BRPI0719264B1 - Processo para a preparação de nebivolol - Google Patents
Processo para a preparação de nebivolol Download PDFInfo
- Publication number
- BRPI0719264B1 BRPI0719264B1 BRPI0719264-9A BRPI0719264A BRPI0719264B1 BR PI0719264 B1 BRPI0719264 B1 BR PI0719264B1 BR PI0719264 A BRPI0719264 A BR PI0719264A BR PI0719264 B1 BRPI0719264 B1 BR PI0719264B1
- Authority
- BR
- Brazil
- Prior art keywords
- formula
- compound
- reaction
- fact
- optionally substituted
- Prior art date
Links
- YFMFEJGRDBJLJY-UHFFFAOYSA-N FC(CC1)C=C(CC2)C1OC2C1OC1 Chemical compound FC(CC1)C=C(CC2)C1OC2C1OC1 YFMFEJGRDBJLJY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/06—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 2
- C07D311/20—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 2 hydrogenated in the hetero ring
Abstract
Description
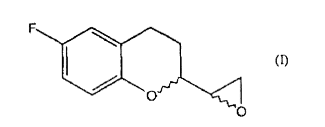





a. a conversão de um composto de fórmula


b. a redução de um composto de fórmula II para dar um composto de fórmula

X é, preferivelmente, um átomo do cloro.
Na presente invenção R é, preferivelmente, um grupo (C1-C6)-alquila ou fenila opcionalmente substituída.


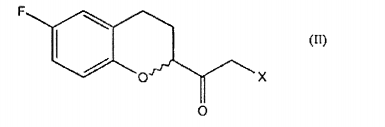
o qual compreende a reação de um composto de fórmula


a. a conversão de um composto de fórmula


b. a redução de um composto de fórmula II para gerar um composto de fórmula
Claims (10)
- Processo para a preparação de um composto de fórmulacaracterizado pelo fato de que compreende

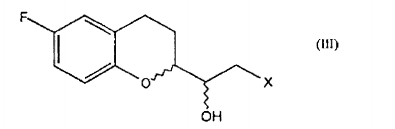
a. a conversão de um composto de fórmulaonde R é um grupo (C1-C6) -alquil, aril opcionalmente substituído ou heteroaril opcionalmente substituído; em um composto de fórmula em que X é halogênio;
em que X é halogênio;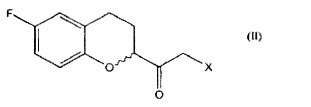
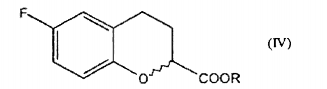
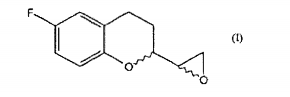
b. a redução de um composto de fórmula II para dar um composto de fórmulac. a reação do referido composto de fórmula III, com uma base para gerar o composto epóxido de Fórmula I.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que a redução é realizada pela reação de um composto de fórmula II com borohidreto de sódio na presença de um solvente alcoólico opcionalmente misturado com água.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que a epoxidação é realizada pela reação de um composto de fórmula III com alcóxidos ou hidróxidos alcalinos na presença de solventes alcoólicos ou éteres opcionalmente na mistura.
- Processo, de acordo com a reivindicação 1, caracterizado pelo fato de que a conversão é realizada pela reação de um composto de fórmula IV com dimetilsulfoxônio metilida para gerar a ceto sulfoxônio ilida correspondente de fórmulaque é transformada em um composto de fórmula II, pela reação com um ácido halogenídrico anidro opcionalmente gerado in situ.

- Processo, de acordo com a reivindicação 4, caracterizado pelo fato de que a dimetilsulfoxônio metilida é preparada in situ a partir do haleto de sulfoxônio correspondente pela reação com uma base na presença de um solvente orgânico.
- Processo, de acordo com a reivindicação 4, caracterizado pelo fato de que o ácido halogenídrico anidro ácido clorídrico anidro gerado in situ pela reação de cloreto de lítio com ácido metanossulfônico na presença de THF.
- Processo para a preparação de um composto de fórmulaem que X é halogênio,

caracterizado pelo fato de que compreende a reação de um composto de fórmulaonde R é um grupo (C1-C6)-alquil, aril opcionalmente substituído ou heteroaril opcionalmente substituído; com dimetilsulfoxônio metilida para gerar a ceto sulfoxônio ilida correspondente de fórmula que é convertida em um composto de fórmula II, por uma reação com ácido halogenídrico anidro opcionalmente gerado in situ.
que é convertida em um composto de fórmula II, por uma reação com ácido halogenídrico anidro opcionalmente gerado in situ.
- Processo para a síntese de Nebivolol caracterizado pelo fato de que a preparação de um composto de fórmulacompreende
a. a conversão de um composto de fórmulaonde R é um grupo (C1-C6) -alquil, aril opcionalmente substituído ou heteroaril opcionalmente substituído; em um composto de fórmula em que X é halogênio;
em que X é halogênio;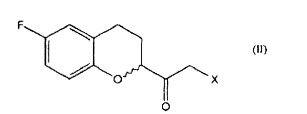
b. a redução de um composto de fórmula II para dar um composto de fórmulac. a reação do referido composto de fórmula III, com uma base para dar o composto epóxido de Fórmula I.
- Processo, de acordo com qualquer uma das reivindicações 1, 2, 3, 4, 5, 6, 7 ou 8, caracterizado pelo fato de que X é um átomo de cloro.
- Composto caracterizado pelo fato de que possui a fórmula dimetilsulfoxônio-2-(6-fluoro-3,4-dihidro-2H-chromen-2-il)-2-oxoetilida.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI2006A001889 | 2006-10-03 | ||
| IT001889A ITMI20061889A1 (it) | 2006-10-03 | 2006-10-03 | Processo di preparazione di nebivololo |
| PCT/EP2007/008549 WO2008040528A2 (en) | 2006-10-03 | 2007-10-02 | Process for preparing nebivolol |
Publications (4)
| Publication Number | Publication Date |
|---|---|
| BRPI0719264A2 BRPI0719264A2 (pt) | 2014-01-28 |
| BRPI0719264A8 BRPI0719264A8 (pt) | 2017-05-16 |
| BRPI0719264B1 true BRPI0719264B1 (pt) | 2021-03-23 |
| BRPI0719264B8 BRPI0719264B8 (pt) | 2021-05-25 |
Family
ID=39030986
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0719264A BRPI0719264B8 (pt) | 2006-10-03 | 2007-10-02 | processo para a preparação de nebivolol |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US7960572B2 (pt) |
| EP (1) | EP2078005B1 (pt) |
| JP (1) | JP5108017B2 (pt) |
| CN (1) | CN101522656B (pt) |
| AU (1) | AU2007304419B2 (pt) |
| BR (1) | BRPI0719264B8 (pt) |
| CA (1) | CA2663339C (pt) |
| ES (1) | ES2424362T3 (pt) |
| HR (1) | HRP20130685T1 (pt) |
| IL (1) | IL197510A (pt) |
| IT (1) | ITMI20061889A1 (pt) |
| PL (1) | PL2078005T3 (pt) |
| PT (1) | PT2078005E (pt) |
| SI (1) | SI2078005T1 (pt) |
| WO (1) | WO2008040528A2 (pt) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITMI20080547A1 (it) * | 2008-03-31 | 2009-10-01 | Zach System Spa | Processo di preparazione di nebivololo |
| EP2163551B1 (en) | 2008-09-08 | 2011-11-16 | Cadila Pharmaceuticals Ltd. | An improved process for the preparation of nebivolol hydrochloride |
| WO2010034927A1 (fr) * | 2008-09-24 | 2010-04-01 | Zach System | Procede de preparation de nebivolol |
| IT1395354B1 (it) | 2009-07-23 | 2012-09-14 | Zach System Spa | Processo di preparazione di nebivololo |
| CN102127061B (zh) * | 2010-01-15 | 2016-02-17 | 浙江华海药业股份有限公司 | 一种制备6-氟-3,4-二氢-2h-1-苯并吡喃-2-环氧乙烷的改进方法 |
| DE102010005953A1 (de) | 2010-01-27 | 2011-07-28 | Corden PharmaChem GmbH, 68305 | Verfahren zur Herstellung von Nebivolol |
| CN102190647A (zh) * | 2010-03-12 | 2011-09-21 | 浙江海翔药业股份有限公司 | 一种奈比洛尔的中间体的制备方法 |
| CN102180855B (zh) * | 2011-03-01 | 2015-11-25 | 浙江华海药业股份有限公司 | 一种奈必洛尔中间体的提纯方法 |
| ITRM20110418A1 (it) | 2011-08-02 | 2013-02-03 | Menarini Int Operations Lu Sa | Processo per la preparazione di epossidi quali intermedi per la sintesi del nebivololo. |
| CN102408402A (zh) * | 2011-12-27 | 2012-04-11 | 上海立科药物化学有限公司 | (r)或(s)- 6-氟 -3 ,4-二 氢- 2h-1-苯并吡喃-2-甲酸的合成方法 |
| CN104016954B (zh) * | 2014-06-06 | 2016-01-27 | 常州市第四制药厂有限公司 | 奈必洛尔中间体的制备及纯化方法 |
| ITUB20160227A1 (it) * | 2016-01-21 | 2017-07-21 | Menarini Int Operations Luxembourg Sa | Processo per la sintesi di intermedi di Nebivololo |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1337429C (en) | 1983-12-05 | 1995-10-24 | Guy Rosalia Eugene Van Lommen | Derivatives of 2,2'-iminobisethanol |
| CA1337432C (en) * | 1988-03-23 | 1995-10-24 | Raymond M. Xhonneux | Method of lowering the blood pressure |
| US7349481B2 (en) | 2002-07-01 | 2008-03-25 | Qualcomm Incorporated | Communication using audible tones |
| HU227236B1 (en) * | 2002-11-06 | 2010-12-28 | Egis Gyogyszergyar Nyilvanosan Muekoedoe Reszvenytarsasag | New process for the production of racemic and the pure [2s[2r*[r[r*]]]]-and [2r[2s*[s[s*]]]]-enantiomers of nebivolol |
| EP1741712B1 (en) * | 2004-07-30 | 2011-06-15 | Torrent Pharmaceuticals Ltd | amorphous form of nebivolol hydrochloride and its preparation |
| ATE465157T1 (de) | 2004-08-11 | 2010-05-15 | Hetero Drugs Ltd | Neues verfahren zur herstellung von nebivololzwischenprodukten |
| US7560575B2 (en) * | 2005-12-28 | 2009-07-14 | Acino Pharma Ag | Process for preparation of racemic Nebivolol |
-
2006
- 2006-10-03 IT IT001889A patent/ITMI20061889A1/it unknown
-
2007
- 2007-10-02 WO PCT/EP2007/008549 patent/WO2008040528A2/en active Application Filing
- 2007-10-02 PL PL07818629T patent/PL2078005T3/pl unknown
- 2007-10-02 CA CA2663339A patent/CA2663339C/en active Active
- 2007-10-02 PT PT78186293T patent/PT2078005E/pt unknown
- 2007-10-02 SI SI200731282T patent/SI2078005T1/sl unknown
- 2007-10-02 CN CN2007800369512A patent/CN101522656B/zh active Active
- 2007-10-02 BR BRPI0719264A patent/BRPI0719264B8/pt active IP Right Grant
- 2007-10-02 AU AU2007304419A patent/AU2007304419B2/en active Active
- 2007-10-02 US US12/443,279 patent/US7960572B2/en not_active Expired - Fee Related
- 2007-10-02 ES ES07818629T patent/ES2424362T3/es active Active
- 2007-10-02 EP EP07818629.3A patent/EP2078005B1/en active Active
- 2007-10-02 JP JP2009530793A patent/JP5108017B2/ja active Active
-
2009
- 2009-03-10 IL IL197510A patent/IL197510A/en active IP Right Grant
-
2013
- 2013-07-22 HR HRP20130685TT patent/HRP20130685T1/hr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN101522656B (zh) | 2011-12-07 |
| WO2008040528A3 (en) | 2008-05-22 |
| IL197510A0 (en) | 2009-12-24 |
| HRP20130685T1 (en) | 2013-09-30 |
| ES2424362T3 (es) | 2013-10-01 |
| JP2010505779A (ja) | 2010-02-25 |
| CN101522656A (zh) | 2009-09-02 |
| US20100069652A1 (en) | 2010-03-18 |
| EP2078005A2 (en) | 2009-07-15 |
| IL197510A (en) | 2014-03-31 |
| US7960572B2 (en) | 2011-06-14 |
| AU2007304419B2 (en) | 2012-03-22 |
| BRPI0719264A2 (pt) | 2014-01-28 |
| PL2078005T3 (pl) | 2013-10-31 |
| WO2008040528A2 (en) | 2008-04-10 |
| BRPI0719264B8 (pt) | 2021-05-25 |
| CA2663339A1 (en) | 2008-04-10 |
| PT2078005E (pt) | 2013-08-23 |
| SI2078005T1 (sl) | 2013-12-31 |
| ITMI20061889A1 (it) | 2008-04-04 |
| CA2663339C (en) | 2014-07-08 |
| AU2007304419A1 (en) | 2008-04-10 |
| EP2078005B1 (en) | 2013-05-08 |
| BRPI0719264A8 (pt) | 2017-05-16 |
| JP5108017B2 (ja) | 2012-12-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0719264B1 (pt) | Processo para a preparação de nebivolol | |
| EP2102196B1 (en) | Process for preparing nebivolol | |
| US20070123719A1 (en) | Synthesis of cannabinoids | |
| CS245783B2 (en) | Production method of 4-phenyl-1,3-dioxan-cis-5-ylalkene acids | |
| AU2002253386A1 (en) | Synthesis of cannabinoids | |
| BRPI0919257B1 (pt) | Processo de preparação de nebivolol | |
| CN114057625B (zh) | 一种c2-酰氧基-3-吲哚啉酮衍生物及其制备方法与应用 | |
| CN111072605A (zh) | 一种氟烷基取代的苯并呋喃衍生物或吲哚衍生物的制备方法 | |
| CA2708141C (en) | Intermediates and methods for making zearalenone macrolide analogs | |
| JP2019509293A (ja) | 4−ペンタフルオロチオフェノール類化合物と調製方法及びペンタフルオロサルファー置換ベンゾピラン化合物の調製方法 | |
| CN112745314B (zh) | 一种具有特异性抑制HIF-2α作用的芳香胺化合物的制备合成方法 | |
| HU191708B (en) | Process for preparing sorbinyl by means of the resolution of 6-fluoro-4-ureido-chromane-4-carboxylic acid | |
| CN112645829B (zh) | 麻黄碱关键中间体(s)-2-甲胺基-1-苯基-1-丙酮的手性合成方法 | |
| CN107556269B (zh) | α-炔基取代醚类化合物的合成方法 | |
| CN117510496A (zh) | 一种环丙烷拼接吡咯喹啉并四环类衍生物的制备方法 | |
| CN116102415A (zh) | 一种5H-二苯并[a,d]环庚三烯-5-酮中间体化合物 | |
| CN113620789A (zh) | 一种手性α-氟代烷氧基醇及其制备方法 | |
| CN115710213A (zh) | 一种顺式手性3-氟-4-羟基哌啶及其衍生物的制备方法 | |
| JP5233675B2 (ja) | 光学活性2−(2’−ピペリジニル)酢酸エステルの製法 | |
| CN116444443A (zh) | 一种苯甲酰-喹唑啉酮衍生物及其制备方法与应用 | |
| CN109776612A (zh) | 一种磷杂色原酮衍生物的合成方法 | |
| JP2003335780A (ja) | スピロキラリティーを有する四級アンモニウム塩及びその利用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B08F | Application dismissed because of non-payment of annual fees [chapter 8.6 patent gazette] |
Free format text: REFERENTE A 3A ANUIDADE. |
|
| B08H | Application fees: decision cancelled [chapter 8.8 patent gazette] |
Free format text: REFERENTE AO DESPACHO 8.6 PUBLICADO NA RPI 2277 DE 26/08/2014. |
|
| B25A | Requested transfer of rights approved |
Owner name: F.I.S. - FABBRICA ITALIANA SINTETICI S.P.A. (IT) |
|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] |
Free format text: DE ACORDO COM O ARTIGO 229-C DA LEI NO 10196/2001, QUE MODIFICOU A LEI NO 9279/96, A CONCESSAO DA PATENTE ESTA CONDICIONADA A ANUENCIA PREVIA DA ANVISA. CONSIDERANDO A APROVACAO DOS TERMOS DO PARECER NO 337/PGF/EA/2010, BEM COMO A PORTARIA INTERMINISTERIAL NO 1065 DE 24/05/2012, ENCAMINHA-SE O PRESENTE PEDIDO PARA AS PROVIDENCIAS CABIVEIS. |
|
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B15K | Others concerning applications: alteration of classification |
Free format text: AS CLASSIFICACOES ANTERIORES ERAM: C07D 311/20 , C07D 407/04 Ipc: C07D 311/20 (2006.01), C07D 407/04 (2006.01), A61P |
|
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 23/03/2021, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 02/10/2007 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |