WO2018043007A1 - 触媒の製造方法、及びアクリロニトリルの製造方法 - Google Patents
触媒の製造方法、及びアクリロニトリルの製造方法 Download PDFInfo
- Publication number
- WO2018043007A1 WO2018043007A1 PCT/JP2017/027950 JP2017027950W WO2018043007A1 WO 2018043007 A1 WO2018043007 A1 WO 2018043007A1 JP 2017027950 W JP2017027950 W JP 2017027950W WO 2018043007 A1 WO2018043007 A1 WO 2018043007A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- containing liquid
- silica
- liquid
- mass
- Prior art date
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 325
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 74
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 title claims description 65
- 239000002002 slurry Substances 0.000 claims abstract description 157
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims abstract description 150
- 239000002243 precursor Substances 0.000 claims abstract description 111
- 239000002245 particle Substances 0.000 claims abstract description 105
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims abstract description 95
- 239000000377 silicon dioxide Substances 0.000 claims abstract description 74
- 239000002994 raw material Substances 0.000 claims abstract description 62
- 239000011259 mixed solution Substances 0.000 claims abstract description 43
- 229910052750 molybdenum Inorganic materials 0.000 claims abstract description 41
- 229910052797 bismuth Inorganic materials 0.000 claims abstract description 38
- 238000002156 mixing Methods 0.000 claims abstract description 36
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims abstract description 32
- 239000011733 molybdenum Substances 0.000 claims abstract description 31
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 claims abstract description 28
- 229910052742 iron Inorganic materials 0.000 claims abstract description 27
- 238000001035 drying Methods 0.000 claims abstract description 22
- 238000002360 preparation method Methods 0.000 claims abstract description 20
- 238000001694 spray drying Methods 0.000 claims abstract description 20
- 238000010304 firing Methods 0.000 claims abstract description 18
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims abstract 4
- 239000007788 liquid Substances 0.000 claims description 358
- 239000000203 mixture Substances 0.000 claims description 98
- 238000000034 method Methods 0.000 claims description 94
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 71
- 229910044991 metal oxide Inorganic materials 0.000 claims description 68
- 150000004706 metal oxides Chemical class 0.000 claims description 68
- 230000008569 process Effects 0.000 claims description 39
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 31
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 claims description 30
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 claims description 30
- 239000011777 magnesium Substances 0.000 claims description 25
- 229910021529 ammonia Inorganic materials 0.000 claims description 15
- 239000011651 chromium Substances 0.000 claims description 15
- 229910052749 magnesium Inorganic materials 0.000 claims description 15
- 239000011701 zinc Substances 0.000 claims description 15
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 14
- 229910052759 nickel Inorganic materials 0.000 claims description 13
- 229910017052 cobalt Inorganic materials 0.000 claims description 12
- 239000010941 cobalt Substances 0.000 claims description 12
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims description 12
- 229910052782 aluminium Inorganic materials 0.000 claims description 11
- 229910052788 barium Inorganic materials 0.000 claims description 11
- 229910052791 calcium Inorganic materials 0.000 claims description 11
- 239000011575 calcium Substances 0.000 claims description 11
- 229910052804 chromium Inorganic materials 0.000 claims description 11
- 229910052733 gallium Inorganic materials 0.000 claims description 11
- 229910052738 indium Inorganic materials 0.000 claims description 11
- 229910052712 strontium Inorganic materials 0.000 claims description 11
- 229910052727 yttrium Inorganic materials 0.000 claims description 11
- 229910052725 zinc Inorganic materials 0.000 claims description 11
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims description 10
- 229910052684 Cerium Inorganic materials 0.000 claims description 10
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 claims description 10
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 claims description 10
- 229910052779 Neodymium Inorganic materials 0.000 claims description 10
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 10
- 229910052777 Praseodymium Inorganic materials 0.000 claims description 10
- 229910052772 Samarium Inorganic materials 0.000 claims description 10
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 10
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 10
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 claims description 10
- 229910052792 caesium Inorganic materials 0.000 claims description 10
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 claims description 10
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 claims description 10
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 claims description 10
- 229910052746 lanthanum Inorganic materials 0.000 claims description 10
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 claims description 10
- QEFYFXOXNSNQGX-UHFFFAOYSA-N neodymium atom Chemical compound [Nd] QEFYFXOXNSNQGX-UHFFFAOYSA-N 0.000 claims description 10
- 229910052700 potassium Inorganic materials 0.000 claims description 10
- 239000011591 potassium Substances 0.000 claims description 10
- PUDIUYLPXJFUGB-UHFFFAOYSA-N praseodymium atom Chemical compound [Pr] PUDIUYLPXJFUGB-UHFFFAOYSA-N 0.000 claims description 10
- 229910052701 rubidium Inorganic materials 0.000 claims description 10
- IGLNJRXAVVLDKE-UHFFFAOYSA-N rubidium atom Chemical compound [Rb] IGLNJRXAVVLDKE-UHFFFAOYSA-N 0.000 claims description 10
- KZUNJOHGWZRPMI-UHFFFAOYSA-N samarium atom Chemical compound [Sm] KZUNJOHGWZRPMI-UHFFFAOYSA-N 0.000 claims description 10
- CIOAGBVUUVVLOB-UHFFFAOYSA-N strontium atom Chemical compound [Sr] CIOAGBVUUVVLOB-UHFFFAOYSA-N 0.000 claims description 10
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 claims description 10
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 claims description 9
- 229910001882 dioxygen Inorganic materials 0.000 claims description 8
- 238000003672 processing method Methods 0.000 claims description 7
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 67
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 62
- 229910052751 metal Inorganic materials 0.000 description 60
- HSJPMRKMPBAUAU-UHFFFAOYSA-N cerium(3+);trinitrate Chemical compound [Ce+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O HSJPMRKMPBAUAU-UHFFFAOYSA-N 0.000 description 56
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 54
- 239000000243 solution Substances 0.000 description 49
- 238000006243 chemical reaction Methods 0.000 description 48
- GEVPUGOOGXGPIO-UHFFFAOYSA-N oxalic acid;dihydrate Chemical compound O.O.OC(=O)C(O)=O GEVPUGOOGXGPIO-UHFFFAOYSA-N 0.000 description 46
- 229910004298 SiO 2 Inorganic materials 0.000 description 43
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 38
- 229910017604 nitric acid Inorganic materials 0.000 description 38
- 238000003756 stirring Methods 0.000 description 38
- 239000002184 metal Substances 0.000 description 34
- 239000007921 spray Substances 0.000 description 33
- RXPAJWPEYBDXOG-UHFFFAOYSA-N hydron;methyl 4-methoxypyridine-2-carboxylate;chloride Chemical compound Cl.COC(=O)C1=CC(OC)=CC=N1 RXPAJWPEYBDXOG-UHFFFAOYSA-N 0.000 description 31
- 239000000843 powder Substances 0.000 description 31
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 30
- MVFCKEFYUDZOCX-UHFFFAOYSA-N iron(2+);dinitrate Chemical compound [Fe+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O MVFCKEFYUDZOCX-UHFFFAOYSA-N 0.000 description 30
- 229910003208 (NH4)6Mo7O24·4H2O Inorganic materials 0.000 description 29
- 230000000052 comparative effect Effects 0.000 description 28
- 150000001735 carboxylic acids Chemical class 0.000 description 26
- RTHYXYOJKHGZJT-UHFFFAOYSA-N rubidium nitrate Inorganic materials [Rb+].[O-][N+]([O-])=O RTHYXYOJKHGZJT-UHFFFAOYSA-N 0.000 description 26
- KHAUBYTYGDOYRU-IRXASZMISA-N trospectomycin Chemical compound CN[C@H]([C@H]1O2)[C@@H](O)[C@@H](NC)[C@H](O)[C@H]1O[C@H]1[C@]2(O)C(=O)C[C@@H](CCCC)O1 KHAUBYTYGDOYRU-IRXASZMISA-N 0.000 description 26
- KBJMLQFLOWQJNF-UHFFFAOYSA-N nickel(ii) nitrate Chemical compound [Ni+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O KBJMLQFLOWQJNF-UHFFFAOYSA-N 0.000 description 24
- UFMZWBIQTDUYBN-UHFFFAOYSA-N cobalt dinitrate Chemical compound [Co+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O UFMZWBIQTDUYBN-UHFFFAOYSA-N 0.000 description 23
- 229910001981 cobalt nitrate Inorganic materials 0.000 description 23
- 239000000047 product Substances 0.000 description 20
- 125000004429 atom Chemical group 0.000 description 17
- MEFBJEMVZONFCJ-UHFFFAOYSA-N molybdate Chemical compound [O-][Mo]([O-])(=O)=O MEFBJEMVZONFCJ-UHFFFAOYSA-N 0.000 description 17
- 230000003068 static effect Effects 0.000 description 14
- 229910004631 Ce(NO3)3.6H2O Inorganic materials 0.000 description 13
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 13
- 239000001301 oxygen Substances 0.000 description 13
- 229910052760 oxygen Inorganic materials 0.000 description 13
- 229910002492 Ce(NO3)3·6H2O Inorganic materials 0.000 description 12
- 229910002554 Fe(NO3)3·9H2O Inorganic materials 0.000 description 12
- YIXJRHPUWRPCBB-UHFFFAOYSA-N magnesium nitrate Chemical compound [Mg+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O YIXJRHPUWRPCBB-UHFFFAOYSA-N 0.000 description 12
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 11
- 229910000608 Fe(NO3)3.9H2O Inorganic materials 0.000 description 10
- 238000001354 calcination Methods 0.000 description 10
- 239000007789 gas Substances 0.000 description 10
- QGUAJWGNOXCYJF-UHFFFAOYSA-N cobalt dinitrate hexahydrate Chemical compound O.O.O.O.O.O.[Co+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O QGUAJWGNOXCYJF-UHFFFAOYSA-N 0.000 description 9
- 239000011164 primary particle Substances 0.000 description 8
- 235000006408 oxalic acid Nutrition 0.000 description 7
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 6
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 6
- PHFQLYPOURZARY-UHFFFAOYSA-N chromium trinitrate Chemical compound [Cr+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O PHFQLYPOURZARY-UHFFFAOYSA-N 0.000 description 6
- ONDPHDOFVYQSGI-UHFFFAOYSA-N zinc nitrate Chemical compound [Zn+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O ONDPHDOFVYQSGI-UHFFFAOYSA-N 0.000 description 6
- 239000012018 catalyst precursor Substances 0.000 description 5
- 230000005484 gravity Effects 0.000 description 5
- 238000009616 inductively coupled plasma Methods 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 238000004220 aggregation Methods 0.000 description 4
- 230000002776 aggregation Effects 0.000 description 4
- 238000013019 agitation Methods 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 238000002149 energy-dispersive X-ray emission spectroscopy Methods 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 150000002739 metals Chemical class 0.000 description 4
- 239000004323 potassium nitrate Substances 0.000 description 4
- 235000010333 potassium nitrate Nutrition 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- 150000003839 salts Chemical group 0.000 description 4
- 125000005372 silanol group Chemical group 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- 229910002651 NO3 Inorganic materials 0.000 description 3
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 3
- 238000005299 abrasion Methods 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 3
- 150000003863 ammonium salts Chemical class 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000005336 cracking Methods 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 238000005304 joining Methods 0.000 description 3
- -1 organic acid salt Chemical class 0.000 description 3
- 238000004445 quantitative analysis Methods 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- 238000000889 atomisation Methods 0.000 description 2
- IWOUKMZUPDVPGQ-UHFFFAOYSA-N barium nitrate Chemical compound [Ba+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O IWOUKMZUPDVPGQ-UHFFFAOYSA-N 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- ZCCIPPOKBCJFDN-UHFFFAOYSA-N calcium nitrate Chemical compound [Ca+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O ZCCIPPOKBCJFDN-UHFFFAOYSA-N 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000004993 emission spectroscopy Methods 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- CHPZKNULDCNCBW-UHFFFAOYSA-N gallium nitrate Chemical compound [Ga+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CHPZKNULDCNCBW-UHFFFAOYSA-N 0.000 description 2
- 239000001307 helium Substances 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- VCJMYUPGQJHHFU-UHFFFAOYSA-N iron(3+);trinitrate Chemical compound [Fe+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O VCJMYUPGQJHHFU-UHFFFAOYSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- 150000002823 nitrates Chemical class 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 239000012495 reaction gas Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 238000004611 spectroscopical analysis Methods 0.000 description 2
- DHEQXMRUPNDRPG-UHFFFAOYSA-N strontium nitrate Chemical compound [Sr+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O DHEQXMRUPNDRPG-UHFFFAOYSA-N 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- XURCIPRUUASYLR-UHFFFAOYSA-N Omeprazole sulfide Chemical compound N=1C2=CC(OC)=CC=C2NC=1SCC1=NC=C(C)C(OC)=C1C XURCIPRUUASYLR-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 description 1
- 229940010552 ammonium molybdate Drugs 0.000 description 1
- 235000018660 ammonium molybdate Nutrition 0.000 description 1
- 239000011609 ammonium molybdate Substances 0.000 description 1
- QZPSXPBJTPJTSZ-UHFFFAOYSA-N aqua regia Chemical compound Cl.O[N+]([O-])=O QZPSXPBJTPJTSZ-UHFFFAOYSA-N 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 238000001479 atomic absorption spectroscopy Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000007809 chemical reaction catalyst Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 238000004952 furnace firing Methods 0.000 description 1
- 229940044658 gallium nitrate Drugs 0.000 description 1
- 230000020169 heat generation Effects 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- FYDKNKUEBJQCCN-UHFFFAOYSA-N lanthanum(3+);trinitrate Chemical compound [La+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O FYDKNKUEBJQCCN-UHFFFAOYSA-N 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002923 metal particle Substances 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000010298 pulverizing process Methods 0.000 description 1
- 239000005297 pyrex Substances 0.000 description 1
- 229910052761 rare earth metal Inorganic materials 0.000 description 1
- 150000002910 rare earth metals Chemical class 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 229910021642 ultra pure water Inorganic materials 0.000 description 1
- 239000012498 ultrapure water Substances 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
Images

Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/0009—Use of binding agents; Moulding; Pressing; Powdering; Granulating; Addition of materials ameliorating the mechanical properties of the product catalyst
- B01J37/0027—Powdering
- B01J37/0045—Drying a slurry, e.g. spray drying
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- B01J21/08—Silica
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/887—Molybdenum containing in addition other metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/8876—Arsenic, antimony or bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/887—Molybdenum containing in addition other metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/8878—Chromium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/04—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing carboxylic acids or their salts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/391—Physical properties of the active metal ingredient
- B01J35/394—Metal dispersion value, e.g. percentage or fraction
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/396—Distribution of the active metal ingredient
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/40—Catalysts, in general, characterised by their form or physical properties characterised by dimensions, e.g. grain size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/50—Catalysts, in general, characterised by their form or physical properties characterised by their shape or configuration
- B01J35/51—Spheres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/04—Mixing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/082—Decomposition and pyrolysis
- B01J37/088—Decomposition of a metal salt
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/18—Preparation of carboxylic acid nitriles by reaction of ammonia or amines with compounds containing carbon-to-carbon multiple bonds other than in six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/24—Preparation of carboxylic acid nitriles by ammoxidation of hydrocarbons or substituted hydrocarbons
- C07C253/26—Preparation of carboxylic acid nitriles by ammoxidation of hydrocarbons or substituted hydrocarbons containing carbon-to-carbon multiple bonds, e.g. unsaturated aldehydes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/06—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms of an acyclic and unsaturated carbon skeleton
- C07C255/07—Mononitriles
- C07C255/08—Acrylonitrile; Methacrylonitrile
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/10—Constitutive chemical elements of heterogeneous catalysts of Group I (IA or IB) of the Periodic Table
- B01J2523/13—Potassium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/10—Constitutive chemical elements of heterogeneous catalysts of Group I (IA or IB) of the Periodic Table
- B01J2523/14—Rubidium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/30—Constitutive chemical elements of heterogeneous catalysts of Group III (IIIA or IIIB) of the Periodic Table
- B01J2523/37—Lanthanides
- B01J2523/3712—Cerium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/50—Constitutive chemical elements of heterogeneous catalysts of Group V (VA or VB) of the Periodic Table
- B01J2523/54—Bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/60—Constitutive chemical elements of heterogeneous catalysts of Group VI (VIA or VIB) of the Periodic Table
- B01J2523/68—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/80—Constitutive chemical elements of heterogeneous catalysts of Group VIII of the Periodic Table
- B01J2523/84—Metals of the iron group
- B01J2523/842—Iron
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/80—Constitutive chemical elements of heterogeneous catalysts of Group VIII of the Periodic Table
- B01J2523/84—Metals of the iron group
- B01J2523/845—Cobalt
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/80—Constitutive chemical elements of heterogeneous catalysts of Group VIII of the Periodic Table
- B01J2523/84—Metals of the iron group
- B01J2523/847—Nickel
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Definitions
- the present invention relates to a method for producing a catalyst and a method for producing acrylonitrile.
- a reaction for producing acrylonitrile by reacting propylene and ammonia in the presence of molecular oxygen is known as an “ammoxidation reaction”, and this reaction is used as an industrial production method of acrylonitrile.
- an oxide catalyst is used to achieve a good acrylonitrile yield.
- a catalyst containing Mo—Bi—Fe or Fe—Sb as an essential component is industrially used.
- a catalyst in which other elements are added in addition to the above-mentioned essential components is also known (see, for example, Patent Documents 1 and 2).
- Patent Documents 3, 4, and 5 describe a method of adding a carboxylic acid to a raw material slurry in a method for preparing an ammoxidation catalyst containing molybdenum, bismuth, and iron as essential components.
- JP 2013-169482 A JP 2008-212779 A JP 2013-17917 A JP 2000-5603 A JP 2000-37631 A
- the present invention has been made in view of the above problems, and an object thereof is to provide a method for producing a catalyst exhibiting a high acrylonitrile yield and a method for producing acrylonitrile.
- a method for producing a catalyst comprising: [2] The method for producing a catalyst according to [1], wherein the catalyst includes a metal oxide having a bulk composition represented by the following general formula (1) and silica.
- X represents one or more elements selected from the group consisting of nickel, cobalt, magnesium, calcium, zinc, strontium and barium
- Y represents cerium, chromium, lanthanum, neodymium, yttrium, One or more elements selected from the group consisting of praseodymium, samarium, aluminum, gallium and indium
- Z represents one or more elements selected from the group consisting of potassium, rubidium and cesium
- a, b, c , D, e, and f represent the atomic ratio of each element, and 0.1 ⁇ a ⁇ 3.0, 0.1 ⁇ b ⁇ 3.0, 0.1 ⁇ c ⁇ 10.0, 0,.
- a standard deviation of a value obtained by dividing a ratio of Bi molar concentration to Mo molar concentration on the catalyst particle surface by a ratio of Bi molar concentration to Mo molar concentration of the metal oxide bulk is 0.2 or less.
- the step (II) includes the following steps (i) and (ii): The method for producing a catalyst according to any one of [1] to [3], wherein the drying step includes the following step (iii): (I) preparing a Mo-containing liquid containing at least Mo and a Bi-containing liquid containing at least Bi; (Ii) The Mo-containing liquid is continuously supplied to the first flow path, the Bi-containing liquid is continuously supplied to the second flow path, and the first flow path and the second flow path are A step of mixing the Mo-containing liquid and the Bi-containing liquid to obtain a MoBi-containing liquid by merging the Mo-containing liquid and the Bi-containing liquid downstream from the respective supply locations. (Iii) A step of drying the MoBi-containing liquid.
- mass supply rates of the Mo-containing liquid and the Bi-containing liquid to the first flow path or the second flow path are set to mA (g / min) and mB (g / min) so that 60% by mass or more of the total amount of the total supply amount MA (g) and MB (g) per batch of the Mo-containing liquid and Bi-containing liquid satisfy the formula (2).
- the method for producing a catalyst according to [6], wherein (MB / mA) / (MB / MA) 0.5 to 1.5 (2) [8]
- the molar supply rates of the Mo-containing liquid and the Bi-containing liquid to the first flow path or the second flow path are set to m ⁇ (mol / min) and m ⁇ (mol / min), respectively.
- a catalyst comprising particles containing a metal oxide having a bulk composition represented by the following formula (1) and silica, The standard deviation of the value obtained by dividing the ratio of Bi molar concentration to Mo molar concentration on the catalyst particle surface by the ratio of Bi molar concentration to Mo molar concentration of the metal oxide bulk is 0.2 or less. catalyst.
- X represents one or more elements selected from the group consisting of nickel, cobalt, magnesium, calcium, zinc, strontium and barium
- Y represents cerium, chromium, lanthanum, neodymium, yttrium, One or more elements selected from the group consisting of praseodymium, samarium, aluminum, gallium and indium
- Z represents one or more elements selected from the group consisting of potassium, rubidium and cesium
- a, b, c , D, e, and f represent the atomic ratio of each element, and 0.1 ⁇ a ⁇ 3.0, 0.1 ⁇ b ⁇ 3.0, 0.1 ⁇ c ⁇ 10.0, 0,.
- a process for producing acrylonitrile comprising a step of reacting propylene, molecular oxygen and ammonia in the presence of the catalyst according to [11].
- the present embodiment a mode for carrying out the present invention (hereinafter simply referred to as “the present embodiment”) will be described.
- the present invention is not limited to the following embodiment and is within the scope not departing from the gist thereof.
- Various variations are possible.
- the catalyst production method of the present embodiment includes a preparation step of preparing a precursor slurry containing molybdenum, bismuth, iron, silica, and carboxylic acid, and spray drying the precursor slurry to obtain dry particles.
- the catalyst production method of the present embodiment can obtain a catalyst exhibiting a high acrylonitrile yield (and acrylonitrile selectivity) by having the above-described configuration.
- yield (selectivity) is high refers to a case where an oxide catalyst having the same or similar composition represented by formula (1) described later is used. The yield of acrylonitrile is high.
- the present inventors consider this as follows. Although the active site in the ammoxidation reaction is molybdate containing bismuth, high acrylonitrile selectivity cannot be obtained by itself. It is considered that the molybdate containing bismuth improves the acrylonitrile selectivity by combining with the molybdate containing iron and the molybdate containing other metals.
- Adding carboxylic acid to the precursor slurry improves the dispersibility of the metal component containing bismuth.
- molybdate containing bismuth is likely to be complexed with molybdate containing other metals, and acrylonitrile selectivity is considered to be improved.
- Silica is mainly used as a support for the catalyst for ammoxidation. It is considered that silanol groups present in silica are bonded to metal components in the slurry, grow to active sites that decompose propylene and acrylonitrile, and lower acrylonitrile selectivity.
- the mechanism of action in the present embodiment is not limited to the above.
- the catalyst obtained by the manufacturing method of this embodiment contains molybdenum, bismuth, and iron as essential components.
- Molybdenum plays a role as an adsorption site for propylene and an activation site for ammonia.
- Bismuth also plays a role in activating propylene and extracting ⁇ -position hydrogen to generate ⁇ -allyl species.
- iron plays a role of supplying oxygen present in the gas phase to the catalytic active site by trivalent / divalent redox.
- the optional component that may be contained in the catalyst obtained by the production method of the present embodiment is not particularly limited, but for example, selected from the group consisting of nickel, cobalt, magnesium, calcium, zinc, strontium, and barium.
- elements Z to be mentioned.
- the element X in the catalyst obtained by the production method of the present embodiment forms a molybdate having appropriate lattice defects, and plays a role of facilitating oxygen migration in the bulk. Further, the element Y has a redox function in the catalyst like iron. Furthermore, the element Z plays the role which suppresses the decomposition reaction of a main product and a raw material by blocking the acid site which exists on the catalyst surface.
- the catalyst obtained by the catalyst production method of the present embodiment preferably has a composition represented by the following general formula (1).
- the acrylonitrile selectivity tends to be further improved.
- Mo 12 Bi a Fe b X c Y d Z e O f (1)
- X represents one or more elements selected from the group consisting of nickel, cobalt, magnesium, calcium, zinc, strontium, and barium
- Y represents cerium, chromium, lanthanum, neodymium, yttrium, praseodymium.
- a represents the atomic ratio of bismuth with respect to 12 atoms of molybdenum, 0.1 ⁇ a ⁇ 3.0, preferably 0.15 ⁇ a ⁇ 1.0, and more preferably 0.2 ⁇ a ⁇ 0.7.
- b represents the atomic ratio of iron to 12 atoms of molybdenum, 0.1 ⁇ b ⁇ 3.0, preferably 0.5 ⁇ b ⁇ 2.5, more preferably 1.0 ⁇ b ⁇ 2.0.
- e represents the atomic ratio of Z to molybdenum 12 atoms, and 0.01 ⁇ e ⁇ 2.0, preferably 0.05 ⁇ e ⁇ 1.0.
- f represents the atomic ratio of oxygen to 12 atoms of molybdenum, and is a value that satisfies the balance of valences. Note that f is a value corresponding to the number of oxygen atoms necessary to satisfy the valence requirements of other elements present.
- g and h it is preferable to adjust g and h as follows. That is, when the metal oxide does not contain magnesium, it is preferable that g ⁇ 1.0 or 6.5 ⁇ g and h ⁇ 1.0 or 6.5 ⁇ h.
- the metal oxide contains magnesium
- the composition of the obtained catalyst can be adjusted to the above range by appropriately adjusting the raw materials to be used and the amount of use thereof.
- the catalyst obtained by the production method of the present embodiment preferably has a more uniform particle surface composition.
- the ratio of the Bi molar concentration to the Mo molar concentration on the catalyst particle surface is determined by metal oxidation.
- the standard deviation of the value divided by the ratio of the Bi molar concentration to the Mo molar concentration of the product bulk is preferably 0.2 or less. The standard deviation will be described later.
- a fluidized bed reaction is generally selected in which a catalyst is fluidized by a reaction gas. Therefore, the catalyst preferably has a certain strength or more. From such a viewpoint, the catalyst is supported on a silica support.
- silica as a carrier, it is possible to suppress the decrease in selectivity of acrylonitrile and greatly improve the wear resistance and particle strength of the catalyst.
- carrier component of catalysts other than a silica For example, oxides, such as an alumina, a titania, a zirconia, can also be included.
- the content of the carrier is preferably 30 to 70% by mass and more preferably 35 to 65% by mass with respect to the total mass of the catalyst and the carrier.
- the support content is 30% by mass or more, the wear resistance and particle strength of the catalyst tend to be further improved.
- the carrier content is 70% by mass or less, the acrylonitrile selectivity tends to be further improved.
- the raw material of silica used as the carrier is not particularly limited, but silica sol is preferable.
- the primary particle diameter of the silica contained in the silica sol is not particularly limited, and silicas having different primary particle diameters may be mixed and used.
- the preparation step in the production method of the present embodiment includes a step of mixing a silica raw material and a carboxylic acid to prepare a silica-carboxylic acid mixed solution (also referred to as “step (I)” in this specification).
- a mixed liquid can be obtained by adding carboxylic acid to a silica raw material.
- Silica sol is preferable as the silica raw material.
- the preferred concentration of silica sol in the raw material state in which no other components are mixed is 10 to 50% by mass.
- the carboxylic acid mixed with the silica raw material is not particularly limited, and examples thereof include oxalic acid, tartaric acid, succinic acid, malic acid, and citric acid. Oxalic acid and tartaric acid are preferred, and oxalic acid is more preferred.
- carboxylic acid is a typical coordinating organic compound, and is considered to promote high dispersion of the metal component by bonding with the metal component.
- the purity of the carboxylic acid used is preferably 99.0% or more.
- the carboxylic acid can be added as a solid such as a powder or as an aqueous solution.
- the content of carboxylic acid is preferably 0.01 to 0.10 molar equivalents relative to the total of metal elements constituting the catalyst in the precursor slurry described below. More preferably, it is 0.02 to 0.09 molar equivalent, and still more preferably 0.02 to 0.07 molar equivalent.
- the content of carboxylic acid is 0.01 molar equivalent or more, the acrylonitrile yield of the resulting catalyst tends to be further improved.
- the content of carboxylic acid is 0.10 molar equivalent or less, heat generation due to decomposition or release of carboxylic acid and cracking of catalyst particles are suppressed in the catalyst production stage, and the strength of the resulting catalyst is further improved. Tend to.
- the content of carboxylic acid can be adjusted to the above range depending on the charging ratio of each raw material.
- the step of adding carboxylic acid to the silica raw material is preferably completed quickly. Since there exists a pH region where the silica raw material becomes unstable in water, the stability of the silica raw material tends to be improved if it can pass through the pH region in a short time.
- the rate of addition of the carboxylic acid SiO 2 1 kg per 5 g / sec. Or more, more preferably SiO 2 1kg per 10g / sec. That's it. The addition rate is 5 g / sec. Due to the above, silica aggregation is suppressed, and the selectivity of the acrylonitrile and the wear strength of the resulting catalyst tend to be improved.
- “per 1 kg of SiO 2 ” means “per 1 kg of SiO 2 contained in the silica raw material in terms of solid content”.
- the stirring time of the silica-carboxylic acid mixed solution is preferably 60 minutes or less, more preferably 20 minutes or less. By being 60 minutes or less, silica aggregation is suppressed, and the selectivity of the acrylonitrile and the wear strength of the resulting catalyst tend to be improved.
- the stirring temperature of the silica-carboxylic acid mixed solution is preferably 30 ° C. or higher, more preferably 35 ° C. or higher, from the viewpoint of improving the dispersibility of the particles.
- the pH of the silica-carboxylic acid mixed solution is preferably 6.50 or less, more preferably 3.00 or less, from the viewpoint of improving the dispersibility of the silica sol.
- the pH of the silica-carboxylic acid mixed solution is higher than 6.50, excessive aggregation of the silica sol proceeds, which tends to cause a decrease in acrylonitrile selectivity and wear strength.
- the preparation step in the production method of the present embodiment is a step of mixing the silica-carboxylic acid mixed solution, molybdenum, bismuth, and iron to obtain a precursor slurry (in this specification, “step (II)”). Also called).
- the order of mixing is not particularly limited.
- a precursor slurry can be obtained by charging a solution containing molybdenum into a silica-carboxylic acid mixed solution and then adding a solution containing bismuth and iron.
- the raw material of each component used for the preparation of the raw slurry is preferably a salt soluble in water or nitric acid.
- a slurry that is used to prepare a precursor slurry in the preparation step in the manufacturing method of the present embodiment and that does not contain a carboxylic acid is also referred to as a “raw material slurry”. That is, the raw material slurry in the manufacturing method of this embodiment contains at least one element selected from the group consisting of molybdenum, bismuth, and iron.
- each element of molybdenum, bismuth, and iron For example, ammonium salt, nitrate, hydrochloride, sulfate, organic acid salt, inorganic salt soluble in water or nitric acid is mentioned.
- an ammonium salt is preferable as a raw material for molybdenum.
- each nitrate is preferable as a raw material of bismuth and iron.
- nitrates are also preferred in that they do not cause residual chlorine that occurs when hydrochloride is used or residual sulfur that occurs when sulfate is used.
- Specific examples of raw materials for each component include, but are not limited to, ammonium paramolybdate, bismuth nitrate, and ferric nitrate.
- the drying step is a step in which the precursor slurry obtained in the preparation step is spray-dried to obtain dry particles.
- spherical fine particles suitable for a fluidized bed reaction can be obtained.
- the spray drying device is not particularly limited, and for example, a general device such as a rotary disk type or a nozzle type can be used.
- the particle size of the catalyst can be adjusted by adjusting the spray drying conditions. When used as a fluidized bed catalyst, the particle size of the catalyst is preferably 25 to 180 ⁇ m.
- An example of the conditions for obtaining catalyst particles having a preferred particle size is as follows.
- the inlet air temperature of the dryer is set.
- examples thereof include spray drying performed at 180 to 250 ° C. and an outlet temperature of 100 to 150 ° C.
- the step (II) preferably includes the following steps (i) and (ii), and the drying step preferably includes the following (iii) step.
- the Mo-containing liquid is continuously supplied to the first flow path, the Bi-containing liquid is continuously supplied to the second flow path, and the first flow path and the second flow path are A step of mixing the Mo-containing liquid and the Bi-containing liquid to obtain a MoBi-containing liquid by merging the Mo-containing liquid and the Bi-containing liquid downstream from the respective supply locations.
- At least one of the Mo-containing liquid and the Bi-containing liquid may include a silica-carboxylic acid mixed liquid.
- both the Mo-containing liquid and the Bi-containing liquid do not contain the silica-carboxylic acid mixed liquid, that is, when the silica-carboxylic acid mixed liquid is prepared independently of the Mo-containing liquid and the Bi-containing liquid, the silica A step of mixing the carboxylic acid mixed solution with the Mo-containing solution, Bi-containing solution, or MoBi-containing solution can be performed.
- the MoBi-containing liquid prepared in the step may be continuously supplied to the (iii) step as it is, or may be supplied to the (iii) step after the (iv) step, or a part thereof Alternatively, the entire amount may be temporarily stored (step (v)) and then supplied to the step (iii).
- step (v) When a step is provided and the MoBi-containing liquid is temporarily stored, the total amount of the MoBi liquid prepared in the step (ii) or the total amount of the MoBi-containing liquid prepared by performing the step (iv) after the step (ii) Is preferably stored (batch processing method).
- step (iii) even if a deviation occurs in the supply ratio of the Mo-containing liquid and the Bi-containing liquid in the step (ii), the whole amount of the raw material is mixed in the step (v), that is, the intended composition
- the MoBi-containing liquid that has been homogenized can be sent to step (iii).
- the continuous processing method is used when the catalyst is to be manufactured in a short time, and the batch processing is performed when the allowable range of the supply ratio is widened. It is preferable to select a method.
- Steps (i) and (ii) are steps for preparing a slurry containing a metal oxide raw material.
- a Mo-containing liquid containing at least Mo a liquid containing a Mo raw material that is a component of the metal oxide, hereinafter also referred to as “A liquid”
- Bi containing at least Bi a step of preparing a containing liquid (a liquid containing a Bi raw material serving as a component of a metal oxide, also referred to as “B liquid”), and (ii) a step of mixing both (hereinafter referred to as Mo-containing liquid) Bi-containing liquid is sometimes referred to as “two liquids”).
- a MoBi-containing liquid that is a slurry is formed.
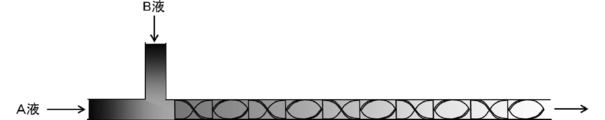
- This MoBi-containing liquid is a precursor of a MoBi-containing metal oxide, and it has been found by the present inventors that the generation conditions of this precursor greatly affect the uniformity of the surface composition of the catalyst particles to be produced. .
- the Mo-containing liquid and the Bi-containing liquid are continuously supplied to the first and second flow paths, respectively, and the first flow path and the second flow path are connected to the Mo-containing liquid and Bi.
- the atomic ratio (bulk composition) of each element in the metal oxide contained in the catalyst finally produced by this method is basically the same as the atomic ratio of the raw material used during production, excluding oxygen atoms. Absent.
- a raw material of Mo a salt soluble in water, nitric acid or the like is preferable, and for example, ammonium molybdate which is an ammonium salt can be mentioned.
- water for nickel, cobalt, magnesium, calcium, zinc, strontium, barium, cerium, chromium, lanthanum, neodymium, yttrium, praseodymium, samarium, aluminum, gallium, indium, potassium, rubidium, and cesium can be used as well.
- Salts soluble in nitric acid are preferred, and examples thereof include nitrates, hydrochlorides, sulfates, and organic acid salts.
- nitrate is preferable from the viewpoint of being easily soluble in water, nitric acid and the like.
- the carrier is not particularly limited, but for example, oxides such as silica (SiO 2 ), alumina, titania, zirconia and the like are preferable.
- oxides such as silica (SiO 2 ), alumina, titania, zirconia and the like are preferable.
- SiO 2 is particularly preferred.
- No particular limitation is imposed on the of SiO 2 material, (., Also known as colloidal silica) silica sol, powdered silica.
- Silica sol is particularly preferable because of easy handling.
- silica sol manufactured by Nalco Japan
- the average primary particle size of silica contained in the silica sol is not particularly limited, but is preferably in the range of 5 to 100 nm, and more preferably in the range of 10 to 50 nm.
- silica sols having different average primary particle diameters can be mixed and used.
- an inorganic acid such as nitric acid, hydrochloric acid, or sulfuric acid may be added for the purpose of increasing the solubility of each metal salt of the raw material metal in addition to the raw material metal described above.
- Nitric acid is particularly preferred because it volatilizes during the calcination step described below and has little residue on the catalyst.
- ammonia water is also preferable to add to the Mo-containing liquid in order to increase the solubility of Mo.
- the Mo-containing liquid is preferably prepared by dissolving or dispersing the Mo raw material in an aqueous medium such as water. It is also preferable to add aqueous ammonia to this solution to increase the solubility of the Mo raw material.
- the Mo atom concentration in the Mo-containing liquid is preferably in the range of 0.1 to 30% by mass.
- the Bi-containing liquid is preferably prepared by dissolving or dispersing the Bi raw material in an aqueous medium such as water, separately from the Mo-containing liquid.
- the Bi-containing liquid is preferably acidic, and it is more preferable that the Bi-containing liquid contains nitric acid because the solubility of the Bi raw material is increased.
- the Bi atom concentration in the Bi-containing liquid is preferably in the range of 0.1 to 10% by mass.
- Raw materials other than Mo raw material and Bi raw material that is, raw materials such as Fe, Ni, Co, alkali metal, Mg, Ca, Sr, Ba, Zn, Mn, rare earth, Cr, In, Ga, Si, Al, Ti, Zr, etc. And in the range which does not produce a precipitate, you may add to any of the said Mo containing liquid and Bi containing liquid, and may add to both. Moreover, you may add to the MoBi containing liquid after mixing the said 2 liquid. It is preferable to add the metal raw material other than Mo to the Bi-containing liquid because it is difficult to cause precipitation. It is preferable to add.
- the Mo-containing liquid and the Bi-containing liquid are continuously supplied to the first and second flow paths, respectively, and the first flow path and the second flow path are Mo-containing.
- the Mo-containing liquid and the Bi-containing liquid are mixed by being merged downstream from the supply locations of the liquid and the Bi-containing liquid.
- the supply of the Mo-containing liquid and the Bi-containing liquid to each flow path is such that the supply molar ratio of Mo and Bi is the atomic ratio of Mo and Bi in the formula (1) of the bulk composition of the metal oxide to be produced, that is, a / 12 is preferably performed so as to be approximately equal to 12.
- the supply molar ratio is preferably in the range of (a / 12) ⁇ 0.8 to (a / 12) ⁇ 1.2.
- the Mo-containing liquid and the Bi-containing liquid are supplied so as to satisfy the following formula (3).
- (M ⁇ / m ⁇ ) / (a / 12) 0.8 to 1.2 (3)
- the Bi-containing liquid with respect to the mass supply rate mA (g / min) to the first channel of the Mo-containing liquid The ratio of mass supply rate mB (g / min) (hereinafter also referred to as “supply ratio”) of the Bi-containing liquid per batch relative to the total supply amount MA (g) per batch of Mo-containing liquid It is preferable to supply so as to be 0.5 to 1.5 times the ratio of supply amount MB (g) (hereinafter also referred to as “mass ratio”).
- the Mo-containing liquid and the Bi-containing liquid are supplied so as to satisfy the following formula (2).
- (MB / mA) / (MB / MA) 0.5 to 1.5 (2)
- the uniformity of the composition of the catalyst particle surface can be further improved.
- (MB / mA) / (MB / MA) is more preferably 0.8 to 1.2, and particularly preferably 1.0.
- the supply ratio (mB / mA) and mass ratio (MB / MA) will be described below with specific examples.
- the necessary amount of raw materials is included and the total amount (MA) is 100 parts by mass
- the Bi-containing liquid for one batch prepared in step (i) contains the necessary amount of Bi raw materials (one of the number of moles of Mo in the Mo-containing liquid / 12)
- the total amount (MB) is 50 parts by mass.
- the mixing of the Mo-containing liquid and the Bi-containing liquid is performed by adding the other liquid to one liquid.
- the concentration of the component to be added is small immediately after the start of mixing, and gradually increases as mixing proceeds.
- the intended atomic ratio is achieved when all of the two liquids have been added, a significant imbalance tends to occur between the added component concentration and the added component concentration in the middle of the addition.
- the imbalance between the Bi and Mo concentrations in the mixed solution at the time of preparing the slurry reduces the uniformity of the surface composition of the catalyst manufactured from the slurry thus prepared later. It turned out to be one of the causes.
- the above-described component concentration imbalance in the mixed liquid can be sufficiently reduced.
- the catalyst composition produced by the atomic ratio of Mo and Bi in the mixed liquid is produced at any stage of the process of mixing the two.
- a slurry is formed in a state close to the atomic ratio.
- the supply ratio (mB / mA) is the mass ratio (MB / MA) as described above. 0.5 to 1.5 times is preferable. However, it is not always necessary to bring the total amount of the two liquids in the range of 0.5 to 1.5 times, and the partial supply of the Mo-containing liquid and the Bi-containing liquid may be outside the above range.
- the merging temperature when the two liquids are merged is preferably 5 ° C. to 98 ° C. 35 ° C to 98 ° C is more preferable, 35 ° C to 80 ° C is more preferable, and 35 ° C to 60 ° C is particularly preferable.
- the merging temperature refers to the liquid temperature at the portion where the two liquids merge and can be measured with a thermometer.
- step (Ii) When two liquids are mixed in step (ii) to form a MoBi-containing liquid (slurry), and then the step of further mixing the MoBi-containing liquid (step (iv)) is performed before the MoBi-containing liquid is dried
- the method of further mixing the produced MoBi-containing liquid is not limited, and examples thereof include passing through a static mixer or stirring using a stirrer.
- the stirring temperature is preferably about the same as the merging temperature at the time of merging the two liquids, specifically, 35 ° C. to 80 ° C. is preferable.
- the configuration of the apparatus used in the production of the catalyst is not limited.
- a quantitative supply device By using a fixed-quantity supply apparatus, two liquids can be supplied at a more accurate ratio.
- the metering supply device is not particularly limited, but a metered liquid pump is preferable.
- the merging portion where the two liquids merge is not particularly limited as long as the two liquids can be merged.
- a Y-shaped or T-shaped tube may be used.
- the cross-sectional shape of the tube may be, for example, a circle (cylinder), and it is also preferable to provide a mechanism or an insert for promoting mixing of the MoBi-containing liquid.
- the angle at which the two liquids merge may be cocurrent, vertical, or opposed.
- a mixer for further mixing the MoBi-containing liquid generated downstream of the junction.
- An example of the mixer is a stirring tank having a stirrer. An example of such a device configuration is shown in FIG.
- each of the above-described devices and the like has a heat retaining and warming structure such as a warm water jacket that can be set to a desired temperature.
- a static mixer as shown in FIG. 2 (mainly a pipe and a twisted plate fixed therein), the fluid supplied into the pipe Are mixed by moving along the flow).
- An example of a static mixer is a static mixer (manufactured by Noritake Company Limited). As shown in FIG. 1, you may combine a static mixer and a stirring tank.
- a catalyst containing a metal oxide having a bulk composition represented by formula (1) By drying the MoBi-containing liquid produced as described above (step (iii)), a catalyst containing a metal oxide having a bulk composition represented by formula (1) can be formed.
- the MoBi-containing liquid preferably has a pH of 3 or less before drying. More preferably, it is 2 or less, More preferably, it is 1 or less.
- the pH can be adjusted by, for example, the amount of acidic solution added to the raw material. If pH is 3 or less, the increase in the viscosity of a MoBi containing liquid can be suppressed, and the supply of the MoBi containing liquid to the next process (iii) drying process can be performed stably.
- the slurry preparation process for forming the catalyst precursor can be performed entirely in liquid form, it is extremely simple compared to a method that requires drying, firing, pulverization operations, etc. for forming the catalyst precursor. I can say that.
- the drying method there is no limitation on the drying method, but it is preferable to obtain the dried particles by spray drying the MoBi-containing liquid.
- spray drying for example, when it is carried out industrially, it is preferable to use a spray dryer.
- the spray drying step first, the MoBi-containing liquid is atomized.
- the atomization method is not particularly limited, and examples thereof include a centrifugal method, a two-fluid nozzle method, and a high-pressure nozzle method. In particular, the centrifugal method is preferable from the viewpoint of not obstructing the nozzle.
- the hot air inlet temperature is preferably 180 ° C. to 250 ° C.
- the hot air outlet temperature is preferably 100 ° C. to 150 ° C.
- the spray device it is preferable to adjust the spray device so that the spray particle size is in a desired range according to the target catalyst particle size.
- the average particle diameter of the catalyst is preferably about 25 to 180 ⁇ m, and therefore the rotation speed of the atomizer (atomizer) is appropriately selected so that the average particle diameter is within this range. .
- the higher the atomizer speed the smaller the average particle size.
- a catalyst having stable catalyst properties and catalyst performance can be rapidly and continuously produced by performing a firing step described later as a series of treatments.
- step and the step (v) are performed simultaneously (that is, the MoBi-containing liquid is stored under stirring).
- the process (v) The amount of storage in is preferably the total amount of the MoBi-containing liquid obtained in step (ii).
- the supply amount of the Mo-containing liquid and the Bo-containing liquid in the step (ii) fluctuates by temporarily storing the entire amount of the MoBi-containing liquid prepared in the step and then supplying it to the drying step ((iii) step).
- the atomic ratio of Mo and Bi in the MoBi-containing liquid supplied to the step (iii) can be made equal to the atomic ratio of both in the formula (1), and the composition of the obtained catalyst particles can be It can be closer to the purpose.
- the storage container is not limited, and for example, a storage container having a stirring function can be used, and the step (v) and the step (iv) can be performed simultaneously.
- the time required for charging the liquid to be charged is within 8 minutes, preferably within 4 minutes, thereby promoting uniform mixing of the two liquids, It can also be manufactured.
- the mass of the input liquid is increased, and due to the synergistic effect of increasing the collision energy with the liquid on the input side and processing in a short time , More uniform slurry mixing tends to be achieved.
- the firing step is a step of firing the dried particles obtained in the drying step.
- the dry particles contain nitric acid
- the denitration treatment is preferably performed at 150 to 450 ° C. for 1.5 to 3 hours. Firing can be performed in an air atmosphere.
- the firing temperature in the firing step is preferably 550 to 650 ° C.
- the calcination temperature is 550 ° C. or higher, crystal growth proceeds sufficiently and the selectivity of the resulting catalyst acrylonitrile tends to be further improved.
- the calcination temperature is 650 ° C. or less, the surface area of the catalyst that can be obtained increases, and the reaction activity of propylene tends to be further improved.
- the firing time is preferably 1 hr to 24 hr, and more preferably 2 hr to 20 hr. Conditions such as calcination temperature and calcination time are appropriately selected so that desired catalyst physical properties and reaction performance can be obtained.
- the catalyst obtained by the production method of the present embodiment is suitable as a catalyst (ammoxidation catalyst) used in the ammoxidation reaction in the production of acrylonitrile described later.
- the catalyst obtained by the manufacturing method of this embodiment mentioned above is not limited to the following, Typically, it has the following characteristics. That is, the catalyst according to the present embodiment is a catalyst including a metal oxide having a bulk composition containing Mo and Bi represented by the following general formula (1) and particles containing silica. Mo 12 Bi a Fe b X c Y d Z e O f (1) (In formula (1), X is one or more elements selected from the group consisting of nickel, cobalt, magnesium, calcium, zinc, strontium and barium, Y is cerium, chromium, lanthanum, neodymium, yttrium, praseodymium, samarium.
- One or more elements selected from the group consisting of aluminum, gallium and indium, Z represents one or more elements selected from the group consisting of potassium, rubidium and cesium, and a, b, c, d and e are: 0.1 ⁇ a ⁇ 2.0, 0.1 ⁇ b ⁇ 3.0, 0.1 ⁇ c ⁇ 10.0, 0.1 ⁇ d ⁇ 3.0, and 0.01 ⁇ e ⁇ 2, respectively. . Is satisfied, and f is the number of oxygen atoms necessary to satisfy the valence requirements of the other elements present.)
- the catalyst of the present embodiment exhibits high acrylonitrile selectivity in the ammoxidation reaction, and has good wear strength that can withstand long-term use as a fluidized bed catalyst. .
- the present inventors consider this as follows. Although the active site in the ammoxidation reaction is molybdate containing bismuth, high acrylonitrile selectivity cannot be obtained by itself. It is considered that the molybdate containing bismuth is uniformly combined with the molybdate containing iron and the molybdate containing other metals at least on the surface of the catalyst particles, so that the acrylonitrile selectivity is improved.
- catalyst particles in which bismuth-containing molybdate and iron-containing molybdate and other metal-containing molybdate are uniformly combined on the surface of the catalyst particles agglomerate each metal component as a single component at the precursor slurry stage.
- the molybdate containing bismuth is considered to be required to be easily combined with the molybdate containing other metals.
- metal particles are supported on a carrier, it is considered that the wear strength of the catalyst is also improved by preventing aggregation of both the metal in the precursor slurry and the particles of the carrier.
- the mechanism of action is not limited to the above.
- the metal oxide constituting the catalyst contains molybdenum, bismuth, and iron as essential components.
- Molybdenum plays a role as an adsorption site for propylene and an activation site for ammonia.
- Bismuth also plays a role in activating propylene and extracting ⁇ -position hydrogen to generate ⁇ -allyl species.
- iron plays a role of supplying oxygen present in the gas phase to the catalytic active site by trivalent / divalent redox.
- metal oxides include one or more elements X selected from the group consisting of nickel, cobalt, magnesium, calcium, zinc, strontium and barium, cerium, chromium, lanthanum, Examples thereof include one or more elements Y selected from the group consisting of neodymium, yttrium, praseodymium, samarium, aluminum, gallium and indium, and one or more elements Z selected from the group consisting of potassium, rubidium and cesium.
- the element X forms a molybdate having moderate lattice defects, and plays a role in facilitating oxygen movement in the bulk.
- the element Y is responsible for the redox function in the catalyst, like iron.
- the element Z plays the role which suppresses the decomposition reaction of a main product and a raw material by blocking the acid site which exists on the catalyst particle surface.
- each element has a role in functioning as a catalyst, such as an element that can replace the function of the element, an element that cannot be replaced, an element that provides an auxiliary action of the function, etc.
- the bulk composition of the metal oxide constituting the catalyst of the present embodiment that is, the ratio of each element constituting the entire metal oxide is represented by the following general formula (1).
- X is one or more elements selected from the group consisting of nickel, cobalt, magnesium, calcium, zinc, strontium and barium
- Y is cerium, chromium, lanthanum, neodymium, yttrium, praseodymium, samarium.
- Z represents one or more elements selected from the group consisting of potassium, rubidium and cesium;
- a represents the atomic ratio of bismuth to 12 atoms of molybdenum, 0.1 ⁇ a ⁇ 2.0, preferably 0.15 ⁇ a ⁇ 1.0, and more preferably 0.2 ⁇ a ⁇ 0. .7
- b represents the atomic ratio of iron to 12 atoms of molybdenum, 0.1 ⁇ b ⁇ 3.0, preferably 0.5 ⁇ b ⁇ 2.5, more preferably 1.0 ⁇ b ⁇ 2.
- c represents the atomic ratio of X with respect to 12 atoms of molybdenum, 0.1 ⁇ c ⁇ 10.0, preferably 3.0 ⁇ c ⁇ 9.0, more preferably 5.0 ⁇ c ⁇ 8. .5, d represents the atomic ratio of Y to 12 atoms of molybdenum, 0.1 ⁇ d ⁇ 3.0, preferably 0.2 ⁇ d ⁇ 2.0, more preferably 0.3 ⁇ d ⁇ 1.
- e represents an atomic ratio of Z to 12 atoms of molybdenum, 0.01 ⁇ e ⁇ 2.0, preferably 0.05 ⁇ e ⁇ 1.0
- f represents the atomic ratio of oxygen to 12 atoms of molybdenum, and is the number of oxygen atoms necessary to satisfy the valence requirements of other elements present.
- g and h when the atomic ratio of nickel is g and the atomic ratio of cobalt is h, it is preferable to adjust g and h as follows. That is, when the metal oxide does not contain magnesium, it is preferable that g ⁇ 1.0 or 6.5 ⁇ g and h ⁇ 1.0 or 6.5 ⁇ h. When the metal oxide contains magnesium, it is preferable that g ⁇ 0.5 or 6.5 ⁇ g and h ⁇ 0.5 or 7.0 ⁇ h.
- the bulk composition (ratio of each element) of the metal oxide is determined based on the raw material charge ratio at the time of synthesis of the catalyst or a well-known elemental quantitative analysis method such as ICP emission spectroscopy, atomic absorption spectrometry, ICP mass spectrometry, It can be determined by fluorescent X-ray analysis or the like.
- the ICP emission spectroscopic analysis method is a method adopted in the examples, and a specific procedure thereof will be described in the examples.
- the catalyst sample can be quantitatively analyzed.
- a fluorescent X-ray apparatus for example, ZSX100e (tube: Rh-4KW, spectral crystal: LiF, PET, Ge, RX25) manufactured by Rigaku
- the value obtained by such an analysis substantially corresponds with the preparation composition on a design.
- the bulk composition can be calculated
- a fluidized bed reaction is generally selected in which a catalyst is fluidized by a reaction gas. Therefore, when using the catalyst of this embodiment as an ammoxidation catalyst, it is preferable that the catalyst has a certain strength or more. From such a viewpoint, in the catalyst of the present embodiment, the metal oxide is supported on a support containing silica.
- silica as a carrier, not only can the selectivity decrease of acrylonitrile be suppressed, but also the wear resistance and particle strength of the catalyst can be greatly improved.
- carrier components other than a silica For example, oxides, such as an alumina, a titania, a zirconia, can be included.
- the content of the carrier is preferably 30 to 70% by mass, more preferably 35 to 65% by mass with respect to 100% by mass of the catalyst.
- the carrier content is 30% by mass or more, the abrasion resistance and particle strength of the ammoxidation reaction catalyst tend to be further improved.
- the carrier content is 70% by mass or less, the acrylonitrile selectivity tends to be further improved.
- the shape of the catalyst particles is not limited, but is preferably spherical.
- the average particle size of the catalyst particles is preferably in the range of 40 to 70 ⁇ m, more preferably in the range of 45 to 65 ⁇ m.
- the amount of catalyst particles having a particle diameter of 5 to 200 ⁇ m is preferably 90 to 100% by mass with respect to the total mass of the catalyst.
- the amount of particles having a particle diameter of 45 ⁇ m or less is preferably 10 to 50% by volume, more preferably 15 to 35% by volume, based on the total volume of the particles.
- the catalyst of this embodiment is excellent in wear resistance strength so that it can be used as a fluidized bed catalyst.
- it has wear resistance that prevents the catalyst particles from being worn or crushed by impacts such as contact between the catalyst particles in the reactor, collision with the reactor wall, contact with gas, etc.
- the catalyst of the present embodiment is a fluidized bed catalyst wear test ("Test Method for Synthetic Fluid Cracking Catalyst" (American Cyanamid Co. Ltd. 6 / 31-4m-1 / 57)).
- the wear loss in the test according to the method described in 1) is preferably 2.5% or less.
- the apparent specific gravity of the catalyst particles of the present embodiment is preferably in the range of 0.85 to 1.15 g / cc.
- the apparent specific gravity of the catalyst particles is 0.85 g / cc or more, the influence of the bulkiness of the catalyst particles when charged into the reactor can be reduced, and the volume of the required reactor tends to be reduced. There is a tendency that it is possible to effectively prevent the occurrence of catalyst loss due to an increase in the amount of catalyst scattered from the reactor to the outside.
- the apparent specific gravity of the catalyst particles is 1.15 g / cc or less, a good flow state of the catalyst particles can be secured, and the reaction performance tends to be effectively prevented from being lowered.
- the apparent specific gravity of the catalyst particles is calculated by calculating the mass per unit volume by placing the sample (catalyst particles) in a constant volume container, measuring the mass of the sample in the container, and calculating the mass per unit volume.
- a 25 ml glass graduated cylinder hereinafter simply referred to as “graduated cylinder” is placed directly under the glass funnel, and the measurement sample (catalyst particles) shaken in advance is dropped onto the glass funnel until the graduated cylinder overflows. .
- the mass of the sample is obtained from the mass of the graduated cylinder containing the sample and the mass of the graduated cylinder measured in advance, and the apparent specific gravity is calculated by dividing it by the volume of the graduated cylinder.
- the ratio (Bi / Mo) surf ) of the Bi molar concentration to the Mo molar concentration on the catalyst particle surface of the catalyst containing the metal oxide having the bulk composition represented by the formula (1) is expressed as metal oxidation.
- the standard deviation of the value divided by the ratio of Bi molar concentration to Mo molar concentration of the product bulk ((Bi / Mo) bulk ) is 0.2 or less.
- the Mo molar concentration and Bi molar concentration on the particle surface can be determined by SEM-EDX (Scanning Electron Microscopes-Energy Dispersive X-ray Spectroscopy) composition analysis.
- the Mo molar concentration and Bi molar concentration of the metal oxide bulk can be obtained by, for example, ICP emission spectroscopy.
- the said standard deviation can be calculated
- Standard deviation ⁇ ((S 1 ⁇ ) 2 + (S 2 ⁇ ) 2 +... + (S 100 ⁇ ) 2 ) / 100 [number] ⁇ 1/2 (4)
- Standard deviation is an indicator of variation. The smaller the standard deviation, the smaller the variation from the average value. Therefore, it means that the smaller the value of the formula (4), the more uniform the surface composition of the particles.
- the value of the formula (4) is 0.2 or less, preferably 0.15 or less, more preferably 0.10 or less, and further preferably 0.07 or less. preferable.
- the method for producing acrylonitrile of the present embodiment is a method for producing acrylonitrile by reacting propylene, molecular oxygen, and ammonia in the presence of the obtained catalyst and the step of obtaining the catalyst by the above-described catalyst production method. Reaction step. Moreover, it can be said that the manufacturing method of acrylonitrile of this embodiment includes the process of making a propylene, molecular oxygen, and ammonia react in presence of the catalyst of this embodiment.
- the production of acrylonitrile by an ammoxidation reaction can be carried out in a fixed bed reactor or a fluidized bed reactor.
- a fluidized bed reactor is preferable from the viewpoint of efficiently removing heat generated during the reaction and increasing the yield of acrylonitrile.
- Propylene and ammonia which are raw materials for the ammoxidation reaction, do not necessarily have high purity, and industrial grade ones can be used.
- the molar ratio of propylene, ammonia and oxygen (propylene / ammonia / oxygen) in the raw material gas is preferably 1.0 / 1.0 to 1.5 / 1.6 to 2.2.
- the gas containing molecular oxygen that can be used in the method for producing acrylonitrile of the present embodiment is not particularly limited.
- air oxygen-enriched air, pure oxygen; helium, argon, carbon dioxide, nitrogen Or an inert gas such as gas diluted with water vapor.
- air when used on an industrial scale, it is preferable to use air for simplicity.
- the reaction temperature is preferably 380 to 480 ° C.
- the reaction pressure is preferably normal pressure to 0.3 MPa.
- the contact time between the raw material gas and the catalyst is preferably 2 to 7 seconds, and more preferably 3 to 6 seconds.
- the present embodiment will be described in more detail with reference to examples.
- the present embodiment is not limited to the examples described below.
- the catalyst composition described in the Example and the comparative example is the same value as the preparation composition of each element.
- the Examples and Comparative Examples describe values and procedures for obtaining 1 kg of catalyst after calcination.
- Example 1 Bulk composition (hereinafter, simply referred to as "composition”.) Is Mo 12.00 Bi 0.50 Fe 1.31 Co 4.05 Ni 3.10 Ce 0.87 Rb 0.10 K 0.08 O f ( The following composition is described by omitting “O f ”.) A catalyst in which the metal oxide represented by 40% by mass of silica was supported by the following procedure was produced.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- Example 2 A catalyst was produced in the same manner as in Example 1 except that the stirring time of the silica-oxalic acid mixture was changed to 30 minutes.
- Example 3 A catalyst was produced in the same manner as in Example 1 except that the stirring time of the silica-oxalic acid mixture was changed to 80 minutes.
- Example 4 Oxalic acid addition rate 8 g / sec. A catalyst was produced in the same manner as in Example 1 except that (per 1 kg of SiO 2 ) was used.
- Example 5 Oxalic acid addition rate 3 g / sec. A catalyst was produced in the same manner as in Example 1 except that (per 1 kg of SiO 2 ) was used.
- Example 6 A catalyst in which a metal oxide represented by a composition represented by Mo 12.00 Bi 0.39 Fe 1.60 Ni 6.97 Mg 0.77 Ce 0.63 Rb 0.17 is supported on 40% by mass of silica is described below. It was manufactured according to the procedure.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 580 ° C. for 2 hours to obtain a catalyst.
- Example 7 A catalyst was produced in the same manner as in Example 6 except that the stirring time of the silica-oxalic acid mixture was 80 minutes.
- Example 8 Oxalic acid addition rate 3 g / sec. A catalyst was produced in the same manner as in Example 6 except that it was changed to (per 1 kg of SiO 2 ).
- Example 9 A catalyst in which a metal oxide represented by Mo 12.00 Bi 1.20 Fe 0.60 Ni 7.80 Cr 1.20 K 0.48 is supported on 60% by mass of silica is produced by the following procedure. did.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- Example 10 A catalyst in which a metal oxide represented by Mo 12.00 Bi 2.40 Fe 2.40 Ni 6.00 Zn 1.80 Ce 0.24 K 0.24 is supported on 60% by mass of silica is described below. It was manufactured according to the procedure.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 570 ° C. for 2 hours to obtain a catalyst.
- Example 11 A catalyst in which a metal oxide represented by Mo 12.00 Bi 0.47 Fe 1.35 Co 4.15 Ni 3.00 Ce 0.91 Rb 0.14 is supported on 40% by mass of silica is described below. It was manufactured according to the procedure.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- Example 12 A metal oxide represented by the composition Mo 12.00 Bi 0.57 Fe 1.01 Co 6.83 Ni 0.98 Mg 0.98 Ce 0.38 Rb 0.12 was supported on 40% by mass of silica.
- the catalyst was prepared by the following procedure.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- Example 13 The silica sol has a pH of 9.00, oxalic acid dihydrate is 25.0 g (0.038 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 1.65.
- a catalyst was produced in the same manner as in Example 12 except that.
- Example 14 The silica sol has a pH of 9.70, oxalic acid dihydrate is 25.0 g (0.038 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 5.30.
- a catalyst was produced in the same manner as in Example 12 except that.
- Example 15 Silica sol has a pH of 10.13, oxalic acid dihydrate is 25.0 g (0.038 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 8.00.
- a catalyst was produced in the same manner as in Example 12 except that.
- Example 16 A catalyst in which a metal oxide represented by a composition Mo 12.00 Bi 0.57 Fe 1.01 Co 2.24 Ni 6.54 Ce 0.38 Rb 0.12 is supported on 40% by mass of silica is described below. It was manufactured according to the procedure.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 600 ° C. for 2 hours to obtain a catalyst.
- Example 17 The silica sol has a pH of 9.00, oxalic acid dihydrate is 25.0 g (0.039 molar equivalents of the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 1.65.
- a catalyst was produced in the same manner as in Example 16 except that.
- Example 18 The silica sol has a pH of 9.70, oxalic acid dihydrate is 25.0 g (0.039 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 5.30.
- a catalyst was produced in the same manner as in Example 16 except that.
- Example 19 The silica sol has a pH of 10.13, oxalic acid dihydrate is 25.0 g (0.039 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 8.00.
- a catalyst was produced in the same manner as in Example 16 except that.
- Example 20 A catalyst in which a metal oxide represented by Mo 12.00 Bi 0.84 Fe 2.06 Co 6.67 Ce 0.56 Rb 0.12 is supported on 40% by mass of silica is produced by the following procedure. did.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 580 ° C. for 2 hours to obtain a catalyst.
- Example 21 The silica sol has a pH of 9.00, oxalic acid dihydrate is 25.0 g (0.040 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 1.65.
- a catalyst was produced in the same manner as in Example 20 except that.
- Example 22 The silica sol has a pH of 9.70, oxalic acid dihydrate is 25.0 g (0.040 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 5.30.
- a catalyst was produced in the same manner as in Example 20 except that.
- Example 23 The silica sol has a pH of 10.13, oxalic acid dihydrate is 25.0 g (0.040 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 8.00.
- a catalyst was produced in the same manner as in Example 20 except that.
- Example 24 A catalyst in which a metal oxide represented by the composition Mo 12.00 Bi 0.27 Fe 0.95 Co 6.69 Ni 2.95 Ce 0.18 Rb 0.13 is supported on 40% by mass of silica Produced according to the procedure.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 595 ° C. for 2 hours to obtain a catalyst.
- Example 25 The silica sol has a pH of 9.00, oxalic acid dihydrate is 25.0 g (0.038 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 1.65.
- a catalyst was produced in the same manner as in Example 24 except that.
- Example 26 The silica sol has a pH of 9.70, oxalic acid dihydrate is 25.0 g (0.038 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 5.30.
- a catalyst was produced in the same manner as in Example 24 except that.
- Example 27 Silica sol has a pH of 10.13, oxalic acid dihydrate is 25.0 g (0.038 molar equivalent to the total amount of metal elements in the precursor slurry), and the silica-oxalic acid mixture has a pH of 8.00.
- a catalyst was produced in the same manner as in Example 24 except that.
- Example 28 A catalyst in which a metal oxide represented by a composition Mo 12.00 Bi 0.82 Fe 1.45 Co 8.14 Ce 0.55 Rb 0.13 is supported on 40% by mass of silica is produced by the following procedure. did.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 585 ° C. for 2 hours to obtain a catalyst.
- Example 29 A catalyst in which the metal oxide represented by the composition Mo 12.00 Bi 1.05 Fe 1.40 Co 8.15 Ce 0.70 Rb 0.13 is supported on 40% by mass of silica is produced by the following procedure. did.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 585 ° C. for 2 hours to obtain a catalyst.
- Example 30 A catalyst in which a metal oxide having a composition represented by Mo 12.00 Bi 0.27 Fe 0.95 Co 8.16 Ni 1.48 Ce 0.18 Rb 0.13 is supported on 40% by mass of silica is described below. It was manufactured according to the procedure.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 585 ° C. for 2 hours to obtain a catalyst.
- Example 31 A catalyst having a composition of Mo 12.00 Bi 0.27 Fe 0.95 Co 7.67 Ni 1.97 Ce 0.18 Rb 0.13 supported on 40% by mass of silica, It was manufactured according to the procedure.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- a metal oxide having a composition of Mo 12.00 Bi 0.50 Fe 1.31 Co 4.05 Ni 3.10 Ce 0.87 Rb 0.10 K 0.08 was supported on 40% by mass of silica.
- the catalyst was prepared by the following procedure. To 1333 g of silica sol containing 30% by mass of SiO 2 , 481.6 g of ammonium paramolybdate [(NH 4 ) 6 Mo 7 O 24 ⁇ 4H 2 O] dissolved in 865.8 g of water was added under stirring.
- oxalic acid dihydrate purity 99.5%
- water 25.0 g of oxalic acid dihydrate (purity 99.5%) dissolved in 200 g of water was added (0.040 molar equivalent to the total amount of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer. At this time, the air temperature at the inlet of the dryer was 230 ° C., and the air temperature at the outlet was 110 ° C. The number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- Example 2 A catalyst was produced in the same manner as in Example 1 except that oxalic acid dihydrate was not added.
- oxalic acid dihydrate purity 99.5%
- 200 g of water was added (0.038 molar equivalent to the total of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 580 ° C. for 2 hours to obtain a catalyst.
- oxalic acid dihydrate purity 99.5%
- 200 g of water was added (0.059 molar equivalent to the total of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- a catalyst in which a metal oxide represented by Mo 12.00 Bi 2.40 Fe 2.40 Ni 6.00 Zn 1.80 Ce 0.24 K 0.24 is supported on 60% by mass of silica is described below. It was manufactured according to the procedure. To 2000 g of silica sol containing 30% by mass of SiO 2 , 271.3 g of ammonium paramolybdate [(NH 4 ) 6 Mo 7 O 24 ⁇ 4H 2 O] dissolved in 487.7 g of water was added under stirring.
- oxalic acid dihydrate purity 99.5%
- 200 g of water was added (0.061 molar equivalent to the total of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 570 ° C. for 2 hours to obtain a catalyst.
- oxalic acid dihydrate purity 99.5%
- water 25.0 g of oxalic acid dihydrate (purity 99.5%) dissolved in 200 g of water was added (0.040 molar equivalent to the total amount of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer. At this time, the air temperature at the inlet of the dryer was 230 ° C., and the air temperature at the outlet was 110 ° C. The number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dry particles were held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- oxalic acid dihydrate purity 99.5%
- water 25.0 g of oxalic acid dihydrate (purity 99.5%) dissolved in 200 g of water was added (0.039 molar equivalent to the total amount of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer. At this time, the air temperature at the inlet of the dryer was 230 ° C., and the air temperature at the outlet was 110 ° C. The number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 600 ° C. for 2 hours to obtain a catalyst.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 580 ° C. for 2 hours to obtain a catalyst.
- oxalic acid dihydrate purity 99.5%
- 200 g of water was added (0.038 molar equivalent to the total of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer. At this time, the air temperature at the inlet of the dryer was 230 ° C., and the air temperature at the outlet was 110 ° C. The number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 595 ° C. for 2 hours to obtain a catalyst.
- oxalic acid dihydrate purity 99.5%
- water 25.0 g of oxalic acid dihydrate (purity 99.5%) dissolved in 200 g of water was added (0.039 molar equivalent to the total amount of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the precursor slurry was dried using a rotary disk type spray dryer.
- the air temperature at the inlet of the dryer was 230 ° C.
- the air temperature at the outlet was 110 ° C.
- the number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 585 ° C. for 2 hours to obtain a catalyst
- oxalic acid dihydrate purity 99.5%
- water 25.0 g of oxalic acid dihydrate (purity 99.5%) dissolved in 200 g of water was added (0.038 molar equivalent to the total of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer. At this time, the air temperature at the inlet of the dryer was 230 ° C., and the air temperature at the outlet was 110 ° C. The number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 585 ° C. for 2 hours to obtain a catalyst.
- oxalic acid dihydrate purity 99.5%
- water 25.0 g of oxalic acid dihydrate (purity 99.5%) dissolved in 200 g of water was added (0.038 molar equivalent to the total of metal elements in the precursor slurry) to prepare a precursor slurry.
- the precursor slurry was dried using a rotary disk type spray dryer. At this time, the air temperature at the inlet of the dryer was 230 ° C., and the air temperature at the outlet was 110 ° C. The number of revolutions of the disk was set to 12,500 revolutions / minute.
- the obtained dried product was held at 200 ° C. for 5 minutes, heated from 200 ° C. to 450 ° C. at 2.5 ° C./min, and denitrated by holding at 450 ° C. for 20 minutes.
- the obtained denitration powder was calcined at 590 ° C. for 2 hours to obtain a catalyst.
- Table 1 shows the production conditions and catalyst compositions of the catalysts obtained in Examples 1-31 and Comparative Examples 1-19.
- reaction tube used for the ammoxidation reaction of propylene a Pyrex (registered trademark) glass tube having an inner diameter of 25 mm and containing 16 10-mesh wire meshes at 1 cm intervals was used.
- the molar ratio of ammonia / propylene and the molar ratio of oxygen / propylene in each example and comparative example were as shown in Table 2.
- the volume ratio of ammonia to propylene was set so that the sulfuric acid basic unit defined by the following formula was 20 ⁇ 2 kg / T-AN.
- the volume ratio of oxygen to propylene was set so that the oxygen concentration in the reactor outlet gas was 0.2 ⁇ 0.02% by volume.
- the contact time defined by the following formula was changed by changing the flow rate of the mixed gas.
- the propylene conversion rate defined by the following formula was set to 99.3 ⁇ 0.2%.
- the yield of acrylonitrile produced by the reaction was defined as the following formula.
- Abrasion resistance strength measurement In accordance with the method described in “Test Method for Synthetic Fluid Cracking Catalyst” (American Cyanamid Co. Ltd. 6 / 31-4m ⁇ 1 / 57) (hereinafter referred to as “ACC method”), the resistance of the catalyst as anti-wear loss. Abrasion strength (attrition strength) was measured. The attrition strength was evaluated by wear loss, and this wear loss was a value defined as follows.
- Wear loss (%) R / (SQ) ⁇ 100
- Q is the mass (g) of the catalyst that is worn and scattered outside during 0 to 5 hours
- R is the mass (g) of the catalyst that is usually worn and scattered outside during 5 to 20 hours
- S is the test It was set as the mass (g) of the catalyst used for.
- Table 2 shows the reaction conditions, reaction results, and attrition strengths of the catalysts obtained in Examples and Comparative Examples.
- AN yield indicates the acrylonitrile yield
- ATT intensity indicates the attrition intensity
- ⁇ indicates that it is not measured.
- the reaction time was 20 hours.
- the wear loss (%) of the catalyst used for the ammoxidation reaction in the fluidized bed is preferably 0 to 2.5%, more preferably 0 to 1.5%.
- Example 32 [Production of catalyst 1] The metal oxide 60 wt% of the raw material charged mass was prepared by adjusting so that the composition is Mo 12 Bi 0.25 Fe 1.4 Ni 3.0 Co 5.8 Ce 0.40 Rb 0.12 O f A catalyst supported on 40% by mass of silica was produced by the following procedure. 666.7 g of an aqueous silica sol containing 30% by mass of SiO 2 having an average primary particle diameter of 12 nm and 500 g of an aqueous silica sol containing 40% by mass of SiO 2 having an average primary particle diameter of 41 nm are mixed into two types. A mixed liquid of silica was obtained.
- a container containing the A1 liquid is connected to one inlet 1 of an empty Y-shaped stainless steel cylindrical tube (inner diameter: 15 mm, length: 30 mm) (merging portion) via the A1 liquid fixed amount feeding pump (first Connected by a channel).
- the container containing the B1 liquid was connected to the other inlet 2 of the Y-tube by a pipe (second flow path) via the B1 liquid fixed amount feeding pump.
- the outlet of the Y-shaped tube was connected to a static mixer having a screw type interior (static mixer), and the outlet of the static mixer was connected to a slurry agitation tank having a stirrer by piping.
- a schematic diagram of the apparatus is shown in FIG.
- the A1 liquid held at 40 ° C. was flowed at a flow rate of 284 (g / min) using an A1 liquid metering pump, and the B1 liquid held at 40 ° C. was 119 (g / min) using a B1 liquid metering pump.
- the two liquids were brought into contact in the parallel direction (cocurrent flow) in the Y-tube and the static mixer. This operation was performed for 10 minutes to prepare a MoBi-containing liquid as a slurry.
- the merging temperature during that time was 40 ° C.
- the entire amount of this MoBi-containing liquid was stored in a slurry agitation tank provided downstream of the static mixer, and stirring was further continued at 40 ° C. for 1 hour after the end of the supply of the two liquids.
- This MoBi-containing liquid had a pH of 1 or less and was in a slurry state before spray drying.
- this MoBi-containing liquid was spray-dried using a spray dryer (Okawara Chemical Industries, model: OC-16) to obtain a dry powder.
- the hot air inlet temperature was 230 ° C. and the hot air outlet temperature was 120 ° C.
- the dried catalyst precursor was pre-baked at 320 ° C. for 2 hours in an air atmosphere using an electric furnace, and then subjected to main baking at 600 ° C. for 2 hours in an air atmosphere to obtain a catalyst. (Catalyst 1).
- the shape of the obtained catalyst 1 was a solid sphere, and the average particle size was 54 ⁇ m.
- the average particle diameter was measured using a laser diffraction / scattering particle size distribution measuring apparatus LA-300 manufactured by Horiba.
- the average particle size of the catalyst in the following examples and reference examples was also measured in the same manner, and was 52 ⁇ m to 55 ⁇ m.
- the Mo concentration and Bi concentration on the surface of 100 particles of the catalyst 1 were measured by SEM-EDX, and the ratio of Bi molar concentration to Bi molar concentration (Bi / Mo) surf was obtained.
- A2 liquid The total amount of A2 liquid was 2917g.
- B2 liquid 16.6% by weight nitric acid 396.7G, bismuth nitrate 43.1g [Bi (NO 3) 3 ⁇ 5H 2 O], iron nitrate 148.0g [Fe (NO 3) 3 ⁇ 9H 2 O], 464.7 g of nickel nitrate [Ni (NO 3 ) 2 .6H 2 O], 45.5 g of magnesium nitrate [Mg (NO 3 ) 2 .6H 2 O], 62.6 g of cerium nitrate [Ce ( NO 3) 3 ⁇ 6H 2 O ], was dissolved rubidium nitrate of 5.89g [RbNO 3].
- B2 liquid The total amount of the B2 liquid was 1166 g.
- Example 34 Its composition is a Mo 12 Bi 0.39 Fe 1.60 Co 4.30 Ni 3.45 Ce 0.68 Rb 0.16 O f and so as to adjust the raw material charged mass metal oxide prepared 58 wt% A catalyst supported on 42% by mass of silica was produced by the following procedure.
- A3 liquid The total amount of A3 liquid was 2927g.
- B3 liquid 16.6 wt% of 36.1g of bismuth nitrate in nitric acid 394.5g [Bi (NO 3) 3 ⁇ 5H 2 O], iron nitrate [Fe (NO 3) of 141.6g 3 ⁇ 9H 2 O ] 275.1 g of cobalt nitrate [Co (NO 3 ) 2 .6H 2 O], 219.9 g of nickel nitrate [Ni (NO 3 ) 2 .6H 2 O], 64.6 g of cerium nitrate [Ce (NO 3) 3 ⁇ 6H 2 O] , was dissolved rubidium nitrate of 5.33g [RbNO 3].
- B3 liquid The total amount of B3 liquid was 1137g.
- A3 liquid and B3 liquid were made to merge by this supply ratio.
- the merging temperature during that time was 40 ° C.
- the entire amount of this MoBi-containing liquid was stored in a slurry agitation tank provided downstream of the static mixer, and stirring was further continued at 40 ° C. for 1 hour after the end of the supply of the two liquids.
- This MoBi-containing liquid had a pH of 1 or less and was in a slurry state before spray drying. Subsequently, this MoBi-containing liquid was spray-dried in the same manner as in Example 32 to obtain a dry powder.
- the hot air inlet temperature was 230 ° C. and the hot air outlet temperature was 110 ° C.
- the dried catalyst precursor is held at 200 ° C.
- Example 35 [Production of catalyst 4]
- the Mo-containing liquid (the same as the A1 liquid. However, hereinafter referred to as “A4 liquid”) and the Bi-containing liquid (the same as the B1 liquid. However, hereinafter “B4 liquid”.
- the total supply amount of the A4 liquid and the B4 liquid was the same as in Example 32. Therefore, the mass ratio in this case was 0.42 as in the Example 32.
- a catalyst was obtained in the same manner as in Example 32 except that the supply ratio of the B4 liquid to the A4 liquid was varied between 0.8 and 1.2 times the mass ratio 0.42 (Catalyst 4). ). All of A4 liquid and B4 liquid (100 mass%) were made to merge by the supply ratio of this range.
- the A4 liquid was simultaneously supplied to the Y-tube through the first flow path at a flow rate of 302 (g / min) and the B4 liquid was simultaneously supplied through the second flow path at a flow rate of 102 (g / min) for 3 minutes.
- the supply flow rates of the A4 solution and the B4 solution were changed, and the A4 solution was supplied simultaneously at a flow rate of 284 (g / min) and the B4 solution at a flow rate of 120 (g / min) for 4 minutes.
- Example 36 [Production of catalyst 5]
- the Mo-containing liquid (the same as the A1 liquid. However, hereinafter referred to as “A5 liquid”) and the Bi-containing liquid (the same as the B1 liquid. However, hereinafter, “B5 liquid”. Prepared).
- the total supply amount of A5 liquid and B5 liquid was the same as in Example 32. Therefore, the mass ratio in this case was 0.42 as in Example 32.
- a catalyst was obtained in the same manner as in Example 32 except that the supply ratio of the B5 liquid to the A5 liquid was varied between 0.5 times and 1.5 times the mass ratio 0.42 (catalyst 5). ). All of A5 liquid and B5 liquid (100 mass%) were made to contact with the supply ratio of this range.
- A5 liquid was supplied to the Y-shaped tube at 334 (g / min) and B5 liquid was simultaneously supplied at 70 (g / min) for 3 minutes.
- the supply flow rates of the A5 liquid and the B5 liquid were changed, and the A5 liquid was supplied simultaneously at 284 (g / min) and the B5 liquid at 120 (g / min) for 5 minutes.
- the supply flow rates of the A5 liquid and the B5 liquid were changed, and the A5 liquid was supplied simultaneously at 248 (g / min) and the B5 liquid at 156 (g / min) for 2 minutes.
- the merging temperature of the A5 liquid and the B5 liquid was 40 ° C.
- the generated MoBi-containing liquid had a pH of 1 or less and was liquid before spray drying.
- Table 4 shows the standard deviation value of the catalyst 5 and the results of the ammoxidation reaction obtained in the same manner as in Example 32.
- Example 37 [Production of catalyst 6]
- the catalyst was prepared in the same manner as in Example 32 except that the Mo-containing liquid and the Bi-containing liquid were mixed with the Y-shaped tube and the static mixer and then directly stored in the slurry agitating tank and directly fed to the spray dryer. Obtained (catalyst 6).
- the joining temperature was 40 ° C.
- the generated MoBi-containing liquid had a pH of 1 or less and was in a slurry state before spray drying.
- Table 4 shows the standard deviation value of the catalyst 6 and the results of the ammoxidation reaction obtained in the same manner as in Example 32.
- Example 38 [Production of catalyst 7]
- the Mo-containing liquid (the same as the A1 liquid. However, hereinafter referred to as “A7 liquid”) and the Bi-containing liquid (the same as the B1 liquid. However, hereinafter “B7 liquid”.
- the mass ratio in this case was 0.42 as in Example 32.
- the supply ratio of the B7 solution to the A7 solution is 0.5 to 1.5 times the mass ratio of 0.36. It went so that it might be in the range. The remaining 40% by mass was performed outside the range of 0.5 to 1.5 times. Otherwise in the same manner as in Example 32, a catalyst was obtained (Catalyst 7).
- the supply flow rates of the A7 solution and the B7 solution were changed, and the A7 solution was supplied simultaneously at a flow rate of 356 (g / min) and the B7 solution at a flow rate of 45 (g / min) for 2 minutes.
- the supply flow rates of the A7 solution and the B7 solution were changed, and the A7 solution was supplied simultaneously at a flow rate of 248 (g / min) and the B7 solution at a flow rate of 156 (g / min) for 2 minutes.
- the merging temperature of the A7 liquid and the B7 liquid was 40 ° C.
- Example 32 Thereafter, a catalyst was obtained in the same manner as in Example 32 (Catalyst 7).
- This MoBi-containing liquid had a pH of 1 or less and was in a slurry state before spray drying.
- Table 4 shows the standard deviation value of the catalyst 7 and the results of the ammoxidation reaction obtained in the same manner as in Example 32.
- the produced MoBi-containing liquid was further stirred at 40 ° C. for 1 hour.
- This MoBi-containing liquid had a pH of 1 or less and was in a slurry state before spray drying.
- a catalyst was obtained in the same manner as in Example 32 (Catalyst 8).
- Table 4 shows the standard deviation value of the catalyst 8 and the results of the ammoxidation reaction obtained in the same manner as in Example 32.
- This MoBi-containing liquid had a pH of 1 or less and was in a slurry state before spray drying. Thereafter, a catalyst was obtained in the same manner as in Example 32 (Catalyst 9).
- Table 4 shows the standard deviation value of the catalyst 9 and the results of the ammoxidation reaction obtained in the same manner as in Example 32.
- the bulk composition of the metal oxide was consistent with the theoretical value calculated from the raw material charge mass (the top 2 in the molar composition expressed on the basis of Mo 12). It was the same as the molar composition calculated from the charged mass to the order of digits).
- Table 3 shows the bulk composition of metal oxides contained in the catalysts obtained in Examples and Reference Examples
- Table 4 shows the standard deviation of (Bi / Mo) surf / (Bi / Mo) bulk on the catalyst particle surface. Furthermore, in Table 4, the manufacturing conditions of each catalyst and the yield at the time of manufacturing acrylonitrile using each catalyst are also shown. The bulk compositions of the metal oxides contained in the catalysts obtained in Examples 32 and 35 to 38 and Reference Examples 1 and 2 were the same.
- the yield of acrylonitrile when the catalysts obtained in Reference Examples 1 and 2 were used was lower than that in Examples 32-38. From the above, it was confirmed that the more uniform the surface composition of the catalyst particles (that is, the smaller the standard deviation), the better the acrylonitrile yield (catalytic performance) in the reaction for producing acrylonitrile by the ammoxidation reaction of propylene. It was.
- the method for producing a catalyst of the present invention has industrial applicability as a method for producing a catalyst used in an ammoxidation reaction of propylene.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
〔1〕
モリブデンと、ビスマスと、鉄と、シリカと、カルボン酸とを含む前駆体スラリーを調製する調製工程と、
前記前駆体スラリーを噴霧乾燥して乾燥粒子を得る乾燥工程と、
前記乾燥粒子を焼成する焼成工程と、
を有し、
前記調製工程が、シリカ原料とカルボン酸を混合し、シリカ-カルボン酸混合液を調製する工程(I)と、前記シリカ-カルボン酸混合液、モリブデン、ビスマス及び鉄を混合する工程(II)とを含む、触媒の製造方法。
〔2〕
前記触媒が、下記一般式(1)で表されるバルク組成を有する金属酸化物と、シリカとを含む、〔1〕に記載の触媒の製造方法。
Mo12BiaFebXcYdZeOf (1)
(式(1)中、Xは、ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム及びバリウムからなる群より選ばれる1種以上の元素を示し、Yは、セリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム及びインジウムからなる群より選ばれる1種以上の元素を示し、Zは、カリウム、ルビジウム及びセシウムからなる群より選ばれる1種以上の元素を示し、a、b、c、d、e及びfは、各元素の原子比を表し、それぞれ、0.1≦a≦3.0、0.1≦b≦3.0、0.1≦c≦10.0、0.1≦d≦3.0、及び0.01≦e≦2.0を満たし、かつ、fは存在する他の元素の原子価要求を満足させるのに必要な酸素の原子数である。)
〔3〕
前記触媒において、触媒粒子表面におけるMoモル濃度に対するBiモル濃度の比率を、金属酸化物バルクのMoモル濃度に対するBiモル濃度の比率で除した値の標準偏差が0.2以下である、〔2〕に記載の触媒の製造方法。
〔4〕
前記工程(II)が、下記(i)工程及び(ii)工程を含み、
前記乾燥工程が、下記(iii)工程を含む、〔1〕~〔3〕のいずれかに記載の触媒の製造方法;
(i)少なくともMoを含むMo含有液と、少なくともBiを含むBi含有液とを用意する工程、
(ii)前記Mo含有液を第一の流路に連続的に供給し、前記Bi含有液を第二の流路に連続的に供給し、かつ第一の流路と第二の流路とを前記Mo含有液及び前記Bi含有液それぞれの供給個所よりも下流で合流させることによって、前記Mo含有液と前記Bi含有液とを混合してMoBi含有液を得る工程、
(iii)前記MoBi含有液を乾燥させる工程。
〔5〕
前記(ii)工程と前記(iii)工程との間に、さらに、(iv)前記MoBi含有液をさらに混合する工程を有する、〔4〕に記載の触媒の製造方法。
〔6〕
前記(ii)工程と前記(iii)工程との間に、さらに、(v)前記MoBi含有液を貯留する工程を有する、〔4〕又は〔5〕に記載の触媒の製造方法。
〔7〕
前記(i)、(ii)及び(v)工程の一連の処理がバッチ処理方式で行われ、(ii)工程で得られたMoBi含有液の一バッチ分全量が(v)工程において貯留され、
前記(ii)工程において、前記Mo含有液及び前記Bi含有液の前記第一の流路又は前記第二の流路への質量供給速度を、各々、mA(g/min)及びmB(g/min)としたときに、前記Mo含有液及び前記Bi含有液の一バッチあたりの全供給量MA(g)及びMB(g)の合計量の60質量%以上が、式(2)を満たすように供給される、〔6〕に記載の触媒の製造方法。
(mB/mA)/(MB/MA)=0.5~1.5 (2)
〔8〕
前記MoBi含有液が、前記(iii)工程へ連続的に供される、〔4〕又は〔5〕に記載の触媒の製造方法。
〔9〕
前記(ii)工程において、前記Mo含有液及び前記Bi含有液の前記第一の流路又は第二の流路へモル供給速度を、各々、mα(mol/min)及びmβ(mol/min)としたときに、前記Mo含有液及び前記Bi含有液が、式(3)を満たすように供給される、〔4〕、〔5〕、又は〔8〕に記載の触媒の製造方法。
(mβ/mα)/(a/12)=0.8~1.2 (3)
〔10〕
〔1〕~〔9〕のいずれかに記載の触媒の製造方法によって触媒を得る工程と、
前記触媒の存在下、プロピレンと、分子状酸素と、アンモニアと、を反応させる工程と、
を含む、アクリロニトリルの製造方法。
〔11〕
下記式(1)で表されるバルク組成を有する金属酸化物及びシリカを含む粒子からなる触媒であって、
触媒粒子表面におけるMoモル濃度に対するBiモル濃度の比率を、金属酸化物バルクのMoモル濃度に対するBiモル濃度の比率で除した値の標準偏差が0.2以下である、
触媒。
Mo12BiaFebXcYdZeOf (1)
(式(1)中、Xは、ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム及びバリウムからなる群より選ばれる1種以上の元素を示し、Yは、セリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム及びインジウムからなる群より選ばれる1種以上の元素を示し、Zは、カリウム、ルビジウム及びセシウムからなる群より選ばれる1種以上の元素を示し、a、b、c、d、e及びfは、各元素の原子比を表し、それぞれ、0.1≦a≦3.0、0.1≦b≦3.0、0.1≦c≦10.0、0.1≦d≦3.0、及び0.01≦e≦2.0を満たし、かつ、fは原子価のバランスを満たす値である。)
〔12〕
〔11〕に記載の触媒の存在下、プロピレンと、分子状酸素と、アンモニアと、を反応させる工程を含む、アクリロニトリルの製造方法。
本実施形態の触媒の製造方法は、モリブデンと、ビスマスと、鉄と、シリカと、カルボン酸とを含む前駆体スラリーを調製する調製工程と、前記前駆体スラリーを噴霧乾燥して乾燥粒子を得る乾燥工程と、前記乾燥粒子を焼成する焼成工程と、を有し、前記調製工程が、シリカ原料とカルボン酸を混合し、シリカ-カルボン酸混合液を調製する工程と、前記シリカ-カルボン酸混合液、モリブデン、ビスマス及び鉄を混合する混合工程とを含む。
本実施形態の製造方法で得られる触媒は、モリブデンと、ビスマスと、鉄と、を必須成分として含む。モリブデンは、プロピレンの吸着サイト及びアンモニアの活性化サイトとしての役割を担っている。また、ビスマスは、プロピレンを活性化させ、α位水素を引き抜いてπアリル種を生成させる役割を担っている。さらに、鉄は、3価/2価のレドックスによって気相に存在する酸素を触媒活性点に供給する役割を担っている。
本実施形態の製造方法で得られる触媒における元素Xは、適度な格子欠陥を有するモリブデートを形成し、酸素のバルク内移動を円滑にする役割を担っている。また、元素Yは鉄と同様に触媒におけるレドックス機能を担っている。さらに、元素Zは、触媒表面に存在する酸点をブロックすることで、主生成物、原料の分解反応を抑制する役割を担っている。
Mo12BiaFebXcYdZeOf (1)
上記式(1)中、Xは、ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム及びバリウムからなる群より選ばれる1種以上の元素を示し、Yはセリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム及びインジウムからなる群より選ばれる1種以上の元素を示し、Zはカリウム、ルビジウム及びセシウムからなる群より選ばれる1種以上の元素を示す。
上記式(1)中、aは、モリブデン12原子に対するビスマスの原子比を示し、0.1≦a≦3.0であり、好ましくは0.15≦a≦1.0であり、より好ましくは0.2≦a≦0.7である。
上記式(1)中、bは、モリブデン12原子に対する鉄の原子比を示し、0.1≦b≦3.0であり、好ましくは0.5≦b≦2.5であり、より好ましくは1.0≦b≦2.0である。
上記式(1)中、cは、モリブデン12原子に対するXの原子比を示し、0.1≦c≦10.0であり、好ましくは3.0≦c≦9.0であり、より好ましくは5.0≦c≦8.5である。
上記式(1)中、dは、モリブデン12原子に対するYの原子比を示し、0.1≦d≦3.0であり、好ましくは0.2≦d≦2.0であり、より好ましくは0.3≦d≦1.5である。
上記式(1)中、eは、モリブデン12原子に対するZの原子比を示し、0.01≦e≦2.0であり、好ましくは0.05≦e≦1.0である。
上記式(1)中、fは、モリブデン12原子に対する酸素の原子比を示し、原子価のバランスを満たす値である。なお、fは、存在する他の元素の原子価要求を満足させるのに必要な酸素の原子数に対応する値である。
さらに、本実施形態において、上記式(1)中、ニッケルの原子比をgとし、コバルトの原子比をhとしたとき、g及びhを次のように調整することが好ましい。すなわち、金属酸化物がマグネシウムを含まない場合は、g<1.0又は6.5<g、かつ、h<1.0又は6.5<hであることが好ましい。また、金属酸化物がマグネシウムを含む場合は、g<0.5又は6.5<g、かつ、h<0.5又は7.0<hであることが好ましい。
本実施形態において、例えば、使用する原料及びその使用量を適宜調整することにより、得られる触媒の組成を上記範囲に調整することができる。
上記の標準偏差については、後述する。
本実施形態の製造方法における調製工程は、シリカ原料とカルボン酸を混合し、シリカ-カルボン酸混合液を調製する工程(本明細書中、「工程(I)」ともいう。)を含む。混合順は特に限定されないが、例えば、シリカ原料にカルボン酸を投入して、混合液を得ることができる。
本実施形態の製造方法における調製工程は、前記シリカ-カルボン酸混合液と、モリブデンと、ビスマスと、鉄とを混合し、前駆体スラリーを得る工程(本明細書中、「工程(II)」ともいう。)を含む。混合順は特に限定されないが、例えば、モリブデンを含む溶液をシリカ-カルボン酸混合液に投入した後、ビスマスと鉄を含む溶液を投入して、前駆体スラリーを得ることができる。
乾燥工程は、調製工程で得られた前駆体スラリーを噴霧乾燥し、乾燥粒子を得る工程である。前駆体スラリーを噴霧乾燥することによって流動層反応に適した球形微粒子を得ることができる。噴霧乾燥装置としては、特に限定されず、例えば、回転円盤式、ノズル式等の一般的なものを用いることができる。噴霧乾燥条件を調節することにより、触媒の粒径を調整することができる。流動層触媒として用いる場合には、触媒の粒径は、好ましくは25~180μmである。好ましい粒径を有する触媒粒子を得るための条件の一例を記載すると、乾燥器上部の中央に設置された、皿型回転子を備えた遠心式噴霧化装置を用い、乾燥器の入口空気温度を180~250℃、出口温度を100~150℃に保持して行う噴霧乾燥が挙げられる。
(i)少なくともMoを含むMo含有液と、少なくともBiを含むBi含有液とを用意する工程、
(ii)前記Mo含有液を第一の流路に連続的に供給し、前記Bi含有液を第二の流路に連続的に供給し、かつ第一の流路と第二の流路とを前記Mo含有液及び前記Bi含有液それぞれの供給個所よりも下流で合流させることによって、前記Mo含有液と前記Bi含有液とを混合してMoBi含有液を得る工程、
(iii)前記MoBi含有液を乾燥させる工程
さらに、本実施形態においては、Mo含有液とBi含有液を混合してMoBi含有液(スラリー)を生成させた後、該MoBi含有液を乾燥させる工程の前(すなわち、(ii)工程と(iii)工程の間)に、MoBi含有液をさらに混合する工程((iv)工程)を設けることができる。
また、(ii)工程と(iii)工程の間に、MoBi含有液を一定量貯留する工程((v)工程)を設けることができる。
(ii)工程で調製したMoBi含有液は、そのまま連続的に(iii)工程に供給してもよいし、(iv)工程を経た後に(iii)工程に供給してもよいし、その一部又は全量を一旦貯留((v)工程)してから(iii)工程に供給してもよい。
(v)工程を設け、MoBi含有液を一旦貯留する場合は、(ii)工程で調製したMoBi液の全量、又は(ii)工程の後に(iv)工程を行って調製したMoBi含有液の全量を貯留することが好ましい(バッチ処理方式)。バッチ処理方式の場合、(ii)工程においてMo含有液とBi含有液との供給比率にずれが生じた場合でも、(v)工程において原料の全量が混合されることになり、すなわち意図する組成に均一化されたMoBi含有液を(iii)工程に送ることができる。
本実施形態において、連続供給方式とバッチ処理方式のいずれを選択するかは特に問わないが、短時間に触媒を製造したい場合は連続処理方式、供給比率のずれの許容幅を広げる場合はバッチ処理方式を選択することが好ましい。
具体的には、(i)少なくともMoを含有するMo含有液(金属酸化物の成分となるMoの原料を含む液であり、以下「A液」とも言う。)と、少なくともBiを含有するBi含有液(金属酸化物の成分となるBiの原料を含む液であり、「B液」とも言う。)を用意する工程と、(ii)両者を混合する工程を含む(以下、Mo含有液とBi含有液のことを「2液」ということもある)。前記2液を混合することにより、スラリーであるMoBi含有液が形成される。
このMoBi含有液は、MoBi含有金属酸化物の前駆体であり、この前駆体の生成条件が、製造する触媒粒子の表面組成の均一性に大きく影響することが本発明者らの研究により分かった。
(ii)工程において、Mo含有液及びBi含有液を、各々、第一及び第二の流路に連続的に供給し、第一の流路と第二の流路とをMo含有液及びBi含有液それぞれの供給個所よりも下流で合流させてMoBi含有液を調製することによって、表面組成の均一性がより高い触媒粒子が得られる傾向にある。
Moの原料としては、水、硝酸などに可溶な塩が好ましく、例えば、アンモニウム塩であるモリブデン酸アンモニウムが挙げられる。
ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム、バリウム、セリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム、インジウム、カリウム、ルビジウム、セシウムの原料としても、上記と同様に、水、硝酸などに可溶な塩が好ましく、例えば、硝酸塩、塩酸塩、硫酸塩、有機酸塩等が挙げられる。特に、水、硝酸などに溶け易い観点から、硝酸塩が好ましい。例えば、硝酸ビスマス、硝酸鉄、硝酸ニッケル、硝酸コバルト、硝酸カリウム、硝酸ルビジウム、硝酸マグネシウム、硝酸カルシウム、硝酸ストロンチウム、硝酸バリウム、硝酸亜鉛、硝酸ランタン、硝酸セリウム、硝酸クロム、硝酸インジウム、硝酸ガリウム等が挙げられる。これらは一種を単独で用いても二種以上を併用してもよい。
シリカゾルに含まれるシリカの平均一次粒子径は特に限定されないが、5~100nmの範囲であることが好ましく、10~50nmの範囲であることがより好ましい。また、異なる平均一次粒子径を有するシリカゾルを混ぜて用いることもできる。
さらに、Mo含有液にはMoの溶解性を増すためにアンモニア水を添加することも好ましい。
また、Bi含有液は、Mo含有液とは別に、上記Biの原料を水等の水性媒体に溶解又は分散させて調製することが好ましい。Bi含有液は酸性であることが好ましく、Bi含有液が硝酸を含むと、Biの原料の溶解度を増すためより好ましい。Bi含有液におけるBi原子濃度は0.1~10質量%の範囲内であることが好ましい。
すなわち、(ii)工程において、Mo含有液及びBi含有液の第一の流路又は第二の流路へのモル供給速度(ただし、各々、Moのモル供給速度、Biのモル供給速度)を、各々、mα(mol/min)及びmβ(mol/min)としたときに、Mo含有液及びBi含有液が、以下の式(3)を満たすように供給されることが好ましい。
(mβ/mα)/(a/12)=0.8~1.2 (3)
すなわち、Mo含有液及びBi含有液が、以下の式(2)を満たすように供給されることが好ましい。
(mB/mA)/(MB/MA)=0.5~1.5 (2)
供給比率が質量比率の0.5倍~1.5倍の範囲であると、触媒の粒子表面の組成の均一性をより高めることができる。(mB/mA)/(MB/MA)は、0.8~1.2がより好ましく、1.0が特に好ましい。
製造する金属酸化物のバルク組成を、式(1)においてa=1であるもの(Mo12原子に対してBi1原子)とし、(i)工程で準備する一バッチ分のMo含有液は、Moの原料を必要量含みその全量(MA)が100質量部であり、(i)工程で準備する一バッチ分のBi含有液はBiの原料を必要量(Mo含有液中のMoのモル数の1/12)含み、その全量(MB)が50質量部とする。
この2液を、単位時間当たりの質量供給量(質量供給速度)(g/min)の比率で表して、Mo含有液:Bi含有液=100:50で供給すると、質量比率、供給比率は、各々、次の通りである。
質量比率(MB/MA)
=50/100=0.5
供給比率(mB/mA)
=50/100=0.5
したがって、その場合が、供給比率が質量比率の1.0倍であることに相当する。
同様に、Mo含有液:Bi含有液=100:25(質量供給速度比)で供給する場合が、供給比率が質量比率(0.5)の0.5倍であることに相当する。Mo含有液:Bi含有液=100:75(質量供給速度比)で供給する場合が(供給比率=75/100=0.75)、供給比率が質量比率(0.5)の1.5倍であることに相当する。
特に好ましい実施形態である、供給比率が質量比率の1.0倍である場合、(ii)工程においては、Mo含有液の一バッチ分全供給量に対するBi含有液の一バッチ分全供給量の比率と同じ比率で2液を供給し、合流させることになり、それは、すなわち、MoとBiの供給モル比率が、製造する金属酸化物のバルク組成式(1)におけるMoとBiのモル比率、すなわちa/12、と一致することになる。
それに対して、本実施形態の製造方法において、2液が合流部に同時に供給する場合、前述のような混合液中の成分濃度のアンバランスを十分に小さくすることができる。
特に、Mo含有液とBi含有液とを特定の比率の範囲内で供給する場合には、両者を混合する工程のどの段階においても、混合液中のMoとBiの原子比率が製造する触媒組成の原子比率に近い状態でスラリーを形成することになる。このような状態でスラリーを調製することによって、最終的に製造された触媒粒子の表面におけるMoとBiの組成比をより均一にすることができる。
具体的には、Mo含有液とBi含有液の一バッチあたりの全供給量の合計量の60質量%以上を、供給比率が質量比率の0.5倍~1.5倍となるように供給することが好ましく、80質量%以上を0.5倍~1.5倍となるように供給することがさらに好ましく、100質量%を(すなわち、(ii)工程の全工程に亘って)0.5倍~1.5倍となるように供給することが特に好ましい。
このことを、上述の、Mo12原子分の原料を含むMo含有液100質量部と、Bi1原子分の原料を含むBi含有液50質量部の例で説明する。この場合、Mo含有液とBi含有液の全供給量の合計量は150質量部であり、その60質量%以上とは90質量部以上に相当する。
すなわち、供給比率が質量比率の0.5倍~1.5倍となるように、2液の全供給量の合計量の60質量%以上を供給するとは、単位時間当たりの供給量(質量供給速度)の比率で表して、Mo含有液:Bi含有液=100:25~100:75の範囲内で2液の合計の90質量部以上を接触させることに相当する。
撹拌温度は、2液を合流させる際の合流温度と同程度とすることが好ましく、具体的には35℃~80℃が好ましい。
第一及び第二の流路へのMo含有液及びBi含有液の供給手段に限定はなく、例えば、定量供給装置を使用することが好ましい。定量供給装置を使用することにより、より正確な比率で2液を供給することができる。該定量供給装置としては、特に限定されないが定量送液ポンプが好ましい。
2液が合流する合流部としては、2液の合流ができれば特に限定されないが、例えばY字状またはT字状等形状の、管でもよい。該管の断面形状は例えば円形(円筒)であってもよく、また内部にMoBi含有液の混合を促す機構や内挿物を設けることも好ましい。2液を合流させる角度は並流であっても、垂直であっても、対向であってもよい。
また混合器の別の例としては、図2に示すような静止型混合器(配管とその中に固定されたねじられた板とを主要部材とし、配管内に供給された流体は、ねじれ面に沿って流れ進んで行くことで混合される)が挙げられる。静止型混合器の一例としては、スタティックミキサー(ノリタケカンパニーリミテド製)等がある。図1に示すように、静止型混合器と攪拌槽を組み合わせてもよい。
MoBi含有液は乾燥前においてpHが3以下であることが好ましい。より好ましくは2以下であり、さらに好ましくは1以下である。pHの調節は、例えば、原料に加える酸性溶液の添加量によって調節できる。pHが3以下であればMoBi含有液の粘度の増加を抑制でき、次工程の(iii)乾燥工程へのMoBi含有液の供給が安定的に行える。
噴霧乾燥工程においては、まず、MoBi含有液を噴霧化する。この噴霧化の方法としては、特に限定されないが、例えば、遠心方式、二流体ノズル方式、及び高圧ノズル方式等が挙げられる。特にノズルの閉塞等を伴わない観点から遠心方式が好ましい。
噴霧化されたMoBi含有液の液滴の乾燥方法に限定はないが、例えば、スプレードライヤー内の熱風によって乾燥されることが好ましい。その際、熱風入口温度は180℃~250℃、熱風出口温度は100℃~150℃とすることが好ましい。
なお、噴霧化の際には、噴霧粒子径が目的とする触媒の粒径に応じた所望の範囲となるよう、噴霧装置を調整することが好ましい。例えば、触媒を流動床反応に用いる場合には、触媒の平均粒子径は25~180μm程度が好ましいため、この範囲の平均粒子径になるように噴霧化装置(アトマイザー)の回転数を適宜選択する。一般にアトマイザー回転数が高いほど、平均粒子径は小さくなる。
また、(i)、(ii)及び(v)工程又は(i)、(ii)、(iv)及び(v)工程の一連の処理がバッチ処理方式で行われる場合には、(v)工程における貯留量は、(ii)工程で得られたMoBi含有液の全量とすることが好ましい。(ii)工程で調製されるMoBi含有液の全量をいったん貯留してから乾燥工程((iii)工程)に供給することにより、(ii)工程においてMo含有液及びBo含有液の供給量が変動する場合においても、(iii)工程に供給されるMoBi含有液中のMoとBiの原子数比を式(1)における両者の原子数比と等しくすることができ、得られる触媒粒子の組成をより目的に近いものとすることができる。
貯留容器に限定はなく、例えば、貯留容器として撹拌機能を有するものを用い、(v)工程と(iv)工程を同時に行うこともできる。
上述した他、例えば、原料スラリー調製において、攪拌混合槽を使い、どちらか一方の液に他方の液を投入するMo含有液とBi含有液とのバッチ混合を採用する場合であっても、1バッチで50kg以上の金属酸化物を製造するようにすると共に、投入する液の投入に掛かる時間を8分以内、好ましくは4分以内とすることで、2液の均一混合を促進し、触媒を製造することもできる。この場合、投入するMo/Bi含有液の量を大きくすることで、投入液の質量を増し、投入される側の液との衝突エネルギーが大きくなることと短時間に処理することの相乗効果により、より均一なスラリー混合が達成される傾向にある。
焼成工程は、乾燥工程で得られた乾燥粒子を焼成する工程である。
乾燥粒子が硝酸を含有する場合は、焼成の前に脱硝処理を行うことが好ましい。脱硝処理は150~450℃で1.5~3時間行うことが好ましい。焼成は空気雰囲気下で行うことができる。
焼成時間は1hr~24hrが好ましく、2hr~20hrがより好ましい。所望する触媒物性、反応性能が得られるように、焼成温度、焼成時間等の条件を適宜選択する。
本実施形態の製造方法により得られる触媒は、後述のアクリロニトリルの製造においてアンモ酸化反応の際に用いる触媒(アンモ酸化用触媒)として好適である。
上述した本実施形態の製法により得られる触媒は、下記に限定されないが、典型的には、次の特徴を有するものである。すなわち、本実施形態に係る触媒は、下記一般式(1)で表されるMoとBiとを含むバルク組成を有する金属酸化物及びシリカを含む粒子からなる触媒である。
Mo12BiaFebXcYdZeOf (1)
(式(1)中、Xは、ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム及びバリウムからなる群より選ばれる1種以上の元素、Yはセリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム及びインジウムからなる群より選ばれる1種以上の元素、Zはカリウム、ルビジウム及びセシウムからなる群より選ばれる1種以上の元素を示し、a、b、c、d及びeは、それぞれ、0.1≦a≦2.0、0.1≦b≦3.0、0.1≦c≦10.0、0.1≦d≦3.0、及び0.01≦e≦2.0を満たし、fは存在する他の元素の原子価要求を満足させるのに必要な酸素の原子数である。)
これについて、本発明者らは、以下のように考察している。アンモ酸化反応における活性点はビスマスを含むモリブデートであるが、それのみでは高いアクリロニトリル選択率を得ることはできない。ビスマスを含むモリブデートが、少なくとも触媒粒子表面において、鉄を含むモリブデートや、その他の金属を含むモリブデートと均一に複合化していることが、アクリロニトリル選択率を向上させると考えられる。
また、ビスマスを含むモリブデートが、鉄を含むモリブデートや、その他の金属を含むモリブデートとその表面において均一に複合化している触媒粒子は、その前駆体スラリーの段階において、各金属成分を単独成分で凝集させずに分散させて、ビスマスを含むモリブデートが、その他の金属を含むモリブデートと複合化しやすい状態にすることが必要であると考えられる。さらに、金属粒子が担体上に担持される場合には、前駆体スラリー中の金属と、担体の粒子、双方の凝集を防ぐことで、触媒の摩耗強度も向上すると考えられる。
ただし、作用機序は上記に限定されない。
本実施形態において触媒を構成する金属酸化物は、モリブデンと、ビスマスと、鉄と、を必須成分として含む。モリブデンは、プロピレンの吸着サイト及びアンモニアの活性化サイトとしての役割を担っている。また、ビスマスは、プロピレンを活性化させ、α位水素を引き抜いてπアリル種を生成させる役割を担っている。さらに、鉄は、3価/2価のレドックスによって気相に存在する酸素を触媒活性点に供給する役割を担っている。
このような組成を有することにより、アクリロニトリル選択率がより向上する傾向にある。
Mo12BiaFebXcYdZeOf (1)
上記式(1)中、Xは、ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム及びバリウムからなる群より選ばれる1種以上の元素、Yはセリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム及びインジウムからなる群より選ばれる1種以上の元素、Zはカリウム、ルビジウム及びセシウムからなる群より選ばれる1種以上の元素を示し、
aは、モリブデン12原子に対するビスマスの原子比を示し、0.1≦a≦2.0であり、好ましくは0.15≦a≦1.0であり、より好ましくは0.2≦a≦0.7であり、
bは、モリブデン12原子に対する鉄の原子比を示し、0.1≦b≦3.0であり、好ましくは0.5≦b≦2.5であり、より好ましくは1.0≦b≦2.0であり、
cは、モリブデン12原子に対するXの原子比を示し、0.1≦c≦10.0であり、好ましくは3.0≦c≦9.0であり、より好ましくは5.0≦c≦8.5であり、
dは、モリブデン12原子に対するYの原子比を示し、0.1≦d≦3.0であり、好ましくは0.2≦d≦2.0であり、より好ましくは0.3≦d≦1.5であり、
eは、モリブデン12原子に対するZの原子比を示し、0.01≦e≦2.0であり、好ましくは0.05≦e≦1.0であり、
fは、モリブデン12原子に対する酸素の原子比を示し、存在する他の元素の原子価要求を満足させるのに必要な酸素の原子数である。
さらに、本実施形態において、上記式(1)中、ニッケルの原子比をgとし、コバルトの原子比をhとしたとき、g及びhを次のように調整することが好ましい。すなわち、金属酸化物がマグネシウムを含まない場合は、g<1.0又は6.5<g、かつ、h<1.0又は6.5<hであることが好ましい。また、金属酸化物がマグネシウムを含む場合は、g<0.5又は6.5<g、かつ、h<0.5又は7.0<hであることが好ましい。
ICP発光分光分析法は実施例において採用した方法であり、その具体的手順については実施例で説明する。また、蛍光X線分析による定量分析の場合は、例えば、蛍光X線装置(例えば、リガク製ZSX100e(管球:Rh-4KW、分光結晶:LiF、PET、Ge、RX25))を用いて、各元素のマトリックス効果を補正する検量線に基づき、触媒サンプルの定量分析を行うことができる。なお、後述する本実施形態の製造方法によって触媒を製造する場合には、このような分析によって得られた値は、設計上の仕込み組成とほぼ一致する。また、これらの元素定量分析によれば、使用前の触媒のほか、使用中の触媒に対しても、そのバルク組成を求めることができる。
なお、触媒粒子の見掛比重は、試料(触媒粒子)を一定容積の容器の中に一定状態で入れ、容器内に入った試料の質量を測定し、単位体積あたりの質量を算出することで求めることができる。
例えば、ガラスロートの真下に25mlガラスメスシリンダー(以下、単に「メスシリンダー」という。)を配置し、予めよく振った測定試料(触媒粒子)をガラスロートに落下させ、メスシリンダーがオーバーフローするまで試料を投入する。メスシリンダーの口より上部に堆積した試料の凸部を摺り切りで除き、またメスシリンダーの外壁に付着した試料を刷毛等で払う。試料入りのメスシリンダーの質量と、事前に測定しておいたメスシリンダーの質量から試料の質量を求め、それをメスシリンダーの容積で割ることにより見掛比重を算出する。
ここで、粒子表面におけるMoモル濃度及びBiモル濃度は、SEM-EDX(Scannning Electron Microscopes-Energy Dispersive X-ray Spectroscopy:走査型電子顕微鏡-エネルギー分散型X線分光法)組成分析により求めることができ、金属酸化物バルクのMoモル濃度及びBiモル濃度は、前述したように、例えば、ICP発光分光分析法で求めることができる。
また、上記標準偏差は、以下の式(4)により求めることができる。
標準偏差={((S1-μ)2+(S2-μ)2+・・・+(S100-μ)2)/100[個数]}1/2 (4)
式(4)中、Sk(k=1~100)は、任意に選ばれた100個粒子中のk個目の粒子の表面におけるMoモル濃度に対するBiモル濃度測定値の比率(Bi/Mo)surfを金属酸化物バルクのMoモル濃度に対するBiモル濃度の比率(Bi/Mo)bulkで除した値であり、μは、100個のSk(k=100)の平均値(=(S1+S2・・・+S100)/100)である。
標準偏差はばらつきの指標である。標準偏差が小さい程、平均値からのばらつきが小さい。したがって、式(4)の値が小さい程、粒子の表面組成がより均一であることを意味する。
本発明者らの研究によれば、プロピレンのアンモ酸化反応によりアクリロニトリルを製造するにあたり、式(4)の値が小さい触媒を用いるとアクリロニトリル収率が高まることが判明した。式(4)の値は、本実施形態においては0.2以下であり、0.15以下であることが好ましく、0.10以下であることがより好ましく、0.07以下であることがさらに好ましい。
本実施形態のアクリロニトリルの製造方法は、前述した触媒の製造方法により触媒を得る工程と、得られた触媒の存在下、プロピレンと、分子状酸素と、アンモニアと、を反応させてアクリロニトリルを製造する反応工程と、を有する。また、本実施形態のアクリロニトリルの製造方法は、本実施形態の触媒の存在下、プロピレンと、分子状酸素と、アンモニアと、を反応させる工程を含むものということもできる。
バルク組成(以下、単に「組成」と記載する。)がMo12.00Bi0.50Fe1.31Co4.05Ni3.10Ce0.87Rb0.10K0.08Of(以降の組成については「Of」を省略して記載する。)で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シリカ-シュウ酸混合液の撹拌時間を30分にしたこと以外は実施例1と同様にして、触媒を製造した。
シリカ-シュウ酸混合液の撹拌時間を80分にしたこと以外は実施例1と同様にして、触媒を製造した。
シュウ酸添加速度8g/sec.(SiO21kgあたり)にしたこと以外は実施例1と同様にして、触媒を製造した。
シュウ酸添加速度3g/sec.(SiO21kgあたり)にしたこと以外は実施例1と同様にして、触媒を製造した。
組成がMo12.00Bi0.39Fe1.60Ni6.97Mg0.77Ce0.63Rb0.17で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シリカ-シュウ酸混合液の撹拌時間を80分にしたこと以外は実施例6と同様にして、触媒を製造した。
シュウ酸添加速度3g/sec.(SiO21kgあたり)にしたこと以外は実施例6と同様にして、触媒を製造した。
組成がMo12.00Bi1.20Fe0.60Ni7.80Cr1.20K0.48で表される金属酸化物を60質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi2.40Fe2.40Ni6.00Zn1.80Ce0.24K0.24で表される金属酸化物を60質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi0.47Fe1.35Co4.15Ni3.00Ce0.91Rb0.14で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi0.57Fe1.01Co6.83Ni0.98Mg0.98Ce0.38Rb0.12で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シリカゾルのpHが9.00、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.038モル当量)、シリカ-シュウ酸混合液のpHが1.65であること以外は実施例12と同様にして、触媒を製造した。
シリカゾルのpHが9.70、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.038モル当量)、シリカ-シュウ酸混合液のpHが5.30であること以外は実施例12と同様にして、触媒を製造した。
シリカゾルのpHが10.13、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.038モル当量)、シリカ-シュウ酸混合液のpHが8.00であること以外は実施例12と同様にして、触媒を製造した。
組成がMo12.00Bi0.57Fe1.01Co2.24Ni6.54Ce0.38Rb0.12で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シリカゾルのpHが9.00、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.039モル当量)、シリカ-シュウ酸混合液のpHが1.65であること以外は実施例16と同様にして、触媒を製造した。
シリカゾルのpHが9.70、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.039モル当量)、シリカ-シュウ酸混合液のpHが5.30であること以外は実施例16と同様にして、触媒を製造した。
シリカゾルのpHが10.13、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.039モル当量)、シリカ-シュウ酸混合液のpHが8.00であること以外は実施例16と同様にして、触媒を製造した。
組成がMo12.00Bi0.84Fe2.06Co6.67Ce0.56Rb0.12で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シリカゾルのpHが9.00、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.040モル当量)、シリカ-シュウ酸混合液のpHが1.65であること以外は実施例20と同様にして、触媒を製造した。
シリカゾルのpHが9.70、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.040モル当量)、シリカ-シュウ酸混合液のpHが5.30であること以外は実施例20と同様にして、触媒を製造した。
シリカゾルのpHが10.13、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.040モル当量)、シリカ-シュウ酸混合液のpHが8.00であること以外は実施例20と同様にして、触媒を製造した。
組成Mo12.00Bi0.27Fe0.95Co6.69Ni2.95Ce0.18Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シリカゾルのpHが9.00、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.038モル当量)、シリカ-シュウ酸混合液のpHが1.65であること以外は実施例24と同様にして、触媒を製造した。
シリカゾルのpHが9.70、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.038モル当量)、シリカ-シュウ酸混合液のpHが5.30であること以外は実施例24と同様にして、触媒を製造した。
シリカゾルのpHが10.13、シュウ酸二水和物が25.0g(前駆体スラリーにおいて金属元素の総和に対し0.038モル当量)、シリカ-シュウ酸混合液のpHが8.00であること以外は実施例24と同様にして、触媒を製造した。
組成がMo12.00Bi0.82Fe1.45Co8.14Ce0.55Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi1.05Fe1.40Co8.15Ce0.70Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi0.27Fe0.95Co8.16Ni1.48Ce0.18Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi0.27Fe0.95Co7.67Ni1.97Ce0.18Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi0.50Fe1.31Co4.05Ni3.10Ce0.87Rb0.10K0.08で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
30質量%のSiO2を含むシリカゾル1333gに、865.8gの水に溶解させた481.6gのパラモリブデン酸アンモニウム[(NH4)6Mo7O24・4H2O]を撹拌下で加え、さらに、16.6質量%の硝酸391.8gに溶解させた55.1gの硝酸ビスマス[Bi(NO3)3・5H2O]、120.3gの硝酸鉄[Fe(NO3)3・9H2O]、268.0gの硝酸コバルト[Co(NO3)2・6H2O]、204.9gの硝酸ニッケル[Ni(NO3)2・6H2O]、85.9gの硝酸セリウム[Ce(NO3)3・6H2O]、3.35gの硝酸ルビジウム[RbNO3]、1.84gの硝酸カリウム[KNO3]を加え、原料スラリーを調製した。その後、水200gに溶解させたシュウ酸二水和物25.0g(純度99.5%)を加えて(前駆体スラリーにおいて金属元素の総和に対し0.040モル当量)前駆体スラリーを調製した。次いで、前駆体スラリーを回転円盤式の噴霧乾燥器を用いて乾燥させた。このとき、乾燥機入口の空気温度は230℃とし、出口の空気温度は110℃とした。また、円盤の回転数は12500回転/分に設定した。得られた乾燥粒子を200℃で5分間保持し、200℃から450℃まで2.5℃/分で昇温し、450℃で20分間保持することで脱硝した。得られた脱硝粉を590℃で2時間焼成して、触媒を得た。
シュウ酸二水和物を添加しないこと以外は実施例1と同様にして、触媒を製造した。
組成がMo12.00Bi0.39Fe1.60Ni6.97Mg0.77Ce0.63Rb0.17で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
30質量%のSiO2を含むシリカゾル1333gに、873.5gの水に溶解させた485.9gのパラモリブデン酸アンモニウム[(NH4)6Mo7O24・4H2O]を撹拌下で加え、さらに、16.6質量%の硝酸396.7gに溶解させた43.1gの硝酸ビスマス[Bi(NO3)3・5H2O]、148.0gの硝酸鉄[Fe(NO3)3・9H2O]、464.7gの硝酸ニッケル[Ni(NO3)2・6H2O]、45.5gの硝酸マグネシウム[Mg(NO3)2・6H2O]、62.6gの硝酸セリウム[Ce(NO3)3・6H2O]、5.89gの硝酸ルビジウム[RbNO3]を加え、原料スラリーを調製した。その後、水200gに溶解させたシュウ酸二水和物25.0g(純度99.5%)を加えて(前駆体スラリーにおいて金属元素の総和に対し0.038モル当量)前駆体スラリーを調製した。次いで、前駆体スラリーを回転円盤式の噴霧乾燥器を用いて乾燥させた。このとき、乾燥機入口の空気温度は230℃とし、出口の空気温度は110℃とした。また、円盤の回転数は12500回転/分に設定した。得られた乾燥粒子を200℃で5分間保持し、200℃から450℃まで2.5℃/分で昇温し、450℃で20分間保持することで脱硝した。得られた脱硝粉を580℃で2時間焼成して、触媒を得た。
シュウ酸二水和物を添加しないこと以外は実施例6と同様にして、触媒を製造した。
組成がMo12.00Bi1.20Fe0.60Ni7.80Cr1.20K0.48で表される金属酸化物を60質量%のシリカに担持した触媒を、以下の手順で製造した。
30質量%のSiO2を含むシリカゾル2000gに、553.7gの水に溶解させた308.0gのパラモリブデン酸アンモニウム[(NH4)6Mo7O24・4H2O]を撹拌下で加え、さらに、16.6質量%の硝酸387.6gに溶解させた84.6gの硝酸ビスマス[Bi(NO3)3・5H2O]、35.2gの硝酸鉄[Fe(NO3)3・9H2O]、329.8gの硝酸ニッケル[Ni(NO3)2・6H2O]、69.8gの硝酸クロム[Cr(NO3)3・9H2O]、7.06gの硝酸カリウム[KNO3]を加え、原料スラリーを調製した。その後、水200gに溶解させたシュウ酸二水和物25.0g(純度99.5%)を加えて(前駆体スラリーにおいて金属元素の総和に対し0.059モル当量)前駆体スラリーを調製した。次いで、前駆体スラリーを回転円盤式の噴霧乾燥器を用いて乾燥させた。このとき、乾燥機入口の空気温度は230℃とし、出口の空気温度は110℃とした。また、円盤の回転数は12500回転/分に設定した。得られた乾燥粒子を200℃で5分間保持し、200℃から450℃まで2.5℃/分で昇温し、450℃で20分間保持することで脱硝した。得られた脱硝粉を590℃で2時間焼成して、触媒を得た。
組成がMo12.00Bi2.40Fe2.40Ni6.00Zn1.80Ce0.24K0.24で表される金属酸化物を60質量%のシリカに担持した触媒を、以下の手順で製造した。
30質量%のSiO2を含むシリカゾル2000gに、487.7gの水に溶解させた271.3gのパラモリブデン酸アンモニウム[(NH4)6Mo7O24・4H2O]を撹拌下で加え、さらに、16.6質量%の硝酸382.2gに溶解させた149.1gの硝酸ビスマス[Bi(NO3)3・5H2O]、124.2gの硝酸鉄[Fe(NO3)3・9H2O]、223.5gの硝酸ニッケル[Ni(NO3)2・6H2O]、68.6gの硝酸亜鉛[Zn(NO3)2・6H2O]、13.3gの硝酸セリウム[Ce(NO3)3・6H2O]、3.11gの硝酸カリウム[KNO3]を加え、原料スラリーを調製した。その後、水200gに溶解させたシュウ酸二水和物25.0g(純度99.5%)を加えて(前駆体スラリーにおいて金属元素の総和に対し0.061モル当量)前駆体スラリーを調製した。次いで、前駆体スラリーを回転円盤式の噴霧乾燥器を用いて乾燥させた。このとき、乾燥機入口の空気温度は230℃とし、出口の空気温度は110℃とした。また、円盤の回転数は12500回転/分に設定した。得られた乾燥粒子を200℃で5分間保持し、200℃から450℃まで2.5℃/分で昇温し、450℃で20分間保持することで脱硝した。得られた脱硝粉を570℃で2時間焼成して、触媒を得た。
組成がMo12.00Bi0.47Fe1.35Co4.15Ni3.00Ce0.91Rb0.14で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
30質量%のSiO2を含むシリカゾル1333gに、864.9gの水に溶解させた481.1gのパラモリブデン酸アンモニウム[(NH4)6Mo7O24・4H2O]を撹拌下で加え、さらに、16.6質量%の硝酸392.0gに溶解させた51.8gの硝酸ビスマス[Bi(NO3)3・5H2O]、123.9gの硝酸鉄[Fe(NO3)3・9H2O]、274.4gの硝酸コバルト[Co(NO3)2・6H2O]、198.1gの硝酸ニッケル[Ni(NO3)2・6H2O]、89.7gの硝酸セリウム[Ce(NO3)3・6H2O]、4.81gの硝酸ルビジウム[RbNO3]を加え原料スラリーを調製した。その後、水200gに溶解させたシュウ酸二水和物25.0g(純度99.5%)を加えて(前駆体スラリーにおいて金属元素の総和に対し0.040モル当量)前駆体スラリーを調製した。次いで、前駆体スラリーを回転円盤式の噴霧乾燥器を用いて乾燥させた。このとき、乾燥機入口の空気温度は230℃とし、出口の空気温度は110℃とした。また、円盤の回転数は12500回転/分に設定した。得られた乾燥粒子を200℃で5分間保持し、200℃から450℃まで2.5℃/分で昇温し、450℃で20分間保持することで脱硝した。得られた脱硝粉を590℃で2時間焼成して、触媒を得た。
組成がMo12.00Bi0.57Fe1.01Co6.83Ni0.98Mg0.98Ce0.38Rb0.12で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シュウ酸二水和物を添加しないこと以外は比較例8と同様にして、触媒を製造した。
組成がMo12.00Bi0.57Fe1.01Co2.24Ni6.54Ce0.38Rb0.12で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シュウ酸二水和物を添加しないこと以外は比較例10と同様にして、触媒を製造した。
組成がMo12.00Bi0.84Fe2.06Co6.67Ce0.56Rb0.12で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シュウ酸二水和物を添加しないこと以外は比較例12と同様にして、触媒を製造した。
組成Mo12.00Bi0.27Fe0.95Co6.69Ni2.95Ce0.18Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
シュウ酸二水和物を添加しないこと以外は比較例14と同様にして、触媒を製造した。
組成がMo12.00Bi0.82Fe1.45Co8.14Ce0.55Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi1.05Fe1.40Co8.15Ce0.70Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi0.27Fe0.95Co8.16Ni1.48Ce0.18Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。
組成がMo12.00Bi0.27Fe0.95Co7.67Ni1.97Ce0.18Rb0.13で表される金属酸化物を40質量%のシリカに担持した触媒を、以下の手順で製造した。

プロピレンのアンモ酸化反応に使用する反応管には、10メッシュの金網を1cm間隔で16枚内蔵した内径25mmのパイレックス(登録商標)ガラス管を使用した。触媒量50cc、反応温度430℃、反応圧力0.17MPaに設定し、プロピレン9容積%の混合ガス(プロピレン、アンモニア、酸素、ヘリウム)を通過させた。各実施例及び比較例におけるアンモニア/プロピレンのモル比及び酸素/プロピレンのモル比は表2に示す値とした。プロピレンに対するアンモニアの容積比は、下記式で定義される硫酸原単位が20±2kg/T-ANとなるように設定した。プロピレンに対する酸素の容積比は、反応器出口ガスの酸素濃度が0.2±0.02容積%になるように設定した。また混合ガスの流速を変更することで、下記式で定義される接触時間を変更した。これによって下記式で定義されるプロピレン転化率が99.3±0.2%となるように設定した。反応によって生成するアクリロニトリル収率は下記式のように定義される値とした。

“Test Method for Synthetic Fluid Cracking Catalyst”(American Cyanamid Co.Ltd.6/31-4m-1/57)に記載の方法(以下「ACC法」と称する。)に準じて、摩耗損失として触媒の耐摩耗性強度(アトリッション強度)の測定を行った。
アトリッション強度は摩耗損失で評価され、この摩耗損失は以下のように定義される値とした。
摩耗損失(%)=R/(S-Q)×100
上記式において、Qは0~5時間の間に外部に摩耗飛散した触媒の質量(g)、Rは通常5~20時間の間に外部に摩耗飛散した触媒の質量(g)、Sは試験に供した触媒の質量(g)とした。

[触媒1の製造]
その組成がMo12Bi0.25Fe1.4Ni3.0Co5.8Ce0.40Rb0.12Ofとなるよう原料仕込質量を調整して製造した金属酸化物60質量%を40質量%のシリカに担持した触媒を、以下の手順で製造した。
一次粒子の平均粒子直径が12nmのSiO2を30質量%含む水性シリカゾル666.7gと、一次粒子の平均粒子直径が41nmのSiO2を40質量%含む水性シリカゾル500gとを混合して、2種シリカの混合液を得た。
次に8質量%しゅう酸水溶液320gを撹拌中の上記シリカゾルの混合液に加え、第1の混合液を得た。次に、水870gに486.2gのパラモリブデン酸アンモニウム〔(NH4)6Mo7O24・4H2O〕を溶解させた液を上記シリカゾルの混合液に加えた(Mo含有液。以降、「A1液」と称す)。その全量は2843gであった。
次いで、16.6質量%濃度の硝酸液400gに、28.09gの硝酸ビスマス〔Bi(NO3)3・5H2O〕、131.1gの硝酸鉄〔Fe(NO3)3・9H2O〕、202.9gの硝酸ニッケル〔Ni(NO3)2・6H2O〕、392.6gの硝酸コバルト〔Co(NO3)2・6H2O〕、39.62gの硝酸セリウム〔Ce(NO3)3・6H2O〕、4.038gの硝酸ルビジウム〔RbNO3〕を溶解させた(Bi含有液。以降、「B1液」と称す)。その全量は1198gであった。
上記Y字管に、40℃に保持したA1液をA1液定量ポンプを用いて284(g/min)の流量、40℃に保持したB1液をB1液定量ポンプを用いて119(g/min)の流量で同時に供給し、Y字管と静止型混合器内において2液を並行方向(並流)に接触させた。この操作を10分間行い、スラリーであるMoBi含有液を調製した。この間のA1液[Mo含有液]に対するB1液[Bi含有液]の供給比率は119(g/min)/284(g/min)=0.42であり、Mo含有液全量に対するBi含有液全量の質量比率1198(g)/2843(g)=0.42の1.0倍であった。この供給比率でA1液とB1液の全部(100質量%)を合流させた。その間の合流温度は40℃であった。
このMoBi含有液を、静止型混合器の下流に設けたスラリー攪拌槽に全量貯留し、2液の供給終了から更に40℃で1時間攪拌を続けた。このMoBi含有液は噴霧乾燥前においてpHが1以下であり、スラリー状であった。
次いで、スプレードライヤー(大川原化工機製、型式:OC-16)を用いてこのMoBi含有液を噴霧乾燥し、乾燥粉末を得た。なお、熱風入口温度は230℃、熱風出口温度120℃とした。
次いで、乾燥した触媒前駆体に電気炉を用いて、空気雰囲気下、320℃で2時間の前焼成を施した後、空気雰囲気下、600℃で2時間の本焼成を施して、触媒を得た(触媒1)。
この触媒1の粒子100個の粒子表面のMo濃度とBi濃度をSEM-EDXで測定し、Moモル濃度に対するBiモル濃度の比率(Bi/Mo)surfを求めた。また、上記で求めた金属酸化物のバルク組成より、(Bi/Mo)bulkを求め、(Bi/Mo)bulkで100個の粒子それぞれの(Bi/Mo)surfを除した値S1・・・S100を求めた。そして、それらの平均値(S1・・・S100の平均値)μを求め、前述した式(4)を用いて標準偏差値を求めた。また、触媒1を用いてプロピレンのアンモ酸化反応によりアクリロニトリルを製造し、収率を算出した。これらの測定方法の詳細は後述する。得られた結果を表4に示す。
[触媒2の製造]
その組成がMo12Bi0.39Fe1.60Ni7.0Mg0.77Ce0.63Rb0.17Ofとなるよう原料仕込質量を調整して製造した金属酸化物60質量%を40質量%のシリカに担持した触媒を、以下の手順で製造した。
次に、16.6質量%の硝酸396.7gに、43.1gの硝酸ビスマス[Bi(NO3)3・5H2O]、148.0gの硝酸鉄[Fe(NO3)3・9H2O]、464.7gの硝酸ニッケル[Ni(NO3)2・6H2O]、45.5gの硝酸マグネシウム[Mg(NO3)2・6H2O]、62.6gの硝酸セリウム[Ce(NO3)3・6H2O]、5.89gの硝酸ルビジウム[RbNO3]を溶解させた。以降、これをB2液と称す。B2液の全量は1166gであった。
このMoBi含有液を、静止型混合器の下流に設けたスラリー攪拌槽に全量貯留し、2液の供給終了から更に40℃で1時間攪拌を続けた。このMoBi含有液は噴霧乾燥前においてpHが1以下であり、スラリー状であった。
次いで、実施例32と同様にこのMoBi含有液を噴霧乾燥し、乾燥粉末を得た。なお、熱風入口温度は230℃、熱風出口温度110℃とした。
次いで、乾燥した触媒前駆体に電気炉を用いて、空気雰囲気下、200℃で5分間保持し、200℃から450℃まで2.5℃/分で昇温し、450℃で20分間保持することで前焼成を施した後、空気雰囲気化、580℃で2時間の本焼成を施して触媒を得た(触媒2)。
実施例32と同様にして得られた、触媒2の標準偏差値及びアンモ酸化反応の結果を表4に示す。
その組成がMo12Bi0.39Fe1.60Co4.30Ni3.45Ce0.68Rb0.16Ofとなるよう原料仕込質量を調整して製造した金属酸化物58質量%を42質量%のシリカに担持した触媒を、以下の手順で製造した。
次に、16.6質量%の硝酸394.5gに36.1gの硝酸ビスマス[Bi(NO3)3・5H2O]、141.6gの硝酸鉄[Fe(NO3)3・9H2O]、275.1gの硝酸コバルト[Co(NO3)2・6H2O]、219.9gの硝酸ニッケル[Ni(NO3)2・6H2O]、64.6gの硝酸セリウム[Ce(NO3)3・6H2O]、5.33gの硝酸ルビジウム[RbNO3]を溶解させた。以降、これをB3液と称す。B3液の全量は1137gであった。
このMoBi含有液を、静止型混合器の下流に設けたスラリー攪拌槽に全量貯留し、2液の供給終了から更に40℃で1時間攪拌を続けた。このMoBi含有液は噴霧乾燥前においてpHが1以下であり、スラリー状であった。
次いで、実施例32と同様にこのMoBi含有液を噴霧乾燥し、乾燥粉末を得た。なお、熱風入口温度は230℃、熱風出口温度110℃とした。
次いで、乾燥した触媒前駆体に電気炉を用いて、空気雰囲気下、200℃で5分間保持し、200℃から450℃まで2.5℃/分で昇温し、450℃で20分間保持することで前焼成を施した後、空気雰囲気化、585℃で2時間の本焼成を施して触媒を得た(触媒3)。
実施例32と同様にして得られた、触媒3の標準偏差値及びアンモ酸化反応の結果を表4に示す。
[触媒4の製造]
実施例1と同様にして、Mo含有液(A1液と同じもの。ただし、以降、「A4液」と称す。)とBi含有液(B1液と同じもの。ただし、以降、「B4液」と称す。)を調製した。A4液、B4液の供給全量は実施例32と同様とし、したがって、この場合の質量比率は、実施例32と同様に0.42であった。
A4液に対するB4液の供給比率を、上記の質量比率0.42の0.8倍から1.2倍の間で変動させた以外は、実施例32と同様にして触媒を得た(触媒4)。この範囲の供給比率でA4液とB4液の全部(100質量%)を合流させた。
具体的には次のように行った。上記Y字管にA4液を第一の流路を通じて302(g/min)の流量、B4液を第二の流路を通じて102(g/min)の流量で3分間同時に供給した。この供給比率は、102(g/min)/302(g/min)=0.34であり、上記の質量比率0.42の0.8倍であった。
次に、A4液、B4液の供給流量を変更し、A4液を284(g/min)の流量、B4液を120(g/min)の流量で4分間同時に供給した。このときの供給比率は、120(g/min)/284(g/min)=0.42であり、上記の質量比率0.42の1.0倍であった。
さらに、A4液、B4液の供給流量を変更し、A4液を268(g/min)の流量、B4液を136(g/min)の流量で3分間同時に供給した。このときの供給比率は、136(g/min)/268(g/min)=0.51であり、上記の質量比率0.42の1.2倍であった。
なお、いずれの段階においても、A4液とB4液の合流温度は40℃であった。生成したMoBi含有液は噴霧乾燥前においてpHが1以下であり、スラリー状であった。
実施例32と同様にして得られた、触媒4の標準偏差値及びアンモ酸化反応の結果を表4に示す。
[触媒5の製造]
実施例32と同様にして、Mo含有液(A1液と同じもの。ただし、以降、「A5液」と称す。)とBi含有液(B1液と同じもの。ただし、以降、「B5液」と称す。)を調製した。A5液、B5液の供給全量は実施例32と同様とし、したがって、この場合の質量比率は、実施例32と同様に0.42であった。
A5液に対するB5液の供給比率を、上記の質量比率0.42の0.5倍から1.5倍の間で変動させた以外は、実施例32と同様にして触媒を得た(触媒5)。この範囲の供給比率でA5液とB5液の全部(100質量%)を接触させた。
具体的には次のように行った。上記Y字管にA5液を334(g/min)、B5液を70(g/min)で3分間同時に供給した。この供給比率は、70(g/min)/334(g/min)=0.21であり、上記の質量比率0.42の0.5倍であった。
次にA5液、B5液の供給流量を変更し、A5液を284(g/min)、B5液を120(g/min)で5分間同時に供給した。この供給比率は、120(g/min)/284(g/min)=0.42であり、上記の質量比率0.42の1.0倍であった。
さらに、A5液、B5液の供給流量を変更し、A5液を248(g/min)、B5液を156(g/min)で2分間同時に供給した。この供給比率は、156(g/min)/248(g/min)=0.63であり、上記の質量比率0.42の1.5倍であった。 なお、いずれの段階においても、A5液とB5液の合流温度は40℃であった。生成したMoBi含有液は噴霧乾燥前においてpHが1以下であり、液状であった。
実施例32と同様にして得られた、触媒5の標準偏差値及びアンモ酸化反応の結果を表4に示す。
[触媒6の製造]
Mo含有液とBi含有液とを上記Y字管および静止型混合器で混合した後、スラリー攪拌槽にいったん貯留せずスプレードライヤーに直接送液した以外は、実施例32と同様にして触媒を得た(触媒6)。なお、合流温度は40℃であった。生成したMoBi含有液は噴霧乾燥前においてpHが1以下であり、スラリー状であった。
実施例32と同様にして得られた、触媒6の標準偏差値及びアンモ酸化反応の結果を表4に示す。
[触媒7の製造]
実施例32と同様にして、Mo含有液(A1液と同じもの。ただし、以降、「A7液」と称す。)とBi含有液(B1液と同じもの。ただし、以降、「B7液」と称す。)を調製した。この場合の質量比率は、実施例32と同様に0.42であった。
A7液に対するB7液の供給比率を、Mo含有液とBi含有液の全供給量の合計の60質量%に当たる量については、上記の質量比率0.36の0.5倍から1.5倍の範囲内となるように行った。残りの40質量%については0.5倍から1.5倍の範囲外となるように行った。それ以外は実施例32と同様にして触媒を得た(触媒7)。
上記Y字管に、A7液を334(g/min)の流量、B7液を70(g/min)の流量で配管を通じて同時に供給を開始し、2分間供給を続けた。このときの供給比率は、70(g/min)/334(g/min)=0.21であり、上記の質量比率0.42の0.5倍であった。
次に、A7液、B7液の供給流量を変更し、A7液を229(g/min)の流量、B7液を192(g/min)の流量で2分間同時に供給した。このときの供給比率は、192(g/min)/229(g/min)=0.84であり、上記の質量比率0.42の2.0倍であった。
さらに、A7液、B7液の供給流量を変更し、A7液を284(g/min)の流量、B7液を119(g/min)の流量で2分間同時に供給した。このときの供給比率は、119(g/min)/284(g/min)=0.42であり、上記の質量比率0.42の1.0倍であった。
続いて、A7液、B7液の供給流量を変更し、A7液を356(g/min)の流量、B7液を45(g/min)の流量で2分間同時に供給した。このときの供給比率は、45(g/min)/356(g/min)=0.13であり、上記の質量比率0.42の0.3倍であった。
最後に、A7液、B7液の供給流量を変更し、A7液を248(g/min)の流量、B7液を156(g/min)の流量で2分間同時に供給した。このときの供給比率は、156(g/min)/248(g/min)=0.63であり、上記の質量比率0.42の1.5倍であった。
なお、いずれの段階においてもA7液とB7液の合流温度は40℃であった。
実施例32と同様にして得られた、触媒7の標準偏差値及びアンモ酸化反応の結果を表4に示す。
[触媒8の製造]
実施例32と同様にしてMo含有液(A1液と同じもの。ただし、以降、「A8液」と称す。)とBi含有液(B1液と同じもの。ただし、以降、「B8液」と称す。)を調製した。
40℃に保持したA8液に、攪拌を行いながら、40℃に保持したB8液を投入しMoBi含有液を調製した。投入には定量送液ポンプを用いた。
具体的には、2843gのA8液全量に、1198gのB8液を599(g/min)の流量で供給した。生成したMoBi含有液をさらに40℃で1時間攪拌した。このMoBi含有液は噴霧乾燥前においてpHが1以下であり、スラリー状であった。
以降は実施例32と同様にして触媒を得た(触媒8)。
実施例32と同様にして得られた、触媒8の標準偏差値及びアンモ酸化反応の結果を表4に示す。
[触媒9の製造]
実施例32と同様にしてMo含有液(A1液と同じもの。ただし、以降、「A9液」と称す。)とBi含有液(B1液と同じもの。ただし、以降、「B9液」と称す。)を調製した。
40℃に保持したB9液に、攪拌を行いながら、40℃に保持したA9液を投入しMoBi含有液を調製した。投入には定量送液ポンプを用いた。
具体的には、1198gのB9液全量に、2843gのA9液を1422(g/min)の流量で供給した。生成したMoBi含有液をさらに40℃で1時間攪拌した。このMoBi含有液は噴霧乾燥前においてpHが1以下であり、スラリー状であった。
以降は実施例32と同様にして、触媒を得た(触媒9)。
実施例32と同様にして得られた、触媒9の標準偏差値及びアンモ酸化反応の結果を表4に示す。
反応によって生成するアクリロニトリル収率は、前述した評価方法に基づいて算出した。
実施例及び参考例における触媒の粒子表面の組成の均一性の評価は次のようにして行い、粒子表面組成の標準偏差として表した。
SEM-EDX(Scanning Electron Microscopes-Energy Dispersive X-ray Spectroscopy:エネルギー分散型X線分光法)組成分析装置(SEM…日立製作所製、型式:SU-70、EDX…堀場製作所製、型式:EMAX・X-max)の試料台に、金属酸化物粒子(焼成粒子)を少量採取し、無作為に選択した粒子画像における真円相当径(面積が等しい真円の直径)が、レーザー回折/散乱式粒度分布測定装置(堀場製作所製レーザー回折/散乱式粒度分布測定装置LA-300)で決定された平均粒子径±10μmである粒子100個について、それぞれの粒子表面の10μm×10μmの面範囲のMoモル濃度とBiモル濃度とを測定し(倍率1500倍、加速電圧20kV)、粒子表面のMo濃度に対するBi濃度の比率(Bi/Mo)を求めた。
次に、同じ触媒粉末から20mgを量り取り、これを210℃の熱王水で溶解し、さらに超純水で測定範囲に薄め、ICP発光分光分析(セイコーインスツル株式会社製、SPS3500DD)で、各構成元素の濃度を測定し、触媒に含まれる金属酸化物のバルク組成を算出すると共に、金属酸化物のMo濃度に対するBi濃度の比率(Bi/Mo)bulkを求めた。なお、後述の触媒1~9おいては、金属酸化物のバルク組成は、いずれも、原料仕込質量から計算される理論値と一致していた(Mo12基準で表記したモル組成において、上位2桁まで仕込質量から計算されるモル組成と同一であった)。
この(Bi/Mo)bulkで前述の100個それぞれの粒子の(Bi/Mo)surfを除した値をSk(k=1~100)とし、100個のSkの平均値μを算出し、式(4)から標準偏差を求めた。
標準偏差={((S1-μ)2+(S2-μ)2+・・・+(S100-μ)2)/100[個数]}1/2 (4)
なお、実施例32、35~38、参考例1~2で得られた触媒に含まれる金属酸化物のバルク組成は同一であった。


一方、Mo含有液(全量)に対してBi含有液を投入した参考例1、及び、Bi含有液(全量)に対してMo含有液を投入した参考例2においては、実施例32~38に比べて、得られた触媒粒子表面におけるMoとBiの組成比の均一性が低かった。また、参考例1及び2において得られた触媒を用いた場合のアクリロニトリルの収率は、実施例32~38よりも低かった。
以上より、触媒粒子の表面組成が均一である(すなわち、標準偏差が小さい)ほど、プロピレンのアンモ酸化反応によりアクリロニトリルを製造する反応においてより優れたアクリロニトリル収率(触媒性能)を示すことが確認された。
Claims (12)
- モリブデンと、ビスマスと、鉄と、シリカと、カルボン酸とを含む前駆体スラリーを調製する調製工程と、
前記前駆体スラリーを噴霧乾燥して乾燥粒子を得る乾燥工程と、
前記乾燥粒子を焼成する焼成工程と、
を有し、
前記調製工程が、シリカ原料とカルボン酸を混合し、シリカ-カルボン酸混合液を調製する工程(I)と、前記シリカ-カルボン酸混合液、モリブデン、ビスマス及び鉄を混合する工程(II)とを含む、触媒の製造方法。 - 前記触媒が、下記一般式(1)で表されるバルク組成を有する金属酸化物と、シリカとを含む、請求項1に記載の触媒の製造方法。
Mo12BiaFebXcYdZeOf (1)
(式(1)中、Xは、ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム及びバリウムからなる群より選ばれる1種以上の元素を示し、Yは、セリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム及びインジウムからなる群より選ばれる1種以上の元素を示し、Zは、カリウム、ルビジウム及びセシウムからなる群より選ばれる1種以上の元素を示し、a、b、c、d、e及びfは、各元素の原子比を表し、それぞれ、0.1≦a≦3.0、0.1≦b≦3.0、0.1≦c≦10.0、0.1≦d≦3.0、及び0.01≦e≦2.0を満たし、かつ、fは存在する他の元素の原子価要求を満足させるのに必要な酸素の原子数である。) - 前記触媒において、触媒粒子表面におけるMoモル濃度に対するBiモル濃度の比率を、金属酸化物バルクのMoモル濃度に対するBiモル濃度の比率で除した値の標準偏差が0.2以下である、請求項2に記載の触媒の製造方法。
- 前記工程(II)が、下記(i)工程及び(ii)工程を含み、
前記乾燥工程が、下記(iii)工程を含む、請求項1~3のいずれか一項に記載の触媒の製造方法;
(i)少なくともMoを含むMo含有液と、少なくともBiを含むBi含有液とを用意する工程、
(ii)前記Mo含有液を第一の流路に連続的に供給し、前記Bi含有液を第二の流路に連続的に供給し、かつ第一の流路と第二の流路とを前記Mo含有液及び前記Bi含有液それぞれの供給個所よりも下流で合流させることによって、前記Mo含有液と前記Bi含有液とを混合してMoBi含有液を得る工程、
(iii)前記MoBi含有液を乾燥させる工程。 - 前記(ii)工程と前記(iii)工程との間に、さらに、(iv)前記MoBi含有液をさらに混合する工程を有する、請求項4に記載の触媒の製造方法。
- 前記(ii)工程と前記(iii)工程との間に、さらに、(v)前記MoBi含有液を貯留する工程を有する、請求項4又は5に記載の触媒の製造方法。
- 前記(i)、(ii)及び(v)工程の一連の処理がバッチ処理方式で行われ、(ii)工程で得られたMoBi含有液の一バッチ分全量が(v)工程において貯留され、
前記(ii)工程において、前記Mo含有液及び前記Bi含有液の前記第一の流路又は前記第二の流路への質量供給速度を、各々、mA(g/min)及びmB(g/min)としたときに、前記Mo含有液及び前記Bi含有液の一バッチあたりの全供給量MA(g)及びMB(g)の合計量の60質量%以上が、式(2)を満たすように供給される、請求項6に記載の触媒の製造方法。
(mB/mA)/(MB/MA)=0.5~1.5 (2) - 前記MoBi含有液が、前記(iii)工程へ連続的に供される、請求項4又は5に記載の触媒の製造方法。
- 前記(ii)工程において、前記Mo含有液及び前記Bi含有液の前記第一の流路又は第二の流路へモル供給速度を、各々、mα(mol/min)及びmβ(mol/min)としたときに、前記Mo含有液及び前記Bi含有液が、式(3)を満たすように供給される、請求項4、5、又は8に記載の触媒の製造方法。
(mβ/mα)/(a/12)=0.8~1.2 (3) - 請求項1~9のいずれか一項に記載の触媒の製造方法によって触媒を得る工程と、
前記触媒の存在下、プロピレンと、分子状酸素と、アンモニアと、を反応させる工程と、
を含む、アクリロニトリルの製造方法。 - 下記式(1)で表されるバルク組成を有する金属酸化物及びシリカを含む粒子からなる触媒であって、
触媒粒子表面におけるMoモル濃度に対するBiモル濃度の比率を、金属酸化物バルクのMoモル濃度に対するBiモル濃度の比率で除した値の標準偏差が0.2以下である、
触媒。
Mo12BiaFebXcYdZeOf (1)
(式(1)中、Xは、ニッケル、コバルト、マグネシウム、カルシウム、亜鉛、ストロンチウム及びバリウムからなる群より選ばれる1種以上の元素を示し、Yは、セリウム、クロム、ランタン、ネオジム、イットリウム、プラセオジム、サマリウム、アルミニウム、ガリウム及びインジウムからなる群より選ばれる1種以上の元素を示し、Zは、カリウム、ルビジウム及びセシウムからなる群より選ばれる1種以上の元素を示し、a、b、c、d、e及びfは、各元素の原子比を表し、それぞれ、0.1≦a≦3.0、0.1≦b≦3.0、0.1≦c≦10.0、0.1≦d≦3.0、及び0.01≦e≦2.0を満たし、かつ、fは原子価のバランスを満たす値である。) - 請求項11に記載の触媒の存在下、プロピレンと、分子状酸素と、アンモニアと、を反応させる工程を含む、アクリロニトリルの製造方法。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17846016.8A EP3508272A4 (en) | 2016-08-31 | 2017-08-01 | PROCESS FOR PRODUCTION OF CATALYST AND PROCESS FOR PRODUCTION OF ACRYLONITRILE |
| CN201780023820.4A CN109070070B (zh) | 2016-08-31 | 2017-08-01 | 催化剂的制造方法及丙烯腈的制造方法 |
| RU2019105288A RU2709012C1 (ru) | 2016-08-31 | 2017-08-01 | Способ получения катализатора и способ получения акрилонитрила |
| JP2018537053A JP6467115B2 (ja) | 2016-08-31 | 2017-08-01 | 触媒の製造方法、及びアクリロニトリルの製造方法 |
| MYPI2019000723A MY190157A (en) | 2016-08-31 | 2017-08-01 | Method for producing catalyst and method for producing acrylonitrile |
| US16/320,428 US10328418B1 (en) | 2016-08-31 | 2017-08-01 | Method for producing catalyst and method for producing acrylonitrile |
| KR1020187029196A KR102029613B1 (ko) | 2016-08-31 | 2017-08-01 | 촉매의 제조 방법 및 아크릴로니트릴의 제조 방법 |
| SA519401140A SA519401140B1 (ar) | 2016-08-31 | 2019-02-20 | طريقة لإنتاج حفاز وطريقة لإنتاج أكريلونتريل |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-169708 | 2016-08-31 | ||
| JP2016169708 | 2016-08-31 | ||
| JP2017-123320 | 2017-06-23 | ||
| JP2017123320A JP6914114B2 (ja) | 2017-06-23 | 2017-06-23 | 金属酸化物触媒及びその製造方法ならびにそれを用いたアクリロニトリルの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018043007A1 true WO2018043007A1 (ja) | 2018-03-08 |
Family
ID=61301498
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/027950 WO2018043007A1 (ja) | 2016-08-31 | 2017-08-01 | 触媒の製造方法、及びアクリロニトリルの製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10328418B1 (ja) |
| EP (1) | EP3508272A4 (ja) |
| KR (1) | KR102029613B1 (ja) |
| CN (1) | CN109070070B (ja) |
| MY (1) | MY190157A (ja) |
| RU (1) | RU2709012C1 (ja) |
| SA (1) | SA519401140B1 (ja) |
| TW (1) | TWI655968B (ja) |
| WO (1) | WO2018043007A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019172592A (ja) * | 2018-03-27 | 2019-10-10 | 旭化成株式会社 | アクリロニトリルの製造方法 |
| CN110612158A (zh) * | 2018-04-13 | 2019-12-24 | 旭化成株式会社 | 催化剂、催化剂的制造方法、丙烯腈的制造方法 |
| CN111744497A (zh) * | 2019-03-28 | 2020-10-09 | 中国石油化工股份有限公司 | 一种氨氧化催化剂颗粒、其制造方法及其应用 |
| RU2761968C1 (ru) * | 2018-08-23 | 2021-12-14 | Асахи Касеи Кабусики Кайся | Способ производства катализатора для аммоксидирования и способ производства акрилонитрила |
| WO2023163067A1 (ja) * | 2022-02-25 | 2023-08-31 | 旭化成株式会社 | 触媒及びその製造方法、並びにアクリロニトリルの製造方法 |
| US11772080B2 (en) | 2019-04-15 | 2023-10-03 | Asahi Kasei Kabushiki Kaisha | Catalyst, method for producing catalyst, and method for producing acrylonitrile |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020039985A1 (ja) * | 2018-08-24 | 2020-02-27 | 旭化成株式会社 | アンモ酸化用触媒の製造方法、及び、アクリロニトリルの製造方法 |
| CN109772356B (zh) * | 2019-03-07 | 2020-06-02 | 营口市向阳催化剂有限责任公司 | 一种丙烯腈催化剂及其制备方法和应用 |
| CN114829001A (zh) * | 2020-07-29 | 2022-07-29 | 株式会社Lg化学 | 用于丙烯的氨氧化催化剂、该催化剂的制造方法、使用该催化剂的氨氧化方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62144752A (ja) * | 1985-11-27 | 1987-06-27 | イ−・アイ・デユポン・ドウ・ヌム−ル・アンド・カンパニ− | 摩擦抵抗触媒、触媒前駆物質、および触媒支持体 |
| JPH04118051A (ja) * | 1990-09-10 | 1992-04-20 | Nitto Chem Ind Co Ltd | 鉄・アンチモン・モリブデン含有酸化物触媒組成物およびその製法 |
| JP2010519202A (ja) * | 2007-02-16 | 2010-06-03 | イネオス ユーエスエイ リミテッド ライアビリティ カンパニー | 混合金属酸化物触媒を用いたプロパン及びイソブタンのアンモ酸化方法 |
| JP2010172851A (ja) * | 2009-01-30 | 2010-08-12 | Asahi Kasei Chemicals Corp | アクリロニトリルの製造用触媒及びアクリロニトリルの製造方法 |
| JP2011518659A (ja) * | 2008-04-09 | 2011-06-30 | ビーエーエスエフ ソシエタス・ヨーロピア | モリブデン、ビスマスおよび鉄を含有する多金属酸化物を含むシェル触媒 |
| JP2012077039A (ja) * | 2010-10-04 | 2012-04-19 | Asahi Kasei Chemicals Corp | 不飽和酸又は不飽和ニトリルの製造方法 |
| JP2016120468A (ja) * | 2014-12-25 | 2016-07-07 | 旭化成ケミカルズ株式会社 | アンモ酸化用触媒及びその製造方法、並びに、アクリロニトリルの製造方法 |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4766232A (en) * | 1973-06-04 | 1988-08-23 | The Standard Oil Company | Production of unsaturated nitriles using catalysts containing boron, gallium or indium |
| JPS5245692B2 (ja) * | 1973-10-16 | 1977-11-17 | ||
| US4212766A (en) * | 1978-04-21 | 1980-07-15 | Standard Oil Company | Process for forming multi-component oxide complex catalysts |
| US5093299A (en) * | 1990-01-09 | 1992-03-03 | The Standard Oil Company | Catalyst for process for the manufacture of acrylonitrile and methacrylonitrile |
| JP3680115B2 (ja) | 1998-04-23 | 2005-08-10 | ダイヤニトリックス株式会社 | 不飽和ニトリル製造用触媒組成物 |
| JP3720625B2 (ja) | 1998-05-21 | 2005-11-30 | ダイヤニトリックス株式会社 | モリブデン−ビスマス−鉄含有複合酸化物触媒の調製法 |
| KR100513664B1 (ko) * | 2002-05-16 | 2005-09-07 | 주식회사 엘지화학 | 프로필렌의 부분 산화 반응용 촉매의 제조 방법 |
| US7071140B2 (en) | 2002-12-02 | 2006-07-04 | The Standard Oil Company | Catalyst for the manufacture of acrylonitrile |
| US7348291B2 (en) * | 2002-12-02 | 2008-03-25 | Ineos Usa Llc | Catalyst for the manufacture of acrylonitrile |
| JP4242197B2 (ja) * | 2003-04-18 | 2009-03-18 | ダイヤニトリックス株式会社 | アクリロニトリル合成用触媒 |
| JP5188005B2 (ja) | 2004-08-30 | 2013-04-24 | 旭化成ケミカルズ株式会社 | 金属酸化物触媒、及びその触媒の製造方法、並びにニトリルの製造方法 |
| JP4588533B2 (ja) | 2005-05-24 | 2010-12-01 | ダイヤニトリックス株式会社 | アクリロニトリル合成用触媒 |
| US7531681B2 (en) | 2007-02-16 | 2009-05-12 | Ineos Usa Llc | Process for the ammoxidation of propane and isobutane |
| JP4954750B2 (ja) | 2007-02-28 | 2012-06-20 | ダイヤニトリックス株式会社 | モリブデン、ビスマス、鉄、シリカ含有複合酸化物触媒の製造方法 |
| US9018128B2 (en) | 2007-09-14 | 2015-04-28 | Res Usa Llc | Promoted, attrition resistant, silica supported precipitated iron catalyst |
| JP5011167B2 (ja) | 2008-03-03 | 2012-08-29 | ダイヤニトリックス株式会社 | アクリロニトリル製造用触媒およびアクリロニトリルの製造方法 |
| JP5371692B2 (ja) | 2008-10-24 | 2013-12-18 | 旭化成ケミカルズ株式会社 | 共役ジオレフィンの製造方法 |
| US8258073B2 (en) * | 2010-03-23 | 2012-09-04 | Ineos Usa Llc | Process for preparing improved mixed metal oxide ammoxidation catalysts |
| JP6118016B2 (ja) | 2011-07-07 | 2017-04-19 | 旭化成株式会社 | アンモ酸化用触媒及びこれを用いたアクリロニトリルの製造方法 |
| US9633407B2 (en) | 2011-07-29 | 2017-04-25 | Intel Corporation | CPU/GPU synchronization mechanism |
| JP5895546B2 (ja) | 2012-01-17 | 2016-03-30 | 三菱化学株式会社 | 複合酸化物触媒及び共役ジエンの製造方法 |
| JP5919870B2 (ja) | 2012-02-17 | 2016-05-18 | 三菱レイヨン株式会社 | アクリロニトリル製造用触媒の製造方法および該アクリロニトリル製造用触媒を用いたアクリロニトリルの製造方法 |
| JP6722284B2 (ja) * | 2016-06-14 | 2020-07-15 | 旭化成株式会社 | アンモ酸化用触媒の製造方法、及びアクリロニトリルの製造方法 |
-
2017
- 2017-08-01 RU RU2019105288A patent/RU2709012C1/ru active
- 2017-08-01 CN CN201780023820.4A patent/CN109070070B/zh active Active
- 2017-08-01 US US16/320,428 patent/US10328418B1/en active Active
- 2017-08-01 MY MYPI2019000723A patent/MY190157A/en unknown
- 2017-08-01 KR KR1020187029196A patent/KR102029613B1/ko active IP Right Grant
- 2017-08-01 WO PCT/JP2017/027950 patent/WO2018043007A1/ja active Application Filing
- 2017-08-01 EP EP17846016.8A patent/EP3508272A4/en active Pending
- 2017-08-02 TW TW106126026A patent/TWI655968B/zh active
-
2019
- 2019-02-20 SA SA519401140A patent/SA519401140B1/ar unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62144752A (ja) * | 1985-11-27 | 1987-06-27 | イ−・アイ・デユポン・ドウ・ヌム−ル・アンド・カンパニ− | 摩擦抵抗触媒、触媒前駆物質、および触媒支持体 |
| JPH04118051A (ja) * | 1990-09-10 | 1992-04-20 | Nitto Chem Ind Co Ltd | 鉄・アンチモン・モリブデン含有酸化物触媒組成物およびその製法 |
| JP2010519202A (ja) * | 2007-02-16 | 2010-06-03 | イネオス ユーエスエイ リミテッド ライアビリティ カンパニー | 混合金属酸化物触媒を用いたプロパン及びイソブタンのアンモ酸化方法 |
| JP2011518659A (ja) * | 2008-04-09 | 2011-06-30 | ビーエーエスエフ ソシエタス・ヨーロピア | モリブデン、ビスマスおよび鉄を含有する多金属酸化物を含むシェル触媒 |
| JP2010172851A (ja) * | 2009-01-30 | 2010-08-12 | Asahi Kasei Chemicals Corp | アクリロニトリルの製造用触媒及びアクリロニトリルの製造方法 |
| JP2012077039A (ja) * | 2010-10-04 | 2012-04-19 | Asahi Kasei Chemicals Corp | 不飽和酸又は不飽和ニトリルの製造方法 |
| JP2016120468A (ja) * | 2014-12-25 | 2016-07-07 | 旭化成ケミカルズ株式会社 | アンモ酸化用触媒及びその製造方法、並びに、アクリロニトリルの製造方法 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019172592A (ja) * | 2018-03-27 | 2019-10-10 | 旭化成株式会社 | アクリロニトリルの製造方法 |
| JP7166770B2 (ja) | 2018-03-27 | 2022-11-08 | 旭化成株式会社 | アクリロニトリルの製造方法 |
| CN110612158A (zh) * | 2018-04-13 | 2019-12-24 | 旭化成株式会社 | 催化剂、催化剂的制造方法、丙烯腈的制造方法 |
| EP3778013A4 (en) * | 2018-04-13 | 2021-06-23 | Asahi Kasei Kabushiki Kaisha | CATALYST, CATALYST PRODUCTION PROCESS, AND ACRYLONITRILE PRODUCTION PROCESS |
| CN110612158B (zh) * | 2018-04-13 | 2023-09-01 | 旭化成株式会社 | 催化剂、催化剂的制造方法、丙烯腈的制造方法 |
| RU2761968C1 (ru) * | 2018-08-23 | 2021-12-14 | Асахи Касеи Кабусики Кайся | Способ производства катализатора для аммоксидирования и способ производства акрилонитрила |
| US11701648B2 (en) | 2018-08-23 | 2023-07-18 | Asahi Kasei Kabushiki Kaisha | Method for producing catalyst for ammoxidation, and method for producing acrylonitrile |
| CN111744497A (zh) * | 2019-03-28 | 2020-10-09 | 中国石油化工股份有限公司 | 一种氨氧化催化剂颗粒、其制造方法及其应用 |
| CN111744497B (zh) * | 2019-03-28 | 2023-07-07 | 中国石油化工股份有限公司 | 一种氨氧化催化剂颗粒、其制造方法及其应用 |
| US11772080B2 (en) | 2019-04-15 | 2023-10-03 | Asahi Kasei Kabushiki Kaisha | Catalyst, method for producing catalyst, and method for producing acrylonitrile |
| WO2023163067A1 (ja) * | 2022-02-25 | 2023-08-31 | 旭化成株式会社 | 触媒及びその製造方法、並びにアクリロニトリルの製造方法 |
| JP7385086B1 (ja) | 2022-02-25 | 2023-11-21 | 旭化成株式会社 | 触媒及びその製造方法、並びにアクリロニトリルの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102029613B1 (ko) | 2019-10-08 |
| TWI655968B (zh) | 2019-04-11 |
| CN109070070B (zh) | 2020-09-15 |
| TW201815473A (zh) | 2018-05-01 |
| RU2709012C1 (ru) | 2019-12-13 |
| US10328418B1 (en) | 2019-06-25 |
| MY190157A (en) | 2022-03-31 |
| US20190168191A1 (en) | 2019-06-06 |
| KR20190022452A (ko) | 2019-03-06 |
| SA519401140B1 (ar) | 2022-07-25 |
| EP3508272A1 (en) | 2019-07-10 |
| CN109070070A (zh) | 2018-12-21 |
| EP3508272A4 (en) | 2019-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2018043007A1 (ja) | 触媒の製造方法、及びアクリロニトリルの製造方法 | |
| JP6722284B2 (ja) | アンモ酸化用触媒の製造方法、及びアクリロニトリルの製造方法 | |
| JP5832785B2 (ja) | アンモ酸化用触媒、その製造方法及びアクリロニトリル又はメタクリロニトリルの製造方法 | |
| JP2016120468A (ja) | アンモ酸化用触媒及びその製造方法、並びに、アクリロニトリルの製造方法 | |
| JP6467115B2 (ja) | 触媒の製造方法、及びアクリロニトリルの製造方法 | |
| EP3626338A1 (en) | Catalyst for ammoxidation, method for producing same and method for producing acrylonitrile | |
| JP6328864B2 (ja) | アンモ酸化用触媒の製造方法、及び、アクリロニトリルの製造方法 | |
| JP6914114B2 (ja) | 金属酸化物触媒及びその製造方法ならびにそれを用いたアクリロニトリルの製造方法 | |
| US10940463B2 (en) | Catalyst, method for producing catalyst, and method for producing acrylonitrile | |
| JP5409100B2 (ja) | アクリロニトリル製造用触媒及びその製造方法 | |
| EP3842144A1 (en) | Method of producing ammoxidation catalyst and method of producing acrylonitrile | |
| JP7101310B2 (ja) | 触媒、触媒の製造方法、アクリロニトリルの製造方法 | |
| JP2018187579A (ja) | 触媒の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018537053 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 20187029196 Country of ref document: KR Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17846016 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2017846016 Country of ref document: EP Effective date: 20190401 |