WO2017104308A1 - 偏光板及び前記偏光板を含む画像表示装置 - Google Patents
偏光板及び前記偏光板を含む画像表示装置 Download PDFInfo
- Publication number
- WO2017104308A1 WO2017104308A1 PCT/JP2016/083291 JP2016083291W WO2017104308A1 WO 2017104308 A1 WO2017104308 A1 WO 2017104308A1 JP 2016083291 W JP2016083291 W JP 2016083291W WO 2017104308 A1 WO2017104308 A1 WO 2017104308A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- film
- polarizing plate
- acrylate
- meth
- acid
- Prior art date
Links
Images



Classifications
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
- G02B5/3083—Birefringent or phase retarding elements
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B23/00—Layered products comprising a layer of cellulosic plastic substances, i.e. substances obtained by chemical modification of cellulose, e.g. cellulose ethers, cellulose esters, viscose
- B32B23/04—Layered products comprising a layer of cellulosic plastic substances, i.e. substances obtained by chemical modification of cellulose, e.g. cellulose ethers, cellulose esters, viscose comprising such cellulosic plastic substance as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B23/08—Layered products comprising a layer of cellulosic plastic substances, i.e. substances obtained by chemical modification of cellulose, e.g. cellulose ethers, cellulose esters, viscose comprising such cellulosic plastic substance as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L1/00—Compositions of cellulose, modified cellulose or cellulose derivatives
- C08L1/08—Cellulose derivatives
- C08L1/10—Esters of organic acids, i.e. acylates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/60—Additives non-macromolecular
- C09D7/61—Additives non-macromolecular inorganic
- C09D7/62—Additives non-macromolecular inorganic modified by treatment with other compounds
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B1/00—Optical elements characterised by the material of which they are made; Optical coatings for optical elements
- G02B1/04—Optical elements characterised by the material of which they are made; Optical coatings for optical elements made of organic materials, e.g. plastics
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B1/00—Optical elements characterised by the material of which they are made; Optical coatings for optical elements
- G02B1/10—Optical coatings produced by application to, or surface treatment of, optical elements
- G02B1/14—Protective coatings, e.g. hard coatings
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
- G02B5/3025—Polarisers, i.e. arrangements capable of producing a definite output polarisation state from an unpolarised input state
- G02B5/3033—Polarisers, i.e. arrangements capable of producing a definite output polarisation state from an unpolarised input state in the form of a thin sheet or foil, e.g. Polaroid
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/133—Constructional arrangements; Operation of liquid crystal cells; Circuit arrangements
- G02F1/1333—Constructional arrangements; Manufacturing methods
- G02F1/1335—Structural association of cells with optical devices, e.g. polarisers or reflectors
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/133—Constructional arrangements; Operation of liquid crystal cells; Circuit arrangements
- G02F1/1333—Constructional arrangements; Manufacturing methods
- G02F1/1335—Structural association of cells with optical devices, e.g. polarisers or reflectors
- G02F1/13363—Birefringent elements, e.g. for optical compensation
Definitions
- the present invention relates to a polarizing plate and an image display device including the polarizing plate.
- LCD Liquid crystal display
- polarizing plates particularly polarizers
- a viewer wears glasses for viewing a stereoscopic image, and parallax images of a right-eye image and a left-eye image are alternately switched at high speed in time series and displayed on an LCD screen.
- opening and closing of the left and right liquid crystal shutters of the stereoscopic image viewing glasses are switched. By switching in this way, the two-dimensional image displayed on the screen can be recognized three-dimensionally.
- ⁇ / 4 is respectively provided on the viewing side of the LCD (the outermost surface side) and on the side far from the eyes of the stereoscopic image viewing glasses. A film is placed. By disposing the ⁇ / 4 film at such a position, the deterioration of the image quality of the LCD can be improved.
- Patent Document 1 Japanese Patent Laid-Open No. 2014-95890 discloses a configuration in which a hard coat film is disposed on the outermost surface of a polarizing plate.
- This hard coat film contains a compound having a cyclic aliphatic hydrocarbon group and having three or more ethylenically unsaturated double bond groups in the molecule, and a polymerization initiator.
- JP 2014-95890 A Japanese Patent No. 5521732
- the degree of polarization of the polarizing plate tends to decrease. Further, in a high temperature dry environment, the polarizer was subjected to stress and warped, whereby the retardation of the polarizer fluctuated and light leakage was likely to occur.
- the present invention has been made in view of the above situation, and an object of the present invention is to provide a polarizing plate in which the degree of polarization of a polarizer does not easily decrease and light leakage does not occur even when exposed to a high-temperature drying environment. It is.
- the polarizing plate of the present invention has a ⁇ / 4 film having a hard coat layer on the surface on at least one surface, and the ⁇ / 4 film has an acetyl group average substitution degree of 1.8 or more and 2.2.
- the dimensional change rate in the direction parallel to the slow axis of the ⁇ / 4 film of the polarizing plate before and after humidity conditioning for 24 hours in an environment is ⁇ 0.5% or more and ⁇ 0.1% or less. To do.
- the present invention is also an image display device including the polarizing plate, and the surface of the polarizing plate on the hard coat layer side is disposed on the viewing side.
- the inventors of the present invention have made extensive studies on the mechanism by which the degree of polarization of the polarizing plate decreases, and found that the degree of polarization of the polarizing plate decreases by different mechanisms in a high temperature and high humidity environment and in a high temperature drying environment. . That is, in a high-temperature and high-humidity environment, the polarizer is hydrolyzed, whereby the iodine trimer contained in the polarizer is decomposed and the degree of polarization is lowered.
- the inventors of the present invention have completed the present invention by earnestly studying the configuration for avoiding the decrease in the degree of polarization. According to the present invention, it is possible to provide a polarizing plate in which the degree of polarization of a polarizer is not easily lowered even when exposed to a high-temperature dry environment, and light leakage does not easily occur.
- FIG. 1 is a schematic diagram of a polarizing plate of the present invention.
- the polarizing plate C of the present invention is a laminate of a ⁇ / 4 film C1 and a polarizer C2.
- the polarizing plate C of the present invention may further include a polarizer protective film C3 on the surface of the polarizer C2 opposite to the side in contact with the ⁇ / 4 film C1.
- the hard coat layer C0 is provided on the surface of the ⁇ / 4 film C1 opposite to the side in contact with the polarizer C2. This makes it difficult for scratches to enter the surface side (viewing side) of the polarizing plate C.
- the polarizer C2 since the polarizer C2 hardly absorbs moisture in the atmosphere, the polarizing plate is hardly warped even in a high-temperature drying environment.
- the polarizing plate C may be further bonded with a protective film on one surface of the polarizing plate C and a separate film on the opposite surface.
- the protective film and the separate film are used for the purpose of protecting the polarizing plate at the time of shipping the polarizing plate and at the time of product inspection.
- the polarizing plate C of the present invention is a ⁇ / 4 film of a polarizing plate before and after being stored for 1000 hours in a temperature environment of 90 ° C. and then conditioned for 24 hours in an environment of 23 ° C. and 55% RH.
- the dimensional change rate in the direction parallel to the slow axis is ⁇ 0.5% or more and ⁇ 0.1% or less.
- the dimensional change rate is preferably ⁇ 0.4% or more and ⁇ 0.15% or less, and more preferably ⁇ 0.35% or more and ⁇ 0.2% or less.
- the dimensional change rate measurement method employs a value calculated as follows.
- each polarizing plate is cut into a size of 150 mm in the width direction and 120 mm in the longitudinal direction, and the surface of the polarizing plate is sharpened with two razors at 100 mm intervals in a direction parallel to the slow axis of the ⁇ / 4 film. Mark the cross with a knife.
- the polarizing plate is conditioned for 24 hours or more in an environment of 23 ° C. and 55% RH, and a mark distance L1 in a direction parallel to the slow axis of the ⁇ / 4 film before processing is measured with a microscope.
- this polarizing plate is arrange
- the dimensional change rate is calculated by obtaining the change rate before and after this processing by the following equation.
- the dimensional change rate of the polarizing plate is the thickness of the ⁇ / 4 film C1, the acetyl group average substitution degree and the butyryl group average substitution degree of the cellulose ester contained in the ⁇ / 4 film C1, the stretching ratio and stretching of the ⁇ / 4 film C1. It can be adjusted by changing various parameters such as the direction, the type and amount of various additives contained in the ⁇ / 4 film C1, and the thickness and composition of the polarizer C2.
- the direction parallel to the slow axis of the ⁇ / 4 film is the same direction as the stretching direction of the ⁇ / 4 film C1.
- the polarizing plate C of the present invention can be produced by a general method, and can be produced by laminating a ⁇ / 4 film C1 and a polarizer C2.
- the polarizing plate C of the present invention includes the polarizer protective film C3, as shown in FIG. 1, the polarizing plate is formed by laminating and bonding in the configuration of ⁇ / 4 film C1 / polarizer C2 / polarizer protective film C3.
- C can be produced.
- the polarizing plate C Since the polarizing plate C is continuously supplied with a long ⁇ / 4 film C1 and a long polarizer C2 and bonded together in a roll-to-roll state, it is wound up in a roll state. Can be made. From the viewpoint of performance and production efficiency, it is preferable that the ⁇ / 4 film C1 is bonded to one surface of the polarizer C2 and the polarizer protective film C3 is bonded to the other surface of the polarizer C2. By laminating in this way, a polarizing plate C in a roll state can be obtained.
- the surface of the ⁇ / 4 film C1 on the side to be bonded to the polarizer C2 is subjected to alkali saponification treatment.
- the surface is then saponified by washing with water and drying.
- the polarizer C2 is immersed in a polyvinyl alcohol adhesive tank.
- the saponified surface of the surface of the ⁇ / 4 film C1 and the adhesive-side surface of the polarizer C2 are overlapped and bonded together.
- the laminated body thus bonded is dried in a dryer for 2 minutes to produce a polarizing plate C in which the ⁇ / 4 film C1 and the polarizer C2 are integrated.
- each layer which comprises the polarizing plate C is demonstrated.
- the ⁇ / 4 film C1 used for the polarizing plate C of the present invention has a cellulose ester having an acetyl group average substitution degree of 1.8 to 2.2 and a butyryl group average substitution degree of 0.1 to 0.4. It is characterized by containing. Presumably by having such an average degree of substitution of butyryl groups, the degree of polarization of polarizers (especially iodine pentamers) is probably lowered because butyryl groups exert a plastic effect even in high-temperature dry environments. Can be suppressed.
- the average degree of acetyl group substitution in the cellulose ester is preferably 1.85 to 2.15, more preferably 1.9 to 2.0.
- the average degree of butyryl group substitution is preferably from 0.15 to 0.35, more preferably from 0.2 to 0.32.
- the average degree of acetyl group substitution is a hydroxyl group (hydroxyl group) substituted (acetylated) with an acetyl group (CH 3 CO) among the three hydroxyl groups (hydroxyl groups) of each anhydroglucose constituting the cellulose ester. Means the average value of the numbers and takes a value in the range of 0-3.
- the average substitution degree of butyryl group is the hydroxyl group (hydroxyl group) substituted with a butyryl group (C 3 H 7 CO) among the three hydroxyl groups (hydroxyl group) of each anhydroglucose constituting cellulose. Means the average value of numbers and takes a value in the range of 0-3.
- acetyl group average substitution degree and butyryl group average substitution degree were determined by the methods prescribed in ASTM-D817-96 (testing method for cellulose ester, etc.).
- the acetyl group average substitution degree is 1.8 or more, deterioration of the film surface quality due to increase in the dope viscosity can be suppressed, and increase in haze due to increase in stretching tension can be suppressed. Moreover, a required phase difference can be obtained because the total acetyl group average substitution degree is 2.2 or less.
- the average degree of butyryl group substitution is 0.1 or more, the butyryl group exhibits a plastic effect even in a high-temperature dry environment, and suppresses a decrease in the degree of polarization of the polarizer (particularly, iodine pentamer). be able to.
- the butyryl group average substitution degree is 0.4 or less, the quality of the film surface is excellent.
- the cellulose ester used for the ⁇ / 4 film C1 can be produced by a known method.
- cellulose is mixed with a raw material cellulose, a predetermined organic acid (such as acetic acid), an acid anhydride (such as acetic anhydride), a catalyst (such as sulfuric acid), and esterified (acetylated and butyrylated).
- a predetermined organic acid such as acetic acid
- an acid anhydride such as acetic anhydride
- a catalyst such as sulfuric acid
- esterified acetylated and butyrylated
- the reaction is allowed to proceed until cellulose triesters (acetylated and butyrylated) are formed.
- triesters acetylation and butyrylation
- the three hydroxyl groups (hydroxyl groups) of the glucose unit are substituted with acetyl groups or butyryl groups of organic acids.
- a cellulose ester having a desired acetyl group average substitution degree and butyryl group average substitution degree is synthesized by hydrolyzing the cellulose triester. Thereafter, a cellulose ester is produced through steps such as filtration, precipitation, washing with water, dehydration, and drying.
- the ⁇ / 4 film may be appropriately mixed with polymer components other than cellulose ester.
- the polymer component to be mixed is preferably one having excellent compatibility with the cellulose ester, and the transmittance when formed into a film is preferably 80% or more, more preferably 90% or more, and further preferably 92% or more.
- Additives added to the dope when producing a ⁇ / 4 film include plasticizers, UV absorbers, retardation modifiers, antioxidants, deterioration inhibitors, peeling aids, surfactants, dyes, There are fine particles.
- additives other than fine particles may be added during the preparation of the cellulose ester solution, or may be added during the preparation of the fine particle dispersion. It is preferable to add a plasticizer, an antioxidant, an ultraviolet absorber, or the like that imparts heat and moisture resistance to the polarizing plate used in the liquid crystal image display device. The additive will be described below.
- Plasticizer In the present invention, a compound known as a so-called plasticizer is added to the ⁇ / 4 film for the purpose of improving mechanical properties, imparting flexibility, imparting water absorption resistance, reducing water vapor permeability, and adjusting retardation. Is preferred.
- plasticizers include polyester plasticizers, polyhydric alcohol ester plasticizers, polycarboxylic acid ester plasticizers (including phthalate ester plasticizers), glycolate plasticizers, ester plasticizers ( Citrate ester plasticizers, fatty acid ester plasticizers, phosphate ester plasticizers, trimellitic ester plasticizers, and the like). These may be used alone or in combination of two or more.
- the plasticizer that can be used for the ⁇ / 4 film is preferably selected from compounds having a normal temperature, normal pressure, liquid, and a boiling point of 200 ° C. or higher.
- Specific examples of the compound name include aliphatic dibasic acid ester type, phthalic acid ester type, and polyolefin type.
- the addition amount of the plasticizer is preferably 0.5 to 40.0% by mass, more preferably 1.0 to 30.0% by mass, and more preferably 3.0 to 20% with respect to the cellulose ester. Particularly preferred is 0.0 mass%.
- the added amount of the plasticizer is 0.5% by mass or more, the plasticizing effect is sufficient and the processability is improved. Further, when the content is 40% by mass or less, separation and elution of the plasticizer can be suppressed when it is aged for a long time, and optical unevenness, contamination to other parts, and the like can be more reliably suppressed.
- the ⁇ / 4 film used for the polarizing plate of the present invention can contain an ultraviolet absorber.
- ultraviolet absorbers include oxybenzophenone compounds, benzotriazole compounds, salicylic acid ester compounds, benzophenone compounds, cyanoacrylate compounds, nickel complex compounds, triazine compounds, and the like.
- a benzotriazole-based compound with little coloring is preferable.
- ultraviolet absorbers described in JP-A-10-182621, JP-A-8-337574, JP-A-2001-72782, JP-A-6-148430, JP-A-2002-31715, JP-A-2002-169020, Polymer ultraviolet absorbers described in JP-A-2002-47357, JP-A-2002-363420, and JP-A-2003-113317 are also preferably used.
- an ultraviolet absorber from the viewpoint of preventing the deterioration of polarizers and liquid crystals, it is excellent in the ability to absorb ultraviolet rays having a wavelength of 370 nm or less, and from the viewpoint of liquid crystal display properties, the absorption of visible light having a wavelength of 400 nm or more is small. Is preferred.
- UV absorbers useful in the present invention include 2- (2'-hydroxy-5'-methylphenyl) benzotriazole, 2- (2'-hydroxy-3 ', 5'-di-tert-butylphenyl) ) Benzotriazole, 2- (2'-hydroxy-3'-tert-butyl-5'-methylphenyl) benzotriazole, 2- (2'-hydroxy-3 ', 5'-di-tert-butylphenyl)- 5-chlorobenzotriazole, 2- (2′-hydroxy-3 ′-(3 ′′, 4 ′′, 5 ′′, 6 ′′ -tetrahydrophthalimidomethyl) -5′-methylphenyl) benzotriazole, 2,2-methylenebis ( 4- (1,1,3,3-tetramethylbutyl) -6- (2H-benzotriazol-2-yl) phenol), 2- (2'-hydroxy-3 ' tert-butyl-5'-methylphenyl) -5-chlorobenzotriazo
- TINUVIN 109 As commercially available products, TINUVIN 109, TINUVIN 171, TINUVIN 326, and TINUVIN 928 (all manufactured by BASF Japan) can be preferably used.
- An example of the polymeric ultraviolet absorber is a reactive ultraviolet absorber RUVA-93 manufactured by Otsuka Chemical Co., Ltd.
- benzophenone compounds include 2,4-dihydroxybenzophenone, 2,2'-dihydroxy-4-methoxybenzophenone, 2-hydroxy-4-methoxy-5-sulfobenzophenone, bis (2-methoxy-4-hydroxy-) 5-benzoylphenylmethane) and the like, but are not limited thereto.
- the ultraviolet absorber described above preferably used in the present invention is preferably a benzotriazole-based ultraviolet absorber or a benzophenone-based ultraviolet absorber, which has high transparency and is excellent in the effect of preventing the deterioration of the polarizing plate and the liquid crystal element, and unnecessary coloring.
- a benzotriazole-based ultraviolet absorber with a lower content is particularly preferably used.
- an ultraviolet absorber is a good solvent for cellulose esters such as methylene chloride, methyl acetate, dioxolane, or a mixed organic of a good solvent and a poor solvent such as a lower aliphatic alcohol (methanol, ethanol, propanol, butanol, etc.)
- a method of dissolving in a solvent and adding it to a cellulose ester solution as an ultraviolet absorber solution to form a dope is preferable.
- the content of the ultraviolet absorber is 0.01 to 5% by mass, particularly 0.5 to 3% by mass.
- retardation adjuster As the retardation adjusting agent added for adjusting the retardation, an aromatic compound having two or more aromatic rings as described in the specification of European Patent 911,656A2 can be used.
- the aromatic ring of the aromatic compound includes an aromatic hetero ring in addition to the aromatic hydrocarbon ring. Particularly preferred is an aromatic heterocycle, and the aromatic heterocycle is generally an unsaturated heterocycle. Of these, a 1,3,5-triazine ring is preferred.
- a heterocyclic group having a free valence on a nitrogen atom, a sugar ester compound, an aromatic ester compound, an aliphatic ester compound, or the like described in JP2012-173677A can be used.
- aromatic ester compound it is preferable to use an aromatic terminal ester plasticizer represented by the following general formula (1).
- B- (GA) nGB (Wherein B is a benzene monocarboxylic acid residue, G is an alkylene glycol residue having 2 to 12 carbon atoms, an aryl glycol residue having 6 to 12 carbon atoms, or an oxyalkylene glycol residue having 4 to 12 carbon atoms, A represents an alkylene dicarboxylic acid residue having 4 to 12 carbon atoms or an aryl dicarboxylic acid residue having 6 to 12 carbon atoms, and n represents an integer of 1 or more.)
- benzene monocarboxylic acid component of the aromatic terminal ester plasticizer used in the present invention examples include, for example, benzoic acid, para-tert-butylbenzoic acid, orthotoluic acid, metatoluic acid, p-toluic acid, dimethylbenzoic acid, ethylbenzoic acid, There are normal propyl benzoic acid, aminobenzoic acid, acetoxybenzoic acid and the like, and these can be used as one kind or a mixture of two or more kinds, respectively.
- alkylene glycol component having 2 to 12 carbon atoms of the aromatic terminal ester plasticizer used in the present invention examples include ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol, 1,2-butanediol, , 3-butanediol, 2-methyl-1,3-propanediol, 1,4-butanediol, 1,5-pentanediol, 2,2-dimethyl-1,3-propanediol (neopentyl glycol), 2, 2-diethyl-1,3-propanediol (3,3-dimethylolpentane), 2-n-butyl-2-ethyl-1,3-propanediol (3,3-dimethylolheptane), 3-methyl-1 , 5-pentanediol 1,6-hexanediol, 2,2,4-trimethyl 1,3-pentanediol, 2-ethyl 1,
- Examples of the oxyalkylene glycol component having 4 to 12 carbon atoms of the aromatic terminal ester include diethylene glycol, triethylene glycol, tetraethylene glycol, dipropylene glycol, tripropylene glycol, and the like. It can be used as a mixture of two or more.
- aryl glycol component having 6 to 12 carbon atoms of the aromatic terminal ester examples include hydroquinone, resorcin, bisphenol A, bisphenol F, bisphenol, etc., and these glycols are used as one kind or a mixture of two or more kinds. it can.
- alkylene dicarboxylic acid component having 4 to 12 carbon atoms of the aromatic terminal ester examples include succinic acid, maleic acid, fumaric acid, glutaric acid, adipic acid, azelaic acid, sebacic acid, and dodecanedicarboxylic acid. These are used as one kind or a mixture of two or more kinds.
- aryl dicarboxylic acid component having 6 to 12 carbon atoms examples include phthalic acid, terephthalic acid, 1,5-naphthalenedicarboxylic acid, 1,4-naphthalenedicarboxylic acid, and the like.
- the number average molecular weight of the aromatic terminal ester plasticizer is preferably 300 to 2000, and more preferably 500 to 1500.
- the acid value is 0.5 mgKOH / g or less, the hydroxyl (hydroxyl) value is 25 mgKOH / g or less, more preferably the acid value is 0.3 mgKOH / g or less, and the hydroxyl (hydroxyl) value is 15 mgKOH / g or less. Is preferred.
- the acid value means the number of milligrams of potassium hydroxide necessary for neutralizing the acid (carboxyl group present at the molecular end) contained in 1 g of the sample.
- the acid value and hydroxyl (hydroxyl group) value are measured according to JIS K0070 (1992).
- Example No. 1 (Aromatic terminal ester sample)> A reaction vessel is charged with 820 parts (5 moles) of terephthalic acid, 608 parts (8 moles) of 1,2-propylene glycol, 610 parts (5 moles) of triyl acid and 0.30 parts of tetraisopropyl titanate as a catalyst. While stirring in an air stream, a reflux condenser was attached to reflux excess monohydric alcohol, and heating was continued at 130 to 250 ° C. until the acid value became 2 or less, and water produced was continuously removed.
- Example No. 2 (Aromatic terminal ester sample)> A sample was used except that 500 parts (3.5 moles) of adipic acid, 305 parts (2.5 moles) of benzoic acid, 583 parts (5.5 moles) of diethylene glycol, and 0.45 parts of tetraisopropyl titanate as a catalyst were used in the reaction vessel. No. In the same manner as in No. 1, an aromatic terminal ester having the following properties was obtained. Viscosity (25 ° C., mPa ⁇ s); 90 Acid value: 0.05
- Example No. 3 (Aromatic terminal ester sample)> Sample No. except that 410 parts (2.5 moles) of phthalic acid, 610 parts (5 moles) of benzoic acid, 737 parts (5.5 moles) of dipropylene glycol and 0.40 parts of tetraisopropyl titanate as the catalyst were used in the reaction vessel. . In the same manner as in No. 1, an aromatic terminal ester plasticizer having the following properties was obtained.
- Viscosity 25 ° C., mPa ⁇ s); 43400 Acid value: 0.2
- this invention is not limited to this.
- the content of the aromatic terminal ester plasticizer used in the present invention is preferably 1 to 20% by mass, more preferably 3 to 11% by mass in the ⁇ / 4 film.
- the ⁇ / 4 film is a vinyl ester having a cellulose ester and a substituent selected from a carboxyl group, a hydroxyl group, an amino group, an amide group, and a sulfonic acid group and having a weight average molecular weight in the range of 500 to 200,000. It is preferable to contain a polymer or oligomer of a system compound.
- the mass ratio of the content of the cellulose ester and the polymer or oligomer is preferably in the range of 95: 5 to 50:50.
- the ⁇ / 4 film contains, for example, an antioxidant, a peroxide decomposer, a radical polymerization inhibitor, a metal deactivator, an acid scavenger, amines and the like as a deterioration inhibitor. Can do.
- Degradation inhibitors are described in, for example, JP-A-3-199201, JP-A-5-97073, JP-A-5-194789, JP-A-5-271471, JP-A-6-107854.
- the amount of the deterioration inhibitor added is a cellulose solution used for the production of a ⁇ / 4 film from the viewpoint of the effect of the addition of the deterioration inhibitor and suppressing bleeding out of the deterioration inhibitor to the film surface (exudation).
- the dope is preferably 0.01 to 1% by mass, and more preferably 0.01 to 0.2% by mass.
- BHT butylated hydroxytoluene
- TBA tribenzylamine
- fine particles can be contained in the ⁇ / 4 film as a matting agent.
- the ⁇ / 4 film is a long film, it can be easily conveyed and taken up by the matting agent.
- the particle size of the matting agent is preferably 10 nm to 0.1 ⁇ m primary particles or secondary particles.
- a substantially spherical matting agent having a primary particle acicular ratio of 1.1 or less is preferably used.
- silicon dioxide is particularly preferable.
- Preferred fine particles of silicon dioxide for the present invention include, for example, Aerosil R972, R972V, R974, R812, 200, 200V, 300, R202, OX50, TT600 (Nippon Aerosil Co., Ltd.) manufactured by Nippon Aerosil Co., Ltd. What is marketed by a brand name can be mentioned, Aerosil 200V, R972, R972V, R974, R202, R812 can be used preferably.
- Examples of polymer fine particles include silicone resin, fluorine resin, and acrylic resin. Silicone resins are preferable, and those having a three-dimensional network structure are particularly preferable. Examples include Tospearl 103, 105, 108, 120, 145, 3120, and 240 (manufactured by Toshiba Silicone Co., Ltd.). Can do.
- the silicon dioxide fine particles preferably have a primary average particle diameter of 20 nm or less and an apparent specific gravity of 70 g / L or more.
- the average diameter of the primary particles is more preferably 5 to 16 nm, further preferably 5 to 12 nm. A smaller primary particle average diameter is preferred because haze is low.
- the apparent specific gravity is preferably 90 to 200 g / L or more, and more preferably 100 to 200 g / L or more. Higher apparent specific gravity makes it possible to produce a high-concentration fine particle dispersion, which is preferable because no haze or aggregates are generated.
- the addition amount of the matting agent in the present invention preferably lambda / 4 film 1 m 2 per 0.01 ⁇ 1.0 g is more preferably 0.03 ⁇ 0.3 g, more preferably 0.08 ⁇ 0.16 g.
- thermal stabilizers such as inorganic fine particles such as kaolin, talc, diatomaceous earth, quartz, calcium carbonate, barium sulfate, titanium oxide, and alumina, and alkaline earth metal salts such as calcium and magnesium may be added.
- a surfactant, a peeling accelerator, an antistatic agent, a flame retardant, a lubricant, an oil agent and the like may be added.
- the ⁇ / 4 film C1 is produced by stretching sequentially or simultaneously in the longitudinal direction (MD direction) and the lateral direction (TD direction), and an in-plane retardation Ro 550 at a wavelength of 550 nm is 80 nm or more. It is preferable that it is 160 nm or less.
- the slow axis b of the ⁇ / 4 film C1 has an angle ⁇ with respect to the longitudinal direction (MD direction) of the ⁇ / 4 film C1 of more than 0 ° and less than 90 °.
- the “long direction” means the transport direction with respect to the width of the ⁇ / 4 film.
- the ⁇ / 4 film C1 is stretched in an oblique direction so as to have a slow axis in a direction that is neither the conveying direction of the long original film nor the orthogonal direction thereof.
- the ⁇ / 4 film C1 is stretched at an angle of more than 0 ° and less than 90 ° with respect to the conveyance direction of the ⁇ / 4 film C1.
- the angle with respect to the transport direction of the ⁇ / 4 film is an angle ⁇ between the longitudinal direction (MD direction) of the ⁇ / 4 film C1 and the slow axis b of the ⁇ / 4 film C1.
- the slow axis of the ⁇ / 4 film means an axis in which the phase is delayed and the traveling speed of the light is slowed when light propagates through the ⁇ / 4 film.
- a ⁇ / 4 film having the slow axis is produced by stretching at an angle of more than 0 ° and less than 90 ° with respect to the extending direction of the original film. Can do.
- the angle ⁇ formed by the conveyance direction of the ⁇ / 4 film and the slow axis can be arbitrarily set to a desired angle of more than 0 ° and less than 90 °, more preferably 30 ° to 60 °, still more preferably Is from 35 ° to 55 °, particularly preferably from 40 to 50 °.
- the variation of the in-plane retardation Ro of the ⁇ / 4 film (stretched film) is 5 nm or less, preferably 3 nm or less, at least 1300 mm in the width direction.
- the in-plane retardation Ro 550 at a wavelength of 550 nm is preferably 80 nm or more and 160 nm or less, and more preferably 100 to 145 nm.
- Such a range of the in-plane retardation Ro 550 can be adjusted by adjusting a draw ratio using a resin that develops a retardation by stretching, or by adding a retardation adjusting agent during the production of a ⁇ / 4 film. it can.
- the ⁇ / 4 film is preferably a retardation plate having a retardation of approximately 1 ⁇ 4 of the wavelength in the visible light wavelength range in order to obtain almost perfect circularly polarized light in the visible light wavelength range. .
- “Retardation of approximately 1/4 in the wavelength range of visible light” is expressed by the following formula (i) measured at a wavelength of 450 nm, which has reverse wavelength dispersion with a longer retardation at wavelengths from 400 nm to 700 nm.
- the retardation value Ro 450 and the retardation value Ro 550 measured at a wavelength of 550 nm preferably satisfy 1 ⁇ Ro 550 / Ro 450 ⁇ 1.6.
- the in-plane retardation Ro ⁇ of the ⁇ / 4 film is represented by the following formula (i).
- the value of the phase difference can be calculated by measuring the birefringence at each wavelength in an environment of 23 ° C. and 55% RH using, for example, Axoscan manufactured by Axometrics.
- n x, n y is, 23 °C ⁇ 55% RH, 450nm, say 550 nm, the maximum of the refractive index in the plane of the refractive index n x (film in each of the 650 nm, also the slow axis direction of the refractive index. ), Ny (refractive index in the direction orthogonal to the slow axis in the film plane), and d is the thickness (nm) of the film.
- substantially 45 ° means 40 ° or more and 50 ° or less.
- the angle between the slow axis in the plane of the ⁇ / 4 film and the absorption axis of the polarizer is preferably 30 ° or more and 60 ° or less, more preferably 35 ° or more and 55 ° or less, and 43 ° or more.
- the angle is more preferably 47 ° or less, and most preferably 44 ° or more and 46 ° or less.
- an obliquely stretched ⁇ / 4 film having a hard coat layer coated thereon is used.
- the ⁇ / 4 film may contain a resin having a positive intrinsic birefringence value or a resin having a negative intrinsic birefringence value in addition to the cellulose ester.
- “Resin having a positive intrinsic birefringence value” refers to a resin having an increased refractive index in the stretched direction
- “resin having a negative intrinsic birefringence” refers to a refractive index in a direction perpendicular to the stretched direction. This refers to a resin that increases.
- the resin having a positive intrinsic birefringence value include cellulose acylate resins, cyclic olefin resins, and polycarbonate resins.
- Examples of the resin having negative intrinsic birefringence include polyester resins, acrylic resins, styrene resins, and copolymers of these polymers. These raw materials may be used by mixing with cellulose ester.
- the ⁇ / 4 film may be a single layer film or a multilayer film.
- an unstretched film is mainly used as a raw film, but it may be a film in which any one of longitudinal stretching, lateral stretching, and oblique stretching has already been carried out alone or multiple times.
- the ⁇ / 4 film used for the polarizing plate of the present invention may be referred to as a cellulose ester film.
- Cellulose ester has a tendency to weaken the reverse wavelength dispersibility while lowering the degree of substitution to relatively improve the retardation development.
- the substitution degree of the cellulose ester is increased, the reverse wavelength dispersibility is increased, but the retardation development property is decreased.
- the film thickness is increased. Accordingly, the retardation development and wavelength dispersion are enhanced by adding various additives such as a retardation adjusting agent and a wavelength dispersion adjusting agent to the cellulose ester.
- the raw material cellulose of the cellulose ester used in the present invention may be wood pulp or cotton linter, and the wood pulp may be softwood or hardwood, but softwood is more preferable.
- a cotton linter is preferably used from the viewpoint of peelability during film formation.
- the cellulose ester made from these can be mixed suitably or can be used independently.
- the ratio of cellulose ester derived from cellulose linter: cellulose ester derived from wood pulp (coniferous): cellulose ester derived from wood pulp (hardwood) is 100: 0: 0, 90: 10: 0, 85: 15: 0, 50:50: 0, 20: 80: 0, 10: 90: 0, 0: 100: 0, 0: 0: 100, 80:10:10, 85: 0: 15, 40:30:30.
- cellulose ester 1 g is added to 20 ml of pure water (electric conductivity 0.1 ⁇ S / cm or less, pH 6.8), and the pH is 6 to 6 when stirred in a nitrogen atmosphere at 25 ° C. for 1 hr. 7.
- the electrical conductivity is preferably 1 to 100 ⁇ S / cm.
- the residual sulfuric acid content in the cellulose ester used in the present invention is preferably in the range of 0.1 to 40 ppm in terms of elemental sulfur. It is considered that elemental sulfur is contained in the form of salt. If the residual sulfuric acid content exceeds 40 ppm, the deposit on the die lip during heat melting increases, such being undesirable. Moreover, since it becomes easy to fracture
- the residual sulfuric acid content in the cellulose ester is preferably as low as possible. However, if it is less than 0.1, the burden of the cellulose ester washing step becomes too large, and it may be easily broken, which is not preferable. The increase in the number of washings may affect the resin, but it is not well understood. Furthermore, the range of 0.1 ppm or more and 30 ppm or less is preferable.
- the residual sulfuric acid content can be similarly measured by ASTM-D817-96.
- the total residual acid amount including other (such as acetic acid) residual acid is preferably 1000 ppm or less, more preferably 500 ppm or less, and further preferably 100 ppm or less.
- a poor solvent such as methanol or ethanol, or, as a result, a mixed solvent of a poor solvent and a good solvent can be used as long as it is a poor solvent. Organic impurities can be removed.
- cellulose ester In order to improve the heat resistance, mechanical properties, optical properties, etc. of cellulose ester, it can be dissolved in a good solvent of cellulose ester and then reprecipitated in a poor solvent to remove low molecular weight components and other impurities of cellulose ester. it can. Furthermore, another polymer or a low molecular weight compound may be added after the cellulose ester reprecipitation treatment.
- the cellulose ester used by this invention is a thing with few bright spot foreign materials when it is made into a film.
- Bright spot foreign matter means that two polarizing plates are placed orthogonally (crossed Nicols), a ⁇ / 4 film (cellulose ester film) is placed between them, and light from the light source is applied from one side to the other. When the cellulose ester film is observed from the surface, the light from the light source appears to leak (foreign matter).
- the number of bright spots having a diameter of 0.01 mm or more is preferably 200 pieces / cm 2 or less.
- the polarizing plate used for the evaluation is desirably composed of a protective film having no bright spot foreign matter, and a polarizing plate using a glass plate for protecting the polarizer is preferably used.
- One of the causes of the bright spot foreign matter is considered to be unacetylated or low-acetylated cellulose contained in the cellulose ester.
- the ⁇ / 4 film used for the polarizing plate of the present invention is a long film stretched in an oblique direction so as to have an in-plane slow axis in the oblique direction with respect to the conveying direction of the long film.
- the film before oblique stretching is referred to as “raw film”.
- the raw fabric film may be produced by either a solution casting method or a melt casting method. A method for producing a solution casting method and an obliquely stretched ⁇ / 4 film will be described below.
- the production of the raw film according to the present invention is a step of preparing a dope by dissolving an additive such as cellulose ester and the plasticizer in a solvent, and a step of casting the dope on a belt-shaped or drum-shaped metal support.
- the raw film according to the present invention preferably contains 60 to 95% by mass of cellulose ester in the solid content.
- the concentration of cellulose ester in the dope is preferably higher because the drying load after casting on the metal support can be reduced. However, if the concentration of cellulose ester is too high, the load during filtration increases and the filtration accuracy increases. Becomes worse.
- the concentration for achieving both of these is preferably 10 to 35% by mass, more preferably 15 to 25% by mass.
- Organic solvents that dissolve cellulose esters and are useful for forming cellulose ester solutions or dopes include chlorinated organic solvents and non-chlorinated organic solvents.
- Methylene chloride methylene chloride
- non-chlorine organic solvent examples include methyl acetate, ethyl acetate, amyl acetate, acetone, tetrahydrofuran, 1,3-dioxolane, 1,4-dioxane, cyclohexanone, ethyl formate, 2,2,2-trifluoroethanol, 2,2,3,3-hexafluoro-1-propanol, 1,3-difluoro-2-propanol, 1,1,1,3,3,3-hexafluoro-2-methyl-2-propanol, 1, Examples include 1,1,3,3,3-hexafluoro-2-propanol, 2,2,3,3,3-pentafluoro-1-propanol, and nitroethane.
- a dissolution method at room temperature can be used, but an insoluble material can be obtained by using a dissolution method such as a high-temperature dissolution method, a cooling dissolution method, or a high-pressure dissolution method. Can be reduced, which is preferable.
- a dissolution method such as a high-temperature dissolution method, a cooling dissolution method, or a high-pressure dissolution method. Can be reduced, which is preferable.
- methylene chloride can be used, but methyl acetate, ethyl acetate, and acetone are preferably used. Particularly preferred is methyl acetate.
- an organic solvent having good solubility with respect to the cellulose ester is referred to as a good solvent, and has a main effect on dissolution, and an organic solvent used in a large amount among them is a main (organic) solvent or a main ( Organic) solvent.
- the dope used for producing the ⁇ / 4 film preferably contains 1 to 40% by mass of an alcohol having 1 to 4 carbon atoms in addition to the organic solvent.
- the alcohol having 1 to 4 carbon atoms casts the dope onto the metal support and then the solvent starts to evaporate.
- the dope film (web) gels and the web becomes strong. It is used as a gelling solvent that makes it easy to peel off.
- the proportion of the alcohol having 1 to 4 carbon atoms is small, it also has a role of promoting the dissolution of the cellulose ester of the non-chlorine organic solvent.
- Examples of the alcohol having 1 to 4 carbon atoms include methanol, ethanol, n-propanol, iso-propanol, n-butanol, sec-butanol, and tert-butanol. Of these, ethanol is preferred because it has excellent dope stability, has a relatively low boiling point, and has good drying properties. These organic solvents are called poor solvents because they are not soluble in cellulose esters alone.
- the concentration of the cellulose ester in the dope is adjusted to 15 to 30% by mass and the dope viscosity is set to a range of 100 to 500 Pa ⁇ s.
- a general method can be used. When heating and pressurization are combined, it is possible to heat above the boiling point at normal pressure. It is preferable to stir and dissolve while heating at a temperature that is equal to or higher than the boiling point of the solvent at normal pressure and that the solvent does not boil under pressure, in order to prevent the generation of massive undissolved materials called gels and macos. Moreover, after mixing a cellulose ester with a poor solvent and making it wet or swell, the method of adding a good solvent and melt
- Pressurization may be performed by a method of injecting an inert gas such as nitrogen gas or a method of increasing the vapor pressure of the solvent by heating. Heating is preferably performed from the outside.
- a jacket type is preferable because temperature control is easy.
- the heating temperature with the addition of the solvent is preferably higher from the viewpoint of the solubility of the cellulose ester, but if the heating temperature is too high, the required pressure increases and the productivity deteriorates.
- a preferred heating temperature is 45 to 120 ° C, more preferably 60 to 110 ° C, and still more preferably 70 ° C to 105 ° C. The pressure is adjusted so that the solvent does not boil at the set temperature.
- a cooling dissolution method is also preferably used, whereby the cellulose ester can be dissolved in a solvent such as methyl acetate.
- the cellulose ester solution is filtered using an appropriate filter medium such as filter paper.
- the filter medium it is preferable that the absolute filtration accuracy is small in order to remove insoluble matters and the like. However, if the absolute filtration accuracy is too small, there is a problem that the filter medium is likely to be clogged. For this reason, a filter medium with an absolute filtration accuracy of 0.008 mm or less is preferable, a filter medium with 0.001 to 0.008 mm is more preferable, and a filter medium with 0.003 to 0.006 mm is more preferable.
- the material of the filter medium there are no particular restrictions on the material of the filter medium, and ordinary filter media can be used.
- plastic filter media such as polypropylene and Teflon (registered trademark), and metal filter media such as stainless steel do not drop off fibers. preferable. It is preferable to remove and reduce impurities, particularly bright spot foreign matter, contained in the raw material cellulose ester by filtration.
- the dope can be filtered by a normal method, but the method of filtering while heating at a temperature not lower than the boiling point of the solvent at normal pressure and in a range where the solvent does not boil under pressure is the filtration pressure before and after filtration.
- the increase in the difference (referred to as differential pressure) is small and preferable.
- a preferred temperature is 45 to 120 ° C., more preferably 45 to 70 ° C., and still more preferably 45 to 55 ° C.
- the filtration pressure is preferably 1.6 MPa or less, more preferably 1.2 MPa or less, and further preferably 1.0 MPa or less.
- the metal support in the casting process is preferably a mirror-finished surface, and a stainless steel belt or a drum whose surface is plated with a casting is preferably used as the metal support.
- the cast width can be 1 to 4 m.
- the surface temperature of the metal support in the casting step is set to ⁇ 50 ° C. to below the temperature at which the solvent boils and does not foam. A higher temperature is preferable because the web can be dried faster, but if it is too high, the web may foam or the flatness may deteriorate.
- a preferable support temperature is appropriately determined at 0 to 100 ° C., and more preferably 5 to 30 ° C.
- the web is gelled by cooling and peeled from the drum in a state containing a large amount of residual solvent.
- the method for controlling the temperature of the metal support is not particularly limited, and there are a method of blowing warm air or cold air, and a method of contacting hot water with the back side of the metal support. It is preferable to use warm water because heat transfer is performed efficiently, so that the time until the temperature of the metal support becomes constant is short.
- warm air considering the temperature drop of the web due to the latent heat of vaporization of the solvent, while using warm air above the boiling point of the solvent, there may be cases where wind at a temperature higher than the target temperature is used while preventing foaming. .
- the residual solvent amount when peeling the web from the metal support is preferably 10 to 150% by mass, more preferably 20 to 40% by mass or 60 to 130% by mass. %, Particularly preferably 20 to 30% by mass or 70 to 120% by mass.
- the temperature at the peeling position on the metal support is preferably ⁇ 50 to 40 ° C., more preferably 10 to 40 ° C., and most preferably 15 to 30 ° C.
- the amount of residual solvent is defined by the following formula.
- Residual solvent amount (% by mass) ⁇ (MN) / N ⁇ ⁇ 100 Note that M is the mass of a sample collected during or after the production of the web or film, and N is the mass after heating M at 115 ° C. for 1 hour.
- the web is peeled off from the metal support, further dried, and dried until the residual solvent amount is 0.5% by mass or less.
- a roll drying method (a method in which a plurality of rolls arranged at the top and bottom are alternately passed through the web and dried) or a tenter method is used while drying the web.
- the web When peeling from the metal support, the web is stretched in the longitudinal direction due to the peeling tension and the subsequent conveying tension. Therefore, in the present invention, when peeling the web from the casting support, the peeling and conveying tensions are reduced as much as possible. It is preferable to carry out in the state. Specifically, for example, it is effective to set it to 50 N / m or more and 170 N / m or less. At that time, it is preferable to apply a cold air of 20 ° C. or less to fix the web rapidly.
- the dried raw film can be stretched to a desired angle by the above-described oblique stretching tenter to form a ⁇ / 4 film.
- the ⁇ / 4 film preferably has an in-plane retardation Ro 550 measured at a light wavelength of 550 nm in the range of 80 to 160 nm, and the web formed as described above is stretched.
- the in-plane phase difference Ro can be given.
- the stretching method applicable to the present invention is not particularly limited.
- Both ends of the web are fixed with clips and pins, and the distance between the clips and pins is extended in the longitudinal direction according to the direction of travel, similarly the method of extending in the horizontal direction and extending in the horizontal direction, or the vertical and horizontal directions are expanded at the same time.
- stretching to both directions is employable individually or in combination.
- the film may be stretched in the transverse direction, in the longitudinal direction, or in both directions, and when stretched in both directions, simultaneous stretching or sequential stretching may be used. May be.
- driving the clip portion by the linear drive method is preferable because smooth stretching can be performed and the risk of breakage and the like can be reduced.
- the film is usually stretched in the width direction (TD direction) and contracted in the transport direction (MD direction), but when contracted, it is easy to match the main chain direction when transported in an oblique direction. In addition, the phase difference effect is even greater.
- the shrinkage rate can be determined by the transport angle.
- the oblique stretching step is a step of stretching the formed long film in a direction oblique to the longitudinal direction.
- a film can be manufactured to desired arbitrary length by manufacturing a film continuously.
- the film after film formation may be continuously supplied to the oblique stretching process from the film forming process without winding up the film. It is preferable to perform the film forming step and the oblique stretching step continuously, because the film forming conditions can be changed by feeding back the film thickness after stretching and the optical value result, and a desired long stretched film can be obtained.
- a long stretched film having a slow axis at an angle of more than 0 ° and less than 90 ° with respect to the longitudinal direction of the ⁇ / 4 film is produced.
- the angle with respect to the longitudinal direction of the film is an angle in the film plane. Since the slow axis in the film plane appears in the stretching direction, in the manufacturing method according to the present embodiment, stretching is performed at an angle of more than 0 ° and less than 90 ° with respect to the extending direction of the ⁇ / 4 film. A long stretched film having such a slow axis can be produced.
- the angle formed between the longitudinal direction of the long stretched film and the slow axis that is, the orientation angle ⁇ can be arbitrarily set to a desired angle within a range of more than 0 ° and less than 90 °.
- an oblique stretching apparatus In order to impart an oblique orientation to the long film subjected to stretching in this embodiment, an oblique stretching apparatus is used.
- the oblique stretching apparatus used in the present embodiment can freely set the orientation angle of the ⁇ / 4 film by variously changing the path pattern of the gripping tool traveling support tool, and further the orientation axis of the ⁇ / 4 film. It is preferable that the film stretching apparatus be capable of highly accurately orienting horizontally in the film width direction and capable of controlling the film thickness and retardation with high accuracy.
- FIG. 2 is a schematic diagram for explaining oblique stretching used in the method for producing a long stretched film of the present embodiment.
- this is an example, and the present invention is not limited to this.
- the feeding direction D1 of the long film is different from the winding direction D2 of the elongated film after stretching, and forms a feeding angle ⁇ i.
- the feeding angle ⁇ i can be arbitrarily set to a desired angle in the range of more than 0 ° and less than 90 °.
- the long film has left and right grippers (a pair of grips) at the entrance of the oblique stretching apparatus (the gripping tool is a grip start point for gripping the long film, and a straight line connecting the grip start points is indicated by reference symbol A). Of the gripping tool) and travels as the gripping tool travels.
- the gripping tool pair is composed of left and right gripping tools Ci and Co that are opposed to a direction substantially perpendicular to the traveling direction of the long film (feeding direction D1) at the entrance of the oblique stretching apparatus.
- the left and right gripping tools Ci and Co travel along an asymmetrical path, respectively, and the position at the end of stretching (the gripping release point at which the gripping tool releases the gripping, and the straight line connecting the gripping release points is denoted by reference symbol B.
- the long stretched film gripped in (shown) is released.
- the left and right gripping tools facing each other at the entrance of the oblique stretching apparatus travel along the inner gripping tool travel support tool Ri and the outer gripping tool travel support tool Ro which are asymmetrical to the left and right, respectively.
- the gripping tool Ci traveling on the inner gripping tool travel support tool Ri has a positional relationship that advances relative to the gripping tool Co traveling on the outer gripping tool travel support tool Ro.
- the long film is obliquely stretched in the direction of ⁇ L.
- substantially vertical indicates that the angle is in a range of 90 ⁇ 1 °.
- the method for producing a ⁇ / 4 film used for the polarizing plate of the present invention is performed using a stretching apparatus capable of oblique stretching.
- This stretching apparatus is an apparatus that heats a long film to an arbitrary temperature at which stretching can be performed and stretches the film in an oblique manner.
- This stretching apparatus is configured to support the traveling of the heating tool Z and a plurality of gripping tools Ci and Co that are gripped on both sides of the long film, and the gripping tools Ci and Co. And a gripping tool travel support tool.
- the gripping tool traveling support tool having the gripping tool has an endless continuous track, and the gripping tool that has released the grip of the long stretched film at the exit of the stretching device is sequentially returned to the gripping start point by the gripping tool travel support tool. It is supposed to be.
- the gripping tool travel support tool may be, for example, a form in which an endless chain whose path is regulated by a guide rail or a gear is provided with a gripping tool, or a form in which an endless guide rail is provided with a gripping tool. It may be. That is, in the present invention, the gripping tool travel support tool may be, for example, an endless guide rail provided with an endless chain, or may be an endless guide rail provided with an endless chain. An endless guide rail without a chain may be used.
- the gripper travel support tool does not include a chain, the gripper travels along the path of the gripper travel support tool itself.
- the gripper travel support tool includes the chain, the gripper travel support tool travels along the path of the gripper travel support tool. Run.
- the gripping tool travels along the path of the gripping tool travel support tool
- the gripping tool is connected via a chain provided with the gripping tool. You may drive
- the gripping tool traveling support of the stretching apparatus has an asymmetric shape on the left and right, and the path pattern is manually or automatically set according to the orientation angle, stretch ratio, etc. given to the long stretched film to be manufactured. It can be adjusted with.
- the path of each gripper travel support tool can be freely set and the path pattern of the gripper travel support tool can be arbitrarily changed.
- the difference in travel speed between at least a gripping tool gripping the film is usually 1% or less, preferably 0.5% or less, more preferably 0.1% or less of the travel speed. It is constant speed. This is because if there is a difference in running speed between the left and right of the long stretched film at the exit of the stretching process, wrinkles and misalignment will occur at the exit of the stretching process, so the speed difference between the left and right gripping tools constituting the gripping tool pair is substantially This is because it is required to be constant speed.
- the ⁇ / 4 film is provided with a hard coat layer on the surface thereof.
- the ⁇ / 4 film has a surface treatment layer other than the hard coat layer on its surface, such as an antistatic layer, a backcoat layer, an antireflection layer, a slippery layer, an adhesive layer, an antiglare layer, and a barrier layer.
- a sex layer can be provided.
- the hard coat layer preferably includes a hardened layer containing a resin containing an alicyclic hydrocarbon and fine particles coated with a polymer silane coupling agent.
- the hard coat layer preferably further includes a buffer layer formed between the cured layer and the ⁇ / 4 film.
- the buffer layer preferably contains a resin different from the resin contained in the cured layer and fine particles coated with a polymer silane coupling agent.
- the cured layer of this embodiment contains an active energy ray-curable resin having an alicyclic structure (hereinafter also simply referred to as a curable resin) and fine particles coated with a polymer silane coupling agent. It is provided in order to avoid the formation of scratches on the surface.
- the alicyclic structure include norbornyl, tricyclodecanyl, tetracyclododecanyl, pentacyclopentadecanyl, adamantyl, diamantanyl and the like.
- An active energy ray is defined as an energy ray that can decompose an active species-generating compound (photopolymerization initiator) to generate an active species.
- active energy rays include light energy rays such as visible light, ultraviolet rays (UV), electron beams (EB), infrared rays, X rays, ⁇ rays, ⁇ rays, and ⁇ rays.
- UV ultraviolet rays
- EB electron beams
- infrared rays X rays
- ⁇ rays ⁇ rays
- ⁇ rays ⁇ rays
- the active energy ray curable resin preferably has an ethylenically unsaturated double bond.
- the ethylenically unsaturated double bond group include polymerizable functional groups such as (meth) acryloyl group, vinyl group, styryl group and allyl group. Among them, (meth) acryloyl group and —C (O) OCH ⁇ CH 2 is preferred.
- the active energy ray-curable resin having an alicyclic structure is preferably composed of a hydrocarbon group having an alicyclic structure and a group having an ethylenically unsaturated double bond bonded via a linking group.
- the linking group include a single bond, an alkylene group, an amide group, a carbamoyl group, an ester group, an oxycarbonyl group, an ether group, or a group obtained by combining these.
- polyols such as diols and triols having an alicyclic structure, carboxylic acids having (meth) acryloyl groups, vinyl groups, styryl groups, allyl groups, carboxylic acid derivatives, epoxy derivatives, isocyanate derivative compounds, etc.
- polyols such as diols and triols having an alicyclic structure, carboxylic acids having (meth) acryloyl groups, vinyl groups, styryl groups, allyl groups, carboxylic acid derivatives, epoxy derivatives, isocyanate derivative compounds, etc.
- R1 is a hydrogen atom or an alkyl group having 1 to 3 carbon atoms
- R2 is an alkylene group or alkylene oxide group having 1 to 5 carbon atoms
- R3 is a hydrogen atom or an alkyl group having 1 to 3 carbon atoms
- n is 1) Or an integer of 2.
- R1 represents a hydrogen atom or an alkyl group having 1 to 3 carbon atoms, preferably a hydrogen atom, a methyl group, or an ethyl group.
- R2 represents an alkylene group having 1 to 5 carbon atoms or an alkylene oxide group, and preferably represents a methylene group, an ethylene group, a methylene oxide group, or an ethylene oxide group.
- R3 represents a hydrogen atom or an alkyl group having 1 to 3 carbon atoms, and preferably represents a hydrogen atom, a methyl group, or an ethyl group.
- Examples of commercially available compounds represented by the above general formulas (I) and (II) include NK ester A-DCP (tricyclodecane dimethanol diacrylate, manufactured by Shin-Nakamura Chemical Co., Ltd.). However, it is not limited to these.
- L and L ′ each independently represent a divalent or higher valent linking group and are not divalent simultaneously.
- n represents an integer of 1 to 3.
- L and L ′ each independently represent a divalent or higher linking group and are not divalent simultaneously.
- n represents an integer of 1 to 2.
- L and L ′ each independently represent a divalent or higher linking group and are not divalent simultaneously.
- n represents an integer of 1 to 2.
- L, L ′ and L ′′ each independently represent a divalent or higher valent linking group.
- L and L ′ each independently represent a divalent or higher linking group, and are not divalent simultaneously.
- the cured layer preferably contains 30% by mass or more of active energy ray-curable resin having an alicyclic structure, and more preferably 50% by mass or more.
- the cured layer preferably contains a photopolymerization initiator to accelerate the curing of the actinic radiation curable resin.
- Specific examples of the photopolymerization initiator include alkylphenone series, acetophenone, benzophenone, hydroxybenzophenone, Michler's ketone, ⁇ -amyloxime ester, thioxanthone and the like, and derivatives thereof. It is not something.
- Commercially available products may be used as the photopolymerization initiator, and preferred examples include Irgacure 184, Irgacure 907, and Irgacure 651 manufactured by BASF Japan.
- the hardened layer may contain fine particles. Although it does not restrict
- the silica fine particles may be hollow particles having cavities inside. Among these, fine particles coated with a polymer silane coupling agent are particularly preferable because they give an appropriate hardness to the cured layer and exhibit good mechanical properties.
- the polymer silane coupling agent refers to a reaction product of a polymerizable monomer and a silane coupling agent (reactive silane compound).
- a polymer silane coupling agent can be obtained, for example, according to the method for producing a reaction product of a polymerizable monomer and a reactive silane compound disclosed in JP-A-11-116240.
- polymerizable monomer examples include (meth) acrylic acid, methyl (meth) acrylate, ethyl (meth) acrylate, (meth) acrylic acid-n-propyl, (meth) acrylic acid isopropyl, (meth) -N-butyl, isobutyl (meth) acrylate, (meth) acrylic acid-n-hexyl, (meth) acrylic acid cyclohexyl, (meth) acrylic acid-n-heptyl, (meth) acrylic acid-n-octyl, ( 2-ethylhexyl (meth) acrylate, nonyl (meth) acrylate, decyl (meth) acrylate, dodecyl (meth) acrylate, phenyl (meth) acrylate, toluyl (meth) acrylate, benzyl (meth) acrylate , 2-methoxyethyl (meth) acrylate
- an organosilicon compound represented by the following formula (2) is preferably used as the reactive silane compound.
- R represents an organic group having 1 to 10 carbon atoms selected from a substituted or unsubstituted hydrocarbon group.
- X represents a (meth) acryloyl group, an epoxy group (glycid group), a urethane group, an amino group, One or more functional groups selected from fluoro groups.
- Specific examples of the organosilicon compound represented by the formula (2) include 3,3,3-trifluoropropyltrimethoxysilane, methyl-3,3,3-trifluoropropyldimethoxysilane, ⁇ - (3, 4-epoxycyclohexyl) ethyltrimethoxysilane, ⁇ -glycidoxymethyltrimethoxysilane, ⁇ -glycidoxymethyltriethoxysilane, ⁇ -glycidoxyethyltrimethoxysilane, ⁇ -glycidoxyethyltriethoxysilane, ⁇ -glycidoxyethyltriethoxys
- Polymeric silane coupling agent is prepared by reacting a polymerizable monomer with a reactive silane compound. Specifically, an organic solvent solution in which a reactive silane compound is mixed in an amount of 0.5 to 20 parts by weight, further 1 to 10 parts by weight with respect to 100 parts by weight of the polymerizable monomer is prepared, and polymerization is started. It can be obtained by adding an agent and heating.
- the polymer silane coupling agent-coated fine particles can be prepared by adding a polymer silane coupling agent to a fine particle organic solvent dispersion and coating the fine particles with the polymer silane coupling agent in the presence of an alkali.
- the average particle size of the resulting polymer silane coupling agent-coated fine particles is preferably 5 to 500 nm, more preferably 10 to 200 nm, from the viewpoint of securing optical properties when used for a ⁇ / 4 film.
- the content of the polymer silane coupling agent-coated fine particles in the cured layer is preferably 0.5 to 80 parts by mass, more preferably 1 to 60 parts by mass as the solid content, from the viewpoint of securing the film strength of the cured layer. .
- the hardened layer may contain a conductive agent in order to impart antistatic properties.
- Preferred conductive agents include metal oxide particles or ⁇ -conjugated conductive polymers.
- An ionic liquid is also preferably used as the conductive compound.
- the cured layer may contain a fluorine-siloxane graft compound, a fluorine compound, a silicone compound, or a compound having an HLB value of 3 to 18 from the viewpoint of improving the coating property.
- the hydrophilicity can be easily controlled by adjusting the types and amounts of these additives.
- the HLB value is Hydrophile-Lipophile-Balance, that is, a hydrophilic-lipophilic balance, and is a value indicating the hydrophilicity or lipophilicity of a compound. The smaller the HLB value, the higher the lipophilicity, and the higher the value, the higher the hydrophilicity.
- the HLB value can be obtained by the following calculation formula.
- HLB 7 + 11.7Log (Mw / Mo)
- Mw represents the molecular weight of the hydrophilic group
- Mo represents the molecular weight of the lipophilic group
- Mw + Mo M (molecular weight of the compound).
- HLB value 20 ⁇ total formula weight of hydrophilic part / molecular weight (J. Soc. Cosmetic Chem., 5 (1954), 294) and the like.
- Emulgen 109P (13.6), Emulgen 120 (15.3), Emulgen 123P (16.9), Emulgen 147 (16.3), Emulgen 210P (10.7), Emulgen 220 (14.2) , Emulgen 306P (9.4), Emulgen 320P (13.9), Emulgen 404 (8.8), Emulgen 408 (10.0), Emulgen 409PV (12.0), Emulgen 420 (13.6), Emulgen 430 (16.2), Emulgen 705 (10.5), Emulgen 707 (12.1), Emulgen 7 9 (13.3), Emulgen 1108 (13.5), Emulgen 1118S-70 (16.4), Emulgen 1135S-70 (17.9), Emulgen 2020G-HA (13.0), Emulgen 2025G (15.
- Emulgen LS-106 (12.5), Emulgen LS-110 (13.4), Emulgen LS-114 (14.0), manufactured by Nissin Chemical Industry Co., Ltd .: Surfynol 104E (4), Surfynol 104H (4), Surfinol 104A (4), Surfinol 104BC (4), Surfinol 104DPM (4), Surfinol 104PA (4), Surfinol 104PG-50 (4), Surfinol 104S (4), Surfi Knoll 420 (4), Surfynol 440 (8), Surfynol 46 (13), Surfynol 485 (17), Surfynol SE (6), Shin-Etsu Chemical Co., Ltd.: X-22-4272 (7), X-22-6266 (8).
- the fluorine-siloxane graft compound refers to a copolymer compound obtained by grafting polysiloxane and / or organopolysiloxane containing siloxane and / or organosiloxane alone on at least a fluorine resin.
- a fluorine-siloxane graft compound can be prepared by a method as described in Examples described later.
- examples of commercially available products include ZX-022H, ZX-007C, ZX-049, and ZX-047-D manufactured by Fuji Chemical Industry Co., Ltd.
- fluorine-based compound examples include Megafac series (F-477, F-487, F-569, etc.) manufactured by DIC Corporation, OPTOOL DSX, OPTOOL DAC, etc. manufactured by Daikin Industries, Ltd.
- silicone compounds are Shin-Etsu Chemical Co., Ltd .: KF-351, KF-352, KF-353, KF-354L, KF-355A, KF-615A, KF-945, KF-618, KF-6011, KF. -6015, KF-6004, manufactured by Big Chemie Japan KK: BYK-UV3576, BYK-UV3535, BYK-UV3510, BYK-UV3505, BYK-UV3500, BYK-UV3510, and the like. These components are preferably added in a range of 0.005 parts by mass or more and 10 parts by mass or less with respect to the solid component in the cured layer composition. Two or more kinds of these components may be added as long as the total additive amount is in the range of 0.005 parts by mass or more and 10 parts by mass or less.
- the hardened layer may contain the ultraviolet absorber described in the above ⁇ / 4 film.
- the cured layer is preferably provided by diluting the above-mentioned components forming the cured layer with a solvent to obtain a cured layer composition, which is applied onto a ⁇ / 4 film by the following method, dried and cured.
- Solvents include ketones (methyl ethyl ketone, acetone, etc.) and / or acetate esters (methyl acetate, ethyl acetate, butyl acetate, etc.), alcohols (ethanol, methanol, normal propanol, isopropanol), propylene glycol monomethyl ether, cyclohexanone, methyl isobutyl ketone. Etc. are preferable.
- the coating amount of the cured layer composition is suitably an amount that results in a wet film thickness of 0.1 to 80 ⁇ m, and preferably an amount that results in a wet film thickness of 0.5 to 30 ⁇ m.
- the dry film thickness is in the range of an average film thickness of 0.01 to 20 ⁇ m, preferably in the range of 1 to 15 ⁇ m. More preferably, it is in the range of 2 to 12 ⁇ m.
- a known method such as a gravure coater, a dip coater, a reverse coater, a wire bar coater, a die coater, or an ink jet method can be used.
- a cured layer composition may be applied onto a buffer layer, which will be described later, and then dried and cured (irradiated with active rays (also referred to as UV curing treatment)), and if necessary, heat treatment may be performed after UV curing.
- the heat treatment temperature after UV curing is preferably 60 ° C. or higher, more preferably 100 ° C. or higher, and particularly preferably 120 ° C. or higher.
- drying process changes from a constant state to a gradually decreasing state when drying starts.
- a section in which the drying speed is constant is called a constant rate drying section, and a section in which the drying speed decreases is called a decreasing rate drying section.
- Drying is preferably performed at a temperature of 30 ° C. or higher in the drying section. More preferably, the temperature in the drying section is 50 ° C. or higher.
- any light source that generates ultraviolet rays can be used without limitation.
- a low pressure mercury lamp, a medium pressure mercury lamp, a high pressure mercury lamp, an ultrahigh pressure mercury lamp, a carbon arc lamp, a metal halide lamp, a xenon lamp, or the like can be used.
- Irradiation conditions vary depending on each lamp, but the irradiation amount of active rays is usually in the range of 50 to 1000 mJ / cm 2 , preferably in the range of 50 to 300 mJ / cm 2 .
- oxygen removal for example, replacement with an inert gas such as nitrogen purge
- the cured state of the surface can be controlled by adjusting the removal amount of the oxygen concentration.
- the tension to be applied is preferably 30 to 300 N / m.
- the method for applying tension is not particularly limited, and tension may be applied in the conveying direction on the back roller, or tension may be applied in the width direction or biaxial direction by a tenter. Thereby, a film having further excellent flatness can be obtained.
- the buffer layer contains a resin different from the resin contained in the hardened layer, and is provided to alleviate interference unevenness between the hardened layer and the ⁇ / 4 film.
- the buffer layer resin preferably includes an acrylic material, and more preferably includes a urethane acrylate resin and fine particles coated with a polymer silane coupling agent.
- Acrylic materials are synthesized from monofunctional or polyfunctional (meth) acrylate compounds such as (meth) acrylic acid esters of polyhydric alcohols, diisocyanates and polyhydric alcohols, and hydroxy esters of (meth) acrylic acid. Such a polyfunctional urethane (meth) acrylate compound can be used.
- polyether resins having an acrylate functional group polyester resins, epoxy resins, alkyd resins, spiroacetal resins, polybutadiene resins, polythiol polyene resins, and the like can be used.
- (meth) acryl means both “acryl” and “methacryl”
- “(meth) acrylate” means both “acrylate” and “methacrylate”
- “( “Meth) acryloyl” refers to both “acryloyl” and “methacryloyl”.
- urethane (meth) acrylate” indicates both “urethane acrylate” and “urethane methacrylate”.
- Examples of the monofunctional (meth) acrylate compound include 2-hydroxyethyl (meth) acrylate, 2-hydroxypropyl (meth) acrylate, 2-hydroxybutyl (meth) acrylate, n-butyl (meth) acrylate, isobutyl ( (Meth) acrylate, t-butyl (meth) acrylate, glycidyl (meth) acrylate, acryloylmorpholine, N-vinylpyrrolidone, tetrahydrofurfuryl acrylate, cyclohexyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, isobornyl (meth) ) Acrylate, isodecyl (meth) acrylate, lauryl (meth) acrylate, tridecyl (meth) acrylate, cetyl (meth) acrylate, stearyl (meth) acrylate, benz
- bifunctional (meth) acrylate compound examples include ethylene glycol di (meth) acrylate, diethylene glycol di (meth) acrylate, butanediol di (meth) acrylate, hexanediol di (meth) acrylate, and nonanediol di (meth).
- Examples of the tri- or higher functional (meth) acrylate compound include trimethylolpropane tri (meth) acrylate, ethoxylated trimethylolpropane tri (meth) acrylate, propoxylated trimethylolpropane tri (meth) acrylate, and tris 2-hydroxyethyl.
- tri (meth) acrylate such as isocyanurate tri (meth) acrylate and glycerol tri (meth) acrylate
- pentaerythritol tri (meth) acrylate dipentaerythritol tri (meth) acrylate, ditrimethylolpropane tri (meth) acrylate
- Functional (meth) acrylate compounds pentaerythritol tetra (meth) acrylate, ditrimethylolpropane tetra (meth) acrylate, dipentaerythritol tet Trifunctional or more polyfunctional (meta) such as (meth) acrylate, dipentaerythritol penta (meth) acrylate, ditrimethylolpropane penta (meth) acrylate, dipentaerythritol hexa (meth) acrylate, ditrimethylolpropane hexa (meth) acrylate
- UV curable acrylate resins UV curable acrylate resins, UV curable urethane acrylate resins, UV curable polyester acrylate resins, UV curable epoxy acrylate resins, UV curable polyol acrylate resins, or UV curable epoxy resins are preferred.
- an ultraviolet curable acrylate resin is preferable.
- polyfunctional acrylate is preferable.
- the polyfunctional acrylate is preferably selected from the group consisting of pentaerythritol polyfunctional acrylate, dipentaerythritol polyfunctional acrylate, pentaerythritol polyfunctional methacrylate, and dipentaerythritol polyfunctional methacrylate.
- the polyfunctional acrylate is a compound having two or more acryloyloxy groups or methacryloyloxy groups in the molecule.
- the polyfunctional acrylate monomer include ethylene glycol diacrylate, diethylene glycol diacrylate, 1,6-hexanediol diacrylate, neopentyl glycol diacrylate, trimethylolpropane triacrylate, trimethylolethane triacrylate, and tetramethylolmethane triacrylate.
- a monofunctional acrylate may also be used.
- Monofunctional acrylates include isobornyl acrylate, 2-hydroxy-3-phenoxypropyl acrylate, isostearyl acrylate, benzyl acrylate, ethyl carbitol acrylate, phenoxyethyl acrylate, lauryl acrylate, isooctyl acrylate, tetrahydrofurfuryl acrylate, behenyl Examples thereof include acrylate, 4-hydroxybutyl acrylate, 2-hydroxyethyl acrylate, 2-hydroxypropyl acrylate, and cyclohexyl acrylate. Such acrylates can be obtained from Nippon Kasei Kogyo Co., Ltd., Shin-Nakamura Chemical Co., Ltd., Osaka Organic Chemical Co., Ltd., etc.
- the desired molecular weight and molecular structure can be designed, the buffer layer can have an appropriate hardness, and the physical properties of the formed buffer layer can be easily balanced.
- polyfunctional urethane acrylate can be preferably used.
- the urethane acrylate is obtained by reacting a polyhydric alcohol, a polyvalent isocyanate, and a hydroxyl group-containing acrylate.
- the solvent contained in the buffer layer forming composition used for forming the buffer layer is preferably a solvent that dissolves or swells the ⁇ / 4 film.
- the composition for forming the buffer layer easily penetrates from the surface of the ⁇ / 4 film into the inside, and improves the adhesion between the ⁇ / 4 film and the buffer layer. Can do.
- a layer in which the resin component of the ⁇ / 4 film and the resin component of the buffer layer are mixed is formed in the vicinity of the surface layer of the ⁇ / 4 film, and the refractive index of the ⁇ / 4 film and the buffer layer is formed by the action of this layer. Can be inclined, and the occurrence of uneven interference can be prevented.
- examples of the solvent for dissolving or swelling the surface of the ⁇ / 4 film include dibutyl ether, dimethoxymethane, dimethoxyethane, diethoxyethane, propylene oxide, 1,4- Ethers such as dioxane, 1,3-dioxolane, 1,3,5-trioxane, tetrahydrofuran, anisole and phenetole, acetone, methyl ethyl ketone, diethyl ketone, dipropyl ketone, diisobutyl ketone, cyclopentanone, cyclohexanone, methylcyclohexanone, And ketones such as methylcyclohexanone, ethyl formate, propyl formate, n-pentyl formate, methyl acetate, ethyl acetate, methyl propionate, ethyl propionat
- the buffer layer preferably contains a photopolymerization initiator.
- the photopolymerization initiator include 2,2-ethoxyacetophenone, 1-hydroxycyclohexyl phenyl ketone, dibenzoyl, benzoin, benzoin methyl ether, benzoin ethyl ether, p-chlorobenzophenone, p-methoxybenzophenone, Michler ketone, acetophenone, 2 -Chlorothioxanthone and the like. You may use these individually or in combination of 2 or more types.
- the buffer layer preferably contains a photosensitizer.
- the photosensitizer include tertiary amines such as triethylamine, triethanolamine and 2-dimethylaminoethanol, alkylphosphine series such as triphenylphosphine, and thioether series such as ⁇ -thiodiglycol. These may be used alone or in combination of two or more.
- the buffer layer preferably contains a leveling agent.
- a leveling agent it is most preferable to use an acrylic leveling agent.
- the leveling agent By using the leveling agent, it is possible to prevent defects such as film thickness unevenness and coating liquid repellency that may occur when the buffer layer is formed.
- an acrylic leveling agent compared to the case of using a fluorine-based or silicone-based leveling agent, recoatability when a cured layer is laminated on the buffer layer, and adhesion between the buffer layer and the cured layer are improved. Deterioration can be prevented.
- quaternary ammonium cations or conductive metal fine particles may be added to the buffer layer to impart conductivity to the buffer layer.
- the coating method for the buffer layer forming composition includes dip coating method, spin coating method, flow coating method, spray coating method, roll coating method, gravure roll coating method, air doctor coating method, plate coating method, wire doctor coating.
- a method such as a method, a knife coating method, a reverse coating method, a transfer roll coating method, a micro gravure coating method, a kiss coating method, a cast coating method, a slot orifice coating method, a calendar coating method, a die coating method, or the like can be employed.
- the microgravure coating method is preferable when a uniform thin film layer is formed
- the die coating method is preferable when a thick film layer needs to be formed.
- the buffer layer contains the same fine particles as those contained in the cured layer, that is, fine particles coated with a polymer silane coupling agent.
- the ratio a / b between the content a of the fine particles contained in the cured layer and the content b of the fine particles contained in the buffer layer may be 1, but is 2 or more and 10 or less. It is desirable.
- the buffer layer may contain the same additive as the hardened layer in addition to the fine particles.
- the buffer layer can be formed on the ⁇ / 4 film by the same method as the method for forming the cured layer.
- the coating amount of the buffer layer composition is suitably an amount that gives a wet film thickness of 5 to 20 ⁇ m, and preferably an amount that gives a wet film thickness of 5 to 15 ⁇ m.
- the dry film thickness is in the range of an average film thickness of 0.5 to 6 ⁇ m, preferably in the range of 0.5 to 4 ⁇ m.
- Examples of the polarizer C2 preferably used for the polarizing plate C of the present invention include a polyvinyl alcohol polarizing film, which includes a polyvinyl alcohol film dyed with iodine and a dichroic dye dyed. .
- a polyvinyl alcohol film a modified polyvinyl alcohol film modified with ethylene is preferably used.
- a polyvinyl alcohol aqueous solution is formed into a film and dyed by uniaxial stretching or dyed or uniaxially stretched and then preferably subjected to a durability treatment with a boron compound. Stretching is preferably performed uniaxially in the film forming direction or stretched in an oblique 45 ° direction with respect to the film forming direction.
- the film thickness of the polarizer C2 is 5 to 40 ⁇ m, preferably 5 to 30 ⁇ m, and particularly preferably 5 to 20 ⁇ m.
- a cellulose ester-containing film is preferably used.
- the polarizing plate of this invention is preferable at the point from which the performance excellent in visibility is exhibited by using it for an image display apparatus.
- an image display device a reflection type, a transmission type, a transflective type liquid crystal display device, a liquid crystal display device of various drive systems such as a TN type, STN type, OCB type, VA type, IPS type, ECB type, etc., a touch panel display device And organic EL display devices and plasma displays.
- a liquid crystal display device is preferable because of its high visibility.
- the image display device includes a sheet-like polarizing plate formed by cutting the long polarizing plate.
- the hard-coat layer is formed in the outermost surface of the said polarizing plate, it is preferable to arrange
- the polarizing plate of the present invention is preferably applied to a liquid crystal display device among image display devices, and the liquid crystal display device is preferably a stereoscopic image display device. That is, the polarizing plate of the present invention can be used in various modes in a stereoscopic image display device.
- the polarizing plate in which the hard-coat layer is formed in a liquid crystal display device it is preferable to bond the side in which the hard-coat layer of a polarizing plate is not formed to a liquid crystal cell.
- the polarizing plate due to the mode and configuration of the polarizing plate, it is possible to reduce luminance reduction and color change in the state of wearing polarized sunglasses, maintain excellent visibility with respect to the use environment, and use environment In contrast, a stereoscopic image display device with higher durability can be obtained.
- FIG. 3 is a schematic cross-sectional view of an example of the configuration of the image display device of the present invention.
- the image display device 200 includes an image display device main body 210, a polarizing plate C of the present invention that is configured as a part of the image display device main body 210, and a front window 220.
- a polarizing plate C is disposed on the viewing side surface of the image display apparatus main body 210.
- the hard coat layer C0 surface of the ⁇ / 4 film C1 of the polarizing plate C and the front window 220 are bonded to each other through a gap filler (photo-curing resin) 230.
- the front window 220 is not particularly limited, and a conventionally known transparent substrate such as an acrylic plate or a glass plate or a touch panel module can be used.
- the material, thickness, and the like of the front window 220 can be appropriately selected according to the application of the image display device.
- Example 1 ⁇ Production of ⁇ / 4 film> ⁇ Fine particle dispersion 1> Fine particles (Aerosil R972V manufactured by Nippon Aerosil Co., Ltd.) 11 parts by mass Ethanol 89 parts by mass The above was stirred and mixed with a dissolver for 50 minutes, and then dispersed with Manton Gorin.
- Fine particle addition liquid 1 The fine particle dispersion 1 was slowly added to the dissolution tank containing methylene chloride with sufficient stirring. Further, the particles were dispersed by an attritor so that the secondary particles had a predetermined particle size. This was filtered through Finemet NF manufactured by Nippon Seisen Co., Ltd. to prepare a fine particle additive solution 1.
- a main dope solution having the following composition was prepared. First, methylene chloride and ethanol were added to the pressure dissolution tank. The cellulose ester was added to the pressure dissolution tank containing the solvent while stirring. This was heated and stirred and completely dissolved, and Azumi Filter Paper No. Azumi Filter Paper No. The main dope solution was prepared by filtration using 244.
- the solvent was evaporated until the amount of residual solvent in the cast (cast) film reached 75%, and then peeled off from the stainless steel belt support with a peeling tension of 130 N / m.
- the temperature is 170 ° C.
- the magnification is 1.5 times
- the slow axis is 45 ° with respect to the film width direction.
- the film was stretched in an oblique direction so as to form
- drying was terminated while the drying zone was conveyed by a number of rolls.
- the drying temperature was 130 ° C. and the transport tension was 100 N / m.
- a ⁇ / 4 film with a hard coat layer was produced by providing a hard coat layer on the surface of the obtained ⁇ / 4 film.
- the hard coat layer was prepared by first forming a buffer layer by applying and curing the buffer layer composition, and then forming a cured layer by applying and curing the cured layer composition on the buffer layer. .
- the polymer silane coupling agent-coated fine particles contained in the buffer layer composition and the cured layer composition those prepared by the following method were used. Below, the procedure which forms a buffer layer and a hardening layer is demonstrated.
- silica sol manufactured by JGC Catalysts & Chemicals Co., Ltd .: Si-45P, SiO 2 concentration 30% by weight, average particle size 45 nm, dispersion medium: water
- silica sol manufactured by JGC Catalysts & Chemicals Co., Ltd .: Si-45P, SiO 2 concentration 30% by weight, average particle size 45 nm, dispersion medium: water
- the solvent of water was replaced with ethanol to prepare 100 g of an ethanol dispersion of silica fine particles (SiO 2 concentration 30 wt%).
- silica fine particle ethanol dispersion and 1.5 g of the polymer silane coupling agent are dispersed in 20 g (25 ml) of acetone, and 20 mg of aqueous ammonia having a concentration of 29.8% by weight is added thereto, followed by stirring at room temperature for 30 hours.
- the polymer silane coupling agent was adsorbed on the silica fine particles.
- silica particles having an average particle diameter of 5 ⁇ m are added and stirred for 2 hours to adsorb the unadsorbed polymer silane coupling agent in the solution to the silica particles, and then the polymer silane coupling that has not been adsorbed by centrifugation.
- Silica particles having an average particle diameter of 5 ⁇ m adsorbing the agent were removed. 1000 g of ethanol is added to the silica fine particle dispersion adsorbing the polymer silane coupling agent, and the silica fine particles are precipitated, separated, dried under reduced pressure, and then dried at 25 ° C.
- the obtained polymer silane coupling agent-coated silica (1) had an average particle size of 57 nm. The average particle size was measured with a laser particle size measuring device.
- the buffer layer composition was prepared by stirring and mixing the polymer silane coupling agent-coated silica (1) prepared above and the following compound.
- Polymer silane coupling agent-coated silica (1) 60 parts by mass (active ray curable resin) Urethane acrylate (U-4H, Shin-Nakamura Chemical Co., Ltd.) 35 parts by mass (photopolymerization initiator) Irgacure 184 (manufactured by BASF Japan) 5 parts by mass (additive) KF-642 (polyether-modified silicone oil, manufactured by Shin-Etsu Chemical Co., Ltd.) 2 parts by mass (solvent) Propylene glycol monomethyl ether 80 parts by weight Methyl acetate 20 parts by weight [Preparation of cured layer composition]
- the polymer silane coupling agent-coated silica (1) prepared above was stirred and mixed with the following compound to prepare a cured layer composition.
- Polymer silane coupling agent-coated silica 60 parts by mass (active ray curable resin) NK ester A-DCP (tricyclodecane dimethanol diacrylate, manufactured by Shin-Nakamura Chemical Co., Ltd.) 35 parts by mass (photopolymerization initiator) Irgacure 184 (manufactured by BASF Japan) 5 parts by mass (additive) KF-642 (polyether-modified silicone oil, manufactured by Shin-Etsu Chemical Co., Ltd.) 2 parts by mass (solvent) Propylene glycol monomethyl ether 80 parts by weight Methyl acetate 20 parts by weight
- the buffer layer composition is applied to the A side of the ⁇ / 4 film produced above (the side not in contact with the casting belt) using an extrusion coater, and the constant rate drying zone temperature is 50 ° C. After drying at an interval temperature of 50 ° C., while purging with nitrogen so that the atmosphere has an oxygen concentration of 1.0% by volume or less, the illuminance of the irradiated part is 100 mW / cm 2 using an ultraviolet lamp, and the irradiation amount is 0.25 J. The coating layer was cured at / cm 2 to form a buffer layer (lower layer) having a dry film thickness of 4 ⁇ m.
- the cured layer composition prepared above was applied onto the buffer layer using a micro gravure coater, and after drying at a constant rate drying zone temperature of 50 ° C. and a reduced rate drying zone temperature of 50 ° C., the oxygen concentration was While purging with nitrogen to an atmosphere of 1.0% by volume or less, using an ultraviolet lamp, the illuminance of the irradiated part is 100 mW / cm 2 , the irradiation amount is 0.3 J / cm 2 , the coating layer is cured, and a dry film A cured layer (upper layer) having a thickness of 2 ⁇ m was formed, wound up in a roll shape, and a ⁇ / 4 film with a hard coat layer was produced.
- a polarizer, the ⁇ / 4 film 101, and a polarizer protective film were bonded to the back side to prepare a polarizing plate 101.
- Step 1 Soaked in a 2 mol / L sodium hydroxide solution at 60 ° C. for 90 seconds, then washed with water and dried to obtain a ⁇ / 4 film 101 saponified on the side to be bonded to the polarizer.
- Step 2 The polarizer was immersed in a polyvinyl alcohol adhesive tank having a solid content of 2% by mass for 1 to 2 seconds.
- Step 3 Excess adhesive adhered to the polarizer in Step 2 was gently wiped off, and this was placed on the ⁇ / 4 film 101 processed in Step 1.
- Step 4 The ⁇ / 4 film 101, the polarizer and the polarizer protective film laminated in Step 3 were bonded at a pressure of 20 to 30 N / cm 2 and a conveying speed of about 2 m / min.
- Step 5 A sample obtained by bonding the polarizer, the ⁇ / 4 film 101, and the polarizer protective film prepared in Step 4 in a dryer at 80 ° C. is dried for 2 minutes, and the polarizing plate 101 including the ⁇ / 4 film 101 is obtained. Was made.
- Example 2 to 6 and Comparative Examples 1 to 6 the acetyl group average substitution degree, propionyl group average substitution degree and butyryl group average substitution degree of the cellulose ester contained in the ⁇ / 4 film are shown in Table 1 below.
- a polarizing plate was produced in the same manner as in Example 1 except that the above changes were made.
- the stretching temperature when producing the ⁇ / 4 film was changed from 170 ° C. to 190 ° C.
- Comparative Example 5 a cellulose ester containing a cellulose diacetate (DAC) is used as the cellulose ester contained in the ⁇ / 4 film.
- DAC cellulose diacetate
- Comparative Example 6 a cellulose acetate is used as the cellulose ester contained in the ⁇ / 4 film.
- the one containing propionate (CAP) was used.
- Example 7 A polarizing plate was produced in the same manner as in Example 1 except that the buffer layer was not provided on the hard coat layer.
- Example 7 A polarizing plate was produced in the same manner as in Example 1 except that the hard coat layer was not provided.
- A-DCP in Table 1 is a resin component (tricyclodecane dimethanol diacrylate (manufactured by Shin-Nakamura Chemical Co., Ltd.)) contained in the cured layer.
- “Urethane acrylate” in Table 1 is a resin component (U-4H, manufactured by Shin-Nakamura Chemical Co., Ltd.) contained in the buffer layer.
- each polarizing plate is cut into a size of 150 mm in the width direction and 120 mm in the longitudinal direction, and the surface of the polarizing plate is sharpened with two razors at 100 mm intervals in a direction parallel to the slow axis of the ⁇ / 4 film. A cross-shaped mark was made with a knife.
- the polarizing plate was conditioned for 24 hours or more in an environment of 23 ° C. and 55% RH, and the inter-mark distance L1 in the direction parallel to the slow axis of the ⁇ / 4 film before processing was measured with a microscope.
- the polarizing plate is placed in an electric thermostat for 1000 hours in a temperature environment of 90 ° C., and the high-temperature and high-humidity-treated sample is again conditioned in an environment of 23 ° C. and 55% RH for 24 hours.
- the distance L2 between the marks in the direction parallel to the slow axis of the ⁇ / 4 film after processing with a microscope is measured, and the rate of change in the direction parallel to the slow axis of the ⁇ / 4 film is obtained by calculating the rate of change before and after the processing.
- the dimensional change rate was calculated.
- ⁇ Performance evaluation> The front contrast change rate ( ⁇ CR) before and after placing the liquid crystal display device produced above in a high-temperature dry environment of 90 ° C. was evaluated.
- ⁇ CR front contrast change rate
- EZ-Contrast 160D manufactured by ELDIM was used, the luminance from the normal direction of the display screen of white display and black display was measured with a liquid crystal display device, and the ratio was defined as the front contrast.
- Front contrast (brightness of white display measured from normal direction of display device) / (brightness of black display measured from normal direction of display device)
- the front contrast of arbitrary 5 points of the liquid crystal display device was measured, and the average value of 3 points excluding the maximum value and the minimum value among the measured values was calculated.
- each liquid crystal display device is placed in a temperature environment of 90 ° C. for 1000 hours (this process is referred to as “high temperature drying process”), and then returned to an environment of 23 ° C. and a relative humidity of 55% RH.
- the front contrast was measured in the same manner as described above.
- the change rate ((DELTA) CR) of the front contrast of the liquid crystal display device after a high temperature drying process with respect to the front contrast of the liquid crystal display device before a high temperature drying process was computed. Specifically, it was calculated by the following calculation.
- ⁇ CR [(Front contrast of liquid crystal display device before high temperature drying treatment) ⁇ (Front contrast of liquid crystal display device after high temperature drying treatment)] / (Front contrast of liquid crystal display device before high temperature drying treatment) ⁇ 100
- Table 1 shows that the smaller the contrast change rate ( ⁇ CR), the lower the polarization degree of the polarizing plate, and the light leakage of the polarizing plate does not occur even when exposed to a high temperature dry environment. Yes. If the contrast change rate ( ⁇ CR) exceeds 30%, it is evaluated that the product cannot be used as a product. If the contrast change rate ( ⁇ CR) is 20% or more and 30% or less, it is evaluated as good, and the contrast change rate. If ( ⁇ CR) is less than 20%, it is evaluated as better.
- Example 1 and Comparative Example 7 produce polarizing plates under the same conditions except that a hard coat layer is provided or not.
- a hard coat layer is provided or not.
- the rate of change ( ⁇ CR) can be suppressed. From this result, it has become clear that by providing a hard coat layer on the surface of the polarizing plate, it is possible to suppress a decrease in contrast of the polarizing plate before and after being exposed to a high temperature drying environment.
- the polarizing plates are produced under the same conditions as in Examples 1 and 7, except that a buffer layer is provided on the hard coat layer.
- a buffer layer is provided on the hard coat layer as in Example 1 so that it is parallel to the slow axis of the ⁇ / 4 film of the polarizing plate before and after being exposed to a high temperature drying environment.
- the dimensional change rate in the direction was lowered and the change rate in contrast was lowered. From this result, it has been clarified that by providing a buffer layer in the hard coat layer, it is possible to suppress a decrease in contrast of the polarizing plate before and after being exposed to a high temperature drying environment.
- Examples 1 to 3 and Comparative Examples 1 to 2 have the same conditions except that the acetyl group average substitution degree of acetyl cellulose contained in the ⁇ / 4 film is changed in the range of 1.6 or more and 2.4 or less.
- a polarizing plate is produced.
- the average acetyl group substitution degree of acetyl cellulose contained in the ⁇ / 4 film as in Examples 1 to 3 in the range of 1.8 to 2.2, the polarizing plate before and after being exposed to a high-temperature drying environment
- the dimensional change rate in the direction parallel to the slow axis of the ⁇ / 4 film can be in the range of ⁇ 0.5% or more and ⁇ 0.1% or less, and the contrast change rate ( ⁇ CR) can be kept low. is made of.
- Examples 1, 4 and 5 and Comparative Examples 3 to 4 were the same under the same conditions except that the average degree of butyryl group substitution of acetylcellulose contained in the ⁇ / 4 film was changed in the range of 0 to 0.6.
- a polarizing plate is produced. Polarization before and after exposure to a high-temperature dry environment by setting the average degree of butyryl group substitution of acetylcellulose contained in the ⁇ / 4 film as in Examples 1, 4 and 5 in the range of 0.1 to 0.4.
- the rate of dimensional change in the direction parallel to the slow axis of the ⁇ / 4 film of the plate can be in the range of ⁇ 0.5% or more and ⁇ 0.1% or less, and the contrast before and after exposure to a high-temperature drying environment
- the rate of change ( ⁇ CR) can be kept low.
- the acetyl group average substitution degree is 1.8 or more and 2.2 or less
- a ⁇ / 4 film containing a cellulose ester having a butyryl group average substitution degree of 0.1 or more and 0.4 or less stored in a temperature environment of 90 ° C. for 1000 hours, and 55% RH at a temperature of 23 ° C.
Landscapes
- Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Optics & Photonics (AREA)
- General Physics & Mathematics (AREA)
- Organic Chemistry (AREA)
- Nonlinear Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Medicinal Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Crystallography & Structural Chemistry (AREA)
- Mathematical Physics (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Inorganic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Polarising Elements (AREA)
- Liquid Crystal (AREA)
- Surface Treatment Of Optical Elements (AREA)
Abstract
Description
図1は、本発明の偏光板の模式図である。本発明の偏光板Cは、図1に示すように、λ/4フィルムC1と、偏光子C2とを積層したものである。本発明の偏光板Cは、図1に示すように、偏光子C2のλ/4フィルムC1と接する側と反対側の面に偏光子保護フィルムC3をさらに有していてもよい。図1には示されていないが、λ/4フィルムC1の偏光子C2と接する側とは反対側の面にハードコート層C0を設ける。これにより偏光板Cの表面側(視認側)にキズが入りにくくなる。しかも、偏光子C2が大気中の水分を吸湿しにくくなることにより偏光板を高温乾燥環境下においても反りが生じにくい。
L1:処理前の印間距離
L2:処理後の印間距離
上記の寸法変化率の偏光板を用いることにより、高温乾燥環境下に偏光板を晒した後にも偏光板Cの応力緩和状態を作ることができる。これにより偏光子保護フィルムC3(「T2フィルム」と記すこともある)の位相差変動を抑制し、以って偏光子保護フィルムC3に起因する光漏れを抑制することができる。
本発明の偏光板Cは、一般的な方法で作製することができ、λ/4フィルムC1と偏光子C2とを貼り合わせることにより製造することができる。本発明の偏光板Cが偏光子保護フィルムC3を含む場合、図1に示すように、λ/4フィルムC1/偏光子C2/偏光子保護フィルムC3の構成で積層して貼り合わせることにより偏光板Cを作製することができる。
本発明の偏光板Cに用いるλ/4フィルムC1は、アセチル基平均置換度が1.8以上2.2以下であり、ブチリル基平均置換度が0.1以上0.4以下であるセルロースエステルを含有することを特徴とする。このようなブチリル基平均置換度を有することにより、推測ではあるがおそらく、高温乾燥環境下においてもブチリル基が可塑的な効果を発揮するので偏光子(特にヨウ素五量体)の偏光度の低下を抑制することができる。上記セルロースエステルにおけるアセチル基平均置換度は1.85~2.15であることが好ましく、より好ましくは1.9~2.0である。またブチリル基平均置換度は0.15以上0.35以下であることが好ましく、より好ましくは0.2以上0.32以下である。
λ/4フィルムを作製するときのドープ中に添加される添加剤としては、可塑剤、紫外線吸収剤、リターデーション調整剤、酸化防止剤、劣化防止剤、剥離助剤、界面活性剤、染料、微粒子等がある。本発明において、微粒子以外の添加剤についてはセルロースエステル溶液の調製の際に添加してもよいし、微粒子分散液の調製の際に添加してもよい。液晶画像表示装置に使用する偏光板には耐熱耐湿性を付与する可塑剤、酸化防止剤、紫外線吸収剤等を添加することが好ましい。以下に添加剤を説明する。
本発明において、λ/4フィルムには、所謂可塑剤として知られる化合物を、機械的性質向上、柔軟性を付与、耐吸水性付与、水蒸気透過率低減、リターデーション調整等の目的で添加することが好ましい。
本発明の偏光板に用いるλ/4フィルムには、紫外線吸収剤を含有させることができる。使用し得る紫外線吸収剤としては、例えば、オキシベンゾフェノン系化合物、ベンゾトリアゾール系化合物、サリチル酸エステル系化合物、ベンゾフェノン系化合物、シアノアクリレート系化合物、ニッケル錯塩系化合物、トリアジン系化合物等を挙げることができるが、着色の少ないベンゾトリアゾール系化合物が好ましい。また、特開平10-182621号、特開平8-337574号、特開2001-72782号記載の紫外線吸収剤、特開平6-148430号、特開2002-31715号、特開2002-169020号、特開2002-47357号、特開2002-363420号、特開2003-113317号記載の高分子紫外線吸収剤も好ましく用いられる。紫外線吸収剤としては、偏光子や液晶の劣化防止の観点から、波長370nm以下の紫外線の吸収能に優れており、かつ、液晶表示性の観点から、波長400nm以上の可視光の吸収が少ないものが好ましい。
リターデーションを調整するために添加するリターデーション調整剤は、欧州特許911,656A2号明細書に記載されているような、二つ以上の芳香族環を有する芳香族化合物を使用することができる。
(式中、Bはベンゼンモノカルボン酸残基、Gは炭素数2~12のアルキレングリコール残基又は炭素数6~12のアリールグリコール残基又は炭素数が4~12のオキシアルキレングリコール残基、Aは炭素数4~12のアルキレンジカルボン酸残基又は炭素数6~12のアリールジカルボン酸残基を表し、またnは1以上の整数を表す。)
一般式(1)中、Bで示されるベンゼンモノカルボン酸残基とGで示されるアルキレングリコール残基又はオキシアルキレングリコール残基又はアリールグリコール残基、Aで示されるアルキレンジカルボン酸残基又はアリールジカルボン酸残基とから構成されるものであり、通常のポリエステル系可塑剤と同様の反応により得られる。
酸価とは、試料1g中に含まれる酸(分子末端に存在するカルボキシル基)を中和するために必要な水酸化カリウムのミリグラム数をいう。酸価及びヒドロキシル(水酸基)価はJIS K0070(1992)に準拠して測定したものである。
反応容器に、テレフタル酸820部(5モル)、1,2-プロピレングリコール608部(8モル)、トリイル酸610部(5モル)及び触媒としてテトライソプロピルチタネート0.30部を一括して仕込み窒素気流中で攪拌下、還流凝縮器を付して過剰の1価アルコールを還流させながら、酸価が2以下になるまで130~250℃で加熱を続け生成する水を連続的に除去した。次いで200~230℃で6.65×103Pa~最終的に4×102Pa以下の減圧下、留出分を除去し、この後濾過して次の性状を有する芳香族末端エステルを得た。
粘度(25℃、mPa・s);19815
酸価 ;0.4
反応容器に、アジピン酸500部(3.5モル)、安息香酸305部(2.5モル)、ジエチレングリコール583部(5.5モル)及び触媒としてテトライソプロピルチタネート0.45部を用いる以外はサンプルNo.1と全く同様にして次の性状を有する芳香族末端エステルを得た。
粘度(25℃、mPa・s);90
酸価 ;0.05
反応容器にフタル酸410部(2.5モル)、安息香酸610部(5モル)、ジプロピレングリコール737部(5.5モル)及び触媒としてテトライソプロピルチタネート0.40部を用いる以外はサンプルNo.1と全く同様にして次の性状を有する芳香族末端エステル系可塑剤を得た。
酸価 ;0.2
以下に、本発明に用いられる芳香族末端エステル系可塑剤の具体的化合物を示すが、本発明はこれに限定されない。


λ/4フィルムは、セルロースエステルと、カルボキシル基、ヒドロキシル基、アミノ基、アミド基、及びスルホン酸基から選ばれる置換基を有しかつ重量平均分子量が500~200,000の範囲内であるビニル系化合物のポリマー又はオリゴマーとを含有することが好ましい。当該セルロースエステルと、当該ポリマー又はオリゴマーとの含有量の質量比が、95:5~50:50の範囲内であることが好ましい。
本発明において、λ/4フィルムには、劣化防止剤として、例えば、酸化防止剤、過酸化物分解剤、ラジカル重合禁止剤、金属不活性化剤、酸捕獲剤、アミン類等を含有させることができる。
本発明では、マット剤として微粒子をλ/4フィルム中に含有させることができる。、λ/4フィルムが長尺状フィルムの場合、マット剤によって搬送や巻き取りをしやすくすることができる。
この他、カオリン、タルク、ケイソウ土、石英、炭酸カルシウム、硫酸バリウム、酸化チタン、アルミナ等の無機微粒子、カルシウム、マグネシウム等のアルカリ土類金属の塩等の熱安定剤を加えてもよい。更に界面活性剤、剥離促進剤、帯電防止剤、難燃剤、滑剤、油剤等も加えてもよい。
式(ii):Rt={(nx+ny)/2-nz}×d
式中、nx、nyは、23℃・55%RH、450nm、550nm、650nmの各々における屈折率nx(フィルムの面内の最大の屈折率、遅相軸方向の屈折率ともいう。)、ny(フィルム面内で遅相軸に直交する方向の屈折率)であり、dはフィルムの厚さ(nm)である。
本発明の偏光板に用いるλ/4フィルムは、長尺状フィルムの搬送方向に対して斜め方向に面内遅相軸を有するように斜め方向に延伸した長尺状フィルムである。斜め延伸前のフィルムを「原反フィルム」と呼称する。原反フィルムは、溶液流延法、又は溶融流延法のいずれの方法で製造されてもよい。以下溶液流延法および斜め延伸のλ/4フィルムの製造方法について説明をする。
なお、Mはウェブ又はフィルムを製造中又は製造後の任意の時点で採取した試料の質量で、NはMを115℃で1時間の加熱後の質量である。
上記のように乾燥して得たウェブをドラム又はバンドから剥離する。ここで剥離したウェブを延伸する。
(斜め延伸工程)
斜め延伸工程は、製膜された長尺フィルムを長尺方向に対して斜めの方向に延伸する工程である。長尺フィルムの製造方法では、フィルムを連続的に製造することにより、所望の任意の長さにフィルムを製造し得る。なお、長尺延伸フィルムの製造方法は、長尺フィルムを製膜した後に一度巻芯に巻き取り、巻回体(原反ともいう)にしてから斜め延伸工程に供給するようにしてもよいし、製膜後のフィルムを巻き取ることなく、製膜工程から連続して斜め延伸工程に供給してもよい。製膜工程と斜め延伸工程を連続して行うことは、延伸後の膜厚や光学値の結果をフィードバックして製膜条件を変更し、所望の長尺延伸フィルムを得ることができるので好ましい。
本実施形態における延伸に供される長尺フィルムに斜め方向の配向を付与するために、斜め延伸装置を用いる。本実施形態で用いられる斜め延伸装置は、把持具走行支持具の経路パターンを多様に変化させることにより、λ/4フィルムの配向角を自在に設定でき、さらに、λ/4フィルムの配向軸をフィルム幅方向に渡って左右均等に高精度に配向させることができ、かつ、高精度でフィルム厚みやリターデーションを制御できるフィルム延伸装置であることが好ましい。
本発明において、λ/4フィルムは、その表面にハードコート層が設けられている。また、λ/4フィルムは、その表面にハードコート層以外の表面加工層として、帯電防止層、バックコート層、反射防止層、易滑性層、接着層、防眩層、バリアー層等の機能性層を設けることができる。
本実施形態の硬化層は、脂環構造を有する活性エネルギー線硬化性樹脂(以下、単に硬化性樹脂とも記載する)と、ポリマーシランカップリング剤で被覆されてなる微粒子とを含有し、偏光板の表面にキズが形成されるのを避ける為に設けられる。脂環構造としては、具体的には、ノルボルニル、トリシクロデカニル、テトラシクロドデカニル、ペンタシクロペンタデカニル、アダマンチル、ジアマンタニル等が挙げられる。


一般式(I)及び一般式(II)において、R1は、水素原子または炭素数1~3のアルキル基を表し、好ましくは、水素原子、メチル基、エチル基を表す。R2は炭素数1~5のアルキレン基またはアルキレンオキサイド基を表し、好ましくは、メチレン基、エチレン基、メチレンオキサイド基、エチレンオキサイド基を表す。R3は、水素原子または炭素数1~3のアルキル基を表し、好ましくは、水素原子、メチル基、エチル基を表す。

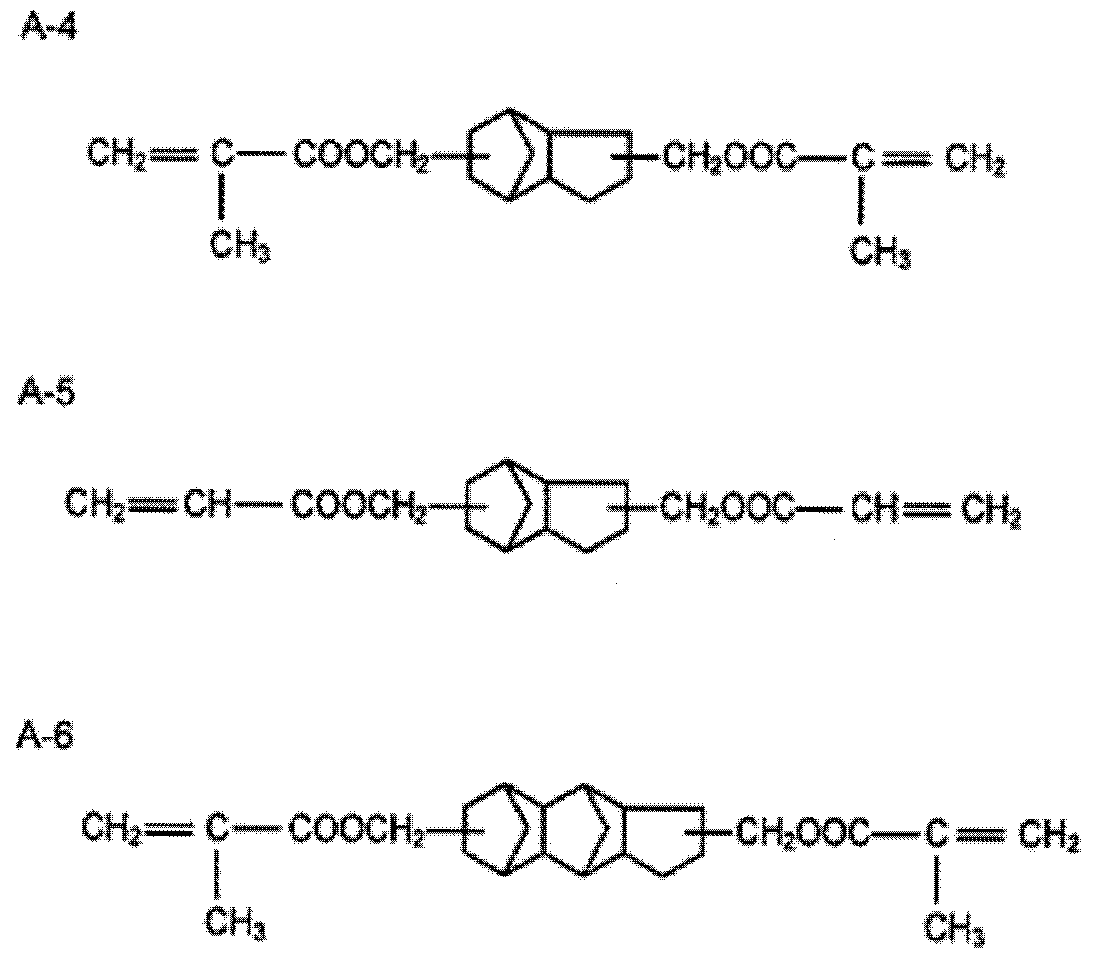








硬化層は、活性線硬化樹脂の硬化促進のため、光重合開始剤を含有することが好ましい。光重合開始剤の含有量は、質量比で、光重合開始剤:活性線硬化樹脂=20:100~0.01:100となる含有量であることが好ましい。光重合開始剤としては、具体的には、アルキルフェノン系、アセトフェノン、ベンゾフェノン、ヒドロキシベンゾフェノン、ミヒラーケトン、α-アミロキシムエステル、チオキサントン等、およびこれらの誘導体を挙げることができるが、特にこれらに限定されるものではない。光重合開始剤としては市販品を用いてもよく、例えば、BASFジャパン(株)製のイルガキュア184、イルガキュア907、イルガキュア651などが好ましい例示として挙げられる。
硬化層は微粒子を含有しても良い。微粒子としては、特に制限されないが、シリカ、アルミナ、ジルコニア、酸化チタン、五酸化アンチモン等が挙げられ、好ましくはシリカである。シリカ微粒子は、内部に空洞を有する中空粒子でも良い。中でも、ポリマーシランカップリング剤で被覆されてなる微粒子が、硬化層に適度な硬度を与え、良好な機械特性を発揮することから特に好ましい。含有量については、微粒子:活性線硬化樹脂=0.1:100~400:100となる含有量が好ましい。
ポリマーシランカップリング剤とは、重合性モノマーとシランカップリング剤(反応性シラン化合物)との反応物をいう。このようなポリマーシランカップリング剤は、例えば、特開平11-116240号公報に開示された重合性モノマーと反応性シラン化合物との反応物の製法に準じて得ることができる。
(式中、Rは、置換または非置換の炭化水素基から選ばれる炭素数1~10の有機基を表す。Xは(メタ)アクロイル基、エポキシ基(グリシド基)、ウレタン基、アミノ基、フルオロ基から選ばれる1種または2種以上の官能基。)
式(2)で表される有機ケイ素化合物として、具体的には、3,3,3-トリフルオロプロピルトリメトキシシラン、メチル-3,3,3-トリフルオロプロピルジメトキシシラン、β-(3,4-エポキシシクロヘキシル)エチルトリメトキシシラン、γ-グリシドキシメチルトリメトキシシラン、γ-グリシドキシメチルトリエキシシラン、γ-グリシドキシエチルトリメトキシシラン、γ-グリシドキシエチルトリエトキシシラン、γ-グリシドキシプロピルトリメトキシシラン、γ-グリシドキシプロピルトリメトキシシラン、γ-グリシドキシプロピルトリエトキシシラン、γ-グリシドキシプロピルトリエトキシシラン、γ-(β-グリシドキシエトキシ)プロピルトリメトキシシラン、γ-(メタ)アクリロオキシメチルトリメトキシシラン、γ-(メタ)アクリロオキシメチルトリエキシシラン、γ-(メタ)アクリロオキシエチルトリメトキシシラン、γ-(メタ)アクリロオキシエチルトリエトキシシラン、γ-(メタ)アクリロオキシプロピルトリメトキシシラン、γ-(メタ)アクリロオキシプロピルトリメトキシシラン、γ-(メタ)アクリロオキシプロピルトリエトキシシラン、γ-(メタ)アクリロオキシプロピルトリエトキシシラン、3-ウレイドイソプロピルプロピルトリエトキシシラン、パーフルオロオクチルエチルトリメトキシシラン、パーフルオロオクチルエチルトリエトキシシラン、パーフルオロオクチルエチルトリイソプロポキシシラン、トリフルオロプロピルトリメトキシシラン、N-β(アミノエチル)γ-アミノプロピルメチルジメトキシシラン、N-β(アミノエチル)γ-アミノプロピルトリメトキシシラン、N-フェニル-γ-アミノプロピルトリメトキシシラン等およびこれらの混合物が挙げられる。
ポリマーシランカップリング剤被覆微粒子は、具体的には、微粒子の有機溶媒分散液にポリマーシランカップリング剤を加え、アルカリ存在下にポリマーシランカップリング剤で微粒子を被覆することによって調製できる。得られるポリマーシランカップリング剤被覆微粒子の平均粒子径の範囲は、5~500nm、さらには10~200nmであることが、λ/4フィルムに用いた際の光学特性を確保できる点で好ましい。
硬化層には、帯電防止性を付与するために導電剤が含まれていても良い。好ましい導電剤としては、金属酸化物粒子又はπ共役系導電性ポリマーが挙げられる。また、イオン液体も導電性化合物として好ましく用いられる。
硬化層には、塗布性を良好にする観点から、フッ素-シロキサングラフト化合物、フッ素系化合物、シリコーン系化合物やHLB値が3~18の化合物が含まれていても良い。これら添加剤の種類や添加量を調整することで、親水性を制御しやすい。HLB値とは、Hydrophile-Lipophile-Balance、つまり、親水性-親油性のバランスのことであり、化合物の親水性又は親油性の大きさを示す値である。HLB値が小さいほど親油性が高く、値が大きいほど親水性が高くなる。また、HLB値は以下のような計算式によって求めることができる。
式中、Mwは親水基の分子量、Moは親油基の分子量を表し、Mw+Mo=M(化合物の分子量)である。或いはグリフィン法によれば、HLB値=20×親水部の式量の総和/分子量(J.Soc.Cosmetic Chem.,5(1954),294)等が挙げられる。
硬化層は、上述のλ/4フィルムで説明した紫外線吸収剤を含有しても良い。紫外線吸収剤の含有量としては、質量比で、紫外線吸収剤:硬化性樹脂=0.01:100~20:100となる含有量であることが好ましい。
硬化層は、上記した硬化層を形成する成分を溶剤で希釈して硬化層組成物とし、これを以下の方法でλ/4フィルム上に塗布し、乾燥、硬化して設けることが好ましい。
後述する緩衝層上に硬化層組成物を塗布した後、乾燥し、硬化(活性線を照射(UV硬化処理とも言う))し、更に必要に応じて、UV硬化後に加熱処理しても良い。UV硬化後の加熱処理温度 は60℃以上が好ましく、更に好ましくは100℃以上であり、特に好ましくは120℃以上である。このような高温でUV硬化後の加熱処理を行うことで、膜強度に優れた硬化層を得ることができる。
緩衝層は、硬化層に含まれる樹脂とは異なる樹脂を含有し、硬化層とλ/4フィルムの干渉ムラを緩和するために設けられる。緩衝層の樹脂としては、アクリル系材料を含んでいることが好ましく、ウレタンアクリレート樹脂と、ポリマーシランカップリング剤で被覆されてなる微粒子とを含んでいることがより好ましい。アクリル系材料としては、多価アルコールの(メタ)アクリル酸エステルのような単官能または多官能の(メタ)アクリレート化合物、ジイソシアネートと多価アルコールおよび(メタ)アクリル酸のヒドロキシエステル等から合成されるような多官能のウレタン(メタ)アクリレート化合物を使用することができる。また、これらの他にも、アクリレート系の官能基を有するポリエーテル樹脂、ポリエステル樹脂、エポキシ樹脂、アルキッド樹脂、スピロアセタール樹脂、ポリブタジエン樹脂、ポリチオールポリエン樹脂等を使用することができる。
本発明の偏光板Cに好ましく用いられる偏光子C2としては、ポリビニルアルコール系偏光フィルムが挙げられ、これはポリビニルアルコール系フィルムにヨウ素を染色させたものと二色性染料を染色させたものがある。ポリビニルアルコール系フィルムとしては、エチレンで変性された変性ポリビニルアルコール系フィルムが好ましく用いられる。偏光子は、ポリビニルアルコール水溶液を製膜し、これを一軸延伸させて染色するか、染色した後一軸延伸してから、好ましくはホウ素化合物で耐久性処理を行ったものが用いられている。延伸は、フィルム製膜方向に一軸延伸を行うか、又はフィルム製膜方向に対して斜め45°方向に延伸することが好ましい。
本発明の偏光板Cに使用される偏光子保護フィルムC3としては、セルロースエステル含有フィルムが好適に用いられ、たとえば、市販のセルロースエステルフィルム(たとえば、コニカミノルタタックKC8UX、KC5UX、KC4UX、KC8UCR3、KC4SR、KC4BR、KC4CR、KC4DR、KC4FR、KC4KR、KC8UY、KC6UY、KC4UY、KC4UE、KC8UE、KC4CT、KC2CT、KC8UY-HA、KC2UA、KC4UA、KC6UAKC、2UAH、KC4UAH、KC6UAH、以上、コニカミノルタ(株)製、フジタックT40UZ、フジタックT60UZ、フジタックT80UZ、フジタックTD80UL、フジタックTD60UL、フジタックTD40UL、フジタック4NRT、フジタック2NRT、フジタックR02、フジタックR06、以上、富士フイルム(株)製)、シクロオレフィンフィルム(例えば、日本ゼオン製のゼオノアフィルム、JSR製のアートンフィルム)が好ましく用いられる。偏光子保護フィルムC3の厚さは特に限定されず、10~200μm程度とすることができ、好ましくは10~100μmであり、より好ましくは10~70μmである。
本発明の偏光板は、画像表示装置に使用することで、視認性に優れた性能が発揮される点で好ましい。画像表示装置としては、反射型、透過型、半透過型液晶表示装置又は、TN型、STN型、OCB型、VA型、IPS型、ECB型等の各種駆動方式の液晶表示装置、タッチパネル表示装置、有機EL表示装置やプラズマディスプレイ等が挙げられる。これら画像表示装置の中でも液晶表示装置が、高い視認性に優れる点で好ましい。
<λ/4フィルムの作製>
〈微粒子分散液1〉
微粒子(アエロジル R972V 日本アエロジル(株)製)
11質量部
エタノール 89質量部
以上をディゾルバーで50分間攪拌混合した後、マントンゴーリンで分散を行った。
メチレンクロライドを入れた溶解タンクに十分攪拌しながら、微粒子分散液1をゆっくりと添加した。さらに、二次粒子の粒径が所定の大きさとなるようにアトライターにて分散を行った。これを日本精線(株)製のファインメットNFで濾過し、微粒子添加液1を調製した。
微粒子分散液1 5質量部
下記組成の主ドープ液を調製した。まず加圧溶解タンクにメチレンクロライドとエタノールを添加した。溶剤の入った加圧溶解タンクにセルロースエステルを攪拌しながら投入した。これを加熱し、攪拌しながら、完全に溶解させたものを安積濾紙(株)製の安積濾紙No.244を使用して濾過し、主ドープ液を調製した。
メチレンクロライド 340質量部
エタノール 64質量部
アセチル基平均置換度が2.0で、かつブチリル基平均置換度が0.2のセルロースアセテートブチレート 100質量部
ショ糖ベンゾエート(平均置換度7.5) 10.0質量部
サンプルNo.1 2.5質量部
紫外線吸収剤(チヌビン928(チバ・ジャパン(株)製))
2.3質量部
微粒子添加液1 1質量部
以上を密閉容器に投入し、攪拌しながら溶解してドープ液を調製した。次いで、無端ベルト流延装置を用い、ドープ液を温度33℃、2000mm幅でステンレスベルト支持体上に均一に流延した。ステンレスベルトの温度は30℃に制御した。
得られたλ/4フィルムの面上に、ハードコート層を設けることによりハードコート層付きλ/4フィルムを作製した。ハードコート層の作製は、まず緩衝層組成物を塗布して硬化させることによって緩衝層を形成し、次に、緩衝層上に硬化層組成物を塗布して硬化させることによって硬化層を形成した。緩衝層組成物及び硬化層組成物に含まれるポリマーシランカップリング剤被覆微粒子は、下記の方法で調整したものを用いた。以下に、緩衝層及び硬化層を形成する手順を説明する。
容器に、メタクリル酸メチル(共栄社化学(株)製:ライトエステルM)30ml、3-メルカプトプロピルトリメトキシシラン(信越化学(株)製:KBM-803)1ml
と、溶媒としてテトラヒドロフラン100ml、重合開始剤としてアゾイソブチロニトリル(関東化学(株)製:AIBN)50mgを添加し、N2ガスで置換した後、80℃で3時間加熱してポリマーシランカップリング剤を調製した。得られたポリマーシランカップリング剤の分子量は16,000であった。なお、分子量の測定は、ゲルパーミエーションクロマトグラフィー装置で測定した。
上記で作製したポリマーシランカップリング剤被覆シリカ(1)と、下記の化合物とを攪拌混合して、緩衝層組成物を調整した。
ポリマーシランカップリング剤被覆シリカ(1) 60質量部
(活性線硬化樹脂)
ウレタンアクリレート(U-4H、新中村化学工業(株)製)
35質量部
(光重合開始剤)
イルガキュア184(BASFジャパン(株)製) 5質量部
(添加剤)
KF-642(ポリエーテル変性シリコーンオイル、信越化学工業株式会社製) 2質量部
(溶剤)
プロピレングリコールモノメチルエーテル 80質量部
酢酸メチル 20質量部
[硬化層組成物の調製]
上記で作製したポリマーシランカップリング剤被覆シリカ(1)と、下記の化合物とを攪拌混合して、硬化層組成物を調整した。
ポリマーシランカップリング剤被覆シリカ(1) 60質量部
(活性線硬化樹脂)
NKエステルA-DCP(トリシクロデカンジメタノールジアクリレート、新中村化学工業社製) 35質量部
(光重合開始剤)
イルガキュア184(BASFジャパン(株)製) 5質量部
(添加剤)
KF-642(ポリエーテル変性シリコーンオイル、信越化学工業株式会社製) 2質量部
(溶剤)
プロピレングリコールモノメチルエーテル 80質量部
酢酸メチル 20質量部
上記で作製したλ/4フィルムのA面(流延ベルトに接していない面)上に、上記緩衝層組成物を、押し出しコーターを用いて塗布し、恒率乾燥区間温度50℃、減率乾燥区間温度50℃で乾燥の後、酸素濃度が1.0体積%以下の雰囲気になるように窒素パージしながら、紫外線ランプを用い照射部の照度が100mW/cm2で、照射量を0.25J/cm2として塗布層を硬化させ、ドライ膜厚4μmの緩衝層(下層)を形成した。
続いて、緩衝層上に、上記で作製した硬化層組成物を、マイクログラビアコーターを用いて塗布し、恒率乾燥区間温度50℃、減率乾燥区間温度50℃で乾燥の後、酸素濃度が1.0体積%以下の雰囲気になるように窒素パージしながら、紫外線ランプを用い照射部の照度が100mW/cm2で、照射量を0.3J/cm2として塗布層を硬化させ、ドライ膜厚2μmの硬化層(上層)を形成して、ロール状に巻き取り、ハードコート層付きのλ/4フィルムを作製した。
厚さ30μmのポリビニルアルコールフィルムを、一軸延伸(温度110℃、延伸倍率5倍)した。
実施例2~6及び比較例1~6においては、λ/4フィルムに含有されるセルロースエステルの、アセチル基平均置換度、プロピオニル基平均置換度及びブチリル基平均置換度を下記の表1に記すように変更したことが異なる他は実施例1と同様にして偏光板を作製した。なお、実施例6においては、λ/4フィルムを作製するときの延伸温度を170℃から190℃に変更した。なおまた比較例5においてはλ/4フィルムに含有されるセルロースエステルとして、セルロースジアセテート(DAC)を含むものを用い、比較例6においてはλ/4フィルムに含有されるセルロースエステルとして、セルロースアセテートプロピオネート(CAP)を含むものを用いた。
ハードコート層に緩衝層を設けなかったことが異なる他は実施例1と同様にして偏光板を作製した。
ハードコート層を設けなかったことが異なる他は実施例1と同様にして偏光板を作製した。

21.5インチの液晶表示装置(IPS226V-PN、LGエレクトロニクスジャパン(株)製)に液晶層を挟んで設置されている2対の偏光板のうち、観察者側の片面の偏光板を剥がし、各実施例及び各比較例で作製した偏光板を、偏光板のハードコート層側の面が視認側となるようにして、光学粘着剤で液晶セルガラスとを貼合して液晶表示装置を作製した。観察者側の偏光板の透過軸とバックライト側の偏光板の透過軸とが直交するように配置した。
上記で作製した液晶表示装置を90℃の高温乾燥環境に配置する前後の正面コントラストの変化率(ΔCR)を評価した。まず、23℃の温度で相対湿度55%RHの環境で、各々の液晶表示装置のバックライトを1週間連続点灯した後、測定を行った。測定にはELDIM社製EZ-Contrast160Dを用いて、液晶表示装置で白表示と黒表示の表示画面の法線方向からの輝度を測定し、その比を正面コントラストとした。
液晶表示装置の任意の5点の正面コントラストを測定し、その測定値のうちの最大値と最小値を除く3点の平均値を算出した。
上記の計算によって得られた結果を下記の表1の「ΔCR」の欄に示す。なお、表1において、コントラストの変化率(ΔCR)が小さいほど、偏光板の偏光度が低下しておらず、高温乾燥環境下に晒しても偏光板の光漏れが生じていないことを示している。コントラストの変化率(ΔCR)が30%を超えると製品として使用できるレベルではないと評価され、コントラストの変化率(ΔCR)が20%以上30%以下であれば良好と評価され、コントラストの変化率(ΔCR)が20%未満であればさらに良好と評価される。
Claims (6)
- 表面にハードコート層を有するλ/4フィルムを少なくとも一方の面に有する偏光板であって、
前記λ/4フィルムは、アセチル基平均置換度が1.8以上2.2以下であり、ブチリル基平均置換度が0.1以上0.4以下であるセルロースエステルを含有し、
90℃の温度環境下に1000時間保管し、23℃の温度で55%RHの相対湿度の環境下に24時間調湿した前後の前記偏光板の前記λ/4フィルムの遅相軸に平行な方向の寸法変化率が-0.5%以上-0.1%以下であることを特徴とする偏光板。 - 前記ハードコート層は、脂環式炭化水素を含む樹脂と、ポリマーシランカップリング剤で被覆されてなる微粒子とを含む硬化層を含むことを特徴とする請求項1に記載の偏光板。
- 前記ハードコート層は、前記硬化層と前記λ/4フィルムとの間に形成される緩衝層をさらに有し、
前記緩衝層は、前記硬化層に含まれる樹脂とは異なる樹脂と、ポリマーシランカップリング剤で被覆されてなる微粒子とを含むことを特徴とする請求項2に記載の偏光板。 - 前記緩衝層に含まれる樹脂は、ウレタンアクリレート樹脂であることを特徴とする請求項3に記載の偏光板。
- 請求項1~4のいずれか一項に記載の偏光板を含む画像表示装置。
- 請求項1~4のいずれか一項に記載の偏光板を含む画像表示装置であって、
前記偏光板のハードコート層側の面が視認側に配置されている、画像表示装置。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017556414A JPWO2017104308A1 (ja) | 2015-12-14 | 2016-11-09 | 偏光板及び前記偏光板を含む画像表示装置 |
| KR1020187013172A KR102035441B1 (ko) | 2015-12-14 | 2016-11-09 | 편광판 및 상기 편광판을 포함하는 화상 표시 장치 |
| CN201680072930.5A CN108369306A (zh) | 2015-12-14 | 2016-11-09 | 偏振片及含有上述偏振片的图像显示装置 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-243491 | 2015-12-14 | ||
| JP2015243491 | 2015-12-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017104308A1 true WO2017104308A1 (ja) | 2017-06-22 |
Family
ID=59056016
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/083291 WO2017104308A1 (ja) | 2015-12-14 | 2016-11-09 | 偏光板及び前記偏光板を含む画像表示装置 |
Country Status (4)
| Country | Link |
|---|---|
| JP (1) | JPWO2017104308A1 (ja) |
| KR (1) | KR102035441B1 (ja) |
| CN (1) | CN108369306A (ja) |
| WO (1) | WO2017104308A1 (ja) |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000347003A (ja) * | 1999-06-02 | 2000-12-15 | Dainippon Printing Co Ltd | 光学フィルム |
| JP2005307055A (ja) * | 2004-04-22 | 2005-11-04 | Fuji Photo Film Co Ltd | セルロースアシレートの製造方法、セルロースアシレートフイルム、それを用いた光学機能性シート、偏光板、液晶表示装置 |
| JP2008083307A (ja) * | 2006-09-27 | 2008-04-10 | Konica Minolta Opto Inc | 偏光板、偏光板の製造方法、及び液晶表示装置 |
| JP2010170128A (ja) * | 2008-12-26 | 2010-08-05 | Fujifilm Corp | セルロースアシレートフィルム、偏光板及び液晶表示装置 |
| JP2012234164A (ja) * | 2011-04-22 | 2012-11-29 | Nitto Denko Corp | 光学積層体 |
| JP2013064813A (ja) * | 2011-09-16 | 2013-04-11 | Konica Minolta Advanced Layers Inc | 偏光板保護フィルム、偏光板保護フィルムの製造方法、偏光板及び液晶表示装置 |
| WO2013065587A1 (ja) * | 2011-10-31 | 2013-05-10 | コニカミノルタアドバンストレイヤー株式会社 | 粘着層付き有機エレクトロルミネッセンス用円偏光板、それを具備する有機エレクトロルミネッセンス表示装置 |
| JP2015087710A (ja) * | 2013-11-01 | 2015-05-07 | コニカミノルタ株式会社 | 光学フィルム、偏光板及び画像表示装置 |
| JP2016177238A (ja) * | 2015-03-23 | 2016-10-06 | コニカミノルタ株式会社 | 光学フィルム、円偏光板、及びタッチパネル表示装置 |
| JP2016206540A (ja) * | 2015-04-27 | 2016-12-08 | コニカミノルタ株式会社 | 位相差フィルム、偏光板、垂直配向型液晶表示装置及び位相差フィルムの製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5521832A (en) | 1978-07-31 | 1980-02-16 | Matsushita Electronics Corp | Electron gun for color picture tube |
| JP4196686B2 (ja) * | 2002-10-31 | 2008-12-17 | コニカミノルタホールディングス株式会社 | 光学フィルムの製造方法 |
| JP4530144B2 (ja) | 2004-03-03 | 2010-08-25 | 富士フイルム株式会社 | セルロースアセテートフィルム、偏光板及び液晶表示装置 |
| JP4686351B2 (ja) | 2004-12-15 | 2011-05-25 | 富士フイルム株式会社 | セルロースアシレートフィルム、セルロースアシレートフィルムの製造方法、並びにそれを用いた偏光板及び液晶表示装置 |
| JP5040651B2 (ja) * | 2005-06-08 | 2012-10-03 | コニカミノルタアドバンストレイヤー株式会社 | セルロースエステルフィルム、偏光板及び液晶表示装置 |
| WO2009084295A1 (ja) * | 2007-12-28 | 2009-07-09 | Konica Minolta Opto, Inc. | アクリル樹脂含有フィルム、それを用いた偏光板及び表示装置 |
| JP5914440B2 (ja) | 2012-09-28 | 2016-05-11 | 富士フイルム株式会社 | ハードコートフィルム、ハードコートフィルムの製造方法、反射防止フィルム、偏光板、及び画像表示装置 |
| JPWO2015133160A1 (ja) * | 2014-03-03 | 2017-04-06 | コニカミノルタ株式会社 | 機能性フィルム、偏光板および表示装置 |
-
2016
- 2016-11-09 CN CN201680072930.5A patent/CN108369306A/zh active Pending
- 2016-11-09 KR KR1020187013172A patent/KR102035441B1/ko active IP Right Grant
- 2016-11-09 JP JP2017556414A patent/JPWO2017104308A1/ja active Pending
- 2016-11-09 WO PCT/JP2016/083291 patent/WO2017104308A1/ja active Application Filing
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000347003A (ja) * | 1999-06-02 | 2000-12-15 | Dainippon Printing Co Ltd | 光学フィルム |
| JP2005307055A (ja) * | 2004-04-22 | 2005-11-04 | Fuji Photo Film Co Ltd | セルロースアシレートの製造方法、セルロースアシレートフイルム、それを用いた光学機能性シート、偏光板、液晶表示装置 |
| JP2008083307A (ja) * | 2006-09-27 | 2008-04-10 | Konica Minolta Opto Inc | 偏光板、偏光板の製造方法、及び液晶表示装置 |
| JP2010170128A (ja) * | 2008-12-26 | 2010-08-05 | Fujifilm Corp | セルロースアシレートフィルム、偏光板及び液晶表示装置 |
| JP2012234164A (ja) * | 2011-04-22 | 2012-11-29 | Nitto Denko Corp | 光学積層体 |
| JP2013064813A (ja) * | 2011-09-16 | 2013-04-11 | Konica Minolta Advanced Layers Inc | 偏光板保護フィルム、偏光板保護フィルムの製造方法、偏光板及び液晶表示装置 |
| WO2013065587A1 (ja) * | 2011-10-31 | 2013-05-10 | コニカミノルタアドバンストレイヤー株式会社 | 粘着層付き有機エレクトロルミネッセンス用円偏光板、それを具備する有機エレクトロルミネッセンス表示装置 |
| JP2015087710A (ja) * | 2013-11-01 | 2015-05-07 | コニカミノルタ株式会社 | 光学フィルム、偏光板及び画像表示装置 |
| JP2016177238A (ja) * | 2015-03-23 | 2016-10-06 | コニカミノルタ株式会社 | 光学フィルム、円偏光板、及びタッチパネル表示装置 |
| JP2016206540A (ja) * | 2015-04-27 | 2016-12-08 | コニカミノルタ株式会社 | 位相差フィルム、偏光板、垂直配向型液晶表示装置及び位相差フィルムの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102035441B1 (ko) | 2019-10-22 |
| KR20180067607A (ko) | 2018-06-20 |
| JPWO2017104308A1 (ja) | 2018-09-27 |
| CN108369306A (zh) | 2018-08-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5038625B2 (ja) | 延伸セルロースエステルフィルム、ハードコートフィルム、反射防止フィルム及び光学補償フィルム、並びにそれらを用いた偏光板及び表示装置 | |
| TWI569967B (zh) | A polarizing plate and a liquid crystal display device using the same | |
| WO2007102327A1 (ja) | 偏光板及び液晶表示装置 | |
| TW200808514A (en) | Process for producing optical film with uneven structure, optical film, wire grid polarizer, and retardation film | |
| WO2014203637A1 (ja) | 偏光板及び液晶表示装置 | |
| WO2018070132A1 (ja) | 偏光板および液晶表示装置 | |
| JP7088279B2 (ja) | 偏光板、偏光板の製造方法及び液晶表示装置 | |
| JP5996163B2 (ja) | 光学フィルムの製造方法、偏光板及び画像表示装置 | |
| KR20160111453A (ko) | 기능성 필름, 편광판 및 표시 장치 | |
| KR101777532B1 (ko) | 광학 필름, 편광판 및 화상 표시 장치 | |
| JP2004277581A (ja) | セルロースエステルフィルム、偏光板、液晶表示装置、セルロースエステルフィルムの製造方法、偏光板の製造方法 | |
| JPWO2015019701A1 (ja) | セルロース誘導体及びその製造方法、光学フィルム、円偏光板及び有機エレクトロルミネッセンス表示装置 | |
| JP2015179204A (ja) | ハードコートフィルム、偏光板および画像表示装置 | |
| JP6164050B2 (ja) | 光学フィルム、偏光板及びそれらの製造方法、並びに画像表示装置 | |
| JP6710967B2 (ja) | 光学フィルム、光学フィルムの製造方法及び偏光板 | |
| WO2013133276A1 (ja) | 偏光板およびこれを用いた表示装置 | |
| JP5845702B2 (ja) | 位相差フィルムの製造方法 | |
| KR102157451B1 (ko) | 편광판 및 액정 표시 장치 | |
| WO2017104308A1 (ja) | 偏光板及び前記偏光板を含む画像表示装置 | |
| JP6676930B2 (ja) | 光学フィルム | |
| JP2017106995A (ja) | 偏光板及び画像表示装置 | |
| JP6728656B2 (ja) | 偏光板保護フィルム及びそれを具備した偏光板 | |
| JP4389517B2 (ja) | 光学フィルムの製造方法 | |
| JP2009075314A (ja) | 位相差フィルムの製造方法、位相差フィルム、それを用いた偏光板、及び液晶表示装置 | |
| JP2017167373A (ja) | 光学用積層フィルム、偏光板、円偏光板及び画像表示装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16875292 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017556414 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20187013172 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16875292 Country of ref document: EP Kind code of ref document: A1 |