BRPI0920834B1 - processo para preparação de derivados protegidos de l-alanina - Google Patents
processo para preparação de derivados protegidos de l-alanina Download PDFInfo
- Publication number
- BRPI0920834B1 BRPI0920834B1 BRPI0920834-8A BRPI0920834A BRPI0920834B1 BR PI0920834 B1 BRPI0920834 B1 BR PI0920834B1 BR PI0920834 A BRPI0920834 A BR PI0920834A BR PI0920834 B1 BRPI0920834 B1 BR PI0920834B1
- Authority
- BR
- Brazil
- Prior art keywords
- formula
- compound
- process according
- fact
- acid
- Prior art date
Links
- 0 C[C@](C(C=O)O)[C@@](C1)([C@]1C1C(C#C)=CC(Cl)=CC1*)NC(OC(C)(C)C)=O Chemical compound C[C@](C(C=O)O)[C@@](C1)([C@]1C1C(C#C)=CC(Cl)=CC1*)NC(OC(C)(C)C)=O 0.000 description 5
- JTHVTKBYEKZELM-FOUAAFFMSA-N C[C@H](C1)C(Br)=C(C)CC1[Ne] Chemical compound C[C@H](C1)C(Br)=C(C)CC1[Ne] JTHVTKBYEKZELM-FOUAAFFMSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/14—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof
- C07C227/16—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof by reactions not involving the amino or carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/06—Preparation of carboxylic acid amides from nitriles by transformation of cyano groups into carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/06—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups by reactions not involving the formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/02—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/04—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C229/06—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one amino and one carboxyl group bound to the carbon skeleton
- C07C229/10—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one amino and one carboxyl group bound to the carbon skeleton the nitrogen atom of the amino group being further bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings
- C07C229/12—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one amino and one carboxyl group bound to the carbon skeleton the nitrogen atom of the amino group being further bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings to carbon atoms of acyclic carbon skeletons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/14—Preparation of carboxylic acid amides by formation of carboxamide groups together with reactions not involving the carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/64—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings
- C07C233/65—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/16—Preparation of carboxylic acid nitriles by reaction of cyanides with lactones or compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/28—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reaction of hydroxy compounds with sulfonic acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F3/00—Compounds containing elements of Groups 2 or 12 of the Periodic System
- C07F3/06—Zinc compounds
Abstract
PROCESSO PARA PREPARAÇÃO DE DERIVADOS PROTEGIDOS DE L-ALANINA. A presente invenção refere-se a um processo inovador destinado à preparação de derivados protegidos de L-alanina, úteis como intermediários na síntese de compostos úteis como moduladores de mu/delta opioides.
Description
[001] A presente invenção refere-se a um processo inovador destinado à preparação de derivados protegidos de L-alanina, úteis como intermediários na síntese de compostos úteis como moduladores de mu/delta opioides.
[002] Os receptores opioides foram identificados em meados dos anos 1970, e foram rapidamente categorizados em três subconjuntos de receptores (mu, delta e kappa). Mais recentemente, os três tipos originais de receptores foram adicionalmente divididos em subtipos. É sabido, também, que a família de receptores opioides são membros da superfamília (GPCR) de receptor acoplado à proteína G. Mais fisiologi-camente pertinentes são os fatos bem estabelecidos de que os receptores opioides são encontrados ao longo do sistema nervoso periférico e central de muitas espécies de mamífero, incluindo seres humanos, e que a modulação dos receptores respectivos pode produzir inúmeros, embora diferentes, efeitos biológicos, tanto desejáveis quanto indesejáveis (D.S. Fries, "Analgesics", em Principles of Medicinal Chemistry, 4a. Edição.; W.O. Foye, T.L. Lemke, e D.A. Williams, Eds.; Williams e Wilkins: Baltimore, Md., 1995; páginas. 247 a 269; J.V. Aldrich, "Analgesics", Burger’s Medicinal Chemistry and Drug Discovery, 5a Edição, Volume 3: Therapeutic Agents, John Wiley & Sons, Inc., 1996, páginas. 321 a 441). Na literatura mais atual, a probabilidade da heterodi-merização das subclasses de receptores opioides foi relatada, com as respostas fisiológicas respectivas ainda indeterminadas (Pierre J.M. Riviere e Jean-Louis Junien, "Opioid receptors: Targets for new gastrointestinal drug development", Drug Development 2000, páginas. 203 a 238).
[003] Os efeitos biológicos identificados nos moduladores de opi-oides levaram a muitos agentes medicinais úteis. Mais significativos são os muitos moduladores do agonista opioide mu que atuam de maneira central comercializados como agentes analgésicos para atenuar a dor (por exemplo, morfina), bem como agonistas mu que atuam peri-fericamente para regular a motilidade (por exemplo, loperamida). Atualmente, os estudos clínicos continuam a avaliar a utilidade medicinal de moduladores de delta, mu, e kappa seletivos, bem como dos compostos que possuem a modulação do subtipo combinado. Prevê-se que essas investigações podem levar a agentes com novas utilidades, ou agentes com efeitos colaterais adversos minimizados relacionados a agentes atualmente disponíveis (exemplos de efeitos colaterais relacionados à morfina incluem constipação, depressão respiratória, e potenciais adicionais). Algumas áreas Gl novas onde os moduladores de opioides misturados ou seletivos estão sendo atualmente avaliados incluem um tratamento em potencial para várias síndromes diarreicas transtornos de motilidade (íleo pós-operatório, constipação), e dor visceral (dor pós-operatória, síndrome do intestino irritável, e transtornos inflamatórios do intestino) (Pierre J. M. Riviere e Jean-Louis Junien, "Opioid receptors: Targets for new gastrointestinal drug development", Drug Development 2000, páginas. 203 a 238).
[004] Quase ao mesmo tempo, os receptores opioides foram identificados, as encefalinas foram identificadas como um conjunto de ligantes opioides endógenos (D.S. Fries, "Analgesics", em Principles of Medicinal Chemistry, 4a. Edição; W.O. Foye; T.L. Lemke, e D.A. Williams, Eds.; Williams e Wilkins: Baltimore, Md., 1995; páginas 247 a 269). Schiller verificou que o truncamento do pentapeptídeo original encefalina em dipeptídeos simplificados rendeu uma série de compostos que mantiveram a atividade opioide (Schiller, P. WO 96/06855).
Entretanto, uma desvantagem em potencial mencionada com relação a esses compostos é a possibilidade de sua instabilidade inerente (P.W. Schiller et al., Int. J. Pept. Protein Res. 1993, 41 (3), páginas 313 a 316).
Entretanto, uma desvantagem em potencial mencionada com relação a esses compostos é a possibilidade de sua instabilidade inerente (P.W. Schiller et al., Int. J. Pept. Protein Res. 1993, 41 (3), páginas 313 a 316).
[005] Mais recentemente, uma série de pseudopeptídeos opioi-des contendo núcleos heteroalifáticos ou heteroaromáticos foi apresentada, entretanto, essa série é relatada mostrando um perfil funcional diferente do que foi descrito nos trabalhos de Schiller. (L.H. Lazarus et al., Peptides 2000, 21, páginas 1663 a 1671)
[006] Adicionalmente, os trabalhos sobre as estruturas relacionadas a morfina foram relatados por Wentland, et al, onde os derivados de carboxamido morfina e seus análogos foram preparados (M.P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, páginas 1717 a 1721; M.P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, páginas 623 a 626). Wentland constatou que a substituição quanto à porção fenol das estruturas relacionadas a morfina por uma carboxamida primária gerou desde atividades iguais até atividades reduzidas em 40 vezes, dependendo do receptor opioide e da carboxamida. Revelou-se, também, que quaisquer N substituições adicionais sobre carboxamida diminuíram significativamente a atividade de ligação desejada.
[007] Os moduladores do receptor opioide, agonistas ou antagonistas são úteis no tratamento e prevenção de vários estados doentios dos mamíferos, por exemplo, dores e transtornos gastrointestinais, como síndromes diarreicas transtornos de motilidade, incluindo íleo pós-operatório e constipação, e dor visceral, incluindo dor pós-operatória, síndrome do intestino irritável, e transtornos inflamatórios do intestino.
[008] Breslin, H.J., et al., na publicação de patentes U.S. n° 2005/0203143 A1, publicada em 15 de setembro de 2005, que está expressamente incorporada na presente invenção por referência em sua totalidade apresenta moduladores do receptor opioide, composições farmacêuticas incluindo esses modulatores, e métodos de tratamento com o uso desses moduladores. A presente invenção refere-se a processos para a preparação de intermediários úteis na síntese dos moduladores do receptor opioide conforme descrito na publicação de patente U.S. n° 2005/0203143 A1.
[009] A presente invenção refere-se a um processo para a preparação de compostos de fórmula (I)
[0010] sendo que
[0011] PG1 é um grupo protetor de nitrogênio;
[0012] R0 é selecionado do grupo que consiste em hidrogênio, alquila e C1-4benzila;
[0013] R6 é selecionado do grupo consistindo em hidrogênio e C1-6 alquila
[0014] R4 é arila ou heteroarila; em que arila ou heteroarila é, op- cionalmente, substituído por um a cinco substituintes independentemente selecionados do grupo consistindo em C1-6alquila, C1-6alcóxi, aril C1-6alcóxi, aril C1-6alquila carbonilóxi, heteroaril C1-6alquila car-bonilóxi, heteroarila, hidróxi, halogênio, aminossulfonila, formilamino, aminocarbonila, C1-6alquilaminocarbonila, di(C1- 6alquila)aminocarbonila, heterociclilcarbonila, carbóxi, e ciano; sendo que C1-6alquila é, opcionalmente, substituído por amino, C1-6alquilamino, ou (C1-6alquila)2amino; e sendo que a porção arila de aril C1-6alquila carbonilóxi é, opcionalmente, substituída por um a quatro substituintes independentemente selecionados do grupo consistindo em C1-6alquila, C1-6alcóxi, halogênio, ciano, amino e hidróxi;
[0015] e enantiômeros farmaceuticamente aceitáveis, diastereô-meros farmaceuticamente aceitáveis, racematos farmaceuticamente aceitáveis e sais farmaceuticamente aceitáveis dos mesmos; compreendendo, consistindo em e/ou consistindo essencialmente em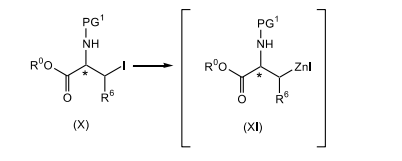
[0016] reagir um composto de fórmula (X), sendo que PG1 é um grupo de proteção de nitrogênio, com zinco; na presença de uma fonte de iodo; em um primeiro solvente orgânico ou uma mistura de solventes orgânicos, sendo que o primeiro solvente orgânico é não-reativo em relação ao iodo da fonte; para produzir o composto correspondente de fórmula (XI);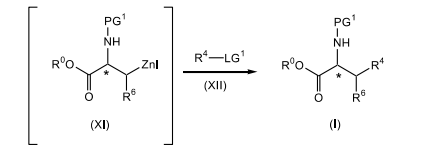
[0017] reagir o composto de fórmula (XI) com um composto de fórmula (XII), em que LG1 é um grupo de saída; na presença de um sistema de catalisador paládio e ligante fosfina; em um segundo solvente orgânico ou uma mistura de solventes orgânicos; para produzir o composto correspondente de fórmula (I).
[0018] A presente invenção refere-se, adicionalmente, a um processo para a preparação de um composto de fórmula (l-B)
[0019] Compreendendo, consistindo em e/ou consistindo essencialmente em
[0020] reagir um composto de fórmula (X-B) com zinco; na presença de uma fonte de iodo; em um primeiro solvente orgânico ou mistura, uma mistura de solventes orgânicos, em que o primeiro solvente orgânico é não-reativo ao iodo da fonte; para produzir o composto correspondente de fórmula (Xl-B); 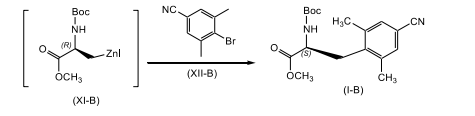
[0021] reagir o composto de fórmula (Xl-B) com um composto de fórmula (XIl-B); na presença de um sistema de catalisador paládio e ligante fosfina; em um segundo solvente orgânico ou uma mistura de solventes orgânicos; para produzir o composto correspondente de fórmula (l-B).
[0022] A presente invenção refere-se, adicionalmente, a um processo para a preparação de um composto de fórmula (ll-B)
[0023] ou um sal farmaceuticamente aceitável do mesmo; que compreende, que consiste em e/ou consistindo essencialmente em
[0024] reagir um composto de fórmula (l-B) com um agente oxi-dante; na presença de uma base inorgânica; em um terceiro solvente orgânico; para produzir o composto correspondente de fórmula (ll-B).
[0025] A presente invenção refere-se, adicionalmente, a um produto preparado de acordo com quaisquer dos processos aqui descritos. De preferência, os compostos preparados de acordo com os processos da presente invenção são substancialmente puros.
[0026] A presente invenção refere-se a um processo inovador para a preparação de compostos de fórmula (I)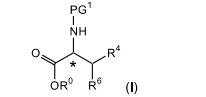
[0027] em que PG1, R0, R4 e R6 são, conforme definido no presente documento, enantiômeros farmaceuticamente aceitáveis, diaste-reômeros, racematos bem como sais dessas substâncias. Os compostos de fórmula (I) são úteis como intermediários na preparação de moduladores de receptor opioide conforme revelado na publicação de patente U.S. n° US2005/0203143 A1, publicada em 15 de setembro de 2005, a qual está aqui incorporada a titulo de referência, em sua totalidade.
[0028] Em uma modalidade, a presente invenção refere-se a um processo para a preparação de um composto de fórmula (l-A) 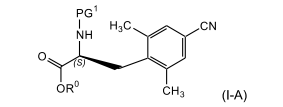
[0029] e, adicionalmente, a um processo para a preparação de um composto de fórmula (l-B)
[0030] também chamado de éster metílico de ácido (S)-2-terc-butoxicarbonilamino-3-(4-ciano-2,6-dimetil-fenil)-propiônico)
[0031] A presente invenção refere-se adicionalmente a um processo para a preparação de um composto de fórmula (ll-A) 
[0032] ou um sal farmaceuticamente aceitável do mesmo; e, adicionalmente, a um processo para a preparação de um composto de fórmula (ll-B)
[0033] também chamado de ácido (S)-2-terc-butoxicarbonilamino-3-(4-carbamoíla-2,6-dimetil-fenil)-propiônico, ou um sal farmaceutica-mente aceitável do mesmo.
[0034] Em uma modalidade da presente invenção, PG1 é selecionado do grupo que consiste em Boc e Cbz. Em outra modalidade da presente invenção, PG1 é Boc.
[0035] Em uma modalidade da presente invenção, R0 é selecionado do grupo que consiste em C1-4alquila e benzila. Em outra modalidade da presente invenção R0 é selecionado do grupo que consiste em metila, etila, isopropila, t-butila e benzila. Em outra modalidade da presente invenção, R0 é metila ou benzila. Em outra modalidade da presente invenção R0 é metila. Em outra modalidade da presente invenção, R0 é outro elemento além de hidrogênio.
[0036] Em uma modalidade da presente invenção, R6 é selecionado do grupo que consiste em hidrogênio e metila. Em outra modalidade da presente invenção, R6 é hidrogênio.
[0037] Em uma modalidade da presente invenção, R4 é selecionado do grupo que consiste em C6-10arila e heteroarila; sendo que heteroarila é selecionada do grupo que consiste em furila, tienila, pirro-lila, oxazolila, tiazolila, imidazolila, pirazolila, piridinila, pirimidinila, pira-zinila, indolila, isoindolila, indolinila, benzofurila, benzotienila, benzimi-dazolila, benzotiazolila, benzoxazolila, quinozinila, quinolinila, isoquino-linila e quinazolinila; e sendo que R4 é, opcionalmente substituído por um a três substituintes independentemente selecionados do grupo consistindo em C1-6alquila (sendo que C1-6alquila é, opcionalmente, substituído por amino, C1-6alquilamino, ou di(C1-6alquil)amino); C1-6alcóxi; fenilC1-6alcóxi; fenil C1-6alquila carbonilóxi (sendo que a porção C1-6alquila é, opcionalmente substituída por amino; e sendo que a porção fenila de fenilC1-6alquila carbonilóxi é, opcionalmente, substituída por C1-6alquila, C1-6alcóxi, halogênio, ciano, amino, ou hidróxi); heteroaril C1-6alquila carbonilóxi fundida com 5 membros; heteroarila com 5 membros não fundida; hidróxi; halogênio; aminossulfonila; for-milamino; aminocarbonila; C1-6alquilaminocarbonila (sendo que a porção C1-6alquila é, opcionalmente, substituída por amino, C1-6alquilamino, ou (C1-6alquil)2amino); di(C1-6alquil)aminocarbonila (sendo que cada porção C1-6alquila é, opcionalmente, substituída por amino, C1-6alquilamino, ou (C1-6alquila)2amino); heterociclilcarbonila (sendo que heterociclila é um anel contendo nitrogênio com 5 a 7 membros, e sendo que a dita heterociclila é fixada ao carbono da car-bonila por meio de um átomo de nitrogênio); carbóxi; e ciano.
[0038] Em outra modalidade da presente invenção, R4 é C6-10arila opcionalmente substituída por um a três substituintes independentemente selecionados do grupo consistindo em (C1-3)alquila, (C1-6)alcóxi, fenil(C1-6)alcóxi; hidróxi; halogênio; formilamino; aminocarbonila; C1-6alquilaminocarbonila; (C1-6alquil)2aminocarbonila; heterociclilcarbonila, sendo que heterociclila é um anel contendo nitrogênio de 5 a 7 membros e a dita heterociclila é fixada ao carbono da carboni-la por meio de um átomo de nitrogênio; carbóxi; e ciano; desde que não mais que um dos substituintes seja formilamino, aminocarbonila, C1-6alquilaminocarbonila, (C1 -6alquila)2aminocarbonila, heterociclilcarbonila, hidróxi, carbóxi, ou um substituinte contendo fenila.
[0039] Em outra modalidade da presente invenção, R4 é fenila substituída por um a três substituintes selecionados independentemente do grupo consistindo em (C1-3)alquila, (C1-3)alcóxi, fenila(C1-3)alcóxi, hidróxi, C1-6alquilaminocarbonila, e aminocarbonila; desde que não mais que um dos substituintes seja aminocarbonila, C1-6alquilaminocarbonila, hidróxi, ou um substituinte contendo fenila.
[0040] Em outra modalidade da presente invenção, R4 é fenila substituída na posição 4 por hidróxi, C1-3alquilaminocarbonila, ou aminocarbonila, e adicional e opcionalmente substituído por um ou dois substituintes independentemente selecionados do grupo consistindo em metila, metóxi, e benzilóxi. Em outra modalidade da presente invenção, R4 é fenila substituída na posição 4 por hidróxi, C1-3alquilaminocarbonila, ou aminocarbonila, e adicional e opcionalmente substituído por um ou dois substituintes metila. Em outra modalidade da presente invenção, R4 é fenila substituída na posição 4 por hidróxi, C1-3alquilaminocarbonila, ou aminocarbonila, e adicionalmente substituído nas posições 2 e 6 por substituintes metila.
[0041] Em uma modalidade, a presente invenção refere-se a um processo para a preparação de um composto de fórmula (I), em que o centro estéreo, conforme indicado pelo "*" está presente em um excesso enantiomérico do enantiômero (R). Em outra modalidade, a presente invenção refere-se a um processo para a preparação de um composto de fórmula (I), sendo que o centro estéreo, conforme indicado pelo "*" está presente em um excesso enantiomérico do enantiômero (S).
[0042] Conforme usado no presente documento, exceto onde especificado em contrário, o termo "alquila" sendo usado sozinho ou como parte de um grupo substituinte refere-se a cadeias carbônicas lineares ou ramificadas que têm de 1 a 8 átomos de carbono ou inúmeros átomos de carbono nos pontos finais dessa faixa. O termo "alcóxi" re-fere-se a um grupo substituinte Oalquila, sendo que alquila é conforme definido acima. Uma cadeia alquila e alcóxi podem ser substituída em um único átomo de carbono. Em grupos substituintes com múltiplos grupos alquila como di(C1-6alquil)amino- os grupos C1-6alquila do dialquilamino podem ser iguais ou diferentes.
[0043] O termo "heterociclila" refere-se a um anel cíclico não-aromático com 5 a 7 membros em que de 1 a 2 membros são nitrogênio, ou um anel cíclico não-aromático com 5 a 7 membros em que zero, um ou dois membros são nitrogênio e até dois membros são oxigênio ou enxofre; sendo que, opcionalmente, o anel contém de zero a uma ligações insaturadas e, opcionalmente, quando o anel tiver 6 ou 7 membros, ele conterá duas ligações insaturadas. O termo "heterociclila" inclui um anel heterocíclico, monocíclico com 5 a 7 membros fundido a um anel benzeno (heterociclila benzofundida), um anel de hetero-arila com 5 ou 6 membros (contendo um dentre O, S ou N e, opcionalmente, um nitrogênio adicional), anel de cicloalquila ou cicloalqueni-la com 5 a 7 membros, um anel de heterociclila com 5 a 7 membros (com a mesma definição acima, mas ausente da opção de um anel fundido adicional) ou fundido com o carbono de ligação de um anel de cicloalquila, cicloalquenila ou heterociclila para formar uma porção espiro. Para os compostos da presente invenção, os membros de anel de átomo de carbono que formam o anel de heterociclila são completamente saturados. Outros compostos da invenção podem ter um anel de heterociclila parcialmente saturado. O termo "heterociclila" inclui, também, um heterociclo monocíclico de ponte com 5 a 7 membros para formar anéis bicíclicos. Esses compostos não são considerados como sendo completamente aromáticos e não são denominados como compostos de heteroarila. Exemplos de grupos heterociclila incluem, e não se limitam a, pirrolinila (incluindo 2H-pirrol, 2-pirrolinila ou 3-pirrolinila), pirrolidinila, 2- imidazolinila, imidazolidinila, 2- pirazolinila, pirazolidinila, piperidinila, morfolinila, tiomorfolinila e piperazinila.
[0044] O termo "arila" refere-se a um anel monocíclico aromático insaturado com 6 membros de carbono ou a um anel policíclico aromático insaturado com 10 a 14 membros de carbono. Exemplos desses anéis de arila incluem fenila, naftalenila, ou antracenila. Os grupos ari-la preferenciais para a prática desta invenção são fenila e naftalenila.
[0045] O termo "heteroarila" refere-se a um anel aromático com 5 ou 6 membros, sendo que o anel consiste em átomos de carbono e tem pelo menos um membro heteroátomo. Os heteroátomos adequados incluem N, O, ou S. No caso de anéis de 5 membros, o anel de heteroarila contém um membro dentre N, O, ou S e, além disso, pode conter até três nitrogénios adicionais. No caso de anéis com 6 membros, o anel de heteroarila pode conter de um a três átomos de nitrogênio. Para o caso em que o anel de 6 membros tem três átomos de nitrogênio, no máximo dois átomos de nitrogênio são adjacentes. Opcionalmente, o anel de heteroarila é fundido a um anel benzeno (heteroarila benzofundida), um anel de heteroarila com 5 ou 6 membros (contendo um dentre O, S, ou N e, opcionalmente, um nitrogênio adicional), um anel de cicloalquila com 5 a 7 membros ou um anel heteroci-clo com 5 a 7 membros (conforme definido acima, mas ausente da opção de um anel fundido adicional). Exemplos de grupos heteroarila incluem, porém não se limitam a furila, tienila, pirrolila, oxazolila, tiazolila, imidazolila, pirazolila, Isoxazolila, isotiazolila, oxadiazolila, triazolila, ti-adiazolila, piridinila, piridazinila, pirimidinila e pirazinila; grupos de heteroarila fundida incluem indolila, isoindolila, indolinila, benzofurila, ben-zotienila, indazolila, benzimidazolila, benzotiazolila, benzoxazolila, benzisoxazolila, benzotiadiazolila, benzotriazolila, quinozinila, quinolini-la, isoquinolinila, e quinazolinila.
[0046] O termo "arilalquila" significa um grupo alquila substituído por um grupo arila (por exemplo, benzila e fenetila). De maneira similar, o termo "arilalcóxi" indica um grupo alcóxi substituído por um grupo arila (por exemplo, benzilóxi).
[0047] O termo "halogênio" refere-se a flúor, cloro, bromo e iodo. Os substituintes que são substituídos por múltiplos halogênios são substituídos de uma maneira que forneçam compostos que são estáveis.
[0048] Toda vez que o termo "alquila" ou "arila" ou qualquer de suas raízes de prefixo aparecer em um nome de um substituinte (por exemplo, arilalquila, alquilamino) isso deve ser interpretado como incluindo aquelas limitações dadas acima para "alquila" e "arila". Os números designados de átomos de carbono (por exemplo, C1-C6) se referem independentemente ao número de átomos de carbono em uma porção alquila ou à porção alquila de um substituinte maior em que alquila aparece como sua raiz de prefixo. Para alquila e substituintes alcóxi, o número designado de átomos de carbono inclui todo o membro independente incluído na faixa individualmente especificada e toda a combinação de faixas dentro da faixa especificada. Por exemplo, C1-6alquila poderia incluir metila, etila, propila, butila, pentila e hexila individualmente, bem como, subcombinações dos mesmos (por exemplo, C1-2, C1-3, C1-4, C1-5, C2-6, C3-6, C4-6, C5-6, C2-5, etc.).
[0049] Quando um grupo particular é "substituído" (por exemplo, alquila, cicloalquila, arila, heteroarila, heterocicloalquila, etc.), esse grupo pode ter um ou mais substituintes, de preferência, de um a cinco substituintes, com mais preferência, de um a três substituintes, com a máxima preferência, de uma dois substituintes, independentemente selecionados a partir da lista de substituintes.
[0050] Com referência aos substituintes, o termo "independentemente" significa que, quando for possível mais de um desses substituintes, os mesmos podem ser iguais ou diferentes entre si.
[0051] Para uso na presente invenção, a notação "*" deve significar a presença de um centro estereogênico. Onde os compostos têm, de acordo com esta invenção, pelo menos um centro quiral, os mesmos podem, consequentemente, existir como enantiômeros. Onde os compostos possuem dois ou mais centros quirais, eles podem adicionalmente existir como diastereômeros. Deve-se entender que todos estes isômeros e misturas dos mesmos estão no escopo da presente invenção. Sendo que, de preferência, o composto está presente como um enantiômero, o enantiômero está presente como um excesso enantiomérico maior que ou igual cerca de 80%, mais preferivelmente, em um excesso enantiomérico maior que ou igual cerca de 90%, ainda mais preferivelmente, e um excesso enantiomérico maior que ou igual cerca de 95%, ainda mais preferivelmente, em um excesso enantiomérico maior que ou igual cerca de 98%, com a máxima preferência, em um excesso enantiomérico maior que ou igual cerca de 99%. Similarmente, em que o composto está presente como um diastereômero, o diastereômero está presente como um excesso diastereomérico maior que ou igual cerca de 80%, mais preferivelmente, em um excesso diastereomérico maior que ou igual cerca de 90%, ainda mais preferivelmente, em um excesso diastereomérico maior que ou igual cerca de 95%, ainda mais preferivelmente, em um excesso diastereomérico maior que ou igual cerca de 98%, com a máxima preferência, em um excesso diastereomérico maior que ou igual cerca de 99%.
[0052] Mais adicionalmente, algumas das formas cristalinas para os compostos da presente invenção podem existir como polimorfos e são previstos a estarem incluídos na presente invenção. Além disso, alguns dos compostos da presente invenção podem formar solvatos com água (isto é, hidratos) ou solventes orgânicos comuns, e que tais solvatos são previstos a estarem situados dentro do escopo desta invenção.
[0053] Sob nomenclatura padrão usada ao longo desta descrição, a porção terminal da cadeia lateral é primeiramente descrita, seguida pela funcionalidade adjacente em direção ao ponto de fixação. Assim, por exemplo, um substituinte "fenil C1-C6 alquilaminocarbonil C1-C6 alquila" refere-se a um grupo com a fórmula
[0054] As abreviações usadas no relatório descritivo, particular-mente nos esquemas e nos exemplos, são as seguintes:
[0055] Conforme usado no presente documento, exceto onde especificado em contrário, o termo "composto substancialmente puro" deverá significar que o mol por cento de impurezas no composto isolado é menor que cerca de 5 mol por cento, de preferência, menor que cerca de 2 mol por cento, com mais preferência, menor que cerca de 0,5 mol por cento e, com a máxima preferência, menor que cerca de 0,1 mol por cento. Em uma modalidade da presente invenção, o composto de fórmula (I) é preparado como um composto substancialmente puro. Em uma modalidade da presente invenção, o composto de fórmula (l-A) é preparado como um composto substancialmente puro. Em outra modalidade da presente invenção, o composto de fórmula (l-B) é preparado como um composto substancialmente puro. Em uma modalidade da presente invenção, o composto de fórmula (ll-A) é preparado como um composto substancialmente puro. Em outra modalidade da presente invenção, o composto de fórmula (ll-B) é preparado como um composto substancialmente puro.
[0056] Conforme usado no presente documento, exceto onde especificado em contrário, o termo "substancialmente isento de uma(s) forma(s) de sal correspondente(s)" quando usado para descrever o composto de fórmula (I) deverá significar que o mol por cento da(s) forma(s) de sal correspondente(s) na base isolada de fórmula (I) é menor que cerca de 5 mol por cento, de preferência, menor que cerca de 2 mol por cento, com mais preferência, menor que cerca de 0,5 mol por cento e, com a máxima preferência, menor que cerca de 0,1 mol por cento. Em uma modalidade da presente invenção, o composto de fórmula (I) é preparado em uma forma que seja substancialmente isenta de forma(s) de sal correspondentes. Em uma modalidade da presente invenção, o composto de fórmula (ll-A) é preparado em uma forma que seja substancialmente isenta de forma(s) de sal correspon-dente(s). Em outra modalidade da presente invenção, o composto de fórmula (ll-B) é preparado em uma forma que seja substancialmente isenta de forma(s) de sal correspondente(s).
[0057] Conforme apresentado mais extensivamente nesta descrição escrita, termos como "reagir" e "reagido" são usados na presente invenção em referência a uma entidade química que é qualquer uma dentre: (a) a forma realmente mencionada dessa entidade química, e (b) qualquer uma das formas dessa entidade química no meio em que o composto estiver sendo considerado, ao ser mencionado.
[0058] O versado na técnica irá reconhecer que, onde não for especificado de outro modo, a(s) etapa(s) de reação será(ão) executa-da(s) sob condições adequadas, de acordo com métodos conhecidos, para fornecer o produto desejado. O versado na técnica reconhecerá adicionalmente que, no relatório descritivo e reivindicações conforme apresentados no presente documento, em que um reagente ou ti-po/classe de reagente (por exemplo, base, solvente, etc.) é relatado em mais de uma etapa de um processo, os reagentes individuais são independentemente selecionados para cada etapa de reação e podem ser iguais ou diferentes entre si. Por exemplo, onde duas etapas de um processo relatam uma base orgânica ou inorgânica como um reagente, a base orgânica ou inorgânica selecionada para a primeira etapa pode ser igual a ou diferente da base orgânica ou inorgânica da segunda etapa. Adicionalmente, o versado na técnica irá reconhecer que, onde uma etapa de reação da presente invenção puder ser executada em uma gama de solventes ou sistemas solventes, a dita etapa de reação poderá, também, ser executada em uma mistura dos solventes adequados ou sistemas solventes. O versado na técnica reconhecerá adicionalmente que, em que duas etapas de processo ou de reação consecutivas são executadas sem isolamento do produto intermediário (isto é, o produto da primeira dentre as duas etapas de processo ou de reação consecutivas), então, a primeira e a segunda etapas de processo ou de reação podem ser executadas no mesmo solvente ou sistema de solvente; ou, alternativamente, podem ser executadas em solventes diferentes ou sistemas de solvente seguindo a troca de solvente, que pode ser concluída de acordo com métodos conhecidos.
[0059] Para fornecer uma descrição mais concisa, algumas das expressões quantitativas dadas no presente documento não estão qualificadas com o termo "cerca de". Deve-se compreender que se o termo "cerca de" for usado explicitamente ou não, cada quantidade dada na presente invenção irá se referir ao dado valor real e também irá se referir à aproximação para tal valor dado que poderia, de modo aceitável, ser inferido com base na habilidade comum na técnica, incluindo aproximações devido às condições experimentais e/ou de medição para tal valor dado.
[0060] Para proporcionar uma descrição mais concisa, algumas das expressões quantitativas da presente invenção são referidas como uma faixa de aproximadamente uma quantidade X para aproximadamente uma quantidade Y. Entende-se que, onde uma faixa é referida, a faixa não se limita aos limites superiores e inferiores referidos, mas inclui, de preferência, a faixa total de aproximadamente uma quantidade X até aproximadamente uma quantidade Y, ou qualquer faixa nesse intervalo.
[0061] Exemplos de solventes, bases, temperaturas de reação e outros parâmetros e componentes de reação adequados são fornecidos nas descrições detalhadas que seguem na presente invenção. O versado na técnica irá reconhecer que a lista dos ditos exemplos não pretende, e não deve ser entendida como limitadora, de qualquer forma, da invenção estabelecida nas reivindicações que seguem depois disso.
[0062] Para uso na presente invenção, exceto onde especificado em contrário, o termo "grupo de saída" significará um átomo ou grupo carregado ou descarregado que se afasta durante uma reação de substituição ou deslocamento. Os exemplos adequados incluem, mas não se limitam a, Cl, Br, I, mesilato, tosilato e similares.
[0063] Durante quaisquer desses processos para a preparação dos compostos da presente invenção, pode ser necessário e/ou desejável proteger os grupos sensíveis ou reativos em quaisquer das moléculas relacionadas. Isso pode ser obtido por meio de grupos protetores convencionais, como aqueles descritos em Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; e T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991. Os grupos protetores podem ser removidos em um estágio subsequente conveniente com o uso de métodos conhecidos na técnica.
[0064] Conforme usado no presente documento, exceto onde especificado em contrário, o termo "grupo de proteção de nitrogênio" se refere a um grupo que pode ser fixado a um átomo de nitrogênio para proteger o átomo de nitrogênio de participar em uma reação e que pode ser prontamente removido após a reação. Os grupos de proteção de nitrogênio adequados incluem carbamatos - grupos com a seguinte fórmula -C(O)O-R, em que R é, por exemplo, metila, etila, t-butila, benzila, feniletila, CH2=CH-CH2-, e similares; amidas - grupos com a seguinte fórmula -C(O)-R’, em que R é, por exemplo, metila, fenila, tri-fluorometila, e similares; derivados de N-sulfonil - grupos com a seguinte fórmula -SO2-R”, em que R” é, por exemplo, tolila, fenila, trifluo-rometila, 2,2,5,7,8-pentametilcroman-6-ila-, 2,3,6-trimetil-4-metoxibenzeno e similares. Outros grupos protetores de nitrogênio adequados podem ser encontrados em textos como T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991.
[0065] O versado na técnica reconhecerá que onde uma etapa de reação da presente invenção pode ser executada em uma variedade de solventes ou sistemas solvente, a dita etapa de reação também pode ser executada em uma mistura dos solventes adequados ou sistemas de solvente.
[0066] Onde os processos para a preparação dos compostos, de acordo com a invenção, dão origem a uma mistura de estereoisôme-ros, esses isômeros poderão ser separados por meio de técnicas convencionais, como cromatografia preparatória. Os compostos podem ser preparados em forma racêmica, ou enantiômeros individuais podem ser preparados por síntese enantioespecífica ou por resolução. Os compostos podem, por exemplo, ser separados em seus enantiômeros de componente por técnicas-padrão, como, a formação de pares diastereoméricos através da formação de sal com um ácido opti-camente ativo, como, ácido (-)-di-p-toluoil-D-tartárico e/ou ácido (+)-di-p-toluoil-L-tartárico seguido de cristalização fracionai e regeneração da base livre. Os compostos podem também ser separados através da formação de ésteres ou amidas diastereoméricas, seguida de separação cromatográfica e remoção do auxiliar quiral, Alternativamente, os compostos podem ser separados com o uso de uma coluna HPLC quiral.
[0067] Adicionalmente, a HPLC quiral contra um padrão pode ser usada para determinar o percentual de excesso enantiomérico (%ee). O excesso enantiomérico pode ser calculado da seguinte forma
[ (Rmoles-Smoles)/(Rmoles+Smoles) ] X 100%
[ (Rmoles-Smoles)/(Rmoles+Smoles) ] X 100%
[0068] onde Rmoles e Smoles são as frações molares de R e S na mistura de tal modo que Rmoles+Smoles = 1. O excesso enantiomérico pode, alternativamente, ser calculado a partir de rotações específicas do enantiômero desejado e da mistura preparada da seguinte forma:
ee = ([a-obs] / [a-max]) X 100.
ee = ([a-obs] / [a-max]) X 100.
[0069] Para o uso em medicina, os sais dos compostos desta invenção se referem a "sais farmaceuticamente aceitáveis" não-tóxicos. Outros sais podem, porém, ser úteis na preparação de compostos de acordo com esta invenção ou de seus sais farmaceuticamente aceitáveis. Os sais farmaceuticamente adequados dos compostos incluem sais de adição de ácido que podem, por exemplo, ser formados pela mistura de uma solução do composto com uma solução de um ácido farmaceuticamente aceitável, como, ácido clorídrico, ácido sulfúrico, ácido fumárico, ácido maleico, ácido succínico, ácido acético, ácido benzoico, ácido cítrico, ácido tartárico, ácido carbônico e ácido fosfórico. Adicionalmente, em que os compostos da invenção transportam uma porção ácida, sais farmaceuticamente aceitáveis adequados da mesma podem incluir sais de metais alcalinos, por exemplo, sais de sódio e potássio; sais de metais alcalino-terrosos, por exemplo, sais de cálcio e magnésio; e sais formados com ligantes orgânicos adequados, por exemplo, sais de amónio quaternário. Dessa forma, sais farmaceuticamente aceitáveis representativos incluem acetato, benze-nossulfonato, benzoato, bicarbonato, bissulfato, bitartrato, borato, brometo, edetato de cálcio, camsilato, carbonato, cloreto, clavulanato, citrato, dicloridrato, edetato, edisilato, estolato, esilato, fumarato, glu-ceptato, gluconato, glutamato, glicolilarsanilato, hexilresorcinato, hi-drabamina, bromidrato, cloridrato, hidroxinaftoato, iodeto, isotionato, lactato, lactobionato, laurato, malato, maleato, mandelato, mesilato, metilbrometo, metilnitrato, metilsulfato, mucato, napsilato, nitrato, sal de amónio N-metilglucamina, oleato, pamoato (embonato), palmitato, pantotenato, fosfato/difosfato, poligalacturonato, silicilato, estearato, sulfato, subacetato, succinato, tanato, tartarato, teoclato, tosilato, trie-tiodeto e valerato.
[0070] Os ácidos representativos que podem ser usados nas preparação de sais farmaceuticamente aceitáveis incluem: ácidos incluindo ácido acético, ácido 2,2-dicloroacético, aminoácidos adiados, ácido adípico, ácido algínico, ácido ascórbico, ácido L-aspártico, ácido ben-zenossulfônico, ácido benzoico, ácido 4-acetamidobenzoico, ácido (+)-canfórico, ácido canforsulfônico, ácido (+)-(1S)-canfor-10-sulfônico, ácido cáprico, ácido caproico, ácido caprílico, ácido cinâmico, ácido cítrico, ácido ciclâmico, ácido dodecilssulfúrico, ácido etano-1,2-dissulfônico, ácido etanossulfônico, ácido 2-hidróxi-etanossulfônico, ácido fórmico, ácido fumárico, ácido galactárico, ácido gentísico, ácido glucoheptônico, ácido D-glucônico, ácido D-glucorônico, ácido L-glutâmico, ácido a-oxo-glutárico, ácido glicólico, ácido hipúrico, ácido bromídrico, ácido clorídrico, ácido (+)-L-láctico, ácido (±)-DL-láctico, ácido lactobiônico, ácido maleico, ácido (-)-L-málico, ácido malônico, ácido (±)-DL-mandélico, ácido metanossulfônico, ácido naftaleno-2-sulfônico, ácido naftaleno-1,5-dissulfônico, ácido 1-hidróxi-2-naftoico, ácido nicotínico, ácido nítrico, ácido oleico, ácido orótico, ácido oxálico, ácido palmítico, ácido pamoico, ácido fosfórico, ácido L-piroglutâmico, ácido salicílico, ácido 4-amino-salicílico, ácido sebáico, ácido esteárico, ácido succínico, ácido sulfúrico, ácido tânico, ácido (+)-L-tartárico, ácido tiociânico, ácido p-toluenossulfônico e ácido undecilênico.
[0071] As bases representativas que podem ser usadas na preparação de sais farmaceuticamente aceitáveis incluem: bases incluindo, amónia, L-arginina, benetamina, benzatina, hidróxido de cálcio, colina, deanol, dietanol amina, dietilamina, 2-(dietilamino)-etanol, etanolami-na, etilenodiamina, N-metil-glucamina, hidrabamina, 1H-imidazol, L-lisina, hidróxido de magnésio, 4-(2-hidroxietil)-morfolina, piperazina, hidróxido de potássio, 1-(2-hidroxietil)-pirrolidina, amina secundária, hidróxido de sódio, trietanol amina, trometamina e hidróxido de zinco.
[0072] A presente invenção é direcionada a um processo para a preparação de compostos de fórmula (I) conforme descrito em detalhe no Esquema 1 abaixo.
[0073] Em conformidade, um composto de fórmula (X) adequadamente substituído, um composto conhecido ou composto preparado por métodos conhecidos, em que PG1 é um grupo de proteção de nitrogênio adequadamente selecionado como Boc, Cbz e similares, de preferência, Boc; é reagido com zinco, de preferência, pó de zinco; em que o zinco está presente, de preferência, em uma quantidade na faixa de cerca de 0,5 a cerca de 3,0 equivalentes molares, com mais preferência, presente em uma quantidade na faixa de cerca de 0,5 a cerca de 1,5 equivalentes molares, com mais preferência, cerca de 1,1 equivalentes molares; na presença de uma fonte de iodo, de preferência iodo; em que a fonte de iodo está presente, de preferência, em uma quantidade na faixa de de cerca de 0,1 a cerca de 1,0 equivalentes molares, com mais preferência, em uma quantidade na faixa de cerca de 0,1 a cerca de 0,5 equivalentes molares, com mais preferência cerca de 0,3 equivalentes molares, com mais preferência em uma quantidade catalítica suficiente para ativar o zinco; em um primeiro solvente orgânico ou mistura desse material, em que o primeiro solvente orgânico é não-reativo em relação ao iodo de fonte, como, DMAc, uma mistura de DMAc e 2-metil-THF, THF, tolueno, DMF e similares, com mais preferência DMAc; de preferência em uma temperatura na faixa de cerca de -20°C a 10°C, com mais preferência, em uma temperatura menor que cerca de 10°C, com mais preferência a cerca de -8°C; para render o composto de fórmula (XI) correspondente. De preferência, o composto de fórmula (XI) não é isolado. De preferência, o zinco e a fonte de iodo são misturados antes da adição ao composto de fórmula (X), a fim de ativar o zinco.
[0074] O composto de fórmula (XI) é reagido com um composto de fórmula adequadamente substituído (XII), em que LG1 é um grupo de saída adequadamente selecionado tal como, Cl, Br, I e similares, de preferência, Br; em que o composto de fórmula (XII) está presente, de preferência, em uma quantidade na faixa de cerca de 0,1 a cerca de 3.0 equivalentes molares, com mais preferência, em uma quantidade na faixa de de cerca de 0,25 a cerca de 1,0 equivalentes molares, com mais preferência, em uma quantidade na faixa de cerca de 0,5 a sobre 1.1 equivalentes molares; na presença de um sistema de catalisador de paládio e ligante fosfina tal como Pd2(dba)3 em combinação com P(o-tol)3, cloreto de paládio em combinação com PPh3, Pd(PPh3)2CI2, Pd(PPh3)4, e similares, com mais preferência Pd2(dba)3 em combinação com P(o-tol)3, em que o sistema de catalisador de paládio e ligante fosfina está presente, de preferência, em uma quantidade catalítica; em um segundo solvente orgânico ou mistura desse material como, DMAc, uma mistura de DMAc e 2-metil-THF, THF, DMF, tolueno e similares, com mais preferência DMAc; de preferência no mesmo solvente conforme usado na etapa anterior; de preferência em uma temperatura na faixa de cerca de 50°C a cerca de 100°C, com mais preferência, a cerca de 80°C; para render o composto de fórmula (I) correspondente. De preferência, o composto de fórmula (XI) é adicionado a uma mistura do composto de fórmula (XII), ao catalisador de paládio e ao agente de fosfina.
[0075] A presente invenção é direcionada adicionalmente a um processo para a preparação de um composto de fórmula (l-A) conforme descrito em maiores detalhes no Esquema 2, abaixo. 
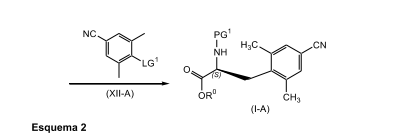
[0076] Em conformidade, um composto de fórmula (X-A) adequa-damente substituído, um composto conhecido ou composto preparado por métodos conhecidos, em que PG1 é um grupo de proteção de nitrogênio adequadamente selecionado tal como Boc, Cbz, e similares, de preferência, Boc; é reagido com zinco, de preferência pó de zinco; em que o zinco está presente, de preferência, em uma quantidade na faixa de de cerca de 0,5 a cerca de 3,0 equivalentes molares, com mais preferência, presente em uma quantidade na faixa de de cerca de 0,5 a cerca de 1,5 equivalentes molares, com mais preferência cerca de 1,1 equivalentes molares; na presença de uma fonte de iodo, de preferência iodo; em que a fonte de iodo está presente, de preferência, em uma quantidade na faixa de de cerca de 0,1 a cerca de 1,0 equivalentes molares, com mais preferência em uma quantidade na faixa de de cerca de 0,1 a cerca de 0,5 equivalentes molares, com mais preferência cerca de 0,3 equivalentes molares, com mais preferência em uma quantidade catalítica suficiente para ativar o zinco; em um primeiro solvente orgânico ou misturas do mesmo, em que o primeiro solvente orgânico é não-reativo em relação ao iodo da fonte, como, DMAc, uma mistura de DMAc e 2-metil-THF, THF, tolueno, DMF e similares, com mais preferência DMAc; de preferência em uma temperatura na faixa de cerca de -20°C a cerca de 10°C, com mais preferência a uma temperatura menor do que cerca de 10°C, com mais preferência a cerca de -8°C; para render o composto de fórmula (Xl-A) correspondente. De preferência, o composto de fórmula (Xl-A) não é isolado. De preferência, o zinco e fonte de iodo são misturados antes da adição ao composto de fórmula (V-A), a fim de ativar o zinco.
[0077] O composto de fórmula (Xl-A) é reagido com um composto de fórmula (Xll-A) adequadamente substituído, em que LG1 é um grupo de saída adequadamente selecionado, como, Cl, Br, I e similares, de preferência Br; em que o composto de fórmula (Xll-A) está presente, de preferência, em uma quantidade na faixa de cerca de 0,1 a cerca de 3,0 equivalentes molares, com mais preferência, em uma quantidade na faixa de cerca de 0,25 a cerca de 1,0 equivalentes molares, com mais preferência, em uma quantidade na faixa de cerca de 0,5 a cerca de 1,1 equivalentes molares; na presença de um sistema de catalisador de paládio e ligante de fosfina como Pd2(dba)3 em combinação com P(o-tol)3, cloreto de paládio em combinação com PPh3, Pd(PPh3)2CI2, Pd(PPh3)4, e similares, com mais preferência Pd2(dba)3 em combinação com P(o-tol)3, em que o sistema de catalisador de paládio e ligante fosfina está presente, de preferência, em uma quantidade catalítica; em um segundo solvente orgânico ou mistura do mesmo, como, DMAc, uma mistura de DMAc e 2-metil-THF, THF, DMF, tolueno, e similares, com mais preferência DMAc; de preferência no mesmo solvente que aquele usado na etapa anterior; de preferência em uma temperatura na faixa de cerca de 50°C a cerca de 100°C, com mais preferência a cerca de 80°C; para render o composto de fórmula (l-A) correspondente. De preferência, o composto de fórmula (Xl-A) é adicionado a uma mistura do composto de fórmula (Xll-A), ao catalisador de paládio e ao agente fosfina.
[0078] A presente invenção é direcionada adicionalmente a um processo para a preparação de um composto de fórmula (l-B), conforme descrito em maiores detalhes no Esquema 3, abaixo. 
[0079] Em conformidade, um composto de fórmula (X-B) adequadamente substituído, um composto conhecido ou composto preparado por métodos conhecidos, é reagido com zinco, de preferência, pó de zinco; em que o zinco está presente, de preferência, em uma quantidade na faixa de cerca de 0,5 a cerca de 3,0 equivalentes molares, com mais preferência, presente em uma quantidade na faixa de cerca de 0,5 a cerca de 1,5 equivalentes molares, com mais preferência cerca de 1,1 equivalentes molares; na presença de uma fonte de iodo, de preferência iodo; em que a fonte de iodo está presente, de preferência, em uma quantidade na faixa de cerca de 0,1 a cerca de 1,0 equivalentes molares, com mais preferência em uma quantidade na faixa de cerca de 0,1 a cerca de 0,5 equivalentes molares, com mais preferência cerca de 0,3 equivalentes molares, com mais preferência em uma quantidade catalítica suficiente para ativar o zinco; em um primeiro solvente orgânico ou mistura do mesmo, em que o primeiro solvente orgânico é não-reativo em relação ao iodo da fonte, como, DMAc, uma mistura de DMAc e 2-metil-THF, THF, tolueno, DMF e similares, com mais preferência DMAc; de preferência em uma temperatura na faixa de cerca de -20°C a cerca de 10°C, com mais preferência a uma temperatura menor que cerca de 10°C, com mais preferência a cerca de -8°C; para render o composto de fórmula (Xl-B) correspondente. De preferência, o composto de fórmula (Xl-B) é não isolado. De preferência, o zinco e fonte de iodo são misturados antes da adição ao composto de fórmula (V-B), para ativar o zinco.
[0080] O composto de fórmula (Xl-B) é reagido com um composto de fórmula (Xll-B) adequadamente substituído, em que o composto de fórmula (Xll-B) está presente, de preferência, em uma quantidade na faixa de cerca de 0,1 a cerca de 3,0 equivalentes molares, com mais preferência, em uma quantidade na faixa de cerca de 0,25 a cerca de 1,0 equivalentes molares, com mais preferência em uma quantidade na faixa de cerca de 0,5 a cerca de 1,1 equivalentes molares; na presença de um sistema de catalisador de paládio e ligante fosfina como Pd2(dba)3 em combinação com P(o-tol)3, cloreto de paládio em combinação com PPh3, Pd(PPh3)2CI2, Pd(PPh3)4, e similares, com mais preferência Pd2(dba)3 em combinação com P(o-tol)3, em que o sistema de catalisador de paládio e ligante fosfina está presente, de preferência, em uma quantidade catalítica; em um segundo solvente orgânico ou mistura do mesmo, como, DMAc, uma mistura de DMAc e 2-metil-THF, THF, DMF, tolueno, e similares, com mais preferência DMAc; de preferência no mesmo solvente que aquele usado na etapa anterior; de preferência em uma temperatura na faixa de cerca de 50°C a cerca de 100°C, com mais preferência a cerca de 80°C; para render o composto de fórmula (l-B) correspondente. De preferência, o composto de fórmula (Xl-B) é adicionado a uma mistura do composto de fórmula (Xll-B), ao catalisador de paládio e ao agente fosfina.
[0081] A presente invenção é direcionada adicionalmente a um processo para a preparação de um composto de fórmula (ll-A), conforme descrito em maiores detalhes no Esquema 4, abaixo.
[0082] Em conformidade, um composto de fórmula (l-A) adequa-damente substituído, em que R0 é, de preferência, diferente de hidrogênio e em que PG1 é um grupo de proteção de nitrogênio adequadamente selecionado como Boc, Cbz, e similares, de preferência PG1 é Boc, é reagido com um agente oxidante adequadamente selecionado, como, peróxido de hidrogênio, LiOH, LiOOH, e similares, de preferência 30% de peróxido de hidrogênio; em que o agente oxidante está presente, de preferência, em uma quantidade excessiva; na presença de uma base inorgânica, como, carbonato de potássio, carbonato de sódio, percarbonato sódio, e similares, de preferência, carbonato de potássio; em que a base inorgânica está presente, de preferência, em uma quantidade na faixa de cerca de 1,0 a cerca de 3,0 equivalentes molares, com mais preferência, em uma quantidade de cerca de 1,6 equivalentes molares; em um terceiro solvente orgânico, como, DMSO, DMF, DMAc, NMP, e similares, de preferência DMSO; em uma temperatura na faixa a partir da temperatura ambiente a cerca de 60°C, de preferência, a cerca de 45°C; para render o composto de fórmula (ll-A) correspondente.
[0083] Em uma modalidade, a presente invenção é direcionada a um processo para a preparação de um composto de fórmula (ll-B), conforme descrito em maiores detalhes no Esquema 5, abaixo. 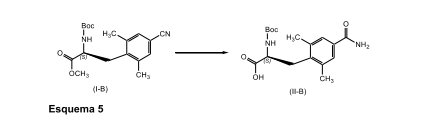
[0084] Em conformidade, um composto de fórmula (l-B) adequadamente substituído é reagido com um agente oxidante adequadamente selecionado, como, peróxido de hidrogênio, LiOH, LiOOH, e similares, de preferência cerca de 30% de peróxido de hidrogênio; em que o agente oxidante está presente, de preferência, em uma quantidade excessiva, com mais preferência em que o agente oxidante está em um quantidade excessiva de cerca de 30% de peróxido de hidrogênio; na presença de uma base inorgânica, como, carbonato de potássio, carbonatos de sódio, percarbonato sódio, e similares, de preferência, carbonato de potássio; em que a base inorgânica está presente, de preferência, em uma quantidade na faixa de cerca de 1,0 a cerca de 3,0 equivalentes molares, com mais preferência em uma quantidade de cerca de 1,6 equivalentes molares; em um terceiro solvente orgânico como DMSO, DMF, DMAc, NMP, e similares, de preferência DMSO; em uma temperatura na faixa a partir da temperatura ambiente a cerca de 60°C, de preferência, a cerca de 45°C; para render o composto de fórmula (ll-B) correspondente.
[0085] Os exemplos a seguir são demonstrados para auxílio no entendimento da invenção, e não têm intenção e não devem ser interpretados para limitar em nenhum aspecto a invenção exposta nas reivindicações que seguem.
[0086] Nos exemplos a seguir, alguns produtos de síntese são listados como tendo sido isolados como um resíduo. Será compreendido pelo de habilidade comum na técnica que o termo "resíduo" não limita o estado físico em que o produto foi isolado e pode incluir, por exempio, um sólido, um óleo, uma espuma, uma goma, um xarope, e similares.
Exemplo 1
Preparação de metil éster de ácido 2-terc-butoxicarbonilamino-3-(4-carbamoil-2.6-dimetil-fenil)-propiônico
Exemplo 1
Preparação de metil éster de ácido 2-terc-butoxicarbonilamino-3-(4-carbamoil-2.6-dimetil-fenil)-propiônico
[0087] DM Ac seco (300 ml), 2-Me-THF (150 ml), l2 (25,4 g, 0,10 mol) e pó de zinco (294,3 g, 4,5 mol), foram adicionados sob nitrogênio em um frasco de fundo redondo 3 L de quatro gargalos equipado com um funil de adição, agitador mecânico, camisa de aquecimento, condensador e termopar. A pasta aquosa resultante foi agitada até que a cor vermelha de l2 desaparecesse (cerca de 2 minutos). Durante a adição, um aumento da temperatura foi observado (de 23°C para 43°C). A mistura resultante foi resfriada com o uso de um banho de gelo/NaCI a cerca de -5°C a -2°C. Durante essa temperatura, uma solução de Boc-β-iodo-alanina-OCH3 (também chamada de metil éster de ácido 2-terc-butoxicarbonilamino-3-iodo-propiônico, 658,3 g, 2,0 mol) em uma mistura de DMAc (250 ml) e 2-Me-THF (500 ml) foi adicionada lentamente ao longo de um período de 2 horas. A temperatura da mistura resultante foi mantida abaixo de 10°C e a mistura envelheceu por um período de cerca de 1 a 2 horas no banho de gelo, então, aquecida para cerca de 15°C a fim de render uma mistura. A mistura resfriada resultante foi usada na etapa seguinte sem manipulação adicional.
[0088] 4-iodo-3,5-dimetil-benzamida (275 g, 1,0 mol), 2-Me-THF (500 ml) e DMA (200 ml), foram adicionados a um frasco de fundo arredondado de 5 L com quatro gargalos equipado com agitador mecânico, camisa de aquecimento, condensador, termopar e entrada de nitrogênio. P(o-tol)3 (24,5 g, 0,08 mol) e Pd2(dba)3 (36,6 g, 0,04 mol) foram adicionados à suspensão e a pasta aquosa resultante foi aquecida para 45 a 50°C. Durante essa temperatura, a mistura preparada na ETAPA A foi adicionada através de cânula ao longo de um período de cerca de 1,5 a 2 horas. A mistura resultante foi resfriada à temperatura ambiente. Foi adicionada sílica (275 g) e a pasta aquosa foi agitada por cerca de 30 minutos. O bloco de sílica foi lavado com 2-Me-THF (3X 500 ml) e EtOAc (3X1 L). A solução resultante foi bruscamente arrefecida com 2 L de HCI aquoso a 1,0 N e as camadas foram separadas. A camada ácida foi novamente extraída com EtOAc (2X1 L). A camada orgânica foi concentrada a cerca de 5,0 L em um rotoevapo-rador e enxaguada com água (3X1 L), e com 50% de salmoura (2,0 L). Os solventes foram removidos pelo rotoevaporador a fim de render um sólido esbranquiçado.
[0089] O composto do título foi cristalizado a partir de EtOAc (2 L) e heptano (2 L) conforme a seguir. Após 16 horas, a mistura resultante foi resfriada em um banho de gelo por 2 horas e mais heptano (500 ml) foi adicionado para completar a precipitação. O sólido foi filtrado e seco em forno a vácuo a 55°C por 48 horas a fim de render o composto do título como um sólido branco.
Exemplo 2
Preparação de metil éster de ácido (S)-2-terc-butoxicarbonilamino-3-(4-ciano-2,6-dimetil-fenil)-popiônico
Exemplo 2
Preparação de metil éster de ácido (S)-2-terc-butoxicarbonilamino-3-(4-ciano-2,6-dimetil-fenil)-popiônico
[0090] Um frasco com fundo arredondado de 50 ml com três gargalos equipado com um funil de adição, agitador magnético, camisa de aquecimento e termopar foi carregado sob nitrogênio, DMAc seco (2 ml), I2 (38,1 mg, 0,15 mmol) e pó de zinco ativado (lavado com 10% de HCI, enxaguado com H2O e acetona) (393 mg, 6 mmol). A mistura resultante foi agitada a 23°C até que a cor vermelha de I2 desaparecesse (2 minutos). Uma solução de metil éster de Boc-β-iodo-L-alanina (1 g, 3 mmol) em DMAc (2 ml) foi adicionada lentamente, (alteração da temperatura de 21°C para 29°C) e a mistura resultante foi agitada a 80°C por 0,5 a 1 hora, então, foi resfriada a 35°C. À mistura resultante foram adicionados, sucessivamente, 4-bromo-3,5-dimetil-benzonitrila (315 mg, 1,5 mmol) em DMAc (6 ml), P(o-tol)3 (36,5 mg, 0,12 mmol) e Pd2(dba)3 (55 mg, 0,06 mmol). A mistura resultante foi aquecida a 70°C, com agitação por 1 hora, então, resfriada à temperatura ambiente. A mistura resultante foi diluída com EtOAc (15 ml) e filtrada com STAND SUPER-CEL 815520. A solução EtOAc foi bruscamente arrefecida com HCI a 1 N (40 ml) e extraída com acetato de etila (20 ml). As fases orgânicas combinadas foram lavadas com H2O (2 x 50 ml) e, então, com 50% de salmoura, foram submetidas à secagem com Na2SO4, filtradas e evaporadas até a secura a vácuo para render um sólido castanho. O composto do título foi cristalizado a partir de EtOAc (5 ml) e de heptano (40 ml) para render um sólido branco.
Exemplo 3
Preparação de metil éster de ácido (S)-2-terc-butoxicarbonilamino-3-(4-ciano-2,6-dimetil-fenil)-propiônico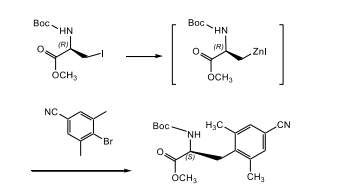
Exemplo 3
Preparação de metil éster de ácido (S)-2-terc-butoxicarbonilamino-3-(4-ciano-2,6-dimetil-fenil)-propiônico
[0091] Um frasco de fundo arredondado com 4 gargalos de 2 L equipado com uma entrada de nitrogênio, um agitador mecânico, um funil de adição e um termopar foi carregado com DMAc anidro (500 ml) e iodo (16,8 g, 0,06 mol) para render uma solução vermelha. À solução agitada foi, então, adicionado pó de zinco (143,9 g, 2,2 mol). A cor vermelha da mistura resultante foi observada até desaparecer em cerca de 2 minutos e uma exoterma (22°C a cerca de 36°C) foi observada. A mistura resultante foi resfriada a -8°C e, então, uma solução de metil éster N-(ferc-butoxicarbonil)-3-iodo-L-alanina (658 g, 2,0 mol) em DMAc anidro (500 ml) foi adicionada lentamente ao longo de cerca de 2 horas, mantendo a temperatura da mistura abaixo de cerca de 10°C, sem agitação. A mistura resfriada resultante foi usada na etapa seguinte sem manipulação adicional.
[0092] Um frasco de fundo arredondado de 5 L com três gargalos equipado com uma entrada de nitrogênio, um agitador mecânico, um funil de adição e um termopar foi carregado com 4-bromo-3,5-dimetil-benzonitrila (210 g, 1,0 mol) e DMAc (750 ml). A suspensão resultante foi agitada e aquecida a 35°C para dissolver os sólidos. À mistura resultante foi, então, adicionado P(o-tol)3 (6,0 g, 0,02 mol), Pd2(dba)3 (9,2 g, 0,01 mol) e a mistura resultante aquecida a cerca de 75 a 80°C. A mistura resfriada preparada na ETAPA A acima foi adicionada através de cânula à mistura de reação em uma taxa que manteve a temperatura a cerca de 75 a 80°C (cerca de 2 horas). A suspensão resultante foi resfriada à temperatura ambiente, então, envelhecida de um dia para o outro com agitação moderada. A suspensão resultante foi, então, aquecida a cerca de 35 a 40°C e filtrada com sílica (540 g). O leito de sílica foi lavado com DMAc (400 ml x 2), as soluções de DMAc combinadas foram resfriadas a cerca de 0 a 5°C e, então, adicionadas lentamente a uma mistura de gelo e água desionizada. A mistura resultante foi mantida fria por 2 horas, ao longo desse tempo, foi observada a precipitação de um sólido branco. A mistura resultante foi, então, aquecida à temperatura ambiente e envelhecida de um dia para o outro. O precipitado sólido foi resfriado por filtragem a vácuo com o uso de um funil de Buchner. O bolo do filtro foi enxaguado com água desionizada (1 L x 3), submetido à secagem a ar de um dia para o outro, então, seca em um forno a vácuo de um dia para o outro. MeOH (1 L) foi adicionado ao sólido e a pasta aquosa resultante foi resfriada a cerca de 0 a 5°C, então, agitada a essa temperatura por 1 hora, com agitação. O sólido foi coletado por filtragem, lavado com metanol frio (400 ml) e seco em um forno a vácuo a 45°C para render o composto do título como um sólido esbranquiçado.
Exemplo 4
Preparação de 4-Bromo-3,5-dimetil-benzonitrila
Exemplo 4
Preparação de 4-Bromo-3,5-dimetil-benzonitrila
[0093] 4-Bromo-3,5-dimetilfenol (50,0 g, 0,25 mol de Aldrich a 99%) e piridina (250 ml), foram adicionados a um frasco de fundo arredondado de 2,0 L com 3 gargalos equipado com funil de adição, agitador mecânico e termopar. A solução resultante foi resfriada a 0°C e anidrido trifluorometanossulfônico (anidrido tríflico) (80,5 g, 0,285 mol de Aldrich a 99%) foi adicionado por gotejamento ao longo de um período de 2 horas. Após a adição, a mistura resultante foi mantida a 0°C por 15 minutos, então, deixada de um dia para o outro à temperatura ambiente. Após 16 horas, a mistura resultante foi resfriada em um banho de gelo e bruscamente arrefecida com H2O (1,7 I), e EtOAc (1,7 I). As camadas da mistura bifásica resultante foram separadas e a camada orgânica foi tratada com HCI 2N (2 x 1,0 L), então, enxaguada uma vez com água (1,0 L) e uma vez com 50% de salmoura. A camada orgânica foi submetida à secagem com Na2SO4, então, concentrada até a secura pelo rotavapor a fim de render 4-bromo-3,5-dimetil-fenil éster de ácido trifluorometanossulfônico como óleo espesso.
[0094] 4-bromo-3,5-dimetil-fenil éster de ácido trifluorometanossulfônico (79,8 g, 0,24 mol) e AcCN (500 ml) foram adicionados a um frasco de fundo arredondado 2,0 L com 3 gargalos equipado com agitador mecânico, adaptador para entrada de nitrogênio, camisa de aquecimento e termopar. À solução resultante foram adicionados Pd(PPh3)4 (27,7 g, 0,024 mol), Cul (9,2 g, 0,048 mol) e Zn(CN)2 (79,8 g, 0,24 mol). A mistura resultante foi agitada por 45 minutos a 50°C, DMAc (150 ml) foi adicionado e a temperatura foi elevada para 80 a 88°C e a mistura envelheceu nessa temperatura de um dia para o outro. A mistura resultante foi resfriada à temperatura ambiente, diluída com EtOAc (200 ml) e filtrada com STAND SUPER-CEL 815520. O bolo SUPER-CEL foi enxaguado com EtOAc (200 ml x 6). As soluções de EtOAc foram combinadas e resfriadas bruscamente com uma mistura de 4:1:4 de NH4CI saturado: NH4OH concentrado: H2O (240 ml: 60 ml: 240 ml). As camadas foram separadas e a camada orgânica foi enxaguada uma vez com água (500 ml) e uma vez com salmoura (500 ml), então, concentradas até a secura a vácuo para render um óleo vermelho espesso. O composto do título foi cristalizado de EtOAc (135 ml) e de heptano (500 ml) para render um cristal branco amarelado.
Exemplo 5
Preparação de ácido (S)-2-terc-butoxicarbonilamino-3-(4-carbamoil-2,6-dimetil-fenil)-propiônico
Exemplo 5
Preparação de ácido (S)-2-terc-butoxicarbonilamino-3-(4-carbamoil-2,6-dimetil-fenil)-propiônico
[0095] Um frasco de fundo arredondado de 50 ml com três gargalos equipado com agitador magnético e termopar foi carregado sob nitrogênio com metil éster de ácido (S)-2-terc-butoxicarbonilamino-3-(4-ciano-2,6-dimetil-fenil)-propiônico (166,2 mg, 0,5 mmol), DMSO (5,0 ml), e K2CO3 (75 mg, 0,5 mmol) e a mistura resultante foi resfriada em um banho de gelo. À mistura resultante foi, então, adicionado 30% de H2O2 (110 ml), por gotejamento via uma seringa. Permitiu-se, então, que a mistura resultante aquecesse à temperatura ambiente e foi observada a dissolução dos sólidos para render uma solução limpa. Após agitação por 2 horas a 45 a 50°C, adicionou-se água (10 ml), o resfriamento foi aplicado e um produto precipitado isolado por filtragem. O sólido branco isolado foi lavado com água (2 x 25 ml), então, seco por 24 horas em bomba de alto vácuo para render o composto do título como um sólido branco.
Exemplo 6
Preparação de ácido 2-terc-butoxicarbonilamino-3-(4-carbamoil-2,6-dimetil-fenil)-propiônico
Exemplo 6
Preparação de ácido 2-terc-butoxicarbonilamino-3-(4-carbamoil-2,6-dimetil-fenil)-propiônico
[0096] Metil éster de ácido 2-terc-butoxicarbonilamino-3-(4-carbamoil-2,6-dimetil-fenil)-propiônico (250 g, 0,713 mol), DMSO (750 ml) e 30% de H202 (250 ml) foram adicionados a um frasco de fundo arredondado de 5 L com três gargalos equipado com funil de adição, agitador mecânico, camisa de aquecimento, condensador de refluxo, termopar e entrada de nitrogênio. O carbonato de potássio (158 g, 1,14 mol, 1,6 eq) foi dissolvido em água (750 ml) e adicionado por go-tejamento ao longo de 30 minutos. Durante a adição, um aumento da temperatura foi observado (de 23°C a 34°C). A mistura resultante foi aquecida a cerca de 42 a 45°C e o progresso da reação monitorado por HPLC. Após 3 horas, foi adicionado à mistura aquecida carvão ativado (ECOSORB-941) (37,5 g, 15% por peso). A pasta aquosa resultante foi submetida a condições de refluxo por 1 hora e, então, filtrada a quente através de Celite®. O bloco de Celite® foi enxaguado com H20 (1,5 L). A mistura resultante foi resfriada a cerca de 10°C e bruscamente arrefecida com 2,ON HCI (pH 2, 1,22 L), para render uma mistura que compreende um sólido branco precipitado. A mistura foi envelhecida sob agitação durante um período de cerca de 4 horas em um banho de gelo e, então, filtrada e seca por 48 horas em um forno a vácuo para render o composto do título como um sólido cristalino branco.
[0097] Embora o relatório descritivo anteriormente mencionado ensine os princípios da presente invenção, com os exemplos fornecidos com o propósito de ilustração, ficará compreendido que a prática da invenção abrange todas as variações, adaptações e/ou modificações usuais de acordo com o escopo das reivindicações a seguir e seus equivalentes.
Claims (24)
- Processo para a preparação de um composto da fórmula (I)em que

PG1 é um grupo protetor de nitrogênio;
R0 é selecionado do grupo consistindo em hidrogênio, C1-4alquila e benzila;
R6 é selecionado do grupo consistindo em hidrogênio e C1-6alquila;
R4 é arila; sendo que a arila é, opcionalmente substituída por um a cinco substituintes independentemente selecionados do grupo consistindo em C1-6alquila, C1-6alcóxi, aril C1-6alcóxi, aril C1-6alquilcarbonilóxi, heteroaril C1-6alquilcarbonilóxi, heteroarila, hidróxi, halogênio, aminossulfonila, formilamino, aminocarbonila, C1-6alquil aminocarbonila, di(C1-6alquil)aminocarbonila, heterociclilcarbonila, car-bóxi, e ciano; sendo que o C1-6alquila é, opcionalmente substituído por amino, C1-6alquilamino, ou (C1-6alquil)2amino; e sendo que a porção arila de aril C1-6alquilcarbonilóxi é, opcionalmente substituída por um a quatro substituintes independentemente selecionados do grupo consistindo em C1-6alquil, C1-6alcóxi, halogênio, ciano, amino e hidróxi;
e enantiômeros farmaceuticamente aceitáveis, diastereô-meros farmaceuticamente aceitáveis, racematos farmaceuticamente aceitáveis e um sal farmaceuticamente aceitável do mesmo;
caracterizado pelo fato de que compreendereagir um composto de fórmula (X), sendo que PG1 é um grupo protetor de nitrogênio, com zinco; na presença de uma fonte de iodo; em um primeiro solvente orgânico, ou uma mistura de solventes orgânicos, sendo que o primeiro solvente orgânico é não-reativo à fonte de iodo; de modo a produzir o composto correspondente de fórmula (XI); reagir o composto de fórmula (XI) com um composto de fórmula (XII), sendo que LG1 é um grupo bromo; na presença de um catalisador de paládio e um sistema de ligação de fosfina; em um segundo solvente orgânico ou uma mistura de solventes orgânicos; de modo a produzir o composto correspondente de fórmula (I).
reagir o composto de fórmula (XI) com um composto de fórmula (XII), sendo que LG1 é um grupo bromo; na presença de um catalisador de paládio e um sistema de ligação de fosfina; em um segundo solvente orgânico ou uma mistura de solventes orgânicos; de modo a produzir o composto correspondente de fórmula (I).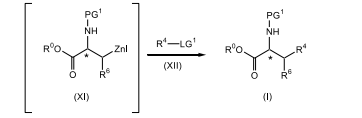
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que PG1 é terc-butóxicarbonila (Boc).
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que R0 é metila.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o zinco é pó de zinco.
- Processo, de acordo com a reivindicação 4, caracterizado pelo fato de que o pó de zinco está presente em uma quantidade na faixa de 0,5 a 1,5 equivalentes molares.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que a fonte de iodo é iodo.
- Processo, de acordo com a reivindicação 6, caracterizado pelo fato de que o iodo está presente em uma quantidade na faixa de 0,1 a 0,5 equivalentes molares.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o primeiro solvente orgânico é dimetilacetamida (DMAc).
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o composto de fórmula (X) é reagido com o zinco a uma temperatura menor que 10°C.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o zinco e a fonte de iodo são misturados antes da adição ao composto de fórmula (X).
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o composto de fórmula (XII) está presente em uma quantidade na faixa de 0,25 a 1,0 equivalentes molares.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o catalisador de paládio e o sistema de ligação de fosfina consistem em uma combinação de Pd2(dba)3 e P(o-tol)3.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o segundo solvente orgânico é dimetilacetamida (DMAc).
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o composto de fórmula (X) é reagido com o composto de fórmula (XII) a uma temperatura na faixa de 50°C a 100°C.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que o composto de fórmula (XI) é adicionado a uma mistura do composto de fórmula (XII), do catalisador de paládio e do sistema de ligação de fosfina.
- Processo, de acordo com qualquer uma das reivindicações 1 e 4 a 15, caracterizado pelo fato de ser para a preparação de um composto de fórmula (l-B)reagir um composto de fórmula (X-B) com zinco; na presença de uma fonte de iodo; em um primeiro solvente orgânico, ou uma mistura de solventes orgânicos, sendo que o primeiro solvente orgânico é não-reativo à fonte de iodo; de modo a produzir o composto correspondente de fórmula (Xl-B);
 reagir o composto de fórmula (Xl-B) com um composto de fórmula (XIl-B); na presença de um catalisador de paládio e um sistema de ligação de fosfina; em um segundo solvente orgânico ou uma mistura de solventes orgânicos; de modo a produzir o composto correspondente de fórmula (l-B).
reagir o composto de fórmula (Xl-B) com um composto de fórmula (XIl-B); na presença de um catalisador de paládio e um sistema de ligação de fosfina; em um segundo solvente orgânico ou uma mistura de solventes orgânicos; de modo a produzir o composto correspondente de fórmula (l-B).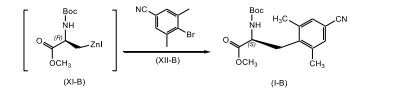
- Processo para a preparação de um composto de fórmula (ll-B)ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que compreende
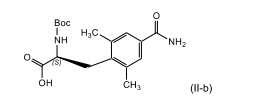 preparar um composto de fórmula (l-B), como definido na reivindicação 16, e reagir o referido composto de fórmula (l-B) com um agente oxidante; na presença de uma base inorgânica; em um terceiro solvente orgânico; de modo a produzir o composto correspondente de fórmula (ll-B).
preparar um composto de fórmula (l-B), como definido na reivindicação 16, e reagir o referido composto de fórmula (l-B) com um agente oxidante; na presença de uma base inorgânica; em um terceiro solvente orgânico; de modo a produzir o composto correspondente de fórmula (ll-B).
- Processo, de acordo com a reivindicação 17, caracterizado pelo fato de que o agente oxidante é selecionado do grupo que consiste em peróxido de hidrogênio, LiOH e LiOOH.
- Processo, de acordo com a reivindicação 18, caracterizado pelo fato de que o agente oxidante é peróxido de hidrogênio.
- Processo, de acordo com a reivindicação 19, caracterizado pelo fato de que o agente oxidante é 30% de peróxido de hidrogênio e está presente em uma quantidade em excesso.
- Processo, de acordo com a reivindicação 17, caracterizado pelo fato de que a base inorgânica é carbonato de potássio.
- Processo, de acordo com a reivindicação 17, caracterizado pelo fato de que a base inorgânica está presente em uma quantidade na faixa de 1,0 a 3,0 equivalentes molares.
- Processo, de acordo com a reivindicação 17, caracterizado pelo fato de que o terceiro solvente orgânico é dimetilsulfóxido (DMSO).
- Processo, de acordo com a reivindicação 17, caracterizado pelo fato de que o composto de fórmula (l-B) é reagido com o agente oxidante a uma temperatura na faixa de temperatura ambiente a 60°C.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10864908P | 2008-10-27 | 2008-10-27 | |
| US61/108,649 | 2008-10-27 | ||
| PCT/US2009/062191 WO2010062590A2 (en) | 2008-10-27 | 2009-10-27 | Process for the preparation of protected l-alanine derivatives |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| BRPI0920834A2 BRPI0920834A2 (pt) | 2019-04-09 |
| BRPI0920834B1 true BRPI0920834B1 (pt) | 2020-11-24 |
Family
ID=41796530
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0920834-8A BRPI0920834B1 (pt) | 2008-10-27 | 2009-10-27 | processo para preparação de derivados protegidos de l-alanina |
Country Status (25)
| Country | Link |
|---|---|
| US (3) | US8710256B2 (pt) |
| EP (1) | EP2362868B1 (pt) |
| JP (2) | JP5592893B2 (pt) |
| KR (1) | KR101690195B1 (pt) |
| CN (2) | CN102264691B (pt) |
| AR (1) | AR075291A1 (pt) |
| AU (1) | AU2009320156B2 (pt) |
| BR (1) | BRPI0920834B1 (pt) |
| CA (1) | CA2741790C (pt) |
| CL (1) | CL2011000936A1 (pt) |
| CO (1) | CO6361989A2 (pt) |
| CR (1) | CR20110286A (pt) |
| EA (1) | EA018913B1 (pt) |
| EC (1) | ECSP11011008A (pt) |
| ES (1) | ES2657676T3 (pt) |
| HN (1) | HN2011001186A (pt) |
| HU (1) | HUE038302T2 (pt) |
| IL (1) | IL212464A (pt) |
| MX (1) | MX2011004393A (pt) |
| NI (1) | NI201100078A (pt) |
| NZ (1) | NZ592415A (pt) |
| PE (2) | PE20141698A1 (pt) |
| TW (1) | TWI468375B (pt) |
| UA (1) | UA110600C2 (pt) |
| WO (1) | WO2010062590A2 (pt) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI468375B (zh) * | 2008-10-27 | 2015-01-11 | Janssen Pharmaceutica Nv | 製備經保護之l-丙胺酸衍生物之方法 |
| TWI579296B (zh) | 2011-12-28 | 2017-04-21 | Chugai Pharmaceutical Co Ltd | Cyclization of Peptide Compounds |
| BR112016017993A2 (pt) | 2014-02-03 | 2017-08-08 | Quadriga Biosciences Inc | Gama-aminoácidos beta-substituídos e análogos como agentes quimioterapêuticos |
| NZ723940A (en) | 2014-02-03 | 2017-12-22 | Quadriga Biosciences Inc | Beta-substituted beta-amino acids and analogs as chemotherapeutic agents |
| WO2017024009A1 (en) | 2015-08-03 | 2017-02-09 | Quadriga Biosciences, Inc. | Beta-substituted beta-amino acids and analogs as chemotherapeutic agents and uses thereof |
| WO2017043626A1 (ja) * | 2015-09-11 | 2017-03-16 | 株式会社カネカ | 光学活性4-カルバモイル-2,6-ジメチルフェニルアラニン誘導体の製造法 |
| CN107129444B (zh) * | 2016-02-29 | 2018-08-31 | 尚科生物医药(上海)有限公司 | (s)-2-叔丁氧羰基氨基-3-(4-氨甲酰基-2,6-二甲基苯基)丙酸制备方法 |
| CN105777584B (zh) * | 2016-03-28 | 2018-01-02 | 成都伊诺达博医药科技有限公司 | 丙氨酸衍生物的制备方法 |
| CN105693554B (zh) * | 2016-04-06 | 2017-08-08 | 成都伊诺达博医药科技有限公司 | 丙氨酸衍生物的制备方法 |
| US10479769B2 (en) | 2016-09-20 | 2019-11-19 | Sun Pharmaceutical Industries Limited | Processes for the preparation of eluxadoline |
| CN106636241B (zh) * | 2016-12-28 | 2020-04-03 | 尚科生物医药(上海)有限公司 | 一种酶法制备艾沙度林中间体的方法 |
| CN106636246B (zh) * | 2016-12-28 | 2020-03-24 | 尚科生物医药(上海)有限公司 | 生物法制备(s)-1-(5-苯基-1h-咪唑-2-基)乙胺 |
| CN106866463B (zh) * | 2017-01-24 | 2018-08-28 | 富乐马鸿凯(大连)医药有限公司 | 艾沙度林中间体的制备方法 |
| KR20230169447A (ko) | 2017-06-09 | 2023-12-15 | 추가이 세이야쿠 가부시키가이샤 | N-치환 아미노산을 포함하는 펩타이드의 합성 방법 |
| US11492369B2 (en) | 2017-12-15 | 2022-11-08 | Chugai Seiyaku Kabushiki Kaisha | Method for producing peptide, and method for processing bases |
| CN110092735B (zh) * | 2018-01-31 | 2021-05-11 | 尚科生物医药(上海)有限公司 | 一种l-丙氨酸衍生物的制备方法 |
| WO2019197274A1 (en) | 2018-04-09 | 2019-10-17 | Quimica Sintetica, S. A. | Process for the preparation of opioid modulators |
| CN113056475A (zh) | 2018-11-30 | 2021-06-29 | 中外制药株式会社 | 用于肽化合物或酰胺化合物的脱保护方法和在固相反应中的树脂脱除方法以及用于生产肽化合物的方法 |
| US20220144762A1 (en) | 2019-03-15 | 2022-05-12 | Chugai Seiyaku Kabushiki Kaisha | Method for preparing aromatic amino acid derivative |
| CN111620876B (zh) * | 2020-05-07 | 2021-09-28 | 普济生物科技(台州)有限公司 | 瑞德西韦关键中间体的合成方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2780372B2 (ja) | 1989-08-29 | 1998-07-30 | 株式会社日立製作所 | デイスク制御装置のキヤツシユ組込制御方法 |
| JP2881473B2 (ja) * | 1990-03-30 | 1999-04-12 | 日産化学工業株式会社 | α―置換シクロペンテノン誘導体及びその製造法並びに該誘導体を得るための有機亜鉛試剤 |
| US5554753A (en) * | 1993-08-25 | 1996-09-10 | Indiana University Foundation | Catalytic enantioselective synthesis of α-amino acid derivatives by phase-transfer catalysis |
| SE9402880D0 (sv) | 1994-08-30 | 1994-08-30 | Astra Ab | New peptide derivatives |
| JP4228587B2 (ja) * | 2002-05-22 | 2009-02-25 | 昭和電工株式会社 | アミノメチル基含有ベンズアミド化合物の製造方法 |
| EP1567534B1 (en) | 2002-12-04 | 2009-07-01 | Ciba Holding Inc. | Process for the synthesis of cycloorganylphosphanes and di(alkali metal/alkaline earth metal) oligophosphanediides |
| SI1725537T1 (sl) | 2004-03-15 | 2011-11-30 | Janssen Pharmaceutica Nv | Nove spojine kot modulatorji opioidnih receptorjev |
| CA2601674A1 (en) * | 2005-03-14 | 2006-09-21 | Janssen Pharmaceutica N.V. | Process for the preparation of opioid modulators |
| TWI468375B (zh) * | 2008-10-27 | 2015-01-11 | Janssen Pharmaceutica Nv | 製備經保護之l-丙胺酸衍生物之方法 |
-
2009
- 2009-10-26 TW TW98136147A patent/TWI468375B/zh not_active IP Right Cessation
- 2009-10-27 AU AU2009320156A patent/AU2009320156B2/en active Active
- 2009-10-27 EP EP09752026.6A patent/EP2362868B1/en active Active
- 2009-10-27 CN CN200980153642.2A patent/CN102264691B/zh active Active
- 2009-10-27 MX MX2011004393A patent/MX2011004393A/es active IP Right Grant
- 2009-10-27 ES ES09752026.6T patent/ES2657676T3/es active Active
- 2009-10-27 NZ NZ592415A patent/NZ592415A/xx unknown
- 2009-10-27 PE PE2014000876A patent/PE20141698A1/es not_active Application Discontinuation
- 2009-10-27 KR KR1020117011699A patent/KR101690195B1/ko active IP Right Grant
- 2009-10-27 JP JP2011534675A patent/JP5592893B2/ja active Active
- 2009-10-27 HU HUE09752026A patent/HUE038302T2/hu unknown
- 2009-10-27 CA CA2741790A patent/CA2741790C/en active Active
- 2009-10-27 EA EA201170618A patent/EA018913B1/ru unknown
- 2009-10-27 CN CN201710219076.7A patent/CN107382781A/zh active Pending
- 2009-10-27 BR BRPI0920834-8A patent/BRPI0920834B1/pt active IP Right Grant
- 2009-10-27 UA UAA201106621A patent/UA110600C2/uk unknown
- 2009-10-27 PE PE2011000939A patent/PE20110417A1/es active IP Right Grant
- 2009-10-27 US US12/606,730 patent/US8710256B2/en active Active
- 2009-10-27 WO PCT/US2009/062191 patent/WO2010062590A2/en active Application Filing
- 2009-10-27 AR ARP090104133A patent/AR075291A1/es active IP Right Grant
-
2011
- 2011-04-26 CL CL2011000936A patent/CL2011000936A1/es unknown
- 2011-04-26 IL IL212464A patent/IL212464A/en active IP Right Grant
- 2011-04-27 NI NI201100078A patent/NI201100078A/es unknown
- 2011-04-27 EC EC2011011008A patent/ECSP11011008A/es unknown
- 2011-04-27 CO CO11051582A patent/CO6361989A2/es active IP Right Grant
- 2011-04-27 HN HN2011001186A patent/HN2011001186A/es unknown
- 2011-05-27 CR CR20110286A patent/CR20110286A/es unknown
-
2014
- 2014-01-23 US US14/162,150 patent/US8853445B2/en active Active
- 2014-07-31 JP JP2014155710A patent/JP2015013862A/ja active Pending
- 2014-09-05 US US14/478,253 patent/US9187408B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0920834B1 (pt) | processo para preparação de derivados protegidos de l-alanina | |
| CN100369894C (zh) | 氧化氮合酶抑制剂磷酸盐 | |
| DE69734773T2 (de) | Nicht-peptidische Bombesin-Rezeptor-Antagonisten | |
| HRP20130893A2 (hr) | Novi spojevi kao modulatori opioidnog receptora | |
| ES2538135T3 (es) | Proceso para la preparación de derivados sustituidos de pirimidina | |
| ES2558856T3 (es) | Síntesis de compuestos de pirrolidina | |
| TWI770841B (zh) | 作為組蛋白脫乙醯基酶6抑制劑之1,3,4-㗁二唑衍生物化合物及包含其的醫藥組合物 | |
| ES2432216T3 (es) | Derivados de 2-(1,2-Bencisoxazol-3-il)bencilaminas | |
| UA79537C2 (en) | Indazolamides possessing analgesic activity | |
| ES2811271T3 (es) | Método para la producción de praziquantel y sus precursores | |
| CN1128989A (zh) | N-取代的氮杂环羧酸及其酯 | |
| CN103025722B (zh) | 具有2-(杂芳基)氨基取代基的氯苯吩嗪及其抗微生物活性 | |
| AU2014202596B2 (en) | Process for the preparation of protected L-alanine derivatives | |
| KR20210147873A (ko) | 설폰아미드 유도체 및 이를 유효성분으로 함유하는 정신질환의 예방 또는 치료용 약학적 조성물 | |
| BR112018007289B1 (pt) | Compostos de derivado de oxadiazol amina como inibidor de histona desacetilase 6 e a composição farmacêutica que compreende os mesmos |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 24/11/2020, OBSERVADAS AS CONDICOES LEGAIS. |