分析用与实验例17同样的方法进行。结果表示在表11中。
【表11】

实施例10~15示出的结果表明通过将具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸的能力的微生物在培养基中进行培养,可以使二肽在该培养基中生成、蓄积,和与具有生产1种氨基酸能力的上述微生物相比,具有生产2种氨基酸能力的上述微生物一方的二肽生产能力高。
【实施例16】
利用具有生产L-Ala能力,而且肽酶基因和二肽转运蛋白质基因缺失的微生物发酵生产L-Ala-L-Ala
用pTrS30、实施例2得到的pPE56或pPE86转化实验例16(4)得到的pepD基因、pepN基因和dpp操纵子基因缺失的大肠杆菌JPNDDP36株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过限制酶处理确认取得了保持pTrS30、pPE56或pPE86的大肠杆菌JPNDDP36株(以下分别称为大肠杆菌JPNDDP36/pTrS30、大肠杆菌JPNDDP36/pPE56和大肠杆菌JPNDDP36/pPE86)。
将上述的转化株分别用与实施例10同样的方法进行培养,表12示出了通过与实验例17同样的方法对培养上清中的生成物分析的结果。
【表12】

【实施例17】
利用具有生产L-Ala和L-Gln能力,而且肽酶基因和二肽转运蛋白质基因缺失的微生物发酵生产L-Ala-L-Gln
(1)具有生产L-Ala和L-Gln能力,而且肽酶基因和二肽转运蛋白质基因缺失的微生物的制作
通过使用实验例16(4)得到的大肠杆菌JPNDDP36株,进行与实施例1相同的操作,向大肠杆菌JPNDDP36株导入glnE基因缺失和glnB基因缺失,取得了具有生产L-Ala和L-Gln能力、而且肽酶基因和二肽转运蛋白质基因缺失的微生物大肠杆菌JPNDDPGBE1株。
(2)L-Ala-L-Gln的发酵生产
使用与实施例16同样的方法用pTrS30、pPE56或pPE86转化上述(1)得到的大肠杆菌JPNDDPGBE1株,取得了保持该质粒的大肠杆菌JPNDDPGBE1株(以下分别称为大肠杆菌JPNDDPGBE1/pTrS30、大肠杆菌JPNDDPGBE1/pPE56和大肠杆菌JPNDDPGBE1/pPE86)。
将大肠杆菌JPNDDPGBE1/pTrS30、大肠杆菌JPNDDPGBE1/pPE56和大肠杆菌JPNDDPGBE1/pPE86用与实施例10同样的方法进行培养,表13示出了通过与实验例17同样的方法对培养上清中的生成物分析的结果。
【表13】

【实施例18】
使用具有生产L-Ala和L-Tyr能力、而且肽酶基因和二肽转运蛋白质基因缺失的微生物发酵生产L-Ala-L-Tyr
用实施例2取得的pPE86转化实验例16中取得的JPNDDP36株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过限制酶处理确认取得了保持pPE86的大肠杆菌JPNDDP36株,将该株命名为大肠杆菌JPNDDP36/pPE86。通过同样的方法,也取得了保持pTrS30的大肠杆菌JPNDDP36株(以下称为大肠杆菌JPNDDP36/pTrS30)、保持pPE56的大肠杆菌JPNDDP36株(称为大肠杆菌JPNDDP36/pPE56)。
然后通过用实施例4取得的pTY2分别转化上述得到的转化株,取得保持pTY2的转化株大肠杆菌JPNDDP36/pTrS30/pTY2、大肠杆菌JPNDDP36/pPE56/pTY2和大肠杆菌JPNDDP36/pPE86/pTY2。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素的实施例10所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC法分析用与实验例17同样的方法进行。结果表示在表14中。
【表14】

【实施例19】
使用具有生产L-Ala和L-Val能力,而且肽酶基因和二肽转运蛋白质基因缺失的微生物发酵生产L-Ala-L-Val
使用实验例16中取得的肽酶基因和编码肽转运蛋白质的操纵子缺失突变株作为亲本株,根据实施例7所述的方法,制作缺失ilvL基因,且ilvG基因的移码突变回复的大肠杆菌JPNDDPILG+1株。
使用实施例2中取得的pPE86转化大肠杆菌JPNDDPILG+1株后,涂布于含有50μg/ml的氨苄青霉素的LB琼脂培养基,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过对质粒进行限制酶处理确认取得了保持质粒pPE86的大肠杆菌JPNDDPILG+1株(以下称为大肠杆菌JPNDDPILG+1/pPE86)。同样取得了保持质粒pTrS30的大肠杆菌JPNDDPILG+1株(以下称为大肠杆菌JPNDDPILG+1/pTrS30)、保持质粒pPE56的大肠杆菌JPNDDPILG+1株(以下称为大肠杆菌JPNDDPILG+1/pPE56)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素的培养基[16g/L磷酸氢二钾、14g/L磷酸二氢钾、5g/L硫酸铵、1g/L柠檬酸(无水)、5g/LCasamino acid(Difco公司生产)、10g/L葡萄糖、10mg/L维生素B1、25mg/L硫酸镁·7水合物、50mg/L硫酸铁·7水合物、用10mol/L的氢氧化钠调整到pH7.2、葡萄糖、维生素B1、硫酸镁·7水合物、硫酸铁·7水合物分别蒸气灭菌后添加]的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC法分析用与实验例17同样的方法进行。结果表示在表15中。
【表15】

【实施例20】
使用具有生产L-Ala和L-Ile能力、而且肽酶基因和二肽转运蛋白质基因缺失的微生物发酵生产L-Ala-L-Ile
以实验例16中取得的肽酶基因和编码肽转运蛋白质的操纵子的缺失突变株大肠杆菌JPNDDP36株作为亲本株,根据实施例7和8所述的方法制作ilvL基因缺失、ilvG基因的移码突变回复、以及ilvA基因置换为抗反馈型ilvA基因的大肠杆菌JPNDDPILG+IA1株。
使用实施例2中取得的pPE86转化大肠杆菌JPNDDPILG+IA1株后,涂布在含有50μg/ml的氨苄青霉素的LB琼脂培养基上,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过限制酶处理确认取得了保持质粒pPE86的大肠杆菌JPNDDPILG+IA1株,将该株命名为大肠杆菌JPNDDPILG+IA1/pPE86。用同样方法取得了保持pTrS30的大肠杆菌JPNDDPILG+IA1株(以下、称为大肠杆菌JPNDDPILG+IA1/pTrS30)、保持pPE56的大肠杆菌JPNDDPILG+IA1株(称为大肠杆菌JPNDDPILG+IA1/pPE56)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素的实施例10所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC法分析用与实验例17同样的方法进行。结果表示在表16中。
【表16】

【实施例21】
使用具有生产L-Ala和L-Leu能力、而且肽酶基因和二肽转运蛋白质基因缺失的微生物发酵生产L-Ala-L-Leu
用实施例2中取得的pPE86转化以实验例16中取得的肽酶基因和编码肽转运蛋白质的操纵子的缺失突变株大肠杆菌JPNDDP36株作为亲本株,根据实施例7和9所述的方法制作ilvL基因缺失、ilvG基因的移码突变回复、以及leuA基因置换为突变型leuA基因的大肠杆菌JPNDDPILG+LA1后,涂布在含有50μg/ml的氨苄青霉素的LB琼脂培养基上,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过限制酶处理确认取得了保持pPE86的大肠杆菌JPNDDPILG+LA1株,将该株命名为大肠杆菌JPNDDPILG+LA1/pPE86。用同样的方法也取得了保持pTrS30的大肠杆菌JPNDDPILG+LA1株(以下称为大肠杆菌JPNDDPILG+LA1/pTrS30)、保持pPE56的大肠杆菌JPNDDPILG+LA1株(称为大肠杆菌JPNDDPILG+LA1/pPE56)。
将上述的转化株分别接种于加入了含有50μg/ml的氨苄青霉素的8ml的LB培养基的试管,于28℃下培养17小时。将该培养液1%接种于加入了8ml含有100μg/ml的氨苄青霉素的实施例10所述的生产培养基的试管,于30℃下培养24小时后,对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC法分析用与实验例17同样的方法进行。结果表示在表17中。
【表17】

【实施例22】
使用具有生产L-Ser和L-Phe能力,而且肽酶基因和二肽转运蛋白质基因缺失的微生物发酵生产L-Ser-L-Phe
将作为编码实验例16取得的肽酶基因和肽转运蛋白质的操纵子的缺失突变株的大肠杆菌JPNDDP36株用实施例6取得的pSE15或pPE212转化后,涂布在含有50μg/ml的氨苄青霉素的LB琼脂培养基上,于30℃下培养过夜。通过众所周知的手法从生长繁殖的菌株提取质粒,通过限制酶处理确认取得了保持pSE15的大肠杆菌JPNDDP36株、和保持pPE212的大肠杆菌JPNDDP36株,分别命名为大肠杆菌JPNDDP36/pSE15和大肠杆菌JPNDDP36/pPE212。
再通过对上述得到的转化株分别用实施例3取得的来自大肠杆菌的抗反馈型pheA基因和抗反馈型aroF基因表达质粒pPHEAF2进行转化,取得保持pPHEAF2的大肠杆菌JPNDDP36/pSE15、和大肠杆菌JPNDDP36/pPE212,它们分别被命名为大肠杆菌JPNDDP36/pSE15/pPHEAF2、和大肠杆菌JPNDDP36/pPE212/pPHEAF2。
将大肠杆菌JPNDDP36/pSE15/pPHEAF2、和大肠杆菌JPNDDP36/pPE212/pPHEAF2分别接种在加入了各含50μg/ml的氨苄青霉素和30μg/ml的氯霉素的8ml的LB培养基的试管,于28℃下培养17小时。将1%该培养液接种在加入了含有100μg/ml的氨苄青霉素和50μg/ml的氯霉素的实施例4所述生产培养基8ml的试管,于30℃下培养24小时后,通过对该培养液进行离心分离取得培养上清。
将培养上清中的培养生成物通过F-moc化法衍生后,使用HPLC对该生成物进行分析。HPLC分析用与实验例17同样的方法进行。结果表示在表18中。
【表18】

实施例16~22示出的结果表明通过对具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力、具有生产该1种以上的氨基酸中的至少1种氨基酸的能力,而且1种以上肽酶和1种以上肽转运蛋白质的活性丧失,或3种以上的肽酶活性丧失的微生物在培养基中进行培养,可以使二肽在该培养基中生成、蓄积,还有该微生物具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力,和具有生产该1种以上的氨基酸中的至少1种氨基酸的能力,与没有丧失肽酶和肽转运蛋白质的活性的微生物相比,显然二肽的生产能力高。
【产业上利用的可能性】
通过本发明可以有效地制造各种二肽。
【序列表FREE TEXT】
序列号19-人工序列的说明:合成DNA
序列号20-人工序列的说明:合成DNA
序列号21-人工序列的说明:合成DNA
序列号22-人工序列的说明:合成DNA
序列号23-人工序列的说明:合成DNA
序列号24-人工序列的说明:合成DNA
序列号25-人工序列的说明:合成DNA
序列号26-人工序列的说明:合成DNA
序列号27-人工序列的说明:合成DNA
序列号28-人工序列的说明:合成DNA
序列号29-人工序列的说明:合成DNA
序列号30-人工序列的说明:合成DNA
序列号31-人工序列的说明:合成DNA
序列号32-人工序列的说明:合成DNA
序列号33-人工序列的说明:在数据库检索中使用的氨基酸序列
序列号34-人工序列的说明:在数据库检索中使用的氨基酸序列
序列号35-人工序列的说明:在数据库检索中使用的氨基酸序列
序列号41-人工序列的说明:合成DNA
序列号42-人工序列的说明:合成DNA
序列号54-人工序列的说明:合成DNA
序列号64-人工序列的说明:合成DNA
序列号65-人工序列的说明:合成DNA
序列号66-人工序列的说明:合成DNA
序列号67-人工序列的说明:合成DNA
序列号68-人工序列的说明:合成DNA
序列号69-人工序列的说明:合成DNA
序列号70-人工序列的说明:合成DNA
序列号71-人工序列的说明:合成DNA
序列号72-人工序列的说明:合成DNA
序列号73-人工序列的说明:合成DNA
序列号74-人工序列的说明:合成DNA
序列号75-人工序列的说明:合成DNA
序列号76-人工序列的说明:合成DNA
序列号77-人工序列的说明:合成DNA
序列号78-人工序列的说明:合成DNA
序列号79-人工序列的说明:合成DNA
序列号80-人工序列的说明:合成DNA
序列号81-人工序列的说明:合成DNA
序列号82-人工序列的说明:合成DNA
序列号83-人工序列的说明:合成DNA
序列号84-人工序列的说明:合成DNA
序列号85-人工序列的说明:合成DNA
序列号86-人工序列的说明:合成DNA
序列号87-人工序列的说明:合成DNA
序列号88-人工序列的说明:合成DNA
序列号89-人工序列的说明:合成DNA
序列号90-人工序列的说明:合成DNA
序列号91-人工序列的说明:合成DNA
序列号92-人工序列的说明:合成DNA
序列号93-人工序列的说明:合成DNA
序列号94-人工序列的说明:合成DNA
序列号95-人工序列的说明:合成DNA
序列号96-人工序列的说明:合成DNA
序列号97-人工序列的说明:合成DNA
序列号98-人工序列的说明:合成DNA
序列号99-人工序列的说明:合成DNA
序列号100-人工序列的说明:合成DNA
序列号101-人工序列的说明:合成DNA
序列号102-人工序列的说明:合成DNA
序列号103-人工序列的说明:合成DNA
序列号104-人工序列的说明:合成DNA
序列号105-人工序列的说明:合成DNA
序列号106-人工序列的说明:合成DNA
序列号107-人工序列的说明:合成DNA
序列号108-人工序列的说明:合成DNA
序列号109-人工序列的说明:合成DNA
序列号110-人工序列的说明:合成DNA
序列号111-人工序列的说明:合成DNA
序列号112-人工序列的说明:合成DNA
序列号113-人工序列的说明:合成DNA
序列号114-人工序列的说明:合成DNA
序列号115-人工序列的说明:合成DNA
序列号116-人工序列的说明:合成DNA
序列号117-人工序列的说明:合成DNA
序列号118-人工序列的说明:合成DNA
序列号119-人工序列的说明:合成DNA
序列号120-人工序列的说明:合成DNA
序列号121-人工序列的说明:合成DNA
序列号122-人工序列的说明:合成DNA
序列号123-人工序列的说明:合成DNA
序列号124-人工序列的说明:合成DNA
序列号125-人工序列的说明:合成DNA
序列号126-人工序列的说明:合成DNA
序列号127-人工序列的说明:合成DNA
序列号128-人工序列的说明:合成DNA
序列号129-人工序列的说明:合成DNA
序列号130-人工序列的说明:合成DNA
序列号131-人工序列的说明:合成DNA
序列号132-人工序列的说明:合成DNA
本说明书还包括以下内容:
1.二肽的制造方法,其特征是将具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸的能力的微生物在培养基中进行培养,使二肽在该培养基中生成、蓄积,从该培养基回收二肽。
2.项目1所述的制造方法,其中具有由1种以上的氨基酸生成二肽的活性的蛋白质是选自以下[1]~[11]中的蛋白质:
[1]具有序列号1~8中任一个表示的氨基酸序列的蛋白质,
[2]由在序列号1~8中的任一个表示的氨基酸序列缺失、置换或附加1个以上的氨基酸的氨基酸序列构成、而且具有由1种以上的氨基酸生成二肽的活性的蛋白质,
[3]由与序列号1~8中的任一个表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[4]具有与序列号17表示的氨基酸序列有80%以上同源性的氨基酸序列,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质,
[5]具有由序列号37或38表示的氨基酸序列的蛋白质,
[6]由在序列号37或38表示的氨基酸序列中缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质,
[7]由与序列号37或38表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质
[8]具有非核糖体肽合成酶(以下称为NRPS)活性的蛋白质,
[9]具有序列号43表示的氨基酸序列的蛋白质,
[10]由在序列号43表示的氨基酸序列中缺失、置换或附加1个以上的氨基酸的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质,
[11]由与序列号43表示的氨基酸序列具有65%以上同源性的氨基酸序列构成,而且具有由1种以上的氨基酸生成二肽的活性的蛋白质。
3.项目1或2所述的制造方法,其中具有由1种以上的氨基酸生成二肽的活性的蛋白质是由选自以下[1]~[8]中的DNA编码的蛋白质:
[1]具有序列号9~16和36中的任一个表示的碱基序列的DNA,
[2]和具有与序列号9~16和36中的任一个表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA,
[3]和具有与序列号18表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA,
[4]具有序列号39或40表示的碱基序列的DNA,
[5]和具有与序列号39或40表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA,
[6]编码具有NRPS活性的蛋白质的DNA,
[7]具有序列号44表示的碱基序列的DNA,
[8]和具有与序列号44表示的碱基序列互补的碱基序列的DNA在严格的条件下杂交,而且编码具有由1种以上的氨基酸生成二肽活性的蛋白质的DNA。
4.项目1所述的制造方法,其中具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力的微生物是带有重组DNA的微生物,该重组DNA含有选自项目3的[1]~[8]中的DNA。
5.项目1~4中任一项所述的制造方法,其中生产氨基酸的能力是通过选自以下[1]~[5]中的方法获得的:
[1]缓和或解除至少一个调控该氨基酸生物合成的结构的方法,
[2]对至少一个参与该氨基酸生物合成的酶表达增强的方法,
[3]使至少一个参与该氨基酸生物合成的酶基因的拷贝数增加的方法,
[4]对至少一个由该氨基酸生物合成途径到该氨基酸以外的代谢产物的旁路的代谢途径进行弱化或阻断的方法,
[5]与野生型株相比,选择对该氨基酸的类似物的抗性高的细胞株的方法。
6.项目1~5中的任一项所述的制造方法,其中微生物是属于埃希氏菌属(Escherichia)、棒杆菌属(Corynebacterium)、芽孢杆菌属(Bacillus)、沙雷氏菌属(Serratia)、假单孢菌属(Pseudomonas)或链霉菌属(Streptomyces)的微生物。
7.项目6所述的制造方法,其中属于埃希氏菌属(Escherichia)、棒杆菌属(Corynebacterium)、芽孢杆菌属(Bacillus)、沙雷氏菌属(Serratia)、假单孢菌属(Pseudomonas)或链霉菌属(Streptomyces)的微生物是大肠杆菌(Escherichia coli)、谷氨酸棒杆菌(Corynebacteriumglutamicum)、产氨棒杆菌(Corynebacterium ammoniagenes)、乳酸发酵棒杆菌(Corynebacterium lactofermentum)、黄色棒杆菌(Corynebacterium flavum)、Corynebacterium efficiens、枯草芽孢杆菌(Bacillus subtilis)、巨大芽孢杆菌(Bacillus megaterium)、粘质沙雷氏菌(Serratia marcescens)、恶臭假单胞菌(Pseudomonas putida)、铜绿假单胞菌(Pseudomonas aeruginosa)、天蓝色链霉菌(Streptomycescoelicolor)或浅青紫链霉菌(Streptomyces lividans)。
8.项目1~5中的任一项所述的制造方法,其中微生物是1种以上的肽酶和具有1种以上肽转运活性的蛋白质(以下也称为肽转运蛋白质)的活性低下或丧失的微生物。
9.项目1~5中的任一项所述的制造方法,其中微生物是3种以上肽酶的活性低下或丧失的微生物。
10.项目8或9所述的制造方法,其中肽酶是具有序列号45~48中的任一个表示的氨基酸序列的蛋白质、或是具有与序列号45~48中的任一个表示的氨基酸序列有80%以上同源性的氨基酸序列,而且具有肽酶活性的蛋白质。
11.项目8或10所述的制造方法,其中肽转运蛋白质是具有序列号49~53中的任一个表示的氨基酸序列的蛋白质、或是由与序列号49~53中的任一个表示的氨基酸序列有80%以上同源性的氨基酸序列构成的蛋白质,而且具有肽转运活性的蛋白质。
12.项目8~11中的任一项所述的制造方法,其中微生物是属于埃希氏菌属(Escherichia)、芽孢杆菌属(Bacillus)或棒杆菌属(Corynebacterium)的微生物。
13.项目12所述的制造方法,其中属于埃希氏菌属(Escherichia)、芽孢杆菌属(Bacillus)或棒杆菌属(Corynebacterium)的微生物是大肠杆菌(Escherichia coli)、谷氨酸棒杆菌(Corynebacteriumglutamicum)、产氨棒杆菌(Corynebacterium ammoniagenes)、乳酸发酵棒杆菌(Corynebacterium lactofermentum)、黄色棒杆菌(Corynebacterium flavum)、Corynebacterium efficiens、枯草芽孢杆菌(Bacillus subtilis)或巨大芽孢杆菌(Bacillus megaterium)。
14.项目1~13中的任一项所述的制造方法,其中氨基酸是选自L-丙氨酸、L-谷氨酰胺、L-谷氨酸、甘氨酸、L-缬氨酸、L-亮氨酸、L-异亮氨酸、L-脯氨酸、L-苯丙氨酸、L-色氨酸、L-蛋氨酸、L-丝氨酸、L-苏氨酸、L-半胱氨酸、L-天冬酰胺、L-酪氨酸、L-赖氨酸、L-精氨酸、L-组氨酸、L-天冬氨酸、L-α-氨基丁酸、L-4-羟基脯氨酸、L-3-羟基脯氨酸、L-鸟氨酸和L-瓜氨酸的氨基酸。
15.项目1~14中的任一项所述的制造方法,其中二肽是式(I)表示的二肽:
R1-R2(I)
(式中、R1和R2可以相同或不同,表示选自L-丙氨酸、L-谷氨酰胺、L-谷氨酸、甘氨酸、L-缬氨酸、L-亮氨酸、L-异亮氨酸、L-脯氨酸、L-苯丙氨酸、L-色氨酸、L-蛋氨酸、L-丝氨酸、L-苏氨酸、L-半胱氨酸、L-天冬酰胺、L-酪氨酸、L-赖氨酸、L-精氨酸、L-组氨酸、L-天冬氨酸、L-α-氨基丁酸、L-4-羟基脯氨酸、L-3-羟基脯氨酸、L-鸟氨酸和L-瓜氨酸的氨基酸)。





































































Claims (7)
1.二肽的制造方法,其特征是将具有生产具有由1种以上的氨基酸生成二肽活性的蛋白质的能力,而且具有生产该1种以上的氨基酸中的至少1种氨基酸的能力的微生物在培养基中进行培养,使二肽在该培养基中生成、蓄积,从该培养基回收二肽,
其中在微生物只能生产1种氨基酸时,所述培养基是添加另外1种氨基酸的培养基,
其中微生物是属于埃希氏菌属(Escherichia)的微生物,以及
其中具有由1种以上的氨基酸生成二肽的活性的蛋白质是由序列号9~16和36中的任一个表示的碱基序列构成的DNA编码的蛋白质,二肽是式R1-R2表示的二肽,式中,
R1为L-Ala时,R2是L-Cys、L-Lys、L-αAB或L-Cit;
R1为L-Gly时,R2是L-Trp、L-Cys、L-Lys、L-αAB或L-Cit;
R1为L-Gln时,R2是L-Phe;
R1为L-Ser时,R2是L-αAB;
R1为L-Cys时,R2是L-Gln或L-Met;
R1为L-Met时,R2是L-Lys或L-His;以及
R1为L-αAB时,R2是L-Gln、L-Thr、L-Arg或L-αAB。
2.权利要求1所述的制造方法,其中生产氨基酸的能力是通过选自以下[1]~[5]中的方法获得的:
[1]缓和或解除至少一个调控该氨基酸生物合成的结构的方法,
[2]对至少一个参与该氨基酸生物合成的酶表达增强的方法,
[3]使至少一个参与该氨基酸生物合成的酶基因的拷贝数增加的方法,
[4]对至少一个由该氨基酸生物合成途径到该氨基酸以外的代谢产物的旁路的代谢途径进行弱化或阻断的方法,
[5]与野生型株相比,选择对该氨基酸的类似物的抗性高的细胞株的方法。
3.权利要求1所述的制造方法,其中属于埃希氏菌属(Escherichia)的微生物是大肠杆菌(Escherichia coli)。
4.权利要求1~3中的任一项所述的制造方法,其中微生物是选自下述[1]的1种以上的肽酶和选自下述[2]的具有1种以上肽转运活性的蛋白质的活性低下或丧失的微生物,
[1]肽酶是由序列号45~48中的任一个表示的氨基酸序列构成的蛋白质、
[2]由序列号49~53中的任一个表示的氨基酸序列构成的蛋白质。
5.权利要求1~3中的任一项所述的制造方法,其中微生物是选自下述[1]的3种以上肽酶的活性低下或丧失的微生物,
[1]肽酶是由序列号45~48中的任一个表示的氨基酸序列构成的蛋白质。
6.权利要求4或5所述的制造方法,其中微生物是属于埃希氏菌属(Escherichia)的微生物。
7.权利要求6所述的制造方法,其中属于埃希氏菌属(Escherichia)的微生物是大肠杆菌(Escherichia coli)。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004189011 | 2004-06-25 | ||
| JP2004-189011 | 2004-06-25 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200580020690.6A Division CN1973042B (zh) | 2004-06-25 | 2005-06-24 | 二肽的制造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103642882A true CN103642882A (zh) | 2014-03-19 |
Family
ID=35063090
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200580020690.6A Active CN1973042B (zh) | 2004-06-25 | 2005-06-24 | 二肽的制造方法 |
| CN201310593697.3A Pending CN103642882A (zh) | 2004-06-25 | 2005-06-24 | 二肽的制造方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200580020690.6A Active CN1973042B (zh) | 2004-06-25 | 2005-06-24 | 二肽的制造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US7514243B2 (zh) |
| EP (1) | EP1627884B9 (zh) |
| JP (1) | JP4796495B2 (zh) |
| CN (2) | CN1973042B (zh) |
| AT (1) | ATE474847T1 (zh) |
| DE (1) | DE602005022387D1 (zh) |
| ES (1) | ES2347875T3 (zh) |
| HK (1) | HK1083345A1 (zh) |
| WO (1) | WO2006001379A1 (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108531465A (zh) * | 2018-04-04 | 2018-09-14 | 南京农业大学 | 一种环二肽合成酶及其应用 |
| CN114072492A (zh) * | 2019-06-17 | 2022-02-18 | Cj第一制糖株式会社 | 生产l-酪氨酸的微生物及利用其生产l-酪氨酸的方法 |
| CN116622795A (zh) * | 2023-07-20 | 2023-08-22 | 凯莱英生命科学技术(天津)有限公司 | L-氨基酸连接酶的制备方法及其在二肽合成上的应用 |
| CN117659116A (zh) * | 2023-12-05 | 2024-03-08 | 珠海瑞德林生物有限公司 | 酪氨酸寡肽的制备方法 |
| CN117721165A (zh) * | 2024-02-08 | 2024-03-19 | 天津凯莱英生物科技有限公司 | Atp再生体系和多肽的合成方法 |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8257943B2 (en) * | 2004-06-25 | 2012-09-04 | Kyowa Hakko Bio Co., Ltd. | Process for producing dipeptides or dipeptide derivatives |
| US8211688B2 (en) * | 2004-06-25 | 2012-07-03 | Kyowa Hakko Bio Co., Ltd. | Process for producing L-glutamine using Escherichia coli with deficient glnB and glnE function |
| DE602006010990D1 (de) * | 2005-03-18 | 2010-01-21 | Kyowa Hakko Bio Co Ltd | Verfahren zur dipeptidherstellung |
| WO2007074858A1 (ja) * | 2005-12-27 | 2007-07-05 | Kyowa Hakko Kogyo Co., Ltd. | ジペプチドの製造法 |
| JPWO2008001837A1 (ja) * | 2006-06-28 | 2009-11-26 | 協和発酵バイオ株式会社 | オリゴペプチドの精製方法 |
| WO2008126783A1 (ja) | 2007-04-06 | 2008-10-23 | Kyowa Hakko Bio Co., Ltd. | ジペプチドの製造法 |
| US20100248307A1 (en) * | 2007-08-31 | 2010-09-30 | Kyowa Hakko Bio Co., Ltd. | Peptide production method |
| EP2212419A1 (en) * | 2007-10-31 | 2010-08-04 | Kyowa Hakko Bio Co., Ltd. | Cyclodipeptide synthases (cdss) and their use in the synthesis of linear dipeptides |
| EP2395096B1 (en) | 2009-02-09 | 2014-04-09 | Kyowa Hakko Bio Co., Ltd. | Process for producing L-amino acids |
| US20130130317A1 (en) | 2010-08-02 | 2013-05-23 | Kyowa Hakko Kirin Co., Ltd | Method for producing substance |
| CN102428807A (zh) * | 2011-09-13 | 2012-05-02 | 上海博琛生物科技有限公司 | 农业有机废弃物阶段发酵及其在设施栽培中的应用 |
| RU2012129311A (ru) | 2012-07-11 | 2014-01-20 | Закрытое акционерное общество " Научно-исследовательский институт Аджиномото-Генетика" (ЗАО "АГРИ") | Днк, кодирующая дипептид-синтезирующий фермер (варианты), бактерия рода escherichia и способ получения дипептидов с ее использованием |
| CN105510497A (zh) * | 2015-11-30 | 2016-04-20 | 精晶药业股份有限公司 | L-丙氨酰-l-丙氨酸的定量检测方法 |
| CN106565822A (zh) * | 2016-10-17 | 2017-04-19 | 湖北泓肽生物科技有限公司 | 手性l‑丙氨酰‑苯丙氨酸的制备方法 |
| CN106967658B (zh) * | 2017-02-23 | 2020-09-15 | 郑州大学 | 一种提高Fab抗体表达量的方法 |
| CN109136158A (zh) * | 2017-06-27 | 2019-01-04 | 中国科学院青岛生物能源与过程研究所 | 一种以生物质水解液为原料合成苯乙烯的基因工程菌及其构建方法与应用 |
| JP2019014663A (ja) * | 2017-07-04 | 2019-01-31 | 天津科技大学 | ペプチドの合成方法 |
| CN108486133B (zh) * | 2018-06-29 | 2021-06-01 | 江南大学 | 一种l-丝氨酸转运蛋白的应用方法 |
| JP2022550349A (ja) | 2019-09-25 | 2022-12-01 | 味の素株式会社 | 細菌の発酵による2-メチル酪酸の製造方法 |
| CN112831451B (zh) * | 2019-11-25 | 2022-09-16 | 中国科学院微生物研究所 | 产丙谷二肽的工程菌及其构建方法与应用 |
| CN114058560B (zh) * | 2020-07-31 | 2023-09-12 | 中国科学院天津工业生物技术研究所 | 甘氨酸的生产方法 |
| KR20240113674A (ko) * | 2023-01-13 | 2024-07-23 | 씨제이제일제당 (주) | 신규 카르노신 합성 효소 및 이를 이용한 카르노신 생산 방법 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2545078B2 (ja) | 1987-04-06 | 1996-10-16 | 協和醗酵工業株式会社 | 核酸関連物質の製造法 |
| JPH03164297A (ja) | 1989-11-24 | 1991-07-16 | Ebaa Kooto Kk | 葉書の製造方法及びその装置 |
| JP4032441B2 (ja) * | 1995-08-30 | 2008-01-16 | 味の素株式会社 | L−アミノ酸の製造方法 |
| AU6077499A (en) | 1998-07-10 | 2000-02-01 | Institut Fur Pflanzengenetik Und Kulturpflanzenforschung | Genes for the biosynthesis of anticapsin and bacilysine, their isolation and their use |
| EP1096011A1 (en) * | 1999-05-10 | 2001-05-02 | Holland Sweetener Company V.o.F. | Microbiological production method for alpha-l-aspartyl-l-phenylalanine |
| US20020064834A1 (en) * | 1999-03-29 | 2002-05-30 | Sascha Doekel | Microbiological production method for alpha-L-aspartyl-L-phenylalanine |
| FR2841260B1 (fr) | 2002-06-21 | 2004-10-22 | Commissariat Energie Atomique | Polynucleotides et polypeptides codes par lesdits polynucleotides impliques dans la synthese de derives des dicetopiperazines |
| JP4447467B2 (ja) * | 2002-12-26 | 2010-04-07 | 協和発酵バイオ株式会社 | ジペプチドの製造法 |
| US20050106703A1 (en) * | 2003-11-05 | 2005-05-19 | Kyowa Hakko Kogyo Co., Ltd. | Microorganisms producing dipeptides and process for producing dipeptides using the microorganisms |
-
2005
- 2005-06-24 WO PCT/JP2005/011636 patent/WO2006001379A1/ja active Application Filing
- 2005-06-24 AT AT05013734T patent/ATE474847T1/de active
- 2005-06-24 DE DE602005022387T patent/DE602005022387D1/de active Active
- 2005-06-24 ES ES05013734T patent/ES2347875T3/es active Active
- 2005-06-24 CN CN200580020690.6A patent/CN1973042B/zh active Active
- 2005-06-24 CN CN201310593697.3A patent/CN103642882A/zh active Pending
- 2005-06-24 US US11/165,211 patent/US7514243B2/en active Active
- 2005-06-24 JP JP2006528630A patent/JP4796495B2/ja active Active
- 2005-06-24 EP EP05013734A patent/EP1627884B9/en active Active
-
2006
- 2006-04-10 HK HK06104302.9A patent/HK1083345A1/xx unknown
-
2009
- 2009-03-10 US US12/400,996 patent/US7939302B2/en active Active
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108531465A (zh) * | 2018-04-04 | 2018-09-14 | 南京农业大学 | 一种环二肽合成酶及其应用 |
| CN114072492A (zh) * | 2019-06-17 | 2022-02-18 | Cj第一制糖株式会社 | 生产l-酪氨酸的微生物及利用其生产l-酪氨酸的方法 |
| CN114072492B (zh) * | 2019-06-17 | 2024-04-30 | Cj第一制糖株式会社 | 生产l-酪氨酸的微生物及利用其生产l-酪氨酸的方法 |
| CN116622795A (zh) * | 2023-07-20 | 2023-08-22 | 凯莱英生命科学技术(天津)有限公司 | L-氨基酸连接酶的制备方法及其在二肽合成上的应用 |
| CN116622795B (zh) * | 2023-07-20 | 2023-10-13 | 凯莱英生命科学技术(天津)有限公司 | L-氨基酸连接酶的制备方法及其在二肽合成上的应用 |
| CN117659116A (zh) * | 2023-12-05 | 2024-03-08 | 珠海瑞德林生物有限公司 | 酪氨酸寡肽的制备方法 |
| CN117659116B (zh) * | 2023-12-05 | 2024-05-10 | 珠海瑞德林生物有限公司 | 酪氨酸寡肽的制备方法 |
| CN117721165A (zh) * | 2024-02-08 | 2024-03-19 | 天津凯莱英生物科技有限公司 | Atp再生体系和多肽的合成方法 |
| CN117721165B (zh) * | 2024-02-08 | 2024-05-03 | 天津凯莱英生物科技有限公司 | Atp再生体系和多肽的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| DE602005022387D1 (de) | 2010-09-02 |
| US7939302B2 (en) | 2011-05-10 |
| US20050287626A1 (en) | 2005-12-29 |
| CN1973042A (zh) | 2007-05-30 |
| US7514243B2 (en) | 2009-04-07 |
| WO2006001379A1 (ja) | 2006-01-05 |
| JPWO2006001379A1 (ja) | 2008-04-17 |
| ES2347875T3 (es) | 2010-11-22 |
| US20090269806A1 (en) | 2009-10-29 |
| HK1083345A1 (en) | 2006-06-30 |
| CN1973042B (zh) | 2014-09-17 |
| ATE474847T1 (de) | 2010-08-15 |
| JP4796495B2 (ja) | 2011-10-19 |
| EP1627884B1 (en) | 2010-07-21 |
| EP1627884A1 (en) | 2006-02-22 |
| EP1627884B9 (en) | 2010-10-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1973042B (zh) | 二肽的制造方法 | |
| CN1973037B (zh) | 二肽或二肽衍生物的制造方法 | |
| US20100120126A1 (en) | Microorganisms producing dipeptides and process for producing dipeptide using the microorganisms | |
| CN101151369B (zh) | 二肽的结晶及其制备方法 | |
| WO2010090330A1 (ja) | L-アミノ酸の製造法 | |
| JP5424530B2 (ja) | ジペプチドまたはジペプチド誘導体の製造法 | |
| CN101715484B (zh) | 二肽的制造方法 | |
| JP4731327B2 (ja) | ジペプチドの製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20140319 |
|
| RJ01 | Rejection of invention patent application after publication |