CN101516954B - 软质聚氨酯泡沫塑料的制造方法 - Google Patents
软质聚氨酯泡沫塑料的制造方法 Download PDFInfo
- Publication number
- CN101516954B CN101516954B CN2007800352530A CN200780035253A CN101516954B CN 101516954 B CN101516954 B CN 101516954B CN 2007800352530 A CN2007800352530 A CN 2007800352530A CN 200780035253 A CN200780035253 A CN 200780035253A CN 101516954 B CN101516954 B CN 101516954B
- Authority
- CN
- China
- Prior art keywords
- polyvalent alcohol
- urethane foam
- polyoxyalkylene polyol
- initiator
- flexible urethane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4866—Polyethers having a low unsaturation value
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4804—Two or more polyethers of different physical or chemical nature
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/76—Polyisocyanates or polyisothiocyanates cyclic aromatic
- C08G18/7607—Compounds of C08G18/7614 and of C08G18/7657
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/04—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers only
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
- C08G65/2603—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds the other compounds containing oxygen
- C08G65/2615—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds the other compounds containing oxygen the other compounds containing carboxylic acid, ester or anhydride groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
- C08G65/2642—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds characterised by the catalyst used
- C08G65/2645—Metals or compounds thereof, e.g. salts
- C08G65/2648—Alkali metals or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
- C08G65/2642—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds characterised by the catalyst used
- C08G65/2645—Metals or compounds thereof, e.g. salts
- C08G65/2663—Metal cyanide catalysts, i.e. DMC's
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2101/00—Manufacture of cellular products
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2110/00—Foam properties
- C08G2110/0008—Foam properties flexible
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2110/00—Foam properties
- C08G2110/0041—Foam properties having specified density
- C08G2110/0058—≥50 and <150kg/m3
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2110/00—Foam properties
- C08G2110/0083—Foam properties prepared using water as the sole blowing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2350/00—Acoustic or vibration damping material
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Toxicology (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Polyurethanes Or Polyureas (AREA)
- Polyethers (AREA)
Abstract
本发明提供可使用来自天然油脂的原料很好地成形具备良好的缓冲性的软质聚氨酯泡沫塑料的软质聚氨酯泡沫塑料的制造方法。该方法具备在催化剂(C)及发泡剂(D)的存在下使包含第1聚氧化烯多元醇(A1)的聚氧化烯多元醇(A)和多异氰酸酯化合物(B)反应的步骤。所述第1聚氧化烯多元醇(A1)通过在聚合催化剂(a)的存在下使烯化氧(c)开环聚合于引发剂(b)而制得。聚合催化剂(a)为选自配位阴离子聚合催化剂及阳离子聚合催化剂的1种以上,引发剂(b)是利用化学反应对天然油脂赋予羟基而形成的引发剂,是羟值为20~250mgKOH/g且以聚苯乙烯换算的重均分子量对数均分子量的比率(Mw/Mn)为1.2以上的来自天然油脂的多元醇。
Description
技术领域
本发明涉及用来自天然油脂的原料制造软质聚氨酯泡沫塑料的方法。
背景技术
一般作为软质聚氨酯泡沫塑料的原料使用的聚醚多元醇通过使例如环氧乙烷及环氧丙烷等烯化氧开环聚合于具有活性氢原子的引发剂而制得。
这些聚醚多元醇及通过该聚醚多元醇和异氰酸酯化合物的反应而得的软质聚氨酯泡沫塑料是来源于石油的化学产品,因此如果实施最终焚烧处理,则会增加空气中的二氧化碳气体。
近年,基于对地球温室效应的考虑,要求形成为即使经过废弃处理也不会增加自然界的二氧化碳气体的产品。
例如,已知如果以作为将空气中的二氧化碳气体固定化而得的化合物的动植物油为原料来制造聚氨酯产品,则在对该产品进行焚烧处理时,来自动植物的碳燃烧而产生的那部分的二氧化碳气体不会增加自然界的二氧化碳气体。
天然动植物油中具备羟基的油脂只有蓖麻油。下述专利文献1揭示了在复合金属氰化物配位催化剂的存在下,将蓖麻油及/或改性蓖麻油作为引发剂使单环氧化物开环聚合而制造聚醚类的方法。但是,由于蓖麻油的价格高,因此难以实用化。
所以,提出了通过化学反应对不具备羟基的天然油脂加成羟基的方法。
例如,在下述专利文献2中揭示了以下方法:通过导入氧及/或空气而对天然油脂的双键赋予羟基,从而改性获得含羟基高分子量化合物及其衍生物,使该化合物及其衍生物与异氰酸酯化合物反应而制得聚氨酯产品。
这里,将通过氧及/或空气的注入而赋予羟基的改性大豆油(一般也被称为曝气大豆油)直接用于与异氰酸酯化合物的反应,未记载对曝气大豆油加成烯化氧而使用的方法。
下述专利文献3中,作为制造官能团有所增加的多元醇的方法,记载了通过氧及/或空气的注入对天然油脂赋予羟基后,用胺类或氢氧化钾等金属催化剂将烯化氧开环聚合于酯改性而得的改性多元醇的方法。此外,虽然有关于软质聚氨酯泡沫塑料的制造的记载,但对于表示该泡沫塑料的特性的物性值完全没有记载。
下述专利文献4中揭示了植物油(Vegetable oil)的精制方法。记载了将未精制油分离为油相和胶质相后,通过对油相注入空气的方式进行精制的方法,所得的精制油被认为可用于聚氨酯泡沫塑料,但对于该泡沫塑料的制造方法没有具体的记载。
下述专利文献5中记载了软质聚氨酯泡沫塑料、半硬质聚氨酯泡沫塑料、硬质聚氨酯泡沫塑料的制造方法。但是,实施例中例示的软质聚氨酯泡沫塑料未使用石油系多元醇而是仅使用植物油作为多元醇来制造聚氨酯泡沫塑料。此外,明确记载所得软质聚氨酯泡沫塑料的密度达到非常高的192~720kg/m3,如果密度降至48~64kg/m3,则机械强度劣化,但对于机械强度并没有具体的评价。
下述专利文献6中记载了在金属催化剂的存在下使一氧化碳及氢与植物油反应而赋予羟基的改性植物油。但是,没有记载对植物油加成烯化氧而使用的方法。
下述专利文献7中记载了软质聚氨酯泡沫塑料的制造方法:以羟基加成的环氧大豆油为引发剂,采用作为阴离子聚合引发剂的氢氧化钾为聚合催化剂,使环氧丙烷及环氧乙烷共聚于该引发剂而获得多元醇,再使该多元醇和多异氰酸酯反应。所述羟基加成的环氧大豆油通过在过量的醇的存在下将环氧化大豆油开环而赋予羟基。
专利文献1:日本专利特开平5-163342号公报
专利文献2:日本专利特表2002-524627号公报
专利文献3:美国专利申请公开第2003/0191274号说明书
专利文献4:美国专利第6476244号说明书
专利文献5:美国专利第6180686号说明书
专利文献6:国际公开第2005/033167号文本
专利文献7:日本专利特开2005-320431号公报
发明的揭示
专利文献2、3、4所记载的通过氧及/或空气的注入而赋予羟基的改性大豆油(曝气大豆油)或专利文献7所记载的环氧化大豆油都是比蓖麻油价廉的原料,特别是曝气大豆油可以低成本制得。
但是,在制造软质聚氨酯泡沫塑料时,即使将以往的来源于石油的原料的一部分用来源于天然油脂的原料替换,也有无法很好地成形的情况或即使成形特性也发生劣化的情况。
献7的[0045]记载了通过模制发泡制造软质聚氨酯泡沫塑料的实施例,但作为所得泡沫塑料的缓冲性指标的回弹性为33~44%左右,根据用途有进一步提高缓冲性的要求。
本发明是鉴于以上情况完成的发明,其目的是提供可用来自天然油脂的原料成形具有良好的缓冲性的软质聚氨酯泡沫塑料的软质聚氨酯泡沫塑料的制造方法。
本发明的技术内容以下所述。
(1)软质聚氨酯泡沫塑料的制造方法,它是具备在催化剂(C)及发泡剂(D)的存在下使聚氧化烯多元醇(A)和多异氰酸酯化合物(B)反应的步骤的软质聚氨酯泡沫塑料的制造方法,所述聚氧化烯多元醇(A)包含第1聚氧化烯多元醇(A1),所述第1聚氧化烯多元醇(A1)通过在聚合催化剂(a)的存在下使烯化氧(c)开环聚合于引发剂(b)而制得,
其特征在于,所述聚合催化剂(a)是不促进来自天然油脂的甘油酯结构的水解的催化剂,所述引发剂(b)是利用化学反应对天然油脂赋予羟基而形成的引发剂,是羟值为20~250mgKOH/g且以聚苯乙烯换算的重均分子量对数均分子量的比率(Mw/Mn)为1.2以上的来自天然油脂的多元醇。
(2)上述(1)记载的软质聚氨酯泡沫塑料的制造方法,其中,所述聚合催化剂(a)为选自配位阴离子聚合催化剂及阳离子聚合催化剂中的一种以上的聚合催化剂。
(3)上述(1)或(2)记载的软质聚氨酯泡沫塑料的制造方法,其中,所述聚氧化烯多元醇(A)还包括所述第1聚氧化烯多元醇(A1)以外的第2聚氧化烯多元醇(A2),该第2聚氧化烯多元醇(A2)的平均官能团数为2~8且羟值为20~160mgKOH/g。
(4)上述(1)~(3)中任一项记载的软质聚氨酯泡沫塑料的制造方法,其中,所述聚氧化烯多元醇(A)中的所述第1聚氧化烯多元醇(A1)和第2聚氧化烯多元醇(A2)的质量比(A1)/(A2)为10/90~90/10。
(5)上述(1)~(4)中任一项记载的软质聚氨酯泡沫塑料的制造方法,其中,所述烯化氧(c)包含环氧丙烷。
(6)上述(1)~(5)中任一项记载的软质聚氨酯泡沫塑料的制造方法,其中,所述引发剂(b)是利用(1)通过在天然油脂中注入空气或氧而生成羟基的方法或/及(2)通过将天然油脂环氧化后将环氧环开环而生成羟基的方法获得的来自天然油脂的多元醇。
(7)上述(1)~(6)中任一项记载的软质聚氨酯泡沫塑料的制造方法,其中,所述引发剂(b)来自大豆油。
(8)上述(1)~(7)中任一项记载的软质聚氨酯泡沫塑料的制造方法,其中,所述多异氰酸酯化合物(B)含有选自甲苯二异氰酸酯、二苯甲烷二异氰酸酯、多亚甲基多苯基异氰酸酯及它们的改性体中的一种以上的多异氰酸酯。
(9)上述(1)~(8)中任一项记载的软质聚氨酯泡沫塑料的制造方法,其中,所述催化剂(C)含有选自胺化合物、反应型胺化合物及有机锡化合物中的一种以上的化合物。
(10)上述(1)~(9)中任一项记载的软质聚氨酯泡沫塑料的制造方法,其中,用水作为所述发泡剂(D)。
通过本发明,可采用来自天然油脂的原料来成形具有良好的缓冲性的软质聚氨酯泡沫塑料。
实施发明的最佳方式
本发明中,数均分子量(Mn)及重均分子量(Mw)是聚苯乙烯换算分子量。具体是通过以下的方法测定的值。对于作为分子量测定用标准试样销售的聚合度不同的数种单分散聚苯乙烯聚合物的凝胶渗透色谱(GPC)采用市售的GPC测定装置进行测定,基于聚苯乙烯的分子量和保留时间(retention time)的关系制作校准曲线。采用该校准曲线对待测定的试样化合物的GPC谱进行计算机分析,藉此求得该试样化合物的数均分子量及重均分子量。该测定方法是公知的。
<聚氧化烯多元醇(A)>
本发明的聚氧化烯多元醇(A)(以下也简称为多元醇(A))至少包含下述第1聚氧化烯多元醇(A1)(以下也简称为第1多元醇(A1))。
多元醇(A)优选含有该第1多元醇(A1)和下述第2聚氧化烯多元醇(A2)(以下也简称为第2多元醇(A2))。第1多元醇所含的多元醇不包含于第2多元醇。
<第1聚氧化烯多元醇(A1)>
本发明的第1多元醇(A1)是在下述特定的聚合催化剂(a)的存在下使烯化氧(c)开环聚合于下述由来自天然油脂的多元醇形成的引发剂而制得的聚氧化烯多元醇。
作为引发剂(b)使用的来自天然油脂的多元醇是采用化学反应对天然油脂赋予羟基而得的高分子量体。
天然油脂可采用本来不具有羟基的油脂,优选使用蓖麻油及精制植物甾醇以外的天然油脂。植物甾醇是来自植物的甾醇,在大豆油、菜籽油等植物油中含有微量。该范围内的混入是被允许的。
此外,天然油脂优选包含具有不饱和双键的脂肪酸的甘油酯。作为该具有不饱和双键的天然油脂的优选例,可例举亚麻仁油、红花油、大豆油、桐油、罂粟子油、菜籽油、芝麻油、米油、山茶油、橄榄油、妥尔油、棕榈油、棉籽油、玉米油、鱼油、牛油、猪油等。
此外,为了利用不饱和键来赋予羟基,碘值高的油脂反应性高且可导入更多的羟基,因此优选。所以,较好的是碘值为50以上的油脂,作为具体例,可例举亚麻仁油、红花油、大豆油、桐油、罂粟子油、菜籽油、芝麻油、米油、山茶油、橄榄油、妥尔油、棉籽油、玉米油、鱼油、猪油等。更好的是碘值为100以上的油脂,作为具体例,可例举亚麻仁油、红花油、大豆油、桐油、罂粟子油、菜籽油、芝麻油、米油、妥尔油、棉籽油、玉米油、鱼油等。从价廉的角度考虑,特好的是大豆油。
作为引发剂(b)使用的来自天然油脂的多元醇的羟值为20~250mgKOH/g。蓖麻油的羟值通常为155~177mgKOH/g,由于蓖麻油及植物甾醇以外的天然油脂不具备羟基,因此羟值在10mgKOH/g以下。可通过化学反应对不具备羟基的天然油脂赋予羟基,使羟值达到20~250mgKOH/g。该羟值如果不足20mgKOH/g,则缺乏交联反应性,可能无法显现足够的物性。另一面方,即使将全部的双键转换为羟基时,也无法使羟值达到碘值以上。碘值的最大值为亚麻仁油的190,但反应时发生水解等,可能有来自作为甘油酯的构成醇的甘油的羟基生成。但是,该值明显变大表示甘油酯键被破坏,可能会造成分子量下降、变为高极性、相溶性或物性下降。此外,羟值如果过高,则因导入大量交联剂而导致柔软性的下降或植物原料的用量的减少。基于这些理由考虑,本发明的来自天然油脂的多元醇的羟值为250mgKOH/g以下,更好为30~200mgKOH/g的范围。
该来自天然油脂的多元醇的作为分子量分布的指标的重均分子量(Mw)和数均分子量(Mn)的比率(Mw/Mn)为1.2以上。蓖麻油及植物甾醇的Mw/Mn为1.1以下,但如果通过化学反应对蓖麻油及植物甾醇以外的天然油脂赋予羟基,则该Mw/Mn会达到1.2以上,要小于该值,以目前的技术很难实现。对于该Mw/Mn的上限值无特别限定,从确保流动性的角度考虑,较好为20以下,特好为15以下。
从相溶性或力学物性的角度考虑,本发明的来自天然油脂的多元醇的重均分子量(Mw)较好为1500以上,更好为1700以上,进一步更好为2000以上。对来自该天然油脂的多元醇的Mw的上限无特别限定,较好为50万以下,从粘度低流动性好的角度考虑,更好为10万以下。
通过化学反应对天然油脂赋予羟基来制造来自天然油脂的多元醇的方法可采用公知方法。其具体例有以下的(1)~(5)的方法。(1)通过在天然油脂中注入空气或氧而生成羟基的方法(以下也称为注入法),(2)通过将天然油脂环氧化后将环氧环开环而生成羟基的方法(以下也称为环氧化后羟基赋予法),(3)在特殊的金属催化剂的存在下使一氧化碳和氢与天然油脂的双键反应而生成羰基后,再使氢反应来导入伯(日文:1級)羟基的方法,(4)在所述(1)之后进行(2)或(3)的方法,(5)在所述(2)或(3)后进行(1)的方法。
这些方法中,单独实施的(1)和(2)的方法有利于成本控制,因此优选。
以下,对(1)、(2)的方法进一步进行说明。
[(1)注入法]
通过对天然油脂注入空气或氧,在使不饱和双键间发生氧化交联的同时使其具备羟基的方法。此外,可通过酯交换反应而导入多元醇。该方法中,根据作为原料使用的油脂的种类及注入时的氧化状态,生成物(来自天然油脂的多元醇)的分子量及羟值可变。
将大豆油作为原料通过该方法制得的来自天然油脂的多元醇的重均分子量(Mw)一般为1500以上,较好为5000~500000,更好为10000~100000。Mw/Mn一般为2以上,更好为3~15。重均分子量的值如果过低,则氧化聚合和羟基的生成不充分,交联性劣化,如果过高,则流动性下降。
作为通过注入法赋予大豆油羟基而获得的来自天然油脂的多元醇(曝气大豆油)的例子,可例举美国USSC公司(Urethane Soy Systems Co.)制的商品名:Soyol系列的产品。
[(2)环氧后羟基赋予法]
使氧化剂作用于天然油脂的不饱和双键而环氧化后,用阳离子聚合催化剂在醇的存在下使环氧环开环而具备羟基的方法。作为氧化剂,可使用过乙酸等过氧化物。
被环氧化的天然油脂的环氧当量可通过作为原料使用的油脂的碘值和对应于该碘值的氧化剂的用量及反应率等来控制。利用该环氧化天然油脂的环氧当量可控制生成物(来自天然油脂的多元醇)的羟值。该生成物(来自天然油脂的多元醇)的分子量根据羟基赋予时的作为开环引发剂的醇的量而变化。醇明显较多时,分子量可能变小,但反应效率低,不利于成本控制。醇少时,环氧化大豆油分子间发生开环聚合反应,分子量急剧变大,存在凝胶化的可能性。
例如,将大豆油环氧化而得的环氧化大豆油可从市售品中获得,具体可例举旭电化工业株式会社制商品名:Adecasizer-O-130P等。阳离子聚合引发剂可采用与使烯化氧(c)开环聚合于引发剂(b)时所用的聚合催化剂(a)同样的阳离子聚合催化剂。例如,可使用三氟化硼乙醚络合物(BF3Et2O)。作为醇,例如可使用脱水甲醇。使环氧化大豆油开环赋予羟基的反应可通过在阳离子聚合引发剂和醇的混合溶液中滴加环氧化大豆油后利用吸附过滤除去催化剂的方法来实施。
以环氧化大豆油为原料通过该方法制得的来自天然油脂的多元醇的重均分子量(Mw)一般为1500以上,较好为1800~5000。Mw/Mn一般为1.2~1.9。
<烯化氧>
本发明所用的烯化氧只要是可开环聚合的烯化氧即可,无特别限定。
作为具体例,可例举环氧乙烷(以下也称为EO)、环氧丙烷(以下也称为PO)、氧化苯乙烯、环氧丁烷、环己烯化氧、缩水甘油醚及丙烯酸缩水甘油酯等缩水甘油基化合物、氧杂环丁烷。
本发明中,可以仅使用1种烯化氧,也可并用2种以上的烯化氧。并用2种以上的烯化氧时,可采用嵌段聚合及无规聚合中的任一种聚合法,也可组合使用嵌段聚合和无规聚合来制造一种第1多元醇(A1)。
作为本发明中的烯化氧(c),较好是至少使用环氧丙烷,更好的是使用环氧乙烷及环氧丙烷。环氧丙烷/环氧乙烷的摩尔比较好为100/0~20/80(即,质量比100/0~25/75)。环氧丙烷/环氧乙烷的摩尔比更好为100/0~40/60(即,质量比47/53),环氧丙烷/环氧乙烷的摩尔比进一步更好为100/0~50/50(即,质量比57/43),环氧丙烷/环氧乙烷的摩尔比特好为99/1~60/40(即,质量比99/1~66/34)。
与仅使用环氧丙烷的情况相比,如果并用环氧丙烷和环氧乙烷,则所得的第1多元醇(A1)的末端伯羟基的比例更大。第1多元醇(A1)中,相对于1分子多元醇中的羟基的总数的末端伯羟基的比例较好为1~60摩尔%。
该环氧丙烷和环氧乙烷的合计质量中,如果环氧乙烷为75质量%以下,则反应性适度,从成形性方面考虑比较理想。
多元醇(A1)中的非石油系成分的含有比例(以下也称为生质度(日文:バイオマス度),相对于引发剂(b)和烯化氧(c)的合计质量的引发剂(b)的质量比例)大于85%,较好为87%以上,更好为90%以上。通过使生质度大于85%,可增加天然成分的含量。
<其它环状化合物>
在制造第1多元醇(A1)时,可使反应体系中存在烯化氧(c)以外的其它环状化合物形成的单体。
作为该环状化合物,可例举ε-己内酯及丙交酯等环状酯、碳酸亚乙酯、碳酸异丙烯酯、碳酸新戊酯等环状碳酸酯类。它们可以进行无规聚合也可进行嵌段聚合。
特别是如果使用由来自植物的糖类发酵而得的乳酸衍生的丙交酯,则可进一步提高第1多元醇(A1)中的生质度,因此优选。
<聚合催化剂(a)>
聚合催化剂(a)是不促进来自天然油脂的甘油酯结构的水解的催化剂,较好是采用选自配位阴离子聚合催化剂及阳离子聚合催化剂的1种以上。更好的是配位阴离子聚合催化剂。
[配位阴离子聚合催化剂]
配位阴离子聚合催化剂可采用公知的催化剂。特好的是具有有机配体的复合金属氰化物配位催化剂(以下也称为DMC(双金属氰化物)催化剂)。
具有有机配体的复合金属氰化物的络合物可采用公知制造方法制得。例如,可通过日本专利特开平2003-165836号公报、特开平2005-15786号公报、特开平7-196778号公报、特表2000-513647号公报中记载的方法制得。
具体来讲,可通过以下方法制得。(1)在水溶液中使金属卤化物和碱金属氰基金属酸盐反应,使有机配体在所得反应生成物中配位后,分离固体成分,再用有机配体水溶液对分离的固体成分进行洗涤的方法;(2)在有机配体水溶液中使金属卤化物和碱金属氰基金属酸盐反应,分离所得的反应生成物(固体成分),再用有机配体水溶液对分离的固体成分进行洗涤的方法等。
将该(1)或(2)的方法中对所述反应生成物洗涤、过滤分离而得的滤饼(固体成分)再次分散于含有相对于滤饼为3质量%以下的聚醚化合物的有机配体水溶液,然后通过蒸除挥发成分,也可制得浆状的复合金属氰化物配位催化剂。为了以高活性制造分子量分布狭窄的第1多元醇(A1),特好的是使用该浆状催化剂。
作为用于调制该浆状催化剂的所述聚醚化合物,优选聚醚多元醇或聚醚一元醇。具体来讲优选每一分子的平均羟基数为1~12、重均分子量为300~5000的聚醚一元醇或聚醚多元醇,该聚醚一元醇或聚醚多元醇是用碱催化剂或阳离子催化剂使烯化氧开环聚合于选自一元醇及多元醇的引发剂而制得。
作为DMC催化剂,优选六氰基钴酸锌络合物。
作为DMC催化剂中的有机配体,可使用醇、醚、酮、酯、胺、酰胺等。
作为优选有机配体,可例举叔丁醇、正丁醇、异丁醇、叔戊醇、异戊醇、N,N-二甲基乙酰胺、乙二醇单叔丁基醚、乙二醇二甲醚(glyme)、二甘醇二甲醚(diglyme)、三甘醇二甲醚(triglyme)、异丙醇或二噁烷。二噁烷可以是1,4-二噁烷也可以是1,3-二噁烷,优选1,4-二噁烷。有机配体可以单独使用1种也可2种以上组合使用。
其中,作为有机配体,优选具有叔丁醇。因此,优选使用作为有机配体的至少一部分具有叔丁醇的复合金属氰化物配位催化剂。具有该有机配体的复合金属氰化物配位催化剂可获得高活性,能够制得总不饱和度低的第1多元醇(A1)。少量使用高活性的复合金属氰化物配位催化剂将烯化氧(c)开环聚合而得的精制前的聚醚类的催化剂残渣少,因此可进一步减少精制后的聚氧化烯多元醇的催化剂残渣。
[阳离子聚合催化剂]
作为阳离子聚合催化剂,可例举四氯化铅、四氯化锡、四氯化钛、三氯化铝、氯化锌、三氯化钒、三氯化锑、金属乙酰丙酮化物、五氟化磷、五氟化锑、三氟化硼、三氟化硼络合物(例如三氟化硼乙醚络合物、三氟化硼丁醚络合物、三氟化硼二噁烷络合物、三氟化硼乙酸酐、三氟化硼三乙胺络合物等),高氯酸、高氯酸乙酰酯、高氯酸叔丁酯、羟基乙酸、三氯乙酸、三氟乙酸、对甲苯磺酸、三氟甲磺酸等无机及有机酸,有机酸的金属盐,三乙基氧鎓四氟硼酸盐、三苯基甲基六氟锑酸盐、烯丙基重氮鎓六氟磷酸盐、烯丙基重氮鎓四氟硼酸盐等复合盐化合物,二乙基锌、三乙基铝、氯化二乙基铝等烷基金属盐,杂多酸、同多酸,MoO2(二酮)Cl、MoO2(二酮)OSO2CF3,具有至少1个含氟元素的芳烃基或含氟元素的芳烃氧基的铝或硼化合物等。
其中,特好的是MoO2(二酮)Cl、MoO2(二酮)OSO2CF3,三氟甲磺酸、三氟化硼、三氟化硼乙醚络合物、三氟化硼丁醚络合物、三氟化硼二噁烷络合物、三氟化硼乙酸酐、三氟化硼三乙胺络合物等三氟化硼配位化合物。
此外,作为本发明中的阳离子聚合催化剂,优选具有至少1个含氟元素的芳烃基或含氟元素的芳烃氧基的铝或硼化合物。作为含氟元素的芳烃基,较好是选自五氟苯基、四氟苯基、三氟苯基、3,5-双(三氟甲基)三氟苯基、3,5-双(三氟甲基)苯基、β-全氟萘基、2,2’,2”-全氟联苯基的1种以上。作为含氟元素的芳烃氧基,优选所述含氟芳烃基上结合有氧元素的烃氧基。
作为具有至少1个含氟元素的芳烃基或含氟元素的芳烃氧基的铝或硼化合物,较好是例如在日本专利特开2000-344881号公报、特开2005-82732号公报或国际公开03/000750号文本中记载的作为路易斯酸的硼化合物、铝化合物。或者较好是在日本专利特开2003-501524号公报或特开2003-510374号公报中记载的作为鎓盐的硼化合物、铝化合物。
作为所述路易斯酸的具体例,可例举三(五氟苯基)硼烷、三(五氟苯基)铝、三(五氟苯氧基)硼烷、三(五氟苯氧基)铝等。其中,三(五氟苯基)硼烷对于烯化氧的开环聚合的催化活性较高,因此是特别优选的催化剂。
作为鎓盐的平衡阳离子,优选三苯甲基阳离子或苯胺阳离子,作为鎓盐,特好的是三苯甲基四(五氟苯基)硼酸盐或N,N’-二甲基苯胺四(五氟苯基)硼酸盐。
[其它的不促进来自天然油脂的甘油酯结构的水解的催化剂]
作为以上的配位阴离子聚合催化剂及阴离子聚合催化剂以外的不促进来自天然油脂的甘油酯结构的水解的催化剂,可例举磷腈鎓催化剂。磷腈鎓催化剂可通过公知方法,例如日本专利特开平11-106500号公报中记载的方法获得。
具体可例举氢氧化四[三(二甲基氨基)正膦亚基氨基]鏻等。
<第1多元醇(A1)的制造方法>
反应容器内在聚合催化剂(a)的存在下,将烯化氧(c)开环聚合于引发剂(b)制得第1多元醇(A1)。烯化氧的开环聚合反应可适当采用公知方法来实施。
具体来讲,首先在具备搅拌机和冷凝套管的耐压反应器中投入引发剂,添加聚合催化剂。然后,在引发剂和聚合催化剂的混合物中投入烯化氧使反应进行,藉此制得第1多元醇(A1)。
本发明中,可以使1种烯化氧均聚于引发剂,也可使2种以上的烯化氧发生嵌段聚合及/或无规聚合。
用于聚合反应的聚合催化剂的量只要是烯化氧的开环聚合所需的量,可以是任意的量,但尽可能地少量。
以下,分别对(1)使用DMC催化剂等配位阴离子聚合催化剂作为聚合催化剂的情况和(2)使用阳离子聚合催化剂作为聚合催化剂的情况进行说明。
(1)使用配位阴离子聚合催化剂的情况下,用于聚合反应的聚合催化剂的量越少,越能够减少作为生成物的聚氧化烯多元醇所含的聚合催化剂的量。藉此,可减少聚合催化剂对于通过聚合反应获得的第1多元醇(A1)与多异氰酸酯化合物(B)的反应性及将该第1多元醇(A1)作为原料使用而制得的聚氨酯产品或功能性油剂等的物性的影响。
通常在烯化氧对引发剂的开环聚合反应后实施从所得的聚氧化烯多元醇除去聚合催化剂的操作,但如上所述,残存于聚氧化烯多元醇的聚合催化剂的量少且之后不会有不良影响产生的情况下,可以不实施除去聚合催化剂的工序而直接将所得的聚氧化烯多元醇用于其后的工序,因此可提高聚氧化烯多元醇的生产效率。
进行烯化氧(c)的聚合反应时的聚合催化剂(a)的用量较好设定为使聚合催化剂中的固体催化剂成分(浆料催化剂中的除去了聚醚化合物及过量的配体等的成分)在刚聚合完毕后的聚合物中存在10~150ppm的量,更好是20~120ppm。使该聚合物中所含的聚合催化剂的固体催化剂成分达到10ppm以上,可获得足够的聚合催化活性,而且由于150ppm以下就可获得足够的催化活性,因此使用更大量的催化剂成分并不经济。但是,使用含有相对于所得的聚合物为150ppm以上的固体催化剂成分的催化剂并无问题。
烯化氧的开环聚合温度较好为30~180℃,更好为70~160℃,特好为90~140℃。聚合温度如果为30℃以下,则烯化氧的开环聚合有时无法开始,如果为180℃以上,则有时聚合催化剂的聚合活性下降。
烯化氧(c)的聚合反应结束后,在除去所得反应生成物中所包含的聚合催化剂时,优选的方法是用选自合成硅酸盐(硅酸镁、硅酸铝等)、离子交换树脂及活性白土等的吸附剂吸附催化剂,再通过过滤将吸附剂除去的方法。另外,可例举用选自碱金属氢氧化物、有机酸及无机酸的中和剂对催化剂进行中和,再通过过滤除去的方法等。从不发生水解的角度考虑,更好的是前一种使用吸附剂的方法。
(2)用阳离子聚合催化剂制造聚氧化烯多元醇的方法中,特别是(2-1)烯化氧的碳数为3以上时,较好的是作为阳离子聚合催化剂,使用选自具有至少1个氟取代苯基或氟取代苯氧基的铝或硼化合物的1种以上的化合物的方法。
该(2-1)的方法中,阳离子聚合催化剂的用量相对于引发剂较好为10~120ppm,更好为20~100ppm。从所得聚氧化烯多元醇的精制及成本的角度考虑,催化剂的用量越少越好,但通过使阳离子催化剂用量为10ppm以上,可获得合适的烯化氧聚合速度。
特好的是平均有1~30、较好是1~20个、特好是2~15个烯化氧开环聚合于引发剂的1个羟基。通过使加成于引发剂的每1个羟基的烯化氧数达到2个以上,易使所得聚氧化烯多元醇的全部末端羟基中伯羟基所占的比例高于45%,且可进一步减少聚合体副产物的量。
该(2-1)的方法中,最好通过冷却反应容器并调节烯化氧向反应容器内的供给速度来将反应容器内温保持于所希望的温度,并同时实施反应。该反应容器内的温度通常为-15~140℃,较好为0~120℃,特好为20~90℃。聚合时间通常为0.5~24小时,较好为1~12小时。
以上(1)使用配位阴离子聚合催化剂的情况及(2)使用阳离子聚合催化剂的情况都最好在良好的搅拌条件下实施烯化氧的聚合反应。采用使用一般的搅拌翼的搅拌法时,在不会向反应液中导入大量气相部的气体引发搅拌效率下降的前提下,最好尽可能地加快搅拌翼的旋转速度。此外,烯化氧的聚合反应中,从缩窄所得聚合物(第1多元醇(A1))的分子量分布的角度考虑,最好尽可能地减慢烯化氧向反应容器内的供给速度。另一方面,如果过慢,则生产效率下降,因此最好权衡比较后来设定烯化氧的供给速度。
烯化氧的聚合反应也可采用反应溶剂来实施。作为优选的反应溶剂,可例示己烷、庚烷及环己烷等脂肪烃,苯、甲苯及二甲苯等芳烃以及氯仿及二氯甲烷等卤素类溶剂。此外,对于溶剂的用量无特别限定,可使用所希望量的溶剂。
另外,可在所得的聚氧化烯多元醇(第1多元醇(A1))中添加抗氧化剂、防腐剂等以防止长期保存中的劣化。
本发明中,第1多元醇的重均分子量较好为1500~50万,更好为1500~30万,特好为2000~10万。
以上所得的第1多元醇(A1)是采用来自天然油脂的引发剂(b)制得的多元醇,对于环境无不利影响。此外,作为引发剂(b),由于采用利用化学反应对天然油脂赋予羟基而得的引发剂,因此可降低原料成本。所以,可以较低的成本制得含有天然油脂产物的第1多元醇(A1)。
<第2聚氧化烯多元醇(A2)>
第2多元醇(A2)是第1多元醇(A1)以外的聚氧化烯多元醇,其平均官能团数为2~8且羟值为20~160mgKOH/g。
作为该第2多元醇(A2),可从作为聚氨酯的原料公知的来自石油的聚氧化烯多元醇中适当选用满足以上特性的多元醇。
第2多元醇(A2)的平均官能团数如果不足2,则泡沫塑料的耐久性、乘坐舒适感下降,如果超过8,则制得的软质泡沫塑料有发硬、拉伸等机械物性劣化的倾向,因此不理想。
第2多元醇(A2)的羟值如果不足20,则粘度提高,作业性劣化,如果超过160,则制得的软质泡沫塑料有发硬、拉伸等机械物性劣化的倾向,不理想。
本发明中,第2多元醇(A2)的重均分子量较好为700~22000,更好为1500~2万,特好为2000~15000。
作为该第2多元醇(A2)的例子,优选在开环聚合催化剂的存在下使环状醚化合物开环聚合于引发剂而得的聚酯多元醇或聚碳酸酯多元醇。
作为被用于第2多元醇(A2)的调制的开环聚合催化剂,可例举例如钠类催化剂、钾类催化剂、铯类催化剂等碱金属化合物催化剂,阳离子聚合催化剂,复合金属氰化物配位催化剂、磷腈鎓催化剂等。
作为引发剂,可例举例如乙二醇、二甘醇、丙二醇、二丙二醇、新戊二醇、1,4-丁二醇、1,6-己二醇、甘油、三羟甲基丙烷、季戊四醇、二甘油、葡萄糖、蔗糖、双酚A、乙二胺以及分子量低于将烯化氧加成于以上化合物而得的目的物的聚氧化烯多元醇等。
作为环状醚化合物,较好例如为碳数2以上的烯化氧,具体可例举环氧乙烷、环氧丙烷、1,2-环氧丁烷、2,3-环氧丁烷、氧化苯乙烯等。优选使用环氧丙烷或环氧乙烷。使用环氧乙烷时,第2多元醇(A2)中环氧乙烷的含量较好为30质量%以下,更好为25质量%以下。该环氧乙烷的含量如果为30质量%以下,则反应性适度,成形性良好。
作为钠、钾类催化剂,可例举钠金属、钾金属,甲醇钠、乙醇钠、丙醇钠、甲醇钾、乙醇钾、丙醇钾等钠或钾的醇盐,氢氧化钠、氢氧化钾,碳酸钠、碳酸钾等。
此外,作为铯类催化剂,可例举例如铯类金属,甲醇铯、乙醇铯、丙醇铯等铯的醇盐,氢氧化铯及碳酸铯等。
复合金属配位催化剂可使用与所述作为聚合催化剂(a)例举的复合金属配位催化剂同样的催化剂。
阳离子聚合催化剂可使用与所述作为聚合催化剂(a)例举的阳离子聚合催化剂同样的催化剂。
作为第2多元醇(A2)的聚酯多元醇,可例举乙二醇、丙二醇等碳数2~10的二元醇,甘油、三羟甲基丙烷、三羟甲基乙烷等碳数2~10的三元醇,季戊四醇、二甘油等四元醇,山梨糖醇、蔗糖等糖类等低分子多元醇和琥珀酸、己二酸、马来酸、富马酸、邻苯二甲酸、间苯二甲酸等碳数2~10的二羧酸,琥珀酸酐、马来酸酐、邻苯二甲酸酐等碳数2~10的酸酐等羧酸缩合而得的聚酯多元醇,还可例举ε-己内酯开环聚合物、β-甲基-δ-戊内酯开环聚合物等内酯类多元醇。
作为第2多元醇(A2)的聚碳酸酯多元醇,可例举例如通过被用于所述聚酯多元醇的合成的低分子醇类和光气的脱盐酸反应或所述低分子醇类和碳酸二亚乙酯、碳酸二甲酯、碳酸二苯酯等的酯交换反应而得的多元醇。
第2多元醇(A2)可使用1种聚氧化烯多元醇,也可混合使用2种以上的聚氧化烯多元醇。混合使用2种以上的聚氧化烯多元醇时,混合的各聚氧化烯多元醇的平均官能团数、羟值及重均分子量最好分别在以上的优选范围内。
本发明中,作为多元醇(A)并用第1多元醇(A1)和第2多元醇(A2)时,该多元醇(A)中的第1多元醇(A1)和第2多元醇(A2)的质量比(A1)/(A2)较好在10/90~90/10的范围内,更好的范围是15/85~80/20。如果两者的合计质量中第2多元醇(A2)的用量为10质量%以上,则软质聚氨酯泡沫塑料的成形性很好地提高,从防止地球温室化的角度考虑,较好为90质量%以下。
<聚合物微粒分散多元醇>
本发明中,作为第1多元醇(A1),也可使用以该多元醇(A1)为基质多元醇的聚合物微粒分散多元醇。
此外,作为第2多元醇(A2),也可使用以该第2多元醇(A2)为基质多元醇的聚合物微粒分散多元醇。
或者,也可以在获得以第1多元醇(A1)为基质多元醇的聚合物微粒分散多元醇后,与第2多元醇(A2)混合,形成为聚合物微粒稳定分散的多元醇(A)。此外,同样地也可以在获得以第2多元醇(A2)为基质多元醇的聚合物微粒分散多元醇后,与第1多元醇(A1)混合,形成为聚合物微粒稳定分散的多元醇(A)。
聚合物微粒分散多元醇是作为基质多元醇(分散介质)的聚氧化烯多元醇中稳定地分散有聚合物微粒(分散质)的分散系。作为聚合物微粒的聚合物,可例举加聚系聚合物、缩聚系聚合物。作为具体例,可例举丙烯腈、苯乙烯、甲基丙烯酸酯、丙烯酸酯、其它的乙烯基单体的均聚体、共聚物等加聚系聚合物,聚酯、聚脲、聚氨酯、三聚氰胺等缩聚系聚合物。由于该聚合物微粒的存在,使聚合物微粒分散多元醇整体的羟值一般要比基质多元醇的羟值低。
聚合物微粒分散多元醇中的聚合物微粒的含量较好为50质量%以下。聚合物微粒的量无需特别多。如果过多,除了成本提高以外并没有坏处。一般优选3~50质量%,更好为3~35质量%。使聚合物微粒分散于基质多元醇中有利于提高泡沫塑料的硬度、通气性及其它物性。聚合物微粒分散多元醇的质量用于计算时,不包括聚合物微粒的质量。
作为第1多元醇(A1)使用聚合物微粒分散多元醇时,涉及所述第1多元醇(A1)的重均分子量的数值是表示基质多元醇的数值。
作为第2多元醇(A2)使用聚合物微粒分散多元醇时,涉及所述第2多元醇(A2)的平均官能团数、羟值及重均分子量的数值是表示基质多元醇的数值。
<其它高分子量活性氢化合物>
作为与多异氰酸酯化合物(B)反应的化合物,可并用所述多元醇(A)和其它高分子量活性氢化合物。
该其它高分子量活性氢化合物具体包括具有2个以上的伯氨基或仲氨基的高分子量聚胺或具有1个以上的伯氨基或仲氨基且具有1个以上的羟基的高分子量化合物或哌嗪类多元醇等。
该其它高分子量活性氢化合物的每官能团的分子量较好为400以上,更好为800以上。此外,每1分子的官能团数较好为2~8。每官能团的分子量较好为5000以下。
作为该其它高分子量活性氢化合物,包括使所述聚氧化烯多元醇(第1多元醇(A1)或第2多元醇(A2))的羟基的一部分或全部转换为氨基而获得的化合物、将所述聚氧化烯多元醇和过量的多异氰酸酯化合物反应而得的末端具有异氰酸酯基的预聚物的异氰酸酯基水解转换为氨基而获得的化合物。
此外,所述哌嗪类多元醇是将烯化氧开环聚合于哌嗪类而获得的聚氧化烯多元醇。本发明所述的哌嗪类不仅指哌嗪,还表示哌嗪中的氢原子被烷基或氨基烷基等有机基团取代而得的取代哌嗪。该哌嗪类必须具有至少2个可与烯化氧反应的活性氢。所述使烯化氧开环聚合而得的哌嗪类多元醇中,构成哌嗪环的2个氮原子为叔氨基。
作为哌嗪类的具体例,可例举哌嗪,2-甲基哌嗪、2-乙基哌嗪、2-丁基哌嗪、2-己基哌嗪、2,5-、2,6-、2,3-或2,2-二甲基哌嗪、2,3,5,6-或2,2,5,5-四甲基哌嗪等成环碳原子上所结合的氢原子被低级烷基取代的烷基哌嗪类,N-(2-氨基乙基)哌嗪等成环氮原子上所结合的氢原子被氨基烷基取代的N-氨基烷基哌嗪类。这些哌嗪类中,优选取代哌嗪类,更好的是用氨基烷基等取代氢而形成的哌嗪这样的分子中具有3个以上的氮原子的取代哌嗪类。此外,取代哌嗪类中优选N-取代哌嗪,更好的是N-氨基烷基哌嗪类,特好的是N-(氨基乙基)哌嗪。
作为开环聚合于哌嗪类的烯化氧,优选碳数2以上的烯化氧,具体可例举环氧乙烷、环氧丙烷、1,2-环氧丁烷、2,3-环氧丁烷、氧化苯乙烯等。
本发明中,所述多元醇(A)与其它高分子量活性氢化合物并用时,该其它高分子量活性氢化合物的用量相对于两者的合计量较好为20质量%以下。该用量如果超过20质量%,则反应性大增,成形性等有劣化的可能性。
<多异氰酸酯化合物(B)>
作为多异氰酸酯化合物(B)(以下也简称为多异氰酸酯(B)),可例举具有2个以上的异氰酸酯基的芳香族多异氰酸酯化合物或2种以上的该化合物的混合物,以及将它们改性而得的改性多异氰酸酯等。具体可例举甲苯二异氰酸酯(TDI)、二苯基甲烷二异氰酸酯(MDI)、多亚甲基多苯基异氰酸酯(统称为粗MDI)等多异氰酸酯或它们的预聚物型改性体、脲酸酯改性体、脲改性体、碳二亚胺改性体等。
多异氰酸酯(B)较好是多异氰酸酯成分中的二苯基甲烷二异氰酸酯类多异氰酸酯及/或多亚甲基多苯基异氰酸酯类多异氰酸酯为0质量%以上100质量%以下。特好是5质量%以上80质量%以下,进一步更好是10质量%以上60质量%以下。二苯基甲烷二异氰酸酯类多异氰酸酯及/或多亚甲基多苯基异氰酸酯类多异氰酸酯如果为80质量%以下,则耐久性等物性及泡沫塑料的触感等良好。
多异氰酸酯(B)也可以是预聚物。具体来讲可以是甲苯二异氰酸酯、二苯基甲烷二异氰酸酯类多异氰酸酯或多亚甲基多苯基异氰酸酯类多异氰酸酯和用化学反应对天然油脂赋予羟基而得的天然油脂多元醇、将烯化氧加成于天然油脂多元醇而得的含天然油脂的聚氧化烯多元醇或所述多元醇(A)的预聚物。
多异氰酸酯(B)的用量以相对于所述多元醇(A)、所述其它高分子量活性氢混合物、交联剂及水等的全部的活性氢的合计的异氰酸酯基的数的100倍表示(通常将该以100倍表示的数值称为异氰酸酯指数),较好在80~125的范围内,特好在85~120的范围内。
<交联剂>
本发明中可以根据需要使用交联剂。作为交联剂,较好是含活性氢的基团数为2~8、羟值为200~2000mgKOH/g的交联剂。作为交联剂,可例举具有2个以上的选自羟基、伯氨基及仲氨基的官能团的化合物等。交联剂可以使用1种也可并用2种以上。
具有羟基的交联剂较好是具有2~8个羟基,可例举多元醇、烯化氧加成于多元醇而得的低分子量聚氧化烯多元醇或具有叔氨基的多元醇等多元醇等。
作为具有羟基的交联剂的具体例,可例举乙二醇、1,4-丁二醇、新戊二醇、1,6-己二醇、二甘醇、三甘醇、二丙二醇、一乙醇胺、二乙醇胺、三乙醇胺、甘油、N-烷基二乙醇、双酚A-烯化氧加成物、甘油-烯化氧加成物、三羟甲基丙烷-烯化氧加成物、季戊四醇-烯化氧加成物、山梨糖醇-烯化氧加成物、蔗糖-烯化氧加成物、脂肪族胺-烯化氧加成物、脂环式胺-烯化氧加成物、杂环多胺-烯化氧加成物、芳香族胺-烯化氧加成物等,但并不仅限于此。优选二乙醇胺。使用了该化合物时的滞后损耗良好。
杂环多胺-烯化氧加成物通过将烯化氧加成于哌嗪、2-甲基哌嗪、2-乙基哌嗪、2-丁基哌嗪、2-己基哌嗪、2,5-、2,6-、2,3-或2,2-二甲基哌嗪、2,3,5,6-或2,2,5,5-四甲基哌嗪等短链烷基取代哌嗪或1-(2-氨基乙基)哌嗪等氨基烷基取代哌嗪等而获得。
作为具有伯氨基或仲氨基的胺类交联剂,包括芳香族多胺、脂肪族多胺、脂环式多胺等。
作为芳香族多胺,优选芳香族二胺。作为芳香族二胺,优选结合氨基的芳香环上具有选自烷基、环烷基、烷氧基、烷硫基、吸电子基团的1个以上的取代基的芳香族二胺,特好的是二氨基苯衍生物。较好的是结合氨基的芳香环上结合了2~4个除吸电子基团以外的所述取代基,特好的是在氨基结合部位的1个以上的邻位结合,更好的是全部结合。
较好是在氨基结合的芳香环上结合了1或2个吸电子基团。也可以在1个芳香环上结合有吸电子基团和其它取代基。烷基、烷氧基及烷硫基的碳数较好为4以下,环烷基较好为环己基。作为吸电子基团,优选卤素原子、三卤代甲基、硝基、氰基、烷氧基羰基等,特好的是氯原子、三氟甲基及硝基。
作为脂肪族多胺,包括碳数6以下的二氨基链烷或聚亚烷基多胺、低分子量聚氧化烯多元醇的羟基的一部分或全部转换为氨基而得的多胺等。此外,还可使用具有2个以上的氨基烷基的芳香族化合物、合计具有2个以上的氨基烷基的芳香族化合物及具有以上所述的取代基的所述芳香族化合物等具有芳香环的多胺。
作为脂环式多胺,包括具有2个以上的氨基及/或氨基烷基的环烷。
作为胺类交联剂的具体例,可例举3,5-二乙基-2,4(或2,6)-二氨基甲苯(DETDA)、2-氯-对苯二胺(CPA)、3,5-二甲硫基-2,4(或2,6)-二氨基甲苯、1-三氟甲基-3,5-二氨基苯、1-三氟甲基-4-氯-3,5-二氨基苯、2,4-甲苯二胺、2,6-甲苯二胺、双(3,5-二甲基-4-氨基苯基)甲烷、4,4-二氨基二苯基甲烷、乙二胺、间二甲苯二胺、1,4-二氨基己烷、1,3-双(氨基甲基)环己烷、异佛尔酮二胺等,但并不限于此。
特好的是二乙基甲苯二胺[即,3,5-二乙基-2,4(或2,6)-二氨基甲苯的1种或2种以上的混合物]、二甲硫基甲苯二胺、一氯二氨基苯、三氟甲基二氨基苯等二氨基苯衍生物。
交联剂的用量相对于100质量份多元醇(A),较好为0.1~10质量份。
<催化剂(C)>
使多元醇(A)和多异氰酸酯(B)反应时,使用催化剂(C)。
作为催化剂(C),只要是促进氨基甲酸酯化反应的催化剂即可,无特别限定。例如较好为胺化合物、有机金属化合物、反应型胺化合物、羧酸金属盐等。反应型胺化合物是指为使胺化合物的结构的一部分与异氰酸酯基反应而实施了羟基化或氨基化的化合物。此外,还可根据目的使用羧酸金属盐等使异氰酸酯基之间发生反应的聚合物化催化剂。催化剂可以单独使用1种也可2种以上组合使用。
作为催化剂(C),更好的是胺化合物、有机金属化合物、反应型胺化合物,有机金属化合物较好为有机锡化合物。
作为胺化合物的具体例,可例举三亚乙基二胺、双-((2-二甲基氨基)乙基)醚的二丙二醇溶液、吗啉类等脂肪族胺类。
作为反应型胺化合物的具体例,可例举二甲基乙醇胺、三甲基氨基乙基乙醇胺、二甲基氨基乙氧基乙氧基乙醇等。
胺化合物催化剂及反应型胺化合物催化剂的用量相对于所述多元醇(A)及所述其它高分子量活性氢化合物的合计100质量份,较好为2.0质量份以下,更好为0.005~1.5质量份。
作为有机金属化合物催化剂,包括有机锡化合物或有机铋化合物、有机铅化合物、有机锌化合物等,作为具体例,可例举氧化二正丁基锡、二月桂酸二正丁基锡、二正丁基锡、二乙酸二正丁基锡、氧化二正辛基锡、二月桂酸二正辛基锡、三氯化一丁基锡、二烷基硫醇二正丁基锡、二烷基硫醇二正辛基锡、2-乙基己酸锡(辛酸锡)等。有机金属化合物类催化剂的用量相对于所述多元醇(A)及所述其它高分子量活性氢化合物的合计100质量份,较好为2.0质量份以下,更好为0.05~1.5质量份。
<发泡剂(D)>
本发明中,发泡剂(D)较好是选自水和惰性气体的至少1种。作为发泡剂(D),更好的是至少使用水。作为惰性气体,具体可例示空气、氮气、液化二氧化碳。对于这些发泡剂的使用量没有特别限定。作为发泡剂只使用水的情况下,相对于所述多元醇(A)及所述其它高分子量活性氢化合物的合计100质量份,较好为10质量份以下,更好为0.1~8质量份。
可以根据发泡倍数等要求适量使用除水及惰性气体以外的其它发泡剂。
<其它成分>
此外,为了形成良好的气泡,还可使用泡沫稳定剂。作为泡沫稳定剂,可例举例如有机硅类泡沫稳定剂或氟类泡沫稳定剂等。泡沫稳定剂的用量相对于所述多元醇(A)及所述其它高分子量活性氢化合物的合计100质量份,较好为0.1~10质量份。作为其它可任意使用的掺入剂,可例举例如填充剂、稳定剂、着色剂、阻燃剂、破泡剂等。作为破泡剂,优选平均官能团数2~8、羟值20~100mgKOH/g、环氧乙烷含量为50~100质量%的多元醇。从软质聚氨酯泡沫塑料的成形性,具体来讲是从减少独立气泡的角度考虑,最好使用破泡剂。
<软质聚氨酯泡沫塑料的制造方法>
作为本发明的软质聚氨酯泡沫塑料的制造方法,可以是将反应性混合物注入密闭的模具内进行发泡成形的方法(模具法),也可以是在开放体系中使反应性混合物发泡的方法(平板(スラブ)法)。
利用模具发泡制造软质聚氨酯泡沫塑料的情况下,优选通过将以上各成分混合而得的反应性混合物直接注入模具的方法(即反应注塑成形法)或者通过将以上各成分混合而得的反应性混合物注入开放状态的模具内后密闭的方法来实施。例如,优选通过用低压发泡机或高压发泡机将反应性混合物注入模具的方法,即,通过将反应性混合物注入开放状态的模具后密闭的方法来实施。高压发泡机优选通常的二液混合型,其中一液为多异氰酸酯(B),另一液使用除多异氰酸酯(B)以外的所有原料的混合物。可根据情况形成催化剂(C)或破泡剂(通常分散或溶解于一部分的高分子量多元醇中使用)作为其它单独成分的总计3种成分的反应性混合物并注入。
反应性混合物的温度较好为10~40℃。低于10℃时,原料的粘度大幅度上升,反应液的液混情况劣化。高于40℃时,反应性大增,成形性等劣化。
对于注入时的模具温度无特别限定,较好为10℃~80℃,特好为30℃~70℃。
对于固化(cure)时间无特别限定,较好为3~20分钟,特好为3~10分钟,进一步更好为1~7分钟。固化时间如果为20分钟以上,则从生产性方面考虑,不理想,如果在1分钟以下,则存在固化性不足的问题。
通过平板发泡制造软质聚氨酯泡沫塑料时,可通过一步发泡成形法、半预聚物法、预聚物法等公知方法来实施。软质聚氨酯泡沫塑料的制造中可采用常用的制造装置。
通过本发明的制造方法,如后述的实施例所示,可使用来自天然油脂的多元醇(第1多元醇(A1))很好地成形为软质聚氨酯泡沫塑料。通过本发明的制造方法,可获得回弹性高、缓冲性良好的软质聚氨酯泡沫塑料。
本发明制得的软质聚氨酯泡沫塑料适用于汽车的内装饰材料,特别是可用于座垫、座椅靠背、枕头、扶手等。但并不限于此,作为其它的使用领域,可例举例如火车的内装饰材料、寝具、垫子、衬层等。
实施例
以下,通过实施例及比较例具体说明本发明,但本发明并不限于此。
[调制例:引发剂(b)的调制]
作为以大豆油为原料通过注入法制得的来自天然油脂的多元醇,将美国USSC公司制的商品名:Soyol R2-052F作为引发剂(b1),将R3-170G作为引发剂(b2)。引发剂(b1)的实测的羟值为45.3[mgKOH/g],酸值为4.3[mgKOH/g],Mn(数均分子量)为1578,Mw(重均分子量)为6562,Mw/Mn的比值为4.16。引发剂(b2)的实测的羟值为170[mgKOH/g],酸值为0.93[mgKOH/g],Mn(数均分子量)为940,Mw(重均分子量)为11753,Mw/Mn的比值为12.50。
[调制例2:含聚合催化剂(a)的浆料催化剂的调制]
作为聚合催化剂(a),使用叔丁醇配位的六氰基钴酸锌络合物(DMC催化剂),该DMC催化剂和多元醇P混合而得的混合物浆料(DMC-TBA催化剂)通过下述方法调制。该浆料中包含的DMC催化剂(固体催化剂成分)的浓度(有效成分浓度)为5.33质量%。
[DMC-TBA催化剂的制造]
将10.2g氯化锌和10g水形成的水溶液注入500mL烧瓶中。以300rpm(转速/分钟)搅拌的同时用30分钟的时间将由4.2g六氰基钴酸钾(K3Co(CN)6)和75g水形成的水溶液滴入所述烧瓶内的氯化锌水溶液中。在这期间将烧瓶内的混合溶液的温度保持为40℃。六氰基钴酸钾水溶液的滴加结束后,再对烧瓶内的混合物搅拌30分钟,然后添加80g叔丁醇(以下简称为TBA)、80g水及0.6g下述多元醇P形成的混合物,于40℃搅拌30分钟后再于60℃搅拌60分钟。
所述多元醇P是用KOH催化剂将环氧丙烷加聚于丙二醇后脱碱精制而得的羟基当量为501的聚氧丙烯二醇。
用直径125mm的圆形滤板和微粒用定量滤纸(日本研华(ADVANTEC)公司制的No.5C)在加压下(0.25MPa)对以上所得的混合物进行过滤,分离出含复合金属氰化物的络合物的固体(滤饼)。
然后,将所得的含复合金属氰化物的络合物的滤饼移入烧瓶,添加36g的TBA和84g水的混合物并搅拌30分钟后,在与以上同样的条件下进行加压过滤得到滤饼。将所得滤饼移入烧瓶,再添加108g的TBA和12g水的混合物并搅拌30分钟,获得TBA-水的混合溶剂中分散有复合金属氰化物配位催化剂(DMC催化剂)的液体(浆料)。在该浆料中添加120g所述多元醇P并混合后,减压下于80℃、3小时,接着于115℃、3小时蒸除挥发性成分,获得浆状DMC催化剂(DMC-TBA催化剂)。
[调制例3:第1多元醇(A1-1)的调制]
作为引发剂(b)使用所述引发剂(b1),作为聚合催化剂(a)使用调制例2中获得的含DMC催化剂的浆料催化剂,按照表1所示配比制得多元醇。
即,将带搅拌机的不锈钢制500ml耐压反应器作为反应器使用,在反应器内投入248.2g引发剂(b1)和682mg(固体催化剂成分为36mg)以上的调制例2调制的浆料催化剂。对反应器内部进行氮置换后升温至120℃,实施2小时的真空脱水。然后,用40分钟向反应器内供给24.1g环氧丙烷(PO)和12.2g环氧乙烷(EO)的混合液,再继续搅拌2小时30分钟,确认不再有压力下降。在这期间将反应器的内温保持为120℃、搅拌速度保持为500rpm来进行反应。
藉此获得第1多元醇(A1-1)。所得多元醇的外观在常温下呈透明液状。该多元醇(A1-1)的特性值(Mw、Mn、Mw/Mn、羟值及生质度)示于表1。
第1多元醇(A1)的生质度是该多元醇中的非石油系成分的含有比例的指标,以下的例子中,作为相对于构成第1多元醇(A1)的原料(引发剂(b)和单体)的合计质量的引发剂(b)的质量的比例(单位:%)算出。该值越大表示天然成分的含有比例越高。
[调制例4:第1多元醇(A1-2)的调制]
作为引发剂(b)使用所述引发剂(b1),作为聚合催化剂(a)使用调制例2中获得的含DMC催化剂的浆料催化剂,按照表1所示配比制得第1多元醇(A1)。本例与调制例3的最大不同点在于作为烯化氧未使用环氧乙烷而是仅使用了环氧丙烷。
在与调制例3相同的反应器内投入237.4g引发剂(b1)和660mg(固体催化剂成分为35mg)与调制例3相同的浆料催化剂。对反应器内部进行氮置换后升温至120℃,实施2小时的真空脱水。然后,向反应器内供给38.1g环氧丙烷,继续搅拌2.5小时,确认不再有压力下降。在这期间将反应器的内温保持为120℃、搅拌速度保持为500rpm来进行反应。
藉此获得第1多元醇(A1-2)。所得多元醇的外观在常温下呈透明液状。该第1多元醇(A1-2)的特性值(Mw、Mn、Mw/Mn、羟值及生质度)示于表1。
[调制例5:第1多元醇(A1-3)的调制]
作为引发剂(b)使用所述引发剂(b1),作为聚合催化剂(a)使用调制例2中获得的含DMC催化剂的浆料催化剂,按照表1所示配比在与调制例3同等的条件下进行反应,制得第1多元醇(A1-3)。
在与调制例3相同的反应器内投入242.2g引发剂(b1)和560mg(固体催化剂成分为30mg)以上的调制例2调制的浆料催化剂。对反应器内部进行氮置换后升温至120℃,实施2小时的真空脱水。然后,用40分钟向反应器内供给31.4g环氧丙烷(PO)和5.9g环氧乙烷(EO)的混合液,再继续搅拌2.5小时,确认不再有压力下降。在这期间将反应器的内温保持为1 20℃、搅拌速度保持为500rpm来进行反应。
所得多元醇的外观在常温下呈透明液状。该第1多元醇(A1-3)的特性值(Mw、Mn、Mw/Mn、羟值及生质度)示于表1。
[调制例6:第1多元醇(A1-4)的调制]
作为引发剂(b)使用所述引发剂(b1),作为聚合催化剂(a)使用调制例2中获得的含DMC催化剂的浆料催化剂,按照表1所示配比在与调制例3同等的条件下进行反应,制得第1多元醇(A1-4)。
在与调制例3相同的反应器内投入252.2g引发剂(b1)和672mg(固体催化剂成分为36mg)以上的调制例2调制的浆料催化剂。对反应器内部进行氮置换后升温至120℃,实施2小时的真空脱水。然后,用40分钟向反应器内供给22.8g环氧丙烷(PO)和4.3g环氧乙烷(EO)的混合液,再继续搅拌2.5小时,确认不再有压力下降。在这期间将反应器的内温保持为120℃、搅拌速度保持为500rpm来进行反应。
所得多元醇的外观在常温下呈透明液状。该第1多元醇(A1-4)的特性值(Mw、Mn、Mw/Mn、羟值及生质度)示于表1。
[调制例7:第1多元醇(A1-5)的调制]
作为引发剂(b)使用所述引发剂(b1),作为聚合催化剂(a)使用调制例2中获得的含DMC催化剂的浆料催化剂,按照表1所示配比在与调制例3同等的条件下进行反应,制得第1多元醇(A1-5)。
在与调制例3相同的反应器内投入251.6g引发剂(b1)和672mg(固体催化剂成分为36mg)以上的调制例2调制的浆料催化剂。对反应器内部进行氮置换后升温至120℃,实施2小时的真空脱水。然后,用40分钟向反应器内供给25.6g环氧丙烷(PO)和2.2g环氧乙烷(EO)的混合液,再继续搅拌2.5小时,确认不再有压力下降。在这期间将反应器的内温保持为120℃、搅拌速度保持为500rpm来进行反应。
所得多元醇的外观在常温下呈透明液状。该第1多元醇(A1-5)的特性值(Mw、Mn、Mw/Mn、羟值及生质度)示于表1。
(比较调制例1:比较多元醇1的调制)
作为引发剂(b)使用所述引发剂(b2),作为聚合催化剂使用KOH,按照表1所示配比制得聚氧化烯多元醇。
在与实施例1相同的反应器内投入198.2g引发剂(b2)和2.0g聚合催化剂KOH(浓度95质量%(作为杂质含水)),升温至120℃,实施2小时的真空脱水,进行醇盐化。然后,用0.8小时向反应器内供给88.4g环氧丙烷,再于120℃反应2小时,确认不再有压力下降。用0.45小时向反应器内供给105.6g环氧乙烷,再于120℃反应1小时,确认不再有压力下降。反应结束后,以除去催化剂为目的添加生成量的5质量%的所述キョウワ-ド 600S(商品名,合成氧化镁碱吸附剂),于120℃真空蒸除水分的同时用2小时吸附除去催化剂。
所得聚氧化烯多元醇(比较多元醇1)的特性值示于表1。
(比较调制例2:比较多元醇2的调制)
作为聚合引发剂使用所述引发剂(b1),作为聚合催化剂使用KOH,按照表1所示配比和反应条件制得聚氧化烯多元醇。
在与调制例3相同的反应器内投入215.8g引发剂(b1)和6.25g聚合催化剂KOH(浓度95质量%),升温至120℃,实施2小时的真空脱水,进行醇盐化。然后,用3小时向反应器内供给20.9g环氧丙烷和10.6g环氧乙烷的混合液,再于120℃反应7小时,确认不再有压力下降。反应结束后,以除去催化剂为目的添加生成量的5质量%的所述キョウワ-ド 600S(商品名,合成氧化镁碱吸附剂),于120℃真空蒸除水分的同时用2小时吸附除去催化剂。
所得聚氧化烯多元醇(比较多元醇2)的特性值示于表1。
[表1]

[实施例及比较例]
按照表2、4、6所示的配比制造软质聚氨酯泡沫塑料,测定该泡沫塑料的物性。测定结果示于表3、5、7。表2、4、6中,多异氰酸酯以外的成分的掺合量的单位为质量份。
下述表2、4、6所示的原料如下所述。
·多元醇(A2-1):平均官能团数为4、羟值为28mgKOH/g、末端含有13质量%聚氧乙烯基的聚氧丙烯氧乙烯多元醇。
·多元醇(A2-2):平均官能团数为3、羟值为28mgKOH/g、末端含有17质量%聚氧乙烯基的聚氧丙烯氧乙烯多元醇。
·多元醇(A2-3):平均官能团数为3、羟值为34mgKOH/g、末端含有14.5质量%聚氧乙烯基的聚氧丙烯氧乙烯多元醇中使丙烯腈聚合而得的聚合物分散多元醇,聚合物分散多元醇的羟值为28mgKOH/g,微粒聚合物量为20质量%。
·多元醇(A2-4):平均官能团数为3、羟值为34mgKOH/g、末端含有14.5质量%聚氧乙烯基的聚氧丙烯氧乙烯多元醇中使丙烯腈与苯乙烯共聚而得的聚合物分散多元醇,聚合物分散多元醇的羟值为23.5mgKOH/g,微粒聚合物量为35质量%。
·交联剂1:二乙醇胺。
·交联剂2:平均官能团数为6、羟值为445mgKOH/g、末端含有28质量%聚氧乙烯基的聚氧丙烯氧乙烯多元醇。
·破泡剂:平均官能团数为3、羟值为48mgKOH/g、环氧丙烷/环氧乙烷以质量比20/80的比例进行无规共聚而得的聚氧丙烯氧乙烯多元醇。
·催化剂(C-1):三亚乙基二胺的33%二丙二醇(DPG)溶液(商品名:TEDAL33,东曹(东ソ一)株式会社制)。
·催化剂(C-2):双(2-二甲基氨基乙基)醚的70%DPG溶液(商品名:TOYOCAT ET,东曹株式会社制)。
·催化剂(C-3):2-乙基己酸锡(商品名:Dabco T-9,美国空气化工产品有限公司(Air Products and Chemicals)制)。
·泡沫稳定剂1:有机硅类泡沫稳定剂(商品名:SF-2962,日本东丽道康宁株式会社制)。
·泡沫稳定剂2:有机硅类泡沫稳定剂(商品名:L-3601,日本东丽道康宁株式会社制)。
·泡沫稳定剂3:有机硅类泡沫稳定剂(商品名:SZ-1325,日本东丽道康宁株式会社制)。
·泡沫稳定剂4:有机硅类泡沫稳定剂(商品名:L-5740S,日本东丽道康宁株式会社制)。
·发泡剂(D):水。
·多异氰酸酯化合物(B-1):TDI-80和粗MDI的质量比80/20的混合物(商品名:コロネ-ト1021,日本聚氨酯工业株式会社制)。
·多异氰酸酯化合物(B-2):TDI-80(2,4-TDI/2,6-TDI=80/20质量%的混合物)(商品名:コロネ-トT-80,日本聚氨酯工业株式会社制)。
多异氰酸酯化合物的用量以异氰酸酯指数(当量比的100倍)表示。
(泡沫塑料物性的测定方法)
评价泡沫塑料物性:总密度、芯密度、25%硬度(ILD硬度)、通气性、回弹性、芯部分的回弹性、撕裂强度、抗拉强度、拉伸率、压缩永久变形、湿热压缩永久变形、滞后损耗。
关于芯密度、芯部分的回弹性,使用从泡沫塑料的中央部去除表面部分并切成纵横100mm、高50mm的尺寸的试样进行测定。
基于JIS K6400(1997年版),对总密度、芯密度、25%硬度、回弹性、撕裂强度、抗拉强度、拉伸率、压缩永久变形、湿热压缩永久变形、滞后损耗进行测定。
(振动特性)
评价振动特性:共振频率(单位Hz)、共振倍率(绝对位移测定)和6Hz的传递率。测定基于JAS0 B407-87进行。作为振动特性测定条件,加压板使用铁研形(荷重:490N),助振总振幅为5mm。
(实施例1~4,比较例1~3)
使用调制例3、4获得的第1多元醇(A1-1)及(A1-2)以及比较调制例1、2获得的比较多元醇,按照表2所示的配比通过模具发泡制得软质聚氨酯泡沫塑料。
此外,作为比较例3,用引发剂(b1)替代第1多元醇(A1),按照表2所示配比通过模具发泡制得软质聚氨酯泡沫塑料。
用于发泡的组合物的生质度示于表2。该组合物的生质度是组合物中的非石油系成分的含有比例的指标,在以下的实施例及比较例中,作为构成该组合物的原料的合计质量中该原料所含的引发剂(b1)的质量比例(单位:%)算出。第1多元醇(A1-1)和(A1-2)以及比较多元醇1和2分别包含的引发剂(b1)的质量通过[该多元醇的用量(质量)×该多元醇的生质度(%)]算出。
实施例1~3及比较例1~3中,通过以下的方法制得软质聚氨酯泡沫塑料。
首先,将除多异氰酸酯化合物以外的所有原料的混合物(含多元醇的混合物)调整至液温30±1℃。与此不同,将多异氰酸酯化合物调整至液温25±1℃。
然后,在所述含多元醇的混合物中加入所述多异氰酸酯化合物,使其达到规定的指数,以高速混合机(3000rpm)搅拌混合5秒钟后立即注入加温至60℃的模具中并密闭。该模具为内部尺寸是纵横400mm、高100mm或70mm的铝制模具。
接着,于60℃固化7分钟后,从模具取出软质聚氨酯泡沫塑料,压陷后于室内(温度23℃、相对湿度50%)放置24小时,再进行各种泡沫塑料物性的测定。
压陷是指从模具取出软质聚氨酯泡沫塑料后连续压缩直至泡沫塑料厚度的75%为止的工序。
实施例4中,通过以下的方法制造软质聚氨酯泡沫塑料。
使用2成分系高压发泡机(佳能株式会社制,头部:FPL18φ-L型),将所述含多元醇的混合物填入一个槽内,将液温调整为25℃±2℃。在另一个槽内填入多异氰酸酯化合物,调整至25℃±2℃。然后,将它们混合而成的原料注入加温至60℃的模具中并密闭。使用模具内部尺寸是纵横400mm、高100mm或70mm的铝制模具。
然后,于60℃固化7分钟后,从模具取出软质聚氨酯泡沫塑料,压陷后于室内(温度23℃、相对湿度50%)放置24小时,再进行各种泡沫塑料物性的测定。
[表2]
[表3]

从表3的结果可看出,使用了第1多元醇(A1-1)及(A1-2)的实施例1~4获得了良好的软质聚氨酯泡沫塑料,该软质聚氨酯泡沫塑料的回弹性高,缓冲性良好。特别是使用了第1多元醇(A1-1)的实施例1~3获得的软质聚氨酯泡沫塑料的缓冲性良好。
此外,关于作为汽车座椅的舒适感的评价指标的振动特性,比较例1~3的共振频率为4Hz以上,对应于此,实施例1~4的该值为4Hz以下,获得良好的结果。即,如果共振频率值为4Hz以下,则人能感觉到的频率范围内的振动被有效减弱,可获得高舒适度。该共振频率越小越好。此外,共振倍率及6Hz的传递率越小舒适度越高。
对应于此,作为聚氧化烯多元醇(A)使用了比较多元醇的比较例1,2的回弹性低,滞后损耗大,缓冲性劣化。该比较多元醇通过使用与第1多元醇(A1-1)及(A1-2)相同的来自大豆油的多元醇作为聚合引发剂、使用阴离子聚合催化剂KOH作为聚合催化剂而制得。
此外,用未加成烯化氧的来自大豆油的多元醇(引发剂(b1))替代第1多元醇(A1-1)及(A1-2)的比较例3的湿热压缩永久变形及干热压缩永久变形明显劣化,与实施例相比回弹性小,缓冲性劣化。此外,撕裂强度明显劣化。
(实施例5、比较例4和5)
作为实施例5,使用调制例3获得的第1多元醇(A1-1),按照表4所示的配比通过平板发泡制得软质聚氨酯泡沫塑料。
比较例4和5是分别用引发剂(b1)替代实施例5中的第1多元醇(A1-1)的例子。用于发泡的组合物的生质度示于表4。
首先,除去作为由有机锡化合物形成的催化剂的催化剂(C-3)和多异氰酸酯化合物后的所有原料的混合物(含多元醇的混合物)的液温调整为23±1℃。与此不同,将多异氰酸酯化合物的液温调整为22±1℃。
然后,在所述含多元醇的混合物中加入催化剂(C-3),以高速混合机(3000rpm)搅拌混合5秒钟后,加入所述多异氰酸酯化合物达到规定的指数,同样地搅拌混合5秒钟。接着于室温立即注入上部开放的模具中。作为模具,使用内部尺寸纵横高都为250mm的木箱的内表面铺满了乙烯树脂的模具。
注入结束2分钟后,从模具取出软质聚氨酯泡沫塑料,于室内(温度23℃、相对湿度50%)放置24小时,再进行各种泡沫塑料物性的测定。
[表4]

[表5]
从表5的结果可看出,使用了第1多元醇(A1-1)的实施例5获得了良好的软质聚氨酯泡沫塑料。另一方面,替代第1多元醇使用了引发剂(b1)的比较例4、5中,掺入了与实施例5中的第1多元醇相同份数的引发剂(b1)的比较例5中,泡沫塑料内部发生破裂,成形性劣化。由于比较例5中泡沫塑料发生破裂,因此无法进行泡沫塑料物性的测定。另外,实施例5和比较例4相比,回弹性低,低回弹性良好。
(实施例6~11)
使用调制例5~7获得的第1多元醇(A1-3)~(A1-5),按照表6所示的配比通过模具发泡制得软质聚氨酯泡沫塑料。用于发泡的组合物的生质度示于表6。
软质聚氨酯泡沫塑料的制造工序与实施例1相同。作为模具,使用内部尺寸是纵横400mm、高100mm的铝制模具。对于所得的软质聚氨酯泡沫塑料,与实施例1同样进行各种泡沫塑料物性的测定。
[表6]
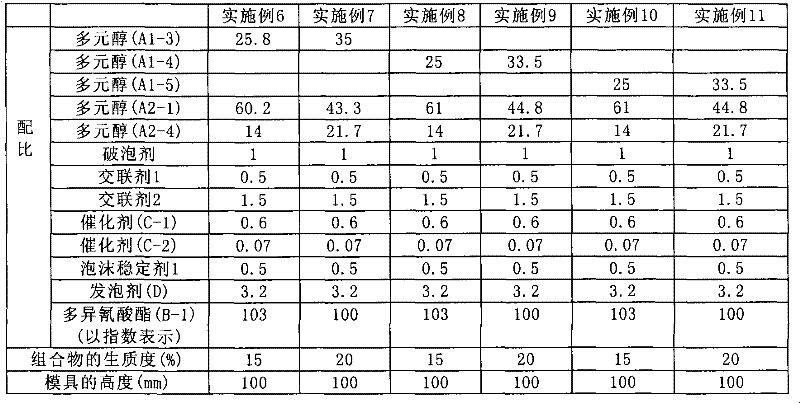
[表7]

如表7的结果所示,使用了第1多元醇(A1-3)~(A1-5)的实施例6~11获得了良好的软质聚氨酯泡沫塑料。该软质聚氨酯泡沫塑料的回弹性高,缓冲性良好,关于振动特性,共振频率为4Hz以下,良好。
产业上利用的可能性
本发明提供使用来自天然油脂的原料,制造回弹性高、具有良好的缓冲性的软质聚氨酯泡沫塑料的方法。
这里引用2006年9月27日提出申请的日本专利申请2006-263134号的说明书、权利要求书和摘要的全部内容作为本发明的说明书的揭示。
Claims (10)
1.软质聚氨酯泡沫塑料的制造方法,它是具备在催化剂(C)及发泡剂(D)的存在下使聚氧化烯多元醇(A)和多异氰酸酯化合物(B)反应的步骤的软质聚氨酯泡沫塑料的制造方法,所述聚氧化烯多元醇(A)包含第1聚氧化烯多元醇(A1),所述第1聚氧化烯多元醇(A1)通过在具有有机配体的复合金属氰化物配位催化剂作为聚合催化剂(a)的存在下使烯化氧(c)开环聚合于引发剂(b)而制得,
其特征在于,所述聚合催化剂(a)是不促进来自天然油脂的甘油酯结构的水解的催化剂,所述引发剂(b)是利用化学反应对天然油脂赋予羟基而形成的引发剂,是羟值为20~250mgKOH/g且以聚苯乙烯换算的重均分子量对数均分子量的比率(Mw/Mn)为1.2以上的来自天然油脂的多元醇。
2.如权利要求1所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述聚氧化烯多元醇(A)还包括所述第1聚氧化烯多元醇(A1)以外的第2聚氧化烯多元醇(A2),该第2聚氧化烯多元醇(A2)的平均官能团数为2~8且羟值为20~160mgKOH/g。
3.如权利要求2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述聚氧化烯多元醇(A)中的所述第1聚氧化烯多元醇(A1)和第2聚氧化烯多元醇(A2)的质量比(A1)/(A2)为10/90~90/10。
4.如权利要求1或2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述烯化氧(c)包含环氧丙烷。
5.如权利要求1或2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述引发剂(b)是利用(1)通过在天然油脂中注入空气或氧而生成羟基的方法或/及(2)通过将天然油脂环氧化后将环氧环开环而生成羟基的方法获得的来自天然油脂的多元醇。
6.如权利要求1或2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述引发剂(b)来自大豆油。
7.如权利要求1或2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述多异氰酸酯化合物(B)含有选自甲苯二异氰酸酯、二苯甲烷二异氰酸酯、 多亚甲基多苯基异氰酸酯及它们的改性体中的一种以上的多异氰酸酯,其中所述改性体是预聚物型改性体、脲酸酯改性体、脲改性体、碳二亚胺改性体。
8.如权利要求1或2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述催化剂(C)含有选自胺化合物及有机锡化合物中的一种以上的化合物。
9.如权利要求1或2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,所述催化剂(C)含有反应型胺化合物。
10.如权利要求1或2所述的软质聚氨酯泡沫塑料的制造方法,其特征在于,用水作为所述发泡剂(D)。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP263134/2006 | 2006-09-27 | ||
| JP2006263134 | 2006-09-27 | ||
| PCT/JP2007/068714 WO2008038678A1 (fr) | 2006-09-27 | 2007-09-26 | Procédé servant à produire une mousse de polyuréthane souple |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101516954A CN101516954A (zh) | 2009-08-26 |
| CN101516954B true CN101516954B (zh) | 2011-07-20 |
Family
ID=39230114
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2007800352530A Expired - Fee Related CN101516954B (zh) | 2006-09-27 | 2007-09-26 | 软质聚氨酯泡沫塑料的制造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20090239964A1 (zh) |
| EP (1) | EP2067804A4 (zh) |
| JP (1) | JP5239864B2 (zh) |
| KR (1) | KR20090068216A (zh) |
| CN (1) | CN101516954B (zh) |
| AU (1) | AU2007301112A1 (zh) |
| TW (1) | TW200835706A (zh) |
| WO (1) | WO2008038678A1 (zh) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW200801060A (en) * | 2006-02-28 | 2008-01-01 | Asahi Glass Co Ltd | Flexible polyurethane foam and process for producing the same |
| EP2125958A4 (en) * | 2007-03-02 | 2012-08-29 | Proprietect Lp | DISAPPEARED ISOCYANATE BASED POLYMER |
| WO2009001783A1 (ja) * | 2007-06-22 | 2008-12-31 | Asahi Glass Company, Limited | ポリマー分散ポリオールおよび軟質ポリウレタンフォームの製造方法 |
| JP5505921B2 (ja) * | 2007-08-01 | 2014-05-28 | 株式会社イノアックコーポレーション | 軟質ポリウレタン発泡体 |
| JP5229229B2 (ja) * | 2007-09-28 | 2013-07-03 | 旭硝子株式会社 | 軟質ポリウレタンフォームおよび熱プレス成形品の製造方法 |
| US8076001B2 (en) * | 2008-09-02 | 2011-12-13 | Ppg Industries Ohio, Inc | Crosslinked coatings comprising lactide |
| WO2011043348A1 (ja) * | 2009-10-05 | 2011-04-14 | 旭硝子株式会社 | 軟質ポリウレタンフォームの製造方法およびシート |
| CN102549022A (zh) * | 2009-10-05 | 2012-07-04 | 旭硝子株式会社 | 聚合物分散多元醇及软质聚氨酯泡沫塑料的制造方法 |
| TW201120077A (en) * | 2009-10-05 | 2011-06-16 | Asahi Glass Co Ltd | Soft polyurethane foam, method for producing the same, and seat cushion for automobile |
| PL2563834T3 (pl) * | 2010-04-29 | 2019-07-31 | Dow Global Technologies Llc | Hybrydowe poliestropolieteropoliole |
| US9382372B2 (en) | 2010-08-06 | 2016-07-05 | Mitsui Chemicals & Skc Polyurethanes Inc. | Polyol, polyol composition, and flexible polyurethane foam using the same |
| JP2012214779A (ja) * | 2011-03-30 | 2012-11-08 | Asahi Glass Co Ltd | 軟質ポリウレタンフォームの製造方法およびシート |
| KR101909320B1 (ko) * | 2011-08-25 | 2018-10-17 | 다우 글로벌 테크놀로지스 엘엘씨 | 이중 금속 시아니드 촉매의 존재 하에 에틸렌 카르보네이트의 중합에 의한 옥시에틸렌 단위를 갖는 폴리에테르 알콜의 제조 방법 |
| MY184782A (en) * | 2011-11-03 | 2021-04-21 | Malaysian Palm Oil Board | A method to produce copolymers of tetrahydrofuran and epoxidized natural oils |
| CN104271546B (zh) | 2012-02-28 | 2017-06-09 | 马来西亚国家石油公司 | 酯的生产方法及其用途 |
| CN104321302A (zh) * | 2012-02-28 | 2015-01-28 | 马来西亚国家石油公司 | 用于聚氨酯应用的物质多元醇的组合物 |
| WO2013129909A1 (en) | 2012-02-28 | 2013-09-06 | Petroliam Nasional Berhad | Lubricant composition of matter and methods of preparation |
| US9587155B2 (en) * | 2012-05-16 | 2017-03-07 | Ecolab Usa Inc. | Emulsion breakers including polyester functionalities |
| MY169226A (en) | 2013-02-28 | 2019-03-19 | Petroliam Nasional Berhad | Preparation of biopolyol esters for lubricant application |
| EP3077437A1 (de) | 2013-11-27 | 2016-10-12 | Covestro Deutschland AG | Mischungen von polyethercarbonatpolyolen und polyetherpolyolen zur herstellung von polyurethanweichschaumstoffen |
| CN104479100A (zh) * | 2014-12-03 | 2015-04-01 | 舟山市银岱汽车零部件有限公司 | 一种富含papi聚氨酯高回弹泡沫材料 |
| WO2016119049A1 (en) * | 2015-01-26 | 2016-08-04 | Trent University | Methods of making triacylglycerol polyols from fractions of metathesized natural oils and uses thereof |
| ES2931312T3 (es) * | 2015-08-17 | 2022-12-28 | Evonik Operations Gmbh | Producción de espumas blandas de poliuretano con dureza mejorada |
| JP6735773B2 (ja) | 2015-12-16 | 2020-08-05 | 株式会社ブリヂストン | シート用パッド |
| CN111690110B (zh) * | 2020-06-22 | 2022-03-11 | 上海墨梵新材料科技有限公司 | 包括含活性基团的聚氨酯催化剂的组合聚醚多元醇及其制备方法 |
| CN115612059B (zh) * | 2022-11-02 | 2023-06-20 | 东莞市腾崴塑胶制品有限公司 | 一种生物基海绵 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1400231A (zh) * | 2001-07-18 | 2003-03-05 | 旭硝子株式会社 | 多元醇和聚合物分散多元醇的制造方法 |
| EP1666514A1 (en) * | 2003-09-19 | 2006-06-07 | Asahi Glass Company Ltd. | Flexible polyurethane foam and process for producing the same |
| CN1802396A (zh) * | 2003-04-25 | 2006-07-12 | 陶氏环球技术公司 | 从含羟甲基的聚酯多元醇生产的聚氨酯泡沫 |
Family Cites Families (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60177013A (ja) * | 1984-02-22 | 1985-09-11 | Ito Seiyu Kk | ポリオ−ル組成物 |
| WO1990004613A1 (en) * | 1988-10-25 | 1990-05-03 | Asahi Glass Company Ltd. | Elastic polyurethane foam and process for its production |
| JP3053937B2 (ja) * | 1991-11-13 | 2000-06-19 | 旭硝子株式会社 | ポリウレタンフォームの製造方法 |
| JPH05163342A (ja) | 1991-12-11 | 1993-06-29 | Asahi Glass Co Ltd | ポリエーテル類の製造方法 |
| US5470813A (en) | 1993-11-23 | 1995-11-28 | Arco Chemical Technology, L.P. | Double metal cyanide complex catalysts |
| JP3680339B2 (ja) * | 1995-02-28 | 2005-08-10 | 旭硝子株式会社 | 高弾性ポリウレタンフォームの製造方法 |
| JPH08253547A (ja) * | 1995-03-16 | 1996-10-01 | Asahi Glass Co Ltd | ポリウレタン溶液組成物 |
| JP3694951B2 (ja) * | 1996-01-16 | 2005-09-14 | 旭硝子株式会社 | 硬質発泡合成樹脂の製造方法 |
| US5639705A (en) | 1996-01-19 | 1997-06-17 | Arco Chemical Technology, L.P. | Double metal cyanide catalysts and methods for making them |
| JP3905638B2 (ja) | 1997-05-28 | 2007-04-18 | 三井化学株式会社 | ポリオキシアルキレンポリオール及びその誘導体、並びに、該ポリオキシアルキレンポリオールの製造方法 |
| US6072021A (en) | 1997-10-24 | 2000-06-06 | E. I. Du Pont De Nemours And Company | Copolymerization of formaldehyde and cyclic ethers using initiators based upon tetraphenyl borates |
| JP3076032B1 (ja) | 1998-07-10 | 2000-08-14 | 三洋化成工業株式会社 | 新規ポリオキシアルキレンポリオールおよび開環重合体の製造法 |
| WO2000002951A1 (fr) * | 1998-07-10 | 2000-01-20 | Asahi Glass Company Ltd. | Catalyseur de polymerisation par decyclisation d'un oxyde d'alkylene, et son procede de fabrication et d'utilisation |
| US20030191274A1 (en) | 2001-10-10 | 2003-10-09 | Kurth Thomas M. | Oxylated vegetable-based polyol having increased functionality and urethane material formed using the polyol |
| US6180686B1 (en) | 1998-09-17 | 2001-01-30 | Thomas M. Kurth | Cellular plastic material |
| ATE291594T1 (de) | 1999-06-04 | 2005-04-15 | Goodrich Corp | Katalysator und verfahren zur herstellung von oligomeren von alpha-methyl- styrol |
| US6476244B2 (en) | 2000-02-23 | 2002-11-05 | South Dakota Soybean Processors | Process for preparing blown vegetable oil |
| WO2003000750A1 (en) | 2001-06-20 | 2003-01-03 | Colorado State University Research Foundation | Polymerization processes using a highly active catalyst |
| JP4122874B2 (ja) * | 2001-07-18 | 2008-07-23 | 旭硝子株式会社 | ポリオールおよびポリマー分散ポリオールの製造方法 |
| EP1283231A1 (en) * | 2001-08-06 | 2003-02-12 | Asahi Glass Company Ltd. | Flexible polyurethane foam, its production method and material system for its production |
| JP2003165836A (ja) | 2001-11-29 | 2003-06-10 | Asahi Glass Co Ltd | スラリー状複合金属シアン化物錯体触媒およびその製造方法、ならびにこれを用いたポリエーテルポリオールまたはポリエーテルモノオールの製造方法 |
| ATE367411T1 (de) * | 2001-11-30 | 2007-08-15 | Asahi Glass Co Ltd | Polyurethanweichschaum und verfahren zur herstellung |
| ATE430770T1 (de) * | 2001-12-21 | 2009-05-15 | Asahi Glass Co Ltd | Polyurethanweichschaumstoff mit geringer rückprallelastizität und herstellungsverfahren dafür |
| DE10240186A1 (de) * | 2002-08-28 | 2004-03-11 | Basf Ag | Verfahren zur Herstellung von emissionsarmen Polyurethan-Weichschaumstoffen |
| JP3906464B2 (ja) * | 2002-12-10 | 2007-04-18 | アイカ工業株式会社 | ウレタン樹脂組成物 |
| JP4556496B2 (ja) | 2003-06-04 | 2010-10-06 | 旭硝子株式会社 | 複合金属シアン化物錯体触媒、その製造方法およびその利用 |
| JP2005082732A (ja) | 2003-09-10 | 2005-03-31 | Sanyo Chem Ind Ltd | 開環重合体の製造法 |
| JP4617793B2 (ja) * | 2003-09-19 | 2011-01-26 | 旭硝子株式会社 | 自動車内装材用の防音材 |
| US8293808B2 (en) * | 2003-09-30 | 2012-10-23 | Cargill, Incorporated | Flexible polyurethane foams prepared using modified vegetable oil-based polyols |
| CN100473675C (zh) * | 2003-11-26 | 2009-04-01 | 旭硝子株式会社 | 软质聚氨酯泡沫塑料及其制造方法、使用其的汽车用座椅 |
| JP4008888B2 (ja) * | 2004-01-30 | 2007-11-14 | 三洋化成工業株式会社 | 軟質ポリウレタンフォームの製造方法 |
| JP2005320431A (ja) * | 2004-05-10 | 2005-11-17 | Honda Motor Co Ltd | 大豆油由来の軟質ポリウレタンフォームからなる自動車シート用クッション |
| US20060041155A1 (en) * | 2004-08-23 | 2006-02-23 | Biobased Chemical | Method of preparing a hydroxy functional vegetable oil |
| CN101056911B (zh) * | 2004-11-18 | 2010-11-24 | 旭硝子株式会社 | 软质聚氨酯泡沫塑料的制造方法 |
| EP1816151A4 (en) * | 2004-11-24 | 2012-07-25 | Asahi Glass Co Ltd | POLYURETHANE-SOFT FOAM, MANUFACTURING METHOD AND CAR SEAT |
| JP4673104B2 (ja) | 2005-03-24 | 2011-04-20 | 株式会社ユニバーサルエンターテインメント | 弾球遊技機 |
| US20060229375A1 (en) * | 2005-04-06 | 2006-10-12 | Yu-Ling Hsiao | Polyurethane foams made with alkoxylated vegetable oil hydroxylate |
| EP1889861A4 (en) * | 2005-04-21 | 2012-03-28 | Asahi Glass Co Ltd | LOW RESILIENCE POLYURETHANE FLEXIBLE FOAM AND PRODUCTION METHOD THEREOF |
| CN101180338B (zh) * | 2005-05-25 | 2011-05-04 | 旭硝子株式会社 | 软质聚氨酯泡沫塑料及其制造方法、利用该软质聚氨酯泡沫塑料的汽车用座垫 |
| JP5145935B2 (ja) * | 2005-05-25 | 2013-02-20 | 旭硝子株式会社 | 軟質ポリウレタンフォームおよびその製造方法、該軟質ポリウレタンフォームを用いた自動車用シート |
| KR20080017325A (ko) * | 2005-05-25 | 2008-02-26 | 아사히 가라스 가부시키가이샤 | 연질 폴리우레탄폼 및 그 제조 방법, 그 연질폴리우레탄폼을 이용한 자동차용 시트 |
| JP5526476B2 (ja) * | 2005-08-05 | 2014-06-18 | 旭硝子株式会社 | 軟質ポリウレタンフォーム、その製造方法および自動車用シート |
| DE102005056432A1 (de) * | 2005-11-26 | 2007-05-31 | Bayer Materialscience Ag | Verfahren zur Herstellung von Polyolen auf Basis natürlicher Öle |
| TW200801060A (en) * | 2006-02-28 | 2008-01-01 | Asahi Glass Co Ltd | Flexible polyurethane foam and process for producing the same |
| US9284401B2 (en) * | 2006-11-13 | 2016-03-15 | Bayer Materialscience Llc | Process for the preparation of polyether-ester polyols |
-
2007
- 2007-09-26 EP EP07828460A patent/EP2067804A4/en not_active Withdrawn
- 2007-09-26 WO PCT/JP2007/068714 patent/WO2008038678A1/ja active Application Filing
- 2007-09-26 JP JP2008536400A patent/JP5239864B2/ja not_active Expired - Fee Related
- 2007-09-26 AU AU2007301112A patent/AU2007301112A1/en not_active Abandoned
- 2007-09-26 KR KR1020097006135A patent/KR20090068216A/ko not_active Application Discontinuation
- 2007-09-26 CN CN2007800352530A patent/CN101516954B/zh not_active Expired - Fee Related
- 2007-09-27 TW TW096136057A patent/TW200835706A/zh unknown
-
2009
- 2009-03-27 US US12/412,416 patent/US20090239964A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1400231A (zh) * | 2001-07-18 | 2003-03-05 | 旭硝子株式会社 | 多元醇和聚合物分散多元醇的制造方法 |
| CN1802396A (zh) * | 2003-04-25 | 2006-07-12 | 陶氏环球技术公司 | 从含羟甲基的聚酯多元醇生产的聚氨酯泡沫 |
| EP1666514A1 (en) * | 2003-09-19 | 2006-06-07 | Asahi Glass Company Ltd. | Flexible polyurethane foam and process for producing the same |
Non-Patent Citations (3)
| Title |
|---|
| JP昭60-177013A 1985.09.11 |
| JP特开2004-189816A 2004.07.08 |
| JP特开平9-194561A 1997.07.29 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101516954A (zh) | 2009-08-26 |
| EP2067804A1 (en) | 2009-06-10 |
| JP5239864B2 (ja) | 2013-07-17 |
| US20090239964A1 (en) | 2009-09-24 |
| EP2067804A4 (en) | 2012-10-31 |
| KR20090068216A (ko) | 2009-06-25 |
| AU2007301112A1 (en) | 2008-04-03 |
| WO2008038678A1 (fr) | 2008-04-03 |
| JPWO2008038678A1 (ja) | 2010-01-28 |
| TW200835706A (en) | 2008-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101516954B (zh) | 软质聚氨酯泡沫塑料的制造方法 | |
| JP5201141B2 (ja) | ポリマー分散ポリオールおよび軟質ポリウレタンフォームの製造方法 | |
| CN101563386A (zh) | 软质聚氨酯泡沫塑料的制造方法 | |
| CN102574976B (zh) | 软质聚氨酯泡沫塑料的制造方法及片材 | |
| CN102741309B (zh) | 基于天然油的多元醇的多元醇预聚物 | |
| CN102341420A (zh) | 高弹性柔性聚氨酯泡沫体 | |
| JP5169752B2 (ja) | 軟質ポリウレタンフォームの製造方法 | |
| JP4798309B2 (ja) | ポリマー分散ポリオールおよび軟質ポリウレタンフォームの製造方法 | |
| CN102574974A (zh) | 软质聚氨酯泡沫塑料及其制造方法以及汽车用座垫 | |
| GB2482176A (en) | Production of polyols for use in low ball rebound polyurethane foams | |
| AU2001263136A1 (en) | Polyurethanes containing dispersed crystalline polyesters | |
| JP5609753B2 (ja) | ポリオキシアルキレンポリオール、ポリマー分散ポリオール、および軟質ポリウレタンフォーム、ならびにこれらの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20110720 Termination date: 20150926 |
|
| EXPY | Termination of patent right or utility model |