WO2017188042A1 - 一本鎖核酸分子用モノマーの製造方法 - Google Patents
一本鎖核酸分子用モノマーの製造方法 Download PDFInfo
- Publication number
- WO2017188042A1 WO2017188042A1 PCT/JP2017/015435 JP2017015435W WO2017188042A1 WO 2017188042 A1 WO2017188042 A1 WO 2017188042A1 JP 2017015435 W JP2017015435 W JP 2017015435W WO 2017188042 A1 WO2017188042 A1 WO 2017188042A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- solvent
- formula
- enantiomer
- alkali metal
- Prior art date
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 19
- 108020004707 nucleic acids Proteins 0.000 title abstract description 11
- 150000007523 nucleic acids Chemical class 0.000 title abstract description 11
- 102000039446 nucleic acids Human genes 0.000 title abstract description 11
- 239000000178 monomer Substances 0.000 title abstract description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 86
- 239000002904 solvent Substances 0.000 claims abstract description 63
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims abstract description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 24
- 239000012190 activator Substances 0.000 claims abstract description 23
- 230000008878 coupling Effects 0.000 claims abstract description 23
- 238000010168 coupling process Methods 0.000 claims abstract description 23
- 238000005859 coupling reaction Methods 0.000 claims abstract description 23
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 claims abstract description 15
- 239000002585 base Substances 0.000 claims abstract description 13
- RKVHNYJPIXOHRW-UHFFFAOYSA-N 3-bis[di(propan-2-yl)amino]phosphanyloxypropanenitrile Chemical compound CC(C)N(C(C)C)P(N(C(C)C)C(C)C)OCCC#N RKVHNYJPIXOHRW-UHFFFAOYSA-N 0.000 claims abstract description 10
- IWHLYPDWHHPVAA-UHFFFAOYSA-N 6-hydroxyhexanoic acid Chemical compound OCCCCCC(O)=O IWHLYPDWHHPVAA-UHFFFAOYSA-N 0.000 claims abstract description 10
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 10
- 239000000654 additive Substances 0.000 claims abstract description 7
- 230000000996 additive effect Effects 0.000 claims abstract description 7
- 239000011541 reaction mixture Substances 0.000 claims abstract description 4
- -1 bicyclic amidine compound Chemical class 0.000 claims description 19
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 claims description 15
- PFYXSUNOLOJMDX-UHFFFAOYSA-N bis(2,5-dioxopyrrolidin-1-yl) carbonate Chemical compound O=C1CCC(=O)N1OC(=O)ON1C(=O)CCC1=O PFYXSUNOLOJMDX-UHFFFAOYSA-N 0.000 claims description 15
- 238000000034 method Methods 0.000 claims description 11
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical group Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 claims description 9
- CIXHNHBHWVDBGQ-UHFFFAOYSA-N n-propan-2-ylpropan-2-amine;2h-tetrazole Chemical group C1=NN=NN1.CC(C)NC(C)C CIXHNHBHWVDBGQ-UHFFFAOYSA-N 0.000 claims description 9
- 229910052757 nitrogen Inorganic materials 0.000 claims description 9
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 claims description 7
- 229910052783 alkali metal Inorganic materials 0.000 claims description 6
- 229910000288 alkali metal carbonate Inorganic materials 0.000 claims description 6
- 150000008041 alkali metal carbonates Chemical class 0.000 claims description 6
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 claims description 5
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Substances C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 claims description 5
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 claims description 5
- 125000001453 quaternary ammonium group Chemical group 0.000 claims description 5
- GXGKKIPUFAHZIZ-UHFFFAOYSA-N 5-benzylsulfanyl-2h-tetrazole Chemical compound C=1C=CC=CC=1CSC=1N=NNN=1 GXGKKIPUFAHZIZ-UHFFFAOYSA-N 0.000 claims description 4
- LCFXLZAXGXOXAP-DAXSKMNVSA-N ethyl (2z)-2-cyano-2-hydroxyiminoacetate Chemical compound CCOC(=O)C(=N/O)\C#N LCFXLZAXGXOXAP-DAXSKMNVSA-N 0.000 claims description 4
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 claims description 3
- XGDRLCRGKUCBQL-UHFFFAOYSA-N 1h-imidazole-4,5-dicarbonitrile Chemical compound N#CC=1N=CNC=1C#N XGDRLCRGKUCBQL-UHFFFAOYSA-N 0.000 claims description 3
- GPIQOFWTZXXOOV-UHFFFAOYSA-N 2-chloro-4,6-dimethoxy-1,3,5-triazine Chemical compound COC1=NC(Cl)=NC(OC)=N1 GPIQOFWTZXXOOV-UHFFFAOYSA-N 0.000 claims description 3
- HJBLUNHMOKFZQX-UHFFFAOYSA-N 3-hydroxy-1,2,3-benzotriazin-4-one Chemical compound C1=CC=C2C(=O)N(O)N=NC2=C1 HJBLUNHMOKFZQX-UHFFFAOYSA-N 0.000 claims description 3
- GONFBOIJNUKKST-UHFFFAOYSA-N 5-ethylsulfanyl-2h-tetrazole Chemical compound CCSC=1N=NNN=1 GONFBOIJNUKKST-UHFFFAOYSA-N 0.000 claims description 3
- 239000000908 ammonium hydroxide Substances 0.000 claims description 3
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 claims description 3
- LKPFBGKZCCBZDK-UHFFFAOYSA-N n-hydroxypiperidine Chemical compound ON1CCCCC1 LKPFBGKZCCBZDK-UHFFFAOYSA-N 0.000 claims description 3
- PAQZWJGSJMLPMG-UHFFFAOYSA-N propylphosphonic anhydride Substances CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 claims description 3
- 239000013638 trimer Substances 0.000 claims description 3
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 claims description 2
- 150000003949 imides Chemical class 0.000 claims 1
- 238000002156 mixing Methods 0.000 abstract description 4
- 108090000623 proteins and genes Proteins 0.000 abstract description 4
- 150000003839 salts Chemical class 0.000 abstract description 2
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 57
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 57
- 239000000203 mixture Substances 0.000 description 49
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 48
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 32
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 27
- 239000000243 solution Substances 0.000 description 27
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 24
- 239000012044 organic layer Substances 0.000 description 21
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 16
- 235000017557 sodium bicarbonate Nutrition 0.000 description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- 239000012299 nitrogen atmosphere Substances 0.000 description 15
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 13
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000009835 boiling Methods 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical class C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- 230000018044 dehydration Effects 0.000 description 5
- 238000006297 dehydration reaction Methods 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- 235000013877 carbamide Nutrition 0.000 description 4
- 150000002170 ethers Chemical class 0.000 description 4
- 150000002825 nitriles Chemical class 0.000 description 4
- 150000003672 ureas Chemical class 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 150000008282 halocarbons Chemical class 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 150000008300 phosphoramidites Chemical class 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 150000003512 tertiary amines Chemical class 0.000 description 3
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- FRXWSJKQUOUMOH-UHFFFAOYSA-N 1-nitrotriazole Chemical compound [O-][N+](=O)N1C=CN=N1 FRXWSJKQUOUMOH-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 2
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 2
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 2
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 238000006482 condensation reaction Methods 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 2
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 2
- LCPDWSOZIOUXRV-UHFFFAOYSA-N phenoxyacetic acid Chemical compound OC(=O)COC1=CC=CC=C1 LCPDWSOZIOUXRV-UHFFFAOYSA-N 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- ZPGDWQNBZYOZTI-SFHVURJKSA-N (2s)-1-(9h-fluoren-9-ylmethoxycarbonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 ZPGDWQNBZYOZTI-SFHVURJKSA-N 0.000 description 1
- 0 *c1ccc(C(c2ccccc2)(c2ccc(*)cc2)OCCCCNC([C@]2NCCC2)=O)cc1 Chemical compound *c1ccc(C(c2ccccc2)(c2ccc(*)cc2)OCCCCNC([C@]2NCCC2)=O)cc1 0.000 description 1
- DMSNKRPEUQVMNF-UHFFFAOYSA-N 1,1-dioxo-1,2-benzothiazol-3-olate 1-methyl-1H-imidazol-1-ium Chemical class CN1C=CN=C1.C1=CC=C2C(=O)NS(=O)(=O)C2=C1 DMSNKRPEUQVMNF-UHFFFAOYSA-N 0.000 description 1
- HSNJERRVXUNQLS-UHFFFAOYSA-N 1-(4-tert-butylphenyl)propan-2-one Chemical compound CC(=O)CC1=CC=C(C(C)(C)C)C=C1 HSNJERRVXUNQLS-UHFFFAOYSA-N 0.000 description 1
- JBWYRBLDOOOJEU-UHFFFAOYSA-N 1-[chloro-(4-methoxyphenyl)-phenylmethyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC(OC)=CC=1)C1=CC=CC=C1 JBWYRBLDOOOJEU-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 1
- UTFVRHWVFZFUNL-UHFFFAOYSA-N 1-hydroxy-4-nitrobenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1[N+]([O-])=O UTFVRHWVFZFUNL-UHFFFAOYSA-N 0.000 description 1
- DGIBHCWBCOAPDN-UHFFFAOYSA-N 1-hydroxy-6-(trifluoromethyl)benzotriazole Chemical compound C1=C(C(F)(F)F)C=C2N(O)N=NC2=C1 DGIBHCWBCOAPDN-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- RXUWFHRBKJDCMV-UHFFFAOYSA-N 1-methylsulfanyltetrazole Chemical compound CSN1C=NN=N1 RXUWFHRBKJDCMV-UHFFFAOYSA-N 0.000 description 1
- HNFVMKAUQPHQCY-UHFFFAOYSA-N 1-phenyl-1h-imidazol-1-ium;2,2,2-trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.C1=NC=C[NH+]1C1=CC=CC=C1 HNFVMKAUQPHQCY-UHFFFAOYSA-N 0.000 description 1
- JLTBOEOEGVJJCU-UHFFFAOYSA-N 1h-benzimidazol-3-ium;2,2,2-trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.C1=CC=C2[NH+]=CNC2=C1 JLTBOEOEGVJJCU-UHFFFAOYSA-N 0.000 description 1
- JAAIPIWKKXCNOC-UHFFFAOYSA-N 1h-tetrazol-1-ium-5-thiolate Chemical compound SC1=NN=NN1 JAAIPIWKKXCNOC-UHFFFAOYSA-N 0.000 description 1
- UFBJCMHMOXMLKC-UHFFFAOYSA-N 2,4-dinitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O UFBJCMHMOXMLKC-UHFFFAOYSA-N 0.000 description 1
- BHAAQCPWCCKULK-UHFFFAOYSA-N 2-benzyl-1h-imidazole-4,5-dicarbonitrile Chemical compound N1C(C#N)=C(C#N)N=C1CC1=CC=CC=C1 BHAAQCPWCCKULK-UHFFFAOYSA-N 0.000 description 1
- WMPWSWMSTJAAPF-UHFFFAOYSA-N 2-bromo-1h-imidazole-4,5-dicarbonitrile Chemical compound BrC1=NC(C#N)=C(C#N)N1 WMPWSWMSTJAAPF-UHFFFAOYSA-N 0.000 description 1
- ZYGBKVIYSKFTGQ-UHFFFAOYSA-N 2-bromo-4,5-diethylimidazole-1-carboxylic acid Chemical compound CCC=1N=C(Br)N(C(O)=O)C=1CC ZYGBKVIYSKFTGQ-UHFFFAOYSA-N 0.000 description 1
- MLIREBYILWEBDM-UHFFFAOYSA-M 2-cyanoacetate Chemical compound [O-]C(=O)CC#N MLIREBYILWEBDM-UHFFFAOYSA-M 0.000 description 1
- QDFXRVAOBHEBGJ-UHFFFAOYSA-N 3-(cyclononen-1-yl)-4,5,6,7,8,9-hexahydro-1h-diazonine Chemical compound C1CCCCCCC=C1C1=NNCCCCCC1 QDFXRVAOBHEBGJ-UHFFFAOYSA-N 0.000 description 1
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- BLFRQYKZFKYQLO-UHFFFAOYSA-N 4-aminobutan-1-ol Chemical compound NCCCCO BLFRQYKZFKYQLO-UHFFFAOYSA-N 0.000 description 1
- MIUOBAHGBPSRKY-UHFFFAOYSA-N 5-(4-nitrophenyl)-2h-tetrazole Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=NNN=N1 MIUOBAHGBPSRKY-UHFFFAOYSA-N 0.000 description 1
- ZIIGSRYPZWDGBT-UHFFFAOYSA-N 610-30-0 Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O ZIIGSRYPZWDGBT-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 1
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 1
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- DJJCXFVJDGTHFX-UHFFFAOYSA-N Uridinemonophosphate Natural products OC1C(O)C(COP(O)(O)=O)OC1N1C(=O)NC(=O)C=C1 DJJCXFVJDGTHFX-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 1
- LNQVTSROQXJCDD-UHFFFAOYSA-N adenosine monophosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(CO)C(OP(O)(O)=O)C1O LNQVTSROQXJCDD-UHFFFAOYSA-N 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 150000001412 amines Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- JHXKRIRFYBPWGE-UHFFFAOYSA-K bismuth chloride Chemical compound Cl[Bi](Cl)Cl JHXKRIRFYBPWGE-UHFFFAOYSA-K 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 1
- IERHLVCPSMICTF-XVFCMESISA-N cytidine 5'-monophosphate Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(O)=O)O1 IERHLVCPSMICTF-XVFCMESISA-N 0.000 description 1
- IERHLVCPSMICTF-UHFFFAOYSA-N cytidine monophosphate Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(COP(O)(O)=O)O1 IERHLVCPSMICTF-UHFFFAOYSA-N 0.000 description 1
- ZLRAAUUPULJGTL-UHFFFAOYSA-N diaminophosphinous acid Chemical compound NP(N)O ZLRAAUUPULJGTL-UHFFFAOYSA-N 0.000 description 1
- 229960005215 dichloroacetic acid Drugs 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- PVBRXXAAPNGWGE-LGVAUZIVSA-L disodium 5'-guanylate Chemical compound [Na+].[Na+].C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP([O-])([O-])=O)[C@@H](O)[C@H]1O PVBRXXAAPNGWGE-LGVAUZIVSA-L 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229940029575 guanosine Drugs 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 1
- ZUSSTQCWRDLYJA-UHFFFAOYSA-N n-hydroxy-5-norbornene-2,3-dicarboximide Chemical compound C1=CC2CC1C1C2C(=O)N(O)C1=O ZUSSTQCWRDLYJA-UHFFFAOYSA-N 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 239000005373 porous glass Substances 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- NRTYMEPCRDJMPZ-UHFFFAOYSA-N pyridine;2,2,2-trifluoroacetic acid Chemical compound C1=CC=NC=C1.OC(=O)C(F)(F)F NRTYMEPCRDJMPZ-UHFFFAOYSA-N 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical class C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- KWGGNXPSFLENPI-UHFFFAOYSA-N toluene;2,2,2-trichloroacetic acid Chemical compound CC1=CC=CC=C1.OC(=O)C(Cl)(Cl)Cl KWGGNXPSFLENPI-UHFFFAOYSA-N 0.000 description 1
- QVOFCQBZXGLNAA-UHFFFAOYSA-M tributyl(methyl)azanium;hydroxide Chemical compound [OH-].CCCC[N+](C)(CCCC)CCCC QVOFCQBZXGLNAA-UHFFFAOYSA-M 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 239000005051 trimethylchlorosilane Substances 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- DJJCXFVJDGTHFX-XVFCMESISA-N uridine 5'-monophosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(O)=O)O[C@H]1N1C(=O)NC(=O)C=C1 DJJCXFVJDGTHFX-XVFCMESISA-N 0.000 description 1
- DUNKXUFBGCUVQW-UHFFFAOYSA-J zirconium tetrachloride Chemical compound Cl[Zr](Cl)(Cl)Cl DUNKXUFBGCUVQW-UHFFFAOYSA-J 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/572—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2330/00—Production
- C12N2330/30—Production chemically synthesised
Definitions
- the present invention relates to a method for producing a monomer used for producing a single-stranded nucleic acid molecule capable of suppressing the expression of a target gene.
- Example A3 has formula (3)
- compound (3) (Hereinafter referred to as compound (3)) and its enantiomer preparation.
- the yield of the single-stranded nucleic acid molecule was not always sufficient when the compound (3) obtained by the method described in US2012 / 0035246 was used.
- the present invention provides a method for producing a compound (3) capable of producing a single-stranded nucleic acid molecule with high yield.
- the base is an alkali metal hydroxide.
- the condensing agent is 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide, N, N′-dicyclohexylcarbodiimide, N, N ′ [1] or [2] which is diisopropylcarbodiimide, 1,1-carbonyldiimidazole, 1-propylphosphonic anhydride cyclic trimer or 2-chloro-4,6-dimethoxy-1,3,5-triazine
- the manufacturing method as described in.
- the additive is 1-hydroxybenzotriazole, 1-hydroxy-7-azabenzotriazole, N-hydroxysuccinimide, ethyl (hydroxyimino) cyanoacetate or N, N′-disuccinimidyl carbonate [1] ] To [3].
- the coupling activator is diisopropylamine tetrazole salt, 1H-tetrazole, 5- (ethylthio) -1H-tetrazole, 5- (benzylthio) -1H-tetrazole or 4,5-dicyanoimidazole. 4].
- Compound (1) used in the production method of the present invention can be obtained by the method described in US2012 / 0035246. More specifically, it can be obtained by Reference Example 4 described later.
- Compound (1) in a solvent is 1-hydroxybenzotriazole, 1-hydroxy-7-azabenzotriazole, N-hydroxysuccinimide, ethyl (hydroxyimino) cyanoacetate, N, N′-disuccinimidyl carbonate, N The group consisting of -hydroxyphthalimide, N-hydroxypiperidine, 3-hydroxy-4-oxo-3,4-dihydro-1,2,3-benzotriazine and N-hydroxy-5-norbornene-2,3-dicarboxylic imide After reacting with 6-hydroxyhexanoic acid in the presence of an additive selected from the above and a condensing agent, water, an alkali metal hydroxide, an alkali metal carbonate, a bicyclic amidine compound, A base selected from the group consisting of alkali metal alkoxides and quaternary ammonium hydroxides; Mixed and describes a process of producing a compound (2).
- solvents inert to the dehydration condensation reaction for example, halogenated hydrocarbons such as dichloromethane and chloroform, aromatic hydrocarbons such as toluene and xylene, tetrahydrofuran, 2-methyltetrahydrofuran, methyl tert-butyl ether, and cyclopentyl.
- halogenated hydrocarbons such as dichloromethane and chloroform
- aromatic hydrocarbons such as toluene and xylene
- tetrahydrofuran 2-methyltetrahydrofuran
- methyl tert-butyl ether cyclopentyl
- Ethers such as methyl ether, 1,4-dioxane and diethyl ether, ketones such as acetone, 2-butanone and methyl isobutyl ketone, esters such as methyl acetate and ethyl acetate, dimethylformamide, dimethylacetamide and N-methylpyrrolidone Amides such as 1,3-dimethyl-2-imidazolidinone, ureas such as N, N′-dimethylpropyleneurea, nitriles such as acetonitrile and propionitrile, and sulfur-containing substances such as dimethylsulfoxide and sulfolane Compound class, and the like.
- the amount of the solvent used is generally 0.5 to 100 times by weight of the compound (1).
- 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide, N, N′-dicyclohexylcarbodiimide, N, N′—
- Condensing agent used for dehydration condensation reaction such as diisopropylcarbodiimide, 1,1-carbonyldiimidazole, 1-propylphosphonic anhydride cyclic trimer or 2-chloro-4,6-dimethoxy-1,3,5-triazine Is mentioned.
- the additive is generally used in a proportion of 0.1 to 20 mol and the condensing agent is generally used in a proportion of 1 to 10 mol with respect to 1 mol of the compound (1).
- a tertiary amine may be added to the reaction.
- the tertiary amine include triethylamine and diisopropylethylamine.
- the amount of tertiary amine added is generally in the range of 0.1 to 30 moles per mole of compound (1).
- the reaction temperature is usually in the range of 10 ° C. to 70 ° C. when using a solvent having a boiling point of 70 ° C. or higher, and is usually in the range of 10 ° C. to the reflux temperature of the solvent when using a solvent having a boiling point of less than 70 ° C. Is within.
- the reaction time is usually 0.5 to 150 hours.
- the solvent is usually distilled off and concentrated. Moreover, after adding another solvent to the residue obtained by distilling a solvent off, you may mix with water and a base.
- the obtained reaction mixture is concentrated as it is or by distilling off the solvent to obtain water and alkali metal hydroxide, alkali metal carbonate, Mix with a base selected from the group consisting of bicyclic amidine compounds, alkali metal alkoxides and quaternary ammonium hydroxides.
- a base selected from the group consisting of bicyclic amidine compounds, alkali metal alkoxides and quaternary ammonium hydroxides.
- halogenated hydrocarbons, aromatic hydrocarbons, esters, ketones or sulfur-containing compounds are used as a solvent, the residue concentrated by distilling off the solvent of the obtained reaction mixture is converted to ethers.
- alkali metal hydroxide examples include lithium hydroxide, sodium hydroxide, and potassium hydroxide.
- alkali metal carbonate examples include sodium carbonate, potassium carbonate, and cesium carbonate.
- bicyclic amidine compound examples include diazabicycloundecene and diazabicyclononene.
- alkali metal alkoxide examples include sodium methoxide and potassium tert-butoxide.
- Examples of the quaternary ammonium hydroxide include benzyltrimethylammonium hydroxide, triethylmethylammonium hydroxide, and tributylmethylammonium hydroxide.
- the amount of the base used is generally 0.1 to 10 mol per 1 mol of compound (1).
- the amount of water used is generally 0.1 to 10 times the weight of the compound (1).
- the order of mixing the compound (1), water and base is not limited, and the base and water may be mixed and then mixed with the compound (1).
- the mixture is usually held for 0.5 to 150 hours.
- the mixture When a solvent having a boiling point of 70 ° C. or higher is used, the mixture is usually maintained within a range of 10 ° C. to 70 ° C., and when a solvent having a boiling point of less than 70 ° C. is used, it is usually within a range of 10 ° C. to the reflux temperature of the solvent. The mixture is retained.
- the compound (2) can be isolated by extracting with an organic solvent and concentrating.
- the obtained compound (2) is reacted with 2-cyanoethyl-N, N, N ′, N′-tetraisopropyl phosphorodiamidite in an inert solvent in the presence of a coupling activator. It describes about the process of obtaining a compound (3).
- the solvent examples include halogenated hydrocarbons such as dichloromethane and chloroform, aromatic hydrocarbons such as toluene and xylene, tetrahydrofuran, 2-methyltetrahydrofuran, methyl tert-butyl ether, cyclopentylmethyl ether, 1,4-dioxane, Ethers such as diethyl ether, ketones such as acetone, 2-butanone and methyl isobutyl ketone, esters such as methyl acetate and ethyl acetate, amides such as dimethylformamide, dimethylacetamide and N-methylpyrrolidone, 1,3- Examples include ureas such as dimethyl-2-imidazolidinone and N, N′-dimethylpropyleneurea, nitriles such as acetonitrile and propionitrile, and sulfur-containing compounds such as dimethyl sulfoxide and sulfolane.
- the coupling activator means a coupling activator used for oligonucleotide synthesis by the phosphoramidite method, and is described in, for example, Tetrahedron, 69, 2013, 3615-3637. Specific examples of the coupling activator are the following (i), (ii) and (iii).
- the reaction temperature is usually in the range of 10 ° C. to 70 ° C. when using a solvent having a boiling point of 70 ° C. or higher, and is usually in the range of 10 ° C. to the reflux temperature of the solvent when using a solvent having a boiling point of less than 70 ° C. It is.
- the reaction time is usually 0.5 hours to 150 hours.
- the compound (3) can be isolated by washing with saturated aqueous sodium bicarbonate and then concentrating the organic layer.
- the obtained compound (3) can be purified by chromatography or the like.
- Single-stranded nucleic acid molecules can be produced using compound (3) based on the phosphoramidite method.
- the above production method of compound (3) can be applied to the enantiomer of compound (3).
- the enantiomer of compound (1) as a raw material can be easily prepared from commercially available raw materials in the same manner as compound (1).
- the enantiomers of compound (3) are also useful as described in US2012 / 0035246.
- Example 1 Compound (1) 16.94 mmol (8.28 g), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride 20.33 mmol (3.90 g), 1-hydroxybenzotriazole 40.62 mmol (6.22 g) ), 61.00 mmol (8.50 ml) of triethylamine and anhydrous dichloromethane (160 mL). To this mixture was further added 20.0 mmol (2.64 g) of 6-hydroxyhexanoic acid at room temperature under a nitrogen atmosphere, and then stirred at room temperature for 3 hours under a nitrogen atmosphere.
- the resulting mixture was diluted with 800 mL of dichloromethane and washed 3 times with 1000 mL of saturated brine.
- the obtained organic layer was dried over sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain 10.40 g of a residue.
- 5.20 g of the obtained residue, 10.4 mL of tetrahydrofuran and 5.2 ml of water were mixed, to which 17.25 mmol (0.72 g) of lithium hydroxide monohydrate was added, and the mixture was stirred at 40 ° C. to 50 ° C. for 6 hours. did.
- the obtained mixture was diluted with 50 mL of dichloromethane, washed with 100 mL of saturated aqueous sodium bicarbonate three times, and then washed with 100 mL of saturated brine.
- the obtained organic layer was dried over sodium sulfate, and then the solvent was distilled off under reduced pressure.
- Example 2 To a mixture of 6.00 g of the compound (1) obtained by the method described in Reference Example 4 and 120 mL of chloroform, 2.83 g of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, 1-hydroxybenzotriazole 4.51 g and triethylamine 4.47 g were added. To the obtained mixture, 1.95 g of 6-hydroxyhexanoic acid was added, and the mixture was stirred at room temperature for 3 hours under a nitrogen atmosphere. The obtained mixture was diluted with chloroform, and washed with a 5% aqueous sodium chloride solution three times. The solvent of the organic layer was distilled off under reduced pressure.
- Example 3 9.42 g of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, 15.04 g of 1-hydroxybenzotriazole and 14.91 g of triethylamine were added to a mixture of 20.00 g of compound (1) and 200 mL of chloroform. . To the obtained mixture, 6.49 g of 6-hydroxyhexanoic acid was added and stirred at room temperature for 3 hours under a nitrogen atmosphere. The obtained mixture was diluted with chloroform, and washed with a 5% aqueous sodium chloride solution three times. The solvent of the organic layer was distilled off under reduced pressure to obtain 29.87 g of residue.
- Example 4 9.42 g of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, 15.04 g of 1-hydroxybenzotriazole and 14.91 g of triethylamine were added to a mixture of 20.00 g of compound (1) and 200 mL of chloroform. . To the obtained mixture, 6.49 g of 6-hydroxyhexanoic acid was added and stirred at room temperature for 3 hours under a nitrogen atmosphere. The obtained mixture was diluted with chloroform, and washed with a 5% aqueous sodium chloride solution three times. The solvent of the organic layer was distilled off under reduced pressure to obtain 29.87 g of residue.
- Example 5 9.42 g of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, 15.04 g of 1-hydroxybenzotriazole and 14.91 g of triethylamine were added to a mixture of 20.00 g of compound (1) and 200 mL of chloroform. . To the obtained mixture, 6.49 g of 6-hydroxyhexanoic acid was added and stirred at room temperature for 3 hours under a nitrogen atmosphere. The obtained mixture was diluted with chloroform, and washed with a 5% aqueous sodium chloride solution three times. The solvent of the organic layer was distilled off under reduced pressure to obtain 29.87 g of residue.
- Example 6 9.42 g of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, 15.04 g of 1-hydroxybenzotriazole and 14.91 g of triethylamine were added to a mixture of 20.00 g of compound (1) and 200 mL of chloroform. . To the obtained mixture, 6.49 g of 6-hydroxyhexanoic acid was added and stirred at room temperature for 3 hours under a nitrogen atmosphere. The obtained mixture was diluted with chloroform, and washed with a 5% aqueous sodium chloride solution three times. The solvent of the obtained organic layer was distilled off under reduced pressure to obtain 29.87 g of a residue.
- Synthesis was performed from the 3 'side to the 5' side using NTS M-4MX-E (manufactured by Nippon Techno Service Co., Ltd.) as a nucleic acid synthesizer.
- NTS M-4MX-E manufactured by Nippon Techno Service Co., Ltd.
- uridine EMM amidite described in Example 2 of US2012 / 0035246, cytidine EMM amidite described in Example 3, adenosine EMM amidite described in Example 4, and guanosine EMM amidite described in Example 5 were used.
- a compound (3) or an enantiomer thereof capable of producing a single-stranded nucleic acid molecule capable of suppressing the expression of a target gene with high yield can be produced.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

しかし、US2012/0035246に記載の方法で得られる化合物(3)を使用した場合の一本鎖核酸分子の収量は必ずしも充分なものではなかった。
〔1〕 式(1)


前記工程で得られた式(2)で示される化合物又はその鏡像異性体と2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトとを、カップリングアクチベーターの存在下、溶媒中で反応させて、化合物(3)又はその鏡像異性体を得る工程
を含む化合物(3)又はその鏡像異性体の製造方法。
〔2〕 塩基がアルカリ金属水酸化物である、〔1〕に記載の製造方法。
〔3〕 縮合剤が1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド、N,N’-ジシクロヘキシルカルボジイミド、N,N’-ジイソプロピルカルボジイミド、1,1-カルボニルジイミダゾール、1-プロピルホスホン酸無水物環状三量体又は2-クロロ-4,6-ジメトキシ-1,3,5-トリアジンである〔1〕又は〔2〕に記載の製造方法。
〔4〕 添加剤が1-ヒドロキシベンゾトリアゾール、1-ヒドロキシ-7-アザベンゾトリアゾール、N-ヒドロキシコハク酸イミド、エチル(ヒドロキシイミノ)シアノアセタート又は炭酸N,N’-ジスクシンイミジルである〔1〕~〔3〕のいずれかに記載の製造方法。
〔5〕 カップリングアクチベーターがジイソプロピルアミンテトラゾール塩、1H-テトラゾール、5-(エチルチオ)-1H-テトラゾール、5-(ベンジルチオ)-1H-テトラゾール又は4,5-ジシアノイミダゾールである〔1〕~〔4〕のいずれかに記載の製造方法。
アルカリ金属アルコキシドとしては、例えばナトリウムメトキシド及びカリウムtert-ブトキシドが挙げられる。
化合物(1)、水及び塩基の混合順序は限定されず、塩基と水とを混合した後、化合物(1)と混合してもよい。
(i)アゾールカップリングアクチベーター
1H-テトラゾール;
5-(4-ニトロフェニル)-1H-テトラゾール、5-(ビス-3,5-トリフルオロメチルフェニル)-1H-テトラゾール、5-エチルチオ-1H-テトラゾール、5-ベンジルチオ-1H-テトラゾール、5-メチルチオ-1H-テトラゾール、5-メルカプト-1H-テトラゾール等の修飾テトラゾールカップリングアクチベーター;
4,5-ジシアノイミダゾール、2-ブロモ-4,5-ジシアノイミダゾール、2-ベンジル-4,5-ジシアノイミダゾール、2-ブロモ-4,5-ジエチル-カルボキシルイミダゾール等のイミダゾールカップリングアクチベーター;
1-ヒドロキシ-ベンゾトリアゾール、6-トリフルオロメチル-1-ヒドロキシ-ベンゾトリアゾール、4-ニトロ-1-ヒドロキシ-ベンゾトリアゾール等の1-ヒドロキシベンゾトリアゾールカップリングアクチベーター;
3-ニトロトリアゾール等の3-ニトロトリアゾールカップリングアクチベーター;
(ii)塩錯体カップリングアクチベーター
ピリジン塩酸塩、トリフルオロ酢酸ピリジン塩等のピリジニウム塩錯体カップリングアクチベーター;
ベンズイミダゾールトリフルオロ酢酸塩、N-フェニルイミダゾールトリフルオロ酢酸塩等のアゾリウム塩錯体カップリングアクチベーター;
サッカリン-1-メチルイミダゾール塩等のサッカリン塩錯体カップリングアクチベーター;
ジアルキル(シアノメチル)アンモニウムテトラフルオロボレート、ジイソプロピルアミンテトラゾール塩等のアンモニウム塩錯体カップリングアクチベーター;
(iii)ホスホロアミダイト法によるオリゴ核酸合成に用いられるその他のカップリングアクチベーター
トリクロロ酢酸、トリフルオロ酢酸、ジクロロ酢酸、2,4-ジニトロ安息香酸等のカルボン酸カップリングアクチベーター;
塩化鉄(III)、塩化アルミニウム(III)、三フッ化ホウ素ジエチルエーテル錯体、塩化ジルコニウム(IV)、塩化ビスマス(III)等のルイス酸カップリングアクチベーター;
トリメチルクロロシラン;
2,4-ジニトロフェノール;
反応には、化合物(2)1モルに対して2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトが一般に1モル~10モルの割合、カップリングアクチベーターが一般に0.1モル~10モルの割合で使用される。
化合物(1)16.94mmol(8.28g)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩20.33mmol(3.90g)、1-ヒドロキシベンゾトリアゾール40.62mmol(6.22g)、トリエチルアミン61.00mmol(8.50ml)及び無水ジクロロメタン(160mL)を混合した。この混合液に、さらに、窒素雰囲気下室温で6-ヒドロキシヘキサン酸20.0mmol(2.64g)を加え、その後、窒素雰囲気下室温で3時間撹拌した。得られた混合液をジクロロメタン800mLで希釈し、飽和食塩水1000mLで3回洗浄した。得られた有機層を硫酸ナトリウムで乾燥した後、溶媒を減圧下で留去して10.40gの残渣を得た。
得られた残渣5.20g、テトラヒドロフラン10.4mL及び水5.2mlを混合し、そこに水酸化リチウム一水和物17.25mmol(0.72g)を加え、40℃~50℃で6時間撹拌した。得られた混合物を室温まで放冷した後、トルエン26mL及び水52mLを加え、室温で15分撹拌した。分取した有機層を重曹水(炭酸水素ナトリウム2.76gと水52mLとを混合)で2回洗浄し、減圧下で溶媒を留去した。残渣をアセトニトリル31.2mLに溶解した後、減圧下で溶媒を留去して化合物(2)を得た(純度93%、収率85%)。
得られた化合物(2)7.13mmol(4.30g)を無水アセトニトリルと混合し、室温で3回共沸脱水を行った。得られた残渣にジイソプロピルアミンテトラゾール塩8.53mmol(1.46g)を加え、減圧下で脱気した後窒素ガスを充填した。得られた混合物に無水アセトニトリル5mLを加え、さらに、2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト8.56mmol(2.58g)の無水アセトニトリル溶液3.5mLを加えた。この混合物を、窒素雰囲気下室温で2時間撹拌した。得られた混合物をジクロロメタン50mLで希釈し、飽和重曹水100mLで洗浄を3回行った後、飽和食塩水100mLで洗浄した。得られた有機層を硫酸ナトリウムで乾燥した後、溶媒を減圧下で留去した。得られた残渣を、充填剤としてアミノシリカゲルを用いたカラムクロマトグラフィー(展開溶媒 ヘキサン:酢酸エチル=1:3、0.05%ピリジン含有)に供し、化合物(3)を得た(収率82.4%)。
参考例4に記載の方法で得られた化合物(1)6.00g及びクロロホルム120mLの混合物に、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩2.83g、1-ヒドロキシベンゾトリアゾール4.51g及びトリエチルアミン4.47gを加えた。得られた混合物に6-ヒドロキシヘキサン酸1.95gを加え、窒素雰囲気下、室温で3時間撹拌した。得られた混合物をクロロホルムで希釈し、5%塩化ナトリウム水溶液で洗浄を3回行った。有機層の溶媒を減圧下で留去した。この残渣にテトラヒドロフラン15mLを加えた後、水酸化リチウム0.59gと水7.4mLとの混合物を加えた。得られた混合物を室温で22時間撹拌した後、トルエンを加え、水で洗浄して得られた有機層を、5%炭酸水素ナトリウム水溶液で洗浄を3回行った後、溶媒を減圧下で留去した。残渣をアセトニトリルと混合し、再び減圧下で溶媒を留去することにより、化合物(2)を25.80g得た。
得られた化合物(2)20.64gにアセトニトリル30mLを加えた後、ジイソプロピルアミンテトラゾール塩2.02g及び2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト3.55gを加え、窒素雰囲気下、室温で1時間撹拌した。反応終了後、得られた混合物をトルエンで希釈し、5%炭酸水素ナトリウム水溶液で洗浄を3回行った。得られた有機層を硫酸マグネシウムで乾燥した後、溶媒を減圧下で留去した。得られた残渣の一部をトリエチルアミンで処理したシリカゲルを用いてカラムクロマトグラフィー(展開溶媒 ヘプタン:酢酸エチル=50:50)を行い、化合物(3)を得た。
化合物(1)20.00g及びクロロホルム200mLの混合物に、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩9.42g、1-ヒドロキシベンゾトリアゾール15.04g及びトリエチルアミン14.91gを加えた。得られた混合物に6-ヒドロキシヘキサン酸6.49gを加え、窒素雰囲気下室温で3時間撹拌した。得られた混合物をクロロホルムで希釈し、5%塩化ナトリウム水溶液で洗浄を3回行った。有機層の溶媒を減圧下で留去し、残渣29.87gを得た。残渣4.98gにアセトニトリル10mLを加えた後、水酸化ナトリウム0.55gと水5.0mLとの混合物を加えた。得られた混合物を30℃で18時間撹拌した後、トルエンを加え、水で洗浄して得られた有機層を、5%炭酸水素ナトリウム水溶液で2回洗浄した後、溶媒を減圧下で留去した。残渣をアセトニトリルと混合し、再び減圧下で溶媒を留去することにより、化合物(2)を5.03g得た。
得られた化合物(2)5.03gにアセトニトリル21mLを加えた後、ジイソプロピルアミンテトラゾール塩1.40g及び2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト2.47gを加え、窒素雰囲気下室温で1.5時間撹拌した。得られた混合物をトルエンで希釈し、5%炭酸水素ナトリウム水溶液で洗浄を3回行った。有機層を硫酸マグネシウムで乾燥した後、溶媒を減圧下で留去した。得られた残渣の一部をトリエチルアミンで処理したシリカゲルを用いてカラムクロマトグラフィー(展開溶媒 ヘプタン:酢酸エチル:トリエチルアミン=40:60:10)を行い、化合物(3)を得た。
化合物(1)20.00g及びクロロホルム200mLの混合物に、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩9.42g、1-ヒドロキシベンゾトリアゾール15.04g及びトリエチルアミン14.91gを加えた。得られた混合物に6-ヒドロキシヘキサン酸6.49gを加え、窒素雰囲気下室温で3時間撹拌した。得られた混合物をクロロホルムで希釈し、5%塩化ナトリウム水溶液で洗浄を3回行った。有機層の溶媒を減圧下で留去し、残渣29.87gを得た。残渣4.98gにテトラヒドロフラン10mLを加えた後、ジアザビシウクロウンデセン2.08gと水1.0mLとの混合物を加えた。得られた混合物を50℃で1872時間撹拌した後、トルエンを加え、水で洗浄して得られた有機層を、5%炭酸水素ナトリウム水溶液で2回洗浄した後、溶媒を減圧下で留去した。残渣をアセトニトリルと混合し、再び減圧下で溶媒を留去することにより、化合物(2)を8.78g得た。
得られた化合物(2)5.03gにアセトニトリル21mLを加えた後、ジイソプロピルアミンテトラゾール塩1.40g及び2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト2.47gを加え、窒素雰囲気下室温で1.5時間撹拌した。得られた混合物をトルエンで希釈し、5%炭酸水素ナトリウム水溶液で洗浄を3回行った。有機層を硫酸マグネシウムで乾燥した後、溶媒を減圧下で留去した。得られた残渣の一部をトリエチルアミンで処理したシリカゲルを用いてカラムクロマトグラフィー(展開溶媒 ヘプタン:酢酸エチル:トリエチルアミン=40:60:10)を行い、化合物(3)を得た。
化合物(1)20.00g及びクロロホルム200mLの混合物に、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩9.42g、1-ヒドロキシベンゾトリアゾール15.04g及びトリエチルアミン14.91gを加えた。得られた混合物に6-ヒドロキシヘキサン酸6.49gを加え、窒素雰囲気下室温で3時間撹拌した。得られた混合物をクロロホルムで希釈し、5%塩化ナトリウム水溶液で洗浄を3回行った。有機層の溶媒を減圧下で留去し、残渣29.87gを得た。残渣4.98gにテトラヒドロフラン10mLを加えた後、カリウムtert-ブトキシド1.53gと水5.0mLとの混合物を加えた。得られた混合物を30℃で22時間撹拌した後、トルエンを加え、水で洗浄して得られた有機層を、5%炭酸水素ナトリウム水溶液で2回洗浄した後、溶媒を減圧下で留去した。残渣をアセトニトリルと混合し、再び減圧下で溶媒を留去することにより、化合物(2)を4.78g得た。
得られた化合物(2)4.78gにアセトニトリル21mLを加えた後、ジイソプロピルアミンテトラゾール塩1.40g及び2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト2.47gを加え、窒素雰囲気下室温で1.5時間撹拌した。反応終了後、得られた混合物をトルエンで希釈し、5%炭酸水素ナトリウム水溶液で3回洗浄した。得られた有機層を硫酸マグネシウムで乾燥した後、溶媒を減圧下で留去した。得られた残渣の一部をトリエチルアミンで処理したシリカゲルを用いてカラムクロマトグラフィー(展開溶媒 ヘプタン:酢酸エチル:トリエチルアミン=40:60:10)を行い、化合物(3)を得た。
化合物(1)20.00g及びクロロホルム200mLの混合物に、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩9.42g、1-ヒドロキシベンゾトリアゾール15.04g及びトリエチルアミン14.91gを加えた。得られた混合物に6-ヒドロキシヘキサン酸6.49gを加え、窒素雰囲気下、室温で3時間撹拌した。得られた混合物をクロロホルムで希釈し、5%塩化ナトリウム水溶液で洗浄を3回行った。得られた有機層の溶媒を減圧下で留去し、残渣29.87gを得た。残渣4.98gにテトラヒドロフラン10mLを加えた後、水酸化ベンジルトリメチルアンモニウム水溶液(40wt%)5.70gと水1.6mLとの混合物を加えた。得られた混合物を30℃で6時間撹拌した後、トルエンを加え、水で洗浄して得られた有機層を、5%炭酸水素ナトリウム水溶液で2回洗浄した後、溶媒を減圧下で留去した。残渣をアセトニトリルと混合し、再び減圧下で溶媒を留去することにより、化合物(2)を10.21g得た。
得られた化合物(2)10.21gにアセトニトリル21mLを加えた後、ジイソプロピルアミンテトラゾール塩1.40g及び2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイト2.47gを加え、窒素雰囲気下室温で1.5時間撹拌した。得られた混合物をトルエンで希釈し、5%炭酸水素ナトリウム水溶液で洗浄を3回行った。有機層を硫酸マグネシウムで乾燥した後、溶媒を減圧下で留去した。得られた残渣の一部をトリエチルアミンで処理したシリカゲルを用いてカラムクロマトグラフィー(展開溶媒 ヘプタン:酢酸エチル:トリエチルアミン=40:60:10)を行い、化合物(3)を得た。
実施例1で製造した化合物(3)を用いて、US2012/0035246の実施例(B1)に記載の一本鎖核酸分子(式(A)

で示される核酸分子。以下、化合物(A)と記す。)を製造した。
US2012/0035246の実施例(A3)の(3)及び(4)の記載にしたがって化合物(3)を製造した(収率85.5%)。得られた化合物(3)を、上記参考例1の記載にしたがって化合物(A)を得た。
参考例3
上記参考例2に記載の化合物(3)、実施例3~6で製造した化合物(3)を用い、上記参考例1の記載に準じて化合物(A)を得た。結果を下記表1に示す。参考例3の結果に見られるように、本発明の製造方法で得られる化合物(3)を使用することにより、従来の製造方法で得られる化合物(3)を使用する場合に比べて化合物(A)が高い収量で得られる。

窒素置換した反応容器にN-[(9H-フルオレン-9-イルメトキシ)カルボニル]-L-プロリン30.00gとアセトニトリル420mLとを混合し、1-ヒドロキシベンゾトリアゾール32.68gを加えた。得られた混合物に4-アミノ-1-ブタノール9.51gを加え、さらにジシクロヘキシルカルボジイミド22.02gのアセトニトリル溶液210mLを室温で滴下した後、室温で1時間撹拌した。反応終了後、生成した沈殿を濾過により除去し、得られた濾液の溶媒を減圧下で留去した。残渣にトルエンを加え、10%炭酸水素ナトリウム水溶液で洗浄した。得られた有機層の溶媒を減圧下で留去することにより、式(a)

で示される化合物を得た。
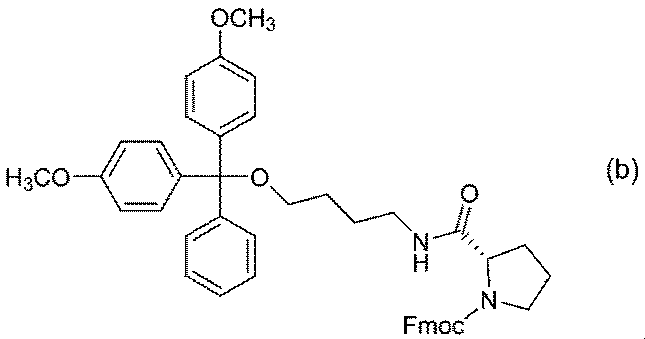
で示される化合物を得た。
Claims (5)
- 式(1)
で示される化合物又はその鏡像異性体を溶媒中、1-ヒドロキシベンゾトリアゾール、1-ヒドロキシ-7-アザベンゾトリアゾール、N-ヒドロキシコハク酸イミド、エチル(ヒドロキシイミノ)シアノアセタート、炭酸N,N’-ジスクシンイミジル、N-ヒドロキシフタルイミド、N-ヒドロキシピペリジン、3-ヒドロキシ-4-オキソ-3,4-ジヒドロ-1,2,3-ベンゾトリアジン、及びN-ヒドロキシ-5-ノルボルネン-2,3-ジカルボン酸イミドからなる群より選ばれる添加剤並びに縮合剤の存在下、6-ヒドロキシヘキサン酸と反応させた後、得られた反応混合物と、水と、アルカリ金属水酸化物、アルカリ金属炭酸塩、二環式アミジン化合物、アルカリ金属アルコキシド及び第4級アンモニウム水酸化物からなる群より選ばれる塩基とを混合して式(2)
で示される化合物又はその鏡像異性体を製造する工程、及び
前記工程で得られた式(2)で示される化合物又はその鏡像異性体と2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトとを、カップリングアクチベーターの存在下、溶媒中で反応させて、式(3)
で示される化合物又はその鏡像異性体を得る工程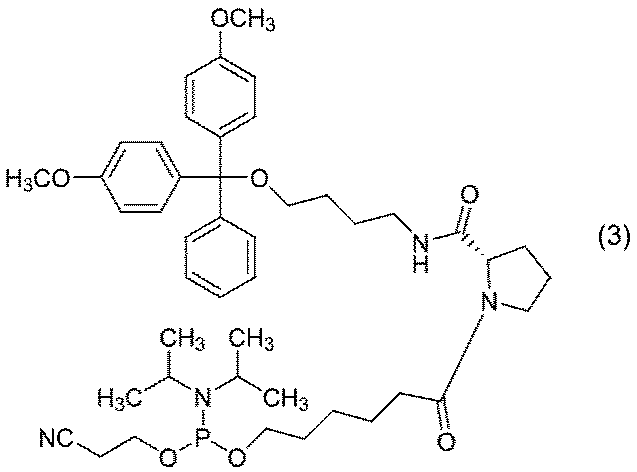
を含む、式(3)で示される化合物又はその鏡像異性体の製造方法。 - 塩基がアルカリ金属水酸化物である請求項1に記載の製造方法。
- 縮合剤が1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド、N,N’-ジシクロヘキシルカルボジイミド、N,N’-ジイソプロピルカルボジイミド、1,1-カルボニルジイミダゾール、1-プロピルホスホン酸無水物環状三量体又は2-クロロ-4,6-ジメトキシ-1,3,5-トリアジンである請求項1又は2に記載の製造方法。
- 添加剤が1-ヒドロキシベンゾトリアゾール、1-ヒドロキシ-7-アザベンゾトリアゾール、N-ヒドロキシコハク酸イミド、エチル(ヒドロキシイミノ)シアノアセタート又は炭酸N,N’-ジスクシンイミジルである請求項1又は2に記載の製造方法。
- カップリングアクチベーターがジイソプロピルアミンテトラゾール塩、1H-テトラゾール、5-(エチルチオ)-1H-テトラゾール、5-(ベンジルチオ)-1H-テトラゾール又は4,5-ジシアノイミダゾールである請求項1又は2に記載の製造方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201780024730.7A CN109071579B (zh) | 2016-04-26 | 2017-04-17 | 单链核酸分子用单体的制造方法 |
| KR1020187030258A KR102362675B1 (ko) | 2016-04-26 | 2017-04-17 | 단일쇄 핵산 분자용 모노머의 제조 방법 |
| ES17789327T ES2853732T3 (es) | 2016-04-26 | 2017-04-17 | Método de producción de un monómero para una molécula de ácido nucleico monocatenario |
| US16/085,330 US10308672B2 (en) | 2016-04-26 | 2017-04-17 | Method for producing monomer for single-stranded nucleic acid molecule |
| JP2018514506A JP6787398B2 (ja) | 2016-04-26 | 2017-04-17 | 一本鎖核酸分子用モノマーの製造方法 |
| EP17789327.8A EP3450439B8 (en) | 2016-04-26 | 2017-04-17 | Method for producing monomer for single-stranded nucleic acid molecule |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-087783 | 2016-04-26 | ||
| JP2016087783 | 2016-04-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017188042A1 true WO2017188042A1 (ja) | 2017-11-02 |
Family
ID=60161424
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/015435 WO2017188042A1 (ja) | 2016-04-26 | 2017-04-17 | 一本鎖核酸分子用モノマーの製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10308672B2 (ja) |
| EP (1) | EP3450439B8 (ja) |
| JP (1) | JP6787398B2 (ja) |
| KR (1) | KR102362675B1 (ja) |
| CN (1) | CN109071579B (ja) |
| ES (1) | ES2853732T3 (ja) |
| WO (1) | WO2017188042A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019208571A1 (ja) | 2018-04-24 | 2019-10-31 | 住友化学株式会社 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
| WO2020202953A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体、及びこれを用いた核酸の製造方法 |
| WO2020202951A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体、及びこれを用いた核酸の製造方法 |
| WO2020202950A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体及びこれを用いた核酸の製造方法 |
| WO2020202952A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体、及びこれを用いた核酸の製造方法 |
| WO2020202949A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | Rnaの製造方法 |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021162070A1 (ja) * | 2020-02-14 | 2021-08-19 | 東レ株式会社 | 核酸製造用モノマーの製造方法 |
| WO2022064908A1 (ja) | 2020-09-24 | 2022-03-31 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2023182274A1 (ja) | 2022-03-23 | 2023-09-28 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110115963B (zh) * | 2019-04-18 | 2020-12-22 | 通用生物系统(安徽)有限公司 | 一种引物合成用偶联活化剂的制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012017919A1 (ja) * | 2010-08-03 | 2012-02-09 | 株式会社ボナック | 含窒素脂環式骨格を有する一本鎖核酸分子 |
| JP2013055913A (ja) * | 2011-09-09 | 2013-03-28 | Bonac Corp | 遺伝子発現制御のための一本鎖rna分子 |
| WO2013133221A1 (ja) * | 2012-03-04 | 2013-09-12 | 株式会社ボナック | microRNA阻害剤 |
| WO2013180038A1 (ja) * | 2012-05-26 | 2013-12-05 | 株式会社ボナック | デリバリー機能を有する遺伝子発現制御用の一本鎖核酸分子 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8691782B2 (en) | 2010-08-03 | 2014-04-08 | Bonac Corporation | Single-stranded nucleic acid molecule having nitrogen-containing alicyclic skeleton |
| BR112014004313A8 (pt) | 2011-08-25 | 2023-02-07 | Bonac Corp | Composto glicosídeo, métodos de produção de um tioéter, de éter e de composto glicosídeo, e, éter. |
| WO2013077446A1 (ja) | 2011-11-26 | 2013-05-30 | 株式会社ボナック | 遺伝子発現制御のための一本鎖核酸分子 |
| US9225789B2 (en) | 2013-10-10 | 2015-12-29 | Pushd, Inc. | Automated mobile positional social media method and system |
-
2017
- 2017-04-17 KR KR1020187030258A patent/KR102362675B1/ko active IP Right Grant
- 2017-04-17 ES ES17789327T patent/ES2853732T3/es active Active
- 2017-04-17 JP JP2018514506A patent/JP6787398B2/ja active Active
- 2017-04-17 CN CN201780024730.7A patent/CN109071579B/zh active Active
- 2017-04-17 EP EP17789327.8A patent/EP3450439B8/en active Active
- 2017-04-17 WO PCT/JP2017/015435 patent/WO2017188042A1/ja active Application Filing
- 2017-04-17 US US16/085,330 patent/US10308672B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012017919A1 (ja) * | 2010-08-03 | 2012-02-09 | 株式会社ボナック | 含窒素脂環式骨格を有する一本鎖核酸分子 |
| JP2013055913A (ja) * | 2011-09-09 | 2013-03-28 | Bonac Corp | 遺伝子発現制御のための一本鎖rna分子 |
| WO2013133221A1 (ja) * | 2012-03-04 | 2013-09-12 | 株式会社ボナック | microRNA阻害剤 |
| WO2013180038A1 (ja) * | 2012-05-26 | 2013-12-05 | 株式会社ボナック | デリバリー機能を有する遺伝子発現制御用の一本鎖核酸分子 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3450439A4 * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112020507A (zh) * | 2018-04-24 | 2020-12-01 | 住友化学株式会社 | 酰胺化合物及使用该化合物的多核苷酸的制造方法 |
| CN112020507B (zh) * | 2018-04-24 | 2023-10-10 | 住友化学株式会社 | 酰胺化合物及使用该化合物的多核苷酸的制造方法 |
| US11401296B2 (en) | 2018-04-24 | 2022-08-02 | Sumitomo Chemical Company, Limited | Amidite compound and method for producing polynucleotide using said compound |
| WO2019208571A1 (ja) | 2018-04-24 | 2019-10-31 | 住友化学株式会社 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
| WO2020202950A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体及びこれを用いた核酸の製造方法 |
| WO2020202949A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | Rnaの製造方法 |
| WO2020202952A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体、及びこれを用いた核酸の製造方法 |
| WO2020202951A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体、及びこれを用いた核酸の製造方法 |
| WO2020202953A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体、及びこれを用いた核酸の製造方法 |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021162070A1 (ja) * | 2020-02-14 | 2021-08-19 | 東レ株式会社 | 核酸製造用モノマーの製造方法 |
| WO2022064908A1 (ja) | 2020-09-24 | 2022-03-31 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2023182274A1 (ja) | 2022-03-23 | 2023-09-28 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| ES2853732T3 (es) | 2021-09-17 |
| EP3450439A1 (en) | 2019-03-06 |
| KR20180136953A (ko) | 2018-12-26 |
| KR102362675B1 (ko) | 2022-02-11 |
| CN109071579A (zh) | 2018-12-21 |
| JP6787398B2 (ja) | 2020-11-18 |
| EP3450439B8 (en) | 2021-06-30 |
| US20190100540A1 (en) | 2019-04-04 |
| EP3450439B1 (en) | 2021-01-20 |
| CN109071579B (zh) | 2021-05-14 |
| US10308672B2 (en) | 2019-06-04 |
| EP3450439A4 (en) | 2019-10-23 |
| JPWO2017188042A1 (ja) | 2019-02-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6787398B2 (ja) | 一本鎖核酸分子用モノマーの製造方法 | |
| US10053466B2 (en) | Process for preparing chiral dipeptidyl peptidase-IV inhibitors | |
| US20150196655A1 (en) | Carbohydrate conjugated rna agents and process for their preparation | |
| JP6985367B2 (ja) | 新規化合物および方法 | |
| KR20180038460A (ko) | 세포독성 벤조다이아제핀 유도체의 제조 방법 | |
| KR20180011848A (ko) | 함질소 복소환 화합물의 제조 방법 및 그 중간체 | |
| US9580457B2 (en) | Process for the preparation of (1-{9-[(4S, 2R, 3R, 5R)-3, 4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl)-6-aminopurin-2-yl}pyrazole-4-yl)-N-methylcarboxamide | |
| US10961220B2 (en) | Method for preparing substituted imidazolyl carboxyamides | |
| KR20090061127A (ko) | 판토프라졸 나트륨 쎄스키히드레이트의 제조방법 | |
| JP2015044856A (ja) | ビフェニルイミダゾール化合物の調製方法 | |
| JP4356111B2 (ja) | N−(2−アミノ−1,2−ジシアノビニル)ホルムアミジンの製造方法 | |
| WO2021162070A1 (ja) | 核酸製造用モノマーの製造方法 | |
| US11136287B2 (en) | Method for producing n-benzyl-2-bromo-3-methoxypropionamide and intermediates thereof | |
| JP7423533B2 (ja) | 配糖体化合物の製造方法 | |
| CN111386274B (zh) | 用于合成除草剂吡唑烷二酮化合物的化学方法 | |
| US20030022862A1 (en) | Process for selective N-acylation of purine nucleosides | |
| EP3313821B1 (en) | Process for the preparation of carbamoylamino pyrazole derivatives | |
| JP2002155058A (ja) | 1位置換ヒダントイン類の製造方法 | |
| JP2023179354A (ja) | H-ホスホネート法を用いたモルフォリノ核酸の製造方法 | |
| JP2022035954A (ja) | N-Boc-ラクタム誘導体及びその製造方法、並びに、環状アミン誘導体の製造方法 | |
| CN113454088A (zh) | 用于制备(6S)-3-[(4S)-4-氰基-2-氧代-吡咯烷-1-基]-6-甲基-N-(3,4,5-三氟苯基)-6,7-二氢-4H-吡唑并[1,5-a]吡嗪-5-甲酰胺的方法 | |
| CN115244044A (zh) | 含氟嘧啶化合物及其制造方法 | |
| KR20070117381A (ko) | 로사탄의 새로운 제조방법 | |
| JP2023528200A (ja) | アンドロゲン受容体アンタゴニストおよびその中間体の製造方法 | |
| JP2019135220A (ja) | L−カルノシン及びその誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2018514506 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20187030258 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017789327 Country of ref document: EP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17789327 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017789327 Country of ref document: EP Effective date: 20181126 |