WO2019196891A1 - 多环氨基甲酰基吡啶酮衍生物、药物组合物及其用途 - Google Patents
多环氨基甲酰基吡啶酮衍生物、药物组合物及其用途 Download PDFInfo
- Publication number
- WO2019196891A1 WO2019196891A1 PCT/CN2019/082195 CN2019082195W WO2019196891A1 WO 2019196891 A1 WO2019196891 A1 WO 2019196891A1 CN 2019082195 W CN2019082195 W CN 2019082195W WO 2019196891 A1 WO2019196891 A1 WO 2019196891A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- hydrogen
- acid
- group
- polycyclic
- Prior art date
Links
- 0 C[C@](CC[C@]1*(*(C=CC2=O)C3=C2OC)[C@]2(*)c(ccc(*)c4*)c4C(*)=*c4c2cccc4)C*1C3=O Chemical compound C[C@](CC[C@]1*(*(C=CC2=O)C3=C2OC)[C@]2(*)c(ccc(*)c4*)c4C(*)=*c4c2cccc4)C*1C3=O 0.000 description 6
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/196—Carboxylic acids, e.g. valproic acid having an amino group the amino group being directly attached to a ring, e.g. anthranilic acid, mefenamic acid, diclofenac, chlorambucil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Definitions
- the present invention relates to polycyclic carbamoylpyridone derivatives, pharmaceutical compositions and uses thereof.
- the flu is an acute viral infection that can easily spread in people, especially during the winter months in temperate regions. Because influenza viruses are prevalent worldwide and can infect anyone of any age group, often causing serious complications and deaths in high-risk populations, is currently a serious public health problem. According to statistics, the annual influenza epidemic is estimated to cause 3 to 5 million serious cases, and the death toll is about 250,000 to 500,000.
- the messenger RNA (mRNA) of influenza virus needs to have both a 5' cap (CAP) structure and a 3'-poly (A) tail structure which are recognized by the host cell translation system.
- CAP 5' cap
- A 3'-poly
- the 5' cap structure is "robbed” from the 5' end of the host cell precursor mRNA by the endonuclease activity of the PA subunit in the influenza virus RNA polymerase complex. This method, called “CAP-snatching”, captures the CAP cap structure of the host mRNA for viral self-mRNA transcription and is required for the transcription initiation of influenza virus.
- the present invention provides a polycyclic carbamoylpyridone derivative represented by the formula (VII), a stereoisomer, a tautomer, a hydrate, a solvate, an active metabolite, a crystalline form, and a pharmaceutically acceptable form.
- Acceptable salt or its prodrug :
- R 1 is hydrogen or helium
- R 2 is hydrogen or helium
- R 3 is hydrogen or helium
- R 1 , R 2 and R 3 are deuterium;
- R 4 is hydrogen or halogen (preferably chlorine);
- R 5 is hydrogen or halogen (preferably fluorine);
- R 6 is hydrogen or halogen (preferably fluorine);
- A is C or O.
- R 7 and R 8 are both hydrogen or both methyl; when A is O, R 7 and R 8 are absent;
- R 9 is hydrogen or methyl
- R 10 is hydrogen or methyl
- R 4 , R 5 and R 6 are not hydrogen at the same time.
- the polycyclic carbamoyl pyridone derivative represented by the formula (VII) preferably satisfies any of the following conditions:
- R 4 is hydrogen; R 5 is fluorine; R 6 is fluorine; A is O; R 9 is hydrogen; and R 10 is hydrogen.
- R 4 is hydrogen; R 5 is fluorine; R 6 is hydrogen; A is O; R 9 is hydrogen; R 10 is a methyl group and the carbon atom directly attached to R 10 is (S).
- R 4 is chlorine; R 5 is hydrogen; R 6 is hydrogen; A is O; R 9 is hydrogen; and R 10 is hydrogen.
- R 4 is hydrogen; R 5 is fluorine; R 6 is fluorine; A is C; R 7 and R 8 are both methyl; R 9 is hydrogen; and R 10 is hydrogen.
- R 4 is hydrogen; R 5 is fluorine; R 6 is fluorine; A is C; R 7 and R 8 are both hydrogen; R 9 is hydrogen and the carbon atom directly attached to R 9 is (R) ; R 10 is hydrogen.
- R 4 is hydrogen; R 5 is fluorine; R 6 is hydrogen; A is C; R 7 and R 8 are both hydrogen; R 9 is hydrogen; and R 10 is hydrogen.
- the alkyl group is preferably a C 1-4 alkyl group, and further preferably a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, an isobutyl group, a t-butyl group. .
- the present invention provides a polycyclic carbamoyl pyridone derivative represented by the formula (I), a stereoisomer, a tautomer, a hydrate, a solvate, an active metabolite, a crystalline form, and a pharmaceutically acceptable form.
- Acceptable salt or its prodrug :
- R 1 is hydrogen or helium
- R 2 is hydrogen or helium
- R 3 is hydrogen or helium
- R 1 , R 2 and R 3 is deuterium.
- the polycyclic carbamoyl pyridone derivative represented by the formula (I) is preferably selected from the group consisting of the following compounds:
- the present invention provides a polycyclic carbamoyl pyridone derivative represented by the formula (II), a stereoisomer, a tautomer, a hydrate, a solvate, an active metabolite, a crystalline form, and a pharmaceutically acceptable form.
- Acceptable salt or its prodrug :
- R 1 is hydrogen or helium
- R 2 is hydrogen or helium
- R 3 is hydrogen or helium
- R 1 , R 2 and R 3 is deuterium.
- the polycyclic carbamoyl pyridone derivative represented by the formula (II) is preferably selected from the group consisting of the following compounds:
- the present invention provides a polycyclic carbamoyl pyridone derivative represented by the formula (III), a stereoisomer, a tautomer, a hydrate, a solvate, an active metabolite, a crystalline form, and a pharmaceutically acceptable form.
- Acceptable salt or its prodrug :
- R 1 is hydrogen or helium
- R 2 is hydrogen or helium
- R 3 is hydrogen or helium
- R 1 , R 2 and R 3 is deuterium.
- the polycyclic carbamoyl pyridone derivative represented by the formula (III) is preferably selected from the group consisting of the following compounds:
- the present invention provides a polycyclic carbamoylpyridone derivative represented by the formula (IV), a stereoisomer, a tautomer, a hydrate, a solvate, an active metabolite, a crystalline form, and a pharmaceutically acceptable form.
- Acceptable salt or its prodrug :
- R 1 is hydrogen or helium
- R 2 is hydrogen or helium
- R 3 is hydrogen or helium
- R 1 , R 2 and R 3 is deuterium.
- the polycyclic carbamoyl pyridone derivative represented by the formula (IV) is preferably selected from the group consisting of the following compounds:
- the present invention provides a polycyclic carbamoyl pyridone derivative represented by the formula (V), a stereoisomer, a tautomer, a hydrate, a solvate, an active metabolite, a crystalline form, and a pharmaceutically acceptable form.
- Acceptable salt or its prodrug :
- R 1 is hydrogen or helium
- R 2 is hydrogen or helium
- R 3 is hydrogen or helium
- R 1 , R 2 and R 3 is deuterium.
- the polycyclic carbamoyl pyridone derivative represented by the formula (V) is preferably selected from the group consisting of the following compounds:
- the present invention provides a polycyclic carbamoyl pyridone derivative represented by the formula (VI), a stereoisomer, a tautomer, a hydrate, a solvate, an active metabolite, a crystalline form, and a pharmaceutically acceptable form.
- Acceptable salt or its prodrug :
- R 1 is hydrogen or helium
- R 2 is hydrogen or helium
- R 3 is hydrogen or helium
- R 1 , R 2 and R 3 is deuterium.
- the polycyclic carbamoyl pyridone derivative represented by the formula (VI) is preferably selected from the group consisting of the following compounds:
- alkyl is used to mean a straight or branched saturated hydrocarbon group which may be monovalent (e.g., methyl), divalent (e.g., methylene), or polyvalent (e.g., methine).
- alkyl group include methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, s-butyl). , t-butyl), pentyl (eg, n-pentyl, isopentyl, neopentyl) and the like.
- the compounds of the invention are generally employed in the form of the free acid or the free base. Alternatively, the compounds of the invention may be used in the form of an acid or base salt.
- the acid addition salts of the free amino compounds of the present invention can be prepared by methods well known in the art and can be prepared from organic and inorganic acids.
- Suitable organic acids include maleic acid, fumaric acid, benzoic acid, ascorbic acid, succinic acid, methanesulfonic acid, acetic acid, trifluoroacetic acid, oxalic acid, propionic acid, tartaric acid, salicylic acid, citric acid, gluconic acid, Lactic acid, mandelic acid, phenylacetic acid, aspartic acid, stearic acid, palmitic acid, glycolic acid, glutamic acid, and benzenesulfonic acid.
- Suitable inorganic acids include hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid and nitric acid.
- the basic salt includes a salt formed with a carboxylate anion, and includes an organic and inorganic cation formed such as an alkali metal ion, an alkaline earth metal ion (for example, lithium, sodium, potassium, magnesium, barium, calcium) and an ammonium ion. a salt, and a substituted derivative thereof (for example, dibenzylammonium, benzylammonium, 2-hydroxyethylammonium, etc.). Therefore, the terms "pharmaceutically acceptable salts" of the general formula (I), formula (II), formula (III), formula (IV), formula (V), formula (VI) or formula (VII) shall include and Acceptable salt form.
- prodrugs are also included in the scope of the present invention.
- a prodrug is any covalently bound carrier which, when administered to a patient, is released in the body to receive formula (I), formula (II), formula (III), formula (IV), formula (V). a compound of formula (VI) or formula (VII).
- Prodrugs are typically prepared by modifying the functional groups in such a way that the modifications can be resolved by conventional exchange or in vivo to give the parent compound.
- Prodrugs include, for example, a compound of the invention in which a hydroxy, amino or thiol group is bonded to any group, wherein the group is detached when administered to a patient to provide a hydroxy, amino or thiol group.
- prodrugs include, but are not limited to, Formula (I), Formula (II), Formula (III), Formula (IV), Formula (V), Formula (VI) or Formula (VII) Derivatives of acetates, formates and benzoate salts of alcohol and amine functional groups of the compounds.
- a carboxylic acid (-COOH) esters such as a methyl ester, an ethyl ester or the like may be included.
- a mixed acid anhydride such as a methoxy group, an ethoxy group, a propoxy group, a t-butoxy group or the like can be included.
- compounds of formula (I), formula (II), formula (III), formula (IV), formula (V), formula (VI) or formula (VII) may have a chiral center, and It can exist as a racemate, a racemic mixture, and as individual enantiomers or diastereomers. All isomeric forms are included within the invention, including mixtures thereof. Furthermore, certain crystalline forms of the compounds of formula (I), formula (II), formula (III), formula (IV), formula (V), formula (VI) or formula (VII) may exist in the form of polymorphs, It is also included in the present invention.
- any compound may contain an unnatural proportion of atomic isotopes at one or more of the atoms that make up the compound.
- the deuterated means that the atom at the relevant site of the compound contains a ruthenium atom exceeding a natural ratio (ie, exceeding the natural abundance of ruthenium). . Therefore, any polycyclic carbamoylpyridone derivative containing a ruthenium atom in a ratio above the natural abundance of ruthenium at the relevant site is within the scope of the present invention.
- the corresponding polycyclic amino group having a corresponding deuteration rate or hydrazine content obtained by introducing a ruthenium atom using a commercially available deuteration reagent by the same or similar chemical synthesis means as shown in the examples of the present invention is understood.
- the acylpyridone derivatives are all within the scope of the present invention.
- the chemical synthesis means and deuterated reagents herein are not limited to those exemplified in the examples, but it should be understood that all synthetic methods or routes that can be used in the art to obtain the compounds of the present invention, and all may be combined with the aforementioned synthetic methods or A deuterated reagent that introduces a ruthenium atom into a target molecule.
- the invention further provides a formula (VII), preferably as shown in formula (I), formula (II), formula (III), formula (IV), formula (V), or formula (VI) Polycyclic carbamoyl pyridone derivatives, stereoisomers, tautomers, hydrates, solvates, active metabolites, crystalline forms, pharmaceutically acceptable salts or prodrugs thereof, in the preparation of 5' caps Use in dependent endonuclease inhibitors.
- a formula (VII) preferably as shown in formula (I), formula (II), formula (III), formula (IV), formula (V), or formula (VI)
- the invention further provides a formula (VII), preferably as shown in formula (I), formula (II), formula (III), formula (IV), formula (V), or formula (VI) Polycyclic carbamoylpyridone derivatives, stereoisomers, tautomers, hydrates, solvates, active metabolites, crystalline forms, pharmaceutically acceptable salts or prodrugs thereof for the preparation of prophylaxis and treatment And/or use in a medicament for alleviating a disease associated with a 5' cap-dependent endonuclease.
- a formula (VII) preferably as shown in formula (I), formula (II), formula (III), formula (IV), formula (V), or formula (VI)
- the disease associated with the 5′ cap-dependent endonuclease refers to a symptom and/or disease which is prevented, treated and/or alleviated by inhibiting the level of 5′ cap-dependent endonuclease in vivo, specifically Refers to symptoms and/or diseases caused by influenza virus infection. More specifically, the influenza virus includes, but is not limited to, alpha (A), ethyl (B), and ethyl (C).
- the symptoms include, but are not limited to, for example, cold symptoms such as fever, chills, headache, muscle pain, body burnout, or respiratory tract inflammation, abdominal pain, runny nose, cough, phlegm, etc. Gastrointestinal symptoms such as vomiting and diarrhea, followed by complications of secondary infections such as acute encephalopathy and pneumonia.
- the invention further provides a method of preventing, treating and/or ameliorating a disease associated with a 5' cap-dependent endonuclease, the method comprising administering to a subject in need thereof a therapeutically effective amount of, as in formula (VII) (preferably, a polycyclic carbamoyl pyridone derivative represented by formula (I), formula (II), formula (III), formula (IV), formula (V), or formula (VI))
- the diseases associated with the 5' cap-dependent endonuclease are specifically as described above.
- the invention further provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a formula (VII) (preferably such as formula (I), formula (II), formula (III), formula (IV), formula (V) Or a polycyclic carbamoylpyridone derivative represented by formula (VI)), a stereoisomer, tautomer, hydrate, solvate, active metabolite, crystalline form thereof, pharmaceutically acceptable a salt or a prodrug thereof, and a pharmaceutically acceptable carrier.
- a formula (VII) preferably such as formula (I), formula (II), formula (III), formula (IV), formula (V) Or a polycyclic carbamoylpyridone derivative represented by formula (VI)
- a stereoisomer, tautomer, hydrate, solvate, active metabolite, crystalline form thereof, pharmaceutically acceptable a salt or a prodrug thereof, and a pharmaceutically acceptable carrier a pharmaceutically acceptable carrier.
- the invention further provides a pharmaceutical composition
- a pharmaceutical composition comprising not only a therapeutically effective amount of formula (VII) (preferably, formula (I), formula (II), formula (III), formula (IV), formula (V) Or a polycyclic carbamoyl pyridone derivative represented by formula (VI)), a stereoisomer, tautomer, hydrate, solvate, active metabolite, crystalline form thereof, pharmaceutically acceptable
- the salt or a prodrug thereof, and a pharmaceutically acceptable carrier further comprise other pharmaceutically active ingredients.
- the other pharmaceutically active ingredient is selected from the group consisting of a neuraminidase inhibitor (eg, oseltamivir, zanamivir, peramivir, and Inavir, etc.), an RNA-dependent RNA polymerase inhibitor (eg, Favipiravir).
- a neuraminidase inhibitor eg, oseltamivir, zanamivir, peramivir, and Inavir, etc.
- an RNA-dependent RNA polymerase inhibitor eg, Favipiravir
- M2 protein inhibitor amantadine
- PB2Cap binding inhibitor VX-787
- anti-HA antibody MHAA4594A
- immunological drug Naitazoxanide
- the compounds of the present invention can be used in combination with other pharmaceutical preparations in order to enhance the efficacy of the compound or to reduce the amount of the compound used.
- a neuraminidase inhibitor eg, oseltamivir, zanamivir, peramivir, and Inavir, etc.
- an RNA-dependent RNA polymerase inhibitor such as Favipiravir
- M2 RNA-dependent RNA polymerase inhibitor
- a protein inhibitor amantadine
- a PB2Cap binding inhibitor VX-787
- an anti-HA antibody MHAA4594A
- an immunologically active drug Neitazoxanide
- the pure form or suitable pharmaceutical composition of a compound of the present invention or a pharmaceutically acceptable salt thereof can be administered by any acceptable mode of administration of a similarly useful agent.
- the pharmaceutical compositions of the present invention can be prepared by combining a compound of the present invention with a suitable pharmaceutically acceptable carrier, diluent or excipient, and can be formulated into a solid, semi-solid, liquid or gaseous form, such as a tablet. , capsules, powders, granules, ointments, solutions, suppositories, injections, inhalants, gels, microspheres and aerosols.
- Typical routes of administration of the pharmaceutical compositions include, but are not limited to, oral, topical, transdermal, inhalation, parenteral, sublingual, buccal, rectal, vaginal and intranasal administration.
- parenteral includes subcutaneous injection, intravenous, intramuscular, intrasternal injection or infusion techniques.
- the pharmaceutical compositions of the present invention are formulated to allow the cleansing ingredients contained therein to be bioavailable after administration of the composition to a patient.
- composition to be administered will in any event contain a therapeutically effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof, for the treatment of a disease or condition of interest in accordance with the teachings of the present invention.
- the pharmaceutical compositions of the invention may be in solid or liquid form.
- the carrier is a microparticle such that the composition is, for example, in the form of a tablet or powder.
- the carrier can be a liquid, and the composition is, for example, an oral syrup, an injectable liquid, or an aerosol suitable for administration, for example, by inhalation.
- the pharmaceutical compositions are preferably in solid or liquid form, wherein the semi-solid, semi-liquid, suspension and gel forms are included in the form considered solid or liquid herein.
- the pharmaceutical compositions can be formulated in the form of powders, granules, compressed tablets, pills, capsules, chewable tablets, powders, and the like.
- Such solid compositions typically contain one or more inert diluents or edible carriers.
- binders such as carboxymethylcellulose, ethylcellulose, microcrystalline cellulose, huangwa gum or gelatin
- excipients such as starch, lactose or paste a disintegrating agent such as alginic acid, sodium alginate, Primogel, corn starch, etc.
- a lubricant such as magnesium stearate or hydrogenated vegetable oil (Sterotex); a glidant such as colloidal silica
- a sweetener For example, sucrose or saccharin
- flavoring agents such as peppermint, methyl salicylate or sweet orange flavoring
- coloring agents such as peppermint, methyl salicylate or sweet orange flavoring.
- the pharmaceutical composition When the pharmaceutical composition is in the form of a capsule, such as a gelatin capsule, it may contain, in addition to materials of the above type, a liquid carrier such as polyethylene glycol or oil.
- a liquid carrier such as polyethylene glycol or oil.
- the pharmaceutical compositions may be in liquid form such as elixirs, syrups, solutions, emulsions or suspensions. This liquid can be administered orally, or delivered by injection, as two examples.
- the composition When intended for oral administration, it is preferred that the composition contains, in addition to the compound of the present invention, one or more of a sweetener, a preservative, a dye/colorant, and a flavor enhancer.
- One or more of a surfactant, a preservative, a wetting agent, a dispersing agent, a suspending agent, a buffering agent, a stabilizer, and an isotonic agent may be included in the composition to be administered by injection.
- the liquid pharmaceutical composition of the present invention may comprise one or more of the following adjuvants: a sterile diluent, such as water for injection, a physiological saline solution, preferably physiological saline, Ringer.
- a sterile diluent such as water for injection
- a physiological saline solution such as physiological saline, Ringer.
- Ringer's solution isotonic sodium chloride, fixed oil (such as synthetic mono or diglyceride, which can be used as a solvent or suspension medium), polyethylene glycol, glycerin, propylene glycol and other solvents; antibacterial agents, for example Benzyl or methylparaben; an antioxidant such as ascorbic acid or sodium bisulfite; a chelating agent such as ethylenediaminetetraacetic acid; a buffer such as acetate, citrate or phosphate, and a tension regulating Reagents such as sodium oxide or dextrose.
- the parenteral formulation can be enclosed in ampoule, disposable syringes or multiple dose vials made of glass or plastic. Saline is the preferred adjuvant.
- the injectable pharmaceutical compositions are preferably sterile.
- the liquid pharmaceutical composition of the present invention to be administered parenterally or orally should contain a certain amount of the compound of the present invention so that a suitable dose can be obtained.
- the pharmaceutical compositions of the invention may be intended for topical administration, in which case the carrier preferably comprises a solution, emulsion, ointment or gel base.
- the matrix may comprise one or more of the following: paraffinic oil, lanolin, polyethylene glycol, beeswax, mineral oil, diluents (such as water and alcohol), and emulsifiers and stabilizers. Thickeners may be present in the pharmaceutical compositions for topical administration. If transdermal administration is desired, the composition can include a transdermal patch or an iontophoresis device.
- compositions for rectal administration may be intended for rectal administration, in the form of a suppository, which will melt in the rectum and release the drug.
- Compositions for rectal administration may contain an oily base as a suitable non-irritating excipient.
- the matrix includes, but is not limited to, lanolin, cocoa butter, and polyethylene glycol.
- compositions of the present invention may include various materials that modify the physical form of the solid or liquid dosage unit.
- the composition can include a substance that forms a coated outer shell around the active ingredient.
- the material forming the outer envelope is generally inert and may be selected, for example, from sugars, shellac and other enteric coating agents.
- the active ingredient can be enclosed in a gelatin capsule.
- compositions of the invention in solid or liquid form may include an agent that binds to a compound of the invention and thereby aids in the delivery of the compound.
- Suitable agents with this ability include monoclonal or polyclonal antibodies, proteins or liposomes.
- compositions of this invention may be comprised of dosage units which may be administered in the form of an aerosol.
- aerosol is used to refer to a variety of systems from colloidal species to systems consisting of pressurized packaging. Delivery can be by liquefied or compressed gas or by a suitable pump system that dispenses the active ingredients. Aerosols of the compounds of the invention may be delivered in a single phase, two phase or three phase system to deliver the active ingredient.
- the side delivery of the aerosol includes the necessary containers, actuators, valves, sub-containers, etc., which together form a kit.
- One skilled in the art can determine a preferred aerosol without undue experimentation.
- compositions of the invention can be made using methods well known in the pharmaceutical art.
- a pharmaceutical composition to be administered by injection can be prepared by combining a compound of the present invention with sterile distilled water to form a solution.
- Surfactants may be added to facilitate the formation of a homogeneous solution or suspension.
- Surfactants are compounds that non-covalently interact with the compounds of the invention, thereby promoting the dissolution or uniform suspension of the compound in an aqueous delivery system.
- a compound of the invention, or a pharmaceutically acceptable salt thereof, is administered in a therapeutically effective amount which will vary depending on a variety of factors, including the activity of the particular compound employed, the metabolic stability of the compound, and the length of action, the patient Age, weight, general state of health, gender and diet, mode of administration and timing, rate of excretion, combination of drugs, severity of a particular condition or condition, and the individual receiving the therapy.
- the compounds of the invention, or pharmaceutically acceptable derivatives thereof, may also be administered simultaneously with, before or after administration of one or more additional therapeutic agents.
- the combination therapy comprises administering a single pharmaceutical formulation containing a compound of the invention and one or more additional active agents, as well as a separate pharmaceutical formulation for administration of the compound of the invention and each active agent.
- a compound of the invention may be administered to a patient together with another active agent in a single oral administration composition (e.g., a tablet or capsule), or each agent may be administered as a separate oral administration formulation.
- the compound of the invention and one or more additional active agents can be administered substantially at the same time (i.e., simultaneously) or at separately staggered times (i.e., sequentially), and combination therapy should be understood as Includes all of these programs.
- the pharmaceutical composition of the present invention will be adjusted depending on the disease state, the route of administration, the age of the patient or the body weight.
- oral administration to an adult, it is usually from 0.1 to 100 mg/kg/day, preferably from 1 to 20 mg/kg/day.
- the suitable dosage for administration of the present invention needs to be set in consideration of the age, body weight, condition, administration route, and the like of the patient, and oral administration is usually in the range of 0.05 to 100 mg/kg/day, preferably 0.1 to 10 mg/kg/day. .
- the pharmaceutical composition of the present invention is administered in a dose which varies widely depending on the administration route, but is usually in the range of 0.005 to 10 mg/kg/day, preferably 0.01 to 1 mg/kg/day.
- the dose of the compound of the present invention varies depending on the administration method, the age, body weight, state of the patient, and the type of the disease, but usually, in the case of oral administration, the dose per adult per day is about 0.05 mg to 3000 mg. Preferably, it is about 0.1 mg to 1000 mg, and it can be administered separately as needed. Further, in the case of parenteral administration, the dosage per adult per day is about 0.01 mg to 1000 mg, preferably about 0.05 mg to 500 mg. It will be appreciated that in the present invention, combinations of substituents and/or variables of the formula are permissible only if they result in a stable compound.
- the present invention relates to a substituted polycyclic carbamoylpyridone derivative having a 5' cap structure (CAP)-dependent endonuclease inhibitory activity, a prodrug thereof, a deuterated compound thereof, and a pharmaceutical composition containing the same And methods of using the composition for inhibiting proliferation of influenza virus.
- CAP 5' cap structure
- the compound of the present invention is a prodrug, it has the following advantages: high oral absorption, good bioavailability, good clearance, and high lung metastasis. Therefore, it can form an excellent drug.
- the parent compound of the compound of the present invention has a high inhibitory activity against a 5' cap structure-dependent endonuclease, and since it is a virus-specific enzyme, it has a high selectivity and can be a drug with reduced side effects. Further, the compound of the present invention and/or the parent compound of the compound of the present invention have the advantages of high metabolic stability, high solubility, high oral absorbability, good bioavailability, good clearance, High lung metastasis, long half-life, high non-protein binding rate, low hERG channel inhibition, low inhibition of liver drug enzymes, inhibition of CytoPathic Effect (CPE) inhibition, and/or in phototoxicity test, Ames test and The genotoxicity test shows negative or no hepatotoxicity, and the like, and therefore the compound of the present invention is preferably pharmaceutically acceptable.
- CPE CytoPathic Effect
- the compounds of the invention and/or the compounds of the invention are useful for the symptoms and/or diseases caused by influenza viruses.
- flu symptoms such as fever, chills, headache, muscle pain, general fatigue, or respiratory symptoms such as sore throat, runny nose, stuffy nose, cough, phlegm, etc., abdominal pain, vomiting, diarrhea
- respiratory symptoms such as sore throat, runny nose, stuffy nose, cough, phlegm, etc.
- abdominal pain, vomiting, diarrhea Further, it is effective to treat and/or prevent complications and improve symptoms of secondary infections such as acute encephalopathy and pneumonia.
- the compounds of the invention are either by hand or Software naming, commercially available compounds using the supplier catalog name.
- the synthetic route is as follows:
- Example Compound 1A (5.0 g) was added with DMA (25 ml), and chloromethyl methyl carbonate (2.4 g) and potassium carbonate (2.9 g), and potassium iodide (1.7 g) were added, and the mixture was warmed to 50 ° C and stirred for 6 hours. Additional DMA (5 ml) was added and stirring was continued for 6 hours. The reaction solution was cooled to room temperature, DMA (25 ml) was added, and the mixture was stirred at 50 ° C for 5 minutes and filtered. To the filtered droplets, 1 mol/L hydrochloric acid water (50 ml) and water (15 ml) were added at 0 to 5 ° C and stirred for 1 hour.
- Example Compound 1A (4.8 g), DMA (30 ml) was added and stirred, and bromohexane (1.7 g) and potassium carbonate (2.9 g), potassium iodide (1.7 g) were added, and the mixture was heated to 50 ° C and stirred for 6 hours. Additional DMA (5 ml) was added and stirring was continued for 6 hours. The reaction solution was cooled to room temperature, DMA (30 ml) was added, and the mixture was stirred at 50 ° C for 5 minutes and filtered. To the filtered droplets, 1 mol/L hydrochloric acid water (50 ml) and water (25 ml) were added at 0 to 5 ° C and stirred for 1 hour. The solid obtained was filtered, dried under reduced pressure at 60 °C, in Example 1C to obtain the compound in a yield 79% .MS: ESI 569.2 [M + H] +.
- the synthetic route is as follows:
- intermediate 21 For the preparation process of intermediate 21, refer to the preparation of intermediate 15, except that lithium tetrahydrogen aluminum-D 4 is replaced with lithium aluminum hydride, and intermediate 14 is replaced with intermediate 20.
- Example Compound 2A (10.0 g), DMA (50 ml) was added and stirred, and chloromethylmethyl carbonate (4.8 g) and potassium carbonate (6.0 g) and potassium iodide (3.4 g) were added, and the mixture was heated to 50 ° C and stirred for 6 hours. Additional DMA (10 ml) was added and stirring was continued for 6 hours. The reaction solution was cooled to room temperature, DMA (50 ml) was added, and the mixture was stirred at 50 ° C for 5 minutes and filtered. To the filtered droplets, 1 mol/L hydrochloric acid water (100 ml) and water (30 ml) were added at 0 to 5 ° C and stirred for 1 hour.
- the synthetic route is as follows:
- the following table compounds and their individual enantiomers were prepared to prepare prodrugs of esters corresponding to the following table compounds, such as orally administered, using drug metabolizing enzymes, hydrolases , digestive juice, or bacteria in the digestive tract, etc., and converted into active metabolites with hydroxyl groups.
- the corresponding ester of the table compound is preferably, but not limited to, methyl methyl carbonate.
- RNP is prepared from viral particles according to a defined method (Reference: VIROLOGY (1976) 73, pages 327-338. LGA M. ROCHOVANSKY). Specifically, 200 ⁇ L of 1 ⁇ 10 3 PFU/mL A/WSN/33 virus was inoculated into 10-day-old developing chicken eggs, cultured at 37° C. for 2 days, and then the allantoic fluid of chicken eggs was recovered.
- the virus particles were purified by ultracentrifugation using 20% sucrose, the virions were lysed using Triton X-100 and lysolecithin, and then the RNP fraction was collected by ultracentrifugation using a 30-70% glycerol density gradient ( 50 to 70% glycerol fraction) was used as an enzyme solution (containing about 1 nM of PB1 ⁇ PB2 ⁇ PA complex).
- enzymatic reaction solution composition: 53 mM Tris hydrochloride (pH 7.8), 1 mM MgCl 2 , 1.25 mM dithiothreitol, 80 mM NaCl, 12.5% glycerol, was dispensed on a 384-well plate made of polypropylene. 0.15 ⁇ L of enzyme solution).
- DMSO a positive control (PC), and a negative control (NC), and mixed well.
- the solution after the termination of the reaction was heated at 85 ° C for 5 minutes, quenched on ice for 2 minutes, and then analyzed by an ABIPRIZ M3730 Genetic Analyzer.
- the peak of the cap-dependent endonuclease product was quantified by the analysis software ABI Genemapper, and the fluorescence intensity of PC and NC was used as 0% inhibition and 100% inhibition, and the CEN reaction inhibition rate (%) of the test compound was determined.
- the IC 50 values were then determined using curve fitting software.
- MDBK cells adjusted to the appropriate number of cells (3X 10 5 /mL) with 2% FCS E-MEM.
- MDCK cells washed twice with HBSS and then adjusted to the appropriate number of cells ( 5 ⁇ 10 5 /mL) with 0.5% BSA E-MEM.
- Trypsin (SIGMA) from porcine pancreas was dissolved in PBS(-) and filtered through a 0.45 ⁇ m filter.
- the culture solution 2% FCS E-MEM was used when MDBK cells were used, and 0.5% BSA E-MEM was used when MDCK cells were used.
- FCS E-MEM 0.5% FCS E-MEM was used when MDCK cells were used.
- BSA E-MEM 0.5% BSA E-MEM was used when MDCK cells were used.
- the same culture solution is used for dilution of the virus, cell, and test sample.
- test sample was diluted with a culture solution to an appropriate concentration in advance, and a 2-5-fold serial dilution series (50 ⁇ L/well) was prepared on a 96-well plate. Two pieces for measuring anti-influenza activity and measuring cytotoxicity were prepared. Triplicate assays were performed for each drug.
- influenza virus was diluted with the culture solution to an appropriate concentration in advance, and each was dispensed at 50 ⁇ L/well into a 96-well plate to which the test sample was added.
- the culture solution was dispensed into the cytotoxicity assay plate at 50 ⁇ L/well.
- the anti-influenza activity assay and the cytotoxicity assay were all cultured for 3 days.
- the 96-well plate cultured for 3 days was observed under a naked eye and a microscope, and the supernatant was removed from the plate in such a manner that cells were not aspirated.
- the WST-8 kit was diluted 10-fold with the culture solution, and the WST-8 solution was dispensed into each well at 100 ⁇ L each. Mix with a plate mixer and incubate for 1 to 3 hours in a CO 2 incubator.
- the absorbance was measured with EnVision at a dual wavelength of 450 nm / 620 nm.
- the calculation is performed using a Microsoft Excel program based on the following calculation formula.
- Example 4 The measurement results of Example 4 and Example 5 are shown in Table 2 for the test substance (Example compound) as the parent compound.
- the compound of the example can prepare a preventive/therapeutic drug for use as a symptom/disease induced by influenza virus infection.
- Administration of administration amount and grouping Oral administration or intravenous administration is carried out using a predetermined administration amount. Set the group as follows. (The amount of each compound administered varies)
- Oral administration is suspension and intragastric administration.
- the intravenous administration is a solution and a tail vein administration.
- Example Compound Number# Example compound BA% Intravenous administration half-life T 1/2 hour 1A 7.1 4.0 2A 5.0 5.0 3A 7.8 5.2 S-033188A 4.7 3.8 1B 18.4 -- 2B 15.9 -- 3B 19.8 -- S-033188B 15.3 --
- the prodrugs (1B, 2B, and 3B) have improved bioavailability as compared with the parent compound (1A, 2A, and 3A).
- the compounds of the examples 1A, 2A, and 3A have a greater degree of improvement than the S-033188A; the compounds of the examples 1B, 2B, and 3B have a greater degree of bioavailability than the S-033188B.
- Intravenous administration of the elimination half-life of the example compounds also has varying degrees of prolongation.
- the compound of the present invention is more excellent in oral absorption than S-033188A/S-033188B, and can form a drug for use as a therapeutic and/or prophylactic agent for symptoms and/or diseases caused by infection with influenza virus, indicating a dose to be administered. Can be smaller and have fewer side effects.
- HEK293 cells expressing the human ether-a-go-go-related gene (hERG) channel were used to study delayed rectifier K + current (I Kr ) , which plays an important role in ventricular repolarization. The role of ).
- the cells were maintained at a membrane potential of -80 mV by whole-cell patch clamp method, and a depolarization stimulus of +50 mV was given for 2 seconds, and a repolarization of -50 mV was further given for 2 seconds. I Kr caused by stimuli .
- the extracellular solution of the test substance was dissolved at the target concentration (NaCl: 137 mmol/L, KCl: 4 mmol/L, CaCl 2 : 1.8 mmol/L, MgCl 2 -6H 2 O: 1 mmol/ L, glucose: 10 mmol/L, HEPES: 10 mmol/L, pH 7.4) Apply to cells for 10 minutes at room temperature. From the obtained I Kr , the absolute value of the maximum tail current was measured using the analysis software on the basis of the current value of the static membrane potential. Further, the inhibition rate of the maximum tail current before application of the test substance was calculated, and the influence of the test substance on I Kr was evaluated in comparison with the medium application group (0.1% DMSO solution).
- mice BALB/cAnNCrlCrlj 6 to 7 week old mice were used for the test.
- A/WS/33, A/Victoria/3/75 or B/Maryland/1/59 were passaged in the lungs of mice to prepare a mouse domesticated virus.
- the cryopreserved mouse domesticated virus solution was rapidly thawed and diluted with DPBS to form an infectivity titer (A/WS/33: 800-4000 TC ID501 mice, A/Victoria/3/75). : 750 TCID 50 /mouse, B/Maryland/1/59: 100 TCID 50 / mouse).
- test sample was suspended in a 0.5% methylcellulose solution at a suitable concentration.
- mice For the mice immediately after the virus infection or after a certain period of time, 200 ul of the diluted test sample was orally administered.
- the animals were fed for 14 days, and the dose ED 50 (mg/kg/day) for each day required for 50% lethal inhibition was calculated and compared with the control group, and the virus inhibitory effect was evaluated.
- Table 5 shows the ED 50 values per administration.
- the (Invitrogen) kit detects the toxic effects of drugs on cells. It is a redox indicator that produces changes in absorbance and fluorescence signals based on metabolic activity. Soluble in water, its oxidized form enters cells and is reduced by mitochondrial enzymes to produce measurable fluorescence and color changes. It is suitable for quantitative analysis of cell viability and cell proliferation and in vitro cytotoxicity studies. Normal cells with metabolic activity are capable of converting reagents into strong fluorescence and color changes, while damaged or inactive cells have lower natural metabolic activity and corresponding signals are lower. Therefore, the fluorescence signal is strong and can reflect the level of cell activity.
- MDCK cells were seeded in 96-well cell culture plates, and the cells were allowed to adhere after use.
- the drug was diluted 8 times in a 3-fold serial gradient from 2 times the highest test concentration in DMEM medium.
- the drug was added to the cells and cultured in a CO 2 incubator at 37 °C.
- the drug-induced cytopathic effect (CPE) was observed under a microscope, and the cell survival rate was measured by adding alamarBlue.
- CPE drug-induced cytopathic effect
- the drug has a toxic effect on MDCK cells at very high concentrations, and the concentration causing a 50% toxic effect is indicated by CC 50, as shown in the table below.
- Example Compound 1A was significantly less toxic than S-033188A. It is indicated that compound 1A has higher safety than S-033188A.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Virology (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- Pulmonology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description






























| 实施例化合物编号# | 实施例化合物BA% | 静脉给药半衰期T 1/2小时 |
| 1A | 7.1 | 4.0 |
| 2A | 5.0 | 5.0 |
| 3A | 7.8 | 5.2 |
| S-033188A | 4.7 | 3.8 |
| 1B | 18.4 | -- |
| 2B | 15.9 | -- |
| 3B | 19.8 | -- |
| S-033188B | 15.3 | -- |
| 实施例化合物编号# | 抑制率% |
| 1A | 3.1 |
| 2A | 7.8 |
| 3A | 3.5 |
| S-033188A | 8.2 |
| 实施例化合物编号# | ED 50mg/kg/day |
| 1B | 0.41 |
| 2B | 0.71 |
| 3B | 0.33 |
| S-033188B | 0.78 |
| 实施例化合物编号# | CC 50nM |
| 1A | 48285 |
| S-033188A | 29882 |
Claims (15)
- 一种如式(VII)所示的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药,
 其中,R 1为氢或氘;R 2为氢或氘;R 3为氢或氘;L为氢、烷基、或(甲氧羰基)氧甲基;且R 1、R 2和R 3中至少有一个为氘;R 4为氢或卤素;R 5为氢或卤素;R 6为氢或卤素;A为C或O,当A为C时,R 7和R 8均为氢或均为甲基;当A为O时,R 7和R 8不存在;R 9为氢或甲基;R 10为氢或甲基;且R 4、R 5和R 6不同时为氢。
其中,R 1为氢或氘;R 2为氢或氘;R 3为氢或氘;L为氢、烷基、或(甲氧羰基)氧甲基;且R 1、R 2和R 3中至少有一个为氘;R 4为氢或卤素;R 5为氢或卤素;R 6为氢或卤素;A为C或O,当A为C时,R 7和R 8均为氢或均为甲基;当A为O时,R 7和R 8不存在;R 9为氢或甲基;R 10为氢或甲基;且R 4、R 5和R 6不同时为氢。 - 如权利要求1所述的如式(VII)所示的多环氨基甲酰基吡啶酮衍生物,其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药,其中,所述的如式(VII)所示的多环氨基甲酰基吡啶酮衍生物满足如下任一条件:条件1:R 4为氢;R 5为氟;R 6为氟;A为O;R 9为氢;R 10为氢;条件2:R 4为氢;R 5为氟;R 6为氢;A为O;R 9为氢;R 10为甲基且与R 10直接相连的碳原子构型为(S);条件3:R 4为氯;R 5为氢;R 6为氢;A为O;R 9为氢;R 10为氢;条件4:R 4为氢;R 5为氟;R 6为氟;A为C;R 7和R 8均为甲基;R 9为氢;R 10为氢;条件5:R 4为氢;R 5为氟;R 6为氟;A为C;R 7和R 8均为氢;R 9为氢且与R 9直接相连的碳原子构型为(R);R 10为氢;条件6:R 4为氢;R 5为氟;R 6为氢;A为C;R 7和R 8均为氢;R 9为氢;R 10为氢。
- 一种如式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)所示的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药,
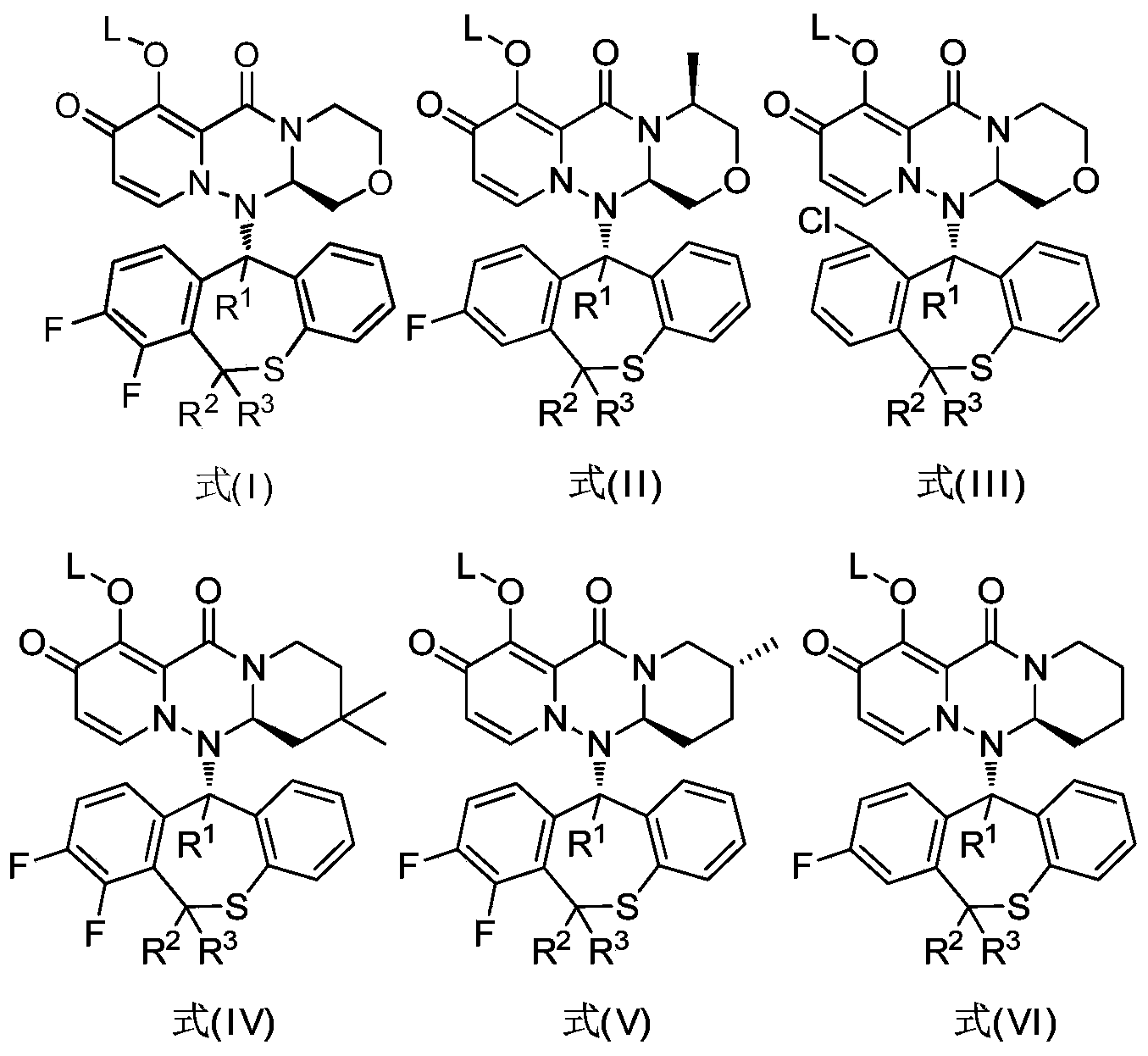 所述的式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)的结构式中,R 1各自独立地为氢或氘;R 2各自独立地为氢或氘;R 3各自独立地为氢或氘;L各自独立地为氢、烷基或(甲氧羰基)氧甲基;且,所述式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)各结构式中,R 1、R 2和R 3中至少有一个为氘。
所述的式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)的结构式中,R 1各自独立地为氢或氘;R 2各自独立地为氢或氘;R 3各自独立地为氢或氘;L各自独立地为氢、烷基或(甲氧羰基)氧甲基;且,所述式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)各结构式中,R 1、R 2和R 3中至少有一个为氘。 - 如权利要求3所述的如式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)所示的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药,其中,所述如式(I)所示的多环氨基甲酰基吡啶酮衍生物选自:
 或,所述如式(II)所示的多环氨基甲酰基吡啶酮衍生物选自:
或,所述如式(II)所示的多环氨基甲酰基吡啶酮衍生物选自: 或,所述如式(III)所示的多环氨基甲酰基吡啶酮衍生物选自:
或,所述如式(III)所示的多环氨基甲酰基吡啶酮衍生物选自: 或,所述如式(IV)所示的多环氨基甲酰基吡啶酮衍生物选自:
或,所述如式(IV)所示的多环氨基甲酰基吡啶酮衍生物选自: 或,所述如式(V)所示的多环氨基甲酰基吡啶酮衍生物选自:
或,所述如式(V)所示的多环氨基甲酰基吡啶酮衍生物选自: 或,所述如式(VI)所示的多环氨基甲酰基吡啶酮衍生物选自:
或,所述如式(VI)所示的多环氨基甲酰基吡啶酮衍生物选自:
- 如权利要求1-4中至少一项所述的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药,其中,所述的其药学上可接受的盐为所述的如式(VII)、式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)所示的多环氨基甲酰基吡啶酮衍生物与有机酸或无机酸所形成的酸加合盐;其中,所述的有机酸选自马来酸、反丁烯二酸、安息香酸、抗坏血酸、琥珀酸、甲磺酸、乙酸、三氟乙酸、草酸、丙酸、酒石酸、水杨酸、柠檬酸、葡萄糖酸、乳酸、扁桃酸、苯乙酸、天冬氨酸、硬脂酸、棕榈酸、乙二醇酸、谷氨酸和苯磺酸中的一种或多种;所述的无机酸选自盐酸、氢溴酸、硫酸、磷酸和硝酸中的一种或多种;或,所述的其药学上可接受的盐为所述的如式(VII)、式(I)、式(II)、式(III)、式(IV)、式(V)、或式(VI)所示的多环氨基甲酰基吡啶酮衍生物与选自碱金属离子、碱 土金属离子、铵离子的有机和无机阳离子中的一种或多种所成的盐;其中,所述的碱金属选自锂、钠、或钾;所述的碱土金属选自镁、钡、或钙。
- 一种药物组合物,其包含治疗有效量的如权利要求1-5中至少一项所述的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药,和药学上可接受的载体。
- 如权利要求6所述的药物组合物,其还包含其他药物活性成分,所述的其他药物活性成分选自神经胺酸酶抑制剂、RNA依赖性RNA聚合酶抑制剂、M2蛋白质抑制剂、PB2Cap结合抑制剂、抗HA抗体、或免疫作用药。
- 如权利要求7所述的药物组合物,所述的神经胺酸酶抑制剂为奥司他韦、扎那米韦、帕拉米韦、或Inavir;所述的RNA依赖性RNA聚合酶抑制剂为Favipiravir;所述的M2蛋白质抑制剂为金刚烷胺;所述的PB2Cap结合抑制剂为VX-787;所述的抗HA抗体为MHAA4594A;所述的免疫作用药为Nitazoxanide。
- 一种如权利要求1-5中至少一项所述的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药在制备5′帽依赖性核酸内切酶抑制剂中的应用。
- 一种如权利要求1-5中至少一项所述的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药在制备预防、治疗和/或缓解与5′帽依赖性核酸内切酶相关的疾病的药物中的应用。
- 如权利要求10所述的应用,其中,所述的与5′帽依赖性核酸内切酶相关的疾病是指由流感病毒感染引发的症状和/或疾病。
- 如权利要求11所述的应用,其中,所述的流感病毒选自甲型、乙型、或丙型。
- 如权利要求11所述的应用,其中,所述的症状选自伴有发热、发冷、头痛、肌肉痛、全身感到倦怠等的类感冒症状、或咽痛、流鼻涕、鼻塞、咳嗽、痰的呼吸道炎症、腹痛、呕吐、腹泻的胃肠症状、进而伴有急性脑病、肺炎二次感染的并发症。
- 一种预防、治疗和/或缓解疾病的方法,所述的方法包括给予需要其的个体治疗有效量的如权利要求1-5中至少一项所述的多环氨基甲酰基吡啶酮衍生物、其立体异构体、互变异构体、水合物、溶剂化物、活性代谢物、结晶形式、药学上可接受的盐或其前药,以抑制该个体体内的5′帽依赖性核酸内切酶水平。
- 如权利要求14所述的方法,其中,所述的疾病是指由流感病毒感染引发的症状和/或疾病;具体地,所述的流感病毒选自甲型、乙型、或丙型;所述的症状选自伴有发热、发冷、头痛、肌肉痛、全身感到倦怠等的类感冒症状、或咽痛、流鼻涕、鼻塞、咳嗽、痰 的呼吸道炎症、腹痛、呕吐、腹泻的胃肠症状、进而伴有急性脑病、肺炎二次感染的并发症。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/044,079 US20210093640A1 (en) | 2018-04-11 | 2019-04-11 | Polycyclic Carbamoylpyridone Derivatives, Pharmaceutical Compositions and Use Thereof |
| RU2020136675A RU2770096C1 (ru) | 2018-04-11 | 2019-04-11 | Полициклические производные карбамоилпиридона, фармацевтические композиции и их применение |
| EP19784679.3A EP3778608A4 (en) | 2018-04-11 | 2019-04-11 | POLYCYCLIC CARBAMOYL PYRIDONE DERIVATIVES, PHARMACEUTICAL COMPOSITIONS AND THEIR USES |
| AU2019252208A AU2019252208B2 (en) | 2018-04-11 | 2019-04-11 | Polycyclic carbamoylpyridone derivatives, pharmaceutical compositions and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810323355.2 | 2018-04-11 | ||
| CN201810323355.2A CN108440564B (zh) | 2018-04-11 | 2018-04-11 | 被取代的多环氨基甲酰基吡啶酮衍生物及其前药 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019196891A1 true WO2019196891A1 (zh) | 2019-10-17 |
Family
ID=63199693
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2019/082195 WO2019196891A1 (zh) | 2018-04-11 | 2019-04-11 | 多环氨基甲酰基吡啶酮衍生物、药物组合物及其用途 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20210093640A1 (zh) |
| EP (1) | EP3778608A4 (zh) |
| CN (1) | CN108440564B (zh) |
| AU (1) | AU2019252208B2 (zh) |
| RU (1) | RU2770096C1 (zh) |
| WO (1) | WO2019196891A1 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112409379A (zh) * | 2020-09-28 | 2021-02-26 | 长沙晶易医药科技有限公司 | 氘代的二氢二苯并硫杂卓化合物以及包含该化合物的药物组合物 |
| CN112778332A (zh) * | 2020-12-31 | 2021-05-11 | 重庆医科大学 | 一种巴洛沙韦酯中间体多环氨基甲酰基吡啶酮的合成方法 |
| WO2021093860A1 (zh) * | 2019-11-13 | 2021-05-20 | 南京知和医药科技有限公司 | 取代的双三环化合物及其药物组合物和用途 |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3008607A1 (en) | 2015-12-15 | 2017-06-22 | Shionogi & Co., Ltd. | A medicament for treating influenza characterized by combining a cap-dependent endonuclease inhibitor and an anti-influenza drug |
| WO2018030463A1 (ja) | 2016-08-10 | 2018-02-15 | 塩野義製薬株式会社 | 置換された多環性ピリドン誘導体およびそのプロドラッグを含有する医薬組成物 |
| CN109721615B (zh) * | 2017-09-18 | 2020-12-29 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其用途 |
| CN109503625A (zh) * | 2018-01-19 | 2019-03-22 | 赵蕾 | 一种多环吡啶酮化合物及其药物组合和用途 |
| CN108440564B (zh) * | 2018-04-11 | 2019-08-06 | 安帝康(无锡)生物科技有限公司 | 被取代的多环氨基甲酰基吡啶酮衍生物及其前药 |
| KR102501180B1 (ko) | 2018-04-24 | 2023-02-21 | 시오노기세야쿠 가부시키가이샤 | 안정성이 우수한 고형 제제 |
| WO2020020267A1 (zh) * | 2018-07-27 | 2020-01-30 | 深圳市塔吉瑞生物医药有限公司 | 一种取代的多环性吡啶酮化合物及其前药 |
| CN109721585B (zh) * | 2018-12-19 | 2020-09-29 | 浙江工业大学 | 一种巴洛沙韦关键中间体的制备方法 |
| CN109516998B (zh) * | 2018-12-25 | 2021-06-25 | 深圳市华先医药科技有限公司 | 一种巴洛沙韦中间体的合成方法 |
| CN111410661B (zh) * | 2019-01-04 | 2023-05-05 | 周雨恬 | 帽依赖性内切核酸酶抑制剂及其用途 |
| CN110105372B (zh) * | 2019-06-05 | 2022-05-10 | 南京焕然生物科技有限公司 | 一种r-7-(苄氧基)-四氢-1h-恶嗪并吡啶并-三嗪-6,8-二酮的制备方法 |
| CN110128366A (zh) * | 2019-06-05 | 2019-08-16 | 南京焕然生物科技有限公司 | 一种(r)-3-甲氧基-4-吗啉羧酸-2-丙烯-1-基酯的制备方法 |
| CN111057070A (zh) * | 2019-10-30 | 2020-04-24 | 浙江工业大学 | 一种巴洛沙韦关键中间体的合成方法 |
| CN112876510A (zh) * | 2020-02-10 | 2021-06-01 | 石家庄迪斯凯威医药科技有限公司 | 一种磷酸酯类多环化合物及其药物组合物和用途 |
| CN111233891B (zh) * | 2020-03-04 | 2021-05-04 | 江苏柯菲平医药股份有限公司 | 一种稠环吡啶酮衍生物及其制备方法和用途 |
| CN113527296A (zh) * | 2020-03-13 | 2021-10-22 | 北京凯因科技股份有限公司 | 一种流感病毒抑制剂 |
| WO2021180147A1 (zh) * | 2020-03-13 | 2021-09-16 | 北京凯因科技股份有限公司 | 一种流感病毒抑制剂 |
| WO2022128934A1 (en) * | 2020-12-15 | 2022-06-23 | Synthon B.V. | Solid forms of baloxavir, salts of baloxavir and cocrystals of baloxavir |
| CN112898312B (zh) * | 2021-01-29 | 2021-11-12 | 湖南南新制药股份有限公司 | 一种稠合多环吡啶酮衍生物及其用途 |
| CN113717199B (zh) * | 2021-08-13 | 2022-04-01 | 北京北朋科技有限公司 | 巴洛沙韦的氘代衍生物及其在抗流感病毒中的应用 |
| CN116082361B (zh) * | 2023-04-11 | 2023-06-13 | 和鼎(南京)医药技术有限公司 | 一种制备玛巴洛沙韦中间体和玛巴洛沙韦的方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018030463A1 (ja) * | 2016-08-10 | 2018-02-15 | 塩野義製薬株式会社 | 置換された多環性ピリドン誘導体およびそのプロドラッグを含有する医薬組成物 |
| CN107709321A (zh) * | 2015-04-28 | 2018-02-16 | 盐野义制药株式会社 | 经取代的多环性吡啶酮衍生物及其前药 |
| CN108440564A (zh) * | 2018-04-11 | 2018-08-24 | 朱孝云 | 被取代的多环氨基甲酰基吡啶酮衍生物及其前药 |
| CN109503625A (zh) * | 2018-01-19 | 2019-03-22 | 赵蕾 | 一种多环吡啶酮化合物及其药物组合和用途 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2013003139A (es) * | 2010-09-24 | 2013-06-18 | Shionogi & Co | Profarmaco de derivado de carbamoilpiridona policiclica substituida. |
| JOP20170169A1 (ar) * | 2016-08-29 | 2019-01-30 | Novartis Ag | مركبات بيريدازين ثلاثية الحلقة مندمجة تفيد في علاج العدوى بفيروس أورثوميكسو |
| WO2020020267A1 (zh) * | 2018-07-27 | 2020-01-30 | 深圳市塔吉瑞生物医药有限公司 | 一种取代的多环性吡啶酮化合物及其前药 |
-
2018
- 2018-04-11 CN CN201810323355.2A patent/CN108440564B/zh active Active
-
2019
- 2019-04-11 US US17/044,079 patent/US20210093640A1/en active Pending
- 2019-04-11 WO PCT/CN2019/082195 patent/WO2019196891A1/zh unknown
- 2019-04-11 EP EP19784679.3A patent/EP3778608A4/en active Pending
- 2019-04-11 AU AU2019252208A patent/AU2019252208B2/en active Active
- 2019-04-11 RU RU2020136675A patent/RU2770096C1/ru active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107709321A (zh) * | 2015-04-28 | 2018-02-16 | 盐野义制药株式会社 | 经取代的多环性吡啶酮衍生物及其前药 |
| WO2018030463A1 (ja) * | 2016-08-10 | 2018-02-15 | 塩野義製薬株式会社 | 置換された多環性ピリドン誘導体およびそのプロドラッグを含有する医薬組成物 |
| CN109503625A (zh) * | 2018-01-19 | 2019-03-22 | 赵蕾 | 一种多环吡啶酮化合物及其药物组合和用途 |
| CN108440564A (zh) * | 2018-04-11 | 2018-08-24 | 朱孝云 | 被取代的多环氨基甲酰基吡啶酮衍生物及其前药 |
Non-Patent Citations (4)
| Title |
|---|
| LGA M.ROCHOVANSKY, VIROLOGY, vol. 73, 1976, pages 327 - 338 |
| RAJ SNER ET AL., COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, vol. 47, no. 1, 1987, pages 65 - 71 |
| See also references of EP3778608A4 |
| WANG, SHIZHEN: "Part VI Research of Nuclear Medicine", MOLECULAR NUCLEAR MEDICINE (2ND EDITION, 30 April 2004 (2004-04-30), pages 417 - 418, XP009524204 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021093860A1 (zh) * | 2019-11-13 | 2021-05-20 | 南京知和医药科技有限公司 | 取代的双三环化合物及其药物组合物和用途 |
| CN115175913A (zh) * | 2019-11-13 | 2022-10-11 | 南京知和医药科技有限公司 | 取代的双三环化合物及其药物组合物和用途 |
| CN115175913B (zh) * | 2019-11-13 | 2023-11-28 | 南京知和医药科技有限公司 | 取代的双三环化合物及其药物组合物和用途 |
| CN112409379A (zh) * | 2020-09-28 | 2021-02-26 | 长沙晶易医药科技有限公司 | 氘代的二氢二苯并硫杂卓化合物以及包含该化合物的药物组合物 |
| WO2022063016A1 (zh) * | 2020-09-28 | 2022-03-31 | 长沙晶易医药科技有限公司 | 氘代的二氢二苯并硫杂卓化合物以及包含该化合物的药物组合物 |
| CN112409379B (zh) * | 2020-09-28 | 2023-07-28 | 长沙晶易医药科技股份有限公司 | 氘代的二氢二苯并硫杂卓化合物以及包含该化合物的药物组合物 |
| CN112778332A (zh) * | 2020-12-31 | 2021-05-11 | 重庆医科大学 | 一种巴洛沙韦酯中间体多环氨基甲酰基吡啶酮的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2019252208A1 (en) | 2020-11-19 |
| EP3778608A1 (en) | 2021-02-17 |
| RU2770096C1 (ru) | 2022-04-14 |
| AU2019252208B2 (en) | 2022-03-03 |
| CN108440564B (zh) | 2019-08-06 |
| US20210093640A1 (en) | 2021-04-01 |
| EP3778608A4 (en) | 2021-06-23 |
| CN108440564A (zh) | 2018-08-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019196891A1 (zh) | 多环氨基甲酰基吡啶酮衍生物、药物组合物及其用途 | |
| CN114426568B (zh) | 2-氧代-3-吡咯烷基丙腈类化合物及其药物组合物和用途 | |
| JP6516705B2 (ja) | Hbv感染に対する抗ウイルス剤としてのスルファモイルベンズアミド誘導体 | |
| EP3391888B1 (en) | Medicine for treating influenza characterized by comprising combination of cap-dependent endonuclease inhibitor with anti-influenza drug | |
| CN111548384A (zh) | 用于抗新型冠状病毒治疗的被取代的n4-羟基胞苷衍生物及其前药 | |
| WO2020020267A1 (zh) | 一种取代的多环性吡啶酮化合物及其前药 | |
| TW201542553A (zh) | 氮雜吡啶酮化合物及其用途 | |
| US20140308282A1 (en) | Pharmaceutical compounds | |
| NZ719729A (en) | Inhibitors of influenza viruses replication | |
| TW201305163A (zh) | 用於治療呼吸道融合病毒感染之化合物 | |
| WO2020241759A1 (ja) | インフルエンザウイルス感染症またはコロナウイルス感染症の予防および/または治療剤 | |
| CN111410661A (zh) | 帽依赖性内切核酸酶抑制剂及其用途 | |
| US20230233573A1 (en) | Treating influenza using substituted polycyclic pyridone derivatives and prodrugs thereof | |
| JP2022507117A (ja) | 呼吸器疾患の処理のための新規な化合物 | |
| JP2021514967A (ja) | ピリジノイミダゾール系化合物の結晶型、塩型及びその製造方法 | |
| JP6034960B2 (ja) | フッ素置換(3r、4r、5s)−5−グアニジノ−4−アセトアミド−3−(ペンタン−3−イルオキシ)シクロヘキセン−1−カルボン酸、そのエステル及びその使用 | |
| CN112771048A (zh) | 流感病毒复制抑制剂及其中间体和用途 | |
| TW202204337A (zh) | 截短側耳素類之醫療用途 | |
| TWI821343B (zh) | 流感病毒複製之抑制劑 | |
| WO2024098856A1 (zh) | 一种抗流感病毒衍生物及其用途 | |
| CN113979936A (zh) | 2-芳脲基-n-[3-(4-吗啉基)丙基]烟酰胺类化合物及其应用 | |
| CN113979935A (zh) | 2-芳脲基-n-(4-氟苄基)烟酰胺类化合物及其应用 | |
| US20230151034A1 (en) | Peptidomimetic n5-methyl-n2-(nonanoyl-l-leucyl)-l-glutaminate derivatives, triazaspiro[4.14]nonadecane derivatives and similar compounds as inhibitors of norovirus and coronavirus replication | |
| CN114057635A (zh) | 2-芳脲基-n-[2-(2-甲氧基苯氧基)乙基]烟酰胺类化合物及其应用 | |
| WO2023137875A1 (zh) | 一种药物组合物及其抗病毒用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19784679 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019252208 Country of ref document: AU Date of ref document: 20190411 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019784679 Country of ref document: EP Effective date: 20201111 |