BRPI0410037B1 - Inibidores de fosfatidilinositol 3-cinase, sua composição farmacêutica, e seu uso - Google Patents
Inibidores de fosfatidilinositol 3-cinase, sua composição farmacêutica, e seu uso Download PDFInfo
- Publication number
- BRPI0410037B1 BRPI0410037B1 BRPI0410037-9A BRPI0410037A BRPI0410037B1 BR PI0410037 B1 BRPI0410037 B1 BR PI0410037B1 BR PI0410037 A BRPI0410037 A BR PI0410037A BR PI0410037 B1 BRPI0410037 B1 BR PI0410037B1
- Authority
- BR
- Brazil
- Prior art keywords
- methyl
- thiazol
- ethyl
- alkyl
- pyrimidin
- Prior art date
Links
- JPQRYIALQBCIFL-UHFFFAOYSA-N Cc1nnc(CCC(O)=O)[s]1 Chemical compound Cc1nnc(CCC(O)=O)[s]1 JPQRYIALQBCIFL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Oncology (AREA)
- Child & Adolescent Psychology (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Communicable Diseases (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Dermatology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
"inibidores de fosfatidilinositol 3-cinase". a presente invenção refere-se a compostos de fórmula i na forma livre ou de sal, em que r^ 1^, r^ 2^, r^ 3^ e r^ 4^ possuem os significados como no relatório descritivo, são úteis para o tratamento de condições que são mediadas pela fosfatidilinositol 3-cinase. composições farmacêuticas que contêm os compostos e um processo para a preparação dos compostos são também descritos.
Description
(54) Título: INIBIDORES DE FOSFATIDILINOSITOL 3-CINASE, SUA COMPOSIÇÃO
FARMACÊUTICA, E SEU USO (51) lnt.CI.: C07D 417/04; C07D 417/14; A61K 31/506; A61K 31/4436; A61P 29/00; A61P 9/10; A61P 3/04; A61P 11/06; A61P 19/02 (30) Prioridade Unionista: 02/05/2003 GB 03 10234.0 (73) Titular(es): NOVARTIS AG (72) Inventor(es): IAN BRUCE; BARBARA VALADE; JUDY HAYLER; EMMA BUDD; BERNARD CUENOUD; THOMAS HUGO KELLER; GAYNOR ELIZABETH PILGRIM; NICOLA PRESS; DARREN MARK LE GRAND; CATHY RITCHIE
Relatório Descritivo da Patente de Invenção para INIBIDORES DE JSFATIDiUNOSlTOL 3_C|NASE SUA COMPOSIÇÃO FARMACÊUTICA E :U USO.
A presente invenção refere-se a compostos orgânicos, suf sparação e seu uso como produtos farmacêuticos.
Em um primeiro aspecto, a presente invenção fornece composto de mula I forma livre ou de sal, em que é Ci-Ce-alquilcarbonila opcionalmente substituída por halo, hidróxi, ciano lina, carbóxi, Ci-C8-alquila, Ci-C8-alcóxi, Ci-C8-haloalquila, Ci-C8 luilamino, di(Ci-C8-alquil)amino, di(Ci-C8-alquil}aminocarbonila, CrCe luilcarbonila, Ci-Ca-alcoxicarbonila, um Ca-Cis-carbooíoiico, ou por um ane terocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de ane lecionados do grupo consistindo em oxigênio, nitrogênio e enxofre,
R1 é um anel heterocíclico de 5 ou 6 membros tendo um ou mais teroátomos de anel selecionados do grupo consistindo em oxigênio rogênío e enxofre,
R1 é aminocarbonila opcionalmente substituída por um C3-Ci5-carbocíclicí por um anel heterocíclico de 5 ou 6 membros tendo um ou maií teroátomos de anel selecionados do grupo consistindo em oxigênio rogênío e enxofre,
R1 é -CO-NRXR1', onde R* e Ry juntamente com o nitrogênio a qual eles sãc ados formam um anel N-heterocíclico de 5 a 12 membros opcional mentí Guindo um ou mais heteroátomos de anel selecionados do grupo consistindc i oxigênio, nitrogênio e enxofre,
R1 é CrCa-alquilaminocarbonila ou CrCs-cidoalquilaminocarbonila em cadí so opcionalmente substituída no grupo de alquila por halo, hidróxi, ciano ina, carbóxi, Ci-Ce-alquila, Gi-Ca-alcóxi, Ci-Cg-alcóxi substituído por hidróxi
-Cs-haloalquila, Gi-Ga-alquii amino, di(Ci-Ca-aIquii)amíno, di(Ci
C8-alquil)amino-carbonila, C^-Cg-alcoxicarbonila, um C3-C15-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou O^Cg-alquilaminocarbonila opcionalmente substituída por hidróxi, ou R1 é CfCg-alquilaminocarbonila ou C3-Cs-cicloalquilamÍnocarbonila em cada caso opcionalmente substituída por aminocarbonila opcionalmente substituída por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é hidrogênio;
R2 é C^-Cg-alquila;
Y é carbono ou nitrogênio; e quando R1 for C^Cg-alquilcarbonila não substituída e Y for carbono então R3 é halo, hidróxi, ciano, amina, carbóxi, -SO2NH2, Ó-Cg-alquila, C,-C8alcóxi, Qj-Cg-haloalquila, amino-C^Cg-alquila, amino-C^Cg-alcóxi, C,-C8alquilaminocarbonifa, difC^Cg-alquiOamino, di(CrC8-alquil)aminocarbonila, di(C1-C8-alquil)amino-C1-C8-alquila, di(C1-C8-alquil)amino-C1-C3-alcóxi, aminocarbonila, CT^-alcoxicarbonila, carbóxi-C^C^alquila, carbóxi-C,-^alcóxi, um C3-C15-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C^C^alquilamino opcionalmente substituído por hidróxi ou di(C.,-C8-alquil)amino, e R4 é hidrogênio, halo, hidróxi, ciano, amina, carbóxi, -SO2NH2 C^Cgalquila, C-rCg-alcóxi, CfC^haloalquila, amino-C^Cg-alquila, amino-C^C^ alcóxi, C,-C8-alquilaminocarbonila, di^-Cg-alquipamino, di^-Cgalquil)aminocarbonila, diíCfCg-alquiQamino-Ó-Cg-alquila, difC^-Cgalquiljamino-C^Ca-alcóxi, aminocarbonila, CfCg-alcoxicarbonila, carbóxi-Cf Cg-alquila, carbóxi-C^-Cg-alcóxi, um C3-C15-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C^ C8-alquilamino opcionalmente substituído por hidróxi ou di^-Cgalquil)amino, de outro modo R3 e R4 são cada um independentemente hidrogênio, halo, hidróxi, ciano, amina, carbóxi, C^Cg-alquiisulfanila, C^Cg-alquiísulfinila, Cr C8-alquilsulfonila, -SO2NH2 C^Cg-alquila, Ç-Cg-haloalquila, amino-CpCgalquifa, amino-CfCg-alcóxi, C^Cg-alquilaminocarbonila, diíC^Cgalquil)aminocarboni!a, diíCçCg-alquiOamino-CfCg-alquila, di(C,-C8alquilJamino-CfCg-alcóxi, Ç-Cg-acilamino, aminocarbonila, C^Cgalcoxicarbonila, carbóxi-C1-C8-alquila, carbóxi-C^Cg-alcóxi, um C3-C1Scarbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, CfCg-alquilamino ou di(C1-C8-alquil)amino cada um sendo opcionalmente substituído por amino, hidróxi, diíC^Cg-alquiljamino ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou Ç-Cg-alcóxi opcionalmente substituído por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre.
Os termos usados no relatório descritivo possuem os seguintes significados:
Opcionalmente substituído como aqui usado significa que o grupo referido pode ser substituído em uma ou mais posições por qualquer um ou qualquer combinação dos radicais listados daqui em diante.
Aminocarbonila como aqui usado significa amino ligado atrevés do átomo de nitrogênio a um grupo de carbonila.
Halogênio ou halo como aqui usado pode ser flúor, cloro, bromo ou iodo; preferivelmente é flúor ou cloro.
CfCg-alquila como aqui usado significa alquila de cadeia reta ou ramificada tendo de 1 a 8 átomos de carbono. Preferivelmente, a CrC3alquila é CfC^alquila.
Grupo C3-C15-carbocíclico como aqui usado significa um grupo carbocíclico tendo de 3 a 15 átomos de carbono do anel, por exemplo um grupo monocíclico, ou cicloalifático, tal como uma C3-C8-cicloalquila, por exemplo ciclopentila, ciclohexila, cicloheptila ou ciclooctila, ou aromático tal como fenila, ou um grupo bicíclico tal como biciclooctíla, biciclononila incluindo indanila e indenila, e biciclodecila incluindo naftila. Preferivelmente o grupo C3-C15-carbocíclico é um grupo C3-C10-carbocíclico, por exemplo ciclopropila, fenila ou naftila. O grupo C3-C15-carbocíclico pode ser substituído ou não substituído. Os substituintes preferidos incluem halo, ciano, amina, nitro, carbóxi, Ç-Cg-alquila, halo-Ç-Cg-aiquila, CrC8-alcóxi, CçCg-alquilcarbonila, CrC3-alquilsulfonila, -SO2NH2, um grupo C3-C15-carbocíclico e um grupo heterocíclico de 5 a 12 membros tendo pelo menos um heteroátomo do anel selecionado de nitrogênio, oxigênio e enxofre.
C3-Cs-cicloalquila como aqui usado significa cicloalquiía tendo de 3 a 8 átomos de carbono do anel, por exemplo um grupo monocíclico tal como uma ciclopropila, ciclobutila, ciclopentila, ciclohexila, cicloheptila ou ciclooctila, qualquer uma das quais pode ser substituída por um ou mais, geralmente um ou dois, grupos de Ç-C^-alquila, ou um grupo bicíclico tal como bicicloheptila ou biciclooctila. Preferivelmente, C3-Cs-cicloalquila é C3C5-cicloalquila isto é ciclopropila, ciclobutila ou ciclopentila.
C1-C8-alquilsulfanila (ou CfCg-alquiltio) como aqui usado significa C^Cgalquila como mais acima definido ligada a -S-. Preferivelmente C,-C3alquilsulfanila é C,-C4- alquilsulfanila, especíalmente metilsulfanila. CfCg-alquilsulfinila como aqui usado significa C^Cg-alquila como mais acima definido ligada a -S(=O)-. Preferivelmente CfCg-alquilsulfinila é alquilsulfinila, especialmente metilsulfinila.
CfCg-alquiisulfonila como aqui usado significa Cj-Cg-alquila como mais acima definido ligada a -SO2-. Preferivelmente Ç-Cg-alquilsulfonila é CrC4“ alquiisulfonila, especíalmente metilsulfonila.
CfCg-alcóxi como aqui usado significa alcóxi de cadeia reta ou ramificada tendo de 1 a 8 átomos de carbono. Preferivelmente, CfCg-alcóxi é Cj-CX,alcóxi.
CrCg-haloalquila como aqui usado significa Ç-Cg-alquila como mais acima definido substituída por um ou mais átomos de haiogênio, preferivelmente um, dois ou três átomos de halogênio, preferivelmente átomos de flúor ou cloro. Preferivelmente, Ç-Cg-haloalquila é Ç-C^alquila substituída por um, dois ou três átomos de fíúor ou cloro.
Amino-CpCg-alquila e amino-Ç-Cg-alcóxi como aqui usados significam amino ligado por um átomo de nitrogênio à Ç-Cg-alquila ou CfCg-alcóxi respectivamente como mais acima definido. Preferivelmente, amino-C^Cgalquila e amino-Ç-Cg-alcóxi são respectivamente amino-CpC^alquila e amino-Ç-Cg-alcóxi.
Carbóxi-C^Cg-alquila e ’'carbóxi-C1-C8-alcóxi como aqui usados significam carbóxi ligado por um átomo de carbono à Ç-Cg-alquila ou C1-Cs-alcóxi respectivamente como mais acima definido. Preferivelmente, carbóxi-Ç-Cgalquila e carbóxi-Ç-Cg-alcóxi são respectivamente carbóxi-C1-C4-alquila e carbóxí-C^Cí-alcóxi.
CfCg-alquilcarbonila, CfCg-alcoxicarbonila e Ç-Cg-haloalquilcarbonila como aqui usados significam Ç-Cg-alquila, CfCg-alcóxi ou Ç-Cg-haloalquila respectivamente como mais acima definidos ligados por um átomo de carbono a um grupo de carbonila. Preferivelmente, C,-C8-alquilcarbonila, C,C8-alcoxicarbonila e Ç-Cg-haloalquilcarbonila são respectivamente CrC4alquilcarbonila, C^C^alcoxicarbonila e C1-C4-haloalquil-carbonila. CfCg-alquilamino, diíC^Cg-alquiOamino e Cg-Cg-cicloalquilamino” como aqui usado significam Ç-Cg-alquila, CfCg-alcóxi e C3-C8-cicloalquila respectivamente como mais acima definidos ligados por um átomo de carbono a um grupo de amino. Os grupos de Cfi-Cg-alquila em diíÇ-Cgalquila)-amino podem ser os mesmos ou diferentes. Preferivelmente, ΟΓΟ8alquilamino, di(C.,-Ca-alquil)amino e C3-CB-cicloalquilamino são respectivamente CfC^alquilamino, di(C,-C4-alquil)amino e C3-C5cicloalquilamino.
Ç-Cg-alquilaminocarbonila, d^CfCg-alquiljaminocarbonilá1 e C3-C8cicloalquilamino-carbonila como aqui usados significam Ç-Cg-alquilamino, di(CrCs-alquil)amino e C3-C8 cicloalquilamino respectivamente como mais acima definido ligados por um átomo de nitrogênio ao átomo de carbono de um grupo de carbonila. Preferivelmente, CfCg-alquilaminocarbonila, di(C·,C8-alquila)-anninocarbonila e C3-C8-cicloalquilaminocart>onila são respectivamente C1-C4-alquilaminocarbonila, di/C^C^-aiquila)aminocarbonila e C3-C5-cicloalquilaminocarbonila.
difC^Cg-alquiOamino-C-j-Cg-alquila'’ e dKC^Cg-alquiOamino-C^Cg-alcóxi como aqui usados significam d^CfCg-alqu^amino como mais acima definido ligados por um átomo de nitrogênio ao átomo de carbono de um C,C8-alquila ou um grupo de CfCg-alcóxi respectivamente. Preferivelmente, di(Cl-C8-alquil)amino-C1-C8-alquila e di(G1-C3-alquil)amino-C1-C3-alcóxi são respectivamente di^-C^alquiOamino-CpC^alquil e di(C,-C4-alquil)aminoCfC^alcóxi.
,lC1-C8-acilamino como aqui usado significa amino substituído por C.,-C6alquilcarbonila como mais acima definido. Preferivelmente CfCg-acilamino é C^C^acilamino, especialmente acetilamino.
Anel heterocíclico de 5 ou 6 membros contendo pelo menos um heteroátomo de anel selecionado do grupo consistindo em nitrogênio, oxigênio e enxofre como aqui usado pode ser, por exemplo, furano, pirrol, pirrolidina, pirazol, imidazol, triazol, isotriazol, tetrazol, tiadiazol, isotiazol, oxadiazol, piridina, piperidina, pirazina, oxazol, isoxazol, pirazina, piridazina, pirimidina, piperazina, pirrolidina, morfolina, triazina, oxazina ou tiazoi. Os anéis heterocíclicos preferidos incluem piperazina, pirrolidina, morfolina, imidazol, isotriazol, pirazol, tetrazol, tiazoi, tiadiazol, piridina, piperidina, pirazina, furano, oxazol, isoxazol, oxadiazol e azetidina. O anel heterocíclico de 5 ou 6 membros pode ser não substituído ou substituído. Os substituintes preferidos incluem halo, ciano, oxo, hidróxi, carboxi, nítro, Ο,-Cg-alquila, Cr C8-alquilsulfonila, aminocarbonila, CfCg-alquilcarbonila e CfCg-alcóxi opcionalmente substituídos por aminocarbonila. Os substituintes especiaimente preferidos incluem halo, oxo, hidróxi, C.,-C4-alquila, CfC^ alquilsulfonila, CfC4-alquilcarbonila e aminocarbonila.
Anel N-heterocíclico de 5 a 12 membros opcionalmente incluindo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre como aqui usado pode ser, por exemplo, azetidina, pirrolidina, imidazolidina, piperidina, piperazina, morfolina ou tetrahidroimidazo-piridina. O anel N-heterocíclico de 5 a 12 membros é preferivelmente um anel N-heterocíclico de 5 a 9 membros. Os anéis Nheterocíclicos de 5 a 12 membros preferidos incluem pirrolidina, morfolina e tetrahidro-imidazo-piridina. O anel N-heterocíclico de 5 a 12 membros pode ser não substituído ou substituído. Os substituintes preferidos incluem halo, cíano, oxo, hidróxi, carbóxi, nitro, Cq-Cg-alquilcarbonila, -SO2-CH3, e CrC3alquila ou CfCg-alcóxi em cada caso opcionalmente substituído por carbóxi, aminocarbonila, C^Cg-alcóxi-carbonila, ou C,-C5-alquilaminocarbonila ou diíC^C^alquiOaminocarbonila em cada caso sendo opcionalmente substituído por hidróxi. Os substituintes especialmente preferidos incluem hidróxi, -SO2-CH3 e aminocarbonila.
Em todo este relatório descritivo e nas reivindicações que seguem, a não ser que o contexto exija de outra maneira, a palavra compreendem, ou variações tais como compreende ou compreendendo, será compreendida por implicar a inclusão de um número inteiro mencionado ou intervalo ou grupo de números inteiros ou intervalos, mas não a exclusão de qualquer outro número inteiro ou intervalo ou grupo de números inteiros ou intervalos.
Em um segundo aspecto, a presente invenção fornece compostos de fórmula I
R‘ fina forma livre ou de sal, em que
R1 é Cg-Cg-alquilcarbonila opcionalmente substituída por halo, hidróxi, ciano, amina, carbóxi, C^Cg-alquila, C^Cg-alcóxi, C^Cg-haloalquila, C^Cgalquilamino, dijCyCg-alquiljamino, diíC^Cg-alquiljaminocarbonila, C^Cgalcoxicarbonila, um C3-C15-carbocíclico, ou por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é aminocarbonila opcionalmente substituída por um C3-C15carbocíclico ou por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é C^Cg-alquilaminocarbonila ou C3-C8-cicloalquilaminocarbonila em cada caso opcionalmente substituída no grupo de alquila por halo, hidróxi, ciano, amina, carbóxi, C^Cg-alquila, C^Cg-alcóxi, C^Cg-haloalquila, C^-Cgalquifamino, diíC^Cg-alquiljamino, di(CrC8-alquil)aminocarbonila, C^Cgalcoxicarbonila, um C3-C15-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre;
R2 é CrC3-alquila;
Y é carbono ou nitrogênio; e quando R1 for C,-C8-alquilcarbonila não substituída e Y for carbono então R3 é [não hidrogênio] halo, hidróxi, ciano, amina, carbóxi, -SO2NH2 C.,-C8aiquila, CfCg-alcóxi, C^-haloalquila, amino-CpCg-alquila, amino-C^Cgalcóxi, C^Cg-alquilaminocarbonila, d^Cg-Cg-alquiljamino, diíCfCgalquil)aminocarbonila, diíC^Cg-alquiOamino-C^Cg-alquila, di(CrCg“ alquilJamino-C^Cs-alcóxi, aminocarbonila, C,-Ca-alcoxicarbonila, carbóxi-Cr C8-alquila, carbóxi-CpCg-alcóxi, um C3-C15-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C,C8-alquilamino opcionalmente substituído por hidróxi ou όί(Ο,<8“ alquil)amino, e R4 é hidrogênio, halo, hidróxi, ciano, amina, carbóxi, -SO2NH2 C^Cgalquila, C^Cg-alcóxi, C^Cg-haloalquila, amino-CfCg-alquila, amino-CrC8alcóxi, C^Cg-alquilaminocarbonila, diíCfCg-alquiljamino, di^-Cgalquil)aminocarbonila, diíÇ-Cg-alquiOamino-C^Cg-alquila, <1ϊ(0,-08alquiljamino-CfCg-alcóxi, aminocarbonila, CfC8-alcoxicarbonila, carbóxi-C·,C8-alquila, carbóxi-CfCg-alcóxi, um C3-C15-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou 0,Cs-alquilamino opcionalmente substituído por hidróxi ou di(CrC8alquil)amino, de outro modo R3 e R4 são cada um independentemente hidrogênio, halo, hidróxi, ciano, amina, carbóxi, -SO2NH2i G^Gg-alquila, C1“C8-haloalquila, amino-CfCg-alquila, amino-C^Cg-alcóxi, C^Cg-alquilaminocarbonila, di(Cr C8-alquii)amino, di^-Cg-alquiljaminocarbonila, di(C1-C3-alquil)amino-C1-C8alquila, di(C1-C8“alquil)amino-C1-C8-alcóxi, aminocarbonila, C^Cgalcoxicarbonila, carbóxi-C^-alquila, carbóxi-CfCg-alcóxi, um C3-C15carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, C^Cg-alquilamino opcionalmente substituído por hidróxi ou di(C.,“C8-alquil)arnino.
Os compostos preferidos da presente invenção incluem os compostos de fórmula I na forma livre ou de sal, em que R1 é C^Cg-alquilcarbonila opcionalmente substituída por difC^Cgalquiljamino, C^Cg-alquilcarbonila ou C^Cg-alcoxicarbonila, ou R1 é um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é -CO-NR*Ry, onde R* e Ry juntamente com o nitrogênio a qual eles são ligado formam um anel N-heterocíclico de 5 a 12 membros opcionalmente incluindo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é C^Cg-alquilaminocarbonila opcionalmente substituída no grupo de alquila por hidróxi, C^Cg-alcóxi, C.,-C8-alcóxi substituído por hidróxi, di(CrC8alquil)amino, diCCfCg-alquiljamíno-carbonila, C^Cg-alcoxicarbonila, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C,Cg-alquilaminocarbonila opcionalmente substituída por hidróxi, ou R1 é C1-C8-alquilaminocarbonila opcionalmente substituída por aminocarbonila opcionalmente substituída por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é hidrogênio:
R2 é CfCa-alquila;
Y é carbono ou nitrogênio: e quando R1 for CfCg-alquilcarbonila não substituída e Y for carbono então R3 será halo, Ç-Cg-alquila ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anei selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, e R4 é hidrogênio ou C^Cg-alquila, de outro modo R3 e R4 são cada um independentemente hidrogênio, halo, ciano, C^Cg-alquilsulfanila, Ç-Cg-alquilsulfinila, Ç-Cg-alquilsulfonila, Ç-Cgalquila, Ç-Cg-acilamino, um C3-C15-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, CrCg-alquilamino ou diíCTCg-alquiljamino cada um sendo opcionalmente substituído por amino, hidroxi, diíCg-Cg-alquiljamino ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou G^Cg-alcóxi opcionalmente substituído por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre.
Os compostos preferidos da presente invenção também incluem compostos de fórmula I na forma livre ou de sal, em que R1 é CfCg-alquilcarbonila ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é C^Cg-alquilaminocarbonila opcionalmente substituída no grupo de alquila por C^Cg-alcoxicarbonila, di^-Cg-alquiOaminocarbonila ou por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre;
R2 é CfCg-alquila;
Y é carbono ou nitrogênio; e quando R1 for C^C^alquilcarbonila não substituída e Y for carbono então R3 será halo, C^Cg-alquila ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, e R4 é hidrogênio ou CfCg-alquila, de outro modo R3 e R4 são cada um independentemente hidrogênio, halo, CfCa-alquila, C3-C8-cicloalquila, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C^Cg-alquilamino opcionalmente substituído por hidroxi ou di(CrC8-alquil)amino.
Os compostos especialmente preferidos da presente invenção incluem compostos de fórmula I na forma livre ou de sal, em que R1 é CpC^-alquilcarbonila opcionalmente substituída por di(C1-C4alquil)amino, CfC^aiquilcarbonila ou C^C^alcoxicarbonila, ou R1 é um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é -CO-NRxRy, onde Rx e Ry juntamente com o nitrogênio a qual eles são ligado formam um anel N-heterocíclico de 5 a 9 membros opcionalmente incluindo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é C.,-C4-alquilaminocarbonila opcionalmente substituída no grupo de alquila por hidroxi, C^C^alcóxi, CfC^alcóxi substituído por hidroxi, di(CfC4alquil)amino, di(C1-C4-alquil)amino-carbonila, C,-C4-alcoxicarbonila, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, CrC4alquilaminocarbonila opcionalmente substituída por hidroxi, ou por C^alcóxi, ou R1 é CrC4-alquilaminocarbonila opcionalmente substituída por aminocarbonila opcionalmente substituída por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é hidrogênio:
R2 é Ç-Gj-alquila;
Y é carbono ou nitrogênio; e quando R1 for CfC^alquilcarboníia não substituída e Y for carbono então R3 será halo, C^Qalquila ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, e R4 é hidrogênio ou CfC^alquila, de outro modo R3 e R4 são cada um independentemente hidrogênio, halo, ciano, C^-alquilsulfanila, CcC^alquilsulfinila, C^C^alquilsulfonila, C,-C4alquila, Ç-C^-acilamino, um C3-C10-carbocíclico, um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, Ç-C^alquilamino ou di^-QalquilJamino cada um sendo opcionalmente substituído por amino, hidróxi, dil^-C^alquiOamino ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C,-C4-alcóxi opcionalmente substituído por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre.
Os compostos especialmente preferidos da presente invenção também incluem compostos de fórmula I na forma livre ou de sal, em que R1 é Ç-C^alquilcarbonila ou um anel N-heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou R1 é Ci-Cralquilaminocarbonila opcionalmente substituída no grupo de alquila por CfC4-alcoxicarboniIa, di(C1-C4-alquii)aminocarbonila ou por um anel N-heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre;
R2 é C^Cj-alquila;
Y é carbono ou nitrogênio; e quando R1 for C^-alquilcarbonila não substituída e Y for carbono então
R3 será halo, C^-alquila ou um anel N-heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, e R4 é hidrogênio ou CfC^alquila, de outro modo R3 e R4 são cada um independentemente hidrogênio, halo, C,-C4-alquila, C3-C5-cicloalquila, um anel N-heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C^C^alquilamino opcionalmente substituída por hidróxi ou di(C1-C4-alquÍI)amino.
Muitos dos compostos representados pela fórmula I são capazes de formar sais de adição de ácido, particularmente sais de adição de ácido farmaceuticamente aceitáveis. Os sais de adição de ácido farmaceuticamente aceitáveis do composto de fórmula I incluem aqueles de ácidos inorgânicos, por exemplo, ácidos haiídricos tais como ácido fluorídrico, ácido clorídrico, ácido bromídrico ou ácido iodídrico, ácido nítrico, ácido sulfúrico, ácido fosfórico; e ácidos orgânicos, por exemplo, ácidos monocarboxílicos alifáticos tais como ácido fórmico, ácido acético, ácido trifluoroacético, ácido propiônico e ácido butírico, ácidos hidróxi alifáticos tais como ácido láctico, ácido cítrico, ácido tartárico ou ácido málico, ácidos dicarboxílicos tais como ácido maléico ou ácido succínico, ácidos carboxílicos aromáticos tais como ácido benzóico, ácido p-clorobenzóico, ácido difenilacético ou ácido trifenilacético, ácidos hidróxi aromáticos tais como ácido o-hidroxibenzóico, ácido p-hidroxibenzóico, ácido 1hidroxinaftaleno-2-carboxílico ou ácido 3-hidroxinaftaleno-2-carboxílico, e ácidos sulfônicos tais como ácido metanossulfônico ou ácido benzenossulfônico. Estes sais podem ser preparados a partir dos compostos de fórmula I mediante procedimentos de formação de sal conhecidos.
Os compostos de fórmula I que contêm grupos acídicos, por exemplo, carboxila, são também capazes de formar sais com bases, em particular bases farmaceuticamente aceitáveis tais como aquelas bem conhecidas na técnica; tais sais adequados incluem sais de metal, particularmente sais de metal alcalino ou de metal alcalino terroso tais como saís de sódio, potássio, magnésio ou cálcio, ou sais com amônia ou aminas orgânicas farmaceuticamente aceitáveis ou bases heterocíclicas tais como etanolaminas, benzilaminas ou piridina. Estes sais podem ser preparados a partir de compostos de fórmula I mediante os procedimentos de formação de sal.
Os compostos preferidos específicos de fórmula I são descritos em seguida nos Exemplos.
A invenção fornece, em um outro aspecto, um processo para a preparação de um composto de fórmula I na forma livre ou de sal que compreende as etapas de:
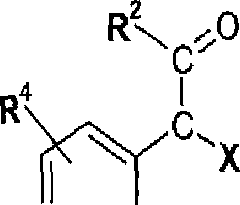
(D (A) reagir um composto de fórmula II
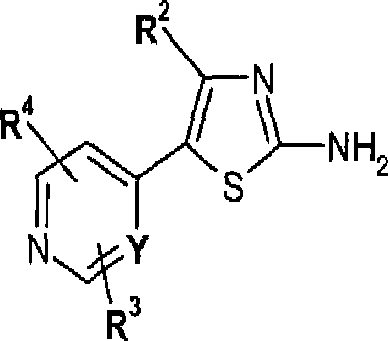
my3 em que R2, R3, R4 e Y são como mais acima definidos e X é halogênio, com um composto de fórmula III nh2 s''C't< I
R1 em que R1 é como mais acima definido;
(B) para a preparação de compostos de fórmula I onde R3 é um anel Nheterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, reagir um composto de fórmula I em que R1, R2, R4 e Y são como mais acima definidos eR5é cloro ou bromo, com um composto de fórmula IV R\
N-H IV Re/ em que Rs e R0 juntos formam um anel N-heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre;
(C) para a preparação de compostos de fórmula I onde R3 é CrC8alquilamino opcionalmente substituído por hidróxi ou di(C-|-Cs-alquil)amirio, reagir um composto de fórmula I em que R1, R2, R4e Y são como mais acima definidos e R3 é cloro ou bromo, com um composto de fórmula V R7—NH2 V em que R7 é C^Cj-alquila opcionalmente substituída por hidróxi ou diCC^-Cgalquil)amino;
(D) para a preparação de compostos de fórmula I onde R1 é C^Cgalquilcarbonila opcionalmente substituída por halo, hidróxi, ciano, amina, carbóxi, CfCj-alquila, C^Cg-alcóxi, CrC8-haloalquila, C^Ce-alquilamino, di^-Cg-alquiOamino, di(CrC8-alquila)-aminocarbonila, CfCg-alquilcarbonila, C^Cg-alcoxicarbonila, um C3-C15-carbocíclico, ou por um anel heterociclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, reagir um composto de fórmula VI
em que R2, R3, R4 e Y são como mais acima definidos, com um composto de fórmula VII
HO-C-R1 VII ou um derivado de formação de amida deste tal como um haleto ou anidrido ácido em que R1 é C-rCg-alquilcarboriila opcionalmente substituída por halo, hidróxi, ciano, amina, carbóxi, C^Cg-alquila, (VCg-alcóxi, C^Cg-haloalquila, C^Cg-alquilamino, di(C1-C8-alquil)amino, difCfCg-alquilaJ-aminocarbonila, CrC8-alquilcarbonila, C^-Cg-alcoxicarbonila, um C3-C15-carbocíclico, ou por um anel heterociclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre;
(E) para a preparação de compostos de fórmula I onde R1 é C^Cgalquilamino-carbonila opcionalmente substituída por halo, hidróxi, ciano, amina, carbóxi, C^Cg-alquila, CfCg-alcóxi, CpC^alcóxi substituído por hidróxi, CfCg-haloalquila, C^Cg-alquilamino, di^-Cg-alquilJamino, dKCq-Cgalquil)aminocarbonila, C^Cg-alcoxicarbonila, um C3-C15-carbocíclico, ou por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou CrC8-alquilaminocarbonila opcionalmente substituída por hidróxi, reagir um composto de fórmula VI em que R2, R3, R4 e Y são como mais acima definidos, com um composto de fórmula VIII o=c=n—R8 VIII em que R8 é CfCg-alquila opcionalmente substituída por halo, hidróxi, ciano, amina, carbóxi, CrC8-alquila, C^Cg-alcóxi, C^Cg-aicóxi substituído por hidróxi, C^Cg-haloalquila, C^Cg-alquilamino, di^-Cg-alquilJamino, di(C.,-Csalquil)aminocarbonila, C^Cg-alcóxi-carbonila, um C8-C16-carbocíclico, um anei heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou C^Cg-alquilaminocarboniia opcionalmente substituída por hidróxi;
(F) para a preparação dos compostos de fórmula ! onde R1 é C^Cgalquilamino-carbonila opcionalmente substituída por halo, hidróxi, ciano, amina, carbóxi, CfCg-alquila, CfCg-alcóxi, CfCg-atcóxi substituído por hidróxi, CfCg-haloalquila, C^Cg-alquilamino, difo^Cg-alquilJamino, di(C.,-C8alquil)aminocarbonila, CfCg-alcoxicarbonila, um C3-C15-carbocíclico, ou por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou Cj-Cg-alquilaminocarbonila opcionalmente substituída por hidróxi, reagir um composto de fórmula IX

em que R2, R3, R4 e Y são como mais acima definido e T1 é um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, com um composto de fórmula X r—NH2 X em que R9 é C^Cg-alquila opcionalmente substituída por halo, hidróxi, ciano, amina, carbóxi, CfCa-alquila, CrC8-alcóxi, C^Cg-alcóxi substituído por hidróxi, CrCg-haloalquila, Ç-Cg-alquilamino, difCfCg-alquilJamino, di^-Cgalquil)aminocarbonila, Ç-Cg-alcoxicarbonila, um C3-C15-carbocíclico, ou por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou Ç-Cg-alquilamínocarbonila opcionalmente substituída por hidróxi;
(G) para a preparação de compostos de fórmula I onde R3 é Ο,-Οθalquilsulfinila ou C^Cg-alquilsulfonila, oxidar a CfCg-alquilsulfanila ou C^Cgalquilsulfinila correspondente respectivamente;
(H) para a preparação de compostos de fórmula I onde R3 é di(C,-Cgalquil)amino opcionalmente substituído por amino, hidróxi, difC^Cgalquil)amino ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, reagir o composto correspondente onde R3 é ΟΓ(38alquilsulfinila ou CrC8-alquilsulfonila com um composto de fórmula Xa H
N Xa
Rm/ V ou uma forma protegida deste onde Rm e Rn são independentemente C^Cgalquila opcionalmente substituída por amino, hidróxi, di{C.,-C3-alquil)amino ou um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre;
(I) para a preparação dos compostos de fórmula I onde R3 é CrC8-alcóxi, reagir o composto correspondente onde R3 é CfCg-alquilsulfinila com um C,Cg-alcóxido de metal alcalino;
{J) para a preparação de compostos de fórmula I onde R3 é C,-C8-alcóxi substituído por um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, reagir o composto correspondente onde R3 é C^Cgalquilsulfintla com um composto de fórmula Xb
HO—V—T2 Xb onde V é Ç-Cg-alquila e T2 é um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre na presença de uma base; ou (K) para a preparação de compostos de fórmula I onde R3 é ciano, reduzir o composto correspondente onde R3 é Ç-Cg-alquilsulfonila com um cianeto de metal alcalino; e (li) remover quaisquer grupos de proteção e recuperar o composto resultante de fórmula I na forma livre ou de sal.
A variante do processo (A) pode ser realizada usando os procedimentos conhecidos para a preparação de aminotiazóis, ou analogamente, por exemplo como em seguida descrito nos Exemplos. O halogênio X é preferivelmente bromo. A reação pode ser realizada em um solvente orgânico, por exemplo um álcool tal como etanol. A temperatura de reação pode ser a partir da temperatura ambiente até a temperatura de refluxo do solvente, mas convenientemente de cerca de 50°C a cerca de 70 °C.
A variante do processo (B) pode ser realizada usando os procedimentos conhecidos para a reação de haletos com anéis Nheterocíclicos nucleofílicos, ou analogamente, por exemplo como em seguida descrito nos Exemplos. A reação pode ser realizada em um solvente orgânico, por exemplo dimetilsulfóxido (DMSO). A temperatura de reação pode ser a partir da temperatura ambiente até a temperatura de refluxo do solvente, mas convenientemente de cerca de 80°C a cerca de 150°C. A temperatura pode ser alcançada mediante o aquecimento convencional ou mediante a irradiação de microondas.
A variante do processo (C) pode ser realizada usando os procedimentos conhecidos para a reação de haletos heterocíclicos com aminas, ou analogamente, por exemplo, como em seguida descrito nos Exemplos. A reação pode ser realizada em um solvente orgânico, por exemplo dimetilsulfóxido (DMSO). A temperatura de reação pode ser a partir da temperatura ambiente até a temperatura de refluxo do solvente, mas convenientemente de cerca de 80° C a cerca de 150° C. A temperatura pode ser obtida por aquecimento convencional ou por irradiação de microondas.
A variante do processo (D) pode ser realizada usando os procedimentos conhecidos para a reação de aminas com ácidos carboxílicos ou um derivado de formação de amida destes tal como um haleto ácido ou anidrido, ou analogamente, por exemplo, como em seguida descrito nos Exemplos. A reação pode ser realizada em um solvente orgânico, por exemplo, diclorometano (DCM). É preferivelmente realizada na presença de uma base, por exemplo diisopropiletilamina (DIPEA). Quando a amina for reagida com um ácido carboxílico é preferivelmente realizada na presença de um agente de acoplamento de peptídeo, por exemplo, 1hidroxibenzotriazol (HOBT). A temperatura de reação pode ser a partir da temperatura ambiente até a temperatura de refluxo do solvente, mas convenientemente de cerca de 60°C a cerca de 80° C.
A variante do processo (E) pode ser realizada usando os procedimentos conhecidos para a reação de aminas com isocianatos, ou analogamente, por exemplo, como em seguida descrito nos Exemplos. A reação pode ser realizada em um solvente orgânico, por exemplo DCM ou dimetilformamida (DMF), preferivelmente na presença de uma base, por exemplo diisopropiletilamina (DIPEA). A temperatura de reação pode ser a partir da temperatura ambiente até a temperatura de refluxo do solvente, mas convenientemente de cerca de 50°C a cerca de 70°C.
A variante do processo (F) pode ser realizada usando os procedimentos conhecidos para a reação de intermediários de carbonil díeterocíclicos (por exemplo acilimidazolídeos quando T for imidazol) com aminas para formar uréias, ou analogamente, por exemplo como em seguida descrito nos Exemplos, A reação pode ser realizada em um solvente orgânico, por exemplo dimetilformamida (DMF). A temperatura de reação pode ser de cerca de 10° C a cerca de 50° C, mas convenientemente na temperatura ambiente.
A variante do processo (G) pode ser realizada usando os procedimentos conhecidos para a oxidação dos grupos de sulfanila para formar grupos de sulfinila ou para a oxidação de grupos de sulfinila para formar grupos de sulfonita ou analogamente por exemplo como em seguida descrito nos Exemplos. O agente oxidante usado é preferivelmente um ácido perbenzóico, especialmente ácido meta-cloroperoxibenzóico (mCPBA). A reação é convenientemente realizada em um solvente orgânico tal como diclorometano (DCM). A temperatura de reação pode ser, por exemplo, de 0 a 30 °C, preferivelmente na temperatura ambiente.
A variante do processo (H) pode ser realizada usando os procedimentos conhecidos para a reação de grupos de sulfinila ou grupos de sulfoniia com aminas secundárias para formar di(alquila)aminas ou analogamente por exemplo como em seguida descrito nos Exemplos. A reação é convenientemente realizada em um solvente orgânico tal como DMF. A temperatura de reação pode ser, por exemplo, de 60 a 100 °C, preferivelmente de 70 a 90 °C.
A variante do processo (I) pode ser realizada usando os procedimentos conhecidos para a reação de grupos de alquilsulfinila com alcóxidos de metal alcalino para formar grupos de alcóxi ou analogamente por exemplo como em seguida descrito nos Exemplos. O alcóxido de metal alcalino é preferivelmente um alcóxido de sódio. A reação é convenientemente realizada em um solvente orgânico tal como metanol. A temperatura de reação pode ser, por exemplo, de 0 a 40 °C, preferivelmente na temperatura ambiente.
A variante do processo (J) pode ser realizada usando os procedimentos conhecidos para a reação de grupos de alquilsulfinila com álcoois primários substituídos para formar grupos de alcóxi substituídos ou analogamente por exemplo como em seguida descrito nos Exemplos. A base é preferivelmente uma base forte tal como hidreto de sódio. A reação é convenientemente realizada em um solvente orgânico tal como DCM. A temperatura de reação pode ser, por exemplo, de 60 a 80 °C, mas preferivelmente ao redor de 70 °C.
A variante do processo (K) pode ser realizada usando os procedimentos conhecidos para a redução dos grupos de alquilsulfonila em um grupo de ciano ou analogamente por exemplo como em seguida descrito nos Exemplos. O cianeto de metal alcalino é preferivelmente cianeto de sódio. A reação é convenientemente realizada em um solvente orgânico tai como dimetilsulfóxido (DMSO). A temperatura de reação pode ser, por exemplo, de 40 a 60 °C, mas preferivelmente ao redor de 50 °C.
Os compostos de fórmula I na forma livre ou de sal podem ser recuperados das misturas de reação e purificados em uma maneira convencional. Misturas isoméricas podem ser separadas em isômeros individuais, por exemplo enantiômeros, em uma maneira convencional, por exemplo, por cristalização fracionária.
Os compostos de fórmula II podem ser preparados mediante a reação de um composto de fórmula XI .0
XI em que R2, R3, R4 e Y são como mais acima definidos, com um agente de halogenação, por exemplo, bromo, ou analogamente, por exemplo como descrito nos Exemplos. A reação pode ser realizada em um solvente orgânico, por exemplo dioxano. A temperatura de reação pode ser de cerca de 0°C a cerca de 30° C, mas convenientemente ao redor de 10°C.
Os compostos de fórmula III, IV e V são comercialmente disponíveis ou podem ser preparados por métodos conhecidos.
Os compostos de fórmula VI onde Y é carbono ou nitrogênio podem ser preparados pela reação de um composto de fórmula II em que R2, R\ R4 e X são como mais acima definidos e Y é carbono ou nitrogênio com tiouréia, ou analogamente, por exemplo como descrito nos Exemplos ou como descrito no relatório descritivo da patente Européia EP 117082 A. A reação pode ser realizada em um solvente orgânico, por exemplo, um álcool tal como etanol. A temperatura de reação pode ser a partir da temperatura ambiente até a temperatura de refluxo do solvente, mas convenientemente de cerca de 50° C a cerca de 70° C.
Os compostos de fórmula VI onde Y é nitrogênio podem ser preparados mediante a reação de um composto de fórmula XII

XII em que R2, R3 e R4 são como mais acima definido com um ácido, por exemplo, ácido trifluoroacético, ou analogamente, por exemplo, como descrito nos Exemplos. A reação pode ser realizada em um solvente polar, por exemplo, água. A temperatura de reação pode ser de cerca de 0 a 100 °C, mas preferivelmente ao redor de 75 °C.
Os compostos de fórmula VII são comercialmente disponíveis ou podem ser preparados por métodos conhecidos.
Os compostos de fórmula VIII são comercialmente disponíveis ou podem ser preparados por métodos conhecidos.
Os compostos de fórmula IX pode ser preparados pela reação de um composto de fórmula VI em que R2, R3, R4, e Y são como mais acima definidos com um composto de fórmula XIII
XIII em que cada T1, que pode ser o mesmo ou diferente, é um anel heterocíclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou analogamente, por exemplo, como descrito nos Exemplos. O composto de fórmula XIII é preferivelmente carbonil diimidazol (CDI). A reação pode ser realizada em um solvente orgânico, por exemplo diclorometano (DCM). A temperatura de reação pode ser de 20°C a cerca de 60 °C, mas convenientemente ao redor de 40 °C.
Os compostos de fórmula X, Xa ou Xb são comercialmente disponíveis ou podem ser preparados por métodos conhecidos.
Os compostos de fórmula XI são comercialmente disponíveis ou podem ser preparados mediante a reação de um composto de fórmula XIV
XIV em que R3, R4 e Y são como mais acima definidos com uma base, tal como butilítio (n-BuLi) ou diisopropil amida de lítio (LDA), depois adição de um composto de fórmula XV
XV
I
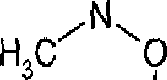
(uma amida Weinreb) em que R2 é como mais acima usando os procedimentos conhecidos para a reação de compostos alquil-substituídos aromáticos com amidas Weinreb, ou analogamente, por exemplo como descrito nos Exemplos. A reação pode ser realizada em um solvente orgânico, por exemplo tetrahídrofurano (THF). A temperatura de reação pode ser de -20° C a cerca de 10° C, mas convenientemente ao redor de 0° C.
Alternativamente, os compostos de fórmula XI em que R3, R4 e Y são como mais acima definidos e R2 é metiia podem ser preparados mediante a reação de um composto de fórmula XIII em que R3, R4 e Y são como mais acima definidos com uma base tal como butilítio (n-BuLi) ou diisopropil amida de lítio (LDA), depois adição de acetato de etila usando os métodos conhecidos para a reação de compostos alquil-substituídos aromáticos com ésteres, ou analogamente, por exemplo como descrito nos Exemplos. A reação pode ser realizada em um solvente orgânico, por exemplo, tetrahídrofurano (THF). A temperatura de reação pode ser de -10° C a cerca de 10°C, mas convenientemente ao redor de 0°C.
Os compostos de fórmula XII podem ser preparados pela reação
XVI de um composto de fórmula XVI

em que R2 e R4 são como mais acima definidos, com um composto de fórmula XVII

XVII em que R3 é como mais acima definido usando o procedimento descrito no relatório descritivo de patente internacional WO 01/72745, ou analogamente, por exemplo, como descrito nos Exemplos.
Os compostos de fórmula XIII, XIIV e XV são comercialmente disponíveis ou podem ser preparados por métodos conhecidos.
Os compostos de fórmula XVI podem ser preparados pela reação de um composto de fórmula XVill

em que R2 é como mais acima definido, com um composto de fórmula XIX CH, CH, v
R4Z| h3c xch3
XIX em que R4 é como mais acima definido usando o procedimento descrito no relatório descritivo de patente internacional WO 01/72745, ou analogamente, por exemplo, como descrito nos Exemplos.
Os compostos de fórmula XVII são comercialmente disponíveis ou podem ser preparados por métodos conhecidos.
Os compostos de fórmula XVIII podem ser preparados pela reação de um composto de fórmula XX
I χχ h3cx zc\ 3 cz XCI
II em que R2 é como mais acima definido com 2-tiouréla ou um forma protegida deste (por exemplo 1-(4-metoxibenzil)- 2-tiouréia) na presença de piridina em metanol como descrito relatório descritivo de patente internacional WO 01/72745, ou analogamente, por exemplo como descrito nos Exemplos.
Os compostos de fórmula XIX e XX são comercialmente disponíveis ou podem ser preparados por métodos conhecidos.
Onde referência é feita aqui aos grupos funcionais protegidos ou aos grupos de proteção, os grupos de proteção podem ser escolhidos de acordo com a natureza do grupo funcional, por exemplo, como descrito em Protective Groups in Organic Synthesis, T.W. Greene and P.G.M. Wuts, John Wiley & Sons Inc, Third Edition, 1999, cuja referência também descreve os procedimentos adequados para a substituição dos grupos de proteção por hidrogênio.
Os compostos de fórmula I na forma livre podem ser convertidos na forma de sal, e vice versa, em uma maneira convencional.Os compostos na forma livre ou de sal podem ser obtidos na forma de hidratos ou solvatos contendo um solvente usado para a cristalização. Os compostos de fórmula I podem ser recuperados das misturas de reação e purificados em uma maneira convencional. Os isômeros, tais como enatiômeros, podem ser obtidos em uma maneira convencional, por exemplo, por cristalização fracionária ou síntese assimétrica dos materiais de partida assimetricamente substituídos correspondentes, por exemplo, optícamente ativos.
Os compostos de fórmula I e seus sais farmaceuticamente aceitáveis, em seguida referidos altemativamente como agentes da invenção, são úteis como produtos farmacêuticos. Em particular, eles apresentam inibição das enzimas fosfatidillnositol 3-cinase (Pi3 cinase), especialmente a gama isoforma (ρ110γ), que são responsáveis de gerar produtos de sinalização fosforilados. As propriedades inibidoras dos compostos de fórmula I podem ser demonstradas nos seguintes procedimentos de teste:
Baculovírus expressando diferente fragmentos de PI3Ky fundidos ao GST foram anteriormente descritos por Stoyanova, S., Bulgarelli-Leva, G„ Kirsch, C., Hanck, T., Klinger, R., Wetzker, R., Wymann, M.P. (1997) Lipid- and protein kinase activities of G protein-coupled PI 3-kinase g: structure-activity analysis and ínteractions with wortmannin. Biochem. J., 324:489. Os resíduos 38-1102 da ΡΙ3Κγ humana são subclonados nos sítios BamH1 e EcoR1 do vetor de transferência pAcG2T (Pharmingen) para criar uma GSTΡΙ3Κγ sem os primeiros 37 resíduos de PI3Ky. Para expressar a proteína recombinante, células de inseto Sf9 (Spodoptera frugiperda 9) são rotineiramente mantidas em densidades de entre 3 X 105 e 3 X 10® cétulas/ml em soro contendo meio TNMFH (Sigma). As células Sf9 cells, em uma densidade de 2 X 10® são infectadas com baculovírus GST-PI3KyA34 humano em uma multiplicidade de infecção (m.o.i.) de 1 por 72 horas. As células infectadas são colhidas por centrifugação em 1400 g por 4 minutos a 4°C e os glóbulos de célula são congelados a -80 °C. Células tanto Sf9 quanto Sf21 funcionam igualmente bem. As células Sf9 (1X109) são colocadas novamente em suspensão em 100 ml de tamponante de lise gelado (4 °C) (50 mM Tris-HCI pH 7,5, 1 % Triton X-100, 150 mM NaCI, 1 mM NaF, 2 mM DTT) e inibidores da protease. As células são incubadas em gelo por 30 minutos depois centrifugadas a 15000 g por 20 minutos a 4 °C. A purificação da amostra sobrenadante é realizada a 4°C por cromatografia de afinidade usando glóbulos de gel agarose SEPHAROSE® acoplados a glutationa (da Amersham Pharmacia Biotech). Uma relação de lisato de célula/resina GST de 50:1 é usada. A resina GST é em primeiro lugar préenxaguada para remover o conservante de etanol e depois equilibrada com tamponante de líse. O lisato de célula (sobrenadante) é adicionado (geralmente como 50 ml de lisato a 1 ml de resina GST em tubos de 50 ml) e suavemente girado em um misturador a 4°C por 2 a 3 horas. O fluxo solto através da amostra é coletado por centrifugação a 1000 g por 5 minutos a 4°C usando uma centrífuga DENLEY®. A resina GST de 1 ml contendo material ligado é transferida para um tubo de centrífuga FALCON® de 15 ml para subsequente etapas de lavagem e eluição. Primeiro uma série de 3 ciclos de lavagens (misturar por inversão suave) é executada com 15 ml de Tampão A de lavagem gelado (50 mM Tris-HCl pH 7,5,1% Triton X-100, 2 mM DTT) entremeados com centrifugação a 1000 g por 5 minutos a 4 °C. Uma etapa de lavagem isolada final é executada com 15 ml de Tampão B de lavagem gelado (50 mM Tris-HCl pH 7,5, 2 mM DTT) e depois centrifugada a 1000 g por 5 minutos a 4 °C. A resina GST lavada é finalmente eluída com 4 ciclos de 1 ml de tamponante de eluição gelado (50 mM Tris-HCl pH 7,5,10 mM glutationa reduzida, 2 mM DTT, 150 mM NaCI, mM NaF, 50 % etileno glicol e inibidores da protease) entremeados com centrifugação em 1000 g por 5 minutos a 4 °C. As amostras são dividas em alíquotas e armazenadas a -20 °C.
Um ensaio de cinase in vitro é estabelecido que mede a transferência do fosfato terminal de trifosfato de adenosina para fosfatidilinositol. A reação de cinase é executada em uma placa de microtitulo de 96 cavidades em branco como um Ensaio de Proximidade por Cintilação. Cada cavidade contém 10 μΙ de composto de teste em 5 % dimetilsulfóxido e 20 μΙ de mistura de ensaio (40 mM Tris, 200 mM NaCI, 2 mM ácido etilenoglicol-aminoetil-tetraacético (EGTA), 15 pg/ml de fosfatidilinositol, 12,5 pM de trifosfato de adenosina (ATP), 25 mM MgCI2, 0,1 pCi (33P]ATP). A reação é iniciada pela adição de 20 pl de mistura de enzima (40 mM Tris, 200 mM NaCI, 2 mM EGTA contendo GST-pUOy recombinante). A placa é incubada em temperatura ambiente por 60 minutos e a reação concluída mediante a adição de 150 pl de solução de interrupção WGA-glóbulo (40 mM Tris, 200 mM NaCI, 2 mM EGTA, 1,3 mM ácido etileno diamina tetraacético (EDTA), 2,6 pM ATP e 0,5 mg de glóbulos de Aglutinina-SPA de Germe de Trigo (Amersham Biosciences) a cada cavidade. A placa é lacrada, incubada em temperatura ambiente por 60 minutos, centrifugada a 1200 rpm e depois contada durante 1 minuto usando um contador de cintilação. A atividade total é determinada pela adição de 10 pl de 5 % dimetilsulfóxido (DMSO) e a atividade não específica é determinada pela adição de 10 pl de 50 mM EDTA em vez do composto de teste.
Os compostos dos Exemplos mais abaixo possuem valores de 1C5Q abaixo de 0,5 μΜ no ensaio anteriormente mencionado. Por exemplo, os compostos dos Exemplos 1, 6, 11, 17, 22, 27, 33, 56, 67, 82, 91, 108, 120 e 133 possuem valores de IC50 de 0,075, 0,165, 0,093, 0,106, 0,050, 0,017, 0,073, 0,127, 0,016, 0,164, 0,025, 0,005, 0,008 e 0,057 respectivamente.
Tendo consideração a sua inibição de enzimas fosfatidilinositol 3-cinase, os compostos de fórmula I na forma livre ou de sal farmaceuticamente aceitável, em seguida alternativamente referidos como agentes da invenção, são úteis no tratamento das condições que são mediadas pela ativação das enzimas Pi3 cinase, particularmente as condições inflamatórias ou alérgicas. O tratamento de acordo com a invenção pode ser sintomático ou profifático.
Conseqüentemente, os agentes da invenção são úteis no tratamento de doenças inflamatórias ou obstrutivas das vias aéreas, resultando, por exemplo, na redução de dano do tecido, inflamação das vias aéreas, hiperatividade brônquica, remodelação ou progressão da doença. As doenças das vias aéreas inflamatórias ou obstrutivas as quais a presente invenção é aplicável incluem asma de qualquer tipo ou gênese incluindo tanto a asma intrínseca (não alérgica) quanto a asma extrínseca (alérgica), e asma branda, asma moderada, asma severa, asma brônquica, asma induzida por exercício, asma ocupacional e asma induzida seguinte a infecção bacteriana. O tratamento de asma deve ser também entendido como tratamento abrangente de indivíduos, por exemplo, de menores do que 4 ou 5 anos de idade, apresentando sintomas de respiração difícil e diagnosticado ou diagnosticável como crianças ofegantes, uma categoria de paciente estabelecida de solicitude médica principal e agora frequentemente identificada como asmáticos incipientes ou de fase prematura. (Por conveniência esta condição asmática particular é referida como síndrome da criança ofegante).
A eficácia profifãtica no tratamento de asma será evidenciada pela freqüência reduzida ou gravidade do ataque sintomático, por exemplo, ataque asmático agudo ou broncoconstritor, melhora na função pulmonar ou hiperatividade das vias aéreas melhorada. Pode ainda ser evidenciada pela necessidade reduzida para outras terapias sintomáticas, isto é, terapia para ou destinada a restringir ou abortar o ataque sintomático quando ele ocorre, por exemplo, antiinflamatória (por exemplo, corticosteróide) ou broncodilatadora. O benefício profilático na asma pode em particular ser evidente em indivíduos propensos ao mergulho matinal”. O mergulho matinal é uma síndrome asmática reconhecida, comum a uma porcentagem substancial de asmáticos e caracterizado pelo ataque de asma, por exemplo, entre as horas de cerca de 4 a 6 am, isto é, em um momento normal substancialmente distante de qualquer terapia de asma sintomática anteriormente administrada.
Outras doenças e condições das vias aéreas inflamatórias ou obstrutivas as quais a presente invenção é aplicável incluem lesão pulmonar aguda (ALI), síndrome da angústia respiratória do adulto/aguda (ARDS), doença pulmonar, das vias aéreas ou do pulmão obstrutiva crônica (COPD, COAD ou COLD), incluindo a bronquite crônica ou dispinéia associada com ela, enfisema, assim como exarcebação da hiperreatividade das vias aéreas consequente a outra terapia de medicamento, em particular outra terapia a medicamento inalada. A invenção é também aplicável ao tratamento de . bronquite de qualquer tipo ou gênese incluindo, por exemplo, a bronquite aguda, araquídica, catarral, crupal, crônica ou ftinóide. Outras doenças das vias aéreas inflamatórias ou obstrutivas as quais a presente invenção é aplicável incluem pneumoconiose (uma doença inflamatória, comumente ocupacional, dos pulmões, freqüentemente acompanhada pela obstrução das vias aéreas, quer crônica quer aguda, e ocasionada pela inalação repetida de poeiras) de qualquer tipo ou gênese, incluindo, por exemplo, aluminose, antracose, asbestose, calicose, ptilose, siderose, silícose, tabacose e bissinose.
Tendo consideração a sua atividade antiinflamatória, em particular em relação à inibição da ativação eosinófila, os agentes da invenção são também úteis no tratamento de distúrbios relacionados com eosinófilo, por exemplo, eosinofilia, em particular os distúrbios relacionados com eosinófilo das vias aéreas (por exemplo, envolvendo a infiltração eosinofílica mórbida dos tecidos pulmonares) incluindo hipereosinifilia quando afeta as vias aéreas e/ou pulmões, assim como, por exemplo, os distúrbios relacionados com eosinófilo das vias aéreas consequente ou concomitante à síndrome de Lòffler, pneumonia eosinofílica, infestação parasítica (em particular metazoárío, incluindo eosinofilia tropical), aspergilose broncopulmonar, poliarterite nodosa (incluindo a síndrome de Churg-Strauss), granuloma eosinofílica e distúrbios relacionados com eosinófilo que afetam as vias aéreas ocasionados pela reação ao medicamento.
Os agentes da invenção são também úteis no tratamento de condições inflamatórias ou alérgicas da peie, por exemplo, psoríase, dermatite de contato, dermatite atópica, alopecia em áreas, eritema multiforme, dermatite herpetiforme, escleroderma, vitiligo, angiite por hípersensibilidade, urticária, penfigóide bolhosa, lúpus eritematoso, pênfigo, epidermólise bolhosa adquirida, e outras condições inflamatórias ou alérgicas da pele, O tratamento de acordo com a invenção pode ser sintomática ou profilática.
Os agentes da invenção podem também ser usados para o tratamento de outras doenças ou condições, em particular doenças ou condições tendo um componente inflamatório, por exemplo, tratamento de doenças e condições dos olhos tais como conjuntivite, ceratoconjuntivite seca e conjuntivite primaveril, doenças que afetam o nariz incluindo rinite alérgica, e doença inflamatória em que as reações auto-imunes são implicadas ou tendo um componente auto-imune ou etiologia, incluindo os distúrbios hematológicos auto-imunes (por exemplo, anemia hemolítica, anemia aplásica, anemia pura de hemácias e trombocitopenia idiopática), lúpus sistêmico eritematoso, policondrite, esclerodoma, granulomatose de Wegener, dermatomiosite, hepatite ativa crônica, miastenia grave, síndrome de Steven-Johnson, espru idiopática, doença intestinal inflamatória autoimune (por exemplo, colite ulcerativa e doença de Crohn), oftalmopatia endócrina, doença de Grave, sarcoidose, alveolite, pneumonite por hipersensibilidade crônica, esclerose múltipla, cirrose biliar primária, uveíte (anterior e posterior), ceratoconjuntivite seca e ceratoconjuntivite primaveril, fibrose pulmonar intersticial, artrite psoriática e glomerulonefrite (com ou semsíndrome nefrótica, por exemplo, incluindo síndrome nefrótica idiopática ou nefropatiade mudança minai).
Outras doenças ou condições que podem ser tratadas com agentes da invenção incluem choque séptico, artrite reumatóide, osteoartrite, doenças proliferativas tais como câncer, aterosclerose, rejeição a aloenxerto seguinte a transplante, acidente vascular cerebral, obesidade, restenose, diabetes, por exemplo, diabetes melito tipo I (diabetes juvenil) e diabetes melito tipo II, doenças diarréicas, lesão isquêmica/reperfusão, retinopatia, tal como retinopatia diabética ou retinopatia induzida por oxigênio hiperbárico, e condições caracterizadas por pressão intraocular elevada ou secreção do humor aquoso ocular, tal como glaucoma.
A eficácia de um agente da invenção na inibição das condições inflamatórias, por exemplo, nas doenças aéreas inflamatórias, pode ser demonstrada em um modelo animal, por exemplo, um modelo de camundongo ou rato, de inflamação das vias aéreas ou outras condições inflamatórias, por exemplo, como descrito por Szarka et al, J. Immunol. Methods (1997) 202:49-57; Renzi et al, Am. Rev. Respir. Dis. (1993) 148:932-939; Tsuyuki et al., J. Clin. Invest. (1995) 96:2924-2931; e Cernadas et al (1999) Am. J. Respir. Cell Mol. Biol. 20:1-8.
Os agentes da invenção são úteis na fabricação de um medicamento para o tratamento de uma doença mediada por fosfatidilinositol 3-cinase. Mais especificamente os agentes da invenção são úteis na fabricação de um medicamento para o tratamento de doenças respiratórias, alergias, artrite reumatóide, aosteoartrite, distúrbios reumáticos, psoríase, colite ulcerativa, doença de Crohn, choque séptico, distúrbios proliferativos tais como câncer, aterosclerose, rejeição a aloenxerto seguinte a transplante, diabetes, acidente vascular cerebral, obesidade ou restenose. O tratamento de acordo com a invenção pode ser sintomático ou profilátíco.
Os agentes da invenção são também úteis como agentes coterapêuticos para uso em combinação com outras substâncias medicamentosas tais como substâncias medicamentosas antiinflamatórias, broncodilatadoras ou anti-histaminicos, particularmente no tratamento de doenças das vias aéreas obstrutivas ou inflamatórias tais como aquelas mencionadas mais acima, por exemplo, como potencializadores da atividade terapêutica de tais medicamentos ou como um meio de reduzir a dosagem requerida ou efeitos colaterais potenciais de tais medicamentos. Um agente da invenção pode ser misturado com a outra substância medicamentosa em uma composição farmacêutica fixa ou pode ser administrado separadamente, antes, simultaneamente ou após a outra substância medicamentosa. Conseqüentemente a invenção inclui uma combinação de um agente da invenção como mais acima descrito com uma substância medicamentosa antiinflamatória, broncodilatadora, anti-histamínica, antitussiva, dito agente da invenção e dita substância medicamentosa estando na mesma ou diferente composição farmacêutica.
Tais medicamentos antiinfiamatórios incluem esteróides, em particular glicocorticosteróides tais como glicocorticosteróides tais como budesonídeo, dipropionato de beclametasona, propionato de fluticasona, ciclesonídeo ou furoato de mometasona, ou os esteróides descritos na WO 02/88167, WO 02/12266, WO 02/100879, WO 02/00679 (especialmente aqueles dos Exemplos 3,11, 14, 17, 19, 26, 34, 37, 39, 51, 60, 67, 72, 73, 90, 99 e 101), WO 03/035668, WO 03/048181, WO 03/062259, WO 03/064445, WO 03/072592, agonistas receptores da glicocorticóide não esteroidal tais como aqueles descritos nas WO 00/00531, WO 02/10143, WO 03/082280, WO 03/082787, WO 03/104195, WO 04/005229; antagonistas LTB4 tais como aqueles descritos na US 5451700; antagonistas LTD4 tais como montelukast e zafirlukast; inibidores PDE4 tais como cilomilast (Ariflo® GlaxoSmitKline), Roflumilast (Byk Gulden),V33
11294Α (Napp), BAY19-8004 (Bayer), SCH-351591 (Schering-Plough), Arofylline (Almirall Prodesfarma), PD189659 (Parke-Davis), AWD-12-281 (Asta Medica), CDC-801 (Celgene), SelCID(TM) CC-10004 (Celgene), KW4490 (Kyowa Hakco Kogyo), WO 03/104204, WO 03/104205, WO
04/000814, WO 04/000839 e WO 04005258 (Merck), assim como aqueles descritos nas WO 98/18796 e WO 03/39544; agonistas A2a tais como aqueles descritos na EP 1052264, EP 1241176, EP 409595A2, WO 94/17090, WO 96/02543, WO 96/02553, WO 98/28319, WO 99/24449, WO
99/24450, WO 99/24451, WO 99/38877, WO 99/41267, WO 99/67263, WO
99/67264, WO 99/67265, WO 99/67266, WO 00/23457, WO 00/77018, WO
00/78774, WO 01/23399, WO 01/27130, WO 01/27131, WO 01/60835, WO
01/94368, WO 02/00676, WO 02/22630, WO 02/96462, e WO 03/086408; antagonistas A2b tais como aqueles descritos na WO 02/42298; e agonistas beta-2 adrenoceptor tais como albuterol (salbutamol), metaproterenol, terbutaline, salmeterol, fenoterol, procaterof, e especialmente, formoterol e sais farmaceuticamente aceitáveis destes, e compostos (na forma livre ou de sal ou de solvato), de fórmula I da WO 00/75114, cujo documento é aqui incorporado por referência, preferivelmente os compostos dos Exemplos

e sais farmaceuticamente aceitáveis deste, assim como compostos (na forma livre ou de sal ou de solvato) de fórmula I da WO 04/16601.
Tais medicamentos broncodilatadores incluem agentes antocolinérgicos ou antimuscarínicos, em particular brometo de ipratrópio, brometo de oxitrópio, sais de tiotrópio e CHF 4226 (Chiesi), mas também aqueles descritos nas WO 01/04118, WO 02/51841, WO 02/53564, WO 03/00840, WO 03/87094, WO 04/05285, WO 02/00652, WO 03/53966, EP 424021, US 5171744, US 3714357 e WO 03/33495.
Tais substâncias medicamentosas de anti-histamina coterapêuticas incluem cloridrato de cetirizina, acetaminofen, fumarato de clemastina, prometazina, loratidina, desioratidina, difenidramina e cloridrato de fexofenadina.
As combinações de agentes da invenção e esteroides, beta-2 agonistas, inibidores PDE4 ou antagonistas LTD4 podem ser usadas, por exemplo, no tratamento de COPD ou, particularmente, asma. As combinações de agentes da invenção e agentes anticolinérgicos ou antimuscarínicos, inibidores PDE4, agonistas receptores de dopamina ou antagonistas LTB4 podem ser usadas, por exemplo, no tratamento de asma ou, particularmente, COPD.
Outras combinações úteis de agentes da invenção com medicamentos antiinflamatórios são aquelas com antagonistas de receptores da quimiocina, por exemplo, CCR-1, CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 e CCR10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, particularmente antagonistas CCR-5 tais como antagonistas Schering-Plough SC-351125, SCH-55700 e SCH-D, antagonistas Takeda tais como cloreto de N-[[4-[[[6,7-dihidro-2-(4metilfenila)-5H-benzo-cíclohepten-8-ilaIcarbonila]amino]feníla]metil]tetrahidro-N,N-dimetií-2H-piran-4-amínio (TAK-770), e antagonistas CCR-5 descritos na US 6166037 (particularmente reivindicações 18 e 19), WO 0066558 (particularmente reivindicação 8) e WO 0066559 (particularmente reivindicação 9).
Os agentes da invenção podem ser administrados por qualquer via apropriada, por exemplo, oralmente, por exemplo, na forma de um tablete ou cápsula; parenteralmente, por exemplo, intravenosamente; por inalação, por exemplo, no tratamento de doença das vias aéreas inflamatória ou obstrutiva; intranasalmente, por exemplo, no tratamento de riníte alérgica; topicamente na pele, por exemplo, no tratamento de dermatite atópica; ou retalmente, por exemplo, no tratamento de doença intestinal inflamatória.
A presente invenção também fornece uma composição farmacêutica que compreende um composto de fórmula I na forma livre ou na forma de um sal farmaceuticamente aceitável, opcionalmente junto com um diluente ou portador farmaceuticamente aceitável para este. A composição pode conter um agente co-terapêutico tal como um medicamento antiinflamatório, broncodilatador ou anti-hrstamínico como mais acima descrito. Tais composições podem ser preparadas usando diiuentes ou excipientes convencionais e técnicas conhecidas no ofício galènico. Desta maneira, as formas de dosagem oral podem incluir comprimidos e cápsulas. As formulações para a administração tópica podem tomar a forma de cremes, ungüentos, géis ou sistemas de liberação transdérmicos, por exemplo, emplastros. As composições para inalação podem compreender aerossol ou outras formulações atomizáveis ou formulações em pó seco.
Quando a composição compreende uma formulação de aerossol, ela preferivelmente contém, por exemplo, um propulsor de hidroflúor-alcano (HFA) tal como HFA134a ou HFA227 ou uma mistura destes, e pode conter um ou mais co-solventes conhecidos na técnica tais como etanol (até 20 % em peso), e/ou um ou mais tensoativos tais como ácido oiéico ou trioleato de sorbitano, e/ou um ou mais agentes de volume tais como lactose. Quando a composição compreende uma formulação de pó seco, preferivelmente contém, por exemplo, o composto de fórmula I tendo um diâmetro de partícula de até 10 mícrons, opcionalmente junto com um diluente ou portador, tal como lactose, da distribuição de tamanho de partícula desejada e um composto que ajuda a proteger contra a deterioração do desempenho do produto devido a umidade. Quando a composição compreende uma formulação nebulosa, preferivelmente contém, por exemplo, o composto de fórmula I ou dissolvido, ou colocado em suspensão, em um veículo contendo água, um co-solvente tal como etanol ou propileno glicol e um estabilizante, que pode ser um tensoativo.
A invenção inclui (A), um agente da invenção na forma inalável, por exemplo, em um aerossol ou outra composição pulverizável ou em partículado inalável, por exemplo, forma micronizada, (B) um medicamento inalável que compreende um agente da invenção na forma inalável; (C) um produto farmacêutico que compreende um tai agente da invenção na forma inalável em associação com um dispositivo de inalação; e (D) um dispositivo de inalação contendo um agente da invenção na forma inalável.
As dosagens de agentes da invenção empregadas na prática da presente invenção naturalmente variará dependendo, por exemplo, da condição particular a ser tratada, o efeito desejado e o modo de administração. Em geral, as dosagens diárias adequadas para a administração oral são da ordem de 0,1 a 10 mg/kg.
EXEMPLOS
Os compostos especíalmente preferidos de fórmula I incluem compostos fórmula XXI

onde R1, Y, Ra e Rb são como mostrados na Tabela 1 abaixo, os métodos de preparação sendo descritos em seguida. A tabela também mostra dos dados de espectrometria de massa. Os exemplos estão na forma livre. TABELA I
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 1 | Ό N | c | H | H | 269,9 |
| 2 | T; N | c | Cl | H | 304,0 |
| 3 | Ό N | c | I NH VH | H | 342,7 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 4 | Y3 N | C | ,NH ,CH3 | H | 397,9 |
| 5 | Y: N | C | 0 | H | 355,0 |
| 6 | Y: N | C | ό | H | 336,2 |
| 7 | Ό N | C | Cl | H | 303,0 |
| 8 | Ν—N H | C | Cl | H | 292,1 |
| 9 | 0 II / XCH, | C | Cl | H | 268 |
| 10 | N-N H | C | ό | H | 324,2 |
| 11 | N-N H | C | 0 | H | 343,2 |
| 12 | T. N | c | Y | H | 354 |
| 13 | 0 II x CH3 | c | ò | H | 319,2 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 14 | 0 Â 0 H,C^ | c | H | 420,3 | |
| 15 | Q o o- -½. | c | 0 | H | 434,3 |
| 16 | 0 II /cv X NH ) I ii ch3 0 | c | H | H | 321 |
| 17 | 0 S NH 0 SA | c | H | H | 335,2 |
| 18 | 0 II X NH I ch3 | c | H | H | 192,1 |
| 19 | 0 II /A / ch.. | c | 1 o n; io | -ch3 | 262,2 |
| 20 | íí ^CH3 /CxNH oA AV | c | -CH3 | -ch3 | 386,1 |
| 21 | 0 II X NH CH, Si | c | H | H | 363,2 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 22 | 0 Λ |H í? lzCH3 n;c ca, | C | H | H | 328,1 |
| 23 | II Γ- C4 /CxNH Ο’-'λ | C | H | H | 358,1 |
| 24 | 0 II /C'NH J) I ch3 | C | H | H | 334,1 |
| 25 | 0 II x CH3 | N | c | H | 320,1 |
| 26 | íí /ΟχΝΗ Ο-Ά | N | 0 | H | 444,1 |
| 27 | 0 II x CH3 | N | Á | H | 275,1 |
| 28 | 0 II x CH3 | N | ! H3C/CxCH3 | H | 277,1 |
| 29 | 0 II / cn3 | N | -ch3 | H | 249,1 |
| 30 | 0 II x CH3 | N | ά | H | 312,1 |
Preparação de certos materiais de partida
2-(5-Etil-oxazol-2-i])-etilamina)
a) Éster benzílico de ácido [2-(2-Hidróxi“butilcarbamoil)-etil]-carbâmico:
Uma mistura que compreende Z-Beta-Ala-OH (9,0 g, 40,3 mmoles), EDCI.HCI (10,0 g, 52,4 mmoies), hidroxibenzotriazoi (5,45 g, 40,3 mmoles), trietilamina (7,3 ml, 52,4 mmoles) em DCM (150 ml) é agitada a 0° C por 30 minutos. 1-amino-2-butanol (4,2 ml, 44,3 mmoles) é adicionado de uma vez e a agitação contínua por 1 hora. A mistura de reação é diluída com água (150 m!) e extraída com diclorometano (2 x 150 ml). As camadas orgânicas são combinadas, secadas por MgSO4, filtradas e concentradas in vacuo para produzir um sólido branco bruto. O produto é purificado por cromatografia em sílica eluindo com acetato de etanol-etila (1:10) para dar o composto do título.
b) Éster benzílico de ácido [2-(2-Oxo-butilcarbamoil)-etil]-carbâmico:
A uma solução agitada de cloreto de oxalila (2M em DCM) (13,35 ml, 26,5 mmoles) em DCM seco a -78°C é adicionado por gotejamento DMSO (2,5 ml, 35,4 mmoles). Após agitação por 15 minutos, a mistura de reação é tratada com uma solução de éster benzílico de ácido [2(2-Hidróxi-butilcarbamoil)-etil]-carbâmico (etapa 1) (6,5 g, 22,1 moles) em DCM seco (40 ml). Trietilamina (13 ml) é adicionada após 1 hora e após agitação a -78° C por 90 minutos, a mistura de reação é deixada se aquecer para a temperatura ambiente. A reação é diluída com DCM (100 ml) e lavada com HCI (1 M, 200 ml), solução saturada de bicarbonato de sódio (200 ml), água (200 ml) e salmoura (200 ml). A parte orgânica é secada por MgSO4, filtrada e concentrada in vacuo para produzir o composto do título como um sólido branco.
c) Éster benzílico de ácido [2-(5-Etil-oxazol-2-il)-etil]-carbâmico:
A uma suspensão agitada de trifenilfosfeno sustentado por polímero (19,6g, 58,9 mmoles) em DCM (250 ml) é adicionado iodo (14,95 g, 58,9 mmoles). Após agitação na temperatura ambiente por 10 minutos, a mistura é tratada com trietilamina (16,4 ml, 117,5 mmoles) seguido por uma solução de éster benzílico de ácido [2-(2-Oxo-butilcarbamoil)-etil]-carbâmico (Etapa 2) (6,88 g, 23,5 mmoles) em DCM (50 ml). A mistura de reação é agitada durante a noite e depois filtrada através de material de filtro Celite®, lavada completamente com DCM (500 ml) e o solvente removido in vacuo para produzir o composto do titulo como um sólido marrom.
d) 2-(5-Etil-oxazol-2-il)-etílamina (sal de cloridrato):
Uma solução de éster benzílico de ácido [2-(5-Etil-oxazol-2-il)etil]-carbâmico (etapa 3) (0,41 g, 1,49 mmol), 2M HCI (0,75 ml) em etanol (40 ml) é agitada sob hidrogênio na presença de 10 % Pd em Carbono (0,041 g) por 5 horas. A mistura de reação é filtrada e concentrada in vacuo para produzir o composto do título. Este é neutralizado usando trietiiamina para produzir 2-(5-etil-oxazol-2-il)-etilamina.
3-Amino-N,N-dimetil-propionamida
a) 3-Amino-N,N-dimetil-propionamida:
Éster benzílico de ácido (2-Dimetilcarbamoil-etil)-carbâmico:
A uma solução esfriada (0 0 C) e agitada de Z-Beta-Ala-OH (1,784 g, 8,0 mmoles) em dioxano (20 ml) é adicionado EDCI.HCI (2,145 g, 11,2 mmoles), hidroxibenzotriazol (1,08 g, 8,0 mmoles) e trietiiamina (1,56 ml, 11,2 mmoles). Após agitação a 0 0 C por 30 minutos, dimetilamina (0,397 g, 8,8 mmoles) é adicionada e a agitação continuou por mais uma hora. A mistura de reação foi deixada se aquecer para a temperatura ambiente e o solvente é removido in vacuo. A purificação inicial do resíduo bruto é realizada por cromatografia instantênea de sílica eluindo com acetato de etila. As frações orgânicas combinadas são lavadas com água (3 x 20 ml), salmoura (1 x 50 ml) e secadas por MgSO4. O solvente é removido in vacuo para produzir o composto do título como um sólido amarelo claro.
b) 3-Amino-N,N-dimetil-propionamida:
A uma solução de éster benzílico de ácido (2-dimetilcarbamoiletil)-carbâmico (0,9 g, 3,6 mmoles) em etanol (35 ml) é adicionado 10 % Pd em Carbono (0,09 g). A solução foi agitada na presença de um fluxo constante de hidrogênio [em série com um hidróxido de sódio (4N) purificado], A mistura de reação é filtrada e concentrada in vacuo para produzir o composto do título como um óleo amarelo claro.
Preparação dos Exemplos Específicos
Exemplo 1 (4-Metil'5-piridin-4-il-tiazol-2-il)-pirazin-2-il-amina
1a) Pirazin-2-il-tiouréia:
Aminopirazina (2 g, 21,03 mmoles) é dissolvida em etanol (20 ml) e benzoilisotiocianato (2,82 ml) é adicionado por gotejamento. A mistura é aquecida para 80° Ccom agitação por 10 minutos depois deixada esfriar para a temperatura ambiente. O solvente é removido in vacuo e o sólido resultante dissolvido em 1M hidróxido de sódio (30 ml) e aquecido sob refluxo por 1 hora. A suspensão resultante é filtrada e o sólido lavado com água e um pouco metanol gelado. O sólido é secado in vacuo para produzir o composto do título, p.f. 239 - 239,5°C, MH+ (AP+): 138 (M+- NH3). Outras tiouréias usadas são ou comercialmente disponíveis ou preparadas em uma maneira análoga a partir da amina de partida apropriada.
b) (4-Metil-5-pirid in-4-il-tiazol-2-il)-pirazin-2-il-amina:
A uma solução agitada de 1-piridin-4-il-propan-2-ona (0,1 g, 0,812 mmol) em dioxano (7 ml) é adicionado bromo (0,029 ml, 0,569 mmol) por gotejamento. Após 45 minutos, o solvente é removido in vacuo e o produto bruto é dissolvido em etanol (15 ml). A solução é tratada com pirazin-2-il-tiouréia (0,125 g, 0,812 mmol) e a mistura de reação é aquecida a 60° C por 3 horas. O solvente é removido in vacuo e purificação por cromatografia em sílica, eiuindo com acetato de etil-metanol proporciona o composto do título.
Exemplo 2 [5-(2-Cloro-piridin-4-il)-4-metil-tiazol-2-ila]-pirazin-2-il-amina
2a) 1 -(2-C!oro-piridin-4'i l)-propan-2-ona:
A uma solução agitada de diisopropilamina (14,22 ml, 101,43 mmoles) esfriada para -78°C é adicionado por gotejamento butillítio (60 ml). A mistura de reação é agitada e deixada se aquecer para 0°C por 15 minutos após cujo tempo 2-cloro-4-metilpiridina é adicionada à solução agitada. Após 1 hora, acetato de etila (18,88 ml) e THF (78 ml) são adicionados durante 45 minutos seguido por ácido acético (11,4 ml, 193,2 mmoles) e a agitação continuou por 20 minutos. A mistura de reação é concentrada in vacuo e o produto bruto dissolvido em com água (200 ml). O aquoso é extraído com clorofórmio (3 x 300 ml) e as partes orgânicas combinadas lavadas com água (200 ml), salmoura (200 ml), secadas por MgSO4 e o solvente removido in vacuo para produzir o produto bruto como um óleo marrom. Purificação por cromatografia em sílica, eluindo com acetato de etil-hexano proporciona o composto do título.
2b) [5-{2-Cloro-piridin-4-il)-4-metil-tiazol-2-iia]-pirazin-2-il“amina:
O composto do título é preparado por um procedimento análogo à (4-Metil-5-piridin-4-il-tiazol-2-il)-pirazin-2-il-amina (Exemplo 1) mediante a substituição 1-piridin-4-il-propan-2-ona (1b) neste procedimento com 1-(2cloro-piridin-4-il)-propan-2-ona.
Exemplo 3
3-{4-[4-Metil-2-(pirazin-2-ilamino)-tiazol-5-ila]-piridin-2-ilamino}-propan-1 -ol [5-(2-Cloro-piridin-4-il)-4-metil-tiazol-2-ila]-pirazin-2-il-amina (Exemplo 2b, 0,077 g, 0,254 mmol) é colocada em suspensão em 3-aminopropanol (3 ml) e aquecida para 150° C usando aquecimento de microondas (forno de microondas Prolabo synthewave® s402). Após 1 hora, a mistura de reação é concentrada in vacuo e diluída com água (50 ml). O produto aquoso é extraído com acetato de etila (2 x 100 ml). O extrato orgânico combinado é secado (MgSO4) e concentrado para proporcionar o composto do título que é purificado por filtração e recristalizado a partir de diclorometano.
Exempío 4
N.N-Dietil-N^-^-metil^-Ípirazin^-ilaminoj-tiazol-S-ilaj-piridin^-ila}propano-1,3-diamina
O composto do título é preparado seguindo o mesmo caminho como o Exemplo 3 mediante a substituição de 3-amino-propanol com N,Ndietilpropilamina.
Exemplo 5 [4-Metil-5-(2-morfolin-4-il-piridin-4-i!)-tiazol-2-ila]-pirazin-2-il-amina
Este composto é preparado seguindo o mesmo caminho como o
Exemplo 3 mediante a substituição de 3-amino-propanol com morfolina. Exemplo 6 [5-(2-lmidazol-1-il-piridin-4-il)-4-metil-tiazol-2-ila]-pirazin-2-il-amina
Uma solução agitada de [5-(2-Cloro-piridin-4-il)-4-metil-tiazoí-2ila]-pirazin-2-il-amína (Ex. 2b, 0,4 g, 1,32 mmol) em DMSO (10 ml) é tratada com imidazol (0,18 g, 2,64 mmoies) seguido por carbonato de césio (0,86 g, 2,64 mmoies). A mistura de reação é aquecida para 140° C por 48 horas e depois deixada esfriar para a temperatura ambiente. A mistura é diluída com água (100 ml) e extraída com acetato de etila (4 x 100 ml). Os orgânicos são combinados, secados por MgSO4 e o solvente removido in vacuo para produzir o produto bruto como um óleo amarelo. Purificação por cromatografia em sílica, eluindo com acetato de etil-metanol (9:1) proporciona o composto do título.
Exemplos 7 a 9
Estes compostos, a saber, [5-(2-Cloro-piridin-4-il)-4-metil-tiazol2-ila]-piridin-3-il-amina, [5-(2-Cloro-piridin-4-il)-4-metil-tiazol-2-ila]-(1 Hpirazol-3-il)-amina e N-[5'(2-Cloro-piridin-4-il)-4-metil-tiazol-2-íla]-acetamida respectivamente, são preparados usando o procedimento de Exemplo 2 a partir de 1-(2-Cloro-piridin-4-il)-propan-2-ona e a tiouréia apropriada.
Exemplo 10 [5-(2-lmidazol-1-il-piridin-4-ÍI)-4-metil-tiazol-2-ila]-(1Hpirazol-3-il)-amina
Este composto é preparadoo seguindo o mesmo caminho como o Exemplo 6 mediante a substituição de [5-(2-Cloro-piridin-4-il)-4-metil-tiazol2-ila]-pirazin-2-il-amina (Exemplo 2) com [5-(2-Cloro-piridin-4-il)-4-metiltiazol-2-ila]-(1 H-pirazol-3-il)-amina (Exemplo 8).
Exemplo 11 [4-Metii-5-(2-morfolin-4-il-piridin-4-il)-tiazol-2-ila]-(1 H-pirazol-3-il)-amina
Este composto é preparado seguindo o mesmo caminho como o Exemplo 5 mediante a substituição de [5-(2-Cloro-piridin-4-il)-4-metil-tiazol“2ila]-pirazin-2-ií-amina (Exemplo 2) com [5-(2-Cloro-piridin-4-il)-4-metil-tiazol2-ila]-(1H-pirazol-3-il)-amina (Exemplo 8).
Exemplo 12 [4-Metil-5-(2-morfolin-4-il-piridin-4-il)-tiazol-2-ila]-piridin-3-il-amina
Este composto é preparado seguindo o mesmo caminho como o Exemplo 5 mediante a substituição de [5-(2-Cloro-piridin-4-il)-4-metil-tiazol-2ila]-pirazin-2-il-amina (Exemplo 2) com [5-(2-Cloro-piridin-4-il)-4-metil-tiazol2-ila]-piridin-3-il-amina (Exemplo 7).
Exemplo 13
N-[4-Metil-5-(2-morfolin-4-il-piridin-4-il)-tiazol-2-ila]-acetamida
13a) 1-(2-Morfolin-4-il-piridin-4-il)-propan-2“Ona:
Uma solução agitada de 1-(2-cloro-piridin-4-il)-propan-2-ona (1,6 g, 9,47 mmoles) em morfolina é aquecida para 105°C por 3 dias. A morfolina é removida in vacuo para produzir o produto bruto. Purificação por cromatografia em sílica, eluindo com 1:1 acetato de etil-hexano proporciona o composto do título.
13b) Bromidrato de 4-Metil-5-(2-morfolin-4-il“Piridin-4-il)-tiazol-2-ilamina:
O composto do título é preparado por um procedimento análogo à (4-metil-5-piridin-4-il-tiazol-2-il)-pirazin-2-il-amína (Exemplo 1) mediante a substituição 1 -piridin-4-il-propan-2-ona (1b) com 1-(2-morfolin-4-il-piridin-4il)-propan-2-ona (13a) e pirazin-2-ii-tiouréia (1a) com N-acetiltiouréia.
13c) N-[4-Metil-5-(2-morfolÍn-4-il-piridin-4-il)-tiazol-2-ila]-acetamida:
Uma suspensão agitada de bromidrato de 4-metil-5-(2-morfolin4-il-piridin-4“il)-tiazol-2-ilamina (13b) (0,035 g, 0,098 mmol) e etildiisopropilamina (0,034 ml, 0,196 mmol) em anidrido acético (2 ml) é aquecida para 75 0 C por 2 horas. A mistura de reação é esfriada para a temperatura ambiente e o solvente é removido in vacuo. Acetato de etila é adicionado e um subproduto sólido branco é extraído por filtração. O filtrado é concentrado in vacuo para proporcionar o composto do título como um sólido branco.
Exemplo 14
Éster etílico de ácido 3-{3-[4-Metil-5-(2-morfolin-4-il-piridin-4-il)-tiazol-2-ila]ureído}-propiônico
A uma suspensão agitada de bromidrato de 4-Metil-5-(2morfolin-4-il-piridin-4-il)-tiazol-2-ilamina (13b) (0,04 g, 0,112 mmol) em DCM (3 ml) é adicionado etildiisopropilamina (0,039 ml, 0,224 mmol) para proporcionar uma solução. A mistura de reação é depois tratada com 3isocianatopropionato de etila (0,015 ml, 0,112 mmol) e aquecida enquanto se agita em 60 ’ C por 5 horas em um vaso de reação lacrado. A solução é diluída com DCM e ácido clorídrico (30 ml, 1N HCI) e as camadas separadas. O produto aquoso é ajustado para pH 8/9 e extraído com acetato de etila (3 x 25 ml). Os produtos orgânicos são combinados e secados (MgSO4), filtrados e concentrados in vacuo para produzir um óleo transparente. Trituração com éter produz o composto do título como um sólido branco.
Exemplo 15
Éster etílico de ácido 4-{3-[4-Metil-5-(2-morfolin-4-il-piridín-4-il)-tiazol-2-ila]ureídoj-butírico
O composto do título é preparado por um procedimento análogo ao éster etílico de ácido 3-{3-[4-Metil-5-(2-morfolin-4-il-piridin-4-il)~tiazol-2ila]-ureído}-propiônico (Exemplo 14) mediante a substituição de 3isocianatopropionato de etila com éster etílico de ácido 4-isocianato-butírico. Exemplo 16
Éster etílico de ácido [3-(4-MetÍI-5-piridin-4-il-tiazol-2-il)-ureído]“acético
Uma solução agitada de 4-metil-5-piridin-4-il-tiazol-2-ilamina (preparada de acordo com o método descrito no relatório descritivo da patente Européia EP 117082 A2) (0,068 g, 3,56 mmoles) em DMF (5 ml) é tratada com isocianatoacetato de etila (0,043 ml, 3,916 mmoles) e a solução é agitada durante a noite. O solvente é removido in vacuo e o produto bruto é dividido entre acetato de etila (50 ml) e água (50 ml). As camadas são separadas e a parte aquosa é extraída com acetato de etila (2 x 50 ml). As partes orgânicas combinadas são lavadas com água, secadas (MgSO4), filtradas e concentradas in vacuo para produzir um óleo. Trituração com acetato de etila produz o composto do título.
Exemplo 17
Éster etílico de ácido 3-[3-(4-Metil-5-piridin-4-il-tiazol-2-il)-ureído]-propiônico
Este composto é preparado por um procedimento análogo ao éster etílico de ácido 3-{3-[4-Metil-5-(2-morfolin-4-il-pindin-4-il)-tiazol-2-ila]ureídoj-propiônico (Exemplo 14) mediante a substituição de bromidrato de 4Metil-5-(2-morfolin-4-il-piridin-4-il)-tiazol-2-ilamina (13b) com 4-metii-5-piridin4-il-tiazo!-2-ilamina (ver Exemplo 16 para referência).
Exemplo 18
1- Metil-3-(4-metil-5-piridin-4-il-tiazol-2-il)-uréia
18a) (4-metil-5-piridin-4-il-tiazol-2-il)-amida de ácido imidazol-1-carboxílico:
A uma solução agitada de carbonildiimidazol (3,91 g, 24,2 mmoles) em DCM (150 ml) é adicionado 4-metil-5-piridin-4-il-tiazol-2-ilamina (preparada de acordo com o método descrito em EP 117082 A2) (3,08 g, 16,1 mmoles) de uma vez. A suspensão é agitada por2,5 horas a 40° C e a mistura de reação é depois filtrada e lavada com DCM para proporcionar o composto do título como um sólido.
18b) 1-Metií-3-(4-metil-5-piridin-4-il-tiazol-2-il)-uréia:
Metilamina (0,030 ml de um 40 % p/p/ solução em água, 0,351 mmol) é adicionada a uma suspensão agitada de (4-metil-5-piridin-4-il-tiazol2- il)-amida de ácido imidazol-1 -carboxílico (18a) (0,1 g, 0,351 mmol) em DMF (3 ml). A mistura de reação é agitada em temperatura ambiente por 1 hora e depois o solvente é removido in vacuo. O produto bruto é dissolvido em THF/DMF (10:1, 3 ml) e passado através de uma resina de isocianato sustentada por polímero (0,9 g, 1,1 mmol/g de carga) e lavado completamente com THF. A solução é concentrada in vacuo e o resíduo lavado com acetato de etila e metanol para proporcionar o composto do título.
Exemplo 19
N-[5-(2,6-Dimetil-piridin-4-il)-4“metil-tiazol-2-ila]-acetamida
O composto do título é preparado por um procedimento análogo à (4-Metil“5-piridin-4-il-tiazol-2-il)-pirazin-2-il-amina (Exemplo 1) mediante a substituição de 1 -piridin-4-il-propan-2-ona (1b) neste procedimento com 1(2,6-dimetii-piridin-4-il)-propan-2-ona e mediante a substituição de pirazin-2il-tiouréia (1a) com N-acetiltiouréia. 1-(2,6-dimetil-piridin-4-il)-propan-2-ona foi preparada de acordo com um método descrito em Tetrahedron Letters,
Voí,25, No, 5, pp 515-518,1984. (Autores: Cíaude Erre et al.)
Exemplo 20
1-[5-{2,6-Dimetil-piridin-4-ii)-4-metil-tiazol-2-ila]-3-[2-(5-etil-oxazol-2-il)-etil]uréia
20a) Bromidrato de 5-(2,6-Dimetií-piridin-4-il)-4-metil-tiazol-2-iiamina:
A uma solução agitada de 1-(2,6-dimetil-piridin-4-il)-propan-2ona (0,8 g, 4,9 mmoles) em dioxano (20 ml) esfriada para 5 a 10 0 C é adicionado por gotejamento bromo (0,25 mi, 4,9 mmoles) em DCM (1 ml). Após a adição estar completa, a mistura de reação é concentrada in vacuo e o produto bruto é dissolvido em etanol (20 ml). A solução é tratada com tiouréia (0,373 g, 49 mmoles) e a mistura de reação é aquecida a 60° C por 30 minutos. A mistura é filtrada e o precipitado amarelo é lavado com etanol, éter dietílico e secado in vacuo para produzir o composto do título.
20b) 5-(2,6-Dimetil-piridin-4-il)-4-metil-tiazol-2-ilamina:
A uma solução agitada de bromidrato de 5-(2,6-Dimetil-piridin-4il)-4-metil-tiazol-2-ilamina (3,29 g, 11 mmoles) em metanol (30 ml) é adicionado metóxido de sódio (2 ml de um 30% solução em metanol, 11 mmoles). A mistura resultante é filtrada através de material de filtro Celite™ e concentrada in vacuo para produzir o composto do título.
20c) [5-(2,6-dÍmeti!-piridin-4-il)-4-metil-tiazol-2-ila]-amida de ácido imidazol-1 carboxílico:
O composto do título é preparado por um procedimento análogo à (4-metil-5-piridin-4-il-tiazol-2-il)-amida de ácido imidazol-1-carboxílico (exemplo 18a) mediante a substituição 4-metil-5-piridin-4-il-tiazol-2-ilamina com 5-(2,6-Dimetil-pirÍdin-4-il)-4-metil-tiazol-2-ilamina.
20d) 1-[5-(2,6-Dimetil-piridin-4-il)-4-metil-tiazol-2-ila]-3-[2-(5-etil-oxazol-2-il)etilj-uréia
O composto do título é preparado por um procedimento análogo à 1-metil-3-(4-metil-5-piridin-4-il-tiazol-2-il)-uréia (exemplo 18b) mediante a substituição de (4-metil-5-piridin-4-il-tiazol-2-il)-amida de ácido imidazol-1carboxílico (exemplo 18a) com [5-(2,6-dimetil-piridin-4-il)-4-metil-tiazol-2-ila]amida de ácido imidazol-1-carboxílico e mediante a substituição metiiamina com 2-(5-etil-oxazol-2-il)-etilamina. A preparação de 2-(5-etil-oxazol-2-il)etilamina é descrita anteriormente.
Exemplos de 21 a 23
Estes compostos, a saber, 1-(4-metil-5-piridin-4-il-tiazoE-2-il)-3(1 -metil-1 H-pirrol-2-ilmetil)-uréia, éster terc-butílico de ácido 3-[3-(4-metil-5piridin-4-il-tiazol-2-il)-ureído]-propiônico e 1-[2-(5-etil-oxazol-2-il)-etil]-3-(4metil-5-piridin-4-ii-tiazol-2-il)-uréia, são preparados pelo mesmo procedimento como o exemplo 18 substituindo metilamina (parte 18b) neste exemplo com a amina apropriada.
Exemplo 24
N,N-Dimetil-3-[3-(4-metil-5-piridin-4-il-tiazol-2-il)-ureído]-propionamida
A uma suspensão agitada de (4-metil-5-piridin-4-il-tiazol-2-il)amida de ácido imidazol-1-carboxílico (exemplo 18a) (0,15 g, 0,526 mmol) em dioxano (10 ml) é adicionado 3-amino-N,N-dimetil-propionamida (0,046 g, 0,526 mmol) de uma vez. A mistura de reação é aquecida em refluxo por 2 horas. A mistura de reação é deixada esfriar para a temperatura ambiente e o solvente é removido in vacuo. Trituração com éter/acetato de etila produz o composto do título como um sólido amarelo claro.
Exemplo 25
N-[4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ila]-acetamida
a) 1-[2“(4-Metóxi-benzilamino)-4-metil-tiazol-5-ila]-etanona:
Uma suspensão agitada de 3-cloro,2,4-pentanediona (1,0 g, 7,43 mmoles) e 1-(4-metóxi benzila)-2-tiouréia (1,46 g, 7,43 mmoles) em metanol (10 ml) é tratada com piridina (0,6 ml). A mistura de reação é agitada em temperatura ambiente por 2 horas e depois o solvente é removido in vacuo. O resíduo bruto é triturado com éter para produzir o composto do título como um sólido branco.
b) (E)-3-Dimetilamino-1-[2-(4-metóxi-benzilamino)-4-metil-tiazol-5-ila]propenona:
Uma suspensão agitada de 1-[2-(4-Metóxi-benzilamino)-4-metiltiazol-5-ila]-etanona (25a) (1,0 g, 3,62 mmoles) em DMFOMA (10 ml) é aquecida para 100° C durante a noite. A mistura de reação é concentrada in vacuo para produzir um óleo marrom, que seguinte a trituração com acetato de etila proporciona o composto do título como um sólido laranja.
c) (4-Metóxi-benzila)-[4-metii-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ilajamina:
A uma suspensão agitada de (E)-3-dimetilamino-1-[2-(4-metóxibenzilamino)-4-metil-tiazol-5-ila]-propenona (25b) (0,585 g, 1,77 mmol) e bromidrato de morfolinaformamidina (0,557 g, 2,65 mmoles) em 2metoxietanol (10ml) é adicionado hidróxido de sódio (0,142 g, 3,54 mmoles). A mistura de reação é agitada e aquecida para 115° C por 12 horas. O solvente é removido in vacuo e triturado com acetato de etila para proporcionar um sólido laranja claro. Outra purificação por cromatografia em sílica eluindo com acetato de etil-hexano (1:1) produz o composto do título.
d) 4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ilamina:
Uma solução que compreende (4-metóxi-benzila)-[4-metil-5-(2morfolin-4-il-pirimidin-4-il)-tiazol-2-ila]-amina (25c) (0,45 g, 1,13 mmol) em ácido trifluoroacético : água (95:5) (10 ml) é aquecida a 75° C por dias. O solvente é removido in vacuo e o resíduo bruto é dissolvido em acetato de etila. O pH é ajustado para pH 12 usando 2N hidróxido de sódio e as camadas são separadas. A camada aquosa é extraída com acetato de etila (2 x 30 ml). As partes orgânicas são combinadas e lavadas com salmoura (50 ml), secadas por MgSO4 e concentradas in vacuo para produzir o composto do título como um sólido marrom.
e) N-[4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ila]-acetamida:
Anidrido acético (3 ml) é adicionado à 4-metil-5-(2-morfolín-4-ilpirimidin-4-il)-tiazol-2-ilamina (25d) (0,045 g, 0,162 mmol) e a mistura de reação é aquecida para 60° C por 1 hora. O solvente é removido in vacuo e o resíduo bruto é dissolvido em acetato de etila (50 ml) e água (50 ml). As camadas são separadas e a camada orgânica é lavada com solução de carbonato de sódio, salmoura, secada por MgSO4 e concentrada in vacuo para produzir um óleo marrom. O resíduo bruto é purificado por cromatografia em sílica eluindo com acetato de etil-hexano (3:2) para proporcionar o composto do título.
Exemplo 26
1- [2-(5-Etil-oxazof-2-il)-etil]-3-[4-metil-5-(2-morfolin-4-il-pirimidin-4-il)-’tiazol-2ila]-uréia
a) [4-metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ila]-amida de ácido imidazol-1-carboxílico:
O composto do título é preparado por um procedimento análogo à (4-metil-5-piridin-4-il-tiazol-2-il)-amida de ácido imidazol-1-carboxílico (exemplo 18a) mediante a substituição de 4-metil-5-piridin-4-il-tiazol-2ilamina (ver exemplo 16 para referência) com 4-metil-5-(2-morfolin-4-il· pirimidin-4-il)-tiazol-2-ilamina (25d).
b) 1-[2-(5-Etil-oxazol-2-il)-etil]-3-[4-metil-5-(2-morfolin-4-il-pirímidin-4-il)-tiazol2- ila]-uréia:
O composto do título é preparado por um procedimento análogo à 1-metil-3-(4-metil-5-piridin-4-il-tiazol-2-il)-uréia (exemplo 18b) mediante a substituição de (4-metil-5-pirÍdin-4-il-tiazol-2-il)-amida de ácido imidazol-1carboxílico (exemplo 18a) com [4-metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol2-ila]-amida de ácido imidazol-1-carboxílico (26a) e mediante a substituição de metilamina com 2-(5-etil-oxazol-2-il)-etilamina. A preparação de 2-(5-etiloxazol-2-il)-etilamina é descrita no exemplo 20d etapas de 1 a 4.
Exemplo 27
N-[5-(2-Ciclopropil-pirimidin-4-il)-4-metil-tiazol-2-ila]-acetamida
O composto do titulo é preparado por um procedimento análogo à N-[4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ila]-acetamida (25e) mediante a substituição de bromidrato de morfolinaformamidina (parte 25c) com cloridrato de ciclopropanocarboxamidina.
Exemplo 28
N-[5-(2-lsopropil-pÍrimidin-4-il)-4-metil-tiazol-2-ila]-acetamida
O composto do título é preparado por um procedimento análogo à N-[4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-i!a]-acetamida (25e) mediante a substituição bromidrato de morfolinaformamidina (parte 25c) com cloridrato de isobutiramidina.
Exemplo 29
N-[4-Metil-5-(2-metil-pirimidin-4-il)-tiazol-2-ila]-acetamida composto do título é preparado por um procedimento análogo à N“[4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ila]-acetamida (25e) mediante a substituição de bromidrato de morfolinaformamidina (parte 25c) com cloridrato de acetamidina.
Exemplo 30
N-[4-Metil-5-(2-piridin-3-il-pirimidin-4-il)-tiazol-2-ila]-acetamida
O composto do titulo é preparado por um procedimento análogo à N-[4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol“2-ila]'acetamida (25e) 10 mediante a substituição de bromidrato de morfolinaformamidina (parte 25c) com cloridrato de nicotinamidina.
Os compostos especialmente preferidos de fórmula I também incluem compostos de fórmula XXI onde R1, Y, Ra e Rb são como mostrados na Tabela 2 abaixo, os métodos de 15 preparação sendo descritos em seguida, A tabela também mostra os dados de espectrometria de massa. Os Exemplos estão na forma livre.
TABELA 2
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 31 | )? / NH 0 1 CH, | c | -ch3 | -ch3 | 362,2 |
| 32 | 0 1) CH, 1 k A Y “· 1 ° | c | -ch3 | -ch3 | 348,2 |
| 331 rCHj / NH 0' \\ YA? | c | Cl | H | 392,1 | |
| 34 § / Ah yA aO | c | Cl | H | 391,0 _ |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 35 | 0 ü O N CH, V? | c | Cl | H | 405,1 |
| 36 | H,C ° y-cH! /%4H u? | c | Cl | H | 278,0 |
| 37 | í? ^ch3 UQ* | c | Cl | H | 393,2 |
| 38 | í? <-CHs /GxNH 0\ | N | I «A | -ch3 | 419,2 |
| 39 | 0 II / xch3 | N | l HA A 3 'N 'CH, I 3 CHj | -ch3 | 349,2 |
| 40 | íí r-CH, /C'NH Ο-'Α υν | N | I h3cz% | -ch3 | 435,1 |
| 41 | íí w | N | I o=s=o I ch3 | -ch3 | 451,2 |
| 42 | í? /C'NH OA u> | N | I /N\ h3c ch3 | -ch3 | 416,2 |
| 43 | ÍÍ A^ /C'''NH OA w | N | I HA /X/A 3 N CH, l J ch3 | -ch3 | 473,3 |
| 44 | íí ach» /CxNH OA U-? | N | 6 | -ch3 | 439,9 |
| Ξχ. | R1 | Y | Ra | Rb | M/s MH+ |
| 45 | ίί r'CH'> /&ΧΝΗ Ο-λ Uv | N | rp.ÍH | -CB, | 501,2 |
| 46 | ίί /-CH3 w | N | ch3 ch3 | -ch3 | 487,3 |
| 47 | ft r-CH3 /C'NH Ο-Ά w | N | I h,,c'''nxx-xN‘ch, | -ch3 | 501,2 |
| 48 | fí r-™, /ΟχΝΗ Ο^λ | N | I h3c'%h3 | -ch3 | 473,3 |
| 49 | ÍÍ f-CH3 /CxNH O-lí w | N | I p^NH ô | -ch3 | 496,2 |
| 50 | í? r~CH3 /CxNH O-A w | N | <Άη3 h3c ch3 | -ch3 | 487,2 |
| 51 | ÍÍ f-CH3 -/C'NH Ο-Λ w | N | I H3C'^''N'X'xzNH J h3c | -ch3 | 487,2 |
| 52 | í? r-cH3 w | N | ^'CH, NH, | -ch3 | 459,2 |
| 53 | H r-CH3 Uv | N | I p^NH ΰ N ch3 | -ch3 | 528,3 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 54 | íí -''''“'NH (W w | N | -ch3 | 515,2 | |
| 55 | íí ach‘! /C'NH oAÍ | N | I .NH ÓH | -ch3 | 471,3 |
| 56 | íí aCHs /CvNH CÍ ua | N | h3A° | -ch3 | 403,2 |
| 57 | í? rCH3 | N | I pN^° <U | -ch3 | 502,2 |
| 58 | ÍÍ ach’ /%h | N | i h3c 0 | -ch3 | 434,2 |
| 59 | ÍÍ ACHa UÁ | N | h’%^ch3 I 3 ch3 | -ch3 | 472,3 |
| 60 | 0 II ΛΛ r\ CH3 UÁ | N | H3%^ch3 I 3 ch3 | -ch3 | 486,3 |
| 61 | íí V™3 UÁ | N | l 3 ch3 | -ch3 | 486,3 |
| 62 | § 3 u> | N | I c III N | -ch3 | 411,2 |
| 63 | 8 /-A /CxNH N\ UQ | N | I h3c'% | -ch3 | 436,1 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 64 | S ^Ck kA> | N | I H3C/NxCH3 | -ch3 | 417,2 |
| 65 | fí rCHí hC... 2 X NH Uk> | N | H,C,. Á ’ Y 'CH, ch3 | -ch3 | 474,2 |
| 66 | 0 CHg II I 3 0 | N | I H,ç-S | -ch3 | 381,1 |
| 67 | 9 CH, II | 3 /θχ /Á Η iT 0 | N | I H,CkS'O | -ch3 | 398,1 |
| 68 | íí 7· 0 | N | H,C,. ,/v Jk 3 CH3 CHj | -ch3 | 436,2 |
| 69 | íí r-CH3 /CxNH 0--¾ w | N | •y—L, CH, | H | 459,2 |
| 70 | í? /-CH3 kA? | N | ô | H | 482,2 |
| 71 | íí '\JL> | N | l η3Λ | H | 482,2 |
| 72 | ÍÍ ^CH3 /CxNH kJ? | N | h>%-Ahs ch3 | H | 458,3 |
| 73 | 0 II Av. x CH3 | N | °γΝΗ ch3 | H | 292,1 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 74 | 0 II x CH3 | N | I .NH h3<A | H | 264,1 |
| 75 | 0 II x A | N | I η3ΑΝΑη3 | H | 278,1 |
| 76 | 0 II x CH3 | N | Q ch3 | H | 332,1 |
| 77 | 0 II z CH3 | N | H | 311,2 | |
| 78 | 0 II x CH3 | N | I o=s=o I ch3 | H | 313,1 |
| 79 | 0 II /A z A | N | I A ch3 | H | 265,1 |
| 80 | 0 II /A / ch3 | N | ó | H | 312,1 |
| 81 | 0 II /A / ch. | N | ό | H | 312,1 |
| 82 | 0 II /C''NH 0 LA fA XC„. h3c \h3 | N | - | H | 404,2 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 83 | 8 r-CH3 /CxNH O-<\ | N | k | H | 399,2 |
| 84 | 0 II xc-, ' NH 0 lzCH3 h3c \h3 | N | I h3cx%h3 | H | 407,2 |
| 85 | 8 /ch3 | N | I h3cx\h3 | H | 402,2 |
| 86 | 0 /CNH ON | N | I H3C/NxCH3 | H | 403,2 |
| 87 | 0 II x CH3 | N | -ch3 | -ch3 | 262,3 |
| 88 | 8 /CxNH 0^ w | N | -ch3 | -ch3 | 387,1 |
| 89 | 8 f-CH’ ^Sin r\ | N | -ch3 | •ch3 | 386,2 |
| 90 | 0 ii o N CH, x NH rfA 3 u> | N | -ch3 | -ch3 | 400,3 |
| 91 | N | H,C \ CH, ch3 | H | 249,1 | |
| 92 | 0 II / xch3 | N | I /θχ H,C \ CH, ch3 | H | 291,2 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 93 | 0 0 | N | I χθχ H,C \ CH, ch3 | H | 390,5 |
| 94 | 0 CH, II I 3 /C N x/ xch3 | N | I /A H.C \ CH, ch3 | H | 334,2 |
| 95 | /C\z/\f(/CH3 0 | N | Á H,C \ CH, ch3 | H | 347,2 ί |
| 96 | 0 0 | N | I /Cx H.C \ CH, CH, | H | 361,2 |
| 97 | 0 II O-1! | N | /<k H.C \ CH, CH, | H | 414,2 |
| 98 | 0 0 | N | À H,C \ CH, CH, | H | 406,2 |
| 99 | 0 II / V '/ CH H 3 | N | ,<L H,C \ CH, ch3 | H | 378,2 |
| 100 | 0 CH, II I 3 / N^Y CH, 0 | N | I /Cx H,C \ CH, ch3 | H | 377,2 |
| 101 | 0 0 ΗΓ A/XC | N | I XCX H,C \ CH, CH3 | H | 435,2 |
| 102 | 0 KcAA CH, II 3 I I 3 H | N | /L H,C \ CH, ch3 | H | 422,2 |
| Ex. | R1 | Y | Ra | Rb | M/s | |
| MH+ | ||||||
| 103 | 0 0 À-xA H | O^CH, | N | ZCX H,C \ CH, ch3 | H | 377,2 |
| 104 | 0 / 'N X H | 'OH | N | I xcx H,C \ CH, ch3 | H | 350,2 |
| 105 | 0 | N | l -C. H.C \ CH, CHS | H | 383,2 | |
| 106 | 0 II | -^N J | N | Á H.C \ CH, ch3 | H | 383,2 |
| 107 | 0 II | A x^N | N | I /cx H,C \ CH, ch3 | H | 383,2 |
| 108 | 0 II /'x/ H | Οχ ch3 | N | /Οχ H,C \ CH, ch3 | H | 350,2 |
| 109 | 0 II ζ \ H | -OH | N | I /Οχ H.C \ CH, ch3 | H | 336,2 |
| 110 | 0 II Cx /χ / / N X/ H | x-OH | N | /Οχ H,C \ CH, ch3 | H | 364,2 |
| 111 | 0 II H | CH. I 3 ,NX ch3 | N | /Οχ H.C \ CH, ch3 | H | 363,2 |
| 112 | 0 II H | 0 | N | I /Οχ H,C \ CH, ch3 | H | 389,2 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 113 | s Π H | N | 1 /<k H,C \ CH, ch3 | H | 403,2 |
| 114 | 0 II H 0 | N | f xcx H.C \ CH, CH, 3 | H | 419,2 |
| 115 | /CH, 0 ó II I X. /-.., -Nx .CH, / V '-z 3 H | N | 1 H’C k?1· | H | 391,2 |
| 116 | ÂV 0 | N | 1 /C. H,C \ CH, CH, | H | 376,1 |
| 117 | H \ ch3 | N | 1 /C. H.C \ CH, ch3 | H | 403,2 |
| 118 | 0 X.„ x\ /°x / / '/ OH H | N | 1 /Cx H.C \ CH, ch3 3 | H | 380,2 |
| 119 | Í? CH, II i 3 Λ^τ> | N | 1 /Cx H3C \ CH, ch3 | H | 385,2 |
| 120 | 0 II .Cx /\ .0,., .CH, H | N | Á H,C \ CH, ch3 | H | 364,2 |
| 121 | 0 CH, II Ξ 3 .C, .OH / 7/ H | N | 1 /cx H.C \ CH, ch3 | H | 350,2 |
| 122 | 0 CH, II I 3 /%A^oh H | N | I /Cx H.C \ CH, ch3 | H | 364,2 |
| Ex. | R1 | Y | Ra | Rb | M/s | |
| MH+ | ||||||
| 123 | Λ | CH, 1γ\/Η5 0 | N | I ,C. H,C \ CH, ch3 | H | 392,2 |
| 124 | 0 II Ay | Y /CL /CH.. °x^CH, | N | /θχ H,C \ CH, ch3 | H | 408,2 |
| 125 | 0 II / 'N Η | CH, -Ac„, | N | Á H.C \ CH, ch3 | H | 392,2 |
| 126 | 0 II / / Y H | N | Á H.C \ CH, ch3 | H | 378,2 | |
| 127 | 0 II | /\ /OH N Y H I OH | N | Á H,C \ CH, ch3 | H | 366,1 |
| 128 | ° V /CY | YCHs X^O GHS lí /'oh, 0 H3C | N | Á H.C \ CH, ch3 | H | 434,2 |
| 129 | 0 II | >rCH-7 0 | N | Á H,C \ CH, ch3 | H | 424,1 |
| 130 | À H | ~x> | N | á H,C \ CH, ch3 | H | 397,2 |
| 131 | s | H3C CH, -Χ> H | N | Á H,C \ CH, 3 CH3 | H | 364,2 |
| 132 | A- | ΑΆ° 0 | N | Á H,C \ CH, ch3 | H | 447,2 |
| Ex. | R1 | Υ | Ra | Rb | M/s MH+ |
| 133 | 0 A^NH2 II ? | Ν | I H.C \ CH, ch3 | H | 389,2 |
| 134 | Λα> Η | Ν | I H,C \ CH, Α | H | 398,2 |
| 135 | 0 II X 'Ν^^ιΙΟΗ | Ν | ] /A π,ο /η, | H | 362,2 |
| 136 | 0 λΟ-οη | Ν | I χΆ H,C \ CH, ch3 | H | 362,2 |
| 137 | Η | Ν | /Á H,C \ CH, ch3 | H | 358,2 |
| 138 | 0 II I Ν /ca^-na> Η | Ν | I /A H,C \ CH3 ch3 | H | 386,2 |
| 139 | 0 Ν==\ CH, | Ν | xÁ H,C \ CH, ch3 | H | 429,3 |
| 140 | 0 0 Η Η h/cH | Ν | I ^A H,C \ CH, 3 ch3 | H | 449,17 |
| 141 | 0 II ΌΗ | Ν | I /A H.C \ CH, ch3 | H | 366,26 |
| 142 | 0 ό. ,ΌΗ / γ Η ΌΗ | Ν | /Á H.C \ CH, ch3 | H | 366,3 |
| Ex. | R1 | Y | Ra | Rb | M/s MH+ |
| 143 | 0 P λ n CIL n-n7 CH3 | N | /<L H,C \ CH, ch3 | H | |
| 144 | Á) | N | H,C \ CH, ch3 | H | 327,2 |
| 145 | n-n X Sr | N | h3c \ ch, ch3 | H | 413,1 |
| 146 | kk | N | H,C \ CH, ch3 | H | 340,2 |
Preparação de certos materiais de partida
As abreviações usadas são como se segue: CDI é 1,1’carbonilimidazol, DCM é diclorometano, DIPEA é diisopropiletilamina, DMF é Dimetilformamida, THF é tetrahidrofurano, HPLC é Cromatografia Líquida de Desempenho Elevado, DMF-DMA é N,N-Dimetiiformamida dimetilacetal, DMSO é sulfóxido de dimetila, NMP é 1-Metil-2-pirrolidina, HCI é Ácido clorídrico, TFA é Ácido trifluoroacético, m-CPBA é ácido metacloroperbenzóico.
(a) Intermediários de aminotiazóis
Os seguintes intermediários de aminotiazol de fórmula (A)

são mostrados na Tabela 3 abaixo, seus métodos de preparação sendo descritos em seguida.
TABELA 3
| Intermediário | Y | Ra | Rb | M/s MH+ |
| AA | C | -CHg | -CHS | 220,19 |
| AB | C | Cl | H | |
| AC | N | -SCH, | -ch3 | 253,02 |
| AD | N | -SOCH;, | -ch3 | - |
| AE | N | -SCH, | H | 239,03 |
| AF | N | -ch3 | -CH, | 221,05 |
| AG | N | -C(CH3)3 | H | 249,12 |
Intermediário AA
5-(2,6-Dimetil-piridin-4-il)-4-metil-tiazol-2-ilamina
Esta é preparada como descrito no Exemplo 20(b).
Intermediário AB
5-(2-Cloro-piridin-4-il)-4-metil-tiazo!-2-ilamina AB1) 1 -Bromo-1 -(2-cloro-p i rid in-4-i I )-pro pan-2-ο na:
Bromo (1,36 ml, 26 mmoles) é adicionado por gotejamento a uma solução agitada de 1-(2-cloro-piridin-4-il)-propan-2-ona (Exemplo 2a,
5,0 g, 29,5 mmoles) em dioxano (150 ml) por 45 minutos a 5-10 °C. Após 20 minutos na temperatura ambiente o solvente é removido in vacuo para proporcionar o composto do título como um sólido amarelo.
AB2) 5-(2-Cloro-piridin-4-il)-4-meti]-tiazol-2-ilamina:
Tiouréia (1,8 g, 29,4 mmoles) é adicionado a uma solução agitada de 1-bromo-1-(2-cloro-piridin-4-il)-propan-2-ona (7,3 g, 29,4 mmoles) em etanol (150 ml). A mistura é aquecida a 60°C por 3 horas depois deixada esfriar para a temperatura ambiente e repousar por 18 h. O sal de bromidrato do composto do título se precipita durante este tempo e é removido por filtração e lavado com éter dietílico.
Intermediário AC
4-Metil-5-(6-metil-2-metilsulfanil-pirimidin-4-il)-tiazol-2-ilamina AC1) 4,6-Di metil-2-metilsu Ifan il-piri mid ina:
4,6-Dimetil-pirimidina-2-tiol (20 g, 142 mmoles) é adicionado lentamente a uma solução de hidróxido de sódio (6,3 g, 156 mmoles) em etanol (120 ml) e água (60 ml), lodeto de metila (9,8 ml, 156 mmoles) é adicionado por gotejamento e a mistura é agitada na temperatura ambiente por 1 hora. Os solventes são removidos in vacuo e o resíduo é dividido entre éter dietílico (200 ml) e água (200 mi). O extrato orgânico é secado (MgSO4) e o solvente é removido para dar o composto do título.
AC2) 1-(6-Metil-2-metilsulfanil-pirimidin-4-il)-propan-2-ona:
n-Butillítio (1,6 M em hexanos, 67 ml, 107 mmoles) é adicionado por gotejamento a uma solução agitada de diisopropilamina (15 ml, 107 mmoles) em THF seco (90 ml) sob argônio a -78 °C. Após 15 minutos em 78°C a 50°C a mistura é esfriada para -78°C e uma solução de 4,6-dimetil-2metilsulfanil-pirimidina (15 g, 97,4 mmoles) em THF seco (45 ml) é adicionada por gotejamento. A reação é agitada por 2,5 horas a -78°C depois N-metóxi-N-metilacetamida (10,4 ml, 97,4 mmoles) é adicionada por gotejamento. A reação é deixada se aquecer para a temperatura ambiente por 1 hora seguido por resfriamento brusco com solução aquosa saturada de cloreto de amônio (10 ml). A mistura é concentrada para remover a maioria do THF depois dividida entre água (200 ml) e DCM (200 ml). O extrato orgânico é separado, secado (MgSOJ, e o solvente é removido para proporcionar o composto do título. O material é uma mistura de formas ceto e enol como observado por 1H RMN (CDCI3)
AC3) 1-Bromo-1-(6-metil-2-metilsulfanil-pirimidin-4“il)-propan-2-ona:
Bromo (1,46 ml, 28,5 mmoles) é adicionado a um solução esfriada (5-10 °C) rapidamente agitada de 1-(6-metil-2-metilsulfanilpírimidin-4-il)-propan-2-ona (7,0 g, 35 mmoles) em dioxano (100 ml) por 30 minutos. A reação é deixada se aquecer para a temperatura ambiente e o solvente é removido in vacuo para produzir o composto do titulo que é usado imediatamente na próxima etapa.
AC4) 4-Metil-5-(6-metil-2-metilsulfanil-pirimidin-4-il)-tiazol-2-ilamina:
Tiouréia (2,7 g, 35 mmoles) é adicionada a uma solução agitada de 1-bromo-1-(6-metil-2-metilsulfanil-pirimidin-4-il)-propan-2-ona (AC3) em etanol (100 ml) a 60 °C. Após 30 minutos a reação é deixada esfriar para a temperatura ambiente. Após repousar por 18 horas o sal de bromidrato do composto do título é removido por filtração e lavado com éter dietílico. O produto é dissolvido em água e 2M hidróxido de sódio é adicionado para precipitar o composto do título como a base livre.
Intermediário AD
5-{2-Metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ilamina m-CPBA (57 - 86 % de pureza, 6,0 g, 20-30 mmoles) é adicionado em porções a um solução/suspensão com agitação rápida de 4metil-5-(6-metil-2-metilsulfanil-pirimidin-4-il)-tiazol-2-ilamina (6,0 g, 23,8 mmoles) em diclorometano seco (200 ml) a 0 °C. Após a adição (15 minutos) a reação é deixada se aquecer lentamente para a temperatura ambiente. A mistura é cuidadosamente adicionada à solução saturada de bicarbonato de sódio (300 ml), agitada, e o extrato orgânico é separado e secado (MgSO4). Este primeiro extrato contém uma mistura 1:1 de sulfóxido e sulfona. A fase aquosa é depois dividida com clorofórmio para extrair o composto do título como sólido amarelo.
Intermediário AE
4- Metil-5-(2-metilsulfanil-pirimidin-4-il)-tiazol-2-ilamina
Este material é preparado pelo procedimento delineado para o intermediário AC, substituindo 4,6-dimetil-pirimidina-2-tiol na primeira etapa (AC1) com 4-metil-2-meti Isulfanil-pirimidina.
Intermediário AF
5- (2,6-Dimetil-pirimidin-4-il)-4-metil-tiazo[-2-ilamina
AF1) 2,4,6-Trimetil-pirimidina:
Este material é preparado seguindo os protocolos descritos em Helvetica Chimica Acta, Vol. 64, No, 1, pp 113-152, 1981. (Autores: K. Burdeska, H. Fuhrer, G. Kabas and A.E. Siegrist)
AF2) 1-Bromo-1-(2,6-dimetil-pirimidin-4-il)-propan-2-ona:
Este material é preparado a partir de 2,4,6-trimetil-pirimidina seguindo o protocolo da etapa 2 usado para preparar 1-bromo-1-(6-metil-2metilsulfanil-pirimidin-4-il)-propan-2-ona (AC3) a partir de 4,6-dimetil-2metilsulfanil-pirimidina (AC1).
AF3) 5-(2,6-Dimetil-pirimidin-4-il)-4-metil-tiazol-2-ilamina:
Este material é preparado pela reação de 1-bromo-1-(2,6dimetil-pirimidin-4-il)-propan-2-ona com tiouréia seguindo o procedimento descrito por intermediário AC4.
Intermediário AG
5-(2-terc-Butil-pirimidin-4-il)-4-metil-tiazol-2-ilamina
Este composto é preparados por dois diferentes métodos:
Método a:
O composto do títuio é preparado por um procedimento análogo à N-[4-Metil-5-(2-morfolin-4-il-pirimidin-4-il)-tiazol-2-ila]-acetamida (25e) mediante a substituição bromidrato de morfolinaformamidina (parte 25c) com cloridrato de terc-butilcarbamida.
Método b:
AG1) 2-terc-B uti l-4-meti l-piri mid i na:
Seguindo um protocolo descrito em Helvetica Chimica Acta, Vol,64, No,1, pp 113-152,1981. (Autores: K. Burdeska, H. Fuhrer, G. Kabas e A.E. Siegrist), metóxido de sódio (30 % em peso em metanol, 28 ml, 146 mmoles) é adicionado por gotejamento durante 2 horas a uma solução agitada de cloridrato de terc-butilcarbamida (10 g, 73 mmoles) e acetilacetaldeído dimetil acetal (10,75 ml, 80 mmoles) em metanol a 60 °C. Após agitação por um adicional de 6 horas a 50°C o solvente é removido in vacuo e o resíduo é diluído com água (500 ml). A solução é levada para pH 7,0 mediante a adição de 6M HCI e extraída com diclorometano (3 x 300 ml). Após secagem (MgSO4) o solvente é removido para dar o produto como um óleo.
AG2) 1 -(2-terc-Buti l-pi ri m id in-4-i I )-propan-2-ona:
Este material é preparado a partir de 2-terc-butil-4-metilpirimidina seguindo o protocolo delineado para o intermediário AC2.
AG3) 1-Bromo-1-(2-terc-butil-pirimidin-4-il)-propan-2-ona:
Bromo (3,25 g, 20,7 mmoles) em clorofórmio (20 ml) é adicionado por gotejamento durante 5 horas a uma solução agitada de 1-(2terc-butil-pirimidin-4-il)-propan-2-ona (4,0 g, 20,7 mmoles) em dioxano (300 ml) mantida em 10 a 15 °C, Quando a adição estiver completa o solvente é removido in vacuo para dar o composto do título como um sal de bromidrato. AG4) 5-(2-terc-Butil-pirimidin-4-il)-4-metil-tiazol-2-ilamina:
1-Bromo-1-(2-terc-butil-pirimidin-4-il)-propan-2-ona (1 g, 37 mmoies) é agitada em etanol (40 ml) a 70°C. Tiouréia (280 mg, 37 mmoies) é adicionada e a agitação continuou por 1 hora a 70°C. Após esfriamento para a temperatura ambiente a mistura é filtrada para proporcionar o composto do título como o sal de bromidrato. Se exigido, o produto é dissolvido em 1M ácido clorídrico aquoso e a solução é levada para pH 8 mediante a adição de solução de hidróxido de sódio para precipitar o composto do título como a base livre.
(b) Intermediários de imidazol-uréia
Os seguintes intermediários de imidazol-uréia de fórmula (B)

são mostrados na Tabela 4 abaixo, cujo método de preparação sendo descrito em seguida.
TABELA 4
| Intermediário | Y | Ra | Rb | Material de Partida | Método |
| BA | C | -ch3 | -ch3 | AA | Ba |
| BB | C | Cl | H | AB | Bb |
| BC | N | -sch3 | -ch3 | AC | Bb |
| BD | N | -SOCH, | -ch3 | AD | Bb |
| BE | N | -SCH3 | H | AE | Bb |
| BF | N | -ch3 | -ch3 | AF | Ba |
| BG | N | -C(CH3)3 | H | AG | Ba |
Método (Ba)
Uma suspensão do aminotiazol (13,4 mmoies) e 1,1’carbonildiimidazol (2,4 g, 14,7 mmol, 1,1 equivalente) em CH2CI2 (75 ml) é aquecida a 40°C - refluxo sob argônio até que nenhum material de partida permaneça (30 minutos a 5 horas) como determinado por HPLC e RMN. Quando esfriado o precipitado sólido é removido por filtração. Este sólido consiste no intermediário de imidazol-uréia (B) juntamente com quantidades variáveis do isocianato e imidazol correspondentes. Este sólido é usado nas etapas subseqüentes visto que o intermediário de imidazol-uréia e o intermediário de isocianato são igualmente adequados como precursores de uréias.
Os seguintes intermediários são preparados por este método: [5-(2,6-dimetil-piridin-4-il)-4-metil-tiazol-2-ila]-amida de ácido imidazol-1 carboxílico (BA), [5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-amida de ácido imrdazoJ-1 -carboxílico (BG), [5-(2,6-dimetil-pirimidin-4-il)-4-metil-tiazol2-ilaj-amida de ácido imidazol-1-carboxílico (BF).
Método (Bb)
Trietilamina ou hidreto de sódio (7,17 mmol, 1,1 equivalente) é adicionado a uma suspensão agitada do aminotiazol (base livre ou sal de bromidrato, 6,53 mmoles) e carbonildiimidazol (1-2 equivalentes) em CH2CI2 seco (40 ml) contendo algumas gotas de DMF para auxiliar a solubilidade se necessário. A reação é aquecida em refluxo sob argônio até que nenhum material de partida reste (18 horas) como determinado por HPLC e RMN. Quando esfriado o precipitado sólido é removido por filtração e lavado com éterdietílico. Este sólido consiste no intermediário de imidazoluréia (B) juntamente com quantidades variáveis do isocianato e imidazol correspondentes. Este sólido é usado nas etapas subseqüentes visto que o intermediário de imidazol-uréia e o intermediário de isocianato são igualmente adequados como precursores de uréias.
Os seguintes intermediários são preparados por este método: [5-(2-cloro-piridin-4-il)-4-metil-tiazol-2-ila]-amída de ácido imidazol-1 carboxílico (BB), [4-metil-5-(6-metil-2-metifsulfanil-pirimidin-4-il)-tiazol-2-ila]amida de ácido imidazol-1-carboxílico (BC), [5-(2-metanossulfinil-6-metilpirimidin-4-il)-4-metil-tiazol-2-ila]-amida de ácido imidazoí-1-carboxílico (BD), e [5-(2-metanossulfinil-pirimidin-4-il)-4-rnetil-tiazo!-2-ila]-amida de ácido imidazol-1-carboxílico (BE).
(c) Intermediário de amina
Muitos dos intermediários de amina (C) que são usados para preparar os compostos finais dos Exemplos na Tabela 2 são comercialmente disponíveis ou são preparados por métodos padrão. A 5 preparação de certos intermediários de amina que não são facilmente disponíveis comercialmente é dada abaixo. Estes são os seguintes intermediários de amina de fórmula (C)

Het onde Het é um dos seguintes:

CA CB CC CD e são como apresentados na Tabela 5 abaixo.
TABELA 5
| Intermediário | Het | R |
| CA | CA | -CH?CH3 |
| CB | CB | -ch?ch3 |
| CC1 | CC | -ch?ch3 |
| CC2 | CC | -ch?ch?ch3 |
| CC3 | CC | -CH(CH3)9 |
| CD | CD | -CH?CH, |
Intermediário CA
2-(5-Etil-oxazol-2-il)-etilamina (Preparada anteriormente)
Etapa 1) Éster benzílico de ácido [2-(2-Hidróxi-butilcarbamoil)-etil]15 carbâmico:
Uma mistura que compreende Z-Beta-Ala-OH (9,0 g, 40,3 mmoles), EDCI.HCI (10,0 g, 52,4 mmoles), hirdoxibenzotriazol (5,45 g, 40,3 mmoles), trietilamina (7,3 ml, 52,4 mmoles) em DCM (150 ml) é agitada a 0 °C por 30 minutos. 1-Amino-2-butanol (4,2 ml, 44,3 mmoles) é adicionado de uma vez e a agitação continua por 1 hora. A mistura de reação é diluída com água (150 ml) e extraída com diclorometano (2 x 150 ml). As camadas orgânicas são combinadas, secadas por MgSO4, filtradas e concentradas in vácuo para produzir um sólido branco bruto. O produto é purificado por cromatografia em sílica eluindo com etanol-acetato de etila (1:10) para dar o composto do título.
Etapa 2) Éster benzílico de ácido [2-(2-Oxo-butilcarbamoil)-etil]-carbâmico:
A uma solução agitada de cloreto de oxalila (2 M em DCM) (13,35 ml, 26,5 mmoles) em DCM seco a -78 °C é adicionado por gotejamento DMSO (2,5 ml, 35,4 mmoles). Após agitação por 15 minutos, a mistura de reação é tratada com uma solução de éster benzílico de ácido [2(2-hidróxi-butilcarbamoil)-etil]-carbâmico (etapa 1) (6,5 g, 22,1 mol) em DCM seco (40 mi). Trietilamina (13 ml) é adicionada após 1 hora e após agitação a -78 °C por 90 minutos, a mistura de reação é deixada se aquecer para a temperatura ambiente. A reação é diluída com DCM (100 ml) e lavada com HCl (1M, 200 ml), solução saturada de bicarbonato de sódio (200 ml), água (200 ml) e salmoura (200 ml). A parte orgânica é secada por MgSO4, filtrada e concentrada in vacuo para produzir o composto do título como um sólido branco.
Etapa 3) Éster benzílico de ácido [2-(5-Etil-oxazol-2-il)-etil]-carbâmico:
A uma suspensão agitada de trifenilfosfeno sustentada por polímero (19,6 g, 58,9 mmoles) em DCM (250 ml) é adicionado iodo (14,95 g, 58,9 mmoles). Após agitação na temperatura ambiente por 10 minutos, a mistura é tratada com trietilamina (16,4 ml, 117,5 mmoles) seguido por uma solução de éster benzílico de ácido [2-(2-oxo-butilcarbamoil)-eti(]-carbâmico (etapa 2) (6,88 g, 23,5 mmoles) em DCM (50 ml). A mistura de reação é agitada durante a noite e depois filtrada através de material de filtrante Celite™, lavada completamente com DCM (500 ml) e o solvente removido in vacuo para produzir o composto do título como um sólido marrom.
Etapa 4) 2-(5-Etil-oxazol-2-il)-etilamina:
Formiato de amônio (0,316 g, 5 mmoles) é adicionado a uma solução de éster benzílico de ácido [2-(5-etil-oxazol-2-íl)-etil]-carbâmico (etapa 3) (1,66 mmol) em metanol (15 ml) e 10 % Pd em Carbono (125 mg) é adicionado sob uma atmosfera inerte. A mistura é agitada na temperatura ambiente por 2 horas. O catalisador é removido por filtração e o filtrado é evaporado. O resíduo é diluído com diclorometano, filtrado para remover sólido não dissolvido e o solvente é removido. O resíduo é dissolvido em DCM e tratado com 1M solução aquosa de hidróxido de sódio (5 ml). O extrato orgânico é separado, secado (MgSO4), filtrado e o solvente é removido. Cristalização a partir de acetato de etila / diclorometano proporciona o composto do título.
Intermediário CB
2-(3-Etil-[1,2,4]oxadiazol-5-il)-etilamina
Etapa 1) N-Hidróxi-propionamidina:
Etanol (100 ml) seguido por cloridrato de hidroxilamina (5,0 g, 72 mmoles) é adicionado a uma solução de K2CO3 (9,93 g, 72 mmoles) em água (25 ml). Propionitrila (5,13 ml, 72 mmoles) é depois adicionada e a mistura é aquecida em refluxo por 18 horas. Após esfriamento, o solvente é removido in vacuo e etanol é adicionado para dissolver o produto. A solução é separada a partir de qualquer sólido não dissolvido e o solvente é removido para deixar ficar o composto do título como um óleo amarelo.
Etapa 2) éster terc-butílico de ácido [2-(3-Etil-[1,2,4]oxadiazol-5-il)-etil]carbâmico:
N-Hidróxi-propionamidina (0,10 g, 1,15 mmol) em DMF (2 ml) é adicionado a uma suspensão agitada de hid reto de sódio (0,05 g de uma dispersão a 60 % em óleo, 1,26 mmol) em DMF (20 ml) na presença de peneiras moleculares (0,1 g). O frasco de reação é depois imerso em um banho de óieo preaquecido a 50°C e a agitação continuou por 5 min. Éster etílico de ácido 3-terc-Butoxicarbonilamino-propiônico (0,25 g, 1,15 mmol) em DMF (2 ml) é adicionado por 5 minutos. Após 3 horas a 50°C a mistura é esfriada para 0°Ce água (3 ml) é adicionada. A mistura é deixada se aquecer para a temperatura ambiente depois filtrada através de material filtrante Celite®l, lavagem com acetato de etila, e o solvente é removido. Purificação por cromatografia, eluindo com hexano:acetato de etila (3:1) proporciona o composto do título.
Etapa 3) 2-(3-Etil-[1,2,4]oxadiazol-5-il)-etilamina:
TFA (0,5 ml) é adicionado a uma solução agitada de éster tercbutílico de ácido [2-(3-propil-[1,2,4]oxadiazol-5-il)-etil]-carbâmico (0,093 g, 0,39 mmol) em DCM (1 ml). Após 1 hora os solventes são removidos para proporcionar o composto do título.
Intermediários CC1, CC2, CC3
Estes compostos, a saber, 2-(1-etil-1 H-imidazol-4-il)-etilaimina (CC1), 2-(1-propil-1H-imidazol-4-il)-etilamina (CC2) e 2-(1-isopropil-1 Himidazol-4-il)-etilamina (CC3) são preparados mediante a alquilação de 7,8dihidro-6H-imidazo[1,5-c]pirimidin-5-ona com o brometo de alquila apropriado seguido por hidrólise como descrito em R. Jain and L. A. Cohen, Tetrahedron, (1996), 52, 5363-5370.
Intermediário CD
2-(2-Etil-1 H-tetrazol-5-il)-etilamina
Etapa 1) Éster terc-butílico de ácido [2-(2-Etil-2H-tetrazol-5-il)-etil]carbâmico:
Uma solução de éster terc-butílico de ácido [2-(1 H-tetrazol-5-il)etii]-carbâmico (preparado pelos protocolos delineados em N. A. Delaney, G. C. Rovnyak and M. Loots, relatório descritivo da patente Européia EP 449523) (1,0 g, 4,69 mmoles) em THF seco (20 ml) é tratada com uma dispersão a 60 % de hidreto de sódio em óleo mineral (0,19 g, 4,69 mmoles) e agitada em temperatura ambiente por 10 minutos. Iodeto de etila (0,375 ml, 4,69 mmoles) é adicionado e a mistura de reação é aquecida em refluxo por 7 horas, depois diluída com acetato de etila e filtrada. O filtrado é evaporado e o resíduo purificado cromatografia em sílica instantênea (eluição 3:2 hexano/acetato de etil) para proporcionar o composto do título, éster terc-butílico de ácido [2-(2-etil-2H-tetrazol-5-il)-etil]-carbâmico, eluído primeiro e éster terc-butílico de ácido [2-(1 -etil-1 H-tetrazol-5-il)-etil]carbâmico, eluído segundo.
Etapa 2) 2-(2-Etil-1 H-tetrazol-5-il)-etilamina:
Éster terc-butílico de ácido [2-(2-Etil-2H-tetrazol-5-il)-etil]75 carbâmico (0,33 g, 1,36 mmol) é dissolvido em diclorometano (3 ml) e tratado com ácido trifluoroacético (1 ml) e agitado na temperatura ambiente por 3 horas. O solvente é removido para proporcionar o composto do título como o sal de TFA.
(d) Intermediários de tiouréia
Os seguintes intermediários de tiouréia de fórmula (D)
H
Het onde Het é um dos seguintes:
Λ, s-^

CH,
Λ
N-N
Λ
N-N
Br
OH

OH
DC
DD
DE

são preparados como descrito em seguida.
Intermediário DA
Pirazin-2-il-tiouréia
A preparação deste material é descrito anteriormente (Exemplo
1a)
Intermediários de DB e DC
A saber, (6-Metil-piridin-3-il)-tiouréia (DB) e (5-Bromo[1,3,4]tiadiazol-2-il)-tiouréia (DC), são preparados por um processo similar à pirazin-2-il-tiouréia (DA) mediante a substituição de aminopirazina no Exemplo 1a com a amina heterocíclica apropriada.
Intermediário DD
Ácido (3-Tioureíd o-pirazol-1 -il)-acético
Etapa 1) 2-(1 H-Pirazol-3-il)-isoindole-1,3-diona:
H-Pirazol-3-ilamina (2 g, 24 mmoles) e éster etílico de ácido 1,3-dioxo-1,3-dihidro-isoindol-2-carboxílico (5,3 g, 24 mmoles) são agitados em THF (70 ml) na temperatura ambiente por 18 horas. A mistura de reação é depois concentrada na metade do volume e filtrada para remover o composto do título que é lavado com metanol seguido por éter dietílico.
Etapa 2) Éster terc-butílico de ácido [3-(1,3-Dioxo-1,3-dihidro-isoindol-2-il)76 pirazol-1-ila]-acético:
Hidreto de sódio (60 % em óleo, 0,538 g, 13,4 mmoles) é adicionado a uma solução agitada de 2-(1 H-pirazol-3-il)-isoindol-1,3-diona (2,39 g, 11,2 mmoles) em DMF seco (25 ml) na temperatura ambiente. Após 10 minutos bromoacetato de terc-butila (1,81 ml, 11,2 mmoles) é adicionado e a reação é agitada por 18 horas. A reação é resfriado bruscamente com água (1 ml), acetato de etila (150 m!) é adicionado e as fase orgânica é lavada com água (3 x 100 ml) seguido por salmoura (1 x 100 ml). O solvente é removido e o produto é purificado por cromatografia em sílica (eluição gradiente: acetato de etila - hexano) para dar o composto do título.
Etapa 3) Éster terc-butílico de ácido (3-Amino-pirazol-1 -il)-acético:
Hidrato de hídrazina (0,27 ml, 6,69 mmoles) é adicionado a uma solução agitada de éster terc-butílico de ácido [3-(1,3-dioxo-1,3-dihidroisoindol-2-il)-pirazoI-1-ila]-acético (2,18 g, 6,69 mmoles) em etanol (12 ml). A reação é agitada a 90°C por 2,5 horas. Quando fria, a reação é diluída com etanol (50 ml) e o precipitado branco é removido por filtração, lavagem com mais etanol (250 ml). O filtrado combinado é evaporado para secura e diclorometano (100 ml) é adicionado. Após filtrar novamente, o filtrado é evaporado para dar o composto do título como um óleo incolor.
Etapa 4) Éster terc-butílico de ácido [3-(3-Benzoil-tioureído)-pirazol-1-ila]acético:
Éster terc-butílico de ácido (3-Amino-pirazol-1-il)-acético (1,05 g, 5,33 mmoles) é adicionado por gotejamento sob argônio a uma solução agitada de isotiocianato de benzoíla (0,754 ml, 5,4 mmoles). Após agitação por 5 minutos a mistura de reação é despejada em água e o composto do título, um precipitado amarelo, é coletado e secado.
Etapa 5) ácido (3-Tioureído-pirazol-1 -il)-acético:
Uma suspensão agitada de éster terc-butílico de ácido [3-(3benzoil-tioureído)-pirazol-1-ila]-acético (0,823 g, 2,28 mmoles) em 2M NaOH (2 ml) é aquecida a 100°C por 30 minutos. Quando fria, a mistura é acidificada para pH 3 com 1M HCI. A mistura é extraída com acetato de etila. A fase orgânica é separadas e a fase aquosa é evaporada para secura para proporcionar o composto do título como um sal de cloridrato misturado com cloreto de sódio.
Intermediário DE
Ácido 3-(3-Tioureído-pirazol-1-il)-propiônico
Este material é preparado por um procedimento análogo ao ácido (3-tioureído-pirazol-1-il)-acético (intermediário DD) mediante a substituição de bromoacetato de terc-butila na etapa 2 com éster tercbutílico de ácido 3-bromo-propiônico.
Intermediário DF
Ácido 3-(5-Tioure ído-[1,3,4]tiadiazol-2-il)-propiônico
Etapa 1) Éster metílico de ácido 3-(5-Amino-[1,3,4]tiadiazol-2-il)-propÍônico:
Éster metílico de ácido 3-clorocarbonil-propiônico (4,4 g, 29 mmoles) é adicionado por gotejamento a uma suspensão agitada de semicarbazida (4,0 g, 44 mmoles) em THF (25 ml) a Q°C. Após agitação na temperatura ambiente por 1 hora, o solvente é removido para dar um sólido branco. Tolueno (30 ml) é adicionado seguido pela adição por gotejamento de ácido metano sulfônico (3,37 ml, 52 mmoles) depois a reação é aquecida a 70°C por 3 horas. A mistura é concentrada in vacuo e metanol (30 ml) é adicionado. Amônia aquosa é depois adicionada com agitação até que uma mistura básica seja obtida. Os solventes são removidos e o resíduo é purificado por cromatografia em sílica eluindo com clorofórmio : metanol (10:1) para dar o produto do título.
Etapa 2) Éster metílico de ácido carbetóxi-3-(5-tioureído-[1,3,4]tiadiazol-2-ÍI)propiônico:
Carbetoxiisocianato (0,49 ml, 3,73 mmoles) em diclorometano seco (10 ml) é adicionado por gotejamento na temperatura ambiente a uma suspensão agitada de éster metílico de ácido 3-(5-amino-[1,3,4]tiadiazol-2il)-propíônico (0,666 g, 3,56 mmoles) em DCM (20 ml). A reação é agitada sob argônio na temperatura ambiente por 18 h depois o solvente é removido para proporcionar o composto do título.
Etapa 3) Ácido 3-(5-tioureído-[1,3,4]tiadiazol-2-il)-propiônico:
Éster metílico de ácido carbetóxi-3-(5-tioureído-[1,3,4]tiadiazol-278 il)-propiônico (0,675 g, 2,91 mmoles) é colocado em suspensão em 2M NaOH (8 ml) e a reação é agitada em refluxo por 3,5 horas. Quando fria, a solução é acidificada para pH 3 com 6M HCÍ e o composto do títuio é removido por filtração e secado.
Preparação dos Exemplos Específicos
Procedimento Geral A para a preparação de uréias mediante a reação dos intermediários de imidazol-uréia de fórmula B com os intermediários de amina de fórmula C:
A amina (0,12 mmol) é adicionada a uma solução I suspensão do intermediário de imidazol uréia (0,11 mmol) em DMF (1,0 ml). Trietilamina pode ser adicionada para intensificar a taxa de reação e especialmente se um ou ambos materiais de partida estiverem presentes como um sal (1,1 equivalente Et3N per equiv. sal). A mistura de reação é submetida a ondas sonoras se necessário até que uma solução transparente fosse obtida. A reação é deixada prosseguir entre a temperatura ambiente e 70°C até que o material de partida seja consumido (30 min a 24 horas). Quando completa, a mistura é concentrada in vacuo para remover o solvente. O produto é convenientemente purificado mediante a dissolução do resíduo bruto em THF (2 ml) e adição deste ao isocianato sustentado por polímero (Argonaut Technologies, 0,5 g, 1,10 mmol) que foi pré-intumescido com THF (2 ml). A mistura de reação é deixada gotejar através da resina sob gravidade e o solvente é removido in vacuo para produzir o composto do título. Alternativamente o produto é purificado por um procedimento padrão, por exemplo, cristalização, cromatografia ou HPLC.
Exemplo 31
3-{3-[5-(2,6-Dimetil-piridin-4-il)-4-metil-tiazol-2-ila]-ureído}-N,N-dimetilpropionamida
A uma solução agitada de [5-(2,6-dimetil-piridin-4-il)-4-metil· tiazol-2-ila]-amida de ácido imidazol-1-carboxílico (Intermediário BA) (0,05 g, 0,16 mmol) em DMF (1,5 ml) sob uma atmosfera inerte é adicionado 3amino-N,N-dimetil-propionamida (Amina) (0,018 g, 0,16 mmol). A mistura de reação é agitada em temperatura ambiente por 18 horas. O solvente é removido in vacuo e o resíduo bruto resultante é dissolvido em THF e passado através de um tampão de resina de isocianato sustentado por polímero (0,5 g, pré-lavado com THF). A solução é concentrada in vacuo e o resíduo bruto é triturado com éter-acetato de etila para proporcionar o composto do título como um sólido amarelo.
Exemplos de 32 a 38
Estes compostos, a saber, 2-{3-[5-(2,6-dimetil-piridin-4-il)-4metil-tiazol-2-ilaJ-ureído}-N,N-dimetil-acetamida, 1-[5-(2-cloro-piridin-4-il)-4metil-tiazol-2-ila]-3-[2-(5-etil-oxazol-2-il)-etil]-uréia, 1-[5-(2-cloro-piridin-4-il)-4metil-tiazol-2-ila]-3-[2-(1 -etil-1 H-imidazol-4-il)-etil]-uréia, 1-[5-(2-cloro-piridin4-il)-4-metil-tiazol-2-ila]-3-[2-(1-propil-1H-imidazol-4-il)-etil]-uréia, 1-(5-(2cloro-piridin-4-il)-4-metil-tiazol-2-ila]-3-[2-(1-isopropil-1H-imidazol-4-il)-etil]uréia, 1-[5-(2-cloro-piridin-4-il)-4-metil-tiazol-2-ila]-3-[2-(2-etil-2H-tetrazol-5-il)etil]-uréia e 1 -(2-(5-etil-oxazol-2-iI)-etíl]-3-[4-metil-5-(6-metil-2-metilsuIfanilpirimidin-4-il)-tiazol“2-ila]-uréia são preparados pelo procedimento geral A usando o intermediário de imrdazol uréia (B) e amina (C) apropriados. Exemplo 39
N-(5-{2-[(2-Dimetilamino-etil)-metil-amino]-6-metil-pirimidin-4-ila}-4-metiltiazol-2-il)-acetamida
Etapa 1) N-[4-(2-Amino-4-metil-tiazol-5-il)-6-metil-pirimidin-2-ila]-N,N,,N'trimetil-etano-1,2-diamina:
N,N,N'-Trimetil-etano-1,2-diamina (0,62 g, 6,16 mmofes) é adicionada a uma solução agitada de 5-(2-metanossulfinil-6-metil-pirimidin4-il)-4-metil-tiazol-2-ilamina (Intermediário AD, 0,33 g, 1,23 mmol) em NMP (15 ml). A reação é aquecida a 70°C por 18 horas depois o solvente é removido in vacuo. O resíduo é purificado por cromatografia de fase reversa (sistema cromatográfico 08 Jones Flashmaster , condições de eluição gradiente MeCN/H2O) para dar o produto do título.
Etapa 2) N-(5-{2-[(2Oimetilamino-etil)-metil-amino]-6-metil-pirimidin-4-ila}-4metil-tiazol-2-il)-acetamida:
N-[4-(2-Amino-4-metil-tiazol-5-il)-6-metil-pirimidin-2-rla]-N,N',N'trimetil-etano-1,2-diamina (0,020 g, 0,065 mmol) em anidrido acético (1 ml) é aquecida a 60°C por 2 horas. Quando fria, o solvente é removido in vacuo e o resíduo é dividido entre acetato de etila (30 ml) e água (30 ml). O extrato orgânico é removido, secado (MgSOJ e o solvente removido para dar o composto do título.
Exemplo 40
1- [2-(5-Etil“Oxazol-2-il)-etil]-3-[5-(2-metanossulfinil-6-metil-pirimidin-4-il)-4metil-tiazol-2-ilaj-uréia
Este composto é preparado pela oxidação de 1 -[2-(5-etil-oxazol2- il)-etil]-3'[4-metíl-5-(6-metil-2-metilsulfanil-pirimidin-4-il)-tiazol-2-ila]-uréia com m-CPBA (ácido meta-cloroperóxi-benzóico) seguindo o protocolo usado para preparar 5-(2-Metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2ilamina (intermediário AD).
Exemplo 41
1-[2-(5-Etil-oxazol-2-il)-etil]-3-[5-(2-metanossulfonil-6-metil-pirimidin-4-il)-4metil-tiazol-2-ila]-uréia
Ácido meta-cloroperoxibenzóico ou m-CPBA (57 a 86 % de pureza, 0,438 g, 1,8 mmol) é adicionado em porções a um solução em agitação rápida de 1-[2-(5-etil-oxazol-2-íl)-etil]-3-[5-(2-metano-sulfinil-6-metilpirimidin-4-il)-4-metil-tíazol-2-ila]-uréia (0,31 g, 0,74 mmol) em diclorometano seco (5 ml) na temperatura ambiente. Após 2 horas a mistura é diluída com diclorometano e lavada com tiossulfito de sódio aquoso e salmoura. O extrato orgânico é separado, secado por MgSO4 e o solvente é removido. Purificação por cromatografia em sílica, eluindo com EtOAc, MeOH (97:3) proporciona o composto do título.
Exemplo 42
1-[5-(2-Dimetilamino-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-[2-(5-etiloxazol-2-il)-etil]-uréia
Uma solução de 1-[2-(5-etil-oxazol-2-il)-etil]-3-[5-(2metanossulfonil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia (0,22 g, 0,049 mmol) e dimetilamina (2M em THF, 0,073 ml, 0,15 mmol) em DMF (1 ml) é aquecida a 70°C por 18 horas. O solvente é removido e o produto é purificado por cromatografia em sílica eluindo com acetato de etila para dar o composto do título.
Exemplo 43
1-(5-{2-[(2-Dimetilamino-etil)-metil-amino]-6-metil-pirimidin-4-ila}-4-metiltiazol-2-il)-3-[2-(5-etil-oxazol-2-il)-etil]-uréia
Uma solução de 1-[2-(5-Etil-oxazol-2-il)-etil]-3-[5-(2metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia (Exemplo 40) (0,25 g, 0,58 mmol) e Ν,Ν,Ν-trimetil-etilenodiamina (0,368 ml, 2,9 mmoles) em DMF (3 ml) é aquecida a 90°C por 2 horas até que nenhum material de partida sobre. O solvente é removido e o resíduo é purificado por cromatografia (sílica, eluição gradiente acetato de etila - metanol) para proporcionar o composto do título.
Exemplo de 44 a 54
Estes compostos, a saber, 1-[2-(5-etil-oxazol-2~il)-etil]-3-[5-(2imidazol-1-il-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia, 1-[2-(5-etiloxazol-2-il)-etil]-3-{4-metil-5-[6-metil-2-(2-morfolin-4-il-etilamino)-pirimidin-4ila]-tiazol-2-ila}-uréia, 1-(5-{2-[(2-dimetilamino-etil)-etil-amino]-6-metilpirimidin-4-ila}-4-metil-tiazol-2-il)-3-[2-(5-etil-oxazol-2-íi)-etil]-uréia, 1-(5-{2-[(2dietilamino-etil)-metil-amino]-6-metil-pirimidin-4-ila}-4-metil-t!azol-2-íl)-3-[2-(5etil-oxazol-2-il)-etil]-uréia, 1-{5-[2-(3-dimetilamino-propilamino)-6-metilpirimidin-4-ila]-4-metil-tÍazol-2-fla}-3-[2-(5-etil-oxazol-2-il)-etil]-uréia, 1-(2-(5etil-oxazol-2-il)-etil]-3-{5-[2-(3-imidazol-1-il-propil-amino)-6-metil-pirimidin-4ila]-4-metil-tiazol-2-ila)-uréia, 1-(5-{2-[(3-dimetilamino-propila)-metil-amino]-6metil-pirimidin-4-ila}-4-metil-tiazol-2-il)-3-[2-(5-etil-oxazol-2-il)-etil]-uréia, 1-{5[2-(2-dietilamino-etilamino)-6-metil-pirimidin-4-ila]-4-metil-tiazol-2-ila}-3-[2-(5etil-oxazol-2-il)-etil]-uréia, 1-(5-{2-[(3-amino-propila)-metil-amino]-6-metilpirimidin-4-ila)-4-metil-tiazol-2-il)-3-[2-(5-etil-oxazol-2-il)-etil]-uréia, 1-(2-(5etil-oxazol-2-il)-etil]-3-(4-metil-5-{6-metil-2-[3-(4-metil-piperazln-1-il)propilamino]-pÍrimidin-4-ila}-tiazol-2-il)-uréia, 1-[2-(5-etil-oxazol-2-il)-etil]-3-(4metil-5-{6-metil-2-[metil-(2-morfolin-4-il-etil)-amino]-pirimidin-4-ila}-tiazol-2-il)uréia são preparados a partir de 1-[2-(5-etil-oxazol-2-il)-etil]-3-[5-(2metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia (Exemplo 40) por um procedimento análogo ao Exemplo 43 usando a amina apropriada.
Exemplo 55
1-[2-(5-Etil-oxazol-2-il)-etil]-3-(4-metil-5-{6-metil-2-[(pirrolidin-2-ilmetil)amino]-pirimidin-4-ila}-tiazol-2-il)-uréia
Etapa 1) Éster terc-butílico de ácido 2-{[4-(2-{3-[2-(5-Etil-oxazol-2-il)-etil]ureído}-4-metil-tiazol-5-il)-6-metil-pirimidin-2-ilamino]-metila}-pirrolidina-1 carboxílico:
Este material é preparado a partir de 1-[2-(5-etil-oxazol-2-il)-etil]3-[5-(2-metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia (Exemplo 40) e éster terc-butílico de ácido 2-aminometil-pirrolidina-1carboxílico por um procedimento anáiogo ao Exemplo 42 mas substituindo DMF por dioxano.
Etapa 2) 1 -[2-(5-Etil-oxazol-2-il)-etil]-3-(4-metil-5-{6-metil-2-[(pirrolidin-2ilmetil)-amino]-pirimidin-4-ila}-tiazol-2-il)-uréia:
Éster terc-butílico de ácido 2-aminometif-pirrolidina-1 -carboxílico (0,299 mmol) é dissolvido em TFA (2,5 ml) na temperatura ambiente. Após 18 horas, a mistura é diluída com NaOH aquoso e o produto é extraído em acetato de etila. O extrato orgânico é separado, secado por MgSO4 e o solvente é removido para dar o composto do título.
Exemplo 56
1- [2-(5-Etil-oxazol-2-il)-etil]-3-[5-(2-metóxi-6-metil-pirimidin-4-il)-4-metil-tiazol2- ila]-uréia
Metóxido de sódio (2M em MeOH, 0,115 ml, 0,23 mmol) é adicionado a uma solução agitada de 1-[2-(5-etil-oxazol-2-il)-etil]-3-[5-(2metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia (Exemplo 40) (0,050 g, 0,115 mmol) em metanol (3 ml) na temperatura ambiente. Após 18 horas, o solvente é removido e o resíduo é dissolvido em DCM e lavado com água. O produto é extraído em 1M HCI e o extrato aquoso é lavado com DCM. A fase aquosa é depois basificada com NaOH aq. e o produto é extraído em DCM. Após secagem (MgSO4) o solvente é removido para dar o composto do título.
Exemplo 57
1-[2-(5-Etil-oxazol-2-il)-etil]-3-{4-metil-5-[6-metil-2-(2-morfolin-4-il-etóxi)83 pirímidin-4-ila]-tiazol-2-ila}-uréia
Hídreto de sódio (0,020 mg, 0,50 mmol) é adicionado a uma solução agitada de 2-morfolin-4-il-etanol (0,084 ml, 0,69 mmol) e 1-[2-(5-etiloxazol-2-iI)-etil]-3-[5-(2-metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2ila]-uréia (Exemplo 40) (0,10 g, 0,23 mmol). A reação é aquecida a 70°C por 8 horas depois deixada esfriar e filtrada. O filtrado é evaporado e o resíduo é dissolvido em DCM (50 ml) e lavado com água (3 x 50 ml). O produto é extraído em 1M HCI e a fase aquosa é lavada com DCM. A fase aquosa é depois basificada com NaOH aq. e o produto é reextraído em DCM. Após secagem (MgSO4) o solvente é removido para dar o composto do título. Exemplo 58
-[2-( 1 -Etil-1 H-imidazol-4-il )-eti l]-3-[5-(2-meta nossulfi nil-6-metil-pirimid in-4-i I)4-metil-tiazol-2-ila]-uréia
Este composto é preparado a partir de [5-(2-metanossulfinil-6metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-amida de ácido imidazol-1-carboxílico (intermediário de imidazol-uréia BD) e 2-(1-etil-1 H-imidazol-4-il)-etilamina (intermediário de tiouréia CC1) usando o procedimento geral A.
Exemplo 59
1-(5-{2-[(2-Dimetilamino-etil)-metil-amino]-6-metil-pirimidin-4-ila}-4-metiltiazol-2-il)-3-[2-(1 -etil-1 H-ímidazol-4-il)-etil]-u réia
Este material é preparado a partir de 1-[2-(1-etil-1 H-imidazol-4il)-etil]-3-[5-(2-metanossulfinil-6-metil-pirimidin-4-il)-4-meti!-tiazol-2-ila]-uréia e N,N,N'-trimetil-etano-1,2-diamina usando o protocolo descrito para o exemplo 42 mas substituindo DMF por dioxano.
Exemplos 60 e 61
Estes compostos, a saber, 1-(5-{2-[(2-dimetilamino-etil)-metilamino]-6-metil-pirimidin-4-ila}-4-metil-tiazol-2-il)-3-[2-(1-propil-1 H-imidazol-4il)-etii]-uréia e 1-(5-{2-[(2-dimetÍlamino-etil)-metil-amino]-6-metil-pirimidin-4ila}-4-metil-tiazol-2-il)-3-[2-(1 -isopropil-1 H-imidazol-4-i!)-eti l]-uréia são preparados por procedimentos análogos à 1-(5-{2-[(2-dimetilamino-etil)metil-amino]-6-metil-pirimidin-4-ila}-4-metil-tiazol-2-il)-3-[2-(1 -etil-1 H-imidazol4-il)-etil]-uréia (Exemplo 59) usando os materiais de partida apropriados.
Exemplo 62
-[5-(2-Ciano-6-metil-pirimid in-4-i!)-4-metíl-tiazol-2-ila]-3-[2-(1 -iso propil-1Himidazol-4-il)-etil]-uréia
Etapa 1) 1-[2-(1-lsopropil-1H-imidazol-4-ii)-etil]-3-[4-metil-5-(6-metil-2metilsulfanil-pirimidin-4-il)-tiazol-2-ila]-uréia:
Este material é preparado a partir de [4-metil-5-(6-metil-2-metilsulfanil-pirimidin-4-il)-tiazol-2-ila]-amida de ácido imidazol-1-carboxílico (intermediário de imidazol-uréia BC) e 2-(1-iso-propil-1 H-imidazol-4-il)etilamina (intermediário de tiouréia CC3) usando o procedimento geral A. Etapa 2) 1-(2-(1-lsopropil-1H-imidazol-4-il)-etil]-3-[5-(2-metanossulfonil-6metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia:
Este material é preparados a partir de 1-(2-(1-isopropil-1 Himidazol-4-il)-etii]-3-[4-metil-5-(6-metil-2-metilsulfanil-pirimidin-4-il)-tiazol-2ila]-uréia pelo mesmo protocolo descrito para 1-[2-(5-etil-oxazol-2-il)-etil]-3[5-(2-metanossulfonil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-iiréia (Exemplo 41).
Etapa 3) 1-[5-(2-Ciano-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-[2-(1isopropil-1 H-imidazol-4-ii)-etil]-uréia:
Cianeto de sódio (0,069 g, 1,4 mmol) é adicionado a uma solução agitada de 1-[2-(1-isopropil-1H-imidazol-4-il)-etil]-3-[5-(2metanossulfonil-6-metil-pirimidin-4-il)-4-metil-tiazol-2-ila]-uréia (0,21 g, 0,47 mmol) em DMSO seco (10 ml), A reação é agitada em 50°C sob argônio por 3 horas depois o solvente é removido. O resíduo é dissolvido em DCM e lavado com água. O extrato orgânico é secado (MgSO4) e o solvente removido para dar o composto do título.
Exemplos de 63 a 72
Estes compostos, a saber, 1-[2-(2-etil-2H-tetrazol-5-il)-etil]-3-[5(2-metanossulfinil-6-metil-pirimidin-4-ií)-4-metil-tiazol-2-ila]-uréia, 1-(5-(2dimetilamino-6-metil-pirimÍdin-4-il)-4-metil-tiazol-2-ila]-3-[2-(2-etil-2H-tetrazol5-il)-etil]-uréia, 1-(5-{2-[(2-dimetilamino-etil)-metil-amino]-6-metil-pirimidin-4ila}-4-metil-tiazol-2-il)-3-[2-(2-etil-2H-tetrazol-5-il)-etil]-uréia, N,N-dimetil-2-{385 [4-metil-5-(6-metil-2-metil-sulfanil-pirimidin-4-il)-tiazol-2-ila]-ureído}acetamida, 2-{3-[5-(2-metanossulfinil-6-metil-pirimidin-4-il)-4-metil-tiazol-2ila]-ureído}-N,N-dimetil-acetamida, 2-[3-(5-{2-[{2-dimetilamino-etif)-metilamino]-6-metil-pirimidin-4-ila}-4-metil-tiazol-2-il)-ureído]-N,N-dimetilacetamida, 1-(5-{2-[(2-dimetilamino-etil)-metil-amino]-pirimidin-4-ila}-4-metiltiazol-2-il)-3-[2-(5-etil-oxazol-2-il)-etil]-uréia, 1-[2-(5-etil-oxazol-2-il)-etil]-3-{5[2-(3-imid azo I-1 -il-propi lamino)-pirímid ίη-4-i la]-4-metil-tiazol-2-ila}-uréia, 1 -[2(1 -etil-1 H-imidazol-4-ií)-etil]-3-[5-(2-metanossulfinil-pirimidin-4-il)-4-metiltiazol-2-iía]-uréia e 1-(5-{2-[(2-dimetilamino-etil)-metil-amino]-pírimidin-4-ila}4-metil-tiazol-2-il)-3-[2-(1 -etil-1 H-imidazol-4-il)-etil]-uréia são preparados a partir dos intermediários apropriados (A, B e C) usando os procedimentos descritos acima para os compostos similares.
Exemplos de 73 a 81
Estes compostos, a saber, N-[5-(2-acetilamino-pirímidin-4-il)-4metil-tiazol-2-ila]-acetamida, N-[4-metil-5-(2-metilamino-pirirnidin-4-il)-tiazol2-ila]-acetamida, N-[5-(2-dimeti!amino-pirimidin-4-il)-4-metil-tiazol-2-ila]acetamida, N-{4-metil-5-[2, (2-metil-tiazol-4-il)-pirimidin-4-ila]-tiazol-2-ila}acetamida, N-[4-metil-5-(2-fenil-pirimidin-4-il)-tiazol-2-ila]-acetamida, N-[5-(2metanossulfonil-pirimidin-4-il)-4-metíl-tiazol-2-ila]-acetamida, N-[5-(2-metóxipirimidín-4-il)-4-metil-tiazol-2-ila]-acetamida, N-[4-metil-5-(2-piridin-4-ilpirimidin-4-il)-tiazol-2-ila]-acetamida e N-[4-metil-5-(2-piridin-2-il-pirimidin-4il)-tiazol-2-i!a]-acetamida são preparados por um procedimento análogo à N[4-metil-5-(2-morfolÍn-4-il-pirimidin-4-il)-tiazol-2-ila]-acetamida (Exemplo 25) mediante a substituição de bromidrato de morfolinaformamidina (parte 25c) com a amidina apropriada.
Exemplos de 82 a 86
Estes compostos, a saber, éster terc-butílico de ácido 3-{3-[5-(2ciclopropil-pirimidin-4-il)-4-metil-tiazol-2-ila]-ureído}-propiônico, 1-[5-(2ciclopropil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-[2-(5-etil-oxazol-2-il)-etil]-uréia, éster terc-butílico de ácido 3-{3-[5-(2-dimetilamino-pirimidin-4-il)-4-metÍItiazol-2-iia]-ureído}-propiônico, 1 -[5-(2-dimetilamino-pirimidin-4-il)-4-metiltiazol-2-ila]-3-[2-(5-etil-oxazol-2-il)-etil]-uréia, 1-[5-(2-dimetilamino-pirrmidin-486 il)-4-metil-tiazol-2-ila]-3-[2-(3-etil-[1,2,4]oxadiazol-5-il)-etil]-uréia são preparados a partir dos aminotiazóis 5-(2-ciclopropil-pirimidin-4-ii)-4-metiltiazol-2-ilamina e [4-(2-amino-4-metil-tiazol-5-il)-pirimidin-2-ila]-dimetii-amina (que é preparada pelo procedimento descrito no Exemplo 25d, 4-metil-5-(2morfolin-4-il-pirimidin-4-il)-tiazol-2-ilamina, mediante a substituição de bromidrato de morfolinaformamidina nesta seqüência pela amidina apropriada) pela reação com 1,Γ-carbonildiimidazol (métodos Ba ou Bb) para dar os intermediários de imidazol-uréia seguido pela reação com a amina apropriada usando o procedimento geral A.
Exemplo 87
N-[5-(2,6-Dimetil-pírimidin-4-il)'4-metil-tiazol-2“ila]-acetamida
Esta é preparada pela acilação de 5-(2,6-dimetil-pirimidin-4-il)-4metil-tiazol-2-ilamina (intermediário AF) usando o procedimento descrito para a N-(5-{2-[(2-dimetilamino-etil)~metil-amino]-6-metil-pirimidin-4-ila}-4metil-tiazol-2-il)-acetamida (Exemplo 39, etapa 2).
Exemplos 88 - 90
Estes compostos, a saber, 1-[5-(2,6-dimetil-pirimidin-4-il)-4-metiltiazol-2-ila]-3-[2-(5-etil-oxazol-2-il)-etill-uréia, 1-[5-(2,6-dimetil-pirimÍdin-4-il)-4metil-tiazol-2-ila]-3-[2-(1-etil-1H-imidazol-4-il)-etil]-uréia e 1-[5-(2,6-dimetilpirimidin-4-il)-4-metil-tiazol-2-ila]-3-[2-(1-propil-1H-imidazol-4-il)-etil]-uréia são preparados a partir da [5-(2,6-dimetil-pirimidin-4-il)-4-metil-tiazol-2-ila]-amida de ácido imidazol-1-carboxílico (BF) usando a amina apropriada (CA, CC1 e CC2).
Exemplo 91
5-(2-terc-Butil-pirÍmidin-4-il)-4-metil-tiazol-2-ilamina
Este composto é preparado como descrito anteriormente (Intermediário AG).
Exemplo 92
N-[5-(2-terc-Butil-pirimidin-4-il)-4-metil-tiazol-2-ifa]-acetamida
Este material é preparado pela acilação de 5-(2-terc-Butilpirimidin-4-il)-4-metil-tiazol-2-ilamina (Intermediário AG) com anidrido acético como descrito para a N-(5-{2-[(2-dimetilamino-etil)-metil-amino]-6-metil87 pirimidin-4-ila)-4-metil-tiazol-2-il)-acetamida (Exemplo 39, etapa 2).
Exemplos de 93 a 96
Estes compostos, a saber, éster etilico de ácido 4-[5-(2-tercbutil-pirimÍdin-4-il)-4-metil-tiazol-2-ilcarbamoíla]-butírico, [5-(2-terc-butilpirimidin-4“il)-4-metil-tiazol-2-ila]-metil-amina, [5-(2-terc-butil-pirimidin-4-il)-4metil-tiazol-2-ila]-amida de ácido 4-oxo-pentanóico e [5-(2-terc-butil-pirimidin4-il)-4-metil-tiazol-2-ila]-amida de ácido 5-oxo-hexanóico são preparados como se segue: Uma mistura de 5-(2-Ιθπ>ΑυύΙ-ρΙπιτ^ίη-4“ϋ)-4-ιτιθυΐ-Ιί3ΖθΙ-2ilamina (intermediário de aminotiazol AG, 0,297 g, 0,12 mmol) e trietilamina (0,25 ml, 1,8 mmol) em DCM (1 ml) é adicionada a uma solução agitada do ácido carboxílico apropriado (0,12 mmol), HOBT (0,0162 g, 0,12 mmol), EDCI.HCI (0,0299 g, 0,156 mmol) e trietilamina (0,025 ml, 0,18 mmol) em DCM (1 ml). Após 18 horas a mistura de reação é filtrada sob gravidade através de um cartucho contendo resina de isocianato sustentado por polímero (0,5 g, pré-lavada com 4 ml THF). Os solventes são removidos para dar os produtos do título que são purificados por HPLC se exigido. Exemplos de 97 a 143
Estes compostos, a saber, 1-[2-(5-terc-butil-oxazol-2-il)-etil]-3-[5(2,6-dimetil-piridin-4'il)-4-metil-tiazol-2-ila]-uréia, éster etilico de ácido 4-{3[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol'2-ila]-ureído}-butírico, 1-[5-(2-tercbutil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(2-propóxi-etil)-uréia, 2-{3-[5-(2-tercbutil-pirimidin-4-il)-4-metil-tiazol-2“ila]-ureído}-N,N-dimetil-acetamida, 3-{3-[5(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-ureído}-N-(2-hidróxi-1,1-dimetiletil)-propionamida, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(3,3dietóxi-propila)-uréia, éster etilico de ácido N-[5-(2-terc-butil-pirimidin-4-il)-4metil-tiazol-2-ila]-succinâmico, 1-[5-(2-terc-butil-pirímidin-4-il)-4-metil-tiazol-2ila]-3-(3-hidróxi-propila)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2ila]-3'piridin-2-ilmetil-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4“metÍI-tiazol-2-ila]3-piridin-3-ilmetil-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3piridin-4-ilmetil-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(2metóxi-eti!)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(2hídróxi-etil)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(4' hidróxi-butila)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(2dimetilamino-etil)-uréia, 1-[5-(2-terc-butil-pirimidin-4-ii)-4-metil-tiazol-2-ila]-3(2-pirrolidin-1-il-etil)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]3- (2-piperidin-1 -i l-etil)-uréia, 1 -[5-(2-terc-butil-pirimid in-4-i I )-4-metil-tiazoi-2ila]-3-(3-morfolin-4-il-propila)-uréia, 1-[5-(2-terc-bittÍI-pirimidin-4-il)-4-metiltiazo!-2-ila]-3-(2-dietilamino-etil)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metiltiazol-2-ila]-3-(2-oxo-tetrahidro-furan-3-il)-uréia, 1-[5-(2-terc-butil-pirimidin-4il)-4-metil-tiazol-2-ila]-3-[2-(1 -metil-pirrolidin-2-il)-etil]-uréia, 1-[5-(2-terc-bulilpirimidin-4-il)-4-metil-tiazol-2-ila]-3-[2-(2-hidróxi-etóxi)-etii]-uréia, 1-[5-(2-tercbutil-pirimidin-4-il)-4-metil-tiazol-2-íla]-3-(1-metil-1 H-pirrol-2-ilmetil)-uréia, 1[5-(2-terc-butil-pirimidin-4-ií)-4-metil-tiazol-2-ila]-3-(2-etóxi-etil)-uréia, 1-(5-(2terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(2-hidróxi-1 -metil-etil)-uréia, 1-[5(2-terc-butil-pirimidin-4'il)-4-metil-tiazol-2-iía]'3-(2-metóxi-1-metil-etil)-uréia, éster etílico de ácido 2-{3-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazoi-2-ila]ureídoj-propiônico, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(2,2dietóxi-etil)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3-(3isopropóxi-propila)-uréia, 1-(5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3(3-etóxi-propila)-uréia, 1-[5-(2-terc-butil-pirimjdin-4-il)-4-metil-tiazol-2-ila]-3(2,3-dihidróxi-propila)-uréia, éster terc-butílico de ácido 2-{3-[5-(2-terc-butilpirimidin-4-il)-4-metil-tiazol-2-ila]-ureído}-2-metil-propiôníco, [5-(2-terc-butilpirimidin-4-il)-4-metil-tiazol-2-ila]-annida de ácido 3-metanossulfonilpirrolid ina-1 -carboxílico, 1 -[5-(2-terc-butil-pirimidin-4-il)-4-metil-tÍazol-2-ila]-3(2-piridin-2-il-etil)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-3(2-hidróxi-1,1-dimetil-etil)-uréia, 3-{3-[5-(2-terc-butil-pirimidin-4-il)-4-metiltiazol-2-ila]-ureído}-N-(2-oxo-tetrahidro-furan-3-il)-propionamida, 1-((5-(2terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-amida} 2-amida de ácido pirrolidina-1,2-dicarboxílico, [5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]amida de ácido 1,4,6,7-tetrahidro-imidazo[4,5-c]piridina-5-carboxílico, [5-(2terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-amida de ácido 3-hidróxipirrolidina-1-carboxílico, [5-(2-terc-butil-pirimidín-4-il)-4-metil-tiazol-2-ila]amida de ácido 3-hidróxi-pirrolídina-1-carboxílico, 1-[5-(2-terc-butil-pirimÍdin4- il)-4-metil-tiazoí-2-ila]-3-(1 H-pirazoi-3-il)-uréia, 1-[5-(2-terc-butil-pírimídin-489 i I )-4-metil-tiazo l-2-ila]-3-(2-i midazo 1-1 -il-etil)-uréia, 1 -[5-(2-terc-b util-p irim idin4-il)-4-metil-tiazol-2-iia]-3-[2-( 1 -isopropil-1 H-imidazol-4-il)-etil]-uréia, 3-{3-[5(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-ureído}-N-(3-hidróxi-2,2-dimetilpropila)-propionamida, 1 -[5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-35 (2,3-dihidróxi-propila)-uréia, 1-[5-(2-terc-butil-pirimidin-4-il)-4-metÍl-tiazol-2ila]-3-(2,3-dihidróxi-propila)-uréia, e 1 -[5-(2-terc-butil-pirimidin-4-il)-4-metiltiazol-2-ila]-3-(5-isopropil-[1,3,4]oxadiazol-2-ilmetil)-uréia são preparados a partir de [5-(2-terc-butil-pirimidin-4-il)-4-metil-tiazol-2-ila]-amida de ácido imidazol-1-carboxílico (intermediário de imidazol-uréia BG) e a amina apropriada (intermediário de amina C) usando o procedimento geral A. Exemplos 144-146
Estes compostos, a saber, [5-(2-terc-butil-pirimidin-4-il)-4-metiltiazol-2-ila]-pirazin-2-il-amina, (5-bromo-[1,3,4]tiadiazol-2-il)-[5-(2-terc-butilpirimidin-4-il)-4-metíl-tiazol-2-ila]-amina e [5-(2-terc-butil-pirimidin-4-il)-415 metil-tiazol-2-ila]-(6-metil-piridin-2-il)-amina são preparados pela reação da intermediário de tiouréia apropriado (DA, DB, DC) com 1-bromo-1-(2-tercbutil-pirimidin-4-il)-propan-2-ona (intermediário de aminotiazol AG3) seguindo o procedimento descrito para a preparação de 5-(2-terc-butilpirimidin-4-il)-4-metÍl-tiazol-2-ilamina (AG4) no Exemplo 91.
Claims (11)
- REIVINDICAÇÕES1. Composto, caracterizado pelo fato de que apresenta a fórmula I na forma livre ou de sal, na qual,R1 é -CO-NR*Ry, sendo que R* e Ry junta mente com o nitrogênio a qual eles são ligados formam um anel N-heterocíclico de 5 a 12 membros substituído ou não-substituído opcionalmente incluindo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, sendo que os substituintes opcionais são selecionados a partir de hidróxi, -SO2-CH3 ou aminocarbonila;R2 é Ci-C3-alquila;Y é carbono ou nitrogênio; eR3 e R4 são cada um índependentemente hidrogênio, halo, hidróxi, ciano, amina, carbóxi, Ct-Ca-alquilsulfanila, CrCs-alquilsulfiníla, Ci-Caalquilsulfonila, -SO2NH2, Ci-C8-alquila, Ci-C8-haloalquila, amino-Ci-Ce-alquila, amino-Ci-Ca-alcóxi, Ci-Cs-alquilaminocarbonila, di(Ci - Cg-a I q u ΪΙ Jaminocarbonila, dí(Ci-Ce-alquil)amíno-Ci-Cs-alquila, di{Ci-Ce-alquü)amino-Ci-Ce-aloóxi, Ci-C$acilamino, aminocarbonila, Ci-C8-alcoxícarbonila, carbóxi-Ci-C8-alquila, carbóxi-CrCg-alcóxi, um C3-Ci5-carbocíclíco, um anel heterociclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, Ci-C8-alquilamino ou di(Ci-C8alquil)amino cada um sendo opcionalmente substituído por amino, hidróxi, di(Ci-Ca-alquil)amino ou um anel heterociclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre, ou Ci-Ce-alcóxi opcionalmente substituído por um anel heterociclico de 5 ou 6 membros tendo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre.
- 2. Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que o anel N-heterocíclico de 5 a 12 membros é substituído por hidróxi,-SO2-CH3 ou aminocarbonila,
- 3. Composto, de acordo com a reivindicação 1 ou 2, caracterizado pelo fato de que R1 é -CO-NRxRy, sendo que R* e Ry juntamente com o5 nitrogênio a qual eles são ligados formam um anel N-heterocíclico de 5 a 12 membros substituído ou não-substituído selecionados do grupo consistindo em pirrolidina, morfolina e tetra-hidro-imidazo-piridina.
- 4. Composto, de acordo com qualquer uma das reivindicações 1 a 3, caracterizado pelo fato de que R1 é -CO-NR*Ry, sendo que Rx e Ry10 juntamente com o nitrogênio a qual eles são ligados formam um anel Nheterociclíco de 5 a 9 membros substituído ou não-substituído opcionalmente incluindo um ou mais heteroátomos de anel selecionados do grupo consistindo em oxigênio, nitrogênio e enxofre.
- 5. Composto, de acordo com a reivindicação 1, caracterizado pelo15 fato de que também é um composto que apresenta a fórmula XXI na qual R1, Y, Ra e Rb são como apresentados na tabela que segue:
R' Y R·1 Rh § - 0 N H,C \ CH., ch3 1 ti 0 II ? 'Ύ) N .<L H.C \ CH, CH, 1 ti ! ÃO- H N C. H.C \ CH. ch3 H R' Y R‘ R1’ 0 x T>i,oh N C. H,C \ CH, CHj 3 íi o II /c-'O_.°h N H,C \ CH, CH, 4 ii - 6. Composto, de acordo com qualquer uma das reivindicações 1 a 5, caracterizado pelo fato de que é para uso como um produto farmacêutico,
- 7. Composto, de acordo com qualquer uma das reivindicações 1 a 5, caracterizado pelo fato de que é para combinar com outra substância de fármaco.
- 8. Composto, de acordo com a reivindicação 7, caracterizado pelo fato de que é para combinar com outra substância de fármaco, que é um antiinflamatório, um broncodilatador, um anti-histamínico, um descongestionante ou uma substância de fármaco de drogas antitussígeno.
- 9. Composição farmacêutica, caracterizada pelo fato de que compreende como ingrediente ativo um composto, como definido em qualquer uma das reivindicações 1 a 5.
- 10. Uso de um composto, como definido em qualquer uma das reivindicações 1 a 5, caracterizado pelo fato de que é para fabricação de um medicamento para o tratamento de uma doença mediada pela fosfatidilínositol 3-cinase.
- 11. Uso de um composto, como definido em qualquer uma das reivindicações 1 a 5, caracterizado pelo fato de que é para fabricação de um medicamento para o tratamento de doenças respiratórias, alergias, artrite reumatóide, osteoartrite, distúrbios reumáticos, psoríase, colite ulcerativa, doença de Crohn, choque séptico, distúrbios proliferativos tais como câncer, aterosclerose, rejeição de aloenxerto seguinte ao transplante, diabetes, acidente vascular cerebral, obesidade ou restenose.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0310234 | 2003-05-02 | ||
| GB0310234.0 | 2003-05-02 | ||
| PCT/EP2004/004603 WO2004096797A1 (en) | 2003-05-02 | 2004-04-30 | Inhibitors of phosphatidylinositol 3-kinase |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0410037A BRPI0410037A (pt) | 2006-04-25 |
| BRPI0410037B1 true BRPI0410037B1 (pt) | 2018-01-16 |
| BRPI0410037B8 BRPI0410037B8 (pt) | 2021-05-25 |
Family
ID=33397059
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0410037A BRPI0410037B8 (pt) | 2003-05-02 | 2004-04-30 | inibidores de fosfatidilinositol 3-cinase, sua composição farmacêutica, e seu uso |
Country Status (28)
| Country | Link |
|---|---|
| US (3) | US20070032487A1 (pt) |
| EP (2) | EP2157091B1 (pt) |
| JP (1) | JP4510807B2 (pt) |
| KR (1) | KR100725885B1 (pt) |
| CN (2) | CN1816549B (pt) |
| AR (1) | AR044519A1 (pt) |
| AT (1) | ATE445614T1 (pt) |
| AU (2) | AU2004234068B2 (pt) |
| BR (1) | BRPI0410037B8 (pt) |
| CA (1) | CA2524401C (pt) |
| CL (1) | CL2004000917A1 (pt) |
| DE (1) | DE602004023602D1 (pt) |
| EC (1) | ECSP056127A (pt) |
| ES (2) | ES2546851T3 (pt) |
| HK (1) | HK1091810A1 (pt) |
| IL (1) | IL171536A (pt) |
| IS (1) | IS8152A (pt) |
| MA (1) | MA27774A1 (pt) |
| MX (1) | MXPA05011740A (pt) |
| NO (1) | NO20055714L (pt) |
| PE (1) | PE20050147A1 (pt) |
| PL (1) | PL1622897T3 (pt) |
| PT (1) | PT1622897E (pt) |
| RU (2) | RU2384580C2 (pt) |
| TN (1) | TNSN05278A1 (pt) |
| TW (1) | TW200519106A (pt) |
| WO (1) | WO2004096797A1 (pt) |
| ZA (1) | ZA200508603B (pt) |
Families Citing this family (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW200519106A (en) * | 2003-05-02 | 2005-06-16 | Novartis Ag | Organic compounds |
| TW200533357A (en) * | 2004-01-08 | 2005-10-16 | Millennium Pharm Inc | 2-(amino-substituted)-4-aryl pyrimidines and related compounds useful for treating inflammatory diseases |
| SE0402735D0 (sv) | 2004-11-09 | 2004-11-09 | Astrazeneca Ab | Novel compounds |
| WO2006066172A1 (en) * | 2004-12-17 | 2006-06-22 | Amgen, Inc. | Aminopyrimidine compounds and methods of use |
| US20060178388A1 (en) | 2005-02-04 | 2006-08-10 | Wrobleski Stephen T | Phenyl-substituted pyrimidine compounds useful as kinase inhibitors |
| WO2006125805A1 (en) * | 2005-05-24 | 2006-11-30 | Laboratoires Serono S.A. | Thiazole derivatives and use thereof |
| EA017166B1 (ru) | 2005-05-24 | 2012-10-30 | Мерк Сероно С.А. | Производные тиазола и их применение |
| WO2006135604A2 (en) * | 2005-06-09 | 2006-12-21 | Merck & Co., Inc. | Inhibitors of checkpoint kinases |
| GB2431156A (en) * | 2005-10-11 | 2007-04-18 | Piramed Ltd | 1-cyclyl-3-substituted- -benzenes and -azines as inhibitors of phosphatidylinositol 3-kinase |
| EA200801716A1 (ru) * | 2006-01-18 | 2009-04-28 | Амген Инк. | Тиазольные соединения и их применение |
| EP1991528A2 (en) * | 2006-01-18 | 2008-11-19 | Siena Biotech S.p.A. | Modulators of alpha7 nicotinic acetylcholine receptors and therapeutic uses thereof |
| CA2635830A1 (en) * | 2006-01-23 | 2007-07-26 | Laboratoires Serono S.A. | Thiazole derivatives and use thereof |
| US8217037B2 (en) * | 2006-04-07 | 2012-07-10 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Thiazole and thiophene analogues, and their use in treating autoimmune diseases and cancers |
| EP2016075A1 (en) * | 2006-05-03 | 2009-01-21 | AstraZeneca AB | Thiazole derivatives and their use as anti-tumour agents |
| GB0612630D0 (en) | 2006-06-26 | 2006-08-02 | Novartis Ag | Organic compounds |
| EP2061787A2 (en) * | 2006-09-01 | 2009-05-27 | Vertex Pharmaceuticals Incorporated | 5-(2-furyl)-1,3-thiazole derivatives useful as inhibitors of phosphatidylinositol 3-kinase |
| GB0620059D0 (en) * | 2006-10-10 | 2006-11-22 | Ucb Sa | Therapeutic agents |
| GB0620818D0 (en) * | 2006-10-19 | 2006-11-29 | Ucb Sa | Therapeutic agents |
| EP2129379B1 (en) * | 2007-02-20 | 2019-04-10 | Novartis AG | Imidazoquinolines as dual lipid kinase and mtor inhibitors |
| TW200906825A (en) * | 2007-05-30 | 2009-02-16 | Scripps Research Inst | Inhibitors of protein kinases |
| EP2166843A4 (en) | 2007-06-01 | 2010-08-11 | Univ Princeton | TREATMENT OF VIRUS INFECTIONS BY MODULATION OF HOST CELL METABOLISMS |
| EP2173728A2 (en) | 2007-07-17 | 2010-04-14 | Amgen Inc. | Heterocyclic modulators of pkb |
| AU2008276512A1 (en) * | 2007-07-17 | 2009-01-22 | Amgen Inc. | Thiadiazole modulators of PKB |
| EP2238134A2 (en) * | 2007-12-20 | 2010-10-13 | Novartis AG | Bis-thiazole derivatives, process for their preparation and their use as medicaments |
| KR20100093129A (ko) * | 2007-12-20 | 2010-08-24 | 노파르티스 아게 | Pi 3 키나제 억제제로서 사용되는 티아졸 유도체 |
| WO2009094224A1 (en) | 2008-01-25 | 2009-07-30 | Millennium Pharmaceuticals, Inc. | Thiophenes and their use as phosphatidylinositol 3-kinase (pi3k) inhibitors |
| CA2716898A1 (en) | 2008-02-27 | 2009-09-03 | Takeda Pharmaceutical Company Limited | Compound having 6-membered aromatic ring |
| UA104147C2 (uk) * | 2008-09-10 | 2014-01-10 | Новартис Аг | Похідна піролідиндикарбонової кислоти та її застосування у лікуванні проліферативних захворювань |
| US8796314B2 (en) | 2009-01-30 | 2014-08-05 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US9090601B2 (en) | 2009-01-30 | 2015-07-28 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
| CA2750935A1 (en) * | 2009-01-30 | 2010-08-12 | Millennium Pharmaceuticals, Inc. | Heteroaryls and their use as pi3k inhibitors |
| JPWO2010125799A1 (ja) * | 2009-04-27 | 2012-10-25 | 塩野義製薬株式会社 | Pi3k阻害活性を有するウレア誘導体 |
| ME02900B (me) * | 2009-04-30 | 2018-04-20 | Glaxo Group Ltd | Indazoli supstituisani oksazolom kao inhibitori pi3-kinaze |
| EP2440556A1 (en) * | 2009-06-10 | 2012-04-18 | Vertex Pharmaceuticals Incorporated | Inhibitors of phosphatidylinositol 3-kinase |
| US8293753B2 (en) | 2009-07-02 | 2012-10-23 | Novartis Ag | Substituted 2-carboxamide cycloamino ureas |
| UY32748A (es) | 2009-07-02 | 2011-01-31 | Novartis Ag | 2-carboxamida-cicloamino-ureas |
| WO2011048936A1 (ja) | 2009-10-19 | 2011-04-28 | 大正製薬株式会社 | アミノチアゾール誘導体 |
| BR112012019459A2 (pt) | 2010-02-03 | 2017-10-17 | Signal Pharm Llc | identificação de mutação de lkb1 como um biomarcador preditivo para sensibilidade para inibidores de tor quinase. |
| MX2013001660A (es) | 2010-08-11 | 2013-06-03 | Millenium Pharmaceuticals Inc | Heteroarilos y usos de los mismos. |
| WO2012021615A1 (en) | 2010-08-11 | 2012-02-16 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| WO2012021611A1 (en) | 2010-08-11 | 2012-02-16 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| WO2012051410A2 (en) | 2010-10-13 | 2012-04-19 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US20130209461A1 (en) * | 2010-11-08 | 2013-08-15 | Novartis Ag | Use of 2-carboxamide cycloamino urea derivatives in the treatment of EGFR dependent diseases or diseases that have acquired resistance to agents that target EGFR family members |
| AU2015203865B2 (en) * | 2010-11-08 | 2016-11-03 | Novartis Ag | Use of 2-carboxamide cycloamino urea derivatives in the treatment of EGFR dependent diseases or diseases that have acquired resistance to agents that target EGFR family members |
| UA112539C2 (uk) * | 2011-03-03 | 2016-09-26 | Новартіс Аг | Спосіб одержання похідних 2-карбоксамідциклоаміносечовини |
| PE20140601A1 (es) * | 2011-04-25 | 2014-05-24 | Novartis Ag | COMBINACION DE UN INHIBIDOR DE CINASA DE FOSFATIDIL-INOSITOL-3 (PI3K) Y UN INHIBIDOR DE mTOR |
| MX359406B (es) | 2011-07-01 | 2018-09-27 | Novartis Ag | Terapia de combinación que comprende un inhibidor de cdk4/6 y un inhibidor de pi3kpara usarse en el tratamiento de cáncer. |
| US20130158023A1 (en) | 2011-08-03 | 2013-06-20 | Signal Pharmaceuticals, Llc | Identification of gene expression as a predictive biomarker for lkb1 status |
| US9926309B2 (en) | 2011-10-05 | 2018-03-27 | The Board Of Trustees Of The Leland Stanford Junior University | Pi-kinase inhibitors with anti-infective activity |
| WO2013148912A1 (en) * | 2012-03-30 | 2013-10-03 | Novartis Ag | Compounds for use in the treatment of neuroblastoma, ewing's sarcoma or rhabdomyosarcoma |
| AU2013203714B2 (en) | 2012-10-18 | 2015-12-03 | Signal Pharmaceuticals, Llc | Inhibition of phosphorylation of PRAS40, GSK3-beta or P70S6K1 as a marker for TOR kinase inhibitory activity |
| EP3251673A1 (en) | 2012-12-13 | 2017-12-06 | IP Gesellschaft für Management mbH | Combination therapy comprising a cdk4/6 inhibitor and a pi3k inhibitor for use in the treatment of cancer |
| TWI628170B (zh) | 2013-02-05 | 2018-07-01 | 先正達合夥公司 | 植物生長調節化合物 |
| BR112015026292B1 (pt) | 2013-04-17 | 2022-04-12 | Signal Pharmaceuticals, Llc | Uso de 1-etil-7-(2-metil-6-(1h-1,2,4-triazol-3-il)piridin-3-il)-3,4-dihidropirazino [2,3-b]pirazin-2(1h)- ona e métodos in vitro |
| KR20160002792A (ko) | 2013-04-17 | 2016-01-08 | 시그날 파마소티칼 엘엘씨 | 암 치료용 tor 키나제 억제제 및 n-(3-(5-플루오로-2-(4-(2-메톡시에톡시)페닐아미노)피리미딘-4-일아미노)페닐)아크릴아미드를 포함하는 병용 요법 |
| CN105392499B (zh) | 2013-04-17 | 2018-07-24 | 西格诺药品有限公司 | 用于治疗癌症的包含tor激酶抑制剂和胞苷类似物的组合疗法 |
| UA115805C2 (uk) | 2013-04-17 | 2017-12-26 | Сігнал Фармасьютікалз, Елелсі | Комбінована терапія, яка включає сполуку дигідропіразинопіразину й антагоніст рецептора андрогену, для лікування раку простати |
| AU2014254050B2 (en) | 2013-04-17 | 2018-10-04 | Signal Pharmaceuticals, Llc | Pharmaceutical formulations, processes, solid forms and methods of use relating to 1-ethyl-7-(2-methyl-6-(1h-1,2,4-triazol-3-yl) pyridin-3-yl) -3,4-dihydropyrazino[2,3-b]pyrazin-2(1h)-one |
| CA2909625C (en) | 2013-04-17 | 2021-06-01 | Signal Pharmaceuticals, Llc | Combination therapy comprising a tor kinase inhibitor and a 5-substituted quinazolinone compound for treating cancer |
| KR102271344B1 (ko) | 2013-04-17 | 2021-07-01 | 시그날 파마소티칼 엘엘씨 | 디하이드로피라지노-피라진을 사용한 암의 치료 |
| CN105407892B (zh) | 2013-05-29 | 2019-05-07 | 西格诺药品有限公司 | 一种化合物的药物组合物、其固体形式及它们的使用方法 |
| NZ714742A (en) | 2014-04-16 | 2017-04-28 | Signal Pharm Llc | Solid forms of 1-ethyl-7-(2-methyl-6-(1h-1,2,4-triazol-3-yl)pyridin-3-yl)-3,4-dihydropyrazino[2,3-b]pyrazin-2(1h)-one, compositions thereof and methods of their use |
| EP3131551A4 (en) | 2014-04-16 | 2017-09-20 | Signal Pharmaceuticals, LLC | SOLID FORMS COMPRISING 1-ETHYL-7-(2-METHYL-6-(1H-1,2,4-TRIAZOL-3-YL) PYRIDIN-3-YL)-3,4-DIHYDROPYRAZINO(2,3-b)PYRAZIN-2(1H)-ONE, AND A COFORMER, COMPOSITIONS AND METHODS OF USE THEREOF |
| CN104803921A (zh) * | 2015-03-14 | 2015-07-29 | 长沙深橙生物科技有限公司 | 一种苯取代嘧啶衍生物的制备方法 |
| CN104744376A (zh) * | 2015-03-14 | 2015-07-01 | 长沙深橙生物科技有限公司 | 一种2-异丙基嘧啶衍生物的制备方法 |
| US11091472B2 (en) | 2016-02-26 | 2021-08-17 | The Regents Of The University Of California | PI-kinase inhibitors with anti-infective activity |
| JP7282045B2 (ja) | 2017-06-22 | 2023-05-26 | セルジーン コーポレイション | B型肝炎ウイルス感染を特徴とする肝細胞癌の治療 |
| US11325900B2 (en) | 2018-01-15 | 2022-05-10 | Aucentra Holdings Pty Ltd | 5-(pyrimidin-4-yl)thiazol-2-yl urea derivatives as therapeutic agents |
| JP7309725B2 (ja) | 2018-08-21 | 2023-07-18 | 杏林製薬株式会社 | 2環性ヘテロ芳香環誘導体 |
| AU2021206651B2 (en) * | 2020-01-07 | 2024-02-22 | Disarm Therapeutics, Inc. | Inhibitors of SARM1 |
Family Cites Families (111)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1219606A (en) | 1968-07-15 | 1971-01-20 | Rech S Et D Applic Scient Soge | Quinuclidinol derivatives and preparation thereof |
| GB8302591D0 (en) * | 1983-01-31 | 1983-03-02 | Fujisawa Pharmaceutical Co | Thiazole derivatives |
| US4649146A (en) | 1983-01-31 | 1987-03-10 | Fujisawa Pharmaceutical Co., Ltd. | Thiazole derivatives and pharmaceutical composition comprising the same |
| DE3703435A1 (de) | 1987-02-05 | 1988-08-18 | Thomae Gmbh Dr K | Neue thiazole, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| EP0373226B1 (en) | 1988-04-15 | 1994-06-15 | Taiho Pharmaceutical Company, Limited | Carbamoyl-2-pyrrolidinone compounds |
| GB8916480D0 (en) | 1989-07-19 | 1989-09-06 | Glaxo Group Ltd | Chemical process |
| GB8923590D0 (en) | 1989-10-19 | 1989-12-06 | Pfizer Ltd | Antimuscarinic bronchodilators |
| CA2038379C (en) | 1990-03-22 | 2002-11-05 | Norma G. Delaney | Trifluoromethyl mercaptan and mercaptoacyl derivatives and method of using same |
| US5451700A (en) | 1991-06-11 | 1995-09-19 | Ciba-Geigy Corporation | Amidino compounds, their manufacture and methods of treatment |
| GB9301000D0 (en) | 1993-01-20 | 1993-03-10 | Glaxo Group Ltd | Chemical compounds |
| GB9414208D0 (en) | 1994-07-14 | 1994-08-31 | Glaxo Group Ltd | Compounds |
| GB9414193D0 (en) | 1994-07-14 | 1994-08-31 | Glaxo Group Ltd | Compounds |
| GB9622386D0 (en) | 1996-10-28 | 1997-01-08 | Sandoz Ltd | Organic compounds |
| EP0946587A2 (en) | 1996-12-16 | 1999-10-06 | Fujisawa Pharmaceutical Co., Ltd. | New amide compounds |
| US6187797B1 (en) * | 1996-12-23 | 2001-02-13 | Dupont Pharmaceuticals Company | Phenyl-isoxazoles as factor XA Inhibitors |
| TW528755B (en) | 1996-12-24 | 2003-04-21 | Glaxo Group Ltd | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| US6166037A (en) | 1997-08-28 | 2000-12-26 | Merck & Co., Inc. | Pyrrolidine and piperidine modulators of chemokine receptor activity |
| EP1027050B1 (en) | 1997-10-27 | 2004-01-14 | Takeda Chemical Industries, Ltd. | 1,3-thiazoles as adenosine a3 receptor antagonists for the treatment of allergy, asthma and diabetes |
| GB9723590D0 (en) | 1997-11-08 | 1998-01-07 | Glaxo Group Ltd | Chemical compounds |
| GB9723566D0 (en) | 1997-11-08 | 1998-01-07 | Glaxo Group Ltd | Chemical compounds |
| GB9723589D0 (en) | 1997-11-08 | 1998-01-07 | Glaxo Group Ltd | Chemical compounds |
| YU44900A (sh) | 1998-01-31 | 2003-01-31 | Glaxo Group Limited | Derivati 2-(purin-9-il)tetrahidrofuran-3,4-diola |
| PE20000270A1 (es) | 1998-02-14 | 2000-05-20 | Glaxo Group Ltd | Derivados de 2-(purin-9-il)-tetrahidrofuran-3,4-diol |
| DE69923681T2 (de) | 1998-06-18 | 2006-01-12 | Bristol-Myers Squibb Co. | Kohlenstoff substituierte aminothiazole als inhibitoren von cyclin-abhängigen kinasen |
| GB9813565D0 (en) | 1998-06-23 | 1998-08-19 | Glaxo Group Ltd | Chemical compounds |
| ATE246701T1 (de) | 1998-06-23 | 2003-08-15 | Glaxo Group Ltd | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivate |
| GB9813535D0 (en) | 1998-06-23 | 1998-08-19 | Glaxo Group Ltd | Chemical compounds |
| GB9813540D0 (en) | 1998-06-23 | 1998-08-19 | Glaxo Group Ltd | Chemical compounds |
| EP1098923B1 (en) | 1998-06-30 | 2004-11-17 | Dow Global Technologies Inc. | Polymer polyols and a process for the production thereof |
| DE69932173T2 (de) | 1998-10-16 | 2007-06-06 | Pfizer Inc. | Adeninderivate |
| GB9913083D0 (en) | 1999-06-04 | 1999-08-04 | Novartis Ag | Organic compounds |
| SK286666B6 (sk) * | 1999-04-23 | 2009-03-05 | Takeda Pharmaceutical Company Limited | 5-Pyridyl-1,3-azolové zlúčeniny, spôsob výroby a použitia |
| EP1659111A3 (en) | 1999-05-04 | 2007-05-09 | Schering Corporation | Piperidine derivatives useful as ccr5 antagonists |
| BR0010304A (pt) | 1999-05-04 | 2002-02-13 | Schering Corp | Derivados de piperazina úteis como antagonistas do ccr5 |
| YU25500A (sh) | 1999-05-11 | 2003-08-29 | Pfizer Products Inc. | Postupak za sintezu analoga nukleozida |
| GB9913932D0 (en) | 1999-06-15 | 1999-08-18 | Pfizer Ltd | Purine derivatives |
| US6322771B1 (en) | 1999-06-18 | 2001-11-27 | University Of Virginia Patent Foundation | Induction of pharmacological stress with adenosine receptor agonists |
| ES2165768B1 (es) | 1999-07-14 | 2003-04-01 | Almirall Prodesfarma Sa | Nuevos derivados de quinuclidina y composiciones farmaceuticas que los contienen. |
| CZ2002861A3 (cs) * | 1999-09-10 | 2002-06-12 | Merck & Co., Inc. | Inhibitory tyrosinkinázy |
| CO5180581A1 (es) | 1999-09-30 | 2002-07-30 | Pfizer Prod Inc | Compuestos para el tratamiento de la isquemia ciones farmaceuticas que los contienen para el tratamiento de la isquemia |
| GB9924361D0 (en) | 1999-10-14 | 1999-12-15 | Pfizer Ltd | Purine derivatives |
| GB9924363D0 (en) | 1999-10-14 | 1999-12-15 | Pfizer Central Res | Purine derivatives |
| GB0003960D0 (en) | 2000-02-18 | 2000-04-12 | Pfizer Ltd | Purine derivatives |
| WO2001072745A1 (en) | 2000-03-29 | 2001-10-04 | Cyclacel Limited | 2-substituted 4-heteroaryl-pyrimidines and their use in the treatmetn of proliferative disorders |
| TWI227240B (en) | 2000-06-06 | 2005-02-01 | Pfizer | 2-aminocarbonyl-9H-purine derivatives |
| PL201530B1 (pl) | 2000-06-27 | 2009-04-30 | L V A T Lab Sa | Karbaminiany pochodne aryloalkiloamin i zastosowanie karbaminianów pochodnych aryloalkiloamin |
| GB0015727D0 (en) | 2000-06-27 | 2000-08-16 | Pfizer Ltd | Purine derivatives |
| GB0015876D0 (en) | 2000-06-28 | 2000-08-23 | Novartis Ag | Organic compounds |
| DE10038639A1 (de) | 2000-07-28 | 2002-02-21 | Schering Ag | Nichtsteroidale Entzündungshemmer |
| AU2001275760B2 (en) | 2000-08-05 | 2005-03-17 | Glaxo Group Limited | 6.alpha., 9.alpha.-difluoro-17.alpha.-'(2-furanylcarboxyl) oxy!-11.beta.-hydroxy-16.alpha.-methyl-3-oxo-androst-1,4,-diene-17-carbothioic acid S-fluoromethyl ester as an anti-inflammatory agent |
| GB0022695D0 (en) | 2000-09-15 | 2000-11-01 | Pfizer Ltd | Purine Derivatives |
| KR100589032B1 (ko) | 2000-10-20 | 2006-06-14 | 에자이 가부시키가이샤 | 질소 함유 방향환 유도체 |
| GB0028383D0 (en) | 2000-11-21 | 2001-01-03 | Novartis Ag | Organic compounds |
| IL156499A0 (en) | 2000-12-22 | 2004-01-04 | Almirall Prodesfarma Ag | Quinuclidine carbamate derivatives and their use as m3 antagonists |
| JP4295985B2 (ja) | 2000-12-28 | 2009-07-15 | ラボラトリオス・アルミラル・ソシエダッド・アノニマ | 新規なキヌクリジン誘導体およびそれを含む医薬組成物 |
| EP1241176A1 (en) | 2001-03-16 | 2002-09-18 | Pfizer Products Inc. | Purine derivatives for the treatment of ischemia |
| ES2314052T3 (es) | 2001-04-30 | 2009-03-16 | Glaxo Group Limited | Derivados de ester de 17beta-carbotioato de androstano con un grupo ester ciclico en la posicion 17alfa. |
| ATE315555T1 (de) | 2001-05-11 | 2006-02-15 | Pfizer Prod Inc | Thiazolderivate und ihre verwendung als cdk- inhibitoren |
| SK14302003A3 (sk) | 2001-05-25 | 2004-08-03 | Kombinácia agonistu adenozín A2a receptora a anticholinergického činidla na ošetrovanie obštrukčných chorôb dýchacích ciest | |
| US20040248867A1 (en) | 2001-06-12 | 2004-12-09 | Keith Biggadike | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| EP2039762A3 (en) | 2001-06-21 | 2009-07-29 | Verenium Corporation | Nitralases |
| CN100496493C (zh) * | 2001-08-13 | 2009-06-10 | 詹森药业有限公司 | 2-氨基-4,5-三取代噻唑基衍生物 |
| WO2003015778A1 (en) | 2001-08-17 | 2003-02-27 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| EP1430051B1 (en) | 2001-09-28 | 2006-12-27 | Cyclacel Limited | N-(4-(4-methylthiazol-5-yl)pyrimidin-2-yl)-n-phenylamines as antiproliferative compounds |
| CA2462980A1 (en) | 2001-10-17 | 2003-04-24 | Ucb, S.A. | Quinuclidine derivatives, processes for preparing them and their uses as m2 and/or m3 muscarinic receptor inhibitors |
| GB0125259D0 (en) | 2001-10-20 | 2001-12-12 | Glaxo Group Ltd | Novel compounds |
| MY130622A (en) | 2001-11-05 | 2007-07-31 | Novartis Ag | Naphthyridine derivatives, their preparation and their use as phosphodiesterase isoenzyme 4 (pde4) inhibitors |
| AU2002356759A1 (en) | 2001-12-01 | 2003-06-17 | Glaxo Group Limited | 17.alpha. -cyclic esters of 16-methylpregnan-3,20-dione as anti-inflammatory agents |
| MXPA04006206A (es) | 2001-12-20 | 2004-12-06 | S A L V A T Lab Sa | Derivados de 1-alquil-azoniabiciclo(2.2.2(octano carbamato y su uso como antagonistas del receptor musrcarinico. |
| WO2003072592A1 (en) | 2002-01-15 | 2003-09-04 | Glaxo Group Limited | 17.alpha-cycloalkyl/cycloylkenyl esters of alkyl-or haloalkyl-androst-4-en-3-on-11.beta.,17.alpha.-diol 17.beta.-carboxylates as anti-inflammatory agents |
| AU2003201693A1 (en) | 2002-01-21 | 2003-09-02 | Glaxo Group Limited | Non-aromatic 17.alpha.-esters of androstane-17.beta.-carboxylate esters as anti-inflammatory agents |
| GB0202216D0 (en) | 2002-01-31 | 2002-03-20 | Glaxo Group Ltd | Novel compounds |
| PE20030968A1 (es) | 2002-02-28 | 2004-01-12 | Novartis Ag | Derivados de 5-feniltiazol como inhibidores de cinasas |
| AU2003230700A1 (en) | 2002-03-26 | 2003-10-13 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| AU2003218342B8 (en) | 2002-03-26 | 2008-07-31 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| EP1496911B1 (en) | 2002-04-10 | 2007-12-19 | University Of Virginia Patent Foundation | Use of a combination comprising a2a adenosine receptor agonists and anti-pathogenic agents for the treatment of inflammatory diseases |
| ES2206021B1 (es) | 2002-04-16 | 2005-08-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de pirrolidinio. |
| DE10224888A1 (de) | 2002-06-05 | 2003-12-24 | Merck Patent Gmbh | Pyridazinderivate |
| US7074806B2 (en) | 2002-06-06 | 2006-07-11 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| DE10225574A1 (de) | 2002-06-10 | 2003-12-18 | Merck Patent Gmbh | Aryloxime |
| DE10227269A1 (de) | 2002-06-19 | 2004-01-08 | Merck Patent Gmbh | Thiazolderivate |
| ES2282667T3 (es) | 2002-06-25 | 2007-10-16 | Merck Frosst Canada Ltd. | Inhibidores de pde4 8-(biaril)quinolinas. |
| JP2005538972A (ja) | 2002-07-02 | 2005-12-22 | メルク フロスト カナダ アンド カンパニー | ジアリール置換エタンピリドンpde4阻害剤 |
| ES2204295B1 (es) | 2002-07-02 | 2005-08-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de quinuclidina-amida. |
| DK1521733T3 (da) | 2002-07-08 | 2014-10-13 | Pfizer Prod Inc | Modulatorer af glucocorticoid receptoren |
| PE20050130A1 (es) | 2002-08-09 | 2005-03-29 | Novartis Ag | Compuestos organicos |
| JP2006514684A (ja) | 2002-10-30 | 2006-05-11 | バーテックス ファーマシューティカルズ インコーポレイテッド | Rockおよび他のプロテインキナーゼとして有用な組成物 |
| WO2004045518A2 (en) | 2002-11-15 | 2004-06-03 | Bristol-Myers Squibb Company | Open chain prolyl urea-related modulators of androgen receptor function |
| GB0305152D0 (en) | 2003-03-06 | 2003-04-09 | Novartis Ag | Organic compounds |
| TW200519106A (en) | 2003-05-02 | 2005-06-16 | Novartis Ag | Organic compounds |
| GB0320197D0 (en) | 2003-08-28 | 2003-10-01 | Novartis Ag | Organic compounds |
| ZA200602755B (en) | 2003-09-06 | 2007-06-27 | Vertex Pharma | Modulators of ATP-binding cassette transporters |
| ZA200605624B (en) | 2004-01-12 | 2007-11-28 | Applied Research Systems | Thiazole derivatives and use thereof |
| SE0402735D0 (sv) | 2004-11-09 | 2004-11-09 | Astrazeneca Ab | Novel compounds |
| EA017166B1 (ru) | 2005-05-24 | 2012-10-30 | Мерк Сероно С.А. | Производные тиазола и их применение |
| WO2006125805A1 (en) | 2005-05-24 | 2006-11-30 | Laboratoires Serono S.A. | Thiazole derivatives and use thereof |
| US7655446B2 (en) * | 2005-06-28 | 2010-02-02 | Vertex Pharmaceuticals Incorporated | Crystal structure of Rho-kinase I kinase domain complexes and binding pockets thereof |
| DE102005048072A1 (de) * | 2005-09-24 | 2007-04-05 | Bayer Cropscience Ag | Thiazole als Fungizide |
| JP2009519346A (ja) | 2005-12-12 | 2009-05-14 | ジェネラブズ テクノロジーズ インコーポレーティッド | N−(5員芳香族環)−アミド抗ウイルス化合物 |
| GB0525671D0 (en) | 2005-12-16 | 2006-01-25 | Novartis Ag | Organic compounds |
| CA2635830A1 (en) | 2006-01-23 | 2007-07-26 | Laboratoires Serono S.A. | Thiazole derivatives and use thereof |
| GB0610243D0 (en) | 2006-05-23 | 2006-07-05 | Novartis Ag | Organic compounds |
| JP2010510246A (ja) | 2006-11-21 | 2010-04-02 | スミスクライン ビーチャム コーポレーション | アミド抗ウイルス化合物 |
| WO2008124000A2 (en) | 2007-04-02 | 2008-10-16 | Ligand Pharmaceuticals Incorporated | Thiazole derivatives as androgen receptor modulator compounds |
| AU2008257559B2 (en) | 2007-05-25 | 2013-10-10 | AbbVie Deutschland GmbH & Co. KG | Heterocyclic compounds as positive modulators of metabotropic glutamate receptor 2 (MGLU2 receptor) |
| US20090004140A1 (en) | 2007-06-26 | 2009-01-01 | Yao-Ling Qiu | 4-substituted pyrrolidine as anti-infectives |
| US20100292236A1 (en) | 2007-07-19 | 2010-11-18 | H. Lundbeck A/S | 5-Membered Heterocyclic Amides And Related Compounds |
| EP2238134A2 (en) * | 2007-12-20 | 2010-10-13 | Novartis AG | Bis-thiazole derivatives, process for their preparation and their use as medicaments |
| KR20100093129A (ko) * | 2007-12-20 | 2010-08-24 | 노파르티스 아게 | Pi 3 키나제 억제제로서 사용되는 티아졸 유도체 |
| UA104147C2 (uk) * | 2008-09-10 | 2014-01-10 | Новартис Аг | Похідна піролідиндикарбонової кислоти та її застосування у лікуванні проліферативних захворювань |
| US8293753B2 (en) * | 2009-07-02 | 2012-10-23 | Novartis Ag | Substituted 2-carboxamide cycloamino ureas |
-
2004
- 2004-04-29 TW TW093111971A patent/TW200519106A/zh unknown
- 2004-04-29 AR ARP040101467A patent/AR044519A1/es not_active Application Discontinuation
- 2004-04-30 CN CN2004800187775A patent/CN1816549B/zh not_active Expired - Fee Related
- 2004-04-30 DE DE602004023602T patent/DE602004023602D1/de not_active Expired - Lifetime
- 2004-04-30 PE PE2004000436A patent/PE20050147A1/es not_active Application Discontinuation
- 2004-04-30 JP JP2006505340A patent/JP4510807B2/ja not_active Expired - Lifetime
- 2004-04-30 RU RU2005137358/04A patent/RU2384580C2/ru not_active IP Right Cessation
- 2004-04-30 PT PT04730527T patent/PT1622897E/pt unknown
- 2004-04-30 AU AU2004234068A patent/AU2004234068B2/en not_active Ceased
- 2004-04-30 ES ES09169157.6T patent/ES2546851T3/es not_active Expired - Lifetime
- 2004-04-30 ES ES04730527T patent/ES2331883T3/es not_active Expired - Lifetime
- 2004-04-30 CL CL200400917A patent/CL2004000917A1/es unknown
- 2004-04-30 KR KR1020057020734A patent/KR100725885B1/ko active IP Right Grant
- 2004-04-30 CA CA2524401A patent/CA2524401C/en not_active Expired - Lifetime
- 2004-04-30 BR BRPI0410037A patent/BRPI0410037B8/pt active IP Right Grant
- 2004-04-30 WO PCT/EP2004/004603 patent/WO2004096797A1/en active Application Filing
- 2004-04-30 PL PL04730527T patent/PL1622897T3/pl unknown
- 2004-04-30 MX MXPA05011740A patent/MXPA05011740A/es active IP Right Grant
- 2004-04-30 EP EP09169157.6A patent/EP2157091B1/en not_active Expired - Lifetime
- 2004-04-30 EP EP04730527A patent/EP1622897B1/en not_active Expired - Lifetime
- 2004-04-30 CN CN200910170714A patent/CN101648949A/zh active Pending
- 2004-04-30 US US10/554,559 patent/US20070032487A1/en not_active Abandoned
- 2004-04-30 AT AT04730527T patent/ATE445614T1/de active
- 2004-04-30 RU RU2009134044/04A patent/RU2481346C2/ru active
-
2005
- 2005-10-23 IL IL171536A patent/IL171536A/en active IP Right Grant
- 2005-10-24 ZA ZA2005/08603A patent/ZA200508603B/en unknown
- 2005-10-28 EC EC2005006127A patent/ECSP056127A/es unknown
- 2005-11-01 TN TNP2005000278A patent/TNSN05278A1/en unknown
- 2005-11-08 MA MA28582A patent/MA27774A1/fr unknown
- 2005-11-25 IS IS8152A patent/IS8152A/is unknown
- 2005-12-02 NO NO20055714A patent/NO20055714L/no not_active Application Discontinuation
-
2006
- 2006-07-31 HK HK06108498.4A patent/HK1091810A1/xx not_active IP Right Cessation
-
2008
- 2008-12-05 AU AU2008255157A patent/AU2008255157B2/en active Active
-
2011
- 2011-01-12 US US13/005,132 patent/US8404684B2/en not_active Expired - Lifetime
-
2013
- 2013-02-22 US US13/774,305 patent/US20130172349A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0410037B1 (pt) | Inibidores de fosfatidilinositol 3-cinase, sua composição farmacêutica, e seu uso | |
| US11773110B2 (en) | Heterocycle amines and uses thereof | |
| ES2297393T3 (es) | Derivados del 5-feniltiazol y su uso como inhibidores de la p13 kinasa. | |
| ES2323000T3 (es) | Derivados de 5-fenil-4-metil-tiazol-2-il-amina como inhibidores de las enzimas fosfatidilinositol 3 quinasa (pi3) para el tratamiento de enfermedades inflamatorias de las vias respiratorias. | |
| JPWO2006054652A1 (ja) | アミド化合物 | |
| US20140315885A1 (en) | Therapeutically Active Thiazolo-Pyrimidine Derivatives | |
| BRPI0619835A2 (pt) | compostos orgánicos |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B15K | Others concerning applications: alteration of classification |
Free format text: PARA INT.CL: C07D 417/04, C07D 417/14, A61K 31/506,A61K 31/4436, A61P 26/00, A61P 11/06, A61P 19/02, A61P 9/10, A61P 3/04 Ipc: C07D 417/04 (2011.01), C07D 417/14 (2011.01), A61K |
|
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] | ||
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 30/04/2004 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |